R 5202:2010
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲 ························································································································· 1
2 引用規格 ························································································································· 1
3 試験の一般的要求事項 ······································································································· 2
3.1 試験回数 ······················································································································ 2
3.2 許容差 ························································································································· 2
3.3 質量,体積,ファクター及び結果の表し方 ·········································································· 2
3.4 強熱 ···························································································································· 2
3.5 ガラス器具 ··················································································································· 2
3.6 その他の一般事項 ·········································································································· 2
4 セメント試料の調製 ·········································································································· 2
5 強熱減量の定量方法 ·········································································································· 3
5.1 要旨 ···························································································································· 3
5.2 高炉セメント及び高炉スラグ以外の場合 ············································································· 3
5.3 高炉セメント及び高炉スラグの場合 ··················································································· 3
5.4 許容差 ························································································································· 4
6 塩酸−炭酸ナトリウム方法による不溶残分の定量方法 ····························································· 4
6.1 要旨 ···························································································································· 4
6.2 試薬 ···························································································································· 4
6.3 試料の量り採り量 ·········································································································· 4
6.4 操作 ···························································································································· 4
6.5 計算 ···························································································································· 5
6.6 許容差 ························································································································· 5
7 二酸化けい素の定量方法 ···································································································· 5
7.1 要旨 ···························································································································· 5
7.2 試薬 ···························································································································· 5
7.3 試料の量り採り量 ·········································································································· 5
7.4 操作 ···························································································································· 5
7.5 計算 ···························································································································· 6
7.6 許容差 ························································································································· 6
8 酸化アルミニウムの定量方法 ······························································································ 6
8.1 要旨 ···························································································································· 6
8.2 試薬 ···························································································································· 6
8.3 光度滴定装置 ················································································································ 7
8.4 操作 ···························································································································· 7
R 5202:2010 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
8.5 空試験 ························································································································· 8
8.6 計算 ···························································································································· 8
8.7 許容差 ························································································································· 8
9 酸化鉄 (III) の定量方法 ···································································································· 8
9.1 要旨 ···························································································································· 8
9.2 試薬 ···························································································································· 8
9.3 装置 ···························································································································· 8
9.4 操作 ···························································································································· 8
9.5 検量線の作成 ················································································································ 9
9.6 計算 ···························································································································· 9
9.7 許容差 ························································································································· 9
10 酸化カルシウムの定量方法 ······························································································· 9
10.1 カルシウム用指示薬を用いる定量方法 ·············································································· 9
10.2 カルセイン指示薬又はカルセイン−PPC指示薬を用いる定量方法 ········································· 12
11 酸化マグネシウムの定量方法 ··························································································· 13
11.1 要旨 ·························································································································· 13
11.2 試薬 ·························································································································· 13
11.3 装置 ·························································································································· 13
11.4 操作 ·························································································································· 13
11.5 検量線の作成 ·············································································································· 14
11.6 計算 ·························································································································· 14
11.7 許容差 ······················································································································· 14
12 三酸化硫黄の定量方法 ···································································································· 14
12.1 要旨 ·························································································································· 14
12.2 試薬 ·························································································································· 14
12.3 操作 ·························································································································· 14
12.4 計算 ·························································································································· 15
12.5 許容差 ······················································································································· 15
13 酸化ナトリウム及び酸化カリウムの定量方法 ······································································ 15
13.1 原子吸光法 ················································································································· 15
13.2 炎光光度法 ················································································································· 17
14 酸化チタン (IV) の定量方法 ···························································································· 18
14.1 要旨 ·························································································································· 18
14.2 試薬 ·························································································································· 18
14.3 操作 ·························································································································· 19
14.4 検量線の作成 ·············································································································· 19
14.5 計算 ·························································································································· 19
14.6 許容差 ······················································································································· 19
15 酸化りん (V) の定量方法 ································································································ 19
R 5202:2010 目次
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
15.1 要旨 ·························································································································· 19
15.2 試薬 ·························································································································· 19
15.3 操作 ·························································································································· 20
15.4 検量線の作成 ·············································································································· 20
15.5 計算 ·························································································································· 20
15.6 許容差 ······················································································································· 20
16 酸化マンガン (II) の定量方法 ·························································································· 20
16.1 原子吸光法 ················································································································· 20
16.2 吸光光度法 ················································································································· 21
17 硫化物硫黄の定量方法 ···································································································· 23
17.1 要旨 ·························································································································· 23
17.2 よう素酸カリウム標準液を用いる直接滴定法 ···································································· 23
17.3 チオ硫酸ナトリウム標準液を用いる逆滴定法 ···································································· 25
17.4 許容差 ······················································································································· 26
18 塩素の定量方法 ············································································································· 26
18.1 電位差滴定法 ·············································································································· 26
18.2 チオシアン酸水銀 (II) による吸光光度法 ········································································· 28
18.3 チオシアン酸アンモニウム溶液による逆滴定法 ································································· 30
附属書A(参考)完全分析によるセメントの主成分の化学分析方法 ··············································· 32
附属書B(参考)塩酸−水酸化カリウム方法による不溶残分の定量方法 ········································· 52
参考文献 ···························································································································· 54
附属書JA(参考)JISと対応国際規格との対比表 ······································································ 55
附属書JB(参考)技術上重要な改正に関する新旧対照表 ···························································· 63
R 5202:2010 目次
(4)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,社団法人セメント
協会 (JCA) から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,日本工業標準調査会
の審議を経て,経済産業大臣が改正した日本工業規格である。
これによって,JIS R 5202:1999は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願,実用新案権又は出願公開後の実用新案登録出願に
抵触する可能性があることに注意を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許
権,出願公開後の特許出願,実用新案権及び出願公開後の実用新案登録出願にかかわる確認について,責
任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
R 5202:2010
セメントの化学分析方法
Methods for chemical analysis of cements
序文
この規格は,1950年に制定され,その後10回の改正を経て今日に至っている。前回の改正は1999年に
行われた。この規格は,セメントの湿式方法による化学分析方法を規定している。
セメントの湿式による化学分析方法の対応国際規格は,2009年3月15日に第1版として発行されたISO
29581-1であるが,対応国際規格ではセメントの主成分[二酸化けい素,酸化アルミニウム,酸化鉄 (III),
酸化カルシウム及び酸化マグネシウム]の分析方法としてセメントを完全に分解する手順を規定している。
しかし,この規格はセメントの品質規格(JIS R 5210,JIS R 5211,JIS R 5212,JIS R 5213及びJIS R 5214)
に引用され,セメントを酸で溶解し,溶解した成分の含有率を測定することを目的としている。
そのため,この規格は,ISO 29581-1で規定されている方法のうち,セメントを酸で溶解する分析方法
を取り入れ,対応国際規格には規定されていない分析方法を追加して作成した日本工業規格である。
なお,この規格で側線又は点線の下線を施してある箇所は,対応国際規格を変更している事項である。
変更の一覧表にその説明を付けて,附属書JAに示す。また,技術上重要な改正に関する新旧対照を,附
属書JBに記載する。
1
適用範囲
この規格は,セメント1) の湿式方法による化学分析方法について規定する。
この規格は,クリンカー及び高炉セメントの製造に用いる高炉スラグの化学分析にも適用することがで
きる。
注記 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 29581-1:2009,Cement−Test methods−Part 1: Analysis by wet chemistry (MOD)
なお,対応の程度を表す記号“MOD”は,ISO/IEC Guide 21-1に基づき,“修正している”
ことを示す。
注1) ここでいうセメントとは,JIS R 5210に規定するポルトランドセメント,JIS R 5211に規定す
る高炉セメント,JIS R 5212に規定するシリカセメント,JIS R 5213に規定するフライアッシ
ュセメント及びJIS R 5214に規定するエコセメントを指す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0113 電位差・電流・電量・カールフィッシャー滴定方法通則
2
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 0115 吸光光度分析通則
JIS K 0121 原子吸光分析通則
JIS K 8005 容量分析用標準物質
JIS K 8102 エタノール (95)(試薬)
JIS P 3801 ろ紙(化学分析用)
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS R 5204 セメントの蛍光X線分析方法
JIS Z 8401 数値の丸め方
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
3
試験の一般的要求事項
3.1
試験回数
試験は,同一試料について2回の繰返しで行う。ただし,品質管理を目的とする場合は1回でもよい。
3.2
許容差
2回の試験結果の差が,許容差より大きい場合,更に1回,再試験を行い,最も近い二つの試験結果を
平均し,分析結果(3.3参照)とする。
3.3
質量,体積,ファクター及び結果の表し方
質量は0.000 1 gまで量る。ビュレットを用いる場合,体積は0.01 mLまで読む。
溶液のファクターは3回の測定値を平均し,決定する。
個々の試験結果は,JIS Z 8401の2. b) によって,塩素 (Cl) を除く分析項目は百分率で小数点以下2け
たに丸める。塩素 (Cl) については百分率で小数点以下3けたに丸める。
分析結果は,2回の試験結果の平均とし,JIS Z 8401の2. c) 規則Aによって,塩素 (Cl) を除く分析項
目は百分率で小数点以下2けたに丸める。塩素 (Cl) については百分率で小数点以下3けたに丸める。
3.4
強熱
強熱は,次のように行う。
あらかじめ空焼きし,風袋を量ったるつぼにろ紙と沈殿(残留物)を入れて乾燥し,酸化雰囲気で炎が
出ないように徐々に加熱してろ紙を灰化した後,規定の温度で強熱する。強熱後,るつぼと内容物をデシ
ケータ中で室温まで放冷した後,質量を量る。
3.5
ガラス器具
分析に使用するガラス器具は,JIS R 3503及びJIS R 3505による。
3.6
その他の一般事項
その他の一般事項は,通常,JIS K 0050,JIS K 0113,JIS K 0115及びJIS K 0121による。
4
セメント試料の調製
試料は,検査単位について平均品質を表すように,セメントを採取し,縮分して約5 kgの代表試料とす
る。その採取方法及び縮分方法は,受渡当事者間の協議によって定める。代表試料を更に縮分器又は四分
法で縮分して,約100 gを採り,JIS Z 8801-1に規定する150 μm又は125 μmのふるいで,ふるう。ふる
い残分中の金属鉄を磁石を使って取り除き,金属鉄を除いた残分をすりつぶして完全に150 μm又は125
μmのふるいを通過させる。試料を気密性のあるきれいな乾いた容器に移し替え,十分に試料が混ざるよ
3
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
うに強く振る。
上記の操作は,試料が周囲の空気にさらされる時間をできるだけ短くするために,手早く行わなければ
ならない。
5
強熱減量の定量方法
5.1
要旨
試料を酸化雰囲気(空気中)で950±25 ℃で恒量になるまで強熱したときの減量を量る。950±25 ℃の
空気中で強熱すると,二酸化炭素と水分とは脱離し,酸化されうる元素は酸化される。金属鉄,酸化鉄 (II)
及び酸化マンガン (II) の酸化による誤差は通常,無視できるので,硫化物の酸化による酸素の増加分を補
正する。
5.2
高炉セメント及び高炉スラグ以外の場合
5.2.1
操作
a) 空焼きをして風袋を量った白金又は磁器るつぼに試料約1 g 2) を0.000 1 gまで正しく量り採る。
b) るつぼに少しすきまを開けてふたをして950±25 ℃に調節した電気炉中に置き,15分間強熱する。
c) るつぼをデシケータの中で放冷し,質量を量る。
d) 15分間ずつ強熱を繰り返し,恒量を求める。強熱前後の質量差が0.000 5 g未満になったときを恒量と
する。
なお,恒量を求めるとき質量が増加した場合は,増加前の質量を採用する。最初の15分間の強熱で
質量が増加した場合は,単に“+”と記す。
注2) 対応国際規格では,量り採り範囲を1.00±0.05 gと規定している。
5.2.2
計算
試料の強熱減量は,次の式によって算出する。
100
×
−
=
m
m'
m
loss
.
ig
ここに, ig.loss: 強熱減量 (%)
m: 試料の質量 (g)
m': 強熱後の試料の質量 (g)
5.3
高炉セメント及び高炉スラグの場合
5.3.1
操作
a) 5.2.1のa)〜d)と同様に操作して,5.2.2の式によって見掛けの強熱減量[ig.loss (ap)]を求める。
b) 強熱前の試料の三酸化硫黄含有率[SO3 (B)]を箇条12によって求める。
c) 強熱後の試料の三酸化硫黄含有率[SO3 (A)]を次の手順に従って求める。
試料2〜3 gをるつぼに量り採り,5.2.1のb)〜d)によって強熱する。ただし,恒量の確認は,強熱前
後の質量差が0.05 %未満になったときとする。この強熱後の試料3) を用いて三酸化硫黄含有率を箇条
12によって求める。
なお,強熱後の三酸化硫黄含有率は,JIS R 5204によって求めたガラスビードの三酸化硫黄の定量
値を用いてもよい。
注3) 強熱後の試料の量り採りに際しては,塊を崩して均一にした後,所定の量を素早く量り採る
とよい。
4
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.3.2
計算
試料の強熱減量は,次の式によって算出する。
]
(B)
(A)
[
0.8
(ap)
3
3
SO
SO
ig.loss
ig.loss
−
×
+
=
ここに,
ig.loss: 強熱減量 (%)
ig.loss (ap): 見掛けの強熱減量 (%)
SO3 (A): 強熱後の試料の三酸化硫黄含有率 (%)
SO3 (B): 強熱前の試料の三酸化硫黄含有率 (%)
5.4
許容差
許容差は0.10 %とする。
6
塩酸−炭酸ナトリウム方法による不溶残分の定量方法
6.1
要旨
この定量方法は,試料を希塩酸で溶解することによって,二酸化けい素の沈殿をできるだけ生成しない
ようにして,セメント中の不溶残分を定量する一般的な方法である。希塩酸溶解の残分は加熱した炭酸ナ
トリウム溶液で処理し,沈殿した二酸化けい素を溶解する。残分は強熱した後,質量を量る。
6.2
試薬
試薬は,次のものを用いる。
a) 塩酸 (1+1)
b) 炭酸ナトリウム溶液 (50 g/L)
c) メチルレッド指示薬 (2 g/L) メチルレッド0.2 gを,JIS K 8102に規定するエタノール100 mLに溶か
す。
6.3
試料の量り採り量
試料は,約1 g 4) を0.000 1 gまで量り採る。
注4) 対応国際規格では,量り採り範囲を1.00±0.05 gと規定している。
6.4
操作
定量操作は,次の手順によって行う。
a) 試料を乾燥したビーカー200 mLに入れ,水約20 mLを加え,ガラス棒でかき混ぜて試料を分散させ
ながら,塩酸 (1+1) 10 mLを加えて溶かす。このとき,必要ならば溶液を少し温め,未溶解の塊は,
ガラス棒の先でよくつぶし,可溶分を完全に溶かす。
b) 温水を加えて50 mLとし,時計皿でふたをして水浴上で10分間加熱する。
c) JIS P 3801に適合したろ紙(5種B,110 mm)を用いてろ過し 5),温水で8回洗浄する。ろ液及び洗
液はビーカー500 mLに受け,室温まで冷却する。その後,全量フラスコ250 mLに洗い移し,標線ま
で水を加えて振り混ぜる。この溶液を試料溶液 (F) とし,三酸化硫黄及び酸化チタン (IV) の定量に
用いる。
なお,酸化チタン (IV) の定量を行わない場合には,ビーカーに受けたろ液及び洗液をそのまま保
存して三酸化硫黄の定量に用いる。
d) ろ紙を漏斗から取り出して元のビーカー200 mLに入れ,炭酸ナトリウム溶液 (50 g/L) 50 mLを加え,
かき混ぜてろ紙をよくほぐし,時計皿でふたをして水浴上で15分間加熱する。
e) メチルレッド指示薬1,2滴を加え,塩酸 (1+1) を少しずつ加えて中和し,溶液の色が黄から赤に変
わってから,更に,2,3滴加える。
f)
JIS P 3801に適合したろ紙(5種B,110 mm)を用いてろ過し,温水で10回洗浄する。

5
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) 残留物をるつぼに入れて乾燥し,徐々に加熱してろ紙を灰化した後,950±25 ℃に調節した電気炉で
30分間強熱し,デシケータ中で放冷した後,質量を量る。
注5) 不溶分が漏れるおそれがある場合には,適量のろ紙粉末を漏斗に入れてろ過するとよい。
6.5
計算
試料中の不溶残分は,次の式によって算出する。
100
.
×
=m
m'
insol
ここに,
insol.: 不溶残分 (%)
m: 試料の質量 (g)
m': 残留物の質量 (g)
6.6
許容差
許容差は,0.10 %とする。
7
二酸化けい素の定量方法
7.1
要旨
試料を過塩素酸で溶解し,砂浴上で加熱して二酸化けい素を脱水して不溶性とした後,塩酸を加え,可
溶性塩類を溶かしてろ過する。沈殿は強熱して質量を量り,二酸化けい素量を求める。
7.2
試薬
試薬は,次のものを用いる。
a) 過塩素酸 (60 %)
b) 塩酸 (1+1)
7.3
試料の量り採り量
試料は,約1 gを0.000 1 gまで量り採る。
7.4
操作
定量操作は,次の手順によって行う。
a) 試料を乾燥したビーカー100 mLに入れ,過塩素酸 (60 %) 10 mLを加え,ガラス棒でかき混ぜて溶解
する。
b) 砂浴上6) で加熱し,内容物がはね飛ばないように注意して水分を蒸発させ,過塩素酸の白煙が出始め
たら時計皿でふたをして7),ビーカーの底を少し砂にうずめるようにして,更に,5分間加熱を続け
る。
c) ビーカーを砂浴から降ろして放冷した後,時計皿を水で洗って取り除き,塩酸 (1+1) 5 mL及び温水
約20 mLを加えてかき混ぜ,ゼリー状の大きな塊をガラス棒でよくつぶす。
d) JIS P 3801に適合したろ紙(5種B,110 mm)を用いてろ過し,温水で10〜12回洗浄する。ろ液はビ
ーカー500 mLに受け,室温まで冷却した後,全量フラスコ250 mLに洗い移し,標線まで水を加えて
振り混ぜる。この溶液を試料溶液 (A) とし,酸化アルミニウムなどの定量に用いる。
e) 沈殿をるつぼに入れて乾燥し,徐々に加熱して炎の出ないように注意しながらろ紙を灰化した後,
1 175±25 ℃8) に調節した電気炉で1時間強熱し,デシケータ中で放冷した後,質量を量る。
注6) 砂浴の温度は,砂の中ほどに温度計を差し込んで,約180 ℃に調節すればよい。
7) 最初からふたをして水分を蒸発させると,時計皿に凝集した水滴が落下し,内容物が飛散す
る場合がある。

6
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8) 強熱は1 050±25 ℃で行ってもよい。
7.5
計算
試料中の二酸化けい素の含有率は,次の式によって算出する。
.
100
2
insol
m
m'
SiO
−
×
=
ここに,
SiO2: 二酸化けい素の含有率 (%)
m': 沈殿の質量 (g)
m: 試料の質量 (g)
insol.: 箇条6で求めた不溶残分 (%)
7.6
許容差
許容差は,0.20 %とする。
8
酸化アルミニウムの定量方法
8.1
要旨
7.4のd)で保存した試料溶液 (A) の一部を分取し,過量のEDTAを加えた後,pHを約3に調節し,煮
沸してEDTA錯化合物を完成させる。冷却した後,pHを5.5に調節し,キシレノールオレンジを指示薬と
して亜鉛標準液で逆滴定する。
8.2
試薬
試薬は,次のものを用いる。
a) EDTA溶液 (0.01 mol/L) エチレンジアミン四酢酸二水素二ナトリウム二水和物3.73 gを水に溶かし
て全量フラスコ1 000 mLに入れ,標線まで水を加えて振り混ぜ,ポリエチレン製の瓶に保存する。
b) 酢酸ナトリウム溶液 (pH 5.5) 酢酸ナトリウム三水和物250 gを水約1 Lに溶かし,酢酸を加えてpH
メーターを用いてpHを5.5に調節する。
c) 酢酸アンモニウム溶液 (200 g/L)
d) キシレノールオレンジ溶液 (1 g/L) 3,3'-ビス[N,N-ジ(カルボキシメチル)-アミノメチル]-o-クレ
ゾールスルホフタレイン0.1 gを水100 mLに溶かし,褐色瓶に入れて冷暗所に保存する。2か月ごと
に新しく調製する。
e) 亜鉛標準液 (0.01 mol/L) JIS K 8005に規定する容量分析用標準物質の亜鉛約0.65 gを0.000 1 gまで
量り採ってビーカー300 mLに入れ,水約10 mL及び塩酸 (1+1) 20 mLを加え,時計皿でふたをして
水浴上で加熱し溶解する。時計皿を水で洗って取り除き,水約100 mL及び酢酸アンモニウム溶液 (200
g/L) を加え,pHメーターを用いてpHを5.5に調節した後,全量フラスコ1 000 mLに移し入れ,標
線まで水を加えて振り混ぜる。この標準液の酸化アルミニウム相当量は,次の式によって算出し,小
数点以下7けたに丸める。
2
96
.
101
000
1
1
39
.
65
)
100
/
(
×
×
×
=
a
m
E
ここに,
E: 亜鉛標準液 (0.01 mol/L) 1 mLの酸化アルミニウム相当量 (g)
m: 亜鉛の質量 (g)
a: 亜鉛の純度 (%)
又は,国家計量標準にトレーサブルな市販の亜鉛標準液V1 mLを分取し,V2 mLの全量フラスコに
移し入れ,標線まで水を加え振り混ぜて,亜鉛として約0.01 mol/Lとなるように調製する。
この標準液の酸化アルミニウム相当量は,次の式によって算出し,小数点以下7けたに丸める。

7
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2
96
.
101
1
39
.
65
)
000
1/
(
2
1
×
×
×
=
V
V
C
E
ここに,
E: 亜鉛標準液 (0.01 mol/L) 1 mLの酸化アルミニウム相当量 (g)
C: 市販の亜鉛標準液の濃度 (mg/mL)
V1: 市販の亜鉛標準液の分取量 (mL)
V2: 全量フラスコの容量 (mL)
8.3
光度滴定装置
滴定槽,検出器及び滴定液添加装置で構成する。
a) 滴定槽 滴定槽は滴定を行う容器であり,滴定中に溶液をかき混ぜることができるもの9)。
b) 検出器 滴定中に溶液の波長530 nm及び650 nm付近における吸光度を測定できるもの。
c) 滴定液添加装置 滴定液を定量的に添加できるもの10)。
注9) 定量操作に用いるビーカーをそのまま使用することが望ましい。
10) ビュレット,自動ビュレットなど。
8.4
操作
8.4.1
目視による場合
a) 7.4のd)で保存した試料溶液 (A) から全量ピペットで25 mLをビーカー300 mLに分取し,試料中の
酸化アルミニウム,酸化鉄 (III),酸化チタン (IV) の含有率に応じて,EDTA溶液 (0.01 mol/L) を表
1に示す添加量,全量ピペットを用いて加え,更に水を加えて液量を約100 mLとする。
b) 酢酸ナトリウム溶液 (pH 5.5) をかき混ぜながら加え,pHメーターを用いて溶液のpHを約3に調節
し11),約5分間煮沸する。
c) 冷却後,酢酸アンモニウム溶液 (200 g/L) を加え,pHメーターを用いて溶液のpHを5.5に調節する。
d) キシレノールオレンジ指示薬1,2滴を滴加し,亜鉛標準液 (0.01 mol/L) で滴定し12),黄色が赤みを
帯びたときを終点とする。
注11) メチルオレンジ指示薬を用いてpHを調節してもよい。
12) 変色が鈍感であるから,終点近くではよくかき混ぜながらゆっくり滴定するとよい。
表1−EDTA溶液 (0.01 mol/L) 添加量
[酸化アルミニウムの含有率]+[酸化鉄 (III) の含有率]+[酸化チタン (IV) の含有率]
%
添加量
mL
5
10
10
20
15
30
8.4.2
光度滴定による場合
a) 8.4.1のa)〜c)と同様に操作する。
b) キシレノールオレンジ指示薬1,2滴を滴加し,溶液をかき混ぜながら亜鉛標準液 (0.01 mol/L) で滴
定を行い,滴定量ごとの波長530 nm付近における吸光度を,溶液の色が赤に変わるまで測定する13)。
c) 滴定量と溶液の吸光度の関係をプロットし,滴定曲線を作成して,変曲点又は滴定曲線の傾斜が最大
の点の横軸の読みを終点とする14)。
なお,空試験も同じ終点判定方法を用いて行う。

8
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注13) 滴定間隔を0.05〜0.10 mL程度で行うとよい。また,空試験は0.02〜0.06 mL程度で行うとよ
い。
14) 滴定曲線の微分量(吸光度の変化量/滴定量の変化量)の最大値を終点として求めてもよい。
8.5
空試験
過塩素酸 (60 %) 10 mLと塩酸 (1+1) 5 mLとをビーカー100 mLに入れ,更に水を約20 mL加えて,よ
くかき混ぜた後,全量フラスコ250 mLに洗い移し,標線まで水を加えて振り混ぜる。これを,試料溶液 (A)
の代わりとし,8.4に従って操作する。
8.6
計算
試料中の酸化アルミニウムの含有率は,次の式によって算出する。
638
.0
)
(
100
25
250
)
(
2
3
2
1
2
3
2
×
+
×
×
×
=
TiO
O
Fe
m
E
V
V
O
Al
−
−
ここに, Al2O3: 酸化アルミニウムの含有率 (%)
V1: 8.4.1のd)又は8.4.2のc)の亜鉛標準液 (0.01 mol/L) の使用量
(mL)
V2: 8.5の亜鉛標準液 (0.01 mol/L) の使用量 (mL)
E: 亜鉛標準液 (0.01 mol/L) 1 mLの酸化アルミニウム相当量 (g)
m: 7.3の試料の質量 (g)
Fe2O3: 箇条9で求めた酸化鉄 (III) の含有率 (%)
TiO2: 箇条14で求めた酸化チタン (IV) の含有率 (%)
8.7
許容差
許容差は,0.20 %とする。
9
酸化鉄 (III) の定量方法
9.1
要旨
7.4のd)で保存した試料溶液 (A) の一部を分取し,原子吸光分析装置を用いて鉄の吸光度を測定する。
9.2
試薬
試薬は,次のものを用いる。
a) 酸化鉄 (III) 標準原液 (Fe2O3:1.0 mg/mL) 金属鉄(99.0 %以上)0.700 gを量り採り,塩酸 (1+1) 約
10 mLを加えて溶かした後,全量フラスコ1 000 mLに移し,標線まで水を加えて振り混ぜる。又は,
国家計量標準にトレーサブルな市販の鉄標準液を水で希釈し,Fe2O3として約1 mg/mLとなるように
調製する。
b) 酸化鉄 (III) 標準液15) 酸化鉄 (III) 標準原液を10倍に希釈して0〜15 mLを段階的に全量フラスコ
100 mLに分取し,過塩素酸 (1+500) を標線まで加えて振り混ぜる。
注15) 酸化鉄 (III) 標準原液,酸化マグネシウム標準原液,酸化アルカリ調合原液及び酸化マンガン
(II) 標準原液を適宜,組み合わせて混合標準液を調製してもよい。
9.3
装置
原子吸光分析装置を用いる。
9.4
操作
定量操作は,次の手順によって行う。
a) 7.4のd)で保存した試料溶液 (A) から一定量16) を全量フラスコ100 mLに分取し,標線まで水を加え
て振り混ぜる。これを試料溶液 (B) とする。
b) 原子吸光分析装置の鉄用光源ランプを用いて,波長248.3 nmにおける試料溶液 (B) の吸光度を測定

