Z 3920:2011
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 2
4 一般事項························································································································· 2
5 定量元素及び定量方法 ······································································································· 2
6 ヒュームの採取方法及び前処理方法 ····················································································· 2
6.1 ヒュームの採取方法 ······································································································· 2
6.2 分析試料の前処理方法 ···································································································· 5
7 鉄定量方法 ······················································································································ 6
7.1 定量方法の区分 ············································································································· 6
7.2 アスコルビン酸還元よう素酸カリウム逆滴定法 ···································································· 7
7.3 塩化物抽出分離スルホサリチル酸吸光光度法 ······································································· 8
7.4 フレーム原子吸光法 ······································································································ 10
7.5 ICP発光分光法 ············································································································ 11
8 マンガン定量方法 ············································································································ 13
8.1 定量方法の区分 ············································································································ 13
8.2 フレーム原子吸光法 ······································································································ 13
8.3 ICP発光分光法 ············································································································ 14
9 銅定量方法 ····················································································································· 16
9.1 定量方法の区分 ············································································································ 16
9.2 フレーム原子吸光法 ······································································································ 16
9.3 電気加熱原子吸光法 ······································································································ 17
9.4 ICP発光分光法 ············································································································ 20
10 ニッケル定量方法 ·········································································································· 21
10.1 定量方法の区分 ··········································································································· 21
10.2 フレーム原子吸光法 ····································································································· 21
10.3 ICP発光分光法 ··········································································································· 23
11 バナジウム定量方法 ······································································································· 24
11.1 定量方法の区分 ··········································································································· 24
11.2 フレーム原子吸光法 ····································································································· 24
11.3 ICP発光分光法 ··········································································································· 26
12 コバルト定量方法 ·········································································································· 27
12.1 定量方法の区分 ··········································································································· 27
12.2 フレーム原子吸光法 ····································································································· 27
Z 3920:2011 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
12.3 ICP発光分光法 ··········································································································· 29
13 鉛定量方法 ··················································································································· 30
13.1 定量方法の区分 ··········································································································· 30
13.2 フレーム原子吸光法 ····································································································· 30
13.3 ICP発光分光法 ··········································································································· 31
14 亜鉛定量方法 ················································································································ 33
14.1 定量方法の区分 ··········································································································· 33
14.2 フレーム原子吸光法 ····································································································· 33
14.3 電気加熱原子吸光法 ····································································································· 34
14.4 ICP発光分光法 ··········································································································· 37
15 アルミニウム定量方法 ···································································································· 38
15.1 定量方法の区分 ··········································································································· 38
15.2 フレーム原子吸光法 ····································································································· 38
15.3 ICP発光分光法 ··········································································································· 40
16 カドミウム定量方法 ······································································································· 41
16.1 定量方法の区分 ··········································································································· 41
16.2 フレーム原子吸光法 ····································································································· 42
16.3 電気加熱原子吸光法 ····································································································· 43
16.4 ICP発光分光法 ··········································································································· 45
17 銀定量方法 ··················································································································· 46
17.1 定量方法の区分 ··········································································································· 46
17.2 フレーム原子吸光法 ····································································································· 46
17.3 電気加熱原子吸光法 ····································································································· 48
17.4 ICP発光分光法 ··········································································································· 50
18 バリウム定量方法 ·········································································································· 51
18.1 定量方法 ···················································································································· 51
18.2 ICP発光分光法 ··········································································································· 51
19 全クロム定量方法 ·········································································································· 53
19.1 定量方法の区分 ··········································································································· 53
19.2 フレーム原子吸光法 ····································································································· 53
19.3 ICP発光分光法 ··········································································································· 54
20 クロム(VI)定量方法 ···································································································· 56
20.1 定量方法の区分 ··········································································································· 56
20.2 分析試料の前処理 ········································································································ 56
20.3 ジフェニルカルバジド吸光光度法 ··················································································· 57
20.4 イオンクロマトグラフ分離ジフェニルカルバジド吸光光度法 ··············································· 58
20.5 フレーム原子吸光法 ····································································································· 61
21 ふっ素定量方法 ············································································································· 63
21.1 定量方法 ···················································································································· 63
Z 3920:2011 目次
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
21.2 熱加水分解分離ランタンアリザリンコンプレキソン吸光光度法 ············································ 63
22 全けい素定量方法 ·········································································································· 66
22.1 定量方法 ···················································································································· 66
22.2 モリブドけい酸青吸光光度法 ························································································· 66
23 結晶質シリカ定量方法 ···································································································· 69
23.1 定量方法 ···················································································································· 69
23.2 X線回折法(基底標準吸収補正法) ················································································ 69
24 りん定量方法 ················································································································ 74
24.1 定量方法 ···················································································································· 74
24.2 モリブドバナドりん酸吸光光度法 ··················································································· 74
Z 3920:2011 目次
(4)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,社団法人日本溶接
協会(JWES)及び財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を改正すべき
との申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格である。
これによって,JIS Z 3920:1991は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
日本工業規格 JIS
Z 3920:2011
溶接ヒューム分析方法
Methods for chemical analysis of welding fumes
序文
この規格は,1978年に制定され,その後2回の改正を経て今日に至っている。前回の改正は1991年に
行われたが,その後の分析技術の進歩,溶接材料の変化に対応するために改正した。
主な改正点は,次のとおり。
a) 定量対象元素として規定できなかったクロム(VI),バリウム及びけい素(全けい素及び結晶質シリ
カ)を追加して規定。
b) 金属元素の定量としてICP発光分光法,アスコルビン酸還元よう素酸カリウム逆滴定法,フレーム原
子吸光法,ジフェニルカルバジド吸光光度法及びイオンクロマトグラフ分離ジフェニルカルバジド吸
光光度法,モリブドけい酸青吸光光度法,X線回折法など分析方法の全般的な見直し。
1
適用範囲
この規格は,溶接時に発生する溶接ヒューム,溶接作業環境中に浮遊する粉じん及び溶接作業者の呼吸
域の粉じん(以下,これらをヒュームという。)に含まれる鉄,マンガン,銅,ニッケル,バナジウム,コ
バルト,鉛,亜鉛,アルミニウム,カドミウム,銀,バリウム,クロム,クロム(VI),ふっ素,けい素,
結晶質シリカ及びりんの定量方法について規定する。
なお,この規格において溶接とは,アーク溶接及びろう付をいう。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS K 0127 イオンクロマトグラフ分析通則
JIS K 0901 気体中のダスト試料捕集用ろ過材の形状,寸法並びに性能試験方法
JIS K 0970 プッシュボタン式液体用微量体積計
JIS K 8005 容量分析用標準物質
JIS K 9901 高純度試薬−硝酸
JIS Z 3001-1 溶接用語−第1部:一般
JIS Z 3001-2 溶接用語−第2部:溶接方法
2
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS Z 3001-3 溶接用語−第3部:ろう接
JIS Z 3001-4 溶接用語−第4部:融接不完全部
JIS Z 3930 アーク溶接のヒューム発生量測定方法
JIS Z 3950 溶接作業環境における浮遊粉じん濃度測定方法
3
用語及び定義
この規格で用いる主な用語及び定義は,JIS Z 3001規格群によるほか,次による。
3.1
分析試料
分析を行うために採取したヒュームの全部又は一部。
3.2
主溶液
分析試料に各種の酸などを加え,加熱分解・抽出などを行った後,液量を一定とした溶液。
3.3
試料溶液
主溶液を分取し,試薬添加,抽出,洗浄,液性調整などの化学処理を行い,分析機器に導入できる状態
に調製した溶液。
3.4
空試験
分析対象成分の含有量ゼロのものを用いて(試料なしで始めることが多い。),全分析操作を忠実に行い,
ゼロであるべき値がどのように出るかを試す試験。通常は,空試験値を実測値から差し引いて真の値とす
る。普通,ブランクテストという。
4
一般事項
この規格において共通な一般事項は,次による。
a) 定量方法に共通な一般事項は,JIS K 0050,JIS K 0115,JIS K 0116,JIS K 0121及びJIS K 0127によ
る。
b) この規格で用いる水は,特に規定がある場合を除き,JIS K 0050の7.1(水及び試薬)に規定するA1
又はA2の水とする。
5
定量元素及び定量方法
定量元素及び定量方法は,表1による。
6
ヒュームの採取方法及び前処理方法
6.1
ヒュームの採取方法
6.1.1
ろ過材
ヒュームの採取に使用するろ過材は,次による。
a) ろ過材は,JIS K 0901の5.2(捕集率試験)に規定する性能試験方法による捕集率が,粒径0.3 μmの
エアロゾルに対して95 %以上であって,かつ,初期圧力損失が低く,ヒューム捕集に伴う圧力損失の
増加が少なく,吸湿性が低いものとする。
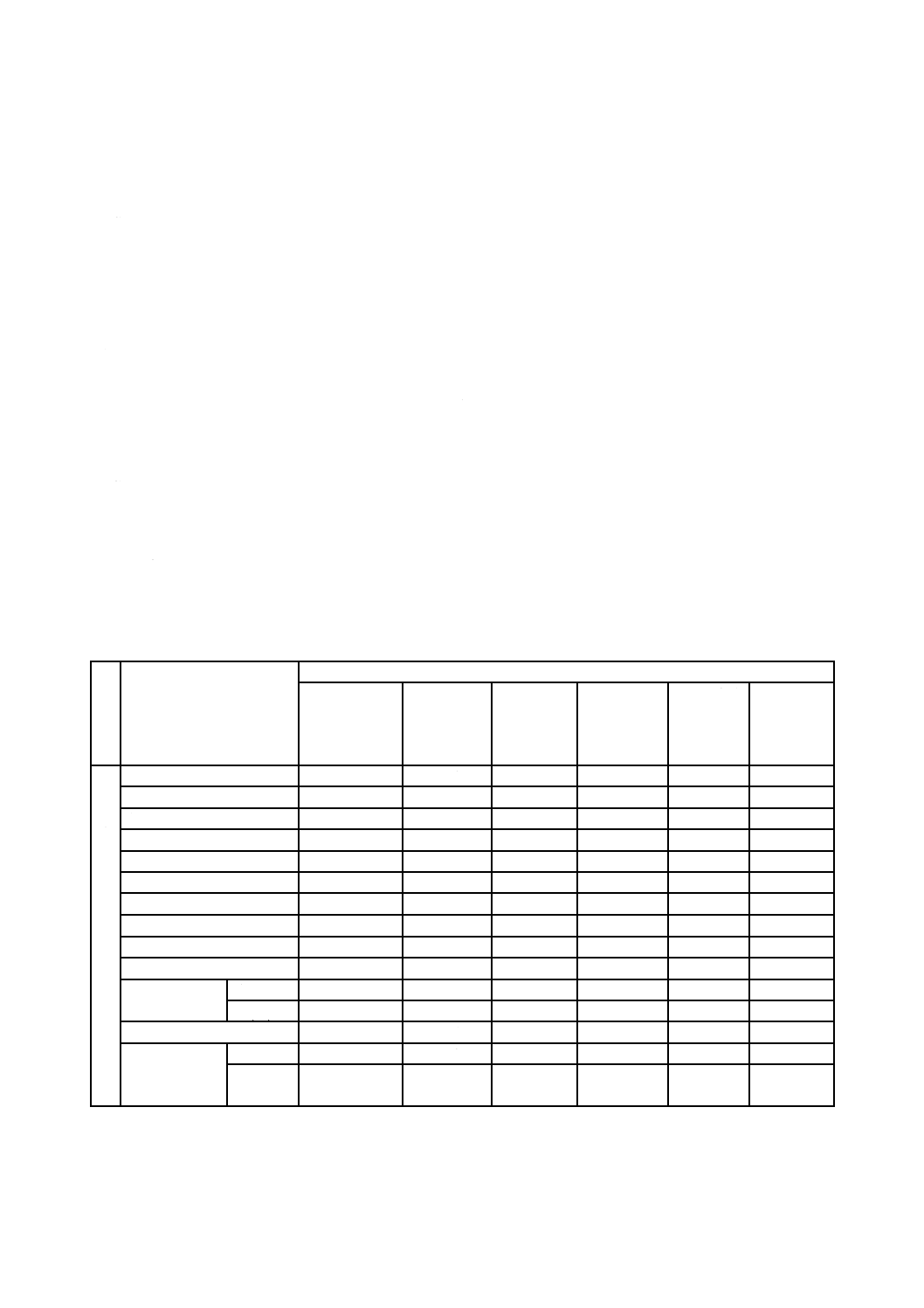
3
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) ヒューム中のクロム(VI),全けい素及び結晶質シリカ以外の元素を定量する場合には,ガラス繊維
製又は石英繊維製のろ過材を使用する。
c) ヒューム中のクロム(VI)を定量する場合に用いるろ過材は,次による。
1) ヒュームの採取を6.1.3 a)又はb)によって行う場合には,ポリ塩化ビニール(PVC)メンブランフィ
ルタ,ポリフルオロエチレン(PFE)メンブランフィルタなどを用いる。
2) ヒュームの採取を6.1.3 c)によって行う場合には,石英繊維製ろ過材を用いる。
3) セルローズを用いているろ過材,ガラス繊維製ろ過材など,クロム(VI)と反応するおそれのある
材質のろ過材は,用いてはならない。
d) ヒューム中の全けい素を定量する場合に使用するろ過材は,次による。
1) ヒュームの採取を6.1.3 a)又はb)によって行う場合には,ポリ塩化ビニール(PVC)メンブランフィ
ルタ又はポリフルオロエチレン(PFE)メンブランフィルタを用いる。
2) ヒュームの採取を6.1.3 c)によって行う場合には,ポリテトラフルオロエチレン(PTFE)ろ紙など
のろ過材を用いる。
3) ガラス繊維製ろ過材,石英繊維製ろ過材など,けい素を含む材質のろ過材は,用いてはならない。
e) ヒューム中の結晶質シリカを定量する場合に使用するろ過材は,次による。
1) ヒュームの採取を6.1.3 a)又はb)によって行う場合には,ふっ素樹脂処理ガラス繊維フィルタを用い
る。
2) ヒュームの採取を6.1.3 c)によって行う場合には,ガラス繊維製又は石英繊維製のろ過材を用いる。
表1−定量元素及び定量方法
溶
接
方
法
定量元素
定量方法
アスコルビン
酸還元よう素
酸カリウム逆
滴定法
吸光光度法
フレーム原
子吸光法
電気加熱原
子吸光法
ICP発光分
光法
X線回折法
ア
ー
ク
溶
接
鉄
○
○a)
○
○
マンガン
○
○
銅
○
○
ニッケル
○
○
バナジウム
○
○
コバルト
○
○
鉛
○
○
亜鉛
○
○
アルミニウム
○
○
バリウム
○
クロム
全Cr
○
○
Cr(VI)
○b) c)
○
ふっ素
○d)
けい素
全けい素
○e)
結晶質シ
リカ
○
4
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
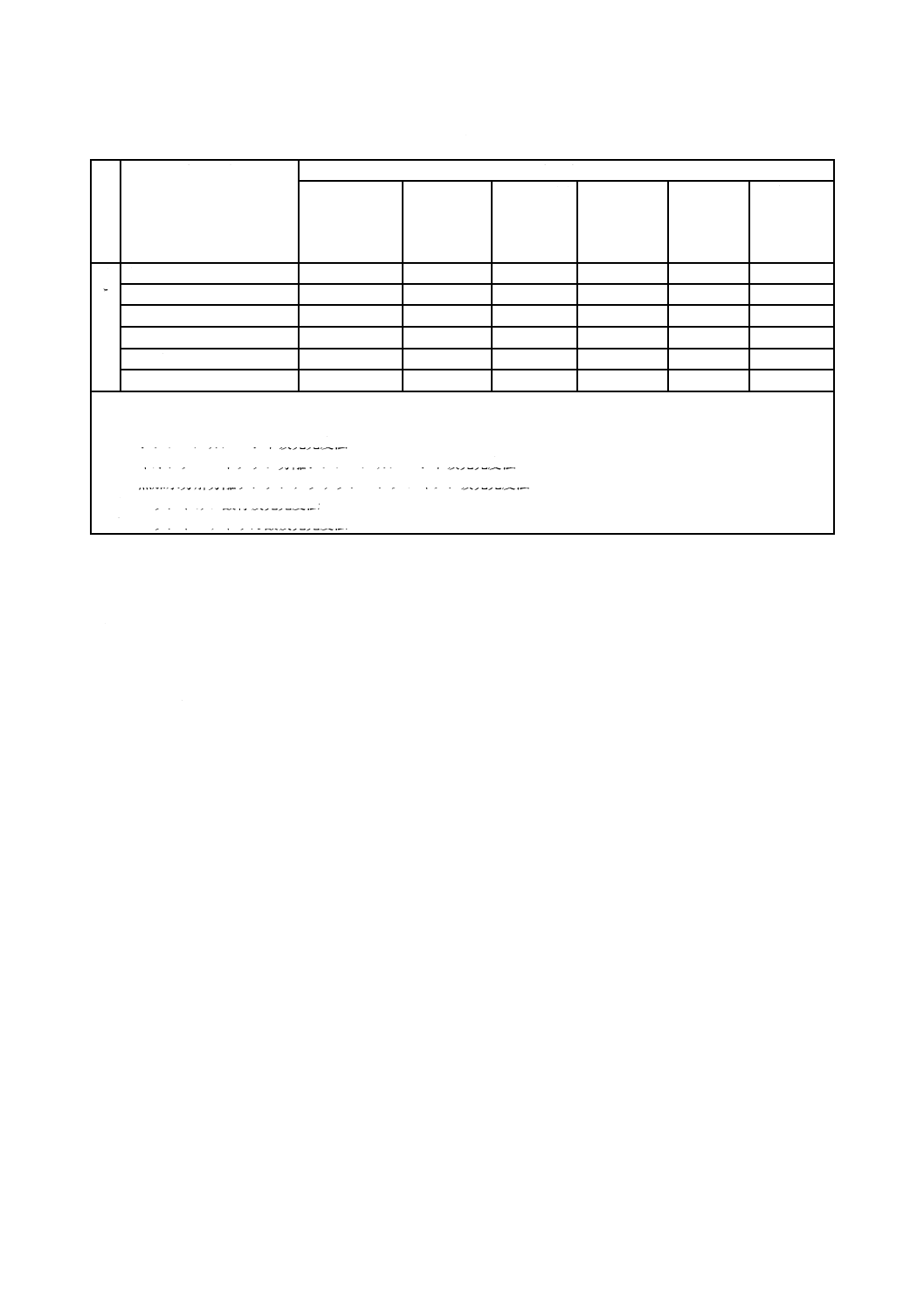
表1−定量元素及び定量方法(続き)
溶
接
方
法
定量元素
定量方法
アスコルビン
酸還元よう素
酸カリウム逆
滴定法
吸光光度法
フレーム原
子吸光法
電気加熱原
子吸光法
ICP発光分
光法
X線回折法
ろ
う
付
銅
○
○
○
亜鉛
○
○
○
カドミウム
○
○
○
銀
○
○
○
ふっ素
○d)
りん
○f)
注記 表中の○は,各元素の定量に適用する定量方法を示している。
注a) 塩化物抽出分離スルホサリチル酸吸光光度法
b) ジフェニルカルバジド吸光光度法
c) イオンクロマトグラフ分離ジフェニルカルバジド吸光光度法
d) 熱加水分解分離ランタンアリザリンコンプレキソン吸光光度法
e) モリブドけい酸青吸光光度法
f) モリブドバナドりん酸吸光光度法
6.1.2
ヒュームの採取量
ヒュームの採取量は,次による。
a) ヒュームの採取を6.1.3 a)又はb)によって行う場合 ヒュームの採取量は,約10 mgとする。ただし,
ろう付の場合のヒューム採取量は,約1 mgとする。
b) ヒュームの採取を6.1.3 c)によって行う場合 ヒュームの採取量は,約100 mgとする。ただし,ろう
付の場合のヒューム採取量は,約10 mgとし,また,7.1 a)の方法によってヒューム中の鉄を定量する
場合のヒューム採取量は,ヒューム中の鉄の含有率に応じて,表2に規定する量の分析試料をはかり
取るのに十分な量とする。
6.1.3
ヒュームの採取
ヒュームの採取は,次のいずれかの手順によって行う。
a) 溶接作業環境中のヒュームの採取
1) JIS Z 3950の5.1.1(分粒装置付きろ過捕集による測定方法)又はJIS Z 3950の5.2(総粉じんの質
量濃度測定方法)に規定するロウボリウムエアサンプラを使用する方法によって,ヒュームを採取
する。
2) ヒュームを採取したろ過材の質量からヒューム採取に使用する前のろ過材の質量を差し引いた質量
を,0.1 mgの桁まで求め,ヒューム採取量とする。
3) 採取したヒュームは,ろ過材を含めて分析試料とする。
b) 個人暴露のヒュームの採取
1) JIS Z 3950の6.3.1(分粒装置付きろ過捕集による測定方法)又はJIS Z 3950の6.4(総粉じんの個
人ばく露質量濃度測定方法)に規定するろ過材を使用する方法によって,ヒュームを採取する。
