2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
H 1411-1996
鉄クロム電熱材分析方法
Methods of chemical analysis for iron chromium electric heating material
1. 適用範囲 この規格は,JIS C 2520に規定された鉄クロム電熱線及び帯の化学成分(クロム,アルミ
ニウム,マンガン,炭素)の分析方法について規定する。
備考 この規格の引用規格を,次に示す。
JIS C 2520 電熱用合金線及び帯
JIS H 0321 非鉄金属材料の検査通則
JIS K 0050 化学分析方法通則
JIS K 0l15 吸光光度分析通則
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS Z 8401 数値の丸め方
2. 一般事項 分析方法に共通な一般事項は,JIS K 0050及びJIS K 0115による。
3. 分析試料の採り方及び取扱い方
3.1
試料の採り方は,JIS H 0321の2.3による。
3.2
鋳込試料を採るときは,その平均品質を代表する試料を得るため,1融解ごとに二つ以上(1融解量
が特に少ないときは一つ)の試料を採る。鋳込試料は,できるだけ完全に製品と同一な品質を得るよう,
特に偏析のないように,注意しなければならない。
3.3
試料の削り方は,次による。
(1) 試料の表面に付着物などがある場合は,紙やすりなどを用いて取り除き清浄にする。
(2) きりそのほかの工具類は,アルコールなどを用いて清浄にする。
(3) 鋳込試料から試料を削り取るときは,中央部及び両端に近い部分などの片面から直角にきりもみして
貫通させるか,両面から少なくとも中心部に達するまできりもみするか,又はそのほか適当な方法に
よる。
(4) 線・帯及び板などの製品試料から試料を削り取るには,きり又は適当な工具を用い,分析操作に適当
な大きさに削り取る。
(5) きりもみするときは,発熱のため削り片の表面が酸化することがあるから,酸化させない程度の圧力
と回転数をきりに与えて行う。この際,油類そのほかの減摩剤を用いたり,冷却のための水などを注
加したりしてはならない。
また削り片に,きりの摩耗粉が混入しないように注意する。
(6) 削り片の大きさは,あまり厚くならない程度とし,長さを約5mm以下とする。
2
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.4
試料の取扱い方
(1) 削り取った試料は,その全部(通常50g以上)を集め,強力な磁石を用いて混入した鉄粉などを注意
深く取り除き,最後に良く混ぜ合わせて分析用試料とする。
(2) 分析試料の採取方法が上記規定によりがたい場合は,受渡当事者間の協定によって別途に定めること
ができる。
(3) 分析用試料はデシケーター中に入れ,1時間以上放置した後はかり取る。
3.5
試料のはかり方 試料のはかり方は,次による。
(1) 分析試料のはかり取りに際しては,試料を良くかき混ぜて平均組成を表すように注意しなければなら
ない。
(2) 分析試料のはかり取りには,原則として化学はかりを用い,規定された量に近い量を分析値の表示け
た数を参考として,必要な位まではかり取る。
4. 分析値の表し方と操作上の注意
4.1
分析値の表し方 分析値は百分率で表し,JIS C 2520に規定された位までにJIS Z 8401によって丸
める。
4.2
分析操作上の注意 分析操作上の注意は,次による。
(1) 分析は同一試料について2回以上行って結果を確かめる。
(2) 分析に当たっては全操作を通じて空試験を行い,測定値を補正しなければならない。
5. クロム定量方法
5.1
方法の区分 クロムの定量方法は,次のいずれかによる。
(1) 過硫酸アンモニウム酸化滴定法
(2) 過塩素酸酸化滴定法
5.2
過硫酸アンモニウム酸化滴定法
5.2.1
要旨 試料を硫酸で分解し硝酸を加えて鉄などを酸化した後,触媒として硝酸銀を加え,更に過硫
酸アンモニウムを加えてクロムを酸化する。これに塩酸を加えて生成した過マンガン酸を分解し,次に硫
酸マンガンを添加する。冷却後,硫酸第一鉄アンモニウム標準溶液を少し過剰に加えて重クロム酸を還元
し,過マンガン酸カリウム標準溶液で過剰の硫酸第一鉄アンモニウムを逆滴定する。
5.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+3)
(2) 硝酸
(3) 硫酸 (1+1, 1+4, 2+100)
(4) 硝酸銀溶液 (5g/l)
(5) 過硫酸アンモニウム溶液 (200g/l) この溶液は,使用の都度調製する。
(6) ピロ硫酸カリウム
(7) 過マンガン酸カリウム溶液 (20g/l)
(8) 硫酸マンガン溶液 硫酸マンガン(4〜6水塩)10gを水100mlに溶解する。
(9) 0.1mol/l硫酸第一鉄アンモニウム標準溶液 結晶硫酸第一鉄アンモニウム(6水塩)40gを水約300ml
に溶解し,これに硫酸 (1+1) 60mlを加え,1 000mlの全量フラスコに移し入れ,水で標線まで薄める。
この標準溶液の力価は,使用の都度0.02mol/l過マンガン酸カリウム標準溶液をもって標定する。も
3
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
し正確に0.02mol/lでないときは0.02mol/lに対する力価を求め,クロム量の算出に際し,硫酸第一鉄
アンモニウム標準溶液の使用量を補正することが必要である。すなわち,ここに調製した硫酸第一鉄
アンモニウム標準溶液25mlを正確に取り,水25ml及びo−フェナントロリン溶液0.25mlを加え,次
の(10)で調製した過マンガン酸カリウム標準溶液で淡緑色を呈するまで滴定し,硫酸第一鉄アンモニ
ウム標準溶液の力価を次の式から求める。
2
2
1
1
V
F
V
F
×
=
ここに,
F1: 硫酸第一鉄アンモニウム標準溶液の力価
F2: 過マンガン酸カリウム標準溶液の力価
V1: 過マンガン酸カリウム標準溶液の使用量 (ml)
V2: 硫酸第一鉄アンモニウム標準溶液の分取量 (25ml)
(10) 0.02mol/l過マンガン酸カリウム標準溶液 過マンガン酸カリウム3.16gを水約1 050mlに溶解し,フ
ラスコを用いて1〜2時間静かに煮沸した後,1夜間暗所に放置し,上澄み液をガラスろ過器 (G4) で
ろ過する(前後に水洗しない)。これを30分間蒸気洗浄した褐色瓶に入れ,暗所にたくわえる。この
溶液の標定は,次のように行う。
150〜200℃で45〜60分間乾燥しデシケーター中で冷却したしゅう酸ナトリウム(JIS K 8005の標準
試薬2〜2.5gを正しくビーカー (500ml) にはかり,水約150mlを加えて溶解した後,250mlの全量フ
ラスコに移し入れ,ビーカーを洗った水とともに標線まで薄める。次にピペットを用いて25mlをビ
ーカー (500ml) に分取し,水200ml及び硫酸10mlを加え,液温を25〜30℃にする。このしゅう酸ナ
トリウム溶液をかくはん棒を用いて緩やかにかき混ぜながら,調製された過マンガン酸カリウム標準
溶液の滴定所要量の約2ml手前まで,ビュレットのコックを全開して滴下する。微紅色が消失した後,
57.5±2.5℃に加温し,引き続きかくはん棒でかき混ぜながら注意深く滴定し,微紅色を保つ点を終点
とし,過マンガン酸カリウム標準溶液の力価を次の式から求める。
2
110
1
G
V
G
F
×
×
=
ここに,
F: 過マンガン酸カリウム標準溶液の力価
G1: しゅう酸ナトリウム(標準試薬)はかり取り量 (g)
V: 過マンガン酸カリウム標準溶液の消費量 (ml)
G2: 正確な0.02mo1/l過マンガン酸カリウム標準溶液1mlに相当
するしゅう酸ナトリウム量 (0.006 702g)
なお,別のビーカー (500ml) に水200ml及び硫酸10mlを取り,57.5±2.5℃に加温して空試験を行
って補正する。
(11) o−フェナントロリン溶液 o−フェナントロリン(1水塩)3gをはかり取り,硫酸第一鉄アンモニウ
ム2.0gを含む水200mlに溶解する。
5.2.3
試料はかり取り量 試料は,0.2gをはかり取る。
5.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り三角フラスコ (500ml) に移し入れ,硫酸 (1+4) 40mlを加えて加熱分解する(1)。分
解後硝酸約3mlを加え,煮沸して第一鉄を酸化するとともに炭化物などを分解し,酸化窒素などを除
去する。
(2) 温水を加えて液量を約150mlとした後,硝酸銀溶液10ml及び過硫酸アンモニウム溶液20〜25mlを加
えて5〜7分間煮沸して(2)クロムを重クロム酸に酸化し,過硫酸アンモニウムを分解させた後,塩酸 (1
4
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
+3) 5mlを加えて過マンガン酸を分解する(3)。次に硫酸マンガン溶液約5mlを加えて(4)2〜3分間煮沸
し,発生した塩素を完全に除去する。
(3) 冷水を用いて常温以下になるまで冷却した後,水を加えて液量を約300mlとし,これにo−フェナン
トロリン溶液0.25mlを指示薬として加え,0.1mol/l硫酸第一鉄アンモニウム標準溶液を試料溶液が赤
褐色を呈し始めた後(5)更に5ml程度が過剰になるよう正確に加え,直ちに0.02mol/l過マンガン酸カリ
ウム標準溶液で滴定し,赤褐色から淡緑色に変わる点を終点とする。
5.2.5
計算 試料中のクロム含有率を,次の式によって算出する。
(
)
100
001734
.0
2
2
1
1
×
×
×
−
×
=
W
F
V
F
V
Cr
ここに,
Cr: 試料中のクロム含有率 [% (m/m)]
V1: 0.1mol/l硫酸第一鉄アンモニウム標準溶液の使用量 (ml)
F1: 0.1mol/l硫酸第一鉄アンモニウム標準溶液の力価
V2: 0.02mol/l過マンガン酸カリウム標準溶液の使用量 (ml)
F2: 0.02mol/l過マンガン酸カリウム標準溶液の力価
W: 試料はかり取り量 (g)
注(1) 酸による分解が不完全な試料にあっては酸で分解した後,ろ紙(5種B)を用いてろ過し,硫酸
(2+100) で数回洗浄し,ろ液及び洗液は主溶液として保存する。不溶解残さはこれをろ紙とと
もに白金るつぼに移し,注意して強熱し,ろ紙を灰化した後,これに約10倍量のピロ硫酸カリ
ウムを混ぜて融解する。冷却後,これをビーカー (300ml) に入れ,少量の水及び硫酸 (1+1) で
溶解した後,水を用いて白金るつぼを洗い出し,ろ紙(5種B)を用いてろ過し,温水で数回洗
浄した後,ろ液及び洗液を先の主溶液に合わせ,以下,5.2.4(2)以降に従って操作し,クロムを
定量する。
(2) もしマンガン含有率が非常に少なくて,過マンガン酸の呈色が現れない場合は,過マンガン酸
カリウム溶液を滴加して紅色を呈するようにする。
(3) 溶液に過マンガン酸の呈色又は二酸化マンガンの沈殿が残存するときは,塩酸 (1+3) を更に2
〜3ml加えた後煮沸して,これを完全に分解する。
(4) 硫酸第一鉄アンモニウム標準溶液だけによる直接滴定を行う場合は,硫酸マンガン溶液の添加
を省略することができる。
(5) 試料溶液が赤褐色を呈した点をもって終点としてもよい。この場合クロムの含有率は,次の式
によって算出する。
100
001734
.0
×
×
×
=
W
F
V
Cr
ここに,
Cr: 試料中のクロム含有率 [% (m/m)]
V: 0.1mol/l硫酸第一鉄アンモニウム標準溶液の使用量 (ml)
F: 0.1mol/l硫酸第一鉄アンモニウム標準溶液の力価
W: 試料はかり取り量 (g)
5.3
過塩素酸酸化滴定法
5.3.1
要旨 試料を過塩素酸,りん酸及びふっ化水素酸で分解後強く加熱し,クロムを重クロム酸に酸化
し,硫酸第一鉄アンモニウム標準溶液の少過剰を加え,過マンガン酸カリウム標準溶液で過剰の硫酸第一
鉄アンモニウムを逆滴定する。
5.3.2
試薬 試薬は,次による。
(1) 混酸 過塩素酸20,りん酸1,ふっ化水素酸1の割合で混合し,ポリエチレン製容器に保存する。
5
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 0.1mol/l硫酸第一鉄アンモニウム標準溶液 5.2.2(9)に従って調製する。
(3) 0.02mol/l過マンガン酸カリウム標準溶液 5.2.2(10)に従って調製する。
(4) o−フェナントロリン溶液 5.2.2(11)に従って調製する。
5.3.3
試料はかり取り量 試料は,0.2gをはかり取る。
5.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取り三角フラスコ (300ml) に移し入れ,混酸20mlを加えて加熱分解し,加熱温度を約
250℃に保ち,白煙が発生して溶液がだいだい色に変わり始めてから約30秒間(6)加熱を続け,クロム
を重クロム酸に酸化する。冷却後,これに温水約50mlを加えて塩類を溶解した後,約2分間煮沸す
る。冷却後,これに水を加えて全容を約150mlに薄める。
(2) この溶液にo−フェナントロリン溶液0.25mlを指示薬として加え,0.1mol/l硫酸第一鉄アンモニウム
