K 2247-2:2009
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 仕様······························································································································· 1
4 試料採取方法 ··················································································································· 2
5 精度及び係争 ··················································································································· 2
附属書A(規定)試料採取方法 ······························································································· 3
附属書B(規定)総窒素量による尿素濃度の定量 ········································································ 5
附属書C(規定)屈折率及び屈折率による尿素濃度の定量 ··························································· 8
附属書D(規定)アルカリ度の定量 ························································································ 11
附属書E(規定)ビウレット濃度の定量··················································································· 13
附属書F(規定)アルデヒド濃度の定量 ··················································································· 17
附属書G(規定)重量法による不溶解分濃度の定量 ··································································· 20
附属書H(規定)吸光光度法によるりん酸濃度の定量 ································································ 22
附属書I(規定)ICP-OES法による微量成分(Al,Ca,Cr,Cu,Fe,K,Mg,Na,Ni及びZn)
濃度の定量 ··················································································································· 26
附属書J(規定)赤外線吸収スペクトルによる定性 ···································································· 31
附属書K(参考)各試験方法の精度 ························································································ 33
附属書JA(規定)ICP-MS法による微量成分(Al,Ca,Cr,Cu,Fe,K,Mg,Na,Ni及びZn)
濃度の定量 ··················································································································· 35
附属書JB(規定)炎光光度法による微量成分(K及びNa)濃度の定量 ········································· 38
附属書JC(参考)JISと対応する国際規格との対比表 ································································ 42
K 2247-2:2009
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第12条第1項の規定に基づき,社団法人自動車技術会(JSAE)から,工業標準
原案を具して日本工業規格を制定すべきとの申出があり,日本工業標準調査会の審議を経て,経済産業大
臣が制定した日本工業規格である。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願,実用新案権又は出願公開後の実用新案登録出願に
抵触する可能性があることに注意を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許
権,出願公開後の特許出願,実用新案権及び出願公開後の実用新案登録出願にかかわる確認について,責
任はもたない。
JIS K 2247の規格群には,次に示す部編成がある。
JIS K 2247-1 第1部:品質要件
JIS K 2247-2 第2部:試験方法
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格
JIS
K 2247-2:2009
ディーゼル機関−NOx還元剤AUS 32−
第2部:試験方法
Diesel engines-NOx reduction agent AUS 32-Part 2: Test methods
序文
この規格は,2006年に第1版として発行されたISO 22241-2を基に作成した日本工業規格であるが,我
が国の実情に合わせるため技術的内容を変更して作成した日本工業規格である。
なお,この規格で側線又は点線の下線を施してある箇所は,対応国際規格を変更している事項である。
変更の一覧表にその説明を付けて,附属書JCに示す。
1
適用範囲
この規格は,JIS K 2247-1に規定したNOx還元剤AUS 32(AUSは,aqueous urea solutionの略語で,尿
素水溶液を表す。)の品質要件への適合性を判定するために必要な試験方法を規定する。
注記1 JIS K 2247(規格群)では,“NOx還元剤AUS 32”を“AUS 32”と略称する。
注記2 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 22241-2:2006,Diesel engines−NOx reduction agent AUS 32−Part 2: Test methods (MOD)
なお,対応の程度を表す記号“MOD”は,ISO/IEC Guide 21-1に基づき,“修正している”
ことを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0133 高周波プラズマ質量分析通則
JIS K 2247-1 ディーゼル機関−NOx還元剤AUS 32−第1部:品質要件
注記 対応国際規格:ISO 22241-1,Diesel engines−NOx reduction agent AUS 32−Part 1: Quality
requirements (MOD)
ISO 3675,Crude petroleum and liquid petroleum products−Laboratory determination of density−Hydrometer
method
ISO 3696,Water for analytical laboratory use−Specification and test methods
ISO 4259,Petroleum products−Determination and application of precision data in relation to methods of test
ISO 12185,Crude petroleum and petroleum products−Determination of density−Oscillating U-tube method
3
仕様
JIS K 2247-1の表1に規定した品質要件への適合性は,この規格の附属書B〜附属書J,附属書JA及び
2
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JBに規定する試験方法によって判定しなければならない。
密度の測定は,ISO 3675又はISO 12185による。
注記 この規格では,物質の質量割合及び体積割合を示すために,それぞれ“質量分率%”及び“体
積分率%”を用いる。
4
試料採取方法
試料採取は,附属書Aによって行う。
5
精度及び係争
この規格で規定しているすべての試験方法は,ISO 4259による精度表示を含んでいる。係争に当たって
は,その解決のために,ISO 4259に規定されている手順に従い,また当該試験方法の精度に基づく試験結
果の解釈を行わなければならない。
この規格の使用者の便宜のために,すべての試験方法に対する精度情報を,附属書Kにまとめて示して
ある。
注記 係争とは,SCRコンバータシステム(以下,SCRコンバータという。)の故障がAUS 32の品質
に起因するものか否かについての訴訟をいう。
3
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(規定)
試料採取方法
序文
この附属書は,試料採取方法について規定する。
A.1 一般
この附属書で規定する試料採取方法は,製造業者から車両のAUS 32タンクまでの流通経路の間で実施
されるすべての試料採取方法に対して有効である。
A.2 原則
JIS K 2247-1に規定しているAUS 32の品質要件に対する上下限値は,試料が分析前に汚染されていな
い場合だけに得られる代表的な分析結果である。
したがって,試料採取に当たっては適切な瓶を用いて,特に微量成分について汚染がないように,また
藻類又はバクテリアの増殖のリスクを最小限度に抑制するようにしなければならない。
注記 この附属書で規定する試料採取方法は,ISO 5667-3に準拠している。
A.3 可能性のある汚染物質
試料採取過程で入り込む異物が,試料を汚染する可能性がある。現実的な条件下では,次の汚染源が大
きな影響を及ぼす可能性がある。
− 試料採取瓶の製造時に用いた加工助剤の残留物
− 空のままで保管している間に瓶の中に付着した汚染物
− 試料採取中の空気中の汚染物:ちり又は周囲からの異物
− 試料採取装置及び試料採取瓶を洗浄したときに用いられた洗剤の残留物
− 燃料
A.4 装置
A.4.1 試料採取瓶 1 000 mLの広口瓶を用いる。試料採取瓶に適した材質としては,高密度ポリエチレ
ン,高密度ポリプロピレン,ポリテトラフルオロエチレン,ポリフッ化ビニリデン及びテトラフルオロエ
チレン−パーフルオロアルキルビニルエーテル共重合体 (PFA) がある。係争時には,PFA瓶を用いるのが
望ましい。
瓶は,最初AUS 32の試料採取に用いる前に洗浄し,最後に脱イオン水ですすいだ後,AUS 32ですすが
なければならない。
A.4.2 ラベル それぞれの瓶には,約10 cm×5 cmの大きさのラベルを付けなければならない。ラベル及
びその上に書かれる文字は,水及びAUS 32によって,ラベルのはがれ及び文字が消えたり,にじんでは
ならない。
4
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.5 試料採取方法
ふた(蓋)をしている広口瓶を開け,ふたは,内側を下向きにして清浄な表面の上に置く。試料採取管
を洗浄した後,瓶に容器からのAUS 32を満たす。最初に満たした分は捨てて,その後直ちにAUS 32を採
取してしっかりとふたをする。次に,瓶にラベルを付ける(A.4.2参照)。
試料を満たしている間は,瓶の中にちり及び液体の汚染物質が入らないように最大限の注意を払う。
