T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 2
3 用語及び定義 ··················································································································· 2
4 品質マネジメントシステムの要素 ························································································ 8
4.1 文書化 ························································································································· 8
4.2 経営者の責任 ················································································································ 8
4.3 製品実現 ······················································································································ 8
4.4 測定,分析及び改善−不適合製品の管理············································································· 8
5 滅菌剤の特性 ··················································································································· 8
5.1 滅菌剤 ························································································································· 8
5.2 微生物殺滅効果の有効性 ································································································· 8
5.3 材質への影響 ················································································································ 9
5.4 環境への配慮 ················································································································ 9
6 プロセス及び設備の特性 ···································································································· 9
6.1 プロセス ······················································································································ 9
6.2 設備 ···························································································································· 9
7 製品の定義 ····················································································································· 10
8 プロセスの定義 ··············································································································· 10
8.1 最大許容線量の確立 ······································································································ 10
8.2 滅菌線量の確立 ············································································································ 11
8.3 滅菌線量及び最大許容線量の規定····················································································· 11
8.4 線源間の最大許容線量,検定線量又は滅菌線量の移転 ·························································· 11
9 バリデーション ··············································································································· 12
9.1 据付適格性の確認(IQ) ································································································ 12
9.2 運転適格性の確認(OQ) ······························································································· 12
9.3 稼働性能適格性の確認(PQ) ························································································· 13
9.4 バリデーションのレビュー及び承認·················································································· 13
10 日常監視及び管理 ·········································································································· 14
11 滅菌からの製品のリリース ······························································································ 15
12 プロセス有効性の維持 ···································································································· 15
12.1 継続的な有効性の立証 ·································································································· 15
12.2 再校正 ······················································································································· 17
12.3 設備のメンテナンス ····································································································· 17
12.4 設備適格性の再確認 ····································································································· 17
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013) 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
12.5 変更の評価 ················································································································· 18
附属書A(参考)この規格の使用上の指針 ··············································································· 19
参考文献 ···························································································································· 32
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,一般社団法人日本
医療機器学会(JSMI)及び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格
を改正すべきとの申出があり,日本工業標準調査会の審議を経て,厚生労働大臣が改正した日本工業規格
である。
これによって,JIS T 0806-1:2010は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
JIS T 0806の規格群には,次に示す部編成がある。
JIS T 0806-1 第1部:医療機器の滅菌プロセスの開発,バリデーション及び日常管理の要求事項
JIS T 0806-2 第2部:滅菌線量の確立
JIS T 0806-3 第3部:線量測定にかかわる指針
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
T 0806-1:2015
(ISO 11137-1:2006,Amd.1:2013)
ヘルスケア製品の滅菌−放射線−
第1部:医療機器の滅菌プロセスの開発,
バリデーション及び日常管理の要求事項
Sterilization of health care products-Radiation-
Part 1: Requirements for development, validation and routine control
of a sterilization process for medical devices
序文
この規格は,2006年に第1版として発行されたISO 11137-1及びAmendment 1 (2013) を基に,技術的内
容を変更することなく作成した日本工業規格である。ただし,追補(amendment)については,編集し,
一体とした。
なお,この規格で点線の下線を施してある箇所は,対応国際規格にはない事項である。
1
適用範囲
1.1
この規格は,医療機器の放射線滅菌プロセスの開発,バリデーション及び日常管理の要求事項につ
いて規定する。
注記 この規格の適用範囲は,医療機器に限定しているが,それ以外の製品及び機器にも適用可能な
要求事項及び指針を規定している。
この規格では,次のいずれかの照射設備を用いる放射線滅菌プロセスを対象としている。
a) 放射性核種 60 Co又は137 Cs
b) 電子線照射設備
c) X線照射設備
1.2
この規格は,スクレイピー,牛海綿状脳症及びクロイツフェルト・ヤコブ病のような海綿状脳症を
引き起こす病原物質の不活化のプロセスに対する開発,バリデーション及び日常管理についての要求事項
は規定しない。
注記 上記の規格は,例えば,ISO 22442-1〜ISO 22442-3で規定されている。
