2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
M 8851-1983
ドロマイトの分析方法
Methods for Chemical Analysis of Dolomite
1. 適用範囲 この規格は,ドロマイトの分析方法について規定する。
引用規格:
JIS K 0050 化学分析通則
JIS K 0115 吸光光度分析のための通則
JIS K 0121 原子吸光分析のための通則
JIS K 8005 容量分析用標準試薬
JIS K 8006 試薬の含量試験中滴定に関する基本事項
JIS R 1306 化学分析用磁器燃焼ボート
JIS R 1307 化学分析用磁器燃焼管
JIS Z 8401 数値の丸め方
JIS Z 8801 標準ふるい
2. 一般事項 分析方法に共通な一般事項は,JIS K 0050(化学分析通則),JIS K 0115(吸光光度分析の
ための通則)及びJIS K 0121(原子吸光分析のための通則)による。
3. 分析項目 この規格で規定する分析項目(1)は,次のとおりとする。
強熱減量 (Ig. loss)
二酸化けい素 (SiO2)
酸化鉄(III) (Fe2O3)
酸化アルミニウム (Al2O3)
酸化カルシウム (CaO)
酸化マグネシウム (MgO)
五酸化りん (P2O5)
全硫黄 (Total S)
注(1) 二酸化けい素(重量法を除く。),酸化鉄 (III),酸化アルミニウム,酸化カルシウム,酸化マグ
ネシウム及び五酸化りんは,それぞれ元素態で定量が行われるが,便宜上,酸化物の形で表示
する。
4. 試料の採り方及び取扱い方
4.1
分析試料は,よくかき混ぜて平均組成を表すように注意し,また,異物が混入していないことを確
かめなければならない。
2
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.2
分析試料は,JIS Z 8801(標準ふるい)の149μmを全量通過させるように粉砕したもの約10gを平
形はかり瓶(直径50mm)に薄く広げ,105〜110℃の空気浴中で1時間以上乾燥した後,デシケーター中
に保存したものからはかり取る。
4.3
分析試料のはかり取りには,化学はかりを用い,規定された量を0.1mgのけたまで読みとる。
5. 分析値のまとめ方
5.1
分析結果は,百分率で表し,JIS Z 8401(数値の丸め方)によって次のように丸める。
(1) 強熱減量,酸化カルシウム及び酸化マグネシウムは,小数点以下第1位
(2) 二酸化けい素,酸化鉄 (III),酸化アルミニウム及び重量法による全硫黄は,小数点以下第2位
(3) 五酸化りん及び熱分解−よう素酸カリウム滴定法による全硫黄は,小数点以下第3位
5.2
分析に当たっては,全操作を通じて空試験を行い,含有率を補正しなければならない。
5.3
分析は,同一試料について原則として2回以上繰り返して行い,その差が表1の許容差 (%) に示す
数値よりも大きいときは改めて2回以上の分析をやり直し,許容差以内のものの平均値を出す。
表1 許容差(2)
単位 %
強熱減量
二酸化けい素
酸化鉄 (III)
過塩素酸脱水重量法
モリブデン青吸光光度法
1,10-フェナントロリン
吸光光度法
原子吸光法
区分
許容差
区分
許容差
区分
許容差
区分
許容差
0.2
0.10以上1.00未満 0.03
0.40未満 0.02
0.10未満 0.01
0.10未満 0.01
0.10以上0.50未満 0.03 0.10以上0.50未満 0.03
1.00以上
0.05 0.40以上1.00未満 0.04 0.50以上1.50未満 0.05 0.50以上1.00未満 0.05
酸化アルミニウム
酸化カル
シウム
酸化マグ
ネシウム
五酸化りん
EDTA-ビスマス逆滴定法
原子吸光法
モリブデン青吸光光度法
区分
許容差
区分
許容差
区分
許容差
0.50未満 0.02
0.50未満 0.02
0.3
0.2
0.050未満 0.002
0.050以上0.100未満 0.006
0.50以上
0.04 0.50以上1.00未満 0.04
0.100以上1.000未満 0.008
五酸化りん
全硫黄
りんバナドモリブデン酸
吸光光度法
熱分解-よう素酸カリウム滴
定法
硫酸バリウム重量法
区分
許容差
区分
許容差
区分
許容差
0.030以上0.100未満 0.004
0.050未満 0.003
0.01以上
0.01
0.100以上
0.007 0.050以上0.100未満 0.005
注(2) 2個の分析値が二つの区分にまたがるときは,2個の分析値の平均値の該当する区分の許容差を適用する。
6. 強熱減量定量方法
6.1
方法の区分 強熱減量の定量方法は,重量法による。
6.2
重量法
6.2.1
要旨 試料を1 050±50℃で恒量になるまで強熱したときの減量をはかる。
6.2.2
試料はかり取り量 試料は,1.0gをはかり取る。
6.2.3
操作 定量操作は,次の手順によって行う。
3
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 試料を白金るつぼ(例えば30番)(3)にはかり取り,少しすき間を開けてふたをし,初めは低温で加熱
し次第に温度を上げ,最後は電気炉中で1 050±50℃で1時間強熱する。ふたを密にしてデシケーター
中で放冷後,その質量をはかる。
(2) 15分間ずつ強熱を繰り返して,恒量(4)になったときの減量を求める。
6.2.4
計算 試料中の強熱減量を,次の式によって算出する。
100
(%)
2
1
×
−
=
W
w
w
強熱減量
ここに, w1: 強熱前の試料の入っている白金るつぼの質量 (g)
w2: 強熱後の試料の入っている白金るつぼの質量 (g)
W: 試料はかり取り量 (g)
注(3) 磁器るつぼを用いてもよい。
(4) 通常は1時間の強熱で恒量が得られる。
7. 二酸化けい素定量方法
7.1
方法の区分 二酸化けい素の定量方法は,次のいずれかによる。
(1) 過塩素酸脱水重量法 この方法は,二酸化けい素含有率0.10%以上の試料に適用する。
(2) モリブデン青吸光光度法 この方法は,二酸化けい素含有率1.00%未満の試料に適用する。
7.2
過塩素酸脱水重量法
7.2.1
要旨 試料を塩酸と過塩素酸で分解し,加熱して白煙を発生させ,けい酸を不溶性としてこし分け
る。沈殿を強熱して質量をはかり,ふっ化水素酸を加え加熱して二酸化けい素を揮散させた後,再び強熱