標準溶液を試料溶液が赤褐色を呈し始めた後(7)更に5mlの過剰を正確に加え,直ちに0.02mol/l過マン
ガン酸カリウム標準溶液で滴定し,赤褐色から淡緑色に変わる点を終点とする。
5.3.5
計算 試料中のクロム含有率を,次の式によって算出する。
(
)
100
001734
.0
2
2
1
1
×
×
×
−
×
=
W
F
V
F
V
Cr
ここに,
Cr: 試料中のクロム含有率 [% (m/m)]
V1: 0.1mol/l硫酸第一鉄アンモニウム標準溶液の使用量 (ml)
F1: 0.1mol/l硫酸第一鉄アンモニウム標準溶液の力価
V2: 過マンガン酸カリウム標準溶液の使用量 (ml)
F2: 過マンガン酸カリウム標準溶液の力価
W: 試料はかり取り量 (g)
注(6) 加熱時間は30秒を超えてはならない。
(7) 試料溶液が赤褐色を呈した点をもって終点としてもよい。この場合クロムの含有率は,次の式
によって算出する。
100
001734
.0
×
×
×
=
W
F
V
Cr
ここに,
Cr: 試料中のクロム含有率 [% (m/m)]
V: 0.1mol/l硫酸第一鉄アンモニウム標準溶液の使用量 (ml)
F: 0.1mol/l硫酸第一鉄アンモニウム標準溶液の力価
W: 試料はかり取り量 (g)
6. アルミニウム定量方法
6.1
方法の区分 アルミニウムの定量方法は,次のいずれかによる。
(1) EDTA滴定法
(2) オキシン滴定法
6.2
EDTA滴定法
6.2.1
要旨 試料を塩酸で分解し,硝酸と過塩素酸でクロムと鉄などを酸化し,塩酸酸性としてメチルイ
ソブチルケトンで鉄及びクロムの大部分を抽出分離する。分離後の酸溶液中の有機物を分解し,水酸化ナ
トリウムを加えてアルカリ性とし,残存する鉄などを沈殿させる。分離後EDTA標準溶液で滴定する。
6.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+1, 2+100)
(2) 硝酸

6
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 過塩素酸
(4) 硫酸 (1+9, 2+100)
(5) アンモニア水 (1+1)
(6) ナトリウム溶液 (100g/l, 10g/l)
(7) 炭酸ナトリウム
(8) 過酸化水素水
(9) 飽和亜硫酸水
(10) 塩化ナトリウム
(11) ピロ硫酸カリウム
(12) 酢酸アンモニウム溶液 (50g/l)
(13) メチルイソブチルケトン
(14) 0.02mol/lエチレンジアミン四酢酸二ナトリウム (EDTA) 標準溶液 エチレンジアミン四酢酸二ナト
リウム(2水塩)7.45gを水に溶解して正しく1lとする。この溶液の力価は,0.02mol/lアルミニウム
標準溶液を用いて標定する。すなわち,0.02mol/lアルミニウム標準溶液10mlを正確にビーカー (300ml)
に取り,水100ml及び酢酸アンモニウム溶液 (50g/l) を加えてpHを3.0±0.2とする。以下6.2.4(7)の
操作に準じて操作し,0.02mol/lEDTA標準溶液1ml当たりのアルミニウム相当量を次の式によって求
める。
2
1
V
V
G
f
×
=
ここに,
f: 0.02mol/lEDTA標準溶液1ml当たりのアルミニウム相当量
(g)
G: 0.02 mol/lアルミニウム標準溶液1ml当たりのアルミニウム
含有量 (g)
V1: 0.02 mol/lアルミニウム標準溶液の使用量 (ml)
V2: 滴定に要したM/50EDTA標準溶液量 (ml)
(15) M/50アルミニウム標準溶液 金属アルミニウム(99.9%以上)0.5396gに塩酸 (1+1) 50mlを加えて加
熱分解し,冷却後1000mlの全量フラスコに移し入れて水で標線まで薄める。
(16) Cu−PAN溶液 1−ピリジルアゾ−2−ナフトール (PAN) 0.1gとCu−EDTAl.3gをジオキサン
(
%
50V
V
) 100mlに溶解する。又は,市販の混合製剤1gをジオキサン (
%
50V
V
) 100mlに溶解する。
6.2.3
試料はかり取り量 試料はアルミニウム含有率に応じ,原則として表1に従ってはかり取る。
表1
アルミニウム含有率%
試料はかり取り量g
2以上 5未満
0.5
5以上
0.25
6.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (300ml) に移し入れ,時計皿で覆い,塩酸 (1+1) 20mlを加えて加熱分解
した後硝酸2mlを加え,煮沸して鉄などを酸化し(8),次に過塩素酸3mlを加えて加熱を続け,濃厚な
白煙が発生してクロムが赤色のクロム酸になるまで加熱する(9)。
(2) 冷却後,硝酸0.5mlを加えた後,塩酸 (1+1) (10)20mlを少量ずつ用いて分液漏斗に移す。メチルイソ
ブチルケトン20mlを加え,約30秒間激しく振り混ぜ(11),鉄,クロムなどを抽出分離する。静置して
2層に分離後,下層の塩酸溶液を別の分液漏斗に移す。更にメチルイソブチルケトン20mlを加えて同
7
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
様に抽出する。2層に分離後,下層の酸溶液を注意してビーカー (300ml) に移す。
(3) これに硝酸5ml及び過塩素酸10mlを加えて加熱蒸発し,濃厚な白煙が発生してクロムが赤色のクロ
ム酸になるまで加熱する。これに塩化ナトリウムを少量ずつ加えてクロムを揮散させる(12)。
(4) これに温水80ml及び水酸化ナトリウム溶液 (100g/l) 10mlを加え,引き続き加熱し,約5分間煮沸し
て,残存する鉄その他を沈殿させる。
(5) 冷却後ろ紙(5種A)を用いてビーカー (500ml) 中にろ過し,水酸化ナトリウム溶液 (10g/l) で洗浄
する。ろ液及び洗液に塩酸 (1+1) を加えて中和し,更にその過剰約5mlを加える。
(6) これに水を加えて液量を約200mlとした後,アンモニア水 (1+1) を加えてpHを約1とする。次に酢
酸アンモニウム溶液を加えてpHを3.0±0.2に調節する。
(7) この溶液を加熱して煮沸する。これにCu−PAN溶液を指示薬として約5滴加え,0.02mol/lEDTA標準
溶液で滴定し,溶液の色が赤紫色から黄色に変化したならば更に加熱し,煮沸する。この間再び赤紫
色を呈したならば,引き続き0.02mol/lEDTA標準溶液で滴定する。この操作を繰り返し,滴定後煮沸
しても赤紫色を呈しなくなった点を終点とする。
6.2.5
計算 試料中のアルミニウム含有率を,次の式によって算出する。
100
×
×
=Wf
V
Al
ここに,
Al: 試料中のアルミニウム含有率 [% (m/m)]
V: 0.02mol/lEDTA標準溶液の使用量 (ml)
f: 0.02mol/lEDTA標準溶液1mlのアルミニウム相当量 (g)
W: 試料はかり取り量 (g)
注(8) 酸による分解不完全な試料では,塩酸及び硝酸で処理した後,水約20mlを加えてろ過し,塩酸
(2+100) で洗い,残さをろ紙とともに白金るつぼに移し,乾燥後強熱灰化した後,約10倍量の
炭酸ナトリウムを混ぜて融解する。冷却後,これを塩酸 (1+1) 及び水に溶解して主溶液に合わ
せ,過塩素酸3mlを加え,加熱蒸発して濃厚な白煙を発生させて,クロムが赤色のクロム酸に
なるまで加熱する。以下,本文に準じてアルミニウムを定量する。
(9) クロムを酸化してクロム酸とし,鉄とともにメチルイソブチルケトンで抽出するために酸化す
る。
(10) 鉄だけを抽出するためには塩酸の濃度が高い方が抽出の速度がほぼ早いが,クロム酸とともに
抽出するためには塩酸の濃度が高いとクロム酸のクロムへの還元が早くなり,抽出率が落ちる。
したがって,鉄の抽出が低下せずに,なるべく塩酸の濃度を低くするため6mol/l程度とする。
(11) 抽出操作は手早く行わないとクロムが還元されるので,できるだけ手早く行う。30秒以上の振
り混ぜは,かえって良くない。2液層に分離後も,できるだけ早く酸溶液を取り出すことが必
要である。
(12) 抽出されずに残ったクロムを除去する。
備考 鉄,クロムの分離は,磁気水銀陰極電解法によって行ってもよい。この場合には試料をビーカ
ー (300ml) にはかり取り,硫酸 (1+9) 20mlを加えて加熱分解し,溶液を40〜50℃に冷却後,
過酸化水素水1〜2mlを滴加して鉄塩その他を酸化又は分解し,過剰の過酸化水素を分解する
ため数分間煮沸した後ろ過し,温硫酸 (2+100) で洗浄する(13)。常温に冷却後,水を加えて
100mlとし,磁気水銀陰極電解装置に移し,10〜15Aの電解電流で電解を行う。電解終了後,
溶液を他のビーカー (300ml) に移し,電解槽内を水で十分洗浄して主溶液に合わせる。この溶
8
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
液に硝酸約1mlを加えて煮沸し,残存する鉄などを酸化する。常温まで冷却後,水酸化ナトリ
ウム溶液 (100g/l) で中和し,更にその過剰10mlを加えて約5分間煮沸して沈殿を完成させる。
以下6.2.4 (5)以降に従って操作し,アルミニウムを定量する。
注(13) 酸による分解不完全な試料では,残さをろ紙と共に白金るつぼに移し,乾燥後,強熱灰化
し,冷却後ピロ硫酸カリウム2gを加えて融解し,主溶液を用いて融解物を溶解し,なるべ
く少量の水でるつぼを洗って主溶液に合わせる。
6.3
オキシン滴定法
6.3.1
要旨 試料を塩酸で分解し,硝酸と過塩素酸でクロムと鉄などを酸化し,塩酸酸性としてメチルイ
ソブチルケトンで鉄及びクロムの大部分を抽出分離する。分離後の酸溶液中の有機物を分解し,水酸化ナ
トリウムを加えて鉄,クロムなどを水酸化物として沈殿分離後,オキシンでアルミニウムを沈殿させ,そ
の沈殿をろ過し,塩酸で溶解する。これに過剰の臭素酸カリウム標準溶液を加え,チオ硫酸ナトリウム標
準溶液で逆滴定する。
6.3.2
試薬 試薬は,次による。
(1)〜(3) 6.2.2(1)〜(3)と同じ。
(4) アンモニア水 (1+1, 1+100)
(5) 水酸化ナトリウム溶液 (100g/l, 10g/l)
(6) 融解合剤(炭酸カリウム1,炭酸ナトリウム1)
(7) 亜硫酸溶液(飽和)
(8) 塩化ナトリウム
(9) よう化カリウム溶液 (100g/l)
(10) 酒石酸溶液 (100g/l)
(11) オキシン酢酸溶液 オキシン(8−オキシキノリン)10gに酢酸30mlを加えて加温溶解し,水で500ml
に薄める。不溶解残さがあればこし分け,褐色瓶に保存する。
(12) メチルイソブチルケトン
(13) 601mol/l臭素酸カリウム標準溶液 臭素酸カリウム2.7837g,臭化カリウム12g及び水酸化カリウム1g
を適量の水に溶かし,これを1 000mlの全量フラスコに移し,水で標線まで薄める。この標準溶液1ml
は,0.225mgのアルミニウムに相当する。
(14) 0.1mol/lチオ硫酸ナトリウム標準溶液 結晶チオ硫酸ナトリウム25gを水に溶解して1lとする。この
溶液は,次のように標定する。
60
1mol/l臭素酸カリウム標準溶液25mlを正確に100ml共栓三角フラスコに取り,よう化カリウム3g
及び塩酸 (2+1) 5mlを加え,直ちに栓をして静かに振り混ぜ,暗所に2〜3分間放置してから,でん
ぷん溶液を指示薬として0.1mol/lチオ硫酸ナトリウム標準溶液で滴定し,チオ硫酸ナトリウム標準溶
液の0.1mol/lに対する力価を次の式から求める。
V
F
25
=
ここに,
F: チオ硫酸ナトリウム標準溶液の0.1mol/lにする力価
V: チオ硫酸ナトリウム標準溶液の滴定消費量 (ml)
(15) インジゴカルミン溶液 インジゴカルミン0.25gを熱水100mlに溶解する。冷却後もし不溶解残さが
あれば,これをろ過して使用する。
(16) でんぷん溶液 可溶性でんぷん1gを少量の水で練り,1lの熱水中にかき混ぜながら注入した後,約1

9
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
分間煮沸して放冷する。この溶液は使用の都度調製する。
なお,よう素のため赤褐色を呈するものは,これを使用してはならない。
6.3.3
試料はかり取り量 試料はアルミニウム含有率に応じ,原則として表2に従ってはかり取る。
表2
アルミニウム含有率%
試料はかり取り量g
2以上 5未満
0.2
5以上
0.1
6.3.4
操作 定量操作は,次の手順によって行う。
(1)〜(3) 6.2.4(1)〜(3)に従う。
(4) 冷却後水100mlを加え,数分間煮沸した後ろ過し,温水で洗浄する。ろ液は亜硫酸溶液(飽和)20ml
を加え,煮沸して過剰の亜硫酸ガスを除去した後,硝酸2〜3mlを加えて更に煮沸する。溶液を冷却
後,水酸化ナトリウム溶液 (100g/l) を加えて中和し,更にその過剰10mlを加えて,しばらく煮沸し
て沈殿を完成させる。
(5) 冷却後ろ過し,沈殿を水酸化ナトリウム溶液 (10g/l) で十分に洗浄する。ろ液及び洗液は合わせてビ
ーカー (300ml) に入れ,塩酸 (1+1) で中和し,更にその5mlを過剰に加える。
(6) これに酒石酸溶液5mlを加えた後,アンモニア水 (1+1) を加えて微アルカリ性とし,50〜60℃に加
