M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,社団法人日本鉄鋼
連盟(JISF)から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,日本工業標準調査
会の審議を経て,経済産業大臣が改正した日本工業規格である。
これによって,JIS M 8514:1995は改正され,この規格に置き換えられる。
改正に当たっては,日本工業規格と国際規格との対比,国際規格に一致した日本工業規格の作成及び日本
工業規格を基礎にした国際規格原案の提案を容易にするために,ISO 9438:1993,Metallurgical-grade
fluorspar - Determination of total phosphorus content - Reduced-molybdophosphate spectrometric
method,ISO 9503:1991,Metallurgical - grade fluorspar - Determination of available fluorspar content
- Modified Willard-Winter method,ISO 4283:1993,All grades of fluorspar - Determination of
carbonate content - Titrimetric method,ISO 9501:1991,Metallurgical - grade fluorspar -
Determination of total sulfur content - Iodometric method after combustion,ISO 9502:1993,
Metallurgical - grade fluorspar - Determination of silica content - Reducedmolybdosilicate
spectrometric method,ISO 9504:1993,Metallurgical - grade flourspar - Determination of antimony
content - Solvent extraction atomic absorption spectrometric method,ISO 9505:1992,All grades of
flourspar - Determination of arsenic content - Silver diethyldithiocarbamate spectrometric method及
びISO 9779:1993,Metallurgical - grade flourspar - Determination of lead content - Solvent extraction
atomic absorption spectrometric methodを基礎として用いた。
JIS M 8514には,次に示す附属書がある。
附属書1(規定)ふっ素定量法−けいふっ化水素酸蒸留分離−硝酸トリウム滴定法
附属書2(規定)ふっ素定量法−塩ふっ化鉛沈殿分離−エチレンジアミン四酢酸ニ水素ナトリウム間
接滴定法
附属書3(規定)けい素定量法−モリブドけい酸青吸光光度法
附属書4(規定)炭酸カルシウム定量方法−二酸化炭素気化分離-塩酸・水酸化ナトリウム滴定法
附属書5(規定)りん定量方法−水酸化鉄共沈分離-モリブドりん酸青吸光光度法
附属書6(規定)硫黄定量方法−熱分解-よう素酸カリウム滴定法
附属書7(規定)ひ素定量方法−三水素化ひ素気化分離-ジエチルジチオカルバミン酸銀吸光光度法
附属書8(規定)鉛定量方法−よう化物抽出原子吸光法
附属書9(規定)アンチモン定量方法−よう化物抽出原子吸光法
附属書10(参考)JISと対応する国際規格との対比表

1
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
M 8514:2003
鉄鋼用ほたる石−分析方法
Metallurgical grade fluorspar - Methods for chemical analysis
序文 この規格は,1991年に第1版として発行されたISO 9438:1993,Metallurgical-grade fluorspar -
Determination of total phosphorus content - Reduced-molybdophosphate spectrometric method,ISO 9503:1991,
Metallurgical - grade fluorspar - Determination of available fluorspar content - Modified Willard-Winter method,
ISO 4283:1993,All grades of fluorspar - Determination of carbonate content - Titrimetric method,ISO 9501:1991,
Metallurgical - grade fluorspar - Determination of total sulfur content - Iodometric method after combustion,ISO
9502:1993,Metallurgical - grade fluorspar - Determination of silica content - Reducedmolybdosilicate spectrometric
method,ISO 9504:1993,Metallurgical - grade flourspar - Determination of antimony content - Solvent extraction
atomic absorption spectrometric method,ISO 9505:1992,All grades of flourspar - Determination of arsenic content
- Silver diethyldithiocarbamate spectrometric method及びISO 9779:1993,Metallurgical - grade flourspar -
Determination of lead content - Solvent extraction atomic absorption spectrometric methodを翻訳し,技術的内容
を変更して作成した日本工業規格である。
なお,この規格で側線又は点線の下線を施してある箇所は,原国際規格を変更している事項である。変
更の一覧表をその説明を付けて,附属書10(参考)に示す。
1. 適用範囲 この、規格は、鉄鋼用ほたる石の分析方法について規定する。
備考 この規格の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide21に基づき,IDT(一致している),MOD(修
正している),NEQ(同等でない)とする。
ISO 9438:1993,Metallurgical-grade fluorspar - Determination of total phosphorus content - Reduced-
molybdophosphate spectrometric method(MOD)
ISO 9503:1991,Metallurgical - grade fluorspar - Determination of available fluorspar content -
Modified Willard-Winter method (MOD)
ISO 4283:1993,All grades of fluorspar - Determination of carbonate content - Titrimetric method
(MOD)
ISO 9501:1991,Metallurgical - grade fluorspar - Determination of total sulfur content - Iodometric
method after combustion (MOD)
ISO 9502:1993,Metallurgical - grade fluorspar - Determination of silica content -
Reducedmolybdosilicate spectrometric method (MOD)
ISO 9504:1993,Metallurgical - grade flourspar - Determination of antimony content - Solvent
extraction atomic absorption spectrometric method (MOD)
ISO 9505:1992,All grades of flourspar - Determination of arsenic content - Silver

2
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
diethyldithiocarbamate spectrometric method (MOD)
ISO 9779:1993,Metallurgical - grade flourspar - Determination of lead content - Solvent extraction
