K 0148:2005 (ISO 14706:2000)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第12条第1項の規定に基づき,財団法人大阪科学技術センター付属ニューマ
テリアルセンター(OSTEC)/財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を制定
すべきとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が制定した日本工業規格である。
制定に当たっては,日本工業規格と国際規格との対比,国際規格に一致した日本工業規格の作成及び日
本工業規格を基礎にした国際規格原案の提案を容易にするために,ISO 14706:2000,Surface chemical
analysis - Determination of surface elemental contamination on silicon wafers by total-reflection X-ray fluorescence
(TXRF) spectroscopyを基礎として用いた。
この規格の一部が,技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の
実用新案登録出願に抵触する可能性があることに注意を喚起する。経済産業大臣及び日本工業標準調査会
は,このような技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の実用新
案登録出願にかかわる確認について,責任はもたない。
JIS K 0148には,次に示す附属書がある。
附属書A(参考)参照試料群
附属書B(参考)相対感度係数
附属書C(参考)参照試料群の調製[5]
附属書D(参考)VPD-TXRF法
附属書E(参考)視射角設定
附属書F(参考)国際共同試験結果
K 0148:2005 (ISO 14706:2000)
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1. 適用範囲 ························································································································ 1
2. 引用規格 ························································································································ 2
3. 定義 ······························································································································ 2
3.1 全反射 ························································································································· 2
3.2 視射角 ························································································································· 2
3.3 臨界角 ························································································································· 2
3.4 VPD-TXRF法 ··············································································································· 2
3.5 擬似ピーク ··················································································································· 2
4. 略語 ······························································································································ 2
5. 原理 ······························································································································ 3
6. 装置 ······························································································································ 3
7. 試料の調製環境及び測定環境 ····························································································· 3
8. 校正用参照試料群 ············································································································ 3
9. 安全 ······························································································································ 4
10. 測定方法 ······················································································································ 4
10.1 測定準備 ····················································································································· 4
10.2 検量線の作成 ··············································································································· 4
10.3 試験試料の測定 ············································································································ 5
11. 結果の表示···················································································································· 5
11.1 計算方法 ····················································································································· 5
11.2 ブランクの補正 ············································································································ 6
12. 精度 ···························································································································· 6
13. 試験報告書 ··················································································································· 6