温し,溶液をかき混ぜながらオキシン酢酸溶液を少過剰に滴加する。再びアンモニア水 (1+1) を滴
加して微アルカリ性とし,50〜60℃に約20分間保ち,アルミニウムオキシン塩の沈殿を完成させる。
(7) この沈殿をろ紙(5種B)を用いてこし分けし,温アンモニア水 (1+100) で洗液に黄色のなくなるま
で洗浄する。ろ紙及び洗液は捨て,ろ紙上のアルミニウムオキシン塩の沈殿は,温塩酸 (1+1) 50ml
をろ紙上から注いで溶解し,熱水で洗浄する。これにインジゴカルミン溶液2〜3滴を加え,かき混ぜ
ながら601 mol/l臭素酸カリウム標準溶液を青色が消失するまで滴加し,更にインジゴカルミン溶液1
滴を加えて標準溶液の過剰の存在を確かめる。これによう化カリウム溶液10mlを加え,遊離したよ
う素を0.1 mol/lチオ硫酸ナトリウム標準溶液で滴定する。よう素の褐色が薄くなったときでんぷん溶
液5mlを加え,よう素でんぷんの青色が消失するまで滴定する。
6.3.5
計算 試料中のアルミニウム含有率を次の式によって算出する。
(
)
100
000225
.0
2
1
×
×
×
−
=
W
F
V
V
Al
ここに,
Al: 試料中のアルミニウム含有率 [% (m/m)]
V1:
60
1mol/l臭素酸カリウム標準溶液の使用量 (ml)
V2: 0.1mol/lチオ硫酸ナトリウム標準溶液の使用量 (ml)
F: 0.1mol/lチオ硫酸ナトリウム標準溶液の力価
W: 試料はかり取り量 (g)
備考 アルミニウムオキシン塩の沈殿を生成後,重量法によって定量してもよい。この沈殿をあらか
じめ乾燥してひょう量したガラスろ過器 (G4) で吸引ろ過し,温水で洗浄した後,107.5±2.5℃
で恒量となるまで乾燥ひょう量し,アルミニウムの含有率を次の式によって算出する。
100
0587
.0
×
×
=
W
w
Al
ここに,
Al: 試料中のアルミニウム含有率 [% (m/m)]
w: アルミニウムオキシン塩 [Al (C9H6ON) 3] の質量 (g)
W: 試料はかり取り量 (g)

10
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7. マンガン定量方法
7.1
方法の区分 マンガンの定量方法は,次のいずれかによる。
(1) 滴定法
(2) 吸光光度法
7.2
滴定法
7.2.1
要旨 試料を過塩素酸で分解し,更に加熱を続けてクロムを酸化した後,塩化ナトリウムを加えて
クロムを塩化クロミルとして除去する。これに混酸及び過硫酸アンモニウムを加えてマンガンを過マンガ
ン酸に酸化し,亜ひ酸ナトリウム標準溶液で滴定する。
7.2.2
試薬 試薬は,次による。
(1) 硝酸
(2) 硝酸 (2+100)
(3) 過塩素酸
(4) 王水 塩酸3,硝酸1の割合で混合する。
(5) 混酸 水435ml中に硫酸150mlを加えて冷却した後,硝酸250ml,りん酸150mlと硝酸銀溶液 (200g/l)
15mlを混合する。
(6) 塩化ナトリウム
(7) 過硫酸アンモニウム溶液 (200g/l) この溶液は,使用の都度調製する。
(8) ピロ硫酸ナトリウム
(9) 標準マンガン溶液 電解マンガン(99.9%以上)0.100gをビーカー (200ml) にはかり取り,硫酸 (1+
4) 50mlを加えて加熱分解し,冷却後500mlの全量フラスコに移し入れ,水で標線まで薄める。この溶
液1mlは,0.2mgのマンガンを含有する。
(10) 亜ひ酸ナトリウム標準溶液 三酸化ひ素 (As2O3) (JIS K 8005の標準試薬)0.5gをビーカー (200ml)
に正しくはかり取り,水酸化ナトリウム溶液 (40g /l) 20mlと水約100mlを加えて加熱溶解し,冷却し
た後,1 000mlの全量フラスコに移す。フェノールフタレインを指示薬として硫酸 (1+35) を加えて
微酸性とし,これに炭酸水素ナトリウム溶液 (50g /l) 20mlを加え,水で標線まで薄める。この標準溶
液のマンガン相当量の決定方法は,次のとおりとする。
試料中に含まれている鉄と同量の純鉄を三角フラスコ (500ml) にはかり取り,混酸を加えて加熱し
て分解した後,これに標準マンガン溶液の一定量を正しく加え,以下,7.2.4(3)以降に従って滴定を行
い,亜ひ酸ナトリウム標準溶液1ml当たりのマンガン相当量を次の式によって算出する。
2
1
0002
.0
V
V
f
×
=
ここに,
f: 亜ひ酸ナトリウム標準溶液1mlのマンガン相当量 (g)
V1: 標準マンガン溶液の使用量 (ml)
V2: 亜ひ酸ナトリウム標準溶液の使用量 (ml)
7.2.3
試料はかり取り量 試料はマンガン含有率に応じ,原則として表3に従ってはかり取る。
表3
マンガン含有率%
試料はかり取り量g
0.05未満
1.0
0.05以上 0.5未満
0.5
0.5以上
0.2
7.2.4
操作 定量操作は,次の手順によって行う。
11
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 試料をはかり取りビーカー (300ml) に移し入れ,時計皿で覆い,過塩素酸を,試料0.5g以下のときは
25ml,1gのときは35mlを加え,静かに加熱して分解する(14)。
(2) 熱板上で過塩素酸の濃厚な白煙を発生させ,液がだいだい色になるまで十分に酸化する。これに塩化
ナトリウム0.5〜1.0gを少量ずつ加え,褐色の煙の発生がやむまで以上の操作を繰り返し,クロムを塩
化クロミルとして揮散させる。クロムを除去した後,再び白煙を発生させる。
(3) 冷却した後,三角フラスコ (500ml) に移し入れ,温水約150mlを加え,数分間煮沸して遊離した塩素
を除き,混酸15mlを加える。
(4) 温水で約250mlに薄めて加熱し,煮沸し始めるとともに過硫酸アンモニウム溶液10mlを少量ずつ加
え,小気泡がなくなり,大気泡になるまで煮沸し,過硫酸アンモニウムを完全に分解するとともにマ
ンガンを十分に酸化して過マンガン酸とした後,直ちに流水中で25℃以下に冷却する。冷却後速やか
に亜ひ酸ナトリウム標準溶液で滴定する。
7.2.5
計算 試料中のマンガン含有率を,次の式によって算出する。
100
×
×
=Wf
V
Mn
ここに,
Mn: 試料中のマンガン含有率 [% (m/m)]
V: 亜ひ酸ナトリウム標準溶液の使用量 (ml)
f: 亜ひ酸ナトリウム標準溶液1mlのマンガン相当量 (g)
W: 試料はかり取り量 (g)
注(14) 過塩素酸による試料分解が困難なときは,王水の適当量を使用して分解し,過塩素酸を加えた
後7.2.4(2)以降に従って操作し,マンガンを定量する。
また,酸による分解が不完全な試料にあっては,残さをこし分け,温硝酸 (2+100) で洗浄し,
残さはろ紙と共に白金るつぼに移し,ろ紙を灰化した後,これにピロ硫酸ナトリウムを混ぜて
融解する。冷却した後,温水でビーカーに洗い移し,次に硝酸で酸性とし,主溶液に合わせる。
7.3
吸光光度法
7.3.1
要旨 試料を王水で分解後,硫酸及びりん酸を加え,加熱して白煙を発生させた後,水で塩類を溶
解する。これに硝酸を加え,次に硝酸銀と過硫酸アンモニウムを加え,マンガンを過マンガン酸に酸化し,
冷却後尿素を加え,水を用いて一定量に薄める。この溶液の一部を採り吸光度を測定した後,亜硝酸ナト
リウムを加えて過マンガン酸の赤紫色を消し,再び吸光度を測定し,前後の差からマンガンを定量する。
7.3.2
試薬 試薬は,次による。
(1) 硝酸
(2) ふっ化水素酸
(3) 硫酸 (1+1, 1+100)
(4) りん酸
(5) 王水 塩酸3,硝酸1の割合で混合する。
(6) 混酸 水40ml中に硫酸20mlを加え,冷却後,硝酸25mlとりん酸15mlを加えて混合する。
(7) 硝酸銀溶液 (20g/l)
(8) 過硫酸アンモニウム溶液 (200g/l) この溶液は,使用の都度調製する。
(9) ピロ硫酸ナトリウム
(10) 亜硝酸ナトリウム溶液 (100g/l)
(11) 尿素溶液 (100g/l)
12
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(12) 標準マンガン溶液 (0.2mgMn/ml) 金属マンガン(99.9%以上)0.100gをビーカー (200ml) にはかり
取り,硫酸 (1+4) 50mlを加えて加熱分解し,冷却後500mlの全量フラスコ中に移し入れ,水で標線
まで薄める。
7.3.3
試料はかり取り量 試料は,0.25gをはかり取る。
7.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (300ml) に移し入れ,時計皿で覆い,王水20mlを加えて加熱分解し,こ
れに硫酸 (1+1) 10ml及びりん酸5mlを加えて引き続き加熱して,硫酸の白煙を発生させる(15)。
(2) 放冷後,水約100mlを加えて加温し,可溶性塩類を溶解する(16)。
(3) この溶液に硝酸5ml及び硝酸銀溶液5mlを加え,加熱して煮沸し始めたときに過硫酸アンモニウム溶
液10mlを少量ずつ加えて引き続き約1分間煮沸し,過硫酸アンモニウムを分解するとともにマンガ
ンを十分に酸化して過マンガン酸とした後,直ちに水を加えて液量を約150mlとし,流水中で25℃以
下に冷却する。
(4) これに尿素溶液10mlを加え,250mlの全量フラスコ中に移し入れ,水を用いて正しく標線まで薄める。
(5) この溶液の一部を光度計の吸収セルに取り,波長530nm付近の吸光度を測定する。
(6) 次に全量フラスコ中の溶液を振り混ぜながら,これに亜硝酸ナトリウム溶液を1滴ずつ滴加して過マ
ンガン酸の赤紫色を消失させた後,再び波長530nm付近の吸光度を測定する。
7.3.5
計算 7.3.4(5)と7.3.4(6)との吸光度の差から,7.3.6で作成した検量線からマンガン量を求め,試料
中のマンガン含有率を次の式によって算出する。
100
×
=WA
Mn
ここに,
Mn: 試料中のマンガン含有率 [% (m/m)]
A: 試料溶液中のマンガン検出量 (g)
W: 試料はかり取り量 (g)
注(15) 塩素イオンを取り除くため,ビーカーの内壁を水洗し,再び加熱して,白煙を発生させること
が望ましい。
(16) この際,けい酸などの沈殿を認めた場合はこし分け,温硫酸 (1+100) で洗浄した後,ろ液及び
洗液は加熱蒸発して約100mlとする。残さが着色している場合は,ろ紙と共に白金るつぼに移
し入れ,強熱して灰化し,ふっ化水素酸処理を行い,ピロ硫酸ナトリウムを混ぜて融解し,温
水で抽出後主溶液に合わせる。
7.3.6
検量線の作成 標準マンガン溶液の各種液量 (0〜20ml) を数個のビーカーに分取し,混酸30mlを
加え,水で液量を約100mlとし,以下7.3.4(3)以降に従って操作し,各々の吸光度を測定する。得た吸光度
とマンガン量との関係線を作成して検量線とする。
8. 炭素定量方法
8.1
方法の区分 炭素の定量方法は,次のいずれかによる。
(1) 重量法
(2) ガス容量法
(3) 中和滴定法
8.2
重量法
13
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.2.1
要旨 試料を酸素気流中で強熱し,炭素を完全に酸化して炭酸ガスとし,これをソーダ石灰,ソー
ダ石綿又は水酸化ナトリウムに吸収させ,その増量をはかる。
8.2.2
試薬 試薬は,次による。
(1) 硫酸
(2) クロム酸飽和硫酸 硫酸(比重1.82, 90%)に重クロム酸カリウムを飽和させ,上澄み液を使用する。
(3) 五酸化りん又は過塩素酸マグネシウム(無水又は3水塩)
(4) 酸素
(5) 金属銅,金属すず,金属鉛又は鉛丹
(6) 白金石綿,パラジウム石綿又は酸化鉄石綿
(7) ソーダ石灰,ソーダ石綿又は水酸化ナトリウム
ソーダ石綿の調製方法:水酸化ナトリウム1kgを水1lに溶解した溶液約500mlにつき,破砕した水
酸化ナトリウム1kgを加えて良く混和し,これに細裂した石綿を少しずつ加えて良く混和し,湿潤状
態をほとんど認めない程度にする。次に,これを150〜180℃で約4時間加熱して水分を除き,この際
加熱によって湿潤状態を増したときには,ときどき石綿を加えて常に初めの状態を保たせる。次に,
これを冷却した後破砕し,目孔約2mmのふるいでふるい分け,ふるいを通ったものを集めて密閉し
て貯蔵する。
(8) 活性アルミナ
(9) ガラス綿
8.2.3
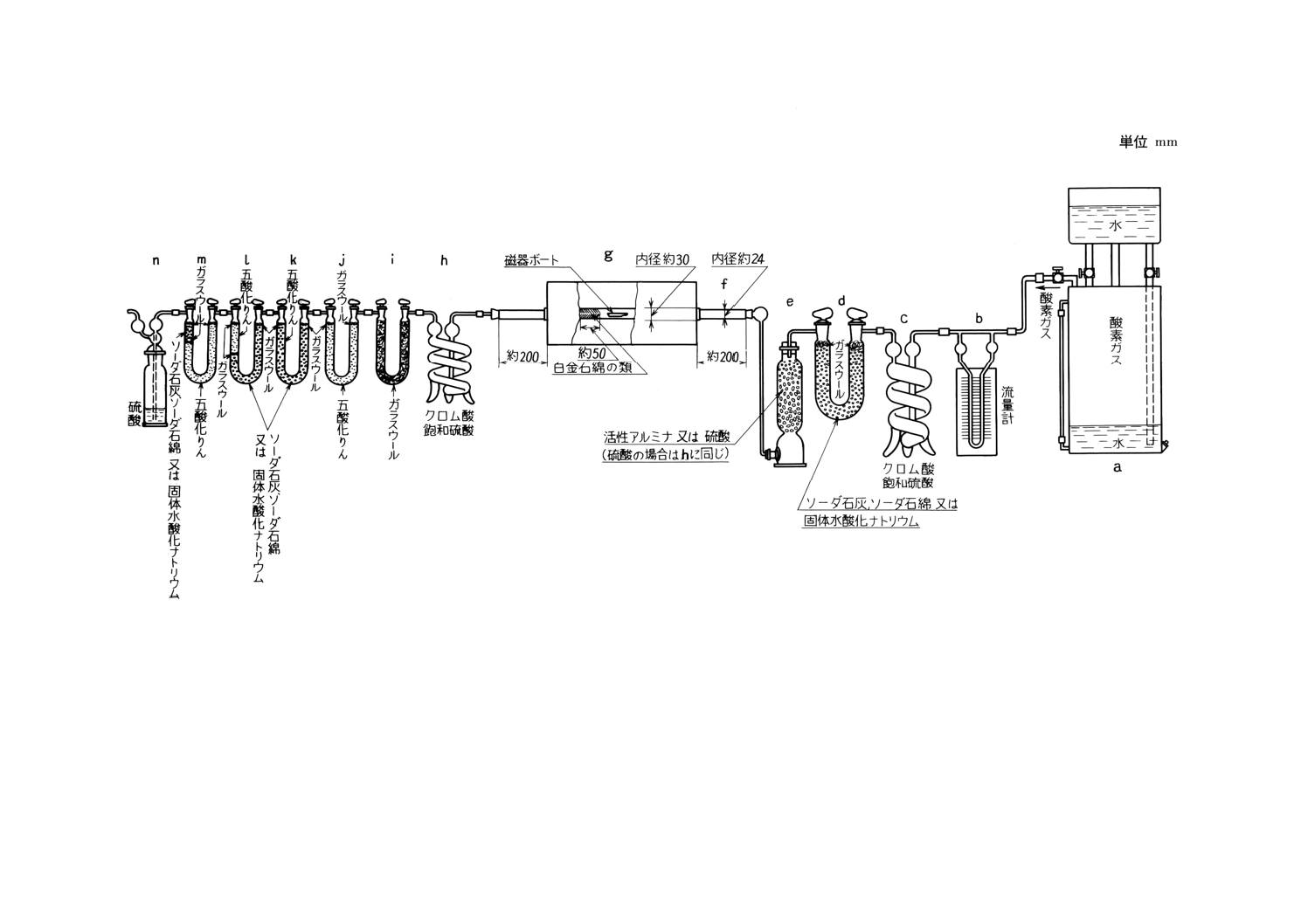
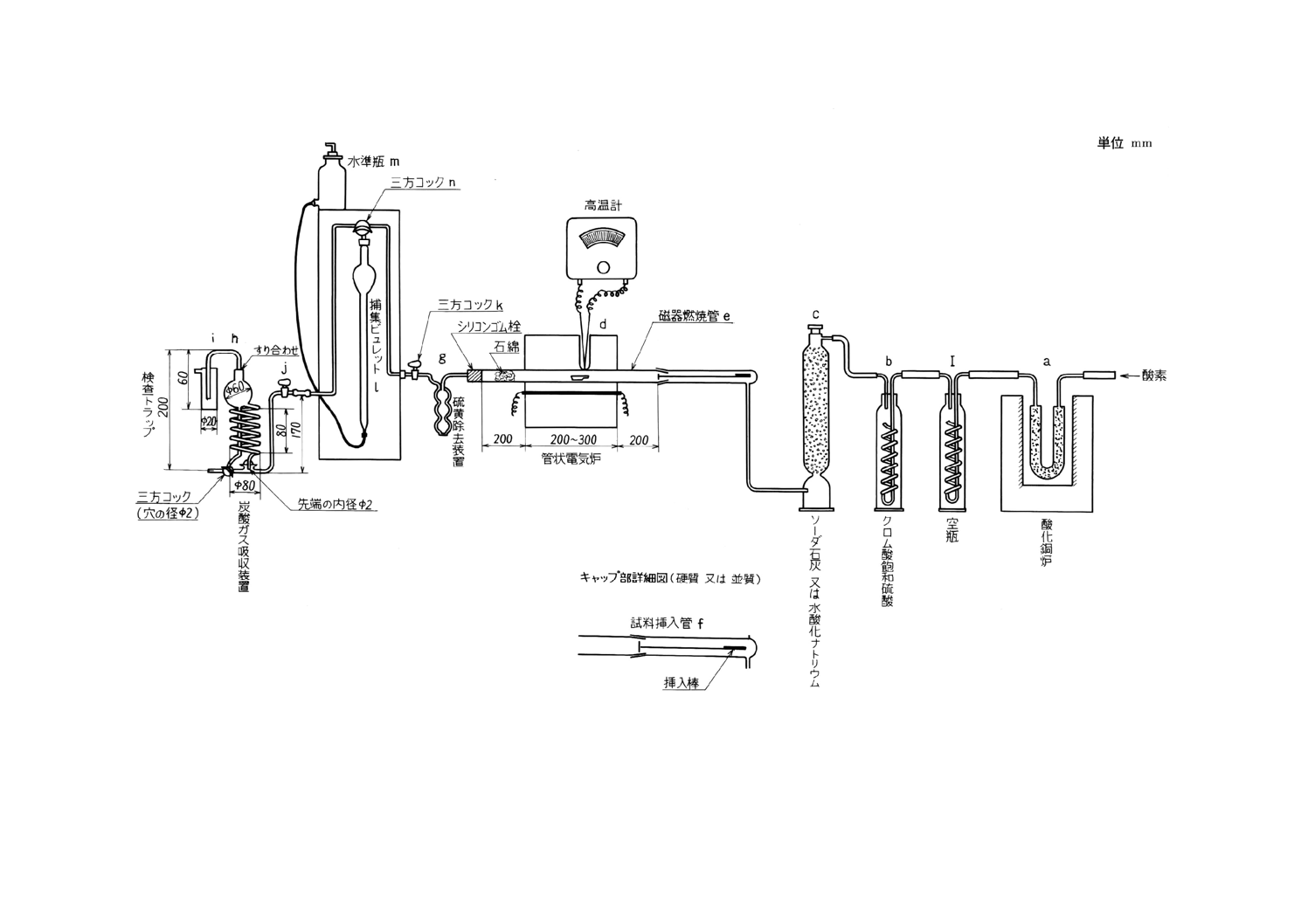
装置及び器具 装置及び器具は,原則として次のものを用いる(付図1参照)。
(1) 酸素清浄装置 この装置は,ガスタンク (a) にたくわえた酸素中に含有する炭酸ガス又は有機性ガス
などを除去し,かつ酸素を清浄・乾燥することを目的とするもので,クロム酸飽和硫酸(17)を入れた洗
瓶 (c) ,ソーダ石灰,ソーダ石綿又は水酸化ナトリウムを詰めた管又は塔 (d) ,硫酸又は活性アルミ
ナを入れた洗瓶又は塔 (e) を順次連結(18)するものとする。
(2) 燃焼炉 燃焼炉は,内径約30mmの管状電気炉 (g) を用い,その中央部において,長さ約150mmを
一定温度に保つことができるものとする。電流を調節して温度を加減し,高温計によって燃焼管の真
