2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
M 8129-1994
鉱石中のコバルト定量方法
Ores−Methods for determination of cobalt
1. 適用範囲 この規格は,鉱石中のコバルト定量方法について規定する。ただし,他の日本工業規格で
コバルト定量方法が規定されている鉱石には適用しない。
備考 この規格の引用規格を,次に示す。
JIS K 0050 化学分析方法通則
JIS K 0113 電位差・電流・電量・カールフィッシャー滴定方法通則
JIS K 0115 吸光光度分析通則
JIS K 0121 原子吸光分析通則
JIS M 8083 ばら積み非鉄金属浮選精鉱のサンプリング方法
JIS M 8101 非鉄金属鉱石のサンプリング,試料調製及び水分決定方法
JIS Z 8401 数値の丸め方
2. 一般事項 定量方法に共通な一般事項は,JIS K 0050,JIS K 0113,JIS K 0115及びJIS K 0121によ
る。
3. 分析試料の採り方及び取扱い方
3.1
試料の採取と調製 試料の採取と調製は,JIS M 8083及びJIS M 8101による。
3.2
試料のはかり方 試料のはかり方は,次による。
(1) 試料のはかり採りに際しては,試料をよくかき混ぜて平均組成を表すように注意し,また,異物が混
入していないことを確かめなければならない。
(2) 試料は,105±5℃に調節されている空気浴に入れて乾燥し,2時間ごとに空気浴から取り出し,デシ
ケーター中で常温まで放冷する。乾燥は,乾燥減量が2時間につき0.1% (m/m) 以下になるまで繰り
返す。ただし,硫化物などを含有するため変質しやすい試料の乾燥条件(温度,時間など)は,受渡
当事者間の協議による。
(3) 試料のはかり採りには,原則として化学はかりを用いる。
4. 分析値の表し方及び操作上の注意
4.1
分析値の表し方 分析値の表し方は,次による。
(1) 分析値は質量百分率で表し,JIS Z 8401によって小数点以下第3位に丸める。
(2) 分析は,同一分析室において2回繰り返して行い,これらの差が室内許容差(以下,許容差という。)
以下のとき,その平均値を求め,JIS Z 8401によって小数点以下第2位に丸めて報告値とする。
(3) 2回繰り返して行った分析値の差が許容差を超えるときは,改めて2回の分析をやり直す。

2
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(4) 許容差は,表1による。
表1 許容差(1)
単位 % (m/m)
定量方法
コバルト含有率の区分
許容差
(繰返し)
1-ニトロソ-2-ナフトール分離
酸化コバルト (III) 重量法
0.5 以上 10
以下
0.150
イオン交換分離電位差滴定法
0.1 以上 3
未満
0.035
3
以上 7
未満
0.075
7
以上 20
以下
0.150
ニトロソR塩吸光光度法
0.01 以上 0.05 未満
0.005
0.05 以上 0.2 未満
0.010
0.2 以上 0.5 以下
0.020
原子吸光法
0.01 以上 0.05 未満
0.005
0.05 以上 0.2 未満
0.010
0.2 以上 1.0 以下
0.020
注(1) 2個の分析値が二つのコバルト含有率の区分にまたがるときは,2個の分析値の
平均値の該当する区分の許容差を適用する。
4.2
分析操作上の注意 分析に当たっては,全操作を通じて空試験を行い,測定値を補正しなければな
らない。
5. 定量方法
5.1
定量方法の区分 鉱石中のコバルト定量方法は,次のいずれかによる。
(1) 1-ニトロソ-2-ナフトール分離酸化コバルト (III) 重量法 この方法は,コバルト含有率0.5% (m/m) 以
上10% (m/m) 以下の試料に適用する。
(2) イオン交換分離電位差滴定法 この方法は,コバルト含有率0.1% (m/m) 以上20% (m/m) 以下の試料
に適用する。
(3) ニトロソR塩吸光光度法 この方法は,コバルト含有率0.01% (m/m) 以上0.5% (m/m) 以下の試料に
適用する。
