19
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
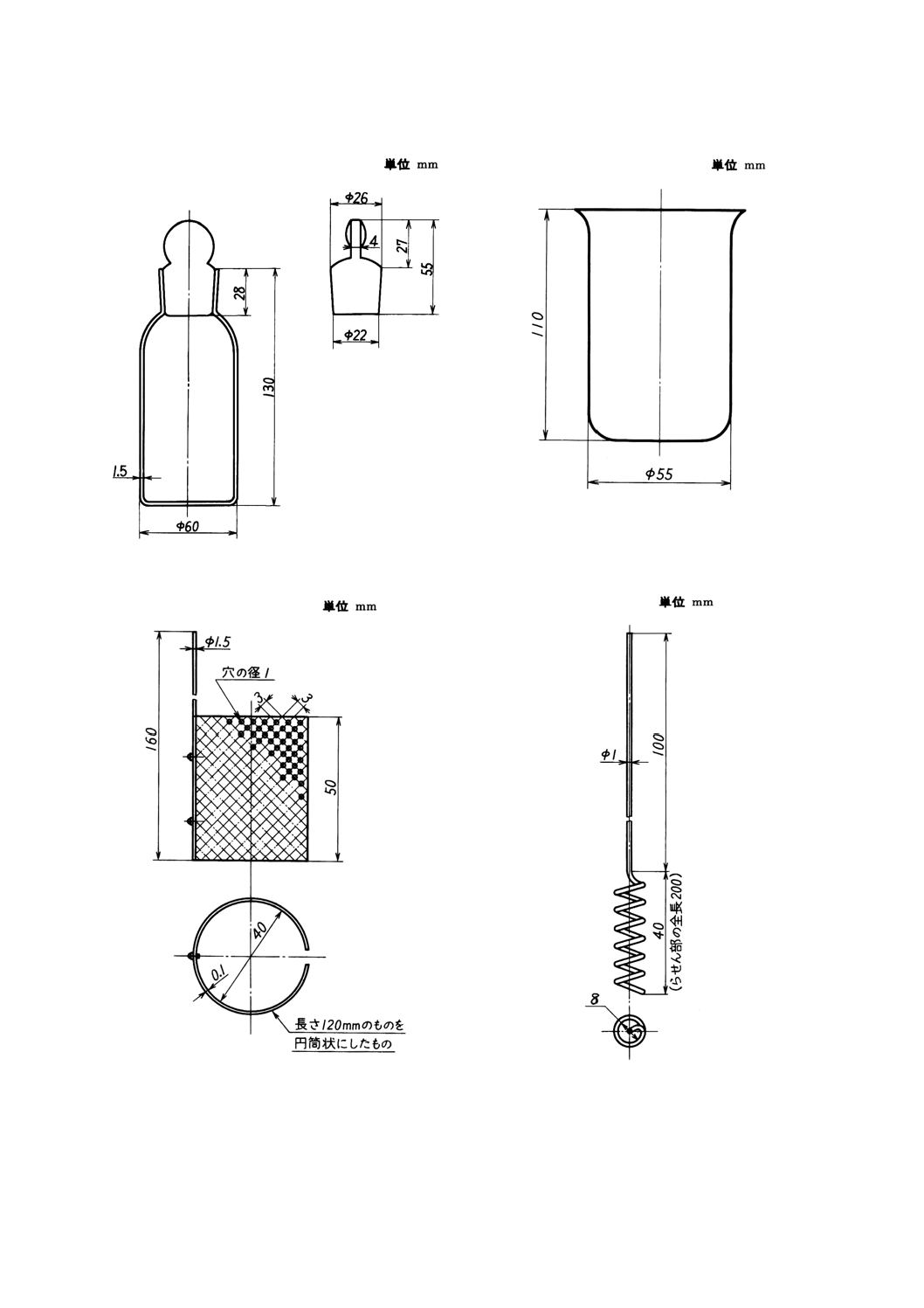
付図1 分解瓶
付図2 電解用ビーカー
付図3 白金電極A
付図4 白金電極B
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
Z 3901-1988
銀ろう分析方法
Methods for Chemical Analysis of
Silver Brazing Filler Metals
1. 適用範囲 この規格は,JIS Z 3261(銀ろう)に規定された銀,銅,ニッケル,亜鉛,カドミウム,
すず,リチウム,鉛及び鉄の定量方法について規定する。
引用規格:
JIS H 2105 鉛地金
JIS K 0050 化学分析方法通則
JIS K 0121 原子吸光分析のための通則
JIS K 8005 容量分析用標準試薬
JIS K 8701 鉛(試薬)
JIS Z 3261 銀ろう
JIS Z 3900 貴金属ろうのサンプリング方法
JIS Z 8401 数値の丸め方
2. 一般事項 分析方法に共通な一般事項は,JIS K 0050(化学分析方法通則)及びJIS K 0121(原子吸
光分析のための通則)による。
3. 分析試料の採り方及び取扱い方
3.1
分析試料の採取と調製 分析試料の採取と調製は,次による。
(1) 分析試料の採取と調製に際しては,平均品質を代表するようにし,特に偏析,汚染などに注意しなけ
ればならない。
(2) 分析試料の採取と調製は,JIS Z 3900(貴金属ろうのサンプリング方法)による。
3.2
試料のはかり方 試料のはかり方は,次による。
(1) 分析試料をはかり取る際には,よくかき混ぜて平均組成を表すように注意し,また,異物が混入して
いないことを確かめなければならない。
(2) 分析試料のはかり取りには,化学はかりを用い,0.1mgのけたまで読み取る。
4. 分析値のまとめ方
4.1
分析回数 分析は,同一試料について2回以上行って結果を確かめる。
4.2
分析値の表示 分析値の表示は,次による。
(1) 分析値は,質量百分率で表す。
2
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) リチウムを除く合金成分の分析値は,小数点以下第2位まで算出した後,二つ以上の分析値を平均し
て,JIS Z 8401(数値の丸め方)によって,小数点以下第1位に丸める。
(3) リチウム及び不純物の分析値は,小数点以下第3位まで算出した後,二つ以上の分析値を平均して,
JIS Z 8401によって,小数点以下第2位に丸める。
5. 分析操作上の注意 分析操作上の注意事項は,次による。
(1) 分析操作に使用する水は,すべて塩化物イオンを含まないものを用いなければならない。
(2) 原子吸光法において吸光度測定の際,フレームに空気・アセチレンを用いる。ただし,精度及び正確
さを満足することを確認してあれば,空気・水素,その他のフレームを用いてもよい。
6. 空試験 分析に当たっては,全操作を通じて空試験を行い,測定値を補正しなければならない。
7. 安全に関する注意 原子吸光法における高圧ガスの取扱い,フレームの点火及び消火には十分に注意
する。
また,燃料ガスにアセチレンを使用しているため試料中の銀などと爆発性のアセチレン化物を作るおそ
れがあるので,使用の都度,バーナーヘッドを水で洗浄するなどの処置をとり,災害の防止に努めなけれ
ばならない。
8. 銀定量方法
8.1
定量方法の区分 銀の定量方法は,次のいずれかによる。
(1) 塩化ナトリウム滴定法(ゲイ・リュサック法) この方法は,BAg-7,7A,7B,18及び21のすずを
含む試料には適用しない。
(2) チオシアン酸アンモニウム滴定法(ホルハルト法) この方法は,BAg-1,1A,2,3,4,5,6,7A,
7B,20,20A及び24の銀含有率51wt%以下の試料に適用する。
(3) 塩化銀重量法
8.2
塩化ナトリウム滴定法
8.2.1
要旨 試料を硝酸で分解し,酸化窒素を除去した後,塩化ナトリウム標準溶液で滴定を行い,塩化
銀による白濁が生じなくなる点を終点とする。
8.2.2
試薬 試薬は,次による。
(1) 硝酸(1+1) 塩化物イオンを含まないもの。
(2) 塩化ナトリウム標準溶液A 塩化ナトリウム[JIS K 8005(容量分析用標準試薬)]5.418gを水に溶解
し,1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。この標準溶液1mlは銀0.01g
に相当し,次の方法によって標定を行う。
銀(99.99wt%以上)1.000gをはかり取って分解瓶に移し入れ,硝酸(1+1)15mlを加え,穏やかに加
熱分解する。以下8.2.5(2)〜(4)の手順に従って操作し,塩化ナトリウム標準溶液で滴定を行い,1ml
に相当する銀量を次の式によって算出する。
10
1.0
2
1
−
+
=
V
V
G
f
ここに,
f: 塩化ナトリウム標準溶液A1mlに相当する銀量 (g)
3
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V1: 塩化ナトリウム標準溶液A使用量 (ml)
V2: 塩化ナトリウム標準溶液B使用量 (ml)
G: 銀はかり取り量 (g)
空試験は,次のように行う。
硝酸(1+1)15mlを分解瓶に取り,8.2.5(1),(2)に準じて操作を行った後,塩化ナトリウム標準溶液B
を滴加し,白濁が生じなくなるまでに要した標準溶液の量をもって空試験値とする。
(3) 塩化ナトリウム標準溶液B (2)で調製した標準溶液Aを水で正確に10倍に薄める。
(4) 標準銀溶液(1mgAg/ml) 銀(99.99wt%以上)0.500gをはかり取ってビーカー (300ml) に移し入れ,硝
酸(1+1)5〜7mlで加熱分解した後,引き続き加熱して酸化窒素を除去し,常温まで冷却した後500ml
の全量フラスコに水を用いて移し入れ,水で標線まで薄める。この溶液は,褐色瓶中に保存する。
8.2.3
器具 分解瓶は,通常付図1のようなものを用いる。良質の試薬瓶 (250ml) を用いてもよい。
8.2.4
試料はかり取り量 試料はかり取り量は,銀含有率に応じ,次の式によって求めた量とする。
なお,銀含有率が未知の場合には,他の方法で大体の銀含有率を調べておく。
100
000
1
×
=
S
m
ここに, m: 計算で求めた試料はかり取り量 (g)
S: 試料中の銀予想含有率 (wt%)
8.2.5
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取って分解瓶に移し入れ,試料1gについて硝酸(1+1)15mlを加え,徐々に温度を上げて
