T 0993-1:2020 (ISO 10993-1:2018)
(1)
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 2
2 引用規格························································································································· 3
3 用語及び定義 ··················································································································· 4
4 医療機器の生物学的評価に適用される一般原則 ······································································ 7
5 医療機器のカテゴリ分類 ··································································································· 12
5.1 一般 ··························································································································· 12
5.2 身体との接触形態によるカテゴリ分類··············································································· 12
5.3 接触期間によるカテゴリ分類 ·························································································· 13
6 生物学的評価のプロセス ··································································································· 14
6.1 生物学的リスク分析のための物理学的及び化学的情報 ·························································· 14
6.2 ギャップ分析及び評価のための生物学的エンドポイントの選択 ·············································· 15
6.3 生物学的試験の実施 ······································································································ 15
7 生物学的評価データの解釈及び総合的な生物学的リスクアセスメント ······································· 21
附属書A(参考)生物学的リスクアセスメントで対処するエンドポイント ······································ 23
附属書B(参考)リスクマネジメントプロセスにおける生物学的評価実施のガイダンス ···················· 28
附属書C(参考)推奨する文献精査の手順 ··············································································· 42
参考文献 ···························································································································· 44
T 0993-1:2020 (ISO 10993-1:2018)
(2)
まえがき
この規格は,産業標準化法第16条において準用する同法第12条第1項の規定に基づき,一般社団法人
日本医療機器テクノロジー協会(MTJAPAN)及び一般財団法人日本規格協会(JSA)から,産業標準原案
を添えて日本産業規格を改正すべきとの申出があり,日本産業標準調査会の審議を経て,厚生労働大臣が
改正した日本産業規格である。これによって,JIS T 0993-1:2012は改正され,この規格に置き換えられた。
なお,この規格の改正公示日から3年間までJIS T 0993-1:2012を適用することができる。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣及び日本産業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
JIS T 0993の規格群には,次に示す部編成がある。
JIS T 0993-1 第1部:リスクマネジメントプロセスにおける評価及び試験
JIS T 0993-7 第7部:エチレンオキサイド滅菌残留物
日本産業規格 JIS
T 0993-1:2020
(ISO 10993-1:2018)
医療機器の生物学的評価−第1部:
リスクマネジメントプロセスにおける評価及び試験
Biological evaluation of medical devices-Part 1: Evaluation and testing
within a risk management process
序文
この規格は,2018年に第5版として発行されたISO 10993-1:2018を基に,技術的内容及び構成を変更す
ることなく作成した日本産業規格である。
なお,この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
この規格の目的は,医療機器の使用によって生じる潜在的な生物学的リスクからヒトを保護することに
ある。この規格は,医療機器の生物学的評価に関する数多くの国際規格,国内規格及び指針をまとめて作
成した。この規格は,医療機器の開発及び評価のプロセスの一つであるリスクマネジメントプロセスにお
ける生物学的評価についての指針である。あらゆる情報源の調査及び既存データの評価,並びに必要に応
じて追加試験の実施を組み合わせる手法をとることで,個々の医療機器に対する生物学的反応と機器の臨
床使用における安全性とを網羅的に評価することが可能になる。“医療機器”という用語は広範囲に及ぶこ
とに注意する。単一の材料で構成される単純な機器もあれば,複数の材料から製造された部材を数多く含
む複雑な機器もある。
この規格は,特定の医療機器を対象にしたものではなく,広く一般的な立場から医療機器の生体組織に
対する影響評価を対象とする。したがって,生物学的評価を完遂するために,医療機器の使用目的から想
定される生体組織との接触形態及び期間によって医療機器をカテゴリに分類し,それぞれに適切と考えら
れる生物学的エンドポイントを定めた。医療機器の定義(3.14)における注記も参照する。
生物学的ハザードの範囲は,広く複雑である。医療機器は,構成材料と生体組織との相互作用だけを考
えて設計することはできない。生体適合性は,医療機器を設計する上で材料選定で考慮する特性の一つに
すぎない。生体適合性が最良となる材料を選択した結果が,機能性の低い医療機器をもたらす可能性もあ
る。生体組織と相互作用することによって機能を発揮する材料については,それも考慮して生物学的評価
を実施する。
材料によって引き起こされる生体反応が,ある局面では副作用と考えられても,異なる状況では副作用
とは考えられない場合もある。生物学的試験はインビトロ試験,エクスビボ試験及び動物モデルを用いた
試験に基づき実施されるので,その生体反応が,医療機器をヒトに使用したときにも想定される反応であ
るかは慎重に判断すべきである。生物学的試験で認められた組織反応が同様にヒトにも起こるとは限らな
いためである。既に医療用途として確立された材料であっても,ある種の患者には副作用をもたらす場合
があるように,同一材料に対する組織反応には個体差があることも認識しておく必要がある。
この規格の第一の役割は,生物学的評価を計画するための枠組みを提供することにある。第二の役割は,
2
T 0993-1:2020 (ISO 10993-1:2018)
生物学的評価の基本原理に関する知識の科学進歩を活用し,インビボ試験と同等の毒性学的評価が可能な
方法が存在する場合には,インビトロモデル並びに化学的,物理学的,形態学的及び表面性状のキャラク
タリゼーション試験を優先することによって,実験動物へのばく露及び使用動物数を最小化することにあ
る。
この規格は,合否の判定基準を含む試験方法を厳格に規定するものではない。それらを厳密に規定した
場合,新しい医療機器の開発及び使用に不必要な制約をもたらし,また,医療機器の一般的な使用におい
ても誤った安全観に陥る可能性がある。医療機器の特性によって特殊な試験方法及び判定基準を定めるこ
とが妥当な場合には,当該製品に特有の個別規格の中で規定することが望ましい。
JIS T 0993規格群は,当該医療機器に関連する全ての因子(使用目的,科学文献の精査,臨床使用実績
などの新しい情報も含む。)を考慮しながら,その要求事項を理解し,当該医療機器に対する生物学的評価
結果を判断できる,十分な教育・経験を積んだ資格のある適切な専門家が用いることを想定して作られて
いる。
附属書Aには,生物学的リスクアセスメントで対処するのがよいエンドポイントを記載している。これ
は人体への接触形態及び接触期間のカテゴリ,並びにそのカテゴリに分類される医療機器の生体適合性を
評価する上で推奨される生物学的エンドポイントを確認するために広く役に立つ。附属書Bは,生物学的
評価を含んだリスクマネジメントプロセスを医療機器に適用するためのガイダンスである。
1
適用範囲
この規格は,次の事項について規定する。
− 医療機器のリスクマネジメントプロセスにおける生物学的評価を管理する一般原則
− 身体との接触形態及び接触期間に基づく医療機器のカテゴリ
− 適切な出典からの既存データの評価
− 要求されるデータセットと既存データとのギャップの特定,その結果に基づくリスク分析
− 医療機器の生物学的安全性の評価に必要な追加データの特定
− 医療機器の生物学的安全性のリスクアセスメント
この規格は,次が予測される材料及び医療機器の評価に適用する。
− 使用目的の対象となる患者の身体に直接又は間接的に接触する。
− 手術用手袋,マスクなど保護器具の場合には,使用者の身体に直接又は間接的に接触する。
この規格は,能動型,非能動型,植込み型及び非植込み型を含む全てのタイプの医療機器に適用可能で
ある。
この規格は,次に起因する生物学的ハザードの評価においても指針となる。
− 医療機器の経時的変化が生物学的安全性に影響するリスク
− 医療機器又は構成部品の破損によって生体組織が新規材料にばく露する場合
ISO 10993のその他の部は,生物学的評価及び関連する試験について規定している。機器に固有の規格
は力学的試験の実施についても記載している。
3
T 0993-1:2020 (ISO 10993-1:2018)
細菌,真菌,酵母,ウイルス,伝達性海綿状脳症(TSE)及びその他の病原体に関連したハザードは,
この規格の対象としない。
注記 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 10993-1:2018,Biological evaluation of medical devices−Part 1: Evaluation and testing within a
risk management process(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”こ
とを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格のうちで,西暦年を付記してあるものは,記載の年の版を適用し,その後の改正版(追補を含む。)
は適用しない。西暦年の付記がない引用規格は,その最新版(追補を含む。)を適用する。
JIS T 0993-7 医療機器の生物学的評価−第7部:エチレンオキサイド滅菌残留物
注記 対応国際規格:ISO 10993-7,Biological evaluation of medical devices−Part 7: Ethylene oxide
sterilization residuals
JIS T 14971:2012 医療機器−リスクマネジメントの医療機器への適用
注記 対応国際規格:ISO 14971:2007,Medical devices−Application of risk management to medical
devices
ISO 10993-2:2006,Biological evaluation of medical devices−Part 2: Animal welfare requirements
ISO 10993-3,Biological evaluation of medical devices−Part 3: Tests for genotoxicity, carcinogenicity and
reproductive toxicity
ISO 10993-4,Biological evaluation of medical devices−Part 4: Selection of tests for interactions with blood
ISO 10993-5,Biological evaluation of medical devices−Part 5: Tests for in vitro cytotoxicity
ISO 10993-6,Biological evaluation of medical devices−Part 6: Tests for local effects after implantation
ISO 10993-9,Biological evaluation of medical devices−Part 9: Framework for identification and
quantification of potential degradation products
ISO 10993-10,Biological evaluation of medical devices−Part 10: Tests for irritation and skin sensitization
ISO 10993-11:2017,Biological evaluation of medical devices−Part 11: Tests for systemic toxicity
ISO 10993-12,Biological evaluation of medical devices−Part 12: Sample preparation and reference materials
ISO 10993-13,Biological evaluation of medical devices−Part 13: Identification and quantification of
degradation products from polymeric medical devices