atomic absorption spectrometric method (MOD)
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0121 原子吸光分析通則
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS R 1306 化学分析用磁器燃焼ボート
JIS R 1307 化学分析用磁器燃焼管
JIS Z 8401 数値の丸め方
JIS Z 8402-1 測定方法及び測定結果の精確さ(真度及び精度)−第1部:一般的な原理及び定義
JIS Z 8402-2 測定方法及び測定結果の精確さ(真度及び精度)−第2部:標準測定方法の併行精度及
び再現精度を求めるための基本的方法
JIS Z 8402-3 測定方法及び測定結果の精確さ(真度及び精度)−第3部:標準測定方法の中間精度
JIS Z 8402-4 測定方法及び測定結果の精確さ(真度及び精度)−第4部:標準測定方法の真度を求め
るための基本的方法
JIS Z 8402-6 測定方法及び測定結果の精確さ(真度及び精度)−第6部:精確さに関する値の実用的
な使い方
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
3. 定義 この規格で用いる主な用語の定義は,JIS K 0050、JIS K 0115、JIS K 0121及びJIS Z 8402-1に
よるほか,次による。
a) 分取 ピペットを用いて正確に分液すること。
b) 正確にVmlを加える ビュレットやピペットを用いてVmlを加えること。
c) Vmlを加える メスシリンダーなどを用いてVmlを加えること。
d) 約Vmlを加える 使用する器具にあらかじめ目盛付きシリンダなどで計量した溶液を入れてみて,同
じ量を目分量で判断して加えること。
e) PTFE 四ふっ化エチレン樹脂
4. 一般事項 定量方法に共通な一般事項は,JIS K 0050, JIS K 0115,JIS K 0121,JISZ 8402-1〜JIS Z
8402-4及びJIS Z 8402-6による。
5. 定量方法の区分 鉄鋼用ほたる石中の各種成分の定量方法は,表1による。

3
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1 鉄鋼用ほたる石中の各種成分の定量方法
成分
定量方法
適用含有率範囲
%(m/m)
附属書
番号
ふっ素
ヘキサフルオロけい酸蒸留分離−硝酸トリウム滴
定法
30 以上 48 以下
1
塩化ふっ化鉛沈殿分離−エチレンジアミン四
酢酸二水素ニナトリウム間接滴定法(2)
30 以上 48 以下
2
けい素
モリブドけい酸青吸光光度法
0.09 以上 14 以下
3
炭酸カルシウム
(1)
二酸化炭素気化分離−塩酸・水酸化ナトリ
ウム滴定法
0.1 以上 10 以下
4
りん
水酸化鉄共沈分離−モリブドりん酸青吸
光光度法
0.002 以上 0.07以下
5
硫黄
熱分解−よう素酸カリウム滴定法
0.01 以上 1 以下
6
ひ素
三水素化ひ素気化分離−ジエチルジチオカル
バミン酸銀吸光光度法
0.0002以上 0.02 以下
7
鉛
よう化物抽出原子吸光法
0.0003以上 0.01 以下
8
アンチモン
よう化物抽出原子吸光法
0.001 以上 0.04 以下
9
注(1) 炭酸カルシウムは,二酸化炭素の定量を行い,換算する。
注(2) アルミニウム0.4%(m/m)以上及び/又は鉄0.6%(m/m)以上を含有する試料には適用しない。
6. 分析試料の採り方及び取扱い方
6.1
分析用原試料 受渡当事者間の協定による方法によって採取調製したもので,JIS Z 8801-1の網ふる
い160μmを全量通過するように粉砕したものを分析用原試料とする。
6.2
分析用試料 分析用原試料を十分に混ぜた後,その約40gを磁器平底蒸発皿などに薄く広げて105
〜110℃の空気浴中で2時間乾燥し,デシケーター中で室温まで放冷して分析用試料とする。
6.3
分析試料のはかり方 分析試料は,分析用試料から化学はかりを用い,各定量方法に規定してある
はかりとり量を0.1mgのけたまで正確にはかりとる。
7. 分析値のまとめ方
7.1
分析回数 分析は,同一分析用試料について室内再現条件(3)で2回行う。
注(3) 同一の分析室における同一試料の分析において,人・日時・装置の一部又はすべてが異なって
いる分析条件。
7.2
分析値の表示 分析値は,はかりとった試料の質量に対する質量百分率で表示し,分析成分によっ
て次に規定する表示の最下位に,JIS Z 8401によって丸める。
a) 小数点以下2けたまで表示する成分 ふっ素,けい素及び炭酸カルシウム。
b) 小数点以下3けたまで表示する成分 りん,硫黄,アンチモン及び含有率が0.001%(m/m)以上のひ素
並びに鉛。
c) 小数点以下4けたまで表示する成分 含有率が0.001%(m/m)未満のひ素及び鉛。
7.3
分析値の精確さの検討 分析値の精確さの検討は,次によって行う。
a) 真度の検討 分析試料と化学的特性とが近似している1個の認証標準物質(4)を分析試料と併行分析
4
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
し,得られた認証標準物質の分析結果と認証値との差の絶対値が,その定量方法規格の対標準物質許
容差(5)を超えなければ,同時に分析して得られた分析試料の分析値の真度は満足できるものと判断す
る。
注(4) 併行分析する認証標準物質は,試料はかりとり量及び定量操作が分析試料と全く同一になるも
のを選ぶ。
(5) 対標準物質許容差は,室間再現許容差計算式のf(n)の代わりに2.0を成分含有率の項に認証値を
入れて求める。
b) 中間精度(室内精度)の検討 同一分析室において,同一分析試料を室内再現条件(3)で2回分析して
得られた2個の分析結果の範囲が,その定量方法規格に規定されている室内再現許容差(6)を超えなけ
れば,この2個の分析結果の間に異常な差はないものと判断する。
注(6) 各定量方法に規定してある室内再現許容差及び室間再現許容差計算式のf(n)の値は,JIS Z
8402-6の表1(許容範囲の係数)による。
参考 n=2では,2.8である。
c) 室間精度の検討 二つの異なる分析室において,同一分析用試料をそれぞれ室内再現条件で2回分析
し,7.4によって得られた各分析室における分析結果の差の絶対値が,その定量方法に規定されてい
る室間再現許容差(6)を超えなければ,この二つの分析室における分析結果の間に異常な差はないもの
と判断する。
7.4
分析値の選択 分析値の選択は,次のいずれかによって行う。
a) 認証標準物質がある場合
1) 分析用試料の再現条件2回の分析結果が7.3b)を,それと併行して行われた認証標準物質の分析結果
が7.3a)を,いずれも満足すれば,この2個の分析結果を平均する。
2) 認証標準物質の分析結果が,7.3a)を満足しない場合は,それと同時に行われた分析試料の分析結果
を捨てて,改めて分析をやり直す。
3) 認証標準物質の分析結果は,7.3a)を満足するが,分析用試料の室内再現条件2回の分析結果が7.3b)
を満足しない場合は,改めて再現条件で2回の分析をやり直し(7),次によって処理する。
注(7) このとき,認証標準物質の分析結果は,7.3a)を満足しなければならない。
3.1) やり直しの結果が7.3b)を満足すれば,やり直しの結果を平均する。最初の分析結果は捨てる。
3.2) やり直しの結果も7.3b)を満足しなければ,最初の2回の分析結果とやり直しの2回の分析結果と
の合計4個のメジアン(8)とする。
注(8) 4個の分析値を大きさの順に並べたときの中央2個の平均値。
b) 認証標準物質がない場合
1) 分析用試料の室内再現条件2回の分析結果が,7.3b)を満足すれば,この2個の分析結果を平均する。
2) 分析用試料の室内再現条件2回の分析結果が,7.3b)を満足しなければ,改めて室内再現条件で2回
の分析をやり直し,a)3)の3.1)又は3.2)によって処理する。
5
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(規定)ふっ素定量法−ヘキサフルオロけい酸蒸留分離
−硝酸トリウム滴定法
1. 要旨 試料に過塩素酸を加えて水蒸気蒸留を行い,ふっ素をヘキサフルオロけい酸として分離する。
緩衝溶液で溶液のpHを調節し,メチルチモールブルーを指示薬として硝酸トリウム標準溶液で滴定する。
2. 試薬 試薬は,次による。
a) 過塩素酸
b) 緩衝溶液(pH3.5) モノクロロ酢酸94.5g及びモノクロロ酢酸ナトリウム207.4gを水に溶解し,1Lとす
る。
c) 標準ふっ素液(10 mgF/ml) 500℃で恒量とし,デシケーター中で常温になるまで放冷したふっ化ナト
リウム22.104gをはかりとって,水に溶解し,溶液を1000mlの全量フラスコに水を用いて移し入れ,
水で標線まで薄めて標準ふっ素液とする。この溶液は,ポリエチレン瓶に移して保存する。
d) 0.025 mol/L硝酸トリウム標準溶液 硝酸トリウム四水和物13.803gをはかりとって,水に溶解し,溶
液を1000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。この溶液のファクターは,
次によって求める。
標準ふっ素液[c) ]を正確に10倍に薄め,この溶液50mlをビーカー(300ml)に正確に取り,水を加
えて液量を約200mlとする。これに緩衝溶液[b) ]5mlを加えた後,メチルチモールブルー溶液を指示
