H 1402 : 2001
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,タングステン・モ
リブデン工業会 (JTMIA) /財団法人日本規格協会 (JSA) から工業標準原案を具して日本工業規格を改正
すべきとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格である。
これによってJIS H 1402-1992は改正され,この規格に置き換えられる。
今回の改正では,不揮発分を形成するカルシウム,けい素,アルミニウム及びマグネシウム並びに硫黄
の定量方法を追加し,改正を行った。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
H 1402 : 2001
タングステン粉及び
タングステンカーバイド粉分析方法
Methods for chemical analysis of tungsten powder and
tungsten carbide powder
1. 適用範囲 この規格は,タングステン粉及びタングステンカーバイド粉中の鉄,モリブデン,カルシ
ウム,けい素,アルミニウム,マグネシウム,酸素,全炭素,遊離炭素,硫黄及び不揮発分の定量方法に
ついて規定する。
なお,それらの定量方法は表1による。
表1 定量方法
成分
定量方法
適用含有率範囲
%(m/m)
鉄
1,10-フェナントロリン吸光光度法
原子吸光法
誘導結合プラズマ (ICP) 発光分光法
0.001以上 0.5以下
0.000 5以上 0.07以下
0.000 5以上 0.07以下
モリブデン
チオシアン酸吸光光度法
原子吸光法
誘導結合プラズマ (ICP) 発光分光法
0.001以上 0.16以下
0.001以上 0.02以下
0.001以上 0.02以下
カルシウム
原子吸光法
誘導結合プラズマ (ICP) 発光分光法
0.000 5以上 0.01以下
0.000 1以上 0.01以下
けい素
四ふっ化けい素気化分離モリブドけい酸青吸光光度法
四ふっ化けい素気化分離誘導結合プラズマ (ICP) 発光分光法
混酸分解誘導結合プラズマ (ICP) 発光分光法
0.000 5以上 0.005以下
0.000 5以上 0.005以下
0.001以上 0.02以下
アルミニウム
陽イオン交換分離原子吸光法
陽イオン交換分離誘導結合プラズマ (ICP) 発光分光法
混酸分解誘導結合プラズマ (ICP) 発光分光法
0.000 2以上 0.01以下
0.000 1以上 0.01以下
0.000 5以上 0.01以下
マグネシウム
原子吸光法
誘導結合プラズマ (ICP) 発光分光法
0.000 5以上 0.005以下
0.000 1以上 0.005以下
酸素
水素還元重量法
酸化重量法
不活性ガス融解−赤外線吸収法
0.01以上
0.01以上
0.01以上
1.2以下
全炭素
焼燃−導電率法
焼燃−電量法
焼燃−熱伝導度法
焼燃−赤外線吸収法(積分法)
焼燃−赤外線吸収法(循環法)
0.001以上
0.001以上
0.001以上
0.001以上
0.001以上
遊離炭素
炭素沈殿分離燃焼−導電率法
炭素沈殿分離燃焼−電量法
炭素沈殿分離燃焼−熱伝導度法
炭素沈殿分離燃焼−赤外線吸収法(積分法)
炭素沈殿分離燃焼−赤外線吸収法(循環法)
0.01以上
0.01以上
0.01以上
0.01以上
0.01以上
硫黄
燃焼−赤外線吸収法(積分法)
0.000 5以上
2
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
成分
定量方法
適用含有率範囲
%(m/m)
不揮発分
揮発分分離重量法
0.002以上
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0016 鉄標準液
JIS K 0037 標準物質−標準液−マグネシウム
JIS K 0038 標準物質−標準液−カルシウム
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS Z 2613 金属材料の酸素定量方法通則
JIS Z 2615 金属材料の炭素定量方法通則
JIS Z 2616 金属材料の硫黄定量方法通則
JIS Z 8401 数値の丸め方
3. 一般事項 分析方法に共通な一般事項は,JIS K 0050,JIS K 0115,JIS K 0116,JIS K 0121,JIS Z 2613,
JIS Z 2615及びJIS Z 2616による。
4. 試料の採り方及び取扱い方
4.1
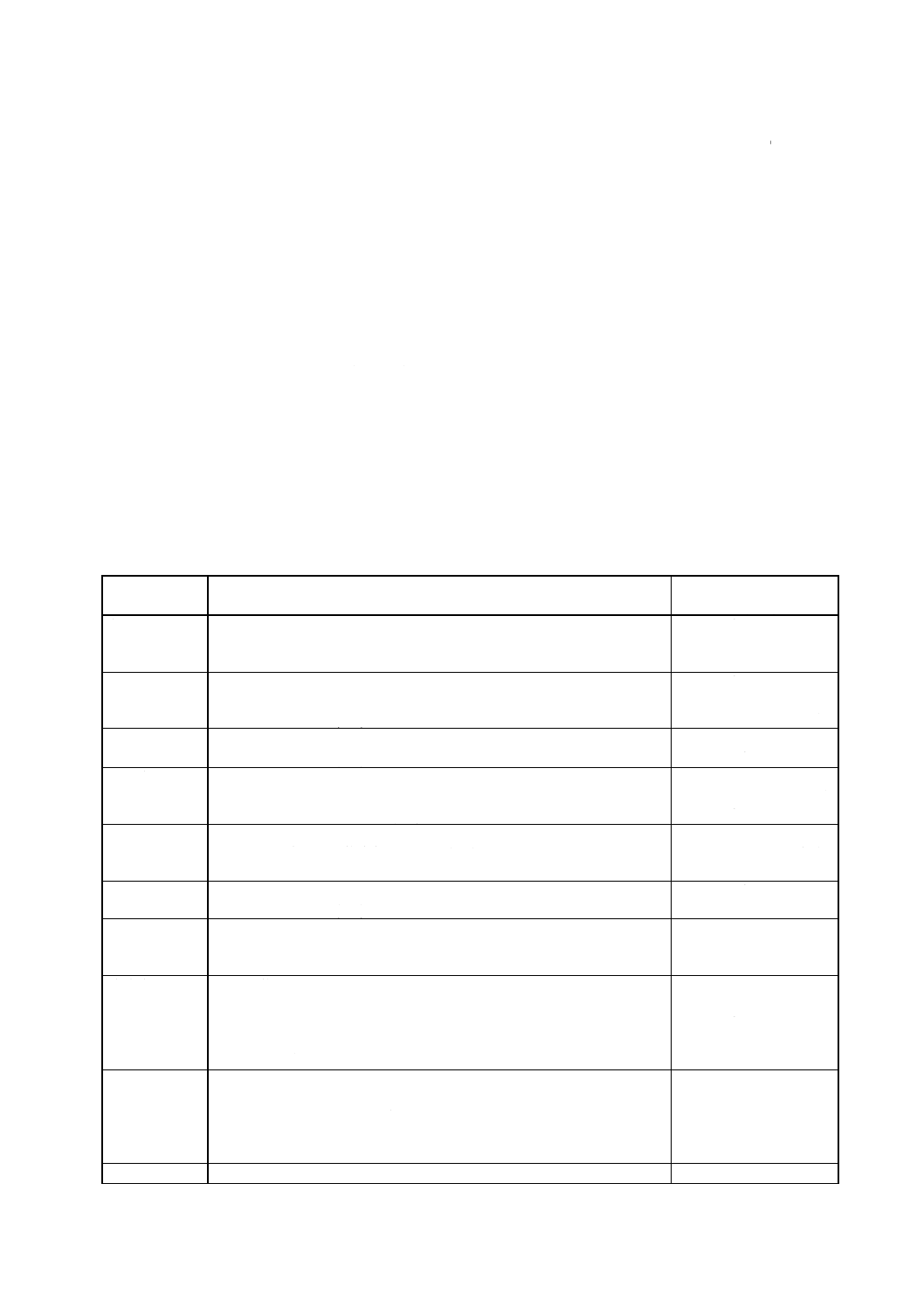
試料の採り方 試料の採り方は,同一製造ロットにつき採取箇所2か所以上をランダムに選び,図1
に示すパイプ形試料採取器を用いて開放部を下に向け,少し傾斜させて容器の底部に達するまで差し込み,
半回転させ,パイプの内部に入った粉末を採取する。
図1 パイプ形試料採取器
3
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.2
試料の取扱い方 試料の取扱い方は,4.1で採取した試料を十分に混合し分析に必要な量を取り,酸
化及び吸湿を防止するために適切な容器に入れ密封し,分析用試料とする。
4.3
試料のはかり方 酸素定量用の試料は,分析用試料開封後速やかにはかり取る。
5. 分析値のまとめ方
5.1
分析回数 通常,同一試料について2回行う。
5.2
空試験 分析に当たっては,空試験を行い,測定値を補正する。
5.3
分析値の表示 分析値は,質量百分率で表し,表1の適用含有率範囲に規定されている定量範囲の
最下位の次のけたまで算出し,JIS Z 8401によって定量範囲の最下位のけたに丸める。ただし,丸めた結
果,小数点以下の有効数字が3けた以上になる場合は,小数点以下有効数字2けたに丸める。
6. 安全衛生に関する注意 分析操作を行うには,常に安全及び衛生に注意しなければならないが,特に
次の事項に注意する。
a) 分析に使用する試薬は,労働安全衛生法有機溶剤中毒予防規則,労働安全衛生法特定化学物質等障害
予防規則並びに毒物及び劇物取締法の基準に従い操作など十分に注意する。
b) 原子吸光法,誘導結合プラズマ (ICP) 発光分光法,並びに酸素,炭素及び硫黄の定量方法における高
圧ガスの取扱いにおいては,高圧ガス取締法及びそれに関連する諸法令の基準に従い,また,運搬,
設置,操作などに十分注意し,火気に十分気をつける。
c) 原子吸光法においては,フレームの点火及び消火に注意し,特に一酸化二窒素・アセチレンフレーム
を使用する場合には逆火及び爆発に注意する。
d) 誘導結合プラズマ (ICP) 発光分光法においては,高電圧,高温,高周波及び強い光が関係する装置の
ため,測定者に対して危険及び健康を阻害する可能性がある。したがって安全機構,危険防止,測定
室環境などに配慮が必要である。
e) 酸素,炭素及び硫黄の燃焼操作においては,高温に加熱されたボート又はるつぼの取扱いは必ずるつ
ぼ挟みなどを使用して火傷などをしないように注意しなければならない。また,火災などが起きない
ように,あらかじめ対策を考慮した所定の場所に置き,十分に冷却したことを確かめてから廃棄する。
f)
不揮発分の定量に,液化塩化水素ボンベを使用するときは,取扱いに十分注意する。また,塩化水素
は十分に処理して排出する。
7. 鉄定量方法
7.1
定量方法の区分 鉄の定量方法は,次のいずれかによる。
a) 1,10-フェナントロリン吸光光度法 この方法は,鉄含有率0.001% (m/m) 以上0.5% (m/m) 以下の試料
に適用する。
b) 原子吸光法 この方法は,鉄含有率0.000 5% (m/m) 以上0.07% (m/m) 以下の試料に適用する。
c) 誘導結合プラズマ (ICP) 発光分光法 この方法は,鉄含有率0.000 5% (m/m) 以上0.07% (m/m) 以下
の試料に適用する。
7.2
1,10-フェナントロリン吸光光度法
7.2.1
要旨 試料を適切な試薬で分解し,酒石酸を加えて,タングステン,鉄などを錯塩とした後pHを
調節し,L (+) -アスコルビン酸で鉄(III)を鉄(II)に還元し,1,10-フェナントロリンを加えて1,10-フェナント
ロリン・鉄(II)錯体を生成させ,光度計を用いてその吸光度を測定する。
7.2.2
試薬 試薬は,次による。
4
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) ほう酸溶液 (50g/L)
b) 混酸A(硝酸1,ふっ化水素酸1,水3)
c) 混酸B(塩酸15,硝酸5,硫酸2)
d) アンモニア水
e) 水酸化ナトリウム溶液 (100g/L)
f)
過酸化水素
g) 酢酸
h) 酒石酸溶液 (500g/L)
i)
L (+) -アスコルビン酸溶液 (50g/L) 使用の都度調製する。
j)
1,10-フェナントロリン溶液 塩化1,10-フェナントロリニウム一水和物0.2gを水に溶解し,水で液量
を100mlとする。
k) 標準鉄溶液 (20μFe/ml) 鉄[99.9% (m/m) 以上]1.000gをはかり取ってビーカー (200ml) に移し入
れ,時計皿で覆い,塩酸(1+1)20mlを加え,穏やかに加熱して分解する。過酸化水素1mlを加え,煮
沸して鉄を酸化するとともに,過剰の過酸化水素を分解する。常温まで冷却した後,時計皿の下面を
水で洗浄して時計皿を取り除き,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線ま
で薄めて原液 (1 000μgFe/ml) とする。又は,JIS K 0016に規定する鉄標準液のFe 1 000を原液とする。
この原液を使用の都度,必要量だけ水で正しく50倍に薄めて標準鉄溶液とする。
l)
p-ニトロフェノール溶液 (4g/L)
7.2.3
試料はかり取り量 試料はかり取り量は,1.0gとし,10mgのけたまではかる。
7.2.4
操作
7.2.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 過酸化水素による分解(1)
注(1) タングステンカーバイド粉には適用しない。
1) 試料をはかり取ってビーカー (100〜200ml) に移し入れる。
2) 数mlの水で試料を湿らせ時計皿で覆い,過酸化水素5〜10mlを加え,放置又は加熱して分解し液
量が約5mlになるまで加熱して蒸発した後,水で薄めて液量を約20mlとする。
3) 室温まで冷却した後,水酸化ナトリウム溶液15mlを加え,数分間放置する。激しい発泡が終わっ
た後,液量が10〜20mlになるまで煮沸し,過剰の過酸化水素を分解し,酒石酸溶液10mlを加え,
煮沸する。
4) 室温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,水で液量を約60mlとする。
b) 混酸Aによる分解
1) 試料をはかり取って白金血(75番又は90番),四ふっ化エチレン樹脂ビーカー (100〜200ml) 又は
ポリエチレンビーカー (100〜200ml) に移し入れる。
2) 白金ふた,四ふっ化エチレン樹脂時計皿又はポリエチレン時計皿で覆い,混酸A10mlを少量ずつ加
え,穏やかに加熱して分解する(2)。引き続き加熱して窒素酸化物などを追い出す。
注(2) ポリエチレンビーカーを用いる場合は,加熱による容器の変形が起こらないように,水浴上又
は水浴中で加熱しながら分解する。
3) 室温まで冷却した後,ふた又は時計皿の下面を水で洗浄してふた又は時計皿を取り除き,酒石酸溶
液10m1及びほう酸溶液20mlをかき混ぜながら加え(3),水で液量を約60mlとする。