2) ヒュームを採取したろ過材の質量からヒューム採取に使用する前のろ過材の質量を差し引いた質量
を,0.1 mgの桁まで求め,ヒューム採取量とする。
3) 採取したヒュームは,ろ過材を含めて分析試料とする。
5
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) ヒューム発生量測定時のヒュームの採取 ヒューム発生量測定時のヒュームの採取は,次のいずれか
による。
1) アーク溶接ヒュームの場合
1.1) JIS Z 3930の4.3(ヒューム捕集装置)に規定するハイボリウムエアサンプラを使用する方法によ
って,ヒュームを採取する。
1.2) ヒュームを採取したろ過材の質量からヒューム採取に使用する前のろ過材の質量を差し引いた質
量を,0.1 mgの桁まで求め,ヒューム採取量とする。
1.3) ろ過材に採取したヒュームの一部(10 mg以上)を,はけなどを用いてろ過材及びはけの一部が混
入しないように注意しながら掃き落とし,デシケータ中で数時間放置した後,分析試料とする。
2) ろう付ヒュームの場合
2.1) JIS Z 3930の4.3(ヒューム捕集装置)に規定するハイボリウムエアサンプラを使用する方法によ
って,ヒュームを採取する。
2.2) ヒュームを採取したろ過材の質量からヒューム採取に使用する前のろ過材の質量を差し引いた質
量を,0.1 mgの桁まで求め,ヒューム採取量とする。
2.3) ろ過材に捕集したヒュームは,ろ過材を含めて分析試料とする。
6.2
分析試料の前処理方法
6.2.1
適用
ここに規定する前処理方法は,表1に規定する各元素を定量するときの分析試料の前処理に適用する。
ただし,鉄[7.1 a)の方法によって定量する場合],クロム(VI),ふっ素,全けい素,結晶質シリカ及びり
んを定量するときの分析試料の前処理方法は,それぞれの定量方法に規定し,ここに規定する前処理方法
は,適用しない。
6.2.2
試薬
6.2.2.1
塩酸(1+1)
6.2.2.2
硝酸(1+1,1+2)
6.2.2.3
過塩素酸
6.2.2.4
ふっ化水素酸
6.2.2.5
炭酸ナトリウム(無水)
6.2.3
主溶液及び空試験溶液の調製
6.2.3.1
主溶液の調製
主溶液の調製は,次のいずれかの手順によって行う。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合
1) アーク溶接ヒュームの場合
1.1) 6.1.3 a) 3)又はb) 3)で得た分析試料を,白金るつぼ(例えば,50番)又は白金皿(例えば,50番)
に移し入れる。硝酸(1+1)6 mL,過塩素酸5 mL及びふっ化水素酸10 mLを加え,穏やかに加
熱してヒューム及びろ過材を分解する。
1.2) 更に加熱を続けて過塩素酸の白煙を発生させ,液量が2〜3 mLになるまで蒸発させる。
1.3) 室温まで放冷した後,塩酸(1+1)10 mLを加えて塩類を溶解し,常温まで冷却する。
なお,不溶解物が認められる場合は,不溶解物をろ紙(5種C)を用いてこし分け,温水で不溶
解物及びろ紙を洗浄し,ろ液及び洗液を合わせて保存する(以下,これをA液という。)。不溶解
物をろ紙とともに白金るつぼ(例えば,20番)に入れ,穏やかに加熱してろ紙を乾燥した後,強
6
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
熱してろ紙を灰化する。炭酸ナトリウム(無水)1.0 gを加え,強熱して融解した後,室温まで放
冷する。少量の水を加え,加熱して融成物を溶解する。注意しながら塩酸(1+1)を滴加して溶
液を微酸性とし,常温まで冷却した後,溶液を保存しておいたA液に少量の水を用いて合わせる。
1.4) 溶液を100 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄めて主溶液とする。
2) ろう付ヒュームの場合
2.1) 6.1.3 a) 3),b) 3)又はc) 2) 2.3)で得た分析試料をビーカー(200 mL)に入れる。硝酸(1+2)40 mL
を加え,時計皿で覆い,穏やかに加熱してヒュームを分解する。
なお,分析試料は,必要ならば,セラミック製はさみを用いてろ過材を切断して小片とした後,
ビーカー(200 mL)に入れる。
2.2) 室温まで冷却した後,時計皿の下面及びビーカーの内壁を水で洗って時計皿を取り除き,溶液を
ろ紙(5種A)を用いてろ過し,ろ過材及びろ紙を水で十分に洗浄し,ろ液と洗液とを合わせる。
2.3) 常温まで冷却した後,溶液を100 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄めて
主溶液とする。
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 6.1.3 c) 1) 1.3)で得た分析試料約10 mgを正確に0.1
mgの桁まではかり取り,白金るつぼ(例えば,50番)又は白金皿(例えば,50番)に移し入れる。
硝酸(1+1)6 mL,過塩素酸5 mL及びふっ化水素酸10 mLを加え,穏やかに加熱して分解する。以
下,a) 1)の1.2)〜1.4)の手順に従って操作する。
6.2.3.2
空試験溶液の調製
空試験溶液の調製は,次のいずれかの手順によって行う。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合 6.1.3 a),b)又はc) 2)でヒュームの採取に
用いたろ過材と同じろ過材を分析試料の代わりに用いて,6.2.3.1 a) 1)又は2)の手順に従って,分析試
料と同じ操作を分析試料と並行して行い,得た溶液を空試験溶液とする。
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 分析試料を用いないで,6.2.3.1 b)の手順に従って,
分析試料と同じ操作を分析試料と並行して行い,得た溶液を空試験溶液とする。
7
鉄定量方法
7.1
定量方法の区分
鉄の定量方法は,次のいずれかによる。
なお,アスコルビン酸還元よう素酸カリウム逆滴定法は,6.1.3 c) 1)によって採取した分析試料にだけ適
用する。
a) アスコルビン酸還元よう素酸カリウム逆滴定法 この方法は,試料溶液中の6 mg以上100 mg以下の
鉄の定量に適用する。
なお,この方法は,6.1.3 c) 1)によって採取した分析試料にだけ適用する。
b) 塩化物抽出分離スルフォサリチル酸吸光光度法 この方法は,試料溶液中の濃度が1 μg/mL以上10
μg/mL以下の鉄の定量に適用する。
c) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.3 μg/mL以上6 μg/mL以下の鉄の定量に適
用する。
d) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上5 μg/mL以下の鉄の定量に適用す
る。
7
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.2
アスコルビン酸還元よう素酸カリウム逆滴定法
7.2.1
要旨
分析試料を塩酸と塩化すず(II)とで分解し,過酸化水素を加えて鉄を鉄(III)に酸化した後,煮沸し
て残留する過酸化水素を分解する。一定量のアスコルビン酸標準液を加えて鉄(III)を鉄(II)に還元し
た後,過剰量のアスコルビン酸をよう素酸カリウム標準液で滴定する。
7.2.2
試薬
7.2.2.1
塩酸(1+1)
7.2.2.2
過酸化水素
7.2.2.3
ふっ化ナトリウム
7.2.2.4
塩化すず(II)溶液
金属すず[99.9 %以上(質量分率)]130 gを塩酸に溶解し,塩酸で液量を1 000 mLとした後,褐色瓶に
入れて保存する。
7.2.2.5
アスコルビン酸標準液
L(+)−アスコルビン酸9 gを水に溶解し,エチレンジアミン四酢酸二水素二ナトリウム二水和物約
100 mg及びぎ酸4 mLを加えた後,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,水で標線まで
薄める。この溶液のファクターを,使用の都度,次の操作によって求める。
アスコルビン酸標準液10.0 mLをビーカー(300 mL)に取り,水で液量を約150 mLとし,塩酸(1+1)
10 mL及び指示薬としてでん粉溶液(7.2.2.7)2 mLを加えた後,よう素酸カリウム標準液(7.2.2.6)で滴
定し,溶液が僅かに青を呈した点を終点としてアスコルビン酸標準液の使用量を求め,次の式によって,
よう素酸カリウム標準液のファクターを求める。
10
V
F=
ここに,
F: アスコルビン酸標準液のファクター
V: よう素酸カリウム標準液の使用量(mL)
7.2.2.6
よう素酸カリウム標準液
あらかじめ120〜140 ℃で90〜120分間乾燥してデシケータ中で常温まで放冷したよう素酸カリウム
3.567 g及びよう化カリウム10 gを水酸化ナトリウム溶液(5 g/L)200 mLに溶解し,溶液を1 000 mLの全
量フラスコに水を用いて移し入れ,水で標線まで薄める。この溶液1 mLは,鉄0.005 585 gに相当する。
7.2.2.7
でん粉溶液
でん粉(溶性)1 gを水約10 mLとかき混ぜた後,100 mLの熱水中にかき混ぜながら加える。溶液を約
1分間煮沸した後,静置して室温まで冷却し,その上澄液を用いる。この溶液は,使用の都度調製する。
7.2.3
操作
7.2.3.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.1.3 c) 1) 1.3)で得た分析試料を,その鉄含有率に応じて,表2に規定する量をはかり取り,ビーカー
(300 mL)に移し入れ,塩酸(1+1)15 mL及び塩化すず(II)溶液(7.2.2.4)5 mLを加え,時計皿
で覆い,沸騰しない程度に加熱し,時々穏やかに振り混ぜながら分解する。
なお,溶液中に不溶解残さがあるとき又は溶液が懸濁しているときは,適量のふっ化ナトリウム(100
mg以下)を加える。
b) 時計皿の下面及びビーカーの内壁を少量の水で洗い,温水を加えて液量を約80 mLとする。過酸化水

8
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
素1 mLを加え,再び加熱し,数分間煮沸して残留している過酸化水素を完全に分解する。時計皿の
下面及びビーカーの内壁を少量の熱水で洗って時計皿を取り除く。
表2−分析試料のはかり取り量及びアスコルビン酸標準液の添加量
分析試料中の鉄含有率
%(質量分率)
分析試料のはかり取り量
mg
アスコルビン酸標準液の添加量
mL
2以上10未満
300
10.0
10以上30未満
200
15.0
30以上60未満
100
15.0
60以上
100
20.0
7.2.3.2
滴定
7.2.3.1 b)で得た試料溶液(熱溶液)に,表2に規定する量のアスコルビン酸標準液(7.2.2.5)を加え,
水で液量を200 mLとした後,約10分間流水中に浸し,室温まで冷却する。次に,でん粉溶液(7.2.2.7)2
mLを指示薬として加え,よう素酸カリウム標準液(7.2.2.6)で滴定し,溶液が僅かに青を呈した点を終点
としてアスコルビン酸標準液の使用量を求める。
7.2.4
空試験
空試験は,行わない。
7.2.5
計算
ヒューム中の鉄含有率を,次の式によって算出する。
100
585
005
.0
)
(
2
1
×
×
−
×
=
m
V
F
V
Fe
ここに,
Fe: 6.1.3 c) 1)で採取したヒューム中の鉄含有率[%(質量分
率)]
V1: 7.2.3.2で添加したアスコルビン酸標準液の量(mL)
F: アスコルビン酸標準液のファクター
V2: 7.2.3.2で得たよう素酸カリウム標準液の使用量(mL)
m: 7.2.3.1 a)ではかり取った分析試料の量(g)
7.3
塩化物抽出分離スルホサリチル酸吸光光度法
7.3.1
要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加え,生成する鉄(III)の塩化物錯体
を酢酸3-メチルブチル・4-メチル-2-ペンタノンで抽出し,水で逆抽出した後,スルホサリチル酸を加えて
スルホサリチル酸鉄錯体を生成させ,光度計を用いて,その吸光度を測定する。
7.3.2
試薬
7.3.2.1
塩酸
7.3.2.2
アンモニア水(1+1)
7.3.2.3
塩化アンモニウム溶液(100 g/L)
7.3.2.4
スルホサリチル酸溶液
5-スルホサリチル酸二水和物50 gを水に溶解し,水で液量を1 000 mLとする。
7.3.2.5
混合溶媒
酢酸3-メチルブチルと4-メチル-2-ペンタノンとを等量ずつ混合する。
7.3.2.6
鉄標準液(100 μg/mL)
鉄[99.9 %(質量分率)以上]を正確に1.00 gはかり取り,ビーカー(200 mL)に移し入れ,塩酸(1
9
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
+1)50 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカー
の内壁を水で洗って時計皿を取り除き,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,水で標線
まで薄めて原液(Fe:1 000 μg/mL)とする。この原液10.0 mLを,使用の都度,100 mLの全量フラスコに
取り,塩酸(1+1)5 mLを加えた後,水で標線まで薄めて鉄標準液とする。
7.3.3
操作
7.3.3.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,鉄量が0.1〜1.0 mgとなるように分取し,分液漏斗(200 mL)
に移し入れる。
b) 塩酸25 mLを加えた後,水を加えて液量を50 mLとする。室温まで放冷した後,混合溶媒(7.3.2.5)
50 mLを加え,3分間激しく振り混ぜる。静置して2相に分離した後,下層の水相を捨てる。分液漏
斗の内壁に付いている水相は,分液漏斗を振り動かして分離し,完全に除去する。
c) 有機相に水30 mLを加え,3分間激しく振り混ぜる。静置して2相に分離した後,下層の水相をビー
カー(100 mL)に移し入れる。分液漏斗に少量の水を加え,分液漏斗の脚部などに残っている水相を,
先の水相が入っているビーカーに洗い移す。有機相は捨てる。
d) 溶液を加熱して水相に混入した混合溶媒を蒸発させ,溶液面に混合溶媒が認められなくなるまで除去
する。常温まで冷却した後,溶液を100 mLの全量フラスコに水を用いて移し入れ,水で液量を約50 mL
とする。
e) 塩化アンモニウム溶液10 mL及びスルホサリチル酸溶液(7.3.2.4)5 mLを加えた後,アンモニア水(1
+1)を溶液の色が赤紫から黄になるまで滴加し,更に過剰に0.2〜0.4 mL加える。常温まで冷却した
後,水で標線まで薄める。
7.3.3.2
吸光度の測定
7.3.3.1 e)で得た溶液の一部を光度計の吸収セル(10 mm)に取り,水を対照液として,波長420 nm付近
の吸光度を測定する。
7.3.4
空試験
6.2.3.2 a)又はb)で得た空試験溶液を,7.3.3.1 a)で分取した主溶液と同量分取し,分液漏斗(200 mL)に
移し入れる。以下,7.3.3.1 b)〜7.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
7.3.5
検量線の作成
鉄標準液(7.3.2.6)0〜10.0 mL(鉄として0〜1.0 mg)を段階的に数個の分液漏斗(200 mL)に取る。以
下,7.3.3.1 b)〜7.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た吸光度と鉄量と
の関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
7.3.6
計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 7.3.3.2及び7.3.4で得た吸光度と7.3.5で作成し
た検量線とから鉄量を求め,次の式によって,ヒューム中の鉄含有率を算出する。
100
100
)
(
1
1
2
1
×
×
−
=
B
m
A
A
Fe
ここに,
Fe: ヒューム中の鉄含有率[%(質量分率)]
A1: 試料溶液中の鉄検出量(mg)
10
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A2: 空試験での鉄検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B1: 7.3.3.1 a)及び7.3.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 7.3.3.2及び7.3.4で得た吸光度と7.3.5で作成した検
量線とから鉄量を求め,次の式によって,ヒューム中の鉄含有率を算出する。
100
100
)
(
1
2
2
1
×
×
−
=
B
m
A
A
Fe
ここに,
Fe: ヒューム中の鉄含有率[%(質量分率)]
A1: 試料溶液中の鉄検出量(mg)
A2: 空試験での鉄検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B1: 7.3.3.1 a)及び7.3.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
7.4
フレーム原子吸光法
7.4.1
要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加えた後,溶液を原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
7.4.2
試薬
7.4.2.1
塩酸(1+1)
7.4.2.2
鉄標準液(Fe:100 μg/mL)
鉄[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)10
mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカーの内壁
を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1+1)40
mLを加えた後,水で標線まで薄める。
7.4.2.3
鉄標準液(Fe:10 μg/mL)
鉄標準液(7.4.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mLを加え
た後,水で標線まで薄める。
7.4.3
操作
7.4.3.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,鉄量が30〜600 μgになるように分取し,100 mLの全量フラ
スコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
7.4.3.2
吸光度の測定
7.4.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長248.3
nmにおける吸光度を測定する。
7.4.4
空試験
6.2.3.2 a)又はb)で得た空試験溶液を,7.4.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコ
に移し入れる。以下,7.4.3.1 b)及び7.4.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
11
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.4.5
検量線の作成
鉄標準液(7.4.2.2)及び/又は鉄標準液(7.4.2.3)の各種液量(鉄として0〜600 μg)を段階的に数個の
100 mLの全量フラスコに取る。以下,7.4.3.1 b)及び7.4.3.2の手順に従って,主溶液と同じ操作を主溶液
と並行して行い,得た吸光度と鉄量との関係線を作成し,その関係線を原点を通るように平行移動して検
量線とする。
7.4.6
計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 7.4.3.2及び7.4.4で得た吸光度と7.4.5で作成し
た検量線とから鉄量を求め,次の式によって,ヒューム中の鉄含有率を算出する。
100
100
)
(
2
1
2
1
×
×
−
=
B
m
A
A
Fe
ここに,
Fe: ヒューム中の鉄含有率[%(質量分率)]
A1: 試料溶液中の鉄検出量(mg)
A2: 空試験での鉄検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B2: 7.4.3.1 a)及び7.4.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 7.4.3.2及び7.4.4で得た吸光度と7.4.5で作成した検
量線とから鉄量を求め,次の式によって,ヒューム中の鉄含有率を算出する。
100
100
)
(
2
2
2
1
×
×
−
=
B
m
A
A
Fe
ここに,
Fe: ヒューム中の鉄含有率[%(質量分率)]
A1: 試料溶液中の鉄検出量(mg)
A2: 空試験での鉄検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B2: 7.4.3.1 a)及び7.4.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
7.5
ICP発光分光法
7.5.1
要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
7.5.2
試薬
7.5.2.1
鉄標準液(Fe:100 μg/mL)
7.4.2.2による。
7.5.2.2
鉄標準液(Fe:10 μg/mL)
7.4.2.3による。
7.5.3
操作
7.5.3.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,鉄量が10〜500 μgになるように分取し,100 mLの全量フラ
12
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
スコに移し入れる。
b) 水を加えて標線まで薄める。
7.5.3.2
発光強度の測定
7.5.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長238.204 nmにおける
発光強度を測定する。
なお,238.204 nmにおける鉄の発光強度が共存元素の影響を受ける場合は,影響を受けない他の測定波
長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又はバック
グラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
7.5.4
空試験
6.2.3.2 a)又はb)で得た空試験溶液を,7.5.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコ
に移し入れる。以下,7.5.3.1 b)及び7.5.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
7.5.5
検量線の作成
鉄標準液(7.5.2.1)及び/又は鉄標準液(7.5.2.2)の各種液量(鉄として0〜500 μg)を段階的に数個の
100 mLの全量フラスコに取る。以下,7.5.3.1 b)及び7.5.3.2の手順に従って,主溶液と同じ操作を主溶液
と並行して行い,得た発光強度と鉄量との関係線を作成し,その関係線を原点を通るように平行移動して
検量線とする。
7.5.6
計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 7.5.3.2及び7.5.4で得た発光強度と7.5.5で作成
した検量線とから鉄量を求め,次の式によって,ヒューム中の鉄含有率を算出する。
100
100
)
(
3
1
2
1
×
×
−
=
B
m
A
A
Fe
ここに,
Fe: ヒューム中の鉄含有率[%(質量分率)]
A1: 試料溶液中の鉄検出量(mg)
A2: 空試験での鉄検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B3: 7.5.3.1 a)及び7.5.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 7.5.3.2及び7.5.4で得た発光強度と7.5.5で作成した
検量線とから鉄量を求め,次の式によって,ヒューム中の鉄含有率を算出する。
100
100
)
(
3
2
2
1
×
×
−
=
B
m
A
A
Fe
ここに,
Fe: ヒューム中の鉄含有率[%(質量分率)]
A1: 試料溶液中の鉄検出量(mg)
A2: 空試験での鉄検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B3: 7.5.3.1 a)及び7.5.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
13
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8
マンガン定量方法
8.1
定量方法の区分
マンガンの定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上50 μg/mL以下のマンガンの定
量に適用する。
b) ICP発光分光法 この方法は,試料溶液中の濃度が0.05 μg/mL以上50 μg/mL以下のマンガンの定量
に適用する。
8.2
フレーム原子吸光法
8.2.1
要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加えた後,溶液を原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
8.2.2
試薬
8.2.2.1
塩酸(1+1)
8.2.2.2
マンガン標準液(Mn:100 μg/mL)
マンガン[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1
+1)10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカー
の内壁を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1
+1)40 mLを加えた後,水で標線まで薄める。
8.2.2.3
マンガン標準液(Mn:10 μg/mL)
マンガン標準液(8.2.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
8.2.3
操作
8.2.3.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,マンガン量が10〜500 μgになるように分取し,100 mLの全
量フラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
8.2.3.2
吸光度の測定
8.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長279.5
nmにおける吸光度を測定する。
8.2.4
空試験
6.2.3.2 a)又はb)で得た空試験溶液を,8.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコ
に移し入れる。以下,8.2.3.1 b)及び8.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
8.2.5
検量線の作成
マンガン標準液(8.2.2.2)及び/又はマンガン標準液(8.2.2.3)の各種液量(マンガンとして0〜500 μg)
を段階的に数個の100 mLの全量フラスコに取る。以下,8.2.3.1 b)及び8.2.3.2の手順に従って,主溶液と
同じ操作を主溶液と並行して行い,得た吸光度とマンガン量との関係線を作成し,その関係線を原点を通
るように平行移動して検量線とする。
8.2.6
計算
計算は,次のいずれかによる。
14
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 8.2.3.2及び8.2.4で得た吸光度と8.2.5で作成し
た検量線とからマンガン量を求め,次の式によって,ヒューム中のマンガン含有率を算出する。
100
100
)
(
4
1
4
3
×
×
−
=
B
m
A
A
Mn
ここに,
Mn: ヒューム中のマンガン含有率[%(質量分率)]
A3: 試料溶液中のマンガン検出量(mg)
A4: 空試験でのマンガン検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B4: 8.2.3.1 a)及び8.2.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 8.2.3.2及び8.2.4で得た吸光度と8.2.5で作成した検
量線とからマンガン量を求め,次の式によって,ヒューム中のマンガン含有率を算出する。
100
100
)
(
4
2
4
3
×
×
−
=
B
m
A
A
Mn
ここに,
Mn: ヒューム中のマンガン含有率[%(質量分率)]
A3: 試料溶液中のマンガン検出量(mg)
A4: 空試験でのマンガン検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B4: 8.2.3.1 a)及び8.2.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
8.3
ICP発光分光法
8.3.1
要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
8.3.2
試薬
8.3.2.1
マンガン標準液(Mn:200 μg/mL)
マンガン[99.9 %(質量分率)以上]0.200 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1
+1)10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカー
の内壁を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1
+1)40 mLを加えた後,水で標線まで薄める。
8.3.2.2
マンガン標準液(Mn:10 μg/mL)
マンガン標準液(8.3.2.1)5.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
8.3.2.3
マンガン標準液(Mn:1.0 μg/mL)
マンガン標準液(8.3.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
8.3.3
操作
8.3.3.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,マンガン量が5〜5 000 μgになるように分取し,100 mLの全
15
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
量フラスコに移し入れる。
b) 水を加えて標線まで薄める。
8.3.3.2
発光強度の測定
8.3.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長257.610 nmにおける
発光強度を測定する。
