K 2501:2003
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,石油連盟 (PAJ)/
財団法人日本規格協会 (JSA) から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,日
本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格である。これによってJIS K 2501:
1992は改正され,この規格に置き換えられる。
今回の改正では,対応国際規格ISO 3771:1994, Petroleum products−Determination of base number−
Perchloric acid potentiometric titration method,ISO 6618:1997, Petroleum products and lublicants−Determination
of acid or base number−Colour-indicator titration method,ISO 6619:1988, Petroleum products and lublicants−
Neutralization number−Potentiometric titration method及びISO 7537:1997, Petroleum products−Determination
of acid number−Semi-micro colour-indicator titration methodを基礎として用いた。
この規格の一部が,技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の
実用新案登録出願に抵触する可能性があることに注意を喚起する。経済産業大臣及び日本工業標準調査会
は,このような技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の実用新
案登録出願にかかわる確認について,責任はもたない。
JIS K 2501には,次に示す附属書がある。
附属書1(参考)指示薬光度滴定法
附属書2(参考)JISと対応する国際規格との対比表
K 2501:2003
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1. 適用範囲 ························································································································ 1
2. 引用規格 ························································································································ 2
3. 定義 ······························································································································ 2
4. 試験方法の種類 ··············································································································· 3
5. 指示薬滴定法 ·················································································································· 3
5.1 試験の原理 ··················································································································· 3
5.2 試薬 ···························································································································· 4
5.3 試験器 ························································································································· 6
5.4 試料採取方法及び調製方法······························································································· 6
5.5 試験の手順 ··················································································································· 7
5.6 計算及び結果 ················································································································ 8
5.7 精度 ···························································································································· 9
5.8 試験結果の報告 ············································································································ 10
6. セミミクロ指示薬滴定法(酸価) ······················································································ 10
6.1 試験の原理 ·················································································································· 10
6.2 試薬 ··························································································································· 11
6.3 酸価試験器(セミミクロ法)··························································································· 12
6.4 試料採取方法及び調製方法······························································································ 15
6.5 試験の手順 ·················································································································· 15
6.6 計算及び結果 ··············································································································· 16
6.7 精度 ··························································································································· 16
6.8 試験結果の報告 ············································································································ 17
7. 電位差滴定法(酸価) ····································································································· 17
7.1 試験の原理 ·················································································································· 17
7.2 試薬 ··························································································································· 18
7.3 試験器 ························································································································ 20
7.4 試料採取方法及び調製方法······························································································ 23
7.5 電位差滴定装置の調整 ··································································································· 23
7.6 試験器の調整 ··············································································································· 24
7.7 試験の手順 ·················································································································· 24
7.8 計算及び結果 ··············································································································· 25
7.9 精度 ··························································································································· 27
7.10 試験結果の報告 ··········································································································· 27
8. 電位差滴定法(塩基価・塩酸法) ······················································································ 27
K 2501:2003 目次
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
8.1 試験の原理 ·················································································································· 27
8.2 試薬 ··························································································································· 28
8.3 試験器 ························································································································ 30
8.4 試料採取方法及び試料調製方法························································································ 32
8.5 電位差滴定装置の調整 ··································································································· 32
8.6 試験器の調整 ··············································································································· 33
8.7 試験の手順 ·················································································································· 33
8.8 結果 ··························································································································· 34
8.9 精度 ··························································································································· 36
8.10 試験結果の報告 ··········································································································· 36
9. 電位差滴定法(塩基価・過塩素酸法) ················································································ 36
9.1 試験の原理 ·················································································································· 36
9.2 試薬 ··························································································································· 36
9.3 試験器 ························································································································ 38
9.4 試料採取方法及び調製方法······························································································ 40
9.5 電位差滴定装置の調整 ··································································································· 40
9.6 試験の手順 ·················································································································· 41
9.7 計算及び結果 ··············································································································· 43
9.8 逆滴定法 ····················································································································· 43
9.9 精度 ··························································································································· 44
9.10 試験結果の報告 ··········································································································· 45
附属書1(参考)指示薬光度滴定法 ························································································· 46
附属書2(参考)JISと対応する国際規格との対比表 ·································································· 53
K 2501:2003
(4)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
白 紙

2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 2501:2003
石油製品及び潤滑油─中和価試験方法
Petroleum products and lublicants─Determination of neutralization number
序文 この規格は,1994年に第2版として発行されたISO 3771,Petroleum products−Determination of base
number−Perchloric acid potentiometric titration method,1997年に第2版として発行されたISO 6618,Petroleum
products and lublicants−Determination of acid or base number−Colour-indicator titration method,1988年に第1
版として発行されたISO 6619,Petroleum products and lublicants−Neutralization number−Potentiometric
titration method及び1997年に第2版として発行された,ISO 7537 Petroleum products−Determination of acid
number−Semi-micro colour-indicator titration methodを元に,対応する部分については,対応国際規格を翻訳
し,技術的内容を変更することなく作成した日本工業規格であるが,対応国際規格に規定されていない規
定項目を日本工業規格として追加している。
なお,この規格で側線又は点線の下線を施してある箇所は,原国際規格を変更している事項又は原国際
規格にはない事項である。変更の一覧表をその説明を付けて附属書2に示す。
1. 適用範囲 この規格は,石油製品及び潤滑油の中和価を指示薬滴定法,電位差滴定法及び指示薬光度
滴定法によって,測定する方法について規定する。
備考1. この規格は,危険な試薬,操作及び装置を使うことがあるが,安全な使用方法をすべてにわ
たって規定しているわけではないので,この試験方法の使用者は,試験に先立って,適切な
安全上及び健康上の禁止事項を決めておかなければならない。
2. 中和価とは,酸価,強酸価,塩基価及び強塩基価の総称をいう。
3. この規格の対応国際規格を表1に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide21に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
表 1 対応国際規格
試験方法
対応国際規格
指示薬滴定法
ISO 6618:1997 Petroleum products and lublicants−Determination of acid or base
number−Colour-indicator titration method (MOD)
セミミクロ指示薬滴定
法(酸価)
ISO 7537:1997 Petroleum products−Determination of acid number Semi-micro
colour-indicator titration method (MOD)
電位差滴定法(酸価)
ISO 6619:1988 Petroleum products and lublicants−Neutralization number−
Potentiometric titration method (MOD)
電位差滴定法
(塩基価・過塩素酸法) ISO 3771:1994 Petroleum products−Determination of base number−Perchloric
acid potentiometric titration method (MOD)
2
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0113 電位差・電流・電量・カールフィッシャー滴定方法通則
JIS K 0557 用水・排水の試験に用いる水
JIS K 1107 高純度窒素
JIS K 2251 原油及び石油製品−試料採取方法
JIS K 2839 石油類試験用ガラス器具
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS K 8102 エタノール (95) (試薬)
JIS K 8121 塩化カリウム(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8223 過塩素酸(試薬)
JIS K 8227 過塩素酸ナトリウム一水和物(試薬)
JIS K 8355 酢酸(試薬)
JIS K 8574 水酸化カリウム(試薬)
JIS K 8603 ソーダ石灰(試薬)
JIS K 8680 トルエン(試薬)
JIS K 8693 p-ナフトールベンゼイン(試薬)
JIS K 8799 フェノールフタレイン(試薬)
JIS K 8809 フタル酸水素カリウム(試薬)
JIS K 8839 2-プロパノール(試薬)
JIS K 8886 無水酢酸(試薬)
JIS K 8893 メチルオレンジ(試薬)
JIS Z 8401 数値の丸め方
JIS Z 8402-6 測定方法及び測定結果の精確さ(真度及び精度)−第6部:精確さに関する値の実用的な
使い方
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
JIS Z 8802 pH測定方法
3. 定義 この規格で用いる主な用語の定義は,次による。
a) 酸価 (acid number) 試料1 g中に含まれる酸性成分を中和するのに要する水酸化カリウムのミリグ
ラム (mg) 数。
b) 強酸価 (strong acid number) 試料1 g中に含まれる強酸性成分を中和するのに要する水酸化カリウ
ムのミリグラム (mg) 数。
c) 塩基価 (base number) 試料1 g中に含まれる塩基性成分を中和するのに要する塩酸又は過塩素酸と
当量の水酸化カリウムのミリグラム (mg) 数。
参考1. グラム当たりの水酸化物のミリ当量で表すこともある。
2. 酸価 [a] を全酸価,塩基価 [c] を全塩基価と呼称することがある。
3
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 強塩基価 試料1 g中に含まれる強塩基性成分を中和するのに要する塩酸と当量の水酸化カリウムの
ミリグラム (mg) 数。
4. 試験方法の種類 試験方法の種類は,表2による。
表 2 試験方法の種類
試験方法
試験項目
特徴(参考)
箇条番号
指示薬滴定法
(ISO 6618に対応)
酸価
強塩基価
強酸価
a) 指示薬による色の変化によって判定するため,淡色油及
び比較的添加剤の少ない油に適している。
b) 簡単な器具で,しかも短時間で測定できる。
5.
セミミクロ指示薬滴定法
(ISO 7537に対応)
酸価
a) 供試試料が少なく,指示薬滴定又は電位差滴定ができな
いものに適している。
b) 試料の希釈比が大きいため,指示薬滴定法に比べて,終
点の判定が容易である。
c) タービンに使用する新油及び使用油の精度は,指示薬滴
定法と同等である。
6.
電位差滴定法
(ISO 6619に対応)
酸価
強酸価
a) 新油及び使用油並びに淡色油及び濃色油の広範囲の油種
に適用できる。
b) 非水溶液の電位差測定のため,応答が遅く測定時間を要
する。
c) 電位差計を用いるため自動化が容易である(手動滴定及
び自動滴定法を規定)。
7.
電位差滴定法
(塩基価・塩酸法)
塩基価
強塩基価
a) 新油及び使用油並びに淡色油及び濃色油の広範囲の油種
に適用できる。
b) 非水溶液の電位差測定のため,応答が遅く測定に時間を
要する。
c) 過塩素酸法に比べ低い値を示すことが多い。
d) 電位差計を用いるため自動化が容易である(手動滴定及
び自動滴定法を規定)。
8.
電位差滴定法
(塩基価・過塩素酸法)
(ISO 3771に対応)
塩基価
a) 滴定溶剤の容量及び試料はかり採り量が異なる2方法(A
法及びB法)があり,少量試料にも適用できる。
b) 新油,使用油及び添加剤ともA法とB法とは同等の結果
になる。
c) 短時間で,しかも精度よく測定できる。
d) 電位差計を用いるため自動化が容易である(手動滴定及
び自動滴定法を規定)。
9.
指示薬光度滴定法
酸価
a) 指示薬の色の変化を透過率としてとらえる自動試験器で
あり,終点の判定が容易である。
b) 室内併行精度及び室間再現精度は,指示薬滴定法と同等
である。
附属書1
(参考)
参考 この規格のほかに酸価試験方法としては,次のものが規定されている。
a) 電気絶縁油のJIS C 2101(電気絶縁油試験方法)
b) 航空タービン燃料油のJIS K 2276(石油製品−航空燃料油試験方法)
5. 指示薬滴定法
5.1
試験の原理 酸価又は強塩基価を試験するには,試料をトルエン,2-プロパノール及び少量の水を
含む滴定溶剤に溶かし,p-ナフトールベンゼインを指示薬として室温で,水酸化カリウムの2-プロパノー
ル溶液又は塩酸の2-プロパノール溶液で滴定する。強酸価を試験するには試料中の強酸成分を熱水で抽出
し,メチルオレンジを指示薬として水酸化カリウムの2-プロパノール溶液で滴定する。
4
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考1. この方法は,トルエンと2-プロパノール混合液に溶解する石油製品及び潤滑油中の酸性成分
又は強塩基性成分を指示薬滴定法によって測定するものである。この方法は,水中の解離定
数が10−9より小さい弱酸性成分又は弱塩基性成分は測定されない。水中の解離定数が10−9
より大きい酸性成分又は塩基性成分の測定に適し,もし,塩類の加水分解定数が10−9より大
きい場合には,その塩類は反応する。
2. 新油及び使用油中の酸性を示す成分としては,有機酸,無機酸,エステル類,フェノール化
合物,ラクトン類,レジン類及び重金属塩類並びに酸化防止剤,清浄剤などの添加剤がある。
また,塩基性を示す成分としては,有機塩基類,無機塩基類,アミノ化合物弱塩の塩類(石
けん),多酸塩基の塩基性塩類及び重金属塩類並びに酸化防止剤,清浄剤などの添加剤がある。
3. この方法は,塩基添加形の潤滑油中の塩基性成分の測定には,不適切であり,このような試
料は電位差滴定法を用いる。
4. この方法は,酸化条件において使用する油の相対的な変化を測定するときに用いることがで
きる。滴定は平衡状態で行われるが,使用条件下における油の性能を予測するために用いら
れる酸性又は塩基性の絶対値を測定するものではない。また,腐食性と酸価又は塩基価との
間には一般的な関係は知られていない。
5. 多くの切削油,さび止め油,混成油又は濃暗色の油のように指示薬による終点のはっきりし
ないものは,電位差滴定法を用いるとよい。この指示薬滴定法によって得られる値は,電位
差滴定法で得られる値と一致するとは限らない。
5.2
試薬 試薬は,次による。
a) 2-プロパノール JIS K 8839に規定するもの。
b) トルエン JIS K 8680に規定するもの。
c) 水 JIS K 0557に規定するA3のもの。
d) フェノールフタレイン溶液 JIS K 8799に規定するフェノールフタレイン0.5 gをJIS K 8102に規定
するエタノール(95)100 mLに溶かしたもの。
e) 滴定溶剤 トルエン500 mL及び水5 mLを2-プロパノール495 mLに加えたもの。
f)
0.1mol/L塩酸の2-プロパノール溶液
1) 調製 2-プロパノール1 LにJIS K 8180に規定する塩酸9 mLを加えて混合する。
備考 市販の0.1 mol/L塩酸の2-プロパノール溶液を用いてもよい。
2) 標定 標定は,次による。
2.1) 0.1 mol/L塩酸の2-プロパノール溶液約8 mLを正しくはかり採り,二酸化炭素を含まない水125 mL
で希釈する。
2.2) 0.1 mol/L水酸化カリウムの2-プロパノール溶液を用い,フェノールフタレインを指示薬として滴
定するか,又は電位差滴定する。
2.3) 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度は,次の式によって算出し,JIS Z 8401の規定に
よって丸めの幅を0.000 1に丸める。
2
1
KOH
HCl
v
v
c
c
×
=
ここに,
cHCl: 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度 (mol/L)
cKOH: 0.1 mol/L水酸化カリウム2-プロパノールの溶液のモル濃
度 (mol/L)
5
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
v1: 終点までの滴定に要した0.1 mol/L水酸化カリウムの2-プ
ロパノール溶液の量 (mL)
v2: 0.1 mol/L塩酸の2-プロパノール溶液のはかり採り量 (mL)
2.4) 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度変化が0.000 5 mol/L以上にならない間隔でときど
き標定し直す。
g) 0.1 mol/L水酸化カリウムの2-プロパノール溶液
1) 調製 三角フラスコ2 000 mLに2-プロパノール約1 Lを採り,これにJIS K 8574に規定する水酸
化カリウム6 gを加え,底にかたまりができないようにかき混ぜながら,15〜20分間静かに煮沸さ
せる。保護管を付けた後,室温まで放冷する。この溶液を二酸化炭素を遮りながら微細なガラスフ
ィルターでろ過する。
溶液は,JIS K 8603に規定するソーダ石灰又はけい酸ソーダなどの乾燥剤を入れた保護管を付け
て大気中の二酸化炭素の侵入を防ぐ。また,溶液が,コルク,ゴム及びけん化するようなグリース
と接触しないようにポリエチレン瓶などに保存する。
備考 市販の0.1 mol/L水酸化カリウムの2-プロパノール溶液を用いてもよい。
2) 標定 フタル酸水素カリウムによる標定は,次による。
2.1) JIS K 8005に規定するフタル酸水素カリウムを105 ℃で2時間乾燥し,その0.1〜0.15 gを0.2 mg
のけたまではかり採り,二酸化炭素を含まない水100 mLに溶かし,フェノールフタレイン溶液を
指示薬として滴定するか,又は電位差滴定する。
2.2) 水100 mLを用いて空試験を行う。
2.3) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度は,次の式によって算出し,JIS Z 8401
の規定によって丸めの幅を0.000 1に丸める。
)
(
23
.
204
000
1
KOH
1b
V
W
c
−
=
ここに, cKOH: 0.1mol/L水酸化カリウムの2-プロパノール溶液のモル濃度
(mol/L)
W: フタル酸水素カリウムのはかり採り量 (g)
V1: 終点までの滴定に要した0.1mol/L水酸化カリウムの2-プロ
パノール溶液の量 (mL)
b: 空試験におけるV1に相当する量 (mL)
204.23: フタル酸水素カリウムの式量
2.4) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度の変化が0.000 5 mol/L以上にならない
間隔でときどき標定し直す。
備考1. アミド硫酸による標定は,次による。
a) JIS K 8005に規定するアミド硫酸の結晶粉末を真空デシケータ中で約48時間乾燥し,その
2.0〜2.5 gを0.1 mgのけたまではかり採り,二酸化炭素を含まない水に溶かし,250 mL全量
フラスコに移し,同じ水を標線まで加える。
b) その25 mLを全量ピペットを用いてはかり採り,水100 mLを加える。JIS K 8001の4.4によ
って調製したブロモチモールブルー溶液を指示薬として0.1 mol/L水酸化カリウムの2-プロ
パノール溶液で滴定する。
c) 水100 mLを用いて空試験を行う。
6
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
)
(
093
.
97
100
KOH
1b
V
W
c
−
=
ここに, cKOH: 0.1mol/L水酸化カリウムの2-プロパノール溶液のモル濃度
(mol/L)
W: アミド硫酸のはかり採り量 (g)
V1: 終点までの滴定に要した0.1mol/L水酸化カリウムの2-プロ
パノール溶液の量 (mL)
b: 空試験におけるV1に相当する量 (mL)
97.093: アミド硫酸の式量
2. 2-プロパノールのような有機溶剤の膨脹係数は,比較的大きいので,0.1 mol/L水酸化カリウ
ムの2-プロパノール溶液の標定は試料の試験温度に近い温度で行う。
h) メチルオレンジ溶液 JIS K 8893に規定するメチルオレンジ0.1 gを水100 mLに溶かしたもの。
i)
p-ナフトールベンゼイン溶液 JIS K 8693に規定するp-ナフトールベンゼイン0.1 gを滴定溶剤100
mLに溶かしたもの。
5.3
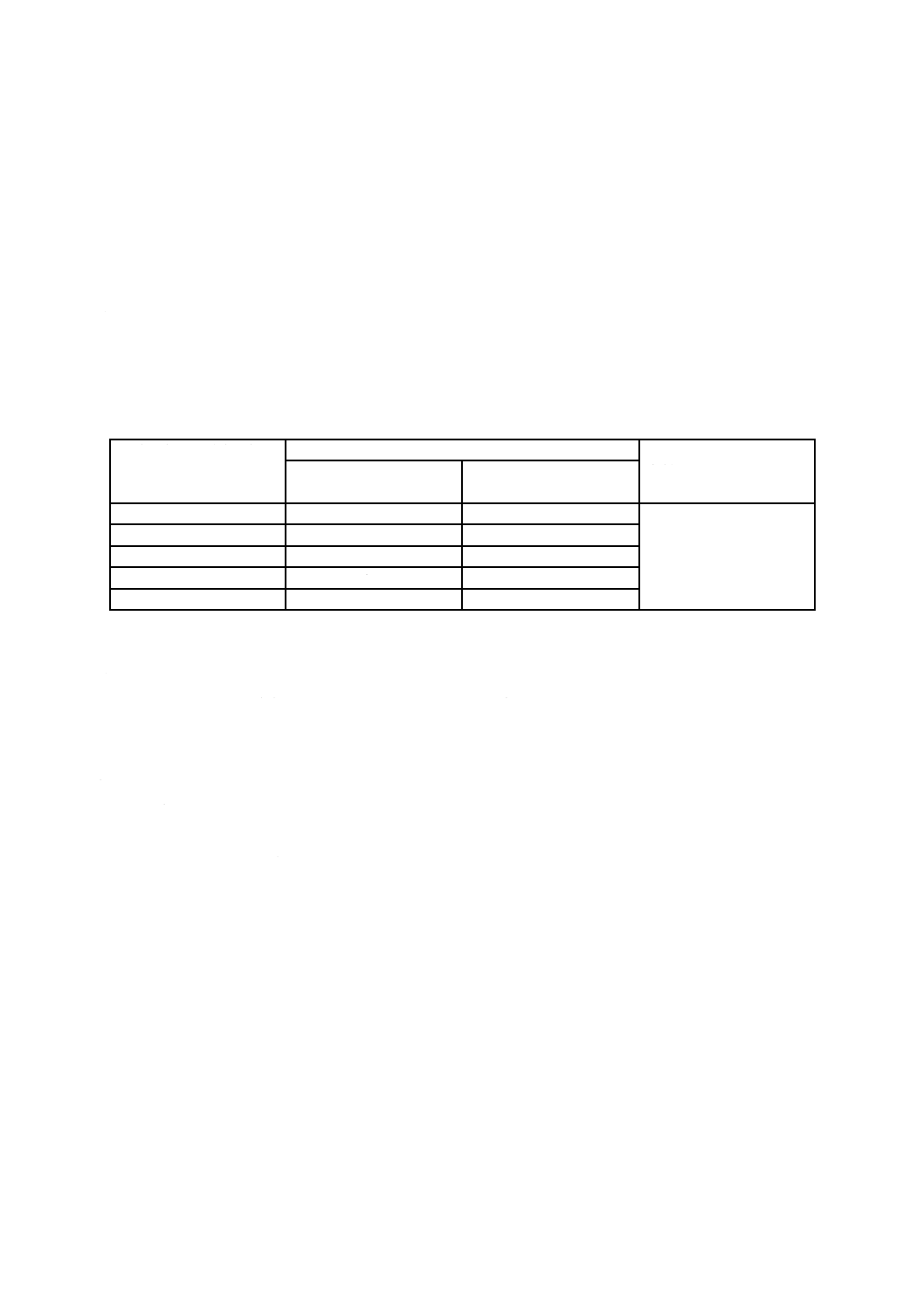
試験器 試験器は,次による。
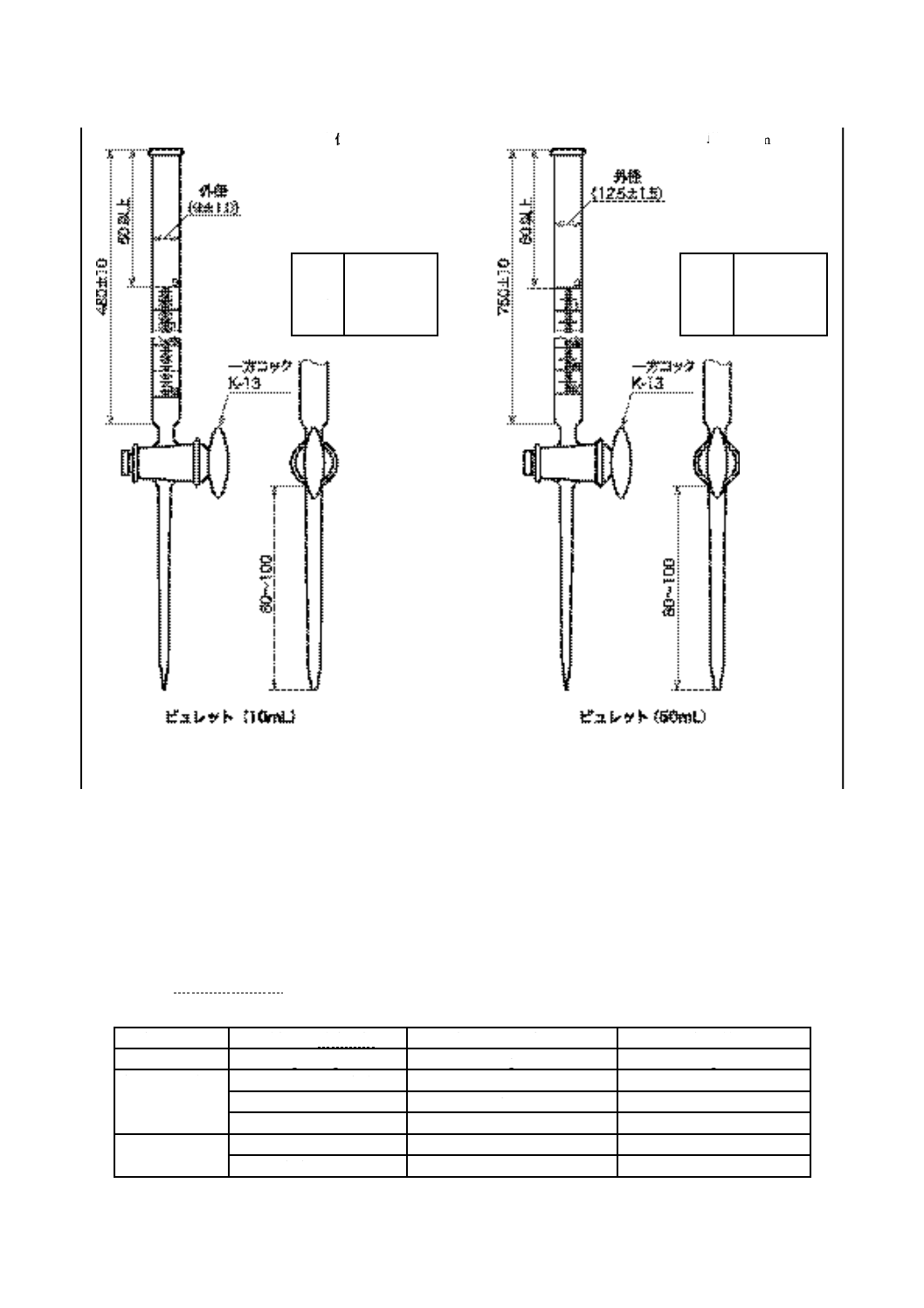
a) ビュレット 図1に示す,0.05 mL目盛付きの10 mL容量又は0.1 mL目盛付きの50 mL容量のもの(1)。
注(1) JIS K 2839に規定する図99及び図100が相当する。材質は,ほうけい酸ガラス-2以上とする。
備考 これと同等の性能をもつ電動ビュレットを用いてもよい。
5.4
試料採取方法及び調製方法 試料採取方法及び調製方法は,次による。
a) 試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法によるか,又はそれらに
準じた方法によって採取及び調製する。
b) 使用油の試料の調製 使用油の試料の調製は,次による。
1) 試料中に沈殿物があるときは,沈殿物自体が酸性又は塩基性であるか,また,それが試料中の酸性
又は塩基性の物質を吸着することがあるから,このような沈殿物を含む試料を採るときは,試料全
体を代表するようによくかき混ぜる。
2) 試料を60 ℃±5 ℃に加熱し,沈殿物を均一にする。沈殿物が含まれていない場合は加熱をしなく
てもよい。
備考 使用油は保存中に変質することがあるので,採取後,できるだけ早く測定する。
3) 試料に沈殿物が目視で観察されるときは,すべての沈殿物を均一にした後,試料をJIS Z 8801-1に
規定する目開き150 μmの金属製網ふるいでろ過し,大きな異物を除去する。
7
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm 単位 mm
備考 図に示すものは一例であり,同一機能であれば別の形状・寸法でもよい。
図 1 ビュレット
5.5
試験の手順 試験の手順は,次による。
5.5.1
酸価
a) 三角フラスコ250 mLに表3に従って試料をはかり採り,滴定溶剤100 mLと指示薬としてp-ナフトー
ルベンゼイン溶液0.5 mLを加え,試料が完全に溶剤に溶けるまで,栓をしないで振り混ぜる。試験溶
液の色が黄とう色を呈したならば,b)に従い操作を進める。もし試験溶液の色が緑又は暗緑色を呈し
たならば,5.5.2に従って強塩基価の測定を行う。
参考 変色が濃く,終点の判定が困難な場合は,p-ナフトールベンゼイン溶液の添加量を0.2 mL程度
にするとよい。
表 3 試料のはかり採り量
試料の種類
酸価又は強塩基価
試料のはかり採り量
はかり採り最小目盛
mgKOH/g
g
g
新油又は淡色油
0を超え 3以下
20 ±2
0.05
3を超え 25以下
2 ±0.2
0.01
25を超え 250以下
0.2 ±0.02
0.001
使用油又は濃色
油
0を超え 25以下
2 ±0.2
0.01
25を超え 250以下
0.2 ±0.02
0.001
目盛
単位 mL
範囲 0〜10
目量 0.05
長線 0.5ごと
数字 1ごと
目盛
単位 mL
範囲 0〜50
目量 0.1
長線 1ごと
数字 1ごと
8
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考 酸価の低い淡色の試料は,正確な結果を得るために20 gを用いてもよい。濃色の試料は,できるだけ色
の濃さによる影響を受けないようにするため規定量を用いる。
b) 直ちに30 ℃以下で0.1 mol/L水酸化カリウムの2-プロパノール溶液で滴定する。終点に近づいたなら
ば溶液中に二酸化炭素が溶け込まないようにして十分に振とうする。酸性の油の場合は,終点に近づ
くにつれて黄とう色が緑又は暗緑色に変化する。終点であるかどうかは,色が15秒間変わったままで
いるか,又は0.1 mol/L塩酸の2-プロパノール溶液を2滴加え,色が元にもどるかどうかで判断する。
備考 色の濃い油は,激しく振とうしてわずかに表面を泡立たせ,台上に置いた白の蛍光灯の光をあ
てながら滴定による色の変化を見る。
c) 空試験は,滴定溶剤100 mLに指示薬としてp-ナフトールベンゼイン溶液0.5 mLを加え,0.1 mol/L水
酸化カリウムの2-プロパノール溶液を0.05〜0.1 mLずつ滴下して行う。
参考 変色が濃く,終点の判定が困難なときは,p-ナフトールベンゼイン溶液の添加量を0.2 mL程度
にするとよい。
5.5.2
強塩基価
a) 5.5.1a)で混合液の色が緑色又は暗緑色を呈したときは,5.5.1 b)の操作に準じ,0.1 mol/L水酸化カリウ
ムの2-プロパノール溶液の代わりに0.1 mol/L塩酸の2-プロパノール溶液を用いて溶液の色が緑色又
は暗緑色から黄とう色に変化するまで滴定する。
b) 空試験は,5.5.1 c)に従って行う。
備考 通常,滴定溶剤には,試料の強塩基性成分と反応する弱酸性不純物を含んでいる。試料の強塩
基価を補正するためには,空試験を行って滴定溶剤中の弱酸性成分に相当する0.1 mol/L水酸化
カリウムの2-プロパノール溶液の量を測定する必要がある。
5.5.3
強酸価
a) 分液漏斗250 mLに試薬約25 gを0.05 gのけたまではかり採り,沸騰水100 mLを加え,十分に振と
うした後,水層を分離し,ビーカ500 mLに保存する。さらに2回,沸騰水50 mLで油中の酸性成分
を抽出し,分離した水層を先のビーカに加える。この抽出液に指示薬としてメチルオレンジ溶液0.1 mL
を加え,ピンク色又は赤を呈したならば0.1 mol/L水酸化カリウムの2-プロパノール溶液で溶液の色
が黄褐色になるまで滴定する。
ピンク色又は赤でない場合は,強酸価を0とする。
b) 空試験は,a)で使用した沸騰水200 mLを三角フラスコ250 mLに採り,指示薬としてメチルオレンジ
溶液0.1 mLを加え,溶液の色が黄とう色を呈したならば,試料の滴定の際に得られた同一の色の濃さ
まで0.1 mol/L塩酸の2-プロパノール溶液で滴定する。もし指示薬の色がピンク色又は赤を呈したな
らば,試料の滴定に用いた同一の終点まで0.1 mol/L水酸化カリウム2-プロパノール溶液で滴定する。
5.6
計算及び結果 計算及び結果は,次による。
次の式によって酸価,強塩基価及び強酸価を算出し,有効数字3けたに丸める。ただし,1 mgKOH/g未
満の場合はJIS Z 8401の規定によって丸めの幅を0.01に丸める。
a) 酸価
m
V
V
c
AN
)
(
KOH
1.