満たした瓶は,可及的速やかに分析室に届けることが望ましい。輸送及び貯蔵期間中は,試料をできる
だけ低い温度(0 ℃〜15 ℃が望ましい。)に維持し,藻類が生えないように日光を遮っておくことが望ま
しい。
試料中のアンモニア濃度が変化する可能性を考慮すると,試料を3週間以内に分析することが望ましい。
A.6 試料の量
試料の最少採取量は,分析の種類による。可能であれば,十分な量(推奨1 000 mL)の試料を確保し,
少なくともAUS 32の品質要件を完全に立証するのに必要な量の2倍を確保する。係争時には,ISO 4259
に従って十分な数の試料を採取しなければならない。
A.7 ラベリング
ラベルは,次の情報を含んでいなければならない。
− 製品名
− 試料を所有する会社名1)
− 試料採取場所の住所1)
− 試料の製造業者名1)
− バッチ又はロット番号
− 試料を採取した容器1)
− 試料を採取した容器の部位(採取点)1)
− 試料採取日及び時刻1)
− 試料の出荷日1)
− 試料採取者の氏名及び署名1)
注1) 係争時だけに必す(須)の項目
5
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(規定)
総窒素量による尿素濃度の定量
序文
この附属書は,総窒素量による尿素濃度の定量方法について規定する。
B.1
一般
この試験方法は,質量分率30 %〜35 %の範囲の尿素濃度の定量に適用することができる。
B.2
原理
試料は,酸素気流中で高温で燃やす。生成した窒素酸化物を窒素に還元し,干渉する他の燃料生成物を
除去した後,窒素を熱伝導度検出器によって測定する。尿素濃度は,定量された総窒素量からビウレット
の窒素量を差し引いて算出する。
B.3
装置
B.3.1 燃焼法に基づく自動窒素分析計
B.3.2 化学天びん 天びんの精度は,用いる分析計の性能及び計量対象物の質量による。分解能は,読み
値の0.1 %又はそれより良いことが望ましい。
B.3.3 補助器具 例えば,
− ピンセット 先端が丸いもの
− マイクロスパチュラ 一方の先端が平らなもの
− ピペット
ピペットは,質量測定に用いるので,校正の必要はない。しかしながら,良好な滴下サイズ(小さな液
滴)の得られることが重要である。固定容積のピペット又は10 μL〜1 000 μLの範囲で容積が調節可能な
ピペット,又は先端の細い使い捨てパスツールピペットを用いてもよい。
B.3.4 一般的な理化学用ガラス器具
B.4
薬品類
B.4.1 脱イオン水 導電率0.1 mS/m未満,ISO 3696の2級のもの。
B.4.2 窒素分析計に適した補助燃焼剤及びその他の装置 次の物質は,単なる例である。利用するシステ
ムに応じて,他の又は類似の物質を用いてもよい。
− すず(錫)製カプセル又は類似の試料容器
− サッカロース,セルロースのような窒素を含まない補助燃焼剤
− 酸化マグネシウムのような窒素を含まない液体吸着剤
B.4.3 窒素定量用の標準物質 できれば窒素濃度が保証されているもの。
例 適切な標準物質としては,次のようなものがある:エチレンジアミン4酢酸(EDTA),ニコチン
酸アミド。
適切な純度の低ビウレット尿素(例えば,結晶性超高純度又は分析級)又は装置製造業者が推奨・提供
6
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
するような,その他の標準物質を用いてもよい。濃度が保証された標準物質が望ましい。
注記 液体標準物質(例えば,尿素水溶液)は,校正目的に対しては適切ではない。
B.4.4 酸素 純度99.995 %以上のもの。
B.4.5 超高純度ガス 窒素分析計に必要な他の超高純度ガス。例えば,ヘリウムでは純度99.996 %以上
のもの。
B.4.6 試薬又は補助剤 装置に応じて必要となる他の試薬又は補助剤。
B.5
手順
B.5.1 一般
試料は,完全に溶解させ,尿素の結晶がないことが望ましい。必要ならば,その後の処置に先立って,
最高40 ℃まで加熱してもよい。
注記 市場には,各種の装置が存在している。その結果としての種々の装置及び操作方法に関する説
明は,この規格の目的ではない。むしろ,操作方法は,各装置の操作マニュアルによるべきも
のである。
B.5.2 検量線の作成
分析計の形式に応じて,かつ,各操作マニュアルに従って(例えば,燃焼チューブ,試薬又は類似品の
交換後),B.5.4に規定している測定を行うことによって,校正を行う。検量線を得るために,それぞれの
装置に見合った適切な点数で,適切な量の標準物質を繰返し測定する。
B.5.3 作動状況及び検量線を確認するための装置の点検
装置の良好な作動状況及び検量線を確認するために,適切な標準物質(1種類)を用いる。濃度が分か
っている尿素標準溶液を用いるのが望ましい。
点検頻度は,分析計の機能による。
B.5.4 試料の測定
試料の測定に用いる窒素分析計で規定する適切な容器(例えば,すず製カプセル)に入れて,試料の質
量をひょう量する。その量は,窒素の絶対量が,検量線の中央領域に位置するような量であることが望ま
しい。
約3倍の燃焼剤(例えば,窒素を含まないセルロース)及び必要に応じて追加する結合剤(例えば,酸
化マグネシウム)を用いる。
液体供給システムを用いるときは,100 μL以上の体積を用いることが望ましい。試料の質量は,ISO 12185
に従って計算された密度を用いて計算する。
分析計(又は制御コンピュータ)に,装置に応じた必要データ(ひょう量した試料量,試料識別番号)
を入力する。分析計にひょう量した試料を導入し,燃焼を開始する。
少なくとも3回の定量を実施する。
B.6
結果
B.6.1 計算
検量線,ベースラインのドリフト又は試料を計算する前に,ブランク試料を用いてブランクの値を求め,
この値を用いてそれぞれの分析結果を補正する。
装置付属のプログラムを用いて,検量線又は試料に対するドリフト補正量を計算する。
試料に対する平均値を計算する。各計算値の間に大きなばらつき(相対標準偏差RSD>1.0 %)が生じ
7
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
た場合には,当該試料の測定を繰り返す。その後,この試料に対する平均値を,すべての測定値から計算
する。
少なくとも3回の窒素量の測定値の平均値から尿素濃度を決定する。
wU=2.143 8×(wN−F×wBi)
ここに,
wU: 尿素濃度(質量分率%)
wN: 窒素濃度の平均値(質量分率%)(0.01 %単位で丸めた値)
wBi: ビウレット濃度の平均値 (%),附属書Eで求めたもの
F: ビウレット濃度を窒素に換算するための係数 (0.407 6)
B.6.2 結果の表示
結果は,少なくとも3回の定量(窒素量の定量)から得られた算術平均値である。尿素濃度の計算結果
は,0.1 %単位に丸める。
B.7
試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(B.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
8
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C
(規定)
屈折率及び屈折率による尿素濃度の定量
序文
この附属書は,屈折率の測定方法及び屈折率による尿素濃度の定量方法について規定する。
C.1 一般
この試験方法は,1.33〜1.39の範囲の屈折率をもつ液体に対して,20 ℃〜30 ℃の温度範囲において適
用することができる。
屈折率の測定結果に基づいたこの方法を尿素濃度の定量に用いる場合には,質量分率の範囲を,30 %〜
35 %としなければならない。
C.2 原理
測定は,温度が決まれば,屈折率が水溶液中の尿素濃度によって決まるという原理に基づいている。
この濃度は,検量線を用いて求める。
注記 この附属書に規定する方法は,ISO 5661に準拠している。
C.3 装置
C.3.1 屈折率計 測定範囲1.330 0〜1.390 0,分解能0.000 1
C.3.2 化学天びん 分解能0.1 mg又はそれより良いもの。
C.3.3 温調器 温度制御精度0.02 ℃
C.3.4 恒温槽
C.3.5 トールビーカ 150 mL
C.3.6 一般的な理化学用ガラス器具
C.4 薬品類
C.4.1 脱イオン水 ISO 3696の3級による導電率0.5 mS/m未満のもの。
C.4.2 尿素 結晶性で,ビウレット含有率が質量分率0.1 %未満のもの。検量線を作成するための尿素を
ひょう量する前に,尿素を105 ℃において2時間乾燥させなければならない。
C.4.3 質量分率32.5 %の尿素標準溶液 標準溶液は,尿素及び脱イオン水を正確にひょう量して作らな
ければならない。目的とする濃度及び許容ばらつきは,10回の測定によって決めなければならない。
この標準溶液は,気密性を確保した上で冷蔵庫で保管し,4週間以内に用いることが望ましい。
C.5 手順
C.5.1 一般
試料は,完全に溶解させ,尿素の結晶がないことが望ましい。必要ならば,その後の処置に先立って,
最高40 ℃まで加熱してもよい。
注記 市場には,各種の装置が存在している。その結果としての種々の装置及び操作方法に関する説
9
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
明は,この規格の目的ではない。むしろ,操作方法は,各装置の操作マニュアルによるべきも
のである。
C.5.2 検量線の作成及び評価係数の計算
ガラスビーカ中の尿素をひょう量し,対応する量の脱イオン水を加えて,次の尿素水溶液を用意する。
30.0 %,31.5 %,32.5 %,33.5 %及び35.0 % (単位は,質量分率)
これらの水溶液の屈折率は,20 ℃±0.02 ℃において測定しなければならない。
作成した線図は,屈折率及び濃度の関係が,直線を示していなければならない。評価係数は,尿素濃度
と屈折率とから,次の式を用いて計算する。
(
)
∑
∑
=
=
−
=
5
1
W
,
U
,
U
5
1
i
i
i
i
n
n
w
F
ここに,
F: 評価係数 (%)
wU,i: i番目の試料の尿素濃度(質量分率%)
nU,i: i番目の試料の屈折率
nW: 水の屈折率で,分解能小数点以下4けたまでの屈
折率計で測定したときの値は,1.333 0。
C.5.3 装置の機能及び検量線の点検
装置の機能は,水又は屈折率標準液を用いて,毎週確認しなければならない。基準値に対して0.000 2
より大きなずれが生じている場合には,製造業者の説明書に基づいて装置を調整する。その後も基準値が
得られないようであれば,それ以降の測定にはその装置を用いることを止めて,製造業者のサービスを要
請しなければならない。
屈折率計の出口側にある温度計の温度を読みながら,温調器の温度設定を所定の値に調節する。所定の
温度が±0.02 ℃の範囲内で維持されるように,水の流量を調節する。
さらに,質量分率32.5 %の尿素標準溶液を用いて,検量線を毎週確認しなければならない。その過程に
おいて,屈折率を測定し,C.6で求めた係数を用いて濃度を計算しなければならない。計算した濃度値が,
所定の値に対して0.1 %(質量分率)より大きい幅でずれている場合には,新しい標準溶液を用いなけれ
ばならない。そのずれが解消しないようであれば,新たに検量線を作成しなければならない。
C.5.4 試料の測定
前処理することなしに,試料そのものを20 ℃±0.02 ℃において測定する。
同一試料について,2回の尿素濃度測定を行う。その差が,0.000 1より大きい場合には,測定を繰り返
す。
10
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
C.6 結果
C.6.1 計算
尿素濃度は,次の式を用いて計算する。
wU=(nP−nW)×F−wBi
ここに,
wU: 尿素濃度(質量分率%)
nP: 試料の屈折率(小数点以下4けた)
nW: 水の屈折率(小数点以下4けた)
F: 評価係数 (%)
wBi: 試料中のビウレット濃度(質量分率%)
(附属書Eによって求めたもの。ビウレットは,尿素と
同じ屈折率をもつ。)
C.6.2 結果の表示
結果は,2回の測定値の算術平均値とする。屈折率の結果は,小数点以下4けたに丸める。尿素濃度の
計算結果は,質量分率0.1 %単位で丸める。