ISO 10993-14,Biological evaluation of medical devices−Part 14: Identification and quantification of
degradation products from ceramics
ISO 10993-15,Biological evaluation of medical devices−Part 15: Identification and quantification of
degradation products from metals and alloys
ISO 10993-16,Biological evaluation of medical devices−Part 16: Toxicokinetic study design for degradation
products and leachables
ISO 10993-17,Biological evaluation of medical devices−Part 17: Establishment of allowable limits for
leachable substances
ISO 10993-18,Biological evaluation of medical devices−Part 18: Chemical characterization of materials
4
T 0993-1:2020 (ISO 10993-1:2018)
ISO/TS 10993-20,Biological evaluation of medical devices−Part 20: Principles and methods for
immunotoxicology testing of medical devices
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
注記 ISO/IECの用語に関するデータベースの情報は,国内では不要なため削除した。
3.1
生体適合性(biocompatibility)
医療機器(3.14)又は材料(3.12)が特定の適用において適切な生体反応を起こし性能を発揮する能力。
3.2
生物学的リスク(biological risk)
医療機器(3.14)又は材料(3.12)との相互作用に関連した有害反応の結果として生じる健康被害の確率
とその被害の重大さとの組合せ。
3.3
生物学的安全性(biological safety)
意図された使用状況下において受容できない生物学的リスク(3.2)が存在しないこと。
3.4
化学成分(chemical constituent)
原料物質,添加剤(抗酸化剤,紫外線安定剤,着色添加物,染料など)及び加工助剤(溶剤,潤滑剤,
消泡剤など)を含む,材料(3.12)及び医療機器(3.14)のいずれか又は両方の製造の過程で使用されるあ
らゆる合成又は天然由来の物質。
3.5
データセット(data set)
様々な情報源から収集された物理学的特性,化学的特性,毒性データなどの医療機器の生物学的反応を
特徴付けるために必要な情報。
3.6
直接接触(direct contact)
生体組織と物理的に接触する医療機器(3.14)又は医療機器の構成部材。
3.7
体内と体外とを連結(externally communicating)
部分的又は全体が体外にあるが,体液及び生体組織のいずれか又は両方と直接又は間接的に接触する医
療機器(3.14)若しくは医療機器の構成部材。
3.8
最終製品(final product)
包装及び該当する場合には滅菌処理を含め,市販前の医療機器に適用される全ての製造工程を終えた医
療機器(3.14)又は医療機器の構成部材。
3.9
形状(geometry)
医療機器(3.14)の幾何学的形状。
5
T 0993-1:2020 (ISO 10993-1:2018)
医療機器の構成(device configuration)
医療機器(3.14)の部品の相対的な配置。
3.10
インプラント(implant)
臨床行為によってヒト体内に埋没されるか,又は上皮表面若しくは目の表面を置き換え,手術後もその
場にとど(留)まることを意図した医療機器(3.14)。
3.11
間接的接触(indirect contact)
医療機器(3.14)又は医療機器の構成部材を通過した液体若しくは気体が生体組織と物理的に接触する
こと(この場合,医療機器又は医療機器の構成部材自体は生体組織と物理的に接触しない。)。
3.12
材料(material)
医療機器(3.14)又はその部分として使用される,合成若しくは天然ポリマー,金属,セラミック,又
は複合体(不活化された生体組織を含む。)。
3.13
材料キャラクタリゼーション(material characterization)
材料の化学組成,構造及びその他の特性,並びに材料特性評価に必要な新規データを対象とした広範囲
かつ一般的な既存情報を収集するプロセス。
注記 この規格では対応英語の“characterization”を“キャラクタリゼーション”とした。キャラクタ
リゼーションとは,生物学的安全性のリスクアセスメントに必要な情報の収集を目的として,
対象物質がもつ性質及び特徴の全容を明らかにすることを意味する。キャラクタリゼーション
の手法としては,既存情報を収集し評価する,新たに試験及び研究を実施して定性的又は定量
的なデータを取得するなどがある。
3.14
医療機器(medical device)
あらゆる機器,装置,用具,器械,器具,インプラント,体外診断薬,ソフトウェア,材料(3.12)又
はその他の類似若しくは関連する物品であって,単独使用又は組合せ使用を問わず,製造業者が人体への
使用を意図し,使用目的が次の一つ以上のもの。
− 疾病の診断,予防,モニタリング,治療又は緩和
− 傷害の診断,モニタリング,治療,緩和又は代償
− 解剖学的構造若しくは生理学的プロセスの検査,代替,調節又は補助
− 生命補助又は維持
− 受胎調節
− 医療機器の殺菌・消毒
− 人体から採取した生体試料の臨床検査による情報提供
なお,体内若しくは体表における薬理学的,免疫学的又は代謝による作用を主な機能とするものではな
い。しかし,意図する機能をそれらの作用が補助する場合がある。また,医療機器には歯科用医療機器も
含まれる。
注記 次のものは,管轄する地域又は国の規制によって,医療機器に含まれない場合がある。
6
T 0993-1:2020 (ISO 10993-1:2018)
− 殺菌・消毒薬
− 身体障害者に対する補助器具
− 動物及びヒトのいずれか又は両方の生体組織を含む医療機器
− 体外受精又は生殖補助医療のための医療機器
(GHTF/SG1/N071:2012の5.1を歯科用機器が含まれることを明白にするために修正)
3.15
ナノマテリアル(nanomaterial)
外形寸法の少なくとも一つがナノスケールであるか又は内部構造若しくは表面構造がナノスケールであ
る材料(3.12)。
(ISO/TR 10993-22:2017の3.7を修正)
注記 ISO/TR 10993-22:2017では,医療機器(3.14)若しくは医療機器の構成部材の劣化,摩耗又は
機械的処理によって,ナノマテリアルに該当する分解物若しくは摩耗粒子が発生する可能性に
ついても考慮している。
3.16
非接触(non-contacting)
医療機器(3.14)又は医療機器の構成部材が直接的にも間接的にも生体組織に接触しないこと。
3.17
物理学的及び化学的情報(physical and chemical information)
生物学的安全性試験又は材料キャラクタリゼーション試験の要否を決定するために使用される,組成,
製造工程,形状及び物理学的性質,身体接触のカテゴリ,並びに臨床使用に関する情報。
3.18
リスク分析(risk analysis)
利用可能な情報を系統的に用いてハザードを特定し,リスクを推定すること。
(JIS T 14971:2012の2.17を修正)
3.19
リスクアセスメント(risk assessment)
リスク分析(3.18)及びリスク評価(3.20)からなる全てのプロセス。
(JIS T 14971:2012の2.18)
3.20
リスク評価(risk evaluation)
判断基準に照らして推定したリスクが受容できるかを判断するプロセス。
(JIS T 14971:2012の2.21)
3.21
リスクマネジメント(risk management)
リスクの分析,評価,コントロール及び監視に対して,管理方針,手順及び実施を体系的に適用するこ
と。
(JIS T 14971:2012の2.22)
3.22
有毒(toxic)
有害な生物学的反応を引き起こすおそれのある性質。
7
T 0993-1:2020 (ISO 10993-1:2018)
3.23
毒性学的ハザード(toxicological hazard)
反応の性質及びその反応を引き起こすために必要とされる用量を考慮した場合の,有害な生物学的反応
を引き起こす化学物質又は材料(3.12)がもつ潜在的危険性。
3.24
毒性学的リスク(toxicological risk)
所定のばく露レベルに応じて,所定の有害反応を生じる蓋然性。
3.25
毒性学的いき(閾)値(toxicological threshold)
耐容摂取量(TI),耐容ばく露量(TE),許容限界(AL),毒性学的懸念のいき値(TTC)などの値を下
回った際に,関連する生物学的エンドポイントに有害作用が現れない限度。
注記 この規格では対応英語の“endpoint”を“エンドポイント”,また,“biological endpoint”を“生
物学的エンドポイント”と定義した。生物学的安全性評価におけるエンドポイントは,評価項
目,評価指標及び最終的な到達目標を示す。
3.26
一過的接触(transitory contact)
医療機器(3.14)又は医療機器の構成部材と生体組織とが非常に短い時間接触すること。
4
医療機器の生物学的評価に適用される一般原則
4.1
ヒトに使用することを目的とした材料又は医療機器の生物学的評価も,図1に示すように,JIS T
14971:2012の附属書Iに基づいたリスクマネジメントプロセスにおける体系的な生物学的評価計画に含め
る。このリスクマネジメントプロセスは,生物学的ハザードの特定,関連する生物学的リスクの推定及び
それらのリスクの受容性の判断を伴う。附属書Bは,このリスクマネジメントプロセスのガイダンスを示
している。生物学的評価は,知識及び経験が豊かな専門家によって計画,実施し,かつ,文書化する。
リスクマネジメントの計画においては,特別な技術的力量を必要とする生物学的評価項目を特定するこ
とが望ましい。さらに,生物学的安全性評価の責任者も特定する。
生物学的評価では,次の事項について利点及び欠点の判断,並びにその妥当性を文書化する。
a) 医療機器の構成(例えば,サイズ,形状及び表面の特性),医療機器の材料の組成一覧など定性的な情
報及び必要な場合には,医療機器の各材料の割合,量などの定量的な情報
b) 種々の材料の構成及びそれらの組成の物理学的,化学的性質
これらの情報が当該機器のリスクマネジメントの下で既に文書化されている場合には,それを参照
してもよい。
注記1 許容事項のため,本文へ移動した。
c) 臨床使用の実績又はヒトばく露データ
これらには,規制当局の承認実績などを含めてもよい。
注記2 許容事項のため,本文へ移動した。
d) 製品及び部品材料,並びに分解生成物及び代謝産物に関する既存の毒性データ及びその他の生物学的
安全性データ
e) 試験の手順
評価には,関連する非臨床及び臨床経験の既存データの調査並びに実際の試験実施の両方を含めること
8
T 0993-1:2020 (ISO 10993-1:2018)
ができる。その結果として,当該材料が,設計中の機器と使用目的及び物理学的形状とが同等で,安全に
使用されてきた実績を十分にもつ場合には改めて試験を行わなくても安全と判断することができる。同等
性評価に有用となる情報が附属書Bに含まれている。材料及び医療機器のいずれか又は両方のリスクアセ
スメントを行うのに十分な情報が活用できる場合は,試験の実施は通常必要ない(附属書C参照)。
4.2
医療機器の製造で使用される材料の選択においては,その材料の特性及び性質の使用目的に対する
適合可否を最初に考慮する。その特性及び性質には,化学的,物理学的,毒性学的,電気的,形態学的及
び機械的性質が含まれる。
4.3
医療機器の生物学的評価を体系的に行うには,次の事項を考慮する。
a) 構成材料(直接的及び間接的に生体組織と接触する全ての材料)
b) 意図的な添加物,製造工程での混入物及び残存物(例えば,エチレンオキサイド残留物の試験はJIS T
0993-7に従って実施する。)
c) 直接的又は間接的に医療機器と接触した後,化学物質が医療機器に移行し,その結果間接的に患者又
は臨床医にばく露する可能性がある包装材料
d) 溶出物(ISO 10993-17及びISO 10993-18参照)
注記1 この規格では対応英語の“leachable substances”及び“leachables”を“溶出物”とした。そ
の定義は,ISO 10993-17に次のように規定されている。“ある医療機器から,水又は機器
の使用に関連した他の液体へ移行する化学物質(例:添加剤,滅菌残留物,製造工程残留
物,分解生成物,溶媒,可塑剤,潤滑剤,触媒,安定剤,抗酸化剤,色素,充塡剤,モノ
マー)”また,“機器の使用に関連した他の液体”とは,輸液などの薬液,血液,胆汁,髄
液などの体液,脂質(脂肪)などが考えられる。
e) 分解生成物(一般原則については,ISO 10993-9を参照。ポリマー,セラミックス及び金属について
はそれぞれ,ISO 10993-13〜ISO 10993-15を参照。)
f)
最終製品中のa)〜e) 以外の成分及びそれらの相互作用
g) 最終製品の性能及び特性
h) 最終製品の物理学的性質(多孔率,粒径,形状,表面形態など)
医療機器の化学成分に関する検証及びISO 10993-18に記載された化学的キャラクタリゼーションに代
表される材料キャラクタリゼーションは,生物学的試験に先立って実施されなければならない(図1参照)。
適切な毒性学的いき値を用いて実施された化学的キャラクタリゼーションは,生物学的安全性試験の要否
判断に使用される(附属書B,ISO 10993-17及びISO 10993-18参照)。
生体適合性への影響が懸念される場合には,医療機器の物理学的特性も考慮する。
注記2 情報については,ISO/TR 10993-19を参照。
ナノマテリアルを含む医療機器,ナノマテリアルを発生させる可能性のある医療機器及びナノマテリア
ル自体が医療機器となる場合には,ナノマテリアルに特有の危険性についても生物学的評価の中で言及す
る(ISO/TR 10993-22参照)。
9
T 0993-1:2020 (ISO 10993-1:2018)
リスク評価には,局所的影響及び全身的影響の両方を考慮しなければならない。
4.4
生物学的評価は,医療機器のカテゴリ分類から始める(箇条5参照)。既存情報の評価は,ギャップ
分析を可能にし,適切な試験項目の選択を容易にする。生物学的評価において最も必要な事項は,主に医
療機器又は材料の生体へのばく露及びハザードの性質,程度,頻度及び期間によって決まる。医療機器又
は材料のリスク評価に十分な情報が活用できる場合,例えば,評価対象となる医療機器の材料キャラクタ
リゼーション(ISO 10993-18及びISO/TR 10993-19参照)によって,既に安全性が確立されている医療機
器又は材料との同等性を示すことができる場合などは,通常,生物学的試験の実施は必要ない(附属書C