注(3) 遊離炭素が認められたときは,ろ紙(5種A)を用いてろ過し,水で3,4回洗浄し,ろ液と洗液
5
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
とを合わせる。
c) 過酸化水素・混酸Bによる分解
1) 試料をはかり取ってビーカー (100〜200ml) に移し入れる。
2) 数mlの水で試料を湿らせ時計皿で覆い,過酸化水素5〜10mlを加え,放置又は加熱して分解し,
タングステン酸が析出するまで加熱して蒸発する。混酸B20mlを少量ずつ加え,加熱し発泡が終わ
った後,時計皿の下面を水で洗浄して時計皿を取り除き,ほとんど乾固するまで加熱を続ける。
3) 室温まで冷却した後,水約5ml及び水酸化ナトリウム溶液20mlを加え,時計皿で覆い,加熱して
塩類を溶解し,酒石酸溶液10mlを加え,煮沸する(3)。
4) 室温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,水で液量を約60mlとする。
7.2.4.2
呈色 呈色は,次の手順によって行う。
a) 7.2.4.1のa)4),b)3)又はc)4)で得た溶液を100mlの全量フラスコに水を用いて移し入れる(4)。
注(4) この溶液の鉄量が500μg以上の場合には,この溶液を100ml全量フラスコに水を用いて移し入れ,
水で標線まで薄めた後,鉄量が500μg未満になるように,溶液を別の100mlの全量フラスコに分
取する。
b) p-ニトロフェノール溶液数滴を指示薬として加えた後,振り混ぜながら溶液の色が黄色になるまでア
ンモニア水を加え(5),次に酢酸を溶液が無色となるまで滴加し,更に数滴を過剰に加える。
注(5) 7.2.4.1 b)で分解した場合,白色の濁りを生じることがあるその場合は7.2.4.1のa)又はc)によっ
て分解を行わなければならない。
c) 常温まで冷却した後,L (+) -アスコルビン酸溶液 [7.2.2. i)] 2ml及び1,10-フェナントロリン溶液 [7.2.2
j)] 10mlを加えて振り混ぜ,水で標線まで薄め,約10分間放置する。
7.2.4.3
吸光度の測定 7.2.4.2 c)で得た溶液の一部を光度計の吸収セル (10mm) に取り,水を対照液とし
て波長510nm付近の吸光度を測定する。
7.2.5
空試験 試料を用いないで試料と同じ操作を試料と並行して行い,得られた溶液の吸光度を空試験
の吸光度とする。
7.2.6
検量線の作成 検量線の作成は,次のいずれかによる。
a) 試料溶液の調製を7.2.4.1 a)によって行う場合 数mlの水及び7.2.4.1 a)2)で加えた量と同量の過酸化
水素を数個のビーカー (100〜200m1) に取り,時計皿で覆い,液量が約5mlになるまで加熱して蒸発
し,7.2.4.1 a)3)に従って操作し,室温まで冷却した後,溶液をそれぞれ100mlの全量フラスコに水を
用いて移し入れ(6),標準鉄溶液 [7.2.2 k)] 0〜25.0ml(鉄として0〜500μg)を段階的に加える。以下,
7.2.4.2のb)〜c)及び7.2.4.3の手順に従って操作した後,得た吸光度と鉄量との関係線を作成し,その
関係線を原点を通るように平行移動して検量線とする。
注(6) 7.2.4.2 a)で注(4)を適用した場合には,これらの溶液をそれぞれ水で標線まで薄めた後,試料溶
液の分取量と同量をそれぞれ100mlの全量フラスコに分取する。
b) 試料溶液の調製を7.2.4.1 b)によって行う場合 混酸A10mlずつを数個の白金皿(75番又は90番),
四ふっ化エチレン樹脂ビーカー (100〜200ml) 又はポリエチレンビーカー (100〜200ml) に取り,酒石
酸溶液10m1及びほう酸溶液30mlをかき混ぜながら加えた後,溶液をそれぞれ100mlの全量フラスコ
に水を用いて移し入れ(6),標準鉄溶液 [7.2.2 k)] 0〜25.0ml(鉄として0〜500μg)を段階的に加える。
以下,7.2.4.2のb)〜c)及び7.2.4.3の手順に従って操作した後,得た吸光度と鉄量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
c) 試料溶液の調製を7.2.4.1 c)によって行う場合 数mlの水及び7.2.4.1 c)2)で加えた量と同量の過酸化水
6
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
素を,数個のビーカー (100〜200ml) に取り,液量が約5mlになるまで加熱して蒸発した後,時計皿
で覆う。混酸B20mlを少量ずつ加え,加熱して発泡が終わった後,時計皿の下面を水で洗浄して時計
皿を取り除きほとんど乾固するまで加熱を続け,7.2.4.1 c)3)に従って操作し,室温まで冷却した後,
溶液をそれぞれ100mlの全量フラスコに水を用いて移し入れ(6),標準鉄溶液 [7.2.2 k)] 0〜25.0ml(鉄
として0〜500μg)を段階的に加える。以下,7.2.4.2のb)〜c)及び7.2.4.3の手順に従って操作した後,
得た吸光度と鉄量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
7.2.7
計算 計算は,次のいずれかによる。
a) 7.2.4.2 a)で分取しない場合 7.2.4.3及び7.2.5で得た吸光度と,7.2.6で作成した検量線とから鉄量を
求め,試料中の鉄含有率を次の式によって算出する。
100
2
1
×
−
=
m
A
A
Fe
ここに, Fe: 試料中の鉄含有率 [% (m/m)]
A1: 試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
m: 試料はかり取り量 (g)
b) 7.2.4.2 a)で分取した場合 7.2.4.3及び7.2.5で得た吸光度と,7.2.6で作成した検量線とから鉄量を求
め,試料中の鉄含有率を次の式によって算出する。
100
100
2
1
×
×
−
=
B
m
A
A
Fe
ここに, Fe: 試料中の鉄含有率 [% (m/m)]
A1: 試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
7.3
原子吸光法
7.3.1
要旨 試料を適切な試薬で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴
霧し,その吸光度を測定する。
7.3.2
試薬 試薬は,次による。
a) りん酸(1+1)
b) 混酸C(塩酸3,硝酸1,水4)
c) タングステン粉 鉄含有率が既知で,かつ,その鉄含有率が試料中の鉄含有率より低いもの。
d) 過酸化水素
e) 標準鉄溶液 (20μgFe/ml) 7.2.2 k)による。
7.3.3
試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
注(7) 試料がタングステンカーバイド粉の場合は,1.07gとする。
7.3.4
操作
7.3.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 過酸化水素による分解(1)
1) 試料をはかり取ってビーカー (100〜200m1) に移し入れる。
2) 数mlの水で試料を湿らせ時計皿で覆い,りん酸(1+1)2m1及び過酸化水素5〜10mlを加え,放置又
7
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
は加熱して分解する。液量が約5mlになるまで加熱して蒸発し常温まで冷却した後,時計皿の下面
を水で洗浄して時計皿を取り除く。
3) 溶液を50m1の全量フラスコに水を用いて移し入れ,水で標線まで薄める(8)。
注(8) この溶液中の鉄量が160μgを超える場合には,鉄量が160μg以下になるように溶液を別の50ml
の全量フラスコに分取し,水で標線まで薄める。
b) 過酸化水素・混酸Cによる分解
1) 試料をはかり取ってビーカー (100〜200ml) に移し入れる。
2) 数mlの水で試料を湿らせ時計皿で覆い,りん酸(1+1)1ml及び過酸化水素5〜10mlを加え,放置又
は加熱して分解する。液量が約5mlになるまで加熱して蒸発し室温まで冷却した後,混酸C10mlを
少量ずつ加え,数分間放置し激しい発泡を終わらせ,約5分間煮沸して常温まで冷却した後,時計
皿の下面を水で洗浄して時計皿を取り除く(3)。
3) 溶液を50mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(8)。
7.3.4.2
吸光度の測定 7.3.4.1のa)3)又はb)3)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸
光光度計の空気・アセチレンフレーム中に噴霧し,波長248.3nmにおける吸光度を測定する。
7.3.5
空試験 7.3.6の検量線の作成操作において得られる標準鉄溶液を添加しない溶液の吸光度を,空
試験の吸光度とする(9)。
注(9) 7.3.4.1のa)3)又はb)3)で注(8)を適用して試料溶液を分取した場合には,空試験液も試料溶液と同
量を分取する。
7.3.6
検量線の作成 検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を7.3.4.1 a)によって行う場合
1) タングステン粉 [7.3.2 c)] を1.0gずつ数個はかり取り,それぞれビーカー (100〜200ml) に移し入
れる。
2) 7.3.4.1 a)2)の操作を試料と並行して行った後,溶液を50mlの全量フラスコに水を用いて移し入れる
(10)。
注(10) 7.3.4.1 a)3)で注(8)を適用した場合には,これらの溶液をそれぞれ水で標線まで薄めた後,試料溶
液の分取量と同量をそれぞれ50mlの全量フラスコに分取する。
3) 標準鉄溶液 [7.3.2 e)] 0〜8.0ml(鉄として0〜160μg)を段階的に加え,水で標線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長248.3nmにおける吸光度を試料と並行して測定し,得た吸光度と鉄量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 試料溶液の調製を7.3.4.1 b)によって行う場合
1) タングステン粉 [7.3.2 c)] を1.0gずつ数個はかり取り,それぞれビーカー (100〜200ml) に移し入
れる。
2) 7.3.4.1 b)2)の操作を試料と並行して行った後,溶液を50mlの全量フラスコに水を用いて移し入れる
(11)。
注(11) 7.3.4.1 b)3)で注(8)を適用した場合には,これらの溶液をそれぞれ水で標線まで薄めた後,試料
溶液の分取量と同量をそれぞれ50mlの全量フラスコに分取する。
3) 7.3.6 a)の3)〜4)の手順に従って操作する。
7.3.7
計算 計算は,次のいずれかによる。
a) 7.3.4.1のa)3)又はb)3)で分取しない場合 7.3.4.2及び7.3.5で得た吸光度と,7.3.6で作成した検量線
8
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
とから鉄量を求め,試料中の鉄含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Fe
ここに, Fe: 試料中の鉄含有率 [% (m/m)]