なお,257.610 nmにおけるマンガンの発光強度が共存元素の影響を受ける場合は,影響を受けない他の
測定波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又は
バックグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
8.3.4
空試験
6.2.3.2 a)又はb)で得た空試験溶液を,8.3.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコ
に移し入れる。以下,8.3.3.1 b)及び8.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
8.3.5
検量線の作成
マンガン標準液(8.3.2.1),マンガン標準液(8.3.2.2)及び/又はマンガン標準液(8.3.2.3)の各種液量
(マンガンとして0〜5 000 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,8.3.3.1 b)及び8.3.3.2
の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た発光強度とマンガン量との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
8.3.6
計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 8.3.3.2及び8.3.4で得た発光強度と8.3.5で作成
した検量線とからマンガン量を求め,次の式によって,ヒューム中のマンガン含有率を算出する。
100
100
)
(
5
1
4
3
×
×
−
=
B
m
A
A
Mn
ここに,
Mn: ヒューム中のマンガン含有率[%(質量分率)]
A3: 試料溶液中のマンガン検出量(mg)
A4: 空試験でのマンガン検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B5: 8.3.3.1 a)及び8.3.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 8.3.3.2及び8.3.4で得た発光強度と8.3.5で作成した
検量線とからマンガン量を求め,次の式によって,ヒューム中のマンガン含有率を算出する。
100
100
)
(
5
2
4
3
×
×
−
=
B
m
A
A
Mn
ここに,
Mn: ヒューム中のマンガン含有率[%(質量分率)]
A3: 試料溶液中のマンガン検出量(mg)
A4: 空試験でのマンガン検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B5: 8.3.3.1 a)及び8.3.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
16
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9
銅定量方法
9.1
定量方法の区分
銅の定量方法は,次のいずれかによる。
なお,電気加熱原子吸光法は,6.2.3.1 a) 2)によって調製した主溶液にだけ適用する。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上5 μg/mL以下の銅の定量に適
用する。
b) 電気加熱原子吸光法 この方法は,試料溶液中の濃度が0.005 μg/mL以上0.1 μg/mL以下の銅の定量に
適用する。ただし,試料溶液中に共存する元素,塩類などが銅の吸光度に影響を及ぼす場合には,こ
の方法で規定する検量線法は,適用してはならない。
c) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上50 μg/mL以下の銅の定量に適用す
る。
9.2
フレーム原子吸光法
9.2.1
要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸,又は硝酸で分解する。塩酸を加えた後,溶液を原子吸光
光度計の空気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
9.2.2
試薬
9.2.2.1
塩酸(1+1)
9.2.2.2
銅標準液(Cu:100 μg/mL)
銅[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)10
mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカーの内壁
を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1+1)40
mLを加えた後,水で標線まで薄める。
9.2.2.3
銅標準液(Cu:10 μg/mL)
銅標準液(9.2.2.2)10.0 mLを使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mLを加えた
後,水で標線まで薄める。
9.2.3
操作
9.2.3.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)若しくは2) 2.3)又は6.2.3.1 b)で得た主溶液を,銅量が10〜500 μgになるように分取し,
100 mLの全量フラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
なお,主溶液の調製を6.2.3.1 a) 2)によって行った場合には,塩酸(1+1)10 mLを加えずに,水で
標線まで薄める。
9.2.3.2
吸光度の測定
9.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長324.8
nmにおける吸光度を測定する。
9.2.4
空試験
6.2.3.2 a)又はb)で得た空試験溶液を,9.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコ
に移し入れる。以下,9.2.3.1 b)及び9.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
17
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.5
検量線の作成
銅標準液(9.2.2.2)及び/又は銅標準液(9.2.2.3)の各種液量(銅として0〜500 μg)を段階的に数個の
100 mLの全量フラスコに取る。以下,9.2.3.1 b)及び9.2.3.2の手順に従って,主溶液と同じ操作を主溶液
と並行して行い,得た吸光度と銅量との関係線を作成し,その関係線を原点を通るように平行移動して検
量線とする。
9.2.6
計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合 9.2.3.2及び9.2.4で得た吸光度と9.2.5で
作成した検量線とから銅量を求め,次の式によって,ヒューム中の銅含有率を算出する。
100
100
)
(
6
3
6
5
×
×
−
=
B
m
A
A
Cu
ここに,
Cu: ヒューム中の銅含有率[%(質量分率)]
A5: 試料溶液中の銅検出量(mg)
A6: 空試験での銅検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B6: 9.2.3.1 a)及び9.2.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 9.2.3.2及び9.2.4で得た吸光度と9.2.5で作成した検
量線とから銅量を求め,次の式によって,ヒューム中の銅含有率を算出する。
100
100
)
(
6
2
6
5
×
×
−
=
B
m
A
A
Cu
ここに,
Cu: ヒューム中の銅含有率[%(質量分率)]
A5: 試料溶液中の銅検出量(mg)
A6: 空試験での銅検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B6: 9.2.3.1 a)及び9.2.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
9.3
電気加熱原子吸光法
9.3.1
要旨
分析試料を硝酸で分解する。硝酸を加えた後,溶液を電気加熱原子吸光光度計の電気加熱炉に注入し,
その吸光度を測定する。
9.3.2
水
試薬の調製及び定量操作に用いる水は,JIS K 0050の7.1(水及び試薬)に規定するA3の水とする。
9.3.3
試薬
9.3.3.1
硝酸(1+1)
硝酸(JIS K 9901)を用いて調製する。
9.3.3.2
銅標準液(Cu:1.0 μg/mL)
銅[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)[硝
酸(JIS K 9901)を用いて調製する。]10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却し
18
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
た後,時計皿の下面及びビーカーの内壁を水(9.3.2)で洗って時計皿を取り除く。溶液を1 000 mLの全量
フラスコに水を用いて移し入れ,硝酸(1+1)[硝酸(JIS K 9901)を用いて調製する。]40 mLを加えた
後,水で標線まで薄め,原液(Cu:100 μg/mL)とする。この原液1.0 mLを,使用の都度,100 mLの全量
フラスコに取り,硝酸(1+1)[硝酸(JIS K 9901)を用いて調製する。]2 mLを加えた後,水(9.3.2)で
標線まで薄める。
9.3.3.3
銅標準液(Cu:0.1 μg/mL)
銅標準液(9.3.3.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)[硝酸(JIS K
9901)を用いて調製する。]2 mLを加えた後,水(9.3.2)で標線まで薄める。
9.3.4
器具
9.3.4.1
マイクロピぺット
JIS K 0970に規定するプッシュボタン式液体用微量体積計,又は自動注入装置を用いる。
9.3.5
操作
9.3.5.1
試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 検量線法によって定量する場合
1) 6.2.3.1 a) 2) 2.3)で得た主溶液を,銅量が0.5〜10 μgになるように分取し,100 mLの全量フラスコに
移し入れる。
2) 硝酸(1+1)(9.3.3.1)10 mLを加えた後,水(9.3.2)で標線まで薄める。
b) 標準添加法によって定量する場合
1) 4個以上の100 mLの全量フラスコを用意し,そのそれぞれに,6.2.3.1 a) 2) 2.3)で得た主溶液を,同
量ずつ分取する。
なお,主溶液の分取量は,分取した主溶液中の銅量が0.5 μg未満とならない量とする。
2) 1個の全量フラスコを除いた他の全量フラスコに,銅標準液(9.3.3.2)及び/又は銅標準液(9.3.3.3)
を,フラスコの溶液中の銅量が0.5〜10 μgとなるように段階的に加える。全ての全量フラスコに硝
酸(1+1)(9.3.3.1)10 mLを加えた後,水(9.3.2)で標線まで薄める。
9.3.5.2
吸光度の測定
吸光度の測定は,次のいずれかの手順によって行う。
a) 検量線法によって定量する場合
1) 9.3.5.1 a) 2)で得た溶液の一部(例えば,10〜50 μL)を,マイクロピペット(9.3.4.1)を用いて電気
加熱原子吸光光度計の電気加熱炉に注入する。
2) 乾燥(100〜120 ℃で30〜40秒間),灰化(600〜1 000 ℃で30〜40秒間)及び原子化(2 000〜2 700 ℃
で4〜6秒間)を行い,波長324.8 nmにおける吸光度を測定する。
なお,吸光度測定時には,バックグラウンド補正を行う。また,乾燥,灰化及び原子化の条件は,
装置,試料溶液の注入量,試料溶液中の塩類濃度などによって異なるので,あらかじめ最適な条件
を求めておく。
b) 標準添加法によって定量する場合
1) 9.3.5.1 b) 2)で得た溶液の一部(例えば,10〜50 μL)を,マイクロピペット(9.3.4.1)を用いて電気
加熱原子吸光光度計の電気加熱炉に注入する。
2) a) 2)の操作を行う。
19
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.3.6
空試験
空試験は,次のいずれかによる。
a) 検量線法によって定量する場合 6.2.3.2 a)で得た空試験溶液を,9.3.5.1 a) 1)で分取した主溶液と同量
分取し,100 mLの全量フラスコに移し入れる。以下,9.3.5.1 a) 2)及び9.3.5.2 a)の手順に従って,主溶
液と同じ操作を主溶液と並行して行う。
b) 標準添加法によって定量する場合 4個以上の100 mLの全量フラスコを用意し,そのそれぞれに,
6.2.3.2 a)で得た空試験溶液を,9.3.5.1 b) 1)で分取した主溶液と同量ずつ分取する。以下,9.3.5.1 b) 2)
及び9.3.5.2 b)の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
9.3.7
検量線の作成
検量線の作成は,次のいずれかによる。
a) 検量線法によって定量する場合 銅標準液(9.3.3.2)及び/又は銅標準液(9.3.3.3)の各種液量(銅と
して0〜10 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,9.3.5.1 a) 2)及び9.3.5.2 a)の
手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た吸光度と銅量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
b) 標準添加法によって定量する場合
1) 試料溶液用検量線 9.3.5.2 b) 2)で得た吸光度と9.3.5.1 b) 2)で銅標準液として添加した銅量との関
係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
2) 空試験用検量線 9.3.6 b)で得た吸光度と9.3.6 b)で銅標準液として添加した銅量との関係線を作成
し,その関係線を原点を通るように平行移動して検量線とする。
9.3.8
計算
計算は,次のいずれかによる。
a) 検量線法によって定量する場合 9.3.5.2 a) 2)及び9.3.6 a)で得た吸光度と9.3.7 a)で作成した検量線と
から銅量を求め,次の式によって,ヒューム中の銅含有率を算出する。
100
100
)
(
7
3
6
5
×
×
−
=
B
m
A
A
Cu
ここに,
Cu: ヒューム中の銅含有率[%(質量分率)]
A5: 試料溶液中の銅検出量(mg)
A6: 空試験での銅検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B7: 9.3.5.1 a) 1)及び9.3.6 a)でそれぞれ分取した主溶液
及び空試験溶液の量(mL)
b) 標準添加法によって定量する場合 9.3.5.2 b) 2)で得た銅標準液を添加しなかった試料溶液の吸光度と
9.3.7 b) 1)で作成した検量線とから,及び9.3.6 b)で得た銅標準液を添加しなかった空試験の吸光度と
9.3.7 b) 2)で作成した検量線とから,それぞれ銅量を求め,次の式によって,ヒューム中の銅含有率を
算出する。
100
100
)
(
8
3
6
5
×
×
−
=
B
m
A
A
Cu
ここに,
Cu: ヒューム中の銅含有率[%(質量分率)]
20
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A5: 試料溶液中の銅検出量(mg)
A6: 空試験での銅検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B8: 9.3.5.1 b) 1)及び9.3.6 b)でそれぞれ分取した主溶液
及び空試験溶液の量(mL)
9.4
ICP発光分光法
9.4.1
要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸,又は硝酸で分解する。溶液を発光分光装置のアルゴンプ
ラズマ中に噴霧し,その発光強度を測定する。
9.4.2
試薬
9.4.2.1
銅標準液(Cu:200 μg/mL)
銅[99.9 %(質量分率)以上]0.200 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)10
mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカーの内壁
を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1+1)40
mLを加えた後,水で標線まで薄める。
9.4.2.2
銅標準液(Cu:10 μg/mL)
銅標準液(9.4.2.1)5.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mLを加え
た後,水で標線まで薄める。
9.4.3
操作
9.4.3.1
試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)若しくは2) 2.3)又は6.2.3.1 b)で得た主溶液を,銅量が10〜5 000 μgになるように分取
し,100 mLの全量フラスコに移し入れる。
b) 水を加えて標線まで薄める。
9.4.3.2
発光強度の測定
9.4.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長324.754 nmにおける
発光強度を測定する。
なお,324.754 nmにおける銅の発光強度が共存元素の影響を受ける場合は,影響を受けない他の測定波
長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又はバック
グラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
9.4.4
空試験
6.2.3.2 a)又はb)で得た空試験溶液を,9.4.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコ
に移し入れる。以下,9.4.3.1 b)及び9.4.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
9.4.5
検量線の作成
銅標準液(9.4.2.1)及び/又は銅標準液(9.4.2.2)の各種液量(銅として0〜5 000 μg)を段階的に数個
の100 mLの全量フラスコに取る。以下,9.4.3.1 b)及び9.4.3.2の手順に従って,主溶液と同じ操作を主溶
液と並行して行い,得た発光強度と銅量との関係線を作成し,その関係線を原点を通るように平行移動し
て検量線とする。
9.4.6
計算
計算は,次のいずれかによる。
21
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合 9.4.3.2及び9.4.4で得た発光強度と9.4.5
で作成した検量線とから銅量を求め,次の式によって,ヒューム中の銅含有率を算出する。
100
100
)
(
9
3
6
5
×
×
−
=
B
m
A
A
Cu
ここに,
Cu: ヒューム中の銅含有率[%(質量分率)]
A5: 試料溶液中の銅検出量(mg)
A6: 空試験での銅検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B9: 9.4.3.1 a)及び9.4.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 9.4.3.2及び9.4.4で得た発光強度と9.4.5で作成した
検量線とから銅量を求め,次の式によって,ヒューム中の銅含有率を算出する。
100
100
)
(
9
2
6
5
×
×
−
=
B
m
A
A
Cu
ここに,
Cu: ヒューム中の銅含有率[%(質量分率)]
A5: 試料溶液中の銅検出量(mg)
A6: 空試験での銅検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B9: 9.4.3.1 a)及び9.4.4でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
10 ニッケル定量方法
10.1 定量方法の区分
ニッケルの定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.3 μg/mL以上10 μg/mL以下のニッケルの定
量に適用する。
b) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上50 μg/mL以下のニッケルの定量に
適用する。
10.2 フレーム原子吸光法
10.2.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加えた後,溶液を原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
10.2.2 試薬
10.2.2.1 塩酸(1+1)
10.2.2.2 ニッケル標準液(Ni:100 μg/mL)
ニッケル[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1
+1)10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカー
の内壁を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1
+1)40 mLを加えた後,水で標線まで薄める。
22
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.2.2.3 ニッケル標準液(Ni:10 μg/mL)
ニッケル標準液(10.2.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
10.2.3 操作
10.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,ニッケル量が30〜1 000 μgになるように分取し,100 mLの全
量フラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
10.2.3.2 吸光度の測定
10.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長232.0
nm又は305.1 nmにおける吸光度を測定する。
10.2.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,10.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,10.2.3.1 b)及び10.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
10.2.5 検量線の作成
ニッケル標準液(10.2.2.2)及び/又はニッケル標準液(10.2.2.3)の各種液量(ニッケルとして0〜1 000
μg)を段階的に数個の100 mLの全量フラスコに取る。以下,10.2.3.1 b)及び10.2.3.2の手順に従って,主
溶液と同じ操作を主溶液と並行して行い,得た吸光度とニッケル量との関係線を作成し,その関係線を原
点を通るように平行移動して検量線とする。
10.2.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 10.2.3.2及び10.2.4で得た吸光度と10.2.5で作
成した検量線とからニッケル量を求め,次の式によって,ヒューム中のニッケル含有率を算出する。
100
100
)
(
10
1
8
7
×
×
−
=
B
m
A
A
Ni
ここに,
Ni: ヒューム中のニッケル含有率[%(質量分率)]
A7: 試料溶液中のニッケル検出量(mg)
A8: 空試験でのニッケル検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B10: 10.2.3.1 a)及び10.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 10.2.3.2及び10.2.4で得た吸光度と10.2.5で作成し
た検量線とからニッケル量を求め,次の式によって,ヒューム中のニッケル含有率を算出する。
100
100
)
(
10
2
8
7
×
×
−
=
B
m
A
A
Ni
ここに,
Ni: ヒューム中のニッケル含有率[%(質量分率)]
A7: 試料溶液中のニッケル検出量(mg)
23
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A8: 空試験でのニッケル検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B10: 10.2.3.1 a)及び10.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
10.3 ICP発光分光法
10.3.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
10.3.2 試薬
10.3.2.1 ニッケル標準液(Ni:200 μg/mL)
ニッケル[99.9 %(質量分率)以上]0.200 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1
+1)10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカー
の内壁を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1
+1)40 mLを加えた後,水で標線まで薄める。
10.3.2.2 ニッケル標準液(Ni:10 μg/mL)
ニッケル標準液(10.3.2.1)5.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
10.3.3 操作
10.3.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,ニッケル量が10〜5 000 μgになるように分取し,100 mLの全
量フラスコに移し入れる。
b) 水を加えて標線まで薄める。
10.3.3.2 発光強度の測定
10.3.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長221.647 nmにおけ
る発光強度を測定する。
なお,221.647 nmにおけるニッケルの発光強度が共存元素の影響を受ける場合は,影響を受けない他の
測定波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又は
バックグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
10.3.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,10.3.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,10.3.3.1 b)及び10.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
10.3.5 検量線の作成
ニッケル標準液(10.3.2.1)及び/又はニッケル標準液(10.3.2.2)の各種液量(ニッケルとして0〜5 000
μg)を段階的に数個の100 mLの全量フラスコに取る。以下,10.3.3.1 b)及び10.3.3.2の手順に従って,主
溶液と同じ操作を主溶液と並行して行い,得た発光強度とニッケル量との関係線を作成し,その関係線を
原点を通るように平行移動して検量線とする。
10.3.6 計算
計算は,次のいずれかによる。
24
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 10.3.3.2及び10.3.4で得た発光強度と10.3.5で
作成した検量線とからニッケル量を求め,次の式によって,ヒューム中のニッケル含有率を算出する。
100
100
)
(
11
1
8
7
×
×
−
=
B
m
A
A
Ni
ここに,
Ni: ヒューム中のニッケル含有率[%(質量分率)]
A7: 試料溶液中のニッケル検出量(mg)
A8: 空試験でのニッケル検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B11: 10.3.3.1 a)及び10.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 10.3.3.2及び10.3.4で得た発光強度と10.3.5で作成
した検量線とからニッケル量を求め,次の式によって,ヒューム中のニッケル含有率を算出する。
100
100
)
(
11
2
8
7
×
×
−
=
B
m
A
A
Ni
ここに,
Ni: ヒューム中のニッケル含有率[%(質量分率)]
A7: 試料溶液中のニッケル検出量(mg)
A8: 空試験でのニッケル検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B11: 10.3.3.1 a)及び10.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
11 バナジウム定量方法
11.1 定量方法の区分
バナジウムの定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が2 μg/mL以上20 μg/mL以下のバナジウムの定
量に適用する。
b) ICP発光分光法 この方法は,試料溶液中の濃度が0.2 μg/mL以上20 μg/mL以下のバナジウムの定量
に適用する。
11.2 フレーム原子吸光法
11.2.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加えた後,溶液を原子吸光光度計の一
酸化二窒素・アセチレンフレーム中に噴霧し,その吸光度を測定する。
11.2.2 試薬
11.2.2.1 塩酸(1+1)
11.2.2.2 バナジウム標準液(V:100 μg/mL)
バナジウム[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,混酸(硫
酸3,硝酸1)10 mLを加え,時計皿で覆い,加熱して分解し,引き続き加熱して,ほとんど乾固するまで
混酸を蒸発させた後,硝酸(1+1)50 mLを加えて塩類を溶解する。常温まで冷却した後,時計皿の下面
及びビーカーの内壁を水で洗って時計皿を取り除き,溶液を1 000 mLの全量フラスコに水を用いて移し入
れ,水で標線まで薄める。
25
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.2.3 操作
11.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,バナジウム量が200〜2 000 μgになるように分取し,100 mL
