2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
Z 3902-1984
黄銅ろう分析方法
Methods for Chemical Analysis of Brass Brazing Filler Metals
1. 適用範囲 この規格は,JIS Z 3262(黄銅ろう)に規定された化学成分(銅,すず,鉄,ニッケル,
鉛,アルミニウム,マンガン,銀,りん,けい素)の分析方法について規定する。
引用規格:
JIS H 0301 地金の試験並びに検査通則
JIS H 0321 非鉄金属材料の検査通則
JIS H 2102 アルミニウム地金
JIS H 2104 ニッケル地金
JIS H 2105 鉛地金
JIS H 2107 亜鉛地金
JIS H 2108 すず地金
JIS H 2121 電気銅地金
JIS H 2141 銀地金
JIS H 4502 電子管陰極用ニッケル板及び条
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析のための通則
JIS K 0121 原子吸光分析のための通則
JIS K 8005 容量分析用標準試薬
JIS K 8701 鉛(試薬)
JIS Z 3262 黄銅ろう
JIS Z 8401 数値の丸め方
2. 一般事項 分析方法に共通な一般事項は,JIS K 0050(化学分析方法通則),JIS K 0115(吸光光度分
析のための通則)及びJIS K 0121(原子吸光分析のための通則)による。
3. 分析試料の採り方及び取扱い方
3.1
分析試料の採取と調製
3.1.1
分析試料の採取と調製に際しては,試料全体の平均品質を代表するようにし,特に偏析,汚染など
に注意しなければならない。
2
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.1.2
分析試料の採取と調製は,JIS H 0321(非鉄金属材料の検査通則)の5.及びJIS H 0301(地金の試
験並びに検査通則)の2.2に準じて行うものとし,1融解ごとに二つ以上(1融解量が特に少ないときは一
つ)の鋳込試料約100gを取り,この試料を削って分析に必要な量の2倍以上の切粉を作り,これを十分に
混合して分析試料とする。
3.1.3
鋳込試料の削り方は,次の手順によって行う。
(1) きり,その他の工具類を,エタノールなどで清浄にする。
(2) 試料の表面に付いている油その他の付着物を除いて清浄にする。
(3) 試料の中央部又は両端に近い部分などを片面から直角にきりもみして貫通させるか,両面から少なく
とも中心部に達するまできりもみする。これらのきりもみができない場合は,適当な方法によって,
削り取る。
(4) 削り試料の大きさは,切粉があまり厚くならない程度にきりもみして長さを約5mm以下にする。も
し,切粉がひも状のときは,清浄なはさみで約5mm以下に切断する。
なお,このとき切粉にきりの摩耗粉が混入しないように注意する。また,試料を冷却するために水
などを注加してはならない。
3.1.4
分析試料の採取と調製が上記の規定によれない場合には,当事者間の協議による。
3.2
分析試料のはかり取り
3.2.1
分析試料をはかり取る際には,良くかき混ぜて平均組成を表すように注意し,また異物が混入して
いないことを確かめなければならない。
3.2.2
分析試料のはかり取りには化学はかりを用い,0.1mgのけたまで読み取る。
4. 分析操作上の注意
4.1
分析は,同一試料について,原則として2回以上行わなければならない。
4.2
分析に当たっては,全操作を通じて空試験を行い,含有率を補正しなければならない。
5. 分析結果の表し方 分析値は百分率で表し,二つ以上の分析値を平均して,JIS Z 3262に規定された
位にJIS Z 8401(数値の丸め方)によって丸める。ただし,亜鉛分については亜鉛以外の化学成分を前記
によって算出した後,各々の総計を100から差し引いた残分とする。
6. 銅定量方法
6.1
方法の区分 銅の定量方法は,電解重量法による。
6.2
電解重量法
6.2.1
要旨 試料を硝酸で分解し,水及び硫酸を加えた後,電解を行い,陰極に析出した銅の質量をはか
る。
6.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+9)
(2) 硝酸 (1+1,1+50,1+100)
(3) 硫酸 (1+1,1+50)
(4) エタノール (95v/v%)
6.2.3
装置及び器具 装置及び器具は,原則として次のものを用いる。
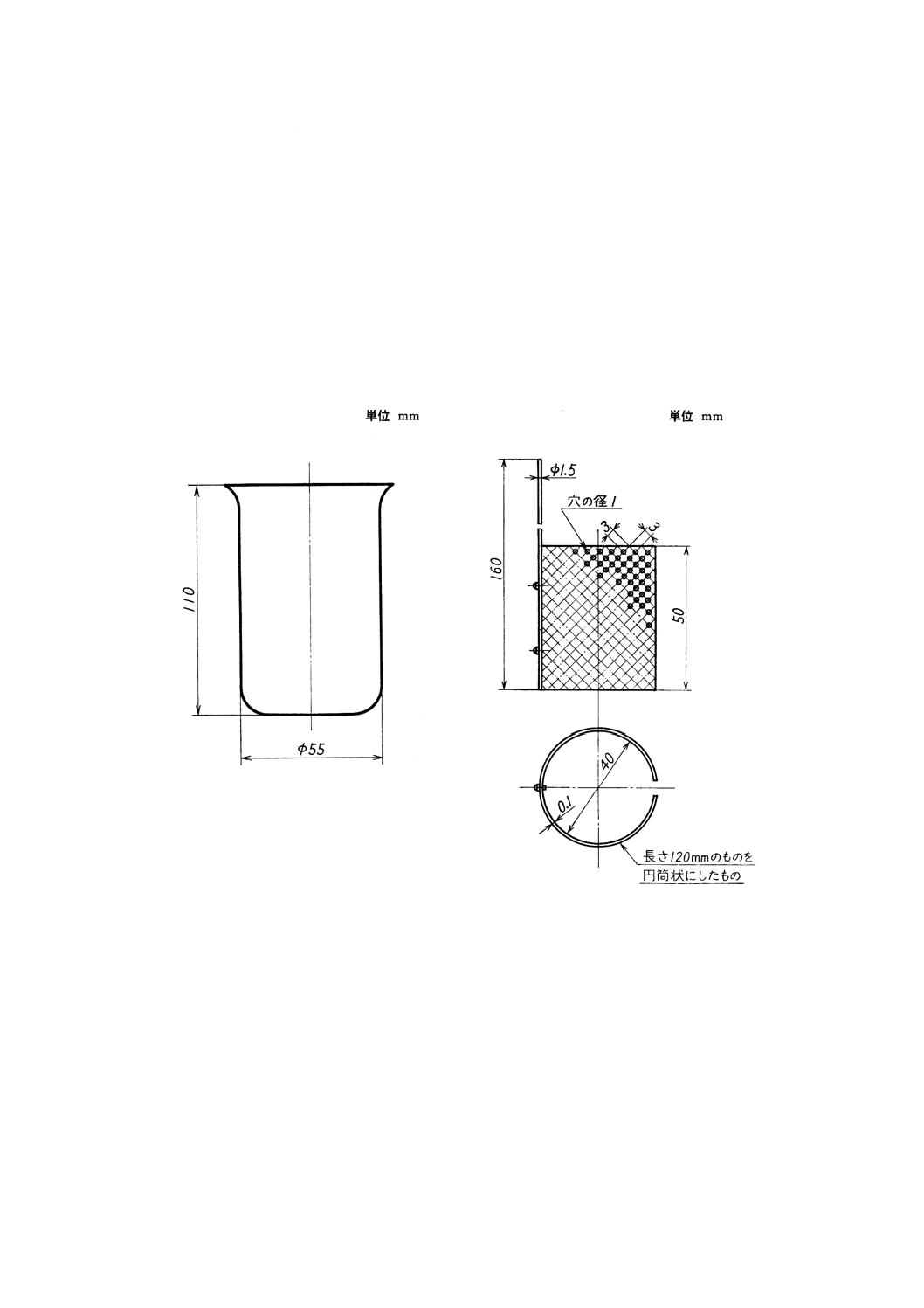
(1) 電解用ビーカー(付図1参照)
3
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 白金電極A(付図2参照)
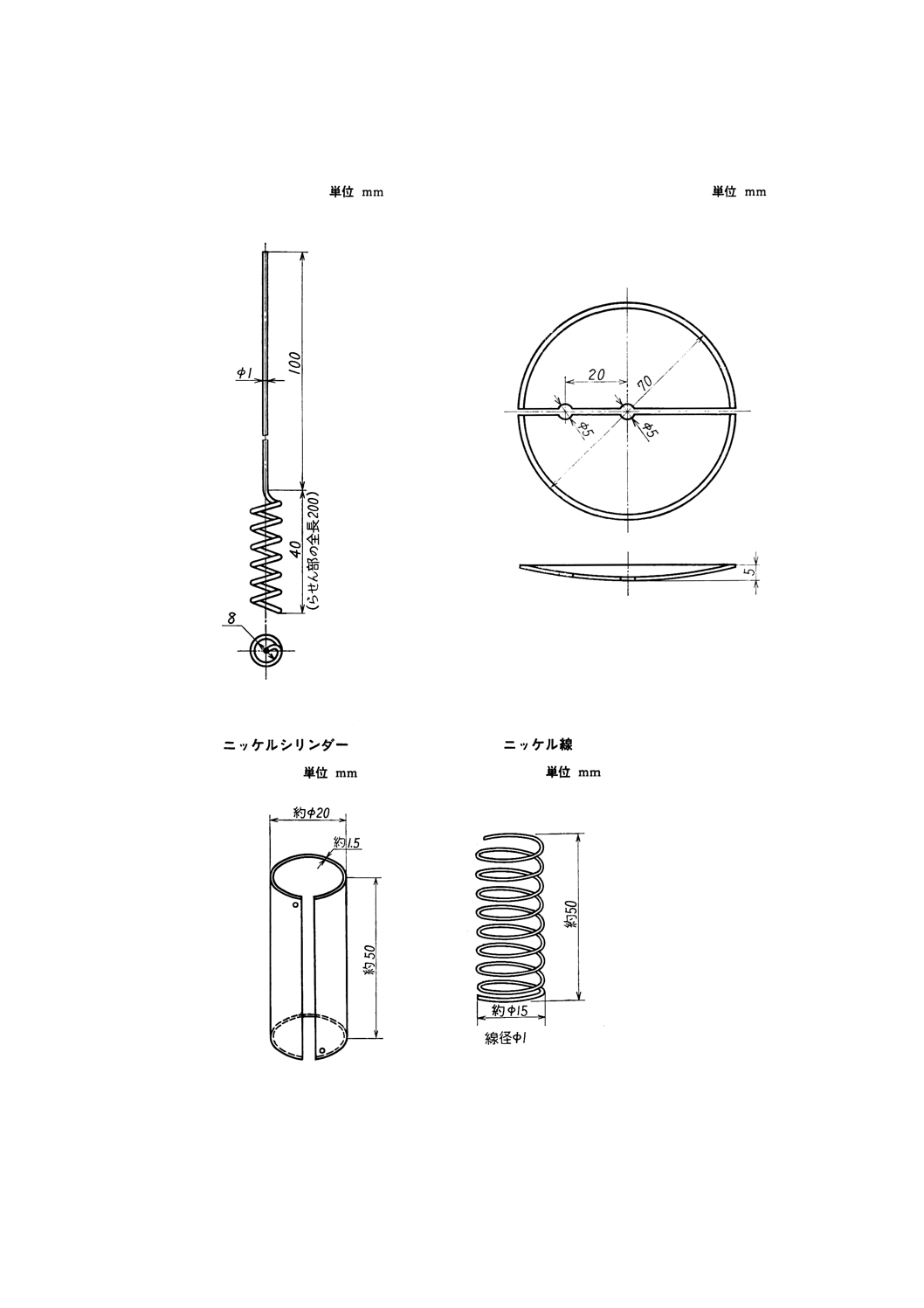
(3) 白金電極B(付図3参照)
(4) 半円形時計皿(付図4参照)
6.2.4
試料はかり取り量 試料は,1gをはかり取る。
6.2.5
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってトールビーカー (300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 10mlを加え静
かに加熱分解し,煮沸して酸化窒素を追い出した後,時計皿の下面及びビーカーの内壁を水洗する。
(2) これに水を加えて液量を約100mlとし,かき混ぜながら,硫酸 (1+1) 10mlを少量ずつ加える(1)。
(3) 溶液は電解用ビーカーに移し入れ,水で約150mlに薄めた後,あらかじめ質量をはかった白金電極A
を陰極とし,白金電極Bを陽極に用い,2個の半円形時計皿で覆い,室温で0.3〜0.5Aの電流を通じ
て1夜間電解する。
(4) 少量の水で時計皿の下面,ビーカーの内壁及び電極の柄の液面に露出した部分を洗い,その洗浄水に
よって電解液面を約5mm上昇させ,更に約30分間電解を続ける。
(5) 新しく電解液にはいった陰極の柄の部分に,もはや銅が析出しなくなれば,電流を流したまま水洗し
ながら両極を引き上げ,最後に手早く新たな水中に浸して陰極を外す。
(6) 陰極は始めは水で,次にエタノール (95v/v%) を用いて洗い,約80℃の空気浴中で速やかに乾燥し,
デシケーター中で約30分間放冷後質量をはかる。
注(1) この際硫酸鉛の沈殿を生じたときは,ろ紙(5種B)を用いてろ過し,硫酸 (1+50) で十分に洗
浄する。
備考1. 試料にけい素を含む場合は,6.2.5(1)の手順に従って操作した後,更に加熱蒸発してシロップ
状とし,これに硝酸 (1+1) 10mlと温水50mlを加え,加熱して可溶性塩類を溶解した後,ろ
紙(5種B)を用いてろ過し,温硝酸 (1十50) を用いて十分に洗浄する。以下,備考4.の操作
に従う。ただし,けい素とともにすずを含む場合は備考2.の操作に従う。
2. 試料にすずを含む場合は,備考1.に従って操作し,可溶性塩類を溶解後更に煮沸するまで加
熱し,温所に1〜2時間静置した後,直ちに少量のろ紙パルプを入れたろ紙(5種B)でろ過
し,温硝酸 (1+50) を用いて十分に洗浄する。以下,備考4.の操作に従う[7.2.5(2)のすずの
定量の際のろ液を用いることができる。]。
3. 試料に銀を含む場合は,備考1.に従って操作し,可溶性塩類を溶解した後,水を加えて液量
を約150mlとし,かき混ぜながら塩酸 (1+9) を滴加して塩化銀の沈殿を生成させる。沈殿
が生じなくなった後,更にその1mlを過剰に加えて約5分間静かに加熱し煮沸する。流水中
で冷却後,ろ紙(5種B)を用いてろ過し,始めは硝酸 (1+100) で,次に冷水で十分に洗浄
する。以下,備考4.の操作に従う(13.2.4における塩化銀をこし分けたろ液を用いることが
できる。)。
4. 備考1.〜3.によった場合は,それぞれのろ液に硫酸 (1+1) 15mlを加え,加熱して硫酸の白煙
が発生するまで蒸発し,放冷後水約50mlを加えて可溶性塩類を溶解し(硫酸鉛の沈殿を認
めたときは注(1)の操作に従って除去する。)。硝酸 (1+1) 5mlを加え,以下6.2.5(3)以降の手
順に従って電解を行う。
6.2.6
計算 試料中の銅含有率を,次の式によって算出する。
()
100
%
×
=Ww
銅
4
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
ω: 陰極に析出した銅の質量 (g)
W: 試料はかり取り量 (g)
