R 2013 : 1998
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が制定した日
本工業規格である。
JIS R 2013には,次に示す附属書がある。
附属書(参考) アルミナ−ジルコニア−シリカ質耐火物中の酸化ハフニウムの蛍光X線分析方法
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
R 2013 : 1998
アルミナ−ジルコニア−シリカ質
耐火物の化学分析方法
Methods for chemical analysis of
refractories containing alumina, zirconia and silica
序文 耐火物の化学分析では,耐火物の各々の材質に合わせた分析方法を用いないと,正しい定量値を得
ることができない。このため,日本工業規格では,各材質についての方法を制定・整備しているので,材
質に合わせて適用しなければならない。
この規格は,このような一連の耐火物の化学分析方法に関する規格整備の一環として,アルミナ−ジルコ
ニア−シリカ質耐火物を対象に制定されたもので,他の材質へは適用できない。
1. 適用範囲 この規格は,アルミナ−ジルコニア−シリカ質耐火物の化学分析方法について規定する。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS K 8001 試薬試験方法通則
JIS R 2001 耐火物用語
JIS R 2551 キャスタブル耐火物の試験試料採取方法
JIS Z 8401 数値の丸め方
JIS Z 8801 試験用ふるい
3. 一般事項 分析方法に共通な一般事項は,JIS K 0050,JIS K 0115,JIS K 0116及びJIS K 0121の規
定による。
4. 定義 この規格で用いる主な用語の定義は,JIS R 2001によるほか,次による。
a) アルミナ−ジルコニア−シリカ質耐火物 化学成分として酸化アルミニウム10〜80mass%,酸化ジル
コニウム(酸化ハフニウムを含む。)5〜50mass%及び酸化けい素(IV)0.1〜45mass%を含有する耐火物。
b) 乾状不定形耐火物 粒及び粉末で構成される耐火物。
c) 湿状不定形耐火物 粒及び粉末に液状物質を加えて構成される耐火物。
2
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5. 分析項目 この規格で規定する分析項目は,次による。
a) 強熱減表 (LOI)
b) 酸化けい素 (IV) (SiO2)
c) 酸化アルミニウム (Al2O3)
d) 酸化鉄 (III)(Fe2O3として全鉄を表す。)
e) 酸化チタン (IV) (TiO2)
f)
酸化カルシウム (CaO)
g) 酸化マグネシウム (MgO)
h) 酸化ナトリウム (Na2O)
i)
酸化カリウム (K2O)
j)
酸化クロム (III) (Cr2O3)
k) 酸化ジルコニウム(酸化ハフニウムを含む。)[ZrO2 (+HfO2)]
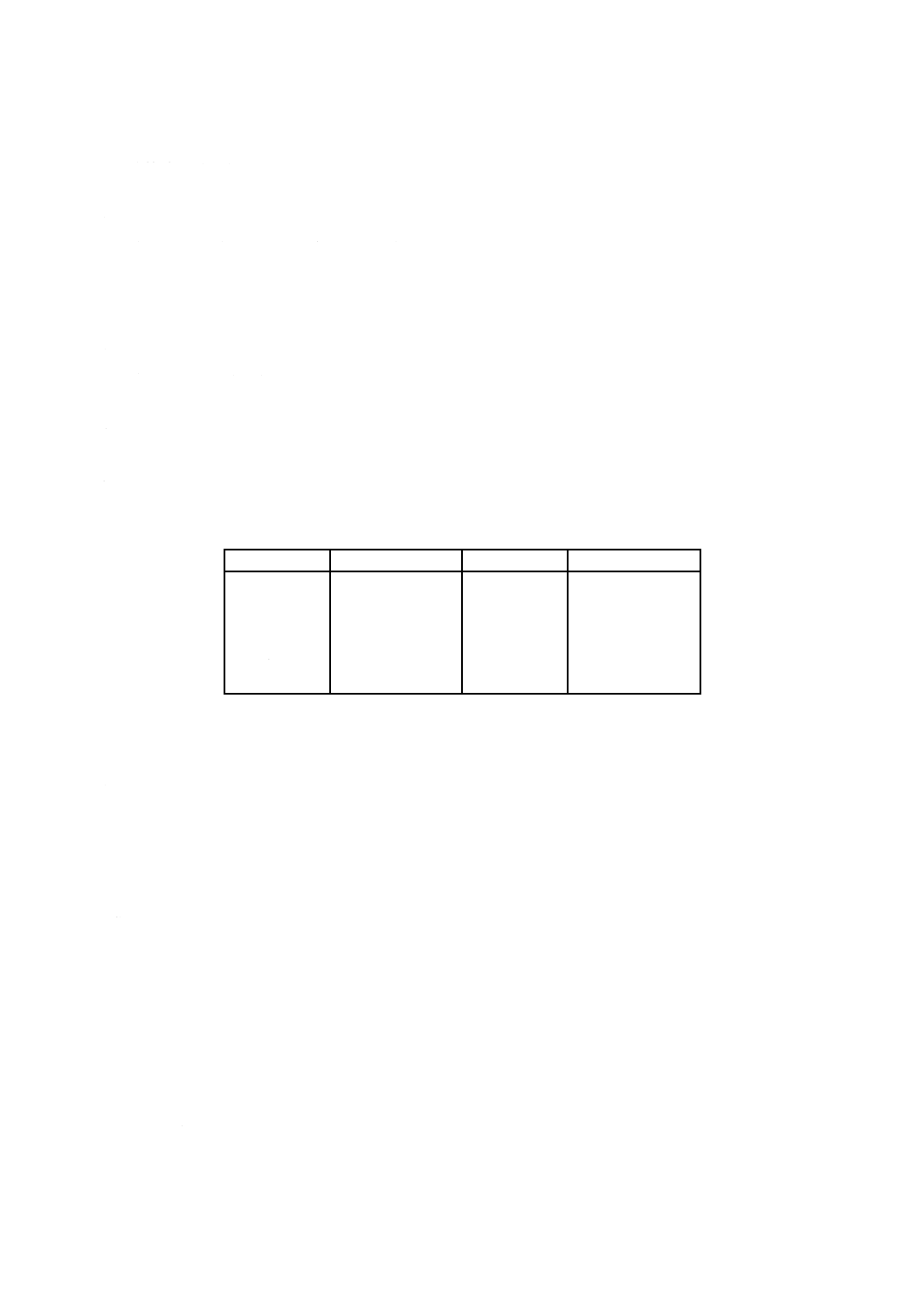
6. 定量範囲 この規格で規定する定量範囲は,表1による。
表1 定量範囲
単位mass%
成分
定量範囲
成分
定量範囲
LOI
−1 〜40
MgO
0.01〜 2
SiO2
0.1 〜45
Na2O
0.01〜 3
Al2O3
10 〜80
K2O
0.01〜 1
Fe2O3
0.01〜 2
Cr2O3
0.01〜 3
TiO2
0.01〜 5
ZrO2 (+HfO2)
5
〜50
CaO
0.01 〜2
7. 試料
7.1
試料の採取及び調製 試料の採取及び調製は,次による。
a) 耐火れんがは,ロットから受渡当事者間の協定に基づく数量の試料をランダムに採取する。採取した
試料は,全量を粉砕してJIS Z 8801に規定する網ふるい6.7mmを通過させ,二分器を用いるか,又は
四分法によって約100gになるまで縮分する。次に,この縮分した全量が網ふるい300μmを通過する
まで粉砕する。
b) 不定形耐火物は,その性状によって乾状と湿状に区分し,次によって試料約100gを調製する。
1) 乾状不定形耐火物は,ロットからランダムに1袋又は50kgを採取し,二分器を用いるか,又は四分
法によって約100gになるまで縮分する。次に,この縮分した全量が網ふるい300μmを通過するま
で粉砕する。
2) 湿状不定形耐火物は,JIS R 2551に規定された一定量を採取し(1),湿状耐火物と反応しない耐熱性
板(例えば,四ふっ化エチレン樹脂板)上に厚みが10mm以下の薄い円盤状になるように広げ,110
±5℃の空気浴中で2時間(2)加熱し,全量を粉砕して網ふるい6.7mmを通過させ,二分器を用いる
か,又は四分法によって約100gになるまで縮分する。次に,この縮分した全量が網ふるい300μm
を通過するまで粉砕する。
注(1) 湿状の耐火モルタルの場合は,1容器全量を採取し,その容器内又は不定形耐火物と反応し
ない清浄な容器に移し,清浄なかき混ぜ機などを用いて均一になるまで十分に混合し,こ
3
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
のうちの1kgを採取する。
(2) 湿状の耐火モルタルの場合は,10時間以上乾燥する。
c) a)又はb)によって得られた試験室試料を,四分法によって縮分して約10gとする。これを網ふるい
106μmを通過する程度まで微粉砕し,平形はかり瓶 (50×30mm) に薄く広げ,110±5℃の空気浴中で
2時間以上乾燥した後,デシケーター中で放冷し保存する。これを分析用試料とする。
7.2
試料の量り方 分析試料は,分析用試料から化学はかりを用いて規定された量を,0.1mgのけたまで
量り取る。
8. 分析値のまとめ方
8.1
分析回数 分析は,日を変えて2回繰り返す。
8.2
空試験 分析に当たっては空試験を行い,測定値を補正する。
8.3
分析値の表示 分析値は乾燥ベースの質量百分率で表し,JIS Z 8401によって次のように丸める。
a) 含有率の整数部が2けたの場合,小数点以下1けた。
b) 含有率の整数部が1けた以下の場合,小数点以下2けた。
8.4
分析値の検討・採択
8.4.1
2個の分析値の差が,表2の許容差を超えないときは,その平均を報告値とする。
8.4.2
2個の分析値の差が許容差を超えるときは,更に2回の分析を繰り返し,その差が許容差を超えな
いときは,その平均を報告値とする。これも許容差を超えるときは,4個の分析値のメジアンを報告値と
する。
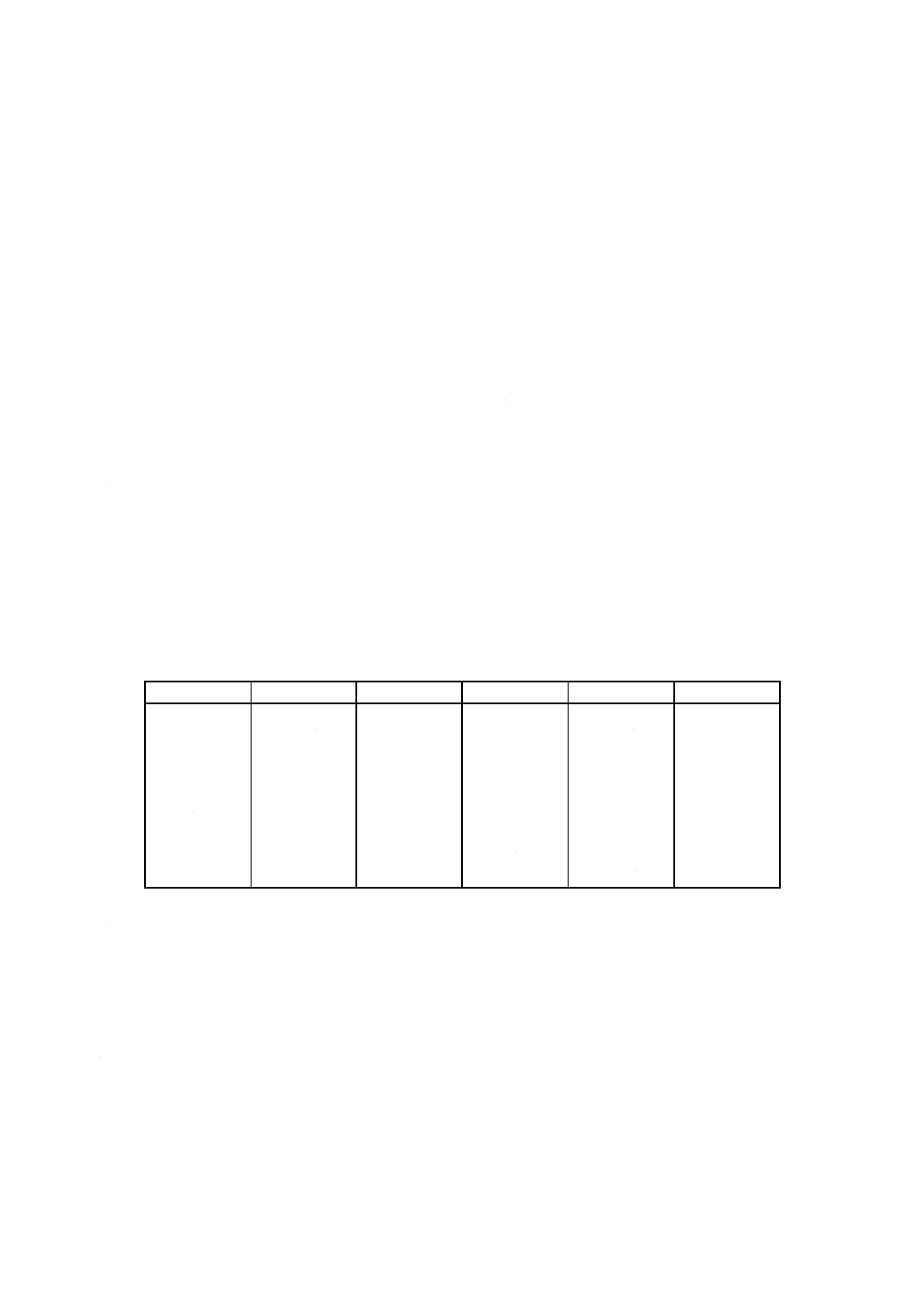
表2 分析値の許容差
単位mass%
成分
含有率
許容差
成分
含有率
許容差
LOI
5未満
0.10
TiO2
2未満
0.10
5以上
0.20
2以上
0.15
SiO2
2未満
0.10
CaO
0.08
2以上8未満
0.20
MgO
0.08
8以上
0.40
Na2O
0.08
Al2O3
40未満
0.30
K2O
0.08
40以上
0.50
Cr2O3
0.10
Fe2O3
0.05
ZrO2 (+HfO2)
20未満
0.20
20以上
0.40
8.5
分析報告 分析報告には,次の事項を記録する。
a) 分析所名
b) 分析年月日
c) 試料名及び試料に関する情報
d) 分析成分名及び分析値(報告値)
9. 強熱減量の定量方法
9.1
定量方法 強熱減量の定量方法は,重量法による。
9.2
重量法
9.2.1
要旨 試料を1 050±25℃で強熱し,質量の増減を測定する。
9.2.2
試料の量り取り量 試料の量り取り量は,1.0gとする。
4
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.3
操作 定量操作は,次の手順によって行う。
a) 白金るつぼ(例えば,20番)又は磁器るつぼ(例えば,B形15ml)を1 050±25℃で一定時間(3)強熱
し,デシケーター中で放冷した後,その質量を量る。
注(3) 白金るつぼの場合は,約15分間,磁器るつぼの場合は,約60分間強熱する。
b) 試料をるつぼの底に薄く広げるように移し入れ,その質量を量る。
c) るつぼにふたをしないで最初は低温で加熱し,次第に温度を上げ,最後は電気炉中で1 050±25℃で約
60分間強熱する。るつぼにふたをしてデシケーター中で放冷した後,ふたを取ってその質量を量る。
9.2.4
計算 試料中の強熱減量は,次の式によって算出する(4)。
100
0
1
2
1
×
−
−
=
m
m
m
m
LOI
ここに, LOI: 強熱減量 (mass%)
m0: 9.2.3 a)で得た質量 (g)
m1: 9.2.3 b)で得た質量 (g)
m2: 9.2.3 c)で得た質量 (g)
注(4) 質量が増加した場合は,“−”(負符号)を付けて表示する。
10. 酸化けい素 (IV) の定量方法
10.1 定量方法の区分 酸化けい素(IV)の定量方法は,次のいずれかによる。
a) 脱水重量吸光光度併用法 酸化けい素 (IV) の含有率4mass%以上の試料に適用する。
b) 凝集重量吸光光度併用法 酸化けい素 (IV) の含有率4mass%以上の試料に適用する。
c) モリブデン青吸光光度法 酸化けい素 (IV) の含有率8mass%未満の試料に適用する。
10.2 脱水重量吸光光度併用法
10.2.1 要旨 試料を炭酸ナトリウムとほう酸で融解後,硫酸に溶かし,蒸発乾固してけい酸を脱水した後,
温水を加えて可溶性塩類を溶かし,ろ過する。沈殿を強熱後,焼成物を炭酸ナトリウムで再融解し,以下,
硫酸溶解,加熱脱水,ろ過及び強熱を繰り返す。焼成物の質量を量り,ふっ化水素酸を加えて酸化けい素
(IV) を揮発させた後,再び強熱して質量を量り,その差から主酸化けい素 (IV) の量を求める。ろ液を分
取してモリブデン青吸光光度法によって溶存酸化けい素 (IV) の量を求める。両者の和から酸化けい素
(IV) の含有率を求める。
10.2.2 試薬 試薬は,次による。プラスチック瓶に保存する。
a) ふっ化水素酸
b) ふっ化水素酸 (1+9)
c) 硫酸 (1+1, 1+15, 1+150)
d) ほう酸
e) ほう酸溶液 (40g/l)
f)
炭酸ナトリウム(5)
注(5) 試薬によっては,カルシウムを微量含むものがある。ICP発光分光法で酸化カルシウムを定量
する場合には,できるだけ高純度の試薬を用いる。
g) 七モリブデン酸六アンモニウム溶液 七モリブデン酸六アンモニウム四水和物20gを水200mlに溶か
し,必要ならろ過する。保存中にモリブデン酸が析出したときは,新しく調製する。
5
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
h) 酒石酸溶液 (100g/l)
i)
L(+)−アスコルビン酸溶液 (100g/λ) 冷暗所に保存する。調製後2週間以上経過したものは,使
用しない方がよい。
j)
酸化けい素 (IV) 標準液 (0.05mgSiO2/ml) 二酸化けい素(99.9mass%以上)を強熱し,デシケーター
中で放冷後,その0.050 0gを白金るつぼに量り取り,炭酸ナトリウム1gと混合した後,加熱融解する。
放冷後,白金るつぼごと水100mlの入ったプラスチックビーカー (200ml) に移し,加熱することなく
融成物を溶解して1 000mlの全量フラスコに移し入れ,水を標線まで加える。使用の都度調製する。
10.2.3 試料の量り取り量 試料の量り取り量は,0.50gとする。
10.2.4 操作 定量操作は,次の手順によって行う。
a) 試料を白金皿(例えば,75番)に量り取り,炭酸ナトリウム3.0g及びほう酸2.0gと混合した後,初
めは低温で加熱し(6),次第に温度を上げ最後は電気炉中で1 100±25℃に加熱し,未分解物が認められ
なくなるまで強熱して融解する(7)。時計皿でふたをして放冷後,ガラス棒でときどきかき混ぜながら,
硫酸 (1+15) 55mlを加え,沸騰水浴上で加熱して溶かす。
注(6) 急激に加熱すると,ほう酸の脱水のために試料が飛散するおそれがある。
(7) 融解時間が長すぎると融成物が硫酸に溶けにくくなる。
b) 時計皿を水洗して除き,引き続き蒸発乾固する。この間,ときどき先端を平らにしたガラス棒で析出