薬として0.025 mol/L硝酸トリウム標準溶液で滴定し,溶液の色が青に変った点を終点とし、
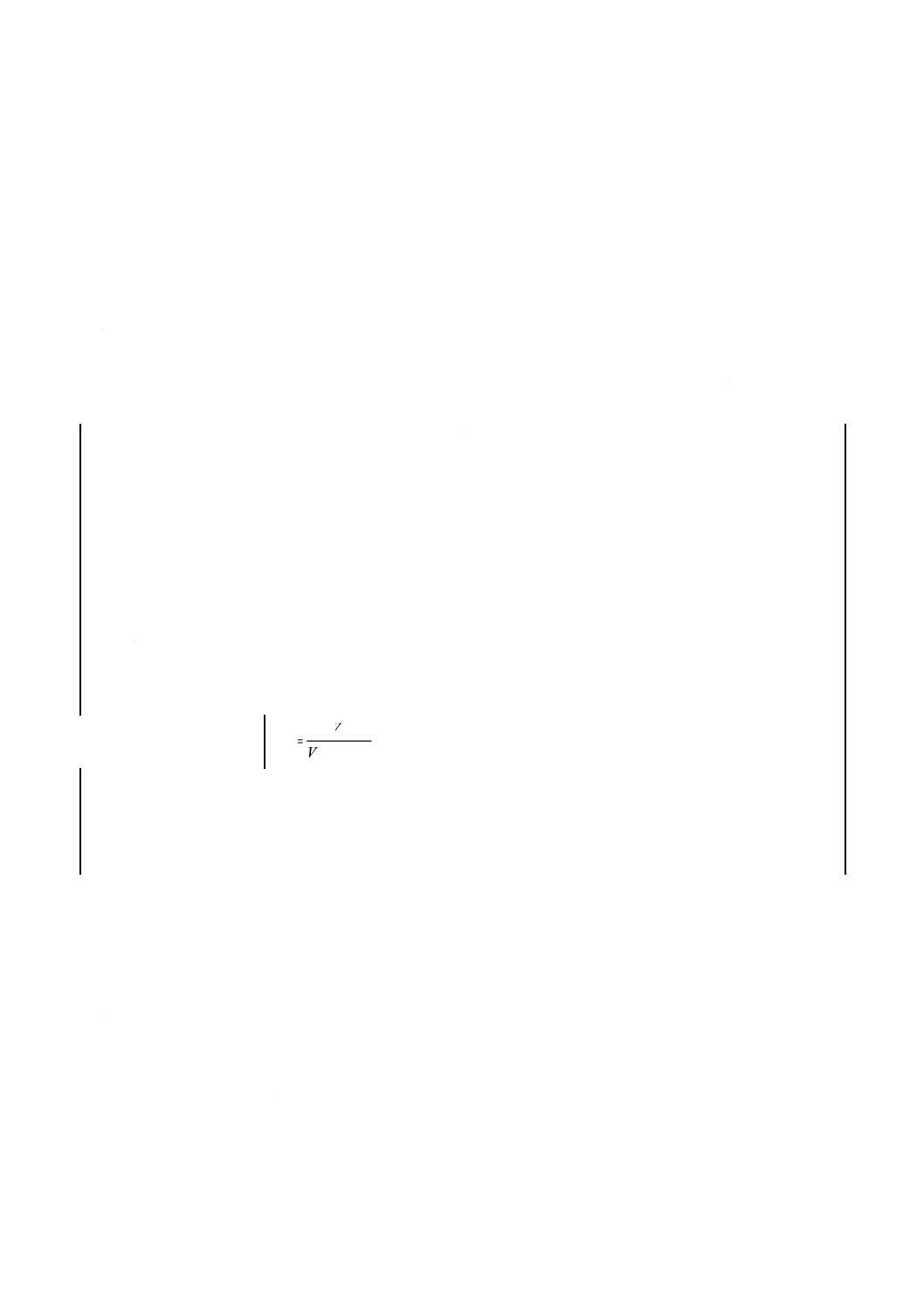
0.025mol/L硝酸トリウム標準溶液のファクターを次の式によって算出する。
1.900
2
1
×
=VV
f
ここに,f:0.025mol/L硝酸トリウム標準溶液のファクター
V1:10倍に薄めた標準ふっ素液の使用量(ml)
V2:0.025 mol/L硝酸トリウム標準溶液の使用量(ml)
e) メチルチモールブルー溶液(10g/L)
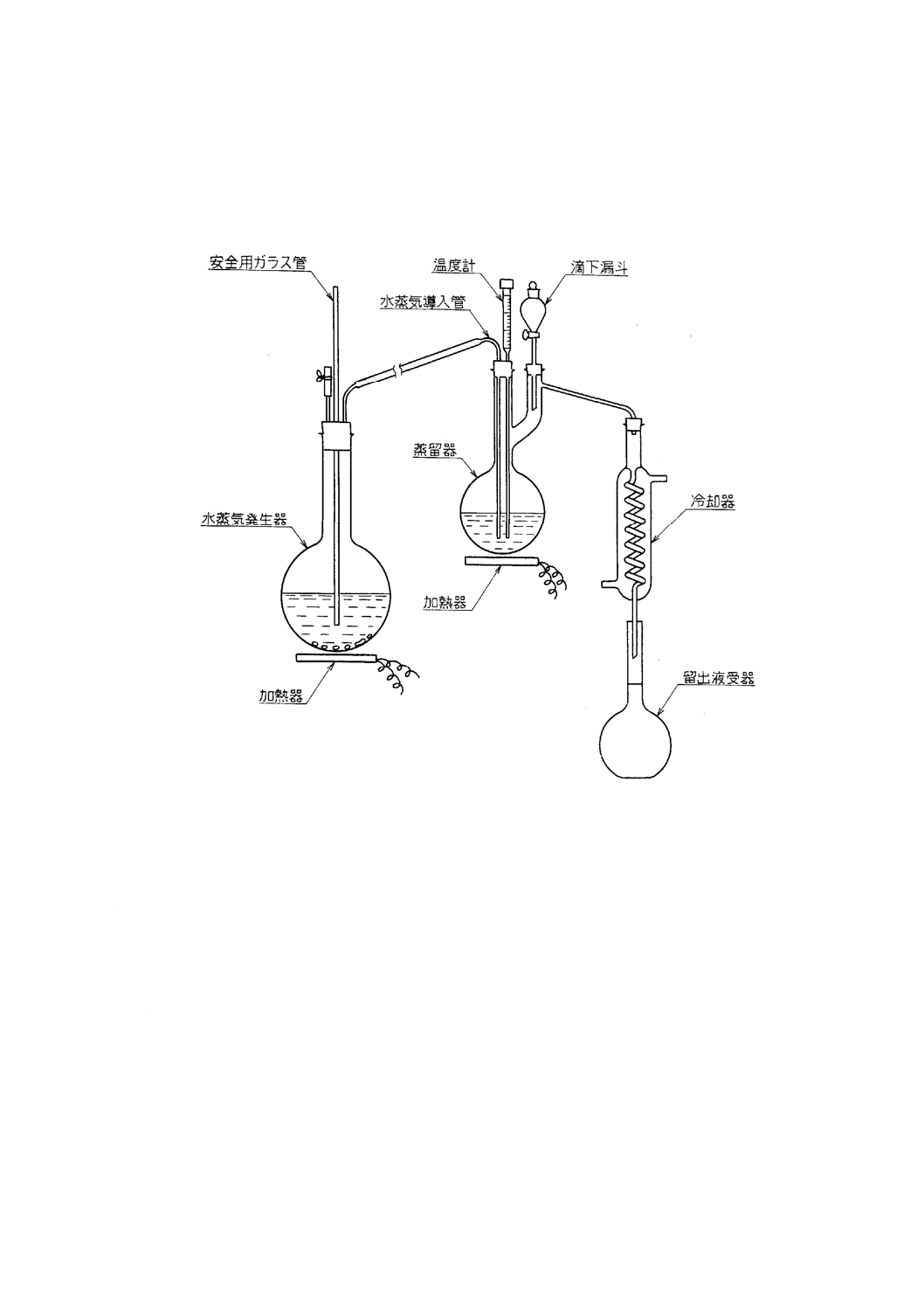
3. 装置及び器具 ふっ素蒸留装置は,一般に次のものを用いる(附属書1図1参照)。
a) 水蒸気発生器 丸底フラスコ(3000〜5000ml)に水及び沸騰石を入れたもの。
b) 蒸留器 主管には水蒸気導入管及び温度計を,側管には滴下漏斗を取り付けた250mlフラスコ。
c) 冷却器 長さ300mlの蛇管冷却器。
d) 加熱器
4. 試料はかりとり量 試料はかりとり量は,0.20gとする。
6
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1 図1 ふっ素蒸留装置の例
5. 操作
5.1 蒸留分離 試料をはかりとって蒸留器へ少量の水で洗い入れ,沸騰石を数個加えた後,更に過塩素酸
35mlを加える。冷却器に冷却水を通じた後,蒸留器を加熱する。蒸留器の温度が140℃になったら,水蒸
気発生器内で発生している水蒸気を蒸留器に導入する(1)。留出液量が毎分4ml程度の速度で60分間蒸留
を続ける。蒸留が終了した後,冷却器内を少量の水で洗浄し留出液と合わせる。
注(1) あらかじめ水蒸気発生器を加熱し,水蒸気を発生させておく。ただし,蒸留器内の温度が140℃
になるまでこの水蒸気を蒸留器内に導入してはならない。
5.2 滴定 5.1で得た溶液に緩衝溶液[2.b]]5mlを加え,更にメチルチモールブルー溶液数滴を指示薬とし
て加え,0.025 mol/L硝酸トリウム標準溶液[2.d]]で滴定し,溶液の色が青になった点を終点とし, 0.025mol/L
硝酸トリウム標準溶液の使用量を求める。

7
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6. 蒸留効率の測定 標準ふっ素液[2.c]]5mlを正確に蒸留器に取り,沸騰石を数個加えた後,更に過塩素
酸35mlを加える。冷却器に冷却水を通じた後,蒸留器を加熱する。蒸留器内の温度が140℃になったら水
蒸気発生器内で発生している水蒸気を蒸留器に導入する(1)。留出液量が毎分4ml程度の速度で60分間蒸
留を続ける。蒸留が終了した後,冷却器内を少量の水で洗浄し留出液と合わせる。以下,5.2の手順に従
って操作する。蒸留効率を次の式によって算出する。
50
1.900
×
×
=
f
R
3
V
ここに,R:ふっ素の蒸留効率
V3:0.025 mol/L硝酸トリウム標準溶液の使用量(ml)
f:0.025mol/L硝酸トリウム標準溶液のファクター
7. 空試験 試料を用いないで5.の全操作を行い,空試験値を求める。
8. 計算
8.1
ふっ素含有率の計算 5.2及び7.で得た滴定量から,試料中のふっ素含有率を次の式によって算出す
る。
(
)
R
m
f
V
V
×
×
×
−
=
001900
.0
5
4
F
ここに, F:試料中のふっ素含有率[%(m/m)]
V4:試料溶液の滴定における0.025 mol/L硝酸トリウム標準溶液の使用量(ml)
V5:空試験溶液の滴定における0.025 mol/L硝酸トリウム標準溶液の使用量(ml)
f:0.025mol/L硝酸トリウム標準溶液のファクター
m:試料はかりとり量(g)
R:ふっ素の蒸留効率
8.2
ふっ化カルシウム含有率の計算 試料中のふっ化カルシウムの含有率は、ふっ素含有率から次の式
によって算出する。
CaF2=2.055×F
ここに,CaF2:試料中のふっ化カルシウム含有率[%(m/m)]
F:8.1で求めた試料中のふっ素含有率[%(m/m)]
9. 許容差 許容差は,附属書1表1による。
附属書1表1 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[0.0035×(ふっ素含有率)+0.0926]
f(n)[−0.0017×(ふっ素含有率)+
0.4766]
参考
この許容差は,ふっ素含有率36.61%(m/m)以上47.77%(m/m)以下の試料を用いて求めたもの
8
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
である。表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場合f(n)=2.8
である。

9
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2(規定)ふっ素定量法−塩化ふっ化鉛沈殿分離
−エチレンジアミン四酢酸ニ水素ニナトリウム間接滴定法
1. 要旨 試料に炭酸ナトリウムを加えて融解し,融成物を硝酸に溶解する。塩化ナトリウム及び硝酸鉛
を加え,生成する塩化ふっ化鉛をこし分ける。沈殿を硝酸で溶解し,キシレノールオレンジを指示薬とし
てエチレンジアミン四酢酸二水素ニナトリウム(以下,EDTA2Naという。)標準溶液で鉛を滴定する。
2. 試薬 試薬は,次による。
a) 塩酸(1+20)
b) 硝酸
c) 硝酸 (1+10)
d) アンモニア水 (1+20)
e) 塩化ナトリウム
f)
炭酸ナトリウム
g) 硝酸鉛溶液 (15g Pb/L) 硝酸鉛(Ⅱ) 24gを水1Lに溶解する。
h) 酢酸アンモニウム溶液(500g/L)
i)
エタノール(99.5)
j)
キシレノールオレンジ溶液 キシノレールオレンジ0.2gを水100mlに溶解する。
k) 0.01 mol/L EDTA2Na標準溶液 調製及び評定方法は,JIS K 8001の4.5(3.3)[0.01mol/Lエチレンジア
ミン四酢酸二水素ニナトリウム溶液(0.01mol/L EDTA2Na溶液)]による。
3. 試料はかりとり量 試料はかりとり量は,0.20gとする。
4. 操作
4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかりとって白金るつぼ(30番)に移し入れる。
b) 別にはかりとった炭酸ナトリウム2gの約半量をるつぼに入れて白金線でかき混ぜ,試料と混合した
後,残りの炭酸ナトリウムで表面を覆う。
c) ガスバーナ上で白金るつぼを回しながら加熱し,炭酸ナトリウムが融解したら白金ふたをかぶせ,約
3分間強熱した後,室温まで放冷する。
d) 水約100ml及び硝酸10mlをビーカー(300ml)に取り,時計皿で覆い,加熱して沸騰させた後,熱源か
ら降ろして4〜5分間静置した液に白金るつぼを横にして入れる。
e) 激しい反応が収まった後,ふたを入れて,4〜5分間放置する。
f)
るつぼ及びふたを水で洗って取り出し,溶液を流水中で常温までに冷却する。
g)時計皿の下面を水で洗って時計皿を取り除き,溶液及び洗液を250mlのPTFE製全量フラスコに水
を用いて移し入れ,水で標線まで薄める。
4.2 沈殿の生成 沈殿の生成は, 次の手順によって行う。
10
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 4.1で得た溶液から25mlを分取してビーカー(200ml)に移し入れ,水を加えて液量を100mlとする。
b) 酢酸アンモニウム溶液0.5ml及び塩化ナトリウム3gを加え,かき混ぜて塩化ナトリウムを溶解する。
c) アンモニア水(1+20)又は硝酸(1+10)を滴加し,pHメータを用いてpHを4.0±0.2に調節する。
d) 溶液をかき混ぜながら,全量ピペットを用いて硝酸鉛溶液[2.g)] 15mlを少量ずつ加え,更に白濁する
まで3分間以上かき混ぜる。
e) 水を加えて液量を200mlとし,時計皿で覆い,室温で3時間(1)以上静置する。
注(1) アルミニウム含有率が0.1%m/m以上の試料の場合は,60時間静置する。
4.3 沈殿の分離及び溶解 沈殿の分離と溶解は, 次の手順によって行う。
a) ろ紙(5種C)を水を用いて漏斗にはり付けた後(2),エタノール(99.5)でろ紙を洗い,水分を完全に
取り除く。
注(2) ろ紙は,エタノール(99.5)ではり付けてもよい。
b) 4.2e)で得た沈殿をa)のろ紙でこし分け,ビーカーの底部に残った沈殿はエタノール(99.5)を用いて
ろ紙に洗い移し,更にろ紙及び沈殿をエタノール(99.5)で3〜4回洗浄する。ろ液及び洗液は捨て,
ビーカー及びガラス棒は,保存する。
c) ろ紙の底部に小穴を開け,ろ紙の上から温めた硝酸(1+10)10mlを滴加して沈殿を溶解した後,温水で
ろ紙を7,8回洗浄する。溶液及び洗液は,b)で保存したビーカーに受け,ビーカー内にはb)で保存
したガラス棒を入れる。時計皿で覆い,穏やかに加熱してビーカーの内壁及びガラス棒に付着した沈
殿を完全に溶解する。
d) 水で液量を約100mlとし,室温まで冷却した後,時計皿の下面を少量の水で洗って時計皿を取り除く。
4.4 滴定 4.3d)で得た溶液に酢酸アンモニウム溶液を1ml加え,pHメータを用いてアンモニア水(1+20)
及び塩酸(1+20)を滴加し,pHを5.20±0.2に調節する。指示薬としてキシレノールオレンジ溶液[2.j]]を4