(4) 原子吸光法 この方法は,コバルト含有率0.01% (m/m) 以上1% (m/m) 以下の試料に適用する。
5.2
1-ニトロソ-2-ナフトール分離酸化コバルト (III) 重量法
5.2.1
要旨 試料を硝酸及び塩酸で分解し,硫酸を加え加熱して濃縮し,硫酸の白煙を発生させる。塩酸
を加えて可溶性塩を溶解し,硫化水素ガスを通じて銅,ひ素などを沈殿させ,ろ過する。ろ液を煮沸して
硫化水素ガスを揮散させ,硝酸で鉄などを酸化した後,アンモニア水及び塩酸で微酸性とし,酸化亜鉛乳
を加えて鉄などを沈殿させ,ろ過する。ろ液を塩酸酸性とした後,1-ニトロソ-2-ナフトール溶液を加え,
コバルトを沈殿させてこし分け,乾燥灰化後,酸化コバルト (III) としてその質量をはかる。
5.2.2
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1)
(3) 硝酸
(4) ふっ化水素酸
(5) 臭化水素酸
(6) 硫酸 (1+1)
3
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(7) 硝硫混酸(水4+硝酸4+硫酸2)
(8) アンモニア水 (1+1)
(9) 臭素
(10) 硫化水素
(11) 塩酸洗浄溶液 塩酸 (1+20) に硫化水素を飽和させる。
(12) 酸化亜鉛乳状液 酸化亜鉛約50gを水約300mlに加え,よく振り混ぜる。
(13) 1-ニトロソ-2-ナフトール溶液 1-ニトロソ-2-ナフトール1gを酢酸25mlに加熱溶解し,水25mlを加
え,約1時間静置後乾燥ろ紙を用いてろ過したもの。この溶液は使用の都度調製する。
(14) メチルオレンジ溶液 (1g/l) メチルオレンジ0.1gを温水100mlに溶解し,冷却後ろ過したもの。
5.2.3
試料はかり採り量 試料はかり採り量は,コバルト含有率に応じて表2によって0.1mgのけたまで
はかる(2)。
表2 試料はかり採り量
コバルト含有率
% (m/m)
試料はかり採り量
g
0.5以上 3未満
0.5 〜2
3 以上 10以下
0.25〜0.5
注(2) 試料は,コバルト量がなるべく10〜30mg程度になるようにはかり採る。
5.2.4
操作
5.2.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採り,ビーカー (200〜300ml) に移し入れ,時計皿で覆い,硝酸10〜20mlを加え,静か
に加熱して分解する(3)。
(2) 激しい反応が終わってから塩酸10〜20mlを加え,引き続き加熱して分解する(4)。
(3) 硫酸 (1+1) 10〜20mlを加えて加熱蒸発し,硫酸の白煙を十分に発生させる(5)。
(4) 放冷後,水約50ml及び塩酸15〜20mlを加え,加熱して可溶性塩を溶解した後,ろ紙(5種B)を用
いてろ過し,温水で十分に洗浄する(6)。
注(3) 酸化鉱又は焼鉱などの場合には,試料をはかり採った後,塩酸10〜20mlを加えて加熱し,さら
に硝酸10〜20mlを加える。
(4) 硫化鉱などを分解した場合,析出した硫黄の分解が不十分な場合には,少量の臭素を加えると
よい。
(5) 試料中に多量のひ素,アンチモン,すず又はセレンを含み,以後の定量操作に対する影響が無
視できないときは,放冷後,水約5ml及び臭化水素酸5〜10mlを加え,加熱蒸発して硫酸の白
煙を十分に発生させる。少し放冷後,硫酸 (1+1) 5〜10ml及び臭化水素酸5〜10mlを加え,再
び加熱蒸発して硫酸の白煙を十分に発生させる。この操作を行う場合には,容量300〜400ml
のビーカーを用い,強熱を避けて飛散しないよう特に注意する必要がある。
(6) 残さ中にコバルトが含まれる場合には,少量の水を用いて残さを白金皿(50番)に洗い移し,
硫酸 (1+1) 約5ml,硝酸約5ml及びふっ化水素酸5〜10mlを加え,加熱して硫酸の白煙を十分
に発生させて二酸化けい素を揮散させ,乾固近くまで濃縮する。放冷後,少量の水を加えて溶
解し,元のろ紙(5種B)を用いてろ過し,温水で十分に洗浄する。ろ液及び洗液は主液に合わ
せるか,又は主液に合わせずに5.4若しくは5.5のいずれかによってコバルトを定量して補正す
る。