した塩類を細かく押しつぶし,粉末にする。放冷後,熱水30mlを加えて水浴上で約5分間加熱して
可溶性塩類を溶かし,ビーカー (300ml) を受け,ろ紙(5種B)を用いてろ過する。熱硫酸 (1+150)
で数回洗浄し,更に熱水で十分に洗浄する。ろ液及び洗液の入ったビーカーは,時計皿で覆い保存す
る。この溶液を保存溶液 (a) とする。
c) 沈殿をろ紙と共に白金皿(例えば,75番)に入れ,硫酸 (1+1) 1滴を加え,ふたをわずかにずらして
覆い,初めは低温で加熱してろ紙を灰化し,ふたを取って1 100±25℃で約10分間強熱する。放冷後,
炭酸ナトリウム3.0gを焼成物を覆うように加え,初めは低温で加熱し,次第に温度を上げ,融解が始
まったらときどき振り混ぜを繰り返して,最後は電気炉中1 100±25℃で5分間加熱して融解する。時
計皿でふたをして放冷後,硫酸 (1+15) 55mlを加え,発泡が穏やかになったら沸騰水浴上に移し,加
熱して溶かす。以下,b)の操作を行い,ここで得た溶液を保存溶液 (b) とする。
d) 沈殿をろ紙と共に白金るつぼ(例えば,30番)に入れ,硫酸 (1+1) 1滴を加えて初めは低温で加熱し
てろ紙を灰化し,1 100±25℃で約60分間強熱する。デシケーター中で放冷した後,その質量を量る。
次いで,るつぼ中の内容物を少量の水で潤し,硫酸 (1+1) 3滴及びふっ化水素酸約10mlを加え,砂
浴上で加熱して蒸発乾固する。1 100±25℃で約5分間加熱し,デシケーター中で放冷した後,その質
量を量り,先の質量との差を求める。るつぼ中の残さは,炭酸ナトリウム1.0g及びほう酸0.3gを加え
て融解し,放冷後,c)の保存溶液 (b) 中に白金るつぼごと入れて加熱して溶かし,必要なら濃縮した
後,b)の保存溶液 (a) と共に500mlの全量フラスコに移し入れ,水を標線まで加える。この溶液を試
料溶液 (A) とし,溶存酸化けい素 (IV),酸化アルミニウム,酸化鉄 (III),酸化チタン (IV),酸化カ
ルシウム,酸化マグネシウム,酸化クロム (III) 及び酸化ジルコニウム(酸化ハフニウムを含む。)の
定量に用いる。
e) 試料溶液 (A) から10mlを正しくプラスチックビーカー (100ml) に分取し,ふっ化水素酸 (1+9) 2ml
を加え,プラスチック棒でかき混ぜて約10分間放置した後,ほう酸溶液50mlを加え,液温を25℃付
近にする。七モリブデン酸六アンモニウム溶液2mlを加えてかき混ぜ,10分間放置する。酒石酸溶液
10mlを加えてかき混ぜ,1分間後にL(+)−アスコルビン酸溶液2mlを加え,100mlの全量フラス
6
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
コに移し入れ,水を標線まで加えて60分間放置する。この溶液の一部を吸収セル (10mm) にとり,
波長650nm付近で水を対照液として吸光度を測定する。
10.2.5 空試験 試料を用いないで10.2.4の操作を行う。ただし,融解操作は省略する。ここで得た試料溶
液 (A) に対応する溶液を空試験液 (A) とする。
10.2.6 検量線の作成 酸化けい素 (IV) 標準液0〜10.0ml[酸化けい素 (IV) として0〜0.5mg]を数個の
プラスチックビーカー (100ml) に段階的にとり,それぞれに10.2.5で得た空試験液 (A) 10mlを加え,10.2.4
e)のふっ化水素酸 (1+9) を添加する以降の操作を行い,吸光度と酸化けい素 (IV) の量との関係線を作成
し,原点を通るように平行移動して検量線とする。
10.2.7 計算 試料中の酸化けい素 (IV) の含有率は,10.2.4 d)で得た主酸化けい素 (IV) の量と,10.2.4 e)
及び10.2.5で得た吸光度と10.2.6で作成した検量線とから求めた溶存酸化けい素 (IV) の量から,次の式
によって算出する。
100
10
500
)
(
)
(
2
1
2
1
2
×
×
−
−
−
=
m
A
A
m
m
SiO
ここに, SiO2: 酸化けい素 (IV) の含有率 (mass%)
m1: 10.2.4 d)で得た質量差 (g)
m2: 10.2.5で得た質量差 (g)
A1: 分取した試料溶液 (A) 中の溶存酸化けい素 (IV) の量 (g)
A2: 分取した空試験液 (A) 中の溶存酸化けい素 (IV) の量 (g)
m: 試料の量り取り量 (g)
10.3 凝集重量吸光光度併用法
10.3.1 要旨 試料を炭酸ナトリウムとほう酸で融解後,硫酸に溶解し,ポリエチレンオキシドを加えてけ
い酸を凝集させた後,ろ過する。沈殿を強熱後,焼成物を炭酸ナトリウムと少量のほう酸で再融解し,以
下,硫酸溶解,凝集,ろ過及び強熱を繰り返す。焼成物の質量を量り,ふっ化水素酸を加えて酸化けい素
(IV) を揮発させた後,再び強熱してその質量を量り,その差から主酸化けい素 (IV) の量を求める。ろ液
を分取してモリブデン青吸光光度法によって溶存酸化けい素 (IV) の量を求める。両者の和から酸化けい
素 (IV) の含有率を求める。
10.3.2 試薬 試薬は,10.2.2 a)〜j)と同じもののほか,次のものを用いる。
a) ポリエチレンオキシド溶液 水200ml中にかき混ぜながら,ポリエチレンオキシド0.1gを少量ずつ加
えて溶かす。調製後2週間を経過したものは使用しない。
10.3.3 試料の量り取り量 試料の量り取り量は,0.50gとする。
10.3.4 操作 定量操作は,次の手順によって行う。
a) 試料を白金皿(例えば,75番)に量り取り,炭酸ナトリウム3.0g及びほう酸2.0gを加えて混合した
後,初めは低温で加熱し(6),次第に温度を上げ,最後は電気炉中で1 100±25℃に加熱して,未分解物
が認められなくなるまで融解する(7)。時計皿でふたをして放冷後,硫酸 (1+15) 55mlを加え,沸騰水
浴上で加熱して溶かす。
b) 時計皿を水洗して除き,引き続き沸騰水浴上で加熱する。液の表面に塩類の被膜が生成したら,ガラ
ス棒でかき混ぜ,シロップ状になるまで蒸発させた後,水10mlを加え,沸騰水浴上で加熱し,ガラ
ス棒でよくかき混ぜて塩類を溶かす。これに適当量(例えば,0.05g)の粉末ろ紙を加えてかき混ぜた
後,ポリエチレンオキシド溶液約10mlを加えてよくかき混ぜ,約5分間放置する。ビーカー (300ml)
を受け,ろ紙(5種B)を用いてろ過し,熱硫酸 (1+150) で数回洗浄し,更に熱水で十分に洗浄する。
7
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ろ液及び洗液の入ったビーカーは,時計皿で覆い保存する。この溶液を保存溶液 (a') とする。
c) 沈殿をろ紙と共に白金皿(例えば,75番)に入れ,硫酸 (1+1) 1滴を加え,ふたをわずかにずらして
覆い,初めは低温で加熱してろ紙を灰化し,ふたを取って1 100±25℃で約10分間強熱する。放冷後,
炭酸ナトリウム3.0gとほう酸0.1gを焼成物を覆うように加え,初めは低温で加熱し,次第に温度を上
げ,融解が始まったらときどき振り混ぜを繰り返して,最後は電気炉中1 100±25℃で5分間加熱して
融解する。時計皿でふたをして放冷後,硫酸 (1+15) 55mlを加え,水浴上で加熱して溶かす。以下,
b)の操作を行い,ここで得た溶液を保存溶液 (b') とする。
d) 沈殿をろ紙と共に白金るつぼ(例えば,30番)に入れ,硫酸 (1+1) 1滴を加え,初めは低温で加熱し
てろ紙を灰化し,1 100±25℃で約60分間強熱する。デシケーター中で放冷した後,その質量を量る。
次いで,るつぼ中の内容物を少量の水で潤し,硫酸 (1+1) 3滴及びふっ化水素酸約10mlを加え,砂
浴上で加熱して蒸発乾固する。1 100±25℃で約5分間強熱し,デシケーター中で放冷した後,その質
量を量り,先の質量との差を求める。るつぼ中の残さは,炭酸ナトリウム1.0g及びほう酸0.3gを加え
て融解し冷却後,c)の保存溶液(b')中にるつぼごと入れて加熱して溶かし,必要なら濃縮した後,b)の
保存溶液 (a') と共に500mlの全量フラスコに移し入れ,水を標線まで加える。この溶液を試料溶液
(A') とし,溶存酸化けい素 (IV),酸化アルミニウム,酸化鉄 (III),酸化チタン (IV),酸化カルシウ
ム,酸化マグネシウム,酸化クロム (III),及び酸化ジルコニウム(酸化ハフニウムを含む。)の定量
に用いる。
e) 以下,10.2.4 e) の手順によって操作する。
10.3.5 空試験 試料を用いないで10.3.4の操作を行う。ただし,融解操作は,省略する。ここで得た試験
溶液 (A') に対応する溶液を空試験液 (A') とする。
10.3.6 検量線の作成 酸化けい素 (IV) 標準液0〜10.0ml[酸化けい素 (IV) として0〜0.5mg]を数個の
プラスチックビーカー (100ml) に段階的に取り,それぞれに10.3.5で得た空試験液 (A') 10mlを加え,10.2.4
e)のふっ化水素酸 (1+9) を添加する以降の操作を行い,吸光度と酸化けい素 (IV) の量との関係線を作成
し,原点を通るように平行移動して検量線とする。
10.3.7 計算 試料中の酸化けい素 (IV) の含有率は,10.3.4 d)で得た主酸化けい素 (IV) の量と,10.3.4 e)
及び10.3.5で得た吸光度と10.3.6で作成した検量線とから求めた溶存酸化けい素 (IV) の量から,次の式
によって算出する。
100
10
500
)
(
)
(
2
1
2
1
2
×
×
−
−
−
=
m
A
A
m
m
SiO
ここに, SiO2: 酸化けい素 (IV) の含有率 (mass%)
m1: 10.3.4 d)で得た質量差 (g)
m2: 10.3.5で得た質量差 (g)
A1: 分取した試料溶液 (A') 中の溶存酸化けい素 (IV) の量 (g)
A2: 分取した空試験液 (A') 中の溶存酸化けい素 (IV) の量 (g)
m: 試料の量り取り量 (g)
10.4 モリブデン青吸光光度法
10.4.1 要旨 試料を炭酸ナトリウムとほう酸で融解し,硫酸で溶解して酸濃度を調節し,七モリブデン酸
六アンモニウムを加え,酒石酸及びL(+)−アスコルビン酸を加えてモリブデン青を呈色させ,吸光度
を測定する。
10.4.2 試薬 試薬は,10.2.2 a),c)及びd)〜j)と同じもののほか,次のものを用いる。

8
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 硫酸 (1+9)
10.4.3 試料の量り取り量 試料の量り取り量は,0.50gとする。
10.4.4 操作 定量操作は,次の手順によって行う。
a) 試料を白金皿(例えば,75番)に量り取り,炭酸ナトリウム3.0g及びほう酸2.0gを加えて初めは低
温で加熱し(6),次第に温度を上げ,最後は電気炉中で1 100±25℃で加熱し,未分解物が認められなく
なるまで強熱して融解する(7)。時計皿でふたをして放冷後,硫酸 (1+9) 55mlを加え,ときどきかき
混ぜながら水浴上で加熱して溶かす。放冷後,少量の水で時計皿を水洗して取り除き,得られた溶液
を500mlの全量フラスコに移し入れ,水を標線まで加える。この溶液を試料溶液 (A") とし,酸化け
い素 (IV),酸化アルミニウム,酸化鉄 (III),酸化チタン (IV),酸化カルシウム,酸化マグネシウム,
酸化クロム (III) 及び酸化ジルコニウム(酸化ハフニウムを含む。)の定量に用いる。
b) この試料溶液 (A") から一定量(8)を正しく2個のプラスチックビーカー (100ml) に分取し,10.4.5の
空試験液 (A") の一定量(8)を加える。ふっ化水素酸 (1+9) 2mlを加え,プラスチック棒でかき混ぜて
約10分間放置した後,ほう酸溶液50mlを加え,水で約80mlに薄めて液温を約25℃にする。七モリ
ブデン酸六アンモニウム溶液5mlを加えてかき混ぜ,10分間放置する。酒石酸溶液20mlを加えかき
混ぜ,1分間後にL(+)−アスコルビン酸溶液10mlを加え,200mlの全量フラスコに移し入れ,水
を標線まで加えて約60分間放置する。この溶液の一部を吸収セル (10mm) にとり,波長650nm付近