上の温度(19)を測定するものとする。炉には,その両端がそれぞれ約200mmを突き出し得る長さをも
つ内径約20〜24mmの磁器燃焼管 (f) を挿入する。
また,その管中に挿入される磁器ボートの位置の後方に,約50mmにわたって,白金石綿,パラジ
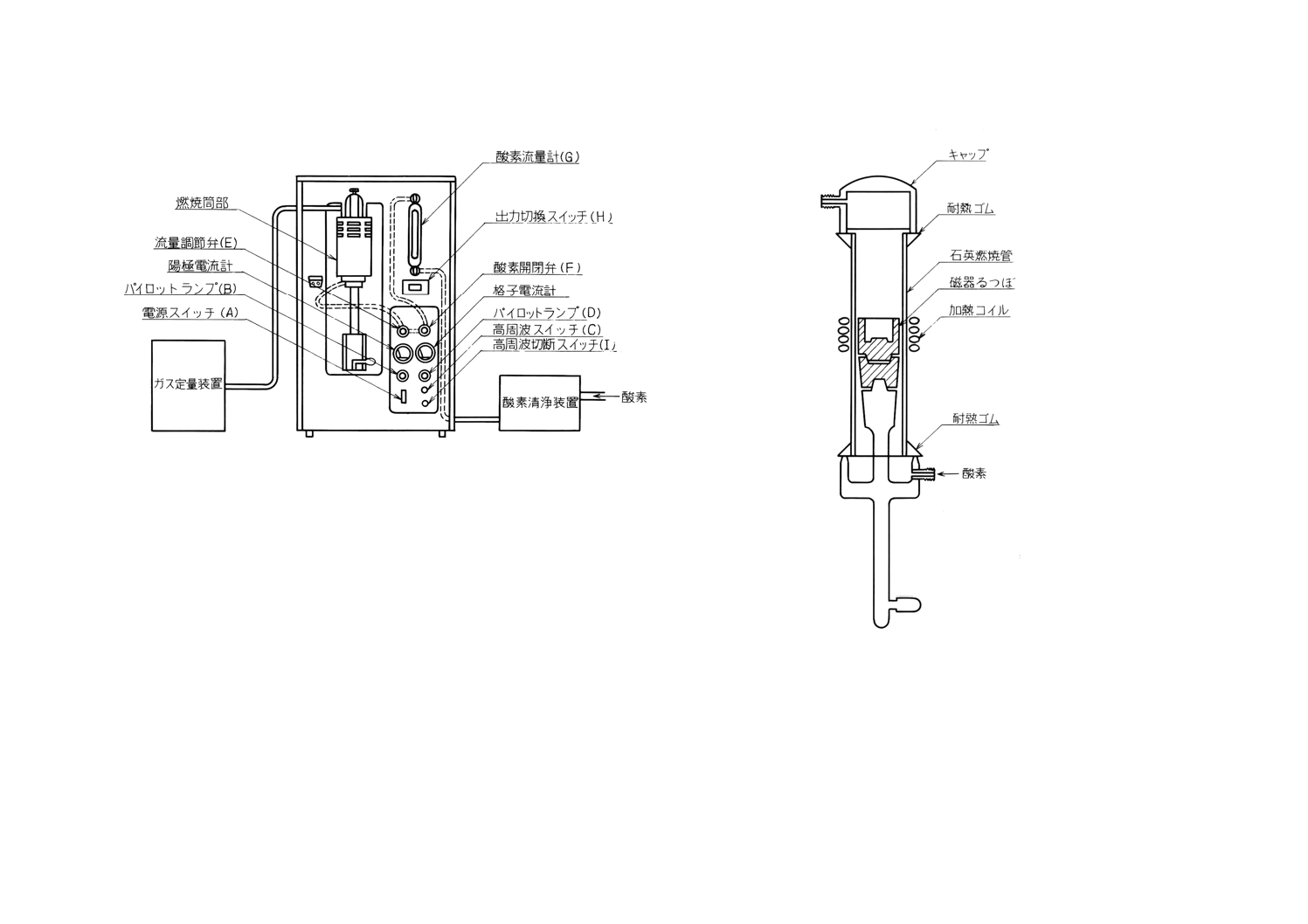
ウム石綿又は酸化鉄石綿を詰めるものとする。管状電気炉の代わりに,高周波誘導加熱装置(付図2)
を使用することができる。この装置は,外径約42mm,内径約37mm,長さ約200mmの石英製縦形燃
焼管と,その中央よりやや下方の外側に巻いた高さ約40〜50mm,巻数3〜4回の加熱コイルと,これ
に高周波電流を供給する高周波発振装置からなり,必要に応じて酸素流量計 (0〜2 000ml/min) ,タイ
ムスイッチ(4〜6分)を附属させるものとする。この装置を使用するときの操作は,備考によるもの
とする。
(3) ガス吸収装置 燃焼炉から出たガスを吸収させるため,次の器具を記載順に順次連結(18)したものをガ
ス吸収装置とする。
クロム酸飽和硫酸を入れた洗瓶 (h) (20),ガラス綿を詰めた管又は塔 (i) ,五酸化りん又は過塩素酸
マグネシウムを入れた管 (j) 及びソーダ石灰(21),ソーダ石綿又は水酸化ナトリウム(22)(23)を入れ,そ
の後に約20mmの厚さに五酸化りん又は過塩素酸マグネシウムを詰めた2個の炭酸ガス吸収管 (k, l) ,
五酸化りん又は過塩素酸マグネシウム及びソーダ石灰,ソーダ石綿又は水酸化ナトリウムを詰めた管
14
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(m) 及び硫酸を入れた洗瓶 (n) を順次連絡(18)するものとする。
(4) 磁器ボート 磁器ボートは,あらかじめ燃焼温度で空焼きしておく。
また磁器ボートには,その半分を覆い,出し入れのじゃまにならない程度の大きさの磁器カバーを
載せるとよい。
8.2.4
試料はかり取り量 試料は,3gをはかり取る。
8.2.5
操作 定量操作は,次の手順によって行う。
(1) 8.2.3の装置を順次に連結し(18)(24),燃焼管 (f) を熱し,その管内温度を1 300〜1 350℃とし,酸素を
毎分約200mlの割合で約20分間通じた後,炭酸ガス吸収管 (k, l) の質量をはかり,各々その変化を
0.000 5g未満になるように装置を整備保持する(25)。
(2) 次に管内温度を1 300〜1 350℃に保った燃焼管の中央部に試料(26)(27)を入れた磁器ボートを挿入して,
直ちに気密に栓をする。
(3) 初めは毎分200mlの割合で酸素を送入し,次に試料の燃焼が始まり燃焼が盛んになったときは,酸素
送入量を増して,燃焼管内の気圧をなるべく外気圧に近くする。燃焼が終われば,酸素を毎分300〜
400mlの割合で10〜15分間送り,発生した炭酸ガスは,これを酸素と共に炭酸ガス吸収管 (k, l) に送
り,完全に吸収させる。
(4) 次に,炭酸ガス吸収管 (k) の炉に近い側から順にコックを全部密閉し,酸素の送入を止め,管 (j) と
管 (m) との連結を解く。
(5) 炭酸ガス吸収管 (k, l) の質量をはかる(28)。
8.2.6
計算 試料中の炭素含有率を,次の式によって算出する。
100
2729
.0
×
×
=
W
A
C
ここに,
C: 試料中の炭素含有率 [% (m/m)]
A: 炭酸ガス吸収管 (k, l) の増量(29) (g)
W: 試料はかり取り量 (g)
注(17) クロム酸飽和硫酸が緑色になれば酸化力もそう失するから,更新しなければならない。
(18) ゴム管は炭酸ガスを吸収するおそれがあるから,装置の各接続にはガラス管を用いてその両端
を密接させ,ゴム管又はビニル管でこれを保持するようにしなければならない。
(19) 高温計指示温度と燃焼管内温度との差に注意して補正する必要がある。
(20) 硫黄含有量0.1%以上の試料にあっては,クロム酸飽和硫酸を入れた洗瓶 (h) を2個連結するの
がよい。
(21) 炭酸ガス吸収管には,ソーダ石灰の粗粒(約3mm)と細粒(約1mm)とを交互に数層に詰め,
ガラス綿又は適当なものでこれを軽く押さえるのがよい。
(22) 炭酸ガス吸収管に水酸化ナトリウムを詰める場合は,水酸化ナトリウムを破砕し,1 000μm(約
16メッシュ)のふるいを通ったもの約20gを詰め,次に少量のガラス綿を置き,更に約5gの
五酸化りん又は過塩素酸マグネシウムを詰めて軽くたたく程度がよい。
(23) 水酸化ナトリウムを破砕するとき,堅い感じがするものには炭酸ガス吸収能力の悪いものが多
いから,このようなものはニッケルるつぼに入れて電気炉で約400℃に加熱融解し,冷却後破
砕して使用するのがよい。
(24) 定量前に必ず気密試験を行い,気密に留意しなければならない。
(25) 日常作業にあっては,作業時間の初期,中期及び終期には,必ず炭素含有量既知の試料を使っ
15
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
て分析し,その日の装置,その他の調子を試験しなければならない。
(26) 試料に油類が付着しているのを認めたときは,アルコール又はエーテルで洗浄し,乾燥した後
使用する。試料を調製するとき,あまり小さい試料や長く巻いた試料にならないように注意す
る。
(27) 試料に助燃剤として金属銅,金属すず,鉛丹又は金属鉛を試料1gにつき0.5〜1gを添加して用
いる。いずれの助燃剤も1gにつき,空試験の増量は0.001g未満でなければならない。
(28) 操作後に試料が完全に融解状態になったかどうかを確かめるとともに,試料が完全に酸化した
かどうかを確かめなければならない。
(29) あらかじめ求めた空試験値を差し引いて補正しなければならない。
備考 高周波誘導加熱装置を使用する場合の操作は,次による。
付図2の装置を連結し(18)(24),燃焼管内にはるつぼを入れずに密栓し,高周波スイッチ (C) を
入れることなく,酸素を毎分300mlの割合で10分間送入した後,炭酸ガス吸収管 (k, l) の質量
をはかり,各々の質量変化を0.000 5g未満となるようにする(25)。
次に試料3gと適当な助燃剤(金属すずなど)を入れたるつぼを燃焼管内に挿入して密栓し,
酸素を毎分300mlの割合で2分間送入して管内の空気を置換した後吸収管を接続する。引き続
き酸素を毎分300mlの割合で2分間送入しながら高周波スイッチ (C) を入れ,試料の燃焼が盛
んになり,クロム酸飽和硫酸 (h) が逆流するおそれがあれば酸素送入量を増して,燃焼管内の
気圧をわずかに外気圧以上にする。高周波スイッチ (C) を入れてから1〜2分間たって試料の
燃焼が終わり,試料の温度が下がり始めたら,高周波切断スイッチ (I) を入れて高周波電流を
断ち,引き続き酸素を毎分300mlの割合で5分間送入して炭酸ガスを酸素と共に炭酸ガス吸収
管 (k, l) に送り,完全に吸収させる。以下8.2.5(4)以降に従って操作し,炭素を定量する(28)。
8.3
ガス容量法
8.3.1
要旨 試料を酸素気流中で強熱し,炭素を完全に酸化して炭酸ガスとし,これを酸素と共にビュレ
ットに捕集して全ガスの容積を測定し,次に炭酸ガスを吸収除去した後,残留ガスの容積を測定する。
8.3.2
試薬 試薬は,次による。
(1) 硫酸 (1+1)
(2) クロム酸飽和硫酸 8.2.2(2)参照。
(3) 水酸化カリウム溶液 (330g/l)
(4) 酸素
(5) 金属銅,金属すず,金属鉛又は鉛丹
(6) マンガン酸カリウム溶液 (50g/l)
(7) 塩化ナトリウム溶液 (260g/l)
(8) 石綿
(9) メチルレッド溶液 メチルレッド0.200gをエチルアルコール (
V
V
95
%) 90mlに溶解し,水を加えて
100mlとする。
8.3.3
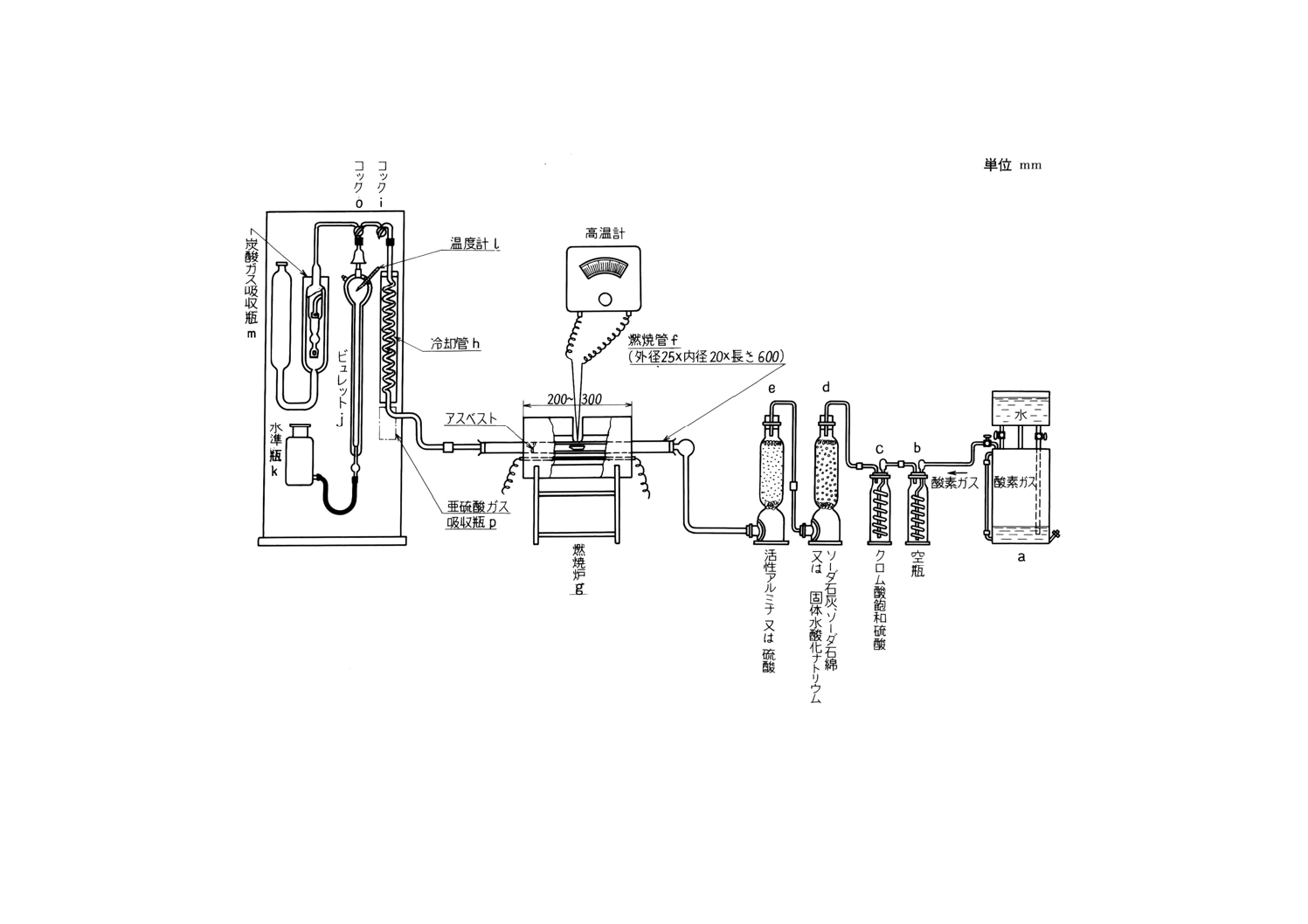
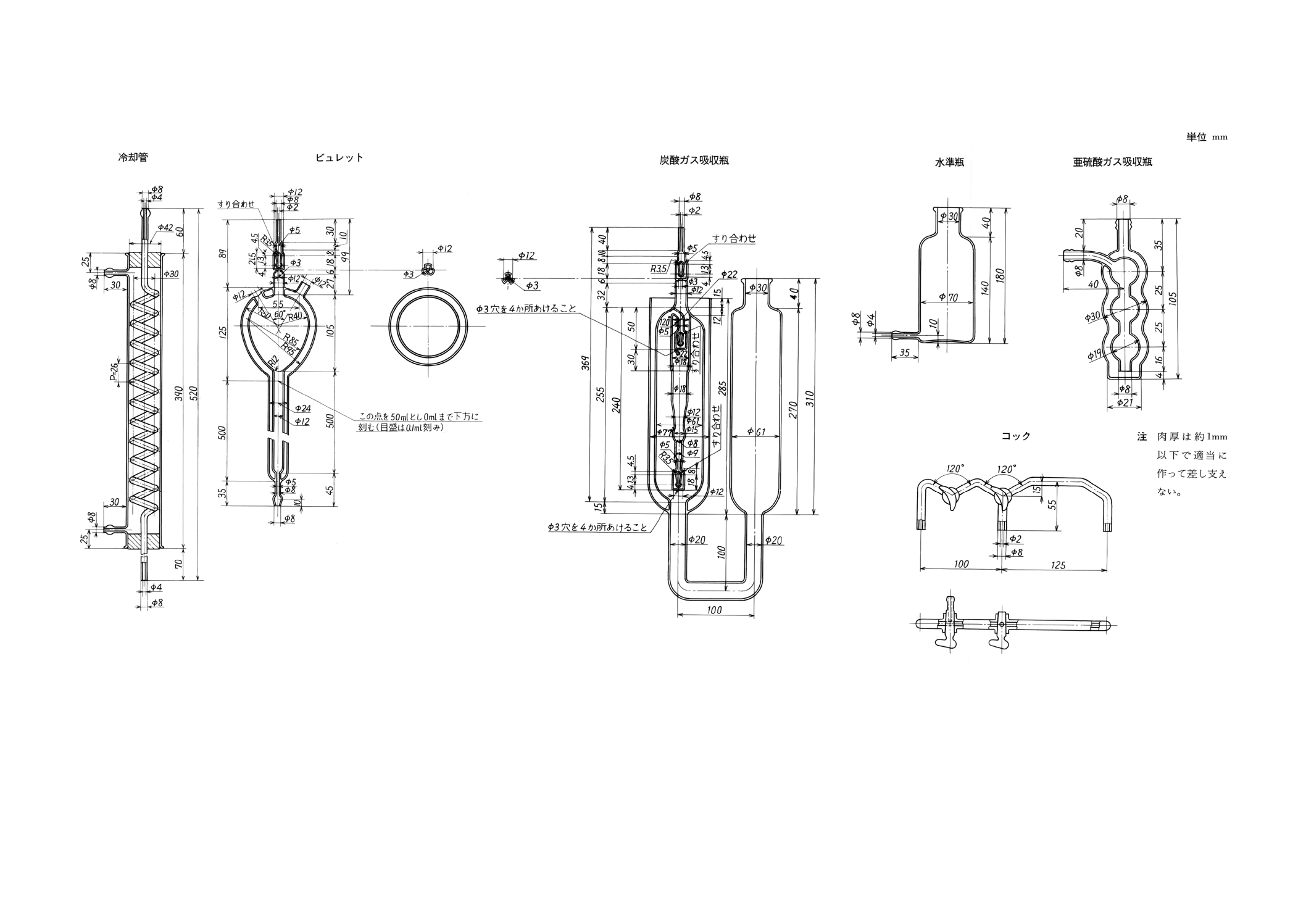
装置及び器具 装置及び器具は,原則として次のものを用いる(付図3参照)。
(1) 酸素清浄装置 8.2.3(1)の酸素清浄装置に準じるものとする。ただし,付図1の流量計 (b) の代わりに
空瓶 (b) を置くものとする。
(2) 燃焼炉 8.2.3(2)の燃焼炉に準じるものとする。ただし燃焼管内には,白金石綿などの代わりに石綿だ
けを詰めるものとする。重量法の場合と同様に,管状電気炉の代わりに高周波誘導加熱装置(付図2)
16
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を使用することができる。この場合の操作は備考による。
(3) ガス分析装置 次のものを順次連結するものとする。
(a) 亜硫酸ガス吸収瓶 (p) 過マンガン酸カリウム溶液 (50g/l) 10mlに硫酸 (1+1) 1mlを加えて酸性と
した溶液を入れておく。
(b) 冷却管 (h)
(c) 三方コック (i) 一方は外気に向けて開けるようにしておく。
(d) 三方コック (o)
(e) ビュレット (j) 塩化ナトリウム溶液 (260g/l) (メチルレッド溶液を指示薬とし,溶液がようやく
赤色となるまで硫酸 (1+1) を滴加して微酸性としたもの)を入れる。ビュレットは,全容約350ml,
目盛部分50ml,0.1ml目盛刻みとする。ただし,この目盛は,16℃,760mmHgを標準として刻むも
のとする。
(f) 水準瓶 (k)
(g) 炭酸ガス吸収瓶 (m) 水酸化カリウム溶液 (330g/l) を入れておく。
(h) 温度計 (l) ビュレット内のガス温度をはかるために取り付けてある。0.1℃まで読み取るものとす
る。
(4) 磁器ボート 磁器ボートは,あらかじめ燃焼温度で空焼きしておく。また磁器ボートにはその半分を
覆い,出し入れにじゃまにならない程度の大きさの磁器カバーを載せるとよい。
8.3.4
試料はかり取り量 試料は,3gをはかり取る。
8.3.5
操作 定量操作は,次の手順によって行う。
(1) 8.3.3の装置を連結し(30),燃焼管を熱して管内温度を1300〜1350℃に上昇させ,装置の気密を確認(31)
した後空試験を行い(32),三方コック (i) 及び (o) を閉じておく(33)。
(2) 次に試料(34)を入れた磁器ボートを燃焼管内の最高温部に挿入し,直ちに気密に栓をした後,酸素タン
クのコックを開いて酸素の送入量を調節しながら約2分間放置し,管内温度を一定させる。
(3) その後水準瓶 (k) をビュレット (j) の球部の位置に置き,三方コック (i) 及び (o) を炉側に開き,三
方コック (i) 又は (o) を調節しながら,炭酸ガス及び酸素の混合ガスを亜硫酸ガス吸収瓶 (p) (35)及び
冷却管 (h) を通じてビュレット (j) に導き,ビュレット (j) の球部の約半分で試料の燃焼を完了させ
る。次に水準瓶 (k) をビュレット (j) の下部目盛以下の位置に置き,約30秒間で混合ガスをビュレ
ット (j) の目盛の下部近くまで捕集した後,三方コック (i) 及び (o) を閉じ,酸素タンクのコックを
閉じて酸素の送入をやめて約1分間放置する。その間,燃焼管の栓を外して,磁器ボートを取り出す
(36)。
(4) 次に水準瓶 (k) をビュレット (j) に沿って上下に動かして,ビュレット (j) と水準瓶 (k) 内の溶液の
水準を合わせてビュレット (j) の目盛を読み,混合ガスの容積とする。
なお,このときの混合ガスの温度を温度計 (l) で測定しておく(37)。
(5) 次に三方コック (o) を炭酸ガス吸収瓶 (m) 側に開き,水準瓶 (k) をビュレット (j) を越えた位置に
上げ,ビュレット (j) の内部に溶液を満たして混合ガスを炭酸ガス吸収瓶 (m) に送り込んで,炭酸ガ
スを水酸化カリウム溶液 (330g/l) 中に吸収させる。次に水準瓶 (k) をビュレット (j) の下部目盛以下
の位置に下げて,残留ガスをビュレット (j) に戻し,三方コック (o) を閉じ,約1分間放置した後水
準瓶 (k) を動かして,ビュレット (j) と水準瓶 (k) 内の溶液の水準を合わせてビュレット (j) の目盛
を読み,残留ガスの容積とする。
なお,このときの残留ガスの温度を温度計 (l) で測定しておく(37)。
17
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(6) (5)の操作を繰り返して,残留ガス容積に変化がなくなったことを確認した後,三方コック (i) を外気
側に開いて,水準瓶 (k) をビュレット (j) を越えた位置に上げて残留ガスを放出させ,三方コック (i)
及び (o) を閉じておく(38)。
8.3.6
計算 混合ガスの容積から,残留ガスの容積と空試験値(33)とを差し引いて,試料による炭酸ガス
の容積(39)とし,付表1の補正係数 (f) (40)(41)を用いて,試料中の炭素含有率を次の式によって算出する。
100
0005027
.0
×
×
×
=
f
W
A
C
ここに,
C: 試料中の炭素含有率 [% (m/m)]
A: 炭酸ガスの容積 (ml)
W: 試料はかり取り量 (g)
f: 補正係数(付表1)
t
b
B
f
+
−
×
=
2.