4
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.2.4.2
コバルトの分離 コバルトの分離は,次の手順によって行う。
(1) ろ液及び洗液はビーカー (300ml) に受け,加熱して液温を約80℃とし,硫化水素ガスを十分に通じ,
銅,ひ素などを沈殿させ,ろ紙(5種B)を用いてろ過し,塩酸洗浄溶液で数回洗浄する。
(2) ろ液及び洗液はビーカー (500ml) に受け,加熱し,硫化水素臭がなくなるまで煮沸を続ける。硝酸5ml
を加え,さらに数分間煮沸した後,水で液量を約200mlとする。
(3) 放冷後かき混ぜながら,アンモニア水を鉄,アルミニウムなどの沈殿が生じる直前まで加え,液量を
水で約300mlとする。
(4) この溶液をかき混ぜながら,酸化亜鉛乳状液を少量ずつ加えて鉄などを完全に沈殿させ,さらに少過
剰加え(7),しばらく放置して沈殿を沈降させた後,ろ紙(5種B)を用いてろ過し,冷水で数回洗浄
する。ろ液及び洗液は合わせてビーカー (500〜800ml) に受け,塩酸5mlを加え,加熱濃縮して主溶
液として保存する。
(5) 沈殿は,温水で元のビーカーに洗い移し,できるだけ少量の温塩酸 (1+1) を加えて加熱溶解し,水
で約200mlとする。この溶液に再び(4)の手順にしたがって酸化亜鉛乳状液を加えて再沈殿操作(8)を行
い,ろ液と洗液は(4)の主液に合わせる。
(6) この全溶液を加熱濃縮(9)して液量約300mlとし,煮沸近くまで加熱する。この溶液をかき混ぜながら,
1-ニトロソ-2-ナフトール溶液20〜60ml(10)を加え,70〜80℃の温所に2時間以上静置して沈殿を熟成
させた後,ろ紙(5種C)を用いてこし分け,温水で数回洗浄する。
(7) 沈殿は,ろ紙と共に磁器るつぼ(PC1又は2)に移し入れて乾燥した後,始めは弱く加熱して炭化物
を焼失させ,次に750〜800℃に強熱して灰化する。
(8) 放冷後,内容物を水でビーカー (500ml) に洗い移し保存する。磁器るつぼには硫酸 (1+1) 5ml及び塩
酸2mlを加え,加熱して十分に白煙を発生させる。磁器るつぼに付着した酸化コバルト (III) を分解
した後放冷し,水約5mlを加え,加熱して可溶性塩を溶解する。この溶液を先に保存しておいたビー
カー (500ml) に洗い移し,硫酸 (1+1) 10〜15ml及び塩酸5mlを加え,加熱して十分に白煙を発生さ
せる。放冷後,少量の水を加え,加熱して可溶性塩を溶解して未分解物のないことを確かめた後(11),
さらに水を加えて液量約50mlとする。
(9) この溶液にメチルオレンジ溶液1, 2滴を指示薬として加え,アンモニア水で溶液の色が赤色から黄色
になるまで中和する。次に塩酸5mlを加え水で液量を約300mlとし,煮沸近くまで加熱する。この溶
液をかき混ぜながら,1-ニトロソ-2-ナフトール溶液を20〜60ml(10)加え,70〜80℃の温所に2時間以
上静置して沈殿を熟成させた後,ろ紙(5種C)を用いてこし分け,冷水で数回,次に塩酸 (1+1) と
冷水とを交互に用いて数回洗浄した後,温水で十分に洗浄する。
注(7) 酸化亜鉛が過剰になると,その上澄み液が白濁を呈する。
(8) 沈殿中にコバルトが残存するおそれのあるときは,さらにこの操作を繰り返すか,又は塩酸に
溶解した後,5.4若しくは5.5のいずれかによってコバルトを定量して補正する。
(9) もし沈殿が認められたときは,ろ紙(5種B)を用いてろ過し,冷水で数回洗浄して除去する。
(10) 1-ニトロソ-2-ナフトール溶液の使用量は,コバルト含有量0.01gにつき通常約20mlの割合とす
る。
(11) 分解が不十分で,未分解物が認められる場合には,さらに硫酸 (1+1) 10〜15ml及び塩酸5ml
を加えて加熱白煙処理を繰り返す。
5.2.4.3
ひょう量 ひょう量は,次の手順によって行う。
(1) 5.2.4.2(9)で得た沈殿は,ろ紙と共に質量既知の磁器るつぼ(12)(PC1又は2)に移し入れ,乾燥した後,
5
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