で水を対照液として吸光度を測定し,2個の測定値(9)を平均する。
注(8) 試料溶液 (A") の分取量及び空試験液 (A") の添加量は,試料中の酸化けい素 (IV) の含有率に
応じて表3による。
表3 試料溶液 (A″) の分取量及び空試験液 (A″) の添加量
酸化けい素 (IV) の含有率
mass%
試料溶液 (A") の分取量
ml
空試験液 (A") の添加量
ml
4未満
20
0
4以上 8未満
10
10
(9) 吸光度の差が,0.005を超えるときは,10.4.4 b) 以降の操作を再び行う。
分光光度計は,吸光度0.5以上の溶液を繰り返し測定したとき,吸光度の差が0.002以内であ
ることが望ましい。
10.4.5 空試験 試料を用いないで10.4.4の操作を行う。ただし,融解操作は,省略する。ここで得た試料
溶液 (A") に対応する溶液を空試験液 (A") とする。
10.4.6 検量線の作成 酸化けい素 (IV) 標準液0〜20.0ml[酸化けい素 (IV) として0〜1mg]を数個のプ
ラスチックビーカー (100ml) に段階的にとり,それぞれに10.4.5の空試験液 (A") 20mlを加え,10.4.4 b)
のふっ化水素酸 (1+9) を添加する以降の操作を行い,吸光度と酸化けい素 (IV) の量との関係線を作成し,
原点を通るように平行移動して検量線とする。
10.4.7 計算 試料中の酸化けい素 (IV) の含有率は,10.4.4 b)及び10.4.5で得た吸光度と10.4.6で作成し
た検量線とから酸化けい素 (IV) の量を求め,次の式によって算出する。
100
500
2
1
2
×
×
−
=
V
m
A
A
SiO
9
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに, SiO2: 酸化けい素 (IV) の含有率 (mass%)
A1: 分取した試料溶液 (A") 中の酸化けい素 (IV) の量 (g)
A2: 分取した空試験液 (A") 中の酸化けい素 (IV) の量 (g)
V: 試料溶液 (A") の分取量 (ml)
m: 試料の量り取り量 (g)
11. 酸化アルミニウムの定量方法
11.1 定量方法の区分 酸化アルミニウムの定量方法は,陽イオン交換カラム分離−CyDTA(シクロヘキ
サンジアミン四酢酸)−亜鉛逆滴定法による。
11.2 陽イオン交換カラム分離−CyDTA−亜鉛逆滴定法
11.2.1 要旨 10.2.4,10.3.4又は10.4.4の試料溶液(A,A'又はA")を分取し,陽イオン交換樹脂カラム
に流し,イオン交換樹脂にアルミニウムなどの陽イオンを吸着させる。カラムに0.8mol/lふっ化水素酸−
0.65mol/lほう酸溶液を流してジルコニウムとチタンを流出させ,次に,ふっ化水素酸 (1+200) を流して
アルミニウムを溶離する。溶離液に硫酸を加えて蒸発し,ほう酸及びふっ化水素酸を除去した後,塩酸に
溶かし,過剰のCyDTAを加え,アンモニア水でpHを調節してアルミニウム−CyDTAキレートを生成さ
せ,ヘキサメチレンテトラミンを加えてpHを再調節した後,キシレノールオレンジを指示薬として過剰
のCyDTAを亜鉛溶液で逆滴定する。
11.2.2 試薬 試薬は,次による。
a) 塩酸 (1+1, 1+2) (5)
b) 硫酸 (1+1)
c) 過塩素酸
d) アンモニア水 (1+1, 1+9)
e) ヘキサメチレンテトラミン(ヘキサミン)
f)
塩化ヒドロキシルアンモニウム溶液 (100g/l)
g) 0.02mol/lCyDTA溶液 シクロヘキサンジアミン四酢酸−水和物7.30gに水酸化ナトリウム溶液
(100g/l) 16ml及び水約150mlを加え,加熱して溶解する。冷却後,水で1 000mlに薄める。
h) 0.02mol/l亜鉛溶液 調製方法及びファクターの計算方法は,JIS K 8001の4.5(滴定用溶液)(1.3) に準
じる。ただし,亜鉛0.66g及び硝酸 (1+1) 10mlを用い,ファクターの計算の分母は,0.653 9とする。
i)
キシレノールオレンジ溶液調製方法及び保存方法は,JIS K 8001の4.4(表8)による。
j)
溶離液A(0.8molふっ化水素酸/l,0.65molほう酸/l) 水960ml中にほう酸40gを加えて溶かした後,
プラスチック容器 (1l) に移し,ふっ化水素酸 (46mass%) 35mlを加え,よくふり混ぜる。
k) 溶離液B プラスチック容器 (1l) に水1 000ml及びふっ化水素酸 (46mass%) 5mlを加え,よくふり混
ぜる。
11.2.3 器具 器具は,次による。
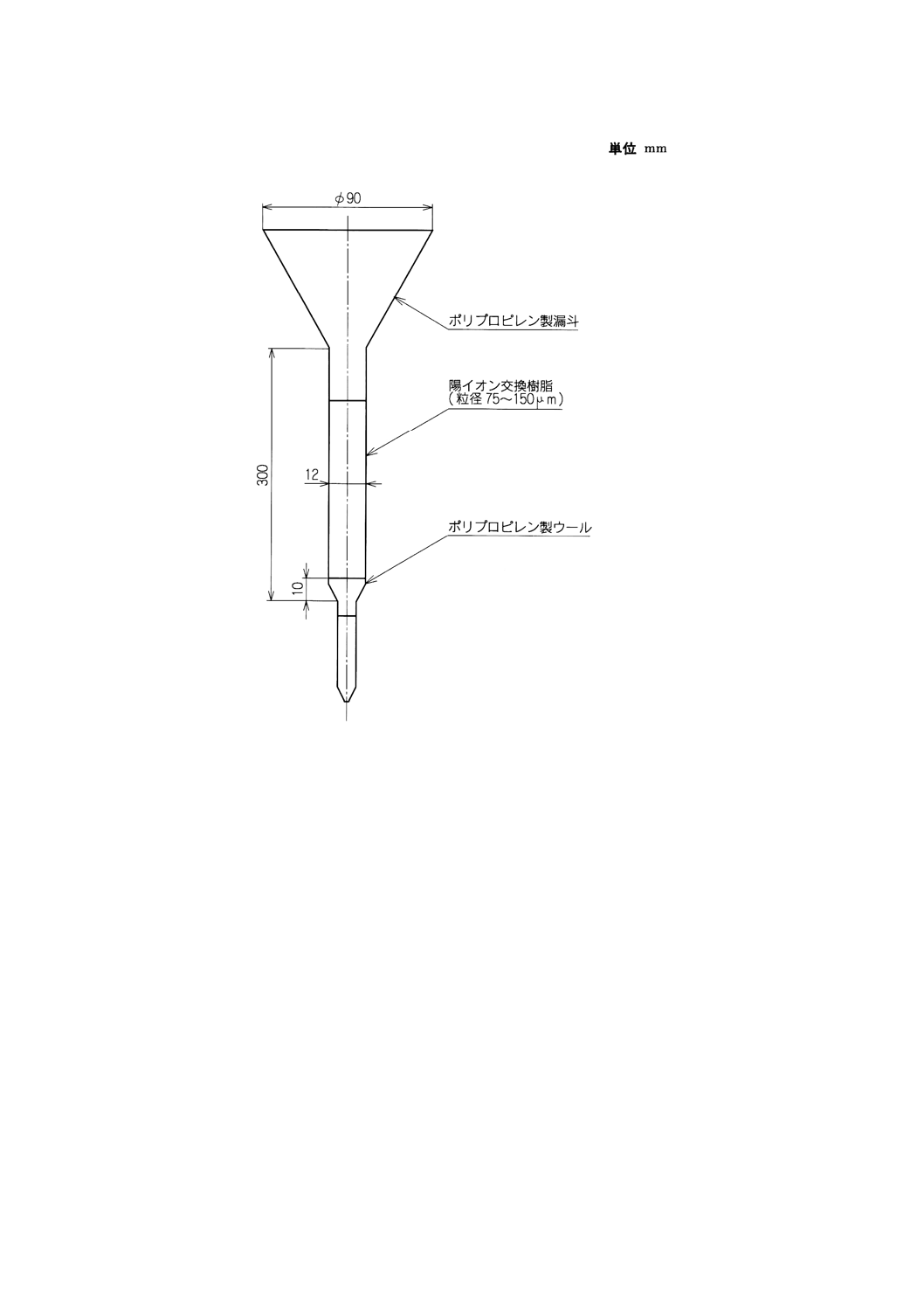
a) 陽イオン交換樹脂カラム 付図1に一例を示す。
上端にプラスチック漏斗を取り付け,下端を細く引き伸ばしたプラスチック管(長さ200mm,内径
12mm)に水でほぐしたプラスチックウールを高さ約10mmになるように詰め,水で湿潤させた強酸
性陽イオン交換樹脂 (DVB8%, 75〜150μm) 約18mlをスラリー状にして流し入れ,沈降させる。ウー
ルの詰め方を加減するなどして流量を毎分1.0〜1.5mlになるように調節した後,塩酸 (1+2) 120ml,
水70mlを順次流しておく。
b) 四ふっ化エチレン樹脂製ビーカー あらかじめ硝酸中で2時間以上加熱した後,水で洗浄して用いる。

10
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.2.4 操作 操作は,次の手順によって行う。
a) 10.2.4 d),10.3.4 d),又は10.4.4 a)で得た試料溶液(A,A'又はA")から50mlを正確に分取し,陽イ
オン交換樹脂カラムに流す(10)。溶離液A10mlずつを2回漏斗の内壁を洗浄しながらカラムに流し,
さらに,60mlを流す。ここまでの流出液は,不要である。
注(10) 前の溶液が,カラムの先端から滴下しなくなってから,次の溶液を流す。以下同じ。
b) 四ふっ化エチレン樹脂製ビーカーを受け,溶離液B10mlずつを2回漏斗の内壁を洗浄しながらカラム
に流す。さらに,溶離液B80mlを流し,アルミニウムを溶出させる。
備考 カラムは,次の手順によって処理して再生する。
(1) プラスチックビーカー (100ml) を受け,塩酸 (1+2) 10mlずつを2回漏斗の内壁を洗浄しな
がらカラムに流し,さらに,塩酸 (1+2) 100mlを流して,鉄,カルシウム,マグネシウムな
どを溶離する(11)。
注(11) この溶液は,白金皿(例えば,150番)に移し,砂浴上で蒸発乾固した後,塩酸 (1+1) 5ml
及び水30mlを加えて沸騰水浴上で溶かし,冷却後,100mlの全量フラスコに移し入れ,水
を標線まで加え,14.及び15.での酸化カルシウム及び酸化マグネシウムの定量に用いること
ができる。
なお,原子吸光法による場合には,100mlに薄める前にランタン溶液10mlを加える。
(2) 水70mlを流して,カラムを再生する。
c) b)の溶出液に硫酸 (1+1) 5ml及び過塩素酸1mlを加え,熱板上で加熱蒸発し,硫酸白煙を発生させる。
放冷後,ビーカーの内壁を少量の水で洗浄し,再び加熱して白煙を発生させる。液量が約2mlになっ
たら放冷し,塩酸 (1+1) 10mlを加えて加熱して溶かし,ビーカー (300ml) に移し入れる。
d) 放冷後,塩化ヒドロキシルアンモニウム溶液2mlを加えてかき混ぜた後,0.02mol/lCyDTA溶液の一定
量(12)を加え,pH計を用いてpHが2.9〜3.1になるまで,アンモニア水 (1+1),次いでアンモニア水 (1
+9) を加える。もし,アンモニア水を加え過ぎたときは,塩酸 (1+1) を加えてpH 3以下に戻してか
ら同様の調節を行う。ヘキサメチレンテトラミンをpH計を用いてpHが5.5〜5.8になるまで加え,指
示薬としてキシレノールオレンジ溶液4,5滴を加えて0.02mol/l亜鉛溶液で滴定する。終点付近にな
ったら,よくかき混ぜながらゆっくり滴定し,黄色がわずかに赤味を帯びた点を終点とする。
注(12) 0.02mol/lCyDTA溶液の添加量は,試料中の酸化アルミニウムの含有率に応じて表4による。
表4 0.02mol/l CyDTA溶液の添加量
酸化アルミニウムの含有率
mass%
添加量
ml
15未満
10
15以上
35未満
20
35以上
55未満
30
55以上
75未満
40
75以上
50
11.2.5 空試験 10.2.5,10.3.5又は10.4.5で得た空試験液(A,A'又はA")を用いて11.2.4の操作を行う。
11.2.6 計算 試料中の酸化アルミニウムの含有率は,次の式によって算出する。
100
50
500
6
019
001
.0
)
(
1
2
3
2
×
×
×
×
−
=
m
F
V
V
O
Al
ここに, Al2O3: 酸化アルミニウムの含有率 (mass%)
V1: 11.2.4 d)の0.02mol/l亜鉛溶液使用量 (ml)
V2: 11.2.5の0.02mol/l亜鉛溶液使用量 (ml)

11
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
F: 0.02mol/l亜鉛溶液のファクター
m: 10.2.4 a),10.3.4 a)又は10.4.4 a)の試料の量り取り量 (g)
備考 陽イオン交換樹脂カラムが用意できない場合には,次のクペロン分離−CyDTA−亜鉛逆滴定法
を用いてもよい。
なお,本法は沈殿の生成からろ過及び洗浄を通じて低温を維持する必要があるので,特に夏
季には温度調節された室内で操作しなければならない。
a) 操作 定量操作は,次による。
1) 10.2.4 d),10.3.4 d) 又は10.4.4 a) の試料溶液(A,A'又はA")から200mlをビーカー (500ml)
に正しく分取し,塩酸20mlを加えて5℃以下(13)に冷却する。適当量(例えば,0.05g)の
粉末ろ紙を加え,かき混ぜながら5℃以下に冷却したクペロン溶液25mlを少しずつ加え,
十分かき混ぜた後,5分間放置する。ビーカー (500ml) を受け,ろ紙(5種B)を用いて
ろ過し,5℃以下に冷却した塩酸 (1+9) で10回洗浄する。以上の操作を通じて,試料溶
液の入ったビーカーは,氷水中に浸すなどして5℃以下に保たなければならない。ろ紙及
び沈殿は,酸化ジルコニウム(酸化ハフニウムを含む。)の定量に用いる。
注(13) できるだけ低温にすることが望ましい。
2) ろ液及び洗液を沸騰水浴上で約20mlになるまで濃縮した後,時計皿で覆い硝酸10ml及び
過塩素酸5mlを加え,砂浴上で約10分間穏やかに煮沸する。時計皿を水洗して除き,引き
続き注意して蒸発して,過塩素酸の白煙が明らかに発生し始めるまで濃縮する。
3) 放冷後,塩酸 (1+1) 10ml及び水約100mlを加え,加熱して塩類を溶かし,冷却後,250ml
の全量フラスコに移し入れ,水を標線まで加える。この溶液100mlを正しく分取し,塩化
ヒドロキシルアンモニウム溶液2mlを加えてかき混ぜた後,0.02mol/lCyDTA溶液の一定量
(14)を加え,pH計を用いてpHが2.9〜3.1になるまで,アンモニア水 (1+1),次いで,ア
ンモニア水 (1+9) を加える。もし,アンモニア水を加え過ぎたときは,塩酸 (1+1) を加
えてpH3以下に戻してから同様の調節を行う。ヘキサメチレンテトラミンを,pH計を用
いてpHが5.5〜5.8になるまで加え,指示薬としてキシレノールオレンジ溶液4,5滴を加
えて0.02mol/l亜鉛溶液で滴定する。終点付近になったらよくかき混ぜながらゆっくり滴定
し,黄色がわずかに赤味を帯びた点を終点とする。
注(14) 0.02mol/lCyDTA溶液の添加量は,試料中の酸化アルミニウムの含有率に応じて表5に
よる。
表5 0.02mol/l CyDTA溶液の添加量
酸化アルミニウムの含有率
mass%
添加量
ml
20未満
20
20以上
35未満
30
35以上
60未満
50
60以上
70
b) 空試験 10.2.5,10.3.5又は10.4.5で得た空試験液(A,A'又はA")を用いてa) の操作を行
う。
c) 計算 試料中の酸化アルミニウムの含有率は,次の式によって算出する。
100
100
250
200
500
6
019
001