1.2.1
この規格では,医療機器が“無菌”であることを表示するための特定の要求事項の詳細は規定しな
い。
注記 医療機器に“無菌”と表示するための国又は地域の規制要求事項に注意を払う必要がある。例
えば,EN 556-1又はANSI/AAMI ST67を参照。
1.2.2
この規格は,医療機器の製造の全ての段階を管理するための品質マネジメントシステムを規定する
ものではない。
注記 この規格では,製造時の完全な品質マネジメントシステムを提供することは要求していないが,
2
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
滅菌処理プロセスを管理するための必要最小限の品質マネジメントシステムの要素はこの文中
に適宜,準拠しなければならない事項として引用している(特に箇条4参照)。滅菌プロセスを
含む医療機器製造の全プロセスに関わる品質マネジメントシステムの規格(JIS Q 13485参照)
に注意しなければならない。
1.2.3
この規格では,バイオロジカルインジケータを放射線滅菌のバリデーション又は監視に使用するこ
と,及び製品のリリースのための薬局方による無菌試験は要求しない。
1.2.4
この規格では,照射施設の設計及び操業に関わる労働安全の要求事項は規定しない。
注記 放射線に関する主な法令として,放射線障害防止法及び電離放射線障害防止規則がある。
1.2.5
この規格では,使用済又は再処理した医療機器の滅菌に関わる要求事項は規定しない。
注記 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 11137-1:2006,Sterilization of health care products−Radiation−Part 1: Requirements for
development, validation and routine control of a sterilization process for medical devices及び
Amendment 1:2013(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”こ
とを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格のうちで,西暦年を付記してあるものは,記載の年の版を適用し,その後の改正版(追補を含む。)
は適用しない。西暦年の付記がない引用規格は,その最新版(追補を含む。)を適用する。
JIS Q 10012 計測マネジメントシステム−測定プロセス及び測定機器に関する要求事項
注記 対応国際規格:ISO 10012,Measurement management systems−Requirements for measurement
processes and measuring equipment(IDT)
JIS Q 13485:2005 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 対応国際規格:ISO 13485:2003,Medical devices−Quality management systems−Requirements
for regulatory purposes(IDT)
JIS T 0806-2:2014 ヘルスケア製品の滅菌−放射線−第2部:滅菌線量の確立
注記 対応国際規格:ISO 11137-2:2013,Sterilization of health care products−Radiation−Part 2:
Establishing the sterilization dose(MOD)
JIS T 11737-1 医療機器の滅菌−微生物学的方法−第1部:製品上の微生物群の測定方法
注記 対応国際規格:ISO 11737-1,Sterilization of medical devices−Microbiological methods−Part 1:
Determination of a population of microorganisms on products(IDT)
JIS T 11737-2 医療機器の滅菌−微生物学的方法−第2部:滅菌プロセスの定義,バリデーション及
び維持において実施する無菌性の試験
注記 対応国際規格:ISO 11737-2,Sterilization of medical devices−Microbiological methods−Part 2:
Tests of sterility performed in the definition, validation and maintenance of a sterilization process
(IDT)
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.1
吸収線量(absorbed dose)
線量(dose)
物質の単位質量当たりに付与された吸収エネルギーの量。
注記1 吸収線量の単位は,グレイ(Gy)。1 Gyは1 J/kgの吸収に相当する。
注記2 この規格では,線量は“吸収線量”の意味で使用する。
3.2
バイオバーデン(bioburden)
製品及び/又は無菌バリアシステムの上又は内部に存在する生育可能な微生物群(ISO/TS 11139参照)。
3.3
バイオロジカルインジケータ(biological indicator)
ある特定の滅菌プロセスに対して,一定の抵抗性を示す生育可能な微生物を含む試験システム(ISO/TS
11139参照)。
3.4
校正(calibration)
計器若しくは測定系の示す値,又は実量器若しくは標準物質の表す値と,標準によって実現される値と
の間の関係を確定する一連の作業(VIM:1993の6.11参照)。
3.5
変更管理(change control)
製品又は手順に対して提案した変更の適切性の評価及び決定(ISO/TS 11139参照)。
3.6
修正(correction)
検出された不適合を除去するための処置(JIS Q 9000参照)。
注記 是正処置(3.7)と合わせて,修正が行われることもある。
3.7
是正処置(corrective action)
検出された不適合又はその他の検出された望ましくない状況の原因を除去するための処置(JIS Q 9000
参照)。
注記1 不適合の要因は,一つ以上である。
注記2 予防処置(3.24)は,発生を未然に防止するためにとるのに対し,是正処置は,再発を防止
するためにとる。
注記3 是正処置は,修正(3.6)とは異なる。
3.8
D値(D value)
D10値(D10 value)
定められた条件下で,試験に用いる微生物数の90 %を不活化するのに要する時間又は放射線量(ISO/TS
11139参照)。
注記 JIS T 0806規格群では,D値は,90 %減少を達成するために必要とされる放射線量をいう。
3.9
開発(development)
4
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
仕様を作り上げる行為(ISO/TS 11139参照)。
3.10
線量分布評価(dose mapping)
あらかじめ定めた条件下で,照射した物質中の線量分布及び変動を測定し,評価すること。
3.11
線量計(dosimeter)
システムとして再現性があり,吸収線量の測定が可能な機器(ISO/TS 11139参照)。
3.12
線量測定(dosimetry)
線量計を使用して吸収線量を測定すること。
3.13
確立(establish)
理論的評価によって決定し,実験によって確認すること(ISO/TS 11139参照)。
3.14
許容外(fault)
あらかじめ定めた許容範囲から一つ又は複数のプロセスパラメータが外れること(ISO/TS 11139参照)。
3.15
ヘルスケア製品[health care product (s)]
体外診断用医療機器を含む医療機器又は生物製剤を含む医薬品(ISO/TS 11139参照)。
3.16
据付適格性の確認,IQ(installation qualification,IQ)
設備がその仕様を満たして提供され,かつ,据え付けられたことの証拠を取得して文書化するプロセス
(ISO/TS 11139参照)。
3.17
照射容器(irradiation container)
照射設備内に搬送される製品を入れるための入れ物。
注記 入れ物にはキャリヤ,カート,トレイ,製品カートン,パレット又はその他の容器がある。
3.18
照射業者(irradiator operator)
製品の照射に責任をもつ会社又は組織。
3.19
最大許容線量(maximum acceptable dose)
その製品の安全性,品質及び性能を損なわない最大の線量として,プロセス仕様で与えられる線量。
3.20
医療機器(medical device)
あらゆる計器,器械,用具,機械,器具,埋込み用具,体外診断薬,検定物質,ソフトウェア,材料又
はその他の同類のもの若しくは関連する物質であって,単独使用か組合せ使用かを問わず,製造業者が人
体への使用を意図し,その使用目的が次の一つ以上であり,
− 疾病の診断,予防,監視,治療又は緩和
− 負傷の診断,監視,治療,緩和又は補助
5
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 解剖学的又は生理学的なプロセスの検査,代替又は修復
− 生命支援又は維持
− 受胎調整
− 医療機器の殺菌
− 人体から採取される標本の体外試験法による医療目的のための情報提供
薬学,免疫学,又は新陳代謝の手段によって体内又は体表において意図したその主機能を達成することは
ないが,それらの手段によって機能の実現を補助するものである(JIS Q 13485参照)。
注記 JIS Q 13485によるこの定義は,医療機器規制国際整合化会議(the Global Harmonization Task
Force:GHTF 2002)によって作成された。
3.21
微生物(microorganism)
細菌,真菌,原虫及びウィルスを包含する微小体(ISO/TS 11139参照)。
注記 ある種の規格によっては,滅菌プロセスのバリデーション及び/又は日常管理において,上記
で定義した全てのタイプの微生物の不活化の滅菌プロセスの有効性を立証することを要求しな
いこともある。
3.22
運転適格性の確認,OQ(operational qualification,OQ)
据え付けた設備をその操作手順に従って用いたとき,あらかじめ定めた限度内で作動する証拠を取得し
文書化するプロセス(ISO/TS 11139参照)。
3.23
稼働性能適格性の確認,PQ(performance qualification,PQ)
操作手順によって据え付けられ,運転されている装置が,あらかじめ定めた判断基準に恒常的に適合し
て稼働し,その結果,仕様に適合する製品を生産することができるという証拠を取得し,文書化するプロ
セス(ISO/TS 11139参照)。
3.24
予防処置(preventive action)
起こり得る不適合又はその他の望ましくない起こり得る状況の原因を除去するための処置(JIS Q 9000
参照)。
注記1 起こり得る不適合の要因は,一つ以上である。
注記2 是正処置(3.7)は,再発を防止するためにとるのに対し,予防処置は,発生を未然に防止す
るためにとる。
3.25
一次製造業者(primary manufacturer)
マーケットに上市したとき医療機器の安全性及び性能だけでなく,その設計及び製造に責任をもつ組織。
3.26
プロセス中断(process interruption)
意図した又は意図しない照射プロセスの一時的停止。
3.27
プロセスパラメータ(process parameter)
あらかじめ定めたプロセス変数の値(ISO/TS 11139参照)。
6
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記 滅菌プロセスの仕様には,プロセスパラメータ及びその許容範囲が含まれる。
3.28
プロセス変数(process variable)
滅菌プロセスの条件で,その変化が微生物の殺滅効果に変動を与えるような条件。
例えば,時間,温度,圧力,濃度,湿度,波長。
3.29
処理カテゴリ(processing category)
一緒に滅菌できる異なる製品又は製品ファミリーの集合。
注記 処理カテゴリは,例えば,組成,密度又は要求線量に基づく。
3.30
製品(product)
プロセスの結果(JIS Q 9000参照)。
注記 この規格においては,製品は有形のものであり,原料,中間品,半組立て品及びヘルスケア製
品でもあり得る。
3.31
製品ファミリー(product family)