C.7 試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(C.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
11
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書D
(規定)
アルカリ度の定量
序文
この附属書は,アルカリ度の定量方法について規定する。
D.1 一般
この試験方法は,アンモニアとして計算されるAUS 32のアルカリ度を,質量分率0.1 %〜0.5 %の範囲
において定量するための方法である。
D.2 原理
測定は,塩酸標準溶液を用いて,試料の遊離アンモニアをpH 5.7の終点まで電位差滴定するという原理
に基づいている。
D.3 装置
D.3.1 化学天びん 分解能0.1 mg又はそれより良いもの。
D.3.2 自動ビュレット
D.3.3 電位差計 分解能0.01 pHで測定可能で,複合ガラス電極をもつもの。
D.3.4 マグネチックスターラ
D.3.5 トールビーカ 150 mL
D.3.6 メスシリンダ 100 mL
D.4 薬品類
D.4.1 一般 分析中は,素性のはっきりしている分析等級の試薬及びISO 3696の3級に相当する導電率
0.5 mS/m未満の蒸留水又は脱イオン水だけを用いる。
D.4.2 塩酸 0.01 mol/Lの標準溶液。
D.4.3 緩衝溶液 アルカリ度の定量には,次の標準緩衝溶液を用いる。
− 標準緩衝溶液,pH 4.008
− 標準緩衝溶液,pH 9.184
− 標準緩衝溶液,pH 8.00
注記 これらの溶液は,市販されている。
D.5 手順
D.5.1 干渉
採取したAUS 32の試料は,アンモニアの生成を避けるために25 ℃以下の温度で保管し,輸送する。
容器は,きつくふたをする。分析に当たっては,アンモニアの蒸発を避けるために,中断によって分析
時間が長くならないようにする。
12
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D.5.2 電位差計システムの点検
電位差計システムは,pH 4.008及びpH 9.180の標準緩衝溶液を用いて,正しく機能していることを確認
しなければならない。
電位差計システムの日常点検には,pH 8.00の標準緩衝溶液を用いる。
D.5.3 予備試験
均質な試料約1 gを0.05 gまでひょう量し(試料質量ms),それを約100 mLの蒸留水又は脱イオン水が
入った150 mLのビーカに入れる。
塩酸標準溶液(0.01 mol/L)で,かくはんしながら終点のpH 5.7まで滴定する。
アンモニア濃度を計算する。
求めたアルカリ度に応じて,次に示す試料をひょう量して,定量に用いる。
− 予備試験で求めたアルカリ度(質量分率%):0.02 0.05 0.1 0.2〜0.5
− 定量のための試料の質量 (g):10 5 2 1
D.5.4 試料の測定
予備試験で求めた均質な試料の質量を0.05 gまでひょう量し(試料質量ms),それを約100 mLの蒸留水
又は脱イオン水が入った150 mLのビーカに入れる。
塩酸標準溶液(0.01 mol/L)で,かくはんしながら,まずは普通の速度でpH 7.5まで滴定し,その後ゆっく
りと終点のpH 5.7まで滴定する。
測定は,2回行う。
D.6 結果
D.6.1 計算
アンモニアの質量分率で表されるアルカリ度は,次の式で得られる。
w(NH3)=( V×0.017)/ ms
ここに,
w(NH3): アンモニアとして計算されたアルカリ度(質量分率%)
V: 滴定に用いた塩酸標準溶液の体積 (mL)
ms: 試料の質量 (g)
D.6.2 結果の表示
2回の測定値の平均値を計算する。その結果は,質量分率0.01 %単位に丸める。
D.7 試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(D.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
13
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書E
(規定)
ビウレット濃度の定量
序文
この附属書は,ビウレット濃度の定量方法について規定する。
E.1
一般
この試験方法は,AUS 32中のビウレット濃度を,吸光光度法によって,質量分率0.1 %〜0.5 %の範囲
において定量するための方法である。この方法は,質量分率1.5 %の濃度まで適用可能であるが,その場
合の精度は確認されていない。
E.2
原理
ビウレットは,酒石酸カリウムナトリウムを含むアルカリ性溶液の中で二価銅と反応して,550 nmにお
いて最大の吸収を示す紫色の錯体を生成する。この有色錯体の550 nmにおける吸光度を分光光度法で読
み取り,標準ビウレット溶液から作成された検量線と照合することによって,ビウレット濃度を求める。
E.3
装置
E.3.1 化学天びん 分解能0.001 gのもの。
E.3.2 減圧ろ過装置 孔径0.45 μmのフィルタに適するもの。
E.3.3 分光光度計 光路長50 mmのセル付き,550 nmで用いる。
E.3.4 メスフラスコ 容量1 000 mL,250 mL,100 mL及び50 mLのもの。
E.3.5 ホールピペット
E.3.6 ロータリエバポレータ
E.3.7 恒温水槽 30 ℃±1 ℃の温度を維持できるもの。
E.4
薬品類
E.4.1 一般 すべての試験において,分析等級の薬品類を用いる。水は,使用前に脱イオン化し,沸騰さ
せて二酸化炭素を除去しなければならない。
E.4.2 飽和炭酸カリウム溶液
E.4.3 硫酸銅溶液 15 gの硫酸銅五水和物(CuSO4・5H2O)を二酸化炭素を除去した水に溶解し,1 000 mL
に薄める。
E.4.4 アルカリ性酒石酸カリウムナトリウム溶液 40 gの水酸化ナトリウムを,1 000 mLのメスフラス
コに入れた500 mLの水に溶解する。冷却後に,50 gの酒石酸カリウムナトリウム四水和物(KNaC4H4O6・
4H2O)を加え,メスフラスコの標線まで水を入れる。使用前に,このフラスコを1日放置する。
E.4.5 0.8 mg/mLのビウレット標準溶液 800 mgの純粋ビウレットを,二酸化炭素を除去した水に溶解
し,1 000 mLに薄める。ビウレットは,使用前に3時間,105 ℃で乾燥させる。
ビウレットは,次の方法で精製してもよい。
14
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 50 gのビウレットを,質量分率25 %のアンモニア溶液500 mLに加え,15分間かくはんする。
− ろ過し,アンモニアを含まない水で洗浄した後,ビウレットを乾燥させる。
− エタノールに溶かし(エタノール1 L/ビウレット10 gの割合),ろ過し,ゆっくり加熱しながら1/4
の体積まで濃縮する。
− 5 ℃まで冷却し,ろ過する。
− 真空乾燥器内で,80 ℃で乾燥させる。
− E.5.5に従って,吸光光度法による純度確認を行う。
エタノールから再結晶化させるステップは,純度の向上が認められなくなるまで繰り返さなければなら
ない。
E.5
手順
E.5.1 干渉
分光光度計による測定は,透明な溶液に対してだけ適用可能である。試料を0.45 μmのフィルタでろ過
し,透明な溶液にする。
アンモニアは二価銅と反応し,550 nmの光を吸収する有色錯体を生成する。この方法は,試料中のアン
モニア濃度が,500 mg/kg未満の場合にだけ適用可能である。
500 mg/kg以上のアンモニアを除去するためには,50.0 gの試料をロータリエバポレータの1 000 mLの
フラスコに入れ,15 mLの飽和炭酸カリウム溶液を加えて,回転速度60 min−1,2 kPa〜3 kPaの減圧下で,
40 ℃で1時間蒸発させ,最終的な体積を約20 mLにする。この溶液を,250 mLのメスフラスコに移す。
E.5.2 検量線の作成
50 mLのメスフラスコを6個用意し,2 mL,5 mL,10 mL,15 mL,20 mL及び25 mLのビウレット標準
溶液を入れ,それぞれのメスフラスコが約25 mLになるまで水を加える。その後,かくはんしながら10 mL
のアルカリ性酒石酸カリウムナトリウム溶液及び10 mLの硫酸銅溶液を加える。それぞれのメスフラスコ
を30 ℃±1 ℃に制御した恒温水槽に浸し,その状態で15分間放置する。
この測定と並行して,同じ手順に従い,また,すべて同量の試薬を用いて(E.5.5参照),ブランク試験
を実施する。
室温まで冷却した後,メスフラスコの標線まで水を加え,十分に混ぜ合わせる。50 mmセルを用いて波
長約550 nmで分光光度計を用いた吸光度測定を行う。参照溶液は,水を使う。
ブランク試験で得られた吸光度をこの測定値から差し引き,検量線を作成する。対象濃度範囲において,
この検量線は,厳密な直線でなければならない。
E.5.3 校正係数の計算
次の式によって,校正係数を計算する。
(
)
(
)
∑
∑
∑
=
=
=
6
1
2
1,
6
1
2
1,
6
1
Bi,
61.6
i
i
i
i
i
i
E
E
E
E
m
F
−
=
−
=
ここに,
F: 校正係数 (mg)
mBi,i: i番目の試料のビウレット質量 (mg)
E1,i: i番目の試料の吸光度
E2: ブランク試験の吸光度
15
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
検量線の作成及び校正係数の計算は毎年行い,その結果を文書にして残しておかなければならない。
E.5.4 デイファクタの計算
デイファクタは,週単位で決めなければならない。
ビウレット標準溶液10 mL(8 mgビウレット)について,E.5.5で規定する測定を実施する。
計算は,次による。
(
)
2
1
D
8
E
E
F
−
=
ここに,
FD: デイファクタ(mg)
E1: 標準溶液の吸光度(2回の測定の平均値)
E2: ブランク試験の吸光度
デイファクタの変動は,校正係数に対して,±5 %以内に収まっていなければならない。試料の測定に
対しては,デイファクタを用いる。
E.5.5 試料の測定
250 mLのメスフラスコに入れた100 gの試料を,0.01 g単位でひょう量する。メスフラスコの標線まで
水を入れて十分に混ぜ合わせる。
その薄めた試料10 mLを,50 mLのメスフラスコに入れ,約25 mLになるまで水を加える。その後,か
き混ぜながら10 mLのアルカリ性酒石酸カリウムナトリウム溶液及び10 mLの硫酸銅溶液を加える。この
メスフラスコを30 ℃±1 ℃に制御した恒温水槽に浸し,約15分間放置する。
試料の測定と並行して,同じ手順に従い,また,すべて同量の試薬を用いて,ブランク試験を実施する。
これを試薬ブランクとする。
室温まで冷却した後,メスフラスコの標線まで水を加え,十分に混ぜ合わせる。50 mmセルを用いて波
長約550 nmで,分光光度計を用いた吸光度測定を行う。参照溶液は,水を使う。
試料自体の吸収を測定するため,薄めた試料10 mLを50 mLのメスフラスコに入れ,標線まで水を加え,
同じ条件でその吸光度を測定する。これを試料ブランクとする。
測定は,2回実施する。
E.6
結果
E.6.1 計算
質量分率%で表されるビウレット濃度は,次の式によって与えられる。
(
)
100
000
1
10
250
S
D
B
S
Bi
×
×
×
×
×
−
=
m
F
E
E
w
ここに,
wBi: ビウレット濃度(質量分率%)
ES: 試料の吸光度
EB: ブランク試験の吸光度(試薬ブランク+試料ブランク)
mS: 薄めた試料を準備するときに用いた試料の質量 (g)
FD: デイファクタ (mg)
E.6.2 結果の表示
結果は,質量分率0.01 %単位で表示する。
16
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
E.7
試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(E.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
17
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書F
(規定)
アルデヒド濃度の定量
序文