.0
)
(
1
2
3
2
×
×
×
×
×
−
=
m
F
V
V
O
Al

12
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに, Al2O3: 酸化アルミニウムの含有率 (mass%)
V1: a) 3)の0.02mol/l亜鉛溶液使用量 (ml)l
V2: b)の0.02mol/l亜鉛溶液使用量 (ml)
F: 0.02mol/l亜鉛溶液のファクター
m: 10.2.4 a),10.3.4 a)又は10.4.4 a)の試料の量り取り量 (g)
12. 酸化鉄 (III) の定量方法
12.1 定量方法の区分 酸化鉄 (III) の定量方法は,次のいずれかによる。
a) 1, 10−フェナントロリン吸光光度法
b) ICP発光分光分析法
12.2 1, 10−フェナントロリン吸光光度法
12.2.1 要旨 10.2.4,10.3.4又は10.4.4の試料溶液(A,A'又はA")を分取し,L(+)−アスコルビン酸
で鉄を還元し,塩化1, 10−フェナントロリニウムを加え,酢酸アンモニウムでpHを調節して鉄を発色さ
せ,吸光度を測定する。
12.2.2 試薬 試薬は,次による。
a) 酒石酸溶液 (100g/l) 10.2.2 h) による。
b) 酢酸アンモニウム溶液 (200g/l)
c) L(+)−アスコルビン酸溶液 (100g/l) 10.2.2 i) による。
d) 塩化1, 10−フェナントロリニウム溶液 塩化1, 10−フェナントロリニウム−水和物1gを水に溶かし
て1 000mlに薄め,冷暗所に保存する。ただし,保存中に着色したときは,新しく調製する。
e) 酸化鉄 (III) 標準液 (1.0mgFe2O3/ml) 鉄(99.9mass%以上)(15)0.6994gを量り取り,ビーカー (200ml) に
移し,ビーカーを時計皿で覆い,塩酸 (1+1) 30mlを加えて沸騰水浴上で加熱して溶かし,冷却後,1
000mlの全量フラスコに移し入れ,水を標線まで加える。
注(15) 表面が酸化している場合には,表面酸化層を塩酸(1+3)で溶解し,水,エタノール (99.5),ジエ
チルエーテルで順次洗浄した後,デシケーターで乾燥させて用いる。
f)
希釈酸化鉄 (III) 標準液 (0.05mgFe2O3/ml) e) の酸化鉄 (III) 標準液を水で正しく20倍に薄める。使用
の都度調製する。
12.2.3 操作 定量操作は,次の手順によって行う。
a) 10.2.4 d),10.3.4 d),又は10.4.4 a)で得た試料溶液(A,A'又はA")から一定量(16)を100mlの全量フラ
スコに分取する。
注(16) 試料溶液(A,A'又はA")の分取量は,試料中の酸化鉄 (III) の含有率に応じて表6による。
表6 試料溶液(A,A'又はA")の分取量
酸化鉄(III)の含有率
mass%
分取量
ml
1未満
50
1以上
25
b) 水で約60mlに薄め,酒石酸溶液5ml及びL(+)−アスコルビン酸溶液2mlを加えて振り混ぜ,塩
化1, 10−フェナントロリニウム溶液10ml及び酢酸アンモニウム溶液10mlを加え,水を標線まで加え,
約30分間放置する。この溶液の一部を吸収セル (10mm) にとり,波長510nm付近で水を対照液にし
て吸光度を測定する。
13
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.2.4 空試験 10.2.5,10.3.5又は10.4.5で得た空試験液(A,A'又はA")を用いて12.2.3の操作を行う。
ただし,空試験液の分取量は,試料溶液の場合と同量とする。
12.2.5 検量線の作成 希釈酸化鉄 (III) 標準液0〜15.0ml[酸化鉄 (III) として0〜0.75mg]を数個の100ml
の全量フラスコに段階的に取り,12.2.3 b)の操作を行い,吸光度と酸化鉄 (III) の量との関係線を作成し,
原点を通るように平行移動して検量線とする。
12.2.6 計算 試料中の酸化鉄 (III) の含有率は,12.2.3 b)及び12.2.4で得た吸光度と12.2.5で作成した検
量線とから酸化鉄 (III) の量を求め,次の式によって算出する。
100
500
2
1
3
2
×
×
−
=
V
m
A
A
O
Fe
ここに, Fe2O3: 酸化鉄 (III) の含有率 (mass%)
A1: 分取した試料溶液(A,A'又はA")中の酸化鉄(III)の量 (g)
A2: 分取した空試験液(A,A'又はA")中の酸化鉄(III)の量 (g)
m: 10.2.4 a),10.3.4 a),又は10.4.4 a)の試料の量り取り量 (g)
V: 12.2.3 a) の試料溶液(A,A'又はA")の分取量 (ml)
12.3 ICP発光分光分析法
12.3.1 要旨 10.2.4,10.3.4又は10.4.4の試料溶液(A,A'又はA")を分取し,ICP発光分光分析装置を用
いて鉄の発光強度を測定する。
12.3.2 試薬 試薬は,次による。
a) 酸化鉄 (III) 標準液 (1.0mgFe2O3/ml) 12.2.2 e)による。
b) 酸化チタン (IV) 標準液 (1.0mgTiO2/ml) チタン(99.9mass%以上)0.599 4gを白金皿(例えば,100
番)にとり,白金皿を四ふっ化エチレン樹脂製の時計皿で覆い,ふっ化水素酸20ml,硫酸 (1+1) 15ml,
及び硝酸0.5mlを加え,沸騰水浴上で加熱して溶かす。時計皿を水で洗って取り除き,砂浴上で硫酸
の濃い白煙が出るまで加熱する。冷却後,白金皿の内壁を少量の水で洗い,再び加熱して白煙を発生
させる。冷却後,水を加え,1 000mlの全量フラスコに移し入れ,水を標線まで加える。
c) 酸化カルシウム標準液 (1.0mgCaO/ml) 炭酸カルシウム(99.9mass%以上)2〜2.5gを白金るつぼ(例
えば,20番)又は磁器るつぼ(例えば,B形15ml)にとり,600±25℃で約60分間加熱した後,デシ
ケーターに入れ放冷する。その1.784 8gを量り取り,ビーカー (200ml) に移し入れ(17),ビーカーを
時計皿で覆い,塩酸 (1+1) 10mlを徐々に加えて溶かし,冷却後,1 000mlの全量フラスコに移し入れ,
水を標線まで加える。
注(17) 例えば,金属製(例えば,白金製)量り取り皿に正しく量り取り,飛散しないように注意して
ビーカーに移し,少量の水で金属製量り取り皿の付着残留物を洗い移す。
d) 酸化マグネシウム標準液 (1.0mgMgO/ml) マグネシウム(99.9mass%以上)(15)0.603 0gを量り取り,
ビーカー (200ml) に移し入れ,ビーカーを時計皿で覆い,塩酸 (1+1) 10mlを徐々に加えて溶解し,
冷却後,1 000mlの全量フラスコに移し入れ,水を標線まで加える。
e) 酸化クロム(III)標準液 (1.0mgCr2O3/ml) 二クロム酸カリウム1.935 6gを量り取り,ビーカー (200ml)
に移し入れ(17),水を加えて液量を50mlとした後,硫酸 (1+1) 5ml及びエタノール (99.5) 5mlを加え
て,溶液の色が緑になるまで水浴上で加熱する。冷却後,1 000mlの全量フラスコに移し入れ,水を
標線まで加える。
f)
酸化ハフニウム標準液 (1.0mgHfO2/ml) (18) 酸化ハフニウム(99.9mass%以上)約1.5gを白金るつぼ
(例えば,20番)にとり,1 050±25℃で1時間強熱した後,デシケーターに入れ,放冷する。その

14
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
0.500 0gを白金皿(例えば,75番)に量り取り,炭酸ナトリウム3.0g及びほう酸2.0gを加えて混合し
た後,電気炉中で1 050±25℃で強熱して融解する。放冷後,時計皿でふたをして硫酸 (1+9) 55mlを
加え,沸騰水浴上で加熱して溶かし,冷却後,500mlの全量フラスコに移し入れ,水を標線まで加え
る。
注(18) 19.の備考によって,酸化ハフニウムを定量する場合だけに用いる。酸化ハフニウムを定量しな
い場合には,不要である。
g) 混合標準液 (0.02mgFe2O3/ml, 0.05mgTiO2/ml, 0.02mgCaO/ml, 0.02mgMgO/ml, 0.03mgCr2O3/ml,
0.03mgHfO2/ml) (19) 1 000mlの全量フラスコに酸化鉄 (III) 標準液,酸化チタン (IV) 標準液,酸化カ
ルシウム標準液,酸化マグネシウム標準液,酸化クロム (III) 標準液及び酸化ハフニウム標準液のそ
れぞれ20ml,50ml,20ml,20ml,30ml及び30ml(19)を正しくとり,水を標線まで加える。この溶液は,
使用の都度調製する。
注(19) 各成分の濃度及び分取量は,試料中の分析成分の含有率によって変えてよい。
h) 添加液 白金皿(例えば,75番)に酸化アルミニウム及び酸化ジルコニウムの一定量(20)(21)を量り取
り,以下,10.2.4,10.3.4又は10.4.4に準じて試料溶液(A,A'又はA")に相当する溶液を調製する(22)。
注(20) 19.の備考によって酸化ハフニウムを定量する場合には,酸化ハフニウムが0.01mass%以下の酸
化ジルコニウムを用いる。
(21) 試料中の酸化アルミニウム及び酸化ジルコニウムの含有率に相当する量(例えば,酸化アルミ
ニウム含有率が50mass%及び酸化ジルコニウムの含有率が30mass%の場合,酸化アルミニウム
を0.25g及び酸化ジルコニウムを0.15g)を量り取る。
(22) 試料と同じ操作法を行って調製する。
i)
検量線用溶液系列I(23) 混合標準液を段階的に数個の100mlの全量フラスコに正しくとり,それぞれ
に添加液20mlを加え,水を標線まで加える。
注(23) 表7に調製例を示す。分析試料の組成及び使用する分析装置の種類・性能に応じて最適な検量線
用溶液系列を調製する。
表7 検量線用溶液系列Iの調製例
検量線用
溶液系列I
添加液
ml
混合標準液
ml
溶液の濃度 (mg/100ml)
Fe2O3
TiO2
CaO
MgO
Cr2O3
HfO2(18)
No.1
20
0
0
0
0
0
0
0
No.2
20
5
0.10
0.25
0.10
0.10
0.15
0.15
No.3
20
10
0.20
0.50
0.20
0.20
0.30
0.30
No.4
20
15
0.30
0.75
0.30
0.30
0.45
0.45
No.5
20
20
0.40
1.00
0.40
0.40
0.60
0.60
12.3.3 操作 定量操作は,次の手順によって行う。
a) 10.2.4 d),10.3.4 d),又は10.4.4 a) で得た試料溶液(A,A'又はA")の20mlを100mlの全量フラスコ
に分取し,水を標線まで加える。この溶液を試料溶液(B)とする。
b) 試料溶液 (B) の一部をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,例えば,波長259.94nm
における発光強度を測定する。
12.3.4 空試験 10.2.5,10.3.5又は10.4.5で得た空試験液(A,A'又はA”)を用いて12.3.3の操作を行う。
試料溶液 (B) に対応する溶液を,空試験液 (B) とする。
12.3.5 検量線の作成 検量線用溶液系列Iを用いて12.3.3 b) の操作を行い(24),発光強度と酸化アルミニ
ウムの量との関係線を作成し,原点を通るように平行移動して検量線とする。
15
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(24) 検量線用溶液系列の測定は,試料溶液及び空試験液の測定との一連の操作として行い,検量線
は,測定ごとに新しいものを作成する。
12.3.6 計算 試料中の酸化鉄 (III) の含有率は,12.3.3及び12.3.4で得た発光強度と12.3.5で作成した検
量線とから酸化鉄 (III) の量を求め,次の式によって算出する。
100
20
500
2
1
3
2
×
×
−
=
m
A
A
O
Fe
ここに, Fe2O3: 酸化鉄 (III) の含有率 (mass%)
A1: 12.3.2の試料溶液 (B) 中の酸化鉄 (III) の量 (g)
