M 8226:2006
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,社団法人日本鉄鋼
連盟(JISF)から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,日本工業標準調査会の
審議を経て,経済産業大臣が改正した日本工業規格である。
今回の改正では,従来のJIS M 8226に規定していたモリブドひ酸青吸光光度法(ISO 7834:1987 Iron ores
- Determination of arsenic content - Molybdenum blue spectrophotometric methodの翻訳規格)に加えて,既に鉄
及び鋼中のひ素定量法としてJISG1257に規定されている電気加熱方式原子吸光法を基礎に、鉄鉱石定量
法として開発した2方法を新たに規定した。
これによって,JIS M 8226:1997は改正され,この規格に置き換えられる。
この規格の一部が,技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の
実用新案登録出願に抵触する可能性があることに注意を喚起する。経済産業大臣及び日本工業標準調査会
は,このような技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の実用新
案登録出願にかかわる確認について,責任はもたない。
JIS M 8226には,次に示す附属書がある。
附属書1(規定)モリブドひ酸青吸光光度法
附属書1A(規定)分析値の採択手順のフローシート
附属書1B(参考)国際共同実験に関する追加情報
附属書1C(参考)国際共同実験で得られた精度データ
附属書2(規定)アルカリ融解−電気加熱方式原子吸光法
附属書3(規定)酸溶解−電気加熱方式原子吸光法
附属書4(参考)JISと対応する国際規格との対比表
M 8226:2006
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1. 適用範囲 ························································································································ 1
2. 引用規格 ························································································································ 1
3. 一般事項 ························································································································ 1
4. 定量方法の区分 ··············································································································· 1
附属書1(規定)モリブドひ酸青吸光光度法 ·············································································· 3
附属書1A(規定)分析値の採択手順のフローシート ·································································· 10
附属書1B(参考)国際共同実験に関する追加情報 ····································································· 11
附属書1C(参考)国際共同実験で得られた精度データ ······························································· 12
附属書2(規定)アルカリ融解−電気加熱方式原子吸光法 ··························································· 13
附属書3(規定)酸溶解−電気加熱方式原子吸光法 ···································································· 18
附属書4(参考)JISと対応する国際規格との対比表 ································································· 22

2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
M 8226:2006
鉄鉱石−ひ素定量方法
Iron ores−Methods for determination of arsenic content
序文 この規格は,1987年に第1版として発行されたISO 7834,Iron ores - Determination of arsenic content
- Molybdenum blue spectrophotometric methodを翻訳し,技術的内容を変更することなく附属書1として作成
した日本工業規格であるが,今回の改正で国際規格にはない定量方法を附属書2及び附属書3として追加
した。この規格とISO 7834との対比表を附属書4に示す。
1. 適用範囲 この規格は,鉄鉱石中のひ素定量方法について規定する。
備考 この規格の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide21に基づき,IDT(一致している),MOD(修
正している),NEQ(同等でない)とする。
ISO 7834:1987,Iron ores−Determination of arsenic content - Molybdenum blue spectrophotometric
method (MOD)
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版(追補を含む。)を適用する。
JIS G 1257 鉄及び鋼−原子吸光分析方法
JIS K 8005 容量分析用標準物質
JIS M 8202 鉄鉱石−分析方法通則
JIS M 8702 鉄鉱石−サンプリング及び試料調製方法
備考 ISO 3082 Iron ores−Sampling and sample preparation proceduresからの引用事項は,この規格
の該当事項と同等である。