この附属書は,アルデヒド濃度の定量方法について規定する。
F.1
一般
この試験方法は,0.5 mg/kg〜10 mg/kgのアルデヒド濃度をもつAUS 32中の,ホルムアルデヒドとして
計算される遊離及び結合アルデヒドの濃度を定量する方法である。
F.2
原理
ホルムアルデヒドは,濃硫酸溶液の中でクロモトロプ酸と反応して,565 nmの波長で最大の吸収を示す
紫色の錯体を生成する。この有色錯体の565 nmにおける吸光度を分光光度法で読み取り,ホルムアルデ
ヒド標準溶液から作成された検量線と照合することによって,アルデヒド濃度を求める。
F.3
装置
F.3.1
化学天びん 分解能0.001 gのもの。
F.3.2
分光光度計 光路長10 mmのセル付き,565 nmで用いる。
F.3.3
メスフラスコ
F.3.4
ホールピペット
F.4
薬品類
F.4.1
一般 すべての試験において,分析等級の薬品類を用いる。
F.4.2
硫酸 質量分率96 %のもの。
F.4.3
クロモトロプ酸(4,5-ジヒドロキシナフタレン‐2,7-ジスルホン酸ナトリウム塩,又は4,5-ジヒドロ
キシナフタレン‐2,7-ジスルホン酸二ナトリウム二水和物) 質量分率15 %の硫酸中に,質量分率3 %
のもの。
この溶液を作るには,41 mLの硫酸を410 mLの水に冷却しながら加え,その後に15 gのクロモトロプ
酸を加えてよくかき混ぜる。
注記 褐色のガラス瓶に保存しておけば,この溶液は,少なくとも3か月は使用可能である。
F.4.4
ホルムアルデヒド標準溶液
− 質量分率37 %の濃度をもつ6.5 g〜7 gのホルムアルデヒド溶液を500 mLのメスフラスコに入れ,標
線まで水を入れてよくかき混ぜる。
− 例えば,ISO 9020:1994の方法によって,溶液中のホルムアルデヒド濃度を定量する。
− この溶液を1 000倍に薄める。メスフラスコにホルムアルデヒドの正確な濃度を書き込む(前段階で
定量したホルムアルデヒド濃度を,1 000で除したもの。)。
18
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
F.5
手順
F.5.1
検量線の作成
50 mLのメスフラスコを6個用意し,0.2 mL,0.5 mL,1 mL,2 mL,5 mL及び10 mLのホルムアルデヒ
ド標準溶液を入れ,それぞれのメスフラスコが約10 mLになるまで水を加える。その後,かくはんしなが
ら1 mLのクロモトロプ酸溶液を加え,その後5分間で20 mLの硫酸を徐々に加える。硫酸を加えている
間の温度上昇は,反応を完全に行うために100 ℃より高いレベルになっていなければならない。それぞれ
のメスフラスコを室温で15分間放置する(追加の冷却は行わない。)。
この測定と並行して,同じ手順に従い,また,すべて同量の試薬を用いて(F.5.4参照),ブランク試験
を実施する。
室温まで冷却した後,メスフラスコの標線まで水を加え,十分に混ぜ合わせる。10 mmセルを用いて波
長約565 nmで分光光度計を用いた吸光度測定を行う。参照溶液は,水を使う。
ブランク試験で得られた吸光度をこの測定値から差し引き,検量線を作成する。対象濃度範囲において,
この検量線は,厳密な直線でなければならない。
F.5.2
校正係数の計算
次の式によって,校正係数を計算する。
(
)
∑
∑
=
=
−
=
6
1
2
,1
6
1
HCHO,
i
i
i
i
E
E
m
F
ここに,
F: 校正係数 (μg)
mHCHO,i: i番目の試料のホルムアルデヒド質量 (μg)
E1,i: i番目の試料の吸光度
E2: ブランク試験の吸光度
検量線の作成及び校正係数の計算は毎年行い,その結果を文書にして残しておかなければならない。
F.5.3
検量線の点検
検量線の点検は,3か月ごとに次のように行わなければならない。
50 mLのメスフラスコを3個用意し,それぞれに2 mLのホルムアルデヒド標準溶液を入れ,総量が約
10 mLになるまで水を加える。F.5.4の手順に従って測定し,F.6の計算式を用いてアルデヒド濃度を計算
する。
その結果を標準溶液の濃度と比較する。その差が,2 %以下であれば,その検量線を用いてもよい。差
が2 %を超えていれば,確認作業を繰り返す。差が再度2 %より大きい場合には,新しい検量線を作成す
る。
F.5.4
試料の測定
50 mLのメスフラスコに入れた5 g〜10 gの試料を,0.01 g単位でひょう量し,総量が約10 mLになるま
で水で薄める。かき混ぜながら1 mLのクロモトロプ酸溶液を加え,更に5分間かけて硫酸20 mLを徐々
に加える。硫酸を加えている間の温度上昇は,反応を完全に行うために,100 ℃より高いレベルになって
いなければならない。それぞれのメスフラスコを室温で15分間放置する(追加の冷却は行わない。)。
試料の測定と並行して,同じ手順に従い,また,すべて同量の試薬を用いて,ブランク試験を実施する。
これを試薬ブランクとする。
19
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
室温まで冷却した後,メスフラスコの標線まで水を加え,十分に混ぜ合わせる。10 mmセルを用いて波
長約565 nmで分光光度計を用いた吸光度測定を行う。参照溶液は,水を使う。
試料自体の吸収を測定するため,試料の測定に用いた質量と同量の試料を50 mLのメスフラスコに入れ,
標線まで水を加え,同じ条件でその吸光度を測定する。これを試料ブランクとする。
F.6
結果
F.6.1
計算
アルデヒド濃度は,次の式で与えられる。
(
)
S
B
S
A
m
F
E
E
w
×
−
=
ここに,
wA: アルデヒド濃度 (mg/kg)
ES: 試料の吸光度
EB: ブランク試験の吸光度(試薬ブランク+試料ブランク)
mS: 用いた試料の質量 (g)
F: 校正係数 (μg)
F.6.2
結果の表示
計算結果は,0.1 mg/kg単位に丸めて表示する。
F.7
試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(F.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
20
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書G
(規定)
重量法による不溶解分濃度の定量
序文
この附属書は,不溶解分の濃度を,重量法を用いて定量する方法について規定する。
G.1
一般
この試験方法は,AUS 32中の1 mg/kg以上の不溶解分の濃度を定量する方法である。
G.2
原理
試料をろ過し,残留物の質量を重量法によって定量する。
G.3
装置
G.3.1 減圧ろ過装置 直径47 mm又は50 mmのメンブランフィルタに適したもの。
G.3.2 メンブランフィルタ 孔径0.8 μmで,セルロース混合エステル製のもの。
G.3.3 シャーレ メンブランフィルタの収納に適したもの(例えば,80 mm×15 mm)。
G.3.4 ピンセット 先端が平らなもの。
G.3.5 化学天びん 分解能が,0.01 mg又はそれより良いもの。
G.3.6 天びん 分解能が,0.01 g又はそれより良いもの。
G.3.7 ビーカ 公称容積が,400 mL(体積目盛の入った背高形状のものが望ましい。)のもの。
G.3.8 恒温槽 温度を105 ℃±2 ℃に維持することができるもの。
G.3.9 デシケータ 乾燥剤を充てんしたもの。
注記 硫酸又は塩化カルシウムは,乾燥剤として適切でない。
G.3.10 一般的な理化学用ガラス器具
G.4
薬品類
G.4.1 脱イオン水 ISO 3696の2級に相当する,導電率0.1 mS/m未満のもの。
G.5
手順
試料は,尿素の結晶がなくなるまで完全に溶解しなければならない。必要ならば,その後の処置をする
前に,最高40 ℃まで加熱しなければならない。
試験に用いるフィルタは,前もって水で洗浄しなければならない。これを行うためには,フィルタを約
100 mLの水を用いて減圧ろ過装置の中で湿らせ,その水をフィルタを通して吸い取らなければならない。
その後,フィルタを質量が安定するまで恒温槽内で乾燥させ,デシケータ中のシャーレに入れて保存しな
ければならない(シャーレ1個にフィルタ1枚)。このメンブランフィルタは,分析を行う直前に,0.01 mg
の精度でひょう量しなければならない。
ひょう量作業中は,フィルタは常にシャーレの中に置いておかなければならない。
試料は,均質になるように十分に振り混ぜる。そのすぐ後で,約100 mL〜150 mLの試料を乾燥したひ
21
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ょう量済みの400 mLのガラスビーカに入れ,0.01 g単位でひょう量し,200 mLの水を加える。試料のひ
ょう量のためにピペットを用いてはならない。
減圧ろ過装置に,準備したメンブランフィルタを取り付ける。フィルタを,減圧せずに少量(1 mL〜2 mL)
の水で湿らせる。用意した試料をろ過容器に入れ,その試料が速やかにフィルタを通って吸引されるよう
に真空度を調節する。
ガラスビーカは,1回当たり約30 mL〜50 mLの水で5回すすぎ洗いをする。すすぎ液もフィルタを通
す(減圧ろ過装置のろ過容器も,すすぎ洗いしなければならない。)。試料は,最初のすすぎ洗いの開始前
に,完全にフィルタを通過していなければならない(フィルタを短時間乾燥させる。)。
ろ過容器を取り外し,フィルタを105 ℃で質量が安定するまで乾燥させる。デシケータ内で室温まで冷
却した後,フィルタを0.01 mg単位で計量する。
尿素の残さが完全になくなるまで,フィルタを完全に洗浄しなければならない。フィルタがシャーレの
ガラス底部にまだ張り付いているようであれば,それは洗浄が十分でなかったことを示している。そのフ
ィルタは廃棄して,分析操作を繰り返さなければならない。
G.6
結果
G.6.1 計算
(
)
000
1
S
FL
FR
ins
×
−
=
m
m
m
w
ここに,
wins: 不溶解分濃度 (mg/kg)
mFL: 使用前の乾燥フィルタの質量 (mg)
mFR: 不溶解分が付着している乾燥フィルタの質量 (mg)
mS: 試料の質量 (g)
G.6.2 結果の表示
個々の計算結果の平均値が,結果として有効である。個々の計算結果の差が,高い方の値の25 %より
大きい場合には,測定をもう一度行わなければならない。結果は,次のように丸める。
10 mg/kg未満:0.1 mg/kg単位
10 mg/kg以上:1 mg/kg単位
G.7
試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(G.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
22
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書H
(規定)
吸光光度法によるりん酸濃度の定量
序文
この附属書は,吸光光度法によるりん酸濃度の定量方法について規定する。
H.1 一般
この試験方法は,AUS 32中の総りん量をりん酸として,0.05 mg/kg〜10 mg/kgの範囲で定量するための
方法である。試料量を変えることによって,測定範囲を拡大することが可能である。
H.2 原理
試料を蒸発させ,炭酸カルシウムと一緒に灰化することによって,りん化合物を無機化する。
この処理の後,試料を塩酸を用いてりん酸からオルトりん酸に変換する。
オルトりん酸イオンは,酸性溶液中でモリブデン酸塩及びアンチモンイオンと反応して,アンチモン・
りんモリブデン酸錯体になる。
この錯体をアスコルビン酸で還元すると,濃い青色のモリブデンブルー錯体になる。この発色の強さが,
オルトりん酸イオンの濃度を示す。
H.3 装置
H.3.1 化学天びん 分解能0.01 g又はそれより良いもの。
H.3.2 灰化皿 プラチナ又は石英ガラス。
H.3.3 ホットプレート又はサンドバス
H.3.4 マッフル炉 700 ℃に設定できるもの。
H.3.5 分光光度計 光路長10 mmのセル付き,800 nmで用いる。
H.3.6 セル 光学ガラス製,光路長10 mm(複数個)
H.3.7 メスフラスコ (複数個)
H.3.8 ホールピペット (複数個)
H.4 薬品類
H.4.1 脱イオン水 ISO 3696の2級相当,導電率0.1 mS/m未満のもの。