の全量フラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
11.2.3.2 吸光度の測定
11.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した原子吸光光度計の一酸化二窒素・アセチレンフレ
ーム中に噴霧し,波長318.4 nmにおける吸光度を測定する。
11.2.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,11.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,11.2.3.1 b)及び11.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
11.2.5 検量線の作成
バナジウム標準液(11.2.2.2)0〜20.0 mL(バナジウムとして0〜2 000 μg)を段階的に数個の100 mLの
全量フラスコに取る。以下,11.2.3.1 b)及び11.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行し
て行い,得た吸光度とバナジウム量との関係線を作成し,その関係線を原点を通るように平行移動して検
量線とする。
11.2.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 11.2.3.2及び11.2.4で得た吸光度と11.2.5で作
成した検量線とからバナジウム量を求め,次の式によって,ヒューム中のバナジウム含有率を算出す
る。
100
100
)
(
12
1
10
9
×
×
−
=
B
m
A
A
V
ここに,
V: ヒューム中のバナジウム含有率[%(質量分率)]
A9: 試料溶液中のバナジウム検出量(mg)
A10: 空試験でのバナジウム検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B12: 11.2.3.1 a)及び11.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 11.2.3.2及び11.2.4で得た吸光度と11.2.5で作成し
た検量線とからバナジウム量を求め,次の式によって,ヒューム中のバナジウム含有率を算出する。
100
100
)
(
12
2
10
9
×
×
−
=
B
m
A
A
V
ここに,
V: ヒューム中のバナジウム含有率[%(質量分率)]
A9: 試料溶液中のバナジウム検出量(mg)
A10: 空試験でのバナジウム検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
26
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B12: 11.2.3.1 a)及び11.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
11.3 ICP発光分光法
11.3.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
11.3.2 試薬
11.3.2.1 バナジウム標準液(V:100 μg/mL)
11.2.2.2による。
11.3.2.2 バナジウム標準液(V:10 μg/mL)
バナジウム標準液(11.3.2.1)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
11.3.3 操作
11.3.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,バナジウム量が20〜2 000 μgになるように分取し,100 mLの
全量フラスコに移し入れる。
b) 水を加えて標線まで薄める。
11.3.3.2 発光強度の測定
11.3.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長309.311 nmにおけ
る発光強度を測定する。
なお,309.311 nmにおけるバナジウムの発光強度が共存元素の影響を受ける場合は,影響を受けない他
の測定波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又
はバックグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
11.3.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,11.3.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,11.3.3.1 b)及び11.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
11.3.5 検量線の作成
バナジウム標準液(11.3.2.1)及び/又はバナジウム標準液(11.3.2.2)の各種液量(バナジウムとして0
〜2 000 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,11.3.3.1 b)及び11.3.3.2の手順に従っ
て,主溶液と同じ操作を主溶液と並行して行い,得た発光強度とバナジウム量との関係線を作成し,その
関係線を原点を通るように平行移動して検量線とする。
11.3.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 11.3.3.2及び11.3.4で得た発光強度と11.3.5で
作成した検量線とからバナジウム量を求め,次の式によって,ヒューム中のバナジウム含有率を算出
する。
27
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
100
)
(
13
1
10
9
×
×
−
=
B
m
A
A
V
ここに,
V: ヒューム中のバナジウム含有率[%(質量分率)]
A9: 試料溶液中のバナジウム検出量(mg)
A10: 空試験でのバナジウム検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B13: 11.3.3.1 a)及び11.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 11.3.3.2及び11.3.4で得た発光強度と11.3.5で作成
した検量線とからバナジウム量を求め,次の式によって,ヒューム中のバナジウム含有率を算出する。
100
100
)
(
13
2
10
9
×
×
−
=
B
m
A
A
V
ここに,
V: ヒューム中のバナジウム含有率[%(質量分率)]
A9: 試料溶液中のバナジウム検出量(mg)
A10: 空試験でのバナジウム検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B13: 11.3.3.1 a)及び11.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
12 コバルト定量方法
12.1 定量方法の区分
コバルトの定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.5 μg/mL以上10 μg/mL以下のコバルトの定
量に適用する。
b) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上50 μg/mL以下のコバルトの定量に
適用する。
12.2 フレーム原子吸光法
12.2.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加えた後,溶液を原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
12.2.2 試薬
12.2.2.1 塩酸(1+1)
12.2.2.2 コバルト標準液(Co:100 μg/mL)
コバルト[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1
+1)10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカー
の内壁を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1
+1)40 mLを加えた後,水で標線まで薄める。
12.2.2.3 コバルト標準液(Co:10 μg/mL)
コバルト標準液(12.2.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
28
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.2.3 操作
12.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,コバルト量が50〜1 000 μgになるように分取し,100 mLの全
量フラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
12.2.3.2 吸光度の測定
12.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長240.7
nmにおける吸光度を測定する。
12.2.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,12.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,12.2.3.1 b)及び12.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
12.2.5 検量線の作成
コバルト標準液(12.2.2.2)及び/又はコバルト標準液(12.2.2.3)の各種液量(コバルトとして0〜1 000
μg)を段階的に数個の100 mLの全量フラスコに取る。以下,12.2.3.1 b)及び12.2.3.2の手順に従って,主
溶液と同じ操作を主溶液と並行して行い,得た吸光度とコバルト量との関係線を作成し,その関係線を原
点を通るように平行移動して検量線とする。
12.2.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 12.2.3.2及び12.2.4で得た吸光度と12.2.5で作
成した検量線とからコバルト量を求め,次の式によって,ヒューム中のコバルト含有率を算出する。
100
100
)
(
14
1
12
11
×
×
−
=
B
m
A
A
Co
ここに,
Co: ヒューム中のコバルト含有率[%(質量分率)]
A11: 試料溶液中のコバルト検出量(mg)
A12: 空試験でのコバルト検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B14: 12.2.3.1 a)及び12.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 12.2.3.2及び12.2.4で得た吸光度と12.2.5で作成し
た検量線とからコバルト量を求め,次の式によって,ヒューム中のコバルト含有率を算出する。
100
100
)
(
14
2
12
11
×
×
−
=
B
m
A
A
Co
ここに,
Co: ヒューム中のコバルト含有率[%(質量分率)]
A11: 試料溶液中のコバルト検出量(mg)
A12: 空試験でのコバルト検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B14: 12.2.3.1 a)及び12.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
29
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.3 ICP発光分光法
12.3.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
12.3.2 試薬
12.3.2.1 コバルト標準液(Co:200 μg/mL)
コバルト[99.9 %(質量分率)以上]0.200 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1
+1)10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカー
の内壁を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1
+1)40 mLを加えた後,水で標線まで薄める。
12.3.2.2 コバルト標準液(Co:10 μg/mL)
コバルト標準液(12.3.2.1)5.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
12.3.3 操作
12.3.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,コバルト量が10〜5 000 μgになるように分取し,100 mLの全
量フラスコに移し入れる。
b) 水を加えて標線まで薄める。
12.3.3.2 発光強度の測定
10.3.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長228.616 nmにおけ
る発光強度を測定する。
なお,228.616 nmにおけるコバルトの発光強度が共存元素の影響を受ける場合は,影響を受けない他の
測定波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又は
バックグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
12.3.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,12.3.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,12.3.3.1 b)及び12.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
12.3.5 検量線の作成
コバルト標準液(12.3.2.1)及び/又はコバルト標準液(12.3.2.2)の各種液量(コバルトとして0〜5 000
μg)を段階的に数個の100 mLの全量フラスコに取る。以下,12.3.3.1 b)及び12.3.3.2の手順に従って,主
溶液と同じ操作を主溶液と並行して行い,得た発光強度とコバルト量との関係線を作成し,その関係線を
原点を通るように平行移動して検量線とする。
12.3.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 12.3.3.2及び12.3.4で得た発光強度と12.3.5で
作成した検量線とからコバルト量を求め,次の式によって,ヒューム中のコバルト含有率を算出する。
30
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
100
)
(
15
1
12
11
×
×
−
=
B
m
A
A
Co
ここに,
Co: ヒューム中のコバルト含有率[%(質量分率)]
A11: 試料溶液中のコバルト検出量(mg)
A12: 空試験でのコバルト検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B15: 12.3.3.1 a)及び12.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 12.3.3.2及び12.3.4で得た発光強度と12.3.5で作成
した検量線とからコバルト量を求め,次の式によって,ヒューム中のコバルト含有率を算出する。
100
100
)
(
15
2
12
11
×
×
−
=
B
m
A
A
Co
ここに,
Co: ヒューム中のコバルト含有率[%(質量分率)]
A11: 試料溶液中のコバルト検出量(mg)
A12: 空試験でのコバルト検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B15: 12.3.3.1 a)及び12.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
13 鉛定量方法
13.1 定量方法の区分
鉛の定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が1 μg/mL以上20 μg/mL以下の鉛の定量に適用
する。
b) ICP発光分光法 この方法は,試料溶液中の濃度が1 μg/mL以上20 μg/mL以下の鉛の定量に適用する。
13.2 フレーム原子吸光法
13.2.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加えた後,溶液を原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
13.2.2 試薬
13.2.2.1 塩酸(1+1)
13.2.2.2 鉛標準液(Pb:100 μg/mL)
鉛[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+4)50
mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカーの内壁
を水で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1+1)40
mLを加えた後,水で標線まで薄める。
13.2.3 操作
13.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,鉛量が100〜2 000 μgになるように分取し,100 mLの全量フ
31
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
13.2.3.2 吸光度の測定
13.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長217.0
nm又は283.3 nmにおける吸光度を測定する。
13.2.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,13.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,13.2.3.1 b)及び13.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
13.2.5 検量線の作成
鉛標準液(13.2.2.2)0〜20.0 mL(鉛として0〜2 000 μg)を段階的に数個の100 mLの全量フラスコに取
る。以下,13.2.3.1 b)及び13.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た吸光
度と鉛量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
13.2.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 13.2.3.2及び13.2.4で得た吸光度と13.2.5で作
成した検量線とから鉛量を求め,次の式によって,ヒューム中の鉛含有率を算出する。
100
100
)
(
16
1
14
13
×
×
−
=
B
m
A
A
Pb
ここに,
Pb: ヒューム中の鉛含有率[%(質量分率)]
A13: 試料溶液中の鉛検出量(mg)
A14: 空試験での鉛検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B16: 13.2.3.1 a)及び13.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 13.2.3.2及び13.2.4で得た吸光度と13.2.5で作成し
た検量線とから鉛量を求め,次の式によって,ヒューム中の鉛含有率を算出する。
100
100
)
(
16
2
14
13
×
×
−
=
B
m
A
A
Pb
ここに,
Pb: ヒューム中の鉛含有率[%(質量分率)]
A13: 試料溶液中の鉛検出量(mg)
A14: 空試験での鉛検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B16: 13.2.3.1 a)及び13.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
13.3 ICP発光分光法
13.3.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
32
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.3.2 試薬
13.3.2.1 鉛標準液(Pb:100 μg/mL)
13.2.2.2による。
13.3.3 操作
13.3.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,鉛量が100〜2 000 μgになるように分取し,100 mLの全量フ
ラスコに移し入れる。
b) 水を加えて標線まで薄める。
13.3.3.2 発光強度の測定
13.3.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長220.351 nmにおけ
る発光強度を測定する。
なお,220.351 nmにおける鉛の発光強度が共存元素の影響を受ける場合は,影響を受けない他の測定波
長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又はバック
グラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
13.3.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,13.3.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,13.3.3.1 b)及び13.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
13.3.5 検量線の作成
鉛標準液(13.3.2.1)1.0〜20.0 mL(鉛として0〜2 000 μg)を段階的に数個の100 mLの全量フラスコに
取る。以下,13.3.3.1 b)及び13.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た発
光強度と鉛量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
13.3.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 13.3.3.2及び13.3.4で得た発光強度と13.3.5で
作成した検量線とから鉛量を求め,次の式によって,ヒューム中の鉛含有率を算出する。
100
100
)
(
17
1
14
13
×
×
−
=
B
m
A
A
Pb
ここに,
Pb: ヒューム中の鉛含有率[%(質量分率)]
A13: 試料溶液中の鉛検出量(mg)
A14: 空試験での鉛検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B17: 13.3.3.1 a)及び13.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 13.3.3.2及び13.3.4で得た発光強度と13.3.5で作成
した検量線とから鉛量を求め,次の式によって,ヒューム中の鉛含有率を算出する。
100
100
)
(
17
2
14
13
×
×
−
=
B
m
A
A
Pb
33
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
Pb: ヒューム中の鉛含有率[%(質量分率)]
A13: 試料溶液中の鉛検出量(mg)
A14: 空試験での鉛検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B17: 13.3.3.1 a)及び13.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
14 亜鉛定量方法
14.1 定量方法の区分
亜鉛の定量方法は,次のいずれかによる。
なお,電気加熱原子吸光法は,6.2.3.1 a) 2)によって調製した主溶液にだけ適用する。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.05 μg/mL以上2 μg/mL以下の亜鉛の定量に
適用する。
b) 電気加熱原子吸光法 この方法は,試料溶液中の濃度が0.001 μg/mL以上0.02 μg/mL以下の亜鉛の定
量に適用する。ただし,試料溶液中に共存する元素,塩類などが亜鉛の吸光度に影響を及ぼす場合に
は,この方法で規定する検量線法は,適用してはならない。
c) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上50 μg/mL以下の亜鉛の定量に適用
する。
14.2 フレーム原子吸光法
14.2.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸,又は硝酸で分解する。塩酸を加えた後,溶液を原子吸光
光度計の空気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
14.2.2 試薬
14.2.2.1 塩酸(1+1)
14.2.2.2 亜鉛標準液(Zn:50 μg/mL)
亜鉛[99.9 %(質量分率)以上]0.200 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)
10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカーの内
壁を水で洗って時計皿を取り除き,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1+1)
40 mLを加えた後,水で標線まで薄めて原液(Zn:200 μg/mL)とする。この原液25.0 mLを,使用の都度,
100 mLの全量フラスコに取り,硝酸(1+1)5 mLを加えた後,水で標線まで薄める。
14.2.2.3 亜鉛標準液(Zn:5.0 μg/mL)
亜鉛標準液(14.2.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mLを
加えた後,水で標線まで薄める。
14.2.3 操作
14.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)若しくは2) 2.3)又は6.2.3.1 b)で得た主溶液を,亜鉛量が5〜200 μgになるように分取
し,100 mLの全量フラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
なお,主溶液の調製を6.2.3.1 a) 2)によって行った場合には,塩酸(1+1)10 mLを加えずに,水で
標線まで薄める。
34
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.2.3.2 吸光度の測定
14.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長213.9
nmにおける吸光度を測定する。
14.2.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,14.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,14.2.3.1 b)及び14.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
14.2.5 検量線の作成
亜鉛標準液(14.2.2.2)及び亜鉛標準液(14.2.2.3)の各種液量(亜鉛として0〜200 μg)を段階的に数個
の100 mLの全量フラスコに取る。以下,14.2.3.1 b)及び14.2.3.2の手順に従って,主溶液と同じ操作を主
溶液と並行して行い,得た吸光度と亜鉛量との関係線を作成し,その関係線を原点を通るように平行移動
して検量線とする。
14.2.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合 14.2.3.2及び14.2.4で得た吸光度と14.2.5
で作成した検量線とから亜鉛量を求め,次の式によって,ヒューム中の亜鉛含有率を算出する。
100
100
)
(
18
3
16
15
×
×
−
=
B
m
A
A
Zn
ここに,
Zn: ヒューム中の亜鉛含有率[%(質量分率)]
A15: 試料溶液中の亜鉛検出量(mg)
A16: 空試験での亜鉛検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B18: 14.2.3.1 a)及び14.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 14.2.3.2及び14.2.4で得た吸光度と14.2.5で作成し
た検量線とから亜鉛量を求め,次の式によって,ヒューム中の亜鉛含有率を算出する。
100
100
)
(
18
2
16
15
×
×
−
=
B
m
A
A
Zn
ここに,
Zn: ヒューム中の亜鉛含有率[%(質量分率)]
A15: 試料溶液中の亜鉛検出量(mg)
A16: 空試験での亜鉛検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B18: 14.2.3.1 a)及び14.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
14.3 電気加熱原子吸光法
14.3.1 要旨
分析試料を硝酸で分解する。硝酸を加えた後,溶液を電気加熱原子吸光光度計の電気加熱炉に注入し,
その吸光度を測定する。
35
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.3.2 水
試薬の調製及び定量操作に用いる水は,JIS K 0050の7.1(水及び試薬)に規定するA3の水とする。
14.3.3 試薬
14.3.3.1 硝酸(1+1)
硝酸(JIS K 9901)を用いて調製する。
14.3.3.2 亜鉛標準液(Zn:1.0 μg/mL)
亜鉛[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)
[硝酸(JIS K 9901)を用いて調製する。]10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷
却した後,時計皿の下面及びビーカーの内壁を水(14.3.2)で洗って時計皿を取り除く。溶液を1 000 mL
の全量フラスコに水を用いて移し入れ,硝酸(1+1)[硝酸(JIS K 9901)を用いて調製する。]40 mLを
加えた後,水で標線まで薄め,原液(Zn:100 μg/mL)とする。この原液1.0 mLを,使用の都度,100 mL
の全量フラスコに取り,硝酸(1+1)[硝酸(JIS K 9901)を用いて調製する。]2 mLを加えた後,水(14.3.2)
で標線まで薄める。