滴加え,よくかき混ぜながら0.01mol/L EDTA2Na標準溶液[2.k]]で滴定し,最後の1滴で溶液の色が鮮や
かな黄になった点を終点とし,0.01mol/L EDTA2Na標準溶液[2.k)]の使用量を求める。
5. 空試験 試料を用いないで,4.1〜4.4の手順に従って試料と同じ操作を試料と併行して行う。
6. 計算
6.1 ふっ素含有率の計算 試料中のふっ素含有率を次の式によって算出する。
(
)
100
10
/1
0001900
.0
2
1
×
×
×
×
−
=
m
f
V
V
F
ここに,F :試料中のふっ素含有率[%(m/m)]
V1 :試料溶液の滴定における0.01mol/L EDTA2Na標準溶液[2.k)]の使用量 (ml)
V2 :空試験液の滴定における0.01mol/L EDTA2Na標準溶液[2.k)]の使用量(ml)
f :0.01mol/L EDTA2Na標準溶液[2.k)]のファクター
m :試料はかりとり量(g)
6.2 ふっ化カルシウム含有率の計算 試料中のふっ化カルシウムの含有率は,ふっ素含有率を次の式によ
って算出する。
CaF2=2.055×F

11
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
F :6.1で求めた試料中のふっ素含有率[%(m/m)]
7. 許容差 許容差は,附属書2表 による。
附属書2表 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[0.182]
f(n)[0.277]
参考1. この許容差は,ふっ素含有率36.62%(m/m)以上43.30%(m/m)以下の試料を用いて求めたも
のである。
表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場合f(n)=2.8で
ある。
12
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3(規定)けい素定量法−モリブドけい酸青吸光光度法
1. 要旨 試料を炭酸ナトリウムで融解し,融成物を温水に溶解する。ほう酸を加え,pHを調節した後,
七モリブデン酸六アンモニウムを加えてけい酸をモリブドけい酸とし,酒石酸と硫酸とを共存させてアス
コルビン酸でモリブドけい酸をモリブドけい酸青に還元し,光度計を用いて,その吸光度を測定する。
2. 試薬 試薬は,次による。
a) 塩酸(10+7)
b) 硫酸(1+1,10+41)
c) ほう酸溶液(40g/L)
d) 炭酸ナトリウム
e) モリブデン酸アンモニウム溶液 七モリブデン酸六アンモニウム四水和物60gを水に溶解し,水で液
量を1Lとする。この溶液は,使用の都度,ろ過して使用する。
f)
酒石酸溶液(100g/L)
g) アスコルビン酸溶液(20g/L) 使用の都度,調製する。
h) 希釈溶液 炭酸ナトリウム4.0gをはかりとって白金るつぼ(30番)に移し入れ,以下,4.1b)〜d)の
手順に従って試料と同じ操作を試料と併行して行い,得た溶液を希釈溶液とする。
i)
標準けい素液(70μgSi/ml) 二酸化けい素(99.9%以上)を1000℃で1時間加熱した後,デシケータ
ー中で常温まで放冷したもの0.0749gをはかりとって白金るつぼ(30番)に移し入れる。炭酸ナトリ
ウム2.5gを加えて混合し,白金るつぼに白金ふたをして,初めは低温で加熱して内容物が融解した後,
温度を1000〜1100℃にあげて15分間以上強熱し,完全に融解する。白金るつぼを熱源から降ろして
室温まで放冷する。温水300mlを入れたビーカー(500ml)中に融成物を白金るつぼごと入れ,穏や
かに加熱して融成物を溶解し,白金るつぼを水で洗って取り出す。溶液を500mlの全量フラスコに水
を用いて移し入れ,水で標線まで薄めて標準けい素液とする。この溶液は,ポリエチレン瓶に移して
保存する。
3. 試料はかりとり量 試料はかりとり量は,0.20gとする。
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかりとって白金るつぼ(30番)に移し入れ,炭酸ナトリウム4.0gを加えて混合する。
b) 白金るつぼに白金ふたをして,初めは低温で加熱して内容物が融解した後,温度を1000〜1100℃にあ
げて15分間以上強熱し,完全に融解する。白金るつぼを熱源から降ろして室温まで放冷する。
c) 温水200mlを入れたPTFE製ビーカー(500ml)中に融成物を白金るつぼごと入れ,温浴上で穏やかに
加熱して融成物を溶解する。白金るつぼを水で洗って取り出す。溶液を室温まで冷却した後,水で液
量を300mlとし,ほう酸溶液20mlを加える。塩酸(10+7)を正確に12ml,少量ずつ加えてよく混合
する。
d) 時計皿で覆い,穏やかに加熱して3分間煮沸した後,ビーカーを熱源から降ろし,常温まで冷却する。
溶液を500mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
13
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.2
呈色 呈色は次の手順によって行う。
a) 4.1d)で得た溶液から,附属書3表1に従って溶液の一部を分取して100mlの全量フラスコに移し入れ,
附属書3表1に従って分取量に応じた希釈溶液[2.h)]を正確に添加し,水40mlを加える。
b) 硫酸(10+41)1mlを正確に加えてよく混合し,モリブデン酸アンモニウム溶液[2.e)]10mlを加え,振り
混ぜて15分間放置する。
c) 酒石酸溶液5mlを加え,振り混ぜて5分間放置する。次に振り混ぜながら硫酸(1+1)10ml,アスコルビ
ン酸溶液[2.g]]2mlを連続して手早く加え,水で標線まで薄め,30分間放置する。
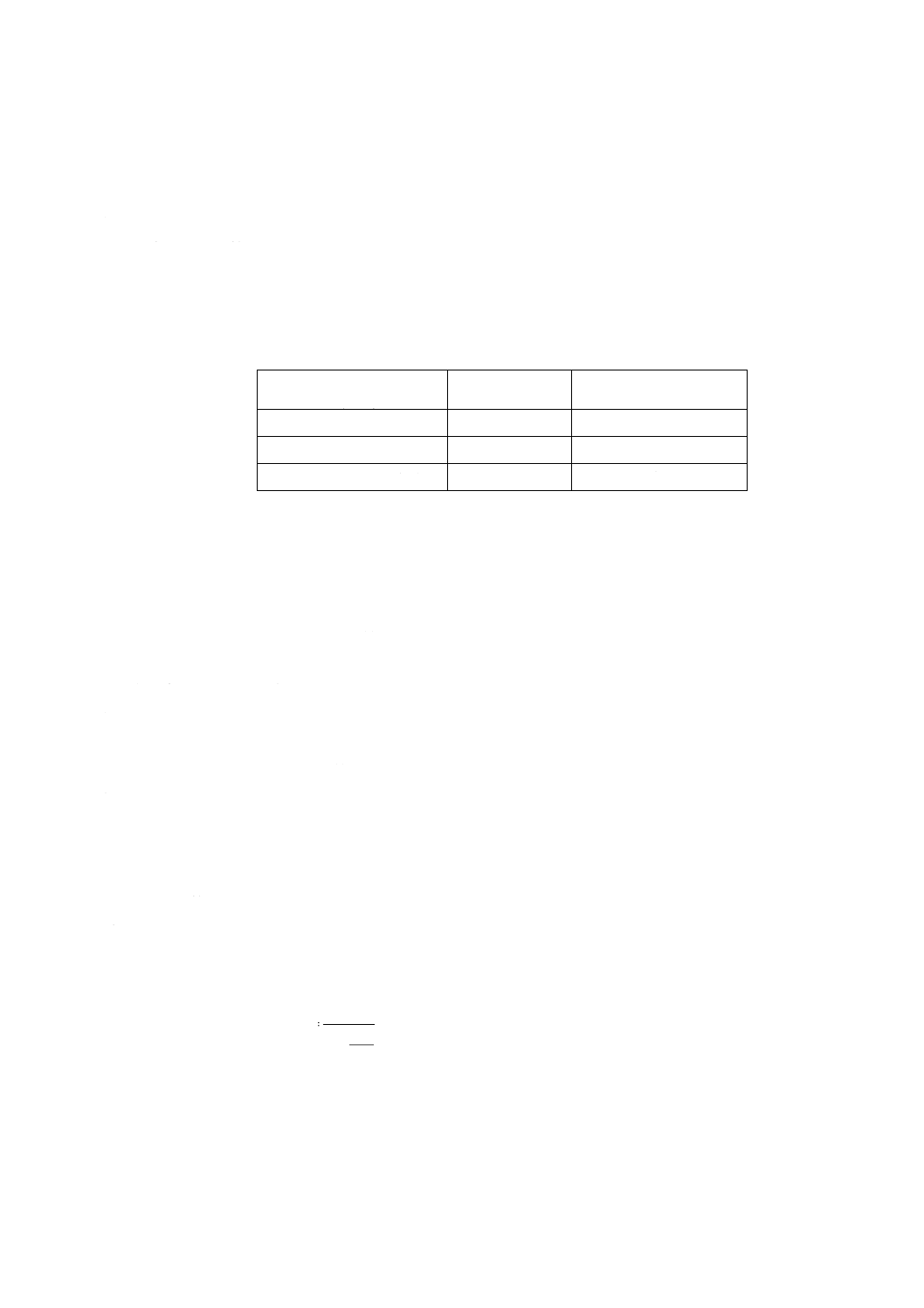
附属書3表 1 分取量及び希釈溶液の添加量
けい素含有率
%(m/m)
分取量
ml
希釈溶液[2.h)]添加量
ml
0.09 以上 2.8 未満
20
0
2.8 以上 5.6 未満
10
10
5.6 以上 14 以下
5
15
4.3
吸光度の測定 4.2c)で得た呈色溶液の一部を光度計の吸収セル(1cm)に取り,水を対照液として波長
650nmにおける吸光度を測定する。
5. 空試験 希釈溶液[2.h)]20mlを100mlの全量フラスコに取り,水40mlを加える。以下,4.2b)〜4.3の
手順に従って試料と同じ操作を試料と併行して行う。
6. 検量線の作成 検量線の作成は,次の手順によって行う。
a) 5個のビーカー(200ml)に標準けい素液[2.i)]0ml,1.0ml,2.0ml,3.0ml及び4.0ml(けい素量として,0
μg,70μg,140μg,210μg及び280μg)を取り,それぞれに希釈溶液[2.h)]20mlを正確に加え、塩酸
(10+7)を用いてpHを2.0に調節する。
b) 100mlの全量フラスコに水を用いて移し入れ,水で液量を60mlとする。
c) 4.2b)〜4.3の手順に従って試料と同じ操作を試料と併行して行い,得た吸光度と呈色溶液中のけい素
量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
7. 計算 計算は,次による。
7.1
けい素含有率の計算 4.3及び5.で得た吸光度と6.で作成した検量線からけい素量を求め,試料中の
けい素含有率を次の式によって算出する。
100
500
2
1
×
×
−
=
B
m
A
A
Si
ここに,Si:試料中のけい素含有率[%(m/m)]
A1 :分取した試料溶液中のけい素検出量(g)
A2 :分取した空試験溶液中のけい素検出量(g)
m :試料はかりとり量(g)

14
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B :試料溶液及び空試験液の分取量(ml)
7.2
2二酸化けい素含有率の計算 試料中の二酸化けい素含有率は,けい素含有率から次の式によって
算出する。
SiO2=2.139×Si
ここに,SiO2:試料中の二酸化けい素含有率[%(m/m)]
Si :7.1で求めた試料中のけい素含有率[%(m/m)]
8. 許容差 許容差は,附属書3表2による。
附属書3表 2 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[0.0038×(けい素含有率)+0.0126]
f(n)[0.0062×(けい素含有率)+0.0186]
参考1. この許容差は,けい素含有率0.68%(m/m)以上9.89%(m/m)以下の試料を用いて求めたもので
ある。
表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場合f(n)=2.8であ
る。
15
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4(規定)炭酸カルシウム定量方法−二酸化炭素気化分離
−塩酸・水酸化ナトリウム滴定法
1. 要旨 試料に塩酸を加え,発生する二酸化炭素を水酸化バリウム溶液に吸収させ,過剰のアルカリを
塩酸で中和する。更に,塩酸の一定量を正確に加えて炭酸バリウムを溶解し,過剰の塩酸を水酸化ナトリ
ウム標準溶液で滴定する。
2. 試薬 試薬は,次による。
a) 塩酸(3+2,1+10)
b) ほう酸
c) 水酸化ナトリウム溶液(40g/L)
d) 窒素
e) 塩化バリウム溶液 塩化バリウム二水和物122gを水に溶解し,水で液量を1Lとする。
f)
