K 0102:2016
(1)
まえがき
この規格は,工業標準化法に基づき,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本
工業規格である。これによって,JIS K 0102:2013は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
K 0102:2016 目次
(2)
目 次
ページ
1. 適用範囲 ························································································································ 1
2. 共通事項 ························································································································ 1
3. 試料 ······························································································································ 4
3.1 試料の採取,試料容器,採水器及び採取操作 ······································································· 4
3.2 試料の取扱い ················································································································ 4
3.3 試料の保存処理 ············································································································· 5
4. 流量 ······························································································································ 6
5. 試料の前処理 ·················································································································· 6
5.1 塩酸又は硝酸酸性で煮沸 ································································································· 6
5.2 塩酸又は硝酸による分解 ································································································· 6
5.3 硝酸と過塩素酸とによる分解···························································································· 7
5.4 硝酸と硫酸とによる分解 ································································································· 7
5.5 フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法及びICP質量分析法を
適用する場合の前処理 ··········································································································· 8
6. 結果の表示 ····················································································································· 9
7. 温度 ······························································································································ 9
7.1 気温 ···························································································································· 9
7.2 水温 ···························································································································· 9
8. 外観 ····························································································································· 10
9. 透視度 ·························································································································· 10
10. 臭気及び臭気強度(TON) ····························································································· 12
10.1 臭気 ·························································································································· 12
10.2 臭気強度(TON) ······································································································· 13
11. 色度 ··························································································································· 14
11.1 刺激値及び色度座標を用いる方法 ··················································································· 15
11.2 三波長を用いる方法 ····································································································· 21
12. pH ····························································································································· 21
12.1 ガラス電極法 ·············································································································· 21
13. 電気伝導率 ·················································································································· 26
14. 懸濁物質及び蒸発残留物 ································································································ 30
14.1 懸濁物質 ···················································································································· 30
14.2 全蒸発残留物 ·············································································································· 31
14.3 溶解性蒸発残留物 ········································································································ 32
14.4 強熱残留物 ················································································································· 32
14.5 強熱減量 ···················································································································· 33
K 0102:2016 目次
(3)
ページ
15. 酸消費量 ····················································································································· 33
15.1 酸消費量(pH4.8) ······································································································ 33
15.2 酸消費量(pH8.3) ······································································································ 34
16. アルカリ消費量 ············································································································ 35
16.1 アルカリ消費量(pH8.3) ····························································································· 35
16.2 アルカリ消費量(pH4.8) ····························································································· 37
16.3 アルカリ消費量(遊離酸) ···························································································· 37
17. 100 ℃における過マンガン酸カリウムによる酸素消費量(CODMn) ······································· 38
18. 欠番 ··························································································································· 41
19. アルカリ性過マンガン酸カリウムによる酸素消費量(CODOH) ············································· 41
20. 二クロム酸カリウムによる酸素消費量(CODCr) ································································ 43
20.1 滴定法による酸素消費量(CODCr) ················································································ 43
20.2 蓋付き試験管を用いた吸光光度法によるCODCr測定法 ······················································· 45
21. 生物化学的酸素消費量(BOD) ······················································································· 47
22. 有機体炭素(TOC) ······································································································ 53
22.1 燃焼酸化-赤外線式TOC分析法 ······················································································ 54
22.2 燃焼酸化-赤外線式TOC自動計測法 ················································································ 57
23. 全酸素消費量(TOD) ··································································································· 57
24. ヘキサン抽出物質 ········································································································· 59
24.1 試料採取 ···················································································································· 59
24.2 抽出法 ······················································································································· 61
24.3 抽出容器による抽出法 ·································································································· 63
24.4 捕集濃縮・抽出法 ········································································································ 64
25. 欠番 ··························································································································· 65
26. 欠番 ··························································································································· 66
27. 欠番 ··························································································································· 66
28. フェノール類 ··············································································································· 66
28.1 フェノール類 ·············································································································· 66
28.2 p-クレゾール類 ··········································································································· 70
29. 欠番 ··························································································································· 72
29.1 欠番 ·························································································································· 72
30. 界面活性剤 ·················································································································· 72
30.1 陰イオン界面活性剤 ····································································································· 72
30.2 非イオン界面活性剤 ····································································································· 78
31. 農薬 ··························································································································· 82
31.1 有機りん農薬 ·············································································································· 82
31.2 ペンタクロロフェノール ······························································································· 89
31.3 エジフェンホス(EDDP) ····························································································· 91
32. 溶存酸素 ····················································································································· 91
K 0102:2016 目次
(4)
ページ
32.1 よう素滴定法 ·············································································································· 91
32.2 ミラー変法 ················································································································· 94
32.3 隔膜電極法 ················································································································· 95
32.4 光学式センサ法 ··········································································································· 97
33. 残留塩素 ····················································································································· 99
33.1 o-トリジン比色法 ······································································································· 100
33.2 ジエチル-p-フェニレンジアンモニウム(DPD)比色法 ······················································· 102
33.3 よう素滴定法 ············································································································· 104
33.4 ジエチル-p-フェニレンジアンモニウム(DPD)吸光光度法 ················································· 106
34. ふっ素化合物 ·············································································································· 108
34.1 ランタン-アリザリンコンプレキソン吸光光度法 ······························································· 108
34.2 イオン電極法 ············································································································· 111
34.3 イオンクロマトグラフ法 ······························································································ 113
34.4 流れ分析法 ················································································································ 114
35. 塩化物イオン(Cl−) ···································································································· 114
35.1 硝酸銀滴定法 ············································································································· 114
35.2 イオン電極法 ············································································································· 116
35.3 イオンクロマトグラフ法 ······························································································ 118
36. よう化物イオン(I−) ·································································································· 123
36.1 よう素抽出吸光光度法 ································································································· 123
36.2 よう素滴定法 ············································································································· 125
37. 臭化物イオン(Br−) ··································································································· 126
37.1 よう素滴定法 ············································································································· 126
37.2 イオンクロマトグラフ法 ······························································································ 128
38. シアン化合物 ·············································································································· 128
38.1 前処理 ······················································································································ 128
38.2 ピリジン-ピラゾロン吸光光度法 ···················································································· 133
38.3 4-ピリジンカルボン酸-ピラゾロン吸光光度法 ··································································· 135
38.4 イオン電極法 ············································································································· 136
38.5 流れ分析法 ················································································································ 138
39. 硫化物イオン(S2−) ···································································································· 138
39.1 メチレンブルー吸光光度法 ··························································································· 139
39.2 よう素滴定法 ············································································································· 142
40. 亜硫酸イオン(SO32−) ································································································· 144
40.1 よう素滴定法 ············································································································· 145
41. 硫酸イオン(SO42−) ···································································································· 147
41.1 クロム酸バリウム吸光光度法 ························································································ 147
41.2 重量法 ······················································································································ 149
41.3 イオンクロマトグラフ法 ······························································································ 150
K 0102:2016 目次
(5)
ページ
42. アンモニウムイオン(NH4+) ························································································ 150
42.1 前処理(蒸留法) ······································································································· 150
42.2 インドフェノール青吸光光度法 ····················································································· 153
42.3 中和滴定法 ················································································································ 154
42.4 イオン電極法 ············································································································· 156
42.5 イオンクロマトグラフ法 ······························································································ 159
42.6 流れ分析法 ················································································································ 159
43. 亜硝酸イオン(NO2−)及び硝酸イオン(NO3−) ······························································· 159
43.1 亜硝酸イオン(NO2−) ································································································ 159
43.2 硝酸イオン(NO3−) ··································································································· 162
44. 有機体窒素 ················································································································· 170
44.1 前処理(ケルダール法) ······························································································ 170
44.2 インドフェノール青吸光光度法 ····················································································· 171
44.3 中和滴定法 ················································································································ 172
45. 全窒素 ······················································································································· 172
45.1 総和法 ······················································································································ 172
45.2 紫外線吸光光度法 ······································································································· 174
45.3 硫酸ヒドラジニウム還元法 ··························································································· 176
45.4 銅・カドミウムカラム還元法 ························································································ 178
45.5 熱分解法 ··················································································································· 180
45.6 流れ分析法 ················································································································ 181
46. りん化合物及び全りん ·································································································· 181
46.1 りん酸イオン(PO43−) ······························································································· 182
46.2 加水分解性りん ·········································································································· 184
46.3 全りん ······················································································································ 185
47. ほう素(B) ··············································································································· 191
47.1 メチレンブルー吸光光度法 ··························································································· 191
47.2 アゾメチンH吸光光度法 ····························································································· 192
47.3 ICP発光分光分析法 ···································································································· 193
47.4 ICP質量分析法 ·········································································································· 194
48. ナトリウム(Na) ········································································································ 196
48.1 フレーム光度法 ·········································································································· 196
48.2 フレーム原子吸光法 ···································································································· 197
48.3 イオンクロマトグラフ法 ······························································································ 197
49. カリウム(K) ············································································································ 200
49.1 フレーム光度法 ·········································································································· 201
49.2 フレーム原子吸光法 ···································································································· 201
49.3 イオンクロマトグラフ法 ······························································································ 202
50. カルシウム(Ca) ········································································································ 202
K 0102:2016 目次
(6)
ページ
50.1 キレート滴定法 ·········································································································· 202
50.2 フレーム原子吸光法 ···································································································· 205
50.3 ICP発光分光分析法 ···································································································· 206
50.4 イオンクロマトグラフ法 ······························································································ 207
51. マグネシウム(Mg) ···································································································· 207
51.1 キレート滴定法 ·········································································································· 208
51.2 フレーム原子吸光法 ···································································································· 209
51.3 ICP発光分光分析法 ···································································································· 210
51.4 イオンクロマトグラフ法 ······························································································ 210
52. 銅(Cu)···················································································································· 210
52.1 ジエチルジチオカルバミド酸吸光光度法 ········································································· 210
52.2 フレーム原子吸光法 ···································································································· 212
52.3 電気加熱原子吸光法 ···································································································· 215
52.4 ICP発光分光分析法 ···································································································· 216
52.5 ICP質量分析法 ·········································································································· 220
53. 亜鉛(Zn) ················································································································· 223
53.1 フレーム原子吸光法 ···································································································· 224
53.2 電気加熱原子吸光法 ···································································································· 224
53.3 ICP発光分光分析法 ···································································································· 225
53.4 ICP質量分析法 ·········································································································· 225
54. 鉛(Pb) ···················································································································· 225
54.1 フレーム原子吸光法 ···································································································· 225
54.2 電気加熱原子吸光法 ···································································································· 226
54.3 ICP発光分光分析法 ···································································································· 227
54.4 ICP質量分析法 ·········································································································· 227
55. カドミウム(Cd) ········································································································ 227
55.1 フレーム原子吸光法 ···································································································· 227
55.2 電気加熱原子吸光法 ···································································································· 228
55.3 ICP発光分光分析法 ···································································································· 229
55.4 ICP質量分析法 ·········································································································· 229
56. マンガン(Mn) ·········································································································· 229
56.1 過よう素酸吸光光度法 ································································································· 229
56.2 フレーム原子吸光法 ···································································································· 231
56.3 電気加熱原子吸光法 ···································································································· 231
56.4 ICP発光分光分析法 ···································································································· 232
56.5 ICP質量分析法 ·········································································································· 232
57. 鉄(Fe) ···················································································································· 232
57.1 フェナントロリン吸光光度法 ························································································ 232
57.2 フレーム原子吸光法 ···································································································· 234
K 0102:2016 目次
(7)
ページ
57.3 電気加熱原子吸光法 ···································································································· 235
57.4 ICP発光分光分析法 ···································································································· 236
58. アルミニウム(Al) ····································································································· 236
58.1 キノリノール吸光光度法 ······························································································ 236
58.2 フレーム原子吸光法 ···································································································· 238
58.3 電気加熱原子吸光法 ···································································································· 239
58.4 ICP発光分光分析法 ···································································································· 240
58.5 ICP質量分析法 ·········································································································· 241
59. ニッケル(Ni) ··········································································································· 241
59.1 ジメチルグリオキシム吸光光度法 ·················································································· 241
59.2 フレーム原子吸光法 ···································································································· 243
59.3 ICP発光分光分析法 ···································································································· 244
59.4 ICP質量分析法 ·········································································································· 244
60. コバルト(Co) ··········································································································· 244
60.1 ニトロソR塩吸光光度法 ····························································································· 244
60.2 フレーム原子吸光法 ···································································································· 245
60.3 ICP発光分光分析法 ···································································································· 246
60.4 ICP質量分析法 ·········································································································· 246
61. ひ素(As) ················································································································· 246
61.1 ジエチルジチオカルバミド酸銀吸光光度法 ······································································ 246
61.2 水素化物発生原子吸光法 ······························································································ 249
61.3 水素化物発生ICP発光分光分析法 ················································································· 253
61.4 ICP質量分析法 ·········································································································· 254
62. アンチモン(Sb) ········································································································ 255
62.1 ローダミンB吸光光度法 ····························································································· 255
62.2 水素化物発生原子吸光法 ······························································································ 257
62.3 水素化物発生ICP発光分光分析法 ················································································· 259
62.4 ICP質量分析法 ·········································································································· 260
63. すず(Sn) ················································································································· 262
63.1 フェニルフルオロン吸光光度法 ····················································································· 262
63.2 ケルセチン吸光光度法 ································································································· 264
63.3 ICP発光分光分析法 ···································································································· 265
63.4 ICP質量分析法 ·········································································································· 266
64. ビスマス(Bi)············································································································ 266
64.1 よう化物抽出吸光光度法 ······························································································ 266
64.2 ICP発光分光分析法 ···································································································· 267
64.3 ICP質量分析法 ·········································································································· 268
65. クロム(Cr) ·············································································································· 268
65.1 全クロム ··················································································································· 268
K 0102:2016 目次
(8)
ページ
65.2 クロム(VI)[Cr(VI)] ····························································································· 272
66. 水銀(Hg)················································································································· 276
66.1 全水銀 ······················································································································ 276
66.2 アルキル水銀(II)化合物 ···························································································· 285
67. セレン(Se) ·············································································································· 287
67.1 3,3'-ジアミノベンジジン吸光光度法················································································ 287
67.2 水素化合物発生原子吸光法 ··························································································· 289
67.3 水素化合物発生ICP発光分光分析法 ·············································································· 291
67.4 ICP質量分析法 ·········································································································· 291
68. モリブデン(Mo) ······································································································· 292
68.1 チオシアン酸吸光光度法 ······························································································ 292
68.2 ICP発光分光分析法 ···································································································· 293
68.3 ICP質量分析法 ·········································································································· 293
69. タングステン(W) ····································································································· 293
69.1 チオシアン酸吸光光度法 ······························································································ 294
69.2 ICP発光分光分析法 ···································································································· 295
69.3 ICP質量分析法 ·········································································································· 295
70. バナジウム(V) ········································································································· 295
70.1 N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法 ··················································· 296
70.2 フレーム原子吸光法 ···································································································· 297
70.3 電気加熱原子吸光法 ···································································································· 297
70.4 ICP発光分光分析法 ···································································································· 298
70.5 ICP質量分析法 ·········································································································· 298
71. 魚類による急性毒性試験 ······························································································· 298
72. 細菌試験 ···················································································································· 302
72.1 欠番 ························································································································· 302
72.2 一般細菌 ··················································································································· 302
72.3 大腸菌群数 ················································································································ 302
72.4 従属栄養細菌 ············································································································· 302
72.5 全細菌 ······················································································································ 302
72.6 レジオネラ ················································································································ 302
73. ウラン(U) ··············································································································· 302
73.1 ICP発光分光分析法 ···································································································· 303
73.2 ICP質量分析法 ·········································································································· 304
附属書1(参考)補足 ·········································································································· 313
附属書2(参考)JISと対応する国際規格との対比表 ································································· 345
日本工業規格 JIS
K 0102:2016
工場排水試験方法
Testing methods for industrial wastewater
1. 適用範囲 この規格は,工場(事業所を含む。以下,同じ。)から排出される排水の試験方法について
規定する。
備考 1. この規格で規定する試験方法のうち,対応国際規格がある場合,その対応国際規格及びその
対応の程度を表す記号を,該当する試験方法ごとに該当箇条に示す。
なお,対応国際規格の技術的内容を変更している箇所は,変更の一覧表に説明を付けて附
属書2に示す。
2. 付表1に示す引用規格は,この規格に引用されることによってこの規格の規定の一部を構成
する。これらの引用規格は,その最新版(追補を含む。)を適用する。
2. 共通事項 共通事項は,次による。
a) 通則 化学分析に共通する一般事項は,JIS K 0050による。
b) 定義 この規格で用いる主な用語の定義は,JIS K 0101,JIS K 0211又はJIS K 0215による。
c) ガスクロマトグラフ法 ガスクロマトグラフ法に共通する一般事項は,JIS K 0114による。
d) 吸光光度法 吸光光度法に共通する一般事項は,JIS K 0115による。
e) 誘導結合プラズマ発光分光分析法 誘導結合プラズマ発光分光分析法(以下,ICP発光分光分析法と
いう。)に共通する一般事項は,JIS K 0116による。
f)
高周波プラズマ質量分析法 高周波プラズマ質量分析法(以下,ICP質量分析法という。)に共通する
一般事項は,JIS K 0133による。
g) 赤外分光法 赤外分光法に共通する一般事項は,JIS K 0117による。
h) 原子吸光法 原子吸光法には,フレーム原子吸光法,電気加熱方式原子吸光法(以下,電気加熱原子
吸光法という。),その他の原子吸光法がある。これらに共通する一般事項は,JIS K 0121による。
i)
イオン電極法 イオン電極法に共通する一般事項は,JIS K 0122による。
j)
イオンクロマトグラフ法 イオンクロマトグラフ法に共通する一般事項は,JIS K 0127による。
k) 流れ分析法 流れ分析法に共通する一般事項は,JIS K 0126による。
l)
定量範囲 それぞれの試験方法の定量範囲は,最終溶液中の質量(mg,μg又はng)で示す。ただし,
原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオンクロマトグラフ法,イ
オン電極法,流れ分析法,有機体炭素(TOC),全酸素消費量(TOD),溶存酸素及び残留塩素の試験
方法においては,最終溶液中の濃度(mg/L又はμg/L)で示す。
なお,アルキル水銀(II)化合物については,試料中の濃度(水銀としての濃度)で示す。
m) 繰返し精度 繰返し精度は,それぞれの試験方法で,定量範囲内で使用する標準液を用い,繰返し試
験で求めた変動係数(%)の概略値で示す。
n) 水 この規格で用いる水は,JIS K 0557に規定するA1〜A4の水とする。ただし,試験項目中で規定
2
K 0102:2016
している場合には,それに従う。溶存酸素を含まない水及び二酸化炭素を含まない水は,次による。
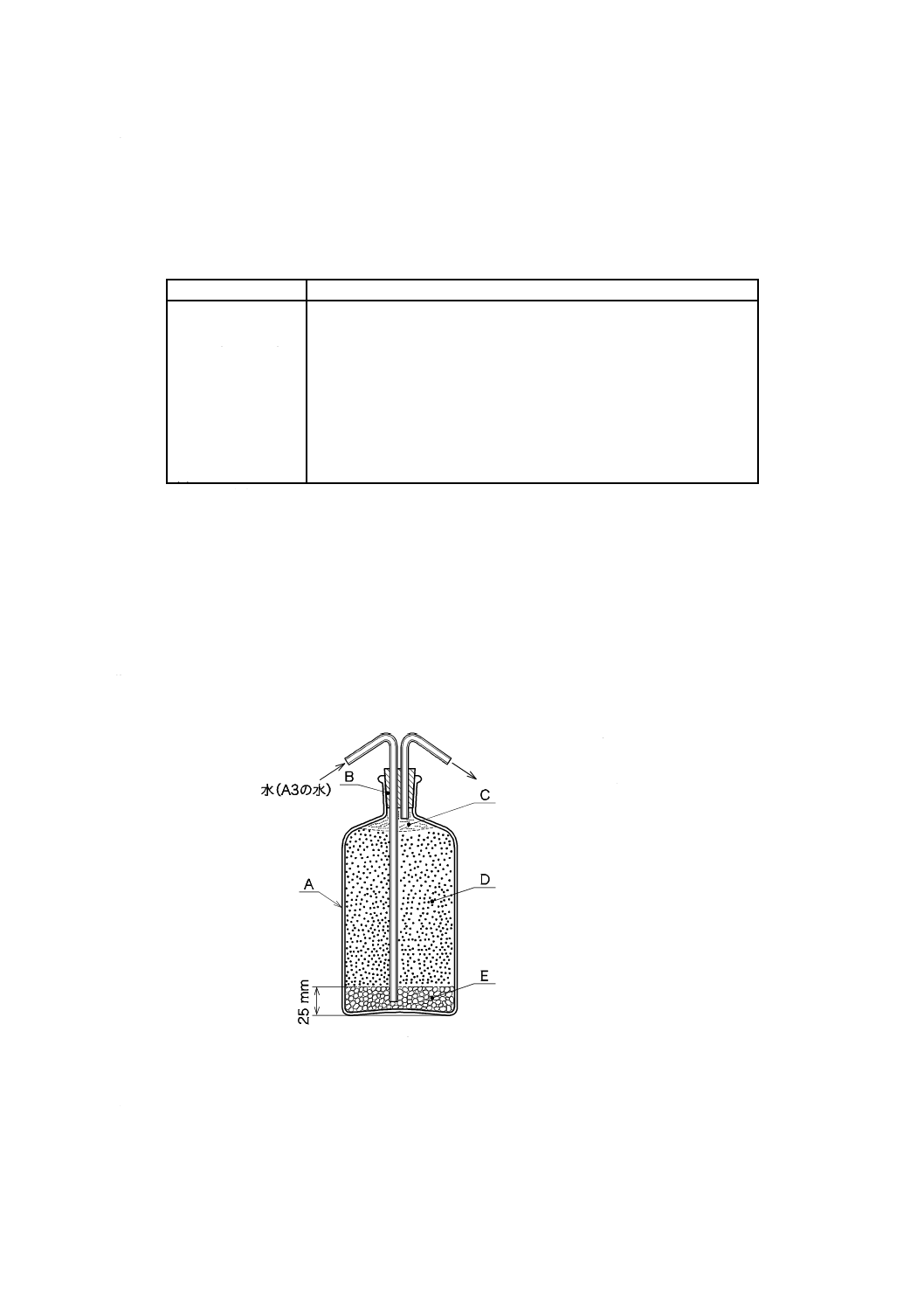
1) 溶存酸素を含まない水[JIS K 0050の附属書E(特殊用途の水の調製方法及び保存方法)参照。] 溶
存酸素を除いた水の調製方法は,次の1.1)〜1.5)のいずれか,又はそれらの二つ以上を組み合わせた
ものを用い,使用時に調製する。保存する場合は,図2.1のようにアルカリ性ピロガロール溶液(1)
を入れたガス洗浄瓶を連結し,空気中の酸素を遮断して保存する。
1.1) JIS K 0557に規定するA2又はA3の水をフラスコに入れ,約5分間煮沸して溶存酸素を除去した
後,図2.1のようにアルカリ性ピロガロール溶液(1)を入れたガス洗浄瓶を連結して,空気中の酸素
を遮断して放冷する[JIS K 0557の4.(種別及び質)備考3.(溶存酸素を含まない水)参照]。
1.2) JIS K 0557に規定するA2又はA3の水をフラスコに入れ,JIS K 1107に規定する窒素2級を約15
分間通気して溶存酸素を除去する[JIS K 0557の4.(種別及び質)備考3.(溶存酸素を含まない
水)参照]。
1.3) JIS K 0557に規定するA2又はA3の水を,酸素分離膜を用いたガス分離管に通水し,溶存酸素を
除去する。
1.4) JIS K 0557に規定するA2又はA3の水を,超音波振動装置で十分脱気を行い,溶存酸素を除去す
る。
1.5) JIS K 0557に規定するA2又はA3の精製直後の水を,JIS K 1107に規定する窒素2級を通じた三
角フラスコに泡立てないように採取したもの。
注(1) JIS K 8780に規定するピロガロール(1,2,3-ベンゼントリオール)6 gを水50 mLに溶かし,着
色瓶に保存する。別に,JIS K 8574に規定する水酸化カリウム30 gを水50 mLに溶かす。使用
時に両液を混合する。この溶液1 mLは,酸素約12 mL(約17 mg)を吸収する。
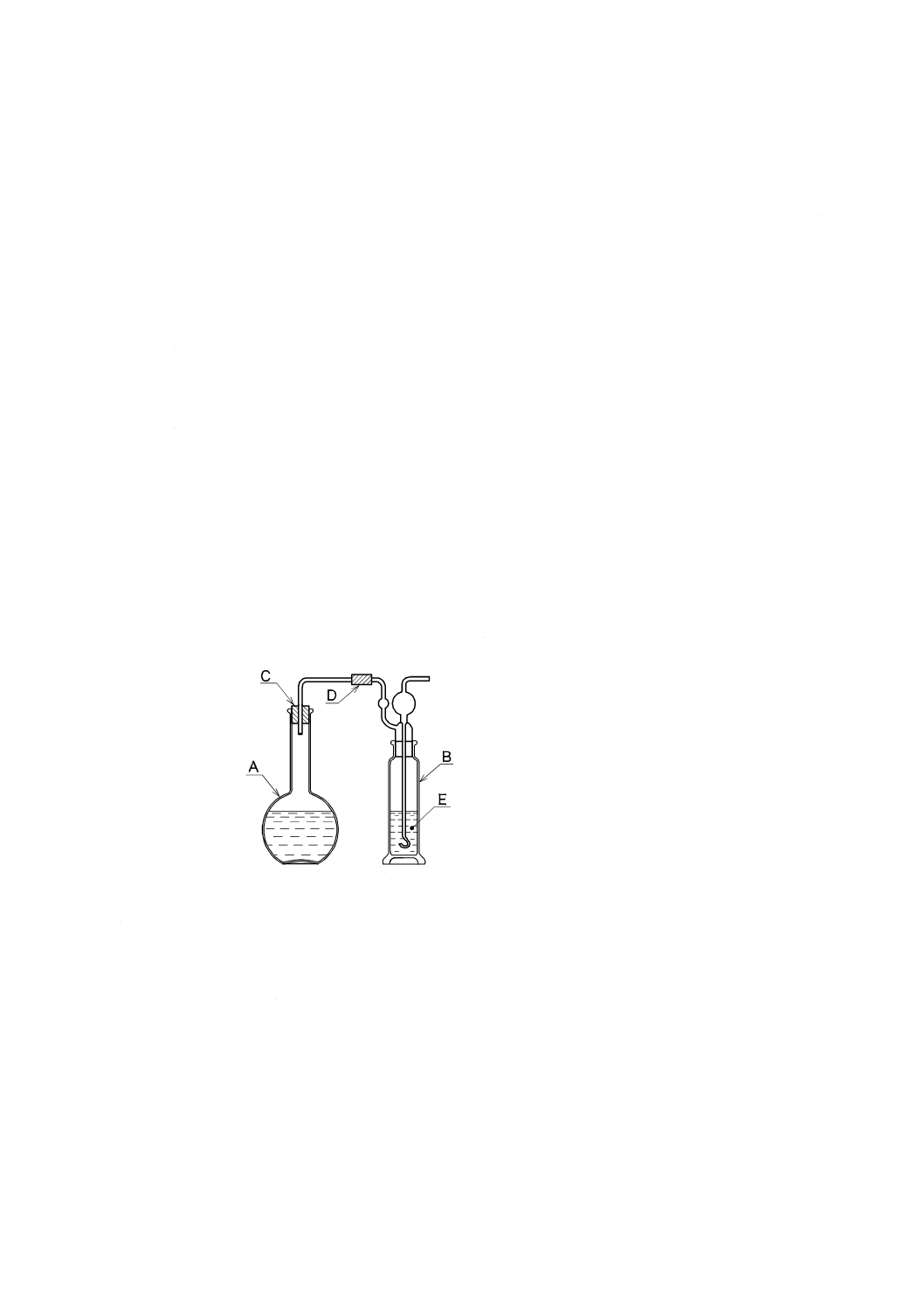
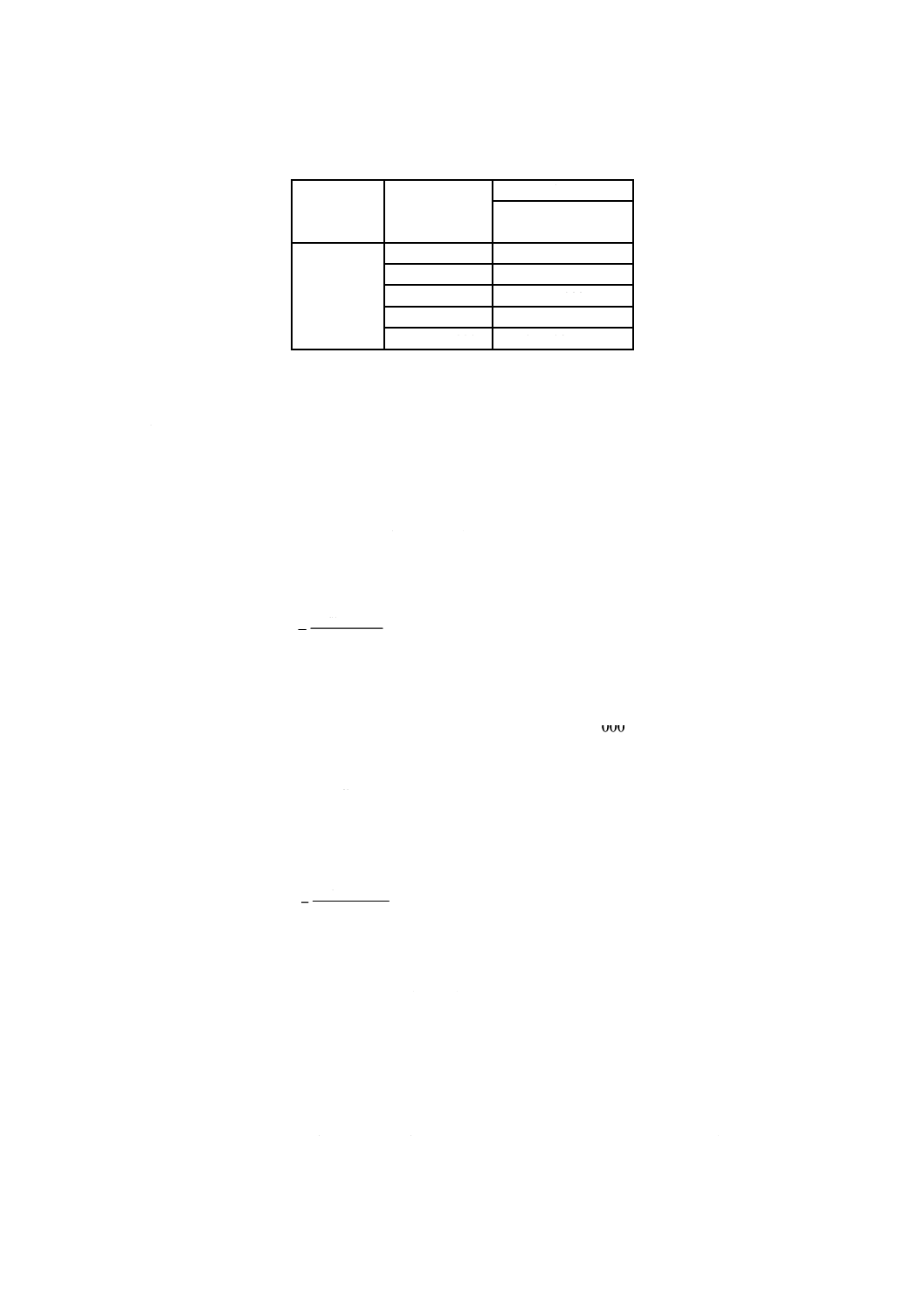
A:平底フラスコ 1 000 mL
B:ガス洗浄瓶 250 mL
C:ゴム栓
D:ゴム管
E:アルカリ性ピロガロール溶液
図2.1 溶存酸素を含まない水の冷却,保存の例
2) 二酸化炭素を含まない水[JIS K 0050の附属書E(特殊用途の水の調製方法及び保存方法)参照。]
二酸化炭素を除いた水の調製方法は,次の2.1)〜2.4)のいずれか,又はそれらの二つ以上を組み合
わせたものを用い,使用時に調製する。保存する場合は,図2.1と同様の装置を用い,ガス洗浄瓶
に水酸化カリウム溶液(250 g/L)(JIS K 8574に規定する水酸化カリウムを用いて調製する。)又は
JIS K 8603に規定するソーダ石灰の二酸化炭素吸収用1号を入れ,空気中の二酸化炭素を遮断して
保存する。
2.1) JIS K 0557に規定するA2又はA3の水をフラスコに入れ,約5分間煮沸して溶存気体及び二酸化
炭素を除去した後,図2.1と同様の装置を用い,ガス洗浄瓶に水酸化カリウム溶液(250 g/L)又
はJIS K 8603に規定するソーダ石灰の二酸化炭素吸収用1号を入れ,空気中の二酸化炭素を遮断
して放冷する[JIS K 0557の4.(種別及び質)備考4.(炭酸を含まない水)参照]。
3
K 0102:2016
2.2) 1.2) と同じ操作を行い,二酸化炭素を除去する。
2.3) JIS K 0557に規定するA2又はA3の水を,二酸化炭素分離膜を用いたガス分離管に通水し,二酸
化炭素を除去する。
2.4) 1.4) と同じ操作を行い,二酸化炭素を除去する。
o) 試薬
1) 試薬は,日本工業規格(以下,JISという。)に規定するもので,試験に支障のないものを用いる。
JISに規定のない場合は,試験に支障のないものを用いる(2)。
滴定液類の標定には,JIS K 8005に規定する容量分析用標準物質のある場合には,それを用いる。
注(2) 電気加熱原子吸光法,ICP質量分析法など,ごく微量の試験には,特に高純度の試薬を用いる。
2) 標準液は,各試験項目で調製方法を規定するもののほか,国家計量標準(計量法第134条)に規定
するトレーサビリティが確保されたもの又はそれを一定濃度に薄めたものを用いる(3)。
注(3) 調製に用いた化合物,添加してある酸などの種類及び濃度が試験に支障のないもの。
参考 トレーサビリティが確保された試薬としては,計量法校正事業者登録制度(Japan Calibration
Service System)によるJCSSマークを付けたものがある。
3) 試薬類の溶液の濃度は,特に断らない限り質量濃度はg/L又はmg/L,モル濃度はmol/L又はmmol/L
で示す。
なお,化合物の質量は,名称の後に括弧で示し,無水物としての値を用いる。
標準液の濃度は,1 mL中の質量(mg/mL,μg/mL又はng/mL)で表す。ただし,残留塩素測定の
塩素標準液,イオン電極法及びフレーム光度法に用いる標準液の濃度は,1 L中の質量(mg/L)で
表す。
4) 試薬類の溶液名称の後に括弧で示す濃度は,標準液以外は,概略の濃度であることを意味する。例
えば,水酸化ナトリウム溶液(0.1 mol/L)は,約0.1 mol/Lの水酸化ナトリウム溶液であることを示
す。また,液体試薬の濃度は,水との混合比[試薬(a+b)]で表す。この表し方は試薬a mLと水
b mLとを混合したことを示す。
なお,溶液名の前に示される濃度は,正確な濃度を意味する。ただし,一般には,端数のない数
値で示し,別にファクターを求めておく。
5) 試薬類の調製に用いる水は,n)の水とする。
6) 標準液を薄めて低濃度の標準液を調製するような場合には,特に断らない限り,10 mL以上の全量
ピペットでとる。
7) 試薬類の名称は,国際純正及び応用化学連合(IUPAC)の無機化学命名法及び有機化学命名法を基
にして,公益社団法人日本化学会が定めた化合物命名法及びJISに規定する試薬の名称と,できる
だけ整合させている。
8) 試薬類,廃液類などによる室内汚染,人体への吸入,付着などに注意する。また,その取扱いにつ
いては,関係法令,規則などに従う。
p) 器具類 この規格で用いるガラス器具,磁器るつぼ,磁器蒸発皿,白金るつぼ,白金皿及びろ紙は,
次による。
1) ガラス器具は,特に断らない限り,JIS R 3503及びJIS R 3505に規定するものを用いる。ただし,
特殊な器具を必要とする場合には,それぞれの試験項目に,その例を図示又は説明する。また,加
熱操作を伴う場合には,JIS R 3503に規定するほうけい酸ガラス−1を用いる。
デシケーターに用いる乾燥剤は,特に断らない限り,JIS Z 0701に規定する包装用シリカゲル乾
4
K 0102:2016
燥剤A形1種を用いる。
2) 磁器るつぼ及び磁器蒸発皿は,JIS R 1301及びJIS R 1302に規定するものを使用する。
3) 白金るつぼ及び白金皿は,JIS H 6201及びJIS H 6202に規定するものを使用する。
4) ろ紙は,JIS P 3801に規定する定量分析用を使用する。ただし,ろ紙の種類は,それぞれの試験項
目で規定する。
備考 シリカ,ほう素,ナトリウム,カリウム,ひ素,亜鉛などを試験する場合には,ほうけい酸ガ
ラスからのこれらの成分の溶出に十分に注意する。
q) ガラス器具類などの洗浄 この規格で用いる全てのガラス器具類,磁器るつぼ,磁器蒸発皿などは,
試験の前に次の洗浄操作を行う。ただし,それぞれの試験項目に規定している場合は,それによる。
1) 金属元素の試験に用いる場合は,A2の水で洗浄した後,硝酸(1.5 mol/L)(JIS K 8541に規定する
硝酸を用いて調製する。)又は塩酸(1.5 mol/L)(JIS K 8180に規定する塩酸を用いて調製する。)に
1日間以上浸し,再びA2の水で洗浄した後,A3の水で洗浄する。
2) 金属元素以外の試験に用いる場合は,A2の水で洗浄した後,更にA3の水で洗浄する。
r) 吸光度の測定(吸光光度法) 吸収セルは,特に記載がない場合には,光路長10 mmのものを用いる。
s)
検量線[吸光光度法,原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオン
クロマトグラフ法,イオン電極法,有機体炭素(TOC),全酸素消費量(TOD),全窒素の熱分解法,
流れ分析法] 検量線の作成には,試験方法に示す定量範囲内を4〜6段階に分け,これに一致するよ
うに標準液をとる。ただし,試験項目で示されている場合は,それに従う。
検量線は,定量範囲内について作成する。
原子吸光法,フレーム光度法,ICP発光分光分析法,ICP質量分析法,イオンクロマトグラフ法,
イオン電極法,有機体炭素(TOC),全酸素消費量(TOD),全窒素の熱分解法及び流れ分析法の試験
においては,新たに作成した検量線を用い,同一項目を多数の試料について連続して試験する場合に
は,試験の途中において,適宜,標準液を用いて濃度指示値の確認を行う。
吸光光度法においては,あらかじめ作成した検量線を用いることができる。
t)
注,備考,図,表及び式 これらは,試験項目ごとに一連の番号を付ける。
u) 試験結果の質の管理 試験結果の質の管理のため,それぞれの試験方法におけるトレーサビリティが
保証された標準物質,又はそれを用いて調製した検量線用標準液を用いて,定期的にその測定値の精
確さ(真度及び精度)を評価することが望ましい。トレーサビリティが保証された標準物質がない場
合は,それぞれの試験方法で使用する標準物質,検量線用標準液を用いる。また,これらの標準物質,
検量線用標準液を用いた添加回収試験などによって定期的にその測定値の精確さを評価することが望
ましい。
3. 試料
3.1
試料の採取,試料容器,採水器及び採取操作 試料とは,試験を行うために採取した水をいう。試
料の採取,試料容器,採水器及び採取操作は,JIS K 0094に従う。
3.2
試料の取扱い 試験は,特に断らない限り,試料中に含まれる全量について行う。このため,試料
に懸濁物がある場合には,十分に振り混ぜて均一にした後,試料を採取して試験に用いる。ただし,陰イ
オンの試験では,特に断らない限り,ろ過した試料を用いる。全量を求める場合には,それぞれの試験項
目で規定する。
溶存状態のものだけを試験する場合には,試料採取後,直ちにろ紙5種C (1)でろ過し,初めのろ液約50
5
K 0102:2016
mLを捨て,その後のろ液を試料とする。
注(1) ろ紙6種又は孔径1 μm以下のろ過材を用いてもよい。ただし,溶存マンガン及び溶存鉄の試験
では,ろ紙5種Cを用いる。その他,ろ過方法が示されている場合は,それに従う。
3.3
試料の保存処理 試験は,特に断らない限り,試料採取後,直ちに行う。直ちに試験ができずに保
存する場合は,JIS K 0094の7.(試料の保存処理)に従って,次のように行い,なるべく早く試験する。0 ℃
付近に保存する場合には,凍結させないようにする。また,試験項目に保存方法が示されている場合には,
それに従う。
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 塩酸(ひ素分析用) JIS K 8180に規定するもの。
3) 硝酸 JIS K 8541に規定するもの。
4) 硫酸 JIS K 8951に規定するもの。
5) りん酸 JIS K 9005に規定するもの。
6) L(+)-アスコルビン酸 JIS K 9502に規定するもの。
7) 水酸化ナトリウム溶液(200 g/L) JIS K 8576に規定する水酸化ナトリウム20 gを水に溶かして,
100 mLとする。
8) 塩基性炭酸亜鉛懸濁液 JIS K 8953に規定する硫酸亜鉛七水和物20 gを水100 mLに溶かし,これ
と等体積の炭酸ナトリウム溶液(100 g/L)(JIS K 8625に規定する炭酸ナトリウムを用いて調製す
る。)とを混合する。使用時に調製する。
9) 硫酸銅(II)五水和物 JIS K 8983に規定するもの。
10) クロロホルム JIS K 8322に規定するもの。
b) 保存処理 保存処理は,次による。
1) 100 ℃における過マンガン酸カリウムによる酸素消費量(CODMn),アルカリ性過マンガン酸カリウ
ムによる酸素消費量(CODOH),二クロム酸カリウムによる酸素消費量(CODCr),生物化学的酸素
消費量(BOD),有機体炭素(TOC),全酸素消費量(TOD),及び界面活性剤の試験に用いる試料
は,0〜10 ℃の暗所に保存する。
2) アンモニウムイオン,有機体窒素及び全窒素の試験に用いる試料は,塩酸又は硫酸を加え,pH2〜3
とし,0〜10 ℃の暗所に保存する。短い日数であれば,保存処理を行わずそのままの状態で0〜10 ℃
の暗所に保存してもよい。
3) 亜硝酸イオン及び硝酸イオンの試験に用いる試料は,試料1 Lにつきクロロホルム約5 mLを加えて
0〜10 ℃の暗所に保存する。短い日数であれば,保存処理を行わずそのままの状態で0〜10 ℃の暗
所に保存してもよい。
4) よう化物イオン及び臭化物イオンの試験に用いる試料は,水酸化ナトリウム溶液(200 g/L)を加え
てpH約10として保存する(試料1 Lにつき水酸化ナトリウム2〜4 粒を加えてもよい。)。
5) シアン化合物及び硫化物イオンの試験に用いる試料は,水酸化ナトリウム溶液(200 g/L)を加えて
pH約12として保存する(試料1 Lにつき水酸化ナトリウム4〜6粒を加えてもよい。)。シアン化合
物の試験に用いる試料で,残留塩素など酸化性物質が共存する場合は,L(+)-アスコルビン酸を
加えて還元した後,pH約12とする。
硫化物イオンの試験には,試料を溶存酸素測定瓶に採取し,試料100 mLにつき塩基性炭酸亜鉛
懸濁液約2 mLを加え,硫化亜鉛として固定して保存してもよい(39.の備考2.参照)。
6
K 0102:2016
6) フェノール類の試験に用いる試料は,りん酸を加えてpH約4とし,試料1 Lにつき硫酸銅(II)五
水和物1 gを加えて振り混ぜ,0〜10 ℃の暗所に保存する。
7) 農薬[パラチオン,メチルパラチオン,EPN,ペンタクロロフェノール及びエジフェンホス(EDDP)]
の試験に用いる試料は,塩酸を加え弱酸性として保存する。
8) りん化合物及び全りんの試験に用いる試料は,試料1 Lにつきクロロホルム約5 mLを加えて0〜
10 ℃の暗所に保存する。短い日数であれば,保存処理を行わずに0〜10 ℃の暗所に保存してもよ
い。ただし,溶存りん化合物の試験に用いる試料は,3.2によってろ過した後,試料1 Lにつきクロ
ロホルム約5 mLを加え,0〜10 ℃の暗所に保存する。短い日数であれば,ろ過後,保存処理を行
わずに0〜10 ℃の暗所に保存してもよい。
全りんの試験に用いる試料は,硫酸又は硝酸を加えてpH約2として保存してもよい。
9) 銅,亜鉛,鉛,カドミウム,マンガン,鉄,アルミニウム,ニッケル,コバルト,ひ素,アンチモ
ン,すず,ビスマス,クロム,水銀,セレン,モリブデン,タングステン,バナジウムなどの金属
元素の試験に用いる試料は,硝酸を加えてpH約1として保存する。
ひ素,アンチモン及びセレンの試験に用いる試料で,有機物及び多量の硝酸イオン並びに亜硝酸
イオンを含まず,試験において硫酸及び硝酸,又は硝酸及び過マンガン酸カリウムによる前処理を
行わない場合には,塩酸(ひ素分析用)を加えてpH約1として保存する。
クロム(VI)の試験に用いる試料は,そのままの状態で0〜10 ℃の暗所に保存する。
溶存状態の金属元素の試験に用いる試料は,3.2によってろ過した後,硝酸を加えてpH約1とし
て保存する。
4. 流量 流量の測定は,JIS K 0094の8.(流量の測定)による。
5. 試料の前処理 試料の前処理操作は,各試験項目で規定するが,金属元素の試験における前処理操作
は,金属元素の種類に関係なく共通するものがほとんどであるため,一括して次に規定する。ただし,金
属元素のうちナトリウム,カリウム,カルシウム,マグネシウム,ひ素,クロム(VI),水銀,溶存マン
ガン及び溶存鉄の試験の前処理は,それぞれの試験項目において規定する。
金属元素の試験の前処理は,主として共存する有機物,懸濁物及び金属錯体の分解を目的としている。
前処理には,試料に各種の酸を加えて加熱する方法を用いるが,試料の状態及び試験の種類によって適
切な方法を選択する。
5.1
塩酸又は硝酸酸性で煮沸 この方法は,有機物及び懸濁物が極めて少ない試料に適用する。
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 硝酸 JIS K 8541に規定するもの。
b) 操作 操作は,次による。
1) 試料(1) 100 mLにつき塩酸5 mL又は硝酸5 mLを加える。
2) 加熱して約10分間静かに煮沸する。
3) 放冷後,必要に応じて水で一定量にする。
注(1) 溶存状態の金属元素を試験する場合には,3.2によってろ過した試料を用いる。
5.2
塩酸又は硝酸による分解 この方法は,有機物が少なく,懸濁物として水酸化物,酸化物,硫化物,
りん酸塩などを含む試料に適用する。
7
K 0102:2016
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 硝酸 JIS K 8541に規定するもの。
b) 操作 操作は,次による。
1) 試料(2)をよく振り混ぜた後,直ちにビーカーにとり,試料100 mLにつき塩酸5 mL又は硝酸5 mL
を加える。
2) 加熱して液量が約15 mLになるまで濃縮する。
3) 不溶解物が残った場合には,ろ紙5種Bでろ過した後,水でよく洗浄する。
4) 放冷後,ろ液と洗液とを適切な容量の全量フラスコに移し入れ,水を標線まで加える。
注(2) 溶存状態の金属元素を試験する場合には,3.2によってろ過した試料を用い,5.1の方法を適用
する。
備考 塩酸と硝酸との混酸による分解が有利な試料の場合には,2)までの操作を行った後,室温まで
放冷する。1)で,塩酸を使用したときは硝酸5 mLを,硝酸を使用したときは塩酸5 mLを加え,
時計皿で覆い,再び加熱し,激しい反応が終わったら時計皿を取り除き,更に加熱して窒素酸
化物を追い出し,約5 mLになるまで濃縮する。この操作で酸が不足している場合は,適量の
塩酸又は硝酸を加え,同じ操作で加熱して溶かす。不溶解物が残った場合は,温水15 mLを加
え,3)及び4)の操作を行う。
5.3
硝酸と過塩素酸とによる分解 この方法は,酸化されにくい有機物を含む試料に適用する。
a) 試薬 試薬は,次による。
1) 過塩素酸 JIS K 8223に規定するもの。
2) 硝酸 JIS K 8541に規定するもの。
b) 操作 操作は,次による。
1) 試料(2)をよく振り混ぜた後,直ちにその適量をビーカー又は磁器蒸発皿にとる。
2) 硝酸5〜10 mLを加え,加熱板上で静かに加熱して約10 mL (3)になるまで濃縮し,放冷する。
3) 硝酸5 mLを加え,次に過塩素酸(4) 10 mLを少量ずつ加え,加熱を続け,過塩素酸の白煙が発生し
始めたら,時計皿で容器を覆い,過塩素酸が器壁を流下する状態に保って有機物を分解する。
4) 有機物が分解しないで残ったときは,更に硝酸5 mLを加えて3)の操作を繰り返す。
5) 放冷後,水を加えて液量を約50 mLに薄め,不溶解物が残った場合には,ろ紙5種Bを用いてろ過
し,水で洗い,ろ液と洗液とを適切な容量の全量フラスコに移し入れ,水を標線まで加える。
注(3) ケルダールフラスコに移して分解してもよい。
(4) 過塩素酸を用いる加熱分解操作は,試料の種類によっては爆発の危険性があるため,次の事項
に注意する。
− 酸化されやすい有機物は,過塩素酸を加える前に,2)の操作によって十分に分解しておく。
− 過塩素酸の添加は,必ず濃縮液を放冷した後に行う。
− 必ず過塩素酸と硝酸とを共存させた状態で,加熱分解を行う。
− 濃縮液を乾固させない。
5.4
硝酸と硫酸とによる分解 この方法は,多種類の試料に適用(5)することができる。
a) 試薬 試薬は,次による。
1) 硝酸 JIS K 8541に規定するもの。
2) 硫酸(1+1) 水1容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
8
K 0102:2016
酸1容を徐々に加える。
b) 操作 操作は,次による。
1) 試料(2)をよく振り混ぜ,直ちにその適量をビーカー又は磁器蒸発皿にとり,硝酸5〜10 mLを加え
る。
2) 加熱して,液量が約10 mL (3)になったら,再び硝酸5 mLと硫酸(1+1)10 mLとを加え,硫酸の
白煙が発生し,有機物が分解するまで加熱する。
3) 有機物の分解が困難な場合は,更に硝酸10 mLを加えて2)の操作を繰り返す。
4) 放冷後,水で液量を約50 mLに薄める。不溶解物(6)が残った場合には,ろ紙5種Bを用いてろ過し,
水で洗い,ろ液と洗液とを適切な容量の全量フラスコに移し入れ,水を標線まで加える。
注(5) 水溶液をそのまま噴霧するフレーム原子吸光法を適用する場合には,好ましくない。
(6) 鉛が含まれていて沈殿を生じる場合には,5.3又は次の操作を行う。
2)の操作を行って溶液をほとんど蒸発乾固し,水約30 mLとJIS K 8180に規定する塩酸15 mL
とを加えて加熱して溶かす。不溶解物がある場合には,ろ紙5種Bを用いてろ過した後,温塩
酸(1+10)(JIS K 8180に規定する塩酸を用いて調製する。)で洗浄する。放冷後,ろ液及び洗
液を適切な容量の全量フラスコに移し入れ,水を標線まで加える。
5.5
フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法及びICP質量分析法を適用する場
合の前処理 試料に含まれている有機物及び懸濁物の量,その存在状態及び適用しようとする原子吸光法,
ICP発光分光分析法,ICP質量分析法などの方法を十分に考慮して5.1〜5.4の方法のうち最適なものを選
択して前処理する(7) (8)。
調製した試料をそのまま噴霧する場合において,フレーム原子吸光法又はICP発光分光分析法を適用す
る場合には,特に断らない限り,試料は塩酸又は硝酸酸性(9),電気加熱原子吸光法及びICP質量分析法を
適用する場合は,硝酸酸性とし,適切な濃度(10)に調節する。
注(7) フレーム原子吸光法又はICP発光分光分析法において,溶媒抽出法を適用する場合の前処理は,
特に断らない限り,各試験項目のとおりとし,妨害する可能性のある有機物その他の妨害物質
を十分に分解する。
フレーム原子吸光法又はICP発光分光分析法において,溶媒抽出法を適用せずに試料を噴霧
する場合には,次に示す前処理によってもよい。
有機物及び懸濁物が極めて少ない試料の場合は,5.1の操作を行う。有機物又は懸濁物を含む
試料の一般的な前処理方法としては,5.3又は5.4を適用する。この場合,白煙を十分に発生さ
せて大部分の硫酸及び過塩素酸を除去しておく。
電気加熱原子吸光法及びICP質量分析法の場合は,酸の種類及び濃度によっては空試験値が
無視できないことがあるので,測定する元素についてあらかじめその影響について調べておく。
いずれの前処理方法を適用するかは,試料に一定量の目的成分を添加して回収試験を行い,
その結果に基づいて判断するとよい。
(8) 2.の注(2)による。高純度の試薬には,JIS K 9901に規定する高純度試薬−硝酸,JIS K 9902に
規定する高純度試薬−塩酸,JIS K 9904に規定する高純度試薬−過塩素酸,JIS K 9905に規定
する高純度試薬−硫酸などがある。
(9) ICP発光分光分析法の場合,硫酸酸性では,試料導入量が少なく感度が悪くなることがあるの
で,5.4の適用はやむを得ない場合だけとする。
(10) フレーム原子吸光法及び電気加熱原子吸光法の場合には,0.1〜1 mol/L,ICP発光分光分析法及
9
K 0102:2016
びICP質量分析法においては,すず及びアンチモンを対象としない場合,0.1〜0.5 mol/Lとする。
また,すず及びアンチモンを対象とする場合には,1〜1.5 mol/Lとする。ただし,いずれの場
合も,検量線作成時の場合とほぼ同じ濃度とする。
6. 結果の表示 試験方法が二つ以上ある場合には,試験方法を付記する。
7. 温度 気温と水温とに分け,試料採取時に測定する。
7.1
気温 気温は,次によって測定する。
a) 器具 器具は,次による。
1) 温度計 JIS B 7411-1に規定する一般用ガラス製棒状温度計の50度温度計
b) 操作 操作は,次による。
1) 採水現場において直射日光及び周囲の強い熱放射を避けた風通しのよい場所で,温度計を地上1.2
〜1.5 mの位置に保ち,感温液の止まるときの目盛を読み取る。
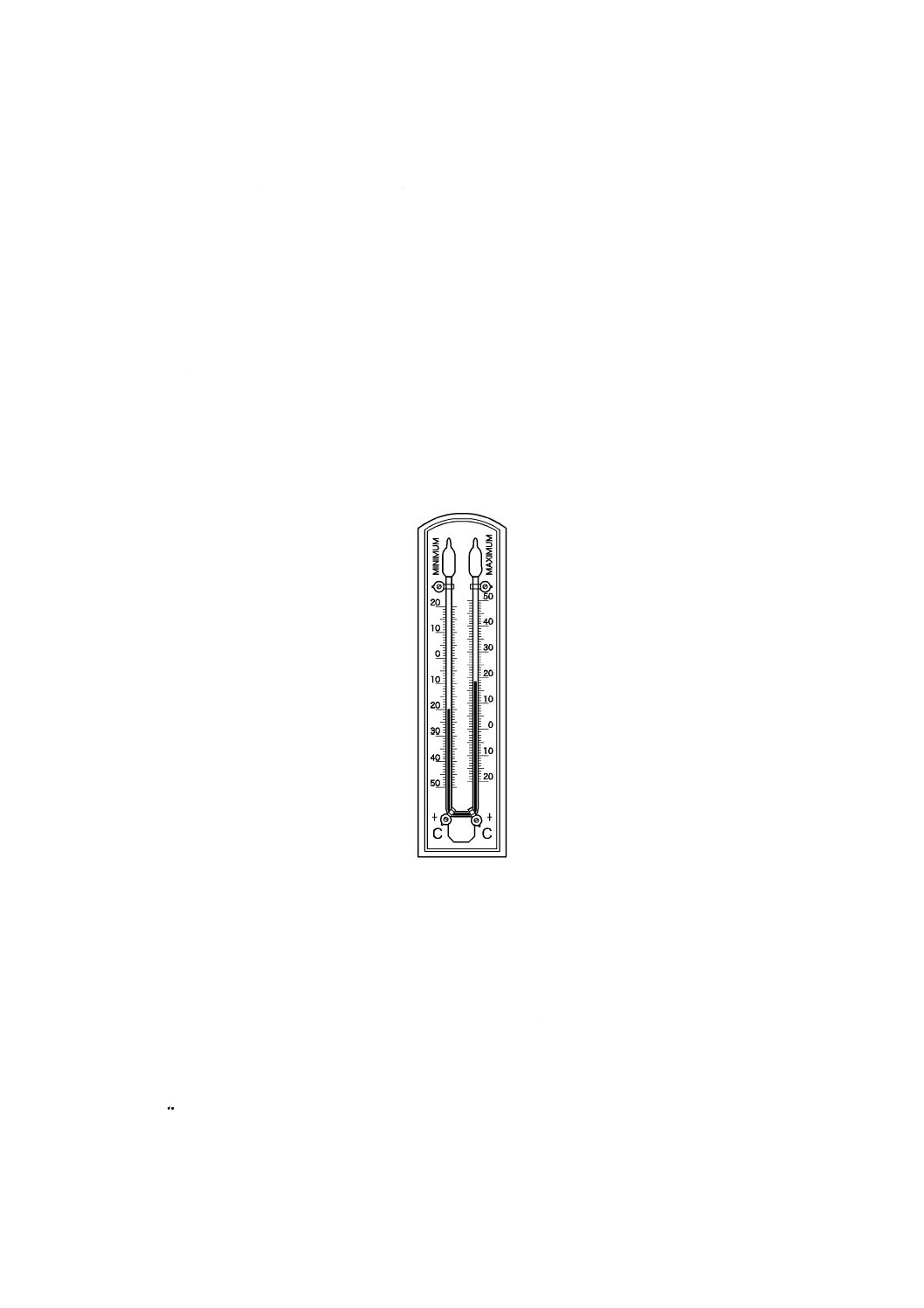
備考 1. 測定期間内の最高温度及び最低温度の測定には,最高最低温度計(図7.1参照)を使用する。
図7.1 最高最低温度計の例
7.2
水温 水温は,次によって測定する。
a) 器具 器具は,次による。
1) 温度計 JIS B 7411-1に規定する一般用ガラス製棒状温度計の50度温度計又は100度温度計
b) 操作 操作は,次による。
1) 温度計を現場の水に直接差し入れるか,採取直後の試料(1)の中に差し入れて感温液の止まる目盛付
近まで浸没した状態に保ち,感温液の止まるときの目盛を読み取る。
注(1) 容器及び外気の温度の影響を避けるため,多量の試料を採取する。
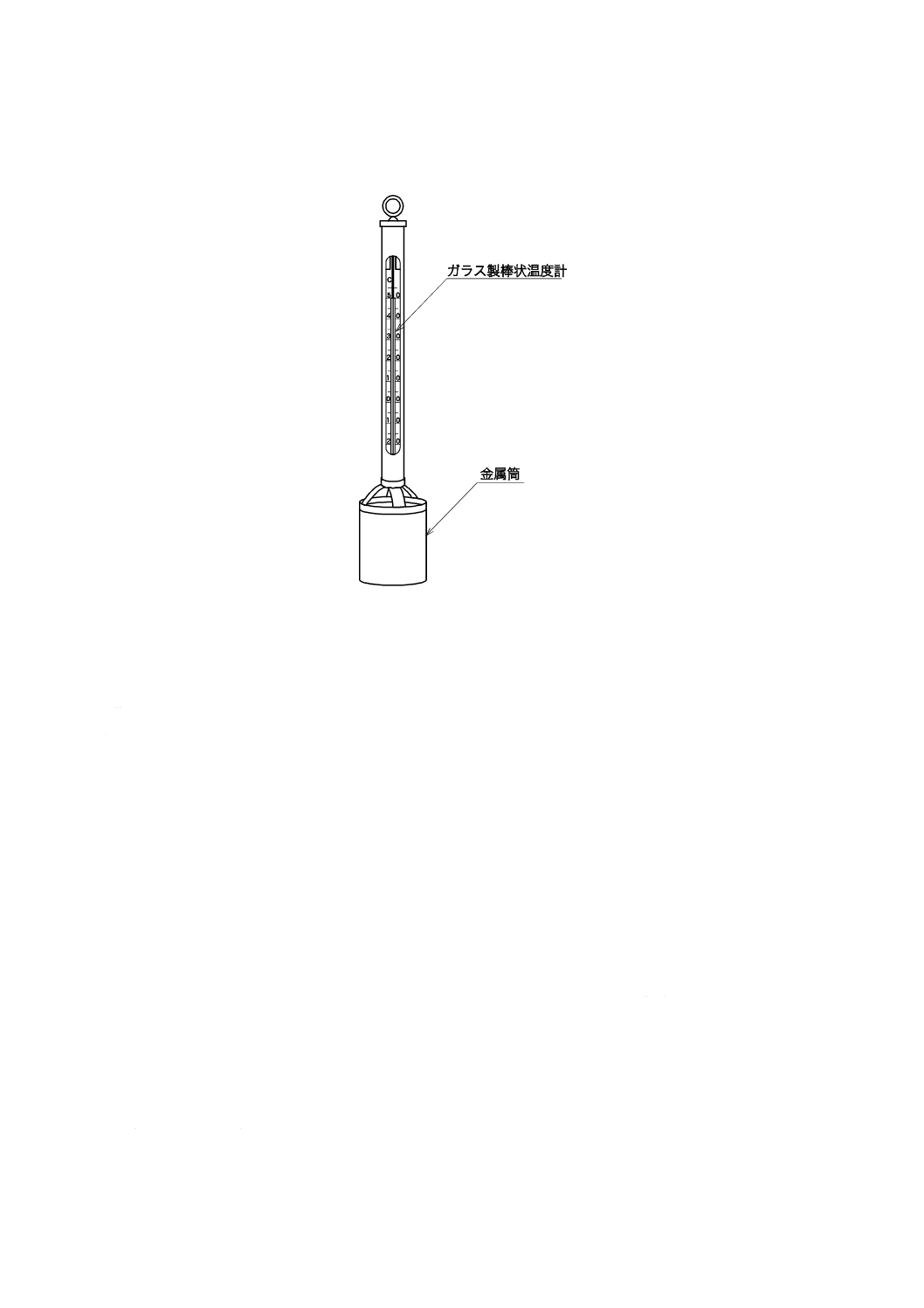
備考 2. ペッテンコーヘル水温計(図7.2参照)を用いる場合には,現場の水に水温計を投入して試
料をくみ上げ,金属筒内に試料を3回入れ替えた後,試料を満たし,感温液の止まるときの
目盛を読み取る。
10
K 0102:2016
備考 3. サーミスター温度計及び金属抵抗温度計は,温度検出部を測定する水中に保ち,指示部の指
針が一定したときの目盛を読み取る。
図7.2 ペッテンコーヘル水温計
8. 外観 外観は,採取直後の試料について観察する。
a) 器具 器具は,次による。
1) ビーカー 300〜500 mL(無色のもの。)
b) 操作 操作は,次による。
1) 採水直後の試料をビーカーにとり,次の事項について肉眼で観察する。
− 試料全体の色の種類及びその程度
− 上澄み液の色の種類及びその程度
− 浮上物,懸濁物などの色の種類及びその量の程度
− 油類,タール類などの状態及びその程度
− その他,試料の泡立ち,臭気など特異な状態
9. 透視度 試料の透明の程度を示すもので,透視度計に試料を入れて上部から透視し,底部に置いた標
識板の二重十字が初めて明らかに識別できるときの水層の高さをはかり,10 mmを1度として表す。
なお,備考2.に示す方法は,1990年に第2版として発行されたISO 7027との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 7027:1990,Water quality−Determination of turbidity(MOD)
測定範囲:1〜30度
a) 器具 器具は,次による。
11
K 0102:2016
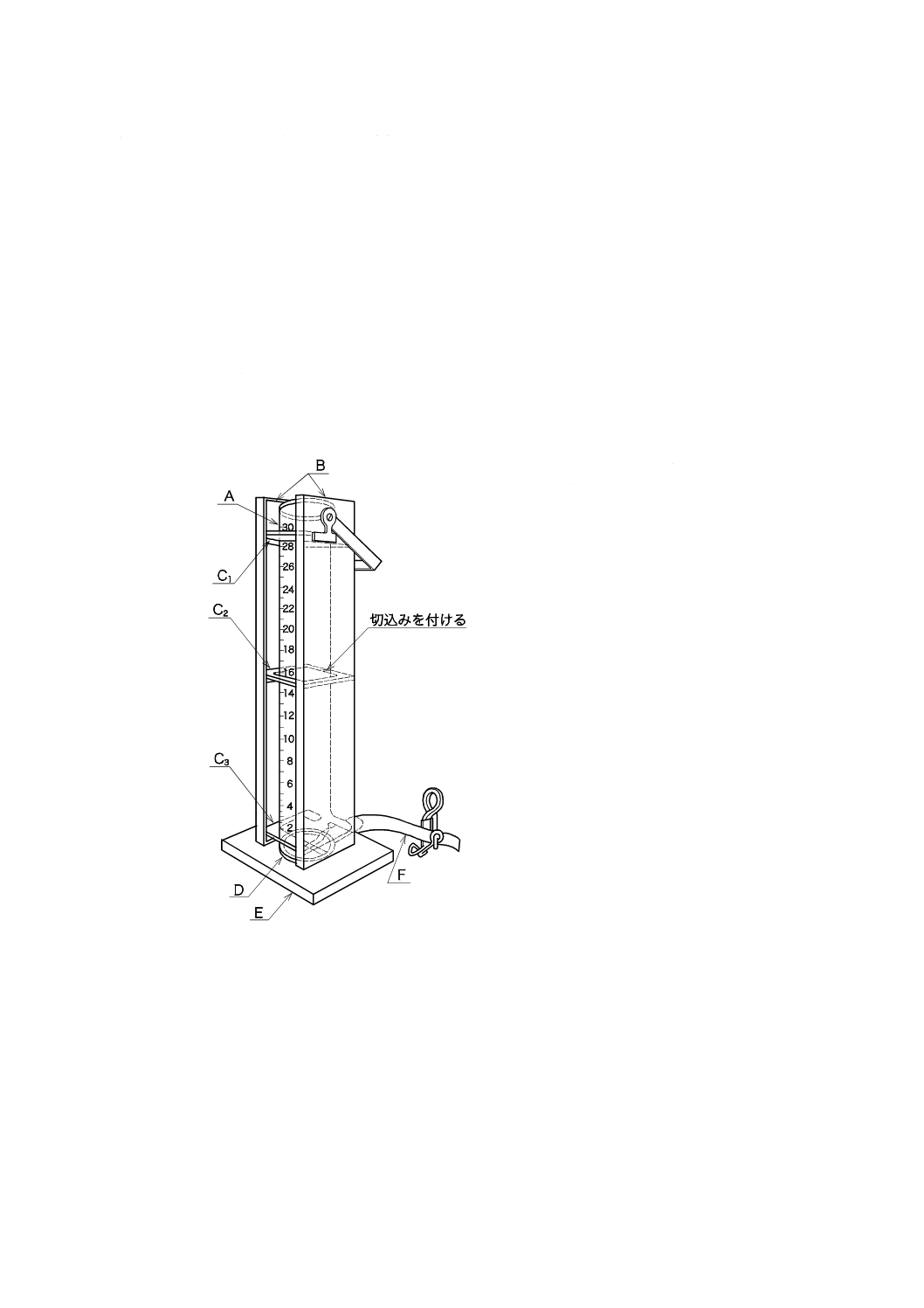
1) 透視度計 図9.1に例を示す。標識板の上側から50 mmの高さまでは5 mmごとに,50〜300 mmま
では10 mmごとに,目盛を施した下口付きのガラス製のもの。底部に図9.2のような標識板を入れ
て用いる。
b) 操作 操作は,次による。
1) よく振り混ぜた試料を透視度計に満たし,上部から底部を透視し,標識板の二重十字が初めて明ら
かに識別できるまで,ゴム管のピンチコックをゆっくり緩めながら下口から試料を速やかに流出さ
せたとき(1)の水面の目盛を読み取る。
2) 1)の操作を2,3回繰り返し,水面の目盛を読み取り,平均値を求め,透視度として度で表す。
注(1) 懸濁物の多い試料の場合には,これが透視度計の底部に沈積することがあり,誤差の原因とな
るので注意する。
備考 1. 同じ照度でも光源の違いによって彩度が異なる場合は,透視度が変わる。光源は昼光とし,
直射日光を避ける。
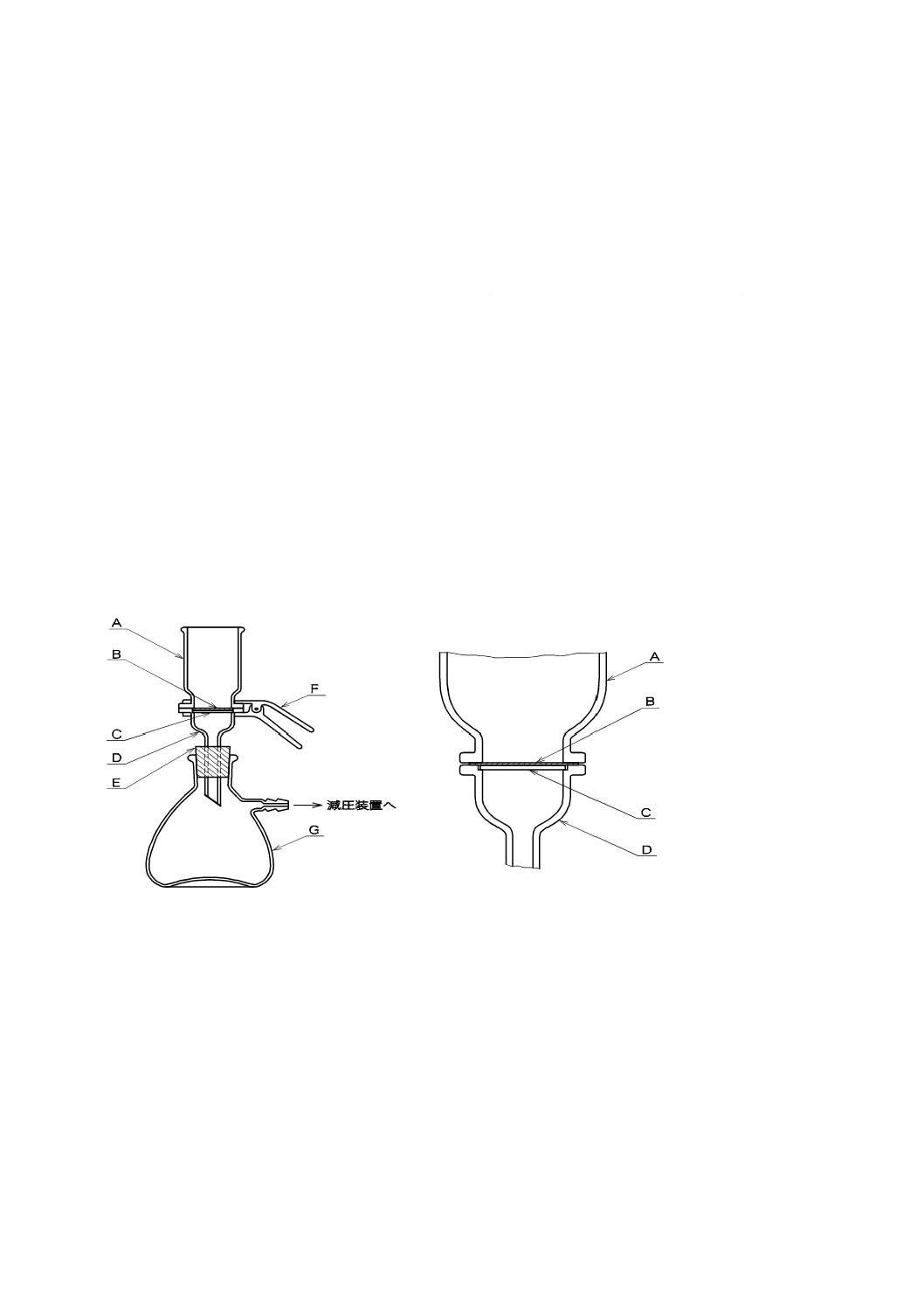
A:
B:
C1〜C3:
D:
E:
F:
下口付きシリンダー
遮蔽用黒板
シリンダー支持枠
標識板
台
ピンチコック付きゴム管
図9.1 透視度計の例
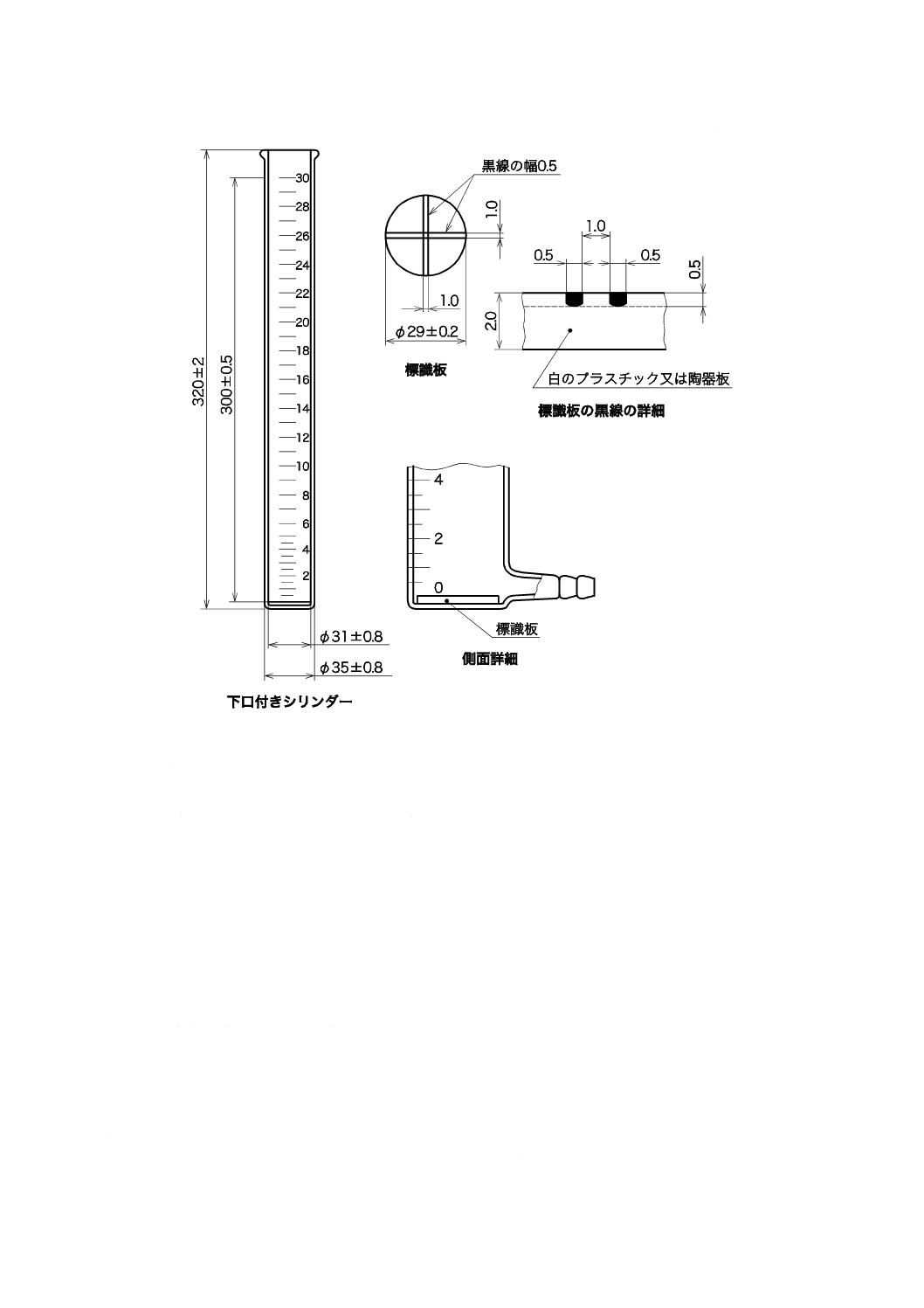
12
K 0102:2016
単位 mm
図9.2 透視度計の詳細図
備考 2. 図9.2に示す下口付きシリンダーのほかに,長さ600±10 mm,内径25±1 mmで,10 mmご
とに目盛を付けたものなどを用いてもよい。
参考 市販品には,材質がアクリル樹脂製のもの,長さが1 mのものもある。
10. 臭気及び臭気強度(TON) 臭気の試験は,臭気と臭気強度(TON)(1)とに区分する。
水の臭気は,細菌,藻類,微生物などの繁殖及び死滅,都市下水,畜舎排水及び工場排水の混入,貯水
槽及び配管系統の内面処理物質の溶出,塩素処理による残留塩素などの影響による。
臭気の試験は,嗅覚によるので,個人差が大きく,さらに,温度,湿度,測定者の食事及び喫煙などに
も影響される。
注(1) TONは,Threshold Odor Numberの略称で,臭気いき(閾)値の希釈倍数,すなわち,明らかに
臭気を感じるときの希釈の倍数値である。
10.1 臭気 試料を約40 ℃に温め,臭気の種類及びその程度を試験する。
a) 器具 器具は,次による。
1) 共栓三角フラスコ 300 mL
b) 操作 操作は,次による。
1) 試料200 mLを共栓三角フラスコ300 mLにとり,軽く栓をして約40 ℃に温める。
13
K 0102:2016
2) 共栓三角フラスコを揺り動かしながら栓をとり,直ちに臭気の有無,臭気の種類及びその程度を試
験する。
3) 臭気の表示は,表10.1に倣い,試料の臭気の種類及びその程度を,概略の理解ができるように表示
する。
表10.1 臭気の分類及び種類の例
臭気の大分類
臭気の種類
(1) 芳香性臭気
メロン臭,すみれ臭,にんにく臭,きゅうり臭,芳香臭,薬味臭など
(2) 植物性臭気
藻臭,青草臭,木材臭,海藻臭など
(3) 土臭,かび臭
土臭,沼沢臭,かび臭など
(4) 魚貝臭
魚臭,肝油臭,はまぐり臭など
(5) 薬品性臭気
フェノール臭,タール臭,油臭,油脂臭,パラフィン臭,塩素臭,硫
化水素臭,クロロフェノール臭,薬局臭,薬品臭など
(6) 金属性臭気
かなけ臭,金属臭など
(7) 腐敗性臭気
ちゅうかい臭,下水臭,豚小屋臭,腐敗臭など
(8) 不快臭
魚臭,豚小屋臭,腐敗臭などが強烈になった不快な臭い
備考 1. 嗅覚の個人差を少なくするため,同一試料を数人で試験するとよい。
2. 試料採取時に試料を温めずにその臭気を試験し,記録しておくとよい(これを,冷
時臭という。)。
10.2 臭気強度(TON) 臭気の強さを表すもので,約40 ℃に保った水に試料を加え,明らかに臭気を感
じるときの希釈の倍数値[臭気いき(閾)値の希釈倍数]で表す。嗅覚の個人差を少なくするため,同一
試料について少なくとも5人,できれば10人程度で試験する。
a) 試薬 試薬は,次による。
1) 臭気のない水 図10.1に示すような装置にJIS K 0557に規定するA3の水を約5 L/(L-活性炭・h)
で通す。
A:ガラス瓶(5 000 mL)
B:ゴム栓
C:ガラスウール
D:粒状活性炭
E:砂利(3〜5 mm)
図10.1 臭気のない水の作り方の例
b) 器具 器具は,次による。
1) 共栓三角フラスコ 300 mL
2) 足長ビュレット 50 mL
3) 水浴 温度調節器の付いたもの。
14
K 0102:2016
c) 予備試験 予備試験は,次による。
1) 試料200 mL,40 mL,10 mL及び4 mLをそれぞれ共栓三角フラスコ300 mLにとり,臭気のない水
を加えて200 mLとし,予備試験の試料とする。
2) 別に,対照水として臭気のない水200 mLを共栓三角フラスコ300 mLにとる。
3) 予備試験の試料と対照水とを水浴上で40〜50 ℃に温めた後,対照水を振り混ぜ,開栓と同時に発
生する臭いを嗅ぐ。
4) 次に,試料の量の少ない方から,同様に操作して予備試験の試料の臭いを対照水と比較し,臭いが
感知できる最少の試料の量(mL)を求める。
d) 操作 操作は,次による。
1) c) 4)で求めた試料の量から表10.2によって試験に用いる試料の量を縦系列に示すmL数として求め
る。
2) 1)で求めた試料の各量をそれぞれ別の共栓三角フラスコ300 mLにとり,臭気のない水を加えて200
mLとし,これをこの試験の試料とする。
3) 次に,c) 2)〜4)と同様に操作して臭いを感知できる最少の試料のmL数を求め,次の式によって臭
気強度(TON)を算出する。
V
TON
200
=
ここに,
V: 試験に用いた試料量(mL)
表10.2 試験に用いる試料の量
単位 mL
予備試験の試料の量
200
40
10
4
試験に用いる試料の量
200
40
10
4.0
100
28.5
8.0
2.9
67
20
6.7
2.0
50
13.3
5.7
1.3
40
10
4.0
1.0
備考 3. 試料の臭気が強すぎるときは,試料を臭気のない水で10倍に薄めてから予備試験及び
試験の操作を行う。
4. 試験に用いる共栓三角フラスコは,あらかじめ,臭気のない水でよく洗浄しておく。
5. 試験は,環境に左右されることが多いので,臭いのない静かな室内で行う。
6. 試験直前の喫煙,喫茶,食事などは避け,更に手に石けん,ローション,香水などの
香りがないようにする。
7. 試験を続けて行うと,4,5回ぐらいで嗅覚が鈍るから,15〜30分間程度休憩する。
8. 臭気度(pO)を求める場合には,次の式による。
TON
TON
pO
log
32
.3
log
2
log
1
×
=
×
=
11. 色度 試料の色を表示する方法として,刺激値及び色度座標を用いる方法並びに三波長を用いる方法
を適用する。
なお,三波長を用いる方法は,1994年に第2版として発行されたISO 7887との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
15
K 0102:2016
(修正している),NEQ(同等でない)とする。
ISO 7887:1994,Water quality−Examination and determination of colour(MOD)
11.1 刺激値及び色度座標を用いる方法 水を対照液として波長400〜700 nmの所定の波長における試料
についての透過パーセントを測定して,三刺激値(1) X,Y,Zを算出し,次に,色度座標x,yを算出し,
試料の色を刺激値Y及び色度座標x,yによって表示する。
注(1) 三刺激値とは,色に対する視神経の感じ方が3種類あると考え,その3種類の視神経に対する
刺激の混合の割合によって色に対する感じ方が変わるとし,それぞれの刺激を与える波長を種
類別にX,Y,Zの系列にまとめ,透過パーセントを集計して求めた値である。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水を孔径約0.1 μmのろ過材を用いてろ過し,初めの約200 mLを
捨てた後のろ液
b) 装置 装置は,次による。
1) 遠心分離器
2) 光度計 分光光度計。吸収セル100 mmを使用できるもので,波長間隔10 nm以下で可視部全域が
測定できるものか,又はこれと同等の性能をもつ色度計
c) 操作 操作は,次による。
1) 試料をろ紙5種C又は孔径1 μm以下のろ過材でろ過するか,又は約30 000 m/s2(約3 000 g)で約
20分間遠心分離して濁りを除去する。
2) この一部を吸収セル100 mm(2)に移し,水を対照液として,表11.1 (3)の各波長における透過パーセ
ントを測定する。
注(2) 色が濃く,吸収セル100 mmで測定できない場合には,適切な光路長の吸収セルを用いて測定
する。
この場合には,所定波長における吸光度を測定し,この値から吸収セル100 mmの吸光度に
換算し,この吸光度から透過パーセントを求めて計算する。
(3) 表11.1は,JIS Z 8719に規定する付表5-1-1{波長間隔20 nmで三刺激値X,Y,Zを計算する
ための重価係数[400-(20)-700]}に従ったものである。
16
K 0102:2016
表11.1 波長間隔20 nmで三刺激値X,Y,Zを計算するための重価係数fX,fY,fZ
波長
λ(nm)
標準の光 C
fX
fY
fZ
400
0.019
−0.003
0.062
420
2.993
0.087
14.387
440
7.634
0.510
38.438
460
6.642
1.382
38.130
480
2.360
3.206
19.545
500
0.068
6.907
5.746
520
1.196
12.876
1.444
540
5.590
18.261
0.356
560
11.751
19.592
0.073
580
16.795
15.991
0.026
600
17.897
10.694
0.013
620
14.021
6.261
0.002
640
7.453
2.901
0.000
660
2.731
1.003
0.000
680
0.756
0.273
0.000
700
0.166
0.059
0.000
合計
98.072
100.000
118.225
色度座標
x=0.310 1
y=0.316 2
z=0.373 7
3) 三刺激値及び色度座標x,yの求め方 表11.1の各所定波長における透過パーセントと対応するfX,
fY,fZそれぞれの値から,次の式によって三刺激値X,Y,Z及び色度座標x,yを算出する。
()()
∑
=
700
400
X
1
λ
τ
λ
f
K
X
()()
∑
=
700
400
Y
1
λ
τ
λ
f
K
Y
()()
∑
=
700
400
Z
1
λ
τ
λ
f
K
Z
Z
Y
X
Y
x
+
+
=
Z
Y
X
Y
y
+
+
=
ここに,
X: 刺激値X
Y: 刺激値Y
Z: 刺激値Z
K: 100.000
fX(λ): 波長λでのfX
fY(λ): 波長λでのfY
fZ(λ): 波長λでのfZ
τ(λ): 波長λでの透過パーセント
4) 刺激値Yの求め方 刺激値Yの値をそのまま刺激値とする。
備考 1. 主波長及び補色主波長の求め方 色度図(図11.1)の中の点Cは,無色の色度座標でx=0.310 1,
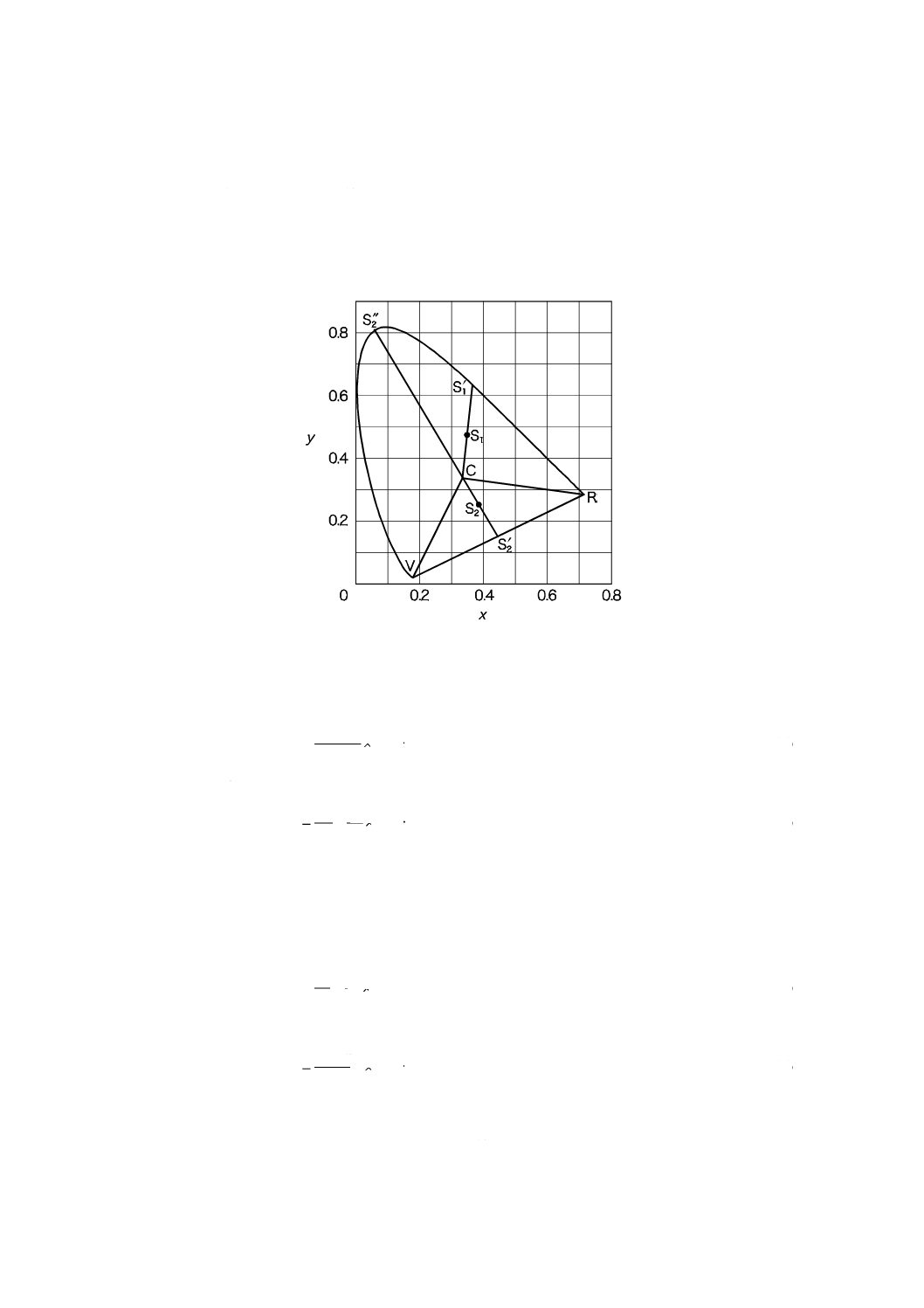
17
K 0102:2016
y=0.316 2。
色度座標が直線RC,直線VC及びスペクトル軌跡によって囲まれた面積内の点S1で表さ
れる色の場合には,直線CS1の延長とスペクトル軌跡との交点S1′に対応する波長を,図11.3
から求める。この波長をその色の主波長といい,記号λdで表す。また,色度座標が三角形CRV
内の点S2で表される色の場合には,直線CS2の延長とスペクトル軌跡との交点S2"に対応す
る波長を,図11.3から求める。この波長をその色の補色主波長といい,記号λcで表す。
図11.1 色度図
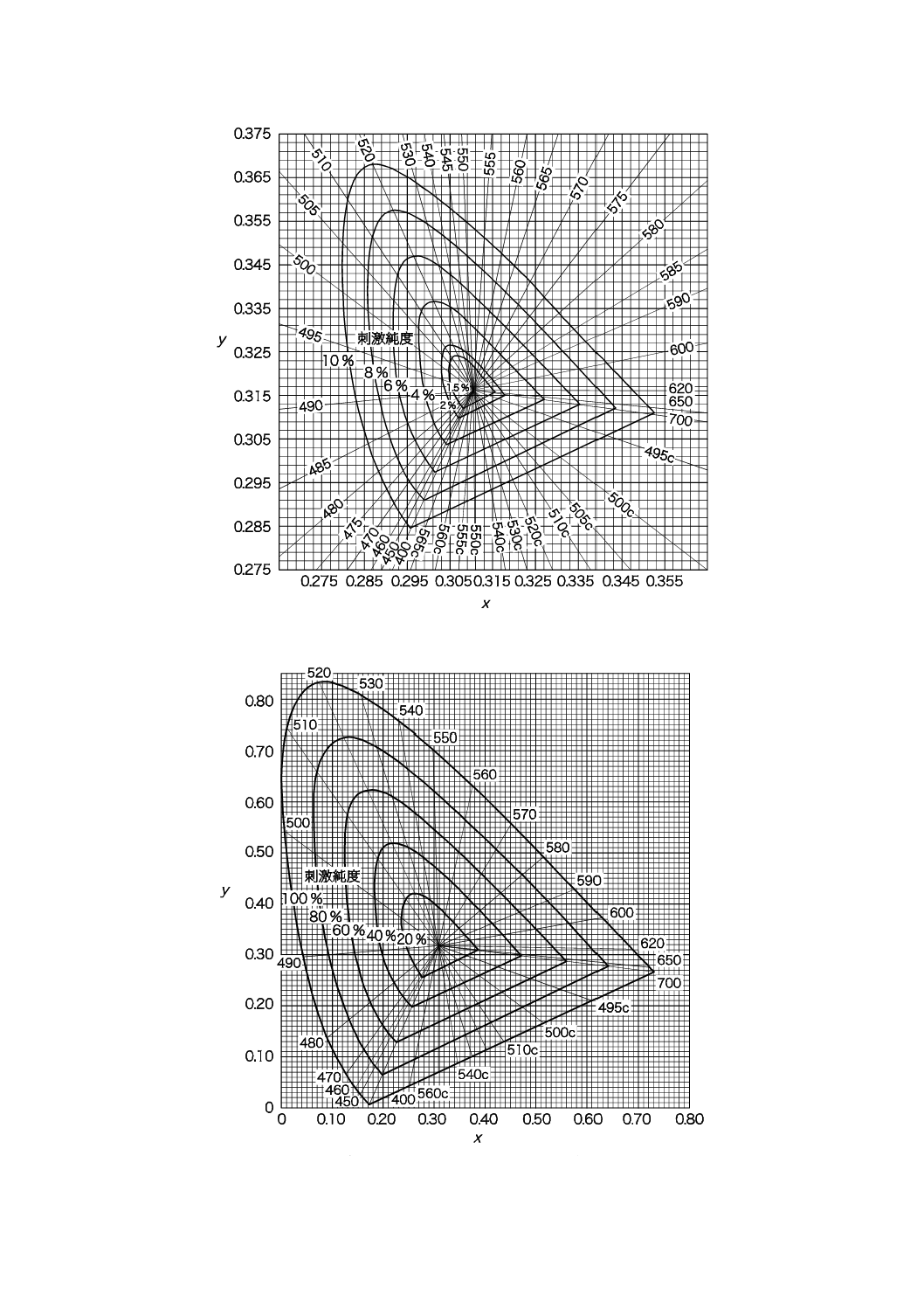
備考 2. 刺激純度の求め方 図11.1において色度座標が点S1によって表される色の場合には,刺激純
度Peは次の式によって求め,その値に%を付ける。
100
c
c×
−
−
=
x
x
x
x
Pe
λ
······································································· (1)
又は
100
c
c×
−
−
=
y
y
y
y
Pe
λ
······································································· (2)
ここに, x,y: 点S1の座標
xc,yc: 点Cの座標
xλ,yλ: 点S1'の座標
また,色度座標が点S2で表される色の場合には,刺激純度Peは,次の式によって求め,
その値に%を付ける。
100
c
p
c×
−
−
=
x
x
x
x
Pe
······································································· (3)
又は
100
c
p
c×
−
−
=
y
y
y
y
Pe
······································································· (4)
ここに, x,y: 点S2の座標
xc,yc: 点Cの座標
xp,yp: 点S2′(直線CS2の延長と純紫軌跡との交点)の座標
ただし,Peを計算するには,分母又は分子の絶対値の大きい式で求める。
18
K 0102:2016
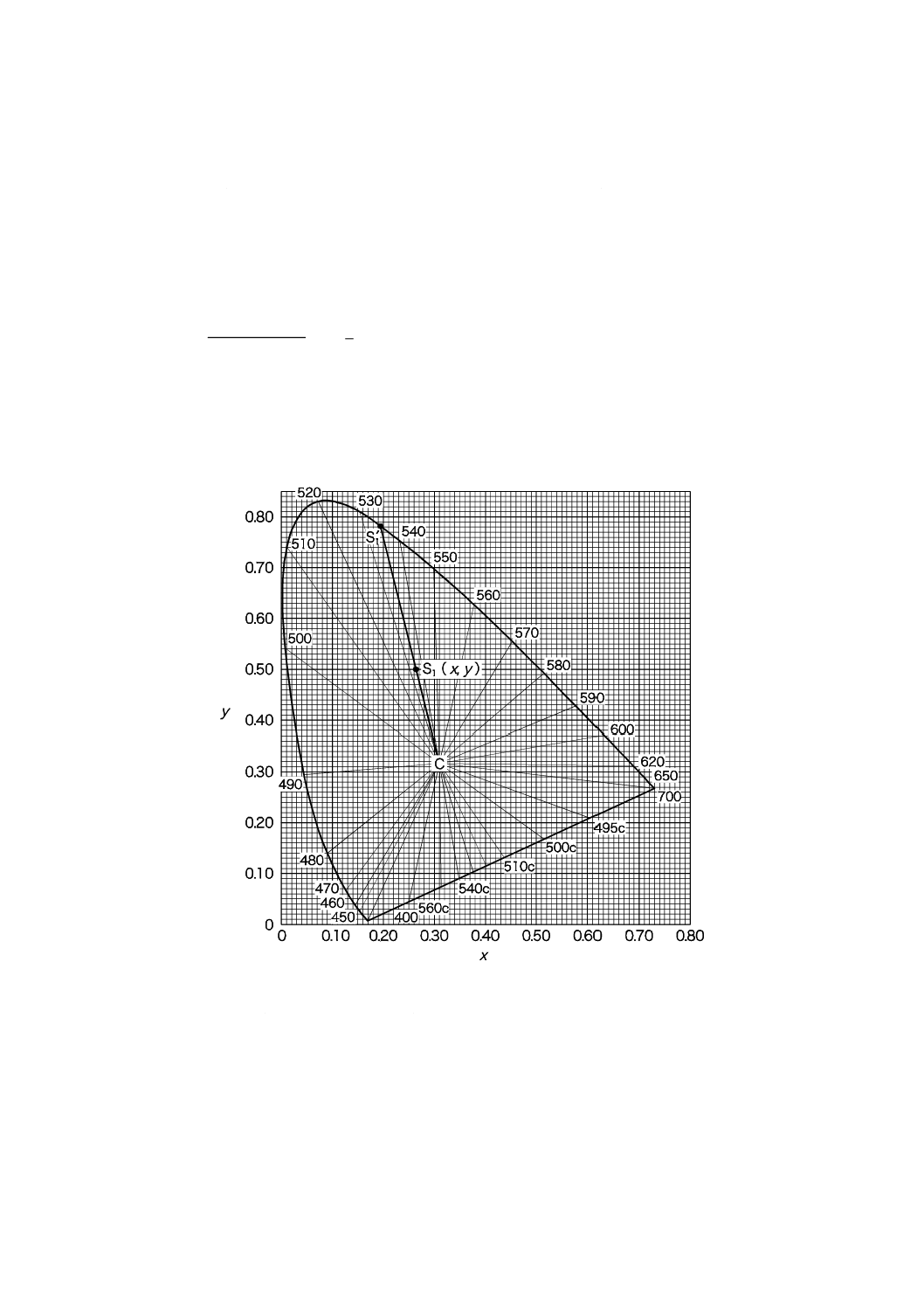
刺激純度及び主波長の求め方の例 試料を測定して得られた三刺激値Xが40.14,Yが76.50,
Zが34.25であったとすれば,色度座標はxが0.266,yが0.507で刺激値Yは76.50である。
この色度座標を図11.2上の点(S1)として求め,次に,無色の点(x=0.310 1,y=0.316 2)
をCとして求める。CとS1とを直線で結び,更に延長してスペクトル軌跡と交わる点をS1′
として求める。S1'は,スペクトル軌跡上の534 nmの位置にある。すなわち,主波長λdは534
nmである。また,図11.2から,S1'の座標xλが0.200,yλが0.785であるので,式(2)によって
刺激純度Peは,
%
70
.
40
100
2
316
.0
785
.0
2
316
.0
507
.0
=
×
−
−
となる。
したがって,試料の色は主波長λd=534 nm,刺激純度Peは40.70 %,刺激値Yは76.50 %と
して表示される。
なお,試料の色がうすい場合には図11.4,また,試料の色が濃い場合には,図11.5を用い
て刺激純度を求めると便利である。
図11.2 2度視野XYZ系による色度図と主波長との関係図
備考 3. 主波長及びその色相 主波長と色相との関係を,表11.2及び図11.3に示す。
波長目盛の入った曲線は,スペクトル軌跡であって,スペクトル軌跡の両端を結ぶ曲線は,
純紫軌跡である。点Cは,標準の光Cの色度座標(x=0.310 1,y=0.316 2)を表す。
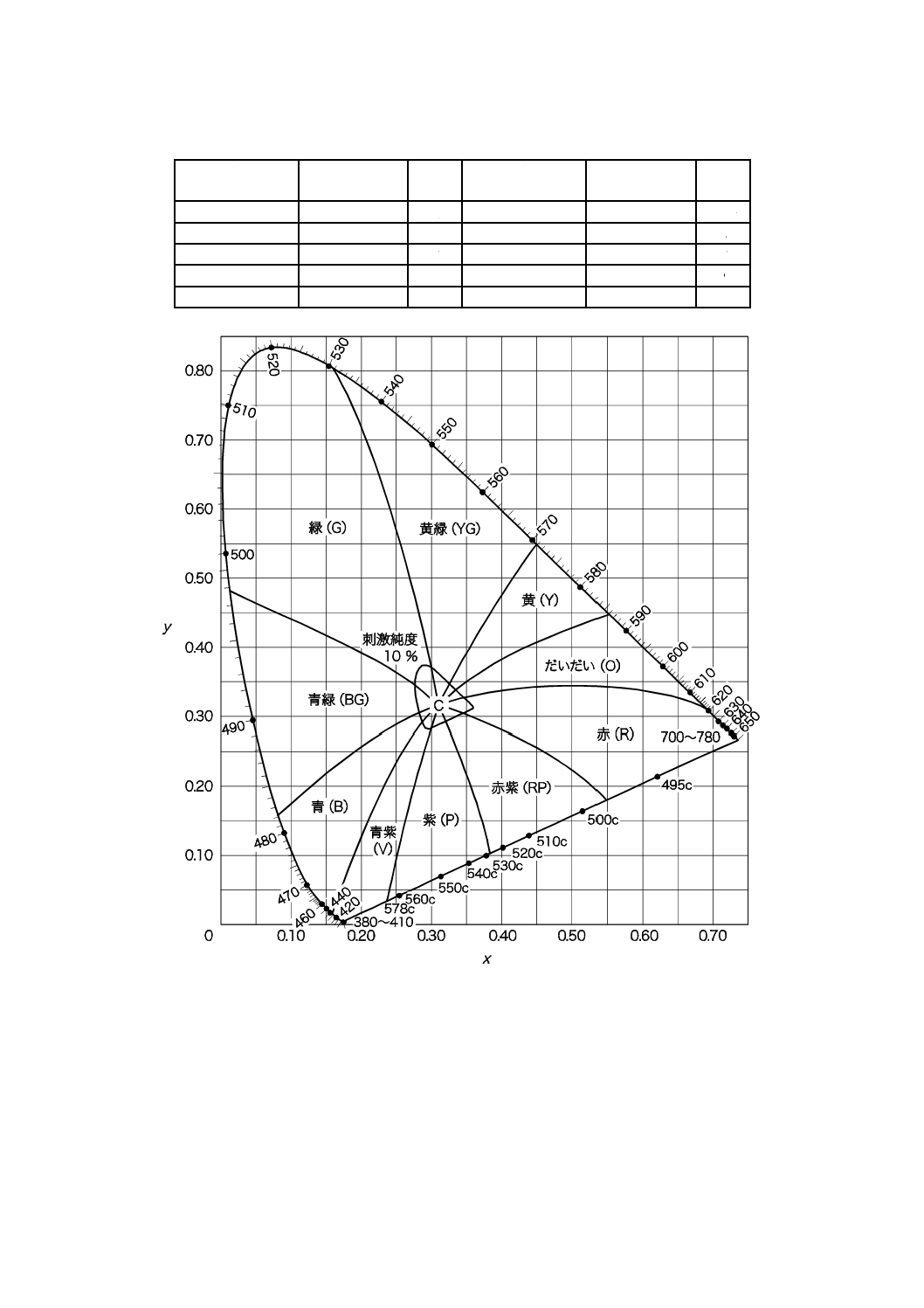
19
K 0102:2016
表11.2 主波長及びその色相名
主波長
nm
色相名
略号
主波長
nm
色相名
略号
498c〜700〜618
赤
R
498〜482
青緑
BG
618〜586
だいだい(橙) O
482〜435
青
B
586〜571
黄色
Y
435〜400〜578c
青紫
V
571〜531
黄緑
YG
578c〜528c
紫
P
531〜498
緑
G
528c〜498c
赤紫
RP
図11.3 2度視野XYZ系による色度図(主波長及びその色相名)
20
K 0102:2016
図11.4 刺激純度(10 %表示)(試料の色がうすい場合)
図11.5 刺激純度(100 %表示)(試料の色が濃い場合)
21
K 0102:2016
11.2 三波長を用いる方法 試料をメンブレンフィルターでろ過し,波長436 nm,525 nm及び620 nmで
吸光度を測定し,それぞれの吸収係数を算出し,それぞれ求めた吸収係数で色を表示する方法。
a) 試薬 試薬は,次による。
1) 水 11.1 a) 1)による。
b) 装置 装置は,次による。
1) 光度計 11.1 b) 2)による。
2) pH計 JIS Z 8802に規定する形式IIを用いる。
3) 温度計 7.2 a) 1)による。
c) 操作 操作は,次による。
1) 試料を孔径0.45 μmのメンブレンフィルターでろ過する。色の測定(4)と並行して,ろ過した試料の
pH及び温度(5)を測定する。
2) この一部を吸収セル(6)に移し,水を対照液として波長436 nm,525 nm及び620 nmの吸光度を測定
する。
3) 得られた吸光度から,次の式によってそれぞれの波長での吸収係数α(λ)(光路長1 m当たりの吸
光度)を算出する。
()
f
d
A×
=
λ
α
ここに, α(λ): 光路長1 m当たりの吸光度(m−1)
A: ろ過後の試料の波長λにおける吸光度
d: 吸収セルの光路長(mm)
f: 光路長1 m当たりの吸光度に換算するための係数(f=1 000)
注(4) 色が濃い場合は,ろ過後の試料を水の一定量で薄める。この場合,α(λ)値の算出には希釈を考慮
する。
(5) pH及び温度は,参考のため結果に付記する。
(6) 吸収係数が10 m−1より小さい場合は,吸収セルの光路長は10 mm以上のものを用いる。
12. pH pH値の測定は,JIS Z 8802に規定するガラス電極法による。
試料のpH値は化学的,物理的,生物化学的作用などによって迅速に変化するため,試料採取後,直ち
に測定する。
なお,この試験方法は,1994年に第1版として発行されたISO 10523との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 10523:1994,Water quality−Determination of pH(MOD)
12.1 ガラス電極法 ガラス電極を用いたpH計によって測定する。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA2又はA3の水。ほう酸塩pH標準液及び炭酸塩pH標準液を調製する
場合は,2. n) 2)の二酸化炭素を含まない水を用いる。
2) 二しゅう酸三水素カリウム二水和物 JIS K 8474に規定するもの。
3) フタル酸水素カリウム JIS K 8809に規定するpH標準液用のもの。
22
K 0102:2016
4) りん酸二水素カリウム JIS K 9007に規定するpH標準液用のもの。
5) りん酸水素二ナトリウム JIS K 9020に規定するpH標準液用のもの。
6) 四ほう酸ナトリウム JIS K 8866に規定するpH標準液用のもの。
7) 炭酸水素ナトリウム JIS K 8622に規定するpH標準液用のもの。
8) 炭酸ナトリウム JIS K 8625に規定するpH標準液用のもの。
b) pH標準液(1) pH標準液は,次の1) 又は2)による。
1) 調製pH標準液 調製pH標準液は,JIS Z 8802の7.3.2(調製方法)による。
2) 認証pH標準液 国家計量標準とのトレーサビリティが確認された認証pH標準液は,第2種を用
いる。
注(1) pH標準液の各温度でのpH値は,表12.1及び表12.2による。
備考 1. 表12.1及び表12.2に記載されていない温度におけるpH値は,補間して求める。
2. 各pH標準液は,保存中にpH値が変化することがあるので長期間保存したものは使用しない。
特に,ほう酸塩pH標準液及び炭酸塩pH標準液は,容易に大気中の二酸化炭素を吸収し,pH
値が低下するので注意する。
3. 各pH標準液は,一度使用したもの及び大気中に開放して放置したものは使用しない。
表12.1 調製pH標準液の各温度におけるpH値の典型値
温度
℃
pH値
しゅう酸塩
フタル酸塩
中性りん酸塩
ほう酸塩
炭酸塩*
0
1.67
4.01
6.98
9.46
10.32
5
1.67
4.01
6.95
9.39
(10.25)
10
1.67
4.00
6.92
9.33
10.18
15
1.67
4.00
6.90
9.27
(10.12)
20
1.68
4.00
6.88
9.22
(10.07)
25
1.68
4.01
6.86
9.18
10.02
30
1.69
4.01
6.85
9.14
(9.97)
35
1.69
4.02
6.84
9.10
(9.93)
38
−
−
−
−
9.91
40
1.70
4.03
6.84
9.07
−
45
1.70
4.04
6.83
9.04
−
50
1.71
4.06
6.83
9.01
−
55
1.72
4.08
6.84
8.99
−
60
1.73
4.10
6.84
8.96
−
70
1.74
4.12
6.85
8.93
−
80
1.77
4.16
6.86
8.89
−
90
1.80
4.20
6.88
8.85
−
95
1.81
4.23
6.89
8.83
−
注*
括弧内の値は,2次補間値を示す。各温度におけるpH値は,JIS Z 8802による。
23
K 0102:2016
表12.2 認証pH標準液のpH値の典型値*(参考)
温度
℃
pH値
しゅう酸塩
フタル酸塩
中性りん酸塩
りん酸塩
ほう酸塩
炭酸塩
第2種
第2種
第2種
第2種
第2種
第2種
0
1.67
4.00
6.98
7.53
9.46
10.32
5
1.67
4.00
6.95
7.50
9.40
10.24
10
1.67
4.00
6.92
7.47
9.33
10.18
15
1.67
4.00
6.90
7.45
9.28
10.12
20
1.68
4.00
6.88
7.43
9.22
10.06
25
1.68
4.01
6.86
7.41
9.18
10.01
30
1.68
4.02
6.85
7.40
9.14
9.97
35
1.69
4.02
6.84
7.39
9.10
9.92
38
1.69
4.03
6.84
7.38
9.08
−
40
1.69
4.04
6.84
7.38
9.07
9.89
45
1.70
4.05
6.83
7.37
9.04
9.86
50
1.71
4.06
6.83
7.37
9.01
9.83
55
1.72
4.08
6.83
−
8.98
−
60
1.72
4.09
6.84
−
8.96
−
70
1.74
4.13
6.84
−
8.92
−
80
1.77
4.16
6.86
−
8.88
−
90
1.79
4.20
6.88
−
8.85
−
95
1.81
4.23
6.89
−
8.83
−
注*
OIML recommendation (R054-e81) に記載の値を小数点以下2桁に丸めたものである。JIS Z 8802による。
c) 器具及び装置 器具及び装置は,次による。
1) pH計 JIS Z 8802に規定する形式IIを用いる(2)。
2) 温度計 JIS B 7411-1に規定する一般用ガラス製棒状温度計の50度温度計又は100度温度計(3)。
注(2) 試験目的に応じてpH計の形式を選択することができる。
JIS Z 8802では,pH計の形式を0〜IIIの4段階に区分し,各形式のpH計の繰返し性は,1
種類のpH標準液を用いてpH値を測定したとき,形式0では±0.005,形式Iでは±0.02,形式
IIでは±0.05及び形式IIIでは±0.1と規定している。
(3) JIS B 7411-1に規定する一般用ガラス製棒状温度計で,各目盛における許容差±1 ℃のもの。
d) pH計の校正 pH計の校正は,次による。
1) 検出部[ガラス電極(4) (5)及び参照電極(6),温度計など]を取り付け,pH計の電源を入れる。
2) 検出部は,水で繰り返し3回以上洗い,きれいな柔らかい紙で拭っておく。
3) 中性りん酸塩pH標準液をビーカーにとり,検出部を浸す。温度補償用ダイヤル又はデジタルスイ
ッチの設定のあるものは目盛値を,中性りん酸塩pH標準液の温度に合わせる(7) (8)。
4) 中性りん酸塩pH標準液の温度に対応するpH値(表12.1又は表12.2)に調節して合わせる。
5) 検出部を水で繰り返し3回以上洗い,きれいな柔らかい紙などで拭っておく。
6) 試料のpH値が7以下の場合は,フタル酸塩pH標準液又はしゅう酸塩pH標準液をビーカーにとり
検出部を浸す。
使用したpH標準液の温度に対応するpH値(表12.1又は表12.2)に調節して合わせる(8)。試料
のpH値が7を超える場合は(9),りん酸塩pH標準液,ほう酸塩pH標準液又は炭酸塩pH標準液を
用い,同じ操作でpH標準液の温度に対応するpH値に合わせる(8)。
24
K 0102:2016
7) 再び2)〜6)の操作を行い,pH値がpH標準液の温度に対応するpH値に±0.05で一致するまでこの
操作を繰り返す。
注(4) 長く乾燥状態にあったガラス電極は,あらかじめ水に浸して平衡に達してから使用する。
(5) ガラス電極が汚れている場合は,必要に応じて洗剤,塩酸(1+20)(JIS K 8180に規定する塩
酸(特級)を用いて調製する。)などで短時間洗い,更に流水で十分に洗う。電極の保守は,備
考4.によるとよい。
(6) 参照電極の汚れの除去はガラス電極と同じ操作で行い,内部液(塩化カリウム溶液)の交換な
どは取扱説明書を参照。液絡部の汚染を避けるため,電解液には2 cm以上の水位差に相当する
静水圧が必要である。
(7) pH標準液の温度は,できるだけ試料の温度に合わせる。
(8) 校正中は,各pH標準液の温度変動は±2 ℃とする。
(9) 試料のpH値が11以上の場合には,備考5.によって測定する。
備考 4. 電極の保守は,次による。
多くの測定を行った後,pH標準液のpH値の読みが平衡になるのに長時間を要するように
なった場合は,次の操作を行って電極系(測定セル)の汚れを除く必要がある。
1) 電極を定期的に(毎日又は毎週)柔らかい紙でよく拭って清浄にする。もし,有機物で
汚染された場合は,エタノール[体積分率70 %][JIS K 8102に規定するエタノール(95)
を用いて調製する。],アセトン(JIS K 8034に規定するもの。),温めた洗剤溶液などで
洗う。
2) 有機溶媒は,ガラス膜表面から水を奪い感度低下をもたらすので,短時間の使用にとど
める。有機溶媒を使用した場合,その後,数時間は電極を水に浸して回復を図らなけれ
ばならない。この場合は,再校正が必要である。
3) 炭酸カルシウムが付着した場合は,うすい塩酸で溶かす。
4) 参照電極及び複合電極は,参照電極用電解液に,ガラス電極は,水に浸しておくとよい。
5. pH値が11以上で,特にアルカリ金属元素の濃度が高い場合には,アルカリ誤差を生じやす
いため,アルカリ誤差の少ない電極を用い,炭酸塩を含まない0.1 mol/L水酸化ナトリウム溶
液[16.1 a) 1) による。]又は水酸化カルシウム溶液(25 ℃における飽和)を用いてpH計の
校正を行う。
0.1 mol/L水酸化ナトリウム溶液又は水酸化カルシウム溶液(25 ℃における飽和)は,大
気中の二酸化炭素を吸収して容易にpH値が低下するので使用の都度調製する。
表12.3に0.1 mol/L水酸化ナトリウム溶液及び水酸化カルシウム溶液(25 ℃における飽和)
の各温度におけるpH値を示す。
25
K 0102:2016
表12.3 0.1 mol/L水酸化ナトリウム溶液及び25 ℃における
飽和水酸化カルシウム溶液の各温度におけるpH値
温度
℃
0.1 mol/L水酸化
ナトリウム溶液
25 ℃における飽和
水酸化カルシウム溶液
温度
℃
0.1 mol/L水酸化
ナトリウム溶液
25 ℃における飽和
水酸化カルシウム溶液
0
13.8
13.43
35
12.6
12.14
5
13.6
13.21
40
12.4
11.99
10
13.4
13.00
45
12.3
11.84
15
13.2
12.81
50
12.2
11.70
20
13.1
12.63
55
12.0
11.58
25
12.9
12.45
60
11.9
11.45
30
12.7
12.30
e) 操作 操作は,次による。
1) 校正したpH計の検出部を水で繰り返し3回以上洗い,きれいな柔らかい紙などで拭っておく。
2) 試料をビーカーにとり,検出部を浸す。温度補償用ダイヤル又はデジタルスイッチの設定のあるも
のは,目盛値を試料の温度(10)に合わせた後,pH値を測定する。
3) 検出部を取り出し,水で繰り返し3回以上洗い,きれいな柔らかい紙などで拭っておく。
4) 再び試料をビーカーにとり,検出部を浸し,pH値を測定する(10)。
5) 再び3)及び4)の操作を行って,3回の測定値が±0.1(11)で一致した測定値を平均して,試料のpH値
を算出する。
注(10) pH値は試料の温度によって異なるので,試料の温度変動は±2 ℃とする。
(11) 緩衝性の低い試料は,容易にpH値が変化するためpH値が±0.1の繰返し性が得られない場合
がある。この場合は,pH値が±0.2で一致する値を平均してpH値を算出する。また,大気中
の二酸化炭素で容易にpH値が変動する場合には,流液形の電極を使用するとよい。
備考 6. イオン強度の低い水(電気伝導率5 mS/m以下)又は緩衝性の低い水のpH値の測定には,特
別な注意が必要である。
7. 試料のpH値が11以上の場合には,溶解性の低いガラス膜をもった電極,すなわち,高アル
カリ電極を用いる。このような電極は,水の測定全般にわたってその使用を勧める。参照電
極はすり合わせスリーブ形で,電解液は塩化カリウム溶液(1 mol/L)(JIS K 8121に規定す
る塩化カリウムを用いて調製する。)を用いる。ガラス電極への塩化カリウムの影響を避ける
ため溶液を静かにかき混ぜる。空気の影響を避けるためには,試料を測定容器に流す。静電
効果を抑制するために,測定容器部分を電磁遮蔽し,試験液中に金属電極を挿入し,接地し
なければならない。
8. 妨害物質 温度,ある種の気体及び有機化合物は,pH値の測定を妨害する。懸濁物は,かな
り大きな誤差をもたらす可能性がある(懸濁効果)。このような場合は,沈降を待って上澄み
部分に電極を挿入するか,又は限外ろ過法を適用する。試料によっては,油脂その他の汚染
物質の電極への付着及び隔膜の汚染が起こる可能性が高い。
参照電極の液絡部は,この汚染を防ぐことができる(6)。隔膜内に沈殿,例えば,硫化銀又
は,たん白フロックを生じるような場合は,試料と参照電極との間に,硝酸カリウム溶液
(1 mol/L)(JIS K 8548に規定する硝酸カリウムを用いて調製する。)の塩橋を用いる必要が
ある。
26
K 0102:2016
13. 電気伝導率 電気伝導率は,溶液がもつ電気抵抗率(Ω・m)の逆数に相当し,(S/m)の単位で表す。
また,電気伝導度は,溶液がもつ電気抵抗(Ω)の逆数に相当し,(S)の単位で表す。
水の試験では,25 ℃の値を用い(S/m)及び(S)の千分の一を単位とし,それぞれ(mS/m)(1)及び(mS)
で表す(2)。
この試験は試料採取後,直ちに行う。直ちに行えない場合には,試料容器に満杯として密栓し,0〜10 ℃
の暗所に保存し,できるだけ早く試験する。
なお,この試験方法は,1985年に第1版として発行されたISO 7888との整合を図ったものである。
注(1) mS/mは,ミリジーメンス毎メートルと読む。
(2) 従来の単位で表した1 μS/cmは,SI単位では0.1 mS/mに相当(1 μS/cm=1×10−6 S/10−2 m=1
×10−4 S/m=0.1 mS/m)する。
備考 1. この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 7888:1985,Water quality−Determination of electrical conductivity(MOD)
2. 従来,水の試験では,電気伝導度及び電気伝導率の単位としてそれぞれ(μS)及び(μS/cm),
また,セル定数の単位として(cm−1)が用いられていた。
電気伝導度としては,(mS)の単位で表した数値を1 000倍すると(μS)の単位で表した
数値となる。
電気伝導率としては,(mS/m)の単位で表した数値を10倍すると(μS/cm)の単位で表し
た数値となる。
また,セル定数としては,(m−1)の単位で表した数値を0.01倍すると(cm−1)の単位で
表した数値となる。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA2又はA3の水。ただし,電気伝導率0.2 mS/m(2 μS/cm)(25 ℃)以
下のもの。調製時に20±2 ℃に調節して用いる。
2) 塩化カリウム JIS K 8121に規定する塩化カリウム(電気伝導率測定用)をめのう乳鉢で粉末にし,
500 ℃で約4時間加熱し,デシケーター中で放冷したもの。
3) 1 mol/kg塩化カリウム標準液 JIS K 0130の7.1(塩化カリウム標準液の調製)による。
4) 0.1 mol/kg塩化カリウム標準液 JIS K 0130の7.1(塩化カリウム標準液の調製)による。
5) 0.01 mol/kg塩化カリウム標準液 JIS K 0130の7.1(塩化カリウム標準液の調製)による。
6) 0.001 mol/kg塩化カリウム標準液 JIS K 0130の7.1(塩化カリウム標準液の調製)による。
これらの塩化カリウム標準液は,ポリエチレン瓶又はほうけい酸ガラス瓶に密栓して保存する。塩
化カリウム標準液の電気伝導率を,表13.1及び表13.2に示す。
なお,これらの塩化カリウム標準液は,市販されているものを用いてもよい。
7) 塩酸(1+1) JIS K 8180に規定する塩酸(特級)を用いて調製する。
8) 硫酸(1+360) 水360容をビーカーにとり,かき混ぜながらJIS K 8951に規定する硫酸1容を徐々
に加える。
9) 電解液 JIS K 8153に規定するヘキサクロロ白金(IV)酸六水和物を30 g及びJIS K 8374に規定す
る酢酸鉛(II)三水和物を0.25 gひょう量し,JIS K 0557に規定する水A2,A3又はA4を用いて全
量1 Lとなるよう調製する。この溶液は必要なときに用いる。
27
K 0102:2016
表13.1 塩化カリウム標準液の電気伝導率(3)
単位 mS/m
温度
℃
電気伝導率の値及びその不確かさ
0.01 mol/kg
塩化カリウム標準液
0.1 mol/kg
塩化カリウム標準液
1 mol/kg
塩化カリウム標準液
CO2
飽和水
電気伝導率
2uc
電気伝導率
2uc
電気伝導率
2uc
電気伝導率
0
77.292
0.023
711.685
0.285
6 348.8
2.5
0.058
5
89.096
0.027
818.370
0.327
7 203.0
2.9
0.068
10
101.395
0.030
929.172
0.372
8 084.4
3.2
0.079
15
114.145
0.034
1043.71
0.42
8 990.0
3.6
0.089
18
121.993
0.037
1114.06
0.45
−
−
0.095
20
127.303
0.038
1161.59
0.46
9 917.0
4.0
0.099
25
140.823
0.042
1282.46
0.51
10 862.0
4.3
0.110
30
154.663
0.046
1405.92
0.56
11 824.0
4.7
0.120
35
168.779
0.051
1531.60
0.61
12 797.0
5.1
0.130
40
183.127
0.055
1659.10
0.66
13 781.0
5.5
0.140
45
197.662
0.059
1788.06
0.72
14 772.0
5.9
0.151
50
212.343
0.064
1918.09
0.77
15 767.0
6.3
0.161
注(3) 米国国立標準技術研究所(NIST)のデータ[Pure Appl.Chem.,vol 73,pp.1783(2001)2ucは拡張不確かさ]
表13.2 0.001 mol/kg塩化カリウム標準液の電気伝導率(4)
塩化カリウム標準液の濃度
mol/kg
温度
℃
電気伝導率
mS/m
0.001 mol/kg
25
14.65
注(4) Shedlovskyのデータ及び密度データから計算で求め,
4桁表示した。[J.Am.Chem.Soc.,54,1411(1932)]
b) 器具及び装置 器具及び装置は,次による。
1) 電気伝導度計 検出部と指示部とから成るもの。検出部は,白金電極面に白金黒めっきを行った電
極を組み入れたセルから成る。セルは,表13.3に示したセル定数のものを用意する。指示部は,ホ
イートストンブリッジ回路などを組み入れたものを用いる。セルは,水中に保存する(5)。
2) 温度計 JIS B 7411-1に規定する一般用ガラス製棒状温度計の50度温度計
3) 恒温槽 25±0.5 ℃に保つことができるもの。
注(5) セル定数を,c) 2)の方法で定期的に確認する。
c) 操作 操作は,次による。
1) 試料の電気伝導率測定 試料の電気伝導率測定は,次による。
1.1) あらかじめ電気伝導度計の電源を入れておく。試料の電気伝導率に応じて表13.3に示すセル定数
をもったセルを用い,水でセルを2,3回洗う。特に汚れている場合には,塩酸(1+100)[JIS K
8180に規定する塩酸(特級)を用いて調製する。]に浸し,更に流水で十分に洗い,最後に水で2,
3回洗う。
1.2) このセルを試料で2,3回洗った後,試料を満たし,25±0.5 ℃(6)に保って電気伝導度(7) (8)の測定
を行う。
測定値が±3 %(9)で一致するまで試料を入れ替えて測定を繰り返し,3回の測定の平均値を電気
伝導度とする。
28
K 0102:2016
1.3) 電気伝導度から次の式によって試料の電気伝導率(mS/m)(25 ℃)を算出する。
L=J×Lx
ここに,
L: 試料の電気伝導率(mS/m)(25 ℃)
J: セル定数(m−1)
Lx: 測定した電気伝導度(mS)
注(6) 精度を特に必要としない場合には,温度補償回路を組み入れた電気伝導度計を用いるか,次の
温度換算式[式(1)]を用いてもよい。電気伝導率は温度によって変化し,1 ℃の上昇で約2 %
大きくなる。ただし,電気伝導率が1 mS/m(10 μS/cm)以下になると,水の解離によって生じ
る水素イオン及び水酸化物イオンの影響が大きくなるので,この換算式は適用できない。
)
25
)(
100
/
(
1
θ
25
−
+
=
θ
α
κ
κ
······························································ (1)
ここに,
κ25: 25 ℃における電気伝導率(mS/m)
κθ: 測定温度θにおける電気伝導率(mS/m)
α: 電気伝導率の温度係数(%)
θ: 測定時の試料の温度(℃)
また,電気伝導率の温度係数(1 ℃当たりの電気伝導率の変化率)αの値は,実験的測定に
よって式(2)から求めることができる。
100
25
1
25
θ
25
25
θ,
×
−
−
×
=
θ
κ
κ
κ
α
·························································· (2)
ここに, αθ,25: κθをκ25に換算するための電気伝導率の温度係数(%)
κ25: 25 ℃における電気伝導率(mS/m)
κθ: 測定温度θにおける電気伝導率(mS/m)
θ: 測定時の試料の温度(℃)
注(7) 電気伝導度計の指示値が電気抵抗(Ω)の場合は,次の式(3)によって電気伝導率(mS/m)を計
算する。
3
10
×
=
x
R
J
κ
·············································································· (3)
ここに,
κ: 試料の電気伝導率(mS/m)(25 ℃)
J: セル定数(m−1)。ただし,電気抵抗率(Ω・m)が直示される
場合は1とする。
Rx: 測定した電気抵抗(Ω)
(8) 電気伝導度計の指示値が電気伝導度(μS)の場合には,次の式(4)によって電気伝導率を算出す
る。
κ=J'×Lx'×0.1 ·········································································· (4)
ここに,
κ: 試料の電気伝導率(mS/m)(25 ℃)
J': セル定数(cm−1)
Lx': 測定した電気伝導度(μS)
(9) 試料の電気伝導率が1 mS/m(25 ℃)未満の場合には,±3 %で一致しないことがあるので,JIS
K 0552に従って試験するか,又は流液形のセルを用いる。
29
K 0102:2016
表13.3 セル定数及び測定範囲
単位 mS/m
区分
セル定数
m−1
測定範囲
検出部材質
(白金黒)
交流2電極
方式
0.1〜1
0.005〜100
1〜10
0.005〜1 000
10〜100
0〜10 000
100〜1 000
0〜100 000
1000〜5 000
0〜500 000
2) セル定数の測定又はセル定数の確認 セル定数の測定又はセル定数の確認は,試料を試験するたび
に行う必要はないが,定期的に表13.1の塩化カリウム標準液を用いてその数値を確かめる。操作は,
次による。
2.1) セルを水で2,3回洗う。次に,塩化カリウム標準液(セル定数に応じ,表13.3の測定範囲に対応
する塩化カリウム標準液を用いる。)で2,3回洗った後,その塩化カリウム標準液を満たす。こ
のセルを25±0.5 ℃に保ち,電気伝導度を測定する。同じ塩化カリウム標準液を数回入れ替えて
測定を行い,測定値が±3 %で一致するまで繰り返す[±3 %で一致しない場合には,白金黒電極
に,3)によって新たに白金黒めっきを行う。]。
2.2) 3回の測定から求めた平均値から,次の式によってセル定数を算出する。
XO
O
H
KC1
2
L
κ
κ
J
+
=
ここに,
J: セル定数(m−1)
LXO: 測定した電気伝導度(mS)。ただし,電気伝導度の指示がμS
になっているときは,μS×000
1
1
の値を用いる。
κKCl: 使用した塩化カリウム標準液のこの温度における電気伝導率
(mS/m)
O
H2
κ
: 塩化カリウム標準液の調製に用いた水のこの温度における電
気伝導率(mS/m)
なお,電気伝導度がμSとなっている場合でセル定数をcm−1の単位で求めたいときは,次の式
による。
'
L
'
κ
'
κ
J'
XO
O
H
KC1
2
+
=
ここに,
J': セル定数(cm−1)
LXO': 測定した電気伝導度(μS)
κKCl': 使用した塩化カリウム標準液のこの温度における電気伝導率
(μS/cm)
O
H2
κ
: 塩化カリウム標準液の調製に用いた水のこの温度における電
気伝導率(μS/cm)
また,表13.1中で濃度の接近している2種類の塩化カリウム標準液を用いてセル定数を測定し,
その値が±1 %で一致しないときは,3)によって,新たに白金黒めっきを行う。
3) 電極の白金黒めっきは,次による。
3.1) 白金黒を電極から取り除くために,塩酸(1+1)中で白金黒電極を陽極として電解する。
3.2) この白金電極をa) 9)電解液に入れ,直流電圧約6 V,電流密度100〜400 A/m2とし,適切な方法で
30
K 0102:2016
電解液をかき混ぜながら,数回極性を切り替え,約10分間を通電する[35〜140 kC/m2]。
3.3) 次に,硫酸(1+360)中で約30分間,ときどき電流の方向を変えて通電し,付着,吸蔵したヘキ
サクロロ白金(IV)酸及び塩素を除く。
14. 懸濁物質及び蒸発残留物 水中に懸濁している物質及び水を蒸発したときの残留物質を,懸濁物質,
全蒸発残留物,溶解性蒸発残留物,強熱残留物及び強熱減量に区分して試験する。
試験は,試料採取後,直ちに行う。直ちに行えない場合には,試料を0〜10 ℃の暗所に保存し,できる
だけ早く試験する。
ここで用いる用語の意味は,次による。
a) 懸濁物質 試料をろ過したとき,ろ過材上に残留する物質。
b) 全蒸発残留物 試料を蒸発乾固したときに残留する物質。
c) 溶解性蒸発残留物 懸濁物質をろ別したろ液を蒸発乾固したときに残留する物質。
d) 強熱残留物 懸濁物質,全蒸発残留物及び溶解性蒸発残留物のそれぞれを600±25 ℃で30分間強熱
したときの残留物で,それぞれの強熱残留物として示す。
e) 強熱減量 d)の測定時における減少量で,それぞれの強熱減量として示す。
14.1 懸濁物質 試料をろ過し,ろ過材上に残留した物質を105〜110 ℃で乾燥し,その質量をはかる。
a) 器具 器具は,次による。
1) ろ過器(分離形) 図14.1に,例を示す。
A:
B:
C:
D:
E:
F:
G:
上部ろ過管
ろ過材
ろ過材保持台
下部ろ過管
ゴム栓
金属性クランプ
吸引瓶
a) ろ過器(分離形)
b) ろ過部の詳細
図14.1 ろ過器(分離形)の例
2) ろ過材 ガラス繊維ろ紙,有機性ろ過膜又は金属性ろ過膜。孔径1 μmで直径25〜50 mm
備考 1. ガラス繊維ろ紙は,目詰まりは少ないがガラス繊維が離脱するおそれがある。有機性ろ過膜
は,種類によって耐薬品性及び耐熱性に差があるので,取扱いに注意する。
b) 操作 操作は,次による。
1) ガラス繊維ろ紙を用いる場合は,あらかじめろ過器に取り付け,水で十分に吸引洗浄した後(1),こ
のろ過材を,時計皿(2)上に置き,105〜110 ℃で約1時間加熱し(3),デシケーター中で放冷した後,
その質量をはかる。
2) ろ過材をろ過器に取り付け,試料(4)の適量(5)をろ過器に注ぎ入れて吸引ろ過する。試料容器及びろ
31
K 0102:2016
過管の器壁に付着した物質は,水でろ過材上に洗い落とし,ろ過材上の残留物質に合わせ,これを
水で数回洗浄する。
3) 残留物は,ろ過材とともにピンセットなどを用いてろ過器から注意して取り外し,1)で用いた時計
皿上に移し,105〜110 ℃で2時間加熱し,先のデシケーター中で放冷した後,その質量をはかる。
4) 次の式によって懸濁物質(mg/L)を算出する。
(
)
V
b
a
S
000
1
×
−
=
ここに,
S: 懸濁物質の濃度(mg/L)
a: 懸濁物質を含んだろ過材及び時計皿の質量(mg)
b: ろ過材及び時計皿の質量(mg)
V: 試料量(mL)
注(1) 合成樹脂によるバインダー処理を行ったガラス繊維ろ紙,有機性ろ過膜及び金属性ろ過膜は洗
浄しなくてよい。
(2) なるべく軽い時計皿を用いるか,又はアルミニウムはくなどの軽い容器を用いる。
(3) 有機性ろ過膜では105〜110 ℃で加熱すると,変形するものがある。この場合は,90 ℃で加熱
する。
(4) 目開き2 mmのふるいを通過した試料を用い,十分に振り混ぜて懸濁物質が均一になってから,
手早く採取する。
(5) 乾燥後の懸濁物質の量が2 mg以上になるように試料をとる。
備考 2. ろ過しにくい試料の場合には,適量をビーカーにとり,その都度よく振り混ぜて,液をろ過
し終わる直前ごとに加え,ろ過速度が極めて遅くなったら試料の追加を止める。ビーカーの
中の残量から試料の量を求める。
3. 試料容器に付着しやすい懸濁物質を含む場合は,適量の試料を採取して,その全量を用いて
試験する。
採取した容器の器壁に付着した懸濁物質は,ポリスマン(ゴム管付きガラス棒)などで落
として,ろ過材上に集める。
4. 懸濁物質中の強熱残留物を定量する場合には,有機性ろ過膜を用いる。
5. 油脂,グリース,ワックスなどを含む試料で,これらを除いた懸濁物質を測定する場合には,
試料をろ過した後,ろ過材を取り外すことなく,ろ過管ごと乾燥し,再び吸引瓶に取り付け
た後,JIS K 8848に規定するヘキサン10 mLずつを数回注ぎ入れ,油脂類を洗って除く。続
いて3)の操作を行う。
14.2 全蒸発残留物 全蒸発残留物は,次によって質量をはかる。
a) 器具 器具は,次による。
1) 蒸発皿 白金製,石英ガラス製又は磁器製
b) 操作 操作は,次による。
1) 適切な材質(6)の蒸発皿を105〜110 ℃で約1時間加熱し,デシケーター中で放冷した後,その質量
をはかる。
加熱,放冷及びひょう量を繰り返して恒量とする。
2) 試料(4)の適量(7)を蒸発皿に入れ(8),注意して蒸発乾固する(9)。
3) この蒸発皿を105〜110 ℃で約2時間加熱し,先のデシケーター中で放冷した後,その質量をはか
32
K 0102:2016
る。
4) 次の式によって全蒸発残留物(mg/L)を算出する。
(
)
V
b
a
R
000
1
×
−
=
ここに,
R: 全蒸発残留物(mg/L)
a: 残留物の入った蒸発皿の質量(mg)
b: 蒸発皿の質量(mg)
V: 試料量(mL)
注(6) 試料の性質によって用いる蒸発皿の材質を選ぶ。
例えば,試料に塩化物イオンと強い酸化性の物質とが共存する場合には,白金皿は用いない。
(7) 乾燥後の全蒸発残留物の質量が,5 mg以上になるように採取する。
(8) 試料の全量が蒸発皿に入らない場合には,数回に分けて入れる。
(9) 蒸発乾固には,加熱板,沸騰水浴,赤外線ランプなどを用い,蒸発中に沸騰させないようにす
る。また,外部からの汚染に注意する。
備考 6. 14.の備考3.による。
7. 引き続き強熱残留物を定量する場合には,蒸発皿はあらかじめ600±25 ℃で加熱して恒量に
したものを用いる。
14.3 溶解性蒸発残留物 溶解性蒸発残留物は,次によって質量をはかる。
a) 器具 器具は,次による。
1) 蒸発皿 14.2 a) 1)による。
2) ろ過器 14.1 a) 1)による。
3) ろ過材 14.1 a) 2)による。
b) 操作 操作は,次による。
1) ろ過材をろ過器に取り付け,試料をろ過する。
2) ろ液について,14.2 b) 1)〜4)に準じて操作する。
備考 8. 14.1の備考1.及び備考4.による。
14.4 強熱残留物
14.4.1 懸濁物質の強熱残留物 懸濁物質の強熱残留物は,次によって質量をはかる。
a) 試薬 試薬は,次による。
1) 硝酸アンモニウム溶液(250 g/L) JIS K 8545に規定する硝酸アンモニウム25 gを水に溶かして100
mLとする。
b) 器具及び装置 器具及び装置は,次による。
1) るつぼ 白金製又は磁器製10〜20 mL
2) 電気炉 600±25 ℃に調節できるもの。
c) 操作 操作は,次による。
1) 600±25 ℃で加熱して恒量にしたるつぼに14.1 b) 3)で得た懸濁物質を,ろ過材とともに移し入れ,