56
0
1−
=
×
×
ここに,
AN: 酸価 (mgK0H/g)
V1: 試料の滴定に要した0.1 mol/L水酸化カリウムの2-プロパ
ノール溶液の量 (mL)
V0: 空試験の滴定に要した0.1 mol/L水酸化カリウムの2-プロ
9
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
パノール溶液の量 (mL)
cKOH: 0.1mol/L水酸化カリウムの2-プロパノール溶液のモル濃度
(mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
b) 強塩基価
m
c
V
c
V
SBN
)
KOH
HCL
(1.
56
0
5
×
×
×
=
ここに,
SBN: 強塩基価 (mgKOH/g)
V5: 試料の滴定に要した0.1 mol/L塩酸の2-プロパノール溶液
の量 (mL)
cHCl: 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度 (mol/L)
V0: 空試験の滴定に要した0.1 mol/L水酸化カリウムの2-プロ
パノール溶液の量 (mL)
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
c) 強酸価
1) 空試験を0.1 mol/L塩酸の2-プロパノール溶液で行った場合
m
c
V
c
V
SAN
)
HCl
KOH
(1.
56
3
2
×
×
×
=
ここに,
SAN: 強酸価 (mgKOH/g)
V2: 5.5.3 a)で得られた抽出液の滴定に要した0.1 mol/L水酸化
カリウムの2-プロパノール溶液の量 (mL)
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
V3: 5.5.3 b)の空試験の滴定に要した0.1 mol/L塩酸の2-プロパ
ノール溶液の量 (mL)
cHCl: 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
2) 空試験を0.1 mol/L水酸化カリウムの2-プロパノール溶液で行った場合
m
V
V
c
SAN
)
(
KOH
1.
56
4
2−
=
×
×
ここに,
SAN: 強酸価 (mgKOH/g)
V2: 5.5.3 a)で得られた抽出液の滴定に要した0.1 mol/L水酸化
カリウムの2-プロパノール溶液の量 (mL)
V4: 5.5.3 b)空試験の滴定に要した0.1 mol/L水酸化カリウムの
2-プロパノール溶液の量 (mL)
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
5.7
精度 この試験方法によって得られた試験結果の許容差(確率0.95)は,次のとおりである。
備考1. この精度は試料の色が非常に濃すぎるものや,指示薬の色の変化が不明確のものには適用し
ない。
2. 試験結果が許容差を外れた場合は,JIS Z 8402-6の規定によって処理する。

10
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 室内併行精度 同一試験室において,同一人が同一試験器で引き続き短時間内に同一試料を2回試験
したとき,試験結果の差の許容差を表4に示す。
表 4 室内併行精度
試料の種類
酸価,強酸価,強塩基価
室内併行許容差
mgKOH/g
mgKOH/g
淡色の鉱油系新油並びに酸化
防止剤入り蒸気タービン油の
新油及び使用油
0.0を超え0.1以下
0.03
0.1を超え0.5以下
0.05
0.5を超え1.0以下
0.08
1.0を超え2.0以下
0.12
備考 その他の油種については,データが不十分であるので精度は規定しない。
b) 室間再現精度 異なる試験室において,別人が別の試験器で同一試料をそれぞれ1回ずつ試験したと
き,2個の試験結果の差の許容差を表5に示す。
表 5 室間再現精度
試料の種類
酸価,強酸価,強塩基価
室間再現許容差
mgKOH/g
mgKOH/g
淡色の鉱油系新油並びに酸化
防止剤入り蒸気タービン油の
新油及び使用油
0.0を超え0.1以下
0.04
0.1を超え0.5以下
0.08
0.5を超え2.0以下
平均値の15%
備考 その他の油種については,データが不十分であるので精度は規定しない。
5.8
試験結果の報告 試験結果には,次の事項を記載する。
a) 試料名,採取場所及び採取年月日
b) JISの規格番号:JIS K 2501
c) 試験の名称・箇条番号及び5.6によって得られた結果
d) 特記事項
6. セミミクロ指示薬滴定法(酸価)
6.1
試験の原理 試料をトルエン,2-プロパノール及び少量の水を含む滴定溶剤に溶かし,p-ナフトール
ベンゼインを指示薬として,室温で窒素雰囲気中において水酸化カリウムの2-プロパノール溶液を用いて
滴定し,酸価を求める。
備考1. この方法は,試料の量が少なく5.及び7.による正確な測定が困難な場合に適用することがで
きる。この方法は,水中の解離定数が10−9を超える酸の測定に適用する。解離定数が10−9
未満の弱酸には適用されない。塩の加水分解定数が10−9より大きい場合には,その塩は反応
する。
2. この方法は,酸化状態で使用中の油に生じる酸価の相対変化を示すために用いることができ
る。滴定は明確な平衡状態で行われるが,この方法は,油の使用状態の性能を予測するため
に用いられる酸の絶対的な性質を測定するものではない。また,腐食性と酸価との間に,一
般的関係は知られていない。
3. この方法は,5.及び7.で必要な試料量より少量ですむため,測定用試料の減少を最小に抑え
られるので,酸化安定度試験等において,定期的に抜き取った酸化油の酸価を試験する方法
に適している。
4. 切削油,さび止め油などの複合物,又は極度に暗色化した石油製品は指示薬の終点があいま
いなので,この方法で試験するには終点の判断が困難な場合がある。このような試料は,十
11
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
分な試料量があれば7.で測定ができる。しかし,この方法と電位差滴定法で得られた結果が,
必ずしも一致するとは限らない。
5. 主として油圧及び蒸気タービンに使用される新油及び酸化油では,この方法を用いて得られ
た結果と5.で得られた結果は,精度の範囲内で一致することが知られている。
6.2
試薬 試薬は,次による。
a) 2-プロパノール JIS K 8839に規定するもの。
b) トルエン JIS K 8680に規定するもの。
c) 水 JIS K 0557に規定するA3のもの。
d) フェノールフタレイン溶液 JIS K 8799に規定するフェノールフタレイン0.10 gをJIS K 8102に規定
するエタノール (95) 50 mL及び水50 mLの混合液に溶解したもの。
e) 0.01 mol/L水酸化カリウムの2-プロパノール溶液
1) 調製 2 Lの三角フラスコに2-プロパノール約1Lを採り,これにJIS K 8574に規定する水酸化カリ
ウム3 gを加え,フラスコの底に固形物が析出するのを防ぐためにかくはんしながら15分間沸騰さ
せる。この溶液を大気から遮り,一夜(約16 h)放置した後,その上澄み液を孔径10 μmの四ふっ
化エチレン樹脂製メンブランフィルタで,不必要に大気にさらされないようにろ過し,このろ液200
mLを2-プロパノールで全量約1Lに希釈し,自動ビュレットの滴定試薬瓶に入れる。
この溶液の標定は,次の操作によって行い,溶液の濃度変化が0.000 3 mol/L以上にならない間隔
でときどき標定し直す。
備考1. 0.01 mol/L水酸化カリウムの2-プロパノール溶液の濃度が0.010 mol/L±0.002 mol/Lの範囲に
なるように希釈する。
2. 有機溶剤類の体積膨張係数は,比較的大きいので,0.01 mol/L水酸化カリウムの2-プロパノ
ール溶液の標定は,使用時の温度に近い温度で行う。
2) 標定 JIS K 8005に規定するフタル酸水素カリウムを110 ℃で1時間以上乾燥し,その約0.1 gを
0.1 mgまではかり採り,水に溶解し,1 Lの全量フラスコに移し,水を標線まで加える。
その40 mLを全量ピペットを用いて滴定ビーカに採り,フェノールフタレイン溶液6滴を加え,
0.01 mol/L水酸化カリウムの2-プロパノール溶液を用いて6.5 b)〜c)によって溶液の色が無色からピ
ンクになるまで滴定する。この操作を2回以上繰り返す。別に用意した水40 mLについて空試験を
行う。
3) 計算 0.01 mol/L水酸化カリウム2-プロパノール溶液の濃度を次の式によって求め,JIS Z 8401の
規定によって丸めの幅0.000 1に丸める。
)
(
23
.
204
40
KOH
2
1
1
V
V
m
c
−
=
ここに,
cKOH: 0.01 mol/L水酸化カリウムの2-プロパノール溶液のモル
濃度 (mol/L)
m1: フタル酸水素カリウムのはかり採り量 (g)
V1: 終点までの滴定に要した0.01 mol/L水酸化カリウムの2-
プロパノール溶液の量の平均値 (mL)
V2: 空試験におけるV1に相当する量 (mL)
204.23: フタル酸水素カリウムの式量
f)
滴定溶剤 トルエン500 mL及び水5 mLを2-プロパノール495 mLに加えたもので,調製後,直ちに
滴定溶剤用ビュレットに入れる。
12
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考 トルエンの蒸気は有毒であるから吸入しないように注意する。特にトルエンが目に入ると危険
であるが,他の有機溶剤の場合でも入らないように注意する。
g) p-ナフトールベンゼイン溶液 JIS K 8693に規定するp-ナフトールベンゼイン1.0 gを滴定溶液100
mLに溶解したもの。
h) 窒素 JIS K 1107に規定する高純度窒素2級以上のもの。
6.3
酸価試験器(セミミクロ法) 図2に示す構造のもので,次のa)〜h)で構成する。
a) 滴定ビュレット 図2に示す形状で0.01 mL目盛付きの容量2 mL以上のミクロビュレットと容量1 L
の暗褐色ガラス製滴定試薬瓶とをすり合わせ面で接合したもの。
ミクロビュレットの足の長さを滴定試薬瓶の底から約20 mm上にし,滴定試薬瓶の底にたい積した
沈殿物をかき乱さないようにする。ミクロビュレット及び滴定試薬瓶の出入口には,大気中の二酸化
炭素及び水を除去する吸収管を取り付ける。
吸収管には二酸化炭素吸収剤としてJIS K 8603に規定するソーダ石灰(元素分析用)を詰め,乾燥
剤として粒度0.84〜2.0 mmの無水硫酸カルシウムなどの乾燥剤を詰める。
備考1. ミクロビュレットと同等の性能をもつ電動ビュレットを用いてもよい。
2. 透明ガラス製滴定試薬瓶を用いる場合は,アルミニウムはくなどで覆い,試薬が光線にさら
されないようにする。
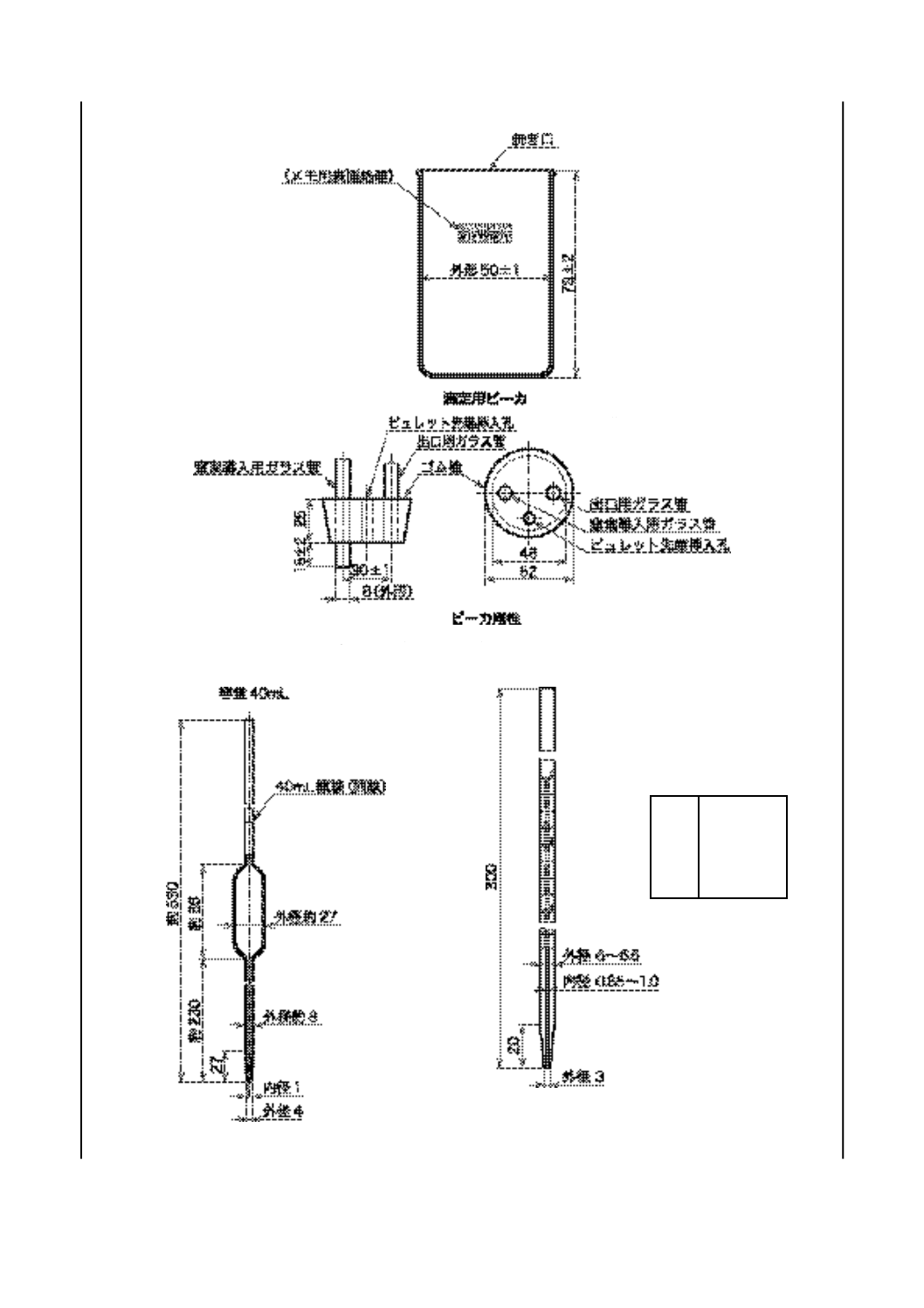
b) 滴定ビーカ 図3に示す形状・寸法のガラス製のもの(2)。
注(2) JIS K 2839に規定する図58のものが相当する。
c) 滴定ビーカ用栓 図3に示す形状・寸法のもので,用いる試薬に侵されず,弾性のあるゴム栓(ネオ
プレンゴム製など)に,窒素導入用ガラス管,出口用ガラス管及び自動ビュレット先端挿入用の孔を
備えたもの。
d) 窒素流量計 流量10 L/h±1 L/hの調節,測定ができるもの。
e) かき混ぜ機 かき混ぜの速さを可変できるマグネチックスターラ,及び四ふっ化エチレン樹脂で被覆
した回転子。
f)
全量ピペット 図4に示す形状・寸法のガラス製のもの(3)。
注(3) JIS K 2839に規定する図229のものが相当する。
g) メスピペット 図5に示す形状・寸法のガラス製のもの(4)。
注(4) JIS K 2839に規定する図230のものが相当する。
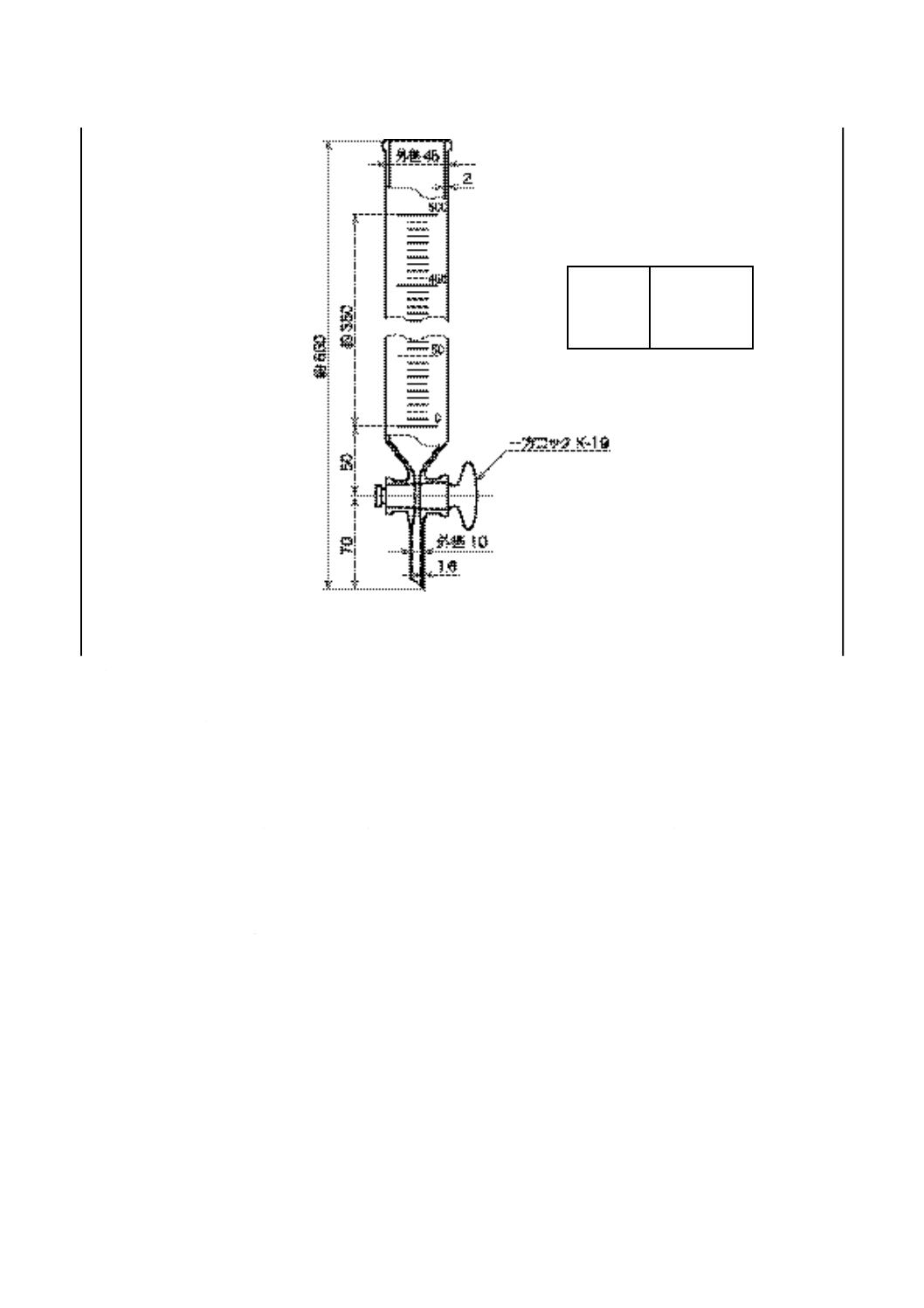
h) 滴定溶剤用ビュレット 図6に示す形状・寸法のガラス製のもの(5)で,ビュレットの上部にゴム栓(ネ
オプレンゴム製など)で連結し,二酸化炭素及び水を除去する吸収管を取り付ける。
注(5) JIS K 2839に規定する図231のものが相当する。
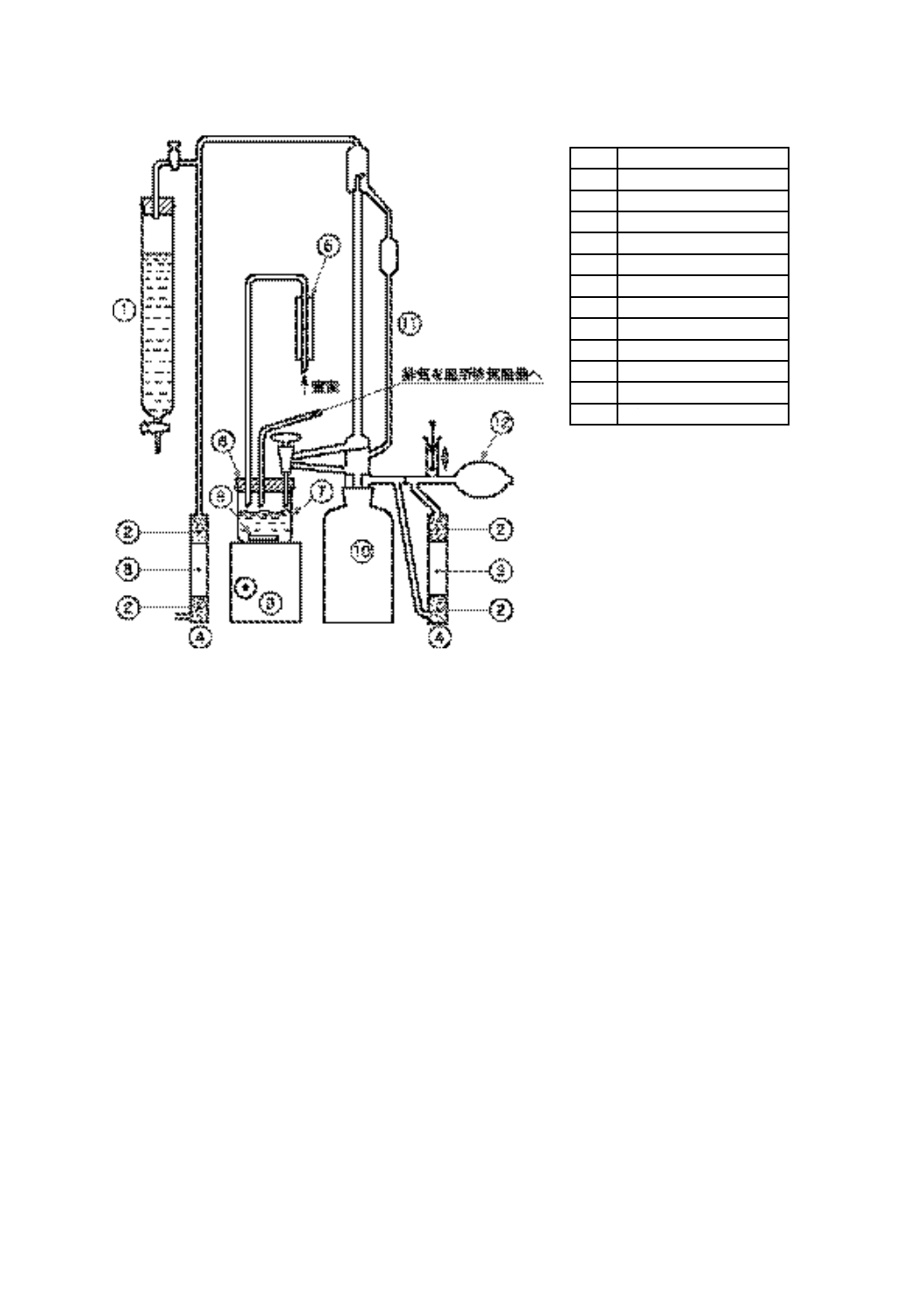
13
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図 2 酸価試験器(セミミクロ法)(一例)
番号
部品名
①
滴定溶剤用ビュレット
②
乾燥剤
③
二酸化炭素吸収剤
④
吸収管
⑤
窒素流量計
⑥
滴定ビーカ用栓
⑦
滴定ビーカ
⑧
回転子
⑨
かき混ぜ機
⑩
滴定試薬瓶
⑪
滴定ビュレット
⑫
ゴム球
14
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図 3 滴定ビーカ及びビーカ用栓(一例)
図 4 全量ピペット
図 5 メスピペット
単位 mm
単位 mm
単位 mm
単位 mm
目盛
単位 mL
範囲 0〜0.1
目量 0.001
長線 0.005ごと
冠線 0.01 ごと
数字 0.01 ごと
15
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図 6 滴定溶剤用ビュレット
6.4
試料採取方法及び調製方法 試料採取方法及び調製方法は,次による。
a) 試料はJIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法によるか,又はそれらに準
じた方法によって採取及び調製する。
6.4.1
試料の前処理 試料中に沈殿物があるような酸化油では,沈殿物が酸性又は塩基性であったり,試
料の塩基性物質又は酸性物質を吸収することがあるので,次の操作を行う。
a) 試料に目視で沈殿物が見られるときは,試料容器内で試料を60 ℃±5 ℃に加熱し全沈殿物が均一に
なるまでかくはんする。
b) 元の試料容器が不透明な材質の場合,又は試料が容器の3/4以上入っている場合には,試料容量の4/3
倍以上の容量がある透明ガラス容器に試料の全量を移し替える。
このとき,元の試料容器を激しくかくはんして沈殿物をすべて移す。
c) ガラス容器に移し替えた試料に沈殿物が目視で観察されるときは,全沈殿物を均一にした後,試料を
JIS Z 8801-1に規定する目開き150 μmの金属製網ふるいでろ過し,大きな異物を除去する。
6.5
試験の手順 試験の手順は,次による。
a) 滴定ビーカに試料を0.1 mg単位まではかり採る。はかり採る試料の量は,表6に示す試料のはかり採
り量を目安とする。
静かに回転子を入れ,滴定溶剤40 mLを全量ピペットで滴定ビーカの側壁についた試料を洗い流す
ようにして加え,次にメスピペットでp-ナフトールベンゼイン溶液0.100 mL±0.002 mLを加える。
単位 mm
目盛
単位 mL
範囲
0〜500
目量
5
長線
50ごと
数字
50ごと

16
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表 6 試料のはかり採り量
予期酸価 (mgKOH/g)
試料のはかり採り量 (g)
0.01以下
5.0〜2.0
0.01〜0.1
2.0〜1.5
0.1 〜0.5
1.5〜1.0
0.5 〜3.0
1.0〜0.2
3.0以上
0.2〜0.1
備考 予期酸価が0.1 mgKOH/g以下で濃色の使用油(酸化油)では,指示薬の変色
が試料の色に影響されないようにはかり採り量を減らす。
b) 滴定ビーカをかき混ぜ機の上に置き,滴定ビーカ用栓でふたをする。滴定ビーカ用栓はあらかじめ図
2に示すように窒素導入用ガラス管と窒素流量計とを接続し,出口用ガラス管には排出管を接続し,
その先端を局所排気設備内に入れる。
自動ビュレットの先端を滴定ビーカ用栓の孔を通して中に入れ,試験溶液の液面上約10 mmになる
ように自動ビュレットの位置を定める。
直ちに窒素を30〜40 L/hの流量で15〜30秒間流し,滴定ビーカ中の空気を排出した後,10 L/h±1 L/h
の流量に調節する。
かき混ぜ機を始動し,試験溶液が飛散しないように,また窒素が試験溶液に入らない程度に,かき
混ぜの強さを調節する。
c) 試験溶液を30 ℃以下の温度で,0.01 mol/L水酸化カリウムの2-プロパノール溶液を用いて滴定する。
終点に近づくにつれ,オレンジ色が緑褐色に変化する。
終点は,緑又は緑褐色が15〜20秒間変化しないで,元に戻らない最初の点とする。
備考 酸価が比較的高い,例えば約3 mgKOH/g以上の試料では,緑褐色に達した後,安定した終点に
達するまでに,滴定液が数滴必要な場合がある。
d) 空試験は,滴定日ごとに滴定溶剤40 mLに,メスピペットでp-ナフトールベンゼイン溶液を0.100 mL
±0.002 mL加え,0.01 mol/L水酸化カリウムの2-プロパノール溶液で滴定する。
6.6
計算及び結果 試料の酸価は次の式によって算出し,有効数字3けたに丸める。ただし,1 mgKOH/g
未満の場合は,JIS Z 8401の規定によって丸めの幅を0.01に丸める。
m
V
V
c
AN
)
(
KOH
1.