加熱し,穏やかに分解する。
(2) 分解した後,瓶を軽く揺り動かし,曲がったガラス管を分解瓶内に挿入し,空気を吹き込んで酸化窒
素を追い出す。この操作を褐色のガスが出なくなるまで数回繰り返し,熱源から下ろして冷却する。
瓶の内壁を洗瓶で水洗し,水を加えて液量を約50mlにする。
(3) 塩化ナトリウム標準溶液Aをピペットで正確に100ml加え,栓をして激しく振り混ぜ沈殿を凝集させ
た後,上澄み液が透明になるまで静置する(1)。
(4) 栓を取り,ビュレットから塩化ナトリウム標準溶液B0.2mlを,瓶の内壁を伝わらせながら静かに加え
る。塩化銀による白濁を生じた後(2),栓をして激しく振り混ぜ静置する。塩化ナトリウム標準溶液
B0.2mlの添加によって白濁が生じなくなるまで上記の操作を繰り返す。
注(1) 生成した塩化銀は日光のために分解されるので,黒い布などで分解瓶を包んでこの操作を行い,
以後の滴定も,このまま光を遮りながら行う。
(2) このとき白濁が生じなければ,溶液を振り混ぜながら標準銀溶液を0.2mlずつ加えて過剰の塩
化物イオンを沈殿させ,白濁が生じなくなってから更にその0.4mlを過剰に加え,以下8.2.5(4)
の手順に従って操作する。ただし,この操作を行った場合には,銀含有率計算の際に添加した
銀の量を差し引いて補正しなければならない。
8.2.6
計算 試料中の銀含有率を,次の式によって算出する(3)。
100
10
1.0
%
2
1
×
×
−
+
=
m
f
V
V
wt
銀
ここに, V1: 塩化ナトリウム標準溶液A使用量 (ml)
V2: 塩化ナトリウム標準溶液B使用量 (ml)(4)
f: 塩化ナトリウム標準溶液A1mlに相当する銀量 (g)
m: 試料はかり取り量 (g)
4
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(3) 空試験は,8.2.2(2)を参照のこと。
(4) 塩化ナトリウム標準溶液Bの使用量には,添加したときの白濁を生じなかった最後の0.2mlを
含めない。
8.3
チオシアン酸アンモニウム滴定法
8.3.1
要旨 試料を硝酸で分解し,酸化窒素を除去した後,硫酸鉄(III)アンモニウムを指示薬として加え,
チオシアン酸アンモニウム標準溶液で滴定を行い,溶液が微赤褐色を呈した点を終点とする。
8.3.2
試薬
(1) 硝酸(1+1) 塩化物イオンを含まないもの。
(2) チオシアン酸アンモニウム標準溶液 チオシアン酸アンモニウム4.0gを水に溶解して1 000mlとする。
この標準溶液1mlは,銀0.005g強に相当し,次の方法によって標定を行う。
銀(99.99wt%以上)0.250gをはかり取ってコニカルビーカー (300ml) に移し入れ,硝酸(1+1)15ml
を加え,徐々に温度を上げて加熱し,静かに分解する。
以下8.3.4(2),(3)の手順に従って操作し,チオシアン酸アンモニウム標準溶液で滴定を行い,1ml
に相当する銀量を次の式によって算出する。
V
G
f=
ここに,
f: チオシアン酸アンモニウム標準溶液1mlに相当する銀量 (g)
G: 銀はかり取り量 (g)
V: チオシアン酸アンモニウム標準溶液使用量 (ml)
(3) 硫酸鉄(III)アンモニウム溶液 硫酸鉄(III)アンモニウム・12水を水に溶解して飽和させ,溶液が黄色
を呈するまで硝酸(1+1)を滴加する。
8.3.3
試料はかり取り量 試料はかり取り量は,0.5gとする。
8.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってコニカルビーカー (300ml) に移し入れ,時計皿で覆い,硝酸(1+1)15mlを加え,
徐々に温度を上げて加熱し,穏やかに分解する。
(2) 引き続き加熱して酸化窒素を追い出し,放冷後時計皿の下面及びビーカーの内壁を水で洗い,水を加
えて約150mlに薄める。
(3) これに硫酸鉄(III)アンモニウム溶液3mlを指示薬として加え,十分にかき混ぜながら,チオシアン酸
アンモニウム標準溶液で滴定を行い,最後の1滴で溶液全体にわたり微赤褐色を呈したときを終点と
する。
8.3.5
計算 試料中の銀含有率を,次の式によって算出する。
100
%
×
×
=
m
f
V
wt
銀
ここに, V: チオシアン酸アンモニウム標準溶液使用量 (ml)
f: チオシアン酸アンモニウム標準溶液1mlに相当する銀量 (g)
m: 試料はかり取り量 (g)
8.4
塩化銀重量法
8.4.1
要旨 試料を硝酸で分解し,沈殿があれば,ろ過した後,塩酸を加えて銀を沈殿させる。ガラスろ
過器を用いてこし分けて乾燥した後,その質量をはかる(ろ液は銅の定量に用いる。)。
8.4.2
試薬 試薬は,次による。
(1) 塩酸 (1+9)
5
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 硝酸(1+1,1+100) 塩化物イオンを含まないもの。
8.4.3
試料はかり取り量 試料はかり取り量は,1.0gとする。
8.4.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (300ml) に移し入れ,時計皿で覆い,硝酸(1+1)10mlを加え,穏やかに
加熱して分解した後,煮沸して酸化窒素を追い出す。時計皿の下面及びビーカーの内壁を水で洗い,
温水を加えて液量を約50mlにする。
このとき,もし沈殿がある場合には,温所に約1時間静置後,少量のろ紙パルプを入れてあるろ紙
(5種B)を用いて手早くろ過し,温硝酸(1+100)で洗浄する。
(2) 水を加えて液量を約150mlとし,かき混ぜながら塩酸(1+9)を少量ずつ加えて塩化銀の沈殿を生成さ
せる。沈殿が生じなくなった後,更にその1mlを過剰に加えて十分にかき混ぜ,約5分間静かに加熱
し煮沸する。
(3) 暗所に1夜間放置した後,あらかじめ恒量としたガラスろ過器 (1G4) を用いてこし分け,初めは硝酸
(1+100)で,次に冷水で十分に洗浄する(ろ液及び洗液は銅の定量に用いる。)。
(4) 沈殿をガラスろ過器と共に約130℃の空気浴中で約1時間乾燥し,デシケーター中で約1時間放冷し
た後,その質量をはかり,恒量となるまでこの操作を繰り返す。
8.4.5
計算 試料中の銀含有率を,次の式によって算出する。
100
6
752
.0
%
0
1
×
×
=
m
m
wt
銀
ここに, m1: 塩化銀の質量 (g)
m0: 試料はかり取り量 (g)
9. 銅定量方法
9.1
定量方法 銅の定量方法は,銅電解重量法による。
9.2
銅電解重量法
9.2.1
要旨 8.4.1で得られたろ液に硫酸を加え,加熱蒸発して白煙を発生させ放冷した後,水及び硝酸