始めは弱く加熱して炭化物を焼失させ,次に750〜800℃(13)で恒量となるまで強熱した後,デシケー
ター中で放冷してその質量をはかる。
注(12) 磁器るつぼは,約800℃で2時間以上強熱し,デシケーター中で放冷してその質量をはかったも
のを用いる。
(13) 強熱温度は,900℃を超えると酸化コバルト (III) が一般化コバルトに変わる傾向があるので注
意を要する。
5.2.5
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
5.2.6
計算 試料中のコバルト含有率を,次の式によって算出する。
100
7342
.0
)
(
2
1
×
×
−
=
m
W
W
CO
ここに, Co: コバルト含有率 [% (m/m)]
W1: 酸化コバルト (III) の入っているるつぼの質量 (g)
W2: るつぼの質量 (g)
m: 試料はかり採り量 (g)
5.3
イオン交換分離電位差滴定法
5.3.1
要旨 試料を硝酸及び塩酸で分解後,硫酸を加え,加熱して硫酸の白煙を発生させる。水で可溶性
塩を溶解し,ろ過した後,加熱して蒸発乾固させる。塩酸を加え,加熱して溶解した後,イオン交換カラ
ムに通してコバルトを吸着させる。次に塩酸を通してマンガンなどを除去した後,希塩酸を通してコバル
トを溶離させる。これに硝酸及び硫酸を加え,加熱蒸発し,硫酸の白煙を発生させる。水を加え,加熱し
て溶解し,くえん酸アンモニウム及びアンモニア水を加えた後,白金及び飽和カロメル電極を用い,ヘキ
サシアノ鉄 (III) 酸カリウム標準溶液で電位差滴定を行う。
5.3.2
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (2+1, 1+2, 1+100)
(3) 硝酸
(4) ふっ化水素酸
(5) 臭化水素酸
(6) 硫酸 (1+1)
(7) アンモニア水
(8) アンモニア水 (7+100)
(9) 臭素
(10) くえん酸アンモニウム溶液 くえん酸(1水塩)300gを水約500mlに溶解し,冷却しながらリトマス
試験紙などを用い,アンモニア水を加えて中和した後,水で1 000mlとしたもの。
(11) M/30ヘキサシアノ鉄 (III) 酸カリウム標準溶液 ヘキサシアノ鉄 (III) 酸カリウム11gを水に溶解し
て1 000mlとしたもの。この溶液の標定は,次のようにして行う。
標準コバルト溶液50mlを正確に,ビーカー (300ml) に分取し,くえん酸アンモニウム溶液100ml
を加えた後5.3.5.3(1)以降の手順にしたがって操作し,電位差滴定を行い,ヘキサシアノ鉄 (III) 酸カ
リウム標準溶液1ml当たりのコバルト相当量を,次の式によって求める。
V
G
f=
6
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
f: ヘキサシアノ鉄 (III) 酸カリウム標準溶液1mlのコバルト相当
量 (g)
G: コバルト量 (g)
V: ヘキサシアノ鉄 (III) 酸カリウム標準溶液使用量 (ml)
(12) M/60ヘキサシアノ鉄 (III) 酸カリウム標準溶液 ヘキサシアノ鉄 (III) 酸カリウム5.5gを水に溶解し
て1 000mlとしたもの。この溶液の標定は,次のようにして行う。
標準コバルト溶液20mlを正確に,ビーカー (300ml) に分取し,(11)に準じて行う。
(13) 標準コバルト溶液 (1mgCo/ml) コバルト[99.9% (m/m) 以上]1.000gを硝酸 (1+1) 20mlで分解し,
冷却後1 000mlの全量フラスコに移し入れ,水で標線まで薄めたもの。
5.3.3
装置及び器具
(1) 電位差滴定装置
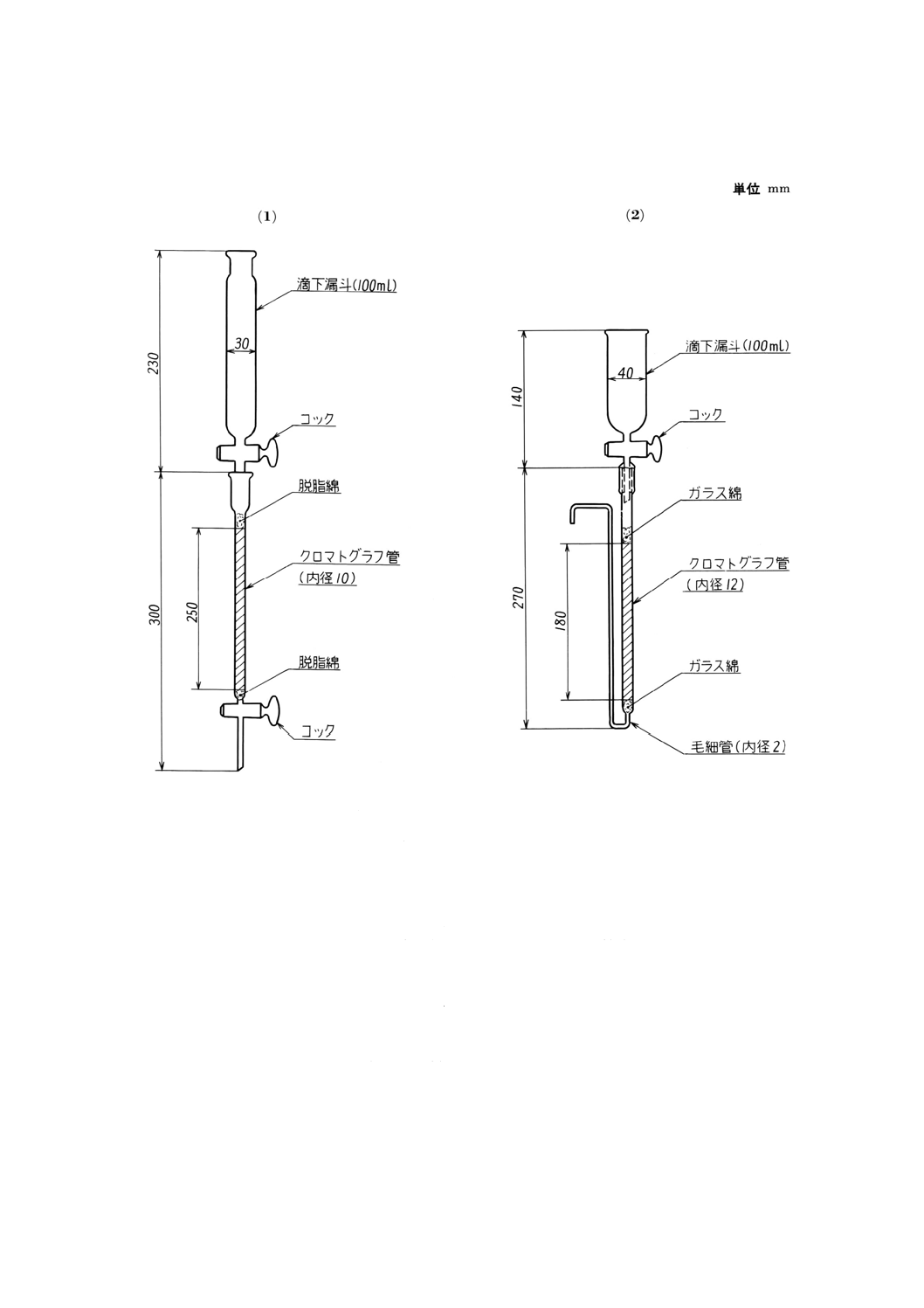
(2) イオン交換カラム イオン交換カラムは,原則として次による(14)。
ガラス製クロマトグラフ管(内径約10mm)に水でほぐした脱脂綿又はガラス綿を約5mmの厚さに
ゆるく詰め,強塩基性陰イオン交換樹脂(C1形149〜74μm分析用)を水で膨潤させる。その約20ml
をスラリー状にして流し入れ,沈降させた後,その上に水でほぐした脱脂綿又はガラス綿を約5mm