56
0
3−
=
×
ここに,
AN: 酸価 (mgKOH/g)
cKOH: 0.01 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
V3: 試料の滴定に要した0.01 mol/L水酸化カリウムの2-プロ
パノール溶液の量 (mL)
V0: 空試験の滴定に要した0.01 mol/L水酸化カリウムの2-プ
ロパノール溶液の量 (mL)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
6.7
精度 この試験方法によって得られた試験結果の許容差(確率0.95)は,次のとおりである。
備考 試験結果が許容差を外れた場合には,JIS Z 8402-6の規定によって処理する。
a) 室内併行精度 同一試験室において,同一人が同一試験器で,同一条件で短時間内に同一試料を2回
試験したときの試験結果の差の許容差を,表7に示す。
b) 室間再現精度 異なる試験室において,別人が別の試験器で同一試料をそれぞれ1回ずつ試験して求
17
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
めた2個の試験結果の差の許容差を,表7に示す。
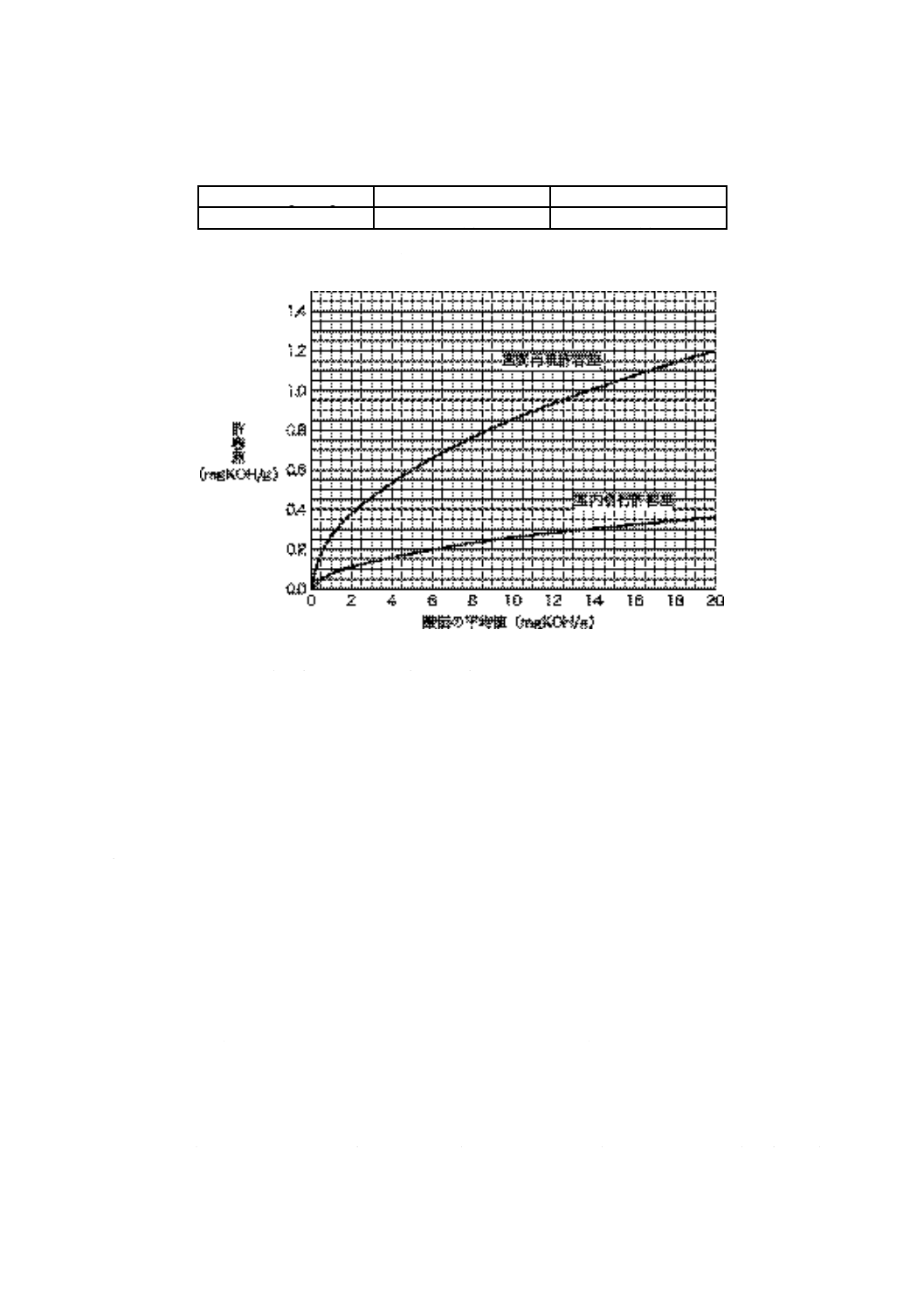
表 7 精度
酸価 mgKOH/g
室内併行許容差
室間再現許容差
0.05を超え20.0以下
0.08 (X) 0.5
0.27 (X) 0.5
備考1. 表中のXは,試験結果の平均値 (mgKOH/g) である。
2. 表7に示す式によって算出した許容差を,図7に示す。
図 7 精度曲線
6.8
試験結果の報告 試験結果には,次の事項を記載する。
a) 試料名,採取場所及び採取年月日
b) JISの規格番号:JIS K 2501
c) 試験方法の名称・箇条番号及び6.6によって得られた結果
d) 特記事項
7. 電位差滴定法(酸価)
7.1
試験の原理 試料をトルエン,2-プロパノール及び少量の水を含む滴定溶剤に溶かし,ガラス電極
と比較電極とを用いて,水酸化カリウムの2-プロパノール溶液で電位差滴定する。メータ(電位差計又は
pH計)の読みと,これに対応する水酸化カリウムの2-プロパノール溶液の滴定量との関係を作図し,滴
定曲線に得られた変曲点を終点とする。明確な変曲点が得られない場合は,非水酸性又は非水塩基性緩衝
液で得られるメータの読みを終点とする。
備考1. この方法は,トルエンと2-プロパノール混合液に溶解するか,又はほとんど溶解する,石油
製品及び潤滑油中の酸性成分を電位差滴定法によって測定するものである。この方法は,水
中の解離定数が10−9より大きい酸性成分の測定に適し,解離定数が10−9より小さい弱酸性
成分は測定できない。もし,塩の加水分解定数が10−9より大きい場合には,その塩は反応す
る。
2. 新油及び使用油中の酸性を示す成分としては,有機酸,無機酸,エステル類,フェノール化
合物,ラクトン類,レジン類,重金属塩類,アンモニアの塩類及びその他の弱塩基成分の塩
18
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
類,多塩基酸の塩類並びに酸化防止剤,清浄剤などの添加剤がある。
3. この方法は,酸化条件において使用する油の相対的な変化を測定するときに用いることがで
き,油の色及び他の物性とは関係がない。滴定は平衡状態で行われるが,使用条件下におけ
る油の性能を予測するための酸性又は塩基性の絶対値を測定するものではない。また,腐食
性と酸価又は塩基価との間に一般的な関係は知られていない。
4. この電位差滴定法で得られる酸価は,指示薬滴定法で得られる値と一致するとは限らない。
5. 新油及び使用油の中には,添加剤として存在するもの,使用中に生成する酸化物などの酸性
成分が含まれている。これらの物質の相対的な量は,塩基で滴定することによって測定でき
る。酸価は,試験条件における油中の酸性物質の量である。この酸価は,潤滑油の品質管理
の指標として用いられる。また,使用時の油の酸化劣化の測定に用いられる。油の使用限界
は,経験をもとに設定しなければならない。
各種の酸化物の多くは,酸価に寄与し,また,有機酸は,腐食性能に大きく影響するが,
試験を使用条件下における油の腐食性の予測に用いてはならない。酸価と金属の腐食性との
間には,一般的な関係は知られていない。
7.2
試薬 試薬は,次による。
a) トルエン JIS K 8680に規定するもの。
b) 水 JIS K 0557に規定するA3のもの。
c) 2-プロパノール JIS K 8839に規定するもの。
d) 滴定溶剤 トルエン500 mL及び水5 mLを2-プロパノール495 mLに加えたもの。
備考 残さ油又はアスファルト性の物質を溶かすには,トルエンの代わりにクロロホルムを用いた溶
剤を使用してもよい。
e) 非水酸性緩衝液 緩衝貯蔵溶液A [g)] 10 mLを滴定溶剤100 mLに加えて混合したもので,1時間以内
に用いる。
f)
非水塩基性緩衝液 緩衝貯蔵溶液B [h)] 10 mLを滴定溶剤100 mLに加えて混合したもので,1時間以
内に用いる。
g) 緩衝貯蔵溶液A 2, 4, 6-トリメチルピリジン(γ-コリジン)24.2 g±0.1 gをはかり採り,2-プロパノー
ル100 mLを入れた1 Lの全量フラスコに移し入れる。かき混ぜながら,0.2 mol/L塩酸の2-プロパノ
ール溶液 [j)] の (150/C1) mL±5 mL(C1は標定で得た0.2 mol/L塩酸の2-プロパノール溶液のモル濃
度)を加えた後,2-プロパノールを標線まで加える。褐色瓶に入れて保存し,1か月以内に用いる。
備考 2, 4, 6-トリメチルピリジン(γ-コリジン)の性状は,沸点が168〜170 ℃,屈折率 (nD20) が1.498
2±0.000 5,また,色は無色である。褐色瓶に入れて保存する。
h) 緩衝貯蔵溶液B m-ニトロフェノール27.8 g±0.1 gをはかり採り,2-プロパノール100 mLを入れた1
Lの全量フラスコに移し入れる。かき混ぜながら,0.2 mol/L水酸化カリウムの2-プロパノール溶液の
(50/C2) mL±1 mL(C2は標定で得た0.2 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度)を加
えた後,2-プロパノールを標線まで加える。褐色瓶に入れて保存し,1か月以内に用いる。
備考 m-ニトロフェノールの性状は,融点が96〜97 ℃また,色は淡黄色である。褐色瓶に入れて保
存する。
i)
0.1mol/L塩酸の2-プロパノール溶液
1) 調製 2-プロパノール1LにJIS K 8180に規定する塩酸9 mLを加えて混合する。
備考 市販の0.1 mol/L塩酸の2-プロパノール溶液を用いてもよい。

19
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 標定 標定は,次による。
2.1) 0.1 mol/L塩酸の2-プロパノール溶液約8 mLを正しくはかり採り,二酸化炭素を含まない水125 mL
で希釈する。
2.2) 0.1 mol/L水酸化カリウムの2-プロパノール溶液を用い,フェノールフタレインを指示薬として滴
定するか,又は電位差滴定する。
2.3) 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度は,次の式によって算出し,JIS Z 8401の規定に
よって丸めの幅を0.000 1に丸める。
2
1
KOH
HCl
v
v
c
c
×
=
ここに,
cHCl: 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度 (mol/L)
cKOH: 0.1 mol/L水酸化カリウム2-プロパノールの溶液のモル濃
度 (mol/L)
v1: 終点までの滴定に要した0.1 mol/L水酸化カリウムの2-プ
ロパノール溶液の量 (mL)
v2: 0.1 mol/L塩酸の2-プロパノール溶液のはかり採り量 (mL)
2.4) 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度変化が0.000 5 mol/L以上にならない間隔でときど
き標定し直す。
j)
0.2mol/L塩酸の2-プロパノール溶液 2-プロパノール1 LにJIS K 8180に規定する塩酸18 mLを加え
て調製し,i) 2)によって標定したもの。
k) 塩化カリウム溶液 JIS K 8121に規定する塩化カリウムを水に溶かした3 mol/L以上の飽和溶液。
l)
0.1mol/L水酸化カリウムの2-プロパノール溶液
1) 調製 三角フラスコ2 000 mLに2-プロパノール約1Lを採り,これにJIS K 8574に規定する水酸化
カリウム6 gを加え,底にかたまりができないようにかき混ぜながら,15〜20分間静かに煮沸させ
る。保護管を付けた後,室温まで放冷する。この溶液を二酸化炭素を遮りながら微細なガラスフィ
ルターでろ過する。
溶液は,JIS K 8603に規定するソーダ石灰又はけい酸ソーダなどの乾燥剤を入れた保護管を付け
て大気中の二酸化炭素の侵入を防ぐ。また,溶液が,コルク,ゴム及びけん化するようなグリース
と接触しないようにポリエチレン瓶などに保存する。
備考 市販の0.1 mol/L水酸化カリウムの2-プロパノール溶液を用いてもよい。
2) 標定 フタル酸水素カリウムによる標定は,次による。
2.1) JIS K 8005に規定するフタル酸水素カリウムを105 ℃で2時間乾燥し,その0.1〜0.15 gを0.2 mg
のけたまではかり採り,二酸化炭素を含まない水100 mLに溶かし,0.1 mol/L水酸化カリウムの
2-プロパノール溶液で電位差滴定する。
2.2) 水100 mLを用いて空試験を行う。
2.3) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度は,次の式によって算出し,JIS Z 8401
の規定によって丸めの幅を0.000 1に丸める。
)
(
23
.
204
000
1
KOH
1b
V
W
c
−
=
ここに, cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
W: フタル酸水素カリウムのはかり採り量 (g)
V1: 終点までの滴定に要した0.1 mol/L水酸化カリウムの2-プ

20
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ロパノール溶液の量 (mL)
b: 空試験におけるV1に相当する量 (mL)
204.23: フタル酸水素カリウムの式量
2.4) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度の変化が0.000 5 mol/L以上にならない
間隔でときどき標定し直す。
備考1. アミド硫酸による標定は,次による。
a) JIS K 8005に規定するアミド硫酸の結晶粉末を真空デシケータ中で約48時間乾燥し,その
2.0〜2.5 gを0.1 mgのけたまではかり採り,二酸化炭素を含まない水に溶かし,250 mL全量
フラスコに移し,同じ水を標線まで加える。
b) その25 mLを全量ピペットを用いてはかり採り,水100 mLを加える。JIS K 8001の4.4によ
って調製したブロモチモールブルー溶液を指示薬として0.1 mol/L水酸化カリウムの2-プロ
パノール溶液で滴定する。
c) 水100 mLを用いて空試験を行う。
)
(
093
.