参照)。
データの解釈には,医療機器又は材料の生体へのばく露及びハザードの性質,程度,頻度及び期間と同
様に,材料の化学的構成も考慮する。
4.5
それぞれの医療機器及び材料について,想定される全ての生物学的ハザードを考慮しなければなら
ないが,それは全ての潜在的ハザードについて試験実施を要求する又は試験が実施可能であるとは限らな
い(箇条5及び箇条6参照)。試験結果で潜在的な生物学的ハザードが全くないことを保証することはでき
ない。そのため生物学的ハザードの検証を行うには,医療機器の臨床使用における予期せぬ人体への有害
作用又は有害事象を注意深く観察しなければならない。
潜在的な生物学的ハザードの範囲は広く,次のものが含まれる。
− 短期的作用(例:急性毒性,皮膚・眼及び粘膜表面に対する刺激性,溶血性,血栓形成性)
− 長期的作用又は特殊毒性作用[例:亜慢性又は慢性毒性,アレルギーを引き起こす感作性,遺伝毒性,
発がん性(催腫瘍性),催奇形性を含む生殖又は発生への作用]
4.6
生物学的試験の実施が必要な場合,いかなるインビトロ試験又はインビボ試験(附属書A参照)も,
使用目的に基づいて選定する。
適切なバリデーションが実施され,実用的で,信頼性及び再現性があるインビトロ試験法が存在する場
合には,インビボ試験に優先して実施を考慮する(ISO 10993-2参照)。初期のリスク評価の所見からイン
ビボ試験の実施が必要となる場合でも,可能であればインビボ試験の実施に先立ち,インビトロ試験での
スクリーニングの実施を考慮する。試験方法の選定と同様に,試験目的の妥当性も示す。また,各試験で
得られたデータは,知識及び経験が豊かな専門家によって評価し,保管する。
標準化されていない試験法及びバリデーションが不十分な試験法の実施が必要となる場合には,試験デ
ザイン及びデータの解釈に対する妥当性について説明することが望ましい。
4.7
製造業者は,医療機器のライフサイクル全体の生物学的安全性を評価する。
4.8
再使用可能な医療機器については,想定される再処理サイクルの最大数を考慮して,生物学的安全
性を評価する。
4.9
医療機器又は材料に次の事項のいずれかが生じた場合は,再び生物学的リスクアセスメントを実施
する。
a) 製品の製造に使用する材料の供給元又は仕様の変更
10
T 0993-1:2020 (ISO 10993-1:2018)
b) 製品の成分・配合,加工,一次包装又は滅菌の変更
c) 保管・貯蔵に関する製造業者の指示又は推奨の変更(例:有効期限及び輸送条件のいずれか又は両方
の変更)
d) 製品の使用目的の変更
e) 製品が人体に使用されたとき,何らかの有害な作用を生じる可能性を想起させる事象
4.10
生物学的評価には,類似の医療機器又は材料の非臨床試験,臨床研究,市販後の臨床実績及び関連
情報を考慮する(附属書B参照)。
4.11
この規格は,旧規格を用いて評価された実績のある製品に対して再試験を行うことは求めない。し
かし,その場合にこの規格への適合確認を行うには,追加の試験実施を不要とした妥当性を示さなければ
ならない。この規格と旧規格とでは,附属書Aに示された生物学的安全性評価のエンドポイントの推奨項
目が異なるが,当該医療機器の臨床実績を基に追加試験の実施を不要とする根拠を文書化することは可能
である。ただし,過去に評価された時点から現在に至るまでに4.9に規定する変更が生じていた場合には,
その変更に関連する生物学的リスクの評価は,この規格に従い実施する。
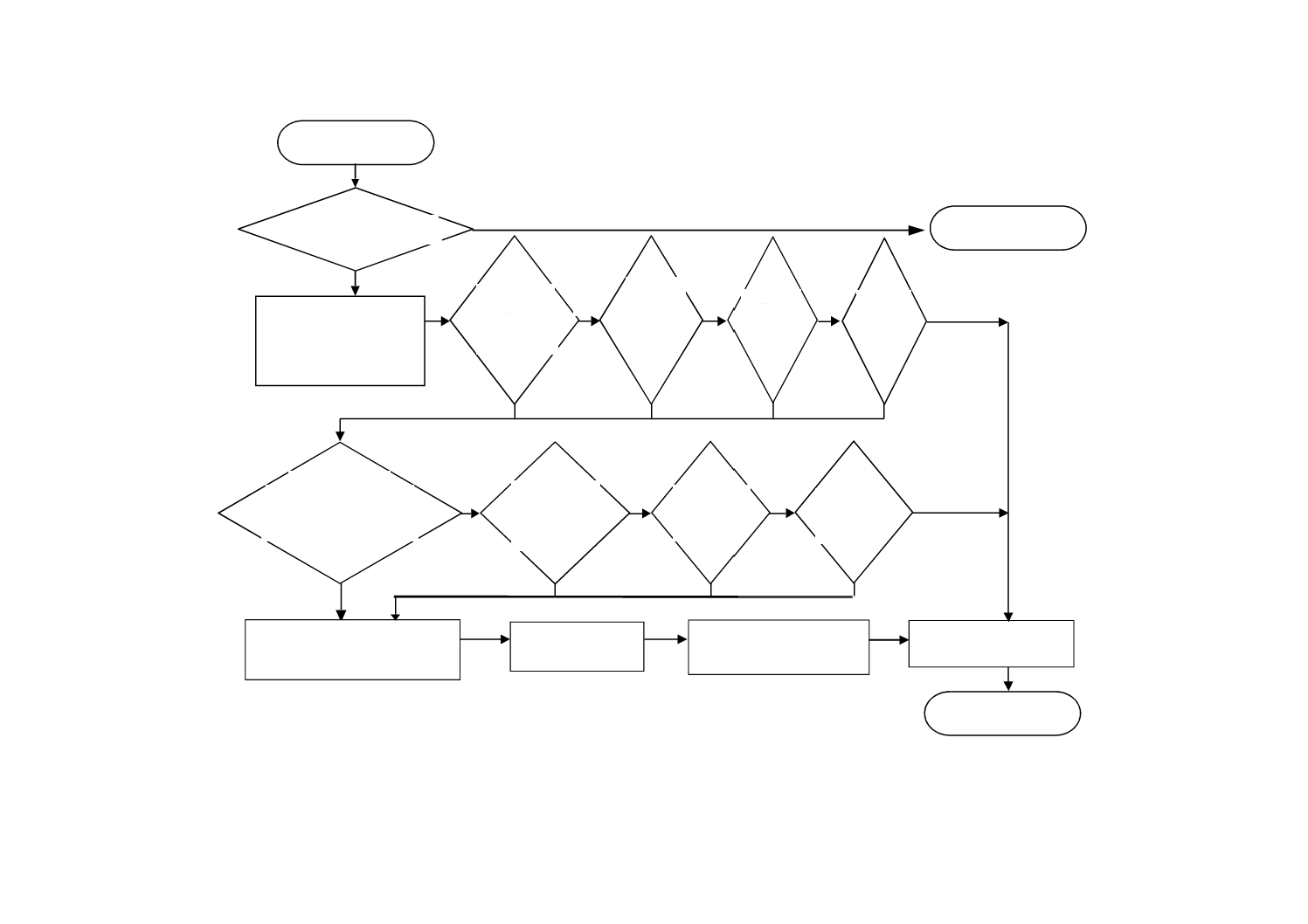
11
T 0993-1:2020 (ISO 10993-1:2018)
図1−リスクマネジメントプロセスの一環として実施する医療機器の生物学的評価の体系的手引
(要約)
いいえ
いいえ
いいえ
いいえ
物理学的・化学的情報を収集
する。必要に応じて,材料キ
ャラクタリゼーション(ISO
10993-18)についても考慮す
る。
はい
4.2,4.3,6.1
市販医療機器
に使用されて
いる材料と同
等(すなわち同
配合組成)か。
4.3,6.1
いいえ
はい
製造方法,
滅菌方法
は,同等か。
(タイプ・
プロセスの
詳細)
4.3,6.1
はい
形状,物理
学的特性
は,同等か。
4.3,6.1
はい
はい
リスクアセスメント
のために十分に正当化できる
根拠及び臨床に関連性のある
データ(化学的,生物学的及び
物理学的)のいずれか又は複数
の組合せがあるか。
6.1,B.4.4
当該医療機器の
全ての化学物質
に十分な毒性学
的データが存在
するか。
6.1,B.4.4
混合された化
学物質に適用
できるデータ
か。
6.1,B.4.4
はい
はい
はい
当該医療機器は患者身体に直
接的又は間接的に接触するか。
箇条1,3.6,3.11
4.2,6.2,7
材料の化学的性質,接触形態及び期間
に基づき,更なる当該医療機器の評価
を実行する。
生物学的エンドポイン
トの選択(附属書A)
毒性学的リスクアセスメン
トを実行する(附属書B)。
試験の実施及び試験省略の正当
化のいずれか又は両方(附属書
A)
はい
いいえ
いいえ
いいえ
いいえ
身体接触
及び臨床
使用は,同
等か。
そのデータの
用量及びばく
露経路は妥当
か。
4.2,6.2,7
5
B.3.1.4,B.4.3.2
開始
生物学的評価の完了
JIS T 0993-1は適用
されない。
はい
6.1,B.4.4
3
T
0
9
9
3
-1
:
2
0
2
0
(I
S
O
1
0
9
9
3
-1
:
2
0
1
8
)
12
T 0993-1:2020 (ISO 10993-1:2018)
5
医療機器のカテゴリ分類
5.1
一般
医療機器は,5.2及び5.3に規定するように,身体との接触形態及び接触期間によってカテゴリに分類す
る。医療機器をカテゴリに分類することによって,適切なデータセットの評価が可能となる(附属書A参
照)。
いずれのカテゴリにも該当しない医療機器を評価する場合にも,この規格の一般原則に従う。複数のカ
テゴリに当てはまる医療機器の場合は,それぞれのカテゴリに対応する適切な評価を実施する。
例1 インプラント部品及びそのデリバリーシステムで構成される医療機器においては,インプラン
ト部品及びデリバリーシステムは個別に評価することが望ましい。
例2 間接的接触によってだけ用いられるガス流路機器を構成する部材は,機器固有の規格に従い生
体適合性の評価を実施することが望ましい(ISO 18562規格群参照)。
5.2
身体との接触形態によるカテゴリ分類
5.2.1
非接触医療機器
身体と接触しない医療機器(又は構成品)は生体適合性の評価は必要ない。診断用ソフトウェア,体外
診断用医療機器1) 及び採血管が非接触医療機器の例である。
注1) 国内では,体外診断用医薬品も含まれる。
5.2.2
表面接触医療機器
表面接触医療機器は,適用部位に従い次のカテゴリに分類する。
a) 皮膚
− 健常皮膚表面と接触する医療機器
例1 電極,人工補てつ(綴)材料,固定用テープ,圧迫包帯及び各種の監視装置
注記 医療機器には,非滅菌環境下で使用者が手袋を着用せずに接触する電子装置のヒューマン
インタフェース(例:コンピュータのキーボード,ダイアル又はボタン,タッチパネル,
SDカード及びUSBメモリ),健常皮膚に直接接触するモニタ又はプログラマのハウジング
(例:携帯電話,タブレットなどの電子機器),滅菌環境下で使用者が手袋を着用した手で
接触する構成部材(例:カテーテル用ハンドル)などが含まれる。これらに該当する機器
の構成部材が,類似する接触形態で広く一般的に利用されている材料から作られている場
合には,更なる生物学的評価を必要としない。
b) 粘膜
− 健常な粘膜と接触する医療機器
例2 コンタクトレンズ,尿道カテーテル,ちつ(膣)内及び腸内医療機器(胃管,S状結腸鏡,
結腸鏡,胃鏡),気管チューブ,気管支鏡,一部の歯科用補てつ材料及び歯列矯正医療機器
c) 損傷表面
− 損傷した身体表面と接触する医療機器
例3 潰瘍,やけど及び肉芽組織のための包帯,創傷治癒機器及び閉塞性パッチ
5.2.3
体内と体外とを連結する医療機器
体内と体外とを連結する医療機器は,適用部位に従い次のカテゴリに分類する。
13
T 0993-1:2020 (ISO 10993-1:2018)
a) 血液流路間接的
− 必ずしも血管又は血液に直接接触しないが,薬剤などの液体用導管として用いる医療機器又は構成
品
例1 輸液セット,延長チューブ,トランスファーセット及び輸血セット
b) 組織,骨又は歯質
− 生体組織,骨,歯髄又は歯質と接触する医療機器若しくは構成品
例2 腹くう(腔)鏡,関節鏡,排液システム,歯科用充塡材料,皮膚縫合針
− 必ずしも直接生体組織又は骨に接触しないが,生体組織又は骨への液体用導管として用いる医療機
器若しくは構成品
例3 洗浄用チューブ,患者に接触する可能性のある流体と接点をもつ医療機器又は構成品
c) 循環血液
− 循環血液と接触する医療機器又は構成品
例4 血管内カテーテル,一時的ペースメーカ電極,人工肺,人工肺用回路及び附属品,透析器,
透析用回路及び附属品,血液成分吸着器及び免疫吸着材
5.2.4
インプラント
インプラントは,適用部位に従い次のカテゴリに分類する。
a) 組織又は骨
− 主として骨と接触する医療機器
例1 整形外科用ピン,整形外科用プレート,人工関節,骨人工補てつ材,骨セメント,骨内医
療機器
− 主として生体組織及び組織液と接触する医療機器
例2 ペースメーカ,薬物注入機器,神経・筋センサ及び刺激装置,人工けん(腱),人工乳房,
人工喉頭,骨膜下インプラント,結さつクリップ,子宮内医療機器(化学的作用によって
機能を発揮するものを除く。)
b) 血液
− 心血管系において主に循環血液と接触する医療機器
例3 ペースメーカ電極,人工動静脈ろう(瘻)フィステル,心臓弁,人工血管,血管内薬物送
液用カテーテル,心室補助医療機器(心室補助人工心臓)
注記 生体組織の多くは循環血液を含むが,このカテゴリは,外科的手技などで一時的な出血を
伴う生体組織に適用される機器(ヘルニア修復用移植片など)は対象としない。
5.3
接触期間によるカテゴリ分類
5.3.1
接触期間及びカテゴリ
医療機器は,想定される接触期間に従い次のカテゴリに分類する。
a) 一時的接触(A):単回,複数又は繰返しの使用による接触期間の累積が,24時間までの医療機器
b) 短期的接触・中期的接触(B):単回,複数又は繰返しの使用による接触期間の累積が,24時間を超え,
30日以内の医療機器
c) 長期的接触(C):単回,複数又は繰返しの使用による接触期間の累積が,30日を超える医療機器
5.3.2
一過的に接触する医療機器
医療機器の中には,身体との接触が非常に短時間となる機器がある(例:使用時間が1分未満のランセ
14
T 0993-1:2020 (ISO 10993-1:2018)
ット,皮下注射針,毛細管チューブ)。これらは,通常,生体適合性の評価を必要としない。ただし,医療
機器が除去された後も生体組織に残存する可能性のあるコーティング又は潤滑剤が使われている製品は生
体適合性の評価が必要となる。累積使用も考慮することが望ましい。
5.3.3
複数の接触期間が該当する医療機器
医療機器の接触期間が複数のカテゴリに該当する場合は,より厳しいカテゴリを適用する。複数回のば
く露が想定される場合には,累積のばく露期間を考慮して,カテゴリに分類する。使用中に適用組織で重
合反応及び生分解のいずれか又は両方が起こり得る医療機器は,それぞれの状態変化が網羅されるよう考
慮して評価する。例えば,適用場所で重合するように意図された生分解性接着剤の場合には,反応開始前
の構成品,中間反応生成物,完全に重合した物質及び分解生成物の評価を行う。
6
生物学的評価のプロセス
6.1
生物学的リスク分析のための物理学的及び化学的情報
図1は,物理学的及び化学的のいずれか又は両方のキャラクタリゼーションと生物学的評価の判断ポイ
ントとの関係を示している。
医療機器又は構成品の物理学的及び化学的情報の収集は,生物学的評価及び材料キャラクタリゼーショ
ンにおいて,重要な第一歩である。収集された情報は図1のフローチャートの材料,製造方法,滅菌方法,
形状,物理学的特性,身体接触及び臨床使用に関する質問に十分に答えられる内容であることが望ましい。
収集が必要な物理学的及び化学的のいずれか又は両方の情報は,材料の組成が明らかとなっているか否か,
非臨床及び臨床においてどの程度安全性及び毒性学的情報が存在するか,医療機器の身体への接触形態,
接触期間などによって異なる。少なくとも,医療機器の化学的成分及び製造過程で使用され残留する可能
性のある加工助剤又は添加物は化学的キャラクタリゼーションの対象としなければならない。また,イン
プラント又は血液と接触する医療機器に関しては物理学的キャラクタリゼーションが必要となる。材料キ
ャラクタリゼーションは,ISO 10993-18に従って実施する。ナノマテリアルのキャラクタリゼーションは,
ISO/TR 10993-22を参照する。
全ての材料,化学物質及び製造過程の組合せが,その医療機器の使用目的において安全に使用されてき
た実績があり,かつ,物理学的特性が変化しなければ,それ以上のキャラクタリゼーション及び追加のデ
ータセット(例:抽出物の化学分析又は生物学的試験)は不要となる。この場合,その根拠を文書化する。
新規材料及び新規化学物質が存在する場合には,それらの化学構造を特定し,定量的な測定を行うこと
が望ましい。
医療機器から抽出及び溶出される物質について,予測されるばく露(量,経路及び頻度)に関して十分
な毒性学的データが得られる場合は,更なる試験を必要としない。溶出物が複数存在する場合は,溶出物
間の相互作用の可能性を考慮することが望ましい。
製造,滅菌,輸送,保管及び使用の条件下で分解する可能性がある場合は,ISO 10993-9及びISO 10993-13
〜ISO 10993-15に従い,分解生成物の有無及び性質を明らかにする。
15
T 0993-1:2020 (ISO 10993-1:2018)
摩耗粒子を生じる可能性のある医療機器及び材料のいずれか又は両方において,その摩耗粒子がナノマ
テリアルに該当する可能性があれば,ISO/TR 10993-22に記載されている事項を考慮することが望ましい。
6.2
ギャップ分析及び評価のための生物学的エンドポイントの選択
入手可能な情報を評価し,医療機器の生物学的評価に必要なデータセットと対比する(箇条4,附属書
A及び附属書C参照)。リスクアセスメントを行う上で追加が必要な情報又は試験項目を特定する。
生物学的エンドポイント(附属書A)の評価と既存の生物学的リスクアセスメント内容とのデータギャ
ップを明らかにし,その重要性を判断する。データギャップを補うために必要なデータセット及び対応策
を特定する。
例えば,日本薬局方収載のプラスチック製医薬品容器試験は,一般に原材料を使った試験である,ISO
10993における医療機器の評価では最終製品を用いることを求めている。そのため,日本薬局方に従い得
られたプラスチック製医薬品容器試験のデータは,適切な根拠がなければ,医療機器評価のデータとして
は不十分である。
特定された化学物質のリスクアセスメントの結果次第では,追加の材料キャラクタリゼーションが必要
となる可能性もある。化学成分の臨床的ばく露の度合いを推定するために,適切な抽出試験を用いること
ができる(ISO 10993-18参照)。評価対象となる溶出物量の受容可否は,ISO 10993-17に従い,医療機器
から抽出される各化合物の量とそれぞれに設定された毒性学的いき値との比較によって確定する。
