T 11737-2:2013 (ISO 11737-2:2009)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 2
3 用語及び定義 ··················································································································· 2
4 品質マネジメントシステムの要素 ························································································ 3
5 製品の選択 ······················································································································ 4
6 無菌性の試験の実施方法 ···································································································· 5
7 無菌性の試験の実施方法の評価 ··························································································· 6
8 無菌性の試験の実施方法の維持 ··························································································· 6
附属書A(参考)滅菌プロセスのバリデーション及び維持において実施する無菌性の試験の指針 ········· 7
参考文献 ···························································································································· 14
T 11737-2:2013 (ISO 11737-2:2009)
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第12条第1項の規定に基づき,一般社団法人日本医療機器学会(JSMI)及
び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を制定すべきとの申出が
あり,日本工業標準調査会の審議を経て,厚生労働大臣が制定した日本工業規格である。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
JIS T 11737の規格群には,次に示す部編成がある。
JIS T 11737-1 第1部:製品上の微生物群の測定方法
JIS T 11737-2 第2部:滅菌プロセスの定義,バリデーション及び維持において実施する無菌性の試
験
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格
JIS
T 11737-2:2013
(ISO 11737-2:2009)
医療機器の滅菌−微生物学的方法−
第2部:滅菌プロセスの定義,バリデーション及び
維持において実施する無菌性の試験
Sterilization of medical devices-Microbiological methods-
Part 2: Tests of sterility performed in the definition,
validation and maintenance of a sterilization process
序文
この規格は,2009年に第2版として発行されたISO 11737-2を基に,技術的内容及び構成を変更するこ
となく作成した日本工業規格である。
なお,この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
この規格は,無菌性の試験を実施する場合の要求事項について規定する。附属書Aに参考として示す事
項は,要求事項ではなく,また監査員のためのチェックリストとして作成されたものでもない。参考とし
て示した事項は,要求事項に適合するための適切な方法を示すものであり,この規格の要求事項への適合
性を達成するために有効な場合は,これらに示す以外の方法を使用してもよい。
1
適用範囲
1.1
この規格は,日常の滅菌処理において用いられる処理と比較して,低減した処理による滅菌剤への
ばく(曝)露を行った医療機器の無菌性の試験の一般的な基準について規定する。これらの試験は,滅菌
プロセスの定義,バリデーション又は維持を行う場合に実施することを意図している。
1.2
この規格は,次のものには適用しない。
a) 滅菌プロセスを経た製品の,日常の出荷のために実施する無菌かどうかの試験
b) 日本薬局方など公定書で定められた無菌試験の実施(3.12参照)
注記1 JIS T 0801-1,JIS T 0806-1,JIS T 0816-1,ISO 14160又はISO 14937では,上記a)又はb)を
要求していない。
c) バイオロジカルインジケータ又は接種した製品の培養
注記2 バイオロジカルインジケータの培養についての指針は,ISO 14161[8]に記載されている。
注記3 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 11737-2:2009,Sterilization of medical devices−Microbiological methods−Part 2: Tests of
sterility performed in the definition, validation and maintenance of a sterilization process(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”
ことを示す。
2
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格のうちで,西暦年を付記してあるものは,記載の年の版を適用し,その後の改正版(追補を含む。)