97
100
KOH
1b
V
W
c
−
=
ここに,
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル
濃度 (mol/L)
W: アミド硫酸のはかり採り量 (g)
V1: 終点までの滴定に要した0.1 mol/L水酸化カリウムの2-
プロパノール溶液の量 (mL)
b: 空試験におけるV1に相当する量 (mL)
97.093: アミド硫酸の式量
2. 2-プロパノールのような有機溶剤の膨張係数は,比較的大きいので,0.1 mol/L水酸化カリウ
ムの2-プロパノール溶液の標定は試料の試験温度に近い温度で行う。
m) 0.2 mol/L水酸化カリウムの2-プロパノール溶液
JIS K 8574に規定する水酸化カリウム12〜13 gを2-プロパノール約1 Lに溶かしたもので,l)1)に
準じて調製し,標定した後,貯蔵する。標定は,JIS K 8005に規定するフタル酸水素カリウム0.2〜0.3
gを0.2 mgのけたまではかり採り,二酸化炭素を含まない水100 mLに溶かしたものを用いる。
7.3
試験器 試験器は,次による。
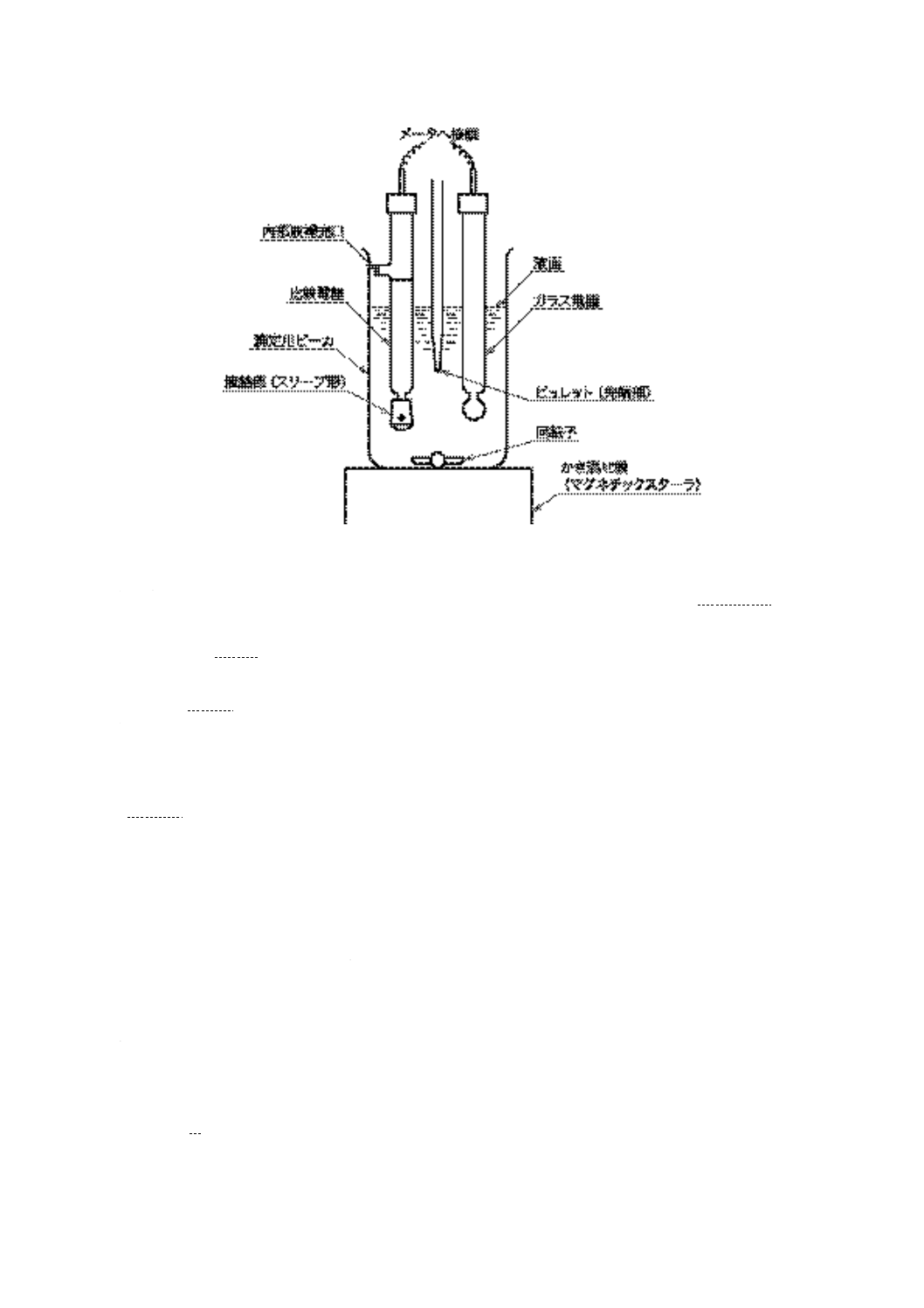
電位差滴定法の試験器の一例を図8に示す。
備考 試験器によっては,静電気に敏感なものがあり,人間が近づくとメータの指針又は記録用ペン
が誤った動きをすることがある。このような場合には,銅製の金網を円筒にして滴定用ビーカ
に巻き電気的に接地する。
21
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図 8 電位差滴定法試験器・滴定部(一例)
a) 手動試験器
1) メータ 2)及び3)に規定するガラス電極と比較電極を用いたとき,少なくとも1 V又は12 pHまで
読むことができるもので,次のいずれかとする。
1.1) 電位差計 JIS K 0113に規定する補償式電位差計,直示式電位差計又はディジタル式電位差計
で,最小目盛5mV以下のもの。
1.2) pH計 JIS Z 8802に規定する形式II以上の性能のpH計で,最小目盛0.1 pH以下のもの。
2) ガラス電極 pH測定範囲0〜14のもの。
3) 比較電極 内部電極として銀・塩化銀電極,飽和カロメル電極などをもち,内部液補充口(栓付き)
及び液絡部があり,7.2 h)に規定する塩化カリウム電極液を満たしたもの。
備考1. 通常,液絡部がスリーブ形(図8参照)のものを用いるが,試料の性状によってはファイバ
ー形,フリット形などを用いてもよい。
2. スリーブを定位置に置き,電極を大気中につるしたときに電極から電解液の漏れが10分間に
1滴を超えてはならない。
3. ガラス電極と比較電極を1本の形にまとめた複合形電極及び銀・塩化銀電極は,応答が遅い
ので,この方法には不適である。
4) 可変速型かき混ぜ器 ガラス製のプロペラ型,又はマグネチックスターラなどで,かき混ぜの強さ
を任意に変えることのできるもの。かき混ぜ器は,あらかじめ接地しておき,滴定操作中にメータ
の読みに狂いが生じないようにする。
5) ビュレット 図9に示す0.05 mL目盛付きの10 mL容量のもので,±0.02 mLの正確度に補正した
もの。
備考1. これと同等の性能をもつ電動ビュレットを用いてもよい。また,ガラス製ビュレットは,JIS
K 2839に規定する図101のものがこれに相当する。
2. 水酸化カリウム溶液用のビュレットは,ソーダ石灰などの二酸化炭素吸収剤を入れた保護管
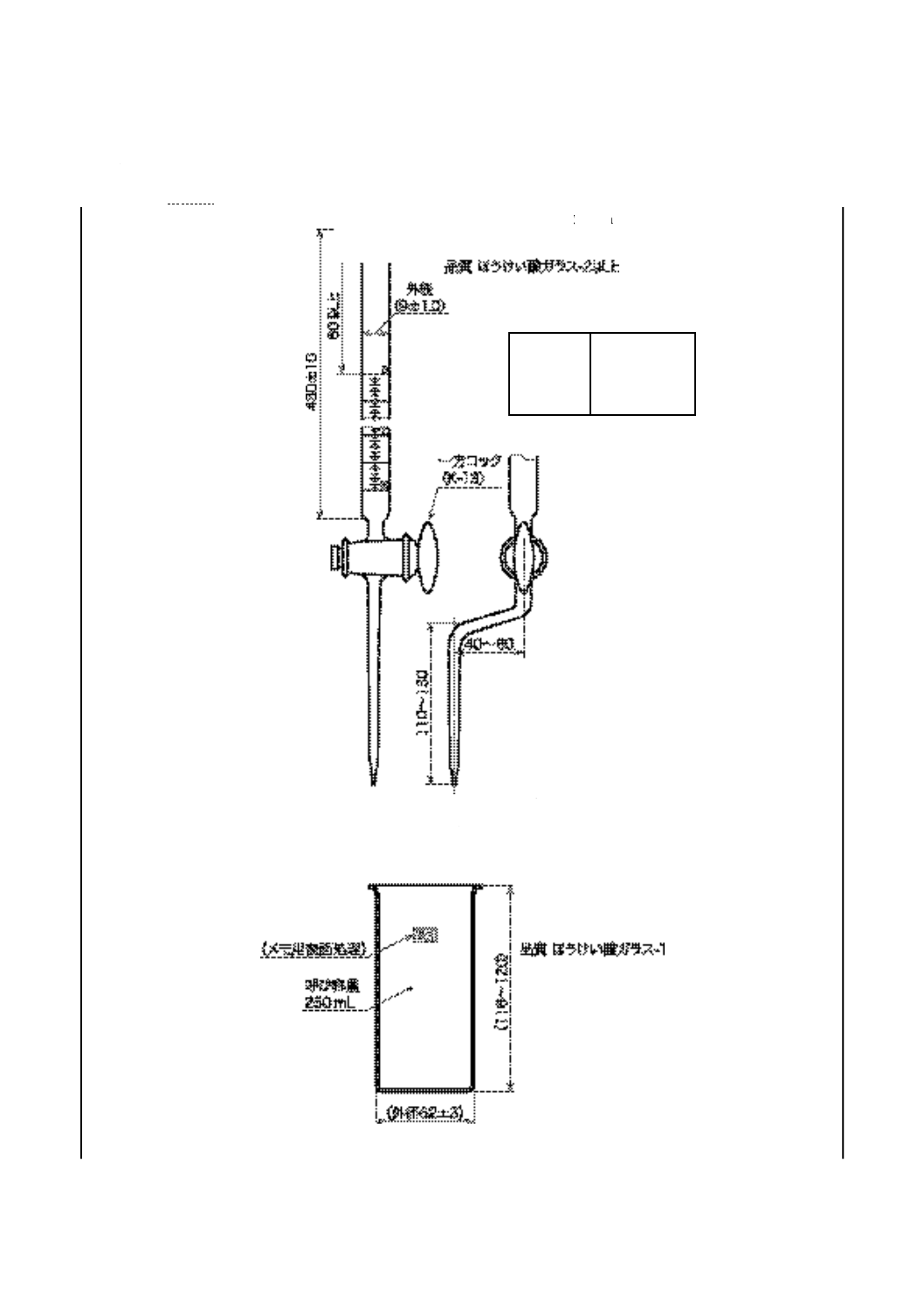
22
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を付ける。
6) 滴定用ビーカ 図10に示すガラス製で容量250 mLのもの。
備考 JIS K 2839に規定する図102のものがこれに相当する。
単位 mm
備考 図に示すものは一例であり,同一機能であれば別の形状・寸法でもよい。
図 9 ビュレット
図 10 滴定用ビーカ
単位 mm
目盛
単位 mL
範囲
0〜10
目量
0.05
長線
0.5ごと
数字
1ごと
23
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) 滴定支持台 ビーカ,電極,かき混ぜ器及びビュレットを支持できる適切なもの。電極,かき混ぜ
器及びビュレットのじゃまにならないようにビーカが取り出せるような配置が望ましい。
b) 自動試験器 自動試験器は,a)に従うもので,次の性能をもつもの。
1) 連続滴定において1分間に0.2 mLの滴定速度,また,変曲点及び緩衝液による終点付近において1
分間に0.05 mLの滴定速度になるように滴定曲線の傾きに対応して滴下速度を自動調節できるもの。
2) 電動ビュレットは,±0.01 mLの正確度のもの。
3) 滴定量及び電位の変化の読みを連続的に記録できるもの。
7.4
試料採取方法及び調製方法 試料の採取方法及び調製方法は,次による。
a) 試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法によるか,又はそれらに
準じた方法によって採取及び調製する。
b) 使用油の試料の調製 使用油の試料の調製は,次による。
1) 試料中に沈殿物があるときは,沈殿物自体が酸性若しくは塩基性であるか,又はそれが試料中の酸
性若しくは塩基性の物質を吸着することがあるから,このような沈殿物を含む試料を採るときは,
試料全体を代表するようによくかき混ぜる。
2) 試料を60 ℃±5 ℃に加熱し,沈殿物を均一にする。沈殿物が含まれていない場合には,加熱をし
なくてもよい。
備考 使用油は保存中に変質することがあるので,採取後,できるだけ早く測定する。
3) 試料に沈殿物が目視で観察されるときは,すべての沈殿物を均一にした後,試料をJIS Z 8801-1に
規定する目開き150 μmの金属製網ふるいでろ過し,大きな異物を除去する。
7.5
電位差滴定装置の調整
7.5.1
電極の準備
a) ガラス電極は,ときどき(連続使用しているときは週1回以上)適切な洗浄剤で洗浄する。次いで,
水の順で洗浄する。
b) 比較電極は塩化カリウム溶液を少なくとも毎週新しいものと取り換える。電極内の塩化カリウム溶液
の液面は常に滴定用ビーカ内の液面より上方に保っておく。
c) 電極を使用しないときは,両電極ともその下半分を水に浸しておく。滴定後は電極を滴定溶剤中に浸
したままにしておいてはならない。電極はこわれやすいからその取扱いには十分注意を要する。
7.5.2
電極の維持
a) 使用の前に,水に浸してあった両電極を取り出し,水ですすぎ,清浄な布又は柔らかな薄い紙でぬぐ
い,余分の水分をふき取る。
b) 比較電極が汚れた場合は,スリーブを緩めて,塩化カリウム溶液を抜き,すり合わせ面を溶剤で洗い,
次いで水で十分洗い,汚れなどの付着物を洗い落とす。次に,比較電極に塩化カリウム溶液を入れ,
すり合わせ面に塩化カリウム溶液が十分にしみわたったら,スリーブをしっかりと締め水ですすぐ。
c) 繰り返し同種類の試料を測定する場合も滴定の都度滴定溶剤で両電極を洗浄し,滴定に先立ち,電極
を少なくとも5分間水に浸し,布又は柔らかい薄い紙でぬぐい,余分の水分をふき取る。
備考1. 試験を始めてからは,液間電位差の変化を防ぐため,比較電極の液絡部のスリーブを動かさ
ないようにする。
2. 使用後は,両電極とも滴定溶剤で十分に洗浄し,水で十分にすすいだ後,水中に浸しておく。
7.5.3
電極の検査
a) 電極を最初に用いる場合には,又は取り換えた場合は,メータと電極との組合せを,次のように検査
24
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
する。その後もときどき検査を行う。
b) 滴定溶剤100 mLと0.1 mol/L水酸化カリウムの2-プロパノール溶液の1.0〜1.5 mLとの混合液に電極
を浸したときのメータの読みと,同じ電極を非水酸性緩衝液中に浸したときのメータの読みとの差は,
480 mV又は8.0 pH以上でなければならない。
備考 電極が汚れていると電位の再現性が悪くなるから,スリーブ接続部に異物がないように電極の
清浄及び定期的に電極の検査を行う。これは,非水緩衝液によって終点を決める場合に特に重
要である。
7.6
試験器の調整 試験器の調整は,次による。
a) 非水緩衝液によるメータの読みの決定 多くの場合,明確な変曲点が滴定曲線に得られないから,酸
及び塩基の終点に相当するメータの読みを決定するため,非水酸性 [7.2 e)] 及び非水塩基性緩衝液
[7.2 f)] でメータの読みを滴定に先立って求めておく。
備考 異なったガラス電極では,水素イオン活量に対する感度,不斉電位差及び比較電極の液間電位
差などが同じではない。したがって,それぞれの電極系について非水酸性及び非水塩基性緩衝
液に相当するメータの読みを求めておかなければならない。
b) メータの読み及び終点 電極を7.5に従って準備し,非水緩衝液の温度を滴定時の液温±1 ℃に保ち
ながらメータの読みの変化が1分間に5 mV又は0.1 pH目盛よりも小さくなった後,メータの指示値
を読み取る。このメータの読みを変曲点が明確でないときの終点とする。
7.7
試験の手順 試験の手順は,次による。
7.7.1
試験の準備
a) 表8に示す量の試料を滴定用ビーカ [7.3 a)6)] にはかり採り,滴定溶剤125 mLを加える。滴定用ビー
カを滴定用支持台の上に置き,7.5に従って準備した電極の位置が約1/2程度溶液に浸るように調節す
る。かき混ぜ器を始動し,ビーカの内容物が飛散しないように,また,空気が入らない程度の強さで
滴定が終わるまでかき混ぜる。
表 8 試料のはかり採り量
酸価又は強酸価
試料のはかり採り量
はかり採り最小目盛
mgKOH/g
g
g
0.05 を超え 1.0 以下
20.0±2.0
0.10
1.0 を超え 5.0 以下
5.0±0.5
0.02
5.0 を超え 20 以下
1.0±0.1
0.005
20
を超え 100 以下
0.25±0.02
0.001
100
を超え 250 以下
0.10±0.01
0.000 5
b) ビュレットに0.1 mol/L水酸化カリウムの2-プロパノール溶液を入れ,その先端が滴定用ビーカの溶
液中に深さ約25 mm浸るようにビュレットの位置を調節する。メータの読みの変化が毎分5 mV又は
0.1 pH目盛よりも小さくなったとき,ビュレット及びメータの読みを記録する。
7.7.2
手動滴定法
a) 0.1 mol/L水酸化カリウムの2-プロパノール溶液を適切に少量ずつ加え,メータの読みが一定となるの
を待ってから再びビュレット及びメータの読みを記録する。
備考 メータの読みの変化が毎分5 mV又は0.1 pH目盛より小さい場合は,メータの読みが一定とみ
なす。
b) 滴定の開始時及びその後は,0.1 mol/L水酸化カリウムの2-プロパノール溶液0.1 mLの滴下によるメ
ータの読みの変化が一様に30 mV又は0.5 pH目盛以上を示すような部分(変曲点)においては,0.1
25
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
mol/L水酸化カリウムの2-プロパノール溶液を0.05 mLずつ加える。
c) 0.1 mol/L水酸化カリウムの2-プロパノール溶液0.1 mLの滴下によるメータの読みの変化が30 mV以
下又は0.5 pH目盛以下を示すような部分(平たん部)においては,メータの読みの変化が30 mV又は
0.5 pH目盛を超えないで,ほぼ等しくなるように0.1 mol/L水酸化カリウムの2-プロパノール溶液の滴
下量を0.1 mLよりも多く加える。
d) このように滴定を続け,メータの読みの変化が0.1 mL当たり5 mV又は0.1 pH目盛を超えることなく,
また,メータの読みが非水塩基性緩衝液よりも塩基性を示すようになったら滴定をやめる。
e) 滴定終了後,滴定した溶液から電極を引き上げ,電極及びビュレットの先端を滴定溶剤,2-プロパノ
ール及び水の順ですすぐ。次の試料を測定する場合には,電極は少なくとも5分間水に浸してから用
いるとよい。電極を使用しない場合は,水に浸しておく。もし,電極が汚れているような場合には,
7.5の操作を行う。
7.7.3
自動滴定法
a) 7.7.2の手動滴定法に規定した滴定条件によって自動滴定を行う。
参考 詳細については,各装置の取扱説明書による。
b) 自動滴定によって滴定曲線を記録する。
c) この方法でメータの読みの変化が0.1 mL当たり5 mV又は0.1 pH目盛を超えることなく,また,メー
タの読みが非水塩基性緩衝液よりも塩基性を示すまで0.1 mol/L水酸化カリウムの2-プロパノール溶
液で滴定を続ける。
d) 滴定終了後,7.7.2 e)に従って電極及びビュレットを洗浄する。
7.7.4
空試験 試験ごとに滴定溶剤125 mLについて,次のように空試験を行う。
酸価の場合は,0.1 mol/L水酸化カリウムの2-プロパノール溶液,強酸価の場合は0.1 mol/L塩酸の2-プ
ロパノール溶液を0.05 mLずつ加え,各滴下ごとにメータの読みが一定になったら,メータの読み及びビ
ュレットの読みを記録する。自動滴定は,7.7.3に従って操作する。
7.8
計算及び結果 計算及び結果は,次による。
7.8.1
手動滴定法
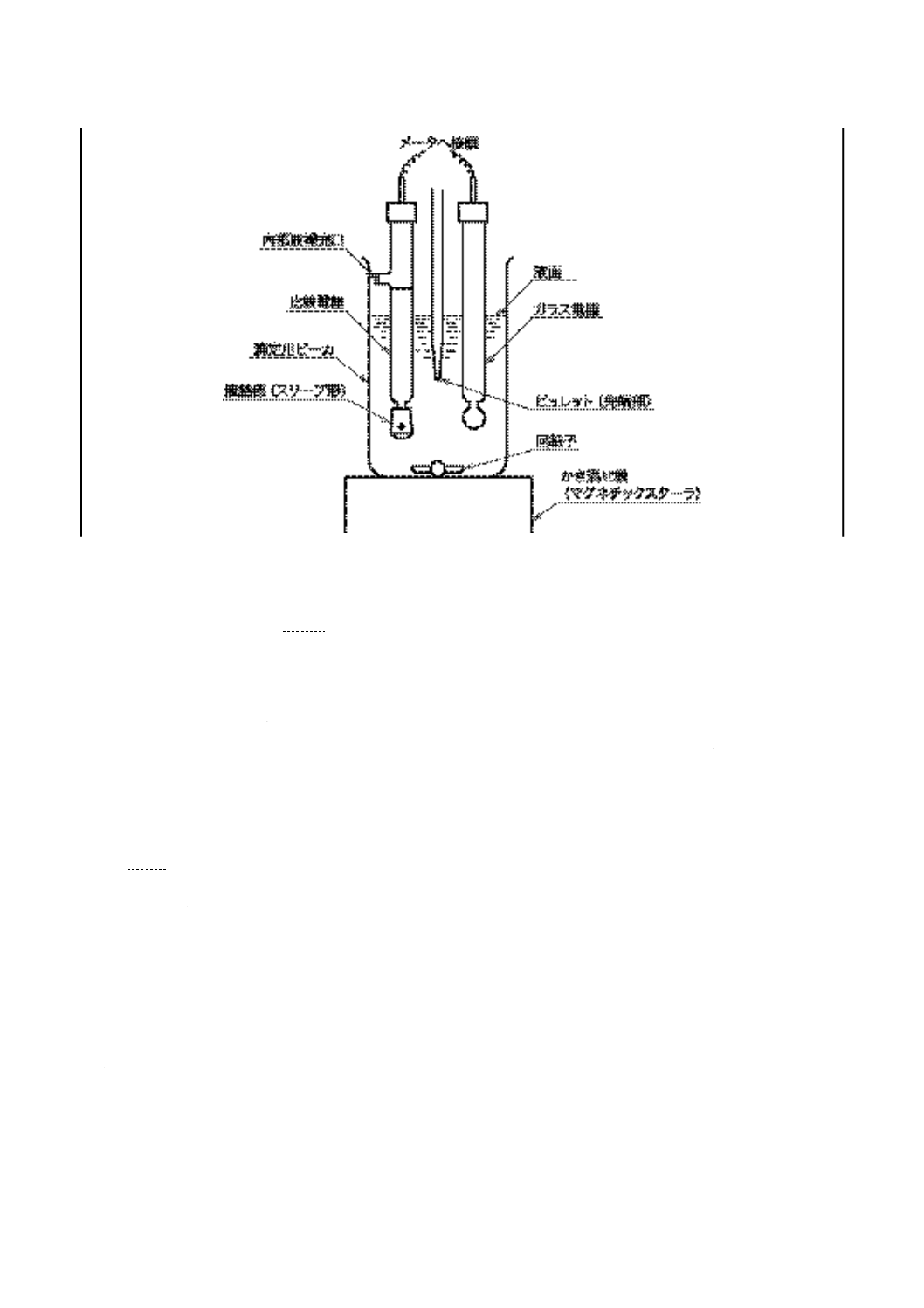
a) メータの読みに対応する0.1 mol/L水酸化カリウムの2-プロパノール溶液の滴定量をプロットし,図
11に示す滴定曲線を作図する。非水酸性又は非水塩基性緩衝液が示すメータの読みの付近に起こる変
曲点を終点として印を付ける。この終点の測定は,JIS K 0113の終点決定方法を用いるが,図11の記
号説明A〜Dを参照する。滴定曲線上に変曲点が見いだせない場合は,非水酸性及び非水塩基性緩衝
液が示すメータの読みを終点とする。
備考 最初のメータの読みが非水酸性及び非水塩基性緩衝液のメータの読みの間にある場合には,試
料は弱酸価と弱塩基価との両方をもつ。最初のメータの読みが非水塩基性緩衝液よりも塩基性
が大きいことを示す場合,又は非水酸性緩衝液よりも酸性が大きいことを示す場合には,試料
はそれぞれ強塩基・弱塩基の混合物又は強酸・弱酸の混合物を含んでいる。
b) 使用油は,一般に非水酸性及び非水塩基性緩衝液が示すメータの読みを終点とする。
備考1. 変曲点は,滴定液を続けて0.05 mLずつ滴下したとき,メータの読みが15 mV目盛以上の変
化を示したとき,又は滴下前の目盛よりも30 %以上の変化を示した点で確認できる。一般に
は,明確な変曲点は,同じ量の滴下を行っている範囲でだけ識別することができる。
2. 共同実験の結果では,新油及び添加剤は明確な変曲点が得られるが,使用油は変曲点が得ら
れない場合が多い。
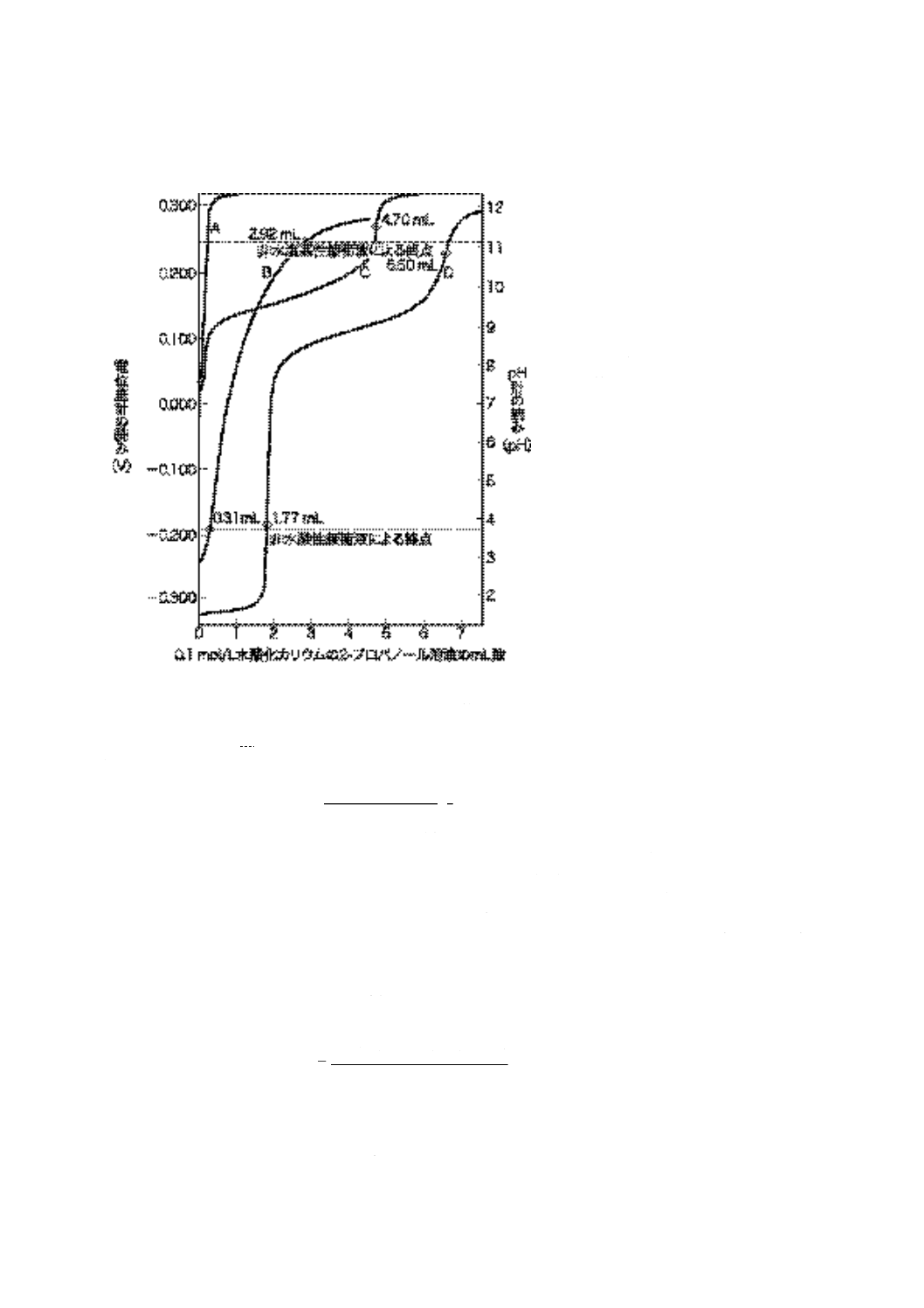
26
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.8.2
自動滴定法 7.8.1に従って自動的に操作を行い,7.7.3 c)で得られる滴定曲線によって終点を求め
る。
図 11 滴定曲線(一例)
7.8.3
計算方法 次の式によって酸価及び強酸価を算出し,有効数字3けたに丸める。ただし,1 mgKOH/g
未満の場合は,JIS Z 8401の規定によって丸めの幅を0.01に丸める。
a) 酸価
m
v
v
c
AN
)
(
KOH
1.
56
0
1−
=
×
ここに,
AN: 酸価 (mgKOH/g)
v1: 試料の滴定に要した0.1 mol/L水酸化カリウムの2-プロパ
ノール溶液の量 (mL)
v0: 空試験の滴定に要した0.1 mol/L水酸化カリウムの2-プロ
パノール溶液の量 (mL)
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
b) 強酸価
m
c
V
c
V
SAN
)
HCl
KOH
(1.
