K 3362:2008
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲 ························································································································· 1
2 引用規格 ························································································································· 1
3 用語及び定義 ··················································································································· 3
4 一般事項 ························································································································· 3
5 試験項目 ························································································································· 3
5.1 試料採取方法 ················································································································ 3
5.2 化学試験 ······················································································································ 3
5.3 物理試験 ······················································································································ 4
5.4 洗浄力評価方法 ············································································································· 4
6 試料採取方法 ··················································································································· 4
6.1 代表試料の採取 ············································································································· 4
6.2 試料の調製 ··················································································································· 5
7 化学試験 ························································································································· 6
7.1 石油エーテル可溶分の定量······························································································· 6
7.2 エタノール可溶分の定量 ································································································· 7
7.3 アニオン界面活性剤の定性及び定量 ··················································································· 8
7.4 カチオン界面活性剤の定性及び定量 ·················································································· 18
7.5 非イオン界面活性剤の定性及び定量 ·················································································· 20
7.6 尿素の定量 ·················································································································· 25
7.7 界面活性剤相当分の定量 ································································································ 27
7.8 カルボキシメチルセルロースナトリウムの定量 ··································································· 27
7.9 過酸化塩の定量 ············································································································ 29
7.10 全りん酸塩の定量 ········································································································ 30
7.11 けい酸塩の定量 ··········································································································· 34
7.12 硫酸塩の定量 ·············································································································· 38
7.13 炭酸塩の定量 ·············································································································· 39
7.14 塩化物の定量 ·············································································································· 45
7.15 ゼオライトの定量 ········································································································ 46
7.16 蛍光増白剤の確認試験 ·································································································· 49
7.17 ひ素 (As) の限度試験 ··································································································· 50
7.18 重金属(Pbとして)の限度試験 ····················································································· 52
7.19 メタノールの限度試験 ·································································································· 53
7.20 エタノールの定量 ········································································································ 54
7.21 水分の定量 ················································································································· 56
K 3362:2008 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
8 物理試験 ························································································································ 58
8.1 粒度 ··························································································································· 58
8.2 見掛け密度 ·················································································································· 59
8.3 pH値·························································································································· 60
8.4 表面張力 ····················································································································· 61
8.5 起泡力及び泡の安定度 ··································································································· 65
8.6 耐硬水性 ····················································································································· 66
9 洗浄力評価方法 ··············································································································· 67
9.1 衣料用合成洗剤の洗浄力評価方法····················································································· 67
9.2 台所用合成洗剤の洗浄力評価方法····················································································· 70
附属書A(参考)衣料用合成洗剤の洗浄力評価例(シェッフェの一対比較法例) ····························· 74
附属書B(参考)台所用合成洗剤の洗浄力評価例 ······································································· 76
K 3362:2008
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,日本石鹸洗剤工業
会(JSDA)及び日本規格協会(JSA)から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,
日本工業標準調査会の審議を経て,経済産業務大臣が改正した日本工業規格である。
この規格は,著作権法で保護対象となっている著作物である。
これによって,JIS K 3362:1998は改正され,この規格に置き換えられた。
この規格の一部が,特許権,出願公開後の特許出願,実用新案権又は出願公開後の実用新案登録出願に
抵触する可能性があることに注意を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許
権,出願公開後の特許出願,実用新案権又は出願公開後の実用新案登録出願に係る確認について,責任は
もたない。
K 3362:2008
(4)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
白 紙
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 3362:2008
家庭用合成洗剤試験方法
Test method of household synthetic detergent
序文
この規格は,1955年に制定され,その後4回の改正を経て今日に至っている。前回の改正は1998年に
行われたが,その後の引用規格の改正に対応するために改正した。
なお,対応国際規格は現時点で制定されていない。
1
適用範囲
この規格は,家庭用合成洗剤の品質の試験方法について規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS H 6201 化学分析用白金るつぼ
JIS K 0050 化学分析方法通則
JIS K 0068 化学製品の水分測定方法
JIS K 0101 工業用水試験方法
JIS K 0114 ガスクロマトグラフ分析通則
JIS K 1408 けい酸ナトリウム(けい酸ソーダ)
JIS K 2241 切削油剤
JIS K 3211 界面活性剤用語
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS K 8013 亜鉛粉末(試薬)
JIS K 8034 アセトン(試薬)
JIS K 8085 アンモニア水(試薬)
JIS K 8101 エタノール(99.5)(試薬)
JIS K 8102 エタノール(95)(試薬)
JIS K 8122 塩化カルシウム二水和物(試薬)
JIS K 8123 塩化カルシウム(試薬)
JIS K 8136 塩化すず(Ⅱ)二水和物(試薬)
JIS K 8150 塩化ナトリウム(試薬)
JIS K 8159 塩化マグネシウム六水和物(試薬)
2
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 8180 塩酸(試薬)
JIS K 8213 1−オクタノール(試薬)
JIS K 8230 過酸化水素(試薬)
JIS K 8247 過マンガン酸カリウム(試薬)
JIS K 8271 キシレン(試薬)
JIS K 8279 キノリン(試薬)
JIS K 8283 くえん酸一水和物(試薬)
JIS K 8288 くえん酸三ナトリウム二水和物(試薬)
JIS K 8295 グリセリン(試薬)
JIS K 8322 クロロホルム(試薬)
JIS K 8355 酢酸(試薬)
JIS K 8359 酢酸アンモニウム(試薬)
JIS K 8361 酢酸エチル(試薬)
JIS K 8372 酢酸ナトリウム(試薬)
JIS K 8374 酢酸鉛(Ⅱ)三水和物(試薬)
JIS K 8410 酸化カルシウム(試薬)
JIS K 8519 しゅう酸二水和物(試薬)
JIS K 8521 しゅう酸アンモニウム一水和物(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8550 硝酸銀(試薬)
JIS K 8566 硝酸ビスマス五水和物(試薬)
JIS K 8574 水酸化カリウム(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8577 水酸化バリウム八水和物(試薬)
JIS K 8593 石油エーテル(試薬)
JIS K 8625 炭酸ナトリウム(試薬)
JIS K 8680 トルエン(試薬)
JIS K 8723 ニトロベンゼン(試薬)
JIS K 8731 尿素(試薬)
JIS K 8747 バナジン(Ⅴ)酸アンモニウム(試薬)
JIS K 8777 ピリジン(試薬)
JIS K 8799 フェノールフタレイン(試薬)
JIS K 8821 ふっ化ナトリウム(試薬)
JIS K 8838 1-プロパノール(試薬)
JIS K 8839 2-プロパノール(試薬)
JIS K 8840 ブロモクレゾールグリーン(試薬)
JIS K 8844 ブロモフェノールブルー(試薬)
JIS K 8847 ヘキサメチレンテトラミン(試薬)
JIS K 8885 二酸化けい素(試薬)
JIS K 8891 メタノール(試薬)
3
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 8897 メチレンブルー(試薬)
JIS K 8905 七モリブデン酸六アンモニウム四水和物(試薬)
JIS K 8906 モリブデン(Ⅵ)酸二ナトリウム二水和物(試薬)
JIS K 8913 よう化カリウム(試薬)
JIS K 8949 硫化ナトリウム九水和物(試薬)
JIS K 8951 硫酸(試薬)
JIS K 8982 硫酸アンモニウム鉄(Ⅲ)・12水(試薬)
JIS K 8987 硫酸ナトリウム(試薬)
JIS K 8997 硫酸マンガン(Ⅱ)五水和物(試薬)
JIS K 9007 りん酸二水素カリウム(試薬)
JIS K 9012 りん酸三ナトリウム・12水(試薬)
JIS K 9019 りん酸水素二ナトリウム・12水(試薬)
JIS K 9512 N,N-ジエチルジチオカルバミド酸銀(試薬)
JIS K 9552 メチルチモールブルー(試薬)
JIS K 9563 キシレノールオレンジ(試薬)
JIS L 0803 染色堅ろう度試験用添付白布
JIS M 8100 粉塊混合物−サンプリング方法通則
JIS P 3801 ろ紙(化学分析用)
JIS R 1301 化学分析用磁器るつぼ
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS R 3703 顕微鏡用スライドガラス
JIS Z 8723 表面色の視感比較方法
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
JIS Z 8802 pH測定方法
ISO 4793 Laboratory sintered (fritted) filters−Porosity grading, classification and designation
3
用語及び定義
この規格で用いる主な用語及び定義は,JIS K 3211によるほか,次による。
3.1
家庭用合成洗剤
石けん以外の合成によって製造された界面活性剤を主成分とする家庭用洗剤。すなわち,衣料用,台所
用,住宅用,家具用などがある。
4
一般事項
試験に共通する一般事項は,JIS K 0050による。
5
試験項目
5.1
試料採取方法
5.2
化学試験
4
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.2.1
石油エーテル可溶分の定量
5.2.2
エタノール可溶分の定量
5.2.3
アニオン界面活性剤の定性及び定量
5.2.4
カチオン界面活性剤の定性及び定量
5.2.5
非イオン界面活性剤の定性及び定量
5.2.6
尿素の定量
5.2.7
界面活性剤相当分の定量
5.2.8
カルボキシメチルセルロースナトリウムの定量
5.2.9
過酸化塩の定量
5.2.10 全りん酸塩の定量
5.2.11 けい酸塩の定量
5.2.12 硫酸塩の定量
5.2.13 炭酸塩の定量
5.2.14 塩化物の定量
5.2.15 ゼオライトの定量
5.2.16 蛍光増白剤の確認試験
5.2.17 ひ素 (As) の限度試験
5.2.18 重金属(Pbとして)の限度試験
5.2.19 メタノールの限度試験
5.2.20 エタノールの定量
5.2.21 水分の定量
5.3
物理試験
5.3.1
粒度
5.3.2
見掛け密度
5.3.3
pH値
5.3.4
表面張力
5.3.5
起泡力及び泡の安定度
5.3.6
耐硬水性
5.4
洗浄力評価方法
5.4.1
衣料用合成洗剤の洗浄力評価方法
5.4.2
台所用合成洗剤の洗浄力評価方法
6
試料採取方法
6.1
代表試料の採取
同一バッチで製造した製品,同一設備で連続製造後の一定時間内の製品など,同一品質とみなされる製
品でロットを形成し,そのロットの容器数に応じて,表1に示す個数を乱数表などの適切な方法によって,
ランダムに規定量の代表試料を採取する。ただし,容器が1 000個を超える場合には,その端数に対して
も表1を適用する。
5
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1−試料抜取数
単位 個
容器数
抜取数
1〜
10
1
11〜
50
2
51〜 100
3
101〜 500
5
501〜1000
10
注記 抜取数については,工程能力に応じて規定
することができる。
6.2
試料の調製
6.1によって採取した代表試料を混合し,製品の形態に応じて試料を調製する。
6.2.1
器具
器具は,次による。
a) 縮分器 JIS M 8100の付図3の6号による(図1参照)。
b) ビーカー ガラス製又はステンレス鋼製で1 000 mLのもの。
単位 mm
図1−縮分器
6
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2.2
操作
操作は,次による。
a) 粉状洗剤又は粒状洗剤のもの 6.1で抜き取った代表試料を粒子自体が破壊しないように注意し,特に
大きな塊状となったものは指でほぐして混合し,全量を縮分器にかけ,適量の試験に用いる試料が得
られるまで繰り返す。
注記1 縮分器の使用に当たっては,目詰まり,試料容器の振り方に注意しなければならない。繰
り返し操作を行う場合,縮分器の片側だけのものを試料とすると,片寄りを生じるおそれ
があるので,いずれを採取するかはランダムに選ばなければならない。
注記2 粉状洗剤又は粒状洗剤には,不均一な性質をもつものがあり,代表的な試料を採取するこ
とが困難なものもある。特に,噴霧乾燥及び冷却工程を経てからほかのものを添加した粉
状洗剤は,物理的混合であるから,振動させただけでも各成分が分離する傾向があるので,
抜き取った代表試料の全部を縮分器にかける。
b) 液状洗剤又はペースト状洗剤のもの 6.1で抜き取った代表試料をビーカーに移し,混合する。液体又
はペーストが透明で均一なときは,これから適量を分取して試料とする。
この場合,不透明であったり沈殿物を含んだりしているときは,気泡が入らないように注意しなが
らガラス棒でかき混ぜ,必要があれば50〜60 ℃のウォーターバス中で加温する1)。均一になってか
ら,かき混ぜながら15〜30 ℃ に冷却し,適量を分取し試料とする。この試料は,共栓付ガラス瓶又
はポリエチレン瓶に入れ,冷暗所に保存する。
なお,加熱する場合は,水の蒸発による損失を考慮し,加熱の前後で質量をはかっておく。
注1) 市販製品には,pH値6.5以下で,50〜60 ℃に加熱することが適切でないものがあるから,
取扱いには十分注意する。
7
化学試験
7.1
石油エーテル可溶分の定量
7.1.1
要旨
石油エーテル可溶分は,試料の水−エタノール溶液を石油エーテルで抽出した場合に,石油エーテルに
抽出される量を求める。
注記 非イオン界面活性剤の中には石油エーテルに抽出されるものがある。
7.1.2
試薬
試薬は,次による。
7.1.2.1 石油エーテル JIS K 8593に規定する石油エーテルを蒸留し,30〜60 ℃で留出する。
7.1.2.2 水−エタノール混液 JIS K 8102に規定するエタノールと水とを等量に混合する。
7.1.2.3 硫酸ナトリウム JIS K 8987の規定による。
7.1.2.4 0.5 mol/L水酸化ナトリウム溶液 JIS K 8001の4.5の(19.2)(0.5 mol/L水酸化ナトリウム溶液)に
規定する溶液とする。
7.1.2.5 フェノールフタレイン溶液 (10 g/L) JIS K 8799に規定するフェノールフタレイン1 gをエタノー
ル (95) 100 mLに溶解したもの。
7.1.3
操作
操作は,次による。
a) 試料約10 gを三角フラスコ300 mLに1 mgまではかりとり,水−エタノール混液200 mLに溶解する。
7
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
このとき不溶分があればろ過する。
b) 0.5 mol/L水酸化ナトリウム溶液5 mLを加え,フェノールフタレイン溶液 (10 g/L) を滴下してアルカ
リ性であることを確認する。
c) 分液漏斗500 mLに移し,石油エーテル50 mLずつで3回抽出する。エマルションが生成したときは,
少量のエタノールを加えて消失させる。
d) 石油エーテル層を合わせ,水−エタノール混液30 mLずつで3回,水30 mLずつで2回洗浄し,硫酸
ナトリウムで脱水した後,乾燥したろ紙を用いて質量既知の三角フラスコ300 mLにろ過し,ろ紙を
少量の石油エーテルで洗う。
e) 水浴上で加熱して石油エーテルを揮散させ,三角フラスコをデシケータ中で室温まで放冷し,三角フ
ラスコ内部に乾燥した空気を送って,残留する石油エーテルの臭いがなくなるまで追い出した後,そ
の質量をはかる。
なお,石油エーテル抽出残液及び洗液は,合わせて保存し,7.3.3に用いる。
7.1.4
計算
石油エーテル可溶分は,次の式によって算出する。
100
×
S
A
C=
ここに, C: 石油エーテル可溶分(質量分率 %)
A: 石油エーテル抽出量 (g)
S: 試料の質量 (g)
7.2
エタノール可溶分の定量
7.2.1
要旨
エタノール可溶分は,試料をエタノールで溶解し,エタノールに溶ける物質の量を求める。
7.2.2
試薬
試薬は,次による。
7.2.2.1 エタノール (95) JIS K 8102の規定による。
7.2.2.2 エタノール (99.5) JIS K 8101の規定による。
7.2.3
装置及び器具
装置及び器具は,次による。
a) 三角フラスコ 300 mL,長さ650 mm以上のガラス管付き。
b) ガラスろ過器 JIS R 3503に規定するろ過板の細孔記号4。
c) ろ過用受器
d) 乾燥器 105±2 ℃に調節できる乾燥器。
7.2.4
操作
操作は,次による。
a) 試料約5 gを三角フラスコ300 mLに1 mgまではかりとり,エタノール100 mLを加え,ガラス管を
付けて水浴上で30分間加熱し,ときどき振り混ぜながら溶解する。
なお,粉状又は粒状試料にはエタノール (95) を用い,液状又はペースト状試料にはエタノール
(99.5) を用いる。
b) 温溶液のままガラスろ過器を用いてろ過し,三角フラスコの残量に再びエタノール (95) 50 mLを加え
て溶解する。温溶液をガラスろ過器を用いてろ過し,熱エタノールで三角フラスコ及びガラスろ過器
8
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
をよく洗浄する。室温まで放冷し,全量フラスコ250 mLにろ液と洗液とを移し,エタノール (95) を
標線まで加える。この中から全量ピペットを用いて100 mLずつ質量既知の2個のビーカー200 mLに
分取する。
c) そのうちの1個を,水浴上で加熱してエタノールを除いた後,105±2 ℃に調節した乾燥器で1時間乾
燥し,デシケータ中で放冷した後,その質量をはかる。
7.2.5
計算
エタノール可溶分は,次の式によって算出する。
なお,ガラスろ過器上の残さは,7.8及び7.10のときの試料とし,分取したビーカーの1個は,7.5.2.1
による非イオン界面活性剤の定量のときの試料とする。界面活性剤成分の赤外吸収スペクトル分析を行う
場合には,定量に用いたエタノール可溶分を試料とする。
S
A
S
A
C
×
×
×
250
100
250
100
=
=
ここに, C: エタノール可溶分(質量分率 %)
A: 乾燥残量 (g)
S: 試料の質量 (g)
7.3
アニオン界面活性剤の定性及び定量
7.3.1
アニオン界面活性剤の定性
7.3.1.1
要旨
石けん以外のアニオン界面活性剤は,メチレンブルーと青の錯体を生じ,石けんは,酸性溶液で脂肪酸
を遊離することによって検出する。
7.3.1.2
試薬
試薬は,次による。
7.3.1.2.1 メチレンブルー溶液 水500 mLにJIS K 8951に規定する硫酸12 gを,かくはんしながら徐々
に加え冷却する。これにJIS K 8897に規定するメチレンブルー0.03 g,JIS K 8987に規定する硫酸ナトリ
ウム50 gを溶解し,水を加えて1 000 mLとする。
7.3.1.2.2 クロロホルム JIS K 8322に規定するクロロホルムをメチレンブルー溶液 (10 g/L) で洗い,JIS
K 8410に規定する酸化カルシウム又はJIS K 8123に規定する塩化カルシウムを加えて蒸留し,JIS K 8987
に規定する硫酸ナトリウムを用いて脱水する。蒸留後3日以内に使用する。
7.3.1.2.3 硫酸 (1+1) JIS K 8951に規定する硫酸を用いて調製する。
7.3.1.3
器具
器具は,次による。
a) 滴定用シリンダー JIS R 3505に規定する呼び容量100 mLのメスシリンダー有栓形。
7.3.1.4
操作
操作は,次による。
a) 石けん以外のアニオン界面活性剤の定性
1) 試験管にメチレンブルー溶液約5 mL,クロロホルム約5 mLを入れ,栓をして激しく振り,静置し
て分層する。
注記 クロロホルム層は普通無色であるが,メチレンブルーの不純物又はエタノールの存在によ
ってわずかな青になることがある。
2) これに試料の約10 g/L(又は,体積分率1 %)溶液を1滴加え,上下に激しく振った後,静置して
9
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
分層させ,クロロホルム層が青になればアニオン界面活性剤の存在を示すことによって検出する。
注記 非イオン界面活性剤の共存は,多少乳化現象のため,分層に時間がかかることはあっても,
定性の障害にはならない。
3) さらに,試料溶液を加え,同じように操作すると,クロロホルム層は,ますます濃い青となること
によって検出する。
b) 石けんの定性
1) 試験管に試料の約10 g/L(又は体積分率1 %)溶液を約5 mL入れる。
2) これに硫酸 (1+1) の2,3滴を加えて振り混ぜ,pH値4以下の酸性にし,静置する。
3) このとき,試料溶液が脂肪酸の遊離によって白濁し,徐々に油状に分離すれば石けんの存在を示す
ことによって検出する。石けんが少量のときは,7.3.3.1によって定量的に分離して検出をしなけれ
ばならない。
7.3.2
アニオン界面活性剤の定量
7.3.2.1
要旨
要旨は,次による。
a) 逆滴定法(A法)及び直接滴定法(B法)の場合 アルキルベンゼンスルホン酸塩,アルキル硫酸塩,
アルキルエトキシ硫酸塩,アルケニルスルホン酸塩及びアルキルスルホン酸塩のような長鎖アルキル
スルホン酸塩又は硫酸塩とメチレンブルーとは,錯化合物を形成し,クロロホルムに溶解して青色に
なる。これに十分な量のカチオン界面活性剤を加えると,アニオン界面活性剤とカチオン界面活性剤
との錯塩が生成し,メチレンブルーが遊離して,アニオン界面活性剤を定量することができる。
b) ISO法(C法)の場合 アルキルベンゼンスルホン酸塩,アルキル硫酸塩,アルキルエトキシ硫酸塩,
アルケニルスルホン酸塩及びアルキルスルホン酸塩のような長鎖アルキルスルホン酸塩又は硫酸塩と
ジミジウムブロマイドとは,錯化合物を形成し,クロロホルムに溶解して赤桃色になる。これに十分
な量のカチオン界面活性剤を加えるとアニオン界面活性剤とカチオン界面活性剤とが錯塩を形成し,
ジミジウムブロマイドが遊離する。遊離したジミジウムブロマイドは,クロロホルム層から水層に移
行し,赤桃色が消失する。同時に過剰のカチオン界面活性剤とアシッドブルー1とは,錯塩を形成し,
クロロホルムに溶解して,青色を呈することによってアニオン界面活性剤を定量することができる。
7.3.2.2
試薬
試薬は,次による。
7.3.2.2.1 アニオン界面活性剤標準液 (0.004 mol/L) 市販のラウリル硫酸ナトリウムの純分換算で約1.2 g
を0.1 mgまではかりとり,水に溶解した後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
そのファクターは,次の式によって算出する。
M
P
S
M
S
f
P
×
×
×
×
5.2
004
.0
100
a
=
=
ここに,
fa: ファクター
S: ラウリル硫酸ナトリウムの質量 (g)
P: ラウリル硫酸ナトリウムの純度(質量分率 %)
M: ラウリル硫酸ナトリウムの化学式量
a) ラウリル硫酸ナトリウムの純度 (P) 及び化学式量 (M) の求め方
1) 純度 (P) の求め方 再結晶によって,精製したラウリル硫酸ナトリウム約5 gを三角フラスコ300
mL中に0.1 mgまではかりとり,これにJIS K 8951に規定する硫酸を用いて調製した0.5 mol/L硫
10
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
酸25 mLを全量ピペットを用いて加え,冷却器を付けてホットプレート又は砂浴上で還流する。発
泡に注意し,ときどき三角フラスコを軽く振り動かしながら還流し,溶液が透明になり,発泡しな
くなってから,更に2時間還流する。冷却後エタノール (99.5) 30 mLを用いて冷却器の内壁を洗い,
次に適量の水で洗った後,冷却器を外す。水を加えて液量を約100 mLにした後,フェノールフタ
レイン溶液 (10 g/L) を数滴加え,JIS K 8576に規定する水酸化ナトリウムで調製した1 mol/L水酸
化ナトリウム溶液で滴定する。同時に空試験を行い,次の式によってラウリル硫酸ナトリウムの純
度を算出する。
S
M
f
B
A
S
M
f
B
A
P
1.0
)
(
100
000
1
)
(
×
×
×
−
×
×
×
×
−
=
=
ここに,
P: ラウリル硫酸ナトリウムの純度(質量分率 %)
A: 試料の滴定に用いた1 mol/L水酸化ナトリウム溶液の量 (mL)
B: 空試験の滴定に用いた1 mol/L水酸化ナトリウム溶液の量
(mL)
f: 1 mol/L水酸化ナトリウム溶液のファクター
M: ラウリル硫酸ナトリウムの化学式量
S: 試料の質量 (g)
2) 化学式量 (M) の求め方 上記純度 (P) を求めるときのフェノールフタレイン溶液 (10 g/L) を指
示薬とし,1 mol/L水酸化ナトリウム溶液で滴定した後の滴定液の50 mLを300 mL分液漏斗に移し,
JIS K 8101に規定するエタノール (99.5) で調製したエタノール (99.5) (2+1) 100 mLを加え,JIS K
8593で規定する石油エーテル50 mLずつで2回抽出する。石油エーテル層を合わせ,水50 mLず
つで2回洗浄し,JIS K 8987で規定する硫酸ナトリウムで脱水した後,適切な濃度に濃縮してガス
クロマトグラフ分析によって炭素数の分布を測定する。ガスクロマトグラフ分析は,JIS K 0114に
従って,最適な測定条件で行い,試料アルコールの各成分パーセントを求める。試料アルコールの
平均分子量を求め,これからラウリル硫酸ナトリウムの化学式量 (M) を求める。
なお,固定相液体は,クロモソルブWにシリコーンSE-30の質量分率10 %のものを用い,カラ
ム槽温度180 ℃,キャリヤーガスは窒素又はヘリウム,検出器は水素炎イオン化検出器で恒温で行
う。
7.3.2.2.2 カチオン界面活性剤標準液(0.004 mol/L塩化ベンゼトニウム溶液) 調製及び標定は,次によ
る。
a) 調製 市販の塩化ベンゼトニウム(1水塩)約1.9 gを水に溶解し1 000 mLとし,カチオン界面活性
剤標準液とする。
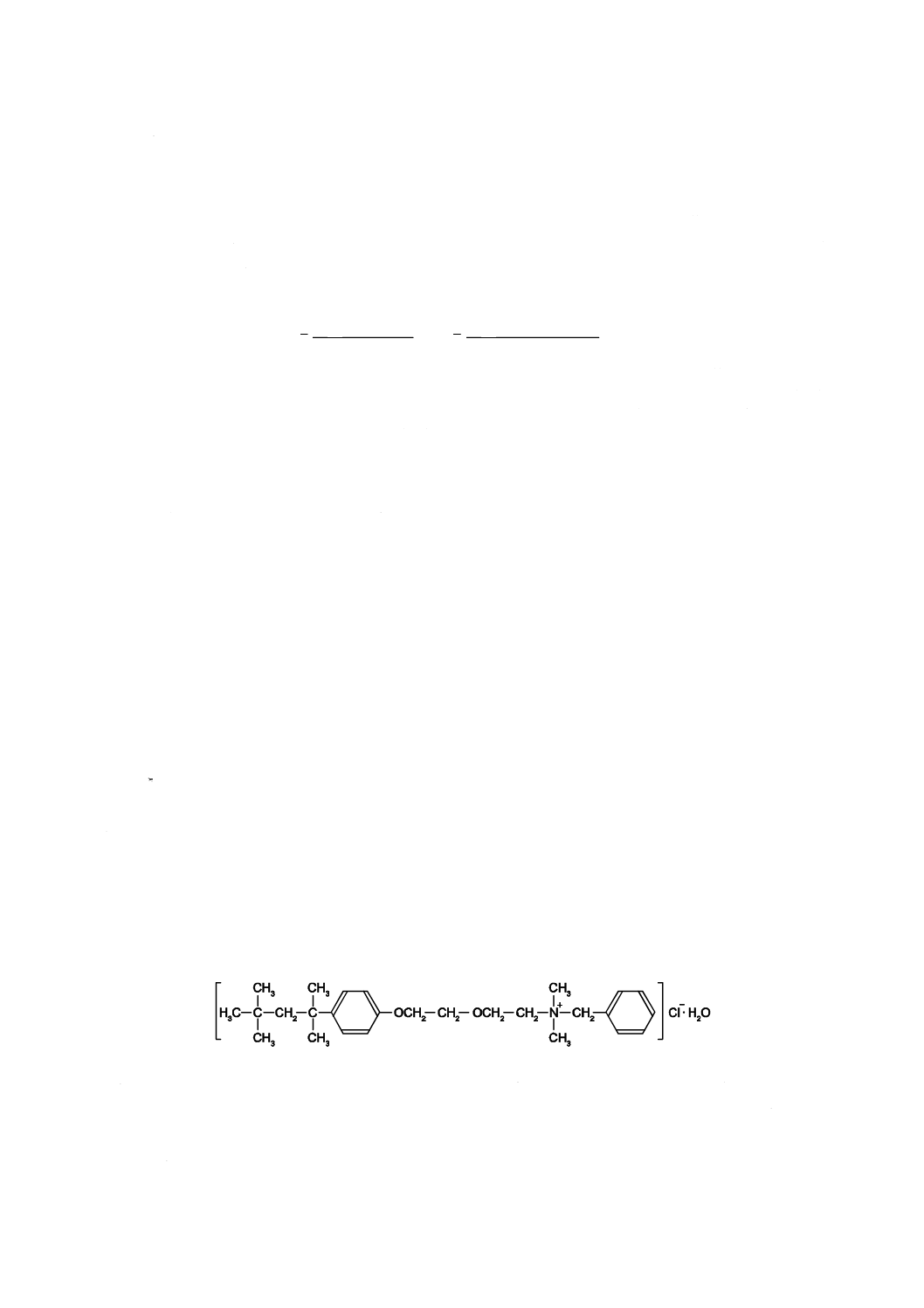
注記 塩化ベンゼトニウム(1水塩)の構造を次に示す。市販にHyamine 1622(Rohm & Haas社)
がある。これは,市販製品の一例である。この情報は,この規格の利用者の便宜のために記
載するもので,この製品を推奨するものではない。
b) 標定
1) 逆滴定法 (A法) 及び直接滴定法 (B法) に用いる場合 アニオン界面活性剤標準液 (0.004 mol/L)
10 mLを,滴定用シリンダーにとりメチレンブルー溶液25 mL及びクロロホルム15 mLを加え,次
に水20 mLを加えた後,カチオン界面活性剤標準液で滴定する。滴定は初め2 mLずつ加え,その
都度栓をして激しく振った後静置する。2層の分離が早くなれば逐次滴定量を減らし,終点近くで
11
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
は1滴 (0.02〜0.03 mL) 刻みで行い,白色板を背景として両層の青さが同一となったときを終点と
する。カチオン界面活性剤標準液のファクターは,次の式によって算出する。
A
f
f
a
b
10×
=
ここに,
fb: カチオン界面活性剤標準液のファクター
A: 滴定に用いたカチオン界面活性剤標準液の量 (mL)
fa: アニオン界面活性剤標準液のファクター
2) ISO法 (C法) に用いる場合 アニオン界面活性剤標準液 (0.004 mol/L) 25 mLを全量ピペットで滴
定用シリンダーにとり,水10 mL,クロロホルム15 mL及び混合指示薬10 mLを加え,カチオン界
面活性剤標準液で滴定する。初め2 mLずつ加え,その都度激しく振った後静置する。下層は赤桃
色を呈する。繰り返し滴定を続け,2層の分離が早くなるに従い,逐次滴定量を減らす。終点近く
では,1滴 (0.02〜0.03 mL) 刻みで行い,クロロホルム層の赤桃色が完全に消失して,薄灰青色に
なる点を終点とする。次の式によってカチオン界面活性剤標準液のファクターを算出する。
A
f
f
a
c
25×
=
ここに, fc: カチオン界面活性剤標準液のファクター
fa: アニオン界面活性剤標準液のファクター
A: 滴定に要したカチオン界面活性剤標準液の量 (mL)
なお,7.3.2.4 a) 1)の逆滴定法(A法)に適用する場合には,カチオン界面活性剤標準液のファク
ターを標定する必要はない。
7.3.2.2.3 メチレンブルー溶液 7.3.1.2.1による。
7.3.2.2.4 クロロホルム 7.3.1.2.2による。
7.3.2.2.5 フェノールフタレイン溶液 (10 g/L) 7.1.2.5による。
7.3.2.2.6 0.5 mol/L硫酸 JIS K 8001の4.5 (26.1)(0.5 mol/L硫酸)の規定による。
7.3.2.2.7 2 mol/L水酸化ナトリウム溶液 JIS K 8576に規定する水酸化ナトリウム約83 gに水を加えて溶
解し,1 000 mLとする。
7.3.2.2.8 2.5 mol/L硫酸溶液 水100 mLをビーカーにとり,JIS K 8951に規定する硫酸15 mLをかき混
ぜながら徐々に加えた後,放冷する。
7.3.2.2.9 1 mol/L水酸化ナトリウム溶液 JIS K 8576に規定する水酸化ナトリウム4 gを水に溶解して100
mLとする。
7.3.2.2.10 混合指示薬 第一のビーカー50 mLにジミジウムブロマイド0.5±0.005 g,第二のビーカー50
mLにアシッドブルー1 0.25±0.005 gをはかりとる。それぞれのビーカーに,JIS K 8101に規定するエタ
ノールを用いて調製した100 g/L熱エタノール20〜30 mLを加えて溶解する。それぞれの溶液を全量フラ
スコ250 mLに移し,100 g/Lエタノールを標線まで加え,これを原液とする。原液20 mLを全量フラスコ
500 mLにとり,水200 mL及び5 mol/L硫酸溶液20 mLを加えて混ぜ合わせ,標線まで水を加える。直射
日光から遮断して貯蔵する。
12
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
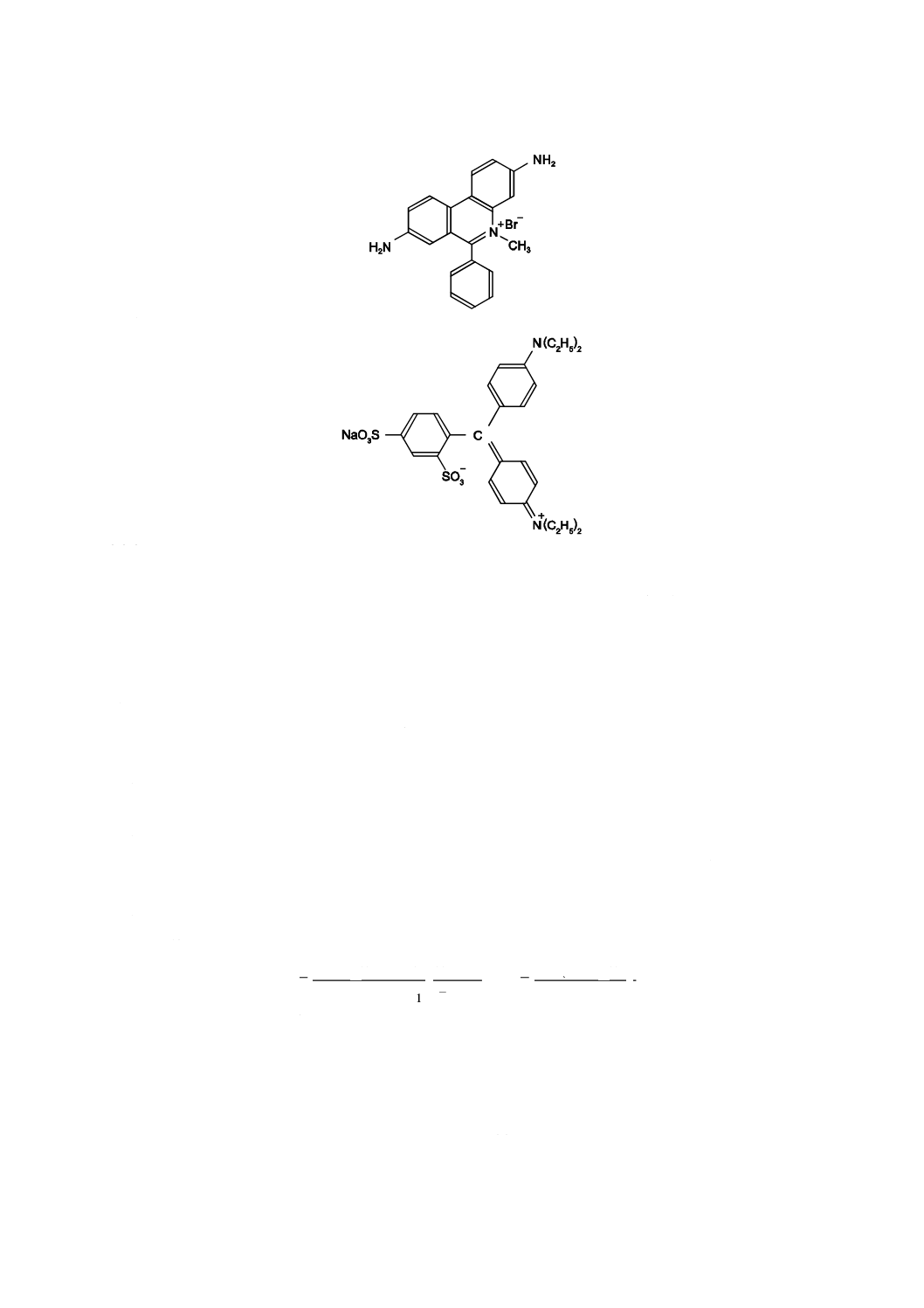
注記1 ジミジウムブロマイド
注記2 アシッドブルー1
7.3.2.3
器具
器具は,次による。
a) 滴定用シリンダー JIS R 3505に規定する呼び容量100 mLのメスシリンダー有栓形。
7.3.2.4
操作
操作は,次による。
a) 全アニオン界面活性剤の結合硫酸の定量
1) 逆滴定法(A法)
1.1) 試料の適量(純分として約1.4 gを含む。)を0.1 mgまではかりとり,水200 mLを加えて加熱溶
解し,冷却した後,全量フラスコ1 000 mLに移し水を加えて1 000 mLとし,試料溶液とする。
1.2) この中から10 mLを滴定用シリンダーにとり,メチレンブルー溶液25 mL及びクロロホルム15 mL
を加え,全量ピペットでカチオン界面活性剤標準液 (0.004 mol/L) 20 mLを加える。
1.3) アニオン界面活性剤標準液を初め2 mLずつ加え,その都度栓をして激しく振った後,静置する。
2層の分離が早くなれば逐次滴定量を減らし,終点近くでは1滴刻みで行い,白色板を背景として
両層の青さが同一となったときを終点とする。
1.4) 同時に空試験を行う。
1.5) 計算 全アニオン界面活性剤の結合硫酸は,次の式によって算出する。
S
f
A
B
S
f
A
B
C
a
a
)
(
20
.3
100
000
1
06
.