JIS Z 8402-6 測定方法及び測定結果の精確さ(真度及び精度)−第6部:精確さに関する値の実用的
な使い方
ISO 648 Laboratory glassware−One-mark pipettes
ISO 1042 Laboratory glassware−One-mark volumetric flasks
ISO 7764 Iron ores−Preparation of predried test samples for chemical analysis
3. 一般事項 定量方法に共通な一般事項は,JIS M 8202による。
4. 定量方法の区分 ひ素の定量方法は,次のいずれかによる。定量方法の選択は,ひ素含有率,要求さ
れる分析精度などを考慮して行う。
a) モリブドひ酸青吸光光度法(ISO 7834) この方法は,ひ素含有率質量分率0.000 1 %以上0.1 %以下
2
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の試料に適用するもので,附属書1による。
b) アルカリ融解−電気加熱方式原子吸光法 この方法は,ひ素含有率質量分率0.000 5%以上0.06 %以下
の試料に適用するもので,附属書2による。
c) 酸溶解−電気加熱方式原子吸光法 この方法は,ひ素含有率質量分率0.000 5 %以上0.02 %以下の試
料に適用するもので,附属書3による。ただし,この方法は,モリブドひ酸青吸光光度法又はアルカ
リ融解−電気加熱方式原子吸光法による定量値と差が生じないことが確認された品種の鉄鉱石だけに
適用できる。
3
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(規定)モリブドひ酸青吸光光度法
序文 この附属書は,1987年第1版として発行されたISO 7834 Iron ores-Determination of arsenic
content-Molybdenum blue spectrophotometric methodを翻訳し,技術的内容及び規格票の様式を変更すること
なく作成したものである。
なお,この附属書及び附属書1Aで下線(点線)を施してある“参考”は,原国際規格にはない事項で
ある。
1. 適用範囲 この附属書は,鉄鉱石中のひ素を蒸留分離モリブドひ酸青吸光光度法によって定量する方
法について規定する。
この方法は,天然鉄鉱石,精鉱,及び焼結鉱などの塊成鉱で,ひ素含有率質量分率0.000 1 %以上0.1 %
(1〜1 000 μg/g)以下の範囲のものに適用する。
参考 この方法は,硫化鉄焼鉱,スケール及びダスト又はこれらの粉粒状のものを加工した団鉱など
の鉄原料にも適用できる。
2. 引用規格 引用規格は,本体に規定している。
参考 原国際規格ではISO 3081,ISO 3082,ISO 3083が引用されているが,2000年の改正でこれら
の規格はISO 3082に統合され,ISO 3082の名称も変更されている。JIS M 8702;2002はISO
3082;2000とMODの関係にある。
3. 原理 試料を,過酸化ナトリウムで焼成して分解し,水及び塩酸で溶解する。
蒸留フラスコに溶液を移し,溶液を濃縮し,臭化カリウム及び硫酸ヒドラジニウムで処理した後,酸度
の調整を行う。三塩化ひ素を蒸留し,蒸留液を硝酸液中に集める。
蒸留液を規定の温度で蒸発乾固し,七モリブデン酸六アンモニウム及び硫酸ヒドラジニウムの混液で処
理し,モリブドひ酸青錯体を呈色させる。
840nm付近の波長で吸光度を測定する。
4. 試薬 分析の際は,分析用試薬(recognized analytical grade)及び脱イオン水又はこれと同等の純度を
もつ水を使用する。
備考 分析試料のひ素含有率が低い(<20 μg/g)場合に精度を上げるためには,20mmセルでの空試験
吸光度測定値が0.025(ひ素の1 μ gに相当)以下になるように試薬を選定するか,又は精製さ
れた試薬を用いるのが望ましい。特に,硝酸は再蒸留を必要とする場合もあり,器具のより厳
重な洗浄操作(5.2)を必要とする場合もある。
4.1
過酸化ナトリウム(Na2O2) 微細粉末
4.2
臭化カリウム(KBr)
4.3
硫酸ヒドラジニウム(N2H4・H2SO4)
4.4
塩酸(密度1.16〜1.19 g/ml)
4.5
硝酸(密度1.4 g/ml)の希釈液(1+1)
4.6
硫酸(密度1.84 g/ml) りんを含まないもの
4
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.7
七モリブデン酸六アンモニウム[(NH4)6Mo7O24・4H2O]溶液(10 g/l)
水400 mlをビーカー(1 l)に入れ,硫酸(4.6)133 mlをかき混ぜながら注意して加える。冷却した後,七
モリブテン酸六アンモニウム四水和物(10±0.1) gを加えて,かき混ぜながら溶解する。1 lの全量フラスコ
又はすり合わせ栓付きメスシリンダに移し入れ,標線まで水で薄めて混合する。
4.8
硫酸ヒドラジニウム(N2H4・H2SO4)溶液(0.15 g/l)
4.9
モリブデン酸―ヒドラジニウム混合液 100 mlの全量フラスコに水70 mlを入れ,七モリブデン酸
六アンモニウム溶液(4.7)(10±0.1) ml及び硫酸ヒドラジニウム溶液(4.8)10 mlを加えて,標線まで水で薄め
て,混合する。この混合液は,使用の都度調製する。
4.10 標準ひ素溶液A,200 μg/ml 三酸化二ひ素(As2O3)数百mgを105 ℃で1時間乾燥してデシケータ
内で放冷する。この乾燥品0.132 gをはかりとって,水酸化ナトリウム溶液(40 g/l)2 mlに溶かし,水30
mlを加え,メチルオレンジ指示薬を用いて硫酸(4.6)の希釈液(1+9)で中和し,次に炭酸水素ナトリウム4 g
を加える。これを500 mlの全量フラスコに移し入れ,水で標線まで薄めて混合する。
この標準溶液Aの1 mlは,ひ素200 μgを含有する。
参考 メチルオレンジ指示薬の調製は,JIS K 8001の4.4表7に記載されている。
4.11 標準ひ素溶液B,5 μg/ml 標準ひ素溶液A(4.10)25 mlを分取し,1 lの全量フラスコに移し入れ,水
で標線まで薄めて混合する。
この標準溶液Bの1 mlは,ひ素5 μgを含有する。
5. 装置 装置は,通常の分析用器具及び次のものを使用する。
備考 ピペット及び全量フラスコは,ISO 648及びISO 1042に規定されている1標線付きピペット及
び1標線付き全量フラスコでなければならない。
5.1
ジルコニウムるつぼ又はガラス質カーボン(vitreous carbon)るつぼ 容量約30 mlのもの。
5.2
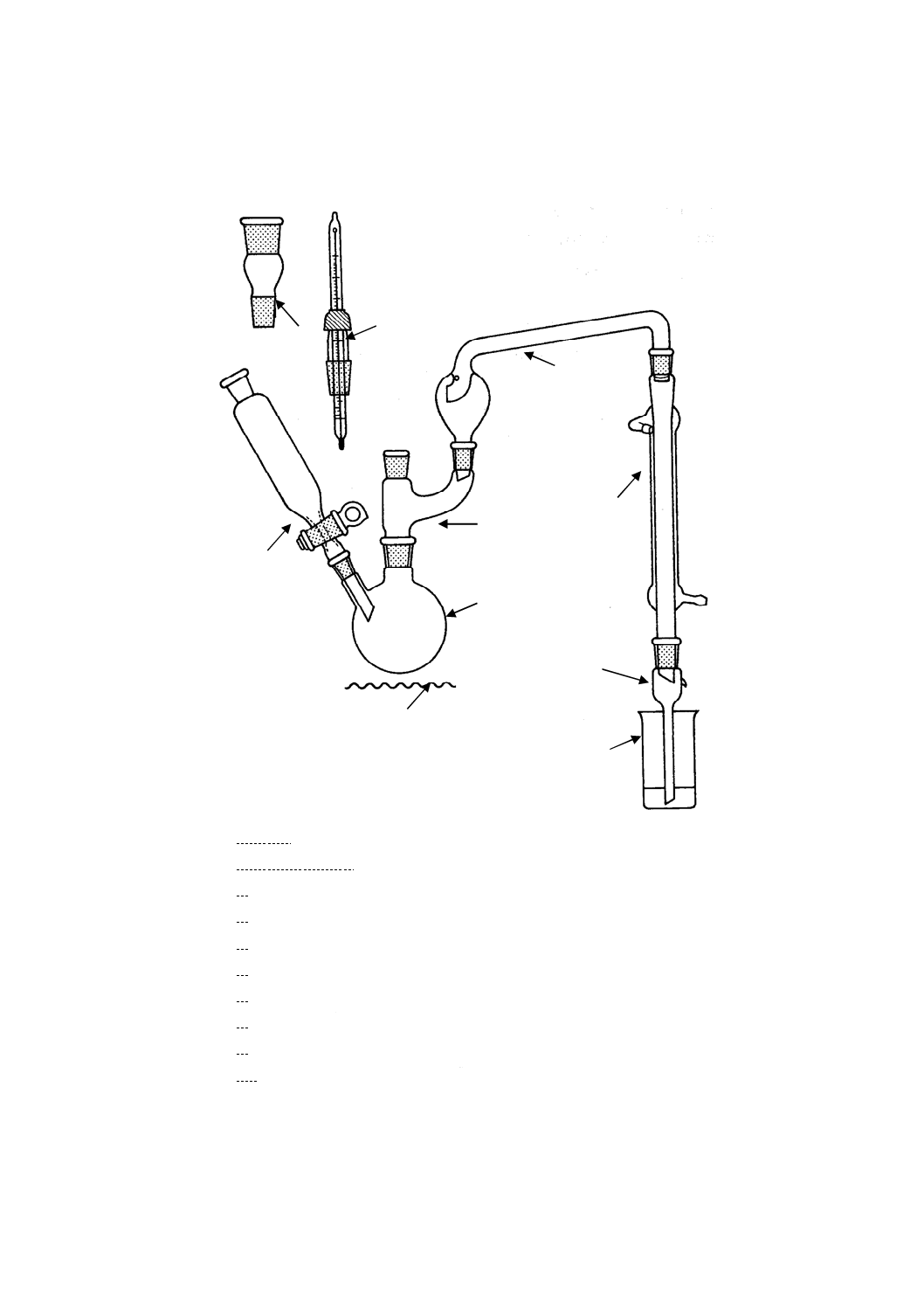