所定のプロセス条件を用いて滅菌が許容できる製品のグループ。
注記 放射線滅菌する製品ファミリーに属する個別製品のバイオバーデンは,類似の微生物の数とタ
イプとで構成されていなければならない。
3.32
適格性の再確認(requalification)
定義した滅菌プロセスが引き続き許容できることを確認するために,バリデーションの一部分を反復実
施すること(ISO/TS 11139参照)。
3.33
サービス(services)
外部から供給を受けるもので,設備が機能を発揮するのに必要なもの。
例えば,電気,水,圧縮空気,排水。
3.34
仕様書(specification)
要求事項を記述した文書。
3.35
あらかじめ定める(specify)
承認を受けた文書の中で詳細を明記すること(ISO/TS 11139参照)。
3.36
無菌(sterile)
生育可能な微生物が存在しないこと(ISO/TS 11139参照)。
3.37
無菌性(sterility)
生育可能な微生物が存在しない状態(ISO/TS 11139参照)。
注記 実際には,そのような微生物が存在しない絶対的な状態を証明することはできない[3.39(滅
7
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
菌)参照]。
3.38
無菌性保証水準,SAL(sterility assurance level,SAL)
滅菌後に,生育可能な1個の微生物が製品上に存在する確率(ISO/TS 11139参照)。
注記 SALは定量値として一般的に,10−3,10−6などと表す。この定量値を無菌性保証に適用すると
きは,SAL 10−6の方がより小さい数値であるが,SAL 10−3よりも高い無菌性保証である。
3.39
滅菌(sterilization)
製品を生育可能な微生物が存在しない状態にするために用いる,バリデートされたプロセス(ISO/TS
11139参照)。
注記 滅菌プロセスでは,微生物の不活化は,指数関数で表現される。したがって,個々の製品に存
在する生育可能な微生物は,確率の観点から表現が可能である。この確率は,非常に低い数に
減らすことはできるが,決してゼロに低減することはできない[3.38(無菌性保証水準)参照]。
3.40
滅菌線量(sterilization dose)
無菌性を達成するためにあらかじめ定めた要求事項を満足するための最小線量。
3.41
滅菌プロセス(sterilization process)
あらかじめ定めた無菌性についての要求事項を達成するための一連の活動又は操作(ISO/TS 11139参照)。
注記 この一連の活動又は操作には,(必要ならば)前処理,あらかじめ定めた条件下での滅菌剤への
ばく(曝)露及び必要な後処理の全てを含むが,滅菌プロセスに先立つ清浄化,消毒又は包装
などの操作は含まない。
3.42
滅菌剤(sterilizing agent)
あらかじめ定めた条件下で無菌性を達成するために十分な殺菌作用をもつ物理的若しくは化学的媒体又
はその組合せ(ISO/TS 11139参照)。
3.43
無菌試験(test for sterility)
滅菌プロセスを経た製品に対して実施する,薬局方で定義された技術的操作(ISO/TS 11139参照)。
3.44
無菌性の試験(test of sterility)
開発,バリデーション又は適格性の再確認の一部として実施する技術的操作で,製品又はその一部分に
生育可能な微生物の存在の有無を判定するために行う試験(ISO/TS 11139参照)。
3.45
トランジット線量(transit dose)
製品又は線源が非照射位置から照射位置までに移動する間に製品が吸収する線量。
3.46
測定の不確かさ(uncertainty of measurement)
測定量に起因すると考えられるばらつきを特徴付ける,測定の結果に関わるパラメータ(VIM:1993参
照)。
8
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.47
バリデーション(validation)
プロセスが,恒常的にあらかじめ定めた仕様に適合する製品を得ることができることを確立するために,
要求される結果を得て,記録し,解釈するための文書化した手順(ISO/TS 11139参照)。
4
品質マネジメントシステムの要素
4.1
文書化
4.1.1
開発,バリデーション,日常管理及び滅菌からの製品リリースの手順は,これらを文書化し,定め
なければならない。
4.1.2
この規格が要求する文書及び記録は,あらかじめ指名した職員(4.2.1参照)によってレビューし,
承認しなければならない。文書及び記録は,JIS Q 13485の該当する箇条によって管理しなければならない。
4.2
経営者の責任
4.2.1
この規格の要求事項を実施し,これに適合するための責任及び権限をあらかじめ定めなければなら
ない。責任は,JIS Q 13485の該当する箇条によって,力量のある職員に割り当てなければならない。
4.2.2
この規格の要求事項を,他の品質マネジメントシステムの組織によって実行する場合は,それぞれ
の組織の責任及び権限をあらかじめ定めなければならない。
4.3
製品実現
4.3.1
購買の手順をあらかじめ定めなければならない。これらの手順は,JIS Q 13485の該当する箇条に
適合しなければならない。
4.3.2
製品の識別及びトレーサビリティの手順をあらかじめ定めなければならない。この手順は,JIS Q
13485の該当する箇条に適合しなければならない。
4.3.3
この規格の要求事項に適合するために,試験用の計器を含む全ての機器の校正について,JIS Q
13485又はJIS Q 10012の該当する箇条に適合したシステムを構築しなければならない。
4.3.4
滅菌プロセスの開発,バリデーション及び日常管理に使用する線量測定機器は,国際又は国家計量
標準にトレースが可能であり,測定の不確かさの水準が分かっていなければならない。
4.4
測定,分析及び改善−不適合製品の管理
不適合と認定した製品の管理,修正,是正処置及び予防処置の手順をあらかじめ定めなければならない。
これらの手順は,JIS Q 13485の該当する箇条に適合しなければならない。
5
滅菌剤の特性
5.1
滅菌剤
5.1.1
滅菌プロセスに使用する放射線の種類を定めなければならない。
5.1.2
電子線又はX線の場合には,電子線のエネルギーを定めなければならない。電子線においてエネ
ルギーが10 MeVを超える場合,又はX線を発生させる電子線のエネルギーが5 MeVを超える場合には,
製品に誘導放射能が発生する可能性を検証し,検証結果及びその決定に至った根拠を文書化しなければな
らない。
5.2
微生物殺滅効果の有効性
放射線で微生物を不活化し,これを滅菌プロセスへ適用することについては,多くの文献がある。これ
らの文献は,微生物の不活化に影響するプロセス変数についての情報を記載している。この規格では,微
生物の不活化に関わるこれらの一般的な研究を参照することは要求しない。
9
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.3
材質への影響
医療機器の製造に使用する広範な材質に対し,放射線が及ぼす影響についての包括的な文献があり,こ
れらの文献は,放射線滅菌する医療機器の設計及び開発に携わる者に役立つ。この規格では,材質への影
響についての研究を行うことは要求しないが,製品自体に対する放射線の影響についての評価は要求する
(8.1参照)。
5.4
環境への配慮
放射線滅菌プロセスを実施することで環境に与える潜在的な影響について評価し,環境を保護する方法
を特定しなければならない。潜在的な影響(ある場合)を含めて評価し,その結果を文書化し,管理のた
めの方法(特定した場合)を定め,実施しなければならない。
6
プロセス及び設備の特性
6.1
プロセス
プロセス変数を特定し,これらを監視及び管理する方法を定めなければならない。
6.2
設備
6.2.1
照射設備及びその運転方法を定めなければならない。照射設備の仕様は,必要に応じて改訂し
(12.5.1参照),照射設備の全使用期間にわたって保管しなければならない(4.1.2参照)。
6.2.2
プロセス管理及び/又は監視を行うソフトウェアは,そのソフトウェアが設計意図に適合している
ことを,文書による証拠として提示できるように,品質マネジメントシステムに基づいて準備しなければ
ならない。
6.2.3
ガンマ線照射設備の場合は,仕様には,少なくとも次の事項を記載しなければならない。
a) 照射設備及びその特徴
b) 放射性核種,ガンマ線源の放射能及び配置
c) 照射設備の位置を含む建屋図面
d) 未照射製品と照射済製品とを分離するための方法(10.3及び10.4参照)
e) 関連するコンベヤシステムの構造及び機能
f)
コンベヤの経路及びコンベヤ速度の範囲
g) 照射容器の寸法,材質及び構造
h) 照射設備及び関連するコンベヤシステムの運転方法及びメンテナンス方法
i)
ガンマ線源の位置を表示する方法
j)
プロセス管理用タイマ又はコンベヤシステムの故障時に,ガンマ線源を自動的に格納位置に戻し,か
つ,自動的にコンベヤを停止させる方法
k) ガンマ線源があらかじめ定めた位置にない場合には,ガンマ線源を自動的に格納位置に戻し,かつ,
自動的にコンベヤを停止させる方法又は影響を受けた製品を判別する方法
6.2.4
電子線照射設備の場合は,仕様には,少なくとも次の事項を記載しなければならない。
a) 照射設備及びその特徴
b) ビーム特性(電子エネルギー,平均ビーム電流,スキャン幅及びスキャン均一度)
c) 照射設備の位置を含む建屋図面
d) 未照射製品と照射済製品とを分離するための方法(10.3及び10.4参照)
e) 関連するコンベヤシステムの構造及び機能
f)
コンベヤの経路及びコンベヤ速度の範囲
10
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) 照射容器の寸法,材質及び構造
h) 照射設備及び関連するコンベヤシステムの運転方法及びメンテナンス方法
i)
照射設備及びコンベヤが稼働していることを示す方法
j)
線量に影響するようなコンベヤの故障時に,照射を停止させる方法
k) ビームが許容外になった場合に,コンベヤを停止させる方法又は影響を受けた製品を判別する方法
6.2.5
X線照射設備の場合は,仕様には,少なくとも次の事項を記載しなければならない。
a) 照射設備及びその特徴
b) ビーム特性(電子又はX線エネルギー,平均ビーム電流,スキャン幅及びスキャン均一度)
c) X線変換器の寸法,材質及び構造
d) 照射設備の位置を含む建屋図面
e) 未照射製品と照射済製品とを分離するための方法(10.3及び10.4参照)
f)
関連するコンベヤシステムの構造及び機能
g) コンベヤの経路及びコンベヤ速度の範囲
h) 照射容器の寸法,材質及び構造
i)
照射設備及び関連するコンベヤシステムの運転方法及びメンテナンス方法
j)
照射設備及びコンベヤが稼働していることを示す方法
k) 線量に影響するようなコンベヤの故障時に,照射を停止させる方法
l)
ビームが許容外になった場合に,コンベヤを停止させる方法又は影響を受けた製品を判別する方法
m) ターゲットの冷却システムが故障した場合に,照射を中止する方法
7
製品の定義
7.1
滅菌する製品は,その包装材料も含めて定義しなければならない。
7.2
製品,包装又は包装内の製品の配置に変更があるときは,これを定義しなければならない(12.5.2
参照)。
7.3
滅菌プロセスの有効性が損なわれないように,バイオバーデンを含めて滅菌しようとする製品の条
件が管理されていることを保証するシステムを定め,かつ,実施しなければならない。このシステムの有
効性を立証し,かつ,JIS T 11737-1に準拠したバイオバーデンの決定を含まなければならない。
7.4
製品ファミリーについて滅菌線量を確立した場合には,JIS T 0806-2:2014に示す製品ファミリーを
定義するための要求事項に適合しなければならない。
7.5
日常処理に処理カテゴリを適用した場合には,対象製品がその処理カテゴリに属するかどうかを文
書化した基準によって評価しなければならない。この評価においては,製品に照射する線量及び処理仕様
に影響を与える製品に関わる変動因子を考慮しなければならない。評価結果は,記録しなければならない
(4.1.2参照)。
7.6
処理カテゴリに含めるための製品の評価についての基準及び処理カテゴリを構成する製品のグルー
プについて,定期的なレビューを実施しなければならない。レビューの結果は,記録しなければならない
(4.1.2参照)。
8
プロセスの定義
8.1
最大許容線量の確立
11
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.1.1