H.4.2 炭酸カルシウム 分析等級のもの。
H.4.3 塩酸 質量分率で25 %の濃度のもの。
H.4.4 硫酸 質量分率で96 %の濃度のもの。
H.4.5 アスコルビン酸 分析等級のもの。
H.4.6 七モリブデン酸六アンモニウム四水和物 分析等級のもの。
H.4.7 酒石酸アンチモニルカリウム0.5水和物
H.4.8 アスコルビン酸溶液 100 g/Lの濃度のもの。
10 gのアスコルビン酸(H.4.5参照)を,100 mLの水(H.4.1参照)に溶かす。
23
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記 この溶液は,冷蔵庫で保管すれば2週間使用可能である。この溶液は,無色である限り使用可
能である。
H.4.9 モリブデン酸塩溶液 13 gの七モリブデン酸六アンモニウム四水和物 (NH4)6Mo7O24・4H2O(H.4.6
参照)を,250 mLの水(H.4.1参照)に溶かす。この溶液に,冷却,かつ,かき混ぜながら150 mLの硫
酸(H.4.4参照)を加える。
その後,0.35 gの酒石酸アンチモニルカリウム0.5水和物K(SbO)C4H4O6・1/2H2O(H.4.7参照)を100 mL
の水(H.4.1参照)に溶解し,それを硫酸モリブデン酸塩溶液に入れて混ぜ合わせる。
注記 褐色ガラス瓶で保管すれば,この溶液は2か月間使用可能である。
H.4.10 りん酸二水素カリウム (KH2PO4) 分析等級,105 ℃で乾燥したもの。
H.4.11 りん酸標準溶液 200 mg/Lの濃度のもの。
286.6 mgのりん酸二水素カリウム(H.4.10参照)をひょう量し,それを1 000 mLのメスフラスコ(H.3.7
参照)に入れて水(H.4.1参照)で溶かす。2 mLの硫酸(H.4.4参照)を加え,メスフラスコの標線まで水
(H.4.1参照)を加え,均質にする。
注記 この溶液は,密封したガラス瓶の中では,少なくとも3か月使用可能である。
H.4.12 りん酸標準溶液 2 mg/Lの濃度のもの。
りん酸標準溶液(H.4.11参照)から,水(H.4.1参照)で100倍に薄めることによって,濃度2 mg/Lの
第2の標準溶液を作る。
H.5 手順
H.5.1 検量線の作成
1 mL,2 mL,5 mL及び10 mLの標準溶液(H.4.12参照)(それぞれ2 μg,4 μg,10 μg及び20 μgのりん
酸イオンに対応。)を別々に50 mLのメスフラスコ(H.3.7参照)に入れ,それを水(H.4.1参照)で薄め
て40 mLにする。それぞれの濃度をH.5.7に従って,10回測定する。
H.5.2 校正係数の計算
次の式によって,校正係数を計算する。
(
)
∑
∑
=
=
−
=
4
1
2
,1
4
1
,
phosphate
i
i
i
i
E
E
m
C
ここに,
C: 校正係数
mphosphate,i: i番目の試料のりん酸イオン質量 (μg)
E1,i: i番目の試料の吸光度
E2: ブランク試験の吸光度
H.5.3 プロセスの点検
H.5.3.1 目的
試験方法が,正しい結果をもたらすかどうかを確認する。
H.5.3.2 原理
標準溶液(H.4.12参照)を,試料と同様に定量する。標準溶液のりん酸量の測定値で検証する。
H.5.3.3 実行
5 mLのりん酸標準溶液をホールピペットで取り,それを50 mLのメスフラスコ(H.3.7参照)に入れて
24
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
分析し(H.5.7参照),りん酸の濃度を計算する(H.6参照)。この手順を,3回繰り返す。
3回の測定値が,規定のりん酸量に対して±2 %未満の誤差であれば,測定は有効である。
H.5.3.4 頻度
この試験方法の検証は,3か月に1度実施することが望ましい。
H.5.4 検量線の点検
H.5.4.1 目的
検量線のこう配が正しいかどうかを,一定の間隔で確認する。
H.5.4.2 原理
りん酸標準溶液を定量し,その結果を検量線から得られた値と比較する。
H.5.4.3 実行
H.5.1と同様に,検量線の測定範囲内の最低3点の濃度を,3回定量する。
測定値の平均値が,規定のりん酸量に対して±2 %未満の誤差であれば,その検量線は有効である。誤
差がそれ以上の場合には,手順を繰り返す。
誤差が規定値(±2 %)以上の場合には,新しい検量線(H.5.1参照)を作成するまで,りん酸濃度を定量
することはできない。
H.5.4.4 頻度
検量線の検証は,少なくとも3年に1度は実施することが望ましい。
H.5.5 試料の調製
試料は,完全に溶解させ,尿素の結晶が残っていてはならない。必要ならば,試料を最高40 ℃まで加
熱してもよい。
H.5.6 試料の灰化
調製された試料(H.5.5参照)を約100 gひょう量し(質量を記録すること。),それを灰化皿(H.3.2参
照)に入れ,100 mgの炭酸カルシウム(H.4.2参照)を加える。それをホットプレート又はサンドバスに
置き,ゆっくり乾燥させる。その後,その試料を700 ℃のマッフル炉(H.3.4参照)の中で完全に分解す
るまで灰化する。試料を冷まして1 mLの塩酸(H.4.3参照)及び20 mL〜30 mLの水(H.4.1参照)を皿の
中に加える。残留物が溶解し,二酸化炭素が除去されるまで沸騰させる。溶液を完全に100 mLのメスフ
ラスコ(H.3.7参照)に移し,標線まで水を加えて均質化する。
H.5.7 分光光度計による測定
(H.3.8のホールピペットを用いて)溶液(H.5.6参照)から正確な量を,50 mLのメスフラスコ(H.3.7
参照)に移す。最大40 mLの試料溶液を用いる。40 mL未満の場合には,40 mLになるまで水(H.4.1参照)
で薄める。
かき混ぜながら1 mLのアスコルビン酸溶液(H.4.8参照)及び2 mLのモリブデン酸塩溶液(H.4.9参照)
を加え,メスフラスコの標線まで水(H.4.1参照)を加えて,均質化する。ブランク試験を,試料溶液を
用いずに同じ方法で行う。
10分〜30分経過してから,試料及びブランクの800 nmにおける吸光度を,分光光度計(H.3.5参照)
を用いて測定する。
H.6 結果
H.6.1 計算
りん酸濃度を,次の式を用いて計算する。
25
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(
)
S
2
1
S
B
S
P
m
F
V
F
V
C
E
E
w
×
×
×
×
×
−
=
ここに,
wP: りん酸濃度 (mg/kg)
ES: 試料の吸光度
EB: ブランク試験の吸光度
C: 校正係数 (μg)
VS: 灰化・溶解後の試料溶液の体積 (mL)
F1: 1 000(kgからgへの換算係数)
V: 吸光度測定に用いた体積 (mL)
F2: 1 000(mgからμgへの換算係数)
mS: 尿素溶液の質量 (g)
H.6.2 結果の表示
結果は,0.01 mg/kg単位に丸める。
H.7 試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(H.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
26
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書I
(規定)
ICP-OES法による微量成分(Al,Ca,Cr,Cu,
Fe,K,Mg,Na,Ni及びZn)濃度の定量
序文
この附属書は,微量成分(Al,Ca,Cr,Cu,Fe,K,Mg,Na,Ni及びZn)濃度の定量方法について規定
する。
I.1
一般
この試験方法は,高周波プラズマ発光分析法による微量成分(アルミニウム,カルシウム,クロム,銅,
鉄,カリウム,マグネシウム,ナトリウム,ニッケル及び亜鉛)濃度の定量方法である。
I.2
原理
微量成分の濃度は,Inductively Coupled Plasma−Optical Emission Spectrometer (ICP-OES)を用いて定量す
る。この方法は,各成分に対して検量線を必要とする。
試料の前処理については,次の二つの異なる分析手順を任意に適用してもよい。
a) (ホットプレート又はマイクロ波マッフル炉を用いて)尿素溶液の乾燥及び灰化を行う。この手順は,
時間がかかるが,感度は良好である。
b) 直接定量,水で5倍に薄めることを含む(基本的手順)。
I.3 装置
I.3.1 灰化法用装置
I.3.1.1
メスフラスコ 公称容量100 mL,クラスA又はクラスBのメスフラスコ。
樹脂製又は石英ガラス製のメスフラスコを用いる。ほうけい酸ガラスを用いてはならない。
I.3.1.2
マッフル炉 温調器は,温度をプログラムできるようになっていることが望ましい。また,マッ
フル炉は,ガス排出口を備えたものであることが望ましい。ガス排出口がない場合には,ブンゼンバーナ
による処理を追加する必要がある。
I.3.1.3
ブンゼンバーナ
注記 灰化温度が高すぎると,アルカリ元素が蒸発する。
I.3.1.4
ホットプレート 500 ℃まで加熱が可能なホットプレート。代わりに,ガス排出口が付いていて,
試料の上方に石英ガラスのあるマイクロ波マッフル炉を用いてもよい。
I.3.1.5
化学天びん 分解能0.1 g又はそれより良いもの。
I.3.1.6
石英ガラス皿 公称容積100 mLのもの。プラチナ皿は,測定結果を小さくする可能性があるの
で,用いてはならない。
I.3.2
直接定量用装置
I.3.2.1
メスフラスコ 公称容量100 mL,クラスA又はクラスBのメスフラスコ。樹脂製又は石英ガラ
ス製のメスフラスコを用いる。ほうけい酸ガラスを用いてはならない。
27
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
I.3.2.2
ピペット 50 μL,100 μL,200 μL,500 μL,1 000 μL及び10 mLのホールピペット又は可変容積
式ピストンピペット。ピペットは,校正したものでなければならない。
I.3.3
測定装置 (ICP-OES) 高塩濃度でもエアロゾルに変換することのできるネブライザシステム(ク
ロスフロー,V溝又は類似品)を用いなければならない。ICPガス(アルゴン)を加湿することが望まし
い。
オートサンプラを用いる場合には,容器,ニードル及び発光分析装置につながる供給ホースは,高分子
材料(高密度ポリエチレン,高密度ポリプロピレン,ポリテトラフルオロエチレンなど)で作られたもの
でなければならない。ほうけい酸ガラス容器を用いてはならない。
I.4 薬品類
I.4.1
一般 特に規定していない限り,少なくとも分析等級の純度レベルに対応する薬品類及び蒸留水/
脱イオン水(ISO 3696の3級に対応する導電率0.5 mS/m未満のもの)を用いなければならない。
測定は,1種類の酸だけを用いて実施する。
I.4.2
灰化法用の薬品類
− 質量分率が最小65 %の硝酸,又はその代替品として,質量分率が最大37 %の塩酸。
− ICP標準溶液,各成分とも1.000 g/L,混合成分でもよい。
注記 濃度が保証されたICP標準溶液であれば,市販品でも使用可能。
I.4.3
直接定量用の薬品類
− 32.5 %尿素溶液,生物学的用途の尿素及び水をひょう量して作ったもの。
− 質量分率が最小65 %の硝酸,又はその代替品として,質量分率が最大37 %の塩酸。
− ICP標準溶液,各成分とも1.000 g/L,混合成分でもよい。
注記 濃度が保証されたICP標準溶液であれば,市販品でも使用可能。
− 多成分標準溶液,各成分とも10 mg/L。それぞれのICP標準溶液からピペットを用いて1 mLを取り,
100 mLのメスフラスコに入れる。メスフラスコの標線まで水を加え振り混ぜる。溶液は,作業日ごと
に新たに用意しなければならない。
I.5 手順
I.5.1 干渉
灰化中に試料が飛散する,又はブンゼンバーナの上若しくはマッフル炉の中で予備的に灰化していると
きの温度が高すぎる(特に,Na及びKの場合)ことによって,測定結果が低めにでることがある。無機
成分の混入(例えば,炉の断熱材の混入)によって,測定結果が高めにでることもある。適切な処置を施
してこれらの誤差を防がなければならない。
灰化手順で,りん成分は定量不能である。なぜなら,ポリりん酸塩が灰化中に生成されて,それは後の
工程で溶解しないからである。