硝酸アンモニウム溶液(250 g/L)を滴加して湿らせた後(10),電気炉に入れ,徐々に温度を上げ600
±25 ℃で約30分間加熱して灰化し,デシケーター中で放冷した後,その質量をはかる。
2) 次の式によって強熱残留物(mg/L)を算出する。
(
)
V
b
a
R
000
1
×
−
=
33
K 0102:2016
ここに,
R: 懸濁物質の強熱残留物(mg/L)
a: 強熱残留物の入ったるつぼの質量(mg)
b: るつぼの質量(mg)
V: 試料量(mL)
注(10) 有機性ろ過膜がニトロ化合物の場合には,JIS K 8839に規定する2-プロパノールを滴加して湿
らせた後,強熱して灰化するとよい。
備考 9. ろ過材にガラス繊維ろ紙を使用した場合には,あらかじめ別の同形のガラス繊維ろ紙を600
±25 ℃の電気炉で加熱して空試験値を求め,ガラス繊維ろ紙の質量を補正する。
14.4.2 全蒸発残留物の強熱残留物 全蒸発残留物の強熱残留物は,次によって質量をはかる。
a) 操作 操作は,次による。
1) 14.2 b) 3)で得た全蒸発残留物について,14.4.1 c)に準じて操作する。
14.4.3 溶解性蒸発残留物の強熱残留物
a) 操作 操作は,次による。
1) 14.3 b)で得た溶解性蒸発残留物について,14.4.1 c)の操作に準じて操作する。
14.5 強熱減量
a) 操作 操作は,次による。
1) 懸濁物質の強熱減量,全蒸発残留物の強熱減量及び溶解性残留物の強熱減量は,次の式によって算
出する。
E=S−R
ここに,
E: それぞれの強熱減量(mg/L)
S: それぞれの蒸発残留物(mg/L)
R: それぞれの強熱残留物(mg/L)
15. 酸消費量 酸消費量は,水に溶けている炭酸水素塩,炭酸塩,水酸化物などのアルカリを所定のpH
に中和するのに要する水素イオンの量(酸の量)を,試料1 Lについてのmmol数で表すか,又は水素イ
オン(酸)に相当する炭酸カルシウムの量に換算して,試料1 Lについてのmg数で表す。
酸消費量を,酸消費量(pH4.8)と酸消費量(pH8.3)とに区分する。
15.1 酸消費量(pH4.8) 酸消費量(pH4.8)は,pH計を用い,0.1 mol/L塩酸で滴定して求める。
a) 試薬 試薬は,次による。
1) 0.1 mol/L塩酸 JIS K 8180に規定する塩酸10 mLを,あらかじめ水100 mLを入れたビーカーに加
えてよくかき混ぜ,水を加えて1 Lとする。
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質の炭酸ナトリウムを600 ℃で約1時間加熱し,デシ
ケーター中で放冷した後,その1.06 gを1 mgの桁まではかりとり,水に溶かして,全量フラス
コ200 mLに移し入れ,水を標線まで加える。
− この20 mLをビーカーにとり,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液
(1) 3〜5滴を加えた後,0.1 mol/L塩酸で滴定する。溶液の色が灰紫を呈するようになったら,
煮沸して二酸化炭素を追い出し,放冷後,溶液の色が灰紫になるまで滴定を続ける。滴定に要
した0.1 mol/L塩酸のmL数から,次の式によって0.1 mol/L塩酸のファクター(f)を算出する。
30
005
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
34
K 0102:2016
ここに,
a: 炭酸ナトリウムの質量(g)
b: 炭酸ナトリウムの純度(質量分率%)
x: 滴定に要した0.1 mol/L塩酸溶液量(mL)
0.005 30: 0.1 mol/L塩酸1 mLに相当する炭酸ナトリウムの質量(g)
注(1) JIS K 8896に規定するメチルレッド20 mgとJIS K 8840に規定するブロモクレゾールグリーン
0.10 gとをJIS K 8102に規定するエタノール(95)100 mLに溶かす。
b) 器具及び装置 器具及び装置は,次による。
1) pH計 JIS Z 8802の形式II
2) マグネチックスターラー 回転による発熱で液温に変化を与えないもの。
c) 操作 操作は,次による。
1) 試料100 mLをビーカーにとり,pH計の電極を入れる。
2) マグネチックスターラーで緩やかにかき混ぜながら0.1 mol/L塩酸で,pH4.8(2)になるまで滴定する。
3) 次の式によって酸消費量(pH4.8)を算出する。
3.1) mmol/Lで表す場合
V
f
a
A
000
1
1.0×
×
×
=
ここに,
A: 酸消費量(pH4.8)(mmol/L)
a: 滴定に要した0.1 mol/L塩酸溶液量(mL)
f: 0.1 mol/L塩酸のファクター
0.1: 0.1 mol/L塩酸1 mLに相当する水素イオンの量(mmol)
V: 試料量(mL)
3.2) CaCO3 mg/Lで表す場合
004
.5
000
1
×
×
×
=
V
f
a
B
ここに,
B: 酸消費量(pH4.8)(CaCO3 mg/L)
a: 滴定に要した0.1 mol/L塩酸溶液量(mL)
f: 0.1 mol/L塩酸のファクター
V: 試料量(mL)
5.004: 0.1 mol/L塩酸1 mLに相当する炭酸カルシウムの質量(mg)
注(2) 必要な場合は,滴定曲線を作成して求める。
備考 1. pH計に代え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合液を用いて滴定し
てもよい。
15.2 酸消費量(pH8.3) 酸消費量(pH8.3)は,pH計を用い,0.1 mol/L塩酸で滴定して求める。
a) 試薬 試薬は,次による。
1) 0.1 mol/L塩酸 15.1 a) 1)による。
b) 器具及び装置 器具及び装置は,次による。
1) pH計 15.1 b) 1)による。
2) マグネチックスターラー 15.1 b) 2)による。
c) 操作 操作は,次による。
1) 試料100 mLをビーカーにとり,pH計の電極を入れる。
2) マグネチックスターラーで緩やかにかき混ぜながら,0.1 mol/L塩酸でpH8.3(2)になるまで滴定する。
3) 次の式によって酸消費量(pH8.3)を算出する。
3.1) mmol/Lで表す場合
35
K 0102:2016
V
f
a
A
000
1
1.0×
×
×
=
ここに,
A: 酸消費量(pH8.3)(mmol/L)
a: 滴定に要した0.1 mol/L塩酸溶液量(mL)
f: 0.1 mol/L塩酸のファクター
0.1: 0.1 mol/L塩酸1 mLに相当する水素イオンの量(mmol)
V: 試料量(mL)
3.2) CaCO3 mg/Lで表す場合
004
.5
000
1
×
×
×
=
V
f
a
B
ここに,
B: 酸消費量(pH8.3)(CaCO3 mg/L)
a: 滴定に要した0.1 mol/L塩酸溶液量(mL)
f: 0.1 mol/L塩酸のファクター
V: 試料量(mL)
5.004: 0.1 mol/L塩酸1 mLに相当する炭酸カルシウムの質量(mg)
備考 2. pH計に代え,指示薬としてフェノールフタレイン溶液(5 g/L)[JIS K 8799に規定するフェ
ノールフタレイン0.5 gをJIS K 8102に規定するエタノール(95)50 mLに溶かし,水を加
えて100 mLとする。]を用いて滴定してもよい。
16. アルカリ消費量 アルカリ消費量は,水に溶けている強酸,炭酸,有機酸及び水酸化物として沈殿す
る金属元素などを所定のpHまで中和するのに要する水酸化物イオンの量(アルカリの量)を試料1 L当
たりのmmol数で表すか,又は水酸化物イオンの量(アルカリの量)に相当する炭酸カルシウムの量に換
算した,試料1 L当たりのmg数で表す。
アルカリ消費量は,アルカリ消費量(pH8.3),アルカリ消費量(pH4.8)及びアルカリ消費量(遊離酸)
に区分する。
16.1 アルカリ消費量(pH8.3) アルカリ消費量(pH8.3)は,pH計を用い,0.1 mol/L水酸化ナトリウム
溶液で滴定して求める。
a) 試薬 試薬は,次による。
1) 0.1 mol/L水酸化ナトリウム溶液 水約30 mLをポリエチレン瓶にとり,冷却しながらJIS K 8576
に規定する水酸化ナトリウム約30 gを少量ずつ加えて溶かし,密栓して4〜5日間放置する。その
上澄み液5 mLをポリエチレン製の気密容器1 Lにとり,2. n) 2)の二酸化炭素を含まない水を加えて
1 Lとし,混合した後,二酸化炭素を遮断して保存する。
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質のアミド硫酸を上口デシケーター中に圧力2 kPa以
下で約48時間放置して乾燥する。その約0.2 gを1 mgの桁まではかりとり,三角フラスコ200
mLに入れ,水約25 mLを加えて溶かす。
− これに指示薬としてブロモチモールブルー溶液(1 g/L)(1) 3〜5滴を加え,この0.1 mol/L水酸
化ナトリウム溶液で滴定し,溶液の色が緑になったときを終点とする。次の式によって0.1
mol/L水酸化ナトリウム溶液のファクター(f1)を算出する。
71
009
.0
1
100
1
×
×
×
=
x
b
a
f
ここに,
a: アミド硫酸の質量(g)
36
K 0102:2016
b: アミド硫酸の純度(質量分率%)
x: 滴定に要した0.1 mol/L水酸化ナトリウム溶液量(mL)
0.009 71: 0.1 mol/L水酸化ナトリウム溶液1 mLに相当するアミド硫
酸の質量(g)
注(1) JIS K 8842に規定するブロモチモールブルー0.1 gをとり,JIS K 8102に規定するエタノール
(95)50 mLに溶かし,水で100 mLとする。
b) 器具及び装置 器具及び装置は,次による。
1) pH計 15.1 b) 1)による。
2) マグネチックスターラー 15.1 b) 2)による。
c) 操作 操作は,次による。
1) 試料100 mLをビーカーにとり,pH計の電極を入れる。
2) マグネチックスターラーで緩やかにかき混ぜながら,0.1 mol/L水酸化ナトリウム溶液でpH8.3(2)に
なるまで滴定する。
3) 次の式によってアルカリ消費量(pH8.3)を算出する。
3.1) mmol/Lで表す場合
V
f
a
A
000
1
1.0
1
×
×
×
=
ここに,
A: アルカリ消費量(pH8.3)(mmol/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液量(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクター
0.1: 0.1 mol/L水酸化ナトリウム溶液1 mLに相当する水酸化物イ
オンの量(mmol)
V: 試料量(mL)
3.2) CaCO3 mg/Lで表す場合
004
.5
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量(pH8.3)(CaCO3 mg/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液量(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクター
V: 試料量(mL)
5.004: 0.1 mol/L水酸化ナトリウム溶液1 mLに相当する炭酸カルシ
ウムの質量(mg)
注(2) 必要な場合は,滴定曲線を作成して求める。
備考 1. pH計に代え,指示薬としてフェノールフタレイン溶液(5 g/L)(15.の備考2.による。)を用
いて滴定してもよい。
2. アミド硫酸による標定の代わりに,JIS K 8005に規定する容量分析用標準物質の炭酸ナトリ
ウムを用いて標定した0.1 mol/L塩酸を使用して,次のように標定してもよい。
標定 標定は,次による。
− 0.1 mol/L塩酸20 mLをビーカーにとり,指示薬としてフェノールフタレイン溶液(5 g/L)
(15.の備考2.による。)3〜5滴を加えた後,この0.1 mol/L水酸化ナトリウム溶液で溶液
の色が微紅色を呈するまで滴定する。
− 滴定に要した0.1 mol/L水酸化ナトリウム溶液のmL数から,次の式によって0.1 mol/L
水酸化ナトリウム溶液のファクター(f1)を算出する。
37
K 0102:2016
x
f
f
×
=20
1
ここに,
f: 0.1 mol/L塩酸のファクター
x: 滴定に要した0.1 mol/L水酸化ナトリウム溶液量(mL)
16.2 アルカリ消費量(pH4.8) アルカリ消費量(pH4.8)は,pH計を用い,0.1 mol/L水酸化ナトリウム
溶液で滴定して求める。
a) 試薬 試薬は,次による。
1) 0.1 mol/L水酸化ナトリウム溶液 16.1 a) 1)による。
b) 器具及び装置 器具及び装置は,次による。
1) pH計 JIS Z 8802の形式II
2) マグネチックスターラー 15.1 b) 2)による。
c) 操作 操作は,次による。
1) 試料100 mLをビーカーにとり,pH計の電極を入れる。
2) マグネチックスターラーで緩やかにかき混ぜながら,0.1 mol/L水酸化ナトリウム溶液でpH4.8(2)に
なるまで滴定する。
3) 次の式によってアルカリ消費量(pH4.8)を算出する。
3.1) mmol/Lで表す場合
V
f
a
A
000
1
1.0
1
×
×
×
=
ここに,
A: アルカリ消費量(pH4.8)(mmol/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液量(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクター
0.1: 0.1 mol/L水酸化ナトリウム溶液1 mLに相当する水酸化物イ
オンの量(mmol)
V: 試料量(mL)
3.2) CaCO3 mg/Lで表す場合
004
.5
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量(pH4.8)(CaCO3 mg/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液量(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクター
V: 試料量(mL)
5.004: 0.1 mol/L水酸化ナトリウム溶液1 mLに相当する炭酸カルシ
ウムの質量(mg)
備考 3. pH計に代え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液[15.の注(1)に
よる。]を用いて滴定してもよい。
16.3 アルカリ消費量(遊離酸) アルカリ消費量(遊離酸)は,水に溶けている硫酸,塩酸,硝酸,強い
有機酸などをしゅう酸カリウムの存在下で0.1 mol/L水酸化ナトリウム溶液で滴定し,pH-水酸化ナトリウ
ム溶液の滴定曲線から求める。
この測定値には,鉄,アルミニウムなどの塩類によるアルカリ消費量は含まれず,遊離酸に対応するア
ルカリ消費量が求められる。
a) 試薬 試薬は,次による。
1) しゅう酸カリウム一水和物 JIS K 8522に規定するもの。
38
K 0102:2016
2) 0.1 mol/L水酸化ナトリウム溶液 16.1 a) 1)による。
b) 器具及び装置 器具及び装置は,次による。
1) pH計 15.1 b) 1)による。
2) マグネチックスターラー 15.1 b) 2)による。
c) 操作 操作は,次による。
1) 試料100 mLをビーカーにとり,液温を約10 ℃に調節する。
2) しゅう酸カリウム一水和物約20 gを加え,かき混ぜて溶かす。
3) マグネチックスターラーで緩やかにかき混ぜ,pH計を用いてpHを測定しながら0.1 mol/L水酸化
ナトリウム溶液で滴定する。
4) 滴定の終点(変曲点)前後では,0.1 mol/L水酸化ナトリウム溶液を0.1 mLずつ加える。
5) pHと0.1 mol/L水酸化ナトリウム溶液との滴定曲線を作成して終点を求め,次の式によってアルカ
リ消費量(遊離酸)を算出する。
5.1) mmol/Lで表す場合
V
f
a
A
000
1
1.0
1
×
×
×
=
ここに,
A: アルカリ消費量(遊離酸)(mmol/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液量(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクター
0.1: 0.1 mol/L水酸化ナトリウム溶液1 mLに相当する水酸化
物イオンの量(mmol)
V: 試料量(mL)
5.2) CaCO3 mg/Lで表す場合
004
.5
000
1
1
×
×
×
=
V
f
a
B
ここに,
B: アルカリ消費量(遊離酸)(CaCO3 mg/L)
a: 滴定に要した0.1 mol/L水酸化ナトリウム溶液量(mL)
f1: 0.1 mol/L水酸化ナトリウム溶液のファクター
V: 試料量(mL)
5.004: 0.1 mol/L水酸化ナトリウム溶液1 mLに相当する炭酸カルシ
ウムの質量(mg)
17. 100 ℃における過マンガン酸カリウムによる酸素消費量(CODMn) 試料を硫酸酸性とし,酸化剤と
して過マンガン酸カリウムを加え,沸騰水浴中で30分間反応させ,そのとき消費した過マンガン酸の量を
求め,相当する酸素の量(O mg/L)で表す。
この試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試
験する。
定量範囲:CODMn O 0.5〜11 mg/L
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA4の水(1) (2)
2) 硫酸(1+2) 水2容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸1容を徐々に加えた後,うすい紅色を呈するまで過マンガン酸カリウム溶液(3 g/L)を加える。
3) 硝酸銀溶液(200 g/L) JIS K 8550に規定する硝酸銀20 gを水に溶かして100 mLとする。着色ガ
39
K 0102:2016
ラス瓶に入れて保存する。
4) しゅう酸ナトリウム溶液(12.5 mmol/L) JIS K 8528に規定するしゅう酸ナトリウム1.8 gを水に溶
かして1 Lとする。ただし,5)の5 mmol/L過マンガン酸カリウム溶液のモル濃度の2.5倍より僅か
に高いモル濃度のものを調製する。
5) 5 mmol/L過マンガン酸カリウム溶液 JIS K 8247に規定する過マンガン酸カリウム0.8 gを平底フ
ラスコにとり,水1 050〜1 100 mLを加えて溶かす。これを1〜2時間静かに煮沸した後,一夜放置
する。
上澄み液を,ガラスろ過器G4を用いてろ過する(ろ過前後に水洗いしない)。ろ液は,約30分
間水蒸気洗浄した着色ガラス瓶に入れて保存する。
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質のしゅう酸ナトリウムをあらかじめ200 ℃で約1時
間加熱し,デシケーター中で放冷する。その約0.42 gを1 mgの桁まではかりとり,少量の水に
溶かして,全量フラスコ250 mLに移し入れ,水を標線まで加える。
− この溶液25 mLを三角フラスコ300 mLにとり,水で約100 mLとし,硫酸(1+2)10 mLを加
える。液温25〜30 ℃で,ビュレットでこの5 mmol/L過マンガン酸カリウム溶液約22 mLを一
度に加え,紅色が消えるまで放置する。
− 次に,50〜60 ℃に加熱し,この5 mmol/L過マンガン酸カリウム溶液で滴定する。終点は微紅
色を約30秒間保つときとする。
− 次の式によって,5 mmol/L過マンガン酸カリウム溶液のファクター(f)(3)を算出する。
675
001
.0
1
250
25
100
×
×
×
×
=
x
b
a
f
ここに,
a: しゅう酸ナトリウムの質量(g)
b: しゅう酸ナトリウムの純度(質量分率%)
x: 滴定に要した5 mmol/L過マンガン酸カリウム溶液量
(mL)
0.001 675: 5 mmol/L過マンガン酸カリウム溶液1 mLに相当するし
ゅう酸ナトリウムの質量(g)
注(1) 水はCODMn値を与える物質を含んでいてはならない。次のようにしてその適否を確認できる。
水100 mLについてc) 1)〜4)を行う。このときの滴定に要した5 mmol/L過マンガン酸カリウ
ム溶液の量をa mLとする。別に,水100 mLについて,加熱を除いたc) 1)〜4)の操作を行う。
このときの滴定に要した5 mmol/L過マンガン酸カリウム溶液の量をb mLとする。(a−b)mL
を求める。この値が0.15〜0.2 mL程度であればよい。これ以上の場合は,水(又は試薬)に有
機物が含まれていることが考えられ,使用に適しない。
(2) 水を蒸留精製する場合は,硫酸(1+2)を加えて微酸性とし,これに過マンガン酸カリウム溶
液(3 g/L)を加えて着色させた後,蒸留するとよい。ただし,蒸留の終わりまで過マンガン酸
の着色している状態を保つ。
(3) ファクターは,なるべく1に近い(0.95〜1.05)ものを使用する。
b) 器具 器具は,次による。
1) 水浴 試料を入れたとき,引き続いて沸騰状態を保てるような,熱容量及び加熱能力が大きなもの。
三角フラスコ300 mLが水浴の底に直接接触しないように,底から離して金網などを設ける。
c) 操作 操作は,次による。
40
K 0102:2016
1) 試料(4)の適量(5)を三角フラスコ300 mLにとり,水を加えて100 mLとし,硫酸(1+2)10 mLを加
え,振り混ぜながら硝酸銀溶液(200 g/L)5 mL(6) (7)を加える。
2) 5 mmol/L過マンガン酸カリウム溶液10 mLを加えて振り混ぜ,直ちに沸騰水浴中に入れ(8) (9),30
分間加熱する(10)。
3) 水浴から取り出し,しゅう酸ナトリウム溶液(12.5 mmol/L)10 mLを加えて振り混ぜ,よく反応さ
せる(11)。
4) 液温を50〜60 ℃で,5 mmol/L過マンガン酸カリウム溶液で僅かに赤い色を呈するまで滴定する。
5) 別に,水100 mLを三角フラスコ300 mLにとり,1)〜4)の操作を行う(12)。
6) 次の式によってCODMn(O mg/L)を算出する。
(
)
2.0
000
1
Mn
×
×
×
−
=
V
f
b
a
COD
ここに, CODMn: 100 ℃における過マンガン酸カリウムによる酸素消費量
(O mg/L)
a: 滴定に要した5 mmol/L過マンガン酸カリウム溶液量(mL)
b: 水を用いた試験の滴定に要した5 mmol/L過マンガン酸カリ
ウム溶液量(mL)
f: 5 mmol/L過マンガン酸カリウム溶液のファクター
V: 試料量(mL)
0.2: 5 mmol/L過マンガン酸カリウム溶液1 mLに相当する酸素
の質量(mg)
注(4) 懸濁物を含む場合には,よく振り混ぜて均一にした後,手早く採取する。
(5) 30分間加熱した後の5 mmol/L過マンガン酸カリウム溶液の残留量が4.5〜6.5 mLになるような
量。ただし,試料のCODMnがO 11 mg/L以下の場合には,100 mLとする。試料の適量は,c)
の操作によって予備試験を行って決める。
CODMnの概略値が分かっている場合には,次の式によって試料の適量(V mL)を求めること
ができる。
(
)
Mn
COD,
2.0
000
1
5.5
~
5.3
5.4
E
V
×
×
=
又は
ここに,
ECOD, Mn: 試料のCODMn予想値(O mg/L)
V: 試料の採取量(mL)
4.5(又は3.5〜5.5): 5 mmol/L過マンガン酸カリウム溶液の反応予
想量(mL)
0.2: 5 mmol/L過マンガン酸カリウム溶液1 mLに
相当する酸素の質量(mg)
(6) 硝酸銀溶液(200 g/L)に代え,これに対応する硝酸銀の粉末を加えてもよい。
(7) 試料中に塩化物イオンが存在する場合は当量になるまで加え,更に5 mLを加える。ただし,塩
化物イオンが多く,硝酸銀溶液(200 g/L)10 mL以上を必要とする場合には,硝酸銀溶液(500
g/L)を用いて当量よりも2 mL過剰に加えるか,又は粉末にした硝酸銀を当量よりも1 g過剰
に加え,更に水5 mLを加える。塩化物イオン1 gに対する硝酸銀(AgNO3)の当量は4.8 gで
ある。
通常の海水[塩化物イオン(18 g/L)]100 mLと当量の硝酸銀は8.64 gで,添加量は9.6 gと
なる。
(8) 多数の試料を一度に入れると,水浴の沸騰が止まるおそれがあるだけでなく,取り出したとき
41
K 0102:2016
のしゅう酸ナトリウム溶液(12.5 mmol/L)の添加操作の所要時間だけ加熱時間のずれが生じる
おそれがある。その所要時間だけの間隔をおいて入れるとよい。
注(9) 三角フラスコが倒れないように,その首に鉛製,鉄製などのリング状のおもりを付ける。
(10) このとき,三角フラスコ300 mL中の試料の液面は,沸騰水浴の水面下になるように保つ。
(11) 塩化銀に酸化マンガン(IV)が混入し,反応にやや時間を要することがある。
(12) 塩化物イオンの多い試料に硝酸銀溶液(200 g/L)5 mL以上を加えた場合も,この操作では,硝
酸銀溶液(200 g/L)5 mLを用いる。
備考 硝酸銀溶液(200 g/L)5 mLに代え,めのう乳鉢でよくすり潰した硫酸銀(JIS K 8965に規定す
る。)の粉末1 gを加えてもよい。ただし,5)でもJIS K 8965に規定する硫酸銀の粉末1 gを用
いる。塩化物イオンの多い試料では,塩化物イオンと当量よりも10 %多い量に,更に1 gを加
えた量を加える。
塩化物イオン1 gと当量の硫酸銀は4.4 gであり,
硫酸銀添加量:[塩化物イオン(g)×4.4×1.1+1](g)=[塩化物イオン(g)×4.84+1](g)
となる。通常の海水[塩化物イオン(18 g/L)]100 mLでは9.7 gとなる。
18. 欠番
19. アルカリ性過マンガン酸カリウムによる酸素消費量(CODOH) 試料をアルカリ性とし,酸化剤とし
て過マンガン酸カリウムを加え,沸騰水浴中で20分間反応させ,そのとき消費した過マンガン酸カリウム
の量を求め,相当する酸素の量(O mg/L)で表す。
この試験は,試料採取後,直ちに行う。直ちに行えない場合は,3.3によって保存し,できるだけ早く試
験する。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA4の水
2) 水酸化ナトリウム溶液(100 g/L) JIS K 8576に規定する水酸化ナトリウムを用いて調製する。
3) 硫酸(2+1) 水1容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸2容を徐々に加える。
4) アジ化ナトリウム溶液(40 g/L) JIS K 9501に規定するアジ化ナトリウム4 gを水に溶かして100 mL
とする。
5) でんぷん溶液(10 g/L) JIS K 8659に規定するでんぷん(溶性)1 gを水約10 mLと混ぜ,次に,
熱水100 mL中によくかき混ぜながら加え,約1分間煮沸した後,放冷する。使用時に調製する。
6) よう化カリウム溶液(100 g/L) JIS K 8913に規定するよう化カリウム10 gを水に溶かして100 mL
とする。使用時に調製する。
7) 過マンガン酸カリウム溶液(2 mmol/L) JIS K 8247に規定する過マンガン酸カリウム0.32 gを平
底フラスコにとり,水1 050〜1 100 mLを加えて溶かす。これを1〜2時間静かに煮沸した後,16時
間以上放置する。上澄み液をガラスろ過器(17G4又は25G4)を用いてろ過する(ろ過前後に水洗
いしない。)。ろ液は,約30分間水蒸気洗浄した着色ガラス瓶に入れて保存する。
8) 0.1 mol/Lチオ硫酸ナトリウム溶液 JIS K 8637に規定するチオ硫酸ナトリウム五水和物26 g及び
JIS K 8625に規定する炭酸ナトリウム0.2 gを2. n) 1)の溶存酸素を含まない水に溶かして1 Lとし,
気密容器に入れて少なくとも2日間放置する。標定は使用時に行う。
42
K 0102:2016
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質のよう素酸カリウムを130 ℃で約2時間加熱し,デ
シケーター中で放冷する。その約0.72 gを1 mgの桁まではかりとり,少量の水に溶かし,全量
フラスコ200 mLに移し入れ,水を標線まで加える。
− この20 mLを共栓三角フラスコ300 mLにとり,JIS K 8913に規定するよう化カリウム2 g及び
硫酸(1+5)(JIS K 8951に規定する硫酸を用いて調製する。)5 mLを加え,直ちに密栓して静
かに混ぜ,暗所に約5分間放置する。
− 水約100 mLを加えた後,遊離したよう素をこのチオ硫酸ナトリウム溶液で滴定し,溶液の黄
色が薄くなってから指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷん
の青い色が消えるまで滴定する。
− 別に,水について同条件で空試験を行って補正したmL数から,次の式によって0.1 mol/Lチオ
硫酸ナトリウム溶液のファクター(f)を算出する。
567
003
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: よう素酸カリウムの質量(g)
b: よう素酸カリウムの純度(質量分率%)
x: 滴定に要した0.1 mol/Lチオ硫酸ナトリウム溶液量(補正
した値)(mL)
0.003 567: 0.1 mol/Lチオ硫酸ナトリウム溶液1 mLに相当するよう素
酸カリウムの質量(g)
9) 10 mmol/Lチオ硫酸ナトリウム溶液 0.1 mol/Lチオ硫酸ナトリウム溶液25 mLを全量フラスコ250
mLにとり,2. n) 1)の溶存酸素を含まない水を標線まで加える。この溶液は使用時に調製し,12時
間以上経過したものは使用しない。
b) 器具 器具は,次による。
1) 共栓三角フラスコ 200 mL
2) 水浴 試料を入れたとき,引き続いて沸騰状態を保つことができる熱容量及び加熱能力が大きなも
の。
c) 操作 操作は,次による。
1) 試料(1)の適量(2)を共栓三角フラスコ200 mLにとり,水を加えて50 mLとし,水酸化ナトリウム溶
液(100 g/L)1 mLを加える。
2) 過マンガン酸カリウム溶液(2 mmol/L)10 mLを加えて振り混ぜ,直ちに沸騰水浴中に入れ,20分
間加熱する。このときフラスコ中の試料の液面は沸騰水浴の水面下で,かつ,共栓三角フラスコが
水浴の底に直接接しないように保つ。
3) 水浴から取り出し,冷水で室温まで冷却した後,アジ化ナトリウム溶液(40 g/L)1 mLを加えて振
り混ぜる。
4) よう化カリウム溶液(100 g/L)1 mL及び硫酸(2+1)0.5 mLを加え(3),栓をして振り混ぜ,暗所
に約5分間放置する。
5) 遊離したよう素を,10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の色がうすい黄色になった
ら,指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が消えるま
で滴定する。
6) 別に,水50 mLを共栓三角フラスコ200 mLにとり,水酸化ナトリウム溶液(100 g/L)1 mLを加え,
43
K 0102:2016
2)〜5)の操作を行う。
7) 次の式によってCODOH(O mg/L)を算出する。
(
)
08
.0
000
1
OH
×
×
×
−
=
V
f
a
b
COD
ここに, CODOH: アルカリ性過マンガン酸カリウムによる酸素消費量(O
mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
b: 水を用いた試験に要した10 mmol/Lチオ硫酸ナトリウム溶
液量(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター(4)
0.08: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する酸素の
質量(mg)
V: 試料量(mL)
注(1) 懸濁物を含む場合は,よく振り混ぜて均一にした後,採取する。
(2) 20分間加熱したときに最初に加えた過マンガン酸カリウム溶液(2 mmol/L)の約1/2が残るよ
うな量。ただし,アルカリ性過マンガン酸カリウムによる酸素消費量が8 mg/L以下の場合には,
50 mLとする。
試料の適量は,c)の操作によって予備試験を行って決める。
CODOHの概略値が分かっている場合には,次の式によって試料の適量(V mL)を求めること
ができる。
OH
COD,
08
.0
000
1
5
E
V
×
×
=
ここに, ECOD, OH: 試料のCODOH予想値(O mg/L)
V: 試料の採取量(mL)
5: 過マンガン酸カリウム溶液(2 mmol/L)の反応予想量(mL)
0.08: 過マンガン酸カリウム溶液(2 mmol/L)1 mLに相当する酸
素の質量(mg)
(3) 鉄を含むときは,硫酸(2+1)の添加前にふっ化カリウム溶液(300 g/L)(JIS K 8815に規定す
るふっ化カリウムを用いて調製する。)1 mLを加える。
(4) a) 8)の0.1 mol/Lチオ硫酸ナトリウム溶液のファクターを用いる。
20. 二クロム酸カリウムによる酸素消費量(CODCr) 二クロム酸カリウムを用いた酸素消費量の定量には,
滴定法又は蓋付き試験管を用いた吸光光度法を適用する。この試験は,試料採取後,直ちに行う。直ちに
行えない場合には,3.3によって保存し,できるだけ早く試験する。
この方法は廃液処理に注意を要する。
なお,蓋付き試験管を用いた吸光光度法によるCODCr測定法は,2002年に第1版として発行されたISO
15705との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 15705:2002,Water-quality−Determination of the chemical oxygen demand index (ST-COD)−
Small-scale sealed-tube method(MOD)
20.1 滴定法による酸素消費量(CODCr) 試料に二クロム酸カリウムと硫酸とを加え,還流冷却器を付け
44
K 0102:2016
て2時間煮沸した後,消費した二クロム酸の量を求め,相当する酸素の量(O mg/L)で表す。
この試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試
験する。
定量範囲:CODCr 1 mg/L以上
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA4の水
2) 硫酸銀-硫酸溶液 JIS K 8965に規定する硫酸銀11 gをJIS K 8951に規定する硫酸1 Lに溶かす。
溶けるまで1〜2日間を要するが,加熱して溶かしてもよい。
3) 硫酸水銀(II)
4) 二クロム酸カリウム溶液( mol/L) JIS K 8517に規定する二クロム酸カリウム1.23 gをとり,
水に溶かして1 000 mLとする。
5) 1,10-フェナントロリン鉄(II)溶液 JIS K 8789に規定する1,10-フェナントロリン一水和物(1) 1.48
gとJIS K 8978に規定する硫酸鉄(II)七水和物0.70 gとを水に溶かして100 mLとする。
6) 25 mmol/L硫酸アンモニウム鉄(II)溶液(2) JIS K 8979に規定する硫酸アンモニウム鉄(II)六水
和物10 gを水約500 mLに溶かし,JIS K 8951に規定する硫酸20 mLをかき混ぜながら徐々に加え
る。冷却した後,水を加えて1 Lとする。使用時に標定する。
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質の二クロム酸カリウムを,あらかじめ,めのう乳鉢
中で砕き,150 ℃で約1時間加熱し,デシケーター中で放冷する。二クロム酸カリウム0.25 g
を1 mgの桁まではかりとり,少量の水に溶かして全量フラスコ200 mLに移し入れ,水を標線
まで加える。
− この溶液20 mLを三角フラスコ300 mLにとり,水を加えて約100 mLとし,JIS K 8951に規定
する硫酸30 mLをかき混ぜながら徐々に加える。
− 冷却した後,指示薬として1,10-フェナントロリン鉄(II)溶液2,3滴を加えて,この25 mmol/L
硫酸アンモニウム鉄(II)溶液で滴定し,溶液の色が青緑から赤褐色に変わった点を終点とす
る。
− 次の式によって25 mmol/L硫酸アンモニウム鉄(II)溶液のファクター( f )を算出する。
226
001
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 二クロム酸カリウムの量(g)
b: 二クロム酸カリウムの純度(質量分率 %)
x: 滴定に要した25 mmol/L硫酸アンモニウム鉄(II)溶液
(mL)
0.001 226: 25 mmol/L硫酸アンモニウム鉄(II)溶液1 mLに相当す
る二クロム酸カリウムの質量(g)
注(1) JIS K 8202に規定する塩化1,10-フェナントロリニウム一水和物を用いる場合には,1.78 gをと
る。
(2) 25 mmol/L硫酸アンモニウム鉄(II)溶液1 mLは,二クロム酸カリウム溶液( mol/L)1 mL
に相当する。
b) 器具及び装置 器具及び装置は,次による。
1) 還流冷却器 共通すり合わせリービッヒ冷却器又は球管冷却器300 mm
240
1
240
1
45
K 0102:2016
2) 丸底フラスコ又は三角フラスコ 容量250〜300 mL。1)の還流冷却器と共通すり合わせのもの。
3) 加熱板又はマントルヒーター
c) 操作 操作は,次による。
1) 試料(3)の適量(4)をあらかじめ硫酸水銀(II)0.4 g (5)を入れた丸底フラスコ又は三角フラスコにとり,
水を加えて20 mLとし,よく振り混ぜる。
2) 二クロム酸カリウム溶液( mol/L)10 mLを加え,注意してよく振り混ぜながら硫酸銀-硫酸溶
液30 mLを加えた後,沸騰石5〜7個を入れる。
3) 還流冷却器を付けて2時間煮沸する。
4) 冷却した後,水約10 mLで還流冷却器を洗って洗液をフラスコに流し入れ,更に水で約140 mLに
薄める。
5) 指示薬として1,10-フェナントロリン鉄(II)溶液2,3滴を加えて,過剰の二クロム酸を25 mmol/L
硫酸アンモニウム鉄(II)溶液で滴定し,溶液の色が青緑から赤褐色に変わった点を終点とする。
6) 別に,水20 mLをとり,1)〜5)の操作を行う。
7) 次の式によってCODCr(O mg/L)を算出する。
2.0
000
1
)
(
Cr
×
×
×
−
=
V
f
a
b
COD
ここに, CODCr: 二クロム酸カリウムによる酸素消費量(O mg/L)
a: 滴定に要した25 mmol/L硫酸アンモニウム鉄(II)溶液(mL)
b: 水を用いた試験の滴定に要した25 mmol/L硫酸アンモニウ
ム鉄(II)溶液(mL)
f: 25 mmol/L硫酸アンモニウム鉄(II)溶液のファクター
V: 試料量(mL)
0.2: 25 mmol/L硫酸アンモニウム鉄(II)溶液1 mLに相当する
酸素の質量(mg)
注(3) 懸濁物を含む場合には,よく振り混ぜて均一にした後,手早く採取する。
(4) 2時間煮沸した後に最初に加えた二クロム酸カリウム溶液( mol/L)の約1/2が残るような
量。
(5) 塩化物イオン40 mgをマスキングするが,海水のように塩化物イオンの濃度が高い場合には,
妨害を除去できないので,この方法は適用できない。
20.2 蓋付き試験管を用いた吸光光度法によるCODCr測定法 この方法は,二クロム酸カリウム溶液,硫
酸水銀(II)溶液,硫酸銀-硫酸溶液の入った蓋付き試験管に試料を入れ,150±5 ℃で2時間加熱した後,
波長600 nmの吸光度を測定し,CODCrの濃度を求めるものである。
定量範囲:CODCr 150〜1 000 mg/L
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA4の水
2) 二クロム酸カリウム溶液(0.1 mol/L) JIS K 8517に規定する二クロム酸カリウムをあらかじめ
105 ℃で2時間加熱し,デシケーター中で放冷する。その29.4 gをはかりとり,水に溶かして約600
mLとし,JIS K 8951に規定する硫酸160 mLをかき混ぜながら徐々に加えて,水で1 000 mLとす
る。
3) 硫酸水銀(II)溶液 硫酸水銀(II)80 gを硫酸(1+9)(JIS K 8951に規定する硫酸を用いて調製
する。)200 mL に溶かす。
240
1
240
1
46
K 0102:2016
4) 硫酸銀-硫酸溶液 20.1 a) 2)による。
5) フタル酸水素カリウム溶液 JIS K 8005に規定する容量分析用標準物質のフタル酸水素カリウムを
105±5 ℃で2時間加熱し,デシケーター内で放冷する。その4.251 gをはかりとり,水で溶かし,
全量フラスコ500 mLに移し入れ,水を標線まで加える。この溶液は,CODCrの10 000 mg/Lに相当
する。検量線作成に用いる。
b) 器具及び装置 器具及び装置は,次による。
1) 蓋付き試験管 耐熱・耐圧・耐酸性のねじ蓋付きガラス製試験管で150±5 ℃,600 kPaで使用でき
るもの。
参考 分解用試薬が入った蓋付き試験管が市販されている。例えば,外径 14 mm,厚さ1 mm,高さ
185 mmのものがある。
2) ブロックヒーター 150±5 ℃を維持できるもので,ヒーターの壁が蓋付き試験管に密着するもの。
内容物を加熱するのに十分な深さがあるものが望ましい。
3) 光度計 分光光度計(6)
注(6) 分解後の試験管をそのまま設置して測定できる光度計を用いてもよい。
c) 操作 操作は,次による。
1) 蓋付き試験管に二クロム酸カリウム溶液(0.1 mol/L)0.5 mL,硫酸水銀(II)溶液0.2 mL及び硫酸
銀-硫酸溶液2.5 mLをとり,静かに振り混ぜる。
2) 1) の蓋付き試験管に,試料(7) (8) (9) 2 mLを加えて蓋をし,静かに振り混ぜる。
3) あらかじめ150±5 ℃に予熱しておいたブロックヒーターで蓋付き試験管を2時間加熱する。
4) ブロックヒーターから取り出し,60 ℃以下まで放置し,蓋付き試験管を振り混ぜた後,更に室温ま
で放置する。
5) 分解・放冷後の溶液(10)の一部を吸収セルに移し(11),波長600 nmの吸光度を測定する。
6) 空試験として水2 mLを用いて,1)〜5)の操作を行い,試料について得た吸光度を補正する。
7) 検量線からCODCr濃度を求める。
注(7) 試料に濁りがある場合は,ホモジナイザー又はミキサーで懸濁物質を均一化する。
(8) 試料の塩化物イオン濃度が1 000 mg/L以上ある場合はこの方法は適用できない。
(9) 試料にマンガンが多量に存在すると着色,沈殿が生じるため,この方法は適用できない。
(10) 分解後の溶液に着色,濁りなどがある場合は滴定法を適用する。
(11) 分解液を移し替える際は,沈殿を含まないよう注意する。
d) 検量線 検量線の作成は,次による。
1) フタル酸水素カリウム溶液15〜100 mLを全量フラスコ1 000 mLに段階的にとり,水を標線まで加
える。
2) この溶液についてc) 1)〜6) の操作を行ってCODCr濃度と吸光度との関係線を作成する。
備考1. CODCr濃度が150 mg/L以下の試料には次の方法を適用する。この場合の定量範囲は,30〜150
mg/Lとする。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA4の水
2) 二クロム酸カリウム溶液(0.015 mol/L) JIS K 8517に規定する二クロム酸カリウムを
あらかじめ105 ℃で2時間加熱し,デシケーター中で放冷する。その4.41 gをはかり
とり,水に溶かして約600 mLとし,JIS K 8951に規定する硫酸160 mLをかき混ぜな
47
K 0102:2016
がら徐々に加えて,水で1 000 mLとする。
3) 硫酸水銀(II)溶液 20.2 a) 3) による。
4) 硫酸銀-硫酸溶液 20.2 a) 4) による。
5) フタル酸水素カリウム溶液 20.2 a) 5) による。
b) 器具及び装置 器具及び装置は,20.2 b) による。
c) 操作 操作は,次による。
1) 蓋付き試験管に二クロム酸カリウム溶液(0.015 mol/L)0.5 mL,硫酸水銀(II)溶液
0.2 mL及び硫酸銀-硫酸溶液2.5 mLをとり,静かに振り混ぜる。
2) 20.2のc) 2)〜4)の操作を行う。
3) 分解・放冷後の溶液(10)の一部を吸収セルに移し(11),波長440 nmの吸光度を測定する。
4) 空試験として水を2 mLを用いて,1)〜3)の操作を行い,試料について得た吸光度を補
正する。
5) 検量線からCODCr濃度を求める。
d) 検量線 検量線の作成は,次による。
1) フタル酸水素カリウム溶液3〜15 mLを全量フラスコ1 000 mLに段階的にとり,水を
標線まで加える。
2) この溶液についてc) 1)〜4)の操作を行ってCODCr濃度と吸光度との関係線を作成する。
21. 生物化学的酸素消費量(BOD) 生物化学的酸素消費量とは,水中の好気性微生物によって消費され
る溶存酸素の量をいう。試料を希釈水で希釈し,20 ℃で5日間放置したとき消費された溶存酸素の量(O
mg/mL)から求める。
この試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試
験する。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水(1)
2) 塩酸(1+11) JIS K 8180に規定する塩酸を用いて調製する。
3) 水酸化ナトリウム溶液(40 g/L) JIS K 8576に規定する水酸化ナトリウム4 gを水に溶かして100 mL
とする。
4) 緩衝液(pH7.2)(A液) JIS K 9017に規定するりん酸水素二カリウム21.75 g,JIS K 9007に規定
するりん酸二水素カリウム8.5 g,JIS K 9019に規定するりん酸水素二ナトリウム・12水44.6 g及び
JIS K 8116に規定する塩化アンモニウム1.7 gを水に溶かして1 Lとする。この緩衝液のpHは7.2
である。
5) 硫酸マグネシウム溶液(B液) JIS K 8995に規定する硫酸マグネシウム七水和物22.5 gを水に溶か
して1 Lとする。
6) 塩化カルシウム溶液(C液) JIS K 8123に規定する塩化カルシウム27.5 gを水に溶かして1 Lとす
る。
7) 塩化鉄(III)溶液(D液) JIS K 8142に規定する塩化鉄(III)六水和物0.25 gを水に溶かして1 L
とする。使用時に調製する。
8) 亜硫酸ナトリウム溶液(12.5 mmol/L) JIS K 8061に規定する亜硫酸ナトリウム1.6 gを水に溶かし
て1 Lとする。使用時に調製する。
48
K 0102:2016
9) よう化カリウム JIS K 8913に規定するもの。
10) 希釈水 水温を20 ℃近くに調節し,ばっ気して溶存酸素を飽和させた水(2) 1 Lに対して,A液,B
液,C液及びD液をそれぞれ1 mLずつ加える。この溶液のpHは7.2である[pH7.2を示さないと
きは,塩酸(1+11)又は水酸化ナトリウム溶液(40 g/L)でpH7.2に調節する。]。希釈水は,培養
瓶に詰めて20 ℃の恒温槽に5日間放置したとき,初めの溶存酸素の量と5日間後の溶存酸素の量
との差がO 0.2 mg/L以下であることをあらかじめ確認しておく(3)。
11) 植種液 下水の上澄み液(4) (5),河川水(6),土壌抽出液(7)などを用いる。
12) 植種希釈水(8) 試験に際し,植種液の適量(9)を希釈水に加えて,植種希釈液を調製する。
注(1) 石英ガラス又はほうけい酸ガラス−1製の蒸留器で精製したもの。又は,同等のもの。
(2) 洗浄して汚染物質を除いた空気を通して,溶存酸素を飽和させるとよい。空気の洗浄は,次に
よる。
空気を活性炭ろ過器[例えば,粒状活性炭をガス乾燥塔(300 mm)に充塡する。]でろ過し,
次に,硫酸酸性にした過マンガン酸カリウム溶液(5 g/L)(JIS K 8247に規定する過マンガン酸
カリウム溶液を用いて調製する。)で洗い,更に水酸化カリウム溶液(250 g/L)(JIS K 8574に
規定する水酸化カリウム溶液を用いて調製する。)で洗う。
(3) 生物化学反応は,含まれている有機物の濃度及び微生物の種類によって異なるため,希釈水に
ついて空試験を行って補正することは困難である。このため,希釈水は,5日間の酸素消費量
がO 0.2 mg/L以下のものを用いる。
(4) 植種液には,家庭生下水がよく用いられる。新鮮な生下水を20 ℃(又は室温)で24〜36時間
放置後,その上澄み液を用いる。下水中に硝化生物(アンモニウムイオン及び亜硝酸イオンを
酸化する生物)の多いもの及び十分な生物化学的平衡に達していない新鮮な下水は好ましくな
い。
(5) 植種液として下水を用いた場合に,正常なBODを示さない試料には,植種液として土壌抽出液
を用いるか,又は試験室でこの試料にならした微生物を培養し,この培養液を用いる。
(6) 常時この試料の放流を受けている河川の放流地点から500〜1 000 m下流の水を植種に用いると
良好な結果を得ることがある。試料中に生物化学的反応に有害な物質が共存しても,その試料
の放流を受けている河川,湖沼などには,耐性をもった生物相が発達していることが多いから
である。
(7) 土壌(植物の生育している土壌)約200 gを水2 L中に加えてかき混ぜた後,その上澄み液を用
いる。
(8) 試料中に好気性の微生物及び細菌が存在しない場合,又はその数が不足している場合に,植種
希釈水を用いる。
試料のBODの試験に植種希釈水を用いるときは,次の操作によって植種希釈水の調製に用い
た植種液についての補正(植種補正)を行う。
植種液を希釈水で適切に希釈して数段階の希釈した植種液を調製し,希釈試料と並行して測
定する。
希釈した植種液の培養前の溶存酸素の量をB1,5日間放置後の溶存酸素の量をB2とするとき
×100が40〜70 %の範囲内にあるものを選び,植種補正値として(B1−B2)×fを用いる
[d) 4)のBODの算出式を参照]。植種希釈水の5日間の溶存酸素の消費量を求めて補正を行っ
てはならない。
(
)
1
2
1
B
B
B−

49
K 0102:2016
注(9) 微生物が正常な活動をするために,植種希釈水のBODがO 0.6〜1 mg/Lになるように加える。
通常,希釈水1 Lに対し,下水の上澄み液では5〜10 mL,河川水では10〜50 mL,土壌抽出液
では20〜30 mL程度である。
b) 器具及び装置 器具及び装置は,次による。
1) 培養瓶 正確に容量の分かっている細口共栓ガラス瓶(100〜300 mL)で,共栓は斜めに切り落と
したもの。図21.1に例を示す。
図21.1 培養瓶の例
2) 恒温器 恒温器は,温度を20±1 ℃に調節できるものを用いる。希釈試料中の藻類による二酸化炭
素同化作用(炭酸同化作用)を防ぐために,光を遮断しておく。同様な仕様の恒温水槽を用いても
よい。
c) 試料の前処理 試料の前処理は,次による。
試料に酸,アルカリ,残留塩素などの酸化性物質,過飽和の溶存酸素又は溶存気体が含まれている
場合には,次の前処理を行う。また,前処理によって液量が増加する場合には,増加分について結果
を補正する。
1) アルカリ又は酸を含む試料 塩酸(1+11)又は水酸化ナトリウム溶液(40 g/L)を加えて試料のpH
を約7にする。
2) 残留塩素などの酸化性物質を含む試料 あらかじめ試料100 mLにJIS K 9501に規定するアジ化ナ
トリウム0.1 gとよう化カリウム1 gとを加えて振り混ぜた後,塩酸(1+1)(JIS K 8180に規定す
る塩酸を用いて調製する。)を加えてpHを約1とし,暗所に数分間放置する。遊離したよう素をで
んぷん溶液を指示薬として亜硫酸ナトリウム溶液(12.5 mmol/L)でよう素でんぷんの青い色が消え
るまで滴定する。別に,同量の試料をとり,先の滴定値から求めた計算量の亜硫酸ナトリウム溶液
(12.5 mmol/L)を加えて残留塩素を還元した後,必要ならば水酸化ナトリウム溶液(40 g/L)又は
塩酸(1+11)を用いてpH約7とする。
3) 溶存酸素又は溶存気体が過飽和の試料 冬季に採取した処理水,河川水などの試料の温度が20 ℃
以下のときは,20 ℃にしたときに溶存酸素及び溶存気体が過飽和になりやすい。また,緑藻類の多
い河川水及び湖沼水では,二酸化炭素同化作用によって酸素が発生するので,溶存酸素が過飽和に
なりやすい。これらの試料では,BOD測定中に溶存酸素が気体となり,培養瓶外に散逸し,結果が
不正確になるから,あらかじめかき混ぜるか,ばっ気するなどの方法によって溶存酸素及び溶存気
体を20 ℃の飽和量近くに減少させておく。
50
K 0102:2016
備考 1. 生物化学的処理が済んだ試料には,炭素質の有機物を分解する好気性細菌のほかにアンモニ
アなどの窒素化合物を酸化(硝化)する硝化細菌が繁殖していることがある。このような試
料では,有機物の酸化分解に消費される酸素と,アンモニアなどの窒素化合物の硝化に消費
される酸素との合量が測定される。この硝化による酸素の消費量は,試料中の窒素化合物の
量に対応するものではなく,硝化細菌の数によって変化する。
硝化作用を抑制した状態の生物化学的酸素消費量を測定するには,次の操作を行う。
d) 1)の希釈試料の調製時に,希釈試料1 LについてN-(2-プロペニル)チオ尿素2 mg(*)
又は2-クロロ-6-(トリクロロメチル)ピリジンの粉末10 mg(**)が含まれるように添加する。
注(*) N-(2-プロペニル)チオ尿素(N-アリルチオ尿素)溶液(1 mg/mL)[N-(2-プロペ
ニル)チオ尿素0.1 gを水に溶かして100 mLとする。0〜10 ℃の暗所に保存する。]
2 mLを加える。
(**) 2-クロロ-6-(トリクロロメチル)ピリジンは,水に溶けにくいので粉末で加える。
添加した後も完全には溶けず一部は浮上することがあるので,培養瓶に移す場合な
ど注意する。水に溶けやすいように,他の試薬と混合したものがあり,これを用い
てもよい。
d) 操作 操作は,次による。
1) 希釈試料の調製 希釈試料の調製は,次による。
− サイホンを用い,泡が入らないように注意して希釈水又は植種希釈水をメスシリンダー(有栓形)
1 000 mL[培養瓶が200 mL以上の場合には,メスシリンダー(有栓形)2 000 mLを用いる。]に
約半分までとる。
− 次に,前処理を行った試料の適量(10) (11) (12)を加え,希釈水又は植種希釈水を1 000 mLの標線[メ
スシリンダー(有栓形)2 000 mLの場合には,2 000 mLの標線]まで加える。栓をして静かに混
ぜ合わせる。
− この溶液を希釈水あるいは植種希釈水で希釈するか又は試料の量を変えて同じ操作を行い,段階
的に希釈倍数の異なる希釈試料4,5種類を調製する(13) (14) (15)。
− 調製したそれぞれの希釈試料について,培養瓶2〜4本を用意し,これらにサイホンを用いて希釈
試料を移し入れ,十分にあふれさせた後,密栓する。
− 希釈倍数の異なる各組の培養瓶のうち1本は,培養前の溶存酸素の定量に用い,ほかは20±1 ℃
に調節した恒温器に入れて5日間培養する(16) (17)。
2) 培養前の希釈試料の溶存酸素量の測定 希釈試料を調製後15分間放置し(18),溶存酸素を32.1,32.2,
32.3又は32.4によって定量する。32.2,32.3又は32.4による場合は,メスシリンダー中に残った希
釈試料を用いて溶存酸素の量を測定してもよい。
3) 培養後の溶存酸素の測定 恒温器の中に5日間培養した希釈試料の溶存酸素の量を2)と同じ方法で
測定する。
4) BODの算出 培養前後の溶存酸素の量から,次の式によって試料のBOD(O mg/L)を算出する(19)。
4.1) 植種を行わない場合
(
)
P
D
D
BOD
2
1−
=
ここに, BOD: 生物化学的酸素消費量(O mg/L)
D1: 希釈試料を調製してから15分間後の溶存酸素の濃度(O mg/L)
51
K 0102:2016
D2: 培養後の希釈試料の溶存酸素の濃度(O mg/L)(20)
P: 希釈試料中の試料の占める割合(試料/希釈試料)
4.2) 植種希釈水を用いた場合(19)
(
)(
)
P
f
B
B
D
D
BOD
×
−
−
−
=
2
1
2
1
ここに, BOD: 生物化学的酸素消費量(O mg/L)
D1: 希釈試料を調製してから15分間後の溶存酸素の濃度(O mg/L)
D2: 培養後の希釈試料の溶存酸素の濃度(O mg/L)(20)
P: 希釈試料中の試料の占める割合(試料/希釈試料)
B1: 植種液のBODを測定する場合の希釈した植種液の培養前の溶
存酸素の濃度(O mg/L)
B2: 植種液のBODを測定する場合の希釈した植種液の培養後の溶
存酸素の濃度(O mg/L)
f:
y
x
x: 試料のBODを測定する場合の希釈試料中の植種液(%)
y: 植種液のBODを測定する場合の希釈した植種液中の植種
液(%)
注(10) 試料の正常なBODを得るための希釈試料の溶存酸素の消費量(D1−D2)は,O 3.6〜6.4 mg/L
の範囲である。希釈不足のため残留する溶存酸素の量がO 1 mg/L以下である,又は逆に希釈し
過ぎて溶存酸素の消費量がO 2 mg/L以下となる場合には,正常なBODを得にくい。
(11) 試料のBODが,経験その他で予想できれば,採取する試料の量は,次のようにして求める。
例えば,20 ℃における溶存酸素の飽和量は,O 9.09 mg/Lであり,これの40 %は約O 3.6 mg/L,
70 %は約O 6.4 mg/Lであるから,希釈水と試料とを合わせて1 Lにする場合には,採取する試
料の量(V mL)は,次の式で得られる。
(
)
BOD
000
1
4.6
~
6.3
E
V
×
=
ここに, EBOD: 試料のBOD予想値(O mg/L)
V: 試料の採取量(mL)
3.6: 20 ℃における溶存酸素の飽和量(O 9.09 mg/L)の40 %相当量
6.4: 20 ℃における溶存酸素の飽和量(O 9.09 mg/L)の70 %相当量
試料のBODがO 5 mg/L以下の場合には,試料の採取量は800 mL以上とする。また,溶存酸
素が十分に含まれていない場合には,ばっ気した後,試験する。
(12) 懸濁物を含む試料の場合には,懸濁物が均一になるように混ぜ合わせた後,適量をとる。
(13) メスシリンダー中に残っている希釈試料を基にして,順次,希釈倍数の高い希釈試料を調製し
続けると,労力及び時間を節約することができる。
(14) 100倍以上に希釈する場合には,一度に希釈せずに,あらかじめ他のメスシリンダー(有栓形)
1 000 mLに試料50〜100 mLをとり,希釈水又は植種希釈水を標線まで加える。この希釈した
試料を用いて1)の希釈試料を調製する。
(15) BODがO 100 mg/L以下の試料の場合には,次の方法によって培養瓶で直接希釈してもよい。
容量の正確に分かっている培養瓶4本を用意し,それぞれにあらかじめ約半量の希釈水又は
植種希釈水を入れておき,次に,希釈倍数に応じて瓶の容量に対する計算量の試料を加え,更
に,瓶内の空間を希釈水又は植種希釈水で満たす。この操作中,泡が入らないように注意する。
(16) 恒温水槽を使用する場合には,培養瓶全体を水に浸す。
52
K 0102:2016
注(17) 培養瓶を水封して恒温器中におく場合には,水封水が蒸発するから,ときどき補充する。
(18) 硫化物,亜硫酸塩,鉄(II)などの還元性物質が共存する場合には,15分間の酸素消費量(Immediate
Dissolved Oxygen Demand,以下,IDODと略記する。)とBODとを区別する。IDODを求めるに
は,次のように操作する。
あらかじめ試料と希釈水との溶存酸素を測定した後,一定の割合で試料を希釈水で薄め,15
分間放置した後,溶存酸素(D1)をはかる。初めに測定した試料と希釈水それぞれの溶存酸素
(O mg/L)とから希釈試料の溶存酸素(Dc)を算出し,次の式によって試料のIDOD(O mg/L)
を算出する。
P
D
D
IDOD
1
c−
=
ここに, IDOD: 15分間の酸素消費量(O mg/L)
Dc: 培養前の希釈試料水の溶存酸素の濃度(O mg/L)=(S×P)+
(Do×p)
S: 試料の溶存酸素の濃度(O mg/L)
Do: 希釈水の溶存酸素の濃度(O mg/L)
p: 希釈試料中の希釈水の占める割合
(希釈水/希釈試料)
P: 希釈試料中の試料の占める割合(試料/希釈試料)
D1: 希釈試料を調製してから15分間放置後の溶存酸素の濃度(O
mg/L)
(19) 次のような方法で算出してもよい。
(
)
(
)
(
)
100
1
1
2
2
1
1
2
1
−
×
×
−
−
×
−
=
n
V
n
B
B
n
D
D
BOD
ここに,
D1: 希釈試料を調製してから15分間後の溶存酸素の濃度(O mg/L)
D2: 培養後の希釈試料の溶存酸素の濃度(O mg/L)
n1: 希釈試料の希釈倍数
試料
希釈試料
n2: 植種液のBOD測定時の希釈倍数
)
測定における植種液(
植種液の
)
植種液(
測定における希釈した
植種液の
mL
mL
BOD
BOD
B1: 植種液のBOD測定における培養前の希釈した植種液の溶存酸
素の濃度(O mg/L)
B2: 植種液のBOD測定における培養後の希釈した植種液の溶存酸
素の濃度(O mg/L)
V: 植種希釈水中に含まれる植種液の体積百分率(%)
通常
(
)
2
2
1
100
6.0
n
B
B
V
×
−
×
>
になるようにする。
(20) 5日間の溶存酸素の消費量(D1−D2)がO 3.6〜6.4 mg/L以内, ×100=40〜70 %の範囲
内の値のものをとり,BODを算出する。この条件の中央値付近になるのが最も望ましい。ただ
し,試料のBODがO 3.6 mg/L以下のときは,希釈しない場合でも5日間の溶存酸素の消費量
は,溶存酸素の飽和量の40 %以上にならない。このような場合には,その値から算出する。
備考 2. 植種液の調製方法 微生物を試料にならすための培養は,次の方法で行うとよい。
1) ガラス水槽(約6 L)に試料5 Lを入れ,塩酸(1+11)又は水酸化ナトリウム溶液(40 g/L)
を用いてpH約7としておく。微生物が豊富に存在する下水,河川水などを植種液とし,
1
2
1
D
D
D−
53
K 0102:2016
その100〜300 mL及び緩衝液(A液)10〜50 mLを加える。よくかき混ぜた後,その一
部をとり,CODMn又は有機体炭素の量を測定しておく。
2) 次に,24〜48時間連続ばっ気した後,再びその一部をとり,CODMn又は有機体炭素の量
を測定する。前後の測定値に顕著な変化がある場合には,試料中に生物化学反応が進行
しているものと判定し,更にばっ気を続けて試料に適応する生物を繁殖させる。
3) もし,顕著な変化が生じない場合には,別に試料をとり,希釈水で適切に希釈した後,
前記と同様に植種を行い,24〜48時間連続ばっ気し,CODMn又は有機体炭素の量の変化,
懸濁物の量の変化などを試験する。その結果,これらに顕著な変化がある場合には,生
物化学反応が活発になっていることの現れである。試料中の有機物の組成によっては,
このような操作を1週間以上試みる必要がある。
4) また,試料を希釈水で10倍以上に希釈して上記の操作を行った場合,CODMn又は有機
体炭素の量に顕著な変化を生じたら,徐々に試料の比率を増加してみることも必要であ
る。このように試料に適応する微生物を培養し,植種液として用いる。
備考 3. 試料操作の確認の方法 植種液,植種希釈水などの使用の適否,又は試験操作を確認するた
めに,次の方法を推奨する。
グルコース-グルタミン酸混合標準液[JIS K 8824に規定するD(+)-グルコース150 mg
及びJIS K 9047に規定するL-グルタミン酸150 mgをとり,水に溶かして全量フラスコ1 000
mLに移し入れ,水を標線まで加える。]5〜10 mLを容量の正確に分かった培養瓶300 mL(培
養瓶が100 mLの場合には,前記の1/3量を用いる。)にとり,植種希釈水を満たして密栓し,
BODを測定する。この標準液のBODは,O 220±10 mg/Lである。もし,この値からの偏差
が著しい場合には,希釈水の水質,植種液の活性度などに疑問がある。
4. 試料中に銅,クロム,水銀,銀,ひ素などの重金属元素が溶存していると正しい測定値が得
られないことがある。このような場合には,これらの重金属元素によくならした植種液を備
考2.によって培養しておく。
22. 有機体炭素(TOC) 有機体炭素とは,水中に存在する有機物中の炭素をいう。これの定量には,燃
焼酸化-赤外線式TOC分析法又は燃焼酸化-赤外線式TOC自動計測法を適用する。
この試験は,試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く
試験する。
試料中に元素状態で存在する炭素の粒子(すす),炭化物,シアン化物イオン,シアン酸イオン及びチオ
シアン酸イオンが存在する場合には,有機体炭素として定量される。
なお,有機体炭素(TOC)の定量は,1999年に第2版として発行されたISO 8245との整合を図ったも
のである。
備考 − この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8245:1999,Water quality−Guidelines for the determination of total organic carbon(TOC)and
dissolved organic carbon(DOC)(MOD)
− 全炭素(TC),無機体炭素(TIC)及び有機体炭素(TOC)の定義は,次による。
1) 全炭素(TC) 水中に存在する有機的に結合した炭素と,無機的に結合した炭素(元素状
54
K 0102:2016
の炭素を含む。)との合量。
2) 無機体炭素(TIC) 水中に存在する無機体の炭素の合量。すなわち,元素状,全二酸化炭
素,一酸化炭素,シアン化物イオン,シアン酸イオン及びチオシアン酸イオン(*)中の炭素
の合量。
3) 有機体炭素(TOC) 水中に存在する有機的に結合した炭素の合量。すなわち,溶存及び
懸濁状で存在する物質中の有機的に結合した炭素,シアン酸イオン,チオシアン酸イオン
中の炭素の合量。
注(*) TOC分析計で,TICを二酸化炭素として測定する場合,そのほとんどは炭酸水素イ
オン及び炭酸イオンだけに起因するとしている。
22.1 燃焼酸化-赤外線式TOC分析法 少量の試料を二酸化炭素を除去した空気又は酸素とともに高温の
全炭素測定管に送り込み,有機物中の炭素及び無機物[無機体炭素(主として炭酸塩類)]中の炭素を二酸
化炭素とした後,その濃度を非分散形赤外線ガス分析計で測定して全炭素(TC)の量を求める。
別に,試料を有機物が分解されない温度に保った無機体炭素測定管に送り込み,生成した二酸化炭素を
測定し,無機体炭素(TIC)の量を求める。
全炭素の量から無機体炭素の量を差し引いて有機体炭素(TOC)の量を算出する。
定量範囲:C 0.5〜25 mg/L,20〜100 mg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3又はA4の水(1) (2) (3)で,二酸化炭素を含まない水(4)を用いる。試薬
の調製及び操作には,この水を用いる。e)によって空試験を行い,使用の適否を確認しておく。
2) TOC標準液(C 1 mg/mL) JIS K 8005に規定する容量分析用標準物質のフタル酸水素カリウムを
120 ℃で約1時間加熱し,デシケーター中で放冷する。その2.125 gをとり,少量の水に溶かして全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。
3) TOC標準液(C 0.1 mg/mL) TOC標準液(C 1 mg/mL)10 mLを全量フラスコ100 mLにとり,水
を標線まで加える。
4) 無機体炭素標準液(C 1 mg/mL) JIS K 8622に規定する炭酸水素ナトリウムをデシケーター中で約
3時間放置し,その3.497 gをとる。別に,JIS K 8005に規定する容量分析用標準物質の炭酸ナトリ
ウムを,あらかじめ600 ℃で約1時間加熱し,デシケーター中で放冷し,その4.412 gをとる。両
者を少量の水に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
5) 無機体炭素標準液(C 0.1 mg/mL) 無機体炭素標準液(C 1 mg/mL)10 mLを全量フラスコ100 mL
にとり,水を標線まで加える。
6) 全炭素測定管 全炭素定量用触媒を充塡したもの。
7) 無機体炭素測定管 無機体炭素定量用触媒を充塡したもの。
8) キャリヤーガス 二酸化炭素を除去した空気又はJIS K 1101に規定する酸素。
注(1) TOCの濃度をできるだけ低くした水を用いる。精製した水は,容器に入れて保存すると徐々に
汚染されてTOCの濃度が高くなることがあるので,精製後は早く使用することが望ましい。
(2) TOCの濃度をできるだけ低くするには,A2又はA3の水を蒸留フラスコにとり,過マンガン酸
カリウム溶液(3 g/L)(JIS K 8247に規定する過マンガン酸カリウムを用いて調製する。)を着
色するまで滴加し,水1 000 mLにつき硫酸(1+1)[5.4 a) 2)による。]2〜3 mLを加えて蒸留
する(蒸留が終わるまで過マンガン酸カリウムによる着色が残るようにする。)。初留分(蒸留
フラスコ中の水量の約1/5に相当する。)を捨て,中間の約1/3に相当する留分を採取する。
55
K 0102:2016
注(3) イオン交換法,蒸留法,逆浸透法,紫外線照射法,活性炭吸着ろ過法,限外ろ過法,精密ろ過
法などを,適宜,組み合わせて精製した水も使用できる。
(4) 2. n) 2)によって精製する。
b) 器具及び装置 器具及び装置は,次による。
1) マイクロシリンジ 20〜150 μL又は自動注入装置
2) TOC分析装置
3) ホモジナイザー又はミキサー 分散した物質の均質化に十分な能力をもつもの。超音波装置,マグ
ネチックスターラーなど。
c) 準備操作 準備操作は,次による。
1) TOC分析装置を作動できる状態にする。
2) TOC標準液(C 1 mg/mL)又はTOC標準液(C 0.1 mg/mL)の一定量(5)(例えば,20 μL)をマイク
ロシリンジでTOC分析装置の全炭素測定管に注入し,指示値(ピーク高さ又はピーク面積)を読み
取る。
3) 2)の操作を5〜7回繰り返して指示値が一定になることを確かめる。
4) 試料をよく振り混ぜて均一にした後,2)と同量をマイクロシリンジで全炭素測定管に注入して,指
示値を読み取り,2)と比較して試料中の概略の全炭素の濃度(C mg/L)を求める。
注(5) 試料の炭素の濃度が低い場合には,注入量は100〜150 μLとし,炭素の濃度が高い場合には,
注入量を少なくするか,又は一定の倍数に薄める。
d) 検量線 検量線の作成は,次による。
1) c) 4)で求めた試料の概略の炭素の濃度がほぼ中央になるようにTOC標準液(C 1 mg/mL)又はTOC
標準液(C 0.1 mg/mL)を全量フラスコ100 mLに段階的にとり,水を標線まで加える。
2) 1)で調製したTOC標準液の最高濃度のものの一定量[例えば,c) 2)と同量]をマイクロシリンジで
全炭素測定管に注入して指示値が最大目盛値の約80 %になるようにTOC分析装置の感度及び標準
液の注入量を調節する。
3) 1)で調製した各濃度のTOC標準液の一定量[2)で定めた量]を,順次,マイクロシリンジで全炭素
測定管に注入して指示値を読み取る。
4) 空試験として,3)と同量の水をマイクロシリンジで全炭素測定管に注入して,指示値を読み取り,
3)の結果を補正し,有機体炭素の量と指示値との関係線を作成して,これを全炭素の検量線とする。
5) 無機体炭素標準液(C 1 mg/mL)又は無機体炭素標準液(C 0.1 mg/mL)を用いて,1)で段階的に調
製したTOC標準液と同量の炭素を含むように無機体炭素標準液を段階的に調製する。
6) 5)で調製した各濃度の無機体炭素標準液の一定量[2)で定めた量]を,順次,マイクロシリンジで
無機体炭素測定管に注入し,指示値を読み取る。
7) 空試験として6)と同量の水をマイクロシリンジで無機体炭素測定管に注入して,指示値を読み取り,
6)の結果を補正し,無機体炭素の量と指示値との関係線を作成して,これを無機体炭素の検量線と
する。
e) 操作 操作は,次による。
1) 試料に懸濁物が含まれている場合には,ホモジナイザー又はミキサーでよくかき混ぜてこれらを均
一に分散させる。
2) 試料の一定量(5)[例えば,d) 2)と同量]をマイクロシリンジで全炭素測定管に注入し,指示値を読
み取る。
56
K 0102:2016
3) 試料の一定量[例えば,d) 6)と同量]をマイクロシリンジで無機体炭素測定管に注入し,指示値を
読み取る。
4) 試料を薄めた場合には,2)及び3)の空試験としてそれぞれ同量の水をマイクロシリンジでとり,2)
及び3)の操作を行って試料について得た結果を補正する。
5) あらかじめ作成した全炭素及び無機体炭素の検量線から注入した試料中の全炭素及び無機体炭素の
量を求め,それぞれの濃度(C mg/L)を算出する。
6) 次の式によって試料の全有機体炭素(TOC)の濃度(C mg/L)を算出する。
TOC=(Ct−Ci)×d
ここに, TOC: 有機体炭素の濃度(C mg/L)
Ct: 注入試料中の全炭素の濃度(C mg/L)
Ci: 無機体炭素の濃度(C mg/L)
d: 注入試料の希釈倍数
備考 1. この方法では,有機体炭素が少なく無機体炭素の多い試料では,誤差が大きくなる。この測
定方法のほか,あらかじめ試料に塩酸を加えてpH 2以下にし,JIS K 1107に規定する窒素2
級を通気して無機体炭素を除去した後,その少量を高温の全炭素測定管に送り込み,炭素の
定量を行ってこれを有機体炭素の量とする方法がある。その方法は,有機体炭素に比べて無
機体炭素が多い試料の場合に優れている。ただし,揮発性の有機物を含む場合には誤差が大
きい。
2. TOC分析装置で有機体炭素を二酸化炭素とする方式には,燃焼酸化法のほかに,高温湿式酸
化法,紫外線酸化法,光触媒酸化法など湿式の酸化法がある。
生成した二酸化炭素の定量には,赤外線分析法のほかに熱伝導度測定法又はガス透過膜式
電気伝導率測定法が用いられる。
3. 検出率の確認の方法 次のいずれかの試薬を調製し,測定範囲の80 %近くなるように希釈し
て測定し,TOCの検出率を確認する方法を推奨する。
1) フタロシアニン四スルホン酸銅(II)四ナトリウム塩0.256 gをとり,水700 mLに溶か
して全量フラスコ1 000 mLに移し入れ,水を標線まで加える。この標準液のTOCは,
100 mg/Lである。
2) JIS K 8532に規定するL(+)-酒石酸をJIS K 8228に規定する過塩素酸マグネシウム(乾
燥用)を入れたデシケーター中で18時間以上放置し,その0.312 5 gをとり,水に溶か
して全量フラスコ1 000 mLに移し入れ,水を標線まで加える。この標準液のTOCは,
100 mg/Lである。
3) JIS K 8789に規定する1,10-フェナントロリン一水和物0.137 6 gをとり,水に溶かして全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。この標準液のTOCは,100 mg/L
である。
4) JIS K 9047に規定するL-グルタミン酸を約80 ℃で約3時間乾燥し,デシケーター中で
放冷し,その0.245 gをとり,水に溶かして全量フラスコ1 000 mLに移し入れ,水を標
線まで加える。この標準液のTOCは,100 mg/Lである。
5) 全量フラスコ50 mLに水約30 mLを入れ,密栓してその質量を測定する。これにJIS K
8839に規定する2-プロパノールの約10.6 mLを速やかに加えて密栓し,その質量を測定
する。次いで水を標線まで加える。この溶液の濃度は,前後の質量の差から求める[2-
57
K 0102:2016
プロパノール1 gは,炭素(C)0.599 gに相当する。]。
この溶液1 mLを全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
22.2 燃焼酸化-赤外線式TOC自動計測法 計測器に供給した試料に酸を加えてpHを2以下にし,通気し
て無機体炭素を除去した後,その一定量をキャリヤーガスとともに高温の全炭素測定管に送り込み,有機
物中の炭素を二酸化炭素とし,その濃度を非分散形赤外線ガス分析計で測定して有機体炭素(TOC)の濃
度を求める。
定量範囲:C 0.05〜150 mg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 22.1 a) 1)による。
2) TOC標準液(C 1 mg/mL) 22.1 a) 2)による。
3) TOC標準液(C 0.1 mg/mL) 22.1 a) 3)による。
4) ゼロ校正液 1)の水を用いる。
5) スパン校正液 TOC標準液(C 0.1 mg/mL)[又はTOC標準液(C 1 mg/mL)]の適量を全量フラス
コにとり,水を標線まで加える。同じ操作で計測器の測定範囲の約80 %に相当するTOCの濃度に
なるように調製する。使用時に調製する。
6) 酸溶液 JIS K 9005に規定するりん酸,JIS K 8180に規定する塩酸又はJIS K 8951に規定する硫酸
でTOCの濃度のできるだけ少ないものを用い,所定の濃度に調製する。
7) キャリヤーガス 22.1 a) 8)による。
b) 装置 装置は,次による。
1) TOC自動計測器 JIS K 0805に規定する測定範囲がC 1 000 μg/L以下又はC 1 mg/L以上の燃焼酸
化-赤外線式TOC自動計測器
c) 準備操作 準備操作は,次による。
1) 酸溶液及びキャリヤーガスを,計測器に供給する。
2) 計測器の暖機運転を行い,各部の機能及び指示記録部を安定させる。
3) ゼロ校正液及びスパン校正液を用いて計測器を校正する。
d) 操作 操作は,次による。
1) 試料を,計測器に供給して指示値が安定したことを確認する。
2) 指示値から試料中の有機体炭素(TOC)の濃度(C mg/L)を求める。
備考 4. TOC自動計測器で有機体炭素を二酸化炭素とする方式には,燃焼酸化方式のほかに酸化剤を
添加して高圧高温下で湿式酸化分解する方式がある。この方式には,試料のpHを2以下の
酸性とし,ばっ気して無機体炭素を除去した後測定する方式と,試料を酸性にし酸化剤を加
えて全炭素を定量し,別に,試料を酸性にし有機物が分解されない温度(約130 ℃)で無機
体炭素を定量して,全炭素の量から無機体炭素の量を差し引いて有機体炭素の量を求める方
式とがある。
5. 備考3.による。
23. 全酸素消費量(TOD) 全酸素消費量とは,試料を燃焼させたとき,試料中の有機物の構成元素であ
る炭素,水素,窒素,硫黄,りんなどによって消費される酸素の量をいう。この試験には,燃焼法を適用
する。
少量の試料を一定量の酸素を含む不活性気体とともに高温の燃焼管に送り込み,有機物などを燃焼させ
58
K 0102:2016
た後,不活性気体中の酸素の濃度を定量し,その減量から全酸素消費量(TOD)を求める。
この試験は,試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く
試験する。
定量範囲:O 10〜500 mg/L,繰返し精度:3〜10 %(装置の形式及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3又はA4の水(1) (2) (3)で,溶存酸素を含まない水(4)を用いる。試薬の
調製及び操作には,この水を用いる。e)によって空試験を行い,使用の適否を確認しておく。
2) TOD標準液(O 1 mg/mL) JIS K 8005に規定する容量分析用標準物質のフタル酸水素カリウムを
120 ℃で約1時間加熱し,デシケーター中で放冷する。その0.851 gをとり,水に溶かして全量フラ
スコ1 000 mLに移し入れ,水を標線まで加える。
3) TOD標準液(O 0.1 mg/mL) TOD標準液(O 1 mg/mL)10 mLを全量フラスコ100 mLにとり,水
を標線まで加える。使用時に調製する。
4) キャリヤーガス JIS K 1107に規定する窒素2級及びJIS K 1101に規定する酸素
注(1) TODの濃度をできるだけ低くした水を用いる。精製した水は,容器に入れて保存すると徐々に
汚染されてTODの濃度が高くなることがあるので,精製後は早く使用することが望ましい。
(2) TODの濃度を低くするには,22.の注(2)の蒸留操作を行う。
(3) 22.の注(3)による。
(4) 2. n) 1)によって精製する。
b) 器具及び装置 器具及び装置は,次による。
1) マイクロシリンジ 10〜20 μL
2) TOD分析装置
c) 準備操作 準備操作は,次による。
1) TOD分析装置を作動できる状態にする。
2) TOD標準液(O 1 mg/mL)又はTOD標準液(O 0.1 mg/mL)の一定量(例えば,20 μL)をマイクロ
シリンジでTOD分析装置に注入し,指示値(ピーク高さ)が最大目盛値の約80 %になるようにTOD
分析装置の感度を調節する。
3) 2)の操作を数回繰り返して指示値が一定になることを確かめる。
4) 試料(5)をよく振り混ぜて均一にした後,その一定量[2)で定めた量]をマイクロシリンジで注入し
て,指示値を読み取り,3)と比較して試料の概略の全酸素消費量を求める。
注(5) TODがO 500 mg/L以上の試料の場合には,水で適切に薄めた後,試験する。
d) 検量線 検量線の作成は,次による。
1) c) 4)で求めた試料の概略のTODの値がほぼ中央になるように,TOD標準液(O 1 mg/mL)又はTOD
標準液(O 0.1 mg/mL)を全量フラスコ100 mLに段階的にとり,水を標線まで加える。
2) 1)で調製したTOD標準液の最高濃度のものの一定量(例えば,20 μL)をマイクロシリンジで注入
し,指示値が最大目盛の約80 %になるようにTOD分析装置の感度及び標準液を調節する。
3) 1)で調製した各濃度のTOD標準液の一定量[2)で定めた量]を,順次,マイクロシリンジで注入し,
指示値を読み取る。
4) 空試験として,3)と同量の水をマイクロシリンジでとり,3)と同様に操作して指示値を読み取り,
3)の指示値を補正してTOD標準液の各酸素相当量と指示値との関係線を作成する。
e) 操作 操作は,次による。
59
K 0102:2016
1) 試料に懸濁物が含まれている場合には,ホモジナイザー又はミキサーでよくかき混ぜてこれらを均
一に分散させる。
2) 試料(6)の一定量[例えば,d) 2)と同量]をマイクロシリンジでTOD分析装置に注入し,指示値を読
み取る。
3) 試料を薄めた場合には,空試験として2)と同量の水をマイクロシリンジでとり,2)の操作を行って
指示値を読み取り,試料について得た結果を補正する。
4) あらかじめ作成した検量線から,注入した試料中の酸素消費量を求め,注入した試料のTOD(O
mg/L)を算出する。
5) 次の式によって,試料中の全酸素消費量(TOD)の濃度(O mg/L)を算出する。
TOD=a×d
ここに, TOD: 全酸素消費量(O mg/L)
a: 注入試料の酸素消費量(O mg/L)
d: 注入試料の希釈倍数
注(6) 試料の全酸素消費量が高い場合には,一定の倍数に薄める。
備考 1. 溶存酸素が共存すると妨害し,特に,全酸素消費量が少ない場合には影響が大きい。この場
合は,別に,試料中の溶存酸素の量を測定して補正する。
2. 試料が酸性で硫酸イオンを含む場合には,高温で加熱すると次のように分解して酸素を生じ
負の誤差となる。
2H2SO4→2H2O+2SO2+O2
ただし,試料が蒸発したとき,硫酸がアルカリ金属塩となるような試料は,この反応は生
じない。このため,硫酸イオンが共存する場合には,水酸化ナトリウム溶液(200 g/L)(JIS
K 8576に規定する水酸化ナトリウムを用いて調製する。)を加えてpHを約11に調節してか
ら試験する。
3. 硝酸イオンが共存する場合には,高温で加熱すると次のように分解して酸素を生じ,負の誤
差となる。
4NaNO3→2Na2O+4NO+3O2又は
2NaNO3→Na2O+N2O+2O2
4. 重金属イオンを含む試料を長時間測定すると燃焼管中の触媒が劣化し,酸化率が低下してく
る。このような場合には,触媒の交換又は再生が必要である。
5. 海水など塩類を多量に含む場合には,指示値が元に戻らないことがあるので,安定した指示
値が得られるまで測定を繰り返すか,又は試料を薄めてから試験する。
24. ヘキサン抽出物質 ヘキサン(n-ヘキサン)抽出物質とは試料を微酸性とし,ヘキサン抽出を行った
後,約80 ℃でヘキサンを揮散させたときに残留する物質をいう。
この試験は,主として揮散しにくい鉱物油及び動植物油脂類の定量を目的とするが,これらのほかヘキ
サンに抽出された揮散しにくいものは,定量値に含まれる。
この試験は,抽出法,抽出容器による抽出法又は捕集濃縮・抽出法を適用する。
24.1 試料採取 試料の採取は,次による。
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
60
K 0102:2016
2) メチルオレンジ溶液(1 g/L) JIS K 8893に規定するメチルオレンジ0.1 gを熱水100 mLに溶かす。
b) 器具 器具は,次による。
1) 試料容器 表層の水及び落下する水の場合は,共栓広口ガラス瓶1〜2 L。下層の水の場合には,採
水器に装着できる共栓ガラス瓶1〜2 L。いずれも使用前にヘキサンでよく洗っておく。
2) 採水器 ハイロート採水器又はこれに類する適切な採水器
c) 採取方法 採取方法は,次による。
1) 落下している水の採取 水路,せき,溝,管などから落下している場合には,試料を直接試料容器
(1)に受け,適切な空間が残る程度に採取をとどめる。
2) 通水状態の配管装置などからの採取 配管,装置などが通水状態の場合には,試料採取弁を開き,
試料採取配管内に滞留している水の約5倍量を約1 L/minの割合で流出させてから試料容器(1)に受
け,適切な空間が残る程度に採取をとどめる(2) (3) (4)。
3) 深い水路及び水槽からの採取 深い水路及び水槽の水を採取する場合には,全層試料を採取できる
採水器を使用し,全層の試料を採取する。ハイロート採水器では,採水器の枠に試料容器を取り付
けて,底部近くに降ろし,採水しながら一定速度で採水器を引き上げ,水面に達したとき適切な空
間が残るように採取する(5)。
4) 貯水池,湖沼,河川などからの採取 試料容器を取り付けた採水器を用い,任意の深さの試料を採
取するか,又は試験目的によって3)に準じて採取する。
注(1) この場合は,試料容器を試料で洗わない。
(2) 高温高圧又は負圧状態にある配管装置などから試料採取する場合には,次による。
高温水の場合は,冷却器を試料採取管に設けて室温以下に冷却する。高圧水(圧力が1.96 MPa
以上)の場合には,減圧器を設けて減圧した後に採取し,高温であれば冷却器を通して室温以
下に冷却する。負圧水の場合には,昇圧器で大気圧にしてから採取する(負圧水で高温の場合
は,昇圧器の前に冷却器を設けて室温にしてから大気圧にする。)[JIS K 0094の4.3(採取弁を
用いる採取)を参照]。
(3) 装置などが停止状態にあるときは,油状物質が配管及び装置中で水と分離していることが多い
ため,通水速度及び通水時間によって油状物質の濃度に変動が生じる。試料採取弁及び配管中
に油状物質が付着しているおそれがある場合には,試料採取弁を全開して約10分間通水してか
ら,約1 L/minの割合で,更に約10分間通水する。この操作を繰り返して洗浄する。
(4) 試料採取直前に流量を変更してはならない。
(5) JIS K 2251に規定する全層試料に準じて採取する。
d) 試料の取扱い 試料の取扱いは,次による。
1) c)によって採取した試料は,他の容器に移し替えたり一部を採取してはならない。試験には全量を
用いる。
2) 試料の量は,試料を入れた容器の質量から試料容器の質量を差し引いて求めるか,又は試料を採取
したときに試料容器の水面の位置に印を付けておき,試験終了時に印のところまで水を入れてその
水の体積を試料の量とする。
3) 試料を保存したり運搬する必要がある場合には,指示薬としてメチルオレンジ溶液(1 g/L)5〜7滴
を加え,溶液の色が赤くなるまで塩酸(1+1)を加えて密栓する(6)。
注(6) 油状物質が浮上している場合には,油状物質が運搬中の振動でにじみやすいので,取扱いに注
意する。
61
K 0102:2016
24.2 抽出法 試料をpH4以下の塩酸酸性にして,ヘキサンで抽出を行い,80 ℃でヘキサンを揮散させて
残留する物質の質量をはかってヘキサン抽出物質を定量する。
定量範囲:5〜500 mg,繰返し精度:10〜20 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
3) 硫酸ナトリウム JIS K 8987に規定するもの。
4) メチルオレンジ溶液(1 g/L) 24.1 a) 2)による。
5) ヘキサン JIS K 8848に規定するもの。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 200 mL及び1 000〜3 000 mLで脚部の短いもの。使用前にヘキサンで洗う。コックにワ
セリンなどの滑剤を塗布しない。
2) 乾燥器 80±5 ℃に温度調節できるもの。
3) 加熱板又はマントルヒーター 80±5 ℃に温度調節できるもの。又は温度調節ができる水浴を用い
てもよい。
4) 蒸留装置 共通すり合わせで,蒸留フラスコ(容量50〜100 mL),トの字形連結管及びリービッヒ
冷却器(長さ300 mm)とを接続できるものを用いる。いずれも使用前にヘキサンでよく洗ってお
く。
5) 蒸発容器 アルミニウムはく皿,白金皿又はビーカー。容量50〜100 mLで,できるだけ質量の小
さいもの。いずれも使用前にヘキサンでよく洗い,80±5 ℃で約30分間加熱し,デシケーター中で
放冷した後,質量を0.1 mgの桁まで求めておく。
c) 操作 操作は,次による。
1) 24.1で採取した試料(7)を分液漏斗1 000〜3 000 mL (8)に移し,指示薬としてメチルオレンジ溶液(1
g/L)2,3滴を加え,溶液の色が赤に変わるまで塩酸(1+1)を滴加する。
2) 試料容器をヘキサン約20 mLずつで2回洗い,洗液を分液漏斗1 000〜3 000 mLに加える。約2分
間激しく振り混ぜ,放置する(9)。
3) 水層は,試料容器に戻し,更に分液漏斗1 000〜3 000 mLを静かに揺り動かして,残った水層をで
きるだけ分離して(10)試料容器に戻す。ヘキサン層は分液漏斗200 mLに移す。
4) 試料容器の水層を1)で使用した分液漏斗1 000〜3 000 mLに入れ,再び2)及び3)の操作を行ってヘ
キサン層と水層とを分離し,ヘキサン層を分液漏斗200 mLに合わせる。
5) 分液漏斗1 000〜3 000 mLを少量のヘキサンで洗い,洗液を分液漏斗200 mLに合わせる。
6) 分液漏斗200 mLを静かに揺り動かして静置し,ヘキサンを損失しないように注意しながら混入し
た水分を十分に分離除去する(10)。
7) ヘキサン層に水約20 mLを加えて約1分間振り混ぜて放置し,水層を捨てる。この洗浄操作を洗液
がメチルオレンジに対して黄色になるまで数回繰り返す。できるだけ水層を除去する。
8) ヘキサン層に硫酸ナトリウム3〜5 gを加えて振り混ぜ,水分を除く(11)。
9) 分液漏斗200 mLの脚部を乾いたろ紙(12)で拭き取り,脱脂綿又はろ紙(12)を用いてヘキサン層をろ過
し,蒸留装置の蒸留フラスコに移し入れる(13)。
10) 分液漏斗200 mLを少量のヘキサンで洗い,この洗液も9)と同じ操作でろ過し,蒸留装置の蒸留フ
ラスコに移し入れる。使用した脱脂綿又はろ紙は,ヘキサン約5 mLずつで2回洗い,この洗液も
62
K 0102:2016
蒸留フラスコに移し入れる。
11) 蒸留フラスコをマントルヒーターに入れ,トの字形連結管及びリービッヒ冷却器を接続して,マン
トルヒーターの温度を約80 ℃に調節し,ヘキサンを毎秒約1滴の留出速度で蒸留し,留出するヘ
キサンを受器に受ける(14)。蒸留は,蒸留フラスコ内の液量が約2 mLになるまで続ける。
12) 蒸留フラスコの残留液を質量既知の蒸発容器に移し入れる。蒸留フラスコを少量のヘキサンで3回
洗い,この洗液も蒸発容器に加える。蒸発容器を約80 ℃に保った加熱板の上又はマントルヒータ
ーの中に置いてヘキサンを揮散させる(15)。
13) 蒸発容器の外側を湿った清浄な布などで拭い,次に,乾いた清浄な布などで拭って,80±5 ℃に調
節した乾燥器中に移し,約30分間加熱する。蒸発容器をデシケーター中で約30分間放冷した後,
その質量を0.1 mgの桁まではかり,蒸発容器の質量を差し引き,ヘキサン抽出物質の質量(mg)
を求める。
14) 空試験として,この試験に使用した全ヘキサンと同量のヘキサンを蒸留フラスコにとり(13),11)〜
13)の操作を行って残留物質の質量(mg)を求める。
15) 次の式によって試料中のヘキサン抽出物質の濃度(mg/L)を算出する。
(
)
V
b
a
P
000
1
×
−
=
ここに,
P: ヘキサン抽出物質の濃度(mg/L)
a: 試験におけるヘキサン抽出物質の質量(mg)
b: 空試験における残留物質の質量(mg)
V: 試料量(mL)
注(7) 試料は通常約1 Lでよいが,ヘキサン抽出物質が5〜500 mgになるように試料を採取し,全量
を用いる。
(8) 試料の量に応じて適切な大きさの分液漏斗を選ぶ。
(9) 試料の性質によっては,エマルションが生成したり,ヘキサン層が濁ったりすることがある。
このような試料では,分液漏斗中の水層をできるだけ元の試料容器に戻し,JIS K 8150に規定
する塩化ナトリウム又はJIS K 8960に規定する硫酸アンモニウム約10 g(ヘキサンに溶ける物
質を含まないもの)を加え,分液漏斗の口に約300 mmの共通すり合わせリービッヒ冷却器又
はジムロート冷却器を取り付け,約80 ℃に保った恒温水浴中に分液漏斗を浸し,約10分間ヘ
キサンを還流させるとエマルションがなくなることがある。この加熱還流のほか,分液漏斗中
のヘキサン層及びエマルション層にJIS K 8150に規定する塩化ナトリウム又はJIS K 8960に規
定する硫酸アンモニウム約10 gを加えて振り混ぜた後,少量の水で遠心分離管に移し,8 000
min −1以上で約5分間遠心分離すると,エマルション層は僅かになり,ヘキサン層の分離を容
易にすることができる。これを分液漏斗に戻し,3)以降の操作に移る。
(10) 分離する水層が約1 mL以下になるまで続ける。試料が多量のグリース類又は固体油脂を含む場
合には,水層を分離する前にヘキサンを追加する。
(11) ヘキサン層が濁っていることがある。このような場合には,水層をできるだけ分離した後,硫
酸ナトリウムを加えて脱水すると透明になることがある。JIS K 8150に規定する塩化ナトリウ
ム又はJIS K 8960に規定する硫酸アンモニウムを使用するほうが効果的な場合もある。ただし,
ヘキサンに溶ける物質を含む試薬は使用しない。
(12) ヘキサンで十分に洗って抽出物質を除いたもので,ろ過のときにはあらかじめ少量のヘキサン
63
K 0102:2016
で潤しておく。
注(13) 蒸留フラスコに一度に入りきらないときは,2,3回に分割してヘキサンを留出させる。
(14) 蒸留によって留出したヘキサンは,再蒸留すれば,再使用できる。
(15) 引火しないように十分に注意をする。溶媒は揮散廃棄せずにできるだけ回収する。ヘキサンを
揮散後,蒸発容器中に水分が認められる場合には,アセトンを加えて蒸発を繰り返すと水分は
除去できる。水分中に塩類が残留すると誤差になるので注意する。もし,塩類が残留する場合
には13)の操作を行い,ヘキサン抽出物質の質量(mg)を求めた後,ヘキサン抽出物質を少量
のヘキサンを加えて溶かし,分離除去する。この操作を繰り返し行い,ヘキサン抽出物質を除
去した後,13)の操作を行って残留物質の質量(mg)を求めて補正する。
備考 濁りが著しい試料又はエマルションが生成しやすい試料の場合は,次のソックスレー抽出法を
適用するとよい。
1) 試料に指示薬としてメチルオレンジ溶液(1 g/L)2,3滴を加え,液の色が赤に変わるまで
塩酸(1+1)を加えてpHを4以下に調節する。
2) 次に,ブフナー漏斗にろ紙5種A 2枚を重ねて敷き,これにけい藻土懸濁液(10 g/L)100 mL
を加えて吸引ろ過し,けい藻土を減圧状態で水約1 Lで洗う。
3) このろ過材に酸性にした試料を加えて吸引ろ過し,十分に吸引した後,ろ過材をろ紙ごと
巻いて円筒ろ紙に移し,漏斗の壁,容器,かき混ぜ棒などをヘキサンで洗ったろ紙で拭き
取り,同じ円筒ろ紙に入れて80±5 ℃の乾燥器中で約30分間加熱する。
4) 円筒ろ紙をソックスレー抽出器に移す。試料容器は十分に乾燥した後,ヘキサン約20 mL
ずつで2回洗い,洗液を円筒ろ紙に注ぐ。次に,抽出操作に移り,抽出を約20回繰り返す。
5) 抽出液を蒸留フラスコに移し入れ,以下,24.2 c) 11)〜15)の操作を行って試料中のヘキサ
ン抽出物質の濃度(mg/L)を算出する。
24.3 抽出容器による抽出法 試料を抽出容器に移し,pH4以下の塩酸酸性にしてヘキサンを加え,かき
混ぜ機によってヘキサン抽出を行い,80 ℃でヘキサンを揮散させ,残留する物質の質量をはかってヘキサ
ン抽出物質を定量する。
定量範囲:5〜200 mg,繰返し精度:10〜20 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
3) 硫酸ナトリウム JIS K 8987に規定するもの。
4) メチルオレンジ溶液(1 g/L) 24.1 a) 2)による。
5) ヘキサン JIS K 8848に規定するもの。
b) 器具及び装置 器具及び装置は,次による。
1) 試料容器 24.1 b) 1)による。ただし,容量1〜5 L
2) 抽出容器 三角フラスコ3 000〜5 000 mL。共通すり合わせのもの。
3) 分液漏斗 200〜500 mLで脚部の短いもの。使用前にヘキサンで洗う。コックにワセリンなどの滑
剤を塗布しない。
4) 乾燥器 24.2 b) 2)による。
5) 加熱板又はマントルヒーター 24.2 b) 3)による。
6) 蒸留装置 24.2 b) 4)による。
64
K 0102:2016
7) 蒸発容器 24.2 b) 5)による。
8) かき混ぜ機 マグネチックスターラー又はかき混ぜ機
c) 操作 操作は,次による。
1) 24.1によって試料容器(1〜5 L)に採取した試料(16)を抽出容器(17)に移し,指示薬としてメチルオレ
ンジ溶液(1 g/L)2,3滴を加え,溶液の色が赤に変わるまで塩酸(1+1)を滴加する。
2) 試料容器をヘキサン25〜50 mL(18)ずつで2回洗い,洗液を抽出容器に入れる。
3) 内容物をかき混ぜ機で約10分間かき混ぜた後,かき混ぜ機を取り除き,静置してヘキサン層を分離
する(19)。
4) 抽出容器の底部にサイホン管を挿入(20)し,水層の大部分を元の試料容器(21)に抜き出す。残ったヘ
キサン層と少量の水とを分液漏斗200〜500 mLに移す。
5) 試料容器の水層を再び抽出容器に移し,試料容器をヘキサン25〜50 mLずつで2回洗い,洗液を抽
出容器に合わせ,再び3)及び4)の操作を行ってヘキサン層と少量の水とを先の分液漏斗200〜500
mLに合わせる。
6) 分液漏斗200〜500 mLを静かに揺り動かし,ヘキサンが損失しないように注意しながら混入した水
分を十分に分離除去する(10)。
7) 以下,24.2 c) 7)〜15)の操作を行い,試料中のヘキサン抽出物質の濃度(mg/L)を求める。
注(16) 試料はヘキサン抽出物質の量が5 mg以上となるように採取し,その全量を用いる。
(17) 抽出容器は試料の量に応じて適切な大きさのものを選ぶ。試料容器はそのまま抽出容器として
用いてもよいが,ガラス瓶の底が平でないため,マグネチックスターラーの回転子がうまく回
転しないことがある。
(18) 試料の量に応じて適切にヘキサンの量を変える。
(19) かき混ぜると白濁し,2層に分離した後も水層が澄明にならないことがある。この場合は,JIS
K 8150に規定する塩化ナトリウム及びJIS K 8987に規定する硫酸ナトリウムを添加すると多少
改善される[注(9)参照]。水層がほぼ澄明になり,ヘキサン層,エマルション層及び水層に分離
できればよい。
(20) 下口ガラスコック付き抽出容器を用いると便利である。
(21) 試料容器を抽出容器とする場合は,別の容器を用意する。
24.4 捕集濃縮・抽出法 試料に捕集剤として塩化鉄(III)を加え,炭酸ナトリウムで水酸化鉄(III)の
沈殿を生成させ,この沈殿にヘキサン抽出物質を捕集濃縮する。水層を捨て,塩酸を加えて沈殿を溶かし,
ヘキサン抽出を行い,80 ℃でヘキサンを揮散させて残留する物質の質量をはかってヘキサン抽出物質を定
量する。
定量範囲:2〜200 mg,繰返し精度:10〜30 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩酸(1+1) 24.3 a) 2)による。
3) 炭酸ナトリウム溶液(200 g/L) JIS K 8625に規定する炭酸ナトリウム20 gを水に溶かして100 mL
とする。
4) 塩化鉄(III)溶液(22) JIS K 8142に規定する塩化鉄(III)六水和物30 gを塩酸(1+11)[21. a) 2)
による。]に溶かして100 mLとする。
5) ヘキサン JIS K 8848に規定するもの。
65
K 0102:2016
注(22) 塩化マグネシウム溶液{JIS K 8159に規定する塩化マグネシウム六水和物40 gを塩酸(1+11)
[21. a) 2)による。]に溶かして100 mLにする。}又は塩化亜鉛溶液[JIS K 8111に規定する塩
化亜鉛20 gを塩酸(1+11)に溶かして100 mLにする。]に代えてもよい。用いる捕集剤中の
ヘキサン抽出物質の量が10 mg/L以上のものを使用してはならない。
硫酸アルミニウム,ポリ塩化水酸化アルミニウムなどを用いると,生成したフロックが塩酸
に溶けにくくなることがある。
b) 器具及び装置 器具及び装置は,次による。
1) 試料容器 24.1 b) 1)による。ただし,容量5 L
2) 分液漏斗 200〜500 mLで脚部の短いもの。使用前にヘキサンで洗う。コックにワセリンなどの滑
剤を塗布しない。
3) 乾燥器 24.2 b) 2)による。
4) 加熱板又はマントルヒーター 24.2 b) 3)による。
5) 蒸留装置 24.2 b) 4)による。
6) 蒸発容器 24.2 b) 5)による。
7) かき混ぜ機 24.3 b) 8)による。
c) 操作 操作は,次による。
1) 24.1によって試料容器(5 L)に試料約4 L(23)を採取し,試料容器に捕集剤として塩化鉄(III)溶液
4 mLを加え,試料容器にかき混ぜ機を入れ,試料をかき混ぜながら,炭酸ナトリウム溶液(200 g/L)
を加えてpH7〜9(24)とする。
2) 約5分間激しくかき混ぜた後,かき混ぜ機を取り除き,沈殿が沈降して完全な澄明層が得られるま
で静置する(25)。
3) 試料容器にサイホン管又は吸引管を挿入し,沈殿を損失しないように上澄み液を抜き出して捨てる。
4) 残った沈殿層に塩酸(1+1)を加えてpHを約1として沈殿を溶かし,分液漏斗200〜500 mLに移
す。
5) 試料容器をヘキサン約20 mLずつで2回洗い,洗液を分液漏斗200〜500 mLに加える。
6) 分液漏斗200〜500 mLを約2分間激しく振り混ぜ,静置してヘキサン層と水層とを分離する(9)。
7) 水層は試料容器に戻し,更に分液漏斗200〜500 mLを静かに振り動かして残った水層をできるだけ
分離して,試料容器に戻す。ヘキサン層は,分液漏斗200 mLに移す。
8) 試料容器の水層を再び分液漏斗200〜500 mLに移し,再び5)〜7)の操作を行い,ヘキサン層を先の
分液漏斗200 mLに合わせる。
9) 分液漏斗200〜500 mLを少量のヘキサンで洗い,洗液を分液漏斗200 mLに合わせる。
10) 分液漏斗200 mLを静かに振り動かし,ヘキサンを損失しないように注意しながら混入した水分を
十分に分離する(10)。
11) 次に,24.2 c) 7)〜15)の操作を行い,試料中のヘキサン抽出物質の濃度(mg/L)を求める。
注(23) 試料がアルカリ性の場合には,あらかじめ中和しておく。
(24) 捕集剤の種類によっては沈殿の生成するpHが異なる。
(25) 沈殿物の層が全液量の1/10以下,通常10〜200 mLになるように捕集剤の添加量を調節すると
よい。
25. 欠番
66
K 0102:2016
26. 欠番
27. 欠番
28. フェノール類 フェノール類とp-クレゾール類とに区分する。
28.1 フェノール類 フェノール類の試験は,試料を前処理(蒸留)後,28.1.2に示す4-アミノアンチピ
リン吸光光度法又は28.1.3に示す流れ分析法を適用し,フェノール標準液を用いて定量した値で表す。
フェノール類は,フェノール分解菌によって分解されやすい。酸化性物質,還元性物質,アルカリなど
の作用も受けやすいので,試験は試料採取後,直ちに行う。直ちに行えない場合は,3.3によって保存し,
できるだけ早く試験する。
この試験で,対象となるフェノール類は,ベンゼン及びその類似体のヒドロキシ誘導体で,規定の方法
によって4-アミノアンチピリンと反応して着色化合物を生成するものをいう。
なお,28.1.1及び28.1.2に示す方法は,1990年に第2版として発行されたISO 6439,流れ分析法は,1999
年に第1版として発行されたISO 14402との整合を図ったものである。
備考 − この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6439:1990,Water quality−Determination of phenol index−4-Aminoantipyrine spectrometric
methods after distillation(MOD)
ISO 14402:1999,Water quality−Determination of phenol index by flow analysis (FIA and CFA)
(MOD)
− この試験で求める“フェノール類”は,ISO 6439によって定義される“フェノール指標”
に相当する。
28.1.1 前処理(蒸留法) りん酸酸性(pH約4)で,硫酸銅(II)の存在の下で加熱蒸留してフェノール
類を留出分離する(1)。
注(1) 試料に色又は濁りがなく,28.1.2の試験の妨害となる物質を含まない場合には,蒸留操作を省
略し,直接28.1.2の試験を行ってよい。ただし,その場合は,りん酸及び硫酸銅(II)の添加に
よる試料の保存は行わず,試料採取後直ちに試験する。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水。ほうけい酸ガラス瓶に保存する。
2) りん酸(1+9) JIS K 9005に規定するりん酸を用いて調製する。
3) 硫酸銅(II)溶液 JIS K 8983に規定する硫酸銅(II)五水和物10 gを水に溶かして100 mLとする。
4) メチルオレンジ溶液(1 g/L) 24.1 a) 2)による。
b) 装置 装置は,次による。
1) 蒸留装置 38.1.1.2 b) 1)による。
c) 蒸留操作 蒸留操作は,次による。
1) 試料250 mL(2)を蒸留フラスコ500 mLにとり,メチルオレンジ溶液(1 g/L)5〜7滴を加え,メチル
オレンジが変色するまでりん酸(1+9)を加えてpHを約4にした後,硫酸銅(II)溶液2.5 mLを
加える(3)。
2) 蒸留フラスコを蒸留装置に取り付け,受器にメスシリンダー(有栓形)250 mLを用いて蒸留する。
67
K 0102:2016
3) メスシリンダー中の留出液が225 mLになったとき,一旦加熱を止める。
4) 蒸留フラスコ中の試料の沸騰がやんだ後,蒸留フラスコに水25 mLを加え,再び蒸留を続けて更に
25 mLを留出させ,全留出液量を250 mLとする(4)。
注(2) 試料中のフェノール類の濃度が50 mg/L以上の場合には,その適量をとり,水を加えて250 mL
とする。
フェノール類の濃度が25 μg/L以下の場合には,試料500 mLを蒸留フラスコ1 000 mLにと
り硫酸銅(II)溶液の添加量は5 mLとして蒸留し,450 mLが留出したとき,一旦加熱を止め,
冷却後,水50 mLを加え,再び蒸留を続けて更に50 mLを留出させ,500 mLとする。
(3) 試料の保存にりん酸及び硫酸銅(II)五水和物の添加を行った場合は,これらの添加は省略する。
(4) 留出液が白濁している場合には,留出液に再びりん酸(1+9)を加えてpHを約4とし,硫酸
銅(II)溶液2.5 mLを加え,蒸留操作を繰り返す。再蒸留を行っても白濁が消えない場合には,
備考1. 4)の油分及びタール類の除去によって処理する。
備考 1. 試料の保存においてりん酸及び硫酸銅(II)五水和物を添加することによって,フェノール
類の生物化学的分解が抑制される。28.1.2及び28.1.3の試験では,酸化性物質,還元性物質,
金属イオン,芳香族アミン類,油分,タール類などは妨害となる。大部分は蒸留操作で取り
除くことができるが,酸化性物質,還元性物質,硫黄化合物,油分及びタール類が試料中に
含まれる場合には,次のように処理する。
1) 酸化性物質 残留塩素のような酸化性物質が含まれている場合,又は試料を酸性にし,よ
う化カリウムを加えるとよう素が遊離する場合は,試料採取直後に,JIS K 9502に規定す
るL(+)-アスコルビン酸の小過剰若しくはJIS K 8978に規定する硫酸鉄(II)七水和物
又はメタ亜ひ酸ナトリウムの小過剰を加える。試料の保存には,これにりん酸を加えてpH
約4とし,試料1 LにつきJIS K 8983に規定する硫酸銅(II)五水和物1 gを加える。
2) 還元性物質 還元性物質が存在する場合には,JIS K 8801に規定するヘキサシアノ鉄(III)
酸カリウムを過剰に加える。試料の保存には,JIS K 9005に規定するりん酸を加えてpH
約4とし,試料1 LにつきJIS K 8983に規定する硫酸銅(II)五水和物1 gを加える。
3) 硫黄化合物 硫化水素及び亜硫酸イオンが含まれている場合には,試料採取直後にりん酸
を加えてpH約4とし,注意して試料に空気を吹き込むか,若しくはかき混ぜて,硫化水
素及び二酸化硫黄を追い出した後,硫酸銅(II)溶液を試料1 Lにつき10 mL加えるか,
又は,試料採取直後に硫酸銅(II)溶液を過剰に加えて硫化銅(I)の沈殿とした後,りん
酸を加えてpH約4とする。
4) 油分及びタール類 油分及びタール類が含まれている場合には,次のいずれかの方法によ
るとよい。
4.1)
試料採取直後に硫酸銅(II)溶液を加えずに,水酸化ナトリウム溶液(100 g/L)[19. a) 2)
による。]を加えてpH12〜12.5とし,分液漏斗に移してJIS K 8322に規定するクロロホ
ルムを加え,油分及びタール類を抽出して,クロロホルム層を捨てる。水層は,沸騰水
浴上で加熱して,残留するクロロホルムを除去する。試料の保存には,これに,りん酸
を加えてpH約4とし,試料1 Lにつき硫酸銅(II)五水和物1 gを加える。
4.2)
蒸留に当たって,試料250 mLをとり,メチルオレンジ溶液(1 g/L)数滴を加え,硫酸
(0.5 mol/L)(JIS K 8951に規定する硫酸を用いて調製する。)で酸性とする。分液漏斗
に移し,JIS K 8150に規定する塩化ナトリウム75 gを加える。クロロホルムを,最初は
68
K 0102:2016
20 mLで,以後12.5 mLずつで4回抽出分離する。クロロホルム層を集めて別の分液漏
斗に入れ,水酸化ナトリウム溶液(100 g/L)[19. a) 2)による。]を,最初は2.0 mL,以
後1.5 mLずつで2回逆抽出する。水層を集め,水浴上でクロロホルムがなくなるまで加
熱する。冷却し,水で250 mLとしてc)の蒸留操作を行う。
5) アミン類 特定の反応条件下では,ある種のアミン類はフェノールとして測定される。こ
の妨害は,pH0.5未満で蒸留することによって最小限に抑えられる。
28.1.2 4-アミノアンチピリン吸光光度法 前処理(蒸留)した試料のpHを約10に調節し,これに4-ア
ミノアンチピリン(4-アミノ-1,2-ジヒドロ-1,5-ジメチル-2-フェニル-3H-ピラゾール-3-オン)溶液とヘキサ
シアノ鉄(III)酸カリウム溶液とを加えて,生成する赤い色のアンチピリン色素の吸光度を波長510 nm
付近で測定し,フェノール標準液による検量線によってフェノール類を定量する。
この方法では,フェノールのほかo-,m-位置に置換基があるフェノール誘導体及び多環式化合物にヒド
ロキシル基が置換したものも4-アミノアンチピリンと反応してアンチピリン色素を生成して定量される。
p-位置に置換基があるフェノール誘導体は,4-アミノアンチピリンと反応しにくいので,ほとんど発色し
ない。アンチピリン色素の発色の強さは,置換基の種類,位置,数などによって差がある。
定量範囲:C6H5OH 50〜500 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水。ほうけい酸ガラス瓶に保存する。
2) 塩化アンモニウム-アンモニア緩衝液(pH10) JIS K 8116に規定する塩化アンモニウム67.5 gをJIS
K 8085に規定するアンモニア水570 mLに溶かし,水で1 Lとする。
3) ヘキサシアノ鉄(III)酸カリウム溶液 JIS K 8801に規定するヘキサシアノ鉄(III)酸カリウムの
大きな結晶9 gをとり,少量の水で表面を洗った後,水に溶かして100 mLとし,必要がある場合,
ろ過する。1週間ごとに調製するが,1週間以内でも色が暗い赤に変わったものは使用しない。
4) 4-アミノアンチピリン溶液(20 g/L) JIS K 8048に規定する4-アミノアンチピリン2.0 gを水に溶
かして100 mLとする。使用時に調製する。
5) フェノール標準液(C6H5OH 1 mg/mL) JIS K 8798に規定するフェノール1.00 gを水に溶かして全
量フラスコ1 000 mLに移し入れ,水を標線まで加え,0〜10 ℃の暗所に保存する。
6) フェノール標準液(C6H5OH 10 μg/mL) フェノール標準液(C6H5OH 1 mg/mL)10 mLを全量フラ
スコ1 000 mLにとり,水を標線まで加える。使用時に調製する。
7) フェノール標準液(C6H5OH 1 μg/mL) フェノール標準液(C6H5OH 10 μg/mL)50 mLを全量フラ
スコ500 mLにとり,水を標線まで加える。使用時に調製する。
8) フェノール標準液(C6H5OH 0.1 μg/mL) フェノール標準液(C6H5OH 10 μg/mL)5 mLを全量フラ
スコ500 mLにとり,水を標線まで加える。使用時に調製する。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 28.1.1の前処理を行った試料(5)の適量(C6H5OHとして50〜500 μgを含む。)をメスシリンダー(有
栓形)100 mLにとり,水を100 mLの標線まで加える。
2) 次に,塩化アンモニウム-アンモニア緩衝液(pH10)3 mLを加えて振り混ぜ,pH10±0.2に調節す
る。
3) 4-アミノアンチピリン溶液(20 g/L)2 mLを加えて振り混ぜ,ヘキサシアノ鉄(III)酸カリウム溶
69
K 0102:2016
液2 mLを加えて十分に振り混ぜた後,約3分間放置する。
4) 別に,空試験として水100 mLについて2)及び3)の操作を行う。
5) 空試験の溶液を対照液として波長510 nm付近の吸光度を測定する。
6) 検量線からフェノールに相当する量を求め,試料中のフェノール類の濃度(C6H5OH mg/L)を算出
する。
注(5) 試料に妨害物質を含まず,直接4-アミノアンチピリン吸光光度法を適用する場合は,採取直後
の試料。
d) 検量線 検量線の作成は,次による。
1) フェノール標準液(C6H5OH 10 μg/mL)5〜50 mLをメスシリンダー(有栓形)100 mLに段階的にと
り,水を100 mLの標線まで加える。
2) c) 2)〜5)の操作を行ってフェノールの量と吸光度との関係線を作成する。
備考 2. c) 3)で得た溶液の色が弱いときは,次の操作を行って,定量してもよい。
1) この溶液の全量を分液漏斗200 mLに移し,JIS K 8322に規定するクロロホルム10 mL
を加えて約1分間激しく振り混ぜた後放置する。
2) クロロホルム層を分離し,乾いたろ紙でろ過するか,ビーカーに移した後,JIS K 8987
に規定する硫酸ナトリウム約1 gを加えて脱水する。
3) 別に,空試験として水100 mLについてc) 2)及びc) 3)の操作並びに1)及び2)の操作を行
う。
4) 空試験のクロロホルム層を対照液とし,2)のクロロホルム層の波長460 nm付近の吸光度
を測定する。
5) 検量線は,フェノール標準液(C6H5OH 1 μg/mL)2.5〜50 mLを用い,c) 2)以降の試料の
場合と同様に操作してフェノールの量と吸光度との関係線を作成する。
3. 試料500 mLを用いて注(2)の操作を行った場合には,次の操作によって定量してもよい。
1) 前処理で得た留出液500 mLを分液漏斗1 000 mLにとり,塩化アンモニウム-アンモニア
緩衝液(pH10)10 mLを加えて振り混ぜ,pH 10±0.2とする。
2) 4-アミノアンチピリン溶液(20 g/L)3 mL,ヘキサシアノ鉄(III)酸カリウム溶液3 mL
を加え,十分に振り混ぜた後約3分間放置し,次いでJIS K 8322に規定するクロロホル
ム20 mLを加えて約1分間激しく振り混ぜた後放置する。
3) 備考2.の2)以降と同様に操作する。ただし,検量線は,フェノール標準液(C6H5OH 1
μg/mL)2.5〜50 mLを分液漏斗1 000 mLに段階的にとり,水を加えて500 mLとし,1)
以下,試料の場合と同様に操作して作成したものを用いる。
4. 備考2.のクロロホルムの代わりに安息香酸メチルを用い,次の操作を行って,定量してもよ
い。
1) c) 3)で得た溶液の全量を分液漏斗200 mLに移し,安息香酸メチル10 mLを加えて約2
分間激しく振り混ぜた後放置する。
2) 安息香酸メチル層を分離し,ビーカーに移した後,JIS K 8987に規定する硫酸ナトリウ
ム約1 gを加えて脱水する。
3) 別に,空試験として水100 mLについてc) 2)及びc) 3)の操作並びに1)及び2)の操作を行
う。
4) 空試験の安息香酸メチル層を対照液とし,2)の安息香酸メチル層の波長465 nm付近の吸
70
K 0102:2016
光度を測定する。
5) 検量線は,フェノール標準液(C6H5OH 1 μg/mL)2.5〜50 mLを用い,試料の場合と同様
にc) 2)及びc) 3)の操作並びに1)〜4)の操作を行ってフェノールの量と吸光度との関係線
を作成する。
備考 5. 次の操作を行って,定量してもよい(アンチピリン色素固相抽出法)。
1) 28.1.1の前処理を行った試料(*)の適量(C6H5OHとして0.5〜15 μgを含む。)をメスシリ
ンダー(有栓形)50 mLにとり,水を50 mLの標線まで加える。
2) 次に,塩化アンモニウム-アンモニア緩衝液(pH10)1.5 mLを加えて振り混ぜ,pH10±
0.2に調節する。
3) 4-アミノアンチピリン溶液(20 g/L)1 mLを加えて振り混ぜ,ヘキサシアノ鉄(III)酸
カリウム溶液1 mLを加えて十分に振り混ぜた後,約10分間放置して,アンチピリン色
素を生成させる。
4) 市販のスチレン・ジビニルベンゼン共重合体充塡カラム,又はODSカラムなどの疎水性
カラムに,JIS K 8032に規定するアセトニトリル10 mL,水10 mLを流して洗浄・コン
ディショニングを行った後,3)の発色溶液を10 mL/minの流量で通液する。
5) 約5分間通気してカラムを乾燥する。
6) JIS K 8032に規定するアセトニトリル5 mLをカラムに流し,アンチピリン色素を溶出
させる。
7) 別に,空試験として水50 mLについて2)〜6)の操作を行う。
8) 空試験のアセトニトリル溶出液を対照液とし,6)のアセトニトリル溶出液の波長475 nm
付近の吸光度を測定する。
9) 検量線からフェノールに相当する量を求め,試料中のフェノール類の濃度(C6H5OH
mg/L)を算出する。
10) 検量線は,フェノールが0.5〜15 μgとなるように,フェノール標準液(C6H5OH 0.1 μg/mL)
又はフェノール標準液(C6H5OH 1 μg/mL)をメスシリンダー(有栓形)50 mLに段階的
にとり,水を50 mLの標線まで加える。続いて,2)〜8)の操作を行ってフェノールの量
と吸光度との関係線を作成する。
注(*) 注(5)による。
6. フェノール標準液(C6H5OH 10 μg/L)を用いて28.1.1 c)及び28.1.2 c)の操作を行い,フェノー
ルの回収率が十分であることを確かめておくとよい。
28.1.3 流れ分析法 試料中のフェノール類を,28.1.1及び28.1.2と同様な原理で蒸留,発色させる流れ分
析法によって定量する。フェノールの種類によっては,生成したアンチピリン色素が水相で時間とともに
退色する場合がある。このため,JIS K 0170-5の試薬濃度,流速等の条件が28.1.2で行った場合と比較し,
整合していることを確認して,その条件で定量する。
定量範囲:C6H5OH:0.01〜1 mg/L,繰返し精度:10 %以下
試験操作などは,JIS K 0170-5による。ただし,JIS K 0170-5の6.3.4(くえん酸蒸留・4-アミノアンチ
ピリン発色CFA法)の方法は除く。
28.2 p-クレゾール類
28.2.1 p-ヒドラジノベンゼンスルホン酸吸光光度法 フェノール類をpH8.0でギブス試薬と反応させてイ
ンドフェノールに変え,アスコルビン酸酸性で水蒸気蒸留して,ギブス試薬と反応しないp-クレゾール類
71
K 0102:2016
を留出させる。留出したp-クレゾール類に,p-ヒドラジノベンゼンスルホン酸と亜硝酸とから生成するジ
アゾ化合物,p-スルホンベンゼンジアゾニウム塩をカップリングさせて生じるアゾ色素の赤い色の吸光度
を測定してp-クレゾール類を定量する。
定量範囲:p-CH3C6H4OH 10〜150 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水。ほうけい酸ガラス瓶に保存する。
2) 硫酸(1+17) 水17容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する
硫酸1容を徐々に加える。
3) 水酸化ナトリウム溶液(100 g/L) JIS K 8576に規定する水酸化ナトリウム10 gを水に溶かして100
mLとする。
4) 炭酸ナトリウム JIS K 8625に規定するもの。
5) 塩化ナトリウム JIS K 8150に規定するもの。
6) メチルオレンジ溶液(1 g/L) 24.1 a) 2)による。
7) クロロホルム又はジエチルエーテル JIS K 8322に規定するクロロホルム又はJIS K 8103に規定す
るジエチルエーテル
8) ギブス試薬 JIS K 8491に規定する2,6-ジブロモ-N-クロロ-p-ベンゾキノンモノイミン(2,6-ジブロ
モキノンクロロイミド)0.5 gをJIS K 8102に規定するエタノール(95)50 mLに溶かす。使用時に
調製する。
9) L(+)-アスコルビン酸 JIS K 9502に規定するもの。
10) アンモニア水(1+7) JIS K 8085に規定するアンモニア水を用いて調製する。
11) p-ヒドラジノベンゼンスルホン酸溶液 次の操作によって,調製する。
− A液 p-ヒドラジノベンゼンスルホン酸0.5水和物1 gとJIS K 8625に規定する炭酸ナトリウム
0.3 gとを水80 mLに加え,沸騰水浴中で加熱して溶かし,これにJIS K 8180に規定する塩酸9 mL
を加え,更に水を加えて100 mLとする。室温では結晶が析出するので,約37 ℃の恒温槽中に保
存する。1週間以上経過したものは使用しない。
− B液 A液4 mLを全量フラスコ100 mLにとり,約10 ℃に冷却した後,亜硝酸ナトリウム溶液
(10 g/L)(JIS K 8019に規定する亜硝酸ナトリウム1 gを水に溶かして100 mLとする。)5 mLを
加えて約10 ℃で3〜5分間放置する。これに,あらかじめ約10 ℃に冷却した水を標線まで加え
る。使用時に調製する。
12) p-クレゾール標準液(p-CH3C6H4OH 1 mg/mL) JIS K 8306に規定するp-クレゾール(p-CH3C6H4OH)
1.00 gをとり,少量の水に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
13) p-クレゾール標準液(p-CH3C6H4OH 0.1 mg/mL) p-クレゾール標準液(p-CH3C6H4OH 1 mg/mL)20
mLを全量フラスコ200 mLにとり,水を標線まで加える。
b) 装置 装置は,次による。
1) 水蒸気蒸留装置 小形,すり合わせのもの。
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料500 mL(p-CH3C6H4OHとして0.1〜1.5 mgを含む。)を分液漏斗1 000 mLにとり,指示薬とし
てメチルオレンジ溶液(1 g/L)2,3滴を加え,溶液の色が赤になるまで硫酸(1+17)を滴加して
酸性[試料が酸性の場合は,水酸化ナトリウム溶液(100 g/L)で溶液の色が黄色になるまで中和し
72
K 0102:2016
た後,硫酸(1+17)を滴加して再び酸性にする。]とし,これに,塩化ナトリウム150 gとクロロ
ホルム(6) 40 mLとを加え,激しく振り混ぜて抽出し,クロロホルム層を別の分液漏斗200 mLに移
す。
2) さらに,クロロホルム25 mLずつを用いて1)と同様の抽出操作を4回繰り返し,クロロホルム層を
先の分液漏斗に合わせる。
3) このクロロホルム層に水酸化ナトリウム溶液(100 g/L)4 mLを加えて逆抽出し,更に3 mLずつで
逆抽出を2回繰り返して,逆抽出液を合わせる。
4) この逆抽出液を沸騰水浴上で加熱して,溶けているクロロホルムを揮散させた後,放冷する。
5) 水約20 mLで全量フラスコ100 mLに移し入れる。次に,炭酸ナトリウム2 gを加え,硫酸(1+17)
を滴加してpH8とし,水約20 mLとギブス試薬5 mLとを加えて約24時間放置する(フェノール類
が共存すれば溶液の色は青になる。)。
6) L(+)-アスコルビン酸1 gを加え,水を標線まで加える。
7) この溶液10 mLを蒸留フラスコにとり,水蒸気蒸留を行い,メスシリンダー(有栓形)50 mLに30
mLを留出させる。
8) 留出液を水で約40 mLに薄めた後,p-ヒドラジノベンゼンスルホン酸溶液のB液5 mLを加えて振
り混ぜ,次に,アンモニア水(1+7)1 mLを加え,水を50 mLの標線まで加えて再び振り混ぜ,
約5分間放置する。
9) 溶液の一部を吸収セルに移し,波長495 nm付近の吸光度を測定する。
10) 空試験として水40 mLをとり,8)及び9)の操作を行って吸光度を求め,試料について得た吸光度を
補正する。
11) 検量線からp-クレゾール類の量を求め,試料中のp-クレゾール類の濃度(p-CH3C6H4OH mg/L)を
算出する。
注(6) クロロホルムの代わりにJIS K 8103に規定するジエチルエーテルを用いてもよい。この場合に
は,塩化ナトリウムを加える必要はない。
d) 検量線 検量線の作成は,次による。
1) p-クレゾール標準液(p-CH3C6H4OH 0.1 mg/mL)1〜15 mLを全量フラスコ100 mLに段階的にとり,
それぞれに硫酸(1+17)を滴加してpH8とし,水約20 mLとギブス試薬5 mLとを加えて約24時
間放置し(フェノール類が共存すれば溶液の色は青になる。),引き続きc) 6)の操作を行う。
2) この溶液10 mLを蒸留フラスコにとり,水蒸気蒸留を行って留出液30 mLを得た後,水で約40 mL
に薄める。次に,p-ヒドラジノベンゼンスルホン酸のB液5 mLを加え,c) 8)〜10)の操作を行って
p-クレゾール(p-CH3C6H4OH)の量と吸光度との関係線を作成する。
29. 欠番
29.1 欠番
30. 界面活性剤 界面活性剤は,陰イオン界面活性剤と非イオン界面活性剤とに区分する。
界面活性剤は,微生物によって容易に分解されるため,試験は,試料採取後,直ちに行う。直ちに試験
が行えない場合は,3.3によって保存し,できるだけ早く試験する。
30.1 陰イオン界面活性剤 陰イオン界面活性剤の定量には,メチレンブルー吸光光度法,エチルバイオ
レット吸光光度法,溶媒抽出-フレーム原子吸光法又はメチレンブルー発色による流れ分析法を適用する。
73
K 0102:2016
陰イオン界面活性剤には,高級アルコール硫酸エステル類,脂肪酸硫酸エステル類及びスルホン酸形陰
イオン界面活性剤{アルキルアリールスルホン酸塩類[直鎖アルキルベンゼンスルホン酸塩類(LAS)],
アルキルスルホン酸塩類,アルケンスルホン酸塩類など}などがある。
なお,メチレンブルー発色による流れ分析法は,2009年に第1版として発行されたISO 16265との整合
を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 16265:2009,Water quality−Determination of the methylene blue active substances (MBAS) index
−Method using continuous flow analysis (CFA)(MOD)
30.1.1 メチレンブルー吸光光度法 陰イオン界面活性剤がメチレンブルー[3,7-ビス(ジメチルアミノ)フ
ェノチアジン-5-イウムクロリド]と反応して生じるイオン対をクロロホルムで抽出して,その吸光度を測
定し,ドデシル硫酸ナトリウムとして表す。
定量範囲:陰イオン界面活性剤[NaO3SO(CH2)11CH3]2〜50 μg,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(1+35) 水35容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する
硫酸1容を徐々に加える。
3) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
4) アルカリ性四ほう酸ナトリウム溶液 JIS K 8866に規定する四ほう酸ナトリウム十水和物9.54 gを
水に溶かした後,水で500 mLとし,これに水酸化ナトリウム溶液(40 g/L)50 mLを加え,水で全
量を1 Lとする。
5) メチレンブルー溶液(0.25 g/L) JIS K 8897に規定するメチレンブルー0.3 gを水に溶かして1 Lと
する。
6) 脱脂綿
7) クロロホルム JIS K 8322に規定するもの。
8) 陰イオン界面活性剤標準液[NaO3SO(CH2)11CH3 1 mg/mL] ドデシル硫酸ナトリウム[NaO3SO
(CH2)11CH3](1)をその100 %に対して1.00 gをとり,水に溶かして全量フラスコ1 000 mLに移し入
れ,水を標線まで加える。
9) 陰イオン界面活性剤標準液[NaO3SO(CH2)11CH3 10 μg/mL] 陰イオン界面活性剤標準液[NaO3SO
(CH2)11CH3 1 mg/mL]10 mLを全量フラスコ1 000 mLにとり,水を標線まで加える。使用時に調製
する。
注(1) 純度及び平均分子量の分かった試薬を用いる。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 200 mL
2) 光度計 分光光度計又は光電光度計
c) 準備操作 準備操作は,次による。
1) 分液漏斗(A)に水50 mL,アルカリ性四ほう酸ナトリウム溶液10 mL及びメチレンブルー溶液(0.25
g/L)5 mLを入れる。
分液漏斗(B)に水100 mL,アルカリ性四ほう酸ナトリウム溶液10 mL及びメチレンブルー溶液
74
K 0102:2016
(0.25 g/L)5 mLを入れる。
2) それぞれにクロロホルム10 mLを加え,約30秒間激しく振り混ぜた後,放置してクロロホルム層
を捨てる。この操作を更に1回繰り返す。
3) それぞれの水層にクロロホルム2〜3 mLを加え,緩やかに振り混ぜた後,放置してクロロホルム層
を捨てる。この操作をクロロホルム層が無色になるまで繰り返して水層中の着色物を除去する。
4) クロロホルムで洗い終わった分液漏斗(B)中の水層に,硫酸(1+35)3 mLを加える。
なお,分液漏斗(A)及び分液漏斗(B)の脚部がぬれているときには,ろ紙などで拭き取る。
d) 操作 操作は,次による。
1) c)の準備操作を行った分液漏斗(A)中の水層に,試料(2)の適量[NaO3SO(CH2)11CH3として2〜50 μg
を含む。]を加える。ただし,全量が100 mLを超えないようにする。
2) クロロホルム10 mLを加えて,緩やかに約1分間振り混ぜて放置し,クロロホルム層をc)の準備操
作を行った分液漏斗(B)に移し入れる。
3) 分液漏斗(B)を緩やかに約1分間振り混ぜた後放置する。分液漏斗の脚部に脱脂綿を詰め,クロ
ロホルム層を全量フラスコ25 mLに移し入れる。
4) 再び分液漏斗(A)にクロロホルム10 mLを加えて,2)及び3)の操作を繰り返して抽出を行い,ク
ロロホルム層を3)と同様に先の全量フラスコ25 mLに合わせ,クロロホルムを標線まで加える。
5) これを吸収セル(3)に移し,クロロホルムを対照液として波長650 nm付近の吸光度を測定する。
6) 空試験として水50 mLを用い,あらかじめc)の準備操作を行った分液漏斗(A)に入れ,2)〜5)の
操作を行って吸光度を測定し,試料について得た吸光度を補正する。
7) 検量線から陰イオン界面活性剤の量を求め,試料中の陰イオン界面活性剤の濃度
[NaO3SO(CH2)11CH3 mg/L]を算出する。
注(2) 酸性の場合には,pH計を用いて水酸化ナトリウム溶液(40 g/L)で,また,アルカリ性の場合
には,硫酸(1+35)でpH約7とする。
(3) 吸収セル50 mmを用いると0.4〜10 μgの陰イオン界面活性剤が定量できる。
e) 検量線 検量線の作成は,次による。
1) 陰イオン界面活性剤標準液[NaO3SO(CH2)11CH3 10 μg/mL]0.2〜5 mLを段階的にとり,あらかじめ
c)の準備操作を行った分液漏斗に入れ,水で全量を約100 mLとする。
2) d) 2)〜6)の操作を行って陰イオン界面活性剤[NaO3SO(CH2)11CH3]の量と吸光度との関係線を作成
する。
備考 1. 硝酸,シアン化物,チオシアン酸などのイオンが多量に存在すると定量を妨害する。
陽イオン界面活性剤は,陰イオン界面活性剤と強く結合するため,その共存量に応じて負
の誤差を与える。しかし,通常の水ではその量は,陰イオン界面活性剤と比較して非常に少
ない。
2. ミズミミズ,イトミミズなどがいる底泥付近の水では,正の誤差が生じやすい。
3. スルホン酸形陰イオン界面活性剤(LASなど)を定量するには,次の操作によってアルコー
ル系などの陰イオン界面活性剤を加水分解し,残ったスルホン酸形陰イオン界面活性剤をd)
の操作で定量して,ドデシル硫酸ナトリウムとして表す。
1) 試料の適量[NaO3SO(CH2)11CH3として4〜100 μgを含む。]をすり合わせ三角フラスコ
にとり,JIS K 8180に規定する塩酸25 mL及び沸騰石5〜7個を加え,水で液量を約50 mL
とした後,還流冷却器を付けて約2時間静かに煮沸する。
75
K 0102:2016
2) 放冷後,指示薬としてフェノールフタレイン溶液(5 g/L)[15.の備考2.による。]5〜7
滴を加え,溶液の色が微紅色になるまで,初めは水酸化ナトリウム溶液(400 g/L)を,
中和点近くなってからは水酸化ナトリウム溶液(40 g/L)[21. a) 3)による。]を加えて中
和し,水で100 mLとする。
3) 以下,c)及びd)の操作を行ってスルホン酸形陰イオン界面活性剤の量を求め,試料中の
スルホン酸形陰イオン界面活性剤の濃度[NaO3SO(CH2)11CH3 mg/L]を算出する。
30.1.2 エチルバイオレット吸光光度法 陰イオン界面活性剤がエチルバイオレット【N-[4-{ビス[4-(ジエ
チルアミノ)フェニル]メチレン}-2,5-シクロヘキサジエン-1-イリデン]-N-エチルエタンアミンイウムクロ
リド】と反応して生じるイオン対をトルエンに抽出して,その吸光度を測定し,ドデシル硫酸ナトリウム
として表す。
定量範囲:陰イオン界面活性剤[NaO3SO(CH2)11CH3]0.5〜12.5 μg,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸ナトリウム溶液(1 mol/L) JIS K 8987に規定する硫酸ナトリウム142 gを水に溶かして1 Lと
する。
3) 酢酸-EDTA緩衝液(pH5) JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二
水和物7.5 gを水に溶かして約700 mLとする。これにJIS K 8355に規定する酢酸12.5 mLを加え,
pH計を用いてpH5になるまで水酸化ナトリウム溶液(2 mol/L)を加えた後,水を加えて1 Lとす
る。
4) エチルバイオレット溶液(1 mmol/L) エチルバイオレット(4) 0.280 gを水に溶かして500 mLとす
る。
5) トルエン JIS K 8680に規定するもの。
6) 陰イオン界面活性剤標準液[NaO3SO(CH2)11CH3 1 mg/mL] 30.1.1 a) 8)による。
7) 陰イオン界面活性剤標準液[NaO3SO(CH2)11CH3 10 μg/mL] 30.1.1 a) 9)による。
8) 陰イオン界面活性剤標準液[NaO3SO(CH2)11CH3 0.5 μg/mL] 陰イオン界面活性剤標準液[NaO3SO
(CH2)11CH3 10μg/mL]10 mLを全量フラスコ200 mLにとり,水を標線まで加える。使用時に調製す
る。
注(4) エチルバイオレットは,塩化亜鉛 モルが付加した複塩を用いる。この複塩以外を用いる場合
には,その濃度が1 mmol/Lになる量をとって調製する。また,c) 7)の空試験の操作を行ったと
きの吸光度の値が大きい(0.04程度以上)場合には,別のロットのエチルバイオレットを用い
て調製し直す。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 200 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料の適量[NaO3SO (CH2)11CH3として0.5〜12.5 μgを含む。]を分液漏斗にとり,水を加えて100 mL
とする。
2) これに硫酸ナトリウム溶液(1 mol/L)5 mL,酢酸-EDTA緩衝液(pH5)5 mL及びエチルバイオレ
ット溶液(1 mmol/L)2 mLを加える。
3) トルエン5 mL(又は10 mL)を加え,約10分間振り混ぜる(5)。
2
1
76
K 0102:2016
4) 静置し,水層約100 mLを捨てる。
5) 更に静置し,トルエン層が完全に分離したら水層を捨てる。
6) トルエン層を吸収セルに移し,トルエンを対照液として波長611 nm付近の吸光度を測定する。
7) 空試験として水100 mLを用い,2)〜6)の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
8) 検量線から陰イオン界面活性剤の量を求め,試料中の陰イオン界面活性剤の濃度
[NaO3SO(CH2)11CH3 mg/L]を算出する。
注(5) 振り混ぜ時間が5分間程度だと吸光度が幾分低くなるので,振り混ぜ時間約10分間を守る。
d) 検量線 検量線の作成は,次による。
1) 陰イオン界面活性剤[NaO3SO (CH2)11CH3 0.5 μg/mL]1〜25 mLを段階的に分液漏斗にとり,水を加
えて100 mLとする。
2) c) 2)〜7)の操作を行って,陰イオン界面活性剤[NaO3SO (CH2)11CH3]の量と吸光度との関係線を作
成する。
備考 4. 海水,又は海水が混入した試料のように多量の塩化物イオンが共存すると,その一部がエチ
ルバイオレットとイオン対を生成し,トルエンに抽出されて吸光度を増加させる。このよう
な試料の場合は,c) 4)及び5)の操作を行った後も,器壁に付着物が残るが,トルエン層を小
形の分液漏斗に移し入れ,エチルバイオレット−硫酸ナトリウム溶液[エチルバイオレット
溶液(1 mmol/L)7.5 mLをとり,JIS K 8987に規定する硫酸ナトリウム5 gを加え,水で500
mLとする。]20 mLを加え,約1分間振り混ぜる。放置した後,水層の大部分を捨て,再び
静置してトルエン層が完全に分離したら水層を捨てる。次に,c) 6)〜8)の操作を行う。
5. 硝酸イオンは,NO3− 1 mg/L程度までは妨害しないが,それ以上共存すると正の誤差を与え
る。
通常の河川水などに含まれるその他のイオンは妨害しない。
陽イオン界面活性剤は,陰イオン界面活性剤と強く結合するため,その共存量に応じて負
の誤差を与える。しかし,通常の水ではその量は,陰イオン界面活性剤と比較して非常に少
ない。
30.1.3 溶媒抽出-フレーム原子吸光法 陰イオン界面活性剤を,カリウムを取り込んだジベンゾ-18-クラウ
ン-6とイオン対とし,これを4-メチル-2-ペンタノンに抽出し,抽出溶液中のカリウムをフレーム原子吸光
法で定量し,ドデシル硫酸ナトリウムとして表示する。
定量範囲:陰イオン界面活性剤[NaO3SO(CH2)11CH3]2.5〜50 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸カリウム(20 mmol/L)-酢酸アンモニウム(50 mmol/L)混合溶液 JIS K 8962に規定する硫
酸カリウム3.5 gとJIS K 8359に規定する酢酸アンモニウム3.9 gとを水に溶かして約700 mLとし,
pH計を用いて硫酸(1+35)[30.1.1 a) 2)による。]を加えpH5として水で1 Lとする。
3) 硫酸カリウム(4 mmol/L)-酢酸アンモニウム(10 mmol/L)混合溶液 硫酸カリウム(20 mmol/L)
-酢酸アンモニウム(50 mmol/L)混合溶液200 mLを水で薄めて1 Lとする。
4) ジベンゾ-18-クラウン-6の4-メチル-2-ペンタノン溶液(0.5 mmol/L) 精製したジベンゾ-18-クラウ
ン-6 (6) 90 mgをJIS K 8903に規定する4-メチル-2-ペンタノン500 mLに溶かす。
5) 陰イオン界面活性剤標準液[NaO3SO(CH2)11CH3 1 mg/mL] 30.1.1 a) 8)による。
77
K 0102:2016
6) 陰イオン界面活性剤標準液[NaO3SO(CH2)11CH3 5 μg/mL] 陰イオン界面活性剤標準液
[NaO3SO(CH2)11CH3 1 mg/mL]5 mLを全量フラスコ1 000 mLにとり,水を標線まで加える。使用
時に調製する。
注(6) ジベンゾ-18-クラウン-6の精製は,次による。
ジベンゾ-18-クラウン-6約2.5 gをJIS K 8858に規定するベンゼン約200 mL中に加え,水浴
上で加熱して溶かす。これをガラスろ過器(1G3)で吸引ろ過する。ろ液が冷えると直ちに結
晶が生成するが,再び水浴上で加熱して溶かし,吸引ろ過する。この操作を結晶が白くなるま
で行った(2,3回)後,ろ液を冷却し,吸引ろ過する。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) フレーム原子吸光分析装置 JIS K 0121に規定するフレーム原子吸光分析装置で,測定対象元素用
の光源を備えたもの。
c) 操作 操作は,次による。
1) 試料の適量[NaO3SO(CH2)11CH3として2.5〜50 μgを含む。]を分液漏斗100 mLにとり,硫酸カリ
ウム(20 mmol/L)-酢酸アンモニウム(50 mmol/L)混合溶液10 mLを加え,水で液量を50 mLと
する。
2) ジベンゾ-18-クラウン-6の4-メチル-2-ペンタノン溶液(0.5 mmol/L)10 mLを加え,約1分間振り
混ぜる。
3) 静置後水層を捨て,硫酸カリウム(4 mmol/L)-酢酸アンモニウム(10 mmol/L)混合溶液25 mLを
加えて振り混ぜ,静置し,水層を捨てる。
4) 4-メチル-2-ペンタノン層をアセチレン-空気フレーム中に導入し,波長766.5 nmの指示値(7)を読み
取る。
5) 検量線から陰イオン界面活性剤の量を求め,試料中の陰イオン界面活性剤の濃度
[NaO3SO(CH2)11CH3 mg/L]を算出する。
注(7) 吸光度又はその比例値。
d) 検量線 検量線の作成は,次による。
1) 陰イオン界面活性剤[NaO3SO(CH2)11CH3 5 μg/mL]0.5〜10 mLを分液漏斗に段階的にとる。
2) c) 1)〜4)の操作を行って陰イオン界面活性剤[NaO3SO(CH2)11CH3]の量と指示値との関係線を作成
する。検量線の作成は,試料測定時に行う。
備考 6. カルシウム及びマグネシウムは,それぞれ500 mg程度まで共存しても影響しない。ナトリウ
ムは50〜70 mgの共存でも,一部がジベンゾ-18-クラウン-6に取り込まれ,陰イオン界面活
性剤とイオン対を作って抽出され,そのまま噴霧すると負の誤差を生じるが,抽出分離後の
溶媒層をc) 3)の硫酸カリウム(4 mmol/L)-酢酸アンモニウム(10 mmol/L)混合溶液と振り
混ぜることによって,ナトリウムはカリウムに置換され,妨害は除かれる。
陽イオン界面活性剤は,陰イオン界面活性剤と強く結合するため,その共存量に応じて負
の誤差を生じる。しかし,通常の水ではその量は,陰イオン界面活性剤と比較して非常に少
ない。非イオン界面活性剤は400 μg程度共存しても妨害しない。
30.1.4 流れ分析法 試料中の陰イオン界面活性剤を,30.1.1と同様な原理で発色させる流れ分析法によっ
て定量する。
定量範囲:陰イオン界面活性剤[NaO3SO(CH2)11CH3]0.02〜5 mg/L
78
K 0102:2016
試験操作などは,JIS K 0170-8による。ただし,JIS K 0170-8の6.3.2(1,2-ジクロロエタン抽出FIA法)
の方法は除く。
30.2 非イオン界面活性剤 非イオン界面活性剤には,ポリオキシエチレンアルキルエーテル類,ポリオ
キシエチレンアルキルフェノールエーテル類,ポリオキシエチレンアルキルエステル類,ポリオキシエチ
レンソルビタンアルキルエステル類などがある。
非イオン界面活性剤の定量には,前処理(イオン交換分離)を行った試料について,テトラチオシアナ
トコバルト(II)酸吸光光度法又はチオシアン酸鉄(III)吸光光度法を適用する。
30.2.1 テトラチオシアナトコバルト(II)酸吸光光度法 非イオン界面活性剤とテトラチオシアナトコバ
ルト(II)酸アンモニウムとの錯体をベンゼンで抽出して,紫外部の吸光度を測定し,ヘプタオキシエチ
レンドデシルエーテルとして表示する。
定量範囲:非イオン界面活性剤[CH3(CH2)11O(CH2CH2O)7H]0.1〜2 mg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩酸(1+11) JIS K 8180に規定する塩酸を用いて調製する。
3) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
4) テトラチオシアナトコバルト(II)酸アンモニウム溶液 JIS K 9000に規定するチオシアン酸アン
モニウム310 gとJIS K 8552に規定する硝酸コバルト(II)六水和物140 gとを水に溶かして500 mL
とする。空試験値が高い場合は,これを分液漏斗1 000 mLに移し,JIS K 8858に規定するベンゼン
50 mLを加えて激しく振り混ぜて放置する。ベンゼン層を捨て,再びベンゼン50 mLを加えて振り
混ぜ,放置する。ベンゼン層を捨て,水層を乾いたろ紙でろ過し,ベンゼンの小滴を除く。
5) 塩化ナトリウム JIS K 8150に規定するもの。
6) 硫酸ナトリウム JIS K 8987に規定するもの。
7) エタノール(95) JIS K 8102に規定するもの。
8) エタノール[体積分率50 %] 水1容に,エタノール(95)1容を加えて調製する。
9) ベンゼン JIS K 8858に規定するもの。
10) 強酸性陽イオン交換樹脂 低架橋度(ジビニルベンゼン含量4〜6 %)で粒子径300〜1 180 μmのも
の。R-Na+形。次のように精製して用いる。
− 強酸性陽イオン交換樹脂250 mLを,内径40〜50 mm,高さ約1 000 mmのカラム(ガラス製又
はアクリル樹脂製)に水とともに流し入れ,気泡が混入しないように充塡する。
− 塩酸(1+11)2 Lを約5 L/(L-樹脂・h)(約20 mL/min)(8)で流した後,水1 Lを同様に流して洗
浄する。
− 次に,水酸化ナトリウム溶液(40 g/L)1 Lを約5 L/(L-樹脂・h)(約20 mL/min)(8)で流し,水1
Lを同様に流して洗浄する。
− さらに,塩酸(1+11)1 Lと水酸化ナトリウム溶液(40 g/L)1 Lとを同様に流して洗浄する。
− 次に,フェノールフタレイン溶液(5 g/L)(15.の備考2.による。)の紅色がほとんど認められな
くなるまで水で洗浄する[約20 L/(L-樹脂・h)(約80 mL/min)(8)で流す。]。
11) 強塩基性陰イオン交換樹脂(I形) 低架橋度(ジビニルベンゼン含量4〜6 %)で粒子径300〜1 180
μmのもの。R-Cl−形。次のように精製して用いる。
− 強塩基性陰イオン交換樹脂(I形)500 mLを,内径40〜50 mm,高さ約1 000 mmのカラム(ガ
ラス製又はアクリル樹脂製)に水とともに流し入れ,気泡が混入しないように充塡する。
79
K 0102:2016
− 水酸化ナトリウム溶液(40 g/L)2 Lを約5 L/(L-樹脂・h)(約40 mL/min)(8)で流した後,水約2
Lを同様に流して洗浄する。
− 次に,塩酸(1+11)2 Lを約5 L/(L-樹脂・h)(約40 mL/min)で流した後,水約2 Lを同様に流
して洗浄する。
− さらに,水酸化ナトリウム溶液(40 g/L)2 Lと塩酸(1+11)2 Lとを同様に流して洗浄する。
− 次に,メチルレッド-ブロモクレゾールグリーン混合溶液[15.の注(1)による。]に対して青い色
になるまで水で洗浄する[約20 L/(L-樹脂・h)(約160 mL/min)(8)で流す。]。
12) 非イオン界面活性剤標準液[CH3(CH2)11O(CH2CH2O)7H 0.1 mg/mL] ヘプタオキシエチレンドデシ
ルエーテル(9)をその100 %に対して0.100 gをはかりとり,水に溶かして全量フラスコ1 000 mLに
移し入れ,水を標線まで加える。使用時に調製する。
注(8) このカラムを用いたときの流量。
(9) 品質を確認する場合には,日本油化学協会で定めた試験方法による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 200 mL
2) イオン交換樹脂カラム 図30.1に例を示す。
2.1) イオン交換樹脂カラムの作り方 イオン交換樹脂カラムの作り方は,次による。
− 強酸性陽イオン交換樹脂と強塩基性陰イオン交換樹脂(I形)とを体積比で1:2になるよう
にとる。
− 水を加えてよく混合しながら,気泡が混入しないように図30.1のガラス管に充塡し,イオン
交換樹脂柱の高さを約200 mmに調節する。
− エタノール[体積分率50 %]100 mLを流す。
− このイオン交換樹脂カラムは,数回繰り返し使用してもよい。
単位 mm
図30.1 イオン交換樹脂カラムの例
80
K 0102:2016
3) 光度計 分光光度計
4) 吸収セル 石英ガラス製又はこれと同等の品質のもの。
c) 前処理 前処理は,次による。
1) 試料(10) 100 mLをとり,エタノール(95)100 mLを加えて振り混ぜる。
2) この溶液をイオン交換樹脂カラムに10〜15 L/(L-樹脂・h)(2.6〜3.9 mL/min)で流し,流出液をビー
カー500 mLに受ける。
3) イオン交換樹脂カラムのイオン交換樹脂柱の上部に液面が近づいたら,エタノール[体積分率50 %]
100 mLを少量ずつ加え,イオン交換樹脂カラム内の試料を流出させる。流出液は2)のビーカー500
mLに合わせる。
4) 流出液を沸騰水浴上で約30 mLになるまで濃縮する。
5) 放冷後,この溶液を全量フラスコ100 mLに移し入れ,水を標線まで加える。
注(10) 酸性の場合には,水酸化ナトリウム溶液(40 g/L)で,また,アルカリ性の場合には,塩酸(1
+11)でpH計を用いてpH約7とする。
d) 操作 操作は,次による。