例えば,臨床使用中に当該化学物質の全量が溶出し,十分な安全域が確保できない場合には,その状況
を考慮した条件にて抽出試験を行ってもよい。
注記 許容事項のため,本文へ移動した。
この規格の第一の目的は,ヒトを保護することである。第二の目的は,動物福祉を確保し,試験動物の
数及びばく露量を最小限にすることである。対象となるインビボ試験にはISO 10993-2を適用する。ただ
し,次の場合には,追加のインビボ試験を実施してはならない。
1) 試験結果が,以前に実施された調査研究から得られる場合。
2) 臨床での使用実績を含め,既存の非臨床データ及び臨床データが生物学的評価の要求事項を満たして
おり,それ以上の動物試験は非倫理的となるような場合。ただし,生物学的評価で既存データを用い
る場合には,評価を行う前に既存データの信頼性を考慮することが望ましい。ISO 10993-18:2005の附
属書Cは,化学的な同等性を判断するための原則を参考として示している。
6.3
生物学的試験の実施
6.3.1
一般
包括的なリスクマネジメントプロセスにおいて医療機器の生物学的試験が必要と判断された場合は,箇
条4に規定する一般原則に加えて,次の事項を適用する。
a) 試験は最終製品又は最終製品を代表する試料若しくは最終機器と同じ方法で処理された材料(必要な
らば,滅菌処理を含む。)を用いて実施する。
b) 試験項目の選択は,次の事項を考慮する。
16
T 0993-1:2020 (ISO 10993-1:2018)
1) 使用目的における医療機器の人体へのばく露又は接触形態,程度,接触期間,頻度及び条件
2) 最終製品の物理学的及び化学的性質
3) 最終製品の処方における化学物質の毒性学的作用
4) 溶出物が存在しない場合又は,溶出物が既知の化学物質であり,かつ,毒性プロファイルが受容可
能であり,ISO 10993-17及びJIS T 14971に従った評価で安全であると判断した場合,追加試験(例
えば,全身毒性試験)は適用しなくてもよい。
5) 医療機器の表面積並びに適用患者の体表面積比及び体重比(例えば,動物モデルを用いた埋植試験
のために医療機器を小形化する。)
6) 文献,使用実績及び非臨床試験に基づく既存の情報
7) 生物学的評価に影響を及ぼすことが考えられる対象試験の感度及び特異性
8) ISO 10993-2:2006の4.4に従い,使用動物の痛み,苦しみ,心理的苦痛又は持続的な有害作用を最
小限にする。
c) 医療機器の抽出物を調製する場合,ISO 10993-12に従い実施する。抽出に使用する溶媒及び抽出条件
は,試験法の妥当性(例:試験目的,根拠,感度,特異性など)と同様に,最終製品の性質及び使用
方法を考慮した適切な条件を選択する。可能であれば,抽出条件は実使用条件より苛酷な条件を選択
する。
d) 必要に応じて,陽性対照群及び陰性対照群を設けることが望ましい。
生物学的評価に用いる試験方法は,感度がよく,精密かつ正確でなければならない。生物学的試験を実
施する場合は,優良試験所規範(Good Laboratory Practice,以下GLPという。)に従って実施する。
注記 JIS Q 17025又は同等のもの。
なお,我が国の医療機器の承認審査においては,生物学的安全性試験はGLPに従って実施す
ることが求められている。
試験方法は,施設間での再現性及び頑健性はもちろんのこと,施設内の再現性も確保されていることが
望ましい。
6.3.2
評価項目に応じた試験の実施
当該医療機器の生物学的評価に必要とされるデータセットをそろえるために,必要に応じて,6.3.2.1〜
6.3.2.15に規定した試験を実施する。既存データで適切な評価が実施可能な場合は,追加試験は必要ない
(附属書A及び附属書C参照)。
医療機器は多様であるため,当該医療機器が該当する医療機器カテゴリで規定された全てのエンドポイ
ントに対して必ずしも試験を要さない又は実行できない場合がある(JIS T 14971参照)。各医療機器の有
用性に基づいた評価が不可欠である。ナノマテリアルは,一般的な医療機器の評価で使用される試験系及
び試験結果の解析に,ナノマテリアル特有の課題(例:分析への干渉)を生じる可能性がある(ISO/TR
10993-22参照)。
表A.1に示されていない追加のエンドポイント(例:生殖毒性,発生毒性,生体内分解性及びトキシコ
キネティクス)が必要になる場合がある。
17
T 0993-1:2020 (ISO 10993-1:2018)
6.3.2.1
細胞毒性
細胞毒性試験は細胞培養技術を用いた手法によって,医療機器及び材料,並びにそれらの抽出物のいず
れか又は複数の組合せによって引き起こされる細胞死(細胞の溶解),細胞増殖の阻害,コロニー形成及び
細胞に対する他の作用を判定する試験である。試験はISO 10993-5に従って実施する。
6.3.2.2
感作性
感作性(遅延型アレルギー)試験は,適切な動物モデルを使用することによって,医療機器及び材料,
並びにそれらの抽出物のいずれか又は複数の組合せの接触感作の潜在性を評価する試験である。試験は
ISO 10993-10に従って実施する。
例え僅かな量の溶出物であっても,繰り返してばく露又は接触することによってアレルギー反応を引き
起こす感作が起こり得るため,注意が必要である。
6.3.2.3
刺激性(皮内反応を含む。)
刺激性試験は,皮膚,眼及び粘膜のような適切な部位を用いて,医療機器及び材料,並びにそれらの抽
出物のいずれか又は複数の組合せの潜在的な刺激性を評価するための試験である。試験は,ISO 10993-10
に従い,適切なばく露又は接触ルート(皮膚,眼,粘膜)及びその接触期間を考慮して実施する。
皮内反応試験は,医療機器の抽出物に対する生体組織の局所的な反応を評価できる。皮膚又は粘膜を対
象とした刺激性の評価が適切でない場合(例:医療機器がインプラントである場合又は血液接触する場合),
また,抽出物の疎水性が高い場合(ISO 10993-10参照)には皮内反応試験を適用する。
6.3.2.4
血液適合性
血液適合性試験は,適切なモデル又はシステムを使用することで,血液と接触する医療機器又は材料に
よる,血液若しくは血液成分への影響を評価する試験である。
血液適合性試験の一つである溶血性試験は,医療機器及び材料,並びにそれらの抽出物のいずれか又は
複数の組合せによる赤血球の溶解程度,及びヘモグロビンの放出をインビトロで評価する試験である。
臨床使用における医療機器又は材料の形状,接触条件及び流体動力学的条件を模擬して,血液と材料又
は医療機器との相互作用を評価できる血液適合性試験を,別に設計してもよい。
全ての血液適合性試験は,ISO 10993-4に従い実施する。
6.3.2.5
材料由来の発熱性
医療機器の生物学的評価における発熱性試験は,医療機器又は材料の抽出物について材料由来の発熱作
用の有無を評価する試験である。単一の試験では,材料由来の発熱反応又はエンドトキシン汚染による発
熱反応のいずれかを識別することは難しい(ISO 10993-11:2017の附属書G参照)。材料由来の発熱反応は
まれ(稀)であるが,生物由来物質を含む医療機器において認められたことがある。
18
T 0993-1:2020 (ISO 10993-1:2018)
6.3.2.6
急性全身毒性
急性全身毒性試験は,医療機器の接触によって有毒な溶出物及び分解生成物が人体にばく露する可能性
がある場合に用いられる。24時間未満の期間における単回又は複数回のばく露による潜在的有害作用を評
価する試験である。試験はISO 10993-11に従い,適切なばく露経路で実施する。
可能であれば,亜急性及び亜慢性毒性試験プロトコル並びに埋植試験プロトコルに急性全身毒性の評価
を含めてもよい。
全身毒性の評価が表A.1に示されている場合,生物学的試験又はリスクアセスメントは,医療機器の臨
床使用に関連する臓器を含めた全身組織における潜在的な生物学的反応を評価する(例:ISO 10993-11:
2017の附属書E)。
6.3.2.7
亜急性及び亜慢性毒性
亜急性及び亜慢性毒性試験は,医療機器及び材料,並びにそれらの抽出物のいずれか又は複数の組合せ
が,24時間以上,かつ,実験動物の全寿命の10 %以下(例えば,ラットでは13週間まで)の期間におい
て単回又は複数回のばく露若しくは接触による全身的な影響を評価する試験である。
評価対象となる材料の慢性毒性に関するデータが存在し,そのデータから亜急性及び亜慢性毒性を十分
に評価できる場合は,亜急性及び亜慢性毒性試験は実施しなくてもよい。試験を実施しない理由は,生物
学的総括評価報告書に記載する。亜急性及び亜慢性毒性試験は,ISO 10993-11に従い,適切な接触形態及
び接触期間で実施する。
可能であれば,局所的作用を評価するための埋植試験プロトコルに亜急性及び亜慢性全身毒性の評価を
含めてもよい。
6.3.2.8
慢性毒性
慢性毒性試験は,医療機器及び材料,並びにそれらの抽出物のいずれか又は複数の組合せが,実験動物
の全寿命の大半(例えば,ラットでは通常6か月)に相当する期間において単回又は複数回ばく露による
全身的な影響を評価する試験である。慢性毒性試験は,ISO 10993-11に従い,適切な接触形態及び接触期
間で実施する。
可能であれば,局所的作用を評価するための埋植試験プロトコルに慢性全身毒性の評価を含めてもよい。
6.3.2.9
埋植による影響
埋植試験は,外科的に埋植又は留置した材料若しくは最終製品の試料が,その適用部位の生体組織へ及
ぼす局所的な病理学的影響を,肉眼的かつ顕微鏡的な視野で観察し,評価する試験である。埋植試験は,
ISO 10993-6に従い,適切な接触形態及び接触期間で実施する。
可能であれば,埋植試験プロトコルに全身毒性の評価(ISO 10993-6参照)又は血液適合性の評価を含
めてもよい(ISO 10993-4)。
19
T 0993-1:2020 (ISO 10993-1:2018)
動物を使用した適切な使用模擬試験は,物理学的及び生物学的リスクの両方(すなわち,毒性学的ハザ
ード及び毒性学的リスクのいずれか又は両方)のエンドポイントの範囲を評価することができる。また,
慢性,亜慢性,亜急性及び急性の全身毒性エンドポイントも,試験に組み入れることができる。
臨床ばく露に相当する量の材料が対象臓器若しくは生体組織に埋植された場合,又は臨床ばく露以上の
過剰量の材料が適用部位以外に埋植された場合には,局所的な作用を評価する埋植試験で全身毒性の評価
が可能である。
6.3.2.10 遺伝毒性
遺伝毒性試験は,医療機器及び材料,並びにそれらの抽出物のいずれか又は複数の組合せによって引き
起こされる遺伝子突然変異,染色体の構造及び数の変化並びにその他のDNA又は遺伝子への潜在的な毒
性を評価する試験である。インビボ試験に先立ち,まずインビトロ試験による評価を検討する。試験はISO
10993-3に従い実施する。
注記 追加情報は,ISO/TR 10993-33を参照する。
いずれかのインビトロ試験が陽性となった場合,そのフォローアップには,不純物,抽出物若しくは溶
出物の化学的同定,又は遺伝毒性試験の追加実施などがある。遺伝毒性リスクの受容可否は,例えば,患
者のばく露,証拠の重み付け(weight of evidence, WOE),及び作用機序(mode of action, MOA)の情報を
含むリスクアセスメントの結果に基づいて判断しなければならない。
6.3.2.11
発がん(癌)性
ISO 10993-3では,医療機器及び材料,並びにそれらの抽出物のいずれか又は複数の組合せが,試験動
物の寿命の大部分の期間にわたり単回又は複数回ばく露する際の発がん性の評価方法を論じている。発が
ん性は,不純物,抽出物又は溶出物の化学的同定,これらの化学物質への患者のばく露,並びにWOE及
びMOAの情報を含むリスクアセスメントで評価できる場合がある。発がん性リスクアセスメントに用い
る情報は文献などから入手可能であるが,評価対象とばく露又は接触の経路及び期間とが合致しているこ
とが望ましい。リスクアセスメントにおいて重大な発がんリスクが認められなかった場合には,通常,当
該医療機器に発がん性試験の実施が適切と考えられることは少ない。しかし,最終製品の発がん性試験が
必要であると判断された場合には,試験動物の全寿命にわたる調査研究又は遺伝子組換えモデルを用いた
試験が必要となる。これらの試験は,OECD Guidelines for the Testing of Chemicalsで記載されているよう
に,一つの試験で慢性毒性及び腫瘍形成性の両方を評価することもできる。
6.3.2.12 生殖及び発生毒性
生殖及び発生毒性試験は,医療機器及び材料,並びにそれらの抽出物のいずれか又は複数の組合せが,
生殖機能,はい(胚)発育(催奇形性)及び胎児発育並びに新生児発育へ悪影響を及ぼす可能性を評価す
る試験である。これらのエンドポイントの評価には,不純物,抽出物又は溶出物の化学的同定,これらの
化学物質への患者のばく露,並びにWOE及びMOAの情報を含むリスクアセスメントが必要になる。生
殖毒性の評価は,医療機器が患者の生殖能に対して悪影響を及ぼす可能性が懸念される場合にだけ実施す
る。妊婦に使用する医療機器又は材料については,発生毒性の評価を検討することが望ましい。
20
T 0993-1:2020 (ISO 10993-1:2018)
新規の材料,生殖又は発生毒性が懸念される既知材料,並びに生殖及び発生毒性に関連の強い母集団
(例:妊婦)に使用する医療機器,及び生殖臓器に局所的に使用する医療機器のいずれか又は複数の組合
せについては,生殖及び発生毒性を評価することが望ましい。
6.3.2.13 生体内分解性
人体内で分解する可能性のある全ての医療機器,生体組織中で残存する医療機器成分又は材料に対して
は生体内分解性を評価する。
次のいずれかの場合,生分解性試験の実施を検討する。
a) 医療機器が,吸収性をもつように設計されている。
b) 最終製品に含まれる材料が,身体との接触の間に,有毒な分解生成物を産生する可能性を示す知見が
得られている。
生分解の速度及び程度に影響するパラメータは,試験報告書の中で必ず記載する。
生体内分解の機序も記載することが望ましい。生体内分解の機序を模擬したインビトロ試験で,分解率
の決定及び有害な分解生成物のばく露量の推定を行うことが望ましい。材料の生体内分解性の評価に,イ
ンビボ試験が必要となる可能性もある。
吸収性医療機器の評価において,既にインビトロとインビボ試験結果との間に相関性が示されており,
インビトロ試験において評価対象機器の生体内分解性が,安全性が確立された既存の医療機器と同程度で
あると判断された場合,インビボ試験は必要ない。微粒子状の分解生成物が産生される際に,これらの粒
子が,臨床実績から安全性が確立されているサイズ分布及び形状と類似している,又は使用目的における
粒子及びそれらの生体内分解性データが既に存在する場合,生分解性試験の実施は必要ない。
生分解性試験に関する一般原則は,ISO 10993-9に示されている。
ポリマー,セラミック及び金属の生体内分解性に関するインビトロ試験は,ISO 10993-13〜ISO 10993-15
でそれぞれ規定されている。
微粒子状の分解生成物が,ナノマテリアルに該当する場合,生分解性試験はISO/TR 10993-22を考慮し
て計画することが望ましい。
6.3.2.14 トキシコキネティクス試験
トキシコキネティクス試験の目的は,化学物質の,吸収,分布,代謝及び排せつ(泄)(Absorption,
Distribution, Metabolizm, Excretionの頭文字を取りADMEと表記される。)を評価することにある。
