2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
M 8121-1997
鉱石中の銅定量方法
Methods for determination of copper in ores
序文 この規格は,1991年に第1版として発行されたISO 9599 (Copper, lead and zinc sulfide concentrates−
Determination of hygroscopic moisture in the analysis sample−Gravimetric method),1994年に第1版として発行
されたISO 10258 (Copper sulfide concentrates−Determination of copper content−Titrimetric methods),及びISO
10469 (Copper sulfide concentrates−Determination of copper content−Electrogravimetric method) を元に作成し
た日本工業規格である。
1. 適用範囲 この規格は,鉱石中の銅定量方法について規定する。ただし,他の日本工業規格で銅定量
方法が規定されている鉱石には適用しない。
備考1. この規格の引用規格を,次に示す。
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS M 8083 ばら積み非鉄金属浮選精鉱のサンプリング方法
JIS M 8101 非鉄金属鉱石のサンプリング,試料調製及び水分決定方法
JIS R 3503 化学分析用ガラス器具
JIS Z 8401 数値の丸め方
JIS Z 8402 分析・試験の許容差通則
2. この規格の対応国際規格を,次に示す。
ISO 9599 : 1991 Copper, lead and zinc sulfide concentrates−Determination of hygroscopic moisture
in the analysis sample−Gravimetric method
ISO 10258 : 1994 Copper sulfide concentrates−Determination of copper content−Titrimetric
methods
ISO 10469 : 1994 Copper sulfide concentrates−Determination of copper content−Electrogravimetric
method
2. 一般事項 分析方法に共通な一般事項は,JIS K 0050,JIS K 0115,JIS K 0116及びJIS K 0121の規
定による。
3. 分析試料の採り方及び取扱い方
3.1
試料の採取と調製 試料の採取と調製は,JIS M 8083又はJIS M 8101の規定による。
2
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.2
試料のはかり方
3.2.1
試料のはかり採り 試料のはかり採りは,化学はかりを用い,はかり方は,次のいずれかによる。
(1) 吸着水分補正法 吸着水分補正法は,附属書1又は附属書2による。
(2) 事前乾燥A法 事前乾燥A法は,次の手順によって行う。
(a) 平形はかり瓶(瓶及びふたの合計質量が20g以下のもの)をそのふたと共に,105±5℃に調節して
ある空気浴に入れて1時間乾燥しデシケーター中で常温まで放冷した後,平形はかり瓶とふたとの
合計質量をはかる(1)。
(b) 各分析方法に規定された質量の分析試料を平形はかり瓶にはかり採り(1),平形はかり瓶のふたと共
にあらかじめ105±5℃に調節してある空気浴に入れて2時間乾燥した後,平形はかり瓶にふたをし
てデシケーター中で常温まで放冷する。平形はかり瓶のふたを少しずらし,直ちに元に戻して試料,
平形はかり瓶及びそのふたの合計質量 (m1) を0.1mgのけたまではかる(2)。
(c) 乾燥した試料を各定量方法に規定されている容器に移しあけた後,直ちに空の平形はかり瓶及びそ
のふたの合計質量 (m2) を0.1mgのけたまではかる。
(d) 試料のはかり採り量は,次の式によって算出する。
m=m1−m2
ここに,
m: 試料のはかり採り量 (g)
m1: 乾燥した試料,平形はかり瓶及びふたの合計質量 (g)
m2: 空の平形はかり瓶及びふたの合計質量 (g)
注(1) ここでは正確にはかる必要はない。
(2) 規定の時間の乾燥で十分であることが確認されていない試料の場合には,試料,平形はかり瓶
及びそのふたの合計質量をはかり,更に2時間乾燥しデシケーター中で放冷後,その質量をは
かる。2時間の乾燥減量が0.5mg以下となるまで乾燥を繰り返す。
(3) 事前乾燥B法 事前乾燥B法は(3),次の手順によって行う。
(a) 分析試料5〜10gを平形はかり瓶(直径約50mmのもの)に移し入れ平らに広げた後,平形はかり
瓶のふたとともにあらかじめ105±5℃に調節してある空気浴に入れて2時間乾燥する。空気浴から
取り出し平形はかり瓶にふたをしてデシケーター中で常温まで放冷する(4)。
(b) デシケーターから平形はかり瓶を取り出し,直ちに各定量方法に規定された質量の試料を0.1mgの
けたまではかり採る(5)。
注(3) この方法は吸湿性の激しい試料には適用できない。
(4) 規定の時間の乾燥で十分であることが確認されていない試料の場合には,試料,平形はかり瓶
及びそのふたの合計質量をはかり,更に2時間乾燥しデシケーター中で放冷後,その質量をは
かる。2時間の乾燥減量が1mg以下となるまで乾燥を繰り返す。
(5) ひょう量の都度,平形はかり瓶をデシケーターから取り出し,平形はかり瓶のふたを取り,速
やかに試料をはかり採る。
4. 分析値のまとめ方 分析値のまとめ方は,次による。
(1) 分析値は,質量百分率で表す。
(2) 分析は,同一試験室において2回繰り返して行い,これらの差が室内再現許容差以下のときは,その
平均値を求め,JIS Z 8401によって小数点以下2けたに丸めて報告値とする。
(3) 2回繰り返して行った分析値の差が室内再現許容差を超えるときは,JIS Z 8402の6.4.2(適用方法A)

3
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の規定による。
(4) 室内再現許容差は,表1による。
表1 室内再現許容差
単位% (m/m)
定量方法
銅含有率の区分
室内再現許容差
電解重量法
15
以上 50 以下
0.150
硫化銅分離チオ硫酸ナトリウム滴定法 5
以上 15 未満
0.080
15
以上 50 以下
0.150
チオ硫酸ナトリウム滴定法
15
以上 50 以下
0.150
原子吸光法
0.01 以上 0.2 未満
0.006
0.2 以上 1 未満
0.040
1
以上 3 以下
0.055
ICP発光分光法
0.01 以上 0.2 未満
0.006
0.2 以上 1 未満
0.040
1
以上 3 未満
0.055
3
以上 10 以下
0.10
アンモニア吸光光度法
0.5 以上 1 未満
0.030
1
以上 5 未満
0.090
5
以上 10 以下
0.150
5. 定量方法の区分 鉱石中の銅定量方法は,次のいずれかによる。
(1) 銅電解重量法 この方法は,銅含有率15% (m/m) 以上50% (m/m) 以下の試料に適用する。
(2) 硫化銅分離チオ硫酸ナトリウム滴定法 この方法は,銅含有率5% (m/m) 以上50% (m/m) 以下の試料
に適用する。
(3) チオ硫酸ナトリウム滴定法 この方法は,銅含有率15% (m/m) 以上50% (m/m) 以下の試料に適用す
る。
(4) 原子吸光法 この方法は,銅含有率0.01% (m/m) 以上3% (m/m) 以下の試料に適用する。
(5) ICP発光分光法 この方法は,銅含有率0.01% (m/m) 以上10% (m/m) 以下の試料に適用する。
(6) アンモニア吸光光度法 この方法は,銅含有率0.5% (m/m) 以上10% (m/m) 以下の試料に適用する。
6. 銅電解重量法
6.1
要旨 試料を硝酸と硫酸とで分解した後,臭化水素酸を加えて加熱し,ひ素,アンチモン,セレン
及びすずを揮散して除去し,水で溶解する。アンモニア水を加えて,妨害元素を水酸化鉄と共沈させて分
離するか,又はチオ硫酸ナトリウムを加えて銅を硫化銅として沈殿させ妨害元素と分離する。硫化銅の沈
殿は,硫酸及び硝酸で分解する。得られた溶液に硫酸及び硝酸を加え,白金電極を用いて電解し陰極に銅
を電着させ,その質量をはかる。
6.2
試薬 試薬は,次による。
(1) 塩酸 (1+1,1+3,1+19)
(2) 硝酸
(3) 硝酸 (1+1)
(4) ふっ化水素酸
(5) 臭化水素酸
(6) 硫酸
4
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(7) 硫酸 (1+1,1+4)
(8) 混酸 硝酸250mlに硫酸250mlを徐々に加える。
(9) アンモニア水
(10) アンモニア水 (1+99)
(11) 臭素
(12) 塩化ナトリウム溶液 (10g/l,0.5g/l)
(13) 硫酸ナトリウム(無水)
(14) チオ硫酸ナトリウム溶液 チオ硫酸ナトリウム五水和物450gを水に溶解して1lとする。