は適用しない。西暦年の付記がない引用規格は,その最新版(追補を含む。)を適用する。
JIS Q 10012 計測マネジメントシステム−測定プロセス及び測定機器に関する要求事項
注記 対応国際規格:ISO 10012,Measurement management systems−Requirements for measurement
processes and measuring equipment(IDT)
JIS Q 13485:2005 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 対応国際規格:ISO 13485:2003,Medical devices−Quality management systems−Requirements
for regulatory purposes(IDT)
JIS T 11737-1:2013 医療機器の滅菌−微生物学的方法−第1部:製品上の微生物群の測定方法
注記 対応国際規格:ISO 11737-1:2006,Sterilization of medical devices−Microbiological methods−Part
1: Determination of a population of microorganisms on products(IDT)
JIS Q 17025:2005 試験所及び校正機関の能力に関する一般要求事項
注記 対応国際規格:ISO/IEC 17025:2005,General requirements for the competence of testing and
calibration laboratories(IDT)
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
好気性微生物(aerobic organism)
代謝のために酸素を必要とする微生物。
3.2
嫌気性微生物(anaerobic organism)
代謝のために酸素を必要としない微生物。
3.3
静菌作用/静真菌作用試験(bacteriostasis/fungistasis test)
無菌性の試験に当たって,微生物の増殖を阻害する物質の存在を検出するために,選択した微生物を使
用して実施する試験。
3.4
バイオバーデン(bioburden)
製品及び/又は包装の上に存在する生育可能な微生物群(ISO/TS 11139:2006の定義2.2参照)。
3.5
培養条件(culture conditions)
微生物の発芽,生育及び/又は増殖を促進するために使用する,培地と培養方法との組合せ。
注記 培養方法には,温度,時間及びその他培養のためにあらかじめ定めた条件を含む場合がある。
(ISO/TS 11139:2006の定義2.10参照)
3.6
通性微生物(facultative organism)
好気性及び嫌気性の両方において代謝できる微生物。
3
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.7
培地性能試験(growth promotion test)
培地が,微生物の増殖を可能にすることを立証するために行う試験。
3.8
医療機器(medical device)
あらゆる計器,器械,用具,機械,器具,埋込み用具,体外診断薬,検定物質,ソフトウェア,材料又
はその他の同類のもの若しくは関連する物質であって,単独使用か組合せ使用かを問わず,製造業者が人
体への使用を意図し,その使用目的が次の一つ以上であり,
− 疾病の診断,予防,監視,治療,又は緩和
− 負傷の診断,監視,治療,緩和,又は補助
− 解剖学的又は生理学的なプロセスの検査,代替,又は修復
− 生命支援又は維持
− 受胎調整
− 医療機器の殺菌
− 人体から採取される標本の体外試験法による医療目的のための情報提供
薬学,免疫学,又は新陳代謝の手段によって体内又は体表において意図したその主機能を達成すること
はないが,それらの手段によって機能の実現を補助するものである(JIS Q 13485:2005の定義3.7参照)。
3.9
製品(product)
プロセスの結果(JIS Q 9000:2006の定義3.4.2参照)。
注記 滅菌規格では,製品は有形のものであり,また原料,中間製品,中間組立物及びヘルスケア製
品でもあり得る。
3.10
分割試料 SIP(sample item portion)
試験を行う医療機器のうちの定義した一部分。
3.11
無菌(sterile)
生育可能な微生物が存在しないこと(ISO/TS 11139:2006の定義2.43参照)。
3.12
無菌試験(test for sterility)
滅菌プロセスを経た製品に対して実施する,各国・地域で定められた薬局方で定義された技術的操作
(ISO/TS 11139:2006の定義2.53参照)。
3.13
無菌性の試験(test of sterility)
開発,バリデーション又は適格性の再確認の一部として実施する技術的操作で,製品又はその一部に生
育可能な微生物の存在の有無を判定するために行う試験(ISO/TS 11139:2006の定義2.54参照)。
4
品質マネジメントシステムの要素
4.1
文書化
4
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.1.1
無菌性の試験を実施するための手順をあらかじめ定めなければならない。
4.1.2
この規格で要求する文書及び記録は,あらかじめ指名した職員(4.2.1参照)によってレビュー及
び承認されなければならない。
文書及び記録は,JIS Q 13485又はJIS Q 17025に従い管理しなければならない。保持する記録には,元
の観察,計算,得たデータ及び最終報告を含まなければならない。記録には,サンプル採取,調製及び試
験に関わった職員の識別を含まなければならない。
4.1.3
計算及びデータ変換は,適切なチェックを受けなければならない。
4.2
経営者の責任
4.2.1
この規格に規定した手順の実行及び実施に関わる責任及び権限を,あらかじめ定めなければならな
い。責任は,JIS Q 13485又はJIS Q 17025によって,力量のある職員に割り当てなければならない。
4.2.2
この規格の要求事項を他の品質マネジメントシステムをもつ組織が実施する場合は,それぞれの責
任及び権限についてあらかじめ定めなければならない。