医療機器,材料及び抽出物(6.3.2.13及びISO 10993-16参照)の溶出物並びに分解生成物の,吸収,分
布,代謝及び代謝のプロセスを測定するためのインビボトキシコキネティクス試験の必要性は,生体内分
解性に関するインビトロ試験の結果を基に考慮する。
21
T 0993-1:2020 (ISO 10993-1:2018)
医療機器の生物学的評価においてトキシコキネティクス試験の要否判断に当たっては,最終製品及び医
療機器の使用目的において,意図された分解生成物,予期せぬ溶出物を含め全ての化学物質を考慮する
(6.3.2.13参照)。
トキシコキネティクス試験が必要な場合,試験の実施に先立ち,理論的な分解機序をインビトロ試験
(例:生体組織,ホモジネート又は細胞)で検証しなければならない。それはISO 10993-2に示す動物の
福祉の理由及びより確実性の高い分解生成物を決定するためにも必要となる。
次のいずれかに該当する場合には,トキシコキネティクス試験の実施を考慮する。
a) 医療機器が吸収性である。
b) 医療機器が長期的に接触するインプラントで,生体内分解又は相当の腐食が起こる若しくは起こる可
能性がある,及び医療機器からの溶出物が発生する可能性があるのいずれか又は複数の条件を満たす。
c) 医療機器から臨床使用中に毒性が懸念される物質又は生体との反応性が高い分解生成物及び溶出物が
発生する若しくは発生する可能性がある。
d) 一定量のナノマテリアルが,臨床使用中に医療機器から発生する又は発生する可能性がある。
e) 医薬品と医療機器とのコンビネーション製品である。
医療機器又は材料からの分解生成物及び溶出物の発生率が,臨床実績を基に安全性が確立された臨床ば
く露レベルであると判断された場合,又は分解生成物及び溶出物に関する十分な毒性学的データ若しくは
毒物動態学データが存在する場合,トキシコキネティクス試験の実施は不要である。
材料が生体内で分解するように設計されている場合を除き,通常,金属及びセラミックスからの溶出物
及び分解生成物の放出は十分に微量である。
分解生成物及び抽出物又は溶出物のトキシコキネティクス試験は,ISO 10993-16に従って実施する。
ナノマテリアルが対象となるトキシコキネティクス試験の具体的な考慮事項は,ISO/TR 10993-22に示
されている。
6.3.2.15 免疫毒性
附属書Aでは明確に扱われていないが,ISO/TS 10993-20では免疫毒性の概要及び医療機器の潜在的な
免疫毒性を取り上げている。材料の化学的性質から免疫毒性作用が示唆される場合,当該化学物質の免疫
毒性が不明である場合などには免疫毒性試験の実施を検討する。免疫毒性試験は,ISO/TS 10993-20に従
い実施する。
ナノマテリアルが対象となる免疫毒性試験の具体的な考慮事項は,ISO/TR 10993-22に示されている。
7
生物学的評価データの解釈及び総合的な生物学的リスクアセスメント
十分な知識及び経験のある専門家が,次の事項を決定し,文書化する。
a) 当該医療機器の生物学的評価についての戦略及び計画内容
22
T 0993-1:2020 (ISO 10993-1:2018)
b) リスクマネジメント計画に沿って,使用目的における当該材料の受容可否を決定する基準
c) 材料特定の適切性
d) 試験の選択及び省略のいずれか又は両方の論拠
e) 既存データ及び試験結果の説明
f)
生物学的評価を完了する根拠となる追加データの必要性判断
g) 当該医療機器についての生物学的安全性の総合的な結論
附属書Aは,個別の医療機器及びカテゴリにおいて考慮することが望ましい一般的なエンドポイントを
示している。
23
T 0993-1:2020 (ISO 10993-1:2018)
附属書A
(参考)
生物学的リスクアセスメントで対処するエンドポイント
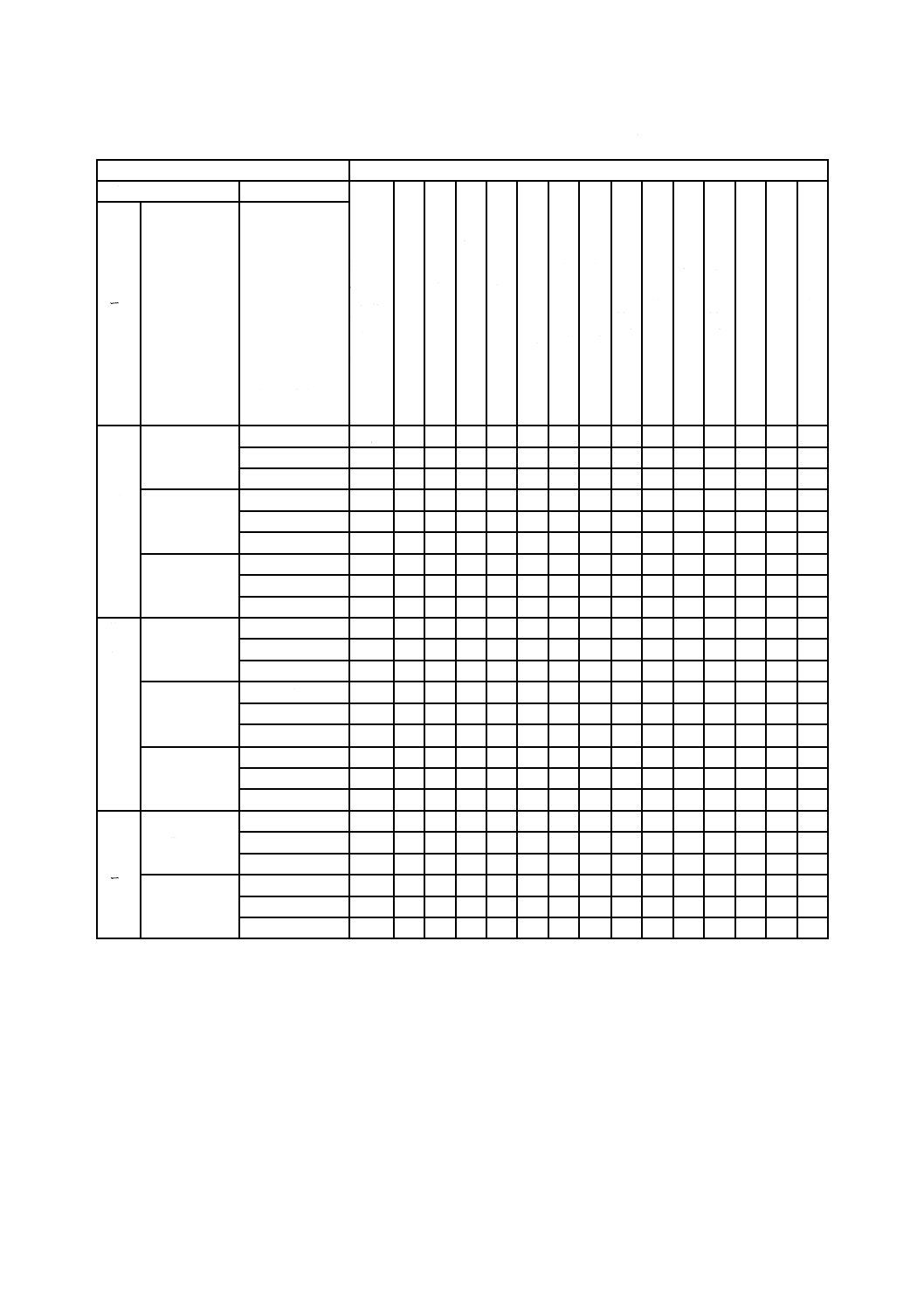
A.1 一般
この附属書の内容は生体適合性評価の骨子であるが,試験実施の照合表ではない。表A.1が示したエン
ドポイントが評価にとって妥当である場合には,まず,追加データの必要性を決定するために,同エンド
ポイントに関連する既存のデータセットを評価することが望ましい。特定の医療機器では,この表に示す
以上又はそれ以下のエンドポイントがふさわしい可能性もある。
表A.1において“X”はリスクアセスメントに先立って必要となる情報を,“E”はリスクアセスメント
で評価されるべきエンドポイントを意味する。このリスクアセスメントとは,既存データの活用,エンド
ポイントに固有な試験の実施,又はエンドポイントの評価に追加データが必要ない根拠の説明のいずれか
が該当する。
生物学的リスクアセスメントにおいては,全ての変更について,その正当性が示されることが望ましい。
医療機器に固有の規格の中に生体適合性に関する要求事項が含まれている場合には,それらを検討するの
がよい。
24
T 0993-1:2020 (ISO 10993-1:2018)
表A.1−生物学的リスクアセスメントで対処するエンドポイント
医療機器のカテゴリ
生物学的評価のエンドポイント
身体との接触形態
接触期間
物
理
学
的
及
び
化
学
的
情
報
の
い
ず
れ
か
又
は
両
方
細
胞
毒
性
感
作
性
刺
激
性
又
は
皮
内
反
応
材
料
由
来
の
発
熱
性
a
)
急
性
全
身
毒
性
b
)
亜
急
性
毒
性
b
)
亜
慢
性
毒
性
b
)
慢
性
毒
性
b
)
埋
植
の
影
響
b
),
c)
血
液
適
合
性
遺
伝
毒
性
d
)
発
が
ん
性
d
)
生
殖
及
び
発
生
毒
性
d
),
e)
生
体
内
分
解
性
f)
カ
テ
ゴ
リ
適用部位
A−一時的
(24時間以内)
B−短・中期的
(24時間超30
日以内)
C−長期的
(30日超)
表
面
接
触
機
器
皮膚
A
X g)
E h) E
E
B
X
E
E
E
C
X
E
E
E
粘膜
A
X
E
E
E
B
X
E
E
E
E
E
E
C
X
E
E
E
E
E
E
E
E
E
損傷表面
A
X
E
E
E
E
E
B
X
E
E
E
E
E
E
E
C
X
E
E
E
E
E
E
E
E
E
E
E
体
内
と
体
外
と
を
連
結
す
る
機
器
血液流路
間接的
A
X
E
E
E
E
E
E
B
X
E
E
E
E
E
E
E
C
X
E
E
E
E
E
E
E
E
E
E
E
E
組織,骨又は
歯質
A
X
E
E
E
E
E
B
X
E
E
E
E
E
E
E
E
C
X
E
E
E
E
E
E
E
E
E
E
E
循環血液
A
X
E
E
E
E
E
E
E j)
B
X
E
E
E
E
E
E
E
E
E
C
X
E
E
E
E
E
E
E
E
E
E
E
E
イ
ン
プ
ラ
ン
ト
組織又は骨i)
A
X
E
E
E
E
E
B
X
E
E
E
E
E
E
E
E
C
X
E
E
E
E
E
E
E
E
E
E
E
血液
A
X
E
E
E
E
E
E
E
E
B
X
E
E
E
E
E
E
E
E
E
C
X
E
E
E
E
E
E
E
E
E
E
E
E

25
T 0993-1:2020 (ISO 10993-1:2018)
表A.1−生物学的リスクアセスメントで対処するエンドポイント(続き)
注a) ISO 10993-11:2017の附属書Gを参照。
b) 十分な動物数及びエンドポイントによって評価されている場合,急性全身毒性,亜急性毒性,亜慢性毒性及
び慢性毒性のいずれか又は複数の組合せを含む埋植試験による評価から得られた情報が適切な場合もある。
急性毒性,亜急性毒性,亜慢性毒性及び慢性毒性を評価するための試験は必ずしも別の試験として行う必要
はない。
c) 適切な埋植部位を考慮する必要がある。例えば,正常な粘膜と接触する機器に関しては,理想的には正常な
粘膜と接触させた試験又は評価を実施するのがよい。
d) 医療機器が発がん性,変異原性及び生殖毒性のいずれか又は複数の組合せをもつことが知られている化学物
質を含む場合には,リスクアセスメントにおいて検討する。
e) 新規の材料,生殖又は発生毒性が懸念される既知材料,並びに生殖及び発生毒性に関連の強い母集団(例:
妊婦)に使用する医療機器,及び生殖臓器に局所的に使用する医療機器のいずれか又は複数の組合せについ
ては,生殖及び発生毒性を評価することが望ましい。
f) 構成部材又は構成材料が患者の体内に残留し,生体内で分解する可能性がある機器については生体内分解性
に関する情報を示すことが望ましい。
g) “X”はリスクアセスメントに先立って必要となる情報を意味する。
h) “E”はリスクアセスメントにおいて評価するエンドポイントを意味する。リスクアセスメントには,既知の
毒性情報を用いた評価,エンドポイントに示された生物学的安全性試験の実施,試験を省略する場合にはそ
の根拠を示すことが含まれる。医療用途として使用されていない新規材料が使用されている場合で,かつ,
文献などで毒性情報が得られない場合には,“E”と記載されていないエンドポイントについても評価の対象
に加える必要がある。機器の特性によって,適切なエンドポイントを選択する場合もある。
i) 組織液及び皮下も生体組織に含める。間接的接触によってだけ用いられるガス回路に用いる機器又は部材に
ついては,その機器に固有の規格を参照する。
j) 体外循環装置に使用される全ての機器。
26
T 0993-1:2020 (ISO 10993-1:2018)
A.2 表A.1に示したエンドポイントの根拠
次のエンドポイントは,旧規格からこの規格への改正で新たに追加されている。それぞれのエンドポイ
ントについて追加の根拠を次に示す。
− 物理学的及び化学的情報のいずれか又は両方(全ての医療機器カテゴリ)
全ての医療機器において,この情報は,更なる生物学的試験の必要性を判断するために使用される。
− 刺激性又は皮内反応(血液流路として間接的かつ長期的に接触する体内と体外とを連結する医療機器)
長期的に血液と間接的に接触する構成機器(例:点滴システム)は,血流中に刺激性物質を放出す
る可能性があるため,生物学的リスクアセスメントの一部として検討することが望ましい。
− 材料由来の発熱性及び急性全身毒性(接触期間を問わず損傷表面に適用する表面接触機器)
抽出物又は溶出物が損傷表面を経由して全身に循環される可能性があるため,材料由来の発熱性及
び急性全身毒性を検討することが望ましい。
− 材料由来の発熱性(全ての体内と体外とを連結する医療機器及び全てのインプラント)
抽出物又は溶出物が全身の血液循環系,リンパ系及び脳脊髄液のいずれか又は複数へ取り込まれる
可能性があるため,材料由来の発熱性を検討することが望ましい。
− 急性全身毒性(粘膜に短・中期的又は長期的に接触する表面接触機器,組織,骨又は歯質に一時的に
接触する体内と体外とを連結する医療機器,及び組織又は骨に一時的に接触するインプラント)
抽出物又は溶出物が粘膜を介して全身の血液循環系,リンパ系及び脳脊髄液のいずれか又は複数へ
取り込まれる可能性があるため,急性全身毒性を検討することが望ましい。
− 亜急性毒性(接触形態を問わず,身体と短・中期的及び長期的に接触する全ての医療機器)
24時間を超えて使用する医療機器又は構成機器は,抽出物又は溶出物が全身の血液循環系,リンパ
系及び脳脊髄液のいずれか又は複数へ取り込まれる可能性があるため,亜急性毒性を検討することが
望ましい。
− 亜慢性毒性及び慢性毒性(接触形態を問わず,身体と長期的に接触する全ての医療機器)
30日を超えて使用する医療機器又は構成機器は,抽出物又は溶出物が全身の血液循環系,リンパ系
及び脳脊髄液のいずれか又は複数へ取り込まれる可能性があるため,亜慢性毒性及び慢性毒性のいず
れか又は両方を検討することが望ましい。
− 埋植の影響(粘膜に短・中期的又は長期的に接触する表面接触機器及び損傷表面に短・中期的又は長
期的に接触する表面接触機器)
このタイプの接触を伴う医療機器又は構成機器は,埋植した際の局所的及び全身的影響を検討する
ことが望ましい。反復使用によって接触期間が一時的接触から短・中期的又は長期的接触へ変更され
る可能性がある医療機器又は構成機器においては,生体組織中への化学物質の蓄積に関する情報が埋
植試験実施要否の判断に有用である。
27
T 0993-1:2020 (ISO 10993-1:2018)
− 埋植の影響(血液流路として間接的かつ長期的に接触する体内と体外とを連結する医療機器)
直接接触を伴う医療機器を併用する場合には,長期的に血液と間接的に接触する構成機器(例:点
滴システム)から血流中に取り込まれた抽出物又は溶出物が,併用される医療機器の直接接触によっ
て引き起こされる炎症反応に影響を及ぼす可能性がある。全ての抽出物又は溶出物の全身毒性に関す
る文献が利用可能な場合及び直接接触する構成機器が存在しない場合には,このカテゴリにおける埋
植の影響評価は不要となる場合もある。
− 遺伝毒性(循環血液に一時的に接触する体内と体外とを連結する医療機器)
体外循環に使用される医療機器又は構成機器の場合には,抽出物又は溶出物が血流内に取り込まれ,
医療機器が抜去された後もそれらが体内に残存する可能性があるため,遺伝毒性を検討することが望
ましい。
− 遺伝毒性(血液に一時的に接触するインプラント)
抽出物又は溶出物が血流内に取り込まれ,医療機器が抜去された後もそれらが体内に残存する可能
性があるため,遺伝毒性を検討することが望ましい。
− 発がん性(損傷表面と長期的に接触する表面接触機器,並びに接触形態を問わず身体と長期的に接触
するインプラント及び体内と体外とを連結する機器)
抽出物又は溶出物が全身の血液循環系,リンパ系及び脳脊髄液のいずれか又は複数へ取り込まれる
可能性があるため,発がん性は,生物学的リスクアセスメントの一部として取り扱うことが望ましい。
28
T 0993-1:2020 (ISO 10993-1:2018)
附属書B
(参考)
リスクマネジメントプロセスにおける生物学的評価実施のガイダンス
B.1
背景情報
B.1.1 一般
この附属書はこの規格の要求事項に従って,医療機器の生物学的評価を実施するためのガイダンスであ
る。この規格は医療機器の生物学的評価の一般的な枠組みを示しているが,実運用に当たっては,より詳
細なガイダンスが有用となるため,この附属書が作成された。当該ガイダンスは,この規格の理解を深め,
要求事項を満たすために活用できる様々な方法及びアプローチを示している。
生物学的評価は,JIS T 14971の要求事項に従い実施されるリスクマネジメントプロセスの中で設定され
た設計検証作業の一つであり,この附属書はJIS T 14971のリスクマネジメントプロセスに,この規格を
適用する際のガイダンスである。この附属書では,医療機器の設計検証の一部として,生物学的評価を行
うためのリスクマネジメントプロセスを確立し,維持する際の考え方及び方法を説明している。
科学技術の進歩に伴い組織反応メカニズムの理解が深まるにつれて,生物学的安全性評価の方法も変化
し,確立された科学的データの検証,物理学的及び化学的キャラクタリゼーション,並びにインビトロ試
験による評価が標準となりつつある。このため,インビボ試験は当該評価では不明な点があり,同問題を
解消するために必要な場合に限り実施する。したがって,この規格は,化学物質を特定するための分析及
びインビトロモデルにおいて,インビボモデルと同等の適切なデータが得られる場合に,当該データを優
先させ,試験動物数及びばく露を最小限にする生物学的安全性評価を計画するための枠組みを示している。
ある特定の医療機器に対する適切なアプローチの選択は,医療機器の特性,利用可能な適切なデータの範
囲及びリスクアセスメントに依存する。