(15) 硫酸アンモニウム鉄 (III) 溶液 硫酸アンモニウム鉄 (III) ・12水43gを硫酸 (1+1) 50ml及び水に溶
かして1 000mlとする。この溶液1mlは鉄約5mgを含む。
(16) 硫酸アンモニウム溶液 (300g/l)
(17) エタノール (99.5)
(18) 標準銅溶液 (100μgCu/ml) 銅 [99.99% (m/m)] 0.100gをはかり採り,ビーカー (300ml) に移し入れ,
時計皿で覆い硝酸 (1+1) 10mlを加えて分解した後,加熱して窒素酸化物を追い出す。水約50mlを加
えて常温まで冷却する。水を用いて1 000mlの全量フラスコに移し入れ,水で標線まで薄める。
6.3
器具 器具は,次による。
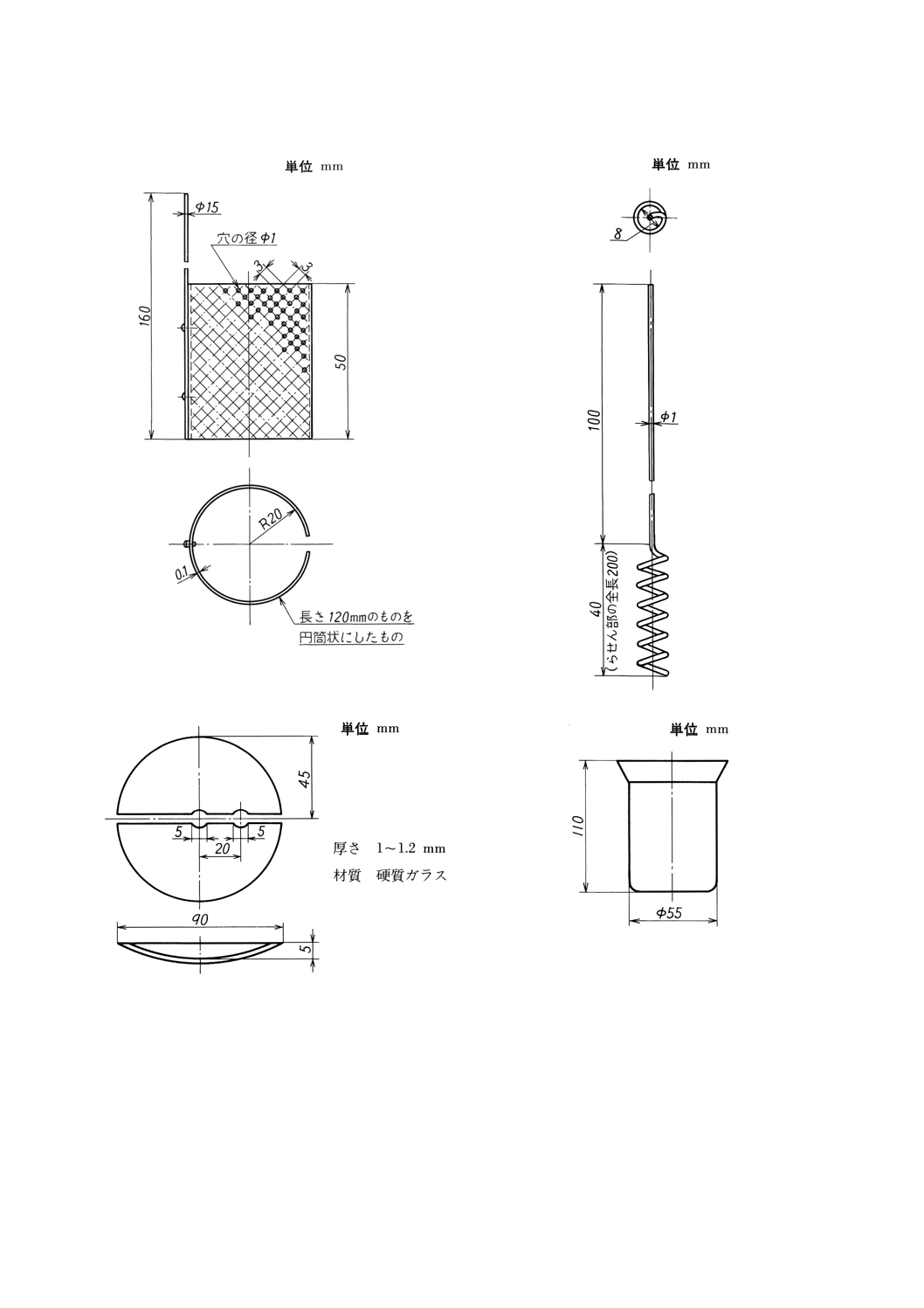
(1) 円筒状白金陰極 原則として図1のものを用いる。
(2) らせん状白金陽極 原則として図2のものを用いる。
(3) 半円形時計皿 原則として図3のものを用いる。
(4) 電解ビーカー 原則として図4のもの又はJIS R 3503のビーカー300mlを用いる。
5
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図1 円筒状白金陰極
図2 らせん状白金陽極
図3 半円形時計皿
図4 電解ビーカー
6.4
試料はかり採り量 試料はかり採り量は,2gとする。
6.5
操作
6.5.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採り,ビーカー (400ml) に移し入れ,水10mlを加えて試料を湿らせた後,硝酸 (1+1)
20mlを加え(6)時計皿で覆い60〜70℃で約10分間加熱する。硫酸 (1+1) 20mlを加えて穏やかに加熱
を続け,激しい反応が終了したら,時計皿の下面を水で洗って時計皿を取り除き,引き続き加熱を続
け硫酸の白煙を十分に発生させる(7)(8)(9)。
6
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 放冷した後,水5ml及び臭化水素酸10mlを注意して加え,加熱して硫酸の白煙を十分に発生させる。
放冷した後,硫酸 (1+1) 5ml及び臭化水素酸10mlを注意して加え,再び加熱して硫酸の白煙を激し
く発生させる(10)。
(3) 放冷した後,水約80mlを加えて加熱して可溶性塩類を溶解し(11),沸騰するまで加熱した後,室温ま
で冷却する。ろ紙(5種B)を用いてろ過して水で十分に洗浄する(12)。ろ液及び洗液はビーカー (A)
(400ml) に受ける。
注(6) 酸化鉱,焼鉱などの場合には,あらかじめ塩酸を加え,徐々に加熱して分解した後硝酸を加え
る。
(7) 不溶解残物の色が黒みを帯びているときは,溶液が熱いうちに混酸 [6.2(8)] を少量ずつ不溶解
残物の色が無色又は青みを帯びた色になるまで加えた後,加熱して硫酸の白煙を十分発生させ
る。
(8) 遊離した硫黄の分解が不十分のときは,硝酸5ml及び臭素1mlを加え,加熱して硫酸の白煙を
十分に発生させる。
(9) 試料中のひ素,アンチモン,すず及びセレンの含有率が各々0.01% (m/m) 以下のときは手順(2)
を省略してもよい。手順(2)を省略した場合で,6.5.2(2)の硫化物分離法を行うときは,ここで残
る硫酸量は6〜8mlが最適である。硫酸の白煙を長時間発生させた場合には,残存する硫酸量が
少なくなるので,引き続き加熱して乾固近くまで濃縮し,放冷後,硫酸 (1+1) 15mlを加える。
(10) 6.5.2(2)の硫化物分離法を行う場合は,ここで残る硫酸量は6〜8mlが最適である。硫酸の白煙
を長時間発生させた場合には残存する硫酸量が少なくなるので,引き続き加熱して乾固近くま
で濃縮し,放冷後,硫酸 (1+1) 15mlを加える。
(11) 試料中に0.01% (m/m) 以上の銀を含むときは,ここで塩化ナトリウム溶液 (10g/l) 1mlを加え塩
化銀として沈殿させて不溶解残分と共にろ過する。
(12) ろ紙上の不溶解残物中に銅が含まれるときは,不溶解残物をろ紙と共に保存し6.5.5又は6.5.6
によって銅を定量する。
6.5.2
銅の分離 銅の分離は,次のいずれかの手順によって行う。
(1) 水酸化物分離法
(a) 6.5.1(3)で得た溶液(13)(14)を加熱して濃縮するか,又は水を加えて約150mlとし,かき混ぜながら,
アンモニア水を一度生成した塩基性銅塩が溶解するまで少量ずつ加え,更に過剰に30mlを加える。
加熱して瞬時沸騰(15)した後,ろ紙(5種B)を用いてろ過し,温アンモニア水 (1+99) で数回洗浄
する。ろ液及び洗液はビーカー (B) (500ml) に受ける。
(b) 沈殿は水を用いて元のビーカー (A) に洗い移し,ろ紙上から温硫酸 (1+4) 15mlを少量ずつ滴加し
てろ紙上の水酸化物の沈殿を溶解した後,ろ紙は温水で数回洗浄する。ろ液及び洗液は主沈殿を洗
い移したビーカー (A) に受け,ろ紙は漏斗と共に保存する。ビーカー (A) に硫酸 (1+1) 10mlを加
え,ふり混ぜて主沈殿を溶解する。水を加えて液量を約100mlとし,かき混ぜながらアンモニア水
を水酸化鉄の沈殿がわずかに生成するまで加え,更に過剰に30mlを加える。加熱して瞬時沸騰(15)
させた後,元のろ紙を用いてろ過し,温アンモニア水 (1+99) で数回洗浄する。ろ液及び洗液は第
1回目のろ液及び洗液の入っているビーカー (B) に受ける。
(c) 沈殿はビーカー (A) に水を用いて洗い移し,ろ紙上から温塩酸 (1+3) を少量ずつ加えて水酸化物
の沈殿を溶解し,更に温塩酸 (1+19) でろ紙を数回洗浄する。塩酸 (1+1) 30mlを加えて沈殿を溶
解し,6.5.5によって銅を定量するために保存する。
7
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(d) ろ液及び洗液は,液量が約150mlとなるまで加熱して濃縮した後(16),硫酸 (1+1) 10mlを加え引き
続き加熱して液量が100mlとなるまで濃縮する。硝酸 (1+1) 5ml(17)を加えて,電解ビーカー [6.3(4)]
に移し入れ水で150〜170mlとする。
注(13) 6.5.1(3)の注(11)を行ったときは,この溶液を加熱して濃縮し硫酸の白煙を十分に発生させ,放冷
した後水で約150mlとする。
(14) 試料中の鉄含有量が約50mg以下のときは,試料溶液中の鉄量が約50mg以上となるように硫酸
アンモニウム鉄 (III) 溶液 [6.2(15)] を加える。
(15) 沸騰したら直ちに加熱を止める。沸騰を続けるとアンモニアの濃度が減少して水酸化鉄中の銅
量が増加し,分析精度が悪くなる。
(16) 液量が約300mlとなるまでは穏やかに加熱する。作業時間を短縮するためにあらかじめ1回目
の沈殿のろ過操作で得たろ液及び洗液を,加熱して濃縮しておくとよい。
(17) 試料中のモリブデン量が0.5mg以上の場合は,更に塩化ナトリウム溶液 (0.5g/l) 10mlを加える。
(2) 硫化物分離法
(a) 6.5.1(3)で得た溶液に,水を加えて約200mlとした後,加熱して70〜90℃とし,かき混ぜながらチオ
硫酸ナトリウム溶液 [6.2(14)] 50mlを加え黄色又は黄褐色のエマルジョンを生成させる。穏やかに
加熱して約10分間煮沸して沈殿を凝縮させた後,直ちにろ紙(5種B)を用いてろ過し温水で数回
洗浄する。ろ液及び洗液は500mlの全量フラスコに受け保存し,6.5.6(1)によって銅を定量する。
(b) 沈殿はろ紙と共に元のビーカー (A) に移し入れ,混酸[6.2(8)] 30mlを加え時計皿で覆い,徐々に加
熱してろ紙及び沈殿を分解し,引き続き加熱して蒸発乾固する(18)。さらに,加熱を続け遊離した硫
黄を分解する。
(c) 硝酸10mlをビーカーの内壁に付着している遊離した硫黄を洗い落としながら加えた後,硫酸 (1+
1) 2mlを加え加熱して硫酸の白煙を十分に発生させる。放冷後,水約100mlを加えて加熱し可溶性
塩類を溶解する。
(d) この溶液に(19)硫酸アンモニウム鉄 (III) 溶液 [6.2(15)] 3mlを加え,かき混ぜながらアンモニア水を
一度生成した塩基性銅塩が溶解するまで少量ずつ加え,更に過剰に10mlを加えた後,沸騰するま
で加熱する。ろ紙(5種B)を用いてろ過し,温アンモニア水 (1+99) で数回洗浄する。ろ液及び
洗液はビーカー (B) (500ml) に受ける。
(e) 沈殿は水を用いて元のビーカー (A) に洗い移し,ろ紙上から温硫酸 (1+4) 15mlを少量ずつ滴加し
てろ紙上の水酸化物の沈殿を溶解した後,ろ紙は温水で数回洗浄する。ろ液及び洗液は主沈殿を洗
い移したビーカー (A) に受け,ふり混ぜて主沈殿を溶解する。水を加きて約100mlとし,かき混ぜ
ながらアンモニア水を水酸化鉄がわずかに生成するまで加え,更に過剰に10mlを加えた後,沸騰
するまで加熱する。もとのろ紙を用いてろ過し,温アンモニア水 (1+99) で数回洗浄する。ろ液及
び洗液は第1回目のろ液及び洗液の入っているビーカー (B) に受ける。
(f) 沈殿は,元のビーカー (A) に水を用いて洗い移し,ろ紙上から温塩酸 (1+3) を少量ずつ加えて水
酸化物の沈殿を溶解し,更に温塩酸 (1+19) でろ紙を数回洗浄する。ろ液及び洗液は,水酸化物の