7. すず定量方法
7.1
方法の区分 すずの定量方法は,次のいずれかによる。
(1) よう素酸カリウム滴定法
(2) 原子吸光法
7.2
よう素酸カリウム滴定法
7.2.1
要旨 試料を硝酸で分解し蒸発乾固し,すずをメタすず酸とした後,温水と硝酸で可溶性塩類を溶
解し,こし分ける。沈殿は硝酸と硫酸に分解後,硫酸白煙を発生させた後塩酸を加え,炭酸水素ナトリウ
ムを入れたキャップを用い,三塩化アンチモンと鉛(又はニッケル)ですずを還元する。でんぷんを指示
薬として,よう素酸カリウム標準溶液で滴定する。
7.2.2
試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸
(3) 硝酸 (1+1,1+50)
(4) 硫酸
(5) 鉛[99.99%以上,JIS H 2105(鉛地金)の特種相当品又はJIS K 8701(鉛)(試薬)の試金用粒状のも
の]
(6) ニッケルシリンダー又はニッケル線 純度 (Ni+Co) 99.8%以上[JIS H 4502(電子管陰極用ニッケル
板及び条)の3種相当品]で,原則として付図5のA又はBの形状とする。このシリンダー又は線は,
繰り返し使用することができる。
(7) 二酸化炭素 酸素を含んでいる場合には,塩化第一銅45gを塩酸 (1+1) 300mlに溶解し,少量の金属
銅削り片を加え,密栓して24時間以上放置した溶液に通して精製する。
(8) 三塩化アンチモン溶液 三塩化アンチモン5gを塩酸 (1+1) 500mlに加熱溶解する。
(9) 炭酸水素ナトリウム溶液(飽和)
(10) 標準すず溶液 (4mg/Sn/ml) すず[99.90%以上,JIS H 2108(すず地金)の1種A相当品]1.000gを
はかり取ってビーカー (300ml) に移し入れ,硫酸20mlを加え,時計皿で覆い加熱分解した後,引き
続いて加熱し,時計皿及びビーカーの内壁に付着した硫黄を揮散させる。冷却後,水及び塩酸 (1+1)
を用いて時計皿及びビーカーの内壁を洗い,塩酸 (1+1) 50mlを加えて可溶性塩類を溶解し,250ml
のメスフラスコに移し入れ,塩酸 (1+1) で標線まで薄める。又は,すず1.000gを水浴上で白金板を
触媒として塩酸50mlで分解し,250mlのメスフラスコに移し入れ,塩酸 (1+1) で標線まで薄めても
よい。
(11) よう素酸カリウム標準溶液 よう素酸カリウム1.784gを水酸化ナトリウム0.5gとよう化カリウム5g
を溶かした水100mlに溶解し,水で1 000mlに薄める。
この溶液は約N/20に相当するが,標定は次のように行う。
標準すず溶液を正確に25ml取り,還元装置の三角フラスコ (500ml) に入れ,水200ml及び塩酸50ml
を加える。以下,7.2.5(4)〜(6)の手順に従って操作し,次の式によって相当するすず量を求める。
V
f
100
.0
=
5
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
f: よう素酸カリウム標準溶液1mlに相当するすず量 (g)
0.100: 標準すず溶液25ml中のすず量 (g)
V: よう素酸カリウム標準溶液使用量 (ml)
(12) よう素標準溶液 よう化カリウム20g及びよう素6.4gをはかり取ってビーカー (300ml) に移し,冷水
約50mlを加え,振り混ぜてよう素を完全に溶解する。水で約150mlに薄め,ガラスろ過器(ブフナ
ー形,17G4)を用いてろ過した後,水を用いて1 000mlとし,褐色瓶に入れて保存する。この溶液1ml
は,すず約3mgに相当するが,標準すず溶液を用いて,使用の都度,標定を行う。標定方法は,(11)
よう素酸カリウム標準溶液に準じる。
(13) でんぷん溶液 でんぷん(溶性)又はでんぷん1gに少量の冷水を加えて泥状とし,100mlの熱水中に
かき混ぜながら加え,約1分間煮沸した後放冷し,不溶解物をろ過する。この溶液は使用の都度調製
し,よう素によって赤褐色となるものを用いてはならない。
7.2.3
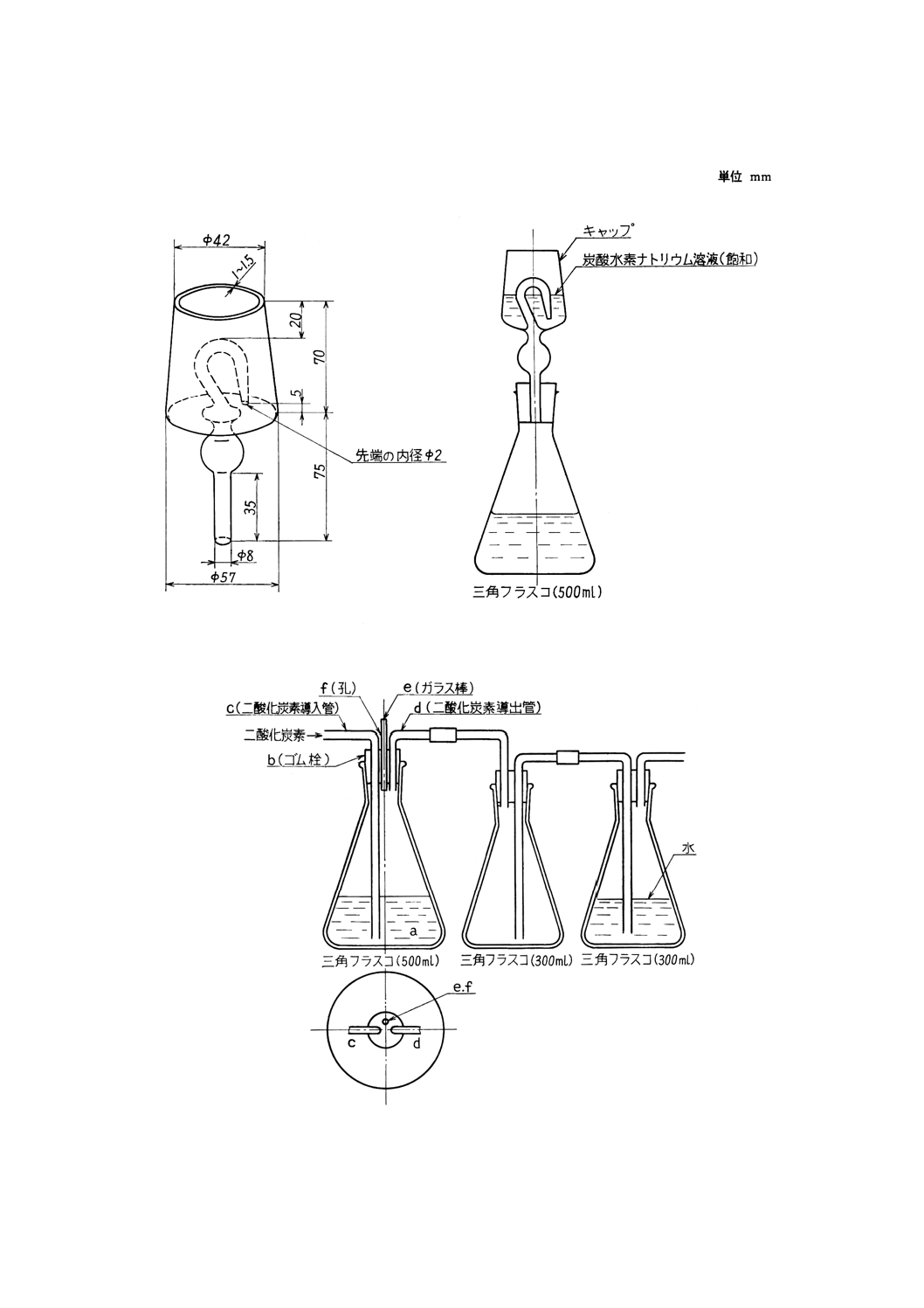
装置及び器具 還元装置は,通常付図6のようなものを用いるが,付図7のものを用いてもよい。
7.2.4
試料はかり取り量 試料は,1gをはかり取る(2)。
7.2.5
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってトールビーカー (300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 15ml(3)を加え,
加熱して分解する。時計皿の下面並びにビーカーの内壁を水で洗浄し,更に水浴上で注意して加熱し,
蒸発乾固する。
(2) 硝酸5ml及び温水約50mlを加え,煮沸して可溶性塩類を溶解し,温所に1〜2時間静置した後,直ち
に少量のろ紙パルプを入れたろ紙(5種C)でこし分け,温硝酸 (1+50) を用いて十分に洗浄する。
(3) 沈殿は,ろ紙と共に元のビーカーに入れ,硝酸15ml及び硫酸10mlを加え,時計皿で覆い,注意して
加熱分解し,更に加熱を続けて硫酸の白煙を十分に発生させる。
もし,この際有機物の分解が不十分のときは,冷却後更に硝酸10mlを加えて加熱分解を繰り返す。
冷却後,時計皿及びビーカーの内壁を水洗し,時計皿を除いて更に加熱蒸発し,硫酸の白煙を十分に
発生させる。
(4) 放冷後,水200ml及び塩酸50mlを加えて塩類を溶解し,溶液を三角フラスコ (500ml) に移し入れ,
三塩化アンチモン溶液10mlと鉛10g,又はニッケルシリンダー若しくは線を加える。三角フラスコに
付図6のキャップを付けたゴム栓を施し,キャップに炭酸水素ナトリウム溶液約50mlを加え,約30
分間静かに煮沸してすずを還元する。
(5) 三角フラスコの熱源を除き,そのまま約5分間放冷する。この間キャップ内の炭酸水素ナトリウム溶
液は三角フラスコ中に逆流するから,溶液が減少して空気を吸入するおそれがあれば追加する。次に
水中に浸して10℃以下になるまで冷却する。
(6) キャップと共にゴム栓を外し(4),直ちにゴム栓の下端及び三角フラスコの内壁をあらかじめ用意して
ある洗浄水(5)で洗浄した後,でんぷん溶液5mlを指示薬として加え,よう素酸カリウム標準溶液(6)で
滴定し,溶液が青紫色に変わった点を終点とする。
7.2.6
計算 試料中のすず含有率を,次の式によって算出する。
()
100
%
×
×
=WV
f
すず
ここに,
f: よう素酸カリウム標準溶液1mlに相当するすず量 (g)
V: よう素酸カリウム標準溶液使用量 (ml)
W: 試料はかり取り量 (g)
注(2) すずの含有量がなるべく20mg以上になるようにはかり取る。
6
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 試料はかり取り量が2〜3gの場合は,25〜30mlを加える。
(4) ニッケルを還元剤として用いた場合は,ニッケルシリンダー又はニッケル線を取り出し,これ
らと共にゴム栓などを水洗する。
(5) 洗浄水は煮沸して空気を追い出し,二酸化炭素を通じながら冷却した水を用いる。
(6) よう素酸カリウム標準溶液の代わりに,よう素標準溶液を用いてもよい。
備考 還元装置に付図7のものを用いる場合は,次のように操作する。
7.2.5(1)〜(3)の手順に従って操作し,硫酸の白煙を十分に発生させる。
放冷後,水200ml及び塩酸50mlを加えて塩類を溶解した後,還元装置の三角フラスコ (a) に
入れ,三塩化アンチモン溶液10mlと鉛10g,又はニッケルシリンダー若しくは線を加え,ゴム
栓 (b) をして二酸化炭素を通じながら約30分間煮沸してすずを還元する。
二酸化炭素を通じながら流水で10℃以下に冷却した後二酸化炭素の送入を止め,二酸化炭素
導入管 (c) 及び導出管 (d) の両端にはめたゴム管をピンチコックで閉じる。ガラス棒 (e) を取
り,孔 (f) からでんぷん溶液5mlを指示薬として加え,更に速やかにビュレットの先端を孔 (f)
に挿入し,よう素酸カリウム標準溶液(6)で滴定する。終点近くになったときゴム栓 (b) を取り
外し,洗浄水(5)で三角フラスコ (a) の内壁ゴム栓 (b) の下端及びガラス管を洗浄して滴定を続
け,溶液が青紫色に変わった点を終点とする。
7.3
原子吸光法
7.3.1
要旨 試料を硝酸とふっ化水素酸との混酸で分解し,ほう酸を加えた後,定容とし,原子吸光光度
計を用いて吸光度を測定する。
7.3.2
試薬 試薬は,次による。
(1) ほう酸溶液 (5w/v%)