4.2.3
あらかじめ定めた試験及び測定を正しく実施するために,必要な全ての機器を利用できるようにし
ておかなければならない。
4.3
製品実現
4.3.1
購買の手順をあらかじめ定めなければならない。これらの手順は,JIS Q 13485又はJIS Q 17025
に適合しなければならない。
4.3.2
この規格の要求事項に適合するために使用する試験用の計器を含む全ての機器を校正するため,
JIS Q 13485,JIS Q 17025又はJIS Q 10012に適合する文書化したシステムについて,あらかじめ定めなけ
ればならない。
4.3.3
試験中に,製品,溶離液又は媒体と接触する機器若しくはその部分は,無菌でなければならない。
4.3.4
適切な品質試験を含め,無菌性の試験に使用する材料の調製及び滅菌の方法をあらかじめ定めなけ
ればならない。
4.4
測定,分析及び改善
異常な結果,予想外の結果,又は仕様外れの結果についての調査並びに修正,是正処置及び予防処置の
手順を規定しなければならない。これらの手順は,JIS Q 13485又はJIS Q 17025に適合しなければならな
い。
5
製品の選択
5.1
一般
5.1.1
無菌性の試験を実施するための製品の選択及び取扱いの手順は,包装材料及びプロセスを含めて,
製品が日常生産を代表するものであることを確実にしなければならない(5.3も参照)。
5.1.2
滅菌プロセスの開発,バリデーション及び日常管理において製品をグループ分けする場合は,製品
をそのグループに含める根拠を記録しなければならない(4.1.2も参照)。その根拠には,試験のために選
択した製品がグループ全体を代表するものであることを確実にしなければならない。
5.2
分割試料(SIP)
5.2.1
適用する規格で,滅菌プロセスの開発,バリデーション及び日常監視のために許容され,また,製
品単位全体を用いるのが実際的でない場合,滅菌方法によって許容される場合は,製品の選択部分[分割
試料(SIP)]で代用してもよい。
5
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.2.2
製品上及び/又は製品内のバイオバーデン分布が分かっていない場合は, SIPは製品が作られて
いる材料のそれぞれをその割合に応じて代表する製品の無作為に選択した部分からなるものでなければな
らない。
バイオバーデン分布が知られており,また,バイオバーデンが製品上及び/又は製品内に均一に分布し
ている場合には,SIPは製品のどのような部分から選択してもよい。
バイオバーデンの分布が既知で,バイオバーデンが製品上及び/又は製品内に均一に分布していない場
合には,SIPは,滅菌プロセスに対して最も過酷なチャレンジであるとみなすことができる製品の部分か
ら選択するか,又は製品を作る材料のそれぞれをその割合に応じて代表する製品から,無作為に選択した
部分から構成するものでなければならない。
5.2.3
選択したSIPの適切さは,立証しなければならない。
注記 放射線滅菌では,SIPの適切さについては,JIS T 0806-2で規定している。
5.3
製品及び分割試料(SIP)の包装
無菌性の試験に使用する製品又はSIPの包装材料及び/又は包装方法が日常生産に使用されているもの
とは異なる場合,包装材料の選択及び包装方法は,次の事項を確実にするものでなければならない。
a) 製品又はSIPが,滅菌剤によって意図した処理を受ける。
b) 製品又はSIPの微生物学的状態が,維持できる。
c) 製品又はSIPへの滅菌剤の到達が,日常生産で使用する包装で達成されるものと同様である。
6
無菌性の試験の実施方法
6.1
無菌性の試験の実施の一般的な方法には,次の二つがある。
a) 製品を培地に直接浸せきするか,又は製品内に培地を加え,その後培養する。
b) 製品から微生物を取り出し,取り出した微生物を培地に移植して,その後培養する。
6.2
個別の製品について,無菌性の試験方法に影響する要素を考慮し,記録しなければならない(4.1.2
参照)。考慮する要素には,少なくとも次の事項を含む。
a) 製品表示で無菌性を標ぼう(榜)する部分。
b) 試験対象の製品の物理的及び/又は化学的性質(6.6参照)。
c) 汚染微生物について,可能性がある種類並びに製品上及び/又は製品内の位置。
6.3
無菌性の試験の実施で,試験結果に影響を与える可能性がある場合には,無菌的操作を確実にしな
ければならない。
6.4
培地に移す前に微生物を洗浄によって製品から取り出す場合,考慮する要素には,次の事項を含め
なければならない。
a) 適切な洗浄液の選択。
b) 汚染微生物を取り出すための洗浄方法の能力(例えば,JIS T 11737-1の7.2参照)。
c) 汚染微生物の生存能力に対する洗浄方法の影響。
6.5
培地に移す前に微生物をろ過によって溶離液又は液体製品から取り出す場合,考慮する要素には,
次の事項も含めなければならない。
a) 適切なろ過システムの選択。
b) 容器,フィルタ及び関連機器を洗い流すための適切な流体の選択(必要な場合)。
6
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.6
試験対象製品の物理的又は化学的性質[6.2 b)参照]が,微生物の増殖に有害な影響を及ぼすような
物質が存在したり,又は放出したりするようなものである場合は,このような物質の影響を中和するか,
取り除くか,又はこれが不可能な場合は,最小限にするようなシステムを用いなければならない。このよ
うなシステムの有効性は立証しなければならない。
6.7
培養条件は,存在が予想される微生物のタイプを考慮した後に選択しなければならない。この考慮
の結果及びその決定の根拠を記録しなければならない(4.1.2参照)。
6.8
製品の滅菌剤へのばく露及び無菌性の試験を実施する間の時間は,可能な限り短くなければならな
い。
6.9
培養後,培地の微生物生育能力を試験し,この試験結果を記録しなければならない(4.1.2参照)。
7
無菌性の試験の実施方法の評価
無菌性の試験の結果を利用する前に,選択した方法の適切さを評価し,その結果を記録しなければなら
ない(4.1.2参照)。
8
無菌性の試験の実施方法の維持
8.1
製品及び/又は製造プロセスに対する修正に当たっては,無菌性の試験の実施方法の変更が必要か