を加えて電解し,陰極に析出した銅の質量をはかる(電解後の残液は,ニッケル,亜鉛及びカドミウムの
定量に用いる。)。
9.2.2
試薬 試薬は,次による。
(1) 硝酸(1+1)
(2) 硫酸(1+1)
(3) エタノール (99.5vol%)
9.2.3
装置及び器具 装置及び器具は,原則として次のものを用いる。
(1) 電解用ビーカー(付図2参照)
(2) 白金電極A(付図3参照)
(3) 白金電極B(付図4参照)
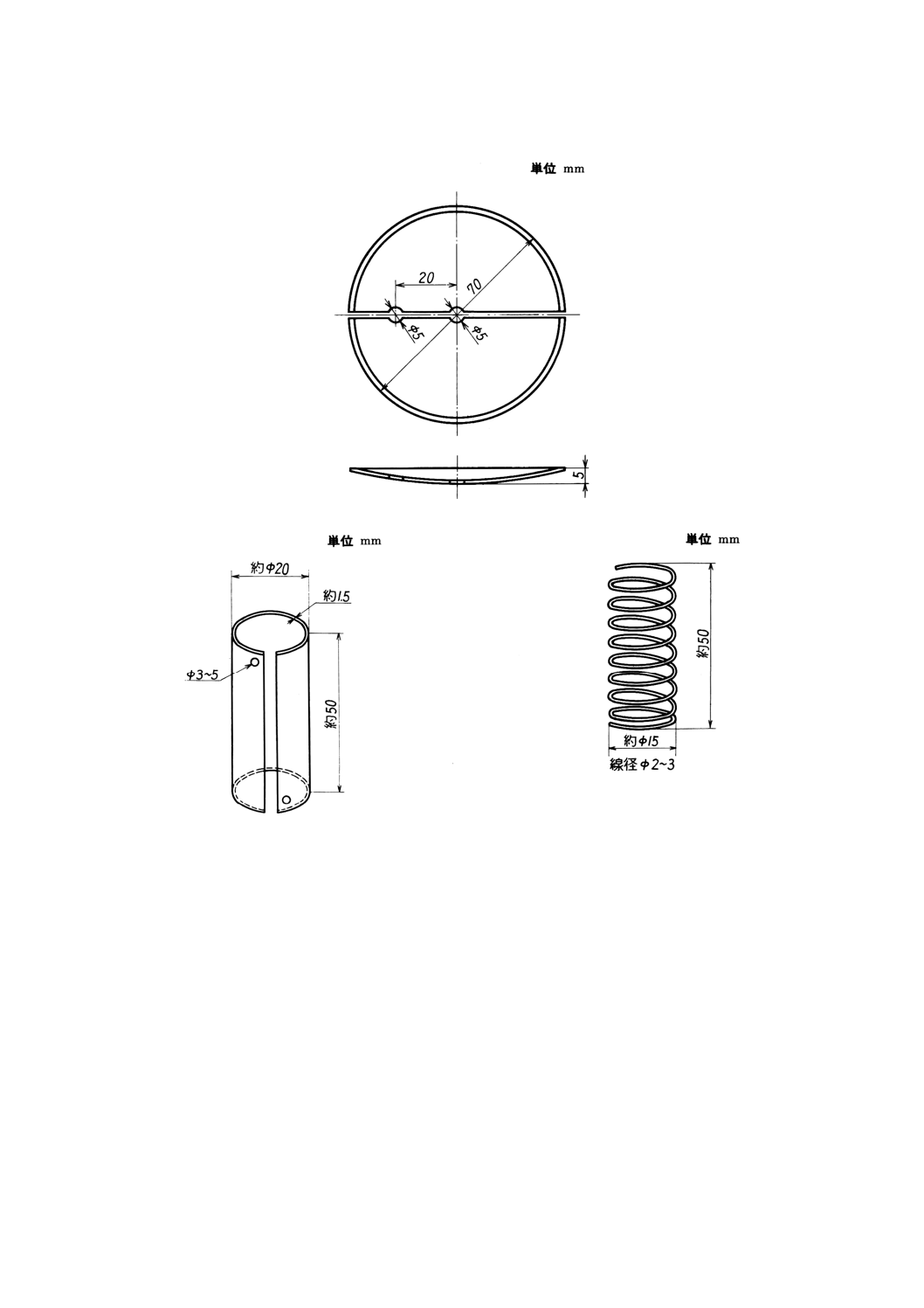
(4) 半円形時計皿(付図5参照)
9.2.4
操作 定量操作は,次の手順によって行う。
(1) 8.4.4(3)で得られたろ液及び洗液に,硫酸(1+1)10mlを加え,加熱蒸発して硫酸の白煙を発生させる。
放冷した後,水約50mlと硝酸(1+1)5mlを加えて可溶性塩類を溶解し,1〜2分間煮沸して酸化窒素を
追い出す。
6
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) この溶液を電解用ビーカーに移し入れ,水を加えて液量を約150mlとし,あらかじめ質量をはかった
白金電極Aを陰極に,白金電極Bを陽極に用い,2個の半円形時計皿で覆い,室温で0.3〜0.5Aの電
流を通じ,7〜8時間電解する。
(3) 少量の水で時計皿の下面,ビーカーの内壁及び両極の柄の液面上に露出した部分を洗い,電解液面を
約5mm上昇させて,更に約30分間電解を続ける。
(4) 新しく電解液中に浸った電極Aの柄に銅が析出しなくなれば,電流を通じたまま水洗しながら両極を
徐々に引き上げ,手早く新たな水中に浸して電極Aを離し,静かに数回上下して水洗する(5)。
エタノールを用いてよく洗った後,直ちに約80℃の空気浴中で速やかに乾燥し,デシケーター中で
約30分間放冷後,その質量をはかる。
注(5) 電解残液及び洗液は合わせ,ニッケル,亜鉛及びカドミウムの定量用に保存する。
9.2.5
計算 試料中の銅含有率を,次の式によって算出する。
100
%
0
1×
=mm
wt
銀
ここに, m1: 電極Aに析出した銅の質量 (g)
m0: 試料はかり取り量 (g)
10. ニッケル定量方法
10.1 定量方法の区分 ニッケルの定量方法は,次のいずれかによる。
(1) ジメチルグリオキシムニッケル重量法
(2) ジメチルグリオキシム分離エチレンジアミン四酢酸二ナトリウム滴定法
(3) 原子吸光法(A法) この方法は,BAg-21以外のすずを含まない試料に適用する。
(4) 原子吸光法(B法) この方法は,BAg-21のすずを含む試料に適用する。
10.2 ジメチルグリオキシムニッケル重量法
10.2.1 要旨 9.2.1で得られた電解残液に,酒石酸及び塩化アンモニウムを加えた後,アンモニア性とし,
ジメチルグリオキシムを加えてニッケルを沈殿させる。この沈殿をガラスろ過器でこし分け,温水で洗浄
した後(ろ液は亜鉛及びカドミウムの定量に用いる。),乾燥し,その質量をはかる。
10.2.2 試薬 試薬は,次による。
(1) アンモニア水
(2) 塩化アンモニウム溶液 (250g/l)
(3) 酒石酸溶液 (250g/l)
(4) ジメチルグリオキシム溶液 ジメチルグリオキシム1.0gをエタノール (95vol%) 100mlに溶解する。
(5) メチルレッド溶液 メチルレッド0.20gをエタノール (95vol%) 90mlに溶解し,水で100mlとする。
10.2.3 操作 定量操作は,次の手順によって行う。
(1) 9.2.4(4)で得られた電解残液を,ビーカー (500ml) に移し入れ,酒石酸溶液20ml及び塩化アンモニウ
ム溶液20mlを加え,メチルレッド溶液を指示薬としてアンモニア水で中和した後,その3mlを過剰
に加え,水で液量を約300mlに薄める。
(2) この溶液を約90℃に加熱し,かき混ぜながらジメチルグリオキシム溶液をニッケル予想含有量10mg
につき7mlの割合で加え,十分にかき混ぜた後,室温まで放冷する。
(3) この沈殿をあらかじめ恒量としたガラスろ過器 (1G4) を用いてこし分け,温水で十分に洗浄する(ろ
液及び洗液は,亜鉛及びカドミウムの定量に用いる。)。
7
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(4) 沈殿をガラスろ過器と共に110〜120℃の空気浴中で約1時間乾燥し,デシケーター中で約1時間放冷
後,その質量をはかり,恒量となるまでこの操作を繰り返す。
10.2.4 計算 試料中のニッケル含有率を,次の式によって算出する。
100
2
203
.0
%
0
1
×
×
=
m
m
wt
ニッケル
ここに, m1: ジメチルグリオキシムニッケルの質量 (g)
m0: 試料はかり取り量 (g)
10.3 ジメチルグリオキシム分離EDTA滴定法
10.3.1 要旨 9.2.1で得られた電解残液を,10.2.1に従って処理し,得られたジメチルグリオキシムニッケ
ルの沈殿を,ろ紙を用いてこし分け(ろ液は亜鉛及びカドミウムの定量に用いる。),塩酸で溶解した後,
エチレンジアミン四酢酸二ナトリウム(以下,EDTAという。)標準溶液の過剰を加え,エリオクロムブラ
ックT(以下,EBTという。)を指示薬としてアンモニア性とし,亜鉛標準溶液で逆滴定する。
10.3.2 試薬 試薬は,次による。
(1) 塩酸(2+1,1+50)
(2) アンモニア水(1+1)
(3) 0.02mol/l亜鉛標準溶液 亜鉛 (JIS K 8005) 1.308gをはかり取り,なるべく少量の塩酸(1+1)で加熱分
解し,常温まで冷却した後,1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
(4) 0.02mol/lEDTA標準溶液 エチレンジアミン四酢酸二ナトリウム二水和物7.45gを水に溶解して1
000mlとする。標定は次のように行う。
0.02mol/lEDTA標準溶液を正確に25ml取り,塩化アンモニウム溶液 (250g/l) 10ml及び指示薬とし
てEBT溶液2〜3滴を加え,水で液量を約100mlに薄めた後,溶液が青色に変わるまでアンモニア水
(1+1)を滴加し,0.02mol/l亜鉛標準溶液で滴定し,溶液が赤紫色に変わった点を終点とし,次の式に
よってファクターを算出する。
25
V
F=
ここに, F: 0.02mol/lEDTA標準溶液のファクター
V: 0.02mol/l亜鉛標準溶液の使用量 (ml)
(5) EBT溶液 EBT0.3gをエタノール (95vol%) 15mlに溶解後,トリエタノールアミン15mlを加える。
10.3.3 操作 定量操作は,次の手順によって行う。
(1) 9.2.4(4)で得られた電解残液を,10.2.3(1),(2)の手順に従って操作する。