沈殿を洗い移してあるビーカー (A) に受け,塩酸 (1+1) 10mlを加えて沈殿を溶解した後,6.5.6(2)
によって銅を定量するために保存する。
(g) ろ液及び洗液は,液量が約150mlとなるまで加熱して濃縮した後(16),硫酸 (1+1) 10mlを加え引き
続き加熱して液量が100mlとなるまで濃縮する。硝酸 (1+1) 5ml(17)を加えて,電解ビーカー [6.3(4)]
に移し入れ水で150〜170mlとする。
8
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(18) ろ紙の分解が不十分のときは,ビーカーが熱いうちに混酸 [6.2(8)] を残物の色が無色又は青み
を帯びた色になるまで少量ずつ滴加し,放冷した後,水20mlを加え再び加熱して硫酸の白煙を
十分発生させる。
(19) 試料中のビスマス及び/又はテルルの含有率が0.01% (m/m) 以下のときは,(d)及び(e)の操作は
省略する。
6.5.3
電解 電解は,次の手順によって行う。
(1) 円筒状白金陰極 [6.3(1)] を約80℃の空気浴中で5〜10分間乾燥し,デシケーター中で約30分間放冷
した後,その質量を0.1mgのけたまではかる。
(2) 6.5.2(1)(d)又は6.5.2(2)(g)で得た溶液に,円筒状白金陰極 [6.3(1)] 及びらせん状白金陽極 [6.3(2)] を挿
入し,電源に接続する。このとき円筒状白金陰極の上部約5mmを電解液面から露出させ,らせん状
白金陽極の下端は陰極の下端より約5mm深くなるように設置する。2個の半円形時計皿 [6.3(3)] で覆
い,液温を15〜30℃に保ち,約0.25Aの電流を通じて約18時間(20)電解する。
(3) 電流を通じたまま半円形時計皿の下面,ビーカーの内壁及び電極の液面に露出している部分を水洗し,
その洗浄水によって電解液面を約5mm上昇させ,更に約1時間電解を続ける。
(4) 電解が終了したら時計皿の下面及びビーカーの内壁を水洗して時計皿を取り除く。電流を通じたまま,
水洗しながら両極を徐々に引き上げ,水を満たしてあるビーカー中に手早く浸して陰極を外す。陰極
を新しい水を満たしてある別のビーカー中に移し数回上下に動かして水洗する。次いでエタノール
(99.5) 中に浸し数回上下に動かし洗浄した後,新しいエタノール (99.5) を満たしてある別のビーカー
中に移し数回上下に動かして水分を除く。約80℃の空気浴中で2〜3分間乾燥し(21),直ちにデシケー
ターに入れ常温まで放冷する。電解残液は,500mlの全量フラスコに移し入れて保存し,6.5.5又は
6.5.6(2)によって銅を定量する。
注(20) 電解時間を短縮したいときは,電解電流を0.5Aまで上げてもよい。
(21) 乾燥温度が高いとき又は乾燥時間が長いときは,電着した銅の表面が酸化し分析結果に高い偏
りを与えるので,温度及び時間に注意して手早く行う。
6.5.4
ひょう量 6.5.3(4)で得た銅を電着した白金陰極の質量を,6.5.3(1)で用いた化学はかりを用いて,
0.1mgのけたまではかる。
6.5.5
水酸化物分離法を用いたときの不溶解残物,水酸化鉄溶解液及び電解残液中の銅の定量 不溶解残
物,水酸化鉄溶解液及び電解残液中の銅の定量は,次の手順によって行う。
(1) 不溶解残物の分解 6.5.1注(12)で保存した不溶解残物をろ紙と共に白金るつぼ(30番)に移し入れ,
乾燥後700〜800℃で加熱してろ紙を灰化する。放冷した後,硫酸 (1+1) 5ml及びふっ化水素酸5〜10ml
を加え加熱してほとんど乾固するまで濃縮する。水5ml及び硫酸 (1+1) 1mlを加えて加熱して塩類を
溶解し,常温まで冷却した後,6.5.3(4)で電解残液を入れてある500mlの全量フラスコに水を用いて移
し入れる。
(2) 水酸化鉄溶解液の処理 6.5.2(1)(c)で保存した水酸化鉄の沈殿を溶解した溶液を,6.5.3(4)で電解残液を
入れてある500mlの全量フラスコに水を用いて移しあける。
(3) 発光強度の測定 6.5.3(4)で得た電解残液と(1)及び(2)で得た溶液の入っている500mlの全量フラスコ
を,水で標線まで薄める。この溶液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長
324.75nmにおける発光強度を測定する。
(4) 検量線の作成 標準銅溶液 [6.2(18)] の各種液量(銅として0〜4mg)を段階的に500mlの全量フラス
コに採り,硫酸 (1+1) 10ml,硝酸 (1+1) 5ml,塩酸 (1+1) 30ml,硫酸アンモニウム溶液130ml及び
9
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試料溶液中に含まれる鉄量とほぼ同じ量の鉄に相当する量の硫酸アンモニウム鉄 (III) 溶液 [6.2(15)]
を加え,水で標線まで薄める。この溶液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,
波長324.75nmにおける発光強度を試料と並行して測定する。得た発光強度と銅量との関係線を作成
し,その関係線を原点を通るように平行移動して検量線とする。
(5) 空試験液の発光強度の測定 試料を用いないで,試料と同じ操作を試料と並行して行った空試験液の
一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nmにおける発光強度を測定す
る。
(6) 銅量の算出 (3)で得た発光強度から(5)で得た発光強度を差し引いた発光強度と(4)で作成した検量線
とから銅量を求める。
6.5.6
硫化物分離法を用いたときのろ液,不溶解残物,水酸化鉄及び電解残液中の銅の定量 ろ液,不溶
解残物,水酸化鉄及び電解残液中の銅の定量は,次の手順によって行う。
(1) 硫化銅を分離したろ液中の銅の定量
(a) 発光強度の測定 6.5.2(2)(a)で500mlの全量フラスコ中に保存したろ液及び洗液は,常温まで冷却し
た後水で標線まで薄める。この溶液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波
長324.75nmにおける発光強度を測定する。
(b) 検量線の作成 標準銅溶液 [6.2(18)] の各種液量(銅として0〜4mg)を段階的に500mlの全量フラ
スコに採り,硫酸 (1+1) 20ml及び硫酸ナトリウム(無水)13gを加えふり混ぜて溶解した後,水で
標線まで薄める。この溶液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nm
における発光強度を試料と並行して測定する。得た発光強度と銅量との関係線を作成し,その関係
線を原点を通るように平行移動して検量線とする。
(c) 空試験液の発光強度の測定 試料を用いないで,試料と同じ操作を試料と並行して行った空試験液
の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nmにおける発光強度を測
定する。
(d) 銅量の算出 (a)で得た発光強度から(c)で得た発光強度を差し引いた発光強度と(b)で作成した検量
線とから銅量を求める。
(2) 不溶解残物,水酸化鉄溶解液及び電解残液中の銅の定量
(a) 不溶解残物の分解 6.5.1注(12)で保存した不溶解残物をろ紙と共に白金るつぼ(30番)に移し入れ,
乾燥後700〜800℃で加熱してろ紙を灰化する。放冷した後,硫酸 (1+1) 5ml及びふっ化水素酸5〜
10mlを加え加熱してほとんど乾固するまで濃縮する。水5ml及び硫酸 (1+1) 1mlを加えて加熱し
て塩類を溶解し,常温まで冷却した後,6.5.3(4)で電解残液を入れてある500mlの全量フラスコに水
を用いて移し入れる。
(b) 水酸化鉄溶解液の処理 6.5.2(2)(f)で保存した水酸化鉄の沈殿を溶解した溶液を,6.5.3(4)で電解残液
を入れてある500mlの全量フラスコに水を用いて移しあける。
(c) 発光強度の測定 (a)及び(b)で得た溶液を入れてある500mlの全量フラスコを常温まで冷却した後,
水で標線まで薄める。この溶液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長
324.75nmにおける発光強度を測定する。
(d) 検量線の作成 標準銅溶液 [6.2(18)] の各種液量(銅として0〜4mg)を段階的に500mlの全量フラ
スコに採り,硫酸 (1+1) 10ml,硝酸 (1+1) 5ml,塩酸 (1+1) 10ml及び硫酸アンモニウム鉄 (III) 溶
液 [6.2(15)] 3mlを加え,水で標線まで薄める。この溶液の一部をICP発光分光装置のアルゴンプラ
ズマ中に噴霧し,波長324.75nmにおける発光強度を試料と並行して測定する。得た発光強度と銅
10
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
(e) 空試験液の発光強度の測定 試料を用いないで,試料と同じ操作を試料と並行して行った空試験液
の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nmにおける発光強度を測
定する。