56
3
2
×
×
×
=
ここに,
SAN: 強酸価 (mgKOH/g)
V2: 試料の滴定に要した0.1 mol/L水酸化カリウムの2-プロパ
ノール溶液の量 (mL)
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
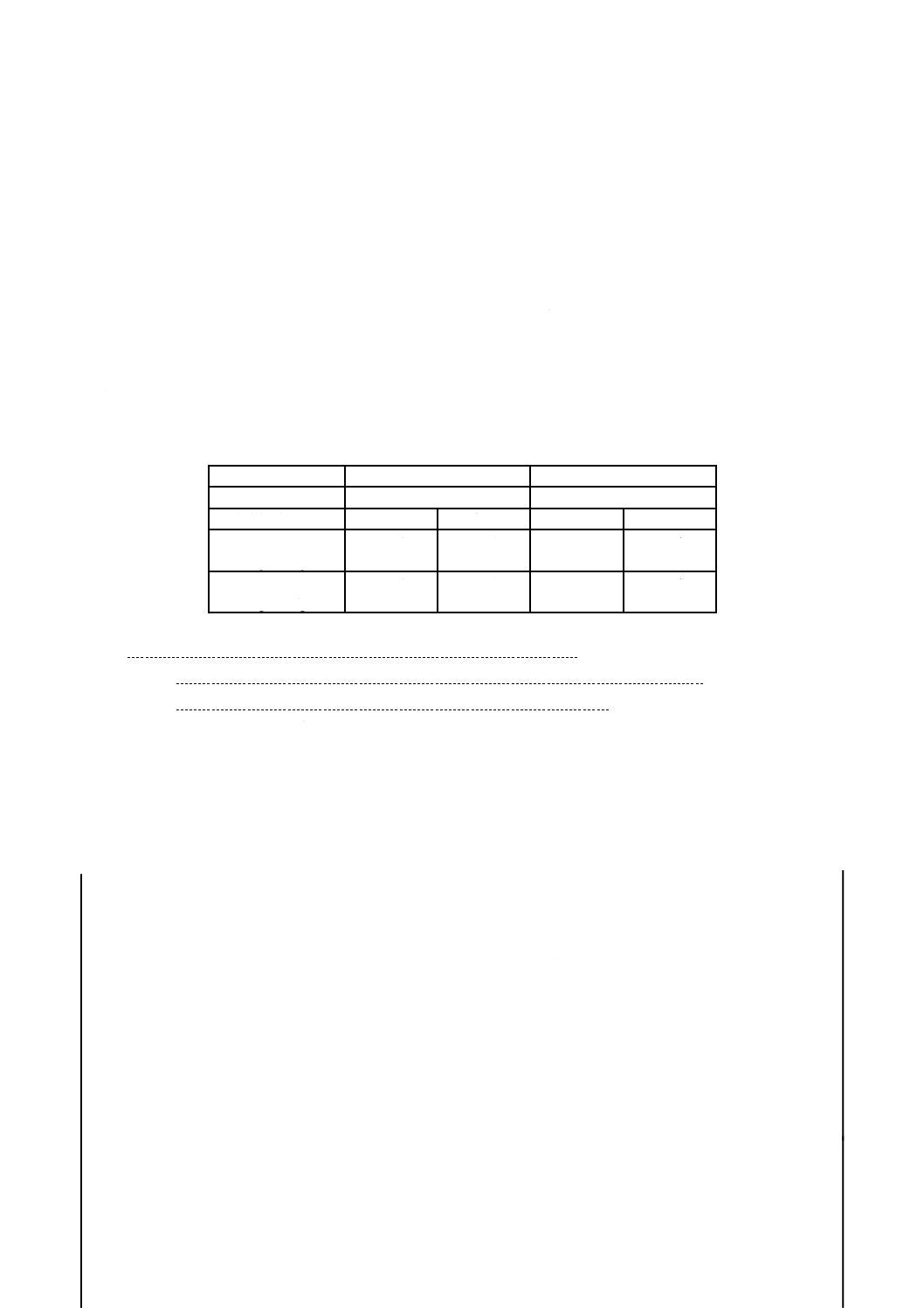
記号説明
A:滴定溶剤についての滴定曲線(空試
験)
B:明確な変曲点を示さない試料の滴定
曲線,両非水緩衝液で得られたメー
タの読みを終点とする。
C:弱酸性成分を含む試料の滴定曲線,
変曲部の最も垂直に近い点を終点と
する。
D:弱酸及び強酸性成分を含む資料の滴
定曲線,二つの変曲部の最も垂直に
近い点を終点とする。
27
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
度 (mol/L)
V3: 空試験の滴定に要した0.1 mol/L塩酸の2-プロパノール溶
液の量 (mL)
cHCl: 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
7.9
精度 この試験方法によって得られた試験結果の許容差(確率0.95)は,次のとおりである。
なお,この精度は,強酸価については規定しない。
備考 試験結果が許容差を外れた場合は,JIS Z 8402-6の規定によって処理する。
a) 室内併行精度 同一試験室において,同一人が同一試験器で引き続き短時間内に同一試料を2回試験
したとき,試験結果の差の許容差を表9に示す。
b) 室間再現精度 異なる試験室において,別人が別の試験器で同一試料をそれぞれ1回ずつ試験したと
き,2個の試験結果の差の許容差を表9に示す。
表 9 精度
試料の種類
新油・添加剤
使用油
終点の求め方
変曲点
緩衝液
滴定操作
手動
自動
手動
自動
室内併行許容差
0.07X
0.06X
0.05X
0.12X
mgKOH/g
室間再現許容差
0.20X
0.28X
0.39X
0.44X
mgKOH/g
備考 Xは試験結果の平均値 (mgKOH/g)
参考 表9で示す以外の試料の精度は,次を目安にするとよい。
a) 新油・添加剤の緩衝液による終点の場合:新油・添加剤の変曲点の許容差
b) 使用油の変曲点による終点の場合:使用油の緩衝液の許容差
7.10 試験結果の報告 試験結果には,次の事項を記載する。
a) 試料名,採取場所及び採取年月日
b) JIS規格番号:JIS K 2501
c) 試験方法の名称・箇条番号及び7.8によって得られた結果
d) 特記事項
8. 電位差滴定法(塩基価・塩酸法)
8.1
試験の原理 試料をトルエン,2-プロパノール及び少量の水を含む滴定溶剤に溶かし,ガラス電極
と比較電極を用いて,塩酸の2-プロパノール溶液で電位差滴定する。メータ(電位差計又はpH計)の読
みと,これに対応する標準液の滴定量との関係を作図し,滴定曲線に得られた変曲点を終点とする。明確
な変曲点が得られない場合は,非水酸性又は非水塩基性緩衝液で得られるメータの読みを終点とする。
備考1. この方法は,石油製品及び潤滑油中に含まれる塩基性成分で,トルエンと2−プロパノール
混合液に溶解するものを電位差滴定法によって測定するものである。この方法によれば,塩
基性の強い化合物類の解離定数が,塩基性が一段弱い化合物類の解離定数の1 000倍以上で
ある場合には,塩基性成分を弱塩基と強塩基のイオン化性をもつグループに区別することが
できる。
2. この方法は,過塩素酸法と区別するために塩酸法と呼ぶことがある。
28
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3. 新油及び使用油中の塩基性を示す成分としては,有機塩基,無機塩基,アミノ化合物,弱酸
塩(石けん),多酸塩基の塩基性塩,重金属塩,酸化防止剤,清浄剤のような添加剤などがあ
る。
4. この方法は,酸化などによる使用油中の塩基価及び強塩基価の変化を測定する場合には,色
又は他の性質に関係なく適用できる。測定を綿密な条件で実施しても,油の使用状態におけ
る性能を予測するための塩基性の絶対値を測定するものではない。例えば,エンジンの腐食
性又は腐食摩耗と塩基価との間には,一般的な関係は知られていない。
5. この[電位差滴定法(塩基価・塩酸法)]は,9.による塩基価と同様な結果を得られるが,過
塩基添加剤を含む油及び窒素重合物を含む油は,過塩素酸法より低い結果が得られることが
ある。
8.2
試薬 試薬は,次による。
a) 2-プロパノール JIS K 8839に規定するもの。
b) トルエン JIS K 8680に規定するもの。
c) 水 JIS K 0557に規定するA3のもの。
d) 非水酸性緩衝液 緩衝貯蔵溶液A 10 mLを滴定溶剤100 mLに加えて混合したもので,1時間以内に用
いる。
e) 非水塩基性緩衝液 緩衝貯蔵溶液B 10 mLを滴定溶剤100 mLに加えて混合したもので,1時間以内に
用いる。
f)
緩衝貯蔵溶液A 2,4, 6-トリメチルピリジン(γ-コリジン)を24.2 g±0.1 gはかり採り,2-プロパノー
ル100 mLを入れた1 Lの全量フラスコに移す。かき混ぜながら0.2 mol/L塩酸の2-プロパノール溶液
(150/C1) mL±5 mL(C1は0.2 mol/L塩酸の2-プロパノール溶液のモル濃度)を加えた後,全量を2-プ
ロパノールで1Lにしたもので,褐色瓶に入れて保存し,1か月以内に用いる。
2, 4, 6-トリメチルピリジン(γ-コリジン)の性状を,次に示す。
沸点
168〜170 ℃
屈折率()
20
D
n
1.498 2±0.000 5
色
無色
g) 緩衝貯蔵溶液B m-ニトロフェノールを27.8 g±0.1 gはかり採り,2-プロパノール100 mLを入れた1
Lの全量フラスコに移す。かき混ぜながら0.2 mol/L水酸化カリウムの2-プロパノール溶液 (50/C2) mL
±1 mL(C2は0.2 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度)を加えた後,全量を2-プ
ロパノールで1 Lにしたもので,褐色瓶に入れて保存し,1か月以内に用いる。
m-ニトロフェノールの性状を,次に示す。
融点
96〜97 ℃
色
淡黄色
h) 0.1 mol/L塩酸の2-プロパノール溶液
1) 調製 2 Lの三角フラスコに2-プロパノール1 Lを採り,JIS K 8180に規定する塩酸9 mLを加えて
混合する。
2) 標定 標定は,次の操作によって行う。
2.1) 0.1 mol/L塩酸の2-プロパノール溶液約8 mLを正しくはかり採り,二酸化炭素を含まない水125 mL
で薄める。

29
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2.2) 0.1 mol/L水酸化カリウムの2-プロパノール溶液を用いてフェノールフタレインを指示薬として滴
定するか,又は電位差滴定する。
2.3) 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度は,次の式によって算出し,JIS Z 8401の規定に
よって丸めの幅を0.000 1に丸める。
2
1
KOH
HCl
v
v
c
c
×
=
ここに,
cHCl: 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度 (mol/L)
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
v1: 終点までの滴定に要した0.1 mol/L水酸化カリウムの2-プ
ロパノール溶液の量 (mL)
v2: 0.1 mol/L塩酸の2-プロパノール溶液のはかり採り量 (mL)
2.4) 0.1 mol/L塩酸の2-プロパノール溶液はモル濃度変化が0.000 5 mol/L以上にならない間隔でときど
き標定し直す。
i)
0.2 mol/L塩酸の2-プロパノール溶液 2-プロパノール1LにJIS K 8180に規定する塩酸18 mLを加え
て混合調製しh)2)に準じて標定したもの。
j)
塩化カリウム電極液 JIS K 8121に規定する塩化カリウムを水に溶かした3 mol/L以上の濃厚液を用
いる。
k) 0.1 mol/L水酸化カリウムの2-プロパノール溶液
1) 調製 2 Lの三角フラスコに2-プロパノール約1 Lを採り,これにJIS K 8574に規定する水酸化カ
リウムを6 g加え,底にかたまりができないようにかき混ぜながら,15〜20分間静かに沸騰させる。
この溶液を二酸化炭素を遮り2日間放置した後,その上澄液を微細なガラスフィルタでろ過してポ
リエチレン瓶などに入れ,ソーダ石灰,乾燥剤などを満たした保護管を付けて保存する。
2) 標定 フタル酸水素カリウムによる標定は,次による。
2.1) JIS K 8809に規定するフタル酸水素カリウムを120 ℃で2時間乾燥し,その0.1〜0.2 gを0.1 mg
のけたまではかり採り,二酸化炭素を含まない水125 mLに溶かし,0.1 mol/L水酸化カリウムの
2-プロパノール溶液を用いて電位差滴定する。
2.2) 水125 mLについて2.1)の操作によって空試験を行う。
2.3) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度は,次の式によって算出し,JIS Z 8401
の規定によって丸めの幅を0.000 1に丸める。
)
(
23
.
204
000
1
KOH
1b
v
W
c
−
=
ここに, cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
W: フタル酸水素カリウムのはかり採り量 (g)
v1: 終点までの滴定に要した0.1 mol/L水酸化カリウムの2-プ
ロパノール溶液の量 (mL)
b: 空試験におけるv1に相当する量 (mL)
204.23: フタル酸水素カリウムの式量
2.4) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度変化が0.000 5 mol/L以上にならない間
隔でときどき標定する。
備考1. アミド硫酸による標定は,次による。
a) JIS K 8005に規定するアミド硫酸の結晶粉末を真空デシケータ中で約48時間乾燥し,その
30
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2.0〜2.5 gを0.1 mgのけたまではかり採り,二酸化炭素を含まない水に溶解し,250 mLの全
量フラスコに移し,水を標線まで加える。
b) その25 mLを全量ピペットを用いてはかり,水100 mLを加える。JIS K 8001によって調製
したブロモチモールブルーを指示薬として0.1 mol/L水酸化カリウムの2-プロパノール溶液
で滴定する。
c) 水100 mLを用いて空試験を行う。
d) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度は,次の式によって算出し,JIS Z
8401の規定によって丸めの幅を0.000 1に丸める。
)
(
093
.
97
000
1
KOH
0
1v
v
W
c
−
=
×
ここに, cKOH: 0.1mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
W: アミド硫酸のはかり採り量 (g)
v1: 終点までの滴定に要した0.1mol/L水酸化カリウムの2-プ
ロパノール溶液の量 (mL)
v0: 空試験におけるv1に相当する量 (mL)
97.093: アミド硫酸の式量
2. 2-プロパノールのような有機溶剤の膨張係数は,比較的大きいので,0.1 mol/L水酸化カリウ
ムの2-プロパノール溶液の標定は試料の測定温度に近い温度で行う。
l)
0.2 mol/L水酸化カリウムの2-プロパノール溶液 JIS K 8574に規定する水酸化カリウム12 gを2-プ
ロパノール約1 Lに溶かし,k)2)に従って調製,標定したもの。
m) 滴定溶剤 JIS K 8680に規定するトルエン500 mL及び水5 mLを2-プロパノール495 mLに加えたも
の。
備考 重質残さ油又はアスファルト物質を十分に溶かすには,トルエンの代わりにクロロホルムを用
いた溶剤を用いてもよい。
8.3
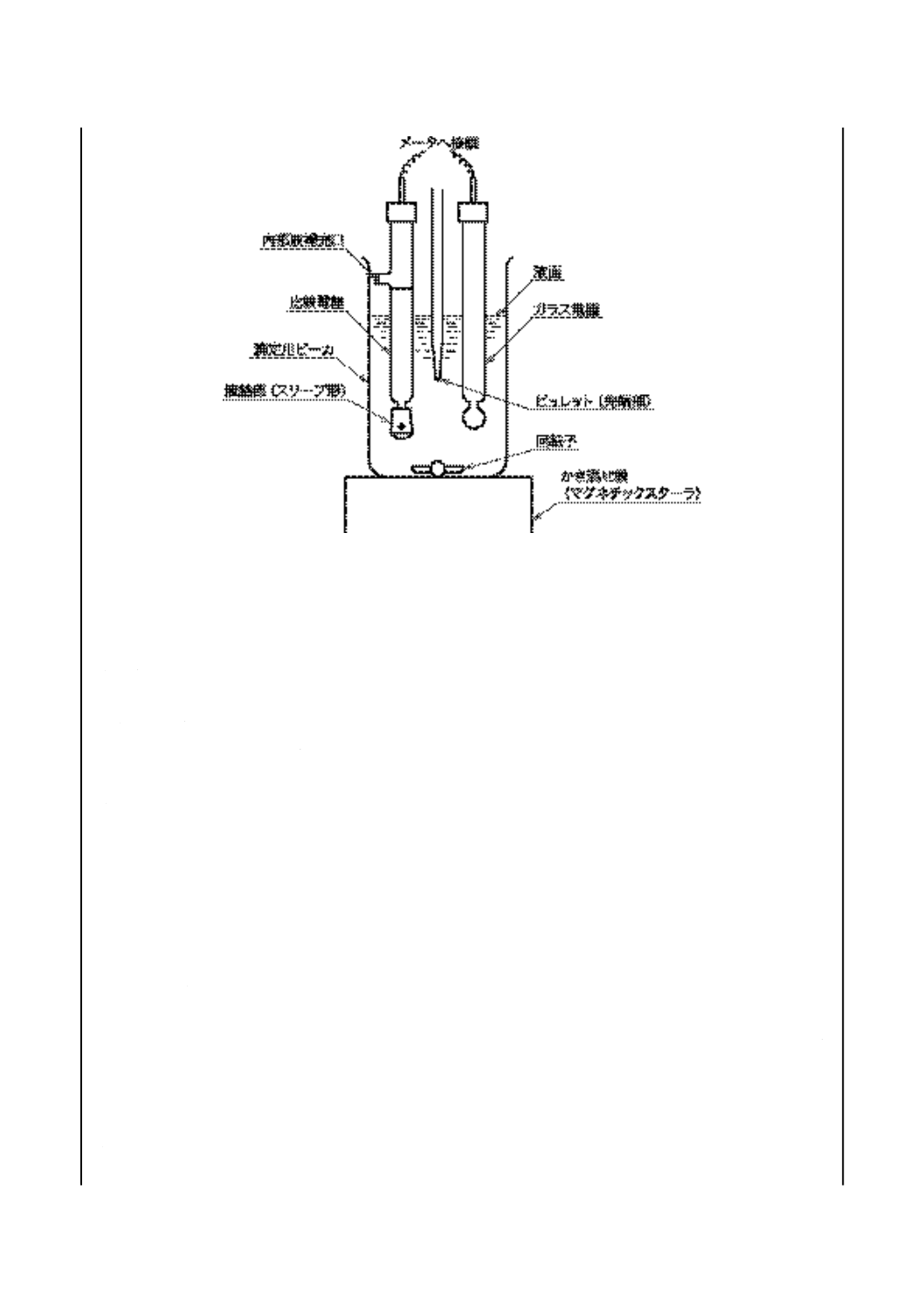
試験器 自動記録式のもの又は手動記録式のもので,滴定部の一例を図12に示す。
31
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考 試験器によっては,静電気に敏感なものがあり,人間が近づくとメータの指針又は記録用
ペンが誤った動きをすることがある。このような場合には,銅製の金網を円筒にして滴定
用ビーカに巻き,電気的に接地する。
図 12 中和価試験器滴定部(電位差滴定法)(一例)
8.3.1
手動試験器
a) メータ b)及びc)に規定するガラス電極と比較電極とを用いたとき,少なくとも1 V又は12 pHまで
読むことができるもので,次のいずれかとする。
1) 電位差計 JIS K 0113に規定する補償式電位差計,直示式電位差計又はディジタル式電位差計で,
最小目盛5 mV以下のもの。
2) pH計 JIS Z 8802に規定する形式II以上の性能のpH計で,最小目盛0.1 pH以下のもの。
b) ガラス電極 pH測定範囲0〜14のもの。
c) 比較電極 内部電極として銀・塩化銀電極,飽和カロメル電極などをもち,内部液補充口(栓付き)
及び液絡部をもち,8.2 j)に規定する塩化カリウム電極液を満たしたもの。
備考 通常,液絡部がスリーブ形(図12参照)のものを用いるが,試料の性状によってはファイバ形,
フリット形などを用いてもよい。
参考 ガラス電極と比較電極とを1本の形にまとめた複合形電極(例えば,ガラス電極/銀・塩化銀
電極)で応答時間が遅い場合は,高い値を示すことがあるので注意しなければならない。
d) かき混ぜ機 マグネチックスターラなどで,かき混ぜの強さを任意に変えることのできるもの。かき
混ぜ機は,あらかじめ接地しておき,滴定操作中にメータの読みに狂いが生じないようにする。
e) ビュレット 図9に示す,0.05 mL目盛付きで容量が10 mL又は20 mLのもので,±0.02 mLの正確
度で校正したもの(6)。
注(6) JIS K 2839に規定する図101のものが相当する。
備考 これと同等の性能をもつ電動ビュレットを用いてもよい。
f)
滴定用ビーカ 図10に示す,ガラス製で容量250 mLのもの(7)。

32
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(7) JIS K 2839に規定する図102のものが相当する。
g) 滴定支持台 ビーカ,電極,かき混ぜ機及びビュレットを支持するのに適切なもので,ビーカが障害
なく取り出せるような配置とする。
8.3.2
自動試験器 自動試験器は,8.3.1に従うもので,次の特性をもつもの。
a) 連続滴定において1分間に0.2 mLの滴定速度,また変曲点及び緩衝液による終点付近において1分間
に0.05 mLの滴定速度で,滴定曲線に対応して滴下速度を自動調節できるもの。
b) 電動ビュレットは,±0.01 mLの正確度のもの。
c) 滴定量と電位の変化の読みを連続的に記録できるもの。
8.4
試料採取方法及び試料調製方法 試料採取方法及び試料調製方法は,次による。
a) 試料はJIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法によるか,又はそれらに準
じた方法によって採取及び調製する。
b) 使用油の試料の調製 使用油の試料の調製は,次による。
1) 試料中に沈殿物があるときは,沈殿物自体が酸性若しくは塩基性であるか,又はそれが試料中の酸
性若しくは塩基性の物質を吸着していることがあるから,このような沈殿物を含む試料をはかり採
るには,試料全体を代表するようによくかき混ぜてとることが重要である。
備考 使用油は保存中に変質することがあるので,はかり採り後,できるだけ速やかに測定する。
2) 試料を60 ℃±5 ℃に加温し,沈殿物を均一にする。
なお,沈殿物が含まれていない場合は加温をしなくともよい。
3) 試料に沈殿物が目視で観察されるときは,全沈殿物を均一にした後,試料をJIS Z 8801-1に規定す
る目開き150 μmの金属製網ふるいでろ過し,大きな異物を除去する。
8.5
電位差滴定装置の調整
8.5.1
電極の準備 電極の準備は,次による。
a) ガラス電極は,ときどき(連続使用しているときは週1回以上)適切な洗剤及び水の順で洗浄した後,
水に浸しておく。
b) 比較電極は塩化カリウム電極液を少なくとも毎週新しいものと取り換える。塩化カリウム電極液面は
常に滴定用ビーカ中の液面より上方に保っておく。
備考 スリーブ形比較電極では塩化カリウム電極液の流出量がスリーブを取り付けたまま垂直に電極
を大気中につるしたとき,10分間に1滴の割合を超えない量とする。
c) 電極を使用しないときは,両電極ともその下半分を水に浸しておく。滴定後は電極を滴定溶剤中に浸
したままにしてはならない。電極は壊れやすいから,その取扱いには十分注意を要する。
8.5.2
電極の維持
a) 使用の前には水に浸してあった両電極を取り出し,水ですすぎ,清浄な布又は柔らかな薄い紙でぬぐ
い,余分の水分をふき取った後,用いる。
b) 使用後は両電極とも溶剤で十分に洗浄し,水で十分にすすいだ後,水に浸しておく。特に比較電極が
汚れた場合にはスリーブを緩めて,すり合わせ面を溶剤で,次いで水で十分洗い,汚れなどの付着物
を洗い落とす。次いで,すり合わせ面に塩化カリウム電極液が十分にしみわたったら,スリーブをし
っかりと締め水ですすぐ。
c) 繰り返し同種類の試料を測定する場合も滴定の都度溶剤で両電極を洗浄し,使用前少なくとも5分間
水に浸してから用いるとよい。
備考 測定を始めてからは,液間電位差の変化を防ぐため,比較電極の液絡部のスリーブを動かさな
33
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
いようにする。
8.5.3
電極の検査
a) 電極を最初に使用する場合又は取り換えた場合には,メータと電極との組合せを次のように検査し,
その後も,ときどき検査を行う。
b) 電極を滴定溶剤100 mLと0.1 mol/L水酸化カリウムの2-プロパノール溶液1.0 mL〜1.5 mLとの混合液
に浸したときのメータの読みと,同じ電極を非水酸性緩衝液中に浸したときのメータの読みとの差は,
0.480 V又は8.0 pH以上でなければならない。
備考 電極が汚れていると電位の再現性に影響するから,電極の清浄,スリーブ接続部の異物除去及
び電極の検査は,十分に注意して行わなければならない。これらのことは,非水緩衝液によっ
て終点を決める場合,特に重要である。
8.6
試験器の調整 試験器の調整は,次による。
a) 非水緩衝液によるメータの続みの決定 多くの場合,明確な変曲点が滴定曲線に得られないから,酸
及び塩基の終点に相当するメータの読みを決定するため,非水酸性及び非水塩基性緩衝液でメータの
読みを滴定に先立って求めておく。
備考 異なったガラス電極では,水素イオン活量に対する感度,不斉電位差,比較電極の液間電位差
などが同じではない。したがって,それぞれの電極系について非水酸性及び非水塩基性緩衝液
に相当するメータの読みを求めておかなければならない。
b) メータの読み及び終点 電極を8.5に従って準備し,非水緩衝液の温度を,滴定時の液温±1 ℃に保
ちながらメータの読みの変化が1分間に5 mV又は0.1 pH目盛よりも小さくなった後,メータを読み
取る。このメータの読みを,変曲点が明確でないときの終点とする。
8.7
試験の手順
8.7.1
試験の準備
a) 滴定用ビーカに表10に示す量の試料をはかり採り,滴定溶剤125 mLを加え,滴定用ビーカを滴定用
支持台の上に置き,8.5に従って準備した電極の位置を,その長さ約1/2が溶液に浸るように調整する。
かき混ぜ機を始動し,ビーカの内容物を飛散させないように,また空気が入らない程度に,激しく滴
定の終わるまでかき混ぜる。
表 10 試料のはかり採り量
塩基価又は強塩基価
試料のはかり採り量
はかり採り最小目盛
mgKOH/g
g
g
0.05を超え
1.0 以下
20.0 ±2.0
0.1
1.0 を超え
5.0 以下
5.0 ±0.5
0.02
5.0 を超え 20 以下
1.0 ±0.1
0.005
20 を超え 100 以下
0.25±0.02
0.001
100 を超え 250 以下
0.10±0.01
0.0005
b) ビュレットに0.1 mol/L塩酸の2-プロパノール溶液を採り,その先端を滴定用ビーカの溶液中に深さ
約25 mm浸るようにビュレットの位置を定め,メータの読みの変化が毎分5 mV又は0.1 pH目盛より
も小さくなったとき,ビュレット及びメータの読みを記録する。
8.7.2
手動滴定法
a) 0.1 mol/L塩酸の2-プロパノール溶液を適当に少量ずつ加え,メータの読みが一定となるのを待ってか
ら再びビュレット及びメータの読みを記録する。
備考 メータの読みの変化が毎分5 mV又は0.1 pH目盛より小さい場合は,メータの読みは一定とみ

34
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なす。
b) 滴定の開始時及びその後,0.1 mol/L塩酸の2-プロパノール溶液0.1 mLの滴下によるメータの読みの
変化が一様に30 mV又は0.5 pH目盛以上を示すような部分(変曲点)においては,0.1 mol/L塩酸の
2-プロパノール溶液を0.05 mLずつ加える。
c) 0.1 mol/L塩酸の2-プロパノール溶液0.1 mLの滴下によるメータの読みの変化が30 mV又は0.5 pH目
盛未満を示すような部分(平たん部)においては,メータの読みの変化が30 mV又は0.5 pH目盛を超
えないで,ほぼ等しくなるように,0.1 mol/L塩酸の2-プロパノール溶液の滴下量を0.1 mLよりも多
く加える。
d) このように滴定を続け,メータの読みの変化が0.1 mL当たり5 mV又は0.1 pH目盛を超えることなく,
またメータの読みが非水酸性緩衝液よりも酸性を示すようになったら滴定をやめる。
e) 滴定終了後試験溶液を取り除き,電極及びビュレットの先端を滴定溶剤,2-プロパノール,水の順で
すすいだ後,水に浸しておく。次の試料を測定する場合は,電極は少なくとも5分間水に浸してから
用いるとよい。電極は使用しない場合は水に浸しておく。もし電極が汚れているような場合には8.5
の操作を行う。
8.7.3
自動滴定法
a) 8.7.2に規定した滴定条件によって自動滴定を行う。
参考 詳細については,各装置の取扱説明書による。
b) 自動滴定によって,滴定曲線を記録する。
c) この方法で,メータの読みの変化が0.1 mL当たり5 mV又は0.1 pH目盛を超えることなく,またメー
タの読みが非水酸性緩衝液よりも酸性を示すまで滴定を続ける。
d) 滴定終了後,8.7.2 e)に従って電極を洗浄する。
8.7.4
空試験
a) 試験ごとに滴定溶剤125 mLについて,次のように空試験を行う。塩基価の場合は0.1 mol/L塩酸の2-
プロパノール溶液,強塩基価の場合は0.1 mol/L水酸化カリウムの2-プロパノール溶液を0.05 mLずつ
加え,各滴下ごとにメータの読みが一定になったら,メータの読み及びビュレットの読みを記録する。
自動滴定は,8.7.3に従って操作する。
8.8
結果
8.8.1
手動滴定法
a) メータの読みに対応する0.1 mol/L塩酸の2-プロパノール溶液の滴定量をプロットし,図13に示す滴
定曲線を作図して非水酸性及び非水塩基性緩衝液が示すメータの読みの付近に起こる変曲点を終点と
して印を付ける。
なお,この終点の測定には,JIS K 0113に規定する終点決定方法を用いるが,図13の記号説明A〜
Dを参照する。滴定曲線上に変曲点が見いだせない場合は,非水酸性及び非水塩基性緩衝液が示すメ
ータの読みを終点とする。
備考1. 最初のメータの読みが非水酸性及び非水塩基性緩衝液のメータの読みの間にある場合には,
試料は弱酸価と弱塩基価との両方をもつ。最初のメータの読みが非水塩基性緩衝液よりも塩
基性が大きいことを示す場合又は非水酸性緩衝液よりも酸性が大きいことを示す場合には,
試料はそれぞれ強塩基・弱塩基の混合物又は強酸・弱酸の混合物を含んでいる。
2. 変曲点は,滴定を0.05 mLずつ続けて滴下しているときにメータの読みの変化が15 mV以上
を示したとき,及び滴下前又は同じ量の滴下が続くメータの読みの変化より30 %以上の変化
35
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を示した点から確認することができる。一般には明確な変曲点は,同じ量を続けて滴下する
ことによって判断できる。
3. この結果では,新油及び添加剤は明確な変曲点が得られることが多く,使用油では変曲点が
得られない場合が多い。
図 13 滴定曲線(一例)
8.8.2
自動滴定法 8.8.1に従って自動的に操作を行い,8.7.3で得られる滴定曲線によって終点を求める。
8.8.3
計算方法 次の式によって塩基価及び強塩基価を算出し,有効数字3けたに丸める。ただし,1
mgKOH/g未満の場合はJIS Z 8401の規定によって丸めの幅を0.01に丸める。
a) 塩基価
m
V
V
c
BN
)
(
HCl
1.