9
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
する。
なお,ランプ電流値,分光器のスリット幅,可燃ガス及び助燃ガスの圧力と流量及び光束位置は,
各装置に応じて適切な条件を設定する。感度が不足する場合は,目盛拡大装置を使用するか,又は装
置に内蔵されている感度調節装置によって調節する。また,感度が必要以上に高い場合は,光束角度
などの調節によって測定感度を減じる。
注16) 分取量は,酸化鉄 (III) 含有率及び使用する装置によって適宜変える。通常,10 mL分取する
のがよい。
9.5
検量線の作成
原子吸光分析装置の鉄用光源ランプを用いて,波長248.3 nmにおける酸化鉄 (III) 標準液のそれぞれの
吸光度を測定し,検量線を作成する。
9.6
計算
9.5で作成した検量線から試料溶液 (B) 中の酸化鉄 (III) 量を求め,試料中の酸化鉄 (III) の含有率は,
次の式によって算出する。
100
250
3
2
×
×
=
v
m
c
O
Fe
ここに, Fe2O3: 酸化鉄 (III) の含有率 (%)
c: 試料溶液 (B) 中の酸化鉄 (III) 量 (g/100 mL)
m: 7.3の試料の質量 (g)
v: 試料溶液 (A) からの分取量 (mL)
9.7
許容差
許容差は,0.10 %とする。
10 酸化カルシウムの定量方法
10.1 カルシウム用指示薬を用いる定量方法
10.1.1 要旨
7.4のd)で保存した試料溶液 (A) の一部を分取し,アンモニア水を加えて中和し,生成した水酸化物の
沈殿をろ過する。ろ液を冷却した後,水酸化カリウム溶液を加えてpHを12.7〜13.2に調節する。次に,
カルシウム用指示薬を加えてEDTA標準液で滴定する。
10.1.2 試薬
試薬は,次のものを用いる。
a) メチルレッド指示薬 (2 g/L) 6.2 c)と同様に調製する。
b) アンモニア水 (1+1)
c) 硝酸アンモニウム溶液 (20 g/L)
d) トリエタノールアミン (1+1)
e) 水酸化カリウム溶液(約3 mol/L) 水酸化カリウム200 gを水に溶かして1 000 mLとする。
f)
カルシウム用指示薬 2-ヒドロキシ-1-(2'-ヒドロキシ-4'-スルホ-1'-ナフチルアゾ)-3-ナフトエ酸0.5 g
と硫酸カリウム50 gとを混合粉砕して均一にし,褐色瓶に保存する。
g) 亜鉛標準液 (0.02 mol/L) JIS K 8005に規定する容量分析用標準物質の亜鉛約0.65 gを0.000 1 gまで
量り採ってビーカー300 mLに入れ,水10 mL及び塩酸 (1+1) 20 mLを加え,時計皿でふたをして水
浴上で溶解する。全量フラスコ500 mLに洗い移し,室温まで冷却した後,標線まで水を加えて振り
混ぜる。次の式によってファクターを算出し,小数点以下4けたに丸める。

10
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
9
653
.0
×
×
=
a
m
f
ここに,
f: 亜鉛標準液 (0.02 mol/L) のファクター
m: 亜鉛の質量 (g)
a: 亜鉛の純度 (%)
h) 緩衝液 (pH 10) 塩化アンモニウム70 gを適量の水に溶かし,アンモニア水570 mLを加え,水を加
えて1 000 mLとする。
i)
EBT指示薬 エリオクロムブラックT 0.2 gをトリエタノールアミン15 mL及びJIS K 8102に規定す
るエタノール5 mLに溶かし,スポイト付き褐色滴瓶に保存する。
j)
EDTA標準液 (0.02 mol/L) エチレンジアミン四酢酸二水素二ナトリウム二水和物7.5 gを適量の水に
溶かして,全量フラスコ1 000 mLに入れ,標線まで水を加えて振り混ぜ,ポリエチレン製の瓶に保存
する。
10.1.3 操作
10.1.3.1 目視による場合
a) EDTA標準液 (0.02 mol/L) を,次のように標定する。
亜鉛標準液 (0.02 mol/L) 25 mLを全量ピペットで分取してビーカー300 mLに入れ,水を加えて約100
mLとし,緩衝液を加えてpHを9.5〜10.0 に調節し17),EBT指示薬2,3滴を加え,タングステンラ
ンプ照明器に載せて,EDTA標準液 (0.02 mol/L) で滴定する。赤紫色から赤みが全く消えて青色とな
ったときを終点とする。
なお,タングステンランプ照明器は,30 W程度のタングステンランプを用い,その光をガラス又は
プラスチック製の乳白板を通して,ビーカー中の溶液を透過させることができる装置とする。この滴
定値から,次の式によってEDTA標準液 (0.02 mol/L) の酸化カルシウム相当量を算出し,小数点以下
7けたに丸める。
v
f
E
×
×
=
25
6
121
001
.0
ここに,
E: EDTA標準液 (0.02 mol/L) 1 mLの酸化カルシウム相当量 (g)
f: 亜鉛標準液 (0.02 mol/L) のファクター
v: EDTA標準液 (0.02 mol/L) の使用量 (mL)
注17) 緩衝液を約3 mL加えれば,規定のpHとなる。
b) 7.4のd)で保存した試料溶液 (A) から全量ピペットで20 mLずつを2個のビーカー300 mLに分取す
る。
c) それぞれに温水を加えて約150 mLとし,煮沸し始めるまで加熱する。
d) メチルレッド指示薬1,2滴を加え,かき混ぜながら溶液の色が赤から黄に変わるまでアンモニア水
(1+1) を徐々に滴加し,更に1,2滴加える。
e) 約1分間煮沸した後,加熱を止め,沈殿が沈むのを待って直ちにJIS P 3801に適合したろ紙(5種A,
110 mm)を用いてろ過し,硝酸アンモニウム温溶液 (20 g/L) で8回洗浄する。ろ液は,ビーカー500
mLに受け,沈殿は捨てる。
なお,使用する硝酸アンモニウム溶液 (20 g/L) は,メチルレッド指示薬2,3滴を加え,溶液の色
が赤から黄に変わるまでアンモニア水 (1+1) を滴加して用いる。この溶液を加熱したときに色が赤
に戻った場合は,更にアンモニア水 (1+1) を滴加して黄色にして用いる。
f)
試料が高炉セメント又は高炉スラグのときは,次の操作によってろ液からマンガンを除く。

11
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ろ液に塩酸 (1+1) を加えて弱酸性とした後,加熱して約150 mLとなるまで濃縮し,これに飽和臭
素水5 mL及びアンモニア水 (1+1) 10 mLを加えて5分間以上煮沸し,沈殿が凝集して溶液が透明と
なった後,JIS P 3801に適合したろ紙(5種B,110 mm)でろ過し,温水で7,8回洗浄する。ろ液に
塩酸 (1+1) を加えて酸性とし,煮沸して臭素を完全に除去する。
g) ろ液を濃縮して約200 mLとし,室温まで冷却する。
h) 一方のろ液にトリエタノールアミン (1+1) 2 mL及び水酸化カリウム溶液(約3 mol/L)を加えてpH
メーターを用いてpHを12.7〜13.2 18) に調節する。
i)
2〜3分間放置した後,カルシウム用指示薬0.1 gを加え,タングステンランプ照明器に載せてEDTA
標準液 (0.02 mol/L) で滴定し,赤紫色から赤みが全く消えて青色となったときを終点とし,これを予
備滴定値とする。
なお,タングステンランプ照明器は,30 W程度のタングステンランプを用い,その光をガラス又は
プラスチック製の乳白板を通して,ビーカー中の溶液を透過させることができる装置とする。
j)
もう一方のろ液にトリエタノールアミン (1+1) 2 mLを加えてかき混ぜ,ビュレットでEDTA標準液
(0.02 mol/L) を加え,i)の予備滴定値より1〜2 mL少ないところで一度止めてかき混ぜる。
k) h)と同量の水酸化カリウム溶液(約3 mol/L)を加えてかき混ぜる。2〜3分間放置した後,カルシウ
ム用指示薬0.1 gを加え,i)と同様にEDTA標準液 (0.02 mol/L) で滴定する。
注18) 溶液のpHが12.7〜13.2の範囲外では,結果が不正確になる。水酸化カリウム溶液(約3 mol/L)
の量は,約7 mLで規定のpHとなる。
10.1.3.2 光度滴定による場合
a) EDTA標準液 (0.02 mol/L) を,8.3に規定する光度滴定装置を用いて次のように標定する。
亜鉛標準液 (0.02 mol/L) 25 mLを全量ピペットで分取してビーカー300 mLに入れ,水を加えて約100
mLとし,緩衝液を加えてpHを9.5〜10.0に調節し17),EBT指示薬2,3滴を加え,溶液をかき混ぜ
ながらEDTA標準液 (0.02 mol/L) で滴定する。滴定量ごとの波長650 nm付近における吸光度を溶液
が完全に青に変わるまで測定する19)。滴定終了後,滴定量と溶液の吸光度の関係をプロットし,滴定
曲線を作成して,変曲点又は滴定曲線の傾斜が最大の点の横軸の読みを終点とする14)。この滴定値か
ら,10.1.3.1のa)の式によってEDTA標準液 (0.02 mol/L) の酸化カルシウム相当量を算出し,小数点
以下7けたに丸める。
b) 10.1.3.1のb)〜j)と同様に操作する。
c) 10.1.3.1のh)と同量の水酸化カリウム溶液(約3 mol/L)を加えてかき混ぜ,2〜3分間放置した後,カ
ルシウム用指示薬を0.1 g加え,溶液をかき混ぜながらEDTA標準液 (0.02 mol/L) で8.3に規定する光
度滴定装置を用いて滴定を行い,滴定量ごとの波長650 nm付近における溶液の吸光度を測定する。
d) 溶液の色が完全に青に変化してから,1 mL程度過滴定を行い測定を終了する20)。
e) 滴定量と溶液の吸光度の関係をプロットし,滴定曲線を作成して,変曲点又は滴定曲線の傾斜が最大
の点の横軸の読みを終点とする14)。
注19) 滴定間隔を0.02〜0.04 mL程度で行うとよい。
20) 滴定間隔を0.04〜0.08 mL程度で行うとよい。
10.1.4 計算
試料中の酸化カルシウムの含有率は,次の式によって算出する。
100
20
250×
×
×
=
m
E
v
CaO

12
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに, CaO: 酸化力ルシウムの含有率 (%)
v: 10.1.3.1のk)又は10.1.3.2のe)で求めたEDTA標準液 (0.02
mol/L) の使用量 (mL)
E: 10.1.3.1のa)又は10.1.3.2のa)で求めたEDTA標準液 (0.02
mol/L) 1 mLの酸化カルシウム相当量 (g)
m: 7.3の試料の質量 (g)
10.1.5 許容差
許容差は,0.25 %とする。
10.2 カルセイン指示薬又はカルセイン−PPC指示薬を用いる定量方法
10.2.1 要旨
7.4のd)で保存した試料溶液 (A) の一部を分取し,水酸化カリウム溶液を加えてpHを12.7〜13.2に調
節する。次にカルセイン指示薬又はカルセイン−PPC指示薬を加えて,EDTA標準液で滴定する。
10.2.2 試薬
試薬は,次のものを用いる。
a) トリエタノールアミン (1+1)
b) 水酸化カリウム溶液(約3 mol/L) 10.1.2のe)と同様に調製する。
c) カルセイン指示薬 カルセイン{3,3'-ビス[N,N-ジ(カルボキシメチル)-アミノメチル]-フルオレセ
イン}0.10 gを硫酸カリウム10 gと共に混合粉砕して均一にし,褐色瓶に保存する。
d) カルセイン−PPC指示薬 カルセイン{3,3'-ビス[N,N-ジ(カルボキシメチル)-アミノメチル]-フ
ルオレセイン}0.08 gと3,3'-ビス[N,N-ジ(カルボキシメチル)-アミノメチル]-フェノールフタレイ
ン0.02 gとを硫酸カリウム10 gと共に混合粉砕して均一にし,褐色瓶に保存する。
e) EDTA標準液 (0.02 mol/L) 10.1.2のj)と同様に調製する。
10.2.3 操作
10.2.3.1 目視による場合
a) 10.1.3.1のa)によってEDTA標準液 (0.02 mol/L) を標定する。
b) 7.4のd)で保存した試料溶液 (A) から,全量ピペットで20 mLずつを2個のビーカー300 mLに分取
し,水を加えて約200 mLとする。
c) 一方の溶液にトリエタノールアミン (1+1) 10 mL及び水酸化カリウム溶液(約3 mol/L)を加えてpH
12.7〜13.2 21) に調節する。
d) 2〜3分間放置した後,カルセイン指示薬又はカルセイン−PPC指示薬約0.05 gを加え,EDTA標準液
(0.02 mol/L) で滴定し22),カルセイン指示薬の場合は,溶液の色が蛍光性黄緑色からオレンジ色(無
蛍光)となったときを終点とし,カルセイン−PPC指示薬の場合は,蛍光性黄緑色から赤紫色(無蛍
光)となったときを終点として,これを予備滴定値とする。
e) もう一方の溶液にトリエタノールアミン (1+1) 10 mLを加えてかき混ぜ,ビュレットでEDTA標準液
(0.02 mol/L) を加え,d)の予備滴定値より1〜2 mL少ないところで一度止めてかき混ぜる。
f)
c)と同量の水酸化カリウム溶液(約3 mol/L)を加えてかき混ぜ,2〜3分間放置する21)。カルセイン
指示薬又はカルセイン−PPC指示薬約0.05 gを加え,d)と同様にEDTA標準液 (0.02 mol/L) で滴定す
る22)。
注21) 溶液のpHが12.7〜13.2の範囲外では結果が不正確になる。
22) 滴定は,黒色紙の上で行うと終点が判別しやすい。
10.2.3.2 光度滴定による場合

13
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 10.1.3.2のa)によってEDTA標準液 (0.02 mol/L) を標定する。
b) 10.2.3.1のb)〜e)と同様に操作する。
c) 10.2.3.1のc)と同量の水酸化カリウム溶液(約3 mol/L)を加えてかき混ぜ,2〜3分間放置した後,カ
ルセイン指示薬又はカルセイン−PPC指示薬を0.05 g加え,溶液をかき混ぜながらEDTA標準液 (0.02
mol/L) で8.3に規定する光度滴定装置を用いて滴定を行い,滴定量ごとの波長530 nm付近における
溶液の吸光度を測定する。
d) カルセイン指示薬の場合は,溶液の色が完全にオレンジ色(無蛍光)に変化してから,カルセイン−
PPC指示薬の場合は,溶液の色が完全に赤紫色(無蛍光)に変化してから,1 mL程度過滴定を行い測
定を終了する20)。
e) 滴定量と溶液の吸光度の関係をプロットし,滴定曲線を作成して,変曲点又は滴定曲線の傾斜が最大
の点の横軸の読みを終点とする14)。
10.2.4 計算
試料中の酸化カルシウムの含有率は,次の式によって算出する。
100
20
250×
×
×
=
m
E
v
CaO
ここに, CaO: 酸化カルシウムの含有率 (%)
v: 10.2.3.1のf)又は10.2.3.2のe)で求めたEDTA標準液 (0.02
mol/L) の使用量 (mL)
E: 10.1.3.1のa)又は10.1.3.2のa)で求めたEDTA標準液 (0.02
mol/L) 1 mLの酸化カルシウム相当量 (g)
m: 7.3の試料の質量 (g)
10.2.5 許容差
許容差は,0.25 %とする。
11 酸化マグネシウムの定量方法
11.1 要旨
7.4のd)で保存した試料溶液 (A) の一部を分取し,原子吸光分析装置を用いてマグネシウムの吸光度を
測定する。
11.2 試薬
試薬は,次のものを用いる。
a) 酸化マグネシウム標準原液 (MgO:1 mg/mL) 金属マグネシウム(99.0 %以上)0.603 gを量り採り,
塩酸 (1+1) 約10 mLを加えて溶かした後,全量フラスコ1 000 mLに移し,標線まで水を加えて振り
混ぜる。又は,国家計量標準にトレーサブルな市販のマグネシウム標準液を水で希釈し,MgOとして
約1 mg/mLとなるように調製する。
b) 酸化マグネシウム標準液15) 酸化マグネシウム標準原液を5倍に希釈して0〜10 mL(MgOとして0
〜2.0 mg)を段階的に全量フラスコ100 mLに分取し,過塩素酸 (1+500) を標線まで加えて振り混ぜ
る。
11.3 装置
原子吸光分析装置を用いる。
11.4 操作
定量操作は,次の手順によって行う。
14
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 7.4のd)で保存した試料溶液 (A) から一定量23) を全量フラスコ100 mLに分取し,標線まで水を加え
て振り混ぜる。これを試料溶液 (C) とする。
b) 原子吸光分析装置のマグネシウム用光源ランプを用いて,波長285.2 nmにおける試料溶液 (C) の吸
光度を測定する。
なお,ランプ電流値,分光器のスリット幅,可燃ガス及び助燃ガスの圧力と流量及び光束位置は,
各装置に応じて適切な条件を設定する。感度が不足する場合は,目盛拡大装置を使用するか,装置に
内蔵されている感度調節装置によって調節する。また,感度が必要以上に高い場合は,光束角度など
の調節によって測定感度を減じる。
注23) 分取量は,酸化マグネシウム含有率と使用する装置によって適宜変える。通常,10 mL分取
するのがよい。
11.5 検量線の作成
原子吸光分析装置のマグネシウム用光源ランプを用いて,波長285.2 nmにおける酸化マグネシウム標準
液のそれぞれの吸光度を測定し,検量線を作成する。
11.6 計算
11.5で作成した検量線から試料溶液 (C) 中の酸化マグネシウム量を求め,試料中の酸化マグネシウムの
含有率は,次の式によって算出する。
100
250×
×
=
v
m
c
MgO
ここに, MgO: 酸化マグネシウムの含有率 (%)
c: 試料溶液 (C) 中の酸化マグネシウム量 (g/100 mL)
m: 7.3の試料の質量 (g)
v: 試料溶液 (A) からの分取量 (mL)
11.7 許容差
許容差は,0.15 %とする。
12 三酸化硫黄の定量方法
12.1 要旨
6.4のc)で保存した試料溶液 (F) の一部又はろ液(洗液を含む)に塩化バリウム溶液を加え,硫酸バリ
ウムの沈殿を生成させる。沈殿をろ別した後,強熱して質量を量る。
12.2 試薬
試薬は,次のものを用いる。
a) 塩化バリウム溶液 (100 g/L) 塩化バリウム100 gを水に溶かして,1 000 mLとする。
12.3 操作
定量操作は,次の手順によって行う。
a) 6.4のc)で保存した試料溶液 (F) から200 mLを全量ピペットで分取して,ビーカー500 mL に入れる。
なお,酸化チタン (IV) の定量を行わない場合には,6.4のc)で保存したろ液及び洗液のすべてを三
酸化硫黄の定量に用いる。
b) 溶液を加熱して5分間煮沸する。入念にかき混ぜながら,溶液を沸点で維持し,塩化バリウム温溶液
(100 g/L) 10 mLをピペットを用いて徐々に滴加し,更に数分間煮沸を続ける。
c) 煮沸に近い温度で約3時間静置する。この間,溶液の量がほぼ200 mLに保たれるように注意し,必
15
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
要ならば適宜温水を加える。
d) 沈殿をJIS P 3801に適合したろ紙(6種,110 mm)を用いてろ過し,温水で8〜10回洗浄する。
e) 沈殿はるつぼに入れて乾燥し,徐々に加熱して炎の出ないように注意しながらろ紙を灰化し,950±
25 ℃の電気炉で30分間強熱し,デシケータ中で放冷した後,質量を量る。
12.4 計算
試料中の三酸化硫黄の含有率は,次の式によって算出する。
100
200
250
343
.0
'
3
×
×
×
=
m
m
SO
ここに, SO3: 三酸化硫黄の含有率 (%)
m: 6.3の試料の質量 (g)
m': 硫酸バリウム(沈殿)の質量 (g)
なお,6.4のc)で保存した溶液をすべて三酸化硫黄の定量に用いた場合には,試料中の三酸化硫黄の含
有率は,次の式によって算出する。
100
343
.0
'
3
×
×
=
m
m
SO
ここに, SO3: 三酸化硫黄の含有率 (%)
m: 6.3の試料の質量 (g)
m': 硫酸バリウム(沈殿)の質量 (g)
12.5 許容差
許容差は,0.10 %とする。
13 酸化ナトリウム及び酸化カリウムの定量方法
13.1 原子吸光法
13.1.1 要旨
7.4のd)で保存した試料溶液 (A) の一部を分取し,原子吸光分析装置を用いて酸化ナトリウム及び酸化
カリウムの吸光度を測定する。
13.1.2 試薬
試薬は,次のものを用いる。
a) カルシウム原液 アルカリ分析用の炭酸カルシウム114.3 gに水300 mLを加え,かき混ぜながら過塩
素酸 (60 %) 600 mLを徐々に加えて溶かす。これを室温まで冷却した後,全量フラスコ1 000 mLに移
し,標線まで水を加えて振り混ぜ,ポリエチレン製の瓶に保存する。この溶液1 mLは,64 mgのCaO
を含む。
b) 塩化ナトリウム(99.5 %以上) 500〜650 ℃に40〜50分間保ち,硫酸デシケータ中で放冷する。
c) 塩化カリウム(99.5 %以上) 110 ℃に2〜3日間保ち,硫酸デシケータ中で放冷する。
d) アルカリ原液 塩化ナトリウム1.886 g及び塩化カリウム1.583 gを適量の水に溶かして全量フラスコ
1 000 mLに移し,標線まで水を加えて振り混ぜ,ポリエチレン製の瓶に保存する。この溶液1 mLは,
それぞれ1 mgのNa2O及びK2Oを含む。又は,国家計量標準にトレーサブルな市販のナトリウム標準
液及びカリウム標準液を水で希釈し,Na2Oとして約1 mg/mL及びK2Oとして約1 mg/mLとなるよう
に調製する。
e) 酸化アルカリ調合原液 表2に従って調製する。カルシウム原液及びアルカリ原液は,全量フラスコ
1 000 mLに全量ピペット又はビュレットで分取し,標線まで水を加えて振り混ぜる。調合原液は,ポ
16
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
リエチレン製の瓶に入れて保存する。
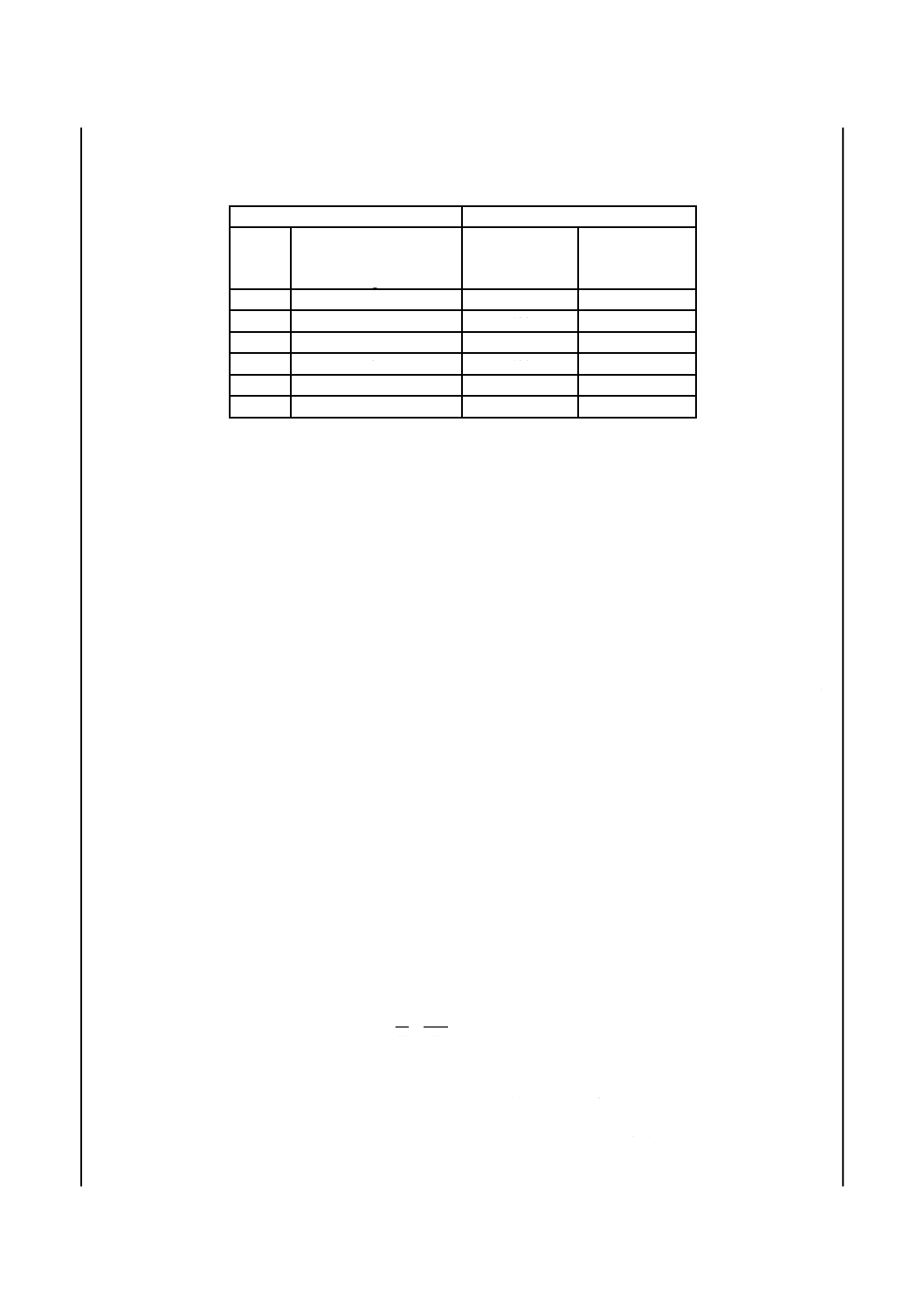
表2−酸化アルカリ調合原液の調合割合
酸化アルカリ調合原液の種類
調合割合
番号
調合原液中のアルカリ量
(Na2O,K2Oとして)
mg/L
カルシウム原液
mL
アルカリ原液
mL
No.1
100
100
100
No.2
75
100
75
No.3
50
100
50
No.4
25
100
25
No.5
10
100
10
No.6
0
100
0
f)
酸化アルカリ標準液15) 酸化アルカリ調合原液を正確に25倍に希釈する。
13.1.3 装置
原子吸光分析装置を用いる。
13.1.4 操作
定量操作は,次の手順によって行う。
a) 7.4のd)で保存した試料溶液 (A) から全量ピペットで10 mLを,全量フラスコ100 mLに分取し,標
線まで水を加えて振り混ぜる。これを試料溶液 (D) とする。
b) 原子吸光分析装置のナトリウム用光源ランプを用いて,波長589.0 nmにおける試料溶液 (D) の吸光
度を測定する。
c) 原子吸光分析装置のカリウム用光源ランプを用いて,波長766.5 nmにおける試料溶液 (D) の吸光度
を測定する。
13.1.5 検量線の作成
原子吸光分析装置のナトリウム及びカリウム用光源ランプを用いて波長589.0 nm及び766.5 nmにおけ
る酸化アルカリ標準液のそれぞれの吸光度を測定し,検量線を作成する。
なお,ランプ電流値,分光器のスリット幅,可燃ガス及び助燃ガスの圧力と流量及び光束位置は,各装
置に応じて適切な条件を設定する。感度が不足する場合は,目盛拡大装置を使用するか,又は装置に内蔵
されている感度調節装置によって調節する。また,感度が必要以上に高い場合は,光束角度などの調節に
よって測定感度を減じる。
13.1.6 計算
13.1.5で作成した検量線から試料溶液 (D) 中の酸化ナトリウム及び酸化カリウム量を求め,試料中の酸
化ナトリウム及び酸化カリウムの含有率は,次の式によって算出する。
100
250
2
2
×
×
=
v
m
c
O
K
O
Na又は
ここに, Na2O又はK2O: 酸化ナトリウム又は酸化カリウムの含有率 (%)
c: 試料溶液 (D) 中の酸化ナトリウム又は酸化カリウ
ムの量 (g/100 mL)
m: 7.3の試料の質量 (g)
v: 試料溶液 (A) からの分取量 (mL)
13.1.7 許容差
17
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
酸化ナトリウムの許容差は,0.03 %とする。
酸化カリウムの許容差は,0.04 %とする。
13.2 炎光光度法
13.2.1 要旨
試料は,塩酸を用いて処理する。溶液中の酸化ナトリウム及び酸化カリウムの含有量は,炎光光度法に
よって測定する。
なお,この方法は,不溶残分量が3 %を超えないセメントについて規定する。
13.2.2 試薬
試薬は,次のものを用いる。
a) 塩酸 (1+9)
b) 緩衝溶液 塩化セシウム50 gと硝酸アルミニウム九水和物250 gとを水に溶かし,約1 Lにする。こ
の溶液は使用するときに調製する。
c) 塩化ナトリウム(99.5 %以上) 500〜650 ℃に40〜50分間保ち,硫酸デシケータ中で放冷する。
d) 塩化カリウム(99.5 %以上) 110 ℃に2〜3日間保ち,硫酸デシケータ中で放冷する。
e) アルカリ保存溶液24) 塩化ナトリウム0.254 2±0.000 5 gと塩化カリウム0.190 7±0.000 5gとを水に溶
かし,1 000 mLの全量フラスコに定容する。
注24) この溶液はNaとして100 mg/L,Kとして100 mg/Lの濃度となる。
13.2.3 装置
十分な安定性があり,波長589 nmのナトリウム線及び波長768 nmのカリウム線の発光強度を測定する
ことが可能な炎光光度計を用いる。
13.2.4 操作
定量操作は,次の手順によって行う。
a) 白金皿又は磁器の蒸発皿にセメント約0.2 g 25) を0.000 1 gまで正しく量り採り,水3 mLで懸濁液と
し,塩酸 (1+9) 20 mLを加えた後,蒸発乾固させる。
b) 残分に温水と塩酸 (1+9) 2 mLとを加え,JIS P 3801に適合したろ紙(5種B,110 mm)を用いてろ過
する。
なお,ろ液は緩衝溶液10 mLを入れた全量フラスコ100 mLに受ける。
c) 全量フラスコの標線近くまで,残分を温水で洗浄する。室温まで冷却した後,標線まで水を加えて振
り混ぜる。
d) 炎光光度計で試料溶液の発光強度を測定し,13.2.5の検量線からNa2OとK2Oとの含有率 (%) を求め
る。
注25) 対応国際規格では,量り採り範囲を0.200±0.005 gと規定している。
13.2.5 検量線の作成
新しいバッチの試薬を用いるときには必ず,この方法によってアルカリ量を求める。
なお,試薬のアルカリ量(13.2.6で試料量を0.2 gとして計算した値)が0.01 %を超えた場合は,違う新
しいバッチで再度,確認するか,新しい検量線溶液を最初から調製する。
検量線の各点の検量線溶液を調製するために,塩酸 (1+9) 20 mLを蒸発乾固させる。塩酸 (1+9) 2 mL
と水3 mLとで蒸発した残分を溶解する。全量フラスコ100 mLに溶液を移し,緩衝溶液10 mLを加える。
ビュレットを用いて全量フラスコに表3に示したアルカリ保存溶液量を加えた後,標線まで水を加えて振
り混ぜる。
18
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3−フラスコに加える保存溶液量
全量フラスコの番号
1
2
3
4
5
6
7
保存溶液 mL
0
1
3
5
10
20
30
フラスコ1から7の各アルカリ量は,試料の量り採り量が0.200 0 gの場合における,セメント中の酸化
ナトリウム又は酸化カリウムの含有率 (%) に換算した値とし,それらの値を表4に示す。
表4−フラスコ中の保存溶液のアルカリ量
全量フラスコの番号
1
2
3
4
5
6
7
Na2O %
0
0.07
0.20
0.34
0.67
1.35
2.02
K2O %
0
0.06
0.18
0.30
0.60
1.20
1.81
炎光光度計の炎光中に検量線溶液を噴霧する。最初にフラスコ1のブランク溶液を噴霧し,装置のゼロ
点補正を行う。
炎光中にアルカリ量の少ない順に検量線溶液(フラスコ2〜7)を噴霧する。589 nmのNa2Oの発光強度
と768 nmのK2Oの発光強度とを測定する。検量線溶液中の酸化ナトリウム及び酸化カリウムの含有率に
対する,測定した強度の検量線を作成する。
13.2.6 計算
13.2.5で作成した検量線から試料量0.200 0 gの場合の仮の酸化ナトリウム及び酸化カリウムの含有率
(%) を求め,試料中の酸化ナトリウム及び酸化カリウムの含有率は,次の式によって補正する。
0
200
.0
2
2
又は
×
=mc
O
K
O
Na
ここに, Na2O又はK2O: 酸化ナトリウム又は酸化カリウムの含有率 (%)
c: 仮の酸化ナトリウム又は酸化カリウムの含有率 (%)
m: 13.2.4 a)の試料の質量 (g)
13.2.7 許容差
酸化ナトリウムの許容差は,0.03 %とする。
酸化カリウムの許容差は,0.04 %とする。
14 酸化チタン (IV) の定量方法
14.1 要旨
6.4のc)で保存した試料溶液 (F) の一部を分取し,塩酸濃度を調節して,アスコルビン酸を加えて鉄を
還元する。次に,ジアンチピリルメタン溶液を加えて試料溶液を呈色させ,吸光度を測定する。
14.2 試薬
試薬は,次のものを用いる。
a) アスコルビン酸溶液 (100 g/L) ポリエチレン製の瓶に入れて冷暗所に保存する26)。
b) ジアンチピリルメタン溶液 (20 g/L) ジアンチピリルメタン2 gを塩酸 (1+5) 30 mLに溶かし,水で
100 mLに希釈する。
c) 酸化チタン (IV) 標準原液 (TiO2:0.20 mg/mL) 酸化チタン (IV)(99.0 %以上)をるつぼに取り,約
1 000 ℃で1時間強熱してデシケータ中で放冷する27)。この中から0.200 gを白金るつぼに量り採り,