1) c) 5)の溶液の適量[CH3(CH2)11O(CH2CH2O)7Hとして0.1〜2 mgを含む。]を分液漏斗200 mLにとり,
水で100 mLとする。
2) テトラチオシアナトコバルト(II)酸アンモニウム溶液15 mLと塩化ナトリウム(11) 35 gとを加えて
約1分間振り混ぜた後,約15分間放置する。
3) ベンゼン(12) 25 mLを加えて約3分間激しく振り混ぜ,放置する。
4) 水層を捨て,ベンゼン層をビーカーに移し,硫酸ナトリウム約5 gを加えて振り混ぜ,脱水する。
5) これを吸収セルに移し,水100 mLについて,2)〜4)の操作を行ったベンゼンを対照液とし,波長
322 nm付近の吸光度を測定する。
6) 空試験として1)と同量のc) 5)の溶液を分液漏斗200 mLにとり,水で100 mLとし,2)のテトラチオ
シアナトコバルト(II)酸アンモニウム溶液15 mLの代わりに水15 mLを用い,2)〜4)の操作を行
った後,ベンゼンを対照液として波長322 nm付近の吸光度を求め,試料について得た吸光度を補
正する。
7) 検量線から非イオン界面活性剤の量を求め,試料中の非イオン界面活性剤の濃度
[CH3(CH2)11O(CH2CH2O)7H mg/L]を算出する。
注(11) 塩化カリウムを用いてもよい。
(12) 1,2-ジクロロエタン又はトルエンを用いてもよい。また,抽出に用いるベンゼン,1,2-ジクロロ
エタン及びトルエンの量を10 mLにしてもよい。ただし,これらの溶媒による場合は,a) 4)の
テトラチオシアナトコバルト(II)酸アンモニウム溶液の処理にベンゼンの代わりにこれらを用
いる。
e) 検量線 検量線の作成は,次による。
1) 非イオン界面活性剤標準液[CH3(CH2)11O(CH2CH2O)7H 0.1 mg/mL]1〜20 mLを分液漏斗200 mLに
段階的にとり,水を加えて100 mLとする。
2) d) 2)〜5)の操作を行って非イオン界面活性剤[CH3(CH2)11O(CH2CH2O)7H]の量と吸光度との関係線
を作成する。
備考 7. ポリエチレングリコールが共存すると非イオン界面活性剤として定量値に含まれて誤差とな
るので,JIS K 8810に規定する1-ブタノール又はJIS K 8900に規定する2-ブタノン(エチル
81
K 0102:2016
メチルケトン)であらかじめ抽出除去した後,c)の前処理を行う。
備考 8. 陰イオン界面活性剤及び陽イオン界面活性剤が共存しない場合には,c)の前処理を省略する
ことができる。
9. 非イオン界面活性剤の濃度がCH3(CH2)11O(CH2CH2O)7H 1 mg/L以下の場合には,次のように
濃縮した後,操作する。
1) 試料500 mLにつき塩化ナトリウム50 gとJIS K 8625に規定する炭酸ナトリウム2.5 g
とを加えて溶かし,分液漏斗1 000 mLに移し,JIS K 8361に規定する酢酸エチル25 mL
を加えて約2分間激しく振り混ぜて放置する。
2) 分離した酢酸エチル層をビーカーに移し,水層には酢酸エチル25 mLを加えて再び抽出
を繰り返す。分離した酢酸エチル層を先のビーカーに合わせる。
3) 酢酸エチル層を水浴上で加熱して酢酸エチルを揮発除去し,少量のメタノールを加えて
溶かし,水を加えて一定体積とした後,c)の前処理を行って定量する。
30.2.2 チオシアン酸鉄(III)吸光光度法 非イオン界面活性剤を塩化ナトリウムの共存下でトルエンに
抽出した後,トルエン層に塩化鉄(III)溶液とチオシアン酸カリウム溶液とを加えて生成する非イオン界
面活性剤とチオシアン酸鉄(III)カリウムとの錯体の吸光度を測定し,ヘプタオキシエチレンドデシルエ
ーテルとして表示する。
定量範囲:非イオン界面活性剤[CH3(CH2)11O(CH2CH2O)7H]0.02〜0.2 mg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩化鉄(III)溶液(1 mol/L) JIS K 8142に規定する塩化鉄(III)六水和物27 gを水に溶かし,水を
加えて全量を100 mLとする。
3) チオシアン酸カリウム溶液(8 mol/L) JIS K 9001に規定するチオシアン酸カリウム77.7 gを水に
溶かし,水を加えて全量を100 mLとする。
4) 塩化ナトリウム溶液(2 mol/L) JIS K 8150に規定する塩化ナトリウム11.7 gを水に溶かし,水を
加えて全量を100 mLとする。
5) 非イオン界面活性剤標準液[CH3(CH2)11O(CH2CH2O)7H 0.01 mg/mL] 30.2.1のa) 12)の非イオン界
面活性剤標準液[CH3(CH2)11O(CH2CH2O)7H 0.1 mg/mL]10 mLを全量フラスコ100 mLにとり,水
を標線まで加える。使用時に調製する。
6) トルエン JIS K 8680に規定するもの。
b) 器具及び装置 器具及び装置は,30.2.1 b)による。
c) 前処理 前処理は,30.2.1 c)による。
d) 操作 操作は,次による。
1) c) の前処理後の溶液の適量[CH3(CH2)11O(CH2CH2O)7Hとして0.02〜0.2 mgを含む。]を分液漏斗
200 mLにとり,水で100 mLとする。
2) 塩化ナトリウム溶液(2 mol/L)5 mLとトルエン10 mLとを加えて約2分間振り混ぜた後,約30分
間放置する。このとき,エマルションなどが生じ,トルエン層と水層との分離が悪い場合は,固体
の塩化ナトリウムを添加する。
3) 水層を捨て,トルエン層にチオシアン酸カリウム溶液(8 mol/L)5 mL,塩化鉄(III)溶液(1 mol/L)
5 mL,塩化ナトリウム溶液(2 mol/L)1 mLとを加えて,約2分間振り混ぜる。約10 分間放置した
後,水層を捨てる。
82
K 0102:2016
4) トルエン層を吸収セルに移し,水100 mLについて,2)〜3)の操作を行ったトルエンを対照液とし,
波長510 nm付近の吸光度を測定する。
5) 検量線から非イオン界面活性剤の量を求め,試料中の非イオン界面活性剤の濃度
[CH3(CH2)11O(CH2CH2O)7H mg/L]を算出する。
e) 検量線 検量線の作成は,次による。
1) 非イオン界面活性剤標準液[CH3(CH2)11O(CH2CH2O)7H 0.01 mg/mL]2〜20 mLを分液漏斗200 mL
に段階的にとり,水を加えて100 mLとする。
2) d) 2)〜4)の操作を行って,非イオン界面活性剤[CH3(CH2)11O(CH2CH2O)7H]の量と吸光度との関係
線を作成する。
備考 10. ポリエチレングリコールが共存する場合は,備考7.による。
11. 陰イオン界面活性剤及び陽イオン界面活性剤が共存しない場合は,備考8.による。
31. 農薬 ここでいう農薬は,有機りん農薬(パラチオン,メチルパラチオン及びEPN),ペンタクロロ
フェノール(PCP)及びエジフェンホス(EDDP)に区分する。
試験は試料採取後,直ちに行う。直ちに行えない場合は,3.3によって保存し,できるだけ早く試験する。
31.1 有機りん農薬 有機りん農薬の試験には,前処理(ヘキサン抽出-カラムクロマトグラフ分離)した
試料について,ガスクロマトグラフ法,ナフチルエチレンジアミン吸光光度法(アベレル-ノリス法),又
はp-ニトロフェノール吸光光度法を適用する。
31.1.1 前処理 試料を塩酸酸性とし,ヘキサンを加えて抽出した後,カラムクロマトグラフによって分離
を行い,妨害物質を除く。
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
2) 塩化ナトリウム JIS K 8150に規定するもの。
3) 硫酸ナトリウム JIS K 8987に規定するもの。
4) 二酸化けい素 カラムクロマトグラフ用
5) 精製けい藻土 カラムクロマトグラフ用
6) ヘキサン JIS K 8848に規定するもの。
7) ニトロメタン JIS K 9523に規定するもの。
8) ヘキサン展開液 ヘキサン400 mLを分液漏斗500 mLにとり,ニトロメタン20 mLを加え,約3
分間激しく振り混ぜた後放置する。上層部のニトロメタン飽和のヘキサンを用いる。
参考1. 精製けい藻土には,市販品がある。
b) 器具 器具は,次による。
1) 分液漏斗 100 mL及び200 mL
2) 濃縮器 クデルナ-ダニッシュ形濃縮器又はロータリーエバポレーター
3) クロマトグラフ管 図31.1に例を示す。
3.1) クロマトグラフ管の作り方 クロマトグラフ管の作り方は,次による。
− 二酸化けい素0.5 gと精製けい藻土0.5 gとを混ぜ,ニトロメタン0.5 mLを加えて十分に混ぜ
合わせ,ヘキサン展開液10 mLを加えてかゆ状にする。
− クロマトグラフ管の底部にヘキサン展開液で湿らせた脱脂綿を詰め,かゆ状にした二酸化け
い素と精製けい藻土との混合物を流し込み,少量のヘキサン展開液でクロマトグラフ管の内
83
K 0102:2016
壁に付着した二酸化けい素及び精製けい藻土を洗い落とし,下のコックを開いてヘキサン展
開液を流出させる。
− 二酸化けい素と精製けい藻土との混合物が沈着し,ヘキサン展開液の液面が二酸化けい素と
けい藻土との混合物の上面の僅かに上になったらコックを閉じる。
単位 mm
図31.1 クロマトグラフ管の例
c) 抽出操作 抽出操作は,次による。
1) 試料100 mL(EPNとパラチオンとの合量又はメチルパラチオン10〜250 μgを含む。)を分液漏斗
200 mLにとり,塩化ナトリウム5 gを加えて溶かした後,塩酸(1+1)を加えて弱酸性とする。
2) ヘキサン20 mLを加えて約3分間激しく振り混ぜた後,放置し,水層は別の分液漏斗200 mLに移
す。
3) 2)の操作を更に2回繰り返す。
4) ヘキサン層を分液漏斗100 mLに合わせ,水層は捨てる。
5) ヘキサン層に水20 mLを加え,約1分間振り混ぜた後放置し,水層は捨てる。この洗浄操作を洗液
が中性になるまで繰り返す。
6) ヘキサン層に硫酸ナトリウム3〜5 gを加えて振り混ぜ,水分を除く。
7) 分液漏斗100 mLの脚部を乾いたろ紙で拭き取り,脱脂綿又はろ紙(1)を用いてヘキサン層をろ過し,
濃縮器に入れる。
8) 分液漏斗100 mLの内壁及び硫酸ナトリウムを少量のヘキサンで洗い,洗液も7)と同じ操作でろ過
し,濃縮器に入れる。
9) 濃縮器を減圧状態にして40 ℃以下でヘキサンの大部分を除去し,更に室温で静かに空気を送って
ヘキサンをほとんど揮散させる。
注(1) ろ過する場合には,あらかじめ少量のヘキサンで潤しておく。
d) 分離操作 分離操作は,次による。
84
K 0102:2016
1) c) 9)の残留物にヘキサン展開液2 mLを加えて溶かす。
2) これをピペットで吸い取り,クロマトグラフ管に静かに加える。
3) ヘキサン展開液2 mLを用いて濃縮器のフラスコの内壁を洗い,ピペットで吸い取り,2)と同様にク
ロマトグラフ管に加える。
4) メスシリンダー(有栓形)を受器とし,コックを開いて流出液をとり,カラム内の溶液の液面が二
酸化けい素とけい藻土との混合物より僅かに上部になったらコックを閉じる(2)。
5) ヘキサン展開液20 mLを静かに加え,コックを開いて流出液をメスシリンダー(有栓形)に受ける。
6) 4)の流出液及び5)の流出液の合量が5 mLとなったらこれを捨て,次の流出液14 mLを集める。こ
の流出部分にEPN及びパラチオンが含まれる。これをEPNとパラチオンとの合量の試験用試料と
する。
7) クロマトグラフ管にヘキサン展開液40 mLを加え,次の流出液24 mLを捨て,これに続く15 mLを
集める。この流出部分にメチルパラチオンが含まれる(3)。これをメチルパラチオンの試験用試料と
する。
注(2) この操作中,ヘキサン展開液の液面は,二酸化けい素とけい藻土との混合物の上面より上部に
保つ。
(3) 二酸化けい素の品質によって,有機りん化合物の流出状態が多少変わることがあるので,標準
液を用いて確かめておく。
備考 1. 分離操作に次の薄層クロマトグラフ分離法を用いてもよい。
1) c) 9)の残留物にJIS K 8034に規定するアセトン2 mLを加えて溶かす。
2) シリカゲル薄層板(*)の下端から約2 cmの高さに左右の両端を2 cmずつあけて,熱風を
緩やかに吹き付けながらこのアセトン溶液1 mLを直線状に添付する。
3) このシリカゲル薄層板の左右の両端には,EPN標準液(C14H14NO4PS 0.5 mg/mL)及びメ
チルパラチオン標準液(C8H10NO5PS 0.5 mg/mL)をそれぞれ2滴ずつ添付する。
4) ヘキサン-アセトン展開液[ヘキサン4容をビーカーにとり,アセトン1容を加えて調製
する。]を入れた展開槽にシリカゲル薄層板を移し,展開液を用いて展開する。
5) 展開距離が10〜12 cmとなったとき,シリカゲル薄層板を取り出して風乾する。
6) 暗所で紫外線(波長253.7 nm)を照射して両端の部分のEPN及びメチルパラチオン標準
液によるスポットを確認し,試料についてのこれに相当する高さのシリカゲル層をかき
とり,それぞれ三角フラスコ100 mLに移す(ガスクロマトグラフ法で定量する場合は,
シリカゲル層を合わせる。)。
7) それぞれにアセトン50 mLを加え,約3分間振り混ぜて有機りん農薬を溶出させる。
8) ろ紙5種Bでろ過し,ろ紙及びろ紙上のシリカゲルをアセトン5 mLで洗う。洗液をろ
液に合わせる。
9) EPNのRfに相当するシリカゲル層から得たアセトン溶液をEPNとパラチオンとの合量
の試験用試料とし,メチルパラチオンのRfに相当するシリカゲル層から得たアセトン溶
液をメチルパラチオンの試験用試料とする。
注(*) 薄層クロマトグラフ用シリカゲル10 gにJIS K 0557に規定するA3の水30 mLを
加え,ガラス板(200×200 mm)に約0.5 mmの厚さに塗る。これを105〜110 ℃
で約3時間加熱した後,デシケーター中で放冷し,保存する。
2. 妨害物質を含まない試料で,31.1.2の方法で定量する場合,又は農薬の合量を定量する場合
85
K 0102:2016
は,分離操作を省略し,d) 1)で得た溶液を試験用試料とすることができる。
備考 3. d)又は備考1.の分離操作においてEPNとパラチオンとは分離できない。また,芳香族ニトロ
化合物及びアミノ化合物が多量に存在すると,有機りん農薬から完全には分離できない。
31.1.2 ガスクロマトグラフ法 有機りん農薬(EPN,パラチオン及びメチルパラチオン)をガスクロマト
グラフ法によって定量する。
定量範囲:EPN(C14H14NO4PS),パラチオン(C10H14NO5PS)及びメチルパラチオン(C8H10NO5PS)
のいずれも1〜20 ng,繰返し精度:5〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) アセトン JIS K 8034に規定するもの。ただし,EPN,パラチオン及びメチルパラチオンの保持時
間に相当する位置にピークのないもの。
2) 窒素 JIS K 1107に規定する窒素2級
3) 水素 JIS K 0512に規定する水素3級
4) EPN標準液(C14H14NO4PS 0.5 mg/mL) EPN(C14H14NO4PS)[フェニルホスホノチオ酸O-エチル
O-(4-ニトロフェニル)エステル]0.050 gをとり,少量のアセトンに溶かし,全量フラスコ100 mL
に移し入れ,アセトンを標線まで加える。
5) EPN標準液(C14H14NO4PS 50 μg/mL) EPN標準液(C14H14NO4PS 0.5 mg/mL)10 mLを全量フラス
コ100 mLにとり,アセトンを標線まで加える。
6) メチルパラチオン標準液(C8H10NO5PS 0.5 mg/mL) メチルパラチオン(C8H10NO5PS)[ホスホロ
チオ酸O,O-ジメチルO-(4-ニトロフェニル)エステル]0.050 gをとり,少量のアセトンに溶かし,
全量フラスコ100 mLに移し入れ,アセトンを標線まで加える。
7) メチルパラチオン標準液(C8H10NO5PS 50 μg/mL) メチルパラチオン標準液(C8H10NO5PS 0.5
mg/mL)10 mLを全量フラスコ100 mLにとり,アセトンを標線まで加える。
8) パラチオン標準液(C10H14NO5PS 0.5 mg/mL) パラチオン(C10H14NO5PS)[ホスホロチオ酸O,O-
ジエチルO-(4-ニトロフェニル)エステル]0.050 gをとり,少量のアセトンに溶かし,全量フラス
コ100 mLに移し入れ,アセトンを標線まで加える。
9) パラチオン標準液(C10H14NO5PS 50 μg/mL) パラチオン標準液(C10H14NO5PS 0.5 mg/mL)10 mL
を全量フラスコ100 mLにとり,アセトンを標線まで加える。
10) 有機りん農薬標準液[(C14H14NO4PS 5 μg,C8H10NO5PS 5 μg,C10H14NO5PS 5 μg)/mL] EPN標準
液(C14H14NO4PS 50 μg/mL),メチルパラチオン標準液(C8H10NO5PS 50 μg/mL)及びパラチオン標
準液(C10H14NO5PS 50 μg/mL)それぞれ10 mLを全量フラスコ100 mLにとり,アセトンを標線ま
で加える。
b) 器具及び装置 器具及び装置は,次による。
1) マイクロシリンジ 5〜10 mL
2) 濃縮器 31.1.1 b) 2)による。
3) ガスクロマトグラフ 次の条件を満たすもので,EPN,パラチオン及びメチルパラチオンの1 ngを
検出できるもの。
− カラム用管 ガラス製内径3〜4 mm,長さ500〜2 000 mm
− カラム充塡剤 けい藻土(粒径180〜250 μm)を酸処理した後,シラン処理した担体(4)にシリ
コーン系固定相液体2〜5 %を含浸させたもの。又はこれと同等の分離性能をもつもの。
− 検出器 炎光光度検出器又はアルカリ熱イオン化検出器
86
K 0102:2016
− キャリヤーガス JIS K 1107に規定する窒素2級を用い,有機りん農薬が3〜15分間で流出す
るように流量を調節する。
− 試料導入部温度 170〜250 ℃
− カラム槽温度 140〜210 ℃
− 検出器槽温度 150〜250 ℃
注(4) けい藻土を主成分とした耐火温度1 100 ℃のれんが。
参考2. カラム充塡剤には,市販品がある。
c) 操作 操作は,次による。
1) 31.1.1 d)で得た,EPNとパラチオンとの合量の試験用試料及びメチルパラチオンの試験用試料又は
備考2.で得た試験用試料を40 ℃以下で減圧濃縮して約5 mLにする。
2) 濃縮物を全量フラスコ10 mL(又は20 mL)に移し,容器を少量のアセトンで数回洗い,洗液を先
の全量フラスコに合わせ,アセトンを標線まで加える。
3) この溶液4 μLをガスクロマトグラフに注入し,ガスクロマトグラムを記録する。
4) メチルパラチオン,パラチオン及びEPNの保持時間に相当する位置のピークについて,指示値(5)
を読み取る。
5) 検量線から,メチルパラチオン,パラチオン及びEPNの量を求め,試料中のメチルパラチオンの濃
度(C8H10NO5PS mg/L),パラチオンの濃度(C10H14NO5PS mg/L)及びEPNの濃度(C14H14NO4PS mg/L)
を算出する。
注(5) ピーク高さ又はピーク面積。
d) 検量線 検量線の作成は,次による。
1) 有機りん農薬標準液[(C14H14NO4PS 5 μg,C8H10NO5PS 5 μg,C10H14NO5PS 5 μg)/mL]0.5〜10 mL
を段階的に全量フラスコ10 mLにとり,アセトンを標線まで加える。
2) c) 3)及び4)の操作を行ってメチルパラチオン(C8H10NO5PS),パラチオン(C10H14NO5PS)及びEPN
(C14H14NO4PS)の量と指示値(5)との関係線をそれぞれ作成する。
31.1.3 ナフチルエチレンジアミン吸光光度法(アベレル-ノリス法) 有機りん農薬(EPN,パラチオン及
びメチルパラチオン)を還元し,亜硝酸でジアゾ化し,過剰の亜硝酸をアミド硫酸アンモニウムで分解し
た後,N-1-ナフチルエチレンジアミンとカップリングさせる。生じた赤紫のアゾ化合物の吸光度を測定し
て,EPNとパラチオンとの合量及びメチルパラチオンを定量する。EPNとパラチオンとの合量は,EPNと
して表す。
定量範囲:EPN(C14H14NO4PS)とパラチオン(C10H14NO5PS)との合量又はメチルパラチオン
(C8H10NO5PS)10〜250 μg,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+1) 31.1.1 a) 1)による。
2) 亜硝酸ナトリウム溶液(2.5 g/L) JIS K 8019に規定する亜硝酸ナトリウム0.25 gを水に溶かして
100 mLとする。1週間以上経過したものは使用しない。
3) アミド硫酸アンモニウム溶液(50 g/L) JIS K 8588に規定するアミド硫酸アンモニウム5 gを水に
溶かして100 mLとする。
4) 亜鉛粉末 JIS K 8013に規定するもの。
5) 二塩化N-1-ナフチルエチレンジアンモニウム溶液(10 g/L) JIS K 8197に規定するN-1-ナフチルエ
チレンジアミン二塩酸塩0.5 gを水に溶かして50 mLとする。使用時に調製する。
87
K 0102:2016
6) エタノール(99.5) JIS K 8101に規定するもの。
7) ヘキサン展開液 31.1.1 a) 8)による。
8) パラフィン
9) EPN標準液(C14H14NO4PS 50 μg/mL) 31.1.2 a) 5)による。
10) メチルパラチオン標準液(C8H10NO5PS 50 μg/mL) 31.1.2 a) 7)による。
b) 器具及び装置 器具及び装置は,次による。
1) 還流冷却器付き三角フラスコ 共通すり合わせリービッヒ冷却器又は球管冷却器200 mm及び共通
すり合わせ三角フラスコ100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 31.1.1 d)で得たEPNとパラチオンとの合量の試験用試料又はメチルパラチオンの試験用試料の適量
[C14H14NO4PS(C10H14NO5PS)又はC8H10NO5PSとして10〜250 μg]を三角フラスコ100 mLにと
り,エタノール(99.5)5 mL,塩酸(1+1)2 mL,水8 mL,亜鉛粉末0.5 g及びヘキサン展開液15
mLを加える。
2) 水浴上でヘキサンを揮散させる。
3) エタノール(99.5)10 mL及びパラフィン0.5 gを加え,還流冷却器を取り付けて水浴上で約5分間
煮沸した後,放冷する。
4) 少量の脱脂綿を用いてろ過し,ろ液を全量フラスコ50 mLに入れる。三角フラスコ100 mLを水約6
mLずつで3回洗って同じ脱脂綿でろ過し,洗液はろ液に合わせる(ろ液と洗液との合量が45 mL
を超えないようにする。)。
5) 塩酸(1+1)1 mL及び亜硝酸ナトリウム溶液(2.5 g/L)1 mLを加えてよく振り混ぜ,約10分間放
置する。
6) アミド硫酸アンモニウム溶液(50 g/L)1 mLを加え,よく振り混ぜて約10分間放置する。
7) 二塩化N-1-ナフチルエチレンジアンモニウム溶液(10 g/L)2 mLを加え,水を標線まで加える。栓
をして振り混ぜ,約20分間放置する。
8) 溶液(6)の一部を吸収セルに移し,波長555 nm付近の吸光度を測定する。
9) 空試験として三角フラスコ100 mLにエタノール(99.5)5 mL,塩酸(1+1)2 mL,水8 mL,亜鉛
粉末0.5 g及びヘキサン展開液15 mLをとり,2)〜8)の操作を行って吸光度を測定し,試料について
得た吸光度を補正する。
10) 検量線からEPNとパラチオンとの合量又はメチルパラチオンの量を求め,試料中の有機りん農薬を
それぞれEPN又はメチルパラチオンの濃度(C14H14NO4PSl又はC8H10NO5PS mg/L)として算出する。
注(6) 溶液が濁っている場合は,乾いたろ紙5種Bでろ過する。
d) 検量線 検量線の作成は,次による。
1) EPN標準液(C14H14NO4PS 50 μg/mL)又はメチルパラチオン標準液(C8H10NO5PS 50 μg/mL)0.2〜5
mLを三角フラスコ100 mLに段階的にとる。
2) c) 1)〜9)の操作を行ってEPN(C14H14NO4PS)又はメチルパラチオン(C8H10NO5PS)の量と吸光度
との関係線を作成する。
31.1.4 p-ニトロフェノール吸光光度法 有機りん農薬(EPN,パラチオン及びメチルパラチオン)をアル
カリで加水分解し,生成した黄色のp-ニトロフェノキシドの吸光度を測定して,EPNとパラチオンとの合
量及びメチルパラチオンを定量する。EPNとパラチオンとの合量はEPNとして表す。
88
K 0102:2016
定量範囲:EPN(C14H14NO4PS)とパラチオン(C10H14NO5PS)との合量がEPNとして50〜600 μg,メ
チルパラチオン(C8H10NO5PS)40〜500 μg,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+4) JIS K 8180に規定する塩酸を用いて調製する。
2) 水酸化カリウム溶液(100 g/L) JIS K 8574に規定する水酸化カリウム22 gを水に溶かして200 mL
とする。
3) 炭酸ナトリウム溶液(10 g/L) JIS K 8625に規定する炭酸ナトリウム5 gを水に溶かして500 mL
とする。
4) 過酸化水素 JIS K 8230に規定するもの。
5) エタノール(99.5) 31.1.3 a) 6)による。
6) ジエチルエーテル JIS K 8357に規定するジエチルエーテル(残留農薬・PCB試験用)
7) パラフィン
8) p-ニトロフェノール標準液(C6H4OHNO2 0.5 mg/mL) JIS K 8721に規定するp-ニトロフェノール
(C6H4OHNO2)0.050 gをエタノール(99.5)に溶かして,全量フラスコ100 mLに移し入れ,エタ
ノール(99.5)を標線まで加える。
9) p-ニトロフェノール標準液(C6H4OHNO2 50 μg/mL) p-ニトロフェノール標準液(C6H4OHNO2 0.5
mg/mL)10 mLを全量フラスコ100 mLにとり,炭酸ナトリウム溶液(10 g/L)を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 還流冷却器付き三角フラスコ 31.1.3 b) 1)による。
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 31.1.1 d)で得たEPNとパラチオンとの合量の試験用試料の適量(C14H14NO4PS又はC10H14NO5PSと
して50〜600 μgを含む。)又はメチルパラチオンの試験用試料の適量(C8H10NO5PSとして40〜500
μgを含む。)を三角フラスコ100 mLにとり,水酸化カリウム溶液(100 g/L)10 mL及びパラフィン
1 gを加え,水浴上でヘキサンを揮散させる。
2) 残留液にエタノール(99.5)10 mLを加え,還流冷却器を取り付け,水浴上で約30分間加熱する。
この間に過酸化水素10 mLを冷却器の先端から少量ずつ加える。
3) 塩酸(1+4)10 mLを加えて冷却する。
4) 少量の脱脂綿を用いてろ過し,ろ液を分液漏斗100 mLに移す。少量の水で三角フラスコ100 mL及
び脱脂綿を洗って洗液をろ液に合わせる。
5) ジエチルエーテル25 mLを加え,よく振り混ぜた後,放置する。
6) 水層を別の分液漏斗100 mLに移し,ジエチルエーテル25 mLを加えて,振り混ぜた後,放置し,
水層を捨てる(7)。
7) ジエチルエーテル層を合わせ,炭酸ナトリウム溶液(10 g/L)10 mLを加え,よく振り混ぜて逆抽出
を行い,放置する。水層を別の分液漏斗100 mLに移す。
8) ジエチルエーテル層に炭酸ナトリウム溶液(10 g/L)10 mL,次に,5 mLを用いて,7)の逆抽出を繰
り返し,水層を先の分液漏斗100 mLに合わせる。ジエチルエーテル層は捨てる。
9) 水層に塩酸(1+4)を加えて酸性とし,ジエチルエーテル25 mLずつで2回抽出し,ジエチルエー
テル層を分液漏斗100 mLに合わせる。水層は捨てる。
10) ジエチルエーテル層に炭酸ナトリウム溶液(10 g/L)10 mL,次に,5 mLを用いて逆抽出を行い,
89
K 0102:2016
水層を全量フラスコ50 mLに移し入れ,炭酸ナトリウム溶液(10 g/L)を標線まで加える。
11) この溶液の一部を吸収セルに移し,波長400 nm付近の吸光度を測定する。
12) 空試験として31.1.1 a) 8)のヘキサン展開液を,EPN及びパラチオンの場合は14 mL,メチルパラチ
オンの場合は15 mLをとり,1)〜11)の操作を行って吸光度を測定し,試料について得た吸光度を補
正する。
13) 検量線からEPNとパラチオンとの合量に相当するp-ニトロフェノールの量(mg)を求め,2.324を
乗じてEPNとパラチオンとの合量(C14H14NO4PS mg)とし,試料中のEPNとパラチオンとの合量
の濃度(C14H14NO4PS mg/L)を算出する。また,検量線からメチルパラチオンに相当するp-ニトロ
フェノールの量(mg)を求め,1.892を乗じてメチルパラチオンの量(mg)とし,メチルパラチオ
ンの濃度(C8H10NO5PS mg/L)を算出する。
注(7) ジエチルエーテル及び炭酸ナトリウム溶液(10 g/L)によって抽出する場合,エマルションが生
じて分離が困難なときは,塩化ナトリウム溶液(飽和)を加えるとよい。
d) 検量線 検量線の作成は,次による。
1) p-ニトロフェノール標準液(C6H4OHNO2 50 μg/mL)0.4〜5 mLを分液漏斗100 mLに段階的にとり,
エタノール(99.5)を加えて10 mLとし,水25 mL及び塩酸(1+4)5 mLを加える。
2) c) 5)〜12)の操作を行ってp-ニトロフェノール(C6H4OHNO2)の量と吸光度との関係線を作成する。
備考 4. 備考2.によって分離操作を省略した場合は,次のように操作して定量する。
1) 31.1.1 d) 1)で得たヘキサン溶液を分液漏斗100 mLに移し,水酸化カリウムのエタノール
溶液[JIS K 8574に規定する水酸化カリウム0.6 gをエタノール(体積百分率 50 %)に
溶かして1 Lとする。]20 mLを加えてよく振り混ぜて放置し,水層を捨てる。
2) この操作を2回繰り返す。2回目の水層が着色しているときは,水層が無色になるまで
この操作を繰り返す。
3) ヘキサン層を三角フラスコ100 mLに移し入れ,c) 1)〜13)の操作を行う。
31.2 ペンタクロロフェノール ペンタクロロフェノールの試験には,前処理した試料について,4-アミ
ノアンチピリン吸光光度法を適用する。
31.2.1 4-アミノアンチピリン吸光光度法 試料をりん酸酸性として蒸留した後,ペンタクロロフェノール
をキシレンで抽出し,4-アミノアンチピリン(4-アミノ-1,2-ジヒドロ-1,5-ジメチル-2-フェニル-3H-ピラゾ
ール-3-オン)と反応させて生じる青い色のアンチピリン色素の吸光度を測定してペンタクロロフェノール
を定量する。
定量範囲:C6Cl5OH 5〜60 μg,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) りん酸(1+9) JIS K 9005に規定するりん酸を用いて調製する。
3) 硫酸銅(II)溶液 28.1.1 a) 3)による。
4) りん酸水素二ナトリウム溶液 JIS K 9019に規定するりん酸水素二ナトリウム・12水24 gを水に溶
かして1 Lとする。
5) 硫酸ナトリウム JIS K 8987に規定するもの。
6) ヘキサシアノ鉄(III)酸カリウム溶液 28.1.2 a) 3)による。
7) 4-アミノアンチピリン溶液(2 g/L) JIS K 8048に規定する4-アミノアンチピリン0.2 gを水に溶か
して100 mLとする。使用時に調製する。
90
K 0102:2016
8) キシレン JIS K 8271に規定するもの。精製を要する場合は,次による。
− JIS K 8951に規定する硫酸の少量を加えて振り混ぜた後,硫酸を分離する。
− この操作を硫酸に色がつかなくなるまで繰り返す。
− 次に,水酸化ナトリウム溶液(10 g/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調製
する。)及び水でそれぞれ数回洗い,JIS K 8123に規定する塩化カルシウムを加えて蒸留する。
9) メチルオレンジ溶液(1 g/L) 24.1 a) 2)による。
10) ペンタクロロフェノール標準液(C6Cl5OH 1 mg/mL) ペンタクロロフェノール0.100 gをとり,水
酸化ナトリウムのメタノール溶液(0.1 mol/L)(JIS K 8576に規定する水酸化ナトリウム0.4 gをJIS
K 8891に規定するメタノールに溶かして100 mLとする。)5 mLに溶かし,全量フラスコ100 mLに
移し入れ,水を標線まで加える。
11) ペンタクロロフェノール標準液(C6Cl5OH 10 μg/mL) ペンタクロロフェノール標準液(C6Cl5OH
1 mg/mL)10 mLを全量フラスコ100 mLにとり,りん酸水素二ナトリウム溶液を標線まで加える。
この溶液10 mLを全量フラスコ100 mLにとり,りん酸水素二ナトリウム溶液を標線まで加える。
着色ガラス瓶に入れる。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 蒸留装置 蒸留フラスコ1 000 mL。共通すり合わせのもの。
2) 分液漏斗 500 mLで脚部の短いもの。
3) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料500 mL(C6Cl5OHとして5〜60 μgを含む。)を蒸留フラスコ1 000 mLにとり,指示薬として
メチルオレンジ溶液(1 g/L)2,3滴を加えた後,りん酸(1+9)を溶液の色が赤になるまで加え,
硫酸銅(II)溶液5 mLを加える。
2) 蒸留フラスコ1 000 mLを蒸留装置に取り付け,受器にメスシリンダー500 mLを用いて蒸留する。
3) 受器中の留出液が450 mLになったとき,一旦加熱を止める。
4) 蒸留フラスコ1 000 mLに水50 mLを加え,再び蒸留を続けて,更に50 mLを留出させる。
5) 留出液を分液漏斗500 mLに移し,塩酸2 mLを加えた後,キシレン10 mLを加え,約5分間激しく
振り混ぜて放置する。
6) 水層を捨て,キシレン層にりん酸水素二ナトリウム溶液10 mL及び4-アミノアンチピリン溶液(2
g/L)2 mLを加えて約30秒間激しく振り混ぜた後,ヘキサシアノ鉄(III)酸カリウム溶液1 mLを
加え,約3分間激しく振り混ぜ,約15分間放置する。
7) 水層を捨て,キシレン層に硫酸ナトリウム1 gを加え,軽く振り混ぜて水分を除去する。
8) キシレン層の一部を吸収セルに移し,キシレンを対照液として波長574 nm付近の吸光度を測定する。
9) 空試験として水500 mLを分液漏斗にとり,5)〜8)の操作を行って吸光度を測定し,試料について得
た吸光度を補正する。
10) 検量線からペンタクロロフェノールの量を求め,試料中のペンタクロロフェノールの濃度(C6Cl5OH
μg/L)を算出する。
d) 検量線 検量線の作成は,次による。
1) ペンタクロロフェノール標準液(C6C15OH 10 μg/mL)0.5〜6 mLを分液漏斗500 mLに段階的にとり,
水で約500 mLとする。
2) c) 5)〜9)の操作を行ってペンタクロロフェノール(C6C15OH)の量と吸光度との関係線を作成する。
91
K 0102:2016
備考 5. 酸化性物質又は還元性物質が共存すると妨害するが,28.の備考1.によって処理すれば妨害を
除くことができる。
6. ペンタクロロフェノール以外のフェノール類も,4-アミノアンチピリンと反応して発色する。
その色調は,ペンタクロロフェノールが青であるのに対し,他のフェノール類は黄色又はだ
いだい黄色であるから,これらがペンタクロロフェノールの10倍量存在しても妨害しない。
しかし,多量に存在すると妨害することがある。キシレン層が著しいだいだい黄色又は赤紫
を呈する場合には,この方法は適用できない。
31.3 エジフェンホス(EDDP) エジフェンホス(EDDP)(ホスホロジチオ酸O-エチル-S,S-ジフェニルエ
ステル)の定量は,JIS K 0128の19.[エジフェンホス(EDDP)]による。
32. 溶存酸素 溶存酸素の定量には,よう素滴定法,ミラー変法,隔膜電極法又は光学式センサ法を適用
する。この試験は,試料採取後,直ちに行う。
なお,よう素滴定法は,1983年に第1版として発行されたISO 5813,隔膜電極法は,2012年に第3版
として発行されたISO 5814との整合を図ったものである。また,光学式センサ法は,2014年に第1版と
して発行されたISO 17289との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 5813:1983,Water quality−Determination of dissolved oxygen−Iodometric method(MOD)
ISO 5814:2012,Water quality−Determination of dissolved oxygen−Electrochemical probe method
(MOD)
ISO 17289:2014,Water quality−Determination of dissolved oxygen−Optical sensor method(MOD)
32.1 よう素滴定法 硫酸マンガン(II)とアルカリ性よう化カリウム-アジ化ナトリウム溶液とを加えて
生成した水酸化マンガン(II)は,溶存酸素によって酸化されて水酸化マンガン(III)となる。次に,硫
酸を加えて水酸化マンガン(III)の沈殿を溶かし,遊離したよう素をチオ硫酸ナトリウム溶液で滴定して
溶存酸素を定量する。
定量範囲:O 0.5 mg/L以上
参考 この方法は,“ウインクラー-アジ化ナトリウム変法”とも呼ばれていた。
a) 試薬 試薬は,次による。
1) 硫酸 JIS K 8951に規定するもの。
2) アルカリ性よう化カリウム-アジ化ナトリウム溶液 JIS K 8574に規定する水酸化カリウム350 g
(又はJIS K 8576に規定する水酸化ナトリウム250 g)とJIS K 8913に規定するよう化カリウム75
gとをそれぞれ水に溶かし,これを混合し,水を加えて500 mLとする。別に,JIS K 9501に規定す
るアジ化ナトリウム5 gを水20 mLに溶かし,これも混合する。遮光したポリエチレン瓶に入れて
暗所に保存する。
3) 硫酸マンガン(II)溶液 JIS K 8997に規定する硫酸マンガン(II)五水和物240 gを水に溶かして
500 mLとする。
4) でんぷん溶液(10 g/L) 19. a) 5)による。
5) 25 mmol/Lチオ硫酸ナトリウム溶液(1) 19. a) 8)の0.1 mol/Lチオ硫酸ナトリウム溶液50 mLを全量
フラスコ200 mLにとり,水を標線まで加える。この溶液は使用時に調製し,12時間以上経過した
92
K 0102:2016
ものは使用しない。
注(1) d)の滴定に10 mmol/Lチオ硫酸ナトリウム溶液を用いる場合は,19. a) 9)による。
b) 器具 器具は,次による。
1) 溶存酸素測定瓶 21. b) 1)の培養瓶を用いる。
c) 試料採取 試料採取は,次のいずれかによって行い,引き続き,採取現地においてd)の操作を行う。
ただし,試料が著しく着色したり,濁りがあったりする場合は,備考1.によって採取する。
1) 直接採取する場合 河川,水路,貯水槽などの表面水を溶存酸素測定瓶で直接採取するには,まず,
試料で溶存酸素測定瓶をよく洗い,溶存酸素測定瓶を水面下に入れ,満水するまで静かに試料を流
し込んで気泡が残らないように密栓する。ばけつなどで採取した場合も,同じ操作で流し入れて密
栓する。
2) 採水器を使用する場合 バンドーン採水器,絶縁採水器などを用いる場合は,採水器の取出口に軟
質塩化ビニル管を接続し,この軟質塩化ビニル管の先端を溶存酸素測定瓶の底まで入れ,気泡が生
じないように注意して試料を溶存酸素測定瓶に1/3ほど手早く流し込み,溶存酸素測定瓶を洗う。
同じ操作で改めて試料を溶存酸素測定瓶に流し入れ,瓶の容量の25〜50 %の試料をあふれさせてか
ら,静かに軟質塩化ビニル管を取り出し,気泡が残らないように密栓する。
3) 配管及び装置類から採取する場合 配管及び装置類に取り付けてある試料採取弁に軟質塩化ビニル
管を接続し,約1 L/minで連続的に通水する。軟質塩化ビニル管の先端を溶存酸素測定瓶の底部ま
で入れ,溶存酸素測定瓶の容量の約5倍量の試料をあふれさせてから,軟質塩化ビニル管を取り出
し,気泡が残らないように密栓する。
d) 操作 操作は,次による。
1) 溶存酸素測定瓶の栓を取り,これに試料100 mLについて硫酸マンガン(II)溶液1 mLとアルカリ
性よう化カリウム-アジ化ナトリウム溶液1 mLとをそれぞれピペットの先端を試料中に挿入して手
早く加え,溶存酸素測定瓶中に空気が残らないように密栓する。
2) 約1分間転倒を繰り返し,生成した沈殿が瓶の全体に広がるように十分に混ぜ合わせる。
3) しばらく静置し,沈殿が沈降したら再び2)の操作を行った後,静置する(2)。
4) 沈殿が沈降し,上澄み液が瓶全体の1/2程度になったら静かに開栓し,瓶の首に沿ってピペットで
試料100 mLについて硫酸1 mLを加え,再び密栓して数回転倒して沈殿を溶かす。
5) この溶液の適量(全量でもよい。)を分取し,三角フラスコに入れる。
6) 25 mmol/Lチオ硫酸ナトリウム溶液(3)で滴定し,溶液の黄色がうすくなってから指示薬としてでん
ぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
7) 次の式によって試料中の溶存酸素の濃度(O mg/L)を算出する(4)。
200
.0
000
1
1
2
1
×
−
×
×
×
=
v
V
V
V
f
a
O
ここに,
O: 溶存酸素の濃度(O mg/L)
a: 滴定に要した25 mmol/Lチオ硫酸ナトリウム溶液量(mL)
V1: 共栓を施したときの溶存酸素測定瓶の容量(mL)
V2: 滴定のため溶存酸素測定瓶から分取した試料量(mL)
v: 加えたアルカリ性よう化カリウム-アジ化ナトリウム溶液と硫
酸マンガン(II)溶液の合計量(mL)
f: 25 mmol/Lチオ硫酸ナトリウム溶液のファクター(5)
0.200: 25 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する酸素の質
量(mg)
93
K 0102:2016
注(2) 3)までの操作を“溶存酸素の固定”と呼ぶ。この固定までの操作を採取した現地で行い,遮光
し,試験室に持ち帰ってもよい。次に,4)以降の操作を行ってよい。この場合もなるべく早く
試験する。
(3) 25 mmol/Lチオ硫酸ナトリウム溶液に代え,注(1)によって調製した,10 mmol/Lチオ硫酸ナトリ
ウム溶液を用いて滴定してもよい。
(4) 注(3)によって滴定した場合は,aには滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
を,また,その1 mLに相当する酸素の質量には0.08(mg)を用いる。
(5) 19. a) 8)の0.1 mol/Lチオ硫酸ナトリウム溶液のファクターを用いる。
備考 1. 著しく着色又は濁りがある試料の採取 c)の操作に準じて試料を共栓ガラス瓶1 Lに採取し,
硫酸カリウムアルミニウム溶液(JIS K 8255に規定する硫酸カリウムアルミニウム・12水10
gを水に溶かし,水を加えて100 mLとする。)10 mLとJIS K 8085に規定するアンモニア水
1〜2 mLとを,ピペットを試料中に挿入して加え,直ちに密栓して転倒させながら約1分間
混合し,静置する。懸濁物が沈降した後,上澄み液を静かに溶存酸素測定瓶に流し入れて満
水とし,気泡が残らないように密栓する。
2. 酸化性物質又は還元性物質の確認 試料50 mLに,硫酸(1+5)(JIS K 8951に規定する硫
酸を用いて調製する。)1 mL,JIS K 8913に規定するよう化カリウム(又はよう化ナトリウ
ム)約0.5 g及びでんぷん溶液(10 g/L)2,3滴を加える。
溶液の色が青に変化したら,酸化性物質が存在する。この場合には,備考3.の操作を行っ
て測定結果を補正する。
溶液が無色のままなら,更によう素溶液(0.005 mol/L)(JIS K 8913に規定するよう化カリ
ウム又はよう化ナトリウム4〜5 gを少量の水に溶かし,JIS K 8920に規定するよう素約130
mgを加える。よう素が溶けた後,水で100 mLとする。)0.2 mLを加え,振り混ぜる。30秒
間放置後,無色ならば還元生物質が存在する。この場合には備考4.の操作によって溶存酸素
の濃度を測定する。
3. 酸化性物質を含む試料の試験 空試験として別の溶存酸素測定瓶を用い,c)の操作によって
試料を採取し,これにアルカリ性よう化カリウム-アジ化ナトリウム溶液1 mLと硫酸1 mL
とをピペットを挿入して加えて密栓し,転倒を繰り返して混合し,次に,硫酸マンガン(II)
溶液1 mLをピペットを挿入して加えて密栓し,転倒を繰り返して混合する。これについて
d) 5)〜6)の操作を行って滴定し,7)の式によって酸化性物質に相当する溶存酸素の濃度を求
め,試料中の溶存酸素の濃度を補正する。
4. 還元性物質を含む試料の試験 c)の操作によって試料を採取し,この試料について,アルカ
リ性よう化カリウム-アジ化ナトリウム溶液の代わりに,よう素-アルカリ性よう化カリウム
溶液を用いてd)の操作を行い,溶存酸素の濃度を算出する。別に空試験として,試料を別の
溶存酸素測定瓶にとり,よう素-アルカリ性よう化カリウム溶液と硫酸とを加え,次に硫酸マ
ンガン(II)溶液を加えた後滴定し,7)の式によって相当する溶存酸素の濃度を求め,試料中
の溶存酸素の濃度を補正する。
よう素-アルカリ性よう化カリウム溶液の調製方法
アルカリ性よう化カリウム溶液(JIS K 8574に規定する水酸化カリウム350 gとJIS K
8913に規定するよう化カリウム75 gとをそれぞれ水に溶かし,これらを混合し,水を加
えて500 mLとする。遮光したポリエチレン瓶に入れて保存する。)約125 mLを全量フラ
94
K 0102:2016
スコ250 mLにとり,よう素溶液(50 mmol/L)(JIS K 8913に規定するよう化カリウム20 g
を少量の水に溶かし,これにJIS K 8920に規定するよう素6.4 gを加えて溶かし,水を加
えて500 mLとする。)10 mLを加えた後,アルカリ性よう化カリウム溶液を標線まで加え
る。使用時に調製する。
この溶液1 mLは,硫化物イオン0.064 mg又は亜硫酸イオン0.16 mgと反応する。必要
があれば亜硫酸イオン,硫化物イオンなどの還元性物質の量を求め,相当するよう素溶液
(50 mmol/L)の量を算出して添加量を求める。
備考 5. 海水試料の試験 海水は微生物を含む場合が多いから,反応を速めて手早く試験する。反応
促進のためにアルカリ性よう化カリウム-アジ化ナトリウム溶液と硫酸マンガン(II)溶液と
をそれぞれ2倍量添加し,次に,硫酸を2倍量加える。
6. 鉄(III)が共存する試料の試験 硫酸の添加前に,試料100 mLについてふっ化カリウム溶
液(300 g/L)[19.の注(3)による。]1 mLを加えれば,鉄(III)100〜200 mg/Lが含まれていて
も妨害しない。
32.2 ミラー変法 流動パラフィンで試料を空気と遮断し,酒石酸ナトリウムカリウム-水酸化ナトリウム
溶液と3,7-ビス(ジメチルアミノ)フェノチアジン-5-イウムクロリド(メチレンブルー)溶液とを加え,
硫酸アンモニウム鉄(II)溶液で滴定し,溶存酸素を定量する。
定量範囲:O 1 mg/L以上
a) 試薬 試薬は,次による。
1) 酒石酸ナトリウムカリウム-水酸化ナトリウム溶液 JIS K 8536に規定する(+)-酒石酸ナトリウ
ムカリウム四水和物350 g及びJIS K 8576に規定する水酸化ナトリウム100 gを水に溶かして,1 L
とする。
2) メチレンブルー溶液 JIS K 8897に規定するメチレンブルー0.1 gを水100 mLに溶かす。
3) 流動パラフィン JIS K 9003に規定するもの。
4) 硫酸アンモニウム鉄(II)溶液 JIS K 8951に規定する硫酸5 mLを水100 mLに加え,これにJIS K
8979に規定する硫酸アンモニウム鉄(II)六水和物5.4 gを加えて溶かし,2. n) 1)の溶存酸素を含ま
ない水を加えて1 Lとする。
標定 標定は,次による。
この溶液の溶存酸素相当量は32.1で溶存酸素の濃度を求めた水を標準とし,c)の操作によってこ
の溶液で滴定し,次の式によって算出する。この標定は,使用時に行う。
b
a
f
1
000
1
50×
×
=
ここに,
f: 硫酸アンモニウム鉄(II)溶液1 mLに相当する溶存酸素の質
量(O mg)
a: 使用した水の溶存酸素の濃度(O mg/L)
b: 滴定に要した硫酸アンモニウム鉄(II)溶液量(mL)
b) 器具 器具は,次による。
1) 試料採取器 注射筒50 mLの先端に内径約1.5 mm,長さ250〜300 mmのガラス管をすり合わせ又
はゴム管で接続したもの。
2) 溶存酸素測定用試験管 外径約30 mm,高さ約200 mmの試験管
3) かき混ぜ棒 直径約3 mm,全長約250 mmのガラス棒で,下部を約10 mm折り曲げたもの又は下
95
K 0102:2016
部をらせん状にしたもの。
4) 足長ビュレット 容量5〜10 mLのもので,足の先端が試験管の下部に達するもの。
c) 操作 操作は,次による。
1) 溶存酸素測定用試験管に,メチレンブルー溶液2滴,酒石酸ナトリウムカリウム-水酸化ナトリウム
溶液5 mL及び流動パラフィン約5 mLを加える。
2) 試料採取器に試料を吸引して2回洗った後,気泡が入らないように徐々に吸引して試料50 mLを採
取する。このときガラス管部分に試料が満たされた状態に保つ。
3) 試料採取器のガラス管の先端を静かに流動パラフィン層の下の水層に入れ,流動パラフィン層が乱
れないように注意しながら,試料50 mLを注入する。
4) かき混ぜ棒を静かに入れ,足長ビュレットの先端を水層に入れる。
5) 足長ビュレットから硫酸アンモニウム鉄(II)溶液を少量ずつ加え,そのたびにかき混ぜ棒で静か
にかき混ぜ,メチレンブルーの青い色が消えるまで滴定する。
6) 次の式によって試料中の溶存酸素の濃度(O mg/L)を算出する。
50
000
1
×
×
=
f
a
O
ここに,
O: 溶存酸素の濃度(O mg/L)
a: 滴定に要した硫酸アンモニウム鉄(II)溶液量(mL)
f: 硫酸アンモニウム鉄(II)溶液1 mLに相当する溶存酸素の質
量(mg)
32.3 隔膜電極法 隔膜電極を試料に浸せき(漬)して溶存酸素濃度を測定する。隔膜電極は酸素透過性
のある隔膜,作用電極,対極,電解液などから構成され,隔膜を透過した酸素が還元されて生じる電流を
測定する。この電流が試料中の溶存酸素量と比例することを利用し,定量する。
定量範囲:O 0.5 mg/L以上,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 亜硫酸ナトリウム溶液 JIS K 8061に規定する亜硫酸ナトリウム約1 gを水に溶かし,水を加えて
500 mLとする。使用時に調製する。この溶液は,ゼロ調節に用いる。
2) 溶存酸素飽和水(6) 水酸化カリウム溶液(250 g/L)(JIS K 8574に規定する水酸化カリウムを用い
て調製する。)で洗浄した空気を,約1 L/minの流量で球形又は板状のガラスろ過器を用いて水に通
気(7)し,溶存酸素を飽和させる。スパン調節操作を行う直前に調製する。
注(6) 溶存酸素飽和水は,試料の温度と±0.5 ℃で一致する温度のものを調製する。この溶液の溶存
酸素の濃度は,表32.1から求めるか,又は32.1によって溶存酸素の濃度を確認する。また,塩
類の濃度の高い試料の溶存酸素の濃度を測定する場合には,試料の塩類のモル濃度に合わせた
量のJIS K 8150に規定する塩化ナトリウムを添加した溶存酸素飽和水を調製する。
(7) 通常,水200 mLの場合には5〜10分間,500 mLの場合には10〜20分間通気する。
b) 器具及び装置 器具及び装置は,次による。
1) 溶存酸素測定容器 ガラス容器100〜300 mLにゴム栓を付け,栓に試料を採取するための注入管及
び排出するための排出管を取り付けたもの(8)。図32.1に例を示す。
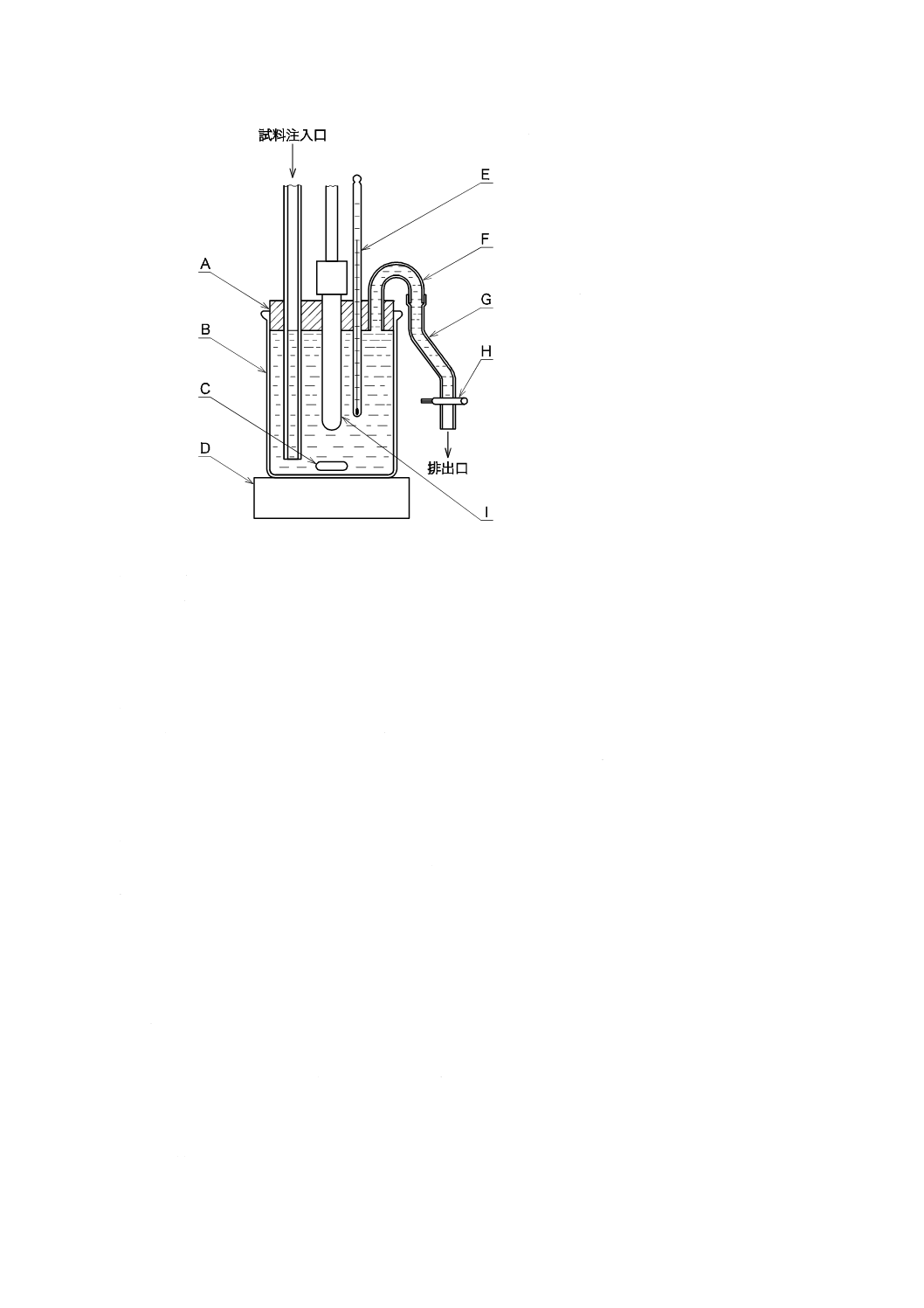
96
K 0102:2016
A:
B:
C:
D:
E:
F:
G:
H:
I:
ゴム栓
ガラス容器
回転子
マグネチックスターラー
温度計
排出管
ガラス管
ピンチコック
電極
図32.1 溶存酸素測定容器の例
2) 温度計 JIS B 7411-1に規定する一般用ガラス製棒状温度計の50度温度計
3) 溶存酸素計 温度補償回路を組み入れたもの。
4) マグネチックスターラー
注(8) 溶存酸素測定瓶を用いてもよい。
c) 準備操作 準備操作は,次による。
1) 溶存酸素計の電極を接続し,約30分間通電しておく。
2) 溶存酸素測定容器に試料と同じ温度にした亜硫酸ナトリウム溶液を注入し,マグネチックスターラ
ーで静かにかき混ぜながら電極を挿入し,指示値が安定してから(9)ゼロ点を確認する。可能であれ
ば,指示値をゼロに合わせる。
3) 電極及び温度計を取り出し,水でよく洗い(10),別の溶存酸素測定容器に挿入する。
4) 注入管の一端から溶存酸素測定容器の底部に静かに溶存酸素飽和水(11)を注入し,溶存酸素測定容器
の容量の25〜50 %を流出させた後,排出管の先端を閉じる。
5) マグネチックスターラーでかき混ぜ(12)ながら,溶存酸素計の指示値が安定するのを待ち,注入した
溶存酸素飽和水の温度と一致していることを確かめ,対応する飽和溶存酸素量を求め,指示値を合
わせる(13)。
6) 2)〜5)の操作を2回又は3回繰り返して,指示値がそれぞれゼロ及び飽和溶存酸素量に合致してい
ることを確かめる。
注(9) 通常2〜5分間を要する。
(10) 準備操作2)から3)に移るときには,電極を特によく洗浄する。
(11) 容器内で通気して溶存酸素飽和水を調製してもよい。
(12) かき混ぜ速度によって指示値に差が生じるので,できるだけ同じ条件に保つ。
(13) 試料温度と気温が同じ場合には,簡易方法として水蒸気飽和させた空気中での校正が可能な計
器もある。
97
K 0102:2016
備考 7. 隔膜電極の扱いについて,使用前及び測定時には,次の事項に注意する。
1) 隔膜の表面に指を触れない。
2) 電解液及び隔膜を交換した場合,又は隔膜を乾燥させてしまった場合は,隔膜を水で湿
らせ,c)の操作を行う前に指示値が安定するように時間をおく。
3) 試料に浸したときに気泡が電極に付着していないことを確認する。
4) 酸素が常に供給されるように,試料は隔膜上を絶えず流れていることが必要である。ま
た,指示値の振れが生じない程度の流量であることを確かめる。
d) 操作 操作は,次による。
1) c) 4)に準じて,試料を溶存酸素測定容器の底部に,気泡が入らないように静かに注入する(14)。
2) マグネチックスターラーでかき混ぜ(12)ながら温度計の目盛を確認し,次に,溶存酸素計の指示値が
安定するのを待って溶存酸素の濃度(O mg/L)を読み取る。測定値に影響を与える試料温度,大気
圧,塩濃度(実用塩分)などを確認する(15)。
注(14) 溶存酸素測定瓶,培養瓶などを測定容器として用いる場合には,サイホンを用いて静かに試料
を容器にとり,直ちに電極及び温度計を挿入して測定する。
(15) 計器によっては,大気圧,塩濃度(実用塩分)などを入力し,濃度計算に反映させる機能をも
つもの,又は,それらを自動で補正する機能をもつものもある。このような自動機能をもたな
い計器を使用する場合は,大気圧,塩濃度の影響を附属書1表6,附属書1表7及び表32.1を
用いて補正する必要がある。
32.4 光学式センサ法 光学式センサを試料に浸せき(漬)して溶存酸素濃度を測定する。光学式センサ
は蛍光物質,りん(燐)光物質などを塗布したセンサキャップ,励起光源,光検出部等から構成され,試
料中で励起された蛍光物質,りん(燐)物質などが発する光を測定する。試料中に酸素が存在すると消光
作用によって発光量が減少するが,この消光作用は溶存酸素量に比例する。光学式センサは発光の位相差
や持続時間等から酸素による消光作用を測定し,溶存酸素濃度を定量する。
定量範囲:O 0.5 mg/L以上,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 亜硫酸ナトリウム溶液 32.3 a) 1) による。この溶液はゼロ調節に用いる。
備考 8. ゼロ調節には次の溶液を用いてもよい。蓋付き容器に水85 mLをとり,JIS K 9502に規定す
るL(+)-アスコルビン酸2 gを溶かす。これに水酸化ナトリウム溶液(40 g/L)(JIS K 8576
に規定する水酸化ナトリウムを用いて調製する。)25 mLを加え,静かにかくはんする。使用
前に3分間待つ。
9. 水蒸気飽和させたJIS K 1107に規定する窒素雰囲気中でゼロ調節を行ってもよい。
2) 溶存酸素飽和水 32.3 a) 2) による。
b) 器具及び装置 器具及び装置は,次による。
1) 溶存酸素測定容器 32.3 b) 1) による。
2) 光学式センサ溶存酸素計 表示・操作部及びプローブから構成される(図32.2参照)。表示・操作
部は溶存酸素の濃度をmg/L単位で,及び/又は酸素の飽和百分率を%単位で表示できるもの。
98
K 0102:2016
図32.2 光学式センサ溶存酸素計の構成例
3) 温度計 JIS B 7411-1に規定する一般用ガラス製棒状温度計の50度温度計(16)。
4) 気圧計 1 hPa単位のもの(17)。
注(16) 通常,溶存酸素計には温度補償回路が組み入れられている。
(17) 装置に気圧計が組み込まれた計器もある。
5) マグネチックスターラー
c) 準備操作 準備操作は,次による。
1) 32.3 c) 2)から5) による(18)。
注(18) プローブは1〜2分間で安定な指示を得る。プローブが異なると安定するまでの時間が異なる場
合があるので,応答時間が最短化されるようにかくはんする。その際,大気から酸素が混入し
ないよう注意する。
備考 10. 計器が校正不能となった場合,応答が不安定又は遅くなった場合などは,センサキャップを
交換する。また,交換は製造業者の取扱説明書を参照して行う。
d) 操作 操作は,次による。
1) 32.3 c) 4) に準じて,試料を溶存酸素測定容器の底部に,気泡が入らないように静かに注入する(19)。
2) マグネチックスターラーでかき混ぜ(12)ながら温度計の目盛を確認し,次に,溶存酸素計の指示値が
安定するのを待って溶存酸素の濃度(O mg/L)を読み取る。測定値に影響を与える試料温度,大気
圧,塩濃度(実用塩分)などを確認する(15)。
注(19) 溶存酸素測定瓶,培養瓶などを測定容器として用いる場合には,サイホンを用いて静かに試料
を容器にとり,直ちにプローブを挿入して測定する。
表32.1 水中の飽和溶存酸素量(1013 hPa)
単位 mg/L
温度
℃
塩濃度(実用塩分 Salinity)(20)
0
9
18
27
36
0
14.62
13.73
12.89
12.11
11.37
1
14.22
13.36
12.55
11.79
11.08
2
13.83
13.00
12.22
11.49
10.80
3
13.46
12.66
11.91
11.20
10.54
4
13.11
12.34
11.61
10.93
10.28
5
12.77
12.03
11.33
10.66
10.04
6
12.45
11.73
11.05
10.41
9.81
7
12.14
11.44
10.79
10.17
9.58
8
11.84
11.17
10.54
9.94
9.37
99
K 0102:2016
表32.1 水中の飽和溶存酸素量(1013 hPa)(続き)
単位 mg/L
温度
℃
塩濃度(実用塩分 Salinity)(20)
0
9
18
27
36
9
11.56
10.91
10.29
9.71
9.16
10
11.29
10.66
10.06
9.50
8.97
11
11.03
10.42
9.84
9.29
8.78
12
10.78
10.19
9.63
9.09
8.59
13
10.54
9.96
9.42
8.90
8.42
14
10.31
9.75
9.22
8.72
8.25
15
10.08
9.54
9.03
8.55
8.09
16
9.87
9.35
8.85
8.38
7.93
17
9.67
9.15
8.67
8.21
7.78
18
9.47
8.97
8.50
8.05
7.63
19
9.28
8.79
8.34
7.90
7.49
20
9.09
8.62
8.18
7.75
7.35
21
8.92
8.46
8.02
7.61
7.22
22
8.74
8.30
7.88
7.47
7.09
23
8.58
8.14
7.73
7.34
6.97
24
8.42
8.00
7.59
7.21
6.85
25
8.26
7.85
7.46
7.09
6.73
26
8.11
7.71
7.33
6.97
6.62
27
7.97
7.58
7.20
6.85
6.51
28
7.83
7.45
7.08
6.73
6.40
29
7.69
7.32
6.96
6.62
6.30
30
7.56
7.20
6.85
6.52
6.20
31
7.43
7.07
6.74
6.41
6.10
32
7.31
6.96
6.63
6.31
6.01
33
7.18
6.84
6.52
6.21
5.92
34
7.07
6.73
6.42
6.11
5.83
35
6.95
6.63
6.32
6.02
5.74
36
6.84
6.52
6.22
5.93
5.65
37
6.73
6.42
6.12
5.84
5.57
38
6.62
6.32
6.03
5.75
5.48
39
6.52
6.22
5.93
5.66
5.40
40
6.41
6.12
5.84
5.58
5.32
41
6.31
6.03
5.75
5.50
5.25
42
6.21
5.94
5.67
5.41
5.17
43
6.12
5.84
5.58
5.33
5.09
44
6.02
5.75
5.50
5.25
5.02
45
5.93
5.67
5.42
5.18
4.95
注(20) 表32.1の実用塩分(Salinity)は,電気伝導率によって定義される値である。1気圧,15 ℃にお
いて,1 kg中に32.435 6 gの塩化カリウムを含む溶液と電気伝導率が等しい海水の塩分を35と
定義している(UNESCO,1981)。
33. 残留塩素 残留塩素とは,塩素剤が水に溶けて生成する次亜塩素酸及びこれがアンモニアと結合して
生じるクロロアミンをいい,前者を遊離残留塩素,後者を結合残留塩素,両者を合わせて残留塩素という。
残留塩素の定量には,濃度が低い場合にはo-トリジン比色法,ジエチル-p-フェニレンジアンモニウム
100
K 0102:2016
(DPD)比色法又はジエチル-p-フェニレンジアンモニウム(DPD)吸光光度法を適用し,濃度が比較的高
い場合には,よう素滴定法を適用する。
この試験は,試料採取後,直ちに行う。
なお,ジエチル-p-フェニレンジアンモニウム(DPD)比色法は,1985年に第1版として発行されたISO
7393-2,よう素滴定法は,1990年に第2版として発行されたISO 7393-3との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 7393-2:1985,Water quality−Determination of free chlorine and total chlorine−Part 2:
Colorimetric method using N,N-diethyl-1,4-phenylenediamine, for routine control purposes
(MOD)
ISO 7393-3:1990,Water quality−Determination of free chlorine and total chlorine−Part 3 : Iodometric
titration method for the determination of total chlorine(MOD)
33.1 o-トリジン比色法 試料に3,3'-ジメチルベンジジン(o-トリジン)溶液を加え,残留塩素との反応で
生じる黄色を,残留塩素標準比色液と比較して残留塩素を定量する方法である。亜ひ酸ナトリウム溶液で
処理し,残留塩素,遊離残留塩素及び結合残留塩素の三つに区別することができる。
定量範囲:Cl 0.01〜2.0 mg/L,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水(1)
2) o-トリジン溶液 二塩化3,3'-ジメチルベンジジニウム(o-トリジン二塩酸塩)0.14 gを水50 mLに
溶かし,塩酸(3+7)(JIS K 8180に規定する塩酸を用いて調製する。)50 mL中にかき混ぜながら
加える。着色瓶に入れて保存する。6か月以上経過したものは使用しない。
3) りん酸塩緩衝液(pH6.5) JIS K 9020に規定するりん酸水素二ナトリウムを110 ℃で約2時間加熱
し,デシケーター中で放冷した後,その22.86 gと,JIS K 9007に規定するりん酸二水素カリウム
46.14 gとを水に溶かして1 Lとする。沈殿が生じた場合には,ろ別する。この溶液200 mLをとり
水で1 Lとする。
4) クロム酸カリウム-二クロム酸カリウム溶液 JIS K 8312に規定するクロム酸カリウム3.63 gとJIS
K 8517に規定する二クロム酸カリウム1.21 gとをりん酸塩緩衝液(pH6.5)に溶かし,全量フラス
コ1 000 mLに移し入れ,りん酸塩緩衝液(pH6.5)を標線まで加える。
5) 残留塩素標準比色液 相当する残留塩素の濃度(Cl mg/L)に応じ,クロム酸カリウム-二クロム酸
カリウム溶液及びりん酸塩緩衝液(pH6.5)を表33.1に示す割合に比色管100 mLにとり,混ぜ合わ
せる。暗所に保存する。沈殿が生じた場合には使用しない。
6) 亜ひ酸ナトリウム溶液(5 g/L) メタ亜ひ酸ナトリウム0.5 gを水に溶かして100 mLとする。
注(1) この試験に用いる水は,残留塩素のないこと及び塩素を消費しないことを,備考3.によって確
かめておく。
b) 器具 器具は,次による。
1) 比色管 100 mL 底部から200±1.5 mmの高さに100 mLの標線を付けた平底のもの。
2) 比色管立 100 mL用 底部及び側面に乳白板を付けたもの。
101
K 0102:2016
表33.1 残留塩素標準比色液(液層200 mm用)
残留塩素
Cl mg/L
クロム酸カリウム-
二クロム酸カリウム溶液
mL
りん酸塩緩衝液
pH6.5
mL
残留塩素
Cl mg/L
クロム酸カリウム-
二クロム酸カリウム溶液
mL
りん酸塩緩衝液
pH6.5
mL
0.01
0.18
99.82
0.70
7.48
92.52
0.02
0.28
99.72
0.80
8.54
91.46
0.05
0.61
99.39
0.90
9.60
90.40
0.07
0.82
99.18
1.00
10.66
89.34
0.10
1.13
98.87
1.10
12.22
87.78
0.15
1.66
98.34
1.20
13.35
86.65
0.20
2.19
97.81
1.30
14.48
85.52
0.25
2.72
97.28
1.40
15.60
84.40
0.30
3.25
96.75
1.50
16.75
83.25
0.35
3.78
96.22
1.60
17.84
82.16
0.40
4.31
95.69
1.70
18.97
81.03
0.45
4.84
95.16
1.80
20.09
79.91
0.50
5.37
94.63
1.90
21.22
78.78
0.60
6.42
93.58
2.00
22.34
77.66
c) 操作 操作は,次による。
1) 比色管にo-トリジン溶液5 mLをとり,これに試料(2)の適量(残留塩素0.2 mg以下を含む。)を加え,
更に水を100 mLの標線まで加え,手早く栓をして振り混ぜる。
2) 5分間(3)暗所に放置する。
3) 上方から透視して残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当
する残留塩素の濃度a(Cl mg/L)を記録する。
4) 別の比色管にo-トリジン溶液5 mLをとり,これに1)の操作と同量の試料を加え,手早く栓をして
振り混ぜる。
5) 5秒間以内に亜ひ酸ナトリウム溶液(5 g/L)5 mLを加えて振り混ぜ,更に,水を100 mLの標線ま
で加えて振り混ぜる。
6) 残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当する残留塩素の濃
度b(Cl mg/L)を記録する。
7) 空試験として比色管100 mLに亜ひ酸ナトリウム溶液(5 g/L)5 mLをとり,これに1)の操作と同量
の試料を加えて振り混ぜる。
8) o-トリジン溶液5 mLを加えて振り混ぜ,更に,水を100 mLの標線まで加えて振り混ぜる。
9) 5秒間以内に残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,これに相当する
残留塩素の濃度c1(Cl mg/L)を記録する。
10) さらに,5分間暗所に放置後,残留塩素標準比色液と比較し,該当する残留塩素標準比色液を求め,
これに相当する残留塩素の濃度c2(Cl mg/L)を記録する。
11) 次の式によって残留塩素,遊離残留塩素及び結合残留塩素の濃度を算出する。
(
)
V
c
a
A
100
2×
−
=
(
)
V
c
b
B
100
1×
−
=
B
A
C
−
=
102
K 0102:2016
ここに,
A: 残留塩素の濃度(Cl mg/L)
a: 3)で求めた残留塩素の濃度(Cl mg/L)
c2: 10)で求めた残留塩素の濃度(Cl mg/L)
V: 試料量(mL)
B: 遊離残留塩素の濃度(Cl mg/L)
b: 6)で求めた残留塩素の濃度(Cl mg/L)
c1: 9)で求めた残留塩素の濃度(Cl mg/L)
C: 結合残留塩素の濃度(Cl mg/L)
注(2) 試料がアルカリ性の場合には,pH計を用い,塩酸(1+5)を加えてpHを約7にする。また,
発色時のpHは常に1.3以下とする。
(3) 残留塩素のうち結合残留塩素は,最高発色に達するのに0 ℃で6分間,20 ℃で3分間,25 ℃
で2分30秒間が必要である。
備考 1. 空試験を行わない場合には,鉄0.3 mg/L以上,マンガン10 μg/L以上又は亜硝酸イオン0.3
mg/L以上が含まれていると妨害する。鉄及びマンガンの妨害を防ぐには,試料100 mLにつ
き1,2-シクロヘキサンジアミン四酢酸溶液(10 g/L)(trans-1,2-シクロヘキサンジアミン四酢
酸一水和物1.05 gを水に溶かして100 mLとする。)3 mLを添加する。
2. 市販の残留塩素測定器を用いる場合には,あらかじめ残留塩素標準比色液と比較して,誤り
のないことを確認しておく。
3. 試験に用いる水に残留塩素がないこと及び塩素を消費しないことを確認する方法は,次のい
ずれかによる。
1) 残留塩素が存在しないことの確認 水[a) 1)]約45 mLを比色管50 mLにとり,JIS K 8913
に規定するよう化カリウム約0.5 gを加えて振り混ぜ,約1分間後,りん酸塩緩衝液
(pH6.5)[33.2 a) 4)による。]2 mL,DPD希釈粉末[33.2 a) 3)による。]0.5 gを加えて振
り混ぜる。発色が認められないことを確認する。
又は33.1 c)の1)及び2)の操作を行って発色が認められないことを確認する。
2) 塩素を消費しないことの確認 水[a) 1)]約45 mLを比色管50 mLにとり,次亜塩素酸
ナトリウム溶液(有効塩素0.1 g/L)[42.2 a) 4)の次亜塩素酸ナトリウム溶液(有効塩素
10 g/L)を100倍に薄めて調製する。]1,2滴を加えて振り混ぜ,約2分間放置する。以
下,1)のりん酸塩緩衝液(pH6.5)以降の操作を行って発色が認められることを確認する。
33.2 ジエチル-p-フェニレンジアンモニウム(DPD)比色法 硫酸N,N-ジエチル-p-フェニレンジアンモニ
ウム(DPD)を比色管にとり,これに試料を加え,残留塩素との反応で生じる桃色から桃紅色を,残留塩
素標準比色液と比較して定量する。
定量範囲:Cl 0.05〜2 mg/L,繰返し精度:5〜10 %
a) 試薬 試薬は,次による。
1) 水 33.1 a) 1)による。
2) よう化カリウム JIS K 8913に規定するもの。
3) DPD希釈粉末 硫酸N,N-ジエチル-p-フェニレンジアンモニウム(N,N-ジエチル-p-フェニレンジア
ミン硫酸塩)1.0 gをめのう乳鉢中で粉砕する。これにJIS K 8987に規定する硫酸ナトリウム24 g
を加えてよく混合し,着色ガラス瓶に入れ,湿気を避けて,0〜10 ℃の暗所に保存する。着色した
ものは使用しない。
4) りん酸塩緩衝液(pH6.5) りん酸二水素カリウム溶液(0.2 mol/L)(JIS K 9007に規定するりん酸
103
K 0102:2016
二水素カリウム27.2 gを水に溶かして1 Lとする。)100 mLをとり,水酸化ナトリウム溶液(0.2
mol/L)(JIS K 8576に規定する水酸化ナトリウム8 gを水に溶かして1 Lとする。)をpH計を用い
てpH6.5になるまで加え,これにtrans-1,2-シクロヘキサンジアミン四酢酸一水和物0.13 gを加えて
溶かす。
5) C. I. Acid Red 265溶液 C. I. Acid Red 265[1-(4-メチルベンゼンスルホンアミド)-7-(2-メチルフェニ
ルアゾ)-8-ヒドロキシ-3,6-ナフタレンジスルホン酸二ナトリウム]を105〜110 ℃で3〜4時間加熱
し,デシケーター中で放冷する。その0.329 gを1 mgの桁まではかりとり,少量の水に溶かして全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。この溶液50 mLを全量フラスコ500 mLに
とり,水を標線まで加える。0〜10 ℃の暗所に保存し,6か月間以上経過したものは使用しない。
6) DPD残留塩素標準比色液 C. I. Acid Red 265溶液を表33.2によって全量フラスコ50 mLにとり,水
を標線まで加える。これを比色管50 mLにそれぞれ移す。密栓して0〜10 ℃の暗所に保存する。6
か月間以上経過したものは使用しない。
表33.2 DPD残留塩素標準比色液(50 mL中)
残留塩素
Cl mg/L
C. I. Acid Red 265 溶液
mL
0.05
0.5
0.1
1.0
0.2
2.0
0.3
3.0
0.4
4.0
0.5
5.0
0.6
6.0
0.7
7.0
0.8
8.0
0.9
9.0
1.0
10.0
1.2
12.0
1.4
14.0
1.6
16.0
1.8
18.0
2.0
20.0
b) 器具 器具は,次による。
1) 比色管 50 mL 底部から150±1 mmの高さに50 mLの標線を付けた平底のもの。
2) 比色管立 50 mL用 底部及び側面に乳白板を付けたもの。
c) 操作 操作は,次による。
1) りん酸塩緩衝液(pH6.5)2.5 mLを比色管50 mLにとり,これにDPD希釈粉末0.5 gを加える。次
に,試料(4)の適量(残留塩素0.1 mg以下を含む。)を加え,更に水を標線まで加える。
2) 栓をしてよく振り混ぜ,1分間(5)以内にその発色を側面から透視して,DPD残留塩素標準比色液と
比較する。該当するDPD残留塩素標準比色液から,これに相当する残留塩素の濃度(Cl mg/L)を
求め,これを遊離残留塩素として,試料中の遊離残留塩素の濃度(Cl mg/L)を算出する。
3) 2)の操作が終了したら,よう化カリウム約0.5 gを加え,栓をして振り混ぜて溶かし,約2分間放置
後,その発色を2)と同様に残留塩素標準比色液と比較する。該当する残留塩素標準比色液からこれ
104
K 0102:2016
に相当する残留塩素の濃度(Cl mg/L)を求め,試料中の残留塩素の濃度を算出する。
4) 結合残留塩素の濃度(Cl mg/L)は,次の式によって算出する。
結合残留塩素の濃度(Cl mg/L)=残留塩素(Cl mg/L)−遊離残留塩素(Cl mg/L)
注(4) 試料の酸性又はアルカリ性が強い場合には,炭酸ナトリウム溶液(50 g/L)(JIS K 8625に規定
する炭酸ナトリウムを用いて調製する。)又は塩酸(1+11)[21. a) 2)による。]を用いてpHを
約6.5に調節する。
(5) 振り混ぜ時間を含める。DPD希釈粉末中の硫酸ナトリウムは完全には溶けなくてもよい。
備考 4. 市販の残留塩素測定器又はガラス色標準スケールを用いる場合には,あらかじめDPD残留塩
素標準比色液と比較して,誤りのないことを確認しておく。
5. 試料中のマンガン酸化物による妨害の補正方法は,次による。
1) 試料100 mLをとり,メタ亜ひ酸ナトリウム溶液(2 g/L)(メタ亜ひ酸ナトリウムを用い
て調製する。)又はチオアセトアミド(エタンチオアミド)溶液(2.5 g/L)1 mLを加え,
振り混ぜる。
2) この溶液を用いて,c) 1)及び2)の操作を行い,マンガン酸化物による発色を残留塩素の
濃度として求める。
3) 得られた値を用いて,遊離残留塩素の濃度及び残留塩素の濃度を補正する。
6. 二酸化塩素(IV)は,残留塩素及び遊離残留塩素の値に含まれる。
7. DPDの酸化は,塩素化合物によるものだけではない。反応は,他の酸化剤によっても生じる。
これには臭素,よう素,ブロモアミン類,ヨードアミン類,オゾン,過酸化水素,クロム酸
塩,マンガン酸化物,亜硝酸塩,鉄(III)イオン及び銅イオンが挙げられる。ただし,この
方法では,銅イオン2 mg/L,鉄(II)イオン3 mg/L,アルミニウム4 mg/L及び亜硝酸イオン
4 mg/Lまではそれぞれ妨害しない。
33.3 よう素滴定法 残留塩素とよう化カリウムとが反応して遊離するよう素をチオ硫酸ナトリウム溶液
で滴定し,残留塩素を定量する。よう素を遊離させる酸化性物質が共存すると,残留塩素として定量され
る。
定量範囲:Cl 0.1 mg以上
a) 試薬 試薬は,次による。
1) 水 33.1 a) 1)による。
2) よう化カリウム 33.2 a) 2)による。
3) 酢酸(1+1) JIS K 8355に規定する酢酸を用いて調製する。
4) でんぷん溶液(10 g/L) 19. a) 5)による。
5) 10 mmol/Lチオ硫酸ナトリウム溶液 19. a) 9)による。
b) 操作 操作は,次による。
1) 試料(2)の適量(Clとして0.1〜7 mgを含む。)を共栓三角フラスコ500 mLにとり,水を加えて約300
mLとし,よう化カリウム1 g及び酢酸(1+1)5 mLを加える。
2) 栓をして振り混ぜ,暗所に約5分間放置する。
3) 遊離したよう素を,10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,
指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴
定する。
4) 空試験として水100 mLをとり,1)〜3)の操作を行う。
105
K 0102:2016
5) 次の式によって試料中の残留塩素の濃度(Cl mg/L)を算出する。
(
)
5
354
.0
000
1
×
×
×
−
=
V
f
b
a
A
ここに,
A: 残留塩素の濃度(Cl mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
b: 空試験に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター
V: 試料量(mL)
0.354 5: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する残留塩
素の質量(mg)
備考 8. 試料の着色又は濁りが著しく試験が困難な場合には,次の方法で残留塩素を分離し測定して
もよい。
1) 試料の適量(Clとして2 mg以上を含む。)を図33.1の蒸留フラスコ200 mLにとり,硫
酸(1+15)を加えてpHを0.9〜1.0に調節し,水で約80 mLとし,蒸留装置に接続する。
2) 10 mmol/Lチオ硫酸ナトリウム溶液を,ガス洗浄瓶(H)には20 mL,ガス洗浄瓶(I)
には5 mLを加え,それぞれ水を加えて50 mLとし,更に酢酸緩衝液(pH3.5)[JIS K 8371
に規定する酢酸ナトリウム三水和物240 gを水約300 mLに溶かし,酢酸460 mLを加え
た後,水で1 Lとする。]4 mLとよう化カリウム0.1 gとを加えて振り混ぜる。
3) 蒸留フラスコを40 ℃の恒温槽中(J)に入れ,緩やかに約40分間通気する。ガス洗浄
瓶中の溶液を三角フラスコ300 mLに移し,水でガス洗浄瓶の内部を洗い洗液を前の溶
液に合わせる。これに,よう素溶液(JIS K 8913に規定するよう化カリウム12 gを少量
の水に溶かし,JIS K 8920に規定するよう素4 gを加えて溶かし,水を加えて1 Lとす
る。)10 mLを加える。
4) 過剰のよう素を10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなった
ら指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が
消えるまで滴定する。
5) 空試験として10 mmol/Lチオ硫酸ナトリウム溶液25 mLを三角フラスコ300 mLにとり,
酢酸塩緩衝液(pH3.5)8 mL,よう化カリウム0.2 g及び水約120 mLを加えて振り混ぜ,
これによう素溶液10 mLを加え,試料と同様に10 mmol/Lチオ硫酸ナトリウム溶液で滴
定する。試料中の残留塩素の濃度(Cl mg/L)は,b) 5)の式によって算出する。
単位 mm
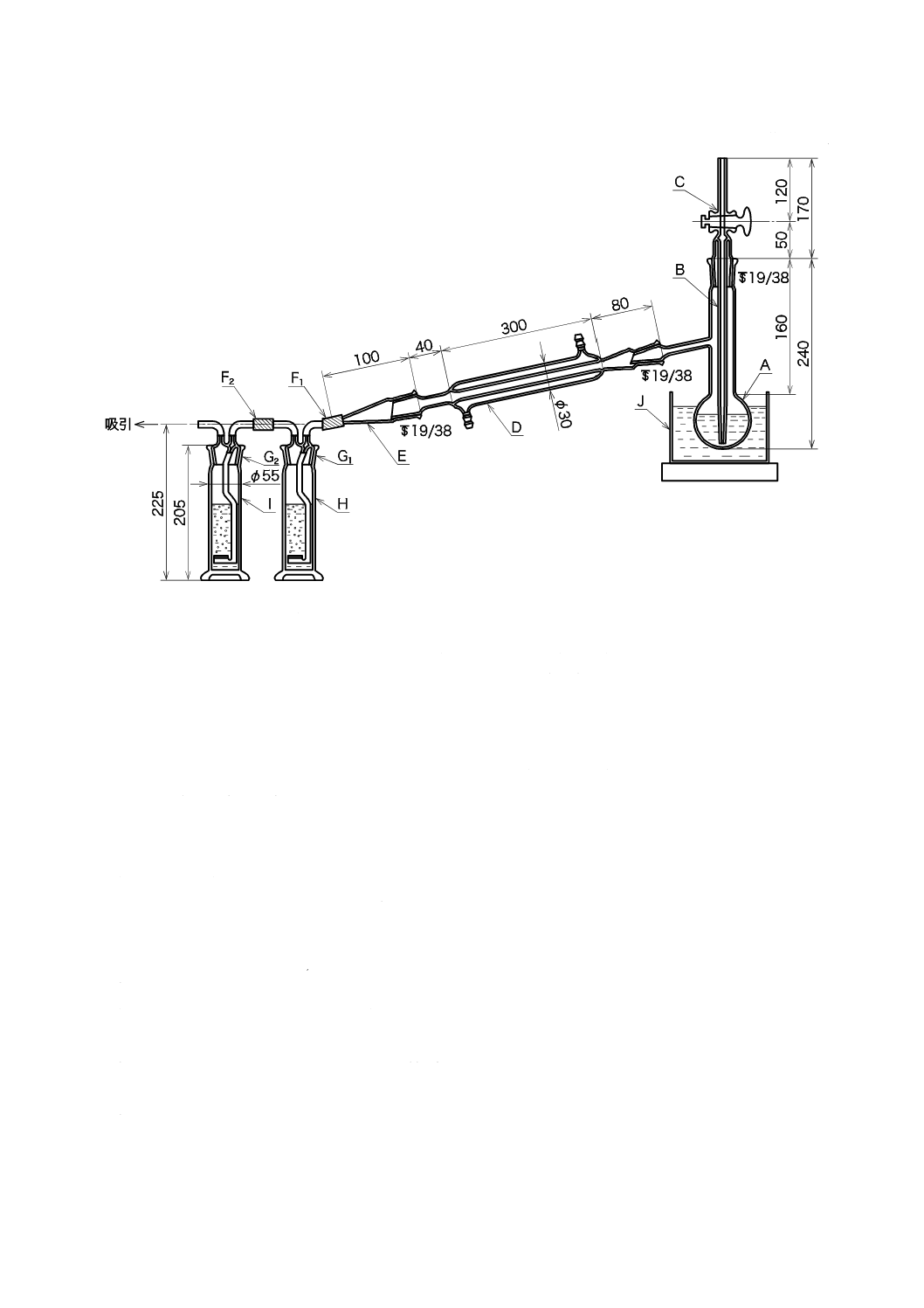
106
K 0102:2016
単位 mm
A:
B:
C:
D:
E:
蒸留フラスコ(共通すり合わせ枝付き)200 mL
中管(先端の内径約1 mm)
一方コック
共通すり合わせリービッヒ冷却器(200〜300 mm)
共通すり合わせアダプター
F1,F2:
G1,G2:
H,I:
J:
ゴム管
共通すり合わせ
共通すり合わせろ過板付きガス洗浄瓶
(250 mL)
恒温槽
図33.1 蒸留装置(共通すり合わせ)の例
33.4 ジエチル-p-フェニレンジアンモニウム(DPD)吸光光度法 試料に硫酸N,N-ジエチル-p-フェニレン
ジアンモニウム(DPD)を加え,残留塩素との反応で生じる桃色から桃紅色を,波長510 nm(又は555 nm)
付近の吸光度を測定して定量する。
定量範囲:Cl 2.5〜150 μg,繰返し分析精度:5〜10 %
a) 試薬 試薬は,次による。
1) 水 33.1 a) 1)による。
2) 希釈水 水1 Lに対し塩素水(濃度約50 mg/L)約3 mLを加え1時間放置した後,煮沸するか又は
紫外線を照射して残留塩素を除く。
3) よう化カリウム 33.2 a) 2)による。
4) DPD希釈粉末 33.2 a) 3)による。
5) りん酸塩緩衝液(pH6.5) 33.2 a) 4)による。
6) 0.1 mol/Lチオ硫酸ナトリウム溶液 19. a) 8)による。
7) 10 mmol/Lチオ硫酸ナトリウム溶液 19. a) 9)による。ファクターは,19. a) 8)の0.1 mol/Lチオ硫酸
ナトリウム溶液のものを用いる。12時間以上経過したものは使用しない。
8) でんぷん溶液(10 g/L) 19. a) 5)による。
9) 塩素標準液 塩素水の調製,有効塩素濃度の測定及び塩素標準液の調製は,次による。
塩素標準液は,使用の都度その有効塩素濃度を測定する。
107
K 0102:2016
9.1) 塩素水の調製 次亜塩素酸ナトリウム溶液[有効塩素7〜12 %(質量百分率)]を水に溶かす。又
は,他の方法によって塩素水を調製してもよい。
9.2) 有効塩素濃度の測定
9.2.1) 9.1)で調製した塩素水100 mLを共栓三角フラスコ200 mLにとり,よう化カリウム1 g及び酢酸
(1+1)(JIS K 8355に規定する酢酸を用いて調製する。)5 mLを加える。
9.2.2) 栓をして振り混ぜ,暗所に5分間放置する。
9.2.3) 遊離したよう素を,0.1 mol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,
指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じた青い色が消えるまで滴定する。
9.2.4) 次の式によって,塩素水に含まれる有効塩素の濃度(Cl mg/L)を算出する。
545
.3
000
1
1
×
×
×
=
V
f
a
C
ここに,
C: 有効塩素の濃度(Cl mg/L)
a: 滴定に要した0.1 mol/Lチオ硫酸ナトリウム溶液量(mL)
f1: 0.1 mol/Lチオ硫酸ナトリウム溶液のファクター
V: 9.2.1)で用いた塩素水(mL)
3.545: 0.1 mol/Lチオ硫酸ナトリウム溶液1 mLに相当する塩素の質
量(mg)
9.3) 塩素標準液(Cl 50 μg/mL) 塩素25 mgに相当するように9.2)で有効塩素の濃度を定量した塩素水
を全量フラスコ500 mLにとり,希釈水を標線まで加える。正確な濃度の標定は,次による。
この溶液100 mLをとり,9.2.1)〜9.2.3)の操作を行う。ただし,滴定には10 mmol/Lチオ硫酸ナ
トリウム溶液を用いる。次の式によって,塩素標準液(Cl 50 μg/mL)の正確な濃度を算出する。
5
354
.0
000
1
1
×
×
×
=
V
f
a
C
ここに,
C: 有効塩素の濃度(Cl mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
f1: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター
V: 滴定に用いた塩素標準液(Cl 0.05 mg/mL)量(mL)
0.354 5: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する塩素の
質量(mg)
9.4) 塩素標準液(Cl 5 μg/mL) 塩素標準液(Cl 50 μg/mL)20 mLを全量フラスコ200 mLにとり,希
釈水を標線まで加える。この溶液は,使用時に調製する。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料(4)の適量について,33.2のc) 1)の操作を行う。
2) 栓をしてよく振り混ぜ,1分間(5)以内に,この溶液の一部を吸収セルにとり,水を対照液として波
長510 nm(又は555 nm)付近の吸光度を測定する。
3) 検量線から塩素の量(Cl μg)を求め,これを遊離残留塩素として試料中の遊離残留塩素の濃度(Cl
mg/L)を算出する。
4) 引き続き2)の残りの溶液によう化カリウム約0.5 gを加え,栓をして振り混ぜて溶かす。約2分間
放置した後,この溶液の一部を吸収セルにとり,水を対照液として波長510 nm(又は555 nm)付
近の吸光度を測定する。
108
K 0102:2016
5) 検量線から塩素の量(Cl μg)を求め,これを残留塩素として試料中の残留塩素の濃度(Cl mg/L)
を算出する。
6) 33.2 c) 4)によって結合残留塩素の濃度(Cl mg/L)を算出する。
d) 検量線 検量線の作成は,次による。
1) 塩素標準液(Cl 5 μg/mL)0.5〜30 mLについて,c) 1)及び2)の操作を行い,吸光度を測定し,塩素
(Cl)の量と吸光度との関係線を作成する。
34. ふっ素化合物 ふっ素化合物は,ふっ化物イオン,金属ふっ化物などの総称であり,ふっ化物イオン
として表す。ふっ化物イオンの定量には,ランタン-アリザリンコンプレキソン吸光光度法,イオン電極法,
イオンクロマトグラフ法又はランタン-アリザリンコンプレキソン発色による流れ分析法を適用する。
なお,イオン電極法は,1992年に第1版として発行されたISO 10359-1,蒸留操作は,1994年に第1版
として発行されたISO 10359-2,イオンクロマトグラフ法は,1992年に第1版として発行されたISO 10304-1
との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 10359-1:1992,Water quality−Determination of fluoride−Part 1: Electrochemical probe method
for potable and lightly polluted water(MOD)
ISO 10359-2:1994,Water quality−Determination of fluoride−Part 2: Determination of inorganically
bound total fluoride after digestion and distillation(MOD)
ISO 10304-1:1992,Water quality−Determination of dissolved fluoride, chloride, nitrite,
orthophosphate, bromide, nitrate and sulfate ions, using liquid chromatography of ions−Part 1:
Method for water with low contamination(MOD)
34.1 ランタン-アリザリンコンプレキソン吸光光度法 ふっ素化合物を蒸留分離し,ランタン(III)とア
リザリンコンプレキソンとの錯体を加え,これがふっ化物イオンと反応して生じる青い色の複合錯体の吸
光度を測定して,ふっ化物イオンを定量し,ふっ素化合物とする。
定量範囲:F− 4〜50 μg,繰返し精度:3〜10 %
備考 1. この方法は,陰イオンの影響は少ないが,陽イオンの影響が多い。特に,アルミニウム,カ
ドミウム,コバルト,鉄,ニッケル,ベリリウム,鉛などが妨害するので,あらかじめ蒸留
してふっ化物イオンを分離する。
a) 試薬 試薬は,次による。
1) 過塩素酸 JIS K 8223に規定するものを,加熱して白煙を発生させた後,放冷したもの。
2) りん酸 JIS K 9005に規定するもの。
3) 水酸化ナトリウム溶液(100 g/L) 28.2.1 a) 3)による。
4) 二酸化けい素 JIS K 8885に規定する二酸化けい素(1)
5) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
6) ランタン-アリザリンコンプレキソン溶液(2) 調製は,次による。
− アリザリンコンプレキソン(1,2-ジヒドロキシアントラキノン-3-イルメチルアミン-N,N-二酢酸
二水和物)0.192 gを,アンモニア水(1+10)(JIS K 8085に規定するアンモニア水を用いて調
製する。)4 mL及び酢酸アンモニウム溶液(200 g/L)(JIS K 8359に規定する酢酸アンモニウム
109
K 0102:2016
を用いて調製する。)4 mLに溶かす。
− これを酢酸ナトリウム溶液(JIS K 8371に規定する酢酸ナトリウム三水和物41 gを水400 mL
に溶かし,JIS K 8355に規定する酢酸24 mLを加えたもの。)中にかき混ぜながら加える。
− この溶液をかき混ぜながらJIS K 8034に規定するアセトン400 mLを徐々に加え,更にランタ
ン溶液[酸化ランタン(III)0.163 gを塩酸(1+5)(JIS K 8180に規定する塩酸を用いて調製す
る。)10 mLに加え,加熱して溶かしたもの。]を加えてかき混ぜる。放冷後,酢酸又はJIS K 8085
に規定するアンモニア水でpH計を用いてpHを約4.7に調節した後,水を加えて1 Lとする。
7) ふっ化物イオン標準液(F− 0.1 mg/mL) JIS K 8005に規定する容量分析用標準物質のふっ化ナト
リウムを白金皿にとり,500 ℃で約1時間加熱し,デシケーター中で放冷する。NaF 100 %に対し
てその0.221 gをとり,少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加え
る。ポリエチレン瓶に入れて保存する。
8) ふっ化物イオン標準液(F− 2 μg/mL) ふっ化物イオン標準液(F− 0.1 mg/mL)10 mLを全量フラ
スコ500 mLにとり,水を標線まで加える。標準液は,ポリエチレン瓶に貯蔵し,1か月間は使用で
きる。
注(1) 結晶質のもので粒径100〜150 μm程度のものを用いる。品質が分からない場合には,白金るつ
ぼ中で1 150 ℃以上で約1時間加熱し,デシケーター中で放冷したものを用いる。この場合,
ふっ化物イオン標準液(F− 2 μg/mL)50 mLをとり,c) 2)〜5)及びd) 1)〜5)を行って回収率を
確認する。
(2) 市販品を用いてもよい。
参考 市販のアルフッソン(商品名)を用いる場合は,その2.5 gを水に溶かして50 mLとする。使
用時に調製する。この情報は,この規格の利用者の便宜を図って記載するもので,この製品を
推奨するものではない。
b) 器具及び装置 器具及び装置は,次による。
1) 蒸留装置 図34.1に例を示す。
2) 光度計 分光光度計又は光電光度計
c) 蒸留操作 蒸留操作は,次による。
1) 試料の適量(3)(F−として30 μg以上を含む。)を磁器蒸発皿又はビーカーにとり,フェノールフタレ
イン溶液(5 g/L)2,3滴を加え,水酸化ナトリウム溶液(100 g/L)を滴加して微アルカリ性とし
た後,加熱して約30 mLに濃縮する。
2) 図34.1の蒸留フラスコ中に水約10 mLで洗い移す。次に,二酸化けい素約1 g,りん酸1 mL及び過
塩素酸40 mL[又はJIS K 8951に規定する硫酸(4) 30 mL]を加える(5)。受器の全量フラスコ250 mL
には水20 mL(6)を加え,逆流止めの先端は水面下に保つ。
3) 蒸留フラスコを直接加熱(7)し,蒸留フラスコ内の液温が約140 ℃に達してから,水蒸気を通す。
4) 蒸留温度を145±5 ℃,留出速度を3〜5 mL/minに調節し,受器の液量が約220 mLになるまで蒸留
を続ける。
5) 冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器
に加え,更に水を標線まで加える。
注(3) 溶存のふっ化物イオンを試験するときは,3.2でろ過した試料を用いる。
(4) JIS K 8951に規定する硫酸をビーカーに入れ,加熱して盛んに白煙を発生させた後,放冷した
もの。
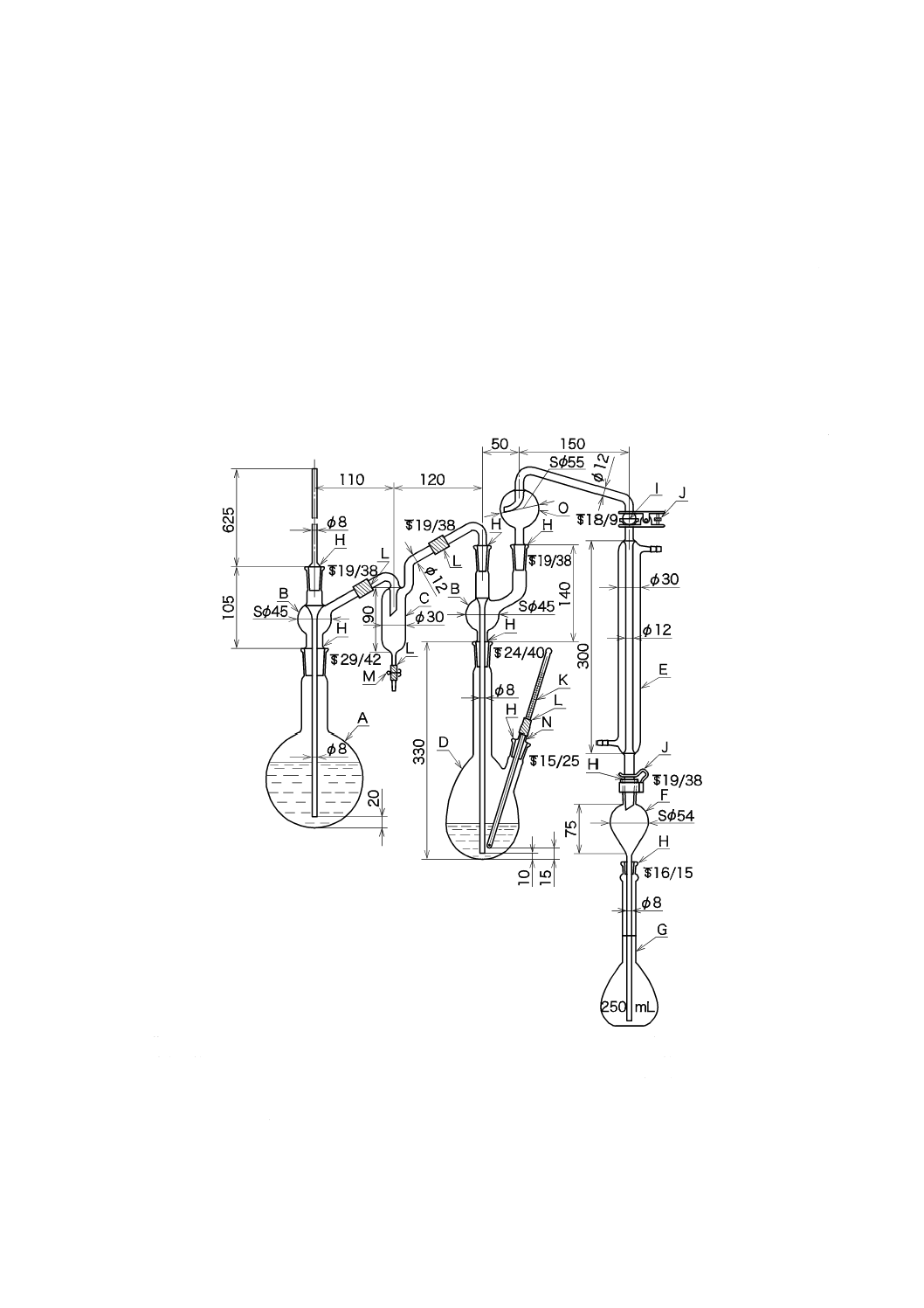
110
K 0102:2016
注(5) 蒸留フラスコには,沸騰石(粒径2〜3 mm)約10個を入れる。
(6) 試料中にふっ化物イオン以外のハロゲン化物が多量に含まれる場合には,水酸化ナトリウム溶
液(40 g/L)[21. a) 3)による。]4〜5滴とフェノールフタレイン溶液(5 g/L)2〜3滴とを加え
ておく。受器中の溶液は蒸留が終わるまで微紅色を保つように,必要に応じて水酸化ナトリウ
ム溶液(40 g/L)を滴加する。
なお,この場合は蒸留が終わった後,留出液に硫酸(1+35)[30.1.1 a) 2)による。]を微紅色
が消えるまで滴加し,以下,5)の操作を行う。蒸留後の定量に,34.3のイオンクロマトグラフ
法を適用する場合は,フェノールフタレイン溶液(5 g/L)を加えず,pH試験紙によって液性を
判別する。
(7) 蒸留フラスコ中の液面まで加熱できるようにフレームを調節する。油浴,グリセリン浴などを
用いてもよい。
単位 mm
A:
B:
C:
D:
E:
水蒸気発生フラスコ1 000 mL
連結導入管
トラップ
蒸留フラスコ500 mL
リービッヒ冷却器300 mm
F:
G:
H:
I:
J:
逆流止め(約50 mL)
受器(全量フラスコ250 mL)
共通すり合わせ
共通球面すり合わせ
押さえばね
K:
L:
M:
N:
O:
温度計200 ℃
ゴム管
ピンチコック
温度計差し込み栓
トラップ球(ケルダール球)
図34.1 蒸留装置の例
111
K 0102:2016
d) 操作 操作は,次による。
1) c)の蒸留操作で得た留出液から30 mL以下の適量(F−として4〜50 μgを含む。)を全量フラスコ50
mLにとる。
2) ランタン-アリザリンコンプレキソン溶液(8) 20 mLを加え,更に水を標線まで加えて振り混ぜ,約1
時間放置する。
3) 別に,水30 mLを全量フラスコ50 mLにとり,2)の操作を行う。
4) 試料について2)で得た溶液の一部を吸収セルに移し,3)の溶液を対照液として波長620 nm付近の吸
光度を測定する。
5) 検量線からふっ化物イオンの量を求め,試料中のふっ化物イオンの濃度(F− mg/L)を算出する。
注(8) 参考で調製したアルフッソン溶液を用いる場合には,その5 mLとJIS K 8034に規定するアセ
トン10 mLとを試料溶液に加えた後,水を標線まで加える。
e) 検量線 検量線の作成は,次による。
1) ふっ化物イオン標準液(F− 2 μg/mL)2〜25 mLを全量フラスコ50 mLに段階的にとる。
2) d) 2)〜4)の操作を行って吸光度を測定し,ふっ化物イオン(F−)の量と吸光度との関係線を作成す
る。
34.2 イオン電極法 ふっ素化合物を前処理して蒸留分離し,緩衝液(イオン強度調節液)を加えてpHを
5.2±0.2に調節し,ふっ化物イオン電極を指示電極として電位を測定し,ふっ化物イオンを定量する。
定量範囲:F− 0.1〜100 mg/L,繰返し精度:5〜20 %
備考 2. 妨害物質を含まない清浄な試料の溶存ふっ化物イオンを定量する場合は,蒸留に代え,試料
をろ過し,備考4.の操作で定量することができる。ただし,この方法では,溶存のふっ化物
イオン及び容易にふっ化物イオンとなる錯イオンが定量される。また,この前処理方法は,
汚濁のある排水には適用できない。
a) 試薬 試薬は,次による。
1) 緩衝液(pH5.2) JIS K 8150に規定する塩化ナトリウム58 gとJIS K 8284に規定するくえん酸水
素二アンモニウム1 gとを水500 mLに加えて溶かし,JIS K 8355に規定する酢酸50 mLを加え,水
酸化ナトリウム溶液(200 g/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調製する。)を滴
加して,pH計を用いてpHを5.2に調節した後,水を加えて1 Lとする。
2) ふっ化物イオン標準液(F− 100 mg/L) 34.1 a) 7)による。標準液は,プラスチック製容器で貯蔵し,
1か月間は使用できる。
3) ふっ化物イオン標準液(F− 10 mg/L) ふっ化物イオン標準液(F− 100 mg/L)20 mLを全量フラス
コ200 mLにとり,水を標線まで加える。使用時に調製する。
4) ふっ化物イオン標準液(F− 1 mg/L) ふっ化物イオン標準液(F−10 mg/L)20 mLを全量フラスコ 200
mLにとり,水を標線まで加える。使用時に調製する。
5) ふっ化物イオン標準液(F− 0.1 mg/L) ふっ化物イオン標準液(F− 1 mg/L)20 mLを全量フラスコ
200 mLにとり,水を標線まで加える。使用時に調製する。
備考 3. 標準液は,測定する濃度によって,濃度範囲を狭めるなど,適切な濃度のものを調製し,検
量線を作成する。
b) 器具及び装置 器具及び装置は,次による。
1) 電位差計 0.1 mV又はそれ以下の電位差を読み取れるもの。高入力抵抗電位差計(例えば,デジタ
ル式pH-mV計,拡大スパン付きpH-mV計,イオン電極用電位差計など)
112
K 0102:2016
2) 指示電極 ふっ化物イオン電極,標準液を用いた起電力の応答は,25 ℃におけるふっ化物イオン濃
度の10倍濃度変化当たり55 mV以上のもの。
3) 参照電極 銀-塩化銀電極を用いる(9) (10)。
4) 測定容器 試料100 mLで扱えるもの。ポリプロピレン製で,恒温ジャケットが取り付けられてい
るもの。
5) 恒温槽 測定容器のジャケットに水温25±0.2 ℃の水を供給できるもの。
6) マグネチックスターラー 四ふっ化エチレン樹脂(PTFE)で被覆した回転子付きのものを用いる。
注(9) 液間電位差の小さい単一液絡形のスリーブ形の電極が望ましい。
(10) 参照電極の内部液に塩化カリウム溶液(飽和)を使用する場合には,液温が低下すると塩化カ
リウムの結晶が析出し,固着して抵抗が大きくなることがあるので注意する。
c) 検量線 検量線の作成は,次による。
1) ふっ化物イオン標準液(F− 0.1 mg/L)100 mLを測定容器にとり,緩衝液(pH5.2)10 mLを加える
(11)。
2) 恒温槽から水を送り,この測定容器の溶液を25±0.5 ℃に保つ。
3) 指示電極(12) (13)と参照電極(10) (14)とを浸し固定した後,回転子を入れ,マグネチックスターラー(15)
を用いて,泡が電極に触れない程度に強くかき混ぜる(16)。
4) 液温を確認し,電位差計で電位を測定する(17)。
5) ふっ化物イオン標準液(F− 1 mg/L),ふっ化物イオン標準液(F− 10 mg/L)及びふっ化物イオン標
準液(F− 100 mg/L)のそれぞれ100 mLを測定容器にとり,それぞれに緩衝液(pH5.2)10 mLを
加える(11)。
6) 2)〜4)の操作を行って,それぞれのふっ化物イオン標準液の電位を測定する(18) (19)。
7) 横軸にふっ化物イオンの濃度の対数を,縦軸に電位をとり,ふっ化物イオンの濃度(mg/L)と電位
との関係線を作成する(18)。
注(11) 緩衝液(pH5.2)の添加によってpH5.2±0.2に調節し,イオン強度を一定にする。
(12) 指示電極(ふっ化物イオン電極)は,使用時にふっ化物イオン標準液(F− 0.1 mg/L)に浸し,
指示値が安定してから使用する。
(13) 指示電極の感応膜にきずがつくと,検量線の勾配(電位勾配)が小さくなり,応答速度も遅く
なるので注意する。また,指示電極の感応膜が汚れると,応答速度が遅くなるので,エタノー
ル(95)を含ませた脱脂綿又は柔らかい紙で汚れを拭き取り,水で洗浄する。
(14) 参照電極は,抵抗の小さいものを選ぶ。一般にスリーブ形又はセラミックス形を用いる。
スリーブ形は,抵抗も小さく最適であるが,スリーブを締め過ぎると抵抗が大きくなり,緩
すぎると液の流出が多くなるので,適度の締付けが必要である。
セラミックス形は抵抗の大きい製品もあるので,イオン電極用を用いる。セラミックス形は
乾燥したり,汚れると抵抗が大きくなるので注意する。
参照電極は,内部液と同じ溶液中に浸しておく。スリーブ形は,使用時にスリーブの締付け
を調節する。
(15) マグネチックスターラーを長時間使用すると,発熱して液温に変化を与えることがあるので,
液温の変化に注意する。
(16) かき混ぜ速度で電位差計の指示が不安定になる場合には,参照電極の抵抗が大きくなっている
ことが多い。
113
K 0102:2016
かき混ぜ速度は,約180〜200 min−1に調節するとよい。
注(17) ふっ化物イオン電極の応答時間は,液温10〜30 ℃の場合には,ふっ化物イオンの濃度が0.1
mg/Lで約1分間,1 mg/L以上では約30秒間である。
セルの電位が,5分間で0.5 mV以上変わらなくなったら,マグネチックスターラーのスイッ
チを切る。少なくとも15秒間後に得られた値を記録する。
(18) ふっ化物イオン標準液(F− 1 mg/L)とふっ化物イオン標準液(F− 100 mg/L)との電位の差は,
110〜120 mV(25 ℃)の範囲に入り,ふっ化物イオンの濃度F−0.1〜100 mg/Lの間の検量線は
直線になる。
(19) 次の測定を開始する前に,回転子,電極などを,次に測定する溶液ですすぐ。
測定は,濃度のうすいものから順に行う。高濃度の試料を測定した場合は,注(12),(13)の操作
を行った後,測定を続ける。
d) 操作 操作は,次による。
1) 34.1 c)の蒸留操作で得た留出液から100 mLを測定容器にとり,緩衝液(pH5.2)10 mLを加える(11)。
2) c) 2)〜4)の操作を行って(19),検量線からふっ化物イオンの濃度を求め,試料中のふっ化物イオンの
濃度(F− mg/L)を算出する。
備考 4. 蒸留操作を行わず,ろ過による処理で測定する場合は,3.2に従って試料をろ過する。緩衝液
(pH5.2)(TISAB) 25 mLを測定容器にとり,ろ過した試料25 mLを加える。次に,d) 2)
の操作を行う。検量線は,同じ操作で作成する。
緩衝液(pH5.2)(TISAB) JIS K 8150に規定する塩化ナトリウム58 g及びJIS K 8355に
規定する酢酸57 mLを,水500 mLを入れたビーカー1 000 mLに加える。溶けるまでかき
混ぜる。水酸化ナトリウム溶液(5 mol/L)(JIS K 8576に規定する水酸化ナトリウムを用
いて調製する。)150 mLとtrans-1,2-シクロヘキサンジアミン四酢酸一水和物4 gとを加え
る。固形物が全て溶けるまでかき混ぜ,pH計を用い,溶液を水酸化ナトリウム溶液(5 mol/L)
でpH5.2に調節する。
5. イオン濃度計の場合には,ふっ化物イオン標準液(F− 1 mg/L)と,ふっ化物イオン標準液
(F− 100 mg/L)とを用い,c) 2)及び3)の操作を行って,イオン濃度計の指示値を1 mg/L及
び100 mg/Lになるように調節する。さらに,ふっ化物イオン標準液(F− 0.1 mg/L)とふっ
化物イオン標準液(F− 10 mg/L)とを用いてイオン濃度計の指示値を確認する。
6. イオン電極法では,ふっ化物イオンだけが測定できるので,あらかじめふっ素化合物を蒸留
操作で全てふっ化物イオンにしてから測定する。
主な共存物質の許容限度を濃度の最大比率で次に示す。
HCO3−,Cl−,NO3−,I−,Br−,HPO42−: 103
SO42−: 104
水酸化物イオン,アルミニウムイオン及び鉄(III)イオンは,いずれも測定を妨害するが,
蒸留分離によって除去されるため影響はない。
7. ふっ化物イオン電極による電位差滴定法 34.1 c)の蒸留操作で得た留出液から100 mLをビ
ーカーにとり,c) 2)〜4)の操作に準じて電位を測定しながら 〜 mol/Lの硝酸ランタン
(III)溶液で滴定して滴定曲線を作図し,滴定終点を求め,ふっ化物イオンの量を算出する。
mol/L硝酸ランタン(III)溶液1 mLは,F− 1.899 mgに相当する。
34.3 イオンクロマトグラフ法 試料をろ過した後,試料中のふっ化物イオンをイオンクロマトグラフ法
300
1
30
1
30
1
114
K 0102:2016
によって定量する。この方法を用いる場合には,試料採取後直ちに試験する。直ちに行えない場合には,0
〜10 ℃の暗所に保存し,できるだけ早く試験する。この方法は,清浄な試料に適用する。溶存のふっ化物
イオン及び容易にふっ化物イオンとなる錯イオンが定量される。
備考 8. 試料に妨害物質が含まれる場合は,34.1 c)の蒸留操作を行った後に適用する。また,ハロゲ
ン化物が多量に含まれる場合は,注(6)第3文(なお書きの部分)を除いた34.1 c)の蒸留操作
を行った後に適用する。
蒸留操作を行った場合は,試料のふっ素化合物が定量される。
a) 試験操作などは,35.3による。
34.4 流れ分析法 試料中のふっ素化合物を,34.1と同様な原理で発色させる流れ分析法によって定量す
る。
定量範囲:F− 0.08〜10 mg/L,繰返し精度:10 %以下
試験操作などは,JIS K 0170-6による。ただし,JIS K 0170-6の6.3.2(ランタン-アリザリンコンプレキ
ソン発色FIA法)による場合は,34.1 c)の蒸留操作を行った後に適用する。
備考 9. 妨害物質,ハロゲン化物又はハロゲン化水素などが多量に含まれる試料にJIS K 0170-6の
6.3.3(蒸留・ランタン-アリザリンコンプレキソン発色CFA法)を適用する場合は,試料に
一定量のふっ化物イオンを添加して試験操作を行ったときに得られる指示値の増加分と,同
量のふっ化物イオンを含む検量線用標準液について同様の操作を行ったときに得られる指示
値とを比較することによって添加回収率を求め,その値が80〜120 %の範囲にあることを確
認し,試料の分析値を添加回収率で補正する。添加回収率がこの範囲の外にある場合は適用
できない。
35. 塩化物イオン(Cl−) 塩化物イオンの定量には,硝酸銀滴定法,イオン電極法又はイオンクロマトグ
ラフ法を適用する。
なお,硝酸銀滴定法は,1989年に第1版として発行されたISO 9297,イオンクロマトグラフ法は,1992
に第1版として発行されたISO 10304-1及び1995年に第1版として発行されたISO 10304-2との整合を図
ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 9297:1989,Water quality−Determination of chloride−Silver nitrate titration with chromate
indicator(Mohr's method)(MOD)
ISO 10304-1:1992,Water quality−Determination of dissolved fluoride, chloride, nitrite,
orthophosphate, bromide, nitrate and sulfate ions, using liquid chromatography of ions−Part 1:
Method for water with low contamination(MOD)
ISO 10304-2:1995,Water quality−Determination of dissolved anions by liquid chromatography of ions
−Part 2: Determination of bromide, chloride, nitrate, nitrite, orthophosphate and sulfate in waste
water(MOD)
35.1 硝酸銀滴定法 試料のpHを約7に調節し,2',7'-ジクロロフルオレセイン二ナトリウム[9-(2-カル
ボキシフェニル)-2,7-ジクロロ-6-ヒドロキシ-3H-キサンテン-3-オン二ナトリウム塩]又はウラニン(フル
オレセインナトリウム)溶液を指示薬として,硝酸銀溶液で滴定して塩化物イオンを定量する。
115
K 0102:2016
定量範囲:Cl− 5 mg以上
備考 1. 臭化物イオン,よう化物イオン,シアン化物イオンなどが共存すると,塩化物イオンとして
定量される。亜硫酸イオン,チオ硫酸イオン及び硫化物イオンのいずれも妨害するが,あら
かじめ過酸化水素で酸化しておけば妨害しない。
a) 試薬 試薬は,次による。
1) 硝酸(1+65) JIS K 8541に規定する硝酸を用いて調製する。
2) 炭酸ナトリウム溶液(50 g/L) JIS K 8625に規定する炭酸ナトリウム5 gを水に溶かして100 mL
とする。
3) ジクロロフルオレセインナトリウム溶液(2 g/L) 2',7'-ジクロロフルオレセイン二ナトリウム0.2 g
を水に溶かして100 mLとする(1)。
4) デキストリン溶液 JIS K 8646に規定するデキストリン水和物2 gを水に溶かして100 mLとする。
使用時に調製する。
5) 40 mmol/L硝酸銀溶液 JIS K 8550に規定する硝酸銀6.8 gを水に溶かして1 Lとし,着色ガラス瓶
に保存する。
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質の塩化ナトリウムをあらかじめ600 ℃で約1時間加
熱し,デシケーター中で放冷する。NaCl 100 %に対してその0.47 gを1 mgの桁まではかりとり,
少量の水に溶かし,全量フラスコ200 mLに移し入れ,水を標線まで加える。
− この20 mLをビーカーにとり,水を加えて液量を約50 mLとし,これにデキストリン溶液5 mL
とジクロロフルオレセインナトリウム溶液(2 g/L)1,2滴とを加え,静かにかき混ぜながらこ
の硝酸銀溶液で滴定する。黄緑の蛍光が消失して僅かに赤くなったときを終点とする。次の式
によって40 mmol/L硝酸銀溶液のファクター(f)を算出する。
7
337
002
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 塩化ナトリウムの質量(g)
b: 塩化ナトリウムの純度(質量分率%)
x: 滴定に要した40 mmol/L硝酸銀溶液量(mL)
0.002 337 7: 40 mmol/L硝酸銀溶液1 mLに相当する塩化ナトリウム
の質量(g)
注(1) ジクロロフルオレセインナトリウム溶液(2 g/L)に代えて,JIS K 8830に規定するウラニン(フ
ルオレセインナトリウム)[9-(2-カルボキシフェニル)-6-ヒドロキシ-3H-キサンテン-3-オン二
ナトリウム]を用い,0.2 gを水100 mLに溶かしたものを用いてもよい。
b) 器具 器具は,次による。
1) ビュレット 10〜50 mLの適切な容量のものを用いる。
2) ビーカー又は磁器皿 50〜100 mLの適切な容量のものを用いる。磁器皿を用いる場合は,白い色の
ものを用いる。
c) 操作 操作は,次による。
1) 試料(2) 50 mL(Cl− 20 mg以上を含む場合には適量をとり,水を加えて50 mLとする。)をビーカー
又は磁器皿にとる。
2) 試料が酸性の場合には,炭酸ナトリウム溶液(50 g/L)で,また,アルカリ性の場合には,硝酸(1
+65)を用いてpHを約7に調節する。
116
K 0102:2016
3) デキストリン溶液5 mL及びジクロロフルオレセインナトリウム溶液(2 g/L)(1) 1,2滴を加えてか
き混ぜる。
4) 静かにかき混ぜながら40 mmol/L硝酸銀溶液で滴定する。黄緑の蛍光が消失して僅かに赤くなった
ときを終点とする。
5) 次の式によって試料中の塩化物イオンの濃度(Cl− mg/L)を算出する。
418
.1
000
1
×
×
×
=
V
f
a
C
ここに,
C: 塩化物イオンの濃度(Cl− mg/L)
a: 滴定に要した40 mmol/L硝酸銀溶液量(mL)
f: 40 mmol/L硝酸銀溶液のファクター
V: 試料量(mL)
1.418: 40 mmol/L硝酸銀溶液1 mLに相当する塩化物イオンの質量
(mg)
注(2) 試料に著しい濁りが認められる場合は,ろ紙5種C(又はろ紙6種)でろ過し,初めのろ液約
50 mLを捨てた後,その後のろ液50 mLをとる。
備考 2. 塩化物イオンの濃度が低い場合には,試料中の塩化物イオン濃度が5 mg(50 mL中)以上と
なるように35.2 a) 2)の塩化物イオン標準液(Cl− 1 000 mg/L)(例えば,5 mL)を試料50 mL
に加え,滴定すると終点がより明瞭になる。この場合,水について空試験を行って滴定値を
補正する。
35.2 イオン電極法 試料に酢酸塩緩衝液を加えてpH約5に調節し,塩化物イオン電極を指示電極として
電位を測定し,塩化物イオンを定量する。
備考 3. この方法では,硫化物イオンなどが妨害する。
定量範囲:Cl− 5〜1 000 mg/L,繰返し精度:5〜20 %
a) 試薬 試薬は,次による。
1) 酢酸塩緩衝液(pH5) JIS K 8548に規定する硝酸カリウム100 gとJIS K 8355に規定する酢酸50 mL
とを水500 mLに加えて溶かし,これに水酸化ナトリウム溶液(100 g/L)[19. a) 2)による。]を加え
て,pH計を用いてpH5に調節し,水を加えて1 Lとする。
2) 塩化物イオン標準液(Cl− 1 000 mg/L) JIS K 8005に規定する容量分析用標準物質の塩化ナトリウ
ムをあらかじめ600 ℃で約1時間加熱し,デシケーター中で放冷する。NaCl 100 %に対してその
1.648 gをとり,少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
3) 塩化物イオン標準液(Cl− 100 mg/L) 塩化物イオン標準液(Cl− 1 000 mg/L)20 mLを全量フラス
コ200 mLにとり,水を標線まで加える。
4) 塩化物イオン標準液(Cl− 10 mg/L) 塩化物イオン標準液(Cl− 100 mg/L)20 mLを全量フラスコ
200 mLにとり,水を標線まで加える。使用時に調製する。
5) 塩化物イオン標準液(Cl− 5 mg/L) 塩化物イオン標準液(Cl− 100 mg/L)10 mLを全量フラスコ
200 mLにとり,水を標線まで加える。使用時に調製する。
備考 4. 34.の備考3.による。
b) 器具及び装置 器具及び装置は,次による。
1) 電位差計 34.2 b) 1)による。
2) 指示電極 塩化物イオン電極
3) 参照電極 34.2 b) 3)による。ただし,二重液絡形のもので,外筒液には硝酸カリウム溶液(100 g/L)
117
K 0102:2016
(JIS K 8548に規定する硝酸カリウムを用いて調製する。)を用いる。
4) 測定容器 34.2 b) 4)による。ただし,ガラス製のものでよい。
5) 恒温槽 34.2 b) 5)による。
6) マグネチックスターラー 34.2 b) 6)による。ただし,回転子は,ガラス被覆のものでもよい。
c) 検量線 検量線の作成は,次による。
1) 塩化物イオン標準液(Cl− 5 mg/L)100 mLを測定容器200 mLにとり,酢酸塩緩衝液(pH5)10 mL(3)
を加える。
2) 恒温水槽から水を送り,測定容器の溶液を25±0.5 ℃にする。
3) 指示電極(4) (5)と参照電極(6) (7) (8)とを浸し,固定した後,回転子を入れ,マグネチックスターラー
(9)で泡が電極に触れない程度に強くかき混ぜる(10)。
4) 液温を確認し,電位差計で電位を測定する(11)。
5) 塩化物イオン標準液(Cl− 10 mg/L)100 mL,塩化物イオン標準液(Cl− 100 mg/L)100 mL及び塩
化物イオン標準液(Cl− 1 000 mg/L)100 mLをそれぞれ測定容器にとり,これに酢酸塩緩衝液(pH5)
10 mL(3)を加える。
6) 2)〜4)の操作を行う(12) (13)。
7) 横軸に塩化物イオンの濃度の対数を,縦軸に電位をとり,塩化物イオンの濃度(Cl− mg/L)と電位
との関係線を作成する(12)。
注(3) 酢酸塩緩衝液(pH5)の添加によって,pH約5に調節し,イオン強度を一定にする。
(4) 指示電極(塩化物イオン電極)は,使用時に塩化物イオン標準液(Cl− 5 mg/L)に浸し,指示
値が安定してから使用する。
(5) 34.の注(13)による。
(6) 34.の注(14)による。
(7) 内筒液に塩化カリウム溶液(飽和)を用いる場合には,液温の低下で塩化カリウムの結晶が析
出し,固着して電気抵抗が大きくなることがあるので注意する。
(8) 外筒液の硝酸カリウム溶液(100 g/L)に内筒液の塩化カリウム溶液が混入してくるので,外筒
液も定期的に取り替える。
(9) 34.の注(15)による。
(10) 34.の注(16)による。
(11) 34.の注(17)による。また,塩化物イオン電極の応答時間は,液温10〜30 ℃の場合,塩化物イオ
ンの濃度がCl− 5 mg/L以上ならば1分間以内である。
(12) 塩化物イオン標準液(Cl− 10 mg/L)と塩化物イオン標準液(Cl− 1 000 mg/L)との電位の差は,
110〜120 mV(25 ℃)の範囲に入り,塩化物イオンの濃度Cl− 5〜1 000 mg/Lの間の検量線は,
直線になる。
(13) 34.の注(19)による。
d) 操作 操作は,次による。
1) 試料100 mL(14) (15)を測定容器にとり,酢酸塩緩衝液(pH5)10 mLを加え,液温をc) 4)の液温の±1 ℃
に調節する。
2) c) 2)〜4)の操作を行って(13),検量線から塩化物イオンの濃度を求め,試料中の塩化物イオンの濃度
(Cl− mg/L)を算出する。
注(14) 試料が酸性の場合には,水酸化ナトリウム溶液(40 g/L)[21. a) 3)による。],アルカリ性の場合

118
K 0102:2016
には,酢酸(1+10)(JIS K 8355に規定する酢酸を用いて調製する。)で,あらかじめpH約5
に調節する。
注(15) 試料に硫化物イオンが含まれている場合には,あらかじめ,酢酸亜鉛溶液(100 g/L)(JIS K 8356
に規定する酢酸亜鉛二水和物12 gを水に溶かして100 mLとする。)を加え,硫化物イオンを固
定してろ紙でろ過し,ろ液のpHを約5に調節する。
備考 5. イオン濃度計の場合には,塩化物イオン標準液(Cl− 10 mg/L)と塩化物イオン標準液(Cl−
1 000 mg/L)とを用い,c) 2)及び3)の操作を行ってイオン濃度計の指示値をCl− 10 mg/L及
びCl− 1 000 mg/Lになるように調節する。さらに,その他の塩化物イオン標準液(Cl− 5 mg/L)
と塩化物イオン標準液(Cl− 100 mg/L)とを用いて,イオン濃度計の指示値を確認する。
6. 主な共存物質の許容限度を最大比率で次に示す。
NO3−,SO42−,PO43−: 104
F−: 102
Br−: 10−2
I−,CN−,S2−: 10−3
7. イオン電極による電位差滴定法 試料100 mLをビーカーにとり,試料のpHを約7に調節し,
塩化物イオン電極又は銀イオン電極を用い,c) 2)〜4)の操作に従って電位を測定しながら10
〜100 mmol/L硝酸銀溶液で滴定し,滴定曲線を作図して終点を求める。ハロゲン化物イオン
が共存するとき滴定曲線の変曲点は,よう化物イオン,臭化物イオン及び塩化物イオンの順
になる。それぞれの変曲点から終点を求め,各イオンの濃度を算出することができる。10
mmol/L硝酸銀溶液1 mLはよう化物イオン1.269 mg,臭化物イオン0.799 mg又は塩化物イオ
ン0.354 5 mgに相当する。
35.3 イオンクロマトグラフ法 試料中の塩化物イオンをイオンクロマトグラフ法によって定量する。こ
の方法によって,ふっ化物イオン,亜硝酸イオン,硝酸イオン,りん酸イオン,臭化物イオン及び硫酸イ
オンも同時に又は単独に定量できる。ただし,亜硝酸イオン,硝酸イオン,りん酸イオン又は臭化物イオ
ンを定量する場合には,試料採取後,3.3の保存処理を行わず,試験は直ちに行う。直ちに行えない場合
には,0〜10 ℃の暗所に保存し,できるだけ早く試験する。
試料マトリックスの影響によって,測定の正確さ及び精度は,イオン種ごとに異なるのが普通である。
この確認には,標準液を添加し,回収率を測定するとよい。
表35.1 イオンクロマトグラフ法による陰イオンの定量範囲の例
陰イオン
サプレッサーあり
mg/L
サプレッサーなし
mg/L
塩化物 (Cl−)
0.1〜25
0.5〜25
ふっ化物 (F−)
0.05〜20
0.1〜20
亜硝酸 (NO2−)
0.1〜25
0.5〜25
硝酸 (NO3−)
0.1〜50
0.5〜50
りん酸 (PO43−)
0.1〜50
0.5〜50
臭化物 (Br−)
0.1〜50
0.5〜50
硫酸 (SO42−)
0.2〜100
1〜100
備考 測定範囲は,検出器,試料注入量,カラムの交換容量などによって変わる。
a) 試薬 試薬は,次による。
119
K 0102:2016
1) 水 JIS K 0557に規定するA2又はA3の水
2) 溶離液 溶離液(16)は,装置の種類及び分離カラムに充塡した陰イオン交換体の種類によって異なる
ので,あらかじめふっ化物イオン,塩化物イオン,亜硝酸イオン,硝酸イオン,りん酸イオン,臭
化物イオン及び硫酸イオンのそれぞれが分離度1.3以上で分離できるものを用いる。分離度の確認
は,備考8.による。
溶離液は,脱気するか,又は脱気した水を用いて調製するとよい。操作中,溶離液に新たな気体
が溶け込むのを避けるための対策を講じる。
3) 再生液 再生液(17)は,サプレッサーを用いる場合に使用するが,装置の種類及びサプレッサーの種
類によって異なる。あらかじめ分離カラムと組み合わせて備考8.の操作を行い,再生液の性能を確
認する。
4) 塩化物イオン標準液(Cl− 1 mg/mL) 35.2 a) 2)による。
5) 塩化物イオン標準液(Cl− 0.1 mg/mL) 塩化物イオン標準液(Cl− 1 mg/mL)10 mLを全量フラス
コ100 mLにとり,水を標線まで加える。
6) ふっ化物イオン標準液(F− 1 mg/mL) JIS K 8005に規定する容量分析用標準物質のふっ化ナトリ
ウムを白金皿にとり,500 ℃で約1時間加熱し,デシケーター中で放冷する。NaF 100 %に対して
その2.210 gをとり,少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
ポリエチレン瓶に入れて保存する。
7) ふっ化物イオン標準液(F− 0.01 mg/mL) ふっ化物イオン標準液(F− 1 mg/mL)1 mLを全量フラ
スコ100 mLにとり,水を標線まで加える。
8) 亜硝酸イオン標準液(NO2− 1 mg/mL) JIS K 8019に規定する亜硝酸ナトリウムを105〜110 ℃で
約4時間加熱し,デシケーター中で放冷した後,亜硝酸ナトリウムの純度を求める(18)。NaNO2 100 %
に対して1.500 gに相当する亜硝酸ナトリウムをとり,少量の水に溶かして,全量フラスコ1 000 mL
に移し入れ,水を標線まで加える。使用時に調製する。
9) 亜硝酸イオン標準液(NO2− 0.1 mg/mL) 亜硝酸イオン標準液(NO2− 1 mg/mL)10 mLを全量フラ
スコ100 mLにとり,水を標線まで加える。使用時に調製する。
10) 硝酸イオン標準液(NO3− 1 mg/mL) JIS K 8548に規定する硝酸カリウムをあらかじめ105±2 ℃
で約2時間加熱し,デシケーター中で放冷する。その1.631 gをとり,少量の水に溶かして全量フラ
スコ1 000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
11) 硝酸イオン標準液(NO3− 0.1 mg/mL) 硝酸イオン標準液(NO3− 1 mg/mL)10 mLを全量フラスコ
100 mLにとり,水を標線まで加える。使用時に調製する。
12) りん酸イオン標準液(PO43− 1 mg/mL) JIS K 9007に規定するりん酸二水素カリウム(pH標準液
用)を105±2 ℃で約2時間加熱し,デシケーター中で放冷する。その1.433 gをとり,水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加え,0〜10 ℃の暗所に保存する。
13) りん酸イオン標準液(PO43− 0.1 mg/mL) りん酸イオン標準液(PO43− 1 mg/mL)10 mLを全量フ
ラスコ100 mLにとり,水を標線まで加える。
14) 臭化物イオン標準液(Br− 1 mg/mL) JIS K 8506に規定する臭化カリウムを105 ℃で約4時間加
熱し,デシケーター中で放冷する。その1.489 g(臭素として1.00 g)をとり,少量の水に溶かし,
全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
15) 臭化物イオン標準液(Br− 0.1 mg/mL) 臭化物イオン標準液(Br− 1 mg/mL)10 mLを全量フラス
コ100 mLにとり,水を標線まで加える。この溶液は使用時に調製する。
120
K 0102:2016
16) 硫酸イオン標準液(SO42− 1 mg/mL) JIS K 8962に規定する硫酸カリウムをあらかじめ約700 ℃
で約30分間加熱し,デシケーター中で放冷する。その1.815 gをとり,少量の水に溶かして全量フ
ラスコ1 000 mLに移し入れ,水を標線まで加える。
17) 硫酸イオン標準液(SO42− 0.1 mg/mL) 硫酸イオン標準液(SO42− 1 mg/mL)10 mLを全量フラス
コ100 mLにとり,水を標線まで加える。使用時に調製する。
注(16) 紫外吸収検出器を用いる場合には,紫外部に吸収のないものを用いる。ただし,サプレッサー
を用いる方式では,炭酸塩系の溶離液も使用できる。
(17) 例として,硫酸(12.5 mmol/L)[硫酸(0.5 mmol/L)(JIS K 8951に規定する硫酸30 mLを少量
ずつ水500 mL中に加え,冷却した後,水で1 Lとする。)25 mLを水で1 Lとする。]を用いる。
(18) 純度の求め方は,JIS K 8019による。
b) 器具及び装置 器具及び装置は,次による。
1) イオンクロマトグラフ イオンクロマトグラフには,分離カラムとサプレッサー(19)とを組み合わせ
た方式のもの,分離カラム単独の方式のものいずれでもよいが,次に掲げる条件を満たすもので,
ふっ化物イオン,塩化物イオン,亜硝酸イオン,硝酸イオン,りん酸イオン,臭化物イオン,硫酸
イオンなどが分離定量できるもの。
1.1) 分離カラム ステンレス鋼製又は合成樹脂製(20)のものに,強塩基性陰イオン交換体(表層被覆形,
全多孔性シリカ形など)を充塡したもの(21)。
1.2) 検出器 電気伝導率検出器又は紫外吸収検出器。ただし,紫外吸収検出器は,亜硝酸イオン,硝
酸イオン及び臭化物イオンの個別又は同時測定において用いる。
1.3) データ処理部 JIS K 0127による。
2) マイクロシリンジ 50〜200 μLの適切なもの。又は自動注入装置。
注(19) 溶離液中の陽イオンを水素イオンに変換するためのもので,溶離液中の陽イオンの濃度に対し
て十分なイオン交換容量をもつ陽イオン交換膜(膜形及び電気透析形がある。)又は同様な性能
をもった陽イオン交換体を充塡したもの。再生液と組み合わせて用いる。ただし,電気透析形
の場合は,再生液として検出器からの流出液(検出器から排出される溶液)を用いる。
(20) 例えば,四ふっ化エチレン樹脂製,ポリエーテルエーテルケトン製などがある。
(21) 備考8.による。
備考 8. イオンクロマトグラフの性能として分離度(R)は1.3以上なければならない。定期的に確認
するとよい。分離度を求めるには,溶離液を一定の流量(例えば,1〜2 mL/min)で流す。ク
ロマトグラムのピーク高さがほぼ同程度となるような濃度の陰イオン混合溶液を調製して,
クロマトグラムを作成し,次の式によって算出する。
2
1
1
2
)
(
2
W
W
t
t
R
R
R
+
−
×
=
ここに,
tR1: 第1ピークの保持時間(s)
tR2: 第2ピークの保持時間(s)
W1: 第1ピークのピーク幅(s)
W2: 第2ピークのピーク幅(s)
c) 準備操作 準備操作は,次による。
1) 試料を3.2によってろ過する。
2) 試料の電気伝導率が10 mS/m(100 μS/cm)(25 ℃)以上の場合には,電気伝導率が10 mS/m以下に
121
K 0102:2016
なるように,水で一定の割合に薄める。
d) 操作 操作は,次による。
1) イオンクロマトグラフを作動できる状態にし,分離カラムに溶離液を一定の流量(例えば,1〜2
mL/min)で流しておく。サプレッサーを必要とする装置では,再生液を一定の流量で流しておく。
2) c)の準備操作を行った試料の一定量(例えば,50〜200 μLの一定量)を,マイクロシリンジ(22)を用
いてイオンクロマトグラフに注入してクロマトグラムを記録する。
3) クロマトグラム上の定量対象の各イオンに相当するピーク(23)について,指示値(24)を読み取る。
4) 試料を薄めた場合には,空試験として試料及び同量の水について,1)〜3)の操作を行って試料につ
いて得た指示値(24)を補正する。
5) 検量線から各イオンの量を求め,試料中の各イオンの濃度(mg/L)を算出する。
注(22) 検量線作成時と同じものを用いる。
(23) 陰イオン混合標準液[例えば(F− 5 μg,Cl− 10 μg,NO2− 10 μg,NO3− 10 μg,PO43− 10 μg,
Br− 10 μg及びSO42− 10 μg)/mL]の一定量(50〜200 μL)を用いて,クロマトグラムを記録
し,各イオンの保持時間に相当するピークの位置を確認しておく。
(24) ピーク高さ又はピーク面積。
e) 検量線 検量線の作成は,次による。
1) a) 4)〜17)からそれぞれ適切な量をとり,測定対象とするイオンを含み,測定濃度範囲よりも高い濃
度の混合希釈標準液を調製する(25)。
2) この混合希釈標準液を4〜5段階に希釈し,検量線作成用の混合希釈標準液を調製する。
3) 各種検量線作成用の混合希釈標準液についてd) 1)〜4)の操作を行い,それぞれの各イオンに相当す
るピークについて指示値(24)を読み取る(26)。
4) 別に,空試験として水についてd) 1)〜4)の操作を行ってそれぞれの各イオンに相当する指示値を補
正した後,各イオンについて横軸にそのイオンの濃度,縦軸にそのイオンの指示値をとり,関係線
を作成する(27)。
注(25) 単独のイオンの測定又は限られた複数のイオンだけの測定の場合は,必要な混合標準液と必要
な陰イオンを含む混合標準液とを調製するとよい。
(26) 各イオンにおけるそれぞれの妨害イオンの許容割合の例は,表35.2による。
(27) 溶離液の組成及び測定対象イオン種によって検量線は,必ずしも直線関係を示さない。
備考 9. 妨害物質
a) モノカルボン酸,ジカルボン酸などの有機酸は,無機陰イオンの定量を妨害することが
ある。
b) 緩衝性溶離液(例えば,炭酸塩/炭酸水素塩)を用いれば,試料のpHが2〜9の範囲の
場合は影響されない。
c) 陰イオン(ふっ化物イオン,塩化物イオン,亜硝酸イオン,硝酸イオン,りん酸イオン,
臭化物イオン及び硫酸イオン)間の濃度の大きな相違があると,不完全分離による典型
的な交差感度妨害を引き起こすことがある。塩化物イオンの定量は,ふっ化物イオンの
濃度が高いと妨害を受けやすい。
d) 各イオンに対する共存イオンの影響
1) 塩化物イオンの濃度が1 mg/Lのとき,亜硝酸イオンは200 mg/L以下ならば妨害しな
い。
122
K 0102:2016
2) 亜硝酸イオンの濃度が1 mg/Lのとき,塩化物イオンは50 mg/L以下,臭化物イオン
200 mg/L以下及び硫酸イオン500 mg/L以下ならば妨害しない。
3) 硝酸イオンの濃度が1 mg/Lのとき,臭化物イオン200 mg/L以下及び硫酸イオン500
mg/L以下ならば妨害しない。
4) 臭化物イオンの濃度が1 mg/Lのとき,亜硝酸イオンは200 mg/L以下ならば妨害しな
い。
5) 硫酸イオンの濃度が1 mg/Lのとき,臭化物イオン200 mg/L以下及び硝酸イオン400
mg/L以下ならば妨害しない。
e) 硫酸イオンの定量は,高濃度のよう化物イオン又はチオ硫酸イオンによって妨害されや
すい。
f)
硫化物イオンは,硫酸イオンの定量誤差の原因になるので,酢酸亜鉛溶液を加えて沈殿
させ,ろ別する。
備考 10. 分離カラムは,使用を続けると性能が低下するので,定期的に備考8.の操作を行って確認す
る。
性能が低下した場合,溶離液の約10倍の濃度のものを調製し,分離カラムに注入して洗浄
した後,備考8.の操作で確認し,性能が回復しない場合には,新品と取り替える。
試料中の懸濁物,有機物(たん白質,油類,界面活性剤など)などによって汚染され性能
が徐々に低下するので,懸濁物を含む試料はc)の準備操作で除去した後に試験する。また,
有機物を含む試料は限外ろ過膜でろ過し,有機物をできるだけ除去した後,試験する。
試料中に分離カラムの充塡剤と親和力の強い陰イオン(例えば,よう化物イオン,クロム
酸イオンなど)が存在すると,これらが充塡剤に吸着され,分離性能が徐々に低下するので,
溶離液の5〜10倍の濃度のものを調製し,試料と同様に分離カラムに注入し洗浄する。
その他酸化性物質又は還元性物質が共存すると,分離カラムの分離性能を低下させる。こ
のような場合には,試料を水で一定の割合に薄めて試験すれば,ある程度は影響を防ぐこと
ができる。
11. 亜硝酸イオン,硝酸イオン及びりん酸イオンの濃度を,亜硝酸体窒素,硝酸体窒素及びりん
酸体りんで表示する場合は,次の換算式を用いる。
a) 亜硝酸体窒素(NO2−-N mg/L)=亜硝酸イオン(NO2− mg/L)×0.304 5
b) 硝酸体窒素(NO3−-N mg/L)=硝酸イオン(NO3− mg/L)×0.225 9
c) りん酸体りん(PO43−-P mg/L)=りん酸イオン(PO43− mg/L)×0.326 1
123
K 0102:2016
表35.2 陰イオンの感度交差の例
[検出:電気伝導率(CD)及び直接紫外吸光]
質量濃度比
溶質/妨害イオン
妨害イオンの
最大許容濃度*
mg/L
Br−/Cl−
1 :
500
Cl−
500
Br−/PO43−
1 :
100
PO43−
100
Br−/NO3−
1 :
50
NO3−
100
Br−/SO42−
1 :
500
SO42−
500
Br−/SO32−
1 :
50
Cl−/NO2−
1 :
50
NO2−
5
Cl−/NO3−
1 :
500
NO3−
500
Cl−/SO42−
1 :
500
SO42−
500
NO3−/Br−
1 :
100
Br−
100
NO3−/Cl−
1 :
500(CD) Cl−
500
1 :
2000(UV) Cl−
500
NO3−/SO42−
1 :
500(CD) SO42−
500
1 :
1000(UV) SO42−
500
NO3−/SO32−
1 :
50
NO2−/Cl−
1 :
250(CD) Cl−(CD)
100
1 : 10000(UV) Cl−(UV)
500
NO2−/PO43−
1 :
50
PO43−
20
NO2−/NO3−
1 :
500
NO3−
500
NO2−/SO42−
1 :
500(CD) SO42−
500
1 :
1000(UV) SO42−
500
PO43−/Br−
1 :
100
Br−
100
PO43−/Cl−
1 :
500
Cl−
500
PO43−/NO3−
1 :
500
NO3−
400
PO43−/NO2−
1 :
100
NO2−
100
PO43−/SO42−
1 :
100
SO42−
500
PO43−/SO32−
1 :
50 **
SO42−/Cl−
1 :
500
Cl−
500
SO42−/NO3−
1 :
500
NO3−
400
SO42−/SO32−
1 :
50 **
SO42−/S2O32−
1 :
500
SO42−/I−
1 :
500
注*
妨害物質の濃度が限度を超えるときは,試料を薄める。
** SO32−は,存在すると常に妨害する。
36. よう化物イオン(I−) よう化物イオンの定量には,よう素抽出吸光光度法又はよう素滴定法を適用す
る。
36.1 よう素抽出吸光光度法 よう化物イオンを硫酸酸性で亜硝酸イオンと反応させ,遊離したよう素を
クロロホルムで抽出してその吸光度を測定してよう化物イオンを定量する。
定量範囲:I− 0.1〜5 mg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 硫酸(1+1) 5.4 a) 2)による。
2) 亜硝酸ナトリウム JIS K 8019に規定するもの。
3) 尿素溶液(10 g/L) JIS K 8731に規定する尿素1 gをとり,水に溶かして100 mLとする。
124
K 0102:2016
4) 硫酸ナトリウム JIS K 8987に規定するもの。
5) クロロホルム JIS K 8322に規定するもの。
6) よう化物イオン標準液(I− 1 mg/mL) JIS K 8913に規定するよう化カリウム1.310 gをとり,少量
の水に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
7) よう化物イオン標準液(I− 0.1 mg/mL) よう化物イオン標準液(I− 1 mg/mL)20 mLを全量フラ
スコ200 mLにとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料の適量(1) (2)(I−として0.1〜5 mgを含む。)を分液漏斗100 mLにとり,水を加えて約50 mLと
する。
2) 硫酸(1+1)[試料がアルカリ性の場合には,硫酸(1+1)を加えて中和しておく。]1 mLと亜硝酸
ナトリウム0.5 gとを加えて振り混ぜる。
3) クロロホルム10 mLを加え,約2分間激しく振り混ぜた後放置する。
4) クロロホルム層を別の分液漏斗100 mLに移す。再び水層にクロロホルム10 mLを加えて抽出し,
クロロホルム層は先のクロロホルム層に合わせる。
5) クロロホルムを入れた分液漏斗に尿素溶液(10 g/L)50 mLを加え,約2分間激しく振り混ぜてクロ
ロホルム層を洗浄する。
6) 約5分間放置した後,クロロホルム層を硫酸ナトリウム約1 gを入れた共栓三角フラスコ50 mLに
移し,振り混ぜて脱水する。
7) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長515 nm付近の吸光度を
測定する。
8) 空試験として水50 mLをとり,2)〜7)の操作を行って試料について得た吸光度を補正する。
9) 検量線からよう化物イオンの量を求め,試料中のよう化物イオンの濃度(I− mg/L)を算出する。
注(1) よう化物イオンの濃度が2 mg/L以下の場合には,試料の適量をとり,水酸化ナトリウム溶液(200
g/L)(JIS K 8576に規定する水酸化ナトリウム20 gを水に溶かして100 mLとする。)を加えて
アルカリ性として,静かに加熱して濃縮する。濃縮中に濁りが生じた場合には,ろ過し,1)以
降の操作を行う。
(2) 有機物が多量に共存する場合には,試料200 mLをとり,硫酸カリウムアルミニウム溶液(JIS K
8255に規定する硫酸カリウムアルミニウム・12水5 gを水に溶かして100 mLとする。)2〜3 mL
を加えた後,水酸化ナトリウム溶液(50 g/L)(JIS K 8576に規定する水酸化ナトリウム5 gを
水に溶かして100 mLとする。)を水酸化アルミニウムの沈殿が生成するまで加える。約5分間
放置した後,ろ過し,そのろ液の適量をとり,1)以降の操作を行う。
d) 検量線 検量線の作成は,次による。
1) よう化物イオン標準液(I− 0.1 mg/mL)1〜50 mLを段階的に分液漏斗100 mLにとる。
2) c) 1)〜8)の操作を行って吸光度を測定し,よう化物イオン(I−)の量と吸光度との関係線を作成す
る。
備考 1. よう素酸イオンが含まれると,硫酸酸性としたとき,よう化物イオンと反応してよう素を生
じるため,よう素酸イオンの一部又は全部がよう化物イオンとして定量される。臭化物イオ
125
K 0102:2016
ンは妨害しない。
36.2 よう素滴定法 よう化物イオンをpH1.3〜2.0で次亜塩素酸で酸化し,よう素酸イオンとする。過剰
の次亜塩素酸を,pH3〜7でぎ酸ナトリウムで分解した後,よう化カリウムを加え,遊離するよう素をチオ
硫酸ナトリウム溶液で滴定してよう化物イオンを定量する。
定量範囲:I− 0.1 mg以上
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
2) 塩酸(1+11) JIS K 8180に規定する塩酸を用いて調製する。
3) 次亜塩素酸ナトリウム溶液(有効塩素35 g/L) 次亜塩素酸ナトリウム溶液(有効塩素7〜12 %)の
有効塩素を定量し(3),有効塩素が35 g/Lになるように水で薄める。使用時に調製する。
4) ぎ酸ナトリウム溶液(400 g/L) JIS K 8267に規定するぎ酸ナトリウム40 gを水に溶かして100 mL
とする。
5) よう化カリウム JIS K 8913に規定するもの。
6) メチルオレンジ溶液(1 g/L) 24.1 a) 2)による。
7) でんぷん溶液(10 g/L) 19. a) 5)による。
8) 10 mmol/Lチオ硫酸ナトリウム溶液 19. a) 9)による。
注(3) 次亜塩素酸ナトリウム溶液(有効塩素7〜12 %)10 mLを全量フラスコ200 mLにとり,水を標
線まで加える。この10 mLを共栓三角フラスコ300 mLにとり,水を加えて約100 mLとする。
よう化カリウム1〜2 g及び酢酸(1+1)[33.4 a) 9.2.1)による。]6 mLを加えて密栓し,よく振
り混ぜて暗所に約5分間放置した後,50 mmol/Lチオ硫酸ナトリウム溶液[19. a) 8)の0.1 mol/L
チオ硫酸ナトリウム溶液を2倍に薄めて調製する。]で滴定する。溶液の黄色が薄くなったら,
指示薬としてでんぷん溶液(10 g/L)l mLを加え,生じたよう素でんぷんの青い色が消えるま
で滴定する。別に,空試験として水10 mLをとり,同じ操作を行って滴定値を補正する。次の
式によって有効塩素量を算出する。
773
001
.0
000
1
10
200
×
×
×
×
=
V
f
a
N
ここに,
N: 有効塩素量(g/L)
a: 滴定に要した50 mmol/Lチオ硫酸ナトリウム溶液量(mL)
f: 50 mmol/Lチオ硫酸ナトリウム溶液のファクター[19. a)
8)の0.1 mol/Lチオ硫酸ナトリウム溶液のファクターを用
いる。]
0.001 773: 50 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する有効
塩素の質量(g)
V: 次亜塩素酸ナトリウム溶液(有効塩素7〜12 %)量(mL)
b) 操作 操作は,次による。
1) 試料の適量(1) (2)(I−として0.1〜5 mgを含む。)を共栓三角フラスコ300 mLにとり,指示薬として
メチルオレンジ溶液(1 g/L)1,2滴を加え,溶液の色が微赤になるまで塩酸(1+11)を滴加した
後,水を加えて約50 mLとする。
2) 次亜塩素酸ナトリウム溶液(有効塩素35 g/L)1 mLを加え,塩酸(1+11)を加えてpH1.3〜2.0に
調節し,沸騰水浴中に約5分間浸す。
3) ぎ酸ナトリウム溶液(400 g/L)5 mLを加え(4),再び沸騰水浴中に約5分間浸して過剰の次亜塩素酸
126
K 0102:2016
を分解する。
4) 放冷後,よう化カリウム1 gと塩酸(1+1)6 mLとを加え,密栓して振り混ぜ,暗所に約5分間放
置する。
5) 遊離したよう素を10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,指
示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴定
する。
6) 空試験として水50 mLを共栓三角フラスコ300 mLにとり,1)〜5)の操作を行う。
7) 次の式によって試料中のよう化物イオンの濃度(I− mg/L)を算出する。
(
)
5
211
.0
000
1
×
×
×
−
=
V
f
b
a
C
ここに,
C: よう化物イオンの濃度(I− mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
b: 空試験の滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液
量(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター(5)
V: 試料量(mL)
0.211 5: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当するよう化
物イオンの質量(mg)
注(4) ぎ酸ナトリウム溶液(400 g/L)による次亜塩素酸の分解は,pH3〜7で行う。pH2.7以下になる
と,よう素酸イオンが還元され,pH7以上になると次亜塩素酸の分解が不完全になる。
(5) 19. a) 8)の0.1 mol/Lチオ硫酸ナトリウム溶液のファクターを用いる。
備考 2. この方法では鉄(II,III)は,妨害する。マンガン0.2 mg以上及びひ酸イオン1 mg以上は,
妨害する。硫化水素及び多量の有機物も妨害する。妨害の除去は,次による。
1) 鉄,マンガン 試料に水酸化ナトリウム溶液(200 g/L)[36.の注(1)による。]を加えてア
ルカリ性とし,約1時間放置した後,ろ過する。ろ液と洗液とを合わせ,30〜50 mLに
なるまで沸騰水浴上で濃縮する。濁り及び沈殿物があるときは,ろ紙5種Aでろ過して
洗浄する。ろ液と洗液とを合わせ,b) 1)以降の操作を行う。
2) ひ酸イオン 試料500 mLにつき塩化鉄(III)溶液(Fe 10 mg/mL)[JIS K 8142に規定す
る塩化鉄(III)六水和物5 gを塩酸(1+1)10 mLに溶かし,水で100 mLとする。]1 mL
を加え,次に,1)と同様に操作する。ただし,水酸化ナトリウム溶液(200 g/L)に代え,
アンモニア水(1+1)を用いる。
3) 硫化水素 試料500 mLにつき硫酸亜鉛溶液(JIS K 8953に規定する硫酸亜鉛七水和物
10 gを水に溶かして100 mLとする。)2〜3 mLを加え,よくかき混ぜ,次に,1)と同様
に操作する。
3. この方法では,よう素酸イオンもよう化物イオンとして定量される。
37. 臭化物イオン(Br−) 臭化物イオンの定量には,よう素滴定法又はイオンクロマトグラフ法を適用す
る。
37.1 よう素滴定法 臭化物イオンをpH6.5〜8.0で次亜塩素酸で酸化し,臭素酸イオンとする。過剰の次
亜塩素酸をpH3〜7でぎ酸ナトリウムで分解した後,よう化カリウムを加え,遊離するよう素をチオ硫酸
ナトリウム溶液で滴定して臭化物イオンを定量する。よう化物イオンも同じ反応をするので,別に定量し
127
K 0102:2016
て差し引く。
定量範囲:Br− 0.1 mg以上
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
2) 塩酸(1+11) JIS K 8180に規定する塩酸を用いて調製する。
3) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
4) りん酸二水素ナトリウム溶液(500 g/L) JIS K 9009に規定するりん酸二水素ナトリウム二水和物
65 gを水に溶かして100 mLとする。
5) 次亜塩素酸ナトリウム溶液(有効塩素35 g/L) 36.2 a) 3)による。
6) ぎ酸ナトリウム溶液(400 g/L) 36.2 a) 4)による。
7) よう化カリウム JIS K 8913に規定するもの。
8) メチルオレンジ溶液(1 g/L) 24.1 a) 2)による。
9) でんぷん溶液(10 g/L) 19. a) 5)による。
10) 10 mmol/Lチオ硫酸ナトリウム溶液 19. a) 9)による。
b) 操作 操作は,次による。
1) 試料の適量(1)(Br−として0.1〜3 mgを含む。)を共栓三角フラスコ300 mLにとり,指示薬としてメ
チルオレンジ溶液(1 g/L)1滴を加え,溶液の色が僅かに赤くなるまで塩酸(1+11)を滴加した後,
水を加えて約50 mLとする。
2) りん酸二水素ナトリウム溶液(500 g/L)2 mLと次亜塩素酸ナトリウム溶液(有効塩素35 g/L)3 mL
とを加え,水酸化ナトリウム溶液(40 g/L)又は塩酸(1+11)を用いてpH6.5〜8.0に調節した後,
約10分間煮沸する。
3) ぎ酸ナトリウム溶液(400 g/L)3 mL (2)を加え,約5分間煮沸し,過剰の次亜塩素酸を分解する。
4) 放冷後,よう化カリウム1 gと塩酸(1+1)6 mLとを加え,密栓して振り混ぜ,暗所に約5分間放
置する。
5) 遊離したよう素を10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなってから,指
示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴定
する。
6) 空試験として水50 mLを共栓三角フラスコ300 mLにとり,1)〜5)の操作を行う。
7) 別に,36.1又は36.2によって試料中のよう化物イオンの濃度(I− mg/L)を定量する。
8) 次の式によって試料中の臭化物イオンの濃度(Br− mg/L)を算出する。
(
)
6
629
.0
2
133
.0
000
1
×
−
×
×
×
−
=
C
V
f
b
a
B
ここに,
B: 臭化物イオンの濃度(Br− mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
b: 空試験の滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液
量(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター(3)
0.133 2: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する臭化物
イオンの質量(mg)
V: 試料量(mL)
C: よう化物イオンの濃度(I− mg/L)
0.629 6: よう化物イオンの量を臭化物イオン相当量に換算する場合
128
K 0102:2016
の係数
90
.