14.3.3.3 亜鉛標準液(Zn:0.1 μg/mL)
亜鉛標準液(14.3.3.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)[硝酸(JIS
K 9901)を用いて調製する。]2 mLを加えた後,水(14.3.2)で標線まで薄める。
14.3.4 器具
14.3.4.1 マイクロピぺット
JIS K 0970に規定するプッシュボタン式液体用微量体積計,又は自動注入装置を用いる。
14.3.5 操作
14.3.5.1 試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 検量線法によって定量する場合
1) 6.2.3.1 a) 2) 2.3)で得た主溶液を,亜鉛量が0.1〜2 μgになるように分取し,100 mLの全量フラスコ
に移し入れる。
2) 硝酸(1+1)(14.3.3.1)10 mLを加えた後,水(14.3.2)で標線まで薄める。
b) 標準添加法によって定量する場合
1) 4個以上の100 mLの全量フラスコを用意し,そのそれぞれに,6.2.3.1 a) 2) 2.3)で得た主溶液を,同
量ずつ分取する。
なお,主溶液の分取量は,分取した主溶液中の亜鉛量が0.1 μg未満とならない量とする。
2) 1個の全量フラスコを除いた他の全量フラスコに,亜鉛標準液(14.3.3.2)及び/又は亜鉛標準液
(14.3.3.3)を,フラスコの溶液中の亜鉛量が0.1〜2 μgとなるように段階的に加える。全ての全量
フラスコに硝酸(1+1)(14.3.3.1)10 mLを加えた後,水(14.3.2)で標線まで薄める。
14.3.5.2 吸光度の測定
吸光度の測定は,次のいずれかの手順によって行う。
a) 検量線法によって定量する場合
1) 14.3.5.1 a) 2)で得た溶液の一部(例えば,10〜50 μL)を,マイクロピペット(14.3.4.1)を用いて電
気加熱原子吸光光度計の電気加熱炉に注入する。
2) 乾燥(100〜120 ℃で30〜40秒間),灰化(約600 ℃で30〜40秒間)及び原子化(約2 000 ℃で4
〜6秒間)を行い,波長213.9 nmにおける吸光度を測定する。
36
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なお,吸光度測定時には,バックグラウンド補正を行う。また,乾燥,灰化及び原子化の条件は,
装置,試料溶液の注入量,試料溶液中の塩類濃度などによって異なるので,あらかじめ最適な条件
を求めておく。
b) 標準添加法によって定量する場合
1) 14.3.5.1 b) 2)で得た溶液の一部(例えば,10〜50 μL)を,マイクロピペット(14.3.4.1)を用いて電
気加熱原子吸光光度計の電気加熱炉に注入する。
2) a) 2)の操作を行う。
14.3.6 空試験
空試験は,次のいずれかによる。
a) 検量線法によって定量する場合 6.2.3.2 a)で得た空試験溶液を,14.3.5.1 a) 1)で分取した主溶液と同量
分取し,100 mLの全量フラスコに移し入れる。以下,14.3.5.1 a) 2)及び14.3.5.2 a)の手順に従って,主
溶液と同じ操作を主溶液と並行して行う。
b) 標準添加法によって定量する場合 4個以上の100 mLの全量フラスコを用意し,そのそれぞれに,
6.2.3.2 a)で得た空試験溶液を,14.3.5.1 b) 1)で分取した主溶液と同量ずつ分取する。以下,14.3.5.1 b) 2)
及び14.3.5.2 b)の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
14.3.7 検量線の作成
検量線の作成は,次のいずれかによる。
a) 検量線法によって定量する場合 亜鉛標準液(14.3.3.2)及び/又は亜鉛標準液(14.3.3.3)の各種液量
(亜鉛として0〜2 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,14.3.5.1 a) 2)及び
14.3.5.2 a)の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た吸光度と亜鉛量との関係
線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 標準添加法によって定量する場合
1) 試料溶液用検量線 14.3.5.2 b) 2)で得た吸光度と14.3.5.1 b) 2)で亜鉛標準液として添加した亜鉛量
との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
2) 空試験用検量線 14.3.6 b)で得た吸光度と14.3.6 b)で亜鉛標準液として添加した亜鉛量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
14.3.8 計算
計算は,次のいずれかによる。
a) 検量線法によって定量する場合 14.3.5.2 a) 2)及び14.3.6 a)で得た吸光度と14.3.7 a)で作成した検量線
とから亜鉛量を求め,次の式によって,ヒューム中の亜鉛含有率を算出する。
100
100
)
(
19
3
16
15
×
×
−
=
B
m
A
A
Zn
ここに,
Zn: ヒューム中の亜鉛含有率[%(質量分率)]
A15: 試料溶液中の亜鉛検出量(mg)
A16: 空試験での亜鉛検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B19: 14.3.5.1 a) 1)及び14.3.6 a)でそれぞれ分取した主溶
液及び空試験溶液の量(mL)
37
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 標準添加法によって定量する場合 14.3.5.2 b) 2)で得た亜鉛標準液を添加しなかった試料溶液の吸光
度と14.3.7 b) 1)で作成した検量線とから,及び14.3.6 b)で得た亜鉛標準液を添加しなかった空試験の
吸光度と14.3.7 b) 2)で作成した検量線とから,それぞれ亜鉛量を求め,次の式によって,ヒューム中
の亜鉛含有率を算出する。
100
100
)
(
20
3
16
15
×
×
−
=
B
m
A
A
Zn
ここに,
Zn: ヒューム中の亜鉛含有率[%(質量分率)]
A15: 試料溶液中の亜鉛検出量(mg)
A16: 空試験での亜鉛検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B20: 14.3.5.1 b) 1)及び14.3.6 b)でそれぞれ分取した主溶
液及び空試験溶液の量(mL)
14.4
ICP発光分光法
14.4.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸,又は硝酸で分解する。溶液を発光分光装置のアルゴンプ
ラズマ中に噴霧し,その発光強度を測定する。
14.4.2 試薬
14.4.2.1 亜鉛標準液(Zn:200 μg/mL)
14.2.2.2で調製した原液(Zn:200 μg/mL)を亜鉛標準液とする。
14.4.2.2 亜鉛標準液(Zn:10 μg/mL)
亜鉛標準液(14.4.2.1)5.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mLを加
えた後,水で標線まで薄める。
14.4.3 操作
14.4.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)若しくは2) 2.3)又は6.2.3.1 b)で得た主溶液を,亜鉛量が10〜5 000 μgになるように分
取し,100 mLの全量フラスコに移し入れる。
b) 水を加えて標線まで薄める。
14.4.3.2 発光強度の測定
14.4.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長213.856 nmにおけ
る発光強度を測定する。
なお,213.856 nmにおける亜鉛の発光強度が共存元素の影響を受ける場合は,影響を受けない他の測定
波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又はバッ
クグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
14.4.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,14.4.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,14.4.3.1 b)及び14.4.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
38
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.4.5 検量線の作成
亜鉛標準液(14.4.2.1)及び/又は亜鉛標準液(14.4.2.2)の各種液量(亜鉛として0〜5 000 μg)を段階
的に数個の100 mLの全量フラスコに取る。以下,14.4.3.1 b)及び14.4.3.2の手順に従って,主溶液と同じ
操作を主溶液と並行して行い,得た発光強度と亜鉛量との関係線を作成し,その関係線を原点を通るよう
に平行移動して検量線とする。
14.4.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合 14.4.3.2及び14.4.4で得た発光強度と
14.4.5で作成した検量線とから亜鉛量を求め,次の式によって,ヒューム中の亜鉛含有率を算出する。
100
100
)
(
21
3
16
15
×
×
−
=
B
m
A
A
Zn
ここに,
Zn: ヒューム中の亜鉛含有率[%(質量分率)]
A15: 試料溶液中の亜鉛検出量(mg)
A16: 空試験での亜鉛検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B21: 14.4.3.1 a)及び14.4.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 14.4.3.2及び14.4.4で得た発光強度と14.4.5で作成
した検量線とから亜鉛量を求め,次の式によって,ヒューム中の亜鉛含有率を算出する。
100
100
)
(
21
2
16
15
×
×
−
=
B
m
A
A
Zn
ここに,
Zn: ヒューム中の亜鉛含有率[%(質量分率)]
A15: 試料溶液中の亜鉛検出量(mg)
A16: 空試験での亜鉛検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B21: 14.4.3.1 a)及び14.4.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
15 アルミニウム定量方法
15.1 定量方法の区分
アルミニウムの定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が2 μg/mL以上30 μg/mL以下のアルミニウムの
定量に適用する。
b) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上50 μg/mL以下のアルミニウムの定
量に適用する。
15.2 フレーム原子吸光法
15.2.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加えた後,溶液を原子吸光光度計の一
酸化二窒素・アセチレンフレーム中に噴霧し,その吸光度を測定する。
39
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
15.2.2 試薬
15.2.2.1 塩酸(1+1)
15.2.2.2 アルミニウム標準液(Al:1.0 mg/mL)
アルミニウム[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(200 mL)に移し入れ,塩酸
(1+1)15 mL及び硝酸(1+1)15 mLを加え,時計皿で覆い,穏やかに加熱して分解する。常温まで冷
却した後,時計皿の下面及びビーカーの内壁を水で洗って時計皿を取り除き,溶液を100 mLの全量フラ
スコに水を用いて移し入れ,硝酸(1+1)5 mLを加えた後,水で標線まで薄める。
15.2.2.3 アルミニウム標準液(Al:100 μg/mL)
アルミニウム標準液(15.2.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)
5 mLを加えた後,水で標線まで薄める。
15.2.3 操作
15.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,アルミニウム量が200〜3 000 μgになるように分取し,100 mL
の全量フラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
15.2.3.2 吸光度の測定
15.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した一酸化二窒素・アセチレンフレーム中に噴霧し,
波長309.3 nmにおける吸光度を測定する。
15.2.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,15.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,15.2.3.1 b)及び15.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
15.2.5 検量線の作成
アルミニウム標準液(15.2.2.2)及びアルミニウム標準液(15.2.2.3)の各種液量(アルミニウムとして0
〜3 000 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,15.2.3.1 b)及び15.2.3.2の手順に従っ
て,主溶液と同じ操作を主溶液と並行して行い,得た吸光度とアルミニウム量との関係線を作成し,その
関係線を原点を通るように平行移動して検量線とする。
15.2.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 15.2.3.2及び15.2.4で得た吸光度と15.2.5で作
成した検量線とからアルミニウム量を求め,次の式によって,ヒューム中のアルミニウム含有率を算
出する。
100
100
)
(
22
1
18
17
×
×
−
=
B
m
A
A
Al
ここに,
Al: ヒューム中のアルミニウム含有率[%(質量分率)]
A17: 試料溶液中のアルミニウム検出量(mg)
A18: 空試験でのアルミニウム検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B22: 15.2.3.1 a)及び15.2.4でそれぞれ分取した主溶液及
40
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 15.2.3.2及び15.2.4で得た吸光度と15.2.5で作成し
た検量線とからアルミニウム量を求め,次の式によって,ヒューム中のアルミニウム含有率を算出す
る。
100
100
)
(
22
2
18
17
×
×
−
=
B
m
A
A
Al
ここに,
Al: ヒューム中のアルミニウム含有率[%(質量分率)]
A17: 試料溶液中のアルミニウム検出量(mg)
A18: 空試験でのアルミニウム検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B22: 15.2.3.1 a)及び15.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
15.3 ICP発光分光法
15.3.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
15.3.2 試薬
15.3.2.1 アルミニウム標準液(Al:200 μg/mL)
アルミニウム標準液(15.2.2.2)20.0 mLを100 mLの全量フラスコに取り,硝酸(1+1)5 mLを加えた
後,水で標線まで薄める。
15.3.2.2 アルミニウム標準液(Al:10 μg/mL)
アルミニウム標準液(15.3.2.1)5.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5
mLを加えた後,水で標線まで薄める。
15.3.3 操作
15.3.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,アルミニウム量が10〜5 000 μgになるように分取し,100 mL
の全量フラスコに移し入れる。
b) 水を加えて標線まで薄める。
15.3.3.2 発光強度の測定
15.3.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長309.271 nmにおけ
る発光強度を測定する。
なお,309.271 nmにおけるアルミニウムの発光強度が共存元素の影響を受ける場合は,影響を受けない
他の測定波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/
又はバックグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
15.3.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,15.3.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,15.3.3.1 b)及び15.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
41
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
15.3.5 検量線の作成
アルミニウム標準液(15.3.2.1)及び/又はアルミニウム標準液(15.3.2.2)の各種液量(アルミニウムと
して0〜5 000 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,15.3.3.1 b)及び15.3.3.2の手順
に従って,主溶液と同じ操作を主溶液と並行して行い,得た発光強度とアルミニウム量との関係線を作成
し,その関係線を原点を通るように平行移動して検量線とする。
15.3.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 15.3.3.2及び15.3.4で得た発光強度と15.3.5で
作成した検量線とからアルミニウム量を求め,次の式によって,ヒューム中のアルミニウム含有率を
算出する。
100
100
)
(
23
1
18
17
×
×
−
=
B
m
A
A
Al
ここに,
Al: ヒューム中のアルミニウム含有率[%(質量分率)]
A17: 試料溶液中のアルミニウム検出量(mg)
A18: 空試験でのアルミニウム検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B23: 15.3.3.1 a)及び15.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 15.3.3.2及び15.3.4で得た発光強度と15.3.5で作成
した検量線とからアルミニウム量を求め,次の式によって,ヒューム中のアルミニウム含有率を算出
する。
100
100
)
(
23
2
18
17
×
×
−
=
B
m
A
A
Al
ここに,
Al: ヒューム中のアルミニウム含有率[%(質量分率)]
A17: 試料溶液中のアルミニウム検出量(mg)
A18: 空試験でのアルミニウム検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B23: 15.3.3.1 a)及び15.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
16 カドミウム定量方法
16.1 定量方法の区分
カドミウムの定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.05 μg/mL以上2 μg/mL以下のカドミウムの
定量に適用する。
b) 電気加熱原子吸光法 この方法は,試料溶液中の濃度が0.001 μg/mL以上0.02 μg/mL以下のカドミウ
ムの定量に適用する。ただし,試料溶液中に共存する元素,塩類などがカドミウムの吸光度に影響を
及ぼす場合には,この方法で規定する検量線法は,適用してはならない。
c) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上30 μg/mL以下のカドミウムの定量
に適用する。
42
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
16.2 フレーム原子吸光法
16.2.1 要旨
分析試料を硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,その吸
光度を測定する。
16.2.2 試薬
16.2.2.1 カドミウム標準液(Cd:50 μg/mL)
カドミウム[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1
+1)10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカー
の内壁を水で洗って時計皿を取り除き,溶液を100 mLの全量フラスコに水を用いて移し入れ,水で標線
まで薄めて原液(Cd:1 000 μg/mL)とする。この原液5.0 mLを,使用の都度,100 mLの全量フラスコに
取り,硝酸(1+1)5 mLを加えた後,水で標線まで薄めてカドミウム標準液とする。
16.2.2.2 カドミウム標準液(Cd:5.0 μg/mL)
カドミウム標準液(16.2.2.1)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
16.2.3 操作
16.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 2) 2.3)で得た主溶液を,カドミウム量が5〜200 μgになるように分取し,100 mLの全量フラ
スコに移し入れる。
b) 水で標線まで薄める。
16.2.3.2 吸光度の測定
16.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長228.8
nmにおける吸光度を測定する。
16.2.4 空試験
6.2.3.2 a)で得た空試験溶液を,16.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコに移
し入れる。以下,16.2.3.1 b)及び16.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
16.2.5 検量線の作成
カドミウム標準液(16.2.2.1)及びカドミウム標準液(16.2.2.2)の各種液量(カドミウムとして0〜200 μg)
を段階的に数個の100 mLの全量フラスコに取る。以下,16.2.3.1 b)及び16.2.3.2の手順に従って,主溶液
と同じ操作を主溶液と並行して行い,得た吸光度とカドミウム量との関係線を作成し,その関係線を原点
を通るように平行移動して検量線とする。
16.2.6 計算
16.2.3.2及び16.2.4で得た吸光度と16.2.5で作成した検量線とからカドミウム量を求め,次の式によって,
ヒューム中のカドミウム含有率を算出する。
100
100
)
(
24
3
20
19
×
×
−
=
B
m
A
A
Cd
ここに,
Cd: ヒューム中のカドミウム含有率[%(質量分率)]
A19: 試料溶液中のカドミウム検出量(mg)
A20: 空試験でのカドミウム検出量(mg)
43
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B24: 16.2.3.1 a)及び16.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
16.3 電気加熱原子吸光法
16.3.1 要旨
分析試料を硝酸で分解する。硝酸を加えた後,溶液を電気加熱原子吸光光度計の電気加熱炉に注入し,
その吸光度を測定する。
16.3.2 水
試薬の調製及び定量操作に用いる水は,JIS K 0050の7.1(水及び試薬)に規定するA3の水とする。
16.3.3 試薬
16.3.3.1 硝酸(1+1)
硝酸(JIS K 9901)を用いて調製する。
16.3.3.2 カドミウム標準液(Cd:1.0 μg/mL)
カドミウム[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1
+1)(16.3.3.1)10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及
びビーカーの内壁を水(16.3.2)で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水(16.3.2)
を用いて移し入れ,硝酸(1+1)(16.3.3.1)40 mLを加えた後,水(16.3.2)で標線まで薄め,原液(Cd:
100 μg/mL)とする。この原液1.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)(16.3.3.1)
2 mLを加えた後,水(16.3.2)で標線まで薄めてカドミウム標準液とする。
16.3.3.3 カドミウム標準液(Cd:0.1 μg/mL)
カドミウム標準液(16.3.3.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)(16.3.3.1)
2 mLを加えた後,水(16.3.2)で標線まで薄める。
16.3.4 器具
器具は,次による。
16.3.4.1 マイクロピぺット
JIS K 0970に規定するプッシュボタン式液体用微量体積計,又は自動注入装置を用いる。
16.3.5 操作
16.3.5.1 試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 検量線法によって定量する場合
1) 6.2.3.1 a) 2) 2.3)で得た主溶液を,カドミウム量が0.1〜2 μgになるように分取し,100 mLの全量フ
ラスコに移し入れる。
2) 硝酸(1+1)(16.3.3.1)10 mLを加えた後,水(16.3.2)で標線まで薄める。
b) 標準添加法によって定量する場合
1) 4個以上の100 mLの全量フラスコを用意し,そのそれぞれに,6.2.3.1 a) 2) 2.3)で得た主溶液を,同
量ずつ分取する。
なお,主溶液の分取量は,分取した主溶液中のカドミウム量が0.1 μg未満とならない量とする。
2) 1個の全量フラスコを除いた他の全量フラスコに,カドミウム標準液(16.3.3.2)及び/又はカドミ
ウム標準液(16.3.3.3)を,フラスコの溶液中のカドミウム量が0.1〜2 μgとなるように段階的に加
44
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
える。全ての全量フラスコに硝酸(1+1)(16.3.3.1)10 mLを加えた後,水(16.3.2)で標線まで薄
める。
16.3.5.2 吸光度の測定
吸光度の測定は,次のいずれかの手順によって行う。
a) 検量線法によって定量する場合
1) 16.3.5.1 a) 2)で得た溶液の一部(例えば,10〜50 μL)を,マイクロピペット(16.3.4.1)を用いて電
気加熱原子吸光光度計の電気加熱炉に注入する。
2) 乾燥(100〜120 ℃で30〜40秒間),灰化(約600 ℃で30〜40秒間)及び原子化(約2 000 ℃で4
〜6秒間)を行い,波長228.8 nmにおける吸光度を測定する。
なお,吸光度測定時には,バックグラウンド補正を行う。また,乾燥,灰化及び原子化の条件は,
装置,試料溶液の注入量,試料溶液中の塩類濃度などによって異なるので,あらかじめ最適な条件
を求めておく。
b) 標準添加法によって定量する場合
1) 16.3.5.1 b) 2)で得た溶液の一部(例えば,10〜50 μL)を,マイクロピペット(16.3.4.1)を用いて電
気加熱原子吸光光度計の電気加熱炉に注入する。
2) a) 2)の操作を行う。
16.3.6 空試験
空試験は,次のいずれかによる。
a) 検量線法によって定量する場合 6.2.3.2 a)で得た空試験溶液を,16.3.5.1 a) 1)で分取した主溶液と同量
分取し,100 mLの全量フラスコに移し入れる。以下,16.3.5.1 a) 2)及び16.3.5.2 a)の手順に従って,主
溶液と同じ操作を主溶液と並行して行う。
b) 標準添加法によって定量する場合 4個以上の100 mLの全量フラスコを用意し,そのそれぞれに,
6.2.3.2 a)で得た空試験溶液を,16.3.5.1 b) 1)で分取した主溶液と同量ずつ分取する。以下,16.3.5.1 b) 2)
及び16.3.5.2 b)の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
16.3.7 検量線の作成
検量線の作成は,次のいずれかによる。
a) 検量線法によって定量する場合 カドミウム標準液(16.3.3.2)及び/又はカドミウム標準液(16.3.3.3)
の各種液量(カドミウムとして0〜2 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,
16.3.5.1 a) 2)及び16.3.5.2 a)の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た吸光度
とカドミウム量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 標準添加法によって定量する場合
1) 試料溶液用検量線 16.3.5.2 b) 2)で得た吸光度と16.3.5.1 b) 2)でカドミウム標準液として添加した
カドミウム量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
2) 空試験用検量線 16.3.6 b)で得た吸光度と16.3.6 b)でカドミウム標準液として添加したカドミウム
量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
16.3.8 計算
計算は,次のいずれかによる。
a) 検量線法によって定量する場合 16.3.5.2 a) 2)及び16.3.6 a)で得た吸光度と16.3.7 a)で作成した検量線
とからカドミウム量を求め,次の式によって,ヒューム中のカドミウム含有率を算出する。
45
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
100
)
(
25
3
20
19
×
×
−
=
B
m
A
A
Cd
ここに,
Cd: ヒューム中のカドミウム含有率[%(質量分率)]
A19: 試料溶液中のカドミウム検出量(mg)
A20: 空試験でのカドミウム検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B25: 16.3.5.1 a) 1)及び16.3.6 a)でそれぞれ分取した主溶
