T 11737-1:2013 (ISO 11737-1:2006)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 2
4 品質マネジメントシステムの要素 ························································································ 4
4.1 文書化 ························································································································· 4
4.2 経営者の責任 ················································································································ 4
4.3 製品実現 ······················································································································ 4
4.4 測定,分析及び改善−不適合製品の管理············································································· 4
5 製品の選択 ······················································································································ 4
5.1 一般 ···························································································································· 5
5.2 分割試料(SIP) ············································································································ 5
6 バイオバーデンの測定及び微生物学的特性付けの方法 ····························································· 5
6.1 バイオバーデンの測定 ···································································································· 5
6.2 バイオバーデンの微生物学的特性付け················································································ 6
7 バイオバーデンの測定方法のバリデーション ········································································· 6
8 日常のバイオバーデン測定及びデータの解釈 ········································································· 7
9 バイオバーデンの測定方法の維持 ························································································ 7
9.1 製品及び/又は製造プロセスの変更··················································································· 7
9.2 バイオバーデンの測定方法の変更······················································································ 7
9.3 バイオバーデンの測定方法の再バリデーション ···································································· 7
附属書A(参考)製品上の微生物群の測定についての指針 ··························································· 8
附属書B(参考)バイオバーデン測定法の指針 ·········································································· 20
附属書C(参考)バイオバーデン測定方法のバリデーション························································ 28
参考文献 ···························································································································· 31
T 11737-1:2013 (ISO 11737-1:2006)
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第12 条第1 項の規定に基づき,一般社団法人日本医療機器学会(JSMI)及
び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を制定すべきとの申出が
あり,日本工業標準調査会の審議を経て,厚生労働大臣が制定した日本工業規格である。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
JIS T 11737の規格群には,次に示す部編成がある。
JIS T 11737-1 第1部:製品上の微生物群の測定方法
JIS T 11737-2 第2部:滅菌プロセスの設定,バリデーション及び維持において実施する無菌性の試
験
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格
JIS
T 11737-1:2013
(ISO 11737-1:2006)
医療機器の滅菌−微生物学的方法−
第1部:製品上の微生物群の測定方法
Sterilization of medical devices-Microbiological methods-Part 1:
Determination of a population of microorganisms on products
序文
この規格は,2006年に第2版として発行されたISO 11737-1を基に,技術的内容及び対応国際規格の構
成を変更することなく作成した日本工業規格である。
なお,この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
この規格は,バイオバーデンを測定するときの要求事項について規定する。附属書A〜附属書Cに参考
として示す事項は,要求事項ではなく,また,監査員のためのチェックリストとして作成されたものでも
ない。参考として示した事項は,要求事項に適合するための適切な方法を示すものであり,この規格の要
求事項への適合性を達成することに有効な場合は,これらに示す以外の方法を使用してもよい。
1
適用範囲
この規格は,医療機器,構成部品,原料又は包装上若しくはその内部における生育可能な微生物群の計
測及び特性付けのための要求事項について規定し,指針を示す。
注記1 微生物学的特性付けの性質及び範囲は,バイオバーデンデータの使用目的による。
この規格では,ウイルス又は原生動物系の汚染物質の計測又は特定についての要求事項は,規定しない。
この規格は,スクレピー,牛海綿脳症及びクロイツウェルト・ヤコブ病のような,海綿脳症の病原物質
の除去及び検出には適用しない。
この規格は,医療機器を製造する環境の微生物学的モニタリングについては,適用しない。
注記2 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 11737-1:2006,Sterilization of medical devices−Microbiological methods−Part 1:
Determination of a population of microorganisms on products(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”
ことを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格のうちで,西暦年を付記してあるものは,記載の年の版を適用し,その後の改正版(追補を含む。)
は適用しない。西暦年の付記がない引用規格は,その最新版(追補を含む。)を適用する。
JIS Q 10012 計測マネジメントシステム−測定プロセス及び測定機器に関する要求事項
2
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記 対応国際規格:ISO 10012,Measurement management systems−Requirements for measurement
processes and measuring equipment(IDT)
JIS Q 13485:2005 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 対応国際規格:ISO 13485:2003,Medical devices−Quality management systems−Requirements
for regulatory purposes(IDT)
JIS Q 17025:2005 試験所及び校正機関の能力に関する一般要求事項
注記 対応国際規格:ISO/IEC 17025:2005,General requirements for the competence of testing and
calibration laboratories(IDT)
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
バイオバーデン(bioburden)
製品,及び/又は無菌バリアシステムの上又は内部に存在する生育可能な微生物群(ISO/TS 11139:2006,
定義2.2参照)。
3.2
修正(correction)
検出された不適合を除去するための処置。
注記 是正処置(3.4)と併せて,修正が行われることもある。
(JIS Q 9000:2006,定義3.6.6参照)
3.3
補正係数(correction factor)
製品及び/又は微生物培養からの不十分な取出しを補正するために適用する数値。
3.4
是正処置(corrective action)
検出された不適合又はその他の検出された望ましくない状況の原因を除去するための処置。
注記1 不適合の原因は,一つ以上のことがあり得る。
注記2 予防処置(3.9)は発生を未然に防止するためにとるのに対し,是正処置は再発を防止するた
めにとる。
注記3 修正(3.2)と是正処置とは異なる(JIS Q 9000:2006,定義3.6.5参照)。
3.5
培養条件(culture conditions)
微生物の発芽,生育及び/又は増殖を促進するために使用する,培地と培養方法との組合せ。
注記 培養方法には,温度,時間及びその他培養のためにあらかじめ定めた条件を含む場合がある。
(ISO/TS 11139:2006,定義2.10参照)
3.6
確立(establish)
理論的評価によって決定し,実験によって確認すること(ISO/TS 11139:2006,定義2.17参照)。
3.7
医療機器(medical device)
3
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
あらゆる計器,器械,用具,機械,器具,埋込み用具,体外診断薬,検定物質,ソフトウェア,材料又
はその他の同類のもの若しくは関連する物質であって,単独使用か組合せ使用かを問わず,製造業者が人
体への使用を意図し,その使用目的が次の一つ以上であり,
− 疾病の診断,予防,監視,治療,又は緩和
− 負傷の診断,監視,治療,緩和,又は補助
− 解剖学的又は生理学的なプロセスの検査,代替,又は修復
− 生命支援又は維持
− 受胎調整
− 医療機器の殺菌
− 人体から採取される標本の体外試験法による医療目的のための情報提供
薬学,免疫学,又は新陳代謝の手段によって体内又は体表において意図したその主機能を達成すること
はないが,それらの手段によって機能の実現を補助するものである(JIS Q 13485,定義3.7参照)。
3.8
微生物学的特性付け(microbial characterization)
微生物をカテゴリごとにグループ分けするプロセス。
注記 カテゴリは,大きく分けて,例えば,選択培地,コロニ若しくは細胞の形態,染色性又はその
他の特性のいずれかに基づいてもよい(ISO/TS 11139:2006,定義2.25参照)。
3.9
予防処置(preventive action)
起こり得る不適合又はその他の望ましくない起こり得る状況の原因を除去するための処置。