80
004
.0
)
(
000
110
×
−
×
×
×
×
×
×
−
=
=
ここに,
C: 全アニオン界面活性剤の結合硫酸(SO3として)(質量分
率%)
B: 空試験の滴定に用いたアニオン界面活性剤標準液の量 (mL)
A: 試料溶液の滴定に用いたアニオン界面活性剤標準液の量
(mL)
fa: アニオン界面活性剤標準液のファクター
S: 試料の質量 (g)
80.06: SO3の化学式量
13
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 直接滴定法(B法)
2.1) 1)の試料溶液10 mLを滴定用シリンダーにとり,メチレンブルー溶液25 mL,クロロホルム15 mL
及び水20 mLを加える。
2.2) カチオン界面活性剤標準液を,初め2 mLずつ加え,その都度栓をして激しく振った後静置する。
2層の分離が早くなれば逐次滴定量を減らし,終点近くでは1滴刻みで行い,白色板を背景として
両層の青さが同一となったときを終点とする。
2.3) 計算 全アニオン界面活性剤の結合硫酸は,次の式によって算出する。
S
f
D
S
f
D
C
b
b
20
.3
100
000
1
06
.
80
004
.0
000
110
×
×
×
×
×
×
×
×
=
=
ここに,
C: 全アニオン界面活性剤の結合硫酸(SO3として)(質量分
率%)
D: 試料溶液の滴定に用いたカチオン界面活性剤標準液の量
(mL)
fb: カチオン界面活性剤標準液のファクター
S: 試料の質量 (g)
80.06: SO3の化学式量
3) ISO法(C法)
3.1) 試料の適量(純分として約1.4 gを含む。)をビーカー300 mLに0.1 mgまではかりとり,水200 mL
を加えて加熱溶解する。
3.2) 室温まで冷却した後,フェノールフタレイン溶液数滴を加え,薄桃色になるまで,水酸化ナトリ
ウム溶液又は硫酸溶液で中和する。
3.3) この溶液を全量フラスコ1 000 mLに移し,標線まで水を加え混合する。この溶液25 mLを全量ピ
ペットで滴定用シリンダーにとり,水10 mL,クロロホルム15 mL及び混合指示薬10 mLを加え,
カチオン界面活性剤標準液で滴定する。
3.4) 初め2 mLずつ加え,その都度激しく振った後静置する。下層は赤桃色を呈する。繰り返し,滴定
を続け,2層の分離が早くなるに従い,逐次滴定量を減らす。終点近くでは,1滴 (0.02〜0.03 mL)
刻みで行い,クロロホルム層の赤桃色が完全に消失して,薄灰青色になる点を終点とする。
3.5) 計算 全アニオン界面活性剤の結合硫酸は,次の式によって算出する。
S
f
D
S
f
D
C
281
.1
100
000
1
06
.
80
004
.0
c
c
000
125
×
×
×
×
×
×
×
×
=
=
ここに,
C: 全アニオン界面活性剤の結合硫酸(SO3として)(質量分
率%)
fc: カチオン界面活性剤標準液のファクター
D: 滴定に用いたカチオン界面活性剤標準液の量 (mL)
S: 試料の質量 (g)
注記1 ハイドロトロープとして,トルエンスルホン酸塩のような比較的低分子量のスルホン
酸塩が含まれていても,150 g/L以下であれば妨害にならない。
注記2 過ほう酸塩,過炭酸塩以外の漂白剤が含まれている場合は,分析前に分解する。
注記3 逆性及び両性の界面活性剤を含む場合には,配合量よりも小さい値を示す。
b) アルキル硫酸塩,アルキルエトキシ硫酸塩などの結合硫酸の定量
1) 試料の適量(純分として約1.4 gを含む。)を0.1 mgまではかりとり,水200 mLを加えて加熱溶解
し,冷却した後,全量フラスコ250 mLに移し,水を標線まで加える。
14
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) この中から50 mLを三角フラスコ300 mLにとり,全量ピペットを用いて0.5 mol/L硫酸50 mLを加
え,長さ650 mm以上のガラス管を付けてホットプレート又は砂浴上で3時間加熱分解する。
3) 冷却後,ガラス管上部から少量の水を注いで内壁を洗った後,フェノールフタレイン溶液 (10 g/L)
を指示薬として2 mol/L水酸化ナトリウム溶液を加えて中和した後,水を加えて200 mLとし,これ
を試料溶液として1)に準じて操作する。
4) 計算 アルキル硫酸塩,アルキルエトキシ硫酸塩などの結合硫酸は,次の式によって求める。
CA=C−Cu
ここに,
CA: アルキル硫酸塩の結合硫酸(SO3として)(質量分率%)
C: 全アニオン界面活性剤の結合硫酸(SO3として)(質量分
率%)
Cu: 未分解アニオン界面活性剤の結合硫酸(SO3として)(質量分
率%)
試料中のアニオン界面活性剤の化学式量が既知の場合は,次の式によって純分を算出する。
なお,市販の家庭用合成洗剤に通常配合されるアニオン界面活性剤の化学式量を,表2に示す。
06
.
80
M
C
P
×
=
ここに,
P: 純分(質量分率%)
C: 結合硫酸(SO3として)(質量分率%)
M: アニオン界面活性剤の化学式量
80.06: SO3の化学式量
表2−アニオン界面活性剤の化学式量
アニオン界面活性剤
化学式量の範囲
代表値
アルキルベンゼンスルホン酸ナトリウム
330〜370
348(炭素数12)
アルキル硫酸ナトリウム
270〜380
288(炭素数12)
アルキルエトキシ硫酸ナトリウム
400〜520
420(炭素数12,EO 3 mol)
アルケニルスルホン酸ナトリウム
300〜370
326(炭素数16)
アルキルスルホン酸ナトリウム
300〜380
328(炭素数16)
7.3.3
石けん分の定量
7.3.3.1
中和滴定法
7.3.3.1.1
要旨
石けん分は,7.1.3で得られた水層部分を酸分解し,生じた脂肪酸を石油エーテルで抽出してその量を求
め,次に脂肪酸の中和価を測定して求める。
7.3.3.1.2
試薬
試薬は,次による。
7.3.3.1.2.1 0.25 mol/L硫酸 JIS K 8001の4.5 (26.1)(0.5 mol/L硫酸)による。この場合,硫酸15 mLを
はかりとる。標定は,JIS K 8001の4.5 (26.1) (b)によって行う。この場合,炭酸ナトリウム0.6〜0.8 gを
0.1 mgのけたまではかりとる。
7.3.3.1.2.2 メチルオレンジ溶液 JIS K 8001の4.4(指示薬)による。
7.3.3.1.2.3 石油エーテル 7.1.2.1による。
7.3.3.1.2.4 硫酸ナトリウム JIS K 8987の規定による。
7.3.3.1.2.5 0.5 mol/L水酸化カリウム・エタノール溶液 JIS K 8574に規定する水酸化カリウム33 gをは
15
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
かりとり,二酸化炭素を含まない水25 mLを加えて溶解した後,JIS K 8102に規定するエタノール(95)を
加えて1 Lとしたもの。標定は,0.5 mol/L 塩酸25 mLをコニカルビーカー200 mLに正確にはかりとり,
二酸化炭素を含まない水50 mLを加え, 指示薬としてフェノールフタレイン溶液数滴を加えて,調製し
た液で滴定する。終点は,液のうすい紅色が約30秒間残る点とする。標定は使用時に行う。
7.3.3.1.2.6 中性エタノール JIS K 8102に規定するエタノール (95) をフェノールフタレイン溶液 (10
g/L) を指示薬として0.5 mol/L水酸化カリウム溶液で中和したもの。使用直前に調製する。
7.3.3.1.2.7 フェノールフタレイン溶液 (10 g/L) 7.1.2.5による。
7.3.3.1.3
操作
操作は,次による。
a) 7.1.3の石油エーテル抽出残液及び洗液を集め,メチルオレンジ溶液を指示薬として酸性になるまで
0.25 mol/L硫酸を加える。
b) 分液漏斗に移し,石油エーテル100 mLを加えて振った後,静置して2層に分離し,下層を別の分液
漏斗に移し,更にこれを石油エーテル25 mLずつで3回抽出する。
c) 石油エーテル層を合わせ,指示薬としてメチルオレンジ溶液を用い,水100 mLずつで中性になるま
で洗浄し,硫酸ナトリウムで脱水した後,質量既知の三角フラスコ300 mLに乾燥したろ紙を用いて
ろ過し,石油エーテルで洗う。
d) 水浴上で大部分の石油エーテルを留去した後,その内部に乾燥空気を送って残った石油エーテルを追
い出した後,三角フラスコをデシケータ中で放冷し,その質量をはかる。
e) 計算 脂肪酸量は,次の式によって算出する。
100
×
S
A
F=
ここに,
F: 脂肪酸量(質量分率%)
A: 石油エーテル抽出量 (g)
S: 7.1.3の試料の質量 (g)
f)
三角フラスコに中性エタノール50 mLを加えて,加温溶解し,指示薬としてフェノールフタレイン溶
液 (10 g/L) 0.5 mLを加え,0.5 mol/L水酸化カリウム溶液で滴定する。30秒間微紅色を保つときを終
点とする。
7.3.3.1.4
計算
脂肪酸の中和価は,次の式によって算出する。
A
f
B
V
N
×
×
05
.
28
.
.=
ここに, N.V.: 脂肪酸の中和価
B: 滴定に用いた0.5 mol/L水酸化カリウム溶液の量 (mL)
f: 0.5 mol/L水酸化カリウム溶液のファクター
A: 石油エーテル抽出量 (g)
28.05: (水酸化カリウムの化学式量)×1/2
石けん分は,次の式によって,ナトリウム塩として算出する。
+
552
2
.
.
1
V
N
F
C=
ここに,
C: 石けん分(質量分率 %)
F: 脂肪酸量(質量分率 %)
N.V.: 脂肪酸の中和価
16
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2 552:
008
.1
990
.
22
106
56
−
56 106: 水酸化カリウムの化学式量56.106×1 000
22.990−1.008: (ナトリウムの原子量)−(水素の原子量)
なお,ここでは,試料中の石けん分を脂肪酸ナトリウム塩として算出しているが,カリウム塩又はトリ
エタノールアミン塩であることが明らかな場合には,上の式中の2 552の代わりに,カリウム塩のときに
1 473を,トリエタノールアミン塩のときに376.1を用いる。
7.3.3.2
アルカリ分相滴定法
7.3.3.2.1
要旨
石けん分は,アルカリ水溶液において,アニオン界面活性剤の性質を示し,その物質の量を分相滴定に
よって求める。この方法では,脂肪酸も定量できる。
7.3.3.2.2
試薬
試薬は,次による。
7.3.3.2.2.1 1-プロパノール JIS K 8838の規定による。
7.3.3.2.2.2 クロロホルム 7.3.1.2.2による。
7.3.3.2.2.3 0.1 mol/L水酸化ナトリウム溶液 JIS K 8576に規定する水酸化ナトリウム0.4 gを水に溶解し
て100 mLとする。
7.3.3.2.2.4 りん酸塩緩衝液 JIS K 9019に規定するりん酸水素二ナトリウム・12水69.84 gを水3 000 mL
に溶解した溶液と,JIS K 9012に規定するりん酸三ナトリウム・12水24.71 gを水1 000 mLに溶解した溶
液を混合し,0.1 mol/L水酸化ナトリウム溶液でpH値を11.6に調整する。
7.3.3.2.2.5 BCG溶液 JIS K 8840に規定するブロモクレゾールグリーン94.0 mgをりん酸塩緩衝液2 500
mLに溶解する。これに,1-プロパノール500 mLを加え,3 000 mLとする。
7.3.3.2.2.6 アニオン界面活性剤標準液 (0.004 mol/L) 7.3.2.2.1による。
7.3.3.2.2.7 カチオン界面活性剤標準液(0.004 mol/L塩化ベンゼトニウム溶液) 調製は,7.3.2.2.2a)によ
る。ただし標定は,次による。
a) 標定 アニオン界面活性剤標準液 (0.004 mol/L) 10 mLを全量ピペットを用いて滴定用シリンダーに
とり,クロロホルム20 mLを加える。さらに,BCG溶液25 mLを全量ピペットを用いて加え,カチ
オン界面活性剤標準液で滴定する。滴定は,初め1 mLずつ加え,その都度栓をして激しく振った後
静置し観察する。2層の分離が早くなれば逐次滴定量を減らして滴定を続ける。終点近くでは,1滴
(0.02〜0.03 mL) 刻みで行い,30秒間振り,2分間静置し,白色板を背景に色調の変化を観察する。色
調は,最初,水層が青色を示しているが,終点付近から青色が徐々にクロロホルム層に移行する。青
色が完全に移行し,水層が無色となった点を終点とする。カチオン界面活性剤標準液のファクターは,
次の式によって算出する。
A
f
f
a
10
d
×
=
ここに,
fd: カチオン界面活性剤標準液のファクター
A: 滴定に用いたカチオン界面活性剤標準液の量 (mL)
fa: アニオン界面活性剤標準液のファクター
17
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.3.3.2.3
器具
器具は,次による。
a) ビーカー 50 mL
b) 全量フラスコ 250 mL
c) 全量ピペット 10 mL及び25 mL
d) 滴定用シリンダー 7.3.2.3a)による。
e) ビュレット 20 mL
f)
メスシリンダー 50 mL,1 000 mL及び3 000 mL
7.3.3.2.4
操作
操作は,次による。
a) 試料の適量(アニオン界面活性剤として0.2〜0.3 g)をビーカー50 mLに0.1 mgまではかりとり,水
25 mLを加えて溶解する。
なお,石けん分として質量分率50 %以上含む試料については,1−プロパノール25 mLを加えて溶
解する。この溶液を全量フラスコ250 mLに移し,ビーカーは水20 mLずつで数回洗い,洗液も全量
フラスコに移した後,水を加えて標線に合わせる。これを試料溶液とする。
b) 試料溶液10 mLを全量ピペットを用いて滴定用シリンダーにとる。さらに,クロロホルム20 mL,次
いでBCG溶液25 mLを全量ピペットを用いて加える。カチオン界面活性剤標準液を,初め1 mLずつ
加え,その都度栓をして激しく振った後,静置し観察する。2層の分離が早くなれば逐次滴定量を減
らして滴定を続ける。終点付近では1滴刻みとし,30秒間振り,2分間静置し,白色板を背景に色調
の変化を観察する。色調は,最初,水層が青色を示しているが終点付近から青色が徐々にクロロホル
ム層に移行する。青色が完全に移行し,水層が無色となった点を終点とする。
c) 同時に空試験を行う。
7.3.3.2.5
計算
石けん分は,次の式によって算出する。
S
M
f
B
A
S
M
f
B
A
C
01
.0
)
(
100
250
10
000
1
004
.0
)
(
d
d
×
×
×
−
×
×
×
×
×
×
−
=
=
ここに,
fd: カチオン界面活性剤標準液のファクター
A: 試料溶液の滴定に用いたカチオン界面活性剤標準液の量 (mL)
B: 空試験に用いたカチオン界面活性剤標準液の量 (mL)
M: 石けんの化学式量
S: 試料の質量 (g)
a) 市販家庭用の合成洗剤に配合される石けんの化学式量(M)を,表3に示す。

18
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3−石けんの化学式量
石けん
分子式
化学式量
ラウリン酸ナトリウム
C12H23O2Na
222
ミリスチン酸ナトリウム
C14H27O2Na
250
パルミチン酸ナトリウム
C16H31O2Na
278
ステアリン酸ナトリウム
C18H35O2Na
306
オレイン酸ナトリウム
C18H33O2Na
304
リノール酸ナトリウム
C18H31O2Na
302
注記1 本法では,pH値が11.6であるので,エステル形非イオン界面活性剤共存下でも,その加
水分解を抑えることができるため,定量に影響を与えない。
注記2 逆性の界面活性剤を含む場合には,配合量よりも小さい値を示す。
注記3 アルカリ性でアニオン性を示す両性界面活性剤を含む場合には,配合値よりも大きい値
を示す。
注記4 石けんの炭素数が10以下のものについては,実際の配合値よりも小さい値を示す。
注記5 スルホン酸塩,硫酸エステル塩を含む場合は,これらも同時に定量されるため,更に,
7.3.2.4 a)によって全アニオン界面活性剤の結合硫酸を求め,次の式によって石けん分だけ
を算出する。
7.3.3.2で得た結果を,次の式によって全アニオン界面活性剤の結合硫酸(SO3として)
(質量分率 %)に換算する。
S
f
B
A
S
f
B
A
C
6
800
.0
)
(
100
000
1
06
.
80
004
.0
)
(
d
d
250
10
1
×
×
−
×
×
×
×
×
×
−
=
=
石けん分は,次の式によって算出する。
ここに,
C1: 7.3.3.2で得た結果を,全アニオン界面活性剤の結合硫酸(SO3として)
(質量分率 %)に換算した値
fd: カチオン界面活性剤標準液のファクター
A: 試料溶液の滴定に用いたカチオン界面活性剤標準液の量 (mL)
B: 空試験に用いたカチオン界面活性剤標準液の量 (mL)
S: 試料の質量 (g)
80.06: SO3の化学式量
06
.