して質量をはかり前後の質量差を求める。
7.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+1,1+100)
(2) 過塩素酸 (60%)
(3) ふっ化水素酸
7.2.3
試料はかり取り量 試料は,1.00gをはかり取る。
7.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をビーカー (200ml) にはかり取り(5),少量の水を加えてスラリー状とし,時計皿で覆って塩酸 (1
+1) 5mlをビーカーの縁から少量ずつ加える。激しい反応が終わったら,時計皿を少量の水で洗って
取り除く。
(2) 過塩素酸5mlを加え,砂浴上で加熱し,過塩素酸の濃い白煙が出始めたら再び時計皿で覆い,引き続
き10分間加熱する。
(3) 少し冷却した後,塩酸 (1+1) 5ml及び温水約50mlを加えて振り混ぜ,不溶解物がほぼ沈降したら直
ちにろ紙(5種B)でこし分け,熱塩酸 (1+100) で数回洗浄し,更に熱水で十分に洗浄する(6)。ろ液
及び洗液はビーカー (300ml) に受け,保存する。
(4) 不溶解物はろ紙と共に白金るつぼ(例えば30番)に入れて乾燥し,徐々に加熱して炎の出ないように
ろ紙を灰化した後,1 050±50℃で1時間強熱し,デシケーター中で放冷後質量をはかり,恒量(4)とな
るまで強熱を繰り返す。
(5) 不溶解物を過塩素酸2,3滴(7)で湿し,ふっ化水素酸5mlを加え,砂浴上で加熱して蒸発乾固する。1
050±50℃で10分間強熱し,デシケーター中で放冷後,質量をはかる。
4
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なお,残留物の付着している白金るつぼは,そのまま保存する。
7.2.5
計算 試料中の二酸化けい素含有率を,次の式によって算出する。
100
(%)
2
1
×
−
=
W
w
w
二酸化けい素
ここに, w1: 7.2.4(4)ではかった質量 (g)
w2: 7.2.4(5)ではかった質量 (g)
W: 試料はかり取り量 (g)
注(5) 有機物を多量に含む試料の場合は,はかり取った試料をあらかじめ磁器るつぼ中で500〜600℃
に加熱し,有機物を分解してからビーカーに移す。
(6) 洗浄が不十分であると,ろ紙の灰化時に爆発飛散するおそれがある。特に不溶解物の多い場合
は,注意する必要がある。
(7) 不溶解物の量が多いときは,適宜増量する。
備考 二酸化けい素が極めて少量であって,特にこれの定量を必要としないときは,7.2.4(5)の操作を
省略して酸不溶解残さとして表示してもよい。この場合は,不溶解物の強熱に磁器るつぼを使
用しても差し支えない。
7.3
モリブデン青吸光光度法
7.3.1
要旨 試料を炭酸ナトリウムとほう酸で融解し,塩酸に溶解して定容とする。この溶液の一定量を
分取し,モリブデン酸アンモニウムを加えてけいモリブデン酸とし,しゅう酸と硫酸を共存させL−アス
コルビン酸を加えてモリブデン青を呈色させ,吸光度を測定する。
7.3.2
試薬 試薬は,次による。ただし,(4)〜(7)はプラスチック瓶に保存する。
(1) 塩酸 (1+9)
(2) 硫酸 (1+3)
(3) 混合融剤[炭酸ナトリウム(無水)2,ほう酸1]
(4) モリブデン酸アンモニウム溶液 モリブデン酸アンモニウム(四水和物)25gを温水に溶かして500ml
とする。必要ならばろ過する。ただし,保存中にモリブデン酸が析出したときは,新しく調製する。
(5) しゅう酸溶液 しゅう酸(二水和物)25gを水に溶かして500mlに薄める。
(6) L−アスコルビン酸溶液 (5w/v%) 冷暗所に保存する。ただし,保存中に着色したときは,新しく調
製する。
(7) 標準二酸化けい素溶液 (1.0mg SiO2/ml) 二酸化けい素(無水けい酸,沈降製)を磁器るつぼに取り,
1050±50℃で約1時間強熱後,デシケーター中で放冷したもの0.250gを白金るつぼ(例えば30番)
にはかり取り,炭酸ナトリウム(無水)2.0gと混合した後,ふたをして初めは低温で加熱し,次第に
温度を上げて最後は1 000℃以上に強熱して融解する。放冷後,温水約100mlを入れたプラスチック
ビーカー (200ml) に白金るつぼを入れ,ときどき揺り動かして融成物を溶解する。白金るつぼ及びふ
たを水洗して取り出し,冷却後,溶液を250mlのメスフラスコに移し入れ,水で標線まで薄める。直
ちにプラスチック瓶に移し入れる。
(8) 試薬ブランク溶液 混合融剤3.0gをはかり,白金るつぼに入れ,ふたをして初めは低温で加熱し,次
第に温度を上げて融解し,2〜3分間引き続き加熱する。以下,7.3.4(2)と同様に操作する。
7.3.3
試料はかり取り量 試料は,0.25gをはかり取る。
7.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料を白金るつぼ(例えば30番)にはかり取り(8),混合融剤3.0gと混合した後,ふたをして,初め
5
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
は低温で加熱し,次第に温度を上げて融解し,2〜3分間引き続き加熱する。
(2) 放冷後,熱塩酸 (1+9) 150mlを入れたビーカー (300ml) 中に白金るつぼを入れ,水浴上で加熱して融
成物を溶解する。白金るつぼ及びふたは水洗して取り出し,溶液を常温まで冷却した後,250mlのメ
スフラスコに移し入れ,水で標線まで薄める。
(3) この溶液25mlを100mlのメスフラスコに正確に分取し,モリブデン酸アンモニウム溶液10mlを加え
て振り混ぜ,5分間放置する。
(4) 次に,振り混ぜながらしゅう酸溶液10ml,硫酸 (1+3) 5ml及びアスコルビン酸溶液2mlを連続して
手早く加え,1分間放置後,水で標線まで薄める。
(5) 5分間放置後,この溶液の一部を光度計の吸収セルに取り,波長650nm付近で吸光度を測定する。
7.3.5
計算 7.3.6で作成した検量線から二酸化けい素量を求め,試料中の二酸化けい素含有率を次の式
によって算出する。
100
25
250
10
(%)
3
×
×
×
=
−
W
a
二酸化けい素
ここに,
a: 7.3.4(5)から得られた二酸化けい素検出量 (mg)
W: 試料はかり取り量 (g)
7.3.6
検量線の作成 標準二酸化けい素溶液を水で正しく20倍に薄め,その0〜5.0ml(二酸化けい素と