(2) この沈殿を,ろ紙(5種A)を用いてこし分け,温水で十分に洗浄する(ろ液及び洗液は,亜鉛及び
カドミウムの定量に用いる。)。ろ紙上の沈殿は温水及び塩酸(2+1)10mlを注いで元のビーカーに洗い
落とし,ろ紙は温水及び温塩酸(1+50)で数回洗浄する。
(3) この溶液に,ニッケル予想含有量10mgについて0.02mol/lEDTA標準溶液を正確に10mlの割合で加え
た後,更にその5mlを過剰に加える。2〜3回振り混ぜ,EBT溶液2〜3滴を指示薬として加えた後,
溶液が青色に変わるまでアンモニア水(1+1)を滴加し(6),直ちに0.02mol/l亜鉛標準溶液で滴定し,溶
液が赤紫色に変わった点を終点とする。
注(6) このときpHを約8.0に調節することが必要であり,この操作によってpHは約8.0となる。
10.3.4 計算 試料中のニッケル含有率を,次の式によって算出する。
8
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(
)
100
174
001
.0
%
2
1
×
×
−
=
m
V
V
wt
ニッケル
ここに, V1: 0.02mol/lEDTA標準溶液使用量 (ml)
V2: 0.02mol/l亜鉛標準溶液使用量 (ml)
m: 試料はかり取り量 (g)
10.4 原子吸光法(A法)
10.4.1 要旨 試料を硝酸で分解した後,原子吸光光度計の空気・アセチレンフレーム中に噴霧し,その吸
光度を測定する。
10.4.2 試薬 試薬は,次による。
(1) 硝酸(1+1)
(2) 銀(99.99wt%以上)
(3) 銅(99.96wt%以上)
(4) 亜鉛(99.99wt%以上)
(5) カドミウム(99.99wt%以上)
(6) 標準ニッケル溶液 (0.5mgNi/ml) ニッケル(99.95wt%以上)1.000gを硝酸(1+1)30mlで加熱分解し,
常温まで冷却した後,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄め,原液
(10mgNi/ml) とする。この原液を使用の都度,必要量だけ水で正しく20倍に薄め,標準ニッケル溶液
とする。
10.4.3 試料はかり取り量 試料はかり取り量は,1.0gとする。
10.4.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (200ml) に移し入れ,時計皿で覆い,硝酸(1+1)20mlを加えて加熱分解
する。分解が終われば,時計皿の下面及びビーカーの内壁を水洗する。
(2) 常温まで冷却した後,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。この溶液
から正しく10mlを分取し,別の100mlの全量フラスコに入れ,水で標線まで薄める(7)。
(3) この溶液の一部を,原子吸光光度計の空気・アセチレンフレーム中に噴霧して波長341.5nm(7)におけ
る吸光度を測定する。
注(7) 使用する原子吸光光度計の種類によって試料溶液の分取比を変えてもよい。
また,測定波長として232.0nmを用いてもよい。いずれの場合でも10.4.5検量線の作成では
同一の条件を用いて作成する。
10.4.5 検量線の作成 試料1g中に含まれるそれぞれの量とほぼ同じになるように銀,銅,亜鉛,カドミ
ウムをはかり取り,ビーカー (200ml) に移し入れ,10.4.4(1)の時計皿で覆い以降の手順に従って操作する。
常温まで冷却した後,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。この溶液から
正しく10mlずつを分取して数個の100mlの全量フラスコに入れ,標準ニッケル溶液0〜8.0ml(ニッケル
として0〜4.0mg)を段階的に加えた後,水で標線まで薄める。以下,10.4.4(3)の手順に従って操作し,試
料と並行して測定した吸光度とニッケル量との関係線を作成して検量線とする。
10.4.6 計算 10.4.5で作成した検量線からニッケル量を求め,試料中のニッケル含有率を,次の式によっ
て算出する。
100
%
×
×
=
B
m
A
wt
ニッケル
ここに, A: 分取した試料溶液中のニッケル検出量 (g)
m: 試料はかり取り量 (g)
9
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B: 分取比
10.5 原子吸光法(B法)
10.5.1 要旨 試料を硝酸とふっ化水素酸との混酸で分解した後,原子吸光光度計の空気・アセチレンフレ
ーム中に噴霧し,その吸光度を測定する。
10.5.2 試薬 試薬は,次による。
(1) ほう酸溶液 (50g/l)
(2) 混酸 硝酸(2+1)1lにふっ化水素酸25mlを加える。
(3) 銀(99.99wt%以上)
(4) 銅(99.96wt%以上)
(5) すず(99.90wt%以上)
(6) 標準ニッケル溶液10.4.2(6)に従って調製する。
10.5.3 試料はかり取り量 試料はかり取り量は,1.0gとする。
10.5.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取って100mlのポリエチレン製全量フラスコ(8)に移し入れ,混酸10mlを加えて室温で
放置して分解する(9)。分解が終わった後,常温まで冷却し,水で標線まで薄める。この溶液から正し
く10mlを分取し,別の100mlのポリエチレン製全量フラスコ(8)に入れ,水で標線まで薄める(7)。
(2) この溶液の一部を原子吸光光度計の空気・アセチレンフレーム中に噴霧して波長341.5nm(7)における
吸光度を測定する。
注(8) ポリエチレン製全量フラスコ(ポリプロピレン製全量フラスコを用いてもよい。)は,JIS K 0050
の9.3(2)によって補正を行って使用する。
注(9) 室温及び混酸の液温が低い場合には,20℃以上に加熱して分解を行う。
備考 10.5.4(1)の操作を,次のように行ってもよい。
試料をはかって,ポリエチレン製ビーカー(100ml〉に移し入れ,混酸10mlを加えて室温で
放置して分解する(9)。これにほう酸溶液10mlを加えた後,100mlのガラス製全量フラスコに移
し入れ,水で標線まで薄める。この溶液から正しく10mlを分取し,別の100mlのガラス製全
量フラスコに入れ,水で標線まで薄める。以下,10.5.4(2)の手順に従って操作し,ニッケルを
定量する。ただし,この場合の検量線は,この操作を用いて作成する。
10.5.5 検量線の作成 試料1g中に含まれるそれぞれの量とほぼ同じになるように銀,銅,すずをはかり
取り,100mlのポリエチレン製全量フラスコ(8)に移し入れ,混酸10mlを加えて室温で放置して分解する(9)。