注記1 起こり得る不適合の原因は,一つ以上のことがある。
注記2 是正処置は再発を防止するためにとるのに対し,予防処置は発生を未然に防止するためにと
る(JIS Q 9000:2006,定義3.6.4参照)。
3.10
製品(product)
プロセスの結果(JIS Q 9000:2006,定義3.4.2参照)。
3.11
公的微生物保存機関(recognized culture collection)
特許手続上の微生物の寄託の国際的承認に関わるブタペスト条約に基づく国際的保存機関(ISO/TS
11139:2006,定義2.38参照)。
注記 日本では,独立行政法人製品評価技術基盤機構バイオテクノロジー本部・生物遺伝資源部門
(NBRC)など。
3.12
回収率(recovery efficiency)
製品から微生物を取出し及び/又はこれを培養するためにあらかじめ定めた技術能力の尺度。
3.13
分割試料(sample item portion)
SIP
試験を行う医療機器のうちの定義した一部分。
4
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.14
あらかじめ定める(specify)
承認を受けた文書の中で詳細を明記すること(ISO/TS 11139 :2006,定義 2.42参照)。
3.15
バリデーション(validation)
プロセスが,恒常的にあらかじめ定めた仕様に適合する製品を得ることができることを確立するために,
要求される結果を得て,記録し,解釈するための文書化した手順(ISO/TS 11139:2006,定義2.55参照)。
注記 バイオバーデンの測定については,“プロセス”とは試験手順であり,また,“製品”とは試験
結果である。バイオバーデン測定技術のバリデーションは,試験方法の有効性及び再現性を評
価するための一連の調査からなる。
4
品質マネジメントシステムの要素
4.1
文書化
4.1.1
バイオバーデンを測定する手順をあらかじめ定めなければならない。
4.1.2
この規格で要求する文書及び記録は,あらかじめ指名した職員によってレビュー及び承認されなけ
ればならない(4.2.1参照)。文書及び記録は,JIS Q 13485又はJIS Q 17025によって管理しなければなら
ない。
4.1.3
保持する記録には,最初の観測,計算,得たデータ及び最終報告を含まなければならない。記録に
は,サンプル採取,調製及び試験に関わった職員の識別を含まなければならない。
4.1.4
計算及びデータ変換は,適切なチェックを受けなければならない。
4.2
経営者の責任
4.2.1
この規格に規定した手順の実行及び実施に関わる責任及び権限をあらかじめ定めなければならな
い。責任は,JIS Q 13485又はJIS Q 17025によって,力量のある職員に割り当てなければならない。
4.2.2
この規格の要求事項を他の品質マネジメントシステムをもつ組織が実施する場合は,それぞれの責
任及び権限についてあらかじめ定めなければならない。
4.2.3
あらかじめ定めた試験及び測定を正しく実施するために,必要な全ての機器が利用できるようにし
ておかなければならない。
4.3
製品実現
4.3.1
購買の手順をあらかじめ定めなければならない。これらの手順は,JIS Q 13485又はJIS Q 17025
に適合しなければならない。
4.3.2
この規格の要求事項に適合するために使用する試験用の計器を含む全ての機器を校正するため,
JIS Q 13485,JIS Q 17025又はJIS Q 10012に適合する文書化したシステムについてあらかじめ定めなけれ
ばならない。
4.3.3
バイオバーデンを測定するための材料の調製及び滅菌のための試験方法を,適切な品質試験を含め
てあらかじめ定めなければならない。
4.4
測定,分析及び改善−不適合製品の管理
仕様外れとなった結果の調査,修正,是正処置及び予防処置についての手順をあらかじめ定めなければ
ならない。これらの手順は,JIS Q 13485又はJIS Q 17025に適合しなければならない。
5
製品の選択
5
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.1
一般
5.1.1
バイオバーデンを測定するための製品の選択及び取扱いの手順は,選択した製品が,包装材料及び
プロセスを含む日常の製造を代表するものであることを確実にするものでなければならない。
5.1.2
製品をバイオバーデンの測定のためにグループ化する場合は,その製品をグループに含める根拠を
記録しなければならない(4.1.2参照)。この根拠には,そのグループから選択した製品について求めたバ
イオバーデンが,グループ全体を代表するものであることを確実にするための判断基準を含めなければな
らない。
5.1.3
バイオバーデンの測定結果は,時間の経過によって変化するため,サンプリングからバイオバーデ
ン測定までの時間を考慮しなければならない。
5.2
分割試料(SIP)
製品上及び/又は内部にバイオバーデンが均等に分布している場合,SIPは,製品試料のいずれの部分
から選んでもよい。バイオバーデンが均等に分布していない場合には,SIPは,製品を構成する各原材料
を比例的に代表するよう無作為に選んだ製品の部分としなければならない。バイオバーデンの分布が既知
の場合には,SIPを滅菌プロセスに対して最も滅菌が困難であるとみなせる製品の部分から選んでもよい。
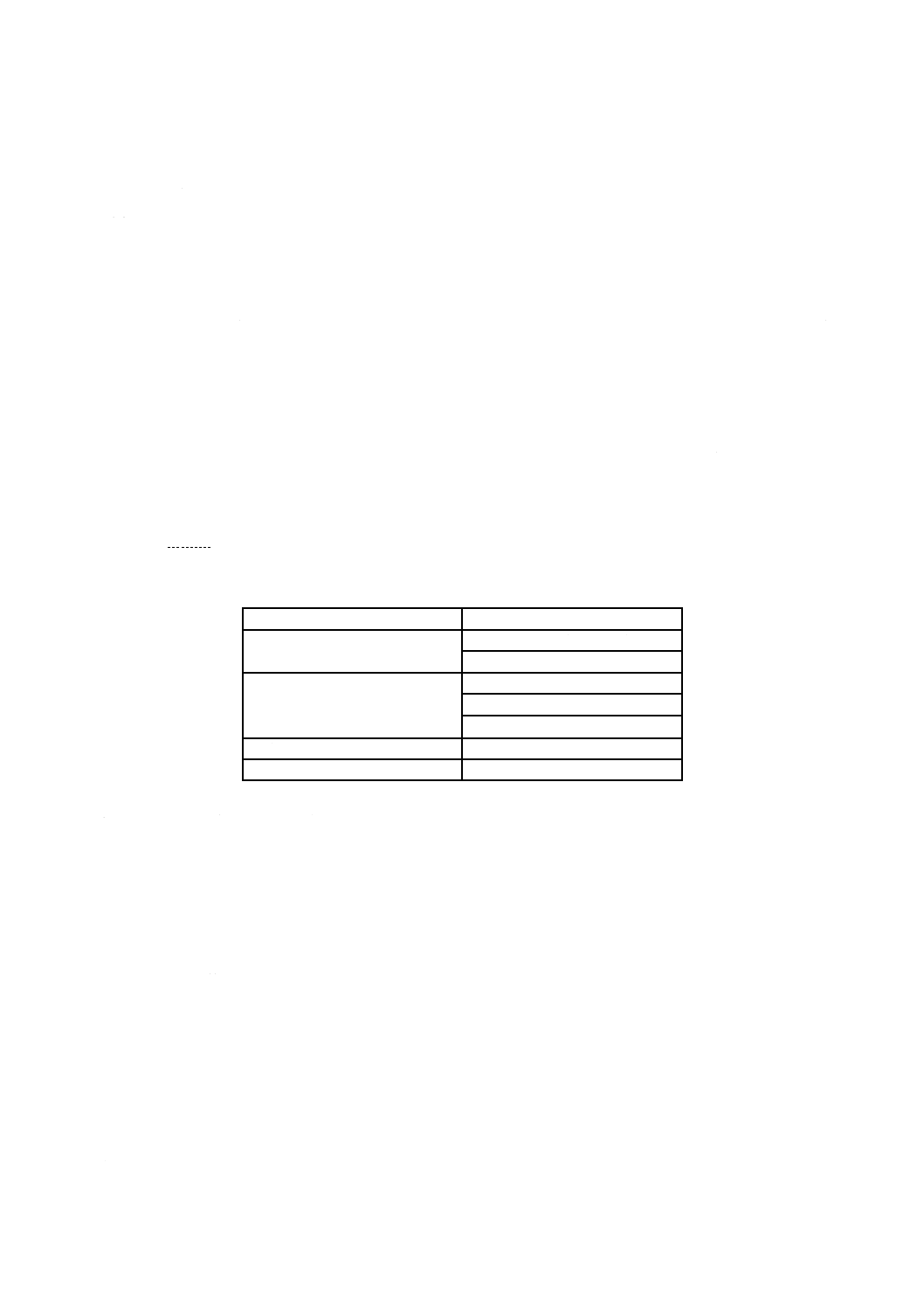
SIPの値は,長さ,質量,容積又は表面積から計算が可能である(例えば,表1を参照。)。
注記 JIS T 0806-2では,SIPの妥当性についての基準を規定している。
表1−SIP計算例
SIPの基準
製品
表面積
インプラント(非吸収性)
チューブ(多様な口径)
質量
粉体
ガウン
インプラント(吸収性)
長さ
チューブ(口径一定)
容積
液体
6
バイオバーデンの測定及び微生物学的特性付けの方法
6.1
バイオバーデンの測定
6.1.1
適切な方法の選択
バイオバーデンを測定するための適切な方法を選択しなければならない。この方法には,次の技術を含
まなければならない。
a) 微生物の取出し(該当する場合)
b) 微生物の培養
c) 菌数測定
精度を決めなければならない。それは,データの使用目的に適したものでなければならない。
6.1.2
微生物の取出し
6.1.2.1
製品から生育可能な微生物を取り出す場合は,取出しの効率を考慮しなければならない。また,
この考慮の結果を記録しなければならない(4.1.3参照)。少なくとも,次の事項を考慮しなければならな
い。
a) 微生物を取り出す方法の能力
6
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 推定される製品上にある微生物のタイプ及び位置
c) 取出しの方法が微生物の生存に与える影響
d) 試験対象の製品の物理的又は化学的性質
6.1.2.2
製品からの生育可能な微生物の取出しを行わない場合は,菌数測定の効率を考慮しなければなら
ない。また,この考慮の結果を記録しなければならない(4.1.3参照)。これには,少なくとも次の事項を
入れなければならない。
a) 推定される製品上にある微生物のタイプ及び位置
b) 試験対象の製品の物理的又は化学的性質
c) in-situ培養によって単一コロニを形成する細胞集合体。
注記 in-situは,“本来の場所での”という意味で,その微生物が存在する製品上及び/又は製品内の
本来あるべき場所での試験を意味する。微生物が周囲からの影響を受けているような場合に,
このような条件での試験が必要となる。
6.1.2.3
製品が,検出する微生物の数又はタイプに悪影響を与える物質を放出するような物理的又は化学
的性質をもつ場合は,こうした放出物質の影響を中和又は除去を行わなければならない。これが不可能な
場合には,最小限に抑えるシステムを採用しなければならない。このシステムの有効性を立証しなければ
ならない。
注記 附属書Bに,殺菌物質又は静菌物質の放出を評価するために使用できる技術を記載する。
6.1.3
微生物の培養
培養条件は,存在する可能性のある微生物のタイプを考慮して選択しなければならない。こうした考慮
の結果及び条件測定の根拠は,記録しなければならない(4.1.2参照)。
6.1.4
菌数測定
菌数測定の方法は,存在する可能性のある微生物のタイプを考慮して選択しなければならない。こうし
た考慮の結果及び条件決定の根拠は,記録しなければならない(4.1.2参照)。
6.2
バイオバーデンの微生物学的特性付け
6.2.1
微生物学的特性付けには,適切な方法を選択しなければならない。
注記 微生物学的特性付けは,バイオバーデンデータの利用(例えば,滅菌プロセスの確立)の幾つ
かの面に影響を与えるような製品の微生物相の変化を検出するために必要である。
6.2.2
微生物学的特性付けは,次の一つ又は複数を用いて行わなければならない。
a) 染色特性
b) 細胞形態
c) コロニ形態
d) 選択培養の利用
e) 生化学的特性
f)
十分なデータベースがある遺伝子配列データ
7
バイオバーデンの測定方法のバリデーション
7.1
バイオバーデンの測定方法は,バリデートし,文書化しなければならない。
7.2
バリデーションでは,次のことを行わなければならない。
a) 製品から微生物を取り出す場合は,取り出す方法の妥当性の評価
b) 補正係数を得るための回収率の決定
7
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 培養条件及び微生物計数方法を含む,菌数測定の妥当性の評価
d) 微生物学的特性付けの技術の妥当性の評価
8
日常のバイオバーデン測定及びデータの解釈
8.1
サンプルサイズ及びサンプリング頻度を定めた文書化したサンプリング計画に従い,日常のバイオ
バーデン測定を行わなければならない。
8.2
製品又は製品グループについてあらかじめ定めた方法を用いて,バイオバーデンを測定しなければ
ならない(5.1.2参照)。
8.3
バイオバーデンの微生物学的特性付けは,バイオバーデンの測定から得られるデータの使用目的に
応じて実施しなければならない(6.2参照)。
微生物学的特性付けにおいて,通常の微生物そう(叢)には見られない分離菌を回収した場合は,これ
らの分離菌の特性の評価を考慮するのがよい。
8.4
滅菌プロセスを確立するためにバイオバーデンデータを使用する場合は,滅菌プロセスの開発,バ
リデーション及び日常管理に関わる適切な規格で規定しているバイオバーデンデータの利用に関わる該当
する要求事項を満たさなければならない。
8.5
医療機器の上又は内部における,バイオバーデンの許容限度値をあらかじめ定めなければならない。
この仕様は,前もって得たデータに基づいたものでなければならない。これらの限度値を超える場合は,
適切な処置をとらなければならない(4.4参照)。
8.6
一定期間にわたって測定したバイオバーデンから得たデータは,傾向を明らかにするために使用し
なければならない。許容限度値をレビューし,必要に応じて改訂しなければならない。
8.7
サンプルサイズ,サンプリング頻度及び/又は許容限度値を定めるために用いる統計的手法は,JIS
Q 13485に適合しなければならない。
9
バイオバーデンの測定方法の維持
9.1
製品及び/又は製造プロセスの変更
製品及び/又は製造プロセスを変更する場合は,バイオバーデンが変化する可能性があるかどうかを判
断するためにレビューしなければならない。レビューの結果は,記録しなければならない(4.1.2参照)。
バイオバーデンが変化する可能性がある場合は,その変化の範囲及び性質を評価するためのバイオバーデ
ンの測定を行わなければならない。
9.2
バイオバーデンの測定方法の変更
日常のバイオバーデンの測定方法についての全ての変更は,評価しなければならない。この評価には,
次の事項を含まなければならない。
a) 変更が測定結果に与える影響の評価
b) 変更した方法の回収率の確立
注記 変更の評価によって,これまでのバリデーション及び回収率が,まだ適用可能であることが明
らかになることがある。
9.3
バイオバーデンの測定方法の再バリデーション
最初のバリデーションデータ(7.2参照)及びそれ以降の再バリデーションデータを,文書化した手順に
従いあらかじめ定めた間隔でレビューしなければならない。再バリデーションを行う範囲を定めなければ
ならない。レビューの結果及び実施した再バリデーションの結果は,記録しなければならない(4.1.3参照)。
8
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
製品上の微生物群の測定についての指針
注記 参照に便利なように,この附属書の箇条番号は,この規格の本文に対応している。
A.1 適用範囲
この附属書は,この規格で規定している要求事項の実施に関わる手引を示す。ここに示す手引は,網羅
的なものではないが,考慮するとよい重要な点を強調するためのものである。
この附属書に記載する以外の方法を採用してもよいが,それらの代替方法は,この規格の要求事項への
適合を達成するのに有効であることを立証するのがよい。
この附属書は,この規格の要求事項への適合を評価するためのチェックリストを意図していない。
A.2 引用規格
引用規格として含まれる文書の要求事項は,この規格の規定部分で引用している範囲内においてだけ,
この規格の要求事項となる。引用は,規格全体の場合もあるし,また,特定の箇条に限定される場合もあ
る。
A.3 用語及び定義
指針はない。
A.4 品質マネジメントシステムの要素