19
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ピロ硫酸カリウム4 gを加えて融解する。冷却後,白金るつぼをビーカーに入れ,硫酸 (1+9) を加え
て温め,50 ℃以下で溶解する。白金るつぼを取り出して水で洗浄した後,溶液を全量フラスコ1 000 mL
に移し,硫酸 (1+9) を標線まで加えて振り混ぜる。又は,市販のチタン標準液を水で希釈し,TiO2
として約0.2 mg/mLとなるように調製する。
d) 酸化チタン (IV) 標準液 (TiO2:0.01 mg/mL) 酸化チタン (IV) 標準原液を使用の都度,水で20倍に
希釈する。
注26) 調製後1か月を経過したものは,使用しないほうがよい。
27) 酸化チタン (IV) は,水分を含む場合があるので,この操作を行う。
14.3 操作
定量操作は,次の手順によって行う。
a) 6.4のc)で保存した試料溶液 (F) の一部を全量フラスコ50 mLに分取し28),塩酸 (1+1) 5 mL及びア
スコルビン酸溶液 (100 g/L) 3 mLを加えて1分間放置した後,ジアンチピリルメタン溶液 (20 g/L) 20
mLを加え,標線まで水を加えて振り混ぜ,1時間放置する。
b) 溶液の一部を吸収セル10 mmに分取し,水を対照液として波長390 nm付近の吸光度を吸光光度計を
用いて測定する。
注28) 分取量は,試料中の酸化チタン (IV) 含有率に応じて加減する。酸化チタン (IV) の含有率が
0.5 %未満の場合は,10 mLが適量である。
14.4 検量線の作成
酸化チタン (IV) 標準液0〜20.0 mL(TiO2として0〜0.200 mg)を段階的に全量フラスコ50 mLに分取
し,14.3のa)及びb)と同様に操作し,検量線を作成する。
14.5 計算
14.4で作成した検量線から呈色溶液中の酸化チタン (IV) 量を求め,試料中の酸化チタン (IV) の含有率
は,次の式によって算出する。
100
250
2
×
×
=
v
m
a
TiO
ここに, TiO2: 酸化チタン (IV) の含有率 (%)
a: 呈色溶液中の酸化チタン (IV) 量 (g/50 mL)
m: 6.3の試料の質量 (g)
v: 試料溶液 (F) の分取量 (mL)
14.6 許容差
許容差は,0.02 %とする。
15 酸化りん (V) の定量方法
15.1 要旨
7.4のd)で保存した試料溶液 (A) の一部を分取し,p-ニトロフェノールを指示薬として水酸化ナトリウ
ム溶液及び硫酸でpHを調節した後,モリブデン酸アンモニウム溶液及びアスコルビン酸溶液を加え,沸
騰水中で呈色させ,冷却後,吸光度を測定する。
15.2 試薬
試薬は,次のものを用いる。
a) p-ニトロフェノール指示薬 (2 g/L)

20
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 水酸化ナトリウム溶液 (100 g/L)
c) 硫酸 (1+1)
d) モリブデン酸アンモニウム溶液 (20 g/L) 七モリブデン酸六アンモニウム四水和物2 gを温水約20
mLに溶かし,硫酸 (1+1) 60 mLを加え,水で100 mLに希釈する。
e) アスコルビン酸溶液 (100 g/L) 14.2のa)の溶液を用いる。
f)
酸化りん (V) 標準原液 (P2O5:0.1 mg/mL) りん酸二水素カリウム(99.0 %以上)を105〜110 ℃で2
時間乾燥し,デシケータ中で放冷したものを0.191 7 g量り採り,水に溶かして全量フラスコ1 000 mL
に移し,標線まで水を加えて振り混ぜる。又は,国家計量標準にトレーサブルな市販のりん酸イオン
標準液を水で希釈し,P2O5として約0.1 mg/mLとなるように調製する。
g) 酸化りん (V) 標準液 (P2O5:0.01 mg/mL) 酸化りん (V) 標準原液を水で10倍に希釈する。
15.3 操作
定量操作は,次の手順によって行う。
a) 7.4のd)で保存した試料溶液 (A) から全量ピペットで50 mLを,全量フラスコ100 mLに分取する。
なお,測定した試料溶液の吸光度が検量線の範囲外となった場合には,試料溶液 (A) の分取量を最
小で5 mLまで少なくしてもよい。
b) p-ニトロフェノール指示薬 (2 g/L) 1滴を加え,次に溶液が黄色を呈するまで水酸化ナトリウム溶液
(100 g/L) を滴加した後,硫酸 (1+1) を滴加し,溶液を無色とする。
c) モリブデン酸アンモニウム溶液 (20 g/L) 10 mL及びアスコルビン酸溶液 (100 g/L) 2 mLを加え,標線
近くまで水を加える。
d) 沸騰水中に15分間放置した後,流水中で室温まで冷却し,標線まで水を加えて振り混ぜる。
e) 溶液の一部を吸収セル10 mmに分取し,水を対照液として波長830 nm付近の吸光度を吸光光度計を
用いて測定する。
15.4 検量線の作成
酸化りん (V) 標準液 (P2O5:0.01 mg/mL) 0〜20 mL(P2O5として0〜0.20 mg)を段階的に全量フラスコ
100 mLに分取し,15.3のb)〜e)と同様に操作し,検量線を作成する。
15.5 計算
15.4で作成した検量線から呈色溶液中の酸化りん (V) 量を求め,試料中の酸化りん (V) の含有率は,
次の式によって算出する。
100
250
5
2
×
×
=
v
m
a
O
P
ここに, P2O5: 酸化りん (V) の含有率 (%)
a: 呈色溶液中の酸化りん (V) 量 (g/100 mL)
m: 7.3の試料の質量 (g)
v: 試料溶液 (A) からの分取量 (mL)
15.6 許容差
許容差は,0.02 %とする。
16 酸化マンガン (II) の定量方法
16.1 原子吸光法
16.1.1 要旨
7.4のd)で保存した試料溶液 (A) の一部を分取し,原子吸光分析装置を用いてマンガンの吸光度を測定

21
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
する。
16.1.2 試薬
試薬は,次のものを用いる。
a) 酸化マンガン (II) 標準原液 (MnO:0.2 mg/mL) 金属マンガン(99.0 %以上)0.154 gを量り採り,
水30 mLと過塩素酸 (60 %) 5 mLとを加えて,水浴上で加熱し,溶解する。室温まで冷却した後,全
量フラスコ1 000 mLに移し,標線まで水を加えて振り混ぜる。又は,国家計量標準にトレーサブルな
市販のマンガン標準液を水で希釈し,MnOとして約0.2 mg/mLとなるように調製する。
b) 酸化マンガン (II) 標準液15) 酸化マンガン (II) 標準原液を10倍に希釈して0〜10 mL(MnOとして
0〜0.2 mg)を段階的に全量フラスコ100 mLに分取し,塩酸 (1+10) 2 mLを加えた後,過塩素酸 (1
+500) を標線まで加えて振り混ぜる。
16.1.3 装置
原子吸光分析装置を用いる。
16.1.4 操作
定量操作は,次の手順によって行う。
a) 7.4のd)で保存した試料溶液 (A) から一定量を全量フラスコ100 mLに分取し,標線まで水を加えて
振り混ぜる29)。これを試料溶液 (E) とする。
b) 原子吸光分析装置のマンガン用光源ランプを用いて波長279.5 nmにおける試料溶液 (E) の吸光度を
測定する。
注29) 分取量は,酸化マンガン (II) 含有率と使用する装置によって適宜変える。通常,10 mL分取
するのがよい。
16.1.5 検量線の作成
原子吸光分析装置のマンガン用光源ランプを用いて波長279.5 nmにおける酸化マンガン (II) 標準液の
吸光度をそれぞれ測定し,検量線を作成する。
16.1.6 計算
16.1.5で作成した検量線から試料溶液 (E) 中の酸化マンガン (II) 量を求め,試料中の酸化マンガン (II)
の含有率は,次の式によって算出する。
100
250×
×
=
v
m
c
MnO
ここに, MnO: 酸化マンガン (II) の含有率 (%)
c: 試料溶液 (E) 中の酸化マンガン (II) 量 (g/100 mL)
m: 7.3の試料の質量 (g)
v: 試料溶液 (A) からの分取量 (mL)
16.1.7 許容差
許容差は,0.02 %とする。
16.2 吸光光度法
16.2.1 要旨
試料中のマンガンを,過よう素酸カリウムを用いてMnO4−に酸化する。紫色を呈した溶液の吸光度を測
定する。鉄 (III) イオンは過マンガン酸イオンの形成を助け,溶液の色を安定させるリン酸と反応し錯体
を作る。
16.2.2 試薬
22
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試薬は,次のものを用いる。
a) 無水硫酸マンガン 硫酸マンガン水和物 (MnSO4・xH2O) を250±10 ℃で質量が変化しなくなるまで
乾燥する。乾燥後の組成は,MnSO4となる。
b) マンガン (II) 標準液 無水硫酸マンガン約2.75 gを0.000 1 gまで正しく量り採り,水に溶かして全
量フラスコ1 000 mLに入れ,標線まで水を加えて振り混ぜる。この標準液のマンガン量Gは,次の
式によって算出し,小数点以下3けたに丸める。
5
748
2.
m
G=
ここに,
G: マンガン (II) 標準液のマンガン量 (mg/mL)
m: 無水硫酸マンガンの質量 (g)
c) 硝酸
d) りん酸
e) 過よう素酸カリウム
f)
アンモニア水
16.2.3 装置
吸光光度計を用いる。
16.2.4 操作
試料中のマンガンの含有率によって,セメントを0.1 gから1 gの間で0.000 1 gまで正しく量り採り,ビ
ーカー200 mLに入れる(マンガンの含有率が0.10 %未満の場合,試料は1 g程度にして,適切なマンガン
濃度になるように試料量を変えることが望ましい。)。水約75 mLを加え,試料を分散させながら注意深く
硝酸15 mLを加え,硫化水素 (H2S) 臭がなくなり,未溶解のセメントがなくなるまで加熱する。
JIS P 3801に適合したろ紙(5種B,110 mm)を用いてろ過し,ろ液はビーカー300 mLに受ける。ろ液
が120 mLとなるまで残分を温水で洗浄する。このろ液にりん酸10 mLを加えて混合し,過よう素酸カリ
ウム1.5 gを加える。過マンガン酸塩に特有なピンク色が現れるまで煮沸する。呈色しなければ,アンモニ
ア水を呈色するまで数滴加える。呈色したら,穏やかに30分間煮沸を続ける。溶液を冷却した後,全量フ
ラスコ200 mLに洗い移す。室温まで冷却し,標線まで水を加えて振り混ぜる。
水を対照液として波長525 nm付近の吸光度を吸光光度計を用いて測定する。吸光度は小数点以下3け
たまで測定する。
16.2.5 検量線の作成
全量フラスコ500 mL (No.1) 及び全量フラスコ1 000 mL (No.2) それぞれにマンガン (II) 標準液20 mL
を全量ピペットで分取し,標線まで水を加えて振り混ぜる。全量フラスコ200 mL (No.3),全量フラスコ500
mL (No.4) 及び全量フラスコ1 000 mL (No.5) それぞれにフラスコNo.2の溶液100 mLを全量ピペットで分
取し,標線まで水を加えて振り混ぜる。
フラスコNo.1〜No.5のそれぞれの溶液100 mLを全量ピペットでビーカー300 mLに分取する。硝酸20
mL,過よう素酸カリウム1.5 gとりん酸10 mLとを加え,加熱して30分間穏やかに煮沸する。20 ℃に冷
却後,ビーカーの溶液を全量フラスコ200 mLに移し,標線まで水を加えて振り混ぜる。
溶液の一部を吸収セルに取り,水を対照液として波長525 nm付近の吸光度を吸光光度計を用いて測定
する。
吸収セルは,適切な光路長のものを使用する。吸光度は,小数点以下3けたまで測定する。
標準液 (E1〜E5) の吸光度を各標準液の濃度 (mg/200 mL) に対してプロットし,検量線を作成する。表
23
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5に各標準液のマンガンの濃度を示す。16.2.2のb)で求めたGが1.000の場合,各標準液の濃度は表5に
示す値となる。Gが1.000ではない場合,表5に示すマンガンの濃度にGを乗じて,マンガンの濃度を求
める。
表5−マンガンの検量線溶液の濃度
検量線溶液
E1
E2
E3
E4
E5
マンガンの濃度 (mg/200 mL)
4.0
2.0
1.0
0.4
0.2
16.2.6 計算
16.2.5で作成した検量線から試料中のマンガン (II) 量を求め,試料中の酸化マンガン (II) の含有率は,
次の式によって算出する。
m
C
.
m
C
Mn
×
=
×
×
=
1
0
000
1
100
Mn
.
MnO
×
=29
1
ここに,
Mn: マンガンの含有率 (%)
C: マンガン (II) の濃度 (mg/200 mL)
m: 16.2.4の試料の質量 (g)
MnO: 酸化マンガン (II) の含有率 (%)
16.2.7 許容差
許容差は,0.02 %とする。
17 硫化物硫黄の定量方法
17.1 要旨
試料を塩酸で溶解し,発生した硫化水素を亜鉛アンミン溶液中で硫化亜鉛の沈殿として捕集する。捕集
した硫化亜鉛は,でんぷん溶液を指示薬として,よう素酸カリウム標準液で滴定する。又はよう素酸カリ
ウム溶液を過剰に加え,遊離したよう素をチオ硫酸ナトリウム標準液で逆滴定する。
17.2 よう素酸カリウム標準液を用いる直接滴定法
17.2.1 試薬
試薬は,次のものを用いる。
a) 亜鉛アンミン溶液 硫酸亜鉛七水和物50 gをアンモニア水 (7+3) 500 mLに溶かし,一夜静置した後,
ろ過する。
b) 塩化すず (II) 溶液 塩化すず (II) 二水和物10 gをビーカー100 mLに入れ,塩酸 (1+1) 7 mLを加え
て静かに加熱して溶解する。冷却した後,水95 mLを加える。この溶液は,使用するときに調製する。
c) でんぷん溶液 でんぷん(水溶性)1 gを水5 mLに懸濁させ,これを煮沸に近い熱水100 mL中にか
き混ぜながら注ぎ込む。冷却した後,水酸化ナトリウム1 gを水10 mLに溶かした溶液を加え,次に,
よう化カリウム3 gを加えてよくかき混ぜる。
d) よう素酸カリウム(容量分析用標準物質) 120〜140 ℃に90〜120分間保ち,硫酸デシケータ中で放
冷する。
e) よう素酸カリウム標準液 よう素酸カリウム1.112 gとよう化カリウム12 gとを水に溶かして全量フ
ラスコ1 000 mLに入れ,標線まで水を加えて振り混ぜる。
f)
酢酸鉛溶液 酢酸鉛 (II) 三水和物13.2 gを水約1 Lに溶かす。
24
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
17.2.2 装置
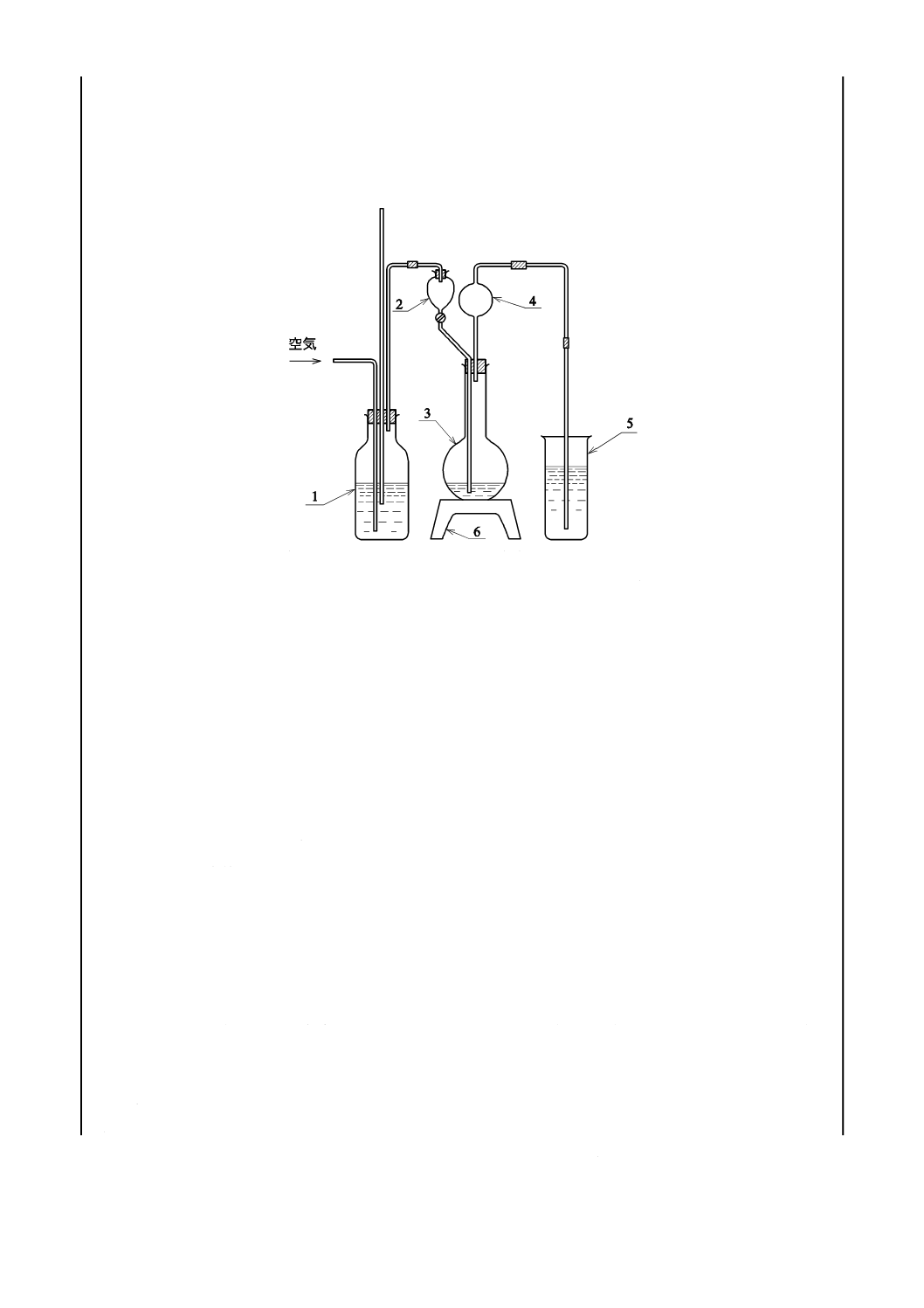
装置は,図1のとおりとする。ゴム栓及びゴム管は,あらかじめ温希塩酸でよく洗浄して用いる。
1
空気洗浄瓶
2
分液漏斗
3
ガス発生用フラスコ500 mL
4
連結球
5
トールビーカー500 mL
6
加熱器
図1−装置
17.2.3 試料の量り採り量
試料は,約5 gを0.000 1 gまで正しく量り採る。
なお,高炉セメント及び高炉スラグの場合,試料は,約1 gを0.000 1 gまで正しく量り採る。
17.2.4 操作
定量操作は,次の手順によって行う。
a) 試料を乾燥したガス発生用フラスコ500 mL中に入れ,次にトールビーカー500 mLに亜鉛アンミン溶
液15 mL及び水285 mLを入れる。
b) フラスコに水約20 mLを加え,静かに振り混ぜて試料をよく分散させ,フラスコの栓を気密にし,連
結球に連なるガス導通管の下端をトールビーカーの溶液中に深く入れる。この操作は,フラスコ中の
試料が固まらないように手早く行う。
c) 分液漏斗を通して塩化すず (II) 溶液25 mLを注入し,直ちに分液漏斗のコックを閉じてフラスコの内
容物を振り混ぜる。場合によって,硫化鉄の溶解を助けるために,金属クロムを0.1 g入れてもよい。
d) 塩酸 (1+3) 100 mLを漏斗から注入し,直ちに漏斗のコックを閉じてフラスコの内容物を振り混ぜる。
漏斗の口を空気洗浄瓶に連結し,分液漏斗のコックを開いて静かに空気を通しながらフラスコを徐々
に加熱し,内容物を5〜6分間緩やかに煮沸した後,加熱を止め,引き続き3〜4分間空気を通す。
なお,空気中に硫化水素又は二酸化硫黄が存在する疑いのある場合には,空気洗浄瓶中に酢酸鉛溶
液を入れておく。
e) ガス導通管を外してそのままビーカー中に残し,かき混ぜ棒として用いる。
f)
ビーカー中の溶液を室温まで冷却した後,でんぷん溶液4 mL及び塩酸 (1+1) 40 mLを加え,直ちに