製品の最大許容線量を確立しなければならない。最大許容線量で処理した場合でも,製品はあらか
じめ定めた有効期間中,機能的な要求事項に適合しなければならない。
8.1.2
最大許容線量を確立するための基本的な技術的要求事項には,次の事項を含めなければならない。
a) 製品の意図する機能について評価可能な施設
b) 日常的に生産する代表的な製品
c) 要求した線量を正確,かつ,精度よく照射できる適切な線源(8.4.1参照)
8.2
滅菌線量の確立
8.2.1
製品の滅菌線量を確立しなければならない。
8.2.2
滅菌線量の確立に当たっては,次のa) 又はb) のいずれかの方法によらなければならない。
a) バイオバーデンの数及び/又は放射線抵抗性についての知見を得た上で,それに基づいて滅菌線量を
設定する。
注記1 滅菌線量の設定方法及びそれらの方法を適用する環境については,JIS T 0806-2:2014に詳
しく規定している。
b) 25 kGy,15 kGy又はVDmaxSDの滅菌線量を選定し,実証する。一次製造業者が,25 kGy,15 kGy又は
VDmaxSDの滅菌線量を実証する場合には,選定した滅菌線量が無菌性についてあらかじめ定めた要求
事項を達成できるという証拠がなければならない(1.2.2参照)。
注記2 VDmax25法,VDmax15法及びVDmaxSD法で滅菌線量を実証する方法及びこれらの方法を適用
する詳細は,JIS T 0806-2:2014に規定している。VDmax25法,VDmax15法及びVDmaxSD法で
は,10−6の無菌性保証水準を達成することになる。
注記3 この規格では,ISO/TS 13004を翻訳して規定しているJIS T 0806-2の附属書JAのVDmaxSD
法を適用している。
8.2.3
滅菌線量を確立するための基本的な技術的要求事項には,次の事項を含めなければならない。
a) JIS T 11737-1に準拠するバイオバーデンの決定及びJIS T 11737-2に準拠する無菌性の試験を実施す
る能力をもつ微生物検査施設
b) 日常的に生産する代表的な製品
c) 要求した線量を正確,かつ,精度よく照射できる適切な線源(8.4.2参照)
注記 放射線滅菌の線量測定に関わる指針を,JIS T 0806-3に示す。
8.3
滅菌線量及び最大許容線量の規定
製品に対して滅菌線量及び最大許容線量を定めなければならない。
8.4
線源間の最大許容線量,検定線量又は滅菌線量の移転
8.4.1
最大許容線量の移転
最大許容線量を当初確立した線源から別種の線源に移転する場合には,二つの線源間における照射条件
の差異が線量の有効性に影響を及ぼさないことを立証しなければならない。結果は文書化し,記録しなけ
ればならない(4.1.2参照)。
8.4.2
検定線量又は滅菌線量の移転
8.4.2.1
次のいずれかの場合を除き,検定線量又は滅菌線量を当初確立した線源から別種の線源に移転し
てはならない。
a) 二つの線源間の運転条件の差異が,微生物殺滅効果に影響を与えないことを立証するデータがある場
合。
b) 8.4.2.2又は8.4.2.3を適用する場合。
12
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.4.2.2
液状の水分を含まない製品の場合には,次に示すいずれかの施設間では,検定線量又は滅菌線量
の移転は可能である。
a) ガンマ線照射設備から別のガンマ線照射設備
b) 電子線照射設備から別の電子線照射設備
c) X線照射設備から別のX線照射設備
8.4.2.3
液状の水分を含む製品の場合には,次に示すいずれかの施設間では,検定線量又は滅菌線量の移
転は可能である。
a) ガンマ線照射設備から別のガンマ線照射設備
b) 同一の運転条件で運転する二つの電子線照射設備
c) 同一の運転条件で運転する二つのX線照射設備
9
バリデーション
9.1
据付適格性の確認(IQ)
9.1.1
照射設備及び関連するコンベヤの運転手順を定めなければならない。
9.1.2
プロセス及び関連するソフトウェアを含む付帯設備は,設計仕様に従った運転ができるか検証する
ために試験しなければならない。試験方法は,文書化し,結果は記録しなければならない(4.1.2参照)。
9.1.3
据付時に行った照射設備の変更は,文書化しなければならない(6.2.1参照)。
9.1.4
ガンマ線照射設備の場合には,線源の放射能及び個々の線源の位置を記録しなければならない
(4.1.2参照)。
9.1.5
電子線照射設備の場合には,ビーム特性(電子エネルギー,平均ビーム電流,スキャン幅及びスキ
ャン均一度)を決定し,記録しなければならない(4.1.2参照)。
9.1.6
X線照射設備の場合には,ビーム特性(電子又はX線エネルギー,平均ビーム電流,スキャン幅
及びスキャン均一度)を決定し,記録しなければならない(4.1.2参照)。
9.2
運転適格性の確認(OQ)
9.2.1
OQに先立ち,監視,制御,表示又は記録に使用する試験計器を含む全ての測定機器が校正してあ
ること確認しなければならない(4.3.3参照)。
9.2.2
OQでは,製品を代表する均一な物質を照射することで,設備があらかじめ定めた滅菌プロセスが
要求する範囲の線量を与える能力を備えていることを立証しなければならない(箇条8参照)。OQでは,
設置した照射設備が,あらかじめ定められた許容基準範囲内で適切な線量を照射し,運転できる能力があ
ることを立証しなければならない。
9.2.3
線量の分布(9.2.4参照)及び変動(9.2.5参照)に関わる照射設備の特性を把握するために,線量
分布評価を実施しなければならない。
注記 線量分布評価の指針を,JIS T 0806-3に示す。
9.2.4
線量分布評価は,1個の照射容器に均一な密度の物質を設計仕様の上限まで載荷して実施しなけれ
ばならない。線量を決定するため,物質中の様々な場所に線量計を配置しなければならない。線量分布評
価を実施している期間中,照射設備が最大負荷となるように線量分布評価を行っている照射容器と同じ物
質を,設計仕様の上限まで載荷した十分な数の照射容器を同時に照射しなければならない。
9.2.5
線量分布評価は,照射容器間の線量の分布及び変動を決定できるように,十分な数の照射容器を用
いて実施しなければならない。
13
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.6
コンベヤの経路が二つ以上ある場合には,線量分布評価は,製品処理に使用する経路ごとに実施し
なければならない。
9.2.7
プロセス中断による線量への影響を決定し,記録しなければならない(4.1.2参照)。
9.2.8
線量分布評価の記録には,照射容器,照射設備の運転条件,使用した物質,線量測定値及び判定結
果を含まなければならない(4.1.2参照)。
9.2.9
ガンマ線照射設備の場合には,タイマ設定及び/又はコンベヤ速度と線量との関係を確立しなけれ
ばならない。
9.2.10 電子線及びX線照射設備の場合には,線量分布評価を実施している期間中のビーム特性(9.1.5又
は9.1.6参照)の変動は,照射設備の仕様限度範囲内でなければならない(6.2.4又は6.2.5参照)。
9.2.11 電子線及びX線照射設備の場合には,ビーム特性(9.1.5及び9.1.6参照),コンベヤ速度及び線量
との関係を確立しなければならない。
9.3
稼働性能適格性の確認(PQ)
9.3.1
線量分布評価は,次のことを実施するためにあらかじめ定めた載荷形態に従って製品を載荷した照
射容器を用いて実施しなければならない。
a) 最大線量及び最小線量の位置及び変動の特定
b) 最大線量及び最小線量並びに日常監視点での線量との関係の決定
9.3.2
製品を滅菌プロセスで処理する方法を定めなければならない。これには,次の事項を含めなければ
ならない。
a) こん(梱)包製品の寸法及び密度
b) こん(梱)包内の製品の並べ方
c) 照射容器についての事項(照射設備内で複数の形式の照射容器を使用する場合)
d) コンベヤ経路についての事項(照射設備内で複数のコンベヤ経路を使用する場合)
9.3.3
線量分布評価は,各処理カテゴリについて実施しなければならない(7.5参照)。
9.3.4
日常の処理で製品を部分的に載荷した照射容器を用いる場合には,この部分載荷による影響が,次
の事項に与える影響を決定し,記録しなければならない(4.1.2参照)。
a) 照射容器内の線量分布
b) 照射設備内に存在する他の照射容器の線量及び線量分布
9.3.5
線量分布評価は,照射容器間の線量の変動を決定できるよう十分な数の照射容器について実施しな
ければならない。
9.3.6
線量分布評価は,製品を処理するために使用するそれぞれのコンベヤ経路について実施しなければ
ならない。
9.3.7
ガンマ線及びX線照射設備では,線量分布評価をしている製品と同時に処理可能な製品(適用し
ている場合は処理カテゴリ)を特定するために線量分布評価を実施しなければならない。どの製品が同時
に処理可能であるかを明確にするために,照射設備内に存在する異なった密度の製品が線量に与える影響
を決定しなければならない。
9.3.8
線量分布評価の記録には,照射容器,載荷形態,コンベヤ経路,照射設備の運転条件,線量測定値
及び結論を含めなければならない(4.1.2参照)。
9.4
バリデーションのレビュー及び承認
9.4.1
据付適格性の確認(IQ),運転適格性の確認(OQ)及び稼働性能適格性の確認(PQ)で得られた
情報は,レビューし,その結果を,記録しなければならない(4.1.2参照)。
14
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.4.2
情報の考察及びレビューによって,製品のプロセス仕様を設定しなければならない(4.1.2参照)。
9.4.3
ガンマ線照射設備の場合には,プロセス仕様に次の事項を含めなければならない。
a) こん(梱)包製品の寸法,密度,こん(梱)包内の製品の並べ方(箇条7及び9.3.2参照)及びそれら
の許容変動範囲を含むこん(梱)包製品に関わる事項
b) 照射容器内の製品の載荷形態(9.3.1参照)
c) 使用するコンベヤ経路(9.3.6参照)
d) 最大許容線量(8.1参照)
e) 滅菌線量(8.2参照)
f)
微生物が生育可能な状態にある製品では,製造から照射完了までの最大時間
g) 日常監視線量計の配置位置
h) 日常監視線量計の線量と最小及び最大線量との関係(9.3.1参照)
i)
照射域において複数回の照射が必要な製品では,照射ごとに要求される製品再配置
9.4.4
電子線及びX線照射設備の場合には,プロセス仕様に次の事項を含めなければならない。
a) こん(梱)包製品の寸法,密度,こん(梱)包内の製品の並べ方(箇条7及び9.3.2参照)及びそれら
の許容変動範囲を含むこん(梱)包製品に関わる事項
b) 照射容器内の製品の載荷形態(9.3.1参照)
c) 使用するコンベヤ経路(9.3.6参照)
d) 最大許容線量(8.1参照)
e) 滅菌線量(8.2参照)
f)
微生物が生育可能な状態にある製品では,製造から照射完了までの最大時間
g) 日常監視線量計の配置位置
h) 日常監視線量計の線量と最小及び最大線量との関係(9.3.1参照)
i)
照射設備の運転条件及びその限度(例えば,ビーム特性及びコンベヤ速度)
j)
照射域において複数回の照射が必要な製品では,照射ごとに要求される製品再配置
10 日常監視及び管理
10.1 照射前,照射中及び照射後の製品の完全性を維持するため,製品の取扱いの手順を定めなければな
らない。
10.2 製品受領,載荷,荷卸,取扱い及び出荷までの全プロセスを通じて製品を数え,その製品数を確認
するシステムを構築し,実施しなければならない。いかなる数量の不一致も照射前及び/又は出荷前まで
に解決しなければならない。
10.3 未照射製品と照射済製品とは分離しなければならない。
10.4 放射線に反応して色調変化するインジケータは,適正に放射線処理を実施したことの証明又は未照
射製品と照射済製品とを判別するための唯一の方法として使用してはならない。
10.5 製品は,プロセス仕様(9.4.3又は9.4.4参照)に従って照射容器に載荷しなければならない。
10.6 線量計は,あらかじめ定めた日常監視点に配置しなければならない。照射後に線量計の測定,結果
の記録(4.1.2参照)及び評価を行わなければならない。
10.7 線量計の配置頻度は,プロセスが管理されていることを確認するのに十分な数でなければならない。
頻度及びその根拠を定めなければならない。
10.8 ガンマ線照射設備の場合には,次の事項を実施しなければならない。
15
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) タイマ設定及び/又はコンベヤ速度は,線源の減衰を考慮して文書化した手順に基づき調整する。