80
)
(
2
1C
C
M
C
−
×
=
ここに,
C: 石けん分(質量分率 %)
C1: 7.3.3.2で得た結果を,全アニオン界面活性剤の結合硫酸(SO3と
して)(質量分率 %) に換算した値
C2: 7.3.2.4 a)で求めた全アニオン界面活性剤の結合硫酸(SO3として)
(質量分率 %)
M: 石けんの化学式量
7.4
カチオン界面活性剤の定性及び定量
7.4.1
カチオン界面活性剤の定性
7.4.1.1
要旨
カチオン界面活性剤は,一般に脂肪族アミン塩,第四級アンモニウム塩,アルキルピリジニウム塩など
がある。これらは,7.3.1と同じ化学反応を利用して定性を行う。
7.4.1.2
試薬
試薬は,次による。
7.4.1.2.1 メチレンブルー溶液 7.3.1.2.1による。
7.4.1.2.2 クロロホルム 7.3.1.2.2による。
19
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.4.1.2.3 アニオン界面活性剤標準液 7.3.2.2.1による。
7.4.1.2.4 ブロモフェノールブルー溶液 JIS K 8844に規定するブロモフェノールブルー67 mgを0.1
mol/L水酸化ナトリウム溶液1 000 mLに溶解する。
7.4.1.3
器具
器具は,次による。
a) 滴定用シリンダー JIS R 3505に規定する呼び容量100 mLのメスシリンダー有栓形。
7.4.1.4
操作
操作は,次による。
a) 7.3.1.4 a)と同様に試験管にメチレンブルー溶液及びクロロホルムを入れ,アニオン界面活性剤標準液
1,2滴を入れて振り,クロロホルム層を鮮やかな青にする。
b) これに試料の約10 g/L(又は体積分率1 %)溶液を数滴加えて上下に激しく振り,クロロホルム層の
状態を観察する。クロロホルム層の青は薄くなり,試料溶液の量が増すに従って無色となるときはカ
チオン界面活性剤の存在を示すことによって定量する。
注記 第四級アンモニウム系カチオン界面活性剤だけを検知する方法 ブロモフェノールブルー溶
液5 mLとクロロホルム約5 mLとを試験管にとり,これに試料の10 g/L(又は体積分率1 %)
溶液を添加して激しく振り,分離したクロロホルム層を観察する。クロロホルム層が青にな
れば,第四級アンモニウム系カチオン界面活性剤の存在を示す。
7.4.2
カチオン界面活性剤の定量
7.4.2.1
要旨
カチオン界面活性剤は,7.4.1と同じ化学反応を利用して定量を行う。
7.4.2.2
試薬
試薬は,次による。
7.4.2.2.1 アニオン界面活性剤標準液 7.3.2.2.1による。
7.4.2.2.2 メチレンブルー溶液 7.3.1.2.1による。
7.4.2.2.3 クロロホルム 7.3.1.2.2による。
7.4.2.3
器具
器具は,次による。
a) 滴定用シリンダー 7.4.1.3a)による。
7.4.2.4
操作
操作は,次による。
a) 試料の適量(純分として約2 gを含む。)を0.1 mgまではかりとり,水に溶解して全量フラスコで1 000
mLとし,試料溶液とする。
b) この中から全量ピペットを用いて10 mLを滴定用シリンダーにとり,メチレンブルー溶液25 mL及び
クロロホルム15 mLを加える。
c) アニオン界面活性剤標準液を,初め2 mLずつ加え,その都度栓をして激しく振った後,静置する。2
層の分離が早くなれば逐次滴定量を減らし,終点近くでは1滴刻みで行い,白色板を背景として両層
の青さが同一となったときを終点とする。同時に空試験を行う。
7.4.2.5
計算
カチオン界面活性剤は,次の式によって算出する。
20
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S
M
f
B
A
C
10
004
.0
)
(
×
×
×
×
−
=
ここに,
C: カチオン界面活性剤(質量分率 %)
A: 試料溶液の滴定に用いたアニオン界面活性剤標準液の量 (mL)
B: 空試験の滴定に用いたアニオン界面活性剤標準液の量 (mL)
f: アニオン界面活性剤標準液のファクター
M: カチオン界面活性剤の化学式量
S: 試料の質量 (g)
7.5
非イオン界面活性剤の定性及び定量
7.5.1
非イオン界面活性剤の定性
7.5.1.1
要旨
この方法は,ポリオキシエチレン系非イオン界面活性剤だけを対象とする。この方法は,アニオン界面
活性剤が共存しても妨害を受けないが,カチオン界面活性剤が共存する場合は,妨害を受けるので適用し
ない。
なお,7.2のエタノール可溶分の水溶液が7.3.1によって,アニオン性及び7.4.1によってカチオン性のい
ずれの反応も示さないときは,非イオン界面活性剤単独とみなす。
7.5.1.2
試薬
試薬は,次による。
7.5.1.2.1 りんモリブデン酸ナトリウム溶液 (100 g/L) 市販のりんモリブデン酸ナトリウム10 gを水に溶
解して100 mLとする。
7.5.1.2.2 塩化バリウム溶液 (100 g/L) JIS K 8001の4.2(試薬溶液)の規定による。
7.5.1.2.3 塩酸 (1+10) JIS K 8180に規定する塩酸を用いて調製した塩酸。
7.5.1.3
操作
操作は,次による。
a) 試料の10 g/L(又は体積分率1 %)溶液5 mLを試験管にとり,塩酸 (1+10) 10 mL及び塩化バリウ
ム溶液 (100 g/L) 10 mLを加えて加熱する。
b) 濁り又は沈殿を生じたときは,これをろ過し,ろ液にりんモリブデン酸ナトリウム溶液 (100 g/L) 1 mL
を加える。
c) 非イオン界面活性剤が存在すると,うすい黄の沈殿が生じる。
7.5.2
非イオン界面活性剤の定量
7.5.2.1
イオン交換クロマトグラフ法
7.5.2.1.1
要旨
アニオン交換樹脂及びカチオン交換樹脂を用いて,アニオン界面活性剤及びカチオン界面活性剤を分離
し,溶出する非イオン界面活性剤を定量する。
7.5.2.1.2
試薬
試薬は,次による。
7.5.2.1.2.1 エタノール (95) 7.2.2.1による。
7.5.2.1.2.2 強酸性カチオン交換樹脂 強酸性低架橋度イオン交換樹脂(H形)
7.5.2.1.2.3 強塩基性アニオン交換樹脂 強塩基性低架橋度イオン交換樹脂(OH形)
注記 イオン交換樹脂には,次のようなものが市販されている。
− 強酸性カチオン交換樹脂 ダウエックス50 WX 2,ダウエックス50 WX 4,アンバーライ
21
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
トIR-120 B,ダイヤイオンSK 102,SK 104などがある。粒度は,297〜149 μm。
− 強塩基性アニオン交換樹脂 ダウエックス1 X 2,ダウエックス1 X 4,アンバーライト1
RA-401,ダイヤイオンSA 11 Bなどがある。粒度は297〜149 μm。
これらは,市販製品の一例である。この情報は,この規格の利用者の便宜のために記載する
もので,この製品を推奨するものではない。
7.5.2.1.3
装置及び器具
装置及び器具は,次による。
a) カラム用管 内径10 mm,長さ約300 mmのカラム。底部にはガラスウールを詰めておく。
b) ビーカー 500 mL
c) 分液漏斗 100 mL
d) 乾燥器 7.2.3 d)による。
7.5.2.1.4
カラムの準備
使用するイオン交換樹脂をそれぞれエタノール (95) でカラム用管に流し込んで充てんし,150〜200 mm
の高さにする。強酸性カチオン交換樹脂を充てんしたカラムを上に,強塩基性アニオン交換樹脂を充てん
したカラムを下に組み合わせる。
7.5.2.1.5
操作
操作は,図2によって行う。
a) 7.2で得たエタノール溶液100 mLをビーカーから分液漏斗に移し入れ,分液漏斗からカラムに徐々に
流し,溶出液をビーカー500 mLに受ける。
b) エタノール (95) 200〜250 mLを流してカラムの中を洗い,その洗液を溶出液に合わせ,水浴上で加熱
してエタノールを除き,105±2 ℃に調節した乾燥器に入れて恒量になるまで乾燥し,その質量をはか
る。
7.5.2.1.6
計算
非イオン界面活性剤は,次の式によって算出する。
D
S
A
D
S
A
C
−
×
−
×
×
250
100
100
250
=
=
ここに,
C: 非イオン界面活性剤(質量分率%)
A: 溶出量 (g)
S: 7.2.5の試料の質量 (g)
D: 石油エーテル可溶分(質量分率%)
22
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図2−分離系統図A(イオン交換クロマトグラフ法)
7.5.2.2
アルミナカラムクロマトグラフ法
7.5.2.2.1
要旨
合成洗剤をアルミナカラムクロマトグラフ法によって,吸着物質と非吸着物質とに分離し,非吸着物質
のうち,クロロホルム可溶分を非イオン界面活性剤の量とする。
7.5.2.2.2
試薬
試薬は,次による。
7.5.2.2.2.1 混合溶剤 JIS K 8361に規定する酢酸エチルとJIS K 8891に規定するメタノールを体積比で
1 : 1に混合する。
7.5.2.2.2.2 活性アルミナ クロマトグラフ用 (43〜74 μm)
7.5.2.2.3
器具
器具は,次による。
a) カラム 内径25 mm,長さ350 mmのガラスコック付きカラム
b) 分液漏斗 300 mL
c) 三角フラスコ 100 mL,500 mL
d) 乾燥器 7.2.3 d) による。
e) アルミナカラム 次によってアルミナカラムを調製する。
カラムの先端に脱脂綿又はガラスウールを詰め2),充てん剤が流れ出ないようにする。活性アルミ
ナ80〜100 gをビーカーにとり,混合溶剤で懸濁させて,気泡が入らないようにカラム用管に流し込
み,カラム用管の60〜70 %の高さまで充てんする。活性アルミナが沈降した後,その上部をろ紙又
は綿栓で押さえ,コックを開き徐々に混合溶剤を流出させ,気泡が生じない程度に混合溶剤を上部に
少量残す。
合成洗剤エタノール溶液
定 性
イオン交換樹脂[強酸性低架橋度イオン交換樹脂(H形)]
吸 着
(尿素,アニオン界面
活性剤の対イオン)
通 過
確 認
イオン交換樹脂[強塩基性低架橋度イオン交換樹脂(OH形)]
非イオン界面活性剤定量
通 過
濃 縮
吸 着
(アニオン界面活性剤)
23
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注2) 脱脂綿又はガラスウールを詰めた後,海砂を5 mm程度載せてから活性アルミナを流し込む
と充てんしやすい。
7.5.2.2.4
操作
操作は,図3によって行う。
a) 試料2〜3 gを0.1 mgまではかり,できるだけ少量の混合溶剤に溶解しカラムに移す。
試料が粉状及び粒状のときは乳鉢でよくつぶし,液体のときは,水浴上でエタノールと共沸するな
どによって,水分をできるだけ除去する。
試料にアニオン界面活性剤が共存する場合には,アニオン界面活性剤として700 mg以下になるよう
に試料をとる。
混合溶剤に不溶物があるときは,懸濁させてからカラムに移す。
b) コックを開き,上部に気泡を生じない程度に混合溶剤を流出させる。少量の混合溶剤でビーカーを洗
い,この操作を2回繰り返す。
c) 次に,混合溶剤300 mLを入れた分液漏斗をカラムに連結し,0.3〜0.5 mL/minの速度で流出させ,三
角フラスコ500 mLに受ける。
試料をカラムに移した後,全操作にわたる溶出液を集める。
d) 水浴上で加熱して混合溶剤を除去し,質量既知の三角フラスコ100 mLに移し,再び水浴で加熱して
混合溶剤を除去した後,105±2 ℃に調節した乾燥器に入れて恒量になるまで乾燥し,その質量をはか
る。
e) 次に,カラム溶出物にクロロホルム約40 mLを加えて加熱し,不溶物が生じた場合には7.6.1によっ
てクロロホルム可溶物の質量を求める。
7.5.2.2.5
計算
非イオン界面活性剤は,次のように算出する。
次の式によってアルミナカラム溶出分(質量分率%)を算出する。
100
E
E
×
S
A
C=
ここに,
CE: アルミナカラム溶出分(質量分率%)
AE: 7.5.2.2.4 d)で求めたアルミナカラム溶出量 (g)
S: 試料の質量 (g)
さらに,次の式によって,非イオン界面活性剤(質量分率%)を算出する。
a) クロロホルム不溶物が生じない場合
C=CE−D
ここに,
C: 非イオン界面活性剤(質量分率%)
CE: アルミナカラム溶出分(質量分率%)
D: 石油エーテル可溶分(質量分率%)
b) クロロホルム不溶物が生じた場合
D
S
A
C
−
×100
=
ここに,
C: 非イオン界面活性剤(質量分率%)
A: クロロホルム可溶物の質量 (g)
S: 試料の質量 (g)
D: 石油エーテル可溶分(質量分率%)
合成洗剤試料溶液
活性アルミナ
通 過
吸 着
(アニオン界面活性剤)
24
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図3−分離系統図B(アルミナカラムクロマトグラフ法)
7.5.3
非イオン界面活性剤の確認
7.5.3.1
要旨
イオン交換クロマトグラフ法又はアルミナカラムクロマトグラフ法によって得られた溶出物を薄層クロ
マトグラフ法によって分離し,ポリオキシエチレン系非イオン界面活性剤及びアルキロールアミド系非イ
オン界面活性剤の有無を確認する。
7.5.3.2
試薬
試薬は,次による。
7.5.3.2.1 薄層クロマトグラフ用シリカゲル 硫酸カルシウム (CaSO4・21H2O) 質量分率 約13 %を含有
するシリカゲル。
7.5.3.2.2 発色試薬(ドラッゲンドルフ変法試薬) 次によって調製したものとする。
a) 酢酸(体積分率30 %) JIS K 8355に規定する酢酸を用いて調製する。
b) 塩基性硝酸ビスマス0.85 gを酢酸(体積分率30 %)100 mLに水40 mLを加えて溶解する。
c) JIS K 8913に規定するよう化カリウム8 gを水20 mLに溶解する。
d) a),b),c)及び水を体積比1 : 1 : 4 : 10で使用直前に混合する。
7.5.3.2.3 展開溶剤 JIS K 8361に規定する酢酸エチル,JIS K 8034に規定するアセトン及び水を体積比
55 : 35 : 10で混合した溶剤を展開槽の中に入れ,槽内を溶剤蒸気で飽和する。
7.5.3.2.4 薄層プレート 厚さ0.3 mmの薄層プレートを調製し,空気中で固化するまで乾燥した後,120 ℃
の乾燥器中で1.5時間活性化を行って乾燥剤を入れた密閉容器中に保存する。
7.5.3.3
操作
操作は,次による。
a) 7.5.2.1又は7.5.2.2で得られた溶出物をエタノール (95) で約5 g/100 mL液とし,薄層プレートの下端
から約15〜20 mmの位置に,この溶液約5 μLをマイクロピペットでスポットする。
25
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 風乾後,展開槽中に入れ,原点から約170 mmの高さまで展開する。展開温度は,25 ℃前後が望まし
い。
c) 展開後のプレートは,風乾又は乾燥器の中で乾燥後,発色試薬を均一に噴霧し観察する。
7.5.3.4
判定
判定は,次による。
a) 黄色の地に,だいだい色又は赤みのだいだい色に発色した多くのスポットを検出したときは,ポリオ
キシエチレン系非イオン界面活性剤の存在が予想される。
注記 酸化エチレン付加数が大きいものは分離が不十分で原点付近でテーリングが認められる。
なお,酸化エチレンの平均付加数が1.2 mol以下のものについての相互分離性はよい。
b) プレートの上方にだいだい色のスポット(通常単一スポット)を検出したときは,アルキロールアミ
ド系非イオン界面活性剤の存在が予想される。
注記 アルキロールアミド系非イオン界面活性剤のスポットは,一般にポリオキシエチレン系非イ
オン界面活性剤のスポットよりうすい。また,アルキロールアミド系非イオン界面活性剤の
中には数成分のスポットが検出されるものもある。
c) 一方,発色試薬で発色しない場合は,ポリオキシエチレン系非イオン界面活性剤及びアルキロールア
ミド系非イオン界面活性剤は存在しないと考えてよい。
注記 ポリオキシエチレン系非イオン界面活性剤及びアルキロールアミド系非イオン界面活性剤以
外の非イオン界面活性剤,多価アルコール,未反応物質(高級アルコール,アルキルベンゼ
ンなど)などについては,更に,別の展開溶剤を用いて分析するか,又はその他の分析方法
で分析すれば成分の推定が可能である。
7.6
尿素の定量
尿素の定量は,アルミナカラムクロマトグラフ法又は酵素分解法の2種類とし,そのいずれかによる。
7.6.1
アルミナカラムクロマトグラフ法
7.6.1.1
要旨
7.5.2.2アルミナカラムクロマトグラフ法で溶出する非吸着物質のうち,クロロホルム不溶分を尿素の量
とする。
7.6.1.2
試薬
試薬は,次による。
7.6.1.2.1 クロロホルム JIS K 8322の規定による。
7.6.1.3
装置
装置は,次による。
a) 乾燥器 105±2 ℃に調節できる乾燥器。
7.6.1.4
操作
操作は,次による。
a) 7.5.2.2で得られた300 mLフラスコ中のアルミナカラム溶出物の質量をはかった後,クロロホルム40
mLを加え水浴上で50 ℃に加熱溶解し,約10分間氷冷した後,上澄み液をろ過し,ろ液を質量既知
の三角フラスコ200 mLに受ける。
b) 300 mLフラスコ中の不溶物にクロロホルム20 mLを加え,再び水浴上で50 ℃に加熱溶解した後,氷
冷し,クロロホルム層をろ過し,先の三角フラスコ200 mLに合わせる。
c) クロロホルムを除去し,105±2 ℃に調節した乾燥器に入れ,30分間乾燥し,その質量をはかる。
26
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.6.1.5
計算
尿素は,次の式によって算出する。
100
E
×
−
S
A
A
C=
ここに,
C: 尿素(質量分率%)
AE: アルミナカラム溶出物の質量 (g)
A: クロロホルム可溶物の質量 (g)
S: 試料の質量 (g)
7.6.2
酵素分解法
7.6.2.1
要旨
試料中の尿素に尿素分解酵素(ウレアーゼ)を作用させ,分解によって生じた炭酸アンモニウムを,酸
を用いて滴定して尿素の量を求める。
7.6.2.2
試薬
試薬は,次による。
7.6.2.2.1 ウレアーゼ ウレアーゼ0.5 g以下で,JIS K 8731に規定する尿素0.25 gを40〜45 ℃ 1時間で
完全に分解するもの。
7.6.2.2.2 0.1 mol/L塩酸 JIS K 8001の4.5 (5.4)(0.1 mol/L塩酸)の規定による。
7.6.2.2.3 メチルオレンジ溶液 JIS K 8001の4.4(指示薬)の規定による。
7.6.2.2.4 トルエン JIS K 8680の規定による。
7.6.2.3
器具
器具は,次による。
a) 共栓付き三角フラスコ 250 mL
b) ビュレット 50 mL
7.6.2.4
操作
操作は,次による。
a) 試料約0.5 gを共栓付き三角フラスコ250 mLに1 mgまではかりとり,水50 mLを加えて溶解し,メ
チルオレンジ溶液を指示薬として酸又はアルカリを用いて中和する。
b) これに細かく粉砕したウレアーゼ0.2 gを加え,共栓にトルエンを浸して密栓し,50 ℃で30分以上と
きどき振り混ぜながら放置する。冷却後,0.1 mol/L塩酸で滴定し,同時に空試験を行う。
7.6.2.5
計算
尿素は,次の式によって算出する。
S
f
B
A
S
f
B
A
C
3
300
.0
)
(
100
206
.
60
1.0
000
11
)
(
×
×
−
×
×
×
×
−
=
=
ここに,
C: 尿素(質量分率%)
A: 滴定に用いた0.1 mol/L塩酸の量 (mL)
B: 空試験に用いた0.1 mol/L塩酸の量 (mL)
f: 0.1 mol/L塩酸のファクター
S: 試料の質量 (g)
60.06: 尿素の化学式量
7.7
界面活性剤相当分の定量
7.7.1
要旨
27
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
合成洗剤に配合された界面活性剤の総量を,エタノール可溶分から石油エーテル可溶分と尿素とを差し
引いた量で界面活性剤相当分として表す。
注記 界面活性剤相当分は,界面活性剤の他に有機化合物も含まれ,実際の界面活性剤の量よりも高
い数値を示すことがある。
7.7.2
計算
界面活性剤相当分は,7.1,7.2及び7.6で得られた結果を基に,次の式によって算出する。
P=A−(B+C)
ここに,
P: 界面活性剤相当分(質量分率%)
A: 7.2で得られたエタノール可溶分(質量分率%)
B: 7.1で得られた石油エーテル可溶分(質量分率%)
C: 7.6で得られた尿素(質量分率%)
7.8
カルボキシメチルセルロースナトリウムの定量
カルボキシメチルセルロースナトリウムの定量は,ナフタレンジオール法又はアントロン法の2種類と
し,そのいずれかによる。
7.8.1
ナフタレンジオール法
7.8.1.1
要旨
カルボキシメチルセルロースナトリウムは硫酸中で加熱するとき分解して,定量的に2,7-ジヒドロキシ
ナフタレンによって赤紫色となる。この吸光度を測定し,カルボキシメチルセルロースナトリウムの含量
を求める。
7.8.1.2
試薬
試薬は,次による。
7.8.1.2.1 カルボキシメチルセルロースナトリウム標準液 カルボキシメチルセルロースナトリウム[エ
ーテル化度0.5〜0.7,純分(無水物として)質量分率95 %以上,塩化ナトリウム分質量分率1.5 %以下,
水分質量分率10 %以下,粘度25 ℃で70〜130 mPa・s(無水物換算で質量分率1 %水溶液)]の粉末を105
±2 ℃で,4時間乾燥し,この約250 mgを全量フラスコ1 000 mLに0.1 mgまではかりとり,水に溶解し
た後,水を標線まで加える。この溶液1 mLは,カルボキシメチルセルロースナトリウム約0.25 mgを含む。
7.8.1.2.2 ナフタレンジオール試薬 市販の2,7-ジヒドロキシナフタレン0.1 gをJIS K 8951に規定する硫
酸1 000 mLに溶解して冷暗所に放置し,黄色が消えてから,褐色瓶に蓄える。
7.8.1.3
装置及び器具
装置及び器具は,次による。
a) 光電光度計又は分光光度計
b) 全量フラスコ 50 mL,200 mL及び1 000 mL
7.8.1.4
操作
操作は,次による。
a) 検量線の作成
1) カルボキシメチルセルロースナトリウム標準液を1 mLの全量ピペットを用いて0 mL,0.1 mL,0.2
mL,0.3 mL,0.5 mL及び1 mLを,それぞれ全量フラスコ50 mLにとる。
2) それぞれにナフタレンジオール試薬20 mLを加え,水浴中で3.5時間加熱する。
3) 室温まで放冷した後,水を標線まで加える。これらを10 mmセルに移し,空試験液を対照液として,
波長530 nmの吸光度を測定し,検量線を作成する。
28
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.8.1.5
測定
測定は,次による。
a) 7.2.3 b)のガラスろ過器上の残分に熱水約150 mLを徐々に注いで溶出した後,全量フラスコ200 mLに
移し,水を標線まで加える。
b) この溶液1 mLを全量ピペットを用いて全量フラスコ50 mLにとり,検量線の作成と同様に操作して
吸光度を測定し,検量線からカルボキシメチルセルロースナトリウムの質量 (mg) を求める。
7.8.1.6
計算
カルボキシメチルセルロースナトリウムは,次の式によって算出する。この場合,試料中のカルボキシ
メチルセルロースナトリウムの量は,標準液の調製に用いたカルボキシメチルセルロースナトリウムと同
じ置換度のものに換算した値とする。
S
A
S
A
C
20
100
000
1
1
200
×
×
×
×
=
=
ここに,
C: カルボキシメチルセルロースナトリウム(質量分率%)
A: 検量線から求めたカルボキシメチルセルロースナトリウムの
質量 (mg)
S: 7.2の試料の質量 (g)
7.8.2
アントロン法
7.8.2.1
要旨
アントロン(アントラキノン還元誘導体)の濃硫酸溶液を炭水化物と熱すると青緑色になる反応を利用
して,カルボキシメチルセルロースナトリウムを定量する。この反応は非常に鋭敏であるが,でんぷん,
しょ糖エステルなど及びその誘導体が共存するときは適用できない。
7.8.2.2
試薬
試薬は,次による。
7.8.2.2.1 硫酸 JIS K 8951の規定による。
7.8.2.2.2 硫酸 (3+2) JIS K 8951に規定する硫酸を用いて調製する。
7.8.2.2.3 カルボキシメチルセルロースナトリウム標準液 置換度既知のカルボキシメチルセルロースナ
トリウムの粉末約100 mgを全量フラスコ200 mLに0.1 mgまではかりとり,硫酸 (3+2) に溶解し,更に,
硫酸 (3+2) を標線まで加える。この溶液1 mLは,カルボキシメチルセルロースナトリウム0.5 mgを含
む。
7.8.2.2.4 アントロン溶液 試薬のアントロン0.2 gを全量フラスコ200 mLにはかりとり,硫酸約50 mL
に溶解し,更に硫酸を標線まで加える。この溶液は調製後,4時間放置し使用前にこの溶液60 mLを約40
mLの水を入れた全量フラスコ100 mLに徐々に入れ,水で冷却した後,冷水を標線まで加える。ただし,
24時間以内に使用する。
7.8.2.2.5 けい藻土
7.8.2.3
装置及び器具
装置及び器具は,次による。
a) 光電光度計又は分光光度計
b) 全量フラスコ 50 mL,200 mL及び1 000 mL
7.8.2.4
操作
29
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
操作は,次による。
a) 検量線の作成
1) カルボキシメチルセルロースナトリウム標準液を全量ピペットを用いて0 mL,0.5 mL,1 mL,2 mL,
3 mL及び5 mLをそれぞれ全量フラスコ50 mLにとる。
2) それぞれにアントロン溶液30 mLを加えて混合する。この混合液を水浴中で正しく15分間加熱し,
冷却後,硫酸 (3+2) を標線まで加える。
3) この溶液を10 mmセルに移し,空試験液を対照液として波長625 nmでの吸光度を測定し,検量線
を作成する。
7.8.2.5
測定
測定は,次による。
a) 試料1 gを1 mgまではかりとり,硫酸 (3+2) に溶解し,全量フラスコ100 mLに移し,硫酸 (3+2) を
標線まで加える。
b) けい藻土をろ過助剤として,この溶液を乾燥したガラスろ過器でろ過する。
c) ろ液5 mLとアントロン溶液30 mLとを全量フラスコ50 mLに入れて混合する。
d) この混合液を検量液作成の場合と同様に操作して吸光度を測定し,検量線からカルボキシメチルセル
ロースナトリウムの質量 (mg) を求める。
7.8.2.6
計算
カルボキシメチルセルロースナトリウムは,次の式によって算出する。この場合,試料中のカルボキシ
メチルセルロースナトリウムの質量は,標準液の調製に用いたカルボキシメチルセルロースナトリウムと
同じ置換度のものに換算した値とする。
S
A
S
A
C
2
100
000
1
5
100
×
×
×
×
=
=
ここに,
C: カルボキシメチルセルロースナトリウム(質量分率%)
A: 検量線から求めたカルボキシメチルセルロースナトリウム
の質量 (mg)
S: 試料の質量 (g)
7.9
過酸化塩の定量
7.9.1
要旨
合成洗剤の中に配合された過ほう酸ナトリウムなどの過酸化塩は,水に溶解したときに過酸化水素を遊
離するので,これを過マンガン酸カリウム溶液で滴定し,その消費量から定量する。ただし,過酸化塩以
外の酸性で過マンガン酸カリウムと反応する物質を含む場合には,適用できない。しかし,過マンガン酸
カリウムと同じように反応するエチレンジアミン四酢酸 (EDTA) 又は同じタイプのキレート剤を含んで
いても,その濃度が10 g/L以下であれば,この方法を適用できる。
7.9.2
試薬
試薬は,次による。
7.9.2.1 硫酸アルミニウム
7.9.2.2 硫酸ビスマスマンガン溶液 JIS K 8566に規定する硝酸ビスマス2 gとJIS K 8997に規定する硫
酸マンガン5.5 gとをJIS K 8951に規定する硫酸を用いて調製した2.5 mol/L硫酸1 000 mLに溶解する。
7.9.2.3 硫酸アルミニウム・ビスマスマンガン溶液 硫酸アルミニウム50 g,JIS K 8566に規定する硝酸
30
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ビスマス5 g及びJIS K 8997に規定する硫酸マンガン7 gを2.5 mol/L硫酸1 000 mLに溶解する。
7.9.2.4 0.02 mol/L過マンガン酸カリウム溶液 JIS K 8001の4.5 (7)(0.02 mol/L過マンガン酸カリウム溶
液)に規定するもの。
7.9.3
装置及び器具
装置及び器具は,次による。
a) 全量フラスコ 1 000 mL
b) コニカルフラスコ 500 mL
c) かき混ぜ機
7.9.4
操作
操作は,次による。
a) 試料約10 gを0.01 gまではかりとり,ビーカー2 000 mLに移す。
b) 全量フラスコ1 000 mLに水を標線まで満たし,ビーカー2 000 mLに加える。
c) かき混ぜ機で3分間激しくかき混ぜ,試料を溶解させる。
d) 別にコニカルフラスコに硫酸ビスマスマンガン溶液50 mLを入れ,絶えずかき混ぜながらうすい紅色
が残るようになるまで,0.02 mol/L過マンガン酸カリウム溶液を加える。
e) 全量ピペットを用いてc) の溶液100 mLをとり,d) のコニカルフラスコに移す。
f)
うすい紅色が少なくとも15秒間残るようになるまで,0.02 mol/L過マンガン酸カリウム溶液で滴定す
る。
g) 終点が明りょうでないときは,硫酸アルミニウム1 g又は硫酸アルミニウム・ビスマスマンガン溶液
20 mLを加えて再測定する。
7.9.5
計算
過酸化塩は,次の式によって算出する。
S
K
f
A
S
K
f
A
C
×
×
×
×
×
×
×
=
=
100
000
1
000
1100
ここに,
C: 過酸化塩(質量分率%)
A: 滴定に用いた0.02 mol/L過マンガン酸カリウム溶液の量 (mL)
f: 0.02 mol/L過マンガン酸カリウム溶液のファクター
S: 試料の質量 (g)
K: 過ほう酸ナトリウム(質量分率%) として求める場合には7.7
を用い,過炭酸ナトリウム(質量分率%) として求める場合
には5.3を用い,有効酸素 (O)(質量分率%) として求める
場合には0.8を用いる。
a) 定量する前の過酸化塩の定性 試料約1 gを水50 mLに溶解し,硫酸 (1+3) 約5 mLを加え,よう化
カリウム約0.5 gを加えて振り混ぜ,でんぷん溶液 (10 g/L) 約1 mLを加える。
b) このとき,過酸化塩を含む試料では,よう素でんぷんが生じ青色を呈する。
7.10 全りん酸塩の定量
全りん酸塩の定量は,吸光光度法又は質量法(ISO法)のいずれかによる。
7.10.1 吸光光度法
7.10.1.1 要旨
試料中に含有される各形態のりん酸塩を硝酸で加水分解した後,これにモリブドバナジン酸塩溶液を加
31
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
えて生成した黄の錯塩の吸光度を測定し,全りん酸塩を求める。けい素,鉄,カルボキシメチルセルロー
スナトリウム及びほう酸などは妨害しない。
7.10.1.2 試薬
試薬は,次による。
7.10.1.2.1 モリブドバナジン酸塩溶液 JIS K 8747に規定するバナジン (V) 酸アンモニウム1.12 gを200
〜300 mLの水に溶解し,JIS K 8541に規定する硝酸250 mLを加え,かき混ぜながら別にJIS K 8905に規
定する七モリブデン酸六アンモニウム四水和物27 gを水約100 mLに溶解したものを少量ずつ注加する。
注加後,更に水を加えて全量を1 000 mLとし,着色瓶に入れて保存する。ただし,保存中に沈殿を生じた
ものは用いてはならない。
7.10.1.2.2 りん酸塩標準液 (P2O5として0.3 mg/mL) 硫酸デシケータ中で24時間以上乾燥したJIS K
9007に規定するりん酸二水素カリウム19.174 gを全量フラスコ1 000 mLにとり,水に溶解して1 000 mL
とする。全量ピペットを用い,この溶液3 mLを全量フラスコ100 mLにとり,硝酸2 mLを加えた後,水
を標線まで加える。
7.10.1.2.3 0.02 mol/L過マンガン酸カリウム溶液 JIS K 8247に規定する過マンガン酸カリウム0.33 gを
全量フラスコ200 mLにとり,水約100 mLを加えて溶解する。次に,1〜2時間静かに煮沸した後,一夜
間暗所に放置する。上澄み液をガラスろ過器でろ過し,褐色瓶に入れて暗所に保存する。
7.10.1.2.4 しゅう酸溶液 JIS K 8519に規定するしゅう酸二水和物10 gとJIS K 8997に規定する硫酸マン
ガン1 gとを約60 ℃の水約60 mLに溶解し,JIS K 8951に規定する硫酸を用いて調製した硫酸 (1+4) 20
mLを加える。溶液が透明になった後,水を加えて100 mLとする。
7.10.1.2.5 硝酸 JIS K 8541の規定による。
7.10.1.3 装置及び器具
装置及び器具は,次による。
a) 光電光度計又は分光光度計
7.10.1.4 試験液の調製
7.2.4のガラスろ過器上の残分に熱水約150 mLを徐々に注いで溶出した後,全量フラスコ200 mLに移
し,水を標線まで加える。この溶液の適量(P2O5として30〜150 mgを含む。)を三角フラスコ300 mLに
とり,JIS K 8541に規定する硝酸10 mL及び水100 mLを加えて約15分間静かに煮沸して分解する。冷却
後,全量フラスコ200 mLに移し,水を標線まで加え,JIS P 3801に規定する定性ろ紙でろ過し,試験液と
する。
7.10.1.5 操作
操作は,次による。
a) 検量線の作成
1) りん酸塩標準液 (P2O5として0.3 mg/mL) の0 mL,1 mL,2 mL,3 mL及び5 mLを全量ピペットを
用い,それぞれを全量フラスコ100 mLにとり,それぞれに水50 mLを加える。
2) これらにモリブドバナジン酸塩溶液20 mLを添加した後,水を標線まで加え,約30分間放置する。
3) この溶液を10 mmセルに移し,空試験液を対照液として波長400 nmにおける吸光度を測定し,検
量線を作成する。
7.10.1.6 測定
測定は,次による。
a) 試料中に過酸化物を含まない場合 試験液2 mL(りん酸塩として0.3〜1.5 mgを含む。)を全量フラス
32
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
コ100 mLにとり,水約50 mL及びモリブドバナジン酸塩溶液20 mLを加え,以下,検量線作成と同
様に操作して吸光度を測定し,検量線からりん酸塩の質量 (mg) を求め,全りん酸塩は,次の式によ
って算出する。
B
S
A
S
B
A
C
×
×
×
×
×
×
000
2
100
000
1
2
200
200
=
=
ここに,
C: 全りん酸塩(P2O5として)(質量分率%)
A: 検量線から求めたP2O5の質量 (mg)
B: エタノール不溶分の水溶液の分取量 (mL)
S: 7.2.5 の試料の質量 (g)
b) 試料中に過酸化物を含む場合 試験液2 mL(りん酸塩として0.3〜1.5 mgを含む。)を全量ピペットを
用いて,全量フラスコ100 mLにとり,硫酸 (1+4) 1 mLを加え,次に0.02 mol/L過マンガン酸カリウ
ム溶液を滴下して振り混ぜ,1分間以上うすい紅色を保持するまで滴下する。次に,振り混ぜながら
無色になるまで,しゅう酸溶液を滴下する。水約50 mL及びモリブドバナジン酸塩溶液20 mLを加え
てよく振り混ぜ,検量線の作成と同様に操作し,全りん酸塩(P2O5として)(質量分率%)を求める。
1) 非イオン界面活性剤が配合されていないことが明らかな場合には,アルコール可溶分を除く操作が
省略できるので,試験液を次のように調製してもよい。
1.1) 試料約5 gをビーカー200 mLに1 mgまではかりとり,水100 mLを加え,水浴上で加熱し,ガラ
ス棒でかき混ぜながら溶解する。室温まで冷却した後,全量フラスコ200 mLに移し,水を標線ま
で加えろ過する。以下,試験液の調製と同様に操作する。
7.10.2 質量法(ISO法)
7.10.2.1 要旨
試料中に含有する各形態のりん酸塩を硝酸で加水分解した後,アセトン溶液中でりん酸塩をりんモリブ
デン酸キノリンの形で沈殿させる。沈殿を乾燥し,重さをはかり全りん酸塩として求める。
7.10.2.2 試薬
試薬は,次による。
7.10.2.2.1 硝酸 JIS K 8541の規定による。
7.10.2.2.2 キノリン JIS K 8279の規定による。
7.10.2.2.3 アセトン JIS K 8034の規定による。
7.10.2.2.4 シトロモリブデン酸試薬 次によって調製する。
a) JIS K 8906に規定するモリブデン (VI) 酸二ナトリウム二水和物70 gをビーカー400 mL中で水150
mLに溶解する。
b) JIS K 8283に規定するくえん酸一水和物60 gをビーカー1 000 mL中で水150 mLに溶解し,硝酸85 mL
を加える。
c) b) のくえん酸溶液をかくはんしながら,これに,a) のモリブデン酸ナトリウム溶液に注ぐ。
d) ビーカー400 mLに水100 mLを入れ,硝酸35 mLを加える。次にキノリン5 mLを加える。
e) c) の溶液をかくはんしながら,これにd) のキノリン溶液を注ぎ,一夜間静置する。全量をろ過るつ
ぼを通じて全量フラスコ1 000 mLに移す。このとき,水ですすがない。
f)
e) の溶液にアセトン280 mLを加え,標線まで水を加えて,よく混ぜる。
7.10.2.3 器具
33
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
器具は,次による。
a) ポリエチレン製ビーカー 400 mL,600 mL,1 000 mL及び2 000 mL
けい酸塩分も同時に定量する場合には,活性溶液がガラスと接触するのを避けなければならないの
で,ガラスビーカーよりポリエチレンビーカーの方がよい。
b) 全量フラスコ 1 000 mL及び100 mL
c) 全量ピペット 5 mL,10 mL,20 mL,25 mL及び50 mLで定期的に検定したピペット。
d) ろ過フラスコ 500 mL
e) ろ過るつぼ 多孔率P=16(孔径インデックス10〜16 μm)の焼結ガラス円盤付きでISO 4793に適合
するもの。
このるつぼは使用前に,260±20 ℃に調節した乾燥器で1時間乾燥し,デシケータ中で放冷する。
f)
乾燥器 260±20 ℃に調節できる乾燥器。
g) かくはん機
7.10.2.4 操作
操作は,次による。
a) 測定用試料 試料約10 gを0.01 gまで正しくはかりとる。
b) 測定
1) 測定用試料をビーカー2 000 mLに移す。全量フラスコ1 000 mLの標線まで35〜40 ℃の水を満たし,
これを測定用試料に加え,全量フラスコで計量した水をすべてビーカーに移す。かくはん機で3分
間激しくかくはんし,少量の不溶性けい酸塩は別として測定用試料を溶解する(L1溶液)。
2) 全量ピペット25 mLを用いて試料溶液 (L1) 25.0 mLをとり,全量フラスコ100 mLに移し標線まで
水を満たす(L2溶液)。
3) 全量ピペットを用いて,P2O5として5〜20 mgを含む量(例えば,P2O5として質量分率約20 %を含
む粉末洗剤では20 mL)のL2溶液をトールビーカー600 mLにとる。
4) この溶液に,硝酸15 mLを加え,約100 mLに希釈する。ビーカーを時計皿で覆い,内容物を静か
に沸騰させ,30分間静かに沸騰を続ける。必要があれば沈殿したけい酸を除くために,別のトール
ビーカー600 mLにろ過し,加水分解中に蒸発した量とほぼ同量の水を用いてろ紙を洗う。
5) 次に,このビーカーをドラフト中に置き,かくはんしないでシトロモリブデン酸試薬50 mLを加え
る。時計皿で覆い,直ちに適切な方法(炎を用いない)で加熱し,10〜15分で約75 ℃(沸騰し始
める)の温度にする。30秒を超えない時間この温度に保つ。
ビーカーをヒータから除き,室温まで冷却する。冷却している間に3〜4回かき混ぜる。沈殿が生
じたら沈降させる。
6) あらかじめ質量をはかったろ過るつぼをろ過フラスコに取り付け,上澄液をろ過るつぼに移しろ過
する。このとき,沈殿はできるだけ多くビーカー中に残す。沈殿は1回につき水30 mLを用い,デ
カンテーションによって6回洗浄する。次いで,洗瓶を用いて沈殿を定量的にろ過るつぼに移す。
そのためにはビーカー内の沈殿を少量の水で,ろ過るつぼに洗い移す。ろ過るつぼの水が全部なく
なってから,同様に沈殿を洗い移す。この操作を更に2回繰り返し,沈殿を全部ろ過るつぼに移す。
ろ過るつぼの水がなくなってから,ろ過るつぼの内側を洗浄瓶を用い,少量の水で洗い流した後
十分に吸引する。
7) ろ過るつぼをろ過フラスコから外し,260±20 ℃に調節した乾燥器に入れ,1時間加熱する。ろ過
るつぼを乾燥器から取り出し,デシケータ中で放冷し,質量をはかる。引き続き加熱,冷却,ひょ
34
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
う量の操作を繰り返し,連続する2回の測定値の差が0.001 g以下になるまで続ける(沈殿が入った
ろ過るつぼの質量をはかるとき,ろ過るつぼをデシケータ中で放冷する時間は,あらかじめ,空の
ろ過るつぼの質量をはかるときに放冷した時間と同じにするとよい。)。
c) 空試験 定量と並行して空試験を行う。空試験は同じ操作で行い,また,試料以外のすべての試薬の
量を同じにする。沈殿の質量は1.5 mgを超えてはならない。もし超えていれば新たに試薬を作り直し,
操作及び空試験を行う。
d) 計算 全りん酸塩は,次の式によってP2O5(質量分率%)として算出する。
0
2
1
0
2
1
8.