分解が終わった後常温まで冷却し,水で標線まで薄める。この溶液から10.0mlずつを分取して数個の100ml
のポリエチレン製全量フラスコ(8)に入れ,標準ニッケル溶液0〜8.0ml(ニッケルとして0〜4.0mg)を段階
的に加えた後,水で標線まで薄める。以下,10.5.4(2)の手順に従って操作し,試料と並行して測定した吸
光度とニッケル量との関係線を作成して検量線とする。
10.5.6 計算 10.5.5で作成した検量線からニッケル量を求め,試料中のニッケル含有率を,次の式によっ
て算出する。
100
%
×
×
=
B
m
A
wt
ニッケル
ここに, A: 分取した試料溶液中のニッケル検出量 (g)
m: 試料はかり取り量 (g)
B: 分取比
10
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11. 亜鉛定量方法
11.1 定量方法の区分 亜鉛の定量方法は,次のいずれかによる。
(1) EDTA滴定法(A法) この方法は,BAg-1,1A,2,5,6,7,7A,7B,20,20Aのニッケルを含ま
ない試料に適用する。
(2) EDTA滴定法(B法) この方法は,BAg-3,4,24のニッケルを含む試料に適用する。
11.2 EDTA滴定法(A法)
11.2.1 要旨 9.2.1で得られた電解残液に酒石酸を加え,水酸化ナトリウムで中和後,pHを10にし,EBT
を指示薬としてEDTA標準溶液で滴定する。カドミウムを含む試料の場合は,この滴定残液を12.2.1に従
ってカドミウム量を求め,前後の滴定値の差から亜鉛量を求める。
11.2.2 試薬 試薬は,次による。
(1) 水酸化ナトリウム溶液 (200g/l)
(2) 緩衝溶液 (pH10) 塩化アンモニウム70gを水に溶解し,アンモニア水570mlを加え,水で1 000ml
とする。
(3) 酒石酸溶液 (250g/l)
(4) 0.02mol/lEDTA標準溶液 10.3.2(4)に従って調製及び標定を行う。
(5) メチルオレンジ溶液 (1g/l)
(6) EBT溶液 10.3.2(5)に従って調製する。
11.2.3 操作 定量操作は,次の手順によって行う。
(1) 9.2.4(4)の電解残液に酒石酸溶液20ml及び指示薬としてメチルオレンジ溶液1〜2滴を加え,水酸化ナ
トリウム溶液で中和する。この溶液を500mlの全量フラスコに移し入れ,水で標線まで薄める。
(2) この溶液から正確に100mlをビーカー (300ml) に分取し,40〜50℃に加熱した後,緩衝溶液2mlを加
え,EBT溶液3〜4滴を指示薬として加え,直ちに0.02mol/lEDTA標準溶液で滴定し,溶液が完全に
青色に変わった点を終点(10)とする (V1ml) (滴定残液は,BAg-1,1A,2のカドミウムの定量に用い
る。)。
注(10) しばらくすると元の赤味を帯びた色に変化する場合があるが,初めに完全に青色に変わった点
を終点とする。
11.2.4 計算 試料中の亜鉛含有率を,次の式によって算出する。
(1) カドミウムを含まない場合(BAg-5,6,7,7A,7B,20,20Aの試料)
100
5
1
308
001
.0
%
1
×
×
×
=
m
V
wt
亜鉛
(2) カドミウムを含む場合(BAg-1,1A,2の試料)
(
)
100
5
1
308
001
.0
%
2
1
×
×
×
−
=
m
V
V
wt
亜鉛
ここに, V1: 0.02mol/lEDTA標準溶液使用量 (ml)
V2: 12.2.3(2)で得られた0.02mol/l硫酸マグネシウム標準溶液使用
量 (ml)
m: 試料はかり取り量 (g)
11
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.3 EDTA滴定法(B法)
11.3.1 要旨 10.2.1又は10.3.1で得られたろ液を塩酸で酸性とした後,水酸化ナトリウムで中和後,11.2.1
に従って操作する。
11.3.2 試薬 試薬は,次による。
(1) 塩酸(1+1)
(2) 水酸化ナトリウム溶液 (200g/l)
(3) メチルオレンジ溶液 (1g/l)
11.3.3 操作 定量操作は,次の手順によって行う。
(1) 10.2.3(3)又は10.3.3(2)のろ洗液及び洗液に,メチルオレンジ溶液1〜2滴を指示薬として加え,塩酸(1
+1)で弱酸性とした後,水酸化ナトリウム溶液で中和し,500mlの全量フラスコに水を用いて移し入
れ,水で標線まで薄める。
(2) 以下11.2.3(2)の手順に従って操作する(滴定残液は,BAg-3のカドミウムの定量に用いる。)。
11.3.4 計算 試料中の亜鉛含有率を,次の式によって算出する。
(1) カドミウムを含まない場合(BAg-4の試料)
100
5
1
308
001
.0
%
1
×
×
×
=
m
V
wt
亜鉛
(2) カドミウムを含む場合(BAg-3の試料)
(
)
100
5
1
308
001
.0
%
2
1
×
×
×
−
=
m
V
V
wt
亜鉛
ここに, V1: 0.02mol/lEDTA標準溶液使用量 (ml)
V2: 12.2.3(2)で得られた0.02mol/l硫酸マグネシウム標準溶液使用
量 (ml)
m: 試料はかり取り量 (g)
12. カドミウム定量方法
12.1 定量方法 カドミウムの定量方法は,ジエチルジチオカルバミン酸ナトリウム(以下,DDTCとい
う。)分離EDTA滴定法による。この方法は,BAg-1〜3に適用する。
12.2 DDTC分離EDTA滴定法
12.2.1 要旨 11.2.1又は11.3.1で得られた滴定残液に,DDTCを加えてカドミウムを沈殿させ,遊離した
EDTAを硫酸マグネシウム標準溶液で滴定する。
12.2.2 試薬 試薬は,次による。
(1) 塩酸(1+4,1+10)
(2) 水酸化ナトリウム溶液 (200g/l)
(3) 緩衝溶液 (pH10) 11.2.2(2)に従って調製する。
(4) DDTC溶液 DDTC10gを水100mlに加熱溶解する。
(5) 洗浄溶液 緩衝溶液20mlにDDTC溶液10mlを加え,水で1 000mlとする。
(6) 0.02mol/l硫酸マグネシウム標準溶液 硫酸マグネシウム七水和物4.93gを水に溶解して1 000mlとす
る。
12
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
標定は,次のように行う。
0.02mol/lEDTA標準溶液を正確に25ml取り,水を加えて約100mlとし,緩衝溶液2ml及び指示薬と
してEBT溶液数滴を加えて0.02mol/l硫酸マグネシウム標準溶液で滴定し,溶液が青色から完全に赤
紅色に変わった点を終点とし,次の式によってファクターを算出する。
V
F
F
1
2
00
.