附属書A(参考)参照試料群 ·································································································· 7
附属書B(参考)相対感度係数 ······························································································· 8
附属書C(参考)参照試料群の調製[5] ················································································· 11
附属書D(参考)VPD-TXRF法 ····························································································· 14
附属書E(参考)視射角設定 ································································································· 16
附属書F(参考)国際共同試験結果 ························································································· 19
参考文献 ···························································································································· 21
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 0148:2005
(ISO 14706:2000)
表面化学分析−全反射蛍光X線分析法(TXRF)に
よるシリコンウェーハ表面汚染元素の定量方法
Surface chemical analysis - Determination of surface elemental
contamination on silicon wafers by total-reflection X-ray fluorescence (TXRF)
spectroscopy
序文 この規格は,2000年に第1版として発行されたISO 14706,Surface chemical analysis - Determination of
surface elemental contamination on silicon wafers by total-reflection X-ray fluorescence (TXRF) spectroscopyを翻
訳し,技術的内容及び規格票の様式を変更することなく作成した日本工業規格である。
なお,この規格で点線の下線を施してある“参考”は,原国際規格にはない事項である。
また,まえがき,目次,本体及び附属書の用語及び文章の後の[ ]内の数字は,参考文献番号を示す。
ISO 14706は,ASTM F 1526,SEMI規格M33及び半導体基盤技術研究会のUCS(Ultra Clean Society)
規格の既存3規格を基に,シリコンウェーハ表面の汚染元素を測定する方法を定めたものである。
全反射蛍光X線分析法(TXRF)で定量分析を行うには,標準物質を必要とする。しかし,1010 atoms/cm2
という低い表面原子濃度の認証標準物質は存在しない。例えそのような認証標準物質を用いることができ
たとしても,分析環境,試料保存環境などで汚染され,その寿命は短いものになってしまう。
したがって,TXRF用の標準物質は,分析を行う適切な機関で調製され,分析,値付けされなければな
らない。このため二つの規格が必要となる。一つはTXRFによる測定手順に関するものであり,もう一つ
は標準物質の調製に関するものである。この規格は,前者に関するものである。
1. 適用範囲 この規格は,シリコン鏡面ウェーハ又はエピタキシャルウェーハの表面原子濃度を,全反
射蛍光X線分析法(TXRF)によって定量する方法について規定する。
この方法は,次の元素分析に適用する。
− 原子番号が16 (S)から92 (U)までの元素
− 表面原子濃度が1 × 1010 atoms/cm2から1 × 1014 atoms/cm2までの汚染元素
− VPD試料前処理法を用いる場合は,表面原子濃度が5 × 108 atoms/cm2から5 × 1012 atoms/cm2までの汚
染元素
備考 この規格の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 14706:2000,Surface chemical analysis - Determination of surface elemental contamination on
silicon wafers by total-reflection X-ray fluorescence (TXRF) spectroscopy (IDT)
2
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版(追補を含む。)を適用する。
JIS B 9920 クリーンルームの空気清浄度の評価方法
備考 ISO 14644-1:1999,Cleanrooms and associated controlled environments - Part 1: Classification of air
cleanlinessが,この規格と一致している。
JIS Z 8402-2 測定方法及び測定結果の精確さ(真度及び精度)−第2部:標準測定方法の併行精度及
び再現精度を求めるための基本的方法
備考 ISO 5725-2:1994,Accuracy (trueness and precision) of measurement methods and results - Part 2:
Basic method for the determination of repeatability and reproducibility of a standard measurement
methodが,この規格と一致している。
3. 定義 この規格で用いる主な用語の定義は,次による。
3.1
全反射 入射線が物質の界面で完全に反射する現象。
備考 入射するX線に対するシリコンの屈折率は,1よりも小さい。表面に対してすれすれの角度で
入射するX線は,その視射角と同一の角度で表面から全反射する。
3.2
視射角 入射X線が試料表面となす角度。
参考 視射角が正しい用語であるが,一般的には入射角も使われる。
3.3
臨界角 試料本体からの蛍光X線強度の視射角に対する依存性をプロットした曲線において,最初
の変曲点に当たる視射角。
3.4
VPD-TXRF法 表面の汚染元素をいわゆるVPD法によって回収した後乾燥し,生じた乾燥こん(痕)
をTXRFで分析する方法。ここで,VPD法とは,ウェーハ表面の酸化物を酸分解し,残留した不揮発性物
質を液滴(通常は超高純度ふっ化水素酸)を用いて回収する方法である。
なお,すべての工程において,環境からの汚染は最小限になるよう配慮する。
3.5
擬似ピーク シリコンウェーハ表面の汚染元素に起因しないにもかかわらず検出されるピーク。
参考 見掛け上の不純物ピークとも呼ばれている。
備考 擬似ピークは,検出器又はX線光路に使用される材料中に含まれる元素に起因する。擬似ピー
クは,入射X線が直接散乱したり,反射したりしたX線が予期せぬ元素に当たることによって
発生する。この現象は,測定誤差を増大させる。擬似ピークは,汚染レベルが1011 atoms/cm2
以下の試料を分析する際に深刻な影響を及ぼす。
4. 略語
FWHM (full width at half maximum) 半値全幅
RM
(reference material) 標準物質
SSD
(solid state detector) 半導体検出器
TXRF
(total-reflection X-ray fluorescence) 全反射蛍光X線
参考 TXRFは,一般にはtotal-reflection X-ray fluorescence spectroscopy までの略語として用いられ,
全反射蛍光X線分析法を示す。
VPD
(vapor phase decomposition) 気相分解
参考 当初は,酸蒸気を用いた気相分解法であったが,蒸気を使わず直接液滴で酸化膜を溶かしなが