273
8936
.2
B: 気圧読み取り値に温度及び重力の補正を行った値 (kPa)
b: t℃における塩化ナトリウム溶液 (260g/l) の水蒸気圧
(kPa)
t: ビュレット内のガスの温度 (℃)
注(30) ゴム管は炭酸ガスを吸収するおそれがあるから,装置の各接続にはガラス管を用い,その両端
を密接させ,ゴム管又はビニル管でこれを保持するようにしなければならない。
(31) 定量前には必ず次のような気密試験を行い,気密に留意しなければならない。酸素タンクのコ
ックを閉じ,水準瓶 (k) をビュレット (j) の下部目盛以下の位置に置き,三方コック (i) 及び
(o) を炉側に開く。次に酸素タンクのコックを徐々に開いて酸素をビュレット (j) の目盛部の
10〜20mlの位置まで捕集し,酸素タンクのコックを閉じ,しばらく放置してビュレット (j) 内
の液面に変化のないことを確かめる。もし,この際液面が下がるようなことがあれば,酸素タ
ンクのコックからビュレット (j) までの間に気密の悪いところがあることを示す。次に三方コ
ック (o) を炉側に,三方コック (i) を外気側に開き,水準瓶 (k) をビュレット (j) を越えた位
置に上げて,ビュレット (j) 内の酸素を外気に放出させ,三方コック (i) 及び (o) を閉じてお
く。
(32) 空試験値は,試料に添加するのと同量の助燃剤をはかり取った磁器ボートを用いて試料の分析
操作と同様に処理し,炭酸ガスの容量を求めておく。助燃剤は金属銅,金属すず,金属鉛又は
鉛丹を試料1gにつき0.5〜1gを添加し混合して用いる。いずれの助燃剤も,1gにつき空試験値
は,炭酸ガス量として0.2ml以下でなければならない。
(33) 日常作業にあっては,作業時間の初期,中期,終期には,必ず炭素含有量既知の標準試料を分
析して,その日の装置その他の調子を試験しなければならない。
(34) 試料に油類が付着しているのを認めたときは,アルコール又はエーテルで洗浄し,乾燥した後
使用する。試料を調製するとき,あまり小さい試料や長く巻いた試料にならないように注意し
なければならない。
(35) この吸収瓶中の溶液に濁りを認めたときには,新しいものと取り替えなければならない。過マ
ンガン酸カリウム溶液 (50g/l) の代わりに無水クロム酸溶液 (50g/l) 10mlを用いてもよい。この
場合に溶液が青みを帯びたなら,新しいものと取り替えなければならない。
なお亜硫酸ガス吸収瓶 (p) を使用したときは,この吸収液中に炭酸ガスが残ることがあるか
18
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ら注意する必要がある。
(36) 取り出した磁器ボートは冷却後,試料の融解状態と酸化状態とを確かめなければならない。
(37) 混合ガスの温度と残留ガスの温度とに差があってはならない。冬期においてビュレット内の溶
液及び炭酸ガス吸収瓶内の溶液の温度が低い場合には温度差を生じないように,あらかじめ酸
素の空通しや標準試料による試験を行っておく必要がある。
(38) 念のためもう一度,燃焼管内のガスをビュレットに捕集し分析して,炭酸ガスが残留しないこ
とを確かめる必要がある。
(39) 定量前には必ず気密試験を行い,気密に留意しなければならない。気密試験は,注(31)に準じる。
(40) 操作後に試料が完全に融解状態になったかを確かめるとともに,試料が完全に酸化したかどう
かを確かめなければならない。
(41) 助燃剤の空試験値は,いずれもその1gにつき,炭酸ガスの容積として0.2ml未満であることが
必要である。
備考 高周波誘導加熱装置を使用する場合の操作は,次のとおりとする。
付図2の装置を連結し(30),気密を確認した後(39)空試験を行い(32),三方コック (o) を閉じ,
三方コック (i) を外気に向けて開いておく。次に試料3gと適当な助燃剤(金属すずなど)を入
れたるつぼを燃焼管内に挿入して密栓し,酸素開閉弁 (F) 及び流量調節弁 (E) を全開して酸素
を送入し,燃焼管内の空気を追い出した後三方コック (i) を閉じ,高周波スイッチ (C) を入れ,
10〜20秒たってから三方コック (o) を炉側に開き,更に三方コック (i) をわずかに炉側に開い
て,混合ガスを亜硫酸ガス吸収瓶 (p) (35)及び冷却管 (h) を通して徐々にビュレット (j) に導き,
1分ぐらいたって試料の燃焼がほぼ終わってから混合ガスを急速にビュレット (j) の目盛の下
部近くまで捕集した後,三方コック (i) 及び (o) を閉じ,高周波切断スイッチ (I) を入れて高
周波電流を断ち,酸素の送入をやめて約1分間放置する。その間,燃焼管内の磁器るつぼを取
り出す(36)(40)。
ビュレット (j) 内の溶液の水準を合わせて目盛を読み,混合ガスの容積とする。
なお,このとき混合ガスの温度も測定しておく(34)。以下,8.3.5(5)以降に従って炭素を定量す
る(38)。
8.4
中和滴定法
8.4.1
要旨 試料を酸素気流中で強熱し,炭素を完全に酸化して炭酸ガスとして捕集し,これを酸素と共
にあらかじめ一定量の水酸化ナトリウム標準溶液を入れてある炭酸ガス吸収装置に導いて炭酸ガスを吸収
させた後,硫酸標準溶液で滴定する。
8.4.2
試薬 試薬は,次による。
(1) クロム酸飽和硫酸 硫酸(比重1.82, 90%)に重クロム酸カリウムを飽和させ,上澄み液を使用する。
(2) ソーダ石灰又は水酸化ナトリウム
(3) 金属銅,金属すず,金属鉛又は鉛丹
(4) 酸素
(5) 過マンガン酸カリウム溶液 過マンガン酸カリウムの飽和溶液(約50g/l)10mlに硫酸 (1+1) 1mlを
加えて酸性とする。
(6) 水酸化バリウム溶液(飽和,約40g/l)
(7) 酸化銅(粒状)
(8) 石綿(精製品)
19
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(9) 0.005mol/l硫酸標準溶液 硫酸0.3mlを水で1lに薄めて調製し,その力価は,無水炭酸ナトリウム(JIS
K 8005の標準試薬)を標準として決定する(JIS K 8001参照)。
(10) 0.01mol/l水酸化ナトリウム溶液 水酸化ナトリウム0.4gをあらかじめ酸素などを通して炭酸ガスを除
去した水に溶解して1lとして調製する。空気中から炭酸ガスなどの酸性ガスが入らないように,ソー
ダ石灰又は水酸化ナトリウムを詰めた吸収管を付けて保存し,自動ビュレットを用いて取り出すよう
にする。この標準溶液の力価は,決める必要がない。
(11) フェノールフタレイン溶液 フェノールフタレイン0.5gをエチルアルコール (
V
V
95
%) 100mlに溶解
する。
8.4.3
装置及び器具 装置及び器具は,原則として次のものを用いる(付図4参照)。
(1) 酸素清浄装置 酸素中に含まれる炭酸ガス又は有機性ガスなどを除去し,かつ酸素を清浄乾燥するこ
とを目的とするもので,粒状酸化銅を詰めた管及びこれを800〜850℃に加熱する炉 (a) ,空瓶 (I) 及
びクロム酸飽和硫酸を入れた洗瓶(42) (b) ,ソーダ石灰又は水酸化ナトリウムを詰めた管又は塔 (c) を
順次連結するものとする(43)。ただし,純良な酸素を用い,酸化銅管 (a) を使用する必要が認められな
いときは,省いてもよい。
(2) 燃焼炉 燃焼炉は,内径約30mm,長さ200〜300mmの管状電気炉 (d) を用い,その中央部において,
長さ約150mm以上を一定温度に保つことができるものとする。電流を調節して温度を加減し,高温
計によって炉の中央部の燃焼管の真上の温度を測定するものとする(44)。炉には,その両端がそれぞれ
約200mmずつ突き出し得る長さをもつ内径約24mmの磁器燃焼管 (e) を挿入する。
また管の入口には,空気中から炭酸ガスなどの酸性ガスの侵入を防ぐため,試料挿入管 (f) を備え,
また管内には磁器ボート挿入位置の後方約50mmにわたって酸化鉄の飛散を防ぐための精製石綿を詰
め,更に管の後には,硫黄酸化物を吸収除去し,併せて一酸化炭素を酸化するために,過マンガン酸
カリウム溶液を入れた硫黄除去装置 (g) を連結するものとする。管状電気炉の代わりに高周波誘導加
熱装置(付図2)を使用することができる。この装置は,外径約42mm,内径約37mm,長さ約200mm
の石英製縦形燃焼管と,その中央よりやや下方の外側に巻いた高さ約40〜50mm,巻数3〜4回の加熱
コイルと,これに高周波電流を供給する高周波発振装置からなり,必要に応じて酸素流量計 (0〜2
000ml/min) ,タイムスイッチ(0〜6分)を附属させるものとする。この装置を使用するときの操作は,
備考によるものとする。
(3) 捕集装置 燃焼炉から出たガスを捕集するため,次の順で連結する。三方コック (n) ,捕集ビュレッ
ト (l) 及び水準瓶 (m) を取り付ける。捕集ビュレットと水準瓶はゴム管で連結し,この中に塩化ナト
リウム溶液(260g/l)[メチルレッドを指示薬とし,溶液がようやく赤色となるまで硫酸(1+1)を滴加し
て微酸性とする]を満たす。
(4) 炭酸ガス吸収装置 捕集装置から出た炭酸ガスを吸収するために,0.01mol/l水酸化ナトリウム溶液の
一定量を入れた内径約4mm,長さ1400〜1600mm,巻径約80mm,巻数約6,高さ約80mmの蛇管状
の吸収管 (h) を主体とし,この中を混合ガスが小気泡となって通過する間に炭酸ガスは吸収され,溶
液は循環する。この吸収管 (h) の後には,吸収管で吸収されずに逃げる炭酸ガスを監視するため,水
酸化バリウム溶液(飽和,約40g/l)約5mlを入れた検査トラップ (i) を連結するものとする。
(5) 磁器ボート 空試験値を下げるため,磁器ボートは,なるべく高温でしかも長時間空焼きした後冷却
し,ピンセットで取り出して,グリースなどを塗らないデシケーター中に保存する。長時間保存した
ものは空試験値が高くなることが多いから使用は避け,再度空焼きするのがよい。
また磁器ボートには,ボートの約半分を覆い,出し入れのじゃまにならない程度の大きさの磁器カ

20
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
バーを載せるとよい。
8.4.4
試料はかり取り量 試料は炭素含有率に応じ,原則として表4に従ってはかり取る。
表4
炭素含有率%
試料はかり取り量g
0.005 以上 0.03 未満
3
0.03 以上 0.05 未満
2
0.05 以上 0.1 未満
1
8.4.5
操作 定量操作は,次の手順によって行う。
(1) 8.4.3の装置を順次に連結して(43)(45)酸素を通じながら燃焼管を加熱して,管内温度を1 300〜1 350℃
に上昇させた後,試料(46)及び助燃剤(47)をはかり取った磁器ボートを燃焼管の端部に入れ,試料挿入
管 (f) をかぶせ,気密にする。三方コック (k) を外気に向けて開き,酸素を毎分200〜300mlの割合
で約5分間通じて燃焼管内の空気を完全に排除してから,三方コック (k) を捕集ビュレット (l) 側に,
三方コック (n) を炭酸ガス吸収装置 (h) 側に開き,コック (j) を調節して酸素を少しずつ通しながら,
炭酸ガス吸収装置 (h) に0.01mol/l水酸化ナトリウム溶液30mlを正確に取り,フェノールフタレイン
溶液3〜4滴を指示薬として加え,検査トラップ (i) を付け,コック (j) を閉じ,三方コック (n) を
捕集ビュレット側に開いておく。
(2) 次に,挿入棒を外部から磁石を用いて操作し,磁器ボートを管内最高温度の部分に押し入れ,挿入棒
は直ちに元に戻す。試料が燃焼し始めても管内が減圧にならないように酸素流量を調節しながら,約
2分間予熱する。
(3) 次に酸素流量を増加して試料を完全に燃焼させ,捕集ビュレット (l) (48)の球部容積の約半分で試料の
燃焼を完了させた後,水準瓶 (m) を下げて炭酸ガスと酸素の混合ガスを捕集ビュレット (l) の最下部
近くまで捕集し,三方コック (k) を閉じ,酸素の送入を止める。
(4) 次に水準瓶 (m) を捕集ビュレット (l) の上位に置き,三方コック (n) を炭酸ガス吸収装置側に開い
てコック (j) を調節し,毎分約40mlの割合で捕集ガスを炭酸ガス吸収装置 (h) の中に導入し,検査
トラップ (i) 中の水酸化バリウム溶液に白濁が生じないように注意しながら炭酸ガスを完全に吸収さ
せる。
(5) 次に酸素を通じながら検査トラップ (i) を除いた後,吸収管の口へビュレットの先端を挿入し,炭酸
ガス吸収装置 (h) を揺り動かしながら0.005mol/l硫酸標準溶液で滴定し,吸収装置の球部にたまって
いる溶液の色が薄いピンク色になったら滴加をやめる。
(6) 蛇管内の溶液が球部に戻り,一様なピンク色になったら次の1滴を加えるようにして滴定を続け,溶
液に薄いピンク色が残る点を終点とする(49)。
(7) 空の磁器ボートを用いて,上記(1),(2),(3)及び(4)の操作に準じて空試験を行う。
8.4.6
計算 試料中の炭素含有率を,次の式によって算出する。
(
)
100
000120
.0
×
×
×
−
=
W
F
B
A
C
ここに,
C: 試料中の炭素含有率 [% (m/m)]
A: 本試験における0.005 mol/l硫酸標準溶液の使用量 (ml)
B: 空試験における0.005 mol/l硫酸標準溶液の使用量 (ml)
F: 使用した硫酸標準溶液の0.005 mol/lに対する力価
W: 試料はかり取り量 (g)
注(42) クロム酸飽和硫酸が緑色になれば酸化力を喪失するから,更新しなければならない。
21
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(43) ゴム管は炭酸ガスを吸収するおそれがあるから,装置の各接続にはガラス管を用い,その両端
を密接させ,シリコーンゴム管又はビニル管でこれを保持するようにしなければならない。
(44) 高温計指示温度と燃焼管内温度との差に注意して補正する必要がある。
(45) 定量前に必ず気密試験を行い,気密に留意しなければならない。
(46) 試料は特に油類が付着しないことを要し,必要があればアルコール又はエーテルで十分に洗浄
し,手あかなどが付着しないよう,ピンセット又は金さじで取り扱う。
また試料は,ボール箱,紙袋などに入れておくと紙片が混入して思わぬ正誤差を与えること
があるから,清浄なガラス瓶に入れておく。
(47) 金属銅,金属すず,金属鉛又は鉛丹を試料と同量ずつ添加し混合して用いる。
(48) 炭酸ガスの捕集を完全にするため,全容約450mlのものを用いるほうがよい。
(49) かすかにピンク色が残る状態は,0.005mol/l硫酸標準溶液を追加してもしばらく続くから,少し
慣れると終点の判定に不便はない。
備考1. 高周波誘導加熱装置を使用する場合の操作は,次のとおりとする。
付図2の装置を連結し(43),気密を確認した後(44)試料(50)及び適当な助燃剤(すずなど)を
入れた磁器るつぼ(あらかじめ空焼きしたもの)を燃焼管に入れ気密にする。
三方コック (k) を外気側に向けて開き,酸素を毎分200〜300mlの割合で約5分間通して,
燃焼管内の空気を完全に排除してから三方コック (k) を捕集ビュレット側に,捕集装置の三
方コック (n) 及びコック (j) を炭酸ガス吸収装置 (h) 側に開き,コック (j) を調節して酸素
を少しずつ通しながら,炭酸ガス吸収装置 (h) に0.01mol/l水酸化ナトリウム標準溶液30ml
を正確に取り,フェノールフタレイン溶液3〜4滴を指示薬として加え,検査トラップ (i) を
付け,コック (j) ,三方コック (k) を閉じておく。高周波スイッチ (C) を入れ,試料が燃焼
し始めてから三方コック (k) を捕集ビュレット (l) 側に開き,三方コック (n) を捕集ビュレ
ット側に導通させ,酸素を導入しながら水準瓶を徐々に下げ,試料の燃焼が捕集ビュレット
(l) の球部容積の約半分で燃焼を完了させるように調節し,燃焼が終わってから混合ガスを急
速に捕集ビュレット (l) の下部近くまで捕集した後三方コック (k) を閉じ,高周波切断スイ
ッチ (I) を入れ高周波電流を断ち,酸素の送入をやめて約1分間放置する。その間に燃焼管
内の磁器るつぼを取り出す。水準瓶を捕集ビュレット (l) の上位部に置き,三方コック (n)
を炭酸ガス吸収装置側に開いておく。以下8.4.5(4)以降に従って炭素を定量する。
注(50) 試料のはかり取り量は,8.4.4に準じる。
2. 次の電量定量法によって炭素を定量してもよい。
電量定量法 装置(51)に酸素を通し,通電して安定した後,1 300〜1 350℃に加熱した燃焼管
の中央に試料0.5gをはかり取り燃焼させる。生成した炭酸ガスを吸収液(52)に吸収させ,この
炭酸を中和するのに要するアルカリを吸収液中の中性塩を電解して生成させ(53),その電量滴
定が完了したときの指示値から,炭素を定量(54)する。
注(51) 装置は酸素定圧装置,燃焼炉,亜硫酸ガス吸収装置,吸収セル(炭酸ガスの吸収が完
全で,電解に要する電量を精密に指示するものを用いる。)を組み合わせた装置によっ
て行う。吸収セルの電解槽は,隔膜として陽極槽には電解液を,陰極槽には吸収液を
入れておく。
(52) 吸収液は,過塩素酸バリウム溶液 (50g/l) にイソプロピルアルコール (
V
V
3
2〜
%) を
加える。
22
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(53) 電解液は,炭酸バリウムに過塩素酸バリウム溶液 (300g/l) を加える。
(54) 試薬,装置及び器具のほか,分析についての共通事項は,この規格に定められた他の
方法に準じる。
2
3
H
1
4
11
-1
9
9
6
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図1 炭素定量装置(8.2重量法,8.2.3装置及び器具)
注 この図は各部の連結の要領を示すもので,各器具の形状は,この規格に抵触しない限り,適宜選択することができる。
2
4
H
1
4
11
-1
9
9
6
付図2(その1) 炭素定量装置(高周波誘導加熱炉の一例)
付図2(その2) 燃焼筒部詳細図(一例)
2
5
H
1
4
11
-1
9
9
6
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図3(その1) 炭素定量装置(8.3ガス容量法,8.3.3装置及び器具)
26
H 1411-1996
付図3(その2) 炭素定量装置(容量法)詳細図
2
7
H
1
4
11
-1
9
9
6
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図4 微量炭素定量装置(8.4中和滴定法,8.4.3装置及び器具)

28
H 1411-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付表1 炭素定量値補正係数 (f) 表[ただしこの表は,塩化ナトリウム溶液 (26%) を用いた場合に使用する。]
ガスの
温度
℃
補正
気圧 (B)
kPa {mmHg}
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
ガスの
温度
℃
補正
気圧 (B)
kPa {mmHg}
98.658 {740}
1.019 1.015 1.011 1.007 1.003 0.999 0.995 0.990 0.986 0.982 0.978 0.973 0.969 0.965 0.961 0.956 0.952 0.948 0.943 0.939 0.934 0.930 0.925 0.920 0.916 0.911 0.906 0.901 0.896 0.892 0.887 0.881 0.876 0.871 0.866 0.860 98.658 {740}
98.792 {741}