A1: 試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
A3: タングステン粉 [7.3.2 c)] 1.0g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
b) 7.3.4.1のa)3)又はb)3)で分取した場合 7.3.4.2及び7.3.5で得た吸光度と,7.3.6で作成した検量線と
から鉄量を求め,試料中の鉄含有率を次の式によって算出する。
100
50
50
3
5
4
×
×
×
−
−
=
B
m
B
A
A
A
Fe
ここに, Fe: 試料中の鉄含有率 [% (m/m)]
A4: 試料溶液中の鉄検出量 (g)
A5: 空試験液中の鉄検出量 (g)
A3: タングステン粉 [7.3.2 c)] 1.0g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
7.4
誘導結合プラズマ (ICP) 発光分光法
7.4.1
要旨 試料を過酸化水素・混酸で分解した後,溶液を誘導結合プラズマ (ICP) 発光分光装置のア
ルゴンプラズマ中に噴霧し,その発光強度を測定する。
7.4.2
試薬 試薬は,次による。
a) りん酸(1+1)
b) 混酸C(塩酸3,硝酸1,水4)
c) タングステン粉 7.3.2 c)による
d) 過酸化水素
e) 標準鉄溶液 (100μgFe/ml) 7.2.2 k)の原液 (1 000μgFe/ml) を使用の都度,必要量だけ水で正しく10
倍に薄めて標準鉄溶液とする。
7.4.3
試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
7.4.4
操作
7.4.4.1
試料溶液の調製 試料溶液の調製は,7.3.4.1 b)による(12)。
注(12) 注(8)は適用しない。
7.4.4.2
発光強度の測定 7.4.4.1で得た溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴン
プラズマ中に噴霧し,波長259.940nmにおける発光強度を測定する(13)。
注(13) 精度及び正確さを確認してあれば,他の波長を用いて測定してもよい。また,高次のスペクト
ル線が使用可能な装置では,高次のスペクトル線を用いてもよい。バックグラウンド補正機構
が付いている装置では,バックグラウンド補正機構を用いてもよい。
なお,この注を適用した場合は,検量線の作成においても同様に行う。
7.4.5
空試験 7.4.6の検量線の作成操作において得られる標準鉄溶液を添加しない溶液の発光強度を,
空試験の発光強度とする。
9
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.4.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) タングステン粉 [7.4.2 c)] を1.0gずつ数個はかり取り,それぞれビーカー (100〜200ml) に移し入れ
る。
b) 7.3.4.1 b)2)に従って操作した後,溶液を50mlの全量フラスコに水を用いて移し入れる。
c) 標準鉄溶液 [7.4.2 e)] 0〜7.0ml(鉄として0〜700μg)を段階的に加え,水で標線まで薄める。
d) これらの溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,波長
259.940nmにおける発光強度を試料と並行して測定し,得た発光強度と鉄量との関係線を作成し,そ
の関係線を原点を通るように平行移動して検量線とする。
7.4.7
計算 7.4.4.2及び7.4.5で得た発光強度と,7.4.6で作成した検量線とから鉄量を求め,試料中の鉄
含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Fe
ここに, Fe: 試料中の鉄含有率 [% (m/m)]
A1: 試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
A3: タングステン粉 [7.4.2 c)] 1.0g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
8. モリブデン定量方法
8.1
定量方法の区分 モリブデンの定量方法は,次のいずれかによる。
a) チオシアン酸吸光光度法 この方法は,モリブデン含有率0.001% (m/m) 以上0.16% (m/m) 以下の試
料に適用する。
b) 原子吸光法 この方法は,モリブデン含有率0.001% (m/m) 以上0.02% (m/m) 以下の試料に適用する。
c) 誘導結合プラズマ (ICP) 発光分光法 この方法は,モリブデン含有率0.001% (m/m) 以上0.02% (m/m)
以下の試料に適用する。
8.2
チオシアン酸吸光光度法
8.2.1
要旨 試料を適切な試薬で分解し,くえん酸及び塩酸を加えて酸性溶液とし,鉄(III),チオシアン
酸カリウム及び酢酸エチルを加え,塩化すず(II)でモリブデン(IV)を還元し,生成するモリブデン・チオシ
アン酸錯体を酢酸エチルに抽出し,光度計を用いてその吸光度を測定する。
8.2.2
試薬 試薬は,次による。
a) 塩酸
b) 硝酸
c) 混酸A(硝酸1,ふっ化水素酸1,水3)
d) 混酸B(塩酸15,硝酸5,硫酸2)
e) 水酸化ナトリウム溶液 (100g/L)
f)
タングステン粉 モリブデン含有率が試料中のモリブデン含有率より低いもの。
g) タングステンカーバイド粉 モリブデン含有率が試料中のモリブデン含有率より低いもの。
h) 過酸化水素
i)
鉄(III)溶液 硫酸アンモニウム鉄(III)・12水0.87gを硫酸 (0.1mol/L) 100m1に溶解する。この溶液1ml
は,鉄として約1mgを含む。
10
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
j)
チオシアン酸カリウム溶液 チオシアン酸カリウム150gを水350mlに溶解する。
k) 塩化すず(II)溶液 塩化すず(II)二水和物150gを塩酸100m1に加熱して溶解し,室温まで冷却した後,
水250mlを加え,少量のすずを加えて褐色瓶に保存する。
l)
くえん酸溶液 くえん酸一水和物500gを水800mlに溶解する。
m) 酢酸エチル
n) 標準モリブデン溶液 (10μgMo/ml) 三酸化モリブデン(VI)[99.5% (m/m) 以上]1.500gをはかり取っ
てビーカー (200ml) に移し入れ,時計皿で覆い,水酸化ナトリウム溶液 (100g/L) 10mlを加え,加熱
して分解する。
常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,溶液を1 000mlの全量フラ
スコに水を用いて移し入れ,水で標線まで薄めて原液 (1 000μgMo/ml) とする。この原液を使用の都
度,必要量だけ水で正しく100倍に薄めて標準モリブデン溶液とする。
8.2.3
試料はかり取り量 試料はかり取り量は,表2により,10mgのけたまではかる。
表2 試料はかり取り量
モリブデン含有率
% (m/m)
試料はかり取り量
g
0.001以上 0.015未満
1.00
0.015以上 0.16以下
0.10
8.2.4
操作
8.2.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 過酸化水素による分解(1)
1) 試料をはかり取ってビーカー (100〜200ml) に移し入れる。
2) 数mlの水で試料を湿らせ時計皿で覆い,過酸化水素5〜10mlを加え,放置又は加熱して分解し,
液量が約5mlになるまで加熱して蒸発した後,水で薄めて液量を約20mlとする。室温まで冷却し
た後,水酸化ナトリウム溶液15mlを加え,数分間放置する。激しい発泡が終わった後,液量が10
〜20mlになるまで煮沸し,過剰の過酸化水素を分解する。
3) 常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,溶液を100mlの全量フラス
コに水を用いて移し入れ,水で標線まで薄める。
b) 混酸Aによる分解
1) 試料をはかり取って白金皿(75番又は90番)に移し入れる。
2) 白金ふたで覆い,混酸A10mlを少量ずつ加え,穏やかに加熱して分解する。白金ふたの下面を水で
洗浄して白金ふたを取り除き,引き続き加熱して蒸発し,軽く乾固する。更に硝酸約1mlを加え再
び乾固し室温まで冷却する。水酸化ナトリウム溶液15mlを加え,白金ふたで覆い加熱して塩類を
溶解する。
3) 常温まで冷却した後,白金ふたの下面を水で洗浄して白金ふたを取り除き(3),溶液を100mlの全量
フラスコに水を用いて移し入れ,水で標線まで薄める。
c) 過酸化水素・混酸Bによる分解
1) 試料をはかり取ってビーカー (100〜200ml) に移し入れる。
2) 数mlの水で試料を湿らせ時計皿で覆い,過酸化水素5〜10mlを加え,放置又は加熱して分解し,
タングステン酸が析出するまで加熱して蒸発する。混酸B20mlを少量ずつ加え,加熱し発泡が終わ
った後,時計皿の下面を水で洗浄して時計皿を取り除き,ほとんど乾固するまで加熱を続ける。室
11
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
温まで冷却した後,水約5ml及び水酸化ナトリウム溶液20mlを加え,時計皿で覆い,加熱して塩
類を溶解する。
3) 常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き(3),溶液を100mlの全量フラ
スコに水を用いて移し入れ,水で標線まで薄める。
8.2.4.2
抽出 抽出は,次の手順によって行う。
a) 8.2.4.1のa)3),b)3)又はc)3)で得た溶液から正しく50mlを分液漏斗 (200ml) に分取する。
b) くえん酸溶液 [8.2.2 l)] 14ml,塩酸6ml及び鉄(III)溶液 [8.2.2 i)] 1mlを加え常温まで冷却した後,チオ
シアン酸カリウム溶液 [8.2.2 j)] 4ml,酢酸エチル15m1及び塩化すず(II)溶液 [8.2.2 k)] 4mlを順次加え,
約1分間激しく振り混ぜる。約2分間放置(14)した後,水相を捨てる。
注(14) 2〜60分間の範囲で呈色は安定である。
8.2.4.3
吸光度の測定 8.2.4.2 b)で得た有機相の一部を光度計の吸収セル (10mm) に取り,水を対照液と
して波長500nm付近の吸光度を測定する。
8.2.5
空試験 空試験は,行わない。
8.2.6
検量線の作成 検量線の作成は,次のいずれかによる。
a) 試料溶液の調製を8.2.4.1 a)によって行う場合 タングステン粉 [8.2.2 f)] を8.2.4.1 a)1)ではかり取っ
た試料の量と同量ずつ数個はかり取り,それぞれビーカー (100〜200ml) に移し入れ,8.2.4.1 a)2)の操
作を行う。常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,溶液を100mlの全
量フラスコに水を用いて移し入れ,標準モリブデン溶液 [8.2.2 n)] 0〜16.0ml(モリブデンとして0〜
160μg)を段階的に加え,水で標線まで薄める。これらの溶液を正しく50mlずつ分取し,それぞれを
分液漏斗 (200ml) に移し入れる。以下,8.2.4.2 b)及び8.2.4.3の手順に従って操作し,得た吸光度と標
準モリブデン溶液として加えたモリブデン量の21の量との関係線を作成し,この関係線を原点を通る