して0〜0.25mg)を,あらかじめ試薬ブランク溶液25mlずつを入れた数個の100mlメスフラスコに段階的
に正しく取り,モリブデン酸アンモニウム溶液10mlを加えて振り混ぜ,5分間放置する。以下,7.3.4(4)
及び(5)の手順に従って操作し,得た吸光度と二酸化けい素含有量との関係線を作成して検量線とする。
注(8) 試料中に有機物,硫化物等の還元性物質を含む場合は,500〜600℃で加熱して分解する。
8. 酸化鉄 (III)
8.1
方法の区分 酸化鉄 (III) の定量方法は,次のいずれかによる。
(1) 1,10−フェナントロリン吸光光度法 この方法は,酸化鉄 (III) 含有率1.50%未満の試料に適用する。
(2) 原子吸光法 この方法は,酸化鉄 (III) 含有率1.00%未満の試料に適用する。
8.2
1,10−フェナントロリン吸光光度法
8.2.1
要旨 7.2.4で保存した白金るつぼの残留物を,混合融剤で融解して塩酸に溶かし,7.2.4で保存し
たろ液及び洗液に加えて定容とする。この一部を分取して,アスコルビン酸で鉄を還元し,1,10−フェナ
ントロリンを加え,酢酸アンモニウムでpHを調節して呈色させ,吸光度を測定する。
8.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+1)
(2) 混合融剤[炭酸ナトリウム(無水)3,ほう酸1]
(3) 酢酸アンモニウム溶液 (20w/v%)
(4) L−アスコルビン酸溶液 (5w/v%) 7.3.2(5)と同じものを用いる。
(5) 1,10−フェナントロリン溶液 1, 10−フェナントロリン塩酸塩(一水和物)1gを水に溶かし,1lに薄
め,冷暗所に保存する。保存中に着色したときは,新しく調製する。
(6) 標準酸化鉄 (III) 溶液−I (0.10mg Fe2O3ml) 硫酸鉄 (III) アンモニウム(12水和物)0.604gをはかり
取り,水約100ml及び硫酸 (1+1) 10mlを加えて溶解した後,1 000mlのメスフラスコに移し,水で標
線まで薄める。
8.2.3
操作 定量操作は,次の手順によって行う。

6
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 7.2.4(5)で保存した白金るつぼに混合融剤2gを加え,ふたをして初めは低温で加熱し,次第に温度を
上げて融解する。
(2) 放冷後,白金るつぼをビーカー (100ml) に入れ,熱水を加えて加熱し,融成物を溶解した後,時計皿
で覆って塩酸 (1+1) 5〜7mlを少量ずつ加える。白金るつぼ及びふたを水洗して取り出し,煮沸して
二酸化炭素を追い出す。冷却後,7.2.4(3)で保存したビーカー (300ml) 中のろ液及び洗液に加える。
(3) この溶液を250mlのメスフラスコに移し入れ,水で標線で薄め試料溶液とし,酸化鉄 (III),酸化アル
ミニウム,酸化カルシウム,酸化マグネシウム及びモリブデン青吸光光度法による五酸化りんの定量
に用いる。
(4) 試料溶液から一定量(9)を100mlのメスフラスコに正確に分取する。
(5) アスコルビン酸溶液2mlを加えて振り混ぜ,1,10−フェナントロリン溶液10ml及び酢酸アンモニウム
溶液10mlを加え,標線まで水を加えて振り混ぜ,30分間放置する。
(6) この溶液の一部を光度計の吸収セルに取り,波長510nm付近で吸光度を測定する。
8.2.4
計算 8.2.5で作成した検量線から酸化鉄 (III) 量を求め,試料中の酸化鉄 (III) 含有率を,次の式
によって算出する。
100
250
10
)(%)
(
3
×
×
×
=
−
V
W
a
III
酸化鉄
ここに,
a: 8.2.3(6)から得られた酸化鉄 (III) 検出量 (mg)
W: 7.2.3の試料はかり取り量 (g)
V: 試料溶液の分取量 (ml)
8.2.5
検量線の作成 標準酸化鉄 (III) 溶液−Iを水で正しく10倍に薄め,その0〜30.0ml[酸化鉄 (III) と
して0〜0.30mg]を100mlのメスフラスコに段階的に取り,8.2.3(5)及び(6)の手順に従って操作し,得た吸
光度と酸化鉄 (III) 量との関係線を作成して検量線とする。
注(9) 試料溶液の分取量は,酸化鉄 (III) 含有率に応じて表2による。
表2 試料溶液の分取量
酸化鉄 (III) 含有率 %
分取量 ml
0.15未満
50
0.15以上
0.30未満
25
0.30以上
0.75未満
10
0.75以上
1.50未満
5
8.3
原子吸光法
8.3.1
要旨 8.2.3の試料溶液の一部を用いて,原子吸光光度計により,鉄の吸光度を測定する。
8.3.2
試薬 試薬は,次による。
(1) 塩酸 (1+1)
(2) マトリックス溶液−I 炭酸カルシウム3.25g,金属マグネシウム0.54g及び8.2.2(2)の混合融剤10gを
ビーカー (300ml) に取り,少量の水を加え,時計皿で覆って塩酸 (1+1) 50mlをビーカーの縁から少
量ずつ加えて分解し,煮沸して二酸化炭素を追い出す。冷却後,時計皿を水洗して除き,250mlのメ
スフラスコに移し,水で標線まで薄める。
(3) マトリックス溶液−II マトリックス溶液−I 50mlを250mlのメスフラスコに取り,塩酸 (1+1) 5ml
を加えて,水で標線まで薄める。
(4) 標準酸化鉄 (III) 溶液−II (1.0mg Fe2O3/ml) 金属鉄(99.9%以上)0.350gをビーカー (200ml) には
かり取り,時計皿で覆い,塩酸 (1+1) 20ml及び硝酸 (1+1) 1mlを加え,穏やかに加熱して分解する。
7
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
冷却後,時計皿を水洗して除き,500mlのメスフラスコに移し,水で標線まで薄める。
8.3.3
操作 定量操作は,次の手順によって行う。
(1) 8.2.3(3)の試料溶液の一部(10)を,原子吸光光度計の鉄用中空陰極ランプを用いて,空気−アセチレンフ
レーム中に噴霧し,波長248.3nmにおける吸光度を測定する(11)。
8.3.4
計算 8.3.5で作成した検量線から酸化鉄 (III) 濃度を求め,試料中の酸化鉄 (III) 含有率を次の式
によって算出する。
100
100
250
10
)(%)
(
3
×
×
×
=
−
W
C
III
酸化鉄
ここに,
C: 試料溶液中の酸化鉄 (III) 濃度 (mg/100ml)