25
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
よう素酸カリウム標準液を用いて手早く滴定し,溶液が青色になったときを終点とする。
17.2.5 計算
試料中の硫化物硫黄の含有率は,次の式によって算出する。
100
50
000
.0
×
×
=
m
v
S
ここに,
S: 硫化物硫黄の含有率 (%)
v: よう素酸カリウム標準液の使用量 (mL)
m: 試料の質量 (g)
0.000 50: よう素酸カリウム標準液1 mLの硫化物硫黄相当量 (g)
17.3 チオ硫酸ナトリウム標準液を用いる逆滴定法
17.3.1 試薬
試薬は,次のものを用いる。
a) 亜鉛アンミン溶液 硫酸亜鉛七水和物50 gをアンモニア水 (7+3) 500 mLに溶かし,一夜静置した後,
ろ過する。
b) 塩化すず (II) 溶液 塩化すず (II) 二水和物10 gをビーカー100 mLに入れ,塩酸 (1+1) 7 mLを加え
て静かに加熱して溶解する。冷却した後,水95 mLを加える。この溶液は,使用するときに調製する。
c) でんぷん溶液 でんぷん(水溶性)1 gに,よう化カリウム (KI) 1 gを加え,これを水に溶かして100
mLにする。2週間以内に使用する。
d) よう素酸カリウム(容量分析用標準物質) 120〜140 ℃に90〜120分間保ち,硫酸デシケータ中で放
冷する。
e) よう素酸カリウム標準液(約0.016 67 mol/L) よう素酸カリウム約3.6 g 30) を0.000 1gまで正しく量
り採り,全量フラスコ1 000 mLに入れる。水酸化ナトリウム0.2 g及びよう化カリウム25 gを加え,
沸騰させて冷却した新鮮な水に溶かす。これを,同じ水を標線まで加えて振り混ぜる。
よう素酸カリウム標準液のファクターFは,次の式によって算出し,小数点以下4けたに丸める。
4
567
3.
m
F=
ここに,
F: よう素酸カリウム標準液(約0.016 67 mol/L)のファクター
m: よう素酸カリウムの質量 (g)
f)
チオ硫酸ナトリウム標準液(約0.1 mol/L) チオ硫酸ナトリウム (Na2S2O3・5H2O) 24.82 gを水に溶か
して,1 000 mLにする。この溶液のファクターfを次の方法に従って求める。
よう素酸カリウム標準液(約0.016 67 mol/L)20 mLを全量ピペットで三角フラスコ500 mLに分取
し,水約150 mLで希釈する。塩酸 (1+1) 25 mLを加えて酸性にしてから,調製したチオ硫酸ナトリ
ウム標準液(約0.1 mol/L)で,溶液の黄色が薄くなるまで滴定する。次にでんぷん溶液2 mLを加え
て滴定を続け,色が青から無色となったときを終点とする。
チオ硫酸ナトリウム標準液(約0.1 mol/L)のファクターf は,次の式によって算出し,小数点以下
4けたに丸める。
V
F
V
.
F
.
.
f
×
=
×
×
×
×
=
20
8
566
3
01
214
67
016
0
20
ここに,
f: チオ硫酸ナトリウム標準液(約0.1 mol/L)のファクター
F: よう素酸カリウム標準液(約0.016 67 mol/L)のファクター
V: チオ硫酸ナトリウム標準液(約0.1 mol/L)の使用量 (mL)
3.566 8: KIO3溶液(約0.016 67 mol/L)1 L中に含まれるよう素酸カ
26
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
リウムの質量 (g)
214.01: よう素酸カリウムの分子量
注30) 対応国際規格では,量り採り範囲を3.6±0.1 gと規定している。
17.3.2 装置
装置は,17.2.2による。
17.3.3 試料の量り採り量
試料は,約5 g 31) を0.000 1 gまで正しく量り採る。
なお,高炉セメント及び高炉スラグの場合,試料は,約1 gを0.000 1 gまで正しく量り採る。
注31) 対応国際規格では,試料の量り採り量は,1 gを規定しているが硫化物硫黄の含有率が少ないこ
とから5 gとした。
なお,対応国際規格では,量り採り範囲を1.00±0.05 gと規定している。
17.3.4 操作
定量操作は,次の手順によって行う。
a) 17.2.4のa)〜e)と同様に操作し,試料溶液を調製する。
b) ビーカー中の溶液を室温まで冷却した後,正確によう素酸カリウム標準液(約0.016 67 mol/L)10 mL
と塩酸25 mLとを加える。チオ硫酸ナトリウム標準液(約0.1 mol/L)で,溶液の黄色が薄くなるまで
滴定する。次にでんぷん溶液2 mLを加えて滴定を続け,色が青から無色になったときを終点とする。
17.3.5 計算
試料中の硫化物硫黄の含有率は,次の式によって算出する。
m
f
V
F
V
S
×
×
×
×
−
×
=
000
1
100
3
60
.1
)]
(
)
[(
2
1
m
f
V
F
V
.
)
(
)
(
×
−
×
×
=
2
1
3
160
0
ここに,
S: 硫化物硫黄の含有率 (%)
V1: よう素酸カリウム標準液(約0.016 67 mol/L)の添加量 (mL)
F: よう素酸カリウム標準液(約0.016 67 mol/L)のファクター
V2: 滴定に要したチオ硫酸ナトリウム標準液(約0.1 mol/L)の
使用量 (mL)
f: チオ硫酸ナトリウム標準液(約0.1 mol/L)のファクター
m: 試料の質量 (g)
17.4 許容差
硫化物硫黄の許容差は,0.04 %とする。
18 塩素の定量方法
18.1 電位差滴定法
18.1.1 要旨
試料を硝酸で溶解し,塩化物イオン標準液を加え,次に共存する妨害成分を酸化するため過酸化水素水
を添加して加熱する。室温まで冷却した後,塩化物イオン電極を用いた電位差滴定装置によって滴定する。
同様に操作して空試験値を差し引く。
18.1.2 試薬
試薬は,次のものを用いる。
a) 硝酸(比重1.38)
27
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 硝酸(約2 mol/L) 硝酸(比重1.38)145 mLを1 000 mLに希釈する。
c) 過酸化水素水(約30 %)
d) 塩化ナトリウム(容量分析用標準物質) 500〜650 ℃に40〜50分間保ち,硫酸デシケータ中で放冷す
る。
e) 塩化物イオン標準液 (0.005 mol/L) 塩化ナトリウム0.292 2 gを適量の水に溶かして全量フラスコ1
000 mLに移し,標線まで水を加えて振り混ぜる。又は,国家計量標準にトレーサブルな市販の塩化物
イオン標準液を水で希釈し,Clとして約0.005 mol/Lとなるように調製する32)。
f)
硝酸銀標準液 (0.005 mol/L) 硝酸銀0.85 gを適量の水に溶かして全量フラスコ1 000 mLに移し,標
線まで水を加えて振り混ぜる。
この標準液は,次のようにして標定する。
塩化物イオン標準液 (0.005 mol/L) 10 mLを全量ピペットでビーカー200 mLに分取し,水を加えて
約100 mLとする。これに,硝酸(約2 mol/L)5 mLを加える。電位差滴定装置にセットし,硝酸銀標
準液 (0.005 mol/L) で滴定する。
塩化ナトリウムを用いて塩化物イオン標準液を調製した場合は,次の式によってファクターを算出
し,小数点以下4けたに丸める。
v
f
10
=
ここに,
f: 硝酸銀標準液 (0.005 mol/L) のファクター
v: 塩化物イオン標準液 (0.005 mol/L) の滴定に要した硝酸銀標
準液 (0.005 mol/L) の使用量 (mL)
市販の塩化物イオン標準液を用いて塩化物イオン標準液を調製した場合は,次の式によってファク
ターを算出し,小数点以下4けたに丸める。
000
1
1
45
.
35
2
1×
×
=
v
v
a
C
ここに,
C: 塩化物イオン標準液 (0.005 mol/L) の濃度 (mol/L)
a: 市販の塩化物イオン標準液の濃度 (mg/L)
v1: 市販の塩化物イオン標準液の分取量 (mL)
v2: 市販の塩化物イオン標準液を定容した量 (mL)
v
C
f
×
=
000
2
ここに,
f: 硝酸銀標準液 (0.005 mol/L) のファクター
C: 塩化物イオン標準液 (0.005 mol/L) の濃度 (mol/L)
v: 塩化物イオン標準液 (0.005 mol/L) の滴定に要した硝酸銀標
準液 (0.005 mol/L) の使用量 (mL)
注32) 例えば,塩化物イオン標準液 (1 000 mg/L) を用いる場合は,塩化物イオン標準液を20 mL分
取し,100 mLに定容すると,0.005 6 mol/Lとなる。
18.1.3 装置
装置は,次のものを用いる。
a) 電位差滴定装置
b) マグネチックスターラ
18.1.4 試料の量り採り量
試料は,約5 g 33) を0.000 1 gまで正しく量り採る。
28
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注33) 対応国際規格では,量り採り量を5.000 gと規定している。
18.1.5 操作
定量操作は,次の手順によって行う。
a) 試料を乾燥したビーカー200 mLに入れ,水20 mLを加え,ガラス棒でかき混ぜて試料を分散させな
がら,硝酸(比重1.38)12 mLを加える。必要ならば加熱して溶かす34)。
b) 温水を加えて100 mLとし,塩化物イオン標準液 (0.005 mol/L) をビュレットで2.00 mL加えるか,又
は塩化物イオン標準液 (0.005 mol/L) を正確に5倍に希釈した溶液を全量ピペットで10 mLを分取し
て加え,過酸化水素水(約30 %)2 mLを加える。
c) 時計皿でふたをし,加熱して1〜2分間静かに煮沸する。室温まで冷却した後,時計皿とガラス棒とを
水で洗って取り除く。
d) 電位差滴定装置にセットし,硝酸銀標準液 (0.005 mol/L) で滴定する。
注34) 二酸化けい素の含有量の高いスラグ類などでは,溶液にゲル状物質を析出することがあるが,
測定値には影響しない。
18.1.6 空試験
空試験操作は,次の手順によって行う。
a) ビーカー200 mLに塩化物イオン標準液 (0.005 mol/L) をビュレットで2.00 mLを入れるか,又は塩化
物イオン標準液 (0.005 mol/L) を正確に5倍に希釈した溶液を全量ピペットで10 mLを分取して,温
水を加えて100 mLとする。
b) 時計皿でふたをし,18.1.5のc)及びd)の操作を行う。
18.1.7 計算
試料中の塩素の含有率は,次の式によって算出する。
100
3
177
000
.0
)
(
2
1
×
×
×
=
m
f
v
v
Cl
−
ここに,
Cl: 塩素の含有率 (%)
m: 試料の質量 (g)
v1: 硝酸銀標準液 (0.005 mol/L) の使用量 (mL)
v2: 空試験による硝酸銀標準液 (0.005 mol/L) の使用量 (mL)
f: 硝酸銀標準液 (0.005 mol/L) のファクター
18.1.8 許容差
塩素の許容差は,0.003 %とする。
18.2 チオシアン酸水銀 (II) による吸光光度法
18.2.1 要旨
試料を硝酸で溶解し,炭酸カルシウムを用いて中和する。ろ液に硫酸アンモニウム鉄 (III) 硝酸溶液及
びチオシアン酸水銀 (II) エタノール溶液を加えて発色させ,吸光度を測定する。
18.2.2 試薬
試薬は,次のものを用いる。
a) 硝酸(約6 mol/L) 硝酸(比重1.38)225 mLに水を加えて500 mLとする。
b) 硝酸 (1+2)
c) 過酸化水素水(約30 %)
d) 炭酸カルシウム35)
e) 硫酸アンモニウム鉄 (III) 硝酸溶液 (100 g/L) 硫酸アンモニウム鉄 (III)・12水50 gを硝酸(約6

29
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
mol/L)に溶かし,500 mLとする。濁りがあればJIS P 3801に適合したろ紙(5種A,110 mm)36) を
用いてろ過し,褐色瓶に入れて保存する。
f)
チオシアン酸水銀 (II) エタノール溶液 (3 g/L) チオシアン酸水銀 (II) 1.5 gをJIS K 8102に規定する
エタノールに溶かし,500 mLとする。濁りがあればJIS P 3801に適合したろ紙(5種A,110 mm)36)
を用いてろ過し,褐色瓶に入れて保存する。
g) 塩化ナトリウム(容量分析用標準物質) 500〜650 ℃に40〜50分間保ち,硫酸デシケータ中で放冷す
る。
h) 塩化物イオン標準原液 (Cl:0.5 mg/mL) 塩化ナトリウム0.824 gを量り採り,適量の水に溶かして全
量フラスコ1 000 mLに移し,標線まで水を加えて振り混ぜる。又は,国家計量標準にトレーサブルな
市販の塩化物イオン標準液を水で希釈し,Clとして約0.5 mg/mLとなるように調製する。
i)
塩化物イオン標準液 (Cl:0.02 mg/mL) 塩化物イオン標準原液 (Cl:0.5 mg/mL) を使用の都度,水で
正しく25倍に希釈する。
注35) 炭酸カルシウムは,塩素含有量の少ないものを用いることが望ましい。
36) 使用するろ紙からの汚染を除くため,ろ過に際しあらかじめろ紙を洗浄しておくことが望ま
しい。
18.2.3 装置
吸光光度計を用いる。
18.2.4 試料の量り採り量
試料は,約2.0 gを0.000 1gまで正しく量り採る。
18.2.5 操作
定量操作は,次の手順によって行う。
a) 試料を乾燥したビーカー200 mLに入れ,水10 mLを加え,ガラス棒でかき混ぜて分散させながら,
硝酸 (1+2) 14 mLを加えて溶解する。このとき,必要ならば少し温めて未溶解の塊をガラス棒の先で
よくつぶして可溶分を完全に溶かす。
なお,フライアッシュセメントB種及びC種並びにシリカセメントB種及びC種の場合は,硝酸 (1
+2) の使用量を10 mLとする。
b) 過酸化水素水(約30 %)2 mLを加え,時計皿でふたをして,濃い赤褐色が消えるまで加熱し,更に
数秒間静かに煮沸する。
c) 室温まで冷却した後,炭酸カルシウム1.5 gを加える。
d) ふたをしたまま再び穏やかに加熱し,約1分間煮沸する。
e) 漏斗にJIS P 3801に適合したろ紙(5種B,110 mm)を付け,ろ紙からの汚染を除くため,温水を漏
斗に満たして4回洗浄する。この洗液は廃棄する。
f)
試料溶液をろ過し,温水で8回洗浄する。ろ液37) は,全量フラスコ200 mLに直接受ける。
g) 室温まで冷却した後,硫酸アンモニウム鉄 (III) 硝酸溶液 (100 g/L) 20 mL及びチオシアン酸水銀 (II)
エタノール溶液 (3 g/L) 15 mLを全量ピペット又はビュレットを用いて加える。
h) 標線まで水を加えて振り混ぜ,10分間放置する。
i)
溶液の一部を光路長50 mmの吸収セルに分取し,水を対照液として波長460 nm付近の吸光度を吸光
光度計を用いて測定する。
注37) ろ液及び洗液の合量は,通常,約150 mLになる。
18.2.6 空試験