蒸留装置 蒸留装置は,注入漏斗付きの側管をもつ250 ml蒸留フラスコ,Y字形連結管,飛まつ除
去管(SH)及び水冷コンデンサで構成される(附属書1図1)。
蒸留フラスコの容量45 ml及び50 mlの位置にマークを付ける。
装置のすり合せ接合部は,ボールジョイントを用いてもよい。
備考1. 最初に装置を使用する際は,蒸留装置,受器アダプタ及び受器をよく洗浄して(すべての内
壁が薄膜状にぬれることを確認するのがよい。),この状態を必要な限り維持するのがよい。
ガラスすり合せ接合部は,有機潤滑剤をきれいに洗浄し除去した後,必要最少限の硫酸(4.6)
で滑らかにする。これとは別に,ポリテトラフルオロエチレン(PTFE)製スリーブを用いて
もよい。
2. 蒸留段階後に使用する器具(受器,ピペット,100 mlの全量フラスコなど)は,次の方法で
特別に洗浄処理をするのがよい。
最初に使用する際は,よく洗浄して,硝酸(1+10)を数時間,容器中に入れておく。これら
の容器はひ素定量用専用に保存し,また,ひ素定量用とラベルで表示しておく。常時使用の
場合は,希硝酸を入れておく時間は30分間に短縮できる。りん酸塩の混入をまねく洗剤は,
この操作では使用しないことが望ましい。
参考 原文ではクロム酸混液又は相当品による洗浄をするようにと規定しているが,クロム酸は環境
上有害なため,使用しないようにその部分は削除した。
5
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3. ヒータは,少なくとも2 ml/minの割合で蒸留できるものを使用するのがよい。困難な場合に
は,適切な断熱テープを用いて飛まつ除去管(図1の7)を断熱する。
1 熱源
2 蒸留フラスコ
3 Y字形連結管
4 注入漏斗
5 試薬注入用アダプタ(TA)
6 温度計
7 飛まつ除去管(SH)
8 水冷コンデンサ
9 受器アダプタ
10 受器[トールビーカー(200 ml)]
附属書1図 1 ひ素蒸留装置
7
1
3
2
10
8
5
4
6
9
6
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.3
分光光度計 840nm付近の吸光度の測定に適したもの。
6. サンプリング及び試料
6.1
分析用試料(laboratory sample)分析には,JIS M 8702に従って採取し,調製した粒度100 μm以下
の分析用試料を用いる。化合水又は酸化しやすい化合物の含有率が著しく高い鉱石の場合には,粒度160
μm以下の試料を用いる。
備考 化合水及び酸化しやすい化合物の著しく高い含有率についてのガイドラインは,ISO 7764に記
載されている。
参考 粒度160 μm以下の試料を用いる鉄鉱石の区分は,JIS M 8202の5.1(分析用試料)のb)による。
6.2
事前乾燥試料(predried test samples)の調製 分析用試料を十分に混合し,容器の全量を代表する
ように数インクリメントで試料を採取する。試料をISO 7764に従って(105±2) ℃で乾燥する(これを事
前乾燥試料という。)。
7. 操作
7.1
分析回数 分析は,事前乾燥試料1個について,附属書1Aに従って少なくとも独立に2回の分析を
行う。
備考 “独立に”という表現は,2度目又は続いて行った分析結果が以前の結果によって影響を受け
ないことを意味する。特にこの分析方法では,この条件は操作の繰返しが同一人が異なった時
間に,又は異なった人によって,いずれの場合も適切な検量線の再校正を含めて行われなけれ
ばならないことを意味する。
7.2
空試験及びチェック試験 一連の定量ごとに,1回の空試験と,同一種類の鉄鉱石認証標準物質の1
個を,1分析試料(1個又は数個)と併行して同一条件で分析しなければならない。認証標準物質の事前乾
燥試料は,6.2に従って調製しなければならない。
同時に数試料を分析する場合,操作が同じで同一の試薬瓶からの試薬を使うときには,1個の空試験値
で代表することができる。
同時に同一種類の鉱石の数試料を分析する場合は,1個の認証標準物質の分析値を使用することができ
る。
備考 認証標準物質は,分析試料と同一種類で,両者の性質が分析操作の変更を必要としない程度に
よく類似したものでなければならない。
7.3
分析試料(test portion) 分析試料は,6.2に従って得られた事前乾燥試料から数インクリメントを
とって,約1 gを0.001 gのけたまではかる。
備考 分析試料は,水分の再吸収を防ぐために迅速にはかりとるのがよい。
7.4
定量
7.4.1
試料の分解 ジルコニウムるつぼ又はガラス質カーボンるつぼ(5.1)に過酸化ナトリウム(4.1)3 gを
入れる。直ちにはかりとった分析試料(7.3)を入れて,薄い金属スパチュラ又はガラス棒でよく混合する。
(420±10) ℃のマッフル炉に1時間入れて焼成する。
るつぼを完全に室温まで放冷し(必要な場合,るつぼを金属ブロック上に置いて冷却してもよい。),時
計皿で覆い,わずかに時計皿を持ち上げ,焼成物の外側周辺に水0.5 mlを加える。反応が終了した後(数
分後),同じ方法で更に水1 mlを加える。数分後,水15 mlを加え,反応が再び終了したら,加熱して焼
成物を完全に分解する。蒸留フラスコの主開口部にY字形連結管及び試薬注入用アダプタ(附属書1図1
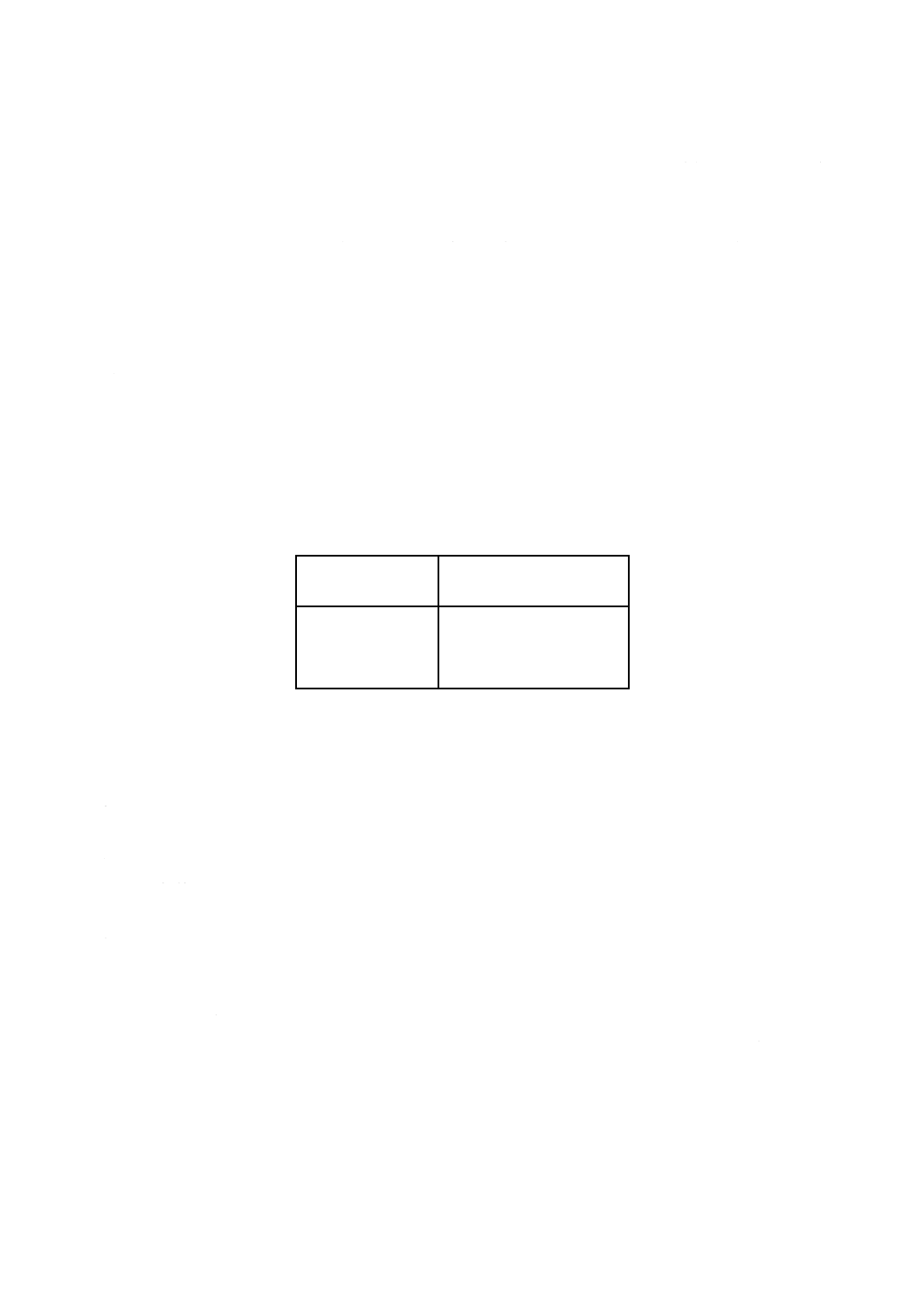
7
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の5)を取り付け,蒸留フラスコにるつぼの内容物を移す。水15 ml及び塩酸(4.4)10 mlをるつぼに加え,
残留物が溶解するまで煮沸して,蒸留フラスコに移し,水20〜25 mlで洗浄する。穏やかに煮沸して,塩
素を取り除き,45〜50 mlに濃縮する。溶液を約50 ℃に冷却する。
7.4.2
三塩化ひ素の蒸留 蒸留装置を完全に連結し(附属書1図1),注入漏斗へ塩酸(4.4)55 mlを加える。
受器アダプタを取り付け,75 mlの位置にマークを付けた受器[トールビーカー(200ml)]に硝酸(4.5)10 mlを
入れ,先端を浸す。乾いた試薬注入用アダプタ(附属書1図1の5)を用いて臭化カリウム(4.2)及び硫酸
ヒドラジニウム(4.3)1 gを加える。試薬注入用アダプタに代えて,温度計を取り付け,蒸留フラスコに塩酸
(4.4)を加える。
頭部の蒸留温度を約108 ℃に保持し,受器に全量が75 mlになるまで蒸留する(約2 ml/minの割合で蒸
留)。
備考 必要に応じて沸騰石を用いてもよい。
蒸留液を100 mlの全量フラスコに移し入れ,標線まで薄めて混合する(この液を試料原液と呼ぶ。)。
予想ひ素含有率によって,附属書1表1に従って必要分取量を決定する。ひ素含有率が60 μg/g未満の