注記 完全な品質マネジメントシステムをもつことがこの規格の要求事項ではないが,滅菌の対象と
なる医療機器のバリデーション及び監視で使用するバイオバーデンの測定の管理に最低限必要
な品質マネジメントシステムの要素は,本文中の該当する箇所(特に,箇条4参照)で規定し
ている。医療機器の製造又は再処理の全ての段階を管理する品質マネジメントシステム(JIS Q
13485参照)に関わる規格に注意を払う必要がある。医療機器製造業者には,薬事法令によっ
て品質マネジメントシステム及び規制当局又は第三者機関によるシステム評価に対する要求事
項が存在することに注意を払う必要がある。
A.4.1 文書化
JIS Q 13485の文書化の要求事項は,文書(仕様書及び手順書を含む。)及び記録の作成及び管理に関係
している。
データの直接的及び間接的収集,処理及び/又は保存のために,試験施設でコンピュータを利用しても
よい。このような用途で使用するハードウェア及びソフトウェアは,管理するのがよい。
使用するコンピュータシステムは,ハードウェア及びソフトウェアを明確にし,これらのいずれかに変
更を加えた場合は,そのことを文書化し,適切な承認を受ける。
電子的データ処理技術によって計算を行う場合は,使用前にソフトウェア(例えば,スプレッドシート
計算)の妥当性を確認し,このバリデーション結果を保存する。
ソフトウェアについては,次の内容を文書化するとよい。
9
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− コンピュータシステムで実行されるアプリケーションソフトウェア
− オペレーションソフトウェア
− 使用するデータパッケージ
全てのソフトウェアは,使用に先立って,使用の可否のためのテストをする。
コンピュータソフトウェアを社内で開発した場合は,次の事項を確実にするための適切な手順を作成す
る。
− ソースコードを含む開発関連文書の保存
− 使用の可否のためのテスト記録の保存
− プログラムに対する修正の文書化
− 機器における変更の文書化及び使用前の正式なテストの実施
これらの管理事項は,市販のソフトウェアパッケージの修正又はカスタマイズにも適用するのがよい。
ソフトウェアプログラムに対する,未承認の変更を検出する又は防止する手順を設ける。
データのとりまとめ,作表及び/又はデータを統計的若しくは他の数学的処理を行うためのソフトウェ
アプログラム,又はその他の電子的に保存したデータの操作若しくは分析を行うソフトウェアプログラム
は,元の入力データを復元できるようにするのがよい。コンピュータデータをアーカイブに保存するため
の特別な手順が必要な場合は,これらの手順を文書化しておく。
文書及び記録を管理するための要求事項は,JIS Q 13485の4.2.3(文書管理)及び4.2.4(記録の管理),
又はJIS Q 17025の4.3(文書管理)及び4.13(記録の管理)で規定している。
技術的記録に関わる要求事項は,JIS Q 17025の4.13.2(技術的記録)及び5.4(試験・校正の方法及び
方法の妥当性確認)で規定している。
コンピュータソフトウェアへの品質マネジメントシステムの適用に関わる手引については,ISO/IEC
90003も参照。
A.4.2 経営者の責任
JIS Q 13485の経営者の責任についての要求事項は,経営者のコミットメント,顧客重視,品質方針,計
画,責任,権限及びコミュニケーション並びにマネジメントレビューに関わるものである。
バイオバーデン測定から得られたデータが信頼でき,かつ,再現可能であるためには,管理された条件
下で測定を実施することが重要である。したがって,医療機器の製造業者の製造現場であれ,離れた場所
であれ,測定で使用した試験施設を,文書化された品質システムに従って管理し,使用するのがよい。
バイオバーデンの測定には,幾つかの別々の組織が関係することがあり,それぞれが方法又は手順の特
定の要素について責任をもつことがある。この規格では,組織が受け入れたそれぞれの責任を定め,その
定められた責任を文書化することを要求している。この定めた権限及び責任は,それぞれの組織の品質マ
ネジメントシステムの中で文書化する。各要素に責任をもつ組織は,これらの要素について,適切な訓練
及び資格認定によってその能力が立証された力量のある職員に割り当てることを要求している。
医療機器の製造業者の直接管理下にある試験施設でバイオバーデンの測定を行う場合は,製造業者の品
質マネジメントシステムによって試験施設を運用する。外部の試験施設を利用する場合には,その試験施
設は,該当する規格(例えば,JIS Q 17025)の認証を受けているのがよい。
10
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
あらゆる試験施設は,品質サービスの提供をコミットすることが望ましく,また,このようなコミット
メントは,品質方針として文書化しておくのがよい。試験施設の組織内の権限及び責任の系統を正式に確
立し,文書化する。試験施設の品質システムの確立に責任をもつ個人を任命し,その人に,システムを実
行することを確実にするための権限を与える。
試験施設の運用は,定期的な内部監査の対象とするのがよい。監査結果は,試験施設の管理者が文書化
し,レビューするのがよい。JIS Q 17025の4.14(内部監査)を参照。
責任及び権限に関わる要求事項は,JIS Q 13485の5.5(責任,権限及びコミュニケーション)に,また,
人的資源に関わる要求事項は,JIS Q 13485の6.2(人的資源)に規定されている。
資源の提供に関わる要求事項は,JIS Q 13485の6.2に,また,機器に関わる要求事項は,JIS Q 17025
の5.5(設備)に規定されている。
A.4.3 製品実現
JIS Q 13485の製品実現についての要求事項は,顧客の要求事項の決定,設計・開発,購買,製造管理,
並びに監視及び測定機器の校正からなる製品ライフサイクルが関連している。
各試験機器について,メンテナンス要求事項を明確にするシステムがあるとよい。校正を必要としない
機器は,明確に識別するのがよい。
試験中に製品,回収液,培地などと接触する機器又はその部品は,無菌であるのがよい。製品から微生
物を取り出すために使用する培地又は回収液は,その無菌状態を確実にするような方法で調製する。
適切な品質試験には,培地性能試験を含むのがよい。一般的に,培地性能試験は,培地ロットごとに選
択した微生物の少数(10〜100 CFU)を接種して実施する。培地性能試験に適した微生物が日本薬局方に
記載されている。培地の品質管理のための,他の認知された定量的及び半定量的方法も利用できる。
購買についての要求事項は,JIS Q 13485の7.4(購買)に規定されている。特に,JIS Q 13485の7.4.3
(購買製品の検証)についての要求事項が,外部の組織から受け取る全ての製品及びサービスに適用され
ることに注意する。
監視及び測定装置の校正についての要求事項は,JIS Q 13485の7.6(監視機器及び測定機器の管理)に
規定している。装置及び測定のトレーサビリティに関わる要求事項は,JIS Q 17025の5.5(設備)及び5.6
(測定のトレーサビリティ)で規定されている。
A.4.4 測定,分析及び改善−不適合製品の管理
JIS Q 13485では,測定,分析及び改善の要求事項は,プロセス監視,不適合製品の管理,データの分析
及び改善(是正処置及び予防処置を含む。)に関連している。
全ての仕様を超えた,又は悪化傾向を示すバイオバーデン結果は,調査が必要となる。調査の最初の段
階では,結果が真実であるか又は間違っているかを評価するのがよい。次の事項は,間違いの原因となる
ため,注意する。
− 不適切なサンプル(例えば,代表的でない,均一でない,不合格材料)
− 不適切なサンプリング材料(例えば,スワブ,容器,包装)
− 不適切な輸送,取扱い又は包装
− 不適切な試験器具(例えば,保管,ピペット,ろ過器)
− 間違った取扱い又は試験方法
− 不適切な培地又は希釈剤
− 不適切な試験環境
− 不適切な培養環境
11
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 計算違い又は書き間違い
結果が間違いによるものである場合は,限度を超えるバイオバーデン結果は,同一製品バッチからのサ
ンプルを用いた再測定を行って検証するのがよい。製品の中で微生物が増殖する可能性がある場合,又は
同一バッチを利用できなくなった場合には,新しいバッチを使用してもよい。
最初の結果が正しいことが確認された場合は,調査の第二段階では,少なくとも,次の事項を考慮する。
a) 滅菌プロセスの有効性について結果がもつ意味
b) サンプルサイズ及び/又は頻度を増やす必要性
c) 製造プロセスの評価の結果。評価では,次の事項に注意するとよい。
1) 原料及び構成部品(供給者,変更)
2) 洗浄,潤滑及び製造用の液体
3) 輸送及び保管容器
4) 作業表面
5) 職員の服装,衛生及び行動
6) 取扱い及び組立
7) 環境条件及びモニタリングの結果(全ての季節的要因を含む。)
8) 包装材料及び手順
9) 貯蔵条件
d) 次の事項を含む回収された微生物学的特性付け
1) 潜在的な発生源
2) 以前の分離株との比較
調査の結果に基づき,是正処置が必要になることがある。是正処置が必要な場合は,その有効性を立証
する必要がある。
是正処置の手順は,JIS Q 13485の8.5.2(是正処置)及びJIS Q 17025の4.11(是正処置)に規定されて
いる。
A.5 製品の選択
A.5.1 一般
A.5.1.1 製品のサンプルを選択し,取り扱う場合は,不注意によってサンプル中の微生物の数及びタイプ
に重大な変化を引き起こすような汚染が起きないようにし,実施するのがよい。サンプリング方法は,一
定期間にわたるバイオバーデンを比較できるように,一貫性があるものがよい。
バイオバーデンの測定のために製品のサンプルを選定するに当たっては,次の二つの方法がある。
a) 無作為に製品を選ぶ。
b) 廃棄する製品,又は他の不合格のため販売に適さない製品を選ぶ。
この選定は,幾つかの要因に依存することがあるが,選択した製品がその製品を代表するバイオバーデ
ンをもっていることが前提となる。不合格品を利用する決定をした場合は,実施する洗浄及び包装を含め
たその製品の全ての基本的な生産段階を経てきたものがよい。その意味では,a)で採取した製品がより望
ましいサンプルとなる。
バイオバーデンを測定するためにサンプリングする場合は,製品を通常の包装に収めるのがよい。
A.5.1.2 バイオバーデンの測定のための製品のグループ分けにおいては,次の事項を考慮する。
12
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 微生物の数
b) 微生物のタイプ
c) 製品のサイズ
d) 構成部品の数
e) 製品の複雑さ
f)
製造プロセスで採用している自動化の程度
g) 製造環境
バイオバーデンの測定のためにグループから選択する製品は,1)無作為に採取する,又は2)計画的なス
ケジュールに従って採取するのがよい。
A.5.1.3 バイオバーデン測定から得たデータを滅菌プロセスの確立のために使用する場合は,製品のサン
プルの採取からバイオバーデン測定までの時間は,最終製造ステップから製品滅菌までの時間を反映する
のがよい。
A.5.2 分割試料(SIP)
バイオバーデンの測定では,可能な場合は,製品全体を利用するのがよいが,利用できる試験用ガラス
容器に製品を収納できない場合には不可能である。分割試料(SIP)とする場合は,できるだけ製品の大き
な部分を使用することが望ましく,また,使用する製品の部分が測定対象の製品全体を代表するバイオバ
ーデンとみなせるのがよい。したがって,手術衣又は体外ドレナージキットのような大形製品を試験する
場合は,SIPの選択には注意が必要である。
SIP作製に当たっては,製品の取扱いに注意するのがよい。SIPを製品から分離するときには,汚染を加
えないように,管理された環境(例えば,ラミナーフローキャビネット内)において,清浄な条件下で実
施するのがよい。
A.6 バイオバーデンの測定及び微生物学的特性付けの方法
A.6.1 バイオバーデンの測定
A.6.1.1 適切な方法の選択
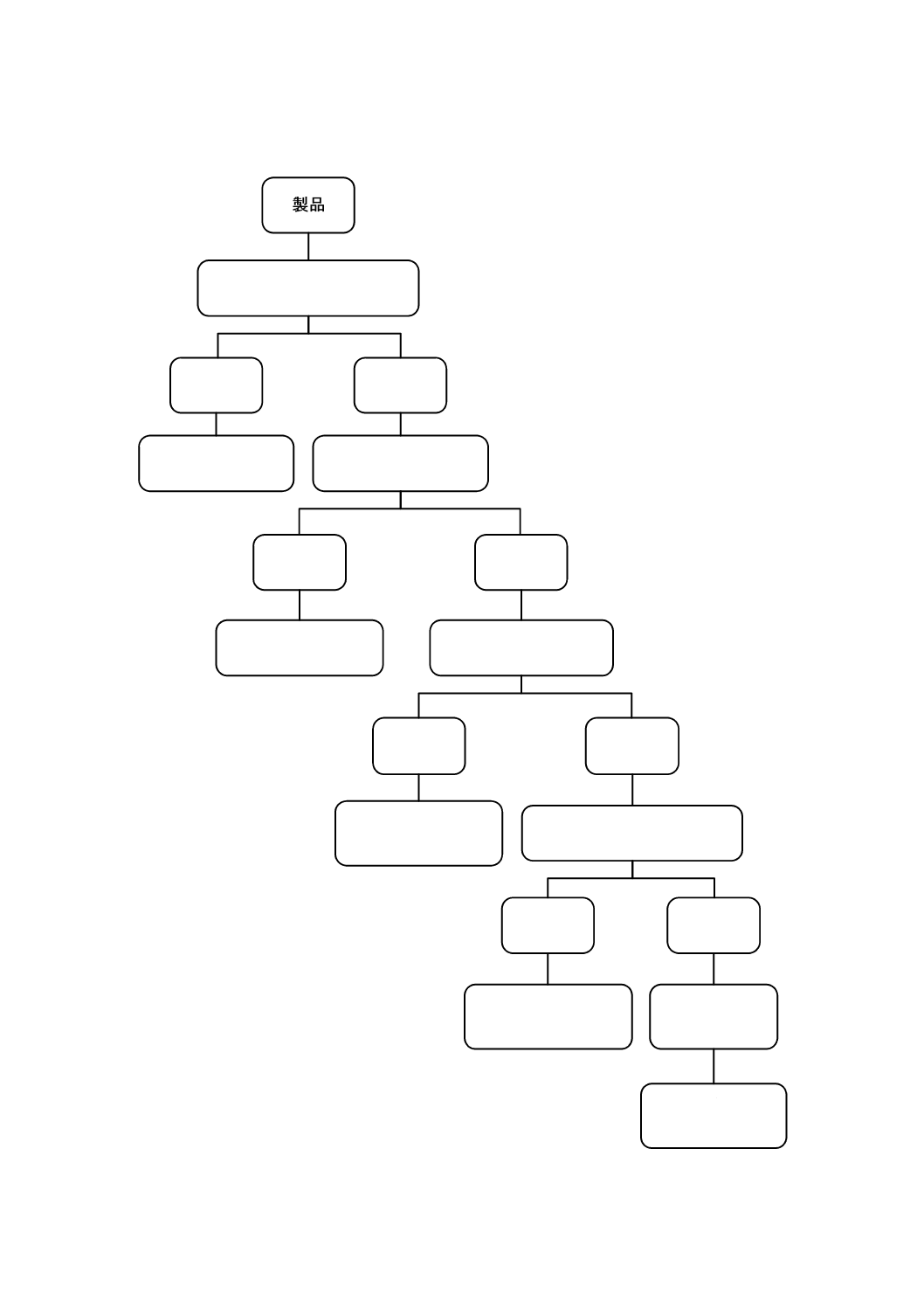
適切な場合は,DNA塩基配列決定法のような分子タイピング法を使用して,培養法の補足としてもよ
い。図A.1は,バイオバーデン測定方法を選択する初期の段階で一般的に適用されるデシジョンツリーを
図示したものである。
A.6.1.2 微生物の取出し
附属書Bを参照。
A.6.1.3 微生物の培養
培地及び培養条件を選択するときには,原料の性質,製造時の製造方法及び条件を考慮する必要がある。
扱いにくい微生物が存在し得る場合以外は,非選択性の培地及び培養条件が適している。
選択性の培地及び培養条件を使用する場合は,少なくとも,次の事項に考慮する。
a) 培地及び培養条件の一つの組合せで,全ての微生物が生育できるものではない。
b) バリデーションのための試験では,日常使用する場合よりも広範な培地及び培養条件を使用すること
が必要になることがある。
c) 選択培地への直接接種では,生理学的なストレス又は損傷を受けた微生物は生育できないことがある。
d) 一部の汚染源は季節的に変動することがあるので,検出する可能性のある微生物汚染源及び微生物の
タイプに留意する。
13
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
培地及び培養条件の例を,表A.1に示す。
ろ過できる液体か?