ように平行移動して検量線とする。
b) 試料溶液の調製を8.2.4.1b)によって行う場合 タングステン粉 [8.2.2 f)] 又はタングステンカーバイ
ド粉 [8.2.2 g)](15)を8.2.4.1 b)1)ではかり取った試料の量と同量ずつ数個はかり取り,それぞれ白金皿
(75番又は90番)に移し入れ8.2.4.1 b)2)の操作を行う。常温まで冷却した後,白金ふたの下面を水
で洗浄して白金ふたを取り除き,溶液を100mlの全量フラスコに水を用いて移し入れ,標準モリブデ
ン溶液 [8.2.2 n)] 0〜16.0ml(モリブデンとして0〜160μg)を段階的に加え,水で標線まで薄める。こ
れらの溶液を正しく50mlずつ分取し,それぞれを分液漏斗 (200ml) に移し入れる。以下,8.2.4.2 b)
及び8.2.4.3の手順に従って操作し,得た吸光度と標準モリブデン溶液として加えたモリブデン量の21
の量との関係線を作成し,この関係線を原点を通るように平行移動して検量線とする。
注(15) 試料がタングステン粉の場合にはタングステン粉 [8.2.2 f)] を用い,試料がタングステンカーバ
イド粉の場合にはタングステンカーバイド粉 [8.2.2 g)] を用いる。
c) 試料溶液の調製を8.2.4.1 c)によって行う場合 タングステン粉 [8.2.2 f)] 又はタングステンカーバイ
ド粉 [8.2.2 g)](15)を8.2.4.1 c)1)ではかり取った試料の量と同量ずつ数個はかり取り,それぞれビーカー
(100〜200ml) に移し入れ,8.2.4.1 c)2)の操作を行う。常温まで冷却した後,時計皿の下面を水で洗浄
して時計皿を取り除き,溶液を100mlの全量フラスコに水を用いて移し入れ,標準モリブデン溶液
[8.2.2 n)] 0〜16.0ml(モリブデンとして0〜160μg)を段階的に加え,水で標線まで薄める。これらの
溶液を正しく50mlずつ分取し,それぞれを分液漏斗 (200ml) に移し入れる。以下,8.2.4.2 b)及び8.2.4.3
の手順に従って操作し,得た吸光度と標準モリブデン溶液として加えたモリブデン量の21の量との関
係線を作成し,この関係線を原点を通るように平行移動して検量線とする。
12
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.2.7
計算 8.2.4.3で得た吸光度と8.2.6で作成した検量線とからモリブデン量を求め,試料中のモリブ
デン含有率を,次の式によって算出する。
100
2
1×
×
=
m
A
Mo
ここに, Mo: 試料中のモリブデン含有率 [% (m/m)]
A: 分取した試料溶液中のモリブデン検出量 (g)
m: 試料はかり取り量 (g)
8.3
原子吸光法
8.3.1
要旨 試料を適切な試薬で分解した後,溶液を原子吸光光度計の一酸化二窒素・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
8.3.2
試薬 試薬は,次による。
a) りん酸(1+1)
b) 混酸C(塩酸3,硝酸1,水4)
c) タングステン粉 モリブデン含有率が既知で,かつ,そのモリブデン含有率が試料中のモリブデン含
有率より低いもの。
d) 過酸化水素
e) 標準モリブデン溶液 (50μgMo/ml) 8.2.2 n)の原液 (1 000μgMo/ml) を使用の都度,必要量だけ水で正
しく20倍に薄めて標準モリブデン溶液とする。
8.3.3
試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
8.3.4
操作
8.3.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 過酸化水素による分解 7.3.4.1 a)による(12)。
b) 過酸化水素・混酸Cによる分解 7.3.4.1 b)による(12)。
8.3.4.2
吸光度の測定 8.3.4.1のa)又はb)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光
度計の一酸化二窒素・アセチレンフレーム中に噴霧し,波長313.3nmにおける吸光度を測定する。
8.3.5
空試験 8.3.6の検量線作成操作において得られる標準モリブデン溶液を添加しない溶液の吸光度
を,空試験の吸光度とする。
8.3.6
検量線の作成 検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を8.3.4.1 a)によって行う場合
1) タングステン粉 [8.3.2 c)] を1.0gずつ数個はかり取り,それぞれビーカー (100〜200ml) に移し入
れる。
2) 7.3.4.1 a)2)の操作を試料と並行して行った後,溶液を50mlの全量フラスコに水を用いて移し入れる。
3) 標準モリブデン溶液 [8.3.2 e)] 0〜4.0ml(モリブデンとして0〜200μg)を段階的に加え,水で標線
まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の一酸化二窒素・アセチレン
フレーム中に噴霧し,波長313.3nmにおける吸光度を試料と並行して測定し,得た吸光度とモリブ
デン量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 試料溶液の調製を8.3.4.1 b)によって行う場合
1) タングステン粉 [8.3.2 c)] を1.0gずつ数個はかり取り,それぞれビーカー (100〜200ml) に移し入
13
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
れる。
2) 7.3.4.1 b)2)の操作を試料と並行して行った後,溶液を50mlの全量フラスコに水を用いて移し入れる。
3) 8.3.6 a)の3)〜4)の手順に従って操作する。
8.3.7
計算 8.3.4.2及び8.3.5で得た吸光度と,8.3.6で作成した検量線とからモリブデン量を求め,試料
中のモリブデン含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Mo
ここに, Mo: 試料中のモリブデン含有率 [% (m/m)]
A1: 試料溶液中のモリブデン検出量 (g)
A2: 空試験液中のモリブデン検出量 (g)
A3: タングステン粉 [8.3.2 c)] 1.0g中に含まれるモリブデン量
(g)
m: 試料はかり取り量 (g)
8.4
誘導結合プラズマ (ICP) 発光分光法
8.4.1
要旨 試料を過酸化水素・混酸で分解した後,溶液を誘導結合プラズマ (ICP) 発光分光装置のア
ルゴンプラズマ中に噴霧し,その発光強度を測定する。
8.4.2
試薬 試薬は,次による。
a) りん酸(1+1)
b) 混酸C(塩酸3,硝酸1,水4)
c) タングステン粉 8.3.2 c)による
d) 過酸化水素
e) 標準モリブデン溶液 (50μgMo/ml) 8.3.2 e)による。
8.4.3
試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
8.4.4
操作
8.4.4.1
試料溶液の調製 試料溶液の調製は8.3.4.1 b)による。
8.4.4.2
発光強度の測定 8.4.4.1で得た溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴン
プラズマ中に噴霧し,波長277.540nmにおける発光強度を測定する(13)。
8.4.5
空試験 8.4.6の検量線の作成操作において得られる標準モリブデン溶液を添加しない溶液の発光
強度を,空試験の発光強度とする。
8.4.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) タングステン粉 [8.4.2 c)] を1.0gずつ数個はかり取り,それぞれビーカー (100〜200ml) に移し入れ
る。
b) 7.3.4.1 b)2)に従って操作した後,溶液を50m1の全量フラスコに水を用いて移し入れる。
c) 標準モリブデン溶液 [8.4.2 e)] 0〜4.0ml(モリブデンとして0〜200μg)を段階的に加え水で標線まで
薄める。
d) これらの溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,波長
277.540nmにおける発光強度を試料と並行して測定し,得た発光強度とモリブデン量との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
8.4.7
計算 8.4.4.2及び8.4.5で得た発光強度と,8.4.6で作成した検量線とからモリブデン量を求め,試
料中のモリブデン含有率を次の式によって算出する。
14
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Mo
ここに, Mo: 試料中のモリブデン含有率 [% (m/m)]
A1: 試料溶液中のモリブデン検出量 (g)
A2: 空試験液中のモリブデン検出量 (g)
A3: タングステン粉 [8.4.2 c)] 1.0g中に含まれるモリブデン量
(g)
m: 試料はかり取り量 (g)
9. カルシウム定量方法
9.1
定量方法の区分 カルシウムの定量方法は,次のいずれかによる。
a) 原子吸光法 この方法は,カルシウム含有率0.000 5% (m/m) 以上0.01% (m/m) 以下の試料に適用する。
b) 誘導結合プラズマ (ICP) 発光分光法 この方法は,カルシウム含有率0.000 1% (m/m) 以上0.01%
(m/m) 以下の試料に適用する。
9.2
原子吸光法
9.2.1
要旨 試料を適切な試薬で分解した後,溶液を原子吸光光度計の一酸化二窒素・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
9.2.2
試薬 試薬は,次による。
a) 塩酸(1+1,1+2)
b) りん酸(1+1)
c) 混酸C(塩酸3,硝酸1,水4)
d) タングステン粉 カルシウム含有率が既知で,かつ,そのカルシウム含有率が試料中のカルシウム含
有率より低いもの。
e) 過酸化水素
f)
標準カルシウム溶液 (20μgCa/ml) あらかじめ110℃で乾燥し,デシケーター中で放冷した炭酸カル
シウム[99.5% (m/m) 以上]0.250gをはかり取り,ビーカー (200ml) に移し入れ,時計皿で覆い,塩
酸(1+1)20mlを少量ずつ加え分解した後,加熱して二酸化炭素などを追い出す。常温まで冷却した後,
時計皿の下面を水で洗浄して時計皿を取り除き,溶液を100mlの全量フラスコに水を用いて移し入れ,
水で標線まで薄めて原液 (1 000μgCa/ml) とする。又は,JIS K 0038に規定するカルシウム標準液の
Ca 1 000を原液とする。この原液を使用の都度,必要量だけ水で正しく50倍に薄めて標準カルシウム
溶液とする。
9.2.3
試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
9.2.4
操作(16)
注(16) 使用する器具は,塩酸(1+1)で洗浄した後,水で洗浄して用いる。
9.2.4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 過酸化水素による分解(1)
1) 試料をはかり取って四ふっ化エチレン樹脂ビーカー又は石英ビーカー (100〜200ml) に移し入れる。
2) 数mlの水で試料を湿らせ四ふっ化エチレン樹脂時計皿又はポリエチレン時計皿で覆い,りん酸(1