場合は,分取せず,試料原液のままとし,ひ素含有率が60 μg/g以上の場合は分取した液を100 mlの全量
フラスコに移し,標線まで薄めて混合する。
附属書1表1 蒸留液の分取量
ひ素含有率範囲
μg/g
試料原液からの分取量
ml
1〜 60
分取なし(試料原液)
50〜 200
25
150〜 500
10
300〜 1000
5
試料原液及び空試験液,又は試料原液及び空試験液の分取希釈液を別々にトールビーカー(200ml)に移し
入れる。
7.4.3
吸光度の測定 吸光度の測定は,次による。
a) 試料原液又は分取希釈液を130 ℃以下の温度で熱板又は水浴によって蒸発乾固する。
b) 乾燥残さの入ったビーカーを,(130±5) ℃の乾燥器中に30分間放置する。
備考 乾燥器の代わりに,最低温度125 ℃が得られる熱板を用いてもよい。
c) 冷却した後,モリブデン酸−ヒドラジニウム混合液(4.9)20 mlを加え,熱板上で(95±5) ℃で25〜30
分間加熱する。
備考 熱板の代わりに水浴を用いてもよい。
d) 室温まで放冷した後,25 mlの全量フラスコに移し入れ,モリブデン酸−ヒドラジニウム混合液(4.9)
を用いてビーカーを洗浄後,洗浄液を全量フラスコに移し入れ,標線までモリブデン酸−ヒドラジニ
ウム混合液(4.9)で薄めて混合する。
e) 10 mmの光路長のセルを用い,840 nm付近の最大吸収波長で,モリブデン酸−ヒドラジニウム混合液
(4.9)を対照液としてゼロ合せをして試料溶液,及び空試験溶液又は希釈空試験溶液の吸光度を測定す
る。試料溶液の吸光度は空試験溶液又は希釈空試験溶液の吸光度を差し引いて補正する。
備考 10 mmセルで吸光度が0.025未満の場合は,20 mmセルを使用するのが望ましい。この場合,
空試験溶液及び検量線シリーズも20 mmセルで測定する。
8
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.5
検量線の作成 標準ひ素溶液B(4.11)を各々0 ml,1 ml,2 ml,5 ml及び12 ml分取し,順次蒸留装置
に移し入れ,水で50 mlに薄め,塩酸(4.4)2.5 mlを加えて,以下7.4.2及び7.4.3に従って操作を続ける。
ひ素の量とひ素無添加溶液で得られた吸光度を差し引いて補正した吸光度との関係をプロットし,8.1
に定義する検量線のこう配(Z)を算出する。Z値は,10 mmセルで約76となるのが望ましい。
8. 結果の表示
8.1
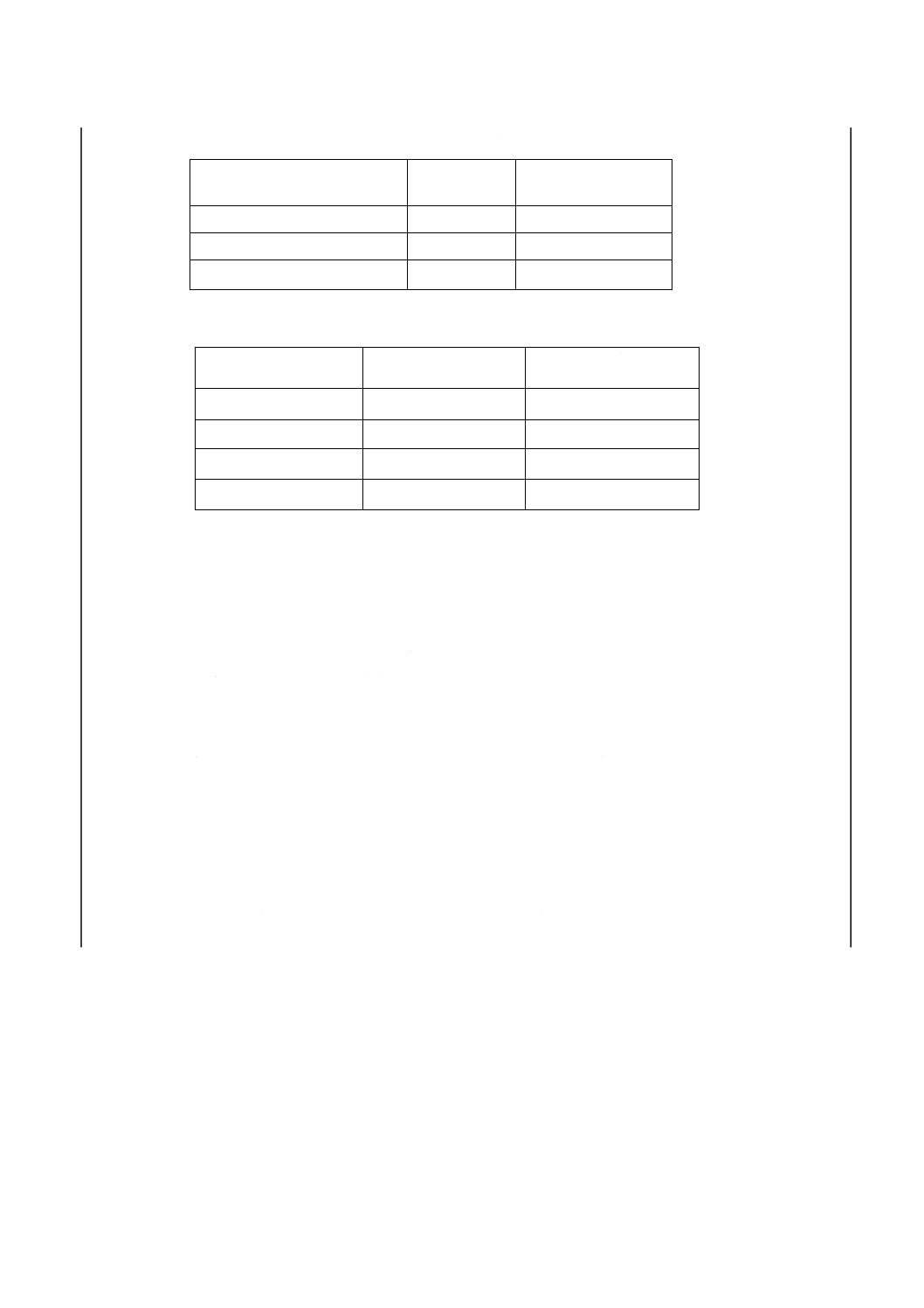
ひ素含有率の計算 ひ素含有率WAs(μg/g)は,式(1)によって計算する。
m
D
Z
E
W
×
×
=
As
····················································································· (1)
ここに, m:はかりとり試料の質量(g)
E:モリブデン酸−ヒドラジニウム混合液(4.9)を対照液として測定し
た空試験溶液又は希釈空試験溶液の吸光度を差し引いて補正した
試料溶液の吸光度
Z:検量線こう配で,次の式によって表す。
吸光度
μg
中ひ素量
25ml
呈色溶液
D:希釈率[全蒸留液を使用した場合はD=1,
分取した場合はD=100/分取量(ml)]
8.2
結果の一般的処理
8.2.1
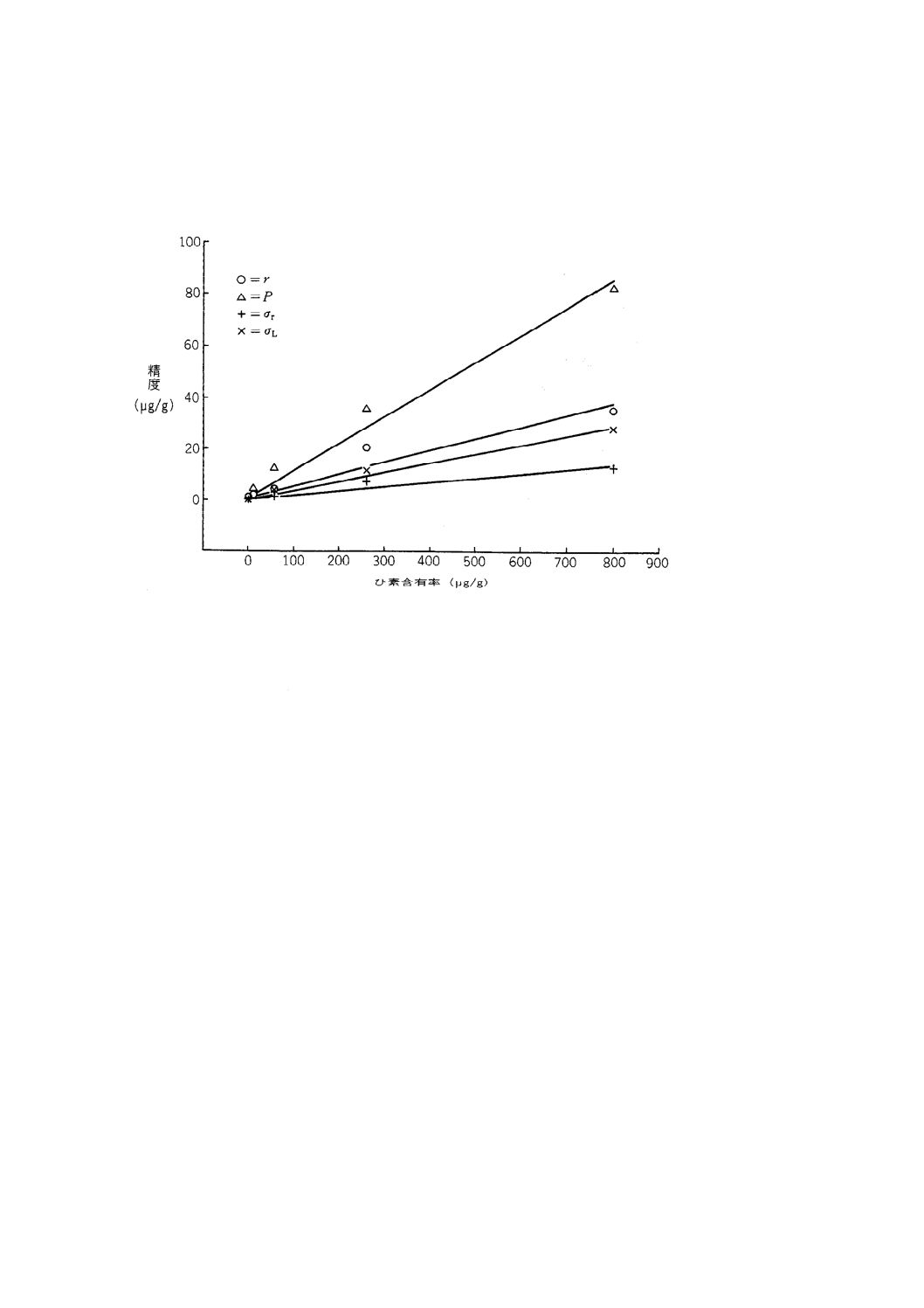
精度及び許容差 この分析方法の精度(μg/g)は,次の回帰式で求める(1)。
注(1) 追加の情報は,附属書1B及び附属書1Cに示す。
r =0.046 0 X+0.90 ··············································································· (2)
P =0.105 8 X+0.83 ··············································································· (3)
σr =0.016 3 X+0.32 ··············································································· (4)
σL =0.035 6 X+0.21 ··············································································· (5)
ここに, X:分析試料のひ素(μg/g)
― 室内計算式[式(2)及び式(4)]:2個の分析値の算術
平均
― 室間計算式[式(3)及び式(5)]:2分析室の最終分析
値(8.2.3)の算術平均
r:室内許容差
P:室間許容差
σr:室内標準偏差
σL:室間標準偏差
8.2.2
分析値の採択 認証標準物質で求めた結果について,その分析結果と認証標準物質の認証値との間
に統計的に有意差が認められてはならない。10か所以上の分析室で,真度(accuracy)及び精度(precision)
ともにこの方法と同レベルの分析方法により分析した認証標準物質に対しては,有意差の検定に式(6)を
用いる。
9
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
n
N
n
s
s
A
A
2
r
2
L
c
c
W
2Wc
2Lc
c
2
σ
+
σ
+
+
≤
−
······················································· (6)
ここに,Ac:認証値
A:認証標準物質を分析して得られた結果又はその平均値
sLc:認証値を決定した分析室の室間標準偏差
sWc:認証値を決定した分析室の室内標準偏差
nWc:認証値を決定した分析室の分析回数の平均
Nc:認証値を決定した分析室の数
n:認証標準物質の分析回数(ほとんどの場合n=1)
σr及びσL:8.2.1の定義による
式(6)の左辺が右辺より小さいか又は等しい場合は,差|Ac−A|は統計的に有意ではなく,逆の場合
は,統計的に有意である。
差が有意であるときは,試料の分析と同時に認証標準物質の分析を繰り返す。差が再び有意である場合
は,同じ種類で別の認証標準物質を用いて同じ操作を繰り返さなければならない。
分析試料の二つの分析値の範囲が8.2.1の式(2)で計算されたrの限度を超えるときは,附属書1Aの
フローシートに従って,更にもう一度同じ種類の認証標準物質と共に分析試料の分析を行わなければなら
ない。
備考 認証標準物質の情報が不十分なときには,次の手順を用いる。
a) 室間標準偏差を推定するのに十分なデータがあれば,
Wc
2Wcn
/
s
を削除して,Lc
sを室平均値
の標準偏差とみなす。
b) 認証標準物質の認証が1分析室だけで行われている場合,又は室間の分析結果がない場合に