25
×
=
ここに, F2: 0.02mol/l硫酸マグネシウム標準溶液のファクター
F1: 0.02mol/lEDTA標準溶液のファクター
V: 0.02mol/l硫酸マグネシウム標準溶液使用量 (ml)
(7) 0.02mol/lEDTA標準溶液 10.3.2(4)に従って調製及び標定を行う。
(8) メチルオレンジ溶液 (1g/l)
(9) EBT溶液 10.3.2(5)に従って調製する。
12.2.3 操作 定量操作は,次の手順によって行う。
(1) 11.2.3(2)又は11.3.3(2)で得られた滴定残液に,DDTC溶液25〜30mlを加え,激しくかき混ぜ,約50℃
に加熱してカドミウムの沈殿を凝縮させ,ろ紙(5種c)を用いてろ過し,ろ液をビーカー (500ml) に
受け,沈殿を洗浄溶液で十分に洗浄する。
(2) ろ液及び洗液に,緩衝溶液5mlを加えた後,EBT溶液4〜5滴を指示薬として加え,0.02mol/l硫酸マ
グネシウム標準溶液で滴定し,溶液が完全に赤紅色に変わった点を終点とする (V2ml)。
12.2.4 計算 試料中のカドミウム含有率を,次の式によって算出する。
100
5
1
248
002
.0
%
2
×
×
×
=
m
V
wt
カドミウム
ここに, V2: 0.02mol/l硫酸マグネシウム標準溶液使用量 (ml)
m: 試料はかり取り量 (g)
備考 ジエチルジチオカルバミン酸のカドミウム錯体の沈殿からカドミウムを定量することができる。
この場合は次のように操作する。
12.2.3(1)の手順によって生じた沈殿を,ろ紙と共に元のビーカーに移し入れ,塩酸(1+4)50ml
を加えて煮沸し溶解する。これをコニカルビーカー (500ml) 中にろ紙(5種A)を用いてろ過
し,温塩酸(1+10)で数回,次に温水で十分に洗浄する。ろ液及び洗液はメチルオレンジ溶液1
〜2滴を加えて水酸化ナトリウム溶液で中和した後,緩衝溶液5mlを加え,EBT溶液4〜5滴を
指示薬として加え0.02mol/lEDTA標準溶液で滴定し,溶液が完全に青色に変わった点を終点と
し,試料中のカドミウム含有率を,次の式によって算出する。
100
5
1
248
002
.0
%
3
×
×
×
=
m
V
wt
カドミウム
ここに, V3: 0.02mol/lEDTA標準溶液使用量 (ml)
m: 試料はかり取り量 (g)
13. すず定量方法
13.1 定量方法の区分 すずの定量方法は,次のいずれかによる。この方法は,BAg-7,7A,7B,18,21
に適用する。
13
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) よう素酸カリウム滴定法
(2) 酸化すず(IV)重量法
(3) 原子吸光法 この方法は,BAg-18には適用しない。
13.2 よう素酸カリウム滴定法
13.2.1 要旨 試料を硝酸で分解し,すずをメタすず酸とした後,硝酸と温水で可溶性塩類を溶解し,こし
分ける。沈殿は,硝酸と硫酸で分解後,硫酸白煙を発生させた後,塩酸に溶解し,三角フラスコに入れ,
炭酸水素ナトリウムを入れたキャップ付きゴム栓を施し,塩化アンチモン(III)及び鉛などですずを還元し,
でんぷん溶液を指示薬として,よう素酸カリウム標準溶液で滴定する。
13.2.2 試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸
(3) 硝酸(1+1,1+50)
(4) 硫酸
(5) 鉛 JIS H 2105(鉛地金)の特種相当品〔JIS K 8701[鉛(試薬)]の試金用粒状のものを用いると便
利である。〕。
(6) ニッケルシリンダー又はニッケル線 純度 (Ni+Co) 99.5wt%以上で,原則として付図6のA又はB
の形状とする。このシリンダー又は線は,繰り返し使用することができる。
(7) 二酸化炭素 酸素を含んでいる場合には,塩化銅(I)45gを塩酸(1+1)300mlに溶解し,少量の金属銅削
片を加え,密栓して24時間以上放置した溶液に二酸化炭素を通じて精製する。
(8) 炭酸水素ナトリウム溶液(飽和)
(9) 塩化アンチモン(III)溶液 塩化アンチモン(III)5gを塩酸(1+1)500mlに加熱溶解する。
(10) 標準すず溶液 (4mgSn/ml) すず(99.90wt%以上)1.000gをはかり取ってトールビーカー (300ml) に
移し入れ,硫酸20mlを加え,時計皿で覆い,加熱分解した後,引き続き加熱し,時計皿及びビーカ
ーの内壁に付着した硫黄を揮散させる。冷却後,水及び塩酸(1+1)を用いて時計皿及びビーカーの内
壁を洗い,塩酸(1+1)50mlを加えて可溶性塩類を溶解し,250mlの全量フラスコに塩酸(1+1)を用いて
移し入れ,塩酸(1+1)で標線まで薄める。又は,すず1.000gを水浴上で白金板を触媒として塩酸50ml
で分解し,250mlの全量フラスコに塩酸(1+1)を用いて移し入れ,塩酸(1+1)で標線まで薄めてもよい。
(11) 0.05Nよう素酸カリウム標準溶液 よう素酸カリウム1.784gを水酸化ナトリウム溶液 (5g/l) 40mlに溶
解し,よう化カリウム5gを加え,水で1 000mlとする。この溶液の標定は次のように行う。
標準すず溶液を正確に25ml取り,還元装置の三角フラスコに入れる。以下13.2.5(4)〜(6)の手順に
従って操作し,1mlに相当するすず量を次の式によって求める。
V
f
100
.0
=
ここに,
f: 0.05Nよう素酸カリウム標準溶液1mlに相当するすず量 (g)
0.100: 標準すず溶液25ml中のすず量 (g)
V: 0.05Nよう素酸カリウム標準溶液使用量 (ml)
(12) でんぷん溶液 でんぷん(溶性)又はでんぷん1gに少量の冷水を加えて泥状とし,100mlの熱水中に
かき混ぜながら加え,約1分間煮沸した後放冷し,不溶解物をろ過する。この溶液は使用の都度調製
し,よう素によって赤褐色となるものを用いてはならない。
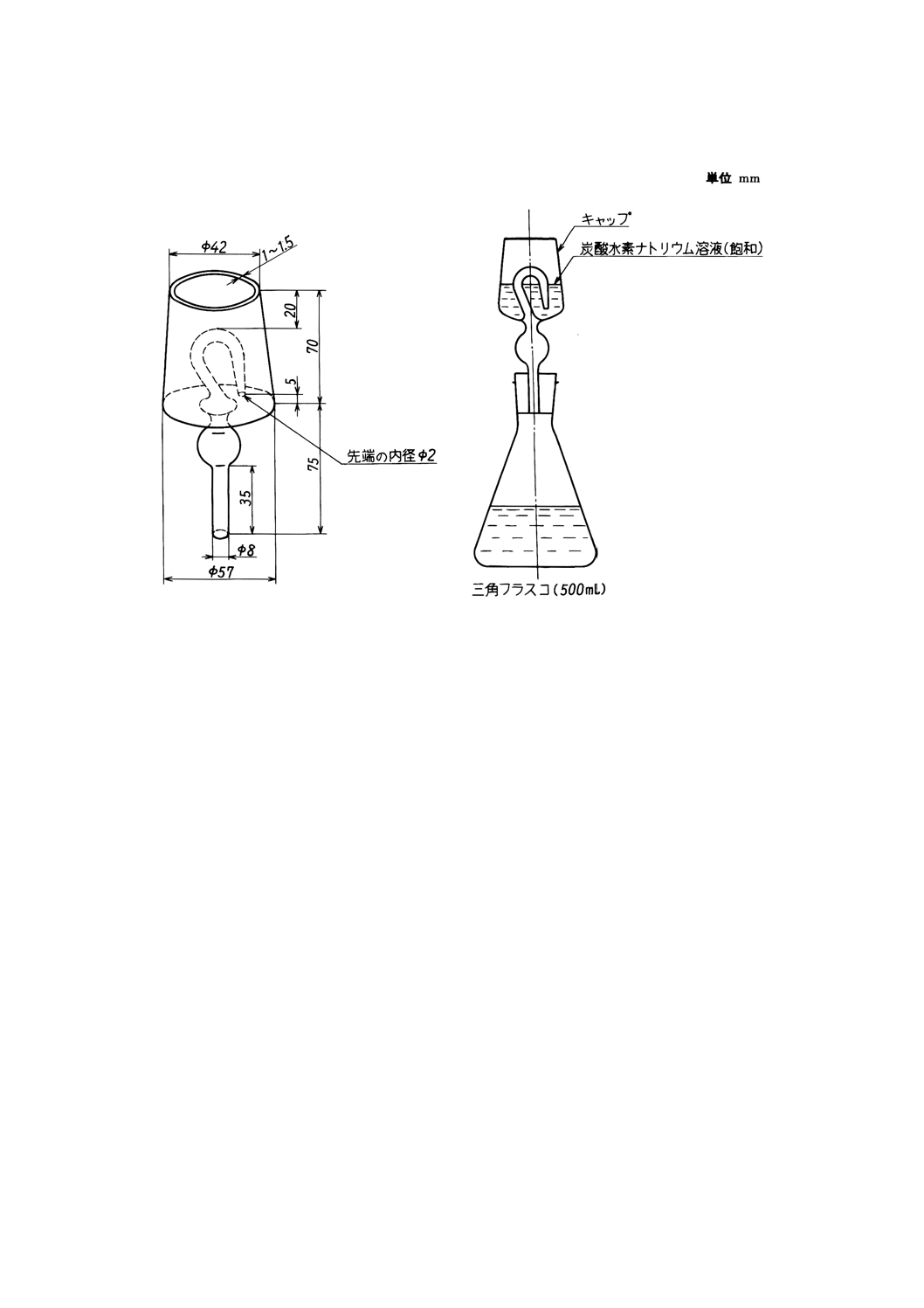
13.2.3 装置及び器具 還元装置は,通常付図7のようなものを用いる。
14
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.2.4 試料はかり取り量 試料はかり取り量は,1.0gとする。
13.2.5 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってトールビーカー (300ml) に移し入れ,時計皿で覆い,硝酸(1+1)15mlを加え,加
熱して分解する。時計皿の下面及びビーカーの内壁を水で洗浄し,更に水浴上で注意して加熱し,蒸
発乾固する。
(2) 硝酸5ml及び温水約50mlを加え,煮沸して可溶性塩類を溶解し,温所に1〜2時間静置した後,直ち
に少量のろ紙パルプを入れたろ紙(5種C)で沈殿をこし分け,温硝酸(1+50)を用いて十分に洗浄す
る。
(3) 沈殿は,ろ紙と共に元のビーカーに入れ,硝酸15ml及び硫酸10mlを加え,時計皿で覆い,注意して
加熱分解し,更に加熱を続けて硫酸の白煙を十分に発生させる。
もし,この際有機物の分解が不十分のときは,冷却後更に硝酸10mlを加えて加熱分解を繰り返す。
冷却後,時計皿の下面及びビーカーの内壁を水洗した後,時計皿を取り去り,更に加熱蒸発して硫酸
の白煙を十分に発生させる。
(4) 放冷後,水200ml及び塩酸50mlを加えて塩類を溶解し,溶液を三角フラスコ (500ml) に移し入れ,
塩化アンチモン(III)溶液10mlと鉛10g(又はニッケルシリンダー若しくはニッケル線)を加える。
三角フラスコに付図7のキャップを付けたゴム栓を施し,キャップに炭酸水素ナトリウム溶液約
50mlを加え,約30分間静かに煮沸してすずを還元する。
(5) 三角フラスコの熱源を除き,そのまま約5分間放冷する。この間キャップ中の炭酸水素ナトリウム溶
液は三角フラスコ中に流入するから,溶液が減少して空気を吸入するおそれがあれば追加する。次に