1.021 1.017 1.012 1.008 1.004 1.000 0.996 0.992 0.987 0.983 0.979 0.975 0.970 0.966 0.962 0.958 0.953 0.949 0.944 0.940 0.936 0.931 0.926 0.922 0.917 0.912 0.907 0.903 0.898 0.893 0.888 0.883 0.877 0.872 0.867 0.862 98.792 {741}
98.925 {742}
1.022 1.018 1.014 1.010 1.006 1.001 0.997 0.993 0.989 0.985 0.980 0.976 0.972 0.968 0.963 0.959 0.955 0.950 0.946 0.941 0.937 0.932 0.928 0.923 0.918 0.914 0.909 0.904 0.899 0.894 0.889 0.884 0.879 0.873 0.868 0.863 98.925 {742}
99.058 {743}
1.024 1.019 1.015 1.011 1.007 1.003 0.999 0.994 0.990 0.986 0.982 0.977 0.973 0.969 0.965 0.960 0.956 0.951 0.947 0.943 0.938 0.933 0.929 0.924 0.920 0.915 0.910 0.905 0.900 0.895 0.890 0.885 0.880 0.875 0.869 0.864 99.058 {743}
99.192 {744}
1.025 1.021 1.017 1.012 1.008 1.004 1.000 0.996 0.991 0.987 0.983 0.979 0.974 0.970 0.966 0.962 0.957 0.953 0.948 0.944 0.939 0.935 0.930 0.926 0.921 0.916 0.911 0.906 0.902 0.897 0.892 0.886 0.881 0.876 0.871 0.865 99.192 {744}
99.325 {745}
1.026 1.022 1.018 1.014 1.010 1.006 1.001 0.997 0.993 0.989 0.984 0.980 0.976 0.972 0.967 0.963 0.958 0.954 0.950 0.945 0.941 0.936 0.931 0.927 0.922 0.918 0.913 0.908 0.903 0.898 0.893 0.888 0.882 0.877 0.872 0.866 99.325 {745}
99.458 {746}
1.028 1.023 1.019 1.015 1.011 1.007 1.003 0.998 0.994 0.990 0.986 0.981 0.977 0.973 0.969 0.964 0.960 0.955 0.951 0.946 0.942 0.937 0.933 0.928 0.923 0.919 0.914 0.909 0.904 0.899 0.894 0.889 0.884 0.878 0.873 0.868 99.458 {746}
99.592 {747}
1.029 1.025 1.021 1.016 1.012 1.008 1.004 1.000 0.996 0.991 0.987 0.983 0.978 0.974 0.970 0.966 0.961 0.957 0.952 0.948 0.943 0.939 0.934 0.929 0.925 0.920 0.915 0.910 0.905 0.900 0.895 0.890 0.885 0.880 0.874 0.869 99.592 {747}
99.725 {748}
1.030 1.026 1.022 1.018 1.014 1.010 1.005 1.001 0.997 0.993 0.988 0.984 0.980 0.975 0.971 0.967 0.962 0.958 0.954 0.949 0.945 0.940 0.935 0.931 0.926 0.921 0.916 0.911 0.907 0.902 0.897 0.891 0.886 0.881 0.876 0.870 99.725 {748}
99.528 {749}
1.032 1.028 1.023 1.019 1.015 1.011 1.007 1.003 0.998 0.994 0.990 0.985 0.981 0.977 0.973 0.968 0.964 0.959 0.955 0.950 0.946 0.941 0.937 0.932 0.927 0.923 0.918 0.913 0.908 0.903 0.898 0.893 0.887 0.882 0.877 0.871 99.528 {749}
99.992 {750}
1.033 1.029 1.025 1.021 1.016 1.012 1.008 1.004 1.000 0.995 0.991 0.987 0.982 0.978 0.974 0.969 0.965 0.961 0.956 0.952 0.947 0.943 0.938 0.933 0.928 0.924 0.919 0.914 0.909 0.904 0.899 0.894 0.889 0.883 0.878 0.873 99.992 {750}
100.12 {751}
1.035 1.030 1.026 1.022 1.018 1.014 1.009 1.005 1.001 0.997 0.992 0.988 0.984 0.979 0.975 0.971 0.966 0.962 0.957 0.953 0.948 0.944 0.939 0.935 0.930 0.925 0.920 0.915 0.910 0.905 0.900 0.895 0.890 0.885 0.879 0.874 100.12 {751}
100.26 {752}
1.036 1.032 1.028 1.023 1.019 1.015 1.011 1.007 1.002 0.998 0.994 0.989 0.985 0.981 0.977 0.972 0.968 0.963 0.959 0.954 0.950 0.945 0.940 0.936 0.931 0.926 0.921 0.916 0.912 0.907 0.902 0.896 0.891 0.886 0.880 0.875 100.26 {752}
100.39 {753}
1.037 1.033 1.029 1.025 1.021 1.016 1.012 1.008 1.004 0.999 0.995 0.991 0.986 0.982 0.978 0.973 0.969 0.964 0.960 0.956 0.951 0.946 0.942 0.937 0.932 0.928 0.923 0.918 0.913 0.908 0.903 0.898 0.892 0.887 0.882 0.876 100.39 {753}
100.52 {754}
1.039 1.035 1.030 1.026 1.022 1.018 1.014 1.009 1.005 1.001 0.996 0.992 0.988 0.983 0.979 0.975 0.970 0.966 0.961 0.957 0.952 0.948 0.943 0.938 0.934 0.929 0.924 0.919 0.914 0.909 0.904 0.899 0.893 0.888 0.883 0.878 100.52 {754}
100.66 {755}
1.040 1.036 1.032 1.027 1.023 1.019 1.015 1.011 1.006 1.002 0.998 0.993 0.989 0.985 0.980 0.976 0.972 0.967 0.963 0.958 0.954 0.949 0.944 0.940 0.935 0.930 0.925 0.920 0.915 0.910 0.905 0.900 0.895 0.889 0.884 0.879 100.66 {755}
100.79 {756}
1.042 1.037 1.033 1.029 1.025 1.020 1.016 1.012 1.008 1.003 0.999 0.995 0.990 0.986 0.982 0.977 0.973 0.968 0.964 0.959 0.955 0.950 0.946 0.941 0.936 0.932 0.926 0.922 0.917 0.912 0.907 0.901 0.896 0.891 0.885 0.880 100.79 {756}
100.92 {757}
1.043 1.039 1.034 1.030 1.026 1.022 1.018 1.013 1.009 1.005 1.000 0.996 0.992 0.987 0.983 0.979 0.974 0.970 0.965 0.961 0.956 0.952 0.947 0.942 0.937 0.933 0.928 0.923 0.918 0.913 0.908 0.903 0.897 0.892 0.887 0.881 100.92 {757}
101.06 {758}
1.044 1.040 1.036 1.032 1.027 1.023 1.019 1.015 1.010 1.006 1.002 0.997 0.993 0.989 0.984 0.980 0.976 0.971 0.967 0.962 0.958 0.953 0.948 0.943 0.939 0.934 0.929 0.924 0.919 0.914 0.909 0.904 0.898 0.893 0.888 0.882 101.06 {758}
101.19 {759}
1.046 1.041 1.037 1.033 1.029 1.025 1.020 1.016 1.012 1.007 1.003 0.999 0.994 0.990 0.986 0.981 0.977 0.972 0.968 0.963 0.959 0.954 0.949 0.945 0.940 0.935 0.930 0.925 0.920 0.915 0.910 0.905 0.900 0.894 0.889 0.884 101.19 {759}
101.32 {760}
1.047 1.043 1.039 1.034 1.030 1.026 1.022 1.017 1.013 1.009 1.004 1.000 0.996 0.991 0.987 0.983 0.978 0.974 0.969 0.965 0.960 0.955 0.951 0.946 0.941 0.937 0.932 0.927 0.922 0.917 0.912 0.906 0.901 0.896 0.890 0.885 101.32 {760}
101.46 {761}
1.049 1.044 1.040 1.036 1.031 1.027 1.023 1.019 1.014 1.010 1.006 1.001 0.997 0.993 0.988 0.984 0.979 0.975 0.970 0.966 0.961 0.957 0.952 0.947 0.943 0.938 0.933 0.928 0.923 0.918 0.913 0.908 0.902 0.897 0.892 0.886 101.46 {761}
101.59 {762}
1.050 1.046 1.041 1.037 1.033 1.029 1.024 1.020 1.016 1.011 1.007 1.003 0.998 0.994 0.990 0.985 0.981 0.976 0.972 0.967 0.963 0.958 0.953 0.949 0.944 0.939 0.934 0.929 0.924 0.919 0.914 0.909 0.903 0.898 0.893 0.887 101.59 {762}
101.72 {763}
1.051 1.047 1.043 1.038 1.034 1.030 1.026 1.021 1.017 1.013 1.008 1.004 1.000 0.995 0.991 0.987 0.982 0.978 0.973 0.969 0.964 0.959 0.955 0.950 0.945 0.940 0.935 0.930 0.925 0.920 0.915 0.910 0.905 0.899 0.894 0.889 101.72 {763}
101.86 {764}
1.053 1.048 1.044 1.040 1.036 1.031 1.027 1.023 1.018 1.014 1.010 1.005 1.001 0.997 0.992 0.988 0.983 0.979 0.974 0.970 0.965 0.961 0.956 0.951 0.946 0.942 0.937 0.932 0.927 0.922 0.917 0.911 0.906 0.901 0.895 0.890 101.86 {764}
101.99 {765}
1.054 1.050 1.045 1.041 1.037 1.033 1.028 1.024 1.020 1.015 1.011 1.007 1.002 0.998 0.994 0.989 0.985 0.980 0.976 0.971 0.967 0.962 0.957 0.952 0.948 0.943 0.938 0.933 0.928 0.923 0.918 0.913 0.907 0.902 0.897 0.891 101.99 {765}
102.12 {766}
1.055 1.051 1.047 1.043 1.038 1.034 1.030 1.026 1.021 1.017 1.012 1.008 1.004 0.999 0.995 0.991 0.986 0.981 0.977 0.972 0.968 0.963 0.958 0.954 0.949 0.944 0.939 0.934 0.929 0.924 0.919 0.914 0.908 0.903 0.898 0.892 102.12 {766}
102.26 {767}
1.057 1.053 1.048 1.044 1.040 1.035 1.031 1.027 1.022 1.018 1.014 1.009 1.005 1.001 0.996 0.992 0.987 0.983 0.978 0.974 0.969 0.965 0.960 0.955 0.950 0.946 0.940 0.935 0.930 0.925 0.920 0.915 0.910 0.904 0.899 0.894 102.26 {767}
102.39 {768}
1.058 1.054 1.050 1.045 1.041 1.037 1.033 1.028 1.024 1.019 1.015 1.011 1.006 1.002 0.998 0.993 0.989 0.984 0.980 0.975 0.970 0.966 0.961 0.956 0.951 0.947 0.942 0.937 0.932 0.927 0.922 0.916 0.911 0.906 0.900 0.895 102.39 {768}
102.52 {769}
1.060 1.055 1.051 1.047 1.042 1.038 1.034 1.030 1.025 1.021 1.016 1.012 1.008 1.003 0.999 0.995 0.990 0.985 0.981 0.976 0.972 0.967 0.962 0.958 0.953 0.948 0.943 0.938 0.933 0.928 0.923 0.918 0.912 0.907 0.901 0.896 102.52 {769}
102.66 {770}
1.061 1.057 1.052 1.048 1.044 1.040 1.035 1.031 1.027 1.022 1.018 1.013 1.009 1.005 1.001 0.996 0.991 0.987 0.982 0.978 0.973 0.968 0.964 0.959 0.954 0.949 0.944 0.939 0.934 0.929 0.924 0.919 0.913 0.908 0.903 0.897 102.66 {770}
102.79 {771}
1.062 1.058 1.054 1.049 1.045 1.041 1.037 1.032 1.028 1.023 1.019 1.015 1.010 1.006 1.002 0.997 0.993 0.988 0.984 0.979 0.974 0.970 0.965 0.960 0.955 0.951 0.945 0.940 0.936 0.930 0.925 0.920 0.915 0.909 0.904 0.898 102.79 {771}
102.92 {772}
1.064 1.059 1.055 1.051 1.047 1.042 1.038 1.034 1.029 1.025 1.020 1.016 1.012 1.007 1.003 0.998 0.994 0.989 0.985 0.980 0.976 0.971 0.966 0.961 0.957 0.952 0.947 0.942 0.937 0.932 0.927 0.921 0.916 0.911 0.905 0.900 102.92 {772}
103.06 {773}
1.065 1.061 1.056 1.052 1.048 1.044 1.039 1.035 1.031 1.026 1.022 1.017 1.013 1.009 1.005 1.000 0.995 0.991 0.986 0.982 0.977 0.972 0.967 0.963 0.958 0.953 0.948 0.943 0.938 0.933 0.928 0.923 0.917 0.912 0.906 0.901 103.06 {773}
103.19 {774}
1.067 1.062 1.058 1.054 1.049 1.045 1.041 1.036 1.032 1.028 1.023 1.019 1.014 1.010 1.006 1.001 0.996 0.992 0.987 0.983 0.978 0.973 0.969 0.964 0.959 0.954 0.949 0.944 0.939 0.934 0.929 0.924 0.918 0.913 0.908 0.902 103.19 {774}
備考1. 表中の気圧は,水銀気圧計の読取値に温度及び重力補正を行った値である。なお,補正気圧の単位及び数値で{ }を付けて表したものは従来単位によるものであって,参考として併記したものである。
温度及び重力の補正は,例えば次の式によって行う。
B=B'(1−0.000 163t−0.002 6cos2φ−0.000 000 2H)
ここに,B:補正した気圧の値 (kPa)
t:気圧計に付いている温度計の読み (℃)
B':気圧計の読み (kPa)
ψ:気圧計のある場所の緯度 (°)
H:海面からの高さ (m)
分析者は,あらかじめB'(0.000 163t+0.002 6cos2ψ−0.000 000 2H)の値を各種の気圧・温度について求めて表にしておけば,使用に便利である。ただし高さの項は,微少であるから省いてもよい。
29
H 1411-1996
2. この表の使用例 試料のはかり取り量を1gとすれば
1mlCO2=0.050 27%C
例えばビュレットの読取値=14.3ml,ガスの温度=29℃,気圧計に付いている温度計の読み (t) : 25℃,気圧計の読み (B') =101.7246kPaであった場合は,東京では気圧の温度及び重力補正値は−0.533 2kPaとなるから,補
正した気圧の値 (B) =101.724 6−0.533 2=101.191 4
したがって,補正気圧101.191 4kPa,ガスの温度29℃のときの補正係数は付表1から0.940であるから,求める炭素量 (%) は次のようになる。
(%)
676
.0
1
940
.0
05027
.0
3.