液及び空試験溶液の量(mL)
b) 標準添加法によって定量する場合 16.3.5.2 b) 2)で得たカドミウム標準液を添加しなかった試料溶液
の吸光度と16.3.7 b) 1)で作成した検量線とから,及び16.3.6 b)で得たカドミウム標準液を添加しなか
った空試験の吸光度と16.3.7 b) 2)で作成した検量線とから,それぞれカドミウム量を求め,次の式に
よって,ヒューム中のカドミウム含有率を算出する。
100
100
)
(
26
3
20
19
×
×
−
=
B
m
A
A
Cd
ここに,
Cd: ヒューム中のカドミウム含有率[%(質量分率)]
A19: 試料溶液中のカドミウム検出量(mg)
A20: 空試験でのカドミウム検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B26: 16.3.5.1 b) 1)及び16.3.6 b)でそれぞれ分取した主溶
液及び空試験溶液の量(mL)
16.4 ICP発光分光法
16.4.1 要旨
分析試料を硝酸で分解した後,溶液を発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測
定する。
16.4.2 試薬
16.4.2.1 カドミウム標準液(Cd:200 μg/mL)
16.2.2.1で調製した原液(Cd:1 000 μg/mL)5.0 mLを100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
16.4.2.2 カドミウム標準液(Cd:10 μg/mL)
カドミウム標準液(16.4.2.1)5.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加えた後,水で標線まで薄める。
16.4.3 操作
16.4.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 2) 2.3)で得た主溶液を,カドミウム量が10〜3 000 μgになるように分取し,100 mLの全量フ
ラスコに移し入れる。
b) 水を加えて標線まで薄める。
46
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
16.4.3.2 発光強度の測定
16.4.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長214.438 nmにおけ
る発光強度を測定する。
なお,214.438 nmにおけるカドミウムの発光強度が共存元素の影響を受ける場合は,影響を受けない他
の測定波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又
はバックグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
16.4.4 空試験
6.2.3.2 a)で得た空試験溶液を,16.4.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコに移
し入れる。以下,16.4.3.1 b)及び16.4.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
16.4.5 検量線の作成
カドミウム標準液(16.4.2.1)及び/又はカドミウム標準液(16.4.2.2)の各種液量(カドミウムとして0
〜3 000 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,16.4.3.1 b)及び16.4.3.2の手順に従っ
て,主溶液と同じ操作を主溶液と並行して行い,得た発光強度とカドミウム量との関係線を作成し,その
関係線を原点を通るように平行移動して検量線とする。
16.4.6 計算
16.4.3.2及び16.4.4で得た発光強度と16.4.5で作成した検量線とからカドミウム量を求め,次の式によっ
て,ヒューム中のカドミウム含有率を算出する。
100
100
)
(
27
3
20
19
×
×
−
=
B
m
A
A
Cd
ここに,
Cd: ヒューム中のカドミウム含有率[%(質量分率)]
A19: 試料溶液中のカドミウム検出量(mg)
A20: 空試験でのカドミウム検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B27: 16.4.3.1 a)及び16.4.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
17 銀定量方法
17.1 定量方法の区分
銀の定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上5 μg/mL以下の銀の定量に適
用する。
b) 電気加熱原子吸光法 この方法は,試料溶液中の濃度が0.001 μg/mL以上0.02 μg/mL以下の銀の定量
に適用する。ただし,試料溶液中に共存する元素,塩類などが銀の吸光度に影響を及ぼす場合には,
この方法で規定する検量線法は,適用してはならない。
c) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上50 μg/mL以下の銀の定量に適用す
る。
17.2 フレーム原子吸光法
17.2.1 要旨
分析試料を硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,その吸
47
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
光度を測定する。
17.2.2 試薬
17.2.2.1 銀標準液(Ag:100 μg/mL)
銀[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)10
mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカーの内壁
を水で洗って時計皿を取り除き,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1+1)40
mLを加えた後,水で標線まで薄める。
17.2.2.2 銀標準液(Ag:10 μg/mL)
銀標準液(17.2.2.1)10.0 mLを100 mLの全量フラスコに取り,硝酸(1+1)5 mLを加えた後,水で標
線まで薄める。
17.2.3 操作
17.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 2) 2.3)で得た主溶液を,銀量が10〜5 000 μgになるように分取し,100 mLの全量フラスコに
移し入れる。
b) 水を加えて標線まで薄める。
17.2.3.2 吸光度の測定
17.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した空気・アセチレンフレーム中に噴霧し,波長328.0
nmにおける吸光度を測定する。
17.2.4 空試験
6.2.3.2 a)で得た空試験溶液を,17.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコに移
し入れる。以下,17.2.3.1 b)及び17.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
17.2.5 検量線の作成
銀標準液(17.2.2.1)及び銀標準液(17.2.2.2)の各種液量(銀として0〜5 000 μg)を段階的に数個の100
mLの全量フラスコに取る。以下,17.2.3.1 b)及び17.2.3.2の手順に従って,主溶液と同じ操作を主溶液と
並行して行い,得た吸光度と銀量との関係線を作成し,その関係線を原点を通るように平行移動して検量
線とする。
17.2.6 計算
17.2.3.2及び17.2.4で得た吸光度と17.2.5で作成した検量線とから銀量を求め,次の式によって,ヒュー
ム中の銀含有率を算出する。
100
100
)
(
28
3
22
21
×
×
−
=
B
m
A
A
Ag
ここに,
Ag: ヒューム中の銀含有率[%(質量分率)]
A21: 試料溶液中の銀検出量(mg)
A22: 空試験での銀検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B28: 17.2.3.1 a)及び17.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
48
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
17.3 電気加熱原子吸光法
17.3.1 要旨
分析試料を硝酸で分解する。硝酸を加えた後,溶液を電気加熱原子吸光光度計の電気加熱炉に注入し,
その吸光度を測定する。
17.3.2 水
試薬の調製及び定量操作に用いる水は,JIS K 0050の7.1(水及び試薬)に規定するA3の水とする。
17.3.3 試薬
17.3.3.1 硝酸(1+1)
硝酸(JIS K 9901)を用いて調製する。
17.3.3.2 銀標準液(Ag:1.0 μg/mL)
銀[99.9 %(質量分率)以上]0.100 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)(17.3.3.1)
10 mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカーの内
壁を水(17.3.2)で洗って時計皿を取り除く。溶液を1 000 mLの全量フラスコに水(17.3.2)を用いて移し
入れ,硝酸(1+1)(17.3.3.1)40 mLを加えた後,水(17.3.2)で標線まで薄めて原液(Ag:100 μg/mL)
とする。この原液1.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)(17.3.3.1)2 mL
を加えた後,水(17.3.2)で標線まで薄めて銀標準液とする。
17.3.3.3 銀標準液(Ag:0.1 μg/mL)
銀標準液(17.3.3.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)(17.3.3.1)
2 mLを加えた後,水(17.3.2)で標線まで薄める。
17.3.4 器具
17.3.4.1 マイクロピぺット
JIS K 0970に規定するプッシュボタン式液体用微量体積計,又は自動注入装置を用いる。
17.3.5 操作
17.3.5.1 試料溶液の調製
試料溶液の調製は,次のいずれかの手順によって行う。
a) 検量線法によって定量する場合
1) 6.2.3.1 a) 2) 2.3)で得た主溶液を,銀量が0.1〜2 μgになるように分取し,100 mLの全量フラスコに
移し入れる。
2) 硝酸(1+1)(17.3.3.1)10 mLを加えた後,水(17.3.2)で標線まで薄める。
b) 標準添加法によって定量する場合
1) 4個以上の100 mLの全量フラスコを用意し,そのそれぞれに,6.2.3.1 a) 2) 2.3)で得た主溶液を,同
量ずつ分取する。
なお,主溶液の分取量は,分取した主溶液中の銀量が0.1 μg未満とならない量とする。
2) 1個の全量フラスコを除いた他の全量フラスコに,銀標準液(17.3.3.2)及び/又は銀標準液(17.3.3.3)
を,フラスコの溶液中の銀量が0.1〜2 μgとなるように段階的に加える。全ての全量フラスコに硝
酸(1+1)(17.3.3.1)10 mLを加えた後,水(17.3.2)で標線まで薄める。
17.3.5.2 吸光度の測定
吸光度の測定は,次のいずれかの手順によって行う。
a) 検量線法によって定量する場合
1) 17.3.5.1 a) 2)で得た溶液の一部(例えば,10〜50 μL)を,マイクロピペット(17.3.4.1)を用いて電
49
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
気加熱原子吸光光度計の電気加熱炉に注入する。
2) 乾燥(100〜120 ℃で30〜40秒間),灰化(約800 ℃で30〜40秒間)及び原子化(約2 300 ℃で4
〜6秒間)を行い,波長328.0 nmにおける吸光度を測定する。
なお,吸光度測定時には,バックグラウンド補正を行う。また,乾燥,灰化及び原子化の条件は,
装置,試料溶液の注入量,試料溶液中の塩類濃度などによって異なるので,あらかじめ最適な条件
を求めておく。
b) 標準添加法によって定量する場合
1) 17.3.5.1 b) 2)で得た溶液の一部(例えば,10〜50 μL)を,マイクロピペット(17.3.4.1)を用いて電
気加熱原子吸光光度計の電気加熱炉に注入する。
2) a) 2)の操作を行う。
17.3.6 空試験
空試験は,次のいずれかによる。
a) 検量線法によって定量する場合 6.2.3.2 a)で得た空試験溶液を,17.3.5.1 a) 1)で分取した主溶液と同量
分取し,100 mLの全量フラスコに移し入れる。以下,17.3.5.1 a) 2)及び17.3.5.2 a)の手順に従って,主
溶液と同じ操作を主溶液と並行して行う。
b) 標準添加法によって定量する場合 4個以上の100 mLの全量フラスコを用意し,そのそれぞれに,
6.2.3.2 a)で得た空試験溶液を,17.3.5.1 b) 1)で分取した主溶液と同量ずつ分取する。以下,17.3.5.1 b) 2)
及び17.3.5.2 b)の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
17.3.7 検量線の作成
検量線の作成は,次のいずれかによる。
a) 検量線法によって定量する場合 銀標準液(17.3.3.2)及び/又は銀標準液(17.3.3.3)の各種液量(銀
として0〜2 μg)を段階的に数個の100 mLの全量フラスコに取る。以下,17.3.5.1 a) 2)及び17.3.5.2 a)
の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た吸光度と銀量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
b) 標準添加法によって定量する場合
1) 試料溶液用検量線 17.3.5.2 b) 2)で得た吸光度と17.3.5.1 b) 2)で銀標準液として添加した銀量との
関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
2) 空試験用検量線 17.3.6 b)で得た吸光度と17.3.6 b)で銀標準液として添加した銀量との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
17.3.8 計算
計算は,次のいずれかによる。
a) 検量線法によって定量する場合 17.3.5.2 a) 2)及び17.3.6 a)で得た吸光度と17.3.7 a)で作成した検量線
とから銀量を求め,次の式によって,ヒューム中の銀含有率を算出する。
100
100
)
(
29
3
22
21
×
×
−
=
B
m
A
A
Ag
ここに,
Ag: ヒューム中の銀含有率[%(質量分率)]
A21: 試料溶液中の銀検出量(mg)
A22: 空試験での銀検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
50
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B29: 17.3.5.1 a) 1)及び17.3.6 a)でそれぞれ分取した主溶
液及び空試験溶液の量(mL)
b) 標準添加法によって定量する場合 17.3.5.2 b) 2)で得た銀標準液を添加しなかった試料溶液の吸光度
と17.3.7 b) 1)で作成した検量線とから,及び17.3.6 b)で得た銀標準液を添加しなかった空試験の吸光
度と17.3.7 b) 2)で作成した検量線とから,それぞれ銀量を求め,次の式によって,ヒューム中の銀含
有率を算出する。
100
100
)
(
30
3
22
21
×
×
−
=
B
m
A
A
Ag
ここに,
Ag: ヒューム中の銀含有率[%(質量分率)]
A21: 試料溶液中の銀検出量(mg)
A22: 空試験での銀検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B30: 17.3.5.1 b) 1)及び17.3.6 b)でそれぞれ分取した主溶
液及び空試験溶液の量(mL)
17.4 ICP発光分光法
17.4.1 要旨
分析試料を硝酸で分解した後,溶液を発光分光分析装置のアルゴンプラズマ中に噴霧し,その発光強度
を測定する。
17.4.2 試薬
17.4.2.1 銀標準液(Ag:200 μg/mL)
銀[99.9 %(質量分率)以上]0.200 gをはかり取り,ビーカー(100 mL)に移し入れ,硝酸(1+1)10
mLを加え,時計皿で覆い,加熱して分解する。常温まで冷却した後,時計皿の下面及びビーカーの内壁
を水で洗って時計皿を取り除き,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1+1)40
mLを加えた後,水で標線まで薄める。
17.4.2.2 銀標準液(Ag:10 μg/mL)
銀標準液(17.4.2.1)5.0 mLを100 mLの全量フラスコに取り,硝酸(1+1)5 mLを加えた後,水で標線
まで薄める。
17.4.3 操作
17.4.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 2) 2.3)で得た主溶液を,銀量が10〜5 000 μgになるように分取し,100 mLの全量フラスコに
移し入れる。
b) 水を加えて標線まで薄める。
17.4.3.2 発光強度の測定
17.4.3.1 b)で得た溶液の一部を,発光分光分析装置のアルゴンプラズマ中に噴霧し,波長328.068 nmに
おける発光強度を測定する。
なお,328.068 nmにおける銀の発光強度が共存元素の影響を受ける場合は,影響を受けない他の測定波
長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又はバック
グラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
51
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
17.4.4 空試験
6.2.3.2 a)で得た空試験溶液を,17.4.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコに移
し入れる。以下,17.4.3.1 b)及び17.4.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
17.4.5 検量線の作成
銀標準液(17.4.2.1)又は銀標準液(17.4.2.2)の各種液量(銀として0〜5 000 μg)を段階的に数個の100
mLの全量フラスコに取る。以下,17.4.3.1 b)及び17.4.3.2の手順に従って,主溶液と同じ操作を主溶液と
並行して行い,得た発光強度と銀量との関係線を作成し,その関係線を原点を通るように平行移動して検
量線とする。
17.4.6 計算
17.4.3.2及び17.4.4で得た発光強度と17.4.5で作成した検量線とから銀量を求め,次の式によって,ヒュ
ーム中の銀含有率を算出する。
100
100
)
(
31
3
22
21
×
×
−
=
B
m
A
A
Ag
ここに,
Ag: ヒューム中の銀含有率[%(質量分率)]
A21: 試料溶液中の銀検出量(mg)
A22: 空試験での銀検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B31: 17.4.3.1 a)及び17.4.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
18 バリウム定量方法
18.1 定量方法
バリウムの定量方法は,ICP発光分光法による。この方法は,試料溶液中の濃度が0.05 μg/mL以上5 μg/mL
以下のバリウムの定量に適用する。
18.2 ICP発光分光法
18.2.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
18.2.2 試薬
18.2.2.1 バリウム標準液(Ba:20 μg/mL)
炭酸バリウム約1 gを白金るつぼに入れ,約120 ℃で約1時間加熱した後,デシケータ中で室温まで放
冷し,その0.288 gをはかり取ってビーカー(100 mL)に移し入れる。時計皿で覆い,硝酸(1+1)10 mL
を少量ずつ加えて分解する。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水で洗って時計皿
を取り除き,溶液を1 000 mLの全量フラスコに水を用いて移し入れ,硝酸(1+1)40 mLを加えた後,水
で標線まで薄めて原液(Ba:200 μg/mL)とする。この原液10.0 mLを,100 mLの全量フラスコに取り,
硝酸(1+1)5 mLを加えた後,水で標線まで薄めてバリウム標準液とする。
18.2.2.2 バリウム標準液(Ba:1.0 μg/mL)
バリウム標準液(18.2.2.1)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,硝酸(1+1)5 mL
を加え,水で標線まで薄める。
52
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
18.2.3 操作
18.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,バリウム量が5〜500 μgになるように分取し,100 mLの全量
フラスコに移し入れる。
b) 水を加えて標線まで薄める。
18.2.3.2 発光強度の測定
18.2.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長455.403 nmにおけ
る発光強度を測定する。
なお,455.403 nmにおけるバリウムの発光強度が共存元素の影響を受ける場合は,影響を受けない他の
測定波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又は
バックグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
18.2.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,18.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,18.2.3.1 b)及び18.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
18.2.5 検量線の作成
バリウム標準液(18.2.2.1)及び/又はバリウム標準液(18.2.2.2)の各種液量(バリウムとして0〜500 μg)
を段階的に数個の100 mLの全量フラスコに取る。以下,18.2.3.1 b)及び18.2.3.2の手順に従って,主溶液
と同じ操作を主溶液と並行して行い,得た発光強度とバリウム量との関係線を作成し,その関係線を原点
を通るように平行移動して検量線とする。
18.2.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 18.2.3.2及び18.2.4で得た発光強度と18.2.5で
作成した検量線とからバリウム量を求め,次の式によって,ヒューム中のバリウム含有率を算出する。
100
100
)
(
32
1
24
23
×
×
−
=
B
m
A
A
Ba
ここに,
Ba: ヒューム中のバリウム含有率[%(質量分率)]
A23: 試料溶液中のバリウム検出量(mg)
A24: 空試験でのバリウム検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B32: 18.2.3.1 a)及び18.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 18.2.3.2及び18.2.4で得た発光強度と18.2.5で作成
した検量線とからバリウム量を求め,次の式によって,ヒューム中のバリウム含有率を算出する。
100
100
)
(
32
2
24
23
×
×
−
=
B
m
A
A
Ba
ここに,
Ba: ヒューム中のバリウム含有率[%(質量分率)]
A23: 試料溶液中のバリウム検出量(mg)
53
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A24: 空試験でのバリウム検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B32: 18.2.3.1 a)及び18.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
19 全クロム定量方法
19.1 定量方法の区分
全クロムの定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,試料溶液中の濃度が0.2 μg/mL以上5 μg/mL以下の全クロムの定
量に適用する。
b) ICP発光分光法 この方法は,試料溶液中の濃度が0.1 μg/mL以上50 μg/mL以下の全クロムの定量に
適用する。
19.2 フレーム原子吸光法
19.2.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。塩酸を加えた後,溶液を原子吸光光度計の一
酸化二窒素・アセチレンフレーム又は空気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
19.2.2 試薬
19.2.2.1 塩酸(1+1)
19.2.2.2 クロム標準液(Cr:100 μg/mL)
二クロム酸カリウム(JIS K 8005に規定する容量分析用標準試薬)を正確に0.283 gはかり取り,水に溶
解する。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
19.2.2.3 クロム標準液(Cr:10 μg/mL)
クロム標準液(19.2.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,水で標線まで薄める。
19.2.3 操作
19.2.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,全クロム量が20〜500 μgになるように分取し,100 mLの全
量フラスコに移し入れる。
b) 塩酸(1+1)10 mLを加えた後,水で標線まで薄める。
19.2.3.2 吸光度の測定
19.2.3.1 b)で得た溶液の一部を,水でゼロ点を調整した一酸化二窒素・アセチレンフレーム又は空気・ア
セチレンフレーム中に噴霧し,波長357.9 nmにおける吸光度を測定する。
19.2.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,19.2.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,19.2.3.1 b)及び19.2.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
19.2.5 検量線の作成
クロム標準液(19.2.2.2)及びクロム標準液(19.2.2.3)の各種液量(クロムとして0〜500 μg)を段階的
に数個の100 mLの全量フラスコに取る。以下,19.2.3.1 b)及び19.2.3.2の手順に従って,主溶液と同じ操
作を主溶液と並行して行い,得た吸光度とクロム量との関係線を作成し,その関係線を原点を通るように
54
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
平行移動して検量線とする。
19.2.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 19.2.3.2及び19.2.4で得た吸光度と19.2.5で作
成した検量線とからクロム量を求め,次の式によって,ヒューム中の全クロム含有率を算出する。
100
100
)
(
33
1
26
25
×
×
−
=
B
m
A
A
Cr
ここに,
Cr: ヒューム中の全クロム含有率[%(質量分率)]
A25: 試料溶液中のクロム検出量(mg)
A26: 空試験でのクロム検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B33: 19.2.3.1 a)及び19.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 19.2.3.2及び19.2.4で得た吸光度と19.2.5で作成し
た検量線とからクロム量を求め,次の式によって,ヒューム中の全クロム含有率を算出する。
100
100
)
(
33
2
26
25
×
×
−
=
B
m
A
A
Cr
ここに,
Cr: ヒューム中の全クロム含有率[%(質量分率)]
A25: 試料溶液中のクロム検出量(mg)
A26: 空試験でのクロム検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B33: 19.2.3.1 a)及び19.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
19.3 ICP発光分光法
19.3.1 要旨
分析試料を,硝酸・過塩素酸・ふっ化水素酸で分解する。溶液を発光分光装置のアルゴンプラズマ中に
噴霧し,その発光強度を測定する。
19.3.2 試薬
19.3.2.1 クロム標準液(Cr:200 μg/mL)
二クロム酸カリウム(JIS K 8005に規定する容量分析用標準試薬)を正確に0.566 gはかり取り,水に溶
解する。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
19.3.2.2 クロム標準液(Cr:10 μg/mL)
クロム標準液(19.3.2.1)5.0 mLを,使用の都度,100 mLの全量フラスコに取り,水で標線まで薄める。
19.3.3 操作
19.3.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 6.2.3.1 a) 1) 1.4)又はb)で得た主溶液を,全クロム量が10〜5 000 μgになるように分取し,100 mLの全
量フラスコに移し入れる。
b) 水を加えて標線まで薄める。
55
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
19.3.3.2 発光強度の測定
19.3.3.1 b)で得た溶液の一部を,発光分光装置のアルゴンプラズマ中に噴霧し,波長206.149 nmにおけ
る発光強度を測定する。
なお,206.149 nmにおけるクロムの発光強度が共存元素の影響を受ける場合は,影響を受けない他の測
定波長を選定する。また,必要に応じてバックグラウンド補正を行う。他の測定波長の選定及び/又はバ
ックグラウンド補正が必要かどうかは,あらかじめ予備測定を行って決定する。
19.3.4 空試験
6.2.3.2 a)又はb)で得た空試験溶液を,19.3.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラス
コに移し入れる。以下,19.3.3.1 b)及び19.3.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行
う。
19.3.5 検量線の作成
クロム標準液(19.3.2.1)及び/又はクロム標準液(19.3.2.2)の各種液量(クロムとして0〜5 000 μg)
を段階的に数個の100 mLの全量フラスコに取る。以下,19.3.3.1 b)及び19.3.3.2の手順に従って,主溶液
と同じ操作を主溶液と並行して行い,得た発光強度とクロム量との関係線を作成し,その関係線を原点を
通るように平行移動して検量線とする。
19.3.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 19.3.3.2及び19.3.4で得た発光強度と19.3.5で
作成した検量線とからクロム量を求め,次の式によって,ヒューム中の全クロム含有率を算出する。
100
100
)
(
34
1
26
25
×
×
−
=
B
m
A
A
Cr
ここに,
Cr: ヒューム中の全クロム含有率[%(質量分率)]
A25: 試料溶液中のクロム検出量(mg)
A26: 空試験でのクロム検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B34: 19.3.3.1 a)及び19.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 19.3.3.2及び19.3.4で得た発光強度と19.3.5で作成
した検量線とからクロム量を求め,次の式によって,ヒューム中の全クロム含有率を算出する。
100
100
)
(
34
2
26
25
×
×
−
=
B
m
A
A
Cr
ここに,
Cr: ヒューム中の全クロム含有率[%(質量分率)]
A25: 試料溶液中のクロム検出量(mg)
A26: 空試験でのクロム検出量(mg)
m2: 6.2.3.1 b)ではかり取った分析試料の量(mg)
B34: 19.3.3.1 a)及び19.3.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
56
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
20 クロム(VI)定量方法
20.1 定量方法の区分
クロム(VI)の定量方法は,次のいずれかによる。
a) ジフェニルカルバジド吸光光度法 この方法は,試料溶液中の0.04 μg/mL以上1 μg/mL以下のクロム
(VI)の定量に適用する。
b) イオンクロマトグラフ分離ジフェニルカルバジド吸光光度法 この方法は,試料溶液中の0.002 μg/mL
以上0.8 μg/mL以下のクロム(VI)の定量に適用する。
c) フレーム原子吸光法 この方法は,試料溶液中の0.5 μg/mL以上5 μg/mL以下のクロム(VI)の定量
に適用する。
20.2 分析試料の前処理
20.2.1 水
水は,JIS K 0050の7.1(水及び試薬)に規定するA3の水とする。
なお,あらかじめ空試験を行って,使用しても支障がないことを確認しておく。
20.2.2 試薬
20.2.2.1 クロム(VI)抽出液
水酸化ナトリウム20 g及び炭酸ナトリウム30 gを水(20.2.1)に溶解し,水(20.2.1)で液量を1 000 mL
とする。この溶液は,使用の都度,窒素又はアルゴンを通じて30分間以上バブリングし,溶存酸素を除去
した後,使用する。
20.2.3 装置
装置は,次による。
20.2.3.1 超音波槽
超音波槽の媒体には,水を用いる。
20.2.4 主溶液の調製
主溶液の調製は,次のいずれかの手順によって行う。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合
1) 6.1.3 a) 3)又はb) 3)で得た分析試料をねじ蓋付きガラス容器(10〜30 mL)に入れ,クロム(VI)抽
出液(20.2.2.1)5 mLを加える。ヒュームがサンプラの内面に沈着・付着しているときは,クロム
(VI)抽出液(20.2.2.1)を用いてガラス容器中に洗い移す。
2) クロム(VI)抽出液(20.2.2.1)をフィルタが完全に沈むまで加える。必要ならば,フィルタがクロ
ム(VI)抽出液中に完全に沈むまでガラス棒で押し下げる。
3) ガラス容器をねじ蓋で閉じ,超音波槽(20.2.3.1)に入れ,槽の水位をガラス容器中の抽出液のレベ
ルを超えるように調節した後,超音波を1時間発振させる。
4) ガラス容器を超音波槽から取り出し,ねじ蓋を取り外し,ねじ蓋の内面及びろ過材を少量の水
(20.2.1)で洗浄し,溶液と洗液とを25 mLの全量フラスコに水(20.2.1)を用いて移し入れ,水(20.2.1)
で標線まで薄め,主溶液とする。
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 6.1.3 c) 1) 1.3)で得た分析試料約10 mgを正確に0.1
mgの桁まではかり取り,ねじ蓋付きガラス容器(10〜30 mL)に移し入れ,クロム(VI)抽出液(20.2.2.1)
5 mLを加える。以下,a) 3)及び4)の手順に従って操作する。
20.2.5 空試験溶液の調製
空試験溶液の調製は,次のいずれかの手順によって行う。
57
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 6.1.3 a)又はb)でヒュームの採取に用いたろ過
材と同じろ過材を分析試料の代わりに用いて,20.2.4 a) 1)〜4)の手順に従って,分析試料と同じ操作を
分析試料と並行して行い,得た溶液を空試験溶液とする。
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 分析試料を用いないで,20.2.4 b)の手順に従って,
分析試料と同じ操作を分析試料と並行して行い,得た溶液を空試験溶液とする。
20.3 ジフェニルカルバジド吸光光度法
20.3.1 要旨
分析試料中のクロム(VI)を水酸化ナトリウム・炭酸ナトリウム溶液で抽出し,硫酸を加えて酸性とし
た後,ジフェニルカルバジドを加えてジフェニルカルバジドクロム(VI)錯体を生成させ,光度計を用い
て,その吸光度を測定する。
20.3.2 水
20.2.1による。
20.3.3 試薬
20.3.3.1 硫酸(1+5,1+70)
20.3.3.2 ジフェニルカルバジド溶液
1,5-ジフェニルカルボノヒドラジド0.50 gをアセトン100 mLに溶解し,水(20.3.2)100 mLを加える。
この溶液は,使用の都度調製し,褐色瓶に保存する。
20.3.3.3 クロム(VI)標準液[Cr(VI):5.0 μg/mL]
二クロム酸カリウム(JIS K 8005に規定する容量分析用標準試薬)を正確に0.283 gはかり取り,水
(20.3.2)に溶解する。溶液を1 000 mLの全量フラスコに移し入れ,水(20.3.2)で標線まで薄めて原液
[Cr(VI):100 μg/mL]とする。この原液5.0 mLを,使用の都度,100 mLの全量フラスコに取り,水(20.3.2)
で標線まで薄めてクロム(VI)標準液とする。
20.3.3.4 クロム(VI)標準液[Cr(VI):1.0 μg/mL]
クロム(VI)標準液(20.3.3.3)20.0 mLを,使用の都度,100 mLの全量フラスコに取り,水(20.3.2)
で標線まで薄める。
20.3.4 操作
20.3.4.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 20.2.4 a) 4)又はb)で得た主溶液(主溶液中に不溶解物などがあるときは,その上澄液)を,クロム(VI)
量が1〜25 μgとなるように分取し,25 mLの全量フラスコに移し入れ,水(20.3.2)を加えて液量を
約20 mLとする。
b) 溶液をかるく振り混ぜながら,硫酸(1+5)を,二酸化炭素の発生(泡の発生)が認められなくなる
まで,滴加する。
c) ジフェニルカルバジド溶液(20.3.3.2)0.5 mLを加えた後,硫酸(1+70)を標線まで加え,振り混ぜ
る。
20.3.4.2 吸光度の測定
20.3.4.1 c)で得た溶液の一部を光度計の吸収セル(10 mm)に取り,水を対照液として波長540 nm付近
の吸光度を測定する。
20.3.5 空試験
20.2.5 a)又はb)で得た空試験溶液を,20.3.4.1 a)で分取した主溶液と同量分取し,25 mLの全量フラスコ
58
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に移し入れ,水(20.3.2)を加えて液量を約20 mLとする。以下,20.3.4.1 b)〜20.3.4.2の手順に従って,
主溶液と同じ操作を主溶液と並行して行う。
20.3.6 検量線の作成
クロム(VI)標準液(20.3.3.3)及びクロム(VI)標準液(20.3.3.4)の各種液量[クロム(VI)として0
〜25 μg]を段階的に数個の25 mLの全量フラスコに取り,水(20.3.2)を加えて液量を約20 mLとする。
以下,20.3.4.1 c)及び20.3.4.2の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得た吸光度と
クロム(VI)量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
20.3.7 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 20.3.4.2及び20.3.5で得た吸光度と20.3.6で作
成した検量線とからクロム(VI)量を求め,次の式によって,ヒューム中のクロム(VI)含有率を算
出する。
100
25
)
(
)
(
35
1
28
27
×
×
−
=
B
m
A
A
VI
Cr
ここに,
Cr(VI): ヒューム中のクロム(VI)含有率[%(質量分率)]
A27: 試料溶液中のクロム(VI)検出量(mg)
A28: 空試験でのクロム(VI)検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B35: 20.3.4.1 a)及び20.3.5でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 20.3.4.2及び20.3.5で得た吸光度と20.3.6で作成し
た検量線とからクロム(VI)量を求め,次の式によって,ヒューム中のクロム(VI)含有率を算出す
る。
100
25
)
(
)
(
35
4
28
27
×
×
−
=
B
m
A
A
VI
Cr
ここに,
Cr(VI): ヒューム中のクロム(VI)含有率[%(質量分率)]
A27: 試料溶液中のクロム(VI)検出量(mg)
A28: 空試験でのクロム(VI)検出量(mg)
m4: 20.2.4 b)ではかり取った分析試料の量(mg)
B35: 20.3.4.1 a)及び20.3.5でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
20.4 イオンクロマトグラフ分離ジフェニルカルバジド吸光光度法
20.4.1 要旨
分析試料中のクロム(VI)を水酸化ナトリウム・炭酸ナトリウム溶液で抽出する。溶液をイオンクロマ
トグラフに注入し,クロム(VI)を分離カラムで分離した後,混合器でジフェニルカルバジドと反応させ,
生成するジフェニルカルバジドクロム(VI)錯体を吸光度検出器に導き,その吸光度を測定する。
20.4.2 水
20.2.1による。
20.4.3 試薬
20.4.3.1 クロム(VI)抽出液
59
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
20.2.2.1による。
20.4.3.2 溶離液
硫酸アンモニウム264 gを水(20.4.2)500 mLに溶解し,アンモニア水65 mLを加えた後,溶液を1 000
mLの全量フラスコに移し入れ,水(20.4.2)で標線まで薄めて原液とする。この原液を使用の都度,必要
量だけ水(20.4.2)で正確に10倍に薄めて溶離液とする。
20.4.3.3 ジフェニルカルバジド溶液
1,5-ジフェニルカルボノヒドラジド0.25 gをメタノール50 mLに溶解し,硫酸(1+6)100 mLを加えた
後,水(20.4.2)を加えて液量を500 mLとする。この溶液は,使用の都度調製し,褐色瓶に保存する。
20.4.3.4 クロム(VI)標準液[Cr(VI):10 μg/mL]
二クロム酸カリウム(JIS K 8005に規定する容量分析用標準試薬)を正確に0.283 gはかり取り,水
(20.4.2)に溶解する。溶液を1 000 mLの全量フラスコに移し入れ,水(20.4.2)で標線まで薄めて原液
[Cr(VI):100 μg/mL]とする。この原液10.0 mLを,使用の都度,100 mLの全量フラスコに取り,水(20.4.2)
で標線まで薄めてクロム(VI)標準液とする。
20.4.3.5 クロム(VI)標準液[Cr(VI):1.0 μg/mL]
クロム(VI)標準液(20.4.3.4)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,水(20.4.2)
で標線まで薄める。
20.4.3.6 クロム(VI)標準液[Cr(VI):0.1 μg/mL]
クロム(VI)標準液(20.4.3.5)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,水(20.4.2)
で標線まで薄める。
20.4.4 装置
20.4.4.1 イオンクロマトグラフ
イオンクロマトグラフは,JIS K 0127による。イオンクロマトグラフは,通常,溶離液槽,ジフェニル
カルバジド溶液槽,送液ポンプ,インジェクタ,保護カラム,分離カラム,混合器,分光光度検出器など
で構成し,それぞれ次による。イオンクロマトグラフの構成の例を,図1に示す。
a) 溶離液槽及びジフェニルカルバジド溶液槽 ガラス又はポリエチレン製のねじ口瓶で,ポリエチレン
管を用いて送液ポンプと結合する。
b) 送液ポンプ 溶離液及びジフェニルカルバジド溶液を所定の流量でカラムに送るためのポンプで,溶
離液及びジフェニルカルバジド溶液と接触する部分に金属材料を用いていないもの。
c) インジェクタ JIS K 0127の4.2 c)[試料導入部(インジェクタ)]の1)に規定する構造のもので,試
料溶液,溶離液及びジフェニルカルバジド溶液と接触する部分に金属材料を用いてなく,かつ,サン
プルループの容積が約100 μLのもの。
d) 保護カラム 分離カラムと同じ充塡剤を充塡したもの。
e) 分離カラム 充塡剤としてpH14まで使用可能な高容量陰イオン交換樹脂を充塡したもの。
f)
混合器 内径約0.1 mm,長さ約1 mのポリエタノールエーテルケトン樹脂管,ポリエチレン管などを
用いる。
なお,溶液の混合を促進するために,管内部に乱流を発生させる突起などを設けたり,ビーズなど
を入れるなどした管を用いる場合には,長さ1 m以下の管を用いてもよい。
g) 分光光度検出器 光度計,積分器,コンピュータ,記録計などで構成する。光度計の吸光度測定部(吸
収セル)は,通常,光路長が10 mmのものを用いる。
60
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1 溶離液槽 5 保護カラム
2 ジフェニルカルバジド溶液槽 6 分離カラム
3 送液ポンプ 7 混合器
4 インジェクタ 8 分光光度検出器
図1−イオンクロマトグラフの構成の例
20.4.5 操作
20.4.5.1 試料溶液の調製
20.2.4 a) 4)又はb)で得た主溶液(主溶液中に不溶解物などがあるときは,その上澄液)を,クロム量が
0.2〜80 μgになるように分取し,100 mLの全量フラスコに移し入れ,水(20.4.2)で標線まで薄める。
20.4.5.2 クロム(VI)の分離及び吸光度の測定
クロム(VI)の分離及び吸光度の測定は,次の手順によって行う。
a) イオンクロマトグラフ(20.4.4.1)を稼働させ,分光光度検出器,溶離液(20.4.3.2)の流量,ジフェニ
ルカルバジド溶液(20.4.3.3)の流量,流出液のpHなどを調節し,装置を安定した状態にする。
なお,溶離液及びジフェニルカルバジド溶液の総流量は,分光光度検出器の最大許容流量以下,溶
離液とジフェニルカルバジド溶液との流量比は,通常,3:1に保持し,流出液のpHは,2以下とす
る。
注記 流出液のpHを2以下に調節するには,必要ならば,溶離液とジフェニルカルバジド溶液と
の流量比又はジフェニルカルバジド溶液の硫酸濃度を調節するとよい。
b) 20.4.5.1で得た試料溶液の一部(装置で指定されている量)を,イオンクロマトグラフのインジェクタ
に注入する。
c) 波長543 nm付近における分光光度検出器の出力信号を測定・記録し,吸光度に対応した出力信号の
ピーク高さ又はピーク面積を測定する。
20.4.6 空試験
20.2.5 a)又はb)で得た空試験溶液を,20.4.5.1で分取した主溶液と同量分取し,100 mLの全量フラスコ
に移し入れ,水(20.4.2)で標線まで薄める。この溶液の一部[20.4.5.2 b)でイオンクロマトグラフ装置に
注入した試料溶液と同量]を,イオンクロマトグラフ装置のインジェクタに注入し,20.4.5.2 c)の操作を試
料と並行して行う。
20.4.7 検量線の作成
検量線の作成は,次の手順によって行う。
61
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) クロム(VI)抽出液(20.4.3.1)を,20.2.4 a)又はb)で主溶液の調製に用いた量と同量取って25 mLの
全量フラスコに入れ,水(20.4.2)で標線まで薄める。この溶液を,20.4.5.1で分取した主溶液と同量
ずつ数個の100 mLの全量フラスコに取り,クロム(VI)標準液(20.4.3.4),クロム(VI)標準液(20.4.3.5)
及びクロム(VI)標準液(20.4.3.6)の各種液量(クロムとして0〜80 μg)を段階的に加え,水(20.4.2)
で標線まで薄める。
b) 各溶液の一部[20.4.5.2 b)でイオンクロマトグラフ装置に注入した試料溶液と同量]を,イオンクロマ
トグラフ装置のインクジェッタに注入し,20.4.5.2 c)の操作を試料溶液と並行して行い,得た出力信号
のピーク高さ又はピーク面積とクロム(VI)量との関係線を作成し,その関係線を原点を通るように
平行移動して,検量線とする。
20.4.8 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 20.4.5.2 c)及び20.4.6で得た出力信号のピーク
高さ又はピーク面積と20.4.7で作成した検量線とからクロム(VI)量を求め,次の式によって,ヒュ
ーム中のクロム(VI)含有率を算出する。
100
25
)
(
)
(
36
1
28
27
×
×
−
=
B
m
A
A
VI
Cr
ここに,
Cr(VI): ヒューム中のクロム(VI)含有率[%(質量分率)]
A27: 試料溶液中のクロム(VI)検出量(mg)
A28: 空試験でのクロム(VI)検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B36: 20.4.5.1及び20.4.6でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 20.4.5.2 c)及び20.4.6で得た出力信号のピーク高さ
又はピーク面積と20.4.7で作成した検量線とからクロム(VI)量を求め,次の式によって,ヒューム
中のクロム(VI)含有率を算出する。
100
25
)
(
)
(
36
4
28
27
×
×
−
=
B
m
A
A
VI
Cr
ここに,
Cr(VI): ヒューム中のクロム(VI)含有率[%(質量分率)]
A27: 試料溶液中のクロム(VI)検出量(mg)
A28: 空試験でのクロム(VI)検出量(mg)
m4: 20.2.4 b)ではかり取った分析試料の量(mg)
B36: 20.4.5.1及び20.4.6でそれぞれ分取した主溶液及び
空試験溶液の量(mL)
20.5 フレーム原子吸光法
20.5.1 要旨
分析試料中のクロム(VI)を水酸化ナトリウム・炭酸ナトリウム溶液で抽出し,溶液を原子吸光光度計
の一酸化二窒素・アセチレンフレーム又は空気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
62
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
20.5.2 試薬
20.5.2.1 希釈液
クロム(VI)抽出液(20.2.2.1)200 mLを水で薄めて,液量を1 000 mLとする。
20.5.2.2 クロム(VI)標準液[Cr(VI):100 μg/mL]
二クロム酸カリウム(JIS K 8005に規定する容量分析用標準試薬)を正確に0.283 gはかり取り,水に溶
解する。溶液を100 mLの全量フラスコに移し入れ,水で標線まで薄めて原液[Cr(VI):1 000 μg/mL]と
する。この原液10.0 mLを,使用の都度,100 mLの全量フラスコに取り,クロム(VI)抽出液(20.2.2.1)
20 mLを加えた後,水で標線まで薄めてクロム(VI)標準液とする。
20.5.2.3 クロム(VI)標準液[Cr(VI):10 μg/mL]
クロム(VI)標準液(20.5.2.2)10.0 mLを,使用の都度,100 mLの全量フラスコに取り,希釈液(20.5.2.1)
で標線まで薄める。
20.5.3 操作
20.5.3.1 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 20.2.4 a) 4)又はb)で得た主溶液(主溶液中に不溶解物などがあるときは,その上澄液)を,クロム(VI)
量が50〜500 μgになるように分取し,100 mLの全量フラスコに移し入れる。
b) 希釈液(20.5.2.1)を加えて標線まで薄める。
20.5.3.2 吸光度の測定
20.5.3.1 b)で得た溶液の一部を,水でゼロ点を調整した一酸化二窒素・アセチレンフレーム又は空気・ア
セチレンフレーム中に噴霧し,波長357.9 nmにおける吸光度を測定する。
20.5.4 空試験
20.2.5 a)又はb)で得た空試験溶液を,20.5.3.1 a)で分取した主溶液と同量分取し,100 mLの全量フラスコ
に移し入れる。以下,20.5.3.1 b)及び20.5.3.2の手順に従って,主溶液と同じ操作を主溶液と並行して行う。
20.5.5 検量線の作成
クロム(VI)標準液(20.5.2.2)及びクロム(VI)標準液(20.5.2.3)の各種液量(クロムとして0〜500 μg)
を段階的に数個の100 mLの全量フラスコに取る。以下,20.5.3.1 b)及び20.5.3.2の手順に従って,主溶液
と同じ操作を主溶液と並行して行い,得た吸光度とクロム(VI)量との関係線を作成し,その関係線を原
点を通るように平行移動して検量線とする。
20.5.6 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 20.5.3.2及び20.5.4で得た吸光度と20.5.5で作
成した検量線とからクロム(VI)量を求め,次の式によって,ヒューム中のクロム(VI)含有率を算
出する。
100
25
)
(
)
(
37
1
28
27
×
×
−
=
B
m
A
A
VI
Cr
ここに,
Cr(VI): ヒューム中のクロム(VI)含有率[%(質量分率)]
A27: 試料溶液中のクロム(VI)検出量(mg)
A28: 空試験でのクロム(VI)検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B37: 20.5.3.1 a)及び20.5.4でそれぞれ分取した主溶液及
63
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 20.5.3.2及び20.5.4で得た吸光度と20.5.5で作成し
た検量線とからクロム(VI)量を求め,次の式によって,ヒューム中のクロム(VI)含有率を算出す
る。
100
25
)
(
)
(
37
4
28
27
×
×
−
=
B
m
A
A
VI
Cr
ここに,
Cr(VI): ヒューム中のクロム(VI)含有率[%(質量分率)]
A27: 試料溶液中のクロム(VI)検出量(mg)
A28: 空試験でのクロム(VI)検出量(mg)
m4: 20.2.4 b)ではかり取った分析試料の量(mg)
B37: 20.5.3.1 a)及び20.5.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
21 ふっ素定量方法
21.1 定量方法
ふっ素の定量方法は,熱加水分解分離ランタンアリザリンコンプレキソン吸光光度法による。この方法
は,試料溶液中の0.1 μg/mL以上1 μg/mL以下のふっ素の定量に適用する。
21.2 熱加水分解分離ランタンアリザリンコンプレキソン吸光光度法
21.2.1 要旨
分析試料に銅及び二酸化けい素を加え,水蒸気・酸素気流中で強熱し,発生するけいふっ化水素を水酸
化ナトリウムに吸収させて捕集する。溶液のpHを調節した後,アセトン及びランタンアリザリンコンプ
レキソンを加えてランタンアリザリンコンプレキソンふっ素錯体を生成させ,光度計を用いて,その吸光
度を測定する。
21.2.2 試薬
21.2.2.1 水酸化ナトリウム溶液
水酸化ナトリウム4.0 gを水に溶解し,水で液量を1 000 mLとし,ポリエチレン瓶に入れて保存する。
この溶液は,使用の都度調製することが望ましい。
21.2.2.2 銅
99 %(質量分率)以上で,径が1〜5 mmの薄い切削片状のもの又は1〜3 mmの粒状のもの。
21.2.2.3 酸素
99.5 %(体積分率)以上の酸素を使用する。
21.2.2.4 二酸化けい素
98 %(質量分率)以上で,粒径が150 μm以下のもの。
21.2.2.5 緩衝溶液
酢酸ナトリウム三水和物100 gを水100 mLに溶解し,酢酸11 mLを加えてかき混ぜる。酢酸を滴加して
溶液のpHを5.2に調節した後,水で液量を1 000 mLとする。
21.2.2.6 ランタンアリザリンコンプレキソン溶液
ランタンアリザリンコンプレキソン3.0 gを水に溶解し,水で液量を100 mLとする。この溶液は,使用
の都度調製する。
64
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
21.2.2.7 アセトン
21.2.2.8 ふっ素標準液(F:10 μg/mL)
ふっ化ナトリウムを白金皿に取り,500〜550 ℃で40〜50分間加熱し,デシケータ中で常温まで放冷し
た後,その2.21 gをはかり取り,水に溶解する。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,
水で標線まで薄めて原液(F:1 000 μg/mL)とし,ポリエチレン瓶に入れて保存する。この原液を使用の
都度,必要量だけ水で正確に100倍に薄めてふっ素標準液とする。
21.2.3 装置及び器具
21.2.3.1 熱加水分解装置
熱加水分解装置は,酸素導入部,水蒸気発生部,反応部及びふっ素化合物吸収部で構成する。図2に熱
加水分解装置の構成の例を示す。
なお,図2は,各部の連結の要領を示すもので,各器具などの形状は,この規格の規定に抵触しない限
り,適宜選択してもよい。
21.2.4 操作
21.2.4.1 分析試料の前処理
21.2.4.1.1 主溶液の調製
主溶液の調製は,次のいずれかの手順によって行う。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合
1) 熱加水分解装置(21.2.3.1)を組み立てる(図2参照)。
2) ヒーター及び管状電気抵抗加熱炉の電源を入れ,水蒸気発生フラスコ中の水を穏やかに沸騰させ,
反応管内中央部の温度が1 400±50 ℃に保持されるように調節する。捕集器に水70 mLを入れ,捕
集補助フラスコに水30 mLを入れた後,捕集器に水酸化ナトリウム溶液(21.2.2.1)2.0 mLを加え
て振り混ぜる。
3) 6.1.3 a) 3),b) 3)又はc) 2) 2.3)で得た分析試料をアルミナボートに入れ,銅(21.2.2.2)1 g及び二酸
化けい素(21.2.2.4)0.5 gを分析試料を覆うようにして加えた後,反応管の中央部に挿入し,直ちに
シリコーンゴム栓をして酸素(21.2.2.3)を流量150〜200 mL/minで20分間流す。
なお,分析試料は,必要ならば,セラミック製はさみを用いてろ過材を切断して小片とした後,
アルミナボートに入れる。
4) 捕集器及び捕集補助フラスコ中の溶液を,常温まで冷却した後,200 mLの全量フラスコに水を用い
て移し入れ,水で標線まで薄め,主溶液とする。
65
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1 流量計
2 ゴム管
3 逆流防止瓶
4 ヒーター
5 水蒸気発生フラスコ(1 L)
6 水滴除去用フラスコ
7 シリコーンゴム栓
8 反応管:内径25 mm,長さ650 mmの石英管
9 管状電気抵抗加熱炉:長さ300〜350 mm。炉端から反応管を
60〜80 mm突き出し,管の先端に石英製キャップをはめ,こ
れをばねで炉壁に固定する。
10 指示熱電温度計
11 アルミナボート:長さ100 mm,外幅20 mm,高さ10 mm
12 石英製キャップ
13 捕集器:容積約130 mLの球を2個連結したもの
14 捕集補助フラスコ:三角フラスコ(500 mL)
図2−熱加水分解装置の構成の例
b) ヒュームの採取を6.1.3 c) 1)によって行った場合
1) a) 1)及びa) 2)の手順に従って操作する。
2) 6.1.3 c) 1) 1.3)で得た分析試料約10 mgを正確に0.1 mgの桁まではかり取り,アルミナボートに入れ,
銅(21.2.2.2)1 g及び二酸化けい素(21.2.2.4)0.5 gを分析試料を覆うようにして加えた後,反応管
の中央部に挿入し,直ちにシリコーンゴム栓をして酸素を流量150〜200 mL/minで20分間流す。
3) a) 4)の操作を行う。
21.2.4.1.2 空試験溶液の調製
空試験溶液の調製は,次のいずれかの手順によって行う。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合 6.1.3 a),b)又はc) 2)でヒュームの採取に
用いたろ過材と同じろ過材を分析試料の代わりに用いて,21.2.4.1.1 a) 2)及び3)の手順に従って,分析
試料と同じ操作を分析試料と並行して行い,得た溶液を空試験溶液とする。
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 分析試料を用いないで,21.2.4.1.1 b) 2)及び3)の手
順に従って,分析試料と同じ操作を分析試料と並行して行い,得た溶液を空試験溶液とする。
21.2.4.2 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 21.2.4.1.1 a) 4)又はb) 3)で得た主溶液を,ふっ素量が10〜100 μgとなるように分取し,100 mLの全量
フラスコに移し入れる。
b) 緩衝溶液(21.2.2.5)10 mL,アセトン35 mL及びランタンアリザリンコンプレキソン溶液(21.2.2.6)