(2) 混酸 硝酸 (2+1) 1lふっ化水素酸25mlを加える。
(3) 銅[99.96%以上,JIS H 2121(電気銅地金)相当品]
(4) 亜鉛[99.99%以上,JIS H 2107(亜鉛地金)の特種相当品]
(5) 標準すず溶液 (5mgSn/ml) すず(99.90%以上,JIS H 2108の1種A相当品)1.250gをはかり取って
ポリエチレンビーカー (200ml) に移し入れ,硝酸 (2+1) 10mlとふっ化水素酸2mlを加えて水浴上で
静かに加熱分解する。常温に冷却後,250mlのポリエチレン製メスフラスコ(7)に移し入れ,水で標線
まで薄める。
7.3.3
試料はかり取り量 試料は,1gをはかり取る。
7.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってポリエチレンビーカー (100ml) に移し入れ,混酸10mlを加えて,室温で放置し
て分解する(8)。分解が終わった後,ほう酸溶液10mlを加える。
(2) 常温まで冷却した後,100mlのメスフラスコに移し入れ,水で標線まで薄める。
(3) 原子吸光光度計を用いて,この溶液を空気−アセチレンフレーム中に噴霧して,波長286.3nmにおけ
る吸光度を測定する。
7.3.5
計算 7.3.6で作成した検量線からすず量を求め,試料中のすず含有率を,次の式によって算出す
る。
()
100
%
×
=WA
すず
ここに,
A: 試料溶液中のすず検出量 (g)
7
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
W: 試料はかり取り量 (g)
7.3.6
検量線の作成 試料に含有する銅及び亜鉛を試料含有率とほぼ同率で,全量が1gとなるようには
かり取り,これを1組とし,数組をはかり取って,それぞれをポリエチレンビーカー (100ml) に移し入れ,
混酸10mlを加えて室温で放置して分解する(8)。これに標準すず溶液0〜10.0ml(すずとして0〜0.05g)を
段階的に加え,更にほう酸溶液10mlを加える。以下,7.3.4(2),(3)の手順に従って操作し,試料と並行し
て測定した吸光度とすず量との関係線を作成して検量線とする。
注(7) ポリエチレン製メスフラスコ(ポリプロピレン製メスフラスコを用いてもよい)はJIS K 0050
の11.(4)によって補正を行って使用する。
(8) 室温及び混酸の液温が低い場合には,20℃以上に加熱して分解を行う。
備考 7.3.4(1)の操作を,次のように行ってもよい。
試料をはかり取って100mlのポリエチレン製メスフラスコ(7)に移し入れ,混酸10mlを加え
て室温で放置して分解する(8)。分解が終わった後,常温まで冷却し,水で標線まで薄める。以
下,7.3.4(3)の手順に従って操作し,すずを定量する。ただし,この場合の検量線は,この操作
を用いて作成する。
8. 鉄定量方法
8.1
方法の区分 鉄の定量方法は,次のいずれかによる。
(1) スルホサリチル酸吸光光度法
(2) 原子吸光法(A法) この方法は,すずを含まないBCuZn-0及びBCuZn-4の試料に適用する。
(3) 原子吸光法(B法) この方法は,すずを含むBCuZn-3及びBCuZn-5の試料に適用する。
8.2
スルホサリチル酸吸光光度法
8.2.1
要旨 試料を硝酸で分解し,アンモニア水を過剰に加えて,銅,亜鉛,ニッケル及び銀などを錯体
として溶解するとともに,鉄などを沈殿させこし分ける。沈殿を塩酸に溶解し,アンモニア水と硝酸を用
いてpHを調節した後,スルホサリチル酸を加えて呈色させて,その吸光度を測定する。
8.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+3,1+50)
(2) 硝酸 (1+1,1+2,1+100)
(3) アンモニア水 (1+1,1+20)
(4) スルホサリチル酸溶液 スルホサリチル酸10gを水約80mlに溶解した後,アンモニア水 (1+1) でpH
を2.0〜2.5とし,水で100mlに薄める。
(5) 標準鉄溶液 (100μgFe/ml) 鉄(99.5%以上)0.100gを塩酸 (1+1) 15mlで加熱分解し,過酸化水素水 (1
+1) 2mlを少量ずつ注意して滴加後,煮沸して過剰の過酸化水素を分解する。冷却後1 000mlのメス
フラスコに移し入れ,水で標線まで薄める。
(6) チモールブルー試験紙 良好なろ紙をチモールブルーのエタノール溶液 (0.1w/v%) に浸した後乾燥し
て用いる。
8.2.3
試料はかり取り量 試料は,1gをはかり取る(9)。
8.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 15mlを加え,静かに
加熱して分解する。時計皿の下面及びビーカーの内壁を水で洗い,引き続き加熱して酸化窒素を追い
出した後(10),水で約60mlに薄める。
8
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) アンモニア水 (1+1) を一度生成した銅,亜鉛などの沈殿が溶解するまで加えて強アンモニア性溶液
とし,加熱して1〜2分間煮沸する。しばらく静置後ろ紙(5種B)を用いてこし分け,温アンモニア
水 (1+20) で十分に洗浄する。
(3) 漏斗下に元のビーカーを受け,ろ紙上から温塩酸 (1+3) のなるべく少量を注いで沈殿を溶解した後,
温塩酸 (1+50) で数回洗浄する(11)。
(4) ろ液及び洗液に水を加えて液量を約60mlとし,アンモニア水 (1+1) を加えてほぼ中和した後,硝酸
(1+2) 及びアンモニア水 (1+1) を用いてpHを2.0〜2.5(12)とする。
(5) 常温まで冷却した後100mlのメスフラスコに移し入れ,スルホサリチル酸溶液3mlを加え,水で標線
まで薄め,よく振り混ぜて呈色させる。
(6) この溶液の一部を光度計の吸収セルに取り,波長545nm付近の吸光度を測定する。
8.2.5
計算 8.2.6で作成した検量線から鉄量を求め,試料中の鉄含有率を次の式によって算出する。
()
100
%
×
=WA
鉄
ここに,
A: 試料溶液中の鉄検出量 (g)
W: 試料はかり取り量 (g)
8.2.6
検量線の作成 標準鉄溶液0〜20.0ml(鉄として0〜2.0mg)を段階的に数個のビーカー (200ml) に
取り,8.2.4(4)〜(6)の手順に従って操作し,得た吸光度と鉄量との関係線を作成して,検量線とする。
注(9) 鉄の含有量がなるべく2mg以下となるように採取量を加減する。
(10) この際けい素,すずなどの沈殿を認めたときは,温所に約1時間静置後,直ちに少量のろ紙パ
ルプを加えたろ紙(5種B)でろ過し,温硝酸 (1+100) を用いて十分に洗浄した後,ろ液及び
洗液を加熱蒸発して約60mlとする。
(11) ろ液に塩化銀の沈殿を認めた場合は,元のろ紙を用いてろ過し,温塩酸 (1+50) で十分に洗浄
した後,ろ液及び洗液を加熱蒸発して約60mlとする。
また,ろ液が銅イオンの呈色を示すときは,8.2.4(2)の操作を行って再沈殿を行い,こし分け
洗浄する。
(12) チモールブルー試験紙を用いることができる。この場合,試験紙が微赤色を呈する程度とする。
8.3
原子吸光法(A法)
8.3.1
要旨 試料を硝酸で分解し,定容とした後,原子吸光光度計を用いて吸光度を測定する。
8.3.2
試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) 銅[7.3.2(3)による。]
(3) 亜鉛[7.3.2(4)による。]
(4) 標準鉄溶液 (25μgFe/ml) 鉄(99.5%以上)0.100gを硝酸 (1+1) 20mlで加熱分解し,常温に冷却後,
100mlのメスフラスコに移し入れ,水で標線まで薄め原液とする。使用の都度,この原液の一定量を
水で正しく40倍に薄め,標準鉄溶液とする。
8.3.3
試料はかり取り量 試料は,1gをはかり取る。
8.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (200ml) に移し入れ,時計皿で覆い,硝酸 (1±1) 20mlを加えて加熱分
解する。分解が終われば時計皿の下面及びビーカーの内壁を水洗する。
(2) 常温まで冷却した後,100mlのメスフラスコに移し入れ,水で標線まで薄める。
9
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 原子吸光光度計を用いて,この溶液を空気−アセチレンフレーム中に噴霧して波長248.3nm(13)におけ