はい
いいえ
ろ過法/混釈培養法
/平板塗沫法を使用
柔軟性のない固体か?
はい
いいえ
超音波法/振とう法を
行い,ろ過/植え付け
繊維質/多孔/柔軟か?
はい
いいえ
ストマック法
又は振とう法を行い,
ろ過/植え付け
半固体又は粉体か?
はい
いいえ
ボルテックス法又は
振とう法を行い,
ろ過/植え付け
上のすべてが
使えない場合
スワブ法/
コンタクトプレート法
図A.1−バイオバーデン測定法のデシジョンツリー
全てが
植付け
植付け
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
植付け

14
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表A.1−培地及び培養条件の例a)
微生物のタイプ
固体培地
液体培地
培養条件b)
培養が容易な好
気性菌,通性嫌
気性菌
ソイビーン・カゼイン・ダイジェスト寒
天培地
(トリプトン大豆寒天)
ニュートリエント寒天培地
血液寒天培地
ブドウ糖トリプトン寒天培地
ソイビーン・カゼイン・ダイジェスト培
地
(トリプトン大豆ブロス)
ニュートリエント培地
30〜35 ℃
3〜7日
酵母及び糸状菌
サブロー・ブドウ糖寒天培地
麦芽エキス寒天培地
ローズベンガル寒天培地
クロラムフェニコール寒天培地
ソイビーン・カゼイン・ダイジェスト寒
天培地
(トリプトン大豆寒天)
ポテトデキストロース寒天培地
サブロー・ブドウ糖培地
麦芽エキスブロス
ソイビーン・カゼイン・ダイジェスト培
地
(トリプトン大豆ブロス)
20〜25 ℃
5〜7日
嫌気性菌
強化クロストリジウム寒天培地c)
シェドラー寒天培地c)
前還元血液寒天培地c)
嫌気性菌用寒天培地c)
ウィルキンス−チャルグレン寒天培地c)
ロバートソンクックドミート培地
液体チオグリコール酸培地
30〜35 ℃
3〜7日
注a) この表は,全てを網羅したものではない。
b) 記載した培養条件は,示した微生物のタイプに通常使用するものである。
c) 嫌気条件下で培養。
全ての非選択培地による嫌気性培養法では,通性嫌気性微生物が生育することがあるので注意するとよ
い。
A.6.1.4 菌数測定
B.6を参照。
A.6.2 バイオバーデンの微生物学的特性付け
製品のバイオバーデンに対してどこまで詳しい微生物学的特性付けが必要になるかは,データの使用目
的による。
医療機器の上又は内部のバイオバーデンの微生物学的特性付けには,様々な方法を利用することができ
る。通常,形態,グラム及び芽胞染色並びに単純な生化学反応(例えば,カタラーゼ,オキシダーゼ,イ
ンドール)などの古典的な試験に基づく分離株の同定によって,微生物が属する科又は属をある程度同定
できる。更に複雑な生化学的,血清学的及び分子的な試験を行うことによって,分離株を属又は種のレベ
ルまで同定することができる。
表A.2は,一般的な分類方法を示したものである。
表A.2−一般的なバイオバーデン分類方法
方法
例
選択性
染色
グラム染色,芽胞染色
低〜中
細胞形態
かん(桿)状,球状
低〜中
コロニ形態
形状,色,組織
低〜中
選択培地の使用
芽胞へのヒートショック,選択性真菌培地 中〜高
分離株の同定
属及び種
高
15
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.7 バイオバーデン測定方法のバリデーション
A.7.1 一般的に,周知の標準的な微生物学的方法では,職員の力量の検証,及び該当する場合は,陽性対
照及び陰性対照の微生物並びに陽性対照及び陰性対照の化学品の適切性の確立の外には,特別なバリデー
ションをする必要はない。通常,これらの活動でバイオバーデン測定の妥当性が十分確認できる。
一般的に,試験施設は,薬局方,ISO,AOAC,ASTMなどが発行する国家及び国際規格が公表している
方法を採用することが奨励されている。規格又は標準試験のような認知された方法は,試験施設間比較を
受けてきたものである。こうした認知された方法を用いる試験施設は,その施設における使用条件下にお
ける方法の正確さ及び信頼性を検証するだけで十分である。
バリデーション時に対照として使用する全ての試験微生物株は,公的微生物保存機関から入手するのが
よい。
A.7.2 医療機器からの試験微生物の回収率のバリデーションには,基本的に次のアプローチのいずれかに
よる。
− サンプル品の反復処理
− 既知レベルの微生物を製品に接種して行う回収量の定量的評価。
これらのアプローチのうち,前者は,自然に発生する微生物を利用するという長所をもつが,通常,比
較的高い初期バイオバーデンが必要である。後者のアプローチは,試験目的のモデルシステムを作成する
ものである。こうしたシステムを使用すると,自然の状況に適用できるのかという疑問が生じるが,この
アプローチは,バイオバーデンのレベルが低い製品には使用できる。
バイオバーデンの測定で使用するために選択した培養条件(例えば,培地及び培養条件)は,潜在的な
微生物を全て検出できるとは限らない。したがって,実際には,バイオバーデンを必然的に過小評価する
ことになる。いずれにせよ,適切な培養条件について決定しなければならない。
培養条件を評価する一つの方法として,製造プロセス,環境及び材料についての知見に基づき培養条件
を選択し,次に,こうした培養条件で算定した微生物を,これに代わる培地及び培養条件の組合せで検出
したものと比較する方法がある。このアプローチによって,低い比率のバイオバーデンが算定されている
ことが分かれば,予定した培養条件を再検討して,測定の最適化を図るのがよい。しかし,このアプロー
チは,低レベルのバイオバーデンをもつ製品にしか使用できない。
菌数測定に関わる指針については,B.6を参照。
バイオバーデンの測定は,単に製品のバイオバーデンを推定しているだけにすぎない。個別の方法の回
収率を向上させることで,バイオバーデンの測定の正確さを高めることができる。微生物取出しのために
は様々な方法が利用でき,個別の方法を使用して得た回収率を基に,採用する方法を決定する。回収率が
50 %未満であった場合には,その方法の改善又は代替方法の利用を検討するのがよいが,状況によっては,
回収率を50 %以上にすることができない場合がある。
環境微生物及び工業的な微生物の微生物学的特性付けで採用する方法を選択する場合,試験施設の職員
は,次の事項に留意する。
− 臨床施設で使用するために開発された試験及びキットの妥当性。
− 生化学試験で最低限の代謝活性を示す微生物(例えば,非発酵性のグラム陰性菌)について,職員が
結果を解釈する能力。
16
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.8 日常のバイオバーデン測定及びデータの解釈
A.8.1 微生物学的品質の効果的な管理を実施し維持していることを立証するために,製品及び/又は構成
部品を監視するプログラムを開発するのがよい。
日常のバイオバーデンレベルの監視では,通常,3〜10個のサンプルサイズとすることが多い。
サンプルサイズは,主として,次の二つの要因を考慮して合理的に選択する。
a) 検出したいバイオバーデンの変化
注記 これは,バイオバーデンレベルの変化(増加又は減少)の重要性及びバイオバーデンの情報を
どのように利用するかによる。平均バイオバーデンレベルの僅かな変化を早期に検出するため
には,大きなサンプルサイズにすることが必要になる場合がある。
b) 個々の試料に存在する,生育可能な微生物数の推定値のばらつき
注記 このような変動の程度によって,変化を検出するために必要なサンプルサイズが決まる。変動
を検出するために必要なサンプルサイズは,試料ごとの変動が僅かな場合は,試料ごとの変動
が大きい場合に比べて,小さなサンプルサイズでよい。
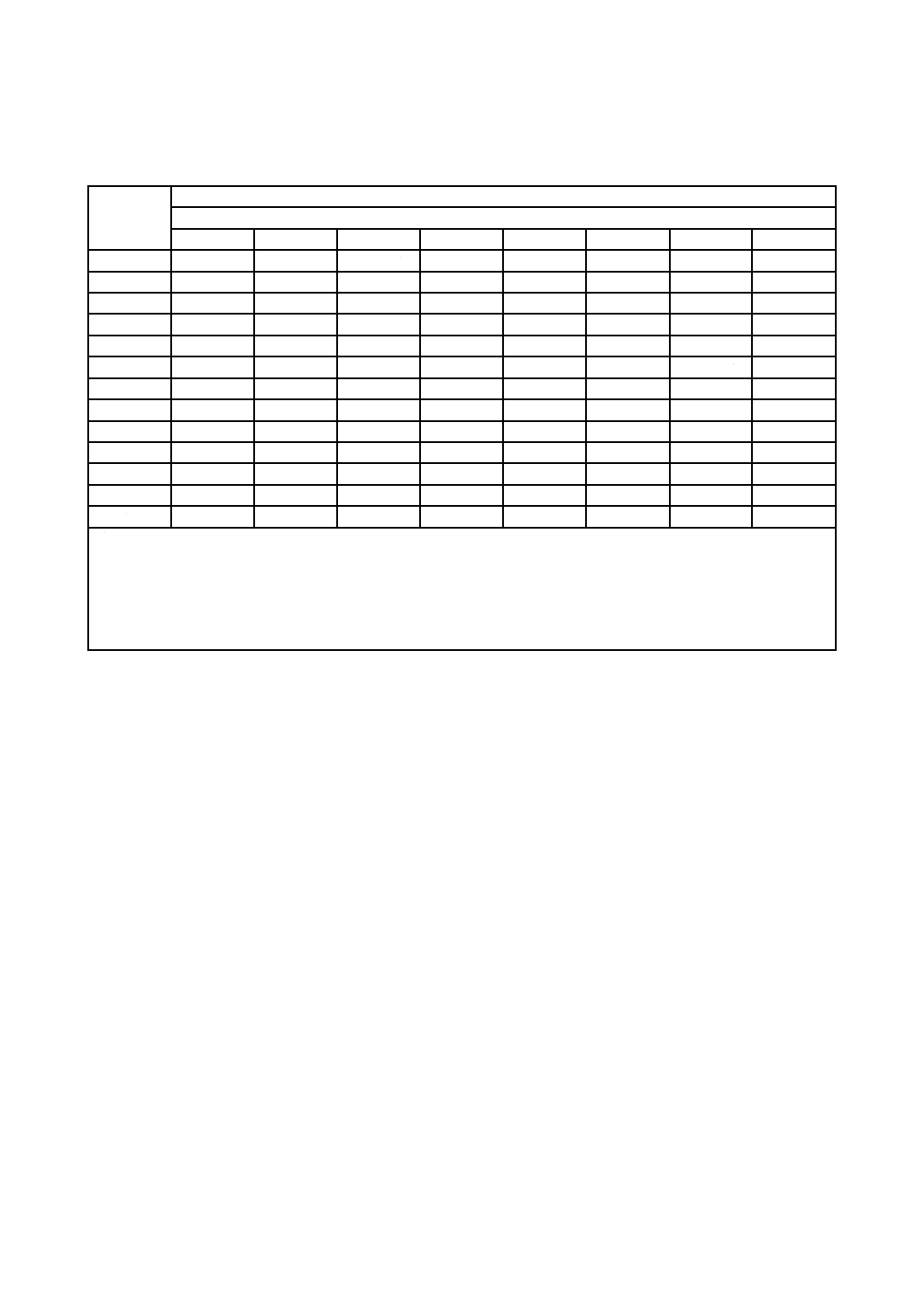
表A.3は,サンプルサイズ及びバイオバーデンの変動が,バイオバーデン数の変化を検出する能力にど
のような影響を与えるかを,例として示したものである。明らかに,大きなサンプルサイズの方が,有意
な変化を検出する信頼性が高くなる。
バイオバーデンデータの利用方法によって,検出が必要な変化の大きさ及び検出の信頼性が変わる。検
出しなければならないバイオバーデン変化量の選択及び検出できる確率の根拠があるとよい。
モニタリング頻度は,次の事項を含む様々な要素を考慮して,合理的に選択する。
− 過去のデータ
− データ採取の目的
− 製造プロセスの性質
− 製品の生産頻度
− バイオバーデン変化をタイムリに検出することの重要性
− 季節的及び環境的な変動
サンプリングは,時間(例えば,毎月)又は生産量(例えば,1バッチおき)に基づく頻度で行っても
よい。しかし,ベースライン値を確立するためには,新製品の初期生産段階では高い頻度でバイオバーデ
ンを測定し,バイオバーデンについての知識が増えるにつれて,この回数を減らしていくのが普通である。
バイオバーデンの測定頻度は,例えば,季節的変動,製造上の変更又は材料の変更によるバイオバーデ
ンの変化を検出できるのがよい。
A.8.2 バイオバーデンの測定方法の選択においては,製品上又はその内部でのバイオフィルム発生の可能
性を考慮する。細胞組織を含む医療機器は,バイオフィルムが発生する可能性がある。バイオフィルムは,
液体と接触する製品上又はその内部で形成されることがある。
A.8.3 A.6.2を参照。
A.8.4 指針はない。
A.8.5 あらかじめ定めた限度を超えた場合は,あらかじめ定めた処置をとる必要がある。是正処置によっ
て,バイオバーデンに影響を与えるプロセス変化が生じたときは,新しいデータを取得し,製品について
新しい限度を確立する。
バイオバーデンの限度は,製品に関わる履歴データに基づいている。このような履歴データがない場合
は,その製品の最初の3バッチを評価した時点で,暫定的な限度値を設定することができる。引き続く試
17
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
験結果に基づき,一定期間の後にこの限度を再評価し,設定した限度が適切かどうかを検証するのがよい。
ある製品のバイオバーデン測定から得たデータが,広く認知されている数学的分布にそのまま従わない
ことがある。例えば,多くのゼロの結果と少数の高い値とを示す場合は,バイオバーデンデータを表すヒ
ストグラムは,右方向への長いテーリングを示す。数学的分布をそのデータセットに適用できる場合は,
データの上限もこれに応じて設定できる。したがって,生産プロセスが履歴データを得たときと同じ方法
で継続するなら,バイオバーデンの上限確率を選択することができる(確率限界は,95 %又は99 %となる
だろう。)。
数学的分布に適合しない場合は,管理図法をバイオバーデン測定から得たデータに適用することが可能
であり,これによって限度を設定することができる。管理図法は,基礎となる数学的な分布に厳密には依
存しない。管理図には,平均値及び標準偏差(又は範囲)をプロットする。平均値は,個別の値より対称
的な分布を示す傾向がある。生データを変換すると,標準シューハート管理図の適用性が更に増す。経験
的に,このようなデータセットを分布に適用する場合は,次の二つの方法による計数値の変換が適切であ
るといえる。
a) 全体的な平均バイオバーデンが10 CFU/単位未満の製品の場合は,対称性を向上させる変換として,
次の式を推奨する。
)1
/
/
(
log10
+
+
=
N
x
N
x
N
Y
··················································· (A.1)
ここに,
Y: 変換した値