126
904
.
79
注(1) 臭化物イオンの濃度が2 mg/L以下の場合には,36.の注(1)と同様に操作する。
(2) ぎ酸ナトリウム溶液(400 g/L)による次亜塩素酸の分解は,pH3〜7で行う。pH2.7以下になる
と,臭素酸イオンが還元される。また,pH7以上になると次亜塩素酸の分解が不完全になる。
(3) 19. a) 8)の0.1 mol/Lチオ硫酸ナトリウム溶液のファクターを用いる。
備考 1. この方法では鉄(II,III)は妨害する。マンガン0.2 mg以上及びひ酸イオン1 mg以上がそれ
ぞれ共存すると妨害する。硫化水素及び多量の有機物も妨害する。妨害の除去は,36.の備考
2.による。
37.2 イオンクロマトグラフ法 35.3による。
38. シアン化合物 シアン化合物は,水中のシアン化物イオン,シアノ錯体などを総称し,シアン化物イ
オンと全シアンとに区分する。
シアン化合物は,前処理でシアン化物イオンとし,定量には,ピリジン-ピラゾロン吸光光度法,4-ピリ
ジンカルボン酸-ピラゾロン吸光光度法,イオン電極法又は4-ピリジンカルボン酸-ピラゾロン発色による
流れ分析法を適用する。
シアン化合物は変化しやすいので,試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によ
って保存し,できるだけ早く試験する。
なお,流れ分析法は,2002年に第1版として発行されたISO 14403との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 14403:2002,Water quality−Determination of total cyanide and free cyanide by continuous flow
analysis(MOD)
38.1 前処理 試料を微酸性として通気又は加熱蒸留し,発生するシアン化水素を捕集する。
38.1.1 シアン化物 この前処理では,シアン化物イオン及び錯生成定数の小さい亜鉛,カドミウムなどの
シアノ錯体からはほぼ完全に,また,ニッケル,銅などのシアノ錯体からは一部シアン化水素を発生する。
鉄(II)及び鉄(III)のシアノ錯体からは,シアン化水素は発生しない。
38.1.1.1 通気法(pH5.0で発生するシアン化水素) 試料のpHを5.0に調節し,恒温水槽で40 ℃に保持
しながら,約1.2 L/minで通気し,発生したシアン化水素を水酸化ナトリウム溶液に捕集する。
a) 試薬 試薬は,次による。
1) 酢酸(1+1) JIS K 8355に規定する酢酸を用いて調製する。
2) 酢酸(1+49) JIS K 8355に規定する酢酸を用いて調製する。
3) 水酸化ナトリウム溶液(200 g/L) JIS K 8576に規定する水酸化ナトリウム20 gを水に溶かして100
mLとする。
4) 水酸化ナトリウム溶液(20 g/L) 水酸化ナトリウム溶液(200 g/L)を水で10倍に薄める。
b) 装置 装置は,次による。
1) 通気装置 図38.1に例を示す。
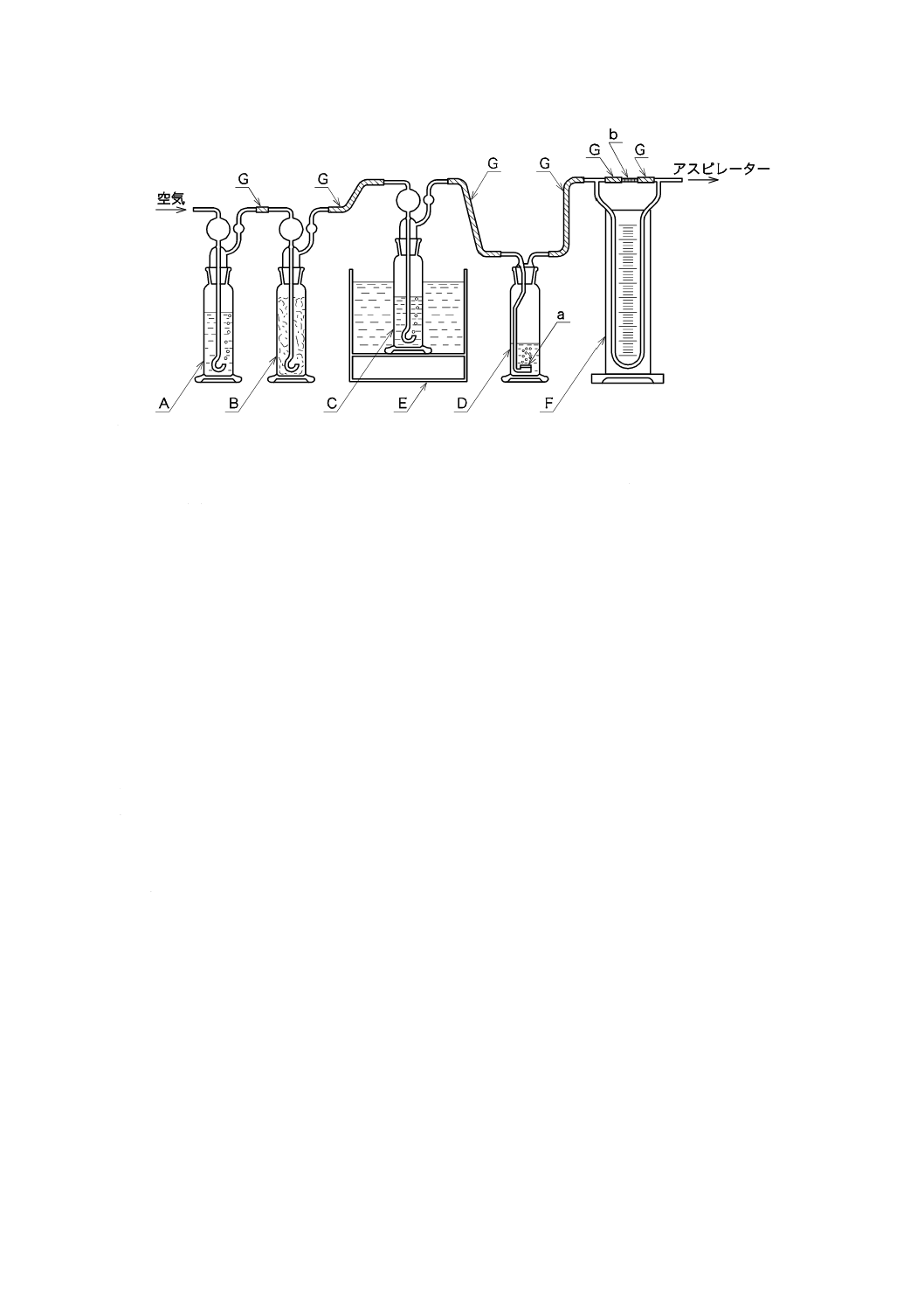
129
K 0102:2016
A:
B:
C:
D:
ガス洗浄瓶250 mL
水酸化ナトリウム溶液(200 g/L)100 mLを入れる。
ガス洗浄瓶250 mL
ガラスウールを軽く詰めておく。
ガス洗浄瓶250 mL(試料用)
ろ過板付きガス洗浄瓶250 mL(シアン化水素吸収用)
E:
F:
G:
a:
b:
恒温水槽(40±2 ℃)
流量計
軟質塩化ビニル管又はシリコーンゴム管
ガラスろ過板G2
毛管
図38.1 通気装置の例
c) 通気操作 通気操作は,次による。
1) 通気装置を図38.1のように組み立て,ろ過板付きガス洗浄瓶(D)には,シアン化水素吸収用とし
て水40 mLと水酸化ナトリウム溶液(20 g/L)20 mLとを入れる。
2) あらかじめ試料100 mLをビーカー300 mLにとり,pH計を用いてpH5.0±0.2になるまで酢酸(1+
1)及び酢酸(1+49),又は水酸化ナトリウム溶液(20 g/L)を滴加し,その量を求める。
3) 試料100 mL(1) (2)をガス洗浄瓶(C)に入れ,2)の操作で求めた,酢酸(1+1)及び酢酸(1+49)
又は水酸化ナトリウム溶液(20 g/L)の量を加え,図38.1のように連結する。
4) 恒温水槽(E)を40±2 ℃に保持して,約1.2 L/minで1時間通気する。
5) 通気後,ろ過板付きガス洗浄瓶(D)の中の水酸化ナトリウム溶液(吸収液)を全量フラスコ100 mL
に移し入れ,ろ過板付きガス洗浄瓶(D)を水で洗い,洗液も移し入れて水を標線まで加える。
注(1) 試料の採取量は,38.2〜38.4のそれぞれの方法に規定した定量範囲から求めた最適量。
(2) 試料中に,油脂類,残留塩素などの酸化性物質,又は硫化物などの還元性物質が含まれている
場合には,あらかじめ備考1.〜3.に示す方法によって除去する。
備考 1. 試料中に多量の油脂類が含まれている場合には,あらかじめ酢酸又は水酸化ナトリウムを加
えてpHを6〜7に調節し,分液漏斗に移し入れる。試料の体積百分率約2 %量のヘキサン又
はクロロホルムを加えて,静かに振り混ぜ,放置して油脂類を分離した後,38.1.1.1の操作を
行う。
2. 試料中に残留塩素などの酸化性物質が含まれている場合には,L(+)-アスコルビン酸溶液
(100 g/L)[JIS K 9502に規定するL(+)-アスコルビン酸10 gを水に溶かして100 mLと
する。]又は亜ひ酸ナトリウム溶液(100 g/L)(メタ亜ひ酸ナトリウム10 gを水に溶かして
100 mLとする。)を加えて還元する。
3. 硫化物が含まれている場合には,あらかじめ酢酸亜鉛溶液(100 g/L)[38.1.1.2 a) 4)による。]
130
K 0102:2016
2 mLを加える。酢酸亜鉛溶液(100 g/L)1 mLは,硫化物イオン約14 mgに相当する。
38.1.1.2 加熱蒸留法(pH5.5で酢酸亜鉛の存在下で発生するシアン化水素) 試料に酢酸亜鉛を加え,pH5.5
に調節して加熱蒸留し,発生するシアン化水素を水酸化ナトリウム溶液に捕集する。
a) 試薬 試薬は,次による。
1) 酢酸(1+1) 38.1.1.1 a) 1)による。
2) 酢酸(1+49) 38.1.1.1 a) 2)による。
3) 水酸化ナトリウム溶液(20 g/L) 38.1.1.1 a) 4)による。
4) 酢酸亜鉛溶液(100 g/L) JIS K 8356に規定する酢酸亜鉛二水和物12 gを水に溶かして100 mLと
する。
b) 装置 装置は,次による。
1) 蒸留装置 図38.2に例を示す。
131
K 0102:2016
単位 mm
図38.2 蒸留装置の例
c) 蒸留操作 蒸留操作は,次による。
1) 試料が強アルカリ性の場合には,試料500 mL (1)をビーカー1 000 mLにとり,酢酸(1+1)を滴加
し,pH計を用いてpH約7とし,この中和に必要な添加量を求める。
2) これに酢酸亜鉛溶液(100 g/L)20 mLを加え,再び酢酸(1+49)を滴加し,pH計を用いてpH5.5
に調節する。この酢酸(1+49)の添加量を求める(3)。
3) 試料500 mLを蒸留フラスコ1 000 mLにとり,沸騰石(4)(粒径2〜3 mm)約10個を入れる。
A:蒸留フラスコ1 000 mL(又は500 mL)
B:連結導入管
C:すり合わせコック
D:注入漏斗
E:トラップ球(ケルダール球)
F:リービッヒ冷却器300 mm
G:逆流止め(約50 mL)
H:受器[メスシリンダー(有栓形)250 mL(又は100 mL)]
I:共通すり合わせ
J:共通球面すり合わせ
K:押さえばね
132
K 0102:2016
4) これに1)で求めた酢酸(1+1)を加え,蒸留フラスコを図38.2のように蒸留装置に接続する。
5) 蒸留装置の受器には,メスシリンダー(有栓形)250 mLを用い,これに水酸化ナトリウム溶液(20
g/L)20 mLを入れ,受器を図38.2のように接続する。
6) 次に,注入漏斗から,酢酸亜鉛溶液(100 g/L)20 mLを加え,更に2)で求めた酢酸(1+49)を加
える。
7) 蒸留フラスコを加熱し,留出速度(5)を2〜3 mL/minに調節し,受器の液量が約230 mLになるまで
蒸留する(6)。
8) 冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器
に加え,更に水を250 mLの標線まで加える。
注(3) 各試薬の添加量は,できるだけ正確にする。
(4) 毛細管の一端を封じたものでもよい。
(5) 留出速度は,3 mL/min以上にしない。シアン化水素の回収率が低下する。
(6) 逆流止めの先端は,常に液面下約15 mmを保つように,メスシリンダー(有栓形)250 mLの
高さを調節する。
備考 4. 試料に多量の油脂類が含まれている場合には,備考1.と同じ操作を行う。
5. 試料に残留塩素などの酸化性物質が含まれている場合には,備考2.と同じ操作を行う。
6. 試料に硫化物などの還元性物質が含まれている場合には,c) 1)〜7)を行って得た,留出液に
対し,次のような酸化処理を行った後,再び蒸留操作を行って除去する。
− 蒸留操作を行った受器中の留出液と洗液とを再び蒸留フラスコに移し,指示薬としてフ
ェノールフタレイン溶液(5 g/L)[15.の備考2.による。]2,3滴を加え,酢酸(1+1)
で中和し,更に硝酸(50 mmol/L)(JIS K 8541に規定する硝酸3.8 mLを水に溶かして1
Lとする。)約30 mLを加える。
− 次に,過マンガン酸カリウム溶液(3 g/L)(JIS K 8247に規定する過マンガン酸カリウ
ムを用いて調製する。)を滴加し,過マンガン酸の微赤になる点又は酸化マンガン(IV)
の褐色の濁りが生成した点から更に過剰に1 mLを加え,水を加えて約300 mLとする。
− 蒸留フラスコを図38.2のように蒸留装置に接続し,受器にはメスシリンダー(有栓形)
100 mLを用い,これに水酸化ナトリウム溶液(20 g/L)20 mLを入れ,受器を図38.2の
ように接続する。
− 蒸留フラスコを加熱し,留出速度を2〜3 mL/minに調節し,受器の液量が約90 mLにな
ったら蒸留を止める。冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内
外を少量の水で洗い,洗液も受器中に加えた後,水を100 mLの標線まで加える。
38.1.2 全シアン(pH2以下で発生するシアン化水素) 試料にりん酸を加えてpH2以下にし,エチレンジ
アミン四酢酸二水素二ナトリウムを加えて加熱蒸留し,発生したシアン化水素を水酸化ナトリウム溶液に
捕集する。
備考 7. 前処理によってシアン化物イオン及びほとんどのシアノ錯体中のシアンは,留出する。酸化
性物質が共存する状態で蒸留すると,チオシアン酸,2-プロペンニトリル(アクリロニトリ
ル)などが分解してシアン化水素が発生するので,あらかじめ酸化性物質を還元しておく。
a) 試薬 試薬は,次による。
1) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
2) 水酸化ナトリウム溶液(20 g/L) 38.1.1.1 a) 4)による。
133
K 0102:2016
3) アミド硫酸アンモニウム溶液(100 g/L) JIS K 8588に規定するアミド硫酸アンモニウム10 gを水
に溶かして100 mLとする。
4) EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物10 gを水
に溶かし,水酸化ナトリウム溶液(20 g/L)5〜7滴を加えて微アルカリ性とし,水を加えて100 mL
とする。
5) りん酸 JIS K 9005に規定するもの。
b) 装置 装置は,次による。
1) 蒸留装置 図38.2に例を示す。
c) 蒸留操作 蒸留操作は,次による。
1) 試料50 mLを蒸留フラスコ500 mLにとり,水を加えて約250 mLとする。沸騰石(4)(粒径2〜3 mm)
約10 個を入れる。指示薬としてフェノールフタレイン溶液(5 g/L)1滴を加える。
2) アルカリ性の場合には,溶液の紅色が消えるまで,りん酸を滴加する(7)。
3) 次に,アミド硫酸アンモニウム溶液(100 g/L)1 mL (8)を加える。
4) 蒸留フラスコを図38.2のように接続し,受器にはメスシリンダー(有栓形)100 mLを用い,これ
に水酸化ナトリウム溶液(20 g/L)20 mLを入れ,図38.2のように接続する。
5) 注入漏斗から蒸留フラスコにりん酸10 mLを加え,次に,EDTA溶液10 mLを加え,少量の水で注
入漏斗を洗い,洗液を蒸留フラスコに加える。
6) 数分間放置した後,蒸留フラスコを加熱し,留出速度(5) 2〜3 mL/minで受器の液量が約90 mLにな
るまで蒸留する(6)。
7) 冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器
に加えた後,更に水を100 mLの標線まで加える。
注(7) りん酸を加えて弱酸性になればよい。
(8) アミド硫酸アンモニウム溶液(100 g/L)は,試料中の亜硝酸イオンの妨害を除くために加える。
これを加えない場合には,亜硝酸イオンが存在すると,加熱蒸留時にEDTAと反応してシアン
化水素を生成する。アミド硫酸アンモニウム溶液(100 g/L)1 mLは,亜硝酸イオン約40 mgに
相当する。亜硝酸イオンが40 mg以上共存する場合には,その量に応じて添加量を増加する。
特殊の試料では,亜硝酸イオン以外にもEDTAとの反応によってシアン化水素を生成し,ア
ミド硫酸アンモニウム溶液(100 g/L)の添加によってもその妨害を除けないものもある。添加
したEDTAが関与すると考えられる場合は,EDTA溶液の添加を除いて1)〜7)の操作を行う。
なお,EDTA以外に類似の反応をする有機物もある。
備考 8. 油脂類の除去は,備考1.の操作を行う。
9. 残留塩素などの酸化性物質が含まれている場合には,備考2.の操作を行う。
10. 試料に硫化物などの還元性物質が含まれている場合には,全シアンの蒸留操作を行って得た
留出液について備考6.の操作を行う。
38.2 ピリジン-ピラゾロン吸光光度法 前処理して得られたシアン化物イオン溶液の一部をとり,酢酸で
中和した後,クロラミンT溶液を加えて塩化シアンとし,これにピリジン-ピラゾロン溶液を加える。この
とき生じる青い色の吸光度を測定してシアン化物イオンを定量する。
定量範囲:CN− 0.5〜9 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 酢酸(1+8) JIS K 8355に規定する酢酸を用いて調製する。
134
K 0102:2016
2) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
3) りん酸塩緩衝液(pH6.8) JIS K 9007に規定するりん酸二水素カリウム17.0 gとJIS K 9020に規定
するりん酸水素二ナトリウム17.8 gとを水に溶かして500 mLとする。
4) クロラミンT溶液(10 g/L) JIS K 8318に規定するp-トルエンスルホンクロロアミドナトリウム三
水和物(クロラミンT)0.62 gを水に溶かして50 mLとする。使用時に調製する。
5) ピリジン-ピラゾロン溶液 JIS K 9548に規定する3-メチル-1-フェニル-5-ピラゾロン0.25 gをJIS K
8777に規定するピリジン20 mLに溶かし,これにJIS K 9545に規定するビス(3-メチル-1-フェニ
ル-5-ピラゾロン)20 mgを溶かし,更に水100 mLを加えて混ぜる。10 ℃以下であれば,1 週間は
使用できる。
6) 0.1 mol/L硝酸銀溶液 JIS K 8550に規定する硝酸銀17 gを水に溶かして1 Lとする。着色ガラス瓶
に保存する。
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質の塩化ナトリウムを600 ℃で約1時間加熱し,デ
シケーター中で放冷する。NaCl 100 %に対してその1.17 gを1 mgの桁まではかりとり,少量
の水に溶かして全量フラスコ200 mLに移し入れ,水を標線まで加える。
− この20 mLをとり,水を加えて液量を約50 mLとし,デキストリン溶液[35.1 a) 4)による。]
5 mL及び指示薬としてジクロロフルオレセインナトリウム溶液(2 g/L)[35.1 a) 3)による。]
3,4滴を加え,0.1 mol/L硝酸銀溶液で滴定し,黄緑の蛍光が消え,僅かに赤くなるときを終
点とする。次の式によって0.1 mol/L硝酸銀溶液のファクター(f)を算出する。
844
005
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 塩化ナトリウムの質量(g)
b: 塩化ナトリウムの純度(質量分率%)
x: 滴定に要した0.1 mol/L硝酸銀溶液量(mL)
0.005 844: 0.1 mol/L硝酸銀溶液1 mLに相当する塩化ナトリウムの質
量(g)
7) シアン化物イオン標準液(CN− 1 mg/mL) JIS K 8443に規定するシアン化カリウム0.63 gを少量
の水に溶かし,水酸化ナトリウム溶液(20 g/L)2.5 mLを加え,水で250 mLとする。この溶液は使
用時に調製し,その濃度は,次の方法で求める。
この溶液100 mLをとり,指示薬としてp-ジメチルアミノベンジリデンロダニンのアセトン溶液
(0.2 g/L)[JIS K 8495に規定するp-ジメチルアミノベンジリデンロダニン[5-(4-ジメチルアミノ
ベンジリデン)-2-チオキソ-4-チアゾリジノン]20 mgをJIS K 8034に規定するアセトン100 mLに
溶かす。]0.5 mLを加え,0.1 mol/L硝酸銀溶液で滴定し,溶液の色が黄色から赤になったときを終
点とする。次の式によってシアン化物イオン標準液の濃度(CN− mg/mL)を算出する。
100
1
204
.5
×
×
×
=
f
a
C
ここに,
C: シアン化物イオン標準液の濃度(CN− mg/mL)
a: 滴定に要した0.1 mol/L硝酸銀溶液量(mL)
f: 0.1 mol/L硝酸銀溶液のファクター
5.204: 0.1 mol/L硝酸銀溶液1 mLに相当するシアン化物イオンの質
量(mg)
8) シアン化物イオン標準液(CN− 1 μg/mL) シアン化物イオン標準液(CN− 1 mg/mL)10 mLを全
135
K 0102:2016
量フラスコ1 000 mLにとり,水酸化ナトリウム溶液(20 g/L)100 mLを加えた後,水を標線まで加
える。その10 mLを全量フラスコ100 mLにとり,水を標線まで加える。使用時に調製する。この
溶液の濃度は,シアン化物イオン標準液(CN− 1 mg/mL)の濃度から算出する。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 38.1の前処理で得られたシアン化物イオン溶液から10 mL(CN−として0.5〜9 μgを含む。)を全量
フラスコ50 mLにとる。
2) 指示薬としてフェノールフタレイン溶液(5 g/L)1滴を加え,静かに振り混ぜながら溶液の紅色が
消えるまで酢酸(1+8)を滴加する。
3) りん酸塩緩衝液(pH6.8)10 mLを加え(9),密栓して静かに振り混ぜる。
4) これにクロラミンT溶液(10 g/L)0.25 mLを加え,直ちに密栓して静かに振り混ぜ,約5分間放置
する。
5) ピリジン-ピラゾロン溶液15 mLを加え,更に水を標線まで加え,密栓して静かに振り混ぜる。
6) 約25 ℃の水浴中に約30分間(10)浸し,溶液の色がうすい紅から紫を経て安定な青になるまで発色(11)
させる。
7) 溶液の一部を吸収セルに移し,波長620 nm付近の吸光度を測定する。
8) 空試験として水10 mLを全量フラスコ50 mLにとり,3)〜7)の操作を行って吸光度を測定し,試料
について得た吸光度を補正する。
9) 検量線からシアン化物イオンの量を求め,試料中のシアン化物イオンの濃度(CN− mg/L)を算出
する。
注(9) 前処理して得られたシアン化物イオン溶液のpHは約13になっており,この溶液10 mLを中和
するのに必要な酢酸(1+8)は約0.5 mLで,これにりん酸塩緩衝液(pH6.8)10 mLを加える
とpH6.8になる。発色時のpH5〜8の範囲に入らなければならない。
(10) 20 ℃以下では,十分に発色せず,30 ℃以上では発色も早いが退色も早くなる。
(11) この条件で発色した場合は,発色後約1時間は安定である。
d) 検量線 検量線の作成は,次による。
1) シアン化物イオン標準液(CN− 1 μg/mL)0.5〜9 mLを全量フラスコ50 mLに段階的にとり,水を
加えて約10 mLとする。
2) c) 2)〜8)の操作を行ってシアン化物イオン(CN−)の量と吸光度との関係線を作成する。
38.3 4-ピリジンカルボン酸-ピラゾロン吸光光度法 前処理して得られたシアン化物イオン溶液の一部を
とり,酢酸で中和した後,クロラミンT溶液を加えて塩化シアンとし,これに4-ピリジンカルボン酸-ピ
ラゾロン溶液を加え,生成する青い色の吸光度を測定してシアン化物イオンを定量する。
定量範囲:CN− 0.5〜9 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 酢酸(1+8) JIS K 8355に規定する酢酸を用いて調製する。
2) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
3) りん酸塩緩衝液(pH7.2) JIS K 9020に規定するりん酸水素二ナトリウム17.8 gを水約300 mLに
溶かし,りん酸二水素カリウム溶液(200 g/L)(JIS K 9007に規定するりん酸二水素カリウムを用
いて調製する。)をpH7.2になるまで加え,水で500 mLとする。
136
K 0102:2016
4) クロラミンT溶液(10 g/L) 38.2 a) 4)による。
5) 4-ピリジンカルボン酸-ピラゾロン溶液 JIS K 9548に規定する3-メチル-1-フェニル-5-ピラゾロン
0.3 gを,JIS K 8500に規定するN,N-ジメチルホルムアミド20 mLに溶かす。別に,4-ピリジンカル
ボン酸1.5 gを水酸化ナトリウム溶液(40 g/L)[21. a) 3)による。]約20 mLに溶かし,塩酸(1+10)
(JIS K 8180に規定する塩酸を用いて調製する。)を滴加してpHを約7とする(12)。両液を合わせ,
水を加えて100 mLとする。この溶液は,10 ℃以下の暗所に保存し,20日間以上経過したものは使
用しない。
6) シアン化物イオン標準液(CN− 1 μg/mL) 38.2 a) 8)による。
注(12) この溶液に代え,4-ピリジンカルボン酸ナトリウム1.8 gを水約50 mLに溶かした溶液を用いて
もよい。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 38.1の前処理で得られたシアン化物イオン溶液から10 mL(CN−として0.5〜9 μgを含む。)を全量
フラスコ50 mLにとる。
2) 指示薬としてフェノールフタレイン溶液(5 g/L)1滴を加え,静かに振り混ぜながら酢酸(1+8)
を滴加して中和した後,りん酸塩緩衝液(pH7.2)10 mL (13)を加える。
3) クロラミンT溶液(10 g/L)0.5 mLを加え,約25 ℃の水浴中に約5分間放置する。
4) 4-ピリジンカルボン酸-ピラゾロン溶液10 mLを加え,更に水を標線まで加え,密栓して静かに振り
混ぜた後,約25 ℃の水浴中で約30分間放置する。
5) この一部を吸収セルに移し,波長638 nm付近の吸光度を測定する。
6) 空試験として水10 mLを全量フラスコ50 mLにとり,りん酸塩緩衝液(pH7.2)10 mLを加えた後,
3)〜5)の操作を行って吸光度を測定し,試料について得た吸光度を補正する。
7) 検量線からシアン化物イオンの量を求め,試料中のシアン化物イオンの濃度(CN− mg/L)を算出
する。
注(13) 発色時のpH7〜8の範囲に入らなければならない。
d) 検量線 検量線の作成は,次による。
1) シアン化物イオン標準液(CN− 1 μg/mL)0.5〜9 mLを全量フラスコ50 mLに段階的にとり,水を
加えて約10 mLとする。
2) c) 2)〜6)の操作を行ってシアン化物イオン(CN−)の量と吸光度との関係線を作成する。
38.4 イオン電極法 前処理して得られたシアン化物イオン溶液(pH12〜13)について,シアン化物イオ
ン電極を指示電極として電位を測定し,シアン化物イオンを定量する。
定量範囲:CN− 0.1〜100 mg/L,繰返し精度:5〜20 %
a) 試薬 試薬は,次による。
1) 水酸化ナトリウム溶液(0.1 mol/L) JIS K 8576に規定する水酸化ナトリウム4 gを水に溶かして1
Lとする。
2) シアン化物イオン標準液(CN− 100 mg/L) 38.2 a) 7)のシアン化物イオン標準液(CN− 1 mg/mL)
20 mLを全量フラスコ200 mLにとり,水酸化ナトリウム溶液(0.1 mol/L)を標線まで加える(14)。
使用時に調製する。
この溶液の濃度は,シアン化物イオン標準液(CN− 1 mg/mL)の濃度から算出する。
137
K 0102:2016
3) シアン化物イオン標準液(CN− 10 mg/L) シアン化物イオン標準液(CN− 100 mg/L)20 mLを全
量フラスコ200 mLにとり,水酸化ナトリウム溶液(0.1 mol/L)を標線まで加える(14)。使用時に調
製する。この溶液の濃度は,シアン化物イオン標準液(CN− 100 mg/L)の濃度から算出する。
4) シアン化物イオン標準液(CN− 1 mg/L) シアン化物イオン標準液(CN− 10 mg/L)20 mLを全量
フラスコ200 mLにとり,水酸化ナトリウム溶液(0.1 mol/L)を標線まで加える(14)。使用時に調製
する。この溶液の濃度は,シアン化物イオン標準液(CN− 10 mg/L)の濃度から算出する。
5) シアン化物イオン標準液(CN− 0.1 mg/L) シアン化物イオン標準液(CN− 1 mg/L)20 mLを全量
フラスコ200 mLにとり,水酸化ナトリウム溶液(0.1 mol/L)を標線まで加える(14)。使用時に調製
する。この溶液の濃度は,シアン化物イオン標準液(CN− 1 mg/L)の濃度から算出する。
注(14) 各シアン化物イオン標準液のpHは,約13になる。
b) 器具及び装置 器具及び装置は,次による。
1) 電位差計 34.2 b) 1)による。
2) 指示電極 シアン化物イオン電極
3) 参照電極 34.2 b) 3)による。
4) 測定容器 35.2 b) 4)による。
5) 恒温槽 34.2 b) 5)による。
6) マグネチックスターラー 34.2 b) 6)による。
c) 検量線 検量線の作成は,次による。
1) シアン化物イオン標準液(CN− 0.1 mg/L)100 mLを測定容器にとる。
2) 恒温槽から水を送り,測定容器の溶液を25±0.5 ℃にする。
3) 指示電極(15) (16)と参照電極(17)とを浸し,マグネチックスターラー(18)で泡が電極に触れない程度に強
くかき混ぜる(19)。
4) 液温を確認し,電位差計で電位を測定する(20)。
5) シアン化物イオン標準液(CN− 1 mg/L)100 mL,シアン化物イオン標準液(CN− 10 mg/L)100 mL
及びシアン化物イオン標準液(CN− 100 mg/L)100 mLをそれぞれ測定容器にとる。
6) 2)〜4)の操作を行って電位を測定する(21) (22)。
7) 横軸にシアン化物イオンの濃度の対数を,縦軸に電位をとり,シアン化物イオンの濃度(CN− mg/L)
と電位との関係線を作成する(21)。
注(15) 指示電極(シアン化物イオン電極)は,使用時にシアン化物イオン標準液(CN− 0.1 mg/L)に
浸し,指示値が安定してから使用する。シアン化物イオン電極の応答時間は,液温10〜30 ℃
の場合,シアン化物イオンの濃度が0.1 mg/Lで約1分間,1 mg/L以上であれば約30秒間であ
る。
(16) 34.の注(13)による。
(17) 34.の注(14)による。
(18) 34.の注(15)による。
(19) 34.の注(16)による。
(20) シアン化物イオン電極には,よう化銀を用いたものが多いので,直射日光を受けると電位が大
幅に変動して,正の誤差を与える。室内照明の影響は少ない。
(21) シアン化物イオン標準液(CN− 0.1 mg/L)とシアン化物イオン標準液(CN− 10 mg/L)との電
位の差は,110〜120 mV(25 ℃)の範囲に入り,シアン化物イオンの濃度0.1〜100 mg/Lの間
138
K 0102:2016
の検量線は直線になる。
注(22) 34.の注(19)による。
d) 操作 操作は,次による。
1) 38.1の前処理で得られたシアン化物イオン溶液から100 mLを測定容器にとる。
2) c) 2)〜4)の操作を行って(22),検量線からシアン化物イオンの濃度を求め,試料中のシアン化物イオ
ンの濃度(CN− mg/L)を算出する。
備考 11. イオン濃度計の場合には,シアン化物イオン標準液(CN− 0.1 mg/L)及びシアン化物イオン
標準液(CN− 10 mg/L)を用い,c) 2)〜4)の操作を行ってイオン濃度計の指示値をCN− 0.1
mg/L及びCN− 10 mg/Lになるように調節する。さらに,その他のシアン化物イオン標準液
(CN− 1 mg/L)及びシアン化物イオン標準液(CN− 100 mg/L)を用いて,イオン濃度計の
指示を確認する。
12. 硫化物イオン及びメルカプト酢酸(チオグリコール酸)の妨害は,前処理で除く。亜硫酸イ
オンは,シアン化物イオンの103倍以内であれば妨害しないので,備考6.の酸化処理を省略
することができる。ホルムアルデヒドは負の誤差を与える。
主な共存物質の許容限度を最大比率で次に示す。
Cl−,F−,NO3−,CrO42−,K+,Na+: 104
Br−,SCN−,HCO3−,CO32−,SO32−,SO42−,PO43−: 103
S2O32−,Ag+: 10
I−: 0.1
13. イオン電極による電位差滴定法 38.1の前処理で得られたシアン化物イオン溶液のうち100
mLを測定容器にとり,指示電極(シアン化物イオン電極又は銀イオン電極)を用い,c) 2)
〜4)の操作に準じて電位を測定しながら1〜100 mmol/L硝酸銀溶液で滴定し,滴定曲線を作
図する。滴定曲線から滴定終点を求め,シアン化物イオンの濃度を算出する。100 mmol/L硝
酸銀溶液1 mLは,シアン化物イオン5.204 mgに相当する。
38.5 流れ分析法 試料中のシアン化合物イオンを,38.3と同様な原理で発色させる流れ分析法によって
定量する。試料を,38.1.1.1,38.1.1.2又は38.1.2の操作で前処理した後に適用する。この場合は,試料中
に懸濁物が含まれても,蒸留前処理で除去可能である。
定量範囲:CN− 0.01〜1 mg/L,繰返し精度:10 %以下
試験操作などは,JIS K 0170-9で規定されたシアン化物に関する規定による。ただし,JIS K 0170-9の
7.3.3[蒸留(pH3.8)−4-ピリジンカルボン酸・ジメチルバルビツール酸発色CFA法]及び7.3.4[ガス拡
散(pH3.8)−4-ピリジンカルボン酸・ジメチルバルビツール酸発色CFA法]の方法は除く。
39. 硫化物イオン(S2−) 硫化物イオンの定量には,メチレンブルー吸光光度法又はよう素滴定法を適用
する。
硫化物イオンは不安定で,酸化されたり,硫化水素として空気中に散逸したりするので,試料採取後直
ちに試験を行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く試験する。
なお,この試験方法の備考3.に示す溶存の硫化物については,1992年に第1版として発行されたISO
10530との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
139
K 0102:2016
(修正している),NEQ(同等でない)とする。
ISO 10530:1992,Water quality−Determination of dissolved sulfide−Photometric method using
methylene blue(MOD)
39.1 メチレンブルー吸光光度法 硫化物イオンが,鉄(III)イオンの存在の下でN,N-ジメチル-p-フェニ
レンジアミンと反応して生成するメチレンブルー[3,7-ビス(ジメチルアミノ)フェノチアジン-5-イウム]
の吸光度を測定して硫化物イオンを定量する。
定量範囲:S2− 5〜40 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 硫酸(1+1) 5.4 a) 2)による。
2) N,N-ジメチル-p-フェニレンジアンモニウム溶液 JIS K 8193に規定する二塩化N,N-ジメチル-p-フ
ェニレンジアンモニウム0.8 gに硫酸(1+1)を加えて100 mLとする。使用時に調製する。
3) 塩化鉄(III)溶液 JIS K 8142に規定する塩化鉄(III)六水和物10 gを水に溶かして100 mLとす
る。
4) りん酸水素二アンモニウム溶液(400 g/L) JIS K 9016に規定するりん酸水素二アンモニウム40 g
を水に溶かして100 mLとする。
5) 硫化物イオン標準液(S2− 1 mg/mL) JIS K 8949に規定する硫化ナトリウム九水和物の結晶7.6 g
をとり,少量の水で表面を洗い,これをろ紙上にとって水を除いた後,2. n) 1)の溶存酸素を含まな
い水に溶かして1 Lとする。気密容器に保存し,使用時に標定する。
標定 標定は,次による。
− よう素溶液(50 mmol/L)(1) 20 mLをとり,共栓三角フラスコ300 mLに入れ,塩酸(1+1)
[24.1 a) 1) による。]0.5 mLを加える。次に,この硫化物イオン標準液20 mLを全量ピペッ
トでとり,ピペットの先端をこのよう素溶液中に入れて加える(2)。直ちに密栓して振り混ぜ
て数分間放置する。
− 0.1 mol/Lチオ硫酸ナトリウム溶液(3)で滴定し,溶液の黄色が薄くなってから,指示薬として
でんぷん溶液(10 g/L)(4) 1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴定す
る。
− 別に,よう素溶液(50 mmol/L)20 mLを共栓三角フラスコ300 mLにとり,塩酸(1+1)0.2
mLを加えた後,同様に0.1 mol/Lチオ硫酸ナトリウム溶液で滴定する。次の式によって硫化
物イオン標準液の濃度(S2− mg/mL)を算出する。
(
)
603
.1
20
1×
×
×
−
=
f
a
b
S
ここに,
S: 硫化物イオン標準液の濃度(S2− mg/L)
a: 滴定に要した0.1 mol/Lチオ硫酸ナトリウム溶液量(mL)
b: よう素溶液(50 mmol/L)20 mLに相当する0.1 mol/Lチオ硫
酸ナトリウム溶液量(mL)
f: 0.1 mol/Lチオ硫酸ナトリウム溶液のファクター
1.603: 0.1 mol/Lチオ硫酸ナトリウム溶液1 mLに相当する硫化物イ
オンの質量(mg)
6) 硫化物イオン標準液(S2− 10 μg/mL) 使用時に硫化物イオン標準液(S2− 1 mg/mL)10 mLを全量
フラスコ1 000 mLにとり,2. n) 1)の溶存酸素を含まない水を標線まで加える。ただし,この溶液の
濃度は,5)の硫化物イオン標準液(S2− 1 mg/mL)の濃度から算出する。
140
K 0102:2016
注(1) JIS K 8913に規定するよう化カリウム45 gを水約100 mLに溶かし,これにJIS K 8920に規定
するよう素13 gを加えて溶かし,水を加えて1 Lとする。着色ガラス瓶に保存する。
注(2) 塩酸酸性にしたよう素溶液に硫化物イオン標準液(S2− 1 mg/mL)を加える。
(3) 19. a) 8)による。
(4) 19. a) 5)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料の適量(5) (6)(S2−として5〜40 μgを含む。)をメスシリンダー(有栓形)50 mLにとり,2. n) 1)
の溶存酸素を含まない水を加えて約40 mLとした後,硫酸(1+1)1 mL (7)を加え,緩やかに振り混
ぜる。
2) N,N-ジメチル-p-フェニレンジアンモニウム溶液0.5 mLを加えて振り混ぜた後,塩化鉄(III)溶液1 mL
を加え,再び振り混ぜ,約1分間放置する。
3) りん酸水素二アンモニウム溶液(400 g/L)1.5 mLを加えた後,メスシリンダー(有栓形)50 mLの
標線まで水を加え,振り混ぜた後,約5分間放置する。
4) 別に,メスシリンダー(有栓形)50 mLに硫酸(1+1)1 mLをとり,水を加えて約40 mLとし,緩
やかに振り混ぜた後,2)及び3)の操作を行う。
5) 3)の溶液を吸収セルにとり,4)の溶液を対照液として波長670 nm付近の吸光度を測定する。
6) 検量線から硫化物イオンの量を求め,試料中の硫化物イオンの濃度(S2− mg/L)を算出する。
注(5) 溶存の硫化物イオンを定量する場合には,備考3.によるろ過操作を行う。
(6) 試料採取後,直ちに試験が行えない場合には,3.3によって試料の保存処理を行うか,備考2.
による硫化亜鉛として固定する保存処理を行う。備考2.による保存処理を行った場合には,39.2
c)によって,硫化亜鉛から硫化水素として分離した後,試験操作を行う。ただし,この場合の
硫化水素の吸収には,酢酸亜鉛溶液に代え,水酸化ナトリウム溶液(20 mmol/L)[38.4 a) 1)の
水酸化ナトリウム溶液(0.1 mol/L)を水で5倍に薄める。]を用いる。
(7) 発色の強さは,pH0.4〜1.0のときに最高になる。試料がアルカリ性の場合には,硫酸(1+1)
で中和した後,更に硫酸(1+1)1 mLを加える。
d) 検量線 検量線の作成は,次による。
1) 硫化物イオン標準液(S2− 10 μg/mL)0.5〜4 mLをメスシリンダー(有栓形)50 mLに段階的にとる。
2) c) 1)〜5)の操作を行って硫化物イオン(S2−)の量と吸光度との関係線を作成する。
備考 1. この方法は,共存物質の妨害が比較的少ないが,酸化性物質及び還元性物質が妨害する。亜
硫酸イオン及びチオ硫酸イオンは,それぞれ10 mg/L以上で妨害する。チオシアン酸イオン
は,少量でも妨害する。
2. 硫化物イオンを硫化亜鉛とする固定及び固定後の定量操作は,次による。
JIS K 8953に規定する硫酸亜鉛七水和物20 gを水100 mLに溶かした溶液と炭酸ナトリウ
ム溶液(100 g/L)とを用意し,使用時にその等体積ずつを混合する。その後,塩基性炭酸亜
鉛の懸濁液を調製する。
試料容器には,21. b) 1)の培養瓶を用い,気泡が残らないように注意して試料を採取した後,
塩基性炭酸亜鉛の懸濁液を試料100 mLにつき約2 mLの割合で試料の液面下に加え,気泡が
残らないように注意して密栓し,転倒して混ぜ合わせ,硫化物イオンを硫化亜鉛の沈殿とし
141
K 0102:2016
て固定する。
この沈殿をろ紙5種Cでろ過するか又は遠心分離によって分離し,沈殿について注(6)に示
す試験操作を行う。
塩基性炭酸亜鉛の懸濁液10 mLは,硫化物イオン約50 mgを固定できる。また,この操作
によって試料中に共存する亜硫酸イオン,チオ硫酸イオンなどと分離定量できる。
なお,この方法を行った場合は,試料中の硫化物イオンの濃度の算出には,試料量は,培
養瓶の容量(mL)から塩基性炭酸亜鉛の懸濁液の添加量(mL)を差し引いた値を用いる。
備考 3. 溶存の硫化物を定量する場合の,試料のろ過操作は,次のいずれかによる。
1) 試料採取後,直ちにろ紙5種C(又はろ紙6種)を用いてろ過し,初めのろ液50 mLを
捨て,その後のろ液を用いる。
2) アスコルビン酸溶液[JIS K 9502に規定するL(+)-アスコルビン酸10 gを水90 mL
に溶かし,水酸化ナトリウム(10 mol/L)(JIS K 8576に規定する水酸化ナトリウムを用
いて調製する。)でpH10±0.1に調製したもの。]5 mLを全量フラスコ50 mLにとる。3
リング付きピストンシリンジ(容量50 mL)に試料を引き込み,孔径0.45 μmのろ過材
を用いた一方向ろ過器を取り付け(図39.1に例を示す。),全量フラスコ50 mLの標線ま
でろ過する。
なお,この操作を行った場合は,c)の操作に用いた試料の量として,c) 1)で分取した
試料の適量に を乗じた値を用いる。39.2のよう素滴定法に用いる場合は,39.2 d)の操
作に用いた試料の量として,39.2 c) 2)で分取した試料の適量に を乗じた値を用いる。
硫化物イオンを分離して定量するには,このろ液について39.2 c)の分離操作を行う。
ただし,このメチレンブルー吸光光度法の場合の硫化水素の吸収には,酢酸亜鉛溶液に
代え,水酸化ナトリウム溶液(20 mmol/L)[注(6)による。]を用いる(定量を39.2のよ
う素滴定法による場合は,酢酸亜鉛溶液を用いる。)。
3) アスコルビン酸溶液[2)による。]5 mLを全量フラスコ50 mLにとる。全量フラスコと
加圧ろ過器(図39.2に例を示す。)に窒素(JIS K 1107に規定する窒素2級)を約10分
間通して空気を追い出す。試料をろ過器に満たし,0.2 MPaを超えない窒素圧力で,全
量フラスコの標線までろ過する(*) (**)。この方法はろ過しにくい試料に適用できる。
この操作を行った場合の試料の量の補正は,2)と同じ。
注(*) 加圧ろ過器と全量フラスコとの接続は,空気の出入りができるだけ少なくなるよう
にする。
(**) ろ過時間は,5分間を超えないようにする。
50
45
50
45
142
K 0102:2016
単位 mm
図39.1 3リング付きピストンシリンジの例
図39.2 加圧ろ過装置の例
39.2 よう素滴定法 硫化物イオン又は硫化物を含む溶液に一定過剰量のよう素溶液と塩酸とを加え,で
んぷん溶液を指示薬として,残ったよう素をチオ硫酸ナトリウム溶液で滴定して硫化物イオンを定量する。
定量範囲:S2− 0.2 mg以上
備考 4. 試料に直接滴定操作を行うと,亜硫酸イオン,チオ硫酸イオンなどの還元性物質も硫化物イ
オンと同様に反応して滴定されるので,あらかじめ硫化物イオンの分離を行ってから操作す
る。
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 硫酸(1+1) 5.4 a) 2)による。
3) 塩化ヒドロキシルアンモニウム溶液(100 g/L) JIS K 8201に規定する塩化ヒドロキシルアンモニ
ウム10 gを水に溶かして100 mLとする。
4) 酢酸亜鉛溶液 JIS K 8356に規定する酢酸亜鉛二水和物24 gを水に溶かして100 mLとする。
5) でんぷん溶液(10 g/L) 19. a) 5)による。
6) よう素溶液(5 mmol/L) 注(1)のよう素溶液(50 mmol/L)50 mLをとり,水で500 mLとする。着
色ガラス瓶に保存する。
7) 10 mmol/Lチオ硫酸ナトリウム溶液 19. a) 9)による。
8) 窒素 JIS K 1107に規定する窒素2級
b) 装置 装置は,次による。
1) 硫化水素発生及び吸収装置 図39.3に例を示す(8)。
143
K 0102:2016
単位 mm
A:
B:
C:
D:
E:
共通すり合わせ丸底フラスコ300 mL又は1 000 mL
連結導入管
枝付連結管
注入漏斗
すり合わせコック
F:
G1,G2:
H:
I:
トラップ球(ケルダール球)
共通すり合わせ三角フラスコ100 mL
ゴム管
共通すり合わせ
図39.3 硫化水素発生及び吸収装置の例
注(8) フラスコ(A)は,試料から直接硫化水素を発生させる場合には,1 000 mLのものを用い,硫
化亜鉛として固定したものから硫化水素を発生させる場合には,300 mLのものを用いる。
c) 分離操作 分離操作は,次による。
1) 酢酸亜鉛溶液5 mLを水で100 mLに薄め,三角フラスコ(G1)及び(G2)にそれぞれ50 mLずつ入
れる。
2) 試料(5) (9)の適量(通常500 mL)をフラスコ(A)に入れ,鉄(III)が含まれている場合には,塩化
ヒドロキシルアンモニウム溶液(100 g/L)1 mL又はJIS K 9502に規定するL(+)-アスコルビン
酸0.1 gを加える。次に,上部の注入漏斗から硫酸(1+1)100 mLを加える。
3) フラスコ(A)を約50 ℃に加熱し,窒素又は二酸化炭素をゆっくりと約20分間通し,硫化水素を
追い出して酢酸亜鉛溶液に吸収させる。
注(9) 備考2. によって硫化亜鉛として固定し,沈殿を分離したものから硫化水素を発生させる場合に
144
K 0102:2016
は,沈殿をろ紙ごと水でフラスコ(A)に入れ,水約50 mLを加える。注入漏斗から塩酸(1+
1)[a) 1)の塩酸を用いて調製する。]50 mLを加え,続いて3)の操作を行う。
備考 5. 泡立ちが激しい試料の場合には,ジフェニルエーテルなどの消泡剤を加えるとよい。
6. 二酸化炭素を約1時間通じれば,加熱しなくても硫化水素を追い出すことができる。
7. 試料から直接硫化水素を発生させる方法では,チオ硫酸イオンのほか,鉄,亜鉛などの金属
元素から硫化物イオンを分離できるが,亜硫酸イオンの分離は完全でない。注(9)の方法では,
亜硫酸イオン及びチオ硫酸イオンは,あらかじめ分離されている。
d) 操作 操作は,次による。
1) 硫化水素を吸収した図39.3の三角フラスコ(G1)及び(G2)によう素溶液(5 mmol/L)を過剰にな
るように一定量を加える。大部分の硫化水素は三角フラスコ(G1)に吸収されているから,よう素
溶液(5 mmol/L)の大部分は三角フラスコ(G1)に加えるようにする。
2) 三角フラスコ(G1)及び(G2)にそれぞれ塩酸(10) 2.5 mLを加えて振り混ぜた後,内容物を三角フ
ラスコ500 mLに移し入れる。水で三角フラスコ(G1)及び(G2)をよく洗い,洗液も合わせる。
3) 10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,よう素の黄色が薄くなったら,指示薬としてでんぷ
ん溶液(10 g/L)約1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
4) 空試験として三角フラスコ500 mLに水100 mLをとり,酢酸亜鉛溶液5 mL及び試験に用いたのと
同量のよう素溶液(5 mmol/L)を加え,更に塩酸5 mLを加えて3)の操作を行う。
5) 次の式によって試料中の硫化物イオンの濃度(S2− mg/L)を算出する。
(
)
3
160
.0
000
1
×
×
×
−
=
V
f
a
b
S
ここに,
S: 硫化物イオンの濃度(S2− mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
b: 空試験に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター
V: 試料量(mL)
0.160 3: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する硫化物
イオンの質量(mg)
注(10) よう素溶液(5 mmol/L)を加えてから塩酸を加える。操作の順序を逆に行うと硫化水素として
損失するおそれがある。
備考 8. 妨害物質の少ない試料で,備考2.によってあらかじめ硫化亜鉛として固定した場合には,c)
の分離操作を行わないで滴定することができる。この場合の操作は,次による。
1) 固定で生じた沈殿をろ紙5種Cでろ別して水で洗浄し,ろ紙とともに三角フラスコ300
mLに移し,水約100 mLを加える。
2) これによう素溶液(5 mmol/L)の一定量を加え,次に,塩酸5 mLを加えてよく振り混
ぜて反応させ,次に,d) 3)〜5)を行って試料中の硫化物イオンの濃度を求める。
3) なお,硫化亜鉛として固定したときの沈殿が着色している場合には,金属元素の共存が
考えられ,滴定の妨害となることが多いから,注(9)の分離操作によって硫化水素として
分離する。
9. 備考3. 2)による。
40. 亜硫酸イオン(SO32−) 亜硫酸イオンの定量には,よう素滴定法を適用する。
145
K 0102:2016
40.1 よう素滴定法 一定量のよう素溶液に酢酸-酢酸ナトリウム緩衝液を加えた後,試料を加え,次に,
過剰のよう素をでんぷん溶液を指示薬としてチオ硫酸ナトリウム溶液で滴定する。別に,同量の試料をと
り,酸性として煮沸して亜硫酸イオンを二酸化硫黄として追い出した後,同一の滴定操作を行い,これを
空試験値として,チオ硫酸イオンなどの還元性物質の影響を補正する。亜硫酸イオンは空気によって酸化
されるので,試験は試料採取後,直ちに行う。
定量範囲:SO32− 0.2 mg以上
a) 試薬 試薬は,次による。
1) 硫酸(1+35) 水35容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する
硫酸1容を徐々に加える。
2) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
3) 酢酸-酢酸ナトリウム緩衝液(pH3.9) JIS K 8371に規定する酢酸ナトリウム三水和物75 gを酢酸
(1+2)(JIS K 8355に規定する酢酸を用いて調製する。)500 mLに溶かす。
4) でんぷん溶液(10 g/L) 19. a) 5)による。
5) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
6) よう素溶液(5 mmol/L) 39.2 a) 6)による。
7) 10 mmol/Lチオ硫酸ナトリウム溶液 19. a) 9)による。
8) 窒素 JIS K 1107に規定する窒素2級
b) 操作 操作は,次による。
1) 三角フラスコによう素溶液(5 mmol/L)20 mLをとり,これに酢酸-酢酸ナトリウム緩衝液(pH3.9)
10 mLを加える。
2) 試料の適量(SO32−として0.2〜10 mgを含む。)をピペットでとり,先端をフラスコの溶液中に浸し,
静かに注入して混ぜ合わせる。
3) 10 mmol/Lチオ硫酸ナトリウム溶液で滴定し,溶液の黄色が薄くなったら,指示薬としてでんぷん
溶液(10 g/L)約1 mLを加え,生じたよう素でんぷんの青い色が消えるまで滴定する。
4) 空試験として試験に用いたのと同量の試料を三角フラスコにとり,硫酸(1+35)6〜7 mLを加え,
窒素(1)を液面に通しながら数分間静かに煮沸して,二酸化硫黄を追い出す。窒素を通したまま冷却
する。
5) 冷却後,指示薬としてフェノールフタレイン溶液(5 g/L)2,3滴を加え,水酸化ナトリウム溶液(40
g/L)で中和する。これに,酢酸-酢酸ナトリウム緩衝液(pH3.9)10 mLを加え,静かに振り混ぜた
後,よう素溶液(5 mmol/L)20 mLを加え,3)の操作によって10 mmol/Lチオ硫酸ナトリウム溶液
で滴定する。
6) 次の式によって試料中の亜硫酸イオンの濃度(SO32− mg/L)を算出する。
(
)
3
400
.0
000
1
×
×
×
−
=
V
f
a
b
S
ここに,
S: 亜硫酸イオンの濃度(SO32− mg/L)
a: 滴定に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
b: 空試験に要した10 mmol/Lチオ硫酸ナトリウム溶液量(mL)
f: 10 mmol/Lチオ硫酸ナトリウム溶液のファクター
V: 試料量(mL)
0.400 3: 10 mmol/Lチオ硫酸ナトリウム溶液1 mLに相当する亜硫酸
イオンの質量(mg)
146
K 0102:2016
注(1) 窒素中の酸素の除去は,水酸化ナトリウム溶液(600 g/L)(JIS K 8576に規定する水酸化ナトリ
ウム60 gを水に溶かして100 mLとする。)75 mLと,水15 mLにJIS K 8780に規定するピロガ
ロール(1,2,3-ベンゼントリオール)5 gを溶かした溶液との混液をガス洗浄瓶に入れ,通気し
て行う。
備考 1. 硫化物イオンは,よう素を消費して妨害し,b) 4)及び5)の空試験を行っても補正することが
できない。硫化物イオンの妨害を除くには39.の備考2.によって硫化物イオンを硫化亜鉛とし
て固定後,ろ紙5種Cでろ過(又は遠心分離)し,そのろ液から亜硫酸イオンを定量する。
2. 鉄(III)イオン及び銅(II)イオンは,よう化物イオンを酸化して妨害する。
3. 配管又は装置から試料を採取する場合には,図40.1に示すような試料採取器を用いると便利
である。この場合には,次のように操作して試料を採取し試験する。
− 2個の試料採取器の下端に軟質塩化ビニル管とY字管とで試料採取用配管に接続する。
試料採取器の出口を上方に向け,出口には何も接続しない。試料の流量は試料採取用
配管出口で調節する。試料の温度が高い場合には,室温より1〜2 ℃低くなるように冷
却する。
− 2個の試料採取器とも同時に8〜12秒間程度で満たされるように流量を調節し,配管及
び試料採取器が試料で十分に洗浄され,内容物が完全に入れ換わるように連続して試料
を十分に流す。
− 次に,2個の試料採取器の上端にあるコックを閉じ,直ちに下端のコックも閉じ,接続
管を外し,試料採取器を逆にして気泡がないことを確かめる。もし,気泡があれば両方
とも試料を捨てて新しく採取する。2個の試料採取器のうち1個を試験用に,他を空試
験用とする。
− あらかじめ,よう素溶液(5 mmol/L)20 mLをビーカーにとり,これに酢酸-酢酸ナトリ
ウム緩衝液(pH3.9)10 mLを入れ,試験用の試料採取器の両端にある足の部分の試料を
捨て,水で洗い,両方の足にJIS K 8102に規定するエタノール(95)2 mLずつを満た
す。
− 試料採取器の下方のエタノールをできるだけ捨てないようにして,試料採取器の足をビ
ーカーの溶液中に浸して上方のコックも開き,静かに下方のコックも開きながら試料を
注入する。
− 以下,b) 3)以降によって試験する。
147
K 0102:2016
単位 mm
A,B:すり合わせコック
図40.1 試料採取器の例
41. 硫酸イオン(SO42−) 硫酸イオンの定量には,クロム酸バリウム吸光光度法,重量法又はイオンクロ
マトグラフ法を適用する。
41.1 クロム酸バリウム吸光光度法 試料にクロム酸バリウムの酸懸濁液を加えて硫酸バリウムを沈殿さ
せ,次に,カルシウムイオンを含むアンモニア水とエタノールとを加え,過剰のクロム酸バリウムを沈殿
させ,遠心分離する。
硫酸イオンと置換して生じたクロム酸イオンの黄色の吸光度を測定して硫酸イオンを定量する。
定量範囲:SO42− 50〜500 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) クロム酸バリウムの酸懸濁液 クロム酸バリウム2.5 gを,酢酸(1+15)(JIS K 8355に規定する酢
酸を用いて調製する。)100 mLと塩酸(1+500)(JIS K 8180に規定する塩酸を用いて調製する。)
100 mLとの混合溶液200 mLに加え,よく振り混ぜて懸濁液を作り,ポリエチレン瓶に保存する。
クロム酸バリウムは,次の方法で調製する。
− JIS K 8312に規定するクロム酸カリウム8 gを水約800 mLに溶かし,酢酸(6 mol/L)(JIS K 8355
に規定する酢酸35 mLを水に溶かして100 mLとする。)10 mLを加えて約70 ℃に温める。
− この溶液を激しくかき混ぜながら,約70 ℃に温めた塩化バリウム溶液(JIS K 8155に規定す
る塩化バリウム二水和物10 gを水に溶かして100 mLとする。)100 mLを滴加してクロム酸バ
リウムを沈殿させ,放置する。上澄み液を捨て,温水約500 mLずつで4回デカンテーション
する。
− 沈殿を遠沈管に移し,遠心分離によって冷水で2,3回洗浄する。この沈殿をガラスろ過器に移
148
K 0102:2016
して吸引ろ過し,105〜110 ℃で約1時間加熱し,デシケーター中で放冷した後,めのう乳鉢で
すり潰す。
2) カルシウムを含むアンモニア水 JIS K 8122に規定する塩化カルシウム二水和物1.85 gを,アンモ
ニア水(3+4)(JIS K 8085に規定するアンモニア水を用いて調製する。)500 mLに溶かし,ポリエ
チレン瓶に入れ,空気中の二酸化炭素が入らないような方法で保存する。図41.1のように保存する
と便利である。
A:
B:
C:
D:
E:
F:
ポリエチレン瓶500 mL
ビュレット(枝付き)5 mL
一方コック
二酸化炭素吸収管
(ソーダ石灰を詰める。)
ゴム栓
ゴム管
図41.1 カルシウムを含むアンモニア水の保存の例
3) エタノール(95) JIS K 8102に規定するもの。
4) 硫酸イオン標準液(SO42− 1 mg/mL) 35.3 a) 16)による。
5) 硫酸イオン標準液(SO42− 0.1 mg/mL) 硫酸イオン標準液(SO42− 1 mg/mL)10 mLをとり,全量
フラスコ100 mLに入れ,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 遠心分離器
2) 遠沈管 共栓付き20〜30 mL
3) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料の適量(SO42−として50〜500 μgを含む。)を遠沈管にとり,水で10 mLとし,20〜30 ℃(1)に
保つ。これに20〜30 ℃に保ったクロム酸バリウムの酸懸濁液4 mLを加えて振り混ぜ,2〜3分間
放置する。
149
K 0102:2016
2) カルシウムを含むアンモニア水の上澄み液1 mLを図41.1のビュレット又はピペットで静かに加え
て混ぜ,更にエタノール(95)10 mLを加えて1分間振り混ぜた後,約10分間放置する。
3) これを遠心分離して,その上澄み液を吸収セルにとり,波長370 nm付近の吸光度を測定する。
4) 空試験として水10 mLをとり,1)〜3)の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
5) 検量線から硫酸イオンの量を求め,試料中の硫酸イオンの濃度(SO42− mg/L)を算出する。
注(1) 反応時の液温が20〜30 ℃の範囲で同一の検量線が得られる。
d) 検量線 検量線の作成は,次による。
1) 硫酸イオン標準液(SO42− 0.1 mg/mL)0.5〜5 mLを遠沈管に段階的にとる。
2) 水で10 mLとした後,c) 1)〜4)の操作を行って硫酸イオン(SO42−)の量と吸光度との関係線を作成
する。
備考 1. 硝酸,炭酸及び炭酸水素の各イオンは,それぞれ50 mg/L以上共存すると妨害する。りん酸,
ひ酸,セレン酸及びバナジン酸の各イオン並びに鉛は,微量でも妨害する。
炭酸イオン及び炭酸水素イオンの除去は,塩酸を加えて煮沸して行う。あらかじめ,試料
の一部をとり,メチルレッド-ブロモクレゾールグリーン混合液を指示薬として,塩酸(1+
100)(JIS K 8180に規定する塩酸を用いて調製する。)で中和して中和に必要な塩酸(1+100)
の量を求め,試料に同量の塩酸(1+100)を加える。塩酸が過剰にならないようにする。
りん酸イオンを含む場合には,試料10 mLに,塩化カルシウム溶液(11 g/L)(JIS K 8123
に規定する塩化カルシウムを用いて調製する。)2 mLを加える。次に水酸化ナトリウム溶液
(10 g/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調製する。)と炭酸ナトリウム
溶液(13 g/L)(JIS K 8625に規定する炭酸ナトリウムを用いて調製する。)との等体積混合
液1 mLを加える。約10分間放置後,遠心分離し,その上澄み液の一定量をとり,塩酸(1
+100)で中和した後,沸騰水浴中で10分間加熱して二酸化炭素を除く。冷却後,水で20 mL
に薄め,その10 mLをとり,c)の操作によって硫酸イオンを定量する。このりん酸イオンの
除去操作を行った場合の検量線は,硫酸イオン標準液について同様に操作して作成する。
41.2 重量法 硫酸イオンを硫酸バリウムとして沈殿させ,その質量をはかって硫酸イオンを定量する。
定量範囲:SO42− 10 mg以上,繰返し精度:2 %
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 塩酸(1+50) JIS K 8180に規定する塩酸を用いて調製する。
3) 塩化バリウム溶液(100 g/L) JIS K 8155に規定する塩化バリウム二水和物11.7 gを水に溶かして
100 mLとする。
4) 硝酸銀溶液(10 g/L) JIS K 8550に規定する硝酸銀1 gを水に溶かして100 mLとする。
b) 操作 操作は,次による。
1) 試料の適量(SO42−として10 mg以上を含む。)を磁器蒸発皿にとり,塩酸3 mLを加えた後,沸騰
水浴上で蒸発乾固し,更に約20分間加熱する。
2) 放冷後,塩酸2 mLで湿らせ,次に,温水20〜30 mLを加え,数分間加熱した後,ろ紙5種Bでろ
過し,塩酸(1+50)で数回洗う。
3) ろ液に水を加えて100 mLとし,沸騰水浴上で加熱し,絶えずかき混ぜながら,これに温塩化バリ
ウム溶液(100 g/L)を滴加し,沈殿が生じなくなったら,更に添加量の20〜50 %を過剰に加える。
150
K 0102:2016
4) 沸騰水浴上で20〜30分間加熱した後,3〜4時間放置する。
5) ろ紙5種C(又は6種)を用いてろ過し,ろ液に塩化物イオンの反応を認めなくなるまで水で洗う
[硝酸銀溶液(10 g/L)で確かめる。]。
6) 沈殿はろ紙とともに,あらかじめ800 ℃で恒量とした磁器るつぼに入れ,乾燥後,徐々に加熱して
ろ紙を一旦炭化した後,灰化する。
7) 引き続き,約800 ℃で約30分間加熱し,デシケーター中で放冷した後,その質量をはかる。
8) 7)の操作を繰り返して恒量とする。
9) 次の式によって試料中の硫酸イオンの濃度(SO42− mg/L)を算出する。
6
411
.0
000
1
×
×
=
V
a
S
ここに,
S: 硫酸イオンの濃度(SO42− mg/L)
a: 硫酸バリウムの質量(mg)
V: 試料量(mL)
0.411 6: 硫酸バリウム1 mgに相当する硫酸イオンの質量(mg)
41.3 イオンクロマトグラフ法 35.3による。
42. アンモニウムイオン(NH4+) アンモニウムイオンの定量には,インドフェノール青吸光光度法,中
和滴定法,イオン電極法,イオンクロマトグラフ法又はインドフェノール青発色による流れ分析法を適用
する。
アンモニウムイオンは変化しやすいから,試験は試料採取後,直ちに行う。直ちに行えない場合には,
3.3によって保存し,できるだけ早く試験する。
なお,蒸留法及び中和滴定法は,1984年に第1版として発行されたISO 5664,イオン電極法は,1984
年に第1版として発行されたISO 6778,イオンクロマトグラフ法は,1998年に第1版として発行された
ISO 14911,流れ分析法は,2005年に第2版として発行されたISO 11732との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 5664:1984,Water quality−Determination of ammonium−Distillation and titration method(MOD)
ISO 6778:1984,Water quality−Determination of ammonium−Potentiometric method(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+, Na+, NH4+, K+, Mn2+, Ca2+, Mg2+, Sr2+
and Ba2+ using ion chromatography−Method for water and waste water(MOD)
ISO 11732:2005,Water quality−Determination of ammonium nitrogen−Method by flow analysis
(CFA and FIA) and spectrometric detection(MOD)
備考 1. インドフェノール青吸光光度法,中和滴定法,イオン電極法及びインドフェノール青発色流
れ分析法は,試料を蒸留処理してアンモニウムイオンを共存物から分離した後,適用する。
イオン電極法及びインドフェノール青発色流れ分析法では,妨害物質を含まない試料の場合
は,蒸留処理を省略できる。また,イオンクロマトグラフ法は,妨害物質を含まない試料に
適用し,3.3の保存処理は行わず,試料採取後,直ちに試験する。
42.1 前処理(蒸留法) 試料に酸化マグネシウムを加えて弱いアルカリ性とし,蒸留を行い,留出したア
ンモニアを硫酸(25 mmol/L)に吸収捕集する。
151
K 0102:2016
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(25 mmol/L) JIS K 8951に規定する硫酸約1.4 mLをあらかじめ水100 mLを入れたビーカー
に加えてよくかき混ぜ,水を加えて1 Lとする。
3) 硫酸(1+35) 水35容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する
硫酸1容を加える。
4) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
5) 酸化マグネシウム JIS K 8432に規定する酸化マグネシウムを使用前に600 ℃で約30分間加熱し,
デシケーター中で放冷する。
6) ブロモチモールブルー溶液(1 g/L) 16.1の注(1)による。
b) 装置 装置は,次による。
1) 蒸留装置 図42.1に例を示す。ガラス器具類は,使用前に水でよく洗う。
152
K 0102:2016
単位 mm
図42.1 蒸留装置の例
c) 操作 操作は,次による。
1) 試料(1)の適量(2)をとり,中性でない場合には,ブロモチモールブルー溶液(1 g/L)5〜7滴を加え,
水酸化ナトリウム溶液(40 g/L)又は硫酸(1+35)でpHを6.0(黄色)〜7.4(青)に調節する。
2) 蒸留フラスコに移し入れ,酸化マグネシウム0.25 g,沸騰石(粒径2〜3 mm)約10個及び水を加え
て液量を約350 mLとする。
3) 蒸留装置を図42.1のように組み立て,受器のメスシリンダー(有栓形)200 mL(3)に硫酸(25 mmol/L)
50 mLを入れる。
A:蒸留フラスコ500 mL
B:連結導入管
C:すり合わせコック
D:注入漏斗
E:トラップ球(ケルダール球)
F:リービッヒ冷却器300 mm
G:逆流止め(約50 mL)
H:受器[メスシリンダー(有栓形)200 mL)]
I:共通すり合わせ
J:共通球面すり合わせ
K:押さえばね
153
K 0102:2016
4) 蒸留フラスコを加熱し,留出速度5〜7 mL/minで蒸留を行う(4)。
5) 約140 mLが留出したら蒸留を止める。
6) 冷却器及び逆流止めを外し,冷却器の内管及び逆流止めの内外を少量の水で洗う。洗液は,受器の
メスシリンダー(有栓形)200 mLに入れ(5),水を200 mLの標線まで加える。
注(1) 試料中に残留塩素が存在するときは,蒸留操作の前に,チオ硫酸ナトリウムの小結晶を加えて
除去しておくとよい。
(2) インドフェノール青吸光光度法で定量する場合にはNH4+として40 μg以上,中和滴定法の場合
には0.3〜40 mg,イオン電極法の場合には40 μg以上,イオンクロマトグラフ法の場合は20 μg
以上を含むようにとる。
(3) 留出液を中和滴定法に用いる場合には,受器には三角フラスコ500 mLを用い,これに硫酸(25
mmol/L)50 mLを正しく加え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液
[15.の注(1)による。]3〜5滴を加えておく。
(4) 冷却器の管の先端は,常に液面下約15 mmを保つようにする。
(5) 留出液を中和滴定法に用いる場合には,冷却器の内管及び逆流止めの内外の洗液は,三角フラ
スコ500 mLに合わせ,全量を滴定に用いる。
備考 2. 蒸留法として水蒸気蒸留法を用いてもよい。この場合は,図42.1の蒸留フラスコに水蒸気を
送るように装置を組み立て,蒸留フラスコを加熱する。沸騰し始めたら,水蒸気を蒸留フラ
スコに送り,留出速度3〜5 mL/minで蒸留し,約140 mLが留出したら蒸留を止める。
3. 妨害物質 蒸留法においても,脂肪族アミン,芳香族アミン類なども留出するので,これら
の共存は妨害となる。
尿素,アセトアミド,ペプトン,アスパラギンなどの窒素を含む有機化合物は,蒸留する
とその一部が加水分解してアンモニアとなり正の誤差を生じる。その程度は,蒸留時のpH
が高くなるほど大きくなる。
42.2 インドフェノール青吸光光度法 アンモニウムイオンが次亜塩素酸イオンの共存の下で,フェノー
ルと反応して生じるインドフェノール青の吸光度を測定してアンモニウムイオンを定量する。
定量範囲:NH4+ 5〜100 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 水酸化ナトリウム溶液(200 g/L) 38.1.1.1 a) 3)による。使用時に調製する。
3) ナトリウムフェノキシド溶液 水酸化ナトリウム溶液(200 g/L)55 mLをビーカーにとり,冷水中
で冷却しながらJIS K 8798に規定するフェノール25 gを少量ずつ加えて溶かす。放冷後,JIS K 8034
に規定するアセトン6 mLを加え,水で200 mLとする。10 ℃以下の暗所に保存し,5日間以上経
過したものは使用しない。
4) 次亜塩素酸ナトリウム溶液(有効塩素10 g/L) 次亜塩素酸ナトリウム溶液(有効塩素7〜12 %)の
有効塩素の濃度を求め(6),有効塩素が約10 g/Lになるように水で薄める。使用時に調製する。
5) アンモニウムイオン標準液(NH4+ 1 mg/mL) JIS K 8116に規定する塩化アンモニウムをデシケー
ター[JIS K 8228に規定する過塩素酸マグネシウム(乾燥用)を入れたもの。]中に16時間以上放
置し,その2.97 gをとり,水に溶かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
6) アンモニウムイオン標準液(NH4+ 10 μg/mL) アンモニウムイオン標準液(NH4+ 1 mg/mL)10 mL
を全量フラスコ1 000 mLにとり,水を標線まで加える。使用時に調製する。
154
K 0102:2016
注(6) 36.の注(3)による。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 使用前に水でよく洗う。
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 42.1の留出液の適量(NH4+として5〜100 μgを含む。25 mL以下)を全量フラスコ50 mLにとり,
水を加えて約25 mLとする。
2) ナトリウムフェノキシド溶液10 mLを加えて振り混ぜる。
3) 次亜塩素酸ナトリウム溶液(有効塩素10 g/L)5 mLを加え,水を標線まで加えた後,栓をして振り
混ぜる。
4) 液温を20〜25 ℃に保って,約30分間(7)放置する。
5) この溶液の一部を吸収セルに移し,波長630 nm付近の吸光度を測定する。
6) 空試験として水25 mLをとり,2)〜5)の操作を行って吸光度を測定し,試料について得た吸光度を
補正する。
7) 検量線からアンモニウムイオンの量を求め,試料中のアンモニウムイオンの濃度(NH4+mg/L)を
算出する。
注(7) 液温が20〜25 ℃のとき約30分間で発色は最高となり,その後約30分間は安定である。
d) 検量線 検量線の作成は,次による。
1) アンモニウムイオン標準液(NH4+ 10 μg/mL)0.5〜10 mLを段階的に全量フラスコ50 mLにとり,
水を加えて約25 mLとする。
2) c) 2)〜6)の操作を行って吸光度を測定し,アンモニウムイオン(NH4+)の量と吸光度との関係線を
作成する。
備考 4. 微量のアンモニウムイオンを定量する場合には,c) 2)の操作でナトリウムフェノキシド溶液
10 mLに続き,ペンタシアノニトロシル鉄(III)酸ナトリウム溶液[JIS K 8722に規定する
ペンタシアノニトロシル鉄(III)酸ナトリウム二水和物0.15 gを水に溶かして100 mLとす
る。]1 mLを加えてもよい。
この場合の定量範囲は,NH4+として2.5〜50 μgとなる。検量線は,同一操作で作成する。
5. アンモニウム体窒素で表示する場合は,次の換算式を用いる。
アンモニウム体窒素(NH4+-N mg/L)=アンモニウムイオン(NH4+ mg/L)×0.776 6
また,アンモニアへの換算は,次の換算式を用いる。
アンモニア(NH3 mg/L)=アンモニウムイオン(NH4+ mg/L)×0.944 1
6. 脂肪族アミン類は妨害しないが,芳香族アミン類の一部は,次亜塩素酸塩によって酸化され
て着色物質を生じるので妨害する。p-アミノフェノールのような物質は,アルカリ性溶液中
でフェノールと反応してインドフェノール青を生じるので妨害する。p-ヒドロキノンは,妨
害しない。ヒドロキシルアミンも妨害するが,JIS K 8230に規定する過酸化水素の当量を加
えて酸化すれば,妨害を除くことができる。
42.3 中和滴定法 前処理(蒸留)を行って留出したアンモニアを一定量の硫酸(25 mmol/L)中に吸収さ
せた溶液について,50 mmol/L水酸化ナトリウム溶液で,残った硫酸を滴定してアンモニウムイオンを定
量する。
定量範囲:NH4+ 0.3〜40 mg,繰返し精度:3〜10 %
155
K 0102:2016
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(25 mmol/L) 42.1 a) 2)による。
3) メチルレッド-ブロモクレゾールグリーン混合溶液 15.の注(1)による。
4) 50 mmol/L水酸化ナトリウム溶液 水約30 mLをポリエチレン瓶にとり,冷却しながらJIS K 8576
に規定する水酸化ナトリウム約35 gを少量ずつ加えて溶かし,密栓して4〜5日間放置する。その
上澄み液2.5 mLをポリエチレン製の気密容器1 Lにとり,2. n) 2)の二酸化炭素を含まない水を加え
て1 Lとし,混合した後,二酸化炭素を遮断して保存する。
4.1) 標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質のアミド硫酸を,上口デシケーター中に圧力2 kPa
以下で約48時間放置して乾燥する。その約1 gを1 mgの桁まではかりとり,少量の水に溶
かして全量フラスコ200 mLに移し入れ,水を標線まで加える。
− その20 mLを三角フラスコ300 mLにとり,指示薬としてブロモチモールブルー溶液(1 g/L)
[16.の注(1)による。]2,3滴を加え,この50 mmol/L水酸化ナトリウム溶液で滴定し,溶液
の色が緑になったときを終点とする。
− 次の式によって50 mmol/L水酸化ナトリウム溶液のファクター(f)を算出する。
855
004
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: アミド硫酸の質量(g)
b: アミド硫酸の純度(質量分率%)
x: 滴定に要した50 mmol/L水酸化ナトリウム溶液量(mL)
0.004 855: 50 mmol/L水酸化ナトリウム溶液1 mLに相当するアミド
硫酸の質量(g)
b) 操作 操作は,次による。
1) 42.1 c)の前処理で得た留出液の全量を用い,50 mmol/L水酸化ナトリウム溶液で溶液の色が灰紫
(pH4.8)になるまで滴定する。
2) 別に,硫酸(25 mmol/L)50 mLを正しく三角フラスコ500 mLにとり,メチルレッド-ブロモクレゾ
ールグリーン混合溶液5〜7滴を加え,50 mmol/L水酸化ナトリウム溶液で溶液の色が灰紫(pH4.8)
になるまで滴定し,硫酸(25 mmol/L)50 mLに相当する50 mmol/L水酸化ナトリウム溶液のmL数
を求める。
3) 次の式によって試料中のアンモニウムイオンの濃度(NH4+ mg/L)を算出する。
(
)
902
.0
000
1
×
×
×
−
=
V
f
a
b
A
ここに,
A: アンモニウムイオンの濃度(NH4+ mg/L)
b: 硫酸(25 mmol/L)50 mLに相当する50 mmol/L水酸化ナトリ
ウム溶液量(mL)
a: 滴定に要した50 mmol/L水酸化ナトリウム溶液量(mL)
f: 50 mmol/L水酸化ナトリウム溶液のファクター
V: 試料量(mL)
0.902: 50 mmol/L水酸化ナトリウム溶液1 mLに相当するアンモニ
ウムイオンの質量(mg)
備考 7. 42.1 c) 3)の硫酸(25 mmol/L)の代わりに,ほう酸溶液(20 g/L)を用いてもよい。この場合
は,次のように操作する。
156
K 0102:2016
1) 三角フラスコ500 mLに,ほう酸溶液(20 g/L)(JIS K 8863に規定するほう酸を用いて
調製する。)50 mLを加え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合
溶液5〜7滴を加え,42.1 c) 4)〜6)の操作を行う。
2) 次に,25 mmol/L硫酸(*)で溶液の色が灰紫(pH4.8)になるまで滴定する。
3) 別に,空試験としてほう酸溶液(20 g/L)50 mLを三角フラスコ500 mLにとり,水150 mL
を加え,指示薬としてメチルレッド-ブロモクレゾールグリーン混合溶液5〜7滴を加え
る。次に,試料の場合と同様に滴定を行う。
4) 次の式によって試料中のアンモニウムイオンの濃度(NH4+ mg/L)を算出する。
(
)
902
.0
000
1
×
×
×
−
=
V
f
b
a
A
ここに,
A: アンモニウムイオンの濃度(NH4+ mg/L)
a: 滴定に要した25 mmol/L硫酸溶液量(mL)
b: 空試験に要した25 mmol/L硫酸溶液量(mL)
f: 25 mmol/L硫酸のファクター
V: 試料量(mL)
0.902: 25 mmol/L硫酸1 mLに相当するアンモニウムイオンの質量
(mg)
注(*) 25 mmol/L硫酸の調製方法 42.1 a) 2)の硫酸(25 mmol/L)を標定して用いる。
標定
− JIS K 8005に規定する容量分析用標準物質の炭酸ナトリウムを600 ℃で約1時
間加熱した後,デシケーター中で放冷する。その0.53 gを1 mgの桁まではかり
とり,水に溶かして全量フラスコ200 mLに移し入れ,水を標線まで加える。
− この20 mLをビーカーにとり,指示薬としてメチルレッド-ブロモクレゾールグ
リーン混合溶液3〜5滴を加えた後,この硫酸(25 mmol/L)で滴定する。
− 溶液の色が灰紫になったら,煮沸して二酸化炭素を追い出し,放冷後,溶液の
色が灰紫になるまで滴定を続ける。
− 次の式によって25 mmol/L硫酸のファクター(f)を算出する。
650
002
.0
1
200
20
100
×
×
×
×
=
x
b
a
f
ここに,
a: 炭酸ナトリウムの質量(g)
b: 炭酸ナトリウムの純度(質量分率%)
x: 滴定に要した硫酸(25 mmol/L)溶液量(mL)
0.002 650: 25 mmol/L硫酸1 mLに相当する炭酸ナトリウムの質量
(g)
42.4 イオン電極法 前処理を行った試料に水酸化ナトリウム溶液を加え,pHを11〜13に調節してアン
モニウムイオンをアンモニアに変え,アンモニア電極を指示電極として電位を測定し,アンモニウムイオ
ンを定量する。電極を汚染したり,電位応答を妨害するような共存物質が存在しなければ蒸留操作を省く
ことができる。
定量範囲:NH4+ 0.1〜100 mg/L,繰返し精度:5〜20 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 水酸化ナトリウム溶液(100 g/L) 19. a) 2)による。
157
K 0102:2016
3) 塩化アンモニウム溶液(0.1 mol/L) JIS K 8116に規定する塩化アンモニウム5.4 gを水約800 mL
に溶かし,水で1 Lとする。
4) アルカリ性緩衝液 JIS K 8576に規定する水酸化ナトリウム40 g及びJIS K 8107に規定するエチレ
ンジアミン四酢酸二水素二ナトリウム二水和物(EDTA二ナトリウム塩)37 gを水約800 mLに溶か
し(8),水で1 Lとする。この溶液は,ポリエチレン瓶に貯蔵する。
5) アンモニウムイオン標準液(NH4+ 100 mg/L) 42.2 a) 5)のアンモニウムイオン標準液(NH4+ 1
mg/mL)20 mLを全量フラスコ200 mLにとり,水を標線まで加える。
6) アンモニウムイオン標準液(NH4+ 10 mg/L) アンモニウムイオン標準液(NH4+ 100 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。
7) アンモニウムイオン標準液(NH4+ 1 mg/L) アンモニウムイオン標準液(NH4+ 10 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。使用時に調製する。
8) アンモニウムイオン標準液(NH4+ 0.1 mg/L) アンモニウムイオン標準液(NH4+ 1 mg/L)20 mL
を全量フラスコ200 mLにとり,水を標線まで加える。使用時に調製する。
注(8) アンモニウムイオン0.5 mg/L以下の低濃度の定量では,この溶液は約20分間煮沸し,冷却後,
水で1 Lに薄めるとよい。
b) 器具及び装置 器具及び装置は,次による。
1) 電位差計 34.2 b) 1)による。
2) 指示電極 アンモニア電極(隔膜形)
3) 参照電極 塩化カリウム溶液(飽和)を充塡した銀-塩化銀電極を用いる。
4) 測定容器 35.2 b) 4)による。
5) 恒温槽 34.2 b) 5)による。
6) マグネチックスターラー 35.2 b) 6)による。
c) 検量線 検量線の作成は,次による。
1) アンモニウムイオン標準液(NH4+ 0.1 mg/L)100 mLを測定容器にとり(9),水酸化ナトリウム溶液
(100 g/L)1 mLを加える(10)。
2) 恒温槽から水を送り,測定容器の溶液を25±0.5 ℃にする。
3) 指示電極(11) (12)と参照電極(13)とを浸し,マグネチックスターラー(14)で泡が電極に触れない程度に強
くかき混ぜる(15)。
4) 液温を確認し,電位差計で電位を測定する(16)。
5) アンモニウムイオン標準液(NH4+ 1 mg/L)100 mL,アンモニウムイオン標準液(NH4+ 10 mg/L)
100 mL及びアンモニウムイオン標準液(NH4+ 100 mg/L)100 mLをそれぞれ三角フラスコ200 mL(9)
にとり,水酸化ナトリウム溶液(100 g/L)1 mLを加える(10)。
6) 2)〜4)の操作を行って,それぞれのアンモニウムイオン標準液の電位を測定する(17)。
7) 横軸にアンモニウムイオンの濃度(NH4+ mg/L)の対数,縦軸に電位(18)をとって,アンモニウムイ
オンの濃度と電位との関係線を作成する(19)。
注(9) アンモニアは揮散しやすいため,できるだけ口の狭い容器を用いる。必要に応じて密閉できる
容器を用いるとよい。
(10) pH約12となる。pH11以上で,アンモニウムイオンはアンモニアとなる。アンモニアは揮散し
やすいので,水酸化ナトリウム溶液(100 g/L)は電位測定の直前に加える。
(11) 隔膜電極は,内部ガラス電極のガラス膜面に隔膜を強く押し付けると隔膜にきずがつくので注
158
K 0102:2016
意する。また,ガラス膜面と隔膜とが離れ過ぎると,応答時間が長くなる。
注(12) 電極の隔膜が汚れると,電位が不安定になり,応答時間も長くなる。
(13) 34.の注(14)による。
(14) かき混ぜが強すぎると,泡が膜を覆い誤差を生じるので注意する。
(15) 34.の注(16)による。
(16) アンモニア電極の応答時間は,液温10〜30 ℃の場合,アンモニウムイオン標準液(NH4+ 0.1
mg/L)では3〜5分間,アンモニウムイオン標準液(NH4+ 1 mg/L以上)では2〜3分間である。
(17) 電位の測定は,低濃度から順に行う。高濃度から低濃度へ移ると,応答速度は遅くなるので,
水で洗ってアンモニアを除き,次に,c) 1)の水酸化ナトリウム溶液(100 g/L)1 mLを加えたア
ンモニウムイオン標準液(NH4+ 0.1 mg/L)に浸し,指示値が安定してから測定する。
なお,水酸化ナトリウムを加えた標準液は,5〜10分間経過するとアンモニアを消失してい
るので,次の校正には新しい標準液を用いる。
(18) 電位は,ネルンスト式に従って,アンモニウムイオンの濃度が1桁の変化当たり約60 mV変化
する。
(19) アンモニウムイオンの濃度NH4+ 0.1 mg/L付近からNH4+ 100 mg/Lまで検量線は直線になる。
備考 8. 試料中のアンモニウムイオンの濃度が想定できる場合は,その濃度範囲を含む標準液を調製
して検量線を作成してもよい。
d) 操作 操作は,次による。
1) 42.1の前処理を行った試料の適量(NH4+として0.02〜20 mgを含む。)をとり,水で約100 mLとし
た後,水酸化ナトリウム溶液(100 g/L)を加えてpHを約8.3に調節し,全量フラスコ200 mLに移
し入れ,水を標線まで加える。この溶液100 mLを三角フラスコ200 mLにとり,水酸化ナトリウム
溶液(100 g/L)1 mLを加える(10)。
2) c) 2)〜4)の操作を行って,検量線からアンモニウムイオンの濃度(NH4+ mg/L)を求め,試料中の
アンモニウムイオンの濃度(NH4+ mg/L)を算出する。
備考 9. イオン濃度計の場合には,アンモニウムイオン標準液(NH4+ 0.1 mg/L)とアンモニウムイオ
ン標準液(NH4+ 10 mg/L)とを用い,c) 1)〜4)の操作を行ってイオン濃度計の指示値を
NH4+ 0.1 mg/L及びNH4+ 10 mg/Lになるように調節する。さらに,アンモニウムイオン標
準液(NH4+ 1 mg/L)とアンモニウムイオン標準液(NH4+ 100 mg/L)とを用いてイオン濃
度計の指示値を確認する。
10. 妨害物質がなく,蒸留処理をせず,ろ過による処理で測定する場合は,3.2に従って試料をろ
過する。ろ液の50 mLを測定容器にとり,アルカリ性緩衝液5 mLを加える。次いで,c) 2)
〜4)の操作を行う。検量線は,同じ操作で作成する。
11. アンモニア電極の保管方法 アンモニア電極を長い期間(例えば,一夜)保管するには,先
端を塩化アンモニウム溶液(0.1 mol/L)に浸しておく。使用前に,十分にすすぐ。
12. 妨害物質
1) 試料中のアンモニウムイオンの濃度が0.1 mg/Lの場合,ヒドラジニウムイオン(N2H5+)
は1 mg/L以下では影響しないが,10 mg/L及び100 mg/L共存すれば,それぞれ+35 %,
+100 %の誤差を生じる。
2) 試料中のアンモニウムイオンの濃度が1 mg/L以上の場合には,ヒドラジニウムイオン
100 mg/Lが共存しても影響しない。試料中のアンモニウムイオンの濃度が0.1 mg/Lの場

159
K 0102:2016
合,モルホリン(テトラヒドロ-1,4-オキサジン,C4H8ONH)は10 mg/Lまで共存しても
影響しないが,100 mg/L共存すると,+100 %の誤差を生じる。
3) また,試料中のアンモニウムイオンの濃度が1 mg/L以上の場合には,モルホリンが100
mg/L共存しても影響しない。
4) アンモニア電極は,アンモニウムイオンの濃度が50 mg/Lを超える試料の測定に連続的
に使用すると,満足な応答をしない。このような水に対しては,測定試料をこの濃度未
満に薄めるのがよい。
5) アミン類は正の妨害を与える。表42.1に示す妨害物質としてヒドラジン,シクロヘキシ
ルアミン,モルホリン,オクタデシルアミン,メタノールアミン,尿素などがある。
6) 界面活性剤及びある種の有機溶媒は,アンモニア電極の膜の寿命を短くする。このため
電極の手入れを必要とする頻度が増加する。この影響はかなり重大で,これらの妨害物
質の濃度が高い試料では,アンモニア電極の劣化を早めることになる。
表42.1 妨害物質の例
妨害物質
妨害物質濃度
mg/L
アンモニウムイオンの濃度ρN=
1 mg/Lにおける見掛けの増加
mg/L
ヒドラジン
4
0.06
シクロヘキシルアミン
1
0.03
モルホリン
10
0.03
オクタデシルアミン
0.4
0.14
メタノールアミン
3.4
0.15 *
尿素
11
0.01
注*
アンモニウムイオンの濃度ρN=0.5 mg/Lにおける見掛けの増加。
備考 13. アンモニウムイオン濃度をアンモニア体窒素で表示する場合は備考5.による。
42.5 イオンクロマトグラフ法 試料中のアンモニウムイオンをイオンクロマトグラフ法によって定量す
る。この方法によって,表48.1に示す陽イオンが同時定量できる。この方法を適用する場合には,3.3の
保存処理を行わず,試料採取後,直ちに試験する。直ちに行えない場合は,0〜10 ℃の暗所に保存し,で
きるだけ早く試験する。
試験操作などは,48.3による。
備考 14. 備考5. による。
42.6 流れ分析法 試料中のアンモニウムイオンを,42.2と同様な原理で発色させる流れ分析法によって
定量する。試料を,42.1の操作でアンモニウムイオンを共存物から分離した後,適用する。ただし,妨害
物質を含まない試料は,蒸留操作を省略してもよい。この場合は懸濁物の多い試料には適用できない。
備考 15. アンモニウムイオン濃度をアンモニア体窒素で表示する場合は,備考5.による。
定量範囲:NH4+ 0.06〜13 mg/L(アンモニア体窒素として0.05〜10 mg/L),繰返し精度:10 %以下
試験操作などは,JIS K 0170-1による。ただし,JIS K 0170-1の6.2(ガス拡散・pH指示薬変色FIA法)
及び6.4(サリチル酸によるインドフェノール青発色CFA法)の方法は除く。
43. 亜硝酸イオン(NO2−)及び硝酸イオン(NO3−)
43.1 亜硝酸イオン(NO2−) 亜硝酸イオンの定量には,ナフチルエチレンジアミン吸光光度法,イオン
160
K 0102:2016
クロマトグラフ法又はナフチルエチレンジアミン発色による流れ分析法を適用する。亜硝酸イオンは変化
しやすいので,試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,できる
だけ早く試験する。ただし,イオンクロマトグラフ法を適用する場合には,3.3の保存処理を行わず,試
料採取後,直ちに試験する。
なお,ナフチルエチレンジアミン吸光光度法は,1984年に第1版として発行されたISO 6777,イオンク
ロマトグラフ法は,1995年に第1版として発行されたISO 10304-2,ナフチルエチレンジアミン発色によ
る流れ分析法は,1996年に第1版として発行されたISO 13395との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6777:1984,Water quality−Determination of nitrite−Molecular absorption spectrometric method
(MOD)
ISO 10304-2:1995,Water quality−Determination of dissolved anions by liquid chromatography of ions
−Part 2: Determination of bromide,chloride,nitrate,nitrite,orthophosphate and sulfate in waste
water(MOD)
ISO 13395:1996,Water quality−Determination of nitrite nitrogen and nitrate nitrogen and the sum of
both by flow analysis (CFA and FIA) and spectrometric detection(MOD)
43.1.1 ナフチルエチレンジアミン吸光光度法 試料に,スルファニルアミド(4-アミノベンゼンスルホン
アミド)を加え,これを亜硝酸イオンによってジアゾ化し,N-1-ナフチルエチレンジアミン二塩酸塩(二
塩化N-1-ナフチルエチレンジアンモニウム)を加えて生じる赤い色のアゾ化合物の吸光度を測定して亜硝
酸イオンを定量する。
定量範囲:NO2− 0.6〜6 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 4-アミノベンゼンスルホンアミド溶液 JIS K 9066に規定するスルファニルアミド(4-アミノベン
ゼンスルホンアミド)2 gを,JIS K 8180に規定する塩酸60 mLと水80 mLとに溶かし,更に水を
加えて200 mLとする。
3) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 JIS K 8197に規定するN-1-ナフチルエチレンジ
アミン二塩酸塩(二塩化N-1-ナフチルエチレンジアンモニウム)0.2 gを水に溶かして200 mLとす
る。着色ガラス瓶に入れて保存し,1週間以上経過したものは使用しない。
4) 亜硝酸イオン標準液(NO2− 1 mg/mL) JIS K 8019に規定する亜硝酸ナトリウムを105〜110 ℃で
約4時間加熱し,デシケーター中で放冷する。その亜硝酸ナトリウムの純度を求め(1),NaNO2 100 %
に対して1.50 gに相当する亜硝酸ナトリウムをとり,少量の水に溶かして全量フラスコ1 000 mLに
移し入れ,水を標線まで加える。使用時に調製する。
5) 亜硝酸イオン標準液(NO2− 20 μg/mL) 亜硝酸イオン標準液(NO2− 1 mg/mL)10 mLを全量フラ
スコ500 mLにとり,水を標線まで加える。使用時に調製する。
6) 亜硝酸イオン標準液(NO2− 2 μg/mL) 亜硝酸イオン標準液(NO2− 20 μg/mL)20 mLを全量フラ
スコ200 mLにとり,水を標線まで加える。使用時に調製する。
注(1) 亜硝酸ナトリウムの純度は,JIS K 8019による。
b) 装置 装置は,次による。
161
K 0102:2016
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 3.2でろ過した試料(2)の適量(NO2−として0.6〜6 μgを含む。)をメスシリンダー(有栓形)10 mL
にとり,水を加えて10 mLとする。
2) 4-アミノベンゼンスルホンアミド溶液1 mLを加えて振り混ぜ,約5分間放置した後,二塩化N-1-
ナフチルエチレンジアンモニウム溶液1 mLを加えて振り混ぜ,室温で約20分間放置する。
3) 溶液の一部を吸収セルに移し,波長540 nm付近の吸光度を測定する。
4) 空試験として水10 mLをメスシリンダー(有栓形)10 mLにとり,2)及び3)の操作を行って吸光度
を測定し,試料について得た吸光度を補正する。
5) 検量線から亜硝酸イオンの量を求め,試料中の亜硝酸イオンの濃度(NO2− mg/L)を算出する。
注(2) ろ過しても,色又は濁りが残る場合には,JIS K 0101の36.1.1 (3.1)の硫酸亜鉛による凝集沈殿,
又は次の硫酸アルミニウム凝集沈殿法によって除去する。
− 試料100 mLにつき硫酸カリウムアルミニウム溶液(JIS K 8255に規定する硫酸カリウム
アルミニウム・12水5 gを水に溶かして100 mLとする。)2 mL及び水酸化ナトリウム溶液
(40 g/L)[21. a) 3)による。]を加えて水酸化アルミニウムのフロックを生成させ,数分間
放置した後,ろ過(初めのろ液約20 mLは捨てる。)して澄明な溶液とする。
− 凝集沈殿処理すると水酸化アルミニウムに亜硝酸イオンが一部吸着されて発色が低下する
ので,別に,亜硝酸イオン標準液(NO2− 2 μg/mL)を段階的にとり,同様に処理したもの
を用いて検量線を作成して定量する。
d) 検量線 検量線の作成は,次による。
1) 亜硝酸イオン標準液(NO2− 2 μg/mL)3〜30 mLを段階的に全量フラスコ100 mLにとり,水を標線
まで加える。その中からそれぞれ10 mLをメスシリンダー(有栓形)10 mLにとる。
2) c) 2)〜4)の操作を行って亜硝酸イオン(NO2−)の量と吸光度との関係線を作成する。
備考 1. 一般に,亜硝酸イオンは,残留塩素などの酸化性物質と共存することはないが,亜硝酸イオ
ンが存在しなくても残留塩素,クロロアミン類(モノクロロアミン,ジクロロアミン及び三
塩化窒素)などが存在すると赤に発色して亜硝酸イオンとして誤認されることがある。
試料中の酸化性物質の存在を確認するには,次のように操作する。
1) 試料100 mLをとり,ふっ化カリウム溶液(300 g/L)[19.の注(3)による。]1 mLとJIS K
9501に規定するアジ化ナトリウム0.5 gとを加える。
2) 塩酸(1+1)[24.3 a) 2)による。]を加えて酸性(pH約1)とし,JIS K 8913に規定する
よう化カリウム1 gを加えてかき混ぜた後,暗所に数分間放置する。
3) これに指示薬としてでんぷん溶液(10 g/L)[19. a) 5)による。]2 mLを加える。よう素で
んぷんの青い色が認められる場合は,酸化性物質が存在する。
4) このような試料について亜硝酸イオンを試験するには,この操作を行った後,よう素で
んぷんの青い色が消えるまで,亜硫酸ナトリウム溶液(50 mmol/L)(JIS K 8061に規定
する亜硫酸ナトリウム0.63 gを水に溶かして100 mLとする。)で滴定し,その消費量か
ら,試料中の酸化性物質の量に相当する亜硫酸ナトリウム溶液(50 mmol/L)の量を求め,
この量を試料に添加した後,c) 1)〜5)の操作を行う。
2. 次の操作によって亜硝酸イオンを定量してもよい。
1) 試料を3.2によってろ過し,その適量(NO2−として3〜30 μgを含む。)を全量フラスコ
162
K 0102:2016
50 mLにとり,水で約40 mLとし,発色試薬1 mLを加えた後,水を標線まで加える。
2) 約20分間放置後,波長540 nm付近の吸光度を測定する。
3) 水について空試験を行い,吸光度を補正する。
4) 検量線は,亜硝酸イオン標準液(NO2− 1 μg/mL)3〜30 mLを段階的にとって作成する。
5) 発色試薬の調製 JIS K 9066に規定するスルファニルアミド(4-アミノベンゼンスルホ
ンアミド)4 gを,JIS K 9005に規定するりん酸10 mLと水50 mLとの混合溶液に溶か
す。これにJIS K 8197に規定するN-1-ナフチルエチレンジアミン二塩酸塩0.2 gを加え
て溶かし,全量フラスコ100 mLに入れ,水を標線まで加える。褐色ガラス瓶に入れ,0
〜10 ℃の暗所に保存すれば,1か月間は安定である。
備考 3. 亜硝酸イオンの濃度を亜硝酸体窒素で表示する場合は,35.の備考11.による。
43.1.2 イオンクロマトグラフ法 試料中の亜硝酸イオンをイオンクロマトグラフ法で定量する。この方法
によって表35.1に示すイオンも同時定量できる。亜硝酸イオンを定量する場合には,3.3の保存処理を行
わず,試料採取後,直ちに試験する。
試験操作などは,35.3による。
43.1.3 流れ分析法 試料中の亜硝酸イオンを,43.1.1と同様な原理で発色させる流れ分析法によって定量
する。
備考 4. 亜硝酸イオンの濃度を亜硝酸体窒素で表示する場合は,35.の備考11. による。
定量範囲:NO2− 0.03〜3 mg/L(亜硝酸体窒素として0.01〜1 mg/L),繰返し精度:10 %以下
試験操作などは,JIS K 0170-2で規定された亜硝酸体窒素に関する規定による。
43.2 硝酸イオン(NO3−) 硝酸イオンの定量には,還元蒸留-インドフェノール青吸光光度法,還元蒸留-
中和滴定法,銅・カドミウムカラム還元-ナフチルエチレンジアミン吸光光度法,ブルシン吸光光度法,イ
オンクロマトグラフ法又はカドミウム還元-ナフチルエチレンジアミン発色による流れ分析法を適用する。
この試験は,試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,できるだけ早く
試験する。ただし,イオンクロマトグラフ法を適用する場合には,3.3の保存処理を行わず,試料採取後
直ちに試験する。
なお,イオンクロマトグラフ法は,1995年に第1版として発行されたISO 10304-2,カドミウム還元-ナ
フチルエチレンジアミン発色による流れ分析法は,1996年に第1版として発行されたISO 13395との整合
を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 10304-2:1995,Water quality−Determination of dissolved anions by liquid chromatography of ions
−Part 2: Determination of bromide,chloride,nitrate,nitrite,orthophosphate and sulfate in waste
water(MOD)
ISO 13395:1996,Water quality−Determination of nitrite nitrogen and nitrate nitrogen and the sum of
both by flow analysis (CFA and FIA) and spectrometric detection(MOD)
43.2.1 還元蒸留-インドフェノール青吸光光度法 試料に水酸化ナトリウムを加えて蒸留を行い,アンモ
ニウムイオン及び一部の有機窒素化合物の分解で生じたアンモニアを除去した後,デバルダ合金を加えて
亜硝酸イオン及び硝酸イオンをアンモニアに還元し,蒸留し,留出したアンモニアを硫酸(25 mmol/L)に
吸収させる。次に,この留出液中のアンモニウムイオンをインドフェノール青吸光光度法によって定量し,
163
K 0102:2016
硝酸イオンと亜硝酸イオンとの合量を求め,その値から,別に,亜硝酸イオンを定量して差し引き,硝酸
イオンの量を算出する。
定量範囲:NO3− 17〜340 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(25 mmol/L) 42.1 a) 2)による。
3) 硫酸(1+35) 水35容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する
硫酸1容を徐々に加える。
4) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
5) 水酸化ナトリウム溶液(300 g/L) JIS K 8576に規定する水酸化ナトリウム30 gを水に溶かして100
mLとする。使用時に調製する。
6) デバルダ合金 JIS K 8653に規定するもの。
7) ナトリウムフェノキシド溶液 42.2 a) 3)による。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 使用前に水でよく洗う。
2) 蒸留装置 42.1 b) 1)による。使用前に水でよく洗う。
3) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料の適量(NO3−として0.14 mg以上を含む。)をとり,試料が中性でない場合には,水酸化ナト
リウム溶液(40 g/L)又は硫酸(1+35)でpH約7に調節する。
2) これを水で蒸留フラスコに洗い移し,水酸化ナトリウム溶液(300 g/L)10 mLと沸騰石(粒径2〜3
mm)5〜7個とを加え,水を加えて約350 mLとし,42.1 c) 3)〜5)の蒸留操作を行って(3)アンモニア
を除去し,冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内外を水でよく洗う。
3) 蒸留フラスコ中の残留液を放冷する。
4) 受器に別のメスシリンダー(有栓形)200 mLを用い,硫酸(25 mmol/L)50 mLを入れる。
5) 蒸留フラスコ中の残留液にデバルダ合金3 gを手早く加え,水を加えて約350 mLとした後,装置を
組み立てる。
6) 42.1 c) 4)〜6)の操作を行う(4)。
7) 6)で得た留出液の適量(NH4+として5〜100 μgを含む。)を全量フラスコ50 mLに分取し,水を加
えて約25 mLとする。
8) 42.2 c) 2)〜5)の操作を行って吸光度を測定する。
9) 空試験として水約100 mLを蒸留フラスコにとり,水酸化ナトリウム溶液(300 g/L)10 mLを加え
た後,4)〜6)の操作を行う。
10) 9)で得た留出液について,7)と同量を全量フラスコ50 mLに分取し,8)の操作を行って吸光度を測
定し,試料について得た吸光度を補正する。
11) 42.2 d)の検量線から,分取した留出液中のアンモニウムイオンの量(NH4+ mg)を求める。
12) 別に,43.1によって試料中の亜硝酸イオンの濃度(NO2− mg/L)を求める。
13) 次の式によって試料中の硝酸イオンの濃度(NO3− mg/L)を算出する。
348
.1
200
000
1
437
.3
2
1
×
−
×
×
×
=
C
V
V
a
N
164
K 0102:2016
ここに,
N: 硝酸イオンの濃度(NO3− mg/L)
a: 11)で求めた留出液中のアンモニウムイオンの質量(NH4+
mg)
3.437: アンモニウムイオンを硝酸イオンの相当量に換算するとき
の係数
04
.
18
00
.
62
V1: 試料量(mL)
V2: 7)で分取した留出液量(mL)
C: 12)で求めた試料中の亜硝酸イオンの濃度(NO2− mg/L)
1.348: 亜硝酸イオンを硝酸イオンの相当量に換算するときの係数
01
.
46
00
.
62
注(3) この操作では,受器には硫酸(25 mmol/L)に代え,水を入れておいてもよい。
(4) 蒸留の始めに泡立ちが激しいときは加熱を弱め,約10分間経過して泡立ちが静まってから再び
蒸留する。
備考 5. c) 2)の操作で留出したアンモニアには,試料中に存在したアンモニウムイオンのほか,有機
窒素化合物の分解によって生じたものを含む場合があるので,この留出液を用いて試料中の
アンモニウムイオンの定量を行ってはならない。
6. 硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11.による。
43.2.2 還元蒸留-中和滴定法 試料に水酸化ナトリウムを加えて蒸留を行い,アンモニウムイオン及び一
部の有機窒素化合物の分解で生じたアンモニアを除去した後,デバルダ合金を加えて亜硝酸イオン及び硝
酸イオンをアンモニアに還元し,蒸留し,留出したアンモニアを一定量の硫酸(25 mmol/L)中に吸収させ,
中和滴定法によって亜硝酸イオン及び硝酸イオンに相当する量を求める。別に,亜硝酸イオンを定量して
差し引き,硝酸イオンの量を算出する。
定量範囲:NO3− 1〜140 mg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(25 mmol/L) 42.1 a) 2)による。
3) 硫酸(1+35) JIS K 8951に規定する硫酸を用いて調製する。
4) 50 mmol/L水酸化ナトリウム溶液 42.3 a) 4)による。
5) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
6) 水酸化ナトリウム溶液(300 g/L) 43.2.1 a) 5)による。
7) デバルダ合金 43.2.1 a) 6)による。
8) メチルレッド-ブロモクレゾールグリーン混合溶液 15.の注(1)による。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 使用前に水でよく洗う。
2) 蒸留装置 42.1 b) 1)による。使用前に水でよく洗う。
c) 操作 操作は,次による。
1) 試料の適量(NO3−として1 mg以上,NO2−及びNO3−の合量がNO3−として140 mg以下を含む。)
をとり,試料が中性でない場合には,水酸化ナトリウム溶液(40 g/L)又は硫酸(1+35)でpHを
約7に調節する。
2) これを水で蒸留フラスコに洗い移し,水酸化ナトリウム溶液(300 g/L)10 mLと沸騰石(粒径2〜3
165
K 0102:2016
mm)5〜7個とを加え,水を加えて約350 mLとし,42.1 c) 3)〜5)の蒸留操作を行って(3)アンモニア
を除去し,冷却器と逆流止めを取り外し,冷却器の内管及び逆流止めの内外を水でよく洗う。
3) 蒸留フラスコ中の残留液を放冷する。
4) 受器として三角フラスコ500 mLを用い,これに硫酸(25 mmol/L)50 mLを正確に加え,指示薬と
してメチルレッド-ブロモクレゾールグリーン混合溶液5〜7滴を加えておく。
5) 蒸留フラスコ中の残留液にデバルダ合金3 gを手早く加え,水を加えて約350 mLとした後,装置を
組み立てる。
6) 42.1 c) 4)〜6)の操作を行う(4)。
7) 留出液の全量を用い,42.3 b) 1)の滴定操作を行う。
8) 空試験として水約100 mLを蒸留フラスコにとり,水酸化ナトリウム溶液(300 g/L)10 mLを加え
た後,4)〜6)の操作を行って得られた留出液について7)と同様に滴定操作を行う。
9) 別に,43.1によって試料中の亜硝酸イオンの濃度(NO2− mg/L)を求める。
10) 次の式によって試料中の硝酸イオンの濃度(NO3− mg/L)を算出する。
(
)
348
.1
100
.3
000
1
×
−
×
×
×
−
=
C
V
f
a
b
N
ここに,
N: 硝酸イオンの濃度(NO3− mg/L)
a: 滴定に要した50 mmol/L水酸化ナトリウム溶液量(mL)
b: 空試験に要した50 mmol/L水酸化ナトリウム溶液量(mL)
f: 50 mmol/L水酸化ナトリウム溶液のファクター
3.100: 50 mmol/L水酸化ナトリウム溶液1 mLに相当する硝酸イオ
ンの質量(mg)
V: 試料量(mL)
C: 9)で求めた試料中の亜硝酸イオンの濃度(NO2− mg/L)
1.348: 亜硝酸イオンを硝酸イオンの相当量に換算するときの係数
01
.
46
00
.
62
備考 7. 硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11.による。
43.2.3 銅・カドミウムカラム還元-ナフチルエチレンジアミン吸光光度法 試料中の硝酸イオンを,銅・
カドミウムカラムによって還元して亜硝酸イオンとし,ナフチルエチレンジアミン吸光光度法によって定
量し,硝酸イオンの濃度を求める。
定量範囲:NO3− 0.8〜8 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次のものを用いる。
1) 水 JIS K 0557に規定するA3の水
2) 塩酸(1+11) JIS K 8180に規定する塩酸を用いて調製する。
3) 塩化アンモニウム-アンモニア溶液 JIS K 8116に規定する塩化アンモニウム100 gを水約700 mL
に溶かした後,JIS K 8085に規定するアンモニア水50 mLを加え,更に水を加えて1 Lとする。
4) カラム活性化液 水約700 mLに水酸化ナトリウム溶液(80 g/L)(JIS K 8576に規定する水酸化ナ
トリウムを用いて調製する。)70 mLを加えたものに,JIS K 8107に規定するエチレンジアミン四酢
酸二水素二ナトリウム二水和物38 g及びJIS K 8983に規定する硫酸銅(II)五水和物12.5 gを溶か
し,更に水酸化ナトリウム溶液(80 g/L)を滴加して溶液のpHを7とした後,水を加えて1 Lとす
る。
5) 銅・カドミウムカラム充塡剤 粒状カドミウム(粒径0.5〜2 mm)約40 gを三角フラスコ300 mL
166
K 0102:2016
にとり,塩酸(1+5)(JIS K 8180に規定する塩酸を用いて調製する。)約50 mLを加えて振り混ぜ
て,カドミウムの表面を洗浄し,洗液を捨て,水約100 mLずつで5回洗浄する。次に,硝酸(1+
39)(JIS K 8541に規定する硝酸を用いて調製する。)約50 mLを加えて振り混ぜてカドミウムの表
面を洗浄し,洗液を捨てる。この操作を2回行った後,水約100 mLずつで5回洗浄する。次に,
カラム活性化液200 mLを加えて約24時間放置し,カドミウムの表面に銅の皮膜を形成させる。こ
の銅-カドミウムカラム充塡剤は,このまま密栓して保存することができる。
なお,この方法で調製したものに代え,市販の銅-カドミウムカラム充塡剤を用いてもよい。
6) カラム充塡液 塩化アンモニウム-アンモニア溶液を水で10倍に薄める。
7) 硝酸イオン標準液(NO3− 1 mg/mL) JIS K 8548に規定する硝酸カリウムをあらかじめ105〜110 ℃
で約2時間加熱し,デシケーター中で放冷する。その1.63 gをとり,水に溶かして全量フラスコ1 000
mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
8) 硝酸イオン標準液(NO3− 10 μg/mL) 硝酸イオン標準液(NO3− 1 mg/mL)10 mLを全量フラスコ
1 000 mLにとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 銅・カドミウムカラム 図43.1 a) に示すようなガラス管の底部にJIS K 8251に規定するガラスウ
ールを詰め,カラム充塡液を満たした後,銅・カドミウムカラム充塡剤を空気に触れないように流
し入れる。上部にガラスウールを詰め,円筒形滴下漏斗を取り付ける。次に,円筒形滴下漏斗から,
カラム充塡液100 mL,硝酸イオン標準液(NO3− 1 mg/mL)をカラム充塡液で100倍に薄めた溶液
200 mL,更にカラム充塡液100 mLの順で,流量約10 mL/minで流下させる。このとき,カラム内
の溶液の液面は,充塡剤より僅かに上部になるようにする。
なお,銅・カドミウムカラムを使用しないときは,銅・カドミウムカラム充塡剤が空気に触れな
いように,上部までカラム充塡液を入れておく。銅・カドミウムカラムは使用に伴って劣化し,硝
酸イオンの亜硝酸イオンへの還元率が低下するので,必要に応じ,カラム活性化液を用いて再生す
る。再生には,銅・カドミウムカラムにカラム活性化液を満たし,2〜3時間放置した後,カラム充
塡液で洗浄する(5)。
2) 光度計 分光光度計又は光電光度計
注(5) 試料について,15〜20回使用するごとにカラム活性化液約20 mLを銅・カドミウムカラムに流
し,次に,カラム充塡液約100 mLで洗浄すれば,硝酸イオンの還元率の低下を防ぐことができ
る。
167
K 0102:2016
単位 mm
a) 銅・カドミウムカラム
b) 円筒形滴下漏斗
b1,b2:
C:
D:
E:
ガラスウール
銅・カドミウムカラム充塡剤
カラム充塡液
一方コック
図43.1 銅・カドミウムカラムの例
c) 操作 操作は,次による。
1) 3.2でろ過した試料(6) (7)の適量(NO3−として8 μg以上,NO2−とNO3−との合量がNO3−として80 μg
以下を含む。)を全量フラスコ100 mLにとる。
2) これに塩化アンモニウム-アンモニア溶液10 mLを加え,更に水を標線まで加えて還元用溶液とする。
3) 上部の円筒形滴下漏斗に還元用溶液を入れ,銅・カドミウムカラム内の液面を銅・カドミウムカラ
ム充塡剤より僅かに上部に保ちながら約10 mL/minで流下させ,流出液約30 mLを捨てる。還元用
溶液を追加し,同様に流下させ,その後の30 mLをメスシリンダー50 mLに集める。
4) この流出液から10 mLを共栓試験管にとり,43.1.1 c) 2)及び3)の操作を行う。
5) 空試験として水を全量フラスコ100 mLにとり,2)〜4)の操作を行って吸光度を測定し,試料につい
て得た吸光度を補正する。
6) 検量線から4)での流出液10 mL中の硝酸イオンの量を求め,試料中の亜硝酸イオンと硝酸イオンと
の合量の濃度(硝酸イオン換算量)(NO3− mg/L)を算出する。
7) 別に,43.1.1によって試料中の亜硝酸イオンの濃度(NO2− mg/L)を求める。
8) 次の式によって試料中の硝酸イオン濃度(NO3− mg/L)を算出する。
N=a−b×1.348
ここに,
N: 硝酸イオンの濃度(NO3− mg/L)
a: 6)で算出した試料中の亜硝酸イオンと硝酸イオンとの合量
168
K 0102:2016
(NO3− mg/L)
b: 7)で求めた試料中の亜硝酸イオンの濃度(NO2− mg/L)
1.348: 亜硝酸イオンを硝酸イオンの相当量に換算するときの係数
01
.
46
00
.
62
注(6) 注(2)による。ただし,凝集沈殿処理しても色が残る場合には,この方法を適用することはでき
ない。このような場合には,43.2.1又は43.2.2によって定量する。
(7) 酸化性物質及び還元性物質は,妨害するのであらかじめ除去する。残留塩素などの酸化性物質
が共存する場合には,当量の亜硫酸ナトリウム溶液(6.3 g/L)(JIS K 8061に規定する亜硫酸ナ
トリウム0.63 gを水に溶かして100 mLとする。)又は亜ひ酸ナトリウム溶液[JIS K 8044に規
定する三酸化二ひ素0.5 gを水酸化ナトリウム溶液(40 g/L)[21. a) 3)による。]5 mLに溶かし
た後,塩酸(1+11)6 mLを加え,水で100 mLとする。]を加えた後,試験する。また,亜硫
酸イオンなどの還元性物質が共存する場合には,弱アルカリ性にして当量の過酸化水素(1+
100)(JIS K 8230に規定する過酸化水素を用いて調製する。)を加えた後,試験する。
d) 検量線 検量線の作成は,次による。
1) 硝酸イオン標準液(NO3− 10 μg/mL)0.8〜8 mLを全量フラスコ100 mLに段階的にとり,c) 2)〜5)
の操作を行って硝酸イオン(NO3−)の量と吸光度との関係線を作成する。
備考 8. 硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11. による。
43.2.4 ブルシン吸光光度法 強い硫酸酸性で,硝酸イオンとブルシンとの反応によって生じる黄色の化合
物の吸光度を測定して硝酸イオンを定量する。
定量範囲:NO3− 5〜100 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(20+3) 水75 mLをビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する
硫酸500 mLを徐々に加え,放冷し,密栓して保存する。
3) ブルシン・4-アミノベンゼンスルホン酸溶液 JIS K 8832に規定するブルシンn水和物(2,3-ジメト
キシストリキニジン-10-オンn水和物)1 gとJIS K 8586に規定するスルファニル酸(4-アミノベン
ゼンスルホン酸)0.1 gとをJIS K 8180に規定する塩酸3 mLに溶かし,水で100 mLとする。
4) 硝酸イオン標準液(NO3− 1 mg/mL) 43.2.3 a) 7)による。
5) 硝酸イオン標準液(NO3− 0.1 mg/mL) 硝酸イオン標準液(NO3− 1 mg/mL)20 mLを全量フラスコ
200 mLにとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 安全ピペット 1 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 3.2でろ過した試料(6) (7) (8)から2 mL(NO3−として5〜100 μgを含む。)ずつを2個のビーカー50 mL
(9)(A1),(B1)にとる。
2) ビーカー(A1)にはブルシン・4-アミノベンゼンスルホン酸溶液1 mLを安全ピペットを用いて加え,
ビーカー(B1)にはブルシン・4-アミノベンゼンスルホン酸溶液の代わりに水1 mLを加える。
3) 別に,2個のビーカー50 mL(A2),(B2)に硫酸(20+3)10 mL (10) ずつをとり,ビーカー(A2)
にビーカー(A1)の溶液を注意して移し,よく振り混ぜる。次に,ビーカー(A2)の溶液をビーカ
169
K 0102:2016
ー(A1)に移し,再びよく振り混ぜる。この操作を5回繰り返した後,暗所(11)に約10分間放置す
る。
4) ビーカー(B2)にはビーカー(B1)の溶液を注意して移し,よく振り混ぜる。次に,ビーカー(B2)
の溶液をビーカー(B1)に移し,再びよく振り混ぜる。この操作を5回繰り返した後,暗所に約10
分間放置する。
5) ビーカー(A2)及び(B2)に水10 mLをそれぞれ加えて振り混ぜ,器壁を洗った後,洗液をそれぞ
れビーカー(A1)及び(B1)の溶液に合わせて振り混ぜる。0〜10 ℃の暗所に約30分間放置する。
6) ビーカー(A1)の溶液の一部を吸収セルにとり,ビーカー(B1)の溶液を対照液として波長410 nm
付近の吸光度を測定する。
7) 空試験として水2 mLずつを2個のビーカー50 mL(9)(A1),(B1)にとり,2)〜6)の操作を行って吸
光度を測定し,試料について得た吸光度を補正する。
8) 検量線から硝酸イオンの量を求め,試料中の硝酸イオンの濃度(NO3− mg/L)を算出する。
注(8) アルカリ性が強い場合は,硫酸(1+5)(JIS K 8951に規定する硫酸を用いて調製する。)を加
えてpHを約7に調節してから試験する。
(9) この試験には,検量線作成時とほぼ同形のビーカー50 mL 4個を用いる。
(10) 自動ビュレット10 mLを用いると操作しやすい。
(11) ビーカーにボール紙箱,木箱などをかぶせておくとよい。
d) 検量線 検量線の作成は,次による。
1) 硝酸イオン標準液(NO3− 0.1 mg/mL)2.5〜50 mLを全量フラスコ100 mLに段階的(12)にとり,水を
標線まで加える。
2) その中からそれぞれ2 mLをとり,c) 2)〜7)の操作を行って硝酸イオン(NO3−)の量と吸光度との
関係線を作成する。
注(12) 硝酸イオンとブルシンとの反応によって生じた黄色は,厳密にはランバート−ベアの法則に従
わず,上方に湾曲するので,検量線作成時の硝酸イオン標準液の濃度の間隔を細かくする必要
がある。
備考 9. 鉄(II),鉄(III)及びマンガン(II)が共存すると正の誤差を生じるが,これらの濃度がそ
れぞれ1 mg/L以下ならば差し支えない。
10. 硝酸イオンとブルシンとの反応は,温度の上昇とともに速くなる。したがって,検量線作成
時と同一温度で操作する。
43.2.5 イオンクロマトグラフ法 試料中の硝酸イオンをイオンクロマトグラフ法で定量する。この方法に
よって表35.1に示すイオンも同時定量できる。硝酸イオンを定量する場合には,3.3の保存処理を行わず,
試料採取後,直ちに試験する。
試験操作などは,35.3による。
備考 11. 硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11. による。
43.2.6 流れ分析法 試料中の硝酸イオンを,43.2.3と同様な原理で還元発色させる流れ分析法によって定
量する。
定量範囲:NO3− 0.09〜88 mg/L(硝酸体窒素として0.02〜20 mg/L),繰返し精度:10 %以下
試験操作などは,JIS K 0170-2で規定された硝酸体窒素に関する規定による。
備考 12. 硝酸イオンの濃度を硝酸体窒素で表示する場合は,35.の備考11. による。
170
K 0102:2016
44. 有機体窒素 試料を前処理して有機物を分解し,有機体窒素をアンモニウムイオンに変え,蒸留分離
した後,インドフェノール青吸光光度法又は中和滴定法でアンモニウムイオンを定量し,別に,処理前の
試料中のアンモニウムイオンを定量して差し引き,有機体窒素を求める。
有機体窒素は変化しやすいので,試験は直ちに行う。直ちに行えない場合には,3.3によって保存し,で
きるだけ早く試験する。
なお,この試験方法は,1984年に第1版として発行されたISO 5663との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 5663:1984,Water quality−Determination of Kjeldahl nitrogen−Method after mineralization with
selenium(MOD)
44.1 前処理(ケルダール法) 試料に,硫酸銅,硫酸及び硫酸カリウムを加え,加熱して有機物を分解す
る。次に,水酸化ナトリウムを加えてアルカリ性とした後,蒸留し,留出したアンモニアを硫酸(25 mmol/L)
に吸収させる。
備考 1. この方法では,アミノ酸,ポリペプチド,たん白質などは分解しやすいが,ニトロ,ニトロ
ソ,アゾ複素環式化合物(特に,ピリジン環をもつ化合物)などは完全には分解できない。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3又はA4の水
2) 硫酸 JIS K 8951に規定するもの。
3) 硫酸(1+35) JIS K 8951に規定する硫酸を用いて調製する。
4) 硫酸(25 mmol/L) 42.1 a) 2)による。
5) 水酸化ナトリウム溶液(500 g/L) JIS K 8576に規定する水酸化ナトリウム50 gを水に溶かして100
mLとする。使用時に調製する。
6) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
7) 硫酸カリウム JIS K 8962に規定するもの。
8) 硫酸銅(II)五水和物 JIS K 8983に規定するものを粉末にしたもの。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 使用前に水でよく洗う。
2) ケルダールフラスコ 200 mL。使用前に水でよく洗う。
3) 蒸留装置 42.1 b) 1)による。使用前に水でよく洗う(1)。
注(1) 数日間以上装置を使用していないときは,使用前に次の操作を行う。
− 蒸留フラスコに水約350 mLと沸騰石5〜7個とを加え,装置を組み立て,少なくとも100 mL
を蒸留する。留出液及び蒸留フラスコ中の残留物を捨てる。
c) 操作 操作は,次による。
1) 試料の適量(2)をビーカー500 mLにとり,硫酸(1+35)を加えて弱酸性とし,加熱して約30 mLに
なるまで濃縮する。
2) 放冷後,少量の水で内容物をケルダールフラスコ200 mLに洗い移す。
3) 硫酸10 mL,硫酸カリウム5 g及び硫酸銅(II)五水和物2 gを加え,加熱して硫酸の白煙を発生さ
せ,引き続き約30分間強熱して有機物を分解(3)する。
4) 放冷後,少量の水を加え,加熱して溶かし,水で蒸留フラスコに洗い移して約300 mLとする。
171
K 0102:2016
5) 蒸留フラスコを図42.1のように連結し,受器にはメスシリンダー(有栓形)200 mLを用い,硫酸
(25 mmol/L)50 mLを入れておく(4)。
6) 蒸留フラスコ上部の注入漏斗から水酸化ナトリウム溶液(500 g/L)40 mLを加えた後,42.1 c) 4)〜
6)の操作を行う。
7) 空試験として水30 mLをとり,3)〜6)の操作を行う。
注(2) インドフェノール青吸光光度法で定量する場合には,有機体窒素をNとして32 μg以上,中和
滴定法の場合には0.23 mg以上とする。いずれも,有機体窒素とアンモニウムイオンとの合量
をNとして30 mg以下を含むようにする。
(3) フラスコ中の溶液は,無色又は淡黄色になる。
(4) 中和滴定法の場合には,メスシリンダー(有栓形)200 mLの代わりに三角フラスコ500 mLを
用いて,42.1 a) 2)の硫酸(25 mmol/L)50 mLを正しく加え,指示薬としてメチルレッド-ブロモ
クレゾールグリーン混合溶液[15.の注(1)による。]5〜7滴を加える。
備考 2. この方法では,硝酸イオン及び亜硝酸イオンは,有機体窒素の定量の妨害にならない。
3. 試料中のアンモニウムイオンをあらかじめ除去した後,有機体窒素を定量する場合には,42.1
c) 1)〜5)を行い,この残留液についてc) 1)〜6)の前処理を行う。
44.2 インドフェノール青吸光光度法 前処理(ケルダール法)による留出液についてインドフェノール
青吸光光度法によってアンモニウムイオンを定量して,試料中に含まれるアンモニウムイオンと有機体窒
素から生じたアンモニウムイオンとの合量を求め,別に,試料中のアンモニウムイオンを定量して差し引
き,有機体窒素を算出する。
定量範囲:N 4〜80 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,42.2 a)による。
b) 器具及び装置 器具及び装置は,42.2 b)による。
c) 操作 操作は,次による。
1) 44.1 c) 6)で得た留出液の適量(Nとして4〜80 μgを含む。)を全量フラスコ50 mLにとり,水を加
えて約25 mLとした後,42.2 c) 2)〜5)の操作を行って吸光度を測定する。
2) 空試験として44.1 c) 7)で得た留出液から1)と同量を分取し,1)の操作を行って吸光度を測定し,試
料について得た吸光度を補正する。
3) 42.2 d)の検量線から,分取した留出液中のアンモニウムイオンの量(mg)を求める。
4) 別に,42.2によって試料中のアンモニウムイオンの濃度(NH4+ mg/L)を求める。
5) 次の式によって試料中の有機体窒素の濃度(N mg/L)を算出する。
6
776
.0
200
000
1
2
1
×
−
×
×
=
A
V
V
a
N
ここに,
N: 有機体窒素の濃度(N mg/L)
a: 3)の留出液中のアンモニウムイオンの質量(mg)
V1: 44.1 c) 1)で用いた試料量(mL)
V2: 1)で分取した留出液量(mL)
A: 4)で求めた試料中のアンモニウムイオンの濃度(NH4+
mg/L)
0.776 6: アンモニウムイオンを窒素の相当量に換算するときの係数
04
.