の厚さにゆるく詰め,塩酸 (2+1) 約50mlを通し,樹脂柱に満たしておく。
なお,流出液の流速は,毎分1ml以下とする。
イオン交換カラムの例を付図1に示す。
注(14) イオン交換カラムは,クロマトグラフ管の内径,イオン交換樹脂の粒径,容量などによって流
出液の流出が異なるので,あらかじめ毎分1ml以下となるように調節しておく。
また,イオン交換樹脂の種類,流出液の流出などによって,吸着,溶離などの状況が幾分異
なるので,あらかじめ5.3.5.2(2)の操作によって,マンガンなどが洗浄除去できることを確認す
るとともに,溶離曲線を求めて,コバルトの吸着,溶離状況を把握し,コバルトが定量的に吸
着されるとともに,所定量の塩酸 (1+2) でコバルトが定量的に溶離できることを確認しておく。
イオン交換分離操作において,多量のマンガンが共存しても,その影響が無視できる程度ま
で分離され,コバルトの吸着及び溶離には影響しない。銅が多量に共存するとコバルトの吸着
を妨害し,鉄が多量に共存するとコバルトの溶離を妨害する傾向がある。通常,マンガン共存
量約300mg以下,銅共存量約200mg以下,鉄共存量約400mg以下のときは,コバルトは定量
的に吸着及び溶離できるが,イオン交換カラムによって幾分異なるので,使用するイオン交換
カラムについて,あらかじめ,マンガン,銅及び鉄の許容量を求めておく。
5.3.4
試料はかり採り量 試料はかり採り量は,原則として表3による(15)。
表3 試料はかり採り量
コバルト含有率
% (m/m)
試料はかり採り量
g
0.1以上 3未満
0.5 〜2
3 以上 20以下
0.25〜0.5
注(15) 試料は,コバルト量が50mgを以下となるようにはかり採る。
また,はかり採った試料中のマンガン,銅及び鉄は,注(14)で求めた許容量以下でなければな
らない。ただし,試料中のマンガン量が0.1mg未満で,注(16)を適用してイオン交換分離の操作
を省略する場合には,銅許容量は500mg以下,鉄許容量は200mg以下となる。
5.3.5
操作

7
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.3.5.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採り,ビーカー (200〜300ml) に移し入れ,時計皿で覆い,硝酸10〜20mlを加え,静か
に加熱して分解する(3)。
(2) 激しい反応が終わってから塩酸10〜20mlを加え,引き続いて加熱して分解する(4)。
(3) 硫酸 (1+1) 10〜20mlを加えて加熱蒸発し,硫酸の白煙を十分に発生させる(5)。
(4) 放冷後,水約50mlを加え,加熱して可溶性塩を溶解した後,ろ紙(5種B)を用いてろ過し,温水で
十分に洗浄する(6)(16)。
注(16) この溶液中のマンガン共存量が0.1mg未満で,銅共存量500mg以下,鉄共存量200mg以下の場
合には,この溶液にアンモニア水を加えて大部分の硫酸を中和し,加熱濃縮して液量を約70ml
とした後,5.3.5.3(1)以降の手順に従って操作し,イオン交換分離の操作を省略することができ
る。
5.3.5.2
コバルトの分離 コバルトの分離は,次の手順によって行う。
(1) ろ液及び洗液は,ビーカー (200〜300ml) に受け,加熱して蒸発させ乾固させる。これに塩酸 (2+1) 約
20 mlを加え,少し加熱して溶解した後,放冷する。
(2) この溶液を毎分1ml以下の流速で,イオン交換カラムに通す。樹脂上に溶液がなくなってから塩酸 (2
+1) 80mlを数回に分けて,ビーカーを洗浄し,洗浄の都度カラムに通す。これまでの流出液は,すべ
て捨てる。次に塩酸 (1+2) 約50mlを毎分1ml以下の流速で通し,コバルトを溶離させ(17),流出液
はビーカー (300ml) に受ける(18)。
(3) この溶液に硝酸5ml及び硫酸 (1+1) 5mlを加え,加熱して硫酸の白煙を発生させ,乾固近くまで蒸発
する。放冷後,水約70mlを加え,加熱して溶解し,室温に冷却する。
注(17) イオン交換カラムは,塩酸 (1+100),水,アンモニア水 (7+100),水のそれぞれ約50mlずつを