は,この認証標準物質はこの規格には適用しない。その使用が避けられない場合は,次の式
を用いる。
n
A
A
2r
2L
c
2
2
σ
σ+
≤
−
··················································································· (7)
8.2.3
最終結果の計算 分析結果は,分析試料の採択し得る値の算術平均か,又は附属書1Aに規定した
手順によって求め,100 μg/g未満のひ素の含有率については小数点以下1けたに丸め,ひ素含有率100 μg/g
以上の場合は整数値に丸める。
最終結果は,質量分率%で表す。ただし,注文者の要求がある場合は μg/gで表す。
9. 試験結果の報告 試験結果の報告には,次の情報を記載する。
a) 分析所の名称及び所在地
b) 試験報告書の発行日付
c) この規格及び附属書の番号(JIS M 8226附属書1)
d) 試料の識別に必要な事項
e) 分析結果
f)
分析結果の参照番号
g) 定量時に気が付いた特記事項,及びこの附属書に規定がない操作で分析試料又は認証標準物質の分析
結果に影響を与えたおそれのある操作
10
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
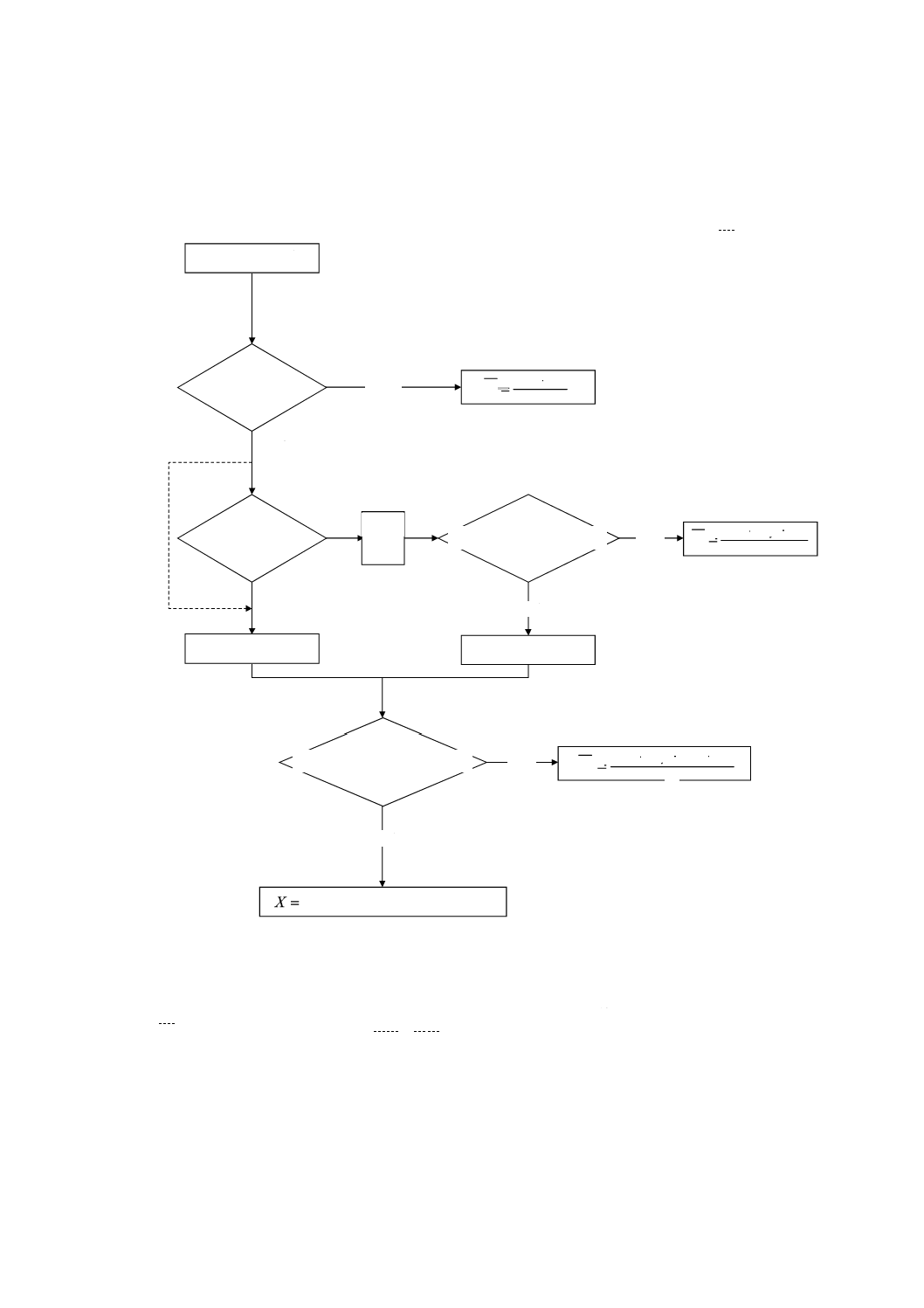
附属書1A(規定)分析値の採択手順のフローシート
参考 この採択手順は、附属書1だけに適用する。他の附属書での分析値の採択はJIS M 8202による。
rは,附属書1の8.2.1で規定。
参考1. 偶数個に対するメジアンは,数値を大きさの順に並べたときの中央2個の平均値。
2. 1.2rで検定せずに,直ちに
3
X,
4
Xを実施してもよい(破線部)。
X1,X2を実施
|X1−X2|≦r
|X1−X2|≦1.2r
いいえ
X3,X4を実施
X3を
実施
2
2
1
X
X
X
+
=
X1,X2,X3の範囲≦1.2r
X4を実施
はい
いいえ
X1,X2,X3,X4の範囲≦1.3r
いいえ
はい
のメジアン
,
,
,
4
3
2
1
X
X
X
X
X~=
3
3
2
1
X
X
X
X
+
+
=
はい
4
X
X
4
3
2
1
+
+
+
=
X
X
X
はい
いいえ

11
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1B(参考)国際共同実験に関する追加情報
8.2.1の回帰式は,6か国20の分析室で5種類の鉄鉱石試料(附属書1B表1参照)を用い,1979年〜1980
年に実施した国際共同分析実験の結果から求めている。
精度データは,附属書1C図1に図示してある。
附属書1B表1 共同実験用試料
試料
ひ素含有率
μg/g
Brazilian ore
Brazil 77-3
2.3
Mt. Newman ore
Australia 97-12
11.8
Baeckegruvan ore
Sweden 79-3
56.4
Lorraine ore
France 79-4
261
Vivaldi ore
Spain 79-11
798
備考1. この国際共同分析実験及び統計的解析の結果報告書(文書ISO/TC 102/SC 2 N605 E,1980年
8月)は,ISO/TC 102/SC 2事務局又はISO/TC 102の事務局で入手できる。
2. この統計的解析は,ISO 5725で規定している手順に従って行った。
12
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1C(参考)国際共同分析実験で得られた精度データ
附属書1C図 1 ひ素含有率X(μg/g)に対する精度の最小二乗法による回帰線
備考 この図は,附属書1の8.2.1の式をグラフ表示したものである。

13
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2(規定)アルカリ融解−電気加熱方式原子吸光法
1. 要旨 試料を無水四ほう酸ナトリウム,炭酸ナトリウム及び炭酸カリウムの混合融剤で融解し,放冷
後塩酸を加えて反応生成物を溶解する。一定量に薄めた後,溶液及びマトリックスモディファイヤを原子
吸光分析装置の電気加熱方式の原子化部に注入して加熱し,その吸光度を測定する。
2. 試薬 試薬は次による。
a) 塩酸
b) 塩酸(1+3)
c) 硝酸
d) 硝酸(5+95)
e) 純酸化鉄 純度の高い酸化鉄で,ひ素を含まないか,又はひ素の含有率が少なく既知であるもの。
f)
混合融剤 無水四ほう酸ナトリウム(Na2B4O7),炭酸ナトリウム(Na2CO3),及び炭酸カリウム(K2CO3)
をそれぞれ4:3:3の割合で混合したもの。
g) 標準ひ素溶液(As 1 μg/ml) 三酸化二ひ素(JIS K 8005) 0.660 2 gをはかりとり,水酸化ナトリウム溶液
(50g/l) 30 mlに溶解して,水で500 mlに薄める。次いで硫酸(1+5) 8 mlを加えて煮沸し,過マンガン酸
カリウム溶液(20 g/l)を微紅色が持続するまで滴加する。さらにしばらく煮沸した後,冷却して1000 ml
の全量フラスコに移し,水で標線まで薄め標準ひ素原液(As 500 μg/ml)とする。使用の都度この原液を
正確に500倍に薄め,標準ひ素溶液とする。
h) マトリックスモディファイヤ マグネシウム及びパラジウムの濃度がそれぞれ50 μg/mlになるように
硝酸マグネシウム溶液及び硝酸パラジウム溶液を混合し,調製したもの。硝酸濃度は(5+95)となるよ
うに調製する。
参考 市販の溶液から所定の濃度となるよう調製して用いてよい。マグネシウム溶液及びパラジウム
溶液を独自に調製する場合の調製例を次に示す。
マグネシウム溶液(Mg 1mg/ml):硝酸マグネシウム六水和物[Mg(NO3)2・6H2O]1.05gをはかりとり
水を加えて溶解し,100mlの全量フラスコに移し入れ標線まで薄めて原液(Mg 1mg/ml)とする。
パラジウム溶液(Pd 1mg/ml):硝酸パラジウム(Ⅱ)二水和物[Pd(NO3)2・2H2O]0.25gをはかりと
り硝酸(1+1)を加えて溶解し,100mlの全量フラスコに硝酸(1+1)を用いて移し入れ,標線まで薄
めて原液(Pd 1mg/ml)とする。
3. 装置及び器具
3.1
原子吸光分析装置 電気加熱方式の原子化部を備えた原子吸光分析装置。電気加熱方式の原子化部
の発熱体は,黒鉛製とし,バックグラウンド補正部及び高速記録計又はコンピュータによる読取装置を備
えたもの。光源には中空陰極ランプ又は無電極放電ランプを用いる。
3.1.1
装置基準 使用する原子吸光分析装置は,5.2に従って調整した後,次に示す装置基準を満足しな