硫酸銅溶液 硫酸銅五水和物100gを水に溶解し,水で液量を1Lとする。
g) 1-ブタノール
h) 0.1mol/L塩酸標準溶液 調製及び標定方法は,JIS K 8001の4.5(5.5)(0.1mol/L塩酸)による。
i)
0.1mol/L標準水酸化ナトリウム溶液 調製及び標定方法は,JIS K 8001の4.5(19.4)(0.1mol/L水酸化ナ
トリウム溶液)による。
j)
メチルオレンジ溶液(1g/L)
k) メチルオレンジ・キシレンシアノールFF溶液 メチルオレンジ0.40gとキシレンシアノールFF0.56g
とを,水とエタノール(95)との等量混合液200mlに溶解する。この溶液は,褐色ガラス瓶に保存する。
l)
フェノールフタレイン溶液 フェノールフタレイン0.025gを水とエタノール(95)との等量混合液
100mlに溶解する。
3. 装置及び器具 二酸化炭素発生・吸収装置は,次のものを用いる(附属書4図1参照)。
a) 洗浄瓶 水酸化カリウム溶液(200g/L)約100mlを入れた,ガラスろ過板(G1)付きのガス洗浄瓶(250ml)。
b) 分解フラスコ 滴下漏斗及び冷却器を取り付けた容量500mlのガラス製三つ口フラスコ
c) 吸収瓶(ドレッセル式) ガス出口から液面までの高さが10mm以上となるガラス製のガス洗浄瓶を2
本連結する。
d) 加熱器(フラスコ用)
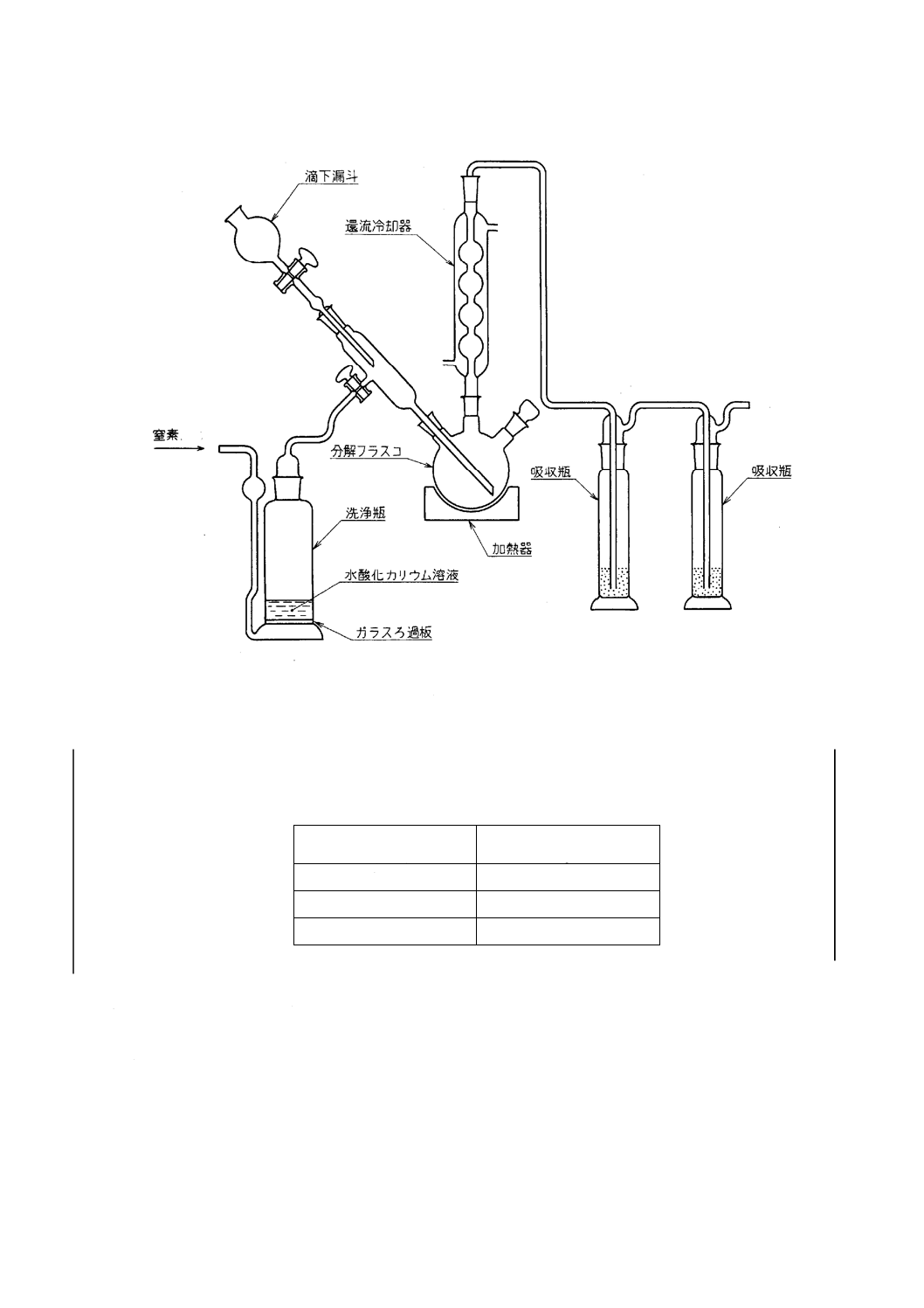
16
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4図 1 二酸化炭素発生・吸収装置の例
4. 試料はかりとり量 試料はかりとり量は,附属書4表1による。
附属書4表 1 試料はかりとり量
炭酸カルシウム含有率
%(m/m)
試料はかりとり量
g
0.1 以上 2 未満
5.0
2 以上 5 未満
2.0
5 以上 10 以下
1.0
5. 操作
5.1
二酸化炭素の発生及び吸収 二酸化炭素の発生及び吸収は,次の手順によって行う。
a) 試料をはかりとって約100mlの水で分解フラスコに移し入れ,ほう酸4g及び硫酸銅溶液[2.f)]5mlを加
える。
b) 分解フラスコの口を閉じ,窒素を40〜50ml/minで分解フラスコに通す。窒素を通じながら,水酸化ナ
トリウム溶液10ml,塩化バリウム溶液[2.e)]10ml,フェノールフタレイン溶液[2.l)]1ml,1-ブタノール
1ml及び水20mlを入れた吸収瓶2本を直列に連結する。
c) 滴下漏斗を通して塩酸(3+2)30mlを分解フラスコに加え (1),滴下漏斗のコックを閉じる。分解フラス

17
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
コを徐々に加熱して穏やかに45分間煮沸させた後,10分間放冷する。この間,窒素は中断させては
ならない。
注(1) 必要ならばゴム球などで加圧して入れる。
5.2
滴定 滴定は, 次の手順によって行う。
a) 装置から第二の吸収瓶を取り外し,ガス導入管の浸せき部を水で洗い,洗浄液を吸収瓶に集める。吸
収瓶内の溶液を塩酸(1+10)で終点近くまで滴定(2)する。次に0.1mol/L塩酸標準溶液[2.h)]で終点を超え
ないように注意しながら,フェノールフタレインの色がちょうど消えるまで滴定する。更に,0.1mol/L
塩酸標準溶液[2.h)]の一定量を正確に加え,沈殿を完全に溶解する。ガス導入ガラス管の浸せき部をこ
の溶液に入れて,付着している炭酸バリウムを溶かした後,再び水で洗浄し,吸収瓶に集める。
注(2) 吸収瓶内の過剰の水酸化ナトリウムを滴定している間の,大気中の二酸化炭素の吸収を避ける
ため,吸収瓶内の溶液上に窒素を流して空気の置換及び侵入の防止を図る。
b) メチルオレンジ溶液又はメチルオレンジ-キシレンシアノールFF溶液[2.k)]を指示薬として数滴加え,
0.1mol/L水酸化ナトリウム溶液[2.i)]で過剰の塩酸を滴定する。
c) 第一の吸収瓶について,a)及びb)の操作を行う。
6. 空試験 試料を用いないで5.1及び5.2の手順に従って試料と同じ操作を行う。
7. 計算 試料中の炭酸カルシウム含有率を,次の式によって算出する。
(
)
100
005005
.0
2
1
×
×
−
=
m
V
V
3
CaCO
ここに, CaCO3:試料中の炭酸カルシウム含有率[%(m/m)]
V1 :両方の吸収瓶中の炭酸バリウムを溶解するために使用した空試験滴定量補
正後の0.1mol/L塩酸標準溶液[2.h)]の量(ml)
V2 :両方の吸収瓶中の塩酸の逆滴定に使用した空試験滴定量補正後の0.1mol/L
水酸化ナトリウム標準溶液[2.i)]の量(ml)
m :試料はかりとり量(g)
8. 許容差 許容差は,附属書4表2による。
附属書4表 2 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[0.0092×(炭酸カルシウム含有率)+0.0107]
f(n)[0.0643×(炭酸カルシウム含有率)+0.0159]
参考1. この許容差は,炭酸カルシウム含有率0.09%(m/m)以上1.08%(m/m)以下の試料を用いて求め
たものである。表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場合
f(n)=2.8である。

18
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書5(規定)りん定量方法−水酸化鉄共沈分離
−モリブドりん酸青吸光光度法
1. 要旨 試料を炭酸ナトリウム及び四ほう酸ナトリウムで融解し,融成物を硝酸に溶解する。アンモニ
ア水を加えてりん酸鉄を沈殿させ,こし分ける。沈殿を硝酸及び過塩素酸に溶解し,過塩素酸の白煙処理
をしてろ過し,ろ液の一部を亜硫酸水素ナトリウムを加え,鉄などを還元した後,七モリブデン酸六アン
モニウム及び硫酸ヒドラジニウム(2+)を加えて生じるモリブドりん酸青を生成させ,光度計を用いて,そ
の吸光度を測定する。
2. 試薬 試薬は,次による。
a) 塩酸(1+1)
b) 硝酸
c) 硝酸(1+1)
d) 過塩素酸
e) アンモニア水
f)
アンモニア水(1+50)
g) 亜硫酸水素ナトリウム溶液(100g/L)
h) ふっ化カルシウム 粉状のものでできるだけ純度が高くりんを含有しないか,又はりん含有率が既知
で,できるだけ低いもの。
i)
融解合剤[炭酸ナトリウム2,四ほう酸ナトリウム(無水)1]
j)
鉄溶液 りん含有率の極めて低い鉄0.30gをビーカー(200ml)にはかりとり,塩酸10mlを加えて時計
皿で覆い,穏やかに加熱して分解する。硝酸3mlを加えて鉄を酸化した後,しばらく煮沸を続けて窒
素酸化物などを追い出す。室温まで冷却した後,水を加えて液量を100mlとする。
k) 呈色試薬溶液 モリブデン酸アンモニウム溶液25ml,硫酸ヒドラジニウム(2+)溶液(1.5g/L)10ml及び水
65mlを使用の都度混合する。
モリブデン酸アンモニウム溶液の調製は,次による。
七モリブデン酸六アンモニウム四水和物20gを温水100mlに溶解し,硫酸(1+1)600mlを加え,室温
まで冷却した後,水で液量を1Lとする。
l)
標準りん液(50μgP/ml) りん酸二水素カリウムを105℃で加熱して恒量とし,デシケーター中で
常温まで放冷したもの0.2197gをビーカー(200ml)にはかりとり,溶液を1000mlの全量フラスコに水
を用いて移し入れ,水で標線まで薄めて標準りん液とする。
3. 試料はかりとり量 試料はかりとり量は,0.50gとする。
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかりとって白金るつぼ(30番)に移し入れ,融解合剤[2.i)]5.0gを加えて混合する。

19
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 白金るつぼに白金ふたをし,初めは低温で加熱し,次第に温度を高めて1000〜1050℃で融解する。室
温まで放冷した後,白金るつぼをビーカー(500ml)に入れ,白金るつぼの中に硝酸(1+1)25mlを加え,
時計皿で覆い,融成物を溶解する。白金るつぼを水で洗って取り出し,鉄溶液[2.j)]10mlを加え,温水
で液量を約300mlとする。
c) 溶液を沸騰直前まで加熱した後,振り混ぜながらアンモニア水を少量ずつ加えて中和し,更に過剰に
加えて約1分間煮沸する。水酸化鉄などの沈殿を静置して沈降させた後,ろ紙(5種A)を用いてこし分
け,アンモニア水(1+50)で数回洗浄する。ろ液及び洗液は,捨てる。
d) 沈殿をろ紙ごと石英ビーカー(200ml)に移し,元のビーカー内壁に付着した沈殿は,塩酸(1+1)で溶解し,
少量の水を用いて石英ビーカーに移し入れる。硝酸5ml及び過塩素酸10mlを加えて乾固直前まで加
熱する。室温まで放冷した後,温水約30mlを加えて塩類を溶解する。溶液をろ紙(5種B)を用いてろ
過した後,温水で十分に洗浄し,ろ液及び洗液を100mlの全量フラスコに集める。常温まで冷却した
後,水で標線まで薄める。
4.2
呈色 呈色は, 次の手順によって行う。
a) 4.1d)で得た溶液から25mlを分取して100mlの全量フラスコ(1)に移し入れる。亜硫酸水素ナトリウム
溶液10mlを加え,沸騰水中に浸して溶液の色が変化しなくなるまで加熱して鉄などを還元する。
注(1) 新しい全量フラスコを使用するときは,水を標線まで入れて沸騰水中で約10分間加熱した後,
常温まで冷却する。この操作を数回繰り返してから使用する。