る吸光度を測定する。
8.3.5
計算 8.3.6で作成した検量線から鉄量を求め,試料中の鉄含有率を,次の式によって算出する。
()
100
%
×
=WA
鉄
ここに,
A: 試料溶液中の鉄検出量 (g)
W: 試料はかり取り量 (g)
8.3.6
検量線の作成 試料に含有する銅及び亜鉛を試料含有率とほぼ同率で全量が1gとなるようにはか
り取り,これを1組として数組をはかり取り,それぞれをビーカー (200ml) に入れ,8.3.4(1)の時計皿で覆
い,以降の手順に従って操作した後,これに標準鉄溶液0〜8.0ml(鉄として0〜200μg)を段階的に加える。
以下,8.3.4の(2)及び(3)の手順に従って操作し,試料と並行して測定した吸光度と鉄量との関係線を作成
して検量線とする。
8.4
原子吸光法(B法)
8.4.1
要旨 試料を硝酸とふつ化水素酸との混酸で分解し,ほう酸を加えた後定容とし,原子吸光光度計
を用いて吸光度を測定する。
8.4.2
試薬 試薬は,次による。
(1) ほう酸溶液 (5w/v%)
(2) 混酸 硝酸 (2+1) 1lにふっ化水素酸25mlを加える。
(3) 銅[7.3.2(3)による。]
(4) 亜鉛[7.3.2(4)による。]
(5) 標準鉄溶液 (250μgFe/ml) 8.3.2(4)で調製した原液の一定量を水で正しく4倍に薄める。
(6) 標準鉄溶液 (25μgFe/ml) 8.3.2(4)によって調製する。
8.4.3
試料はかり取り量 試料は,BCuZn-3については0.2g,BCuZn-5については1gをはかり取る。
8.4.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってポリエチレンビーカー (100ml) に移し入れ,混酸10mlを加えて室温で放置して
分解する(8)。分解が終わった後,水約40ml及びほう酸溶液10mlを加える。
(2) 常温まで冷却した後,100mlのメスフラスコに移し入れ,水で標線まで薄める。
(3) 原子吸光光度計を用いて,この溶液を空気−アセチレンフレーム中に噴霧して波長248.3nm(13)におけ
る吸光度を測定する。
8.4.5
計算 8.4.6で作成した検量線から鉄量を求め,試料中の鉄含有率を,次の式によって算出する。
()
100
%
×
=WA
鉄
ここに,
A: 試料溶液中の鉄検出量 (g)
W: 試料はかり取り量 (g)
10
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.4.6
検量線の作成 試料に含有する銅及び亜鉛を試料含有率とほぼ同率で,かつ試料はかり取り量と同
一となるようにはかり取り,これを1組として数組をはかり取り,それぞれをポリエチレンビーカー
(100ml) に移し入れ,混酸10mlを加えて室温で放置して分解する(8)。これにBCuZn-3の試料の場合には,
標準鉄溶液 (250μgFe/ml) 0〜12.0ml(鉄として0〜3mg),BCuZn-5の試料の場合には,標準鉄溶液
(25μgFe/ml) 0〜8.0ml(鉄として0〜200μg)を段階的に加え,更にほう酸溶液10mlを加える。以下,8.4.4(2),
(3)の手順に従って操作し,試料と並行して測定した吸光度と鉄量との関係線を作成して検量線とする。
注(13) 波長372.0nmを用いてもよい。
備考 8.4.4(1)の操作を,すず定量方法の7.3原子吸光法の備考に準じて操作し,鉄を定量してもよい。
9. ニッケル定量方法
9.1
方法の区分 ニッケルの定量方法は,次のいずれかによる。
(1) ジメチルグリオキシム重量法
(2) ジメチルグリオキシム分離EDTA滴定法
9.2
ジメチルグリオキシム重量法
9.2.1
要旨 試料を,6.2.1に従って電解し,銅などを分離する。電解残液に酒石酸及び塩化アンモニウ
ムを加えた後,アンモニア性とし,ジメチルグリオキシムを加えてニッケルを沈殿させる。この沈殿をガ
ラスろ過器でこし分け,温水で洗浄した後,乾燥し,その質量をはかる。
9.2.2
試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) 過塩素酸
(3) 硫酸 (1+1,1+100)
(4) アンモニア水
(5) 塩化アンモニウム溶液 (25w/v%)
(6) アルミニウム 99.5%以上,JIS H 2102(アルミニウム地金)の2種相当品で,ニッケルを含まないも
の。削片として用いる。
(7) 酒石酸溶液 (25w/v%)
(8) ジメチルグリオキシム溶液 ジメチルグリオキシム1.0gを,エタノール (95v/v%) 100mlに溶解する。
(9) メチルレッド溶液 メチルレッド0.10gをエタノール (95v/v%) 90mlに溶解し,水で100mlとする。
9.2.3
試料はかり取り量 試料は,0.5gをはかり取る。
9.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってトールビーカー (300ml) に移し入れ,6.2.5(1)〜(5)の手順に従って電解を行う。
(2) 電解残液[6.2.5(5)の電解残液を用いてもよい(14)]をビーカー (500ml) に移し入れ,酒石酸溶液10ml
及び塩化アンモニウム溶液20mlを加え,メチルレッド溶液を指示薬としてアンモニア水で中和した
後,その3mlを過剰に加え,液量を約300mlに薄める。
(3) この溶液を約90℃に加熱し,かき混ぜながらジメチルグリオキシム溶液をニッケル予想含有量10mg
につき7mlの割合で加えてニッケルを沈殿させ,更にその5mlを過剰に加え,十分にかき混ぜた後,
室温まで放冷する。
(4) この沈殿を,あらかじめ恒量としたガラスろ過器 (1G3) を用いてこし分け,温水で十分に洗浄した後
110〜120℃の空気浴中で約1時間乾燥し,デシケーター中で放冷後その質量をはかり,恒量となるま
でこの操作を繰り返す。
11
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.5
計算 試料中のニッケル含有率を,次の式によって算出する。
()
100
2
203
.0
=
%
×
×
W
w
ニッケル
ここに,
w: ニッケルジメチルグリオキシムの質量 (g)
W: 試料はかり取り量 (g)
注(14) この溶液中のニッケル含有量が30mgを超えるときは,残液を250mlのメスフラスコに移し入れ,
水で標線まで薄めた後,ニッケル含有量がなるべく30mg程度となるように,その一定量をビー
カー (500ml) に分取する。
備考 電解法の代わりに,次のようにアルミニウム還元法によって銅を分離することができる。この
場合は次のように操作する。試料をはかり取ってビーカー (300ml) に移し入れ,時計皿で覆い,
硝酸 (1+1) 10mlを加え,静かに加熱分解する。
硫酸 (1+1) 15ml又は過塩素酸10mlを加え,加熱蒸発して白煙を発生させる。放冷後,水約
150mlを加えて可溶性塩類を溶解した後,アルミニウムの削片3gを加え,静かに煮沸して銅を
析出させる。水約50mlを加え,直ちにろ紙(5種A)を用いてビーカー (500ml) 中にろ過し,
温硫酸 (1+100) で洗浄する。
以下,9.2.4(2)の酒石酸溶液添加以降の手順に従って操作し,ニッケルを定量する。
9.3
ジメチルグリオキシム分離EDTA滴定法
9.3.1
要旨 試料を9.2.1に従って処理し,得られたニッケルジメチルグリオキシムの沈殿を,ろ紙を用
いてこし分け,塩酸で溶解した後,EDTAの過剰を加え,EBTを指示薬としてアンモニア性とし,亜鉛標
準溶液で逆滴定する。
9.3.2
試薬 試薬は,次による。
(1) 塩酸 (2+1,1+50)
(2) アンモニア水 (1+1)
(3) M/50亜鉛標準溶液 亜鉛[JIS K 8005(容量分析用標準試薬)]1.307gをはかり取り,なるべく少量
の塩酸 (1+1) で加熱分解し,冷却後,1 000mlのメスフラスコに移し入れ,水で標線まで薄める。
(4) M/50エチレンジアミン四酢酸二ナトリウム (EDTA) 標準溶液 エチレンジアミン四酢酸二ナトリウ
ム(2水塩)7.45gを水に溶解して1 000mlとする。標定は,次のように行う。
M/50EDTA標準溶液を正確に25ml取り,塩化アンモニウム溶液 (25w/v%) 10ml及びEBT溶液2〜3
滴を加え,水で液量を約100mlに薄めた後,溶液が青色に変わるまでアンモニア水 (1+1) を滴加し,
M/50亜鉛標準溶液で滴定し,溶液が赤紫色に変わった点を終点とし,次の式によってファクターを算
出する。
00
.