W: 7.2.3の試料はかり取り量 (g)
8.3.5
検量線の作成 標準酸化鉄 (III) 溶液−IIを水で正しく10倍に薄め,その0〜20.0ml[酸化鉄 (III)
として0〜2.0mg]を100mlのメスフラスコに段階的に取り,マトリックス溶液−I 20ml及び塩酸 (1+1) 2ml
をそれぞれに加えて,水で標線まで薄める。次に,原子吸光光度計の鉄用中空陰極ランプを用い,空気−
アセチレンフレーム中に噴霧し,波長248.3nmにおける吸光度を測定し,得た吸光度と酸化鉄 (III) 濃度
との関係線を作成して検量線とする。
注(10) 酸化鉄 (III) 含有率が0.50%以上1.00%未満の場合は,試料溶液の一部を取ってマトリックス溶
液−IIで正しく2倍に薄める。なお,この場合の計算は,次の式を用いる。
100
100
250
2
10
)(%)
(
3
×
×
×
×
=
−
W
C
III
酸化鉄
ここに,
C: 試料溶液中の酸化鉄 (III) 濃度 (mg/100ml)
W: 7.2.3の試料はかり取り量 (g)
(11) ランプ電流値,分光器のスリット幅,燃料ガス及び助燃ガスの圧力と流量,フレーム中を透過
する光束の位置は,各装置に応じて適当な条件を設定する。感度が不足する場合は,目盛拡大
装置を使用するか,装置に内蔵されている感度調節装置により調節する。また,感度が必要以
上に高い場合は,バーナー角度などの調節により測定感度を減ずる。
9. 酸化アルミニウム
9.1
方法の区分 酸化アルミニウムの定量方法は,次のいずれかによる。
(1) EDTA−ビスマス逆滴定法 この方法は,酸化鉄 (III) +酸化アルミニウム含有率0.10%以上の試料に
適用する。
(2) 原子吸光法 この方法は,酸化アルミニウム含有率1.00%未満の試料に適用する。
9.2
EDTA−ビスマス逆滴定法
9.2.1
要旨 8.2.3の試料溶液の一部を分取し,EDTAの一定量を加えた後pHを調節する。煮沸してアル
ミニウム−EDTAキレートを生成させ,冷却後,キシレノールオレンジを指示薬として加え,M/100ビス
マス標準溶液で過剰のEDTAを逆滴定する。
9.2.2
試薬 試薬は,次による。
(1) 硝酸 (1+1,1+4)
(2) 緩衝溶液 酢酸アンモニウム200gを適量の水に溶解し,酢酸100mlを加え,水で500mlに薄める。
(3) EDTA溶液 (0.01M) エチレンジアミン四酢酸二ナトリウム(二水和物)3.73gを水に溶かして1lと
する。プラスチック瓶に保存する。

8
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(4) M/100ビスマス標準溶液 ビスマス(99.9%以上)2.090gを0.1mgまで正しくビーカー (200ml) には
かり取り,硝酸 (1+1) 50mlを加え,加熱して分解し,二酸化窒素を除去する。冷却後,水で正しく1
000mlに薄める。この溶液1mlは,酸化アルミニウム0.000 509 8gに相当する。
(5) キシレノールオレンジ溶液 (0.1w/v%) 褐色瓶に入れて冷暗所に保存する。1,2か月ごとに新しく調
製する。
9.2.3
操作 定量操作は,次の手順によって行う。
(1) 8.2.3(3)の試料溶液から一定量(12)をビーカー (300ml) に正確に分取する。EDTA溶液10mlを正確に加
え,液量が100mlに満たない場合には水で約100mlに薄める。
(2) 緩衝溶液20mlを加え,pH計を用いて硝酸 (1+1) を滴加してpH約3.5とした後,硝酸 (1+4) を滴
加してpH3.0〜3.2に調節する(13)。時計皿で覆って5分間煮沸した後,常温まで冷却する。
(3) 時計皿を水洗して除き,キシレノールオレンジ溶液4,5滴を指示薬として加え,M/100ビスマス標準
溶液で滴定し,溶液が黄色から赤みを帯びた点を終点とする。
(4) 空試験溶液から(1)の試料溶液と同一容量をビーカー (300ml) に正確に分取し,EDTA溶液10mlを正
確に加え,液量が100mlに満たない場合には水で約100mlに薄める。以下,(2)及び(3)と同様に操作し
てM/100ビスマス標準溶液で滴定する。
9.2.4
計算 試料中の酸化アルミニウム含有率を,次の式によって算出する。
638
.0
100
250
8
509
000
.0
)
(%)
3
2
1
2
×
−
×
×
×
−
=
O
Fe
V
W
υ
υ
(
酸化アルミニウム
ここに,
v1: 9.2.3(3)のM/100ビスマス標準溶液使用量 (ml)
v2: 9.2.3(4)のM/100ビスマス標準溶液使用量 (ml)
W: 7.2.3の試料はかり取り量 (g)
V: 試料溶液の分取量 (ml)
Fe2O3: 8.2又は8.3で求めた酸化鉄 (III) 含有率 (%)
注(12) 試料溶液の分取量は,酸化鉄 (III) +酸化アルミニウム含有率に応じて表3による。
表3 試料溶液の分取量
酸化鉄 (III) +酸化アルミニウム含有率% 分取量 ml
0.10以上 1.00未満
100
1.00以上 2.50未満
50
2.50以上
25
(13) pH3.0以下になった場合は,緩衝溶液を滴加する。
9.3
原子吸光法
9.3.1
要旨 8.2.3の試料溶液の一部を用いて,原子吸光光度計により,アルミニウムの吸光度を測定す
る。
9.3.2
試薬 試薬は,次による。
(1) 塩酸 (1+1)
(2) 過塩素酸 (1+1)
(3) マトリックス溶液−I 8.3.2(2)と同じものを用いる。
(4) 標準酸化アルミニウム溶液 (1.0mg Al2O3/ml) 金属アルミニウム(99.9%以上)0.265gを白金皿(例
えば100番)にはかり取り,時計皿で覆い,塩酸 (1+1) 20mlを加えて加熱し,分解する。放冷後,
時計皿を水洗して取り除き,500mlのメスフラスコに移し,水で標線まで薄める。
9.3.3
操作 定量操作は,次の手順によって行う。
9
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 8.2.3の試料溶液の一部を,原子吸光光度計のアルミニウム用中空陰極ランプを用い,酸化二窒素−ア
セチレンフレーム中に噴霧し,波長309.3nmにおける吸光度を測定する(11)。
9.3.4
計算 9.3.5で作成した検量線から酸化アルミニウム濃度を求め,試料中の酸化アルミニウム含有
率を次の式によって算出する。
100
100
250
10
(%)
3
×
×
×