(f) 銅量の算出 (c)で得た発光強度から(e)で得た発光強度を差し引いた発光強度と(d)で作成した検量
線とから銅量を求める。
6.6
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
6.7
計算 試料中の銅含有率を,次の式によって算出する。
H
m
C
G
G
G
G
Cu
−
+
−
−
−
=
100
100
100
)
(
)
(
2
1
2
1
×
×
′
′
ここに, Cu: 試料中の銅含有率 [% (m/m)]
G1: 6.5.4で得た電解後の白金陰極の質量 (g)
G2: 6.5.3(1)で得た電解前の白金陰極の質量 (g)
G1': 6.6で得た電解後の白金陰極の質量 (g)
G2': 6.6で得た電解前の白金陰極の質量 (g)
C: 6.5.5又は6.5.6で得た水酸化鉄,電解残液などの中の銅検出量
(g)
m: 試料はかり採り量 (g)
H: 吸着水分率 [% (m/m)] ただし,事前乾燥A法又は事前乾燥B
法を用いたときは,H=0とする。
7. 硫化銅分離チオ硫酸ナトリウム滴定法
7.1
要旨 試料を硝酸と硫酸とで分解した後,臭化水素酸を加えて加熱し,ひ素,アンチモン,セレン
及びすずを揮散して除去し,水で溶解する。チオ硫酸ナトリウムを加えて銅を硫化銅として沈殿させて,
こし分ける。沈殿を硫酸と硝酸とで分解し,加熱して乾固した後水で溶解する。炭酸ナトリウムを加えて
微アルカリ性とし,次いで酢酸で微酸性とした後,ふっ化水素アンモニウムを加えて共存する鉄の影響を
隠ぺいする。よう化カリウムを加えて銅と当量のよう素を遊離させ,遊離したよう素をチオ硫酸ナトリウ
ム標準溶液で滴定する。
7.2
試薬 試薬は,次による。
(1) 硝酸
(2) 硝酸 (1+1)
(3) ふっ化水素酸
(4) 臭化水素酸
(5) 硫酸 (1+1)
(6) 混酸 硝酸250mlに硫酸250mlを徐々に加える。
(7) 臭素
(8) 臭素水(飽和)
(9) ふっ化水素アンモニウム溶液 (250g/l)
(10) 炭酸ナトリウム溶液 (20g/l)
(11) 硫酸ナトリウム(無水)
(12) チオ硫酸ナトリウム溶液 チオ硫酸ナトリウム五水和物200gを水に溶解して1lとする。
(13) チオシアン酸カリウム溶液 (100g/l)

11
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(14) よう化カリウム
(15) 酢酸 (1+3)
(16) 0.08mol/lチオ硫酸ナトリウム標準溶液 チオ硫酸ナトリウム五水和物20gを新しく煮沸して冷却した
水1lに溶解する。炭酸ナトリウム0.2gを加えかき混ぜて溶解し1日間以上放置する。この溶液1ml
は銅約5mgに相当するが,標定は,次のようにして行う(22)。
銅[99.99% (m/m) 以上]の小片を温酢酸 (1+3) に浸して表面の酸化物を除去した後,水次いでエ
タノール (95) で洗浄し乾燥する。この小片から試料中に含まれると思われる銅量とほぼ同じ質量を
0.1mgのけたまで3個はかり採り (M1, M2, M3) それぞれビーカー (400ml) に移し入れる。硝酸 (1+1)
10mlを加えて分解した後,硫酸 (1+1) 5mlを加えて加熱して乾固する。放冷後水40mlを加えて加熱
して可溶性塩類を溶解し放冷する。以下,7.4.4(1)又は(2)の操作に従って操作し,それぞれのチオ硫酸
ナトリウム標準溶液1ml当たりの銅相当量を,次の式によって求める。
この標準溶液は褐色瓶に入れて保存し,少なくとも5日ごとに標定する。
i
i
i
V
M
f=
ここに,
fi: チオ硫酸ナトリウム標準溶液1ml当たりの銅相当量 (mg)
Mi: 銅はかり採り量 (mg)
Vi: チオ硫酸ナトリウム標準溶液の使用量 (ml)
f1,f2及びf3の範囲が0.01mgCu/ml以下のときは,3個の平均値を算出し,これを小数点以下第3位
に丸めて,チオ硫酸ナトリウム標準溶液1mlの銅相当量 (mg) の平均値(F)とする。f1,f2及びf3の範
囲が0.01mgCu/mlを超えたときは,改めて初めから標定を行わなければならない。
(17) 標準銅溶液 (100μgCu/ml) 6.2(18)による。
(18) でんぷん溶液 (2g/l) でんぷん(溶性)1gに水10mlを加えて,よくかき混ぜた後,約500mlの熱水
中にかき混ぜながら少量ずつ加える。約1分間煮沸し放冷する。この溶液は,使用の都度調製する。
注(22) チオ硫酸ナトリウム標準溶液の銅相当量は,標定のときの溶液量,溶液温度,銅量及びよう化
カリウムの添加量によって変化する。したがって,チオ硫酸ナトリウム標準溶液の標定をする
ときと試料溶液を滴定するときは,これらの要因をそろえなくてはならない。
7.3
試料はかり採り量 試料はかり採り量は,銅含有率に応じて,表2による。
表2 試料はかり採り量
銅含有率
% (m/m)
試料はかり採り量
g
5以上15未満
1.0
15以上25未満
0.8
25以上50以下
0.4
7.4
操作
7.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採り,ビーカー (400ml) に移し入れ,水10mlを加えて試料を湿らせた後,硝酸 (1+1)
20mlを加えて(6)時計皿で覆い60〜70℃で約10分間加熱する。硫酸 (1+1) 15mlを加えて穏やかに加
熱を続け,激しい反応が終了したら,時計皿の下面を水で洗って時計皿を取り除き,引き続き加熱を
続け,硫酸の白煙を十分に発生させる(7)(8)。
(2) 放冷した後(9),水5ml及び臭化水素酸10mlを注意して加え,加熱して硫酸の白煙を十分に発生させ
る。放冷した後,硫酸 (1+1) 5ml及び臭化水素酸10mlを注意して加え,再び加熱して硫酸の白煙を
12
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
激しく発生させる(23)。
(3) 放冷した後,水約80mlを加えて加熱して可溶性塩類を溶解し,沸騰するまで加熱を続ける。ろ紙(5
種B)を用いてろ過し温水で十分洗浄し(24),ろ液及び洗液はビーカー (400ml) に受ける。
注(23) このときに残存する最適硫酸量は,6〜8mlである。硫酸の白煙を長時間発生させた場合には残
存する硫酸量が少なくなるので,引き続き加熱して乾固近くまで濃縮し,放冷後,硫酸 (1+1)
15mlを加える。
(24) ろ紙上の不溶解残物中に銅が含まれるときは,不溶解残物をろ紙と共に保存し7.4.5によって銅
を定量する。
7.4.2
銅の分離 7.4.1(3)で得た溶液に,水を加えて約200mlとした後,加熱して70〜90℃とし,かき混
ぜながらチオ硫酸ナトリウム溶液 [7.2(12)] 40mlを加えて黄色又は黄褐色のエマルジョンを生成させる。
加熱して徐々に温度を上げ煮沸して沈殿を凝縮させる。直ちにろ紙(5種B)を用いてろ過し温水で数回
洗浄する。ろ液及び洗液は500mlの全量フラスコに受けて保存し,7.4.5によって銅を定量する。
7.4.3
硫化銅の分解 硫化銅の分解は,次のいずれかの手順によって行う。
(1) 硫化銅だけをビーカーに移しあけて分解するとき
(a) 7.4.2で得た沈殿を,水を用いて元のビーカーに洗い移し,ろ紙上に残った沈殿はろ紙をひろげてろ
紙上から臭素溶液(飽和)と硝酸を3〜5mlずつ交互に滴加して分解し,温水で十分に洗浄する(25)。
これらの溶液は,主沈殿を洗い移した元のビーカーに受ける。
(b) 硫酸 (1+1) 2ml及び硝酸10mlを加えて徐々に加熱して沈殿を分解し,引き続き加熱を続け蒸発乾
固する。さらに,加熱を続け遊離した硫黄を分解する。
(c) 硝酸10mlをビーカーの内壁に付着している遊離した硫黄を洗い落としながら加えた後,硫酸 (1+
1) 2mlを加えて加熱し硫酸の白煙を十分発生させる。
(d) 放冷後,水40mlを加えて加熱し可溶性塩類を溶解する。
注(25) ろ紙上の銅の沈殿が完全に分解しないときは,ろ紙を保存し7.4.5によって銅を定量する。
(2) 硫化銅をろ紙と共に分解するとき
(a) 7.4.2で得た沈殿は,ろ紙と共に元のビーカーに移し入れ,混酸 [7.2(6)] 30mlを加え,時計皿で覆い
徐々に加熱してろ紙及び沈殿を分解し,引き続き加熱して蒸発乾固する(26)。さらに,加熱を続け遊
離した硫黄を分解する。
(b) 硝酸10mlを時計皿の下面及びビーカーの内壁に付着している遊離した硫黄を洗い落としながら加
えた後,時計皿の下面を水を洗って時計皿を取り除く。硫酸 (1+1) 2mlを加えて加熱し硫酸の白煙
を十分発生させる。
(c) 放冷後,水40mlを加えて加熱し可溶性塩類を溶解する。
注(26) ろ紙の分解が不十分のときは,ビーカーが熱いうちに混酸 [7.2(6)] を少量ずつ残物の色が無色
又は青みを帯びた色になるまで加え,放冷した後,水20mlを加えて再び加熱し蒸発乾固する。
7.4.4
滴定 滴定は,次のいずれかの手順によって行う。
(1) チオシアン酸カリウムを用いないとき
(a) 7.4.3の(1)(d)又は(2)(c)で得た溶液に,炭酸ナトリウム溶液を銅の沈殿が生成し始めるまで加える。
酢酸 (1+3) を加えて銅の沈殿を消失させた後,更に3〜5mlを加え,次いでふっ化水素アンモニウ
ム溶液1mlを加える。
(b) よう化カリウム15gを加えて振り混ぜて溶解した後,直ちにチオ硫酸ナトリウム標準溶液 [7.2(16)]