14
=
×
×

30
H 1411-1996
参考
鉄クロム電熱材中のけい素,コバルト,モリブデン,
チタン,ニオブの定量方法
序文 次に記載するけい素,コバルト,モリブデン,チタン,ニオブの各定量方法は,参考のために示す
ものであって,規格の一部ではない。
1. けい素定量方法
1.1
方法の区分 けい素の定量方法は,重量法による。
1.2
重量法
1.2.1
要旨 試料を王水で分解し,過塩素酸を加えて加熱蒸発し,けい素を不溶性けい酸とし,こし分け
た後強熱して恒量となし,次にふっ化水素酸を加えてけい酸を蒸発揮散させ,その量をはかる。
1.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+10)
(2) 過塩素酸
(3) ふっ化水素酸
(4) 硫酸 (1+3)
(5) 王水 塩酸3,硝酸1の割合で混合する。
(6) 酒石酸アンモニウム溶液 酒石酸15gを水500mlに溶解し,アンモニア水15mlを加え,水で1lに薄
める。
1.2.3
試料はかり取り量 試料はけい素含有率に応じ,原則として参考表1に従ってはかり取る。
参考表1
けい素含有率%
試料はかり取り量g
1未満
3
1以上
1
1.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (300ml) に移し入れ,時計皿で覆い,王水を試料1gのときは20ml,3gの
ときは40mlを徐々に加えて静かに加熱して完全に分解した後,過塩素酸を試料1gのときは20ml,3g
のときは40mlを加え,強く加熱して蒸発した過塩素酸の蒸気がビーカー壁を伝わって逆流する程度
に15〜20分間加熱を続ける。
(2) しばらく放冷後,温水約120mlを加え,かき混ぜて可溶性塩類を溶解し,直ちにろ紙(5種B)を用
いてろ過し,ビーカー壁に付着したけい酸は,ゴム管付ガラス棒を用いてすり落とし,ろ紙上に移し,
初めは温水で,次に温塩酸 (1+10) で,最後に温水で,ろ液に鉄イオンの反応がなくなるまで洗う(1)。
(3) 残さはろ紙と共に白金るつぼに移し入れ,注意して加熱灰化し,1 100℃以上で強熱して恒量とし,デ
シケーター中で室温まで放冷して第1回のひょう量をする。
(4) 次に残さを硫酸 (1+3) で潤し,ふっ化水素酸(2)3〜5mlを加え,注意して加熱し,けい酸及び硫酸を
揮散させた後,1 100℃以上で強熱して恒量とし,デシケーター中で室温まで放冷して第2回のひょう
31
H 1411-1996
量をする。
1.2.5
計算 試料中のけい素含有率を,次の式によって算出する。
(
)
100
4674
.0
2
1
×
×
−
=
W
w
w
Si
ここに,
Si: 試料中のけい素含有率 [% (m/m)]
w1: 第1回のひょう量 (g)
w2: 第2回のひょう量 (g)
W: 試料はかり取り量 (g)
注(1) けい酸の洗浄には,温水と温塩酸の代わりに温酒石酸アンモニウム溶液を用い,ろ紙に付着し
た鉄塩の黄色が消えるまで約3〜4回洗浄するようにしてもよい。
(2) 使用した量と同量のふっ化水素酸及び硫酸の強熱残さを求めて,操作中の揮散減量に加算しな
ければならない。ただし,ふっ化水素酸は,その1mlにつき残さ量が0.04mgを超えてはならな
い。
2. コバルト定量方法
2.1
方法の区分 コバルトの定量方法は,次のいずれかによる。
(1) 直接吸光光度法 この方法は,コバルト含有率0.01%以上0.4%未満の試料に適用する。
(2) 抽出吸光光度法 この方法は,コバルト含有率0.001%以上0.5%未満の試料に適用する。
2.2
直接吸光光度法
2.2.1
要旨 試料を混酸で分解し,硝酸を加えて酸化する。これを水で一定量とし,その中から一定量を
2個のビーカーに分取する。1個に酢酸ナトリウム及びニトロソR塩を加えた後,硝酸を加えて煮沸し,
試料液とする。他の1個には酢酸ナトリウム及び硝酸を加えた後,ニトロソR塩を加え,これを対照液と
して試料液の吸光度を測定する。
2.2.2
試薬 試薬は,次による。
(1) 硝酸
(2) 混酸 硫酸3,りん酸3,水14の割合に混合する。
(3) 酢酸ナトリウム溶液 (500g/l)
(4) ニトロソR塩溶液 (20g/l)
(5) 標準コバルト溶液 (0.1mgCo/ml) 高純度金属コバルト1gを硝酸 (1+1) 30mlで分解した後,水で正
確に1lとする。その中から一定量を正確に分取し,水で正しく10倍量に薄めて標準コバルト溶液と
する。
2.2.3
試料はかり取り量 試料は,0.5gをはかり取る。
2.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (100ml) に移し入れ,時計皿で覆い,混酸20mlを加えて加熱分解し,硝
酸3mlを加えて引き続き加熱する。もし炭化物など未分解残さを認めた場合は,硫酸白煙処理を行い,
完全に分解する。冷却後少量の水を加えて塩類を溶解し,50mlの全量フラスコに入れ,水で標線まで
薄める。
(2) これから2個のビーカー (200ml) にコバルト含有量に応じ,一定量(3)を分取し,水を加えて液量を約
25mlとする。このうち1個のビーカーには酢酸ナトリウム溶液(4)及びニトロソR塩溶液10mlを加え,
加熱して1〜2分間煮沸する。次に硝酸10mlを加え,更に約1分間煮沸して冷却する。他の1個のビ
32
H 1411-1996
ーカーには酢酸ナトリウム溶液20mlを加え,次に硝酸10ml(5)を加えて沈殿を溶解し,加熱して1〜2
分間煮沸した後,ニトロソR塩溶液10mlを加え,更に約1分間煮沸して冷却する。これを対照液と
する。
(3) この両液をそれぞれ100mlの全量フラスコに移し入れ,水で標線まで薄める。その一部を光度計の吸
収セルに取り,対照液を対照として530nm付近の吸光度を測定する。
2.2.5
計算 2.2.6で作成した検量線からコバルト量を求め,試料中のコバルト含有率を次の式によって
算出する。
100
×
×
=
B
W
A
Co
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
A: 試料溶液中のコバルト検出量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
2.2.6
検量線の作成 標準コバルト溶液の各種液量 (0〜5ml) をビーカー (100ml) に分取し,酢酸ナトリ
ウム溶液及び硫酸 (1+3) を用いて,pHを6±0.2に調節し,これにニトロソR塩溶液 (20g/l) 10mlを加え
て加熱し,1〜2分間煮沸する。次に硝酸10mlを加え,引き続き約1分間煮沸し,冷却後,溶液を100ml
の全量フラスコに入れ,標線まで水を加える。
他のビーカー (100ml) にはコバルトを加えず,試薬だけを上記の操作に従って処理し,正しく100ml
とし,これを対照液とする。以下2.2.4(3)に準じて各々の吸光度を測定し,得た吸光度とコバルト量との関
係線を作成して検量線とする。
注(3) 溶液の分取量はコバルト含有率に応じ,原則として参考表2に従ってはかり取る。
参考表2
コバルト含有率%
試料溶液分取量ml
0.01 以上
0.05 未満
20
0.05 以上
0.1 未満
10
0.1 以上
0.4 未満
5
(4) 酢酸ナトリウム溶液の使用量は,試料溶液分取量に応じ,原則として参考表3による。
参考表3
試料溶液分取量ml
酢酸ナトリウム溶液使用量ml
5
20
10
30
20
40
(5) コバルトの呈色に適当な条件では,ニトロソR塩によってコバルト以外の元素によっても呈色
又は沈殿するが,呈色後過剰の硝酸を加えるとコバルトニトロソR塩だけが残り,他の錯塩な
どは分解する。しかし,あらかじめ過剰の硝酸酸性溶液にしておくと,コバルトはもちろん他
の元素による呈色もない。ニトロソR塩自身も波長530nm付近にはかなりの吸収があるので,
この操作を行ったものを対照液とすれば,試薬及び試料の着色イオンによる吸収が補正される。
2.3
抽出吸光光度法

33
H 1411-1996
2.3.1
要旨 試料を塩酸及び硝酸で分解し,過塩素酸を加え,加熱して白煙を発生させクロムを酸化する。
これを塩酸に溶解してメチルイソブチルケトンによって,鉄及びクロムの大部分を抽出除去する。次にア
ンモニアアルカリ性として,チタン及びニオブを沈殿させて除去する。これを蒸発して濃縮した後,塩酸
チオシアン酸アンモニウム及び塩化第一すずを加え,コバルトのチオシアン酸錯塩を生成させ,メチルイ
ソブチルケトンで抽出し,吸光度を測定する。
2.3.2
試薬 試薬は,次による。
(1) 塩酸 (5+1, 1+1)
(2) 硝酸
(3) 過塩素酸
(4) アンモニア水
(5) 塩化第一すず(2水塩)
(6) チオシアン酸アンモニウム溶液 (600g/l)
(7) メチルイソブチルケトン
(8) 標準コバルト溶液 2.2.2(5)に従って調製する。
2.3.3
試料はかり取り量 試料はコバルト含有率に応じ,原則として参考表4に従ってはかり取る。
参考表4
コバルト含有率%
試料はかり取り量g
0.001 以上 0.01未満
1〜2
0.01 以上 0.1 未満
0.5〜1
0.1 以上 0.5 未満
0.1〜0.5
2.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (100ml) に移し入れ,時計皿で覆い,塩酸 (1+1) 10〜20ml及び硝酸3〜
10ml(6)で分解し,これに過塩素酸3〜10mlを加え,更に加熱して白煙を発生させ,引き続き加熱して
クロムを十分に酸化する。この場合,乾固する必要はない(7)。
(2) これに硝酸0.5〜1mlを加えた後,塩酸 (1+1) 10〜30mlを少量ずつ用いて手早く分液漏斗に洗い移す
(8)。直ちにこれと同量のメチルイソブチルケトンを加えて手早く約30秒間(9)振り混ぜる。2液層に分
離後,直ちに下層の酸溶液を別の分液漏斗に移し,再び同量のメチルイソブチルケトンで同様に抽出
する。
(3) 下層の酸溶液をビーカーに取り,液量が10ml以上のときは蒸発して約10ml(10)とする。これに水を10
〜30ml程度加えて薄め,アンモニア水を過剰に加えてアルカリ性(11)とする。
(4) 沈殿をろ紙でろ過し,水で数回洗浄する。ろ液を約5mlに蒸発濃縮した後,塩酸 (5+1) 0.5ml(12)を加
え,分液漏斗に移し,チオシアン酸アンモニウム溶液(13)5mlを少量ずつ用いてビーカーを洗い,分液
漏斗に移す。次に塩化第一すず(14)2gを固体のまま加え,振り混ぜて溶解した後,メチルイソブチル
ケトン10mlを加えて約30秒間振り混ぜる。
(5) 2液層に分離後,上層のメチルイソブチルケトン層を取り,630nm(15)付近の吸光度を測定する。
なお,試薬だけを用いて同様に処理して空試験を行い(16),吸光度を補正する。
2.3.5
計算 2.3.6で作成した検量線からコバルト量を求め,試料中のコバルト含有率を次の式によって
算出する。
100
×
=WA
Co

34
H 1411-1996
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
A: 試料溶液中のコバルト検出量 (g)
W: 試料はかり取り量 (g)
2.3.6
検量線の作成 標準コバルト溶液の各種液量 (0〜3ml) を数個の分液漏斗に取り,塩酸 (5+1)
0.5mlと約3mlの水を加えた後,チオシアン酸アンモニウム溶液5ml及び水を加えて10mlとし,塩化第一
すず2gを固体のまま加え,振り混ぜて溶解した後,メチルイソブチルケトン10mlをビュレットから加え,
約30秒間振り混ぜて静置し,2液層に分離する。メチルイソブチルケトン層を取り,630nm付近の吸光度
を測定し,得た吸光度とコバルト量との関係線を作成して検量線とする。
注(6) 塩酸 (1+1) の添加量は,試料はかり取り量に応じ,原則として参考表5に従う。硝酸は塩酸 (1
+1) の量に従い,3〜10mlの間で適宜に加える。
参考表5
試料はかり取り量g
塩酸 (1+1) ml
0.1 以上 0.5 未満
10
0.5 以上 1 未満
15
1 以上 2 未満
20
(7) 過塩素酸は,次に行う抽出に影響しないので,全部を追い出す必要はない。クロムはクロム酸
にならないと抽出されないので,完全に酸化する必要がある。クロム酸の抽出率は最高90%程
度である。
(8) 硝酸の少量を加えた後,塩酸 (1+1) に溶解するのは,クロム酸をできるだけ還元しないで溶解
するためである。塩酸は更に薄い方が安全であるが,鉄の抽出率が下がるので,鉄とクロムの
両方になるべく適するように塩酸 (1+1) とした。塩類の溶解及び分液漏斗への洗い移しは,で
きるだけ手早く行わないと,クロムの抽出がうまくゆかないので注意する必要がある。
(9) 試料の塩酸溶液にメチルイソブチルケトンを加えると,更にクロム酸の還元が速くなるため,
直ちにメチルイソブチルケトンを加えて30秒以内振り混ぜて手早く抽出する。30秒以上の振
り混ぜは,かえってクロムの抽出に対してはよくない。2液層に分かれた後も,直ちに分ける
ことが必要である。2回目の抽出では,クロムの抽出はあまり期待できず,鉄の抽出を目的と
している。
(10) 塩酸溶液が多いとき,そのまま次のアンモニア水でアルカリ性にすると,生成するアンモニウ
ム塩の量が多すぎて次の抽出の際に種々の不都合が生じるから,塩酸の一部を除去するため
10ml程度に濃縮する。
(11) チタン及びニオブは,いずれもチオシアン酸錯塩がコバルト錯塩と共に抽出されるので分離す
る必要がある。これらはメチルイソブチルケトン抽出によって除去されないので,鉄及びクロ
ムの大部分を抽出除去した後,塩酸溶液にアンモニア水を過剰に加え,塩化アンモニウムの存
在でアンモニアアルカリ性とし,チタン及びニオブを沈殿させて分離する。
(12) コバルトのチオシアン酸錯塩の抽出において塩酸の濃度は0.5mol/l以下が適当であるので,塩
酸 (5+1) 0.5mlを加える。
(13) 抽出にはチオシアン酸アンモニウムの濃度が4mol/l以上が適当であるので,5mlを加えて全量
10mlとする。
(14) 以上の操作で鉄はほとんど除去されているが,分離後に試薬などからくる鉄のブランクを除去
するために,塩化第一すずによって還元を行う。これによって鉄はチオシアン酸錯塩として抽
出されず,コバルトだけが抽出される。塩化第一すずは酸化されやすいものであるから注意す

35
H 1411-1996
る必要がある。
(15) 可視部における最大吸収は625〜635nmにあるので,中心の630nmを用いる。この波長におけ
る分子吸光係数は1 750である。
(16) 普通は空試験値は極めて少なく,吸光度で0〜0.015程度である。
3. モリブデン定量方法
3.1
方法の区分 モリブデンの定量方法は,次のいずれかによる。
(1) 重量法 この方法は,モリブデン含有率0.03%以上の試料に適用する。
(2) 吸光光度法 この方法は,モリブデン含有率0.03%以上0.5%未満の試料に適用する。
3.2
重量法
3.2.1
要旨 試料を王水と過塩素酸で処理分解し,過塩素酸の白煙を発生させる。放冷後,水を加えて可
溶性塩類を溶解し,こし分けた後,ろ液に亜硫酸水を加え,煮沸してクロムを還元する。これを10℃以下
に冷却し,α−ベンゾインオキシムを加えてモリブデンを沈殿させた後こし分け,乾燥後低温で灰化し,
放冷後その質量をはかる。次にアンモニア水を加えて酸化モリブデンを溶解し,こし分けた後,不溶解残
さを強熱し,放冷後質量をはかり,両質量の差からモリブデンを定量する。
3.2.2
試薬 試薬は,次による。
(1) 過塩素酸
(2) 硫酸 (2+100)
(3) 王水 塩酸3,硝酸1の割合で混合する。
(4) アンモニア水
(5) 臭素溶液(飽和)
(6) 亜硫酸溶液(飽和)
(7) α−ベンゾインオキシム溶液 α−ベンゾインオキシム10gをメチルアルコール500mlに溶解する。
(8) α−ベンゾインオキシム洗浄液 α−ベンゾインオキシム溶液25〜50mlを硫酸 (1+99) 1l中に加えて
調製する。この洗浄液は使用の都度調製し,10℃以下に冷却して用いる。
3.2.3
試料はかり取り量 試料はモリブデン含有率に応じ,原則として参考表6に従ってはかり取る。
参考表6
モリブデン含有率%
試料はかり取り量g
0.03以上 1未満
2
1 以上 3未満
1
3 以上
0.5
3.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (300ml) に移し入れ,時計皿で覆い,王水50ml及び過塩素酸30mlを加え
て分解し,濃厚な白煙を発生してクロムが赤色のクロム酸になるまで濃縮する。
(2) 放冷後,水約100mlを加えて煮沸し,可溶性塩類を溶解した後,ろ紙(5種B)でろ過し,温硫酸 (2
+100) で洗浄する。
(3) ろ液に亜硫酸溶液(飽和)30mlを加え,再び煮沸してクロムを還元した後,溶液を10℃以下に冷却
し(17),かき混ぜながら徐々にα−ベンゾインオキシム溶液10mlを加え,更にモリブデン含有量0.01g
に対し5mlを過剰に加え,かき混ぜながら溶液が黄色を呈するまで臭素溶液(飽和)を加え(18),次に
なお少量のα−ベンゾインオキシム溶液(19)を加える。

36
H 1411-1996
(4) ビーカーを冷却して溶液を10℃以下に保ちながら,ときどきかき混ぜて約10分間放置後,水に浸し
た少量のろ紙パルプを加え,ろ紙(5種A)を用いてろ過する。
(5) 沈殿はα−ベンゾインオキシム洗浄液で十分に洗浄した後(20),ろ紙と共に磁器るつぼに移し入れ,乾
燥後約500℃で注意して加熱し,ろ紙を灰化し,デシケーター中で放冷後その質量をはかり,これを
不純酸化モリブデン量(21)とする。
(6) るつぼ中にアンモニア水5mlを加えて不純酸化モリブデンを溶解し,これをろ紙(5種B)でこし分
け,温水で十分に洗浄し,乾燥後強熱灰化し,デシケーター中で放冷後質量をはかり,この質量を不
純酸化モリブデン量から差し引いて得た質量を酸化モリブデン (MoO3) とする。
3.2.5
計算 試料中のモリブデン含有率を次の式によって算出する。
100
6666
.0
×
×
=
W
w
Mo
ここに,
Mo: 試料中のモリブデン含有率 [% (m/m)]
w: 差し引いて得た酸化モリブデンの質量 (g)
W: 試料はかり取り量 (g)
注(17) 10℃以下で試薬を添加する理由は,モリブデン酸 (H2MoO4) の還元を防ぐのが第一目的であり,
併せて鉄の吸着を防ぐことができる。
(18) 臭素溶液を加える理由も,α−ベンゾインオキシムによるモリブデン酸の還元を防ぐためである。
(19) 試薬の過剰量は,沈殿をこし分けた際,ろ液を放置して針状結晶が析出すれば十分である。
(20) 沈殿の生成,ろ過,洗浄の操作は,すべて10℃以下で行う。
(21) 灰化物が白色の場合は,これを酸化モリブデンとみなし,以下の操作を省略することができる。
3.3
吸光光度法
3.3.1
要旨 試料を過塩素酸で分解し,塩化ナトリウム処理を行い,クロムを塩化クロミルとして揮散さ
せる。冷却した後,硫酸,チオシアン酸ナトリウム及び塩化第一すずを加えて呈色させ,チオシアン酸モ
リブデン錯塩の吸光度を測定する。
3.3.2
試薬 試薬は,次による。
(1) 過塩素酸
(2) 硫酸 (1+3)
(3) 塩化ナトリウム
(4) チオシアン酸ナトリウム溶液 (100g/l)
(5) 塩化第一すず溶液 塩化第一すず(2水塩)50gをビーカー (300ml) にはかり取り,塩酸 (1+1) 200ml