=
−
W
C
酸化アルミニウム
ここに,
C: 試料溶液中の酸化アルミニウム濃度 (mg/100ml)
W: 7.2.3の試料はかり取り量 (g)
9.3.5
検量線の作成 標準酸化アルミニウム溶液0〜10.0ml(酸化アルミニウムとして0〜10.0mg)を数
個のビーカー (200ml) に段階的に分取する。それぞれにマトリックス溶液−I 20mlずつを正確に加え,砂
浴上で穏やかに蒸発乾固する。放冷後,それぞれに塩酸 (1+1) 2ml,過塩素酸 (1+1) 3mlを加え,加熱し
て可溶性塩を溶解し,それぞれ100mlのメスフラスコに移し,水で標線まで薄める。次に,原子吸光光度
計のアルミニウム用中空陰極ランプを用い,酸化二窒素−アセチレンフレーム中に噴霧し,波長309.3nm
における吸光度を測定し,得た吸光度と酸化アルミニウム濃度との関係線を作成して検量線とする。
10. 酸化カルシウム
10.1 方法の区分 酸化カルシウムの定量方法は,EDTA滴定法による。
10.2 EDTA滴定法
10.2.1 要旨 8.2.3の試料溶液を分取し,トリエタノールアミン,硫化ナトリウムを加えて妨害イオンを
マスキングし,pHを約13に調節した後,カルセインを指示薬としてEDTA標準溶液で予備滴定する。次
に,再び8.2.3の試料溶液を分取し,妨害イオンをマスキングした後,予備滴定量よりも1〜2ml少ない量
のEDTA標準溶液を加え,水で薄める。水酸化カリウムを加えてpHを約13に調節した後,カルセインを
指示薬としてEDTA標準溶液で滴定する。
10.2.2 試薬 試薬は,次による。ただし,(1)(2)及び(4)は,プラスチック瓶に保存する。
(1) 水酸化カリウム溶液 水酸化カリウム250gを水に溶かして1lとする。
(2) 硫化ナトリウム溶液 硫化ナトリウム(九水和物)10gを水に溶かして100mlとする。使用の都度調
製することが望ましい。
(3) トリエタノールアミン (1+2)
(4) M/50エチレンジアミン四酢酸二ナトリウム (EDTA) 標準溶液 エチレンジアミン四酢酸二ナトリウ
ム(二水和物)7.5gを適量の水に溶かした後,水で1 000mlに薄める。この溶液の標定方法は,JIS K
8006(試薬の含量試験中滴定に関する基本事項)の2.(20)に従う。
(5) カルセイン指示薬 3,3'−ビス[N,N'-ジ(カルボキシメチル)アミノメチル]フルオレセイン0.1gを,
硫酸カリウム10gと粉砕混合する。
備考だけで使用する試薬
(6) 塩酸 (1+1)
(7) アンモニア水 (1+1)
(8) 臭素水
10.2.3 操作 定量操作は,次の手順によって行う。
(1) 8.2.3(3)の試料溶液から10mlをビーカー (300ml) に正確に分取し,水で約200mlに薄める。
(2) トリエタノールアミン (1+2) 5ml及び硫化ナトリウム溶液1mlを加えた後,水酸化カリウム溶液を加
10
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
えてpH12.7〜13.2になるように調節し,かき混ぜて2〜3分間放置する。
(3) カルセイン指示薬約0.05gを加え,溶液の蛍光性緑色が消え,だいだい色に変わるまでM/50EDTA標
準溶液で滴定する(14)。
(4) 次に,再び8.2.3(3)の試料溶液から10mlをビーカー (500ml) に正確に分取し,水で約300mlに薄める。
(5) トリエタノールアミン (1+2) 5ml及び硫化ナトリウム溶液1mlを加えた後,(3)で滴定したM/50EDTA
標準溶液の使用量より1〜2ml少ない量を加えてかき混ぜる。水酸化カリウム溶液を加えてpH12.7〜
13.2になるように調節し,かき混ぜて2〜3分間放置する。
(6) カルセイン指示薬約0.05gを加え,よくかき混ぜながらM/50EDTA標準溶液でゆっくり滴定し,蛍光
性緑色が消えてだいだい色になった点を終点とする(14)。
10.2.4 計算 試料中の酸化カルシウム含有率を,次の式によって算出する。
100
10
250
6
121
001
.0
(%)
1
×
×
×
×
=
W
F
υ
酸化カルシウム
ここに, v1: 10.2.3(5)及び(6)のM/50EDTA標準溶液使用量 (ml)
F: M/50EDTA標準溶液のファクター
W: 7.2.3の試料はかり取り量 (g)
注(14) 黒色紙又は黒色板上で行うと,終点が判別しやすい。
備考 試料中に酸化マンガン0.1%以上を含む場合は,次のように操作する。
8.2.3(3)の試料溶液から50mlをビーカー (200ml) に正確に分取し,臭素水5mlを加え,アン
モニア水 (1+1) を滴加して溶液を絶えずアルカリ性に保ちながら5分間以上煮沸する。沈殿
が凝集して溶液が澄明になった後,小形ろ紙(5種B)でろ過し,温水で十分に洗浄する。ろ
液及び洗液はビーカー (300ml) に受け,塩酸 (1+1) を加えて酸性とし,煮沸して過剰の臭素
を完全に追い出し,80ml以下になるまで加熱して蒸発を続ける。冷却後,100mlのメスフラス
コに移し,水で標線まで薄める。これから20mlをビーカー (300ml) に正確に分取し,水で約
200mlに薄め,以下,10.2.3(2)及び(3)の手順に従って操作する。次に,この溶液から20mlをビ
ーカー (500ml) に正確に分取し,水で約300mlに薄める。以下,10.2.3(5)及び(6)の手順に従っ
て操作する。
11. 酸化マグネシウム
11.1 方法の区分 酸化マグネシウムの定量方法は,EDTA滴定法による。
11.2 EDTA滴定法
11.2.1 要旨 8.2.3の試料溶液を分取し,塩酸ヒドロキシルアミンを加えて鉄を還元し,トリエタノール
アミン及び硫化ナトリウムを加えて妨害イオンをマスキングした後,緩衝溶液を加えてpHを約10に調節
する。EBTを指示薬としてEDTA標準溶液で酸化カルシウムと酸化マグネシウムの合量を滴定する。これ
から10.2で滴定した酸化カルシウムを差し引いて,酸化マグネシウムを求める。
11.2.2 試薬 試薬は,次による。
(1) 緩衝溶液 (pH10) 塩化アンモニウム70gを適量の水に溶かし,アンモニア水500mlを加えて水で1l
に薄める。プラスチック瓶に保存する。
(2) 塩酸ヒドロキシルアミン溶液 (5w/v%)
(3) エリオクロムブラックT (EBT) 溶液 調製方法は,JIS K 8006の3.による。
(4) その他の試薬は,10.2.2(2)〜(4)と同じものを用いる。