ら残留不揮発成分を回収する方法も,広義でVPDと呼ばれる。
3
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5. 原理 試料にX線を照射すると,試料を構成する元素から元素特有のエネルギーをもつ蛍光X線が発
生する。この蛍光X線の強度は,試料中の各元素の量に比例する。
X線を全反射条件で試料に入射させると,散乱X線の強度が減少する。このとき,シリコンウェーハ表
面部分に存在する元素(表面上に付着した原子を含む)の蛍光X線はより強く励起する。これによって,
信号/バックグラウンド比(S/B)及び信号/ノイズ比(S/N)のよい蛍光X線スペクトルを得ることができる。
検出限界は,原子番号,励起エネルギー,光子フラックス,検出器分解能,励起されたX線のエネルギ
ーバンド幅,装置固有のバックグラウンドレベル,積分時間及びブランク値に依存する。一定条件で分析
したときの検出限界値は,分析対象となる元素の原子番号によって2けた以上変化する。
備考 測定深さは視射角によって変化するが,フィルム状汚染の場合には通常5 nm以下である。測定
領域は直径約10 mmであるが,X線検出器と試料との相対位置関係によって変化する。粒子状
汚染の場合の蛍光X線収率は,粒子の大きさ,分布及び構成元素によって変化する。
6. 装置
6.1
TXRF装置として最低限必要な構成は,次による。
X線源,モノクロメータ,3次元に移動可能な試料台,X線検出器(SSD)及び信号処理用コンピュー
タ。
6.2
入射X線として用いる単色X線。
6.3
蛍光X線検出器は,Mn-K-LII,IIIラインをFWHM 200 eV以下の分解能で分析できるもの。
参考 Mn-K-LII,IIIはMnKαとも呼ばれる。
6.4
試料台は,視射角を0 mrad (0°)から8.7 mrad (0.5°)の範囲で,± 0.17 mrad (0.01°)以内の精度で制御で
きるもの。
6.5
試料室は,雰囲気を必要に応じて減圧,又はヘリウム若しくは窒素で置換できるもの。
7. 試料の調製環境及び測定環境
7.1
試料の調製及び測定を行う環境(例えば,浮遊微粒子,温度,湿度)は,JIS B 9920 クラス4 と同等
以上でなければならない。
備考 測定したい元素と同じ元素を含む浮遊微粒子が,試料表面に付着すると測定誤差の増大の原因
となる。
7.2
装置を設置してある場所の機械的振動はできるだけ小さくし,5 × 10‒3 m/s2 (0.5 Gal)を上回ってはな
らない。
備考 機械的振動は.検出系のエネルギー分解能を悪化させ,結果として検出限界及びピーク分離能
を低下させる。
8. 校正用参照試料群
8.1
信頼性の高い校正を行うための参照試料群(RMs)として,参照元素で故意に汚染させた故意汚染参
照試料群と,参照元素を付着させないブランク参照試料の2種類を用いる。ブランク参照試料は,故意汚
染参照試料調製の汚染レベルを決定するために用いる(附属書A参照)。
4
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.2
故意汚染参照試料群は,シリコンウェーハ表面に参照元素を一定量均一に付着させて調製する。故
意汚染参照元素は,Ni又はFeとする。故意汚染参照試料の参照元素の表面原子濃度は,約1 × 1012 atoms/cm2
とする(附属書C参照)。
8.3
参照元素の故意汚染参照試料群表面への付着形態は,アングルスキャン測定で確認する(附属書E参
照)。
8.4
故意汚染参照試料群の表面の参照元素量は,信頼性の高い定量分析法で決定しなければならない(附
属書C参照)。
8.5
ブランク参照試料は,鏡面ウェーハ又はエピタキシャルウェーハでなければならない。ブランク参
照試料の表面汚染元素は,検出限界以下でなければならない。ブランク参照試料の結晶方位は,故意汚染
参照試料群と同じでなければならない。
8.6
故意汚染参照試料群とブランク参照試料とは,同じ容器内に保管する。
9. 安全 この測定方法では,X線を使用するため,装置から発生するX線を人体のいかなる部分にも照
射しないようにすることが必す(須)である。特に,X線の光路に手及び指をさらさないこと並びに散乱し
た二次X線照射から目を防護することが重要である。X線に関する安全法規及び基準を遵守する。
10. 測定方法
10.1 測定準備
10.1.1 測定試料は,すべて鏡面ウェーハ又はエピタキシャルウェーハでなければならない。
10.1.2 VPD-TXRF法では,環境からの汚染を最小限にして,試料表面汚染元素を酸の液滴で回収し,乾
燥する(附属書D参照)。
10.1.3 校正用の測定を含めた一連の測定において,試料はTXRF装置の試料台上で同一の結晶方位でなけ
ればならない。
10.1.4 視射角は,臨界角の25 %から75 %の間に設定する。
備考1. 粒子状汚染では,その形状によっては,視射角が低すぎると誤差が大きくなる。
2. 臨界角は,入射X線のエネルギーによって決まる。9.67 keV (W-LII-MIV)では3.20 mrad (0.18°),
17.4 keV (Mo-K-LII,III)では1.78 mrad (0.10°),11.4 keV (Au-LII-MIV)では2.72 mrad (0.16°)となる。
参考 W-LII-MIV,Mo-K-LII,III及びAu-LII-MIVは,それぞれWLβ1,MoKα及びAuLβ1とも呼ばれる。
10.1.5 次の測定条件を設定する。
a) 回転対陰極を使用する場合は,X線の励起電圧は30 kV以上,励起電流は200 mA以上及び積分時間
は500 s以上とする。
b) Mo又はW陽極封入X線管球の場合は,X線の励起電圧は30 kV以上,励起電流は40 mA以上及び積
分時間は500 s以上とする。
c) もし,X線の検出強度が強すぎる場合には,励起電流を下げて適切なカウント数になるように調節し
なければならない。
10.1.6 試料の中心を検出器の中心に合わせる。試料を回転させて蛍光X線を測定し,擬似ピークが最小
になる入射方位を探す。その後の測定は,できるだけ同じ入射方位で実施する。試料中心部以外の試料位
置で,擬似ピークを最小にする最適な入射方位を設定できないTXRF装置の場合には,表面マッピングデ
ータの解釈には十分注意する必要がある。
10.2 検量線の作成
5
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.2.1 ブランク参照試料を測定し,蛍光X線積分強度を求める。
10.2.2 故意汚染参照試料を10.2.1と同一条件で測定する。VPD-TXRF法では,故意汚染参照試料の乾燥
こん(痕)の中心を検出器の中心直下に置く。
10.2.3 ブランク参照試料の参照元素に相当するピークの蛍光X線の積分強度が,故意汚染参照試料群の
参照元素の蛍光X線積分強度に対して10 %未満であることを確認する。
もし,ブランク参照試料の積分強度が,故意汚染参照試料群の積分強度の10 %以上になっていれば,
故意汚染参照試料及びブランク参照試料の両方を廃棄して,新たな故意汚染参照試料及びブランク参照試
料を調製する。
なお,この確認は適切な間隔で行う。
10.2.4 次のa)又はb)の手順によって参照元素からのX線積分強度を求める。
a) 測定されたディジタル信号を数値処理にてスムージングを行う。
測定値からバックグラウンドを差し引いたX線積分強度を求める。
b) 測定値に最もよく一致するガウス関数を求める。次にガウス関数のピーク高さとFWHMとから積分
強度を求める。
10.2.5 検量線(参照元素のX線積分強度に対する表面原子濃度のプロット)を引く。検量線は,原点を
通らなければならない。
10.3 試験試料の測定
10.3.1 10.1で規定する方法によって試験試料を測定する。VPD-TXRF法を使った場合には,回収液の乾
燥こん(痕)の中心を検出器の中心直下に置く。
10.3.2 10.2で規定する方法で,汚染元素からのX線積分強度を求める。
2本以上の蛍光X線ピークが重なった場合には,ピーク分離を行って対象元素のX線の積分強度を求め
る。
備考1. 目的元素の測定の繰返し性及び再現性は,使用した入射X線の種類によって異なる。
2. 入射X線の視射角が設定値からずれると測定誤差が大きくなる。
3. 試料の表面粗さが大きくなると測定誤差が増大する。
4. VPD乾燥こん(痕)から得られる値は,乾燥こん(痕)の形状及び含まれる元素に大きく依
存する。
11. 結果の表示
11.1 計算方法 各汚染元素ごとの表面原子濃度Cmは,10.の測定方法によって得られた結果並びに式(1)
及び式(2)を用いて計算する。
s
s
I
C
K=
················································································ (1)