282
1
)
(
25
10
100
000
1
07
032
.0
)
(
m
V
m
m
m
V
m
m
C
×
×
−
×
×
×
×
×
×
−
=
=
ここに,
C: 全りん酸塩(P2O5として)(質量分率%)
m0: 測定試料の質量 (g)
m1: 測定によって求めたりんモリブデン酸キノリン沈殿の
質量 (g)
m2: 空試験で求めた沈殿の質量 (g)
V: 測定に用いたL2溶液の量 (mL)
0.032 07: りんモリブデン酸キノリン [(C9H7N) 3H3 (PO4・12 moO3)]
1 gに相当するP2O5の質量 (g)
1) 同一試料について,同一測定者が同じ装置を用いて同時に,又は連続して測定した二つの値の差が
P2O5含量質量分率18〜30 %の場合で0.5 %を超えてはならない。
2) 同一試料について二つの異なる実験室で測定した値の差が,P2O5含量質量分率18〜30 %の場合で
1.1 %を超えてはならない。
7.11 けい酸塩の定量
けい酸塩の定量方法は,吸光光度法又は質量法のいずれかによる。
吸光光度法は,質量法より迅速である。この方法では,SiO2分の定量によってけい酸塩(質量分率%)
が得られる。けい酸ナトリウム (Na2O・nSiO2) は,その種類に応じ,求めたSiO2分に次の数値を乗じて求
める。
けい酸ナトリウム1号:1.49
けい酸ナトリウム2号:1.41
けい酸ナトリウム3号:1.33
7.11.1 吸光光度法
7.11.1.1 要旨
試料にモリブデン酸アンモニウムを加えて生成した,けいモリブデン酸錯塩の吸光度を測定して,けい
酸塩(質量分率:SiO2として)を求める。
7.11.1.2 試薬
試薬は,次による。
7.11.1.2.1 モリブデン酸アンモニウム溶液 (100 g/L) JIS K 8905に規定する七モリブデン酸六アンモニ
ウム四水和物21.2 gを水に溶解して200 mLとする。ろ過して冷暗所に保存する。
なお,著しく変色をした場合又は明らかに認められる程度の沈殿が生じた場合には,用いてはならない。
7.11.1.2.2 くえん酸溶液 (100 g/L) JIS K 8283に規定するくえん酸一水和物10.9 gを水に溶解して,100
mLとする 。
なお,かびが生じるので,1週間以上経過したものは用いてはならない。
35
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.11.1.2.3 けい酸塩標準液 (SiO2として0.1 mg/mL) JIS K 8885に規定する粉末二酸化けい素を精製脱水
後,その0.5 gを白金るつぼにはかりとり,JIS K 8625に規定する炭酸ナトリウム4 gを加え,十分に混ぜ
合わせて加熱し,30分間融解する。冷却後,水に溶解して全量フラスコ500 mLに移す。白金るつぼは,
水でよく洗い,洗液も全量フラスコに加え,水を標線まで加える。この溶液は,ポリエチレン瓶に保存す
る。
この溶液の一定量をとり,7.11.2によって定量し,この溶液1 mLに含まれるけい酸塩を求め,水を加え
て1 mL中にけい酸塩として約0.1 mgを含むように調製する。
7.11.1.2.4 硫酸 (1+4) JIS K 8951に規定する硫酸を用いて調製する。
7.11.1.2.5 0.02 mol/L過マンガン酸カリウム溶液 7.10.1.2.3 による。
7.11.1.2.6 しゅう酸溶液 7.10.1.2.4による。
7.11.1.3 装置及び器具
装置及び器具は,次による。
a) ビーカー ステンレス鋼製,1 000 mL又はそれ以上のビーカー。
b) かくはん棒 ステンレス鋼製
c) 白金るつぼ JIS H 6201の30番
d) 光電光度計又は分光光度計
7.11.1.4 操作
操作は,次による。
a) 検量線の作成
1) けい酸塩標準液 (SiO2として0.1 mg/mL)の0 mL,2 mL,4 mL,6 mL及び8 mLを全量ピペットを
用いてそれぞれ全量フラスコ100 mLにとり,それぞれに水50 mL,硫酸 (1+4) 1 mL及びモリブデ
ン酸アンモニウム溶液 (100 g/L) 5 mLを加えてよく振り混ぜ,約3分間放置する。
2) くえん酸溶液 (100 g/L) 5 mLを加え,水を標線まで加えた後,再び振り混ぜ,直ちに10 mmセルに
移し,空試験液を対照液として波長420 nm付近における吸光度を測定し,検量線を作成する。
注記 くえん酸は,すべてのりん酸塩による発色を防止する。
b) 試験液の調製
1) 試料約1 gを0.1 mgまではかりとり,ステンレス鋼製ビーカーに移し,熱水約800 mLを加え,時
計皿でふたをして,水浴上でステンレス鋼製のかくはん棒でときどきかき混ぜながら加熱する。
2) 室温に冷却した後,硬質ガラス製全量フラスコ1 000 mLに水で洗い移し,水を標線まで加えた後,
よく振り混ぜて試験液とする。
3) 試料中にゼオライトを含む場合は,遠心分離するか又はろ紙でろ過することによって,これを分離
除去する。この操作を行っても,溶液が濁っている場合は,更に0.45 μm以下の細孔のろ過器でろ
過する。この液は,15分間以内に使用する。
c) 測定
1) 試料中に過酸化物を含まない場合
1.1) 試験液の適量(けい酸塩として0.3〜0.8 mgを含む。)を全量フラスコ100 mLにとり,水を加えて
約50 mLとし,硫酸 (1+4) 1 mL及びモリブデン酸アンモニウム溶液5 mLを加えて,よく振り混
ぜる。以下,検量線の場合と同様に操作し,空試験液を対照液として吸光度を測定し,検量線か
らけい酸塩の質量を求める。
1.2) 計算 けい酸塩は,次の式によって算出する。
36
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
×
×V
S
A
C=
ここに,
C: けい酸塩(質量分率 %:SiO2として)
A: 検量線から求めたけい酸塩の質量(mg:SiO2として)
S: 試料の質量 (g)
V: 試験液の採取量 (mL)
2) 試料中に過酸化物を含む場合
2.1) 試験液の適量(けい酸塩として0.3〜0.8 mgを含む。)を全量フラスコ100 mLにとり,硫酸 (1+
4) 1 mLを加え,次に過マンガン酸カリウム溶液を滴下して振り混ぜ,1分間以上うすい紅色を保
つまで滴下する。
なお,通常,過マンガン酸カリウム溶液は,1,2滴で十分である。過酸化物があれば,過モリ
ブデン酸塩を生成し,けいモリブデン酸塩と同様な発色をして,吸光度が過大に出る原因となる
ので,過酸化物を分解しておく必要がある。
2.2) 振り混ぜながら無色になるまで,しゅう酸溶液を滴下する。モリブデン酸アンモニウム溶液5.0 mL
を加えてよく振り混ぜる。以下c) 1.2)と同様に測定し,けい酸塩 (SiO2) を求める。
7.11.2 質量法
7.11.2.1 要旨
試料中のエタノール可溶分を,エタノールを用いて抽出除去する。エタノール不溶分から水不溶分を除
き,これから塩酸可溶分を除去し,残さからけい酸塩を求める。この方法は,すべてのタイプの市販粉末
洗剤のけい酸塩の定量に適用するが,けい酸塩以外に酸不溶物質を含む洗剤には適用しない。
7.11.2.2 試薬
試薬は,次による。
7.11.2.2.1 エタノール (99.5) JIS K 8101の規定による。
7.11.2.2.2 塩酸 JIS K 8180の規定による。
7.11.2.2.3 硝酸銀溶液 (5 g/L) JIS K 8550に規定する硝酸銀0.5 gを水に溶解して100 mLとする。
7.11.2.2.4 軽石 粒径2〜4 mm又は沸騰石として用いられる相当品。
7.11.2.3 装置及び器具
装置及び器具は,次による。
a) ソックスレー抽出器 フラスコ容量500 mL,抽出管容量200 mL
b) 抽出用ガラス円筒ろ紙 多孔率P=1.6 (1.6 μm),直径約36 mm,長さ約95 mm
c) ガラスろ過器 JIS R 3503に規定するろ過板の細孔記号4。
d) 乾燥器 105±2 ℃に調節できるもの。
e) ろ過るつぼ 磁製,多孔率P=4 (1.6〜4 μm) のろ過るつぼ。
f)
白金るつぼ
g) 電気炉 900〜960 ℃に調節できる電気炉。
h) ろ紙 JIS P 3801の規定による。
7.11.2.4 操作
操作は,次による。
a) 測定用試料 ビーカー600 mL又は抽出用ガラス円筒ろ紙に,試料約10 gを0.01 gまではかりとる。
b) 有機物の除去 有機物の除去は,ソックスレー抽出又はビーカー処理のいずれかによる。
37
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) ソックスレー抽出
1.1) ソックスレー抽出器のフラスコ中に,エタノール (99.5) 300 mL及び軽石数個を入れる。
1.2) ソックスレー抽出器の抽出管に,試料を入れた抽出用ガラス円筒ろ紙を入れ,装置(フラスコ抽
出管,冷却管)を組み立てる。
1.3) 抽出を開始し,最初に液が吸い上げられてから2時間30分の間は速い速度で抽出を続ける。放冷
した後,抽出器に残留しているエタノール層をフラスコに移し,エタノール可溶分を捨てる。
2) ビーカー処理による抽出
2.1) ビーカー内の試料にエタノール (99.5) 約250 mLを加える。
2.2) 時計皿で覆い,加熱し,かき混ぜ機又はマグネチックスターラを用いてかき混ぜながら5分間沸
騰させる。
2.3) ビーカーを放冷し,不溶物質を沈殿させる。中位の目のろ紙を用い,エタノール層をろ過する。
2.4) 同じろ紙を用いて,新たにエタノール (99.5) を加えて,2回この抽出を繰り返す。
2.5) 不溶物の入っているビーカーに,熱エタノール (99.5) (50〜60 ℃) 約75 mLを加え,硬い塊が残
っているときはガラス棒を用いて砕く。不溶物を沈殿させ,同じろ紙を用いてろ過する。この操
作を2回繰り返す。
2.6) ろ紙の底に穴をあけ,残留物を熱水約100 mLを用いて,不溶物を入れたビーカー中に洗い流す。
c) 測定
1) b) 1)で抽出後,ソックスレー抽出器から円筒ろ紙を取り出し,熱水100 mLを用いて内容物をビー
カー400 mLに移す。又はb) 2)によってビーカー600 mL中にエタノール不溶物を得る。
2) 時計皿でふたをして約30分間水浴上で,ガラス棒でときどきかき混ぜながら加熱する。
3) 温溶液のままガラスろ過器又はろ紙を用いてろ過し,ろ液を500 mLビーカーにとる。残分に熱水
約50 mLを加えて別のビーカーにとり,水浴上で同様に加熱する。温溶液を前と同様にろ過し,ろ
液は先のろ液と合わせる。少量の熱水でビーカー及びガラスろ過器又はろ紙をよく洗浄しながらろ
過し,ろ液を同様に合わせる。
4) このろ液に塩酸30 mLを加え,ガラス棒でかき混ぜ,蒸気浴上で蒸発・乾固させる。
5) 水35〜40 mLを加えてときどきかき混ぜながら10分間加熱する。再び塩酸10 mLを加えて,かき
混ぜ,4)と同様にして蒸発し,乾固させる。
6) 塩酸10 mLを加え,残留物を溶解し,3回蒸発し乾固させる。ビーカー及び残留物を105±2 ℃に
保った乾燥器中に1時間放置する。熱水50 mL及び塩酸10 mLを加える。ときどきかき混ぜながら
蒸気浴上で10分間加熱する。
7) 900 ℃に調整した電気炉で加熱し,デシケータ中で放冷後,あらかじめ質量をはかったろ過るつぼ
を用いて吸引ろ過するか,又はJIS P 3801に規定する5種Bのろ紙を通してろ過する。
8) 熱水を用いてフィルター上の沈殿を洗浄し,その洗液に硝酸銀溶液1,2滴を加えて試験し,塩化物
の反応がなくなるまで,洗浄を続ける。
9) ろ紙の場合には,ろ紙を白金るつぼに入れる。あらかじめ900 ℃に調整した電気炉で加熱し,デシ
ケータ中で放冷した後質量をはかっておく。
10) 白金るつぼと内容物は900 ℃まで徐々に加熱してから,900〜960 ℃に調整した電気炉中に30分間
放置する。デシケータ中で放冷後その質量を1 mgまではかる。
d) 計算 けい酸塩は,次の式によって算出する。
38
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
×
S
W
C=
ここに,
C: けい酸塩(SiO2として)(質量分率%)
W: 残留物の質量 (g)
S: 試料の質量 (g)
7.12 硫酸塩の定量
試料中の有機物をn-アミルアルコールで抽出した後,塩化バリウムを加えて無機硫酸塩を硫酸バリウム
として沈殿,ろ過し,硫酸ナトリウムとして求める。
7.12.1 試薬
試薬は,次による。
7.12.1.1 塩化ナトリウム JIS K 8150の規定による。
7.12.1.2 n-アミルアルコール
7.12.1.3 塩酸 JIS K 8180の規定による。
7.12.1.4 メチルオレンジ溶液 (1 g/L) JIS K 8001の4.4(指示薬)の規定による。
7.12.1.5 塩化バリウム溶液 (100 g/L) JIS K 8001の4.2(試薬溶液)の規定による。
7.12.1.6 硝酸銀溶液 (5 g/L) 7.11.2.2.3 による。
7.12.2 器具
器具は,次による。
a) ビーカー 100 mL及び500 mL
b) 分液漏斗 250 mL
c) るつぼ JIS R 1301に規定するPC1A形30 mL
7.12.3 操作
操作は,次による。
a) 試料の適量(Na2SO4として0.1〜0.3 gを含む。)をビーカー100 mL中に0.1 mgまではかりとる。ただ
し,固体試料では5 g以下,液体試料では15 g以下をはかりとる。
b) 水50 mLを加え,試料が溶解するまでかき混ぜ,必要があれば50 ℃を超えない程度に加熱する。こ
れに塩化ナトリウム15 gを加え,よくかき混ぜる。
c) この溶液を分液漏斗250 mLに移し,n-アミルアルコール30 mLずつで3回抽出する。
d) 水層をビーカー500 mL中に移す。元のビーカー100 mLに塩化ナトリウム5 gを加え,これを少量の
水で溶解し,この溶液を用いてn-アミルアルコール抽出液を洗浄し,洗浄液はビーカー500 mL中の水
層に加える。
e) この水層にメチルオレンジ溶液 (1 g/L) を数滴加え,塩酸で酸性にしてから,更に過剰の塩酸10〜20
mLを加え,必要があればろ過する。
f)
これに水を加えて約300 mLとした後,沸点近くまで加熱し,熱塩化バリウム溶液 (100 g/L) 10 mLを
滴下しながらかき混ぜる。
g) 約2時間水浴上で加熱し,その後,室温に1時間以上放置する。JIS P 3801に規定するろ紙5種Cで
ろ過し,洗液に硝酸銀溶液1,2滴を加えて,塩化物の反応が認められなくなるまで温水で洗浄する。
h) このろ紙を乾燥し,質量既知のるつぼに移し,完全に灰化するまで強熱し,デシケータ中で放冷した
後,その質量をはかる。
7.12.4 計算
39
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
硫酸塩 (Na2SO4) は,次の式によって算出する。
S
A
S
A
C
86
.