で滴定する(22)(27)。終点に近づいて溶液の色が黄褐色から淡黄色になったら(28),指示薬としてでん
13
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ぷん溶液 [7.2(18)] 5mlを加えて滴定を続け,最後の1滴で青色が消失した点を終点とし,チオ硫酸
ナトリウム標準溶液の使用量を求める。
注(27) 銅量が多いときは,遊離したよう素が揮散しやすいので,よう化カリウムを加えた後速やかに
滴定する。
また,滴定の初めは,ビーカーを激しく振り混ぜてはいけない。銅量が少ないときは,よう
化カリウムと銅イオンの反応が遅いので,十分に振り混ぜた後滴定する。
(28) 銀,ビスマス,水銀及び/又は鉛が含まれるときは,溶液の変色が不明りょうになる。このと
きは,溶液が淡褐色になったらでんぷん溶液 [7.2(18)] を加える。
(2) チオシアン酸カリウムを用いるとき
(a) 7.4.3の(1)(d)又は(2)(c)で得た溶液に,炭酸ナトリウム溶液を銅の沈殿が生成し始めるまで加える。
酢酸 (1+3) を加えて銅の沈殿を消失させた後,更に3〜5mlを加え,次いでふっ化水素アンモニウ
ム溶液1mlを加える。
(b) よう化カリウム3gを加えて振り混ぜて溶解した後,直ちにチオ硫酸ナトリウム標準溶液 [7.2(16)]
で滴定する(22)(27)。終点に近づいて溶液の色が黄褐色から淡黄色になったら(28),指示薬としてでん
ぷん溶液 [7.2(18)] 5mlを加え溶液が淡青色となるまで滴定を続ける。
(c) チオシアン酸カリウム溶液5mlを加えて,引き続き滴定を続け,最後の1滴で青色が消失した点を
終点とし,チオ硫酸ナトリウム標準溶液の使用量を求める。
7.4.5
不溶解残物,ろ紙及びろ液中の銅の定量 不溶解残物,ろ紙及びろ液中の銅の定量は,次の手順に
よって行う。
(1) 不溶解残物の分解 7.4.1(3)の注(24)で保存した不溶解残物は,ろ紙と共に白金るつぼ(30番)に移し
入れ,乾燥後700〜800℃で加熱してろ紙を灰化する。放冷した後,硫酸 (1+1) 5ml及びふっ化水素酸
5〜10mlを加えて加熱しほとんど乾固するまで濃縮する。水5ml及び硫酸 (1+1) 1mlを加えて加熱し
て塩類を溶解し,常温まで冷却した後,7.4.2でろ液及び洗液を入れてある500mlの全量フラスコに水
を用いて移し入れる。
(2) ろ紙の分解 7.4.3(1)(a)の注(25)で保存したろ紙をビーカー (300ml) に移し入れ,混酸 [7.2(6)] 30mlを
加えて加熱し蒸発乾固する。ろ紙の分解が不十分のときは,混酸の添加と蒸発乾固の操作を繰り返す。
水5ml及び硫酸 (1+1) 1mlを加え加熱して溶解し,7.4.2でろ液及び洗液を入れてある500mlの全量
フラスコに水を用いて移し入れる。
(3) 発光強度の測定 (1)及び(2)で得た溶液の入っている500mlの全量フラスコを常温まで冷却した後,水
で標線まで薄める。この溶液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nm
における発光強度を測定する。
(4) 検量線の作成 標準銅溶液 [7.2(17)] の各種液量(銅として0〜3mg)を段階的に500mlの全量フラス
コに採り,硫酸 (1+1) 3ml及び硫酸ナトリウム(無水)13gを加えて,水で標線まで薄める。この溶
液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nmにおける発光強度を試
料と並行して測定する。得た発光強度と銅量との関係線を作成し,その関係線を原点を通るように平
行移動して検量線とする。
(5) 空試験液の発光強度の測定 試料を用いないで,試料と同じ操作を試料と並行して行った空試験液の
一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nmにおける発光強度を測定す
る。
(6) 銅量の算出 (3)で得た発光強度から(5)で得た発光強度を差し引いた発光強度と(4)で作成した検量線
14
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
とから銅量を求める。
7.5
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
7.6
計算 試料中の銅含有率を,次の式によって算出する。
H
m
C
F
V
V
Cu
−
+
−
=
100
100
100
)
(
2
1
×
×
×
ここに, Cu: 試料中の銅含有率 [% (m/m)]
V1: 7.4.4の(1)(b)又は(2)(c)で得たチオ硫酸ナトリウム標準溶液の
使用量 (ml)
V2: 7.5で得たチオ硫酸ナトリウム標準溶液の使用量 (ml)
F: 7.2(16)で得たチオ硫酸ナトリウム標準溶液1ml当たりの銅相当
量 (mg) の平均値
C: 7.4.5(6)で得た不溶解残物及びろ液中の銅検出量 (mg)
m: 試料はかり採り量 (mg)
H: 吸着水分率 [% (m/m)] ただし,事前乾燥A法又は事前乾燥B
法を用いたときは,H=0とする。
8. チオ硫酸ナトリウム滴定法
8.1
要旨 試料を硝酸と硫酸とで分解した後,臭化水素酸を加えて加熱しひ素,アンチモン,セレン及
びすずを揮散して除去し乾固する。硫酸を加えて塩類を溶解した後,ふっ化水素アンモニウムを加えて共
存する鉄の影響を隠ぺいする。よう化カリウムを加えて銅と当量のよう素を遊離させ,遊離したよう素を
チオ硫酸ナトリウム標準溶液で滴定する。
8.2
試薬 試薬は,次による。
(1) 硝酸
(2) 硝酸 (1+1)
(3) ふっ化水素酸
(4) 臭化水素酸
(5) 硫酸 (1+1,1+1 000)
(6) 混酸 硝酸250mlに硫酸250mlを徐々に加える。
(7) 臭素
(8) ふっ化水素アンモニウム
(9) よう化カリウム
(10) 0.08mol/lチオ硫酸ナトリウム標準溶液 チオ硫酸ナトリウム五水和物20gを新しく煮沸して冷却した
水1lに溶解する。炭酸ナトリウム0.2gを加えかき混ぜて溶解し1日間以上放置する。この溶液1ml
は銅約5mgに相当するが,標定は,次のようにして行う(22)。
銅[99.99% (m/m) 以上]の小片を温酢酸 (1+3) に浸して表面の酸化物を除去した後,水次いでエ
タノール (95) で洗浄し乾燥する。この小片から試料中に含まれると思われる銅量とほぼ同じ質量を
0.1mgのけたまで3個はかり採り (M1, M2, M3) それぞれビーカー (400ml) に移し入れる。硝酸 (1+1)
10mlを加えて分解した後,硫酸 (1+1) 5mlを加えて加熱して乾固する。放冷後水40mlを加えて加熱
して可溶性塩類を溶解し放冷する。以下8.4.2の(1)又は(2)に従って操作し,それぞれのチオ硫酸ナト
リウム標準溶液1ml当たりの銅相当量を,次の式によって求める。
この標準溶液は褐色瓶に入れて保存し,少なくとも5日ごとに標定する。

15
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
i
i
i
V
M
f=
ここに,
fi: チオ硫酸ナトリウム標準溶液1ml当たりの銅相当量 (mg)
Mi: 銅はかり採り量 (mg)
Vi: チオ硫酸ナトリウム標準溶液の使用量 (ml)
f1,f2及びf3の範囲が0.01mgCu/ml以下のときは,3個の平均値を算出し,これを小数点以下第3位
に丸めて,チオ硫酸ナトリウム標準溶液1mlの銅相当量 (mg) の平均値 (F) とする。f1,f2及びf3の
範囲が0.01mgCu/mlを超えたときは,改めて初めから標定を行わなければならない。
(11) 標準銅溶液 (100μgCu/ml) 6.2(18)による。
(12) でんぷん溶液 (2g/l) 7.2(18)による。
8.3
試料はかり採り量 試料はかり採り量は,銅含有率に応じて,表3による。
表3 試料はかり採り量
銅含有率
% (m/m)
試料はかり採り量
g
15以上25未満
0.8
25以上50以下
0.4
8.4
操作
8.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採ってビーカー (400ml) に移し入れ,水10mlを加えて試料を湿らせた後,硝酸 (1+1)
20mlを加え(6)時計皿で覆い60〜70℃で約10分間加熱する。硫酸 (1+1) 15mlを加えて穏やかに加熱
を続ける。激しい反応が終了したら,時計皿の下面を水で洗って時計皿を取り除き,引き続き加熱を
続け硫酸の白煙を十分に発生させる(7)(8)。
(2) 放冷した後(9)水5ml及び臭化水素酸10mlを注意して加え,加熱して硫酸の白煙を十分に発生させる。
放冷した後,硫酸 (1+1) 5ml及び臭化水素酸10mlを注意して加え,再び加熱して硫酸の白煙を激し
く発生させ蒸発乾固する(29)。
(3) ビーカーの内壁を水で洗浄した後再び加熱して乾固する。硫酸 (1+1 000) 40ml加えて加熱し可溶性塩
類を溶解する。放冷後,ふっ化水素アンモニウム3gを加え振り混ぜて溶解する。
注(29) 不溶解残物中に銅が含まれるときは,放冷した後水20ml及び硫酸 (1+1) 1mlを加え加熱して可
溶性塩類を溶解し,ろ紙(5種B)を用いてろ過し温水で十分洗浄する。ろ液及び洗液はビーカ