R
m
m
S
I
K
C
×
=
·········································································· (2)
ここに, K: 10.2.5で求めた検量線の傾き
CS: 参照元素の表面原子濃度,atoms/cm2
IS: 参照元素からの蛍光X線の積分強度,counts/s (cps)
Cm: 試験試料の汚染元素の表面原子濃度,atoms/cm2
Im: 試験試料の汚染元素からの蛍光X線の積分強度,counts/s (cps)
SR: 相対感度係数,これは各元素間の感度の差を補正
6
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考 参照元素以外の相対感度係数SRを正確に求めるため,これらと異なる2種類以上の元素をもつ
試料の測定がよく行われる。この結果を用いると,理論的な相対感度係数を用いずに直接その
元素の検量線を引くことができる。
相対感度係数は,参照試料群を用いた前記検量線から求めるか,又は附属書Bに記載の方法
によって求めることができる。
11.2 ブランクの補正 1011 atoms/cm2未満の表面原子濃度での測定では,装置固有のブランクが無視でき
なくなることがある。実際の表面原子濃度は,試験試料から求められた表面原子濃度Cmから同じ結晶方
位で汚染のないウェーハを使用して,10.1及び10.2に規定の測定条件と11.1の計算手順で求めた表面原子
濃度C0とを差し引いて補正しなければならない。
12. 精度 この規格に規定する手順を基にして国際共同試験が,日本,ヨーロッパ及び米国の15機関によ
って実施された。配布された試料は4試験試料と1参照試料とからなる。15機関から17組の測定結果が
得られた。併行精度と再現精度は JIS Z 8402-2に則して計算され,結果は附属書Fに記載した。
13. 試験報告書 試験報告書の記載項目は,次による。
a) 試験試料を特定できる情報
b) 使用したX線源の種類,例 W回転対陰極
c) 使用した励起X線,例 W-LII-MIV
d) X線源の励起電圧,例 30 kV
e) X線源の励起電流,例 200 mA
f)
使用した視射角,例 1.8 mrad (0.10°)
g) 積分時間,例 500 s
h) 参照試料群の調製方法,例 SC1浸せき法(附属書A参照)
i)
参照試料群元素名(Ni又はFe)及びその表面原子濃度,例 Ni 1.05 × 1012atoms/cm2
j)
試験試料の測定位置,例 ウェーハ中心
k) 使用した校正方法,例 10.2.4のa)又は10.2.4のb)
l)
試験試料から検出された各元素名及びその表面原子濃度
7
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A(参考)参照試料群
この附属書は,本体に関連する事柄を補足するもので,規定の一部ではない。
A.1 本体の故意汚染参照試料群は,SC1(standard cleaning solution 1:水,アンモニア水及び過酸化水素
からなるシリコンウェーハ洗浄液)浸せき法又はスピンコート法によって,Ni又はFeを表面原子濃度約
1012 atoms/cm2に付着させて調製する(附属書C参照)。
A.2 清浄度の高いクリーンルームに測定装置を設置している場合は,SC1浸せき法によって調製したFe
参照試料群を用いることが望ましい。
A.3 日常の測定においては,Ni参照試料群の使用が一般的である。
A.4 参照試料群は,各ロット又は各バッチから1枚又はそれ以上調製する。
A.5 同一ロット又は同一バッチの試料は,表面原子濃度が同一であるとみなす。
A.6 故意汚染参照試料群の校正方法については,附属書Cに記載する。
A.7 VPD-TXRF測定方法の参照試料群は,既知量の汚染元素を含む液滴を,疎水性の鏡面ウェーハ又は
エピタキシャルウェーハ上に載せて乾燥し,乾燥こん(痕)としたものを用いる。
A.8 VPDの回収率は,ほぼ100 %とみなす。
8
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B(参考)相対感度係数
この附属書は,本体に関連する事柄を補足するもので,規定の一部ではない。
B.1
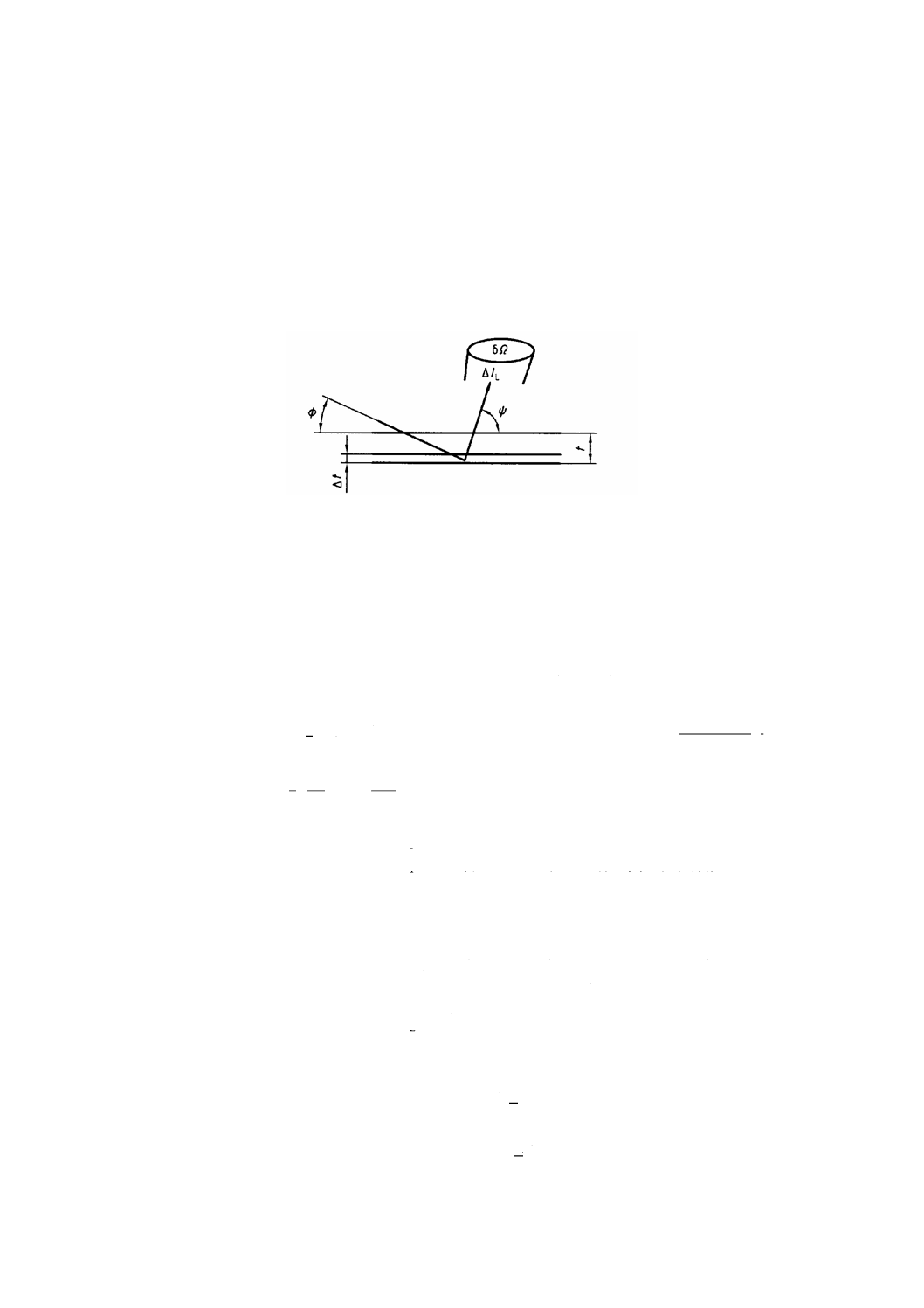
相対感度係数は,図B.1に示したモデルから計算できる。
ここに,
Ω: 立体角
φ: 視射角
Ψ: 取出し角
t: X線侵入深さ
図 B.1 TXRF概念図
薄い微小領域Δtから発生する元素Aの測定スペクトルLのX線強度⊿ILは,次の式による。
]}
cosec
)
/
[(
{
exp
4
1
)
/
(
)
/
(
cosec
)
/
]}(
cosec
)
/
[(
{
exp
L
M
L
A
A
A
P
M
P
,
A
A
P
M
P
M
P
,0
L
ψ
ρ
ρ
μ
π
δ
ω
γ
γ
ρ
μ
ρ
μ
φ
ρ
ρ
μ
φ
ρ
ρ
μ
λ
λ
λ
λ
λ
λ
t
Ω
g
w
t
t
I
I
,
,
,
,
−
−
×
∆
−
=
∆
·· (B.1)
ここに,
I0,λP: 波長λPの入射X線強度
(μ/ρ)M,λP: 試料Mに入射するX線の質量吸収係数[2]
ρ: 試料Mの密度
wA: 試料中の元素Aの質量分率
(μ/ρ)A,λP: 元素Aに入射するX線の質量吸収係数
γA: 元素Aの当該殻の吸収端におけるジャンプ比[2]
ωA: 元素Aの当該殻の蛍光収率[2, 3, 4]
gL: 測定されたスペクトルLの相対遷移確率[5]
(μ/ρ)M,λL: 試料Mに対する波長λLにおける測定スペクトル
Lの質量吸収係数
深さtの値が非常に小さいと仮定すると,
1
]}
cosec
)
/
[(
{
exp
P
M
=
−
φ
ρ
ρ
μ
λ
t
,
1
]}
cosec
)
/
[(
{
exp
L
M
=
−
ψ
ρ
ρ
μ
λ
t
,
9
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
全反射における入射と反射の二重励起を考慮すると式(B.1)は,次のように表される。
φ
ρ
π
δ
ω
γ
γ
ρ
μ
λ
λ
cosec
4
1
)
/
(
2
L
A
A
A
,
A
A
,0
L
P
P
t
Ω
g
w
I
I
−
=
······················ (B.2)
深さtの値が非常に小さいと仮定しているので,
wAρt = CA [Ar,A/NA] ·································································· (B.3)
ここに,
CA: 元素Aの表面原子濃度
Ar,A: 元素Aの原子質量
NA: アボガドロ数
式(B.2)及び式(B.3)から
L
A
A
A
A
A
A
r,
,0
A
L
A
1
)
/
(
cosec
)
N
/
(
4
2
P
P
g
A
Ω
I
C
I
S
ω
γ
γ
ρ
μ
φ
π
δ
λ
λ
−
=
=
,
したがって,相対感度係数SRは,次の式による。
RM
RM
r,
RM
RM
RM
,
RM
A
A
r,
L
A
A
A
,A
RM
A
R
RM
1
)
/
(
1
)
/
(
P
P
E
A
g
E
A
g
S
S
S
−
−
=
=
λ
λ
λ
ω
γ
γ
ρ
μ
ω
γ
γ
ρ
μ
·············· (B.4)
ここに,
Ar,RMは,参照元素の相対原子質量
EA, ERMは,半導体検出器内のそれぞれの波長λLとλRMに対する減衰係数
上の式は,試料が均一な密度と平滑な表面とをもち,発散のない単色X線が使用され,また,存在する
ほかの元素による多重散乱や励起がないという仮定に基づいている。
B.2
これらのパラメータが未知である場合,又は概略の表面原子濃度だけが要求される場合には,参照
元素をNiとしてまとめた表B.1又は表B.2を用いる。
B.3
表B.1は,入射X線としてW-LII-MIVと12.5 µm厚のBe窓を付けた半導体検出器とを用いて計算し
たSRである。表B.2は,入射X線としてMo-K-LII,IIIと12.5 µm厚のBe窓を付けた半導体検出器とを用い
て計算したSRである。これらと異なった条件(例えば,Au陽極,カーボンフィルタを用いるなど)では,
式(B.4)からSRを計算しなければならない。
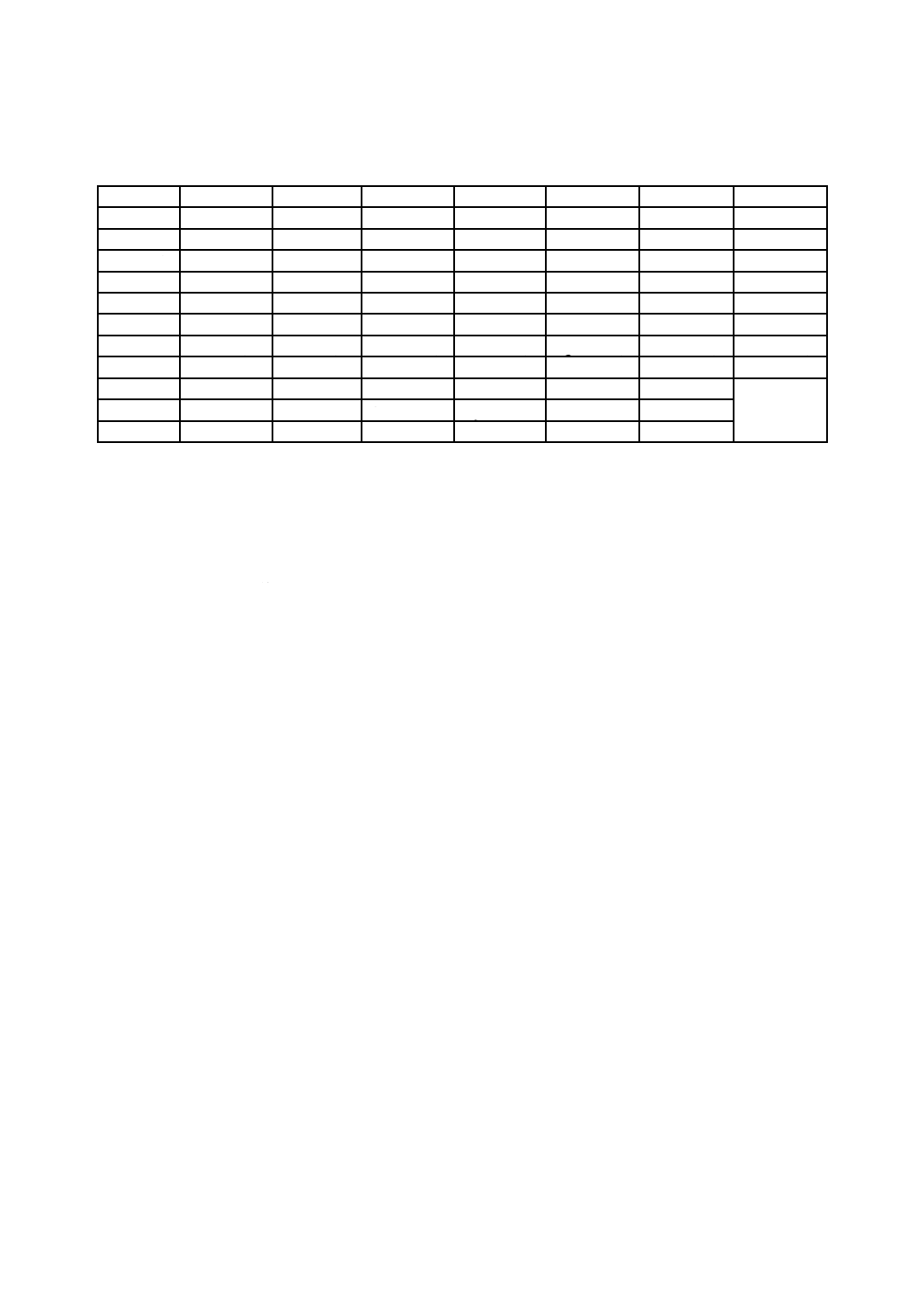
表 B.1 W-LII-MIVの各元素の相対感度係数表
原子番号
16
17
18
19
20
21
22
スペクトル
S-K-LII,III
Cl-K-LII,III
Ar-K-LII,III
K-K-LII,III
Ca-K-LII,III
Sc-K-LII,III