x: 変換前の値
N: 不偏分散に基づく係数で,平均値の2乗/(不偏分散−
平均値)
履歴平均値が履歴(グループ内)不偏分散を超える場合は,係数Nが未定義になるので変換を無視
する。
b) 全体平均バイオバーデンが10 CFU/単位を超える製品の場合も,分布は,右方向の長いテールをもっ
て非対称的になる傾向がある。こうしたデータは,実際には対数分布で最もよく近似することができ
る。実際に個別の計数値の対数をとると,データは,ほぼ正規分布に近似する。バイオバーデン数が
ゼロということがあるので,対数をとる前に,全ての計数値に定数の0.1を加算するのがよい。この
場合に推奨する変換は,次の式(A.2)のようになる。
Y=log10(x+0.1) ······································································ (A.2)
ここに,
Y: 変換した値
x: 変換前の値
平均バイオバーデンの標準シューハート管理図に対する管理限度値は,その製品について十分な履歴を
収集した後で確定することができる(管理図作成手順については,JIS Z 9020-1,JIS Z 9021及びISO/TR
7871を参照)。その製品について十分な履歴データが利用できない場合は,少数の測定値を用いた暫定的
な管理限度値を設定してもよい。利用できるデータの数が増えたら,限度値を変更すればよい。該当する
場合は,限度値を設定するときに季節変動を考慮するのがよい。外れ値としたデータは,限度値設定時に
破棄するのがよい。
異常に大きいか又は小さいとされたデータについては,調査する。これらのデータが,試験エラー又は
製造プロセスで発生する突発的な高値とみなせる場合は,データを外れ値と判断し,バイオバーデンの監
視のための限度の設定に当たっては計算から除外してもよい。バイオバーデンデータを品質関連の判断に
利用するために分析する場合は,“生育なし”,“多すぎて計数不能(TNTC)”などの個別の試験結果を分
18
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
析に含める。
A.8.6 実際の傾向をサンプリング変動から区別する場合に,経時的に収集したデータをグラフ化すると役
立つ。バイオバーデン値が事前に設定した限度内にある場合でも,グラフ化することによって微生物数に
有意な変化が生じていることを示すことができる。
特に多くの観測値が記録されている場合は,バイオバーデン測定から得たデータについて統計的な計算
を実施する前に,有意な特徴が明らかになるようにデータを処理する必要がでてくることがある。データ
処理は,測定値をグループ分けし,度数分布表及びヒストグラムを作成し,定性的に行うことができる。
完了後,データについて傾向を調べることができる。
バイオバーデンに適用できる傾向分析手法は,幾つかある。これらの傾向分析手法として,シューハー
ト管理図,範囲に基づく管理(BOR),累積和管理図(ISO/TR 7871参照)などがあるが,これらに限定し
ない。それぞれの手法が,通常のランダムな結果の分散からのずれを予想し,仕様外の結果を際立せるた
めに使用できる。
幾つかの例において,利用可能なデータセットに基づく処置をとるべきかどうか,又は追加データが必
要かを判断するために,これらの手法を複数利用するのがよいことがある。
A.8.7 JIS Q 13485の8.1(一般)では,適切な統計的手法の選択を含め,適切な測定及び分析方法を計画
し実施することを要求している。広範囲の製品についてのバイオバーデン測定から得たデータを調査する
ことで,このようなデータの変動を明らかにできる。一つのグループからの測定値は,試料グループ内で
変動することがあるので,データの分析では平均値を利用するのが普通である。これらの平均値が,高値,
中間値又は低値を取り得ることは明らかであり,また,平均値は,時間とともに変動する。さらに,バイ
オバーデンを構成する微生物のタイプも変動する。
バイオバーデンの測定から得たデータの頻度分布は,分布が非常に非対称的になっており,かつ,非常
に長いテールを示すことが多いという特性が共通してみられる。低いデータ又は中間データについては,
最頻値はゼロになる。これらの状況では,バイオバーデンは一般に低くなるが,有効な管理を実施しても,
ときおり高値が発生することがある。
これらの頻度分布が極端に非対称的になるということは,対称的分布に基づいて確立された品質管理手
法が,必ずしも適切ではないことを意味している。次のいずれかの統計的手法を個々の事例に合わせて開
発することが必要な場合がある。
a) データ分布を対称的にするための変換を行い,標準的な手法を適用する。
b) 非対称的分布に特別に適合させた新しい手法の開発。
A.9 バイオバーデンの測定方法の維持
指針はない。
19
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表A.3−サンプルサイズの変化に伴う及びサンプルの変動範囲内における,バイオバーデンレベルの
10倍の変化を検出するバイオバーデンデータに対するシューハート管理図の確率
サンプル
サイズ
サンプル内変動
標準偏差
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
3
0.997 23
0.908 26
0.678 71
0.454 92
0.299 57
0.201 88
0.141 08
0.102 41
4
0.999 88
0.977 25
0.841 34
0.630 56
0.443 20
0.308 54
0.218 35
0.158 66
5
1.000 00
0.995 20
0.929 51
0.766 32
0.577 06
0.418 82
0.303 11
0.222 45
6
1.000 00
0.999 11
0.971 22
0.860 48
0.691 21
0.524 66
0.390 37
0.290 98
7
1.000 00
0.999 85
0.989 03
0.920 67
0.782 20
0.620 65
0.475 97
0.361 58
8
1.000 00
0.999 98
0.996 06
0.956 74
0.850 97
0.703 86
0.556 74
0.431 89
9
1.000 00
1.000 00
0.998 65
0.977 25
0.900 73
0.773 37
0.630 56
0.500 00
10
1.000 00
1.000 00
0.999 56
0.988 41
0.935 43
0.829 67
0.696 25
0.564 46
11
0.958 90
0.874 06
0.753 37
0.624 24
12
0.974 34
0.908 26
0.802 06
0.678 71
13
0.984 25
0.934 09
0.842 83
0.727 59
14
0.990 49
0.953 24
0.876 44
0.770 85
15
0.994 34
0.967 21
0.903 77
0.808 66
注記 表A.3は,次に基づいて作成した。
− バイオバーデン計数値は,対数変換した。
− 標準シューハート管理図を使用。
− 変換したデータは,正規分布に従う。
− バイオバーデンは,10倍増加した。
− サンプルの平均値は,3σ管理限界よりも大きい値をもつ。
20
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(参考)
バイオバーデン測定法の指針
B.1 一般
B.1.1 バイオバーデンの測定は,様々な状況で行われる。こうした測定の責任者は,サンプリング率,培
地の性質及び培養条件,並びに方法の開発及びバリデーションの範囲を含めて,測定を実施するそれぞれ
の状況を考慮に入れる。
B.1.2 バイオバーデンを測定するプロセスの主要なステップの順序を,図B.1に示す。こうした測定に当
たる責任者は,原料,構成部品,製造環境,製造プロセス及び製品の性質についての知識を利用して,そ
れぞれのステップにあった適切な手法を選択するのがよい。
サンプルの選択(A.5.1参照)
↓
試料の収集(A.5.1参照)
↓
試験施設への輸送(B.2.1.7参照)
↓
処理手法(B.2及びB.3参照)
↓
培地への輸送(B.4参照)
↓
培養(B.5参照)
↓
菌数測定(B.6参照)及び特性付け(A.6.2参照)
↓
データの解釈(A.8参照)
図B.1−バイオバーデンの測定プロセスの主要なステップの流れ
B.2 微生物の取出しを行う場合の方法
B.2.1 一般
B.2.1.1 この附属書に記載する幾つかの方法を,微生物の検出数を増やし,変動を減らすために組み合わ
せてもよい。
B.2.1.2 微生物が表面に付着する度合いは,表面,含まれる微生物及びその他の物質(例えば,潤滑油)
の性質によって変動する。汚染源も,付着の度合いに影響を与える。微生物を取り出すために用いる処理
は,何らかの物理的な力を伴った洗浄又は直接的な表面サンプリングから構成してもよい。回収を向上さ
せるために界面活性剤を使用してもよいが,高濃度の界面活性剤は,微生物の生育を阻害することがある
ので注意する。
21
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.2.1.3 無菌でない流体と接触する材料の場合,微生物は,バイオフィルムの形で発生することがある。
バイオフィルムは,表面に強力に付着する基質中に微生物が封入されている組織である。バイオフィルム
中の微生物は,滅菌プロセスに対して強い抵抗力を示すことがある。バイオフィルムは,数分以内に生成
を始め,細胞組織を含む医療機器上又は使用済みの医療機器で非常に大きく成長する。こうした場合は,
バイオフィルム形成の可能性を考慮するのがよく,B.2.2に概要を示す処理によって微生物をバイオフィル
ムから完全に取り出せるとは考えない方がよい。取出し手法のバリデーションにおいて,反復回収の間,
高い微生物数が繰り返して記録される場合は,バイオフィルムの存在を示している。
B.2.1.4 バイオバーデン測定に使用する処理方法は,再現性があるのがよい。また,過剰なキャビテーシ
ョン,せん断力,温度上昇又は浸透圧変化のような,微生物の生存能力に影響を与える可能性のある条件
は避ける。
B.2.1.5 管理が他より容易な幾つかの処理がある。処理の選択及び適切な処理条件の考案に当たっては,
条件及びその管理方法を考慮する。例えば,処理において,微生物取出し量を増すために,時間を延長す
る又は機械的かくはん(撹拌)の種類若しくは条件を変更してもよい。
B.2.1.6 一部の処理方法においては,試験対象の製品を解体(例えば,混合法,ストマック法及びボルテ
ックス法)することがある。解体した材料が存在すると,菌数測定が困難になることがある。例えば,解
体した材料を回収液から分離するなどの追加処理が必要になることがある。求めた計数値が代表的な値で
あることを確実にするために,注意を払う。
B.2.1.7 試料は,可能な限り速やかに試験施設に輸送する。輸送の遅れが避けられない場合も試料を保管
する条件を選んで,微生物数に変化が生じないようにする。最長保管時間をあらかじめ定めるのがよい。
乾燥は,微生物数を大幅に減少させる原因となることがあるので,保管条件及び保管時間の選択に当たっ
ては考慮する。
B.2.2 取出し方法
B.2.2.1 ストマック法
B.2.2.1.1 試料及び既知量の回収液を,無菌のストマックバッグに入れる。往復動パドルがバッグ上で動
作して,回収液を試料に浸透させ,かつ,これを囲むようにする。