幾つかの成分の直接定量は,共存成分に含まれる炭素のために,妨害されるかもしれない。干渉は,用
いるネブライザシステムによっても起こる可能性がある。直接定量の間に装置に関係する問題が起きた場
合には,試料は,I.2 a) に従って処理しなければならない。
定量対象の微量成分が,樹脂製容器(試料瓶,メスフラスコなど)の内面に付着している可能性もある。
それゆえに,内面は使用前に酸(HCl,HNO3)で常に洗浄しておかなければならない。

28
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
I.5.2
試料の前処理
I.5.2.1
灰化法用試料の前処理
試料100 gを0.1 gの誤差範囲内でひょう量し,石英ガラス皿に入れる。ホットプレートの上でゆっくり
蒸発させながら濃縮し,最終的に乾燥させる。試料が飛散しなくなるまで乾燥したら,その試料をマッフ
ル炉の中に入れ,350 ℃から開始して2時間以内に700 ℃まで上昇させ,完全に灰化させる。少なくとも
30分間は,700 ℃を維持する。
ガス排出口のある温度制御可能なマッフル炉がない場合には,試料をブンゼンバーナ(ドラフト中で)
で大部分を灰化しておき,その後にマッフル炉の中で700 ℃で灰化する。
マイクロ波マッフル炉を用いて灰化する場合には,次の作業手順に従わなければならない。
− 室温から開始する。
− 30分以内に,200 ℃まで直線的に昇温させる。
− 200 ℃で,10分間保持する。
− 120分以内に,700 ℃まで直線的に昇温させる。
− 700 ℃で,30分以上保持する。
試料が室温まで冷えるのを待って,5 mLの硝酸(又は塩酸)及び約20 mLの水を用いて,加熱しなが
ら残留物を溶解する。その溶液をすべて,100 mLのメスフラスコに移す。このメスフラスコが室温まで冷
えるのを待って,標線まで水を加えてよく振る。
I.5.2.2
直接定量のための試料前処理
適切なネブライザ及び個々の成分に対して,十分に低い検出下限をもつ発光分析装置を用いる場合には,
試料は,次のように前処理しなければならない。
− 20 gの試料を0.01 gの誤差範囲内でひょう量し,100 mLのメスフラスコに入れる。
− 約50 mLの水及び5 mLの硝酸(又は塩酸)を順番に加える。
− メスフラスコの標線まで水を加え,その溶液を均質化する。
I.5.3
検量線の作成
検量線作成の頻度は,用いる発光分析装置(装置製造業者の仕様及びガイドライン)による。検量線を
点検し,そのドリフトを修正するために,作業日ごとに最低濃度及び最高濃度の標準溶液を測定すること
が望ましい。表I.1及び表I.2に規定する成分濃度を用いることが望ましい。
個々の成分の発光強度は,(通常は,ICPコンピュータソフトの助けを借りて)検量線を用いて変換され
る。
灰化法については,表I.1を参照する。
表I.1−検量線作成用溶液(灰化法)
溶液
各成分濃度
mg/L
酸添加量
mL/L
0
0
50
1
0.010
2
0.030
3
0.100
4
0.300
5
1.000
6
5.000
29
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
直接定量については,表I.2を参照する。
表I.2−検量線作成用溶液(直接定量)
溶液
各成分濃度
mg/L
酸添加量
mL/L
32.5 %尿素溶液
mL/L
0
0
50
200
1
0.005
2
0.010
3
0.020
4
0.050
5
0.100
6
0.200
7
0.500
I.5.4
試料の測定
発光強度測定に当たっては,表I.3に規定する波長を用いなければならない。
表I.3−測定波長
単位 nm
成分
波長
Ca
396.85,317.93又は393.37
Cr
205.56又は267.72
Fe
259.94又は239.56
K
766.49
Cu
324.75又は327.39
Mg
279.55又は285.21
Na
588.99又は589.59
Ni
352.45,231.60,227.07又は221.65
Zn
213.85,206.20又は202.55
Al
396.15,394.40又は167.08
前処理した各溶液の測定は,少なくとも3回繰り返して行うものとするが,試料交換の都度十分な洗浄
時間を確保しなければならない。中間の洗浄については,質量分率3 %の硝酸(又は塩酸)溶液を用いる
とよい。
I.6
結果
I.6.1
計算
測定値がmg/Lの単位で出力される場合には,後で対応する試料の濃度 (mg/kg) に変換する。
I.6.2
結果の表示
成分ごとの結果は,全測定値の算術平均値とする。その結果は,正確に有効数字2けたまで表示する。
I.7
試験報告書
報告書は,次のデータを含まなければならない。
30
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(I.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
31
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書J
(規定)
赤外線吸収スペクトルによる定性
序文
この附属書は,赤外線吸収スペクトルによる定性分析方法について規定する。
J.1
一般
この試験方法は,AUS 32試料の赤外線吸収スペクトルを同定するための手順を規定する。質量分率で
10 %を超える濃度をもつすべての尿素水溶液は,同じ特徴的なピークをもつ赤外線吸収スペクトルを示す。
この方法を用いてAUS 32試料を既知の試料と比較すれば,赤外線吸収スペクトルが同等かどうかを判
定することができる。この方法では,濃度の違い又は不純物の有無の違いを判定することはできない。
J.2
原理
光が尿素水溶液の薄い層を通過するとき,赤外線が特異的に吸収される。記録されたスペクトルから,
尿素の赤外線吸収スペクトルとの同等性を判定することができる。代わりに,適切な全反射法(ATR法)
を用いてもよい。
J.3
装置
J.3.1
600 cm−1〜4 000 cm−1の波数範囲で,スペクトルを記録することのできるフーリエ変換赤外分光光
度計(FTIR)又は任意の赤外分光光度計。分解能は,4 cm−1又はそれより良いものを選ぶのが望ましい。
J.3.2
水溶液に適した光学セル,例えばKRS5 (TlBr/TlI),ZnSeなど,光路長約100 μm。代わりに,液体
に適したATR装置を用いてもよい。
警告 KRS5の窓材は,極端な毒性をもつ。
J.4
手順
空気の泡が混ざらないように注意しながら,分析すべき試料を透過セルに満たす。このセルを赤外分光
光度計のセルホルダに設置し,赤外線吸収スペクトルを記録できるようにする。代わりに,試料をATRプ
リズムの上に置いてもよい。
得られたスペクトルとAUS 32の既知の基準スペクトルとを目視で比較する。
J.5
結果の表示
定性の結果として,次のいずれかを表示する。
− Yes (基準と同じ場合)
− No (基準と異なる場合)
32
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
J.6
例
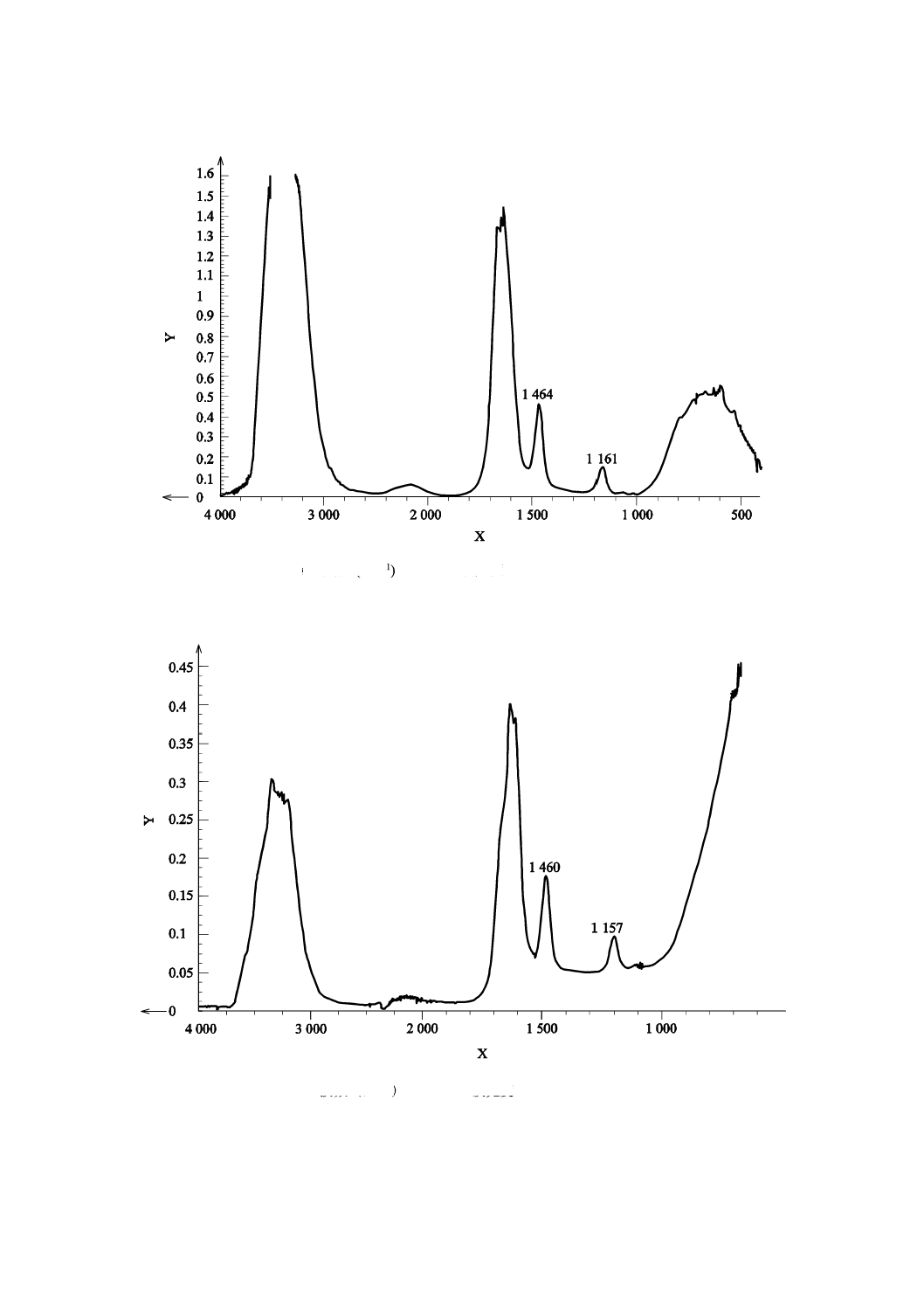
X:波数 (cm−1) Y:吸光度
図J.1−32.5 %尿素水溶液の基準スペクトルの例(透過法の場合)
X:波数(cm−1) Y:吸光度
図J.2−32.5 %尿素水溶液の基準スペクトルの例(ATR法の場合)
33
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書K
(参考)
各試験方法の精度
序文
この附属書は,附属書B〜附属書JBの各試験方法の精度をまとめたもので,規定の一部ではない。
K.1 一般
ISO 4259に準拠する統計手法で決定された試験方法の精度は,表K.1に記載する。また,この規格で提
示している試験精度の統計的有意性は,K.2及びK.3で記載していて,“xx(単位)”は,その併行精度及
び室間再現精度を示す。
K.2 併行精度,r
同一測定者が,同じ装置を用いて一定の操作条件の下で,同一の試料を測定したときの2回の測定結果
の差が,rを超える確率は,正常な試験方法で適正に実施した場合,究極的に5 %になる。
K.3 室間再現精度,R
異なる測定者が,異なる試験室において同一の試料をお互いに関与することなく,1回測定したときの
二つの測定結果の差が,Rを超える確率は,正常な試験方法で適正に実施した場合,究極的に5 %になる。
表K.1−併行精度及び室間再現精度

34
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
特性及び単位
併行精度
r
室間再現精度
R
備考
尿素濃度(総窒素量)
質量分率 %
0.4
1.0
質量分率30 %〜35 %
尿素濃度(屈折率)
質量分率 %
0.1
1.0
質量分率30 %〜35 %
屈折率
−
0.000 1
0.001
屈折率1.33〜1.39
ISO 3675による密度
kg/m3
0.5
1.2
−
ISO 12185による密度
kg/m3
0.2
0.5
−
アルカリ度(NH3換算)
質量分率 %
0.01
0.2×x
質量分率0.1 %〜0.5 %
ビウレット
質量分率 %
0.01
0.04
質量分率0.1 %〜0.5 %
アルデヒド
mg/kg
0.14
0.5×x
0.5 mg/kg〜10 mg/kg
不溶解分
mg/kg
0.23×x
0.38×x
>1 mg/kg
りん酸(PO4)
mg/kg
0.02
0.03
0.1 mg/kg〜1 mg/kg
ICP-OES法
カルシウム
mg/kg
0.02
0.1×x
−
鉄
mg/kg
0.01
0.3×x
−
銅
mg/kg
0.01
0.2×x
−
亜鉛
mg/kg
0.01
0.3×x
−
クロム
mg/kg
0.01
0.3×x
−
ニッケル
mg/kg
0.01
0.3×x
−
アルミニウム
mg/kg
0.02
0.3×x
−
マグネシウム
mg/kg
0.02
0.3×x
−
ナトリウム
mg/kg
0.03
0.5×x
−
カリウム
mg/kg
0.03
0.5×x
−
ICP-MS法
カルシウム
mg/kg
0.03
−
−
鉄
mg/kg
0.01
−
−
銅
mg/kg
0.01
−
−
亜鉛
mg/kg
0.02
−
−
クロム
mg/kg
0.01
−
−
ニッケル
mg/kg
0.02
−
−
アルミニウム
mg/kg
0.02
−
−
マグネシウム
mg/kg
0.02
−
−
ナトリウム
mg/kg
0.02
−
−
カリウム
mg/kg
0.03
−
−
炎光光度法
ナトリウム
mg/kg
0.01
−
−
カリウム
mg/kg
0.01
−
−
xは,平均値。
注記1 密度定量のための試験方法の精度は,現存するISO 3675及びISO 12185から引用したものである。
注記2 この規格に記載されるICP-MS法及び炎光光度法を除く,すべての試験方法の精度は,2004年にオ
ーストリア,ドイツ及びオランダの18のラボが参加して実施したラボ間試験プログラムにおいて
得られたものである。得られたデータは,ISO 4259に従って評価された。
注記3 屈折率の試験方法の精度は,分解能0.000 01の屈折率計で得られたものである。
35
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JA
(規定)
ICP-MS法による微量成分(Al,Ca,Cr,Cu,
Fe,K,Mg,Na,Ni及びZn)濃度の定量
序文
この附属書は,微量成分(Al,Ca,Cr,Cu,Fe,K,Mg,Na,Ni及びZn)濃度の定量方法について規
定する。
JA.1 一般
この試験方法は,高周波プラズマ質量分析法による微量成分(アルミニウム,カルシウム,クロム,銅,
鉄,カリウム,マグネシウム,ナトリウム,ニッケル及び亜鉛)濃度の定量方法である。
JA.2 原理
微量成分の濃度は,Inductively Coupled Plasma−Mass Spectrometer (ICP-MS)を用いて定量する。この方法
は,各成分に対して検量線を必要とする。
試料の前処理については,マイクロ波分解装置を用いて,尿素溶液の分解を行う。
JA.3 装置
JA.3.1 分解用装置