ー (400ml) に受け加熱して乾固する。ビーカーの内壁を水で洗浄し再び加熱して乾固する。以
下8.4.1の(3)に従って操作する。ろ紙上の不溶解残物は,ろ紙と共に保存し8.4.3によって銅を定
量する。
8.4.2
滴定 滴定は,次のいずれかの手順によって行う。
(1) チオシアン酸カリウムを用いないとき 8.4.1(3)で得た溶液によう化カリウム15gを加えふり混ぜて溶
解した後,直ちにチオ硫酸ナトリウム標準溶液 [8.2(10)] で滴定する(22)(27)。終点に近づいて溶液の色
が黄褐色から淡黄色になったら(28),指示薬としてでんぷん溶液 [8.2(12)] 5mlを加え,引き続き滴定を
続け,最後の1滴で青色が消失した点を終点とし,チオ硫酸ナトリウム標準溶液の使用量を求める。
(2) チオシアン酸カリウムを用いるとき 8.4.1(3)で得た溶液によう化カリウム3gを加えふり混ぜて溶解
した後,直ちにチオ硫酸ナトリウム標準溶液 [8.2 (10)] で滴定する(22)(27)。終点に近づいて溶液の色が
黄褐色から淡黄色になったら(28),指示薬としてでんぷん溶液 [8.2(12)] 5mlを加えて溶液が青色となる
まで滴定を続ける。チオシアン酸カリウム溶液5mlを加えて,引き続き滴定を続け,最後の1滴で青
16
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
色が消失した点を終点とし,チオ硫酸ナトリウム標準溶液の使用量を求める。
8.4.3
不溶解残物中の銅の定量 不溶解残物中の銅の定量は,次の手順によって行う。
(1) 不溶解残物の分解 8.4.1(2)の注(29)で保存した不溶解残物は,ろ紙と共に白金るつぼ(30番)に移し
入れ,乾燥後700〜800℃に加熱してろ紙を灰化する。放冷した後,硫酸 (1+1) 5ml及びふっ化水素酸
5〜10mlを加え加熱しほとんど乾固するまで濃縮する。水5ml及び硫酸 (1+1) 1mlを加えて加熱して
塩類を溶解し,常温まで冷却した後,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄
める。
(2) 発光強度の測定 (1)で得た溶液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長
324.75nmにおける発光強度を測定する。
(3) 検量線の作成 標準銅溶液 [8.2(11)] の各種液量(銅として0〜0.3mg)を段階的に100mlの全量フラ
スコに採り,硫酸 (1+1) 1mlを加えて,水で標線まで薄める。この溶液の一部をICP発光分光装置の
アルゴンプラズマ中に噴霧し,波長324.75nmにおける発光強度を試料と並行して測定する。得た発
光強度と銅量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
(4) 空試験液の発光強度の測定 試料を用いないで,試料と同じ操作を試料と並行して行った空試験液の
一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nmにおける発光強度を測定す
る。
(5) 銅量の算出 (2)で得た発光強度から(4)で得た発光強度を差し引いた発光強度と(3)で作成した検量線
とから銅量を求める。
8.5
空試験 試料を用いないで試料と同じ操作を試料と並行して行う。
8.6
計算 試料中の銅含有率を,次の式によって算出する。
H
m
C
F
V
V
Cu
−
+
−
=
100
100
100
)
(
2
1
×
×
×
ここに, Cu: 試料中の銅含有率 [% (m/m)]
V1: 8.4.2の(1)又は(2)で得たチオ硫酸ナトリウム標準溶液の使用量
(ml)
V2: 8.5で得たチオ硫酸ナトリウム標準溶液の使用量 (ml)
F: 8.2(10)で得たチオ硫酸ナトリウム標準溶液1ml当たりの銅相当
量 (mg) の平均値
C: 8.4.3(5)で得た不溶解残物の中の銅検出量 (mg)
m: 試料はかり採り量 (mg)
H: 吸着水分率 [% (m/m)] ただし,事前乾燥A法又は事前乾燥B
法を用いたときは,H=0とする。
9. 原子吸光法
9.1
要旨 試料を硝酸と塩酸とで分解し,硫酸を加えて加熱し蒸発乾固する。塩酸を加えて塩類を溶解
しろ過する。ろ液を空気・アセチレンフレーム中に噴霧し,原子吸光光度計を用いて,その吸光度を測定
する。
9.2
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1,1+19)
(3) 硝酸
(4) 過塩素酸

17
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(5) ふっ化水素酸
(6) 硫酸 (1+1)
(7) 標準銅溶液 (10μgCu/ml) 銅 [99.99% (m/m)] 0.100gをはかり採り,ビーカー (300ml) に移し入れ,
時計皿で覆い硝酸 (1+1) 10mlで分解した後,加熱して窒素酸化物を追い出す。水50mlを加え常温ま
で冷却する。水を用いて1 000mlの全量フラスコに移し入れ,水で標線まで薄める。この溶液を原液
(100mgCu/ml) とする。この原液を使用の都度必要量だけ水で正確に10倍に薄めて標準銅溶液とする。
9.3
試料はかり採り量 試料はかり採り量は,0.2gとする。
9.4
操作
9.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採りビーカーに移し入れ,塩酸10ml及び硝酸5mlを加えて(6),時計皿で覆い,徐々に
加熱して分解し,放冷した後硫酸 (1+1) 5mlを加えて(30),引き続き加熱し蒸発乾固する。
(2) 放冷した後,塩酸 (1+1) 10ml及び水30mlを加えて塩類を溶解した後,ろ紙(5種B)を用いてろ過
し,温水で十分に洗浄する(31)。ろ液及び洗液は常温まで冷却した後,100mlの全量フラスコに水を用
いて移し入れ,水で標線まで薄める(32)。
注(30) 硫酸 (1+1) 5mlの代わりに,過塩素酸5mlを用いてもよい。ただし,過塩素酸は有機物が共存
すると,激しい爆発をする危険があるので,このようなときは過塩素酸は使用してはならない。
(31) 残物中に銅が含まれる場合は,残物は少量の水を用いて白金るつぼ(30番)に移し入れ,硫酸
(1+1) 5ml及びふっ化水素酸5〜10mlを加えて加熱しほとんど乾固するまで濃縮する。少量の
水を加えて溶解し,ろ紙(5種B)を用いてろ過し温水で数回洗浄する。ろ液及び洗液は主液に
合わせる。
(32) 試料中の銅含有率が0.2% (m/m) 以上の場合は,銅含有率に応じて表4に規定された量を100ml
の全量フラスコに分取し,塩酸 (1+19) を加えた後,水で標線まで薄める。
表4 銅含有率と分取量
銅含有率
% (m/m)
分取量
ml
0.2以上1.0未満
25
1.0以上2.0未満
10
2.0以上3.0以下
5
9.4.2
吸光度の測定 9.4.1(2)で得た溶液の一部を水を用いてゼロ点を調節してある原子吸光光度計の空
気−アセチレンフレーム中に噴霧し,波長324.8nmにおける吸光度を測定する。
9.5
空試験 試料を用いないで試料と同じ操作を試料と並行して行う。
9.6
検量線の作成 標準銅溶液 [8.2(11)] の各種液量(銅として0〜0.5mg)を段階的に100mlの全量フ
ラスコに採り,塩酸 (1+1) 10mlを加えて水で標線まで薄める。この溶液の一部を水を用いてゼロ点を調
節してある原子吸光光度計の空気・アセチレンフレーム中に噴霧し,波長324.8nmにおける吸光度を試料
と並行して測定する。得た吸光度と銅量との関係線を作成し,その関係線を原点を通るように平行移動し
て検量線とする。
9.7
計算 計算は,次のいずれかの手順によって行う。
(1) 9.4.1(2)注(32)を行わなかった場合 9.4.2及び9.5で得た吸光度と9.6で作成した検量線とからそれぞれ
の銅量を求め,試料中の銅含有率を,次の式によって算出する。
18
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
H
m
A
A
Cu
−
−
=
100
100
100
2
1
×
×
ここに, Cu: 試料中の銅含有率 [% (m/m)]
A1: 試料溶液中の銅検出量 (mg)
A2: 空試験溶液中の銅検出量 (mg)
m: 試料はかり採り量 (mg)
H: 吸着水分率 [% (m/m)] ただし,事前乾燥A法又は事前乾燥
B法を用いたときは,H=0とする。
(2) 9.4.1(2)注(32)で分取をした場合 9.4.2及び9.5で得た吸光度と9.6で作成した検量線とからそれぞれの
銅量を求め,試料中の銅含有率を,次の式によって算出する。
H
B
m
A
A
Cu
−
−
=
100
100
100
100
2
1
×
×
×
ここに, Cu: 試料中の銅含有率 [% (m/m)]
A1: 試料溶液中の銅検出量 (mg)
A2: 空試験溶液中の銅検出量 (mg)
B: 試料溶液の分取量 (ml)
m: 試料はかり採り量 (mg)
H: 吸着水分率 [% (m/m)] ただし,事前乾燥A法又は事前乾燥B
法を用いたときは,H=0とする。
10. ICP発光分光法
10.1 要旨 試料を硝酸と塩酸とで分解し,硫酸を加えて加熱して蒸発乾固する。塩酸を加えて塩類を溶