当ガイダンスの適用可否を判断するためには,法的規制及び法的ガイダンスを考慮することが望ましい。
当ガイダンスを使用する組織体は,任意に当ガイダンスの全て又は一部を自身のリスクマネジメントプ
ロセスに取り込むことができる。
当ガイダンスは,リスクマネジメントプロセスの評価者,適合性認証機関及び規制当局のための背景情
報として有用である。
B.1.2 その他の基準,ガイダンス文書及び法的規制との関係
この規格及び附属書と,医療機器の生物学的安全性評価の基準及び一般的なリスクマネジメントとの関
係は,次のように要約される。
− この附属書は,この規格の運用ガイダンスを提供する。
− 生物学的評価はリスクマネジメントの構成要素の一つであり,この附属書は生物学的評価の実施に関
29
T 0993-1:2020 (ISO 10993-1:2018)
するJIS T 14971の運用ガイダンスを含んでいる。
この附属書はこの規格に新たな要求事項を追加したり,この規格の要求事項を変更するものではない。
この附属書は,規制当局の調査又は認証検査の基準となる要求事項を含んでいない。
B.2
リスクマネジメントの実践としての生物学的評価
B.2.1 一般
B.2及びB.3は,製造業者が医療機器に関連する生物学的ハザードの特定,リスク推定及び分析,リス
クコントロール及びその有効性の監視などの一連のプロセスを示している。医療機器のリスクと効用とを
十分に考慮した生物学的評価の計画を適切に実行することによって,患者の安全が守られることが望まし
い。潜在リスクを受容できなければ,医療機器の使用によって患者は効用が得られない。医療機器のリス
クはその特性及び用途によって多様である。医療機器で受容されるリスクのレベルは,その使用によって
もたらされる効用に依存する。
生物学的リスクの検証は,医療機器のリスクアセスメントの一つにすぎない。したがって,医療機器の
リスク評価はあらゆる側面から考慮することが望ましい。場合によっては,生物学的安全性とは異なる側
面から材料の利点を考慮することもある。例えば,生物学的に最も安全な材料であっても力学的強度が受
容できない場合もある。その場合は,目的の力学的強度をもつ材料が生物学的安全性においても受容可否
を検証する必要がある。これは,医療機器の設計,開発において求められるリスクマネジメントプロセス
の一部として行われる生物学的評価の基本である。
材料選択及びリスク分析は,医療機器の設計プロセスで必要不可欠な要素である。材料選択は生物学的
安全性の評価に重要な要素であり,体系的に取り組むことで適切なデータ収集が可能となる。JIS Q 13485
及びJIS T 14971に従って,受容される生物学的リスクの判断基準は設計プロセスの最初に定められるこ
とが望ましい。出発材料及び配合並びに包装,輸送及び保存期間を含む様々な条件が最終製品の生体適合
性に影響を及ぼす可能性があるため,全てをリスクアセスメントに組み込み検証することが望ましい。生
物学的評価は,リスク分析の結果又は同材料の臨床使用実績に基づいて,一定の安全性を満たすことを立
証する目的で計画,実行する。この評価は,医療機器に関する全てのハザード特定及びリスク推定を含め
たリスクマネジメント計画の構成要素の一つとなる。適切なリスクアセスメントには,医療機器への生物
学的反応と同様に,毒物学的ハザード及びばく露経路の明確化が必要である。
ハザード特定に重要な要素は,材料キャラクタリゼーションである(ISO 10993-18及びISO/TR 10993-19
を参照)。次の事項について明確化する。
− 代用できる適切な材料も含めそれぞれの材料を定め,特徴を示す。
− 材料,添加剤,加工助剤などに存在するハザードを特定する。
− 下流の工程(例:材料の構成成分間の化学的相互作用,最終製品の滅菌)が最終製品に存在する化学
物質に与える潜在的影響を特定する。
− 製品の使用中に放出される化学物質(例:生体内分解性インプラントの中間又は最終分解物)を特定
する。
30
T 0993-1:2020 (ISO 10993-1:2018)
− ばく露量(医療機器に含まれる全量又は臨床使用での溶出量)を推定する。
− 書籍を含む利用可能な毒性及びその他の生物学的安全性のデータを検証する。
生物学的安全性の評価で検証する情報を次に示す。
− 関連性のある部材成分,混合部材の毒性データ
− 関連性のある部材成分,混合部材の臨床使用実績から得た情報
− 生物学的試験のデータ
まずハザードを特定し,そこから想定されるリスクを評価する。この段階では,材料に由来する重篤な
毒性学的リスクの有無を判断する。
既存データからリスクが受容されると結論付けられる場合には,更なる生物学的安全性試験の実施は必
要ない。リスクが受容できない場合は試験を実施しないことが望ましい。既存データが不十分な場合には,
追加データを取得する。生物学的安全性試験の目的は,リスク評価を行う上での追加データの取得にある。
したがって,試験を実施する根拠は,既存データの適切なリスク分析に基づく。
全ての試験結果を評価することが望ましい。報告書には,根拠を明文化し,得られた知見に対する評価
及びその受容可否の判断を記載する。
評価者は,入手した情報が生物学的安全性評価に利用可能か否かを検討する。利用可能と判断した場合
には,安全性に関する結論を導く上で必要な全ての判断の根拠,評価に影響を与える試験結果,その他の
情報などを報告書に記載する。
評価報告書には,全ての根拠に関する出典及び重要性を示し,全体の結論は科学的原理に重点を置き,
正確,明瞭かつ分かりやすく記載することが望ましい。結論を導くための要因が,判断するための論理的
根拠並びに判断の根底にある不確実性の特定及び考察も含め,十分に検討されていることが重要である。
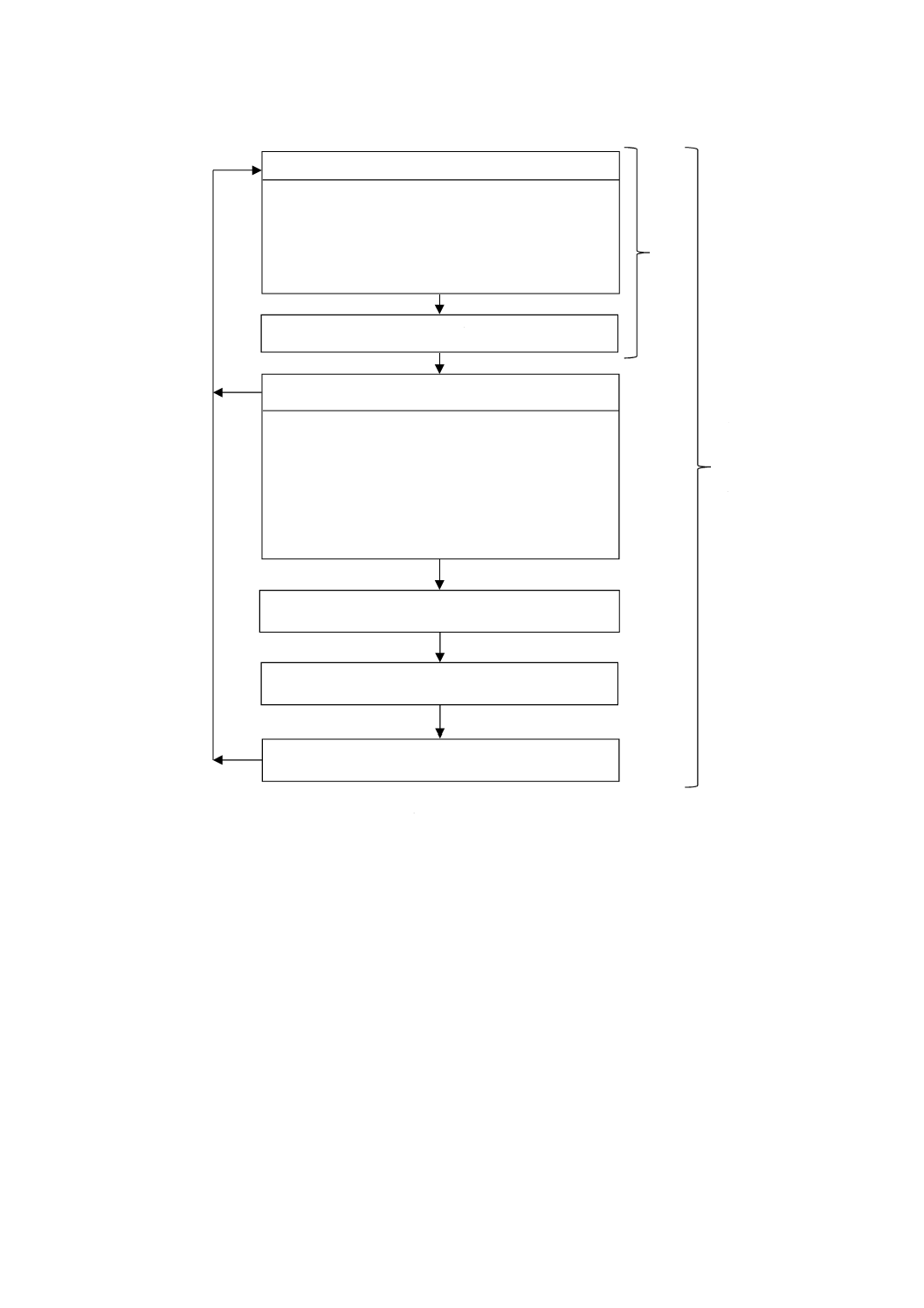
リスクマネジメントの構成要素は,図B.1に集約されている(JIS T 14971:2012から引用)。リスクマネ
ジメントプロセスでは,生物学的評価以外の様々な要因も検証される。
要約すると,医療機器の生物学的評価は医療機器のリスクマネジメントの一つであり,生物学的評価は
この規格及びJIS T 14971の要求事項を満たすことを目的に実施することが望ましい。
31
T 0993-1:2020 (ISO 10993-1:2018)
図B.1−リスクマネジメントプロセスの概要(JIS T 14971:2012から引用)
リスク分析
・ 使用目的及び医療機器の安全に関する特質の明確
化
・ ハザードの特定
・ 個々のハザードに対するリスク推定
リスク評価
リスクコントロール
・ リスクコントロール手段の選択
・ リスクコントロール手段の実施
・ 残留リスクの評価
・ リスク/効用 分析
・ リスクコントロール手段から生じるリスク
・ リスクコントロールの完了
製造及び製造後の情報
残留リスクの全体的な受容可能性の評価
リスクマネジメント報告書
リ
ス
ク
ア
セ
ス
メ
ン
ト
リ
ス
ク
マ
ネ
ジ
メ
ン
ト
32
T 0993-1:2020 (ISO 10993-1:2018)
B.2.2 生物学的評価計画
JIS T 14971:2012の3.4は,事前にリスクマネジメント計画を立てるよう求めている。生物学的評価もリ
スクマネジメント計画の一部となるため,同様に生物学的評価計画書の作成が必要となる。単に附属書A
で特定した全てのエンドポイントに対する試験の実施を計画することはJIS T 14971又はこの規格の要求
事項を満たしたことにはならない。当ガイダンスの医療機器への適用事例は,ISO 18562-1にも示されて
いる。
生物学的評価計画は,次の事項を最低限考慮し,知識及び経験が豊富な専門家チームによって作成する
ことが望ましい。
− リスク分析を行うために,公表されている文献(情報源及び調査方法を含む。),内部及び提供された
データ並びにその他の文書から,適用できる情報の収集
− 医療機器の使用に関わる全ての技術的な性能要件に対する評価の実施有無
− 設計管理プロセスにおける精査及び承認の体制
− 評価の最終結論及び追加試験の必要性
− 生物学的リスクアセスメント結果に対する最終精査及び承認の体制。生物学的リスクアセスメントに
は,適用するリスクコントロールの手段,全ての残留リスクに対する文書化及び製品表示などによる
残留リスクの開示を含む。
B.3
リスクマネジメントに関するガイダンス
B.3.1 リスクアセスメント
B.3.1.1 序文
リスクアセスメントは,リスクを特定及び推定するリスク分析と,リスクコントロールによる軽減措置
をとるべきリスクを特定するリスク評価との組合せである。
B.3.1.2 リスク分析
リスク分析は特定のハザードを明確化し,その重大さを評価するプロセスである。生物学的評価におい
て重要な検討事項には,構成材料の潜在的毒性及びそのばく露経路がある。また,物理学的特性が生物学
的反応に及ぼす影響も重要な検討事項となる。リスク分析では,当該ばく露経路における原料及び成分の
いずれか又は両方に由来する毒性学的影響を基に体系的にリスクを推定することが望ましい。したがって,
リスク分析は間接的又は直接的に生体組織と接触する医療機器の原料及び成分の特定及びキャラクタリゼ
ーションから始まる。その作業は,製造用の添加剤,加工助剤,滅菌残留物などの潜在的な混入物質の存
在を考慮し,医療機器の製造過程での最終形態を対象に行うことが望ましい。材料加工時の材料組成及び
化学的性質(内部及び表面への影響を含む。)の影響も考慮することが望ましい。特に,反応性の強い又は
危険な成分が使われる場合,製造,加工,保管又は原料の分解によってそのような成分が生成される可能
性がある場合は,これら毒性物質の残留を考慮することが望ましい。包材からの混入物質による相互作用
又は混入物質自体のばく露も考慮することが望ましい。
物理学的及び化学的な材料特性は生物学的安全性と密接に関連するため,次に関連する事項は,リスク
分析の段階で特定することが求められる。
− 摩耗,応力,疲労[例:特に,全置換型人工関節など荷重がかかる医療機器及び医療機器から発生す
33
T 0993-1:2020 (ISO 10993-1:2018)
る微粒子(ナノマテリアルを含む。)又は材料の分解]
− 摩擦及びそれに関連した刺激(例:カテーテルの使用)
− 材料の組合せによる相互作用(化学的相互作用)(例:柔軟性の違い,ガルバニック腐食,摩耗)
− 熱(例:熱による分解又は熱によって誘発された材料の変化)
− 製造工程(例:発生した内部応力が環境応力,亀裂,形態の変化又は分解を助長する。)
− 環境との相互作用[例:内視鏡(胃酸),包帯(外部の環境),紫外線,洗剤,除菌及び滅菌工程]
− 電気(例:短絡,分解,加熱,筋肉刺激)
− 成分間の潜在的な相互作用
− 物理学的形状の影響(例:ナノマテリアルを含む微粒子)
− 再処理
− 輸送,経時的変化
材料の情報は,文献精査,供給元からの情報提供,内部で取得したデータ又は製造工程及び配合組成が
同等な市場販売品との比較を通して得ることができる。
注記1 附属書Cは,文献精査に関するガイダンスを示す。
化学的キャラクタリゼーションに続き,材料の成分に関する毒性学的影響と用量反応との関係を考慮す
ることが望ましい。
毒性学的影響の範囲は広い。ばく露経路及び期間に応じて考慮すべき毒性学的影響のガイダンスを箇条
5及び附属書Aに示す。
抽出物及び溶出物のキャラクタリゼーションに加え,形状,剛性など生物学的に悪影響を及ぼす可能性
がある医療機器の物理学的特性を考慮することが望ましい。
注記2 微粒子のキャラクタリゼーション及び試験において,ナノマテリアルには特有の注意が必要
である。サブミクロンの成分からなる材料は,同じ材料でも汎用サイズの場合と異なる挙動
をとることがあり,汎用サイズで得られたデータから外挿することが適切でないケースもあ
る。
B.3.1.3 リスク推定
材料の化学的な毒性に対してリスクの推定を行う場合は,毒性が懸念される化学物質のばく露量を考慮
する。例えば,溶出物又は溶解する成分の生物学的利用能(ISO 10993-17を参照)。材料の物性に対して
リスクの推定を行う場合は,医療機器の使用方法を考慮する。
リスクは通常,危害が起こる確率及びその重大さを明らかにすることによって推定される。一般的な毒
物学的条件の場合,危害が起こる確率は,有毒な成分の生物学的利用能及び関連組織での用量反応の知見
から推定する。重大さは毒性の性質から評価する。材料の物性に対するリスク推定では,危害が起こる確
率を摩耗片の物理学的試験の結果から推定する。重大さは文献又は動物試験から推定される生物学的反応
の性質から評価する。
34
T 0993-1:2020 (ISO 10993-1:2018)
公表された文献,内部で保有するデータ及び当該医療機器又は材料の臨床使用実績から入手した情報が,
ハザードの推定又は定量化に不十分である場合には,リスク推定を目的とした化学的若しくは物理学的キ
ャラクタリゼーション,又は動物試験の実施が必要となる。その場合はISO 10993の適切な部に従い実施
することが望ましい。
リスク推定を目的とする試験項目は,現行のデータセットを精査し,不足項目に対して適切に選択する
ことが望ましい(附属書C参照)。
リスク分析に求められるデータの量及び分析項目は,使用目的によって様々であり,生体組織との接触
形態及び接触期間に依存する。通常,患者と間接的に接触する材料,健常皮膚とだけ接触する医療機器,
及び生体組織,投与媒体,粘膜又は損傷表面と直接的に接触しない医療機器については要求されるデータ
の量が比較的少ない。
B.3.1.4 リスク評価
リスク評価はリスク分析の中で特定されたリスクの重要性を評価し,軽減するための要件及び機会(リ
スクコントロール)を特定するステップとなる。医療機器を構成する全ての材料に対して体系的に評価す
ることが望ましい。
材料の生体適合性は,使用目的及び接触形態などの条件と関連付けて考える必要がある。例えば,抽出
物又は溶出物の毒性に関する考察は,ばく露経路,期間,体内利用率などを考慮することが望ましい。特
に,臨床使用実績又は類似した適用におけるヒトのばく露データについて考察することが重要となる。例
えば,最終製品が非刺激性であることを示した臨床研究は,動物を用いた刺激性試験を省略する正当な根
拠として使用することができる。しかし,生物学的な悪影響が材料の組合せに起因することもあるため,
インプラント材料を用いた臨床試験の結果を最終製品の埋植試験を省略する根拠とするには不十分である。
体系的な生物学的リスク評価には,適切な評価方針を決定できる専門知識及び入手したデータを厳格に
評価し,追加試験の要否を的確に判断する能力をもった専門家が求められる(箇条7参照)。
B.3.2 リスクコントロール
リスクコントロールはリスクを軽減させる対策を特定し,実行するプロセスである。生物学的安全性に
関する,リスクコントロールには設計の変更作業を伴うこともある。リスクコントロールの事例を次に示
す。
− より危険なばく露経路を避ける又はばく露時間を短くする設計変更
− 血流低下時の血栓形成を最小限にすることを目的に,幾何学的表面特性を最適化する設計変更
− 有害な生物学的反応の原因となる医療機器の不具合(例:微粒子発生又は塗装コート材の剝離)を防
ぐための設計変更
− 組成変更又は材料変更による毒性の軽減
− 危険な残留物若しくは加工助剤を減少又は除去を目的とした製造工程の変更
リスクは,ワーストケースを想定して取得したデータよりも,より的確にリスクを推測できるデータを
35
T 0993-1:2020 (ISO 10993-1:2018)
示すことで軽減することも可能である。その場合は,対処が必要な不確実要素を特定した当初のリスク分