B.2.2.1.2 処理時間を定める。
B.2.2.1.3 この方法は,軟質,繊維質及び/又は吸収性の材料に特に適しているが,袋を破裂させるよう
な材料(例えば,針又は硬質部品をもつ試料。)には適さない。
B.2.2.1.4 この方法は,使用する回収液が比較的大量の場合は,低濃度の微生物を含む懸濁液が生じるこ
とがある。可能なら,回収液をフィルタにかけるとよい。
B.2.2.2 超音波法
B.2.2.2.1 試料を,適切な容器内に入った既知量の回収液に漬ける。容器及び内容物を超音波浴内で処理
するか,又は超音波プローブを回収液に漬ける。微生物は,高エネルギーの超音波処理によって不活化す
ることがあり,また,プローブを使用すると超音波浴以上に不活化する。超音波処理方法は,B.9によっ
てバリデーションをするのがよい。
B.2.2.2.2 超音波処理の公称周波数及び処理時間を定めるのがよい。さらに,対象物を超音波浴内に置く
位置も定めるとよい。一部の超音波は,シールドによって低減することがあるので,同時処理する試料数
の制限を考慮するのがよい。
B.2.2.2.3 この方法は,非浸透性の固体試料及び形状が複雑な試料に特に適している。埋込みパルス発生
器のような電子部品をもつ医療機器の場合は,破壊することがある。
22
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.2.2.2.4 超音波処理エネルギー及び超音波処理時間は,微生物の破壊及び死滅又は回収液の過熱を引き
起こさないのがよい。
B.2.2.3 振とう法(機械又は手動)
B.2.2.3.1 試料を,適切な容器に入れた既知量の回収液に漬け,微生物の取出しを助けるために機械式振
とう機(例えば,往復動,回転又はスナップ動作)上で振動を加える。手動による振動でもよいが,その
効果は人によって異なることがある。
B.2.2.3.2 振動の時間及び振動数を定める。
B.2.2.3.3 一定の大きさのガラスビーズを加えて表面摩擦を大きくし,それによって回収率を高めるよう
にしてもよい。ここで加える振動時間及び振動数並びにガラスビーズのサイズは,微生物の過熱及び/又
はこれに損傷を与える可能性がないものがよい。
注記 ガラスビーズを追加することで,微生物が付着できる表面積を増すことになる。
B.2.2.4 ボルテックス法
B.2.2.4.1 試料を,ボルテックスミキサの回転パッド上に渦巻き流が発生するように置いた密閉容器内の,
既知量の回収液に漬ける。このようにして発生する渦巻き流は,手で押しつける力によって変動する。渦
巻き流が変動すると,取出しがばらつくことがある。
B.2.2.4.2 使用する容器,混合時間及びミキサの設定速度を定める。
B.2.2.4.3 この方法は,短時間で容易に実施できるが,主として小さな試料に適している。
B.2.2.5 フラッシング法
B.2.2.5.1 回収液を,試料の内くう(腔)に通す。回収液は,重力又は加圧によって流す。もう一つの方
法として,製品に回収液を満たし,封をしてかくはんしてもよい。
B.2.2.5.2 機器と回収液との接触時間,フラッシング速度及び液体量を定める。
B.2.2.5.3 機器の構成及び内くうのサイズによっては,内部表面から微生物を完全に取り出すために必要
とする物理的な力が制限されることがある。
B.2.2.6 混合法(ホモジナイズ/分解)
B.2.2.6.1 試料を,適切な容器内の既知量の回収液に漬ける。この試料を,あらかじめ定めた時間,混合
するか又は裁断する。
B.2.2.6.2 この時間は,試料及びブレンダーによって左右されるが,回収液の過熱及び微生物の損傷の可
能性が生じるほど長くすることは望ましくない。
B.2.2.6.3 この方法は,微生物数を平板培養によって算定できるように,試料を十分に小さな片に分割す
る方法である。
B.2.2.7 スワブ法
B.2.2.7.1 スワブは,通常,棒又は柄に取り付けた吸着材からなる。可溶性の吸着材でもよい。
B.2.2.7.2 通常の使用方法では,スワブに回収液を浸し,試料表面を拭う。回収率は,最初に表面を湿潤
化させ,次に乾燥したスワブで拭き取ることで向上することがある。スワブを回収液に移し,かくはんし
ながら微生物をスワブから取り出す。可溶性のスワブの場合は,スワブを回収液中で溶解させてもよい。
B.2.2.7.3 スワブは,不規則な形状又は比較的アクセスしにくい部分へのサンプリングに役立つ。また,
大きな面積からのサンプリングにも有用である。
B.2.2.7.4 この方法は,スワブ操作の変動によって,誤差を生じやすい。また,表面上の全ての微生物が
スワブによって収集されるとは限らない。収集された微生物の一部が,スワブ自体に捕集され,その結果
検出されないこともある。
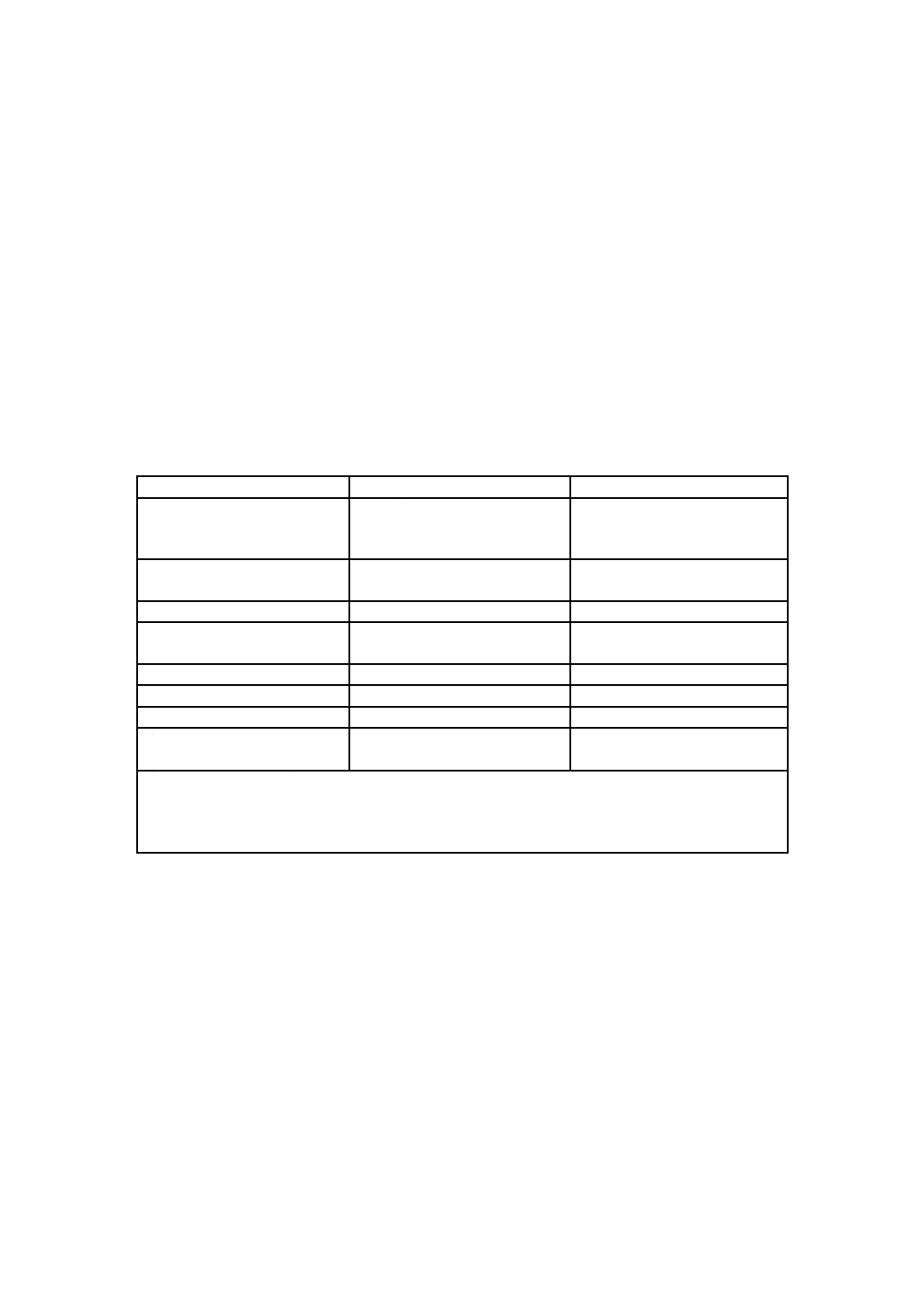
23
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.2.2.7.5 スワブには,殺菌剤又は静菌剤が存在しないものがよい。
B.2.3 回収液,希釈剤及び輸送培地
B.2.3.1 バイオバーデンの測定において,製品から微生物を取り出すのに回収液を使用することができる。
菌数測定のために取り出した微生物を輸送するために輸送培地を使用することができ,また,希釈剤は,
計数可能な数の微生物を含む懸濁液を得るために使用することができる。
B.2.3.2 回収液及び希釈剤の性質は,採用した方法の全体的効率に影響を与えることがある。希釈剤又は
回収液を選択する場合は,その組成(例えば,成分及びその濃度,浸透圧及びpH)を考慮するのがよい。
組成は,微生物の増殖又は不活化を起こさないものがよい。
B.2.3.3 微生物を固体表面から取り出すために液体を使用する場合は,界面活性剤を入れることを検討し
てもよい。
B.2.3.4 通常使用される回収液及び希釈剤として,表B.1に示すものがある。
表B.1−回収液及び希釈剤の例
溶液
濃度
用途
ペプトンバッファ水
りん酸塩 0.067 M
塩化ナトリウム 0.43 %
ペプトン 0.1 %
一般
カルゴンリンゲル液
1/4強度
アルギン酸カルシウムスワブの
溶解
ペプトン水
0.1〜1.0 %
一般
りん酸塩緩衝生理食塩液
りん酸塩 0.2 M
塩化ナトリウム 0.9 %
一般
リンゲル液
1/4強度
一般
塩化ナトリウム
0.25〜0.9 %
一般
チオ硫酸塩リンゲル液
1/4強度
残留塩素の中和
水
該当せず
水性サンプルの希釈。計数前に
可溶性材料を調製する。
注記 このリストは,全てを網羅したものではない。ポリソルベート80のような界面活性剤を,回収
液及び希釈剤に追加してもよい。通常,0.01〜0.1 %の濃度を採用するが,用途によって異なる。
個別の処理で使用するときの適切な濃度では,発泡が起きる可能性があるので注意が必要であ
る。
B.3 回収液による微生物の取出しを行わない場合の方法
B.3.1 コンタクトプレート法
B.3.1.1 コンタクトプレート又はスライドは,生育可能な微生物を培地表面に付着させるために,固形培
地を表面に押しつける方法である。プレート又はスライドを培養して,コロニを発生させる。
B.3.1.2 このようなシステムは,使いやすいという利点がある。結果は,培地と接触した面積と直接関連
付ける。
B.3.1.3 表面上での微生物の自然凝集,寒天表面でのコロニの広がり,寒天の乾燥,嫌気性の場所が存在
する可能性などが,潜在的短所として挙げられる。
B.3.1.4 この方法は,一般的に効率が低いので,他の方法が適用できない場合にだけ採用するのがよい。
コンタクトプレート及びスライドは,通常,平たん(坦)面又は少なくとも凹凸の少ない表面にだけ使用
できる。
24
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.3.2 寒天重層法
B.3.2.1 寒天重層法は,製品の表面を溶融寒天培地(温度は約45 ℃)でコーティングし,固まらせ,そ
の後培養し,コロニを生成させる。この方法は,バイオバーデンが低く,かつ,製品の形状が適している
場合にだけ適用することができる。
B.3.2.2 表面上での微生物の自然凝集,寒天界面でのコロニの広がり,寒天の乾燥,嫌気性の場所が存在
する可能性などが,潜在的短所として挙げられる。
B.3.3 MPN法(最確数法)
B.3.3.1 MPN法は,微生物がランダムに分布している製品の生育可能な微生物数を推定するための,十
分に確立された,かつ,完全に文書化された方法である。主要な用途は,食品業界及び水道業界であり,
これらの業界では,この方法を液体,粉末及び半固体の製品又は原料に使用している。この方法は,低い
平均バイオバーデン数をもつ製品に特に適している。
B.3.3.2 この方法は,生育可能な微生物を,容量当たり又は質量当たり同数含む複数のサンプルを製品か
ら採取し,これを液体培地で培養し,陽性数を調べて菌数を推定する方法である。十分な量の試験液が利
用できる場合は,接種した培地のいずれかの希釈段階が陽性にならなくなるまで希釈し,ニュートリエン
ト培地などに接種することもできる。1組の希釈段階で陽性が発生する頻度から,サンプル中又はサンプ
ルを採取した製品中に存在する生育可能な微生物の数を推定する。推定値についての95 %信頼限界は,比
較的広い。推定値及びその信頼限界は,出版されているMPN表(DeMan[20])から得ることができる。こ
の表は,サンプル中に存在する生育可能な微生物の数がポアソン分布に従って分布しているという仮定に
基づいて作成されたものである。
MPN法を適用するための重要な要求事項は,調査対象の製品全体における微生物数がランダムに分布し
ていることである。したがって,MPN法は,液体医療機器,高粘性流体,粉末又は製品からの回収液につ
いてバイオバーデンの測定をする場合に有益となる。しかし,固体の医療機器の微生物数についてバイオ
バーデンの測定をする場合は,この方法を一般的に適用できるかどうかを決めるのは難しい。低い平均バ
イオバーデンをもつ機器では,微生物数の分布は,通常,ランダムではない。このような場合,大部分の
サンプルは,検出可能な微生物をもたず,少数の製品に大量の微生物が存在する“スパイク”として微生
物が検出され,これが平均バイオバーデンに大きく寄与している。MPN法では,微生物数にかかわらず,
スパイクは陽性1として記録され,したがって,求められた平均値は形式的なものであり,このため,バ
イオバーデンを過小に評価するという望ましくない結果が生じる可能性がある。
B.3.3.3 MPN法は,実行が簡単であり,統計的な意味では,正確な測定というよりは,総括的な評価に
向いている。
B.3.3.4 殺菌物質又は静菌物質がある場合は,B.8で示す事項を考慮する。
B.4 培地への輸送
B.4.1 一般
B.4.1.1 処理をすると,通常,微生物の懸濁液が生じる。懸濁液中の菌数測定は,次に記載する方法の一
つを用いて行うことができる。
B.4.1.2 培地への輸送に先立って,過小評価を避けるために微生物の集合体を分散させるための追加的処
理が必要になることがある。場合によっては,試料から微生物を取り出すための処理によって,このよう
な集合体が分散されることがある。
B.4.1.3 殺菌物質又は静菌物質が存在する場合は,これらが培養方法の選択に影響を与えることがある。
25
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
回収液中に殺菌物質又は静菌物質が存在する場合は,希釈,ろ過による取出し又は化学的な不活化によっ
て,これらを無効な濃度まで低減できる場合がある。
B.4.2 膜ろ過法