+1)1ml及び過酸化水素5〜10mlを加え,放置又は加熱して分解する。液量が約5mlになるまで加
熱して蒸発し常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除く。
3) 溶液を50mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
15
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 過酸化水素・混酸Cによる分解
1) 試料をはかり取って,四ふっ化エチレン樹脂ビーカー又は石英ビーカー (100〜200ml) に移し入れ
る。
2) 数mlの水で試料を湿らせ四ふっ化エチレン樹脂時計皿又はポリエチレン時計皿で覆い,りん酸(1
+1)1ml及び過酸化水素5〜10mlを加え,放置又は加熱して分解する。液量が約5mlになるまで加
熱して蒸発し室温まで冷却した後,混酸C10mlを少量ずつ加え,数分間放置し激しい発泡を終わら
せ,約5分間煮沸して常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除く(17)。
注(17) 遊離炭素が認められたときは,ろ紙(5種A)を用いてろ過する。使用するろ紙は漏斗に装着し
てから温塩酸(1+2)で数回洗浄し,さらに,温水で洗浄してから用いる。
3) 溶液を50mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
9.2.4.2
吸光度の測定 9.2.4.1のa)3)又はb)3)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸
光光度計の一酸化二窒素・アセチレンフレーム中に噴霧し,波長422.7nmにおける吸光度を測定する。
9.2.5
空試験 9.2.6の検量線の作成操作において得られる標準カルシウム溶液を添加しない溶液の吸光
度を,空試験の吸光度とする。
9.2.6
検量線の作成 検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を9.2.4.1 a)によって行う場合
1) タングステン粉 [9.2.2 d)] を1.0gずつ数個はかり取り,それぞれ四ふっ化エチレン樹脂ビーカー又
は石英ビーカー (100〜200ml) に移し入れる。
2) 9.2.4.1 a)2)の操作を試料と並行して行った後,溶液を50mlの全量フラスコに水を用いて移し入れる。
3) 標準カルシウム溶液 [9.2.2 f)] 0〜5.0ml(カルシウムとして0〜100μg)を段階的に加え,水で標線ま
で薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の一酸化二窒素・アセチレン
フレーム中に噴霧し,波長422.7nmにおける吸光度を試料と並行して測定し,得た吸光度とカルシ
ウム量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 試料溶液の調製を9.2.4.1 b)によって行う場合
1) タングステン粉 [9.2.2 d)] を1.0gずつ数個はかり取り,それぞれ四ふっ化エチレン樹脂ビーカー又
は石英ビーカー (100〜200ml) に移し入れる。
2) 9.2.4.1 b)2)の操作を試料と並行して行った後,溶液を50mlの全量フラスコに水を用いて移し入れる。
3) 9.2.6 a)の3)〜4)の手順に従って操作する。
9.2.7
計算 9.2.4.2及び9.2.5で得た吸光度と,9.2.6で作成した検量線とからカルシウム量を求め,試料
中のカルシウム含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Ca
ここに, Ca: 試料中のカルシウム含有率 [% (m/m)]
A1: 試料溶液中のカルシウム検出量 (g)
A2: 空試験液中のカルシウム検出量 (g)
A3: タングステン粉 [9.2.2 d)] 1.0g中に含まれるカルシウム量
(g)
m: 試料はかり取り量 (g)
9.3
誘導結合プラズマ (ICP) 発光分光法
16
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.3.1
要旨 試料を過酸化水素・混酸で分解した後,溶液を誘導結合プラズマ (ICP) 発光分光装置のア
ルゴンプラズマ中に噴霧し,その発光強度を測定する。
9.3.2
試薬 試薬は,次による。
a) 塩酸(1+1)
b) りん酸(1+1)
c) 混酸C(塩酸3,硝酸1,水4)
d) タングステン粉 9.2.2 d)による
e) 過酸化水素
f)
標準カルシウム溶液 (20μgCa/ml) 9.2.2 f)による。
9.3.3
試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
9.3.4
操作(16)
9.3.4.1
試料溶液の調製 試料溶液の調製は,9.2.4.1 b)による。
9.3.4.2
発光強度の測定 9.3.4.1で得た溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴン
プラズマ中に噴霧し,波長393.366nmにおける発光強度を測定する(13)。
9.3.5
空試験 9.3.6の検量線の作成操作において得られる標準カルシウム溶液を添加しない溶液の発光
強度を,空試験の発光強度とする。
9.3.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) タングステン粉 [9.3.2 d)] を1.0gずつ数個はかり取り,それぞれ四ふっ化エチレン樹脂ビーカー又は
石英ビーカー (100〜200ml) に移し入れる。
b) 9.2.4.1 b)2)に従って操作した後,溶液を50mlの全量フラスコに水を用いて移し入れる。
c) 標準カルシウム溶液 [9.3.2 f)] 0〜5.0ml(カルシウムとして0〜100μg)を段階的に加え,水で標線まで
薄める。
d) これらの溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,波長
393.366nmにおける発光強度を試料と並行して測定し,得た発光強度とカルシウム量との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
9.3.7
計算 9.3.4.2及び9.3.5で得た発光強度と,9.3.6で作成した検量線とからカルシウム量を求め,試
料中のカルシウム含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Ca
ここに, Ca: 試料中のカルシウム含有率 [% (m/m)]
A1: 試料溶液中のカルシウム検出量 (g)
A2: 空試験液中のカルシウム検出量 (g)
A3: タングステン粉 [9.3.2 d)] 1.0g中に含まれるカルシウム量
(g)
m: 試料はかり取り量 (g)
10. けい素定量方法
10.1 定量方法の区分 けい素の定量方法は,次のいずれかによる。
a) 四ふっ化けい素気化分離モリブドけい酸青吸光光度法 この方法は,けい素含有率0.000 5% (m/m) 以
上0.005% (m/m) 以下の試料に適用する。
b) 四ふっ化けい素気化分離誘導結合プラズマ (ICP) 発光分光法 この方法は,けい素含有率0.000 5%
(m/m) 以上0.005% (m/m) 以下の試料に適用する。
17
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 混酸分解誘導結合プラズマ (ICP) 発光分光法 この方法は,けい素含有率0.001% (m/m) 以上0.02%
(m/m) 以下の試料に適用する。
10.2 四ふっ化けい素気化分離モリブドけい酸青吸光光度法
10.2.1 要旨 試料を過酸化水素で分解し,硫酸を加え酸性とした溶液に,ふっ化水素酸を加えた後,窒素
(又は酸素)を通気してけい素を四ふっ化けい素として気化分離し,ほう酸に吸収させる。七モリブデン
酸六アンモニウムを加え,けい素をモリブドけい酸とした後,しゅう酸及びL (+) -アスコルビン酸を加え
てモリブドけい酸青を生成させ,光度計を用いてその吸光度を測定する。
10.2.2 試薬 試薬は,次による。
a) 塩酸(1+1)
b) ふっ化水素酸(1+19)
c) 硫酸(3+1)
d) 吸収溶液 ほう酸5gを水に溶解し,水で液量を1 000mlとする。
e) 窒素(又は酸素) 99.9% (v/v) 以上のもの。
f)
過酸化水素
g) 七モリブデン酸六アンモニウム溶液 七モリブデン酸六アンモニウム四水和物10gを水に溶解し,水
で液量を100mlとする。
h) しゅう酸溶液 しゅう酸二水和物10gを水に溶解し,水で液量を100mlとする。
i)
L (+) -アスコルビン酸溶液 (30g/L) 使用の都度調製する。
j)
標準けい素溶液 (10μgSi/ml) あらかじめ1 000℃で強熱し,デシケーター中で室温まで放冷した二酸
化けい素[99.9% (m/m) 以上]0.214gをはかり取って白金るつぼ(30番)に移し入れ,炭酸ナトリウ
ム2.5gを加えて混合し,加熱して融解する。放冷した後,温水約50mlを入れたポリエチレンビーカ
ー (200ml) 中に浸し,水浴上で温めて融成物を溶解した後,白金るつぼを水洗して取り出す。常温ま
で冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原液 (1
000μgSi/ml) とし,ポリエチレン瓶中に保存する。この原液を使用の都度,必要量だけ水で正しく100
倍に薄めて標準けい素溶液とする。
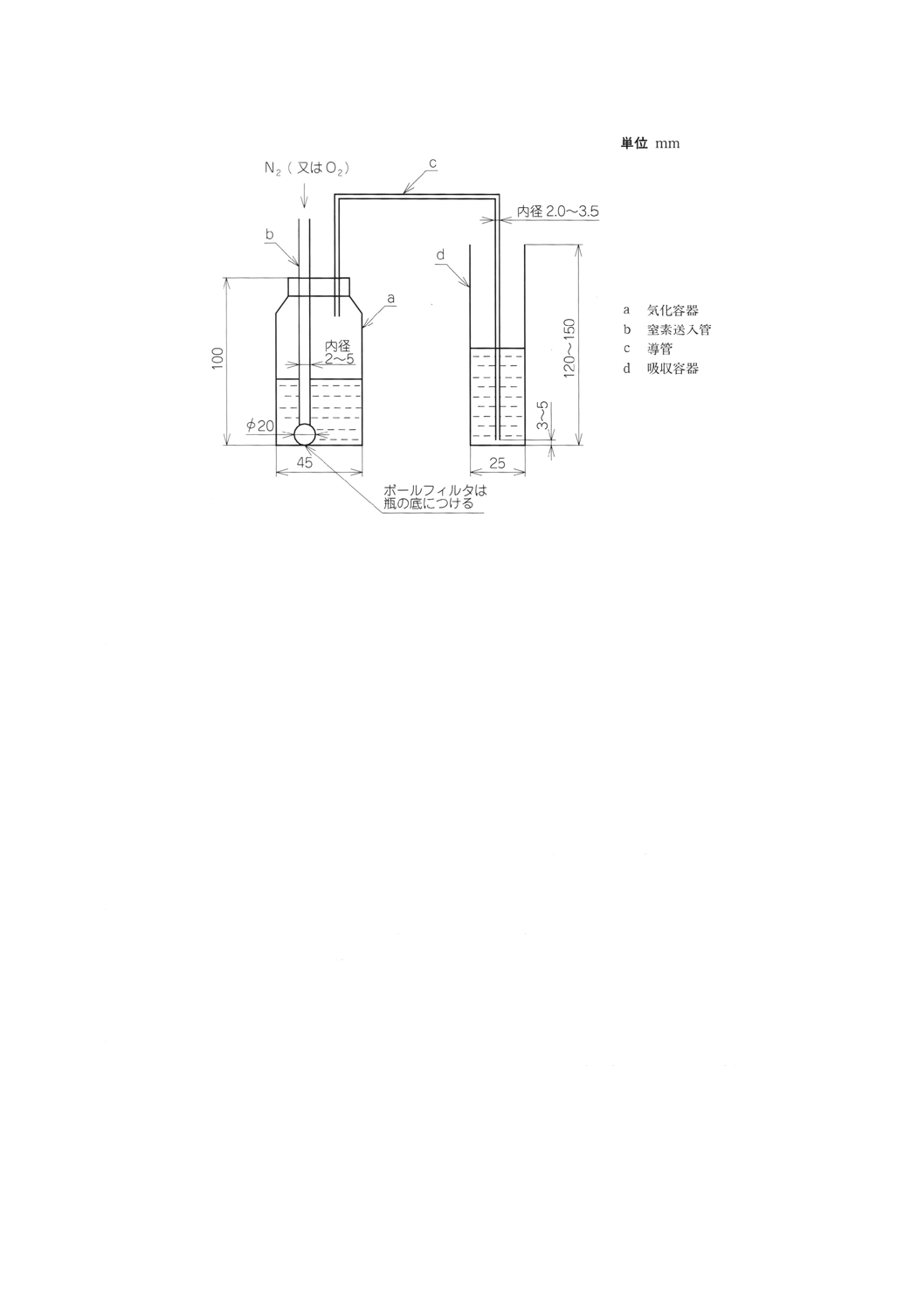
10.2.3 装置 四ふっ化けい素気化装置は,気化容器,窒素送入管,導管及び吸収容器からなる。装置は,
通常図2に示すものを用いる。
a) 気化容器 容量150mlの四ふっ化エチレン樹脂ふた付き瓶。
b) 窒素送入管 直径約20mmのボールフィルタの付いた内径2〜5mmの四ふっ化エチレン樹脂管又はポ
リエチレン管。
c) 導管 内径2〜3.5mmの四ふっ化エチレン樹脂管又はポリエチレン管。
d) 吸収容器 内径約25mm,高さ120〜150mmで25ml標線入りの四ふっ化エチレン樹脂容器又はポリエ
チレン容器。
18
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図2 四ふっ化けい素気化装置の例
10.2.4 試料はかり取り量 試料はかり取り量は,1.0gとし,10mgのけたまではかる。
10.2.5 操作
10.2.5.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取って,気化容器 (a) に移し入れる。
b) 数mlの水で試料を湿らせ,過酸化水素5〜10mlを加え,放置又は加熱して分解する。液量が約10ml
になるまで加熱して蒸発した後,室温まで冷却する。
c) 水で冷却しながら硫酸(3+1)40mlを加え(18),再び室温まで冷却する。
注(18) 容器の壁に付着した水滴に,ふっ化けい素が吸収されるのを防ぐため容器を回転させながら内
壁に沿って少量ずつ添加する。
10.2.5.2 四ふっ化けい素の気化分離及び吸収 四ふっ化けい素の気化分離及び吸収は,次の手順によって
行う。
a) 10.2.5.1 c)で得た気化容器を図2のように組み立て,吸収容器 (d) に吸収溶液 [10.2.2d)] 25mlを入れ