ければならない。b)の検量線の直線性について基準を満足できない場合は,基準を満足するように附属書
2表3又は附属書2表4に示す標準ひ素溶液添加量を変えて検量線を作成してもよい。
a) 1%吸収質量 ひ素の1 %吸収質量は,30 pg以下でなければならない。1 %吸収質量の求め方は,JIS G
1257附属書31付録AのA.1(1 %吸収質量mcの求め方)による。

14
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 検量線の直線性 検量線の上部側20 %又は25 %範囲のこう配(吸光度の変化で表す。)の,同じ方法
で算出した下部側20 %又は25 %範囲のこう配に対する比が0.95以上でなければならない。
3.2
マイクロピペット 5〜50 μlの溶液を採取できるもの。装置に備えられた自動サンプラを用いても
よい。
4. 試料はかりとり量 試料はかりとり量は,0.20 gとする。
5. 操作
5.1
試料溶液の調製 試料溶液の調製は,次の手順による。
a) 試料をはかりとって白金るつぼ(30番)に移し入れ,混合融剤[2.f)] 1.5 gを加えて試料と融剤を混合する。
b) ブンゼンバーナを用いて,徐々に温度を上げながら試料を融解する。
c) 室温まで放冷した反応生成物の入ったるつぼを,ビーカー(200 ml)に入れ,塩酸(1+3) 50 mlを加えてビ
ーカーを時計皿で覆い,熱板の低温部で沸騰しない程度に穏やかに加熱(約90 ℃)して反応生成物
を溶解する。
d) るつぼを少量の水で4,5回洗浄して取り出した後,ビーカーの液を100 mlの全量フラスコに移し入
れ,水で標線まで薄める。
e) この溶液から附属書2表1に示すひ素含有率に対応した量を分取し(1),100 mlの全量フラスコに移し
入れて水で標線まで薄める。
注(1) 分取量は,原子吸光分析装置の感度,分光干渉及び検量線の直線性を考慮して変更してもよい。
5.2 原子吸光分析装置の調整 装置製造業者の指示書に従って必要とする装置のパラメータを調節し(2),
電気加熱方式の原子化部を接続する。装置のゼロ点調節を行い,記録計のベースラインを設定する。加熱
プログラムを作動させて原子化部の黒鉛管(3)を加熱し,ゼロ点の安定性,ベースラインの安定性及び原子
化機構内に分光干渉のないことを確認する。
新しい黒鉛管は,あらかじめ空試験値が安定するまで数回空焼きして使用する。
注(2) 操作上のパラメータに対する最適条件は,装置ごとに異なる。
(3) プラットホームを用いてもよい。
5.3
吸光度の測定 吸光度の測定は,次の手順による。
a) 5.1e)で得た溶液から附属書2表1に示すひ素含有率に対応した試料溶液採取量(4)及びマトリックスモ
ディファイヤ[2.h)]5 μlを,マイクロピペット又は自動サンプラーを用いて5.2で調整した原子吸光分
析装置の原子化部の黒鉛管に注入する。
注(4) 試料溶液採取量は,原子吸光分析装置の感度,分光干渉,検量線の直線性を考慮して,5〜20
μlの範囲内で変更してもよい。
b) 黒鉛管を乾燥・灰化・原子化のプログラムで逐次加熱し(5),波長193.7 nmにおけるひ素の吸光度を測
定する(6)。同一試料溶液を3回ずつ原子化し平均値を採用する。
注(5) 黒鉛管の加熱条件の例を附属書2表2に示す。
(6) 記録計で得られたピーク面積又はピーク高さを読みとる。
15
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2表1 分取量及び試料溶液採取量
試料中のひ素含有率
(質量分率%)
分取量
ml
試料溶液採取量
μl
0.0005以上0.010未満
25
10
0.010以上0.024未満
25
5
0.024以上0.06以下
10
5
附属書2表2 黒鉛管の加熱条件の例
加熱プログラム
加熱温度
℃
加熱時間
秒
乾燥
130
30
灰化
1200
20
原子化
2400
3
クリーン化
2600
3
6. 空試験 7.の検量線作成操作において得られる標準ひ素溶液を添加しない溶液の吸光度を空試験の吸
光度とする。
7. 検量線の作成 検量線の作成は次の手順による。
a) ひ素含有率質量分率0.010 %未満の検量線作成の場合は6個,ひ素含有率質量分率0.010 %以上0.024 %
未満の検量線作成の場合及びひ素含有率質量分率0.024 %以上0.06 %以下の検量線作成の場合は5個
のビーカー(200 ml)を用意し,それぞれに純酸化鉄[2.e)]0.20 gをはかりとり,5.1a)〜5.1d)の手順に従
って試料と同じ操作を試料と併行して行う。
b) a)で得た溶液から試料の分取量と同量の液量を分取して,100 mlの全量フラスコに移し入れ,試料中
のひ素含有率に応じて附属書2表3又は附属書2表4に従い,標準ひ素溶液[2.g)]を添加して(7)水で標
線まで薄める。
注(7) 標準ひ素溶液添加量は,原子吸光分析装置の感度,分光干渉,検量線の直線性を考慮して変更
してもよい。
c) 5.3の手順に従って試料と同じ操作を試料と併行して行い,得た吸光度と添加したひ素量との関係線を
作成し,その関係線を原点を通るように平行移動して検量線とする。
16
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2表3 検量線溶液調製−ひ素含有率質量分率0.010 %未満
標準ひ素溶液[2.g)]
添加量
ml
溶液中のひ素濃度
ng/ml
溶液10 μl中の
ひ素量
pg
25 ml分取,10 μl溶液採取で
の試料において相当する
ひ素含有率
(質量分率%)
0
0
0
0
1
10
100
0.002
2
20
200
0.004
3
30
300
0.006
4
40
400
0.008
5
50
500
0.010
附属書2表4 検量線溶液調製−ひ素含有率質量分率0.010 %以上0.024 %未満,又は0.024 %以上
0.06 %以下
標準ひ素溶液
[2.g)]添加量
ml
溶液中のひ
素濃度
ng/ml
溶液5 μl中の
ひ素量
pg
25 ml分取,5 μl溶液採取
での試料において相当す
るひ素含有率
(質量分率%)
10 ml分取,5 μl溶液採取で
の試料において相当する
ひ素含有率
(質量分率%)
0
0
0
0
0
3
30
150
0.006
0.015
6
60
300
0.012
0.030
9
90
450
0.018
0.045
12
120
600
0.024
0.060
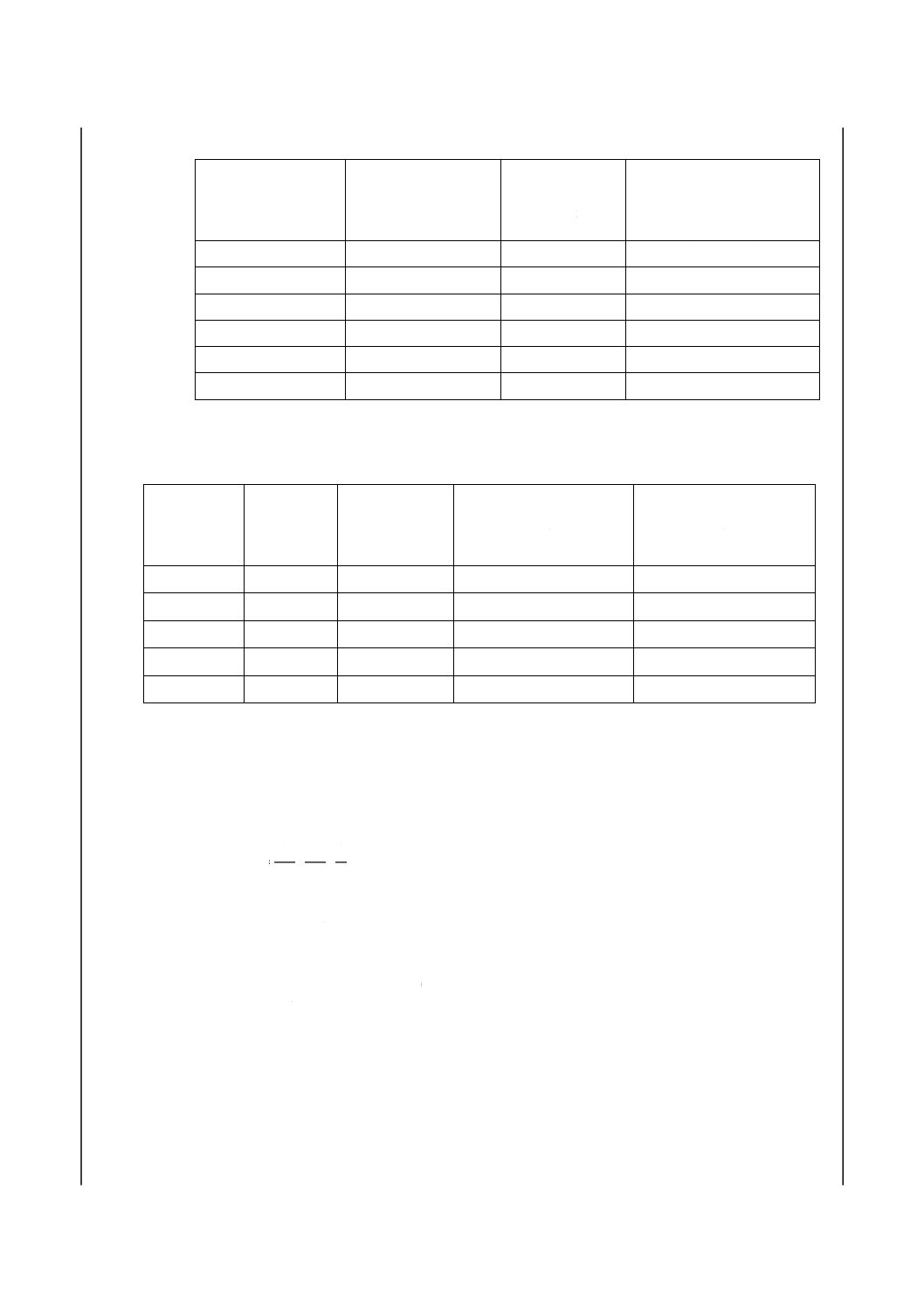
8. 計算 5.及び6.で得た吸光度と7.で作成した検量線とからひ素量を求め,試料中のひ素含有率を次の
式によって算出する。なお、分析値の最終表示は,ひ素含有率質量分率が0.010 %未満では小数点以下4
けたまで表し,0.010 %以上では小数点以下3けたまで表す。