A2: 12.3.3の空試験液 (B) 中の酸化鉄 (III) の量 (g)
m: 10.2.4 a),10.3.4 a) 又は10.4.4 a) の試料の量り取り量 (g)
13. 酸化チタン (IV) の定量方法
13.1 定量方法の区分 酸化チタン (IV) の定量方法は,次のいずれかによる。
a) ジアンチピリルメタン吸光光度法
b) ICP発光分光分析法
13.2 ジアンチピリルメタン吸光光度法
13.2.1 要旨 10.2.4,10.3.4又は10.4.4の試料溶液(A,A'又はA")を分取し,酸の濃度を調節した後,
CyDTAを加えて加熱し,ジルコニウムをマスキングする。L(+)−アスコルビン酸を加えて鉄を還元し,
ジアンチピリルメタンで発色させ,吸光度を測定する。
13.2.2 試薬 試薬は,次による。
a) 塩酸 (1+1)
b) L(+)−アスコルビン酸溶液 (100g/l) 10.2.2 i)による。
c) CyDTA溶液 シクロヘキサンジアミン四酢酸一水和物2gに水酸化ナトリウム溶液 (100g/l) 4.6mlを加
えて加熱溶解し,冷却後,水で250mlに薄める。
d) ジアンチピリルメタン溶液 (15g/l) ジアンチピリルメタン一水和物1.5gを塩酸 (1+5) 45mlに溶か
し,水で100mlに薄める。
e) 希釈酸化チタン (IV) 標準液 (0.01mgTiO2/ml) 12.3.2 b)の酸化チタン (IV) 標準液を水で正しく100
倍に薄める。使用の都度調製する。
13.2.3 操作 定量操作は,次の手順によって行う。
a) 10.2.4 d),10.3.4 d) 又は10.4.4 a) で得た試料溶液(A,A'又はA")の正しく一定量(25)を50mlの全量
フラスコに分取する。塩酸 (1+1) 5ml及びCyDTA溶液1mlを加えて振り混ぜた後,沸騰水浴中に浸
し,5分間加熱して流水中で常温まで冷却する。
注(25) 試料溶液(A,A'又はA”)の分取量は,試料中の酸化チタン (IV) の含有率に応じて表8による。

16
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表8 試料溶液(A,A'又はA")の分取量
酸化チタン(IV)の含有率
mass%
分取量
ml
1未満
25
1以上
2.5未満
10
2.5以上
5
b) L(+)−アスコルビン酸溶液2mlを加え,1分間放置した後,ジアンチピリルメタン溶液10mlを加
え,水を標線まで加え,約90分間放置する。この溶液の一部を吸収セル (10mm) に取り,波長390nm
付近で水を対照液として吸光度を測定する。
13.2.4 空試験 10.2.5,10.3.5又は10.4.5で得た空試験液(A,A'又はA")を用いて13.2.3の操作を行う。
ただし,空試験液の分取量は,試料溶液の場合と同量とする。
13.2.5 検量線の作成 13.2.2e)の希釈酸化チタン (IV) 標準液0〜25.0ml[酸化チタン (IV) として0〜
0.25mg]を数個の50mlの全量フラスコに段階的に取り,13.2.3 a) の塩酸 (1+1) 5mlを添加する以降の操
作を行い,吸光度と酸化チタン (IV) の量との関係線を作成し,原点を通るように平行移動して検量線と
する。
13.2.6 計算 試料中の酸化チタン (IV) の含有率は,13.2.3 b)及び13.2.4で得た吸光度と13.2.5で作成し
た検量線とから酸化チタン (IV) の量を求め,次の式によって算出する。
100
500
2
1
2
×
×
−
=
V
m
A
A
TiO
ここに, TiO2: 酸化チタン (IV) の含有率 (mass%)
A1: 分取した試料溶液(A,A'又はA")中の酸化チタン(IV)の量 (g)
A2: 分取した空試験液(A,A'又はA")中の酸化チタン(IV)の量 (g)
m: 10.2.4 a),10.3.4 a),又は10.4.4 a) の試料の量り取り量 (g)
V: 13.2.3 a)の試料溶液(A,A'又はA")の分取量 (ml)
13.3 ICP発光分光分析法
13.3.1 要旨 12.3.3の試料溶液 (B) を取り,ICP発光分光分析装置を用いてチタンの発光強度を測定する。
13.3.2 操作 12.3.3 a) で得た試料溶液 (B) の一部をICP発光分光分析装置のアルゴンプラズマ中に噴霧
し,例えば,波長334.94nmにおける発光強度を測定する。
13.3.3 空試験 12.3.4で得た空試験液 (B) を用いて,13.3.2の操作を行う。
13.3.4 検量線の作成 12.3.2 i) の検量線用溶液系列Iを用いて,13.3.2の操作を行い(24),得た発光強度と
酸化チタン (IV) の量との関係線を作成し,原点を通るように平行移動して検量線とする。
13.3.5 計算 試料中の酸化チタン(IV)の含有率は,13.3.2及び13.3.3で得た発光強度と13.3.4で作成した
検量線とから酸化チタン (IV) の量を求め,次の式によって算出する。
100
20
500
2
1
2
×
×
−
=
m
A
A
TiO
ここに, TiO2: 酸化チタン (IV) の含有率 (mass%)
A1: 13.3.2の試料溶液 (B) 中の酸化チタン (IV) の量 (g)
A2: 13.3.3の空試験液 (B) 中の酸化チタン (IV) の量 (g)
m: 10.2.4 a),10.3.4 a)又は10.4.4 a)の試料の量り取り量 (g)
17
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14. 酸化カルシウム定量方法
14.1 定量方法の区分 酸化カルシウムの定量方法は,次のいずれかによる。
a) 原子吸光法
b) ICP発光分光分析法
14.2 原子吸光法
14.2.1 要旨 試料をふっ化水素酸,過塩素酸及び硝酸を用いて,加熱分解する。蒸発乾固した後,塩酸に
溶かして一定液量とする。この溶液の一部を取り,原子吸光分析装置を用いてカルシウムの原子吸光を吸
光度で測定する。
14.2.2 試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸
c) 過塩素酸
d) ふっ化水素酸
e) ランタン溶液 酸化ランタン (III) 50gをビーカー (1 000ml) に量り取り,塩酸 (1+1) 200mlを加えて
加熱分解し,水で1 000mlに薄める。
f)
酸化ナトリウム標準液 (0.5mgNa2O/ml) 塩化ナトリウム1〜1.5gを白金るつぼ(例えば,30番)に
とり,600±25℃で約60分間加熱した後,デシケーターに入れ放冷する。その0.942 9gを量り取り,
ビーカー (200ml) に移し入れ(17),水約100mlを加えて溶かし,1 000mlの全量フラスコに移し入れ,
水を標線まで加える。
g) 酸化カリウム標準液 (0.5mgK2O/ml) 塩化カリウム1〜1.5gを白金るつぼ(例えば,30番)にとり,
600±25℃で約60分間加熱した後,デシケーターに入れ放冷する。この中から0.791 4gを量り取り,
ビーカー (200ml) に移し入れ(17),水約100mlを加えて溶解し,1 000mlの全量フラスコに移し入れ,
水で標線まで薄める。
h) マトリックス溶液 数個の白金皿(例えば,150番)のそれぞれに,試料0.20g中の酸化ジルコニウム
量 (g) 及び酸化アルミニウム量 (g) に相当する酸化ジルコニウム(99.9mass%以上),及び酸化アルミ
ニウム(99.99mass%以上)を量り取り,以下14.2.4 a)の操作を行う。
i)
混合標準液II (0.1mgCaO/ml, 0.1mgMgO/ml, 0.02mgNa2O/ml, 0.02mgK2O/ml) 1 000mlの全量フラスコ
に12.3.2 c)の酸化カルシウム標準液,12.3.2 d)の酸化マグネシウム標準液,酸化ナトリウム標準液及び
酸化カリウム標準液をそれぞれ100ml,100ml,40ml及び40ml取り,水を標線まで加える。
j)
検量線用溶液系列II(26) h)の数個のマトリックス溶液をそれぞれ100mlの全量フラスコに移し入れ,
混合標準液IIの一定量を段階的に加え,ランタン溶液10mlを加えた後,水を標線まで加える。直ち
にプラスチック瓶に移し入れる。
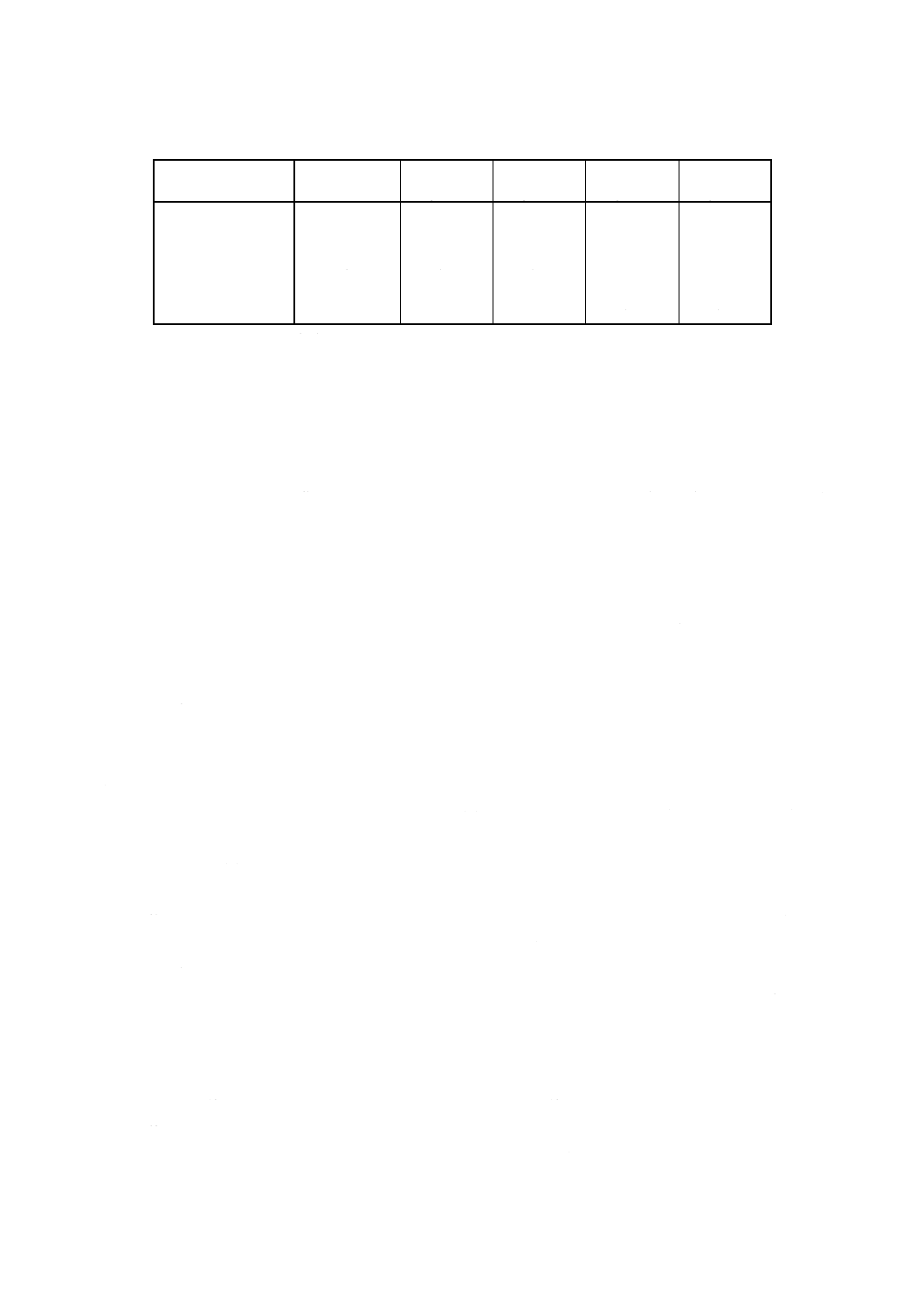
注(26) 表9に調製例を示す。分析試料の組成及び使用する分析装置の種類・性能に応じて最適な検量線
用溶液系列を調製する。
18
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表9 検量線用溶液系列IIの調製例
検量線用溶液系列II
混合標準液II
ml
CaO
mg/100ml
MgO
mg/100ml
Na2O
mg/100ml
K2O
mg/100ml
No.1
0
0
0
0
0
No.2
5
0.5
0.5
0.1
0.1
No.3
10
1.0
1.0
0.2
0.2
No.4
15
1.5
1.5
0.3
0.3
No.5
20
2.0
2.0
0.4
0.4
No.6
25
2.5
2.5
0.5
0.5
14.2.3 試料の量り取り量 試料の量り取り量は,0.20gとする。
14.2.4 操作 定量操作は,次の手順によって行う。
a) 試料を白金皿(例えば,150番)に量り取り,水で潤し,過塩素酸5ml,硝酸2ml,及びふっ化水素酸
10mlを加え,よくかき混ぜ,砂浴上で注意して加熱分解し(27),過塩素酸の白煙を激しく発生させて
蒸発乾固する。放冷後,白金皿の内壁を少量の水で洗い,再び過塩素酸3ml,硝酸2ml及びふっ化水
素酸5mlを加え,砂浴上で蒸発乾固する。放冷後,白金皿の内壁を少量の水で洗い,過塩素酸3mlを
加え,砂浴上で加熱し,蒸発乾固して残留するふっ化物を揮散させる。放冷後,塩酸 (1+1) 5.0ml及
び水約20mlを加え,時計皿で覆い,沸騰水浴上で加熱して溶かし(28),プラスチックビーカー (200ml)
を受け,プラスチック漏斗及びろ紙(5種B)を用いてろ過し,熱水で十分に洗浄する(29)。
注(27) 白金皿内容物のかき混ぜには,太目の白金合金(例えば,白金−ロジウム)線の先端を折り曲
げたもの,白金製さじ,四ふっ化エチレン樹脂製棒又はさじなどが利用できる。加熱していく
と試料が白金皿の底に固化して試薬と反応しにくくなるので,砂浴から降ろし放冷後,固化物
を白金皿の底からはがし,よくつぶすとよい。加熱分解を続け,液量が少なくなり,過塩素酸
の白煙が発生する直前になると試料によっては激しく反応し,飛散することがあるので注意す
る。
(28) 塩酸が揮発するので,できるだけ短時間で溶解する。
(29) 溶液中に微粒子が漏れることがあるが,測定上差し支えない。
b) 放冷後,ランタン溶液10mlを加え,100mlの全量フラスコに移し入れ,水を標線まで加えた後,直ち
にプラスチック瓶に移す。この溶液を試料溶液 (C) とし,原子吸光法による酸化カルシウム,酸化マ
グネシウム,酸化ナトリウム,及び酸化カリウムの定量に用いる。
c) この試料溶液 (C) (30)の一部を取り,原子吸光分析装置のアセチレン−酸化二窒素フレーム中に噴霧し,
波長422.7nmにおける原子吸光を吸光度で測定する。
注(30) 試料溶液 (C) 中の濃度が原子吸光分析装置の定量上限を超えるときは,試料溶液 (C) から正し
く一定量 (xml) を100mlの全量フラスコにとり,塩酸 (1+1) (5.0−5.0×x/100) ml及びランタン
溶液 (10−10×x/100) mlを加え,水を標線まで加え,この溶液について測定する。
14.2.5 空試験 試料を用いないで14.2.4の操作を行う(31)。ここで得た試料溶液 (C) に対応する溶液を空
試験液 (C) とする。
注(31) 注(30)によるときは,空試験液 (C) も試料溶液と同様に調製する。
14.2.6 検量線の作成 検量線用溶液系列II(32)を用いて14.2.4 c)の操作を行い(24),吸光度と酸化カルシウ
ムの量との関係線を作成し,原点を通るように平行移動して検量線とする。
注(32) 注(30)によるときは,検量線用溶液系列IIについても酸化ジルコニウム及び酸化アルミニウムの