析の結果に基づき,適切な試験を選択することが望ましい。不確実要素を伴うリスクを軽減するには添付
文書による警告,禁忌など,試験以外の方法で対処することも可能である。
リスクコントロールによって新しいハザード又はより高いレベルのリスクが発生した場合は,再試験が
必要な場合もある。
リスク軽減のための動物試験の実施は,既知の知見の精査,化学的又は物理学的キャラクタリゼーショ
ン,インビトロ評価など全ての代替手段をとることができない場合にだけ検討することが望ましい。
B.3.3 残留リスクの受容可能性の評価
リスク分析,リスク評価及びリスクコントロールを実施した後,それらの作業結果を精査し,残留リス
クを文書化する。さらに,適切な表示及び添付文書による警告,注意など,残留リスクの開示要否を判断
する。
B.3.4 製造後監視
リスクアセスメントは,生物学的試験が必要となる場合もあるが,入手した情報の判断を行うプロセス
である。このアセスメントは,必要に応じて医療機器の市販後調査及び臨床使用における不具合情報から
入手できる新しい情報と併せて,更新することが望ましい。監視は,評価対象の医療機器に関連する有害
事象の発生状況及び類似した医療機器又は材料に関する最新の知見を考慮することが望ましい。また,監
視に関連する科学文献の継続的な精査も実施することが望ましい。
B.4
生物学的評価における具体的な留意事項
B.4.1 材料キャラクタリゼ−ション
B.4.1.1 化学的キャラクタリゼ−ション
化学的キャラクタリゼーションのデータは,次のような場合における生物学的アセスメントにおいて最
も有用である。
− 知的所有権の問題を解決する場合
− 医療機器を構成する化学物質のうち,一つ以上の成分を変更する場合
− 化学物質の毒性データが容易に入手可能な場合
− 抽出又は化学分析が容易に行える場合
B.4.1.2 生物学的評価における化学的キャラクタリゼーションデータの使用
この規格は,生物学的評価の対象となる医療機器に対する化学的キャラクタリゼーションの実施を製造
業者に求めている。例えば4.3では,医療機器の生物学的評価に影響を与える意図的な添加物,製造工程
での混入物,残存物,及び溶出物を考慮するよう製造業者に指示している。しかし,生物学的安全性評価
を行う際に,これらの情報の取扱いを具体的に明示したガイダンスはない。
ハザード特定の見地から,適切な生物学的試験を選択する際,医療機器から遊離する化学物質の情報は
有用である。例えば,腎毒性をもつ化学物質の存在が判明している場合には,ISO 10993-11に記載された
36
T 0993-1:2020 (ISO 10993-1:2018)
急性毒性又は亜慢性毒性試験を行う上で,腎への影響評価に注意を払うことが可能となる。このように,
化学物質の情報は,臨床上で最も関連深いエンドポイントに対応した生物学的試験の立案に有用である。
化学的キャラクタリゼーションのデータはリスク推定にも有用である。毒性学的いき値又は化学物質に
特有の限度値(ISO 10993-17及びISO 10993-18を参照)が入手できる場合には,使用環境を模した条件下
で医療機器から遊離する化学物質の量及び速度から,ばく露量及び副作用の可能性を評価することができ
る。
B.4.1.3 材料組成の知的所有権
機密性の高い知的所有権に関する情報であるなどの理由で,製造業者が必要なデータ(例:全組成デー
タ)を入手できない場合は,材料供給元に,材料の使用用途に関連した生物学的評価結果の有無を照会す
ることが望ましい。場合によっては,材料供給元が機密性を保持したい材料組成に関する情報(マスター
ファイル)を,製造業者のもつ生物学的評価結果と分けて,審査機関又は規制当局へ提出することも可能
である。これによって審査機関又は規制当局は,材料供給元の情報を参照でき,製造業者が申請した医療
機器の申請書の審査と並行して,機密情報を製造業者に開示することなく審査できる。
B.4.1.4 物理学的キャラクタリゼーション
微粒子及びナノマテリアルが医療機器に使用される場合には,物理学的キャラクタリゼーションが必要
である(ISO/TR 10993-22)。また,物理学的形状(形状,粒子サイズ,多孔性,表面の粗さ)が医療機器
の生物学的反応に多大な影響を与えたり,安全性に影響を与える可能性もある。物理学的特性が影響を及
ぼす可能性は,リスク評価の一部として検討することが重要である。科学文献又はその他の情報源から入
手したデータがリスクの推定に不十分である場合,適切な機能モデルを用いた追加試験又は物理学的形状
による影響の研究が必要となる。
例を次に示す。
− 血流及び血液適合性に関する形状の評価
− 組織侵入に対する多孔性の評価
− 局所及び遠位部の組織反応に対する摩耗粒子の放出の評価
− 細胞接着,形質発現及び増殖に対する表面組成(微細構成)の評価
B.4.1.5 製造工程の影響
添加剤の使用又は混入物質の有無とともに,製造条件が材料に与える影響も考慮することが重要である。
一般的に,生物学的安全性評価を目的とした材料の試験は,当該医療機器の最終製品と同等の方法で加工
(滅菌する製品であれば滅菌工程も含む。)した試験材料で行うことが望ましい。試験に用いた材料と最終
製品との製造方法に違いがある場合,その違いが生物学的安全性評価に重大な影響を及ぼさない正当な根
拠を示す必要がある。特に次に示す事項を検討することが望ましい。
− 材料特性において原料又は表面の変化を起こす工程(例:成形,表面処理,溶接,機械加工)
− 触媒,酸化防止剤,顔料,表面処理剤及びその他意図的に加えた添加剤又は加工助剤
− 工程における潜在的な混入物質(例:洗浄・消毒・殺菌薬,エッチング剤,離型剤,切削油及び切削
37
T 0993-1:2020 (ISO 10993-1:2018)
粉,潤滑油又は材料加工時の残留物などの製造機械に由来する混入物質)
− 製造及び加工,並びに臨床使用及び保存期間中の分解生成物
− 化学物質及び添加剤の加工における潜在的な残留物
B.4.2 既存データの収集
ギャップ分析を行う前に,次の事項についてデータの収集範囲を決定しておくことが望ましい。
− 構成材料,成分又は他の関連する化合物の毒性データ(附属書C参照)
− 構成材料又は製品の既存生物学的安全性データ
− 臨床使用実績又はヒトへのばく露データ
B.4.3 機器の試験の検討
B.4.3.1 生物学的試験への段階的アプローチ
リスク評価に当たり,追加試験の実施が必要であると判明した場合には,段階的なアプローチをとるこ
とが望ましい。試験は化学的及び物理学的キャラクタリゼーション並びにインビトロ試験によるスクリー
ニングから始めるのがよい。動物試験を進める前に,キャラクタリゼーション及びインビトロ試験の結果
を精査することが望ましい。
B.4.3.2 長期試験(慢性毒性,生殖毒性,生体内分解性及び発がん性)の実施時期
長期試験の必要性は,想定された用途に応じた科学的に妥当な根拠に基づいて判断する。
患者への化学物質のばく露が,毒性学的いき値を下回る非常に低いレベルであることが確認でき,適切
にリスクアセスメントが実施された場合,長期試験を不要と判断することは妥当である。次の要素は,長
期試験を不要とする科学的根拠の説明に必要である。
− ばく露量(患者一人当たりの医療機器又は材料の総量)
− ばく露時間
− 生物学的利用能
次に該当する場合は,長期試験が必要となる可能性がある。
− 材料の量及びばく露時間から長期間の毒性学的影響の懸念がある。
− 構成する化学物質に毒性があることが知られている,又は懸念されている。
− 評価対象の材料(又は類似の材料)に関する長期使用のデータが不十分である。
− 特別な化学的理由がある(例:特定の慢性的な毒性の影響を示すアラート構造をもつ。)。
− 短期のスクリーニングで毒性を懸念する結果が得られている(例:インビトロ遺伝性試験)。
− 当該材料の生体安定性に既知の懸念があり,基礎的な検証データが不十分である(例:検討中の材料
又は配合について,適切なモデルを用いた加速試験データによる検証)。
長期試験の選択については,いまだに議論に結論が得られていないこと,及び国ごとに試験の要求事項
に相違があることにも注意することが望ましい。
38
T 0993-1:2020 (ISO 10993-1:2018)
B.4.3.3 吸収性材料のインビトロ試験系のpH及び浸透圧の補正
生体内で吸収されることが意図されている重合体,金属又はセラミック材料は,可溶性の成分又は分解
生成物を発生する。材料の分解速度が速い場合,発生した生成物の濃度が高くなることによって,インビ
トロ試験系のpH及び浸透圧のいずれか又は両方が変化する可能性がある。生体内ではかん(潅)流及び
炭酸塩の平衡などが複合的に働くため,吸収性を意図した材料を評価する際には,生理学的に適切な条件
を維持するため,インビトロ試験系のpH及び浸透圧のいずれか又は両方の補正が必要である。報告書に
は,pH及び浸透圧のいずれか又は両方を補正せずに実施したインビトロ試験との比較によって補正の効果
及び化学的な妥当性も記載する。補正によって重要な考慮事項が見過ごされる可能性があるため,補正及
び非補正下で実施した分析の結果を比較することが望ましい。
B.4.4 生物学的安全性アセスメント
B.4.4.1 臨床データのリスクアセスメントへの使用
図1は,評価対象となる医療機器の化学組成及び物性(例:形状及び表面の特性),並びに接触形態及び
期間が既存の医療機器と同じである場合において,リスクアセスメントに必要な臨床データ(物理学的,
化学的及び生物学的)の有無及びその妥当性について判断する際の考え方を示している。
リスクアセスメントに係る臨床データの充足性有無の判断は,医療機器を構成する全ての材料の長期に
わたる安全な使用実績の有無を考慮する。最終製品の材料が既存の医療機器と同一であり(配合及び製造
工程を含む。),ばく露特性も同じで,かつ,入手した臨床データが生体適合性のエンドポイントを満たす
場合,材料キャラクタリゼーションの結果に基づいて実施したリスクアセスメントは,生物学的安全性評
価として妥当である。
注記 臨床使用実績及び医療機器不具合情報の使用に関するガイダンスは,米国毒性物質疾病登録庁
(ATSDR)及び日本のガイドラインに示されている。
B.4.4.2 投与量及び経路の妥当性をもつ“十分な毒性データ”
化学的キャラクタリゼーションによって医療機器から遊離する化学物質の種類を特定することは可能で
あるが,医療機器の臨床使用方法に合致したばく露経路での化学物質の毒性データが入手できない場合も
ある。
6.3.2.14に規定する生理学的薬物動態モデリングを含め,投与用量と投与経路との違いからばく露量を外
挿することは可能であるが,その外挿に当たっては慎重に行うとともに,投与局所への作用も考慮するこ
とが望ましい。
臨床使用で想定されるばく露量に比べ,過度に高い投与用量を適用した試験の結果から毒性学的影響を
判断する場合は,注意が必要である。また,吸収性材料のインビトロ試験を実施する場合には,試験系が
生体内を模擬した条件となるようサンプル濃度などを調整する必要がある(吸収性材料のpH及び浸透圧
の補正に関するガイダンスであるB.4.3.3を参照)。
動物実験のデータを臨床使用条件に外挿するために検討することが望ましい要素は,ISO 10993-17で解
説されている。
39
T 0993-1:2020 (ISO 10993-1:2018)
B.4.4.3 ISO 10993-17に従った溶出物の受容可能性(許容限度)の決定
ISO 10993-17に示されているように,リスクのキャラクタリゼーションでは,患者又は臨床医がばく露
する化学物質の量と“安全な”量又は許容摂取量(TI)とを比較する。ばく露量をTIで除した値(dose/TI)
が1を超える場合,ばく露された患者に副作用が起こる可能性が高くなる。しかし,dose/TIを溶出物の受
容可否を判断する“明白な境界”としない方がよい。dose/TIが高いほど,患者及び使用者のいずれか又は
両方に副作用が起こる可能性も高くなる。しかし,TI算出の基となった試験で認められた副作用の重症さ,
化学物質の薬物動態,医療機器から化学物質を抽出する条件,TI算出に用いた不確実係数なども考慮に入
れることが重要である。ISO 10993-17では,許容限界(AL)の設定及び医療機器からの溶出物の受容可能
性を評価するために必要な臨床使用実績及び利用可能な情報の詳細を示している。
B.4.4.4 毒性学的懸念のいき値(TTC)
材料中の低濃度毒性成分を検証する場合又はTIを文献から導き出すことができない場合は,“毒性学的
懸念のいき値”の概念を考慮することが望ましい。当該物質の既知の毒性影響,特に毒性用量を参照する
ことによって,当該物質の量が,毒性を発現する用量に比べて十分に少ないことを立証することができる。
B.4.4.5 リスクアセスメントにおける混合物に関するガイダンス
ISO 10993-17において,患者又は臨床医は,医療機器の使用によって単一の化学物質ではなく,通常,
医療機器から生成される多数の化学物質にばく露されることが示されている。多数の化学物質への同時ば
く露は,化学物質が単独で投与された場合と比較して,ばく露物質の毒性を増強又は減弱させる可能性が
ある。
図1では,患者又は臨床医が化学物質の混合物にばく露された場合にも,個々の化学物質毒性データの
適用可否を考慮するよう求めている。
化学物質の混合ばく露に関する毒性データの入手は困難であることから,この規格では医療機器の生物
学的評価のための単一化学物質での毒性データを使用するよう記載している。化学構造が類似した化学物
質に同時にばく露される場合は,互いに付加的な毒性作用を示す可能性もある。化学構造に類似性がなけ
れば,付加的又は抑制性の毒性作用の有無を検証することはできない。また,化学物質が化学的に相互作
用し,類似又は新規の毒性学的リスクをもたらす可能性がある。化学物質の混合物に対するリスクアセス
メントの方法は,ISO 10993-17:2002の附属書Bに記載されている。
B.4.5 一般的なガイダンス
B.4.5.1 生物学的安全性の再評価が必要となる変更
従来の医療機器の設計業務では,設計変更に伴い,リスクアセスメントを再度実施することが必要にな
る。設計変更によって医療機器の生物学的性能が変化する可能性があるため,変更によって及ぼされる影
響を評価することが重要である。変更に伴うリスクを適切に特定,評価,アセスメントし,かつ,コント
ロールすることが望ましい。リスクが受容できないと判明した場合,再試験の実施に先立って,追加の情
報を入手することが望ましい。再試験は,評価に必要と判断された場合だけに実施されることが望ましい。
再試験を実施する根拠は,既存データのリスク分析の結果に基づくことが望ましい。
材料を変更する場合には再評価が必要となるが,その再評価の範囲は変更内容に対して適切であること
40
T 0993-1:2020 (ISO 10993-1:2018)
が望ましく,変更された特定の材料,医療機器の特長及び使用法,並びに潜在的な相互作用に着目するこ
とが望ましい。
試験の実施が必要と判断された場合には,次に示す段階的なアプローチを踏むことが望ましい。
a) 物理学的及び化学的キャラクタリゼーション
b) インビトロ試験
c) 動物試験
動物試験は最終的な手段であり,キャラクタリゼーション及びインビトロ試験から十分な情報が得られ
なかった場合にだけ実施することが望ましい。材料又は最終製品の生物学的性能に影響を及ぼす代表的な
設計変更の事例を次に示す。
− 加工(例:滅菌,洗浄,表面処理,溶接,射出成型,機械加工,一次包装)
− 材料の供給元(例:新しい供給元,新しい設備)