水中に浸して10℃以下になるまで冷却する。
(6) キャップと共にゴム栓を外し(11),直ちにゴム栓の下端及び三角フラスコの内壁を水洗(あらかじめ煮
沸して空気を追い出した後,二酸化炭素を通しながら冷却した水を用いる。)した後,でんぷん溶液
5mlを指示薬として加え,0.05Nよう素酸カリウム標準溶液で滴定し,溶液が青紫色に変わった点を
終点とする。
注(11) ニッケルを還元剤として用いた場合は,ニッケルシリンダー又はニッケル線を取り出し,これ
らと共にゴム栓などを水洗する。
13.2.6 計算 試料中のすず含有率を,次の式によって算出する。
100
%
×
×
=
m
f
V
wt
すず
ここに, V: 0.05Nよう素酸カリウム標準溶液使用量 (ml)
f: 0.05Nよう素酸カリウム標準溶液1mlに相当するすず量 (g)
m: 試料はかり取り量 (g)
13.3 酸化すず(IV)重量法
13.3.1 要旨 試料を硝酸で分解して蒸発乾固し,硝酸と温水を加えて可溶性塩類を溶解した後,メタすず
酸の沈殿をこし分ける。沈殿は強熱して酸化すずとし,その質量をはかる。
13.3.2 試薬 試薬は,次による。
(1) 硝酸 塩化物イオンを含まないもの
(2) 硝酸(1+1,1+50) 塩化物イオンを含まないもの
13.3.3 試料はかり取り量 試料はかり取り量は,1.0gとする。
13.3.4 操作 定量操作は,次の手順によって行う。
15
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 試料(12)をはかり取ってトールビーカー (300ml) に移し入れ,時計皿で覆い,硝酸(1+1)15mlを加え,
加熱して分解する。時計皿の下面及びビーカーの内壁を水で洗浄し,更に水浴上で注意して加熱し,
蒸発乾固する。
(2) 硝酸5ml及び温水約50mlを加え,煮沸して可溶性塩類を溶解し,温所に1〜2時間静置した後,直ち
に少量のろ紙パルプを入れたろ紙(5種C)でこし分け,沈殿を完全にろ紙上に移し,温硝酸(1+50)
を用いて十分に洗浄し,洗液に銀イオンの反応を認めなくなるまでとする。
(3) 沈殿はろ紙と共に約100℃の空気浴中で乾燥後,あらかじめ質量をはかった磁器るつぼ中にもみ落と
す。ろ紙は別に灰化した後,前記のるつぼ中に入れ,硝酸2〜3滴で潤す。初めは徐々に加熱し,次第
に温度を高め,最後は約1 000℃で約1時間強熱して恒量とし,デシケーター中で約1時間放冷後,質
量をはかる。
注(12) メタすず酸の沈殿に銀,銅などが吸着,混入するのを防ぐため,試料はなるべく細かく削るこ
とが望ましい。
13.3.5 計算 試料中のすず含有率を,次の式によって算出する。
100
6
787
.0
%
0
1
×
×
=
m
m
wt
すず
ここに, m1: 酸化すず(IV)の質量 (g)
m0: 試料はかり取り量 (g)
13.4 原子吸光法
13.4.1 要旨 試料を硝酸とふっ化水素酸との混酸で分解した後,原子吸光光度計の空気・アセチレンフレ
ーム中に噴霧し,その吸光度を測定する。
13.4.2 試薬 試薬は,次による。
(1) ほう酸溶液 (50g/l)
(2) 混酸 硝酸(2+1)1lにふっ化水素酸25mlを加える。
(3) 銀(99.99wt%以上)
(4) 銅(99.96wt%以上)
(5) 亜鉛(99.99wt%以上)
(6) 標準すず溶液 (0.005gSn/ml) すず(99.90wt%以上)1.250gをはかり取ってポリエチレンビーカー
(200ml) に移し入れ,硝酸(2+1)10mlとふっ化水素酸2mlを加えて水浴上で穏やかに加熱分解する。
常温に冷却した後,250mlのポリエチレン製全量フラスコ(8)に水を用いて移し入れ,水で標線まで薄
める。
13.4.3 試料はかり取り量 試料はかり取り量は,1.0gとする。
13.4.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取って100mlのポリエチレン製全量フラスコ(8)に移し入れ,混酸10mlを加えて室温で
放置して分解する(9)。分解が終わった後,常温まで冷却し,水で標線まで薄める。
(2) この溶液の一部を原子吸光光度計の空気・アセチレンフレーム中に噴霧して,波長286.3nmにおける
吸光度を測定する。
備考 13.4.4(1)の操作を,次のように行ってもよい。
試料をはかり取ってポリエチレン製ビーカー (100ml) に移し入れ,混酸10mlを加えて室温
で放置して分解する(9)。これにほう酸溶液10mlを加えた後,100mlのガラス製全量フラスコに
水を用いて移し入れ,水で標線まで薄める。以下,13.4.4(2)の手順に従って操作し,すずを定
16
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
量する。ただし,この場合の検量線は,この操作を用いて作成する。
13.4.5 検量線の作成 銀0.56g,銅0.22g及び亜鉛0.17gを1組とし,数組をはかり取って,それぞれを
100mlのポリエチレン製全量フラスコ(8)に移し入れ,混酸10mlを加えて室温で放置して分解する(9)。これ
に標準すず溶液0〜12.0ml(すずとして0〜0.06g)を段階的に加えた後,常温まで冷却して水で標線まで
薄める。以下,13.4.4(2)の手順に従って操作し,試料と並行して測定した吸光度とすず量との関係線を作
成して検量線とする。
13.4.6 計算 13.4.5で作成した検量線からすず量を求め,試料中のすず含有率を,次の式によって算出す
る。
100
%
×
=mA
wt
すず
ここに, A: 試料溶液中のすず検出量 (g)
m: 試料はかり取り量 (g)
14. リチウム定量方法
14.1 定量方法 リチウムの定量方法は,原子吸光法による。この方法はBAg-8Aに適用する。
14.2 原子吸光法
14.2.1 要旨 試料を硝酸で分解した後,原子吸光光度計の空気・アセチレンフレーム中に噴霧してその吸
光度を測定する。
14.2.2 試薬 試薬は,次による。
(1) 硝酸(1+1)
(2) 銀・銅混液 銀(99.99wt%以上)1.80g及び銅(99.96wt%以上)0.70gをはかり取ってビーカー (300ml)
に移し入れ,硝酸(1+1)50mlを加えて加熱分解した後,数分間静かに煮沸して酸化窒素を追い出す。
常温まで冷却した後,硝酸(1+1)50mlを加え,500mlの全量フラスコに水を用いて移し入れ,水で標
準まで薄める。
(3) 標準リチウム溶液 (15μgLi/ml) 120℃で恒量となるまで乾燥した硝酸リチウム0.745 0gを水に溶かして
正しく100mlに薄めて原液 (750μgLi/ml) とする。この原液を使用の都度,必要量だけ水で50倍に薄
めて標準リチウム溶液とする。
14.2.3 試料はかり取り量 試料はかり取り量は,0.50gとする。
14.2.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってコニカルビーカー (300ml) に移し入れ,時計皿で覆い,硝酸(1+1)20mlを加え,
静かに加熱して分解する。時計皿の下面及びビーカーの内壁を水で洗い,引き続き数分間煮沸して酸
化窒素を追い出す。
(2) 常温まで冷却した後,1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
(3) この溶液の一部を,原子吸光光度計の空気・アセチレンフレーム中に噴霧して波長670.8nmにおける
吸光度を測定する。
14.2.5 検量線の作成 銀・銅混液10.0mlずつをはかり取って,数個の100mlの全量フラスコに入れ,標
準リチウム溶液0〜10.0ml(リチウムとして0〜150μg)を段階的に加えた後,水で標線まで薄める。以下
14.2.4(3)の手順に従って操作し,試料と並行して測定した吸光度とリチウム量との関係線を作成して検量