11
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.2.3 操作 定量操作は,次の手順によって行う。
(1) 8.2.3(3)の試料溶液から10mlをビーカー (300ml) に正確に分取し(15),水で約100mlに薄める。
(2) 塩酸ヒドロキシルアミン溶液5ml,トリエタノールアミン (1+2) 5ml及び硫化ナトリウム溶液1mlを
加え,次に緩衝溶液を加えてpH10.0±0.2に調節する。
(3) EBT溶液2,3滴を指示薬として加え,M/50EDTA標準溶液で滴定し,終点付近になったらよくかき
混ぜながらゆっくり滴定し,赤みが全く消えて鮮明な青色となった点を終点とする。
11.2.4 計算 試料中の酸化マグネシウム含有率を,次の式によって算出する。
100
10
250
1
806
000
.0
)
(
(%)
1
2
×
×
×
×
−
=
W
F
υ
υ
酸化マグネシウム
ここに, v1: 10.2.3(6)のM/50EDTA標準溶液使用量 (ml)
v2: 11.2.3(3)のM/50EDTA標準溶液使用量 (ml)
F: M/50EDTA標準溶液のファクター
W: 7.2.3の試料はかり取り量 (g)
注(15) 試料中に酸化マンガン0.1%以上を含む場合は,10.2.4の備考に従って操作した100mlメスフラス
コ中の溶液20mlを正確に分取し,水で約100mlに薄め,以下,11.2.3(2)及び(3)に従って操作す
る。
ただし,この場合は塩酸ヒドロキシルアミン溶液の添加を必要としない。
12. 五酸化りん
12.1 方法の区分 五酸化りんの定量方法は,次のいずれかによる。
(1) モリブデン青吸光光度法 この方法は,五酸化りん含有率1.000%未満の試料に適用する。
(2) りんバナドモリブデン酸吸光光度法 この方法は,五酸化りん含有率0.030%以上の試料に適用する。
12.2 モリブデン青吸光光度法
12.2.1 要旨 8.2.3の試料溶液の一部を分取し,水酸化ナトリウムと硫酸で酸濃度を調節した後,モリブ
デン酸アンモニウムとアスコルビン酸を加え加熱して呈色させ,吸光度を測定する。
12.2.2 試薬 試薬は,次による。
(1) 硫酸 (1+1)
(2) 水酸化ナトリウム溶液 (10w/v%) プラスチック瓶に保存する。
(3) モリブデン酸アンモニウム硫酸溶液 モリブデン酸アンモニウム(四水和物)2gを温水約20mlに溶
かし,必要ならばろ過し,硫酸 (1+1) 60mlを加えて水で100mlに薄める。
(4) L−アスコルビン酸溶液 (5w/v%) 7.3.2(6)と同じものを用いる。
(5) 標準五酸化りん溶液 (0.10mg P2O5/ml) りん酸二水素カリウムを105〜110℃で3時間乾燥し,デシ
ケーター中で放冷したもの0.192gをはかり取り,水に溶かして1 000mlのメスフラスコに移し,水で
標線まで薄める。
(6) p−ニトロフェノール溶液 (0.2w/v%)
12.2.3 操作 定量操作は,次の手順によって行う。
(1) 8.2.3(3)の試料溶液から一定量(16)を100mlのメスフラスコに正確に分取し,p−ニトロフェノール溶液
1滴を指示薬として加え,溶液が黄色になるまで水酸化ナトリウム溶液を滴加し,次に硫酸 (1+1) を
滴加して無色とした後,更に2,3滴過剰に加える。
(2) これにモリブデン酸アンモニウム硫酸溶液10ml及びアスコルビン酸溶液2mlを加え,水で標線まで
12
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
薄める。沸騰水浴中で15分間加熱した後,流水中で室温まで冷却する。
(3) この溶液の一部を光度計の吸収セルに取り,波長830nm付近で吸光度を測定する。
12.2.4 計算 12.2.5で作成した検量線から五酸化りん量を求め,試料中の五酸化りん含有率を,次の式に
よって算出する。
100
250
10
(%)
3
×
×
×
=
−
V
W
a
五酸化りん
ここに, a: 12.2.3(3)から得られた五酸化りん検出量 (mg)
W: 7.2.3の試料はかり取り量 (g)
V: 試料溶液の分取量 (ml)
12.2.5 検量線の作成 標準五酸化りん溶液を水で正しく10倍に薄め,その0〜20.0ml(五酸化りんとして
0〜0.20mg)を100mlのメスフラスコに段階的に取り,12.2.3(2)及び(3)の手順に従って操作し,得た吸光度
と五酸化りん量との関係線を作成して検量線とする。
注(16) 試料溶液の分取量は,試料中の五酸化りん含有率に応じて表4による。
表4 試料溶液の分取量
五酸化りん含有率 %
分取量 ml
0.200未満
25
0.200以上0.500未満
10
0.500以上
5
12.3 りんバナドモリブデン酸吸光光度法
12.3.1 要旨 試料を過塩素酸で分解し,加熱して白煙を発生させ,けい酸を不溶性としてろ過する。ろ液
にバナジン酸アンモニウム及びモリブデン酸アンモニウムを加えてりんバナドモリブデン酸を呈色させ,
吸光度を測定する。
12.3.2 試薬 試薬は,次による。
(1) 過塩素酸 (60%)
(2) モリブデン酸アンモニウム溶液 7.3.2(4)と同じものを用いる。
(3) バナジン酸アンモニウム溶液 バナジン酸アンモニウム5gを熱水500mlに溶かし,冷却後,過塩素
酸20mlを加えて水で1lに薄める。
(4) 標準五酸化りん溶液 (0.10mg P2O5/ml) 12.2.2(5)と同じものを用いる。
12.3.3 試料はかり取り量 試料のはかり取り量は,試料中の五酸化りん含有率に応じて表5による。
表5 試料はかり取り量
五酸化りん含有率 %
試料はかり取り量 g
0.500未満
1
0.500以上1.000未満
0.5
1.000以上2.000未満
0.25
12.3.4 操作 定量操作は,次の手順によって行う。
(1) 試料をビーカー (100ml) にはかり取り(5),少量の水を加えてスラリー状とし,時計皿で覆って過塩素
酸10mlをビーカーの縁から徐々に加える。激しい反応が終わったら,時計皿を少量の水で洗浄して
取り除く。
(2) 砂浴上で加熱し,過塩素酸の濃い白煙が出始めたら再び時計皿で覆い,引き続き10分間加熱する。放
冷後,水約30mlを加え,直ちにろ紙(5種B)でろ過し,水で数回洗浄する。ろ液及び洗液は100ml
のメスフラスコに受ける。
13