25
V
F=
ここに, F: M/50EDTA溶液のファクター
V: M/50亜鉛標準溶液の使用量 (ml)
(5) エリオクロムブラックT (EBT) 溶液 エリオクロムブラックT0.3gをエタノール (95v/v%) 15mlに溶解
後,トリエタノールアミン15mlを加える。
9.3.3
試料はかり取り量 試料は,0.5gをはかり取る。
9.3.4
操作 定量操作は,次の手順による。
(1) 試料をはかり取ってトールビーカー (300ml) に移し入れ,以下,9.2.4(1)〜(3)の手順に従って操作す

12
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。
(2) この沈殿を,ろ紙(5種A)を用いてこし分け,温水で十分に洗浄する。ろ紙上の沈殿を温水及び熱
塩酸 (2+1) を注いで元のビーカーに洗い落とし,ろ紙は温水及び温塩酸 (1+50) で数回洗浄する。
(3) この溶液に,ニッケル予想含有量10mgについてM/50EDTA標準溶液を正確に10mlの割合で加えた
後,更にその5mlを過剰に加える。2〜3回振り混ぜ,EBT溶液2〜3滴を指示薬として加えた後,溶
液が青色に変わるまでアンモニア水 (1+1) を滴加し(15),直ちにM/50亜鉛標準溶液で滴定し,溶液
が赤紫色に変わった点を終点とする。
9.3.5
計算 試料中のニッケル含有率を,次の式によって算出する。
()(
)
100
174
001
.0
%
2
1
×
×
W
V
V−
=
ニッケル
ここに, V1: M/50EDTA標準溶液の使用量 (ml)
V2: M/50亜鉛標準溶液の使用量 (ml)
W: 試料はかり取り量 (g)
注(15) このときpHを約8.0に調節することが必要であり,この操作によってpHは約8.0となる。
10. 鉛定量方法
10.1 方法の区分 鉛の定量方法は,次のいずれかによる。
(1) 電解重量法
(2) 原子吸光法(A法) この方法は,すずを含まないBCuZn-0,1,4,6の試料に適用する。
(3) 原子吸光法(B法) この方法は,すずを含むBCuZn-2,3,5の試料に適用する。
10.2 電解重量法
10.2.1 要旨 試料を硝酸で分解した後硝酸酸性溶液で電解を行い,鉛を酸化鉛 (IV) として陽極に析出さ
せ,その質量をはかる。
10.2.2 試薬 試薬は,次による。
(1) 硝酸 (1+1,1+3,1+100)
(2) エタノール (95v/v%) 陽極に析出した酸化鉛 (IV) はエタノールの存在で遊離酸に容易に溶解するか
ら遊離酸を含む場合は水酸化ナトリウム又は水酸化バリウムの少量を加え中和した後,蒸留して用い
る。
10.2.3 装置及び器具 装置及び器具は,原則として次のものを用いる。
(1) 電解用ビーカー(付図1参照)
(2) 白金電極A(付図2参照)
(3) 白金電極B(付図3参照)
(4) 半円形時計皿(付図4参照)
10.2.4 試料はかり取り量 試料は鉛含有率に応じ,原則として次の表に従ってはかり取る。
種 類
(記号)
試料はかり取り量
g
BCuZn 0,4,5
1.0
BCuZn 1,2,3,6
3.0
10.2.5 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (500ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 30ml(16)を加え加熱分
解し,静かに煮沸して酸化窒素を追い出した後,時計皿の下面及びビーカーの内壁を水洗する(17)。

13
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 溶液を電解用ビーカーに移し入れ,水で約150mlに薄めた後(18),あらかじめ質量をはかった白金電極
Aを陽極とし,白金電極Bを陰極に用いて2個の半円形時計皿で覆い,液温を15〜30℃として0.2〜
0.3Aの電流を通じて約1時間電解する。少量の水で時計皿の下面,ビーカーの内壁及び電極の柄の液
面に露出した部分を洗い,電解液面を約5mm上昇させ,更に約30分間電解を続ける。
(3) 新しく電解液中にはいった陽極にもはや鉛の析出が認められなければ,電流を通じたまま水洗しなが
ら両極を引き上げ,最後は手早く新たな水中に浸して陽極を外す。
(4) 陽極は始めは水で,次にエタノール (95v/v%) を用いて洗い,約200℃の空気浴中で約15分間乾燥し
た後,デシケーター中で約30分間放冷し,その増量を酸化鉛 (IV) としてはかる。
10.2.6 計算 試料中の鉛含有率を,次の式によって算出する。
()
100
2
866
.0
%
×
×
W
w
=
鉛
ここに,
w: 酸化鉛 (IV) の質量 (g)
W: 試料はかり取り量 (g)
注(16) 試料はかり取り量1gのときは30mlを使用するが,3gの場合は50mlとする。
(17) 試料にすず,けい素を含む場合は6.2.5の備考1.又は備考2.に準じて操作し,ろ過分離する。
(18) 試料にマンガンを含む場合は,この際遊離硝酸量が20mlとなるように硝酸を追加する。
10.3 原子吸光法(A法)
10.3.1 要旨 試料を硝酸で分解し,定容とした後,原子吸光光度計を用いて吸光度を測定する。
10.3.2 試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) 銅[7.3.2(3)による。]
(3) 亜鉛[7.3.2(4)による。]
(4) ニッケル[99.95%以上,JIS H 2104(ニッケル地金)の特種相当品]
(5) 標準鉛溶液(500μgPb/ml及び25μgPb/ml) 鉛(99.99%以上,JIS H 2105の特種相当品)0.500gを硝
酸 (1+1) 20mlで加熱分解し,常温に冷却後100mlのメスフラスコに移し入れ,水で標線まで薄め,
原液とする。使用の都度,この原液の一定量を水で正しく10倍及び200倍に薄め,標準鉛溶液とする。
10.3.3 試料はかり取り量 試料は,鉛含有率に応じ,原則として次の表に従ってはかり取る。
種類
(記号)
試料はかり取り量
g
BCuZn 0,4
0.5
BCuZn 1,6
1.0
10.3.4 操作 定量操作は,次の手順によって行う。
(1) 8.3.4(1)の手順に従って操作する。
(2) 8.3.4(2)の手順に従って操作する(19)。
(3) 原子吸光光度計を用いて,この溶液を空気−アセチレンフレーム中に噴霧して波長217.0nm又は
283.3nmにおける吸光度を測定する。
10.3.5 計算 10.3.6で作成した検量線から鉛量を求め,試料中の鉛含有率を,次の式によって算出する。
()
100
%
×
=WA
鉛
ここに,
A: 試料溶液中の鉛検出量 (g)
W: 試料はかり取り量 (g)
14
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.3.6 検量線の作成 試料に含有する銅,亜鉛及びニッケルを試料含有率とほぼ同率で,全量が試料はか
り取り量と同一となるようにはかり取り,これを1組として数組をはかり取り,それぞれをビーカー
(200ml) に入れ,8.3.4(1)の時計皿で覆い,以降の手順に従って操作した後,BCuZn-0及び4の場合には標
準鉛溶液 (500μgPb/ml) 0〜5.0ml(鉛として0〜2.5mg),BCuZn-1及び6の場合には標準鉛溶液 (25μgPb/ml)
0〜8.0ml(鉛として0〜200μg)を段階的に加える。以下,8.3.4(2)及び10.3.4(3)の手順に従って操作し,試
料と並行して測定した吸光度と鉛量との関係線を作成して検量線とする。
注(19) 溶液中にけい酸の沈殿を認めた場合(BCuZn-6の場合)には,乾燥ろ紙(5種B)で乾燥ビーカ
ーに必要な量だけろ過して測定に用いる。
10.4 原子吸光法(B法)
10.4.1 要旨 試料を硝酸とふっ化水素酸との混酸で分解し,ほう酸を加えた後定容とし,原子吸光光度計
を用いて吸光度を測定する。
10.4.2 試薬 試薬は,次による。
(1) ほう酸溶液 (5w/v%)
(2) 混酸 硝酸 (2+1) 1lにふっ化水素酸25mlを加える。
(3) 銅[7.3.2(3)による。]
(4) 亜鉛[7.3.2(4)による。]
(5) 標準鉛溶液 10.3.2(5)によって調製する。
10.4.3 試料はかり取り量 試料は,BCuZn-2及び3については1g,BCuZn-5については0.5gをはかり取
る。
10.4.4 操作 定量操作は,次の手順によって行う。
(1) 8.4.4(1)の手順に従って操作する。
(2) 8.4.4(2)の手順に従って操作する。
(3) 10.3.4(3)の手順に従って操作する。
10.4.5 計算 10.4.6で作成した検量線から鉛量を求め,試料中の鉛含有率を,次の式によって算出する。
()
100
%
×
=WA
鉛
ここに,
A: 試料溶液中の鉛検出量 (g)
W: 試料はかり取り量 (g)
10.4.6 検量線の作成 試料に含有する銅及び亜鉛を試料含有率とほぼ同率で,全量が試料はかり取り量と
同一となるようにはかり取り,これを1組として数組をはかり取り,それぞれをポリエチレンビーカー
(100ml) に移し入れ,8.4.4(1)の手順に従って操作した後,BCuZn-2及び3の場合には標準鉛溶液
(25μgPb/ml) 0〜8.0ml(鉛として0〜200μg),BCuZn-5の場合には標準鉛溶液 (500μgPb/ml) 0〜5.0ml(鉛と
して0〜2.5mg)を段階的に加える。以下,8.4.4(2)及び10.3.4(3)の手順に従って操作し,試料と並行して測
定した吸光度と鉛量との関係線を作成して検量線とする。
備考 10.4.4(1) の操作を,すず定量方法の7.3原子吸光法の備考に準じて操作し,鉛を定量してもよ
い。
11. アルミニウム定量方法
11.1 方法の区分 アルミニウムの定量方法は,次のいずれかによる。
(1) オキシン抽出吸光光度法
15
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 原子吸光法(A法) この方法は,すずを含まないBCuZn-1及び6の試料に適用する。
(3) 原子吸光法(B法) この方法は,すずを含むBCuZn-2及び3の試料に適用する。
11.2 オキシン抽出吸光光度法
11.2.1 要旨 試料を硝酸で分解した後,すず,けい素を含む場合はろ過し,これに硫酸を加え加熱蒸発し
て硫酸塩とし,水銀陰極電解を行い,銅,亜鉛及びその他の金属を分離する。電解残液に酒石酸,亜硫酸
ナトリウム及びシアン化カリウムを順次に加えて鉄を還元した後pHを調節し,オキシンを加え,ベンゼ
ンを用いてアルミニウムを抽出し,その吸光度を測定する。
11.2.2 試薬 試薬は,次による。
(1) 塩酸 (1+1)
(2) 硝酸 (1+1)
(3) 硫酸 (1+1,1+9,1+50)
(4) アンモニア水 (1+1)