る。
b) 試料溶液に,窒素送入管 (b) からふっ化水素酸(1+19)1mlを加えた後,窒素(又は酸素)を通気し,
徐々に流量を上げ,毎分約1 000mlの流量で約30分間通気する。
c) 吸収容器 (d) を外し,水で標線まで薄める。
10.2.5.3 呈色 呈色は,次の手順によって行う。
a) 10.2.5.2 c)で得た溶液から正しく10mlを25mlポリエチレン全量フラスコに分取する。
b) 塩酸(1+1)0.5ml及び七モリブデン酸六アンモニウム溶液 [10.2.2 g)] 1.5mlを加え振り混ぜた後,10分
間放置する。次に,しゅう酸溶液 [10.2.2 h)] 2mlを加え振り) 混ぜ,直ちに(30秒以内)L (+) -アス
コルビン酸溶液 [10.2.2 i)] 2mlを加え振り混ぜ,水で標線まで薄める。
10.2.5.4 吸光度の測定 10.2.5.3 b)で得た溶液の一部を光度計の吸収セル (10mm) に取り,水を対照液と
して波長810nm付近の吸光度を測定する。
19
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.2.6 空試験 試料を用いないで試料と同じ操作を試料と並行して行い,得られた溶液の吸光度を空試験
の吸光度とする。
10.2.7 検量線の作成 数個の気化容器 (a) に,標準けい素溶液 [10.2.2 j)] 0〜5.0ml(けい素として0〜
50μg)を段階的に加え,水で液量を10mlとした後,水で冷却しながら硫酸(3+1)40mlを加え (18) ,室温
まで冷却する。気化容器を図2のように組み立て,吸収容器 (d) に吸収溶液 [10.2.2 d)] 25mlを入れる以
下,10.2.5.2 b)〜10.2.5.4の手順に従って操作し,得た吸光度とけい素量との関係線を作成し,その関係線
を原点を通るように平行移動して検量線とする。
10.2.8 計算 10.2.5.4及び10.2.6で得た吸光度と,10.2.7で作成した検量線とからけい素量を求め,試料
中のけい素含有率を次の式によって算出する。
100
25
10
2
1
×
×
−
=
m
A
A
Si
ここに, Si: 試料中のけい素含有率 [% (m/m)]
A1: 分取した試料溶液中のけい素検出量 (g)
A2: 空試験液中のけい素検出量 (g)
m: 試料はかり取り量 (g)
10.3 四ふっ化けい素気化分離誘導結合プラズマ (ICP) 発光分光法
10.3.1 要旨 試料を過酸化水素で分解し,硫酸を加え酸性とした溶液に,ふっ化水素酸を加えた後,窒素
(又は酸素)を通気してけい素を四ふっ化けい素として気化分離し,ほう酸に吸収させ,溶液を誘導結合
プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定する。
10.3.2 試薬 試薬は,次による。
a) ふっ化水素酸(1+19)
b) 硫酸(3+1)
c) 吸収溶液 ほう酸5gを水に溶解し,水で液量を1 000mlとする。
d) 窒素(又は酸素) 99.9% (v/v) 以上のもの。
e) 過酸化水素
f)
標準けい素溶液 (10μgSi/ml) 10.2.2 j)による。
10.3.3 装置 装置は,10.2.3による。
10.3.4 試料はかり取り量 試料はかり取り量は,1.0gとし,10mgのけたまではかる。
10.3.5 操作
10.3.5.1 試料溶液の調製 試料溶液の調製は,10.2.5.1による
10.3.5.2 四ふっ化けい素の気化分離及び吸収 四ふっ化けい素の気化分離及び吸収は,10.2.5.2による。
10.3.5.3 発光強度の測定 10.2.5.2 c)で得た溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアル
ゴンプラズマ中に噴霧し,波長251.611nmにおける発光強度を測定する(13)。
10.3.6 空試験 試料を用いないで試料と同じ操作を試料と並行して行い,得られた溶液の発光強度を空試
験の発光強度とする。
10.3.7 検量線の作成 検量線の作成は,次の手順によって行う。
a) 数個の気化容器 (a) に,標準けい素溶液 [10.3.2f)] 0〜5.0m1(けい素として0〜50μg)を段階的に加
え,水で液量を10mlとした後,水で冷却しながら硫酸(3+1)40mlを加え(18),室温まで冷却する。
b) 気化容器を図2のように組み立て,吸収容器 (d) に吸収溶液 [10.3.2 c)] 25mlを入れる以下,10.2.5.2 b)
20
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
〜c)の手順に従って操作する。
c) これらの溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,波長
251.611nmにおける発光強度を試料と並行して測定し,得た発光強度とけい素量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
10.3.8 計算 10.3.5.3及び10.3.6で得た発光強度と,10.3.7で作成した検量線とからけい素量を求め,試
料中のけい素含有率を次の式によって算出する。
100
2
1
×
−
=
m
A
A
Si
ここに, Si: 試料中のけい素含有率 [% (m/m)]
A1: 試料溶液中のけい素検出量 (g)
A2: 空試験液中のけい素検出量 (g)
m: 試料はかり取り量 (g)
10.4 混酸分解誘導結合プラズマ (ICP) 発光分光法
10.4.1 要旨 試料を過酸化水素・混酸で分解した後,溶液を誘導結合プラズマ (ICP) 発光分光装置のア
ルゴンプラズマ中に噴霧し,その発光強度を測定する。
10.4.2 試薬 試薬は,次による。
a) りん酸(1+1)
b) 混酸C(塩酸3,硝酸1,水4)
c) タングステン粉 けい素含有率が既知で,かつ,そのけい素含有率が試料中のけい素含有率より低い
もの。
d) 過酸化水素
e) 標準けい素溶液 (50μgSi/ml) 10.2.2 j)の原液 (1 000μgSi/ml) を使用の都度,必要量だけ水で正しく
20倍に薄めて標準けい素溶液とする。
10.4.3 試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
10.4.4 操作
10.4.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取って,四ふっ化エチレン樹脂ビーカー (100〜200ml) に移し入れる。
b) 数mlの水で試料を湿らせ四ふっ化エチレン樹脂時計皿で覆い,りん酸(1+1)1m1及び過酸化水素5〜
10mlを加え,放置又は加熱して分解する。液量が約5mlになるまで加熱して蒸発し室温まで冷却した
後,混酸C10mlを少量ずつ加え,数分間放置し激しい発泡を終わらせ,約5分間煮沸して常温まで冷
却した後,時計皿の下面を水で洗浄して時計皿を取り除く(3)。
c) 溶液を50mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
10.4.4.2 発光強度の測定 10.4.4.1 c)で得た溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアル
ゴンプラズマ中に噴霧し,波長250.690nmにおける発光強度を測定する(13)。
10.4.5 空試験 10.4.6の検量線の作成操作において得られる標準けい素溶液を添加しない溶液の発光強
度を,空試験の発光強度とする。
10.4.6 検量線の作成 検量線の作成は,次の手順によって行う。
a) タングステン粉 [10.4.2 c)] を1.0gずつ数個はかり取り,それぞれ四ふっ化エチレン樹脂ビーカー
(100〜200ml) に移し入れる。
b) 10.4.4.1 b)に従って操作した後,溶液を50mlの全量フラスコに水を用いて移し入れる。
21
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 標準けい素溶液 [10.4.2 e)] 0〜4.0ml(けい素として0〜200μg)を段階的に加え,水で標線まで薄める。
d) これらの溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,波長
250.690nmにおける発光強度を試料と並行して測定し,得た発光強度とけい素量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
10.4.7 計算 10.4.4.2及び10.4.5で得た発光強度と,10.4.6で作成した検量線とからけい素量を求め,試
料中のけい素含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Si
ここに, Si: 試料中のけい素含有率 [% (m/m)]
A1: 試料溶液中のけい素検出量 (g)
A2: 空試験液中のけい素検出量 (g)
A3: タングステン粉 [10.4.2 c)] 1.0g中に含まれるけい素量 (g)
m: 試料はかり取り量 (g)
11. アルミニウム定量方法
11.1 定量方法の区分 アルミニウムの定量方法は,次のいずれかによる。
a) 陽イオン交換分離原子吸光法 この方法は,アルミニウム含有率0.000 2% (m/m) 以上0.01% (m/m) 以
下の試料に適用する。
b) 陽イオン交換分離誘導結合プラズマ (ICP) 発光分光法 この方法は,アルミニウム含有率0.000 1%
(m/m) 以上0.01% (m/m) 以下の試料に適用する。
c) 混酸分解誘導結合プラズマ (ICP) 発光分光法 この方法は,アルミニウム含有率0.000 5% (m/m) 以
上0.01% (m/m) 以下の試料に適用する。
11.2 陽イオン交換分離原子吸光法
11.2.1 要旨 試料を過酸化水素で分解し,硝酸を加えた後,陽イオン交換カラムに通して,アルミニウム
を吸着させ,タングステンを流出させる。希塩酸で洗浄して残存するタングステンを除いた後,塩酸でア
ルミニウムを溶離する。溶出液に硝酸及び過塩素酸を加え乾固し,塩酸を加えて溶解した後,溶液を原子
吸光光度計の一酸化二窒素・アセチレンフレーム中に噴霧し,その吸光度を測定する。
11.2.2 試薬 試薬は,次による。
a) 塩酸(1+1,1+3,1+50)
b) 硝酸(1+1)
c) 過塩素酸(1+1)
d) 過酸化水素
e) 標準アルミニウム溶液 (20μgAl/ml) アルミニウム[99.9% (m/m) 以上]1.000gをはかり取ってビー
カー (200ml) に移し入れ,時計皿で覆い,塩酸(1+1)10m1及び硝酸(1+1)1mlを加え,穏やかに加熱
して分解する。常温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除き,溶液を1 000ml
の全量フラスコに水を用いて移し入れ,水で標線まで薄めて原液 (1 000μgAl/ml) とする。この溶液を
使用の都度,必要量だけ水で正しく50倍に薄めて標準アルミニウム溶液とする。
11.2.3 器具 器具は,通常次による。
陽イオン交換カラム 一端を細くしたポリエチレン管(長さ250mm,内径10mm)に水でほぐしたポリ
エチレンウールを約5mmの厚さに緩く詰め,水で膨潤させた強酸性陽イオン交換樹脂(74〜149mm,交
換容量1.9meq/ml以上のもの)約10mlをスラリー状にして流し入れ,沈降させた後,その上に水でほぐし
22