b) 呈色試薬溶液[2.k)]25mlを加え再び沸騰水中で10分間加熱を続け,溶液を常温まで冷却した後,水で
標線まで薄める。
4.3
吸光度の測定 4.2b)で得た呈色溶液の一部を光度計の吸収セル(1cm又は2cm)(2)に取り,水を対照液
として波長825nmにける吸光度を測定する。
注(2) 試料中のりん含有率が0.03%(m/m)未満の場合は2cmの吸収セルを,りん含有率が0.03%(m/m)
以上の場合は,1cmの吸収セルを用いるとよい。
5. 空試験 試料を用いないで,4.1及び4.2の手順に従って試料と同じ操作を試料と併行して行う。
6. 検量線の作成 検量線の作成は,次の手順によって行う。
a) 5個の白金るつぼ(30番)を用意し,ふっ化カルシウム[2.h)]0.500g及び融解合剤[2.i)]5gずつをはかりと
って,それぞれに加えてよく混合する。
b) 白金るつぼに白金ふたをし,初めは低温で加熱し,次第に温度を高めて1000〜1050℃で融解する。室
温まで放冷した後,5個の白金るつぼを5個のビーカー(500ml)にそれぞれ入れ,白金るつぼの中に硝
酸(1+1)25mlを加えて時計皿で覆い,融成物を溶解する。白金るつぼを水で洗って取り出した後,標
準りん液[2.l)]0ml, 1.0ml, 3.0ml, 5.0ml及び7.0ml(りんとして0μg,50μg,150μg,250μg及び350μ
g)を加え,更に鉄溶液[2.j)]10mlを加えて温水で液量を約300mlとする。
c) 4.1c)〜4.3の手順に従って試料と同じ操作を試料と併行して行い,得た吸光度と呈色溶液中のりん量
との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
7. 計算 4.3及び5.で得た吸光度と6.で作成した検量線からりん量を求め,試料中のりん含有率を次の式
によって算出する。
20
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
200
25
2
1
×
×
−
=
m
A
A
P
ここに,P :試料中のりん含有率[%(m/m)]
A1 :分取した試料溶液中のりん検出量(g)
A2 :分取した空試験液中のりん検出量(g)
m :試料はかりとり量(g)
8. 許容差 許容差は,附属書5表1による。
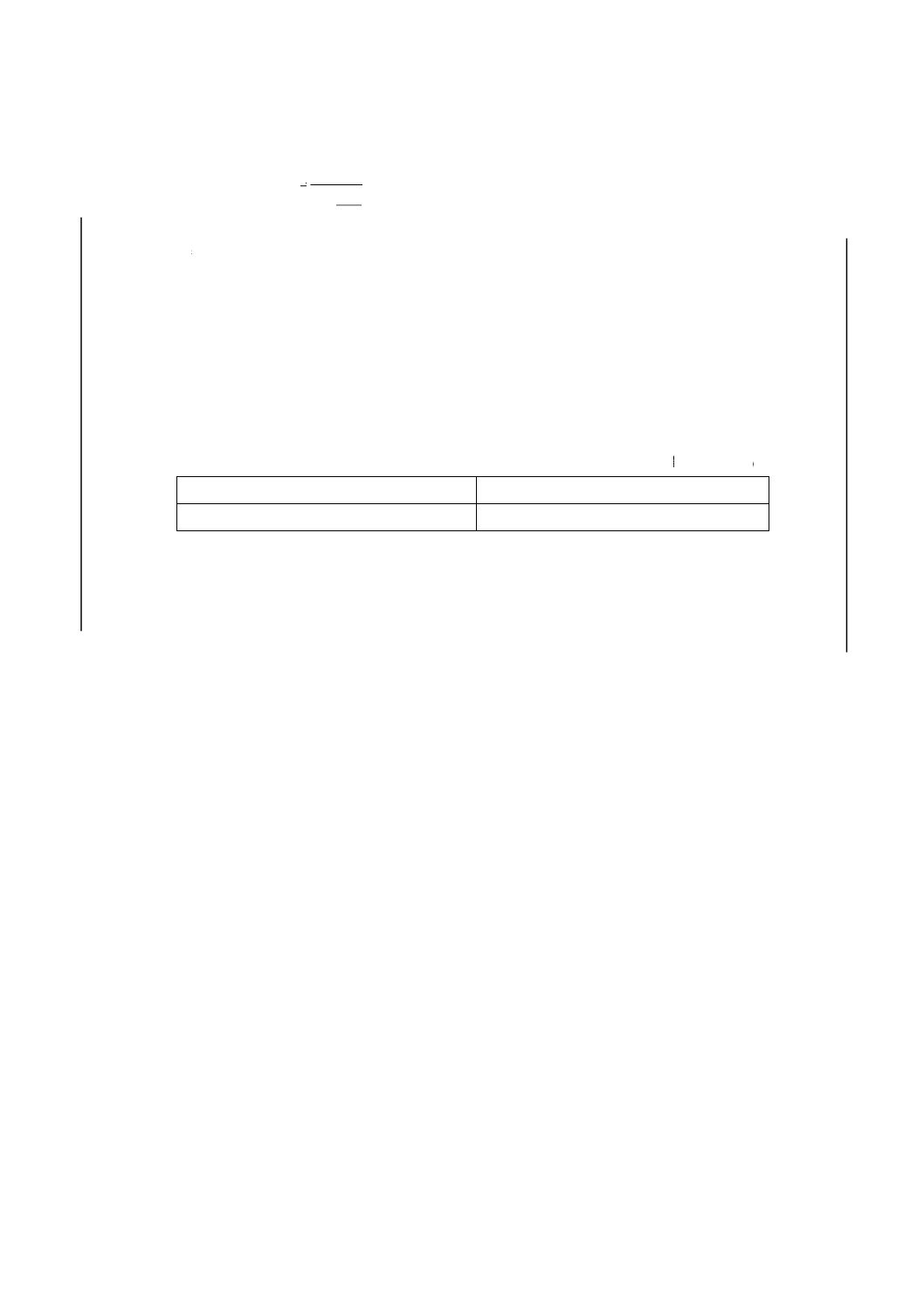
附属書5表 1 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[0.0030×(りん含有率)+0.0032]
f(n)[0.0165×(りん含有率)+0.0036]
参考1. この許容差は,りん含有率0.004%(m/m)以上0.043%(m/m)以下の試料を用いて求めたもので
ある。表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場合f(n)=2.8
である。
21
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書6(規定)硫黄定量方法−熱分解-よう素酸カリウム滴定法
1. 要旨 試料を酸化タングステン(Ⅵ)と混合し,窒素気流中で高温に加熱して硫黄を二酸化硫黄とする。
これを塩酸を含むよう化カリウム溶液に吸収させ,でんぷんを指示薬としてよう素酸カリウム標準溶液で
滴定する。
2. 試薬 試薬は,次による。
a) 窒素
b) 過塩素酸マグネシウム 粒径0.5〜2mmのもの。
c) 酸化タングステン(Ⅵ) 粉末状のもの。
d) ソーダ石灰又は水酸化ナトリウム 粒径0.5〜2mmのもの。
e) 吸収液 塩酸(3+197)80ml,よう化カリウム溶液(30g/L)1ml及びでんぷん溶液1mlを混合する。この溶
液は,使用の都度,調製する。
でんぷん溶液の調製は,次による。
でんぷん(溶性)2gをはかりとってビーカー(200ml)に移し入れ,水10mlでのり状に練り,約50mlの
沸騰水中に注ぎ入れ,約1分間煮沸して冷却し,水で100mlに薄める。
f)
ふっ化カルシウム 粉状で,できるだけ純度が高く,硫黄を含有しないか,又は硫黄含有率が既知で,
できるだけ低いもの。
g) よう素酸カリウム標準溶液 よう素酸カリウム(JIS K 8005)0.2225gを水約100mlに溶解し,溶液を
1000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
h) 標準硫黄液(5 mgS/ml) 105℃で加熱して恒量とし,デシケーター中で常温になるまで放冷した硫酸
カリウム13.587gをはかりとって,水に溶解し,溶液を500mlの全量フラスコに水を用いて移し入れ,
水で標線まで薄めて標準硫黄液とする。
3. 装置,器具及び材料 硫黄定量装置は,一般に次のものを用いる(附属書6図1及び附属書6図2参
照)。
a) 窒素清浄装置(記号a) 窒素を清浄乾燥するためのもので,ソーダ石灰又は水酸化ナトリム[2.d)]と過
塩素酸マグネシウム[2.b)]を詰めた塔。
b) 熱分解炉(記号b) 一般に次のものを用いる。
1) 管状電気抵抗加熱炉(長さ約300mm)は,電流を調節して温度を加減し,炉の中央部の長さ約150mm
を1200±25℃の一定温度に保つことができるもの。
2) 炉内には長さ約600〜700mm,内径24mmで1200±25℃に耐える磁器燃焼管(記号c)(JIS R 1307の
CT 0又はCT 1)を挿入する。燃焼管(記号c)の出口部は,炉壁から100〜150mm突き出させ,出口部
にはテーパを付けてすり合わせガラス製キャップ(記号g)をはめ,ばね(記号h)で炉壁に締め付ける。
3) ガラス製キャップ(記号g)と炉壁との間に遮熱板(記号i)を置き,炉体からの熱が吸収瓶(記号e)に当
たらないようにする。
4) 炉中央部の燃焼管(記号c)の真上の温度を熱電高温度計で測定する。熱電高温度計の指示値は,一般
に燃焼管(記号c)内の温度と異なるので,その差を求めておき,必要がれば指示値を補正して燃焼管
(記号c)内の温度を求める。
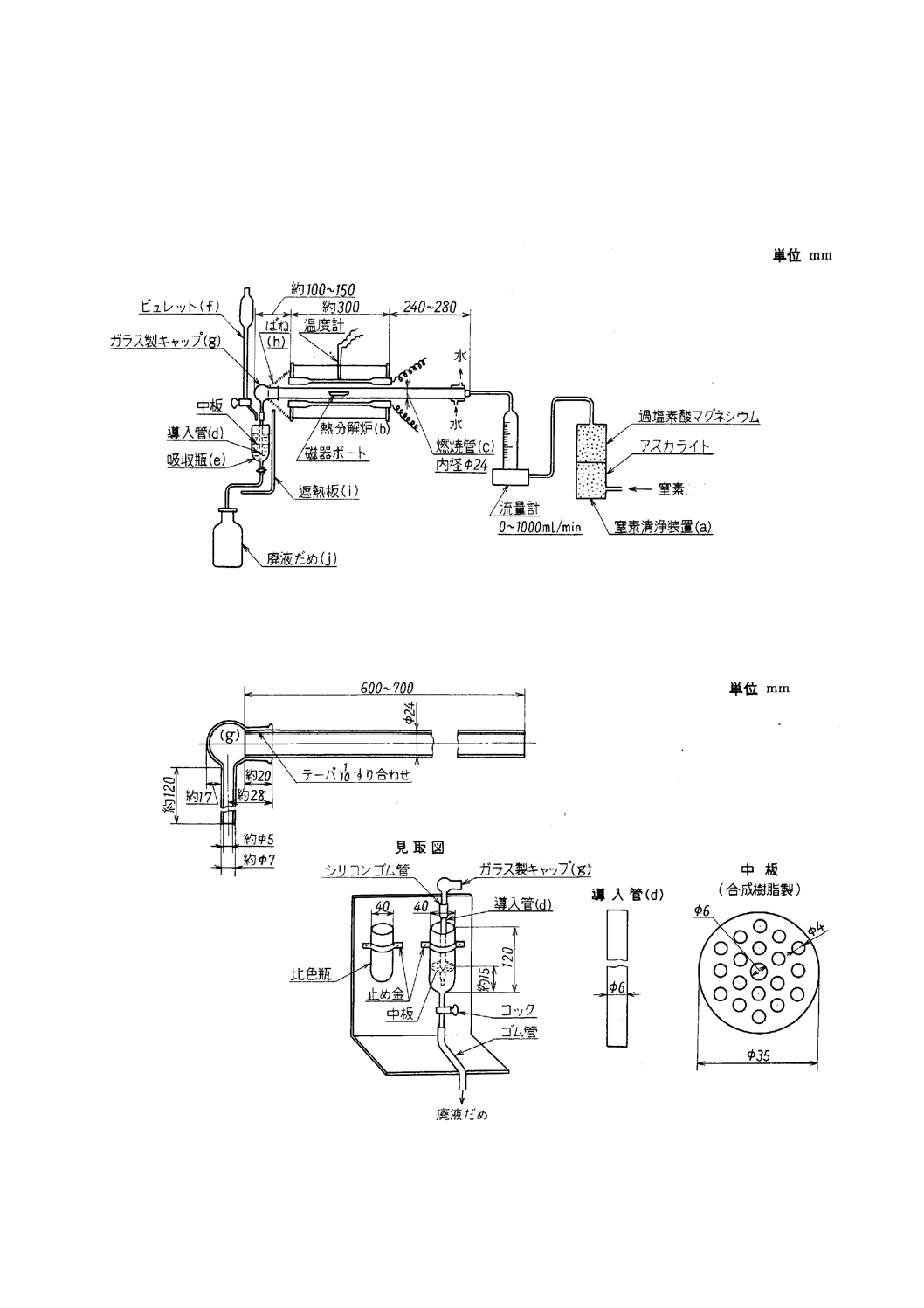
22
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5) 燃焼管(記号c)と窒素清浄装置(記号a)との連結は,すり合わせ又は耐熱性のシリコーンゴム栓を用
いる。
6) 新しい磁器燃焼管を使用するときには,1200℃で30分間以上窒素気流中で空焼きを行う。
附属書6図 1 硫黄定量装置の例
附属書6図 2 燃焼管(c)及び吸収瓶(e)の詳細図
23
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 吸収瓶(記号e) 吸収液[2.e]]約80mlを入れ,ガラス製キャップ(記号g)の先に内径6mmの導入管(ガラ
ス製)(記号d)を取り付け,その先端が吸収液の最下部に達するようにし,この管(記号d)に吸収液の底
部から約15mmの位置に合成樹脂板に多くの小孔をあけた中板を取り付け,中板から液面までの高さ
を60〜80mmとした吸収瓶。
ガラス製キャップ(記号g)と導入管(記号d)との連結は,すり合わせ又は耐熱性のシリコーンゴム栓
を用いる。
d) ビュレット(記号f) 25ml
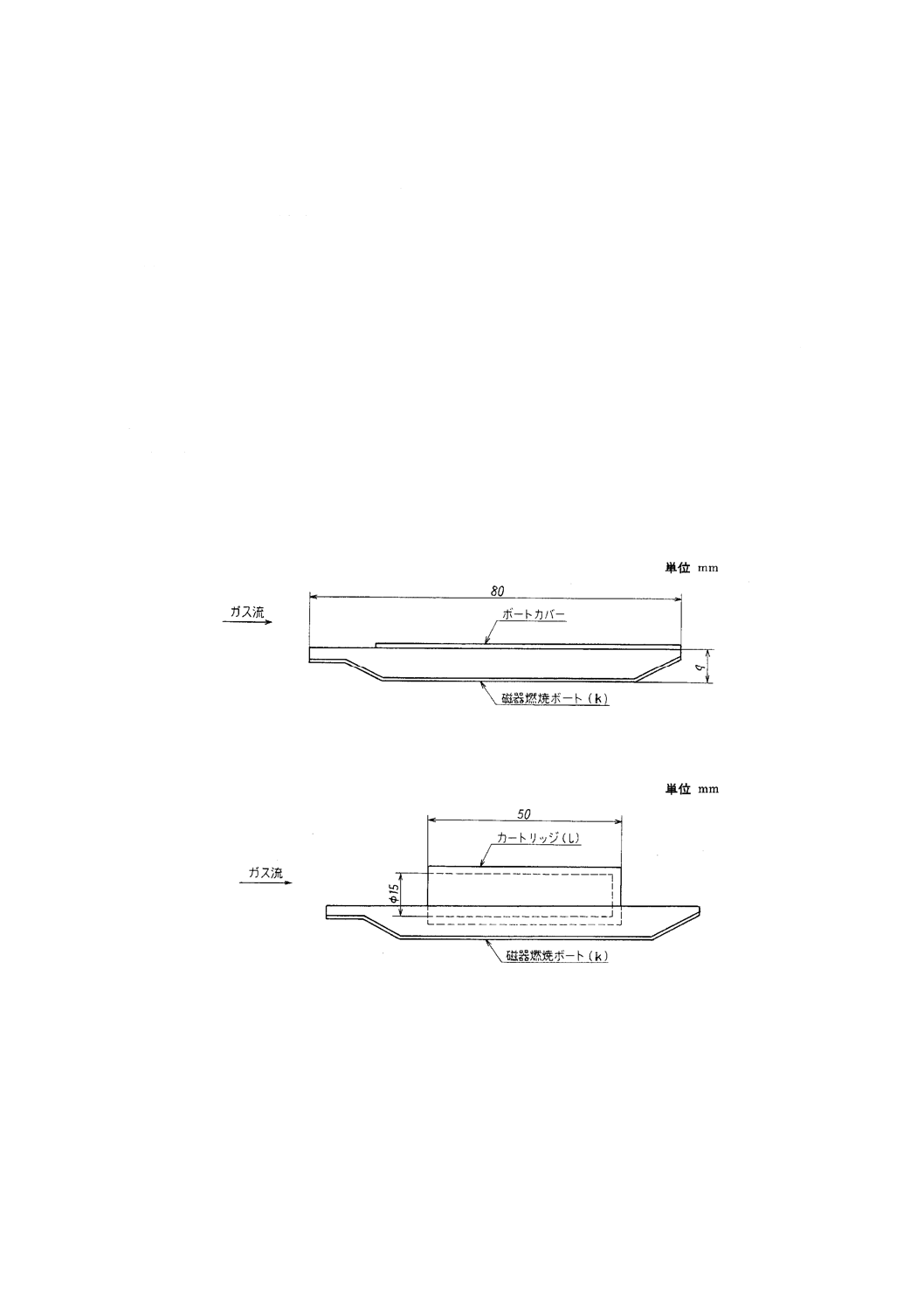
e) 磁器燃焼ボート及び磁器燃焼ボートカバー(以下,ボート及びカバーという。) ボート(記号k)は,
JIS R 1306のCB 1を用い,必要に応じてカバーを使用する。カバーは,JIS R 1306のCBC 1を用い
る。ボート及びカバーは,あらかじめ窒素気流中で1200℃で10分間加熱して,デシケーター中に保
存したものを用いる(1)。カバーの代わりに多孔質のカートリッジ[附属書6図3(記号l)]を用いてもよ
い。
注(1) デシケーターからの出し入れは,ピンセットなどで扱い,直接手で触れてはならない。長時間
保存したものは,空試験値が高くなっているおそれがあるので,再度空焼きを行ってから使用
する。
附属書6図 3 磁器燃焼ボート,磁器燃焼ボートカバー及びカートリッジの例
4. 試料はかりとり量 試料はかりとり量は,附属書6表1による。
24
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書6表 1 試料はかりとり量
硫黄含有率
%(m/m)
試料はかりとり量
g
0.01 以上 0.5 未満
0.50
0.5 以上 1 以下
0.20
5. 操作
5.1
装置の準備 3.の装置を気密に連結し,燃焼管(記号c)を加熱してその管内温度を1200℃とする。吸
収瓶(記号e)に,あらかじめ,よう素酸カリウム標準溶液[2.g]]を硫黄含有率に応じて,予想される全使用
量の80〜90%を滴加しておく。
5.2
二酸化硫黄の発生 試料をはかりとってひょう量瓶に移し入れ,酸化タングステン(Ⅵ)[2.c)]1gを加
えて十分に混合した後,ボートに移し入れてカバーをかぶせ,挿入棒で燃焼管(記号c)の加熱部の中央に挿
入して気密に栓をする。直ちに,窒素を150〜200ml/min(2)の流量で送入する。
注(2) 高硫黄含有率の試料については,窒素送入流量を150/minに調節する。
5.3
吸収及び滴定 発生した二酸化硫黄を窒素と共に吸収瓶(記号e)に導き,吸収液の青色が消えない程
度に,よう素酸カリウム標準溶液[2.g)]を少量ずつ絶えず滴加し,青色が消えなくなってから引き続き5分