B.4.2.1 回収液をろ過した後,適切な培地上でフィルタを培養し,コロニを形成させることで,効果的に
菌数測定できる。通常,微生物を捕集するには,孔径が0.45 μm以下のフィルタが適切である。
B.4.2.2 ろ過には通常,吸引又は加圧が必要になる。メンブレンフィルタの変形又は損傷を招くことがあ
るので,背圧を過大にしないように注意する。
B.4.2.3 繊維製品の残さ(渣)のような,微粒子を含む回収液を膜ろ過する場合は,微粒子が目詰まりを
起こし,ろ過が困難なことがある。
B.4.2.4 培養については,メンブレンフィルタを寒天の表面上又は液体培地を含ませた吸収パッドの上に
置いてもよい。メンブレンフィルタ上に生成したコロニを計数することができ,また,微生物学的特性付
けのために分離できる。
B.4.2.5 膜ろ過法は,微生物濃度が低い懸濁液の場合は,特に有用である。
B.4.2.6 膜ろ過法は,微生物を回収液から分離し,メンブレンフィルタ上で洗浄した後培養するため,回
収液中の物質が殺菌物質又は静菌物質を含んでいる疑いがある場合に有用である。一部の膜は,微生物の
生育を阻害するような物質を吸収又は放出することがあるので,菌数測定に適したメンブレンフィルタを
使用することが重要である。メンブレンフィルタは,回収液に耐性をもつものがよい。
B.4.3 混釈培養法
B.4.3.1 混釈培養法では,分取した懸濁液を約45 ℃の温度で溶融寒天培地と混和する。次に,混合物を
プレート内で固まらせ,これを培養し,その結果得られるコロニを計数する。
B.4.3.2 混釈培養法では,回収液から微生物を分離しない。殺菌物質又は静菌物質が存在する場合は,B.8
に示した事項を考慮する。
B.4.3.3 混釈培養できる回収液の量は限られており,したがって,この方法は,微生物濃度が低い懸濁液
では必要とする感度が得られないことがある。
B.4.4 平板塗沫培養法
B.4.4.1 平板塗沫培養法は,分取した懸濁液を塗沫用具で固体培地の表面に塗沫する。
B.4.4.2 培地の表面に塗沫した懸濁液は,不連続なコロニが形成できるように吸収させなければならな
い。吸収量によって,1枚のプレートで処理できる分取の量が決まる。
B.4.4.3 殺菌物質又は静菌物質が存在する場合は,B.8に示した事項を考慮する。
B.4.4.4 塗沫培養できる回収液の量は限られているため,この方法は,微生物濃度が低い懸濁液では必要
とする感度が得られないことがある。
B.4.5 らせん状培養法
B.4.5.1 らせん状培養法は,自動装置を使用する。この装置は,分取懸濁液を固体培地の表面に滴下させ
る。懸濁液は,培地中心から周辺に向かってらせんを描きながら,速度を落としつつ広がっていく。適切
な培養後,プレート全体又は一部のコロニ数から懸濁液中の生育可能な微生物数を求める。
B.4.5.2 らせん状培養法は,従来の連続希釈及び表面拡散法を使用した場合の結果と非常によい相関関係
をもち,再現可能な結果をもたらすことが示されている。装置の構造及び毛細管の使用並びに小さな容積
のため,らせん状培養法は,十分にかくはんされ,毛細管を詰まらせるような塊がなく,また,高濃度の
微生物を含む懸濁液の培養に主として利用される。
B.4.5.3 殺菌物質又は静菌物質が存在する場合は,B.8に示した事項を考慮する。
26
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.5 培養(培地及び培養条件)
B.5.1 培地及び培養条件の例を,表A.1に示す。
B.5.2 非選択的嫌気性培地は,通性嫌気性微生物が生育することもあるので注意する。
B.6 菌数測定
B.6.1 コロニ計数による菌数測定では,次のような状況のための手順を確立しておく。
a) 小コロニの検出(例えば,立体顕微鏡を使用)
b) 異常なコロニの計数及び記録(例えば,広がったコロニ)
c) 密集コロニの計測及び記録(例えば,多すぎて計数不能)
d) 連続希釈からの計数の記録
B.6.2 コロニ計数による菌数測定では,1枚の平板で生成するコロニ数を考慮する。このコロニ数は,生
育可能な各微生物が,隣接する微生物の悪影響を受けることなく,独立したコロニとして成長できるよう
なものがよい。
B.6.3 標準的なプレート計数法では,通常,1枚の平板におけるコロニ数に下限を設ける。この下限は,
バイオバーデンが低い医療機器におけるバイオバーデン測定に必ずしも適用する必要はない。
B.6.4 測定者間での変動を評価するのがよい。測定者間での変動は,10 %にもなることもある。
B.6.5 繊維が存在すると,独立したコロニの形成を妨げ,それによって菌数測定が難しくなることがある。
B.6.6 自動式の菌数測定では,システムのバリデーションを,JIS Q 17025によって実施するのがよい。
B.7 その他の微生物検出技術
コロニ計数以外の技術をバイオバーデン測定のために採用してもよい。こうした技術には,代謝活性の
測定(例えば,インピーダンス法又は蛍光法)を含む。このような方法は,生育可能な微生物数と相関関
係にあり,コロニ計数を基準として校正する必要があるので“間接法”と呼ばれる。これらの代替技術は,
低レベルの微生物を検出するのに十分な感度をもつのが望ましい。通常,検出数の下限は,100 CFU以上
である。
B.8 バイオバーデン測定に影響を与える物質に関わるスクリーニング
B.8.1 スクリーニングは,製品から懸濁液中に出る物質が,弱い微生物の生育可能性に及ぼす影響を調査
するために行う。これは,その方法が6.1.2.3に適合するか評価するときに使用できるアプローチである。
B.8.2 滅菌済み製品に予定している微生物取出しの方法を適用する。回収液を取出し法に使用する場合
は,B.8.3の手順に従うが,製品を直接培地に導入する場合には,B.8.4がより適している。
B.8.3 回収液は,製品から取り出した微生物の生育を促進することも阻害することも望ましくない。回収
液の影響を求めるために,既知の少数の微生物を製品に植え付け,日常のバイオバーデン測定に予定して
いる時間,この製品を回収液中に放置する。この処理の後,回収した微生物を計数する。使用できる微生
物は,薬局方に記載されている。結果の評価については,B.8.5を参照する。使用する微生物数は,約100
CFUとするのがよい。
B.8.4 製品を回収培地に直接導入する場合(例えば,MPN法の場合は,B.3.3参照。),日本薬局方で定め
ている静菌試験を利用することができる。この試験では,製品を少数の微生物とともに培地に導入し,日
常のバイオバーデン測定で予定しているものと同じ条件で培養する。使用する微生物の数は,約100 CFU
とするのがよい。結果の評価については,B.8.5を参照する。所定の培養期間後,目に見える生育があるか
27
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を調べる。
医療機器が,培地中にゆっくりと放出される静菌物質を含んでいる場合は,培養期間の終了時に,少数
の微生物をもつ製品−培地の組合せを試してみるのもよい。
B.8.5 植え付けた微生物数及び回収数が明らかに異なっている場合,又は静菌試験で微生物の成長が見ら
れない場合には,バイオバーデン測定のための方法を再検討する。阻害物質の低減,不活性化又は除去の
ために,希釈,中和又はろ過を導入する必要な場合がある。
B.9 物理的な力の悪影響に関してのスクリーニング
物理的な力を用いて,製品から微生物を取り出すことができる(B.2.2参照)。これらの力が,バイオバ
ーデン測定に与える影響を評価する。既知の少数の微生物(約100 CFU)を,使用する物理的な力にさら
すとよい。菌数測定によって,物理的な力の影響について程度が分かる。また,製品から取り出した微生
物の生育に対して回収液が与える潜在的影響を考慮する。
28
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C
(参考)
バイオバーデン測定方法のバリデーション
C.1 反復回収を用いたバリデーション
注記 この方法は,バリデーションプロセスにおいて,自然な製品上のバイオバーデンを使用する。
“徹底的な回収”と呼ぶこともある。
C.1.1 バリデーションを開始する前に,微生物の取出し方法を定め,文書化しておく。
C.1.2 回収率の測定対象となる,複数の製品又はその部品を選択する。各製品について,バイオバーデン
を測定するために通常使用する方法を適用する。
C.1.3 バイオバーデンの測定後,同一試料に再度この操作を行い,更に微生物が取り出せるかどうかを調
査する。この操作を,更に反復してもよい。
適切な反復回数は,製品の性質及びバイオバーデンを構成する微生物及び初期汚染レベルを含む幾つか
の要因によって決まる。反復回数を確定するために予備実験を行うことがある。
C.1.4 製品によっては,処理を繰り返した後でも生育可能な微生物が製品上に残っているかどうかを確定
するのがよい場合がある。これは,次のいずれかの方法によって達成できる。
a) 製品の表面を融解した培地でコーティングし,培地を固まらせてから,製品をあらかじめ定めた条件
で培養する。培養後,形成されたコロニを計数する。
b) 製品を液体培地に浸せき(漬)し,あらかじめ定めた培養条件を適用し,成長の有無を調べる。液体
培地に浸せきし,培養した後に,製品の一部で生育可能な微生物の存在が明らかとなった場合は,そ
の結果を利用してMPN法による算出を行うことができる。ただし,全ての結果が生育を示している
場合には,MPN法を適用することができず,バリデーションの方法を再検討する。
C.1.5 1回目の取出しで計数したコロニ数を,繰返しで得られたコロニ総数に対する割合として表す。コ
ロニ総数に占める割合を試験した各試料について計算し,これを使用して回収率を求める。C.2に作業例
を示す。
C.1.6 この方法は,バイオバーデン測定は,回収した微生物の累積数が大きく増加しなくなるまで反復す
るのが本来望ましいということに基づいている。繰返しごとに,回収液を製品又は製品部分から全量を回
収し,菌数測定をする。連続して回収したものから,累積した結果を比較する。ただし,この方法が必ず
しも正確ではないことに注意する。回収した微生物数と実際に製品上にある数との正確な関係は,常に立
証できるものではない。
C.2 補正係数の計算例
C.2.1 この例では,表C.1に反復処理によるバリデーションのためのデータを示す。これらのデータは,
五つの医療機器についてのものである。
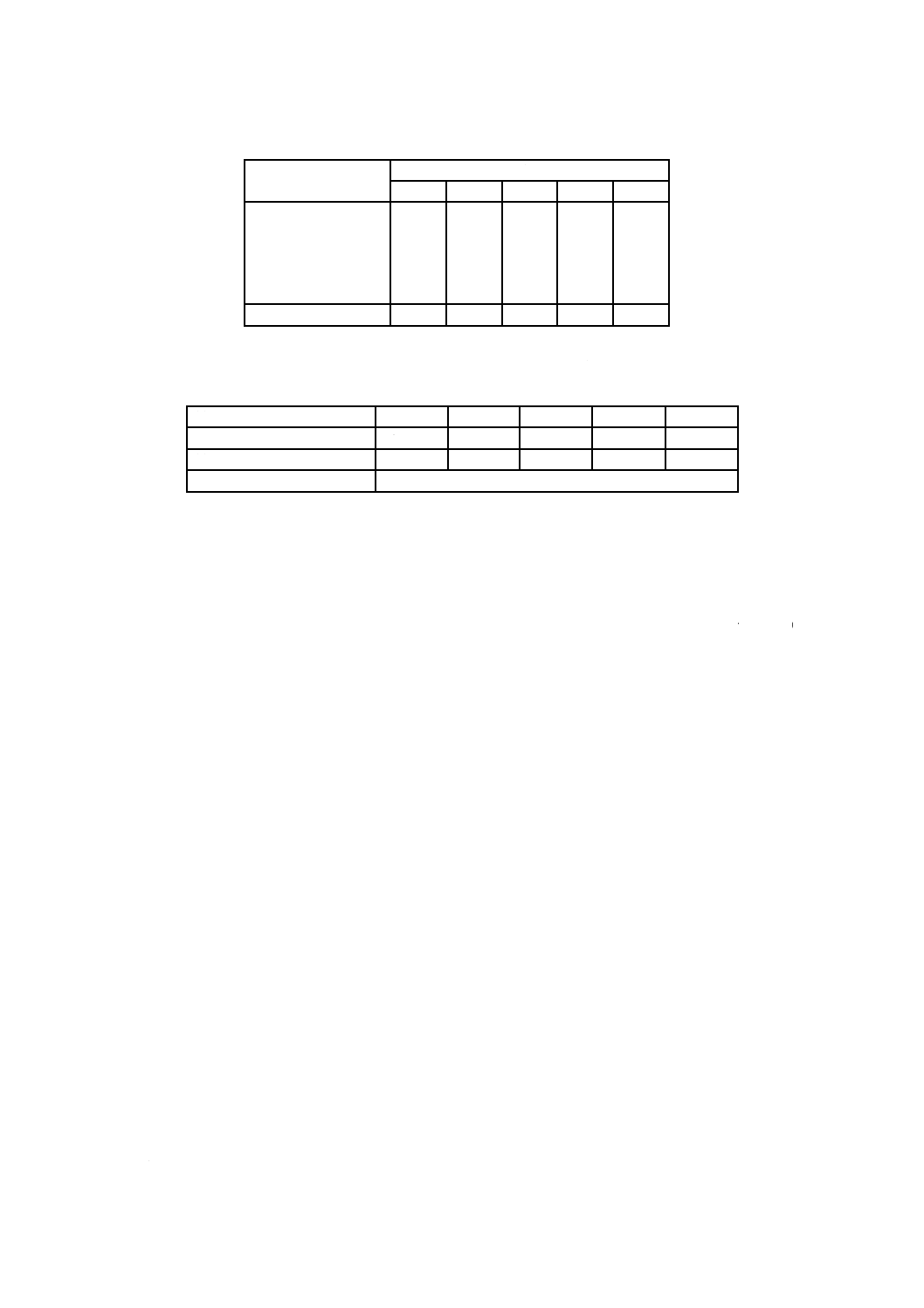
29
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表C.1−複数の医療機器における反復処理から求めたコロニ計数値
処理
試料
1
2
3
4
5
1
60
50
70
55
45
2
10
12
5
2
3
3
1
0
2
0
0
4
0
1
0
0
1
寒天重層
2
1
2
1
0
コロニ計数総数
73
64
79
58
49
C.2.2 表C.1のデータから,回収した微生物の割合を,次のように計算できる。
最初の処理で回収した数
60
50
70
55
45
回収した総数
73
64
79
58
49
最初の処理での回収率
82 %
78 %
89 %
95 %
92 %
最初の処理での平均回収率
87.2 %(範囲:78〜95 %)
寒天重層から求めたコロニ計数値を,計算に含めている。医療機器の性質によっては,寒天重層を利用
できないものがある。
C.2.3 最初の処理による平均回収率を用いると,回収率に対する補正係数は,次の式(C.1)のようになる。
15
.1
2.