(5) 硝酸アンモニウム溶液 (20w/v%)
(6) 亜硫酸ナトリウム溶液(飽和)
(7) シアン化カリウム溶液 (10w/v%)
(8) 緩衝溶液 塩化カリウム0.75gとほう酸0.63gを水に溶解し,水酸化ナトリウム溶液 (1N) 8mlを加え
た後,水で90mlに薄める。
(9) 酒石酸溶液 (10w/v%)
(10) オキシン溶液 オキシン1gを酢酸6mlに溶解した後,温水を加えて約95mlとし,これにアンモニア
水を滴加し,沈殿を生じる直前でやめ,水で100mlとする。必要があれば,ろ過して用いる。
(11) ジンコン溶液 ジンコン0.1gを水酸化ナトリウム溶液 (1N) 2mlに溶解し,水で100mlに薄める。こ
の溶液は調製後2日以上経過したものは用いない方がよい。
(12) ベンゼン
(13) 標準アルミニウム溶液 (10μgAl/ml) アルミニウム(99.9%以上)0.100gをはかり取ってビーカー
(100ml) に移し入れ,塩酸 (1+1) 5mlと硝酸 (1+1) 5mlを加え加熱分解した後,酸化窒素を追い出し,
冷却後1 000mlのメスフラスコに移し入れ,水で標線まで薄める。この溶液を原液とし,使用の都度
その一定量を分取し,水で正しく10倍に薄めて標準アルミニウム溶液とする。
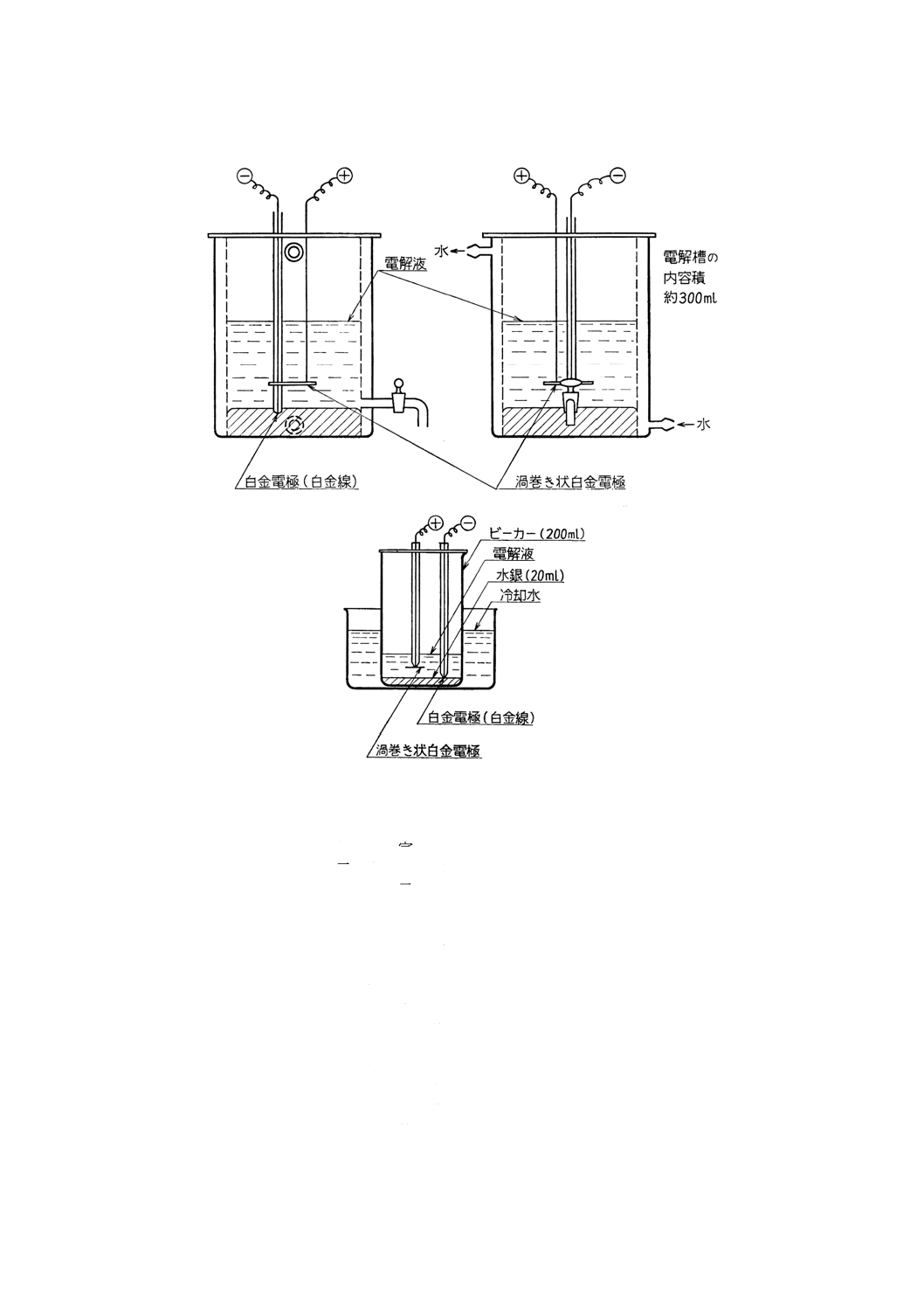
11.2.3 装置 水銀陰極電解装置 電解槽は,原則として付図8のものを用いる。
11.2.4 試料はかり取り量 試料は,0.3gをはかり取る。
11.2.5 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (200ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 10mlを加えて加熱分
解した後,時計皿の下面及びビーカーの内壁を水洗する(20)。
(2) 硫酸 (1+1) 5mlを加え,加熱蒸発して大部分の硫酸を追い出す。
(3) 放冷後硫酸 (1+9) 2mlと水約50mlを加え,加熱して可溶性塩類を溶解した後(21),水銀陰極電解装置
の電解槽に移し入れ,水銀を陰極とし白金を陽極に用い,水で冷却しながら数アンペアの電流で電解
を行う。
(4) 電解が終われば(22)電流を通じたまま水銀と電解液を分離し,電解残液はビーカー (200ml) に移し入
れる。不溶解残物を認めた場合はろ紙(5種B)を用いてろ過し,硫酸 (1+50) で洗浄する。
(5) 溶液は加熱蒸発して液量を20〜30mlとし,酒石酸溶液2ml,亜硫酸ナトリウム溶液2ml及びシアン化
カリウム溶液5mlをこの順序でかき混ぜながら加え,約70℃に1〜2分間加熱する(23)。
16
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(6) 室温に冷却し,硝酸アンモニウム溶液10mlを加えた後,アンモニア水 (1+1) と塩酸 (1+1) を用い
て溶液のpHを8〜9に調節する。
(7) 少量の水を用いて分液漏斗 (200ml) に洗い移し,水で液量を約100mlとした後,オキシン溶液3ml
とベンゼン15mlを加えて約1分間振り混ぜ,アルミニウムのオキシン塩を抽出する。静置後下層を
別の分液漏斗 (200ml) に移し入れ,再びオキシン溶液3mlとベンゼン15mlを加えて抽出を行い,こ
の操作をベンゼン層に着色を認めなくなるまで繰り返す(24)。
(8) 抽出層は合併し,乾燥ろ紙(5種A)を用いて50mlのメスフラスコ中にろ過し,ろ紙をベンゼンで洗
浄後ベンゼンを用いて標線まで薄める。
(9) この溶液の一部を光度計の吸収セルに取り,波長390nm付近の吸光度を測定する。
11.2.6 計算 11.2.7で作成した検量線からアルミニウム量を求め,試料中のアルミニウム含有率を次の式
によって算出する。
()
100
%
×
=WA
アルミニウム
ここに,
A: 試料溶液中のアルミニウム検出量 (g)
W: 試料はかり取り量 (g)
11.2.7 検量線の作成 標準アルミニウム溶液0〜6.0ml(アルミニウムとして0〜60μg)を段階的に数個の
ビーカー (100ml) に取り,11.2.5(5)の酒石酸溶液添加以降の手順に従って操作し,得た吸光度とアルミニ
ウム量との関係線を作成して検量線とする。
注(20) 試料にすず,けい素を含む場合は,6.2.5の備考1.又は備考2.に準じて操作し,ろ過分離する。
(21) この際硫酸鉛の沈殿を認めたときは,ろ紙(5種B)を用いてろ過し,硫酸 (1+50) で十分に
洗浄する。
(22) 電解時間は装置によって異なるが,2〜3Aの電流で1〜2時間で終了する。電解残液の1滴を取
り,これに緩衝溶液2滴とジンコン溶液1滴を加えて亜鉛の存否を検出し,電解の終了を判定
することができる。亜鉛が存在すれば青色を示すが,黄赤色のときは電解が終わったものと見
なす。
(23) この操作は有毒シアン化水素を発生するおそれがあるので,必ずドラフト内で行う。
(24) ベンゼン15mlで1回に抽出されるアルミニウムの量は,約20μgである。
11.3 原子吸光法(A法)
11.3.1 要旨 試料を硝酸で分解し,定容とした後,原子吸光光度計を用いて吸光度を測定する。
11.3.2 試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) 銅[7.3.2(3)による。]
(3) 亜鉛[7.3.2(4)による。]
(4) ニッケル[10.3.2(4)による。]
(5) 標準アルミニウム溶液 (25μgAl/ml) アルミニウム[99.90%以上,JIS H 2102(アルミニウム地金)の
特1種相当品]0.100gを塩酸 (1+1) 5mlと硝酸 (1+1) 5mlを加えて加熱分解した後,酸化窒素を追い
出す。常温に冷却後100mlのメスフラスコに移し入れ,水で標線まで薄め,原液とする。使用の都度,
この原液の一定量を水で正しく40倍に薄め,標準アルミニウム溶液とする。
11.3.3 試料はかり取り量 試料は,2gをはかり取る。
11.3.4 操作 定量操作は,次の手順によって行う。
17
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 8.3.4(1)の手順に従って操作する。
(2) 8.3.4(2)の手順に従って操作する(19)。
(3) 原子吸光光度計を用いて,この溶液を酸化二窒素−アセチレンフレーム中に噴霧して,波長309.3nm
における吸光度を測定する。
11.3.5 計算 11.3.6で作成した検量線からアルミニウム量を求め,試料中のアルミニウム含有率を,次の
式によって算出する。
()
100
%
×
=WA
アルミニウム
ここに,
A: 試料溶液中のアルミニウム検出量 (g)
W: 試料はかり取り量 (g)
11.3.6 検量線の作成 試料に含有する銅,亜鉛及びニッケルを試料含有率とほぼ同率で,全量が2gとな
るようにはかり取り,これを1組として数組をはかり取り,それぞれをビーカー (200ml) に入れ,8.3.4(1)
の時計皿で覆い,以降の手順に従って操作した後,これに標準アルミニウム溶液0〜8.0ml(アルミニウム
として0〜200μg)を段階的に加える。以下,8.3.4(2)及び11.3.4(3)の手順に従って操作し,試料と並行して
測定した吸光度とアルミニウム量との関係線を作成して検量線とする。
11.4 原子吸光法(B法)
11.4.1 要旨 試料を硝酸とふっ化水素酸との混酸で分解し,ほう酸を加えた後,定容とし,原子吸光光度
計を用いて吸光度を測定する。
11.4.2 試薬 試薬は,次による。
(1) ほう酸溶液 (5w/v%)
(2) 混酸 硝酸 (2+1) 1lにふっ化水素酸25mlを加える。
(3) 銅[7.3.2(3)による。]
(4) 亜鉛[7.3.2(4)による。]
(5) 標準アルミニウム溶液 (25μgAl/ml) 11.3.2(5)によって調製する。
11.4.3 試料はかり取り量 試料は,2gをはかり取る。
11.4.4 操作 定量操作は,次の手順によって行う。
(1) 8.4.4(1)の手順に従って操作する。
(2) 8.4.4(2)の手順に従って操作する。
(3) 11.3.4(3)の手順に従って操作する。
11.4.5 計算 11.4.6で作成した検量線からアルミニウム量を求め,試料中のアルミニウム含有率を,次の
式によって算出する。
()
100
%
×
=WA
アルミニウム
ここに,
A: 試料溶液中のアルミニウム検出量 (g)
W: 試料はかり取り量 (g)
18
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.4.6 検量線の作成 試料に含有する銅及び亜鉛を試料含有率とほぼ同率で,全量が2gとなるようには
かり取り,これを1組として数組をはかり取り,それぞれをポリエチレンビーカー (100ml) に入れ,8.4.4(1)
の手順に従って操作した後,これに標準アルミニウム溶液0〜8.0ml(アルミニウムとして0〜200μg)を段
階的に加える。以下,8.4.4(2)及び11.3.4(3)の手順に従って操作し,試料と並行して測定した吸光度とアル
ミニウム量との関係線を作成して検量線とする。
備考 11.4.4(1)の操作を,すず定量方法の7.3原子吸光法の備考に準じて操作し,アルミニウムを定量
してもよい。
12. マンガン定量方法
12.1 方法の区分 マンガンの定量方法は,次のいずれかによる。
(1) 過硫酸アンモニウム酸化亜ひ酸滴定法
(2) 原子吸光法
12.2 過硫酸アンモニウム酸化亜ひ酸滴定法
12.2.1 要旨 試料を混酸で分解し,過硫酸アンモニウムを加えてマンガンを酸化し,冷却後亜ひ酸ナトリ
ウム標準溶液で滴定する。
12.2.2 試薬 試薬は,次による。
(1) 混酸 水435ml中に硫酸150mlを加え,冷却後硝酸250ml,りん酸150ml及び硝酸銀溶液 (20w/v%) 15ml
を混和する。
(2) 過硫酸アンモニウム溶液 (20w/v%) この溶液は使用の都度調製する。
(3) 亜ひ酸ナトリウム標準溶液 三酸化ひ素0.500g (JIS K 8005) をはかり取ってビーカー (200ml) に移
し入れ,水酸化ナトリウム溶液 (4w/v%) 20mlと水約100mlを加えて加熱溶解し,冷却した後1 000ml
のメスフラスコに洗い移す。これにフェノールフタレインを指示薬として硫酸 (1+35) を加えて微酸
性とし,炭酸水素ナトリウム溶液 (5w/v%) 20mlを加えて水で標線まで薄める。この標準溶液の標定方
法は,次のとおりとする。
電解銅と電解ニッケルとを試料中の含有量に近い比率にはかり取り,12.2.4(1)に準じて混酸で分解
した後,これに標準マンガン溶液の定量を正しく加え,以下,12.2.4(2)以降の手順に従って操作し滴
定を行い,次の式によって相当するマンガン量を算出する。