60
100
6
608
.0
×
×
×
=
=
ここに,
C: 硫酸塩(Na2SO4として)(質量分率%)
A: 残分の質量 (g)
S: 試料の質量 (g)
0.608 6: 硫酸バリウムの硫酸ナトリウムへの換算係数
7.13 炭酸塩の定量
炭酸塩の定量は,ガスクロマトグラフ法,中和滴定法又は炭酸ガス電極法のいずれかによる。
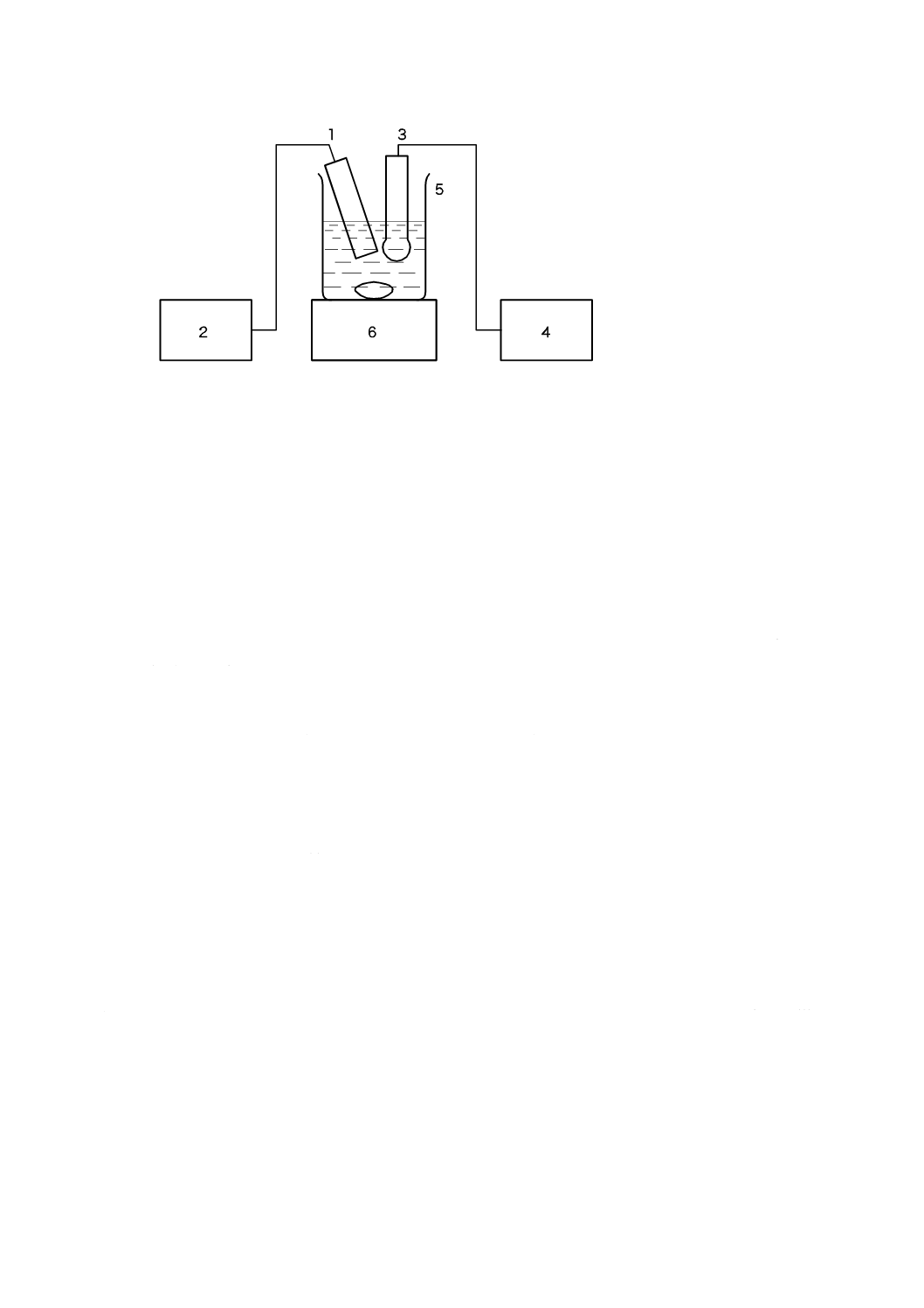
7.13.1 ガスクロマトグラフ法
7.13.1.1 要旨
試料に塩酸を加えて炭酸塩を分解し,発生した二酸化炭素をガスクロマトグラフで分析し,炭酸塩を炭
酸ナトリウム (Na2CO3) として求める。
7.13.1.2 試薬
試薬は,次による。
7.13.1.2.1 塩酸 (1+3) JIS K 8180に規定する塩酸を用いて調製する。
7.13.1.2.2 炭酸ナトリウム JIS K 8625に規定する炭酸ナトリウムを白金るつぼ中で500〜600 ℃に40〜
50分間保ち,硫酸デシケータ中で放冷する。
7.13.1.3 装置及び器具
装置及び器具は,次による。
a) ガスクロマトグラフ
1) カラム管 内径3 mm,長さ1 000 mmのステンレス鋼製
2) 脱水・脱塩化水素管 内径10 mm,長さ100 mmのガラス管
3) 検出器 熱伝導度検出器
4) 記録計 ストリップチャート式ペン書自動平衡記録計及びそれに類する記録計
b) キャリヤーガス ヘリウム
c) 試料導入器 容量5 mL,目盛0.1 mLの注射器
d) 四方切換コック キャリヤーガスとの接触面は,すべてふっ素樹脂製。
e) 反応容器 図4に示すような,耐圧密閉形の耐熱ガラス製で,接続部にボールジョイント(6,7)を用
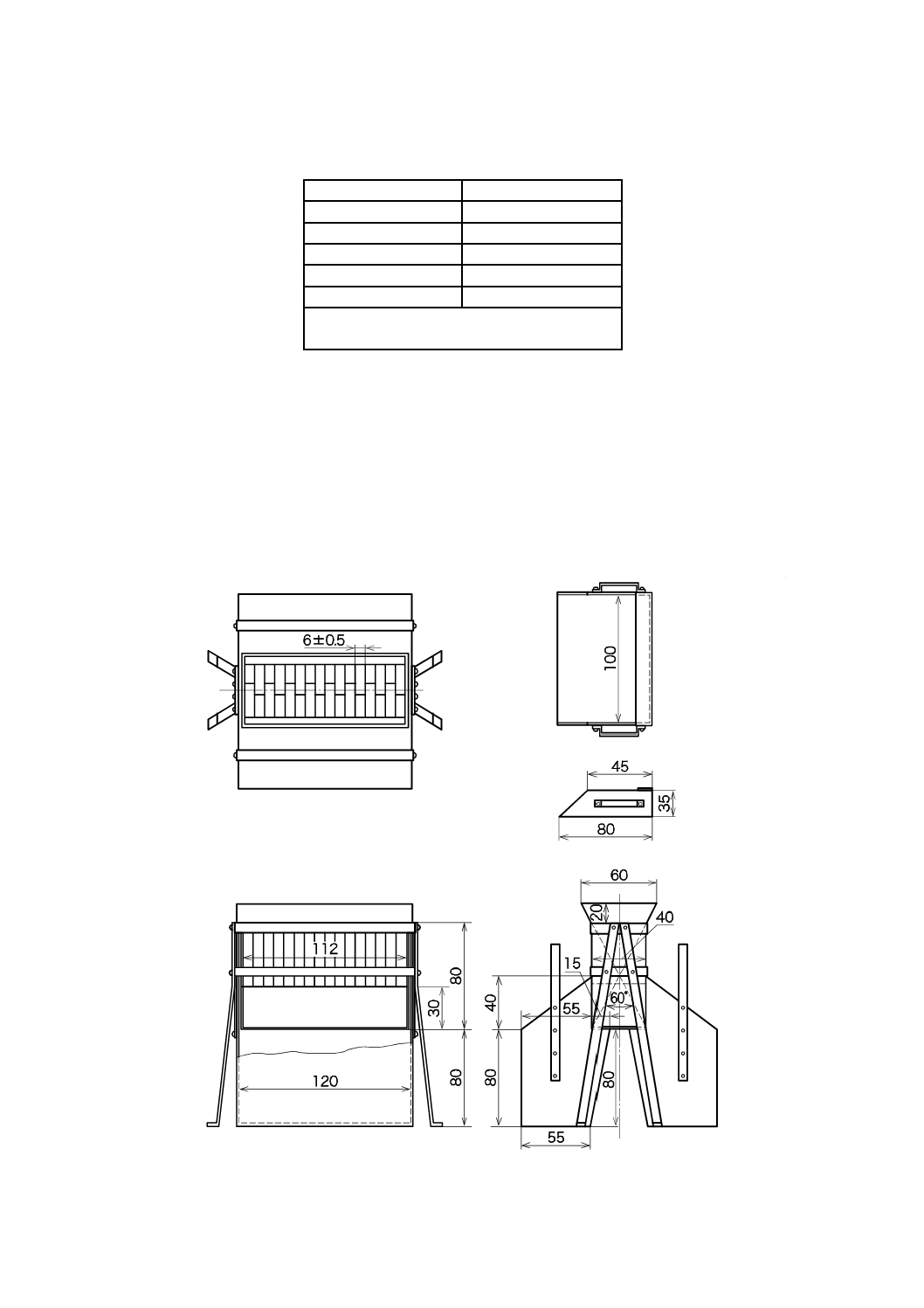
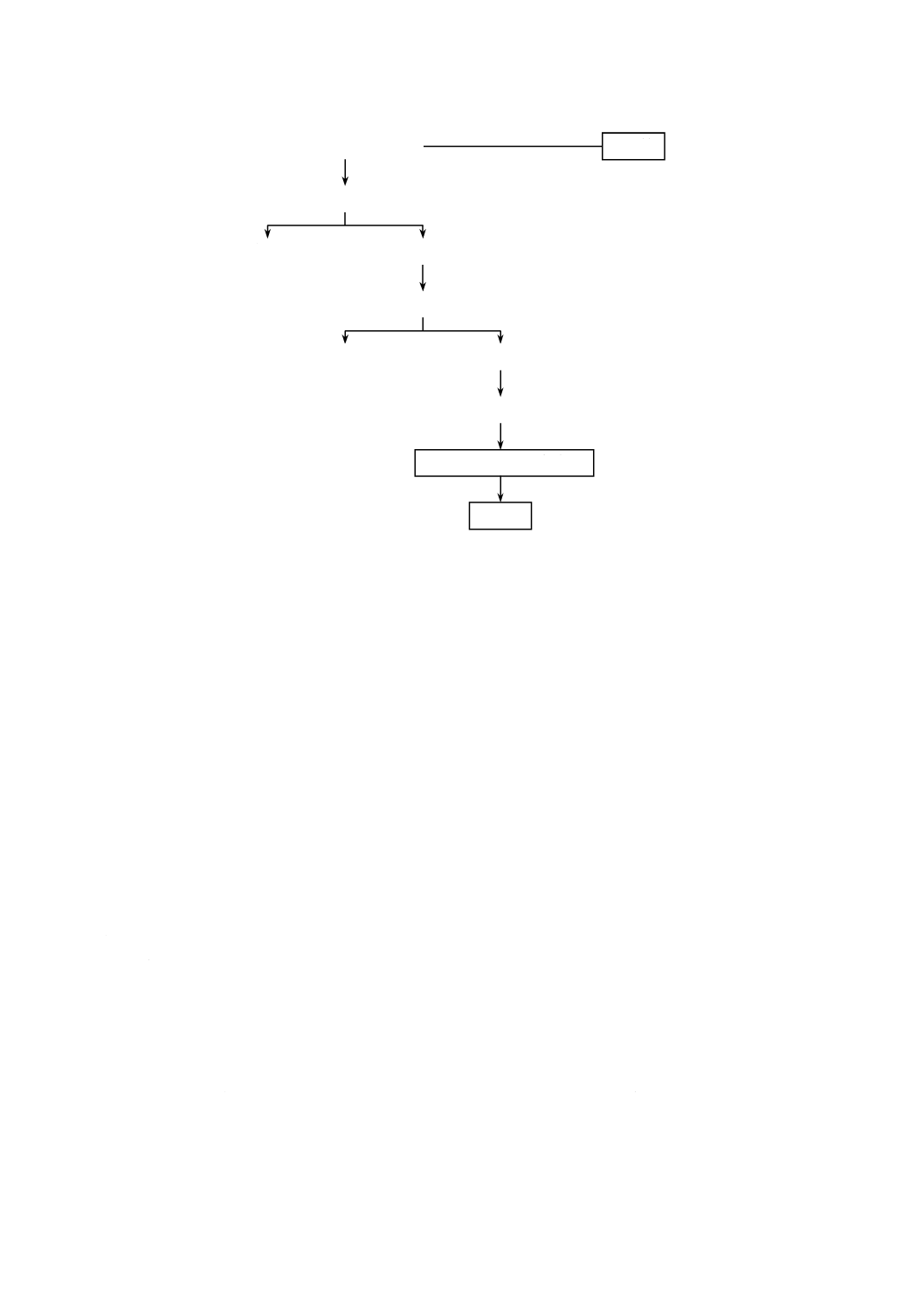
いて,中央にシリコーンゴム栓を装着できる三つ口フラスコとする。
容器の寸法の概要を図4a)に示す。内容積は約25 mLが適切とする。反応容器をマグネチックスタ
ーラ上に設置する。その一例を図4b)に示す。マグネチックスターラの上に設置台を置く。この場合,
この設置台は,コルク板に反応容器の底部が入る穴をあけたものとする。反応容器を設置台上に置き,
容器内にスターラピースを入れる。スターラピースを十分に回転させるには,反応容器の底面がマグ
ネチックスターラ上の5 mm以内にあることが望ましい。
単位 mm
40
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
1 マグネチックスターラ
2 設置台
3 反応容器
4 シリコーンゴム栓
5 スターラピース
6 ボールジョイント(入口)
7 ボールジョイント(出口)
8 試料導入器
a) 寸法の概略
b) 装置の一例
図4−反応容器
7.13.1.4 操作
操作は,次による。
a) 装置の準備
1) カラム ガスクロマトグラフ用シリカゲル(170〜250 μm)を充てんし,200 ℃で5時間空焼きを
行う。
2) 脱水・脱塩化水素管 脱水剤として過塩素酸マグネシウム(元素分析用試薬,粒状)と脱塩化水素
剤として活性炭(粒状)をそれぞれ40 mm充てんする。管の両端及び両剤の間には石英ウールを詰
める。ガスは過塩素酸マグネシウム側から入り,活性炭側へ出る。
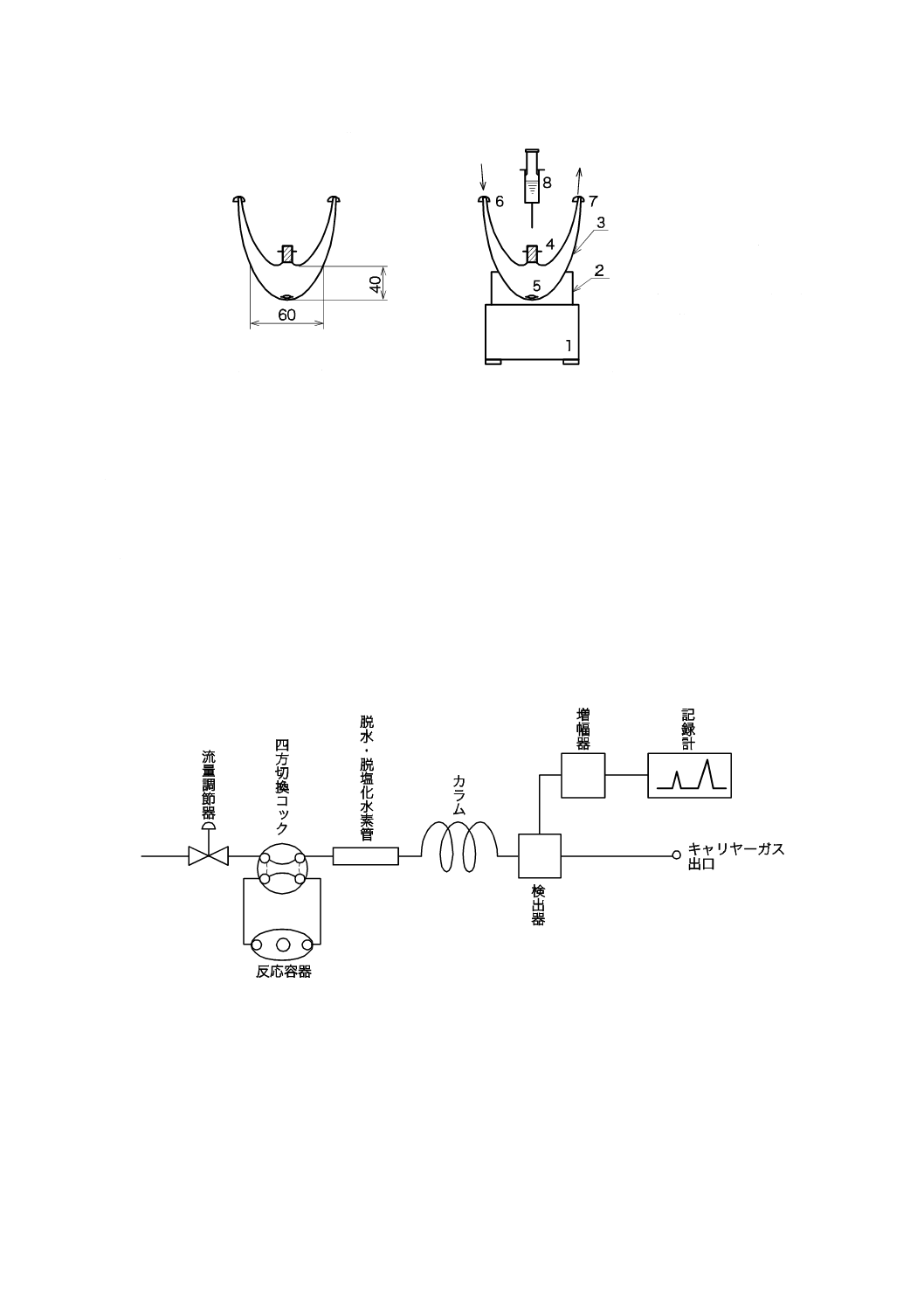
3) 装置の組立 装置の構成例を,図5に示す。装置の組立は図6のように行い,接続部はすべてふっ
素樹脂製チューブ(内径1.0 mm,外径1.5 mm)を用いて気密に接続する。
図5−装置構成(例)
41
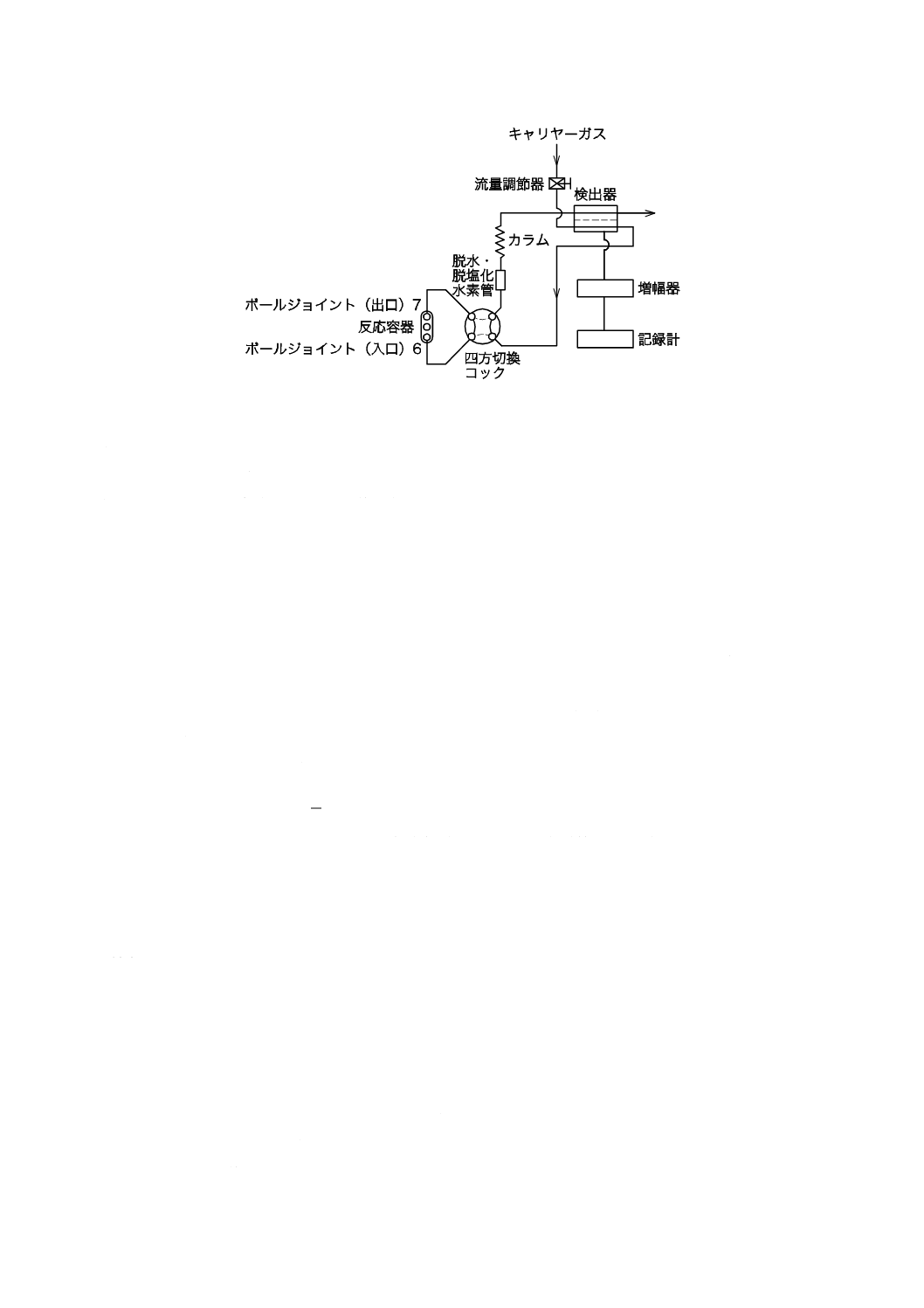
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図6−装置の組立
b) 調整 キャリヤーガスが反応容器を通らないで,直接カラム管へ流れるように四方切換コックを設定
し,キャリヤーガスを60 mL/minの速度で流し,室温で熱伝導検出器を安定させる。
c) 検量線の作成 炭酸ナトリウム(無水)の0 mg,2.5 mg,5 mg,10 mg,15 mg及び20 mgを0.01 mg
まではかりとり,スターラピースを入れた反応容器に入れる。シリコーンゴム栓をし,ボールジョイ
ントをキャリヤーガスの流路に接続する。シリコーンゴム栓を通じて,試料導入器で塩酸 (1+3) 5 mL
を反応容器内に滴下する。試料導入器を抜き取り,マグネチックスターラで30秒間かき混ぜる。
炭酸塩は塩酸によって分解されて二酸化炭素を発生するから,次に四方切換コックを切り換えて,
反応容器内の二酸化炭素を含むガスをキャリヤーガスでカラムに導入する。2分30秒後切換コックを
元に戻し,反応容器を取り外す。得られたクロマトグラムからピーク面積を求め,検量線を作成する。
d) 測定 炭酸ナトリウムとして2.5〜15 mgを含むように試料をはかりとり,以下,検量線の作成と同様
に操作してピーク面積を求め,検量線から炭酸ナトリウムの量 (mg) を求める。
7.13.1.5 計算
炭酸塩は,次の式によって算出する。
100
×
S
A
S=
ここに, C: 炭酸塩(Na2CO3として)(質量分率%)
A: 検量線から求めた炭酸ナトリウム (mg)
S: 試料の質量 (mg)
a) この方法の繰返しの測定精度の相対標準偏差は,炭酸ナトリウム自体の場合は1.5 %程度,洗剤中に
5〜10 %含まれる炭酸ナトリウムの場合は3 %程度である。
7.13.2 中和滴定法
7.13.2.1 要旨
試料に塩酸を加えて炭酸塩を分解し,発生した二酸化炭素を炭酸バリウムとして固定化し,過剰の水酸
化バリウムを1 mol/L塩酸で滴定し,炭酸塩の量を求める。
7.13.2.2 試薬
試薬は,次による。
7.13.2.2.1 1 mol/L塩酸 JIS K 8001の4.5 (5.1)(1 mol/L塩酸)の規定による。
7.13.2.2.2 0.1 mol/L水酸化バリウム溶液 JIS K 8577に規定する水酸化バリウム八水和物31.5 gに水を加
え1 000 mLとし,約70 ℃の水浴中で加熱し溶解する。放冷後ろ過し,ソーダ石灰管を付けた試薬瓶に保
42
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
存する。
7.13.2.2.3 塩酸 (1+5) JIS K 8180に規定する塩酸で調製する。
7.13.2.2.4 1-オクタノール JIS K 8213の規定による。
7.13.2.2.5 過酸化水素溶液(質量分率1 %) JIS K 8230に規定する過酸化水素5 mLを水で150 mLと
する。
7.13.2.2.6 フェノールフタレイン溶液 (10 g/L) JIS K 8001の4.4(指示薬)の規定による。
7.13.2.3 装置及び器具
装置及び器具は,次による。
a) 二酸化炭素固定化装置(図7参照)
b) ガス洗浄瓶 250 mL
c) ビュレット 25 mL
7.13.2.4 操作
操作は,次による。
a) 炭酸塩として0.1〜0.8 gを含むように試料を二酸化炭素発生管(250 mLのガス洗浄瓶)に0.1 mgまで
はかりとり,水100 mLを加えて溶解又は分散させる。
なお,試料が亜硫酸ナトリウムを含む試料の場合には,水90 mL及び過酸化水素溶液(質量分率1 %)
10 mLを加えて約3分間振とうし,溶解させる。
b) 循環するとき,発泡する試料には1-オクタノールを消泡剤として十数滴加え,その影響を除く。
c) 別に,乾燥した二酸化炭素吸収管(250 mLのガス洗浄瓶)に0.1 mol/L水酸化バリウム溶液100 mL
をとり,1-オクタノール十数滴を加える。
d) 図7のように装置を取り付け,すり合わせ部分をよく確認し,空気ポンプによって循環流量約2 L/min
で装置内の空気を循環させながら,二酸化炭素発生管に三方コックを通し,注射器によって塩酸 (1
+5) 20 mLを加える。
e) 三方コックを切り換え,更に5分間循環を続ける。二酸化炭素吸収管を取り外し,内壁を水で洗浄し
た後,フェノールフタレインを指示薬として加え,過剰の水酸化バリウムを1 mol/L塩酸で滴定する。
f)
同時に空試験として0.1 mol/L水酸化バリウム溶液100 mLを滴定する。
7.13.2.5 計算
炭酸塩は,次の式によって算出する。
S
K
f
A
B
S
K
f
A
B
C
1.0
)
(
100
000
1
)
(
×
×
×
−
×
×
×
×
−
=
=
ここに,
C: 炭酸塩(Na2CO3として)(質量分率%)
B: 空試験の滴定に用いた1 mol/L塩酸の量 (mL)
A: 試料の滴定に用いた1 mol/L塩酸の量 (mL)
f: 1 mol/L塩酸のファクター
S: 試料の質量 (g)
k: 係数 Na2CO3のとき53.00
NaHCO3のとき42.05
CaCO3のとき50.05
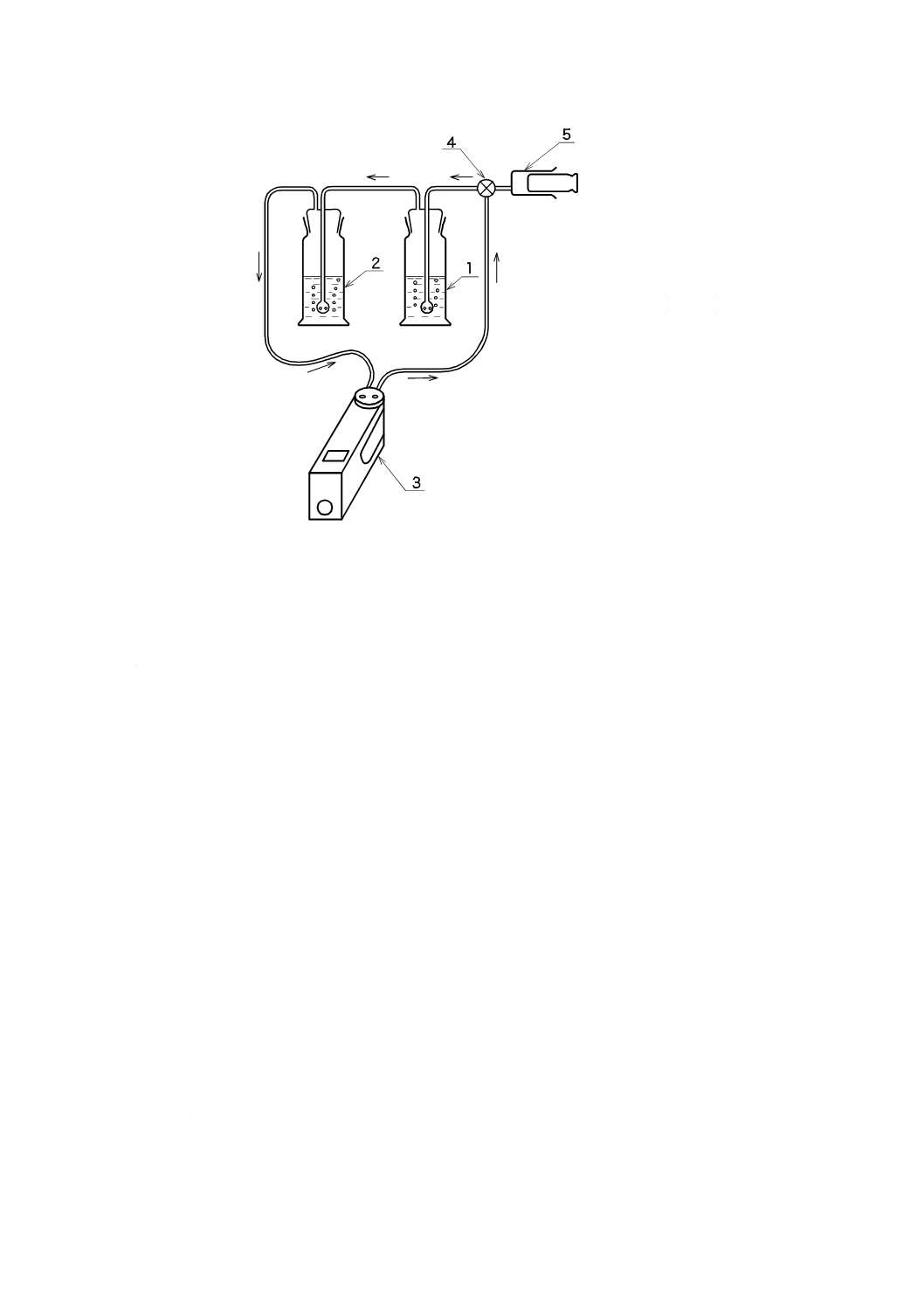
43
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1 二酸化炭素発生管(250 mL)
2 二酸化炭素吸収管(250 mL)
3 空気ポンプ
4 三方コック
5 注射器(50 mL)
図7−二酸化炭素固定化装置
7.13.3 炭酸ガス電極法
7.13.3.1 要旨
試料溶液を酸性にして炭酸塩を分解し,生成した二酸化炭素を炭酸ガス電極を用いて測定し,炭酸塩と
して求める。
7.13.3.2 試薬
試薬は,次による。
7.13.3.2.1 塩酸 JIS K 8180の規定による。
7.13.3.2.2 緩衝液 JIS K 8288に規定するくえん酸三ナトリウム二水和物294 gに水を加えて1 000 mLと
し,JIS K 8180に規定する塩酸を加えてpH値を4.5に調整する。
7.13.3.2.3 溶存気体を含まない水 イオン交換水又は蒸留水をフラスコに入れ,5分間沸騰させ,溶存気
体を除去した後,室温まで冷却する。
7.13.3.2.4 炭酸ナトリウム 7.13.1.2.2 による。
7.13.3.2.5 標準液 炭酸ナトリウム0.963 8 gを水に溶解し,全量フラスコで200 mLとする。この溶液を,
全量ピペットで,2 mL,10 mL及び20 mLをそれぞれ全量フラスコ100 mLにとり,水を標線まで加えて
混合し標準液とする。この溶液1 000 mL中に,二酸化炭素として,ぞれぞれ40 mg,200 mg及び400 mg
を含む。
7.13.3.3 装置及び器具
装置及び器具は,次による。
a) 炭酸ガス電極
b) 電位差計 入力抵抗が1012 Ω以上で,1 mVまで読み取ることができる電位差計。
c) pHメーター
d) マグネチックスターラ
44
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
e) 恒温水槽
7.13.3.4 操作
操作は,次による。
a) 測定の準備
1) 試料の適量(CO2として0.02〜0.20 gを含む。)を0.1 mgまではかりとり,水に溶解して全量フラス
コで500 mLとする。これを試験液とする。
2) 標準液,試験液及び緩衝液を25 ℃の恒温水槽に入れ30分以上放置し,液温を25 ℃にする。
3) 装置を図8のように組み立て,300 mLのビーカーに溶存気体を含まない水を約200 mLとスターラ
ピースを入れ,装置にセットする。スターラを回転させ,電位が一定になるまで待つ。電位が一定
になったとき,その電位を記録し,スターラの回転を止めておく。
b) 検量線の作成
1) ビーカー100 mLに標準液50 mLを全量ピペットでとり,スターラピースを入れて装置にセットす
る。このとき,電極の下端を液面から約10 mm下に浸す。
2) 25 ℃の緩衝液5 mLを全量ピペットで加え,直ちにスターラを回転させ,同時に時間を計測する。
このとき,回転速度は,気泡を巻き込まない程度の速さに調節する。
3) 緩衝液を加え終わってから30秒以内に,少量の塩酸を加えてpH値を5.0に調整し,2分後に電位
を読み取る。
4) 電極を水で洗浄した後,元のビーカーをセットしてスターラを回転させ,a) で記録した電位の90 %
以上回復した後,次の測定を行う。
なお,電位の回復が緩慢になった場合は,溶存気体を含まない水を取り換える。
5) 横軸(対数)に濃度,縦軸(リニア)に電位をとり,検量線を作成する。
c) 測定 ビーカー100 mLに試験液50 mLを全量ピペットでとる。以下,検量線の作成と同様に操作し
て電位を測定し,検量線から二酸化炭素の量を求める。
7.13.3.5 計算
炭酸塩の量は,次の式によって算出する。
S
K
A
K
S
A
C
05
.0
100
000
1
000
1500
×
×
×
×
×
×
=
=
ここに,
C: 炭酸塩(質量分率%)
A: 検量線から求めた二酸化炭素の濃度 (mg/L)
S: 試料の質量 (g)
K: CO2を炭酸塩に換算するための係数
Na2CO3のとき2.41
NaHCO3のとき1.91
注記 酸性下で,炭酸ガス以外の酸性ガスを発生する試料は,定量値が高めになる。
45
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1 炭酸ガス電極
2 電位差計
3 pH電極
4 pHメーター
5 ビーカー
6 マグネチックスターラ
図8−二酸化炭素測定装置
7.14 塩化物の定量
7.14.1 要旨
試料の水溶液に一定過剰量の硝酸銀溶液を加え,過剰の硝酸銀を硫酸アンモニウム鉄 (III) 溶液を指示
薬としてチオシアン酸アンモニウム溶液で逆滴定することによって,塩化物の量を求める。
7.14.2 試薬
試薬は,次による。
7.14.2.1 硝酸 JIS K 8541の規定による。
7.14.2.2 0.1 mol/L硝酸銀溶液 JIS K 8001の4.5 (14)(0.1 mol/L硝酸銀溶液)の規定による。
7.14.2.3 0.1 mol/Lチオシアン酸アンモニウム溶液 JIS K 8001の4.5 (20)(0.1 mol/Lチオシアン酸アンモ
ニウム溶液)の規定による。
7.14.2.4 硫酸アンモニウム鉄 (III) 溶液 (100 g/L) JIS K 8982に規定する硫酸アンモニウム鉄 (III)・12
水10 gを水約50 mLに溶解し,硫酸約5 mLを加えて酸性とし,更に水を加えて100 mLとする。
7.14.2.5 ニトロベンゼン JIS K 8723に規定するニトロベンゼンを,試験直前に蒸留して用いる。
7.14.2.6 1-オクタノール JIS K 8213に規定するもの。
7.14.3 器具
器具は,次による。
a) 共栓付き三角フラスコ 500 mL
7.14.4 操作
操作は,次による。
a) 試料約2 gを共栓付き三角フラスコ500 mLに0.1 mgまではかりとり,水100 mLを加えて溶解する。
b) 硝酸10 mLを加えて約10分間静かに煮沸する。著しく発泡する場合は,少量の1-オクタノールを添
加して泡を消す。
c) 冷却後,0.1 mol/L硝酸銀溶液25 mLを全量ピペットで加え,更にニトロベンゼン5 mLを加えて激し
く振とうする。
d) 硫酸アンモニウム鉄 (III) 溶液 (100 g/L) 5 mLを指示薬として加え,過剰の硝酸銀を0.1 mol/Lチオシ
アン酸アンモニウム溶液でうすい紅色になるまで滴定し,同時に空試験を行う。
7.14.5 計算
塩化物は,次の式によって算出する。
46
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S
f
A
B
S
f
A
B
C
4
584
.0
)
(
100
000
1
1.0
44
.