順次通した後,塩酸 (2+1) 約50mlを通せば再使用できる。
(18) 使用するイオン交換カラムによって幾分異なるが,鉄が約200mg以上共存するときは,コバル
トの溶離が不完全になるので,このような場合には,さらに塩酸 (1+2) 約50mlを通す。
5.3.5.3
電位差滴定 電位差滴定は,次の手順によって行う。
(1) 5.3.5.2(3)で得た溶液にくえん酸アンモニウム溶液100mlを加えた後,速やかに15℃(19)以下に冷却す
る。これにアンモニア水80mlを加え,直ちに(19)白金及び飽和カロメル電極を挿入し,表4のヘキサ
シアノ鉄 (III) 酸カリウム標準溶液で電位差滴定を行い,電位飛躍が最大値を示した点を終点とする。
表4
コバルト量
mg
ヘキサシアノ鉄 (III) 酸カリウム標準溶液
10以上
M/30
20以下
M/60
注(19) 液温が高い場合及び放置時間が長い場合には,空気酸化によって低値を与えるおそれがある。
5.3.6
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
5.3.7
計算 試料中のコバルト含有率を,次の式によって算出する。
100
×
×
=mV
f
CO
ここに, Co: コバルト含有率 [% (m/m)]
f: ヘキサシアノ鉄 (III) 酸カリウム標準溶液1mlのコバルト相当
量 (g)
8
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V: ヘキサシアノ鉄 (III) 酸カリウム標準溶液使用量 (ml)
m: 試料はかり採り量 (g)
5.4
ニトロソR塩吸光光度法
5.4.1
要旨 試料を硝酸及び塩酸で分解後,硫酸を加え,加熱して硫酸の白煙を発生させる。硫酸を加え
て可溶性塩を溶解し,りん酸を加えた後,不溶解物をろ過してろ液を水で一定量に薄め,この溶液の一部
を分取する。これにくえん酸及び酢酸ナトリウムを加え,アンモニアでpHを7.0±0.5の範囲に調節した
後,1-ニトロソ-2-ナフトール-3, 6-ジスルホン酸二ナトリウム(以下,ニトロソR塩と略す。)を加え,加
熱してコバルトを呈色させる。次に,硝酸を加えて加熱し,鉄などの影響を除いた後,水で一定量に薄め
る。次に,ニトロソR塩と硝酸の添加順序を変え,同様の処理を行ってコバルトの呈色をおさえた溶液を
対照液として吸光度を測定する。
5.4.2
試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸
(3) ふっ化水素酸
(4) 臭化水素酸
(5) 硫酸 (1+1, 1+19)
(6) りん酸 (1+1)
(7) アンモニア水 (1+1)
(8) 臭素
(9) くえん酸溶液 くえん酸(1水塩)40gを水に溶解して100mlとしたもの。
(10) 酢酸ナトリウム溶液 酢酸ナトリウム(3水和物)30gを水に溶解して100mlとしたもの。
(11) ニトロソR塩溶液 ニトロソR塩1gを水に溶解して100mlとしたもの。
(12) 標準コバルト溶液 (10 μgCo/ml) コバルト[99.5% (m/m) 以上]1.000gを硝酸 (1+1) 20mlで分解し,
冷却後,1 000mlの全量フラスコに移し入れ,水で標線まで薄めて原液とする。この原液を使用の都
度,必要量だけ水で正確に100倍に希釈して,標準コバルト溶液とする。
5.4.3
試料はかり採り量 試料はかり採り量は,原則として表5による(20)。
表5 試料はかり採り量
コバルト含有率
% (m/m)
試料はかり採り量
g
0.01以上 0.2未満
1 〜2
0.2 以上 0.5以下
0.5〜1
注(20) コバルト量が,なるべく1〜2.5mg程度になるようにはかり採る。
5.4.4
操作
5.4.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採り,ビーカー (200〜300ml) に移し入れ,時計皿で覆い,硝酸10〜20mlを加え,静か
に加熱して分解する(3)。
(2) 激しい反応が終わってから塩酸10〜20mlを加え,引き続き加熱して分解する(4)。