JA.3.1.1 マイクロ波分解装置 試料を密閉系で温度及び圧力をかけて分解する装置であり,温度及び圧力
をプログラムできるようになっていることが望ましい。
なお,装置に用いる容器は,装置の温度及び圧力に耐え得るクラスA又はクラスBの分解容器を用いる。
JA.3.1.2 化学天びん 分解能0.1 g又はそれより良いもの。
JA.3.1.3 メスフラスコ 公称容量100 mL,クラスA又はクラスBのメスフラスコ。樹脂製のメスフラス
コを用いる。
JA.3.1.4 ピペット ホールピペット又は可変容積式ピストンピペット。ピペットは,校正したものでなけ
ればならない。
JA.3.2 測定装置 (ICP-MS) JIS K 0133で規定する装置。試料に含まれる測定対象元素を,高周波プラズ
マによってイオン化する。生成したイオンを質量分析計に誘導し,測定対象元素の質量/電荷数 (m/z)に
おけるイオンの強度(個数)を測定することによって,元素の濃度を測定する装置。
なお,尿素溶液で測定する元素は,ノーマルプラズマ条件下では感度が低く,アルゴンの分子イオンに
干渉されるため,クールプラズマ条件下で測定するのが望ましい。
JA.4 分解用薬品類
JA.4.1 一般 特に規定しない限り,少なくとも試薬特級の純度レベルに対応する薬品類及び超純水(電気
抵抗率0.15 MΩ・m以上又は導電率6.7 μS/m以下のもの)を用いなければならない。
JA.4.2 薬品類
− 質量分率が最低65 %の高純度硝酸。
36
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記 JIS K 9901を満足する硝酸であればよい。
− ICP標準溶液,各成分とも10 mg/L,混合成分でもよい。
注記 濃度が保証されたICP標準溶液であれば,市販品でも使用可能。
− 検量線用多成分標準溶液,混合成分のもの。それぞれのICP標準溶液からピペットを用いて1 mLを
取り,100 mLのメスフラスコに入れる。メスフラスコの標線まで超純水を加え振り混ぜ,各成分とも
100 μg/Lの溶液を作製する。100 μg/L溶液からそれぞれ検量線濃度に応じて規定量を採取し,100 mL
のメスフラスコに入れる。メスフラスコの標線まで超純水を入れ,一定濃度に調製後,よく振り混ぜ
る。
なお,標準溶液は,作業日ごとに新たに用意しなければならない。
調製時の注意事項
a) 用いる質量数にスペクトル干渉が生じないような元素の組合せにする。
b) 検量線用多成分標準溶液と測定用試料溶液との液性は,できるだけ一致させる。液性が酸で
ある場合には,少なくとも酸の濃度を一致させる。
JA.5 手順
JA.5.1 試料の分解
試料1 gを分解容器にひょう量し,硝酸5 mLを加え密閉した状態にして,マイクロ波分解装置で完全に
分解する。
なお,分解の条件は,装置ごとに異なるが,少なくとも2時間を要する。
試料が室温まで下がるのを待ってから分解容器を取り出し,容器内の残留物を100 mLのメスフラスコ
に入れる。分解容器内を2〜3回超純水で洗浄し,100 mLのメスフラスコに入れた後,メスフラスコの標
線まで超純水を加える。
JA.5.2 検量線の作成
検量線の作成は,測定ごとに実施する。作成した検量線を点検し,装置の安定性を確認するためには,
作業日ごとに感度,ブランク及び検量線の相関係数を確認することが望ましい。
検量線用多成分標準溶液の濃度は,各成分とも0 μg/L,1 μg/L及び2 μg/Lに調製する。
この方法は,絶対検量線法であり,共存成分による非スペクトル干渉が無視できるほど小さいときに有
効であるため,検量線の測定領域内で測定することが望ましい。また,装置の長時間連続運転,試料測定
数の累積などによって検量線が変動する場合があるため,正確に定量するには,一定時間ごと又は一定測
定回数ごとに検量線用多成分標準溶液を測定して,検量線を校正しなければならない。
JA.5.3 試料の測定
試料の測定は,少なくとも3回繰り返して行うものとするが,試料交換の都度,十分な洗浄時間を確保
しなければならない。中間の洗浄については,質量分率10 %の硝酸溶液を用いるとよい。装置内に残っ
た元素の洗浄に有効である。
JA.5.4 試料の定量
個々の成分の濃度は,検量線を用いてイオン強度(個数)から自動的に変換される。
なお,試料は,100倍に薄めているため,100を乗じて試料の定量を実施する。
37
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JA.6 結果
JA.6.1 計算
測定値がμg/Lの単位で出力される場合には,後で対応する試料濃度 (mg/kg)に変換する。
JA.6.2 結果の表示
成分ごとの結果は,全測定値の算術平均値とし,その結果は,正確に有効数字2けたまで表示する。
JA.7 試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(JA.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
38
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JB
(規定)
炎光光度法による微量成分(K及びNa)濃度の定量
序文
この附属書は,微量成分(K及びNa)濃度の定量方法について規定する。
JB.1 一般
この試験方法は,炎光光度法による微量成分(カリウム及びナトリウム)濃度の定量方法である。
JB.2 原理
微量成分の濃度は,炎光光度計を用いて定量する。この方法は,各成分に対して検量線を必要とする。
試料の前処理については,次の分析手順を適用する。
ホットプレート又はマイクロ波マッフル炉を用いて,尿素溶液の乾燥及び灰化を行う。
JB.3 装置
JB.3.1 灰化用装置
JB.3.1.1 メスフラスコ 公称容量100 mL,クラスA又はクラスBのメスフラスコ。
樹脂製又は石英ガラス製のメスフラスコを用いる。ほうけい酸ガラスを用いてはならない。
JB.3.1.2 マッフル炉 温調器は,温度をプログラムできるようになっていることが望ましい。また,マッ
フル炉は,ガス排出口を備えたものであることが望ましい。ガス排出口がない場合には,ブンゼンバーナ
による処理を追加する必要がある。
JB.3.1.3 ブンゼンバーナ
注記 灰化温度が高すぎると,アルカリ元素が蒸発する。
JB.3.1.4 ホットプレート 500 ℃まで加熱が可能なホットプレート。代わりに,ガス排出口が付いていて,
試料の上方に石英ガラスのあるマイクロ波マッフル炉を用いてもよい。
JB.3.1.5 化学天びん 分解能0.1 g又はそれより良いもの。
JB.3.1.6 石英ガラス皿 公称容積100 mLのもの。プラチナ皿は,測定結果を小さくする可能性があるの
で,用いてはならない。
JB.3.2 測定装置(炎光光度計) オートサンプラを用いる場合には,容器,ニードル及び炎光光度計につ
ながる供給ホースは,高分子材料(高密度ポリエチレン,高密度ポリプロピレン,ポリテトラフルオロエ
チレンなど)で作られたものでなければならない。ほうけい酸ガラス容器を用いてはならない。
JB.4 薬品類
JB.4.1 一般 特に規定していない限り,少なくとも分析等級の純度レベルに対応する薬品類及び蒸留水/
脱イオン水(ISO 3696の3級に対応する導電率0.5 mS/m未満のもの)を用いなければならない。
測定は,1種類の酸だけを用いて実施する。
JB.4.2 灰化法用の薬品類
− 質量分率が最低65 %の硝酸,又はその代替品として,質量分率が最高37 %の塩酸。
39
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− K及びNa標準溶液,各成分とも1.000 g/L,混合成分でもよい。
注記 濃度が保証されたK及びNa標準溶液であれば,市販品でも使用可能。
JB.5 手順
JB.5.1 干渉
灰化中に試料が飛散する,又はブンゼンバーナの上若しくはマッフル炉の中で予備的に灰化していると
きの温度が高すぎる(特に,K及びNaの場合)ことによって,測定結果が低めにでることがある。無機
成分の混入(例えば,炉の断熱材の混入)によって,測定結果が高めにでることもある。適切な処置を施
してこれらの誤差を防がなければならない。
定量対象の微量成分が,樹脂製容器(試料瓶,メスフラスコなど)の内面に付着している可能性もある。
それゆえに,内面は使用前に酸 (HCl,HNO3)で常に洗浄しておかなければならない。
JB.5.2 試料の前処理
試料100 gを0.1 gの誤差範囲内でひょう量し,石英ガラス皿に入れる。ホットプレートの上でゆっくり
蒸発させながら濃縮し,最終的に乾燥させる。試料が飛散しなくなるまで乾燥したら,その試料をマッフ
ル炉の中に入れ,350 ℃から開始して2時間以内に700 ℃まで上昇させ,完全に灰化させる。少なくとも
30分間は,700 ℃を維持する。
ガス排出口のある温度制御可能なマッフル炉がない場合には,試料をブンゼンバーナ(ドラフト中で)
で大部分を灰化しておき,その後にマッフル炉の中で700 ℃で灰化する。
マイクロ波マッフル炉を用いて灰化する場合には,次の作業手順に従わなければならない。
− 室温から開始する。
− 30分以内に,200 ℃まで直線的に昇温させる。
− 200 ℃で,10分間保持する。
− 120分以内に,700 ℃まで直線的に昇温させる。
− 700 ℃で,30分以上保持する。
試料が室温まで冷えるのを待って,5 mLの硝酸(又は塩酸)及び約20 mLの水を用いて,加熱しなが
ら残留物を溶解する。その溶液をすべて,100 mLのメスフラスコに移す。このメスフラスコが室温まで冷
えるのを待って,標線まで水を加えてよく振る。
JB.5.3 検量線の作成
検量線作成の頻度は,用いる炎光光度計(装置製造業者の仕様及びガイドライン)による。検量線を点
検し,そのドリフトを修正するために,作業日ごとに最低濃度及び最高濃度の標準溶液を測定することが
望ましい。表JB.1に規定される成分濃度を用いることが望ましい。
個々の成分の発光強度は,(通常は,炎光光度計のコンピュータソフトの助けを借りて)検量線を用いて
変換される。
40
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表JB.1−検量線作成用溶液
溶液
各成分濃度
mg/L
酸添加量
mL/L
0
0
50
1
0.010
2
0.030
3
0.100
4
0.300
5
1.000
6
5.000
JB.5.4 試料の測定
発光強度測定に当たっては,表JB.2に規定する波長を用いなければならない。
表JB.2−測定波長
単位 nm
成分
波長
K
766.5
Na
589.0
前処理した各溶液の測定は,少なくとも3回繰り返して行うものとするが,試料交換の都度十分な洗浄
時間を確保しなければならない。中間の洗浄については,質量分率3 %の硝酸(又は塩酸)溶液を用いる
とよい。
JB.6 結果
JB.6.1 計算
測定値がmg/Lの単位で出力される場合には,後で対応する試料の濃度(mg/kg)に変換する。
JB.6.2 結果の表示
成分ごとの結果は,全測定値の算術平均値とする。その結果は,正確に有効数字2けたまで表示する。
JB.7 試験報告書
報告書は,次のデータを含まなければならない。
a) 試験に供した製品のタイプ及び名称
b) この規格の規格番号
c) 用いた試料採取方法
d) 試験結果(JB.6参照)
e) もしあれば,この規格で規定した測定方法からの逸脱事項
f)
試験日
41
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献 JIS K 9901 高純度試薬−硝酸
ISO 5661,Petroleum products−Hydrocarbon liquids−Determination of refractive index
ISO 5667-3,Water quality−Sampling−Part 3: Guidance on the preservation and handling of water
samples
ISO 9020:1994,Binders for paints and varnishes−Determination of free-formaldehyde content of
amino resins−Sodium sulfite titrimetric method