b) 線源位置,タイマ設定及び/又はコンベヤ速度,及び照射容器の移動状況を監視し,記録する(4.1.2
参照)。
10.9 電子線及びX線照射設備の場合には,電子ビーム特性(9.1.5及び9.1.6参照)及びコンベヤ速度を
監視し,記録しなければならない(4.1.2参照)。
10.10
プロセス中断及び/又はプロセスの不具合が発生した場合には,講じた措置とともにその内容を
記録しなければならない(4.1.2参照)。
10.11
照射プロセスの記録には,照射日を記載して,照射バッチにトレース可能なようにしなければな
らない(4.3.2参照)。
11 滅菌からの製品のリリース
11.1 製品の滅菌プロセスからのリリースに先立ち,あらかじめ定めた定期的な試験,校正,メンテナン
ス作業及び必要な適格性の再確認を全て実施し,結果を記録しなければならない(4.1.2参照)。
11.2 記録のレビュー及び滅菌後の製品をリリースする手順を定めなければならない(4.1.2参照)。この手
順書には,測定システムの不確かさを考慮した上で,滅菌プロセスが適正に行われたと認定するための要
求事項(該当する場合には,9.4.3及び9.4.4を参照)を定めなければならない。これらの要求事項に適合
しない場合には,製品は不合格とし4.4に従って取り扱わなければならない。
製品を滅菌済み製品としてリリースし,配送するためには,これに加えて品質マネジメントシステム(JIS
Q 13485参照)に規定された製造記録及び検査記録が必要になる場合もある。
12 プロセス有効性の維持
12.1 継続的な有効性の立証
12.1.1 一般
確立した滅菌線量の有効性が継続していることを,次の事項を実施して立証しなければならない。
a) 製品上に存在する微生物数と,あらかじめ定めたバイオバーデンの限度との関係を監視するためのバ
イオバーデンの決定
b) 製品上に存在するバイオバーデンの放射線抵抗性を監視するための滅菌線量監査
注記 JIS T 0806-2:2014に規定した滅菌線量監査の実施方法には,バイオバーデンの決定を実施す
ることが記載されている。
c) VDmaxSD法の場合は,JIS T 0806-2:2014のJA.4.2.2による。
12.1.2 バイオバーデンの決定頻度
12.1.2.1 平均バイオバーデンが1.5以上の製品の場合には,バイオバーデンの決定の最大間隔は,3か月
としなければならない。
12.1.2.2 平均バイオバーデンが1.5未満の製品で,次のいずれかに該当する場合には,バイオバーデンの
決定の最大間隔は,3か月としなければならない。
a) 滅菌線量を方法2(JIS T 0806-2:2014参照)で設定した場合
b) 25 kGyを滅菌線量として選定した場合(8.2.2参照)
12.1.2.3 平均バイオバーデンが1.5未満の製品で,次のいずれかに該当する場合には,バイオバーデンの
決定の最大間隔は,1か月としなければならない。
a) 滅菌線量を方法1(JIS T 0806-2:2014参照)で設定した場合
16
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 15 kGyを滅菌線量として選定した場合(8.2.2参照)
12.1.2.4 製品バッチの製造間隔が,該当する1か月又は3か月(12.1.2.1〜12.1.2.3参照)よりも長い場合
には,バイオバーデンの決定は,バッチごとに実施しなければならない。
12.1.2.5 バイオバーデンの決定結果があらかじめ定めた限度を超えた場合には,JIS T 11737-1に従って調
査しなければならない。調査によってバイオバーデンの決定結果が正しいと判明した場合には,4.4に規定
された手順を実行し,滅菌線量監査を直ちに実施しなければならない。滅菌線量監査の結果によって,次
のいずれかを実施しなければならない。
a) 滅菌線量監査に失敗した場合には,12.1.3.5に従って作業を実施しなければならない。
b) 滅菌線量監査は合格であって,バイオバーデンがあらかじめ定めた限度を超え続けている場合には,
滅菌線量監査の前に適用していた線量を用いて引き続き滅菌を行わなければならない。また,次の事
項を実施しなければならない。
1) 滅菌線量を方法1(JIS T 0806-2:2014参照)で確立した場合は,バイオバーデンがあらかじめ定め
た限度に戻す,又は滅菌線量が再確立されるまでは,滅菌線量監査の間隔を3か月としなければな
らない。
2) 滅菌線量を方法2(JIS T 0806-2:2014参照)で確立した場合は,12.1.3.2に適合する条件が達成する
までは,滅菌線量監査の間隔を3か月としなければならない。
3) 25 kGyの滅菌線量を選定し,VDmax25法で実証し,かつ,平均バイオバーデンが1 000以下の場合に
は,現在行っている滅菌線量監査の間隔を引き続き適用しなければならない。
4) 25 kGyの滅菌線量を選定し,VDmax25法で実証し,かつ,平均バイオバーデンが1 000を超える場合
には,別の方法を用いて滅菌線量を確立しなければならない。
5) 15 kGyの滅菌線量を選定し,VDmax15法で実証し,かつ,平均バイオバーデンが1.5以下の場合には,
現在行っている滅菌線量監査の間隔を引き続き適用しなければならない。
6) 15 kGyの滅菌線量を選定し,VDmax15法で実証し,かつ,平均バイオバーデンが1.5を超える場合に
は別の方法を用いて滅菌線量を確立しなければならない。
7) VDmaxSD法の場合は,JIS T 0806-2:2014のJA.4による。
12.1.3 滅菌線量監査の頻度
12.1.3.1 最初に滅菌線量監査を実施する間隔を決定するために,次のa) 又はb) のいずれかを実施しなけ
ればならない。
a) 滅菌線量監査の間隔を3か月とする。
b) 最初に選定した滅菌線量監査の間隔の根拠を作成し,文書化する。根拠を作成する場合には,少なく
とも次の事項に関わるレビュー及び得られた結論を記録しなければならない。
1) あらかじめ定めたバイオバーデンの限度
2) バイオバーデンの決定から得られる利用可能なデータ,これらのデータを取得した期間及びバイオ
バーデンを構成する微生物の特徴
注記 例えば,特徴は,コロニー若しくは細胞の形態,染色特性又は選択培地での発育
3) バイオバーデンを構成する微生物の抵抗性に関わる利用可能なデータ
4) 滅菌線量を確立するのに用いた方法及びその方法についての安全度
5) 日常処理で適用する線量と滅菌線量との差及びその差異についての安全度
6) 製品を構成する材料,特に天然由来材料の使用及びその材料の微生物学的品質管理
7) 製造プロセス,特にバイオバーデン又は抵抗性に影響する製造プロセス
17
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8) 製造プロセスでの管理及び監視手順
9) 製品の製造バッチ間隔
10) 製造環境,特に微生物管理及び監視の程度並びに該当期間の製造環境の安定性に関わる利用可能な
データ
11) 製造区域内の作業者の健康,清潔度及び着衣の管理
12) 同一の製品ファミリーに属する他の製品の微生物学的品質に関わる利用可能なデータ
12.1.3.2 滅菌線量監査の実施間隔が延長できるのは,次の条件を全て達成した場合だけである。
a) 前回に選択した間隔で滅菌線量監査を実施し,線量増加及び滅菌線量を再確立することなく,少なく
とも4回連続している。
b) 上記a) と同じ期間にわたりバイオバーデンが安定して限度内にあることを立証するデータが得られ
ている。このデータには,次の事項を含む。
1) バイオバーデンの決定は少なくとも3か月ごとに実施する。ただし,平均バイオバーデンが1.5未
満で滅菌線量を方法1で設定,又は15 kGyの滅菌線量を選択し実証した場合には,1か月ごとに実
施する。
なお,VDmaxSD法の場合は,JIS T 0806-2:2014のJA.4.2.2による。
2) バイオバーデンの特徴(例えば,コロニー又は細胞の形態,染色特性又は選択培地での発育)
c) 製品のバイオバーデンを管理しており,この管理の有効性がJIS Q 13485に規定する滅菌医療機器に
関わる品質マネジメントシステムの各箇条を実施することによって立証されている。
12.1.3.3 12.1.3.4を適用する場合を除き,滅菌線量監査の最大間隔は,12か月としなければならない。
12.1.3.4 製品バッチの製造間隔が,12.1.3.1及び/又は12.1.3.2によって決定した間隔より長い場合には,
滅菌線量監査は,製造バッチごとに実施しなければならない。
12.1.3.5 滅菌線量監査が失敗した場合には,JIS T 0806-2:2014によって処置しなければならない。滅菌線
量監査の頻度は,次の事項が全て達成されるまでは3か月を超えてはならない。
a) 滅菌線量監査の失敗又はバイオバーデン増加の原因を調査し,修正及び/又は是正処置が実施されて
いる。
b) 滅菌線量監査の実施間隔に関わる根拠(12.1.3.1参照)をレビューし,必要ならば新しい間隔を定める。
c) 滅菌線量監査の実施間隔を延長するための基準が12.1.3.2に適合している。
12.2 再校正
滅菌プロセスの制御,指示又は記録に用いる測定機器の正確さ及び信頼性を,4.3.3によって定期的に検
証しなければならない。
12.3 設備のメンテナンス
12.3.1 文書化した手順によって予防メンテナンスを計画し,実施しなければならない。メンテナンス記録
は,保管しなければならない(4.1.2参照)。
12.3.2 メンテナンスの計画,手順及び記録は,指名した職員があらかじめ定めた間隔でレビューし,その
結果を文書化しなければならない。
12.4 設備適格性の再確認
12.4.1 あらかじめ定めた製品及び規定した設備を用いて,滅菌プロセスの適格性の再確認をしなければな
らない。適格性の再確認は,あらかじめ定めた間隔及び全ての変更(12.5参照)を評価した後に実施しな
ければならない。適格性の再確認を実施する範囲は,正当な理由付けをしなければならない。
12.4.2 適格性の再確認の手順を定め,適格性の再確認の記録は,保管しなければならない(4.1.2参照)。
18
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.4.3 適格性の再確認のデータは,文書化した手順に従ってあらかじめ定めた許容基準に対してレビュー
しなければならない。許容基準に適合しなかった場合には,実施した修正及び是正処置の記録とともに保
管しなければならない(4.1.2参照)。
12.5 変更の評価
12.5.1 線量及び線量分布に影響を及ぼす可能性のある照射設備の全ての変更を評価しなければならない。
これらの一方又は両方に影響を与えると判断した場合には,IQ,OQ及び/又はPQの一部又は全てを再
度実施しなければならない(9.1,9.2又は9.3参照)。判定の根拠を含む評価の結果を記録しなければなら
ない(4.1.2参照)。
12.5.2 製品,その包装又は滅菌のための製品配置の変更が,滅菌プロセスの適切性に与える影響を評価し
なければならない。変更の内容に基づいて実施しなければならないプロセスの定義又はPQの内容を決定
しなければならない。決定した根拠を含む評価の結果は,記録しなければならない(4.1.2参照)。
19
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
この規格の使用上の指針
注記1 この附属書に示す指針は,この規格に適合しているかを評価するためのチェックリストとし
て意図したものではない。この指針の意図するところは,規定した要求事項を達成するため
の説明及び可能な方法を提示することで,この規格の統一的な理解及び実施の手助けになる
ことである。この指針で示す以外の方法を適用することも可能であるが,ほかの方法を使用
する場合には,この規格に適合することを立証しなければならない。
注記2 この附属書の番号付けは,参照が容易となるように,この規格の要求事項を示した本体の箇
条と対応している。
A.1 適用範囲
A.1.1 指針はない。
A.1.2 指針はない。
A.1.2.1 指針はない。
A.1.2.2 医療機器の滅菌プロセスの開発,バリデーション及び日常管理のためには,文書化した手順を効
果的に実施することが必要である。これらの手順は,一般に品質マネジメントシステムの要素と考えられ
ている。この規格では,医療機器の品質マネジメントシステムであるJIS Q 13485を準拠することで,効
率的な滅菌の管理に必須である品質マネジメントシステムの要素を確認し,規定している。
A.1.2.3 微生物殺滅のメカニズムと放射線量との関係は,十分に確立したものであるため,バリデーショ