30
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
水10 mLに硝酸 (1+2) 2 mLを加え,以下,18.2.5のb)〜i)に従って操作し,空試験値を求める。
18.2.7 検量線の作成
塩化物イオン標準液 (Cl:0.02 mg/mL) 0〜25 mL(Clとして0〜0.5 mg)を段階的に全量フラスコ200 mL
に分取し,水を加えて約150 mLとした後,以下,18.2.5のg)〜i)に従って吸光度を測定し,検量線を作成
する38)。
注38) この検量線は,直線にならないので,点数を多くとることが必要である。
18.2.8 計算
18.2.7で作成した検量線から呈色溶液中の塩化物イオン量を求め,試料中の塩素含有率は,次の式によ
って算出する。
100
10
)
(
3×
×
=
−
m
b
a
Cl
−
ここに,
Cl: 塩素の含有率 (%)
a: 呈色溶液中の塩化物イオン量 (mg/200 mL)
b: 空試験溶液中の塩化物イオン量 (mg/200 mL)
m: 試料の質量 (g)
注記 この試験に用いた溶液中には水銀が含まれているので,その廃液処理については,十分に注意
する必要がある。
18.2.9 許容差
塩素の許容差は,0.003 %とする。
18.3 チオシアン酸アンモニウム溶液による逆滴定法
18.3.1 要旨
試料を硝酸で溶解し,共存する各種の硫化物イオンを酸化するため,加熱する。硝酸によって溶解した
塩化物は,室温まで冷却した後,硝酸銀溶液を添加して塩化物イオンを沈殿させ,再度,煮沸して沈殿物
をろ別する。
ろ液を25 ℃以下に冷却し,残留する硝酸銀を,硫酸アンモニウム鉄 (III) 溶液を指示薬として,チオシ
アン酸アンモニウム溶液で逆滴定する。
18.3.2 試薬
試薬は,次のものを用いる。
a) 硝酸 (1+2)
b) 硝酸 (1+100)
c) 硝酸銀標準液 (0.05 mol/L) 150±10 ℃で質量が一定となるまで乾燥した硝酸銀8.494 0 g 39) を正し
く量り採り,適量の水に溶かして全量フラスコ1 000 mLに移し,標線まで水を加えて振り混ぜる。褐
色瓶に保存し,遮光する。
d) 硫酸アンモニウム鉄 (III) 溶液 硫酸アンモニウム鉄 (III)・12水の飽和水溶液100 mLに硝酸 (1+2)
10 mLを加える。
e) チオシアン酸アンモニウム溶液(約0.05mol/L) チオシアン酸アンモニウム3.8±0.1 gを適量の水に
溶かして全量フラスコ1 000 mLに移し,標線まで水を加えて振り混ぜる。
注39) 対応国際規格では,量り採り範囲を8.494 0±0.000 5 gと規定している。
18.3.3 試料の量り採り量
試料は,約5 g 40) を0.000 1 gまで正しく量り採る。
31
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注40) 対応国際規格では,量り採り範囲を5.00±0.05 gと規定している。
18.3.4 操作
定量操作は,次の手順によって行う。
a) 試料を乾燥したビーカー500 mLに入れ,水50 mLを加え,ガラス棒でかき混ぜて分散させながら,
硝酸 (1+2) 50 mLを加えて溶解する。
b) このとき,未溶解の塊をガラス棒の先でよくつぶして可溶分を完全に溶かし,溶液をこぼさないよう
に加熱して,1〜2分間煮沸する(硫化物を含む試料の場合は,ドラフトの中で操作する。)。
c) 加熱を止め,硝酸銀標準液 (0.05 mol/L) 5 mLを全量ピペットで加える。
d) ふたたび加熱し,少なくとも1〜2分間煮沸する。溶液をJIS P 3801に適合したろ紙(5種A,110 mm)
を用いてろ過し,ろ液は500 mLビーカーに受ける。残分及びろ紙は硝酸 (1+100) で洗浄する。ビー
カー及びガラス棒を硝酸 (1+100) で洗浄し取り除く。ろ液は約200 mLとなる。
e) ろ液は光を避けるか,又は暗所で25 ℃以下に冷却する。
f)
硫酸アンモニウム鉄 (III) 溶液を指示薬として5 mL加え,チオシアン酸アンモニウム溶液(約0.05
mol/L)をかき混ぜながら滴下し,かすかなピンク色(オレンジ色)が消えなくなるまで滴定する。
g) 滴定に使用したチオシアン酸アンモニウム溶液(約0.05 mol/L)の使用量 (v2) を求める。v2が0.5 mL
より少ない場合には,試料量を半分にして手順をやり直す。
h) 空試験として,乾燥したビーカー500 mLに水50 mLと硝酸 (1+2) 50 mLとを加え,以下,上記のb)
〜f)に従って操作し,滴定に使用したチオシアン酸アンモニウム溶液(約0.05 mol/L)の使用量 (v1) を
求める。
18.3.5 計算
試料中の塩素の含有率は,次の式によって算出する。
100
000
1
)
(5
773
.1
1
2
1
×
×
×
×
=
m
v
v
v
Cl
−
m
v
v
v
×
×
=
1
2
1
)
(
5
0.886
−
ここに,
Cl: 塩素の含有率 (%)
m: 試料の質量 (g)
v1: 空試験によるチオシアン酸アンモニウム溶液(約0.05 mol/L)
の使用量 (mL)
v2: チオシアン酸アンモニウム溶液(約0.05 mol/L)の使用量 (mL)
18.3.6 許容差
塩素の許容差は,0.003 %とする。
32
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
完全分析によるセメントの主成分の化学分析方法
序文
セメントの湿式による化学分析方法の国際規格は,ISO 29581-1 (Cement−Test methods−Part 1:Analysis
by wet chemistry) (セメントの湿式による化学分析方法は,ISO 680:1990として規定されていたが,2009
年3月15日に改正された。その際,規格番号は,ISO 680からISO 29581-1に変更となった。)であるが,
国際規格は,セメントの主成分(二酸化けい素,酸化アルミニウム,酸化第二鉄,酸化カルシウム及び酸
化マグネシウム)の分析方法としてセメントを完全に分解する方法を規定している。一方,この規格の本
体では,セメント中の酸に溶解する成分の化学分析を行うと規定しているため,国際規格で規定している
主成分の分析方法をこの規格の本体に取り入れることができなかった。
そのため,これらの方法を翻訳し,技術的内容を変更することなく附属書として示した。
なお,点線の下線は,規定内容を明確にするため追加した部分である。
A.1 適用範囲
この附属書は,セメントの湿式による化学分析方法について規定する。
この附属書は,標準方法を規定するとともに,分析項目によっては,標準方法と同じ結果が得られると
考えられる代替方法も規定する。ただし,疑義が生じた場合には,標準方法だけを適用する。また,標準
方法又は代替方法と異なる方法で分析してもよい。ただし,それらの方法による分析結果が標準方法によ
る分析結果と同等であることを示す必要があり,疑義が生じた場合には,標準方法だけを適用する。
この附属書は,セメントのほかにセメントの構成物質にも適用できる。
A.2 引用規格
次に掲げる規格は,この附属書に引用されることによって,この附属書の規定の一部を構成する。これ
らの引用規格は,その最新版(追補を含む。)を適用する。
ISO 385,Laboratory glassware−Burettes
ISO 835,Laboratory glassware−Graduated pipettes
A.3 試験の一般的要求事項
A.3.1 試験回数
試験は,同一試料について1回又は2回以上行う。品質管理を目的とする分析の場合の試験回数は,最
低で1回とする。品質管理を目的とする分析でない場合の試験回数は,2回とする(A.3.3参照)。また,
疑義が生じた場合の分析の試験回数は,2回とする(A.3.3参照)。
A.3.2 繰返し精度及び再現精度
A.3.2.1 一般
この附属書における繰返し精度又は再現精度は,例えば,百分率,グラムの単位などで繰返し又は再現
時の標準偏差として示されている。
33
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.3.2.2 繰返し精度
繰返し精度の標準偏差は,同じ条件下(同じ試験者,同じ器具と装置,同じ試験室,そして短時間の間
隔)で同一の試料について同じ試験方法で行われた試験結果間の精度を示す。
A.3.2.3 再現精度
再現精度の標準偏差は,異なる条件下(異なる試験者,異なる器具と装置,異なる試験室)で同一の試
料について同じ試験方法で行われたそれぞれの試験結果間の精度を示す。
A.3.3 質量,体積,ファクター及び結果の表し方
質量は0.000 1 gまで量る。ビュレットの体積は0.05 mLまで読む。
溶液のファクターは3回の測定値を平均し,小数点以下3けたで示す。
試験回数が1回の場合は,試験結果は一般に百分率で小数点以下2けたで示す。
試験回数が2回の場合は,試験結果は2回の定量値の平均とし,一般に百分率で小数点以下2けたで示
す。
2回の定量値の差が,繰返し精度の標準偏差の2倍以上であった場合,再試験を行い,最も近い二つの
定量値の平均とする。すべての個々の試験結果は,記録として残す。
A.3.4 強熱
強熱は,次のように行う。
あらかじめ空焼きし,風袋を量ったるつぼにろ紙と沈殿を入れて乾燥し,酸化雰囲気で炎が出ないよう
に徐々に加熱してろ紙を灰化した後,規定の温度で強熱する。強熱後,るつぼと内容物をデシケータ中で
室温まで放冷した後,質量を量る。
A.3.5 恒量
15分間の強熱を繰り返し行い,そのたび放冷した後,質量を量る。その前後の質量の差が0.000 5 g未満
になったときを,恒量とする。
A.3.6 塩化物イオンの有無の確認(硝酸銀試験)
沈殿を通常5,6回洗浄した後,漏斗の足を2,3滴の水で洗い流す。ろ紙と沈殿を数mLの水で洗浄し,
試験管に受ける。硝酸銀溶液(A.4.2.27参照)を数滴加え,溶液の濁り又は沈殿の有無を調べる。濁り又
は沈殿があった場合は,硝酸銀試験で塩化物イオンが認められなくなるまで,沈殿の洗浄を続ける。
A.4 試薬
A.4.1 一般
試薬は分析用試薬とし,水は電気伝導度が0.5 mS/m以下の蒸留水又はイオン交換水とする。
%はただし書きがない限り,質量百分率を示す。
この規格において使用する試薬の密度 (ρ) (g/cm3,20 ℃) は,次のとおりである。
− 塩酸 1.18〜1.19
− 酢酸 1.05〜1.06
− 硝酸 1.40〜1.42
− りん酸 1.71〜1.75
− 過塩素酸 1.60〜1.67
− アンモニア水 0.88〜0.91
なお,薄めた試薬は,塩酸 (1+2) のように表し,これは容積で塩酸:水=1:2で混合したものを示す。
34
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.4.2 使用する試薬
A.4.2.1 塩酸 (HCl)
A.4.2.2 塩酸 (1+2)
A.4.2.3 塩酸 (1+19)
A.4.2.4 塩酸 (1+99)
A.4.2.5 塩酸 (pH 1.60±0.05) 塩酸 (A.4.2.1) を2 Lの水に5,6滴加え,pHメーター (A.5.16.1) を用い
てpHを1.60±0.05に調節する。溶液は,ポリエチレン製の瓶に保存する。
A.4.2.6 ふっ化水素酸 (HF) (>40 %)
A.4.2.7 ふっ化水素酸 (1+3)
A.4.2.8 硝酸 (HNO3)
A.4.2.9 硫酸 (H2SO4) (>98 %)
A.4.2.10 硫酸 (1+1)
A.4.2.11 ほう酸 (H3BO3)
A.4.2.12 酢酸 (CH3COOH)
A.4.2.13 アミノ酢酸 (NH2CH2COOH)
A.4.2.14 アンモニア水 (NH4OH)
A.4.2.15 アンモニア水 (1+1)
A.4.2.16 アンモニア水 (1+10)
A.4.2.17 水酸化ナトリウム (NaOH)
A.4.2.18 水酸化ナトリウム溶液 (4 mol/L) 水酸化ナトリウム (A.4.2.17) 160 gを水に溶かして,1 000 mL
にする。ポリエチレン製の瓶に保存する。
A.4.2.19 水酸化ナトリウム溶液 (2 mol/L) 水酸化ナトリウム (A.4.2.17) 80 gを水に溶かして,1 000 mL
にする。ポリエチレン製の瓶に保存する。
A.4.2.20 塩化アンモニウム (NH4Cl)
A.4.2.21 塩化すず (II) 二水和物 (SnCl2・2H2O)
A.4.2.22 過酸化ナトリウム (Na2O2) 粉末
A.4.2.23 塩化ナトリウム (NaCl) 110±5 ℃で恒量となるまで乾燥する。
A.4.2.24 炭酸ナトリウム (Na2CO3) 250±10 ℃で恒量となるまで乾燥する。
A.4.2.25 炭酸ナトリウムと塩化ナトリウムの混合物 炭酸ナトリウム (A.4.2.24) 7 gと塩化ナトリウム
(A.4.2.23) 1 gを混合する。
A.4.2.26 硝酸銀 (AgNO3) 150±5 ℃で恒量となるまで乾燥する。
A.4.2.27 硝酸銀溶液 硝酸銀 (A.4.2.26) 5 gを水に溶かし,硝酸 (A.4.2.8) 10 mLを加え,水で1 000 mLに
する。
A.4.2.28 ポリ酸化エチレン溶液 平均分子量が200 000〜600 000のポリ酸化エチレン[(-CH2-CH2-O-)n]
0.25 gをよくかくはんしながら100 mLの水に溶かす。この溶液は,2週間以内に使用する。
A.4.2.29 ほう酸溶液 ほう酸 (A.4.2.11) 約50 gを水に溶かして,1 000 mLにする。
A.4.2.30 くえん酸溶液 くえん酸一水和物 (C6H8O7・H2O) 10 gを水に溶かして,100 mLにする。
A.4.2.31 炭酸カルシウム (CaCO3) (>99.9 %) 200±10 ℃で恒量となるまで乾燥する。
A.4.2.32 モリブデン酸アンモニウム溶液 七モリブデン酸六アンモニウム四水和物[(NH4)6Mo7O24・4H2O]
10 gを水に溶かして,100 mLにする。ポリエチレン製の瓶に入れて保存する。この溶液は,1週間以内に
35
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
使用する。
A.4.2.33 硫酸銅溶液 硫酸銅 (II) 五水和物 (CuSO4・5H2O) 0.45 gを水に溶かして,全量フラスコ50 mLに
移し,標線まで水を加えて振り混ぜる。
A.4.2.34 酢酸アンモニウム溶液 酢酸アンモニウム (CH3COONH4) 250 gを水に溶かして,1 000 mLにす
る。
A.4.2.35 トリエタノールアミン[N(CH2CH2OH)3]溶液 (1+4) トリエタノールアミンの純度は,99 %
を超えるものとする。
A.4.2.36 還元溶液 塩化すず (II) 二水和物 (A.4.2.21) 1 gを,塩酸 (A.4.2.1) 1 mLを加えた水に溶かして,
100 mLにする。この溶液は,1日以内に使用する。
A.4.2.37 緩衝液 (pH 1.40) アミノ酢酸 (A.4.2.13) 7.505±0.001 gと塩化ナトリウム (A.4.2.23) 5.850±
0.001 gを水に溶かして,1 000 mLにする。この溶液300 mLを塩酸 (1+99) (A.4.2.4) で1 000 mLに希釈す
る。
A.4.2.38 二酸化けい素標準液
A.4.2.38.1 二酸化けい素 (SiO2) (>99.9 %)
1 175±25 ℃で恒量となるまで強熱する。
A.4.2.38.2 標準原液
炭酸ナトリウム (A.4.2.24) 2.0±0.1 gを入れた白金るつぼに強熱した二酸化けい素 (A.4.2.38.1) 0.200 0±
0.000 5 gを量り採る。
混合物を加熱し,少なくとも15分間赤熱色の状態で溶融する。室温に冷却した後,融成物をポリエチレ
ン製のビーカーに入れ,水を入れて溶解し,次に全量フラスコ200 mLに移し,標線まで水を加えて振り
混ぜる。
溶液はポリエチレン製の瓶に入れて保存する。
この溶液1 mLは,1 mgの二酸化けい素を含む。
A.4.2.38.3 標準液
標準原液5 mLを全量ピペットを用いて全量フラスコ250 mLに分取し,標線まで水を加えて振り混ぜる。
溶液はポリエチレン製の瓶に入れて保存する。この標準液1 mLは,0.02 mgの二酸化けい素を含む。また,
この標準液は,調製後1週間以内に使用する。
A.4.2.38.4 補正溶液
二酸化けい素の定量方法(A.13.3〜A.13.5参照)に従って,補正溶液を調製する。調製は表A.1の試薬
を水に溶かして,500 mLにする。
36
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表A.1−補正溶液500 mL中の組成
試薬
二重蒸発乾固方法
(A.13.3)
ポリ酸化エチレン方法
(A.13.4)
塩酸−塩化アンモニウム
方法
(A.13.5)
塩酸 (mL)
75
70
15
硫酸 (1+1) (mL)
1
1
−
硝酸 (mL)
−
−
1
ポリ酸化エチレン溶液 (mL)
−
5
−
塩化アンモニウム (g)
−
−
1
炭酸ナトリウム (g)
1.75
1.75
1.75
塩化ナトリウム (g)
0.25
0.25
0.25
過酸化ナトリウム (g)
3
3
−
A.4.2.38.5 検量線の作成
表A.2に示す量の二酸化けい素標準液を,ビュレットを用いてかくはん子の入ったポリエチレン製ビー
カー100 mLに取る。
全量ピペットで補正溶液20 mLを加え,表A.2に示す量の水をビュレットを用いて加え,40 mLにする。
スターラーでかき混ぜながら,ふっ化水素酸 (1+3) (A.4.2.7) を15滴加える。少なくとも1分間かき混ぜ
続ける。次に全量ピペットで,ほう酸溶液 (A.4.2.29) を15 mL加える。
全量ピペットでモリブデン酸アンモニウム溶液 (A.4.2.32) 5 mLを加える。水酸化ナトリウム溶液 (4
mol/L) (A.4.2.18) 又は塩酸 (1+2) (A.4.2.2) を滴下して,溶液のpHを1.60±0.05に調節する。校正する値
に近い緩衝液[例えば,pH 1.40 (A.4.2.37)]で校正したpHメーターを用いる。溶液をpH 1.60±0.05の塩
酸 (A.4.2.5) で全量フラスコ100 mLに洗い移す。20分後に,全量ピペットでくえん酸溶液 (A.4.2.30) 5 mL
を加えてかき混ぜ,5分間静置する。次に全量ピペットで還元溶液 (A.4.2.36) 2 mLを加える。この時点を
発色開始0分とする。
pH 1.60±0.05の塩酸 (A.4.2.5) を標線まで加えて振り混ぜる。発色開始後30分で,吸光光度計 (A.5.8) に
よって吸収セル10 mm (A.5.9) を用いて,同様に調製した空試験液を対照液として,815 nm付近の吸光度
を測定する。表A.2に示した二酸化けい素量と吸光度との関係から検量線を作成する。
検量線から二酸化けい素量 (mgSiO2/100 mL) を求める。
表A.2−検量線用二酸化けい素標準液量及び二酸化けい素含有量
番号
空試験
1
2
3
4
SiO2標準溶液 (mL)
0
2
5
10
20
水 (mL)
20
18
15
10
0
SiO2含有量 (mgSiO2/100 mL)
0
0.04
0.10
0.20
0.40
A.4.2.39 カルシウム標準液(約0.01 mol/L)
炭酸カルシウム (A.4.2.31) 1.00±0.01 gを±0.000 5 gまでビーカー400 mLに量り採り,水約100 mLを加
える。時計皿でビーカーにふたをし,塩酸 (1+2) (A.4.2.2) 約10 mLを徐々に加える。炭酸カルシウムが
完全に溶解するまでガラス棒でかくはんする。その後,溶液中に溶けている二酸化炭素を追い出すために,
煮沸する。室温まで冷却後,溶液を全量フラスコ1 000 mLに洗い移し,標線まで水を加えて振り混ぜる。
37
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.4.2.40 エチレンジアミン四酢酸二水素二ナトリウム二水和物 (EDTA) 溶液(約0.03 mol/L)
A.4.2.40.1 調製
EDTA 11.17±0.01 gを水に溶かし,1 000 mLにする。ポリエチレン製の瓶で保存する。
A.4.2.40.2 標定
カルシウム標準液(約0.01 mol/L) (A.4.2.39) 50 mLを全量ピペットを用いて,光度滴定装置 (A.5.10) に
適したビーカーに分取する。次に,操作に適した量の水を加える。
pHメーター (A.5.16.1) を用いて,水酸化ナトリウム溶液(A.4.2.18又はA.4.2.19)でこの溶液のpHを
12.5±0.2に調節する。
次の二つの方法のうち,いずれかの方法で滴定する。
a) 光度滴定方法(標準方法)
ムレキシド指示薬 (A.4.2.44) 又はカルセイン−メチルチモールブルー混合指示薬 (A.4.2.50) を約0.1 g
加える。光度滴定装置 (A.5.10) にビーカーを置く。
なお,吸光度の測定波長はムレキシド指示薬 (A.4.2.44) を使用する場合は620 nm,カルセイン−メチル
チモールブルー混合指示薬 (A.4.2.50) を使用する場合は520 nmとする。試料溶液をかくはんしながら
EDTA溶液(約0.03 mol/L)で滴定する。EDTA溶液(約0.03 mol/L)の滴定量と吸光度を用い,変色域付
近の滴定曲線を作成する。滴定曲線において変色域付近の最大傾斜線と変色後の吸光度がほとんど変化し
ない部分の線との交点を終点とし,滴定量V3を求める。
EDTA溶液(約0.03 mol/L)のファクターfDは,式(A.1)によって算出する。
3
4
3
4
D
652
.
16
03
.0
09
.
100
50
V
m
V
m
f
×
=
×
×
×
=
············································· (A.1)
ここに,
m4: カルシウム標準液 (A.4.2.39) を調製するために要した炭酸カ
ルシウムの質量 (g)
V3: 滴定に要したEDTA溶液(約0.03 mol/L)の量 (mL)
b) 目視方法(代替方法)
カルコン指示薬 (A.4.2.46) 又はPatton and Reeders指示薬 (A.4.2.51) 約0.1 gを加える。試料溶液をかく
はんしながら,EDTA溶液(約0.03 mol/L)で滴定する。カルコン指示薬の場合は,ピンク色から青色に
変わるまで,Patton and Reeders指示薬の場合は,紫色から青色に変わるまで滴定し,1滴加えても青みが
増さなくなったときを終点とする。
EDTA溶液(約0.03 mol/L)のファクターfDは,式(A.2)によって算出する。
A.4.2.41 銅錯体溶液
硫酸銅溶液 (A.4.2.33) 25 mLを全量ピペットを用いて,ビーカー400 mLに分取し,EDTA溶液(約0.03
mol/L) (A.4.2.40) をビュレットで正確に加える。加えるEDTA溶液(約0.03 mol/L)の量 (V5) は,次のよ
うに求める。
硫酸銅溶液 (A.4.2.33) 10 mLを全量ピペットを用いて,ビーカー600 mLに分取し,水で200 mLに希釈
し,アンモニア水 (A.4.2.14) 10 mLとムレキシド指示薬 (A.4.2.44) 約0.1 gを加える。試料溶液をかくはん
しながら,EDTA溶液(約0.03 mol/L) (A.4.2.40) で滴定する。ピンク色から紫色に変わったときを終点 (V4)
とする。
銅錯体溶液を調製するときに硫酸銅溶液25 mLに加えるEDTA溶液(約0.03 mol/L)の量V5は,式(A.2)
によって算出する。
4
5
5.2 V
V
×
=
··········································································· (A.2)
38
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
V4: 滴定に要したEDTA溶液(約0.03 mol/L)の量 (mL)
A.4.2.42 エチレングリコール−ビス(アミノエチルエーテル)四酢酸 (EGTA) 溶液(約0.03 mol/L)
A.4.2.42.1 調製
EGTA 11.40±0.01 gをビーカー600 mLに量り採り,水400 mLと水酸化ナトリウム溶液 (2 mol/L)
(A.4.2.19) 30 mLを加える。溶液を加熱してEGTAを完全に溶かした後,室温まで冷却する。pHメーター
(A.5.16.1) を用いて,塩酸 (1+2) (A.4.2.2) を滴下しながら溶液のpHを7.0±0.5に調節する。溶液を全量
フラスコ1 000 mLに移し,標線まで水を加えて振り混ぜる。溶液はポリエチレン製の瓶で保存する。
A.4.2.42.2 標定
カルシウム標準液(約0.01 mol/L) (A.4.2.39) 50 mLを全量ピペットを用いて,光度滴定装置 (A.5.10) に
適したビーカーに分取する。次に,光度滴定装置の操作に適した量の水を加える。トリエタノールアミン (1
+4) (A.4.2.35) 25 mLを加える。
pHメーター (A.5.16.1) を用いて,水酸化ナトリウム溶液(A.4.2.18又はA.4.2.19)でこの溶液のpHを
12.5±0.2に調節する。
ムレキシド指示薬 (A.4.2.44) 又はカルセイン指示薬 (A.4.2.45) を約0.1 g加える。光度滴定装置 (A.5.10)
にビーカーを置き,試料溶液をかくはんしながらEGTA溶液(約0.03 mol/L)で滴定する。
なお,吸光度の測定波長はムレキシド指示薬 (A.4.2.44) を使用する場合は620 nm,カルセイン指示薬
(A.4.2.45) を使用する場合は520 nmとする。EGTA溶液(約0.03 mol/L)の滴定量と吸光度を用い,変色
域付近の滴定曲線を作成する。滴定曲線において変色域付近の最大傾斜線と変色後の吸光度がほとんど変
化しない部分の線との交点を終点とし,滴定量V6を求める。
EGTA溶液(約0.03 mol/L)のファクターfGは,式(A.3)によって算出する。
6
5
6
5
G
652
.
16
03
.0
09
.
100
50
V
m
V
m
f
×
=
×
×
×
=
············································· (A.3)
ここに,
m5: カルシウム標準液(約0.01 mol/L)(A.4.2.39) を調製するため
に要した炭酸カルシウムの質量 (g)
V6: 滴定に要したEGTA溶液(約0.03 mol/L)の量 (mL)
A.4.2.43 1,2-ジアミノシクロヘキサン四酢酸一水和物 (DCTA) 溶液(約0.01 mol/L)
A.4.2.43.1 調製
DCTA 3.64±0.01 gをビーカー600 mLに量り採り,水約400 mLと水酸化ナトリウム溶液 (2 mol/L)
(A.4.2.19) 10 mLを加える。溶液を加熱してDCTAを完全に溶かした後,室温まで冷却する。pHメーター
(A.5.16.1) を用いて,塩酸 (1+2) (A.4.2.2) を滴下しながら溶液のpHを7.0±0.5に調節する。溶液を全量
フラスコ1 000 mLに移し,標線まで水を加えて振り混ぜる。溶液はポリエチレン製の瓶で保存する。
A.4.2.43.2 標定
カルシウム標準液(約0.01 mol/L) (A.4.2.39) 50 mLを全量ピペットを用いて,光度滴定装置 (A.5.10) に
適したビーカーに分取する。次に,光度滴定装置の操作に適した量の水を加える。
pHメーター (A.5.16.1) を用いて,アンモニア水 (A.4.2.14) でこの溶液のpHを10.5±0.2に調節する。
ムレキシド指示薬 (A.4.2.44) 又はカルセイン指示薬 (A.4.2.45) を約0.1 g加える。光度滴定装置 (A.5.10)
にビーカーを置き,試料溶液をかくはんしながらDCTA溶液(約0.01 mol/L)で滴定する。
なお,吸光度の測定波長はムレキシド指示薬 (A.4.2.44) を使用する場合は620 nm,カルセイン指示薬
(A.4.2.45) を使用する場合は520 nmとする。DCTA溶液(約0.01 mol/L)の滴定量と吸光度を用い,変色
域付近の滴定曲線を作成する。滴定曲線において変色域付近の最大傾斜線と変色後の吸光度がほとんど変
39
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
化しない部分の線との交点を終点とし,滴定量V7を求める。
DCTA溶液(約0.01 mol/L)のファクターfCは,式(A.4)によって算出する。
7
6
7
6
C
955
.
49
01
.0
09
.
100
50
V
m
V
m
f
×
=
×
×
×
=
············································· (A.4)
ここに,
m6: カルシウム標準液(約0.01 mol/L)(A.4.2.39) を調製するため
に要した炭酸カルシウムの質量 (g)
V7: 滴定に要したDCTA溶液(約0.01 mol/L)の量 (mL)
A.4.2.44 ムレキシド指示薬 ムレキシド (ammonium purpurate, C8H4N5O6NH4) 1.0±0.1 gと塩化ナトリウム
(A.4.2.23) 100±1 gを混合粉砕する。
A.4.2.45 カルセイン指示薬 カルセイン {bis [(bis (carboxymethyl)-amino-methyl)]-2′,7′-fluorescein,
C30H26N2O13} 1.0±0.1 gと硝酸カリウム (KNO3) 100±1 gを混合粉砕する。
A.4.2.46 カルコン指示薬 カルコン [sodium 2-hydroxy-4-(2-hydroxy-1-naphthylazo) naphthalene-1-sulfonate,
C20H13N2NaO5S, EriochromeBlue-Black R] 1.0±0.1 gと無水硫酸ナトリウム (Na2SO4) 100±1 gを混合粉砕す
る。
A.4.2.47 スルホサリチル酸指示薬(5-スルホサリチル酸二水和物)
A.4.2.48 PAN指示薬 PAN[1- (2-pyridylazo)-2-naphthol, C15H11N3O] 0.10±0.01 gをエタノール (C2H5OH, ρ
=0.79 g/cm3) 100±1 mLに溶かす。
A.4.2.49 メチルチモールブルー指示薬 メチルチモールブルー {sodium salt of 3′,3′′-bis-[bis
(carboxy-methyl)-aminomethyl]-thymolsulfophthalein, C37H41N2O13SNa3} 1.0±0.1 gと硝酸カリウム (KNO3)