Ti-K-LII,III
SR
0.0225
0.0369
0.0563
0.0826
0.117
0.168
0.225
原子番号
23
24
25
26
27
28
29
スペクトル
V-K-LII,III
Cr-K-LII,III
Mn-K-LII,III
Fe-K-LII,III
Co-K-LII,III
Ni-K-LII,III
Cu-K-LII,III
SR
0.302
0.402
0.517
0.659
0.850
1.000
1.237
原子番号
30
33
42
46
47
50
スペクトル
Zn-K-LII,III
As-LIII-MIV,V Mo-LIII-MIV,V Pd-LIII-MIV,V Ag-LIII-MIV,V Sn-LIII-MIV,V
SR
1.512
0.0045
0.0440
0.0819
0.0930
0.146
10
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表 B.2 Mo-K-LII,IIIの各元素の相対感度係数表
原子番号
16
17
18
19
20
21
22
スペクトル
S-K-LII,III
Cl-K-LII,III
Ar-K-LII,III
K-K-LII,III
Ca-K-LII,III
Sc-K-LII,III
Ti-K-LII,III
SR
0.0194
0.0321
0.0488
0.0731
0.105
0.153
0.207
原子番号
23
24
25
26
27
28
29
スペクトル
V-K-LII,III
Cr-K-LII,III
Mn-K-LII,III
Fe-K-LII,III
Co-K-LII,III
Ni-K-LII,III
Cu-K-LII,III
SR
0.276
0.373
0.483
0.641
0.813
1.000
1.242
原子番号
30
33
42
46
47
50
73
スペクトル
Zn-K-LII,III
As-K-LII,III
Mo-LIII-MIV,V
Pd-LII-MIV,V
Ag-LIII-MIV,V
Sn-LIII-MIV,V
Ta-LIII-MIV,V
SR
1.538
2.445
0.0412
0.0777
0.0894
0.142
2.253
原子番号
74
78
79
80
82
92
スペクトル W-LIII-MIV,V
Pt-LIII-MIV,V
Au-LIII-MIV,V Hg-LIII-MIV,V
Pb-LIII-MIV,V
U-LIII-MIV,V
SR
2.566
3.643
3.734
4.015
5.023
5.574
B.4
装置条件によっては,補正係数を適用してもよい。
B.5
相対感度係数を用いる代わりに,独自の参照試料を用いた検量線を作ってもよい。
B.6
B.5のような検量線を用いた場合には,適切な間隔で参照試料群を再校正しなければならない。
11
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C(参考)参照試料群の調製[5]
この附属書は,本体に関連する事柄を補足するもので,規定の一部ではない。
C.1 TXRF用参照試料群の要求事項 TXRFは高感度な分析法であり,他の分析法に比べて標準物質上の
元素濃度が極めて低く,わずかな汚染を受けても標準物質は使用不可能になる。このため,永久に使用で
きる標準物質は存在せず,その都度新しい標準物質を調製し校正して用いる必要があるので,容易に調製
及び校正可能な参照試料群(標準物質)が望ましい。
励起X線が照射する試料表面の照射面積及びフラックス密度は,視射角によって大きく変化し,また,
検出器の信号強度は,試料とX線検出器との相対位置関係に依存する[6]。参照試料群の校正に際して,
TXRF分析が行われた位置だけの領域に存在する汚染元素を定量分析する方法はない。したがって,信頼
性の高い参照試料群を調製する方法としては,参照元素をウェーハ全面に均一に付着させる方法が望まし
い。また,これら以外に,長期間にわたる品質の安定も要求される。
上記の要求条件を満足する参照試料群を調製する方法として,SC1浸せき法[IAP法(immersion in alkaline
hydrogen peroxide solution)としても知られている。] [7,8]及びスピンコート法[9]がある。スピンコート法は
元素が粒子状に付着する傾向がある。他の方法としては,マイクロドロップ法[6]も挙げられる。ただし,
マイクロドロップ法は,ウェーハ表面上の一部分に参照元素を付着させる方法であり,従って,試料表面
の参照元素の濃度は面内均一ではない。
C.2 参照試料群用シリコンウェーハ 参照試料群の調製に用いるシリコンウェーハ基板に要求される汚
染表面原子濃度は,1010 atoms/cm2未満とする。16M DRAM (dynamic random-access memory)用又はそれ以
上の品質のウェーハが適切である。
ウェーハの結晶方位が異なれば擬似ピークが生じる条件が変わる[10]。したがって,参照試料群基板と
して用いる鏡面ウェーハ又はエピタキシャルウェーハの結晶方位は,試験試料と同一にすることが望まし
い。
C.3 参照元素 参照元素としては,X線発生効率が高い遷移金属が望ましい。このうちNi及びFeを次
の理由で推奨する。
a) X線源としてW-LII-MIV線を用いた場合,散乱したW-LII-MIV線のエスケープピークがCuピークに重
なる。これはピーク分離過程で誤差が生じる原因になる。
b) X線源としてW-LII-MIV線を用いた場合,散乱したW-LII-MIV線及びコンプトンピークがZnピークに
重なる。これは,ベースラインの引き方によって誤差が生じる原因になる。
c) 国際共同試験の結果から,FeもNiも比較的安定であることが示された。すなわち,FeもNiも均一な
フィルム状に付着し,自然酸化膜中に存在している[11]。
d) Feを含む微粒子が空気中に存在することは一般的であり,これがウェーハ表面に落下したり,検出器
の窓に付着したりすると擬似ピークの発生原因となる。
したがって,Fe参照試料群には,他の元素,例えば,Niよりも慎重な取扱いが要求される。
なお,国際共同試験では,すべての参加機関でFe参照試料群は汚染されていなかったことが確認でき
12
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ている。
e) 参照元素としてCo,V,又はMnを用いることに関しては,それらの元素が自然酸化膜にどのように
影響するか知られていない。さらに,これら元素の自然酸化膜の内外での挙動も知られていないし,
これらの参照試料群の精度は国際共同試験でも検証していない。
C.4 参照試料群の調製方法
C4.1 SC1浸せき法 最初にウェーハ表面原子濃度が1010 atoms/cm2以下になるように洗浄する。次に,参
照試料群元素が自然酸化膜中に一定量付着するように,所定濃度の参照元素のイオンを含むSC1洗浄液(水,
アンモニア水,過酸化水素の体積比が5:0.1〜1:1)にウェーハを浸せきして,参照元素を一定量付着させ
る。
溶液中のアンモニア濃度が適切でないと,例えば,Niなど元素によっては均一に付着できないときがあ
る。均一に表面付着させるには,体積比5:0.25:1を推奨する。
SC1洗浄液は,自然酸化膜のエッチングと生成が同時に進むので,自然酸化膜の厚みが一定に維持され
ながら溶液中の汚染元素が自然酸化膜中に取り込まれるという特徴がある。
C4.2 スピンコート法[9] 最初に,ウェーハ表面を親水性で,表面原子濃度が1010 atoms/cm2以下になるよ
うに洗浄する。次に,参照元素のイオンを一定量含む酸性液滴をウェーハ表面全体に広がるように乗せる。
そのまま参照元素がウェーハ表面に分布するよう一定時間放置してからスピン乾燥を行う。
この方法によって表面汚染元素は,ウェーハ面内に均一に分布する。同時に2元素以上付着させること
もできる。
C4.3 マイクロドロップ法[6] 最初に,ウェーハ表面を疎水性で,表面原子濃度が1010 atoms/cm2以下にな
るように洗浄する。次に,参照元素のイオンを既知量を含む酸性液滴をウェーハ上に滴下して乾燥させる。
付着した元素の表面濃度は,液滴の濃度及び体積によって決まる。蛍光X線強度は,乾燥こん(痕)と
検出器との相対位置関係によって大きく変化することに留意すべきである[6]。また,乾燥こん(痕)の径
も強度に影響する[11](附属書E参照)。
C.5 参照試料群の校正 参照試料群は,調製後できるだけ早く校正を行う。
SC1浸せき法又はスピンコ−ト法で調製したウェーハは,自然酸化膜中に含まれる元素をVPD法で回収
し,AAS, ICP-MS又はその他の適切な方法で校正する(附属書D参照)。
Niも含めて,ウェーハ表面の参照元素を酸化膜中からふっ化水素酸で直接回収すると,ふっ化水素酸液