58
)
(
×
×
−
×
×
×
×
×
−
=
=
ここに,
C: 塩化物(NaClとして)(質量分率%)
B: 空試験の滴定に用いた0.1 mol/Lチオシアン酸アンモニウム
溶液の量 (mL)
A: 試料の滴定に用いた0.1 mol/Lチオシアン酸アンモニウム溶
液の量 (mL)
S: 試料の質量 (g)
F: 0.1 mol/Lチオシアン酸アンモニウム溶液のファクター
58.44: NaClの化学式量
7.15 ゼオライトの定量
7.15.1 要旨
鉱酸酸性でゼオライトが溶解し,生成したアルミニウムイオンをEDTAとの錯体を形成させ,過剰の
EDTAを酢酸亜鉛を用いて逆滴定することによって,アルミニウムを定量し,試料中のゼオライトを求め
る。測定は,キシレノールオレンジ法(A法)又はメチルチモールブルー法(B法)のいずれかによる。
7.15.2 試薬
試薬は,次による。
7.15.2.1 キシレノールオレンジ法(A法) 次の試薬を使用する。
7.15.2.1.1 硝酸 JIS K 8541の規定による。
7.15.2.1.2 水酸化ナトリウム溶液 (200 g/L) JIS K 8576に規定する水酸化ナトリウム20 gを水に溶解し
て100 mLとする。
7.15.2.1.3 酢酸ナトリウム溶液 (82 g/L) JIS K 8372に規定する酢酸ナトリウム8.2 gを水に溶解して100
mLとする。
7.15.2.1.4 酢酸アンモニウム溶液 (77 g/L) JIS K 8359に規定する酢酸アンモニウム7.7 gを水に溶解して
100 mLとする。
7.15.2.1.5 0.01 mol/L EDTA2Na溶液 JIS K 8001の4.5 (3.3)(0.01 mol/L EDTA2Na溶液)の規定による。
7.15.2.1.6 0.01 mol/L酢酸亜鉛溶液 JIS K 8001の4.5 (9)(0.01 mol/L酢酸亜鉛溶液)の規定による。
7.15.2.1.7 キシレノールオレンジ溶液 (1 g/L) JIS K 9563に規定するキシレノールオレンジ0.1 gを水に
溶解して100 mLとし,褐色ガラス瓶に保存する。
7.15.2.1.8 ふっ化ナトリウム JIS K 8821の規定による。
7.15.2.2 メチルチモールブルー法(B法) 次の試薬を使用する。
7.15.2.2.1 塩酸 (1 mol/L) JIS K 8180に規定する塩酸10 mLと水110 mLとを混合する。
7.15.2.2.2 エタノール (95) JIS K 8102の規定による。
7.15.2.2.3 水酸化ナトリウム溶液 (40 g/L) JIS K 8576に規定する水酸化ナトリウム4 gを水に溶解して
100 mLとする。
7.15.2.2.4 水酸化ナトリウム溶液 (200 g/L) 7.15.2.1.2による。
7.15.2.2.5 ヘキサメチレンテトラミン JIS K 8847の規定による。
7.15.2.2.6 メチルオレンジ溶液 JIS K 8001の4.4(指示薬)による。褐色ガラス瓶に保存する。
7.15.2.2.7 0.1 mol/L EDTA2Na溶液 JIS K 8001の4.5 (3.1)(0.1 mol/L EDTA2Na溶液)による。
7.15.2.2.8 0.1 mol/L 酢酸亜鉛溶液 JIS K 8001の4.5 (9)(0.01 mol/L酢酸亜鉛溶液)による。この場合,
酢酸亜鉛二水和物23 g,酢酸2 mLを用いる。標定はJIS K 8001の4.5 (9) (b)によって行う。
47
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.15.2.2.9 メチルチモールブルー混合試薬 JIS K 9552に規定するメチルチモールブルー0.1 gとJIS K
8150に規定する塩化ナトリウム10 gを乳鉢で粉砕混和する。
7.15.3 器具
器具は,次による。
a) キシレノールオレンジ法(A法) 次の器具を使用する。
1) ビーカー 200 mL及び500 mL
2) ビュレット 10 mL
3) 全量ピペット 10 mL及び25 mL
4) 全量フラスコ 500 mL
5) pHメーター
6) マグネチックスターラ
b) メチルチモールブルー法(B法) 次の器具を使用する。
1) ビーカー 200 mL
2) ビュレット 10 mL及び30 mL
3) 全量ピペット 25 mL及び50 mL
4) 全量フラスコ 100 mL
5) ウォーターバス
6) 遠心分離器
7) 沈殿管 容量50 mLで,ガラス製で丸底の沈殿管。
8) pHメーター
9) マグネチックスターラ
7.15.4 操作
操作は,次による。
a) キシレノールオレンジ法(A法)
1) 測定
1.1) 試料の適量(ゼオライトとして約200 mgを含む。)をビーカー500 mLに0.1 mgまではかりとり,
水200 mL及び硝酸25 mLを加え,15分間煮沸した後室温まで冷やし,全量フラスコ500 mLに移
して水を標線まで加える。この溶液をろ過し,ろ液を全量ピペット25 mLを用いてビーカー200 mL
にとり,試料溶液とする。
1.2) 水を約30 mL加えた後,試料溶液のpH値が2.0〜2.5になるようにかき混ぜながら,水酸化ナト
リウム溶液 (200 g/L) を加え,次いで,酢酸ナトリウム溶液 (82 g/L) を加えてpH値を3.0〜3.5
に調整する。
1.3) 0.01 mol/L EDTA2Na溶液を全量ピペット10 mLを用いて加え10分間煮沸し冷却した後,酢酸アン
モニウム溶液 (77 g/L) 20 mLを加え,水を加えて全量を150 mLとし,キシレノールオレンジ溶液
を4,5滴加え,マグネチックスターラでかくはんしながら0.01 mol/L酢酸亜鉛溶液で滴定する。
1.4) 溶液の色が黄から赤に変わる点を終点とする。
1.5) さらに,ふっ化ナトリウム500 mgを加え,約10分間煮沸する。
1.6) 冷却後,再び0.01 mol/L酢酸亜鉛溶液で滴定する。終点は溶液の色が黄から赤に変わる点とする。
2) 計算 ゼオライトは,次の式によって算出する。
48
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S
f
A
S
f
A
C
652
.3
25
500
100
767
.6
98
.
26
100
1
000
11
×
×
×
×
×
×
×
×
×
=
=
ここに,
C: ゼオライト (Na12Al12Si12O48・27H2O)(質量分率%)
A: ふっ化ナトリウム処理後の滴定に用いた0.01 mol/L酢酸亜鉛
溶液の量 (mL)
S: 試料の質量 (g)
f: 0.01 mol/L酢酸亜鉛溶液のファクター
26.98: Alの原子量
6.767: Alからゼオライトへの換算係数
2.1) この方法では,ニトリロトリアセテートのような一部の有機ビルダーの妨害を受ける。
2.2) 鉄,銅などの金属及びEDTAの妨害を除くため,滴定の終了した試料溶液にふっ化ナトリウムを
加えた後,再び0.01 mol/L酢酸亜鉛溶液によって滴定を行うが,一般の洗剤に含まれる重金属類
は微量であり,必ずしもふっ化ナトリウム処理を行う必要はない。この場合,同時に空試験を行
い,a) 1.5)のふっ化ナトリウム処理以降の操作を省略する。
2.3) 空試験を行うときのゼオライトは,次の式によって算出する。
S
f
V
B
S
f
V
B
C
652
.3
)
(
25
500
100
767
.6
98
.
26
1001
000
11
)
(
×
×
−
×
×
×
×
×
×
×
−
=
=
ここに,
C: ゼオライト(質量分率%)
B: 空試験の滴定に用いた0.01 mol/L酢酸亜鉛溶液の量 (mL)
V: 逆滴定に用いた0.01 mol/L酢酸亜鉛溶液の量 (mL)
S: 試料の質量 (g)
f: 0.01 mol/L酢酸亜鉛溶液のファクター
b) メチルチモールブルー法(B法)
1) 測定
1.1) 試料の適量(ゼオライトとして0.2〜0.55 gを含む。)を沈殿管50 mLに0.1 mgまではかりとり,
エタノール (95) 35 mLを加え,ウォーターバスに浸してガラス棒でかくはんしながらエタノール
可溶成分を加温溶解した後,4 000 min−1で遠心分離し,上澄液を傾斜して捨てる。次いで,水35
mLを加えて同様の操作を2回行う。
1.2) 塩酸 (1 mol/L) 35 mLを加え,ウォーターバスに浸してガラス棒でかくはんしながらゼオライトを
加温溶解した後,4 000 min−1で遠心分離し,上澄液を傾斜してビーカー100 mLに移す。この操作
を更に1回繰り返し,溶液を合わせる。
1.3) 室温まで冷却した後,全量フラスコ100 mLに移し標線まで水を加えて試料溶液とする。
1.4) 試料溶液を全量ピペット50 mLでビーカー200 mLにとり,メチルオレンジ溶液を1,2滴加え,
水酸化ナトリウム溶液 (200 g/L) で中和する。
1.5) 0.1 mol/L EDTA2Na溶液を全量ピペット25 mLを用いて加え,塩酸 (1 mol/L) 又は水酸化ナトリウ
ム溶液 (40 g/L) でpH値を3に調整した後,時計皿でふたをし,20分間煮沸して錯体を形成させ
る。
1.6) 室温まで冷却した後ヘキサメチレンテトラミン10 gを加え,水で全量を約100 mLとし,メチル
チモールブルー混合試薬50〜100 mgを添加して,マグネチックスターラでかくはんしながら0.1
49
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
mol/L酢酸亜鉛溶液で滴定する。終点は,桃色を経て青色となった点とする。
1.7) 別に試料溶液の代わりに塩酸 (1 mol/L) 35 mLを用いて空試験を行う。
2) 計算 ゼオライトの量は,次の式によって算出する。
S
f
A
B
S
f
A
B
C
651
.3
)
(
100
100
50
000
1
8.
323
0.
191
2
698
.2
)
(
1
1
×
×
−
×
×
×
×
×
×
−
=
=
ここに,
C: ゼオライト (Na12Al12Si12O48・27H2O)(質量分率%)
B: 空試験の滴定に用いた0.1 mol/L酢酸亜鉛溶液の量 (mL)
A: 試料溶液の滴定に用いた0.1 mol/L酢酸亜鉛溶液の量 (mL)
f1: 0.1 mol/L酢酸亜鉛溶液のファクター
S: 試料の質量 (g)
2.1) 試料採取量は,4 g以下とする。
7.16 蛍光増白剤の確認試験
蛍光増白剤の確認は,染着法又は直接法のいずれかによって試験する。
7.16.1 染着法
7.16.1.1 要旨
試料の水溶液にろ紙を浸し,ろ紙に蛍光増白剤を染着させてそのろ紙に紫外線ランプを照射し,蛍光の
有無を確認する。
7.16.1.2 器具
器具は,次による。
a) 紫外線ランプ 波長360 nm付近の紫外線を発生するランプ。
b) ろ紙片 JIS P 3801に規定する5種Aのろ紙を幅約25 mm,長さ50〜70 mmに切ったろ紙片。
7.16.1.3 操作
操作は,次による。
a) 試験の直前に,水に溶解した試料溶液 (2 g/100 mL) 100 mLを40 ℃に保ち,この中にろ紙片を入れ30
分間浸す。
b) このろ紙片を40 ℃の温水100 mLに5分間浸して洗う。この洗浄操作を2回繰り返した後,ろ紙を取
り出し,別の新しいろ紙に挟んで,ろ紙片の表面の水分を除く。
c) 暗室,又は暗箱内で紫外線ランプを用い,約200〜300 mmの距離から照射し,水に浸したろ紙と比較
し,明るく輝くときは蛍光増白剤が存在する。
7.16.2 直接法
7.16.2.1 要旨
試料に直接紫外線ランプを照射し,蛍光の有無を検出する。この方法は,主に粉状及び粒状合成洗剤に
用いる。
7.16.2.2 器具
器具は,次による。
a) 紫外線ランプ 7.16.1.2 a) による。
7.16.2.3 操作
試料を黒紙上に平らに広げ,暗室又は暗箱内で紫外線ランプを用い,約200〜300 mmの距離から照射す
る。蛍光増白剤が含まれるときは,その部分が明るく輝く。
なお,直接法は,迅速法であるが,天然物の一部にも蛍光を発するものがあるので,蛍光増白剤を含ま
50
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ないものを,含むものと誤認するおそれがある。わずかな蛍光を認めた場合には,染着法で確認する。ま
た,判定を行うときには,紫外線が直接眼に入らないように注意する。
7.17 ひ素 (As) の限度試験
7.17.1 要旨
この方法は,試料中のひ素をひ化水素として発生させ,N,N-ジエチルジチオカルバミド酸銀のピリジン
溶液に吸収させて赤紫に発色させ,その程度をひ素標準液と比較して判定する限度試験法とする。
7.17.2 試薬
試薬は,次による。
7.17.2.1 しゅう酸アンモニウム飽和溶液 JIS K 8521に規定するしゅう酸アンモニウム一水和物を用い
て調製する。
7.17.2.2 アンモニア水 JIS K 8085の規定による。
7.17.2.3 塩酸 (1+2) JIS K 8180に規定するひ素分析用塩酸を用いて調製する。
7.17.2.4 水酸化ナトリウム (200 g/L) JIS K 8576に規定する水酸化ナトリウムを用いて調製する。
7.17.2.5 よう化カリウム溶液 JIS K 8913に規定するよう化カリウム16.5 gを水に溶解して100 mLとし,
遮光して保存する。
7.17.2.6 酸性塩化すず (II) 溶液 JIS K 8136に規定する塩化すず (II) 二水和物4 gをJIS K 8180に規定
する塩酸125 mLに溶解し,水を加えて250 mLとし,共栓瓶に入れ密栓して保存する。調製後1か月以内
に用いる。
7.17.2.7 酢酸鉛 (II) 溶液 JIS K 8374に規定する酢酸鉛 (II) 三水和物11.8 gを水に溶解して100 mLと
し,JIS K 8355に規定する酢酸2滴を加える。密栓して保存する。
7.17.2.8 亜鉛粉末 JIS K 8013に規定するひ素分析用亜鉛(粒度約800 μm)を用いる。
7.17.2.9 N,N-ジエチルジチオカルバミド酸銀ピリジン溶液 (5 g/L) JIS K 9512に規定するN,N-ジエチル
ジチオカルバミド酸銀0.5 gをJIS K 8777に規定するピリジンに溶解して100 mLとする。遮光した共栓瓶
に入れ,冷所に保存する。
7.17.2.10 ひ素標準液 (Asとして0.000 75 mg/mL) JIS K 8005の9.4に規定する酸化ひ素 (Ⅲ) を微細な
粉末とし,105±2 ℃で4時間乾燥し,その0.100 gを水酸化ナトリウム溶液 (200 g/L) 5 mLに溶解する。
この溶液を,JIS K 8951に規定する硫酸を用いて調製した硫酸 (1+15) で中和し,更に硫酸 (1+15) 10
mLを加え,煮沸後,冷却した水を加えて,全量フラスコ1 000 mLに移し入れ,水を標線まで加えて原液
とする。
全量ピペットを用いて,原液10 mLを全量フラスコ1 000 mLにとり,硫酸 (1+15) 10 mLを加え,煮沸
後,冷却した水を標線まで加えてひ素標準液とする。ひ素標準液は使用時に調製する。
なお,この試験及び試験溶液の調製に用いる試薬は,空試験で発色しないか,又はほとんど発色しない
ものを用いる。
7.17.3 装置及び器具
装置及び器具は,次による。
a) ひ素試験装置 図9に示す。
排気管に約30 mmの高さにガラスウールを詰め,酢酸鉛溶液及び水の等容量の混液で均一に潤し,
管の下端から静かに吸引して過剰の液を除く。これをゴム栓に差し込み,小孔がわずかに出るように
して発生瓶に付ける。排気管の上端にはガラス管を垂直に固定したゴム栓を付ける。ガラス管の排気
管側の下端は,ゴム栓の下端と同一平面とする。
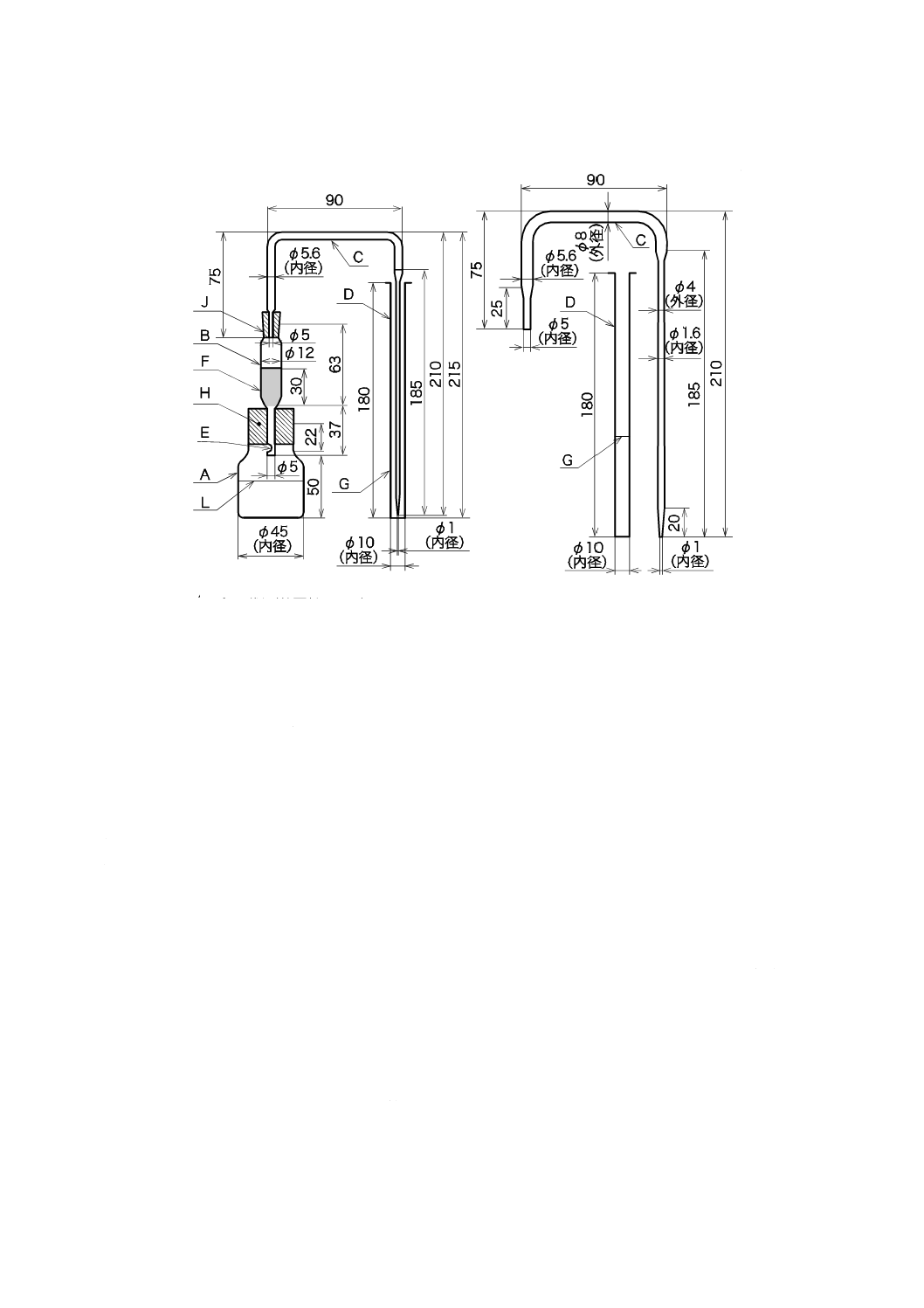
51
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
A:発生瓶(容量約70 mL)
B:排気管
C:ガラス管(内径5.6 mm,吸収管に入れる部分は,先端を内径1 mmに引き伸ばす。)
D:吸収管(内径10 mm)
E:小孔
F:ガラスウール
G:5 mLの標線
H及びI:ゴム栓
L:40 mLの標線
図9−ひ素試験装置
b) 全量フラスコ 50 mL及び20 mL
c) 分解フラスコ ケルダールフラスコ100 mL
7.17.4 操作
操作は,次による。
a) 標準色の調製
1) 発生瓶にひ素標準液 (Asとして0.000 75 mg/mL) 1 mLを入れ,アンモニア水で中和し,塩酸 (1+2)
5 mL及びよう化カリウム溶液5 mLを加えて2〜3分間放置した後,酸性塩化すず (II) 溶液5 mL
を加えて10分間放置する。
2) 次に水を加えて40 mLとし,亜鉛粉末2 gを加え,直ちに排気管及びガラス管を連結したゴム栓を
発生瓶に付ける。ガラス管の細管部の先端はあらかじめN,N-ジエチルジチオカルバミド酸銀ピリジ
ン溶液5 mLを入れたひ化水素吸収管の底まで入れる。
3) 25 ℃の水中に発生瓶を肩まで浸し,1時間放置する。
4) 吸収管を外し,必要があればピリジンを加えて5 mLとし,この溶液の発色を標準色とする。この
52
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
発色は,試料6.7 gの水溶液 (6.7 g/L) 中のひ素として0.05 mg/Lに相当する。
b) 試験溶液の調製 試料0.67 gをはかりとり,少量の水で分解フラスコに移す。これにJIS K 8951に規
定する硫酸10 mLを加え,よく混合し,初めは穏やかに加熱し激しい反応が終わった後放冷する。次
にJIS K 8541に規定する硝酸5 mLを加え,白煙が発生するまで加熱する。溶液がなお褐色になると
きは,冷却後硝酸5 mLを追加して加熱する。この操作を溶液が無色又はうすい黄色となるまで繰り
返す。冷却後,全量フラスコ50 mLに溶液を移し,分解フラスコを少量の水で洗い,全量フラスコに
加え,水を標線まで加える。この溶液を前処理溶液とする。
なお,この前処理溶液は,7.18の重金属の定量にも用いる。この前処理溶液30 mLを分解フラスコ
にとり,しゅう酸アンモニウム飽和溶液10 mLを加え,再び白煙が発生するまで加熱する。冷却後,
全量フラスコ20 mLに移し,分解フラスコを少量の水で洗い標線まで水を加えて試験溶液とする。
c) 測定 発生瓶に試験溶液5 mLをとり,標準液の場合と同様にa) の操作を行う。続いてa) 及びc) の
操作を同時に行う。
7.17.5 判定
7.17.4 c) で得た発色は,7.17.4 a)で得た発色よりも濃くてはならない。
7.18 重金属(Pbとして)の限度試験
7.18.1 要旨
この方法は,試料中の重金属を酢酸酸性で硫化ナトリウム溶液と反応させ,生じた有色硫化物の着色を
鉛標準液のそれと比較して判定する限度試験法とする。
7.18.2 試薬
試薬は,次による。
7.18.2.1 塩酸 (1+3) JIS K 8180に規定する塩酸を用いて調製する。
7.18.2.2 アンモニア水 (1+2) JIS K 8085に規定するアンモニア水を用いて調製する。
7.18.2.3 酢酸 (1+15) JIS K 8355に規定する酢酸を用いて調製する。
7.18.2.4 フェノールフタレイン溶液 (10 g/L) JIS K 8799に規定するフェノールフタレインを用いて調製
する。
7.18.2.5 鉛標準液 (Pbとして0.01 mg/mL) JIS K 8001の4.3(標準液)表5に規定する鉛標準液で,ジ
チゾン用を用いる。原液及び標準液の調製並びに保存には可溶性鉛塩を含まないガラス器具を用いる。
7.18.2.6 硫化ナトリウム溶液 JIS K 8949に規定する硫化ナトリウム九水和物5 gを水10 mL及びJIS K
8295に規定するグリセリン30 mLの混液に溶解する。遮光した小瓶にほとんど全量満たし,密栓して保存
する。調製後3か月以内に使用する。
7.18.3 器具
器具は,次による。
a) 石英製蒸発皿
b) 共栓付きネスラー管 50 mL
7.18.4 操作
操作は,次による。
a) 比較標準液の調整 鉛標準液 (Pbとして0.01 mg/mL) 2 mLを別の共栓付きネスラー管にとり酢酸 (1
+15) 2 mL及び水を加えて50 mLとし,これを比較標準液とする。ただし,鉛標準液2 mLによって
得られる色は,試料6.7 gの水溶液 (6.7 g/L) 中の鉛として1.0 mg/Lに相当する。
b) 試験溶液の調整 7.17.4 b) で得た前処理溶液10 mLを石英製蒸発皿にとり,水浴上で加熱して水分を
53
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
蒸発させた後,直火上で乾固する。乾固が不十分なときは残分にJIS K 8951に規定する硫酸1 mLを
加え,引き続き加熱してほとんど白色となるまで乾固する。冷却後,塩酸2 mL及びJIS K 8541に規
定する硝酸0.5 mLを加え,水浴上で蒸発乾固する。次に,塩酸 (1+3) 1 mL及び水15 mLを加え,加
温して溶解する。冷却後,フェノールフタレイン溶液を1滴加え,液がわずかに紅色に着色するまで
アンモニア水 (1+2) を滴下し,酢酸 (1+15) 2 mLを加え,必要があればろ過し,ろ液を共栓付きネ
スラー管に入れ,水を加えて50 mLとし,試験溶液とする。
注記 pH値を3.5〜4.0に保つためのもので,これよりpH値が低いと硫黄が析出しやすく,高いと
鉄による妨害を受けることがある。
c) 測定 共栓付きネスラー管中の比較標準液及び試験溶液に硫化ナトリウム溶液2滴をそれぞれ加えて
よく混合し,5分間放置後,両管を白色板又は白紙を背景として上方及び側面から観察して液の色を
比較する。
注記 規定時間以上放置すると,硫黄が析出して液が濁り,測定できなくなることがある。
7.18.5 判定
試験溶液での着色は,比較標準液での着色よりも濃くてはならない。
7.19 メタノールの限度試験
7.19.1 要旨
試料中に含まれるメタノールを,多孔性高分子物質を充てん剤として用いたガスクロマトグラフ法で分
析し,同一条件でのメタノール標準液のガスクロマトグラムと比較して判定する限度試験法とする。
7.19.2 試薬
試薬は,次による。
7.19.2.1 2-プロパノール JIS K 8839の規定による。
7.19.2.2 メタノール JIS K 8891の規定による。
7.19.3 装置及び器具
装置及び器具は,次による。
a) ガスクロマトグラフ 次に示す主要部分で構成する。
なお,ここに示す各部以外の構造上の規定については,JIS K 0114による。
1) 試料導入部 カラム槽温度より約50 ℃高く保持できるもの。
2) 検出器 水素炎イオン化検出器。
b) 分離カラム 内径3〜4 mm,長さ2 000〜3 000 mmのガラス管に,充てん剤として多孔性高分子物質
(粒度170〜300 μm)を充てんしたカラム。
注記 多孔性高分子物質としては,ポラパックQ (Porapak−Q) などがある。これは,市販製品の一
例である。この情報は,この規格の利用者の便宜のために記載するもので,この製品を推奨
するものではない。
c) キャリヤーガス 窒素又はヘリウム
d) 試料導入器 容量10 μL,目盛0.1〜1 μLのマイクロシリンジ
e) 全量フラスコ 100 mL及び1 000 mL
7.19.4 操作
操作は,次による。
a) ガスクロマトグラフの調整
1) カラム槽温度を130〜150 ℃,試料導入部をカラム槽より約50 ℃高い温度に,検出器温度をカラム
54
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
槽と同程度から約50 ℃高い温度にそれぞれ設定する。
2) マイクロシリンジに1 μLの2-プロパノール溶液 (100 mg/mL) をとり,試料導入部から素早く注入
し,2-プロパノールのピークが8〜10分で現れるように,キャリヤーガス流量及びカラム槽の温度
を調節する。さらに,2-プロパノールのピークの高さができるだけ大きく,しかも記録紙からスケ
ールアウトしないように感度を調節する。また,ピークの半値幅が10 mm以上で,できるだけ二等
辺三角形になるように,記録計の記録紙送り速度を調節する。
b) 試験溶液及び標準液の調製
1) 試験溶液 液状の洗剤の場合は,試料100 gに2-プロパノール10.0 gを加え,よく混合して試験溶
液とする。
2) 標準液 メタノール10 gを0.01 gまで全量フラスコ1 000 mLにはかりとり,水を標線まで加える。
この溶液10 mLを全量フラスコ100 mLにとり,2-プロパノール10 gを加えた後,水を標線まで加
える。この溶液を標準液とする。
3) 2-プロパノール溶液 (100 mg/mL) 全量フラスコ100 mLに2-プロパノール10.0 gをとり,水を標
線まで加える。
c) 測定
1) マイクロシリンジに1 μLの標準液をとり,試料導入部から素早く注入し,直ちに,記録計の記録紙
送りを開始し,クロマトグラムを記録する。メタノールのピークが現れる部分は,感度が32倍程度
になるように感度を切り換えて,その感度を記入する。標準液と全く同じ条件で試験溶液の測定を
行う。
なお,標準液と試験溶液とは,それぞれ別のマイクロシリンジを用いることが望ましい。また,
試験溶液のメタノール部の減衰比は,標準液の場合と同じにする。
注記 メタノールのピークは,2-プロパノールの保持時間の1/4程度の位置に現れる。
2) 記録されたメタノール及び2-プロパノールのピーク面積をJIS K 0114に規定する半値幅法,又はデ
ータ処理装置を用いる方法によって測定する。2-プロパノールについては減衰比の補正を行う。
注記 通常の液体洗剤には,エタノールが含まれていることが多いので,メタノールと2-プロパ
ノールのピークとの間にエタノールのピークが現れるが,エタノールはこの試験方法とは
関係なく,メタノールの測定を妨害しない。
7.19.5 判定
試験溶液について得られたメタノール/2-プロパノールのピーク面積比は,標準液について得られたメタ
ノール/2-プロパノールのピーク面積比より大きくてはならない。
7.20 エタノールの定量
7.20.1 要旨
試料中に含まれるエタノールを多孔性高分子物質を充てん剤として用いたガスクロマトグラフ法を用い,
内部標準法によって定量する。
7.20.2 試薬
試薬は,次による。
7.20.2.1 2-プロパノール JIS K 8839の規定による。
7.20.2.2 エタノール (99.5) JIS K 8101の規定による。
7.20.3 装置及び器具
装置及び器具は,次による。
55
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) ガスクロマトグラフ 7.19.3 a) による。
b) 分離カラム 7.19.3 b) による。
c) キャリヤーガス 7.19.3 c) による。
d) 試料導入器 7.19.3 d) による。
e) 全量フラスコ 100 mL
7.20.4 操作
操作は,次による。
a) ガスクロマトグラフの調整
1) カラム槽温度を130〜150 ℃,試料導入部をカラム槽より約50 ℃高い温度に,検出器温度をカラム
槽と同程度から約50 ℃高い温度にそれぞれ設定する。
2) マイクロシリンジに1 μLの内部標準物質溶液をとり,試料導入部から素早く注入し,2-プロパノー
ルのピークが8〜10分で現れるように,キャリヤーガスの流量及びカラム槽の温度を調節する。さ
らに,2-プロパノールのピークの高さが,記録紙の30〜60 %の間になるように感度を調節する。
また,ピークの半値幅が10 mm以上で,できるだけ二等辺三角形になるように,記録計の記録紙送
り速度を調節する。
b) 内部標準物質溶液及び試験液の調整
1) 内部標準物質溶液 あらかじめ水5 mLを入れた全量フラスコ100 mLに2-プロパノール約1 gを0.1
mgまではかりとり水を標線まで加える。これを内部標準物質溶液とする。
2) 試験液 共栓付き三角フラスコに試料の適量(エタノール30〜90 mgを含む。)を0.1 mgまではか
りとり,これに内部標準物質溶液5 mLと水15 mLとを加えた後,よくかき混ぜて試験液とする。
c) 検量線の作成 あらかじめ水約5 mLを入れた全量フラスコ100 mLに,エタノール約1 gを0.1 mgま
ではかりとり水を標線まで加える。この溶液2 mL,4 mL,6 mL,8 mL及び10 mLを全量ピペットを
用いて,それぞれ共栓付き三角フラスコにとり,それぞれ内部標準物質溶液5.0 mLを加えた後,水を
加えて20 mLとする。マイクロシリンジにこれらの液1 μLをとり,試料導入部から素早く注入し,
記録計の記録紙送りを開始する。記録されたエタノール及び2-プロパノールのピーク面積を,JIS K
0114に規定する半値幅法,又はデータ処理装置を用いる方法によって測定する。得られたエタノール
の2-プロパノールに対するピーク面積比と,注入した溶液中の両者の質量比から検量線を作成する。
d) 測定 マイクロシリンジに1 mLの試験液をとり,試料導入部から素早く注入し,記録計の記録紙送
りを開始する。検量線の作成と同様にして,エタノールの2-プロパノールに対するピーク面積比を測
定し,あらかじめ作成した検量線を用いて,相対質量を求める。
7.20.5 計算
エタノールは,次の式によって算出する。
100
×
×
S
W
A
C=
ここに,
C: エタノール(質量分率%)
A: 検量線から求めた相対質量比
W: 内部標準物質の添加量 (g)
S: 試料の質量 (g)
56
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.21 水分の定量
水分の定量方法は,加熱減量法,蒸留法及びカールフィッシャー法の3種類とする。水分の測定値は方
法によって異なるから,測定値には加熱減量法,蒸留法及びカールフィッシャー法のいずれによったかを
付記する。
7.21.1 加熱減量法
7.21.1.1 要旨
試料を105±2 ℃で2時間乾燥したときの減量をはかり水分とする。この場合は,水及び揮発成分の含
量が求められる。
7.21.1.2 装置及び器具
装置及び器具は,次による。
a) 熱風循環式乾燥器 105±2 ℃に調節できる乾燥器。
b) 平形はかり瓶 直径50 mm
7.21.1.3 操作
試料約3 gを平形はかり瓶に0.1 mgまではかりとり,105±2 ℃の熱風循環式乾燥器中で2時間乾燥し,
デシケータ中で放冷した後,質量をはかる。
なお,水分の多い液体試料をはかりとるときは,あらかじめ強熱した後,冷却した径3 mmのガラスビ
ーズ又は海砂(1号)を試料の約3倍量加える。
7.21.1.4 計算
水分は,次の式によって算出する。
100
×
S
W
C=
ここに,
C: 水分(質量分率%)
W: 減量 (g)
S: 試料の質量 (g)
7.21.2 蒸留法
7.21.2.1 要旨
試料をキシレンと共に蒸留し,留出分離した水及び水に可溶な揮発分の含量(質量分率%)を求める。
7.21.2.2 試薬
試薬は,次による。
7.21.2.2.1 キシレン JIS K 8271に規定するもの。
7.21.2.3 装置及び器具
水分蒸留装置の例を,図10に示す。
57
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
A:蒸留フラスコ
B:検水管
C:冷却管
図10−水分蒸留装置の例
7.21.2.4 操作
操作は,次による。
a) 表4に示す量の試料を蒸留フラスコにとり,少なくとも同量以上(この場合最低100 mL)のキシレン
を加え,混合した後,装置を組み立てる。
表4−試料採取量
推定水分含量(質量分率%)
試料採取量 (g)
1以下
200
1〜5
100
5以上
留出水分が2〜5 mLになるように試料
をはかりとる。
b) 冷却器の上端からキシレンを蒸留フラスコの方へあふれるまで検水管に流し込む。冷却器の上端を綿
で軽く栓をする。
c) マントルヒータを用いてフラスコを加熱し,1分間約100滴の速度で蒸留し,大部分の水分が留出し
た後,1分間約200滴とする。
d) 検水管に水が留出しなくなってから,30分間蒸留を続けた後,加熱を止め,冷却管及び検水管の内側
に付着した水滴は,ら旋状針金で落とし,約5 mLのキシレンで洗い落とす。
e) 5分間以上放置してキシレン層が透明になった後,20±1.5 ℃で水量を読み取る。
7.21.2.5 計算
水分は,次の式によって算出する。
58
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S
V
S
V
C
8.