(3) 硫酸 (1+1) 10〜20mlを加えて加熱蒸発し,硫酸の白煙を十分に発生させる(21)。
(4) 放冷後,硫酸 (1+19) 30ml及び水約20mlを加え,加熱して可溶性塩類を溶解する。これにりん酸 (1
+1) 10mlを加えた後,二酸化けい素などの不溶解残さをろ紙(5種B)を用いてろ過し,温水で十分
に洗浄する(6)。ろ液及び洗液は,ビーカー (200〜300ml) に受け,冷却した後,200mlの全量フラスコ
9
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に移し入れ,水で標線まで薄める。
注(21) 残さ中にコバルトが含まれる場合には,少量の水を用いて残さを白金皿(50番)に洗い移し,
硫酸 (1+1) 約5ml,硝酸約5ml及びふっ化水素酸5〜10mlを加え,加熱して硫酸の白煙を十分
に発生させ,二酸化けい素を揮散させて乾固近くまで濃縮する。少量の水を加え,加熱して可
溶性塩類を溶解し,元のろ紙(5種B)を用いてろ過し,温水で十分に洗浄する。ろ液及び洗液
は,ビーカー (200ml) に受け,加熱して1〜2mlに濃縮した後,主液に合わせる。
5.4.4.2
呈色 呈色は,次の手順によって行う。
(1) 5.4.4.1(4)で得た溶液を20 mlずつ分取し2個のビーカー (200ml) に移し入れ,それぞれに,くえん酸
溶液5mlを加えてかき混ぜた後,酢酸ナトリウム溶液20mlを加えてかき混ぜる。
(2) pH計を用い,アンモニア水 (1+1) を滴加してpHを7.0±0.5の範囲に調節する。
(3) このうち一方の溶液には,ニトロソR塩溶液10mlを正しく加え,加熱して5〜6分間煮沸を続けた後,
煮沸をやめ,約5分間放置する。これに硝酸10mlを加え,再び加熱して1〜2分間煮沸する。冷却し
た後,100mlの全量フラスコに移し入れ,水で標線まで薄めて測定溶液とする。
(4) 他方の溶液には,加熱して5〜6分間煮沸を続けた後,煮沸をやめ,約5分間放置する。これに硝酸
10mlを加え,再び煮沸するまで加熱した後,ニトロソR塩溶液10mlを正しく加え,さらに加熱して
1〜2分間煮沸する。冷却した後,100mlの全量フラスコに移し入れ,水で標線まで薄めて対照溶液と
する。
(5) 測定溶液の一部を光度計のセルに取り,対照溶液を対照として波長530nm付近の吸光度を測定する。
5.4.5
空試験 5.4.6の検量線の作成手順において得られる,標準コバルト溶液を添加しない溶液の吸光
度を空試験の吸光度とする。
5.4.6
検量線の作成 標準コバルト溶液0〜30ml(Coとして0〜300μg)を段階的に数個のビーカー
(200ml) に正しく取り,硫酸 (1+19) 3ml,りん酸 (1+1) 1ml,くえん酸溶液5ml及び酢酸ナトリウム溶液
20mlを加え,水を加えて液量を50〜60mlとする。以下,5.4.4.2(2)以降の手順に従って操作し,試料と並
行して測定した吸光度とコバルト量との関係線を作成し,検量線とする。
5.4.7
計算 5.4.6で作成した検量線からコバルト量を求め,試料中のコバルト含有率を,次の式によっ
て算出する。
100
100
×
×
=
B
m
A
CO
ここに, Co: コバルト含有率 [% (m/m)]
A: 分取した試料溶液中のコバルト検出量 (g)
B: 分取量 (ml)
m: 試料はかり採り量 (g)
5.5
原子吸光法
5.5.1
要旨 試料を塩酸と硝酸で分解し,硫酸を加え,加熱して蒸発乾固する。塩酸を加えて可溶性塩類
を溶解し,ろ過した後,ランタン溶液を加え,水で一定量に薄め,原子吸光光度計を用いて吸光度を測定
する。
5.5.2
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1, 1+2, 1+5, 1+19)
10
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 硝酸
(4) 硝酸 (1+1)
(5) ふっ化水素酸
(6) 硫酸 (1+1)
(7) ランタン溶液 塩化ランタン(7水和物)10gを水に溶かして100mlとしたもの。この溶液1ml中に
は約37mgのランタンを含む。
(8) 標準コバルト溶液 (10μgCo/ml) 5.4.2(12)による。