2
1
0002
.0
V
V
f
×
=
ここに,
f: 亜ひ酸ナトリウム標準溶液1mlに相当するマンガン量 (g)
V1: 標準マンガン溶液の使用量 (ml)
V2: 亜ひ酸ナトリウム標準溶液の使用量 (ml)
標準マンガン溶液 (200μgMn/ml) の調製 マンガン(99.9%以上)0.100gをはかり取ってビーカー
(200ml) に移し入れ,硝酸 (1+4) 50mlを加え加熱して分解し,冷却後500mlのメスフラスコに移し入
れ,水で標線まで薄める。
12.2.3 試料はかり取り量 試料は,0.5gをはかり取る。
12.2.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取って三角フラスコ (500ml) に移し入れ,混酸30mlを加えて加熱分解し,反応が終わ
ればフラスコの内壁を洗い,再び煮沸して酸化窒素を追い出す。
(2) これに温水約200mlを加えて加熱を続け,煮沸し始めたときに過硫酸アンモニウム溶液10mlを注意
19
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
して加え,小気泡が大気泡となるまで2〜3分間煮沸して過硫酸アンモニウムを分解するとともに,マ
ンガンを十分に酸化して過マンガン酸とした後,流水中で25℃以下に冷却する。
(3) 冷却後速やかに亜ひ酸ナトリウム標準溶液で滴定する。
12.2.5 計算 試料中のマンガン含有率を,次の式によって算出する。
()
100
%
×
×
=Wf
V
マンガン
ここに,
V: 亜ひ酸ナトリウム標準溶液の使用量 (ml)
f: 亜ひ酸ナトリウム標準溶液1mlに相当するマンガン量 (g)
W: 試料はかり取り量 (g)
12.3 原子吸光法
12.3.1 要旨 試料を硝酸とふっ化水素酸との混酸で分解し,ほう酸を加えた後,定容とし,原子吸光光度
計を用いて吸光度を測定する。
12.3.2 試薬 試薬は,次による。
(1) ほう酸溶液 (5w/v%)
(2) 混酸 硝酸 (2+1) 1lにふっ化水素酸25mlを加える。
(3) 銅[7.3.2(3)による。]
(4) 亜鉛[7.3.2(4)による。]
(5) 標準マンガン溶液 (250μgMn/ml) マンガン(99.9%以上)0.500gを硝酸 (1+1) 20mlで加熱分解し,
常温に冷却後,100mlのメスフラスコに移し入れ,水で標線まで薄め,原液とする。使用の都度,こ
の原液の一定量を水で正しく20倍に薄め,標準マンガン溶液とする。
12.3.3 試料はかり取り量 試料は,0.2gをはかり取る。
12.3.4 操作 定量操作は,次の手順によって行う。
(1) 8.4.4(1)の手順に従って操作する。
(2) 8.4.4(2)の手順に従って操作する。
(3) 原子吸光光度計を用いてこの溶液を空気−アセチレンフレーム中に噴霧して,波長279.5nm又は
403.1nmにおける吸光度を測定する。
12.3.5 計算 12.3.6で作成した検量線からマンガン量を求め,試料中のマンガン含有率を,次の式によっ
て算出する。
()
100
%
×
=WA
マンガン
ここに,
A: 試料溶液中のマンガン検出量 (g)
W: 試料はかり取り量 (g)
12.3.6 検量線の作成 銅0.11g及び亜鉛0.08gをはかり取り,これを1組として数組をはかり取り,それ
ぞれをポリエチレンビーカー (100ml) に入れ,8.4.4(1)の手順に従って操作した後,標準マンガン溶液0〜
8.0ml(マンガンとして0〜2.0mg)を段階的に加える。以下,8.4.4(2)及び12.3.4(3)の手順に従って操作し,
試料と並行して測定した吸光度とマンガン量との関係線を作成して検量線とする。
備考 12.3.4(1)の操作を,すず定量方法の7.3原子吸光法の備考に準じて操作し,マンガンを定量して
もよい。
13. 銀定量方法
13.1 方法の区分 銀の定量方法は,次のいずれかによる。
20
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 塩化銀重量法
(2) 原子吸光法
13.2 塩化銀重量法
13.2.1 要旨 試料を硝酸で分解し,塩酸又は塩化ナトリウムを加え1夜間放置した後,ガラスろ過器を用
いてこし分け,乾燥後その質量をはかる(ろ液は銅の定量に用いることができる。)。
13.2.2 試薬 試薬は,次による。
(1) 塩酸 (1+9)
(2) 硝酸 (1+1,1+3,1+100)
(3) 塩化ナトリウム溶液 (10w/v%)
13.2.3 試料はかり取り量 試料は,2gをはかり取る。
13.2.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってトールビーカー (300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 20mlを加え静
かに加熱して分解する。時計皿の下面及びビーカーの内壁を水洗した後,更に加熱してシロップ状と
する。
(2) 硝酸 (1+3) 20mlを加えて塩類を溶解し,ろ紙(5種B)を用いて二酸化けい素などをろ過し,温硝酸
(1+100) で洗浄する。水で約150mlに薄めた後,溶液をかき混ぜながら,塩酸 (1+9) 又は塩化ナト
リウム溶液を塩化銀の白濁を生じなくなるまで滴加し,更にその数滴を過剰に加えて約5分間静かに
煮沸し,暗所に1夜間放置する。
(3) 沈殿はガラスろ過器 (1G4) でこし分け,始めは硝酸 (1+100) で,次に冷水を用いて十分に洗浄し(ろ
液及び洗液を銅の定量に用いることができる。),ガラスろ過器とともに110〜120℃の空気浴中で恒量
となるまで乾燥し,デシケーター中で放冷後,塩化銀としてその質量をはかる。
13.2.5 計算 試料中の銀含有率を,次の式によって算出する。
()
100
6
752
.0
%
×
×
=
W
w
銀
ここに,
w: 塩化銀の質量 (g)
W: 試料はかり取り量 (g)
13.3 原子吸光法
13.3.1 要旨 試料を硝酸で分解し,定容とした後,原子吸光光度計を用いて吸光度を測定する。
13.3.2 試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) 銅[7.3.2(3)による。]
(3) 亜鉛[7.3.2(4)による。]
(4) ニッケル[10.3.2(4)による。]
(5) 標準銀溶液 (250μgAg/ml) 銀[99.99%以上,JIS H 2141(銀地金)の1種相当品]1.00gを硝酸 (1+
1) 20mlで加熱分解し,常温に冷却後,100mlのメスフラスコに移し入れ,水で標線まで薄め,原液と
する。使用の都度,この原液の一定量を水で正しく20倍に薄め,標準銀溶液とする。
13.3.3 試料はかり取り量 試料は,0.1gをはかり取る。
13.3.4 操作 定量操作は,次の手順によって行う。
(1) 8.3.4(1)の手順に従って操作する。
(2) 8.3.4(2)の手順に従って操作する。
21
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 原子吸光光度計を用いて,この溶液を空気−アセチレンフレーム中に噴霧して波長328.1nm又は
338.3nmにおける吸光度を測定する。
13.3.5 計算 13.3.6で作成した検量線から銀量を求め,試料中の銀含有率を,次の式によって算出する。
()
100
%
×
=WA
銀
ここに,
A: 試料溶液中の銀検出量 (g)
W: 試料はかり取り量 (g)
13.3.6 検量線の作成 銅0.05g,亜鉛0.04g及びニッケル0.01gをはかり取り,これを1組として数組をは
かり取り,それぞれをビーカー (200ml) に入れ,8.3.4(1)の時計皿で覆い,以降の手順に従って操作した後,
標準銀溶液0〜5.0ml(銀として0〜1.25mg)を段階的に加える。以下,8.3.4(2)及び13.3.4(3)の手順に従っ
て操作し,試料と並行して測定した吸光度と銀量との関係線を作成して検量線とする。
14. りん定量方法
14.1 方法の区分 りんの定量方法は,りんバナドモリブデン酸吸光光度法による。
14.2 りんバナドモリブデン酸吸光光度法
14.2.1 要旨 試料を混酸で分解し,これに過酸化水素水を加えて煮沸した後,バナジン酸アンモニウムと
モリブデン酸アンモニウムを加えて呈色させ,吸光度を測定する。
14.2.2 試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) 混酸 硝酸320mlと塩酸120mlとに水500mlを加えてよく混和し,冷却後水で1lに薄める。
(3) 過酸化水素水 (1+9)
(4) バナジン酸アンモニウム溶液 バナジン酸アンモニウム2.5gを温水約500mlに溶解し,これに硝酸 (1
+1) 20mlを加え,冷却後水で1lに薄める。
(5) モリブデン酸アンモニウム溶液 モリブデン酸アンモニウム(4水和物)100gを約50℃の温水600ml
に溶解した後水で1lとし,使用の都度ろ過して用いる。
(6) 標準りん溶液 (150μgP/ml) 硫酸を乾燥剤としたデシケーター中に1夜間放置した,りん酸二水素カ
リウム0.6591gをはかり取り,水約200mlを加えて溶解し,硝酸 (1+5) 10mlを加え,1 000mlのメス
フラスコに移し入れ,水で標線まで薄める。
14.2.3 試料はかり取り量 試料は,0.5gをはかり取る。
14.2.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (200ml) に移し入れ,時計皿で覆い,混酸20mlを加えて静かに分解す
る。時計皿の下面及びビーカーの内壁を水で洗い,引き続き加熱蒸発してシロップ状とし,放冷後硝
酸 (1+1) 20mlを加え,加熱して可溶性塩類を溶解した後,ろ紙(5種B)を用いてろ過し,硝酸 (1
+200) で十分に洗浄する。
(2) ろ液に過酸化水素水 (1+9) 3〜5mlを加え,3〜5分間静かに煮沸し,バナジン酸アンモニウム溶液10ml
を加えてよく振り混ぜ,1〜2分間煮沸した後常温まで冷却する。
(3) この溶液を100mlのメスフラスコに移し入れ,モリブデン酸アンモニウム溶液10mlを加え,水で標
線まで薄め,よく振り混ぜて呈色させる。
(4) 約5分間放置した後,溶液の一部を光度計の吸収セルに取り,波長430〜470nmの吸光度を測定する。
22
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.2.5 計算 14.2.6で作成した検量線からりん量を求め,試料中のりん含有率を,次の式によって算出す
る。
()
100
%
×
=WA
りん
ここに,
A: 試料溶液中のりん検出量 (g)
W: 試料はかり取り量 (g)
14.2.6 検量線の作成 銅(99.95%以上)0.25gずつをはかり取って数個のビーカー (200ml) に移し入れ,