18
01
.
14
172
K 0102:2016
44.3 中和滴定法 前処理(ケルダール法)による留出液について中和滴定法によってアンモニアを定量
して,試料中に含まれるアンモニウムイオンと有機体窒素から生じたアンモニウムイオンとの合量を求め,
別に,試料中のアンモニウムイオンを定量して差し引き,有機体窒素を算出する。
定量範囲:N 0.23〜30 mg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 50 mmol/L水酸化ナトリウム溶液 42.3 a) 4)による。
b) 操作 操作は,次による。
1) 44.1 c) 6)で得た留出液の全量を用い,42.3 b) 1)の滴定操作を行う。
2) 空試験として44.1 c) 7)で得た留出液の全量を用いて,1)と同様の滴定操作を行う。
3) 別に,42.3によって試料中のアンモニウムイオンの濃度(NH4+ mg/L)を求める。
4) 次の式によって試料中の有機体窒素の濃度(N mg/L)を算出する。
(
)
6
776
.0
700
.0
000
1
×
−
×
×
×
−
=
A
V
f
a
b
N
ここに,
N: 有機体窒素の濃度(N mg/L)
b: 空試験の滴定に要した50 mmol/L水酸化ナトリウム溶液量
(mL)
a: 滴定に要した50 mmol/L水酸化ナトリウム溶液量(mL)
f: 50 mmol/L水酸化ナトリウム溶液のファクター
0.700: 50 mmol/L水酸化ナトリウム溶液1 mLに相当する窒素の質
量(mg)
V: 44.1 c) 1)で用いた試料量(mL)
A: 3)で求めた試料中のアンモニウムイオンの濃度(NH4+
mg/L)
0.776 6: アンモニウムイオンを窒素の相当量に換算するときの係数
04
.
18
01
.
14
備考 4. 44.1 c) 5)の硫酸(25 mmol/L)の代わりにほう酸溶液(20 g/L)を用いてもよい。その場合は
42.の備考7.の操作を行う。ただし,空試験は,水30 mLを用いて44.1 c) 3)〜6)の操作を行っ
て得られた留出液について試料と同様に滴定した値を用いる。また,試料中の有機体窒素の
濃度の算出には4)の式は適用できない。
45. 全窒素 亜硝酸イオンと硝酸イオンに相当する窒素と,アンモニウムイオンと有機体窒素に相当する
窒素とを求めて合計する総和法,全窒素化合物を硝酸イオンに変えた後の紫外線吸光光度法,硫酸ヒドラ
ジニウム還元法,銅・カドミウムカラム還元法,又は熱分解法,若しくは流れの中で全窒素化合物を硝酸
イオンに変えて紫外線吸光光度法,銅・カドミウムカラム還元法を用いた流れ分析法を適用する。
45.1 総和法 試料に水酸化ナトリウムを加えて蒸留を行い,アンモニウムイオン及び一部の有機窒素化
合物の分解で生じたアンモニアを除いた後,デバルダ合金を加えて亜硝酸イオン及び硝酸イオンを還元し
てアンモニアとし,蒸留によって分離し,インドフェノール青吸光光度法で窒素の量を定量する。別に,
試料に硫酸銅,硫酸カリウム及び硫酸を加えて加熱分解して,有機体窒素をアンモニウムイオンに変えた
後,アルカリ性として蒸留し,試料中に含まれるアンモニウムイオンとともに蒸留分離し,インドフェノ
ール青吸光光度法によってその窒素の量を定量する。先に求めた亜硝酸イオン及び硝酸イオン相当の窒素
173
K 0102:2016
量を合わせて,全窒素の濃度を算出する。
定量範囲:N 8〜160 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸(25 mmol/L) 42.1 a) 2)による。
3) 硫酸(1+35) 42.1 a) 3)による。
4) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
5) 水酸化ナトリウム溶液(300 g/L) 43.2.1 a) 5)による。
6) デバルダ合金 43.2.1 a) 6)による。
7) 硫酸カリウム JIS K 8962に規定するもの。
8) 硫酸銅(II)五水和物 44.1 a) 8)による。
9) ナトリウムフェノキシド溶液 42.2 a) 3)による。
10) 次亜塩素酸ナトリウム溶液(有効塩素10 g/L) 42.2 a) 4)による。
11) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
12) アンモニウムイオン標準液(NH4+ 1 mg/mL) 42.2 a) 5)による。
13) アンモニウムイオン標準液(NH4+ 10 μg/mL) 42.2 a) 6)による。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 使用前に水でよく洗う。
2) ケルダールフラスコ 200 mL。使用前に水でよく洗う。
3) 蒸留装置 42.1 b) 1)による。使用前に水でよく洗う。
4) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料50 mL(1)をとり,中性でない場合には水酸化ナトリウム溶液(40 g/L)又は硫酸(1+35)でpH
約7に調節する。
2) 43.2.1 c) 2)〜6)の操作を行う。
3) 試料について得た留出液から25 mL(2)を全量フラスコ50 mLにとる。
4) 42.2 c) 2)〜5)の操作を行って吸光度を測定する。
5) 空試験として水約50 mLをとり,水酸化ナトリウム溶液(300 g/L)10 mLを加えた後,43.2.1 c) 4)
〜6)の操作を行い,得られた留出液について3)及び4)の操作を行って吸光度を測定し,4)で得た吸
光度を補正する。
6) 42.2 d)の検量線から3)で分取した留出液25 mL中のアンモニウムイオンの量(mg)を求める。
7) 別に,試料50 mL(3)をケルダールフラスコ200 mLにとり,44.1 c) 3)〜6)の操作を行う。
8) この留出液の25 mL(2)を全量フラスコ50 mLにとり,42.2 c) 2)〜5)の操作を行って吸光度を測定す
る。
9) 空試験として水50 mLをとり,7)及び8)の操作を行って吸光度を測定し,8)で得た吸光度を補正す
る。
10) 42.2 d)の検量線から8)で分取した留出液25 mL中のアンモニウムイオンの量(mg)を求める。
11) 次の式によって試料中の全窒素の濃度(N mg/L)を算出する(4)。
6
776
.0
25
200
50
000
1
6
776
.0
25
200
50
000
1
×
×
×
+
×
×
×
=
b
a
N
174
K 0102:2016
ここに,
N: 全窒素の濃度(N mg/L)
a: 6)の操作で得たアンモニウムイオンの質量(mg)
b: 10)の操作で得たアンモニウムイオンの質量(mg)
0.776 6: アンモニウムイオンを窒素の相当量に換算するときの係数
04
.
18
01
.
14
注(1) 全窒素の低濃度のものを定量する必要がある場合は,試料の量を増加する。ただし,この場合
は,11)の式中の
50
000
1
×
a
に代え,
V
a
000
1
×
を用いる。
ここに,
V: 試料量(mL)
(2) 留出液200 mL中のアンモニウムイオンが0.8 mg以上の場合は,留出液の適量(NH4+が0.4 mg
未満になる量)を硫酸(25 mmol/L)25 mLが入った全量フラスコ100 mLにとり,水を標線ま
で加え,これから25 mLを分取する。
(3) 全窒素の低濃度のものを定量する必要がある場合には,試料の量を増加し,44.1 c) 1)及び2)の
操作を行う。ただし,この操作を行った場合は,11)の式中の
50
000
1
×
b
に代え,
V
b
000
1
×
を用い
る。
ここに,
V: 試料量(mL)
(4) 3)又は8)の操作で,注(2)の操作を行った場合には,11)の式中のa又はbに代え,それぞれ
c
a100
×
又は
d
b100
×
を用いる。ただし,c又はdは,それぞれ注(2)の操作において,全量フラス
コ100 mLにとった溶液の量(mL)。
45.2 紫外線吸光光度法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120 ℃に加熱し
て窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液のpHを2〜3とした後,硝酸イ
オンによる波長220 nmの吸光度を測定して定量する。この方法は,試料中の有機物が分解されやすく,
少量であり,また,試験に影響する量の臭化物イオン,クロムなどを含まない場合に適用する。
定量範囲:N 5〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩酸(1+16) JIS K 8180に規定する塩酸を用いて調製する。
3) 塩酸(1+500) JIS K 8180に規定する塩酸を用いて調製する。
4) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 JIS K 8826に規定する水酸化ナトリウム(窒素
測定用)20 gを水500 mLに加えた後,JIS K 8253に規定するペルオキソ二硫酸カリウム(窒素り
ん測定用)15 gを溶かす。使用時に調製する(5)。
5) 窒素標準液(N 0.1 mg/mL) JIS K 8548に規定する硝酸カリウムをあらかじめ105〜110 ℃で約2
時間加熱し,デシケーター中で放冷する。その0.722 gをとり,少量の水に溶かし,全量フラスコ1 000
mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
6) 窒素標準液(N 20 μg/mL) 窒素標準液(N 0.1 mg/mL)50 mLを全量フラスコ250 mLにとり,水を
標線まで加える。使用時に調製する。
注(5) この溶液の窒素含有量は,0.4 mg/L以下でなければならない。
b) 器具及び装置 器具及び装置は,次による。
175
K 0102:2016
1) 分解瓶 耐圧の四ふっ化エチレン樹脂瓶又は耐熱・耐圧のガラス瓶(容量約100 mL)で,高圧蒸気
滅菌器中(約120 ℃)で使用できるもの(6)。
2) 高圧蒸気滅菌器 JIS T 7322又はJIS T 7324に規定するもので,約120 ℃に加熱できるもの。
3) 光度計 分光光度計
4) 吸収セル 石英ガラス製
注(6) ガラス製アンプル(容量約100 mL)で高圧蒸気滅菌器中(約120 ℃)で使用できるものを用い
てもよい。
c) 操作 操作は,次による。
1) 試料(7) 50 mLを分解瓶にとる。
2) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液10 mLを加え,直ちに密栓した後,混合する。
3) 高圧蒸気滅菌器に入れて加熱し,約120 ℃に達してから30分間加熱分解を行う。
4) 分解瓶を高圧蒸気滅菌器から取り出し,放冷する。
5) 上澄み液(8) 25 mLをビーカー50 mLに分取する。
6) 塩酸(1+16)5 mL(9)を加えて溶液のpHを2〜3に調節する。
7) 溶液の一部を吸収セルに移し,波長220 nmにおける吸光度を測定する。
8) 空試験として水50 mLを分解瓶にとり,2)〜7)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
9) 検量線から5)で分取した溶液中の全窒素の量を求め,次の式によって試料中の全窒素の濃度(N
mg/L)を算出する(10)。
50
000
1
25
60×
×
=a
N
ここに,
N: 全窒素の濃度(N mg/L)
a: 5)で分取した溶液25 mL中の全窒素の質量(mg)
注(7) 試料50 mL中の全窒素が0.1 mg以上の場合には,試料の適量(Nとして0.2 mg未満を含む。)
を全量フラスコ100 mLにとり,水を標線まで加えたものを用いる。ただし,試料50 mL中の
全窒素が,0.1 mg以上でpH5〜9の範囲にない場合には,試料の適量(Nとして0.2 mg未満を
含む。)をとり,塩酸(1+11)[21. a) 2)による。]又は水酸化ナトリウム溶液(40 g/L)[21. a) 3)
による。]で中和した後,全量フラスコ100 mLに移し入れ,水を標線まで加えたものを用いる。
(8) 水酸化物の沈殿を含まないように注意する。必要に応じ,孔径1 μm以下のガラス繊維ろ紙を用
いてろ過し,初めのろ液5〜10 mLを捨てた後のろ液を用いる。
(9) 分解後の溶液に水酸化物の沈殿を生じた場合には,注(8)によってろ過したろ液25 mLをとり,
水酸化物の生成量に応じて濃度を低めた塩酸5 mLを添加し,pH2〜3に調節する。
(10) 1)の操作において,注(7)の操作を行った試料を用いた場合には,次の式によって試料中の全窒
素の濃度(N mg/L)を算出する。
V
a
N
100
50
000
1
25
60
×
×
×
=
ここに,
N: 全窒素の濃度(N mg/L)
a: 5)で分取した溶液25 mL中の全窒素の質量(mg)
V: 試料量(mL)
d) 検量線 検量線の作成は,次による。
1) 窒素標準液(N 20 μg/mL)1〜10 mLを段階的に全量フラスコ100 mLにとり,それぞれに水を標線
176
K 0102:2016
まで加える。
2) その25 mLをそれぞれビーカー50 mLにとり,塩酸(1+500)5 mLを加えた後,一部を吸収セルに
移し,波長220 nmの吸光度を測定する。
3) 別に,空試験として水25 mLをビーカー50 mLにとり,塩酸(1+500)5 mLを加えた後,波長220
nmの吸光度を求め,窒素標準液について得た吸光度を補正する。採取した溶液25 mL中の窒素(N)
の量と吸光度との関係線を作成する。
備考 1. 試料50 mL中の全窒素が0.1 mg未満でpH5〜9の範囲にない場合には,試料の適量(50〜100
mL)をとり,塩酸(1+11)又は水酸化ナトリウム溶液(40 g/L)を用いて中和し(中和に用
いた両溶液の量を求めておく。),この溶液から50 mLを分解瓶にとり,c) 2)〜5)の操作を行
う。ただし,この場合は,次の式を用いる。
V
b
V
a
N
+
×
×
×
=
50
000
1
25
60
ここに,
N: 全窒素の濃度(N mg/L)
a: 前処理後に分取した試料25 mL中の全窒素の質量(mg)
b: 中和に要した塩酸(1+11)及び水酸化ナトリウム溶液(40 g/L)
量(mL)
V: 試料量 (mL)
2. c) 7)で吸収セルに移す溶液の全窒素の濃度が0.4 mg/L未満の場合には,吸収セル50 mmを用
いる。ただし,空試験及び検量線の作成には,いずれにも吸収セル50 mmを用いる。検量線
は窒素標準液(N 20 μg/mL)を5倍に薄めた窒素標準液(N 4μg/mL)1〜10 mLをとり,d)
の検量線と同じ操作で作成する。
3. この方法では臭化物イオン10 mg/L,クロム0.1 mg/L程度で妨害することがある。このよう
な試料には,この方法は適用しない。
45.3 硫酸ヒドラジニウム還元法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120 ℃
に加熱して窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液中の硝酸イオンを銅を
触媒として硫酸ヒドラジニウムによって還元して亜硝酸イオンとし,ナフチルエチレンジアミン吸光光度
法によって定量し,全窒素の濃度を求める。この方法は,試料中の有機物が分解されやすく,少量である
場合に適用する。
定量範囲:N 0.33〜3.3 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 45.2 a) 4)による。
3) 銅-亜鉛溶液 JIS K 8983に規定する硫酸銅(II)五水和物0.08 gとJIS K 8953に規定する硫酸亜鉛
七水和物1.76 gとを水に溶かして200 mLとし,その5 mLを水で薄めて250 mLとする。
4) 硫酸ヒドラジニウム溶液(7 g/L) JIS K 8992に規定する硫酸ヒドラジニウム3.5 gを水に溶かして
500 mLとする。
5) 硫酸ヒドラジニウム溶液(0.7 g/L) 硫酸ヒドラジニウム溶液(7 g/L)を水で10倍に薄める。使用
時に調製する。
6) 4-アミノベンゼンスルホンアミド溶液 43.1.1 a) 2)による。
7) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 43.1.1 a) 3)による。
8) 窒素標準液(N 20 μg/mL) 45.2 a) 6)による。
177
K 0102:2016
9) 窒素標準液(N 4 μg/mL) 窒素標準液(N 20 μg/mL)20 mLを全量フラスコ100 mLにとり,水を
標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 分解瓶 45.2 b) 1)による。
2) 共栓試験管 材質及び形状が同じものを用いる。
3) 水浴 35±1 ℃に調節できるもの。
4) 高圧蒸気滅菌器 45.2 b) 2)による。
5) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料(11) (12) 50 mLを分解瓶にとる。
2) 45.2 c) 2)〜4)の操作を行う。
3) 上澄み液(8) (13) 10 mLを共栓試験管にとる。
4) 銅-亜鉛溶液1 mLを加えて振り混ぜた後,硫酸ヒドラジニウム溶液(0.7 g/L)1 mLを加えて振り混
ぜ,35±1 ℃の水浴中に浸す。
5) 2時間後,水浴から取り出し,室温まで冷却する。
6) 43.1.1 c) 2)及び3)の操作を行う。
7) 空試験として水50 mLを分解瓶にとり,2)〜6)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
8) 検量線から分解瓶にとった溶液50 mL中の全窒素の量を求め(14),次の式によって試料中の全窒素の
濃度(N mg/L)を算出する。
50
000
1
×
=a
N
ここに,
N: 全窒素の濃度(N mg/L)
a: 分解瓶にとった溶液50 mL中の全窒素の質量(mg)
注(11) 注(7)による。ただし,試料中の全窒素の濃度(N mg/L)の算出には,次の式を用いる。
V
a
N
100
50
000
1
×
×
=
ここに,
N: 全窒素の濃度(N mg/L)
a: 分解瓶にとった溶液50 mL中の全窒素の質量(mg)
V: 試料量(mL)
(12) 備考1.による。ただし,全窒素の濃度(N mg/L)の算出には,次の式を用いる。
V
b
V
a
N
+
×
×
=
50
000
1
ここに,
N: 全窒素の濃度(N mg/L)
a: 分解瓶にとった溶液50 mL中の全窒素の質量(mg)
b: 中和に要した塩酸(1+11)及び水酸化ナトリウム溶液
(40 g/ L)量(mL)
V: 試料量(mL)
(13) 分解瓶にとった溶液50 mL中の窒素が20 μg以上の場合には,上澄み液の適量(Nとして30 μg
未満となる量)を全量フラスコ100 mLにとり,水酸化ナトリウム溶液(40 g/L)[21. a) 3)によ
る。]5 mLを加えた後,水を標線まで加えたものを用いる。
(14) 3)の操作で注(13)を行った場合には,検量線によって求めた窒素の量(mg)に を乗じること
c
100
178
K 0102:2016
によって,分解瓶にとった試料50 mL中の全窒素の量(mg)を求める。ただし,cは全量フラ
スコ100 mLにとった上澄み液の量(mL)。
d) 検量線 検量線の作成は,次による。
1) 窒素標準液(N 4 μg/mL)1〜10 mLを段階的に全量フラスコ100 mLにとり,それぞれに水を標線ま
で加える。
2) この溶液についてc) 1)〜7)の操作を行って分解瓶にとった溶液50 mL中の窒素(N)の量と吸光度
との関係線を作成する。
備考 4. 試料が海水などの場合は,含まれる無機物が硝酸イオンの還元率に影響するので,次の標準
添加法を行う。
試料40 mLを分解瓶にとり,水10 mLを加える。以下,c) 2)〜7)の操作を行って吸光度を
測定し,次に示す検量線から試料40 mL中の全窒素の量(mg)を求める。別に,空試験とし
て水50 mLを分解瓶にとり,c) 2)〜7)の操作を行って吸光度を測定し,d)の検量線から,相
当する窒素の量(mg)を求める。次の式によって試料中の全窒素の濃度(N mg/L)を算出す
る。
(
)
40
000
1
×
−
=
b
a
N
ここに,
N: 全窒素の濃度(N mg/L)
a: 試料40 mL中の全窒素の質量(mg)
b: 空試験で得た窒素の質量(mg)
検量線 窒素標準液(N 4 μg/mL)1〜8 mLを段階的に分解瓶にとり,それぞれに試料40 mL
を加えた後,水を加えて50 mLとし,c) 2)〜6)の操作を行って吸光度を測定し,その値から
試料40 mLを用いて得た吸光度を差し引いて補正する。
なお,海水など多量のマグネシウムイオンが存在する試料の場合は,分解操作を行った溶
液のpHが低下して,マグネシウムの一部が上澄み液又はろ液に混入し,硝酸イオンの還元
率を低下させる。このため,分解後の溶液に水酸化ナトリウム溶液(40 g/L)[21. a) 3)によ
る。]を加えてpH12.6〜12.8とした後の上澄み液を用いる。この操作を行った場合には,全
窒素の算出式については,希釈による補正を行う。
試料40 mL中の窒素の量が10 μg以上の場合には,試料の適量(Nとして25 μg未満を含
む。)をとり,注(7)に準じた操作を行い,これから40 mLを分解瓶にとる。また,試料40 mL
中の窒素の量が10 μg未満でpH5〜9の範囲にない場合には,備考1.に準じた中和操作を行い,
これから40 mLを分解瓶にとる。これらの操作を行った場合には,検量線の作成においても,
この溶液を用い,全窒素の算出式については,それぞれ希釈に伴う補正を行う。
45.4 銅・カドミウムカラム還元法 試料にペルオキソ二硫酸カリウムのアルカリ性溶液を加え,約120 ℃
に加熱して窒素化合物を硝酸イオンに変えるとともに有機物を分解する。この溶液中の硝酸イオンを銅・
カドミウムカラムによって還元して亜硝酸イオンとし,ナフチルエチレンジアミン吸光光度法によって定
量し,全窒素の濃度を求める。この方法は,試料中の有機物が分解されやすく,少量である場合に適用す
る。
定量範囲:N 0.2〜2 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
179
K 0102:2016
2) 塩酸(1+11) JIS K 8180に規定する塩酸を用いて調製する。
3) 塩化アンモニウム-アンモニア溶液 43.2.3 a) 3)による。
4) 水酸化ナトリウム-ペルオキソ二硫酸カリウム溶液 45.2 a) 4)による。
5) カラム活性化液 43.2.3 a) 4)による。
6) 銅・カドミウムカラム充塡剤 43.2.3 a) 5)による。
7) カラム充塡液 43.2.3 a) 6)による。
8) 4-アミノベンゼンスルホンアミド溶液 43.1.1 a) 2)による。
9) 二塩化N-1-ナフチルエチレンジアンモニウム溶液 43.1.1 a) 3)による。
10) 窒素標準液(N 0.1 mg/mL) 45.2 a) 5)による。
11) 窒素標準液(N 2 μg/mL) 窒素標準液(N 0.1 mg/mL)10 mLを全量フラスコ500 mLにとり,水を
標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 分解瓶 45.2 b) 1)による。
2) 高圧蒸気滅菌器 45.2 b) 2)による。
3) 銅・カドミウムカラム 43.2.3 b) 1)による。
4) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料(15) (16) 50 mLを分解瓶にとる。
2) 45.2 c) 2)〜4)の操作を行う。
3) 分解瓶に塩酸(1+11)10 mLを加えて振り混ぜた後,溶液を全量フラスコ100 mLに移す。
4) 分解瓶の内壁を少量の水で数回洗浄して,洗液を3)の全量フラスコ100 mLに加える。
5) 塩化アンモニウム-アンモニア溶液10 mLを加え,水を標線まで加えて還元用溶液とする(17)。
6) 43.2.3 c) 3)及び4)の操作を行う。
7) 空試験として水50 mLを分解瓶にとり,2)〜6)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
8) 検量線から還元用溶液中の全窒素の量を求め,次の式によって試料中の全窒素の濃度(N mg/L)を
算出する(18)。
50
000
1
×
=a
N
ここに,
N: 全窒素の濃度(N mg/L)
a: 還元用溶液100 mL中の全窒素の質量(mg)
注(15) 注(7)による。ただし,試料中の全窒素の濃度(N mg/L)の算出には,次の式を用いる。
V
a
N
100
50
000
1
×
×
=
ここに,
N: 全窒素の濃度(N mg/L)
a: 還元用溶液100 mL中の全窒素の質量(mg)
V: 試料量(mL)
(16) 備考1.による。ただし,試料中の全窒素の濃度(N mg/L)の算出には,次の式を用いる。
V
b
V
a
N
+
×
×
=
50
000
1
ここに,
N: 全窒素の濃度(N mg/L)
180
K 0102:2016
a: 還元用溶液100 mL中の全窒素の質量(mg)
b: 中和に要した塩酸(1+11)及び水酸化ナトリウム溶液(40 g/
L)量(mL)
V: 試料量(mL)
注(17) 1)で分解瓶にとった溶液50 mL中の全窒素が20 μg以上の場合には,全量フラスコ200〜500 mL
を用い,5)の操作において塩化アンモニウム-アンモニア溶液を最終液量100 mL当たり10 mL
となるように添加した後,水を標線まで加えたものを還元用溶液とする。
(18) 3)の操作において注(17)を行った場合は,算出式に を乗じて補正する。ただし,cは用いた
全量フラスコの容量(mL)。
d) 検量線 検量線の作成は,次による。
1) 窒素標準液(N 2 μg/mL)1〜10 mLを段階的に全量フラスコ100 mLにとり,c) 5)及び6)の操作を行
って吸光度を測定する。
2) 別に,水約50 mLを全量フラスコ100 mLにとり,c) 5)及び6)の操作を行って吸光度を測定し,窒
素標準液(N 2 μg/mL)について得た吸光度を補正し,窒素(N)の量と吸光度との関係線を作成す
る。
45.5 熱分解法 試料中の窒素化合物を熱分解してアンモニア又は窒素を生成させ,それらを定量する。
又は一酸化窒素に変えた後,化学発光法によって窒素を定量し,それぞれ全窒素を求める。
定量範囲:N 1〜200 mg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 全窒素標準液(N 0.2 mg/mL) JIS K 8548に規定する硝酸カリウムをあらかじめ105〜110 ℃で約2
時間加熱し,デシケーター中で放冷する。その1.444 gをとり,少量の水に溶かして全量フラスコ1
000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
b) 器具及び装置 器具及び装置は,次のとおりとする。
1) マイクロシリンジ 20〜150 μL
2) ホモジナイザー又はミキサー
3) 全窒素分析装置
c) 準備操作 準備操作は,次による。
1) 全窒素分析装置を作動できる状態にする。
2) 全窒素標準液(N 0.2 mg/mL)の一定量(例えば,20 μL)をマイクロシリンジで全窒素分析装置の
試料注入部から注入し,指示値(ピーク高さ)が最大目盛値の約80 %になるように全窒素分析装置
の感度を調節する。
3) 2)の操作を繰り返し,指示値が一定になることを確かめる。
4) 試料(19)をよく振り混ぜて均一にした後,その一定量[例えば,2)と同量]をマイクロシリンジで試
料注入部から注入して指示値を読み取り,3)と比較して試料中の概略の全窒素の濃度を求める。
注(19) 全窒素の濃度がN 200 mg/L以上の試料の場合には,水で薄めた後,試験する。
d) 検量線 検量線の作成は,次による。
1) c) 4)で求めた試料中の概略の全窒素の濃度がほぼ中央になるように,全窒素標準液(N 0.2 mg/mL)
を全量フラスコ100 mLに段階的にとり,水を標線まで加えて各濃度の全窒素標準液を調製する。
2) 1)で調製した全窒素標準液の最高濃度のものの一定量[例えば,c) 2)と同量]をマイクロシリンジ
100
c
181
K 0102:2016
で試料注入部に注入し,指示値が最大目盛値の約80 %になるように全窒素分析装置の感度を調節す
る。
3) 順次,1)で調製した各濃度の全窒素標準液の一定量[2)で定めた量]をマイクロシリンジで試料注
入部から注入し,指示値を読み取る。
4) 空試験として3)と同量の水をマイクロシリンジでとり,3)と同様に操作して指示値を読み取り,3)
の指示値を補正して窒素(N)の濃度と指示値との関係線を作成する。
e) 操作 操作は,次による。
1) 試料に懸濁物が含まれている場合には,ホモジナイザー又はミキサーでよくかき混ぜてこれらを均
一に分散させる。
2) 試料の一定量[例えば,d) 2)と同量]をマイクロシリンジで装置の試料注入部に注入し,指示値を
読み取る。
3) 空試験として2)と同量の水をマイクロシリンジでとり,2)と同様に操作して指示値を読み取り,2)
の指示値を補正する。
4) あらかじめ作成した検量線から,注入試料中の全窒素の濃度を求め,試料中の全窒素の濃度(N
mg/L)を算出する。
備考 5. 全窒素分析装置には各種のものがある。アンモニアを生成させて定量する方式としては,試
料を水素気流中で熱分解し,全窒素を触媒によってアンモニアとした後,電量滴定法又は電
気伝導度測定法によるものがある。
窒素を生成させて定量する方式としては,試料をヘリウム気流中で熱分解し,触媒によっ
て全窒素を窒素とした後,熱伝導度測定法によるものがある。また,化学発光方式による方
式としては,試料を酸素気流中で熱分解して全窒素を一酸化窒素とし,更にこれをオゾンと
反応させ,二酸化窒素に酸化するとき生じる化学発光(波長650〜900 nm)を測定するもの
がある。
45.6 流れ分析法 試料中の窒素化合物の酸化分解,その結果生じる硝酸イオンの定量を45.2又は45.4と
同様な原理の流れ分析法によって行い,全窒素を定量する。
定量範囲:N:0.1〜2.0 mg/L[JIS K 0170-3の6.3.3(ペルオキソ二硫酸カリウム分解・カドミウム
還元吸光光度FIA法)及び6.3.5(ペルオキソ二硫酸カリウム分解・カドミウム還元吸光光度CFA法)],
1〜20 mg/L[JIS K 0170-3の6.3.2(ペルオキソ二硫酸カリウム分解・紫外検出FIA法)及び6.3.4(ペ
ルオキソ二硫酸カリウム分解・紫外検出CFA法)],繰返し精度:10 %以下
試験操作などは,JIS K 0170-3による。ただし,JIS K 0170-3の6.3.2及び6.3.4は海水に適用できない。
46. りん化合物及び全りん りん化合物は,りん酸,ポリりん酸,動物質及び植物質中のりんなど,水中
に存在するりん化合物のりんを意味し,りん酸イオン,加水分解性りん及び全りんに区分する。また,ろ
過した試料について試験することによって,それぞれを溶存及び懸濁のものに区別できる。りん化合物は
変化しやすいので,試験は試料採取後,直ちに行う。直ちに行えない場合には,3.3によって保存し,で
きるだけ早く試験する。
なお,モリブデン青吸光光度法,加水分解性りん及び全りんのペルオキソ二硫酸カリウム分解法は,2004
年に第2版として発行されたISO 6878,イオンクロマトグラフ法は,1995年に第1版として発行された
ISO 10304-2,流れ分析法は,2003年に第1版として発行されたISO 15681-1及びISO 15681-2との整合を
図ったものである。
182
K 0102:2016
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6878:2004,Water quality−Determination of phosphorus−Ammonium molybdate spectrometric
method(MOD)
ISO 10304-2:1995,Water quality−Determination of dissolved anions by liquid chromatography of ions
−Part 2: Determination of bromide, chloride, nitrate, nitrite, orthophosphate and sulfate in waste
water(MOD)
ISO 15681-1:2003,Water quality−Determination of orthophosphate and total phosphorus contents by
flow analysis (FIA and CFA)−Part 1: Method by flow injection analysis (FIA)(MOD)
ISO 15681-2:2003,Water quality−Determination of orthophosphate and total phosphorus contents by
flow analysis (FIA and CFA)−Part 2: Method by continuous flow analysis (CFA)(MOD)
46.1 りん酸イオン(PO43−) りん酸イオンの定量には,モリブデン青吸光光度法,イオンクロマトグラ
フ法又はモリブデン青発色による流れ分析法を適用する。
46.1.1 モリブデン青吸光光度法 りん酸イオンが七モリブデン酸六アンモニウム及びタルトラトアンチ
モン(III)酸カリウムと反応して生成するヘテロポリ化合物をL(+)-アスコルビン酸で還元し,生成し
たモリブデン青の吸光度を測定してりん酸イオンを定量する。
定量範囲:PO43− 2.5〜75 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) アスコルビン酸溶液(72 g/L) JIS K 9502に規定するL(+)-アスコルビン酸7.2 gを水に溶かし
て100 mLとする。0〜10 ℃の暗所に保存する。着色した溶液は使用しない。
3) モリブデン酸アンモニウム溶液 JIS K 8905に規定する七モリブデン酸六アンモニウム四水和物6
gとJIS K 8533に規定するビス[(+)-タルトラト]二アンチモン(III)酸二カリウム三水和物0.24
gとを水約300 mLに溶かし,これに硫酸(2+1)(JIS K 8951に規定する硫酸を用いて調製する。)
120 mLを加え,次に,JIS K 8588に規定するアミド硫酸アンモニウム(1) 5 gを加えて溶かした後,
水を加えて500 mLとする。
4) モリブデン酸アンモニウム-アスコルビン酸混合溶液 モリブデン酸アンモニウム溶液とアスコル
ビン酸溶液(72 g/L)とを体積比で5:1の割合になるように混合する。使用時に調製する。
5) りん酸イオン標準液(PO43− 0.1 mg/mL) JIS K 9007に規定するりん酸二水素カリウム(pH標準
液用)を105±2 ℃で約2時間加熱し,デシケーター中で放冷する。その0.143 3 gをとり,水に溶
かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
6) りん酸イオン標準液(PO43− 5 μg/mL) りん酸イオン標準液(PO43− 0.1 mg/mL)10 mLを全量フ
ラスコ200 mLにとり,水を標線まで加える。使用時に調製する。
注(1) 試料中に硝酸イオン及び亜硝酸イオンが共存しない場合には,アミド硫酸アンモニウムを添加
しなくてもよい(備考3.参照)。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料(2) (3)の適量(PO43−として2.5〜75 μgを含む。)をメスシリンダー(有栓形)25 mLにとり,水
183
K 0102:2016
を25 mLの標線まで加える。
2) モリブデン酸アンモニウム-アスコルビン酸混合溶液2 mLを加えて振り混ぜた後,20〜40 ℃(4)で,
約15分間放置する。
3) 溶液の一部を吸収セルに移し,波長880 nm付近 (5)の吸光度を測定する(6)。
4) 空試験として水25 mLをとり,2)及び3)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
5) 検量線からりん酸イオンの量を求め,試料中のりん酸イオンの濃度(PO43− mg/L)を算出する(7)。
注(2) 溶存のりん酸イオンを定量する場合には,3.2によってろ過した試料を用いる。
(3) 試料が酸性の場合は,指示薬としてp-ニトロフェノール溶液(1 g/L)[46.2 a) 7)による。]2,3
滴を加え,水酸化ナトリウム溶液(40 g/L)[21. a) 3)による。]を用いて僅かに黄色になるまで
中和する。ただし,このときアルミニウムなどの水酸化物の沈殿が生じる場合には,沈殿が生
じる直前でとどめる。
(4) 検量線作成時と同じ温度となるようにする。
(5) 波長880 nm付近の吸光度の測定ができない場合には,波長710 nm付近の吸光度を測定する。
(6) 試料に濁り又は色がある場合は,1)と同量の試料をとり,モリブデン酸アンモニウム-アスコル
ビン酸混合溶液2 mLに代えてモリブデン酸アンモニウム溶液2 mLを用いて1)及び2)の操作を
行ってこの溶液を対照液として吸光度を測定するか,この溶液の吸光度を測定して試料につい
て得た吸光度を補正する。ただし,これらの場合は,試料に対しての4)による空試験の補正は
行わない。
なお,この操作による場合,濁りの著しい試料では誤差が大きくなる。
(7) りん酸イオンの濃度をりん酸体りんで表示する場合は,35.の備考11.による。
d) 検量線 検量線の作成は,次による。
1) りん酸イオン標準液(PO43− 5 μg/mL)0.5〜15 mLをメスシリンダー(有栓形)25 mLに段階的に
とり,水を25 mLの標線まで加える。
2) c) 1)〜4)の操作を行ってりん酸イオン(PO43−)の量と吸光度との関係線を作成する。
備考 1. 試料中にひ素(V)が含まれるときは,りん酸イオンと同様に発色するので,次の操作でそ
の妨害を除去する。この操作によってひ素(V)10 mg/Lの妨害が除去できる。
試料20 mLに硫酸(1 mol/L)(JIS K 8951に規定する硫酸5.6 mLを水約80 mL中に加え,
放冷後,水で100 mLとする。)1 mL,チオ硫酸ナトリウム溶液(7.65 g/L)(JIS K 8637に規
定するチオ硫酸ナトリウム五水和物1.2 gを水100 mLに溶かし,保存剤としてJIS K 8625に
規定する炭酸ナトリウム約50 mgを加える。)0.5 mLを加え,5〜10分間放置してひ素(V)
をひ素(III)とする。注(3)に従って中和し,水で25 mLとする。続いてc) 2)〜5)の操作によ
ってりん酸イオンを定量する。
2. 塩化物イオン及び硫酸イオンが多量(40 g/L程度)に共存しても妨害しない。しかし,多量
のアンモニウムイオン及びカリウムが共存すると,濁りが生じて妨害となる。
3. モリブデン酸アンモニウム溶液中にアミド硫酸アンモニウムを添加しない場合は,亜硝酸イ
オン1.5 mg以上存在すると,発色が妨害される。アミド硫酸アンモニウムを添加したモリブ
デン酸アンモニウム溶液を用いるときは,亜硝酸イオン約7 mgまでは妨害しない。それ以上
のときは,別にアミド硫酸アンモニウム溶液を調製して追加する。
4. 鉄(III)約10 mg以上は,発色を妨害する。
184
K 0102:2016
備考 5. りん酸塩が懸濁物として含まれるときは,試薬添加後,約15分間経過しても徐々に吸光度が
増加することがある。
6. りん酸イオンの濃度が低い試料の場合には,試料の量及びモリブデン酸アンモニウム-アスコ
ルビン酸混合溶液の量を増加してモリブデン青を発色させ,2,6-ジメチル-4-ヘプタノン[ジ
イソブチルケトン(DIBK)]で抽出して定量できる。操作は,46.3.1の備考12.による。
7. 次のような操作で発色させ,定量することもできる。
試料の適量(PO43−として5〜150 μgを含む。)を全量フラスコ50 mLにとり,水を加えて
約40 mLとし,モリブデン酸アンモニウム-アスコルビン酸混合溶液3.5 mLを加え,水を標
線まで加えて振り混ぜた後,20〜40 ℃で約15分間放置して発色させ,波長880 nm付近(又
は710 nm付近)の吸光度を測定する。水を用いた空試験を行って吸光度を補正する。
検量線は,りん酸イオン標準液(PO43− 5 μg/mL)1〜30 mLについて試料と同様に操作し
て作成する。
8. a) 3)のモリブデン酸アンモニウム溶液とa) 2)のアスコルビン酸溶液とを,混合せずに,別々
に添加することもできる。例えば,46.1.1の備考7.の操作で,モリブデン酸アンモニウム溶
液3 mL,続いてアスコルビン酸溶液0.5 mLを加え,水で50 mLとする。
46.1.2 欠番
46.1.3 イオンクロマトグラフ法 35.3による(7)。
46.1.4 流れ分析法 試料中のりん酸イオンを,46.1.1と同様な原理で発色させる流れ分析法によって定量
する。
定量範囲:PO43− 0.03〜3 mg/L(Pとして0.01〜1 mg/L),繰返し精度:10 %以下
試験操作などは,JIS K 0170-4で規定されたりん酸イオンに関する規定による。
備考 9. りん酸イオンの濃度をりん酸体りんで表示する場合は,35.の備考11.による。
46.2 加水分解性りん 試料を酸性として煮沸したとき,加水分解によってりん酸イオンとなるものをい
う。
試料に硫酸-硝酸の混酸を加え,煮沸してりん酸イオンとした後,定量し,この値から加水分解前のりん
酸イオンを差し引き,りん酸イオンに換算した値で表示する。
定量範囲:PO43− 2.5〜75 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硫酸-硝酸の混液 JIS K 8951に規定する硫酸300 mLを水約600 mL中に注意してかき混ぜながら
加えた後,放冷する。これにJIS K 8541に規定する硝酸4 mLと水とを加えて全量を1 Lとする。
3) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
4) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
5) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。
6) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
7) p-ニトロフェノール溶液(1 g/L) JIS K 8721に規定するp-ニトロフェノール0.1 gを水に溶かして
100 mLとする。
8) りん酸イオン標準液(PO43− 5 μg/mL) 46.1.1 a) 6)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
185
K 0102:2016
c) 操作 操作は,次による。
1) 試料(3) (8)の適量(PO43−として1 mg以下を含む。)をビーカー200 mLにとり,水を加えて50〜100 mL
とした後,硫酸-硝酸の混酸1 mLを加える。
2) 静かに煮沸する。液量が25 mL以下になったら水を加え,液量を25〜50 mLに保って約90分間煮
沸(9)する。
3) 放冷後,ろ紙5種Bを用いてろ過し,温水で3,4回洗う。
4) ろ液と洗液とを合わせ,指示薬としてp-ニトロフェノール溶液(1 g/L)3〜5滴を加え,溶液が僅か
に黄色になるまで水酸化ナトリウム溶液(40 g/L)を滴加(3)した後,全量フラスコ100 mLに移し入
れ,水を標線まで加える。
5) この溶液の適量をとり,46.1.1によってりん酸イオンの量を求め,試料中のりん酸イオンの濃度
(PO43− mg/L)に換算し,この値から別に46.1.1によって定量した試料中のりん酸イオンの濃度
(PO43− mg/L)を差し引いて加水分解性りんとし,りん酸イオンの濃度(PO43− mg/L)で表す(7)。
注(8) 溶存の加水分解性りんを定量する場合には,注(2)によってろ過した試料を用いる。
(9) 二りん酸イオン,トリポリりん酸イオンなどは約1時間でりん酸イオンになる。
46.3 全りん ペルオキソ二硫酸カリウム分解,硝酸-過塩素酸分解又は硝酸-硫酸分解によって試料中のり
ん化合物などを分解し,生成したりん酸イオンをモリブデン青吸光光度法で定量し,これを全りんとして
りんの濃度で表す。又は一連のペルオキソ二硫酸カリウム分解及びりん酸イオンのモリブデン青吸光光度
法による定量を流れ分析法によって行い,全りん濃度を求める。
46.3.1 ペルオキソ二硫酸カリウム分解法 試料にペルオキソ二硫酸カリウムを加え,高圧蒸気滅菌器中で
加熱して有機物などを分解し,この溶液についてりん酸イオンを定量して全りんの濃度を求める。
定量範囲:P 1.25〜25 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
3) ペルオキソ二硫酸カリウム溶液(40 g/L) JIS K 8253に規定するペルオキソ二硫酸カリウム(窒素・
りん測定用)4 gを水に溶かして100 mLとする。
4) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。ただし,アミド硫酸アンモニウムは加えなく
てもよい(10)。
5) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
6) りん標準液(P 50 μg/mL) JIS K 9007に規定するりん酸二水素カリウム(pH標準液用)を105±2 ℃
で約2時間加熱し,デシケーター中で放冷する。その0.220 gをとり,少量の水に溶かして全量フラ
スコ1 000 mLに移し入れ,水を標線まで加える。0〜10 ℃の暗所に保存する。
7) りん標準液(P 5 μg/mL) りん標準液(P 50 μg/mL)20 mLを全量フラスコ200 mLにとり,水を標
線まで加える。使用時に調製する。
注(10) 全りんの定量では,前処理によって亜硝酸イオンは存在しなくなるから,アミド硫酸アンモニ
ウムを添加する必要がない。
b) 器具及び装置 器具及び装置は,次による。
1) 分解瓶 耐熱・耐圧性のガラス瓶(容量約100 mL)で,高圧蒸気滅菌器中(約120 ℃)で使用で
きるもの(11)。
2) 高圧蒸気滅菌器 JIS T 7322又はJIS T 7324に規定するもので,約120 ℃に加熱できるもの。
186
K 0102:2016
3) 光度計 分光光度計又は光電光度計
注(11) ガラス製アンプル(容量約100 mL)で,高圧蒸気滅菌器中(約120 ℃)で使用できるものを用
いてもよい。
c) 操作 操作は,次による。
1) 試料(12) 50 mLを分解瓶にとる。
2) ペルオキソ二硫酸カリウム溶液(40 g/L)10 mLを加え,密栓して混合する。
3) 高圧蒸気滅菌器に入れて加熱し,約120 ℃に達してから30分間加熱分解する。
4) 分解瓶を取り出し,放冷後(13),上澄み液(14) (15) 25 mLをメスシリンダー(有栓形)25 mLに分取す
る。
5) 46.1.1 c) 2)及び3)の操作を行って吸光度を測定する(16)。
6) 空試験として水50 mLを分解瓶にとり,2)〜5)の操作を行って吸光度を測定し,試料について得た
吸光度を補正する。
7) 検量線から4)で分取した溶液25 mL中のりんの量を求め,次の式によって試料中の全りんの濃度(P
mg/L)を算出する(17)。
50
000
1
25
60×
×
=a
P
ここに,
P: 全りんの濃度(P mg/L)
a: 4)で分取した溶液25 mL中の全りんの質量(mg)
注(12) 試料50 mL中の全りんが60 μg以上の場合には,試料の適量(Pとして0.12 mg未満を含む。)
を全量フラスコ100 mLにとり,水を標線まで加えたものを用いる。ただし,試料50 mL中の
全りんが60 μg以上で,pH5〜9の範囲にない場合には,試料の適量(Pとして0.12 mg未満を
含む。)をとり,硫酸(1+35)[43.2.1 a) 3)による。]又は水酸化ナトリウム溶液(40 g/L)を用
いて中和した後,全量フラスコ100 mLに移し入れ,水を標線まで加えたものを用いる。
(13) 塩化物イオンを多く含む試料の場合は,塩素が生成してモリブデン青の発色を妨害するおそれ
があるので,分解後の溶液に亜硫酸水素ナトリウム溶液(50 g/L)(JIS K 8059に規定する亜硫
酸水素ナトリウムを用いて調製する。)を1 mL加える。又は,塩素臭のなくなるまで煮沸し,
放冷後,水でもとの体積とする。
(14) 上澄み液に濁りが認められる場合には,ろ紙5種C又は孔径1 μm以下のガラス繊維ろ紙を用
いてろ過し,初めのろ液5〜10 mLを捨てた後のろ液を用いる。
(15) 分解後の溶液中に金属水酸化物の沈殿が認められる場合には,これらが溶ける点まで硫酸(1
+35)[及び必要に応じ水酸化ナトリウム溶液(40 g/L)]を加えたものを用いる(加えた両溶液
の量を求めておく。)。
なお,金属水酸化物の沈殿を溶かした後の溶液に濁りが認められる場合には,更に注(14)の操
作を行う。
(16) 4)の操作後の溶液にひ素(V)が含まれる場合には,備考1.に準じ,次の操作でひ素(III)に還
元する。分解後の溶液に硫酸(1 mol/L)3 mL(46.の備考1.による。)及びチオ硫酸ナトリウム
溶液(7.65 g/L)(46.の備考1.による。)1.5 mLを加え,5〜10分間放置してひ素(V)をひ素(III)
とする。注(3)に従って酸を中和した後,この上澄み液25 mLをとり,5)の操作を行う。この操
作を行った場合は,7)の式の に代え, を用いる。ただし,cは注(16)において用いた,硫
酸(1 mol/L),チオ硫酸ナトリウム溶液(7.65 g/L)及び水酸化ナトリウム溶液(40 g/L)の合
25
60
25
60c
+
187
K 0102:2016
量(mL)。
注(17) 1)の操作において,注(12)の操作を行った場合には,次の式によって試料中の全りんの濃度(P
mg/L)を算出する。
V
a
P
100
50
000
1
25
60
×
×
×
=
ここに,
P: 全りんの濃度(P mg/L)
a: 4)で分取した溶液25 mL中の全りんの質量(mg)
V: 試料量(mL)
また,4)において,注(13)の操作,注(15)の操作,又は注(13)と注(15)との操作を併せて行った場
合には,7)の式又は上記の式中の に代えてそれぞれ , 又は を用いる。ただし,
bは注(15)において添加した硫酸(1+35)及び水酸化ナトリウム溶液(40 g/L)(mL)。
d) 検量線 検量線の作成は,次による。
1) りん標準液(P 5 μg/mL)1〜20 mLを段階的に全量フラスコ100 mLにとり,それぞれ水を標線まで
加える。その25 mLをそれぞれメスシリンダー(有栓形)25 mLにとり,46.1.1 c) 2)及び3)の操作
を行って吸光度を測定する。
2) 別に,空試験として水25 mLをメスシリンダー(有栓形)25 mLにとり,46.1.1 c) 2)及び3)の操作
を行って吸光度を測定し,りん標準液(P 5 μg/mL)について得た吸光度を補正する。
3) 採取した溶液25 mL中のりん(P)の量と吸光度との関係線を作成する。
備考 10. 試料のpHが5〜9の範囲にない場合には,注(12)によって中和してから分解操作を行うが,試
料50 mL中の全りんが60 μg未満の場合は,試料の適量(50〜100 mL)をとり,硫酸(1+
35)[43.2.1 a) 3)による。]及び水酸化ナトリウム溶液(40 g/L)で中和した後,この溶液から
50 mLを分解瓶にとる。ただし,この場合は,中和に要した両溶液のmL数を求めておき,
試料中の全りんの濃度(P mg/L)の算出にはc) 7)に代え,次の式を用いる。
V
b
V
a
P
+
×
×
×
=
50
000
1
25
60
ここに,
P: 全りんの濃度(P mg/L)
a: c) 4)で分取した溶液25 mL中の全りんの質量(mg)
b: 中和に要した硫酸(1+35)及び水酸化ナトリウム溶液(40 g/L)
量(mL)
V: 試料量(mL)
11. c) 1)の操作で分解瓶にとった試料中のりんの濃度が低く0.1 mg/L未満の場合には,c) 5)の吸
光度の測定には光路長50 mmの吸収セルを用いる。また,空試験及び検量線の作成における
吸光度の測定にも光路長50 mmの吸収セルを用いる。
なお,この場合,検量線の作成には,a) 7)のりん標準液(P 5 μg/mL)に代え,これを10
倍に薄めて調製したりん標準液(P 0.5 μg/mL)を用いる。
12. りんの濃度が低く十分な定量精度が得にくい試料については,次の加熱濃縮操作を行うか,
又は備考13. のモリブデン青の溶媒抽出を行う。
試料100〜250 mLをビーカー200〜500 mLにとり,硫酸(2+1)(JIS K 8951に規定する硫
酸を用いて調製する。)1,2滴を加えた後,加熱板上で加熱して液量が50 mL以下になるま
で濃縮する。この溶液を水酸化ナトリウム溶液(40 g/L)で中和した後,分解瓶(あらかじ
め50 mLの位置に印を付けたもの。)に移し,水を加えて50 mLとし,c) 2)以下の操作を行
25
60
25
61(
)
25
60b
+
(
)
25
61b
+
188
K 0102:2016
う。ただし,この場合,試料中の全りんの濃度の算出には,c) 7)の式中の に代え, を
用いる。ただし,Vは試料の量(mL)。
備考 13. c)の操作に準じて発色させたモリブデン青を2,6-ジメチル-4-ヘプタノン[ジイソブチルケト
ン(DIBK)]で抽出することによって微量のりんを定量できる。
c) 1)〜3)の操作を行った後,分解瓶中の溶液(12)を分液漏斗100 mLに移し,分解瓶は水10
mLで洗浄し,洗液を分液漏斗に合わせる。これにモリブデン酸アンモニウム-アスコルビン
酸混合溶液5.5 mLを加えて20〜40 ℃(4)で約15分間放置する。分液漏斗に2,6-ジメチル-4-
ヘプタノン5 mLを加えて約5分間振り混ぜる。静置後,水層を捨て,2,6-ジメチル-4-ヘプタ
ノン層(水滴などによる濁りがあれば,乾燥したろ紙で手早くろ過する。)の一部を吸収セル
に移し,波長640 nm付近の吸光度を測定する。
空試験として水50 mLを分解瓶にとり,試料の場合と同じ操作を行って吸光度を測定し,
試料について得た吸光度を補正する。検量線から試料中の全りんの量を求め,次の式によっ
て試料中の全りんの濃度(P mg/L)を算出する。
V
a
P
000
1
×
=
ここに,
P: 全りんの濃度(P mg/L)
a: 測定した全りんの質量(mg)
V: 試料量(mL)
検量線 a) 7)のりん標準液(P 5 μg/mL)を10倍に薄めてりん標準液(P 0.5 μg/mL)を調製
し,その1〜25 mLを段階的に全量フラスコ100 mLにとり,それぞれ水を標線まで加える。
この50 mLをそれぞれ分液漏斗100 mLにとり,水20 mLを加えた後,試料についてと同じ
操作を行って吸光度を測定する。空試験として水70 mLを分液漏斗100 mLにとり,同様の
操作を行って吸光度を測定し,りん標準液(P 0.5 μg/mL)について得た吸光度を補正し,採
取した溶液50 mL中のりん(P)の量と吸光度との関係線を作成する。
46.3.2 硝酸-過塩素酸分解法 試料に硝酸を加えて加熱濃縮後,硝酸及び過塩素酸を加え,再び加熱して
有機物などを分解し,この溶液についてりん酸イオンを定量し,全りんの濃度を求める。この方法は,多
量の有機物を含む試料及び分解しにくい有機りん化合物を含む試料に適用する。
定量範囲:P 1.25〜25 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硝酸 JIS K 8541に規定するもの。
3) 過塩素酸 JIS K 8223に規定するもの。
4) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
5) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
6) 水酸化ナトリウム溶液(200 g/L) 38.1.1.1 a) 3)による。
7) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。
8) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
9) p-ニトロフェノール溶液(1 g/L) 46.2 a) 7)による。
10) りん標準液(P 5 μg/mL) 46.3.1 a) 7)による。
b) 装置 装置は,次による。
50
000
1
V
000
1
189
K 0102:2016
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料50 mL (18)をビーカーにとる。
2) 硝酸を加えて弱酸性とし,加熱板上で静かに加熱して15〜20 mLに濃縮する。
3) これに硝酸2〜5 mLを加えて再び加熱し,約10 mLになるまで濃縮した後,更に硝酸2 mLを加え
て加熱し,約10 mLになるまで濃縮し,放冷する。
4) 過塩素酸5 mL(19)を少量ずつ加える。加熱板上で加熱を続け,過塩素酸の白煙が発生し始めたらビ
ーカーを時計皿で覆い,過塩素酸がビーカーの内壁を還流する状態に保つ(20) (21)。
5) 放冷後,水約30 mLを加える(22)。
6) この溶液に指示薬としてp-ニトロフェノール溶液(1 g/L)3〜5滴を加え,初めに水酸化ナトリウム
溶液(200 g/L)を,次に,水酸化ナトリウム溶液(40 g/L)を加えて溶液が僅かに黄色になるまで
中和する(23)。
7) 溶液を全量フラスコ50 mLに移し入れ,水を標線まで加える。
8) この溶液25 mLを共栓試験管に分取し(24),46.1.1 c) 2)及び3)の操作を行って吸光度を測定する。
9) 空試験として1)で採取した試料と同量の水をビーカーにとり,2)〜8)の操作を行って吸光度を測定
し,試料について得た吸光度を補正する。
10) 検量線から8)で分取した溶液25 mL中のりんの量を求め,次の式によって試料中の全りんの濃度(P
mg/L)を算出する(25)。
V
a
P
000
1
25
50×
×
=
ここに,
P: 全りんの濃度(P mg/L)
a: 8)で分取した溶液25 mL中の全りんの質量(mg)
V: 試料量(mL)
注(18) 試料中の全りんの濃度が低い場合には,50 mL以上としてもよい。多量の塩化物イオンを含む
試料で全りんの濃度が高い場合には,50 mL未満としてもよい。
(19) 試料に多量の塩化物イオンが含まれる場合には,塩化物イオンの当量よりも多い量を更に加え
る。
(20) 過塩素酸を用いる加熱分解操作は,試料の種類によっては爆発の危険性があるため,次のこと
に注意する。
− 酸化されやすい有機物は,過塩素酸を加える前に,2)及び3)の操作によって十分に分解し
ておく。
− 過塩素酸の添加は,必ず濃縮液を放冷した後に行う。
− 必ず過塩素酸と硝酸とを共存させた状態で加熱分解を行う。
− 濃縮液を乾固させない。
(21) この操作によっても有機物が分解されず,溶液に色が残った場合には,硝酸2 mLを加えて加熱
する操作を繰り返す。
(22) 必要に応じ,加熱して可溶性塩を溶かす。加熱しても不溶解物が残った場合には,ろ紙5種C
又は孔径1 μm以下のガラス繊維ろ紙を用いて溶液をろ過し,次に,ろ紙を少量の水で洗浄し,
ろ液と洗液とを合わせる。
(23) 中和するときに金属水酸化物の沈殿が認められる場合には,水酸化ナトリウム溶液(40 g/L)の
190
K 0102:2016
添加は,沈殿の生じる直前でとどめる。必要に応じ硫酸(1+35)を用いて調節する。
注(24) 分取する溶液25 mL中の全りんが25 μg以上になる場合には,この溶液の適量(りん含有量25
μg未満となる量)をメスシリンダー(有栓形)50 mLに分取し,水を加えて25 mLとする。
(25) 8)において注(24)の操作を行った場合には,10)の式中の に代えて を用いる。ただし,bは
メスシリンダー(有栓形)に分取した溶液の量(mL)。
d) 検量線 46.3.1 d) の検量線と同じ操作によって作成する。
46.3.3 硝酸-硫酸分解法 試料に硝酸を加えて加熱濃縮後,硝酸及び硫酸を加え,更に加熱して有機物な
どを分解し,この溶液についてりん酸イオンを定量し,全りんの濃度を求める。この方法は,多量の有機
物を含む試料及び分解しにくい有機りん化合物を含む試料に適用する。
定量範囲:P 1.25〜25 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 硝酸 JIS K 8541に規定するもの。
3) 硫酸(1+1) 5.4 a) 2)による。
4) アスコルビン酸溶液(72 g/L) 46.1.1 a) 2)による。
5) 水酸化ナトリウム溶液(40 g/L) 21. a) 3)による。
6) 水酸化ナトリウム溶液(200 g/L) 38.1.1.1 a) 3)による。
7) モリブデン酸アンモニウム溶液 46.1.1 a) 3)による。
8) モリブデン酸アンモニウム-アスコルビン酸混合溶液 46.1.1 a) 4)による。
9) p-ニトロフェノール溶液(1 g/L) 46.2 a) 7)による。
10) りん標準液(P 5 μg/mL) 46.3.1 a) 7)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 46.3.2 c) 1)及び2)の操作を行う。
2) 1)の操作を行った後の溶液に硫酸(1+1)2 mL(19)及び硝酸2〜5 mLを加え,加熱して硫酸の白煙が
発生するまで濃縮し,ビーカーを時計皿で覆い,更に加熱して硫酸の白煙を短時間強く発生させた
後,放冷する。
3) この溶液に硝酸5 mLを加えて再び加熱し,硫酸の白煙が発生するまで加熱する(21)。
4) 放冷後,水約30 mLを加え,約10分間静かに煮沸する(22)。
5) 以下,46.3.2 c) 6)〜8)の操作を行う。
6) 空試験として1)で採取した試料と同量の水をビーカーにとり,46.3.2 c) 2)の操作を行った後,2)〜5)
の操作を行って吸光度を測定し,試料について得た吸光度を補正する。
7) 検量線から5)で分取した溶液25 mL中のりんの量を求め,46.3.2 c) 10)の式によって試料中の全りん
の濃度(P mg/L)を算出する。
d) 検量線 46.3.1 d) の検量線と同じ操作によって作成する。
46.3.4 流れ分析法 試料中のりん化合物などを,46.3.1と同様な原理で加水分解又は酸化分解してりん酸
イオンとした後,りん酸イオンをモリブデン青吸光光度法によって定量する一連の操作を流れ分析法によ
って行い,全りん濃度を求める。
定量範囲:P− 0.02〜10 mg/L,繰返し精度:10 %以下
25
50
b
50
191
K 0102:2016
試験操作などは,JIS K 0170-4で規定された全りんに関する規定による。ただし,JIS K 0170-4の7.3.2
(UV照射酸化分解前処理モリブデン青発色FIA法)及び7.3.4(UV照射酸化分解前処理モリブデン青発
色CFA法)は除く。
47. ほう素(B) ほう素の定量には,メチレンブルー吸光光度法,アゾメチンH吸光光度法,ICP発光分
光分析法又はICP質量分析法を適用する。
なお,アゾメチンH吸光光度法は,1990年に第1版として発行されたISO 9390,ICP発光分光分析法は,
1996年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 9390:1990,Water quality−Determination of borate−Spectrometric method using azomethine-H
(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
47.1 メチレンブルー吸光光度法 ほう素化合物に硫酸とふっ化水素酸とを加えてテトラフルオロほう酸
イオンとした後,メチレンブルー[3,7-ビス(ジメチルアミノ)フェノチアジン-5-イウムクロリド]を加
え,生成するイオン会合体を1,2-ジクロロエタンで抽出し,その吸光度を測定してほう素を定量する。
定量範囲:B 0.1〜1 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。これらは,ポリエチレン瓶に保存する。
1) 水 JIS K 0557に規定するA3の水(ただし,石英ガラス又は金属製の蒸留器を用いて調製したも
の。)
2) 硫酸(3+97) JIS K 8951に規定する硫酸を用いて調製する。
3) ふっ化水素酸(1+9) JIS K 8819に規定するふっ化水素酸を用いて調製する。
4) 硫酸銀溶液(0.3 g/L) JIS K 8965に規定する硫酸銀0.15 gを水に溶かして500 mLとする。
5) メチレンブルー溶液(0.4 g/L) JIS K 8897に規定するメチレンブルー0.48 gを水に溶かして100 mL
とする。この溶液10 mLを全量フラスコ100 mLにとり,水を標線まで加える。
6) 1,2-ジクロロエタン JIS K 8465に規定するもの。
7) ほう素標準液(B 0.1 mg/mL) JIS K 8863に規定するほう酸0.572 gをとり,水に溶かし,全量フラ
スコ1 000 mLに移し入れ,水を標線まで加える。
8) ほう素標準液(B 1 μg/mL) ほう素標準液(B 0.1 mg/mL)10 mLを全量フラスコ1 000 mLにとり,
水を標線まで加える。
9) ほう素標準液(B 0.1 μg/mL) ほう素標準液(B 1 μg/mL)20 mLを全量フラスコ200 mLにとり,
水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具 石英ガラス又はソーダ石灰ガラス製のもの。
2) 分液漏斗 ポリエチレン製50 mL
3) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料(1) (2) (3)の適量(Bとして0.1〜1 μgを含む。)を分液漏斗にとり,水で15 mLとし,硫酸(3+
192
K 0102:2016
97)3 mL,メチレンブルー溶液(0.4 g/L)3 mL及び1,2-ジクロロエタン10 mLを加えて約1分間振
り混ぜ,放置する(4) (5)。
2) 1,2-ジクロロエタン層を捨て(6),水層にふっ化水素酸(1+9)3 mLを加えて約1時間放置する。
3) 1,2-ジクロロエタン10 mLを加え,約1分間激しく振り混ぜて,放置する。
4) 1,2-ジクロロエタン層を別の分液漏斗に移し,硫酸銀溶液(0.3 g/L)5 mLを加えて約1分間振り混
ぜ,1,2-ジクロロエタン層を洗い,放置する。
5) 1,2-ジクロロエタン層の一部を吸収セルに入れ,1,2-ジクロロエタンを対照液として波長660 nm付
近の吸光度を測定する。
6) 空試験として水15 mLをとり,1)〜5)の操作を行って試料について得た吸光度を補正する。
7) 検量線からほう素の量を求め,試料中のほう素の濃度(B mg/L)を算出する。
注(1) 懸濁物が含まれる場合には,ろ過又は遠心分離によって除去する。
(2) 懸濁物を含まない試料又は懸濁物を除いた試料に多量の有機物が含まれる場合には,試料の一
定量を白金皿にとり,JIS K 8625に規定する炭酸ナトリウム0.1 gを加えて蒸発乾固した後,融
解する。放冷後,水を加え,加熱して融成物を溶かし,硫酸(3+97)を加えて中和した後,水
で液量を一定とする。
この溶液の適量(Bとして0.1〜1 μgを含む。)を分液漏斗にとり,水を加えて15 mLとし,
硫酸(3+97)3 mLとふっ化水素酸(1+9)3 mLとを加えて振り混ぜ,約1時間放置する。次
に,メチレンブルー溶液(0.4 g/L)3 mLを加えて振り混ぜた後,1,2-ジクロロエタン10 mLを
加えて約1分間激しく振り混ぜ,ほう素のイオン会合体を抽出する。以下,c) 4)以降の操作を
行う。
(3) 試料が中性でない場合には,硫酸(3+97)又は水酸化ナトリウム溶液(40 g/L)[21. a) 3)によ
る。]で中和する。
(4) ふっ化物イオンが共存する場合はc) 1)の操作を行うと,ほう素が抽出され失われるから,注(2)
の操作を行う。
(5) 1,2-ジクロロエタン層と水層とが分かれるには,かなりの時間を要する。
(6) この抽出で,試料中の陰イオン界面活性剤などが除かれる。
d) 検量線 検量線の作成は,次による。
1) ほう素標準液(B 0.1 μg/mL)1〜10 mLを分液漏斗50 mLに段階的にとり,c) 1)〜6)の操作を行い,
ほう素(B)の量と吸光度との関係線を作成する。
備考 1. 多量の硝酸イオンは妨害する。クロム酸イオンは妨害するが,過酸化水素(1+100)(JIS K
8230に規定する過酸化水素を用いて調製する。)5〜7滴を加えた後,煮沸して過剰の過酸化
水素を分解すれば妨害しない。
2. 分液漏斗,吸収セルなどにメチレンブルーが付着したときは,JIS K 8102に規定するエタノ
ール(95)で洗う。
47.2 アゾメチンH吸光光度法 ほう酸が,pH約6でアゾメチンH[8-N-(2-ヒドロキシベンジリデン)-ア
ミノ-1-ヒドロキシ-3,6-ナフタレンジスルホン酸]と反応して生成する黄色の錯体の吸光度を測定してほう
素を定量する。
定量範囲:B 5〜25 μg,繰返し精度:3〜10 %
備考 3. この方法は,汚濁の少ない試料に適用する。
a) 試薬 試薬は,次による。これらは,ポリエチレン瓶に保存する。
193
K 0102:2016
1) 水 47.1 a) 1)による。
2) アゾメチンH溶液 アゾメチンH一ナトリウム塩[8-N-(2-ヒドロキシベンジリデン)-アミノ-1-
ヒドロキシ-3,6-ナフタレンジスルホン酸一ナトリウム塩]1.0 gとJIS K 9502に規定するL(+)-
アスコルビン酸3.0 gとを少量の水に溶かした後,全量フラスコ100 mLに移し入れ,水を標線まで
加える。この溶液はポリエチレン瓶に保存する。4〜6 ℃の暗所に保存すれば1週間は安定である。
3) 緩衝液(pH5.9) JIS K 8359に規定する酢酸アンモニウム250 g,JIS K 8951に規定する硫酸15 mL,
JIS K 9005に規定するりん酸5 mL,JIS K 8283に規定するくえん酸一水和物1.0 g及びJIS K 8107
に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物1.0 gを水250 mL中に加え,加熱
して溶かす。
4) アゾメチンH混合溶液 アゾメチンH溶液と緩衝液(pH5.9)の等体積とを混合する。使用時に調
製する。
5) ほう素標準液(B 1 μg/mL) 47.1 a) 8)による。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具 47.1 b) 1)による。
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料(1)の適量(Bとして5〜25 μgを含む。)をポリエチレンビーカー100 mLにとり,水を加えて25
mLとする。
2) アゾメチンH混合溶液10 mLを加え,20 ℃の暗所に約2時間放置する。
3) 溶液の一部を吸収セル(7)に移し,波長410 nm付近の吸光度を測定する。
4) 空試験として水25 mLをポリエチレンビーカー100 mLにとり,2)及び3)の操作を行って吸光度を測
定し,試料について得た吸光度を補正する。
5) 検量線からほう素の量を求め,試料中のほう素の濃度(B mg/L)を算出する。
注(7) 吸収セル50 mmを用いれば,ほう素1〜5 μgが定量できる。
d) 検量線 検量線の作成は,次による。
1) ほう素標準液(B 1 μg/mL)5〜25 mLをポリエチレンビーカー100 mLに段階的にとり,c) 1)〜4)の
操作を行ってほう素(B)の量と吸光度との関係線を作成する。
備考 4. この方法では,ナトリウム,カリウム,カルシウム,マグネシウム,亜鉛,りん酸イオン,
硫酸イオン及び硝酸イオンは妨害しない。
鉄,マンガン,アルミニウム,銅,クロム,ベリリウム,チタン,バナジウム及びジルコ
ニウムは正の誤差を与える。
47.3 ICP発光分光分析法 試料を高周波誘導結合プラズマ中に導入し,ほう素による発光を波長249.773
nmで測定してほう素を定量する。
定量範囲:B 20〜8 000 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。これらは,ポリエチレン瓶に保存する。
1) 水 47.1 a) 1)による。
2) ほう素標準液(B 20 μg/mL) 47.1 a) 7)のほう素標準液(B 0.1 mg/mL)50 mLを全量フラスコ250 mL
にとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具 47.1 b) 1)による。
194
K 0102:2016
2) ICP発光分光分析装置
c) 操作 操作は,次による。
1) 試料(1)を試料導入部を通して発光部に導入し,ほう素(249.773 nm)の発光強度を測定する(8) (9)。
2) 空試験として水について1)の操作を行って試料について得た発光強度を補正する。
3) 検量線からほう素の量を求め,試料中のほう素の濃度(B μg/L)を算出する。
注(8) 塩類の濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116に規定する標準添加
法を用いるとよい。ただし,この場合は,試料の種類によらずバックグラウンド補正を行う必
要がある。
(9) 高次のスペクトル線が使用可能な装置では,高次のスペクトル線を用いて測定してもよい。
また,精度及び正確さを確認してあれば,他の波長を用いてもよい。
d) 検量線 検量線の作成は,次による。
1) ほう素標準液(B 20 μg/mL)0.1〜40 mLを全量フラスコ100 mLに段階的にとり,水を標線まで加
える。この溶液についてc) 1)の操作を行う。
2) 別に,空試験として,水についてc) 1)の操作を行って,標準液について得た発光強度を補正し,ほ
う素(B)の量と発光強度との関係線を作成する。検量線の作成は,試料測定時に行う。
備考 5. 波長の異なる2本以上のスペクトル線の同時測定が可能な装置では,内標準法によることが
できる。操作は,次による。
1) 試料の適量を全量フラスコ100 mLにとり,イットリウム溶液(Y 50 μg/mL)10 mLを加
えた後,水を標線まで加える。
2) この溶液についてc) 1)の操作を行ってほう素(249.773 nm)及びイットリウム(371.029
nm)の発光強度を測定し,ほう素の発光強度とイットリウムの発光強度との比を求める。
3) 空試験として,試料に代えて水を用い,1)及び2)の操作を行い,ほう素の発光強度とイ
ットリウムの発光強度との比を求め,2)で得た発光強度比を補正する。
4) 検量線から,ほう素の量を求め,試料中のほう素の濃度(B μg/L)を算出する。
5) 検量線 全量フラスコ100 mL数個に,ほう素標準液(B 20 μg/mL)0.1〜40 mLを段階
的にとり,イットリウム溶液(Y 50 μg/mL)10 mLを加え,水を標線まで加える。この
溶液について2)の操作を行ってほう素及びイットリウムの発光強度を測定し,ほう素の
発光強度とイットリウムの発光強度との比を求める。別に,空試験として,ほう素標準
液に代えて水を用い,同じ操作を行って,同様にほう素とイットリウムとの発光強度の
比を求め,ほう素標準液でのほう素とイットリウムとの発光強度比を補正し,ほう素の
量と,ほう素とイットリウムとの発光強度比との関係線を作成する。
6) イットリウム溶液(Y 50 μg/mL)の調製 酸化イットリウム(III)0.318 gをとり,JIS K
9901に規定する高純度試薬−硝酸5 mLを加え,加熱して溶かし,煮沸して窒素酸化物
を追い出し,放冷後,全量フラスコ250 mLに移し,水を標線まで加える。この溶液10 mL
を全量フラスコ200 mLにとり,水を標線まで加える。
6. ほう素を含む溶液を発光部に導入した場合には,メモリー効果が他の元素の場合より大きい
ため,次の溶液を噴霧する前に,水を十分な時間噴霧して前の試料の影響を除去する。
47.4 ICP質量分析法 試料に内標準元素を加え,試料導入部を通して高周波プラズマ中に噴霧し,ほう
素及び内標準元素のそれぞれの質量/電荷数における指示値(10)を測定し,ほう素の指示値と内標準元素の
指示値との比を求めてほう素を定量する。
195
K 0102:2016
定量範囲:B 0.5〜500 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
注(10) イオンカウント値又はその比例値。
a) 試薬 試薬は,次による。これらは,ポリエチレン瓶に保存する。
1) 水 47.1 a) 1)による。
2) 内標準液(1 μg/mL) 内標準元素としてイットリウム又はインジウムを用いる。内標準液の調製に
は,次の2.1)〜2.2)に規定する溶液のうち内標準とする元素の溶液2 mLを全量フラスコ100 mLに
とり,硝酸(1+1)(JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。)2 mLを加え,水
を標線まで加える。使用時に調製する。
2.1) イットリウム溶液(Y 50 μg /mL) 備考5.の6)による。
2.2) インジウム溶液(In 50 μg /mL) インジウム0.250 gをとり,JIS K 9901に規定する高純度試薬−
硝酸10 mLを加え,加熱して溶かし,煮沸して窒素酸化物を追い出し,放冷後,全量フラスコ250
mLに移し入れ,水を標線まで加える。この溶液25 mLを全量フラスコ500 mLにとり,硝酸(1
+1)[47.4 a) 2)による。]10 mLを加え,水を標線まで加える。
3) ほう素標準液(B 10 μg/mL) 47.1 a) 7)のほう素標準液(B 0.1 mg/mL)25 mLを全量フラスコ250 mL
にとり,水を標線まで加える。使用時に調製する。
4) ほう素標準液(B 0.5 μg/mL) ほう素標準液(B 10 μg/mL)5 mLを全量フラスコ100 mLにとり,
水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具 47.1 b) 1)による。
2) ICP質量分析装置
備考 7. イオン源として,高周波プラズマと同等の性能をもつものを用いてもよい。
c) 操作 操作は,次による。
1) 試料(1)の適量(Bとして0.05〜50 μgを含む。)を全量フラスコ100 mLにとり,内標準液(1 μg/mL)
1 mLを加え,水を標線まで加える。
2) 1)の溶液を試料導入部を通してイオン化部に導入し,ほう素(質量数11又は10)及び内標準元素
[イットリウム(質量数89)又はインジウム(質量数115)]のそれぞれの質量/電荷数における指
示値を読み取り,ほう素の指示値と内標準元素の指示値との比を求める。
3) 空試験として,水について1)及び2)の操作を行って,試料について得たほう素と内標準元素との指
示値の比を補正する。
4) 検量線からほう素の量を求め,試料中のほう素の濃度(B μg/L)を算出する。
d) 検量線 検量線の作成は,次による。
1) ほう素標準液(B 10 μg/mL)又はほう素標準液(B 0.5 μg/mL)0.1〜5 mLを,全量フラスコ100 mL
に段階的にとり,内標準液(1 μg/mL)1 mLを加え,水を標線まで加える。使用時に調製する。こ
の溶液についてc) 2)の操作を行う。
2) 別に,空試験として,水についてc) 1) 及びc) 2)の操作を行って標準液について得たほう素と内標
準元素との指示値の比を補正し,ほう素の濃度に対する,ほう素の指示値と内標準元素の指示値と
の比の関係線を作成する。検量線の作成は,試料測定時に行う。
備考 8. 主成分元素又は有機物の含有量が少なく,非スペクトル干渉が無視できる試料の場合は,内
標準元素の添加を省略し,検量線法によって定量してもよい。
9. 備考6.による。
196
K 0102:2016
48. ナトリウム(Na) ナトリウムの定量には,フレーム光度法,フレーム原子吸光法又はイオンクロマ
トグラフ法を適用する。
なお,フレーム光度法は,1993年に第1版として発行されたISO 9964-3,フレーム原子吸光法は,1993
年に第1版として発行されたISO 9964-1,イオンクロマトグラフ法は,1998年に第1版として発行された
ISO 14911との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 9964-1:1993,Water quality−Determination of sodium and potassium−Part 1 : Determination of
sodium by atomic absorption spectrometry(MOD)
ISO 9964-3:1993,Water quality−Determination of sodium and potassium−Part 3 : Determination of
sodium and potassium by flame emission spectrometry(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+, Na+, NH4+, K+, Mn2+, Ca2+, Mg2+, Sr2+
and Ba2+ using ion chromatography−Method for water and waste water(MOD)
48.1 フレーム光度法 試料をアセチレン-空気フレーム,水素-酸素フレームなどの中に噴霧し,このとき
生じる波長589.0 nmの輝線の強さを測定してナトリウムを定量する。
定量範囲:Na 30〜300 μg/L,0.3〜3 mg/L,3〜30 mg/L,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) ナトリウム標準液(Na 1 000 mg/L) JIS K 8005に規定する容量分析用標準物質の塩化ナトリウム
を600 ℃で約1時間加熱し,デシケーター中で放冷する。NaCl 100 %に対してその2.542 gをとり,
少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。ポリエチレン瓶に保
存する。
2) ナトリウム標準液(Na 3〜30 mg/L) ナトリウム標準液(Na 1 000 mg/L)を段階的にとり,これを
水で薄めてNa 3〜30 mg/Lの標準液を調製する(1)。
注(1) 低い濃度の測定用には,Na 30〜300 μg/L又はNa 0.3〜3 mg/Lの標準液を調製する。
b) 装置 装置は,次による。
1) フレーム光度計
c) 操作 操作は,次による。
1) ナトリウム標準液(Na 30 mg/L)(2)をフレーム光度計のフレーム中に噴霧し,波長589.0 nmの指示
値が100を示すように調節する。
2) 水を噴霧して指示値がゼロを示すように調節する。
3) ナトリウム標準液(Na 3〜30 mg/L)(1)を順次噴霧し,ナトリウム(Na)の濃度と指示値との関係線
を作成し,検量線とする。
4) 試料(3) (4)(ナトリウムの濃度が30 mg/L以上の場合は薄める。)を噴霧して指示値を読み取り,検
量線から試料中のナトリウムの濃度(Na mg/L)を求める。
注(2) 低い濃度の測定用には,Na 3 mg/L又はNa 0.3 mg/Lの標準液を用いる。
(3) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(4) 試料に干渉物質が含まれる場合には,その影響を無視できる濃度まで薄めて測定するか,又は
試料と同程度の干渉物質を含むナトリウム標準液(Na 3〜30 mg/L)を調製し,検量線を作成す
る。
197
K 0102:2016
備考 1. カリウム及びカルシウムが共存すると正の誤差を生じる。
このような試料には,塩化セシウム溶液(25 g/L)[塩化セシウム25 gをJIS K 8180に規
定する塩酸50 mL及び水450 mLに溶かし,水を加えて1 Lとする。この溶液1 Lは,セシウ
ム(Cs)を約20 gを含む。]を試料40 mLに対して5 mLを加えることで,カリウム,カル
シウムなどの影響を抑制できる。この操作を行った場合は,検量線作成時の操作も塩化セシ
ウム溶液(25 g/L)を試料と同様に加えて行う。また,リチウム,バリウム,遊離酸,りん
酸塩,ほう酸塩,しゅう酸塩,シリカ,グルコース,ゼラチンなどが共存すると負の誤差を
生じる。マグネシウム及び硫酸イオンはほとんど干渉しない。
多量のけい酸塩が共存する場合には,試料の適量を石英ガラスビーカー又は白金皿にとり,
塩酸(1+1)[24.2 a) 2)による。]を加えて酸性とした後,蒸発乾固する。放冷後,塩酸(1
+1)5滴及び少量の水を加え,加熱して溶かし,ろ紙5種Bでろ過し,ろ液を水で一定量と
する。
48.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,ナトリウムによる原子吸光を波
長589.0 nmで測定してナトリウムを定量する。
定量範囲:Na 0.05〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) ナトリウム標準液(Na 0.1 mg/mL) 48.1 a) 1)のナトリウム標準液(Na 1 000 mg/L)10 mLを全量フ
ラスコ100 mLにとり,水を標線まで加える。使用時に調製する。
2) ナトリウム標準液(Na 10 μg/mL) ナトリウム標準液(Na 0.1 mg/mL)20 mLを全量フラスコ200 mL
にとり,水を標線まで加える。使用時に調製する。
b) 装置 装置は,次による。
1) フレーム原子吸光分析装置 JIS K 0121に規定するフレーム原子吸光分析装置で,測定対象元素用
の光源を備えたもの。
c) 操作 操作は,次による。
1) 試料(3)をフレーム中に導入し,波長589.0 nmの指示値(5)を読み取る。
2) 空試験として,水について,1)の操作を行って試料について得た指示値を補正する。
3) 検量線からナトリウムの量を求め,試料中のナトリウムの濃度(Na mg/L)を算出する。
注(5) 吸光度又はその比例値
d) 検量線 検量線の作成は,次による。
1) ナトリウム標準液(Na 10 μg/mL)0.5〜40 mLを全量フラスコ100 mLに段階的にとり,水を標線ま
で加える。この溶液についてc) 1)の操作を行う。
2) 別に,空試験として水についてc) 1)の操作を行って標準液について得た指示値を補正し,ナトリウ
ム(Na)の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
備考 2. 塩化セシウム溶液(25 g/L)を加えることで,カリウム,カルシウムなどの影響を抑制でき
る。添加量などは,備考1.による。
48.3 イオンクロマトグラフ法 試料中の陽イオンをイオンクロマトグラフ法によって定量する。検出器
には電気伝導率検出器を用いる。この方法によって,表48.1に示す陽イオンが同時定量できる。アンモニ
ウムイオンを同時定量する場合は,3.3の保存処理を行わず,試料採取後,直ちに行う。直ちに行えない
場合は,0〜10 ℃の暗所に保存し,できるだけ早く試験する。
それぞれの陽イオンの定量範囲,繰返し精度などの例を,表48.1に示す。
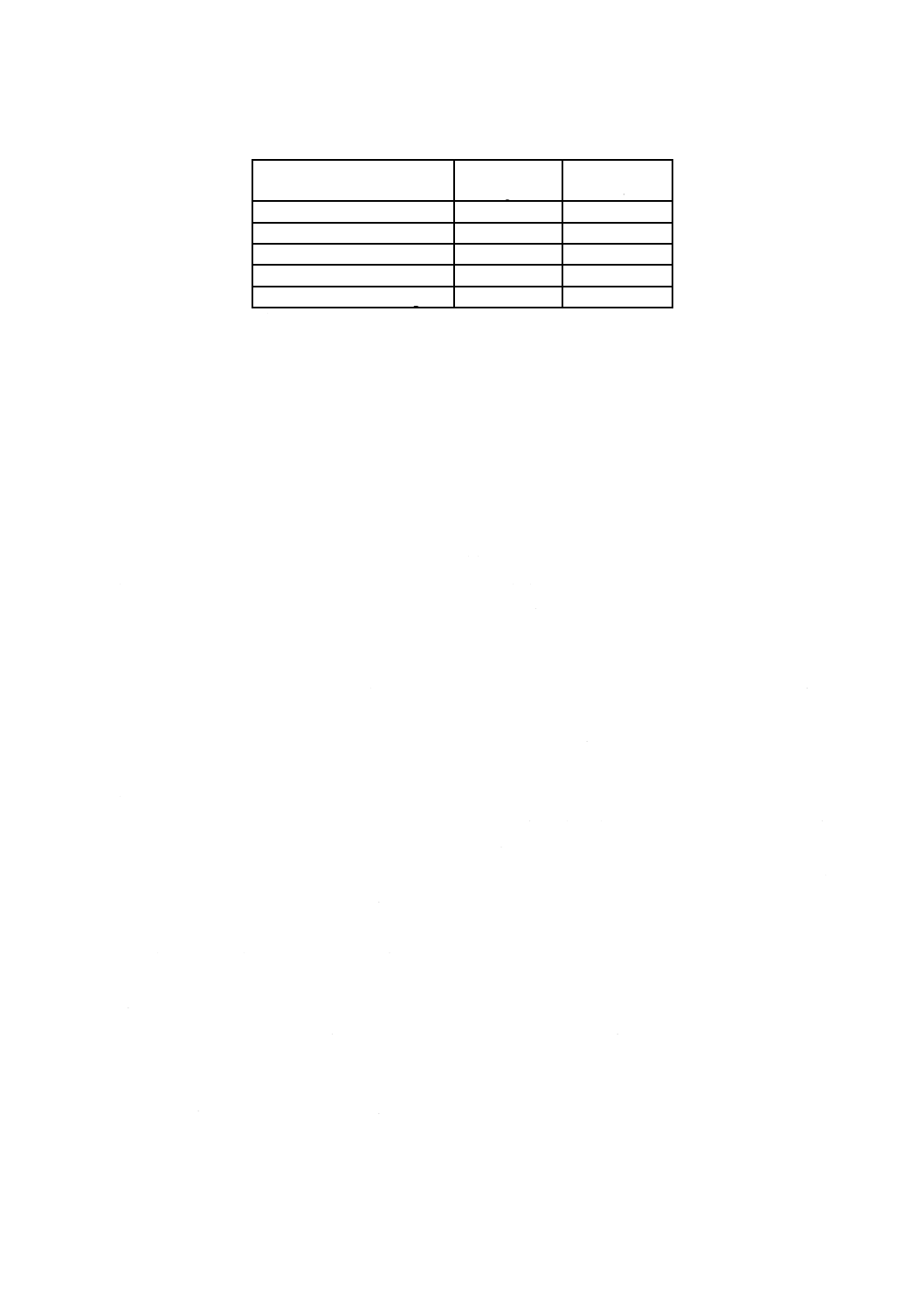
198
K 0102:2016
表48.1 各陽イオンの定量範囲などの例*
対象陽イオン
定量範囲
mg/L
繰返し精度
%
アンモニウムイオン(NH4+)
0.1〜30
2〜10
ナトリウム (Na)
0.1〜30
2〜10
カリウム (K)
0.1〜30
2〜10
カルシウム (Ca)
0.2〜50
5〜10
マグネシウム (Mg)
0.2〜50
5〜10
注*
定量範囲は,検出器,試料注入量,カラムのイオン交換容
量などによって変わる。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA2又はA3の水
2) 溶離液 溶離液は,装置の種類及び分離カラムに充塡した陽イオン交換体の種類によって異なるの
で,あらかじめ,アンモニウムイオン,ナトリウム,カリウム,カルシウム及びマグネシウムの分
離の確認は,備考3.による。
3) 再生液 再生液を必要とする場合,装置の種類及びサプレッサーの種類によって再生液が異なる。
あらかじめ分離カラムと組み合わせて,備考3.の操作を行って再生液の性能を確認する。
4) ナトリウム標準液(Na 1 mg/mL) 48.1 a) 1)を用いる。
5) アンモニウムイオン標準液(NH4+ 1 mg/mL) 42.2 a) 5) を用いる。
6) カリウム標準液(K 1 mg/mL) JIS K 8121に規定する塩化カリウムを500 ℃で約4時間加熱し,
デシケーター中で放冷する。その1.907 gをとり,少量の水に溶かして全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。ポリエチレン瓶に保存する。
7) カルシウム標準液(Ca 1 mg/mL) JIS K 8617に規定する炭酸カルシウムを105±2 ℃で約2時間
加熱し,デシケーター中で放冷する。その2.498 gをとり,水約50 mLに分散させ,これに塩酸(1
+1)[24.2 a) 2)による。]40 mLを加えて溶かす。沸騰しない程度に数分間加熱して二酸化炭素を除
く。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
8) マグネシウム標準液(Mg 1 mg/mL) JIS K 8432に規定する酸化マグネシウムを約800 ℃で約2時
間加熱し,デシケーター中で放冷する。その1.658 gを塩酸(1+1)[24.2 a) 2)による。]40 mLに溶
かして全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
9) 陽イオン混合標準液[(NH4+ 0.1 mg,Na 0.1 mg,K 0.1 mg,Ca 0.2 mg,Mg 0.2 mg)/mL](6) ア
ンモニウムイオン標準液(NH4+ 1 mg/mL)10 mL,ナトリウム標準液(Na 1 mg/mL)10 mL,カリ
ウム標準液(K 1 mg/mL)10 mL,カルシウム標準液(Ca 1 mg/mL)20 mL及びマグネシウム標準液
(Mg 1 mg/mL)20 mLをそれぞれ全量フラスコ100 mLにとり,水を標線まで加える。使用時に調
製する。
10) 陽イオン混合標準液[(NH4+ 10 μg,Na 10 μg,K 10 μg,Ca 20 μg,Mg 20 μg)/mL](6) アンモニ
ウムイオン標準液(NH4+ 1 mg/mL)5 mL,ナトリウム標準液(Na 1 mg/mL)5 mL,カリウム標準
液(K 1 mg/mL)5 mL,カルシウム標準液(Ca 1 mg/mL)10 mL及びマグネシウム標準液(Mg 1 mg/mL)
10 mLをそれぞれ全量フラスコ500 mLにとり,水を標線まで加える。又は陽イオン混合標準液
[(NH4+ 0.1 mg,Na 0.1 mg,K 0.1 mg,Ca 0.2 mg,Mg 0.2 mg)/mL]10 mLを全量フラスコ100 mL
にとり,水を標線まで加える。いずれも使用時に調製する。
注(6) 陽イオンをそれぞれ単独に測定する場合,又はいずれかの同時測定の場合は,この操作に準じ
199
K 0102:2016
て必要な混合標準液を調製して用いてもよい。
b) 器具及び装置 器具及び装置は,次による。
1) イオンクロマトグラフ イオンクロマトグラフは,分離カラムとサプレッサー(7)とを組み合わせた
方式のもの,分離カラム単独の方式のものいずれかで,次に掲げる条件を満たすもので,アンモニ
ウムイオン,ナトリウム,カリウム,カルシウム及びマグネシウムが分離定量できるもの。
1.1) 分離カラム ステンレス鋼製又は合成樹脂製(8)のものに,陽イオン交換体を充塡したもの(9)。
1.2) 検出器 電気伝導率検出器
1.3) データ処理部 JIS K 0127の5.7(データ処理部)による。
2) マイクロシリンジ 10〜200 μLの適切なもの。又は自動注入装置
注(7) 溶離液中の陰イオンを水酸化物イオンに変換するためのもので,溶離液中の陰イオンの濃度に
対して十分なイオン交換容量をもった陰イオン交換膜(膜形,電気透析形がある。)又はこれと
同等な性能をもった陰イオン交換体を充塡したもの。再生液と組み合わせて用いる。
(8) 例えば,四ふっ化エチレン樹脂製,ポリエーテルエーテルケトン製などがある。
(9) 備考3.による。
c) 準備操作 準備操作は,次による。
1) 試料を3.2によってろ過する。
2) 試料の電気伝導率が10 mS/m(100 μS/cm)(25 ℃)以上の場合には,電気伝導率が10 mS/m以下に
なるように,水で一定の割合に薄める。
d) 操作 操作は,次による。
1) イオンクロマトグラフを作動できる状態にし,分離カラムに溶離液を一定の流量(例えば,1〜2
mL/min)で流しておく。再生液を必要とするサプレッサー装置では,再生液を一定の流量で流して
おく。
2) 陽イオン混合標準液[(NH4+ 10 μg,Na 10 μg,K 10 μg,Ca 20 μg,Mg 20 μg)/mL](例えば,20
〜200 μLの一定量)をマイクロシリンジ(10)を用いて,イオンクロマトグラフに注入してクロマトグ
ラムを記録し,各陽イオンの保持時間に相当するピークの位置を確認しておく。
3) c)の準備操作を行った試料の一定量(例えば,20〜200 μLの一定量)をマイクロシリンジ(10)を用い
て,イオンクロマトグラフに注入し,クロマトグラムを記録する。
4) クロマトグラム上の各陽イオンに相当するピークについて,指示値(11)を読み取る。
5) 試料を薄めた場合には,空試験として試料と同量の水について,1)〜4)の操作を行って試料につい
て得た結果を補正する。
6) 検量線から各陽イオンの濃度を求め,試料中の各陽イオンの濃度(mg/L)を算出する(12)。
注(10) 検量線作成時と同じものを用いる。
(11) ピーク高さ又はピーク面積。
(12) 注(6)によった場合は,各陽イオンのそれぞれの量を求め,濃度を算出する。
e) 検量線 検量線の作成は,次による。
1) 陽イオン混合標準液[(NH4+ 0.1 mg,Na 0.1 mg,K 0.1 mg,Ca 0.2 mg,Mg 0.2 mg)/mL]0.1〜30 mL
を段階的に全量フラスコ100 mLにとり,水を標線まで加え,d) 1)〜4)の操作を行って各陽イオンに
相当する指示値を読み取る。
2) 別に,空試験として,水についてd) 1)〜4)の操作を行って各陽イオンに相当する指示値を補正した
後,各陽イオンの量と指示値との関係線を作成する(13)。検量線の作成は測定時に行う。
200
K 0102:2016
検量線は,必ずしも直線関係を示さない。
注(13) 注(6)によった場合は,その陽イオンについて作成する。
備考 3. 溶離液を一定の流量(例えば,1〜2 mL/min)で流し,陽イオン混合標準液[(NH4+ 10 μg,
Na 10 μg,K 10 μg,Ca 20 μg,Mg 20 μg)/mL]の一定量をイオンクロマトグラフに注入し,
クロマトグラムを求め,各陽イオンの分離度(R)が1.3以上に分離できるものを用いる。分
離度(R)は,35.の備考8.の式によって求める。また,定期的に分離カラムの性能を確認す
るとよい。
4. 分離カラムは,使用を続けると性能が低下するので,定期的に備考3.の操作で確認する。性
能が低下している場合には,溶離液の20〜200倍の濃度のものを調製し,分離カラムに注入
し,洗浄した後,性能を確認する。性能が回復しない場合は新品と取り替える。
試料中の懸濁物,有機物などによっても汚染されて性能が徐々に低下する。懸濁物を含む
試料は,c)の準備操作で除去した後,試験する。また,有機物(たん白質,油脂,界面活性
剤など)を含む試料は限外ろ過膜でろ過し,できるだけ有機物を除去した後,試験する。
試料中に分離カラムの充塡剤と親和力の強い陽イオン(例えば,カルシウム,マグネシウ
ムなど)が存在すると,これらが充塡剤に吸着され,分離性能が徐々に低下するので,定期
的に溶離液の20〜200倍の濃度のものを調製し,試料と同様に分離カラムに注入し洗浄する。
その他,酸化性物質又は還元性物質が共存すると,分離カラムの分離性能が低下する。こ
のような場合には,試料を水で一定の割合に薄めて試験すれば,ある程度は影響を防ぐこと
ができる。
5. カルボン酸形の陽イオン交換カラムと,溶離液として,硝酸溶液,メタンスルホン酸溶液,
[2,6-ピリジンジカルボン酸-L(+)-酒石酸]溶液などを用いると,1価陽イオンのほかに
カルシウム,マグネシウムなど2価の陽イオンの溶離及び同時定量が可能になる。
6. 妨害物質
− アミノ酸及び脂肪族アミンのような有機化合物は,無機陽イオンの定量を妨害する可能
性がある。
− 2,6-ピリジンジカルボン酸(PDA)のような強い錯形成剤が溶離液に含まれてなく,サ
プレッサーを用いない場合は,亜鉛,ニッケル及びカドミウムのような陽イオンによる
妨害があり得る。
− マンガンのような他の陽イオンによる妨害の程度は,用いる分離カラムの選択性に依存
する。
− アンモニウムイオン及びナトリウムの定量において,それらの濃度に大きな違いがある
場合,相互の影響があり得る。ナトリウムの濃度が1 mg/Lのときアンモニウムイオン及
びカリウムは,いずれも100 mg/L以下であれば妨害しない。
− 試料中の固形物及び鉱物油・洗剤・フミン酸の有機物は,分離カラムの寿命を短くする
ので除去する。
49. カリウム(K) カリウムの定量には,フレーム光度法,フレーム原子吸光法又はイオンクロマトグラ
フ法を適用する。
なお,フレーム光度法は,1993年に第1版として発行されたISO 9964-3,フレーム原子吸光法は,1993
年に第1版として発行されたISO 9964-1,イオンクロマトグラフ法は,1998年に第1版として発行された
201
K 0102:2016
ISO 14911との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 9964-1:1993,Water quality−Determination of sodium and potassium−Part 1 : Determination of
sodium by atomic absorption spectrometry(MOD)
ISO 9964-3:1993,Water quality−Determination of sodium and potassium−Part 3 : Determination of
sodium and potassium by flame emission spectrometry(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+, Na+, NH4+, K+, Mn2+, Ca2+, Mg2+, Sr2+
and Ba2+ using ion chromatography−Method for water and waste water(MOD)
49.1 フレーム光度法 試料をアセチレン-空気フレーム,水素-酸素フレームなどの中に噴霧し,このとき
生じる波長766.5 nm又は769.9 nmの輝線の強さを測定してカリウムを定量する。
定量範囲:K 40〜400 μg/L,0.4〜4 mg/L,4〜40 mg/L,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) カリウム標準液(K 1 000 mg/L) 48.3 a) 6)による。
2) カリウム標準液(K 4〜40 mg/L) カリウム標準液(K 1 000 mg/L)を段階的にとり,これを水で薄
めてK 4〜40 mg/Lの標準液を調製する(1)。
注(1) 低い濃度の測定用には,K 0.4〜4 mg/L又はK 40〜400 μg/Lの標準液を調製する。
b) 装置 装置は,次による。
1) フレーム光度計
c) 操作 操作は,次による。
1) カリウム標準液(K 40 mg/L)(2)をフレーム光度計のフレーム中に噴霧し,波長766.5 nm又は769.9
nmの指示値が100を示すように調節する。
2) 水を噴霧して指示値がゼロを示すように調節する。
3) カリウム標準液(K 4〜40 mg/L)(1)を順次噴霧し,カリウム(K)の濃度と指示値との関係線を作
成し,検量線とする。
4) 試料(3) (4)(カリウム濃度が40 mg/L以上の場合は薄める。)を噴霧して指示値を読み取り,検量線
から試料中のカリウムの濃度(K mg/L)を求める。
注(2) 低い濃度の測定用には,K 4 mg/L又はK 0.4 mg/Lの標準液を用いる。
(3) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(4) 試料に干渉物質が含まれる場合には,その影響を無視できる濃度まで薄めて測定するか,又は
試料と同程度の干渉物質を含むカリウム標準液(K 4〜40 mg/L)を調製し,検量線を作成する。
備考 1. ナトリウムが共存すると正の誤差を生じる。その他の共存物質による影響は,48.の備考1.