100±1 gを混合粉砕する。
A.4.2.50 カルセイン−メチルチモールブルー混合指示薬 カルセイン (A.4.2.45) 0.20±0.02 g, メチルチモ
ールブルー (A.4.2.49) 0.10±0.01 gと硝酸カリウム (KNO3) 100±1 gを混合粉砕する。
A.4.2.51 Patton and Reeders指示薬 [2-hydroxy-1-(2-hydroxy-4-sulfo-1-napthylazo)-3-napthoic acid,
C21H14N2O7S] 1.0±0.1 gと無水硫酸ナトリウム (Na2SO4) 100±1 gを混合粉砕する。
A.4.2.52 混合指示薬 o-クレゾールフタレインコンプレキソン [o-cresolphthaleindi-(methyliminodi-acetic
acid), C32H32N2O12] 0.10±0.01g,メチルレッド (o-carboxybenzene-azodimethyl-aniline, C15H14N3NaO2) 0.020±
0.001 g,ナフトールグリーンB (C30H15FeN3Na3O15S3) 0.030±0.001 gと塩化ナトリウム (A.4.2.23) 10.0±0.1 g
を混合粉砕する。
A.5 装置
A.5.1 天びん ±0.000 5 gの測定精度をもつものとする。
A.5.2 るつぼ
A.5.2.1 るつぼ 容量20 mL〜25 mLの磁器るつぼ又は白金るつぼ。
注記 この附属書において,白金るつぼを使用することが必要な場合は,明記されている。明記され
ていない場合は,磁器るつぼを使用してもよい。
A.5.2.2 ふた 必要に応じて,るつぼ (A.5.2.1) に合うものを使用する。
A.5.3 セラミック製の断熱材 るつぼの過熱を防ぐために使用する。炉内にるつぼを置いたとき,温度の
平衡を保つために使用する。
A.5.4 磁器の蒸発皿 約200 mLのものとする。
A.5.5 電気炉 500±10 ℃,950±25 ℃及び1 175±25 ℃に設定できるものとする。
40
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.5.6 デシケータ 乾燥剤として無水過塩素酸マグネシウムMg(ClO4)2又はシリカゲルを用いる。
注記 水分の吸収の程度を示す指示薬が含まれているシリカゲルを使用する場合は,毒性のないもの
を推奨する。
A.5.7 乾燥器 110±5 ℃,120±5 ℃,150±5 ℃,200±10 ℃及び250±10 ℃に設定できるものとする。
A.5.8 吸光光度計 525 nm及び815 nm付近における溶液の吸光度を測定できるものとする。
A.5.9 セル 吸光光度計用
A.5.10 光度滴定装置 マグネチックスターラで溶液をかくはんしながら,滴定用ビーカー中の溶液の520
nm及び620 nm付近の吸光度を測定できるものとする。
A.5.11 スターラ 例えば,マグネチックスターラ。かくはん子は,例えばPTFEで被覆された化学的な反
応性のないもの。
A.5.12 蒸発乾固用装置 105±5 ℃に調節できるものとする(例えば,水浴又はホットプレート)。
A.5.13 砂浴又はホットプレート 約400 ℃に調節できるものとする。
A.5.14 ろ紙 灰分のないものとする。
注記 平均孔径が約2 μmのろ紙を細孔ろ紙,約7 μmのろ紙を中孔ろ紙,約20 μmのろ紙を粗孔ろ紙
と呼ぶ。
A.5.15 ガラス製体積計 分析上,十分な精度をもつものとする。例えば,ISO 385及びISO 835で規定さ
れているクラスAのもの。
A.5.16 pH測定器具
A.5.16.1 pHメーター pH=±0.05の測定精度をもつものとする。
A.5.16.2 pH試験紙 pH=0〜14の範囲で測定が可能なものとする。
A.6 セメント試料の調製
化学分析に当たり,次に従って,均質な分析用試料を調製する。
約100 gの代表試料を縮分器又は四分法で縮分して採取し,150 μm又は125 μmのふるいで,ふるう。
ふるい残分中の金属鉄を磁石を使って取り除き,金属鉄を除いた残分をすりつぶして完全に150 μm又は
125 μmのふるいを通過させる。試料を気密性のあるきれいな乾いた容器に移し替え,十分に試料が混ざる
ように強く振る。
上記の操作は,試料が周囲の空気にさらされる時間をできるだけ短くするために,手早く行わなければ
ならない。
注記 品質管理を目的とする分析で,金属鉄の含有が分析値に与える影響がほとんどない場合は,金
属鉄の除去を行う必要はない。
A.7〜A.12 (本体及び附属書Bで記載されているため削除している。)
A.13 主成分の定量方法
A.13.1 要旨
主成分の分析は,試料を完全に溶解した後に行う。塩酸−塩化アンモニウム方法(代替方法)による分
解は不溶残分[塩酸−炭酸ナトリウム方法による(箇条6)]が1.5 %を超えないセメントに対して,用い
てもよい。
試料は,過酸化ナトリウム方法による焼結(融解)又は塩酸−塩化アンモニウム方法による処理によっ
41
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
て分解する。過酸化ナトリウム方法では,焼結物(融成物)を塩酸で溶解した後,二酸化けい素の大部分
を二重蒸発乾固方法又はポリ酸化エチレン方法によって析出させる。塩酸−塩化アンモニウム方法では,
二酸化けい素の大部分が沈殿として分離される。これらの析出した不純物二酸化けい素はふっ化水素酸と
硫酸で処理して二酸化けい素を揮発させる。揮発残分は炭酸ナトリウムと塩化ナトリウムの混合物で融解
した後,融成物を塩酸で溶解し,不純物二酸化けい素をろ別したろ液に加える。
塩酸−塩化アンモニウム方法によって試料を分解したとき,ふっ化水素酸と硫酸で処理して二酸化けい
素を揮発させた後の残分が0.5 %以上であった場合には,この分解方法を適用することはできない。この
場合は,過酸化ナトリウム方法によって試料を分解しなければならない。
上記の操作によって得られた最終溶液を用いて,可溶性二酸化けい素は吸光光度法で定量する。また,
酸化鉄 (III),酸化アルミニウム,酸化カルシウム及び酸化マグネシウムは,錯滴定法によって定量する。
分析操作の概要図を図A.1に示す。
図A.1に示した分析操作によって不純物,純粋及び可溶性二酸化けい素の量は異なるが,どの操作手順
に従っても全二酸化けい素の含有率は同じ値となる。
A.13.2 過酸化ナトリウムによる試料の分解
セメント1.00±0.05 g (m17) を±0.000 5 gまで量り採り,過酸化ナトリウム (A.4.2.22) 約2 gを量り採り,
白金るつぼ (A.5.2.1) にそれらを入れ,へらでよく混ぜる。へらについた試料及び過酸化ナトリウムは,
は(刷)毛で混合物に戻す。約1 gの過酸化ナトリウムで,混合物を覆う。まず電気炉 (A.5.5) を開けた
状態で入り口付近に,ふた (A.5.2.2) をしたるつぼを置き約2分間予熱する。その後500±10 ℃に調節し
た炉内の断熱材 (A.5.3) の上にるつぼを置く。
30分後,電気炉からるつぼを取り出し,室温まで冷却する。このとき,焼結物がるつぼの側面にくっつ
いていない状態が望ましい。焼結物がるつぼの側面にくっついた場合は,焼結温度を10 ℃下げ,焼結操
作をやり直す。
焼結物をビーカー400 mLに移し,水150 mLでるつぼに付着している残留物を洗い移す。
時計皿でビーカーにふたをし,焼結物が完全に分散するまで溶液を加熱する。次に注意深く塩酸
(A.4.2.1) 50 mLを加え,かき混ぜる。このとき,焼結物は完全に溶けなければならない。焼結物が完全に
溶けない場合は,焼結温度を10 ℃上げるか又は焼結時間を2倍にして,焼結操作をやり直す。溶液に硫
酸 (1+1) (A.4.2.10) 1 mLを加え,加熱し30分間煮沸する。
この溶液はA.13.3又はA.13.4の二酸化けい素の沈殿生成に使用する。
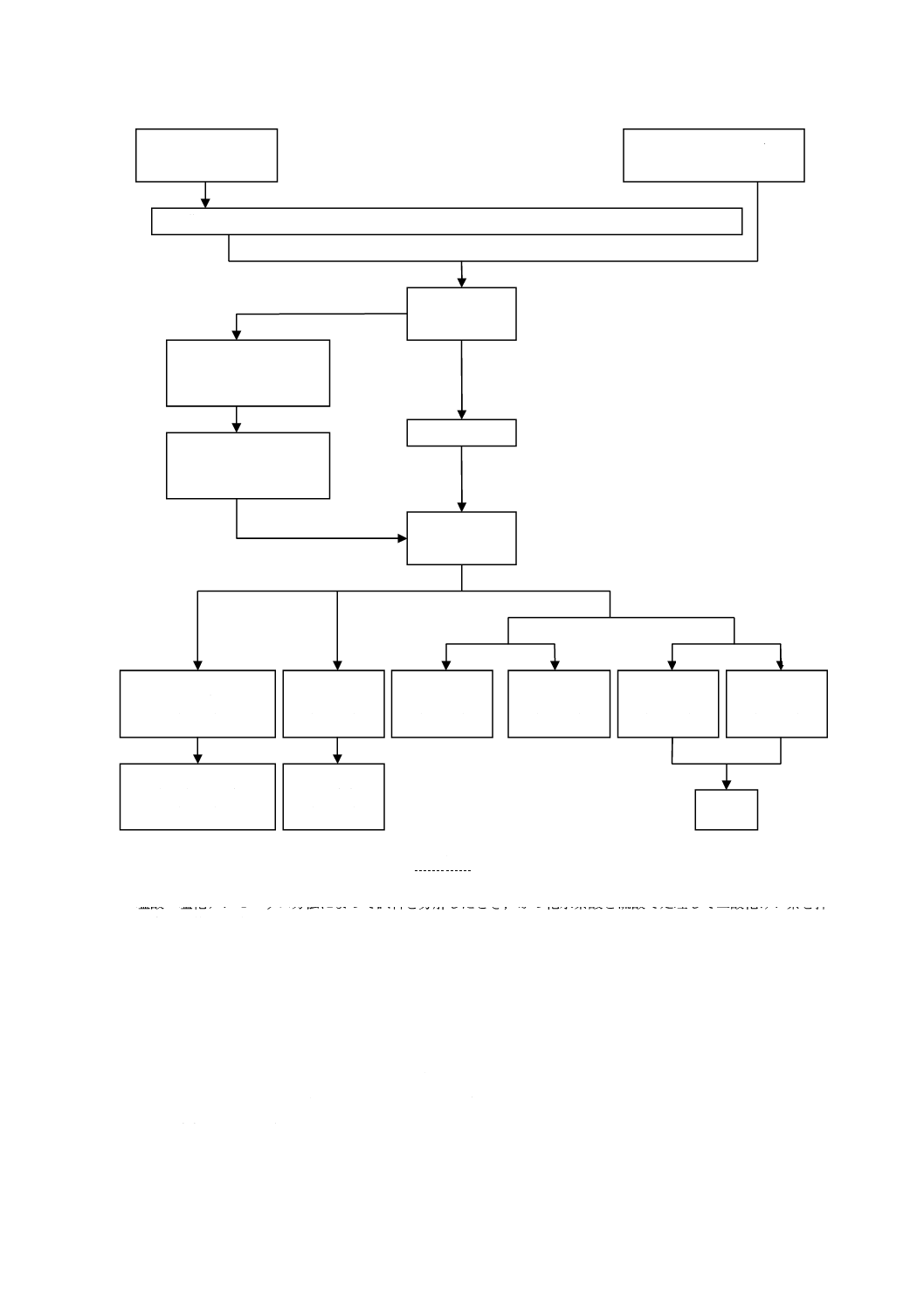
42
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注a) 不溶残分[塩酸−炭酸ナトリウム方法による(箇条6)]が1.5 %を超える場合は,過酸化ナトリウム方法を用い
る。
b) 塩酸−塩化アンモニウム方法によって試料を分解したとき,ふっ化水素酸と硫酸で処理して二酸化けい素を揮
発させた後の残分が0.5 %以上であった場合には,この分解方法を適用することはできない。この場合は,過酸
化ナトリウム分解によって試料を分解しなければならない。
図A.1−主成分の化学分析概要図
A.13.3 二重蒸発乾固方法による二酸化けい素の定量方法(標準方法)
A.13.3.1 操作
105±5 ℃に調節した蒸発乾固用装置 (A.5.12) 上でA.13.2で調製した溶液を蒸発乾固する。蒸発乾固し
た後,乾固物を塩酸 (A.4.2.1) 数滴で湿らし,この温度で1時間放置する。
室温まで冷却した後,塩酸 (A.4.2.1) 10 mLを乾固物に加える。2,3分後に水50 mLを加えて煮沸し,
ただちに中孔ろ紙 (A.5.14) を用いてろ過し,ビーカー又は蒸発皿にろ液を受ける。ろ紙及び残分を温水で
Na2O2による分解
: 試料1 g(A.13.2)
又は
HCl+NH4Clによる分解 a)
: 試料1 g(A.13.5)
二重蒸発乾固方法(A.13.3) 又は ポリ酸化エチレン方法(A.13.4)
不純物
二酸化けい素
HF+H2SO4による
純粋二酸化けい素(I)
の揮発b)(A.13.6)
ろ液
Na2CO3+NaClでの残分の
融解と溶解
(A.13.7)
最終溶液
500 mL
又は
可溶性二酸化けい素(II)
吸光光度法
(A.13.8)
Fe2O3
EDTA滴定法
(A.13.10)
CaO
EGTA滴定法
(A.13.12)
MgO
DCTA滴定法
(A.13.13)
CaO
EDTA滴定法
(A.13.14)
CaO+MgO
EDTA滴定法
(A.13.15)
(I)+(II)
全二酸化けい素
(A.13.9)
Al2O3
EDTA滴定法
(A.13.11)
MgO
差分
20 mL
100 mL
25 mL
50 mL
50 mL
50 mL
43
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3回洗浄する。このろ液と洗浄液を同様に蒸発乾固した後,塩酸 (A.4.2.1) 10 mLと水50 mLを残分に加え
る。その後,煮沸し,1回目のろ過で使用した中孔ろ紙を用いてろ過し,全量フラスコ500 mLにろ液を受
ける。
ろ紙及び残分を硝酸銀試験 (A.3.6) で塩化物イオンが認められなくなるまで温水で洗う。洗浄液は,全
量フラスコ500 mLに受ける。
ろ紙及び沈殿を白金るつぼに入れ,1 175±25 ℃で強熱 (A.3.4) して恒量 (A.3.5) を確認し,質量m18
を量る。
なお,強熱温度は標準の1 175±25 ℃と同じ値となることを示せば,1 050 ℃〜1 175 ℃の範囲としても
よい。
注記 通常,60分間の強熱で恒量となる。
A.13.6に従って残分を揮発させる。次に,A.13.7に従ってA.13.6の揮発残分を溶解した溶液を全量フラ
スコ500 mL中のろ液と洗浄液に加え,室温まで冷却した後,標線まで水を加えて振り混ぜる。この溶液
を最終溶液とする。
最終溶液は,吸光光度法による可溶性二酸化けい素 (A.13.8) 及び錯滴定法による酸化鉄 (III) (A.13.10),
酸化アルミニウム (A.13.11),酸化カルシウム(A.13.12又はA.13.14)及び酸化マグネシウム(A.13.13又
はA.13.15)の定量に用いる。
A.13.3.2 計算
試料中の不純物二酸化けい素の含有率 (
imp
,
SiO2
w
) は,式(A.5)によって算出する。
100
17
18
imp
,
SiO2
×
=m
m
w
································································· (A.5)
ここに,
m17: A.13.2の試料の質量 (g)
m18: A.13.3.1の沈殿の質量 (g)
A.13.4 ポリ酸化エチレン方法による不純物二酸化けい素の定量方法(代替方法)
A.13.4.1 操作
A.13.2で調製(保存)した溶液を蒸発乾固する。蒸発乾固した後,ビーカー又は蒸発皿を放冷し,水5 mL
と塩酸 (A.4.2.1) 10 mLを乾固物に加える。かき混ぜながら,ろ紙粉末を加え,次にポリ酸化エチレン溶液
(A.4.2.28) 5 mLを加える。このとき,沈殿とポリ酸化エチレンを十分に混合し,ビーカーの側面にくっつ
いてしまった沈殿も一緒に混ぜるようにする。その後,水10 mLを加え,軽くかき混ぜ,5分間放置する。
次に沈殿を中孔ろ紙 (A.5.14) を用いてろ過し,温塩酸 (1+19) (A.4.2.3) で洗浄する。このとき,ポリス
マンでビーカーの側面に付いた沈殿も移す。ろ紙及び沈殿の洗浄は温塩酸 (1+19) で少なくとも5回行う。
ろ液は全量フラスコ500 mLに受ける。続いて温水でろ紙上の沈殿が十分にばらばらになるように洗浄し,
硝酸銀試験 (A.3.6) で塩化物イオンが認められなくなるまで洗浄を続ける。
洗浄液は全量フラスコ500 mLの溶液に合わせる。
ろ紙及び沈殿を白金るつぼに入れ,1 175±25 ℃に調節した電気炉で強熱 (A.3.4) して恒量 (A.3.5) を
確認し,質量m19を量る。
なお,強熱温度は標準の1 175±25 ℃と同じ値となることを示せば,1 050 ℃〜1 175 ℃の範囲としても
よい。
注記 通常,60分間の強熱で恒量となる。
44
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.13.6に従って残分を揮発させる。次に,A.13.7に従ってA.13.6の揮発残分を溶解した溶液を全量フラ
スコ500 mL中のろ液と洗浄液に加え,室温まで冷却した後,標線まで水を加えて振り混ぜる。この溶液
を最終溶液とする。最終溶液は,吸光光度法による可溶性二酸化けい素 (A.13.8) 及び錯滴定法による酸化
鉄 (III) (A.13.10),酸化アルミニウム (A.13.11),酸化カルシウム (A.13.12又はA.13.14)及び酸化マグネシ
ウム (A.13.13又はA.13.15)の定量に用いる。
A.13.4.2 計算
試料中の不純物二酸化けい素の含有率 (
imp
,
SiO2
w
) は,式(A.6)によって算出する。
100
17
19
imp
,
SiO2
×
=m
m
w
································································· (A.6)
ここに,
m17: A.13.2の試料の質量 (g)
m19: A.13.4.1の沈殿の質量 (g)
A.13.5 塩酸−塩化アンモニウム方法による不純物二酸化けい素の定量方法(代替方法)
注記 この方法は不溶残分[塩酸−炭酸ナトリウム方法による(箇条6)]が1.5 %を超えない場合に
限り適用が可能である。
A.13.5.1 操作
セメント1.00±0.05 g (m20) を±0.000 5gまで量り採り,ビーカー100 mLに移す。塩化アンモニウム
(A.4.2.20) 約1 gを加え,ガラス棒でよく混ぜる。時計皿でビーカーにふたをし,注意深く塩酸 (A.4.2.1) 10
mLをビーカーの側面を伝わって流れるように加える。泡立ちが止まったら硝酸 (A.4.2.8) 10滴を加え,ガ
ラス棒でかき混ぜる。
時計皿でビーカーにふたをし,水浴上で30分間加熱する。粗孔ろ紙 (A.5.14)(国際規格では粗孔ろ紙と
なっているが,二酸化けい素が通過するおそれがあるので中孔ろ紙で行うほうがよい。)を用いて沈殿をろ
過する。ろ液は,全量フラスコ500 mLに受ける。
この操作においてゲル状の沈殿をできる限りろ紙に移す。また,温塩酸 (1+99) (A.4.2.4) を用いてビー
カーの内壁に付着した沈殿をすべてポリスマンでこすり落として移す。
沈殿を温塩酸 (1+99) (A.4.2.4) で洗浄する。最後に,沈殿及びろ紙を少量の温水で洗浄し,硝酸銀試験
(A.3.6) で塩化物イオンが認められないことを確認する。洗浄液は全量フラスコ500 mLの溶液に合わせる。
ろ紙及び沈殿を白金るつぼに入れ,1 175±25 ℃に調節した電気炉で強熱 (A.3.4) して恒量 (A.3.5) を
確認し,質量m21を量る。
なお,強熱温度は標準の1 175±25 ℃と同じ値となることを示せば,1 050 ℃〜1 175 ℃の範囲としても
よい。
注記 通常,60分間の強熱で恒量となる。
A.13.6に従って残分を揮発させる。
A.13.5.2 計算
試料中の不純物二酸化けい素の含有率 (
imp
,
SiO2
w
) は,式(A.7)によって算出する。
100
20
21
imp
,
SiO2
×
=m
m
w
································································· (A.7)
ここに,
m20: A.13.5.1の試料の質量 (g)
m21: A.13.5.1の沈殿の質量 (g)
45
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.13.6 純粋二酸化けい素の定量方法
A.13.6.1 操作
A.13.3.1,A.13.4.1又はA.13.5.1に従って得た残分を約0.5 mL〜1 mLの水で湿らせ,硫酸 (A.4.2.9) を2
滴加え,次にふっ化水素酸 (A.4.2.6) 約10 mLを加える。ドラフト内の砂浴又はホットプレート (A.5.13) で
揮発させ,次に硫酸の白煙が出なくなるまで加熱を続ける。
1 175±25 ℃に調節した電気炉 (A.5.5) で,揮発残分の入ったるつぼを10分間強熱し,デシケータ中で
室温まで放冷した後,質量を量る (m22)。
なお,強熱温度は標準の1 175±25 ℃と同じ値となることを示せば,1 050 ℃〜1 175 ℃の範囲としても
よい。
揮発残分は,A.13.7に従って分解する。揮発残分が0.5 %を超えた場合は,過酸化ナトリウムによる分
解方法 (A.13.2) に従って,分析を最初からやり直さなければならない。
A.13.6.2 計算
試料中の純粋二酸化けい素の含有率 (
p,
SiO2
w
) は,式(A.8)によって算出する。
100
23
22
24
p,
SiO2
×
−
=
m
m
m
w
··························································· (A.8)
ここに,
m22: A.13.6.1の残分の質量 (g)
m23: A.13.2 (m17) 又はA.13.5.1 (m20) の試料の質量 (g)
m24: A.13.3.1 (m18),A.13.4.1 (m19) 又はA.13.5.1 (m21) の
沈殿の質量 (g)
A.13.7 揮発残分の融解
A.13.6.1に従って得た揮発残分に,炭酸ナトリウムと塩化ナトリウムの混合物 (A.4.2.25) を2 g加え,
ガスバーナーなどを用いて,赤熱色になるまで加熱する。この時,揮発残分を完全に溶かすため,融成物
を頻繁に振り動かす。
るつぼの底に揮発残分が残っていないことを目視で確認する。白金るつぼ及び融成物を冷却し,ビーカ
ー250 mLにるつぼを入れ,水100 mLを加えた後,塩酸 (A.4.2.1) 数mLで酸性にする。融成物が完全に
溶解したら,溶液から白金るつぼを取り除き,水で白金るつぼを洗浄する。
溶液は完全に透明でなければならない。そうでない場合は,中孔ろ紙 (A.5.14) を用いてろ過し,残分を
洗浄する。次に,ろ紙を灰化した後,強熱し,残分を上述の操作で再融解し溶解する。A.13.3.1,A.13.4.1
又はA.13.5.1で保存した不純物二酸化けい素の定量におけるろ液と洗浄液を含む全量フラスコ500 mLの
溶液に,融成物を溶解した溶液を加える。標線まで水を加えて振り混ぜる。
この溶液を最終溶液とする。最終溶液は,吸光光度法による可溶性二酸化けい素 (A.13.8) の定量及び酸
化鉄 (III) (A.13.10),酸化アルミニウム (A.13.11),酸化カルシウム(A.13.12又はA.13.14)及び酸化マグ
ネシウム(A.13.13又はA.13.15)の錯滴定に使用される。
A.13.8 可溶性二酸化けい素の定量方法
A.13.8.1 操作
A.13.7で保存した最終溶液20 mLを,全量ピペットでかくはん子 (A.5.11) を入れたポリエチレン製ビー
カーに分取し,水20 mLを加える。スターラ (A.5.11) でかき混ぜながら,ふっ化水素酸 (1+3) (A.4.2.7) を
15滴加える。少なくとも1分間かき混ぜ続ける。次に,全量ピペットでほう酸溶液 (A.4.2.29) 15 mLを加
える。次いで,全量ピペットでモリブデン酸アンモニウム溶液 (A.4.2.32) 5 mLを加え,pHメーター
(A.5.16.1) を用いて,水酸化ナトリウム溶液 (2 mol/L) (A.4.2.19) 又は塩酸 (1+2) (A.4.2.2) を滴下して溶液
46
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
のpHを1.60±0.05に調節する。
なお,pHメーターはpHが1.6に近いpHの緩衝液[例えば,pH 1.40 (A.4.2.37)]を用いて校正したもの
を使用する。
pH 1.60±0.05の塩酸 (A.4.2.5) を用いて全量フラスコ100 mLに溶液を洗い移す。発色開始(モリブデン
酸アンモニウム溶液を加えた時点)20分後に,全量ピペットでくえん酸溶液 (A.4.2.30) 5 mLを加えてか
き混ぜ,5分間静置する。その後,全量ピペットで還元溶液 (A.4.2.36) 2 mLを加える(この時点を発色時
間0分とする)。
pH 1.60±0.05の塩酸 (A.4.2.5) を標線まで加えて振り混ぜる。発色開始後,正確に30分で,溶液の一部
を吸収セル (A.5.9) に取り,同様に調製した空試験液を対照液として,吸光光度計 (A.5.8) を用いて吸光
度を測定する。
なお,波長及びセルの光路長は検量線の作成 (A.4.2.38.5) 時と同じとする。検量線から二酸化けい素量
c25 (mgSiO2/100 mL) を求める。
A.13.8.2 計算
試料中の可溶性二酸化けい素の含有率 (
,sol
SiO2
w
) は,式(A.9)によって算出する。
23
25
23
25
sol
,
SiO
5.2
000
1
20
100
500
2
m
c
m
c
w
×
=
×
×
×
×
=
············································ (A.9)
ここに,
m23: A.13.2 (m17) 又はA.13.5.1 (m20) の試料の質量 (g)
c25: A.13.8.1で求めた100 mL溶液中の二酸化けい素量
(mgSiO2/100 mL)
A.13.9
二酸化けい素の計算
A.13.9.1
計算
全二酸化けい素の量は,純粋二酸化けい素 (A.13.6) 及び可溶性二酸化けい素 (A.13.8) の含有率の合量
とする。
A.13.9.2
繰返し精度及び再現精度
繰返し精度の標準偏差 0.10 %
再現精度の標準偏差 0.25 %
A.13.10 酸化鉄 (III) の定量方法
A.13.10.1 操作
注記1 チタンの存在は,EDTAによる滴定速度に影響する。この場合,例えば,自動ビュレットを
用いて,ゆっくりと滴定を進めることによって誤差を低減できる。また,硫酸 (1+1)
(A.4.2.10) 2 mLを滴定前に加えることでチタンをマスキングすることが可能である。
注記2 この方法は光度滴定によって終点判定を行う。正確さは劣るが,目視による終点判定も可能
である。この場合,スルホサリチル酸指示薬 (A.4.2.47)(紫色から黄色に変色)が適切であ
る。
A.13.7で保存した最終溶液100 mLを,光度滴定装置 (A.5.10) に適したビーカーに全量ピペットで分取
する。次に,光度滴定装置の操作に適した量の水を加える。
アミノ酢酸 (A.4.2.13) 0.5 g及びスルホサリチル酸指示薬 (A.4.2.47) を0.3 g〜0.4 g加える。
pHメーター (A.5.16.1) を用いて,アンモニア水 (1+1) (A.4.2.15) 及びアンモニア水 (1+10) (A.4.2.16)
で,この溶液のpHを1.5±0.1に調節する。
吸光度の測定波長を520 nmにした光度滴定装置 (A.5.10) にビーカーを置き,溶液をかくはんしながら
47
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
EDTA溶液(約0.03 mol/L)(A.4.2.40) で滴定する。その際,溶液の温度を47.5±2.5 ℃に維持する。EDTA
溶液(約0.03 mol/L)の滴定量と吸光度を用い,変色域付近の滴定曲線を作成する。滴定した総量 (Vtot) を
記録する。終点の滴定量 (V10) は滴定曲線において変色域付近の最大傾斜線と変色後の吸光度がほとんど
変化しない部分の線との交点から求める。EDTA溶液(約0.03 mol/L)の過剰滴定量 (Vex) は (Vtot) から
(V10) を差し引き,求める。
滴定中,溶液の温度は50 ℃を超えてはならない。超えた場合は,再試験を行わなければならない。
この滴定後の溶液は,酸化アルミニウム (A.13.11.1) の定量にも用いる。
A.13.10.2 計算
試料中の酸化鉄 (III) の含有率 (
3
2O
Fe
w
) は,式(A.10)によって算出する。
100
100
000
1
2
500
692
.
159
03
.0
23
D
10
O
Fe
3
2
×
×
×
×
×
×
×
×
=
m
f
V
w
23
D
10
7
197
.1
m
f
V×
×
=
·······················································(A.10)
ここに,
V10: 滴定に要したEDTA溶液(約0.03 mol/L)の量 (mL)
fD: A.4.2.40.2で求めたEDTA溶液(約0.03 mol/L)の
ファクター
m23: A.13.2又はA.13.1.5の試料の質量 (g)
A.13.10.3 繰返し精度及び再現精度
繰返し精度の標準偏差 0.08 %
再現精度の標準偏差 0.15 %
A.13.11 酸化アルミニウムの定量方法
A.13.11.1 操作
A.13.10.1の測定が終わった後の溶液を室温に冷やす。酢酸 (A.4.2.12) 5 mLを加え,pHメーター
(A.5.16.1) を用いて,酢酸アンモニウム溶液 (A.4.2.34) を1滴ずつ加えて,この溶液のpHを3.05±0.05
に調節する。この範囲は厳密に守らねばならず,pHが3.1を超えてはならない。溶液を加熱して煮沸し,
銅錯体溶液 (A.4.2.41) を3滴加え,PAN指示薬 (A.4.2.48) 2 mLを加える。
滴定中,溶液は穏やかに煮沸し続ける。赤紫色から淡い黄色になるまでEDTA溶液(約0.03 mol/L)
(A.4.2.40) で滴定する。赤紫色が再び現れたら,EDTA溶液(約0.03 mol/L)を1滴ずつ加え,少なくとも
1分間黄色が保持するまで滴定する。
A.13.11.2 計算
試料中の酸化アルミニウムの含有率 (
3
2O
Al
w
) は,式(A.11)によって算出する。
100
100
000
1
2
500
961
.
101
03
.0
23
D
11
O
Al
3
2
×
×
×
×
×
×
×
×
=
m
f
V
w
23
D
11
7
764
.0
m
f
V×
×
=
······················································· (A.11)
ここに,
V11: ここで滴定に要したEDTA溶液(約0.03 mol/L)の量と
過剰滴定量 (Vex) (A.13.10.1) の合量 (mL)
fD: A.4.2.40.2で求めたEDTA溶液(約0.03 mol/L)の
ファクター
m23: A.13.2又はA.13.5.1の試料の質量 (g)
A.13.11.3 繰返し精度及び再現精度
繰返し精度の標準偏差 0.10 %
48
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
再現精度の標準偏差 0.25 %
A.13.12 EGTA溶液による酸化カルシウムの定量方法(標準方法)
A.13.12.1 操作
注記1 この方法は光度滴定によって終点判定を行う。正確さは劣るが,目視による終点判定も可能
である。この場合,カルセイン−メチルチモールブルー混合指示薬 (A.4.2.50) (淡い緑色か
らピンク色に変色)が適切である。
注記2 この方法は酸化ストロンチウムも定量され,酸化カルシウム量として含まれる。
A.13.7で保存した最終溶液25 mLを光度滴定装置 (A.5.10) に適したビーカーに全量ピペットで分取し,
水を加えてA.4.2.42.2と同じ量にして,次にトリエタノールアミン溶液 (1+4) (A.4.2.35) 25 mLを加える。
pHメーター (A.5.16.1) を用いて,水酸化ナトリウム溶液 (4 mol/L) (A.4.2.18) でこの溶液のpHを12.5±
0.5に調節する。
ムレキシド指示薬 (A.4.2.44) 又はカルセイン指示薬 (A.4.2.45) を約0.1 g加える。吸光度の測定波長は
カルセイン指示薬を使用する場合は520 nm,ムレキシド指示薬を使用する場合は620 nmとし,光度滴定
装置 (A.5.10) にビーカーを置き,溶液をかくはんしながらEGTA溶液(約0.03 mol/L)(A.4.2.42) で滴定
する。
EGTA溶液(約0.03 mol/L)の滴定量と吸光度を用い,変色域付近の滴定曲線を作成する。終点の滴定
量 (V12) は滴定曲線において変色域付近の最大傾斜線と変色後の吸光度がほとんど変化しない部分の線と
の交点から求める。
A.13.12.2 計算
試料中の酸化カルシウムの含有率 (wCaO) は,式(A.12)によって算出する。
100
25
000
1
500
08
.
56
03
.0
23
G
12
CaO
×
×
×
×
×
×
×
=
m
f
V
w
23
G
12
8
364
.3
m
f
V×
×
=
························································(A.12)
ここに,
V12: 滴定に要したEGTA溶液(約0.03 mol/L)の量 (mL)
fG: A.4.2.42.2で求めたEGTA溶液(約0.03 mol/L)の
ファクター
m23: A.13.2又はA.13.5.1の試料の質量 (g)