線とする。
17
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.2.6 計算 14.2.5で作成した検量線からリチウム量を求め,試料中のリチウム含有率を,次の式によっ
て算出する。
100
%
×
=mA
wt
リチウム
ここに, A: 試料溶液中のリチウム検出量 (g)
m: 試料はかり取り量 (g)
15. 鉛及び鉄の定量方法
15.1 定量方法の区分 鉛及び鉄の定量方法は,次のいずれかによる。
(1) 原子吸光法(A法) この方法は,BAg-7,7A,7B,18,21以外のすずを含まない試料に適用する。
(2) 原子吸光法(B法) この方法は,BAg-7,7A,7B,18,21のすずを含む試料に適用する。
15.2 原子吸光法(A法)
15.2.1 要旨 試料を硝酸で加熱分解した後,原子吸光光度計の空気・アセチレンフレーム中に噴霧してそ
の吸光度を測定する。
15.2.2 試薬 試薬は,次による。
(1) 硝酸(1+1)
(2) 銀(99.99wt%以上)
(3) 銅(99.96wt%以上)
(4) 亜鉛(99.99wt%以上)
(5) カドミウム(99.99wt%以上)
(6) 標準鉛溶液 (25μgPb/ml) 鉛(99.99wt%以上)0.100gを硝酸(1+1)10mlで加熱分解し,常温まで冷却
した後,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄め,原液 (1 000μgPb/ml) とす
る。この原液を使用の都度,必要量だけ水で正しく40倍に薄めて標準鉛溶液とする。
(7) 標準鉄溶液 (25μgFe/ml) 鉄(99.5wt%以上)0.100gを硝酸(1+1)20mlで加熱分解した後,(6)に準じ
て水で薄め,標準鉄溶液を調製する。
15.2.3 試料はかり取り量 試料はかり取り量は,1.0gとする。
15.2.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取ってビーカー (200ml) に移し入れ,時計皿で覆い,硝酸(1+1)20mlを加えて加熱分解
する。分解が終われば,時計皿の下面及びビーカーの内壁を水洗する。
(2) 常温まで冷却した後,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
(3) この溶液の一部を原子吸光光度計の空気・アセチレンフレーム中に噴霧して,鉛については波長
217.0nm又は283.3nm,鉄については波長248.3nmにおける吸光度を測定する。
15.2.5 検量線の作成 試料1g中に含まれるそれぞれの量とほぼ同じになるように銀,銅,亜鉛及びカド
ミウムをはかり取り,これを1組として数組を作製し,それぞれをビーカー (200ml) に移し入れ,15.2.4(1)
の時計皿で覆い,以降の手順に従って操作した後,これに標準鉛溶液0〜8.0ml(鉛として0〜200μg又は
標準鉄溶液0〜8.0ml(鉄として0〜200μg)を段階的に加えた後,15.2.4(2)以降の手順に従って操作し,試
料と並行して測定した吸光度と鉛又は鉄量との関係線を作成して検量線とする。
15.2.6 計算 15.2.5で作成した検量線から鉛又は鉄量を求め,試料中の鉛又は鉄含有率を,次の式によっ
て算出する。
18
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
%
×
=mA
wt
鉄
又は
鉛
ここに, A: 試料溶液中の鉛又は鉄検出量 (g)
m: 試料はかり取り量 (g)
15.3 原子吸光法(B法)
15.3.1 要旨 試料を硝酸とふっ化水素酸との混酸で分解した後,原子吸光光度計の空気・アセチレンフレ
ーム中に噴霧してその吸光度を測定する。
15.3.2 試薬 試薬は,次による。
(1) ほう酸溶液 (50g/l)
(2) 混酸 硝酸(2+1)1lにふっ化水素酸25mlを加える。
(3) 銀[15.2.2(2)による。]
(4) 銅[15.2.2(3)による。]
(5) 亜鉛[15.2.2(4)による。]
(6) すず(99.90wt%以上)
(7) 標準鉛溶液 15.2.2(6)に従って調製する。
(8) 標準鉄溶液 15.2.2(7)に従って調製する。
15.3.3 試料はかり取り量 試料はかり取り量は,1.0gとする。
15.3.4 操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取って100mlのポリエチレン製全量フラスコ(8)に移し入れ,混酸10mlを加えて室温で
放置して分解する(9)。分解が終わった後,常温まで冷却し,水で標線まで薄める。
(2) この溶液の一部を原子吸光光度計の空気・アセチレンフレーム中に噴霧して,鉛については波長
217.0nm又は283.3nm,鉄については波長248.3nmにおける吸光度を測定する。
備考 15.3.4(1)の操作を,次のように行ってもよい。
試料をはかり取って,ポリエチレン製ビーカー (100ml) に移し入れ,混酸10mlを加えて室
温で放置して分解する(8)。これにほう酸溶液10mlを加えた後,100mlのガラス製全量フラスコ
に移し入れ,水で標線まで薄める。以下,15.3.4(2)の手順に従って操作し,鉛又は鉄を定量す
る。ただし,この場合の検量線はこの操作を用いて作成する。
15.3.5 検量線の作成 銀0.56g,銅0.22g,亜鉛0.17g及びすず0.05gを1組として数組をはかり取って,
それぞれを100mlのポリエチレン製全量フラスコ(8)に移し入れ,混酸10mlを加えて室温で放置して分解
する(9)。これに標準鉛溶液0〜8.0ml(鉛として0〜200μg)又は標準鉄溶液0〜8.0ml(鉄として0〜200μg)
を段階的に加えた後,常温まで冷却し,水で標線まで薄める。以下,15.3.4(2)の手順に従って操作し,試
料と並行して測定した吸光度と鉛又は鉄量との関係線を作成して検量線とする。
15.3.6 計算 15.3.5で作成した検量線から鉛又は鉄量を求め,試料中の鉛又は鉄含有率を,次の式によっ
て算出する。
100
%
×
=mA
wt
鉄
又は
鉛
ここに, A: 試料溶液中の鉛又は鉄検出量 (g)
m: 試料はかり取り量 (g)
19
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図1 分解瓶
付図2 電解用ビーカー
付図3 白金電極A
付図4 白金電極B
20
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図5 半円形時計皿
付図6A ニッケルシリンダー
付図6B ニッケル線
21
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図7 還元装置の例
22
Z 3901-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS Z 3901 銀ろう分析方法 改正原案調査作成委員会 構成表
氏名
所属
(委員長)
俣 野 宣 久
川崎製線株式会社
有 賀 正
東海大学
荒 木 誠 司
大蔵省造幣局
内 仲 康 夫
通商産業省基礎産業局
笹 谷 勇
工業技術院標準部
大河内 春 乃
科学技術庁金属材料技術研究所
神 岡 正 男
日本国有鉄道鉄道技術研究所
古 矢 元 佑
清峰金属工業株式会社
松 井 文 夫
日本アイ・ティー・エス株式会社
山 本 博 信
株式会社徳力本店
前 田 修
田中貴金属工業株式会社
吉光寺 博
石福金属興工株式会社
橋 本 謙 三
橋本貴金属工業株式会社
乾 昌 弘
乾庄貴金属化工株式会社
堀 仁
水野ハンディー・ハーマン株式会社
有 井 満
株式会社東芝
岡 島 義 昭
株式会社日立製作所
今 村 孝
三菱電機株式会社
堀 泰 治
東京ラヂエーター製造株式会社
(事務局)
池 原 平 晋
社団法人日本溶接協会