中に存在する汚染元素が妨害することがある。このため自然酸化膜をふっ化水素酸蒸気で溶解して参照元
素を回収する。
また,放射化分析も用いられている。この方法は,放射化後の環境汚染,例えば,VPD試料調製やその
後の分析操作に起因するような汚染の影響を回避することができる。
重イオン後方散乱法を含むラザフォード後方及び前方散乱法は,直接参照試料群を校正することができ
る方法である。
マイクロドロップ法によって調製した参照試料群に付着した元素の表面濃度は,その液滴の濃度及び体
積によって計算できるので校正する必要がない。正確な計算方法については,SEMI規格M33に記載され
ている。
13
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
C.6 推奨する参照試料群 SC1法の場合は,1011 atoms/cm2から1013 atoms/cm2の濃度範囲でウェーハ面内
及び深さ方向分布が良好な故意汚染参照試料を調製することができる。さらに,実質的に同一な故意汚染
参照試料群を一度に25枚調製することができる。
スピンコート法の場合は,付着した元素の深さ方向分布は一定ではない。これは定量結果の誤差の原因
にはなるが,しかし,スピンコート法による試料は,国際共同試験において満足すべき再現性を示した。
マイクロドロップ法の場合は,位置合せ誤差が蛍光X線強度の誤差の原因になる。さらに,故意汚染参
照試料上での参照元素の分布と測定試料の元素の分布とが通常は異なっており,このことも誤差の原因に
なる。しかしながら,この方法は簡単であり低表面原子濃度に対応できる。
C.7 参照試料群の保管及び寿命 故意汚染参照試料群は,ブランク参照試料と同一の容器で,かつ,TXRF
装置と同一の環境で保管しなければならない。
これらの注意にもかかわらず,測定中に参照試料群に浮遊微粒子が付着することがある。
容器に,保管中に自然酸化膜が成長したり炭化水素が付着することがある。
故意汚染参照試料及びブランク参照試料の調製後の偶発的汚染レベルをブランク参照試料のバックグラ
ウンド補正後の定量値から推定する。もし,ブランク参照試料の参照元素の汚染レベルが故意汚染参照試
料群の表面原子濃度の10 %以上になった場合,故意汚染参照試料群及びブランク参照試料が,有効寿命
を超えたとみなして交換すべきである。
なお,通常期待される参照試料群の寿命は6か月以下と考えられる。
C.8 注意事項 シリコンウェーハ表面の汚染には,粒子状汚染とフィルム状汚染とがあることに留意す
べきである。例えば,Znは両方の形態が起り得る。蛍光X線強度は,汚染の物理状態及び深さに依存す
る。粒子状汚染の元素レベルとVPD法による乾燥こん(痕)の汚染量とをフィルム状付着の故意汚染参照
試料を使って定量すると実際よりも高めの数値になる。しかしながら,未知試料の分析に対しては最も妥
当な定量方法である。
14
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書D(参考)VPD-TXRF法
この附属書は,本体に関連する事柄を補足するもので,規定の一部ではない。
D.1 概要 標準TXRF法は,1010 atoms/cm2から1013 atoms/cm2の範囲で有効である。VPD-TXRFを使用
することによって,実用的な測定範囲を5 × 108 atoms/cm2から5 × 1012 atoms/cm2に向上させることができ
る。VPD-TXRF法は,汚染元素をウェーハ表面全体から回収し,それを小さな残さ状に付着させてTXRF
で分析する。
D.2 汚染元素の回収 汚染元素はウェーハ表面の自然酸化膜を分解して回収する。これは,鏡面ウェー
ハ又はエピタキシャルウェーハ表面をVPD容器内でフッ化水素酸蒸気に曝すか,又はウェーハ表面をフッ
化水素酸液滴で走査することによって行われる。
フッ化水素酸処理した表面は疎水性になる。これによって,ウェーハ表面の液滴はウェーハ表面で球状
になり,接触角は1.05 rad (60°)を超える。そのため液滴は集まりやすくなる。別の方法として,1滴のフ
ッ化水素酸又はフッ化水素酸 + 硝酸を容器とウェーハの間に挟み込ませることによって,ウェーハ表面
全体を一気に酸に曝す方法もある[13]。薬品類は,超高純度で作業環境はISO Class 4以上で行わなければ
ならない。得られた液滴は,AAS又はICP-MS法で分析することも可能である。
参考 ICP-MS(高周波プラズマ質量分析法又は誘導結合型プラズマ質量分析法とも言う)は,AAS
(原子吸光分析法)に比べて高感度であるが,分析対象元素であるFeがキャリアガスのArで
スペクトル干渉を受けるので,分析装置及び方法に配慮しなければならない。AASとICP-MS
の分析については,JIS K 0121原子吸光分析通則とJIS K 0133高周波プラズマ質量分析通則が
参考になる。
備考 汚染元素の回収に関する特許は各国に申請されている。JISは,これら特許の存在,有効性及
び適用範囲に関する権威ある又は包括的な情報は与えられない。この規格の使用者は,これら
の特許の存在と侵害の危険性は各自の責任によることを助言する。
D.3 乾燥こん(痕)の作製 回収した液滴は,汚染元素を回収したシリコンウェーハ表面上で乾燥する
か,又は新たに疎水性にした別のウェーハ上で乾燥する。VPD-TXRF法の定量精度は,乾燥こん(痕)の
形状(面積及び高さ)に依存し,小さい程精度はよくなる。よく知られているように,疎水性のシリコン
ウェーハは,クリーンルーム内の浮遊微粒子,化学物質などを強く引き付け付着させやすい性質がある。
液滴の乾燥には,減圧又は常温で清浄な不活性ガス流が取り扱える乾燥装置を使用することを推奨する。
D.4 TXRFによる測定
D.4.1 乾燥こん(痕)の大きさ 乾燥こん(痕)は小さい。国際共同試験で用いられた乾燥こん(痕)の
直径は,350 μm,高さは,50 nmであった。
D.4.2 乾燥こん(痕)位置の検出 乾燥こん(痕)の正確な位置を記録しておかないと乾燥こん(痕)を
検出することが非常に難しくなるので,TXRF装置が搭載しているサーチシステムを使用するか硝酸でス
ポットマークを付けて乾燥こん(痕)の位置を検出しやすくする方法が行われている。
15
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D.4.3 乾燥こん(痕)の形状効果 乾燥こん(痕)が大きいと測定誤差が増大する。さらに,相異なる金
属元素の塩が分離して,これらの元素が不均一に分布することがある。この不均一性は,特に,多元素汚
染の場合に分析精度がより悪化する原因になる。
16
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書E(参考)視射角設定
この附属書は,本体に関連する事柄を補足するもので,規定の一部ではない。
E.1
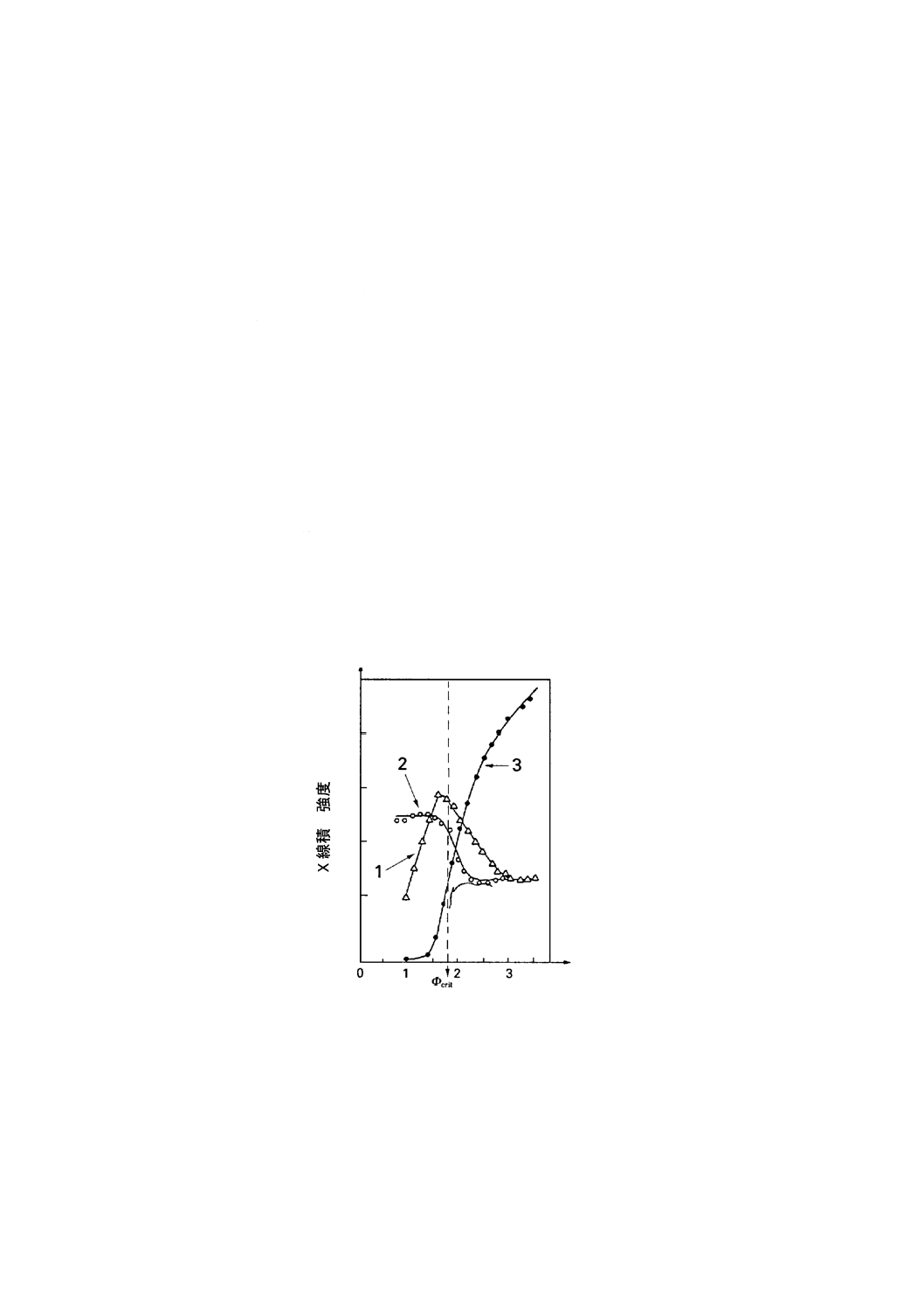
理想試料におけるTXRFアングルスキャン[13] シリコン鏡面ウェーハのような理想的な試料を使
用して,Mo-K-LII,IIIX線を入射させたTXRFアングルスキャン測定例を,図E.1に示す。ここで,特に重
要なのは,表面原子濃度が同一であるにもかかわらず粒子状汚染とフィルム状汚染とでは,アングルスキ
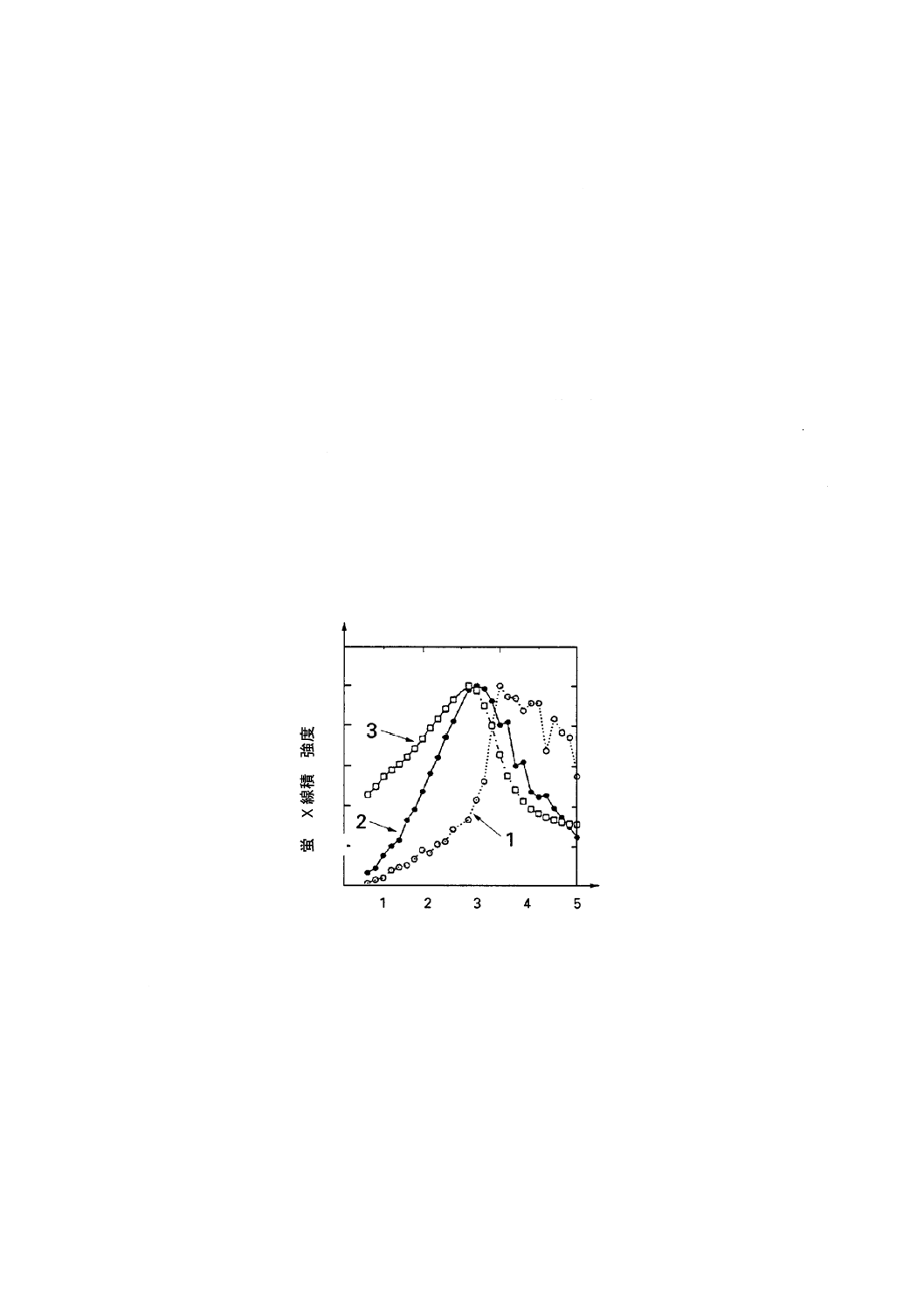
ャン曲線に相違がでることである。約1 mrad (0.06°)では,同一表面原子濃度の粒子状汚染とフィルム状汚
染とでは,蛍光X線強度に非常に大きな差が生じている。1.3 mrad (0.07°)では,両者の間にほとんど差が
ない。この角度は,2種類の汚染タイプのアングルスキャン曲線が交差するので,“クロスオーバー角”と
呼ばれることがある。
このクロスオーバー角の重要性は,参照試料及び未知試料のアングルスキャンのことを考慮すれば明ら
かである。ここで,参照試料のアングルスキャンが図E.1のフィルムタイプであると仮定する。もし,未
知試料のアングルスキャン曲線も図E.1のフィルム状(被膜又はスパッタされた)であれば,定量にどの
入射角を用いてもよい。しかしながら,未知試料が図E.1の粒子状(残さ)であれば,図E.1のクロスオ
ーバー角での測定だけ正確な定量ができることになる。もし,この場合,測定が低角度,例えば,1 mrad (理
論臨界角の56 %)で行えば,図E.1の視射角1 mradにおける信号強度比から見積もると誤差は約2.5倍に
なる。さらに,低角度で測定すると誤差は5倍以上になる。
図中の番号を以下に示す。
1:フィルム状汚染
2:粒子状汚染
3:バルク汚染
図E.1 蛍光X線積分強度の視射角依存性を示す実験曲線
蛍
光
X
線
積
分
強
度
(A
.U
.)