量を希釈後の試料溶液中の濃度と同じようになるよう調節する。
19
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.2.7 計算 試料中の酸化カルシウムの含有率は,14.2.4 c) 及び14.2.5で得た吸光度と14.2.6で作成した
検量線とから酸化カルシウムの量を求め,次の式によって算出する。
100
100
2
1
×
×
−
=
V
m
A
A
CaO
ここに, CaO: 酸化カルシウムの含有率 (mass%)
A1: 試料溶液 (C) 又は希釈試料溶液中の酸化カルシウムの量 (g)
A2: 空試験液 (C) 又は希釈試料溶液中の酸化カルシウムの量 (g)
V: 試料溶液 (C) の分取量 (ml)(分取しない場合は100)
m: 試料の量り取り量 (g)
14.3 ICP発光分光分析法
14.3.1 要旨 12.3.3の試料溶液 (B) を取り,ICP発光分光分析装置を用いてカルシウムの発光強度を測定
する。
14.3.2 操作 12.3.3 a)で得た試料溶液 ( B)の一部をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,
例えば,波長393.37nmにおける発光強度を測定する。
14.3.3 空試験 12.3.4で得た空試験液 (B) を用いて,14.3.2の操作を行う。
14.3.4 検量線の作成 12.3.2 i)の検量線用溶液系列Iを用いて14.3.2の操作を行い(24),得た発光強度と酸
化カルシウムの量との関係線を作成し,原点を通るように平行移動して検量線とする。
14.3.5 計算 試料中の酸化カルシウムの含有率は,14.3.2及び14.3.3で得た発光強度と14.3.4で作成した
検量線とから酸化カルシウムの量を求め,次の式によって算出する。
100
20
500
2
1
×
×
−
=
m
A
A
CaO
ここに, CaO: 酸化カルシウムの含有率 (mass%)
A1: 14.3.2の試料溶液 (B) 中の酸化カルシウムの量 (g)
A2: 14.3.3の空試験液 (B) 中の酸化カルシウムの量 (g)
m: 10.2.4 a),10.3.4 a),又は10.4.4 a)の試料の量り取り量 (g)
15. 酸化マグネシウムの定量方法
15.1 定量方法の区分 酸化マグネシウムの定量方法は,次のいずれかによる。
a) 原子吸光法
b) ICP発光分光分析法
15.2 原子吸光法
15.2.1 要旨 14.2.4の試料溶液 (C) を取り,原子吸光分析装置を用いてマグネシウムの原子吸光を吸光度
で測定する。
15.2.2 操作 14.2.4 b)で得た試料溶液 (C) (30)の一部を原子吸光分析装置のアセチレン−酸化二窒素フレ
ーム又はアセチレン−空気フレーム中に噴霧し,波長285.2nmにおける原子吸光を吸光度で測定する。
15.2.3 空試験 14.2.5で得た空試験液(31)を用いて,15.2.2の操作を行う。
15.2.4 検量線の作成 14.2.2 j)の検量線用溶液系列II(32)を用いて15.2.2の操作を行い(24),吸光度(原子吸
光)と酸化マグネシウムの量との関係線を作成し,原点を通るように平行移動して検量線とする。
15.2.5 計算 試料中の酸化マグネシウムの含有率は,15.2.2及び15.2.3で得た吸光度と15.2.4で作成した
検量線とから酸化マグネシウムの量を求め,次の式によって算出する。
20
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
100
2
1
×
×
−
=
V
m
A
A
MgO
ここに, MgO: 酸化マグネシウムの含有率 (mass%)
A1: 試料溶液 (C) 又は希釈試料溶液中の酸化マグネシウムの量
(g)
A2: 空試験液 (C) 又は希釈試料溶液中の酸化マグネシウムの量
(g)
V: 試料溶液(C)の分取量 (ml) (分取しない場合は100)
m: 14.2.4 a)の試料の量り取り量 (g)
15.3 ICP発光分光分析法
15.3.1 要旨 12.3.3の試料溶液 (B) を取り,ICP発光分光分析装置を用いてマグネシウムの発光強度を測
定する。
15.3.2 操作 12.3.3 a) で得た試料溶液 (B) の一部をICP発光分光分析装置のアルゴンプラズマ中に噴霧
し,例えば,波長279.55nmにおける発光強度を測定する。
15.3.3 空試験 12.3.4で得た空試験液 (B) を用いて,15.3.2の操作を行う。
15.3.4 検量線の作成 12.3.2 i)の検量線用溶液系列Iを用いて15.3.2の操作を行い(24),得た発光強度と酸
化マグネシウムの量との関係線を作成し,原点を通るように平行移動して検量線とする。
15.3.5 計算 試料中の酸化マグネシウムの含有率は,15.3.2及び15.3.3で得た発光強度と15.3.4で作成し
た検量線とから酸化マグネシウムの量を求め,次の式によって算出する。
100
20
100
2
1
×
×
−
=
m
A
A
MgO
ここに, MgO: 酸化マグネシウムの含有率 (mass%)
A1: 15.3.2の試料溶液 (B) 中の酸化マグネシウムの量 (g)
A2: 15.3.3の空試験液 (B) 中の酸化マグネシウムの量 (g)
m: 10.2.4 a),10.3.4 a)又は10.4.4 a)の試料の量り取り量 (g)
16. 酸化ナトリウム定量方法
16.1 定量方法の区分 酸化ナトリウムの定量方法は,次のいずれかによる。
a) 炎光光度法
b) 原子吸光法
16.2 炎光光度法
16.2.1 要旨 14.2.4の試料溶液 (C) をとり,炎光光度計のフレーム中に噴霧し,ナトリウムの発光強度を
測定する。
16.2.2 操作 14.2.4 b)で得た試料溶液 (C) (30)の一部を炎光光度計のフレーム中に噴霧し,波長589.0nm(33)
における発光強度を測定する。
注(33) ナトリウム用フィルターを使用してもよい。
16.2.3 空試験 14.2.5で得た空試験液 (C) (31)を用いて,16.2.2の操作を行う。
16.2.4 検量線の作成 14.2.2 j)の検量線用溶液系列II(32)を用いて16.2.2の操作を行い(24),発光強度と酸化
ナトリウムの量との関係線を作成する。
21
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
16.2.5 計算 試料中の酸化ナトリウムの含有率は,16.2.2及び16.2.3で得た発光強度と16.2.4で作成した
検量線とから酸化ナトリウムの量を求め,次の式によって算出する。
100
2
1
2
×
−
=
m
A
A
O
Na
ここに, Na2O: 酸化ナトリウムの含有率 (mass%)
A1: 試料溶液 (C) 中の酸化ナトリウムの量 (g)
A2: 空試験液 (C) 中の酸化ナトリウムの量 (g)
m: 14.2.4 a)の試料の量り取り量 (g)
16.3 原子吸光法
16.3.1 要旨 14.2.4の試料溶液 (C) を取り,原子吸光分析装置を用いてナトリウムの原子吸光を吸光度で
測定する。
16.3.2 操作 14.2.4 b)で得た試料溶液 (C) (30)の一部を原子吸光分析装置のアセチレン−空気フレーム中
に噴霧し,波長589.0nmにおける原子吸光を吸光度で測定する。
16.3.3 空試験 14.2.5で得た空試験液(30)を用いて,16.3.2の操作を行う。
16.3.4 検量線の作成 14.2.2 j)の検量線用溶液系列II(32)を用いて16.3.2の操作を行い(24),吸光度と酸化ナ
トリウムの量との関係線を作成し,原点を通るように平行移動して検量線とする。
16.3.5 計算 試料中の酸化ナトリウムの含有率は,16.3.2及び16.3.3で得た吸光度と16.3.4で作成した検
量線とから酸化ナトリウムの量を求め,次の式によって算出する。
100
100
2
1
2
×
×
−
=
V
m
A
A
O
Na
ここに, Na2O: 酸化ナトリウムの含有率 (mass%)
A1: 試料溶液 (C) 又は希釈試料溶液中の酸化ナトリウムの量 (g)
A2: 空試験液 (C) 又は希釈試料溶液中の酸化ナトリウムの量 (g)
V: 試料溶液 (C) の分取量 (ml)(分取しない場合は100)
m: 14.2.4 a)の試料の量り取り量 (g)
17. 酸化カリウム定量方法
17.1 定量方法の区分 酸化カリウムの定量方法は,次のいずれかによる。
a) 炎光光度法
b) 原子吸光法
17.2 炎光光度法
17.2.1 要旨 14.2.4の試料溶液 (C) をとり,炎光光度計のフレーム中に噴霧し,カリウムの発光強度を測
定する。
17.2.2 操作 14.2.4 b)で得た試料溶液 (C) (30)の一部を炎光光度計のフレーム中に噴霧し,波長766.5nm(34)
における発光強度を測定する。
注(34) カリウム用フィルターを使用してもよい。
17.2.3 空試験 14.2.5で得た空試験液 (C) (30)を用いて,17.2.2の操作を行う。
17.2.4 検量線の作成 14.2.2 j)の検量線用溶液系列II(32)を用いて17.2.2の操作を行い(24),発光強度と酸化
カリウムの量との関係線を作成して検量線とする。
17.2.5 計算 試料中の酸化カリウムの含有率は,17.2.2及び17.2.3で得た発光強度と17.2.4で作成した検
量線とから酸化カリウムの量を求め,次の式によって算出する。
22
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
2
1
2
×
−
=
m
A
A
O
K
ここに, K2O: 酸化カリウムの含有率 (mass%)
A1: 試料溶液 (C) 中の酸化カリウムの量 (g)
A2: 空試験液 (C) 中の酸化カリウムの量 (g)
m: 14.2.4 a) の試料の量り取り量 (g)
17.3 原子吸光法
17.3.1 要旨 14.2.4の試料溶液 (C) をとり,原子吸光分析装置を用いてカリウムの原子吸光を吸光度で測
定する。
17.3.2 操作 14.2.4 b) で得た試料溶液(C)(30)の一部を原子吸光分析装置のアセチレン−空気フレーム中
に噴霧し,波長766.5nm(35)における原子吸光を吸光度で測定する。
注(35) 試料溶液中の酸化カリウムの濃度が高い場合は,波長769.9nm又は404.4nmを用いてもよい。
17.3.3 空試験 14.2.5で得た空試験液(31)を用いて17.3.2の操作を行う。
17.3.4 検量線の作成 14.2.2 j)の検量線用溶液系列II(32)(36)を用いて17.3.2の操作を行い(24),吸光度と酸
化カリウムの量との関係線を作成し,原点を通るように平行移動して検量線とする。
注(36) 注(35)による場合は,その濃度に合わせた検量線用溶液系列IIを作成し,試料溶液と同じ波長を
用いて検量線を作成する。
17.3.5 計算 試料中の酸化カリウムの含有率は,17.3.2及び17.3.3で得た吸光度と17.3.4で作成した検量
線とから酸化カリウムの量を求め,次の式によって算出する。
100
100
2
1
2
×
×
−
=
V
m
A
A
O
K
ここに, K2O: 酸化カリウムの含有率 (mass%)
A1: 試料溶液 (C) 又は希釈試料溶液中の酸化カリウムの量 (g)
A2: 空試験液 (C) 又は希釈試料溶液中の酸化カリウムの量 (g)
V: 試料溶液 (C) の分取量 (ml) (分取しない場合は100)
m: 14.2.4 a)の試料の量り取り量 (g)
18. 酸化クロム(III)の定量方法
18.1 定量方法の区分 酸化クロムの定量方法は,次のいずれかによる。
a) 原子吸光法
b) ICP発光分光分析法
18.2 原子吸光法
18.2.1 要旨 10.2.4,10.3.4又は10.4.4の試料溶液(A,A'又はA")を分取し,原子吸光分析装置を用い
てクロムの原子吸光を吸光度で測定する。
18.2.2 試薬 試薬は,次による。
a) 酸化クロム (III) 標準液 (1.0mgCr2O3/ml) 二クロム酸カリウム(99.9mass%以上)1.935 6gを量り取
り,ビーカー (200ml) に移し入れ(17),水を加えて液量を50mlとした後,硫酸 (1+1) 5ml及びエタノ