による。
49.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,カリウムによる原子吸光を波長
766.5 nmで測定してカリウムを定量する。
定量範囲:K 0.05〜5 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) カリウム標準液(K 0.1 mg/mL) 48.3 a) 6)のカリウム標準液(K 1 000 mg/L)10 mLを全量フラス
コ100 mLにとり,水を標線まで加える。使用時に調製する。
202
K 0102:2016
2) カリウム標準液(K 10 μg/mL) カリウム標準液(K 0.1 mg/mL)20 mLを全量フラスコ200 mLに
とり,水を標線まで加える。使用時に調製する。
b) 装置 装置は,48.2 b) 1)による。
c) 操作 操作は,次による。
1) 試料(3)をフレーム中に導入し,波長766.5 nmの指示値(5)を読み取る。
2) 空試験として水について,1)の操作を行って試料について得た指示値を補正する。
3) 検量線からカリウムの量を求め,試料中のカリウムの濃度(K mg/L)を算出する。
注(5) 吸光度又はその比例値。
d) 検量線 検量線の作成は,次による。
1) カリウム標準液(K 10 μg/mL)0.5〜50 mLを全量フラスコ100 mLに段階的にとり,水を標線まで
加える。この溶液についてc) 1)の操作を行う。
2) 別に,空試験として,水についてc) 1)の操作を行って標準液について得た指示値を補正し,カリウ
ム(K)の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
備考 2. 塩化セシウム溶液(25 g/L)を加えることで,カルシウムなどの影響を抑制できる。添加量
などは,48.の備考1.による。
49.3 イオンクロマトグラフ法 48.3による。
備考 3. 48.の備考3.による。
4. 48.の備考4.による。
5. 48.の備考5.による。
6. 48.の備考6.による。カリウムの濃度が1 mg/Lのときアンモニウムイオン及びナトリウムは
いずれも100 mg/L以下であれば妨害しない。また,アミン類のうちメタンアミン(モノメチ
ルアミン)が共存するとカリウムのピークと重なり妨害する。
50. カルシウム(Ca) カルシウムの定量には,キレート滴定法,フレーム原子吸光法,イオンクロマト
グラフ法又はICP発光分光分析法を適用する。
なお,キレート滴定法は,1984年に第1版として発行されたISO 6058,フレーム原子吸光法は,1986
年に第1版として発行されたISO 7980,イオンクロマトグラフ法は,1998年に第1版として発行された
ISO 14911,ICP発光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6058:1984,Water quality−Determination of calcium content−EDTA titrimetric method(MOD)
ISO 7980:1986,Water quality−Determination of calcium and magnesium−Atomic absorption
spectrometric method(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+, Na+, NH4+, K+, Mn2+, Ca2+, Mg2+, Sr2+
and Ba2+ using ion chromatography−Method for water and waste water(MOD)
50.1 キレート滴定法 試料をpH12以上とし,指示薬としてHSNN{2-ヒドロキシ-1-[(2'-ヒドロキシ-4'-
スルホ-1'-ナフタレニル)アゾ]-3-ナフタレンカルボン酸(IUPACによる名称)}を加え,エチレンジアミ
203
K 0102:2016
ン四酢酸二水素二ナトリウム溶液で滴定してカルシウムを定量する。
定量範囲:Ca 0.2〜5 mg
a) 試薬 試薬は,次による。
1) 水酸化カリウム溶液 JIS K 8574に規定する水酸化カリウム250 gを水に溶かして500 mLとする。
ポリエチレン瓶に保存する。
2) シアン化カリウム溶液(100 g/L) JIS K 8443に規定するシアン化カリウム10 gを水に溶かして100
mLとする。ポリエチレン瓶に保存する。
3) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 39.2 a) 3)による。
4) HSNN溶液(1) JIS K 8776に規定する2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-
ナフトエ酸0.5 gをJIS K 8891に規定するメタノール100 mLに溶かし,JIS K 8201に規定する塩化
ヒドロキシルアンモニウム0.5 gを加える。着色ガラス瓶に保存する。
5) 10 mmol/L EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和
物3.8 gをとり,水1 Lに溶かした後,ポリエチレン瓶に保存する。
標定 10 mmol/L 亜鉛溶液25 mLをビーカー200 mLにとり,水75 mLを加え,水酸化ナトリウ
ム溶液(100 g/L)[19. a) 2)による。]でpH6〜8に調節する。塩化アンモニウム-アンモニア緩衝
液(pH10)[28.1.2 a) 2)による。]2 mLと指示薬としてEBT溶液(5 g/L)[51.1 a) 4)による。]2,
3滴又はEBT粉末指示薬(JIS K 8736に規定するエリオクロムブラックT 0.10 gとJIS K 8150に
規定する塩化ナトリウム10 gをすり潰し混合する。)約0.05 gとを加え,調製した10 mmol/L EDTA
溶液で滴定する。終点は,溶液の色が赤から青に変わる点とする。
ファクターは,次の式によって算出する。
(
)
V
f
f
25
1×
=
ここに,
f: 10 mmol/L EDTA溶液のファクター
f1: 10 mmol/L 亜鉛溶液のファクター
V: 滴定に要した10 mmol/L EDTA溶液量(mL)
6) 10 mmol/L亜鉛溶液 JIS K 8001に準じ,次の方法で調製する。
− JIS K 8005に規定する容量分析用標準物質の亜鉛約0.5 gを塩酸(1+3)(JIS K 8180に規定する
塩酸を用いて調製する。)で洗い,水洗いし,更にJIS K 8101に規定するエタノール(99.5)及び
JIS K 8103に規定するジエチルエーテルで順次洗った後,直ちに上口デシケーターに入れ,2.0 kPa
以下で数分間保った後,減圧下で約12時間保つ。
− その0.33 gを0.1 mgの桁まではかりとり,硝酸(1+1)(JIS K 8541に規定する硝酸を用いて調
製する。)20 mL中に加え,加熱して溶かし,煮沸して窒素酸化物を追い出し,放冷後,全量フラ
スコ500 mLに移し入れ,水を標線まで加える。
この10 mmol/L 亜鉛溶液のファクターは,次の式による。
×
=
100
0
327
.0
1
A
a
f
ここに,
f1: 10 mmol/L 亜鉛溶液のファクター
a: はかりとった亜鉛の質量(g)
A: 亜鉛の純度(質量分率%)
0.327 0: 10 mmol/L亜鉛溶液500 mL中の亜鉛の相当量(g)
注(1) HSNN溶液に代え,次の方法で調製したHSNN粉末指示薬を用いてもよい。
204
K 0102:2016
− JIS K 8776に規定するHSNN 0.2 gとJIS K 8962に規定する硫酸カリウム10 gとをよく
すり潰し混合する。
− JIS K 8776に規定するHSNN 0.2 gとJIS K 8150に規定する塩化ナトリウム100 gとをよ
くすり潰し混合する。
参考 HSNNは,カルコンカルボン酸という名称でも知られている。
b) 操作 操作は,次による。
1) 試料(2)の適量(Caとして5 mg以下を含む。)をビーカーにとり,水を加えて約50 mLとする。
2) 水酸化カリウム溶液4 mLを加え,よくかき混ぜた後,約5分間放置する(3)。
3) シアン化カリウム溶液(100 g/L)0.5 mL(4)及び塩化ヒドロキシルアンモニウム溶液(100 g/L)0.5 mL
を加えてかき混ぜる。
4) 指示薬としてHSNN溶液(5) 5,6滴を加え(6),10 mmol/L EDTA溶液で,溶液の色が赤紫から青にな
るまで滴定する。
5) 次の式によって試料中のカルシウムの濃度(Ca mg/L)を算出する。
8
400
.0
000
1
×
×
×
=
V
f
a
C
ここに,
C: カルシウムの濃度(Ca mg/L)
a: 滴定に要した10 mmol/L EDTA溶液量(mL)
V: 試料量(mL)
f: 10 mmol/L EDTA溶液のファクター
0.400 8: 10 mmol/L EDTA溶液1 mLに相当するカルシウムの質量
(mg)
注(2) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。また,この試験に影響
を与える有機物及び着色物質を含む場合には,5.の操作を行った後,中和する。ただし,5.4の
方法は適用しない。
(3) 放置したときに生じる沈殿の量が多いと,終点が不明瞭になる。このような場合には,1回目
の滴定で,概略の滴定量を求めておき,別のビーカーに同量の試料をとり,1回目の滴定に要
した10 mmol/L EDTA溶液の量よりも約1 mL少ない量の10 mmol/L EDTA溶液を加え,水酸化
カリウム溶液4 mLを加えて,よく振り混ぜた後,約5分間放置する。次に,シアン化カリウム
溶液(100 g/L)0.5 mL(4)と塩化ヒドロキシルアンモニウム溶液(100 g/L)0.5 mLとを加えて振
り混ぜる。これに指示薬としてHSNN溶液5,6滴を加え(5) (6),再び10 mmol/L EDTA溶液で,
溶液の色が青になるまで滴定する。
(4) 亜鉛,銅などシアン化カリウムによってマスキングする金属類が共存しない場合は,添加しな
くてよい。
(5) HSNN溶液の代わりに注(1)のHSNN粉末指示薬の適量を用いてもよい。ただし,粉末指示薬は
HSNNが溶けるのに時間を要するので注意する。
(6) この指示薬は,添加後放置すると酸化され,終点の変色が不明瞭になる。
備考 1. シアン化カリウムは,有毒である。取扱い及び廃棄には,必要な予防措置をとる。シアン化
カリウムを含む溶液は,酸性にしてはならない。
2. アルミニウム,バリウム,鉛,鉄,コバルト,銅,マンガン,すず及び亜鉛の金属イオンは,
カルシウムと同じように滴定される。また,終点の色の変化を不明瞭にする。
オルトりん酸イオンの濃度が1 mg/L以上の場合は,滴定時のpH条件ではカルシウムと沈
205
K 0102:2016
殿する。滴定に時間がかかった場合,又はカルシウムの含有量が高い場合(100 mg/L又は2.5
mmol/Lより高い)には,炭酸カルシウムの沈殿が生じる。
鉄30 mg/L以下の妨害はシアン化カリウム250 mg又はJIS K 8663に規定する2,2′,2''-ニト
リロトリエタノール(トリエタノールアミン)5〜7 mLを滴定直前に添加することによって,
マスキングできる。シアン化物イオンは,亜鉛,銅及びコバルトの妨害も最小にする。また,
トリエタノールアミンはアルミニウムの妨害を小さくする。シアン化カリウムを加える前に
溶液がアルカリ性であることを確認する。
50.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,カルシウムによる原子吸光を波
長422.7 nmで測定してカルシウムを定量する。
定量範囲:Ca 0.2〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
2) ランタン(III)溶液(La 50 g/L) 酸化ランタン(III)29 gを少量ずつ塩酸(1+1)500 mLに加え
て溶かす。
3) カルシウム標準液(Ca 0.5 mg/mL) 48.3 a) 7)のカルシウム標準液(Ca 5 mg/mL)10 mLを全量フラ
スコ100 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
4) カルシウム標準液(Ca 20 μg/mL) カルシウム標準液(Ca 0.5 mg/mL)10 mLを全量フラスコ250 mL
にとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
b) 装置 装置は,次による。
1) フレーム原子吸光分析装置 JIS K 0121に規定するフレーム原子吸光分析装置で,測定対象元素用
の光源を備え,かつ,バックグラウンド補正が可能なもの。
c) 操作 操作は,次による。
1) 試料(7)の適量(Caとして20〜400 μgを含む。)を全量フラスコ100 mLにとり,塩酸(1+1)2 mL
を加えた後,水を標線まで加える。
2) この溶液10 mLを乾いたビーカーにとり,ランタン(III)溶液(La 50 g/L)1 mLを加える。
3) 2)の溶液をフレーム中に導入し,波長422.7 nmの指示値(8)を読み取る。
4) 空試験として試料と同量の水について,1)〜3)の操作を行って試料について得た指示値を補正する。
5) 検量線からカルシウムの量を求め,試料中のカルシウムの濃度(Ca mg/L)を算出する。
注(7) 懸濁物が含まれている場合には,ろ過又は遠心分離によって除去する。
(8) 吸光度又はその比例値
d) 検量線 検量線の作成は,次による。
1) カルシウム標準液(Ca 20 μg/mL)1〜20 mLを全量フラスコ100 mLに段階的にとり,試料と同じ酸
の濃度になるように塩酸(1+1)を加えた後,水を標線まで加える。この溶液についてc) 2)及び3)
の操作を行う。
2) 別に,空試験として水について試料と同じ酸の濃度になるように塩酸(1+1)を加えた後,c) 2)及
び3)の操作を行って標準液について得た指示値を補正し,カルシウム(Ca)の量と指示値との関係
線を作成する。検量線の作成は,試料測定時に行う。
備考 3. りん酸イオン,硫酸イオン,アルミニウムなどは妨害するが,ランタン(III)溶液(La 50 g/L)
を加えることによって妨害を抑制することができる。
4. アセチレン-一酸化二窒素フレームを用いるときは,塩化セシウム溶液(25 g/L)を用いる。
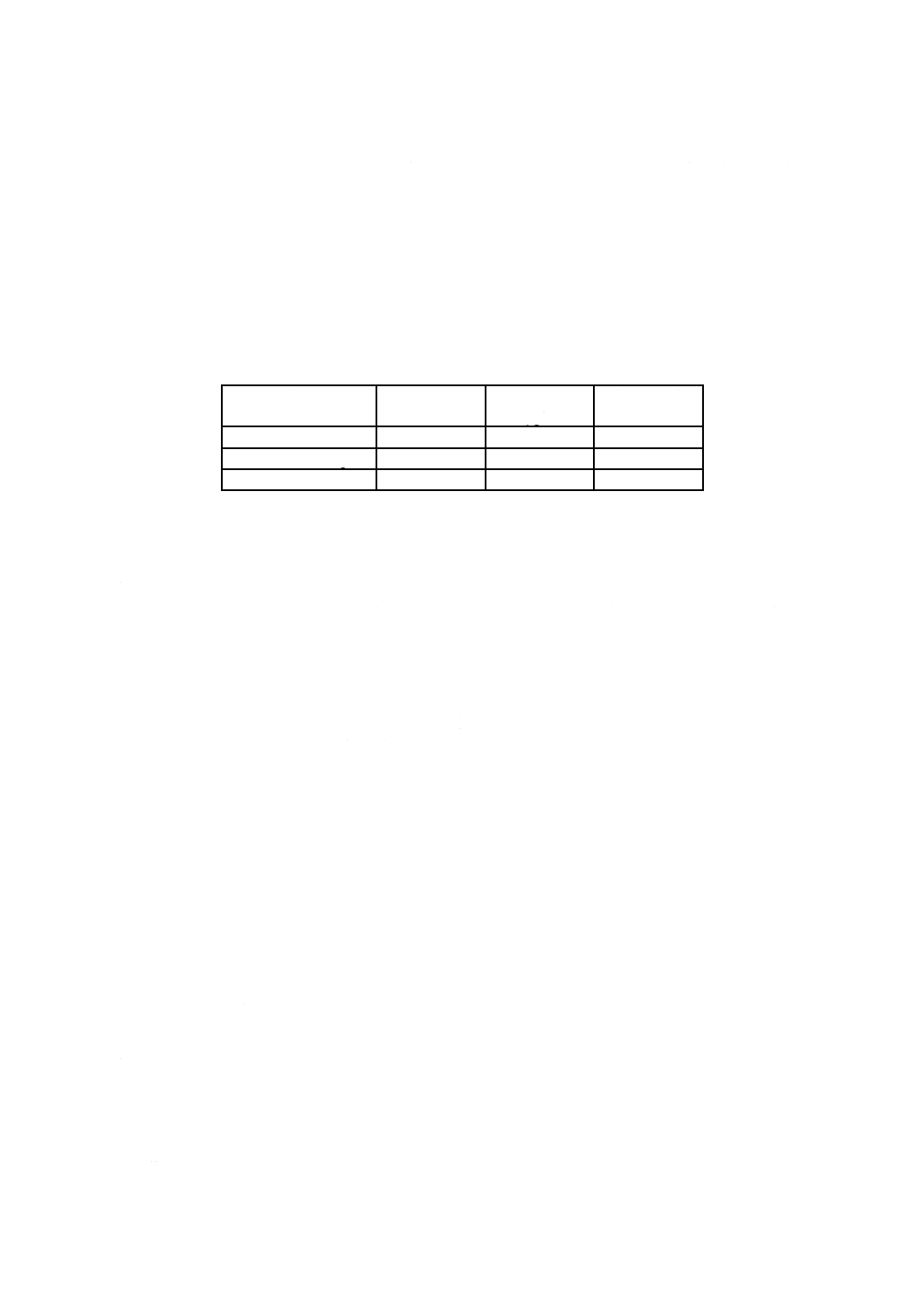
206
K 0102:2016
この溶液の調製は,次による。
塩化セシウム溶液(25 g/L) 塩化セシウム25 g(Csとして約20 g)を塩酸(0.1 mol/L)(JIS
K 8180に規定する塩酸を用いて調製する。)1 Lに溶かす。
備考 5. 多量のマグネシウム(1 000 mg/L以上)の共存は,負の誤差を与える。
50.3 ICP発光分光分析法 試料を試料導入部を通して誘導結合プラズマ中に噴霧し,カルシウムによる
発光を波長393.367 nmで測定してカルシウムを定量する。この方法によって,表50.1に示す元素が同時
定量できる。
それぞれの元素の測定波長,定量範囲,繰返し精度などの例を,表50.1に示す。
表50.1 対象元素の定量範囲などの例*
対象元素
測定波長
nm
定量範囲
μg/L
繰返し精度
%
カルシウム (Ca)
393.367
10〜5 000
2〜10
マグネシウム(Mg)
279.553
5〜3 000
2〜10
イットリウム(Y)**
371.029
−
−
注*
装置及び測定条件によって異なる。
** 内標準元素
a) 試薬 試薬は,次による。
1) 塩酸(1+1) 50.2 a) 1)による。
2) カルシウム標準液(Ca 0.1 mg/mL) 48.3 a) 7)のカルシウム標準液(Ca 1 mg/mL)10 mLを全量フラ
スコ100 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
3) カルシウム標準液(Ca 10 μg/mL) カルシウム標準液(Ca 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
4) マグネシウム標準液(Mg 0.1 mg/mL) 48.3 a) 8)のマグネシウム標準液(Mg 1 mg/mL)10 mLを全
量フラスコ100 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
5) マグネシウム標準液(Mg 1 μg/mL) マグネシウム標準液(Mg 0.1 mg/mL)10 mLを全量フラスコ1
000 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
c) 操作 操作は,次による。
1) 試料(7)の適量(Caとして1〜500 μg,Mgとして0.5〜300 μgを含む。)を全量フラスコ100 mLにと
り,塩酸の濃度が約0.1 mol/Lになるように塩酸(1+1)を加え,水を標線まで加える。
2) 1)の溶液を試料導入部を通して発光部に導入し,カルシウム及びマグネシウムの発光強度を測定す
る(9) (10)。
3) 空試験として塩酸(1+1)5 mLに水を加えて100 mLとした溶液について,2)の操作を行って,2)
で得た発光強度を補正する。
4) 検量線からカルシウム及びマグネシウムの量を求め,試料中のカルシウム及びマグネシウムの濃度
(mg/L)を算出する(11)。
注(9) 47.の注(8)による。
(10) 47.の注(9)による。
(11) 測定対象の元素の中から,必要な元素だけの濃度を算出する。
207
K 0102:2016
d) 検量線 検量線の作成は,次による。
1) a) 2)又はa) 3)のいずれかの一定量及びa) 4)又はa) 5)のいずれかの一定量を適切な容量の全量フラ
スコにとり,水を標線まで加え,カルシウム及びマグネシウムを含み,試料の測定濃度範囲よりも
高い濃度(12)の混合標準液を調製する。
2) 全量フラスコ100 mLに,カルシウム及びマグネシウムの測定濃度範囲を含むように,この混合標
準液を段階的にとり,塩酸(1+1)2 mLを加え,水を標線まで加える。
3) 2)で調製した溶液についてc) 2)の操作を行う。
4) 空試験としてc) 3)の操作を行う。
5) カルシウム及びマグネシウムそれぞれの量とそれぞれの発光強度との関係線を作成する。
注(12) 作成する検量線の最高の濃度の5〜10倍程度。
備考 6. 波長の異なる2本以上のスペクトル線の同時測定が可能な装置を用いるときは,内標準法に
よることができる。操作は,次による。
1) 試料の適量を全量フラスコ100 mLにとり,イットリウム溶液(Y 50 μg/mL)[47.の備考
5. 6)による。]10 mLを加えた後,塩酸の濃度が約0.1 mol/Lになるように塩酸(1+1)
を加え,水を標線まで加える。
2) この溶液について,c) 2)の操作を行ってカルシウム,マグネシウム及びイットリウムの
発光強度を測定し,カルシウム及びマグネシウムそれぞれの発光強度とイットリウムの
発光強度との比を求める。
3) 空試験として,試料に代えて同量の水を用い,1)及び2)の操作を行い,カルシウム及び
マグネシウムそれぞれの発光強度とイットリウムの発光強度との比を求め,2)で得た発
光強度の比を補正する。
4) 検量線から,カルシウム及びマグネシウムの量を求め,試料中のカルシウム(Ca mg/L)
及びマグネシウム(Mg mg/L)の濃度を算出する。
5) 検量線 全量フラスコ100 mL数個にd) 2)に従って混合標準液を段階的にとり,イット
リウム溶液(Y 50 μg/mL)10 mL及び塩酸(1+1)2 mLを加えた後,水を標線まで加え
る。この溶液について,2)の操作を行い,カルシウム,マグネシウム及びイットリウム
の発光強度を測定し,カルシウム及びマグネシウムそれぞれの発光強度とイットリウム
の発光強度との比を求める。別に,空試験として,混合標準液に代えて水を用い,同じ
操作を行って,同様に発光強度の比を求め,混合標準液での発光強度比を補正し,カル
シウム又はマグネシウムの量と,それぞれの発光強度とイットリウムの発光強度との比
との関係線を作成する。
50.4 イオンクロマトグラフ法 48.3による。
51. マグネシウム(Mg) マグネシウムの定量には,キレート滴定法,フレーム原子吸光法,イオンクロ
マトグラフ法又はICP発光分光分析法を適用する。
なお,キレート滴定法は,1984年に第1版として発行されたISO 6059,フレーム原子吸光法は,1986
年に第1版として発行されたISO 7980,イオンクロマトグラフ法は,1998年に第1版として発行された
ISO 14911,ICP発光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
208
K 0102:2016
(修正している),NEQ(同等でない)とする。
ISO 6059:1984,Water quality−Determination of the sum of calcium and magnesium−EDTA
titrimetric method(MOD)
ISO 7980:1986,Water quality−Determination of calcium and magnesium−Atomic absorption
spectrometric method(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
ISO 14911:1998,Water quality−Determination of dissolved Li+,Na+,NH4+,K+,Mn2+,Ca2+,Mg2+,
Sr2+ and Ba2+ using ion chromatography−Method for water and waste water(MOD)
51.1 キレート滴定法 試料に緩衝液を加えてpHを約10に調節し,指示薬としてエリオクロムブラック
T[3-ヒドロキシ-4-[(1-ヒドロキシ-2-ナフタレニル)アゾ]-7-ニトロ-1-ナフタレンスルホン酸ナトリウム
(IUPACの名称による。)]を加え,エチレンジアミン四酢酸二水素二ナトリウム溶液で滴定し,カルシウ
ムとマグネシウムとの合量に対する滴定量を求め,カルシウムに対する滴定量を差し引き,マグネシウム
を定量する。
定量範囲:MgとCaとの合量がCaとして0.15〜5 mg
a) 試薬 試薬は,次による。
1) シアン化カリウム溶液(100 g/L) 50.1 a) 2)による。
2) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 39.2 a) 3)による。
3) 塩化アンモニウム-アンモニア緩衝液(pH10) 28.1.2 a) 2)による。
4) EBT溶液(5 g/L) JIS K 8736に規定するエリオクロムブラックT[1-(1-ヒドロキシ-2-ナフチル
アゾ)-6-ニトロ-2-ナフトール-4-スルホン酸ナトリウム]0.5 gをJIS K 8891に規定するメタノール
100 mLに溶かし,JIS K 8201に規定する塩化ヒドロキシルアンモニウム0.5 gを加える(1)。着色ガ
ラス瓶に入れて保存する。
5) 10 mmol/L EDTA溶液 50.1 a) 5)による。この溶液1 mLは,Mg 0.243 1 mgに相当する。
注(1) 指示薬にメタニル(metanil)塩(メタニルイエロー,4-アニリドアゾベンゼンスルホン酸ナト
リウム塩),{[3-[4-(フェニルアミノ)フェニル]アゾ]ベンゼンスルホン酸ナトリウム}0.17
gを加えると終点の検出が容易になる。この場合には,赤から灰青(pale grey)又は緑に変わる。
b) 操作 操作は,次による。
1) 試料(2)の適量(MgとCaとの合量がCaとして5 mg以下を含む。)をビーカーにとり,水を加えて
約50 mLとする。
2) シアン化カリウム溶液(100 g/L)0.5 mL(3),塩化ヒドロキシルアンモニウム溶液(100 g/L)5〜7
滴及び塩化アンモニウム-アンモニア緩衝液(pH10)1 mLを加える。
3) 指示薬としてEBT溶液(5 g/L)2,3滴(4)を加える。
4) 10 mmol/L EDTA溶液で,溶液の赤みが消えて青になるまで滴定(5)する。
5) 別に,同量の試料をとり,50.1 b) 1)〜4)の操作を行ってカルシウムの量に相当する10 mmol/L EDTA
溶液の滴定量(mL)を求める。
6) 次の式によって試料中のマグネシウムの濃度(Mg mg/L)を算出する。
1
243
.0
000
1
Ca
×
×
×
−
=
f
V
b
V
a
M
ここに,
M: マグネシウムの濃度(Mg mg/L)
209
K 0102:2016
a: 滴定に要した10 mmol/L EDTA溶液量(mL)
b: 50.1 b)で滴定に要した10 mmol/L EDTA溶液量(mL)
V: 試料量(mL)
f: 10 mmol/L EDTA溶液のファクター
VCa: 50.1 b)での試料量(mL)
0.243 1: 10 mmol/L EDTA溶液1 mLに相当するマグネシウムの質量
(mg)
注(2) 50.の注(2)による。
(3) 試料に亜鉛,銅,コバルトなどが共存しない場合は,添加しなくてもよい。
(4) EBT溶液(5 g/L)に代えて,備考1.に示す指示薬を用いてもよい。
(5) エリオクロムブラックTの変色は遅いので,変色点近くではよくかき混ぜながら徐々に滴定す
る。
備考 1. EBT溶液(5 g/L)以外の指示薬の調製方法は,次による。
− JIS K 8736に規定するエリオクロムブラックT0.5 gをJIS K 8663に規定する2,2′,2″-ニト
リロトリエタノール(トリエタノールアミン)100 mLに溶かす。この溶液の粘性を減ら
すため,25 mLまではトリエタノールアミンの代わりにJIS K 8102に規定するエタノー
ル(95)を加えてもよい。
2. 50.の備考2.による。
51.2 フレーム原子吸光法 試料をアセチレン-空気フレーム中に噴霧し,マグネシウムによる原子吸光を
波長285.2 nmで測定してマグネシウムを定量する。
定量範囲:Mg 20〜400 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸(1+1) 50.2 a) 1)による。
2) ランタン(III)溶液(La 50 g/L) 50.2 a) 2)による。
3) マグネシウム標準液(Mg 0.5 mg/mL) 48.3 a) 8)のマグネシウム標準液(Mg 1 mg/mL)50 mLを全
量フラスコ100 mLにとり,塩酸(1+1)5 mLを加えた後,水を標線まで加える。
4) マグネシウム標準液(Mg 2 μg/mL) マグネシウム標準液(Mg 0.5 mg/mL)10 mLを全量フラスコ
100 mLにとり,水を標線まで加える。この溶液10 mLを全量フラスコ250 mLにとり,塩酸(1+1)
5 mLを加えた後,水を標線まで加える。
b) 装置 装置は,50.2 b)による。
c) 操作 操作は,次による。
1) 試料(6)の適量(Mgとして2〜40 μgを含む。)を全量フラスコ100 mLにとり,塩酸(1+1)2 mL
を加えた後,水を標線まで加える。
2) この溶液10 mLを乾いたビーカーにとり,ランタン(III)溶液(La 50 g/L)1 mLを加える。
3) 2)の溶液をフレーム中に導入し,波長285.2 nmの指示値(7)を読み取る。
4) 空試験として試料と同量の水について,1)〜3)の操作を行って試料について得た指示値を補正する。
5) 検量線からマグネシウムの量を求め,試料中のマグネシウムの濃度(Mg μg/L)を算出する。
注(6) 50.の注(7)による。
(7) 50.の注(8)による。
d) 検量線 検量線の作成は,次による。
1) マグネシウム標準液(Mg 2 μg/mL)1〜20 mLを全量フラスコ100 mLに段階的にとり,試料と同じ
210
K 0102:2016
酸の濃度になるように塩酸(1+1)を加えた後,水を標線まで加える。この溶液についてc) 2)及び
3)の操作を行う。
2) 別に,空試験として水について試料と同じ酸の濃度になるように塩酸(1+1)を加えた後,c) 2)及
び3)の操作を行って標準液について得た指示値を補正し,マグネシウム(Mg)の量と指示値との関
係線を作成する。検量線の作成は,試料測定時に行う。
備考 3. アルミニウムは少量(2 mg/L)でも妨害するが,ランタン(III)溶液(La 50 g/L)を添加す
ることによって妨害を抑制することができる。
4. 50.の備考4.による。
51.3 ICP発光分光分析法 50.3による。
51.4 イオンクロマトグラフ法 48.3による。
52. 銅(Cu) 銅の定量には,ジエチルジチオカルバミド酸吸光光度法,フレーム原子吸光法,電気加熱
原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,1996
年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
52.1 ジエチルジチオカルバミド酸吸光光度法 試料中に共存する金属元素のマスキング剤として,くえ
ん酸塩及びエチレンジアミン四酢酸二水素二ナトリウム(EDTA)を加え,アンモニア水でpH約9とした
後,N,N-ジエチルジチオカルバミド酸ナトリウム(ジエチルカルバモジチオ酸ナトリウム)を加え,生成
する黄褐色の銅錯体を酢酸ブチルで抽出し,その吸光度を測定して銅を定量する。
定量範囲:Cu 2〜30 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) アンモニア水(1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
2) 硫酸ナトリウム JIS K 8987に規定するもの。
3) くえん酸水素二アンモニウム溶液(100 g/L) JIS K 8284に規定するくえん酸水素二アンモニウム
10 gを水に溶かして100 mLとする。
くえん酸水素二アンモニウム中に銅が含まれるときは,次の操作によって精製する。
− くえん酸水素二アンモニウム10 gを水80 mLに溶かし,アンモニア水(1+1)を加えてpH約9
とした後,水を加えて100 mLとする。
− これを分液漏斗に入れ,5)のジエチルジチオカルバミド酸ナトリウム溶液(10 g/L)2 mL及び7)
の酢酸ブチル10 mLを加え,激しく振り混ぜて放置する。
− 水層を乾いたろ紙でろ過し,酢酸ブチルの小滴を除いたろ液を用いる。
4) EDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物2 gを水
に溶かして100 mLとする。
211
K 0102:2016
5) ジエチルジチオカルバミド酸ナトリウム溶液(10 g/L) JIS K 8454に規定するN,N-ジエチルジチオ
カルバミド酸ナトリウム三水和物1.3 gを水に溶かして100 mLとする。着色瓶に保存し,2週間以
上経過したものは使用しない。
6) メタクレゾールパープル溶液(1 g/L) JIS K 8889に規定するメタクレゾールパープル0.1 gをJIS K
8102に規定するエタノール(95)50 mLに溶かし,水を加えて100 mLとする。
7) 酢酸ブチル JIS K 8377に規定するもの。
8) 銅標準液(Cu 0.1 mg/mL) JIS K 8005に規定する容量分析用標準物質の銅を塩酸(1+3)(JIS K
8180に規定する塩酸を用いて調製する。)で洗い,水洗いし,JIS K 8101に規定するエタノール(99.5)
で洗う。次に,JIS K 8103に規定するジエチルエーテルで洗った後,直ちに上口デシケーター中に
入れ圧力2 kPa以下で数分間保った後,減圧下で約12時間保つ。Cu 100 %に対してその0.100 gを
とり,硝酸(1+1)(JIS K 8541に規定する硝酸を用いて調製する。)20 mLに溶かし,煮沸して窒
素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又はJIS
K 8983に規定する硫酸銅(II)五水和物0.393 gをとり,硝酸(1+1)20 mLを加えて溶かし,全量
フラスコ1 000 mLに移し入れ,水を標線まで加える。
9) 銅標準液(Cu 1 μg/mL) 銅標準液(Cu 0.1 mg/mL)10 mLを全量フラスコ1 000 mLにとり,硝酸
(1+1)[52.1 a) 8)による。]20 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL又は300 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(1)(Cuとして2〜30 μgを含む。)を分液漏斗にとり,指示薬としてメ
タクレゾールパープル溶液(1 g/L)2,3滴を加えた後,くえん酸水素二アンモニウム溶液(100 g/L)
5 mL及びEDTA溶液1 mLを加える。
2) アンモニア水(1+1)を加えて溶液の色がうすい紫(2)になるまで(pH約9)中和し,水を加えて50
mL(3)とする。
3) ジエチルジチオカルバミド酸ナトリウム溶液(10 g/L)2 mLを加えて混合し,次に,酢酸ブチル(4) 10
mLを加え,約3分間激しく振り混ぜて放置する。
4) 水層を捨て,酢酸ブチル層を硫酸ナトリウム約1 gを入れた共栓試験管に移し,振り混ぜる(5)。
5) その一部を吸収セルに移し,酢酸ブチルを対照液として波長440 nm付近の吸光度を測定する。
6) 空試験として水約20 mLをとり,1)〜5)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
7) 検量線から銅の量を求め,試料中の銅の濃度(Cu mg/L)を算出する。
注(1) 有機物及び濁りを含まない試料で銅の濃度が低い場合には,試料250 mLまでの適量をとり,5.1
を行い,1)〜6)に準じて操作する。この場合は,試料は前処理した全量を用い,試薬は1)〜4)
におけるものと同量を用いる。検量線は試料と同様に操作して作成する。
(2) 1)におけるメタクレゾールパープル溶液(1 g/L)を加えずに,pH計又はpH試験紙を用いても
よい。
(3) 分液漏斗にあらかじめ印を付けておく。
(4) 抽出溶媒としてクロロホルム,ベンゼンなどを用いてもよい。ただし,試料中にある種の陰イ
オン界面活性剤(例えば,スルホン酸形のもの。),タンニンなどが含まれるときは,銅の抽出
212
K 0102:2016
が不完全になる。
注(5) 乾いたろ紙でろ過するか,又は分液漏斗の脚部に乾いた脱脂綿を詰めてろ過してもよい。
d) 検量線 検量線の作成は,次による。
1) 銅標準液(Cu 1 μg/mL)2〜30 mLを分液漏斗に段階的にとり,c) 1)〜6)の操作を行って銅(Cu)の
量と吸光度との関係線を作成する。
備考 1. EDTA溶液を加えない場合には,ジエチルジチオカルバミド酸ナトリウムは多くの金属元素
と反応する。しかし,水銀,ひ素,鉛,すず,アンチモンなど,大部分の金属錯体は無色で
ある。鉄,ニッケル,コバルトなどの錯体は有色であるが,この方法では,EDTA溶液によ
ってマスキングされる。
2. ビスマスは銅とともに抽出され黄色を示すが,銅の量の2倍量以下のときは,ほとんど影響
しない。
2倍量以上を含むときは,c)の操作によって測定した吸光度をA1とし,別に,銅の試験に
用いた試料と同量の試料をとり,c) 1)の後に,シアン化カリウム溶液(50 g/L)(JIS K 8443
に規定するシアン化カリウムを用いて調製する。)3 mLを加え,銅をシアノ錯体とした後,
c) 2)〜6)の操作を行ってビスマス錯体だけを抽出し,その吸光度をA2とする。銅による吸光
度はA1−A2である。
3. 試料を5.1によって前処理する場合,シアン化合物を含むときは十分に加熱する。
52.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,銅による原子吸
光を波長324.8 nmで測定して銅を定量する。
定量範囲:Cu 0.2〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 銅標準液(Cu 10 μg/mL) 52.1 a) 8)の銅標準液(Cu 0.1 mg/mL)50 mLを全量フラスコ500 mLにと
り,硝酸(1+1)[52.1 a) 8)による。]10 mLを加えた後,水を標線まで加える。
b) 装置 装置は,次による。
1) フレーム原子吸光分析装置 JIS K 0121に規定するフレーム原子吸光分析装置で,測定対象元素用
の光源を備え,かつ,バックグラウンド補正が可能なもの。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 4. 銅の濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,次によるか,
又は備考5.若しくは備考6.による。これらの準備操作に使用する試薬及び器具類は,測定対
象元素について空試験を行って使用に支障のないことを確認しておく。これらの準備操作は,
亜鉛,鉛,カドミウム,ニッケル及びコバルトの定量にも使用できる。
1) 試料500 mL(又は100〜500 mLの一定量)をビーカーにとり,JIS K 8180に規定する塩
酸10 mLを加え,約5分間煮沸する。放冷後,分液漏斗1 000 mL(又は200〜500 mL)
に移し入れる。
2) くえん酸水素二アンモニウム溶液(100 g/L)[52.1 a) 3)による。]10 mL及び指示薬とし
てメタクレゾールパープル溶液(1 g/L)[52.1 a) 6)による。]2,3滴を加えた後,アンモ
ニア水(1+1)[52.1 a) 1)による。]を溶液の色が僅かに紫になるまで加える。
3) ジエチルジチオカルバミド酸ナトリウム溶液(10 g/L)[52.1 a) 5)による。]5 mLを加え
て振り混ぜた後,JIS K 8377に規定する酢酸ブチル10〜20 mL(*1)を加え,約1分間激し
213
K 0102:2016
く振り混ぜ,静置する。
4) 酢酸ブチル層を分離し,ビーカー100 mLに入れる。水層に酢酸ブチル5 mLを加えて抽
出操作を繰り返す。抽出した酢酸ブチル層は先のビーカーに合わせる(*2)。
5) 加熱して酢酸ブチルを揮散させた後,JIS K 8541に規定する硝酸2 mLとJIS K 8223に
規定する過塩素酸2 mLとを加えて加熱し,有機物を分解する。ほとんど乾固した後,
放冷する。
6) 残留物を硝酸(1+15)(JIS K 8541に規定する硝酸を用いて調製する。)10 mLに溶かし,
これを銅の定量に用いる。
注(*1) JIS K 8903に規定する4-メチル-2-ペンタノン(メチルイソブチルケトン,MIBK)
又は2,6-ジメチル-4-ヘプタノン(ジイソブチルケトン,DIBK)を用いてもよい。
2,6-ジメチル-4-ヘプタノンは,水との相互溶解がほとんどないので,その添加量
は少なくてもよい。
(*2) 抽出した有機層に抽出に使用した有機溶媒を加えて液量を一定量にしたもの,又
は抽出条件を一定にして,1回抽出を行った有機層をそのまま噴霧して原子吸光
分析することもできる。ただし,検量線は,銅標準液について同じ操作を行って
作成する。
備考 5. この操作は,ISO 8288の第2章の抽出操作との整合を図ったものである。
この準備操作は,亜鉛,鉛,カドミウム,ニッケル及びコバルトの定量にも使用できる。
1) 試料200 mLをとり,備考4. 1)による酸処理をした後,pHを3.5〜4.0とする。
2) 分液漏斗500 mLに移し入れ,硫酸アンモニウム溶液(飽和)(JIS K 8960に規定する硫
酸アンモニウムを用いて調製する。)20 mLを加える。1-ピロリジンカルボジチオ酸アン
モニウム(ピロリジン-N-ジチオカルバミド酸アンモニウム)(APDC)溶液(10 g/L)5 mL
を加え,静かに振り混ぜた後,約3分間放置する。
3) 次に,JIS K 8903に規定する4-メチル-2-ペンタノン10 mLを加え,約3分間激しく振り
混ぜ,光及び熱を遮断して静置する。
4) 有機層を分離し,ビーカー100 mLに入れる。水層に4-メチル-2-ペンタノン5 mLを加え,
抽出操作を繰り返す。抽出した有機層は先のビーカーに合わせる。
5) この有機層を備考4.の5)及び6)と同様に処理し,銅の定量に用いる。
6. キレート樹脂による分離濃縮法 この準備操作は,測定対象元素をキレート樹脂を充塡した
固相で分離濃縮する方法であり,亜鉛,鉛,カドミウム,鉄,ニッケル及びコバルトの定量
にも使用できる。試薬,操作などは次による。
1) 試薬 試薬は,次による。
1.1)
水 JIS K 0557に規定するA3又はA4の水。測定対象元素について空試験を行って使
用に支障のないことを確認しておく。
1.2)
硝酸 JIS K 9901に規定する高純度試薬−硝酸
1.3)
硝酸(2 mol/L),硝酸(1 mol/L) 1.2)の硝酸を水で希釈して調製する。
1.4)
アンモニア水 JIS K 8085に規定するアンモニア水
1.5)
酢酸アンモニウム JIS K 8359に規定する酢酸アンモニウム
1.6)
酢酸アンモニウム溶液(0.5 mol/L) 酢酸アンモニウム38.5 gを水に溶かして1 000 mL
とする(*3)。
214
K 0102:2016
1.7)
酢酸アンモニウム溶液(0.1 mol/L) 酢酸アンモニウム溶液(0.5 mol/L)200 mLを全
量フラスコ1 000 mLに移し入れ,水を標線まで加える(*3)。
注(*3) 酢酸アンモニウム溶液に含まれる測定元素の影響を避けるため,2.1)の調製済み
の固相に溶液を通過させるか,2.1)の調製済みの固相を溶液瓶に入れて一晩静置
するなどを行い,測定元素の量を低減した酢酸アンモニウム溶液を使用する。
2) 器具 器具は,次による。
2.1)
キレート樹脂充塡固相 イミノ二酢酸キレート樹脂を充塡した固相(*4)で,硝酸(2
mol/L),水,酢酸アンモニウム溶液(0.1 mol/L)を流下し,固相の洗浄及び調製を行
ったもの(*5)。
注(*4) 例えば,イミノ二酢酸キレート樹脂[粒子径38〜75 [μm](200〜400[メッシュ])]
1 gをポリプロピレン製カートリッジ(容量8 mL)に充塡したカラムを使用で
きる。また,イミノ二酢酸キレート樹脂を固定化したディスクも使用できる。
キレート樹脂は,イミノ二酢酸キレート樹脂の他に,ポリアミノポリカルボン
酸キレート樹脂など,試料中の測定元素を捕集可能な吸着容量をもったキレー
ト樹脂は使用できる。
(*5) 使用するキレート樹脂によって,硝酸を流下する前にメタノール,アセトニト
リル,アセトンなどの有機溶媒処理が必要なものもある。また,硝酸の濃度,
酢酸アンモニウム溶液の濃度及びpH値,各溶液量及び流下速度についても異
なるものがある。さらに,3.3)〜3.5)の操作についても,キレート樹脂によって,
各溶液の濃度,pH値,各溶液量及び流下速度が異なるものもある。
3) 操作 操作は,次による。
3.1)
試料1 000 mL又はその適量(*6)をビーカーにとり,硝酸を試料1 000 mLにつき10 mL
加え,約10分間煮沸し放冷する。不溶解物が残った場合には,ろ紙5種C(*7)を用い
てろ過し,水で洗い,ろ液と洗液をビーカーに移し入れる。
3.2)
この溶液に酢酸アンモニウムを酢酸アンモニウム溶液として0.1 mol/L(*8)になるよう
に加え,アンモニア水を用いてpH5.6に調節する。
3.3)
この溶液を,2.1)の固相に流下し,測定元素を固相に吸着させる(*9)。
3.4)
酢酸アンモニウム溶液(0.5 mol/L)を流下させて固相を洗浄する(*10)。
3.5)
固相の上端から硝酸(1 mol/L)5 mLを2回流下させて,測定元素を溶出させる。得
られた溶出液は試験管に受ける。
3.6)
この溶出液を全量フラスコ20 mLに移し入れ,標線まで水を加え,測定元素の定量に
用いる(*11)。
注(*6) 測定対象元素の量が各々の定量範囲内になるように調整する。
(*7) ろ紙6種又は1 μm以下のろ過材も使用できる。
(*8) 試料1 000 mLでは酢酸アンモニウムの添加量は,7.7 gになる。
(*9) 流下流量はあらかじめ測定元素が固相に吸着できる範囲を確認しておき,調節
する。固相カラムの場合は,5〜20 mL/minが一般的である。流量調整が必要な
場合は,ポンプ又はガスによる加圧,及びポンプによる吸引を行う。
(*10) 樹脂及び試料条件によっては,酢酸アンモニウム溶液の代わりに水を用いるも
のもある。また,酢酸アンモニウム溶液による洗浄後,更に水による洗浄が必
215
K 0102:2016
要なものもある。
注(*11) カルシウム及びマグネシウムなどの濃度の高い試料の場合,3.6)の溶液中にも高
濃度のカルシウム,マグネシウムが含まれるため,測定の妨害に注意をする。
空試験及び添加回収試験を行い,精確さを確認することが望ましい。
d) 操作 操作は,次による。
1) c)の準備操作を行った試料を,フレーム中に導入し,波長324.8 nmの指示値(6)を読み取る。
2) 空試験としてc)の準備操作での試料と同量の水をとり,試料と同様にc)及びd) 1)の操作を行って試
料について得た指示値を補正する。
3) 検量線から銅の量を求め,試料中の銅の濃度(Cu mg/L)を算出する。
注(6) 吸光度又はその比例値。
e) 検量線 検量線の作成は,次による。
1) 銅標準液(Cu 10 μg/mL)2〜40 mL (7)を全量フラスコ100 mLに段階的にとり,c) 1)を行った試料と
同じ酸の濃度になるように酸を加えた後,水を標線まで加える(8)。
2) この溶液についてd) 1)の操作を行う。
3) 別に,空試験として水についてc) 1)を行った試料と同じ酸の濃度になるように酸を加えた後,d) 1)
の操作を行って標準液について得た指示値を補正し,銅(Cu)の量と指示値との関係線を作成する。
検量線の作成は,試料測定時に行う。
注(7) 準備操作として溶媒抽出を適用するときは,銅標準液(Cu 10 μg/mL)の量を,適宜,減らす。
(8) 備考4.又は備考5.によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層又は2,6-
ジメチル-4-ヘプタノン層をそのまま噴霧する場合の検量線の作成は,次による。
銅標準液(Cu 10 μg/mL)を適切な濃度(Cu 0.1〜1 μg/mL)に薄め,その2〜40 mLを段階的
にとり,500 mL(又は100〜500 mLの一定量)とした後,試料と同様に備考4.並びにd) 1)及び
2)を行って銅(Cu)の量と指示値との関係線を作成する。
52.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,銅による原子吸光を波長324.8
nmで測定して銅を定量する。
定量範囲:Cu 5〜100 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 7. この方法は,共存する酸,塩の種類及び濃度の影響を受けやすいので,備考8.の操作を行わ
ない場合は,これらの影響の少ない試料に適用する。
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水。定量する元素について空試験を行って使用に支障のないこと
を確認しておく。
2) 硝酸(1+1) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) 銅標準液(Cu 1 μg/mL) 52.1 a) 9)による。
b) 器具及び装置 器具及び装置は,次による。
1) マイクロピペット JIS K 0970に規定するピストン式ピペット5〜50 μL又は自動注入装置
2) 電気加熱原子吸光分析装置 JIS K 0121に規定する電気加熱原子吸光分析装置で,測定対象元素用
の光源を備え,かつ,バックグラウンド補正が可能なもの。
3) 発熱体 黒鉛製又は耐熱金属製のもの。
4) フローガス JIS K 1105に規定するアルゴン2級
c) 準備操作 準備操作は,次による。
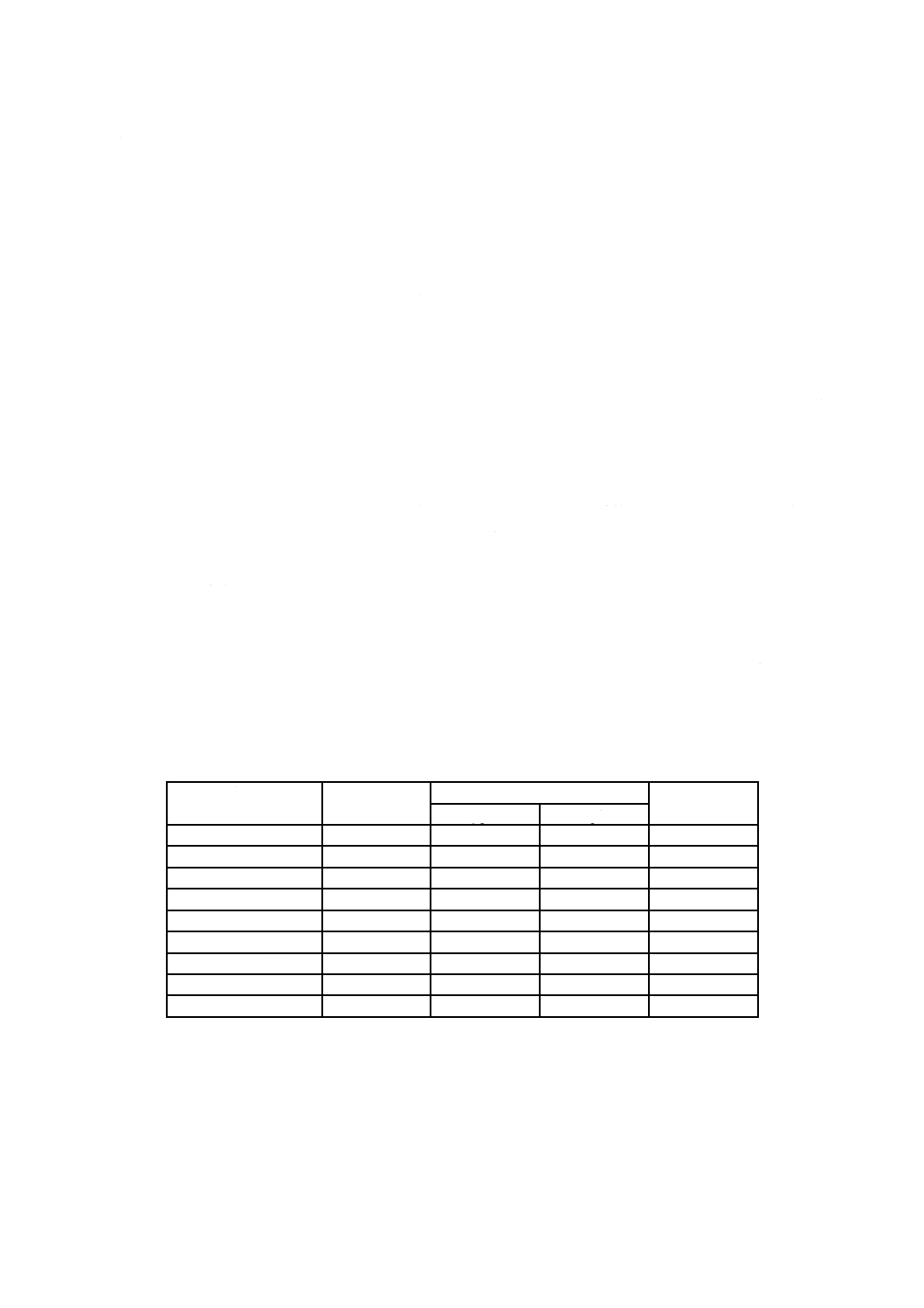
216
K 0102:2016
1) 試料を5.によって処理する。
備考 8. 試料の銅の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イオンなどの共存物
質の濃度が高く,測定を妨害する場合の準備操作は備考6.による。
d) 操作 操作は,次による。
1) c)の準備操作を行った試料の一定量(例えば,10〜50 μl)をマイクロピペットで発熱体に注入し,
乾燥(100〜120 ℃,30〜40秒間)した後,灰化(600〜1 000 ℃,30〜40秒間)し,次に,原子化
(9)(2 200〜2 700 ℃,3〜6秒間)し,波長324.8 nmの指示値(6)を読み取る(10)。
2) 空試験としてc)の準備操作での試料と同量の水をとり,試料と同様にc)及びd) 1)の操作を行い,試
料について得た指示値を補正する。
3) 検量線から銅の量を求め,試料中の銅の濃度(Cu μg/L)を算出する。
注(9) 乾燥,灰化及び原子化の条件は,装置によって異なる。試料の注入量及び共存する塩類の濃度
によっても異なることがある。
(10) 引き続いて少なくとも1)の操作を3回繰り返し,指示値が合うことを確認する。
e) 検量線 検量線の作成は,次による。
1) 銅標準液(Cu 1 μg/mL)0.5〜10 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試料と
同じ酸の濃度になるように酸を加えた後,水を標線まで加える。
2) この溶液について,d) 1)の操作を行う。
3) 別に,空試験として,水についてc) 1)を行った試料と同じ酸の濃度になるように酸を加えた後,d) 1)
の操作を行って標準液について得た指示値を補正し,銅(Cu)の量と指示値との関係線を作成する。
検量線の作成は,試料測定時に行う。
52.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,銅
による発光を波長324.754 nmで測定して銅を定量する。この方法によって表52.1に示す元素が同時定量
できる。それぞれの元素ごとの測定波長,定量範囲及び繰返し精度の例を,表52.1に示す。
表52.1 測定波長,定量範囲及び繰返し精度の例*
対象元素
測定波長
nm
定量範囲
繰返し精度
%
μg/L
mg/L
銅 (Cu)
324.754
20〜5 000
−
2〜10
亜鉛 (Zn)
213.856
10〜6 000
−
2〜10
鉛 (Pb)
220.351
−
0.1〜2
2〜10
カドミウム (Cd)
214.438
10〜2 000
−
2〜10
マンガン (Mn)
257.610
10〜5 000
−
2〜10
鉄 (Fe)
238.204
10〜5 000
−
2〜10
ニッケル (Ni)
221.647
40〜2 000
−
2〜10
コバルト (Co)
228.616
30〜3 000
−
5〜10
イットリウム(Y)**
371.029
−
−
−
注*
装置及び測定条件によって異なる。
** 内標準元素として,イットリウム(Y)のほか,インジウム(In)及びイッテルビウム
(Yb)も使用できる。
a) 試薬 試薬は,次による。
1) 銅標準液(Cu 1 mg /mL) JIS K 8005に規定する容量分析用標準物質の銅を塩酸(1+3)で洗い,
水洗いし,JIS K 8101に規定するエタノール(99.5)で洗い,次に,JIS K 8103に規定するジエチ
217
K 0102:2016
ルエーテルで洗った後,直ちに上口デシケーター中に入れ,2.0 kPa以下で数分間保った後,減圧下
で約12時間保つ。Cu 100 %に対してその1.00 gをとり,硝酸(1+1)30 mL中に加え,煮沸して溶
かし,窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
2) 銅標準液(Cu 10 μg/mL) 銅標準液(Cu 1 mg/mL)5 mLを全量フラスコ500 mLにとり,硝酸(1
+1)10 mLを加えた後,水を標線まで加える。
3) 亜鉛標準液(Zn 1 mg/mL) JIS K 8005に規定する容量分析用標準物質の亜鉛を塩酸(1+3)で洗
い,水洗いし,JIS K 8101に規定するエタノール(99.5)で洗い,次に,JIS K 8103に規定するジ
エチルエーテルで洗った後,直ちに上口デシケーター中に入れ,圧力2 kPa以下で数分間保った後,
減圧下で約12時間保つ。Zn 100 %に対してその1.00 gをとり,硝酸(1+1)30 mLに溶かし,煮沸
して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
4) 亜鉛標準液(Zn 10 μg/mL) 亜鉛標準液(Zn 1 mg/mL)5 mLを全量フラスコ500 mLにとり,硝酸
(1+1)10 mLを加えた後,水を標線まで加える。
5) 鉛標準液(Pb 1 mg/mL) JIS K 8701に規定する鉛(99.9 %以上)1.00 gをとり,硝酸(1+1)30 mL
に溶かし,加熱して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線
まで加える。又はJIS K 8563に規定する硝酸鉛(II)1.60 gをとり,硝酸(1+1)20 mL及び適量
の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
6) 鉛標準液(Pb 10 μg/mL) 鉛標準液(Pb 1 mg/mL)5 mLを全量フラスコ500 mLにとり,硝酸(1
+1)10 mLを加え,水を標線まで加える。
7) カドミウム標準液(Cd 0.1 mg/mL) カドミウム(99.9 %以上)0.100 gをとり,硝酸(1+1)20 mL
に溶かし,煮沸して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線
まで加える。
8) カドミウム標準液(Cd 10 μg/mL) カドミウム標準液(Cd 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
9) マンガン標準液(Mn 1 mg/mL) JIS K 8247に規定する過マンガン酸カリウム2.88 gをとり,水150
mLに硝酸(1+1)10 mLを加えた溶液に溶かす。過酸化水素水(1+9)(JIS K 8230に規定する過
酸化水素10 mLを水で薄めて100 mLとする。)を滴加し,かき混ぜて脱色した後,煮沸して過剰の
過酸化水素を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又はマ
ンガン(99.9 %以上)1.00 gをとり,硝酸(1+3)20 mLに溶かし,煮沸して窒素酸化物を追い出す。
放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
10) マンガン標準液(Mn 10 μg/mL) マンガン標準液(Mn 1 mg/mL)5 mLを全量フラスコ500 mLに
とり,硝酸(1+1)10 mLを加え,水を標線まで加える。
11) 鉄標準液(Fe 1 mg/mL) 鉄(99.5 %以上)1.000 gをとり,塩酸(1+1)30 mL中に入れ,加熱し
て溶かし,放冷後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又はJIS K 8979に
規定する硫酸アンモニウム鉄(II)六水和物[ビス(硫酸)鉄(II)アンモニウム六水和物]7.02 g
をとり,塩酸(1+1)20 mL及び適量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線
まで加える。
12) 鉄標準液(Fe 0.1 mg/mL) 鉄標準液(Fe 1 mg/mL)10 mLを全量フラスコ100 mLにとり,塩酸(1
+1)2 mLを加えた後,水を標線まで加える。
13) 鉄標準液(Fe 10 μg/mL) 鉄標準液(Fe 0.1 mg/mL)10 mLを全量フラスコ100 mLにとり,塩酸(1
+1)2 mLを加えた後,水を標線まで加える。
218
K 0102:2016
14) ニッケル標準液(Ni 0.1 mg/mL) JIS K 9062に規定するニッケル(99.9 %以上)0.100 gをとり,硝
酸(1+1)20 mLに溶かし,煮沸して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移
し入れ,水を標線まで加える。
15) ニッケル標準液(Ni 10 μg/mL) ニッケル標準液(Ni 0.1 mg/mL)10 mLを全量フラスコ100 mLに
とり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。
16) コバルト標準液(Co 0.1 mg/mL) コバルト(99.5 %以上)0.100 gをとり,硝酸(1+1)20 mLに
溶かし,煮沸して窒素酸化物を追い出す。放冷後,全量フラスコ1 000 mLに移し入れ,水を標線ま
で加える。
17) コバルト標準液(Co 10 μg/mL) コバルト標準液(Co 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
備考9. 試料の噴霧に超音波ネブライザー又はこれと同等の性能をもったものを用いてもよい。この
場合は,定量下限値を1桁程度下げることができる。ただし,メモリー効果に注意し,十分
に洗浄を行う。
c) 準備操作 準備操作は,次による。
1) 試料を5.5によって処理する。
備考 10. 試料の銅又は測定対象元素の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イ
オンなどの共存物質の濃度が高く,測定を妨害する場合の準備操作は,次の1)〜3)の操作に
よるか,又は備考6.による。次の準備操作は,亜鉛,鉛,カドミウム,マンガン,鉄,ニッ
ケル,コバルト,モリブデン及びバナジウムの定量にも使用できる。
1) 試料500 mL(又は100〜500 mLの一定量)をビーカーにとり,JIS K 8180に規定する塩
酸5 mLを加え,約5分間煮沸する。
2) 放冷後,酢酸-酢酸ナトリウム緩衝液(pH5)(JIS K 8371に規定する酢酸ナトリウム三水
和物19.2 gとJIS K 8355に規定する酢酸3.4 mLとを水に溶かして1 Lとする。)(*12) 10
mLを加え,アンモニア水(1+1)(JIS K 8085に規定するアンモニア水を用いて調製す
る。)又は硝酸(1+10)(JIS K 8541に規定する硝酸を用いて調製する。)でpHを5.2
に調整する。
注(*12) この操作に用いる酢酸-酢酸ナトリウム緩衝液(pH5)は使用前に1-ピロリジンカ
ルボジチオ酸アンモニウム溶液,ヘキサメチレンアンモニウム-ヘキサメチレンカ
ルバモジチオ酸(ヘキサメチレンアンモニウム-ヘキサメチレンジチオカルバミド
酸)のメタノール溶液,JIS K 8271に規定するキシレンとを加えて振り混ぜ,精
製する。
3) この溶液を分液漏斗1 000 mL(又は200〜500 mL)に移し,1-ピロリジンカルボジチオ
酸アンモニウム溶液(20 g/L)2 mLとヘキサメチレンアンモニウム-ヘキサメチレンカル
バモジチオ酸(HMA-HMDC)のメタノール溶液(20 g/L)2 mLとを加えて混合した後,
JIS K 8271に規定するキシレンの一定量(5〜20 mL)を加えて約5分間激しく振り混ぜ
て静置する。水層を捨てキシレン層を共栓試験管に入れる。定量操作は,備考12.による。
d) 操作 操作は,次による。
1) c) 1)の準備操作を行った試料を試料導入部を通して発光部に導入し(11) (12),各測定対象元素の波長
219
K 0102:2016
の発光強度を測定する(13) (14)。
2) 空試験としてc) 1)の準備操作で用いた試料と同量の水をとり,c) 1)及び1)の操作を行って各測定対
象元素に相当する発光強度を測定し,試料について得た発光強度を補正する。
3) 検量線から各測定対象元素の量を求め,試料中の各元素の濃度(μg/L)を算出する(15)。
注(11) 試料の測定を始める前に,硝酸(1+20)(JIS K 8541に規定する硝酸を用いて調製する。)を噴
霧して測定系を洗い流す。また,各試料測定の間にも洗い流しを行う。
(12) 備考11.による。
(13) 47.の注(8)による。
(14) 47.の注(9)による。
(15) 50.の注(11)による。
e) 検量線 検量線の作成は,次による。
1) a)の測定対象元素の各標準液のそれぞれ一定量をとり,希釈して一定量として混合標準液を調製す
る。この混合標準液は,測定対象の元素を含み(16),測定する濃度範囲よりも高い濃度(17)とする。
2) 全量フラスコ100 mLに,測定濃度範囲を含むように,混合標準液を段階的にとり,c) 1)の試料と同
じ酸の濃度となるように硝酸(1+1)(18)を加えた後,水を標線まで加える。
3) 2)で調製した各溶液についてd) 1)の操作を行う。
4) 空試験として,全量フラスコ100 mLにc) 1)の試料と同じ酸の濃度となるように硝酸(1+1)を加
え,水を標線まで加えた溶液について,d) 1)の操作を行い,3)で得た発光強度を補正する。
5) 各測定対象元素について,それぞれの元素の量と発光強度との関係線を作成する。
注(16) 測定対象の単独又は限られた複数の元素だけでもよい。
(17) 作成する検量線の最高濃度の5〜10倍程度。
(18) 又は塩酸(1+1)。ただし,指定されている場合はそれに従う。
備考 11. 波長の異なる2本以上のスペクトル線の同時測定が可能な装置では,内標準法によることが
できる。操作は,次による。
なお,内標準元素は,イットリウム(Y)のほか,インジウム(In)及びイッテルビウム(Yb)
も使用できる。
1) c) 1)で処理した試料の適量を全量フラスコ100 mLにとり,イットリウム溶液(Y 50
μg/mL)[47.の備考5. 6)による。]10 mLを加えた後,水を標線まで加える。
2) この溶液についてd) 1)の操作を行って各測定対象元素の発光強度及びイットリウムの
発光強度を測定し,各測定対象元素の発光強度とイットリウムの発光強度との比を求め
る。
3) 空試験として,試料に代えて同量の水を用い,c) 1),1)及び2)の操作を行い,各測定対
象元素の発光強度とイットリウムの発光強度との比を求め,2)で得た発光強度比を補正
する。
4) 各測定対象元素についての検量線から,各測定対象元素の量を求め,試料中の各測定対
象元素の濃度(mg/L)を算出する。
5) 検量線 全量フラスコ100 mL数個にe) 2)に従って混合標準液を段階的にとり,イット
リウム溶液(Y 50 μg/mL)10 mL及び1)の試料と同じ酸の濃度となるように硝酸(1+1)
(*)を加えた後,水を標線まで加える。この溶液について2)の操作を行い,各測定対象元
素及びイットリウムの発光強度を測定し,各測定対象元素の発光強度とイットリウムの
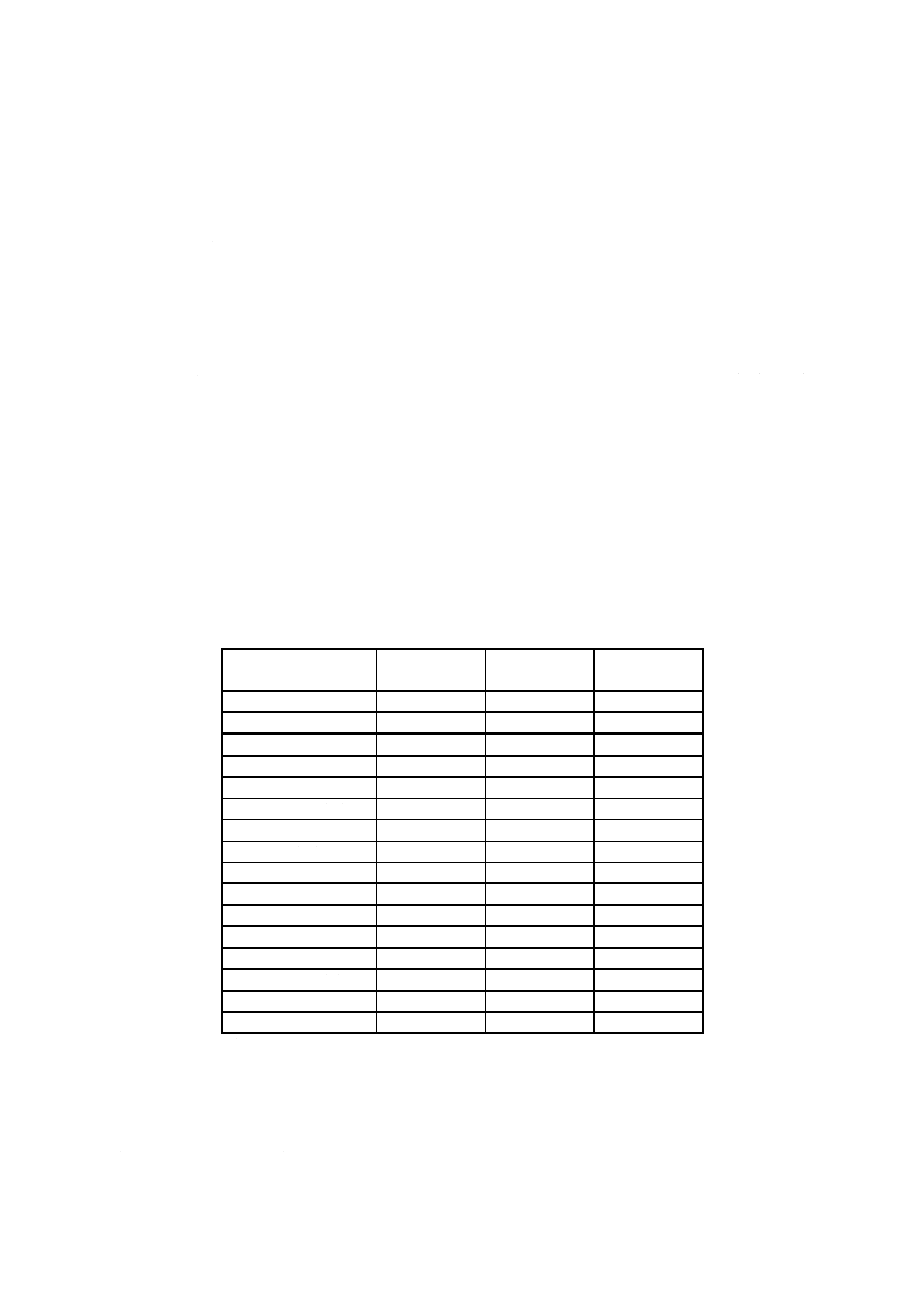
220
K 0102:2016
発光強度との比を求め,別に,空試験として,混合標準液に代えて水を用い,同じ操作
を行って,同様に発光強度の比を求め,混合標準液での各測定対象元素の発光強度比を
補正し,各測定対象元素の量と,各測定対象元素の発光強度とイットリウムの発光強度
との比との関係線を作成する。
注(*) 注(18)による。
備考 12. 備考10. 1)〜3)によって準備操作を行った場合は,キシレン層をそのまま噴霧して測定対象の
各元素の発光強度を測定して定量する。その場合の検量線の作成は,次による。
1) e) 1)に準じ,測定対象元素を含む混合標準液を調製する。ただし,各測定対象元素はe) 1)
の混合標準液よりも低濃度(0.1〜1 μg/mL)とする。
2) 調製した混合標準液を段階的にとり,水で500 mLとし,備考10. 1)〜3)の操作及び各測
定対象元素の発光強度の測定操作を行う。
3) 空試験として,水500 mLを用いて,2)の操作を行い,2)で得た各測定対象元素の発光強
度を補正し,各測定対象元素の量とその発光強度との関係線を作成する。
52.5 ICP質量分析法 試料を前処理した後,内標準元素を加え,試料導入部を通して高周波プラズマ中
に噴霧し,銅及び内標準元素のそれぞれの質量/電荷数における指示値(19)を測定し,銅の指示値と内標準
元素の指示値との比を求めて銅を定量する。この方法によって,表52.2に示す元素が同時定量できる。そ
れぞれの元素ごとの定量範囲,繰返し精度などの例を,表52.2に示す。
注(19) イオンカウント値又はその比例値。
表52.2 定量範囲,繰返し精度及び質量数の例*
対象元素
定量範囲
μg/L
繰返し精度
%
質量数
銅(Cu)
0.5〜500
2〜10
63,65
亜鉛(Zn)
0.5〜500
2〜10
66,68,64
鉛(Pb)
0.3〜500
2〜10
208,206,207
カドミウム(Cd)
0.3〜500
2〜10
111,114
マンガン(Mn)
0.5〜500
2〜10
55
アルミニウム(Al)
0.5〜500
2〜10
27
ニッケル(Ni)
0.5〜500
2〜10
60,58
コバルト(Co)
0.5〜500
2〜10
59
ひ素(As)
0.5〜500
2〜10
75
ビスマス(Bi)
0.3〜500
2〜10
209
クロム(Cr)
0.5〜500
2〜10
53,52,50
セレン(Se)
0.5〜500
2〜10
82,77,78
バナジウム(V)
0.5〜500
2〜10
51
イットリウム(Y)**
−
−
89
インジウム(In)**
−
−
115
ビスマス(Bi)**
−
−
209
注*
装置及び測定条件によって異なる。
** 内標準元素
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
221
K 0102:2016
3) 内標準液(1 μg/mL) 内標準元素としてイットリウム,インジウム又はビスマスを用いる。内標準
液の調製には,次の3.1)〜3.3)に規定する溶液のうち内標準とする元素の溶液2 mLを全量フラスコ
100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調製する(20) (21)。
3.1) イットリウム溶液(Y 50 μg/mL) 47.の備考5. 6)による。
3.2) インジウム溶液(In 50 μg/mL) 47.4 a) 2.2)による。
3.3) ビスマス溶液(Bi 50 μg/mL) 酸化ビスマス(III)0.279 gをとり,硝酸(1+1)10 mLを加え,
加熱して溶かし,放冷後,全量フラスコ250 mLに移し入れ,水を標線まで加える。この溶液25 mL
を全量フラスコ500 mLにとり,硝酸(1+1)10 mLを加え,水を標線まで加える。
4) 混合標準液[(Cu 10 μg,Zn 10 μg,Pb 10 μg,Cd 10 μg,Mn 10 μg,Al 10 μg,Ni 10 μg,Co 10 μg,
As 10 μg,Bi 10 μg,Cr 10 μg,Se 10 μg,V 10 μg)/mL](16) (21) 52.4 a)の1) 銅標準液(Cu 1 mg/mL),
3) 亜鉛標準液(Zn 1 mg/mL),5) 鉛標準液(Pb 1 mg/mL),9) マンガン標準液(Mn 1 mg/mL)のそ
れぞれ5 mL,58.2 a) 2) アルミニウム標準液(Al 0.5 mg/mL)10 mL及び67.1 a) 11) セレン標準液(Se
0.2 mg/mL)25 mL,並びに52.4 a)の7) カドミウム標準液(Cd 0.1 mg/mL),14) ニッケル標準液(Ni
0.1 mg/mL),16) コバルト標準液(Co 0.1 mg/mL),58.4 a)の3) クロム標準液(Cr 0.1 mg/mL),7) バ
ナジウム標準液(V 0.1 mg/mL),61.1 a) 12) ひ素標準液(As 0.1 mg/mL),64.1 a) 5) ビスマス標準
液(Bi 0.1 mg/mL)のそれぞれ50 mLを全量フラスコ500 mLにとり,硝酸(1+1)10 mLを加えた
後,水を標線まで加える(22)。
5) 混合標準液[(Cu 0.5 μg,Zn 0.5 μg,Pb 0.5 μg,Cd 0.5 μg,Mn 0.5 μg,Al 0.5 μg,Ni 0.5 μg,Co 0.5
μg,As 0.5 μg,Bi 0.5 μg,Cr 0.5 μg,Se 0.5 μg,V 0.5 μg)/mL](16) (21) 4)の混合標準液5 mLを全
量フラスコ100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調製する。
注(20) 3種類の内標準元素は,単独又は混合して用いてもよい。ICP質量分析法では,主成分(マトリ
ックス)による非スペクトル干渉の大きさは質量数に依存するため,測定対象元素と比較的質
量数の近いものを内標準元素とするとよい。ここに挙げた3種類以外にも,元の試料に無視で
きる量より少ない量しか含まれていないことが確認できれば,内標準元素として用いてもよい。
ビスマスが測定対象元素であるときは,内標準元素としてビスマスを用いることはできない。
ビスマスの代わりにタリウムを用いることがある。
(21) 定期的に濃度の安定性を,新たに調製した標準液と比較して確認する。特に,濃度の低い標準
液は濃度が低下しやすいため注意する。
(22) 標準液は,混合したときに沈殿を生じないものを用いる。
b) 装置 装置は,次による。
1) ICP質量分析装置
備考 13. イオン源として,高周波プラズマと同等の性能をもつものを用いてもよい。
14. サンプリングコーン及びスキマーコーンの材質からの汚染が認められないことを確認する。
c) 準備操作 準備操作は,次による(23)。
1) 試料を5.5によって処理する。ただし,クロムを定量する場合は,前処理に5.3は用いない。
2) 1)で処理した試料の適量(測定対象元素として0.05〜50 μgを含む。)を全量フラスコ100 mLにと
り,内標準液(1 μg/mL)1 mLを加え,硝酸の最終濃度が0.1〜0.5 mol/Lとなるように硝酸(1+1)
を加えた後,水を標線まで加える。
注(23) 分析者からの汚染がないように注意する。JIS T 9107に規定する使い捨て手術用ゴム手袋(打
粉のないもの)などを用いるとよい。
222
K 0102:2016
備考 15. 試料の銅又は測定対象元素の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イ
オンなどの共存物質の濃度が高く,測定を妨害する場合の準備操作は,備考6.による[ただ
し,3.6)は除く]。得られた液は全量フラスコ20 mLに移し入れ,内標準液(1 μg/mL)0.2 mL
を加え,標線まで水を加える。
d) 操作 操作は,次による(24)。
1) ICP質量分析装置を作動できる状態にし,c) 2)の溶液を試料導入部を通してイオン化部に導入して
測定対象元素及び内標準元素(イットリウム,インジウム又はビスマス)のそれぞれの質量/電荷数
(25)における指示値を読み取り,測定対象元素の指示値と内標準元素の指示値との比をそれぞれ求め
る。
2) 空試験として,c) 1)での試料と同量の水をとり,試料と同様にc)及びd) 1)の操作を行って測定対象
元素の指示値と内標準元素の指示値との比を求め,試料について得た測定対象元素と内標準元素と
の指示値の比を補正する。
3) 検量線から測定対象元素の量を求め,試料中の測定対象元素の濃度(μg/L)を算出する。
注(24) 妨害物質の存在が不明の場合には,定量に先だってICP質量分析計による定性分析を行うこと
によって,測定対象元素及び内標準元素の測定質量数に対する妨害(スペクトル干渉及び非ス
ペクトル干渉)の有無と程度を推定することができる。スペクトル干渉が認められる場合には,
測定質量数の変更,試料の希釈又は前処理を行って妨害の軽減を図る。スペクトル干渉のため,
上記のイットリウム,インジウム又はビスマスを内標準元素として使用できない場合もあるが,
その場合には,他の内標準元素を用いる。スペクトル干渉の例を,表52.3に示す。非スペクト
ル干渉(マトリックス干渉ともいい,検量線の傾きに影響する。)は,一般にこの方法で採用し
ている内標準法によって補正できるが,妨害物質の濃度が高い場合には,補正が不十分となる
ことがある。このような場合には,試料の希釈又は前処理を行った後,内標準法を適用して妨
害の軽減を図る。非スペクトル干渉の程度は,標準液を添加して回収率を求めることによって
推定することができる。すなわち,試料(元の試料又は希釈・前処理後の試料)中の測定対象
元素の濃度が10 ng/mL分だけ(ただし,試料中の測定元素の濃度が高い場合には,増加分が精
度よく測定できるように,試料中と同程度の濃度だけ)増加するように,測定対象元素の標準
液(0.5 μg/mL)の適量を試料に添加後,d)に準じた操作を行って測定対象元素の濃度を求め,
その回収率を求める。回収率が90〜110 %の範囲にあれば,非スペクトル干渉は,ほぼ無視し
得るものと考えられる。
(25) 質量数を設定するには,表52.2及び表52.3を参考にするとよい。複数の安定同位体がある場合,
複数の同位体の質量/電荷数を用いて測定を行うことによって,スペクトル干渉による妨害を推
定することができる。
e) 検量線 検量線の作成は,次による。
1) a) 4)の混合標準液(各元素濃度10 μg/mL)又はa) 5)の混合標準液(各元素濃度0.5 μg/mL)いずれ
か(26)の混合標準液0.1〜5 mLを,全量フラスコ100 mLに段階的にとり,内標準液(1 μg/mL)1 mL
を加え,c) 2)の試料と同じ酸の濃度になるように硝酸(1+1)を加えた後,水を標線まで加える。
使用時に調製する。
2) この溶液についてd) 1)の操作を行う。
3) 別に,空試験として全量フラスコ100 mLに内標準液(1 μg/mL)1 mLを加え,c) 2)の試料と同じ酸
の濃度になるように硝酸(1+1)を加え,水を標線まで加えた後,d) 1)の操作を行って標準液につ

223
K 0102:2016
いて得た指示値の比をそれぞれ補正し,測定対象元素の濃度に対する,測定元素の指示値と内標準
元素の指示値との比の関係線をそれぞれ作成する。検量線の作成は,試料測定時に行う。
注(26) a) 4)の混合標準液の調製に用いた各元素の標準液のうち,測定対象元素の各標準液のそれぞれ
一定量をとり,希釈して一定量として混合標準液を調製して用いてもよい。この混合標準液は,
測定対象の元素を含み,測定する濃度範囲よりも高い濃度とする。
備考 16. 注(24)の操作で,主成分の元素又は有機物の含有量が少なく,非スペクトル干渉が無視できる
試料の場合は,内標準元素の添加を省略し,検量線法によって定量してもよい。
表52.3 スペクトル干渉の例*
元素
質量数
同重体及び
2価イオンの干渉
多原子イオンの干渉
銅(Cu)
63
40Ar23Na,31P16O16O,26Mg37Cl
銅(Cu)
65
32S16O16OH,33S16O16O,32S33S
亜鉛(Zn)
64
Ni+
32S16O16O,32S32S,27Al37Cl,48Ca16O
亜鉛(Zn)
66
Ba++
34S16O16O,32S34S,31P35Cl,54Fe12C
亜鉛(Zn)
68
Ba++,Ce++
40Ar14N14N,36S16O16O,32S36S,36Ar32S,31P37Cl,54Fe14N,56Fe12C
鉛(Pb)
206
鉛(Pb)
207
鉛(Pb)
208
カドミウム(Cd)
111
95Mo16O,94Mo16OH,94Zr16OH
カドミウム(Cd)
114
Sn+
98Mo16O,97Mo16OH
マンガン(Mn)
55
40Ar14NH,38Ar16OH,23Na32S
アルミニウム(Al)
27
12C15N,13C14N
ニッケル(Ni)
58
Fe+
42Ca16O,44Ca14N,23Na35Cl,24Mg34S
ニッケル(Ni)
60
44Ca16O,43Ca16OH,25Mg35Cl,23Na37Cl
コバルト(Co)
59
43Ca16O,42Ca16OH,24Mg35Cl
ひ素(As)
75
Nd++,Sm++
40Ar35Cl,40Ca35Cl
ビスマス(Bi)
209
クロム(Cr)
50
36Ar14N,34S16O
クロム(Cr)
52
40Ar12C,36Ar16O,36S16O,35Cl16OH
クロム(Cr)
53
37Cl16O,36Ar16OH
セレン(Se)
77
40Ar37Cl,36Ar40ArH,40Ca37Cl
セレン(Se)
78
Kr+
38Ar40Ar,43Ca35Cl
セレン(Se)
82
Kr+
40Ar40ArH2,34S16O16O16O,H81Br
バナジウム(V)
51
35Cl16O,37Cl14N,34S16OH,36Ar14NH
イットリウム(Y)**
89
インジウム(In)**
115
Sn+
ビスマス(Bi)**
209
注*
装置及び測定条件によって異なる。
** 内標準元素
53. 亜鉛(Zn) 亜鉛の定量には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又は
ICP質量分析法を適用する。
なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,1996
年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
224
K 0102:2016
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
53.1 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,亜鉛による原子
吸光を波長213.9 nmで測定して亜鉛を定量する。
定量範囲:Zn 0.05〜2 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 亜鉛標準液(Zn 10 μg/mL) 52.4 a) 4)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 1. 亜鉛の濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,52.の備考
4.,備考5.又は備考6.による。
d) 操作 操作は,次による。
1) 52.2 d) 1)の操作を行う。ただし,波長は213.9 nmを用いる。
2) 空試験として52.2 d) 2)の操作を行う。
3) 検量線から亜鉛の量を求め,試料中の亜鉛の濃度(Zn mg/L)を算出する。
e) 検量線 検量線の作成は,次による。
1) 亜鉛標準液(Zn 10 μg/mL)0.5〜20 mL(1)を全量フラスコ100 mLに段階的にとり,c) 1)を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える(2)。
2) 52.2 e) 2)及び3)の操作を行い,亜鉛(Zn)の量と指示値との関係線を作成する。検量線の作成は,
試料測定時に行う。
注(1) 準備操作として溶媒抽出を適用するときは,亜鉛標準液(Zn 10 μg/mL)の量を,適宜,減らす。
(2) 備考1.によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層又は2,6-ジメチル-4-
ヘプタノン層をそのまま噴霧する場合の検量線の作成は,次による。
亜鉛標準液(Zn 10 μg/mL)を適切な濃度(Zn 0.1〜1 μg/mL)に薄め,その0.5〜20 mLを段
階的にとり約100 mLとした後,試料と同様に備考1.並びにd) 1)及び2)を行って亜鉛(Zn)の
量と指示値との関係線を作成する。
53.2 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,亜鉛による原子吸光を波長
213.9 nmで測定して亜鉛を定量する。
定量範囲:Zn 1〜20 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 2. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) 亜鉛標準液(Zn 1 μg/mL) 52.4 a) 4)の亜鉛標準液(Zn 10 μg/mL)10 mLを全量フラスコ100 mLに
とり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。使用時に調製する。
225
K 0102:2016
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 3. 試料の亜鉛の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イオンなどの共存
物質の濃度が高く,測定を妨害する場合の準備操作は52.の備考6.による。
d) 操作 操作は,次による。
1) 52.3 d) 1)の操作を行う。ただし,波長は213.9 nmを用いる。
2) 空試験として52.3 d) 2)の操作を行う。
3) 検量線から亜鉛の量を求め,試料中の亜鉛の濃度(Zn μg/L)を算出する。
e) 検量線 検量線の作成は,次による。
1) 亜鉛標準液(Zn 1 μg/mL)0.1〜2 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試料と
同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてd) 1)の操作を行
う。
2) 52.3 e) 2)及び3)の操作を行い,亜鉛(Zn)の量と指示値との関係線を作成する。検量線の作成は,
試料測定時に行う。
53.3 ICP発光分光分析法 52.4による。
53.4 ICP質量分析法 52.5による。
54. 鉛(Pb) 鉛の定量には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP
質量分析法を適用する。
なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,1996
年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
54.1 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,鉛による原子吸
光を波長283.3 nmで測定して鉛を定量する。
定量範囲:Pb 1〜20 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 鉛標準液(Pb 0.1 mg/mL) 52.4 a) 5)の鉛標準液(Pb 1 mg/mL)10 mLを全量フラスコ100 mLにと
り,硝酸(1+1)[52.1 a) 8)による。]10 mLを加え,水を標線まで加える。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する(1)。
注(1) 水溶液をそのまま噴霧する場合は,硫酸イオンが存在すると鉛に負の誤差を与えるので,硫酸
を用いる前処理は行わない。
226
K 0102:2016
備考 1. 鉛の濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,52.の備考4.
又は備考5.若しくは備考6.による。
d) 操作 操作は,次による。
1) 52.2 d) 1)の操作を行う。ただし,波長は283.3 nmを用いる。
2) 空試験として52.2 d) 2)の操作を行う。
3) 検量線から鉛の量を求め,試料中の鉛の濃度(Pb mg/L)を算出する。
e) 検量線 検量線の作成は,次による。
1) 鉛標準液(Pb 0.1 mg/mL)1〜20 mL(2)を全量フラスコ100 mLに段階的にとり,c) 1)を行った試料と
同じ酸の濃度になるように酸を加えた後,水を標線まで加える(3)。
2) 52.2 e) 2)及び3)の操作を行い,鉛(Pb)の量と指示値との関係線を作成する。検量線の作成は,試
料測定時に行う。
注(2) 準備操作として溶媒抽出を適用するときは,鉛標準液(Pb 0.1 mg/mL)の量を,適宜,減らす。
(3) 備考1.によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層又は2,6-ジメチル-4-
ヘプタノン層をそのまま噴霧する場合の検量線の作成は,次による。
鉛標準液(Pb 0.1 mg/mL)を適切な濃度(Pb 1〜5 μg/mL)に薄め,その1〜20 mLを段階的に
とり,約500 mL(又は100〜500 mLの一定量)とした後,試料と同様にc)の準備操作を行って
鉛の量と指示値との関係線を作成する。
54.2 電気加熱原子吸光法 試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム
(II)を加え,電気加熱炉で原子化し,鉛による原子吸光を波長283.3 nmで測定して鉛を標準添加法によ
って定量する。
定量範囲:Pb 5〜100 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 2. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) 硝酸パラジウム(II)溶液(Pd 10 μg/mL) 硝酸パラジウム(II)0.108 gを硝酸(1+1)10 mLを
加えて溶かし,全量フラスコ500 mLに移し入れ,水を標線まで加える。この溶液20 mLを全量フ
ラスコ200 mLにとり,水を標線まで加える。
4) 鉛標準液(Pb 1 μg/mL) 52.4 a) 6)の鉛標準液(Pb 10 μg/mL)10 mLを全量フラスコ100 mLにとり,
硝酸(1+1)20 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 3. 試料の鉛の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イオンなどの共存物
質の濃度が高く,測定を妨害する場合の準備操作は52.の備考6.による。
d) 操作 操作は,次による。
1) c)の準備操作を行った試料15 mLずつをそれぞれ全量フラスコ20 mLにとり,鉛標準液(Pb 1 μg/mL)
を加えないものと,0.1〜2 mLの範囲で段階的に3段階以上添加したものとを調製し,それぞれの
溶液の酸の濃度が同じになるように硝酸(1+1)を加えた後,水を標線まで加える。
2) 1)の操作を行った試料の100 μL以上の一定量をマイクロピペットで小形の容器にとり,これと同体
227
K 0102:2016
積の硝酸パラジウム(II)溶液(Pd 10 μg/mL)を加え,よく混ぜ合わせる。
3) 52.3 d) 1)の操作を行う。ただし,灰化温度は500〜800 ℃,原子化(4)温度は1 800〜2 500 ℃,波長
は283.3 nmを用いる。
4) 空試験としてc)の準備操作での試料と同量の水をとり,試料と同様にc)の操作を行った後,その15
mLを全量フラスコ20 mLにとる。次に,d) 1)の溶液の酸の濃度と同じになるように,硝酸(1+1)
を加えた後,水を標線まで加える。この溶液について,2)及び3)の操作を行って,試料について得
た指示値を補正する。
5) 鉛の添加量と指示値との関係線を作成し,鉛の量を求め,試料中の鉛の濃度(Pb μg/L)を算出する。
注(4) 52.の注(9)による。
54.3 ICP発光分光分析法 52.4による。
54.4 ICP質量分析法 52.5による。
55. カドミウム(Cd) カドミウムの定量には,フレーム原子吸光法,電気加熱原子吸光法,ICP発光分
光分析法又はICP質量分析法を適用する。
なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288及び1994年に第1版として
発行されたISO 5961,電気加熱原子吸光法は,1994年に第1版として発行されたISO 5961,ICP発光分
光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 5961:1994,Water quality−Determination of cadmium by atomic absorption spectrometry(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
55.1 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,カドミウムによ
る原子吸光を波長228.8 nmで測定してカドミウムを定量する。
定量範囲:Cd 50〜2 000 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) カドミウム標準液(Cd 10 μg/mL) 52.4 a) 8)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 1. カドミウムの濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,52.2
の備考4.又は備考5.若しくは備考6.による。
2. 試料中に多量の鉄又はマンガンが含まれている場合は,次のイオン交換樹脂による方法によ
ってカドミウムを分離濃縮する。
1) 試料の適量にJIS K 8180に規定する塩酸を加えて約2 mol/Lの塩酸酸性とする。これを
イオン交換カラム[I形の強塩基性陰イオン交換樹脂を塩化物イオン形に調製したもの
をカラム(例えば,内径10 mm,長さ200 mm)に充塡したもの。]に約3 mL/minで流
228
K 0102:2016
してカドミウムをクロロ錯体として吸着させた後,塩酸(1+9)(JIS K 8180に規定する
塩酸を用いて調製する。)で洗浄する。
2) 受器を代え,硝酸(1+12)(JIS K 8541に規定する硝酸を用いて調製する。)で溶離し,
溶離液を一定液量とする。
備考 3. 試料中に多量の亜鉛,銅などが含まれている場合には,試料の適量にJIS K 8509に規定する
臭化水素酸を加えて約0.5 mol/Lの臭化水素酸溶液とし,その50 mLに対してN,N-ジオクチ
ル-1-オクタンアミン(トリオクチルアミン)の4-メチル-2-ペンタノン溶液(体積百分率1 %)
10 mLを加えて振り混ぜ,カドミウムを抽出する。抽出した4-メチル-2-ペンタノン層をその
まま噴霧して原子吸光分析に用いる。
d) 操作 操作は,次による。
1) 52.2 d) 1)の操作を行う。ただし,波長は228.8 nmを用いる。
2) 空試験として52.2 d) 2)の操作を行う。
3) 検量線からカドミウムの量を求め,試料中のカドミウムの濃度(Cd μg/L)を算出する。
e) 検量線 検量線の作成は,次による。
1) カドミウム標準液(Cd 10 μg/mL)0.5〜20 mL(1)を全量フラスコ100 mLに段階的にとり,c) 1)を行
った試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える(2)。
2) 52.2 e) 2)及び3)の操作を行い,カドミウム(Cd)の量と指示値との関係線を作成する。検量線の作
成は,試料測定時に行う。
注(1) 準備操作として溶媒抽出を適用するときは,カドミウム標準液(Cd 10 μg/mL)の量を,適宜,
減らす。
(2) 備考1.によって準備操作を行い,酢酸ブチル層,4-メチル-2-ペンタノン層又は2,6-ジメチル-4-
ヘプタノン層をそのまま噴霧する場合の検量線の作成は,次による。
カドミウム標準液(Cd 10 μg/mL)を適切な濃度(Cd 0.1〜1 μg/mL)に薄め,その0.5〜20 mL
を段階的にとり,約100 mLとした後,試料と同様に備考1.並びにd) 1)及び2)を行ってカドミ
ウム(Cd)の量と指示値との関係線を作成する。
備考 4. アルカリ金属のハロゲン化物が多量に存在すると,その分子吸収,光散乱などによって正の
誤差を生じる。このような場合には,バックグラウンド補正を行うか,又はあらかじめカド
ミウムを分離する。
55.2 電気加熱原子吸光法 試料を前処理した後,マトリックスモディファイヤーとして硝酸パラジウム
(II)を加え電気加熱炉で原子化し,カドミウムによる原子吸光を波長228.8 nmで測定してカドミウムを
標準添加法によって定量する。
定量範囲:Cd 0.5〜10 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 5. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) 硝酸パラジウム(II)溶液(Pd 10 μg/mL) 54.2 a) 3)による。
4) カドミウム標準液(Cd 1 μg/mL) 52.4 a) 8)のカドミウム標準液(Cd 10 μg/mL)10 mLを全量フラ
スコ100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
5) カドミウム標準液(Cd 0.1 μg/mL) カドミウム標準液(Cd 1 μg/mL)10 mLを全量フラスコ100 mL
229
K 0102:2016
にとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 6. 試料のカドミウムの濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イオンなど
の共存物質の濃度が高く,測定を妨害する場合の準備操作は52.の備考6.による。
d) 操作 操作は,次による。
1) 54.2 d) 1)の操作を行う。ただし,カドミウム標準液(Cd 0.1 μg/mL)を用いる。
2) 54.2 d) 2)の操作を行う。
3) 52.3 d) 1)の操作を行う。ただし,灰化温度は500〜800 ℃,原子化(3)温度は1 600〜2 200 ℃,波長
は228.8 nmを用いる。
4) 54.2 d) 4)の操作を行う。
5) カドミウムの添加量と指示値との関係線を作成し,カドミウムの量を求め,試料中のカドミウムの
濃度(Cd μg/L)を算出する。
注(3) 52.の注(9)による。
55.3 ICP発光分光分析法 52.4による。
55.4 ICP質量分析法 52.5による。
56. マンガン(Mn) マンガンの定量には,過よう素酸吸光光度法,フレーム原子吸光法,電気加熱原子
吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
56.1 過よう素酸吸光光度法 試料を硫酸酸性とした後,過よう素酸カリウムを加え,加熱して赤紫の過
マンガン酸イオンを生成させ,その吸光度を測定してマンガンを定量する。
定量範囲:Mn 40〜500 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 硫酸(1+1) 5.4 a) 2)による。
2) りん酸 JIS K 9005に規定するもの。
3) 過よう素酸カリウム JIS K 8249に規定するもの。
4) マンガン標準液(Mn 1 mg/mL) 52.4 a) 9)による。
5) マンガン標準液(Mn 20 μg/mL) マンガン標準液(Mn 1 mg/mL)5 mLを全量フラスコ250 mLに
とり,水を標線まで加える。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
230
K 0102:2016
1) 5.の操作を行った(1)試料の適量(2)(Mnとして40〜500 μgを含む。)をとり,硫酸(1+1)10 mL(3)
を加え,加熱して硫酸の白煙を発生させ,ハロゲン化物を除去する。
2) 放冷後,水約20 mLとりん酸1 mLとを加え,加熱して内容物を溶かす。不溶解物がある場合には,
ろ別し,ろ紙と沈殿とを温水で洗い,ろ液と洗液とを合わせ,水を加えて約45 mLにする。
3) 過よう素酸カリウム0.5 g(4)を加え,沸騰水浴中で30分間加熱し(5)発色させる。
4) 流水で冷却した後,全量フラスコ50 mLに移し入れ,水を標線まで加える。
5) この溶液の一部を吸収セルに移し,波長525 nm付近又は545 nm付近の吸光度を測定する。
6) 空試験として水約30 mLをとり,硫酸(1+1)10 mLとりん酸1 mLとを加えた後,3)〜5)の操作を
行って吸光度を測定し,試料について得た吸光度を補正する。
7) 検量線からマンガンの量を求め,試料中のマンガンの濃度(Mn μg/L)を算出する。
注(1) 試料中にこの試験に影響する有機物及び妨害物質が含まれていない場合には,前処理を省略し
てもよい。
(2) 試料は最大500 mLとする。
(3) 前処理操作に硫酸を用いた場合には,硫酸は加えない。ただし,試料中の硫酸は約5 mLになる
ように調節する。
(4) 過よう素酸カリウムの代わりに,硝酸銀溶液(5 g/L)(JIS K 8550に規定する硝酸銀0.5 gを水
に溶かして100 mLとする。)2 mLとペルオキソ二硫酸アンモニウム溶液(200 g/L)(JIS K 8252
に規定するペルオキソ二硫酸アンモニウム20 gを水に溶かして100 mLとする。)5 mLとを加
えて約1分間煮沸して発色させてもよい。この場合は,発色時の硫酸の濃度は約0.5 mol/Lにな
るようにする。この方法で塩化銀の沈殿が生じたら,これが消えるまで硝酸酸性硝酸水銀(II)
溶液[硝酸水銀(II)n水和物5 gを硝酸(1+2)(JIS K 8541に規定する硝酸を用いて調製する。)
20 mLに溶かし,水で100 mLとする。]を滴加し,更に数滴過剰に加えて塩化物イオンをマス
キングする。
(5) 加熱時間が長すぎると,生成した過マンガン酸イオンが分解するおそれがあるから,加熱時間
は正しく守る。
d) 検量線 検量線の作成は,次による。
1) マンガン標準液(Mn 20 μg/mL)2〜25 mLをビーカー100 mLに段階的にとり,水を加えて液量を約
30 mLとし,硫酸(1+1)10 mLとりん酸1 mLとを加えた後,c) 3)〜6)の操作を行ってマンガン(Mn)
の量と吸光度との関係線を作成する。
備考 1. 溶存マンガンを定量する場合には,3.2によってろ過した試料(ただし,ろ過にはろ紙5種C
を用いる。)の適量(Mnとして40〜500 μgを含む。)を用い,c) 1)以降の操作を行う。
2. マンガンの濃度が低い場合には,次の鉄共沈法で濃縮して定量できる。
1) 試料500 mLまでの適量をとり,約90 ℃に加熱し,硫酸アンモニウム鉄(III)溶液(Fe
2 mg/mL)[JIS K 8982に規定する硫酸アンモニウム鉄(III)・12 水1.8 gをとり,硝酸
(1+6)(JIS K 8541に規定する硝酸を用いて調製する。)10 mL及び水に溶かして100 mL
とする。]5 mL及びJIS K 8230に規定する過酸化水素5〜10 mLを加え,この溶液をか
き混ぜながらアンモニア水(1+1)(JIS K 8085に規定するアンモニア水を用いて調製す
る。)又は水酸化ナトリウム溶液(100 g/L)[19. a) 2)による。]を加えて,水酸化鉄(III)
の沈殿を生成させる。
2) 沈殿が沈降した後,ろ紙5種Aを用いてろ過し,温水で洗う。
231
K 0102:2016
3) 沈殿をできるだけ元のビーカーに移し,ろ紙に付着した沈殿は,少量の過酸化水素(1
+10)(JIS K 8230に規定する過酸化水素を用いて調製する。)を加えた硫酸(1+9)(JIS
K 8951に規定する硫酸を用いて調製する。)50 mLで溶かし,ろ紙は水で洗う。
4) ろ液及び洗液は元のビーカーに受け,加熱して沈殿を溶かすとともに過酸化水素を分解
して液量を約40 mLに濃縮し,りん酸1 mLを加えた後,c) 3)〜7)の操作を行ってマンガ
ンを定量する。
56.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,マンガンによる
原子吸光を波長279.5 nmで測定してマンガンを定量する。
定量範囲:Mn 0.1〜4 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) マンガン標準液(Mn 10 μg/mL) 52.4 a) 10)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 3. 溶存マンガンを定量する場合には,3.2によってろ過した試料(ただし,ろ過にはろ紙5種C
を用いる。)の適量をとり,5.5によって処理する。
4. マンガンの濃度が低い場合には,備考2.に準じて処理し,マンガンを濃縮分離する。沈殿は
少量の過酸化水素(1+10)[56.の備考2. 3)による。]を加えた塩酸(1+2)(JIS K 8180に規
定する塩酸を用いて調製する。)の少量に溶かし,ろ紙は温水で洗浄する。ろ液と洗液とを合
わせ,0.1〜1 mol/Lの塩酸酸性溶液の一定量とする。
d) 操作 操作は,次による。
1) 52.2 d) 1)の操作を行う。ただし,波長は279.5 nmを用いる。
2) 空試験として52.2 d) 2)の操作を行う。
3) 検量線からマンガンの量を求め,試料中のマンガンの濃度(Mn mg/L)を算出する。
e) 検量線 検量線の作成は,次による。
1) マンガン標準液(Mn 10 μg/mL)1〜40 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。
2) 52.2 e) 2)及び3)の操作を行い,マンガン(Mn)の量と指示値との関係線を作成する。検量線の作成
は,試料測定時に行う。
備考 5. シリカを多量に含む場合には,干渉抑制剤としてカルシウム(又はマグネシウム)を200 mg/L
程度加えておくとよい。
56.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,マンガンによる原子吸光を波
長279.5 nmで測定してマンガンを定量する。
定量範囲:Mn 1〜30 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 6. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) マンガン標準液(Mn 1 μg/mL) 52.4 a) 10)のマンガン標準液(Mn 10 μg/mL)10 mLを全量フラス
コ100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
232
K 0102:2016
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 7. 溶存マンガンを定量する場合には,備考3.による。
d) 操作 操作は,次による。
1) 52.3 d) 1)の操作を行う。ただし,灰化温度は500〜800 ℃,原子化(6)温度は2 000〜2 700 ℃(4〜6
秒間),波長は279.5 nmを用いる。
2) 空試験として52.3 d) 2)の操作を行う。
3) 検量線からマンガンの量を求め,試料中のマンガンの濃度(Mn μg/L)を算出する。
注(6) 52.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) マンガン標準液(Mn 1 μg/mL)0.1〜3 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてd) 1)の操作
を行う。
2) 52.3 e) 2)及び3)の操作を行い,マンガン(Mn)の量と指示値との関係線を作成する。検量線の作成
は,試料測定時に行う。
56.4 ICP発光分光分析法 52.4による。
備考 8. 溶存マンガンを定量する場合には,備考3.による。
56.5 ICP質量分析法 52.5による。
備考 9. 溶存マンガンを定量する場合には,備考3.によって処理した後,52.5 c) 2)を行う。
57. 鉄(Fe) 鉄の定量には,フェナントロリン吸光光度法,フレーム原子吸光法,電気加熱原子吸光法
又はICP発光分光分析法を適用する。
なお,フェナントロリン吸光光度法は,1988年の第2版として発行されたISO 6332,ICP発光分光分析
法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 6332:1988,Water quality−Determination of iron−Spectrometric method using 1,10-
phenanthroline(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
57.1 フェナントロリン吸光光度法 微酸性溶液中で塩化ヒドロキシルアンモニウムと1,10-フェナントロ
リンとを加えた後,酢酸アンモニウムを加えてpHを4〜5に調節し,生成するだいだい赤の鉄(II)錯体
の吸光度を測定して鉄を定量する。
定量範囲:Fe 20〜500 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
2) 硝酸(1+1) JIS K 8541に規定する硝酸を用いて調製する。
3) アンモニア水(1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
233
K 0102:2016
4) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 39.2 a) 3)による。
5) 1,10-フェナントロリン溶液(1 g/L) JIS K 8202に規定する塩化1,10-フェナントロリニウム一水和
物1.3 gを水に溶かして1 Lとする。又はJIS K 8789に規定する1,10-フェナントロリン一水和物1.1
gをJIS K 8102に規定するエタノール(95)100 mLに溶かし,水を加えて1 Lとする。
6) 酢酸アンモニウム溶液(500 g/L) JIS K 8359に規定する酢酸アンモニウム500 gを水に溶かして1
Lとする。
7) 鉄標準液(Fe 1 mg/mL) 52.4 a) 11)による。
8) 鉄標準液(Fe 10 μg/mL) 52.4 a) 13)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.(1)の操作を行った試料の適量(Feとして20〜500 μgを含む。)をビーカーにとり,硝酸(1+1)1
〜2 mLを加えて煮沸する。
2) 水を加えて50〜100 mLとした後,アンモニア水(1+1)を加えて微アルカリ性とする。これを数
分間煮沸して沈殿を生成させ(2) (3),しばらく放置する。
3) 沈殿が沈降した後,ろ紙5種Aでろ過し,温水で3,4回洗う。沈殿は元のビーカーに洗い入れ,
塩酸(1+1)4 mLを加え,加熱して溶かし,先のろ紙でろ過し,同時にろ紙に付着している水酸化
鉄(III)を溶かす。ろ紙は温水で5〜7回洗う。
4) ろ液と洗液とを合わせ,水を加えて液量を約70 mLとした後,塩化ヒドロキシルアンモニウム溶液
(100 g/L)1 mL(4)を加えて振り混ぜる。
5) 1,10-フェナントロリン溶液(1 g/L)5 mLを加えて振り混ぜ,続いて酢酸アンモニウム溶液(500 g/L)
10 mL(5)を加えて再び振り混ぜ,放冷する。
6) 全量フラスコ100 mLに移し入れ,水を標線まで加えて約20分間放置する。
7) この溶液の一部を吸収セルに移し,波長510 nm付近の吸光度を測定する。
8) 空試験として水約50 mLをとり,硝酸(1+1)1〜2 mLを加えた後,2)〜7)の操作を行って吸光度
を測定し,試料について得た吸光度を補正する。
9) 検量線から鉄の量を求め,試料中の鉄の濃度(Fe mg/L)を算出する。
注(1) 試料に有機物及び懸濁物が少なく,また,妨害物質が共存しない場合には,試料100 mLにつき
塩酸(1+1)4 mLを加えて液量が約2/3になるまで煮沸し,4)以降の操作を行って定量しても
よい。
(2) 鉄の量が極めて微量(Fe 20 μg以下)の場合には,捕集剤としてJIS K 8255に規定する硫酸カ
リウムアルミニウム・12水0.1 gを加えて溶かし,再びアンモニア水(1+1)を加えて微アル
カリ性とし,水酸化アルミニウムを生成させ,鉄を捕集してろ別する。このとき沈殿は塩酸に
溶けにくくなるので,塩酸の添加量を多くし,液量が約5 mLになるまで加熱濃縮する。次に,
水で液量を約70 mLとし,JIS K 8536に規定する(+)-酒石酸ナトリウムカリウム四水和物
0.1 gを加える。以下,4)以降の操作を行う。
(3) 発色を妨害する物質が含まれていない場合には,4)以降の操作を行って定量してもよい(備考
4.参照)。
(4) JIS K 9502に規定するL(+)-アスコルビン酸0.1 gを加えてもよい。
(5) 発色時のpHは約4.8になる。塩酸の濃度が高い場合には,アンモニア水(1+1)で中和し,発
234
K 0102:2016
色時のpHを4〜5に調節する。また,pH調節の操作は,c)の操作順序に従い,1,10-フェナント
ロリン溶液(1 g/L)を加えた後に行う。
d) 検量線 検量線の作成は,次による。
1) 鉄標準液(Fe 10 μg/mL)2〜50 mLを全量フラスコ100 mLに段階的にとり,塩酸(1+1)4 mLを
加え,c) 4)〜8)の操作を行って鉄(Fe)の量と吸光度との関係線を作成する。
備考 1. 溶存鉄を定量する場合には,3.2によってろ過した試料(ただし,ろ過にはろ紙5種Cを用
いる。)の適量(Feとして20〜500 μgを含む。)をとり,硝酸(1+1)1〜2 mLを加えて煮沸
する。以下,c) 2)〜9)によって定量する。妨害物質が共存しない場合には,c) 2)及び3)を省
略し,c) 4)以降の操作を行って定量してもよい。
2. 懸濁鉄を求めるには,鉄(全鉄)から溶存鉄を差し引く。
3. 鉄(II)を定量する場合には,次のように操作する。
1) 3.2によってろ過したろ液の適量(Feとして20〜500 μgを含む。)を全量フラスコ100 mL
にとる。1,10-フェナントロリン溶液(1 g/L)5 mLを加えた後,酢酸アンモニウム溶液
(500 g/L)を加えpHを約5に調節し,2. n) 1)の溶存酸素を含まない水を標線まで加え,
約20分間放置する。
2) 以下,c) 7)〜9)の操作を行って鉄の量を求め,試料中の鉄(II)の濃度[Fe(II)mg/L]
を算出する。
なお,鉄(II)は大気中の酸素によって容易に酸化されるから,この試験は,試料採
取後,直ちに行うが,全操作を直ちに行えない場合には,採取現場で発色までの操作を
行ってもち帰った後,吸光度を測定する。
4. 鉄をあらかじめ水酸化物として分離しないで試験する場合には,水銀,銅,カドミウム,ニ
ッケル,コバルト,亜鉛などが妨害する。ただし,カドミウム50 mg/L,亜鉛10 mg/L,水銀
1 mg/Lまでは妨害しない。pH 3.5で発色させれば,銅は10 mg/L,コバルトは10 mg/Lまで
は妨害しない。ニッケルが10 mg/L程度共存する場合は,EDTA溶液(JIS K 8107に規定す
るエチレンジアミン四酢酸二水素二ナトリウム二水和物3.7 gを水100 mLに溶かす。)5 mL
を添加し,約10分間煮沸すれば妨害しない。また,多量の亜鉛が共存するときは,pHが 9
で1,10-フェナントロリン溶液(1 g/L)を多量に加えて発色させれば,妨害を防ぐことができ
る。りん酸イオンが多量に共存すると妨害するが,発色時のpHを5〜7に調節し,約2時間
放置した後,吸光度を測定すれば妨害は少なくなる。
57.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,鉄による原子吸
光を波長248.3 nmで測定して鉄を定量する。
定量範囲:Fe 0.3〜6 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 鉄標準液(Fe 10 μg/mL) 52.4 a) 13)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 5. 溶存鉄を定量する場合には,3.2によってろ過した試料(ただし,ろ過にはろ紙5種Cを用
いる。)の適量をとり,5.によって処理する。
6. 懸濁鉄は,備考2.による。
235
K 0102:2016
備考 7. 鉄の濃度が低い試料で,しかも干渉物質がほとんど含まれていない場合には,試料100 mL
をとり,JIS K 8541に規定する硝酸2 mLを加えて煮沸した後,57.1 c) 2)及び3)に準じて操
作し,分離濃縮する。
8. 鉄の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イオンなどの共存物質の濃
度が高く,測定を妨害する場合の準備操作は,52.の備考6. による。
d) 操作 操作は,次による。
1) 52.2 d) 1)の操作を行う。ただし,波長は248.3 nmを用いる。
2) 空試験として52.2 d) 2)の操作を行う。
3) 検量線から鉄の量を求め,試料中の鉄の濃度(Fe mg/L)を算出する。
e) 検量線 検量線の作成は,次による。
1) 鉄標準液(Fe 10 μg/mL)3〜60 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試料と同
じ酸の濃度になるように酸を加えた後,水を標線まで加える。
2) 52.2 e) 2)及び3)の操作を行い,鉄(Fe)の量と指示値との関係線を作成する。検量線の作成は,試
料測定時に行う。
備考 9. 56.の備考5.による。
57.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,鉄による原子吸光を波長248.3
nmで測定して鉄を定量する。
定量範囲:Fe 5〜100 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 10. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) 鉄標準液(Fe 1 μg/mL) 52.4 a) 13)の鉄標準液(Fe 10 μg/mL)10 mLを全量フラスコ100 mLにと
り,硝酸(1+1)2 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 11. 溶存鉄を定量する場合には,備考5.による。
12. 懸濁鉄は,備考2.による。
13. 試料の鉄の濃度が低い試料で,アルカリ金属イオン,アルカリ土類金属イオンなどの共存物
質の濃度が高く,測定を妨害する場合の準備操作は52.の備考6. による。
d) 操作 操作は,次による。
1) 52.3 d) 1)の操作を行う。ただし,原子化(6)温度は2 200〜2 800 ℃,波長は248.3 nmを用いる。
2) 空試験として52.3 d) 2)の操作を行う。
3) 検量線から鉄の量を求め,試料中の鉄の濃度(Fe μg/L)を算出する。
注(6) 52.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) 鉄標準液(Fe 1 μg/mL)0.5〜10 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試料と同
じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてd) 1)の操作を行う。
2) 52.3 e) 2)及び3)の操作を行い,鉄(Fe)の量と指示値との関係線を作成する。検量線の作成は,試
236
K 0102:2016
料測定時に行う。
57.4 ICP発光分光分析法 52.4による。
備考 14. 溶存鉄を定量する場合には,備考5.による。
58. アルミニウム(Al) アルミニウムの定量には,キノリノール吸光光度法,フレーム原子吸光法,電気
加熱原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
58.1 キノリノール吸光光度法 微酸性にした試料に,塩化ヒドロキシルアンモニウムと1,10-フェナント
ロリンとを加えて鉄をマスキングした後,8-キノリノール及び酢酸アンモニウムを加え,生成する錯体を
クロロホルムで抽出する。シアン化カリウムを含む塩化アンモニウム溶液で洗浄して,アルミニウムとと
もに抽出された銅,ニッケル,コバルトなどを除去した後,アルミニウム錯体の吸光度を測定してアルミ
ニウムを定量する。
定量範囲:Al 5〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+2) JIS K 8180に規定する塩酸を用いて調製する。
2) アンモニア水(1+2) JIS K 8085に規定するアンモニア水を用いて調製する。
3) 硫酸ナトリウム JIS K 8987に規定するもの。
4) 塩化ヒドロキシルアンモニウム溶液(100 g/L) 39.2 a) 3)による。
5) 酢酸アンモニウム溶液(150 g/L) JIS K 8359に規定する酢酸アンモニウム15 gを水に溶かして100
mLとする。この溶液を分液漏斗に入れ,8-キノリノール-クロロホルム溶液(JIS K 8775に規定す
る8-キノリノール2 gをJIS K 8322に規定するクロロホルム100 mLに溶かす。)5 mLを加え,激
しく振り混ぜた後,放置し,クロロホルム層を捨てる。この操作を,クロロホルム層が着色しなく
なるまで繰り返す。次に,水層にクロロホルム5 mLを加え,激しく振り混ぜた後,放置し,クロ
ロホルム層を捨てる。この操作を,水層に黄色が認められなくなるまで繰り返す。水層は乾いたろ
紙5種Bでろ過し,溶液中のクロロホルムの小滴を除く。
6) シアン化カリウム-塩化アンモニウム溶液 JIS K 8443に規定するシアン化カリウム1.0 gを水に溶
かして500 mLとする。これにJIS K 8116に規定する塩化アンモニウムを少量ずつ溶かし,pHを
9.0〜9.5に調節する。この溶液は5)の酢酸アンモニウム溶液と同様に操作して8-キノリノールクロ
ロホルム溶液及びクロロホルムで洗浄し,精製する。
7) 1,10-フェナントロリン溶液(1 g/L) 57.1 a) 5)による。
8) 8-キノリノール溶液(10 g/L) JIS K 8775に規定する8-キノリノール2 gにJIS K 8355に規定する
酢酸5 mLを加え,僅かに加熱して溶かした後,水を加えて200 mLとする。
9) クロロホルム JIS K 8322に規定するもの。
10) アルミニウム標準液(Al 0.1 mg/mL) JIS K 8255に規定する硫酸カリウムアルミニウム・12水[ビ
237
K 0102:2016
ス(硫酸)カリウムアルミニウム-水(1/12)]1.76 gをとり,塩酸(1+1)[57.1 a) 1)による。]20 mL
に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又はJIS K 8069に規定する
アルミニウム(99.9 %以上)0.100 gをとり,塩酸(1+1)20 mL中に加熱して溶かし,放冷後,全
量フラスコ1 000 mLに移し入れ,水を標線まで加える。
11) アルミニウム標準液(Al 1 μg/mL) アルミニウム標準液(Al 0.1 mg/mL)10 mLを全量フラスコ
1 000 mLにとり,塩酸(1+1)[57.1 a) 1)による。]20 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 200 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った(1)試料の適量(2)(Alとして5〜50 μgを含む。)をとり,塩化ヒドロキシルアンモ
ニウム溶液(100 g/L)1 mLと1,10-フェナントロリン溶液(1 g/L)5 mLとを加えて振り混ぜ,アン
モニア水(1+2)を滴加してpHを約3.5(3)に調節する。
2) 水を加えて液量を約80 mLとした後,約15分間放置する。
3) 8-キノリノール溶液(10 g/L)3 mLと酢酸アンモニウム溶液(150 g/L)10 mLとを加え,アンモニ
ア水(1+2)を滴加してpHを5.2〜5.5(4)に調節する。
4) この溶液を分液漏斗に移し,水を加えて液量を約100 mLとした後,クロロホルム10 mL(又は20 mL)
を加え,約1分間激しく振り混ぜて放置する。
5) クロロホルム層を分離して,別の分液漏斗に移し入れ,シアン化カリウム-塩化アンモニウム溶液
25 mLを加え,振り混ぜて放置する。
6) クロロホルム層を共栓試験管30 mLに入れ,硫酸ナトリウム約1 gを加えて軽く振り混ぜて水分を
除く。
7) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長390 nm付近の吸光度を
測定する。
8) 空試験として水約70 mLをとり,1)〜7)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
9) 検量線からアルミニウムの量を求め,試料中のアルミニウムの濃度(Al mg/L)を算出する。
注(1) 5.のうち5.3の方法は用いない。有機物が少ない試料の場合には,試料100 mLにつきJIS K 8180
に規定する塩酸5 mLを加え,静かに加熱して液量が約1/5になるまで濃縮してもよい。
(2) 一般に試料は50〜100 mLとし,最大500 mLまで用いてもよい。
(3) ブロモフェノールブルー試験紙を用いる。
(4) ブロモクレゾールグリーン試験紙を用いる。pHが5.2〜5.5の範囲にならないときは,塩酸(1
+2)又はアンモニア水(1+2)を用いて調節する。
d) 検量線 検量線の作成は,次による。
1) アルミニウム標準液(Al 1 μg/mL)5〜50 mLを分液漏斗に段階的にとり,水を加えて液量約70 mL
とし,以下,c) 1)〜8)の操作を行ってアルミニウム(Al)の量と吸光度との関係線を作成する。
備考 1. 試料にふっ化物イオンが含まれる場合には,ふっ化物イオン0.5 mgに対し硫酸ベリリウム36
mgを加えておけば,妨害を防ぐことができる。
2. クロムが存在する場合には,pH5.2〜5.5における抽出は,できるだけ低温で行う。氷水で冷
却するとよい。
238
K 0102:2016
備考 3. マンガンが多量に含まれる場合には,錯体を抽出したクロロホルム溶液を塩化ヒドロキシル
アンモニウムを加えたpH7以下の酢酸-酢酸アンモニウム溶液(JIS K 8359に規定する酢酸ア
ンモニウム173 gを水約800 mLに溶かし,この溶液にJIS K 8355に規定する酢酸13 mLを
加え,水で1 Lとする。)で洗浄してマンガンを除去する。
4. チタン,モリブデンなどが含まれている場合には,c) 3)の操作で,銅,ニッケル,コバルト
などを除いた後,pH10のアンモニアアルカリ性塩化アンモニウム溶液(50 g/L)(JIS K 8116
に規定する塩化アンモニウム50 gを水約500 mLに溶かし,この溶液にJIS K 8085に規定す
るアンモニア水をpH約10まで加え,水で1 Lとする。)25 mLにJIS K 8230に規定する過
酸化水素2 mLを加えた溶液でクロロホルム層を洗浄する。
5. この方法では,鉄0.45 mgまでの存在は影響しない。
6. アルミニウム及び鉄を同時に定量する場合には,次のように操作する。
1) c) 1)の塩化ヒドロキシルアンモニウム溶液(100 g/L)1 mLと1,10-フェナントロリン溶
液(1 g/L)5 mLとを加える操作を省略し,以下,c) 2)〜7)の操作を行って波長390 nm
付近の吸光度A及び,470 nm付近の吸光度Bを測定する。
2) 別に,空試験として水約80 mLをとり,c) 3)〜7)の操作を行って試料について得た吸光
度A及び吸光度Bを補正し,それぞれ吸光度A′及び吸光度B′とする。
3) 波長470 nm付近の鉄(III)の検量線から吸光度B′に相当する鉄(III)の量を求め,鉄
の濃度(Fe mg/L)を算出する。
4) また,吸光度B′に相当する鉄(III)の量を波長390 nm付近の鉄(III)の検量線に適用
して,波長390 nm付近での鉄(III)による吸光度Cを求める。吸光度A′から吸光度C
を差し引き,波長390 nm付近のアルミニウムによる吸光度Dを求める。
5) 吸光度Dを用い,波長390 nm付近のアルミニウムの検量線からアルミニウムの量を求
め,試料中のアルミニウムの濃度(Al mg/L)を算出する。
6) 検量線 検量線の作成は,次による。
6.1)
アルミニウム標準液(Al 1 μg/mL)5〜50 mLを分液漏斗に段階的にとり,水を加えて
液量約80 mLとし,c) 3)〜6)の操作を行って波長390 nm付近の吸光度を測定する。
6.2)
別に,鉄(III)標準液(Fe 10 μg/mL)(*)0.5〜10 mLを分液漏斗に段階的にとり,水を
加えて約80 mLとし,c) 3)〜6)の操作を行って波長470 nm付近及び波長390 nm付近
の吸光度を測定する。
6.3)
空試験として水約80 mLをとり,c) 3)〜6)の操作を行って波長470 nm付近及び波長
390 nm付近の吸光度を測定し,アルミニウム標準液及び鉄(III)標準液について得た
吸光度を補正する。アルミニウム(Al)の量と波長390 nm付近の吸光度,鉄(Fe)の
量と波長470 nm付近の吸光度及び波長390 nm付近の吸光度との関係線を作成する。
注(*) 鉄(III)標準液(Fe 10 μg/mL) JIS K 8982に規定する硫酸アンモニウム鉄
(III)・12水[ビス(硫酸)鉄(III)アンモニウム-水(1/12)]8.63 gをとり,
硫酸(1+1)[5.4 a) 2)による。]20 mL及び水を加えて溶かし,全量フラスコ1 000
mLに移し入れ,水を標線まで加える。この溶液を鉄(III)標準液(Fe 1 mg/mL)
とし,その10 mLを全量フラスコ1 000 mLにとり,硫酸(1+1)10 mLを加え
た後,水を標線まで加える。
58.2 フレーム原子吸光法 試料を前処理した後,アセチレン-一酸化二窒素フレーム中に噴霧し,アルミ
239
K 0102:2016
ニウムによる原子吸光を波長309.3 nmで測定してアルミニウムを定量する。
定量範囲:Al 5〜100 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩化カリウム溶液(100 g/L) JIS K 8121に規定する塩化カリウム10 gを水に溶かして100 mLと
する。
2) アルミニウム標準液(Al 0.5 mg/mL) JIS K 8255に規定する硫酸カリウムアルミニウム・12水[ビ
ス(硫酸)カリウムアルミニウム-水(1/12)]8.794 gをとり,塩酸(1+1)[57.1 a) 1)による。]20 mL
に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。又はJIS K 8069に規定する
アルミニウム(99.9 %以上)0.500 gをとり,塩酸(1+1)30 mL中に入れ,加熱して溶かし,放冷
後,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
d) 操作 操作は,次による。
1) c)の準備操作を行った試料の適量(Alとして0.5〜10 mgを含む。)を全量フラスコ100 mLにとり,
JIS K 8180に規定する塩酸1 mLを加え,水を標線まで加える。
2) この溶液50 mLを乾いたビーカーにとり,塩化カリウム溶液(100 g/L)2 mLを加える。
3) 2)の試料をアセチレン-一酸化二窒素フレーム(5)中に導入し,波長309.3 nmにおける指示値(6)を読み
取る。
4) 空試験としてc)の準備操作での試料と同量の水をとり,試料と同様にc)及びd) 1)〜3)の操作を行っ
て指示値を読み取り,試料について得た指示値を補正する。
5) 検量線からアルミニウムの量を求め,試料中のアルミニウムの濃度(Al mg/L)を算出する。
注(5) 多燃料フレームの方が高感度が得られる。
(6) 吸光度又はその比例値。
e) 検量線 検量線の作成は,次による。
1) アルミニウム標準液(Al 0.5 mg/mL)1〜20 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行
った試料と同じ酸の濃度になるように酸及びJIS K 8180に規定する塩酸を加えた後,水を標線まで
加える。
2) この溶液についてd) 2)及び3)の操作を行う。
3) 別に,空試験として水についてc) 1)を行った試料と同じ酸の濃度になるように酸及び塩酸を加えた
後,d) 2)及び3)の操作を行って標準液について得た指示値を補正する。
4) アルミニウム(Al)の量と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
58.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,アルミニウムによる原子吸光
を波長309.3 nmで測定してアルミニウムを定量する。
定量範囲:Al 20〜200 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 7. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) アルミニウム標準液(Al 1 μg/mL) 58.1 a) 10)のアルミニウム標準液(Al 0.1 mg/mL)5 mLを全量
240
K 0102:2016
フラスコ500 mLにとり,硝酸(1+1)10 mLを加え,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
d) 操作 操作は,次による。
1) 52.3 d) 1)の操作を行う。ただし,原子化(7)温度は2 200〜3 000 ℃,波長は309.3 nmを用いる。
2) 空試験として52.3 d) 2)の操作を行う。
3) 検量線からアルミニウムの量を求め,試料中のアルミニウムの濃度(Al μg /L)を算出する。
注(7) 52.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) アルミニウム標準液(Al 1 μg/mL)2〜20 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行っ
た試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてd) 1)
の操作を行う。
2) 52.3 e) 2)及び3)の操作を行い,アルミニウム(Al)の量と指示値との関係線を作成する。検量線の
作成は,試料測定時に行う。
58.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ア
ルミニウムによる発光を波長309.271 nmで測定してアルミニウムを定量する。この方法によって表58.1
に示す元素が同時定量できる。それぞれの元素ごとの測定波長,定量範囲及び繰返し精度の例を,表58.1
に示す。
表58.1 測定波長,定量範囲及び繰返し精度の例*
対象元素
測定波長
nm
定量範囲
μg/L
繰返し精度
%
アルミニウム(Al)
309.271
80〜4 000
2〜10
クロム(Cr)
206.149
20〜4 000
2〜10
モリブデン(Mo)
202.030
40〜4 000
2〜10
バナジウム(V)
309.311
20〜2 000
2〜10
イットリウム(Y)**
371.029
−
−
注*
装置及び測定条件によって異なる。
** 内標準元素。イットリウム(Y)のほか,インジウム(In)及びイッテルビ
ウム(Yb)も使用できる。
a) 試薬 試薬は,次による。
1) 硝酸(1+1) JIS K 8541に規定する硝酸を用いて調製する。
2) アルミニウム標準液(Al 20 μg/mL) 58.2 a) 2)のアルミニウム標準液(Al 0.5 mg/mL)10 mLを全量
フラスコ250 mLにとり,硝酸(1+1)5 mLを加えた後,水を標線まで加える。
3) クロム標準液(Cr 0.1 mg/mL) JIS K 8005に規定する容量分析用標準物質の二クロム酸カリウムを
150 ℃で約1時間加熱し,デシケーター中で放冷する。K2Cr2O7 100 %に対してその0.283 gをとり,
少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
4) クロム標準液(Cr 10 μg/mL) クロム標準液(Cr 0.1 mg/mL)50 mLを全量フラスコ500 mLにとり,
硝酸(1+1)10 mLを加えた後,水を標線まで加える。
5) モリブデン標準液(Mo 0.1 mg/mL) JIS K 8905に規定する七モリブデン酸六アンモニウム四水和
241
K 0102:2016
物0.184 gを少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
6) モリブデン標準液(Mo 20 μg/mL) モリブデン標準液(Mo 0.1 mg/mL)20 mLを全量フラスコ100 mL
にとり,水を標線まで加える。
7) バナジウム標準液(V 0.1 mg/mL) JIS K 8747に規定するバナジン(V)酸アンモニウム0.230 gを
とり,硫酸(1+1)[5.4 a) 2)による。]10 mL及び熱水200 mLに溶かす。放冷後,全量フラスコ1 000
mLに移し入れ,水を標線まで加える。
8) バナジウム標準液(V 10 μg/mL) バナジウム標準液(V 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
b) 装置 装置は,52.4 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.5によって処理する。ただし,クロムを定量する場合は,前処理に5.3は用いない。
備考 8. 準備操作を行った試料溶液のアルカリ金属イオン,アルカリ土類金属イオンなどの濃度が高
くアルミニウムの濃度が低い場合のアルミニウムの定量には,試料100 mLをとり,58.1 c) 1)
〜5)のクロロホルムをJIS K 8051に規定する3-メチル-1-ブタノール(イソアミルアルコール)
に代えた操作を行って,3-メチル-1-ブタノール層を共栓試験管に移し入れる。この場合は,
58.1 c) 6)の硫酸ナトリウムを加えて水分を除く操作を省略してもよい。
d) 操作 52.4 d)による。ただし,表58.1に示すそれぞれの金属元素の波長の発光強度を測定する。
e) 検量線 52.4 e)による(8)。
注(8) 備考8.によって準備操作を行い,3-メチル-1-ブタノール層をそのまま噴霧する場合の検量線の
作成は,次による。
アルミニウム標準液(Al 20 μg/mL)を適切な濃度(Al 1〜4 μg/mL)に薄め,全量フラスコ
100 mLに段階的にとり,水で100 mLとした後,試料と同様に備考8.並びに52.4 d) 1)及び2)の
操作を行ってアルミニウム(Al)の量と発光強度との関係線を作成する。
58.5 ICP質量分析法 52.5による。
59. ニッケル(Ni) ニッケルの定量には,ジメチルグリオキシム吸光光度法,フレーム原子吸光法,ICP
発光分光分析法又はICP質量分析法を適用する。
なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,1996
年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
59.1 ジメチルグリオキシム吸光光度法 試料にくえん酸塩を加え,アンモニア水で微アルカリ性とした
後,2,3-ブタンジオンジオキシム(ジメチルグリオキシム)を加えて生成したニッケル錯体をクロロホル
ムで抽出し,これを希塩酸で逆抽出する。抽出液に臭素及びアンモニア水を加えてニッケルを酸化し,再
び2,3-ブタンジオンジオキシムを加えて生じる赤褐色のニッケル錯体の吸光度を測定してニッケルを定量
242
K 0102:2016
する。
定量範囲:Ni 2〜50 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+20) JIS K 8180に規定する塩酸を用いて調製する。
2) アンモニア水(1+1)及び(1+5) JIS K 8085に規定するアンモニア水を用いて調製する。
3) 臭素水(飽和) JIS K 8529に規定する臭素3〜4 mLを水100 mLに加えて激しく振り混ぜ,放置後
その上澄み液を用いる。
4) くえん酸水素二アンモニウム溶液(100 g/L) JIS K 8284に規定するくえん酸水素二アンモニウム
10 gを水約80 mLに溶かし,アンモニア水(1+1)を滴加してpHを約7に調節した後,水を加え
て100 mLとする。
5) フェノールフタレイン溶液(5 g/L) 15.の備考2.による。
6) ジメチルグリオキシムのエタノール溶液(10 g/L) JIS K 8498に規定するジメチルグリオキシム1 g
をJIS K 8102に規定するエタノール(95)に溶かして100 mLとする。
7) ジメチルグリオキシムの水酸化ナトリウム溶液(10 g/L) JIS K 8498に規定するジメチルグリオキ
シム1 gを水酸化ナトリウム溶液(10 g/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調製
する。)に溶かし,水酸化ナトリウム溶液(10 g/L)を加えて100 mLとする。不溶解物があるとき
はろ過する。
8) クロロホルム JIS K 8322に規定するもの。
9) ニッケル標準液(Ni 0.1 mg/mL) 52.4 a) 14)による。
10) ニッケル標準液(Ni 5 μg/mL) ニッケル標準液(Ni 0.1 mg/mL)50 mLを全量フラスコ1 000 mLに
とり,硝酸(1+1)[58.4 a) 1)による。]20 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(1)(Niとして2〜50 μgを含む。)を分液漏斗にとり,くえん酸水素二
アンモニウム溶液(100 g/L)5 mL及び指示薬としてフェノールフタレイン溶液(5 g/L)(2) 2,3滴
を加え,次に,アンモニア水(1+5)を溶液の色が僅かに紅色になるまで滴加する。さらに,アン
モニア水(1+5)2,3滴及び水を加えて約100 mLとする。
2) ジメチルグリオキシムのエタノール溶液(10 g/L)2 mLとクロロホルム10 mLとを加え,約1分間
激しく振り混ぜて放置した後,クロロホルム層を別の分液漏斗に入れる。水層にクロロホルム5 mL
を加え,約1分間激しく振り混ぜて抽出し,放置後,クロロホルム層をとり,先の分液漏斗に合わ
せる。この操作を更に1回繰り返す。
3) クロロホルム層を入れた分液漏斗にアンモニア水(1+50)(JIS K 8085に規定するアンモニア水を
用いて調製する。)10〜20 mLを加え,約30秒間振り混ぜて放置した後,クロロホルム層を別の分
液漏斗に移す。
4) クロロホルム層を入れた分液漏斗に塩酸(1+20)10 mLを加え,約1分間激しく振り混ぜ,ニッケ
ルを逆抽出する。放置後,クロロホルム層を別の分液漏斗に入れる。クロロホルム層には,再び塩
酸(1+20)5 mLを加え,逆抽出を繰り返す。クロロホルム層は捨て,水層は先の水層に合わせて
全量フラスコ25 mLに入れる。
243
K 0102:2016
5) 臭素水(飽和)2 mLを加えて振り混ぜ,約1分間放置する。次に,アンモニア水(1+1)を加えて
中和し,更にアンモニア水(1+1)2 mLを過剰に加え,流水で室温以下に冷却する。
6) ジメチルグリオキシムの水酸化ナトリウム溶液(10 g/L)2 mLを加えて振り混ぜ,ニッケルを発色
させた後,水を標線まで加える。
7) この溶液の一部を吸収セルに移し,波長450 nm付近の吸光度を測定する。
8) 空試験として水約50 mLをとり,1)〜7)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
9) 検量線からニッケルの量を求め,試料中のニッケルの濃度(Ni μg/L)を算出する。
注(1) 有機物及び濁りを含まない試料でニッケルの濃度が低い場合は,試料500 mLまでの適量をとり,
5.1を行い,1)〜8)に準じて操作して定量できる。この場合には,試料は前処理した全量を用い,
試薬は1)〜6)におけるものと同量を用いる。検量線は,試料と同様に操作して作成する。
(2) リトマス試験紙を用いてもよい。
d) 検量線 検量線の作成は,次による。
1) ニッケル標準液(Ni 5 μg/mL)0.4〜10 mLを全量フラスコ25 mLに段階的にとり,c) 5)〜8)の操作
を行ってニッケル(Ni)の量と吸光度との関係線を作成する。
59.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,ニッケルによる
原子吸光を波長232.0 nmで測定してニッケルを定量する。
定量範囲:Ni 0.3〜6 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) ニッケル標準液(Ni 10 μg/mL) 52.4 a) 15)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
備考 1. ニッケルの濃度が低い試料で,抽出操作を妨害する物質を含まない場合の準備操作は,次に
よるか,又は52.の備考4.,備考5.又は備考6.による。また,次の操作を行い,ニッケルの定
量に用いてもよい。
試料100 mLをビーカーにとり,JIS K 8180に規定する塩酸5 mLを加え,約5分間煮沸し
た後,放冷する。59.1 c) 1)〜3)に準じて操作し,ニッケルをジメチルグリオキシム錯体とし
て,クロロホルム層に抽出する。クロロホルム層を合わせ,塩酸(1+20)[59.1 a) 1)による。]
10 mLを加え,振り混ぜてニッケルを逆抽出する。水層を分離した後,クロロホルム層に塩
酸(1+20)5 mLを加えて逆抽出を行う。逆抽出液を合わせ,全量フラスコ25 mLに移し入
れ,水を標線まで加える。
d) 操作 操作は,次による。
1) 52.2 d) 1)の操作を行う。ただし,波長は232.0 nmを用いる。
2) 空試験として,52.2 d) 2)の操作を行う。
3) 検量線からニッケルの量を求め,試料中のニッケルの濃度(Ni mg/L)を算出する。
e) 検量線 検量線の作成は,次による。
1) ニッケル標準液(Ni 10 μg/mL)3〜60 mL(3)を全量フラスコ100 mLに段階的にとり,c) 1)を行った
試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。
2) 52.2 e) 2)及び3)の操作を行い,ニッケル(Ni)の量と指示値との関係線を作成する。検量線の作成
244
K 0102:2016
は,試料測定時に行う。
注(3) 準備操作として溶媒抽出法を適用するときは,ニッケル標準液の量を,適宜,減らす。
59.3 ICP発光分光分析法 52.4による。
59.4 ICP質量分析法 52.5による。
60. コバルト(Co) コバルトの定量には,ニトロソR塩吸光光度法,フレーム原子吸光法,ICP発光分
光分析法又はICP質量分析法を適用する。
なお,フレーム原子吸光法は,1986年に第1版として発行されたISO 8288,ICP発光分光分析法は,1996
年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
60.1 ニトロソR塩吸光光度法 試料にくえん酸塩を加え,アンモニア水で微アルカリ性とした後,ジエ
チルジチオカルバミド酸ナトリウム(ジエチルカルバモジチオ酸ナトリウム)を加え,生成するコバルト
錯体を酢酸ブチルで抽出する。酢酸ブチル層を蒸発乾固し,残留物を硝酸及び硫酸で分解する。水に溶か
し,酢酸ナトリウムでpHを6に調節し,ニトロソR塩(3-ヒドロキシ-4-ニトロソ-2,7-ナフタレンジスル
ホン酸二ナトリウム)を加えてコバルト錯体を生成させ,その吸光度を測定してコバルトを定量する。
定量範囲:Co 1〜30 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 硝酸 JIS K 8541に規定するもの。
2) 硫酸(1+1) 5.4 a) 2)による。
3) アンモニア水(1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
4) くえん酸水素二アンモニウム溶液(100 g/L) 52.1 a) 3)による。
5) 酢酸ナトリウム溶液(200 g/L) JIS K 8371に規定する酢酸ナトリウム三水和物33.2 gを水に溶か
して100 mLとする。
6) ジエチルジチオカルバミド酸ナトリウム溶液(20 g/L) JIS K 8454に規定するN,N-ジエチルジチオ
カルバミド酸ナトリウム三水和物2.6 gを水に溶かして100 mLとする。着色瓶に保存し,2週間以
上経過したものは使用しない。
7) メタクレゾールパープル溶液(1 g/L) 52.1 a) 6)による。
8) ニトロソR塩溶液(2 g/L) 1-ニトロソ-2-ナフトール-3,6-ジスルホン酸二ナトリウム(ニトロソR
塩)0.2 gを水に溶かして100 mLとする。
9) 酢酸ブチル JIS K 8377に規定するもの。
10) コバルト標準液(Co 0.1 mg/mL) 52.4 a) 16)による。
11) コバルト標準液(Co 2 μg/mL) コバルト標準液(Co 0.1 mg/mL)20 mLを全量フラスコ1 000 mL
にとり,硝酸(1+1)[58.4 a) 1)による。]20 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
245
K 0102:2016
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(Coとして1〜30 μgを含む。)を分液漏斗100 mLにとり,くえん酸
水素二アンモニウム溶液(100 g/L)5 mL及びメタクレゾールパープル溶液(1 g/L)2,3滴を加え,
次に,アンモニア水(1+1)を溶液の色がうすい紫になるまで加えた後,水で液量を約50 mLとす
る。
2) ジエチルジチオカルバミド酸ナトリウム溶液(20 g/L)3 mL (1)を加えて振り混ぜ,次に,酢酸ブチ
ル10 mLを加え,約2分間振り混ぜて放置する。
3) 酢酸ブチル層を分離し,ビーカー100 mLに移し,水層には酢酸ブチル10 mLを加えて再び抽出し,
酢酸ブチル層は,先のビーカー中の酢酸ブチルに合わせる。
4) 酢酸ブチル層を静かに加熱し,軽く蒸発乾固した後,残留物に硝酸3 mL及び硫酸(1+1)1 mLを
加えて加熱し,有機物を分解するとともに大部分の酸を追い出す。
5) 水10 mLを加えて溶かし,酢酸ナトリウム溶液(200 g/L)を加え,pH試験紙を用いてpHを5〜6
に調節する。
6) ニトロソR塩溶液(2 g/L)1 mL (2)を加えて約1分間煮沸する。引き続き硝酸2 mLを加えた後,約
1分間煮沸し,流水で急冷する。
7) 全量フラスコ25 mLに移し入れ,水を標線まで加える。
8) 溶液の一部を吸収セルに移し,波長420 nm付近の吸光度を測定する。
9) 空試験として水10 mLをとり,5)〜8)の操作を行って,試料について得た吸光度を補正する。
10) 検量線からコバルトの量を求め,試料中のコバルトの濃度(Co μg/L)を算出する。
注(1) ニッケル,銅,カドミウム,鉛,ひ素などもジエチルジチオカルバミド酸と反応するから,こ
れらが多量に含まれているときは,ジエチルジチオカルバミド酸ナトリウムを過剰に加える。
また,これらの金属元素もニトロソR塩によって発色して妨害するが,ニッケルはコバルトの
約30倍,銅は20倍程度は影響しない。
(2) コバルトニトロソR錯体は,直射日光によって分解されるおそれがあるから,ニトロソR塩添
加後は直射日光を避けて操作する。
d) 検量線 検量線の作成は,次による。
1) コバルト標準液(Co 2 μg/mL)0.5〜15 mLをビーカー100 mLに段階的にとり,液量を10〜15 mL
にした後,c) 5)〜9)の操作を行って吸光度を測定し,コバルト(Co)の量と吸光度との関係線を作
成する。
60.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレーム中に噴霧し,コバルトによる
原子吸光を波長240.7 nmで測定してコバルトを定量する。
定量範囲:Co 0.5〜10 mg/L,繰返し精度:5〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) コバルト標準液(Co 0.1 mg/mL) 52.4 a) 16)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.5によって処理する。
備考 1. コバルトの濃度が低い試料で抽出操作を妨害する物質を含まない場合の準備操作は,52.の備
246
K 0102:2016
考4.,備考5.又は備考6. による。
d) 操作 操作は,次による。
1) 52.2 d) 1)の操作を行う。ただし,波長は240.7 nmを用いる。
2) 空試験として52.2 d) 2)の操作を行う。
3) 検量線からコバルトの量を求め,試料中のコバルトの濃度(Co mg/L)を算出する。
e) 検量線 検量線の作成は,次による。
1) コバルト標準液(Co 0.1 mg/mL)0.5〜10 mL(3)を全量フラスコ100 mLに段階的にとり,c) 1)を行っ
た試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。
2) 52.2 e) 2)及び3)の操作を行い,コバルト(Co)の量と指示値との関係線を作成する。検量線の作成
は,試料測定時に行う。
注(3) 準備操作として溶媒抽出法を適用するときは,コバルト標準液の量を,適宜,減らす。
60.3 ICP発光分光分析法 52.4による。
60.4 ICP質量分析法 52.5による。
61. ひ素(As) ひ素の定量には,ジエチルジチオカルバミド酸銀吸光光度法,水素化物発生原子吸光法,
水素化物発生ICP発光分光分析法又はICP質量分析法を適用する。
なお,水素化物発生原子吸光法は,1996年に第1版として発行されたISO 11969,ICP発光分光分析法
は,1996年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11969:1996,Water quality−Determination of arsenic−Atomic absorption spectrometric method
(hydride technique)(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
61.1 ジエチルジチオカルバミド酸銀吸光光度法 ひ素を水素化ひ素として発生させ,ジエチルジチオカ
ルバミド酸銀(N,N-ジエチルカルバモジチオ酸銀)のクロロホルム溶液に吸収させ,生成する赤紫の吸光
度を測定してひ素を定量する。
定量範囲:As 2〜10 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するひ素分析用のもの。試薬の調製及び操作にはこれを用いる。
2) 塩酸(1+1) 1)の塩酸を用いて調製する。
3) 硝酸 JIS K 8541に規定するもの。
4) 硫酸 JIS K 8951に規定するもの。
5) 硫酸(1+5) 4)の硫酸を用いて調製する。
6) よう化カリウム溶液(200 g/L) JIS K 8913に規定するよう化カリウム20 gを水に溶かして100 mL
とする。
7) 塩化すず(II)溶液 JIS K 8136に規定する塩化すず(II)二水和物40 gを1)の塩酸に溶かし,こ
の塩酸で100 mLとする。JIS K 8580に規定するすずの粒状2,3粒を加え,着色ガラス瓶に保存す
る。使用時に適量をとり,水で10倍に薄める。
247
K 0102:2016
8) 酢酸鉛(II)溶液(100 g/L) JIS K 8374に規定する酢酸鉛(II)三水和物12 gをJIS K 8355に規
定する酢酸1,2滴及び水に溶かして100 mLとする。
9) 亜鉛 JIS K 8012に規定するひ素分析用のもの。ただし,JIS Z 8801-1に規定する試験用ふるいで
ふるい分け,目開き1.4 mmのふるいを通り,1 mmのふるいに止まるものを用いる。
10) ジエチルジチオカルバミド酸銀溶液 JIS K 9512に規定するN,N-ジエチルジチオカルバミド酸銀
0.25 gとJIS K 8832に規定するブルシンn水和物(2,3-ジメトキシストリキニジン-10-オンn水和物)
0.1 gとをJIS K 8322に規定するクロロホルムに溶かし,クロロホルムで100 mLとする。
11) クロロホルム JIS K 8322に規定するもの。
12) ひ素標準液(As 0.1 mg/mL) JIS K 8005に規定する容量分析用標準物質の酸化ひ素(III)を105 ℃
で約2時間加熱し,デシケーター中で放冷する。As2O3 100 %に対してその0.132 gをとり,水酸化
ナトリウム溶液(40 g/L)2 mLに溶かした後,水を加えて500 mLとし,次に,硫酸(1+10)を加
えて微酸性とし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
13) ひ素標準液(As 1 μg/mL) ひ素標準液(As 0.1 mg/mL)10 mLを全量フラスコ1 000 mLにとり,
水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 水素化ひ素発生装置 図61.1及び図61.2に例を示す。
2) 光度計 分光光度計又は光電光度計
単位 mm
A:
B:
b:
C:
D:
水素化ひ素発生瓶100 mL
導管
酢酸鉛(II)溶液(100 g/L)で湿らせ
たガラスウール
水素化ひ素吸収管(共栓付き)
ゴム栓
図61.1 水素化ひ素発生装置の例
248
K 0102:2016
単位 mm
A:
B:
b:
C:
D:
E:
F:
水素化ひ素発生瓶100 mL
導管
酢酸鉛(II)溶液(100 g/L)で湿らせ
たガラスウール
水素化ひ素吸収管(共栓付き)
亜鉛投入管
平面すり合わせ
押さえばね
図61.2 水素化ひ素発生装置の例
c) 操作 操作は,次による。
1) 試料の適量(Asとして2〜10 μgを含む。)をビーカーにとり,硫酸3 mL及び硝酸5 mLを加えて加
熱板上で加熱して硫酸の白煙を発生させて試料中の有機物などを分解する(1)。
2) 放冷後,ビーカーの器壁を少量の水で洗い,再び硫酸の白煙が発生するまで加熱する。
3) 放冷後,少量の水を加えて溶かし,水素化ひ素発生瓶に洗い移し,液量を約40 mLとする。
4) 塩酸(1+1)2 mL,よう化カリウム溶液(200 g/L)15 mL及び塩化すず(II)溶液5 mLを加えて振
り混ぜ,約10分間放置する。
5) 水素化ひ素発生瓶,導管及びジエチルジチオカルバミド酸銀溶液5 mLを入れた水素化ひ素吸収管
を連結した後,水素化ひ素発生瓶に亜鉛約3 gを手早く投入する(2)。水素化ひ素発生瓶を約25 ℃の
水浴中に入れ,約1時間放置してジエチルジチオカルバミド酸銀溶液に水素化ひ素を吸収,発色さ
せる。
6) この溶液にクロロホルムを加えて5 mLとする。
7) 溶液の一部を吸収セルに移し,クロロホルムを対照液として波長510 nm付近の吸光度を測定する。
8) 空試験として硫酸3 mL及び硝酸5 mLをビーカーにとり,1)〜7)の操作を行って吸光度を測定し,
試料について得た吸光度を補正する。
9) 検量線(3)からひ素の量を求め,試料中のひ素の濃度(As μg/L)を算出する。
注(1) 分解中に硝酸が不足した場合には,硝酸を追加する。
(2) 図61.2の水素化ひ素発生装置を用いるときは,亜鉛投入管に亜鉛約3 gを入れ,吸収管を連結
249
K 0102:2016
した後,亜鉛投入管を回転して亜鉛を試料中に添加する。
注(3) 検量線は傾きが変動するので,必ず試験時に作成する。
d) 検量線 検量線の作成は,次による。
1) ひ素標準液(As 1 μg/mL)2〜10 mLを水素化ひ素発生瓶に段階的にとり,硫酸3 mLを加えた後,
水で約40 mLとし,c) 4)〜8)の操作を試料と同時に行って吸光度を測定し,ひ素(As)の量と吸光
度との関係線を作成する。
備考 1. 試料のひ素の濃度が低く多量の試料を必要とし,ひ素の濃縮及び蒸発に長時間を要する場合
又は水素化ひ素の発生を妨害する物質が含まれている場合は,次の操作によってひ素を濃縮
するとともに分離し定量する。
1) 試料1 Lに硝酸3 mLを加え,過マンガン酸カリウム溶液(3 g/L)[61.2 a) 6)による。]を
滴加して着色させた後,煮沸してひ素(V)に酸化する。このとき過マンガン酸の色が
消えたときは,再び過マンガン酸カリウム溶液(3 g/L)を添加して着色させ煮沸する。
2) できるだけ少量の過酸化水素(1+30)(JIS K 8230に規定する過酸化水素を用いて調製
する。)を滴加し,過剰の過マンガン酸を分解する。
3) これに鉄(III)溶液(Fe 10 mg/mL)[JIS K 8142に規定する塩化鉄(III)六水和物5 g
又はJIS K 8982に規定する硫酸アンモニウム鉄(III)・12水9 gとa) 1)の塩酸5 mLとを
水に溶かして100 mLとする。]5 mL及びメタクレゾールパープル溶液(1 g/L)[52.1 a) 6)
による。]2,3滴を加え,液温約80 ℃でかき混ぜながら,アンモニア水(1+2)(JIS K
8085に規定するアンモニア水を用いて調製する。)を溶液の色が紫(pH約9)になるま
で滴加する。放置して沈殿が沈降した後,小形のろ紙5種Aでろ別し,温水で2,3回
洗浄する。沈殿は温硫酸(1+5)18 mL及び塩酸(1+1)2 mLをろ紙上から滴加して溶
かし,ろ紙は温水で洗浄する。
4) ろ液及び洗液を水素化ひ素発生瓶に入れ,水で約40 mLとする。よう化カリウム溶液(200
g/L)15 mL及び塩化すず(II)溶液5 mLを加えて振り混ぜ,約10分間放置した後,c) 5)
〜9)の操作を行ってひ素を定量する。
この方法を用いた場合には,空試験として鉄(III)溶液(Fe 10 mg/mL)5 mLを水素化ひ
素発生瓶にとり,硫酸(1+5)18 mLと塩酸(1+1)2 mLとを加え,水で約40 mLにした後,
c) 4)〜7)の操作を行って吸光度を測定し,試料について得た吸光度を補正する。
61.2 水素化物発生原子吸光法 試料を前処理してひ素を水素化ひ素とし,水素-アルゴンフレーム中に導
き,ひ素による原子吸光を波長193.7 nmで測定してひ素を定量する。
定量範囲:As 5〜50 μg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸 61.1 a) 1)による。
2) 塩酸(1 mol/L) 1)の塩酸を用いて調製する。
3) 塩酸(1+1) 1)の塩酸を用いて調製する。
4) 硝酸 JIS K 9901に規定する高純度試薬−硝酸による。
5) 硫酸(1+1) JIS K 9905に規定する高純度試薬−硫酸を用いて5.4 a) 2)の操作によって調製する。
6) 過マンガン酸カリウム溶液(3 g/L) JIS K 8247に規定する過マンガン酸カリウム0.3 gを水に溶か
して100 mLとする。
7) よう化カリウム溶液(200 g/L) 61.1 a) 6)による。
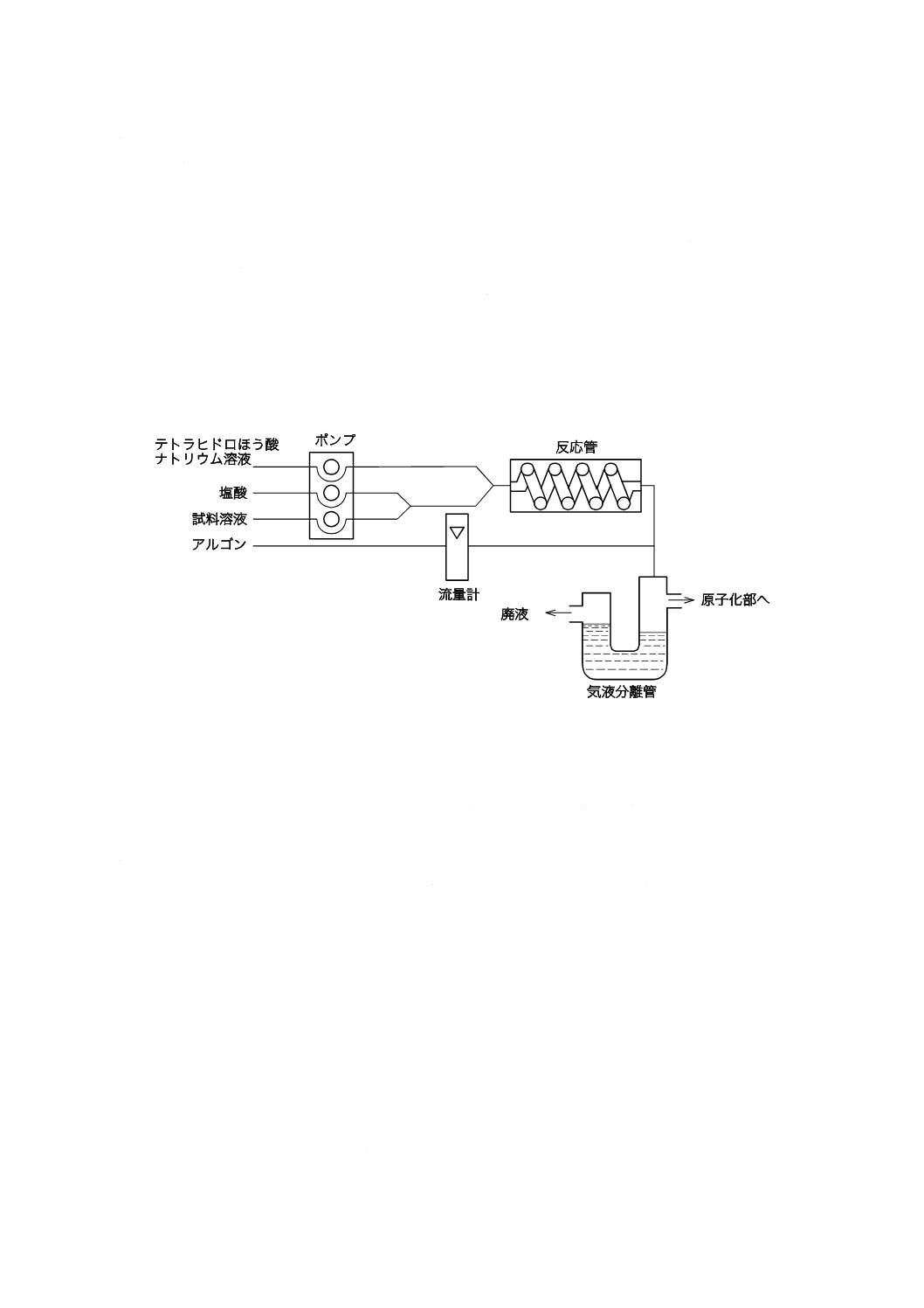
250
K 0102:2016
8) テトラヒドロほう酸ナトリウム溶液(10 g/L) テトラヒドロほう酸ナトリウム5 gを水酸化ナトリ
ウム溶液(0.1 mol/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調製する。)に溶かして500
mLとする。使用時に調製する。
9) アルゴン JIS K 1105に規定するアルゴン2級
10) ひ素標準液(As 0.1 μg/mL) 61.1 a) 13)のひ素標準液(As 1 μg/mL)10 mLを全量フラスコ100 mL
にとり,3)の塩酸(1+1)2 mLを加えた後,水を標線まで加える。
11) アスコルビン酸溶液(100 g/L) JIS K 9502に規定するL(+)-アスコルビン酸10 gを水に溶かし
て100 mLとする。
b) 装置 装置は,次による。
1) 連続式水素化物発生装置 図61.3に例を示す。
2) フレーム原子吸光分析装置 52.2 b) 1)による。
図61.3 連続式水素化物発生装置構成の例
c) 操作 操作は,次による。
1) 試料(4)の適量(Asとして0.1〜1 μgを含む。)をビーカー(5) 100 mLにとり,硫酸(1+1)1 mL及び
硝酸2 mLを加え,更に過マンガン酸カリウム溶液(3 g/L)を溶液が着色するまで滴加する。
2) 加熱板上で加熱(6)して硫酸の白煙を発生させる(7) (8)。
3) 2)の溶液を室温まで放冷した後,水10 mL,塩酸3 mL,よう化カリウム溶液(200 g/L)2 mL及び
アスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置した後,全量フラスコ20 mLに洗い
移し,水を標線まで加える。
4) 連続式水素化物発生装置にアルゴンを流しながら,3)の溶液,テトラヒドロほう酸ナトリウム溶液
(10 g/L)(9)及び塩酸(1 mol/L)(9)を,定量ポンプで連続的に装置内に導入(9)し,水素化ひ素を発
生させる。
5) 発生した水素化ひ素と廃液とを分離した後,水素化ひ素を含む気体を水素-アルゴン(10)フレーム(11)
中に導入し,波長193.7 nmの指示値(12)を読み取る。
6) 空試験として試料と同量の水をとり,1)〜5)の操作を行った後,指示値を読み取り,試料について
得た指示値を補正する。
7) 検量線からひ素の量を求め,試料中のひ素の濃度(As μg/L)を算出する。
注(4) 有機物及び亜硝酸イオンを含まない試料は1)〜3)の代わりに,次のように操作してもよい。
251
K 0102:2016
試料の適量(Asとして0.1〜1 μgを含む。)をビーカー100 mLにとり,塩酸3 mLを加え,沸
騰しない程度に5〜7分間加熱した後,冷却する。次に,よう化カリウム溶液(200 g/L)2 mL
及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置した後,全量フラスコ20 mL
に洗い移し,水を標線まで加える。また,多量の有機物を含む場合は,c) 1)及び2)の代わりに,
次のように操作してもよい。
試料の適量に硫酸(1+1)1 mL,硝酸2 mL及びJIS K 8223に規定する過塩素酸3 mLを加え,
加熱して硫酸白煙を発生させ(*),有機物を分解する。
注(*) 過塩素酸を用いる加熱分解操作は,試料の種類によっては,爆発の危険性があるため,
硝酸が共存する状態で行うように注意する。
注(5) ほうけい酸ガラスには,ひ素を含むものがあるので注意する。四ふっ化エチレン樹脂製ビーカ
ーを用いるとよい。
(6) 加熱中に過マンガン酸の色が消えたときは,過マンガン酸カリウム溶液(3 g/L)を追加する。
(7) 硝酸が残存すると,分解生成物である窒素酸化物によって水素化ひ素の発生が阻害されるので,
十分に硫酸の白煙を発生させて硝酸を除去する。
(8) この白煙は刺激性があり,加熱分解操作はドラフト内で行う。
(9) 装置によって,塩酸及びテトラヒドロほう酸ナトリウム溶液の流量及び濃度は異なる。
(10) JIS K 1107に規定する窒素2級を用いてもよい。
(11) 加熱吸収セル方式のものを用いてもよい。
(12) 吸光度又はその比例値。
d) 検量線 検量線の作成は,次による。
1) ひ素標準液(As 0.1 μg/mL)1〜10 mL(13)を段階的に全量フラスコ20 mLにとり,塩酸3 mL,よう
化カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置
した後,水を標線まで加える。
2) c) 4)及び5)の操作を行う。
3) 別に,空試験として全量フラスコ20 mLに水10 mL,塩酸3 mL,よう化カリウム溶液(200 g/L)2
mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置した後,水を標線まで加えた
ものを用い,c) 4)及び5)の操作を行って,2)で得た指示値を補正し,ひ素(As)の量と指示値との
関係線を作成する。検量線の作成は,試料測定時に行う。
注(13) 注(11)によった場合は,水素-アルゴンフレームに比べて10〜50倍程度(装置及び操作条件によ
って異なる。)感度がよいので,ひ素標準液(As 0.1 μg/mL)の量を,適宜,減らす。
備考 2. c) 1)及び2)の加熱分解操作において,循環式の分解装置を用いて分解操作を行ってもよい。
例を図61.4に示す。この分解装置を用いる場合の分解操作は,次による。
試料50 mLを丸底フラスコにとる。JIS K 9905に規定する高純度試薬−硫酸5 mL,JIS K
8230に規定する過酸化水素5 mL及び沸騰石数個を加え,丸底フラスコを図61.4に示すよう
に装置に連結する。丸底フラスコの内容物を沸騰するまで加熱し,凝縮物を凝縮物受器に集
める。硫酸の白煙が発生するまで加熱を続ける。分解液の外観を見て,濁りがあり,着色し
ていれば,更に過酸化水素5 mLを加え,沸騰を続ける。分解液が無色で濁りがなくなれば,
丸底フラスコ及び内容物を冷却し,凝縮物を丸底フラスコに戻す。
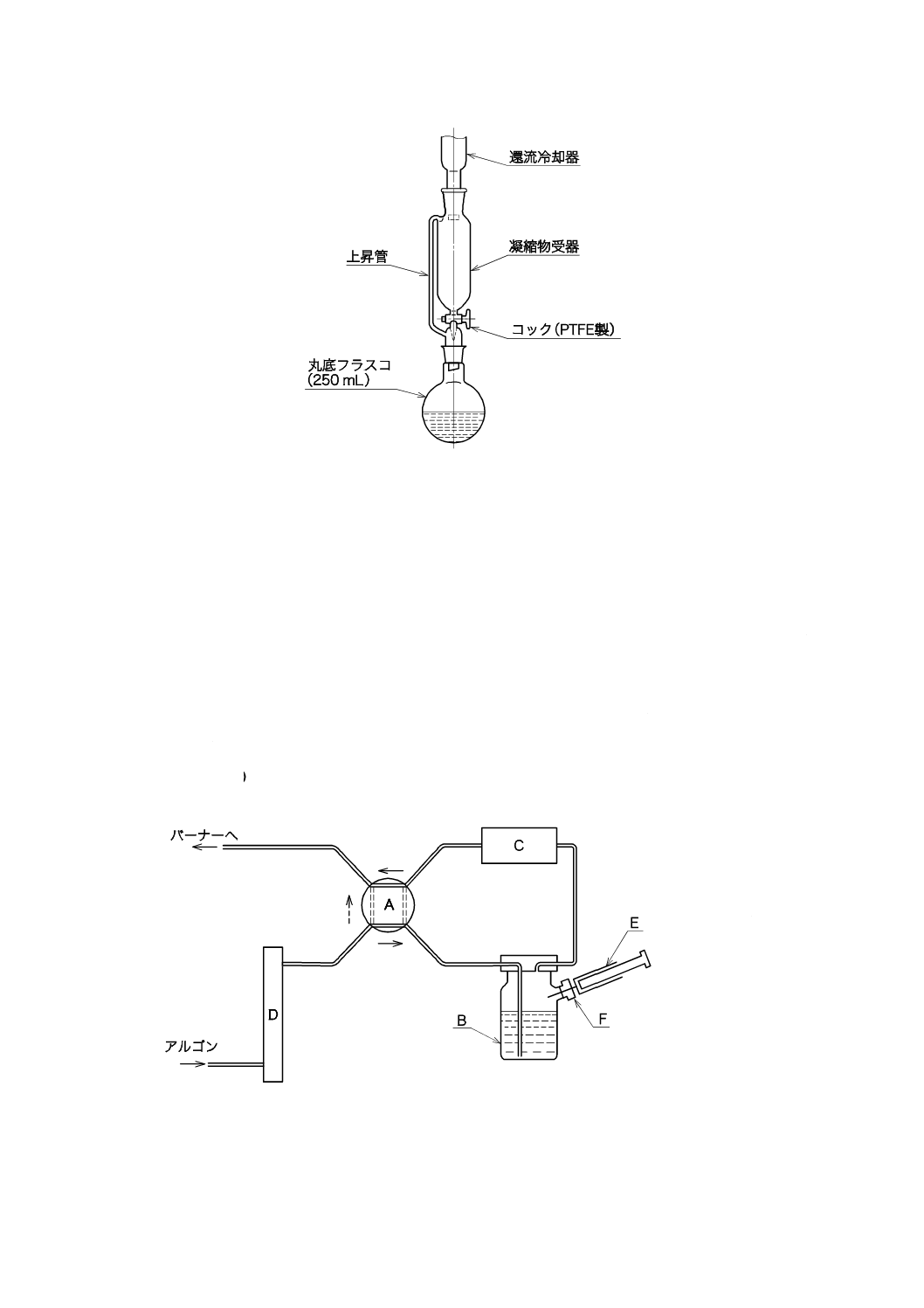
252
K 0102:2016
図61.4 試料分解装置の例
備考 3. 連続式水素化物発生装置の代わりに,図61.5に例を示す水素化物発生装置を用いて定量して
もよい。この場合の操作は,次による。
1) c) 1)〜3)の操作を行った溶液の全量を水素化物発生装置の反応容器に移し入れる。
2) 水素化物発生装置と原子吸光分析装置とを連結し,系内の空気をアルゴン(*)で置換する。
3) コックを回転して,アルゴンで溶液をバブリングする状態にする。
4) セプタムを通してシリンジなどによってテトラヒドロほう酸ナトリウム溶液(10 g/L)2
mLを手早く加え,発生する水素化物を水素-アルゴン(*)フレーム(**)中に導入し,波長
193.7 nmの指示値(***)を読み取る。
5) c) 6)及び7)の操作を行い,試料中のひ素の濃度(As μg/L)を算出する。
注(*) 注(10)による。
(**) 注(11)による。
(***) 注(12)による。
A:コック
B:反応容器
C:貯留器
D:流量計
E:シリンジ
F:セプタム
図61.5 水素化物発生装置構成の例
4. ごく低濃度の水素化物を分析する目的で,水素化物を濃縮する場合には,ガラスビーズなど
を充塡したU字管を液体窒素中に浸したコールドトラップを用いて水素化物を捕集する。捕
253
K 0102:2016
集後,U字管を引き上げて室温に戻し,気化した水素化物をアルゴンでフレーム中に送り込
む。
備考 5. 水素化物発生法は,鉄,ニッケル,コバルト,白金,パラジウムなどの遷移金属によって発
生効率が影響される。また,アンチモン,セレンなどの水素化合物を形成する元素によって
も発生効率が低下する。よう化カリウムは,ひ素をV価からIII価に還元するために用いら
れるが,遷移金属による干渉の低減にも効果がある。
なお,未知試料のように共存物質の影響が不明な場合は,未知試料に一定量の測定対象元
素(ここではひ素)を添加したときに得られる指示値の増加分と,同量の測定元素を含む検
量線用標準液の指示値とを比較することによって,干渉の大きさを知ることができる。共存
物質による干渉がある場合は,標準添加法を適用すると真度が向上するが,干渉が大きい場
合は,水素化物発生法は適用できない。
61.3 水素化物発生ICP発光分光分析法 試料を前処理してひ素を水素化ひ素とし,試料導入部を通して
誘導結合プラズマ中に導入し,ひ素による発光を波長193.696 nmで測定してひ素を定量する。
定量範囲:As 1〜50 μg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,61.2 a)による。
b) 装置 装置は,次による。
1) 連続式水素化物発生装置 61.2 b) 1)による。
2) ICP発光分光分析装置
c) 操作 操作は,次による。
1) 試料(4)の適量(Asとして0.02〜0.2 μgを含む。)をビーカー(5) 100 mLにとり,硫酸(1+1)1 mL
及び硝酸2 mLを加え,更に過マンガン酸カリウム溶液(3 g/L)を溶液が着色するまで滴加する。
2) 61.2 c) 2)〜4)の操作を行う。
3) 発生した水素化ひ素と廃液とを分離した後,水素化ひ素を含む気体を発光部に導入し,波長193.696
nmの発光強度を測定する。
4) 空試験として試料と同量の水をとり,1)〜3)の操作を行い,試料について得た発光強度を補正する。
5) 検量線からひ素の量を求め,試料中のひ素の濃度(As μg/L)を算出する。
d) 検量線 検量線の作成は,次による。
1) ひ素標準液(As 0.1 μg/mL)0.2〜10 mLを段階的に全量フラスコ20 mLにとり,塩酸3 mL,よう化
カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置し
た後,水を標線まで加える。
2) この溶液について,連続式水素化物発生装置にアルゴンを流しながら,1)の溶液,テトラヒドロほ
う酸ナトリウム溶液(10 g/L)(9)及び塩酸(1 mol/L)(9)を,定量ポンプで連続的に装置内に導入(9)
し,水素化ひ素を発生させ,引き続きc) 3)の操作を行う。
3) 別に,空試験として全量フラスコ20 mLに水10 mL,塩酸3 mL,よう化カリウム溶液(200 g/L)2
mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置した後,水を標線まで加えた
ものを用い,61.2 c) 4)及びc) 3)の操作を行って,標準液について得た発光強度を補正する。
4) ひ素(As)の量と発光強度との関係線を作成する。検量線の作成は,試料測定時に行う。
備考 6. 水素化物を発生させるときに副生する水素が発光部に導入されることによって,プラズマが
不安定になる場合があるので,特に導入初期には水素の量が多くなり過ぎないように注意す
る。
254
K 0102:2016
備考 7. 61.の備考5.による。また,標準添加法を用いる場合は,バックグラウンド補正を行う。
8. 有機物及び亜硝酸イオンを含まない試料は,c) 1)及び2)の代わりに,次のように操作しても
よい。試料の適量(Asとして0.02〜1 μgを含む。)をビーカー100 mLにとり,塩酸3 mLを
加え,沸騰しない程度に5〜7分間加熱した後,冷却する。次に,よう化カリウム溶液(200 g/L)
2 mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60分間静置した後,全量フラス
コ20 mLに洗い移し,水を標線まで加え,61.2 c) 4)の操作を行う。また,多量の有機物を含
む場合は,c) 1)及び2)の代わりに,次のように操作してもよい。
試料の適量に硫酸(1+1)1 mL,硝酸2 mL及びJIS K 8223に規定する過塩素酸3 mLを
加え,加熱して硫酸白煙を発生させ,有機物を分解する。冷却後,水10 mL,塩酸3 mL,よ
う化カリウム溶液(200 g/L)2 mL及びアスコルビン酸溶液(100 g/L)0.4 mLを加え,約60
分間静置した後,全量フラスコ20 mLに洗い移し,水を標線まで加え,61.2 c) 4)の操作を行
う。
61.4 ICP質量分析法 52.5による。
備考 9. 塩酸,塩化物イオンなどの塩素を多量に含む試料では,表52.3に示す40Ar35Cl,40Ca35Clなど
の多原子イオンのスペクトル干渉が大きくなるため,これを補正する手法又は低減化する手
法を用いる。40Ar35Clのスペクトル干渉を補正する手法として,35Clと37Clとの同位体比が一
定であることを利用した次の補正式を用いる。
IAs=I75−(A35/A37)×f75/77×I77
ここに,
IAs: ひ素の指示値
I75: m/z 75における指示値
I77: m/z 77における指示値
A35:
35Clの同位体存在度
A37:
37Clの同位体存在度
f75/77: m/z 75と77に対する質量差別効果の係数
なお,f75/77は,装置,測定条件などによって異なるため,試料に含まれる塩素の濃度と同
程度の塩酸又は塩化物イオンだけを含む水溶液を,試料測定の前又は後に測定して求めてお
く。この水溶液のm/z 75とm/z 77とにおける指示値の比をとると,(A35/A37)×f75/77の積の値
が求められる。この値を用いて試料の指示値を補正する。また,セレンが共存する試料に対
しては,次の補正式を用いる。
IAs=I75−(A35/A37)×f75/77×[I77−(A77/A78)×f77/78×I78]
又は
IAs=I75−(A35/A37)×f75/77×[I77−(A77/A82)×f77/82×I82]
ここに,
IAs: ひ素の指示値
I75: m/z 75における指示値
I77: m/z 77における指示値
I78: m/z 78における指示値
I82: m/z 82における指示値
A35:
35Clの同位体存在度
A37:
37Clの同位体存在度
A77:
77Seの同位体存在度
A78:
78Seの同位体存在度
A82:
82Seの同位体存在度
f75/77: m/z 75と77に対する質量差別効果の係数
f77/78: m/z 77と78に対する質量差別効果の係数
255
K 0102:2016
f77/82: m/z 77と82に対する質量差別効果の係数
なお,これらの補正式を用いる方法において,補正量が元の測定値の50 %を超える試料に
ついては正確さが低下するため,補正式を用いる方法は適用できない。
備考 10. スペクトル干渉を低減する手法として,61.3 c)に準じて水素化ひ素を発生させ,イオン化部
に導入してひ素(質量数75)の指示値を測定し,ひ素を定量してもよい。
11. スペクトル干渉を低減する手法として,JIS K 0133の磁場形二重収束質量分析計又はコリジ
ョン・リアクションセルを用いることができる。これらの方法を用いる場合も海水のように
塩濃度が高い試料では,適宜,希釈して適用することによってスペクトル干渉及び非スペク
トル干渉を低減することができる。
62. アンチモン(Sb) アンチモンの定量には,ローダミンB吸光光度法,水素化物発生原子吸光法,水
素化物発生ICP発光分光分析法又はICP質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
62.1 ローダミンB吸光光度法 試料に塩酸及び硫酸を加えた後,アンチモンを硫酸セリウム(IV)で酸
化してアンチモン(V)とし,ジイソプロピルエーテル(2,2′-オキシビスプロパン)で抽出する。抽出し
たアンチモン(V)にローダミンB[9-(2-カルボキシフェニル)-3,6-(ジエチルアミノ)キサンチリウム
クロリド]を加え,生成する赤紫のアンチモンローダミンB錯体の吸光度を測定してアンチモンを定量す
る。
定量範囲:Sb 1〜30 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 塩酸(2+1) 1)の塩酸を用いて調製する。
3) 硝酸 JIS K 8541に規定するもの。
4) 硫酸(1+1) 5.4 a) 2)による。
5) 硫酸セリウム(IV)溶液(30 g/L) JIS K 8976に規定する硫酸セリウム(IV)四水和物3.6 gを硫
酸(1+1)6 mL及び水に溶かして100 mLとする。
硫酸セリウム(IV)四水和物は,品質によっては硫酸(1+1)6 mL及び水に溶けないことがある。
このときは更に硫酸(1+1)を加え,加熱して硫酸の白煙を約30分間発生させる。放冷後,水を加
えて加熱して溶かし,水で100 mLとする。
6) ジイソプロピルエーテル JIS K 9528に規定するもの。
7) ローダミンB溶液(0.2 g/L) JIS K 9038に規定するローダミンB 0.02 gを硫酸(1+1)6 mL及び
水に溶かし,水を加えて100 mLとする。使用時に約80 ℃に加熱し,放冷後使用する。
8) アンチモン標準液(Sb 0.2 mg/mL) JIS K 8080に規定するアンチモン(99.9 %以上)0.200 gをビ
ーカーにとり,硫酸20 mLを加え,加熱して溶かす。放冷後,水約100 mLを加えて溶かし,全量
256
K 0102:2016
フラスコ1 000 mLに移し入れ,硫酸(1+10)を標線まで加える。
9) アンチモン標準液(Sb 2 μg/mL) アンチモン標準液(Sb 0.2 mg/mL)10 mLを全量フラスコ1 000 mL
にとり,硫酸(1+10)[62.1 a) 8)による。]を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(Sbとして1〜30 μgを含む。)をビーカー100 mLにとり,硝酸5 mL
及び硫酸(1+1)5 mLを加え,硫酸の白煙が発生し始めるまで加熱する(1)。
2) 放冷後,塩酸(2+1)10 mLを加えて残留物を溶かし,少量の水で分液漏斗100 mLに洗い移し,液
量を約30 mLとする。
3) これを振り混ぜながら硫酸セリウム(IV)溶液(30 g/L)を溶液の色が黄色になるまで滴加し,更
に0.5 mLを過剰に加え,約5分間放置してアンチモンを酸化する。
4) 塩酸20 mL及び硫酸セリウム(IV)溶液(30 g/L)1 mLを加えて振り混ぜる。
5) ジイソプロピルエーテル30 mL(2)を加え,約2分間激しく振り混ぜた後,放置し,分離した水層を
捨てる。
6) ジイソプロピルエーテル層にローダミンB溶液(0.2 g/L)5 mLを加えて約1分間激しく振り混ぜた
後,放置し,分離した水層を捨てる。
7) ジイソプロピルエーテル層を目盛付き試験管に入れ,約60 ℃の温水中に約1分間浸し,泡が生じ
るまで加熱した後,直ちに流水で冷却する。
8) ジイソプロピルエーテルを加えて30 mLとし,その一部を吸収セルに移し,ジイソプロピルエーテ
ルを対照液として波長550 nm付近の吸光度を測定する。
9) 空試験として水15 mL,硫酸(1+1)5 mL及び塩酸(2+1)10 mLを分液漏斗100 mLにとり,3)
〜8)の操作を行って試料について得た吸光度を補正する。
10) 検量線からアンチモンの量を求め,試料中のアンチモンの濃度(Sb μg/L)を算出する。
注(1) 5.4の操作を行ったときは,残留する硫酸が約2.5 mLになるまで加熱濃縮する。
(2) アンチモンの量が極めて微量のときは,ジイソプロピルエーテル10 mLを用いる。
d) 検量線 検量線の作成は,次による。
1) アンチモン標準液(Sb 2 μg/mL)0.5〜15 mLを分液漏斗100 mLに段階的にとり,次に,硫酸の濃
度をほぼ一定にするためアンチモン標準液の量とは逆に硫酸(1+1)5〜2.5 mLを段階的に加える。
2) 更に塩酸(2+1)10 mL及び水をそれぞれ加えて液量を約30 mLとした後,c) 3)〜9)の操作を行っ
て吸光度を測定し,アンチモン(Sb)の量と吸光度との関係線を作成する。
備考 1. この方法では,鉄2 mgまでは妨害しない。
2. 試料中の有機物が微量で前処理の必要がなく,かつ,アンチモンの濃度が低い場合には,次
の酸化マンガン(IV)共沈法で分離濃縮する。この方法は,妨害物質からの分離に利用でき
る。
1) 試料500 mLまでの適量をとり,試料100 mLにつきJIS K 8541に規定する硝酸3 mL及
び硝酸マンガン(II)溶液[JIS K 8568に規定する硝酸マンガン(II)六水和物16 gを水
に溶かして100 mLとする。]2 mLを加え,静かに加熱して煮沸する。
2) 引き続き,溶液100 mLにつき過マンガン酸カリウム溶液(30 g/L)(JIS K 8247に規定
257
K 0102:2016
する過マンガン酸カリウム3 gを水に溶かして100 mLとする。)2 mLを加え,5〜10分
間静かに煮沸して酸化マンガン(IV)の沈殿を生成させる。約20分間放置した後,ろ紙
5種Bを用いてろ別し,温水で5〜7回洗浄する。
3) 沈殿は元のビーカーに移し入れ,ろ紙に付着した沈殿は硝酸(1+1)[52.1 a) 8)による。]
10 mLと過酸化水素(1+30)[61.1の備考1. 2)による。]とをろ紙上に交互に滴加して溶
かし,ろ紙は温水でよく洗浄する。この洗液は元のビーカーに合わせ,加熱して酸化マ
ンガン(IV)を溶かし,引き続き加熱して過酸化水素を分解した後,水を加えて約200 mL
とする。
4) この溶液に硝酸マンガン(II)溶液[備考2. 1)による。]1 mLを加えて加熱し,静かに
煮沸する。過マンガン酸カリウム溶液(30 g/L)2 mLを加え,煮沸を約5〜10分間続け
て,再び酸化マンガン(IV)の沈殿を生成させる。これをろ別して温水で洗う。
5) 沈殿は元のビーカーに移し入れ,ろ紙に付着している沈殿は,温硫酸(1+3)(JIS K 8951
に規定する硫酸を用いて調製する。)10 mLに過酸化水素(1+30)[61.1の備考1. 2)によ
る。]5 mLを加えた混合溶液を滴加して溶かし,ろ液を元のビーカーに受ける。ろ紙は
水で洗い,洗液はろ液に合わせる。加熱して沈殿を溶かし,硫酸の白煙が発生するまで
濃縮する。
62.2 水素化物発生原子吸光法 試料を前処理した後,アンチモンを水素化アンチモンとし,水素-アルゴ
ンフレーム中に導き,アンチモンによる原子吸光を波長217.6 nmで測定してアンチモンを定量する。
定量範囲:Sb 4〜20 μg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 塩酸(2+1) 1)の塩酸を用いて調製する。
3) 硝酸 JIS K 8541に規定するもの。
4) 硫酸(1+1) 5.4 a) 2)による。
5) チオ尿素溶液(0.1 mol/L) JIS K 8635に規定するチオ尿素0.76 gを水に溶かして100 mLとする。
6) テトラヒドロほう酸ナトリウム溶液(10 g/L) 61.2 a) 8)による。
7) アルゴン 61.2 a) 9)による。
8) アンチモン標準液(Sb 0.1 μg/mL) 62.1 a) 9)のアンチモン標準液(Sb 2 μg/mL)10 mLを全量フラ
スコ200 mLにとり,硫酸(1+10)(JIS K 8951に規定する硫酸を用いて調製する。)を標線まで加
える。
b) 装置 装置は,次による。
1) 連続式水素化物発生装置 61.2 b) 1)による。
2) フレーム原子吸光分析装置 52.2 b) 1)による。
c) 操作 操作は,次による。
1) 試料(3)の適量(Sbとして0.1〜0.5 μgを含む。)をビーカー100 mLにとり,硫酸(1+1)1 mL及び
硝酸2 mLを加える。
2) 加熱板上で乾固する直前まで加熱する。
3) 2)の溶液を室温まで放冷した後,塩酸5 mL及びチオ尿素溶液(0.1 mol/L)3 mLを加え,全量フラ
スコ25 mLに移し入れ,水を標線まで加える。
4) 連続式水素化物発生装置にアルゴンを流しながら,3)の溶液,テトラヒドロほう酸ナトリウム溶液
258
K 0102:2016
(10 g/L)(4)及び塩酸(1 mol/L)(4)を,定量ポンプで連続的に装置内に導入(4)し,水素化アンチモ
ンを発生させる。
5) 発生した水素化アンチモンと廃液とを分離した後,水素化アンチモンを含む気体を水素-アルゴン(5)
フレーム(6)中に導入し,波長217.6 nmの指示値(7)を読み取る。
6) 空試験として試料と同量の水をとり,1)〜5)の操作を行った後,指示値を読み取り,試料について
得た指示値を補正する。
7) 検量線からアンチモンの量を求め,試料中のアンチモンの濃度(Sb μg/L)を算出する。
注(3) 有機物を含まない試料は1)〜3)の代わりに,次のように操作してもよい。
試料の適量(Sbとして0.1〜0.5 μgを含む。)をビーカー100 mLにとり,塩酸5 mLを加え,
沸騰しない程度に5〜7分間加熱した後,冷却する。次に,チオ尿素溶液(0.1 mol/L)3 mLを
加え,全量フラスコ25 mLに移し入れ,水を標線まで加える。また,多量の有機物を含む場合
は,1)及び2)の代わりに,次のように操作してもよい。
試料の適量(Sbとして0.1〜0.5 μgを含む。)に硫酸(1+1)1 mL,硝酸2 mL及びJIS K 8223
に規定する過塩素酸3 mLを加え加熱して,白煙を発生させて有機物を分解する。
(4) 61.の注(9)による。
(5) 61.の注(10)による。
(6) 61.の注(11)による。
(7) 61.の注(12)による。
d) 検量線 検量線の作成は,次による。
1) アンチモン標準液(Sb 0.1 μg/mL)1〜5 mLを全量フラスコ25 mLに段階的にとり,塩酸5 mL及び
チオ尿素溶液(0.1 mol/L)3 mLを加え,水を標線まで加える。
2) c) 4)及び5)の操作を行う。
3) 別に,空試験として水17 mLを用い,塩酸5 mL及びチオ尿素溶液(0.1 mol/L)3 mLを加え,c) 4)
及び5)の操作を行って,2)で得た指示値を補正し,アンチモン(Sb)の量と指示値との関係線を作
成する。検量線の作成は,試料測定時に行う。
備考 3. 61.の備考2.による。
4. 図61.3と同様な装置の代わりに,図61.5に例を示す水素化物発生装置を用いて定量してもよ
い。この場合の操作は,次による。
1) c) 1)〜3)の操作を行った溶液の全量を水素化物発生装置の反応容器に移し入れる。
2) 水素化物発生装置と原子吸光装置とを連結し,系内の空気をアルゴン(*)で置換する。
3) コックを回転して,アルゴンで溶液をバブリングする状態にする。
4) セプタムを通してシリンジなどによってテトラヒドロほう酸ナトリウム溶液(10 g/L)2
mLを手早く加え,発生する水素化物を水素-アルゴン(*)フレーム(**)中に導入し,波長
217.6 nmの指示値(***)を読み取る。
5) c) 6)及び7)の操作を行い,試料中のアンチモンの濃度(Sb μg/L)を算出する。
注(*) 61.の注(10)による。
(**) 61.の注(11)による。
(***) 61.の注(12)による。
5. 水素化アンチモンの濃縮は,61.の備考4.と同じ操作による(水素化ひ素を水素化アンチモン
と読み替える。)。
259
K 0102:2016
62.3 水素化物発生ICP発光分光分析法 試料を前処理してアンチモンを水素化アンチモンとし,試料導
入部を通して誘導結合プラズマ中に導入し,アンチモンによる発光を波長206.833 nmで測定してアンチモ
ンを定量する。
定量範囲:Sb 1〜50 μg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 硝酸 JIS K 8541に規定するもの。
3) 硫酸(1+1) 5.4 a) 2)による。
4) チオ尿素溶液(0.1 mol/L) 62.2 a) 5)による。
5) テトラヒドロほう酸ナトリウム溶液(10 g/L) 61.2 a) 8)による。
6) アルゴン 61.2 a) 9)による。
7) アンチモン標準液(Sb 0.1 μg/mL) 62.2 a) 8)による。
b) 装置 装置は,次による。
1) 連続式水素化物発生装置 61.2 b) 1)による。
2) ICP発光分光分析装置
c) 操作 操作は,次による。
1) 試料(8)の適量(Sbとして0.025〜1.25 μgを含む。)をビーカーにとり,硫酸(1+1)1 mL及び硝酸
2 mLを加える。
2) 62.2 c) 2)〜4)の操作を行う。
3) 発生した水素化アンチモンと廃液とを分離した後,水素化アンチモンを含む気体を発光部に導入し,
波長206.833 nmの発光強度を測定する。
4) 空試験として試料と同量の水をとり,1)〜3)の操作を行い,試料について得た発光強度を補正する。
5) 検量線からアンチモンの量を求め,試料中のアンチモンの濃度(Sb μg/L)を算出する。
注(8) 有機物を含まない試料は,1)及び2)の代わりに,次のように操作してもよい。
試料の適量(Sbとして0.025〜1.25 μgを含む。)をビーカー100 mLにとり,塩酸5 mLを加え,
沸騰しない程度に5〜7分間加熱した後,冷却する。次に,チオ尿素溶液(0.1 mol/L)3 mLを
加え,全量フラスコ25 mLに移し入れ,水を標線まで加える。次に,62.2 c) 4)の操作を行う。
また,多量の有機物を含む場合は,1)及び2)の代わりに,次のように操作してもよい。
試料の適量(Sbとして0.025〜1.25 μgを含む。)に硫酸(1+1)1 mL,硝酸2 mL及びJIS K 8223
に規定する過塩素酸3 mLを加え加熱して,白煙を発生させて有機物を分解する。冷却後,チオ
尿素溶液(0.1 mol/L)3 mLを加え,全量フラスコ25 mLに移し入れ,水を標線まで加える。次
に,62.2 c) 4)の操作を行う。
d) 検量線 検量線の作成は,次による。
1) アンチモン標準液(Sb 0.1 μg/mL)0.25〜12.5 mLを全量フラスコ25 mLに段階的にとり,塩酸5 mL
及びチオ尿素溶液(0.1 mol/L)3 mLを加え,水を標線まで加える。
2) この溶液について,連続式水素化物発生装置にアルゴンを流しながら,1)の溶液,テトラヒドロほ
う酸ナトリウム溶液(10 g/L)(4)及び塩酸(1 mol/L)(JIS K 8180に規定する塩酸を用いて調製する。)
(4)を,定量ポンプで連続的に装置内に導入(4)し,水素化アンチモンを発生させ,引き続きc) 3)の操
作を行う。
3) 別に,空試験として水17 mLを用い,塩酸5 mL及びチオ尿素溶液(0.1 mol/L)3 mLを加え,2)の

260
K 0102:2016
操作を行って標準液について得た発光強度を補正する。
4) アンチモン(Sb)の量と発光強度との関係線を作成する(9)。検量線の作成は,試料測定時に行う。
注(9) 塩類濃度が高い試料で,検量線法が適用できない場合には,JIS K 0116の標準添加法を用いる
とよい。ただし,この場合は試料の種類によらずバックグラウンド補正を行う。
備考 6. 61.の備考6.による。
7. 共存する酸及び塩の影響を受けやすいので注意する。影響の有無は,試料に適量のアンチモ
ンを添加したときに,その指示値の増加分を検量線によって濃度に換算することによって確
認できる。
8. 鉄,クロム(VI)及びバナジウムはそれぞれ1 000倍,ニッケルは200倍,コバルトは500
倍程度共存する場合でも妨害は除去できる。
62.4 ICP質量分析法 試料を前処理した後,内標準元素を加え,試料導入部を通して高周波プラズマ中
に噴霧し,アンチモン及び内標準元素のそれぞれの質量/電荷数における指示値(10)を測定し,アンチモン
の指示値と内標準元素の指示値との比を求めてアンチモンを定量する。この方法によって表62.1に示す元
素が同時定量できる。それぞれの元素ごとの定量範囲,繰返し精度などの例を,表62.1に示す。
注(10) 52.の注(19)による。
表62.1 定量範囲,繰返し精度及び質量数の例*
対象元素
定量範囲
μg/L
繰返し精度
%
質量数
アンチモン(Sb)
0.5〜500
2〜10
121,123
すず(Sn)
0.5〜500
2〜10
118,120
モリブデン(Mo)
0.5〜500
2〜10
95,98
タングステン(W)
0.5〜500
2〜10
182,184
イットリウム(Y)**
−
−
89
インジウム(In)**
−
−
115
ビスマス(Bi)**
−
−
209
注*
装置及び測定条件によって異なる。
** 内標準元素
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) 塩酸(1+1) JIS K 9902に規定する高純度試薬−塩酸を用いて調製する。
4) 内標準液(1 μg/mL) 内標準元素としてイットリウム,インジウム又はビスマスを用いる。内標準
液の調製は,次の4.1)〜4.3)に規定する溶液のうち内標準元素として使用するものの2 mLを全量フ
ラスコ100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調製する(11) (12)。
4.1) イットリウム溶液(Y 50 μg/mL) 47.の備考5. 6)による。
4.2) インジウム溶液(In 50 μg/mL) 52.5 a) 3.2)による。
4.3) ビスマス溶液(Bi 50 μg/mL) 52.5 a) 3.3)による。
5) 混合標準液[(Sb 10 μg,Sn 10 μg,Mo 10 μg,W 10 μg)/mL](12) (13) 62.1 a) 8)アンチモン標準液
(Sb 0.2 mg/mL)25 mL,63.1 a) 11)すず標準液(Sn 0.1 mg/mL),58.4 a) 5)モリブデン標準液(Mo 0.1
mg/mL)及び69.1 a) 7)タングステン標準液(W 0.1 mg/mL)のそれぞれ50 mLを全量フラスコ500 mL
261
K 0102:2016
にとり,塩酸(1+1)(14) 10 mLを加えた後,水を標線まで加える(15)。
6) 混合標準液[(Sb 0.5 μg,Sn 0.5 μg,Mo 0.5 μg,W 0.5 μg)/mL](12) (13) 5)の混合標準液5 mLを
全量フラスコ100 mLにとり,塩酸(1+1)(14) 15 mLを加え,水を標線まで加える。使用時に調製
する。
注(11) 52.の注(20)による。
(12) 52.の注(21)による。
(13) 52.の注(16)による。
(14) 又は硝酸(1+1)。
(15) 52.の注(22)による。
b) 装置 装置は,次による。
1) ICP質量分析装置
備考 9. 52.の備考13.による。
10. 52.の備考14.による。
c) 準備操作 準備操作は,次による(16)。
1) 試料を5.によって処理する。
2) 1)で処理した試料の適量(測定対象元素として0.05〜50 μgを含む。)を全量フラスコ100 mLにと
り,内標準液(1 μg/mL)1 mLを加え,酸の最終濃度が1〜1.5 mol/Lとなるように塩酸(1+1)(14)
を加えた後,水を標線まで加える。
注(16) 52.の注(23)による。
d) 操作 操作は,52.5 d)による(17) (18)。
注(17) 52.の注(24)による。
(18) 52.の注(25)による。質量数を設定するには,表62.1及び表62.2を参考にするとよい。
e) 検量線 検量線の作成は,次による。
1) a) 5)の混合標準液(各元素濃度10 μg/mL)又はa) 6)の混合標準液(各元素濃度0.5 μg/mL)の混合
標準液0.1〜5 mLを,全量フラスコ100 mLに段階的にとり,内標準液(1 μg/mL)1 mLを加え,c)
2)の試料と同じ酸の濃度になるように塩酸(1+1)(14)を加えた後,水を標線まで加える。使用時に
調製する(19)。
2) この溶液についてd)の操作を行う。
3) 別に,空試験として全量フラスコ100 mLに内標準液(1 μg/mL)1 mLを加え,c) 2)の試料と同じ酸
の濃度になるように塩酸(1+1)(14)を加え,水を標線まで加えた後,d)の操作を行って標準液につ
いて得た指示値の比をそれぞれ補正し,測定対象元素濃度に対する,測定元素の指示値と内標準元
素の指示値との比の関係線をそれぞれ作成する。検量線の作成は,試料測定時に行う。
注(19) すず,アンチモンなどの標準液は,酸濃度が低いと加水分解などによって元素濃度が低下しや
すいため注意する。
備考 11. 52.の備考16.による。
262
K 0102:2016
表62.2 スペクトル干渉の例*
元素
質量数
同重体及び2価
イオンの干渉
多原子イオンの
干渉
アンチモン(Sb)
121
アンチモン(Sb)
123
Te+
すず(Sn)
118
すず(Sn)
120
Te+
モリブデン(Mo)
95
79Br16O
モリブデン(Mo)
98
Ru+
タングステン(W)
182
166Er16O
タングステン(W)
184
Os+
168Er16O 168Yb16O
イットリウム(Y)**
89
インジウム(In)**
115
Sn+
ビスマス(Bi)**
209
注*
装置及び測定条件によって異なる。
** 内標準元素
63. すず(Sn) すずの定量には,フェニルフルオロン吸光光度法,ケルセチン吸光光度法,ICP発光分光
分析法又はICP質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
63.1 フェニルフルオロン吸光光度法 すずを過マンガン酸カリウムですず(IV)とし,くえん酸及びポ
リビニルアルコールの存在の下で,フェニルフルオロン(2,3,7-トリヒドロキシ-9-フェニル-6-フルオロン)
を加えて黄色の錯体を生成させ,その吸光度を測定して定量する。
定量範囲:Sn 3〜40 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+11) JIS K 8180に規定する塩酸を用いて調製する。
2) 塩酸(1+4) JIS K 8180に規定する塩酸を用いて調製する。
3) 硫酸(1+1) 5.4 a) 2)による。
4) L(+)-アスコルビン酸 JIS K 9502に規定するもの。
5) くえん酸溶液(100 g/L) JIS K 8283に規定するくえん酸一水和物11 gを水に溶かして100 mLとす
る。
6) アンモニア水(1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
7) 過マンガン酸カリウム溶液(3 g/L) 61.2 a) 6)による。
8) ポリビニルアルコール溶液(5 g/L) JIS K 9550に規定するポリビニルアルコール0.5 gを水に溶か
して100 mLとする。
9) ブロモクレゾールグリーン溶液(0.5 g/L) JIS K 8840に規定するブロモクレゾールグリーン0.05 g
263
K 0102:2016
をJIS K 8102に規定するエタノール(95)20 mLに溶かし,水を加えて100 mLとする。
10) フェニルフルオロン溶液(0.1 g/L) JIS K 9547に規定するフェニルフルオロン0.05 gを,JIS K 8102
に規定するエタノール(95)100 mLに塩酸(1+2)(JIS K 8180に規定する塩酸を用いて調製する。)
10 mLを加えた溶液に溶かし,エタノール(95)を加えて500 mLとする。
11) すず標準液(Sn 0.1 mg/mL) JIS K 8580に規定するすず0.100 gをビーカーにとり,JIS K 8951に
規定する硫酸10 mLを加え,時計皿で覆い,加熱して溶かす。放冷後,塩酸(1+4)200 mLを加
えて溶かし,室温に冷却する。全量フラスコ1 000 mLに移し入れ,塩酸(1+4)を標線まで加える。
12) すず標準液(Sn 2 μg/mL) すず標準液(Sn 0.1 mg/mL)10 mLを全量フラスコ500 mLにとり,塩
酸(1+10)(JIS K 8180に規定する塩酸を用いて調製する。)を標線まで加える。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(Snとして7.5〜100 μgを含む。)をとり,硫酸(1+1)5 mL(1)を加え,
加熱して硫酸の白煙を発生させた後,乾固近くまで濃縮する。
2) 放冷後,塩酸(1+4)10 mLを徐々に加え,加熱して溶かし,室温まで冷却した後,全量フラスコ
50 mLに移し入れ,水を標線まで加える。
3) この溶液20 mLずつを全量フラスコ50 mL及びビーカー100 mLにそれぞれとる。
4) ビーカー中の溶液に水を加えて約50 mLとし,指示薬としてブロモクレゾールグリーン溶液(0.5
g/L)2,3滴を加え,アンモニア水(1+1)で溶液の色が緑になるまで滴定する。中和に要したア
ンモニア水(1+1)の量を記録する。
5) 全量フラスコ50 mL中の溶液に過マンガン酸カリウム溶液(3 g/L)を溶液の色が微赤になるまで滴
加し,約5分間放置してすずを酸化する。
6) L(+)-アスコルビン酸約0.1 gを加えて振り混ぜ,過剰の過マンガン酸を還元する。
7) くえん酸溶液(100 g/L)5 mL,塩酸(1+11)1.5 mL及び4)で中和に要したものと同量のアンモニ
ア水(1+1)を加える(pHは1.5〜2.0になる。)。
8) ポリビニルアルコール溶液(5 g/L)5 mLとフェニルフルオロン溶液(0.1 g/L)5 mLとを加えて振
り混ぜた後,水を標線まで加え,約20分間放置する。
9) 空試験として塩酸(1+4)10 mLを全量フラスコ50 mLにとり,水を標線まで加える。以下,3)〜
8)の操作を行う。
10) 8)で得た溶液の一部を吸収セルに入れ,9)で得た溶液を対照液として波長510 nm付近の吸光度を測
定する。
11) 検量線からすずの量を求め,試料中のすずの濃度(Sn μg/L)を算出する。
注(1) 試料の前処理に硫酸を用いた場合には加えない。
d) 検量線 検量線の作成は,次による。
1) すず標準液(Sn 2 μg/mL)1.5〜20 mLを全量フラスコ50 mL及びビーカー100 mLに段階的にとり,
c) 4)〜10)の操作を行ってすず(Sn)の量と吸光度との関係線を作成する。
備考 1. 有機物及び濁りを含まない試料で,すずの濃度が低い場合には,62.の備考2.によって酸化マ
ンガン(IV)共沈法で分離濃縮する。次に,c) 2)以降の操作を行って定量する。
2. 妨害元素は,ゲルマニウム,ジルコニウム,アンチモン,ビスマス,鉄などである。ただし,
鉄は,L(+)-アスコルビン酸で還元しておけば,約10 mgまでは妨害しない。アンチモン
264
K 0102:2016
は,酸化してV価にしておき,ビスマスは発色させた後,JIS K 8107に規定するエチレンジ
アミン四酢酸二水素二ナトリウム二水和物0.3 gを加えて錯体とすれば,それぞれ0.5 mgま
で妨害しない。
63.2 ケルセチン吸光光度法 すずを過マンガン酸カリウムですず(IV)とし,過剰の過マンガン酸を還
元する。ケルセチン[2-(3,4-ジヒドロキシフェニル)-3,5,7-トリヒドロキシ-4H-ベンゾピラン-4-オン]を
加え,黄色のすず-ケルセチン錯体を生成させ,4-メチル-2-ペンタノンで抽出し,その吸光度を測定して定
量する。
定量範囲:Sn 2〜20 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
2) 硫酸(1+1) 5.4 a) 2)による。
3) 硫酸(1+19) JIS K 8951に規定する硫酸を用いて調製する。
4) L(+)-アスコルビン酸 JIS K 9502に規定するもの。
5) 過マンガン酸カリウム溶液(3 g/L) 61.2 a) 6)による。
6) 硫酸ナトリウム JIS K 8987に規定するもの。
7) チオ尿素溶液(50 g/L) JIS K 8635に規定するチオ尿素10 gを水に溶かして200 mLとする。
8) 4-メチル-2-ペンタノン JIS K 8903に規定するもの。
9) ケルセチン溶液(1 g/L) ケルセチン0.2 gをJIS K 8101に規定するエタノール(99.5)約100 mL
に溶かし,JIS K 8180に規定する塩酸10 mLを加えた後,エタノール(99.5)で200 mLとする。使
用時に調製する。
10) すず標準液(Sn 5 μg/mL) 63.1 a) 11)のすず標準液(Sn 0.1 mg/mL)10 mLを全量フラスコ200 mL
にとり,塩酸(1+10)(JIS K 8180に規定する塩酸を用いて調製する。)を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(Snとして2〜20 μgを含む。)をとり,硫酸(1+1)5 mL(1)を加え,
加熱して硫酸の白煙を発生させた後,乾固近くまで濃縮する。
2) 放冷後,硫酸(1+19)5 mLを加え,加熱して溶かし,過マンガン酸カリウム溶液(3 g/L)を溶液
の色が微赤になるまで滴加し,約5分間放置してすずを酸化する。
3) L(+)-アスコルビン酸0.1 gを加えて振り混ぜ,過剰の過マンガン酸を還元する。
4) 分液漏斗100 mLに少量の水とともに移し入れ,チオ尿素溶液(50 g/L)20 mL及び塩酸(1+1)15
mLを加えて振り混ぜる。ケルセチン溶液(1 g/L)20 mLを加えて再び振り混ぜ,約15分間放置す
る。
5) 4-メチル-2-ペンタノン15 mLを加えて約1分間激しく振り混ぜ,すず-ケルセチン錯体を抽出する。
6) 水層を捨て,有機層に硫酸(1+19)25 mLを加えて約30秒間振り混ぜる。
7) 水層を捨て,有機層に硫酸ナトリウム約5 gを加えて振り混ぜる。
8) 空試験として,硫酸(1+19)5 mLをとり,2)〜7)の操作を行う。
9) 7)の有機層の一部を吸収セルにとり,8)の有機層を対照液として波長440 nm付近の吸光度を測定す
る。
265
K 0102:2016
10) 検量線からすずの量を求め,試料中のすずの濃度(Sn μg/L)を算出する。
d) 検量線 検量線の作成は,次による。
1) すず標準液(Sn 5 μg/mL)0.4〜4 mLを段階的にとり,硫酸(1+19)5 mLを加え,加熱して液量を
約5 mLとし,過マンガン酸カリウム溶液(3 g/L)を溶液の色が微赤になるまで加える。
2) 以下,c) 3)〜9)の操作を行ってすず(Sn)の量と吸光度との関係線を作成する。
備考 3. 有機物及び濁りを含まない試料で,すずの濃度が低い場合には,62.の備考2.によって酸化マ
ンガン(IV)共沈法で分離濃縮し,以下,c) 2)以降の操作を行って定量する。
63.3 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に導入し,す
ずによる発光を波長189.989 nmで測定してすずを定量する。
定量範囲:Sn 0.4〜2 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) すず標準液(Sn 10 μg/mL) 63.1 a) 11)のすず標準液(Sn 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,塩酸(1+10)(JIS K 8180に規定する塩酸を用いて調製する。)を標線まで加える。
b) 装置 装置は,52.4 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
d) 操作 操作は,次による。
1) c)の準備操作を行った試料を試料導入部を通して発光部に導入し(2),波長189.989 nmの発光強度を
測定する(3) (4) (5)。
2) 空試験としてc)の準備操作での試料と同量の水をとり,試料と同様にc)及びd) 1)の操作を行って試
料について得た発光強度を補正する。
3) 検量線からすずの量を求め,試料中のすずの濃度(Sn mg/L)を算出する。
注(2) 52.の注(11)による。
(3) 波長の異なる2本のスペクトル線を同時測定が可能な装置では,内標準法によることができる。
内標準法を用いるときの操作は,次による。
1) 52.の備考11.の1)及び2)の操作を行う。ただし,すずの波長として189.989 nmを用いる。
2) 別に,すず標準液(Sn 10 μg/mL)4〜20 mLを全量フラスコ100 mLに段階的にとり,イッ
トリウム溶液(Y 50 μg/mL)10 mLをそれぞれ加え,d) 1)の試料と同じ酸の濃度になるよ
うに酸を加えた後,水を標線まで加える。
3) この溶液について,d) 1)の噴霧操作を行い,52.の備考11.の2)〜4)の操作を行って試料中
のすずの濃度(Sn mg/L)を算出する。ただし,すずの波長として189.989 nmを用いる。
(4) 47.の注(8)による。
(5) 47.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) すず標準液(Sn 10 μg/mL)4〜20 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試料と
同じ酸の濃度になるように酸を加えた後,水を標線まで加える。
2) この溶液についてd) 1)の操作を行う。
3) 別に,空試験として水について,c) 1)を行った試料と同じ酸の濃度になるように酸を加えた後,d) 1)
の操作を行って,標準液について得た発光強度を補正する。
4) すず(Sn)の量と発光強度との関係線を作成する。検量線の作成は,試料測定時に行う。
266
K 0102:2016
63.4 ICP質量分析法 62.4による。
64. ビスマス(Bi) ビスマスの定量には,よう化物抽出吸光光度法,ICP発光分光分析法又はICP質量分
析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
64.1 よう化物抽出吸光光度法 試料を硫酸酸性とし,ビスマスとよう化物イオンとを反応させ,生成す
るだいだい色のよう化物錯体(テトラヨードビスマス酸)を3-メチル-1-ブタノールで抽出し,その吸光度
を測定してビスマスを定量する。
定量範囲:Bi 3〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 硫酸(1+1) 5.4 a) 2)による。
2) よう化カリウム溶液(20 g/L) JIS K 8913に規定するよう化カリウム2 gを水に溶かして100 mL
とする。
3) ホスフィン酸ナトリウム溶液(250 g/L) ホスフィン酸ナトリウム一水和物30 gを水に溶かして
100 mLとする。
4) 3-メチル-1-ブタノール JIS K 8051に規定するもの。
5) ビスマス標準液(Bi 0.1 mg/mL) ビスマス(99.9 %以上)0.100 g又は酸化ビスマス(III)(99.9 %
以上)0.112 gをとり,硝酸(1+1)[57.1 a) 2) による。]10 mLに溶かし,煮沸して窒素酸化物を追
い出す。放冷後,全量フラスコ1 000 mLに移し入れ,硫酸(1+20)(JIS K 8951に規定する硫酸を
用いて調製する。)を標線まで加える。
6) ビスマス標準液(Bi 2 μg/mL) ビスマス標準液(Bi 0.1 mg/mL)10 mLを全量フラスコ500 mLにと
り,硫酸(1+100)(JIS K 8951に規定する硫酸を用いて調製する。)を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(Biとして3〜50 μgを含む。)をビーカー100 mLにとり,硫酸(1+1)
5 mL(1)を加えた後,加熱して硫酸の白煙を発生させ,乾固近くまで濃縮する。
2) 残留物に硫酸(1+1)4 mL及び少量の水を加え,加熱して溶かし(2),分液漏斗100 mLに少量の水
で洗い移す。
3) よう化カリウム溶液(20 g/L)3 mLを加えて振り混ぜ,次に,ホスフィン酸ナトリウム溶液(250 g/L)
10 mL及び水を加えて液量を約50 mLとし,振り混ぜる。約30分間(3)放置して遊離したよう素を還
元する。
4) 3-メチル-1-ブタノール10 mLを加えて振り混ぜ,よう化物錯体を抽出する。
267
K 0102:2016
5) 3-メチル-1-ブタノール層を分離し,その一部を吸収セルに移し,3-メチル-1-ブタノールを対照液と
して,波長340 nm(4)付近の吸光度を測定する。
6) 空試験として硫酸(1+1)4 mL及び少量の水を分液漏斗100 mLに入れ,3)〜5)の操作を行って吸
光度を測定し,試料について得た吸光度を補正する。
7) 検量線からビスマスの量を求め,試料中のビスマスの濃度(Bi μg/L)を算出する。
注(1) 5.のうち5.4の方法を用いたときは,硫酸を加えない。
(2) 不溶解物は,ろ別し,水洗いして除く。
(3) ホスフィン酸によるよう素の還元は,液温の影響が大きいので注意する。約20 ℃で約30分間
は必要である。
(4) 波長465 nm付近でもよい。ただし,感度は1/2程度になる。
d) 検量線 検量線の作成は,次による。
1) ビスマス標準液(Bi 2 μg/mL)1.5〜25 mLを分液漏斗100 mLに段階的にとり,c) 2)〜6)の操作を行
って吸光度を測定し,ビスマス(Bi)の量と吸光度との関係線を作成する。
備考 1. 試料中の有機物が微量で前処理の必要がない場合は,次のいずれかの方法によって濃縮分離
する。
a) 62.の備考2.によって酸化マンガン(IV)共沈法で分離濃縮し,以下,c) 2)〜7)の操作を
行って定量する。
b) 試料の適量(Biとして3〜50 μgを含む。)をとり,鉄(III)溶液(Fe 10 mg/mL)[61.