99
100
998
.0
×
×
×
=
=
ここに,
C: 水分(質量分率%)
V: 留出した水の量 (mL)
0.998: 20 ℃における水の密度 (g/cm3)
S: 試料の質量 (g)
なお,水分蒸留装置内部は,使用前にあらかじめ,硝酸 (6 mol/L) と過酸化水素水 (30 %) とを体積比
3対1で混合した溶液で洗浄後,水洗,乾燥しておく。また,蒸留フラスコ中の内容物が加熱によって泡
立つ場合は,加熱脱水したオレイン酸又はオレイルアルコールを入れることによって泡立ちを防止できる。
検水管の刻度1目盛の誤差は0.05 mL以下とするが,市販測定装置は校正を行った方がよい。
7.21.3 カールフィッシャー法
カールフィッシャー法は,JIS K 0068による。
8
物理試験
8.1
粒度
8.1.1
要旨
粒度は,規定された網ふるいを用いて試料をふるい分け,そのふるい上の残分及び受け皿上の残分の質
量をはかり,試料に対する百分率で表す。
8.1.2
装置及び器具
装置及び器具は,次による。
a) ふるい JIS Z 8801-1に規定する網ふるい。
b) ふるい分け機械 毎分130〜165回の打振と回転運動とを同時に与えることのできるもの。
c) 化学はかり 0.1 gまではかることができるはかり。
8.1.3
操作
操作は,次のいずれかによる。
a) ふるい分け機械によるふるい分け方法 ふるい目の開きの大きさが大から小の順で異なっている数個
のふるいの一組(以下,組ふるいという。)を用いる。
組ふるいのふるい目の開きの小から大の順に受け皿の上に積み重ね,その最上部のふるいの上に規
定量の試料を入れ,ふたをしてふるい分け機械に取り付け,規定の時間(例:3分30秒±30秒)振動
した後,それぞれのふるいに残留した試料及び受け皿中の試料の質量をはかる。
なお,ふるいの背面に付着している粒子は,はけでよく払い落とす。
b) ふるいによるふるい分け方法 組ふるいを用い,ふるい目の開きの大きいふるいから始め,順次,小
さいふるいを用い,一度に各ふるいごとに次の操作によってふるい分ける。
規定量の試料をふるいの上にとり,受け皿を置き,ふたをして試料が網の面に均等に広がるように
少し傾けて片手で持ち,他方の手で約150回ふるいの縁を打ち当てる。ふるいは25回打ち当てるごと
に約1/6回転させる。
ふるいの上に残った試料の質量を0.1 gまではかる。ふるいを通過した受け皿中の試料は,順次ふる
い目の開きの小さい組ふるいに移して同様の操作を行う。
8.1.4
計算
粒度は各ふるいの網上に残った試料の質量から,次の式によってふるい残分を算出し,それぞれ列記す
59
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。ただし,ふるい分け機械によるふるい分け方法,又は手ぶるいによるふるい分け方法のいずれによっ
たかを記載する。
なお,ふるい上に残った試料及び受け皿中の試料の合計が,初めの試料の質量より2 %未満の減量のと
きは,受け皿中の試料の質量にそれだけ加算する。また,2 %以上減少したときは試験をやり直す。
100
×
S
B
A=
ここに,
A: ふるい残分(質量分率%)
B: ふるい上に残った試料の質量 (g)
S: 試料の質量 (g)
8.2
見掛け密度
8.2.1
要旨
試料の単位容積当たりの質量を測定し,見掛け密度とする。
8.2.2
装置
見掛け密度測定器の例を,図11に示す。漏斗及びスタンドは黄銅製,カップ(受け器)は,アクリル樹
脂製又はガラス製のものを用いる。
8.2.3
操作
操作は,次による。
a) 測定器を安定な台の上に置き,三脚ねじを調節して水平に保ち,乾燥した漏斗をスタンドに垂直に載
せ,下の口にダンパーを軽く当ててふさぐ。
b) 漏斗の直下にあらかじめ洗浄乾燥し質量を0.1 gまではかったカップを置き,次に縮分した試料約120
mLを静かに漏斗内に入れる。
c) ダンパーを手早く全開して,漏斗内の試料をカップ中に自然落下させる。試料が塊状で漏斗に付着す
る場合は,あらかじめガラス棒でよくばらばらにしておく。カップから盛り上がった部分は,ガラス
棒(径約8 mm,長さ約150 mm)ですり落とした後,試料の入ったカップの質量を0.1 gまではかる。
8.2.4
計算
見掛け密度は,次の式によって算出する。
V
W
W
S
1
2−
=
ここに,
S: 見掛け密度 (g/mL)
W2: 試料の入ったカップの質量 (g)
W1: 空のカップの質量 (g)
V: カップの容積 (mL)
60
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図11−見掛け密度測定器
8.3
pH値
8.3.1
要旨
温度0〜95 ℃における規定の濃度の水溶液について,ガラス電極を用いたpH計でpH値を測定する。
8.3.2
装置
JIS Z 8802の4.(pH計の種類及び形式)に規定する形式IIを用いる。
8.3.3
操作
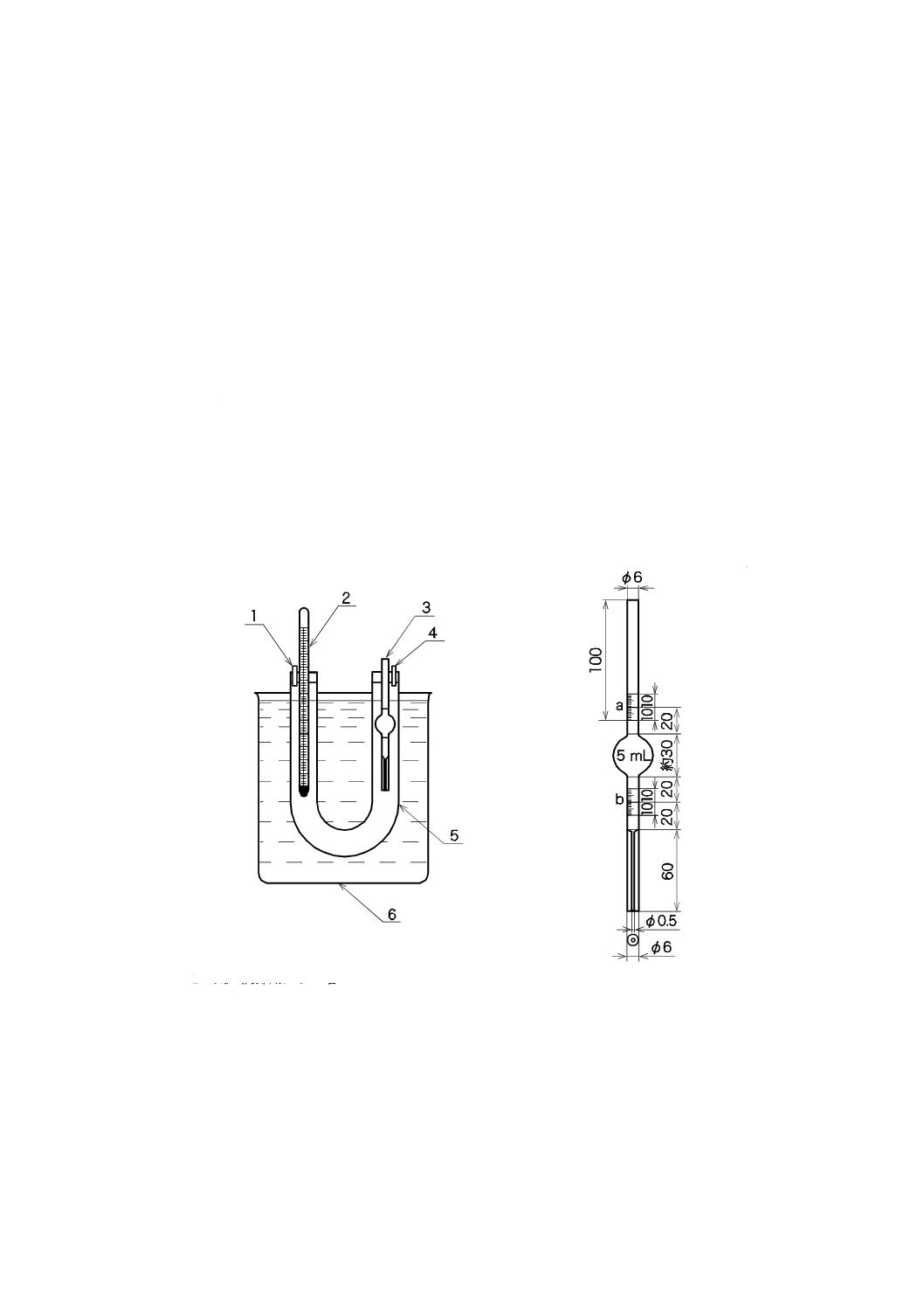
61
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS Z 8802の7.2(測定方法)の手順によって,試料溶液のpH値を測定する。
8.3.4
測定結果の記録
測定値には,試料溶液の温度,濃度及び使用したpH計の形式を記載する。測定値は,小数点以下1け
たまで記録する。
なお,測定に使用する水のpH値は,6.5〜7.0付近でなければならない。
8.4
表面張力
8.4.1
滴容法
8.4.1.1
要旨
円形の管の口から一定量の液体を静かに滴下し,その落下滴数,比重及び水の表面張力から,その液の
表面張力を求める。
8.4.1.2
装置及び器具
装置及び器具は,次による。
a) 滴数計 図12に示す。
b) U字管 図12に示す。
c) 恒温槽 図12に示す。
単位 mm
1 気圧調節用ガラス管
4 気圧調節用ガラス管
2 温度計
5 U字管
3 滴数計
6 恒温槽
図12−滴数計
8.4.1.3
操作
操作は,次による。
a) 試験液の調製 硬質ガラス器具を用いて煮沸後,冷却した水を使用し,室温でその100 mLに規定濃
62
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
度に相当する試料を溶解したものを試験液とする(例えば,無水物0.25 gに相当する試料を溶解して
2.5 g/L溶液とする。)。
試験液は使用の都度調製し,密栓し,あらかじめ試験温度に約20分間保持した後使用する。
b) 測定
1) 滴数計を試験液で2回洗った後,これに試験液を満たし,規定の温度に保った恒温槽中のU字管内
に垂直に保持し,試験液の液面が標線aからbまで(容積約5 mL)降下する間に落下した全滴数を
求める。
2) 落下した試験液が正しく上下標線間の容積に一致しないときは,別に標線上下の目盛と滴数との関
係を求めて,全滴数を上下標線間の容積に補正する。補正のときの精度は0.1滴とする。
3) 毎分12±2滴の落下速度で滴を落下させる。測定温度は±0.2 ℃の範囲内で一定とする。
4) 別に蒸留水を用いて同様の測定を行う。
8.4.1.4
計算
表面張力は,次の式によって小数点以下2けたまで算出する。
0
0σ
n
n
σ
×
=
ここに,
σ: 測定温度における試験液の表面張力 (10−3 N/m)
n0: 蒸留水の滴数
n: 試験液の滴数
σ0: 測定温度における水の表面張力 (10−3 N/m)
なお,σ0の値は,表5に示す。
表5−空気に接する水の表面張力
単位 10−3 N/m
温度 (℃)
表面張力
温度 (℃)
表面張力
温度 (℃)
表面張力
−10
77.10
17
73.20
28
71.47
− 5
76.40
18
73.05
29
71.31
0
75.62
19
72.89
30
71.15
5
74.90
20
72.75
35
70.35
10
74.20
21
72.60
40
69.55
11
74.07
22
72.44
50
67.90
12
73.92
23
72.28
60
66.17
13
73.78
24
72.12
70
64.41
14
73.64
25
71.96
80
62.60
15
73.48
26
71.82
90
60.74
16
73.34
27
71.64
100
58.84
注記 測定値には,試験液の試料濃度及び測定温度を併記する。
滴数計の滴の出口の面はよく磨き,使用前に硝酸 (6 mol/L) と過酸化水素水 (30 %) との
体積比3 : 1の混液で清浄にし,更に水洗乾燥する。
滴の落下速度は,滴数計にゴム管を連結し,その一端に適切な毛細管を付けて調節する。
8.4.2
輪環法
8.4.2.1
要旨
金属の輪を水平に試験液に接触させ,垂直に液面から引き上げる。輪が液面を離れるのに必要な力を,
ねじばかりで測定して,試験液の表面張力を求める。
63
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.4.2.2
装置及び器具
装置及び器具は,次による。
a) 表面張力計(図13参照)
A
鋼線
C
さお
F
支持台
G
白金環
H
試料内容
I
台
J
ねじ
K
目盛
L
指針
M
取っ手
N
取っ手
P
取っ手
S
水平調節ねじ
Z
取っ手
図13−デュヌイ (Du Nouy) 表面張力計
b) 試料容器 ガラス製,内径約45 mmの容器。
c) 白金環 白金線の長さ約40 mm
8.4.2.3
操作
操作は,次による。
a) 装置の調整
1) 支持台Fの上(鋼線Aの真下)に水準器を置き,装置が水平となるように水平調節ねじを用いて調
節する。
2) 白金環をさおの先端につるし,次に目盛のゼロ(零)に指針を合わせる。白金環に紙片を載せ,支
持台の上端と,さおが至近の距離になるように,取っ手Zを用いて調節する。
3) 紙片の上に質量の分かった物体を載せ,再びさおが支持台と至近の距離になるように取っ手Mを調
節し,目盛の指度を読み取る。
この指度が次の式から算出したθと一致していないときは,取っ手Nを調節して一致するまでこ
の操作を繰り返す。指度目盛の1目盛は,10−3 N/mに相当する。
R
WG
π
θ4
=
ここに,
θ: 指度 (10−3 N/m)
W: 白金環に載せた紙片及び物体の全質量 (g)
64
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
G: 重力の定数 (980 cm/s2)
R: 白金環の半径 (cm)
b) 校正(水の表面張力の測定) 必要があればJIS K 2241によって行う。
c) 測定 測定は,次による。
1) 白金環をアセトン,エチルエーテルなどの溶剤及び水で十分に洗浄する。環を乾燥した後,ガス炎
の酸化炎又はアルコールランプで赤熱するまで焼く。これを冷却後,さおの先端につるして再び指
針を目盛のゼロに合わせた後,さおが支持台と至近の距離になるように取っ手Zを調節する。
2) 試料容器に規定温度±1 ℃に保持した規定濃度の試験液を入れて台の上に載せ,取っ手Pを緩め,
台を適切な高さまで上げ,更にねじを回して,白金環を試験液表面上約1 mmぐらいの位置に保ち,
液面と白金環の面が平行であるかどうかを,液面に映る環の像と実物の平行性から確かめ,平行で
なければ,白金環の柄を曲げて環に対して水平にする。水平になった後,ねじを回して,白金環を
試験液表面に静かに触れさせる。
3) 白金環が液面から離れないように注意しながら,ねじを回して容器を台と共に下げると,環は表面
張力のため引っ張られて,さおが下がる。
これを元に戻すため,取っ手Mを回して鋼線をねじり,支持台にさおが至近距離になるようにす
る。ねじと取っ手Mは液面を離れる直前には,極めて徐々に動かして,環が液面を離れたときの指
度 (θ ) を読み取る。
4) 測定は同一試験液について3回行い,それぞれの測定結果と3回の平均値との差が2 %を超えない
ときの平均値 (θ ) をとる。
8.4.2.4
計算
表面張力は,次の式によって算出する。
σ =θ×F
ここに,
σ: 表面張力 (10−3 N/m)
θ: 指度の平均値 (10−3 N/m)
F: 補正ファクター
なお,補正ファクターは,次の式によって求める。
rR
d
D
C
F
679
.1
34
045
.0
)
(
52
014
.0
0
725
.0
2
−
+
−
+
θ
=
ここに,
θ: 指度の平均値 (10−3 N/m)
C: 白金環の円周2πR (cm)
D 3): 25 ℃における下相の密度 (g/mL)
d 3): 25 ℃における上相の密度 (g/mL)
R: 白金環の半径 (cm)
r: 白金線の半径 (cm)
注記 Fを求める式は,白金環によって持ち上げられる液量による影響を補正するものである。
[Zuidema and Waters: Ind and Eng Chem AnaL Ed 13.312 (1941)]。
注3) 合成洗剤の希薄水溶液の測定にあっては,D=水溶液の密度,d=空気の密度となるので,近似
値としてD−d=1 (g/mL) を用いてもよい。
8.4.2.5
精度
別人,別装置の2個の測定値とその平均値との差が5 %を超えない場合,いずれも正しいものとする。
なお,測定値には,試験液の濃度及び測定温度を併記する。また,測定は,振動及び通風のある場所及
65
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
びガスの発生する場所は避けなければならない。
8.5
起泡力及び泡の安定度
8.5.1
要旨
規定濃度の試料水溶液200 mLを規定の温度条件で900 mmの高さから30秒間で液面上に落下させたと
きに生じる泡の高さを測定して起泡力として,その5分後の高さを泡の安定度として測定する。
8.5.2
装置及び器具
起泡力測定装置を,図14に示す。
8.5.3
操作
操作は,次による。
a) 起泡力測定装置の内筒を垂直に立て,規定温度の水をポンプによって外筒に循環させて一定温度に保
つ。
b) 規定濃度の試験液を同温度に保ちながら,その50 mLを内筒の管壁に沿って静かに側面全体を潤すよ
うに流し込む。
c) ピペットAに試験液200 mLをとり,これを図14に示す位置に置き,その上端のコックを開き試料溶
液が約30秒間で流出するようにし,かつ,液滴が内筒液面の中心に落ちるようにして流下させる。
d) すべての溶液が流出した後,直ちに,目視によって泡の高さ(mm)(起泡力)をはかる。さらに,5分
後に,目視によって泡の高さ(mm)(泡の安定度)を測定する。この操作を数回行い,それぞれの測定
値の平均を整数位まで求めて,起泡力及び泡の安定度とする。
なお,測定データ中に試料溶液の濃度が無水物換算又は有姿の別を記載する。
例 無水物換算 2.5 g/L又は体積分率0.25 %
単位 mm
66
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図14−起泡力測定装置
8.6
耐硬水性
8.6.1
要旨
規定濃度の試料溶液に,一定量の硬水をかき混ぜながら加えたときの試料溶液の透明度によって耐硬水
性を評価する。
8.6.2
装置及び器具
装置及び器具は,次による。
a) シリンダー JIS R 3505に規定するメスシリンダー 500 mL
b) 判定板 図15による。
c) 恒温槽 50±3 ℃に調節できる恒温槽。

67
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図15−判定板
8.6.3
試薬
試薬は,次による。
8.6.3.1 硬水 JIS K 8122に規定する塩化カルシウム二水和物約270 mgをはかりとり,水に溶解して全量
フラスコで,1 000 mLにする。この溶液は,JIS K 0101の15.1(全硬度)によって硬度を測定する。
8.6.4
操作
規定濃度の試料100 mLをシリンダーに入れ,50±3 ℃の硬水100 mLをかき混ぜながら注加し,50±3 ℃
の恒温槽中に10分間放置した後取り出し,直ちに判定板の上に置き,判定板上の十字が見えるものを合格
とする。
なお,測定データは試料溶液濃度が無水物換算又は有姿の別を記載する。
例 無水物換算2.5 g/L又は体積分率0.25 %
9
洗浄力評価方法
洗浄力評価方法は,衣料用合成洗剤及び台所用合成洗剤の2種類の洗浄力評価方法とする。
9.1
衣料用合成洗剤の洗浄力評価方法
9.1.1
要旨
襟あか布を試料洗剤で洗浄したときの汚れ落ち程度を目視によって判定し,洗浄力判定用指標洗剤に対
する洗浄力を評価する。
9.1.2
試薬及び試験用材料
試薬及び試験用材料は,次による。
9.1.2.1 直鎖アルキルベンゼンスルホン酸ナトリウム 平均分子量345±5,純分質量分率50 %以上,未
反応油分(原料アルキルベンゼン)質量分率1 %以下,硫酸ナトリウム分質量分率2 %以下及び水分質量
分率50 %以下のもの。純分は7.3.3によって,測定する。
9.1.2.2 ゼオライト 4A型の結晶構造をもち,平均粒径10 μm以下のもの。純分(無水物として)は7.15
によって測定する。
9.1.2.3 けい酸ナトリウム JIS K 1408の規定による。
9.1.2.4 炭酸ナトリウム JIS K 8625の規定による。
9.1.2.5 カルボキシメチルセルロースナトリウム エーテル化度0.5〜0.7,純分(無水物として)質量分
率95 %以上,塩化ナトリウム質量分率0.5 %以下,水分質量分率10 %以下,粘度25 ℃で70〜130 mPa・
s(無水物換算で10 g/L水溶液)。
9.1.2.6 硫酸ナトリウム JIS K 8987の規定による。
9.1.2.7 塩化カルシウム(二水和物) JIS K 8122の規定による。
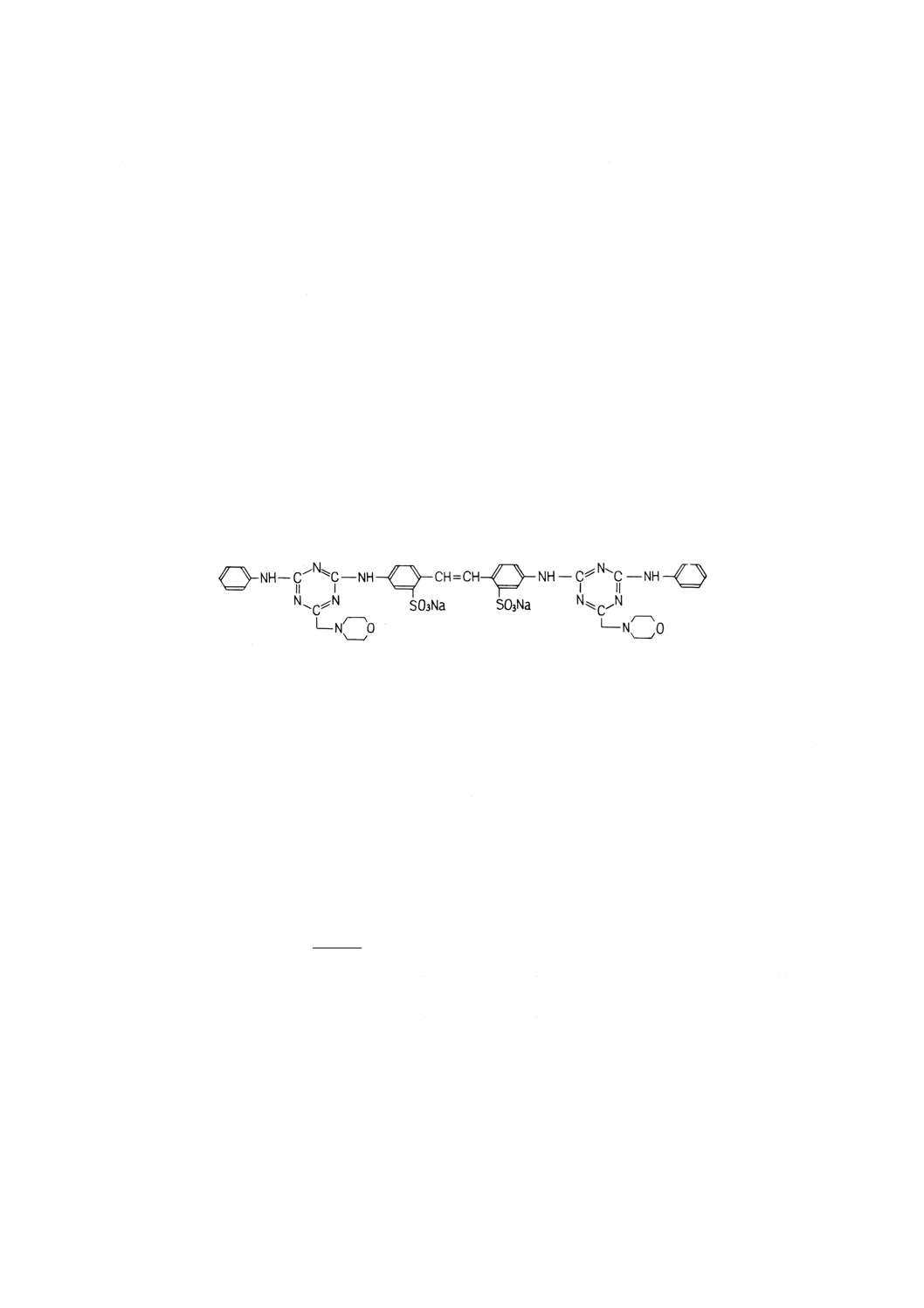
68
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.1.2.8 襟あか布 JIS L 0803に規定する単一繊維布を試験布として用いて,次によって調製したもの。
試験布を110×130 mmに裁断し,裁断布2枚のそれぞれ短辺を布目の方向をそろえて縫い代10 mmを
もって縫い合わせ,襟布 (110×240 mm) を作る。