ール5mlを加えて,溶液が緑色になるまで水浴上で加熱する。冷却後,水で正しく1 000mlに薄める。
b) 希釈酸化クロム (III) 標準液 (0.05mgCr2O3/ml) 酸化クロム (III) 標準液を水で正しく20倍に薄める。
c) 添加液は,12.3.2 h)による。
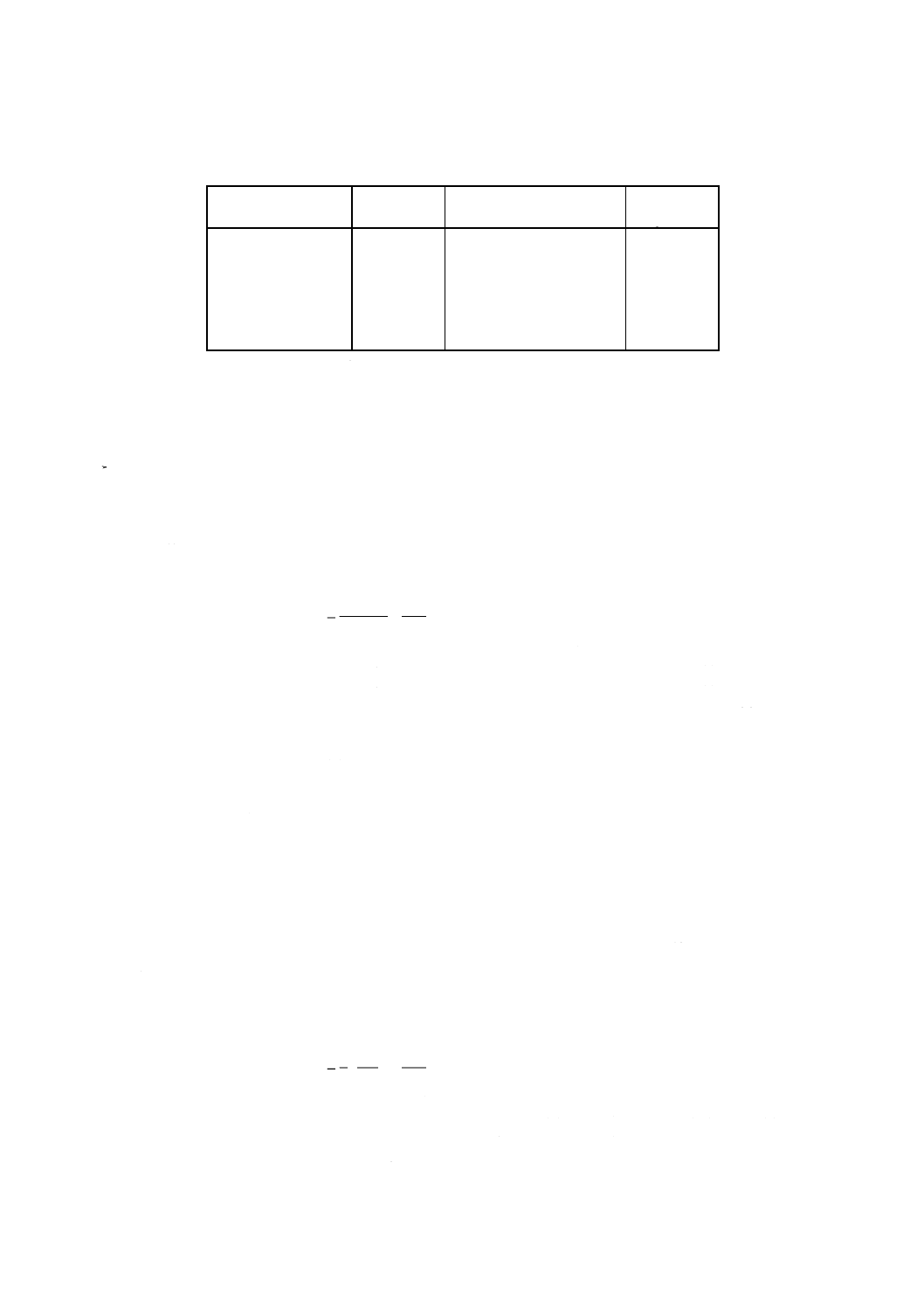
d) 検量線用溶液系列III 希釈酸化クロム (III) 標準液の一定量(37)を数個の100mlの全量フラスコにとり,
それぞれに添加液25mlを加え,水を標線まで加える。
23
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(37) 検量線用溶液系列IIIの調製例を,表10に示す。
表10 検量線用溶液系列IIIの調製例
検量線用溶液系列III
添加液
ml
希釈酸化クロム(III)標準液
ml
Cr2O3
mg/100ml
No.1
25
0
0
No.2
25
5
0.25
No.3
25
10
0.50
No.4
25
15
0.75
No.5
25
20
1.00
No.6
25
25
1.25
18.2.3 操作 10.2.4 d),10.3.4 d)又は10.4.4 a)で得た試料溶液(A,A'又はA")の25mlを100mlの全量フ
ラスコに分取し,水を標線まで加える。この試料溶液(D)の一部を原子吸光分析装置のフレーム中に噴霧し,
波長359.3nmにおける原子吸光を吸光度で測定する。
18.2.4 空試験 10.2.5,10.3.5,又は10.4.5で得た試料溶液(A,A'又はA")を用いて,18.2.3の操作を行
う。
18.2.5 検量線の作成 18.2.2 d)の検量線用溶液系列IIIを用いて18.2.3の操作を行い(24),吸光度と酸化ク
ロム (III) の量との関係線を作成し,原点を通るように平行移動して検量線とする。
18.2.6 計算 試料中の酸化クロム(III)の含有率は,18.2.3及び18.2.4で得た吸光度と18.2.5で作成した検
量線とから酸化クロム(III)の量を求め,次の式によって算出する。
100
25
500
2
1
3
2
×
×
−
=
m
A
A
O
Cr
ここに, Cr2O3: 酸化クロム(III)の含有率 (mass%)
A1: 18.2.3の試料溶液中の酸化クロム (III) の量 (g)
A2: 18.2.4の空試験液中の酸化クロム (III) の量 (g)
m: 10.2.4,10.3.4又は10.4.4の試料の量り取り量 (g)
18.3 ICP発光分光分析法
18.3.1 要旨 12.3.3の試料溶液 (B) の一部をとり,ICP発光分光分析装置を用いてクロムの発光強度を測
定する。
18.3.2 操作 12.3.3 a) で得た試料溶液 (B) の一部をICP発光分光分析装置のアルゴンプラズマ中に噴霧
し,例えば,波長205.55nm(38)における発光強度を測定する。
注(38) 波長267.72nmは,白金の分光干渉を受けるので微量の酸化クロム (III) の定量には用いてはな
らない。
18.3.3 空試験 12.3.4で得た空試験液(B)を用いて18.3.2の操作を行う。
18.3.4 検量線の作成 12.3.2 i)の検量線用溶液系列Iを用いて18.3.2の操作を行い(24),発光強度と酸化ク
ロム (III) の量との関係線を作成し,原点を通るように平行移動して検量線とする。
18.3.5 計算 試料中の酸化クロム (III) の含有率は,18.3.2及び18.3.3で得た発光強度と18.3.4で作成し
た検量線とから酸化クロム (III) の量を求め,次の式によって算出する。
100
25
500
2
1
3
2
×
×
−
=
m
A
A
O
Cr
ここに, Cr2O3: 酸化クロム (III) の含有率 (mass%)
A1: 18.3.2の試料溶液 (B) 中の酸化クロム (III) の量 (g)
A2: 18.3.3の空試験液 (B) 中の酸化クロム (III) の量 (g)
m: 10.2.4 a),10.3.4 a)又は10.4.4 a)の試料の量り取り量 (g)
24
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
19. 酸化ジルコニウム(酸化ハフニウムを含む。)の定量方法
19.1 定量方法 酸化ジルコニウム(酸化ハフニウムを含む。)の定量方法は,マンデル酸重量法による。
19.2 マンデル酸重量法
19.2.1 要旨 10.2.4,10.3.4又は10.4.4の試料溶液(A,A'又はA")に塩酸を加えた後,加熱し,DL−マ
ンデル酸を加えてジルコニウム(ハフニウムを含む。)を沈殿させてろ過する。沈殿を強熱してその質量を
量り,酸化ジルコニウム(酸化ハフニウムを含む。)とする。
19.2.2 試薬 試薬は,次による。
a) 塩酸
b) DL−マンデル酸
c) 洗浄液 DL−マンデル酸10gを水180mlに溶かし,塩酸20mlを加える。
19.2.3 操作 定量操作は,次の手順によって行う。
a) 10.2.4 d),10.3.4 d)又は10.4.4 a)で得た試料溶液(A,A'又はA")の200mlをビーカー (500ml) に分取
し(39),塩酸50mlを加えた後,砂浴又は熱板上で加熱する。液が沸騰したならば,注意してDL−マン
デル酸16〜18gを加え,泡立ち始めたら90℃以上の水浴上に移し,適切量(例えば,0.2g)のろ紙粉
末を加えて十分にかき混ぜ,水浴上で沈殿を熟成する。上澄み液が透明になった後,更に30分以上水
浴上で加熱を続け,適切量(例えば,0.2g)のろ紙粉末を加え,ろ過を通じて沈殿の入ったビーカー
を水浴上で保温しながらろ紙(5種C)を用いてろ過し,温洗浄液で10回洗浄する。
注(39) 含有率の低い試料の場合には,試料溶液(A,A'又はA")の分取量を200ml以上(例えば,400ml)
とし,200mlに濃縮するとよい。必要なら,別に試料溶液を10.に準じて調製してもよい。
b) 沈殿をろ紙と共に白金るつぼ(例えば,30番)に入れ,初めは低温で加熱し,次第に温度を上げてろ
紙を灰化し,1 100±25℃で30分間強熱する。デシケーター中で放冷した後,その質量を量る。
19.2.4 空試験 試料を用いないで19.2.3の操作を行う。
19.2.5 計算 試料中の酸化ジルコニウム(酸化ハフニウムを含む。)の含有率は,19.2.3 b)及び19.2.4の
酸化ジルコニウム(酸化ハフニウムを含む。)の量とから次の式によって算出する。
100
200
500
)
(
2
1
2
2
×
×
−
=
+
m
m
m
HfO
ZrO
ここに,
ZrO2 (+HfO2) : 酸化ジルコニウム(酸化ハフニウムを含む。)
の含有率 (mass%)
m0: 10.2.4 a),10.3.4 a)又は10.4.4 a)の試料の量り取
り量 (g)
m1: 19.2.3 b)で量った沈殿の質量 (g)
m2: 19.2.4で量った沈殿の質量 (g)
参考 酸化クロム (III) を含まない試料には,次のクペロン重量法を適用することもできる。ただし,
結果は,参考値とする。
a) 操作 11.2.6備考a) 1)で得た沈殿をろ紙と共に白金るつぼ(例えば,30番)に入れ,初めは
低温で加熱し(40),次第に温度を上げてろ紙を灰化し,1 100±25℃で30分間強熱する。デシ
ケーター中で放冷した後,その質量を量る。
注(40) 赤外線ランプを使用するとよい。
b) 空試験 11.2.6備考b)で得たろ紙について,a)の操作を行う。
c) 計算 試料中の酸化ジルコニウム(酸化ハフニウムを含む。)の含有率は,a)及びb)で得た混
25
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
合酸化物の量とから次の式によって算出する。
)
(
100
200
500
)
(
2
3
2
2
1
2
2
TiO
O
Fe
m
m
m
HfO
ZrO
+
−
×
×
−
=
+
ここに, ZrO2 (+HfO2) : 酸化ジルコニウム(酸化ハフニウムを含む。)の含
有率 (mass%)
m0: 10.2.4 a),10.3.4 a)又は10.4.4 a)の試料の量り取り
量 (g)
m1: a)で量った沈殿の質量 (g)
m2: b)で量った沈殿の質量 (g)
Fe2O3: 12.で求めた酸化鉄 (III) の含有率 (mass%)
TiO2: 13.で求めた酸化チタン (IV) の含有率 (mass%)
備考 酸化ハフニウムは,次の方法によって定量することができる。
a) 操作 12.3.3 a)で得た試料溶液 (B) の一部をICP発光分光分析装置のアルゴンプラズマ中に
噴霧し,例えば,波長277.34nmにおける発光強度を測定する。
b) 空試験 12.3.4で得た空試験液 (B) を用いてa)の操作を行う。
c) 検量線の作成 12.3.2 i)の検量線用溶液系列Iを用いてa)の操作を行い(24),発光強度と酸化
ハフニウムの量との関係線を作成し,原点を通るように平行移動して検量線とする。
d) 計算 試料中の酸化ハフニウムの含有率は,a)及びb)で得た発光強度とc)で作成した検量線
とから酸化ハフニウムの量を求め,次の式によって算出する。
100
20
500
2
1
2
×
×
−
=
m
A
A
HfO
ここに, HfO2: 酸化ハフニウムの含有率 (mass%)
A1: a)の試料溶液 (B) 中の酸化ハフニウムの量 (g)
A2: b)の空試験液 (B) 中の酸化ハフニウムの量 (g)
m: 10.2.4 a),10.3.4 a)又は10.4.4 a)の試料の量り取り量 (g)
26
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図1 陽イオン交換樹脂カラム
27
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書(参考) アルミナ−ジルコニア−シリカ質耐火物中の
酸化ハフニウムの蛍光X線分析方法
序文 この附属書(参考)は,アルミナ−ジルコニア−シリカ質耐火物中の酸化ハフニウムの蛍光X線分
析方法について記述するものであり,規定の一部ではない。耐火物中の酸化ハフニウムは,一般にその含
有率も低く,耐火物に与える特性も,共存する酸化ジルコニウムと化学的・物理的に類似している。その
ため,酸化ハフニウムだけを単離して定表しないで,酸化ジルコニウムとの合量として定量して“酸化ジ
ルコニウム(酸化ハフニウムを含む。)”として表すことが多く,本体でも,そのように取り扱っている。
一方,蛍光X線分析方法では,ジルコニウムとハフニウムが分離されて計測される。そのため,本体では,
JIS R 2216に使用する標準物質の値付けなどを目的として,19.の備考において,酸化ハフニウムのICP発
光分光分析法を示した。
しかし,ICP発光分光分析装置は必ずしも一般に広く普及しているとはいえない。そこで,この附属書(参
考)では,本体19.の備考を補完する目的で,試薬合成法による酸化ハフニウムの蛍光X線分析方法を示
す。
1. 適用範囲及び定量範囲 この附属書(参考)は,アルミナ−ジルコニア−シリカ質耐火物中の酸化ハ
フニウムの定量に適用する。この附属書による酸化ハフニウムの定量範囲は,おおむね2.0mass%以下とす
る。
2. 引用規格 次に掲げる規格は,この附属書(参考)に引用されることによって,この附属書の一部を
構成する。これら引用規格は,その最新版を適用する。
JIS K 0119 蛍光X線分析方法通則
JIS R 2216 耐火れんが及び耐火モルタルの蛍光X線分析方法
3. 一般事項 分析方法の一般事項は,本体に定められるほか,JIS K 0119及びJIS R 2216による。
4. 定義 この附属書に用いる主な用語の定義は,JIS K 0119及びJIS R 2216による。
5. 定量方法 酸化ハフニウムの定量方法は,共存成分補正蛍光X線分析方法による。
6. 要旨 試料を融剤と共にガラスビード作製容器中で融解後,冷却してガラスビードを作成する。この
ガラスビードについて,蛍光X線分析装置を用いてハフニウムの特性X線を測定し,共存成分補正法によ
って酸化ハフニウムの含有率を求める。
7. 装置及び器具
7.1