混酸20mlを加えて分解し,その各々に標準りん溶液0〜10.0ml(りんとして0〜1.5mg)を段階的に加え,
13.2.4(1)のシロップ状に蒸発(ただし,ろ過操作は省略する。)以降の手順に従って操作し,得た吸光度と
りん量との関係線を作成し,検量線とする。
15. けい素定量方法
15.1 方法の区分 けい素の定量方法は,次のいずれかによる。
(1) 二酸化けい素重量法
(2) モリブデン黄抽出吸光光度法
(3) 原子吸光法
15.2 二酸化けい素重量法
15.2.1 要旨 試料を硝酸で分解した後,過塩素酸を加えて加熱蒸発し白煙を発生させ,放冷後,温水で可
溶性塩類を溶解し,二酸化けい素をこし分け,強熱して恒量とする。これにふっ化水素酸を加えて二酸化
けい素を揮散させ,その減量をはかる。
15.2.2 試薬 試薬は,次による。
(1) 硝酸
(2) 硝酸 (1+1)
(3) 過塩素酸
(4) ふっ化水素酸
(5) 臭化水素酸 (1+1)
(6) 硫酸 (1+5)
15.2.3 試料はかり取り量 試料は,5gをはかり取る。
15.2.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (500ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 50mlを加え,静かに
加熱して分解した後,煮沸して酸化窒素を追い出す。
(2) これに過塩素酸35mlを加え,加熱蒸発して過塩素酸の白煙が発生し始めてから約10分間加熱し,濃
厚な白煙を発生させる。放冷後,温水約150mlを加え,加熱して可溶性塩類を溶解し,直ちにろ紙(5
種B)でこし分け,温水で十分に洗浄する(25)。
(3) 沈殿は,ろ紙とともに白金るつぼに入れ,乾燥後加熱してろ紙を灰化した後,約1 100℃で約30分間
強熱し,デシケーター中で放冷後,その質量をはかり,更に強熱を繰り返して恒量とする。
(4) これにふっ化水素酸約2mlと硫酸 (1+5) 数滴を加え,注意して加熱して乾固した後強熱し,放冷後
減量をはかる。再びふっ化水素酸の処理を繰り返し,もし減量のあるときは,更にこの処理を減量の
なくなるまで繰り返して行い,全減量を二酸化けい素 (SiO2) とする。
15.2.5 計算 試料中のけい素含有率を,次の式によって算出する。
23
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
()(
)
100
4
467
.0
%
2
1
×
×
−
=
W
w
w
けい素
ここに, w1: ふっ化水素酸処理前の質量 (g)
w2: ふっ化水素酸処理後の質量 (g)
W: 試料はかり取り量 (g)
注(25) 試料にすずを含有し,その分解が完全でない場合は,沈殿をろ紙とともに元のビーカーに入れ,
硝酸30mlと過塩素酸20mlを加えて加熱分解し,引き続いて蒸発を行う。もし溶液が黒色を呈し
たときは,硝酸数滴を滴加して透明溶液とし,濃厚な白煙が発生するまで加熱蒸発する。
放冷後,臭化水素酸 (1+1) 30mlを数回に分けて加え,加熱蒸発し,濃厚な白煙を約10分間
発生させて,すずを揮散させる。
放冷後,温水約150mlを加え,加熱して可溶性塩類を溶解し,直ちにろ紙(5種B)でこし
分け,温水で十分に洗浄する。
15.3 モリブデン黄抽出吸光光度法
15.3.1 要旨 試料を硝酸とふっ化水素酸で分解し,ほう酸を加えた後pHを調節し,モリブデン酸アンモ
ニウムを加えて,けい素などを呈色させる。溶液に酒石酸を加え,硫酸を加えて酸濃度を調節した後,イ
ソアミルアルコールを用いてけいモリブデン酸を抽出し,硫酸で洗浄後吸光度を測定する。
15.3.2 試薬 試薬は,次による。
(1) 硝酸 (1+2)
(2) ふっ化水素酸 (1+9)
(3) 硫酸 (1+3,1+10)
(4) ほう酸溶液 水1lにほう酸60gを加え,飽和溶液を作る。
(5) アンモニア水 (1+2)
(6) モリブデン酸アンモニウム溶液 モリブデン酸アンモニウム(4水和物)10gを水に溶解して100ml
とする。
(7) 酒石酸溶液 (10w/v%)
(8) イソアミルアルコール
(9) 混合溶媒(クロロホルム3,1−ブタノール1)
(10) 標準けい素溶液 (10μgSi/ml) あらかじめ1 000℃に強熱し,デシケーター中で放冷した二酸化けい素
(99.9%以上)0.535gを白金るつぼ(30番)にはかり取り,炭酸ナトリウム(無水)2.5gを混和して
融解する。放冷後,温水100mlを入れたポリエチレンビーカー (200ml) 中に浸して融成物を溶解した
後,500mlのメスフラスコに移し入れ,水で標線まで薄め,ポリエチレン容器に保存して原液
(500μgSi/ml) とする。使用の都度,必要量だけ水で正しく50倍に薄め,標準けい素溶液とする。
15.3.3 試料はかり取り量 試料は,1gをはかり取る。
15.3.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってポリエチレンビーカー (200ml) に移し入れ,ポリエチレン時計皿で覆い,硝酸 (1
+2) 20mlを加えて分解後,ふっ化水素酸 (1+9) 3mlを加え,約70℃の温水中に入れ,ときどき振り
混ぜながら約15分間放置する。
(2) ほう酸溶液25mlを加えてよくかき混ぜた後,500mlのメスフラスコに移し入れ,水で標線まで薄める。
この溶液50mlを分取し,アンモニア水 (1+2) と硝酸 (1+2) を用いてpHを1.0〜1.2に調節する。
(3) この溶液にモリブデン酸アンモニウム溶液10mlを加えて約10分間放置し,けいモリブデン酸などを
24
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
完全に呈色させる(26)。
(4) 溶液を分液漏斗 (200ml) に入れ,酒石酸溶液10mlと硫酸 (1+3) 40mlを加えて振り混ぜた後(27),イ
ソアミルアルコール40mlを加えて約1分間激しく振り混ぜ,けいモリブデン酸を抽出する。静置後,
下層の溶液を捨て,アルコール層に硫酸 (1+10) 40mlを加えてよく振り混ぜて洗浄し,洗液を捨てる。
(5) 溶媒層の一部を乾燥ろ紙を用いて光度計の吸収セルにろ過し,波長400nm付近の吸光度を測定する。
15.3.5 計算 15.3.6で作成した検量線からけい素量を求め,試料中のけい素含有率を,次の式によって算
出する。
()
100
10
1
%
×
×
=
W
A
けい素
ここに,
A: 試料溶液中のけい素検出量 (g)
W: 試料はかり取り量 (g)
15.3.6 検量線の作成 標準けい素溶液0〜25.0ml(けい素として0〜250μg)を段階的に数個のポリエチレ
ンビーカー (200ml) に取り,水で約50mlとして,硝酸 (1+2) とアンモニア水 (1+2) を用いてpHを1.0
〜1.2に調節する。分液漏斗 (200ml) に移し入れ,以下,15.3.4(3)〜(5)の手順に従って操作し,得た吸光
度とけい素量との関係線を作成して検量線とする。
注(26) 試料にりんを含む場合は溶液の硝酸濃度を約1Nとし,分液漏斗に移し入れ,混合溶媒50mlを
加えて約1分間激しく振り混ぜる。静置後下層の溶媒を捨て,再び混合溶媒30mlを加えて抽出
を行い,溶媒層にりんモリブデン酸の黄色が認められなくなるまで抽出操作を繰り返す。
(27) この際硫酸濃度は3〜5Nとする。
15.4 原子吸光法
15.4.1 要旨 試料を硝酸とふっ化水素酸との混酸で分解し,ほう酸を加えた後,定容とし,原子吸光光度
計を用いて吸光度を測定する。
15.4.2 試薬 試薬は,次による。
(1) ほう酸溶液 (5w/v%)
(2) 混酸 硝酸 (2+1) 1lにふっ化水素酸25mlを混和する。
(3) 銅[7.3.2(3)による。]
(4) 亜鉛[7.3.2(4)による。]
(5) ニッケル[10.3.2(4)による。]
(6) 標準けい素溶液 (500μgSi/ml) 15.3.2(10)の原液を用いる。
15.4.3 試料はかり取り量 試料は,1gをはかり取る。
15.4.4 操作 定量操作は,次の手順によって行う。
(1) 8.4.4(1)の手順に従って操作する。
(2) 8.4.4(2)の手順に従って操作する。
(3) 原子吸光光度計を用いて,この溶液を酸化二窒素−アセチレンフレーム中に噴霧して波長251.6nmに
おける吸光度を測定する。
15.4.5 計算 15.4.6で作成した検量線からけい素量を求め,試料中のけい素含有率を,次の式によって算
出する。
()
100
%
×
=WA
けい素
25
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
A: 試料溶液中のけい素検出量 (g)
W: 試料はかり取り量 (g)
15.4.6 検量線の作成 試料に含有する銅,亜鉛及びニッケルを試料含有率とほぼ同率で全量が1gとなる
ようにはかり取り,これを1組として数組をはかり取り,それぞれをポリエチレンビーカー (100ml) に入
れ,8.4.4(1) の手順に従って操作した後,標準けい素溶液0〜6.0ml(けい素として0〜3.0mg)を段階的に
加える。以下,8.4.4(2)及び15.4.4(3)の手順に従って操作し,試料と並行して測定した吸光度とけい素量と
の関係線を作成して検量線とする。
備考 15.4.4(1)の操作を,すず定量方法の7.3原子吸光法の備考に準じて操作し,けい素を定量しても
よい。
付図1 電解用ビーカー
付図2 白金電極A
26
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図3 白金電極B
付図4 半円形時計皿
付図5 A
付図5 B
27
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図6 還元装置の例
付図7 還元装置の例
28
Z 3902-1984
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図8 水銀陰極電解用電解槽の例
黄銅ろう分析方法改正原案作成委員会構成表
氏名
所属
(委員会長)
服 部 只 雄
環境計測器サービス株式会社
俣 野 宣 久
川崎製線株式会社
三 上 博
財団法人日本溶接技術センター
古 矢 元 佑
清峰金属工業株式会社
吉 田 信 之
工業技術院標準部材料規格課
田 口 喬
株式会社徳力本店
前 田 修
田中貴金属工業株式会社
吉光寺 博
石福金属興業株式会社
山 田 栄 一
東京芝浦電気株式会社
松 井 文 夫
三菱電機株式会社
乾 昌 弘
乾庄貴金属化工株式会社
椙 原 寿喜男
橋本貴金属工業株式会社
村 田 利 昭
日本ハンディー・ハーマン株式会社
内 藤 正 夫
富士伸銅株式会社
上 田 進
日本伸銅株式会社
(事務局)
長谷川 義 治
社団法人日本溶接協会