87
100=
············································································ (C.1)
場合によっては,ワーストケースを反映させるために,回収率の範囲で最低の値を使用する場合がある。
これは,データの利用方法による。
C.3 製品に接種する方法
C.3.1 接種した製品を用いるバリデーション
C.3.1.1 回収率を確立するために,既知数の選択した微生物を製品上に接種することで,人工的バイオバ
ーデンを形成することができる。微生物は栄養型細胞であってもよいが,最も普通のアプローチでは好気
性微生物芽胞を用いる。乾燥時に生育能力を喪失するので,実際には栄養型細胞を利用するのは困難であ
る。
C.3.1.2 製品に接種するときに使用する微生物懸濁液を調製し,その生菌数を測定する。
製品接種によるバリデーションで使用する微生物を選択する場合は,その微生物が乾燥に耐えられるこ
とが重要であり,通常,好気性微生物芽胞を使用する。
C.3.1.3 この懸濁液の適切な希釈液を調製し,この希釈液の生菌数を接種時に測定する。
適切な希釈を確定するために,予備実験が必要な場合がある。人工的バイオバーデンを構成する微生物
数は,製品の自然汚染と同程度とするのがよい。バイオバーデンが低い品目の場合は,製品上に100 CFU
以下となるようにするとよい。
C.3.1.4 回収率を求める対象の滅菌済みの製品又はその部品を複数個用意する。各製品又は部品に一定量
の微生物懸濁液を接種し,クリーンベンチ内で乾燥させる。
エチレンオキサイドで滅菌した試料の場合は,十分エアレーションし,残留物による影響を減らすのが
よい。製品から溶出した物質の阻害作用を,予備実験で調査するのがよい。
30
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
懸濁液は,自然の汚染を除去するのが最も困難な部分を含むように製品に分布させるのがよい。
微生物接種法は,付着物,懸濁液の付着の有無,接種物の凝集,接種レベルの変動などの問題があるの
で,これらを考慮して製品へ接種するとよい。
吸収性物質でできた製品への接種は,微生物の懸濁液中に浸せきして行う。この方法によって,製品上
に微生物を均等に分布させることができる。
C.3.1.5 バイオバーデンの測定のために定めた方法を用いて微生物数(接種微生物)を求める。
C.3.1.6 取り出した微生物数の製品に接種した数に対する割合を求める。この割合を各製品について計算
し,回収率を確立する。
C.3.1.7 接種法によるバイオバーデン回収のバリデーションで得た結果は,真のバイオバーデンの回収を
そのまま表したものではない。
C.3.2 製品接種を用いた補正係数の計算例
C.3.2.1 バリデーションでは,予備実験からバイオバーデンが非常に低いことが明らかだったため,製品
接種法を選択した。
C.3.2.2 Bacillus atrophaeus (旧Bacillus subtilis var niger)の水懸濁液を調製し,懸濁液の生菌数を最適培
養条件を用いて測定した。
C.3.2.3 0.1 mL当たり100 CFUの芽胞を含むように懸濁液の希釈液を調製した。製品にこの希釈懸濁液を
0.1 mL接種し,クリーンベンチ内で乾燥させた。
C.3.2.4 接種した製品を,選択した方法で取り出した。取り出した芽胞数平均値は35で,25〜40の範囲
であった。
C.3.2.5 したがって,回収の補正係数は,次のようになる。
9.2
35
100=
31
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] ISO/TR 7871:1997,Cumulative sum charts−Guidance on quality control and data analysis using CUSUM
techniques
[2] JIS Z 9020-1:2011 管理図−第1部:一般指針
[3] JIS Z 9021:1998 シューハート管理図
注記 対応国際規格:ISO 8258:1991,Shewhart control charts(MOD)
[4] JIS Q 9000:2006 品質マネジメントシステム−基本及び用語
注記 対応国際規格:ISO 9000:2005,Quality management systems−Fundamentals and vocabulary(IDT)
[5] JIS T 0816-1 ヘルスケア製品の滅菌−湿熱−第1部:医療機器の滅菌プロセスの開発,バリデーショ
ン及び日常管理の要求事項
注記 対応国際規格:ISO 17665-1,Sterilization of health care products−Moist heat−Part 1:
Requirements for the development, validation and routine control of a sterilization process for
medical devices(IDT)
[6] JIS T 0806-1 ヘルスケア製品の滅菌−放射線−第1部:医療機器の滅菌プロセスの開発,バリデーシ
ョン及び日常管理の要求事項
注記 対応国際規格:ISO 11137-1,Sterilization of health care products−Radiation−Part 1: Requirements
for development, validation and routine control of a sterilization process for medical devices(IDT)
[7] JIS T 0806-2 ヘルスケア製品の滅菌−放射線−第2部:滅菌線量の確立
注記 対応国際規格:ISO 11137-2,Sterilization of health care products−Radiation−Part 2: Establishing
the sterilization dose(IDT)
[8] JIS T 0806-3 ヘルスケア製品の滅菌−放射線−第3部:線量測定にかかわる指針
注記 対応国際規格:ISO 11137-3,Sterilization of health care products−Radiation−Part 3: Guidance on
dosimetric aspects(IDT)
[9] JIS T 0801-1 ヘルスケア製品の滅菌−エチレンオキサイド−第1部:医療機器の滅菌プロセスの開発,
バリデーション及び日常管理の要求事項
[10] ISO 11138-2,Sterilization of health care products−Biological indicators−Part 2: Biological indicators for
ethylene oxide sterilization processes
[11] ISO/TS 11139:2006,Sterilization of health care products−Vocabulary
[12] JIS T 11737-2 医療機器の滅菌−微生物学的方法−第2部:滅菌プロセスの定義,バリデーション及
び維持において実施する無菌性の試験
注記 対応国際規格:ISO 11737-2,Sterilization of medical devices−Microbiological methods−Part 2:
Tests of sterility performed in the definition, validation and maintenance of a sterilization process
(IDT)
[13] ISO 14160:1998,Sterilization of single-use medical devices incorporating materials of animal origin−
Validation and routine control of sterilization by liquid chemical sterilants
[14] ISO 14937:2000,Sterilization of health care products−General requirements for characterization of a
sterilizing agent and the development, validation and routine control of a sterilization process for medical
devices
32
T 11737-1:2013 (ISO 11737-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[15] ISO/IEC 90003:2004, Software engineering−Guidelines for the application of ISO 9001:2000 to computer
software
[16] ASTM D4855-97, Standard Practice for Comparing Test Methods
[17] Budapest Treaty on the International Recognition of the Deposit of Microorganisms for the Purposes of Patent
and Procedure, Budapest 28th April, 1977, amended 26th September, 1980
[18] BONALSKY, J.R., A Model system for testing raw materials for microbial content., Pharm. Technol., 1980, vol.
4, No. 2, pp.49 - 51
[19] COLLINS, C.H., LYNE, P.M. and GRANGE, J.M., Collins and Lyneʼs Microbiological Methods, 7th Edition,
Butterworth-Heinemann Ltd, Oxford, 1995
[20] DEMAN, J.C., M.P.N. Tables Corrected. European J. Appl. Microbiol 1983; vol. 17; pp. 301-305
[21] HALLS, NA., et al., The Occurrence of Atypically High Presterilization Microbial Counts (“Spikes”) on
Hypodermic Products., Radiat. Phys. Chem., 1983, vol. 22, No. 3 - 5, pp. 663-666
[22] HITCHENS, A.D. and MISHRA-SZYMANSKI, A., AOAC International Qualitative and Quantitative
Microbiology Guidelines for Methods Validation, Journal of AOAC International, vol. 82, No 2, 1999, pp. 402 -
415
[23] International Conference on Harmonization (ICH) Validation of Analytical Methods: Definitions and
Terminology (CPMP/ICH/381/95)
[24] International Conference on Harmonization (ICH) Validation of Analytical Methods: Methodology
(CPMP/ICH/281/95)
[25] LUNDHOLM, M., Comparison of Methods of Quantitative Determinations of Airborne bacteria and evaluation
of total viable counts. Appl. Environ. Microbiol. 1982; vol. 44, No. 1; pp. 179 - 183
[26] PDA BIOBURDEN RECOVERY VALIDATION TASK FORCE, Technical Report: Bioburden Recovery
Validation, Journal of Parenteral Science & Technology, 1990, vol. 44, No. 6, pp. 324-331
[27] PDA Technical Report No 33, Evaluation, Validation and Implementation of New Microbiological Testing
Methods, PDA Journal of Pharmaceutical Science and Technology Supplement TR33, vol. 54, No. 3, May/June
2000
[28] PULEO, J.R., FAVERO, M.S. and PETERSON, J.J., Use of ultrasonic energy in assessing microbial
contamination on surfaces, Appl. Microbiol., 1967, vol. 15, No. 6, pp. 1345 - 51
[29] SOKOLSKI, W.T. and CHIDESTER, C.G., Improved Viable Counting Method for Petroleum-Based Ointments,
J. Pharm. Sci., 1964 vol. 53, pp. 103 - 107
[30] SHIRTZ, J.T., Sterility Testing, Pharmaceutical Engineering, November/December 1987, pp. 35-37
[31] USP 28/NF 23 2005, General Information, <1225> Validation of Compendial Methods, USP 28/NF 23, United
States Pharmacopeial Convention Inc., Rockville, MD, 2005
[32] 第15改正日本薬局方,一般試験法,4.05 微生物限度試験法