どうかを判定するためにレビューしなければならない。レビューによって,変更が必要な場合は,箇条6
を適用しなければならない。
8.2
無菌性の試験の実施方法の変更に当たっては,試験方法の有効性が,引き続き適切かを評価しなけ
ればならない。この評価結果は,記録しなければならない(4.1.2参照)。
7
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
滅菌プロセスのバリデーション及び維持において実施する
無菌性の試験の指針
注記1 この附属書は,この規格で規定している要求事項の実施に関わる指針を示す。ここに示す指
針は,網羅的なものではないが,考慮するとよい重要な点を強調するためのものである。こ
の附属書に記載する以外の方法を採用してもよいが,それらの方法は,この規格の要求事項
への適合を達成するのに有効であることを立証することが望ましい。この附属書は,この規
格の要求事項への適合を評価するためのチェックリストを意図していない。
注記2 参照を容易にするため,この附属書の箇条番号は,この規格の本体と対応している。
A.1 適用範囲
指針はない。
A.2 引用規格
この規格の引用規格として記載した文書に規定される要求事項は,この規格の規定の部分に引用されて
いる範囲だけが,要求事項となる。引用は,規格全体の場合も又は特定の箇条若しくは細分箇条に限定さ
れる場合もある。
JIS Q 13485の引用については,完全な品質マネジメントシステムを確保することがこの規格の要求事項
ではないことに注意するのがよい。滅菌の対象となる医療機器のバリデーション及び監視で使用するバイ
オバーデンの測定の管理に最低限必要な品質マネジメントシステムの要素は,本文中の該当する箇所(特
に,箇条4参照)で要求事項として規定している。医療機器の製造又は再処理の全ての段階を管理する品
質マネジメントシステム(JIS Q 13485参照)及び試験施設の品質マネジメントシステム(JIS Q 17025参
照)に関わる規格に注意を払う必要がある。医療機器製造業者には,薬事法令によって品質マネジメント
システム及び規制当局又は第三者機関によるシステム評価に対する要求事項がある場合がある。
A.3 用語及び定義
指針はない。
A.4 品質マネジメントシステムの要素
A.4.1 文書化
A.4.1.1 指針はない。
A.4.1.2 JIS Q 13485の文書化の要求事項は,文書(仕様書及び手順書を含む。)及び記録の作成並びに管
理に関係している。
文書及び記録の管理についての要求事項は,JIS Q 13485:2005の4.2.3及び4.2.4又はJIS Q 17025:2005
の4.3,4.13及び5.4に規定している。
A.4.1.3 データの直接的及び間接的収集,処理及び/又は保存のために,試験施設でコンピュータを利用
する場合がある。このような用途で使用するハードウェア及びソフトウェアは,管理するのがよい。
8
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
使用するコンピュータシステムは,ハードウェア及びソフトウェアを明確にし,これらのいずれかに変
更を加えた場合は,そのことを文書化し,適切な承認を受ける(4.1.2及び4.2.1参照)。
電子的データ処理技術によって計算を行う場合は,使用前にソフトウェア(例えば,表計算ソフト)の
妥当性を確認し,このバリデーション結果を保存する。
ソフトウェアについては,次の内容を文書化するとよい。
− コンピュータシステムで実行されるアプリケーションソフトウェア
− オペレーションソフトウェア
− 使用するデータパッケージ
全てのソフトウェアは,使用に先立って,使用の可否のためのテストをする。
コンピュータソフトウェアを社内で開発する場合は,次の事項を確実にするための適切な手順を作成す
る。
− ソースコードを含む開発関連文書の保存
− 使用の可否のためのテスト記録の保存
− プログラムに対する修正の文書化
− 機器における変更の文書化及び使用前の正式なテストの実施
これらの管理事項は,市販のソフトウェアパッケージの修正又はカスタマイズにも適用するのがよい。
ソフトウェアプログラムに対する,未承認の変更を検出する又は防止する手順を設ける。
データのとりまとめ,作表及び/又はデータを統計的若しくは他の数学的処理を行うためのソフトウェ
アプログラム,又はその他の電子的に保存したデータの操作若しくは分析を行うソフトウェアプログラム
は,元の入力データを復元できるようにするのがよい。コンピュータデータをアーカイブに保存するため
の特別な手順が必要な場合は,これらの手順を文書化しておく。
品質マネジメントシステムのコンピュータソフトウェアへの適用の指針については,ISO/IEC 90003[17]
も参照。
A.4.2 経営者の責任
A.4.2.1 JIS Q 13485の経営者の責任についての要求事項は,経営者のコミットメント,顧客重視,品質
方針,計画,資源の提供,責任,権限及びコミュニケーション並びにマネジメントレビューに関わるもの
である。
試験施設は,品質サービスの提供をコミットすることが望ましく,また,このようなコミットメントは,
品質方針として文書化しておくのがよい。試験施設の組織内の権限及び責任の系統を正式に確立し,文書
化する。試験施設の品質システムの確立に責任をもつ個人を任命し,その人に,システムを実行すること
を確実にするための権限を与える。
責任及び権限に関わる要求事項は,JIS Q 13485:2005の5.5に,また,人的資源に関わる要求事項は,JIS
Q 13485:2005の6.2に規定している。
A.4.2.2 無菌性の試験の使用及び実施には,幾つかの別々の組織が関与することがあり,それぞれが方法
又は手順の特定の要素について責任をもつことがある。この規格では,組織が受け入れたそれぞれの責任
を定め,その定められた責任を文書化することを要求している。この定めた権限及び責任は,それぞれの
組織の品質マネジメントシステムの中で文書化する。
各要素に責任をもつ組織は,これらの要素について,適切な訓練及び資格認定によってその能力が立証
された力量のある職員に割り当てることを要求している。
無菌性の試験を医療機器製造業者の直接の管理下にある試験施設で実施する場合,試験施設の活動は製
9
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