C
V
V
m
)
m
m
(
As
+
×
×
×
=
−3
2
1
2
1
10
-
ここに,As:試料中のひ素含有率(質量分率%)
m1:試料溶液中のひ素検出量(pg)
m2:空試験液中のひ素検出量(pg)
m:試料はかりとり量(g)
V1:分取量(ml)
V2:溶液採取量(μl)
C:純酸化鉄[2.e)]中のひ素含有率(質量分率%)
9. 許容差 許容差(8)は,附属書2表5による。
注(8) 許容差計算式中のf(n)の値は,JIS Z 8402-6の表1(許容範囲の係数)による。ここで,nの値
は,室内再現許容差の場合は同一分析室内における分析回数,室間再現許容差の場合は分析に

17
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
関与した分析室数とする。また,(As)は,許容差を求めるひ素含有率(質量分率%)とする。
附属書2表 5 許容差
単位 質量分率%
ひ素含有率
室内再現許容差
室間再現許容差
0.000 5以上0.06以下
f(n)[0.026 8×(As)+0.000 054]
f(n)[0.044 0×(As)+0.000 051]
参考 この許容差は,ひ素含有率質量分率0.0005 %以上0.057 %以下の試料を用い,共同実験した結
果から求めたものである。

18
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3(規定)酸溶解−電気加熱方式原子吸光法
1. 要旨 試料を塩酸と硝酸とで分解し,溶液をろ過する。ろ液を一定量に薄めた後,溶液及びマトリッ
クスモディファイヤを原子吸光分析装置の電気加熱方式の原子化部に注入して加熱し,その吸光度を測定
する。
2. 試薬 試薬は次による。
a) 塩酸
b) 塩酸(2+100)
c) 硝酸
d) 硝酸(5+95)
e) 純酸化鉄 純度の高い酸化鉄で,ひ素を含まないか,又はひ素の含有率が少なく既知であるもの。
f)
標準ひ素溶液(As 1 μg/ml) 三酸化二ひ素(JIS K 8005) 0.660 2 gをはかりとり,水酸化ナトリウム溶液
(50 g/l) 30 mlに溶解して,水で500 mlに薄める。次いで,硫酸(1+5) 8 mlを加えて煮沸し,過マンガ
ン酸カリウム溶液(20 g/l)を微紅色が持続するまで滴加する。さらにしばらく煮沸した後,冷却し1 000
mlの全量フラスコに移し,水で標線まで薄め標準ひ素原液(As 500 μg/ml)とする。使用の都度この原液
を正確に500倍に薄め,標準ひ素溶液とする。
g) マトリックスモディファイヤ マグネシウム及びパラジウムの濃度がそれぞれ50 μg/mlになるように
硝酸マグネシウム溶液及び硝酸パラジウム溶液を混合し,調製したもの。硝酸濃度は(5+95)となるよ
うに調製する。
参考 市販の溶液から所定の濃度となるよう調製して用いてよい。マグネシウム溶液及びパラジウム
溶液を独自に調製する場合の調製例を次に示す。
マグネシウム溶液(Mg 1 mg/ml):硝酸マグネシウム六水和物[Mg(NO3)2・6H2O]1.05gをはかりと
り水を加えて溶解し,100mlの全量フラスコに移し入れ標線まで薄めて原液(Mg 1 mg/ml)とする。
パラジウム溶液(Pd 1 mg/ml):硝酸パラジウム(Ⅱ)二水和物[Pd(NO3)2・2H2O]0.25gをはかりと
り硝酸(1+1)を加えて溶解し,100mlの全量フラスコに硝酸(1+1)を用いて移し入れ,標線まで薄
めて原液(Pd 1 mg/ml)とする。
3. 装置及び器具
3.1
原子吸光分析装置 電気加熱方式の原子化部を備えた原子吸光分析装置。電気加熱方式の原子化部
の発熱体は,黒鉛製とし,バックグラウンド補正部及び高速記録計又はコンピュータ化された読み取り装
置を備えたもの。光源には中空陰極ランプ又は無電極放電ランプを用いる。
3.1.1
装置基準 使用する原子吸光分析装置は,5.2に従って調整した後,次に示す装置基準を満足しな
ければならない。b)の検量線の直線性について基準を満足できない場合は,基準を満足するように附属書
3表3又は附属書3表4に示す標準ひ素溶液添加量を変えて検量線を作成してもよい。
a) 1%吸収質量 ひ素の1 %吸収質量は,30 pg以下でなければならない。1%吸収質量の求め方は,JIS G
1257附属書31付録AのA.1(1 %吸収質量mcの求め方)による。
b) 検量線の直線性 検量線の検量線の上部側20 %範囲のこう配(吸光度の変化で表す。)の,同じ方法
で算出した下部側20 %範囲のこう配に対する比が0.95以上でなければならない。

19
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.2
マイクロピペット 5〜50 μlの溶液を採取できる採取体積が可変のもの。装置に備えられた自動サ
ンプラーを用いてもよい。
4.試料はかりとり量 試料のはかりとり量は,0.50 gとする。
5. 操作
5.1
試料溶液の調製 試料溶液の調製は,次の手順による。
a) 試料をはかりとってビーカー(200 ml)に移し入れ,時計皿で覆い,硝酸5 ml及び塩酸15 mlを加え約
110〜150 ℃の熱板上で溶液を煮沸しない程度に緩やかに加熱して分解する。
b) 室温まで冷却した後,ろ紙(5種B)で100 mlの全量フラスコにろ過し,ビーカー壁に付着している不
溶解残さは,ゴム付きガラス棒を用いてこすり落とし,温水を用いてろ紙上に移す。ろ紙及び不溶解
残さを温めた塩酸(2+100)で塩化鉄(Ⅲ)の黄色が認められなくなるまで数回洗浄し,洗液はろ液に
合わせる。この溶液を常温まで冷却した後,水で標線まで薄める。残さは捨てる。
c) この溶液10 mlを分取して(1)100 mlの全量フラスコに移し入れ,水で標線まで薄める。
注(1) 分取量は,原子吸光分析装置の感度,分光干渉及び検量線の直線性を考慮して変更してもよ
い。
5.2 原子吸光分析装置の調整 装置製造業者の指示書に従って必要とする装置のパラメータを調節し(2),
電気加熱方式の原子化部を接続する。装置のゼロ点調節を行い,記録計のベースラインを設定する。加熱
プログラムを作動させて原子化部の黒鉛管(3)を加熱し,ゼロ点の安定性,ベースラインの安定性及び原子
化機構内に分光干渉のないことを確認する。
新しい黒鉛管は,あらかじめ空試験値が安定するまで数回空焼きして使用する。
注(2) 操作上のパラメータに対する最適条件は,装置ごとに異なる。
(3) プラットホームを用いてもよい。
5.3
吸光度の測定 吸光度の測定は,次の手順による。
a) 5.1c)で得た溶液について附属書3表1に示すひ素含有率に対応した試料液採取量(4)及びマトリックス
モディファイヤ[2.g)]5 μlを,マイクロピペット又は自動サンプラを用いて5.2で調整した原子吸光分