ン及びプロセス監視のためにバイオロジカルインジケータを使用しないほうがよい。
A.1.2.4 指針はない。
A.1.2.5 指針はない。
A.2 引用規格
この規格の引用規格に含まれる要求事項は,この規格の要求事項の部分に引用する範囲だけが要求事項
となる。引用は,規格全体の場合もあれば,特定の箇条に限定される場合もある。
A.3 用語及び定義
指針はない。
A.4 品質マネジメントシステムの要素
注記 A.1.2.2も参照。
A.4.1 文書化
文書化及び記録の管理についての要求事項は,JIS Q 13485の4.2.3及び4.2.4にそれぞれ規定している。
JIS Q 13485では,文書化に対する要求事項は,文書(仕様及び手順を含む。)の作成,管理及び記録につ
いてである。
20
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.4.2 経営者の責任
責任及び権限についての要求事項は,JIS Q 13485の5.5に規定しており,人的資源についての要求事項
は,JIS Q 13485の6.2に規定している。
JIS Q 13485では,経営者の責任に対する要求事項は,経営者のコミットメント,顧客重視,品質方針,
計画,責任,権限,コミュニケーション及びマネジメントレビューに関連している。
滅菌プロセスの開発,バリデーション及び日常管理は,幾つかの別々の組織を包含することがあり,各
組織は特定の要素に責任をもっている。この規格では,組織が受け入れたそれぞれの責任を定め,その定
められた責任を文書化することを要求している。この定めた権限及び責任は,それぞれの組織の品質マネ
ジメントシステムの中で文書化する。各要素に責任をもつ組織は,これらの要素について,適切な訓練及
び資格認定によって,その能力が立証された力量のある職員に割り当てることを要求している。
放射線滅菌では,作業を一次製造業者及び照射業者の二つの基本的な組織で行う場合がある。照射業者
は,滅菌サービスの専門家であり,一般的に一次製造業者及び照射業者は,個別の品質マネジメントシス
テムを運用しており,権限及び責任の範囲を契約書又は技術合意書に記載する。一次製造業者及び照射業
者それぞれが分担する主な責任には,次のようなものがある。
a) 一次製造業者
− 滅菌線量の確立
− 製品ファミリーの開発
− 最大許容線量の確立
− PQ
− 照射業者へ提示する製品の仕様(例えば,製品の密度,配置,寸法)に適合することなどの製造プ
ロセスの管理
− 照射業者へ提示する仕様の改訂
− 処理カテゴリに影響を与える製品関連の変動因子の変更管理
− 滅菌に先立って“無菌”又は“滅菌済み”とラベル表示する製品の管理
− 製品のリリース
b) 照射業者
− IQ
− OQ
− 照射プロセスの管理
− 照射設備の変更管理
− 放射線量の証明
− 処理カテゴリの開発
A.4.3 製品実現
注記 JIS Q 13485では,製品実現に対する要求事項には,顧客要求事項の決定,設計・開発,購買,
製造管理,監視及び測定機器の校正からなる製品ライフサイクルに関連している。
A.4.3.1 購買についての要求事項は,JIS Q 13485の7.4に規定している。特にJIS Q 13485の7.4.3の購
買製品の検証についての要求事項が,外部の組織から受け取る全ての製品及びサービスに適用されること
に注意するとよい。
A.4.3.2 識別及びトレーサビリティについての要求事項は,JIS Q 13485の7.5.3に規定している。
A.4.3.3 監視及び測定機器の校正についての要求事項は,JIS Q 13485の7.6に規定している。
21
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.4.3.4 放射線滅菌の線量測定に関わる指針を,JIS T 0806-3に示す。
A.4.4 測定,分析及び改善−不適合製品の管理
不適合製品の管理及び是正処置の手順は,JIS Q 13485の8.3及び8.5.2にそれぞれ規定している。
JIS Q 13485では,測定,分析及び改善の要求事項は,プロセス監視,不適合製品の管理,データの分析
及び改善(是正処置及び予防処置を含む。)に関連している。
A.5 滅菌剤の特性
A.5.1 滅菌剤
あらかじめ定めたエネルギー以上の電子線又はX線で照射した製品に放射能が誘起される可能性の評価
は,一般に入手可能な文献,誘導放射能の測定及び/又は誘導放射能の模式モデルに基づいて実施するの
が望ましい。
実験的手法及び理論的手法の評価の一例として,Grégoireら[22]の文献がある。7.5 MeVの電子線加速器
で発生したX線で医療機器に使用される様々な材質を50 kGyまで照射したときの誘導放射能の実測値及
び計算値が報告されている。これらの物質は,次のようなものである。
a) 放射化の可能性が極めて少ないもの(非金属,例えば,炭化水素ベースのポリエチレン及びポリスチ
レン)
b) 低水準ではあるが放射化の可能性があるもの(例えば,ステンレス鋼及び黄銅)
c) 比較的高い放射化の可能性があり詳細な検討が必要なもの(例えば,タンタル)
Grégoireら[22]の論文に紹介されていない物質(例えば,銀及び金)も放射化の可能性があるので詳細な
検討が必要である。
A.5.2 微生物殺滅効果の有効性
指針はない。
A.5.3 材質への影響
指針はない。
A.5.4 環境への配慮
環境マネジメントシステムの原則は,放射線滅菌プロセスにも適用できる。JIS Q 14001では,環境マネ
ジメントシステムの仕様を規定している。JIS Q 14040は,ライフサイクルアセスメントの設計の指針を記
載する。爆発性又は引火性の物質を照射する場合にも評価するのが望ましい。
A.6 プロセス及び設備の特性
注記 箇条6の目的は,滅菌プロセスに使用する設備及びその運転を定義することである。
A.6.1 プロセス
指針はない。
A.6.2 設備
指針はない。
A.7 製品の定義
注記 箇条7の目的は,滅菌する製品を定義し,滅菌前の微生物学的な品質を決定することである。
A.7.1 指針はない。
A.7.2 指針はない。
22
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.7.3 この箇条の目的は,滅菌前に材質の性状,製品の包装及び手順を考慮してバイオバーデンが低水準
で安定なものとすることである。これは一般的には,医療機器の製造を通してJIS Q 13485に従った品質
マネジメントシステムを適用することで達成できる。
A.7.4 JIS T 0806-2:2014を参照。
A.7.5 製品を処理カテゴリに含めるかどうかを評価する基準は,放射線滅菌独自のものであり,ほかの滅
菌法(例えば,エチレンオキサイド及び湿熱)に適用するのは必ずしも適切でない場合がある。
ガンマ線又はX線照射設備の場合には,日常の製品処理は,多数の照射容器が存在する照射設備で行わ
れる。照射した線量に対して隣接する照射容器に与える影響をOQの線量分布評価で決定し,同時に処理
できる製品の情報を得る。通常,この線量分布評価の情報は,処理する製品のプロセスを決定する照射業
者が使用する処理カテゴリにその製品を含めるかどうかを評価するのにも使用する。
ガンマ線及びX線照射設備の場合には,ある処理カテゴリに製品が含まれるかどうかを評価する二つの
主な要因は,類似の線量(滅菌線量及び最大許容線量)及び線量吸収特性(例えば,密度及び載荷形態)
である。一般に製品がある処理カテゴリに含まれるのは,その処理カテゴリに属する製品があらかじめ定
めた線量範囲を外れることなく,同一のタイマ設定で製品を処理できることに基づいている。OQで処理
カテゴリに含む可能性のある製品の範囲を決める線量分布評価を行わなかった場合には,処理カテゴリに
含まれる各々の製品について線量分布評価を実施することが望ましい。
電子照射設備の場合には,X線又はガンマ線照射設備に比べPQ時により多くの線量分布評価を行う。
しかし,必要な線量分布評価の回数を減らすために,製品を処理カテゴリにグループ分けしてもよい。
製品のグループ分けは,照射容器内の製品,包装及び製品の載荷形態でその処理カテゴリ内の製品にあら
かじめ定めた線量限度を超えることなく,同一のプロセスパラメータで製品を処理できる結果となった場
合だけ適用できる。照射容器内の製品数,分布及び配置方法並びに製品の密度及び分布を考慮することが
望ましい。
製品に照射する線量及び処理仕様に影響を与える製品関連の変動因子に修正を加えると,製品がその処
理カテゴリに属すると判断した根拠が変更される場合がある。このような場合には,新たな処理カテゴリ
を定義することが望ましい。それらの製品関連の変動因子の例には,次のようなものがある。
a) 照射箱の寸法
b) 製品を入れた照射箱の質量
c) 照射箱内の製品配置
d) 照射箱内の製品数
e) 滅菌線量
f)
最大許容線量
A.7.6 処理カテゴリの定期的な見直しは,通常,1回/年行う。
A.8 プロセスの定義
注記 箇条8の目的は,あらかじめ定めた製品(箇条7参照)に適用する滅菌プロセスに対し,最大
許容線量及び滅菌線量を確立することである。
A.8.1 最大許容線量の確立
A.8.1.1 製品寿命の全期間にわたり製品の品質,安全性及び性能を保証するためのプログラムは,適切な
材質を選定すること(AAMI TIR17 [17])から始めるのがよい。一般に試験計画を設定する場合には,次の
変動因子について評価することが望ましい。
23
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 原材料
− 製造プロセス
− 放射線量
− 放射線の種類
− 照射後の保管条件
この計画には,特定の合格基準の決まった適切な試験によって,機能性及び生物学的な安全性(JIS T
0993-1参照)の評価を含めることが望ましい。
試験計画で導き出した線量は,製品の最大許容線量を決定するのに用いる。
試験計画で次に必要なことは,あらかじめ定めた製品寿命の全期間を通して基準に合格することを裏付
ける証拠を得ることである。この情報を実際にかかる期間より短い期間で得るための一つの方法は,加速
試験を実施することである。放射線による製品に対する悪影響は,高温下でより早く現れるので,実時間
で生じる変化と加熱で生じる変化とを関係付ける提案がなされている(AAMI TIR17 [17]参照)。ただし,
加速試験は,実時間試験の代用にはならない。
線量測定関係の詳細な指針については,JIS T 0806-3の箇条6に示す。
A.8.1.2 放射線滅菌の線量測定に関わる指針を,JIS T 0806-3に示す。
A.8.2 滅菌線量の確立
A.8.2.1 JIS T 0806-2:2014に示す。
A.8.2.2 8.2.2 a) については,この方法によって滅菌線量を確立するために,次のことが適用できる。
1) 平均バイオバーデンが0.1以上の製品の滅菌線量を確立するために,バイオバーデンを構成する微生
物の数及び抵抗性についての知識を適用できる(JIS T 0806-2:2014の方法1参照)。
2) 平均バイオバーデンがいかなる水準にあっても,製品の滅菌線量を確立するためにはバイオバーデン
を構成する微生物の抵抗性についての知識が適用できる(JIS T 0806-2:2014の方法2参照)。
8.2.2 b) については,平均バイオバーデンが1 000以下の製品には25 kGyの実証,平均バイオバーデン
が1.5以下の製品には15 kGyの実証の方法を,JIS T 0806-2:2014で規定している。
A.8.2.3 指針はない。
A.8.3 滅菌線量及び最大許容線量の規定
指針はない。
A.8.4 線源間の最大許容線量,検定線量又は滅菌線量の移転
A.8.4.1 最大許容線量の移転
最大許容線量の有効性を当初設定した線源以外の線源で評価する場合には,線量率及び照射中の製品温
度を考慮することが望ましい。例えば,線量率が高くなるほど,製品材質への望ましくない効果は減少す
る傾向がある。
低線量率の線源(ガンマ線)又は中線量率線源(X線)で適合性を確認した製品は,通常,高線量率の
線源(電子線)では材質の適合性を立証するために,最小限の適合性確認試験で十分である。逆に高線量
率で適合性を確認した製品に低線量率又は中線量率を適用する場合には,詳細な確認試験を必要とする場
合がある。
線量率及び製品温度が同等の場合には,同種の線源間での移転は適切である。
A.8.4.2 検定線量又は滅菌線量の移転
A.8.4.2.1 線量率が大幅に異なる線源間の移転では,微生物殺滅の効力が異なることに注意する。線量率
24
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の違いが微生物殺滅の効力に影響を与えないことを立証することが,移転を認めるための必要なデータと