造業者の品質マネジメントシステム下で実施する。外部の試験施設を使用する場合は,適切な国際規格(例
えば,JIS Q 17025)又は該当する規制要求事項によって認証された試験施設であることが望ましい。
A.4.2.3 資源の提供に関わる要求事項は,JIS Q 13485:2005の6.1に,また,機器に関わる要求事項は,
JIS Q 17025:2005の5.5に規定している。
A.4.3 製品実現
A.4.3.1 JIS Q 13485の製品実現についての要求事項は,顧客の要求事項の決定,設計・開発,購買,製
造管理,並びに監視及び測定機器の校正からなる製品ライフサイクルが関連している。
購買についての要求事項は,JIS Q 13485:2005の7.4に規定されている。特にJIS Q 13485:2005の7.4.3
についての要求事項が,外部の組織から受け取る全ての製品及びサービスに適用されることに注意する。
A.4.3.2 各試験機器について,メンテナンス要求事項を明確にするシステムがあるとよい。
校正を必要としない機器は,明確に識別するのがよい。監視及び測定装置の校正についての要求事項は,
JIS Q 13485:2005の7.6に規定している。
装置及び測定のトレーサビリティに関わる要求事項は,JIS Q 17025:2005の5.5及び5.6に規定している。
A.4.3.3 微生物を取り出すために使用する培地又は回収液は,その無菌状態を確実にするような方法で調
製する。
A.4.3.4 適切な品質試験には,培地性能試験を含むのがよい。一般的に,培地性能試験は,培地のバッチ
ごとに選択した微生物の少数(10〜100 CFU)を接種して実施する。培地性能試験については適切な微生
物が,日本薬局方に記載されている。
A.4.4 測定,分析及び改善
JIS Q 13485では,測定,分析及び改善の要求事項は,プロセス監視,不適合製品の管理,データの分析
及び改善(是正処置及び予防処置を含む。)に関連している。
試験施設の活動は,定期的な内部監査を受けるのがよい。監査結果は文書化し,試験施設マネジメント
によってレビューするとよい。
無菌性の試験において,異常な,予想外の又は仕様外の結果を得た場合は,調査を必要とする。調査の
初期段階には,結果が正しいのか又は誤りなのかの評価を含めるのがよい。次の事項は,誤りの原因とな
るため,注意する。
− 不適切なサンプル(例 代表的でない,均一でない,不合格材料)
− 不適切な輸送,取扱い又は保管
− 不適切な試験器具(例 ピペット,ろ過器)
− 間違った取扱い又は試験方法
− 不適切な培地又は希釈剤
− 不適切な試験環境
− 不適切な培養環境
− 試験結果の解釈の間違い
− 転記の間違い
調査結果に基づき,特定の是正処置が必要になることもある。是正処置が必要になった場合は,その有
効性の立証が必要である。
是正処置の手順は,JIS Q 13485:2005の8.5.2及びJIS Q 17025:2005の4.11に規定している。
A.5 製品の選択
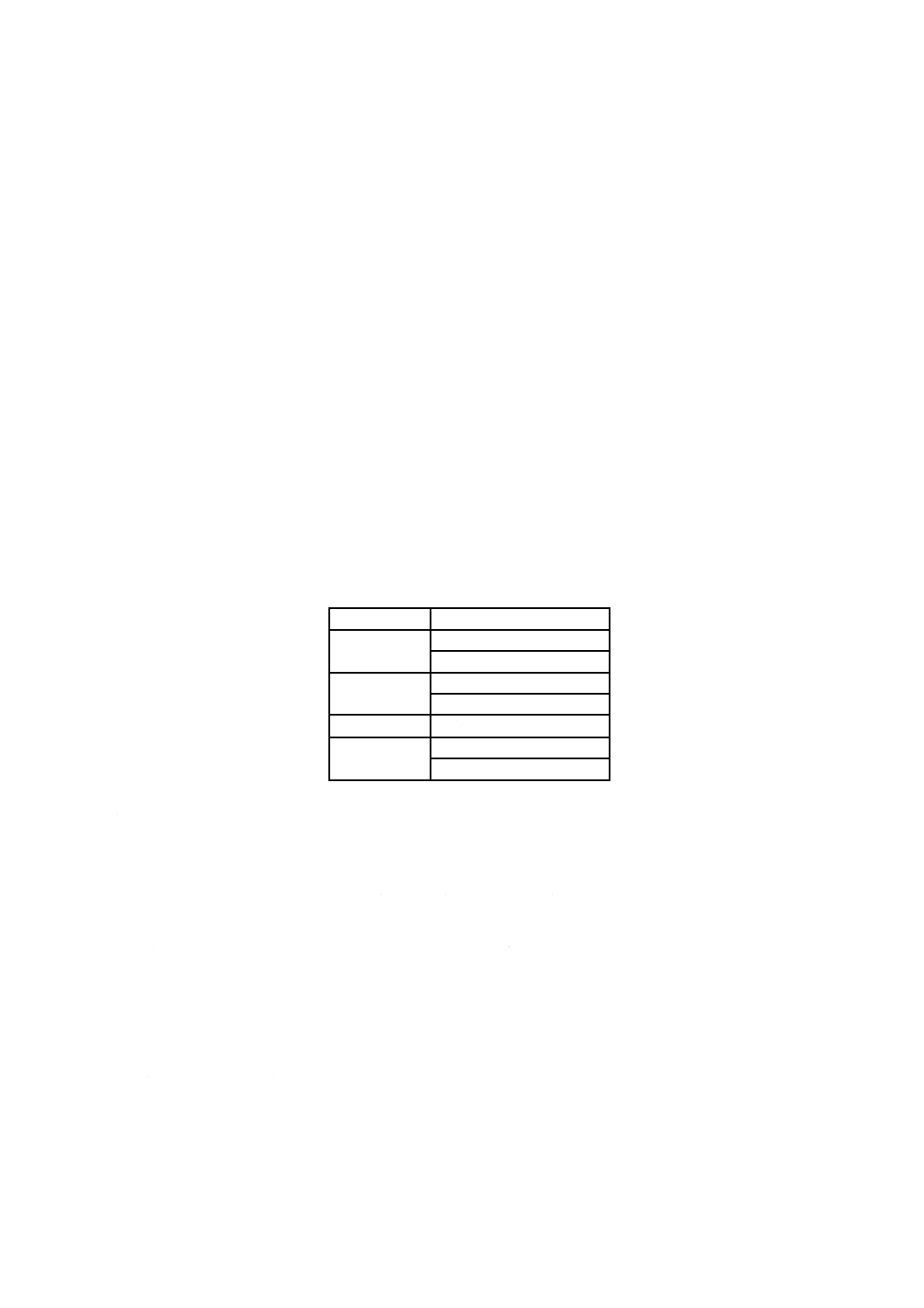
10
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.5.1 一般
A.5.1.1 製品は,日常プロセス手順を代表する条件下で生産されるバッチの製品から選択する。試験のた
めの製品は,無作為に選択する。
選択する製品及びバッチの数は,滅菌プロセスのバリデーション及び日常管理のための要求事項を規定
した関連する規格に規定されている場合がある。
不注意による汚染の混入及び試料上及び/又は製品内の微生物の数及びタイプに対する変更を避けるた
めに,試料の選択及び取扱いの方法を選択する。
試験に用いる製品は,合格品と同じ処理及び条件に従っており,不合格の原因が試験の有効性を損なわ
ない場合は,製造プロセス中で不合格品から選んでもよい。
A.5.1.2 製品のグループ分けについての要求事項は,滅菌プロセスの開発,バリデーション及び日常管理
に関する個別の規格に規定している(例えば,JIS T 0801-1[1]及びJIS T 0806-2 [4]参照)。
A.5.2 分割試料(SIP)
A.5.2.1 実施可能な場合は,製品全体を試験に使用するのがよいが,これは常に可能であるとは限らない
ことが認められている。このような状況では,試験中に取扱いが便利な製品の分割試料(SIP)を選択して
代用してもよい。SIPは,試験施設で容易に操作することができ,できるだけ大きい製品の一部であるの
がよい。SIPは,製品の長さ,質量,容量又は表面積に基づいて選択することができる。表A.1を参照。
表A.1−SIPの選択例
SIPの基準
製品
長さ
チューブ(口径一定)
包帯のロール
質量
粉体
ガウン
体積
液体
表面積
外科用ドレープ
チューブ(多様な口径)
製品又はSIPが試験施設の使用可能な容器中で試験できない場合は,二つ以上の容器に分割し,これら
の容器を一つにまとめて評価してもよい。分割したうちの一つの容器が陽性の結果を得た場合は,製品全
体を陽性とみなす。
流路だけが無菌であると表示している製品の場合,流路だけを製品とみなすのがよい。
A.5.2.2 SIP上の微生物汚染は,滅菌プロセスにかけられる微生物学的チャレンジを代表するものとする。
製品が複雑な場合,SIPは製品の様々な要素のバイオバーデンを代表するものとする。
A.5.2.3 指針はない。
A.5.3 製品及び分割試料の包装
製品は,本来の形状及び包装で滅菌剤にばく露するのがよい。ただし,無菌性の試験を実施するときの
操作の最小化及び/又は単純化並びに汚染から生じる偽陽性の可能性を減らすために,滅菌剤にばく露す
る前に製品を分解し,再包装してもよい。
滅菌剤に対する微生物の反応に対する製品の分解及び再包装が与える影響を考慮することが重要である。
例えば,分解が微生物の化学的な環境を変えてしまう場合がある。
滅菌剤の微生物への到達に対して,製品の分解が与える影響も考慮することが重要である。
11
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
滅菌剤へのばく露前にSIPを調製して包装する場合は,バイオバーデンの変更を最小限にするために選
択した条件下で実施するのがよい。
A.6 無菌性の試験の実施方法
A.6.1 箇条6に示したように,無菌性の試験の実施方法は,次のa)及びb)の二つのカテゴリに分けるこ
とができる。