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
たポリエチレンウールを約5mmの厚さに詰める。この陽イオン交換カラムは,ポリエチレンウールの詰
め方を調節するなどして流出液の流量を毎分1.0〜1.5mlになるようにした後,塩酸(1+3)100ml,水100ml
を順次通しておく。
11.2.4 試料はかり取り量 試料はかり取り量は,3.0gとし,10mgのけたまではかる。
11.2.5 操作
11.2.5.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取って,四ふっ化エチレン樹脂ビーカー又は石英ビーカー (100〜200ml) に移し入れる。
b) 数mlの水で試料を湿らせ,四ふっ化エチレン樹脂時計皿又は石英時計皿で覆い,過酸化水素20〜30ml
を加え,放置又は加熱して分解する。時計皿の下面を水で洗浄して時計皿を取り除き,硝酸(1+1)1ml
を加える(17)。
c) 水で薄めて液量を約100mlとし,陽イオン交換カラムに通す。次に塩酸(1+50)20mlを用いてビーカ
ーを洗浄して陽イオン交換カラムに通し,更に塩酸(1+50)100mlを通し,流出液は捨てる。
d) 塩酸(1+3)100mlを陽イオン交換カラムに通し,溶出液は四ふっ化エチレン樹脂ビーカー (100〜
200ml) に受け,硝酸(1+1)2ml及び過塩素酸(1+1)1mlを加え,加熱して蒸発乾固する。
e) 塩酸(1+1)5mlを加えて塩類を溶解し,25mlの全量フラスコに水を用いて移し入れ,水で標線まで薄
める。
11.2.5.2 吸光度の測定 11.2.5.1 e)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の一
酸化二窒素・アセチレンフレーム中に噴霧し,波長309.3nmにおける吸光度を測定する。
11.2.6 空試験 試料を用いないで試料と同じ操作を試料と並行して行い,得られた溶液の吸光度を空試験
の吸光度とする。
11.2.7 検量線の作成 検量線の作成は,次の手順によって行う。
a) 数個の25mlの全量フラスコに,塩酸(1+1)5mlを加え,標準アルミニウム溶液 [11.2.2 e)] 0〜15.0ml
(アルミニウムとして0〜300μg)を段階的に加え,水で標線まで薄める。
b) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の一酸化二窒素・アセチレンフ
レーム中に噴霧し,波長309.3nmにおける吸光度を試料と並行して測定し,得た吸光度とアルミニウ
ム量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
11.2.8 計算 11.2.5.2及び11.2.6で得た吸光度と,11.2.7で作成した検量線とからアルミニウム量を求め,
試料中のアルミニウム含有率を次の式によって算出する。
100
2
1
×
−
=
m
A
A
Al
ここに, Al: 試料中のアルミニウム含有率 [% (m/m)]
A1: 試料溶液中のアルミニウム検出量 (g)
A2: 空試験液中のアルミニウム検出量 (g)
m: 試料はかり取り量 (g)
11.3 陽イオン交換分離誘導結合プラズマ (ICP) 発光分光法
11.3.1 要旨 試料を過酸化水素で分解し,硝酸を加えた後,陽イオン交換カラムに通してアルミニウムを
吸着させ,タングステンを流出させる。希塩酸で洗浄して残存するタングステンを除いた後,塩酸でアル
ミニウムを溶離する。溶出液に硝酸及び過塩素酸を加え乾固し,塩酸を加えて溶解した後,溶液を誘導結
合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,その発光強度を測定する。
11.3.2 試薬 試薬は,次による。
23
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 塩酸(1+1,1+3,1+50)
b) 硝酸(1+1)
c) 過塩素酸(1+1)
d) 過酸化水素
e) 標準アルミニウム溶液 (20μgAl/ml) 11.2.2 e)による。
11.3.3 器具 器具は,11.2.3による。
11.3.4 試料はかり取り量 試料はかり取り量は,3.0gとし,10mgのけたまではかる。
11.3.5 操作
11.3.5.1 試料溶液の調製 試料溶液の調製は,11.2.5.1による。
11.3.5.2 発光強度の測定 11.3.5.1で得た溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴ
ンプラズマ中に噴霧し,波長396.152nmにおける発光強度を測定する(13)。
11.3.6 空試験 試料を用いないで試料と同じ操作を試料と並行して行い,得られた溶液の発光強度を空試
験の発光強度とする。
11.3.7 検量線の作成 検量線の作成は,次の手順によって行う。
a) 数個の25mlの全量フラスコに,塩酸(1+1)5mlを加え標準アルミニウム溶液 [11.3.2 e)] 0〜15.0ml(ア
ルミニウムとして0〜300μg)を段階的に加え,水で標線まで薄める。
b) これらの溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,波長
396.152nmにおける発光強度を試料と並行して測定し,得た発光強度とアルミニウム量との関係線を
作成し,その関係線を原点を通るように平行移動して検量線とする。
11.3.8 計算 11.3.5.2及び11.3.6で得た発光強度と,11.3.7で作成した検量線とからアルミニウム量を求め,
試料中のアルミニウム含有率を次の式によって算出する。
100
2
1
×
−
=
m
A
A
Al
ここに, Al: 試料中のアルミニウム含有率 [% (m/m)]
A1: 試料溶液中のアルミニウム検出量 (g)
A2: 空試験液中のアルミニウム検出量 (g)
m: 試料はかり取り量 (g)
11.4 混酸分解誘導結合プラズマ (ICP) 発光分光法
11.4.1 要旨 試料を過酸化水素・混酸で分解した後,溶液を誘導結合プラズマ (ICP) 発光分光装置のア
ルゴンプラズマ中に噴霧し,その発光強度を測定する。
11.4.2 試薬 試薬は,次による。
a) りん酸(1+1)
b) 混酸C(塩酸3,硝酸1,水4)
c) タングステン粉 アルミニウム含有率が既知で,かつ,そのアルミニウム含有率が試料中のアルミニ
ウム含有率より低いもの。
d) 過酸化水素
e) 標準アルミニウム溶液 (20μgAl/ml) 11.2.2 e)による。
11.4.3 試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
11.4.4 操作
11.4.4.1 試料溶液の調製 試料溶液の調製は,9.2.4.1 b)による。
24
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.4.4.2 発光強度の測定 11.4.4.1で得た溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴ
ンプラズマ中に噴霧し,波長394.401nmにおける発光強度を測定する(13)。
11.4.5 空試験 11.4.6の検量線の作成操作において得られる標準アルミニウム溶液を添加しない溶液の発
光強度を,空試験の発光強度とする。
11.4.6 検量線の作成 検量線の作成は,次の手順によって行う。
a) タングステン粉 [11.4.2 c)] を1.0gずつ数個はかり取り,それぞれ四ふっ化エチレン樹脂ビーカー又は
石英ビーカー (100〜200ml) に移し入れる。
b) 9.2.4.1 b)2)に従って操作した後,溶液を50mlの全量フラスコに水を用いて移し入れる。
c) 標準アルミニウム溶液 [11.4.2 e)] 0〜5.0ml(アルミニウムとして0〜100μg)を段階的に加え,水で標
線まで薄める。
d) これらの溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,波長
394.401nmにおける発光強度を試料と並行して測定し,得た発光強度とアルミニウム量との関係線を
作成し,その関係線を原点を通るように平行移動して検量線とする。
11.4.7 計算 11.4.4.2及び11.4.5で得た発光強度と,11.4.6で作成した検量線とからアルミニウム量を求め,
試料中のアルミニウム含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Al
ここに, Al: 試料中のアルミニウム含有率 [% (m/m)]
A1: 試料溶液中のアルミニウム検出量 (g)
A2: 空試験液中のアルミニウム検出量 (g)
A3: タングステン粉 [11.4.2 c)] 1.0g中に含まれるアルミニウム量
(g)
m: 試料はかり取り量 (g)
12. マグネシウム定量方法
12.1 定量方法の区分 マグネシウムの定量方法は,次のいずれかによる。
a) 原子吸光法 この方法は,マグネシウム含有率0.000 5% (m/m) 以上0.005% (m/m) 以下の試料に適用
する。
b) 誘導結合プラズマ (ICP) 発光分光法 この方法は,マグネシウム含有率0.000 1% (m/m) 以上0.00 5%
(m/m) 以下の試料に適用する。
12.2 原子吸光法
12.2.1 要旨 試料を適切な試薬で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴
霧し,その吸光度を測定する。
12.2.2 試薬 試薬は,次による。
a) 塩酸(1+1,1+2)
b) りん酸(1+1)
c) 混酸C(塩酸3,硝酸1,水4)
d) タングステン粉 マグネシウム含有率が既知で,かつ,そのマグネシウム含有率が試料中のマグネシ
ウム含有率より低いもの。
e) 過酸化水素
f)
標準マグネシウム溶液 (10μgMg/ml) 金属マグネシウム[99.5% (m/m) 以上]1.000gをはかり取って
25
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ビーカー (200ml) に移し入れ,時計皿で覆い,硝酸(1+1)50mlを少量ずつ加え,穏やかに加熱して分
解した後,煮沸して窒素酸化物などを追い出す。常温まで冷却した後,時計皿の下面を水で洗浄して
時計皿を取り除き,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原液 (1
000μgMg/ml) とする。又は,JIS K 0037に規定するマグネシウム標準液のMg 1000を原液とする。こ
の原液を使用の都度,必要量だけ水で正しく100倍に薄めて標準マグネシウム溶液とする。
12.2.3 試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
12.2.4 操作(16)
12.2.4.1 試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 過酸化水素による分解 9.2.4.1 a)による。
b) 過酸化水素・混酸Cによる分解 9.2.4.1 b)による。
12.2.4.2 吸光度の測定 12.2.4.1のa)又はb)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光
光度計の空気・アセチレンフレーム中に噴霧し,波長285.2nmにおける吸光度を測定する。