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) バナジン酸アンモニウム溶液5.0mlを加え,水で約80mlに薄めて振り混ぜる。これにモリブデン酸ア
ンモニウム溶液10mlを加え,水で標線まで薄め,30分間放置する。
(4) この溶液の一部を光度計の吸収セルに取り,波長460nm付近で吸光度を測定する。
12.3.5 計算 12.3.6で作成した検量線から五酸化りん量を求め,試料中の五酸化りん含有率を,次の式に
よって算出する。
100
10
(%)
3×
×
=
−
W
a
五酸化りん
ここに,
a: 12.3.4(4)から得られた五酸化りん検出量 (mg)
W: 試料はかり取り量 (g)
12.3.6 検量線の作成 標準五酸化りん溶液0〜50.0ml(五酸化りんとして0〜5.0mg)を100mlのメスフラ
スコに段階的に取り,過塩素酸8mlを加え,12.3.4(3)及び(4)の手順に従って操作し,得た吸光度と五酸化
りん量との関係線を作成して検量線とする。
13. 全硫黄
13.1 方法の区分 全硫黄の定量方法は,次のいずれかによる。
(1) 熱分解−よう素酸カリウム滴定法 この方法は,全硫黄含有率0.100%未満の試料に適用する。
(2) 硫酸バリウム重量法 この方法は,全硫黄含有率0.01%以上の試料に適用する。
13.2 熱分解−よう素酸カリウム滴定法
13.2.1 要旨 試料を三酸化タングステンと混合し,窒素気流中で高温に加熱して硫黄を二酸化硫黄とする。
これをよう化カリウム及びでんぷんを含む塩酸吸収液に吸収させ,よう素酸カリウム標準溶液で滴定する。
13.2.2 試薬 試薬は,次による。
(1) 硫酸
(2) 過塩素酸マグネシウム 粒径0.5〜2mmのものを用いる。
(3) 三酸化タングステン 粉末状のものを用いる。
(4) ソーダ石灰 粒径3〜4mmのものを用いる。
(5) クロム酸飽和硫酸 硫酸に重クロム酸カリウムを飽和させる。
(6) 塩酸吸収液 塩酸 (1.5+98.5) 80ml,よう化カリウム溶液 (3w/v%) 1ml及びでんぷん溶液[でんぷん(溶
性)2gに水10mlを加え,十分に振り混ぜ,約50mlの熱水中に注ぎ入れ,約1分間煮沸した後冷却し,
水で100mlに薄める。この溶液は,使用の都度調製する]1mlを混ぜ合わせる。
(7) よう素酸カリウム標準溶液 よう素酸カリウム[JIS K 8005(容量分析用標準試薬)]0.222 5gを水に
溶かし,1 000mlのメスフラスコに移し,水で標線まで薄める。
(8) 窒素ガス(99.5%以上)
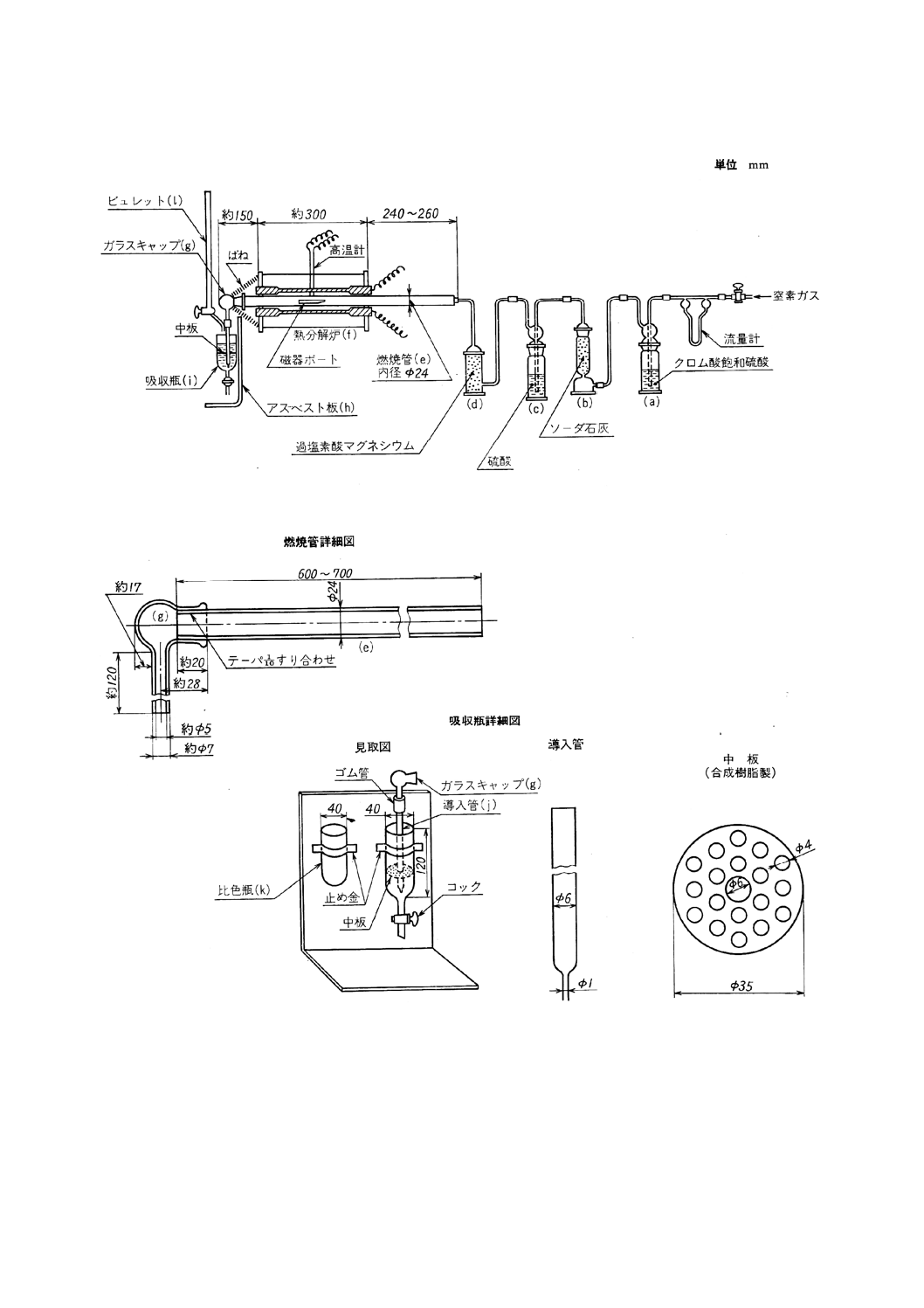
13.2.3 装置,器具及び材料 装置,器具及び材料は,原則として図のものを用いる。
(1) 窒素清浄装置 この装置は,窒素ガスを清浄,乾燥させるためのもので,それぞれ洗気瓶又は洗浄塔
にクロム酸飽和硫酸 (a),ソーダ石灰 (b),硫酸 (c) 及び過塩素酸マグネシウム (d) を詰めたものを
用いる(17)。
(2) 熱分解炉 熱分解炉 (f) は,原則として次のものを用いる。
管状電気炉は,長さ約300mmで,電気抵抗加熱体を用いて加熱し,電流を調節して温度を加減し,
炉の中央部において長さ約150mmの部分を1 250±50℃の一定温度に保つことができるようにする。
炉内には,長さ600〜700mm,内径24mmで,1250±50℃に耐える磁器燃焼管 (e) [JIS R 1307(化
14
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
学分析用磁器燃焼管)のCT0又はCT1]を挿入し,燃焼管の出口部は炉壁から100〜150mm突き出さ
せる。また,出口部にテーパーを付けて,すり合わせガラスキャップ (g) をはめ,ばねで炉壁に締め
付ける。更にガラスキャップと炉壁の間にアスベスト板 (h) を置き,炉体からの熱が吸収瓶に当たら
ないようにする。
炉の中央部の燃焼管の直上の温度を熱電対高温度計で測定する。熱電対高温度計の指示値は,一般
に燃焼管内の温度と異なるので,その差を求めておき,必要があれば指示値を補正して燃焼管内の温
度を求める。
燃焼管と窒素清浄装置との連結は,耐熱性けい素樹脂製栓を用いる。なお,新しい磁器燃焼管を使
用するときは,あらかじめ1 250±50℃で30分間以上窒素気流中で空焼きを行う。
15
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図 硫黄定量装置

16
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 吸収瓶 吸収瓶 (i) には,塩酸吸収液約80mlを入れ,塩酸吸収液が微青色を呈するまで,よう素酸カ
リウム標準溶液を滴加しておく。ガラスキャップの先端に,内径6mmで先端を内径1mmに絞ったガ
ラス製導入管 (j) を吸収瓶の最下部に達するように取り付け,さらに,塩酸吸収液面下約15mmの位
置に,合成樹脂板に多くの小穴を開けた中板を取り付ける(18)。
(4) 比色瓶 比色瓶 (k) には,塩酸吸収液約80mlを入れ,塩酸吸収液が微青色を呈するまで,よう素酸
カリウム標準溶液を滴加しておく。
(5) ビュレット ビュレット (1) は,原則として容量25mlのものを用いる。ただし,全硫黄含有率が
0.010%未満の場合は,最小目盛が0.05mlのセミミクロビュレット又はそれ以上の滴加精度を有する滴
定器具を用いる。なお,ビュレットの先端部は,吸収瓶中において滴定しやすいように細工しておく。
(6) 磁器燃焼ボート及び磁器燃焼ボートカバー(以下,単にボート及びカバーという。) ボートは,JIS R
1306(化学分析用磁器燃焼ボート)のCB1又はCB2を用い,必要に応じてカバーを使用する。ボー
ト及びカバーは,あらかじめ窒素気流中で1 250±50℃で10分間強熱後,デシケーター中で保存した
ものを用いる(19)。
13.2.4 試料はかり取り量 試料はかり取り量は,試料中の全硫黄含有率に応じて表6による。
表6 試料はかり取り量
全硫黄含有率%
試料はかり取り量g
0.050未満
1.00
0.050以上0.100未満
0.50
13.2.5 操作 定量操作は,次の手順によって行う。
(1) 装置を気密に連結し,燃焼管 (e) を加熱して管内温度を1 250±50℃とする。
(2) 試料をはかり取ってはかり瓶に入れ,三酸化タングステン1gを加えて十分混合した後,ボートに移し