なる。移転が微生物殺滅の効力に変化を与えないことの立証は,移転しようとしている線源を使用して,
検定線量試験(JIS T 0806-2:2014参照)に満足することで達成される。
A.8.4.2.2 乾燥状態で照射を行う場合は,微生物殺滅効力は,運転条件に影響を受けないという実験的証
拠があり,移転は差し支えない。
A.8.4.2.3 液状の水分が存在する条件で照射を行う場合は,線源の運転特性によって微生物殺滅効力が影
響を受けることがあるという実験的証拠があり,移転は制限される。
A.9 バリデーション
注記1 この規格の目的のため,バリデーションは,据付適格性の確認(IQ),運転適格性の確認(OQ)
及び稼働性能適格性の確認(PQ)の少なくとも三つの主要な構成要素がある。
注記2 大規模な据付け及び新規設備の場合には,使用者の要求事項を決めて文書化することから開
始するのが一般的である。設備の供給者がほぼ決定した時点で使用者の要求事項及び設備仕
様及び設備配置を検討して,問題点があれば解決する。この段階を一般に設計適格性の確認
(DQ)と名付ける。この規格では,DQについての要求事項は規定しない。
A.9.1 据付適格性の確認(IQ)
IQは,滅菌設備及び付帯設備が仕様どおりに供給され,据付けされたことを示すために行う。
IQは,設計及び据付けに関わる要求事項を記載する文書の作成から始める(A.9の注記2参照)。IQは,
文書化した要求事項に基づくことが望ましい。工事及び据付けは,これらの要求事項に対して評価するこ
とが望ましい。IQの文書化には,図面並びに全ての建築資材,設備の寸法及び許容差,支援サービス及び
動力源の詳細などを含んでいることが望ましい。
IQは,OQを実施する前に完了していることが望ましい。
ISO 11137:1995の発行前に操業開始した放射線施設で,据付け中に照射設備に加えた変更記録がない場
合には,これらの記録を後から作成する必要はない。
A.9.2 運転適格性の確認(OQ)
放射線滅菌の線量測定に関わる指針は,JIS T 0806-3を参照。
A.9.3 稼働性能適格性の確認(PQ)
PQは,特定の製品を用いて行うバリデーションであり,設備があらかじめ定めた範囲内で線量を照射
するために,あらかじめ定めた規準に基づき安定的に運転できていることを示し,製品の無菌性について
の要求事項に適合していることを立証するものである。
放射線滅菌の線量測定に関わる指針は,JIS T 0806-3を参照。
9.3.2 b) については,電子線照射の場合,こん(梱)包内の製品配置が重要となる。さらに,ガンマ線
及びX線照射の場合でも,密度が線量分布に影響する可能性がある場合(例えば,液体容器,金属インプ
ラントなど),製品配置が重要になることもある。
9.3.2 c) については,製品を照射容器内で固定するシステムの場合,固定のために使用する材料及び固
定の方法を仕様書に含むことが望ましい。
A.9.4 バリデーションのレビュー及び承認
この作業には,滅菌処理の適合性を確認し,プロセス仕様を作成し,承認するためのバリデーションデ
ータを検討し,文書化することが含まれる。
25
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.10
日常監視及び管理
注記 日常監査及び管理の目的は,バリデートされ,かつ,あらかじめ定めたとおりの滅菌プロセス
が製品に対して行われたことを立証することである。
A.10.1
指針はない。
A.10.2
JIS Q 13485では,製品の取扱い及び保管に関わる要求事項を規定している。
A.10.3
製品を分離する場合には,次の事項に注意する。
a) 製品の物理的な分離
b) 信頼性ある在庫管理システムの使用
ラベル及び/又はスタンプの使用は,この手順の一部となり得る。
A.10.4
指針はない。
A.10.5
製品が照射容器内で移動して線量分布に影響する可能性がある場合には,処理中の想定外の移動
を避けるため,保持具を用いて製品を固定するのが望ましい。
A.10.6
製品が仕様どおりに処理されたことを確認するため,プロセスパラメータの監視及び日常線量測
定の結果を検討する。この検討には,必要ならば測定結果が範囲外の場合にとるべき措置を含むことが望
ましい。
測定結果が,範囲外となった場合の措置(例えば,再処理,範囲外の測定値の信頼性検討,製品廃棄,
その他必要な措置)を文書化して実施することが望ましい。
電子線照射設備は,設備によってその特性及び監視する方法が異なる。滅菌線量が製品に必ず照射され
たことを保証する運転条件の監視及び日常的な線量測定の寄与の程度は照射設備ごとに異なる。照射業者
は,滅菌処理が適正に実施されたという信頼を与える運転条件の監視及び日常線量測定の実施などの監視
手順を作成することが望ましい。
A.10.7
線量測定に関わる指針は,JIS T 0806-3を参照。
A.10.8
指針はない。
A.10.9
指針はない。
A.10.10 製品が仕様どおりに処理されたことを確認するため,プロセスパラメータの監視及び日常線量測
定の結果をレビューする。このレビューには,必要ならばプロセス中断のときにとるべき措置も含むこと
が望ましい。
正常な運転条件からの逸脱時(例えば,停電又はコンベヤの誤作動)には,すぐにプロセスを中断し,
線源を自動的に安全な位置に格納することが望ましい。これらの原因及びプロセス中断の時間を記録し,
再開の手順を文書化し,これを実施することが望ましい。
照射設備又はコンベヤの故障の場合には,滅菌線量が照射され,かつ,最大許容線量を超えない製品が
提供できることを保証するための文書化した手順を実施することが望ましい。
微生物が生育可能な状態にない製品でプロセス中断が発生した場合には,照射設備内で製品を移動せず
に中断することは,通常,特別な対応策を必要としない。しかし,これらのプロセス中断は記録し,線量
測定の有効性を保証するためにレビューすることが望ましい。
微生物が生育可能な状態にない製品のプロセス中断の場合には,次の事項をプロセス仕様書に記載する。
− 製造完了から滅菌処理完了までの最大経過時間
− この経過時間内の保管及び輸送の条件
最大経過時間及び条件は,製品の微生物的な性質として無菌性を損なわないことを保証できるように選
定する。滅菌中にプロセス中断が起き,それが原因であらかじめ定めた時間を超えて滅菌の完了が遅れた
26
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
場合には,製品の微生物的品質を確認し適切な対応策を講じることが望ましい。この中には製品廃棄も含
まれる。線量測定に関わる指針は,JIS T 0806-3を参照。
あらかじめ定めた線量以下となるようなプロセス変動が発生し,次の事項に該当する場合には,製品を
追加照射してもよい。
a) 製品の微生物の生育可能性を考慮する。
b) 最小線量を達成し,かつ,最大許容線量を超えないことを保証できる方法で線量が照射できる。詳細
な指針については,JIS T 0806-3を参照。
A.10.11 指針はない。
A.11
滅菌からの製品のリリース
指針はない。
A.12
プロセス有効性の維持
A.12.1
継続的な有効性の立証
A.12.1.1
一般
滅菌線量の有効性を維持するためには,微生物の数及びタイプについてバイオバーデンが安定し,管理
された条件下で製品を製造する必要がある。有効性が継続して維持されていることを立証するため,滅菌
線量監査をあらかじめ定めた間隔で実施する。この最大間隔は,次の事項に基づき決定する。
a) 滅菌線量設定時に得た知見
b) 製造プロセス及び材料の変動を検知する必要性及びこれらの変動を調べる頻度に関して受容できるリ
スクの大きさについての共通認識
c) 潜在的な季節的変動又は材料の微生物相の変動又は製造環境
d) 滅菌プロセスで一般に認められた再バリデーションの頻度
A.12.1.2
バイオバーデンの決定頻度
A.12.1.2.1
指針はない。
A.12.1.2.2
指針はない。
A.12.1.2.3
指針はない。
A.12.1.2.4
指針はない。
A.12.1.2.5
滅菌線量が継続して有効であることを立証するためのバイオバーデンの限度を設定する場合
には,無菌性に関する規定の要求事項を達成するために必要なバイオバーデン限度を超過したという事実
に基づいて行うのが望ましい。
A.12.1.3
滅菌線量監査の頻度
A.12.1.3.1
a) バイオバーデンの季節的な変動を検出するため,歴史的慣例として3か月の間隔が適用されてきたが,
管理した条件下で製造する製品では,季節変動は見いだされない可能性がある。バイオバーデンにつ
いては,その数及び微生物のタイプに季節変動がなく,管理ができることを示せる場合には,線量監
査の頻度を減らすことを検討してもよい。この検討を行う場合には,12.1.3に規定するプロセス及び
監視に関わる事項を含めなければならない。全ての関連事項を検討しなければならないが,これらの
事項全てから信頼できる結果が得られたり,同じ重み(すなわち,重要度が同程度)をもっているこ
とではないことに注意する。
27
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 指針はない。
A.12.1.3.2
製品及びその製造に関わる経験が深まるにつれて,滅菌線量監査の実施間隔は3か月,次い
で6か月,最終的に12か月に延ばすという考えが起こってきた。しかしながら滅菌線量監査の実施頻度を
減らすことは,製造プロセスにおける変動を検出する能力の低下をもたらすことを認識しておくことが望
ましい。したがって,実施前に頻度を減らすことによる影響を検討することが必要である。
A.12.1.3.3
指針はない。
A.12.1.3.4
指針はない。
A.12.1.3.5
指針はない。
A.12.2
再校正
指針はない。
A.12.3
設備のメンテナンス
メンテナンス記録をレビューする場合に,メンテナンスの計画及び手順は,機器について得られた情報
に基づき必要に応じて改訂するのが望ましい。
A.12.4
設備適格性の再確認
照射設備の適格性の再確認の期間は,照射設備が常に仕様どおりに稼働しているという保証を与えるよ
うに選定するのが望ましい。ガンマ線照射設備の場合には,通常,適格性の再確認は,線源補充と関連し
て行う。電子線及びX線照射設備の場合には,通常1年ごとに実施し,特定部分の適格性の再確認は,こ
のサイクルより短い間隔で行う。適格性の再確認で照射設備のIQ及び/又はOQの状態が変化したことが
判明した場合には,PQを再度実施しなければならない場合もある。
A.12.5
変更の評価
A.12.5.1
ガンマ線照射設備の場合には,変更の後にOQの実施が必要な例には,次のようなものがある。
− 線源補充
− 線源の配置及び位置の変更
− コンベヤの変更
− コンベヤ経路の変更
− 照射容器の変更
OQの範囲は,変更の種類及び程度によって異なる(表A.1参照)。
電子線照射設備で照射設備の性能に影響を及ぼすと思われる変更を実施した場合には,OQを実施する。
これに該当する変更の例としては,次のようなものがある。
− コンベヤの変更
− 照射容器の最大設計寸法の増加
− スキャンマグネットの修理又は交換
− ベンディングマグネットの修理又は交換
− 平行ビームマグネットの修理又は交換
− 散乱効果を発生する照射設備の部品の変更
OQの範囲は,変更の種類及び程度によって異なる(表A.2参照)。例えば,照射容器の最大寸法を増加
した場合には,完全な適格性の再確認が必要になるのに対して,コンベヤの部品交換の場合には,コンベ
ヤが適正に稼働するかの確認だけでよいこともある。
X線照射設備で設備の性能に影響を及ぼす可能性のある変更を実施した場合には,OQを実施する。
これに該当する変更の例としては,次のようなものがある。
28
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− コンベヤの変更
− 照射容器の最大設計寸法の増加
− スキャンマグネットの修理又は交換
− ベンディングマグネットの修理又は交換
− 平行ビームマグネットの修理又は交換
− 散乱効果を発生する照射設備の部品の変更
− X線ターゲットの変更
OQの範囲は,変更の種類及び程度によって異なる(表A.3参照)。例えば,照射容器の最大寸法を増加
した場合には,完全な適格性の再確認が必要になるのに対して,コンベヤの部品交換の場合には,コンベ
ヤが適正に稼働するかの確認だけでよいこともある。
A.12.5.2
指針はない。
29
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
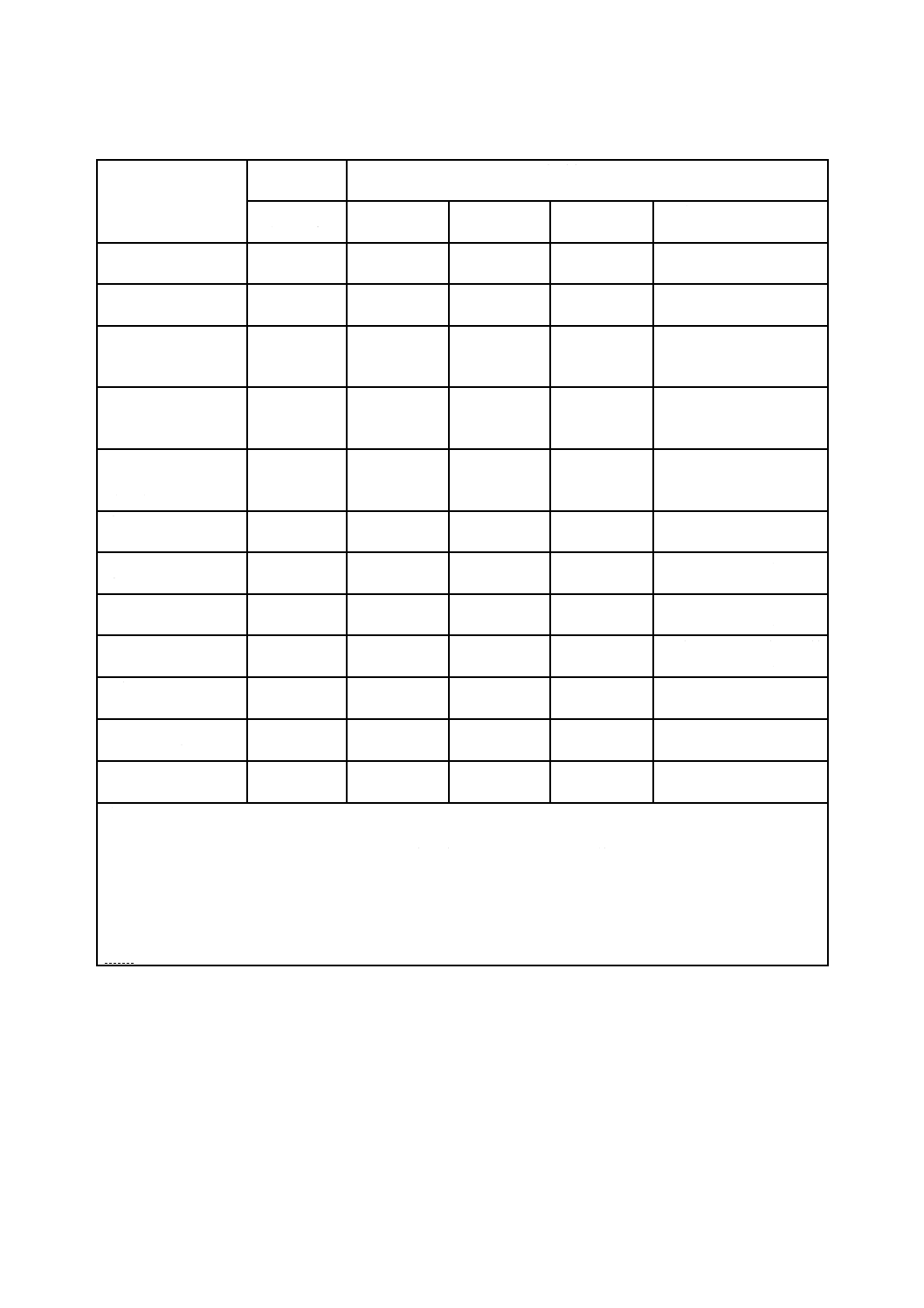
表A.1−ガンマ線照射設備の変更における適格性の再確認に関わる指針a)
照射設備の変更
据付適格性の
確認
運転適格性の確認