5.5.3
試料はかり採り量 試料は,0.2gをはかり採る。
5.5.4
操作
5.5.4.1
試料溶液の調整 試料溶液の調整は,次の手順によって行う。
(1) 試料をはかり採り,ビーカー (200ml) に移し入れ,時計皿で覆い,塩酸10ml及び硝酸5mlを加え,
静かに加熱して分解する(3)。
(2) 放冷後,硫酸 (1+1) 5mlを加え,引き続き加熱して蒸発乾固する。
(3) 放冷後,塩酸 (1+1) 10ml及び水約30ml(22)を加え,加熱して可溶性塩を溶解した後,ろ紙(5種B)
を用いてろ過し,温水で十分に洗浄する(21)。冷却した後,ろ液及び洗液は100mlの全量フラスコに移
し入れ,ランタン溶液10mlを加え,水で標線まで薄める(23)。
注(22) 試料中にアンチモン,すず,ビスマスなどを含み加水分解するおそれのある場合は,水の代わ
りに塩酸 (1+1) 25mlを追加する。
(23) コバルト量が多い場合には,検量線の直線領域で測定精度の良い濃度範囲に入るように,適当
量を分取して100mlの全量フラスコに移し入れ,ランタン溶液をランタン相当量として検量線
とほぼ等量となるように加え,塩酸 (1+19) で標線まで薄める。ただし,注(22)の操作を行った
場合には塩酸 (1+5) で薄める。
5.5.4.2
吸光度の測定 5.5.4.1(3)で得た溶液の一部を,水を用いて零点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長240.7nmの吸光度を測定する。
5.5.5
空試験 5.3.6の検量線の作成手順において得られる,標準コバルト溶液を添加しない溶液の吸光
度を空試験の吸光度とする。
5.5.6
検量線の作成 標準コバルト溶液0〜40ml(Coとして0〜400μg)(24)を段階的に数個の100mlの全
量フラスコに正しく取り,塩酸 (1+1) 10ml(25)及びランタン溶液10mlを加え,水で標線まで薄める。以下,
5.5.4.2の手順に従って試料と同様に操作し,試料と並行して測定した吸光度とコバルト量との関係線を作
成し,検量線とする。
注(24) 使用装置及び測定波長の感度に応じて,濃度範囲を適宜増減する。
(25) 注(22)の操作を行った場合には,塩酸 (1+1) 35mlを加える。
5.5.7
計算 5.5.6で作成した検量線からコバルト量を求め,試料中のコバルト含有率を,次の式によっ
て算出する。
100
100
×
×
=
B
m
A
CO
ここに, Co: コバルト含有率 [% (m/m)]
A: 分取した試料溶液中のコバルト検出量 (g)
B: 分取量 (ml)
11
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m: 試料はかり採り量 (g)
付図1 イオン交換カラムの例
鉱石中のコバルト定量方法改正原案作成委員会 構成表
氏名
所属
(委員長)
奥 谷 忠 雄
日本大学理工学部工業化学教室教授
市 川 五 朗
住友金属鉱山株式会社
稲 垣 勝 彦
日本鉱業協会
尾 上 喬
同和鉱業株式会社
岸 野 忠 信
財団法人日本規格協会
佐 山 恭 正
三菱マテリアル株式会社
丹 野 一 雄
東邦亜鉛株式会社
中 村 靖
株式会社日鉱共石
野 村 紘 一
三菱マテリアル株式会社
宮 本 幸 夫
工業技術院標準部
渡 部 武 雄
三井金属鉱業株式会社
12
M 8129-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
鉱石中のコバルト定量方法改正原案作成委員会 構成表
氏名
所属
(委員長)
中 村 靖
日本鉱業株式会社
池 田 重 司
大蔵省造幣局東京支局
岩 田 晶 夫
住友金属鉱山株式会社
岩 橋 康 夫
日本鉱業協会技術部
岸 肇
三菱金属株式会社
小 林 透
東邦亜鉛株式会社
清 水 博 司
同和鉱業株式会社
田 沼 滉
志村化工株式会社
束 原 巌
古河金属工業株式会社
東 野 徳 夫
工業技術院地質調査所
外 岡 和 夫
古河鉱業株式会社
中 田 勉
日曹金属株式会社
松 井 敬 二
三井金属鉱業株式会社
吉 田 信 之
工業技術院標準部