解しろ過する。ろ液をICP発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度をはかる。
10.2 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1,1+9)
(3) 硝酸
(4) 過塩素酸
(5) ふっ化水素酸
(6) 硫酸 (1+1)
(7) 亜鉛溶液 (10mgZn/ml) 亜鉛[99.9% (m/m) 以上]11.0gを塩酸 (1+1) 20mlで分解し,室温まで冷
却した後,水で100mlとする。
(8) 鉛溶液 (10mgPb/ml) 鉛[99.9% (m/m) 以上]1.0gを硝酸 (1+1) 30mlで分解し,室温まで冷却した
後,水で100mlとする。
(9) 鉄溶液 (20mgFe/ml) 塩化鉄 (III) 六水和物48.4gを塩酸 (1+100) 500mlに溶解する。
(10) 標準銅溶液 (1mgCu/ml) 銅 [99.99% (m/m)] 0.100gをはかり採り,ビーカー (300ml) に移し入れ,
時計皿で覆い硝酸 (1+1) 10mlで分解した後,加熱して窒素酸化物を追い出す。水50mlを加え常温ま
で冷却する。100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
10.3 試料はかり採り量 試料はかり採り量は,0.2gとする。
10.4 操作
10.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採りビーカーに移し入れ,塩酸10ml及び硝酸5mlを加えて(6),時計皿で覆い,徐々に
19
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
加熱して分解し,放冷した後硫酸 (1+1) 5mlを加え(30)再び加熱して蒸発し乾固する。
(2) 放冷した後,塩酸 (1+1) 10ml及び水30mlを加えて可溶性塩類を溶解した後,ろ紙(5種C)を用い
てろ過し,温水で十分に洗浄する(31)。ろ液及び洗液は常温まで冷却した後,100mlの全量フラスコに
水を用いて移し入れ,水で標線まで薄める。
10.4.2 発光強度の測定 10.4.1で得た溶液の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,波
長324.75nmにおける発光強度を測定する。
10.5 空試験 試料を用いないで試料と同じ操作を試料と並行して行う。
10.6 検量線の作成 検量線の作成は,次の手順によって行う。
(1) 試料溶液中に共存する量とほぼ同一量になるように,亜鉛溶液 [10.2(7)],鉛溶液 [10.2(8)] 及び鉄溶
液 [10.2(9)] (33)を数個の100mlの全量フラスコに採り,各々に塩酸 (1+1) 10mlを加える。標準銅溶液
[10.2(10)] の各種液量(銅として0〜20mg)を段階的に加えて水で標線まで薄める。
(2) この溶液の一部を,ICP発光分光装置のアルゴンプラズマ中に噴霧し,波長324.75nmにおける発光強
度を試料と並行して測定する。得た発光強度と銅量との関係線を作成し,その関係線を原点を通るよ
うに平行移動して検量線とする。
注(33) 試料が亜鉛精鉱の場合は亜鉛溶液と鉄溶液を,鉛精鉱の場合は鉛溶液と鉄溶液を,試料溶液と
同じ濃度になるように加える。
10.7 計算 10.4.2及び10.5で得た発光強度と10.6で作成した検量線とからそれぞれの銅量を求め,試料
中の銅含有率を,次の式によって算出する。
H
m
A
A
Cu
−
−
=
100
100
100
2
1
×
×
ここに, Cu: 試料中の銅含有率 [% (m/m)]
A1: 試料溶液中の銅検出量 (mg)
A2: 空試験溶液中の銅検出量 (mg)
m: 試料はかり採り量 (mg)
H: 吸着水分率 [% (m/m)] ただし,事前乾燥A法又は事前乾燥B
法を用いたときは,H=0とする。
11. アンモニア吸光光度法
11.1 要旨 試料を硝酸と塩酸とで分解し,硫酸を加えて加熱して乾固する。塩化アンモニウム及び水を
加えて塩類を溶解した後,アンモニア水を加え,銅のアンモニア錯体を生成させるとともに鉄などを沈殿
させる。ろ過し,光度計を用いてろ液の吸光度を測定する。
11.2 試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸
(3) 過塩素酸
(4) ふっ化水素酸
(5) 硫酸 (1+1)
(6) アンモニア水
(7) 過酸化水素 (1+9)
(8) 臭素
(9) 塩化アンモニウム溶液 (250g/l)

20
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(10) 硫酸アンモニウム鉄 (IIl) 溶液 硫酸アンモニウム鉄 (III) ・12水17gを硫酸 (1+1) 5ml及び水に溶解
して100mlとする。この溶液は,鉄約20mgを含む。
(11) 標準銅溶液 (1mgCu/ml) 銅 [99.99% (m/m)] 0.100gをはかり採り,ビーカー (300ml) に移し入れ,
時計皿で覆い硝酸 (1+1) 10mlで分解した後,硫酸 (1+1) 5mlを加え,加熱して硫酸の白煙を十分に
発生させる。放冷後,水50mlを加え加熱して溶解し常温まで冷却する。100mlの全量フラスコに水を
用いて移し入れ,水で標線まで薄める。
11.3 試料はかり採り量 試料はかり採り量は,銅含有率に応じて,表5による。
表5 試料のはかり採り量(34)
銅含有率
% (m/m)
試料はかり採り量
g
0.5 以上 1 未満
2
1 以上 5 未満
1
5 以上 10 以下
0.5
注(34) はかり採った試料中のニッケルとコバルトとの合計量が3mgを超えるときは,この分析方法を
適用することはできない。試料はかり採り量を減らすか,他の方法を用いる。
11.4 操作
11.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採ってビーカー (300ml) に移し入れ,塩酸10ml及び硝酸5mlを加えて(6),時計皿で覆
い,徐々に加熱して分解し,放冷した後硫酸 (1+1) 5mlを加えて(30),引き続き加熱して蒸発乾固す
る(35)。
(2) 放冷後,硫酸 (1+1) 1ml及び塩化アンモニウム溶液20mlを加え加熱して可溶性塩類を溶解し(36),常
温まで冷却する。水を用いて100mlの全量フラスコに移し入れる。
注(35) 不溶解残物中に銅が含まれるときは,放冷した後水20ml及び硫酸 (1+1) 1mlを加えて加熱し可
溶性塩類を溶解した後,ろ紙(5種B)を用いてろ過し温水で十分洗浄する。ろ液及び洗液はビ
ーカー (300ml) に受ける。不溶解残物は少量の水を用いて白金るつぼ(30番)に移し入れ,硫
酸 (1+1) 5ml及びふっ化水素酸10mlを加えて加熱し,ほとんど乾固するまで濃縮する。少量の
水を加えて溶解し主液にあわせる。溶液量が約50mlとなるまで加熱する。
(36) 試料中にマンガン10mg以上を含むときは,アンモニア水を水酸化鉄の沈殿がわずかに生成す
るまで加えた後,過酸化水素 (1+9) 5m1を加えて,約5分間煮沸してマンガンを沈殿させ,冷
却後沈殿と共に100mlの全量フラスコに移し入れる。さらに,アンモニア水20mlを加えた後,
常温まで冷却し水で標線まで薄める。以下11.4.3による。
11.4.2 呈色 11.4.1(2)で得た溶液を振り混ぜながらアンモニア水を水酸化鉄の沈殿がわずかに生成する
まで加え,更に過剰に20mlを加えた後,常温まで冷却し,水で標線まで薄める。
11.4.3 吸光度の測定 11.4.2で得た溶液の上澄み液を,乾いたろ紙を用いて乾いたビーカーにろ過し,最
初のろ液約30mlは捨て,その後のろ液の一部を光度計の吸収セル (10mm) に移し入れ,波長580nm付近
の吸光度を測定する。
11.5 空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
21
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.6 検量線の作成 (37)標準銅溶液 [11.2(11)] の各種液量(銅として0〜50mg)を段階的に数個の100ml
の全量フラスコに採り,これに塩化アンモニウム溶液20ml及びアンモニア水20mlを加え,常温まで冷却
した後,水で標線まで薄める。この溶液の一部を光度計の吸収セル (10mm) に採り,波長580nm付近の吸
光度を測定する。得た吸光度と銅量との関係線を作成し,その関係線を原点を通るように平行移動して検
量線とする。
注(37) 注(36)の操作を行ったときに用いる検量線は,次による。
標準銅溶液 [11.2(11)] の各種液量(銅として0〜50mg)を段階的に数個のビーカー (200ml) に
採り,それぞれに硫酸アンモニウム鉄 (III) 溶液 [11.2(10)] 10ml及び塩化アンモニウム溶液
20mlを加え更にアンモニア水を水酸化鉄の沈殿がわずかに生成するまで加える。約5分間煮沸
し,常温まで冷却した後100mlの全量フラスコに水を用いて移し入れ,アンモニア水を加え水
で標線まで薄める。この溶液の一部を光度計の吸収セル (10mm) に採り,波長580nm付近の吸