析装置の原子化部の黒鉛管に注入する。
注(4) 試料溶液採取量は,原子吸光分析装置の感度,分光干渉,検量線の直線性を考慮して,5〜20
μlの範囲内で変更してもよい。
b) 黒鉛管を乾燥・灰化・原子化のプログラムで逐次加熱し(5),波長193.7 nmにおけるひ素の吸光度を測
定する(6)。同一試料溶液を3回ずつ原子化し平均値を採用する。
注(5) 黒鉛管の加熱条件の例を附属書3表2に示す。
(6) 記録計で得られたピーク面積又はピーク高さを読みとる。
附属書3表1 試料液採取量
試料中のひ素含有率
(質量分率%)
試料液採取量
μl
0.000 5以上0.010未満
10
0.010以上0.02以下
5
20
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3表 2 モデファイヤを使用した場合の加熱条件の例
加熱プログラム
加熱温度
℃
加熱時間
秒
乾燥
130
30
灰化
1 200
20
原子化
2 400
3
クリーン化
2 600
3
6. 空試験 7.の検量線作成操作において得られる標準ひ素溶液を添加しない溶液の吸光度を空試験の吸
光度とする。
7. 検量線の作成 検量線の作成は次の手順による。
a) 6個のビーカー(200 ml)を用意し,それぞれに純酸化鉄[2.e)]0.50 gをはかりとり,5.1の手順に従って
試料と同じ操作を試料と併行して行う。
b) a)で得た溶液から10 mlを分取して(1)100 mlの全量フラスコに移し入れ,試料中のひ素含有率により
附属書3表3又は附属書3表4に従って標準ひ素溶液[2.f)]を添加して(7)水で標線までに薄める。
注(7) 標準ひ素溶液添加量は,原子吸光分析装置の感度,分光干渉,検量線の直線性を考慮して変更
してもよい。
c) 5.3の手順に従って試料と同じ操作を試料と併行して行い,得た吸光度と添加したひ素量との関係線を
作成し,その関係線を原点を通るように平行移動して検量線とする。
附属書3表 3 検量線溶液調製−ひ素含有率質量分率0.010 %未満
標準ひ素溶液[2.f)]
添加量
ml
溶液中のひ素濃度
ng/ml
溶液10 μl中の
ひ素量
pg
10 ml分取,10 μl溶液採取で
の試料において相当する
ひ素含有率
(質量分率%)
0
0
0
0
1
10
100
0.002
2
20
200
0.004
3
30
300
0.006
4
40
400
0.008
5
50
500
0.010
21
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3表 4 検量線溶液調製−ひ素含有率質量分率0.010 %以上0.02 %以下
標準ひ素溶液[2.f)]
添加量
ml
溶液中のひ素濃度
ng/ml
溶液5 μl中の
ひ素量
pg
10 ml分取,5 μl溶液採取で
の試料において相当する
ひ素含有率
(質量分率%)
0
0
0
0
2
20
100
0.004
4
40
200
0.008
6
60
300
0.012
8
80
400
0.016
10
100
500
0.020
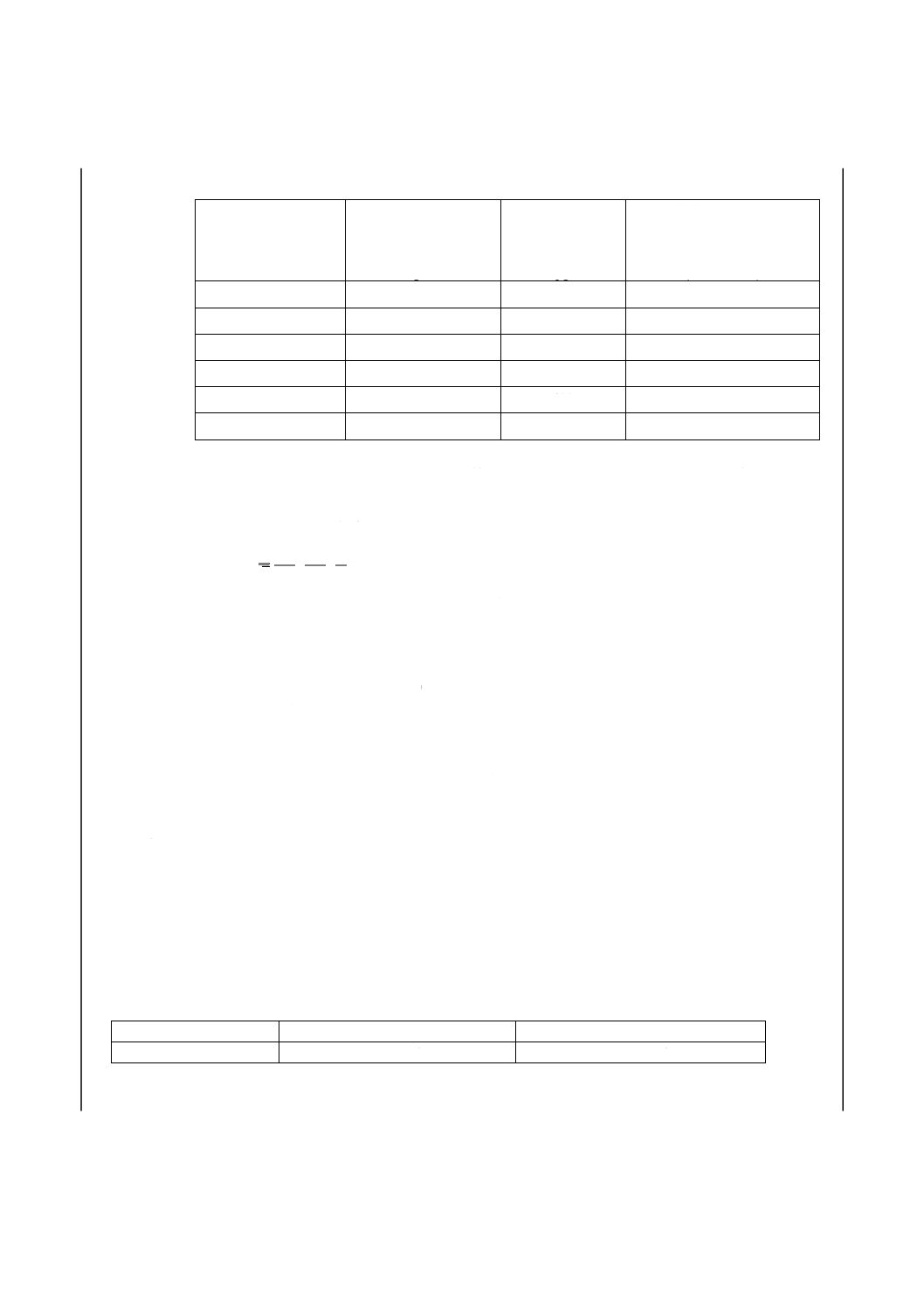
8. 計算 5.及び6.で得た吸光度と7.で作成した検量線とからひ素量を求め,試料中のひ素含有率を次の
式によって算出する。なお,分析値の最終表示は,ひ素含有率質量分率が0.010 %未満では小数点以下4
けたまで表し,0.010 %以上では小数点以下3けたまで表す。
C
V
V
m
)
m
m
(
As
+
×
×
×
=
−3
2
1
2
1
10
-
ここに,As:試料中のひ素含有率(質量分率%)
m1:試料溶液中のひ素検出量(pg)
m2:空試験液中のひ素検出量(pg)
m:試料はかりとり量(g)
V1:分取量(ml)
V2:溶液採取量(μl)
C:純酸化鉄[2.e)]中のひ素含有率(質量分率%)
9. 許容差 許容差(8)は,附属書3表5による。
注(8) 許容差計算式中のf(n)の値は,JIS Z 8402-6の表1(許容範囲の係数)による。ここで,nの値
は,室内再現許容差の場合は同一分析室内における分析回数,室間再現許容差の場合は分析に
関与した分析室数とする。また,(As)は,許容差を求めるひ素含有率(質量分率%)とする。
附属書3表 5 許容差
単位 質量分率(%)
ひ素含有率
室内再現許容差
室間再現許容差
0.000 5以上0.02以下
f(n)[0.041 3×(As)+0.000 069]
f(n)[0.050 2×(As)+0.000 15]
参考 この許容差は,ひ素含有率質量分率0.000 5 %以上0.014 %以下の試料を用い,共同実験した結
果から求めたものである。
22
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
23
M 8226:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
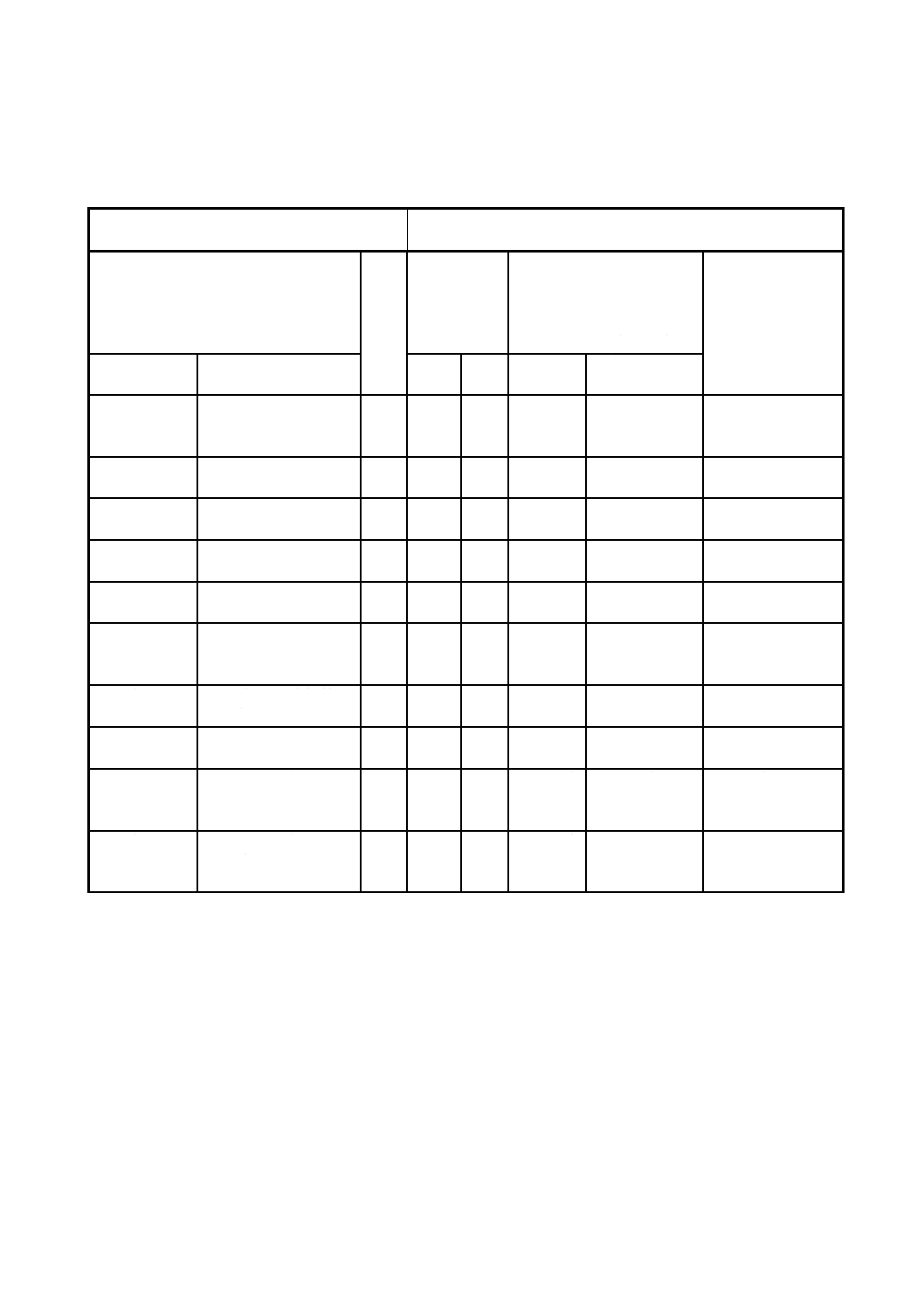
附属書4(参考)JISと対応する国際規格との対比表
JIS M 8226:200X 鉄鉱石−ひ素定量方法
ISO 7834:1987 鉄鉱石−ひ素定量方法−モリブデン青吸光光
度法
(Ⅰ)JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ)国際規
格の規定
(Ⅳ)JISと国際規格との技
術的差異の項目ごとの評
価及びその内容
表示箇所:本体、附属書
表示方法:実線の側線
(Ⅴ)JISと国際規
格との技術的差
異の理由及び今
後の対策
項目番号
内容
項目
番号
内容 項目ごと
の評価
技術的差異の
内容
1.適用範囲
適用範囲を規定。
MOD/追
加
JISの様式に
従って記載。
JIS独自分析法を
追加規定してい
るから。
2.引用規格
引用規格を規定。
MOD/追
加
JISの引用を
追加。
同上
3.一般事項
一般事項を規定。
MOD/追
加
JIS独自規定
である。
同上
4.定量方法の
区分
規定した3定量法の
区分を規定。
MOD/追
加
JIS独自規定
である。
同上
附属書1
モリブドひ酸青吸光
光度法を規定。
本体
IDT
−
−
附属書1A
分析値の採択手順の
フローシートを記
載。
附属
書A
IDT
−
−
附属書1B
国際実験の追加情報
を記載。
附属
書B
IDT
−
−
附属書1C
精度データの図を記
載。
附属
書C
IDT
−
−
附属書2
アルカリ融解−電気
加熱方式原子吸光法
を規定。
MOD/追
加
JIS独自規定
である。
JIS独自分析法を
追加規定してい
るから。
附属書3
アルカリ融解−電気
加熱方式原子吸光法
を規定。
MOD/追
加
JIS独自規定
である。
同上
JISと国際規格との対応の程度の全体評価;MOD