56
0
1−
=
×
×
ここに,
BN: 塩基価 (mgKOH/g)
V1: 試料の滴定に要した0.1 mol/L塩酸の2-プロパノール溶液の
量 (mL)
V0: 空試験の滴定に要した0.1 mol/L塩酸の2-プロパノール溶液
の量 (mL)
cHCl: 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
b) 強塩基価
m
c
V
c
V
SBN
)
KOH
HCL
(1.
56
3
2
×
×
+
=
ここに,
SBN: 強塩基価 (mgKOH/g)
V2: 試料の滴定に要した0.1 mol/L塩酸の2-プロパノール溶液
の量 (mL)
cHCl: 0.1 mol/L塩酸の2-プロパノール溶液のモル濃度 (mol/L)
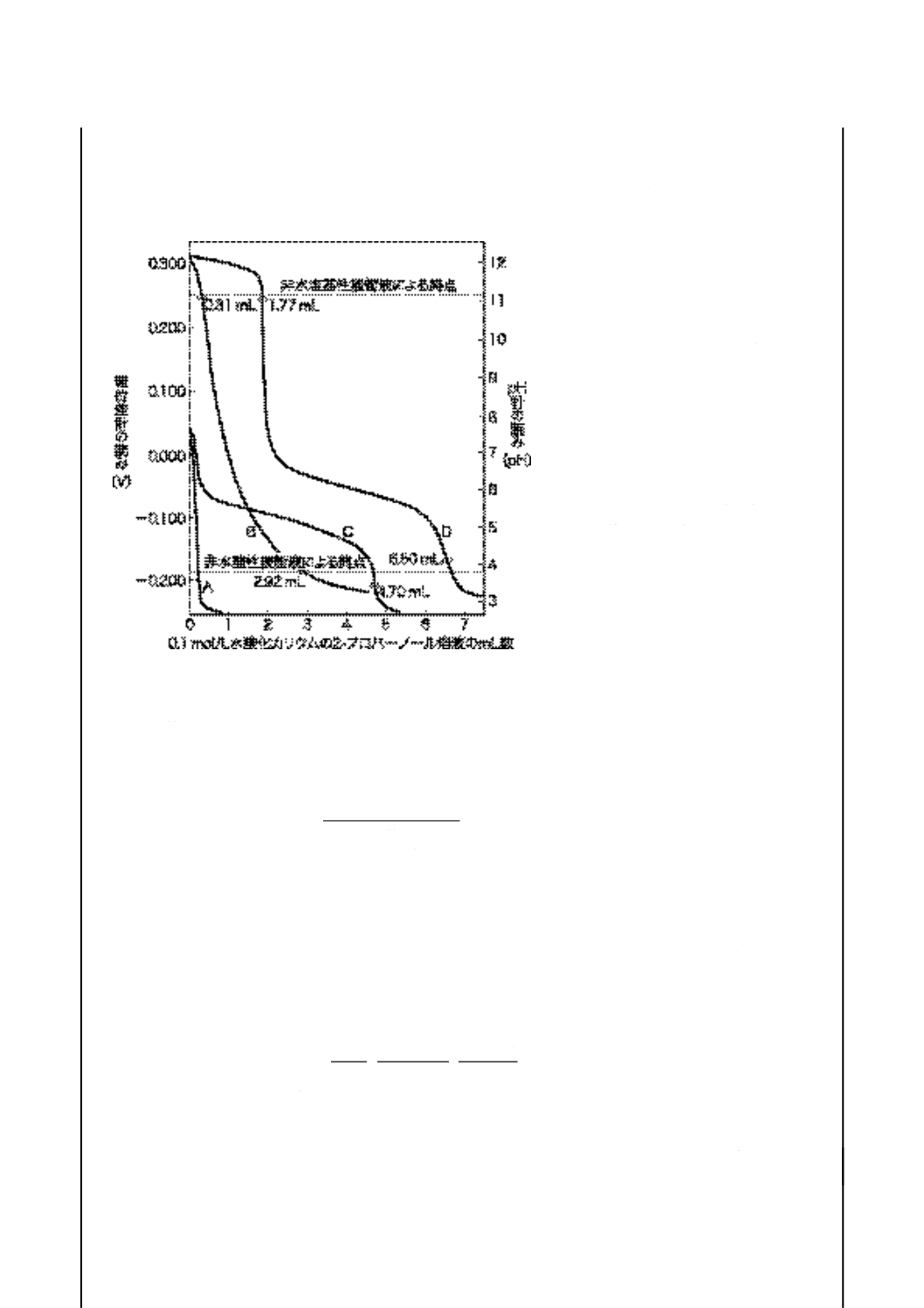
記号説明
A:滴定溶剤についての滴定曲線(空試
験)
B:明確な変曲点を示さない試料の滴定
曲線,両非水緩衝液で得られたメー
タの読みを終点とする。
C:弱酸性成分を含む試料の滴定曲線,
変曲部の最も垂直に近い点を終点と
する。
D:弱酸及び強酸性成分を含む資料の滴
定曲線,二つの変曲部の最も垂直に
近い点を終点とする。
36
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V3: 空試験の滴定に要した0.1 mol/L水酸化カリウムの2-プロ
パノール溶液の量 (mL)
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
8.9
精度 この試験方法によって得られた試験結果の許容差(確率0.95)は,次のとおりである。
備考 試験結果が許容差を外れた場合には,JIS Z 8402-6の規定によって処理する。
a) 室内併行精度 同一試験室において,同一人が同一試験器で,同一条件で短時間内に同一試料を2回
試験したときの試験結果の差の許容差を,表11に示す。
b) 室間再現精度 異なる試験室において,別人が別の試験器で同一試料をそれぞれ1回ずつ試験して求
めた2個の試験結果の差の許容差を,表11に示す。
表 11 精度
塩基価又は強塩基価
mgKOH/g
変曲点が明確な場合
変曲点が不明確な場合室
内併行許容差及び室間再
現許容差
室内併行許容差
mgKOH/g
室間再現許容差
mgKOH/g
0.05 を超え 1.0 以下
0.04
0.08
規定しない
1.0 を超え 5.0 以下
0.1
0.4
5.0 を超え 20 以下
1.0
2
20
を超え100 以下
4
8
100
を超えるもの
10
20
8.10 試験結果の報告 試験結果には,次の事項を記載する。
a) 試料名,採取場所及び採取年月日
b) JISの規格番号:JIS K 2501
c) 試験方法の名称・箇条番号及び8.8によって得られた結果
d) 特記事項
9. 電位差滴定法(塩基価・過塩素酸法)
9.1
試験の原理 試料をクロロベンゼン及び酢酸を含む滴定溶剤に溶かし,ガラス電極と比較電極を用
いて,過塩素酸の酢酸溶液で電位差滴定する。メータ(電位計)の読みと,これに対応する液の滴定量と
の関係を作図し,滴定曲線で得られた変曲点を終点とする。明確な変曲点が得られない場合には酢酸ナト
リウムの酢酸溶液を用いて逆滴定する。
これらの方法で,A法は120 mLの滴定溶剤を用い,B法では60 mLの滴定溶剤を用いた試験ができる。
備考1. この方法は,石油製品及び潤滑油中に含まれる塩基性成分で,酢酸とクロロベンゼンに溶解
するものを過塩素酸の酢酸溶液で電位差滴定法によって測定するものである。
2. 新油及び使用油中の塩基性を示す成分としては,有機塩基,無機塩基,アミノ化合物,弱酸
塩(石けん),多酸塩基の塩基性塩,重金属塩及び酸化防止剤,清浄剤のような添加剤がある。
参考 この[電位差滴定法(塩基価・過塩素酸法)]は,8.による塩基価と同様な結果を得られるが,
過塩基添加剤を含む油及び窒素重合物を含む油は,塩酸法より高い結果が得られることがある。
9.2
試薬 試薬は,次による。
a) 酢酸 JIS K 8355に規定するもの。
備考 酢酸,無水酢酸及びクロロベンゼンは毒性があり,刺激性をもつ。これらの薬品の取扱いはす
37
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
べて十分に換気された場所又はドラフト内で行う。
b) 無水酢酸 JIS K 8886に規定するもの。[a)備考参照]。
c) クロロベンゼン[a)備考参照]
備考 クロロベンゼンの代わりにトルエンを用いてもよい。
d) トルエン JIS K 8680に規定するもの。
e) フタル酸水素カリウム JIS K 8005に規定するものを,120℃で2時間乾燥後,デシケータ中で室温ま
で放冷したもの。
f)
無水炭酸ナトリウム JIS K 8005に規定するもの。
g) 過塩素酸ナトリウム電極液 JIS K 8227に規定する過塩素酸ナトリウムを酢酸中に飽和したもので比
較電極の内部液として用いる。この内部液の底部には,常に過剰の過塩素酸ナトリウムの結晶が残っ
ているようにする。
備考 過塩素酸ナトリウムは毒性があり刺激性をもつ。この試験方法の操作条件では危険はないが,
過塩素酸ナトリウムは加熱されると強力な酸化剤となるので取扱いに注意する。また,こぼし
た場合には,直ちに水で十分に洗浄する。
h) 滴定溶剤 酢酸1容とクロロベンゼン2容とを混合したもの。
i)
0.1 mol/L過塩素酸の酢酸溶液
1) 調製 JIS K 8223に規定する過塩素酸(含量70〜72 %)8.5 mLを,あらかじめ酢酸500 mLと無水
酢酸30 mLとを混合した溶液中にゆっくりと加えて混合した後,更に酢酸を加えて全量を1 Lにす
る。もし過塩素酸(含量60〜62 %)を用いる場合には,過塩素酸は10.2 mL,無水酢酸は38 mLに
する。
備考1. 過塩素酸の酢酸溶液は,この試験方法の操作条件では危険はないが,濃過塩素酸は加熱され
ると強力な酸化剤となるので取扱いに注意する。有機物と一緒に加熱すると爆発性の混合物
を形成するので,有機物との接触を避け,またこぼした場合には水で十分に洗浄する。
2. 滴定時に試験溶液中の試料に含まれる第1級アミン又は第2級アミンのアセチル化を防止す
るため,無水酢酸を過剰に加えてはならない。
2) 標定
2.1) A法 フタル酸水素カリウム0.1〜0.2 gを0.1 mgのけたまではかり採り,温酢酸40 mLに溶かす。
この溶液にクロロベンゼン80 mLを加え,室温まで冷却後,9.6.4又は9.6.5と同様な操作で0.1 mol/L
過塩素酸の酢酸溶液で滴定する。別に用意した酢酸40 mLとクロロベンゼン80 mLとの混合液に
ついて空試験を行う。
2.2) B法 フタル酸水素カリウム0.05〜0.1 gを0.1 mgのけたまではかり採り,温酢酸20 mLに溶かす。
この溶液にクロロベンゼン40 mLを加え,室温まで冷却後,9.6.4又は9.6.5と同様な操作で0.1 mol/L
過塩素酸の酢酸溶液で滴定する。別に用意した酢酸20mLとクロロベンゼン40mLとの混合液につ
いて空試験を行う。
3) 計算 0.1 mol/L過塩素酸の酢酸溶液のモル濃度は,次の式によって算出し,JIS Z 8401の規定によ
って丸めの幅を0.000 1に丸める。
)
(
23
.
204
000
1
0
1
0
V
V
m
C
−
=
×
ここに,
C0: 0.1 mol/L過塩素酸の酢酸溶液のモル濃度 (mol/L)
m: フタル酸水素カリウムのはかり採り量 (g)
38
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V1: 終点までの滴定に要した0.1 mol/L過塩素酸の酢酸溶液の
量 (mL)
V0: 空試験に要した0.1 mol/L過塩素酸の酢酸溶液の量 (mL)
204.23: フタル酸水素カリウムの式量
備考1. 有機溶剤類の膨張係数は比較的大きいので,0.1 mol/L過塩素酸の酢酸溶液の標定時温度と使
用時温度との差は,5 ℃以内でなければならない。
もし,使用時温度が標定時温度より高く,その差が5 ℃を超えるときは,その使用量に係
数 (1−0.001t) を,また使用時温度が標定時温度より低く,その差が5℃を超えるときは係数
(1+0.001t) をそれぞれ乗じる。ここで,tは標定時温度と使用時温度との差(絶対値)をい
う。
2. 0.1 mol/L過塩素酸の酢酸溶液は少なくとも週に1回再標定をする。また,モル濃度が変化し
たと疑われるような場合には,より頻繁に再標定をしなければならない。
j)
0.1 mol/L酢酸ナトリウムの酢酸溶液
1) 調製 JIS K 8005に規定する炭酸ナトリウム(無水)を規定の条件で乾燥した後,その約5.30 gを
300 mLのビーカに0.1 mgのけたまではかり採り,酢酸50 mLを少量ずつ加えて溶かし,これを1L
の全量フラスコに移し入れる。次いでビーカを酢酸で十分に共洗いし,その洗液を全量フラスコに
加えてから,標線まで酢酸を加える。
備考 発熱及び二酸化炭素の発生による激しい泡立ちが起こるので,溶液が飛散しないように十分注
意し,泡立ちを生じなくなってから全量フラスコに移す。
2) 標定
2.1) A法 0.1 mol/L過塩素酸の酢酸溶液8.00 mLをはかり採り,滴定溶剤120 mLに溶かす。この溶液
を9.6.4又は9.6.5と同様の操作で0.1 mol/L酢酸ナトリウムの酢酸溶液で滴定する。
2.2) B法 0.1 mol/L過塩素酸の酢酸溶液4.00 mLをはかり採り,滴定溶剤60 mLに溶かす。この溶液
を9.6.4又は9.6.5と同様の操作で0.1 mol/L酢酸ナトリウムの酢酸溶液で滴定する。
3) 計算 0.1 mol/L酢酸ナトリウムの酢酸溶液のモル濃度は,次の式によって算出し,JIS Z 8401の規
定によって丸めの幅を0.000 1に丸める。
A法の場合
2
0
0
1
)
(8.00
V
C
V
C
−
=
B法の場合
2
0
0
1
)
(4.00
V
C
V
C
−
=
ここに, C1: 0.1 mol/L酢酸ナトリウムの酢酸溶液のモル濃度 (mol/L)
C0: 0.1 mol/L過塩素酸の酢酸溶液のモル濃度 (mol/L)
V0: 空試験に要した0.1 mol/L過塩素酸の酢酸溶液の量 (mL)
V2: 0.1 mol/L酢酸ナトリウムの酢酸溶液の量 (mL)
備考 0.1 mol/L酢酸ナトリウムの酢酸溶液は少なくとも週に1回再標定する。また,モル濃度が変化
したと疑われるような場合には,より頻繁に再標定をしなければならない。
9.3
試験器 自動記録式のもの及び手動記録式のもので,滴定部の一例を図14に示す。
39
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考 試験器によっては,静電気に敏感なものがあり,人間が近づくとメータの指針又は記録用
ペンが誤った動きをすることがある。このような場合には,銅製の金網を円筒にして滴定
用ビーカに巻き,電気的に接地する。
図 14 中和価試験器滴定部(過塩素酸法)(一例)
a) メータ b)及びc)に規定するガラス電極と比較電極を用いたとき,少なくとも1 Vまで読むことがで
きるものでJIS K 0113の規定を満足する補償式電位差計,直示式電位差計又はディジタル式電位差計
で,最小目盛5 mV以下のもの。
b) ガラス電極 pH測定範囲0〜14のもの。
c) 比較電極 内部電極として銀・塩化銀電極,飽和カロメル電極などをもち,内部液補充口(栓付き)
及び液絡部をもち,9.2 g)に規定する過塩素酸ナトリウム電極液を満たしたもの。
備考 通常,液絡部がスリーブ形(図14参照)のものを用いるが,試料の性状によってはファイバー
形,フリット形などを用いてもよい。
参考 ガラス電極と比較電極を1本の形にまとめた複合形電極(例えば,ガラス電極/銀・塩化銀電
極)で応答時間が遅い場合は,高い値を示すことがあるので注意しなければならない。
d) かき混ぜ機 プロペラ付き電動かき混ぜ機,マグネチックスターラなどで,かき混ぜの強さを任意に
変えることのできるもの。
備考1. かき混ぜ機は,あらかじめ接地しておき,滴定操作中にメータの読みに狂いが生じないよう
にする。
2. かき混ぜ機の試験溶液に浸る部分(プロペラ,回転子など)は,試料,試薬などで腐食され
ない材質(例えば,ガラス)のものとする。
e) ビュレット 図9に示す,0.05 mL目盛付きで容量が10 mL又は20 mLのもので,±0.02 mLの正確
度で校正したもの(8)。
注(8) JIS K 2839に規定する図101のものが相当する。
備考 これと同等の性能をもつ電動ビュレットを用いてもよい。
40
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
f)
滴定用ビーカ ガラス製又は試薬に対して不活性なプラスチック製のトールビーカで,A法には図10
に示す容量250 mLのもの(9),B法には電極が60 mLの滴定溶剤に浸るような容量約150 mLのもの。
注(9) JIS K 2839に規定する図102のものが相当する。
g) 滴定支持台 ビーカ,電極,かき混ぜ機及びビュレットを支持するのに適切なもので,ビーカが障害
なく取り出せるような配置とする。
9.4
試料採取方法及び調製方法 試料採取方法及び調製方法は,次による。
a) 試料はJIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法によるか,又はそれらに準
じた方法によって採取及び調製する。
b) 使用油の試料の調製 使用油の試料の調製は,次による。
1) 試料中に沈殿物があるときは,沈殿物自体が酸性若しくは塩基性であるか,又はそれが試料中の酸
性若しくは塩基性の物質を吸着していることがあるから,このような沈殿物を含む試料をはかり採
るには,試料全体を代表するようによくかき混ぜてとることが重要である。
備考 使用油は保存中に変質することがあるので,採取後,できるだけ速やかに測定する。
2) 試料を60 ℃±5 ℃に加温し,沈殿物を均一にする。
なお,沈殿物が含まれていない場合は加温をしなくともよい。
3) 試料に沈殿物が目視で観察されるときは,全沈殿物を均一にした後,試料をJIS Z 8801-1に規定す
る目開き150 μmの金属製網ふるいでろ過し,大きな異物を除去する。
9.5
電位差滴定装置の調整
9.5.1
電極の準備 電極の準備は,次による。
a) ガラス電極を水で十分にすすいだ後,下半分を水中に浸しておく。
備考 新しいガラス電極又は乾燥状態で保存しておいたガラス電極は,使用する前に6時間以上水中
に浸し,電極の非対象電位を安定させてから用いる。
b) 比較電極の内部液として塩化カリウムの水溶液が入っている場合には,次によってこれを過塩素酸ナ
トリウム電極液に換える。
1) 塩化カリウム水溶液を流し出し,電極の外管内を過塩素酸ナトリウム電極液で数回洗った後,この
電極液を内部液補充口まで満たす。
2) スリーブ形比較電極を用いるときは,スリーブを緩めて1)の操作をした後,スリーブのすり合わせ
面を数滴の過塩素酸ナトリウム電極液で洗う。次いで,すり合わせ面に過塩素酸ナトリウム電極液
が十分にしみわたったならば,スリーブをしっかりとはめ,再び外管内に過塩素酸ナトリウム電極
液を満たし,内部液補充口に栓をする。
3) フリット形又はファイバ形比較電極は,1)の操作をした後,この電極を大気中に保持したとき,過
塩素酸ナトリウム電極液の漏れの速さは,わずかににじみ出る程度(1時間に1滴以内)がよい。
備考 比較電極の内部液の交換に際しては,過塩素酸ナトリウム電極液が直接皮膚に触れないように
注意する。このためには,手術用ゴム手袋などを用いるとよい。
c) 比較電極の外管及び液絡部の周囲を水で十分にすすいだ後,下半分を水に浸しておく。その際,過塩
素酸ナトリウム電極液の液面は常に水面より上方に保っておく。
d) 電極を使用するときは,両電極とも清浄な布若しくは柔らかい紙で水分をふき取るか,又は滴定溶剤
で十分に洗って用いる。その際,比較電極の電極液面は,常に滴定用ビーカ中の液面より上方に保ち,
使用中電極液内に汚染物が侵入するのを防ぐ。また,電極を使用している間は,比較電極の内部液補
充口の栓を取り外しておく。
41
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.5.2
電極の検査 電極を最初に使用する場合,又は取り換えた場合には,メータと電極との組合せを次
によって検査し,その後もときどき検査する。
a) A法 酢酸100 mLにフタル酸水素カリウム0.2gを溶かし,よく混合した溶液に電極を浸し,メータ
の読みを記録する。次に,電極をクロロベンゼンで洗った後,酢酸100 mLに0.1 mol/L過塩素酸の酢
酸溶液1.5 mLを加えた溶液に浸し,メータの読みを記録する。得られた二つのメータの読みの差は
0.3 V以上でなければならない。
b) B法 酢酸50 mLにフタル酸水素カリウム0.1gを溶かし,よく混合した溶液に電極を浸し,メータの
読みを記録する。次に,電極をクロロベンゼンで洗った後,酢酸50 mLに0.1 mol/L過塩素酸の酢酸
溶液0.75 mLを加えた溶液に浸し,メータの読みを記録する。得られた二つのメータの読みの差は0.3
V以上でなければならない。
9.5.3
電極の洗浄 電極を使用した後は,クロロベンゼン又は滴定溶剤で十分に洗い,電極に付着した油
状物質を取り除く。次に,水で洗浄し,ガラス電極の特性回復及び比較電極の液絡部の周囲に析出した過
塩素酸ナトリウムの除去を行う。再び,滴定溶剤で両電極を洗う。引き続いて電極を使用しない場合は,
比較電極の内部液補充口に栓をした後,両電極を水に浸しておく。一連の試料の滴定を始める前に,滴定
溶剤の空試験を1回又は2回行い,電極の特性回復ができたかどうかを調べる。
9.5.4
電極の保守 電極の保守は,次による。
a) ガラス電極の電極膜の汚れは,電位こう配の低下,非対象電位差の増加など電極特性を変化させ,試
験誤差の原因となることがあるので,電極膜の表面が9.5.3による洗浄では取り除くことができない汚
れが認められる場合は,次のようにして清浄にする。
なお,このガラス電極の清浄操作を行った後は,9.5.2によって電極の検査を行う。クロロベンゼン
など汚れを落とすのに適した溶剤を湿した脱脂綿で電極膜の表面をこすり,汚れを取り除く。この際,
強くこすり過ぎて,電極膜を破損しないように注意する。次に,石けん水,液状合成洗剤などを十分
に湿した脱脂綿で電極膜の表面を数回ぬぐった後,水で十分にすすいで清浄にする。
備考 ガラス電極の汚れがひどく,上記の方法では清浄にできない場合は,a)の清浄操作後の電極を
硝酸1容と水1容に少量の過酸化水素水を加えた溶液中に約30分間浸した後,水で十分にすす
ぐとよい。
b) 比較電極は,少なくとも週に1回,過塩素酸ナトリウム電極液を新しいものと取り換える。
c) 滴定の後は,わずかの間でも電極を滴定溶液中に浸したままにしてはならず,9.5.3によって電極を洗
浄した後,下半分を水に浸しておく。
備考 電極はこわれやすいので,その取扱いには十分注意する。
9.6
試験の手順
9.6.1
試料のはかり採り量 試料のはかり採り量は,予期塩基価に応じて,その概略量を次の式によって
算出し,表12に規定する精度で滴定用ビーカにはかり採る。ただし,最大はかり採り量は,A法について
は20 g,B法については10 gとする。
A法の場合
e
A
28
B
m=
又は,B法の場合
e
B
10
B
m=

42
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに, m: 概略の試料はかり採り量 (g)
Be: 試料の予期塩基価 (mgKOH/g)
表 12 試料はかり採り量
試料はかり採り量 g
はかり採り最小目盛 g
0.10 を超え
0.25以下
0.0005
0.25 を超え
1.0 以下
0.001
11.0 を超え
5.0 以下
0.005
5.0 を超え
10 以下
0.02
10
を超え
20 以下
0.05
備考 予期塩基価が未知の場合には,次の操作で概略量を求めることが
できる。A法の場合には0.2〜0.3 g,B法の場合には0.1〜0.2 g
の測定試料をはかり採り,終点を570 mVとして滴定する。この
値を変曲点として塩基価を計算し,概略の試料のはかり採り量を
求める。
B法では,特に測定試料のはかり採り量を少なくする必要のあ
る高塩基価石油製品の場合には,正確なはかり採り量を求めるこ
とが重要である。
9.6.2
試験溶液の調製 滴定用ビーカに滴定溶剤120 mL(A法)又は60 mL(B法)を加えて滴定支持
台上に置き,試料が溶解するまで溶液をかき混ぜる。
備考 試料が溶けにくいときは,初めにクロロベンゼン80 mL(A法)又は40 mL(B法)を加えて
溶かし,次に酢酸40 mL(A法)又は20 mL(B法)を加える。
9.6.3
試験の準備
a) 9.5.1によって準備した電極を試験溶液中に浸す。この際,電極の長さの約1/2が溶液に浸るように調
整する。かき混ぜ機を始動し,試験溶液が飛散しないように,また空気が入らない程度にかき混ぜの
強さを調整した後,メータのスイッチを入れる。
b) 滴定に先立って,メータの指針を電位調整用ダイヤルによって右方向に移動させ,適切な位置に設定
する。
備考 通常,700 mVの位置にメータの指針を設定するとよい。メータの指針は滴定中,左方向(試験
機器によっては,右方向になるものもある)に移動していくので,このメータ指針の初期設定
操作をしておかないと,メータの目盛範囲から指針が外れるおそれがある。
c) ビュレットに,0.1 mol/L過塩素酸の酢酸溶液をとり,ビュレットの先端が試料溶液中に深さ約25 mm
浸るようにその位置を定める。最初のビュレットの読み及びメータの読みを記録する。
9.6.4
試料の測定 試料の測定は,次のa)又はb)による。
a) 手動滴定法 0.1 mol/L過塩素酸の酢酸溶液を少量ずつ加え,メータの指針が一定の位置を示すのを待
ってから,再びビュレット及びメータの読みを記録する。滴定の開始時及びその後,0.1 mol/L過塩素
酸の酢酸溶液0.1 mLごとの滴下によるメータの読みの変化が30 mV以上を示すような部分(変曲点)
では,0.1 mol/L過塩素酸の酢酸溶液を0.05 mLずつ加える。0.1 mol/L過塩素酸の酢酸溶液0.1 mLの
滴下によるメータの読みの変化が30 mV未満を示すような部分(平たん部)ではメータの読みの変化
が30 mVを超えないで,ほぼ等しくなるように,0.1 mLよりも多く(ただし,0.25 mLを超えてはな
らない。)加える。
このようにして滴定を続け,メータの読みの変化が0.1 mL当たり5 mVを超えなくなったときに滴
定をやめ,メータのスイッチを切る。
備考 メータの読みの変化が毎分5 mVよりも小さい場合には,メータの読みは一定とみなす。
43
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 自動滴定法 試験器取扱説明書によって試験器を調整し,滴定速度を毎分1.0 mL以下に設定して滴定
する。
備考 a)及びb)の滴定を以下直接滴定法という。
9.6.5
機器の洗浄 滴定が終わったならば,かき混ぜ機を止め,滴定用ビーカを取り除き,電極及びビュ
レットの先端をクロロベンゼン又は滴定溶剤と水と滴定溶剤の順にすすぐ。続いて試験を行わない場合は,
電極はその下半分を水に浸しておく。
9.6.6
空試験 試験ごとに,滴定溶剤120 mL(A法)又は60 mL(B法)について次のように1回の空
試験を行う。手動滴定法の場合は,0.1 mol/L過塩素酸の酢酸溶液を0.05 mLずつ加え,滴下ごとにメータ
の読みが一定になるのを待ってから,メータの読みとビュレットの読みとを記録する。自動滴定法の場合
は,9.6.4 b)による。
9.7
計算及び結果 計算及び結果は,次による。
a) 手動滴定法の場合は,メータの読みに対応する0.1 mol/L過塩素酸の酢酸溶液の滴定量を方眼紙上にプ
ロットし,滴定曲線を作図する。
b) 手動滴定法又は自動滴定法によって得られた滴定曲線上の変曲点を滴定の終点とする。変曲点の決定
はJIS K 0113の規定による。
c) もし変曲点がない場合及び判定が不十分の場合には,9.8に示す逆滴定法によらなければならない。
d) 塩基価は,次の式によって算出し,有効数字3けたに丸める。ただし,1 mgKOH/g未満の場合はJIS Z
8401の規定によって丸めの幅を0.01に丸める。
m
C
V
V
BN
1.