の備考1. 3)による。]1 mLと硝酸(1+1)10 mLとを加えて煮沸する。かき混ぜながら
アンモニア水(1+1)[52.1 a) 1)による。]を水酸化鉄(III)が生成するまで加え,更に
過剰に5 mLとJIS K 8613に規定する炭酸アンモニウム3 gとを加えて,約5分間煮沸
する。沈殿が沈降した後,ろ紙5種Aでろ別した後,水で洗う。ろ紙上の沈殿は温硫酸
(1+4)(JIS K 8951に規定する硫酸を用いて調製する。)10 mLを滴加して溶かし,ろ
紙は水で洗う。ろ液と洗液とを合わせ,c) 3)〜7)の操作を行って定量する。
64.2 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ビ
スマスによる発光を波長223.061又は306.772 nmで測定してビスマスを定量する。
定量範囲:Bi 50〜5 000 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) ビスマス標準液(Bi 0.1 mg/mL) 64.1 a) 5)による。
2) ビスマス標準液(Bi 10 μg/mL) ビスマス標準液(Bi 0.1 mg/mL)10 mLを全量フラスコ100 mLに
とり,硝酸(1+1)[52.1 a) 8)による。]20 mLを加えた後,水を標線まで加える。使用時に調製す
る。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
d) 操作 操作は,次による。
1) 52.4 d) 1)による。ただし,波長223.061 nm(又は306.772 nm)の発光強度を測定する(5) (6) (7)。
2) 52.4 d) 2)及び3)の操作を行い,試料中のビスマスの濃度(Bi μg/L)を算出する。
注(5) 52.の注(11)による。
268
K 0102:2016
(6) 47.の注(8)による。
(7) 47.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) ビスマス標準液(Bi 10 μg/mL)0.1〜50 mLを全量フラスコ100 mLに段階的にとり,引き続き52.4 e)
1)〜3)の操作を行い,ビスマス(Bi)の量と発光強度との関係線を作成する。検量線の作成は,試
料測定時に行う。
64.3 ICP質量分析法 52.5による。
65. クロム(Cr) 全クロムとクロム(VI)とに区分する。
65.1 全クロム 全クロムの定量には,ジフェニルカルバジド吸光光度法,フレーム原子吸光法,電気加
熱原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
なお,フレーム原子吸光法及び電気加熱原子吸光法は,1990年に第1版として発行されたISO 9174,ICP
発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 9174:1990,Water quality−Determination of total chromium−Atomic absorption spectrometric
methods(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
65.1.1 ジフェニルカルバジド吸光光度法 クロム(III)を過マンガン酸カリウムで酸化してクロム(VI)
とした後,1,5-ジフェニルカルボノヒドラジド(ジフェニルカルバジド)を加え,生成する赤紫の錯体の
吸光度を測定して全クロムを定量する。
定量範囲:Cr 2〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 硫酸(1+9) 水9容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸1容を徐々に加える。
2) 過マンガン酸カリウム溶液(3 g/L) 61.2 a) 6)による。
3) 亜硝酸ナトリウム溶液(20 g/L) JIS K 8019に規定する亜硝酸ナトリウム2 gを水に溶かして100
mLとする。使用時に調製する。
4) 尿素溶液(200 g/L) JIS K 8731に規定する尿素20 gを水に溶かして100 mLとする。
5) ジフェニルカルバジド溶液(10 g/L) JIS K 8488に規定する1,5-ジフェニルカルボノヒドラジド(ジ
フェニルカルバジド)1 gをJIS K 8034に規定するアセトン100 mLに溶かし,JIS K 8355に規定す
る酢酸1滴を加えて酸性とする。褐色ガラス瓶に入れ,0〜10 ℃の暗所に保存する。2週間は安定
である。
6) クロム標準液(Cr 0.1 mg/mL) 58.4 a) 3)による。
7) クロム標準液(Cr 2 μg/mL) クロム標準液(Cr 0.1 mg/mL)20 mLを全量フラスコ1 000 mLにとり,
水を標線まで加える。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
269
K 0102:2016
c) 操作 操作は,次による。
1) 5.の操作(1)を行った試料の適量(2)(Crとして2〜50 μgを含む。)をビーカーにとり,硫酸(1+9)3
mL(3)を加え,加熱して硫酸の白煙を軽く発生させる(4) (5)。放冷後,水約30 mLを加え,残留物を
加熱して溶かす。
2) 溶液を静かに加熱し,過マンガン酸カリウム溶液(3 g/L)を1滴ずつ加え着色させる。引き続き加
熱し,溶液の赤い色が消えそうになったら,更に過マンガン酸カリウム溶液(3 g/L)を滴加し,常
に赤い色を保つようにして数分間煮沸を続ける。
3) 流水で冷却し,尿素溶液(200 g/L)10 mLを加え,激しくかき混ぜながら亜硝酸ナトリウム溶液(20
g/L)(6)を1滴ずつ加えて溶液の赤い色を消し,過剰の過マンガン酸及び酸化マンガン(IV)を分解
する。
4) 全量フラスコ50 mLに移し入れ,液温を約15 ℃(7)に保ち,ジフェニルカルバジド溶液(10 g/L)1 mL
を加え,直ちに振り混ぜる。水を標線まで加えて振り混ぜ,約5分間放置する(8)。
5) 溶液の一部を吸収セルに移し,波長540 nm付近の吸光度を測定する。
6) 空試験として水約30 mLをとり,硫酸(1+9)3 mLを加えた後,4)及び5)の操作を行って吸光度を
測定し,試料について得た吸光度を補正する。
7) 検量線からクロムの量を求め,試料中の全クロムの濃度(Cr μg/L)を算出する。
注(1) 5.のうち,5.3の方法は用いない。
(2) クロムの濃度が低く,有機物及び懸濁物がほとんど含まれていない場合には,試料500 mLまで
の適量をとり,試料100 mLにつきJIS K 8951に規定する硫酸2 mLを加えて加熱し,煮沸して
放冷する。硫酸アンモニウム鉄(II)溶液(5 mg/mL)[JIS K 8979に規定する硫酸アンモニウ
ム鉄(II)六水和物3.5 gを硫酸5〜7滴を含む水100 mLに溶かす。]1 mLを加え,よくかき混
ぜ,更にJIS K 8541に規定する硝酸2 mLを加え,煮沸して鉄(II)を酸化する。この溶液を放
置した後,アンモニア水(1+4)(JIS K 8085に規定するアンモニア水を用いて調製する。)を
加えて中和し,アンモニア臭を認めなくなるまで煮沸し,約80 ℃で約20分間保ち,沈殿を熟
成させる。沈殿をろ紙5種Aを用いてろ別し,温硝酸アンモニウム溶液(10 g/L)(JIS K 8545
に規定する硝酸アンモニウムを用いて調製する。)で5〜7回洗った後,硫酸(1+15)(JIS K 8951
に規定する硫酸を用いて調製する。)5 mLを加えて溶かし,ろ紙は温水で洗い,2)〜6)の操作を
行う。ただし,検量線を作成する場合は,標準液にそれぞれ[鉄(III)]溶液(Fe 10 mg/mL)
[61.の備考1. 3)による。]0.5 mLを加える。
(3) ジフェニルカルバジドによるクロム(VI)の発色には,硫酸の濃度は0.1 mol/L程度が適切であ
る。
(4) 前処理で多量の硫酸が添加されている場合には,加熱蒸発し,硫酸の白煙を発生させて硫酸を
除去した後,硫酸(1+9)3 mLを加える。この硫酸の白煙の発生に当たって強熱してはならな
い。硫酸クロム(III)の無水物が生成して不溶解性となる。JIS K 8987に規定する硫酸ナトリ
ウム20 mg程度を加えておくと安全である。
(5) 前処理で硫酸の白煙の発生を行った場合には,この操作は省略してもよい。
(6) 亜硝酸ナトリウム溶液(20 g/L)の代わりにアジ化ナトリウム溶液(50 g/L)(JIS K 9501に規
定するアジ化ナトリウム5 gを水に溶かして100 mLとする。)を用いることができる。この場
合,アジ化ナトリウム溶液(50 g/L)を注意して滴加し,よく振り混ぜて過マンガン酸を分解し,
続いて2〜3分間煮沸して過剰のアジ化ナトリウムを分解する。
270
K 0102:2016
注(7) 液温は,発色に影響するので,約15 ℃に保つことが必要である。
(8) 発色は2〜3分間ほどで最高になり,その後,徐々に退色するが,5〜15分間はほとんど変化し
ない。
d) 検量線 検量線の作成は,次による。
1) クロム標準液(Cr 2 μg/mL)1〜25 mLを段階的にビーカーにとり,それぞれに硫酸(1+9)3 mL
を加え,水を加えて液量を約30 mLとした後,c) 2)〜6)の操作を行ってクロム(Cr)の量と吸光度
との関係線を作成する。
備考 1. 試料が鉄を含むとき,鉄が多量になるに従って吸光度が低くなるが,発色液50 mL中約1 mg
で一定となる(約20 %低い値を示す。)。ただし,ジフェニルカルバジド溶液を加える前に,
二りん酸ナトリウム溶液(JIS K 8785に規定する二りん酸ナトリウム十水和物5 gを水に溶
かして100 mLとする。)2 mLを加えると,鉄2.5 mgまでは影響しない。
鉄がクロムより少ない場合には,無視してもよい。
2. この方法では,モリブデン,水銀,バナジウムなどが影響する。ただし,モリブデンは0.1 mg
まで影響しない。水銀は,塩化物イオンの添加によって妨害しなくなる。また,バナジウム
は発色後,10〜15分間経過してから吸光度を測定すれば,その影響は無視できる。
3. 鉄その他の妨害が多い場合には,次の操作を行う。
1) 試料の適量(Crとして2〜50 μgを含む。)を分液漏斗にとり,試料20 mLにつき硫酸(1
+1)[5.4 a) 2)による。]5 mLを加え,硫酸の濃度を約1.8 mol/Lとし,これに過マンガ
ン酸カリウム溶液(3 g/L)を滴加し,僅かに着色させる。
2) これに,クペロン溶液(50 g/L)[JIS K 8289に規定するクペロン(N-ニトロソ-N-フェニ
ルヒドロキシルアミンアンモニウム塩)(N-ヒドロキシ-N-ニトロソベンゼンアミンアン
モニウム塩)5 gを水に溶かして100 mLとする。]5 mLとJIS K 8322に規定するクロロ
ホルム10 mLとを加えて約30秒間激しく振り混ぜ,鉄その他を抽出し,静置する。
3) クロロホルム層を分離し,水層に再びクペロン溶液(50 g/L)1 mLとクロロホルム10 mL
とを加えて再び抽出し,クロロホルム層を分離する。水層をビーカー100 mLに移し,加
熱蒸発して軽く乾固する。
4) これに少量のJIS K 8951に規定する硫酸とJIS K 8541に規定する硝酸とを加え,再び蒸
発乾固して有機物を分解する。硫酸(1+1)[5.4 a) 2)による。]0.3 mL及び水約30 mL
に溶かす。過マンガン酸カリウム溶液(3 g/L)でクロムを酸化した後,c)によって操作
する。
65.1.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気フレームなどの中に噴霧し,クロム
による原子吸光を波長357.9 nmで測定して全クロムを定量する。
定量範囲:Cr 0.2〜5 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) クロム標準液(Cr 10 μg/mL) 58.4 a) 4)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5. (1)又は備考4.によって処理する。
備考 4. クロムの濃度が低い試料で,有機物及び懸濁物をほとんど含まない場合の準備操作は,次に
よる。
271
K 0102:2016
試料の適量をとり,注(2)に準じて操作し,水酸化鉄(III)にクロムを共沈させる。沈殿を
ろ紙5種Aでろ別し,少量の硝酸(1+2)(JIS K 8541に規定する硝酸を用いて調製する。)
に加熱して溶かし,ろ紙は温水で洗浄する。ろ液と洗液とを合わせ,0.1〜1 mol/Lの塩酸又
は硝酸酸性溶液の一定量とする。
d) 操作 操作は,次による。
1) 52.2 d) 1)の操作を行う(9)。ただし,波長は357.9 nmを用いる。
2) 空試験として52.2 d) 2)の操作を行う。
3) 検量線からクロムの量を求め,試料中の全クロムの濃度(Cr mg/L)を算出する。
注(9) 少燃料のアセチレン-空気,又はアセチレン-一酸化二窒素フレームを用いる。
e) 検量線 検量線の作成は,次による。
1) クロム標準液(Cr 10 μg/mL)2〜50 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試料
と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。
2) 52.2 e) 2)及び3)の操作を行い,クロム(Cr)の量と指示値との関係線を作成する。検量線の作成は,
試料測定時に行う。
備考 5. クロムの濃度が低い試料で,抽出を妨害する物質を含まない場合には,次の操作で定量して
もよい。
1) 試料の適量(Crとして5〜100 μgを含む。)をビーカー100 mLにとり,硫酸(1+2)(JIS
K 8951に規定する硫酸を用いて調製する。)2 mLを加え,過マンガン酸カリウム溶液(3
g/L)5〜7滴を加えて加熱する。過マンガン酸の色が消えそうになったら,更に過マン
ガン酸カリウム溶液(3 g/L)を滴加し,常に溶液の色が微赤を保つように5〜7分間煮
沸を続ける。
2) 流水で冷却し,これを分液漏斗に移し,水を加えて約100 mLとする。N,N-ジオクチルオ
クタンアミン(トリオクチルアミン)の酢酸ブチル溶液(30 g/L)20 mLを加え,約10
分間激しく振り混ぜた後,放置する。酢酸ブチル層をそのままフレーム中に噴霧してク
ロムを定量する。
3) 検量線の作成には,クロム標準液(Cr 10 μg/mL)を適切な濃度に薄めて用いる。JIS K 8377
に規定する酢酸ブチルの代わりにJIS K 8903に規定する4-メチル-2-ペンタノンを用い
てもよい。
6. アセチレン-空気フレームでは,多燃料フレームにすると感度は高くなるが,鉄,ニッケルな
ど共存物による妨害も大きくなる。この場合,硫酸ナトリウム,二硫酸カリウム又はふっ化
水素アンモニウム(二ふっ化水素アンモニウム)を1 %程度共存させるとよい。
アセチレン-一酸化二窒素フレームでは,妨害の大部分は抑制される。
65.1.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,クロムによる原子吸光を波長
357.9 nmで測定して全クロムを定量する。
定量範囲:Cr 5〜100 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 7. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) クロム標準液(Cr 1 μg/mL) 58.4 a) 4)のクロム標準液(Cr 10 μg/mL)10 mLを全量フラスコ100 mL
272
K 0102:2016
にとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5. (1)又は備考4.によって処理する。ただし,最終溶液は0.1〜1 mol/Lの硝酸酸性とする。
d) 操作 操作は,次による。
1) 52.3 d) 1)の操作を行う。ただし,灰化温度は500〜600 ℃,原子化(10)温度は2 400〜2 900 ℃(5〜
10秒間),波長は357.9 nmを用いる。
2) 空試験として52.3 d) 2)の操作を行う。
3) 検量線からクロムの量を求め,試料中の全クロムの濃度(Cr μg/L)を算出する。
注(10) 52.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) クロム標準液(Cr 1 μg/mL)0.5〜10 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試料
と同じ酸の濃度になるように硝酸(1+1)を加えた後,水を標線まで加える。この溶液についてd)
1)の操作を行う。
2) 52.3 e) 2)及び3)の操作を行い,クロム(Cr)の量と指示値との関係線を作成する。検量線の作成は,
試料の測定時に行う。
65.1.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ク
ロムによる発光を波長206.149 nmで測定して全クロムを定量する。この方法によって,表58.1に示す元
素が同時定量できる。それぞれの元素ごとの定量範囲,繰返し精度などの例を,表58.1に示す。
a) 試薬 試薬は,58.4 a)による。
b) 装置 装置は,52.4 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5. (1) 又は備考4.によって処理する。
d) 操作 操作は,58.4 d)による。
e) 検量線 検量線は,58.4 e)による。
備考 8. 備考5.に準じて抽出操作し,有機層をプラズマ中に噴霧してもよい。この場合,プラズマト
ーチとしては,有機溶媒用のトーチを用いる。
65.1.5 ICP質量分析法 52.5による。
65.2 クロム(VI)[Cr(VI)] クロム(VI)の定量には,ジフェニルカルバジド吸光光度法,フレーム
原子吸光法,電気加熱原子吸光法,ICP発光分光分析法,ICP質量分析法又はジフェニルカルバジド発色
による流れ分析法を適用する。
なお,ジフェニルカルバジド吸光光度法は,1994年に第1版として発行されたISO 11083,ICP発光分
光分析法は,1996年に第1版として発行されたISO 11885,ジフェニルカルバジド発色流れ分析法は2006
年に第1版として発行されたISO 23913との整合を図ったものである。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11083:1994,Water quality−Determination of chromium (VI)−Spectrometric method using 1,5-
diphenylcarbazide(MOD)
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
273
K 0102:2016
emission spectroscopy(MOD)
ISO 23913:2006,Water quality−Determination of chromium (VI)−Method using flow analysis (FIA
and CFA) and spectrometric detection(MOD)
65.2.1 ジフェニルカルバジド吸光光度法 試料に1,5-ジフェニルカルボノヒドラジド(ジフェニルカルバ
ジド)を加え,生成する赤紫の錯体の吸光度を測定してクロム(VI)を定量する。
定量範囲:Cr(VI)2〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 硫酸(1+9) 水9容をビーカーにとり,これを冷却し,かき混ぜながらJIS K 8951に規定する硫
酸1容を徐々に加える。
2) エタノール(95) JIS K 8102に規定するもの。
3) ジフェニルカルバジド溶液(10 g/L) 65.1.1 a) 5)による。
4) クロム(VI)標準液[Cr(VI)2 μg/mL] 65.1.1 a) 7)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料の適量[Cr(VI)として2〜50 μgを含む。(例えば,25 mL)]を2個のビーカー(A),(B)に
とり,試料が酸性の場合には,水酸化ナトリウム溶液(40 g/L)[21. a) 3)による。]で,また,アル
カリ性の場合は,硫酸(1+35)[43.2.1 a) 3)による。]で中和する。
2) ビーカー(A)の溶液は,全量フラスコ50 mL(A)に移し入れ,硫酸(1+9)2.5 mLを加える。
3) ビーカー(B)の溶液に硫酸(1+9)2.5 mLを加え,次にエタノール(95)を少量加え,煮沸して
クロム(VI)をクロム(III)に還元し,過剰のエタノールを追い出す。放冷後,全量フラスコ50 mL
(B)に移し入れる。
4) 全量フラスコ(A)及び(B)を約15 ℃に保ち,それぞれにジフェニルカルバジド溶液(10 g/L)1
mLずつを加え,直ちに振り混ぜ,水を標線まで加え,約5分間放置する。
5) 全量フラスコ(A)の一部を吸収セルに移し,全量フラスコ(B)の溶液を対照液として波長540 nm
付近の吸光度を測定する。
6) 検量線からクロム(VI)の量を求め,試料中のクロム(VI)の濃度[Cr(VI)mg/L]を算出する。
d) 検量線 検量線の作成は,次による。
1) クロム(VI)標準液[Cr(VI)2 μg/mL]1〜25 mLを段階的にとり,c) 2)〜4)の操作における全量
フラスコ(A)に対するのと同じ操作を行う。
2) この溶液の一部を吸収セルに移し,水約30 mLについてc) 3)及び4)の操作における全量フラスコ(B)
に対するのと同じ操作を行った溶液を対照液とし,波長540 nm付近の吸光度を測定し,クロム[Cr
(VI)]の量と吸光度との関係線を作成する。
備考 9. 試料を酸性にしたとき,クロム(VI)を還元する物質が共存する場合は,c)の操作では定量
は困難である。ただし,クロム(III)が含まれていない試料は65.1によって定量する。また,
妨害物質には,備考2.に示すもの以外に次のものがある。
− 鉛,バリウム及び銀イオン(塩類)の存在下で,クロム(VI)は,難溶性のクロム酸塩
を生じ,それらに含まれるクロム(VI)は定量されない。
− モリブデン(VI)及び水銀塩類は,試薬によってそれぞれ黄色及び青となるが,吸収強
度はクロム(VI)に比べて著しく低い。鉄(III)は1 mg/Lを超えると黄色となる。また,
274
K 0102:2016
バナジウムは黄色になるが,やがて退色する。
− 妨害金属イオンは,りん酸緩衝液(JIS K 9017に規定するりん酸水素二カリウム348 g
を水1 000 mLに溶かし,pHを調節し,pH9.0±0.2とする。)の存在下で,沈殿助剤の硫
酸アルミニウム溶液(硫酸アルミニウム・18水247 gを水に溶かし,1 Lとする。)を試
料1 Lについて1 mL用いて沈殿させ,ろ別して除く。このとき,クロム(III)も沈殿除
去できる。
− 酸化性又は還元性の物質によるクロムの原子価の変化は,次の前処理によって避けられ
る。
酸化性物質は,中性とした試料に亜硫酸塩を添加して還元する。クロム(VI)はこの
条件下では反応しない。過剰の亜硫酸塩及び他の還元性物質は,次いで次亜塩素酸塩に
よって酸化する。過剰の次亜塩素酸塩及び生成したクロロアミンは,酸性下で塩化ナト
リウムによって分解し,生成した塩素は,空気で追い出す。
− アンモニア体窒素は,500 mg/L未満では妨害しない。しかし,アミン化合物は次亜塩素
酸塩によってクロロアミンに変化し,塩化物イオンの添加によって,必ずしも分解しな
い。この妨害は,1,5-ジフェニルカルバジドの添加時に黄色又は茶色を帯びた色の出現
によって分かる。
− 亜硝酸体窒素は,20 mg/Lを超えると赤紫のクロム(VI)-1,5-ジフェニルカルバゾン錯
体を生成して妨害する。
備考 10. 備考2.による。
65.2.2 フレーム原子吸光法 試料を前処理した後,アセチレン-空気などのフレーム中に噴霧し,クロム
による原子吸光を波長357.9 nmで測定してクロム(VI)を定量する。
定量範囲:Cr(VI) 0.2〜5 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) クロム標準液(Cr 10 μg/mL) 58.4 a) 4)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料の適量(懸濁物を含む場合には,ろ紙5種C又は孔径0.45 μmのろ過材でろ過し,最初のろ液
約50 mLを捨て,その後のろ液を用いる。)をとり,JIS K 8180に規定する塩酸又はJIS K 8541に
規定する硝酸を加えて0.1〜1 mol/Lの酸性溶液とする。ただし,試料中にクロム(III)が含まれて
いる場合には,備考11.のb)によって操作する。
備考 11. クロム(VI)の濃度が低い試料で妨害物質を含まない場合には,次のように操作する。
a) 試料中にクロム(III)が含まれていないときは,備考4.又は備考5.に準じて操作する。
b) 試料中にクロム(III)が含まれるときは,次による。
1) 試料の適量(500 mL以下)をとり,硫酸アンモニウム鉄(III)溶液[JIS K 8982に規
定する硫酸アンモニウム鉄(III)・12 水 5 gを硫酸(1+1)[5.4 a) 2)による。]1 mL
に溶かし,水で100 mLにする。]1 mLを加えてかき混ぜる。
2) アンモニア水(1+4)[65.の注(2)による。]を加えて微アルカリ性とした後,アンモニ
ア臭がほとんどなくなるまで静かに煮沸する。沸騰近くの温度に保って沈殿を熟成さ
せた後,ろ紙5種Aでろ過し,温硝酸アンモニウム溶液(10 g/L)[65.の注(2)による。]
で洗浄する。
275
K 0102:2016
3) ろ液と洗液とを合わせ,塩酸又は硝酸を加えて0.1〜1 mol/Lの酸性溶液とする。
d) 操作 操作は,65.1.2 d)に準じて操作する。
e) 検量線 検量線の作成は,次による。
1) 65.1.2 e)に準じて検量線を作成する。
65.2.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,クロムによる原子吸光を波長
357.9 nmで測定して,クロム(VI)を定量する。
定量範囲:Cr(VI) 5〜100 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 12. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) クロム標準液(Cr 1 μg/mL) 58.4 a) 4)のクロム標準液(Cr 10 μg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
1) 試料の適量をとり,65.2.2 c) 1)又は備考11.によって処理する。ただし,硝酸を用い,最終溶液は,
0.1〜1 mol/Lの硝酸酸性溶液とする。
d) 操作 操作は,65.1.3 d)に準じて操作する。
e) 検量線 検量線の作成は,次による。
1) 65.1.3 e)に準じて検量線を作成する。
65.2.4 ICP発光分光分析法 試料を前処理した後,試料導入部を通して高周波プラズマ中に噴霧し,クロ
ムによる発光を波長206.149 nmで測定してクロム(VI)を定量する。この方法によって表58.1に示す元
素が同時定量できる。それぞれの元素ごとの定量範囲,繰返し精度などの例を,表58.1に示す。
a) 試薬 試薬は,58.4 a)による。
b) 装置 装置は,52.4 b)による。
c) 準備操作 操作は,次による。
1) 試料の適量をとり,65.2.2 c) 1)又は備考11.によって処理する。ただし,硝酸を用い,最終溶液は,
0.1〜0.5 mol/Lの硝酸酸性溶液とする。
d) 操作 操作は,58.4 d)に準じて操作する。
e) 検量線 検量線の作成は,58.4 e)による。
65.2.5 ICP質量分析法 試料を前処理した後,内標準物質を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,クロム及び内標準物質のそれぞれの質量/荷電数におけるイオンの電流を測定し,クロムのイ
オンの電流と内標準物質のイオンの電流との比を求めてクロム(VI)を定量する。この方法によって表52.2
に示す元素が同時定量できる。それぞれの元素ごとの定量範囲,繰返し精度などの例を,表52.2に示す。
a) 試薬 試薬は,52.5 a)による。
b) 装置 装置は,52.5 b)による。
備考 13. 52.の備考13.による。
14. 52.の備考14.による。
c) 準備操作 準備操作は,次による。
1) 試料の適量をとり,65.2.2 c) 1)又は備考11.によって処理する。ただし,硝酸を用い,最終溶液は,
276
K 0102:2016
0.1〜0.5 mol/Lの硝酸酸性溶液とする。
2) 52.5 c) 2)による。
d) 操作 操作は,52.5 d)による。
e) 検量線 検量線の作成は,52.5 e)による。
65.2.6 流れ分析法 試料中のクロム(VI)を,65.2.1と同様な原理で発色させる流れ分析法によって定量
する。
定量範囲:Cr(VI) 5 μg/L〜5 mg/L
試験操作などは,JIS K 0170-7による。
66. 水銀(Hg) 全水銀とアルキル水銀とに区分する。
66.1 全水銀 全水銀の定量には,還元気化原子吸光法,加熱気化原子吸光法又は加熱気化−金アマルガ
ム捕集原子吸光法を適用する。
66.1.1 還元気化原子吸光法 試料を過マンガン酸カリウムで前処理した後,塩化すず(II)で水銀(II)
を還元する。この溶液に通気して発生する水銀蒸気による原子吸光を波長253.7 nmで測定し,水銀を定量
する。
定量範囲:Hg 0.5〜10 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 硝酸 JIS K 8541に規定する微量金属測定用のもの。
2) 硫酸(1+1) 5.4 a) 2)による。ただし,硫酸は水銀の含有量が1 μg/L以下のものを用いる。
3) 過マンガン酸カリウム溶液(50 g/L) JIS K 8247に規定する過マンガン酸カリウム(1) 50 gを水に溶
かしてガラスろ過器(17G4又は25G4)でろ過した後,水で1 Lとする。着色ガラス瓶に保存する。
4) ペルオキソ二硫酸カリウム溶液(50 g/L) JIS K 8253に規定するペルオキソ二硫酸カリウム(特級)
50 gを水に溶かして1 Lとする(2)。
5) 塩化ヒドロキシルアンモニウム溶液(80 g/L) JIS K 8201に規定する塩化ヒドロキシルアンモニウ
ム8 gを水に溶かして100 mLとする。この溶液の水銀の含有量は1 μg/L以下とする。精製の必要
がある場合には,この溶液を分液漏斗に移し,ジチゾン-クロロホルム溶液(50 mg/L)[JIS K 8490
に規定するジチゾン5 mgをJIS K 8322に規定するクロロホルム100 mLに溶かす。ただし,この溶
液中の水銀の含有量は0.5 μg/L以下とする。]を少量加えて振り混ぜて放置した後,クロロホルム層
を捨てる。この操作をクロロホルム層が変色しなくなるまで繰り返す。水層を乾いたろ紙でろ過し
クロロホルムの小滴を除く。
6) 塩化すず(II)溶液 JIS K 8136に規定する塩化すず(II)二水和物(水銀分析用)10 gに硫酸(1
+20)[JIS K 8951に規定する硫酸(水銀の含有量が1 μg/L以下のもの。)を用いて調製する。]60 mL
を加え,かき混ぜながら加熱して溶かす。放冷後,水を加えて100 mLにする。この溶液の水銀の
含有量は1 μg/L以下とする。精製の必要がある場合には,JIS K 1107に規定する窒素2級を通気す
る。1週間以上経過したものは使用しない。
7) L-システイン溶液(10 mg/L) L-システイン(水銀含有量が1 mg/kg以下のもの)10 mgに水及び
硝酸2 mLを加えて溶かした後,水で1 Lとする。
8) 水銀標準液(Hg 0.5 mg/mL) JIS K 8139に規定する塩化水銀(II)0.339 gをとり,少量の水に溶
かし,全量フラスコ500 mLに移し入れ,硝酸(1+1)[1)の硝酸を用いて調製する。]5 mLを加え
た後,水を標線まで加える。ほうけい酸ガラス瓶に保存する。
277
K 0102:2016
9) 水銀標準液(Hg 10 μg/mL) 水銀標準液(Hg 0.5 mg/mL)10 mLを全量フラスコ500 mLにとり,
L-システイン溶液(10 mg/L)を標線まで加える。ほうけい酸ガラス瓶に保存する。1か月間以上経
過したものは使用しない。
10) 水銀標準液(Hg 0.1 μg/mL) 水銀標準液(Hg 10 μg/mL)10 mLを全量フラスコ1 000 mLにとり,
L-システイン溶液(10 mg/L)を標線まで加える。使用時に調製する。
11) 水銀標準液(Hg 0.01 μg/mL) 水銀標準液(Hg 0.1 μg/mL)10 mLを全量フラスコ100 mLにとり,
L-システイン溶液(10 mg/L)を標線まで加える。使用時に調製する。
注(1) 原子吸光分析用試薬など,水銀含有量の少ないものを用いる。
(2) JIS K 8252に規定するペルオキソ二硫酸アンモニウムを用いてもよい。いずれも溶液中の水銀
は1 μg/L以下とする。
b) 器具及び装置 器具及び装置は,次による。
1) 原子吸光分析装置又は水銀専用原子吸光装置
2) 水銀還元気化装置(3) 原子吸光分析装置と併用する。
3) 光源 水銀測定用の光源を用いる。
4) ピストン式ピペット 10〜100 μL及び100〜1 000 μLが分取できるもの。JIS K 0970による。
注(3) 還元容器,吸収セル,空気ポンプ,流量計,乾燥管及び連結管から構成する。図66.1及び図66.2
に構成例を示す。
なお,各構成部分の詳細は,次のとおりである。
− 還元容器 ガラス瓶(又は三角フラスコ)300〜350 mL(250 mLの位置に印を付けておく。)
又は図66.3に示すような分液漏斗形(300 mL)のものを用いる。
− 吸収セル 長さ100〜300 mm程度の石英ガラス製のもの,又はガラス製若しくはプラスチ
ック製(水銀蒸気を吸着しないもの。)で両端に石英ガラス窓を付けたもの。
− 空気ポンプ 送気能力0.5〜3 L/minをもつダイアフラムポンプ又はこれと同等の性能をも
つ空気ポンプ。水銀蒸気に接する部分が金属製の場合は,コロジオンなどを塗布しておく。
− 流量計 流量0.5〜5 L/minが測定できるもの。
− 乾燥管 乾燥管又はU字管。JIS K 8228に規定する過塩素酸マグネシウム(乾燥用),JIS
K 8124に規定する塩化カルシウム(乾燥用)などの乾燥剤を充塡しておくか,又はコール
ドトラップで代用してもよい。吸収セルの部分に小形電球を点灯するなどして吸収セル内
の温度が周囲の温度よりも約10 ℃高くなるようにしておけば,乾燥管を用いなくてもよ
い。
− 連結管 軟質塩化ビニル管
278
K 0102:2016
A:
B:
C:
D:
E:
F:
G:
H:
I:
還元容器
乾燥管
流量計
吸収セル
空気ポンプ
記録計
水銀測定用の光源
原子吸光用検出器
水銀除去装置
※矢印は気体の流れ
方向を示す。
図66.1 密閉循環方式の構成の例
A:
B:
C:
D:
E:
F:
G:
H:
I:
還元容器
乾燥管
流量計
吸収セル
空気ポンプ
記録計
水銀測定用の光源
原子吸光用検出器
水銀除去装置
※矢印は気体の流れ
方向を示す
図66.2 開放送気方式の構成の例
279
K 0102:2016
単位 mm
図66.3 還元容器の例
c) 操作 操作は,次による。
1) 試料(4)の適量(Hgとして0.075〜1.5 μgを含む。)を三角フラスコ300 mL(5)にとり,水を加えて約
150 mLとする(6)。
2) 硫酸(1+1)20 mL,硝酸5 mL及び過マンガン酸カリウム溶液(50 g/L)20 mL(7)を加えて振り混
ぜ,約15分間放置する。
3) 過マンガン酸の色が消えたときは,溶液の赤い色が約15分間持続するまで,過マンガン酸カリウム
溶液(50 g/L)を少量ずつ加える。
4) ペルオキソ二硫酸カリウム溶液(50 g/L)10 mLを加え,約95 ℃の水浴中に三角フラスコ300 mL
を浸して約2時間加熱する。
5) 室温まで冷却し,塩化ヒドロキシルアンモニウム溶液(80 g/L)10 mLを添加して過剰の過マンガン
酸を還元する。
6) 直ちに溶液を還元容器に移し(8) 水で250 mLとした後,通気回路を組み立てる。
7) 手早く塩化すず(II)溶液10 mLを加え,あらかじめ設定した最適流量(9)で空気ポンプを作動し,
空気を循環(10)させる。
8) 波長253.7 nmの指示値(11)を読み取る。
9) バイパスコック(12)を回して,指示値が元に戻るまで通気を続ける。
10) 空試験として試料と同量の水をとり,2)〜4)と同量の試薬を加えた後,5)〜9)の操作を行って指示値
を読み取り,試料について得た指示値を補正する。
11) 検量線から水銀の量を求め,試料中の全水銀の濃度(Hg μg/L)を算出する。
280
K 0102:2016
注(4) 有機物その他の妨害物質が含まれていない試料については1)〜5)を省略し,試料を直接還元容
器にとり,硫酸(1+1)20 mLを加えた後,6)〜11)の操作を行ってもよい。この場合は,検量
線も同様に作成する。
(5) 還元容器用の三角フラスコ又はガラス瓶を用いてもよい。
(6) よう素を0.1 mg/L以上含む場合は負の干渉を与えるため,試料を適宜希釈して使用する。
(7) 有機物の少ない試料では,適宜,減量する。
(8) 分解に還元容器を用いた場合は,そのまま連結する。
(9) 最適流量は装置によって異なるので,あらかじめ求めておくが,通常は1〜1.5 L/minである。
(10) 開放送気方式の場合は,還元容器の通気管にコックを付け,塩化すず(II)溶液を添加後,約2
分間激しく振り混ぜた後,装置に連結し,ポンプの作動と同時にコックを開く。
(11) 吸光度又はその比例値。開放送気方式の場合は,ピーク高さ又はピーク面積を測定する。
(12) 過マンガン酸カリウム溶液(50 g/L)5容に,硫酸(1+4)(JIS K 8951に規定する硫酸を用い
て調製する。)1容の割合で加えた溶液を入れたガス洗浄瓶を通して,水銀を除去する。開放送
気方式の場合は除去後,大気中に放出する。
d) 検量線 検量線の作成は,次による。
1) 水銀標準液(Hg 0.1 μg/mL)1〜20 mLを還元容器300 mLに段階的にとり,硫酸(1+1)20 mL及
び水を加えて250 mLとした後,通気回路を組み立てc) 7)〜9)の操作を行う。
2) 別に,空試験として水230 mLを還元容器300 mLにとり,硫酸(1+1)20 mLを加えた後,通気回
路を組み立て,c) 7)〜9)の操作を行って水銀標準液について得た指示値を補正し,水銀(Hg)の量
と指示値との関係線を作成する。検量線の作成は,試料測定時に行う。
備考 1. 塩化物イオンを多量に含む試料では,過マンガン酸カリウム処理においては塩化物イオンが
酸化されて塩素となり,波長253.7 nmの光を吸収して正の誤差を生じる。この場合は,塩化
ヒドロキシルアンモニウム溶液(80 g/L)を過剰に加え,塩素を十分に還元しておく。還元
容器中に存在する塩素は,窒素などの送入によってあらかじめ追い出しておく。
2. ベンゼン,アセトンなどは波長253.7 nmの光を吸収して正の誤差を生じる。この種の揮発性
有機物を含む試料に対しては,過マンガン酸カリウムによる前処理を行った後,次のいずれ
かの操作を適用する。
a) 少量のヘキサンと振り混ぜて揮発性有機物を抽出除去する。
b) 重水素ランプなどによるバックグラウンド補正を行う。
c) 水銀測定用の光源と重水素ランプを用いて指示値の差を求めておき,次に,塩化すず(II)
溶液の添加を省略して同様の測定を行い,両指示値の差として水銀を定量する。
d) 水銀をジチゾン錯体として抽出分離した後,加熱気化法によって測定する。
3. 定量範囲の下限(0.5 μg/L)以下の測定が必要な場合は,高感度の水銀専用原子吸光装置を用
いて,次の操作によって定量する。この場合の定量範囲は,0.05〜0.5 μg/Lとする。測定は,
次の操作で行う。
1) 試料(*1)の適量(Hgとして0.25〜2.5 ngを含む。)を分解試験管10 mLにとり,水を加え
て,5 mL(*2) (*3)とする。
2) 硫酸(1+1)0.7 mL,硝酸0.2 mL及び過マンガン酸カリウム溶液(50 g/L)0.7 mL(*4)
を加えかくはんし,約15分間放置する。
3) 過マンガン酸の色が消えたときは,溶液の赤い色が約15分間持続するまで,過マンガン
281
K 0102:2016
酸カリウム溶液(50 g/L)を少量ずつ加える。
4) ペルオキソ二硫酸カリウム溶液(50 g/L)を0.3 mLを加え,約95 ℃の水浴中に分解試
験管を浸して約2時間加熱する。
5) 室温まで冷却し,塩化ヒドロキシルアンモニウム溶液(80 g/L)を0.4 mLを添加して過
剰の過マンガン酸を還元する。
6) これに,手早く塩化すず(II)溶液0.4 mLを加え,あらかじめ設定した最適流量(*5)で空
気ポンプを作動し,空気を循環(*6)させる。
7) 波長253.7 nmの指示値(*7)を読み取る。
8) 空試験として試料と同量の水をとり,2)〜7)の操作を行って指示値を読み取り,試料に
ついて得た指示値を補正する。
9) 検量線から水銀の量を求め,試料中の全水銀の濃度(Hg μg/L)を算出する。
10) 検量線の作成は,次による。
a) 11)の水銀標準液をピストン式ピペットを用いて分解試験管に段階的に取り,硫酸(1
+1)0.7 mL及び水を加えて5 mLとした後,6)及び7)の操作を行う。別に空試験として
硫酸(1+1)0.7 mL に水を加えて5 mLとした後,6)及び7)の操作を行って水銀標準液
について得た指示値を補正し,水銀(Hg)の量と指示値との関係線を作成する。検量線
の作成は,試料測定時に行う。
注(*1) 有機物その他の妨害物質が含まれていない試料については1)〜5)を省略し,試料を分解試験管
にとり,硫酸(1+1)0.7 mLを加えた後,6) 及び7) の操作を行ってもよい。この場合は,検
量線も同様に作成する。
(*2) 分解試験管に50 mLのものを用いた場合は,20 mLとする。この場合の2)以降の操作における
試薬の添加量は4倍とする。この場合は,検量線も同様に作成する。
(*3) 注(6)による。
(*4) 注(7)による。
(*5) 最適送気流量は,あらかじめ求めておくが,通常は0.1〜1.0 L/minである。
(*6) 注(10)による。
(*7) 注(11)による。
66.1.2 加熱気化原子吸光法 試料を過マンガン酸カリウムで前処理した後,硫酸酸性溶液から水銀をジチ
ゾン錯体として有機溶媒に抽出する。有機溶媒を揮散させた後,残留物を加熱して水銀蒸気を発生させ,
その原子吸光を波長253.7 nmで測定して,水銀を定量する。
定量範囲:Hg 0.5〜10 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 硝酸 66.1.1 a) 1) による。
2) 硫酸(1+1) 66.1.1 a) 2) による。
3) 過マンガン酸カリウム溶液(50 g/L) 66.1.1 a) 3) による。
4) ペルオキソ二硫酸カリウム溶液(50 g/L) 66.1.1 a) 4) による。
5) 塩化ヒドロキシルアンモニウム溶液(80 g/L) 66.1.1 a) 5) による。
6) ジチゾン-クロロホルム溶液(0.1 g/L) JIS K 8490に規定するジチゾン10 mgをJIS K 8322に規定
するクロロホルムに溶かして100 mLとする。ただし,溶液中の水銀の含有量は1 μg/L以下とする。
7) 2,3-ジメルカプト-1-プロパノール溶液(体積百分率 0.1 %) 2,3-ジメルカプト-1-プロパノール0.1 mL
282
K 0102:2016
をJIS K 8322に規定するクロロホルムに溶かして10 mLとする。使用時にクロロホルムで10倍に
薄める。
8) 水銀標準液(Hg 0.1 μg/mL) 66.1.1 a)10) による。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 500 mL
2) 原子吸光分析装置又は水銀専用原子吸光装置
3) 水銀加熱気化装置 原子吸光分析装置と併用する。図66.4に加熱気化法の装置の構成の例を示す。
4) 光源 水銀測定用の光源を用いる。
A:
B:
C:
D:
E:
F:
G:
H:
I:
J:
K:
加熱管
乾燥管
流量計
吸収セル
空気ポンプ
バーナー
水銀測定用の光源
原子吸光用検出器
水銀除去装置
記録計
磁器ボート
図66.4 加熱気化方式の構成の例
c) 操作 操作は,次による。
1) 試料(4)の適量(Hgとして0.075〜1.5 μgを含む。)を三角フラスコ300 mLにとり,水を加えて約150
mLとする。
2) 66.1.1 c) 2)〜5)の操作を行う。
3) この溶液を分液漏斗500 mLに移し,ジチゾン-クロロホルム溶液(0.1 g/L)5 mLを加えて約2分間
激しく振り混ぜた後,放置する。
4) クロロホルム層を分離し試験管に移す。
5) 水層に再びジチゾン-クロロホルム溶液(0.1 g/L)5 mLを加えて約2分間激しく振り混ぜた後,放
置する。
6) クロロホルム層を先の試験管に合わせ,これに水5 mLを加えて振り混ぜた後,放置する。
7) 水層をスポイトで取り除く。
8) このクロロホルム溶液の一定量(2.5 mL以下)を磁器ボートに移す。
9) 2,3-ジメルカプト-1-プロパノール溶液(体積百分率 0.1 %)0.1 mLを加え,通気してクロロホルム
を揮散させる。
10) 磁器ボートを水銀加熱気化装置の加熱管に挿入し,吸引(13)と加熱とを同時に開始する。
11) 波長253.7 nmの指示値(11)を読み取る。
283
K 0102:2016
12) 空試験として試料と同量の水をとり,2)と同量の試薬を加えた後,3)〜11)の操作を行って試料につ
いて得た指示値を補正する。
13) 検量線から水銀の量を求め,試料中の全水銀の濃度(Hg μg/L)を算出する。
注(13) 最適な吸引量を求めておく。通常は1〜1.5 L/minである。
d) 検量線 検量線の作成は,次による。
1) 水銀標準液(Hg 0.1 μg/mL)1〜20 mLを分液漏斗500 mLに段階的にとり,硫酸(1+1)20 mLを
加え,水で200 mLとした後,c) 3)〜11) の操作を行う。
2) 別に,空試験として水200 mL及び硫酸(1+1)20 mLを分液漏斗500 mLにとり,c) 3)〜11) の操
作を行って水銀標準液について得た指示値を補正し,水銀(Hg)の量と指示値との関係線を作成す
る。検量線の作成は,試料測定時に行う。
66.1.3 加熱気化−金アマルガム捕集原子吸光法 試料を加熱燃焼させ,気化した水銀を捕集管に捕集し,
捕集剤を再度加熱して水銀を気化させる。その原子吸光を波長253.7 nmで測定して,水銀を定量する。
定量範囲:Hg 0.5〜10 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水銀標準液(Hg 10 μg/mL) 66.1.1 a) 9)による。
2) 水銀標準液(Hg 0.1 μg/mL) 66.1.1 a)10)による。
3) 水銀標準液(Hg 0.01 μg/mL) 66.1.1 a)11)による。
b) 器具及び装置 器具及び装置は,次による。
1) 加熱気化−金アマルガム捕集原子吸光分析装置
2) 水銀加熱気化装置(14) 原子吸光分析装置と併用する。
なお,図66.5に金アマルガム捕集管を用いる加熱気化法の装置の構成の例を示す。
3) 光源 水銀測定用の光源を用いる。
4) ピストン式ピペット 66.1.1b) 4)による。
注(14) 試料加熱燃焼管,燃焼ガス処理部,水銀捕集管,吸収セルなどから構成する。
− 試料加熱燃焼管 セラミック又は石英製の管状のもの。
− 燃焼ガス処理部 試料を加熱燃焼したときに発生する干渉ガス成分を湿式又は乾式で吸収
除去するもの。
− 金アマルガム捕集管 珪藻土等の融点600 ℃以上,粒径100〜1 000 μmの耐熱性多孔質粒
子を担持体とし,これに塩化金酸(III)水溶液又は粒子径10〜500 nmの金ナノ粒子分散液
を含浸,乾燥させた後,空気を流しながら約800 ℃で約30分間焼成したものを詰めたも
の。
− 吸収セル 石英ガラス製のもの,又はガラス製若しくはプラスチック製(水銀蒸気を吸着
しないもの。)で両端に石英ガラス窓を付けたもの。
284
K 0102:2016
A:
B:
C:
D:
E:
F:
G:
H:
I:
J:
ガス流量調整器
試料加熱燃焼管
燃焼ガス処理部
金アマルガム捕集管
吸収セル
水銀測定用の光源
原子吸光用検出器
水銀検出部
水銀回収部
試料ボート
図66.5 金アマルガム捕集管を用いる加熱気化方式の構成の例
c) 操作 操作は,次による。
1) 試料200 μL(Hgとして0.1〜2 ngを含む)をピストン式ピペットを用いて,試料ボート(15)に加える。
懸濁物を含む場合は,別途,試料5 mL以上を三角フラスコに採取し,ホモジナイザーによる均一
化又は酸分解による均一化を行った後に試験に用いる。
2) 水銀加熱気化装置にキャリヤーガス(16)を最適流速で流しながら,試料ボートを水銀加熱気化装置の
試料加熱燃焼管に挿入し,500〜900 ℃(装置により最適温度は異なる)で加熱分解して,気化した
水銀を金アマルガム捕集管に捕集する。
3) 金アマルガム捕集管を600〜950 ℃(装置により最適温度は異なる)で加熱して水銀を再び気化さ
せ,波長253.7 nmの指示値(11)を読み取る。
4) 空試験として試料と同量の水をとり,ホモジナイザーによる均一化又は酸分解による均一化を行っ
た場合には,同様の操作を行った後,2)及び3)の操作を行って指示値を読み取り,試料について得
た指示値を補正する。
5) 検量線から水銀の量を求め,試料中の全水銀の濃度(Hg μg/L)を算出する。ただし,3回以上の繰
返し測定の変動係数が10 %以内であることを確認する(17)(18)。
注(15) 硫黄,ハロゲン化合物などを含む試料で,加熱分解時にSO2,ハロゲンガスなど干渉成分の発
生が考えられる場合には,750 ℃,3時間以上加熱処理をした活性アルミナ,炭酸ナトリウム,
水酸化カルシウム等を単独又は混合したものをあらかじめ添加した試料ボードを用いてもよい。
(16) キャリヤーガスは,試料加熱燃焼管に到る流路の途中に金アマルガム捕集管などを設置するこ
とによって水銀を除去した空気又は酸素を用いる。最適送気流量は,あらかじめ求めておくが,
通常は0.1〜1.0 L/minである。
(17) 繰返し試験の結果から求めた変動係数が10 %以上であった場合は,試験をやり直す。
(18) 懸濁物又は溶存物質を多量に含む試料,並びに共存物質が未知の試料に対しては,試料5 mL
を三角フラスコに採取し,水銀標準液0.01 μg/mLを5 mL添加して1)〜3)の操作を行って,添
加回収試験を行い,回収率が90〜110 %の範囲にあることを確認してから試験を行う。
d) 検量線 検量線の作成は,次による。
1) 水銀標準液(Hg 0.01 μg/mL)の適量10〜200 μLをピストン式ピペットを用いて,段階的に試料ボ
ートにとり,水を加えて200 μLとした後,c) 2)及び3)の操作を行う。
285
K 0102:2016
2) 別に,空試験として水200 μLを試料ボートにとり,c) 2)及び3) の操作を行って水銀標準液につい
て得た指示値を補正し,水銀(Hg)の量と指示値との関係線を作成する。検量線の作成は,試料測
定時に行う。
66.2 アルキル水銀(II)化合物 アルキル水銀(II)化合物の定量は,アルキル水銀(II)化合物のうち,
エチル水銀(II)化合物及びメチル水銀(II)化合物を対象とし,水銀の量で表示する。定量にはガスクロ
マトグラフ法を適用する。
66.2.1 ガスクロマトグラフ法 アルキル水銀(II)化合物をトルエンで抽出し,L-2-アミノ-3-メルカプト
プロピオン酸(L-システイン)によって選択的に逆抽出した後,トルエンで再び抽出し,ガスクロマトグ
ラフ法を用いて定量する。
定量範囲:Hg 0.5 μg/L以上(試料換算)
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。ただし,予期保持時間付近にピークを生じないもの。
2) 塩酸(1+1) 1)の塩酸を用いて調製する。
3) アンモニア水 JIS K 8085に規定するもの。ただし,予期保持時間付近にピークを生じないもの。
4) アンモニア水(1+1) 3)のアンモニア水を用いて調製する。
5) 塩化ナトリウム溶液(200 g/L) JIS K 8150に規定する塩化ナトリウム200 gを水に溶かして1 Lと
する。予期保持時間付近にピークを生じないもの。
6) トルエン JIS K 8680に規定するもの。ただし,予期保持時間付近にピークを生じないもの。
7) L-システイン-酢酸ナトリウム混合溶液 JIS K 8470に規定するL-システイン塩酸塩一水和物1 g,
JIS K 8371に規定する酢酸ナトリウム三水和物0.8 g及びJIS K 8987に規定する硫酸ナトリウム12.8
gを水に溶かして100 mLとする。予期保持時間付近にピークを生じないもの。
8) 塩化エチル水銀標準液(Hg 10 mg/mL)又は塩化メチル水銀液(Hg 10 mg/mL) クロロエチル水銀
(II)[塩化エチル水銀(II)]0.132 g又はクロロメチル水銀(II)[塩化メチル水銀(II)]0.125 gを
少量のトルエンに溶かし,全量フラスコ10 mLに移し入れ,トルエンを標線まで加える。
9) 塩化エチル水銀標準液(Hg 0.1 mg/mL)又は塩化メチル水銀標準液(Hg 0.1 mg/mL) 塩化エチル
水銀標準液(Hg 10 mg/mL)又は塩化メチル水銀標準液(Hg 10 mg/mL)1 mLを全量フラスコ100 mL
にとり,トルエンを標線まで加える。
10) 塩化エチル水銀標準液(Hg 1 μg/mL)又は塩化メチル水銀標準液(Hg 1 μg/mL) 塩化エチル水銀
標準液(Hg 0.1 mg/mL)又は塩化メチル水銀標準液(Hg 0.1 mg/mL)1 mLを全量フラスコ100 mL
にとり,トルエンを標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 50 mL及び500 mL。コックにワセリンなどの滑材を塗布しない。
2) 共栓試験管 5〜10 mL
3) マイクロシリンジ 1〜10 μL
4) ガスクロマトグラフ 次に挙げる条件を満たすもの。
4.1) 分離カラム ガラス製管(内径3 mm,長さ400〜1 500 mm)に4.2)のカラム充塡剤を詰めたもの
(19)。
4.2) カラム充塡剤 酸洗浄した後,シラン処理(20)を行った粒径180〜250 μmの耐火れんが(21)にエステ
ル系固定相液体5〜25 %を含浸させたもの。
4.3) 検出器 電子捕獲検出器又はこれと同等の性能をもつもの。
286
K 0102:2016
4.4) キャリヤーガス JIS K 1107に規定する窒素2級 流量30〜80 mL/min
4.5) 試料気化室湿度 140〜240 ℃
4.6) カラム槽温度 130〜180 ℃
4.7) 検出器槽温度 140〜200 ℃
4.8) 装置の感度 4.1)〜4.7)の条件下で塩化メチル水銀(又は塩化エチル水銀)を水銀(Hg)として40
ngを注入したときのS/N比が3以上とする。
注(19) キャピラリーカラムを用いてもよい。ただし,低濃度においてもアルキル水銀のハロゲン化物
の鋭いピークが得られるもの。キャリヤーガスはヘリウムを用い,流量は1〜3 mL/min程度と
する。
(20) ジクロロジメチルシランのトルエン溶液(体積百分率 1 %)中に担体を浸し,水浴上で約1時
間保った後,乾燥する。この処理をした担体が市販されている。また,あらかじめ担体に臭化
カリウム又は塩化ナトリウム(予期保持時間付近にピークを生じないもの。)5〜10 %を含浸さ
せた後,液層を被覆したものを用いると鋭いピークが得られる。
(21) 31.の注(4)による。
参考 カラム充塡剤には市販品がある。
c) 操作 操作は,次による。
1) 試料200 mLを分液漏斗500 mLにとり,中性でない場合は,アンモニア水又は塩酸で中和した後,
塩酸を加えて約2 mol/Lとする(22)。
2) トルエン50 mLを加え,約2分間激しく振り混ぜて放置した後,水層を別の分液漏斗500 mLに移
し,トルエン層は保存する。
3) 水層にトルエン50 mLを加えて約2分間激しく振り混ぜ,放置した後,水層を捨てる。
4) トルエン層を合わせ塩化ナトリウム溶液(200 g/L)20 mLを加え,約1分間激しく振り混ぜ,トル
エン層を洗浄し(23),放置後,水層を捨てる。
5) トルエン層にL-システイン-酢酸ナトリウム混合溶液8 mLを加え,約2分間激しく振り混ぜ,放置
後,水層を分液漏斗50 mLに移す。
6) 塩酸2 mLとトルエン5 mLとを加えて約2分間激しく振り混ぜ,放置後,水層を捨て,トルエン層
を共栓試験管に移す(24)。
7) マイクロシリンジを用い,トルエン層の一定量をガスクロマトグラフに注入し,ガスクロマトグラ
ムを記録する。
8) 塩化エチル水銀(II)又は塩化メチル水銀(II)の保持時間(25)に相当する位置のピークについて,
指示値(26)を読み取る(27)。
9) 先に測定に使用した共栓試験管内のトルエン層の残部1 mLを別の共栓試験管にとり,L-システイ
ン-酢酸ナトリウム混合溶液1 mLを加えて約2分間激しく振り混ぜ,放置する。
10) 上部のトルエン層から,先にガスクロマトグラフに注入したトルエン層と同量をマイクロシリンジ
を用いてガスクロマトグラフに注入する。この結果,先に得られたピークが消滅した場合には,先
のピークはエチル水銀(II)化合物又はメチル水銀(II)化合物によるものと判断する。
11) 空試験として水200 mLをとり,1)〜10)の操作を行って指示値を読み取り,試料について得た指示
値を補正する。
12) 検量線から水銀の量を求め,試料中のアルキル水銀(II)化合物の濃度を水銀の濃度(Hg μg/L)と
して算出する。
287
K 0102:2016
注(22) 試料中に硫化物イオン及びチオシアン酸イオンが含まれているときは,約2 mol/Lの塩酸酸性
とした試料にJIS K 8138に規定する塩化銅(I)の粉末100 mgを加えて十分にかき混ぜ,しば
らく放置する。沈殿をろ別し,ろ紙を塩酸(1+5)[a) 1)の塩酸を用いて調製する。]で2,3回
洗浄する。
注(23) 多量の無機水銀が存在する場合は,電子捕獲検出器を用いたとき,メチル水銀の位置に無機水
銀によるピークを生じることがあるので,洗浄を繰り返す。また,トルエン層に塩酸が残留す
るとL-システインによるアルキル水銀の逆抽出が不完全になるので,洗液が中性になるまで洗
浄を繰り返す。
(24) 水分が存在すると,ガスクロマトグラフに注入したとき異常ピークが生じることがあるので,
JIS K 8987に規定する硫酸ナトリウムなどを用いて脱水する。
(25) 操作において塩酸を使用するため,エチル水銀(II)化合物又はメチル水銀(II)化合物は,そ
れぞれ塩化エチル水銀(II)又は塩化メチル水銀(II)として挙動する。
(26) ピーク高さ又はピーク面積。
(27) 測定時に標準液の一定量を注入して,検出器の感度の経時変化を補正する。また,ガスクロマ
トグラフへの試料の注入量と得られる指示値との関係が直線になる範囲をあらかじめ求めてお
き,測定される指示値がこの範囲内となるように試料の注入量を調節する。
d) 検量線 検量線の作成は,次による。
1) 分液漏斗50 mLにL-システイン-酢酸ナトリウム混合溶液8 mLをとり,これに塩化メチル水銀標準
液(Hg 1 μg/mL)又は塩化エチル水銀標準液(Hg 1 μg/mL)を検出器の感度に応じて段階的に加え,
以下,c) 5)〜8)及び11)の操作を行い,塩化エチル水銀(II)又は塩化メチル水銀(II)に相当する
水銀(Hg)の量と指示値との関係線を作成する。
備考 4. 試料中にアルキル水銀(II)化合物のトルエン抽出を妨害する成分が含まれている場合には,
試料に一定量の塩化エチル水銀又は塩化メチル水銀標準液を加えてその回収率を求め,定量
値を補正する。
67. セレン(Se) セレンの定量には,3,3′-ジアミノベンジジン吸光光度法,水素化合物発生原子吸光法,
水素化合物発生ICP発光分光分析法又はICP質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
67.1 3,3'-ジアミノベンジジン吸光光度法 セレンを水酸化鉄(III)と共沈させて濃縮した後,3,3′-ジアミ
ノベンジジンを加えてセレン錯体を生成させる。溶液のpHを調節してトルエンで錯体を抽出し,その黄
色の吸光度を測定してセレンを定量する。
定量範囲:Se 2〜50 μg,繰返し精度:2〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
288
K 0102:2016
2) 塩酸(1+2) JIS K 8180に規定する塩酸を用いて調製する。
3) アンモニア水(1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
4) アンモニア水(1+2) JIS K 8085に規定するアンモニア水を用いて調製する。
5) 臭化カリウム JIS K 8506に規定するもの。
6) 硫酸アンモニウム鉄(III)溶液 JIS K 8982に規定する硫酸アンモニウム鉄(III)・12水9 gと硫
酸(1+1)[5.4 a) 2)による。]4 mLとを水に溶かして100 mLとする。
7) EDTA溶液(40 g/L) JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物
4.4 gを水に溶かして100 mLとする。
8) トルエン JIS K 8680に規定するもの。
9) ブロモチモールブルー溶液(1 g/L) 16.の注(1)による。
10) 3,3′-ジアミノベンジジン溶液(5 g/L) 四塩化3,3′-ジアミノベンジジニウム二水和物0.92 gを水に
溶かして100 mLとする。使用時に調製する。
11) セレン標準液(Se 0.2 mg/mL) JIS K 8598に規定するセレン(99.5 %以上)0.200 gをとり,硝酸(1
+1)[52.1 a) 8)による。]20 mLに溶かし,煮沸して窒素酸化物を追い出す。放冷後,全量フラスコ
1 000 mLに移し入れ,水を標線まで加える。
12) セレン標準液(Se 2 μg/mL) セレン標準液(Se 0.2 mg/mL)10 mLを全量フラスコ1 000 mLにとり,
塩酸(1+100)(JIS K 8180に規定する塩酸を用いて調製する。)を標線まで加える。使用時に調製
する。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(1)(Seとして2〜50 μgを含む。)をビーカー200 mLにとり,水で約100
mLとし,約50 ℃に加熱する。
2) 臭化カリウム0.5 gを加えて溶かし,次に,硫酸アンモニウム鉄(III)溶液2 mL及びブロモチモー
ルブルー溶液(1 g/L)5〜7滴を加える。
3) 溶液をかき混ぜながら溶液の色が青になるまでアンモニア水(1+1)を加えた後,溶液の色が黄色
になるまで煮沸する。
4) 沈殿が沈降するまで放置し,ろ紙5種Aでろ別し,温水で洗浄する。
5) 沈殿は元のビーカーに洗い落とし,ろ紙に付着した沈殿は,温塩酸(1+1)約2 mLを滴加して溶
かし,沈殿の入っているビーカーに受け,ろ紙は水洗いする。
6) 加熱して沈殿を溶かし,水で液量を約30 mLとする。
7) 放冷後,EDTA溶液(40 g/L)10 mLを加え,アンモニア水(1+2)を滴加してpHを1.5〜2.0 に調
節する。
8) 3,3′-ジアミノベンジジン溶液(5 g/L)2 mLを加えて振り混ぜ,沸騰水浴中で約10分間加熱する。
9) 流水で冷却後,アンモニア水(1+2)を滴加してpHを約6に調節し,分液漏斗100 mLに移し入れ,
水で約60 mLとする。
10) トルエン10 mLを加えて約30秒間振り混ぜた後,放置する。
11) トルエン層の一部を吸収セルに移し,トルエンを対照液として波長420 nm付近の吸光度を測定す
る。
289
K 0102:2016
12) 空試験として硫酸アンモニウム鉄(III)溶液2 mLをビーカーにとり,水で30 mLとした後,7)〜
11)の操作を行って,試料について得た吸光度を補正する。
13) 検量線からセレンの量を求め,試料中のセレンの濃度(Se μg/L)を算出する。
注(1) 試料が濁り及び有機物を含まず,セレンの濃度が高い場合は,試料の適量をビーカーにとり,
約50 ℃に加熱する。臭化カリウム0.5 gを加えて溶かし,7)以降の操作を行ってもよい。
d) 検量線 検量線の作成は,次による。
1) セレン標準液(Se 2 μg/mL)1〜25 mLをビーカー200 mLに段階的にとり,液量を水で約100 mLと
し,約50 ℃に加熱する。c) 2)〜12)の操作を行って吸光度を測定し,セレン(Se)の量と吸光度と
の関係線を作成する。
67.2 水素化合物発生原子吸光法 試料を前処理して,セレンをセレン化水素とし,水素-アルゴンフレー
ム中に導き,セレンによる原子吸光を波長196.0 nmで測定してセレンを定量する。
定量範囲:Se 2〜12 μg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 塩酸(1+1) 1)の塩酸を用いて調製する。
3) 硝酸 JIS K 8541に規定するもの。
4) 硫酸(1+1) 5.4 a) 2)による。
5) テトラヒドロほう酸ナトリウム溶液(10 g/L) 61.2 a) 8)による。
6) アルゴン JIS K 1105に規定するアルゴン2級
7) セレン標準液(Se 0.1 μg/mL) 67.1 a) 12)のセレン標準液(Se 2 μg/mL)5 mLを全量フラスコ100 mL
にとり,硫酸(1+100)(JIS K 8951に規定する硫酸を用いて調製する。)を標線まで加える。使用
時に調製する。
b) 装置 装置は,次による。
1) 連続式水素化合物発生装置 61.2 b) 1)による。
2) フレーム原子吸光分析装置 52.2 b) 1)による。
c) 操作 操作は,次による。
1) 試料(2)の適量(Seとして0.05〜0.3 μgを含む。)をビーカー100 mLにとり,硫酸(1+1)1 mL及び
硝酸2 mLを加える。
2) 加熱板上で乾固する直前まで加熱する。
3) 放冷した後,塩酸(1+1)20 mLを加え,90〜100 ℃で約10分間加熱する。
4) 放冷した後,全量フラスコ25 mLに移し入れ,水を標線まで加える。
5) 連続式水素化合物発生装置にアルゴンを流しながら,4)の溶液,テトラヒドロほう酸ナトリウム溶
液(10 g/L)(3)及び塩酸(1 mol/L)(JIS K 8180に規定する塩酸を用いて調製する。)(3)を,定量ポ
ンプで連続的に装置内に導入(3)し,セレン化水素を発生させる。
6) 発生したセレン化水素と廃液とを分離した後,セレン化水素を含む気体を水素-アルゴン(4)フレーム
(5)中に導入し,波長196.0 nmにおける指示値(6)を読み取る。
7) 空試験として試料と同量の水をとり,1)〜6)の操作を行った後,指示値を読み取り,試料について
得た指示値を補正する。
8) 検量線からセレンの量を求め,試料中のセレンの濃度(Se μg/L)を算出する。
注(2) 有機物を含まない試料は1)〜3)の代わりに,次のように操作してもよい。
290
K 0102:2016
試料の適量(Seとして0.05〜0.3 μgを含む。)をビーカー100 mLにとり,試料と同量の塩酸
を加え,90〜100 ℃で約10分間加熱する。また,多量の有機物を含む場合は1)及び2)の代わり
に,次のように操作してもよい。
試料の適量(Seとして0.05〜0.3 μgを含む。)をとり,硫酸(1+1)1 mL,硝酸2 mL及びJIS
K 8223に規定する過塩素酸3 mLを加えて加熱し(*),白煙を発生させて有機物を分解する。
注(*) 過塩素酸を用いる加熱分解操作は,試料の種類によっては爆発の危険性があるため,硝
酸が共存する状態で行うように注意する。
(3) 61.の注(9)による。
(4) 61.の注(10)による。
(5) 61.の注(11)による。
(6) 61.の注(12)による。
d) 検量線 検量線の作成は,次による。
1) セレン標準液(Se 0.1 μg/mL)0.5〜3 mL(7)を段階的ビーカー100 mLにとり,塩酸(1+1)20 mLを
加え,90〜100 ℃で約10分間加熱する。
2) c) 4)〜6)の操作を行う。
3) 別に,空試験として塩酸(1+1)20 mLをビーカー100 mLにとり,90〜100 ℃で約10分間加熱し
た後,c) 4)〜6)の操作を行って,2)の標準液について得た指示値を補正し,セレン(Se)の量と指
示値との関係線を作成する。検量線の作成は,試料測定時に行う。
注(7) 注(5)によった場合は,水素-アルゴンフレームに比べて10〜50倍程度(装置及び操作条件によ
って異なる。)感度がよいので,セレン標準液(Se 0.1 μg/mL)の量を,適宜,減らす。
備考 1. 61.の備考2.による。
2. 連続式水素化合物発生装置の代わりに,図61.5に例を示す水素化物発生装置を用いて定量し
てもよい。この場合の操作は,次による。
1) c) 1)〜4)の操作を行った溶液の全量を水素化合物発生装置の反応容器に移し入れる。
2) 水素化合物発生装置と原子吸光分析装置とを連結し,系内の空気をアルゴン(*)で置換す
る。
3) コックを回転して,アルゴン(*)でバブリングする状態にする。
4) セプタムを通して,シリンジなどによってテトラヒドロほう酸ナトリウム溶液(10 g/L)
2 mLを手早く加え,発生する水素化合物を水素-アルゴン(*)フレーム(**)中に導入し,波
長196.0 nmの指示値(***)を読み取る。
5) c) 7)及び8)の操作を行い,試料中のセレンの濃度(Se μg/L)を算出する。
注(*) 61.の注(10)による。
(**) 61.の注(11)による。
(***) 61.の注(12)による。
3. 61.の備考4.による。
4. セレンの水素化合物発生法は,銅,ニッケル,コバルト,白金,パラジウムなどの遷移金属
によって発生効率が影響される。また,アンチモン,ひ素などの水素化物を形成する元素に
よっても発生効率が低下する。塩酸はセレンをVI価からIV価に還元するために用いられる
が,一般に,塩酸濃度を高くすると遷移金属による干渉が低減することが多い。
なお,未知試料のように共存物質の影響が不明な場合は,未知試料に一定量の測定対象元
291
K 0102:2016
素(ここでは,セレン)を添加したときに得られる指示値の増加分と,同量の測定元素を含
む検量線用標準液の指示値とを比較することによって,干渉の大きさを知ることができる。
共存物質による干渉がある場合は,標準添加法を適用すると真度が向上するが,干渉が大
きい場合は,水素化合物発生法は適用できない。
なお,この操作では試験溶液の最終の塩酸の濃度は約6 mol/Lとなるが,高濃度の塩酸溶
液が定流量ポンプなどの材質に悪影響を及ぼす場合は,c) 4)において全量フラスコ25 mLの
代わりに全量フラスコ50 mLを用いてもよい。ただし,銅などの干渉が増加する傾向がある
ので,これらを多く含む試料には注意する。
67.3 水素化合物発生ICP発光分光分析法 試料を前処理して,セレンをセレン化水素とし,試料導入部
を通して誘導結合プラズマ中に導入し,セレンによる発光を波長196.026 nmで測定してセレンを定量する。
定量範囲:Se 1〜20 μg/L,繰返し精度:3〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 塩酸(1+1) 1)の塩酸を用いて調製する。
3) 硝酸 JIS K 8541に規定するもの。
4) 硫酸(1+1) 5.4 a) 2)による。
5) テトラヒドロほう酸ナトリウム溶液(10 g/L) 61.2 a) 8)による。
6) アルゴン 67.2 a) 6)による。
7) セレン標準液(Se 0.1 μg/mL) 67.2 a) 7)による。
b) 装置 装置は,次による。
1) 連続式水素化合物発生装置 61.2 b) 1)による。
2) ICP発光分光分析装置
c) 操作 操作は,次による。
1) 試料(2)の適量(Seとして0.025〜0.5 μgを含む。)をビーカーにとる。次に,67.2 c) 1)〜5)の操作を
行う。
2) 発生したセレン化水素と廃液とを分離した後,セレン化水素を含む気体を発光部に導入し,波長
196.026 nmの発光強度を測定する。
3) 空試験として試料と同量の水をとり,1)及び2)の操作を行い,試料について得た発光強度を補正す
る。
4) 検量線からセレンの量を求め,試料中のセレンの濃度(Se μg/L)を算出する。
d) 検量線 検量線の作成は,次による。
1) セレン標準液(Se 0.1 μg/mL)0.25〜5 mLをビーカー100 mLに段階的にとる。次に,67.2 d) 1)〜3)
の操作を行う。ただし,水素-アルゴンフレームを発光部と,また,指示値を発光強度と読み替える。
備考 5. 61.の備考6.による。
6. 備考4.による。また,標準添加法を用いる場合は,バックグラウンド補正を行う。
67.4 ICP質量分析法 52.5による。
備考 7. 塩酸,塩化物イオンなどの塩素を多量に含む試料では,表52.3に示す40Ar37Clなどの多原子
イオンのスペクトル干渉が大きくなるため,質量数77は測定に用いない。また,海水などの
ように塩化物イオンに加えて,比較的高濃度の硫酸イオン,臭化物イオンなどを含む試料で
は,質量数78又は82においても,表52.3に示すCl,S,Brを含む多原子イオンのスペクト
292
K 0102:2016
ル干渉が大きくなり,測定は困難である。このため,スペクトル干渉を低減する手法を用い
る。
備考 8. スペクトル干渉を低減する手法として,67.3 c)に準じてセレン化水素を発生させ,イオン化
部に導入してセレン(質量数77,78又は82)の指示値を測定し,セレンを定量してもよい。
9. 61.の備考11.による。
68. モリブデン(Mo) モリブデンの定量には,チオシアン酸吸光光度法,ICP発光分光分析法又はICP
質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
68.1 チオシアン酸吸光光度法 試料に過塩素酸を加えて加熱蒸発し,モリブデンをモリブデン(VI)に
酸化した後,これにチオシアン酸ナトリウム及び塩化すず(II)を加え,生成するモリブデン錯体を酢酸
ブチルで抽出し,その吸光度を測定してモリブデンを定量する。
定量範囲:Mo 1〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(1+1) JIS K 8180に規定する塩酸を用いて調製する。
2) 過塩素酸 JIS K 8223に規定するもの。
3) 硫酸(1+1) 5.4 a) 2)による。
4) 硫酸(1+9) JIS K 8951に規定する硫酸を用いて調製する。
5) 硫酸アンモニウム鉄(III)溶液 JIS K 8982に規定する硫酸アンモニウム鉄(III)・12水10 gと硫
酸(1+1)5 mLとを水に溶かして100 mLとする。
6) チオシアン酸ナトリウム溶液(200 g/L) JIS K 9002に規定するチオシアン酸ナトリウム20 gを水
に溶かして100 mLとする。
7) 塩化すず(II)溶液 JIS K 8136に規定する塩化すず(II)二水和物12 gに塩酸(1+1)40 mLを
加え,加熱して溶かし,放冷後,水で100 mLとする。JIS K 8580に規定するすずの粒状2,3粒を
加え,着色ガラス瓶に保存する。
8) 酢酸ブチル JIS K 8377に規定するもの。
9) モリブデン標準液(Mo 0.1 mg/mL) 58.4 a) 5)による。
10) モリブデン標準液(Mo 2 μg/mL) モリブデン標準液(Mo 0.1 mg/mL)10 mLを全量フラスコ500 mL
にとり,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(Moとして1〜50 μgを含む。)をビーカー100 mLにとり,過塩素酸3
293
K 0102:2016
mLを加え,加熱蒸発して過塩素酸の白煙を発生させる。
2) 放冷後,硫酸(1+1)10 mLと水10 mLとを加え,加熱して溶かす。不溶解物があればろ紙5種B
でろ過して水洗いする。
3) 放冷後,分液漏斗100 mLに水で洗い入れ,液量を水で約40 mLとする。
4) 硫酸アンモニウム鉄(III)溶液1 mL(1)とチオシアン酸ナトリウム溶液(200 g/L)5 mLとを加えて
振り混ぜる。
5) 塩化すず(II)溶液10 mLを加えて振り混ぜた後,約5分間放置してモリブデン錯体を生成させる。
6) 酢酸ブチル10 mLを加えて約1分間振り混ぜ,放置する。
7) 水層を捨て,酢酸ブチル層に硫酸(1+9)10 mL,チオシアン酸ナトリウム溶液(200 g/L)1 mL及
び塩化すず(II)溶液5 mLを加えて振り混ぜ(2),放置する。
8) 酢酸ブチル層を目盛付きメスシリンダーに入れ,酢酸ブチルを加えて10 mLとする。
9) 酢酸ブチル層の一部を吸収セルに移し,酢酸ブチルを対照液として波長470 nm付近の吸光度を測定
する。
10) 空試験として過塩素酸3 mL及び硫酸(1+1)10 mLを分液漏斗にとり,水で液量を約40 mLとし,
4)〜9)の操作を行って試料について得た吸光度を補正する。
11) 検量線からモリブデンの量を求め,試料中のモリブデンの濃度(Mo μg/L)を算出する。
注(1) 試料中に鉄が10 mg以上含まれている場合には加えない。
(2) 鉄(III)のチオシアン酸錯体を完全に還元する。
d) 検量線 検量線の作成は,次による。
1) モリブデン標準液(Mo 2 μg/mL)0.5〜25 mLを分液漏斗に段階的にとり,過塩素酸3 mL及び硫酸
(1+1)10 mLをそれぞれ加え,更に水で液量を約40 mLとし,c) 4)〜10)の操作を行って吸光度を
測定し,モリブデン(Mo)の量と吸光度との関係線を作成する。
備考 1. 5.のうち5.4の方法を用いた場合には,硫酸の白煙を発生させ,有機物を分解したときの液量
が約5 mLになるよう加熱蒸発又はJIS K 8951に規定する硫酸を加えて約5 mLに調節する。
次に,水20 mLを加え,加熱して溶かし,過マンガン酸カリウム溶液(3 g/L)[61.2 a) 6)に
よる。]を溶液の色が過マンガン酸の微紅色を保つまで滴加し,モリブデン(VI)に十分に
酸化し,過塩素酸3 mLを加えてc) 3)の操作を行う。
2. 試料中にタングステンが含まれている場合には,その大部分は酸処理したときタングステン
酸として沈殿するのでろ別分離ができるが,微量のタングステンは残留して妨害するから,
塩化すず(II)溶液を加える前にJIS K 8532に規定するL(+)-酒石酸1 gを加えて振り混
ぜ,溶かしておく。
68.2 ICP発光分光分析法 58.4による。
備考 3. 準備操作を行った試料のアルカリ金属イオン,アルカリ土類金属イオンなどの濃度が高く,
モリブデンの濃度が低い場合には,52.の備考10. 1)〜3)及び12.に準じた操作を行うとよい。
68.3 ICP質量分析法 62.4による。
69. タングステン(W) タングステンの定量には,チオシアン酸吸光光度法,ICP発光分光分析法又はICP
質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
294
K 0102:2016
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
69.1 チオシアン酸吸光光度法 硫酸及びりん酸溶液中で塩化すず(II)でタングステンを還元し,タング
ステン(V)とし,これにチオシアン酸を加え,生成する黄色のタングステン錯体をジイソプロピルエー
テル(2,2′-オキシビスプロパン)で抽出し,その吸光度を測定してタングステンを定量する。
定量範囲:W 5〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 過塩素酸 JIS K 8223に規定するもの。
2) 硫酸(1+1) 5.4 a) 2)による。
3) りん酸 JIS K 9005に規定するもの。
4) 塩化すず(II)溶液 JIS K 8136に規定する塩化すず(II)二水和物20 gにJIS K 8180に規定する
塩酸を加え,加熱して溶かし,放冷後,塩酸で100 mLとする。JIS K 8580に規定するすずの粒状2,
3粒を加え,着色ガラス瓶に保存する。
5) チオシアン酸ナトリウム溶液(200 g/L) 68.1 a) 6)による。
6) ジイソプロピルエーテル JIS K 9528に規定するもの。
7) タングステン標準液(W 0.1 mg/mL) JIS K 8612に規定するタングステン(VI)酸ナトリウム二水
和物0.180 gを少量の水に溶かし,全量フラスコ1 000 mLに移し入れ,水を標線まで加える。
8) タングステン標準液(W 1 μg/mL) タングステン標準液(W 0.1 mg/mL)10 mLを全量フラスコ1 000
mLに移し入れ,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 5.の操作を行った試料の適量(Wとして5〜50 μgを含む。)をビーカー100 mLにとり,硫酸(1+1)
5 mL,過塩素酸1 mL(1)及びりん酸1 mLを加え,加熱して硫酸の白煙を発生させる。
2) 放冷後,水5 mLを加えて溶かし,塩化すず(II)溶液20 mLを加えて振り混ぜ,60 ℃の水浴中で
約10分間加熱してタングステンを還元した後,水で約40 mLとし,冷却する。
3) 少量の水で分液漏斗100 mLに移し入れ,チオシアン酸ナトリウム溶液(200 g/L)5 mLを加えて振
り混ぜる。
4) ジイソプロピルエーテル10 mLを加えて振り混ぜた後,放置する。
5) ジイソプロピルエーテル層の一部を吸収セルに移し,ジイソプロピルエーテルを対照液として波長
400 nm付近の吸光度を測定する。
6) 空試験としてビーカー100 mLに硫酸(1+1)5 mL及びりん酸1 mLを加えた後,2)〜5)の操作を行
って,試料について得た吸光度を補正する。
7) 検量線からタングステンの量を求め,試料中のタングステンの濃度(W μg/L)を算出する。
注(1) 5.のうち5.3の方法を用いたときは加えない。
d) 検量線 検量線の作成は,次による。
295
K 0102:2016
1) タングステン標準液(W 1 μg/mL)5〜50 mLをビーカー100 mLに段階的にとり,c) 1)〜6)の操作を
行って吸光度を測定し,タングステン(W)の量と吸光度との関係線を作成する。
備考 モリブデンは妨害となるから,次の操作によって分離する。
1) 5.の操作を行った試料約50 mLにJIS K 8532に規定するL(+)-酒石酸1 gとJIS K 8982
に規定する硫酸アンモニウム鉄(III)・12水0.1 g(試料中に鉄が含まれる場合には加えな
い。)とを加えて溶かし,アンモニア水(1+1)[52.1 a) 1)による。]で中和する。次に,硫
酸(1+4)(JIS K 8951に規定する硫酸を用いて調製する。)1.5 mLを加え,水で100 mL
とする。
2) これに硫化水素を通じて硫化モリブデンを沈殿させ,これにJIS K 8103に規定するジエチ
ルエーテル2〜3 mLを加えて激しくかき混ぜた後,ろ紙5種Cでろ過し,硫化水素水(飽
和)で洗浄する。
3) ろ液と洗液とを合わせ,加熱して硫化水素を追い出すとともに液量を少量とし,c) 2)以降
の操作を行う。
69.2 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,タ
ングステンによる発光を波長207.911 nmで測定してタングステンを定量する。
定量範囲:W 50〜5 000 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) タングステン標準液(W 0.1 mg/mL) 69.1 a) 7)による。
2) タングステン標準液(W 10 μg/mL) タングステン標準液(W 0.1 mg/mL)10 mLを全量フラスコ
100 mLにとり,塩酸(1+1)(JIS K 8180に規定する塩酸を用いて調製する。)20 mLを加えた後,
水を標線まで加える。使用時に調製する。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
d) 操作 操作は,次による。
1) 52.4 d) 1)による。ただし,波長207.911 nmの発光強度を測定する(2) (3) (4)。
2) 52.4 d) 2)及び3)の操作を行い,試料中のタングステンの濃度(W μg/L)を算出する。
注(2) 52.の備考11.による。
(3) 47.の注(8)による。
(4) 47.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) タングステン標準液(W 10 μg/mL)0.1〜50 mLを全量フラスコ100 mLに段階的にとり,引き続き
52.4 e) 1)〜3)の操作を行い,タングステン(W)の量と発光強度との関係線を作成する。検量線の
作成は,試料測定時に行う。
69.3 ICP質量分析法 62.4による。
70. バナジウム(V) バナジウムの定量には,N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法,
フレーム原子吸光法,電気加熱原子吸光法,ICP発光分光分析法又はICP質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
296
K 0102:2016
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
70.1 N-ベンゾイル-N-フェニルヒドロキシルアミン吸光光度法 試料を過マンガン酸カリウムで酸化し
てバナジウムをバナジウム(V)とし,これにN-ベンゾイル-N-フェニルヒドロキシルアミン(BPHA)を
加えて生成した赤紫のバナジウム錯体を塩酸酸性溶液からクロロホルムで抽出し,その吸光度を測定して
バナジウムを定量する。
定量範囲:V 2〜50 μg,繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 塩酸(2+1) JIS K 8180に規定する塩酸を用いて調製する。
2) 硝酸 JIS K 8541に規定するもの。
3) 過塩素酸 JIS K 8223に規定するもの。
4) 銅(II)溶液(10 mg/mL) JIS K 8660に規定する銅(99.9 %以上)1 gを硝酸(1+1)[52.1 a) 8)
による。]10 mL中に加え,加熱して溶かし,過塩素酸20 mLを加え,加熱蒸発させて過塩素酸の
白煙を十分に発生させる。放冷後,水で薄めて100 mLとする。
5) BPHAクロロホルム溶液(2 g/L) JIS K 9569に規定するN-ベンゾイル-N-フェニルヒドロキシルア
ミン0.2 gをJIS K 8322に規定するクロロホルム100 mLに溶かす。
6) 過マンガン酸カリウム溶液(3 g/L) 61.2 a) 6)による。
7) バナジウム標準液(V 0.1 mg/mL) 58.4 a) 7)による。
8) バナジウム標準液(V 2 μg/mL) バナジウム標準液(V 0.1 mg/mL)20 mLを全量フラスコ1 000 mL
にとり,硫酸(1+1)[5.4 a) 2)による。]10 mLを加えた後,水を標線まで加える。
b) 器具及び装置 器具及び装置は,次による。
1) 分液漏斗 100 mL
2) 光度計 分光光度計又は光電光度計
c) 操作 操作は,次による。
1) 試料の適量(Vとして2〜50 μgを含む。)をとり,硝酸5 mLと過塩素酸3 mLとを加えて加熱蒸発
し,過塩素酸の白煙を十分に発生させて乾固近くまで蒸発濃縮する。
2) 放冷後,水10 mLを加えて溶かし,銅(II)溶液(10 mg/mL)1 mLを加えた後,過マンガン酸カリ
ウム溶液(3 g/L)を溶液の色がうすい赤となるまで滴加し,更に1滴を過剰に加えて約5分間放置
し,バナジウムを酸化する。
3) 分液漏斗に移し入れ,水で約30 mL(1)とする。
4) BPHAクロロホルム溶液(2 g/L)10 mLを加える。次に,塩酸(2+1)40 mLを加えて,過剰の過
マンガン酸を還元し,直ちに約30秒間振り混ぜてバナジウム錯体を抽出する。
5) 放置した後,クロロホルム層を乾いたろ紙5種Bでろ過する。
6) クロロホルム層の一部を吸収セルに移し,JIS K 8322に規定するクロロホルムを対照液として波長
530 nm付近の吸光度を測定する。
7) 空試験として水約10 mLをとり,2)〜6)の操作を行って試料について得た吸光度を補正する。
297
K 0102:2016
8) 検量線からバナジウムの量を求め,試料中のバナジウムの濃度(V μg/L)を算出する。
注(1) あらかじめ分液漏斗に目印を付けておく。
d) 検量線 検量線の作成は,次による。
1) バナジウム標準液(V 2 μg/mL)1〜25 mLを分液漏斗に段階的にとり,c) 2)〜7)の操作を行ってバナ
ジウム(V)の量と吸光度との関係線を作成する。
70.2 フレーム原子吸光法 試料を前処理した後,アセチレン-一酸化二窒素フレーム中に噴霧し,バナジ
ウムによる原子吸光を波長318.4 nmで測定してバナジウムを定量する。
定量範囲:V 1〜20 mg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 硝酸 JIS K 8541に規定するもの。
2) 硝酸アルミニウム溶液(400 g/L) JIS K 8544に規定する硝酸アルミニウム九水和物70 gをとり,
少量の水を加え,加熱して溶かす。放冷後,水を加えて100 mLにする。
3) バナジウム標準液(V 0.1 mg/mL) 58.4 a) 7)による。
b) 装置 装置は,52.2 b)による。
c) 準備操作 準備操作は,次による。
1) 試料を5.によって処理する。
d) 操作 操作は,次による。
1) c)の準備操作を行った試料の適量(Vとして0.1〜2 mgを含む。)を全量フラスコ100 mLにとり,
硝酸1 mLを加えた後,水を標線まで加える。
2) この溶液50 mLを乾いたビーカーにとり,硝酸アルミニウム溶液(400 g/L)1 mLを加える。
3) 58.2 d) 3)及び4)の操作を行う。ただし,波長は318.4 nmを用いる(2)。
4) 検量線からバナジウムの量を求め,試料中のバナジウムの濃度(V mg/L)を算出する。
注(2) 58.の注(5)による。感度の最も高い部分は,フレームのごく限られた位置であるから,この位置
をあらかじめ確かめておく。
e) 検量線 検量線の作成は,次による。
1) バナジウム標準液(V 0.1 mg/mL)1〜20 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った
試料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。
2) この溶液についてd) 2)〜4)の操作を行い,バナジウム(V)の量と指示値との関係線を作成する。
検量線の作成は,試料測定時に行う。
70.3 電気加熱原子吸光法 試料を前処理した後,電気加熱炉で原子化し,バナジウムによる原子吸光を
波長318.4 nmで測定してバナジウムを定量する。
定量範囲:V 10〜200 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
備考 1. 52.の備考7.による。
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) バナジウム標準液(V 1 μg/mL) 58.4 a) 7)のバナジウム標準液(V 0.1 mg/mL)2 mLを全量フラス
コ200 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。
b) 器具及び装置 器具及び装置は,52.3 b)による。
c) 準備操作 準備操作は,次による。
298
K 0102:2016
1) 試料を5.によって処理する。
d) 操作 操作は,次による。
1) 52.3 d) 1)の操作を行う。ただし,灰化温度は500〜600 ℃,原子化(3)温度は2 400〜3 000 ℃(5〜10
秒),波長は318.4 nmを用いる。
2) 空試験として52.3 d) 2)の操作を行う。
3) 検量線からバナジウムの量を求め,試料中のバナジウムの濃度(V μg/L)を算出する。
注(3) 52.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) バナジウム標準液(V 1 μg/mL)1〜20 mLを全量フラスコ100 mLに段階的にとり,c) 1)を行った試
料と同じ酸の濃度になるように酸を加えた後,水を標線まで加える。この溶液についてd) 1)の操作
を行う。
2) 52.3 e) 2)及び3)の操作を行い,バナジウム(V)の量と指示値との関係線を作成する。検量線の作
成は,試料測定時に行う。
70.4 ICP発光分光分析法 58.4による。
備考 2. 準備操作を行った試料のアルカリ金属イオン,アルカリ土類金属イオンなどの濃度が高く,
バナジウムの濃度が低い場合には,52.の備考10. 1)〜3)及び12.に準じた操作を行うとよい。
70.5 ICP質量分析法 52.5による。
71. 魚類による急性毒性試験 魚類による急性毒性試験としては,LC50(Median lethal concentration)の測
定が行われる。排水のLC50は,魚類を急性毒性物質の含まれている排水の一連の希釈液中に一定時間飼
育し,その間に供試魚の50 %が死ぬような希釈液中の排水濃度で表す。一般には,24時間,48時間及び
96時間のLC50を求める。
備考 1. LC50は魚類への許容濃度ではない。許容濃度は,種々の生物による亜急性毒,慢性毒試験な
どの結果に基づいて慎重に決定するべきものであるが,これを推定するための一つの便法と
して,LC50に適用係数を乗じることが行われている。適用係数として,かつては48時間LC50
に対して0.1を用いたが,96時間LC50に対して0.01前後の値を用いることがある。LC50
は供試魚の種類,健康状態などによって著しく相違するほか,希釈水の水質,水温などによ
っても変動する。しかし,これらの相互関係はまだ明らかでない。このため,放流水域の生
物への影響を知る基礎資料として利用するような場合は,供試魚種,希釈水などは,個々の
水域の自然条件に似たものを選択するのがよい。
これに対し,種々の排水の毒性を比較したり,毒性の異なる理由を検討したりする場合は
供試魚種,その他の条件など測定方法を統一した方が便利である。ここに規定するLC50測
定方法は,測定条件を個々の水域の自然条件に近づけるという主旨を著しく損なうことなし
に,一方において試験結果の再現性,有効性などを確保しようとしたものである。
a) 供試魚及び希釈水 供試魚及び希釈水は,次による。
1) 供試魚 供試魚種は,次の基本条件を考慮して決定する。
なお,試験前に供試魚の種類を正しく査定しておく。
1.1) 水温,餌,取扱いなど,実験室内の飼育管理条件に適している。
1.2) 大きさがそろい,健康であり,一度に多数得られる。
1.3) 供試魚に適している種類として,サケ科(ニジマス,カワマス,ヤマメ,アマゴなど),コイ科(コ
299
K 0102:2016
イ,フナ,タナゴ,オイカワなど),メダカ科(メダカ,ヒメダカなど)及びタップミノー科(グ
ッピー,タップミノーなど)が挙げられる。
1.4) 供試魚を準備する場合の注意点
1.4.1) 同時に使用する魚体の大きさがほぼ一定したものを選び,最大のものの全長が最小のものの1.5
倍以上にならない。
1.4.2) できるだけ平均全長50 mm以下の小さな魚を使用する(1)。
1.4.3) 同一試験に使用する魚は,同一条件で入手する。同じ水域,同じ養魚場などから同じ時期に採取
したものを用いる。入手した供試魚は,適切な池又は水槽に入れ,適切な水温及び水質の下で,
試験に供するときまで飼育しておく。
1.5) 試験を行う場合は,少なくとも1週間(2)順応水槽中で飼育し,試験を行う場合の水温及び水質(希
釈水と同一の水質)に順応させる。順応期間中は,なるべく1日1回餌を与え,試験2日間前か
らは,餌を与えないでおく。
1.6) 試験時には健康な魚を使用し,試験前の4日間において死んだり発病したりする魚が10 %以下の
状態でなければ使用してはならない。この試験には,病気,外見及び行動に異常のある魚は使用
しない。
2) 希釈水 試料を薄めるために使用する水は,流入する水域への影響を検討するのが目的の場合には,
実際に試料が流入する水域の上流の水を使用する。しかし,その水域の上流の水を入手しにくい場
合は,適切な水に必要な化学成分を加え,pH,酸消費量又はアルカリ消費量,塩類などを水域の水
質とほぼ同様になるように調製する。また,他の排水の成分などが混入している場合は,その成分
の添加も必要である。
懸濁物の多い水域の水を使用する場合は,沈降又はろ過によって,その大部分を除いてから使用
する。
特定の水域の問題を考慮しないでよい場合は,希釈水として特に異常な成分を含まない,pHが7
前後の良質の井戸水又は水道水(残留塩素を除いたもの。)を使用する。
希釈水は,よくばっ気して酸素を十分に溶かしておく。
注(1) 供試魚が特に大きい場合は,それに応じて大きな試験水槽を使うことが必要になる。
(2) できれば10日間又はそれ以上。
備考 2. 流入する水域の問題が関連している場合の供試魚の決定には,1)の基本条件のほか,産業的,
生態的な重要性,毒物に対する感受性などを考慮する。
b) 器具及び装置 器具及び装置は,次による。
1) 試験水槽 清浄なガラス製又はステンレス鋼製の水槽。容量30 L(同一形状のもの。)
2) 恒温設備 恒温室又は恒温水槽。12〜28 ℃の指定温度を±2 ℃で保てるもの。
3) 順応水槽 供試魚を飼育しておくために必要な容量をもち(100〜200 L程度のもの。),必要に応じ
て温度調節及び空気の送入ができるもの。
c) 試料の採取及び保存 排水は,瓶に口まで満たすように採取した後,密栓し,温度が採水時よりも上
がらないようにして保存する。また,細菌によって分解しやすい有機物を含む排水では0〜4 ℃に貯
蔵し(この場合,凍結させない。),できるだけ早く試験する。同一試験に使用する排水は,同一時期
に採水する。
d) 試験条件 試験条件は,次による。
1) 水温は,コイ,フナ,メダカ,グッピーなどの温水魚では20〜28 ℃,ニジマス,ヤマメなどの冷
300
K 0102:2016
水魚では12〜18 ℃の範囲の一定温度とする。試験中に水温の変化が生じないように注意し,指定
の温度の±2 ℃とする。
2) 試験水槽中に希釈試料の量は,魚体1 gにつき1 L以上とする。また,各水槽中の供試魚は,少な
くとも7尾以上とする。ただし,同じ希釈水を入れた幾つかの水槽に同数ずつ分けて試験してもよ
い。
3) 希釈水中の試料濃度は,必要に応じ予備試験を行って決定する。予備試験では,試料の濃度範囲の
幅を広くとり,対数値の間隔が等しくなるように等比級数的に試料を薄める(例えば,100,10,1,
0.1 %又は100,32,10,3.2 %のようにする。)。
予備試験での供試魚体数は,7尾以下でも差し支えない。魚体の質量に応じて,試験水槽の大き
さ,水量なども縮小する。
試験に用いる試料の濃度は,24時間までに大部分の魚が死ぬ濃度と,96時間後も大部分魚が生き
残る濃度の範囲内にあるが,1回の予備試験の後,更にその中間の濃度範囲について再び予備試験
を行う。
4) 試験に用いる濃度範囲が求められたら,その範囲内に各対数値の間隔が等しくなるように5〜10 段
階にとり,試験の濃度とする(例えば,100,75,56,42,32,24,18,13.5,10 %のようにする。)。
5) 試験中に希釈試料の毒性の強さが変化することがあるので,予備試験中に検討しておく。
試験中に溶存酸素が不足しないように,希釈試料中の溶存酸素の濃度は,温水魚ではO 4 mg/L以
上,また,冷水魚ではO 5 mg/L以上を保つようにする(3)。
注(3) この場合問題になるのは,溶存酸素の減少及び毒性の減少である。
備考 3. 成分の変化しやすい場合は,試験中希釈試料を交換する必要がある。これは,定濃度流水式
装置による連続的な交換が望ましいが,一定時間(24時間以内)ごとの全量交換を行っても
よい。また,揮発成分の減少を防ぐために,試験水槽の水深が浅すぎないように注意する(例
えば,試験水槽30 Lの水深を200 mm以下にならないようにする。)。
e) 操作 操作は,次による。
1) 5〜10段階の希釈試料をつくり,それぞれの試験水槽に入れる。試料を薄めるとき,懸濁物は均等
に混ぜ,必要以上に空気とかき混ぜない。
2) 希釈試料の準備ができたら,なるべく早く供試魚を水槽に入れる(4) (5)。
3) 試験開始後4時間及び8時間後の供試魚の状態を観察し,横転したり,異常な動作を示したりする
のを記録しておく。
4) 試験中,死魚はなるべく早く水槽から取り除く。供試魚の死は,魚の尾柄部をガラス棒で軽く押さ
え,魚体が反応を示さなくなったことによって認定する。
5) 24時間,48時間及び96時間後の死魚数を記録する。
6) 試験と並行して希釈水だけを用いた対照試験を行う。対照試験において10 %を超える死魚又は不健
全魚があった場合は,試験結果を採用しない。
7) 試験の途中で,希釈試料について,溶存酸素,pH及び主要な毒成分の試験を行う。この水質試験は,
試験開始前及び試験終了時に行う。溶存酸素の測定は,できるだけ度々行う。
なお,水質試験の試料は,なるべく必要最少量を採取する。
8) 図71.1のように横軸に希釈試料の試料濃度の対数を,また,縦軸に死亡率(%)をとり,測定され
た死亡率が50 %より上のものと下のものとで最も50 %に近いものを記入し,この両点を直線で結
び,50 %の線と交わる点に相当する濃度をLC50とする。
301
K 0102:2016
9) LC50は,採用した時間によって24時間LC50,48時間LC50及び96時間LC50に区分し,希釈試
料中の試料濃度で表示する。
10) 試験結果には,次の事項を付記する。
− 各濃度における魚体数及び死亡率
− 供試魚の種類,入手経路,平均全長,平均質量(体重),試験水温
− 希釈試料の交換,その他の管理方法
− 試料の入手,試験までの保管の経路及び水質並びに希釈水の入手経路及び水質(希釈水の水質は,
酸消費量又はアルカリ消費量,pH,カルシウム,マグネシウム,塩化物イオン,透視度など)
− 試験水槽の水量及び水深,試験に使用した各水槽中の供試魚数及びその質量,供試魚を入れた時
間,4時間後及び8時間後又はその他の時期に観察された供試魚の状態
− 試験開始時及び試験終了時に行った水質の試験値,特に溶存酸素及びpH
図71.1 片対数紙上でのLC50算出の例
注(4) 30分間以内に入れる。水槽に入れるまでの時間は記録しておく。
(5) 供試魚の移替えは,魚体を傷めないように,柔らかいたもなどによって行う。乾いたものの表
面に置いたり,又は不必要に長く外気に露出したりしないようにする。移替えの場合に不手際
のあったものは,供試魚から除く。
備考 4. 試験目的によっては,供試魚に海産魚を使用する。試験に適した海産魚としては,入手及び
飼育管理の容易さから,ハゼ類,クサフグ,メジナ,ボラなどの幼魚,及びアミメハギなど
が挙げられる。
海産魚を取り扱う場合には,溶存酸素の減少,魚の排せつ物,食い残しの餌による飼育水
の悪化などに注意が必要である。
試験前に海産魚を順応飼育するには,流水式水槽を用いることが望ましいが,困難な場合
は,循環ろ過方式の飼育水槽を用いる。試験は,淡水魚と同様に止水中で行ってもよいが,
試験水槽中の希釈試料に対し,魚体は,必ず1 Lに0.3 g以下とし,少なくとも24時間ごと
に希釈試料を全量交換する必要がある。
5. e)で用いたLC50の算出方法は,有害な刺激量と生物の反応率(ここでは試料濃度と死亡率)
とが片対数紙上で通常はシグモイド曲線を示し,その曲線の中心部がほぼ直線であることを
302
K 0102:2016
利用したものであり,試料が放流水域の生物に及ぼす影響を論じる目的などには,この方法
で十分な精度が得られる。
備考 6. 各種排水間の毒性の程度を相互的に比較するなど,試験結果をより定量的に使用したいとき
には,死亡率のプロビット変換によってシグモイド曲線が直線となることを利用し,対数確
率紙上にとった各点に近似的に適合する直線を引き,これからLC50を読み取ることによっ
て,更に精度を高めることができる。その方法は,図71.2のように,対数確率紙の縦軸のプ
ロビット目盛に死亡率を,横軸の対数目盛に試料濃度をとり,各点に適合した直線を目測で
引く。この場合,主として死亡率16〜84 %の間に存在する各点に直線を合わせ,また,各点
から直線への縦軸方向の距離を最小にするように努める。直線上において,死亡率50 %の線
と交わる点の試料濃度をLC50とする。
図71.2 対数確率紙上でのLC50算出の例
72. 細菌試験 細菌の試験は,一般細菌,大腸菌群,従属栄養細菌,全細菌及びレジオネラについて行う。
72.1 欠番
72.2 一般細菌 JIS K 0350-10-10の規定による。
72.3 大腸菌群数 JIS K 0350-20-10の規定による。
72.4 従属栄養細菌 JIS K 0350-30-10の規定による。
72.5 全細菌 JIS K 0350-40-10の規定による。
72.6 レジオネラ JIS K 0350-50-10の規定による。
73. ウラン(U) ウランの定量には,ICP発光分光分析法及びICP質量分析法を適用する。
なお,ICP発光分光分析法は,1996年に第1版として発行されたISO 11885との整合を図ったものであ
る。
備考 この試験方法の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21-1に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy(MOD)
303
K 0102:2016
73.1 ICP発光分光分析法 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ウ
ランによる発光を波長385.958 nm(又は367.007 nm),イットリウムによる発光を波長371.029 nmでそれ
ぞれ測定して,ウランとイットリウムとの発光強度比を求め定量する。
定量範囲:U 0.2〜20 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1 mol/L) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
3) 硝酸(2 mol/L) JIS K 9901に規定する高純度試薬−硝酸を用いて調製する。
4) アンモニア水 JIS K 8085に規定するもの。
5) 酢酸アンモニウム JIS K 8359に規定するもの。
6) 酢酸アンモニウム溶液(0.5 mol/L) JIS K 8359に規定する酢酸アンモニウム38.5 gを水で溶かし
て1 000 mLとする。ウランの汚染が測定を妨害することのないことを確認しておく。
7) 酢酸アンモニウム溶液(0.1 mol/L) 酢酸アンモニウム溶液(0.5 mol/L)200 mLを水で薄めて,1 000
mLとする。ウランの汚染が測定を妨害することのないことを確認しておく。
8) CyDTA溶液(0.1 mol/L) trans-1,2-シクロヘキサンジアミン-N,N,N′,N′-四酢酸一水和物3.6 gをと
り,水酸化ナトリウム溶液(1 mol/L)(JIS K 8576に規定する水酸化ナトリウムを用いて調製する。)
に溶かして100 mLとする。
9) ウラン標準液(U 0.1 mg/mL) 酢酸ウラニル(VI)二水和物0.177 3 mgをとり,硝酸(0.2 mol/L)
[3)の硝酸(2 mol/L)を1)の水で10倍に薄める。]に溶かし,全量フラスコ1 000 mLに移し入れ,
硝酸(0.2 mol/L)を標線まで加える。
10) ウラン標準液(U 10 μg/mL) ウラン標準液(U 0.1 mg/mL)20 mLを全量フラスコ200 mLにとり,
硝酸(0.2 mol/L)を標線まで加える。
参考 ウラン濃度が10 μg/mLとして市販されている。
11) イットリウム溶液(Y 0.5 μg/mL) 47.の備考5. 6)のイットリウム溶液(Y 50 μg/mL)10 mLを全量
フラスコ1 000 mLにとり,硝酸(1+1)[52.3 a) 2)による。]1.5 mLを加え,水を標線まで加える。
使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) キレート樹脂充塡固相 52.の備考6.による。
2) ICP発光分光分析装置
c) 準備操作 準備操作は,次による。
1) 試料1 000 mL又はその適量(Uとして0.2〜20 μgを含む。)を5.によって処理する。
2) この溶液に酢酸アンモニウム7.7 g又はその適量を加えて溶かす。続いて,CyDTA溶液(0.1 mol/L)
10 mLを加える。ただし,酢酸アンモニウムの添加量は,1)の試料の量に合わせ,酢酸アンモニウ
ム溶液として約0.1 mol/Lとなるように加える。
3) アンモニア水を用いてpHを5.6に調節した後,キレート樹脂充塡固相ディスクに加圧又は吸引によ
って,流量50〜100 mL/minで(1)流下させる。
4) 酢酸アンモニウム溶液(0.5 mol/L)50 mLを流下させてキレート樹脂充塡固相ディスクを洗浄する。
5) キレート樹脂充塡固相ディスクを硝酸(1 mol/L)5 mLを用いて2回,緩やかに通してウランを溶
出させ,全量フラスコ20 mLに受ける。
6) イットリウム溶液(Y 0.5 μg/mL)2 mLを加えた後,水を標線まで加える。

304
K 0102:2016
注(1) キレート樹脂充塡固相ディスクに代え,キレート樹脂充塡固相カートリッジカラムを用いた場
合は,5〜20 mL/minとする。
d) 操作 操作は,次による。
1) c)の準備操作を行った試料を試料導入部を通して発光部に導入し(2),ウランの波長385.958 nm(又
は367.007 nm),イットリウムの波長371.029 nmでそれぞれの発光強度を測定する(3)。ただし,測
定波長は,試料のスペクトルを観察し,スペクトル干渉のない波長を選択する。
2) ウランとイットリウムとの発光強度比を求める。
3) 空試験として,試料に代えて同量の水を用い,c)の準備操作,1)及び2)の操作を行う。次いで,試
料について2)で得た発光強度比を補正する。
4) 検量線から,ウランの量を求め,試料中のウランの濃度(U μg/L)を算出する。
注(2) 52.の注(11)による。
(3) 47.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) ウラン標準液(U 10 μg/mL)2〜10 mLを全量フラスコ100 mLに段階的にとり,イットリウム溶液
(Y 0.5 μg/mL)10 mLを加え,c) 6)を行った試料と同じ酸の濃度になるように酸を加えた後,水を
標線まで加える。
2) この溶液についてd) 1)及び2)の操作を行う。
3) 空試験として,全量フラスコ100 mLに硝酸(1 mol/L)50 mL,イットリウム溶液(Y 0.5 μg/mL)
10 mLを加えた後,水を標線まで加えた溶液について,d) 1)及び2)の操作を行い,2)で得た発光強
度比を補正する。
4) ウラン(U)の量とウランとイットリウムとの発光強度比の関係線を作成する。検量線の作成は,
試料測定時に行う。
備考 この試験方法で示した定量範囲は,試料1 000 mLで超音波ネブライザーを用いた場合のもので
ある。
超音波ネブライザーを用いる場合,メモリー効果による空試験値が上昇することがあるので,
十分低下したことを確認してから次の試料の測定を行う。
73.2 ICP質量分析法 試料を前処理した後,内標準元素を加え,試料導入部を通して誘導結合プラズマ
中に噴霧し,ウラン及び内標準元素のそれぞれの質量/電荷数における指示値(4)を測定し,ウランの指示値
と内標準元素の指示値との比を求めてウランを定量する。定量範囲,繰返し精度等の例を,表73.1に示す。
注(4) 52.の注(19)による。
表73.1 定量範囲,繰返し精度及び質量数の例*
対象元素
定量範囲
μg/L
繰返し精度
%
質量数
ウラン(U)
0.05〜50
2〜10
238
イットリウム(Y)**
−
−
89
インジウム(In)**
−
−
115
タリウム(Tl)**
−
−
205
ビスマス(Bi)**
−
−
209
注*
装置及び測定条件によって異なる。
** 内標準元素
305
K 0102:2016
a) 試薬 試薬は,次による。
1) 水 52.3 a) 1)による。
2) 硝酸(1+1) 52.3 a) 2)による。
3) 内標準液(0.05 μg/mL) 52.5 a) 3)の内標準液(1 μg/mL)5 mLを全量フラスコ100 mLにとり,硝
酸(1+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(5) (6)。
4) ウラン標準液(U 10 μg/mL) 73.1 a) 10)による(6)。
5) ウラン標準液(U 0.5 μg/mL) ウラン標準液(U 10 μg/mL)5 mLを全量フラスコ100 mLにとり,
硝酸(1+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(6)。
注(5) 52.の注(20)による。
(6) 52.の注(21)による。
b) 装置 装置は,次による。
1) ICP質量分析装置
備考 1. 52.の備考13.による。
2. 52.の備考14.による。
c) 準備操作 準備操作は,52.5 c)による。
d) 操作 操作は,52.5 d)による(7) (8)。
注(7) 52.の注(24)による。
(8) 73.1 c)の操作によって,ウランを共存塩類から分離して測定してもよい。ただし,ウラン(U)
として1〜100 ngを含む量とし,内標準液はイットリウム溶液(Y 0.5 μg/mL)ではなく,a) 3)
内標準液(0.05 μg/mL)を使用する。
e) 検量線 検量線の作成は,52.5 e)による。
備考 3. 52.の備考16.による。
306
K 0102:2016
付表1 引用規格
JIS B 7411-1 一般用ガラス製温度計−第1部:一般計量器
JIS H 6201 化学分析用白金るつぼ
JIS H 6202 化学分析用白金皿
JIS K 0050 化学分析方法通則
JIS K 0094 工業用水・工場排水の試料採取方法
JIS K 0101 工業用水試験方法
JIS K 0114 ガスクロマトグラフィー通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0117 赤外分光分析方法通則
JIS K 0121 原子吸光分析通則
JIS K 0122 イオン電極測定方法通則
JIS K 0126 流れ分析通則
JIS K 0127 イオンクロマトグラフィー通則
JIS K 0128 用水・排水中の農薬試験方法
JIS K 0130 電気伝導率測定方法通則
JIS K 0133 高周波プラズマ質量分析通則
JIS K 0170-1 流れ分析法による水質試験方法−第1部:アンモニア体窒素
JIS K 0170-2 流れ分析法による水質試験方法−第2部:亜硝酸体窒素及び硝酸体窒素
JIS K 0170-3 流れ分析法による水質試験方法−第3部:全窒素
JIS K 0170-4 流れ分析法による水質試験方法−第4部:りん酸イオン及び全りん
JIS K 0170-5 流れ分析法による水質試験方法−第5部:フェノール類
JIS K 0170-6 流れ分析法による水質試験方法−第6部:ふっ素化合物
JIS K 0170-7 流れ分析法による水質試験方法−第7部:クロム(VI)
JIS K 0170-8 流れ分析法による水質試験方法−第8部:陰イオン界面活性剤
JIS K 0170-9 流れ分析法による水質試験方法−第9部:シアン化合物
JIS K 0211 分析化学用語(基礎部門)
JIS K 0215 分析化学用語(分析機器部門)
JIS K 0350-10-10 用水・排水中の一般細菌試験方法
JIS K 0350-20-10 用水・排水中の大腸菌群試験方法
JIS K 0350-30-10 用水・排水中の従属栄養細菌試験方法
JIS K 0350-40-10 用水・排水中の全細菌試験方法
JIS K 0350-50-10 工業用水・工場排水中のレジオネラ試験方法
JIS K 0512 水素
JIS K 0552 超純水の電気伝導率試験方法
JIS K 0557 用水・排水の試験に用いる水
JIS K 0805 有機体炭素(TOC)自動計測器
JIS K 0970 ピストン式ピペット
307
K 0102:2016
JIS K 1101 酸素
JIS K 1105 アルゴン
JIS K 1107 窒素
JIS K 2251 原油及び石油製品−試料採取方法
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS K 8012 亜鉛(試薬)
JIS K 8013 亜鉛粉末(試薬)
JIS K 8019 亜硝酸ナトリウム(試薬)
JIS K 8032 アセトニトリル(試薬)
JIS K 8034 アセトン(試薬)
JIS K 8044 三酸化二ひ素(試薬)
JIS K 8048 4-アミノアンチピリン(試薬)
JIS K 8051 3-メチル-1-ブタノール(試薬)
JIS K 8059 亜硫酸水素ナトリウム(試薬)
JIS K 8061 亜硫酸ナトリウム(試薬)
JIS K 8069 アルミニウム(試薬)
JIS K 8080 アンチモン(試薬)
JIS K 8085 アンモニア水(試薬)
JIS K 8101 エタノール(99.5)(試薬)
JIS K 8102 エタノール(95)(試薬)
JIS K 8103 ジエチルエーテル(試薬)
JIS K 8107 エチレンジアミン四酢酸二水素二ナトリウム二水和物(試薬)
JIS K 8111 塩化亜鉛(試薬)
JIS K 8116 塩化アンモニウム(試薬)
JIS K 8121 塩化カリウム(試薬)
JIS K 8122 塩化カルシウム二水和物(試薬)
JIS K 8123 塩化カルシウム(試薬)
JIS K 8124 塩化カルシウム(乾燥用)(試薬)
JIS K 8136 塩化すず(II)二水和物(試薬)
JIS K 8138 塩化銅(I)(試薬)
JIS K 8139 塩化水銀(II)(試薬)
JIS K 8142 塩化鉄(III)六水和物(試薬)
JIS K 8150 塩化ナトリウム(試薬)
JIS K 8153 ヘキサクロロ白金(IV)酸六水和物(試薬)
JIS K 8155 塩化バリウム二水和物(試薬)
JIS K 8159 塩化マグネシウム六水和物(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8193 二塩化N,N-ジメチル-p-フェニレンジアンモニウム(試薬)
JIS K 8197 N-1-ナフチルエチレンジアミン二塩酸塩(試薬)
308
K 0102:2016
JIS K 8201 塩化ヒドロキシルアンモニウム(試薬)
JIS K 8202 塩化1,10-フェナントロリニウム一水和物(試薬)
JIS K 8223 過塩素酸(試薬)
JIS K 8228 過塩素酸マグネシウム(試薬)
JIS K 8230 過酸化水素(試薬)
JIS K 8247 過マンガン酸カリウム(試薬)
JIS K 8249 過よう素酸カリウム(試薬)
JIS K 8251 ガラスウール(試薬)
JIS K 8252 ペルオキソ二硫酸アンモニウム(試薬)
JIS K 8253 ペルオキソ二硫酸カリウム(試薬)
JIS K 8255 硫酸カリウムアルミニウム・12水(試薬)
JIS K 8264 ぎ酸(試薬)
JIS K 8267 ぎ酸ナトリウム(試薬)
JIS K 8271 キシレン(試薬)
JIS K 8283 くえん酸一水和物(試薬)
JIS K 8284 くえん酸水素二アンモニウム(試薬)
JIS K 8288 くえん酸三ナトリウム二水和物(試薬)
JIS K 8289 クペロン(試薬)
JIS K 8295 グリセリン(試薬)
JIS K 8306 p-クレゾール(試薬)
JIS K 8312 クロム酸カリウム(試薬)
JIS K 8318 p-トルエンスルホンクロロアミドナトリウム三水和物(試薬)
JIS K 8322 クロロホルム(試薬)
JIS K 8355 酢酸(試薬)
JIS K 8356 酢酸亜鉛二水和物(試薬)
JIS K 8357 ジエチルエーテル(残留農薬・PCB試験用)(試薬)
JIS K 8359 酢酸アンモニウム(試薬)
JIS K 8361 酢酸エチル(試薬)
JIS K 8371 酢酸ナトリウム三水和物(試薬)
JIS K 8374 酢酸鉛(II)三水和物(試薬)
JIS K 8377 酢酸ブチル(試薬)
JIS K 8397 サリチル酸ナトリウム(試薬)
JIS K 8432 酸化マグネシウム(試薬)
JIS K 8443 シアン化カリウム(試薬)
JIS K 8454 N,N-ジエチルジチオカルバミド酸ナトリウム三水和物(試薬)
JIS K 8465 1,2-ジクロロエタン(試薬)
JIS K 8470 L-システイン塩酸塩一水和物(試薬)
JIS K 8474 二しゅう酸三水素カリウム二水和物(試薬)
JIS K 8488 1,5-ジフェニルカルボノヒドラジド(試薬)
JIS K 8490 ジチゾン(試薬)
309
K 0102:2016
JIS K 8491 2,6-ジブロモ-N-クロロ-p-ベンゾキノンモノイミン(試薬)
JIS K 8495 p-ジメチルアミノベンジリデンロダニン(試薬)
JIS K 8498 ジメチルグリオキシム(試薬)
JIS K 8500 N,N-ジメチルホルムアミド(試薬)
JIS K 8506 臭化カリウム(試薬)
JIS K 8509 臭化水素酸(試薬)
JIS K 8517 二クロム酸カリウム(試薬)
JIS K 8522 しゅう酸カリウム一水和物(試薬)
JIS K 8528 しゅう酸ナトリウム(試薬)
JIS K 8529 臭素(試薬)
JIS K 8530 臭素酸カリウム(試薬)
JIS K 8532 L(+)-酒石酸(試薬)
JIS K 8533 ビス[(+)-タルトラト]二アンチモン(III)酸二カリウム三水和物(試薬)
JIS K 8536 (+)-酒石酸ナトリウムカリウム四水和物(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8544 硝酸アルミニウム九水和物(試薬)
JIS K 8545 硝酸アンモニウム(試薬)
JIS K 8548 硝酸カリウム(試薬)
JIS K 8550 硝酸銀(試薬)
JIS K 8552 硝酸コバルト(II)六水和物(試薬)
JIS K 8563 硝酸鉛(II)(試薬)
JIS K 8565 硝酸バリウム(試薬)
JIS K 8568 硝酸マンガン(II)六水和物(試薬)
JIS K 8574 水酸化カリウム(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8580 すず(試薬)
JIS K 8586 スルファニル酸(試薬)
JIS K 8588 アミド硫酸アンモニウム(試薬)
JIS K 8598 セレン(試薬)
JIS K 8603 ソーダ石灰(試薬)
JIS K 8612 タングステン(VI)酸ナトリウム二水和物(試薬)
JIS K 8613 炭酸アンモニウム(試薬)
JIS K 8617 炭酸カルシウム(試薬)
JIS K 8622 炭酸水素ナトリウム(試薬)
JIS K 8625 炭酸ナトリウム(試薬)
JIS K 8635 チオ尿素(試薬)
JIS K 8637 チオ硫酸ナトリウム五水和物(試薬)
JIS K 8646 デキストリン水和物(試薬)
JIS K 8653 デバルダ合金(試薬)
JIS K 8659 でんぷん(溶性)(試薬)
310
K 0102:2016
JIS K 8660 銅(試薬)
JIS K 8663 2,2′,2″-ニトリロトリエタノール(試薬)
JIS K 8680 トルエン(試薬)
JIS K 8701 鉛(試薬)
JIS K 8721 p-ニトロフェノール(試薬)
JIS K 8722 ペンタシアノニトロシル鉄(III)酸ナトリウム二水和物(試薬)
JIS K 8731 尿素(試薬)
JIS K 8736 エリオクロムブラックT(試薬)
JIS K 8747 バナジン(V)酸アンモニウム(試薬)
JIS K 8775 8-キノリノール(試薬)
JIS K 8776 2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-ナフトエ酸(試薬)
JIS K 8777 ピリジン(試薬)
JIS K 8780 ピロガロール(試薬)
JIS K 8785 二りん酸ナトリウム十水和物(試薬)
JIS K 8789 1,10-フェナントロリン一水和物(試薬)
JIS K 8798 フェノール(試薬)
JIS K 8799 フェノールフタレイン(試薬)
JIS K 8801 ヘキサシアノ鉄(III)酸カリウム(試薬)
JIS K 8809 フタル酸水素カリウム(試薬)
JIS K 8810 1-ブタノール(試薬)
JIS K 8815 ふっ化カリウム(試薬)
JIS K 8819 ふっ化水素酸(試薬)
JIS K 8824 D(+)-グルコース(試薬)
JIS K 8826 水酸化ナトリウム(窒素測定用)(試薬)
JIS K 8830 ウラニン(試薬)
JIS K 8832 ブルシンn水和物(試薬)
JIS K 8839 2-プロパノール(試薬)
JIS K 8840 ブロモクレゾールグリーン(試薬)
JIS K 8842 ブロモチモールブルー(試薬)
JIS K 8848 ヘキサン(試薬)
JIS K 8858 ベンゼン(試薬)
JIS K 8863 ほう酸(試薬)
JIS K 8866 四ほう酸ナトリウム十水和物(試薬)
JIS K 8885 二酸化けい素(試薬)
JIS K 8889 メタクレゾールパープル(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8893 メチルオレンジ(試薬)
JIS K 8896 メチルレッド(試薬)
JIS K 8897 メチレンブルー(試薬)
JIS K 8900 2-ブタノン(試薬)
311
K 0102:2016
JIS K 8903 4-メチル-2-ペンタノン(試薬)
JIS K 8905 七モリブデン酸六アンモニウム四水和物(試薬)
JIS K 8913 よう化カリウム(試薬)
JIS K 8920 よう素(試薬)
JIS K 8949 硫化ナトリウム九水和物(試薬)
JIS K 8951 硫酸(試薬)
JIS K 8953 硫酸亜鉛七水和物(試薬)
JIS K 8960 硫酸アンモニウム(試薬)
JIS K 8962 硫酸カリウム(試薬)
JIS K 8965 硫酸銀(試薬)
JIS K 8976 硫酸セリウム(IV)四水和物(試薬)
JIS K 8978 硫酸鉄(II)七水和物(試薬)
JIS K 8979 硫酸アンモニウム鉄(II)六水和物(試薬)
JIS K 8982 硫酸アンモニウム鉄(III)・12水(試薬)
JIS K 8983 硫酸銅(II)五水和物(試薬)
JIS K 8987 硫酸ナトリウム(試薬)
JIS K 8992 硫酸ヒドラジニウム(試薬)
JIS K 8995 硫酸マグネシウム七水和物(試薬)
JIS K 8997 硫酸マンガン(II)五水和物(試薬)
JIS K 9000 チオシアン酸アンモニウム(試薬)
JIS K 9001 チオシアン酸カリウム(試薬)
JIS K 9002 チオシアン酸ナトリウム(試薬)
JIS K 9003 流動パラフィン(試薬)
JIS K 9005 りん酸(試薬)
JIS K 9007 りん酸二水素カリウム(試薬)
JIS K 9009 りん酸二水素ナトリウム二水和物(試薬)
JIS K 9016 りん酸水素二アンモニウム(試薬)
JIS K 9017 りん酸水素二カリウム(試薬)
JIS K 9019 りん酸水素二ナトリウム・12水(試薬)
JIS K 9020 りん酸水素二ナトリウム(試薬)
JIS K 9038 ローダミンB(試薬)
JIS K 9047 L-グルタミン酸(試薬)
JIS K 9062 ニッケル(試薬)
JIS K 9066 スルファニルアミド(試薬)
JIS K 9501 アジ化ナトリウム(試薬)
JIS K 9502 L(+)-アスコルビン酸(試薬)
JIS K 9512 N,N-ジエチルジチオカルバミド酸銀(試薬)
JIS K 9523 ニトロメタン(試薬)
JIS K 9528 ジイソプロピルエーテル(試薬)
JIS K 9545 ビス(3-メチル-1-フェニル-5-ピラゾロン)(試薬)
312
K 0102:2016
JIS K 9547 フェニルフルオロン(試薬)
JIS K 9548 3-メチル-1-フェニル-5-ピラゾロン(試薬)
JIS K 9550 ポリビニルアルコール(試薬)
JIS K 9569 N-ベンゾイル-N-フェニルヒドロキシルアミン(試薬)
JIS K 9704 2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール(試薬)
JIS K 9901 高純度試薬−硝酸
JIS K 9902 高純度試薬−塩酸
JIS K 9904 高純度試薬−過塩素酸
JIS K 9905 高純度試薬−硫酸
JIS P 3801 ろ紙(化学分析用)
JIS R 1301 化学分析用磁器るつぼ
JIS R 1302 化学分析用磁器蒸発ざら
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS T 7322 医療用高圧蒸気滅菌器
JIS T 7324 医療用小型高圧蒸気滅菌器
JIS T 9107 使い捨て手術用ゴム手袋
JIS Z 0701 包装用シリカゲル乾燥剤
JIS Z 8719 条件等色指数−照明光条件等色度の評価方法
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
JIS Z 8802 pH測定方法
313
K 0102:2016
附属書1(参考)補足
この参考は,本体の規定に関する事柄を,本体に準じる形で補足したものであって,規定の一部ではな
い。
附属書1の構成
I.