間窒素を流す。燃焼管(記号c)の出口の栓を外し,吸収液でガラス管内を数回洗浄し,再び栓をして窒素を
流してわずかに青色を呈するまで滴定し,終点とする。
6. 空試験 試料を用いないで5.の手順に従って試料と同じ操作を行う。
7. 検量線の作成 ふっ化カルシウム[2.f)]0.5g及び酸化タングステン(Ⅵ)[2.c)]1gをひょう量瓶にはかりと
って,十分に混合した後,ボートに移し入れたものを5個準備し,それぞれに標準硫黄液[2.h)]0ml, 0.10ml,
0.20m, 0.30ml及び0.40ml(硫黄として0μg,500μg,1000μg,1500μg,及び2000μg)を加える。これらのボ
ートを105±5℃で2時間乾燥し,デシケーター中で放冷する。以下,5.1〜5.3の操作を行い,よう素酸カ
リウム標準溶液[2.g)]の全使用量と硫黄量との関係線を求めて検量線とする(3)。
注(3) 検量線は,一連の試料を分析する都度,作成する。
8. 計算 5.3及び6.で得た滴定量と7.で作成した検量線とから硫黄量を求め,試料中の硫黄含有率を次の
式によって算出する。
m
A
A
2
1−
=
S
ここに,S :試料中の硫黄含有率[%(m/m)]
A1:試料吸収液中の硫黄検出量(g)
A2:空試験吸収液中の硫黄検出量(g)
m :試料はかりとり量(g)

25
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9. 許容差 許容差は,附属書6表2による。
附属書6表 2 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[0.0059×(硫黄含有率)+0.0012]
f(n)[0.0165×(硫黄含有率)+0.0040]
参考1. この許容差は,硫黄含有率0.01%(m/m)以上0.826%(m/m)以下の試料を用いて求めたものであ
る。表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場合f(n)=2.8で
ある。
26
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書7(規定)ひ素定量方法−三水素化ひ素気化分離
-ジエチルジチオカルバミン酸銀吸光光度法
1. 要旨 試料を塩酸,硝酸及び臭素で分解し,硫酸を加えて加熱し白煙を発生させた後,ひ素を亜鉛で
還元し,発生した三水素化ひ素をジエチルジチオカルバミン酸銀に吸収させ,光度計を用いてその吸光度
を測定する。
2. 試薬 試薬は,次による。
a) 塩酸
b) 硝酸
c) 硫酸(1+2)
d) 亜鉛 砂粒状で,ひ素を含まないもの。
e) 臭素水[飽和,約3.5%(v/v)]
f)
よう化カリウム
g) 塩化すず(Ⅱ)溶液 塩酸80mlをビーカー(200ml)に入れ,水浴上で加熱しながら塩化すず(Ⅱ)二水和物
40gを少量ずつ加えて溶解し,室温まで冷却した後,塩酸で液量100mlとする。
h) 酢酸鉛溶液 酢酸鉛三水和物12gを水約80mlに溶解し,酢酸1,2滴を加えた後,水で100mlとする。
i)
クロロホルム
j)
吸収液A ジエチルジチオカルバミン酸銀0.25gと2,3-ジメトキシストリキニジン-10-オン・二水和物
0.1gにクロロホルム100mlを加え,よくかき混ぜて溶解する。
k) 吸収液B ジエチルジチオカルバミン酸銀0.5gにピリジン100mlを加え,よくかき混ぜて溶解する。
l)
標準ひ素液(1μgAs/ml) 三酸化二ひ素(JIS K 8005)0.1320gをはかりとってビーカー(500ml)に移
し入れる。これに水酸化ナトリウム溶液(4g/L)2mlを加えて溶解し,水で約400mlとする。硫酸(1+10)
を加えて微酸性とした後,溶液を1000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄め
て原液とする。この原液を,使用の都度,必要量だけ水で正確に100倍に薄めて標準ひ素液とする。
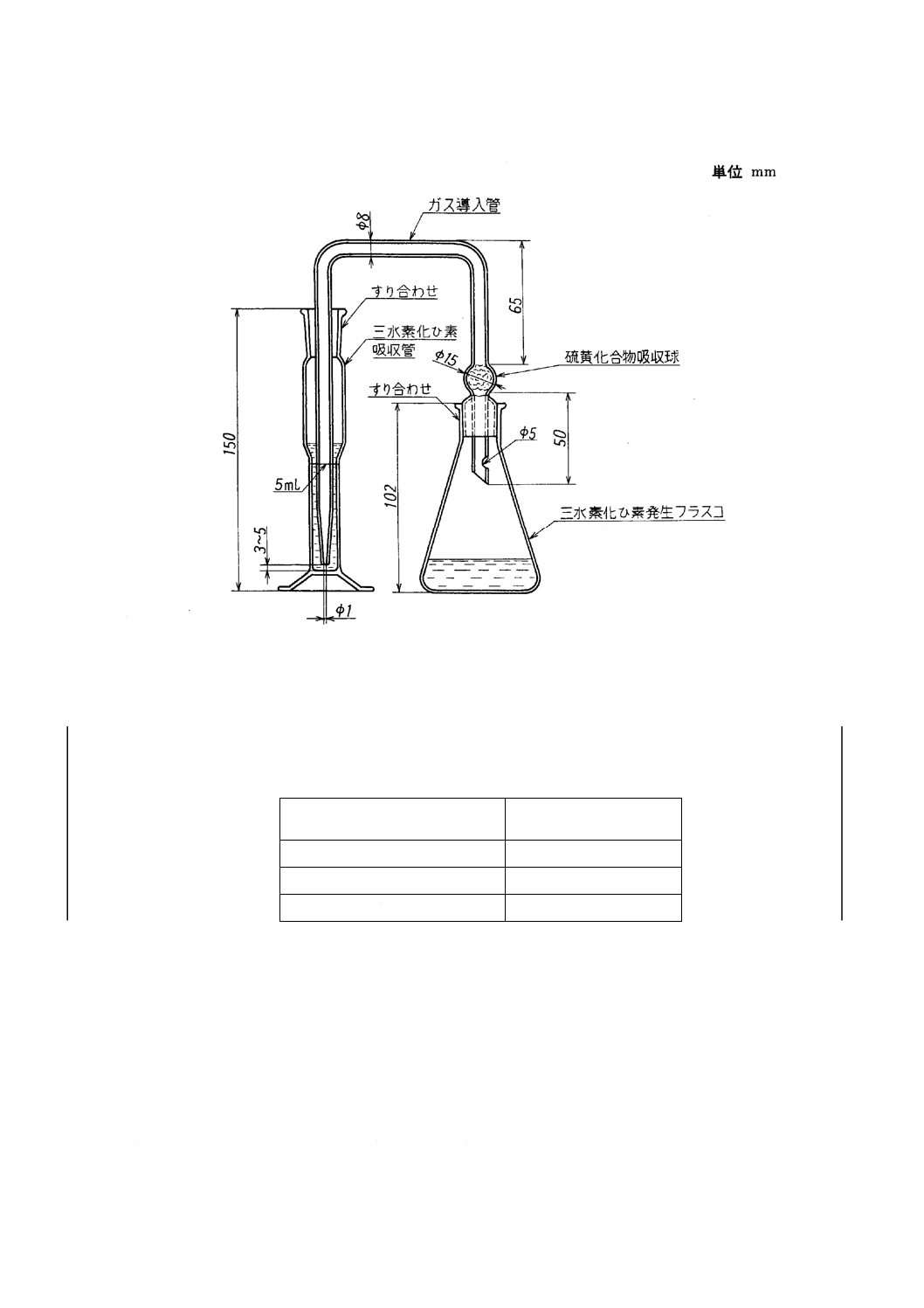
3. 装置及び器具 三水素化ひ素発生装置は,一般に次のものを用いる(附属書7図1参照)。
a) 三水素化ひ素発生フラスコ ガス導入管を取り付けた容量約100mlのガラス製三角フラスコ。
b) ガス導入管 硫黄化合物吸収球をもち,かつ,先を細くしたガラス管。硫黄化合物吸収球には,酢酸
鉛溶液[2.h)]で湿したガラスウールを充てんする。
c) 三水素化ひ素吸収管 5ml目盛付きの容量約15mlのガラス製シリンダー。
27
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書7図 1 三水素化ひ素発生・吸収装置の例
4. 試料はかりとり量 試料はかりとり量は,附属書7表1による。
附属書7表 1 試料はかりとり量
ひ素含有率
%(m/m)
試料はかりとり量
g
0.0002以上0.004未満
0.50
0.004 以上 0.008未満
0.25
0.008 以上 0.02 以下
0.10
5. 操作
5.1
試料溶液の調製 試料をはかりとってPTFE製ビーカー(100ml)に移し入れ,PTFE製時計皿で覆
い,塩酸6ml,硝酸2ml,硫酸(1+2)5ml及び臭素水5mlを加えて加熱する。三酸化硫黄の白煙が発生する
まで加熱を続け,室温まで放冷した後,時計皿の下面を水で洗って時計皿を取り除き,少量の水で塩類を
溶解する。
5.2
三水素化ひ素の発生及び呈色 三水素化ひ素の発生と呈色は, 次の手順によって行う。
a) 5.1で得た溶液を約10mlの水を用いて三水素化ひ素発生フラスコ[3.a)]に移し入れる。
b) 塩酸5ml,よう化カリウム3g及び塩化すず(Ⅱ)溶液[2.g)]3mlを加え,液量が40mlになるまで水を加

28
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
えて振り混ぜ,約15分間放置する。
c) 三水素化ひ素吸収管[3.c)]に吸収液A[2.j)]又は吸収液B[2.k)]のいずれかを正確に5ml加える。
d) 三水素化ひ素発生フラスコに亜鉛[2.d)]3gを加え,直ちにガス導入管で三水素化ひ素吸収管に連結し,
1時間放置し,発生した三水素化ひ素を吸収液に吸収させて呈色させる(1)。
注(1) 吸収液Aを用いた場合は,クロロホルムの一部が揮散して吸収液量が減少するので,吸収させ
た後,クロロホルムを加えて液量を5mlとする。
5.3
光度の測定 5.2d)で得た呈色溶液の一部を光度計の吸収セル(1cm)に取り,吸収液を対照液(2)として
波長530nmにける吸光度を測定する。
注(2) 5.2c)で使用した吸収液と同じ吸収液を用いる。
6. 空試験 試料を用いないで,5.1〜5.3のの手順に従って試料と同じ操作を試料と併行して行う。
7. 検量線の作成 5個の三水素化ひ素発生フラスコ[3.a)]を用意し,それぞれに標準ひ素液[2.l)] 0ml,
4.0ml, 10.0ml, 15.0ml及び20.0ml(ひ素として0μg,5μg,10μg,15μg及び20μg)を取る。以下5.2b)〜
5.3の手順に従って試料と同じ操作を試料と併行して行い,得た吸光度と呈色溶液中のひ素量との関係を作
成し,その関係線を原点を通るように平行移動して検量線とする。
8. 計算 5.3及び6.で得た吸光度と7.で作成した検量線からひ素量を求め,試料中のひ素含有率を次の式
によって算出する。
m
A
A
2
1−
=
As
ここに,As:試料中のひ素含有率[%(m/m)]
A1 :試料溶液中のひ素検出量(g)
A2 :空試験液中のひ素検出量(g)
m :試料はかりとり量(g)
9. 許容差 許容差は,附属書7表2による。
附属書7表 2 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[0.0188×(ひ素含有率)+0.00004]
f(n)[0.0250×(ひ素含有率)+0.00007]
参考
この許容差は,ひ素含有率0.0002%(m/m)以上0.014%(m/m)以下の試料を用いて求めたもので
ある。表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場合f(n)=2.8
である。
29
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書8(規定)鉛定量方法−よう化物抽出原子吸光法
1. 要旨 試料を硝酸,ふっ化水素酸及び過塩素酸で分解した後,乾固させる。これに塩酸を加えて溶解
し,塩酸酸性溶液としてからアスコルビン酸及びよう化カリウムを加え,生成する鉛のよう化物錯体をト
リオクチルフォスフィンオキシド(以下,TOPOという。)・4-メチル-2-ペンタノンで抽出し,有機相を原
子吸光光度計の空気・アセチレンフレーム中に噴霧し,その吸光度を測定する。
2. 試薬 試薬は,次による。
a) 塩酸(1+1)
b) 硝酸
c) 過塩素酸
d) ふっ化水素酸
e) 炭酸カルシウム(アルカリ分析用)
f)
よう化ナトリウム溶液 よう化ナトリウム41.5g及びアスコルビン酸10gを温水約60mlに溶解し,室
温まで冷却した後,塩酸(1+1)30mlを加え,水で液量を100mlとする。
g) アスコルビン酸溶液(150g/L) この溶液は,使用の都度,調製する。
h) TOPO・4-メチル-2-ペンタノン溶液 TOPO 5gを4-メチル-2-ペンタノン100mlに溶解する。
i)
4-メチル-2-ペンタノン
j)
標準鉛液(20μgPb/ml) 鉛[99.9%(m/m)以上]1.000gをはかりとってビーカー(300ml)に移し入れる。