56
)
(
0
3
4
×
×
−
=
ここに,
BN: 塩基価 (mgKOH/g)
V4: 終点までの滴定に要した0.1 mol/L過塩素酸の酢酸溶
液の量 (mL)
V3: 空試験におけるV4に相当する量 (mL)
C0: 0.1 mol/L過塩素酸の酢酸溶液のモル濃度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
参考 塩基価をグラム当たりの水酸化物のミリ当量で表すときは,次の式によって算出する。
m
C
V
V
BNe
0
3
4
)
(
×
−
=
ここに,
BNe: グラム当たりの水酸化物のミリ当量 (mmolOH/g)
V4: 終点までの滴定に要した0.1 mol/L過塩素酸の酢酸溶液の
量 (mL)
V3: 空試験におけるV4に相当する量 (mL)
C0: 0.1 mol/L過塩素酸の酢酸溶液のモル濃度 (mol/L)
m: 試料のはかり採り量 (g)
9.8
逆滴定法 逆滴定法による測定は,次による。
a) 試料を9.6に準じて滴定用ビーカにはかり採る。ただし,最大はかり採り量は,A法については5 g,
B法については2.5 gまでとする。
備考 この試料量で変曲点が得られない場合は,試料量を半分(A法では2.5 g,B法については1.25
g)にして再試験する。
b) 試料をはかり採った滴定用ビーカに,滴定溶剤120 mL(A法)又は60 mL(B法)を加えて滴定支持
台上に置き,試料が溶解するまでかき混ぜる。試料が溶けにくいときは,9.6.2の備考による。

44
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) ビュレット又はピペットを用いて,試験溶液に0.1 mol/L過塩素酸の酢酸溶液8.00 mL(A法)又は4.00
mL(B法)を正しく加える。次いで,滴定用ビーカ内容物が飛散しないように,また,空気が入らな
い程度にかき混ぜの強さを調節し,ビーカ内容物を2〜3分かき混ぜる。
その後は,0.1 mol/L酢酸ナトリウムの酢酸溶液を用いて9.6.3,9.6.4 a)又はb),9.6.5及び9.6.6と同
様な操作で電位差滴定する。
備考 メータの指針の初期設定操作は,次による。
逆滴定法では,試験溶液に0.1 mol/L過塩素酸の酢酸溶液8.00 mL(A法)又は4.00 mL(B
法)が加えられているため,メータのスイッチを入れたとき,通常,メータの指針は極端に左
方向に移動し,目盛範囲から外れることが多い。
このようなときは,電位調整用ダイヤルを回してメータの指針を移動させ,目盛範囲の適切
な位置,例えば,100 mVに設定する。ただし,試験機器によっては指示値が反対になることも
ある。
d) 試料を用いずにb)及びc)と同様の操作で空試験を行う。
e) 塩基価は,次の式によって算出し,有効数字3けたに丸める。ただし,1 mgKOH/g未満の場合には,
JIS Z 8401の規定によって丸めの幅を0.01に丸める。
m
C
V
V
BN
1.
56
)
(
1
5
2
×
×
−
=
ここに,
BN: 塩基価 (mgKOH/g)
V2: 空試験で,終点までの滴定に要した0.1 mol/L酢酸ナトリウ
ムの酢酸溶液の量 (mL)
V5: 試料の滴定におけるV2に相当する量 (mL)
C1: 0.1 mol/L酢酸ナトリウムの酢酸溶液のモル濃度 (mol/L)
m: 試料のはかり採り量 (g)
56.1: 水酸化カリウムの式量
参考 塩基価をグラム当たりの水酸化物のミリ当量で表すときは,次の式によって算出する。
m
C
V
V
BNe
1
5
2
)
(
×
−
=
ここに, BNe: グラム当たりの水酸化物のミリ当量 (mmolOH/g)
V2: 空試験で,終点までの滴定に要した0.1 mol/L酢酸ナトリウ
ムの酢酸溶液の量 (mL)
V5: 試料の滴定におけるV2に相当する量 (mL)
C1: 0.1 mol/L酢酸ナトリウムの酢酸溶液のモル濃度 (mol/L)
m: 試料のはかり採り量 (g)
9.9
精度 この試験方法によって得られた試験結果の許容差(確率0.95)は,次のとおりである。
備考 この精度を外れたときは,JIS Z 8402-6の規定によって処理する。
a) 室内併行精度 同一試験室において,同一人が同一試験器で,同一条件で短時間内に同一試料を2回
試験したときの試験結果の差の許容差を,表13に示す。
b) 室間再現精度 異なる試験室において,別人が別の試験器で同一試料をそれぞれ1回ずつ試験して求
めた2個の試験結果の差の許容差を,表13に示す。
表 13 精度
滴定法
方法
室内併行許容差
室間再現許容差

45
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
直接滴定
A法
0.03X
0.07X
B法
0.05X
0.07X
逆滴定
A法
0.24X
0.32X
備考 表中のXは,試験結果の平均値 (mgKOH/g)
参考 精度設定のため検討した試料の塩基価の範囲は,次のとおりである。
参考表 1 試料の種類及び塩基価
試料の種類
滴定法
塩基価の範囲 mgKOH/g
新油
直接滴定
6〜70
添加剤
直接滴定
5〜300
使用油
直接滴定
5〜27
9.10 試験結果の報告 試験結果には,次の事項を記載する。
a) 試料名,採取場所及び採取年月日
b) JISの規格番号:JIS K 2501
c) 試験方法の名称・箇条番号及び9.7,9.8によって得られた結果並びにA法,B法の別
d) 特記事項

46
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(参考)指示薬光度滴定法
この附属書(参考)は,潤滑油の酸価を試験する方法を参考として示すもので,規定の一部ではない。
1. 適用範囲 この附属書は,潤滑油の酸価を指示薬滴定法を基に,自動光度滴定装置を用いた透過率測
定によって自動的に試験する方法について記述する。
備考1. この規格は,危険な試薬,操作及び装置を使うことがあるが,安全な使用方法をすべてにわ
たって規定しているわけではないので,この試験方法の使用者は,試験に先立って,適切な
安全上及び健康上の禁止事項を決めておかなければならない。
2. この方法は,本体5.指示薬滴定法による酸価を試験する代わりに,自動光度滴定装置を用い
た透過率測定によって自動的に試験する方法であり,試料の着色によって指示薬の変色が目
視で不明りょうな場合にも適用できる。
3. この方法は,指示薬の変色を敏感にとらえることができれば,油種を制限することなく適用
できる。
2. 引用規格 次に掲げる規格は,この附属書に引用されることによって,この附属書の規定の一部を構
成する。これらの引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0557 用水・排水の試験に用いる水
JIS K 2251 原油及び石油製品−試料採取方法
JIS K 2839 石油類試験用ガラス器具
JIS K 8005 容量分析用標準物質
JIS K 8574 水酸化カリウム(試薬)
JIS K 8603 ソーダ石灰(試薬)
JIS K 8680 トルエン(試薬)
JIS K 8693 p-ナフトールベンゼイン(試薬)
JIS K 8839 2-プロパノール(試薬)
JIS Z 8401 数値の丸め方
JIS Z 8402-6 測定方法及び測定結果の精確さ(真度及び精度)−第6部:精確さに関する値の実用的な
使い方
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
3. 定義 この附属書で用いる主な用語の定義は,次による。
a) 酸価 試料1 g中に含まれる酸性成分を中和するのに要する水酸化カリウムのミリグラム (mg) 数。
4. 試験の原理 試料をトルエン,2-プロパノール及び少量の水を含む混合溶剤に溶かし,指示薬として
p-ナフトールベンゼインを加え,水酸化カリウムの2-プロパノール溶液で滴定する。このときの指示薬の
色の変化を光度滴定用液浸型プローブ電極(以下,電極という。)を用いて透過率としてとらえ,これに対
応する滴定溶液の滴定量との関係を作図し,透過率変化の最も大きい部分と,完全に過滴定になり透過率

47
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
がほとんどなくなった部分との交点(B交点)から求めた終点から酸価を求める。
参考 終点の決定方法として,指示薬滴定法と最も一致するBackward交点(変曲点を基準にした接
線と過滴定部との交点。以下,B交点という。)を採用した(附属書1図1参照)。
5. 試薬 試薬は,次による。
a) トルエン JIS K 8680に規定するもの。
b) 水 JIS K 0557に規定するA3のもの。
c) 2-プロパノール JIS K 8839に規定するもの。
d) 滴定溶剤 トルエン500 mL及び水5mLを2-プロパノール495 mLに加えたもの。
e) 0.1mol/L水酸化カリウムの2-プロパノール溶液
1) 調製 三角フラスコ2 000 mLに2-プロパノール約1Lを採り,これにJIS K 8574に規定する水酸化
カリウム6 gを加え,底にかたまりができないようにかき混ぜながら,15〜20分間静かに煮沸する。
この溶液を二酸化炭素を遮り数日間放置した後,その上澄液をポリエチレン瓶などに入れ,JIS K
8603に規定するソーダ石灰及び乾燥剤を満たした保護管を付けて保存する。
備考 市販の0.1 mol/L水酸化カリウムの2-プロパノール溶液を用いてもよい。
2) 標定 標定は,次による。
2.1) JIS K 8005に規定するフタル酸水素カリウムを105 ℃で2時間乾燥し,その0.1〜0.2 gを0.1 mg
のけたまではかり採り,二酸化炭素を含まない水125 mLに溶かし,0.1 mol/L水酸化カリウムの
2-プロパノール溶液を用いて電位差滴定する。
2.2) 水125 mLについて2.1)の操作によって空試験を行う。
2.3) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度は,次の式によって算出し,JIS Z 8401
の規定によって丸めの幅を0.001に丸める。
)
(
23
.
204
000
1
KOH
1b
v
W
c
−
=
ここに, cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
W: フタル酸水素カリウムのはかり採り量 (g)
v1: 終点までの滴定に要した0.1 mol/L水酸化カリウムの2-プ
ロパノール溶液の量 (mL)
b: 空試験におけるv1に相当する量 (mL)
204.23: フタル酸水素カリウムの式量
2.4) 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃度の変化が0.000 5 mol/L以上にならない
間隔でときどき標定し直す。
備考 2-プロパノールのような有機溶剤の膨張係数は,比較的大きいので,0.1 mol/L水酸化カリウム
の2-プロパノール溶液の標定は試験時に近い温度で行う。
f) p-ナフトールベンゼイン溶液 JIS K 8693に規定するp-ナフトールベンゼイン1.0 gを滴定溶剤100
mLに溶かしたもの。
6. 試験器 試験器は,自動式のもので次による。
a) 自動光度滴定装置 自動光度滴定装置は,例えば,次のような測定制御機能を備えたもの。
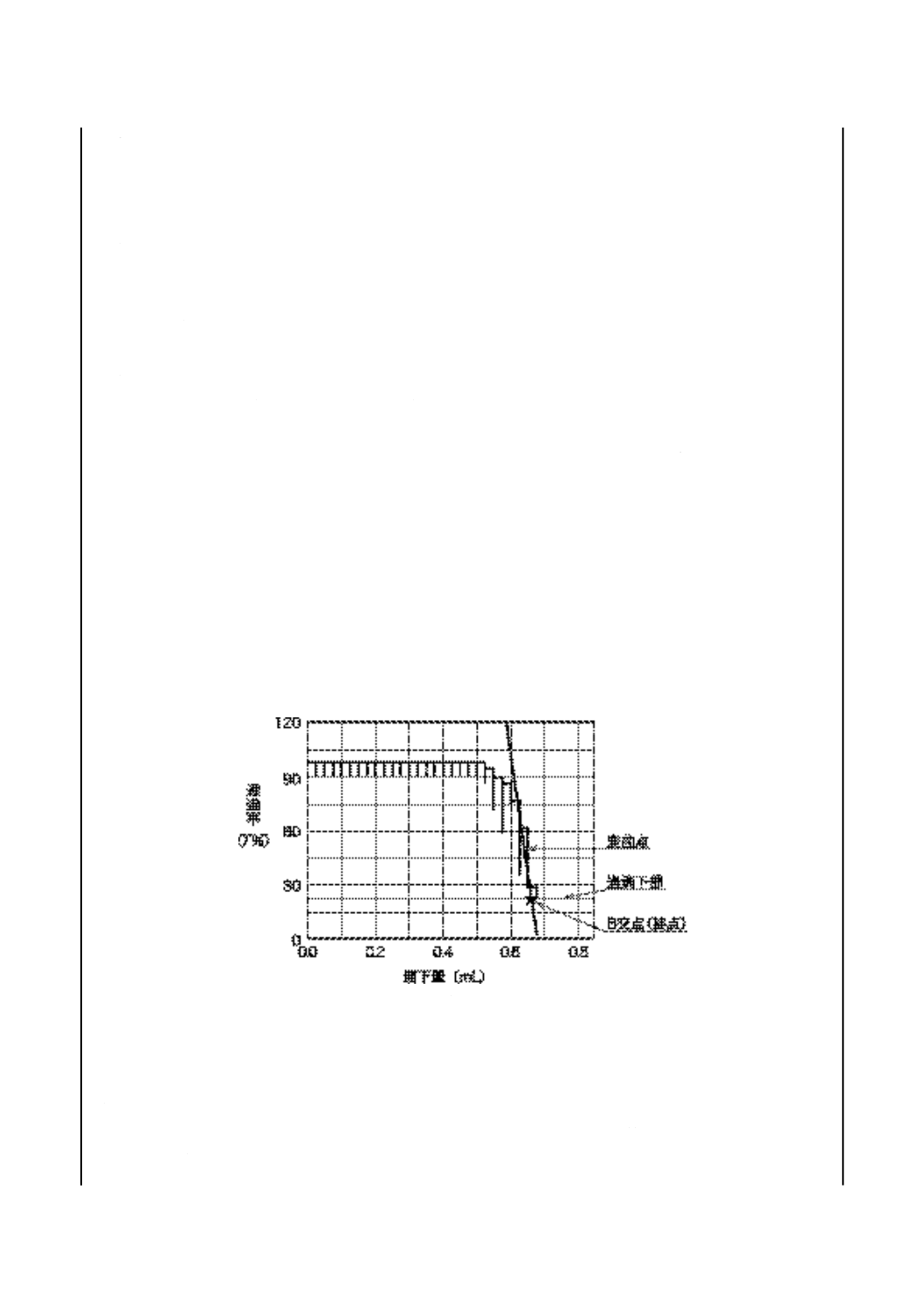
48
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 透過率検出機能 指示薬の変色を透過率で検出し,試薬の滴下量と透過率の関係を記録できるもの。
透過率は,特定の波長を検出するため650 nmの干渉フィルタを用いる。
2) 滴定容量の制御(最少滴下量) 滴定制御装置によって自動電動ビュレットの滴下量を制御でき,
最少滴下量を0.01mLに設定できるもの。
3) 滴下間隔の制御(待ち時間) 滴定試薬を,所定間隔で滴下するためのタイマーを備えたもの。滴
下間隔の基本は10秒間隔とする。
4) 指示薬の変色の戻りを検出する機能(待ち感度) 透過率の変化を検出しながら,試薬の滴下間隔
を調節する機能を備えたもの。すなわち,一定時間に透過率の変化度合が大きいときは,更に滴下
間隔を延長できる機能である。
5) 待ち感度機能の上限を定める機能(待ち感度上限時間) 待ち感度機能で指示薬の色の戻りを検出
し,滴下間隔を伸ばして滴定するが,色の戻りが小刻みに頻繁に起こる場合は,所定時間経過した
ときに,次の滴下を行う機能を備えたもの。
6) 終点検出機能(B交点検出) 指示薬の色がだいだい色→うす緑→濃い緑→青に変化していく過程
を作図した場合に,うす緑→濃い緑の間で最も透過率の変化度合の大きい所(変曲点)の接線と,
更に,試薬の滴下を続け,青に変化しほとんど透過率が変化しなくなった線(過滴下部)との交点
(B交点)を終点とする。終点の判定例を附属書1図1に示す。
6.1) 変曲点の検出 当初100 %程度ある透過率が,滴定試薬を滴下するごとに,指示薬の色の変化に
合わせて減少してくる。試薬の滴下量に対する透過率の変化量の大きさを求めると,最も色の変
化の大きなときと,その前後ではかなり明確に違いがみられる。特に,指示薬の色が,うす緑→
濃い緑に変わるところで顕著に見られる。この最も色の変化の大きいところを変曲点とする。
6.2) 過滴下機能 変曲点検出後,更に滴定を続け指示薬の色が青に変色し,ほぼ一定の透過率を示す
ところまで滴定を行える機能。
附属書1図 1 終点の判定(一例)
6.3) 作図機能:滴定過程における透過率の変化を詳細に作図できる機能を備えたものを用いると便利
である。
b) 電極 附属書1図2に示すような構造で,色の変化を透過率で表すための検出装置。
c) 干渉フィルタ 650 nmの波長の光を選択的に透過することができるもの。
d) かき混ぜ機 マグネチックスターラなどで,かき混ぜの強さを任意に調節できるもの。
e) 滴定用ビーカ ガラス製で容量250 mLのもの。
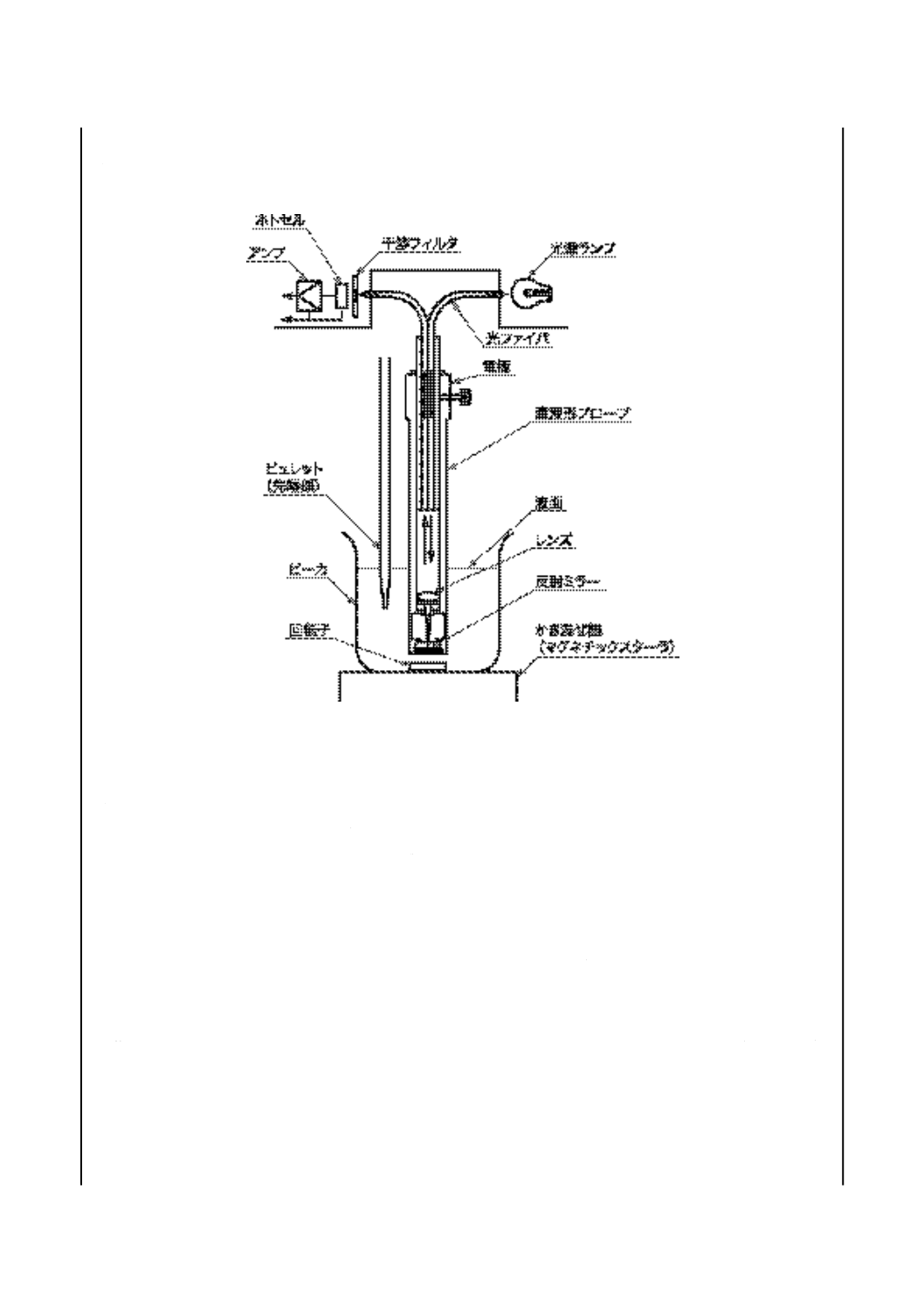
49
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考 JIS K 2839に規定する図102のものが相当する。
f)
滴定支持台 ビーカ,電極,ビュレットなどを支持するのに適切なもの。電極,ビュレットなどに障
害なくビーカが取り出せるような配置が望ましい。
附属書1図 2 指示薬光度滴定法(一例)
7. 試料採取方法及び調製方法 試料採取方法及び調製方法は,次による。
a) 試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法によるか,又はそれらに
準じた方法によって採取及び調製する。
b) 使用油の試料の調製 使用油の試料の調製は,次による。
1) 試料中に沈殿物があるときは,沈殿物自体が酸性若しくは塩基性であるか,又はそれが試料中の酸
性若しくは塩基性の物質を吸着することがあるから,このような沈殿物を含む試料を採るときは,
試料全体を代表するようによくかき混ぜる。
2) 試料を60 ℃±5 ℃に加熱し,沈殿物を均一にする。沈殿物が含まれていない場合は加熱をしなく
てもよい。
備考 使用油は保存中に変質することがあるので,採取後,できるだけ速やかに試験する。
3) 試料に沈殿物が目視で観察される場合は,すべての沈殿物を均一にした後,試料をJIS Z 8801-1に
規定する目開き150 μmの金属製網ふるいでろ過し,大きな異物を除去する。
8. 試験の準備 試験の準備は,次による。
a) 自動測定制御機能の設定 自動測定を行う場合の測定条件制御機能を設定する。それぞれのパラメー
タは,用いる機種によって異なるので,必要な測定条件を次のように定める。この条件を満足する設
50
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定値を所定のパラメータに設定する。附属書1表1に設定値の一例を示す。
附属書1表 1 設定値の一例
終点検出方式
B交点検出
検出波長
650 nm
検出単位
透過率T%
スタートタイマー
60秒
滴下量
空試験の場合0.010 mL
本試験の場合0.025 mL
待ち時間
10秒
待ち感度
透過率1 %
待ち感度上限時間
60秒
検出感度
透過率4 %
過滴下量
0.3〜0.5 mL
b) 透過率T%の校正 透過率検出の感度を透過率0 %〜100 %で調整する。この場合,ランプ又は反射ミ
ラーを清浄にしてから実施する。
c) 自動ビュレットチップの気泡除去 自動ビュレットのチップに気泡だめが付いている場合には,所定
の手順によって気泡を除去する。
d) 電極の洗浄 電極及びビュレットの先端をトルエン又は滴定溶剤ですすいでから試験に用いる。
9. 試験の手順 試験の手順は,次による。
a) 試料はかり採り量 試料のはかり採り量は,予期酸価に応じて,その概略量を次の式によって算出し,
mg単位の精度で滴定用ビーカにはかり採る。ただし,最大はかり採り量は20 gとする。
Ae
m
5
=
ここに,
m: 概略の試料のはかり採り量 (g)
Ae: 試料の予期酸価 (mgKOH/g)
b) 試験溶液の調製 滴定用ビーカに滴定溶剤100 mLを加え,次に,指示薬としてp-ナフトールベンゼ
イン溶液0.15 mLを加えて滴定支持台上に置き,試料が溶解するまでかき混ぜる。
備考1. 滴定溶剤中に,あらかじめ指示薬としてp-ナフトールベンゼイン溶液を滴定溶剤100 mLに
対して0.15 mLに相当する量を加えたものを使用してもよい。
2. 自動サイクラーを用いる場合,測定中の試料の次の試料に滴定溶剤をあらかじめ分注すると
きは,測定待ちのあいだはかき混ぜてはならない(空気中の二酸化炭素を吸収するなど悪影
響が出ることがある。)。
c) 自動測定の開始 準備した電極を試料溶液中に浸す。この際,電極の透過率測定部に気泡が入らない
ように,電極の浸る長さ及びかき混ぜの強さを調節する。自動滴定装置を8. a)の設定条件によって,