a) 製品の直接浸せき
直接浸せきは,医療機器の無菌性の試験を実施するのに好ましい方法である。直接浸せきでは,製
品又はSIPを培地の入った一つの容器(又は複数の容器,A.5.2.1参照)に無菌的に入れ培養する。培
地と製品全体又はSIPとの接触が達成されるよう,十分な量の培地を用いる。さらに,次の事項を考
慮する。
− 滅菌剤へのばく露前の分解(A.5.3も参照)
− 培地に浸せき前の分解及び/又は操作
− 培地に浸せき(漬)後のかくはん(撹拌)
− 製品表面の湿潤化を改善するために,培地への表面活性剤(静菌又は殺菌効果をもたないことが
立証されているもの)の添加
培養中は,培地と製品又はSIPとの接触を維持する。
製品の流路の無菌性の試験を実施する場合は,流路を培地で満たして,製品を培養する。
b) 製品からの微生物取り出し
医療機器が静菌/静真菌作用などの特性をもっており,直接浸せきを使用することが可能でない場
合,微生物の取り出しが必要となる。
この方法を使用する場合は注意が必要である。この方法では,製品から全ての微生物を洗い出せな
いことがある。製品から全ての微生物を取り出せないと,試験が無効になる場合がある。取り出し操
作中に起こる汚染は,偽陽性の結果を招くことがある。
培養に移る前に,物理的処理によって製品から微生物を取り出す手順は,更に次のように分けるこ
とができる。
− 洗い出し及びメンブレンろ過法
− 洗い出し及び洗浄液の培養
これら二つの手順で,初めに行う処理は,製品又はSIPから微生物を取り出すことである。使用す
る方法は,バイオバーデン測定に使用されるものと同一で,JIS T 11737-1:2013のB.2.2に記載されて
いる。同様に,適切な洗浄液の選択に関する考慮事項は,バイオバーデンの測定に使用するものと同
一で,JIS T 11737-1:2013のB.2.3及び表B.1に記載されている。
製品又はSIPから微生物を取り出した後,メンブレンろ過法を用いるか,又は洗浄液全体を培養し,
無菌性の試験を実施してもよい(A.6.4参照)。
A.6.2 指針はない。
A.6.3 無菌性の試験を実施する場合に適用できる無菌操作には,次のものが含まれる。
− 管理された環境内で試験を実施する。
例 バリア分離:環境的に管理された専用の室内に置いた層流フード又はバイオセーフティキャビ
ネット。
注記 管理された環境の詳細は,JIS B 9920[9],JIS B 9919[10]及びJIS B 9917-7 [11]に示されている。
12
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 試験に使用する全ての機器,材料及び器具を滅菌する。
− 試験用具,培地及び試験品を,試験区域に無菌的に導入する。
− 試験品を試験区域に導入する前に,包装の外側の汚染を除く。
− 試験区域内の表面の汚染を除く。
− 試験を実施するために必要な操作を最小限にする。
− 無菌的操作の実施に関する教育訓練。
A.6.4 溶離液の培養によって無菌性の試験を実施するために,培地を溶離液として用い,その後,無菌容
器に移して培養する方法がある。
もう一つの方法は,微生物の生育に影響しない洗浄液を使用し,洗浄後,洗浄液を無菌容器の中で同容
量の2倍の濃度の培地と混合し,培養する。洗浄液の容量が培地の容量の10 %以下の場合は,洗浄液を無
菌容器の中で通常の濃度の培地と混合し,培養してもよい。
A.6.5 ろ過を使って無菌性の試験を実施するためには,孔径が0.45 μm以下の無菌のメンブレンフィルタ
に,真空又は加圧によって溶出液を通す。
溶出液と接触した表面を,更に無菌の溶離液又は中和剤を含んだ溶液ですすぎ(A.6.6参照),そのすす
いだ液体をメンブレンフィルタでろ過する。その後,培地を無菌状態でろ過ユニットに移すか,又はメン
ブレンフィルタを無菌状態で培地に移す。
これらの操作を行った後,培養を行う。
A.6.6 試験を行う製品が,偽陰性(A.7参照)を引き起こすような阻害物質を培地内に放出していないか
調査するのがよい。これは,静菌作用/静真菌作用試験で実施する場合のように,製品を含んだ培地に少
数の代表的な菌を接種して行う。
殺菌物質又は静菌物質が検出された場合,その影響は,次によって最小限にすることができる。
a) 培地又は洗浄液への中和剤の添加
b) ろ過による洗浄液からの殺菌物質又は静菌物質の除去
c) 希釈による,有効でないレベルまでの殺菌物質又は静菌物質の濃度の低減。これは,培地又は洗浄液
の容積を増加させ,また,必要な場合は,製品を幾つかの試験容器に分割することによって達成して
もよい。
一般に,現行の薬局方で示される手順,微生物,力価及び培養時間は適切であるが,培養温度及び培地
については,無菌性の試験を実施するときの条件と同じでなければならない。
A.6.7 滅菌プロセスの開発,バリデーション及び日常管理のための個別の規格では,無菌性の試験に使用
する培養条件を推奨している場合がある。
一般に,滅菌剤にばく露されても生き残る可能性がある好気性及び通性の微生物の培養に最適であると
の仮定のもとに,一つの培養タイプの培地が使われる。ソイビーン・カゼイン・ダイジェスト培地を使用
する場合,通常は30±2 ℃で14日間の培養条件を使用する。他の培地を無菌性の試験で使用する場合は,
他の培養条件を考慮するとよい。
無菌性の試験に推奨される培養温度は,バイオバーデンの測定に推奨される温度よりも低い場合がある。
より低い温度及びより長い培養時間を用いることで,損傷した又はきずついた微生物が回復することがあ
る。
次の場合は,培養条件の選択が必要である。
− 滅菌プロセスの開発,バリデーション及び日常管理のための個別の規格が,使用すべき培地を規定し
ていない。
13
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 滅菌剤にばく露されても生き残る可能性のある微生物(例えば,嫌気性微生物又はマイコバクテリア
の存在)のため,単一の培地条件の使用が妥当でない場合。
これらの事例において培養条件を選択する場合に考慮すべき要素には,次の事項を含む。
− 製品の性質
− 製造方法
− 微生物汚染の潜在的な汚染源
− 汚染の可能性のある微生物のタイプ
JIS T 11737-1に従って実施するバイオバーデンの測定による微生物の種類に関する情報は,培養条件の
選択の手助けとなる場合がある。