据付試験及び
設備の文書化
機器の試験
機器の校正
照射設備の
線量分布評価
線量分布評価の種類
線源の補充,除去又は
再配置
〇
〇
設計限界までの均一物質
キャリヤ又は照射容
器の再設計
〇
〇
〇
設計限界までの均一物質
照射室内の天井コン
ベヤの移動又は再配
置
〇
〇
〇
設計限界までの均一物質
重要な製品経路内の
停止設備の移動又は
再配置
〇
〇
〇
設計限界までの均一物質
重要な製品経路外の
停止設備の移動又は
再配置
〇
〇
線源ケーブルの交換
〇
〇
線源駆動設備の再設
計
〇
〇
トランジット線量
線源及び製品間距離
に影響する再設計
〇
〇
〇
設計限界までの均一物質
トランジット線量
線源ラックの再設計
〇
〇
〇
設計限界までの均一物質
トランジット線量
照射設備サイクルタ
イマの変更
〇
〇
〇
照射設備の安全監視
設備の形式変更
〇
〇
〇
照射設備のプール水
監視設備の形式変更
〇
〇
〇
(該当する場合)
注記1 線源の形状を変更することなく線源を追加した場合には,数学モデル又は改良目的の結果を確認するため
の均一線量分布評価の一部だけを実施するだけでもよい場合がある。一方,形状変更を伴う線源追加の場
合には,中央載荷,部分載荷などある程度の補完調査に加えて,均一線量分布評価を繰り返す必要がある
場合もある。
注記2 稼働試験(例えば,線源位置の検定)で判定できない結果の場合,線源ケーブルを交換した後に線量分布
評価の実施が必要になる場合がある。
注記3 OQ線量分布評価の結果,PQを繰り返すこともある。
注a) ○印は,実施する項目。対応国際規格では,レ印である。
30
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
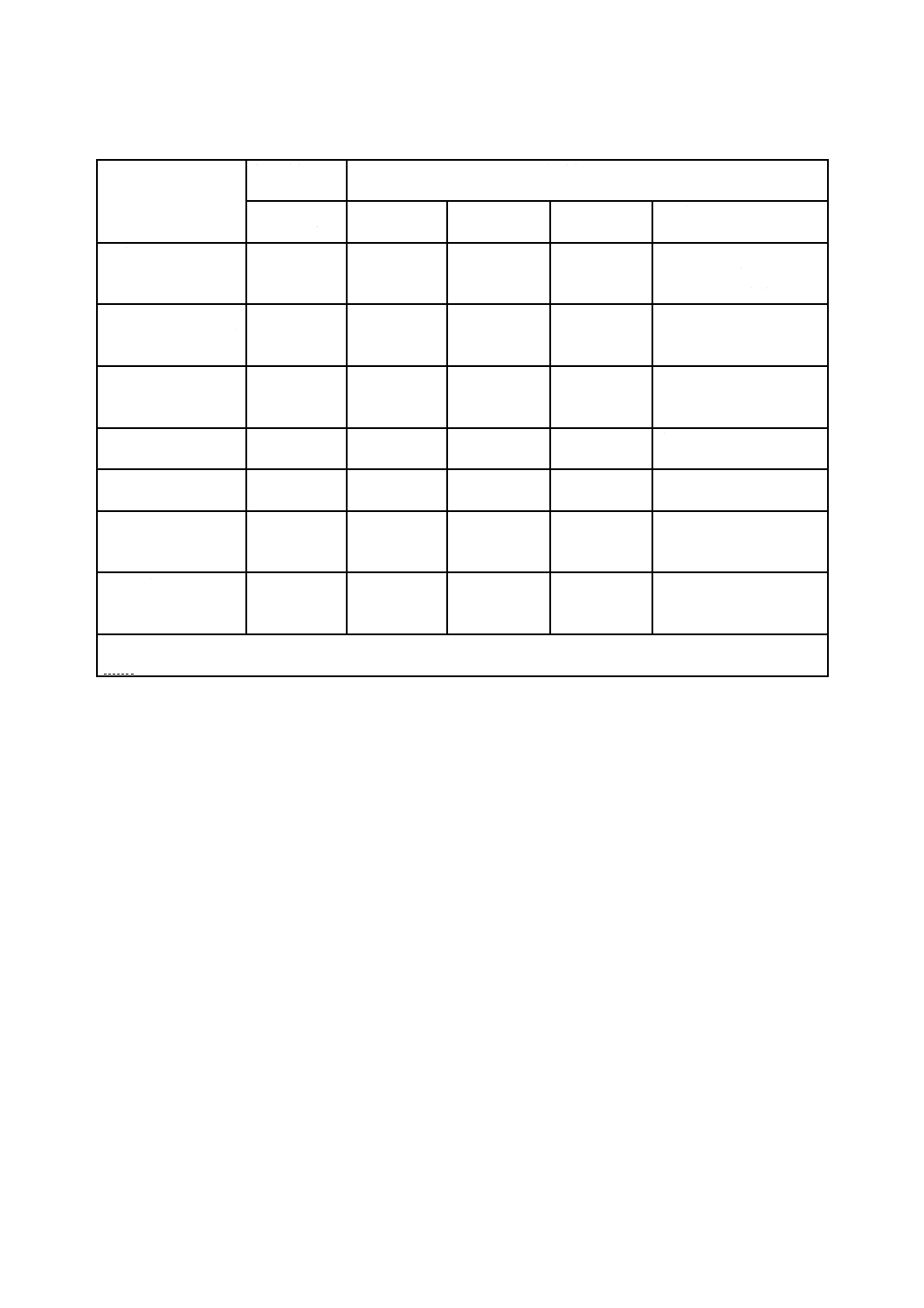
表A.2−電子線照射設備の変更における適格性の再確認に関わる指針a)
照射設備の変更
据付適格性の
確認
運転適格性の確認
据付試験及び
設備の文書化
機器の試験
機器の校正
照射設備の
線量分布評価
線量分布評価の種類
加速器の機械配置
〇
〇
ビームスキャン方向のス
キャン均一度及びビーム
進行方向の深度線量分布
ビームステアリング
又はフォーカスマグ
ネット設備
〇
〇
ビームスキャン方向のス
キャン均一度及びビーム
進行方向の深度線量分布
ベンディングマグネ
ット
〇
〇
〇
ビームスキャン方向のス
キャン均一度及びビーム
進行方向の深度線量分布
ビーム電流監視設備
〇
〇
〇
製品進行方向のスキャン
均一度
スキャンマグネット
〇
〇
〇
ビームスキャン方向のス
キャン均一度
コンベヤ速度監視及
び/又は回路の制御
〇
〇
〇
製品進行方向のスキャン
均一度
プロセス中断試験
コンベヤシステム,モ
ータ,ベルト及びギヤ
設定比
〇
〇
注記 OQ線量分布評価の結果,PQを繰り返すこともある。
注a) ○印は,実施する項目。対応国際規格では,レ印である。

31
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表A.3−X照射設備の変更における適格性の再確認に関わる指針a)
照射設備の変更
据付適格性の
確認
運転適格性の確認
据付試験及び
設備の文書化
機器の試験
機器の校正
照射設備の
線量分布評価
線量分布評価の種類
加速器の機械配置
〇
〇
ビームスキャン方向のス
キャン均一度及びビーム
進行方向の深度線量分布
ビームステアリング
又はフォーカスマグ
ネット設備
〇
〇
ビームスキャン方向のス
キャン均一度及びビーム
進行方向の深度線量分布
ベンディングマグネ
ット
〇
〇
〇
ビームスキャン方向のス
キャン均一度及びビーム
進行方向の深度線量分布
ビーム電流監視設備
〇
〇
〇
製品進行方向のスキャン
均一度
スキャンマグネット
〇
〇
〇
ビームスキャン方向のス
キャン均一度
コンベヤ速度監視及
び/又は回路の制御
〇
〇
〇
製品進行方向のスキャン
均一度
プロセス中断試験
コンベヤシステム,モ
ータ,ベルト及びギヤ
設定比
〇
〇
搬送照射容器の再設
計
〇
〇
〇
製品進行方向のスキャン
均一度
製品進行方向の深度線量
分布
照射室内のコンベヤ
の除去又は移動
〇
〇
〇
製品進行方向のスキャン
均一度
製品進行方向の深度線量
分布
線源製品間距離に影
響する再設計
〇
〇
〇
製品進行方向のスキャン
均一度
ビームスキャン方向のス
キャン均一度
製品進行方向の深度線量
分布
照射設備の安全監視
設備形式の変更
〇
〇
〇
X線ターゲットの交
換,再設計又は再配置
〇
〇
〇
ビームスキャン方向及び
ビーム進行方向のスキャ
ン均一度
製品進行方向のスキャン
均一度
ビーム進行方向の深度線
量分布
注記 OQ線量分布評価の結果,PQを繰り返すこともある。
注a) ○印は,実施する項目。対応国際規格では,レ印である。
32
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] JIS Q 9000:2006 品質マネジメントシステム−基本及び用語
注記 対応国際規格:ISO 9000:2005,Quality management systems−Fundamentals and vocabulary(IDT)
[2] JIS Q 9001:2008 品質マネジメントシステム−要求事項
注記 対応国際規格:ISO 9001:2008,Quality management systems−Requirements(IDT)
[3] JIS Q 14001:2004 環境マネジメントシステム−要求事項及び利用の手引
注記 対応国際規格:ISO 14001:2004,Environmental management systems−Requirements with guidance
for use(IDT)
[4] JIS Q 14040:2010 環境マネジメント−ライフサイクルアセスメント−原則及び枠組み
注記 対応国際規格:ISO 14040:2006,Environmental management−Life cycle assessment−Principles
and framework(IDT)
[5] JIS T 0806-3:2010 ヘルスケア製品の滅菌−放射線−第3部:線量測定にかかわる指針
注記 対応国際規格:ISO 11137-3:2006,Sterilization of health care products−Radiation−Part 3:
Guidance on dosimetric aspects(IDT)
[6] JIS T 0841-1 最終段階で滅菌される医療機器の包装−第1部:材料,無菌バリアシステム及び包装シ
ステムに関する要求事項
注記 対応国際規格:ISO 11607-1,Packaging for terminally sterilized medical devices−Part 1:
Requirements for materials, sterile barrier systems and packaging systems(IDT)
[7] JIS T 0841-2 最終段階で滅菌される医療機器の包装−第2部:成形,シール及び組立プロセスのバリ
デーション
注記 対応国際規格:ISO 11607-2,Packaging for terminally sterilized medical devices−Part 2: Validation
requirements for forming, sealing and assembly processes(IDT)
[8] JIS T 0993-1:2012 医療機器の生物学的評価−第1部:リスクマネジメントプロセスにおける評価及
び試験
注記 対応国際規格:ISO 10993-1:2009,Biological evaluation of medical devices−Part 1: Evaluation and
testing within a risk management process(MOD)
[9] ISO 11137:1995,Sterilization of health care products−Requirements for validation and routine control−
Radiation sterilization
[10] ISO/TS 11139:2006,Sterilization of health care products−Vocabulary
[11] ISO/TS 13004:2013,Sterilization of health care products−Radiation−Substantiation of selected sterilization
dose: Method VDmaxSD
[12] International Vocabulary of Basic and General Terms in Metrology (VIM), BIPM, IEC, IFCC, ISO, IUPAC,
IUPAP, OIML, 2nd ed., 1993 Geneva (1993)
[13] ISO 22442-1,Medical devices utilizing animal tissues and their derivatives−Part 1: Application of risk
management
[14] ISO 22442-2,Medical devices utilizing animal tissues and their derivatives−Part 2: Controls on sourcing,
collection and handling
[15] ISO 22442-3,Medical devices utilizing animal tissues and their derivatives−Part 3: Validation of the
33
T 0806-1:2015 (ISO 11137-1:2006,Amd.1:2013)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
elimination and/or inactivation of viruses and transmissible spongiform encephalopathy (TSE) agents
[16] EN 556-1:2001,Sterilization of Medical Devices−Requirements for medical devices to be designated
“STERILE”−Part 1: Requirements for terminally sterilized medical devices
[17] AAMI TIR17:1997,Radiation sterilization−Material qualification
[18] ANSI/AAMI ST67:2003,Sterilization of Medical Devices−Requirements for Products Labeled “Sterile”
[19] ANSI/HGB N43.10-2001,Safe Design and Use of Panoramic, Wet Source Storage Gamma Irradiators
(Category IV) and Dry Source Storage Gamma Irradiators (Category II), Health Physics Society, McLean, VA,
2001
[20] IAEA Safety Series No. 107, Radiation Safety of Gamma and Electron Irradiation Facilities, Vienna, 1992
[21] Global Harmonization Task Force (GHTF)−Study Group 1 (SG1), Document N029R16:2005−Information
Document concerning the definition of the term “Medical Device”
[22] GRÉGOIRE, O., CLELAND, M.R., MITTENDORFER, J., VANDER DONCKT, M. and MEISSNER, J.
Radiological safety of medical devices sterilized with X-rays at 7.5 MeV, Radiation Physics and Chemistry 67,
Issue 2, June 2003, pp. 149-16