視射角(mrad)
17
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
実際の分析では,未知試料の汚染レベルが低いのでその蛍光X線の強度は非常に小さく,アングルスキ
ャンによって付着形態をチェックするのは困難である。このため低視射角度を用いたからといって定量に
大きな誤差が入ったか否かは必ずしも確認できるわけではない。
理想的な試料では,正確な定量が可能となる唯一の視射角度は,クロスオーバー角であるので,この角
度の使用が推奨される。しかし,実際の参照試料群では,アングルスキャン測定を行っても,常に図E.1
のフィルム状のアングルスキャンと同一の曲線になるとは限らず,いくらかの広がりをもつことが多い。
この場合,クロスオーバー角は範囲をもち,概略1.2 mrad (理論臨界角の67 %)から1.5 mrad (理論臨界角
の84 %)の間となる。
E.2
フィルム状汚染のアングルスキャン[6, 15] 測定深さは,X線強度が1/eに減少する侵入深さと定義
されている。侵入深さはX線エネルギー,視射角及び表面物質の電子密度で決まる。シリコン表面では5 nm
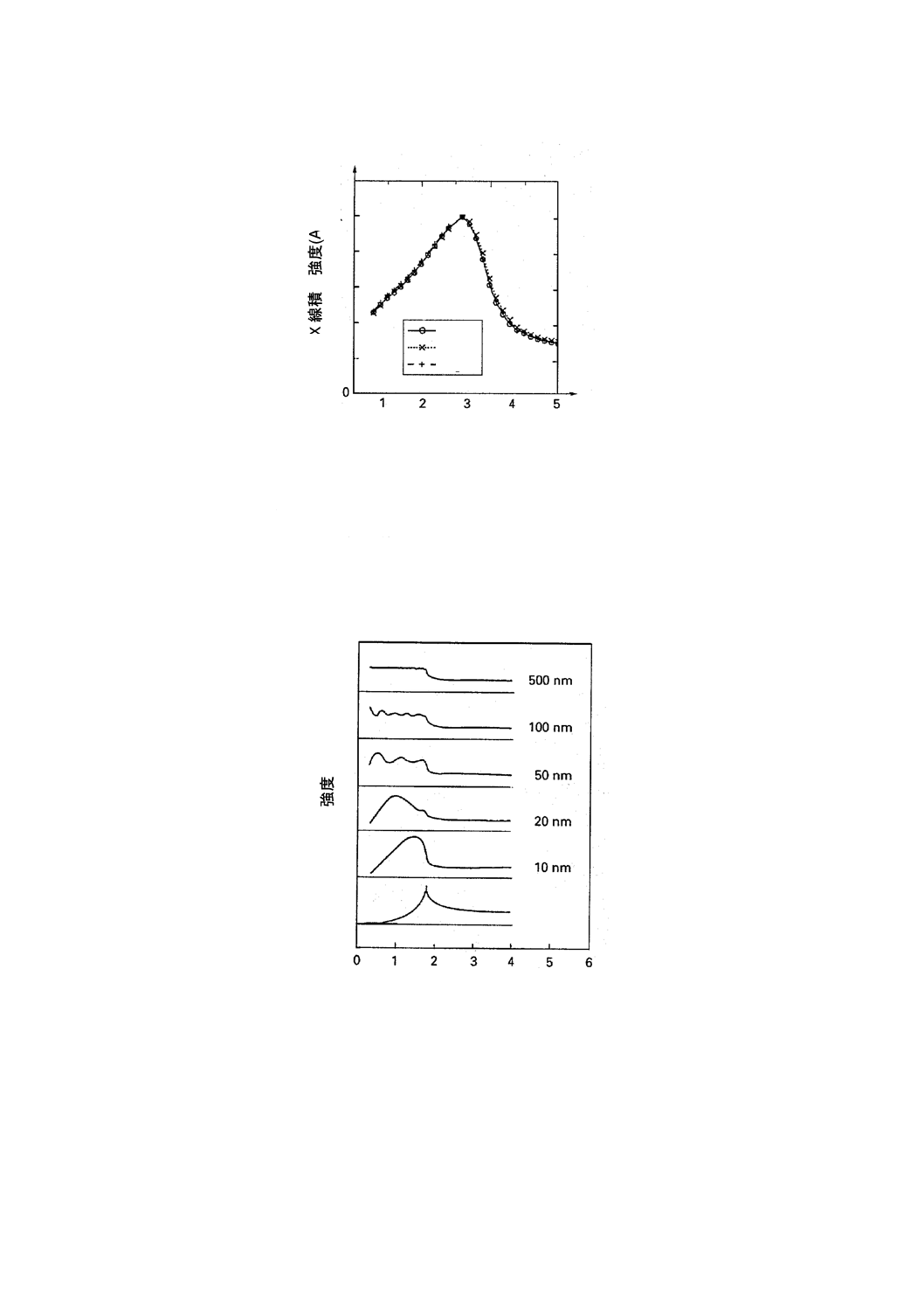
未満である。汚染元素の蛍光X線強度は視射角によって異なる。図E.2は,SC1浸せき法で調製した3種
類のNi参照試料群からの蛍光X線であるNi-K-LII,IIIX線のアングルスキャン曲線である。Ni-1は約1011
atoms/cm2,Ni-2は約1012 atoms/cm2及びNi-3は約1014 atoms/cm2である。アングルスキャン曲線は,各々
の最大値で規格化している。この曲線は付着したフィルム状の中でも差があることを示している。Ni-3は,
NiSi析出物が粒子状に生成しているものと考えられる。図E.3には, 3機関で測定した同一試料を用いた
アングルスキャン測定例を示す。各機関間でのアングルスキャン曲線には差がないことが分かる。
図中の番号を,以下に示す。
1:Ni-1 約1011 atoms/cm2
2:Ni-2 約1012 atoms/cm2
3:Ni-3 約1013 atoms/cm2
図E.2 SC1浸せき法で調製した異なる表面原子濃度をもつNi参照試料群のアングルスキャン曲線
蛍
光
X
線
積
分
強
度
(A
.U
.)
n
視射角(mrad)
18
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図 E.3 アングルスキャン測定時のX線積分強度の再現性
E.3
粒子状汚染のアングルスキャン[12] シリコン表面でのVPD乾燥こん(痕)は,数ミクロンの高さ
がある場合がある。したがって,入射X線は乾燥こん(痕)に侵入し,かつ,シリコンウェーハ表面で反
射する。図E.4は高さが異なる乾燥こん(痕)からの蛍光X線のアングルスキャン曲線である。臨界角以
下での全反射条件での励起では,試料表面上にX線の定在波が視射角に応じた周期で生じることによって,
蛍光X線強度の視射角依存性に種々の振動が生じている。
図E.4 残さ高さの影響
E.4
視射角設定 視射角設定は高角度側,すなわち,臨界角の75 %が望ましく,これは実際のシリコン
表面は,粒子状及びフィルム状の両方で覆われているためである。VPD-TXRFでの視射角設定も通常の
TXRFと同じにする。しかしながら,このことに関して,この規格作成段階では理論的に明快に説明でき
ていない。
蛍
光
X
線
積
分
強
度
(A
.U
.)
e
n
視射角(mrad)
機関I
機関III
機関IX
蛍
光
X
線
積
分
強
度
(A
.U
.)