を加えて加熱溶解し,冷却した後水を用いて500mlに薄める。
(6) 標準モリブデン酸溶液 結晶モリブデン酸アンモニウム(4水塩)0.184gを温水に溶解後,室温に冷
却し,水を用いて正しく1lに薄める。この溶液1mlは,約0.1mgのモリブデンを含有するが,α−ベ
ンゾインオキシム重量法によって,その含有量を決定する。
3.3.3
試料はかり取り量 試料はモリブデン含有率に応じ,原則として参考表7に従ってはかり取る。
参考表7
モリブデン含有率%
試料はかり取り量g
0.03 以上 0.1 未満
1
0.1 以上 0.6 未満
0.5
0.6 以上 1.2 未満
0.25
1.2 以上
0.1
37
H 1411-1996
3.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (200ml) に移し入れ,時計皿で覆い,過塩素酸20mlを加え(22),完全に分
解するまで加熱を続け,更に過塩素酸の濃厚な白煙を発生させ,クロムをクロム酸に酸化する。これ
に塩化ナトリウム0.5〜1gを数回に分けて加え,大部分のクロムを塩化クロミルとして揮散させる。
(2) 放冷後,水20mlを加えて加熱し,約1分間静かに煮沸して可溶性塩類を溶解し(23),室温に冷却し,
100mlの全量フラスコに移し入れ,水を用いて標線まで薄める。
(3) この溶液10mlを50mlの全量フラスコに分取し,硫酸 (1+3) 20ml(24)を加えて振り混ぜ,更にチオシ
アン酸ナトリウム溶液10mlを加えて振り混ぜ,フラスコを振りながら塩化第一すず溶液5mlを滴加
し,水を用いて標線まで薄める(25)。
(4) 5〜20℃で約10分間放置後,溶液の一部を光度計の吸収セルに取り,波長460nm付近の吸光度を測定
する。
3.3.5
計算 3.3.6で作成した検量線から,モリブデンの量を求め,その含有率を次の式によって算出す
る。
100
×
×
=
B
W
A
Mo
ここに,
Mo: 試料中のモリブデン含有率 [% (m/m)]
A: 試料溶液中のモリブデン検出量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
3.3.6
検量線の作成 試料中に含まれる鉄と同量の純鉄をビーカー (200ml) に数個はかり取り,各々に
標準モリブデン溶液の各種液量 (0〜30ml) (試料0.5gに対し0.1〜0.6%Mo相当)と過塩素酸20mlを加え
て加熱分解し,以下,3.3.4(2)以降に準じて操作を行い,吸光度を測定し,得た吸光度とモリブデン量との
関係線を作成して検量線とする。
注(22) 試料の分解には,塩酸と硝酸の少量を加えてもよい。
(23) この際,けい酸などの残さを認めたときは,これをこし分け,なるべく少量の硫酸 (1+100) を
用いて洗浄する。
(24) 硫酸 (1+3) の使用量は,過塩素酸20mlを用いたとき15〜25mlの範囲では,吸光度に影響は
ない。
(25) モリブデン含有率が0.1%以下の場合は,チオシアン酸塩の褐色の呈色を促進するために,溶液
の一部を試験管に取り,沸騰水中に浸して一定時間加温した後,直ちに流水中で冷却し,吸光
度を測定してもよい。ただしこの場合は,同一操作に従って作成した検量線を使用しなければ
ならない。
4. チタン定量方法
4.1
方法の区分 チタンの定量方法は,過塩素酸−過酸化水素吸光光度法による。
4.2
過塩素酸−過酸化水素吸光光度法
4.2.1
要旨 試料を王水で加熱分解し,過塩素酸を加えて白煙処理を行い,塩酸又は塩化ナトリウムを加
えてクロムを除去した後,けい酸などをろ別する。この溶液の吸光度及び過酸化水素水を加えてチタンを
呈色させたときの吸光度を測定し,前後の吸光度の差からチタンを定量する。
4.2.2
試薬 試薬は,次による。

38
H 1411-1996
(1) 塩酸
(2) 過塩素酸
(3) 王水 塩酸3,硝酸1の割合で混合する。
(4) 塩化ナトリウム
(5) 過酸化水素水
(6) 標準チタン溶液 (0.5mgTi/ml) 金属チタン(99%以上)0.250gに塩酸 (1+1) 60mlを加えて加熱分解
し,冷却した後水で500mlに薄める。
4.2.3
試料はかり取り量 試料はチタン含有率に応じ,原則として参考表8に従ってはかり取る。
参考表8
チタン含有率%
試料はかり取り量g
0.1未満
1
0.1以上 0.8未満
0.5
0.8以上 2.0未満
0.2+純鉄0.3
4.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (300ml) に移し入れ,適当量の王水を加えて加熱分解する。過塩素酸(26)
を加え,更に加熱を続けて濃厚な過塩素酸の白煙が発生し,溶液がだいだい色に変わり始めたころか
ら1〜2分間加熱する。
(2) 塩酸又は塩化ナトリウムを少量ずつ加えて,クロムを塩化クロミルとして揮散除去し,クロムが50mg
以下になるようにする。
(3) 放冷後,温水約30mlを加えて振り混ぜ,溶液が黄色となりクロムの存在を認めたときは,過酸化水
素水をクロムが還元されて溶液が青色になるまで滴加する。
(4) 煮沸して過剰の過酸化水素を分解し,室温まで冷却した後,ろ紙(5種A)を用いて100mlの全量フ
ラスコにろ過し,温水を用いて4〜5回洗浄して,けい酸などをろ別する。流水を用いて室温まで冷却
した後,水で標線まで薄める。
(5) この溶液の一部を光度計の吸収セルに取り,波長420nm付近の吸光度を測定する。
(6) 次に全量フラスコ中の残液に過酸化水素水0.5mlを加えてチタンを呈色させ,再び吸光度を測定する。
(7) 前後の吸光度の差から,4.2.5で作成した検量線によってチタンの含有率を求める。
4.2.5
検量線の作成 試料中に含まれる鉄と同量の純鉄をビーカー (300ml) に数個はかり取り,各々に
標準チタン溶液の各種液量 (0〜8ml) を加え,以下4.2.4 (1)以降の操作に準じて処理し,得た吸光度とチタ
ン量との関係線を作成して検量線とする。ただし,試料1g及び0.5g(試料0.2g+純鉄0.3gの場合を含む)
の検量線は,別々に作成しなければならない。
注(26) 過塩素酸使用量は試料はかり取り量に応じ,原則として参考表9に従う。
参考表9
試料はかり取り量g
過塩素酸使用量ml
1
35〜40
0.5
25
0.2+純鉄0.3
25
備考1. モリブデン2mg以上及びバナジウムを含む試料は,次のように操作する。
4.2.4(1)〜(4)及び(6)の操作を行って吸光度を測定する。残液に酸性ふっ化アンモニウムの結
晶を,試料0.5gの場合には1g,試料1gの場合には2gを加えて振り混ぜ,過酸化チタンの黄
色を消し,再び吸光度を測定し,前後の吸光度の差からチタン量を決定する。

39
H 1411-1996
2. ニオブを含む試料は,次のように操作する。
4.2.4(1)の操作を行い,放冷後水約30mlを加えて振り混ぜ塩類を溶解する。ろ紙(5種C)
を用いてろ過し,温水で数回洗浄し,ろ液及び洗液は合わせて保存する。ろ紙及び残さは白
金るつぼ中で強熱灰化後,ふっ化水素酸処理を行い,硫酸水素ナトリウム約2gを加えて融解
後,主溶液で溶解する。この溶液を磁気水銀陰極電解装置に移し,約15Aの電解電流で約30
分間電解する。電解液をビーカーに移し,加熱して約40mlに濃縮した後冷却し,100mlの全
量フラスコに移し入れ,りん酸 (1+2) 3mlを加えた後,水で標線まで薄める。以下4.2.4(5)
以降に従って操作し,チタンを定量する。この場合の検量線は,標準チタン溶液だけで作成
したものを用いる。
5. ニオブ定量方法
5.1
方法の区分 ニオブの定量方法は,次のいずれかによる。
(1) 重量法 この方法は,ニオブ含有率0.1%以上の試料に適用する。
(2) 吸光光度法 この方法は,ニオブ含有率0.01%以上1%以下の試料に適用する。
5.2
重量法
5.2.1
要旨 試料を塩酸,硝酸及び過塩素酸で分解し,これに亜硫酸ナトリウム及びタンニンを加えて煮
沸した後こし分けし,ニオブ(タンタルを含む)を大部分の鉄その他の元素から分離する。この沈殿を灰
化し,硫酸とふっ化水素酸とで溶解した後再沈殿し,これを強熱後ひょう量して,ニオブとタンタルの合
量を求める。この沈殿中のタンタルを吸光光度法によって求めて補正する。
5.2.2
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (2+100)
(3) 硝酸
(4) 過塩素酸
(5) ふっ化水素酸
(6) 硫酸 (1+1, 1+3)
(7) りん酸 (1+4)
(8) 過酸化水素水
(9) 炭酸カリウム
(10) 亜硫酸ナトリウム溶液 (200g/l)
(11) ピロ硫酸カリウム
(12) タンニン
(13) ピロガロール溶液 (300g/l) この溶液は,使用の都度調製する。
(14) しゅう酸アンモニウム溶液 (30g/l)
5.2.3
試料はかり取り量 試料はニオブ含有率に応じ,原則として参考表10に従ってはかり取る。
参考表10
ニオブ含有率%
試料はかり取り量g
1未満
2
1以上 2未満
1
2以上
0.5
40
H 1411-1996
5.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料をはかり取りビーカー (500ml) に移し入れ,試料1gにつき塩酸20ml及び硝酸5ml(27)を加え,加
熱して分解する。これに過塩素酸(28)を加え,加熱を継続して過塩素酸の白煙を発生させ,クロムを完
全に重クロム酸に酸化した後,なお5〜10分間,白煙発生処理を続ける。
(2) 放冷後少量の水及び塩酸15mlを加えて塩類を溶解した後,水で250mlに薄め,亜硫酸ナトリウム溶
液50ml及びタンニン1gを順次に加えて良く振り混ぜ,加熱して約10分間煮沸する。しばらく放置
した後,少量のろ紙パルプを加えて振り混ぜ,ろ紙(5種C)を用いてこし分け,熱塩酸 (2+100) で
十分に洗浄する。
(3) 沈殿をろ紙と共に白金るつぼに移し,乾燥後強熱して灰化する。
(4) 放冷後これに硫酸 (1+3) 3ml及びふっ化水素酸5〜7mlを加え,加熱して沈殿を溶解し,更に加熱を
継続して硫酸の白煙を発生させる。放冷後,あらかじめ過酸化水素水1mlを入れたビーカー (500ml)
中に冷水で洗い移し,水で全容を約250mlとする。
(5) 塩酸15ml,亜硫酸ナトリウム溶液50mlを加え,直ちにタンニンlgを加えて加熱し,10〜15分間煮沸
して沈殿を完成させる。しばらく放置した後少量のろ紙パルプを加えて振り混ぜ,ろ紙(5種C)を
用いてこし分け,温塩酸 (2+100) で十分に洗浄する。
(6) 沈殿をろ紙と共に質量既知のるつぼに移し,乾燥した後灰化させ,約1 000℃で約30分間強熱して恒
量とし,デシケーター中で放冷後ひょう量し,ニオブ(タンタルを含む)酸化物の質量を求める(29)。
(7) これにピロ硫酸カリウム約5gを加え,650〜700℃で約7分間強熱して融解し,放冷後しゅう酸アンモ
ニウム溶液約50mlを入れてあるビーカー (300ml) に移し,静かに加熱して融塊を溶解する。この溶
液を100mlの全量フラスコに,しゅう酸アンモニウム溶液で洗い移し,標線まで薄める。この溶液50ml
を100mlの全量フラスコに分取し,りん酸 (1+4) 10mlを加えて振り混ぜる。次にピロガロール溶液
30mlを加えた後,しゅう酸アンモニウム溶液で標線まで薄める。
(8) この溶液の一部を光度計の吸収セルに取り,波長430nm付近の吸光度を測定する。
5.2.5
計算 5.2.6で作成した検量線からタンタル量を求める。このタンタル量を酸化物に換算し,前記
のひょう量値から差し引き,試料中のニオブを次の式によって算出する。
(
)
100
6990
.0
2
1
×
×
−
=
W
w
w
Nb
ここに,
Nb: 試料中のニオブ含有率 [% (m/m)]
w1: ニオブ(タンタルを含む)酸化物ひょう量値 (g)
w2: 酸化タンタル量 (g) =タンタル量 (g) ×1.221
W: 試料はかり取り量 (g)
5.2.6
タンタル検量線の作成 標準タンタル溶液(五酸化タンタル)0.244 2gを磁器るつぼにはかり取り,
ピロ硫酸カリウム約10gを加えて融解し,冷却後しゅう酸アンモニウム溶液 (30g/l) で融塊を溶解して正
しく1lとする。この溶液(1mlは,タンタル0.2mgを含有する。)の各種液量 (0〜10ml) を数個の100ml
の全量フラスコに分取し,5.2.4(7)のりん酸 (1+4) 10ml添加以降に従って操作し,各々の吸光度を測定し,
得た吸光度とタンタル量との関係線を作成して検量線とする。
注(27) 試料が分解困難なときは,更に塩酸及び硝酸の量を適宜追加して完全に分解する。
(28) 試料のはかり取り量が1g以下では20ml,2gでは25mlを加える。
(29) タンタルが微量で分離定量する必要がない場合には5.2.4(7)の操作を省略し,ニオブ(タンタル
を含む)酸化物の量から,これをすべてニオブとして定量してもよい。

41
H 1411-1996
備考1. モリブデンを含有する試料は,次のように操作する。
5.2.4(1)〜(3)の手順によって操作し,ニオブ及びタンタルの沈殿を強熱灰化する。放冷後硫
酸 (1+3) 2〜3滴,ふっ化水素酸3〜5mlを加え,加熱蒸発して硫酸を完全に揮散させる。こ
れに炭酸カリウム4〜5gを加え,強熱して残さを完全に融解する。冷却した後るつぼ中に少
量の水を加えて,緩く加熱して融塊を溶解する。
水でるつぼ内容物をビーカー (500ml) に移し,全液量を150〜200mlとし,良く振り混ぜ
ながら硫酸マグネシウム溶液(30)25mlを滴加し,1〜4時間放置して沈殿を完成させる。これ
に少量のろ紙パルプを加えて振り混ぜ,ろ紙(5種C)でこし分け,硫酸マグネシウム洗浄
液(31)で十分に洗浄する。沈殿をろ紙と共に元のビーカーに移し,塩酸15mlを加えて振り混
ぜ,水酸化鉄などを溶解した後,水で約300mlとし,タンニン1gを加えて約10分間煮沸し,
ろ紙(5種C)を用いてろ過し,熱塩酸 (2+100) で十分に洗浄する。
以下5.2.4(6)以降に従って操作し,ニオブを定量する。
注(30) 硫酸マグネシウム溶液 硫酸マグネシウム20gと塩化アンモニウム40gとを水に溶解
し,アンモニア水4mlを加え,水で500mlとする。
(31) 硫酸マグネシウム洗浄液 硫酸マグネシウム10g,塩化アンモニウム20g,炭酸カリウ
ム10gを水に溶解し,アンモニア水4mlを加え,水で1lとする。
2. チタンを含有する試料は,次のように操作する。
5.2.4(1)〜(5)の手順によって操作し,5.2.4(3)〜(5)の手順を繰り返し[ただしタンニンの添
加は省略する(32)],ニオブを再沈殿する。以下5.2.4(6)以降に従って操作し,ニオブを定量す
る。
注(32) 熱塩酸 (2+100) 1lに約1gのタンニンを加えた洗浄液を使用する。
5.3
吸光光度法
5.3.1
要旨 試料に塩酸,硫酸及び過塩素酸を加えて分解し,亜硫酸ナトリウム及びタンニンによって得
たニオブ(タンタルを含む)の沈殿を灰化後,ピロ硫酸カリウムで融解してしゅう酸アンモニウム塩とし,
亜硫酸ナトリウムとピロガロールを加えてニオブを呈色させ,その吸光度を測定する。
5.3.2
試薬 試薬は,次による。
(1) 5.2.2(1)〜(6)による。
(2) 亜硫酸ナトリウム溶液 (200g/l)
(3) ピロ硫酸カリウム
(4) タンニン
(5) ピロガロール溶液 (300g/l) この溶液は,使用の都度調製する。
(6) しゅう酸アンモニウム溶液 (30g/l)
(7) 標準ニオブ溶液 (0.1mgNb/ml) 五酸化ニオブ0.1430gを磁器るつぼにはかり取り,ピロ硫酸カリウム
約10gを加えて融解し,冷却後しゅう酸アンモニウム溶液 (30g/l) で浸出して正確に1lとする。
5.3.3
試料はかり取り量 試料はニオブ含有率に応じ,原則として参考表11に従ってはかり取る。
参考表11
ニオブ含有率%
試料はかり取り量g
0.5未満
1
0.5以上
0.5
5.3.4
操作 定量操作は,次の手順によって行う。
42
H 1411-1996
(1) 5.2.4(1)〜(2)の手順によって操作する。
(2) 沈殿をろ紙と共に磁器るつぼに移し,乾燥後強熱して灰化する。
(3) これにピロ硫酸カリウム約5gを加え,650〜700℃で約7分間(33)強熱して融解する。
(4) 冷却後しゅう酸アンモニウム溶液約5mlを入れてあるビーカー (300ml) に移し,静かに加熱して融塊
を溶解した後冷却する。この溶液を100mlの全量フラスコに,しゅう酸アンモニウム溶液で洗い移し,
標線まで薄める。
(5) この溶液の一定量(34)を100mlの全量フラスコに分取し,しゅう酸アンモニウム溶液を加えて液量を約
50mlとし,亜硫酸ナトリウム溶液10mlを加えて良く振り混ぜる。次にピロガロール溶液30mlを加え
た後,しゅう酸アンモニウム溶液で標線まで薄める。
(6) この溶液の一部を光度計の吸収セルに取り,波長430nm付近の吸光度を測定する。
5.3.5
計算 5.3.6で作成した検量線からニオブ量を求め,試料中のニオブ含有率を次の式によって算出
する。
100
×
×
=
B
W
A
Nb
ここに,
Nb: 試料中のニオブ含有率 [% (m/m)]
A: 試料溶液中のニオブ検出量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
5.3.6
ニオブ検量線の作成 標準ニオブ溶液の各種液量 (1〜7ml) を数個の100mlの全量フラスコに分取
し,以下5.3.4(5)〜(6)の手順によって操作し,得た吸光度とニオブ量との関係線を作成して検量線とする。
注(33) 長時間加熱すると硫酸が揮散して融解が不完全になる。
(34) ニオブ含有率0.5%未満では20ml,0.5%以上では10mlを分取する。
備考1. モリブデンを含有する試料は,次のように操作する。
5.2備考1.の熱塩酸 (2+100) で十分に洗浄するまでの操作を行い,モリブデンを除く。次
に5.2.4(5)〜(7)の手順によって操作し,ニオブを定量する。
2. チタンを含有する試料は,次のように操作する。
5.2.4(3)〜(5)の手順を繰り返し[ただし,タンニンの添加は省略し,熱塩酸 (2+100) 1lに
約1gのタンニンを加えた洗浄液を使用する。]ニオブを再沈殿し,チタンを除く。次に5.2.4
(2)〜(7)の手順によって操作し,ニオブを定量する。
43
H 1411-1996
非鉄金属部会 電熱材及び抵抗材分析方法専門委員会 構成表(昭和42年2月1日改正のとき)
氏名
所属
(委員会長)
後 藤 秀 弘
東北大学
俣 野 宣 久
金属材料技術研究所
菅 谷 宏
鉄道技術研究所
服 部 只 雄
古河電気工業株式会社
森 田 義 男
三菱電機株式会社
山 田 栄 一
東京芝浦電気株式会社
従 野 睦 秀
赤羽冶金株式会社
菅 井 三 郎
王子合金株式会社
渡 部 武 利
細川製線株式会社
大 森 茂 生
株式会社東京ワイヤー製作所
角 健 蔵
東海高熱工業株式会社
杉 本 正 勝
日本金属工業株式会社
中 田 重 徳
古河特殊金属工業株式会社
野 崎 松 郎
日立熱器具株式会社
望 月 平 一
日本冶金工業株式会社
(事務局)
石 井 清 次
工業技術院標準部材料規格課
種 橋 誠 治
工業技術院標準部材料規格課
(事務局)
廣 瀬 浩 二
工業技術院標準部材料規格課(平成8年3月1日改正のとき)
斎 藤 充
工業技術院標準部材料規格課(平成8年3月1日改正のとき)