時計皿で覆い,硝酸(1+1)30mlを加え,穏やかに加熱して分解する。水約50mlを加えて再び加熱し,
1〜2分間煮沸して窒素酸化物を追い出す。常温まで冷却した後,時計皿の下面を水で洗って時計皿を
取り除き,溶液を1000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原液とする。
この原液を,使用の都度,必要量だけ水で正確に50倍に薄めて標準鉛液とする。
3. 試料はかりとり量 試料はかりとり量は,1.0gとする。
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかりとって白金皿(100番)に移し入れる。硝酸5ml,ふっ化水素酸10ml及び過塩素酸10ml
を加え,加熱して過塩素酸の白煙が発生しはじめたら熱源より降ろす。室温まで放冷した後,少量の
水で白金皿の縁を洗い,再び加熱し,蒸発して乾固する。
b) 室温まで放冷した後,塩酸(1+1)10mlを加え,加熱して可溶性の塩類を溶解し,室温まで冷却する。
4.2
鉛の抽出 鉛の抽出は,次の手順によって行う。
a) 4.1b)で得た溶液を少量の水を用いて分液漏斗(100ml)に移し入れ,アスコルビン酸溶液[2.g)]10mlを
加え,よく振り混ぜた後,約5分間放置する。
b) よう化ナトリウム溶液[2.f)]10mlを加え,水を加えて液量を約50mlとする。TOPO・4-メチル-2-ペン
タノン溶液[2.h)] を正確に10ml加え,1分間激しく振り混ぜ,靜置して2層に分離した後,下層の水
相を捨てる。
c) 有機相を乾いたろ紙(5種A)を用いて,共栓付き容器(50ml)にろ過して保存する。

30
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.3
吸光度の測定 4.2c)で得た有機相の一部を,4-メチル-2-ペンタノンを用いてゼロ点を調整した原子
吸光光度計の空気・アセチレンフレーム中に噴霧し,波長283.3nmにおける吸光度を測定する。
5. 空試験 試料を用いないで,4.1〜4.3の手順に従って試料と同じ操作を試料と併行して行う。
6. 検量線の作成 検量線の作成は,次の手順によって行う。
a) 炭酸カルシウム1.0gを6個のビーカー(200ml)にはかりとり,,それぞれに標準鉛液[2.j)] 0ml, 1.0ml,
2.0ml, 3.0ml, 4.0ml及び5.0ml(鉛として0μg,20μg,40μg,60μg,80μg及び100μg)を加えた後,
塩酸(1+1)10mlを少量ずつ加えて,炭酸カルシウムを溶解する。1,2分間煮沸し,二酸化炭素の気泡が
発生しなくなったら室温まで冷却する。
b) 溶液を少量の水を用いて分液漏斗(100ml)に移し入れ,アスコルビン酸溶液[2.g)] 10mlを加え,よく
振り混ぜた後、約5分間放置する。以下,4.2b)〜4.3の手順に従って,試料と同じ操作を試料と併行
して行い,得た吸光度と鉛量との関係を作成し,その関係線を,原点を通るように平行移動して検量
線とする。
7. 計算 4.3及び5.で得た吸光度と6.で作成した検量線から鉛量を求め,試料中の鉛含有率を次の式によ
って算出する。
100
×
−
=
m
Pb
2
1A
A
ここに,Pb:試料中の鉛含有率[%(m/m)]
A1 :試料溶液中の鉛検出量(g)
A2 :空試験液中の鉛検出量(g)
m :試料はかりとり量(g)
8. 許容差 許容差は,附属書8表による。
附属書8表1 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[0.0085×(鉛含有率)+0.00007]
f(n)[0.0448×(鉛含有率)+0.00005]
参考1. 参考 この許容差は,鉛含有率0.0002%(m/m)以上0.0043%(m/m)以下の試料を用いて求め
たものである。表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場
合f(n)=2.8である。
31
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書9(規定)アンチモン定量方法−よう化物抽出原子吸光法
1. 要旨 試料を硝酸,ふっ化水素酸及び過塩素酸で分解した後,硫酸を加え,加熱して白煙を発生さ
せる。塩酸を加えて溶解し,アンチモンをアスコルビン酸及びよう化カリウムを加え,生成するアンチ
モンのよう化物錯体をトリオクチルフォスフィンオキシド(以下,TOPOという。)・4-メチル-2-ペンタ
ノンで抽出し,有機相を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,その吸光度を測定す
る。
2. 試薬 試薬は,次による。
a) 塩酸(1+1)
b) 硝酸
c) 過塩素酸
d) ふっ化水素酸
e) 硫酸(1+1)
f)
よう化カリウム溶液 よう化カリウム30g及びアスコルビン酸10gを温水約80mlに溶解し,室温
まで冷却した後,塩酸10mlを加え,水で液量を100mlとする。この溶液は,使用の都度,調製す
る。
g) アスコルビン酸
h) TOPO・4-メチル-2-ペンタノン溶液 TOPO 5gを4-メチル-2-ペンタノン100mlに溶解する。
i)
4-メチル-2-ペンタノン
j)
標準アンチモン液(50μgSb/ml) アンチモン[99.9%(m/m)以上]1.000gをはかりとってビーカー
(200ml)に移し入れる。時計皿で覆い,王水20mlを加え,穏やかに加熱して分解する。常温まで冷
却した後,時計皿の下面を塩酸(1+1)で洗って時計皿を取り除き,1000mlの全量フラスコに塩酸(1+1)
を用いて移し入れ,塩酸(1+1)で標線まで薄めて原液とする。この原液溶液を使用の都度,必要量だ
け塩酸(1+1)で正確に20倍に薄めて標準アンチモン液とする。
3. 試料はかりとり量 試料はかりとり量は,0.50gとする。
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかりとって白金皿(100番)に移し入れる。硝酸5ml,ふっ化水素酸10ml及び過塩素酸10ml
を加えて加熱し,過塩素酸の白煙を発生させて試料を分解する。
b) 硫酸(1+1)10mlを加えて振り混ぜ,加熱を続けて三酸化硫黄の白煙を約10分間発生させる(1)。
注(1) 加熱を続けて白煙の発生が一時弱くなり,再び白煙が強く発生してから10分間とする。
c) 室温まで放冷した後,塩酸(1+1)10mlを加え,2,3分間加熱して可溶性の塩類を溶解(2)した後,室温
まで冷却し,アスコルビン酸5gを加えて約5分間放置する。
注(2) 硫酸カルシウムの沈殿が完全に溶解しなくてもよい。
4.2
アンチモンの抽出
a) 4.1c)で得た溶液を少量の水を用いて分液漏斗(100ml)に移し入れる。
32
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) よう化カリウム溶液[2.f)]15mlを加え,水を加えて液量を約50mlとする。TOPO・4-メチル-2-ペン
タノン溶液[2.h)] を正確に10ml加え,1分間激しく振り混ぜ,靜置して2層に分離した後,下層の
水 相を捨てる。
c) 有機相を乾いたろ紙(5種A)を用いて,共栓付き容器(50ml)にろ過して保存する。
4.3
吸光度の測定 4.2c)で得た有機相の一部を4-メチル-2-ペンタノンを用いてゼロ点を調整した原子
吸光光度計の空気・アセチレンフレーム中に噴霧し,波長217.6nmにおける吸光度を測定する。
5. 空試験 試料を用いないで,4.1〜4.3の手順に従って試料と同じ操作を試料と併行して行う。
6. 検量線の作成 検量線の作成は,次の手順によって行う。
a) 5個のビーカー(100ml)を用意し,それぞれに硫酸(1+1)10mlを加え,附属書9表1に従って標準アン
チモン液[2.j)]及び塩酸(1+1)を加えた後,アスコルビン酸5gを加えて約5分間放置する。
b) 溶液を少量の水を用いて分液漏斗(100ml)に移し入れる。以下,4.2b)〜4.3の手順に従って試料と同
じ操作を試料と併行して行い,得た吸光度とアンチモン量との関係を作成し,その関係線を原点を
通るように平行移動して検量線とする。
附属書9表 1 標準アンチモン液及び塩酸の添加量
単位 ml
標準アンチモン液
塩酸(1+1)
0
10
1.0
9
2.0
8
3.0
7
4.0
6
7. 計算 4.3及び5.で得た吸光度と6.で作成した検量線からアンチモン量を求め,試料中のアンチモン
含有率を次の式によって算出する。
100
2
1
×
−
=
m
A
A
Sb
ここに,Sb:試料中のアンチモン含有率[%(m/m)]
A1 :試料溶液中のアンチモン検出量(g)
A2 :空試験液中のアンチモン検出量(g)
m :試料はかりとり量(g)
8. 許容差 許容差は,附属書9表2による。

33
M 8514:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書9表 2 許容差
単位 %(m/m)
室内再現許容差
室間再現許容差
f(n)[−0.0148×(アンチモン含有率)+0.00023]
f(n)[−0.0107×(アンチモン含有率)+0.00029]
参考1. この許容差は,アンチモン含有率0.001%(m/m)以上0.0034%(m/m)以下の試料を用いて求め
たものである。表におけるf(n)は,JIS Z 8402-6の表1の許容範囲の係数であり,n=2の場
合f(n)=2.8である。