蛍光X線分析装置 蛍光X線分析装置は,JIS K 0119による。使用する装置は,HfMαに十分な測
定感度をもつものでなければならない。ロジウム又はクロムを対陰極にしたX線管を用いることが望まし
い。
28
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.2
ガラスビード作製装置 ガラスビード作製には,1 100〜1 250℃以内の一定温度に昇温できる構造を
備えた電気抵抗炉又は高周波誘導炉を用いる。成分の偏析や気泡がないガラスビードを得るため,自動融
解成形機にあっては融解物のかき混ぜ機構をもつものでなければならない。
また,手操作で作製するものにあっては,炉内部において容易に融解物のかき混ぜができるものでなけ
ればならない。
7.3
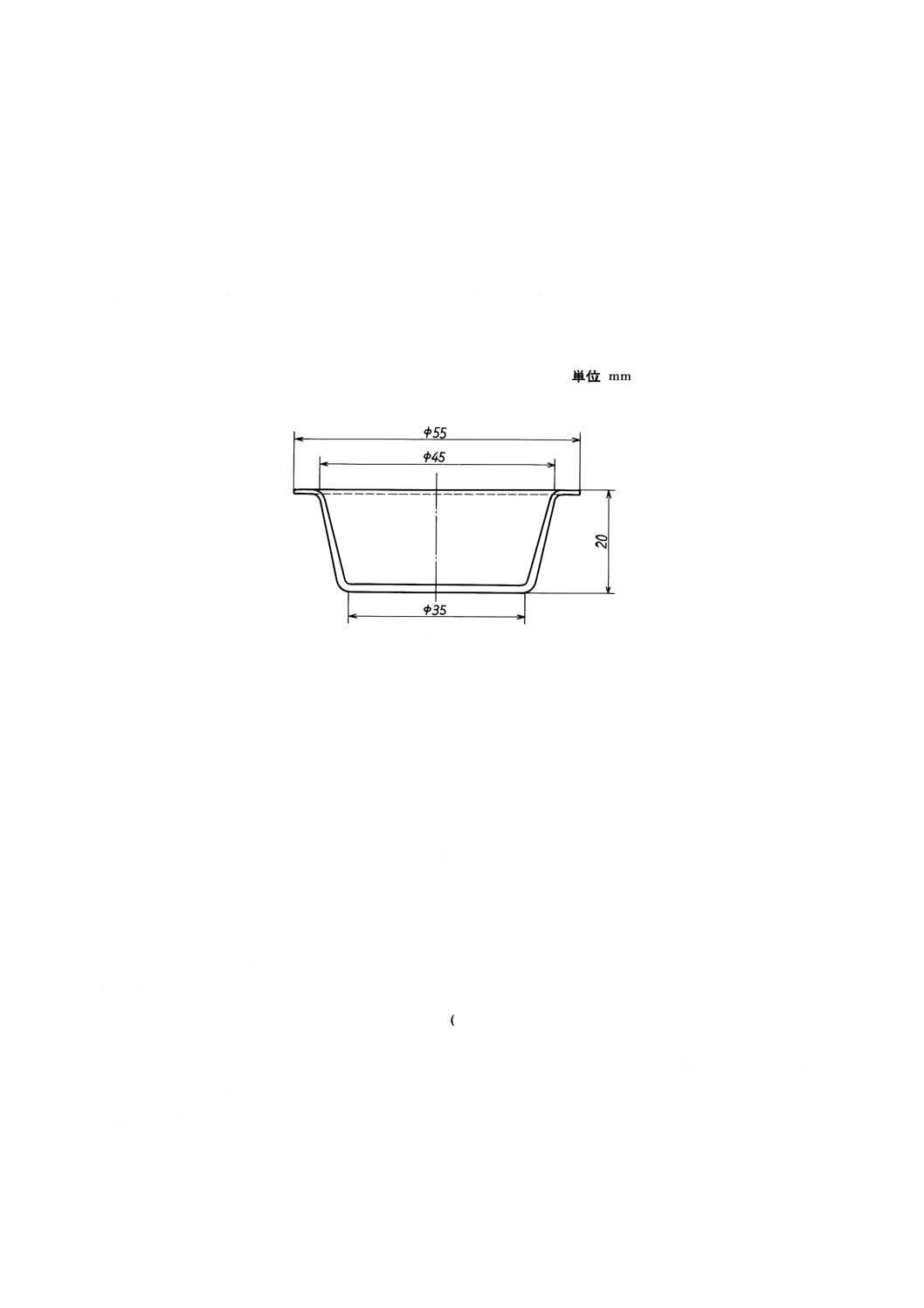
ガラスビード作製容器 ガラスビード作製容器は,内部底面を平滑に仕上げた白金合金(1)で,加熱
融解操作によって変形しにくい構造のものを用いる。常に容器内部底面は,平滑に保つ。ガラスビード作
製容器の例を附属書図1に示す。
注(1) 容器の材質としては,例えば,白金 (95%) −金 (5%) を用いる。
附属書図1 ガラスビード作製容器の例
8. 試薬 試薬は,次による。
a) 四ほう酸リチウム
b) 融剤 四ほう酸リチウムを,白金皿(例えば,150番)に入れ,650〜700℃の電気炉中で約4時間加
熱した後,デシケーター中で放冷し,保存する。
c) よう化リチウム二水和物
d) はく離促進剤 次のいずれかの形で用いる。
1) よう化リチウム含有粉末 ガラスビード作製容器に四ほう酸リチウム5g及びよう化リチウム二水
和物1gを加え,1 100〜1 150℃の電気炉中でできる限り短時間で融解し,冷却後,粉砕したものを,
密閉できるガラス瓶に入れて保存する。
2) よう化リチウム溶液 よう化リチウム二水和物25gを水に溶かして50mlとした後,プラスチック
容器に入れて保存する。
e) 酸化ハフニウム標準液 (1.0mgHfO2/ml) 本体12.3.2 f)による。
f)
希釈酸化ハフニウム標準液 (20μgHfO2/ml) 500mlの全量フラスコに酸化ハフニウム標準溶液10ml
をとり,水を標線まで加える。この溶液は,使用の都度調製する。
g) 酸化ジルコニウム(99.5mass%以上,酸化ハフニウム0.02mass%以下)(2) 酸化ジルコニウム中の酸化
ハフニウムの含有率は,次によって定量する。
1) 6個(3)のガラスビード作製容器のそれぞれに,酸化ジルコニウム0.500 0g,融剤5.000g及びはく離
促進剤の一定量(例えば,よう化リチウム溶液50μl)を量り取り,それぞれについて十分に混合し
29
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
た後,希釈酸化ハフニウム標準液0〜5ml(酸化ハフニウムとして0〜0.1mg,酸化ジルコニウム中
の酸化ハフニウムの含有率として0〜0.02mass%に相当する。)を加え,注意して110±5℃の空気浴
中で約30分間加熱する。
2) 加熱完了後,ガラスビード作製装置に移し,1 100〜1 150℃で一定時間(例えば,10分間)融解す
る。この間,ときどき容器を振り混ぜ,融解物の均質化を図るとともに気泡を放出させる。融解完
了後,放冷してガラスビードを作製する。
3) 得られたガラスビードの底面がX線照射面となるように,蛍光X線分析装置の試料ホルダーに正し
く装着し,蛍光X線分析装置を用いてガラスビードのHfMαのX線強度(4)(5)を測定する。
4) 得られたハフニウムのX線強度と酸化ハフニウムの添加量(酸化ジルコニウム中の酸化ハフニウム
含有率対応)との関係線(一次式)を作成し,関係式からハフニウムのX線強度が零における酸化
ハフニウム量を求め,符号を変えて(6)酸化ジルコニウム中の酸化ハフニウムの含有率とする。
注(2) 超微粉で吸湿しやすいものがある。そのような試薬では,白金るつぼに入れ,1 050±25℃で約
1時間加熱し,デシケーター中で放冷したものを使用する。
(3) ガラスビード作製容器が6個用意できないときは,数回に分けて1)と2)の操作を行い,6個の
ガラスビードを完成させるとよい。
(4) バックグラウンドを測定し,ハフニウムの真のX線強度を求める。測定条件は,次の条件を標
準とする。X線管:端窓型ロジウム管球,印加電圧:50kV,分光結晶:PET,スリット:コー
ス,ゴニオメータ内温度:36.5℃,HfMα2θ:118.88°,バックグラウンド2θ:116.23°及び121.53°,
計測時間:各角度共に100秒間
(5) HfMαで十分な感度が得られないときは,HfLβ1を用いるとよい。
(6) 計算値は,負符号となるので,正符号とする。
h) 酸化ハフニウム(99.9mass%以上)(2)
i)
酸化けい素(IV)(99.9mass%以上)(2)
j)
酸化アルミニウム(99.9mass%以上)(2)
9. 操作 定量操作は,次の手順によって行う。
a) ガラスビード作成容器に試料0.500 0gと融剤5.000g及びはく離促進剤の一定量を量りとり,十分に混
合する。
b) 以下,8. g)の2)及び3)に従って操作し,ハフニウムのX線強度を測定する。
10. 検量線の作成 合量が0.500 0gになるように酸化ジルコニウム及び酸化アルミニウムに酸化ハフニウ
ムを量り取り(7),以下,9.a)融剤の量り取り以降の操作を行い,検量線用ガラスビードのX線強度を測定
し(4),ハフニウムのX線強度と酸化ハフニウムの量(7)との関係線を作成して検量線とする(8)。
注(7) 附属書表1に,検量線用ガラスビード系列の一例を示す。No.6からNo.9は,波長固定型装置の
場合にだけ作製する。
なお,表中のXは,8.g)4)で求めた酸化ジルコニウム中の酸化ハフニウムの含有率を示す。化
学はかりの最小読み取りけたが0.1mgの場合,例えば,酸化ジルコニウムと酸化ハフニウムを
正しく9量対1量の比で量り取り,アルコールを加えて粉砕器中で十分混合し,乾燥したもの
を用いる。
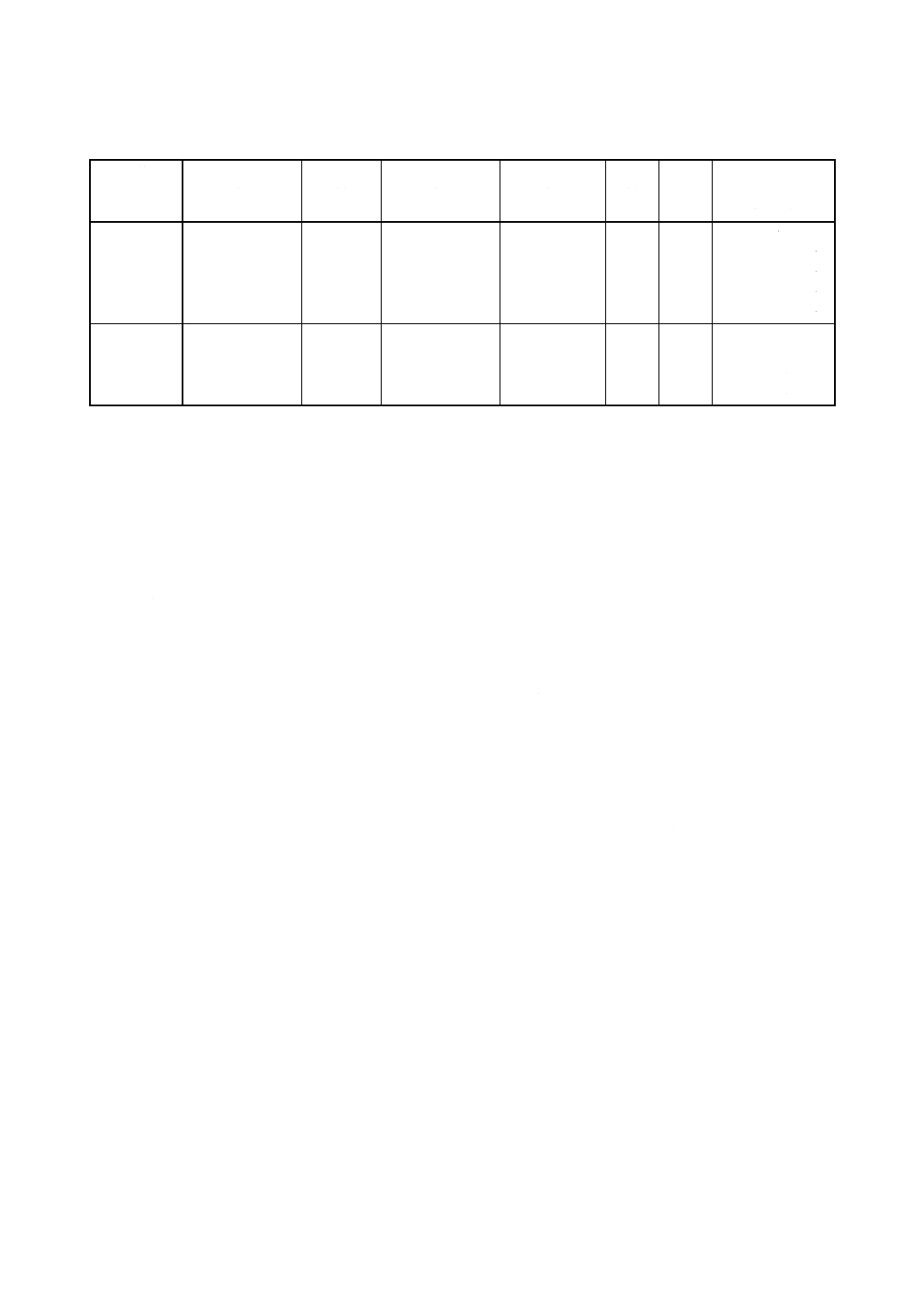
30
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書表1 検量線用ガラスビード系列の一例
検量線用
ガラスビード
系列
酸化ジルコニウム
(g)
酸化けい素
(g)
酸化アルミニウム
(g)
酸化ハフニウム
(g)
融剤
(g)
はく離
溶液
(μl)
酸化ハフニウム
含有率相当量
(mass%)
No.1
0.500 0
0
0
0
5.000
50
X
No.2
0.497 5
0
0
0.002 50
5.000
50
0.500+0.995・X
No.3
0.495 0
0
0
0.005 00
5.000
50
1.000+0.990・X
No.4
0.492 5
0
0
0.007 50
5.000
50
1.500+0.985・X
No.5
0.490 0
0
0
0.010 00
5.000
50
2.000+0.980・X
No.6
0
0.500 0
0
0
5.000
50
0
No.7
0
0
0.500 0
0
5.000
50
0
No.8
0.250 0
0.250 0
0
0
5.000
50
0.5・X
No.9
0.250 0
0
0.250 0
0
5.000
50
0.5・X
(8) 波長固定型装置では,検量線用ガラスビード系列No.1からNo.5を用いて検量線を求め,No.1
及びNo.6からNo.9を用いて,酸化けい素(IV)及び酸化アルミニウムのバックグラウンド補正値
を求める。
なお,走査型装置で,注(4)によってバックグラウンドを測定し差し引いた場合には,バック
グラウンド補正は不要である。蛍光X線分析装置に内蔵の計算ソフトを利用して,検量線及び
バックグラウンド補正値を求めるとよい。
11. 計算 試料中の酸化ハフニウムの含有率は,9.b)で得たハフニウムのX線強度,本体で求めた試料中
の他成分の含有率及び10.の検量線とから,次の式によって算出する(9)(10)。
HfO2= (aI+b) (1−0.000 84・WSiO2+0.000 53・WAl2O3)
ここに,
HfO2: 酸化ハフニウムの含有率 (mass%)
a: 10.で求めた検量線のこう(勾)配
b: 10.で求めた定数
I: 9.b)のハフニウムのX線強度
WSiO2: 本体で求めた酸化けい素の含有率 (mass%)
WAl2O3: 本体で求めた酸化アルミニウムの含有率 (mass%)
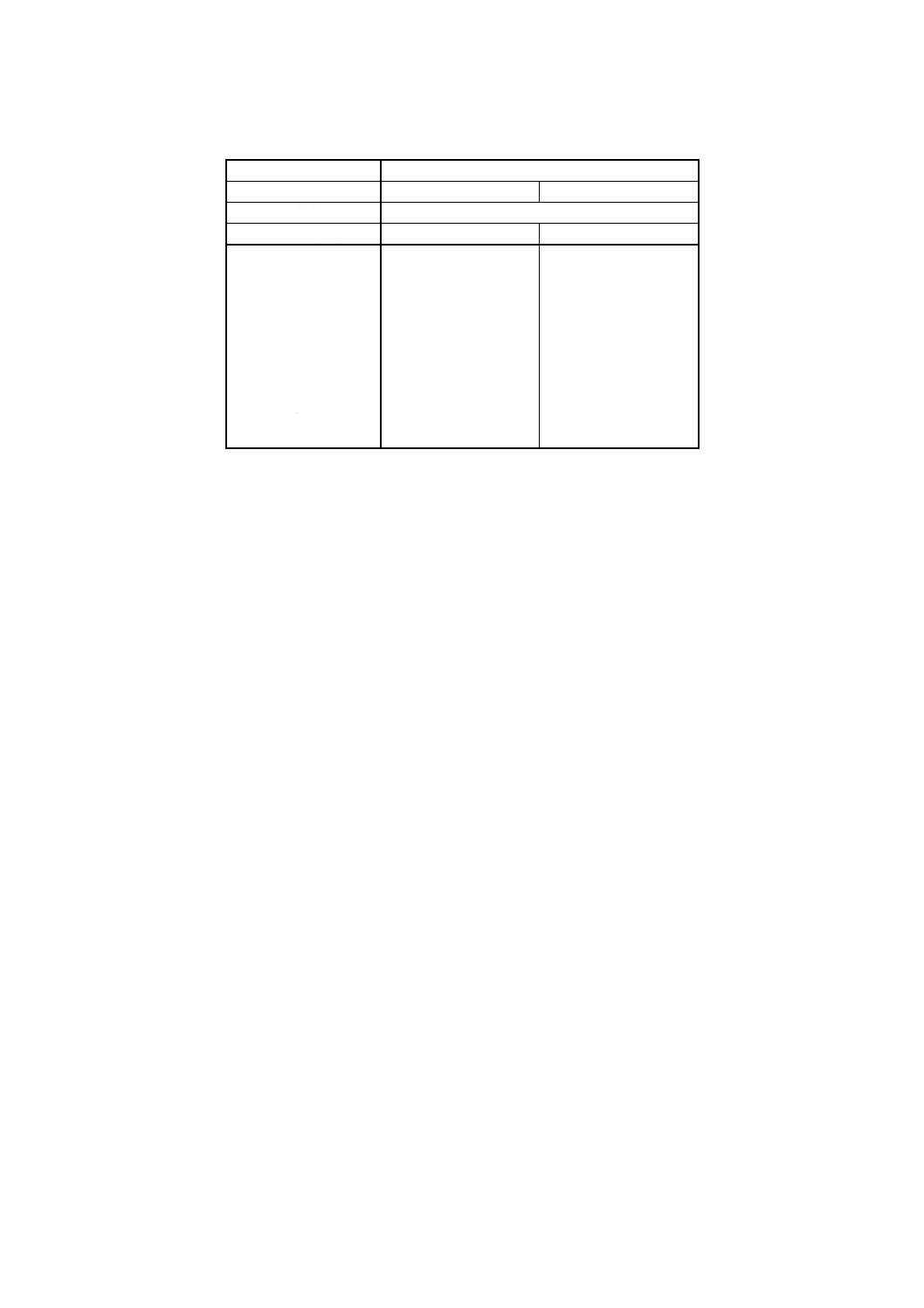
注(9) 酸化ジルコニウム,酸化けい素(IV)及び酸化アルミニウム以外の成分が多く含まれる試料では,
附属書表2に示す理論共存成分補正係数を用いた酸化けい素 (IV) 及び酸化アルミニウム以外
の共存成分補正を行うとよい。
(10) 波長固定型装置の場合には,次の式によって算出する。
HfO2= (aI+b) (1−0.000 76・WSiO2+0.000 69・WAl2O3)+lSiO2・WSiO2+lAl2O3・WAl2O3
ここに,
lSiO2: 10.の注(8)で求めたSiO2のバックグラウンド補正係数
WAl2O3: 10.の注(8)で求めたAl2O3のバックグラウンド補正係数
31
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書表2 理論共存成分補正係数
測定線
HfMα
装置の型式
走査型
波長固定型
X線管
端窓型Rh
管電圧 (kV)
50
40
dSiO2
−0.000 84
−0.000 76
dAl2O3
0.000 53
0.000 69
dFe2O3
0.000 23
0.000 39
dTiO2
−0.000 49
−0.000 34
dCaO
−0.000 66
−0.000 52
dMgO
0.000 37
0.000 53
dNa2O
0.000 22
0.000 37
dK2O
−0.000 82
−0.000 68
dP2O3
−0.000 80
−0.000 71
dCr2O3
−0.000 15
0.000 00
32
R 2013 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS R 2013原案作成委員会 構成表
氏名
所属
(委員長)
中 川 善兵衛
東京工業大学
(幹事)
○ 鹿 野 弘
黒崎窯業株式会社技術研究所
○ 村 田 守
鳴門教育大学
福 水 健 文
通商産業省生活産業局
大 嶋 清 治
工業技術院標準部
山 村 修 蔵
財団法人日本規格協会技術・検査部
藤 貫 正
日本磁気共鳴医学会
多 田 格 三
フジ化学研究所
荒 木 慎 介
耐火物協会
石 井 章 生
新日本製鐵株式会社プロセス技術研究所
高 橋 忠 明
日本鋼管株式会社製鋼技術開発部
熊 谷 正 人
川崎製鉄株式会社千葉製鉄所
鈴 木 隆 夫
住友金属工業株式会社製鋼技術部
海老名 克 己
三菱マテリアル株式会社セメント事業本部
吉 澤 紀 男
株式会社ヨータイ
渡 辺 高
東芝セラミックス株式会社
沓 掛 行 徳
旭硝子株式会社高砂工場
海老沢 律
川崎炉材株式会社管理部
菅 野 登
イソライト工業株式会社
(分科会主査)
○ 三 橋 久
岡山セラミックス技術振興財団
(分科会幹事)
○ 朝 倉 秀 夫
品川白煉瓦株式会社技術研究所
○ 南 政 光
黒崎窯業株式会社測定評価センター
○ 板 倉 正 勝
東芝セラミックス株式会社刈谷製造所
● 吉 田 清 志
川崎炉材株式会社製品開発部
● 森 邦 夫
旭硝子株式会社セラミックス事業部
● 福 井 洋 一
ハリマセラミック株式会社研究開発部
● 平 松 康 宏
株式会社ヨータイ技術研究所
● 江 尻 勉
九州耐火煉瓦株式会社技術管理部
● 戸 松 一 郎
株式会社TYK研究所
● 中 山 信 司
東芝モノフラックス株式会社神崎工場
● 鬼 塚 浩 次
大光炉材株式会社技術研究所
● 宮 脇 正 夫
日本特殊炉材株式会社技術部
● 河 野 久 征
理学電機工業株式会社応用技術センター
(事務局)
細 川 周 明
耐火物技術協会
備考 ○印は,分科会委員併任 ●印は,分科会委員専任を示す。
文責 JIS R 2013原案作成委員会分科会