42
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書JC
(参考)
JISと対応する国際規格との対比表
JIS K 2247-2:2009 ディーゼル機関−NOx還元剤AUS 32−第2部:試験方法
ISO 22241-2:2006 Diesel engines−NOx reduction agent AUS 32−Part 2: Test
methods
(Ⅰ)JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ)国際規格の規定
(Ⅳ)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(Ⅴ)JISと国際規格との技術的差異の
理由及び今後の対策
箇条番号
及び名称
内容
箇条
番号
内容
箇条ごと
の評価
技術的差異の内容
5 精度及び
係争
−
5.1
この規格で引用している
試験精度の統計的特徴は,
5.2及び5.3で規定してい
て,“xx(単位)”は,その
併行精度及び室間再現精
度を示す。
削除
5.1の併行精度及び室間再現
精度についての説明を削除
し,K.1へ移動した。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
注記 係争とは,SCR
コンバータシステム
(以下,SCRコンバー
タという。)の故障が
AUS 32の品質に起因
するものか否かについ
ての訴訟をいう。
−
追加
係争の意味が分かりにくい
ので,注記として説明を追加
したもので技術的差異はな
い。
技術的な差異はないので,このままと
する。
−
5.2
併行精度の説明を記載し
ている。
削除
5.2は削除し,K.2へ移動し
た。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
−
5.3
室間再現精度の説明を記
載している。
削除
5.3は削除し,K.3へ移動し
た。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
4
K
2
2
4
7
-2
:
2
0
0
9
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
43
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(Ⅰ)JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ)国際規格の規定
(Ⅳ)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(Ⅴ)JISと国際規格との技術的差異の
理由及び今後の対策
箇条番号
及び名称
内容
箇条
番号
内容
箇条ごと
の評価
技術的差異の内容
附属書A
(規定)
A.2
注記 この附属書で規
定する試料採取方法
は,ISO 5667-3に準拠
している。
Annex
A
A.2
NOTEこの附属書で規定
する試料採取方法は,ISO
5667-2及びISO 5667-3に
準拠している。
削除
ISO 5667-2は,既に廃止され
ている。
機会をとらえて,国際規格の修正を提
案する。
附属書B
(規定)
−
Annex
B
B.7 Precision
併行精度及び室間再現精
度を規定している。
削除
Annex Kで同じ内容を参考と
してまとめて記載しており,
規格として矛盾している。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
附属書C
(規定)
屈折率の分解能を小数
点以下4けたとし,計
算結果を小数点以下4
けたに丸める。
Annex
C
屈折率の分解能を小数点
以下5けたとし,計算結果
を小数点以下4けたに丸
める。
変更
分解能が小数点以下4けたで
も,濃度換算時の有効けた数
は確保できる。
機会をとらえて,国際規格の修正を提
案する。
−
C.7 Precision
併行精度及び室間再現精
度を規定している。
削除
Annex Kで同じ内容を参考と
してまとめて記載しており,
規格として矛盾している。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
附属書D
(規定)
−
Annex
D
D.7 Precision
併行精度及び室間再現精
度を規定している。
削除
Annex Kで同じ内容を参考と
してまとめて記載しており,
規格として矛盾している。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
4
K
2
2
4
7
-2
:
2
0
0
9
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
44
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(Ⅰ)JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ)国際規格の規定
(Ⅳ)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(Ⅴ)JISと国際規格との技術的差異の
理由及び今後の対策
箇条番号
及び名称
内容
箇条
番号
内容
箇条ごと
の評価
技術的差異の内容
附属書E
(規定)
E.5.5 試料の測定
試薬ブランク及び試料
ブランクの説明を追加
した。
Annex
E
E.5.5
追加
試薬ブランク及び試料ブラ
ンクの説明がないので追加
したもので,技術的差異はな
い。
技術的な差異はないので,このままと
する。
−
E.7 Precision
併行精度及び室間再現精
度を規定している。
削除
Annex Kで同じ内容を参考と
してまとめて記載しており,
規格として矛盾している。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
附属書F
(規定)
F.5.4 試料の測定
試薬ブランク及び試料
ブランクの説明を追加
した。
Annex
F
−
追加
試薬ブランク及び試料ブラ
ンクの説明がないので追加
したもので,技術的差異はな
い。
技術的な差異はないので,このままと
する。
−
F.7 Precision
併行精度及び室間再現精
度を規定している。
削除
Annex Kで同じ内容を参考と
してまとめて記載しており,
規格として矛盾している。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
附属書G
(規定)
−
Annex
G
G.7 Precision
併行精度及び室間再現精
度を規定している。
削除
Annex Kで同じ内容を参考と
してまとめて記載しており,
規格として矛盾している。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
附属書H
(規定)
−
Annex
H
H.7 Precision
併行精度及び室間再現精
度を規定している。
削除
Annex Kで同じ内容を参考と
してまとめて記載しており,
規格として矛盾している。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
4
K
2
2
4
7
-2
:
2
0
0
9
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
45
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(Ⅰ)JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ)国際規格の規定
(Ⅳ)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(Ⅴ)JISと国際規格との技術的差異の
理由及び今後の対策
箇条番号
及び名称
内容
箇条
番号
内容
箇条ごと
の評価
技術的差異の内容
附属書I
(規定)
I.4 薬品類
混合成分でもよいこと
を追加した。
Annex
I
I.4
追加
実際に用いられており,同等
性があるので,混合成分でも
よいことを追加した。
機会をとらえて,国際規格の修正を提
案する。
−
I.7 Precision
併行精度及び室間再現精
度を規定している。
削除
Annex Kで同じ内容を参考と
してまとめて記載しており,
規格として矛盾している。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
附属書J
(規定)
Annex
J
一致
附属書K
(参考)
K.1 一般
Annex
K
−
追加
併行精度及び室間再現精度
についての説明を5.1から削
除し,移動した。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
K.2 併行精度,r
−
追加
5.2を移動した。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
K.3 室間再現精度,R
−
追加
5.3を移動した。
欧州で実施したラボ間試験の結果であ
り,参考とすべきと強く要求していた
が受け入れられなかった経緯がある。
機会をとらえて,国際規格の修正を提
案する。
表K.1
微量成分の試験方法の
精度として,ICP-MS
法及び炎光光度法の精
度を追加した。
微量成分の試験法の精度
として,ICP-OES法の精度
だけを規定している。
追加
ICP-MS法及び炎光光度法
は,ICP-OES法よりも精度が
高く,国内では一般的に用い
られている。
機会をとらえて高周波プラズマ質量分
析法の追加を国際提案する。
4
K
2
2
4
7
-2
:
2
0
0
9
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
46
K 2247-2:2009
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(Ⅰ)JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ)国際規格の規定
(Ⅳ)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(Ⅴ)JISと国際規格との技術的差異の
理由及び今後の対策
箇条番号
及び名称
内容
箇条
番号
内容
箇条ごと
の評価
技術的差異の内容
附属書K
(参考)
(続き)
表K.1
注記3を追加した。
Annex
K
−
追加
屈折率及び屈折率による尿
素濃度の定量のための試験
方法の精度は,分解能
0.000 01の屈折率計で得られ
たものであることを明記し
た。
技術的な差異はないので,このままと
する。
附属書JA
(規定)
ICP-MS法を規定した。
−
−
選択
ICP-MS法は,ICP-OES法よ
りも精度が高く,国内では一
般的に用いられている。
機会をとらえて高周波プラズマ質量分
析法の追加を国際提案する。
附属書JB
(規定)
炎光光度法を規定し
た。
−
−
選択
炎光光度法は,ICP-OES法よ
りも精度が高く,国内では一
般的に用いられている。
機会をとらえて,炎光光度法の追加を
国際提案する。
JISと国際規格との対応の程度の全体評価:ISO 22241-2:2006,MOD
注記1 箇条ごとの評価欄の用語の意味は,次による。
− 一致 ················ 技術的差異がない。
− 削除 ················ 国際規格の規定項目又は規定内容を削除している。
− 追加 ················ 国際規格にない規定項目又は規定内容を追加している。
− 変更 ················ 国際規格の規定内容を変更している。
− 選択 ················ 国際規格の規定内容とは異なる規定内容を追加し,それらのいずれかを選択するとしている。
注記2 JISと国際規格との対応の程度の全体評価欄の記号の意味は,次による。
− MOD ··············· 国際規格を修正している。
4
K
2
2
4
7
-2
:
2
0
0
9
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。