入れ,必要ならばカバーで覆い,挿入棒で燃焼管の加熱部の中央に送入して気密に栓をする。
(3) 直ちに,窒素ガスを毎分150〜200ml(20)の割合で送入し,発生した二酸化硫黄を窒素ガスと共に吸収
瓶 (i) に導き,よう素酸カリウム標準溶液を滴加して塩酸吸収液を呈色させ,二酸化硫黄の流入によ
って青色が消失しないように絶えず滴加を続ける。青色の消失がゆるやかになったら滴加量を調節し,
比色瓶 (k) の呈色と対照しながら,塩酸吸収液がわずかに青色を呈するまで滴定し,終点とする。
(4) 三酸化タングステン1gだけを用いて(1)以降の操作を行い,空試験値を求める。
13.2.6 計算 試料中の全硫黄含有率を,次の式によって算出する。
100
1
000
.0
)
(
(%)
1
2
×
×
−
=
W
υ
υ
全硫黄
ここに, v1: 13.2.5(3)のよう素酸カリウム標準溶液使用量 (ml)
v2: 13.2.5(4)のよう素酸カリウム標準溶液使用量 (ml)
W: 試料はかり取り量 (g)
注(17) 窒素ガスが高純度の場合には,過塩素酸マグネシウムとソーダ石灰を詰めた洗浄塔だけを使用
してもよい。
(18) 内径6mmで先端に径約1mmの小穴を開けたガラス製導入管を吸収瓶の最下部に達するように
取り付け,導入管の先端から塩酸吸収液面までの高さを60〜80mmとしてもよい。
(19) デシケーターからの出し入れは,ピンセットなどで扱い,直接手を触れてはならない。長時間
保存したものは,空試験値が高くなるおそれがあるため,再度空焼きを行ってから使用する。
(20) 全硫黄含有率の高い試料の場合は,窒素ガス送入量を毎分150mlにする。
17
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.3 硫酸バリウム重量法
13.3.1 要旨 試料に臭素水及び塩酸を加えて分解し,加熱して過剰の臭素を除去する。不溶解物をろ過し,
酸濃度を調節した後,塩酸ヒドロキシルアミンで鉄を還元する。熱時に塩化バリウムを加えて硫酸バリウ
ムの沈殿を生成させ,熟成後,沈殿をこし分け,強熱して質量をはかる。
13.3.2 試薬 試薬は,次による。
(1) 塩酸 (1+1)
(2) アンモニア水 (1+1)
(3) 塩化バリウム溶液 塩化バリウム(二水和物)10gを水に溶かし,100mlとする。
(4) 塩酸ヒドロキシルアミン溶液 11.2.2(2)と同じものを用いる。
(5) 臭素水(飽和約4%)
(6) メチルレッド溶液 メチルレッド0.2gをエタノール (95) 90mlに溶かし,水で100mlに薄める。
13.3.3 試料はかり取り量 試料は,5.0gをはかり取る。
13.3.4 操作 定量操作は,次の手順によって行う。
(1) 試料をビーカー (200ml) にはかり取り,少量の水を加えてスラリー状とし,臭素水10mlを加え,時
計皿で覆い,塩酸 (1+1) 25mlをビーカーの縁から少量ずつ加える。激しい反応が終わったら時計皿
で覆ったまま加熱し,約10分間穏やかに煮沸する。
(2) 時計皿を水洗して取り除き,ろ紙(6種)でろ過し,温水で数回洗浄する。ろ液及び洗液は,ビーカ
ー (500ml) に受ける。
(3) メチルレッド溶液2,3滴を指示薬として加え(21),溶液の色が黄色になるまでアンモニア水 (1+1) を
滴加する。直ちに,塩酸 (1+1) 3mlを加えた後,塩酸ヒドロキシルアミン溶液5〜10mlを加え数分間
煮沸する。
(4) 液量を水で約300mlに薄め,沸騰寸前まで加熱して熱源から降ろし,溶液をガラス棒で激しくかき混
ぜながら熱塩化バリウム溶液10mlを滴加する。時計皿で覆って,水浴上で2時間以上加熱して沈殿
を熟成させる(22)。
(5) 沈殿をろ紙(5種C)でこし分け,初めは塩酸 (1+100) で数回,次に水で,洗液に塩化物イオンが認
められなくなるまで洗浄する。
(6) 沈殿はろ紙と共に白金るつぼ(例えば30番)(3)に入れ,初めは低温で加熱し,次第に温度を上げてろ
紙を灰化する。700〜800℃で30分間強熱し,デシケーター中で放冷後,沈殿の質量をはかる。
13.3.5 計算 試料中の全硫黄含有率を,次の式によって算出する。
100
4
137
.0
)
(
(%)
2
1
×
×
−
=
W
w
w
全硫黄
ここに, w1: 硫酸バリウムの入っている白金るつぼの質量 (g)
w2: 白金るつぼの質量 (g)
W: 試料はかり取り量 (g)
注(21) この際,メチルレッドによる赤みが退色するような場合は,臭素が残留しているので,時計皿
で覆って再び加熱し,約10分間穏やかに煮沸する。
(22) 沈殿が少量の場合は,更に常温で一夜間放置する。
1
8
M
8
8
5
1
-1
9
8
3
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
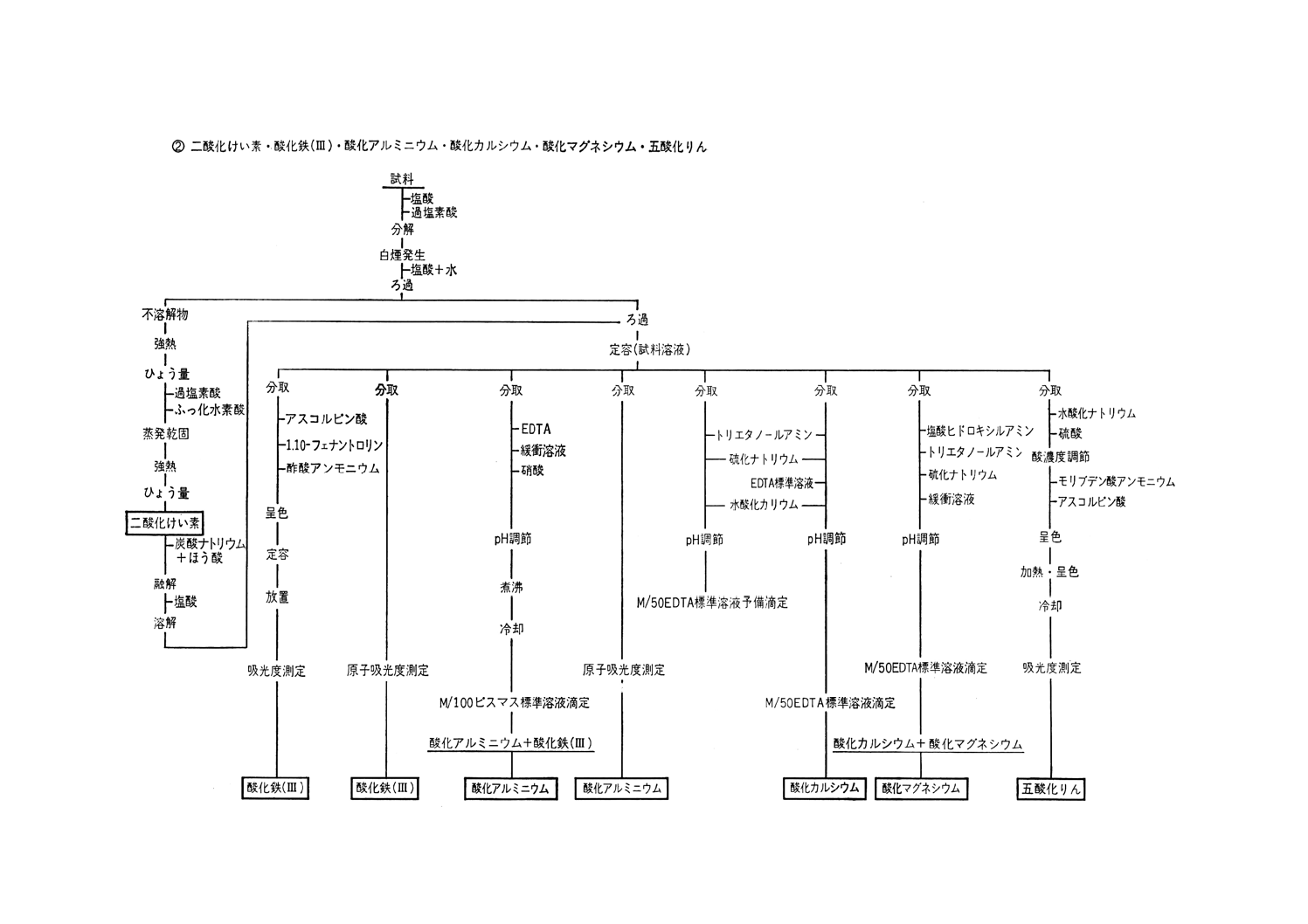
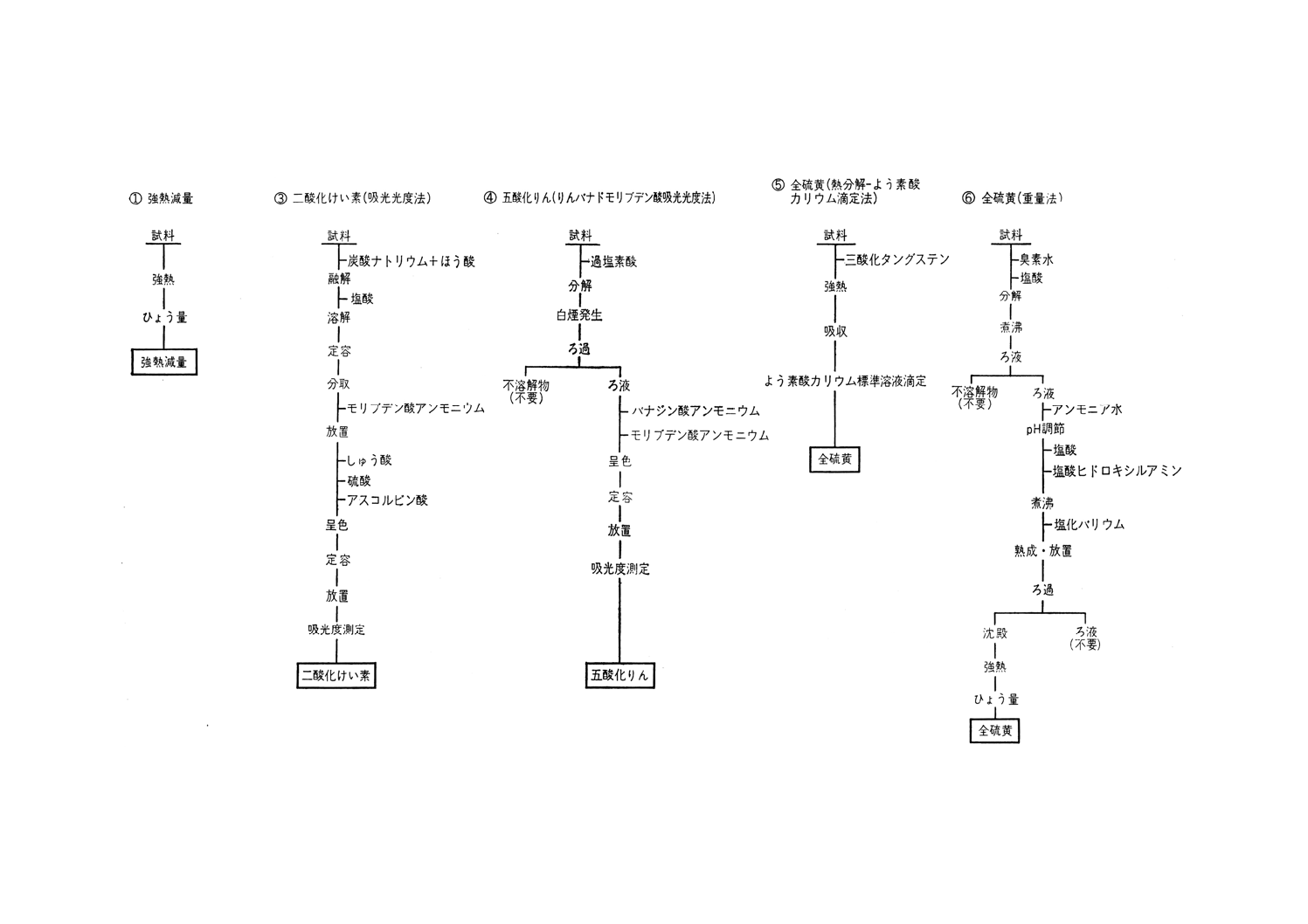
参考 分析系統図
1
9
M
8
8
5
1
-1
9
8
3
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考 分析系統図(続き)
20
M 8851-1983
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ドロマイト分析方法改正原案作成委員会構成表(50音順)
氏名
所属
(委員長)
藤 貫 正
工業技術院地質調査所
池 永 治 郎
三菱鉱業セメント株式会社中央研究所
内 田 清 彦
住友セメント株式会社中央研究所
片 柳 仁
吉澤石灰工業株式会社
加 藤 修
社団法人セメント協会研究所
神 宮 滋
社団法人日本鉄鋼連盟技術管理部
佐 伯 正 夫
新日本製鐵株式会社基礎研究所
富 田 欽 三
日本石灰協会
針間矢 宣 一
川崎製鐵株式会社技術研究所
平 野 裕 市
九州耐火煉瓦株式会社
布 施 美智雄
旭硝子株式会社研究開発部
船 戸 己知雄
日本セメント株式会社中央研究所
丸 田 俊 久
秩父セメント株式会社埼玉事業所
室 屋 千 裕
川崎鉱業株式会社研究室
森 山 恒 雄
日鉄鉱業株式会社技術開発部
吉 田 信 之
工業技術院標準部
渡 辺 彦 祐
石灰石鉱業協会技術部
(審議参加者)
伊 藤 辰 雄
新日本製鐵株式会社堺製鐵所
加 藤 昌 宏
住友セメント株式会社中央研究所
古谷田 武 満
秩父セメント株式会社試験研修センター
中 井 正
川崎製鐵株式会社技術研究所