電気伝導率における温度補正係数
II.
不揮発性鉱物油類及び不揮発性動植物油脂類
III.
フェノール標準液の標定
IV.
ドデシル硫酸ナトリウムの純度及び平均分子量の測定法
V.
陽イオン界面活性剤
VI.
ふっ素化合物における蒸留操作
VII. イオンクロマトグラフ法に用いる溶離液の例
VIII. 硫化物定量用ストリッピング装置を用いる定量法
IX.
硫酸イオンの硫酸バリウム比濁法
X.
サリチル酸塩を用いるインドフェノール青吸光光度法
XI.
イオン電極法によるアンモニウムイオン定量のための標準添加法
XII. ナトリウム(Na)のイオン電極法
XIII. ヘキサメチレンアンモニウム−ヘキサメチレンジチオカルバミド酸(HMA-HMDC)による溶媒抽
出法
XIV. 銀(Ag),バリウム(Ba)及びベリリウム(Be)のICP発光分光分析法
XV.
水銀(Hg),銀(Ag),バリウム(Ba)及びベリリウム(Be)のICP質量分析法
XVI. 薄層クロマトグラフ分離−原子吸光法
XVII. 飽和溶存酸素量と塩濃度,圧力,温度との影響補正
XVIII. JIS K 0102:2013の水中の飽和溶存酸素(表32.1)とこの規格の表32.1との対応
I.
電気伝導率における温度補正係数 電気伝導率測定において,温度補正係数を用い,天然水の電気伝
導率をθ ℃から25.0 ℃へ変換するための例を示す。温度補正係数の例を,附属書1表1に示す。
温度補正係数による換算は,次の式による。
κ25=κθ×fθ,25
ここに,
κ25: 25 ℃における電気伝導率
κθ: θ ℃における電気伝導率
fθ,25: κθをκ25に換算するための温度補正係数
なお,この表は,1985年に第1版として発行されたISO 7888を参考としてている。
備考 ISO 7888:1985,Water quality−Determination of electrical conductivity
314
K 0102:2016
附属書1表1 天然水の電気伝導率をθ ℃から25.0 ℃へ変換するための温度補正係数fθ,25の例
θ
fθ,25
℃
.0
.1
.2
.3
.4
.5
.6
.7
.8
.9
0
1.918
1.912
1.906
1.899
1.893
1.887
1.881
1.875
1.869
1.863
1
1.857
1.851
1.845
1.840
1.834
1.829
1.822
1.817
1.811
1.805
2
1.800
1.794
1.788
1.783
1.777
1.772
1.766
1.761
1.756
1.750
3
1.745
1.740
1.734
1.729
1.724
1.719
1.713
1.708
1.703
1.698
4
1.693
1.688
1.683
1.678
1.673
1.668
1.663
1.658
1.653
1.648
5
1.643
1.638
1.634
1.629
1.624
1.619
1.615
1.610
1.605
1.601
6
1.596
1.591
1.587
1.582
1.578
1.573
1.569
1.564
1.560
1.555
7
1.551
1.547
1.542
1.538
1.534
1.529
1.525
1.521
1.516
1.512
8
1.508
1.504
1.500
1.496
1.491
1.487
1.483
1.479
1.475
1.471
9
1.467
1.463
1.459
1.455
1.451
1.447
1.443
1.439
1.436
1.432
10
1.428
1.424
1.420
1.416
1.413
1.409
1.405
1.401
1.398
1.394
11
1.390
1.387
1.383
1.379
1.376
1.372
1.369
1.365
1.362
1.358
12
1.354
1.351
1.347
1.344
1.341
1.337
1.334
1.330
1.327
1.323
13
1.320
1.317
1.313
1.310
1.307
1.303
1.300
1.297
1.294
1.290
14
1.287
1.284
1.281
1.278
1.274
1.271
1.268
1.265
1.262
1.259
15
1.256
1.253
1.249
1.246
1.243
1.240
1.237
1.234
1.231
1.228
16
1.225
1.222
1.219
1.216
1.214
1.211
1.208
1.205
1.202
1.199
17
1.196
1.193
1.191
1.188
1.185
1.182
1.179
1.177
1.174
1.171
18
1.168
1.166
1.163
1.160
1.157
1.155
1.152
1.149
1.147
1.144
19
1.141
1.139
1.136
1.134
1.131
1.128
1.126
1.123
1.121
1.118
20
1.116
1.113
1.111
1.108
1.105
1.103
1.101
1.098
1.096
1.093
21
1.091
1.088
1.086
1.083
1.081
1.079
1.076
1.074
1.071
1.069
22
1.067
1.064
1.062
1.060
1.057
1.055
1.053
1.051
1.048
1.046
23
1.044
1.041
1.039
1.037
1.035
1.032
1.030
1.028
1.026
1.024
24
1.021
1.019
1.017
1.015
1.013
1.011
1.008
1.006
1.004
1.002
25
1.000
0.998
0.996
0.994
0.992
0.990
0.987
0.985
0.983
0.981
26
0.979
0.977
0.975
0.973
0.971
0.969
0.967
0.965
0.963
0.961
27
0.959
0.957
0.955
0.953
0.952
0.950
0.948
0.946
0.944
0.942
28
0.940
0.938
0.936
0.934
0.933
0.931
0.929
0.927
0.925
0.923
29
0.921
0.920
0.918
0.916
0.914
0.912
0.911
0.909
0.907
0.905
30
0.903
0.902
0.900
0.898
0.896
0.895
0.893
0.891
0.889
0.888
31
0.886
0.884
0.883
0.881
0.879
0.877
0.876
0.874
0.872
0.871
32
0.869
0.867
0.866
0.864
0.863
0.861
0.859
0.858
0.856
0.854
33
0.853
0.851
0.850
0.848
0.846
0.845
0.843
0.842
0.840
0.839
34
0.837
0.835
0.834
0.832
0.831
0.829
0.828
0.826
0.825
0.823
35
0.822
0.820
0.819
0.817
0.816
0.814
0.813
0.811
0.810
0.808
備考 附属書1表1の温度補正係数は,多数の天然水についての測定値の平均である。これらはκ25の値
が約6〜100 mS/mで,地下水,わき水及び表層水の組成と類似の水試料だけに適用する。
陽イオン 陰イオン
主成分
Ca2+
HCO3−
微量成分
Mg2+
SO42−,Cl−,NO3−
II. 不揮発性鉱物油類及び不揮発性動植物油脂類 ヘキサン抽出物質を不揮発性鉱物油類と不揮発性動植
物油脂類とに区分する。
1. 不揮発性鉱物油類 ヘキサン抽出物質として抽出定量したヘキサン抽出物質を再びヘキサンに溶か
315
K 0102:2016
し,活性けい酸マグネシウムカラムに通して不揮発性動植物油脂類などの極性物質を吸着除去し,流出液
を約80 ℃に加熱してヘキサンを揮散させ,残留する物質の質量をはかって不揮発性鉱物油類の濃度を求
める。
定量範囲:2〜100 mg,繰返し精度:10〜30 %
a) 試薬 試薬は,次による。
1) ヘキサン JIS K 8848に規定するもの。
2) 活性けい酸マグネシウム 粒径150〜250 μm 使用前にヘキサンで洗い,150 ℃で約2時間加熱し
てデシケーター中で放冷したもの(1)。
注(1) 活性けい酸マグネシウムには,市販品がある。
b) 器具及び装置 器具及び装置は,次による。
1) 活性けい酸マグネシウムカラム 内径約10 mm,長さ約150 mmのガラス管にコックを付けたもの
にJIS K 8251に規定するガラスウール(又はシリカウール)を詰め,その上120〜130 mmに活性
けい酸マグネシウムを気泡が混入しないようにヘキサンとともに充塡する(2)。コックにワセリンな
どの滑剤を塗布しない。
2) 乾燥器 本体24.2 b) 2)による。
3) 加熱板又はマントルヒーター 本体24.2 b) 3)による。
4) 蒸留装置 本体24.2 b) 4)による。
5) 蒸発容器 本体24.2 b) 5)による。
注(2) 極性基をもつ物質80〜100 mgを吸着する能力がある。使用した活性けい酸マグネシウムは,約
600 ℃で約4時間強熱すれば繰り返し使用できる。
c) 操作 操作は,次による。
1) 本体24.2及び24.3によってヘキサン抽出物質を測定した蒸発容器中に残留するヘキサン抽出物質
に,ヘキサン20〜30 mLを加えて溶かし,全量フラスコ100 mL (3)に移し入れる。
2) 再びヘキサン20〜30 mLを加えて残留物(4)が残らないように溶かし,全量フラスコ100 mLに合わ
せる。
3) ヘキサン10〜20 mLずつで蒸発容器を2,3回洗い,洗液も全量フラスコ100 mLに合わせ,ヘキサ
ンを標線まで加える。
4) このヘキサン溶液を活性けい酸マグネシウムカラムに徐々に注ぎ入れながら,流量1〜2 mL/minで
流出させる。
5) 最初の流出液約20 mL (5)を捨て,その後の流出液50 mLをとり,蒸留装置の蒸留フラスコに移し入
れる。
6) 蒸留フラスコをマントルヒーターに入れ,トの字形連結管及びリービッヒ冷却器を接続して,マン
トルヒーターを約80 ℃に調節し,ヘキサンを毎秒約1滴の留出速度で蒸留し,留出するヘキサン
を受器に受ける。蒸留フラスコ内の液量が約2 mLになるまで蒸留を続ける。トの字形連結管の上
部口からJIS K 1107に規定する窒素2級を室温になるまで送入する。
7) 蒸留フラスコ中の残留液を質量既知の蒸発容器に移し入れる。蒸留フラスコを少量のヘキサンで3
回洗い,この洗液も蒸発容器に加える。蒸発容器を約80 ℃に保った加熱板の上又はマントルヒー
ターの中に置いてヘキサンを揮散させる。
8) 蒸発容器の外側を湿った清浄な布などで拭い,次に,乾いた清浄な布などでよく拭って,80±5 ℃
に調節した乾燥器に入れ,約30分間加熱する。蒸発容器をデシケーター中で約30分間放冷した後,
316
K 0102:2016
その質量を0.1 mgの桁まではかり,蒸発容器の質量を差し引き,流出液中のヘキサン抽出物質の質
量(mg)を求める。
9) 空試験として,ヘキサン50 mLを蒸留フラスコにとり,6)〜8)の操作を行って残留物の質量(mg)
を求める。
10) 次の式によって試料中の不揮発性鉱物油類の濃度(mg/L)を算出する。
(
)
V
b
a
H
000
1
50
100×
×
−
=
ここに,
H: 不揮発性鉱物油類(mg/L)
a: 流出液中のヘキサン抽出物質の質量(mg)
b: 空試験における残留物の質量(mg)
V: 本体24.2 c)における試料量(mL)
注(3) ヘキサン抽出物質中に動植物油脂類などの極性物質が200 mg以上含まれている場合は,容量
100 mL以上の全量フラスコを用いる。ヘキサン100 mL中に動植物油脂類などの極性物質が200
mg以下になるように調製する。
(4) 残留物中に塩類が残留している場合は,ガラス棒などで残留物を細かく砕き,ヘキサンを加え
てヘキサン抽出物質を溶出させる。
(5) 不揮発性炭化水素の流出点は,活性けい酸マグネシウムの粒度と詰め方とで多少異なるので,
あらかじめ確認しておくとよい。
2. 不揮発性動植物油脂類 ヘキサン抽出物質の濃度と不揮発性鉱物油脂類の濃度との差を,不揮発性動
植物油脂類の濃度として求める。
A=P−H
ここに,
A: 不揮発性動植物油脂類(mg/L)
P: ヘキサン抽出物質(mg/L)
H: 不揮発性鉱物油類(mg/L)
III. フェノール標準液の標定 本体28.で調製したフェノール標準液(C6H5OH 1 mg/mL)の濃度の標定に
適用できる。
a) 試薬 試薬は,次による。
1) 臭素酸カリウム溶液( mol/L) JIS K 8530に規定する臭素酸カリウム2.78 gとJIS K 8506に規
定する臭化カリウム10 gとを水に溶かして1 Lとする。
2) 塩酸 JIS K 8180に規定するもの。
3) よう化カリウム JIS K 8913に規定するもの。
4) でんぷん溶液(10 g/L) 本体19. a) 5)による。
5) 0.1 mol/Lチオ硫酸ナトリウム溶液 本体19. a) 8)による。
b) 標定操作 標定操作は,次による。
1) 共栓三角フラスコ500 mLに調製したフェノール標準液(C6H5OH 1 mg/mL)50 mLをとり,水約100
mLを加える。これに臭素酸カリウム溶液( mol/L)50 mL(約40 mLに相当する量が反応する。)
を加え,更に塩酸5 mLを加える(このとき2,4,6-トリブロモフェノールの白い沈殿を生じる。)。密
栓して静かに振り混ぜ,褐色の臭素が遊離した後,約10分間放置する。
2) 次に,よう化カリウム約1 gを加え,遊離したよう素を0.1 mol/Lチオ硫酸ナトリウム溶液で滴定し,
溶液の黄色が薄くなってから,指示薬としてでんぷん溶液(10 g/L)1 mLを加え,生じたよう素で
60
1
60
1
317
K 0102:2016
んぷんの青い色が消えるまで滴定する。これに要した0.1 mol/Lチオ硫酸ナトリウム溶液の滴定mL
数を(b)とする。
3) 別に,水100 mLに臭素酸カリウム溶液( mol/L)20 mLを加えた溶液について,前と同様に操作
して滴定に要する0.1 mol/Lチオ硫酸ナトリウム溶液のmL数(a)を求める。次の式によって,フ
ェノール標準液の濃度(C6H5OH mg/mL)を算出する。
(
)
569
.1
50
1
5.2
×
×
×
−
=
f
b
a
P
ここに,
P: フェノール標準液の濃度(C6H5OH mg/mL)
f: 0.1 mol/Lチオ硫酸ナトリウム溶液のファクター
1.569: 0.1 mol/Lチオ硫酸ナトリウム溶液1 mLに相当するフェノー
ルの質量(mg)
IV. ドデシル硫酸ナトリウムの純度及び平均分子量の測定法 本体30.1の試験において用いるドデシル硫
酸ナトリウムの純度及び平均分子量を求める測定方法である。この測定方法は,必要な場合に行うとよい。
ドデシル硫酸ナトリウムの純度は,ドデシル硫酸ナトリウムの一定量を硫酸酸性で加熱還流後,冷却し,
フェノールフタレインを指示薬として1 mol/L水酸化ナトリウム溶液で微赤まで滴定し,滴定に要した水
酸化ナトリウム溶液の量から算出する。
平均分子量は,ドデシル硫酸ナトリウムの一定量を硫酸酸性で加熱還流後,冷却し,ヘキサンで高級ア
ルコール類を抽出した後,ヘキサン溶液をガスクロマトグラフに注入し,炭素数10〜14の各高級アルコー
ルのモル百分率を求め,算出する。
a) 試薬 試薬は,次による。
1) 硫酸(0.5 mol/L) 水1 L中にJIS K 8951に規定する硫酸30 mLを徐々に加える。
2) エタノール(95) JIS K 8102に規定するもの。
3) フェノールフタレイン溶液(5 g/L) 本体の15.の備考2.による。
4) ヘキサン JIS K 8848に規定するもの。
5) 硫酸ナトリウム JIS K 8987に規定するもの。
6) 1 mol/L水酸化ナトリウム溶液 水約60 mLをポリエチレン瓶などの耐アルカリ容器にとり,冷却
しながらJIS K 8576に規定する水酸化ナトリウム80 g(少量の水で表面を洗う。)を少量ずつ加え
て溶かし,密栓して4〜5日間放置する。その上澄み液50 mLをポリエチレン製の気密容器1 Lにと
り,本体2. n) 2)の二酸化炭素を含まない水を加えて1 Lとし,混合した後,二酸化炭素を遮断して
保存する。
標定 標定は,次による。
− JIS K 8005に規定する容量分析用標準物質のアミド硫酸を上口デシケーター中に2 kPa以下で
約48時間放置して乾燥する。その約2 gを1 mgの桁まではかりとり,三角フラスコ200 mLに
入れ,水約25 mLを加えて溶かす。
− これに指示薬としてブロモチモールブルー溶液(1 g/L)[本体16.の注(1)による。]3〜5滴を加
え,この1 mol/L水酸化ナトリウム溶液で滴定し,溶液の色が緑になったときを終点とする。
次の式によって1 mol/L水酸化ナトリウム溶液のファクター(f)を算出する。
1
097
.0
1
100
×
×
×
=
x
b
a
f
ここに,
a: アミド硫酸の質量(g)
60
1
318
K 0102:2016
b: アミド硫酸の純度(質量分率%)
x: 滴定に要した1 mol/L水酸化ナトリウム溶液(mL)
0.097 1: 1 mol/L水酸化ナトリウム溶液1 mLに相当するアミド硫酸
の質量(g)
7) 高級アルコール混合標準液 1-デカノール0.50 g,1-ドデカノール0.50 g及び1-テトラデカノール
0.50 gをはかりとり,JIS K 8848に規定するヘキサン15 mLに溶かす。
b) 器具及び装置 器具及び装置は,次による。
1) 還流冷却器付き三角フラスコ リービッヒ冷却部200〜300 mmの還流冷却器又は球管冷却器200〜
300 mm,及びすり合わせ三角フラスコ300 mL
2) 分液漏斗 300 mL
3) ガスクロマトグラフ 次の条件を満たすもの。
− カラム用管 ステンレス鋼製内径3〜4 mm,長さ1〜2 m
− カラム充塡剤 粒径150〜250 μmの耐火れんが(1)を担体とし,これにシリコーン系固定相液体
約10 %を含浸させたもの。
− 検出器 水素炎イオン化検出器
− キャリヤーガス JIS K 1107に規定する窒素2級又はヘリウム(体積百分率99.8 %)
− 試料気化室温度 290〜300 ℃
− カラム槽温度 170〜180 ℃
− キャリヤーガスの流量は,高級アルコール類が約30分間で流出するように調節する。
注(1) けい藻土を主成分とした耐火温度1 100 ℃のれんが。
参考 耐火れんがは,けい藻土を主成分とした耐火温度1 100 ℃のものがある。
カラム充塡剤には,市販品がある。
c) 純度の測定 操作は,次による。
1) ドデシル硫酸ナトリウム約4 gを1 mgの桁まではかりとり,三角フラスコ300 mLに入れる。
2) 硫酸(0.5 mol/L)20 mLを加えた後,還流冷却器を付けて沸騰水浴上で加熱する。泡立ちに注意し,
ときどき三角フラスコを軽く揺り動かし,液が透明になるまで加熱する。
3) 引き続き加熱板上で約2時間,加熱還流する。
4) 放冷後,冷却器の上部からエタノール(95)約30 mLを注いで内壁を洗い,適量の水で洗った後,
冷却器を取り除く。
5) 水を加えて液量を約100 mLとし,指示薬としてフェノールフタレイン溶液(5 g/L)5〜7滴を加え,
1 mol/L水酸化ナトリウム溶液で滴定し,溶液の色が微紅色になった点を終点とする。
6) 別に,同一条件で空試験を行い,次の式によってドデシル硫酸ナトリウムの純度(%)を算出する。
(
)
100
000
1
×
×
×
×
−
=
S
M
f
a
b
P
ここに,
P: ドデシル硫酸ナトリウムの純度(質量分率%)
a: 滴定に要した1 mol/L水酸化ナトリウム溶液(mL)
b: 空試験に要した1 mol/L水酸化ナトリウム溶液(mL)
f: 1 mol/L水酸化ナトリウム溶液のファクター
M: ドデシル硫酸ナトリウムの平均分子量
S: ドデシル硫酸ナトリウムの質量(g)
d) ドデシル硫酸ナトリウムの平均分子量の測定 操作は,次による。
1) c) 5)の操作で得られた溶液中から50 mLを分液漏斗300 mLにとる。
319
K 0102:2016
2) エタノール[体積分率66 %]100 mLとヘキサン50 mLとを加えて振り混ぜ,高級アルコール類を
抽出する。
3) 静置してヘキサン層を分離し,水層を別の分液漏斗300 mLに移す。
4) この水層にヘキサン50 mLを加え,振り混ぜて抽出し,静置してヘキサン層を分離する。水層を捨
て,ヘキサン層を前のヘキサン層に合わせる。
5) これに水50 mLを加えて振り混ぜ,静置してヘキサン層を分離し,水層を捨てる。この洗浄操作を
再び行い,水層を完全に分離して捨てる。
6) 硫酸ナトリウム約2 gを加えてヘキサン層を脱水した後,磁器蒸発皿100 mLに移し入れ,沸騰水浴
又は加熱板上でヘキサンを揮散させる。
7) 残留した高級アルコール類にヘキサン15 mLを加えて溶かす(高級アルコール約100 g/Lを含むヘ
キサン溶液になる。)。
8) ガスクロマトグラフをJIS K 0114に従って最適条件に設定し,高級アルコール混合標準液1 μLを
マイクロシリンジでとり,ガスクロマトグラフに注入し,各高級アルコールのクロマトグラムを記
録し,流出位置を確認する。
9) 次に,7)で得た高級アルコール類のヘキサン溶液1 μLをマイクロシリンジでとり,ガスクロマトグ
ラフに注入し,各高級アルコールのクロマトグラムを記録する。
10) 9)の操作を3回繰り返して行い,各高級アルコールのピーク面積を測定(半値幅法などで)し,ピ
ーク面積の平均値を求める。
11) 炭素数10〜14の各高級アルコールのモル百分率を次の式によって求め,ドデシル硫酸ナトリウムの
平均分子量を算出する。
100
14
14
12
12
10
10
10
10
10
×
+
+
=
C
R
C
R
C
R
C
R
m
100
14
14
12
12
10
10
12
12
12
×
+
+
=
C
R
C
R
C
R
C
R
m
100
14
14
12
12
10
10
14
14
14
×
+
+
=
C
R
C
R
C
R
C
R
m
102
100
100
100
14
14
12
12
10
10
+
×
+
×
+
×
=
m
C
m
C
m
C
M
ここに, m10〜m14: 炭素数10〜14の各高級アルコールのモル百分率(%)
C10〜C14: 炭素数10〜14の各高級アルコールの分子量(C10=158.29,
C12=186.34,C14=214.39)
R10〜R14: 炭素数10〜14の各高級アルコールのピーク面積
M: ドデシル硫酸ナトリウムの平均分子量
102: ドデシル硫酸ナトリウムに換算するための補正項
(SO4Naの式量からOHの式量を差し引いた値)
320
K 0102:2016
V. 陽イオン界面活性剤 陽イオン界面活性剤には,脂肪族アミン塩類,第四アンモニウム化合物,アル
キルピリジウム塩類などがある。陽イオン界面活性剤の定量には,オレンジII吸光光度法を適用するが,
通常の水の中では,陽イオン界面活性剤は陰イオン界面活性剤と安定なイオン対を作って存在するため,
直接は定量できない。このため,前処理(イオン交換分離)を行って陰イオン界面活性剤を除去した試料
について,オレンジII吸光光度法を適用する。
オレンジII吸光光度法 陽イオン界面活性剤が陰イオン性のオレンジII[4-(2-ヒドロキシ-1-ナフタレ
ニル)アゾベンゼンスルホン酸ナトリウム]と反応して生じるイオン対をクロロホルムに抽出して,吸光
度を測定し,テトラデシルジメチルベンジルアンモニウムクロリドとして表す。
定量範囲:陽イオン界面活性剤[CH3(CH2)12CH2N(CH3)2CH2C6H5Cl]20〜350 μg,
繰返し精度:3〜10 %
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 塩化ナトリウム JIS K 8150に規定するもの。
3) 硫酸ナトリウム JIS K 8987に規定するもの。
4) 酢酸-酢酸ナトリウム緩衝液(pH3.5) JIS K 8355に規定する酢酸6 mLとJIS K 8371に規定する酢
酸ナトリウム三水和物0.9 gとを水に溶かして1 Lとする。
5) オレンジII溶液 オレンジII[4-(2-ヒドロキシ-1-ナフタレニル)アゾベンゼンスルホン酸ナトリ
ウム五水和物]0.1 gを水に溶かして100 mLとする。
6) メタノール(体積百分率50 %) JIS K 8891に規定するメタノールを用いて調製する。
7) クロロホルム JIS K 8322に規定するもの。
8) 強塩基性陰イオン交換樹脂(I形) 本体30.2.1 a) 11)による。使用時にJIS K 8891に規定するメタ
ノールで3,4回,次に,水で3,4回洗浄する。
9) 陽イオン界面活性剤標準液[CH3(CH2)12CH2N(CH3)2CH2C6H5Cl 0.1 mg/mL] テトラデシルジメチ
ルベンジルアンモニウムクロリドをその100 %に対して0.100 gをはかりとり,水に溶かして全量フ
ラスコ1 000 mLに移し入れ,水を標線まで加える。使用時に調製する。
10) 陽イオン界面活性剤標準液[CH3(CH2)12CH2N(CH3)2CH2C6H5Cl 10 μg/mL] 陽イオン界面活性剤標
準液[CH3(CH2)12CH2N(CH3)2CH2C6H5Cl 0.1 mg/mL]20 mLを全量フラスコ200 mLにとり,水を標
線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) 円筒形滴下漏斗 100 mL
2) 分液漏斗 200 mL
3) 陰イオン交換樹脂カラム 内径約12 mm,長さ約300 mmのコック付きガラス管の下部にJIS K 8251
に規定するガラスウールを詰め,これに強塩基性陰イオン交換樹脂(I形)約25 mLに水を加えて
よく混合しながら,気泡が混入しないように充塡し,上部にガラスウールを詰め,水面がガラスウ
ールの上約10 mmになるように調節する。これを陰イオン交換樹脂カラムとする。上部に円筒形滴
下漏斗を取り付ける。
この陰イオン交換樹脂カラムは,繰り返し約20回使用できる。ただし,再生して再使用はできな
い。
4) 光度計 分光光度計又は光電光度計
321
K 0102:2016
c) 前処理 前処理は,次による。
1) 試料(1) 100 mL[CH3(CH2)12CH2N(CH3)2CH2C6H5Clとして20〜350 μgを含む。]を陰イオン交換樹脂
カラムの上部の円筒形滴下漏斗にとり,陰イオン交換樹脂カラムに流量約1 mL/minで流下させ(2),
流出液を三角フラスコ300 mLに受ける。
2) 陰イオン交換樹脂カラムの上部の円筒形滴下漏斗中の試料がなくなる寸前に流下を止め,メタノー
ル(体積百分率 50 %)50 mLを上部の円筒形滴下漏斗に加え,再び約1 mL/minで流下させ(2),流
出液は,先の三角フラスコ300 mLに合わせる。
注(1) 試料が酸性の場合は,水酸化ナトリウム溶液(40 g/L)[本体21. a) 3)による。]で,また,アル
カリ性の場合は,塩酸(1+11)[本体63.1 a) 1)による。]で中和する。
(2) 液面を陰イオン交換樹脂カラムの上部のガラスウールの上10 mm程度に保つようにする。
d) 操作 操作は,次による。
1) c) 2)で得た流出液を分液漏斗に移し入れ,酢酸-酢酸ナトリウム緩衝溶液(pH3.5)30 mL,塩化ナト
リウム1.5 g及びオレンジII溶液10 mLを加えて振り混ぜる。
2) クロロホルム10 mLを加え,約3分間激しく振り混ぜた後,放置する。
3) クロロホルム層をメスシリンダー(有栓形)20 mLに移す。
4) 水層にクロロホルム10 mLを加え,再び約3分間激しく振り混ぜた後,放置する。
5) クロロホルム層を3)のメスシリンダー(有栓形)20 mLに合わせ,クロロホルムを20 mLの標線ま
で加える。
6) 硫酸ナトリウム約3 gを加えて振り混ぜて水分を除く。
7) クロロホルム層の一部を吸収セルに移し,クロロホルムを対照液として波長485 nm付近の吸光度を
測定する。
8) 空試験として水100 mLとメタノール(体積百分率 50 %)50 mLとを分液漏斗にとり,1)〜7)の操
作を行って,試料について得た吸光度を補正する。
9) 検量線から陽イオン界面活性剤の量を求め,試料中の陽イオン界面活性剤の濃度[CH3(CH2)12CH2N
(CH3)2CH2C6H5Cl mg/L]を算出する。
e) 検量線 検量線の作成は,次による。
1) 陽イオン界面活性剤標準液[CH3(CH2)12CH2N(CH3)2CH2C6H5Cl 10 μg/mL]2〜35 mLを段階的に分液
漏斗200 mLにとり,水を加えて100 mLとし,これにメタノール(体積百分率 50 %)50 mLを加
え,d) 1)〜8)の操作を行って,陽イオン界面活性剤[CH3(CH2)12CH2N(CH3)2CH2C6H5Cl]の量と吸光
度との関係線を作成する。
備考 陽イオン界面活性剤の100倍以上の陰イオン界面活性剤が共存しても分離できる。硫酸イオン,
硝酸イオン,炭酸イオン及びりん酸イオンが共存しても妨害しない。
VI. ふっ素化合物における蒸留操作 ふっ素化合物の濃度が0.2 mg/L以上で,多量に無機物を含む試料中
の全無機ふっ化物の定量における蒸留方法を示す。
なお,この試験方法は,1994年に第1版として発行されたISO 10359-2を参考として記載した。
備考 ISO 10359-2:1994,Water quality−Determination of fluoride−Part 2: Determination of inorganically
bound total fluoride after digestion and distillation
a) 試薬 試薬は,次による。
1) 硫酸(10+3) 水150 mLにJIS K 8951に規定する硫酸500 mLを注意して冷却しながら加える。
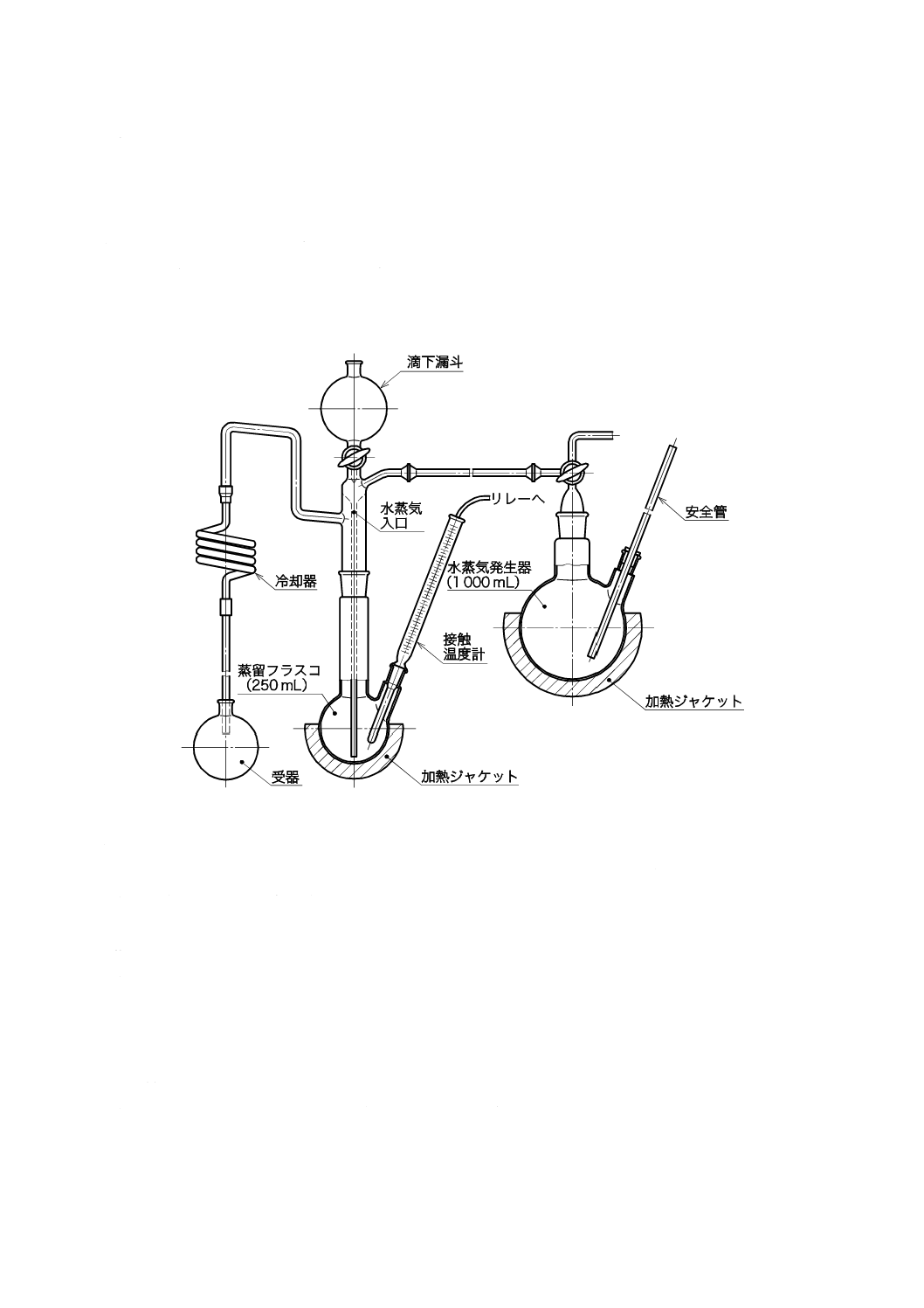
322
K 0102:2016
2) 水酸化ナトリウム溶液(100 g/L) JIS K 8576に規定する水酸化ナトリウム50 gを水に溶かして500
mLとする。
3) メチルレッド溶液 メチルレッドのナトリウム塩0.2 gをJIS K 8102に規定するエタノール(95)
100 mLに溶かす。
b) 器具及び装置 器具及び装置は,次による。
1) ニッケル皿 容量700 mLまでの適切な大きさのもの。
2) るつぼ 磁器又はニッケル製の容量60 mLのもの。
3) 蒸留装置 附属書1図1に例を示す。
附属書1図1 蒸留装置の例
c) 操作 操作は,次による。
1) 試料を振り混ぜ,適量(F−として0.1〜1 000 mgを含む。例えば,500 mL)をニッケル皿にとる。
2) 水酸化ナトリウム溶液(100 g/L)を加えてpHを11〜12に調節し,約30 mLになるまで蒸発濃縮す
る。
3) るつぼに移し入れ,過熱及び飛散を防ぐように,注意して乾固するまで蒸発する。
4) 残留物をJIS K 8576に規定する水酸化ナトリウム2 gで覆い,400〜500 ℃(暗赤熱)で約10分間
加熱した後,放冷し,融成物を少量の水に溶かす。
5) 全量が50 mLを超えないようにして,これを蒸留フラスコに移す。
6) フラスコを蒸留装置に連結し,滴下漏斗を用い,注意して,硫酸(10+3)60 mL,次いで,りん酸
10 mLを加える。
7) 水酸化ナトリウム溶液(100 g/L)20 mLを入れた全量フラスコ500 mLを受器とし,冷却器の内管
の先端を溶液中に浸す。
8) 水蒸気発生器及び蒸留フラスコの加熱ジャケットのスイッチを入れ,加熱し,蒸留フラスコ内容物
323
K 0102:2016
が沸騰し始めたら水蒸気を通す。
9) 蒸留フラスコの内容物が155 ℃に達するまで加熱を続け,この温度をほぼ一定に保つように加熱を
調節する。
10) 留出量が約10 mL/minとなるように水蒸気の供給量を調節する(1)。
11) 留出液の量が約450 mLになったら蒸留を止め,冷却管の内管の内外を少量の水ですすぐ(2)。
12) メチルレッドを指示薬として,全量フラスコの内容物を中和し,水を標線まで加える。
注(1) 多数の試料の分析を行うには,温度計を接触式温度計に代え,加熱をリレーで調節するとよい。
(2) 定量の妨害となるような,蒸留装置のふっ化物による汚染は,次の測定を行う前に,水を蒸留
(空蒸留)することによって防ぐことができる。
VII. イオンクロマトグラフ法に用いる溶離液の例 本体の35.の塩化物イオンなどの定量に用いるイオ
ンクロマトグラフ法においては,いろいろな溶離液が用いられ,その選択は,分離カラム及び検出器の種
類に依存する。そのため,適切な溶離液の組成については,カラムの製造業者の使用説明書に従う。次に,
溶離液の組成の例を示す。
なお,この溶離液の組成の例は,1992年に第1版として発行されたISO 10304-1,1995年に第1版とし
て発行されたISO 10304-2を参考として記載した。
備考 ISO 10304-1:1992,Water quality−Determination of dissolved fluoride,chloride,nitrite,orthophosphate,
bromide, nitrate and sulfate ions, using liquid chromatography of ions−Part 1: Method for water with
low contamination
ISO 10304-2:1995,Water quality−Determination of dissolved anions by liquid chromatography of ions
−Part 2: Determination of bromide, chloride, nitrate, nitrite, orthophosphate and sulfate in waste water
− 溶離液の取扱い 全ての溶離液は,脱気する。又は脱気した水を用いて溶離液を調製する。操作
中,溶離液に新たに気体が溶け込むのを避けるための手段を講じる(例えば,ヘリウム置換によ
る。)。細菌又は藻類の生育を避けるために,溶離液は暗所に貯蔵し,2〜3日ごとに更新する。
− サプレッサー手法を用いるイオンクロマトグラフィーの溶離液の例 サプレッサー手法の適用で
は,溶離液としては水酸化ナトリウム,炭酸ナトリウム−炭酸水素ナトリウム,炭酸水素ナトリ
ウム,四ほう酸ナトリウムなどの弱解離性酸の塩溶液を用いることができる。
− 炭酸ナトリウム−炭酸水素ナトリウム濃厚液 次の濃厚溶離液を試料に添加することは,試
料の前処理及び溶離液の調製に都合がよいことが示されている。
− JIS K 8625に規定する炭酸ナトリウム25.4 g及びJIS K 8622に規定する炭酸水素ナトリ
ウム25.5 gを全量フラスコ1 000 mLにとり,水に溶かし,水を標線まで加える。
− この溶液は,炭酸ナトリウム0.24 mol/L及び炭酸水素ナトリウム0.30 mol/Lを含み,4
〜6 ℃で貯蔵すれば,数箇月間は安定である。
− 炭酸ナトリウム−炭酸水素ナトリウム溶離液 次の溶離液は1回の測定で,臭化物イオン,
塩化物イオン,亜硝酸イオン,オルトりん酸イオン及び硫酸イオンを定量するのに適用でき
る。
− 濃厚液50 mLを全量フラスコ5 000 mLにとり,水を標線まで加える。
− この溶液は,炭酸ナトリウム0.002 4 mol/L及び炭酸水素ナトリウム0.003 mol/Lを含む。
− サプレッサー手法を用いないイオンクロマトグラフィーの溶離液の例 サプレッサーを用いない
イオンクロマトグラフィーには,塩溶液(例えば,フタル酸水素カリウム,p-ヒドロキシ安息香
324
K 0102:2016
酸,四ほう酸ナトリウム-グルコン酸ナトリウム及び安息香酸ナトリウム)が用いられる。これら
の溶液にはいろいろな添加物,例えば,アルコールを添加してもよい。濃度は通常,0.000 5〜0.01
mol/Lの範囲にある。これらのうち,ある種のアルカリ性濃厚液は化学的に安定ではないことに
注意する。
− シリカゲルを基材とした陰イオン交換体のための移動相 シリカゲルを基材とした陰イオン
交換体カラムを用いるイオンクロマトグラフィーには,pH1.5〜6.5の溶離液を使用する。
− フタル酸水素カリウム濃厚液 次の濃厚溶離液の試料への添加は,試料の前処理及び溶離液
の調製に有用であることが明らかにされている。
− フタル酸水素カリウム JIS K 8809に規定するフタル酸水素カリウム20.5 gを全量フラ
スコ1 000 mLにとり,水に溶かし,水を標線まで加える。
− この溶液はフタル酸水素カリウム0.1 mol/Lを含み,4〜6 ℃で保存すれば長期間安定で
ある。
− フタル酸水素カリウム溶離液 1回の測定で塩化物イオン,硝酸イオン,亜硝酸イオン,オ
ルトりん酸イオン及び硫酸イオンを定量するため,次の溶離液がよい結果を与えることが明
らかにされている。
− 濃厚液 100 mLを全量フラスコ5 000 mLにとり,これにJIS K 8891に規定するメタノー
ル 500 mLを加える。水で薄め,水酸化カリウム溶液(0.1 mol/L)(JIS K 8574に規定す
る水酸化カリウムを用いて調製する。)でpHを5に調節し,水を標線まで加える。
− この溶液は,フタル酸水素カリウム0.002 mol/L及びメタノール10 %(体積百分率)を
含む。
− 高分子を基材とする陰イオン交換体のための溶離液 高分子を基材とした陰イオン交換体を用い
たイオンクロマトグラフィーでは,塩基性だけでなく酸性の溶離液を用いることができる。
代表的な酸性溶離液の例として,フタル酸水素カリウムを含む溶液がある。代表的な塩基性溶
離液として,p-ヒドロキシ安息香酸塩又は四ほう酸ナトリウム-グルコン酸ナトリウムを含む溶液
がある。
塩化物イオン,硝酸イオン,オルトりん酸イオン及び硫酸イオンを1回の測定で定量するため,
四ほう酸ナトリウム-グルコン酸ナトリウム溶離液がよい結果を与えることが明らかにされてい
る。次の方法で調製する。
− 四ほう酸ナトリウム 0.85 g(JIS K 8866に規定する四ほう酸ナトリウム十水和物1.61 g)及
び水酸化リチウム 0.22 gを全量フラスコ5 000 mLにとる。これにグルコン酸水溶液(体積
百分率 50 %)0.6 mL,JIS K 8295に規定するグリセリン 3.1 mL及びJIS K 8032に規定する
アセトニトリル 600 mLを加え,水を標線まで加える。
VIII. 硫化物定量用ストリッピング装置を用いる定量法 硫化物定量用ストリッピング装置を用いる,工
場排水中の溶存硫化物の定量について示す。
なお,この試験方法は,1992年に第1版として発行されたISO 10530を参考として記載した。
備考 ISO 10530:1992,Water quality−Determination of dissolved sulfide−Photometric method using
methylene blue
a) 試薬 試薬は,次による。
1) 水 本体2. n) 1)による。
325
K 0102:2016
2) フタル酸塩緩衝液 JIS K 8809に規定するフタル酸水素カリウム80 gを水920 mLに溶かし,水酸
化ナトリウム溶液(40 g/L)[本体21. a) 3)による。]及び塩酸(1 mol/L)[本体21. a) 2)による。]で
pHを4.0に調節する。
3) 酢酸亜鉛溶液 本体39.2 a) 4)による。
4) 発色試薬 JIS K 8193に規定する二塩化N,N-ジメチル-p-フェニレンジアンモニウム2 gを水200 mL
に懸濁させ,JIS K 8951に規定する硫酸200 mLを注意して加え,冷却し,水を加えて1 000 mLと
する。
5) 硫酸アンモニウム鉄(III)溶液 水約300 mLにJIS K 8951に規定する硫酸10 mLを注意して加え,
冷却する。この溶液に,JIS K 8982に規定する硫酸アンモニウム鉄(III)・12水50 gを加えて溶か
し,水を加えて500 mLとする。
b) 操作 操作は,次による。
1) フタル酸塩緩衝液25 mLを反応フラスコにとる。
2) 酢酸亜鉛溶液20 mLを吸収容器にとる。
3) 装置を組み立て,溶液にJIS K 1107に規定する窒素2級を約40 L/hで2分間通気する。
4) 本体39.の備考3.で得たろ液を滴下漏斗から反応フラスコに入れる。滴下漏斗は水で洗い,洗液を反
応フラスコに入れる。
5) 反応フラスコの溶液に窒素[b) 3)による。]を40 L/hで30分間通気する。
6) 吸収容器を取り外し,側部のすり合わせジョイント部分から発色試薬10 mL,次いで硫酸アンモニ
ウム鉄(III)溶液1 mLを加える。
7) 吸収容器に水を満たし,栓をして振り混ぜ,10分間放置する。
8) 溶液を全量フラスコ100 mLに移し,吸収容器を水で洗って洗液も全量フラスコに合わせ,水を標
線まで加える。
9) 水を対照液として,波長665 nm付近の吸光度を測定する。
10) 空試験はろ液と同量の水を用いて,同じ操作を行う。
11) 検量線は,硫化物イオン標準液について,発色操作を行って作成する。
単位 mm
326
K 0102:2016
単位 mm
附属書1図2 溶存硫化物定量用ストリッピング装置
IX. 硫酸イオンの硫酸バリウム比濁法 試料に安定剤及び塩化バリウムを加え,一定の条件の下で硫酸イ
オンと反応させて硫酸バリウムとし,比濁して硫酸イオンを定量する。
定量範囲:SO42− 1〜5 mg,繰返し精度:10 %
a) 試薬 試薬は,次による。
1) グリセリン溶液[体積分率50 %] JIS K 8295に規定するグリセリンを用いて調製する。
2) 塩化ナトリウム溶液 JIS K 8150に規定する塩化ナトリウム240 gとJIS K 8180に規定する塩酸20
mLとを水に溶かして1 Lとする。
3) 塩化バリウム JIS K 8155に規定する塩化バリウム二水和物をふるい分け,JIS Z 8801-1に規定す
る目開き710 μmのふるいを通り,500 μmのふるいに止まるもの。
4) 硫酸イオン標準液(SO42− 1 mg/mL) 本体35.3 a) 16)による。
b) 装置 装置は,次による。
1) 光度計 分光光度計又は光電光度計
2) マグネチックスターラー
c) 操作 操作は,次による。
1) ろ過した試料(1)の適量(SO42−として1〜5 mgを含む。)を2個のコニカルビーカー100 mLにとり,
水を加えてそれぞれ50 mLとする。
2) それぞれにグリセリン溶液[体積分率50 %]10 mLと塩化ナトリウム溶液5 mLとを加え(2),マグ
ネチックスターラーを用いてかき混ぜる。
3) 一方のコニカルビーカーに塩化バリウム0.3 gを加え(2),それぞれ約1分間引き続いてかき混ぜた後,
約4分間放置し,再び15秒間かき混ぜる。
327
K 0102:2016
4) 直ちにこの液を吸収セルに移し,塩化バリウムを加えない方を対照液とし,1分間以内に波長450 nm
付近の吸光度(3)を測定する。
5) 空試験として水50 mLずつを2個のコニカルビーカーにとり,2)〜4)の操作を行って,試料につい
て得た吸光度を補正する。
6) 検量線から硫酸イオンの量を求め,試料中の硫酸イオンの濃度(SO42− mg/L)を算出する。
注(1) 酸性又はアルカリ性の試料の場合は,pHを約7に調節する。
(2) このときのpHは1.4〜1.6になる。
(3) 見掛けの吸光度。
d) 検量線 検量線の作成は,次による。
1) 硫酸イオン標準液(SO42− 1 mg/mL)1〜5 mLを段階的にとり,水を加えて50 mLとし,c) 2)〜5)
の操作を行って硫酸イオン(SO42−)の量と吸光度との関係線を作成する。
X. サリチル酸塩を用いるインドフェノール青吸光光度法 アンモニウムイオンがペンタシアノニトロシ
ル鉄(III)酸ナトリウムの存在下でサリチル酸塩及びジクロロイソシアヌル酸と反応して生じるインドフ
ェノール青の吸光光度法を測定してアンモニウムイオンを定量する。
定量範囲:NH4+ 2〜40 μg,繰返し精度:2〜10 %
なお,この試験方法は,1984年に第1版として発行されたISO 7150-1を参考として記載した。
備考 ISO 7150-1:1984,Water quality−Determination of ammonium−Part 1: Manual spectrometric method
a) 試薬 試薬は,次による。
1) 水 JIS K 0557に規定するA3の水
2) 発色試薬 JIS K 8397に規定するサリチル酸ナトリウム130 g及びJIS K 8288に規定するくえん酸
三ナトリウム二水和物130 gを全量フラスコ1 000 mL中の水に溶かす。水を加えて約950 mLとし,
JIS K 8722に規定するペンタシアノニトロシル鉄(III)酸ナトリウム二水和物0.970 gを加え,固形
物を溶かし,水を標線まで加える。
3) ジクロロイソシアヌル酸ナトリウム溶液 JIS K 8576に規定する水酸化ナトリウム32.0 gを水500
mLに溶かす。溶液を室温に冷却し,ジクロロイソシアヌル酸ナトリウム二水和物2.00 gを加える。
固形物を溶かし,溶液を全量フラスコ1 000 mLに移し,水を標線まで加える。
4) アンモニウムイオン標準液(NH4+ 1 mg/mL) 本体42.2 a) 5)による。
5) アンモニウムイオン標準液(NH4+ 100 μg/mL) アンモニウムイオン標準液(NH4+ 1 mg/mL)10 mL
を全量フラスコ100 mLにとり,水を標線まで加える。使用時に調製する。
6) アンモニウムイオン標準液(NH4+ 1 μg/mL) アンモニウムイオン標準液(NH4+ 100 μg/mL)1 mL
を全量フラスコ100 mLにとり,水を標線まで加える。使用時に調製する。
b) 器具及び装置 器具及び装置は,次による。
1) ガラス器具類 本体42.2 b) 1)による。
2) 分光光度計 光路長10〜50 mmの吸収セルが使用できるもの。
3) 恒温槽 25 ℃±1 ℃に保つ。
c) 操作 操作は,次による。
1) 試料(1)の適量(NH4+として2〜40 μgを含む。)を全量フラスコ50 mLにとり,必要があれば,水を
加えて約40 mLにする。
2) 発色試薬4.0 mLを加え,よく混ぜ合わせた後,ジクロロイソシアヌル酸ナトリウム溶液4.0 mLを
328
K 0102:2016
加え,よく混ぜ合わせる(2)。
3) 水を標線まで加える。全量フラスコをよく振り混ぜ,恒温槽中で25±1 ℃(3)に保つ。
4) 30分間以上経過した後(4),恒温槽から取り出し,この溶液の一部を吸収セルに移し,波長655 nm
付近で水を対照液として吸光度を測定する。
5) 空試験として,水40 mLについて2)〜4)の操作を行って吸光度を測定し,試料について得た吸光度
を補正する。
6) 検量線からアンモニウムイオンの量を求め,試料中のアンモニウムイオンの濃度(NH4+ mg/L)を
算出する。
注(1) 懸濁物を含む試料の場合は,沈降するのを待つか又は洗浄したガラス繊維ろ紙(孔径1 μm)で
自然ろ過する。また,試料が着色していたり,海水など塩類を多量に含む試料に適用する場合
は,あらかじめ本体42.1の蒸留操作を行う。
(2) このとき,pH12.6±0.1となることが望ましい。試料が極端に酸性又はアルカリ性であると誤差
を生じるので,あらかじめ水酸化ナトリウム溶液(40 g/L)[本体21. a) 3)による。]又は硫酸(1
+35)[本体43.2.1 a) 3)による。]で中和しておく。
(3) この方法による発色は,温度の影響を大きく受けるので,発色時の液温は正しく保つ。
(4) 30分間以降,その後60分間は一定の吸光度を示す。
d) 検量線 検量線の作成は,次による。
1) アンモニウムイオン標準液(NH4+ 1 μg/mL)2〜40 mLを段階的に全量フラスコ50 mLにとり,水
を加えて40 mLとする。
2) c) 2)〜5)の操作を行って吸光度を測定し,アンモニウムイオン(NH4+)の量と吸光度との関係線を
作成する。
備考 1. 妨害物質
a) アニリン,2-アミノエタノール(エタノールアミン)などの第一アミンが問題になるが,
通常の試料中には高濃度(1 mg/L程度)に存在することはまれである。
b) 強酸性及び強アルカリ性は,インドフェノール青の生成を妨害する。同様に,次亜塩素
酸を還元するような物質も妨害するが,通常の試料には存在しない。このような試料を
測定する場合は,次の備考2.の操作を行う。
c) 塩類濃度の高い試料では,くえん酸の錯化力を超えた場合にマグネシウムの沈殿が生じ
る。このような試料では,本体42.1の蒸留及び備考2.の操作を行う。
2. 蒸留による分離 試料が著しく着色し又は塩分を含んでいて,吸光度の測定に誤差を与えそ
うな場合,及び高濃度のマグネシウム又は塩化物の妨害が予想される場合は,試料を蒸留す
る。蒸留操作は,本体42.1による。ただし,留出液の捕集には塩酸(1+99)(JIS K 8180に
規定する塩酸を用いて調製する。)50 mLを用いる。次いで,留出液を水酸化ナトリウム溶液
(40 g/L)[本体21. a) 3)]で中和し,一定の体積200 mLとする。
e) 一般 低濃度のアンモニウムイオンの定量では,分析環境中の微量のアンモニアの影響を特に受けや
すい。本体42.に示す指示に十分注意すれば,この影響は少なくなるが,偏った結果の可能性は残る。
誤差の可能性を確かめる二つの方法は,次による。
f)
空試験及び校正標準吸光度値の監視 空試験液及び一連の検量線用溶液について得られた実測吸光
度値(対照セル中の水に対して測定した)を常に記録しておく。この測定値の記録によって,いかな
る偏差も発見できると考えられる。この種の偏差は,空試験液若しくは検量線用溶液のアンモニウム
329
K 0102:2016
汚染,又は一つ以上の試薬の欠陥に由来する。いずれの場合も改善策を講じなければならない。
g) 分析結果の正確さの確認
− この方法を最初に適用する場合,全標準偏差(少なくとも自由度9)の見積りを,最高濃度の検
量線用溶液の約50 %の濃度のコントロール用アンモニウム体窒素標準液について求めておくと
よい。
− このコントロール用標準液は,検量線用に用いてはならない。
− コントロール用標準液の一つについて,全分析操作を行う。測定値は,検量線から求める。この
コントロール用標準液の測定濃度は,次の式の範囲にならなければならない。
ρN2±3s1
ここに, ρN2: 溶液の濃度
s1: コントロール用標準液についてあらかじめ求めた標準偏差
− いずれの分析バッチにおいてもこの基準が達成できないときは,その原因を検討し,試験を繰り
返さなければならない。
− コントロール用標準液について,少なくとも20回以上の測定を行い,前述の基準を満足する全て
の値を用いて,今後の使用に備えてs1を再計算しなければならない。
XI. イオン電極法によるアンモニウムイオン定量のための標準添加法 本体42.4のイオン電極法によるア
ンモニウムイオンの定量法において,ある種の排水試料の試験は,試料中の妨害物質の影響によって複雑
となる。その影響を最小にするため,また,疑わしい結果を確かめるために,次に示す標準添加法を用い
ることができる。ただし,本体42.の備考12.で記載した妨害物質を含む試料の例のように,このような影
響が著しい場合は,試料の蒸留処理を行うとよい。
なお,この方法は,1984年に第1版として発行されたISO 6778を参考として記載した。
備考 ISO 6778:1984,Water quality−Determination of ammonium−Potentiometric method
− 本体42.4の備考10.に従って操作する。ただし,電位測定後,プローブは測定試料中に浸したまま
にしておく。
− 測定試料の予想するアンモニウムイオンの濃度が50〜100 %増加する程度の量のアンモニウムイオ
ン標準液を,測定試料に加える。新しい電位及び添加したアンモニウムイオン標準液の体積を記録
する。次の試料を分析する前に,アンモニア電極を水で十分にすすぐ(1) (2) (3)。
注(1) 試料中のアンモニウムイオンの濃度が予測できないならば,添加するアンモニウムイオン標準
液の体積は,電位変化が少なくとも20 mVを示すのに十分な量とする。
(2) アンモニウムイオン標準液の添加量は,測定試料の希釈を最小にするためできるだけ少なくす
ることが望ましい。
(3) この操作は,プローブの応答が直線となる範囲においてだけ適用する。
− 測定結果の表現 アンモニウムイオンの濃度,ρN(mg/L)は,次の式によって示す。
1
)1
(
)
(
log
2
1
N2
N
−
+
−
=
k
S
E
E
anti
kρ
ρ
ここに, ρN2: 添加に使用したアンモニウムイオン標準液のアンモニウムイ
オンの濃度(NH4+ mg/L)
E1: プローブの最初の電位(mV)
E2: プローブの最終の電位(mV)
330
K 0102:2016
S: アンモニウムイオン濃度10倍の変化当たりのmVで表した検
量線の傾き(k)
0
1
V
V
k=
ここに,
V0: 測定試料の量(mL)
V1: 添加に用いた標準液の量(mL)
XII. ナトリウム(Na)のイオン電極法 試料のpHを10.2〜10.6に調節し,ナトリウムイオン電極を指
示電極として電位を測定し,ナトリウムを定量する。
定量範囲:Na 1〜100 mg/L,繰返し精度:5〜20 %
a) 試薬 試薬は,次による。
1) トリス緩衝液 JIS K 9704に規定する2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール[トリス
(ヒドロキシメチル)アミノメタン]60 gをとり,水に溶かして1 Lとする。
2) ナトリウム標準液(Na 100 mg/L) 本体48.1 a) 1)のナトリウム標準液(Na 1 000 mg/L)20 mLを全
量フラスコ200 mLにとり,水を標線まで加える。
3) ナトリウム標準液(Na 10 mg/L) ナトリウム標準液(Na 100 mg/L)20 mLを全量フラスコ200 mL
にとり,水を標線まで加える。
4) ナトリウム標準液(Na 1 mg/L) ナトリウム標準液(Na 10 mg/L)20 mLを全量フラスコ200 mLに
とり,水を標線まで加える。
b) 装置 装置は,次による。
1) 電位差計 本体34.2 b) 1)による。
2) 指示電極 ナトリウム電極[ガラス膜(1)電極]
3) 参照電極 本体34.2 b) 3)による。ただし,外筒液には硝酸アンモニウム溶液(100 g/L)(JIS K 8545
に規定する硝酸アンモニウムを用いて調製する。)又は硝酸カリウム溶液(100 g/L)(JIS K 8548に
規定する硝酸カリウムを用いて調製する。)を用いる。
4) 測定容器(セル) 本体34.2 b) 4)による。
5) 恒温槽 本体34.2 b) 5)による。
6) マグネチックスターラー 本体34.2 b) 6)による。
注(1) 液体膜のナトリウム電極を用いる場合は,備考3.による。
c) 検量線 検量線の作成は,次による。
1) ナトリウム標準液(Na 1 mg/L)100 mLを測定容器にとり,トリス緩衝液10 mL (2)を加える。
2) この測定容器(セル)のまま水浴を用いて,液温を一定温度(例えば,25±0.5 ℃)にする。
3) 水浴で液温が一定に保たれている測定容器(セル)に,指示電極(3) (4)と参照電極(5) (6)とを浸し,固
定した後,回転子を入れ,マグネチックスターラー(7)を用いて,泡が電極に触れない程度に強くか
き混ぜながら(8),電位差計で電位を測定する(9)。
4) ナトリウム標準液(Na 10 mg/L)及びナトリウム標準液(Na 100 mg/L)100 mLを測定容器にとり,
トリス緩衝液10 mL (2)をそれぞれ加える。2)及び3)の操作を行ってナトリウム標準液(Na 10〜100
mg/L)の電位を測定する(9)。
5) 縦軸にナトリウムの濃度の対数を,横軸に電位(mV)をとって,ナトリウムの濃度(Na mg/L)と
電位との関係線を作成する(10) (11)。
注(2) トリス緩衝液は,測定時のpHを10.2〜10.6に調節し,イオン強度を一定にするためのもので
331
K 0102:2016
ある。
注(3) ナトリウムイオン電極は,使用時にナトリウム標準液(Na l mg/L)に浸し,指針が安定してか
ら電位を測定する。
(4) 本体34.の注(13)による。
(5) 本体34.の注(10)による。
(6) 本体34.の注(14)による。
(7) 本体34.の注(15)による。
(8) 本体34.の注(16)による。
(9) ナトリウムイオン電極の応答時間は,液温10〜30 ℃の場合は2〜3分間である。
(10) ナトリウム標準液Na 1 mg/LとNa 100 mg/Lとの電位の差は,100〜120 mV(25 ℃)の範囲に
入り,ナトリウムの濃度1〜100 mg/Lの間の検量線は直線になる。
(11) 本体34.の注(19)による。
d) 操作 操作は,次による。
1) 試料100 mL(12)を測定容器(セル)にとり,トリス緩衝液10 mL(2)を加える。
2) c) 2)及び3)の操作を行って(11),検量線からナトリウムの濃度を求め,試料中のナトリウムの濃度(Na
mg/L)を算出する。
3) 空試験として,試料の代わりに水を用いてc) 1)及び3)の操作を行って,試料について得たナトリウ
ムの濃度を補正する。
注(12) 試料が酸性又は強アルカリ性で,トリス緩衝液を加えてもpHが10.2〜10.6に入らない場合に
は,あらかじめ試料を全量フラスコ200 mLに入れ,これをアンモニア水(1+1)[本体52.1 a)
1)による。]又は塩酸(1+2)[本体58.1 a) 1)による。]で,pHを約10 に調節し,水を標線ま
で加える。これから100 mLをとり,1)以降の操作を行って測定したナトリウムの濃度(Na mg/L)
を希釈割合に応じて補正し,試料中のナトリウムの濃度(Na mg/L)を求める。
備考 1. イオン濃度計の場合には,ナトリウム標準液(Na 1 mg/L)とナトリウム標準液(Na 100 mg/L)
とを用いて,c) 1)〜3)の操作を行ってイオン濃度計の指針をそれぞれNa 1 mg/LとNa 100
mg/Lとになるように調節する。さらに,ナトリウム標準液(Na 10 mg/L)を用いてイオン濃
度計の指示値を確認する。
2. ナトリウムイオン電極(ガラス膜)における主な共存物質の許容限度を,最大比率で次に示
す。
Li+:4.5×10,K+:4×102,NH4+:1.3×103,Ag+:0.2
3. 液体膜のナトリウムイオン電極を用いる場合は,参照電極の外筒液に酢酸リチウム溶液(1
mol/L)又は酢酸リチウム緩衝液(酢酸リチウム二水和物102 gを水約500 mLに溶かし,こ
れにJIS K 8355に規定する酢酸2 mL及び水を加えて全量1 Lとする。pH6〜7になる。)を
用いる。検量線は,ナトリウム標準液100 mL当たり酢酸リチウム緩衝液10 mLを加え,pH
を6〜7に調節し,c) 2)以降の操作を行って作成する。測定は,検量線の作成と同じ条件にな
るように操作する。
この場合の共存物質の許容限度を,最大比率で次に示す。
K+:1.25×102Li+:6.25×103,NH4+:1.4×103,Ca2+:1.5×104
また,界面活性剤が共存するとドリフトを生じるので注意する。
332
K 0102:2016
XIII. ヘキサメチレンアンモニウム−ヘキサメチレンジチオカルバミド酸(HMA-HMDC)による溶媒抽
出法 この試験方法は,本体52.2 c)の準備操作を行った溶液の銅の濃度が低い場合に適用する。ただし,
この操作は,銅,鉛,カドミウム,コバルト及びニッケルの定量に適用する。
なお,この試験方法は,1986年に第1版として発行されたISO 8288の第3章の抽出操作を参考として
記載した。
備考 ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−
Flame atomic absorption spectrometric methods
a) 試薬 試薬は,次による。
1) HMA-HMDC抽出液(6.8 g/L) ヘキサメチレンアンモニウム−ヘキサメチレンジチオカルバミド
酸(HMA-HMDC)1.7 gをビーカーにとり,JIS K 8271に規定するキシレン74 mLに静かに加熱し
ながら溶かす。放冷後,2,4-ジメチル-3-ペンタノン(ジイソプロピルケトン,DIPK)(bp 124.5 ℃)
を加えて250 mLとする。
2) HMA-HMDCメタノール溶液(55 g/L) HMA-HMDC5.5 gをビーカーにとり,JIS K 8891に規定す
るメタノールを加え,静かに加熱しながら溶かす。放冷後,メタノールを加えて100 mLとする。
3) ぎ酸塩緩衝液 JIS K 8264に規定するぎ酸(純度98.0 %以上)368 g及びJIS K 8283に規定するく
えん酸一水和物14 gを水350 mLに溶かす。この溶液を振り混ぜ,冷却しながらJIS K 8576に規定
する水酸化ナトリウム243 gを徐々に加える。JIS K 8889に規定するメタクレゾールパープル(m-
クレゾールスルホフタレイン)50 mgを加えて,振り混ぜる。この溶液をかき混ぜながら,
HMA-HMDC抽出液(6.8 g/L)で連続2回抽出して精製する。
4) 銅標準液(Cu 1 mg/mL) 本体52.4 a) 1)による。
5) 銅標準液-DIPK(Cu 50 μg/mL) 銅標準液(Cu 1 mg/mL)5 mLを全量フラスコ100 mLにとり,ぎ
酸50 mL及びくえん酸一水和物0.2〜0.5 gを加え,振り混ぜる。DIPKを標線まで加える。
b) 操作 操作は,次による。
1) 試料(通常400 mL)を全量フラスコ500 mLにとり,ぎ酸塩緩衝液20 mLを加える。このとき指示
薬の色は純黄色である。赤が現れたときは,ぎ酸塩緩衝液20 mLを追加する。
2) HMA-HMDCメタノール溶液(55 g/L)2 mLを加え振り混ぜた後,3〜5分間放置する。
3) HMA-HMDC抽出液(6.8 g/L)20 mLを加え,3〜5分間激しく振り混ぜ,10〜15分間静置する。次
に,全量フラスコの首の部分に有機層がくるまで静かに水を加える(1)。
4) 定量操作は,本体52.2 d)及びe)による。ただし,検量線用標準液の調製は,c)による。
注(1) 有機層を噴霧する場合は,全量フラスコの首の部分から直接行ってもよい。また,有機層を長
時間保存するときは,水層が残らないようにピペットでこれを取り出し,0〜10 ℃の暗所に保
存する。
c) 検量線用標準液の調製 銅標準液-DIPK(Cu 50 μg/L)を,本体52.2 e)の検量線と同じ濃度範囲になる
ように全量フラスコ25 mLに段階的にとり(2),HMA-HMDC抽出液(6.8 g/L)を標線まで加える。使
用時に調製する。
注(2) 少なくとも4段階で調製する。また,標準液の採取にマイクロピペット(マイクロピペットの
プラスチックチップを硝酸に3〜5時間浸した後,使用前に水ですすいだもの。40 ℃を超える
温度は避ける。)を用いるとよい。
d) 抽出操作の確認 抽出操作の確認は,次によるとよい。
1) 試料を分析する前に,試料の予想濃度の範囲で銅標準液(Cu 1 mg/mL)を用いて少なくとも2段階
333
K 0102:2016
の濃度で調製する。希釈には硝酸(1+1)[本体52.1 a) 8)による。]を用いる。
2) b) 1)〜3)の抽出操作を行い,本体52.2 d) 1)の操作を行って指示値を読み取る。
3) 銅標準液-DIPK(Cu 50 μg/mL)を用いて1)で調製した20〜50倍濃度のものを少なくとも2段階調
製する。続いて,本体52.2 d) 1)の操作を行って指示値を読み取る。
4) 2)で得た指示値と3)で得た指示値とが同じ吸光度であることを確認する。
e) 妨害物質 妨害物質を,次に示す。
− 鉄を含めて重金属元素は,20 mg/Lまで許容できる。重金属元素の全濃度が20 mg/Lを超える場
合は,測定試料と抽出液との体積比を20:1未満にしなければならない。
− ニトリロ三酢酸の許容濃度は,250 mg/Lである。エチレンジアミン四酢酸(EDTA)はニッケル
の抽出を妨害する。エチレンジアミン四酢酸二ナトリウム二水和物25 mg/Lは,銅,亜鉛,鉛,
カドミウム及びコバルトの抽出には影響しない。
− 天然水中に含まれるフミン酸は,酸性にすると沈殿する。この沈殿は,ろ過によって除去すると
よい。沈殿には,重金属元素は取り込まれない。ろ液中の重金属元素は,b) 3)のヘキサメチレン
ジチオカルバミド酸のジイソプロピルケトン-キシレン抽出によって完全に回収できる。
XIV. 銀(Ag),バリウム(Ba)及びベリリウム(Be)のICP発光分光分析法
試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,これらの金属元素による発光
を所定の波長で測定して定量する。
なお,この方法は,1996年に第1版として発行されたISO 11885を参考として記載した。
備考 ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic
emission spectroscopy
1. 銀(Ag) 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,銀による発光を
波長328.068 nm(又は338.289 nm)で測定して定量する。
定量範囲:Ag 20〜5 000 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) 銀標準液(Ag 0.1 mg/mL) JIS K 8550に規定する硝酸銀0.157 5 gを,全量フラスコ1 000 mLにと
り,硝酸(1+1)[本体52.1 a) 8)による。]20 mLを加えた後,水を標線まで加える(1)。暗所で保存
する。
注(1) 原子吸光用銀標準液(Ag 1 000 mg/L)を用いてもよい。
2) 銀標準液(Ag 10 μg/mL) 銀標準液(Ag 0.1 mg/mL)10 mLを全量フラスコ100 mLにとり,硝酸(1
+1)[本体52.1 a) 8)による。]2 mLを加えた後,水を標線まで加える。使用時に調製する。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
c) 準備操作 準備操作は,次による。
1) 試料を本体5.によって処理する。
d) 操作 操作は,次による。
1) 本体52.4 d) 1)による。ただし,波長328.068 nm(又は338.289 nm)の発光強度を測定する(2) (3) (4)。
2) 本体52.4 d) 2)及び3)の操作を行い,試料中の銀の濃度(Ag μg/L)を算出する。
注(2) 本体52.の備考11.による。
(3) 本体47.の注(8)による。
334
K 0102:2016
注(4) 本体47.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) 銀標準液(Ag 10 μg/mL)0.2〜50 mLを全量フラスコ100 mLに段階的にとり,引き続き本体52.4 e)
1)〜3)の操作を行い,銀(Ag)の量と発光強度との関係線を作成する。検量線の作成は,試料測定
時に行う。
2. バリウム(Ba) 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,バリウム
による発光を波長233.527 nm(又は455.403,493.409 nm)で測定して定量する。
定量範囲:Ba 10〜5 000 μg/L, 繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) バリウム標準液(Ba 0.1 mg/mL) JIS K 8565に規定する硝酸バリウム0.190 3 gを,全量フラスコ1
000 mLにとり,硝酸(1+1)[本体52.1 a) 8)による。]20 mLを加えた後,水を標線まで加える(1)。
注(1) 原子吸光用バリウム標準液(Ba 1 000 mg/L)を用いてもよい。
2) バリウム標準液(Ba 10 μg/mL) バリウム標準液(Ba 0.1 mg/mL)10 mLを全量フラスコ100 mLに
とり,硝酸(1+1)[本体52.1 a) 8)による。]2 mLを加えた後,水を標線まで加える。使用時に調
製する。
b) 装置 装置は,次による。
1) ICP発光分光分析装置
c) 準備操作 準備操作は,次による。
1) 試料を本体5.によって処理する。
d) 操作 操作は,次による。
1) 本体52.4 d) 1)による。ただし,波長233.527 nm(又は455.403,493.409 nm)の発光強度を測定す
る(2) (3) (4)。
2) 本体52.4 d) 2)及び3)の操作を行い,試料中のバリウムの濃度(Ba μg/L)を算出する。
注(2) 本体52.の備考11.による。
(3) 本体47.の注(8)による。
(4) 本体47.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) バリウム標準液(Ba 10 μg/mL)0.1〜50 mLを全量フラスコ100 mLに段階的にとり,引き続き本体
52.4 e) 1)〜3)の操作を行い,バリウム(Ba)の量と発光強度との関係線を作成する。検量線の作成
は,試料測定時に行う。
3. ベリリウム(Be) 試料を前処理した後,試料導入部を通して誘導結合プラズマ中に噴霧し,ベリリ
ウムによる発光を波長313.042 nm(又は234.861,313.107 nm)で測定して定量する。
定量範囲:Be 5〜2 000 μg/L,繰返し精度:2〜10 %(装置及び測定条件によって異なる。)
a) 試薬 試薬は,次による。
1) ベリリウム標準液(Be 0.1 mg/mL) 硫酸ベリリウム四水和物1.966 gを,全量フラスコ1 000 mLに
とり,硝酸(1+1)[本体52.1 a) 8)による。]20 mLを加えた後,水を標線まで加える(1)。
注(1) 原子吸光用ベリリウム標準液(Be 1 000 mg/L又はBe 100 mg/L)を用いてもよい。
2) ベリリウム標準液(Be 10 μg/mL) ベリリウム標準液(Be 0.1 mg/mL)10 mLを全量フラスコ100 mL
にとり,硝酸(1+1)[本体52.1 a) 8)による。]2 mLを加えた後,水を標線まで加える。
b) 装置 装置は,次による。
335
K 0102:2016
1) ICP発光分光分析装置
c) 準備操作 準備操作は,次による。
1) 試料を本体の5.によって処理する。
d) 操作 操作は,次による。
1) 本体52.4 d) 1)による。ただし,波長313.042 nm(又は234.861,313.107 nm)の発光強度を測定す
る(2) (3) (4)。
2) 本体52.4 d) 2)及び3)の操作を行い,試料中のベリリウムの濃度(Be μg/L)を算出する。
注(2) 本体52.の備考11.による。
(3) 本体47.の注(8)による。
(4) 本体47.の注(9)による。
e) 検量線 検量線の作成は,次による。
1) ベリリウム標準液(Be 10 μg/mL)0.1〜40 mLを全量フラスコ200 mLに段階的にとり,引き続き本
体52.4 e) 1)〜3)の操作を行い,ベリリウム(Be)の量と発光強度との関係線を作成する。検量線の
作成は,試料測定時に行う。
XV.
水銀(Hg),銀(Ag),バリウム(Ba)及びベリリウム(Be)のICP質量分析法
試料を前処理した後,内標準元素を加え,試料導入部を通して高周波プラズマ中に噴霧し,測定対象元
素及び内標準元素のそれぞれの質量/電荷数における指示値(1)を測定し,測定対象元素の指示値と内標準元
素の指示値との比を求めて測定対象元素を定量する。この方法によって附属書1表2に示す元素が同時定
量できる。それぞれの元素ごとの定量範囲,繰返し精度などの例を,附属書1表2に示す。
注(1) 本体52.の注(19)による。
附属書1表2 定量範囲,繰返し精度及び質量数の例*
対象元素
定量範囲
μg/L
繰返し精度
%
質量数
水銀(Hg)
0.5〜500
2〜10
202,200
銀(Ag)
0.5〜500
2〜10
107,109
バリウム(Ba)
0.5〜500
2〜10
137,138
ベリリウム(Be)
0.5〜500
2〜10
9
イットリウム(Y)**
−
−
89
インジウム(In)**
−
−
115
ビスマス(Bi)**
−
−
209
注*
装置及び測定条件によって異なる。
** 内標準元素
a) 試薬 試薬は,次による。
1) 水 本体52.3 a) 1)による。
2) 硝酸(1+1) 本体52.3 a) 2)による。
3) 内標準液(1 μg/mL) 本体52.5 a) 3)による(2)。
4) 水銀標準液(Hg 10 μg/mL) 本体66.1.1 a) 9)による(3)。
5) 水銀標準液(Hg 0.5 μg/mL) 水銀標準液(Hg 10 μg/mL)5 mLを全量フラスコ100 mLにとり,硝
酸(1+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(3)。

336
K 0102:2016
6) 銀標準液(Ag 10 μg/mL) 附属書1 XIV.の1. a) 2)による(3)。
7) 銀標準液(Ag 0.5 μg/mL) 銀標準液(Ag 10 μg/mL)5 mLを全量フラスコ100 mLにとり,硝酸(1
+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(3)。
8) バリウム標準液(Ba 10 μg/mL) 附属書1 XIV.の2. a) 2)による(3)。
9) バリウム標準液(Ba 0.5 μg/mL) バリウム標準液(Ba 10 μg/mL)5 mLを全量フラスコ100 mLに
とり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(3)。
10) ベリリウム標準液(Be 10 μg/mL) 附属書1 XIV.の3. a) 2)による(3)。
11) ベリリウム標準液(Be 0.5 μg/mL) ベリリウム標準液(Be 10 μg/mL)5 mLを全量フラスコ100 mL
にとり,硝酸(1+1)2 mLを加えた後,水を標線まで加える。使用時に調製する(3)。
12) 混合標準液[(Hg 10 μg,Ag 10 μg,Ba 10 μg,Be 10 μg)/mL](3) (4) 各元素の標準液(0.1 mg/mL)
(5) 50 mL[水銀の場合は,本体66.1.1 a) 8)水銀標準液(Hg 0.5 mg / mL)10 mL]を全量フラスコ500
mLにとり,硝酸(1+1)10 mLを加えた後,水を標線まで加える。使用時に調製する。
13) 混合標準液[(Hg 0.5 μg,Ag 0.5 μg,Ba 0.5 μg,Be 0.5 μg)/mL](3) (4) 12)の混合標準液5 mLを
全量フラスコ100 mLにとり,硝酸(1+1)2 mLを加え,水を標線まで加える。使用時に調製する。
注(2) 本体52.の注(20)による。
(3) 本体52.の注(21)による。
(4) 本体52.の注(16)による。
(5) 本体52.の注(22)による。
b) 装置 装置は,次による。
1) ICP質量分析装置
備考 1. 本体52.の備考13.による。
2. 本体52.の備考14.による。
c) 準備操作 準備操作は,本体52.5 c)による。
d) 操作 操作は,本体52.5 d)による(6)。
注(6) 本体52.の注(24)による。ただし,スペクトルの干渉の例は,附属書1表3による。
e) 検量線 検量線の作成は,本体52.5 e)による。
備考 3. 本体52.の備考16.による。
附属書1表3 スペクトル干渉の例*
元素
質量数
同重体及び2価
イオンの干渉
多原子イオンの
干渉
水銀(Hg)
200
184W16O
水銀(Hg)
202
銀(Ag)
107
91Zr16O
銀(Ag)
109
93Nb16O,92Zr16OH
バリウム(Ba)
137
バリウム(Ba)
138
La+,Ce+
ベリリウム(Be)
9
イットリウム(Y)**
89
インジウム(In)**
115
Sn+
ビスマス(Bi)**
209
注*
装置及び測定条件によって異なる。
** 内標準元素
337
K 0102:2016
XVI. 薄層クロマトグラフ分離−原子吸光法 アルキル水銀(II)化合物をベンゼンで抽出し,アルミナ
カラムで濃縮する。これを薄層クロマトグラフ法によって分離し,塩化メチル水銀(II)化合物又は塩化
エチル水銀(II)化合物に相当するスポットを剝離し,原子吸光法によって水銀として定量する。
定量範囲:Hg 0.5 μg/L以上
a) 試薬 試薬は,次による。
1) 塩酸 JIS K 8180に規定するもの。
2) 塩酸(1+10) JIS K 8180に規定する塩酸を用いて調製する。
3) 塩酸(1+50) JIS K 8180に規定する塩酸を用いて調製する。
4) アンモニア水(1+1) JIS K 8085に規定するアンモニア水を用いて調製する。
5) 活性アルミナ 活性アルミナ[クロマトグラフ吸着用(50 μm)]100 gに水を加えてかき混ぜる。
メチルレッドのエタノール溶液(1 g/L)[JIS K 8896に規定するメチルレッド0.1 gをJIS K 8102に
規定するエタノール(95)100 mLに溶かす。着色瓶に保存する。]2,3滴を加えた後,塩酸(1+
20)を滴加して中和する。ろ紙でろ別し,水で洗浄した後,ろ紙を剝がし,180〜220 ℃で約2時間
加熱し,デシケーター中に入れて放冷し,保存する。
6) シリカゲル 薄層クロマトグラフ用,硫酸カルシウム5 %を含むもの。
7) クロロホルム JIS K 8322に規定するもの。
8) ベンゼン JIS K 8858に規定するもの。
9) ヘキサン-クロロホルム溶液 (1) JIS K 8848に規定するヘキサン1容にJIS K 8322に規定するクロ
ロホルム9容を加える。
10) ジチゾン-クロロホルム溶液(50 mg/L) 本体 66.1.2 a) 6)のジチゾン-クロロホルム溶液(0.1 g/L)
をJIS K 8322に規定するクロロホルムで2倍に薄める。
11) 塩化エチル水銀標準液(Hg 1 μg/mL)又は塩化メチル水銀標準液(Hg 1 μg/mL) 本体66.2.1 a) 10)
による。
注(1) ヘキサン-アセトン溶液[ヘキサン85容をビーカーにとり,アセトン15容を加えて調製する。]
又は水を飽和した1-ブタノールを用いてもよい。
b) 器具及び装置 器具及び装置は,次による。
1) アルミナカラム 附属書1図3に例を示す。
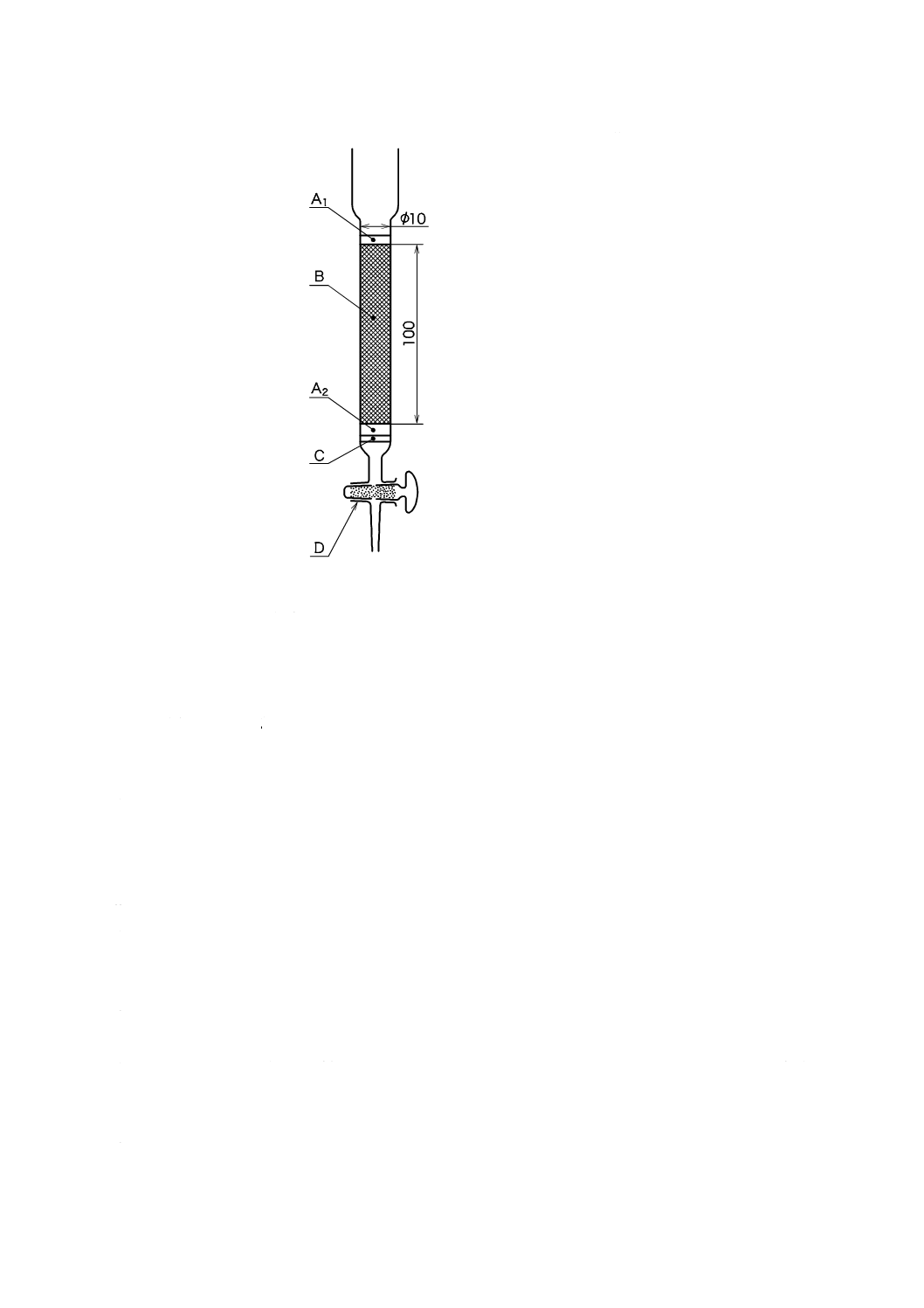
338
K 0102:2016
単位 mm
A1,A2:
B:
C:
D:
ガラスウール
活性アルミナ
ガラスろ過板
すり合わせ
附属書1図3 アルミナカラムの例
2) 分液漏斗 50 mL及び500 mL。コックにワセリンなどの滑剤を塗布しない。
3) シリカゲル薄層板 薄層クロマトグラフ用シリカゲル30 gに水60 mLを加え,ガラス板(200×200
mm)に0.2〜0.25 mmの厚さに均一に塗る。105〜110 ℃で約3時間加熱し,デシケーター中に入れ
放冷し,保存する。
4) マイクロシリンジ 100 mL
5) 分解フラスコ 300 mL
c) 操作 操作は,次による。
1) 試料200 mLを分液漏斗にとり,中性でない場合はアンモニア水又は塩酸で中和した後,塩酸を加
えて約2 mol/Lとする(2)。
2) ベンゼン50 mLを加え,約2分間激しく振り混ぜて放置した後,水層を別の分液漏斗に移し,ベン
ゼン層は保存する。
3) 水層にベンゼン50 mLを加え,約2分間激しく振り混ぜて放置した後,水層を捨てる。
4) ベンセン層を合わせ,塩酸(1+50)20 mLを加えて約1分間振り混ぜて洗浄した後,水層を捨てる。
5) ベンゼン層を乾いたろ紙でろ過して水分を除いた後,アルミナカラムに流し入れ,流量10 mL/min
で流下させる(3)。
6) ベンゼン10 mLを流し入れてアルミナカラムを洗浄した後,上部のガラスウールを取り除き,上部
から約2 cmの厚さの活性アルミナ層を取り出す。
7) この活性アルミナ層を分液漏斗50 mLに入れ,塩酸(1+10)5 mL及びクロロホルム3 mLを加え,
約3分間激しく振り混ぜて放置する。
8) クロロホルム層を取り出し,乾いた小形ろ紙でろ過して水分を除く。
9) このクロロホルム溶液1.5 mLをとり,シリカゲル薄層板の下端から約20 mmの位置に,両端を約
30 mmずつあけて20 mm以上の間隔で数点に分けてマイクロシリンジで添付する(4)。
339
K 0102:2016
10) 同じ薄層板の両端に塩化エチル水銀標準液(Hg 1 μg/mL)又は塩化メチル水銀標準液(Hg 1 μg/mL)
100 mLを同様に添付する。
11) 直ちに薄層板をヘキサン-クロロホルム溶液を展開溶媒(5)とする展開槽に浸し,上昇法によって約
150 mm展開する。
12) 薄層板を風乾し,ジチゾン-クロロホルム溶液(50 mg/L)を噴霧し,エチル水銀(II)化合物又はメ
チル水銀(II)化合物を発色させる。
13) 塩化エチル水銀標準液(Hg 1 μg/mL)又は塩化メチル水銀標準液(Hg 1 μg/mL)の発色添付と同じ
Rf値(6) (7)に相当する展開位置の部分のシリカゲルをガラス板から剝離し,三角フラスコ300 mLに
移して,水で液量を約200 mLとする。
14) 以下,本体66.1.1 c) 2)〜11)によって水銀を定量する(8)。
注(2) 試料中に硫化物イオン及びチオシアン酸イオンが含まれているときは,約2 mol/Lの塩酸酸性
とした試料にJIS K 8138に規定する塩化銅(I)の粉末100 mgを加えて十分にかき混ぜ,しば
らく放置する。沈殿をろ別し,ろ紙を塩酸(1+5)[a) 1)の塩酸を用いて調製する。]で2,3回
洗浄する。
(3) 吸引して流量を調節する。
(4) できるだけ添付を小さくするため,5〜7か所に分けて行う。
(5) 試料中にフェニル水銀(II)が存在すると,エチル水銀(II)との分離が明確にならない場合が
ある。エチル水銀(II)を目的とする場合には,JIS K 8810に規定する1-ブタノールを用いる
とよい。
(6) Rf値は,条件によって著しく異なることがあるので,標準液を必ず同時に展開し,実測によっ
て確認する必要がある。
(7) 肉眼で確認できる量は,1回の添付でHg 0.2 μg以上である。肉眼で確認できない場合には,同
時に展開した標準液と同じRf値の位置に印を付ける。
(8) 剝離したシリカゲルを磁器ボートに入れ,本体66.1.2 c) 10)〜13)によって定量してもよい。
d) 検量線 検量線の作成は,次による。
1) 分液漏斗50 mLに塩化エチル水銀標準液(Hg 1 μg/mL)又は塩化メチル水銀標準液(Hg 1 μg/mL)
を検出器の感度に応じて段階的にとり,以下,c) 7)〜14)の操作を行って,塩化エチル水銀(II)又
は塩化メチル水銀(II)に相当する水銀(Hg)の量と指示値との関係線を作成する。
備考 試料中にアルキル水銀(II)化合物のベンゼン抽出を妨害する成分が含まれている場合には,
試料に一定量の塩化エチル水銀又は塩化メチル水銀標準液を加えてその回収率を求め,定量値
を補正する。
XVII. 飽和溶存酸素量と塩濃度,圧力,温度の影響補正 ある圧力における酸素の溶解度は温度によって
変化する。同様に,ある温度における溶解度は圧力によって変化する。さらに,溶解度は塩濃度が高くな
るにつれて減少する。これらのデータを附属書1表4〜附属書8に示す。
なお,これらのデータは,2014年に発行されたISO 17289を参考として記載した。
a) 電気伝導率と塩濃度(1) 使用する電気伝導率計で塩濃度を測定できない場合は附属書1表4を用いる。
電気伝導率計を使用して20 ℃における電気伝導率を測定し,次に附属書1表4を用いて塩濃度を整
数値で求める。
20 ℃における電気伝導率への換算機能をもたない電気伝導率計を使用する場合は,本体13.の注(4)
340
K 0102:2016
に従い,実験的測定によって求めた電気伝導率の温度係数(1 ℃当たりの電気伝導率の変化率;α)
を用いて,20 ℃の電気伝導率に換算する。附属書1表4はInternational Oceanographic Table(2)から電
気伝導率5.4 S/mまでを計算したものである。
注(1) ここでの塩濃度とは,電気伝導率により定義される値で,実用塩分(Salinity)である。
(2) UNESCO Institute of Oceanographic Sciences, International oceanographic tables. UNESCO, Paris, Vol.
1, 1971
附属書1表4 電気伝導率(20 ℃)と塩濃度(1)との関係
電気伝導率
S/m
塩濃度
電気伝導率
S/m
塩濃度
電気伝導率
S/m
塩濃度
0.5
3
2.0
13
3.5
25
0.6
4
2.1
14
3.6
25
0.7
4
2.2
15
3.7
26
0.8
5
2.3
15
3.8
27
0.9
6
2.4
16
3.9
28
1.0
6
2.5
17
4.0
29
1.1
7
2.6
18
4.2
30
1.2
8
2.7
18
4.4
32
1.3
8
2.8
19
4.6
33
1.4
9
2.9
20
4.8
35
1.5
10
3.0
21
5.0
37
1.6
10
3.1
22
5.2
38
1.7
11
3.2
22
5.4
40
1.8
12
3.3
23
−
−
1.9
13
3.4
24
−
−
b) 標高及び気圧(例) ある標高における実際の気圧を求める際は,附属書1表5の値を用いる。附属
書1表5のデータは,標高0 mの大気圧を1 013 hPaとして求めたものである。附属書1表5から得
た気圧又は地方気象台発表の大気圧を測定器に入力する。
備考 計器が自動的に気圧補正を行う場合,この操作は不要である。
附属書1表5 標高及び気圧(例)
標高
m
気圧
hPa
標高
m
気圧
hPa
0
1013
1 800
815
150
995
1 950
800
300
979
2 100
785
450
960
2 250
771
600
943
2 400
756
750
926
2 550
742
900
910
2 700
728
1050
893
2 850
715
1200
877
3 000
701
1350
861
3 150
688
1500
846
3 300
675
1650
830
−
−

341
K 0102:2016
c) 飽和溶存酸素量と温度・気圧との関係 飽和溶存酸素量と温度・気圧との関係を附属書1表6及び附
属書1表7に示す。
附属書1表6 飽和溶存酸素量と温度・気圧との関係(低気圧側のデータ)
温度
℃
気圧 hPa
733
767
800
833
867
900
933
飽和溶存酸素量(mg/L)
0
10.56
11.04
11.53
12.01
12.49
12.98
13.46
1
10.27
10.74
11.21
11.68
12.15
12.62
13.09
2
9.98
10.44
10.90
11.36
11.82
12.27
12.73
3
9.72
10.16
10.61
11.05
11.50
11.94
12.39
4
9.46
9.89
10.33
10.76
11.20
11.63
12.06
5
9.21
9.64
10.06
10.48
10.91
11.33
11.75
6
8.98
9.39
9.80
10.22
10.63
11.04
11.46
7
8.75
9.16
9.56
9.96
10.37
10.77
11.17
8
8.54
8.93
9.33
9.72
10.11
10.51
10.90
9
8.33
8.72
9.10
9.48
9.87
10.25
10.64
10
8.13
8.51
8.88
9.26
9.64
10.01
10.39
11
7.94
8.31
8.68
9.04
9.41
9.78
10.15
12
7.76
8.12
8.48
8.84
9.20
9.56
9.92
13
7.58
7.94
8.29
8.64
8.99
9.34
9.96
14
7.41
7.76
8.10
8.45
8.79
9.14
9.48
15
7.25
7.59
7.93
8.26
8.60
8.94
9.28
16
7.10
7.43
7.76
8.09
8.42
8.75
9.08
17
6.94
7.27
7.59
7.92
8.24
8.56
8.89
18
6.80
7.12
7.43
7.75
8.07
8.39
8.70
19
6.66
6.97
7.28
7.59
7.91
8.22
8.53
20
6.52
6.83
7.13
7.44
7.75
8.05
8.36
21
6.39
6.69
6.99
7.29
7.59
7.89
8.19
22
6.26
6.56
6.85
7.15
7.45
7.74
8.04
23
6.14
6.43
6.72
7.01
7.30
7.59
7.88
24
6.02
6.31
6.59
6.88
7.16
7.45
7.73
25
5.91
6.19
6.47
6.75
7.03
7.31
7.59
26
5.80
6.07
6.35
6.62
6.90
7.18
7.45
27
5.69
5.96
6.23
6.50
6.77
7.05
7.32
28
5.58
5.85
6.12
6.38
6.65
6.92
7.19
29
5.48
5.74
6.01
6.27
6.53
6.80
7.06
30
5.38
6.64
5.90
6.16
6.42
6.68
6.94
31
5.28
6.54
5.80
6.05
6.31
6.56
6.82
32
5.19
5.44
5.69
5.95
6.20
6.45
6.70
33
5.10
5.35
5.59
5.84
6.09
6.34
6.59
34
5.01
5.25
5.50
5.74
5.99
6.23
6.48
35
4.92
5.16
5.40
5.64
5.89
6.13
6.37
36
4.83
5.07
5.31
5.55
5.79
6.03
6.26
37
4.75
4.98
5.22
5.46
5.69
5.93
6.16
38
4.67
4.90
5.13
5.36
5.60
5.83
6.06
39
4.58
4.81
5.04
5.27
5.50
5.73
5.96
40
4.50
4.73
4.96
5.19
5.41
5.64
5.87
41
4.43
4.65
4.88
5.10
5.32
5.55
5.77
42
4.35
4.57
4.79
5.01
5.24
5.46
5.68
43
4.27
4.49
4.71
4.93
5.15
5.37
5.59
44
4.20
4.41
4.63
4.85
5.07
5.28
5.50
45
4.12
4.34
4.55
4.77
4.98
5.20
5.41

342
K 0102:2016
附属書1表7 飽和溶存酸素量と温度・気圧との関係(高気圧側のデータ)
温度
℃
気圧 hPa
967
1 000
1 013
1 033
1 066
1 100
1 133
飽和溶存酸素量(mg/L)
0
13.94
14.43
14.62
14.91
15.39
15.88
16.36
1
13.56
14.03
14.22
14.50
14.97
15.44
15.91
2
13.19
13.65
13.83
14.10
14.56
15.02
15.48
3
12.84
13.28
13.46
13.73
14.17
14.62
15.06
4
12.50
12.93
13.11
13.37
13.80
14.24
14.67
5
12.18
12.60
12.77
13.02
13.45
13.87
14.29
6
11.87
12.28
12.45
12.69
13.11
13.52
13.93
7
11.57
11.98
12.14
12.38
12.78
13.19
13.59
8
11.29
11.69
11.84
12.08
12.47
12.87
13.26
9
11.02
11.41
11.56
11.79
12.17
12.56
12.94
10
10.76
11.14
11.29
11.51
11.89
12.26
12.64
11
10.51
10.88
11.03
11.25
11.61
11.98
12.35
12
10.27
10.63
10.78
10.99
11.35
11.71
12.07
13
10.04
10.40
10.54
10.75
11.10
11.45
11.80
14
9.82
10.17
10.31
10.51
10.86
11.20
11.54
15
9.61
9.95
10.08
10.29
10.62
10.96
11.30
16
9.41
9.74
9.87
10.07
10.40
10.73
11.06
17
9.21
9.54
9.67
9.86
10.18
10.51
10.83
18
9.02
9.34
9.47
9.66
9.98
10.29
10.61
19
8.84
9.15
9.28
9.46
9.77
10.09
10.40
20
8.66
8.97
9.09
9.28
9.58
9.89
10.19
21
8.49
8.79
8.92
9.10
9.40
9.70
10.00
22
8.33
8.63
8.74
8.92
9.21
9.51
9.80
23
8.17
8.46
8.58
8.75
9.04
9.33
9.62
24
8.02
8.30
8.42
8.59
8.87
9.16
9.44
25
7.87
8.15
8.26
8.43
8.71
8.99
9.27
26
7.73
8.00
8.11
8.28
8.55
8.83
9.11
27
7.59
7.86
7.97
8.13
8.40
8.67
8.94
28
7.45
7.72
7.83
7.99
8.25
8.52
8.79
29
7.32
7.59
7.69
7.85
8.11
8.37
8.64
30
7.20
7.46
7.56
7.71
7.97
8.23
8.49
31
7.07
7.33
7.43
7.58
7.84
8.09
8.35
32
6.95
7.20
7.31
7.46
7.71
7.96
8.21
33
6.84
7.08
7.18
7.33
7.58
7.83
8.08
34
6.72
6.97
7.07
7.21
7.46
7.70
7.95
35
6.61
6.85
6.95
7.09
7.34
7.58
7.82
36
6.50
6.74
6.84
6.98
7.22
7.46
7.70
37
6.40
6.63
6.73
6.87
7.10
7.34
7.57
38
6.29
6.53
6.62
6.76
6.99
7.22
7.46
39
6.19
6.42
6.52
6.65
6.88
7.11
7.34
40
6.09
6.32
6.41
6.55
6.78
7.00
7.23
41
6.00
6.22
6.31
6.45
6.67
6.90
7.12
42
5.90
6.12
6.21
6.35
6.57
6.79
7.01
43
5.81
6.03
6.12
6.25
6.47
6.69
6.91
343
K 0102:2016
XVIII. JIS K 0102:2013の水中の飽和溶存酸素(表32.1)とこの規格の表32.1との対応
JIS K 0102:2013の表32.1水中の飽和溶存酸素量の値はJIS制定当時において陸水学関係で一般的であっ
たTruesdale et.al.,1955の1気圧でのDO濃度の式を用いて飽和度を計算したものである。一方,この規格
の表32.1はISO 5814:2012のTable A.3の飽和溶存酸素の表を採用したものであり,Benson et.al.,1984の圧
力を考慮したDO濃度の式を用いている。そのため,両者の間には最大で3 %の誤差が生じる場合がある。
JIS K 0102:2013の表32.1で校正された溶存酸素計を用いて測定した値を,この規格に対応した数値に変
換する場合は次の式を用いる。
{(この規格の表32.1の飽和溶存酸素量(mg/L)/(JIS K 0102:2013の表32.1の飽和溶存酸素量(mg/L))}
×実測値
実測値はJIS K 0102:2013の表32.1で校正された溶存酸素計を用いて測定した値
参考にJIS K 0102:2013の表32.1を附属書1表8に示す。
附属書1表8 JIS K 0102:2013表32.1の水中の飽和溶存酸素
温度
℃
水中の塩化物イオン Cl−mg/L
塩化物イオン
100 Cl−mg/Lごとに
差し引く溶存酸素
O mg/L
0
5 000
10 000
15 000
20 000
溶存酸素
O mg/L
0
14.16
13.40
12.63
11.87
11.10
0.015 3
1
13.77
13.03
12.29
11.55
10.80
0.014 8
2
13.40
12.68
11.97
11.25
10.52
0.014 4
3
13.04
12.35
11.65
10.95
10.25
0.014 0
4
12.70
12.03
11.35
10.67
9.99
0.013 5
5
12.37
11.72
11.06
10.40
9.74
0.013 1
6
12.06
11.42
10.79
10.15
9.51
0.012 8
7
11.75
11.15
10.52
9.90
9.28
0.012 4
8
11.47
10.87
10.27
9.67
9.06
0.012 0
9
11.19
10.61
10.03
9.44
8.85
0.011 7
10
10.92
10.36
9.79
9.23
8.66
0.011 3
11
10.67
10.12
9.57
9.02
8.47
0.011 0
12
10.43
9.90
9.36
8.82
8.29
0.010 7
13
10.20
9.68
9.16
8.64
8.11
0.010 4
14
9.97
9.47
8.97
8.46
7.95
0.010 1
15
9.76
9.27
8.78
8.29
7.79
0.009 9
16
9.56
9.06
8.60
8.12
7.63
0.009 6
17
9.37
8.90
8.44
7.97
7.49
0.009 4
18
9.18
8.73
8.27
7.82
7.36
0.009 1
19
9.01
8.57
8.12
7.67
7.22
0.008 9
20
8.84
8.41
7.97
7.54
7.10
0.008 7
21
8.68
8.26
7.83
7.40
6.97
0.008 6
22
8.53
8.11
7.70
7.26
6.85
0.008 4
23
8.39
7.98
7.57
7.16
6.74
0.008 2
24
8.25
7.85
7.44
7.04
6.65
0.008 1
25
8.11
7.72
7.32
6.95
6.52
0.007 9
26
7.99
7.60
7.21
6.82
6.42
0.007 8
27
7.87
7.48
7.10
6.71
6.32
0.007 7
344
K 0102:2016
附属書1表8 JIS K 0102:2013表32.1の水中の飽和溶存酸素(続き)
温度
℃
水中の塩化物イオン Cl−mg/L
塩化物イオン
100 Cl−mg/Lごとに
差し引く溶存酸素
O mg/L
0
5 000
10 000
15 000
20 000
溶存酸素
O mg/L
28
7.75
7.37
6.99
6.61
6.22
0.007 6
29
7.64
7.26
6.88
6.51
6.12
0.007 6
30
7.53
7.16
6.78
6.41
6.03
0.007 5
31
7.43
7.06
6.66
6.31
5.93
0.007 5
32
7.32
6.96
6.59
6.21
5.84
0.007 4
33
7.23
6.86
6.49
6.12
5.75
0.007 4
34
7.13
6.77
6.40
6.03
5.65
0.007 4
35
7.04
6.67
6.30
5.93
5.56
0.007 4
備考 塩化物濃度の20 000 mg/LはSalinity 36に相当する。
参考文献
[1] Standard Methods for the Examination of Water and Wastewater, 1998, 20th Edition, pp4-132 and 4-133.
[2] BENSON, B.B and KRAUSE, D. jr. 1980. The concentration and isotopic fractionation of gases dissolved in
fresh water in equilibrium with the atmosphere: I. Oxygen. Limnol. Oceanogr. 25:662.
[3] MORTIMER, C.H. 1981. The oxygen content of air-saturated fresh waters over ranges of temperature and
atmospheric pressure of limnological interest. Int. Assoc. Theoret. Appl. Limnol., Communication No. 22,
Stuttgart, West Germany.
[4] BENSON, B.B. and KRAUSE, D. jr. 1984. The concentration and isotopic fractionation of oxygen dissolved
in freshwater and seawater in equilibrium with the atmosphere. Limnol. Oceanogr. 29:620.
附属書2(参考)JISと対応する国際規格との対比表
JIS K 0102:2016 工場排水試験方法
ISO 5663:1984 Water quality−Determination of Kjeldahl nitrogen−Method after
mineralization with selenium ほか(附属書2付表1参照)
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
1. 適用範
囲
工場から排出される排水
の試験方法について規定。
ISO 5663
ほか,合計
42規格
1
様々な水(具体的には
個別の規格で規定)の
水質試験方法を規定し
ている。
変更
JISは,工場排水の試験方法
に限定して規定。
例えば,ISO規格の適用範囲に含
まれている飲料水の試験方法は,
JISの対象外である。
箇条8〜箇条72の65箇条
について試験方法を規定。
一つの物質を試験対象
とした規格及び多くの
物質を試験対象とした
規格の合計42の試験
方法規格からなる。
変更
JISは,必要な試験方法を一
つの規格とした(したがっ
て,JISは,規格の構成を変
更して採用している。)。
強制法規が,この規格を引用して
いるため。
−
追加
国際規格にない試験方法の
追加。
必要な試験方法規格が,国際規格
にない。
2. 共通事
項
化学分析の通則,用語の定
義,分析法の一般事項,試
薬,器具類,試験方法など
についての共通事項を規
定。
−
−
ISO規格は,これら事
項を個々の規格で規
定。
追加
−
多数のISO規格の試験方法を,一
つのJISとしているため。
3. 試料
試料の採取,試料の取扱い
及び試料の保存処理につ
いて規定。
−
−
−
追加
−
2.と同じ。
4. 流量
流量の測定方法について
規定。
−
−
−
追加
−
2.と同じ。
5. 試料の
前処理
金属元素の試験における
前処理操作を,一括して規
定。
−
−
−
追加
−
2.と同じ。
3
4
5
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
6. 結果の
表示
試験方法が二つ以上ある
場合は,試験方法を付記す
る。
−
−
−
追加
−
2.と同じ。
7. 温度
試料温度の測定方法を規
定。
−
−
−
追加
−
2.と同じ。
8. 外観
試料外観の観察方法を規
定。
−
−
−
追加
−
2.と同じ。
9. 透視度
透視度計による測定法を
規定。
ISO 7027
3〜6
濁度の測定(第2章半
定量法)
変更
ISO規格は,半定量法として,
透視用試験管法,透視用試験
円盤による方法及び光学濁
度計を用いる方法になって
いるが,JISは,透視用試験
管法だけの採用とした。
適用範囲から飲料水を除く。
透視用試験円盤による方法は,透
明度といわれているものでJISに
は規定せず。今後検討。また,光
学濁度計による方法は,JIS K
0101(工業用水試験方法)の濁度
と同等と考えられ,JIS K 0101改
正時に検討する。
飲料水の試験方法は,JISでは規
定できない。
10. 臭気及
び臭気強度
(TON)
10.1 臭気
10.2 臭気強度(TON)
−
−
−
追加
−
対応国際規格がない。
11. 色度
11.1 刺激値及び色度座標
を用いる方法
−
−
−
追加
−
対応国際規格がない。
11.2 三波長を用いる方法
ISO 7887
1〜5
章
色の試験及び策定
変更
適用範囲から飲料水を除く。
規格の構成を変更している
が,実質的な技術的差異はな
い。
飲料水の試験方法は,JISでは規
定できない。
12. pH
12.1 ガラス電極法
ISO 10523
3〜12
pHの測定
変更
定義部分を不採用。
ISO規格におけるpHの定義に整
合性がない。
ISO規格見直し時に,修正提案を
検討する。
3
4
6
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
13. 電気伝
導率
ISO 7888
2〜10
電気伝導率の測定
変更
規格の構成を変更している
が,実質的な技術的差異はな
い。
14. 懸濁物
質及び蒸発
残留物
14.1 懸濁物質
14.2 全蒸発残留物
14.3 溶解性蒸発残留物
14.4 強熱残留物
14.5 強熱減量
−
−
−
追加
−
対応国際規格がない。
15. 酸消費
量
15.1 酸消費量(pH4.8)
15.2 酸消費量(pH8.3)
−
−
−
追加
−
対応国際規格がない。
16. アルカ
リ消費量
16.1 アルカリ消費量
(pH8.3)
16.2 アルカリ消費量
(pH4.8)
16.3 アルカリ消費量(遊
離酸)
−
−
−
追加
−
対応国際規格がない。
17. 100 ℃
における過
マンガン酸
カリウムに
よる酸素消
費量
(CODMn)
−
−
−
追加
−
対応国際規格がない。
18. 欠番
−
−
−
−
−
−
−
19. アルカ
リ性過マン
ガン酸カリ
ウムによる
酸素消費量
(CODOH)
−
−
−
追加
−
対応国際規格がない。
3
4
7
K
0
1
0
2
:
2
0
1
6

(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
20. 二クロ
ム酸カリウ
ムによる酸
素消費量
(CODCr)
20.1 滴定法による酸消費
量(CODCr)
−
−
−
追加
−
20.2 蓋付き試験管を用い
た吸光光度法による
CODCr測定法
ISO 15705
2〜10
規格の構成を変更している
が,実質的な技術的差異はな
い。
21. 生物化
学的酸素消
費量
(BOD)
−
−
−
追加
−
強制法規で引用されている項目。
22. 有機体
炭素(TOC)
22.1 燃焼酸化-赤外線式
TOC分析法
22.2 燃焼酸化-赤外線式
TOC自動計測法
ISO 8245
3〜10
全有機体炭素(TOC)
及び溶存有機体炭素
(DOC)測定方法の指
針
変更
11.2と同じ。
11.2と同じ。
23. 全酸素
消
費
量
(TOD)
−
−
−
追加
−
対応国際規格がない。
24. ヘキサ
ン抽出物質
24.1 試料採取
24.2 抽出法
24.3 抽出容器による抽出
法
24.4 捕集濃縮・抽出法
−
−
−
追加
−
対応国際規格がない。
25. 欠番
−
−
−
−
−
−
−
26. 欠番
−
−
−
−
−
−
−
27. 欠番
−
−
−
−
−
28. フェノ
ール類
28.1 フェ
ノール類
28.1.1 前処理(蒸留法)
28.1.2 4-アミノアンチピリ
ン吸光光度法
ISO 6439
3〜6
フェノール指標の測定
-蒸留−4-アミノアン
チピリン吸光光度法
変更
11.2と同じ。
11.2と同じ。
28.1.3流れ分析法
ISO 14402
JIS K 0170-5:2011附属書JA(参考)JISと対応国際規格との対比表を参照
28.2 p-クレ
ゾール類
28.2.1 p-ヒドラジノベンゼ
ンスルホン酸吸光光度法
−
−
−
追加
−
対応国際規格がない。
3
4
8
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
29. ホルム
アルデヒド
欠番
−
−
−
−
−
−
30. 界面活
性剤
30.1 陰イ
オン界面活
性剤
30.1.2 エチルバイオレッ
ト吸光光度法
30.1.3 溶媒抽出-フレーム
原子吸光法
−
−
−
追加
−
対応国際規格がない。
30.1.4流れ分析法
ISO 16265
JIS K 0170-8:2011附属書JA(参考)JISと対応国際規格との対比表を参照
30.2非イオ
ン界面活性
剤
30.2.1 テトラチオシアナ
トコバルト(II)酸吸光光
度法
−
−
−
追加
−
対応国際規格がない。
31. 農薬
31.1 有機
りん農薬
31.1.2 ガスクロマトグラ
フ法
31.1.3 ナフチルエチレン
ジアミン吸光光度法(アベ
レル-ノリス法)
−
−
−
追加
−
対応国際規格がない。
31.2 ペン
タクロロフ
ェノール
31.2.1 4-アミノアンチピリ
ン吸光光度法
−
−
−
追加
−
対応国際規格がない。
31.3エジフ
ェンホス
(EDDP)
JIS K 0128による。
−
−
−
追加
−
対応国際規格がない。
32. 溶存酸
素
32.1よう素滴定法
ISO 5813
3〜9
溶存酸素の定量−よう
素滴定法
変更
11.2と同じ。
11.2と同じ。
32.2 ミラー変法
−
−
−
追加
−
対応国際規格がない。
32.3 隔膜電極法
ISO 5814
3〜8
溶存酸素の定量−電気
化学的プローブ法
変更
11.2と同じ。
11.2と同じ。
32.4 光学式センサ法
ISO 17289
2〜7
溶存酸素の定量−光学
式センサ法
追加
規格の構成を変更している
が,実質的な技術的差異はな
い。
3
4
9
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
33. 残留塩
素
33.1 o-トリジン比色法
−
−
−
追加
−
対応国際規格がない。
33.2 ジエチル-p-フェニレ
ンジアンモニウム(DPD)
比色法
ISO 7393-2
2〜10
遊離塩素及び全塩素の
定量−第2部:日常管
理のためのN,N-ジエ
チル-1,4-フェニレンジ
アミン比色法
変更
11.2と同じ。
11.2と同じ。
33.3 よう素滴定法
ISO 7393-3
3〜11
遊離塩素及び全塩素の
定量−第3部:全塩素
定量のためのよう素滴
定法
追加
11.2と同じ。
11.2と同じ。
33.4 ジエチル-p-フェニレ
ンジアンモニウム(DPD)
吸光光度法
−
−
−
追加
−
対応国際規格がない。
34. ふっ素
化合物
34.1 ランタン-アリザリン
コンプレキソン吸光光度
法
ISO 10359-1 3〜10
飲料水及び低汚濁水の
電気化学プローブ法
変更
11.2と同じ。
11.2と同じ。
34.2 イオン電極法
ISO 10359-1 3〜10
飲料水及び低汚濁水の
電気化学プローブ法
変更
11.2と同じ。
11.2と同じ。
ISO 10359-2 3〜10
加熱分解及び蒸留後の
無機ふっ化物の定量
(イオン電極法を用い
た定量法)
変更
11.2と同じ。
11.2と同じ。
34.3 イオンクロマトグラ
フ法
ISO 10304-1 3〜11
イオンの液体クロマト
グラフィーによる溶存
陰イオンの定量−第1
部
変更
JISは,ISO規格の,原理及
び校正方法を削除。
JISでは,装置に関する通則を別
途規定し,この通則を引用する体
系となっている。
3
5
0
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
35. 塩化物
イオン
(Cl−)
35.1 硝酸銀滴定法
ISO 9297
3〜8
クロム酸塩を指示薬と
する硝酸銀滴定(モー
ル法)
変更
JISは,ISO規格のクロム酸
指示薬を削除。
環境基準などで規制対象となって
いる物質を含む試薬は,極力使用
しないこととしている。
35.2 イオン電極法
−
−
−
追加
−
対応国際規格がない。
35.3 イオンクロマトグラ
フ法
ISO 10304-1 3〜11
34.3と同じ。
変更
34.3と同じ。
34.3と同じ。
ISO 10304-2 3〜11
イオンの液体クロマト
グラフィーによる溶存
陰イオンの定量−第2
部
変更
34.3と同じ。
34.3と同じ。
36. よう化
物イオン
(I−)
36.1 よう素抽出吸光光度
法
36.2 よう素滴定法
−
−
−
追加
−
対応国際規格がない。
37. 臭化物
イオン
(Br−)
37.1 よう素滴定法
37.2 イオンクロマトグラ
フ法
−
−
−
追加
−
対応国際規格がない。
38. シアン
化合物
38.1 前処理
38.1.1 シアン化物
38.1.2 全シアン(pH2以下
で発生するシアン化水素)
38.2 ピリジン-ピラゾロン
吸光光度法
38.3 4-ピリジンカルボン
酸-ピラゾロン吸光光度法
38.4 イオン電極法
−
−
−
追加
−
強制法規で引用されている項目。
38.5 流れ分析法
ISO 14403
JIS K 0170-9:2011附属書JA(参考)JISと対応国際規格との対比表を参照のこと
39. 硫化物
イオン
(S2−)
39.1 メチレンブルー吸光
光度法
ISO 10530
3〜12
溶存硫化物の定量−メ
チレンブルー吸光光度
法
変更
11.2と同じ。
11.2と同じ。
39.2 よう素滴定法
−
−
−
追加
−
対応国際規格がない。
3
5
1
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
40. 亜硫酸
イオン
(SO32−)
40.1 よう素適定法
−
−
−
追加
−
対応国際規格がない。
41. 硫酸イ
オン
(SO42−)
41.1 クロム酸バリウム吸
光光度法
41.2 重量法
41.3 イオンクロマトグラ
フ法
−
−
−
追加
−
対応国際規格がない。
42. アンモ
ニウムイオ
ン(NH4+)
42.1 前処理(蒸留法)
ISO 5664
2〜9
蒸留後の中和滴定法に
おける蒸留操作
変更
11.2と同じ。
11.2と同じ。
42.2 インドフェノール青
吸光光度法
−
−
−
追加
−
強制法規で引用されている項目。
42.3 中和滴定法
ISO 5664
2〜9
蒸留後の中和滴定法に
おける中和滴定操作
変更
JISは,塩酸標準液(滴定溶
液)を硫酸滴定溶液に変更。
塩酸標準液(滴定溶液)の濃度が
薄く,低濃度域での終点が明確に
判断できるか疑問であるため。
ISO規格見直し時に,修正提案を
検討する。
42.4 イオン電極法
ISO 6778
2〜10
電気プローブ法(イオ
ン電極法)
変更
JISは,ISO規格の測定時の
アルカリ性緩衝液の添加を
削除。
ISO規格見直し時に,修正提案を
検討する。
42.5 イオンクロマトグラ
フ法
ISO 14911
3〜13
イオンクロマトグラフ
法を用いた溶存アンモ
ニウムイオンなどの定
量
変更
適用範囲から飲料水を除く。
対象イオンからリチウムイ
オン,マンガンイオン,スト
ロンチウムイオン及びバリ
ウムイオンを除いた。
11.2と同じ。
42.6流れ分析法
ISO 11732
JIS K 0170-1:2011附属書JA(参考)JISと対応国際規格との対比表を参照
3
5
2
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
43. 亜硝酸
イ
オ
ン
(NO2−)及
び硝酸イオ
ン(NO3−)
43.1 亜硝
酸イオン
(NO2−)
43.1.1 ナフチルエチレン
ジアミン吸光光度法
ISO 6777
2〜11
亜硝酸塩の定量−吸光
光度法
変更
11.2と同じ。
11.2と同じ。
43.1.2 イオンクロマトグ
ラフ法
ISO
10304-2
3〜11
35.3と同じ。
変更
34.3と同じ。
34.3と同じ。
43.2 硝酸
イオン
(NO3−)
43.2.1 還元蒸留-インドフ
ェノール青吸光光度法
43.2.2 還元蒸留-中和滴定
法
43.2.3 銅・カドミウムカラ
ム還元-ナフチルエチレン
ジアミン吸光光度法
43.2.4 ブルシン吸光光度
法
−
−
−
追加
−
対応国際規格がない。
43.2.5 イオンクロマトグ
ラフ法
ISO 10304-2 3〜11
35.3と同じ。
変更
34.3と同じ。
34.3と同じ。
43.2.6流れ分析法
ISO 13395
JIS K 0170-2:2011附属書JA(参考)JISと対応国際規格との対比表を参照
44. 有機体
窒素
44.1前処理(ケルダール
法)
ISO 5663
3〜12
ケルダール窒素の定量
−セレンを用いる無機
質化後の定量
変更
11.2と同じ。
JISは,ISO規格が触媒とし
て使用しているセレンを削
除。
11.2と同じ。
環境基準などで規制対象となって
いる物質を含む試薬は,極力使用
しないこととしている。
44.2インドフェノール青
吸光光度法
44.3 中和滴定法
−
−
−
追加
−
対応国際規格がない。
3
5
3
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
45. 全窒素
45.1 総和法
45.2 紫外線吸光光度法
45.3 硫酸ヒドラジニウム
還元法
45.4 銅・カドミウムカラ
ム還元法
45.5 熱分解法
−
−
−
追加
−
対応国際規格がない。
46. りん化
合物及び全
りん
46.1 りん酸イオン
(PO43−)
46.1.1 モリブデン青吸光
光度法
ISO 6878
2〜8
りんの定量−モリブデ
ン酸アンモニウム吸光
光度法
変更
11.2と同じ。
11.2と同じ。
46.1.3 イオンクロマトグ
ラフ法
ISO
10304-2
3〜11
35.3と同じ。
変更
34.3と同じ。
34.3と同じ。
46.1.4 流れ分析法
ISO 15681-1
ISO 15681-2
JIS K 0170-4:2011附属書JA(参考)JISと対応国際規格との対比表を参照
46.2 加水分解性りん
ISO 6878
2〜8
りんの定量−モリブデ
ン酸アンモニウム吸光
光度法
変更
JISは,ISO規格の分解条件
を削除。
ISO規格見直し時に,修正提案を
検討する。
46.3 全りん
46.3.1 ペルオキソ二硫酸
カリウム分解法
46.3.2 硝酸-過塩素酸分解
法
ISO 6878
2〜8
りんの定量−モリブデ
ン酸アンモニウム吸光
光度法
変更
JISは,ISO規格の分解条件
を削除。
ISO規格見直し時に,修正提案を
検討する
46.3.3 硝酸-硫酸分解法
−
−
−
追加
−
対応国際規格がない。
3
5
4
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
47. ほう素
(B)
47.1 メチレンブルー吸光
光度法
−
−
−
追加
−
対応国際規格がない。
47.2 アゾメチンH吸光光
度法
ISO 9390
3〜9
ほう素の定量−アゾメ
チンH吸光光度法
変更
JISは,ISO規格の分解条件
を削除。
ISO規格見直し時に,修正提案を
検討する。
47.3 ICP発光分光分析法
ISO 11885
3〜12
誘導プラズマ発光分光
分析による元素の定量
変更
JISは,内標準法の適用も可
能としている。また,対象元
素及び混合標準液の構成も
変更した。
適用範囲から飲料水を除く。
ISO規格見直し時に,修正提案を
検討する
飲料水の試験方法は,JISでは規
定できない。
47.4 ICP質量分析法
−
−
−
追加
−
対応国際規格がない。
48. ナトリ
ウム(Na)
48.1 フレーム光度法
ISO 9964-3
3〜9
ナトリウム及びカリウ
ムの定量−第3部:フ
レーム発光法によるナ
トリウム及びカリウム
の定量
変更
11.2と同じ。
11.2と同じ。
48.2 フレーム原子吸光法
ISO 9964-1
3〜9
ナトリウム及びカリウ
ムの定量−第1部:原
子吸光法によるナトリ
ウムの定量
変更
11.2と同じ。
11.2と同じ。
48.3 イオンクロマトグラ
フ法
ISO 14911
3〜13
42.5と同じ。
変更
42.5と同じ。
42.5と同じ。
49. カリウ
ム(K)
49.1 フレーム光度法
ISO 9964-3
3〜9
48.1と同じ。
変更
11.2と同じ。
11.2と同じ。
49.2 フレーム原子吸光法
ISO 9964-1
3〜9
48.2と同じ。
変更
11.2と同じ。
11.2と同じ。
49.3 イオンクロマトグラ
フ法
ISO 14911
3〜13
42.5と同じ。
変更
42.5と同じ。
42.5と同じ。
3
5
5
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
50. カルシ
ウム(Ca)
50.1 キレート滴定法
ISO 6058
3〜11
カルシウムの定量−
EDTA滴定法
変更
11.2と同じ。
11.2と同じ。
50.2 フレーム原子吸光法
ISO 7980
2〜8
カルシウム及びマグネ
シウムの定量
変更
11.2と同じ。
11.2と同じ。
50.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
追加
47.3と同じ。
47.3と同じ。
50.4 イオンクロマトグラ
フ法
ISO 14911
3〜13
42.5と同じ。
変更
11.2と同じ。
11.2と同じ。
51. マグネ
シウム
(Mg)
51.1 キレート滴定法
ISO 6059
3〜11
カルシウム及びマグネ
シウムの合計量の定量
−EDTA滴定法
変更
11.2と同じ。
11.2と同じ。
51.2 フレーム原子吸光法
ISO 7980
2〜8
50.2と同じ。
変更
11.2と同じ。
11.2と同じ。
51.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
51.4 イオンクロマトグラ
フ法
ISO 14911
3〜13
42.5と同じ。
変更
42.5と同じ。
42.5と同じ。
52. 銅(Cu) 52.2 フレーム原子吸光法
ISO 8288
A〜C
法
コバルト,ニッケル,
銅,亜鉛,カドミウム
及び鉛の定量−原子吸
光法
変更
JISは,ISO規格の混合標準
液の調製を削除。
原子吸光法は,個別に金属元素を
測定するものであり,混合標準液
を調製する必要性はない。
ISO規格見直し時に,修正提案を
検討する。
52.3 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
52.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
52.5 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
53. 亜鉛
(Zn)
53.1 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
追加
52.2と同じ。
52.2と同じ。
53.2 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
53.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
53.4 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
3
5
6
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
54. 鉛(Pb) 54.1 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
変更
52.2と同じ。
52.2と同じ。
54.2 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
54.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
54.4 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
55. カドミ
ウム(Cd)
55.1 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
変更
52.2と同じ。
52.2と同じ。
ISO 5961
第2章 原子吸光法によるカド
ミウムの定量
変更
JISは,ISO規格の試料の前
処理法を変更。
ISO規格見直し時に,修正提案を
検討する。
55.2 電気加熱原子吸光法
ISO 5961
第3章 原子吸光法によるカド
ミウムの定量
変更
発熱体を用いた直接濃縮法
を削除。
ISO規格見直し時に,修正提案を
検討する。
55.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
55.4 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
56. マンガ
ン(Mn)
56.1 過よう素酸吸光光度
法
−
−
−
追加
−
対応国際規格がない。
56.2 フレーム原子吸光法
−
−
−
追加
−
対応国際規格がない。
56.3 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
56.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
56.5 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
57. 鉄(Fe) 57.1 フェナントロリン吸
光光度法
ISO 6332
3〜11
鉄の1,10-フェナント
ロリン吸光光度法
変更
JISは,ISO規格の溶存鉄を
不採用。
強制法規には,57.2〜57.4が引用
されている。
57.2 フレーム原子吸光法
−
−
−
追加
−
57.3 電気加熱原子吸光法
−
−
−
追加
−
57.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
追加
47.3と同じ。
58. アルミ
ニウム(Al)
58.1 キノリノール吸光光
度法
−
−
−
追加
−
対応国際規格がない。
58.2 フレーム原子吸光法
−
−
−
追加
−
対応国際規格がない。
58.3 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
58.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
3
5
7
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
59. ニッケ
ル(Ni)
59.1 ジメチルグリオキシ
ム吸光光度法
−
−
−
追加
−
対応国際規格がない。
59.2 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
変更
52.2と同じ。
52.2と同じ。
59.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
59.4 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
60. コバル
ト(Co)
60.1 ニトロソR塩吸光光
度法
−
−
−
追加
−
対応国際規格がない。
60.2 フレーム原子吸光法
ISO 8288
A〜C
法
52.2と同じ。
変更
52.2と同じ。
52.2と同じ。
60.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
60.4 ICP質量分析法
−
−
−
追加
−
対応国際規格がない。
61. ひ素
(As)
61.1 ジエチルジチオカル
バミド酸銀吸光光度法
−
−
−
追加
−
対応国際規格がない。
61.2 水素化物発生原子吸
光法
ISO 11969
3〜12
ひ素の水素化物発生に
よる原子吸光法
変更
11.2と同じ。
11.2と同じ。
61.3 水素化物発生ICP発
光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
61.4 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
62. アンチ
モン(Sb)
62.1 ローダミンB吸光光
度法
−
−
−
追加
−
対応国際規格がない。
62.2 水素化物発生原子吸
光法
−
−
−
追加
−
対応国際規格がない。
62.3 水素化物発生ICP発
光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
63. すず
(Sn)
63.1 フェニルフルオロン
吸光光度法
−
−
−
追加
−
対応国際規格がない。
63.2 ケルセチン吸光光度
法
−
−
−
追加
−
対応国際規格がない。
63.3 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
3
5
8
K
0
1
0
2
:
2
0
1
6

(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
64. ビスマ
ス(Bi)
64.1 よう化物抽出吸光光
度法
−
−
−
追加
−
対応国際規格がない。
64.2 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
65. クロム
(Cr)
65.1 全クロム
65.1.1 ジフェニルカルバ
ジド吸光光度法
−
−
−
追加
−
対応国際規格がない。
65.1.2 フレーム原子吸光
法
ISO 9174
3
全クロムの定量−原子
吸光法
変更
JISは,ISO規格の溶存クロ
ム及び塩化ランタンを不採
用。
当該試験方法は,強制法規に引用
されている。
ISO規格見直し時に,修正提案を
検討する。
65.1.3 電気加熱原子吸光
法
ISO 9174
4
全クロムの定量−電子
加熱原子吸光法
変更
JISは,ISO規格の標準添加
法を削除。
ISO規格見直し時に,修正提案を
検討する。
65.1.4 ICP発光分光分析法 ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
65.1.5 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
65.2 クロム(VI)[Cr(VI)]
65.2.1 ジフェニルカルバ
ジド吸光光度法
ISO 11083
2〜9
クロム(VI)の定量−
1,5-ジフェニルカルバ
ジド吸光光度法
変更
JISは,ISO規格の試料の前
処理(凝集処理)を削除。
当該試験方法は,強制法規に引用
されている。
ISO規格見直し時に,修正提案を
検討する。
65.2.2 フレーム原子吸光
法
−
−
−
追加
−
対応国際規格がない。
65.2.3 電気加熱原子吸光
法
−
−
−
追加
−
対応国際規格がない。
65.2.4 ICP発光分光分析法 ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
65.2.5 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
65.2.6流れ分析法
ISO 23913
JIS K 0170-7:2011附属書JC(参考)JISと対応国際規格との対比表を参照
66. 水銀
(Hg)
66.2 アルキ
ル水銀(II)
化合物
66.2 アルキル水銀(II)化
合物
66.2.1 ガスクロマトグラ
フ法
−
−
−
追加
−
対応国際規格がない。
3
5
9
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
67. セレン
(Se)
67.1 3,3′-ジアミノベンジ
ジン吸光光度法
−
−
−
追加
−
対応国際規格がない。
67.2 水素化合物発生原子
吸光法
67.3 水素化合物発生ICP
発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
67.4 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
68. モリブ
デン(Mo)
68.1 チオシアン酸吸光光
度法
−
−
−
追加
−
対応国際規格がない。
68.2 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
68.3 ICP質量分析法
−
−
−
追加
−
対応国際規格がない。
69. タング
ステン(W)
69.1 チオシアン酸吸光光
度法
−
−
−
追加
−
対応国際規格がない。
69.2 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
70. バナジ
ウム(V)
70.1 N-ベンゾイル-N-フェ
ニルヒドロキシルアミン
吸光光度法
−
−
−
追加
−
対応国際規格がない。
70.2 フレーム原子吸光法
−
−
−
追加
−
対応国際規格がない。
70.3 電気加熱原子吸光法
−
−
−
追加
−
対応国際規格がない。
70.4 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
70.5 ICP質量分析法
−
−
−
追加
−
強制法規で引用されている項目。
71. 魚類に
よる急性毒
性試験
魚類による急性毒性試験
について規定。
−
−
−
追加
−
対応国際規格がない。
72. 細菌試
験
一般細菌,大腸菌群数,従
属栄養細菌,全細菌及びレ
ジオネラの試験について
規定。
−
−
−
追加
−
対応国際規格がない。
3
6
0
K
0
1
0
2
:
2
0
1
6
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇
条ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
項目番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
73. ウラン
(U)
73.1 ICP発光分光分析法
ISO 11885
3〜12
47.3と同じ。
変更
47.3と同じ。
47.3と同じ。
73.2 ICP質量分析法
−
−
−
追加
−
対応国際規格がない。
附属書1
(参考)
補足事項
−
−
−
追加
−
JISと国際規格との対応の程度の全体評価:MOD
備考1. 項目ごとの評価欄の記号の意味は,次のとおりである。
− 追加 ················ 国際規格にない規定項目又は規定内容を追加している。
− 変更 ················ 国際規格の規定内容を変更している。
2. JISと国際規格との対応の程度の全体評価欄の記号の意味は,次による。
− MOD ··············· 国際規格を修正している。
3
6
1
K
0
1
0
2
:
2
0
1
6
附属書2付表1 対応国際規格
JIS K 0102
工場排水試験方法
ISO 5663:1984,Water quality−Determination of Kjeldahl nitrogen−Method after mineralization with selenium
ISO 5664:1984,Water quality−Determination of ammonium−Distillation and titration method
ISO 5813:1983,Water quality−Determination of dissolved oxygen−Iodometric method
ISO 5814:2012,Water quality−Determination of dissolved oxygen−Electrochemical probe method
ISO 5961:1994,Water quality−Determination of cadmium by atomic absorption spectrometry
ISO 6058:1984,Water quality−Determination of calcium content−EDTA titrimetric method
ISO 6059:1984,Water quality−Determination of the sum of calcium and magnesium−EDTA titrimetric method
ISO 6332:1988,Water quality−Determination of iron−Spectrometric method using 1,10-phenanthroline
ISO 6439:1990,Water quality−Determination of phenol index−4-Aminoantipyrine spectrometric methods after distillation
ISO 6777:1984,Water quality−Determination of nitrite−Molecular absorption spectrometric method
ISO 6778:1984,Water quality−Determination of ammonium−Potentiometric method
ISO 6878:2004,Water quality−Determination of phosphorus−Ammonium molybdate spectrometric method
ISO 7027:1990,Water quality−Determination of turbidity
ISO 7393-2:1985,Water quality−Determination of free chlorine and total chlorine−Part 2: Colorimetric method using N,N-diethyl-1,4-phenylenediamine,
for routine control purposes
ISO 7393-3:1990,Water quality−Determination of free chlorine and total chlorine−Part 3: Iodometric titration method for the determination of total chlorine
ISO 7887:1994,Water quality−Examination and determination of colour
ISO 7888:1985,Water quality−Determination of electrical conductivity
ISO 7980:1986,Water quality−Determination of calcium and magnesium−Atomic absorption spectrometric method
ISO 8245:1999,Water quality−Guidelines for the determination of total organic carbon (TOC) and dissolved organic carbon (DOC)
ISO 8288:1986,Water quality−Determination of cobalt, nickel, copper, zinc, cadmium and lead−Flame atomic absorption spectrometric methods
ISO 9174:1990,Water quality−Determination of total chromium−Atomic absorption spectrometric methods
ISO 9297:1989,Water quality−Determination of chloride−Silver nitrate titration with chromate indicator (Mohr's method)
ISO 9390:1990,Water quality−Determination of borate−Spectrometric method using azomethine-H
ISO 9964-1:1993,Water quality−Determination of sodium and potassium−Part 1: Determination of sodium by atomic absorption spectrometry
ISO 9964-3:1993,Water quality−Determination of sodium and potassium−Part 3: Determination of sodium and potassium by flame emission spectrometry
ISO 10304-1:1992,Water quality−Determination of dissolved fluoride, chloride, nitrite, orthophosphate, bromide, nitrate and sulfate ions, using liquid
chromatography of ions−Part 1: Method for water with low contamination
ISO 10304-2:1995,Water quality−Determination of dissolved anions by liquid chromatography of ions−Part 2: Determination of bromide, chloride, nitrate,
nitrite, orthophosphate and sulfate in waste water
ISO 10359-1:1992,Water quality−Determination of fluoride−Part 1: Electrochemical probe method for potable and lightly polluted water
ISO 10359-2:1994,Water quality−Determination of fluoride−Part 2: Determination of inorganically bound total fluoride after digestion and distillation
ISO 10523:1994,Water quality−Determination of pH
ISO 10530:1992,Water quality−Determination of dissolved sulfide−Photometric method using methylene blue
3
6
2
K
0
1
0
2
:
2
0
1
6

附属書2付表1 対応国際規格(続き)
JIS K 0102
工場排水試験方法
ISO 11083:1994,Water quality−Determination of chromium (VI)−Spectrometric method using 1,5-diphenylcarbazide
ISO 11732:2005,Water quality−Determination of ammonium nitrogen−Method by flow analysis (CFA and FIA) and spectrometric detection
ISO 11885:1996,Water quality−Determination of 33 elements by inductively coupled plasma atomic emission spectroscopy
ISO 11969:1996,Water quality−Determination of arsenic−Atomic absorption spectrometric method(hydride technique)
ISO 13395:1996,Water quality−Determination of nitrite nitrogen and nitrate nitrogen and the sum of both by flow analysis (CFA and FIA) and spectrometric
detection
ISO 14402:1999,Water quality−Determination of phenol index by flow analysis (FIA and CFA)
ISO 14403:2002,Water quality−Determination of total cyanide and free cyanide by continuous flow analysis
ISO 14911:1998,Water quality−Determination of dissolved Li+, Na+, NH4+, K+, Mn2+, Ca2+, Mg2+, Sr2+ and Ba2+ using ion chromatography−Method for
water and waste water
ISO 15681-1:2003,Water quality−Determination of orthophosphate and total phosphorus contents by flow analysis (FIA and CFA)−Part 1: Method by flow
injection analysis (FIA)
ISO 15681-2:2003,Water quality−Determination of orthophosphate and total phosphorus contents by flow analysis (FIA and CFA)−Part 2: Method by
continuous flow analysis (CFA)
ISO 15705:2002, Water quality−Determination of the chemical oxygen demand index (ST-COD)−Small-scale sealed-tube method
ISO 16265:2009,Water quality−Determination of the methylene blue active substances (MBAS) index−Method using continuous flow analysis (CFA)
ISO 17289:2014,Water quality−Determination of dissolved oxygen−Optical sensor method
ISO 23913:2006,Water quality−Determination of chromium (VI)−Method using flow analysis (FIA and CFA) and spectrometric detection
3
6
3
K
0
1
0
2
:
2
0
1
6