滴定を行う。
d) 測定結果の判定 作図した滴定チャートから,滴定結果に異状があるか否かを判断し,次の四つに分
類する。
1) 滴定正常 変曲点が透過率の最大変化の位置で検出でき,変曲点の位置と終点がほとんど一致して
いる(指示薬光度滴定法に適した試料の結果でありそのまま報告できる。)[附属書1図3 a)]。
2) 滴定正常(再計算) 変曲点が透過率の最大変化の位置で検出できていない場合であり,変曲点の
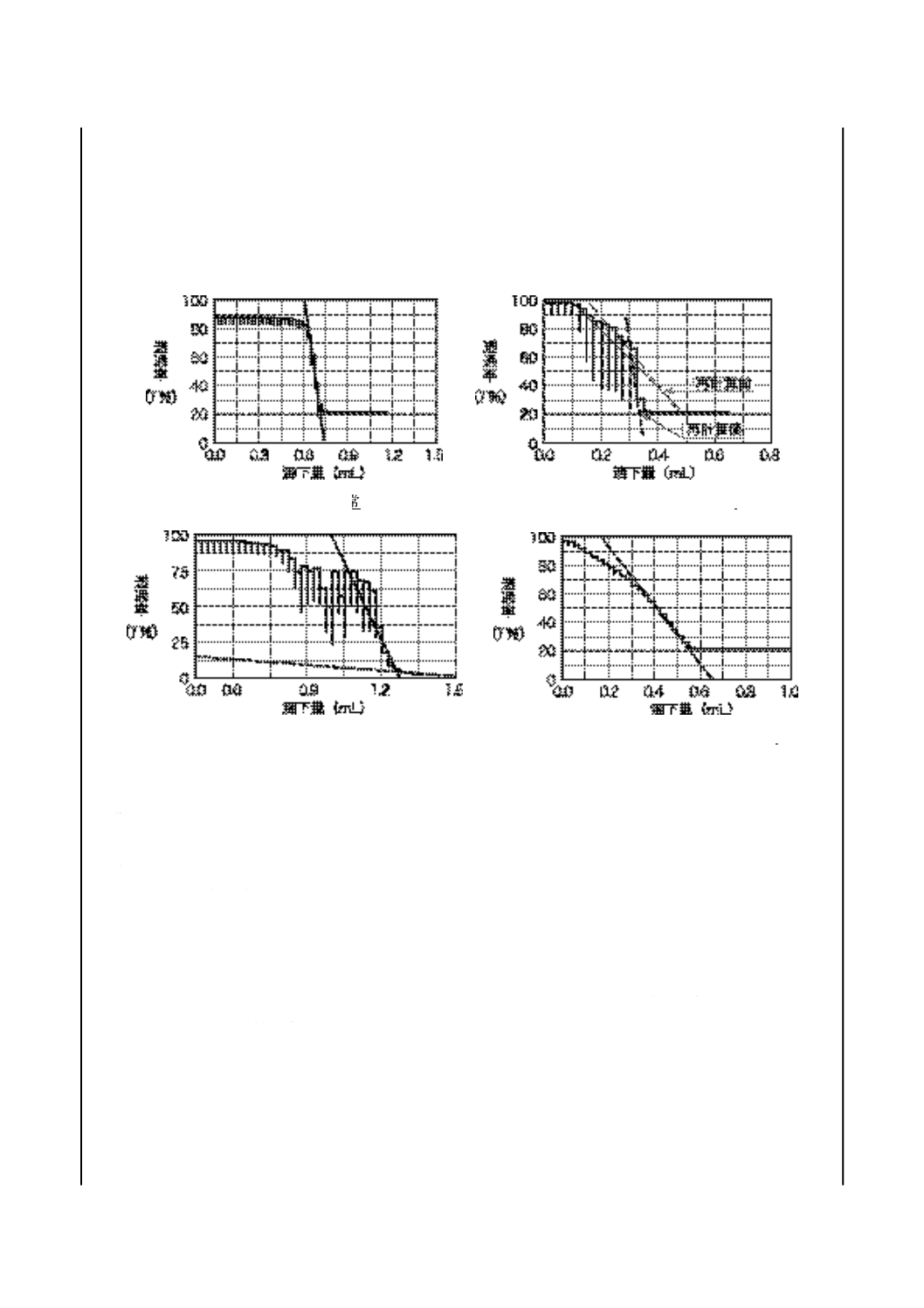
51
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
位置と終点が異なっている場合(指示薬光度滴定法に適した試料の結果であるが,終点検出に異状
があり再計算を要する。)[附属書1図3 b)]。
3) 滴定異常(再試験) 滴定中に一度下がった透過率が再度上がる場合(試験溶液のかき混ぜが不十
分なことが考えられるので,再試験を必要とするもの。)[附属書1図3 c)]。
4) 滴定異常(適用外) 透過率の最大変化の位置が認められず,変曲点が判別できない場合(指示薬
光度滴定法に適さない試料)[附属書1図3 d)]。
a) 滴定正常 b) 滴定正常(再計算)
c) 滴定異常(再計算) d) 滴定異常(適用外)
附属書1図 3 滴定チャート(一例)
e) 再計算又は再試験の実施 d)によって判断した結果,次の再計算又は再試験を実施する。
1) 再計算 変曲点検出位置の異常が認められた場合には,変曲点を透過率の最大変化の位置で検出で
きるようにした後,終点を再検出して再計算を行う。
2) 再試験 試料のはかり採り量を変更して再試験を実施する。再試験後も滴定チャートに異常が認め
られる場合は適用外とする。
備考 この場合の試料はかり採り量は,9. a)の規定にはこだわらない。
f)
機器の洗浄 滴定が終わったならば,かき混ぜ機をとめ,滴定用ビーカを取り除き,電極及びビュレ
ットの先端をトルエン又は滴定溶剤ですすぐ。
g) 空試験 試験ごとに,滴定溶剤100 mLに指示薬としてp-ナフトールベンゼイン溶液0.15 mLを加え
たものについて,最少滴下量を0.01 mLに設定して8. a)の設定条件によって滴定を行い,空試験の滴
下量を求める。
備考 b)〜c)及びf)〜g)の操作を自動光度滴定装置附属の自動サイクラーなどビーカを自動的に交換
でき,また,滴定溶剤を自動的に分注できる装置を使用してもよい。
10. 計算及び結果 計算及び結果は,次による。
52
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
滴下量と透過率との関係を作図して終点を求め,滴定が正しく行われているか,9. d)によって評価した
後,次の式によって酸価を算出し,有効数字3けたに丸める。ただし,1 mgKOH/g未満の場合には,JIS Z
8401によって丸めの幅を0.01に丸める。
m
V
V
c
AN
)
(
KOH
1.
56
0
1−
=
×
ここに,
AN: 酸価 (mgKOH/g)
V1: 試料の滴定に要した0.1 mol/L水酸化カリウムの2-プロパ
ノール溶液の量 (mL)
V0: 空試験の滴定に要した0.1 mol/L水酸化カリウムの2-プロ
パノール溶液の量 (mL)
cKOH: 0.1 mol/L水酸化カリウムの2-プロパノール溶液のモル濃
度 (mol/L)
m: 試料のはかり採り量 (g)
11. 精度 この試験方法によって得られた試験結果の許容差は(確率0.95),次のとおりである。この許容
差は9. d)の測定結果の判定で,滴定正常について適用する。
備考 試験結果が許容差を外れた場合には,JIS Z 8402-6の規定によって処理する。
a) 室内併行精度 同一の試験室において,同一人が同一試験器で,引き続き短時間内に同一試料を2回
試験したときの試験結果の差の許容差を附属書1表2に示す。
b) 室間再現精度 異なる試験室において,別人が別の試験器で,同一試料をそれぞれ1回ずつ試験した
とき,2個の試験結果の差の許容差を附属書1表2に示す。
附属書1表 2 精度
酸価
室内併行許容差
室間再現許容差
mgKOH/g
mgKOH/g
mgKOH/g
0.0を超え0.1以下
0.03
0.04
0.1を超え0.5以下
0.05
0.08
0.5を超え1.0以下
0.08
平均値の15 %
1.0を超え2.0以下
0.12
2.0を超えるもの
平均値の10 %
12. 試験結果の報告 試験結果には,次の事項を記載する。
a) 試料名,採取場所及び採取年月日
b) JISの規格番号:JIS K 2501附属書1
c) 試験方法の名称・箇条番号及び10.によって得られた結果
d) 特記事項
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2(参考)JISと対応する国際規格との対比表
JIS K 2501:2003 石油製品及び潤滑油−中和価試験方法
ISO 6618:1997 石油製品及び潤滑油−中和価試験方法−指示薬滴定法
ISO 7537:1997 石油製品−酸価試験方法−セミミクロ指示薬滴定法
ISO 6619:1988 石油製品及び潤滑油−中和価−電位差滴定法
ISO 3771:1994 石油製品−塩基価の測定−過塩素酸電位差滴定法
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の項目
ごとの評価及びその内容
表示箇所:本体
表示方法:点線の下線又は実線の側線
(V)JISと国際規格との技術的差異
の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
1.適用範囲
中和価とは,酸価,強酸
価,塩基価,強塩基価
ISO 6618
ISO 7537
ISO 6619
ISO 3771
1.
中和価とは,酸価,強酸
価,塩基価
MOD/追加 JISは強塩基価を規定。
国内の実態を考慮し,旧JISの定
義に合わせた。
2.引用規格
引用するJISを規定
ISO 6618
ISO 7537
ISO 6619
2.
MOD/追加 JISは引用しているJISを規定。 試薬,器具,精確さに関する値の実
用的な使い方についてはJISに規
定されているものを用いた。
ISO 3771
ISO 385-1,ISO 3696
MOD/削除
ISO 3771,ISO 6353-2 MOD/削除
ISO 6618,ISO 6619
MOD/削除
ISO 7537を規定
MOD/削除
3.定義
酸価,強酸価,
ISO 6618 3.
酸価,強酸価
IDT
−
塩基価
ISO 7537
塩基価を規定
IDT
−
強塩基価を規定。
ISO 6619
ISO 3771
MOD/追加 JISは強塩基価を規定。
国内の実態を考慮し,旧JISの定
義に合わせた。
4.試験方法の種類
構成する試験方法を記載 −
−
−
MOD/追加 ISOでは規定していない。
JIS Z 8301に基づく様式
5.指示薬滴定法
ISO 6618 −
−
−
−
−
5.1試験の原理
試験の原理を規定
4.
JISに同じ
IDT
−
−
5
3
K
2
5
0
1
:2
0
0
3
54
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の項目
ごとの評価及びその内容
表示箇所:本体
表示方法:点線の下線又は実線の側線
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
5.2試薬
p-ナフトールべンゼイン
指示薬,水酸化カリウム
標準液,2-プロパノール,
トルエン他を規定。
5.
試薬及び試薬調製方法
を規定
MOD/追加 ・JISは塩酸標準溶液及び水酸
化カリウム標準溶液の濃度計
算方法を規定。
・濃度計算式を追加し,計算方
法を分かりやすいようにした。
・JISはアミド硫酸による標定
方法を規定。
・国内の実態に合わせ,旧JIS
の規定を残した。
5.3試験器
ビュレットを規定
6.
JISに同じ
MOD/追加 ビュレットの図を追加。
ビュレットを詳細に示すため,
図を追加した。
5.4試料採取方法及び
調製方法
試料の採取方法及び調製
方法を規定。
7.
JISに同じ
IDT
−
−
5.5試験の手順
試験の手順を規定
8.
9.
10.
JISに同じ
IDT
−
−
5.6計算及び結果
計算方法及び
結果の表し方を規定
11.
計算方法を規定
MOD/変更 構成内容の変更及び報告けた
の変更。
国内の実態に合わせて変更し
た。
12.
結果の表し方を規定
丸めの幅
結 果
JIS
ISO
1未満
0.01
0.1
1 以上
3けた
0.1
5.7精度
精度を規定
13.
JISに同じ
IDT
−
−
5.8試験結果の報告
報告事項を規定
14.
JISに同じ
IDT
−
−
6.セミミクロ指示薬滴
定法(酸値)
ISO 7537 −
−
−
−
−
6.1試験の原理
試験の原理を規定
4.
JISに同じ
IDT
−
−
6.2試薬
p-ナフトールべンゼイン
指示薬,水酸化カリウム
標準液,2-プロパノール,
トルエン他を規定
5.
JISに同じ
IDT
−
−
5
4
K
2
5
0
1
:2
0
0
3
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の項目
ごとの評価及びその内容
表示箇所:本体
表示方法:点線の下線又は実線の側線
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
6.3酸価試験器
滴定ビュレット,滴定用
ビーカ,かき混ぜ機,ピ
ペット,窒素流量計他を
規定
6.
JISに同じ
MOD/追加 滴定ビーカ,ピペット,溶剤用
ビュレット等の図を追加
試験器具を詳細に示すため図を
追加した。
6.4試料採取方法及び
調製方法
試料の採取方法及び調製
方法を規定
7.
JISに同じ
IDT
−
−
6.5試験の手順
試験の手順を規定
8.
JISに同じ
IDT
−
−
6.6計算及び結果
計算方法及び結果の表し
方を規定
9.
計算方法を規定
MOD/変更 構成内容の変更及び報告けた
の変更
国内の実態に合わせて変更し
た。
10.
結果の表し方を規定
丸めの幅
酸 価
JIS ISO
0.01以上0.10未満 0.01 0.01
0.10以上10未満
0.01 0.1
10以上
3けた 0.1
6.7精度
精度を規定
11.
JISに同じ
IDT
−
−
6.8試験結果の報告
報告事項を規定
12.
JISに同じ
IDT
−
−
7.電位差滴定法(酸価)
ISO 6619 −
−
−
−
−
7.1試験の原理
試験の原理を規定
3.
JISに同じ
IDT
−
−
7.2試薬
トルエン,2-プロパノー
ル,塩酸標準溶液,水酸
化カリウム標準溶液,緩
衝溶液他を規定
4.
試薬及び試薬調製方法
を規定
MOD/追加 ・JISは塩酸標準溶液及び水酸
化カリウム標準溶液の濃度計
算方法を規定。
・濃度計算式を追加し,計算方
法を分かりやすいようにした。
・JISはアミド硫酸による標定
方法を規定。
・国内の実態に合わせ,旧JIS
の規定を残した。
7.3試験器
メータ,ガラス電極,比
較電極,ビュレット,滴
定用ビーカ,かき混ぜ機
他を規定。
5.
メータ,ビュレット等を
規定
MOD/追加 ・JISはメータの種類(電位差
計,pH計)を規定。
・分かりやすいようにした。
・JISはビュレット,滴定用ビ
ーカの図を追加
・試験器具を詳細に示すため図
を追加した。
・JISは自動ビュレットを用い
てもよいとした。
・国内の実態に合わせ,自動ビュ
レットを許容した。
5
5
K
2
5
0
1
:2
0
0
3
56
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の項目
ごとの評価及びその内容
表示箇所:本体
表示方法:点線の下線又は実線の側線
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
7.4試料採取方法及び
調製方法
試料の採取方法及び調製
方法を規定
6.
JISに同じ
IDT
−
−
7.5電位差滴定装置の
調整
電極の準備,維持,検査
方法を規定。
7.
JISに同じ
MOD/追加 電極の洗浄方法を詳細に規定
した。
電極の洗浄は重要なため,規定
した。
7.6試験器の調整
試験器の調整方法を規定
8.
JISに同じ
MOD/追加 異なった電極使用上の注意点
を規定した。
電極の違いにより感度等が異な
るため,注意点を規定した。
7.7試験の手順
試験の手順を規定
9.
JISに同じ
MOD/追加 メータの読み等を詳細に規定
した。
メータの読み難しいため,詳細
に規定した。
7.8計算及び結果
計算方法及び
結果の表し方を規定
10.
計算方法を規定
MOD/追加 JIS K 0113の終点決定方法を追
加した。
参考になるJISの規定を追加し
た。
MOD/変更 構成内容の変更及び報告けた
を規定した。
国内の実態に合わせて変更し
た。
丸めの幅
測定値
JIS
ISO
1未満
0.01
規定なし
1以上
3けた 規定なし
7.9精度
精度を規定
11.
JISに同じ
IDT
−
−
7.10試験結果の報告
報告事項を規定
12.
JISに同じ
IDT
−
−
8.電位差滴定法(塩基
価・塩酸法)
対応ISO
規格なし
−
−
−
−
−
8.1試験の原理
試験の原理を規定
−
−
−
−
−
8.2試薬
トルエン,2-プロパノー
ル,塩酸標準溶液,水酸
化カリウム標準液,緩衝
溶液他を規定。
−
−
−
−
−
8.3試験器
メータ,ガラス電極,比
較電極,ビュレット,滴
定用ビーカ,かき混ぜ機
他を規定。
−
−
−
−
−
5
6
K
2
5
0
1
:2
0
0
3
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の項目
ごとの評価及びその内容
表示箇所:本体
表示方法:点線の下線又は実線の側線
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
8.4試料採取方法及び
試料調製方法
試料の採取方法及び調製
方法を規定
−
−
−
−
−
8.5電位差滴定装置の
調整
電極の準備,検査,洗浄
方法,保守を規定
−
−
−
−
−
8.6試験器の調整
試験器の調整方法を規定
−
−
−
−
−
8.7試験の手順
試験の手順を規定
−
−
−
−
−
8.8結果
計算方法及び
結果の表し方を規定
−
−
−
−
−
8.9精度
精度を規定
−
−
−
−
−
8.10試験結果の報告
報告事項を規定
−
−
−
−
−
9.電位差滴定法(塩基
価・過塩素酸法)
ISO 3771 −
−
−
−
−
9.1試験の原理
試験の原理を規定
4.
JISに同じ
IDT
−
−
9.2試薬
酢酸,無水酢酸,過塩素
酸標準液,過塩素酸標準
液,クロロべンゼン,酢
酸ナトリウム標準液他を
規定
5.
JISに同じ
MOD/追加 JISはクロロべンゼンの代わり
にトルエンの使用を許容
毒性のあるクロロべンゼンの代
替としてトルエンの使用を許容
した。
9.3試験器
メータ,ガラス電極,比
較電極,ビュレット,滴
定用ビーカー,かき混ぜ
機他を規定
6.
JISに同じ
MOD/追加 試験器滴定部の一例の図を追
加
試験器滴定部を詳細に示すため
図を追加した。
9.4試料採取方法及び
調製方法
試料の採取方法及び調製
方法を規定
7.
JISに同じ
IDT
−
−
9.5電位差滴定装置の
調整
電極の準備,検査,洗浄
方法,保守を規定
8.
JISに同じ
IDT
−
−
9.6試験の手順
試験の手順を規定
9.
JISに同じ
IDT
−
−
9.7計算及び結果
計算方法及び
結果の表し方を規定
10.
計算方法を規定
MOD/変更 構成内容の変更及び報告けた
の変更
国内の実態に合わせて変更した。
12.
結果の表し方を規定
5
7
K
2
5
0
1
:2
0
0
3
58
K 2501:2003
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)国際規
格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の項目ご
との評価及びその内容
表示箇所:本体
表示方法:点線の下線又は実線の側線
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごとの
評価
技術的差異の内容
丸めの幅
塩基価
JIS ISO
0.01以上0.10未満 0.01 0.1
0.10以上10未満
0.01 0.1
10以上100未満
3けた 0.1
100以上
3けた整数
9.8逆滴定法
逆滴定の手順,計算方法
及び結果の表し方を規
定
11.
逆滴定の手順及び計算
方法を規定
MOD/変更 構成内容の変更及び報告けた
の変更
国内の実態に合わせて変更し
た。
12.
結果の表し方を規定
丸めの幅
塩基価
JIS ISO
0.01以上0.10未満 0.01 0.1
0.10以上10未満
0.01 0.1
10以上100未満
3けた 0.1
100以上
3けた整数
9.9精度
精度を規定
13.
JISに同じ
IDT
−
−
9.10試験結果の報告
報告事項を規定
14.
JISに同じ
IDT
−
−
附属書1(参考)
−
参考のため評
価対象外
ISOに規定されていない。
−
指示薬光度滴定法
JISと国際規格との対応の程度の全体評価:ISO 6618 : 1997 ; MOD,ISO 7537 : 1997 ; MOD,ISO 6619 : 1988 ; MOD,ISO 3771 : 1994 ; MOD
備考1. 項目ごとの評価欄の記号の意味は,次のとおりである。
−IDT…………… 技術的差異がない。
−MOD/削除……国際規格の規定項目又は規定内容を削除している。
−MOD/追加……国際規格にない規定項目又は規定内容を追加している。
−MOD/変更……国際規格の規定内容を変更している。
2. JISと国際規格との対応の程度の全体評価欄の記号の意味は,次のとおりである。
−MOD……………国際規格を修正している。
5
8
K
2
5
0
1
:2
0
0
3