A.6.8 滅菌剤へのばく露及び培養までの時間は,滅菌剤が微生物にもたらす致死的損傷からの修復能力に
影響を与えることがある。滅菌剤へのばく露後,製品又はSIPについて,無菌性の試験をできるだけ迅速
に実施するために,あらゆる努力を尽くすのがよい。移動による遅れが避けられない場合は,微生物の生
存能力の損失又は菌数の変化を防止するために,被験試料を保存する条件を選択するのがよい。
無菌性の試験を実施する前に経過する可能性がある最大の時間間隔を規定する。
A.6.9 通常,培養後の培地の判定は,肉眼で行う。
濁り,臭気,色の変化,皮膜,沈殿物及び凝結は,生育の証拠となる。
目視検査は,濁りの検知を助けるために,背後から照明を当てて行ってもよい。
濁りが,微生物の生育によるものではない可能性もある。
濁りについては,次の事項によって,生育によるものであるかどうかを確認できる。
a) 顕微鏡観察。
b) 濁った培地の部分(それぞれ1 cm3以上)を同じ培地の新しい容器に移し,少なくとも4日間培養す
る。
c) 一般に受け入れられている微生物学的手順(例えば,分離のための線状培養)を使用して,濁った培
地を植え継いで培養する。
A.7 無菌性の試験の実施方法の評価
無菌性の試験の実施方法を評価する場合には,偽陽性又は偽陰性となる不正確な結果の可能性を考慮す
る。
無菌性の試験における偽陽性の発生は,滅菌剤による処理の効果が低いとみなされ,バリデーションで
取得したデータの解釈に影響を与えることがある。特に立証されない限り(A.4.4参照),陽性結果は,滅
菌剤で処理されても生き残った微生物によるものであるとみなす必要がある。無菌性の試験を実施する場
合の偽陽性の発生頻度を評価するために,代表的な無菌の製品を用いたシミュレーションを実施するのが
よい。
偽陰性の発生に影響する要素には,次のものが含まれる。
− 培養条件が微生物の生育に適切でない(A.4.3.3参照)。
− 試験中に製品から放出された殺菌物質及び/又は静菌物質の存在(A.6.6参照)。
− 滅菌剤による処理から培養までの時間(A.6.8参照)。
A.8 無菌性の試験の実施方法の維持
指針はない。
14
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] JIS T 0801-1 ヘルスケア製品の滅菌−エチレンオキサイド−第1部:医療機器の滅菌プロセスの開発,
バリデーション及び日常管理の要求事項
注記 対応国際規格:ISO 11135-1,Sterilization of health care products−Ethylene oxide−Part 1:
Requirements for development, validation and routine control of a sterilization process for medical
devices(IDT)
[2] ISO/TS 11135-2, Sterilization of health care products−Ethylene oxide−Part 2: Guidance on the application of
ISO 11135-1
[3] JIS T 0806-1 ヘルスケア製品の滅菌−放射線−第1部:医療機器の滅菌プロセスの開発,バリデーシ
ョン及び日常管理の要求事項
注記 対応国際規格:ISO 11137-1,Sterilization of health care products−Radiation−Part 1: Requirements
for development, validation and routine control of a sterilization process for medical devices(IDT)
[4] JIS T 0806-2 ヘルスケア製品の滅菌−放射線−第2部:滅菌線量の確立
注記 対応国際規格:ISO 11137-2,Sterilization of health care products−Radiation−Part 2: Establishing
the sterilization dose(IDT)
[5] ISO 11138-2, Sterilization of health care products−Biological indicators−Part 2: Biological indicators for
ethylene oxide sterilization processes
[6] ISO/TS 11139:2006, Sterilization of health care products−Vocabulary
[7] ISO 14160:1998, Sterilization of single-use medical devices incorporating materials of animal origin−
Validation and routine control of sterilization by liquid chemical sterilants
[8] ISO 14161, Sterilization of health care products−Biological indicators−Guidance for the selection, use and
interpretation of results
[9] JIS B 9920 クリーンルームの空気清浄度の評価方法
注記 対応国際規格:ISO 14644-1,Cleanrooms and associated controlled environments−Part 1:
Classification of air cleanliness(MOD)
[10] JIS B 9919 クリーンルームの設計・施工及びスタートアップ
注記 対応国際規格:ISO 14644-4,Cleanrooms and associated controlled environments−Part 4: Design,
construction and start-up(MOD)
[11] JIS B 9917-7 クリーンルーム及び関連制御環境−第7部:隔離装置
注記 対応国際規格:ISO 14644-7,Cleanrooms and associated controlled environments−Part 7:
Separative devices (clean air hoods, gloveboxes, isolators and mini-environments)(IDT)
[12] ISO 14937, Sterilization of health care products−General requirements for characterization of a sterilizing
agent and the development, validation and routine control of a sterilization process for medical devices
[13] JIS T 0816-1 ヘルスケア製品の滅菌−湿熱−第1部:医療機器の滅菌プロセスの開発,バリデーショ
ン及び日常管理の要求事項
注記 対応国際規格:ISO 17665-1,Sterilization of health care products−Moist heat−Part 1:
Requirements for the development, validation and routine control of a sterilization process for
medical devices(IDT)
15
T 11737-2:2013 (ISO 11737-2:2009)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[14] ISO 20857, Sterilization of health care products−Dry heat−Requirements for the development, validation and
routine control of a sterilization process for medical devices
[15] JIS Q 9000:2006 品質マネジメントシステム−基本及び用語
注記 対応国際規格:ISO 9000:2005,Quality management systems−Fundamentals and vocabulary(IDT)
[16] JIS Q 9001:2008 品質マネジメントシステム−要求事項
注記 対応国際規格:ISO 9001:2008,Quality management systems−Requirements(IDT)
[17] ISO/IEC 90003, Software engineering−Guidelines for the application of ISO 9001:2000 to computer software
[18] AKERS, J.D., et al., Survey on Sterility Testing Practices, J. Parenteral Sci. Technol., 41, 6, 1987
[19] ALEXANDER, K. and BRYANS, T., Evaluation of the Sterility Test for Detection of Microbial Contaminants
of Allografts, Cell and Tissue Banking, 7, 1, pp. 23‒28, 2006
[20] Association of Analytical Chemists. Official Methods of Analysis. 15th ed., Arlington, AOAC; pp. 430‒437,
1992
[21] Association of Analytical Chemists. Bacteriological Analytical Manual (BAM). 6th ed., Arlington, AOAC;1984
[22] BLOCK, S.S., Disinfection, Sterilization and Preservation, 5th ed., 2001
[23] GERHARDT, P., et al., Manual of Methods for General Bacteriology, American Society for Microbiology,
Washington, DC, 1981
[24] MATHEWS, A.G., Optimal incubation conditions for sterility tests, Develop. Biol. Stand., 23, pp. 94‒102, 1974
[25] MELTZER, L.L. and ORDAL, Z.J., Thermal Injury and Recovery of Bacillus subtilus, Applied Microbiology,
24, 6, pp. 878‒884, 1972
[26] RUSSELL, A.D., Principles of Antimicrobial Activity, in Block, S.S., (ed.) Disinfection, Sterilization and
Preservation, Lea & Febiger, Philadelphia, PA, 4th edition, p. 27, 1991
[27] SOKOLSKI, W.T. and CHIDESTEY, C.G., Improved viable counting method for petroleum‒based ointments, J.
Pharm. Sci., 53, pp. 103‒107, 1964
[28] STRAKA, R.P. and STOKES, J.L., Rapid destruction of bacteria in commonly used diluents and its elimination.
J. App. Microbiology, 5, p. 21, 1957
[29] The European Pharmacopoeia, 6th ed., European Directorate for the Quality of Medicines (EDQM), Strasbourg,
2008
[30] 日本薬局方
[31] The United States Pharmacopoeia, 31st ed., United States Pharmacopoeial Convention (USP), Rockville, MD