10.0 mLを順次加え,水で標線まで薄めた後,60分間放置する。
21.2.4.3 吸光度の測定
21.2.4.2 b)で得た溶液の一部を光度計の吸収セル(10 mm)に取り,水を対照液として波長620 nm付近
の吸光度を測定する。
66
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
21.2.5 空試験
21.2.4.1.2 a)又はb)で得た空試験溶液を,21.2.4.2 a)で分取した主溶液と同量分取し,100 mLの全量フラ
スコに移し入れる。以下,21.2.4.2 b)及び21.2.4.3の手順に従って,主溶液と同じ操作を主溶液と並行して
行う。
21.2.6 検量線の作成
ふっ素標準液(21.2.2.8)0〜10.0 mL(ふっ素として0〜100 μg)を段階的に数個の100 mLの全量フラス
コに取り,以下,21.2.4.2 b)及び21.2.4.3の手順に従って,主溶液と同じ操作を主溶液と並行して行い,得
た吸光度とふっ素量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
21.2.7 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a),b)又はc) 2)によって行った場合 21.2.4.3及び21.2.5で得た吸光度と21.2.6
で作成した検量線とからふっ素量を求め,次の式によって,ヒューム中のふっ素含有率を算出する。
100
200
)
(
38
3
30
29
×
×
−
=
B
m
A
A
F
ここに,
F: ヒューム中のふっ素含有率[%(質量分率)]
A29: 試料溶液中のふっ素検出量(mg)
A30: 空試験でのふっ素検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B38: 21.2.4.2 a)及び21.2.5でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 21.2.4.3及び21.2.5で得た吸光度と21.2.6で作成し
た検量線とからふっ素量を求め,次の式によって,ヒューム中のふっ素含有率を算出する。
100
200
)
(
38
5
30
29
×
×
−
=
B
m
A
A
F
ここに,
F: ヒューム中のふっ素含有率[%(質量分率)]
A29: 試料溶液中のふっ素検出量(mg)
A30: 空試験でのふっ素検出量(mg)
m5: 21.2.4.1.1 b) 2)ではかり取った分析試料の量(mg)
B38: 21.2.4.2 a)及び21.2.5でそれぞれ分取した主溶液及
び空試験溶液の量(mL)
22 全けい素定量方法
22.1 定量方法
全けい素定量方法は,モリブドバナドりん酸吸光光度法による。この方法は,試料溶液中の0.04 μg/mL
以上3 μg/mL以下のけい素の定量に適用する。
22.2 モリブドけい酸青吸光光度法
22.2.1 要旨
試料に炭酸ナトリウム及びほう酸を加え,加熱して融解した後,融成物を塩酸に溶解する。溶液に七モ
リブデン酸六アンモニウムを加え,けい酸をモリブドけい酸とする。酒石酸アンモニウムを加え,りん,
67
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ひ素などをマスキングした後,アスコルビン酸でモリブドけい酸を還元してモリブドけい酸青を生成させ,
光度計を用いて,その吸光度を測定する。
22.2.2 水
水は,蒸留水を用いる。
22.2.3 試薬
22.2.3.1 塩酸
22.2.3.2 塩酸(1+5)
22.2.3.3 過酸化水素(3+97)
22.2.3.4 融解合剤[炭酸ナトリウム(無水)2,ほう酸1]
22.2.3.5 過マンガン酸カリウム溶液(飽和)
22.2.3.6 亜硝酸ナトリウム溶液(20 g/L)
22.2.3.7 モリブデン酸アンモニウム溶液
七モリブデン酸六アンモニウム四水和物100 gを温水(22.2.2)に溶解し,常温まで冷却した後,水(22.2.2)
で液量を1 000 mLとする。溶液は,ポリエチレン瓶に保存し,使用の都度,乾いたろ紙でろ過して用いる。
22.2.3.8 希釈溶液
融解合剤3.0 gを白金るつぼに入れ,約500 ℃で約5分間加熱し,次いで約1 100 ℃で約10 分間加熱し
て融解する。放冷した後,るつぼを熱水(22.2.2)100 mL及び塩酸10 mLを入れたポリエチレンビーカー
(200 mL)中に入れ,水浴上で穏やかに加熱して融成物を溶解し,るつぼを水(22.2.2)で洗ってビーカ
ーから取り出す。常温まで冷却した後,溶液を200 mLのポリエチレン製全量フラスコに水(22.2.2)を用
いて移し入れ,水(22.2.2)で標線まで薄める。
22.2.3.9 アスコルビン酸
22.2.3.10 酒石酸アンモニウム溶液(100 g/L)
22.2.3.11 けい素標準液(Si:50 μg/mL)
白金るつぼ中で1 000 ℃に強熱し,デシケータ中で室温まで放冷した二酸化けい素(99.95 %以上)0.428
gを白金るつぼ(25番)にはかり取り,炭酸ナトリウム(無水)2.5 gを加え,よく混合した後,加熱して
融解する。放冷した後,白金るつぼを温水(22.2.2)100 mLを入れた白金皿(150番)又はポリエチレン
ビーカー(200 mL)中に浸し,水浴上で穏やかに加熱して融成物を溶解する。常温まで放冷した後,白金
るつぼを水(22.2.2)で洗って取り出し,溶液を1 000 mLの全量フラスコに水(22.2.2)を用いて移し入れ,
水(22.2.2)で標線まで薄めて原液(Si:200 μg/mL)とする。この原液は,ポリエチレン瓶に移し入れて
保存する。この原液25.0 mLを,使用の都度,ポリエチレンビーカー(100 mL)に取り,pH計を用い,塩
酸(1+5)を滴加してpHを5〜7に調節した後,溶液を100 mLの全量フラスコに水(22.2.2)を用いて移
し入れ,水(22.2.2)で標線まで薄めてけい素標準液とする。
22.2.3.12 けい素標準液(Si:2.0 μg/mL)
けい素標準液(22.2.3.11)4.0 mLを,使用の都度,100 mLの全量フラスコに取り,水(22.2.2)で標線
まで薄める。
22.2.4 操作
22.2.4.1 分析試料の前処理
分析試料の前処理は,次のいずれかの手順によって行う。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合
1) 6.1.3 a) 3)又はb) 3)で得た分析試料を白金るつぼ(25番)に入れ,低温で加熱してろ過材を灰化し
68
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
た後,室温まで放冷する。
2) 白金るつぼに融解合剤1.5 gを加え,ヒュームとよく混合した後,約500 ℃で約5分間加熱し,次
いで約1 100 ℃で約10分間加熱してヒュームを融解する。
3) 室温まで放冷した後,白金るつぼを熱水(22.2.2)約50 mL及び塩酸5 mLを入れたポリエチレンビ
ーカー(200 mL)中に入れ,水浴上で穏やかに加熱して融成物を溶解し,るつぼを水(22.2.2)で
洗ってビーカーから取り出す。
なお,溶液が濁っているときは,過酸化水素(3+97)を数滴加え,水浴上で加熱して未溶解物を
溶解した後,溶液の色が僅かに赤紫を呈するまで過マンガン酸カリウム溶液を滴加し,次いで,溶
液の赤紫色が消えるまで亜硝酸ナトリウム溶液を滴加する。
4) 常温まで冷却した後,溶液を100 mLのポリエチレン製全量フラスコに水(22.2.2)を用いて移し入
れ,水(22.2.2)で標線まで薄めて主溶液とする。
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 6.1.3 c) 1) 1.3)で得た分析試料約10 mgを正確に0.1
mgの桁まではかり取り,白金るつぼ(25番)に移し入れる。融解合剤1.5 gを加え,分析試料とよく
混合した後,約500 ℃で約5分間加熱し,次いで約1 100 ℃で約10分間加熱して分析試料を融解す
る。以下,a) 3)及び4)の手順に従って操作する。
22.2.4.2 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 22.2.4.1 a) 4)又はb)で得た主溶液を,けい素量が4〜300 μgになるように,同量ずつ2個の100 mLの
全量フラスコに分取し,水(22.2.2)を加えて液量を約70 mLとする。
なお,主溶液の分取量は,20 mL以下とする。分取量が10 mL未満の場合は,希釈溶液(22.2.3.8)
を加えて10.0 mLとした後,水(22.2.2)を加えて液量を70 mLとする。
b) 第1の全量フラスコだけにモリブデン酸アンモニウム溶液(22.2.3.7)4.0 mLを加え,少量の水(22.2.2)
で全量フラスコの内壁を洗浄し,よく振り混ぜた後,20〜30 ℃で20分間放置する。
c) 第1及び第2の全量フラスコに酒石酸アンモニウム溶液8.0 mLを加え,かるく振り混ぜ,30秒以内
にアスコルビン酸約0.2 gを加え,よく振り混ぜた後,水(22.2.2)で標線まで薄め,10分間放置する。
22.2.4.3 吸光度の測定
22.2.4.2 c)で得た第1及び第2の全量フラスコの溶液の一部をそれぞれ光度計の吸収セル(10 mm)に取
り,第2の全量フラスコの溶液を対照液として,波長810 nm付近又は660 nm付近における第1の全量フ
ラスコの溶液の吸光度を測定する。
なお,吸光度の測定は,22.2.4.2 c)の操作終了後60分以内に行う。
注記 試料溶液中のけい素濃度が0.04〜1 μg/mLの場合には,波長810 nm付近で,0.5〜3 μg/mLの場
合には,波長660 nm付近で吸光度を測定するとよい。
22.2.5 空試験
希釈溶液(22.2.3.8)を,22.2.4.2 a)における主溶液の分取量と同量(ただし,主溶液の分取量が10 mL
未満の場合には,10.0 mL)ずつ2個の100 mLの全量フラスコに取り,水(22.2.2)を加えて液量を約70 mL
とする。以下,22.2.4.2 b)〜22.2.4.3の手順に従って主溶液と同じ操作を主溶液と並行して行う。
22.2.6 検量線の作成
検量線の作成は,次の手順によって行う。
a) けい素標準液(22.2.3.11)及び/又はけい素標準液(22.2.3.12)の各種液量(けい素として4〜300 μg)
を段階的に数個の100 mLの全量フラスコに取り,そのそれぞれに希釈溶液(22.2.3.8)10.0 mLを加
69
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
え,水(22.2.2)で液量を約70 mLとする。
b) モリブデン酸アンモニウム溶液(22.2.3.7)4.0 mLを加え,少量の水(22.2.2)で全量フラスコの内壁
を洗浄し,よく振り混ぜた後,20〜30 ℃で20分間放置する。
c) 酒石酸アンモニウム溶液8.0 mLを加え,かるく振り混ぜ,30秒以内にアスコルビン酸約0.2 gを加え,
よく振り混ぜた後,水(22.2.2)で標線まで薄め,10分間放置する。
d) 各溶液の一部を光度計の吸収セル(10 mm)に取り,水(22.2.2)を対照液として,22.2.4.3と同一の
測定波長における吸光度を試料溶液と並行して測定し,得た吸光度とけい素量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
なお,吸光度の測定は,c)の操作終了後60分以内に行う。
22.2.7 計算
計算は,次のいずれかによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 22.2.4.3及び22.2.5で得た吸光度と22.2.6で作
成した検量線とからけい素量を求め,次の式によって,ヒューム中の全けい素含有率を算出する。
100
100
)
(
39
1
32
31
×
×
−
=
B
m
A
A
Si
ここに,
Si: ヒューム中の全けい素含有率[%(質量分率)]
A31: 試料溶液中のけい素検出量(mg)
A32: 空試験でのけい素検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
B39: 22.2.4.2 a)で分取した主溶液の量(mL)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 22.2.4.3及び22.2.5で得た吸光度と22.2.6で作成し
た検量線とからけい素量を求め,次の式によって,ヒューム中の全けい素含有率を算出する。
100
100
)
(
39
6
32
31
×
×
−
=
B
m
A
A
Si
ここに,
Si: ヒューム中の全けい素含有率[%(質量分率)]
A31: 試料溶液中のけい素検出量(mg)
A32: 空試験でのけい素検出量(mg)
m6: 22.2.4.1 b)ではかり取った分析試料の量(mg)
B39: 22.2.4.2 a)で分取した主溶液の量(mL)
23 結晶質シリカ定量方法
23.1 定量方法
結晶質シリカの定量方法は,X線回折法(基底標準吸収補正法)による。この方法は,メンブランフィ
ルタ上の10 μg/cm2以上1 000 μg/cm2以下の結晶質シリカの定量に適用する。
注記 結晶質シリカとは,石英,クリストバライト及びトリディマイトをいう。
23.2 X線回折法(基底標準吸収補正法)
23.2.1 要旨
分析試料に硝酸又は塩酸を加え,分析試料中の金属粒子を分解した後,残さをこし分けて測定試料とす
る。測定試料をX線回折分析装置の試料台に固定し,測定試料中の結晶質シリカの回折線強度を測定する。
70
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
23.2.2 試薬
23.2.2.1 塩酸(1+1,1+100)
23.2.2.2 硝酸(1+1,1+100)
23.2.2.3 イソプロピルアルコール
23.2.3 装置及び材料
装置及び材料は,次による。
23.2.3.1 X線回折分析装置
X線管球の電圧・電流を約40 kV・40 mAに設定できる普及型の装置又はそれ以上の性能をもつ装置を用
いる。
23.2.3.2 基底標準金属板
X線回折線の吸収補正に用いるもので,金属板の金属には,通常,亜鉛を用いる。
注記 基底標準金属板には,メンブランフィルタの固定専用の金属基底標準試料台が市販されている。
23.2.3.3 超音波洗浄器
23.2.3.4 吸引ろ過器
メンブランフィルタをろ過器に装着したとき,メンブランフィルタの有効ろ過面積が2 cm2となるもの
を用いる。
23.2.3.5 メンブランフィルタ
孔径が0.4 μm以下のポリカーボネート製又は孔径が1.2 μm以下のアセチルアセテート製のものを用い
る。
23.2.3.6 標準物質
結晶質シリカの含有率が既知の石英,クリストバライト及びトリディマイトを用いる。
注記 結晶質シリカの標準物質としては ,現在,表3に示すようなものがある。ただし,この表は,
この規格の利用者の便宜を図って記載するもので,これらの標準物質を推奨するものではない。
同じ結果が得られる場合は,これと同等の他の標準物質を使用してもよい。
なお,表3に示す標準物質で,結晶質シリカの吸入性粉じんとして頒布されている標準物質
は,米国NISTのものである。その他は,吸入性粉じんの粒径の粒子だけから成る標準物質で
はない。例えば,社団法人日本作業環境測定協会の標準石英試料JAWE455は,10 μm付近の粗
い粒子が比較的多く,そのまま定量分析に使用する場合とその中から吸入性粒子を抽出して吸
入性石英試料として使用する場合とが考えられる。後者の吸入性石英試料として使用する場合
は,標準試料をダストボックスの中で発じんさせて,所定の分粒装置を通過した吸入性粒子を
捕集して作製するなどの必要がある。
71
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3−結晶質シリカの標準物質の例
標準物質名
種類
粒径
発行国(機関,会社名)
用途,ほか
JAWE455
石英
<10 μm
日本(日本作業環境測定
協会)
りん酸法用,X線回折定量用
DQ12
石英
<5 μm
ドイツ
石英砂(非晶質を含む,石英含有率=
87 %)
MIN-U-SIL5 石英
<5 μm
米国(US Silica社,ほか) 石英砂,X線回折定量用,動物実験用
CSQZ
石英
吸入性
中国
SRM1878a
石英
1.6 μm(MMD) 米国(NIST)
X線回折定量用
SRM2679a
石英
1.6 μm(平均) 米国(NIST)
フィルタ試料(マトリックス:粘土)
SIKRON
F600
石英
ドイツ(Quartzwerke社) 石英砂
CB-25
クリストバライト 4.1 μm(MMD) 米国(NIOSH)
合成品,X線回折定量用
SRM1879a
クリストバライト 3.5 μm(MMD) 米国(NIST)
合成品,X線回折定量用
TY-27
トリディマイト
3.9 μm(MMD) 米国(NIOSH)
合成品,X線回折定量用
MMD:質量基準メディアン径
23.2.4 操作
23.2.4.1 分析試料の前処理
分析試料の前処理は,次のいずれかの手順によって行う。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合
1) 6.1.3 a) 3)又はb) 3)で得た分析試料を,硝酸(1+1)又は塩酸(1+1)50 mLを入れたコニカルビー
カー(100 mL)中に入れる。
2) 穏やかに約1時間加熱して分析試料に含まれる金属粒子を分解した後,室温まで放冷する。
なお,金属粒子は,できるだけ分解することが望ましいが,完全に分解しなくてもよい。
3) コニカルビーカーを超音波洗浄器に入れて約1分間超音波処理を行った後,水,塩酸(1+100)又
は硝酸(1+100)を用いてろ過材表面を洗浄し,ろ過材に付着している粒子を完全にコニカルビー
カー内に洗い落とし,ろ過材をピンセットを用いてコニカルビーカーから取り出す。
4) 溶液中の不溶解物を,吸引ろ過器(23.2.3.4)に装着した直径25 mmのメンブランフィルタ(23.2.3.5)
を用いて吸引しながらこし分け,コニカルビーカーの内壁に付着している不溶解物を水でメンブラ
ンフィルタ上に完全に洗い移し,水で数回メンブランフィルタを洗浄した後,メンブランフィルタ
をろ過器から取り外し,取り外したままの状態で風乾して測定試料とする。
b) ヒュームの採取を6.1.3 c) 1)によって行った場合
1) 6.1.3 c) 1) 1.3)で得た分析試料1〜10 mgを正確に0.1 mgの桁まではかり取り,硝酸(1+1)又は塩
酸(1+1)50 mLを入れたコニカルビーカー(100 mL)中に入れる。
2) a) 2)の操作を行う。
3) a) 4)の操作を行う。
23.2.4.2 回折線強度の測定
回折線強度の測定は,次の手順によって行う。
a) 23.2.4.1 a) 4)又はb) 3)で得た測定試料を,X線回折分析装置(23.2.3.1)の試料台に装着した基底標準
金属板(23.2.3.2)に固定する。
b) 測定試料中の結晶質シリカ及び基底標準金属板の金属の回折線強度を,所定の回折角度(2θ)でそれ
ぞれ測定する。
72
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記 一次X線にCuKa線を用いた場合,石英の第1回折線は2θで26.6°,第2回折線は20.8°に出
現する。クリストバライトの第1回折線は,22.1°に出現するが,その他の回折線はかなり弱
く,実際には定量用回折線として使用が難しい。トリディマイトは,20.6°,21.6°,23.3°付
近に強い回折線が出現する。また,基底標準金属板に通常使用する亜鉛の回折線は,2θで43.2°
に出現する回折線を使用するとよい。
c) メンブランフィルタ(23.2.3.5)を,X線回折分析装置の試料台に装着した基底標準金属板(23.2.3.2)
に固定し,基底標準金属板の金属の回折線強度を,b)と同一条件で測定する。
d) 吸収補正係数を,次の式によって算出する。
θ
θ
R
T
T
R
K
−
−
=1
ln
f
ここに,
Kf: 吸収補正係数
Rθ: 基底標準金属板の金属の回折角(sinθm)と結晶質
シリカの回折角(sinθs)との比(sinθm/sinθs)
ln: 自然対数
T: 回折線強度の減少率(Im/I 0m)
Im: b)で得た基底標準金属板の金属の回折線強度測定
値
I 0m: c)で得た基底標準金属板の金属の回折線強度測定
値
e) 測定試料中の結晶質シリカの吸収補正回折線強度を,次の式によって算出する。
f
sK
I
I
×
=
ここに,
I: 測定試料中の結晶質シリカの吸収補正回折線強度
Is: b)で得た結晶質シリカの回折線強度測定値
Kf: d)で得た吸収補正係数
23.2.5 空試験
空試験は,行わない。
23.2.6 検量線の作成
検量線の作成は,次の手順によって行う。
a) 結晶質シリカの標準物質(23.2.3.6)を,含まれている結晶質シリカの量が10.0 mgとなるように正確
にはかり取り,ビーカー(500 mL)に移し入れる。イソプロピルアルコール100〜150 mLを加えてか
くはん(攪拌)した後,ビーカーを超音波洗浄器に入れて超音波処理を行い,粒子を十分に分散させ,
懸濁液を1 000 mLの全量フラスコにイソプロピルアルコールを用いて移し入れ,イソプロピルアルコ
ールで標線まで薄めて母液とする。
注記 母液1 mLは,結晶質シリカ0.01 mg(10 μg)に相当する。
b) 母液から1.0 mL,2.0 mL,3.0 mL,5.0 mL,10.0 mL,30.0 mL,50.0 mL,100.0 mL及び200.0 mLを分
取し,9個のコニカルビーカー(300 mL)にそれぞれ移し入れる。分取量が30 mL未満の場合には,
液量が50 mL以上になるようにイソプロピルアルコールを加える。
なお,母液を分取するときは,その都度母液を激しく振り混ぜ,目視で粒子が均一に懸濁している
ことを確認した後,直ちにピペットで吸引する。
c) コニカルビーカーに分取した母液中の標準物質を,吸引ろ過器(23.2.3.4)に装着した直径25 mmの
メンブランフィルタ(23.2.3.5)を用いて吸引しながらこし分け,コニカルビーカーの内壁に付着して
いる標準物質をイソプロピルアルコールでメンブランフィルタ上に完全に洗い移し,水で数回メンブ
73
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ランフィルタを洗浄した後,メンブランフィルタをろ過器から取り外し,取り外したままの状態で風
乾して検量線用試料とする。
d) 検量線用試料(9個)を,X線回折分析装置(23.2.3.1)の試料台に装着した基底標準金属板(23.2.3.2)
に固定し,各検量線用試料ごとに,結晶質シリカ及び基底標準金属板の金属の回折線強度を,それぞ
れ23.2.4.2 b)と同一条件で,順次測定する。
e) 吸収補正係数を,9個の検量線用試料ごとに,次の式によって算出する。
θ
θ
R
T
T
R
K
−
−
=1ln
)n(
)n(f
ここに,
Kf (n): 9個の検量線用試料ごとの吸収補正係数
(n=1,2,3……,9)
Rθ: 基底標準金属板の金属の回折角(sinθm)と結晶質
シリカの回折角(sinθs)との比(sinθm/sinθs)
ln: 自然対数
T(n): 9個の検量線用試料ごとの回折線強度の減少率
(Im (n)/I0m)(n=1,2,3……,9)
Im (n): 9個の検量線用試料ごとにd)で得た基底標準金属
板の金属の回折線強度測定値
I 0m: 23.2.4.2 c)で得た基底標準金属板の金属の回折線強
度測定値
f)
検量線用試料中の結晶質シリカの吸収補正回折線強度を,9個の検量線用試料ごとに,次の式によっ
て求め,得た結晶質シリカの各吸収補正回折線強度と各検量線用試料中の結晶質シリカ量との関係線
を作成し,検量線とする。
)n(f
)n(s
)n(
K
I
I
×
=
ここに,
I (n): 9個の検量用試料ごとの結晶質シリカの吸収補正
回折線強度(n=1,2,3……,9)
Is (n): 9個の検量線用試料ごとにd)で得た結晶質シリカ
の回折線強度測定値(n=1,2,3……,9)
Kf (n): e)で得た9個の検量線用試料ごとの吸収補正係数
(n=1,2,3……,9)
23.2.7 計算
計算は,次のいずれによる。
a) ヒュームの採取を6.1.3 a)又はb)によって行った場合 23.2.4.2 e)で得た測定試料中の結晶質シリカの
吸収補正回折線強度と23.2.6 f)で作成した検量線とから,測定試料中の結晶質シリカ量を求め,次の
式によって,ヒューム中の結晶質シリカ含有率を算出する。
100
1
33
2
×
=m
A
SiO
ここに,
SiO2: ヒューム中の結晶質シリカ含有率[%(質量分率)]
A33: 測定試料中の結晶質シリカ検出量(mg)
m1: 6.1.3 a) 2)又はb) 2)で得たヒューム採取量(mg)
b) ヒュームの採取を6.1.3 c) 1)によって行った場合 23.2.4.2 e)で得た結晶質シリカの吸収補正回折線強
度と23.2.6 f)で作成した検量線とから,測定試料中の結晶質シリカ量を求め,次の式によって,ヒュ
ーム中の結晶質シリカ含有率を算出する。
100
7
33
2
×
=mA
SiO
74
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
SiO2: ヒューム中の結晶質シリカ含有率[%(質量分率)]
A33: 測定試料中の結晶質シリカ検出量(mg)
m7: 23.2.4.1 b) 1)ではかり取った分析試料の量(mg)
24 りん定量方法
24.1 定量方法
りんの定量方法は,モリブドバナドりん酸吸光光度法による。この方法は,試料溶液中の1 μg/mL以上
20 μg/mL以下のりんの定量に適用する。
24.2 モリブドバナドりん酸吸光光度法
24.2.1 要旨
分析試料を硝酸で分解し,溶液をろ過する。ろ液に過マンガン酸カリウムを加え,加熱してりんをりん
(V)に酸化した後,過酸化水素を加えて過剰の過マンガン酸を分解する。バナジン酸アンモニウムを加
え,煮沸して過酸化水素を分解した後,七モリブデン酸六アンモニウムを加えてモリブドバナドりん酸錯
体を生成させ,光度計を用いて,その吸光度を測定する。
24.2.2 試薬
24.2.2.1 硝酸(2+3,1+5)
24.2.2.2 過酸化水素(1+9)
24.2.2.3 過マンガン酸カリウム溶液(10 g/L)
24.2.2.4 バナジン酸アンモニウム溶液
バナジン酸(V)アンモニウム2.50 gを温水500 mLに溶解し,硝酸(1+1)20 mLを加える。常温まで
冷却した後,水で液量を1 000 mLとする。
24.2.2.5 モリブデン酸アンモニウム溶液
七モリブデン酸六アンモニウム四水和物100 gに水600 mLを加え,約50 ℃に加熱して溶解する。常温
まで冷却した後,水で液量を1 000 mLとする。この溶液は,使用の都度,乾いたろ紙でろ過して用いる。
24.2.2.6 りん標準液(P:100 μg/mL)
105 ℃で恒量となるまで乾燥し,デシケータ中で室温まで放冷したりん酸二水素カリウム0.440 gをはか
り取り,水200 mLに溶解する。硝酸(1+5)10 mLを加え,常温まで冷却した後,1 000 mLの全量フラ
スコに水を用いて移し入れ,水で標線まで薄める。
24.2.3 操作
24.2.3.1 分析試料の前処理
24.2.3.1.1 主溶液の調製
主溶液の調製は,次の手順によって行う。
a) 6.1.3 a) 3),b) 3)又はc) 2) 2.3)で得た分析試料をビーカー(100 mL)に入れる。
b) 硝酸(2+3)30 mLを加え,時計皿で覆い,約30分間穏やかに加熱して分解する。室温まで冷却した
後,時計皿の下面及びビーカーの内壁を水で洗って時計皿を取り除き,溶液をろ紙(5種A)を用い
てろ過し,ろ過材及びろ紙を温水で十分に洗浄する。ろ液及び洗液を合わせ,常温まで冷却した後,
溶液を200 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄めて,主溶液とする。
なお,分析試料は,必要ならば,セラミック製はさみを用いてろ過材を切断して小片とした後,ビ
ーカー(100 mL)に入れる。
75
Z 3920:2011
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
24.2.3.1.2 空試験溶液の調製
6.1.3 a),b)又はc) 2)でヒュームの採取に用いたろ過材と同じろ過材を分析試料の代わりに用いて,
24.2.3.1.1 a)及びb)の手順に従って,分析試料と同じ操作を分析試料と並行して行い,得た溶液を空試験溶
液とする。
24.2.3.2 試料溶液の調製
試料溶液の調製は,次の手順によって行う。
a) 24.2.3.1.1 b)で得た主溶液を,りん量が100〜2 000 μgとなるように分取し,ビーカー(200 mL)に移
し入れる。
b) 硝酸(1+5)20 mLを加え,時計皿で覆い,約1分間穏やかに煮沸する。過マンガン酸カリウム溶液
2 mLを加え,再び煮沸し始めるまで穏やかに加熱する。時計皿の下面を水で洗って時計皿を取り除き,
過酸化水素(1+9)1 mLを加え,かき混ぜて過マンガン酸を分解する。
c) バナジン酸アンモニウム溶液(24.2.2.4)10.0 mLを加え,時計皿で覆い,加熱して溶液の色が透明な
青緑となるまで煮沸する。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水で洗って時計
皿を取り除き,溶液を100 mLの全量フラスコに水を用いて移し入れ,水で液量を約80 mLとする。
d) モリブデン酸アンモニウム溶液(24.2.2.5)10.0 mLを加え,水で標線まで薄め,よく振り混ぜた後,5
分間以上放置する。
注記 モリブドバナドりん酸錯体は,モリブデン酸アンモニウム添加後5分以内に完全に呈色し,
少なくとも1時間は安定である。
24.2.3.3 吸光度の測定
24.2.3.2 d)で得た溶液の一部を光度計の吸収セル(10 mm)に取り,水を対照液として波長470 nm付近
の吸光度を測定する。
24.2.4 空試験
24.2.3.1.2で得た空試験溶液を,24.2.3.2 a)で分取した主溶液と同量分取し,ビーカー(200 mL)に移し
入れる。以下,24.2.3.2 b)〜24.2.3.3の手順に従って主溶液と同じ操作を主溶液と並行して行う。
24.2.5 検量線の作成
りん標準液(24.2.2.6)0〜20.0 mL(りんとして0〜2 000 μg)を段階的に数個のビーカー(200 mL)に
取り,以下,24.2.3.2 b)〜24.2.3.3の手順に従って主溶液と同じ操作を主溶液と並行して行い,得た吸光度
とりん量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
24.2.6 計算
24.2.3.3及び24.2.4で得た吸光度と24.2.5で作成した検量線とからりん量を求め,次の式によって,ヒュ
ーム中のりん含有率を算出する。
100
200
)
(
40
3
35
34
×
×
−
=
B
m
A
A
P
ここに,
P: ヒューム中のりん含有率[%(質量分率)]
A34: 試料溶液中のりん検出量(mg)
A35: 空試験でのりん検出量(mg)
m3: 6.1.3 a) 2),b) 2)又はc) 2) 2.2)で得たヒューム採取
量(mg)
B40: 24.2.3.2 a)及び24.2.4でそれぞれ分取した主溶液及
び空試験溶液の量(mL)