注記 酸化ストロンチウムも定量され,酸化カルシウム量として含まれる。
A.13.12.3 繰返し精度と再現精度
繰返し精度の標準偏差 0.18 %
再現精度の標準偏差 0.37 %
A.13.13 DCTA溶液による酸化マグネシウムの定量方法(標準方法)
A.13.13.1 操作
注記 この方法は光度滴定によって終点判定を行う。目視による終点判定も可能である。この場合,
メチルチモールブルー指示薬 (A.4.2.49)(青色から灰色に変色)が適切である。
A.13.7で保存した最終溶液50 mLを光度滴定装置 (A.5. 10) に適したビーカーに全量ピペットで分取し,
トリエタノールアミン溶液 (1+4) (A.4.2.35) 50 mLを加え,EGTA溶液(約0.03 mol/L) (A.4.2.42) V13を
正確に加える。
V13量 (mL) は,式(A.13)によって算出する。
5.1
2
12
13
+
×
=
V
V
····································································(A.13)
49
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
V12: A.13.12.1で求めたEGTA溶液(約0.03 mol/L)の量 (mL)
算出した量のEGTA溶液(約0.03 mol/L)を加えた後,光度滴定装置の操作に適した量の水を加える。
pHメーター (A.5.16.1) を用いて,アンモニア水 (A.4.2.14) でこの溶液のpHを10.5±0.5に調節する。
メチルチモールブルー指示薬 (A.4.2.49) 約0.1 gを加える。
吸光度の測定波長を620 nmにした光度滴定装置 (A.5.10) にビーカーを置き,溶液をかくはんしながら
DCTA溶液(約0.01 mol/L)(A.4.2.43) で滴定する。
DCTA溶液(約0.01 mol/L)の滴定量と吸光度を用い,変色域付近の滴定曲線を作成する。終点の滴定
量 (V14) は滴定曲線において変色域付近の最大傾斜線と変色後の吸光度がほとんど変化しない部分の線と
の交点から求める。
A.13.13.2 計算
試料中の酸化マグネシウムの含有率 (wMgO) は,式(A.14)によって算出する。
100
50
000
1
500
311
.
40
01
.0
23
C
14
MgO
×
×
×
×
×
×
×
=
m
f
V
w
23
C
14
1
403
.0
m
f
V×
×
=
·························································(A.14)
ここに,
V14: 滴定に要したDCTA溶液(約0.01 mol/L)の量 (mL)
fC: A.4.2.43.2で求めたDCTA溶液(約0.01 mol/L)の
ファクター
m23: A.13.2又はA.13.5.1の試料の質量 (g)
A.13.13.3 繰返し精度及び再現精度
繰返し精度の標準偏差 0.15 %
再現精度の標準偏差 0.15 %
A.13.14 EDTA溶液による酸化カルシウムの定量方法(代替方法)
A.13.14.1 試験上の制約
注記1 この方法は光度滴定によって終点判定を行う。正確さは劣るが,目視による終点判定も可能
である。この場合,カルコン指示薬 (A.4.2.46)(ピンク色から青色に変色),カルセイン−メ
チルチモールブルー混合指示薬 (A.4.2.50)(ピンク色から黄色に変色)又はPatton and Reeders
指示薬 (A.4.2.51)(紫色から明るい青色に変色)が適切である。
注記2 この方法は酸化ストロンチウムも定量され,酸化カルシウム量として含まれる。
この方法は,マンガンが共存している場合でも酸化カルシウムの定量に用いることができる。この方法
をEDTA溶液による酸化マグネシウムの定量方法 (A.13.15) と共に用いる場合は,マンガンの定量 (16.2)
を先に行い,A.13.15.1の制約に適合しているかを確認する必要がある。
A.13.14.2 操作
A.13.7で保存した最終溶液50 mLを光度滴定装置 (A.5.10) に適したビーカーに全量ピペットで分取す
る。次に,光度滴定装置の操作に適した量の水を加え,トリエタノールアミン溶液 (1+4) (A.4.2.35) 50 mL
を加える。
pHメーター (A.5.16.1) を用いて,水酸化ナトリウム溶液 (4 mol/L) (A.4.2.18) でこの溶液のpHを12.5
±0.5に調節する。
約0.1 gのムレキシド指示薬 (A.4.2.44),カルセイン指示薬 (A.4.2.45) 又はカルセイン−メチルチモール
ブルー混合指示薬 (A.4.2.50) を加える。吸光度の測定波長はムレキシド指示薬を使用する場合は620 nm,
カルセイン指示薬を使用する場合は520 nmとし,光度滴定装置 (A.5.10) にビーカーを置き,溶液をかく
50
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
はんしながらEDTA溶液(約0.03 mol/L)(A.4.2.40) で滴定する。EDTA溶液(約0.03 mol/L)の滴定量と
吸光度を用い,変色域付近の滴定曲線を作成する。終点の滴定量 (V15) は滴定曲線において変色域付近の
最大傾斜線と変色後の吸光度がほとんど変化しない部分の線との交点から求める。
A.13.14.3 計算
試料中の酸化カルシウムの含有率 (wCaO) は,式(A.15)によって算出する。
100
50
000
1
500
08
.
56
03
.0
23
D
15
CaO
×
×
×
×
×
×
×
=
m
f
V
w
23
D
15
4
682
.1
m
f
V×
×
=
·························································(A.15)
ここに,
V15: 滴定に要したEDTA溶液(約0.03 mol/L)の量 (mL)
fD: A.4.2.40.2で求めたEDTA溶液(約0.03 mol/L)の
ファクター
m23: A.13.2又はA.13.5.1の試料の質量 (g)
注記 酸化ストロンチウムも定量され,酸化カルシウム量として含まれる。
A.13.14.4 繰返し精度及び再現精度
繰返し精度の標準偏差 0.15 %
再現精度の標準偏差 0.43 %
A.13.15 EDTA溶液による酸化マグネシウムの定量方法(代替方法)
A.13.15.1 試験上の制約
注記 この方法は光度滴定によって終点判定を行う。目視による終点判定も可能である。この場合,
カルセイン−メチルチモールブルー混合指示薬 (A.4.2.50)(ピンク色から無色に変色),混合指
示薬 (A.4.2.52)(ピンク色から無色に変色)又はフタレインパープル1 gをNaCl 100gに混合し
たもの(紫色から淡いピンク色に変色)が指示薬として適切である。
三酸化マンガン (Mn2O3) の含有率が0.5 %より多いセメントの場合,DCTA溶液(約0.01 mol/L)
(A.4.2.43) による酸化マグネシウムの定量方法だけが適用できる。
A.13.15.2 操作
A.13.7で保存した最終溶液50 mLを光度滴定装置 (A.5.10) に適したビーカーに全量ピペットで分取し,
次に光度滴定装置の操作に適した量の水及びトリエタノールアミン溶液 (1+4) (A.4.2.35) 50 mLを加える。
pHメーター (A.5.16.1) を用いて,アンモニア水 (1+1) (A.4.2.15) でこの溶液のpHを10.5±0.5に調節
する。
ビュレットを用いて,A.13.14.2であらかじめ求めた酸化カルシウムの滴定に必要なEDTA溶液(約0.03
mol/L)(A.4.2.40) 量V15を加える。
次にメチルチモールブルー指示薬 (A.4.2.49),カルセイン−メチルチモールブルー混合指示薬 (A.4.2.50)
又は混合指示薬 (A.4.2.52) を約0.1 g加える。
吸光度の測定波長を620 nmにした光度滴定装置 (A.5.10) にビーカーを置き,溶液をかくはんしながら,
EDTA溶液(約0.03 mol/L)(A.4.2.40) で滴定する。EDTA溶液(約0.03 mol/L)の滴定量と吸光度を用い,
変色域付近の滴定曲線を作成する。終点の滴定量 (V16) は滴定曲線において変色域付近の最大傾斜線と変
色後の吸光度がほとんど変化しない部分の線との交点から求める。
A.13.15.3 計算
試料中の酸化マグネシウムの含有率 (wMgO) は,式(A.16)によって算出する。
51
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
50
000
1
)
(
500
311
.
40
03
.0
23
D
15
16
MgO
×
×
×
×
−
×
×
×
=
m
f
V
V
w
23
D
15
16
)
(
3
209
.1
m
f
V
V
×
−
×
=
··············································(A.16)
ここに,
V15: A.13.14.2で求めた酸化カルシウムの定量に必要なEDTA溶液
(約0.03 mol/L)の量 (mL)
V16: 滴定に要したEDTA溶液(約0.03 mol/L)の量 (mL)
fD: A.4.2.40.2で求めたEDTA溶液(約0.03 mol/L)の
ファクター
m23: A.13.2又はA.13.5.1の試料の質量 (g)
A.13.15.4 繰返し精度及び再現精度
繰返し精度の標準偏差 0.21 %
再現精度の標準偏差 0.25 %
52
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(参考)
塩酸−水酸化カリウム方法による不溶残分の定量方法
序文
セメントの湿式による化学分析方法の国際規格は,ISO 29581-1 (Cement−Test methods−Part 1:Analysis
by wet chemistry)(セメントの湿式による化学分析方法は,ISO 680:1990として規定されていたが,2009
年3月15日に改正された。その際,規格番号は,ISO 680からISO 29581-1に変更となった。)であり,
その中で不溶残分の定量方法は,塩酸−炭酸ナトリウム方法及び塩酸−水酸化カリウム方法が規定されて
いる。しかし,それらに優先関係が示されていないことから,この規格(本体)では,従来からJIS R 5202
として用いられている塩酸−炭酸ナトリウム方法を規定した。そのため,塩酸−水酸化カリウム方法は翻
訳し,技術的内容を変更することなく附属書として示した。
B.1
適用範囲,引用規格,試験の一般的要求事項及びセメント試料の調製
適用範囲,引用規格,試験の一般的要求事項及びセメント試料の調製は,それぞれ附属書Aによる。
B.2
要旨
この定量方法は,試料を塩酸で溶解し,セメント中の不溶残分を定量する方法である。セメントを塩酸
で溶解し,残分を加熱した水酸化カリウム溶液で処理する。処理後の残分を強熱した後,質量を量る。
B.3
試薬
B.3.1 塩酸
B.3.2 塩酸 (1+3)
B.3.3 塩酸 (1+9)
B.3.4 水酸化カリウム溶液 水酸化カリウム (KOH) 250 gを水に溶かして,1 000 mLにする。ポリエチレ
ン製の瓶に保存する。
B.3.5 硝酸銀 (AgNO3) 150±5 ℃で乾燥する。
B.3.6 硝酸銀溶液 硝酸銀 (B.3.5) 5 gを水に溶かし,硝酸 (HNO3) 10 mLを加え,水で1 000 mLにする。
B.4
装置
B.4.1 磁器の蒸発皿 約200 mLのものとする。
B.4.2 水浴 105±5 ℃に調節できるものとする。
B.4.3 球管冷却器
B.4.4 ろ紙 灰分のないものとし,平均孔径が約7 μmのろ紙。
B.5
操作
セメント1.00±0.05 g (m1) を磁器の蒸発皿 (B.4.1) に量り採り,水25 mLを加え,ガラス棒で試料を分
散させる。塩酸 (B.3.1) 40 mLを加える。緩やかに溶液を温め,先端が平らなガラス棒で試料をつぶし,セ
メントを完全に溶解する。水浴上で蒸発乾固した後,塩酸 (B.3.1) を20 mLとして,同様に蒸発乾固を2
53
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
回繰り返す。
最後の蒸発乾固した残分に塩酸 (1+3) (B.3.2) 100 mLを加えて再度温め,ろ紙 (B.4.4) でろ過し,硝酸
銀試験 (A.3.6) で塩化物イオンが認められなくなるまで,温水で洗浄する。
ろ紙とその中身を球管冷却器付きのコニカルフラスコ250 mLに移し,水酸化カリウム溶液 (B.3.4) 100
mLを加える。
室温で16時間放置した後,還流状態で溶液を4時間煮沸する。
ろ紙 (B.4.4) を用いて不溶残分をろ過し,水で洗浄する。更に塩酸 (1+9) (B.3.3) 100 mLで洗浄し,硝
酸銀試験 (A.3.6) で塩化物イオンが認められなくなるまで温水で洗う。残留物を950±25 ℃で強熱 (A.3.4)
して恒量 (A.3.5) を確認し,質量 (m2) を量る。
注記 通常,30分間の強熱で恒量となる。
B.6
計算
試料中の不溶残分 (wins) は,次の式(B.1)によって算出する。
100
1
2
ins
×
=m
m
w
········································································ (B.1)
ここに,
m1: 試料の質量 (g)
m2: 残留物の質量 (g)
B.7
繰返し精度及び再現精度
繰返し精度の標準偏差 0.15 %
再現精度の標準偏差 0.18 %
54
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
JIS R 5210 ポルトランドセメント
JIS R 5211 高炉セメント
JIS R 5212 シリカセメント
JIS R 5213 フライアッシュセメント
JIS R 5214 エコセメント
55
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JA
(参考)
JISと対応国際規格との対比表
JIS R 5202:2010 セメントの化学分析方法
ISO 29581-1:2009 Cement−Test methods−Part 1: Analysis by wet chemistry
(I)JISの規定
(II)
国際
規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異
の箇条ごとの評価及びその内容
(V)JISと国際規格との技術的差異の理由
及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
1 適用範囲 JISの品質規格で規定
されているセメント。
クリンカー及び高炉セ
メントの製造に用いる
高炉スラグに適用。
1 適用範囲 セメント及びセメント
の構成物質。
変更
国際規格では“セメント
の構成物質”も適用範囲
に含めている。
国際規格は主成分の分析体系が完全分析
であるので,種々な“セメントの構成物
質”も適用範囲に含めている。整合はセ
メントの品質規格が国際規格として制定
されるまで,保留せざるをえない。
2 引用規格
3.1 試験回
数
試験は,同一試料につ
いて2回の繰返しで行
う。ただし,品質管理
を目的とする場合は1
回でもよい。
3.1 試験回
数
JISに同じ。
一致
−
−
3.2 許容差
2回の試験結果の差が
許容差より大きい場
合,更に1回再試験を
行い,最も近い二つの
試験結果を平均し,分
析結果(3.3参照)とす
る。
3.2 繰返し
精度と再現
精度
2回の定量値の差が繰
返し精度の標準偏差の
2倍以上であった場
合,再試験を行い,最
も近い二つの定量値の
平均をとる。
変更
国際規格は繰返し精度の
標準偏差の2倍がJISの
許容差に該当する。
国際規格の繰返し精度の標準偏差の2倍
を許容差とすることは,分析の信頼性の
確保の点から大きい場合があり,また手
順によって変化するのでJIS独自の値を
採用した。
3
R
5
2
0
2
:
2
0
1
0
56
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)
国際
規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異
の箇条ごとの評価及びその内容
(V)JISと国際規格との技術的差異の理由
及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
3.3 質量,
体積,ファ
クター及び
結果の表し
方
・ 質量:0.000 1 gま
で量る。
・ 体積:ビュレット
を用いる場合,
0.01 mLまで読む。
・ ファクターのけた
数:分析項目ごと
に規定。
・ 結果の表し方:Cl
を除く分析項目に
ついて小数点以下
2けた。Clは小数
点以下3けた。
3.3 質量,
体積,ファ
クター及び
結果の表し
方
・ 質量:0.000 1 gま
で量る。
・ 体積:ビュレット
を用いる場合,
0.05 mLまで読む。
・ ファクターのけた
数:小数点以下3
けた。
・ 結果の表し方:原
則,小数点以下2
けた。
変更
・ ビュレットを用いる
場合の最小の読み取
り値が異なる。
・ ファクターのけた数
の決め方が異なる。
・ 国際規格は“原則”
として規定してい
る。
体積の読み取りは分析の信頼性の確保か
らJISの値としている。ファクターのけ
た数は分析精度の観点から一律に決める
ことはできないため,分析項目ごとに規
定。結果の表し方はあいまいさをなくす
ため,JISでは分析項目ごとに規定。
3.4 強熱
灰化,強熱,放冷の手
順を規定。
3.4 強熱
JISに同じ。
一致
−
−
3.5 ガラス
器具
分析に使用するガラス
器具は,JIS R 3503及
びJIS R 3505による。
−
−
追加
−
国際規格では規定していないが,分析に
ついての共通事項として必要なため,JIS
では追加した。
3.6 その他
の一般事項
その他の一般事項は,
通常,JIS K 0050,JIS
K 0113,JIS K 0115及
びJIS K 0121による。
−
−
追加
−
国際規格では規定していないが,分析に
ついての共通事項として必要なため,JIS
では追加した。
−
−
3.5 恒量
15分間の強熱を繰り
返し行い,その前後の
質量の差が0.000 5 g未
満になったとき。
削除
−
箇条5(強熱減量の定量方法)において規
定した。
−
−
3.6 塩化物
イオンの有
無の確認
沈殿の洗浄液について
塩化物イオンの有無を
硝酸銀溶液を用いて確
認する。
削除
−
ろ液を使用して確認試験を行うのは,分
析誤差を生じる原因となるので,分析の
信頼性の確保から採用しない。
3
R
5
2
0
2
:
2
0
1
0
57
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)
国際
規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異
の箇条ごとの評価及びその内容
(V)JISと国際規格との技術的差異の理由
及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
−
−
3.7 空試験
試料を入れずに空試験
を行い,その結果によ
って補正計算を行う。
削除
−
国際規格では一般的共通事項として空試
験を規定しているが,その結果の反映の
仕方があいまいである。よって,JISでは
酸化アルミニウムの定量方法及び塩素の
定量方法においてだけ規定した。
4 セメント
試料の調製
試料は,検査単位につ
いて平均品質を表すよ
うに,セメントを採取
し,縮分して約5 kgの
代表試料とする。
採取方法及び縮分方法
は,受渡当事者間の協
議による。
JIS Z 8801-1に規定す
る150 μm又は125 μm
のふるいでふるう。残
分中の金属鉄を磁石を
用いて取り除く。
6 セメント
試料の調製
150 μm又は125 μm網
ふるいでふるう。残分
中の金属鉄を磁石を用
いて取り除く。
追加
国際規格は代表試料のサ
ンプリング方法が規定さ
れていない。
代表試料のサンプリング方法は,分析の
信頼性にも影響を及ぼすので,JISの規定
を追加した。
5 強熱減量
の定量方法
強熱:950±25 ℃,15
分間。
恒量:強熱前後の質量
の差が0.000 5 g未満に
なったとき。
高炉セメント及び高炉
スラグの場合は,強熱
前後のSO3量を定量
し,補正する。
強熱後のSO3量の定量
はJIS R 5204によっ
て,ガラスビードの定
量値を用いてもよい。
7 強熱減量
の定量方法
強熱:950±25 ℃,15
分間。
恒量:強熱前後の質量
の差が0.000 5 g未満に
なったとき。
硫化物硫黄 (S2−) を
含む試料は強熱前後の
SO3量を定量し,補正
する。
追加
JISでは高炉セメント及
び高炉スラグの場合の強
熱後のSO3量の定量方法
として,ガラスビードの
定量値を用いてもよいこ
とを追加。
蛍光X線分析法は現時点で国際規格とし
て制定されていないが,制定後はJISの
内容を提案する。
3
R
5
2
0
2
:
2
0
1
0
58
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)
国際
規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異
の箇条ごとの評価及びその内容
(V)JISと国際規格との技術的差異の理由
及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
6 塩酸−炭
酸ナトリウ
ム方法によ
る不溶残分
の定量方法
塩酸−炭酸ナトリウム
分解
試料:約1 g
塩酸 (1+1):10 mL,
炭酸ナトリウム:2.5 g
強熱:950±25 ℃,30
分間
9 塩酸−炭
酸ナトリウ
ム方法によ
る不溶残分
の定量方法
塩酸−炭酸ナトリウム
分解
試料:1.00±0.05 g
塩酸:10 mL,炭酸ナ
トリウム:5 g
強熱:950±25 ℃,恒
量を確認
変更
塩酸及び炭酸ナトリウム
の使用量がJISの2倍で
ある。
JISの試薬の使用量は試料の可溶分を溶
解するのに十分である。
−
−
10塩酸−
水酸化カリ
ウム方法に
よる不溶残
分の定量方
法
塩酸−水酸化カリウム
分解
試料:1.00±0.05 g
塩酸:40 mL,
水酸化カリウム溶液
(250 g/L):100 mL
強熱:950±25 ℃,恒
量を確認
削除
−
国際規格において塩酸−炭酸ナトリウム
方法と塩酸−水酸化カリウム方法との使
用の優先関係は示されていない。そのた
め,塩酸−水酸化カリウム方法は附属書B
(参考)とし,国際規格の理解の一助と
した。
7 二酸化け
い素,
8 酸化アル
ミニウム,
9 酸化鉄
(III),
10 酸化カ
ルシウム及
び
11 酸化マ
グネシウム
の定量方法
過塩素酸 (60 %) と塩
酸 (1+1) を用いて二
酸化けい素を析出して
ろ別し,ろ液を用いて
主成分の定量を行う。
13 主成分
の定量
試料の分解は次のいず
れかの方法による。
・(本法)過酸化ナトリ
ウム分解−ポリ酸化エ
チレン法
・(別法)過酸化ナトリ
ウム分解−二重蒸発乾
固法
・(別法)塩酸−塩化ア
ンモニウム分解法
変更
JISは試料を酸で分解す
るが,国際規格は試料を
完全に分解する方法を採
用している。したがって,
酸に対する不溶分の多い
試料についてはJISと国
際規格とでは定量値に差
が生じる。
JISにおけるセメントの品質規格値は酸
可溶分分解によって得られた定量値に基
づいているため,規格の整合性の観点及
び“技術的差異の内容”から完全分解に
よる方法は採用できない。
そのため,国際規格の主成分の定量方法
は附属書A(参考)とし,国際規格の理
解の一助とした。
3
R
5
2
0
2
:
2
0
1
0
59
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)
国際
規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異
の箇条ごとの評価及びその内容
(V)JISと国際規格との技術的差異の理由
及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
12 三酸化
硫黄の定量
方法
試料溶液は塩酸分解溶
液。
硫酸バリウムの沈殿生
成時間:3時間
強熱:950±25 ℃,30
分間
8 三酸化硫
黄の定量方
法
試料溶液は塩酸分解溶
液。
硫酸バリウムの沈殿生
成時間:12〜24時間
強熱:950±25 ℃,恒
量(15分間)
恒量を確認することを
規定。
変更
硫酸バリウムの沈殿生成
におけるpH及び生成時
間が異なる。
硫酸バリウムの生成時間はJIS R 9101(セ
ッコウの化学分析方法)でも3時間と規
定されており,JIS間での整合の観点か
ら,3時間を採用する必要があった。
また,生成時間が長いと鉄の吸着による
誤差が生じる可能性もある。
−
−
17 炎光光
度法による
アルカリの
定量
(標準方
法)
試料の不溶残分量によ
って試料の分解方法が
異なる。不溶残分量が
3 %を超える場合は完
全分解する。3 %以下
の場合は塩酸 (1+19)
で分解するが,不溶分
も含め,溶解後の溶液
をメスフラスコに移
す。そして,炎光光度
計で光度を測定する前
に固形分をろ過する。
削除
JISは酸可溶性アルカリ
の定量方法だけを規定し
ているのに対し,国際規
格は試料を完全分解する
方法も規定している。
JISにおけるセメントの品質規格値は酸
可溶分分解によって得られた定量値に基
づいているため,規格の整合性の観点か
ら完全分解による方法は採用できない。
また,国際規格では3 %以下の場合は塩酸
(1+19) で分解するが,不溶分も含め,溶
解後の溶液を全量フラスコに移すことが
規定されているが,この手順は分析化学
上,許容できない。
13.1 酸化
ナトリウム
及び酸化カ
リウムの定
量方法(原
子吸光法)
試料溶液は試料溶液
(A)(過塩素酸と塩酸を
使用)を希釈。
18.2原子吸
光法による
酸可溶アル
カリの定量
方法(代替
方法)
試料を塩酸 (1+19)
で分解する。
追加
JISでは主成分の分析で
調製した試料溶液 (A)
を希釈して定量するが,
国際規格は別途,試料を
分解する。
主成分の分析体系が整合されるまで,対
応しない。
3
R
5
2
0
2
:
2
0
1
0
60
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)
国際
規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異
の箇条ごとの評価及びその内容
(V)JISと国際規格との技術的差異の理由
及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
13.2 酸化
ナトリウム
及び酸化カ
リウムの定
量方法(炎
光光度法)
国際規格の内容の内,
試料の不溶残分量が
3 %以下の場合の手順
を規定。
18.1炎光光
度法による
アルカリの
定量
(代替方
法)
試料の不溶残分量によ
って試料の分解方法が
異なる。不溶残分量が
3 %を超える場合は硫
酸と塩酸を共存させ,
試料を分解する。3 %
以下の場合は塩酸 (1
+19) で分解する。定
量は炎光光度計を用い
て行う。
変更
JISは酸可溶性アルカリ
の定量方法だけを規定し
ているのに対し,国際規
格は試料を完全分解する
方法も規定している。
JISにおけるセメントの品質規格値は酸
可溶分分解によって得られた定量値に基
づいているため,規格の整合性の観点か
ら完全分解による方法は採用できない。
不溶残分量が3 %以下の場合は,酸可溶分
分解を規定しているので,その方法に限
定し採用した。
14 酸化チ
タン (IV)
の定量方法
ジアンチピリルメタン
吸光光度法
試料溶液は,塩酸分解
溶液を使用する。
−
−
追加
JISとして必要な定量方
法であるため追加した。
−
15 酸化り
ん (V) の
定量方法
モリブデン青吸光光度
法
試料溶液は,試料溶液
(A) の一部を使用す
る。
−
−
追加
JISとして必要な定量方
法であるため追加した。
−
16.1 酸化
マンガン
(II) の定量
方法(原子
吸光法)
フレーム原子吸光法
試料溶液は試料溶液
(A) を希釈。
−
−
追加
JISとして必要な定量方
法であるため追加した。
−
16.2 酸化
マンガン
(II) の定量
方法(吸光
光度法)
過よう素酸吸光光度法
試料溶液は硝酸分解溶
液。
12 吸光光
度法による
マンガンの
定量方法
JISに同じ。
一致
−
−
3
R
5
2
0
2
:
2
0
1
0
61
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)
国際
規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異
の箇条ごとの評価及びその内容
(V)JISと国際規格との技術的差異の理由
及び今後の対策
箇条番号
及び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
17 硫化物
硫黄の定量
方法
よう素酸カリウム標準
液を用いた直接滴定法
及びチオ硫酸ナトリウ
ム標準液を用いた逆滴
定法(国際規格)を規
定した。
11 硫化物
硫黄の定量
方法
チオ硫酸ナトリウム標
準液を用いた逆滴定法
を規定。
選択
JISでは二つの方法を規
定。
−
18 塩素の
定量方法
電位差滴定法,チオシ
アン酸水銀 (II) 吸光
光度法及びチオシアン
酸アンモニウム溶液に
よる逆滴定法(国際規
格)を規定した。
14 塩素の
定量方法
標準方法としてチオシ
アン酸アンモニウム溶
液による逆滴定法を規
定し,代替方法として
電位差滴定法を規定し
ている。
選択
JISでは三つの方法を規
定。
−
−
−
15 二酸化
炭素(標準
方法)
吸収剤(水酸化ナトリ
ウムベース)を用いた
質量方法を規定。
削除
−
品質規格の項目として規定されていない
が,今後,定量精度の検討を行った上で,
規定化の検討を行う。
−
−
16 二酸化
炭素(代替
方法)
吸収剤(水酸化ナトリ
ウムベース)を用いた
質量方法を規定。
削除
−
品質規格の項目として規定されていない
が,今後,定量精度の検討を行った上で,
規定化の検討を行う。
附属書A
(参考)
−
附属書B
(参考)
−
JISと国際規格との対応の程度の全体評価:ISO 29581-1:2009,MOD
3
R
5
2
0
2
:
2
0
1
0