− 材料の仕様(例:許容範囲の拡大,新しい仕様)
− 配合(例:新しい材料,新しい添加剤,許容範囲の変更)
− 保管条件(例:有効期限の延長,許容範囲の拡大,新しい輸送条件)
− 生物学的環境(例:臨床使用での変更)
材料の変更に伴い検討する特性の事例を次に示す。
− 化学組成(例:組成,純度,溶出物の概要)
− 物理学的特性(例:形態,微細構成)
− 機械的特性(例:耐摩耗性,強度)
− 生体内安定性,環境安定性及び化学的安定性
− 電気的性質及び電磁両立性(EMC)の生物学的影響
化学的キャラクタリゼーションのデータは,リスクアセスメントにおいて評価対象である材料と臨床使
用実績をもつ類似した既存材料との毒性学的な同等性を判断するために用いられる。毒性学的な同等性を
判断する際の原則は,ISO 10993-18:2005の附属書Cで説明されている。
B.4.5.2 優良試験所規範(GLP)
生物学的評価を立証する全ての試験は,製造業者の品質マネジメントシステムの必須要件であると考え
られ,品質管理試験と同様に,バリデーション及びトレーサビリティが求められる。開発及び上市決定の
判断材料として,安全性に関する最終結論が適切に導かれたことを示す保証が必要となる。安全性評価は
それを立証するデータであり,評価の構成要素として科学的完全性の立証が必要となる。非臨床試験に適
用する品質システム管理としてGLPがある。GLP試験は,国際的な公的制度によって認定された試験機
関において,その機関が定める品質基準下で実施される試験である。GLP試験は,JIS Q 17025又は同等
の基準に適合した試験機関の品質基準下で実施される。
B.4.5.3 生体適合性評価の文書化
生体適合性評価の文書には,次の事項を含める。
41
T 0993-1:2020 (ISO 10993-1:2018)
− 医療機器の一般的な説明又は図面
− 5.2で定義されている身体に接触する全ての医療機器の材料,構成及び配合に関する定量的な情報,並
びに物理学的特性に関する定性的又は定量的な情報
− 製造において混入物質の発生の可能性がある加工条件の説明
− 5.2で定義されている身体に接触する医療機器の各部品に関連する毒性データ及び臨床使用実績の精
査
− 生物学的試験の報告書
− データの評価
− リスク分析及びリスクコントロールの完了を宣言する陳述書
収集された情報は,設計管理プロセスの一部として医療機器の設計文書とすることが望ましい(例えば,
JIS Q 13485:2018の箇条7)。また,リスクマネジメントファイル(JIS T 14971:2012の2.23参照)の構成
要素として扱うことが望ましい。非臨床試験及び臨床試験は,設計検証及びバリデーションの一部である
(例えば,それぞれJIS Q 13485:2018の7.3.6及び7.3.7)。JIS Q 13485の設計管理に則した製品設計の文
書には,生物学的安全性の要件を含めた設計の要求事項並びに設計された医療機器の要求事項への適否を
検証する非臨床試験,臨床試験及び設計レビューの記録が含まれる。
42
T 0993-1:2020 (ISO 10993-1:2018)
附属書C
(参考)
推奨する文献精査の手順
C.1 一般
医用材料又は医療機器の生物学的評価の計画立案及びその妥当性判断のために,文献の精査及び評価は
必須である。文献精査の目的は,当該生物学的評価の科学的な根拠を構築することにある。また,文献精
査は,医療機器のリスク及び効用の検証に不可欠な情報を提供するとともに,計画した評価をISO 10993-2
が要求する倫理性をもって実行する上でも必要となる。
注記 文献精査は,医療機器の生物学的安全性を示すために,関連データが文献から十分に入手可能
であり,更なる試験の実施を不要と決定するか,又は入手データでは不十分であると結論を導
くために役立てることができる。
文献精査は,厳密性及び客観性をもって科学的に実施することが望ましく,第三者による検証に耐える
内容であることが望ましい。
C.2 手順
C.2.1 概要
文献精査に先立ち,入手可能な全ての研究成果及びデータを特定,選択,照合及び精査する計画を作成
することが望ましい。これらの計画は文書化した後,対象とした科学文献について系統的な精査を実施す
る。
C.2.2 目的
文献精査の目的は,明瞭に定めることが望ましい。医療機器又は材料について既存の知見を考慮した上
で,定めた目的に合致する研究項目を特定する。
C.2.3 文書の選択基準
データの選択又は除外基準の設定は,妥当性のある根拠に基づき実施する。公表データは,定評のある
科学出版物から引用することが望ましい。非GLPデータよりもGLPに適合して取得したデータを優先す
る。出版バイアスを回避するために,未公表の関連データなど入手可能な全てを対象にすることが望まし
い。全てのデータは引用元を明確化する。
精査に使用した文献及びデータの出典のデータベース,一覧などを作成して調査した範囲を明示する。
C.2.4 文書の評価
文献精査においては,医療機器の使用目的を考慮した上で,文書の質,並びに文献と医療機器又は材料
の特徴及び性質との関連性を明確に評価する。
43
T 0993-1:2020 (ISO 10993-1:2018)
考慮するのがよい具体的な事項を次に示す。
a) 適用した文献の妥当性を評価するために,技術内容,主要性能,設計及び動作原理に基づいた考察か
ら,選択した文献が対象とする医療機器又は材料と生物学的安全性評価を行う医療機器との間の同等
性を示す。
b) 選択した文書において試験に用いられた実験動物が,生物学的安全性評価を行う医療機器又は材料に
適切である。
c) 選択した文書が対象とする医療機器又は材料の使用条件が生物学的安全性評価を行う医療機器の使用
目的に合致している。
C.2.5 厳格な文献評価
文献精査においては,試験デザインの異なる試験間,及び公表・未公表データ間の重要度を定量的に評
価するため,重み付けする。未公表データを評価に用いる場合には,その妥当性を文献精査で明らかにす
る必要がある。
考慮すべき要素としては,次の事項が挙げられる。
− 著者の結論が,得られたデータで裏付けられている。
− 文献内容は,現時点の医療実態及び最新技術を反映している。
− 文献は定評ある科学雑誌から引用されている,又は査読が行われる定評のある科学雑誌に掲載されて
いる。
− 公表された文献が,科学的な原理に基づいた試験結果を反映している。
文献精査では,文献の厳格な評価を実施することが望ましい。文書を入手・評価した後に,選択基準と
照合し,文献を採択又は除外した妥当性を明らかにする。文献精査が当該医療機器及びその使用目的に合
致していれば,精査は終了である。最終的に次の内容から構成される文献精査報告書を作成する。
− 材料又は医療機器の簡潔な説明(使用目的を含む。)
− 選択した全ての文献及びデータの解析(有利なデータ及び不利なデータの両方を含む。)
− 有害性並びにそれに伴うリスク及び適切な安全対策の厳格な評価
− 異なる文献の重み付け方法の記載。特に,同一著者の文献を複数引用する場合には,同一試験が過大
に重み付けされないように注意する。
− 評価に用いた引用文献の一覧
− 結論及びその妥当性。当該医療機器の安全性及び性能に関する全ての見地からの情報を充足するため
に,文献精査の目的の達成方法,及び不足根拠の補足方法を明確化する。
− 文献精査の実施者の署名及び日付
44
T 0993-1:2020 (ISO 10993-1:2018)
参考文献
[1] JIS T 6001 歯科用医療機器の生体適合性の評価
注記 原国際規格では,ISO 7405,Dentistry−Evaluation of biocompatibility of medical devices used in
dentistryを記載している。
[2] JIS Q 9000 品質マネジメントシステム−基本及び用語
注記 原国際規格では,ISO 9000,Quality management systems−Fundamentals and vocabularyを記載
している。
[3] JIS Q 9001 品質マネジメントシステム−要求事項
注記 原国際規格では,ISO 9001,Quality management systems−Requirementsを記載している。
[4] JIS Q 9004 品質マネジメント−組織の品質−持続的成功を達成するための指針
注記 原国際規格では,ISO 9004,Quality management−Quality of an organization−Guidance to achieve
sustained successを記載している。
[5] ISO/TR 10993-22,Biological evaluation of medical devices−Part 22: Guidance on nanomaterials
[6] ISO/TR 10993-33,Biological evaluation of medical devices−Part 33: Guidance on tests to evaluate
genotoxicity−Supplement to ISO 10993-3
[7] JIS Q 13485:2018 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 原国際規格では,ISO 13485:2016,Medical devices−Quality management systems−Requirements
for regulatory purposesを記載している。
[8] JIS Q 17025 試験所及び校正機関の能力に関する一般要求事項
注記 原国際規格では,ISO/IEC 17025,General requirements for the competence of testing and
calibration laboratoriesを記載している。
[9] ISO 18562 (all parts),Biocompatibility evaluation of breathing gas pathways in healthcare applications
[10] Previews B.I.O.S.I.S. Ovid Technologies, Inc.
[11] Guideline on the limits of genotoxic impurities, European Medicines Agency Evaluation of Medicines for
Human Use (EMEA)
[12] Black, J., Biological Performance of Materials: Fundamentals of Biocompatibility, CRC Press, 2006
[13] Boutrand J. ed. Biocompatibility and Performance of Medical Devices. Woodhead Publishing, 2012
[14] Bush R.B. A Bibliography of Monographic Works on Biomaterials and Biocompatibility: Update II. J. Biomed.
Mater. Res. 1999, 48 pp. 335-341 [Appl Biomater]
[15] Tinkler J.J.B. Biological Safety and European Medical Device Regulations. Quality First International Press,
London, 2000
[16] Williams D.F. Fundamental aspects of biocompatibility. Biocompatibility. 1 CRC. 1980
[17] Williams D.F. Definitions in Biomaterials. Progress in Biomedical Engineering. 1987, 4 pp. 1-72
[18] EMBASE. Elsevier B.V.
[19] IPCS. World Health Organization
[20] IRIS. U.S. Environmental Protection Agency
[21] PubMed. U.S. National Library of Medicine
[22] SciFinder, American Chemical Society
45
T 0993-1:2020 (ISO 10993-1:2018)
[23] SciSearch®−A Cited Reference Science Database, Dialog, LLC.
[24] Toxnet. U.S. National Library of Medicine
[25] ToxGuides. Agency for Toxic Substances & Disease Registry (ATSDR)
[26] OECD Guidelines for the Testing of Chemicals−Section 4: Health Effects
[27] 医療機器の安全性に関する非臨床試験の実施の基準に関する省令(平成17年厚生労働省令第37号及
び平成20年厚生労働省令第115号)
[28] 和英対訳 医療機器の製造販売承認申請等に必要な生物学的安全性評価の基本的考え方について,第
1版,東京,薬事日報社,2012年11月
[29] USA. GLP reference in the Federal Register
[30] Use of International Standard. ISO 10993-1, “Biological evaluation of medical devices−Part 1: Evaluation
and testing within a risk management process” Guidance for Industry and Food and Drug Administration Staff,
June 16, 2016
[31] ICH Q3A Impurities in New Drug Substances
[32] ICH Q3B Impurities in New Drug Products
[33] ICH Q3C Impurities: Guideline for Residual Solvents
[34] ICH Q3D Impurities: Guidelines for Elemental Impurities
[35] ICH M7 ASSESSMENT AND CONTROL OF DNA REACTIVE (MUTAGENIC) IMPURITIES IN
PHARMACEUTICALS TO LIMIT POTENTIAL CARCINOGENIC RISK.
[36] ISO/TR 10993-19,Biological evaluation of medical devices−Part 19: Physico-chemical, morphological and
topographical characterization of materials
[37] GHTF/SG1/N071:2012,Definition of the Terms 'Medical Device' and 'In Vitro Diagnostic (IVD) Medical
Device'