光度を測定する。得た吸光度と銅量との関係線を作成し,その関係線を原点を通るように平行
移動して検量線とする。
11.7 計算 11.4.3及び11.5で得た吸光度と11.6で作成した検量線とからそれぞれの銅量を求め,試料中
の銅含有率を,次の式によって算出する。
H
m
A
A
Cu
−
−
=
100
100
100
2
1
×
×
ここに, Cu: 試料中の銅含有率 [% (m/m)]
A1: 試料溶液中の銅検出量 (g)
A2: 空試験溶液中の銅検出量 (g)
m: 試料はかり採り量 (g)
H: 吸着水分率 [% (m/m)] ただし,事前乾燥A法又は事前乾燥B
法を用いたときは,H=0とする。
22
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1 分析用試料の吸着水分の測定方法
1. 適用範囲 この附属書は,分析用試料の吸着水分の測定方法について規定する。この方法は,例えば,
ケロシンなどの揮発性成分を含まない吸着水分含有率が0.05% (m/m) 以上2% (m/m) 以下の試料に適用す
る。この方法は,酸化するおそれのある硫化精鉱に用いることはできない。この場合は附属書2による。
2. 要旨 試料を規定された温度で恒量となるまで乾燥し,熱乾燥減量を求める。
3. 装置 装置は,次による。
(1) 平形はかり瓶 ガラス製,石英製又は耐食性金属製の直径約50mmのふた付きのもの。
(2) 平皿 耐食性のもので,試料の厚さが3〜5mmとなる底面積をもつもの。
4. 試料
4.1
実験室用試料 粒径150μm以下の試料を用いる。
4.2
試験試料の調製 化学分析及び吸着水分の測定に十分な量の実験室試料を平皿 [3.(2)] に移し入れ,
3〜5mmの厚さになるように平らに広げる。粉じん(塵)による試料の汚染を防ぐため,試料の上部を空
気が自由に流れるようにしてカバーをする。試験試料を実験室大気に平衡にするため,2時間又は平衡に
達するために十分な時間放置する。試験試料の質量の変化が,放置2時間当たり0.1% (m/m) より少なく
なるまで放置する。
5. 操作
5.1
平形はかり瓶の準備 平形はかり瓶 [3.(1)] 及びそのふたを空気浴中で105±5℃で1時間乾燥する。
平形はかり瓶にふたをしてデシケーター中で常温まで放冷後,デシケーターから取り出してふたをわずか
にずらし,直ちに元に戻して,その質量を0.1mgのけたまではかる (m1)。
5.2
試料のはかり採り 大気と平衡にした試験試料 (4.2) から約10gを5.1で質量をはかった平形はかり
瓶 [3.(1)] にはかり採り,約3〜5mmの厚さになるように平らに広げる。平形はかり瓶,ふた及び試料の
合計質量を0.1mgのけたまではかる (m2) (1)。
注(1) 5分以内に本体に規定された銅の分析に必要な量の試料をはかり採る。
5.3
乾燥及びひょう量 試料をはかり採った平形はかり瓶及びふたを空気浴に入れ,105±5℃で2時間
乾燥する。試料が入っている平形はかり瓶とそのふたを取り出し,ふたをしてデシケーター中で室温まで
放冷後,デシケーターから取り出してふたをわずかにずらし,直ちに元に戻し,その質量を0.1mgのけた
まではかる。再び空気浴中で105±5℃で2時間乾燥しデシケーター中で室温まで放冷後,デシケーターか
ら取り出してふたをわずかにずらし,直ちに元に戻し,その質量を0.1mgのけたまではかる (m3) (2)。
注(2) もし前後の質量の差が±1mgを超えたときは,再び乾燥とひょう量を繰り返す。2時間の乾燥を
3回繰り返しても前後の質量の差が±1mg以下とならないときは,附属書2に規定する方法で吸
着水分を求める。
6. 計算 試料中の吸着水分率を,次の式によって小数点以下2位まで算出する。
23
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
1
2
3
2
×
m
m
m
m
H
−
−
=
ここに,
H: 試料中の吸着水分率 [% (m/m)]
m1: 乾燥した平形はかり瓶とふたの合計質量 (g)
m2: 乾燥前の試料と平形はかり瓶とふたの合計質量 (g)
m3: 乾燥後の試料と平形はかり瓶とふたの合計質量 (g)
24
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2 酸化するおそれがある分析用試料の吸着水分の測定方法
1. 適用範囲 この附属書は,酸化するおそれがある分析用試料の吸着水分の測定方法について規定する。
この方法は,例えば,ケロシンなどの揮発性成分を含まない吸着水分含有率が0.05% (m/m) から2% (m/m)
で,空気中で乾燥するとき酸化するおそれがある試料に適用する。
2. 要旨 試料を,乾燥した窒素中で規定された温度で恒温となるまで乾燥し,熱乾燥減量を求める。
3. 試薬 試薬は,次による。
(1) 窒素 酸素含有率が30μl/l以下の乾燥ガス。
4. 装置 装置は,次による。
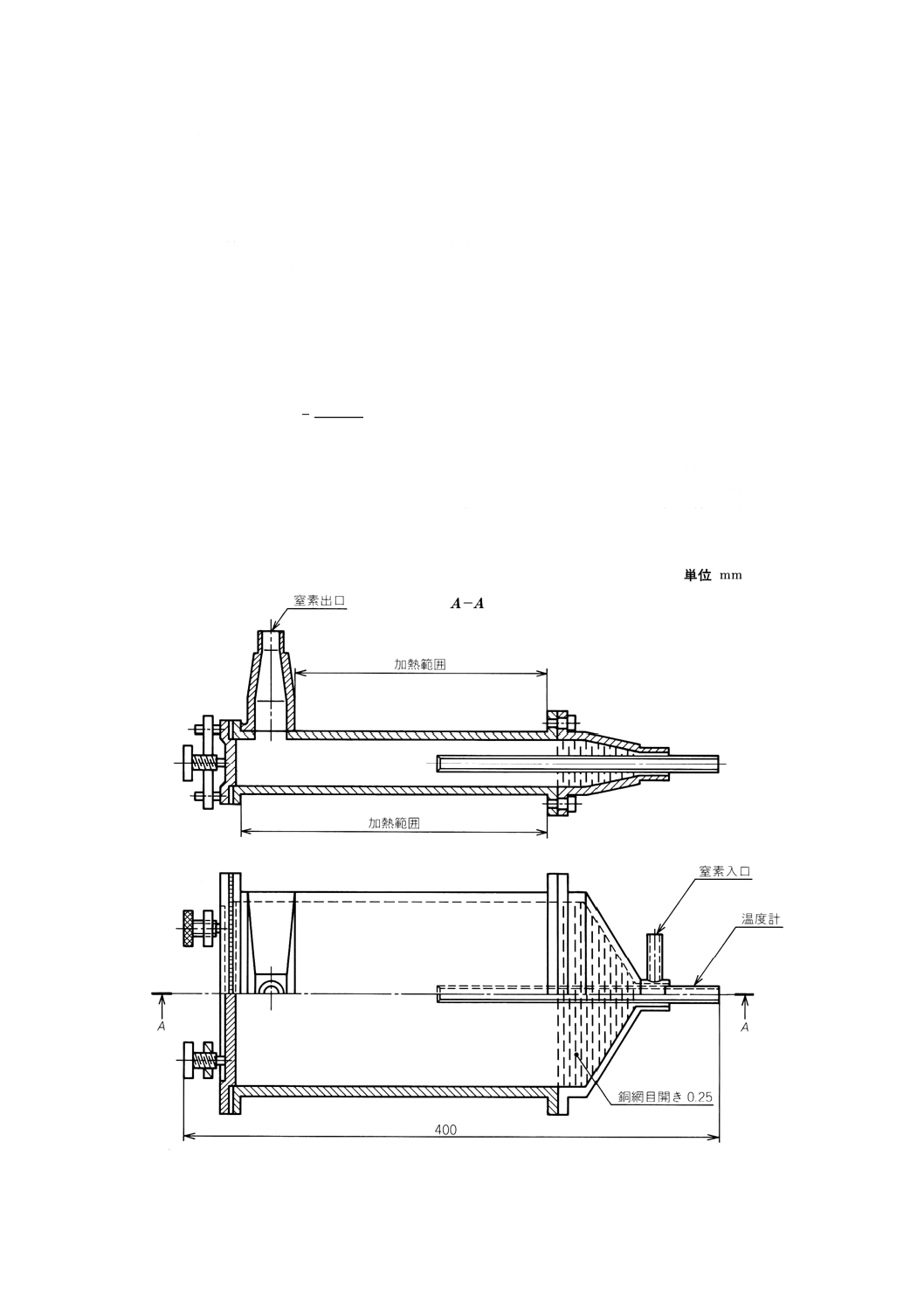
(1) 乾燥器 1時間当たり乾燥器の容量の15〜20倍の予熱した窒素を供給できる装置の付いた,空間容量
が小さく,105±5℃に保持できるもの。一例を附属書2図1に示す。
(2) 平形はかり瓶 ガラス製,石英製又は耐食性金属製の直径約50mmのふた付きのもの。
(3) 平皿 耐食性のもので,試料の厚さが3〜5mmとなる底面積をもつもの。
(4) 乾燥塔 容量約250mlで,窒素を乾燥するための無水過塩素酸マグネシウムを詰めたもの。
5. 試料
5.1
実験室用試料 粒径150μm以下の試料を用いる。
5.2
試験試料の調製 化学分析及び吸着水分の測定に十分な量の実験室試料を平皿 [4.(3)] に移し入れ,
3〜5mmの厚さになるように平らに広げる。粉じんによる試料の汚染を防ぐため,試料の上部を空気が自
由に流れるようにしてカバーをする。試験試料を実験室大気に平衡とするため,2時間又は平衡(1)に達す
るのに十分な時間放置する。
注(1) 試験試料の質量の変化が,放置2時間当たり0.1% (m/m) より少なくなったとき平衡に達したも
のとする。
6. 操作
6.1
平形はかり瓶の準備 平形はかり瓶 [4.(2)] 及びそのふたを乾燥器 [4.(1)] 中で1時間当たり乾燥器
の容量の15〜20倍の予熱した窒素 [3.(1)] を乾燥塔 [4.(4)] を通して供給しながら,105±5℃で1時間乾
燥する。平形はかり瓶にふたをしてデシケーター中で室温まで放冷した後,デシケーターから取り出して
ふたをわずかにずらし,直ちに元に戻し,その質量を0.1mgのけたまではかる (m2)。
6.2
試料のはかり採り 5.2で大気と平衡にした試験試料から約10gを,6.1で質量をはかった平形はか
り瓶 [4.(2)] にはかり採り,約3〜5mmの厚さになるように平らに広げる。平形はかり瓶,ふた及び試料
の合計質量を0.1mgのけたまではかる (m2) (2)。
注(2) 5分以内に本体の各分析方法に規定されている銅の分析に必要な量の試料をはかり採る。
25
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.3
乾燥及びひょう量 試料をはかり採った平形はかり瓶及びふたを乾燥器 [4.(1)] に入れ,1時間当た
り乾燥器の容量の15〜20倍の予熱した窒素 [3.(1)] を乾燥塔 [4.(4)] を通じて供給しながら,105±5℃で
恒量(3)となるまで乾燥する。恒量となったら(4),試料が入っている平形はかり瓶とそのふたを取り出し,
ふたをしてデシケーター中で室温まで放冷後,デシケーターから取り出して,ふたをわずかにずらし直ち
に元に戻し,その質量を0.1mgのけたまではかる (m3)。
注(3) 30分間乾燥した前後の質量の差が±1mg以下のとき恒量となったものとする。
(4) この方法では,1.5時間から3時間で乾燥は終了する。その後105±5℃で更に30分間乾燥すれ
ば恒量となる。
7. 計算 試料中の吸着水分率を,次の式によって小数点以下2位まで算出する。
100
1
2
3
2
×
m
m
m
m
H
−
−
=
ここに,
H: 試料中の吸着水分率 [% (m/m)]
m1: 乾燥した平形はかり瓶とふたの合計質量 (g)
m2: 乾燥前の試料と平形はかり瓶とふたの合計質量 (g)
m3: 乾燥後の試料と平形はかり瓶とふたの合計質量 (g)
附属書2図1 窒素を用いる乾燥器の例
26
M 8121-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS M 8121 原案作成委員会 構成表
氏名
所属
(委員長)
奥 谷 忠 雄
日本大学理工学部
揖 斐 敏 夫
通商産業省資源エネルギー庁鉱業課
天 野 徹
通商産業省工業技術院材料規格課
末 冨 巧
大蔵省造幣局東京支局試験課
因 幸二郎
財団法人日本規格協会技術部
○ 永 井 巌
住友金属鉱山株式会社中央研究所分析センター
○ 丹 野 一 雄
東邦亜鉛株式会社安中製錬所品質保証部
○ 尾 上 喬
同和鉱業株式会社中央研究所分析室
○ 中 村 靖
株式会社ジャパンエナジー分析センター
○ 端 洋 志
三井金属鉱業株式会社総合研究所分析技術研究室
○ 佐 山 恭 正
三菱マテリアル株式会社中央研究所分析・材料試験研究部
(事務局)
稲 垣 勝 彦
日本鉱業協会技術部
備考 ○印が付いている者は,分科会委員も兼ねる。