62
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記1 箇条ごとの評価欄の用語の意味は,次による。
− 一致 ················ 技術的差異がない。
− 削除 ················ 国際規格の規定項目又は規定内容を削除している。
− 追加 ················ 国際規格にない規定項目又は規定内容を追加している。
− 変更 ················ 国際規格の規定内容を変更している。
− 選択 ················ 国際規格の規定内容とは異なる規定内容を追加し,それらのいずれかを選択するとしている。
注記2 JISと国際規格との対応の程度の全体評価欄の記号の意味は,次による。
− MOD ··············· 国際規格を修正している。
3
R
5
2
0
2
:
2
0
1
0
63
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JB
(参考)
技術上重要な改正に関する新旧対照表
現行規格 (JIS R 5202:2010)
旧規格 (JIS R 5202:1999)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
規格名称
セメントの化学分析方法
規格名称
ポルトランドセメントの化学分析方
法
適用範囲において旧規格で準用規定
としていたポルトランド以外の他の
セメント,クリンカー及び高炉スラグ
にも明確に適用できるように改正し
たため。
構成の変更
本体は,従来から国内で用いられてきた試験方法を対
応国際規格であるISO 29581-1と可能な限り整合させ
た。具体的には,ISO 29581-1で規定されている酸可
溶分析の方法を追加した。なお,国内ではほとんど用
いられていない完全分析によるセメントの主成分の化
学分析方法を,附属書A(参考),塩酸−水酸化カリウ
ム方法による不溶残分の定量方法を附属書B(参考)
として示した。
−
本体は,従来から国内で用いられてい
た試験方法を規定し,附属書(規定)
にISO 680と一致した“国際規格によ
るセメントの化学分析方法”を規定。
旧規格ではJISと国際規格との整合の
観点から,国際規格の内容のすべてを
附属書(規定)として規定していた。
しかし,不溶残分量が大きい試料の場
合,旧規格の本体の酸可溶分析と附属
書(規定)の完全分析とでは定量値が
異なるため,“分析方法の報告”の箇
条を設け,本体又は附属書のいずれか
の方法で分析を行ったかを報告する
など,複雑な構成となっていた。
ISO 680がISO 29581-1として2009年
3月に発行され,我が国の意見も一部
採用されたことから,全体構成は,国
際整合化した。ただし,国内での実態
等をかんが(鑑)み,国内でほとんど
用いられていない試験方法は,附属書
(参考)とするなど技術上の一部を変
更した。
3
R
5
2
0
2
:
2
0
1
0
64
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格 (JIS R 5202:2010)
旧規格 (JIS R 5202:1999)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
1 適用範囲
この規格は,セメント1)の湿式方法による化学分析方
法について規定する。
この規格は,クリンカー及び高炉セメントの製造に
用いる高炉スラグの化学分析にも適用することがで
きる。
注1) ここでいうセメントとは,JIS R 5210に規定す
るポルトランドセメント,JIS R 5211に規定す
る高炉セメント,JIS R 5212に規定するシリカ
セメント,JIS R 5213に規定するフライアッシ
ュセメント及びJIS R 5214に規定するエコセメ
ントを指す。
1. 適用範囲
この規格は,ポルトランドセメントの
化学分析方法について規定する。
なお,この化学分析方法は,高炉セ
メント,シリカセメント,フライアッ
シュセメント,クリンカー及び高炉セ
メントの製造に用いる高炉スラグの
化学分析にも準用することができる。
実態に合わせ,他のセメント試験方法
規格と同様に,旧規格において準用規
定としていたポルトランドセメント
以外のセメントに対しても明確に適
用できるようにした。また,クリンカ
ー及び高炉セメントの製造に用いる
高炉スラグに対しても明確に適用で
きるようにした。
2 引用規格
JIS K 0050 化学分析方法通則
JIS K 0113 電位差・電流・電量・カールフィッシャ
ー滴定方法通則
JIS K 0115 吸光光度分析通則
JIS K 0121 原子吸光分析通則
JIS K 8005 容量分析用標準物質
JIS K 8102 エタノール (95)(試薬)
JIS P 3801 ろ紙(化学分析用)
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS R 5204 セメントの蛍光X線分析方法
JIS Z 8401 数値の丸め方
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふる
い
2. 引用規格
JIS K 0018 標準物質−pH標準液−
しゅう酸塩
JIS K 0050 化学分析方法通則
JIS K 0113 電位差・電流・電量・カ
ールフィッシャー滴定方法通則
JIS K 0115 吸光光度分析通則
JIS K 0121 原子吸光分析通則
JIS K 8005 容量分析用標準物質
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS R 5201 セメントの物理試験方法
JIS Z 8401 数値の丸め方
JIS Z 8801 試験用ふるい
最新のJISの制定,改正等の動向を反
映した。
−
−
3. 分析項目
15の分析項目を列記していた。
国際規格では分析項目の箇条がない
ため削除した。
3
R
5
2
0
2
:
2
0
1
0
65
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格 (JIS R 5202:2010)
旧規格 (JIS R 5202:1999)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
3.1 試験回
数
試験は,同一試料について2回の繰返しで行う。ただ
し,品質管理を目的とする場合は1回でもよい。
6.2 許容差
分析は,同一試料について原則として
2回繰り返して行い,その差が許容差
(%) に示す数値よりも大きいときは再
分析し,許容差以内のものの平均値を
とる。
国際規格と整合するため,試験回数
の箇条を設けた。また,品質管理を
目的とする場合の試験回数を規定し
た。
3.2 許容差
2回の試験結果の差が,許容差より大きい場合,更に
1回,再試験を行い,最も近い二つの試験結果を平均
し,分析結果(3.3参照)とする。
内容の変更はないが,構成の変更に
よって箇条の番号を変更した。
3.3 質量,体
積,ファク
ター及び結
果の表し方
質量は0.000 1 gまで量る。ビュレットを用いる場合,
体積は0.01 mLまで読む。
溶液のファクターは3回の測定値を平均し,決定す
る。
個々の試験結果は,JIS Z 8401の2. b)によって,塩
素 (Cl) を除く分析項目は百分率で小数点以下2けた
に丸める。塩素 (Cl) については百分率で小数点以下3
けたに丸める。
分析結果は,2回の試験結果の平均とし,JIS Z 8401
の2. c) 規則A によって,塩素 (Cl) を除く分析項目
は百分率で小数点以下2けたに丸める。塩素 (Cl) に
ついては百分率で小数点以下3けたに丸める。
6.1 分析値
分析値は,百分率で表し,JIS Z 8401
によって次のように丸める。
a) 強熱減量,不溶残分,二酸化けい
素,酸化アルミニウム,酸化第二
鉄,酸化カルシウム,酸化マグネ
シウム,三酸化硫黄,酸化ナトリ
ウム,酸化カリウム,二酸化チタ
ン,五酸化りん,一酸化マンガン
及び硫化物硫黄の14成分は,小数
点以下2けたに丸める。
b) 塩素は,小数点以下3けたに丸め
る。
国際規格に整合させ,“試験の一般
的要求事項”として,質量,体積の
読み取り単位を規定した。また,溶
液のファクターの算出方法を規定し
た。さらに,試験結果及び分析結果
の値のまとめ方を明確にした。
3.4 強熱
強熱は,次のように行う。
あらかじめ空焼きし,風袋を量ったるつぼにろ紙と沈
殿(残留物)とを入れて乾燥し,酸化雰囲気で炎の出
ないように徐々に加熱してろ紙を灰化した後,規定の
温度で強熱する。強熱後,るつぼと内容物とをデシケ
ータ中で室温まで放冷した後,質量を量る。
附属書3.4 強
熱
現行規格に同じ。
国際規格に整合させ,“試験の一般
的要求事項”として規定した。
3
R
5
2
0
2
:
2
0
1
0
66
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格 (JIS R 5202:2010)
旧規格 (JIS R 5202:1999)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
4 セメント試
料の調製
試料は,検査単位について平均品質を表すように,セ
メントを採取し,縮分して約5 kgの代表試料とする。
その採取方法及び縮分方法は,受渡当事者間の協議に
よって定める。代表試料を更に縮分器又は四分法で縮
分して,約100 gを採り,JIS Z 8801-1に規定する
150 μm又は125 μmのふるいで,ふるう。ふるい残分
中の金属鉄を磁石を使って取り除き,金属鉄を除いた
残分をすりつぶして完全に150 μm又は125 μmのふる
いを通過させる。試料を気密性のあるきれいな乾いた
容器に移し替え,十分に試料が混ざるように強く振
る。
上記の操作は,試料が周囲の空気にさらされる時間
をできるだけ短くするために,手早く行わなければな
らない。
4.1 セメント
の試料調整
試料は,JIS R 5201の4.(試料)によ
って採取し,調整したものを用いる。
国際規格と整合した。
ただし,国際規格には代表試料のサ
ンプリング方法が規定されていな
い。
代表試料のサンプリング方法は化学
分析の信頼性にも影響を及ぼすの
で,その方法を追加した。
−
−
7. 分析方法
の報告
分析結果を報告する場合は,分析項目
ごとに本体又は附属書のいずれかの
方法で行ったかを明記する。
なお,報告する分析項目すべての分
析を本体又は附属書のいずれかの方
法で行った場合は,まとめて明記して
もよい。
完全分析によるセメントの主成分の
化学分析方法を附属書(参考)とし
たため,区別して報告する必要がな
くなったので削除した。
5 強熱減量の
定量方法
強熱温度を950±25 ℃と規定。
試料が高炉セメント及び高炉スラグの場合 (5.3) は,
強熱前後(強熱温度は950±25 ℃)の三酸化硫黄含有
率を求め,硫化物の酸化による増量を補正する。
なお,強熱後の三酸化硫黄含有率は,JIS R 5204に
よって求めたガラスビードの三酸化硫黄の定量値を
用いてもよい。
8. 強熱減量
の定量方法
附属書7. 強
熱減量の定量
方法
強熱温度を975±25 ℃と規定。ただ
し,試料が高炉セメント及び高炉スラ
グの場合は,補正方法として補正方法
1(強熱前後の試料中の三酸化硫黄量
から補正する方法)と補正方法2(試
料中の硫化物硫黄量から補正する方
法)を規定。また,注で“700±25 ℃
で強熱してもよい”ことを示してい
た。
JIS R 5211(高炉セメント)の改正に
よって,高炉セメントへの石灰石の
混合が可となった。しかし,石灰石
の脱炭酸と硫化物の酸化が分離可能
な適切な温度はなく,旧規格の補正
方法1を採用し,強熱温度を950±
25 ℃とした。なお,旧規格の補正方
法2は国際規格の改正によって削除
されたため,削除した。
3
R
5
2
0
2
:
2
0
1
0
67
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格 (JIS R 5202:2010)
旧規格 (JIS R 5202:1999)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
6 塩酸−炭酸
ナトリウム方
法による不溶
残分の定量方
法
JIS P 3801に適合したろ紙(5種B,110 mm)を用い
てろ過し,温水で8回洗浄する。ろ液及び洗液はビー
カー500 mLに受け,室温まで冷却する。その後,全
量フラスコ250 mLに洗い移し,標線まで水を加えて
振り混ぜる。この溶液を試料溶液 (F) とし,三酸化硫
黄及び酸化チタン (IV) の定量に用いる。
なお,酸化チタン (IV) の定量を行わない場合には,
ビーカーに受けたろ液及び洗液をそのまま保存して
三酸化硫黄の定量に用いる。
9. 不溶残分
の定量方法
附属書9. 塩
酸−炭酸ナト
リウム方法に
よる不溶残分
の定量方法
ろ紙(5種B,11.0 cm)を用いてろ過
し,温水で8回洗浄する。ろ液は,ビ
ーカー500 mLに受け,そのまま保存
して三酸化硫黄の定量に用いる。
6.4 c)で保存した試料溶液 (F) の一
部を使用して,酸化チタン (IV) の定
量を行えるようにした。
8.2 d) キシレ
ノールオレン
ジ溶液
2か月ごとに新しく調製する。
11.2 d) キシ
レノールオレ
ンジ指示薬
1,2か月ごとに新しく調製する。
冷暗所に正しく保存すれば,2か月は
使用できるため。
8.2 e) 亜鉛標
準液
亜鉛標準液の調製方法として,JIS K 8005に規定する
亜鉛(99.99 %以上)を使用する方法又は国家計量標
準にトレーサブルな市販の亜鉛標準液を使用する方
法を規定。
11.2 e) 亜鉛
標準液
亜鉛標準液の調製方法として,JIS K
8005に規定する亜鉛(99.99 %以上)
を使用する方法を規定。
国家計量標準にトレーサブルな標準
液が市販されており,これを使用で
きるようにした。
8.5 空試験
空試験用溶液を試料溶液 (A) の調製で用いる試薬
[過塩素酸 (60 %) 及び塩酸 (1+1)]を用いて調製
し,試料溶液 (A) の代わりとする。
11.5 空試験
空試験用溶液を試料溶液 (A) の調製
と同じ操作で調製する。
酸化アルミニウムの定量方法は逆滴
定であり,そのための空試験である
ことから,調製方法を簡便化した。
9.2 a) 酸化鉄
(III) 標準原
液
金属鉄(99.0 %以上)又は国家計量標準にトレーサブ
ルな市販の鉄標準液を水で希釈し,Fe2O3として約1
mg/mLとなるように調製する。
12.2 a) 酸化
第二鉄標準原
液
金属鉄(99.9 %以上)を用いて調製す
る。
標準液の調製に用いる金属鉄の純度
を入手可能なものとした。また,国
家計量標準にトレーサブルな標準液
が市販されており,これを使用でき
るようにした。
10.2 カルセイ
ン指示薬又は
カルセイン−
PPC指示薬を
用いる定量方
法
酸化カルシウムの定量において,カルセイン指示薬の
単独使用を認めた。
13.2 カルセ
イン−PPC指
示薬を用いる
定量方法
酸化カルシウムの定量において,カル
セインとPPCの混合指示薬を用いる
ことを規定。
カルセイン指示薬の単独使用によっ
ても,終点の判別に問題のないこと
から,単独使用を追加した。
3
R
5
2
0
2
:
2
0
1
0
68
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格 (JIS R 5202:2010)
旧規格 (JIS R 5202:1999)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
11.2 a) 酸化
マグネシウム
標準原液
金属マグネシウム(99.0 %以上)又は国家計量標準に
トレーサブルな市販のマグネシウム標準液を水で希
釈し,MgOとして約1 mg/mLとなるように調製する。
14.2 a) 酸化
マグネシウム
標準原液
金属マグネシウム(99.9 %以上)を用
いて調製する。
標準液の調製に用いる金属マグネシ
ウムの純度を入手可能なものとし
た。また,国家計量標準にトレーサ
ブルな標準液が市販されており,こ
れを使用できるようにした。
12 三酸化硫
黄の定量方法
6.4 c)で保存した試料溶液 (F) から200 mLを分取し
て,ビーカー500 mL に入れる。
なお,酸化チタン (IV) の定量を行わない場合には,
6.4 c)で保存した溶液をすべて三酸化硫黄の定量に用
いる。
15. 三酸化硫
黄の定量方法
9.4 c)で保存したろ液を水で約200 mL
に薄め,加熱して煮沸しながら塩化バ
リウム温溶液 (100 g/L) 10 mLをピペ
ットを用いて徐々に滴加し,更に数分
間煮沸を続ける。
三酸化硫黄の定量をする際に,6.4 c)
で保存した試料溶液 (F) を200 mL
分取してもよい手順を追加した。
13 酸化ナト
リウム及び酸
化カリウムの
定量方法
13.1として原子吸光法,13.2として炎光光度法を規定。 16. 酸化ナト
リウム及び酸
化カリウムの
定量方法
原子吸光法を規定。
国際規格に炎光光度法が規定されて
いるため,追加した。
13.1.2 b) 塩
化ナトリウム
塩化ナトリウム(99.5 %以上)を規定。
16.2 b) アル
カリ原液
塩化ナトリウム及び塩化カリウムの
純度について規定していない。
標準液の調製に用いる塩化ナトリウム
の純度を規定した。
13.1.2 c) 塩化
カリウム
塩化カリウム(99.5 %以上)を規定。
標準液の調製に用いる塩化カリウムの
純度を規定した。
13.1.2 d) ア
ルカリ原液
塩化ナトリウム及び塩化カリウム,又は国家計量標準
にトレーサブルな市販のナトリウム標準液及びカリ
ウム標準液を水で希釈し,Na2Oとして約1 mg/mL及
びK2Oとして約1 mg/mLとなるように調製する。
16.2 b) アル
カリ原液
塩化ナトリウム及び塩化カリウムを
用いて調製する。
国家計量標準にトレーサブルな標準
液が市販されており,これを使用で
きるようにした。
14 酸化チタ
ン (IV) の定
量方法
6.4 c)で保存した試料溶液 (F) の一部を分取し,定量
する。
17. 二酸化チ
タンの定量方
法
試料を塩酸で溶解し,ろ過した後,定
容する。
酸化チタン (IV) の定量に,6.4 c)で
保存した試料溶液 (F) の一部を使用
できるように規定した。
14.2 c) 酸化
チタン (IV)
標準原液
酸化チタン (IV) (99.0 %以上)又は市販のチタン標
準液を水で希釈し,TiO2として約0.2 mg/mLとなるよ
うに調製する。
17.2 c) 二酸
化チタン標準
原液
二酸化チタンを用いて調製する。
標準液の調製に用いる酸化チタン
(IV) の純度を規定した。また,標準
液が市販されており,これを使用で
きるようにした。
3
R
5
2
0
2
:
2
0
1
0
69
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格 (JIS R 5202:2010)
旧規格 (JIS R 5202:1999)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
15.2 f) 酸化
りん (V) 標
準原液
りん酸二水素カリウム(99.0 %以上)又は国家計量標
準にトレーサブルな市販のりん酸イオン標準液を水
で希釈し,P2O5として約0.1 mg/mLとなるように調製
する。
18.2 f) 五酸
化りん標準原
液
りん酸二水素カリウムを用いて調製
する。また,濃度を1.0 mgP2O5/mLと
規定。
標準液の調製に用いるりん酸二水素
カリウムの純度を規定した。また,
国家計量標準にトレーサブルな標準
液が市販されており,これを使用で
きるようにした。これに伴い,濃度
を変更した。
15.3 a) 操作
試料溶液 (A) 50 mLを,全量フラスコ100 mLに分取
する。
なお,測定した試料溶液の吸光度が検量線の範囲外
となった場合には,試料溶液 (A) の分取量を最小で5
mLまで少なくしてもよい。
18.3 a) 操作
試料溶液 (A) 50 mLを,全量フラスコ
100 mLに分取する。
検量線の範囲外となる試料に対応す
るため。
16 酸化マン
ガン (II) の
定量方法
16.1として原子吸光法を,16.2として吸光光度法を規
定。また,分析値は酸化マンガン (II) で表す。
19. 一酸化マ
ンガンの定量
方法
附属書12. 吸
光光度法によ
るマンガンの
定量方法
19.で原子吸光法を,附属書の12.で吸
光光度法を規定。
附属書の12.で規定していた国際規格
の方法を本体で規定した(構成の変
更を参照)。
16.1.2 a) 酸
化マンガン
(II) 標準原液
金属マンガン(99.0 %以上)又は国家計量標準にトレ
ーサブルな市販のマンガン標準液を水で希釈し,MnO
として約0.2 mg/mLとなるように調製する。
19.2 a) 一酸
化マンガン標
準原液
金属マンガン(99.9 %以上)を用いて
調製する。
標準液の調製に用いる金属マンガン
の純度を入手可能なものとした。ま
た,国家計量標準にトレーサブルな
標準液が市販されており,これを使
用できるようにした。
17 硫化物硫
黄の定量方
法
17.2としてよう素酸カリウム標準液を用いる直接滴
定法を,17.3としてチオ硫酸ナトリウム標準液を用い
る逆滴定法を規定。
20. 硫化物硫
黄の定量方法
附属書11.硫
化物硫黄の定
量方法
20.でよう素酸カリウム標準液を用い
る直接滴定法を,附属書の11.でチオ
硫酸ナトリウム溶液を用いる逆滴定
法を規定。
附属書の11.で規定していた国際規格
の方法を本体で規定した(構成の変
更の欄を参照)。
17.2.1 d)
17.3.1 d) よう
素酸カリウム
容量分析用標準物質の使用を規定。
20.2 d)
附属書4.48
よう素酸カリ
ウム
よう素酸カリウムの純度について規
定していない。
容量分析用標準物質の使用を規定し
た。
3
R
5
2
0
2
:
2
0
1
0
70
R 5202:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
現行規格 (JIS R 5202:2010)
旧規格 (JIS R 5202:1999)
改正理由
箇条番号
及び題名
内容
箇条番号
及び題名
内容
18 塩素の定
量方法
18.1として電位差滴定法を,18.2としてチオシアン酸
水銀 (II) による吸光光度法を,18.3としてチオシア
ン酸アンモニウム溶液による逆滴定法を規定。
21. 塩素の定
量方法
21.1でチオシアン酸水銀 (II) による
で吸光光度法を,21.2で電位差滴定法
を規定。
国際規格の改正によって塩素の定量
方法が追加されたため,追加した。
なお,電位差滴定法は日本から提案
し,国際規格で採用された方法であ
る。
18.1.2 d)
18.2.2 g) 塩
化ナトリウ
ム
塩化ナトリウム(容量分析用標準物質)を規定。
−
塩化ナトリウムの純度について規定
していない。
容量分析用標準物質の使用を規定し
た。
18.1.2 e) 塩
化物イオン
標準液
塩化ナトリウム又は国家計量標準にトレーサブルな
市販の塩化物イオン標準液を水で希釈し,Clとして約
0.005 mol/Lとなるように調製する[18.1.2 f)で国家計
量標準にトレーサブルな市販の塩化物イオン標準液
を用いた場合の硝酸銀標準液 (0.005 mol/L) のファク
ターの計算式を追加]。
21.2.2 d)
0.005 mol/l塩
化物イオン標
準液
塩化ナトリウムを用いて調製する。
国家計量標準にトレーサブルな標準
液が市販されており,これを使用で
きるようにした。
18.2.2 h) 塩
化物イオン
標準原液
塩化ナトリウム又は国家計量標準にトレーサブルな
市販の塩化物イオン標準液を水で希釈し,Clとして約
0.5 mg/mLとなるように調製する。
21.1.2 g) 塩化
物イオン標準
原液
塩化ナトリウムを用いて調製する。
国家計量標準にトレーサブルな標準
液が市販されており,これを使用で
きるようにした。
3
R
5
2
0
2
:
2
0
1
0