表面下
10nm層
視射角(mrad)
残さ高さ
19
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書F(参考)国際共同試験結果
この附属書は,本体に関連する事柄を補足するもので,規定の一部ではない。
F.1
概要 この規格に記載する方法の精度を評価するため,1995年7月から9月にかけて国際共同試験
が行われた。この計画に日本,ヨーロッパ及び米国から合計15機関が参加した。
シリコンウェーハの試験試料4枚及び参照試料1枚を各機関に配布した。各試験試料及び参照試料は各
別々の容器に入れた。偶発的な汚染を防ぐために,スポークアンドハブ法による共同試験とした。15機関
から17組の測定データが得られた。
測定データの統計解析は, JIS Z 8402-2に従って実施した。
参考 スポークアンドハブ法とは,同一と見なされる複数の試料を参加機関に分配し,並行して分析
することで機関間でのクロスチェックを行う方法である。
F.2
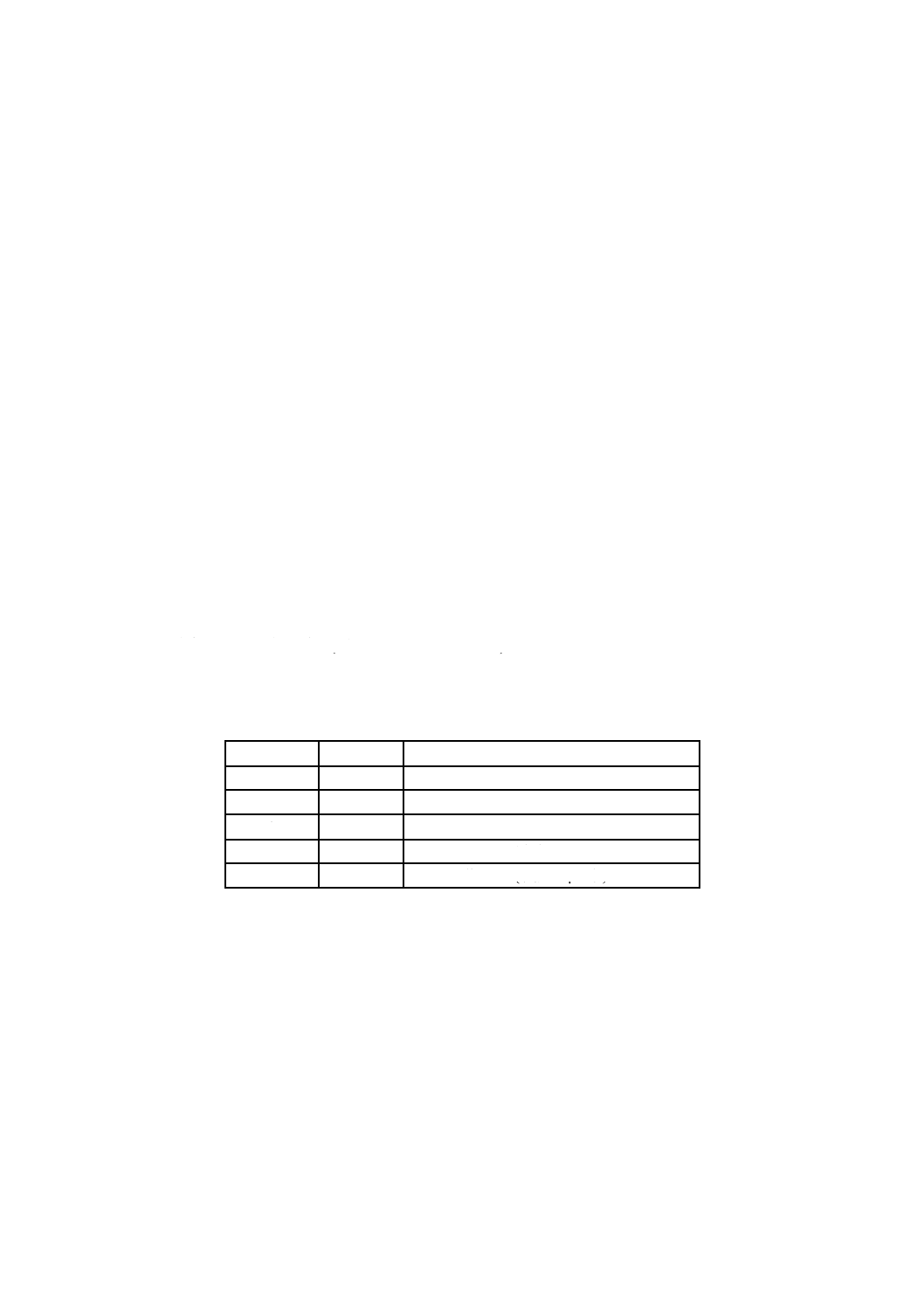
試験試料及び参照試料群 各機関に配布した分析試料の内訳は,SC1浸せき法によってNi又はFe
で故意に汚染した3枚(測定水準1,2,3)及びマイクロドロップ法によってFe (測定水準4)又はNi (測定水
準5)を,同一ウェーハ上に付着させた1枚である。検量線を作成する参照試料はSC1浸漬法によってNi
を付着させて作った。配布に先立ち参照試料と各試験試料はVPD-AAS法によって定量した。
すべての参加機関には参照試料(Ni 0.85 × 1012 atoms/cm2)の表面原子濃度が公開されたが,試験試料の4
枚については公開しなかった。
故意汚染の内容の詳細を,表F.1にまとめた。
表F.1 汚染の詳細
測定水準
元素
不純物の表面濃度
1
Ni
0.11 × 1012 atoms/cm2
2
Fe
0.91 × 1012 atoms/cm2
3
Fe
0.047 × 1012 atoms/cm2
4
Fe
1.08 × 1012 atoms (液滴10 μL中)
5
Ni
1.03 × 1012 atoms (液滴10 μL中)
F.3
TXRF分析手順 最初に,まず参照試料を一回測定して検量線を引く。試験試料は1日3回3日間
測定し定量する。これらのデータは,“定量データ”と称する。参照試料の元素はNiである。参照元素と
してFeの有効性についても検討した。Feを付着した試験試料(測定水準2)を参照試料として定量計算した。
このデータは,以下,“シミュレーションデータ”という。
F.4
統計解析
F4.1 全般 測定の安定性,測定中に偶発的な汚染が発生してないこと及び試料の均一性を確認した後,
JIS Z 8402-2の原則に従って併行精度及び再現精度を計算した。

20
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
F4.2 外れ値の統計解析 JIS Z 8402-2に従って,マンデル(Mandel)の方法によって検定した。この検定
では,5 %外れ値(straggler)又は1 %外れ値(outlier)はなかった。続いて,JIS Z 8402-2に従って,グ
ラッブズ(Grubbs)の方法によって99 %の信頼性で検定した。この検定で,一つの試験試料中2個のデ
ータが1 %外れ値として除外された。
F4.3 併行精度及び再現精度の計算 併行精度及び再現精度を求めるため,定量データ及びシミュレーシ
ョンデータをJIS Z 8402-2に従って処理した。
F.5
統計解析結果
F.5.1
定量データの統計解析結果を表F.2に示す。再現精度のデータには,試験試料間の不均一性が含ま
れることに留意しなければならない。
表F.2 測定データの併行精度及び再現精度
測定水準
機関数
p
平均値
m
(× 1012 atoms/cm2)
併行精度
sr
(× 1012 atoms/cm2)
再現精度
sR
(× 1012 atoms/cm2)
1
15
0.105
0.015
0.037
2
17
1.026
0.120
0.272
3
17
0.094
0.017
0.036
4
15
1.454
0.121
0.897
5
14
1.370
0.106
0.811
F.5.2
シミュレーションデータの統計解析結果を表F.3に示す。機関間再現性のデータには,試験試料間
の不均一性が含まれることに留意しなければならない。
表 F.3 シミュレーションデータの併行精度及び再現精度
測定水準
機関数
p
平均値
m
(× 1012atoms/cm2)
併行精度
sr
(× 1012atoms/cm2)
再現精度
sR
(× 1012atoms/cm2)
1
15
0.114
0.016
0.045
2
17
a
a
a
3
17
0.086
0.013
0.032
4
16
1.260
0.114
0.845
5
16
1.271
0.109
0.764
備考 表中のaは,測定水準 2 (Fe)の試験片を参照試料として使用する。
次のように,測定水準2 (Fe)の試験片を参照試料として使用して得られた定量結果を統計解析した。
a) ある機関でのFe参照試料は,その機関の結果だけに使用する。
b) Fe参照試料の表面原子濃度は,0.91 × 1012 atoms/cm2と仮定する。
c) 相対感度係数は,Ni参照試料による定量値及びFe参照試料測定値から算出する。
d) 検量線は,3日間各3回のFe参照試料の蛍光X線強度の平均値から計算する。
e) 検量線を用いて測定水準1,3,4,5の表面原子濃度を定量する。
21
K 0148:2005 (ISO 14706:2000)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] W. H. MCMASTER, N. K. DEL. GRANDE, J. H. MALLETT and J. H. HUBBELL: Compilation of X-Ray Cross
Sections, Section 2, Rev. 1, University of California Livermore, USA, Atomic Energy Commission Rep.
UCRL-50174 (1969).
[2] M. O. KRAUSE: J. Phys. Chem. Ref. Data 8, 307 (1979).
[3] W. BAMBYNEK, et al.: Rev. Mod. Phys., 44, 716 (1972).
[4] CRC Handbook of Chemistry and Physics, 75th edition, edited by D. R. LIDE, 10-278 (1995).
[5] UCS Standard: Test Method for Measuring Surface Contamination on Silicon Wafers by Total Reflection X-Ray
Fluorescence Spectroscopy, Ultra-Clean Technology, Vol. 8, No. 1, 44-82 (1996).
[6] H. KONDO, J. RYUTA, E. MORITA, T. YOSHIMI and Y. SHIMANUKI: Quantitative Analysis of Surface
Contaminations on Si Wafers by Total Reflection X-ray Fluorescence, Jpn. J. Appl. Phys., 31, L11-13 (1992).
[7] Y. MORI, K. UEMURA, K. SHIMANOE and T. SAKON: Adsorption Species of Transition Metal Ions on Silicon
Wafer in SC-1 Solution, J. Electrochem. Soc., 142, 3104-3109 (1995).
[8] Y. MORI, K. SHIMANOE and T. SAKON: A Standard Sample Preparation Method for the Determination of Metal
Impurities on a Silicon Wafer by Total Reflection X-ray Fluorescence Spectrometry, Anal. Sci., 11, 499-504
(1995).
[9] M. HORAI, T. NARIDOMI, Y. OKA, K. MURAKAMI, S. SUMITA, N. FUJINO and T. SHIRAIWA: A Method of
Quantitative Contamination with Metallic Impurities of the surface a Silicon Wafer, Jpn. J. Appl. Phys., 27,
L2361-2363 (1988).
[10] K. YAKUSHIJI, S. OHKAWA, A. YOSHINAGA and J. HARADA: Main Peak Profiles of Total Reflection X-ray
Fluorescence Analysis of Si (001) Wafers Excited by Monochromatic X-ray Beam W-Lβ (I), Jpn. J. Appl. Phys.,
31, L2872-2876 (1992).
[11] R. KLOCKENKÄMPER, J. KNOTH, A. PRANGE AND H. SCHWENKE: Total-Reflection X-Ray Fluorescence
Spectroscopy, Anal. Chem., Vol. 64, No. 23, 1115A (1992).
[12] M. B. SHABANI, T. YOSHIMI AND H. ABE: Low-Temperature Out-Diffusion of Cu from Silicon Wafers,
J. Electrochemical Soc., Vol. 143, No. 6, 2025-2029 (1996).
[13] W. BERNEIKE, J. KNOTH, H. SCHWENKE AND U. WEISBROD: Surface analysis for Si-wafers using total
reflection X-ray fluorescence analysis, Freseius Z. Anal. Chem., 333, 524 (1989).
[14] P. EICHINGER, H. J. RATH AND H. SCHWENEKE: Applications of Total Reflection X-ray Fluorescence Analysis for
Metallic Trace Impurities on Silicon Wafer Surfaces, Semiconductor Fabrication: Technology and Metrology,
American Society for Testing and Materials, STP 990, 305-313 (1989).