これを作業衣,Yシャツなどの着衣の襟の折目をまたいで,ボタン又は両面テープを用いて固定し,2
〜7日間着用させて襟あか布を作製する。
襟あか布を着衣から取り外し,襟あか布の中から縫い目を中心としてできるだけ左右均等に汚染されて
いるものを選別し,汚れの程度に応じて大汚れ,中汚れ及び小汚れの3段階に区分して,それぞれの段階
の襟あか布を5枚ずつ,計15枚を用意した後,縫い代部の糸を抜いて二組に分け,試験に用いる。
縫い代部の糸を抜く前に,一対の襟あか布が対称であることを示す記号(例えば1,1'のような数字)を
15枚の襟あか布のすべてについて,それぞれの襟あか布の両すみに油性のマーキングペンで記入しておく。
襟あか布を洗浄に用いる場合,汚染から洗浄までの時間は特に考慮しないが,なるべく1か月以内に洗
浄試験に用いる。また,その期間中の襟あか布は,デシケータ中に保存する。
9.1.2.9 蛍光増白剤 4,4'-ビス(4-アニリノ-6-モルホリノ-1,3,5-トリアジン-2-イル)アミノスチルベ
ン-2,2-ジスルホン酸ナトリウムを主成分とする蛍光増白剤。
C.I.40625
構造式
9.1.2.10 洗浄力判定用指標洗剤 次によって調製したもの。
直鎖アルキルベンゼンスルホン酸ナトリウム,けい酸ナトリウム,炭酸ナトリウム,カルボキシメチル
セルロースナトリウム及び硫酸ナトリウムを純分又は無水物として質量比で15 : 5 : 7 : 1 : 55にはかりとり,
乳鉢及び乳棒を用いてよく混合後,磁器蒸発皿に移し105±2 ℃の乾燥器中で乾燥した後,乳鉢及び乳棒
を用いて粉末にし,すべて,ふるいを通過させる。7.21.1によって上記粉末の水分を測定し,この粉末と
ゼオライトを無水物換算で83 : 17の割合で混合し,デシケータ中に保存する。
試験対象物に蛍光増白剤が含まれているときは,洗浄力判定用指標洗剤に蛍光増白剤質量分率0.5 %を
入れてもよい。
直鎖アルキルベンゼンスルホン酸ナトリウムは,7.3.2によって測定したときの純分から,次の式によっ
て算出したものとする。
A
B
100
15×
=
ここに,
A: 直鎖アルキルベンゼンスルホン酸ナトリウムの純分(質量分
率%)
B: 直鎖アルキルベンゼンスルホン酸ナトリウムの質量比の値
9.1.2.11 使用水 JIS K 8122に規定する塩化カルシウム二水和物133 mgをはかりとり,水に溶解し全量
フラスコで1 000 mLとする。
注記 9.1.2.1及び9.1.2.5の品質についての試験方法は,日本油化学会で定めたものがある。
9.1.3
装置及び器具
装置及び器具は,次による。
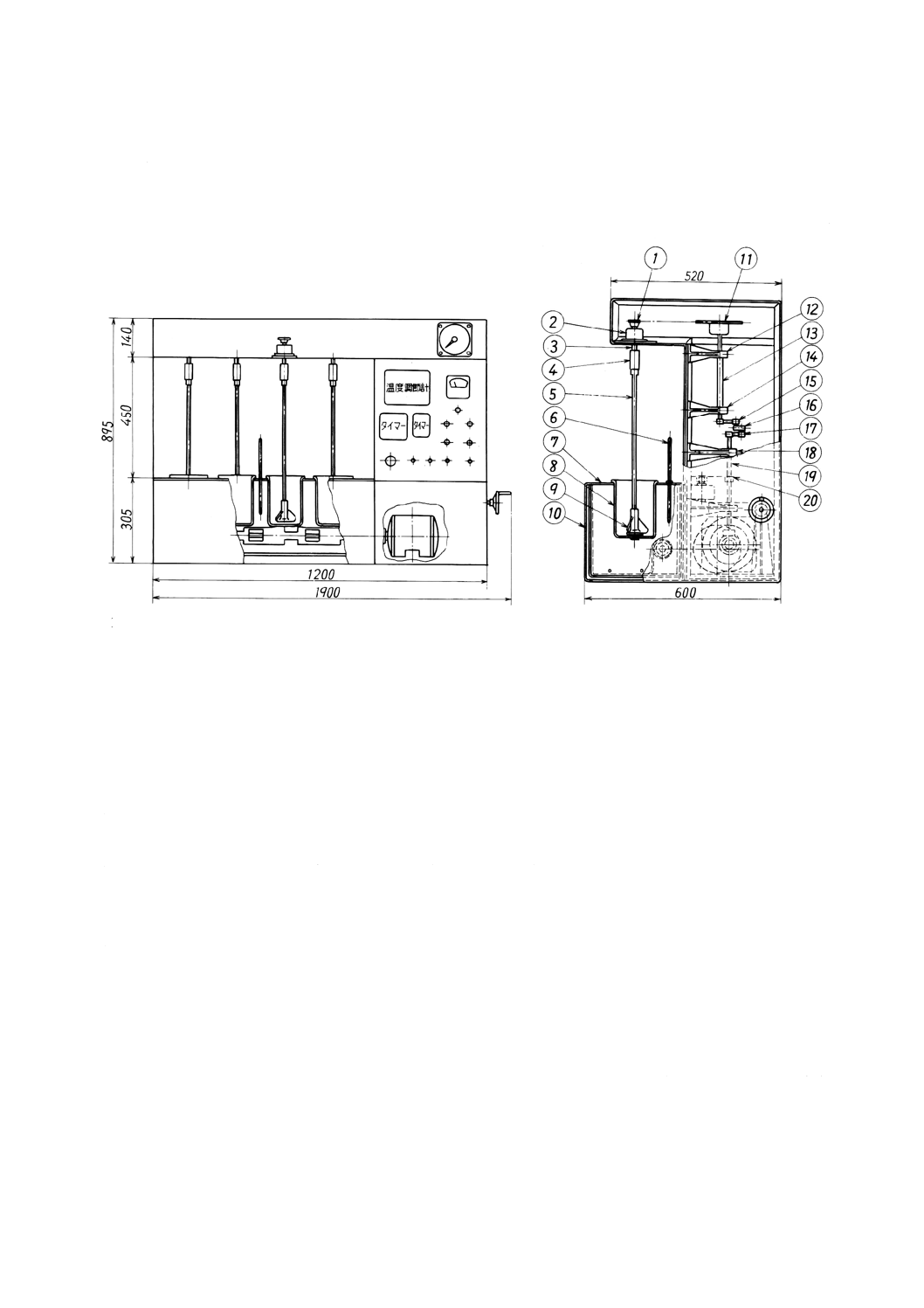
69
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) かき混ぜ式洗浄力試験機 かき混ぜ式洗浄力試験機の例を,図16に示す。
例 Terg-O-Tometerなど
単位 mm
1 スプロケット
2 軸受
3 軸
4 ジョイント
5 回転軸
6 温度計
7 試料カップ取付台
8 試料カップ
9 回転翼
10 恒温水槽
11 スプロケット
12 軸受
13 軸
14 軸受
15 クランク
16 クランク
17 クランク
18 軸受
19 軸
20 プール
図16−かき混ぜ式洗浄力試験機
b) 乾燥器 105±2 ℃ に調節できる乾燥器。
c) 乳鉢及び乳棒
d) 磁器蒸発皿
e) ふるい JIS Z 8801-1に規定する網ふるい(目開き355 μm)を使用する。
9.1.4
操作
操作は,次による。
a) 洗浄力判定用指標洗剤を7.21.1によって水分を測定し,無水物に換算して1.33 gをはかりとり,使用
水1 000 mLに溶解する。この溶液を洗浄力判定用指標洗剤溶液とする。
b) 試料を,その洗剤の標準使用濃度 (g/L) になるようにはかりとり,使用水1 000 mLに溶解する。この
溶液を試料溶液とする。
c) 15枚の襟あか布の一組は30 ℃の洗浄力判定用指標洗剤溶液1 000 mL中に,他の一組は,30 ℃の試
料溶液1 000 mLに入れて,かき混ぜ式洗浄力試験機(回転速度120±5 min−1)を用いて10分間洗浄
する。
d) 同一洗剤溶液の中の襟あか布は洗浄終了後,まとめて含水率が質量分率200 %以下になるよう軽く手
で絞ってから,30 ℃の使用水1 000 mL中に入れ,かき混ぜ式洗浄力試験機(回転速度120±5 min−1)
70
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を用いて3分間すすぐ。この操作を2回繰り返す。
e) すすぎが終了した襟あか布は,風乾後,対称の布を再び短辺部で縫い合わせ,アイロン仕上げを行っ
て,洗浄力の判定に供する。
9.1.5
評価
評価は,次による。
a) 操作が終了した15枚の襟あか布を白色の台紙上に記号順に配列し,JIS Z 8723の6.(色比較のための
照明)によって,一対の襟あか布を左右見比べながら1枚ずつ,15枚の襟あか布について汚れ落ちの
程度を目視で判定する。
この判定は,3人の判定者が行う。
b) 洗浄力判定用指標洗剤で洗浄した襟あか布の汚れ落ちの程度に対して,試料で洗浄した襟あか布の汚
れ落ちの程度を比較し,表6の区分によって評価する。
表6−評価区分
明らかに劣る場合
−2
やや劣る場合
−1
ほとんど差がない場合
0
やや勝る場合
+1
明らかに勝る場合
+2
c) 結果の解析及び評価 15枚の洗浄布について評価された結果は,シェッフェの一対比較法によって解
析し,有意差検定を行い,危険率5 %で有意差がない場合及び有意差のある場合で,評価点の合計が
0以上の場合を指標洗剤と同等以上とする。附属書Aに評価例を示す。
9.2
台所用合成洗剤の洗浄力評価方法
9.2.1
概要
この方法は,モデル汚れを付着させたガラス片を用いて,試料洗剤と洗浄力判定用指標洗剤とで洗浄し,
その汚れ落ちの程度を目視によって比較し,洗浄力を評価する。
9.2.2
試薬,試験用材料試薬及び試験用材料
試薬,試験用材料試薬及び試験用材料は,次による。
9.2.2.1 牛脂 日本薬局方
9.2.2.2 大豆油 日本薬局方
9.2.2.3 モノオレイン 化粧品原料基準の親油型モノオレイン酸グリセリンに規定するもの。
9.2.2.4 オイルレッド(スダンIII) C.I. (Colour Index) −26 100
注記 構造式
9.2.2.5 クロロホルム JIS K 8322の規定による。
9.2.2.6 直鎖アルキルベンゼンスルホン酸ナトリウム 9.1.2.1 による。
9.2.2.7 エタノール (99.5) JIS K 8101の規定による。
9.2.2.8 尿素 JIS K 8731の規定による。
71
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.2.9 水酸化ナトリウム溶液 (50 g/L) JIS K 8576に規定する水酸化ナトリウムを用いて調製する。
9.2.2.10 塩酸 (1+6) JIS K 8180に規定する塩酸を用いて調製する。
9.2.2.11 塩化カルシウム二水和物 JIS K 8122の規定による。
9.2.2.12 塩化マグネシウム六水和物 JIS K 8159の規定による。
9.2.2.13 洗浄力判定用指標洗剤 次によって調製する。
a) 直鎖アルキルベンゼンスルホン酸ナトリウム,エタノール (99.5),尿素及び水を質量比15 : 5 : 5 : 75
で混合する。ただし,水はイオン交換水又は蒸留水を用いる。
b) 指標洗剤のpH値が,7.0±0.5となるように水酸化ナトリウム溶液 (50 g/L) 又は塩酸 (1+6) を用いて
調製する。
9.2.2.14 使用水 塩化カルシウム二水和物59.0 mg及び塩化マグネシウム六水和物27.2 mgをはかりとり,
水に溶解して1 000 mLとする。この使用水は約11 000 mL調製する。
9.2.2.15 汚こう(垢)浴 牛脂及び大豆油を体積比1 : 1で混合した油脂20 g,モノオレイン0.25 g及び
オイルレッド0.1 gを同時にクロロホルム60 mLに溶解して汚こう浴を調製する。
9.2.2.16 モデル汚れガラス片 次によって調製する。
a) モデル汚れガラス片は,清浄なスライドガラスを用いて作る。このガラス片は6枚を一組とし,1 mg
まで質量をはかっておく。
b) 25±1 ℃の汚こう浴中に1枚ずつ約55 mmのところまで1〜2秒間浸し,汚こうを付着させた後取り
出す。スライドガラスの下部に付着した汚こうのたまりは,清浄なガーゼなどの布又はろ紙を用いて
吸いとらせ,汚こうを均一な状態にして,25±1 ℃に調節した恒温槽中で風乾し質量をはかる。
c) 風乾放置時間1時間以上2時間以内にモデル汚れガラス片を試験に用いる。この際,モデル汚れガラ
ス片6枚当たりの汚こう付着量は,0.140±0.010 gになるようにする。
d) モデル汚れガラス片は,6枚を一組として試料洗剤用に三組,指標洗剤用に三組準備する。
e) モデル汚れガラス片の作り方の注意事項
1) 洗浄までの一連の操作は,“スライドガラスに汚こうを付着させる”→“モデルガラス片の放置”→
“モデルガラス片の質量測定”→“洗浄”のように行われるが,モデル汚れガラス片の温度管理は
洗浄効率に重要な影響を与えるので,洗浄までの一連の操作は恒温室で行うことが望ましい。
なお,恒温室の使用が困難な場合には,モデル汚れガラス片を放置するのに恒温槽を用いてもよ
い。その場合には,スライドガラスは汚こうを付着させた後,直ちに恒温槽に入れ,恒温槽から取
り出したモデル汚れガラス片は,速やかに質量をはかり,洗浄に供することが必要である。
2) 汚こう浴中のオイルレッド及び汚こうの濃度は,常に一定になるようにする。
3) 汚こう付着量は,洗浄力に影響を与えるため,一連の試験は規定の付着量になるように注意する。
9.2.3
装置及び器具
装置及び器具は,次による。
a) 比色管 図17に示す比色管 容量100 mL
b) スライドガラス JIS R 3703に規定する標準形。
c) 洗浄力試験器 洗浄力試験器の例として,リーナツ形のものを図18に示す。
d) 恒温槽 25±1 ℃ に調節できる恒温槽。
72
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図17−比色管
単位 mm
1 回転軸
2 回転翼
3 スライドガラスセット上板(厚さ2 mm)
4 スライドガラスセット下板(厚さ2 mm)
5 スライドガラス支持板(厚さ0.5 mm)
6 両板支柱(2-M4ねじ長さ8)
7 ナット
8 平座金
9 洗浴ビーカー(硬質ガラス厚さ2.5 mm)
注記 洗浴ビーカーを除き,構成部品の材質はすべてJIS G 4303に規定するSUS 304を用いる。
図18−洗浄力試験器(例)

73
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.4
操作
モデル汚れガラス片六組について,三組は洗浄力判定用指標洗剤を用い,ほかの三組は共試洗剤を用い
て,それぞれ次による。
a) 洗浄力判定用指標洗剤1.5 gをはかりとり,使用水に溶解して1 000 mLとする。この溶液は約3 000 mL
調製する。
この溶液を洗浄力判定用指標洗剤溶液とする。
b) 供試洗剤を,その洗剤の標準使用濃度 (g/L) になるようにはかりとり,使用水に溶解して1 000 mLと
する。この液は約3 000 mL調製する。この溶液を試料溶液とする。
c) モデル汚れガラス片の一組について,30±1 ℃の洗浄力判定用指標洗剤溶液又は30±1 ℃の試料溶液
700 mL中に入れ,洗浄力試験器(回転速度250±10 min−1)を用い,3分間洗浄する。
d) 洗浄が終了したモデル汚れガラス片は,30±1 ℃の使用水700 mL中に入れて洗浄試験器を用いて,1
分間すすぐ。
e) すすぎが終了したモデル汚れガラス片は,室温で一昼夜風乾後,洗浄力の判定に供する。
9.2.5
評価
洗浄力の評価は,次による。
a) 操作を終了した6枚一組のガラス片に残留したモデル汚れを,それぞれの組ごとにクロロホルムに溶
解して100 mLとする。
b) 試料溶液を用いて操作したクロロホルム溶液と洗浄力判定用指標洗剤溶液を用いて操作したクロロホ
ルム溶液とをそれぞれ比色管に入れ,白色板又は白紙を背景としてその赤の程度を目視で比較し,汚
れ落ちの程度を判定する。この判定は,3人の判定者が行う。
c) 評価基準 洗浄力判定用指標洗剤溶液を用いて操作したクロロホルム溶液の赤の程度(汚れ落ちの程
度)に対して,試料溶液を用いて操作したクロロホルム溶液の赤の程度(汚れ落ちの程度)を比較し,
表7に示す区分によって評価する。
表7−評価区分
明らかに濃い場合(明らかに劣る場合)
−2
やや濃い場合(やや劣る場合)
−1
ほとんど差がない場合
0
やや薄い場合(やや勝る場合)
+1
明らかに薄い場合(明らかに勝る場合)
+2
d) 結果の解析及び評価 三組の試験用ガラス片について評価された結果は,一対比較法に準じた方法に
よって解析し,t検定で有意差検定を行う。
なお,評価点の平均値が0以上の場合には合格とし,t検定を行わない。評価点の平均値が0以下の
場合はt検定によって,5 %危険率で有意差がない場合も合格とする。また,t検定によって,5 %危
険率で有意差がある場合には不合格とする。附属書Bに,評価例を示す。
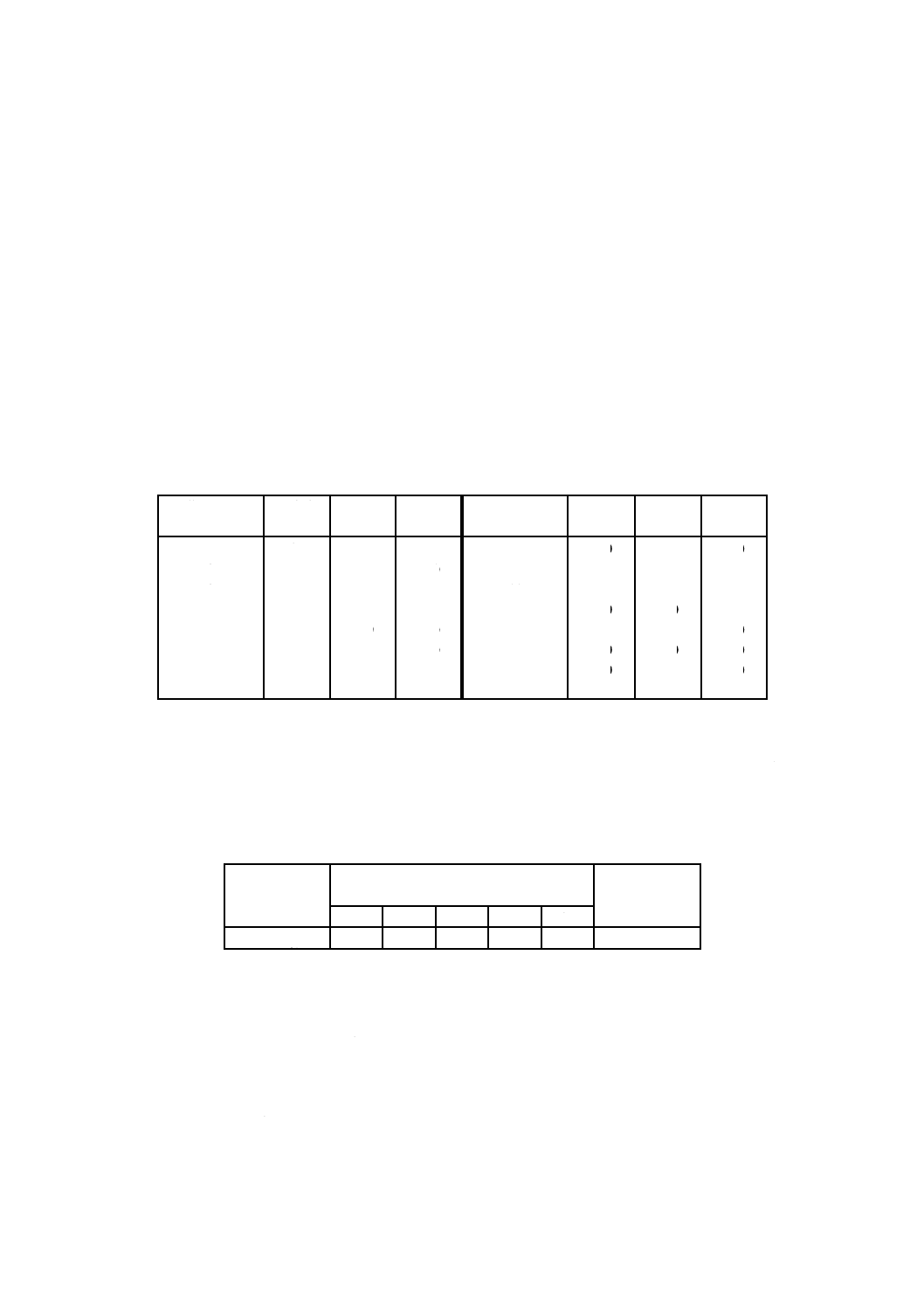
74
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
衣料用合成洗剤の洗浄力評価例
(シェッフェの一対比較法例)
序文
この附属書は,衣料用合成洗剤の洗浄力の評価例について記載するものであって,規定の一部ではない。
A.1 シェッフェの一対比較法での解析例
A洗剤で洗浄した襟あか布の汚れ落ち程度について,3人の判定者がそれぞれB洗剤で洗浄した襟あか
布の汚れ落ち程度を対照として評価し,得られた表A.1の結果について解析する。
表A.1−評価結果
襟あか布
判定者
判定者
判定者
襟あか布
判定者
判定者
判定者
No.
1
2
3
No.
1
2
3
1
+1
+1
+1
9
0
−1
0
2
+1
+1
0
10
+1
+1
+1
3
+1
+1
+1
11
+2
+1
+1
4
0
+1
+1
12
0
0
+1
5
−1
0
0
13
−1
−1
0
6
+1
+1
0
14
0
0
0
7
+1
+2
+2
15
0
−1
0
8
0
+1
+1
A.2 評価点の度数
3人の判定者が判定した15枚の襟あか布はすべて繰返しとみなして,表A.2のように評価点の度数を求
める。
表A.2−評価点の度数
評価点 (N)
評価点の合計
(ΣNifi)
−2
−1
0
+1
+2
度数 (f)
0
5
16
21
3
22
A.3 平方和
平方和は,次の式によって求める。
総平方和 (ST)=ΣNi2fi
ST=(−2)2×0+(−1)2×5+…
+(+2)2×3=38
主効果の平方和 (SA)=(ΣNifi)2/n
SA=(22)2/45=10.76
ただし,n=Σfi
誤差の平方和 (SE)=ST−SA
SE=38−10.76=27.24
75
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.4 自由度
自由度は次の式によって求める。
主効果の自由度 (ΦA)=t−1
ΦA=2−1=1
ただし,t:試料数
誤差の自由度 (ΦE) =t (−1) (n−1) /2
ΦE=2×(2−1)×(45−1) /2=44
A.5 F検定
分散分析表を作り,表A.3のようにF検定を行う。
表A.3−分散分析表
要因
平方和 (S)
自由度 (φ)
不偏分散 (V)
分散比 (F)
主効果 (A)
10.76
1
10.76
17.4
誤差 (E)
27.24
44
0.62
総計 (T)
38.00
45
F144 (0.05) =4.06,F144 (0.01) =7.24
A.6 解析結果
B洗剤に比べて,A洗剤の洗浄力は優れている(有意水準1 %)。
76
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(参考)
台所用合成洗剤の洗浄力評価例
序文
この附属書は,台所用合成洗剤の洗浄力の評価例について記載するものであって,規定の一部ではない。
B.1
一対比較法による解析例
供試洗剤Aで洗浄したモデル汚れ(汚れ落ちの程度)について,3人の判定者がそれぞれ指標洗剤Bで
洗浄したモデル汚れ(汚れ落ちの程度)を対象として評価し,得られた表B.1の評価結果について解析す
る。
表B.1−評価結果
組合せ
判定者1
判定者2
判定者3
指標洗剤
供試洗剤
B1
A1
0
0
+1
B1
A2
0
+1
0
B1
A3
−1
0
−1
B2
A1
−1
0
0
B2
A2
0
0
0
B2
A3
−1
−1
−1
B3
A1
0
0
0
B3
A2
0
0
+1
B3
A3
−1
0
0
注記1 B1,B2,B3:指標洗剤によって繰返し実験を行ったそれぞれのモデル汚れ
注記2 A1,A2,A3:供試洗剤によって繰返し実験を行ったそれぞれのモデル汚れ
B.2
評価点の平均値
3人の判定者が判定した評価結果は,すべて繰返しとみなし,27回の繰返しとして評価点の平均値を表
B.2のように求める。
表B.2−評価点の度数
評価点 (N)
評価点の合計
(ΣNifi)
−2
−1
0
+1
+2
度数 (f)
0
7
17
3
0
−4
評価点の平均値
148
.0
27
4
i
−
−
∑
=
=
=nx
x
ここに,
xi: 評価点
n: 繰返し数
77
K 3362:2008
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B.3
標準偏差
標準偏差は,次の式によって算出する。
(
)
602
.0
1
1
2
i
2
i
=
=
−
−∑
∑
n
x
x
n
σ
B.4 t値
tの値は,次の式によって求める。
280
.1
−
=
=σn
x
t
B.5 t検定
t=(n−1,α)を求め,t検定を行う。
t= (26,0.05)=2.056であるから
|t|<t (26,0.05)
B.6
解析の結果
指標洗剤Bと供試洗剤Aとの洗浄力は同等とみなす。