12.2.5 空試験 12.2.6の検量線の作成操作において得られる標準マグネシウム溶液を添加しない溶液の
吸光度を,空試験の吸光度とする。
12.2.6 検量線の作成 検量線の作成は,次のいずれかの手順によって行う。
a) 試料溶液の調製を12.2.4.1 a)によって行う場合
1) タングステン粉 [12.2.2 d)] を1.0gずつ数個はかり取り,それぞれ四ふっ化エチレン樹脂ビーカー
又は石英ビーカー (100〜200ml) に移し入れる。
2) 9.2.4.1 a)2)の操作を試料と並行して行った後,溶液を50mlの全量フラスコに水を用いて移し入れる。
3) 標準マグネシウム溶液 [12.2.2 f)] 0〜5.0ml(マグネシウムとして0〜50μg)を段階的に加え,水で標
線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長285.2nmにおける吸光度を試料と並行して測定し,得た吸光度とマグネシウム量
との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 試料溶液の調製を12.2.4.1 b)によって行う場合
1) タングステン粉 [12.2.2 d)] を1.0gずつ数個はかり取り,それぞれ四ふっ化エチレン樹脂ビーカー
又は石英ビーカー (100〜200m1) に移し入れる。
2) 9.2.4.1 b)2)の操作を試料と並行して行った後,溶液を50mlの全量フラスコに水を用いて移し入れる。
3) 12.2.6 a)の3)〜4)の手順に従って操作する。
12.2.7 計算 12.2.4.2及び12.2.5で得た吸光度と,12.2.6で作成した検量線とからマグネシウム量を求め,
試料中のマグネシウム含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Mg
ここに, Mg: 試料中のマグネシウム含有率 [% (m/m)]
A1: 試料溶液中のマグネシウム検出量 (g)
A2: 空試験液中のマグネシウム検出量 (g)
A3: タングステン粉 [12.2.2 d)] 1.0g中に含まれるマグネシウム
量 (g)
m: 試料はかり取り量 (g)
12.3 誘導結合プラズマ (ICP) 発光分光法
26
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.3.1 要旨 試料を過酸化水素・混酸で分解した後,溶液を誘導結合プラズマ (ICP) 発光分光装置のア
ルゴンプラズマ中に噴霧し,その発光強度を測定する。
12.3.2 試薬 試薬は,次による。
a) 塩酸(1+1,1+2)
b) りん酸(1+1)
c) 混酸C(塩酸3,硝酸1,水4)
d) タングステン粉 12.2.2 d)による。
e) 過酸化水素
f)
標準マグネシウム溶液 (10μgMg/ml) 12.2.2 f)による。
12.3.3 試料はかり取り量 試料はかり取り量は,1.0g(7)とし,10mgのけたまではかる。
12.3.4 操作(16)
12.3.4.1 試料溶液の調製 試料溶液の調製は,12.2.4.1 b)による。
12.3.4.2 発光強度の測定 12.3.4.1で得た溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴ
ンプラズマ中に噴霧し,波長279.553nmにおける発光強度を測定する(13)。
12.3.5 空試験 12.3.6の検量線の作成操作において得られる標準マグネシウム溶液を添加しない溶液の
発光強度を,空試験の発光強度とする。
12.3.6 検量線の作成 検量線の作成は,次の手順によって行う。
a) タングステン粉 [12.3.2 d)] を1.0gずつ数個はかり取り,それぞれ四ふっ化エチレン樹脂ビーカー又
は石英ビーカー (100〜200m1) に移し入れる。
b) 9.2.4.1 b)2)に従って操作した後,溶液を50mlの全量フラスコに水を用いて移し入れる。
c) 標準マグネシウム溶液 [12.3.2 f)] 0〜5.0ml(マグネシウムとして0〜50μg)を段階的に加え,水で標線
まで薄める。
d) これらの溶液の一部を,誘導結合プラズマ (ICP) 発光分光装置のアルゴンプラズマ中に噴霧し,波長
279.553nmにおける発光強度を試料と並行して測定し,得た発光強度とマグネシウム量との関係線を
作成し,その関係線を原点を通るように平行移動して検量線とする。
12.3.7 計算 12.3.4.2及び12.3.5で得た発光強度と,12.3.6で作成した検量線とからマグネシウム量を求
め,試料中のマグネシウム含有率を次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Mg
ここに, Mg: 試料中のマグネシウム含有率 [% (m/m)]
A1: 試料溶液中のマグネシウム検出量 (g)
A2: 空試験液中のマグネシウム検出量 (g)
A3: タングステン粉 [12.3.2 d)] 1.0g中に含まれるマグネシウム
量 (g)
m: 試料はかり取り量 (g)
13. 酸素定量方法
13.1 定量方法の区分 酸素の定量方法は,次のいずれかによる。
a) 水素還元重量法 この方法は,酸素含有率0.01% (m/m) 以上のタングステン粉の試料に適用する。
b) 酸化重量法 この方法は,酸素含有率0.01% (m/m) 以上のタングステン粉の試料に適用する。
c) 不活性ガス融解−赤外線吸収法 この方法は,酸素含有率0.01% (m/m) 以上1.2% (m/m) 以下の試料
27
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に適用する。
13.2 水素還元重量法
13.2.1 要旨 試料を水素気流中で加熱して還元し,その減量をはかる。
13.2.2 試薬 試薬は,次による。
a) 水素
b) 窒素
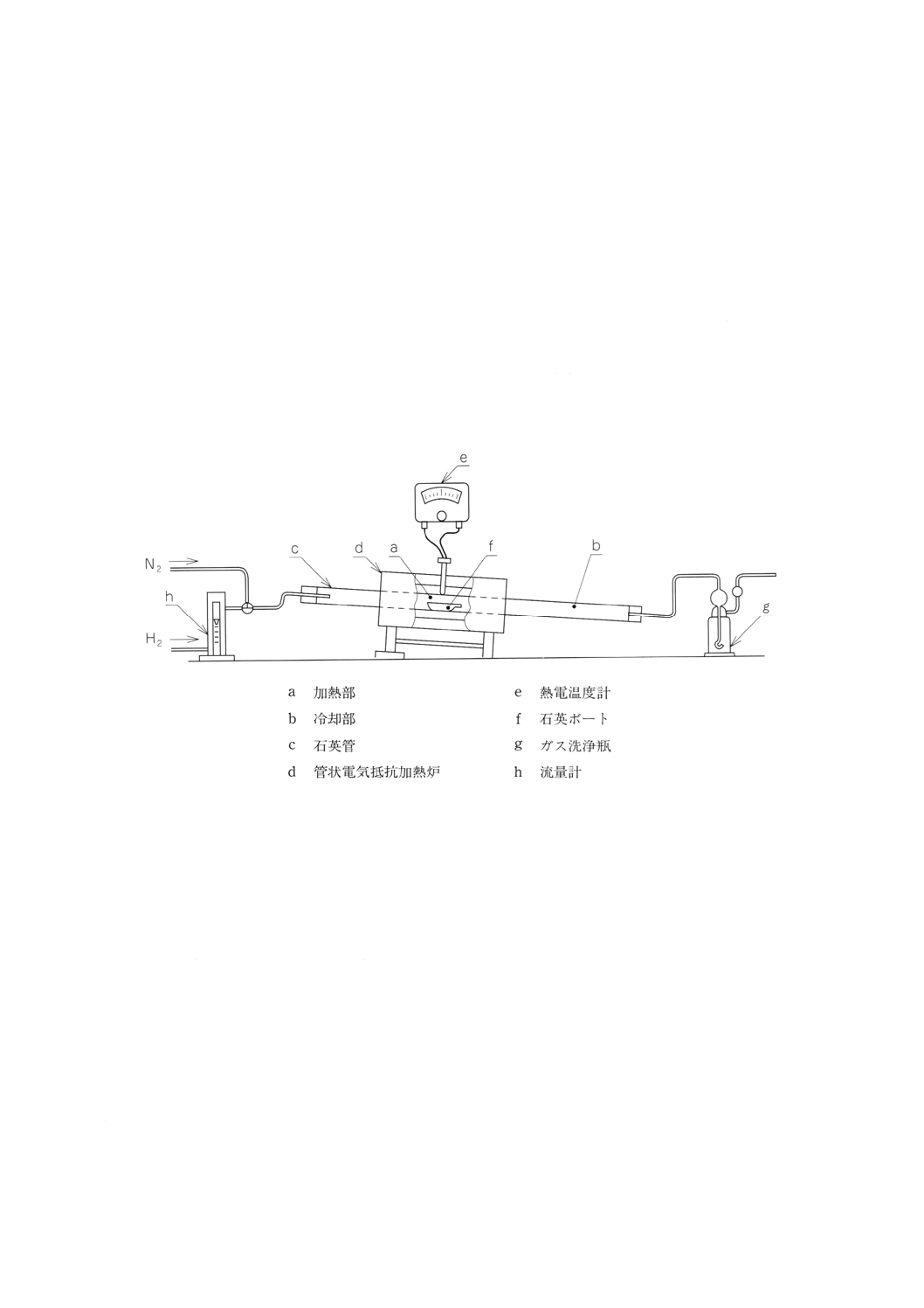
13.2.3 装置 酸素定量装置は,通常図3に示すものを用いる。
装置は,流量計 (h),石英管 (c),管状電気抵抗加熱炉 (d) 及び水を入れたガス洗浄瓶 (g) とからなり(19),
石英管は,試料を挿入し加熱する位置[加熱部 (a)]と,試料を冷却する位置[冷却部 (b)]とがあり,水
素は,一方から導入される。管状電気抵抗加熱炉 (d) は,炉の中央部において約1 000℃の温度を保つこ
とのできるもので,石英管の加熱中央部の温度を熱電温度計 (e) を用いて測定できるもの。
注(19) 石英管を3〜5度傾斜させ,水素出口側を下げる。
図3 酸素定量装置(水素還元重量法)の例
13.2.4 試料はかり取り量 試料はかり取り量は,5.0gとし,0.1mgのけたまではかる。
13.2.5 操作
13.2.5.1 準備操作 準備操作は,次の手順によって行う。
a) 装置に水素を送って内部の空気と完全に置換(20)した後,管状電気抵抗加熱炉 (d) に通電して昇温させ
る。
注(20) 爆鳴試験によって空気の存在を認めなくなってから管状電気抵抗加熱炉 (d) の昇温を行う。
なお,排気水素は,ガス洗浄瓶 (g) を通した後,屋外に排気する。
b) 水素流量を約100ml/分に調節し,加熱部 (a) を980〜1 020℃(21)に保持し,石英ボート (f) を装置に
水素を通じたまま水素出口側から石英管の加熱部 (a) に挿入する。
注(21) 熱電温度計の指示値は,一般に管内の温度と異なるのでその差を求めておき,その差を補正し
て温度計の温度を設定する。
c) 13.2.5.2のb)〜c)の手順に従って処理して恒量とした後,その質量を0.1 mgのけたまではかる。
13.2.5.2 定量操作 定量操作は,次の手順によって行う。
28
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 試料を13.2.5.1 c)で恒量とした石英ボート (f) にはかり取り,これを装置に水素を通じたまま水素出
口側から石英管の加熱部 (a) に挿入する。
b) 980〜1 020℃で約30分間加熱し,石英ボートを水素出口側から石英管の冷却部 (b) に移し,約20分
間放冷する。
c) 石英ボートを取り出し,デシケーター中で約10分間放冷した後,その質量を0.1mgのけたまではか
る。
備考 測定終了後,管状電気抵抗加熱炉 (d) への通電を止め,装置に窒素を送り,次に水素を止めて
内部を窒素で完全に置換することが望ましい。
13.2.5.3 計算 試料中の酸素含有率を,次の式によって算出する。
100
)
(
1
2
0
×
−
−
=
m
m
m
m
O
ここに,
O: 試料中の酸素含有率 [% (m/m)]
m0: 試料はかり取り量 (g)
m2: 13.2.5.2 c)で得た質量 (g)
m1: 13.2.5.1 c)で得た質量 (g)
13.3 酸化重量法
13.3.1 要旨 試料を空気中で加熱してタングステンを完全に三酸化タングステンとし,その増量をはかる。
13.3.2 装置 装置は,約800℃の温度を保つことのできる電気抵抗加熱炉で加熱部中央付近の温度を熱電
温度計を用いて測定できるもの。
13.3.3 試料はかり取り量 試料はかり取り量は,5.0gとし,0.1mgのけたまではかる。
13.3.4 操作
13.3.4.1 準備操作 準備操作は,白金皿(50番又は75番)又は磁器るつぼ(A形50ml)を電気抵抗加熱
炉に挿入し,13.3.4.2 c)の手順に従って処理して恒量とした後,その質量を0.1mgのけたまではかる。
13.3.4.2 定量操作 定量操作は,次の手順によって行う。
a) 試料を13.3.4.1で恒量とした白金皿(50番又は75番)又は磁器るつぼ(A形50ml)にはかり取り,
これを電気抵抗加熱炉に挿入する。
b) 電気抵抗加熱炉の扉を少し開け,温度を徐々に上昇させ500〜600℃で約10分間加熱した後,炉の扉
を閉める。
c) 電気抵抗加熱炉の温度を上昇させ750〜780℃とした後,約1時間保持する白金皿(50番又は75番)
又は磁器るつぼ(A形50ml)を取り出しデシケーター中で約1時間放冷し,その質量を0.1mgのけた
まではかる。この操作を恒量になるまで繰り返す。
13.3.5 計算 試料中の酸素含有率を,次の式によって算出する。
100
0
793
.0
)
(
0
1
2
0
×
×
−
−
=
m
m
m
m
O
ここに,
O: 試料中の酸素含有率 [% (m/m)]
m0: 試料はかり取り量 (g)
m2: 13.3.4.2 c)で得た質量 (g)
m1: 13.3.4.1で得た質量 (g)
13.4 不活性ガス融解−赤外線吸収法
29
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.4.1 要旨 不活性ガス気流中で,黒鉛るつぼを用いて,試料を浴金属とともにインパルス方式によって
加熱して融解し,酸素を一酸化炭素として他のガスとともに抽出する。抽出ガス中の一酸化炭素をそのま
ま赤外線検出器に導くか,加熱した酸化銅(II)で一酸化炭素を二酸化炭素に酸化した後,赤外線検出器に導
き,抽出ガス中の一酸化炭素又は二酸化炭素による赤外線吸収量の変化を測定する。
13.4.2 材料及び試薬 材料及び試薬は,次による。
a) 浴金属 ニッケルのカプセル・バスケット,すず(22)のはく又はカプセル状のもの。
注(22) すずの調製は,JIS Z 2613の5.2(2)(すず)による。
b) 不活性ガス 純度99.99% (v/v) 以上のヘリウム,窒素又はアルゴンを用いる。
c) 黒鉛るつぼ インパルス炉に適合するもの。その例を付図1に示す。
d) 検量線用試料 酸素含有率が既知のもの。
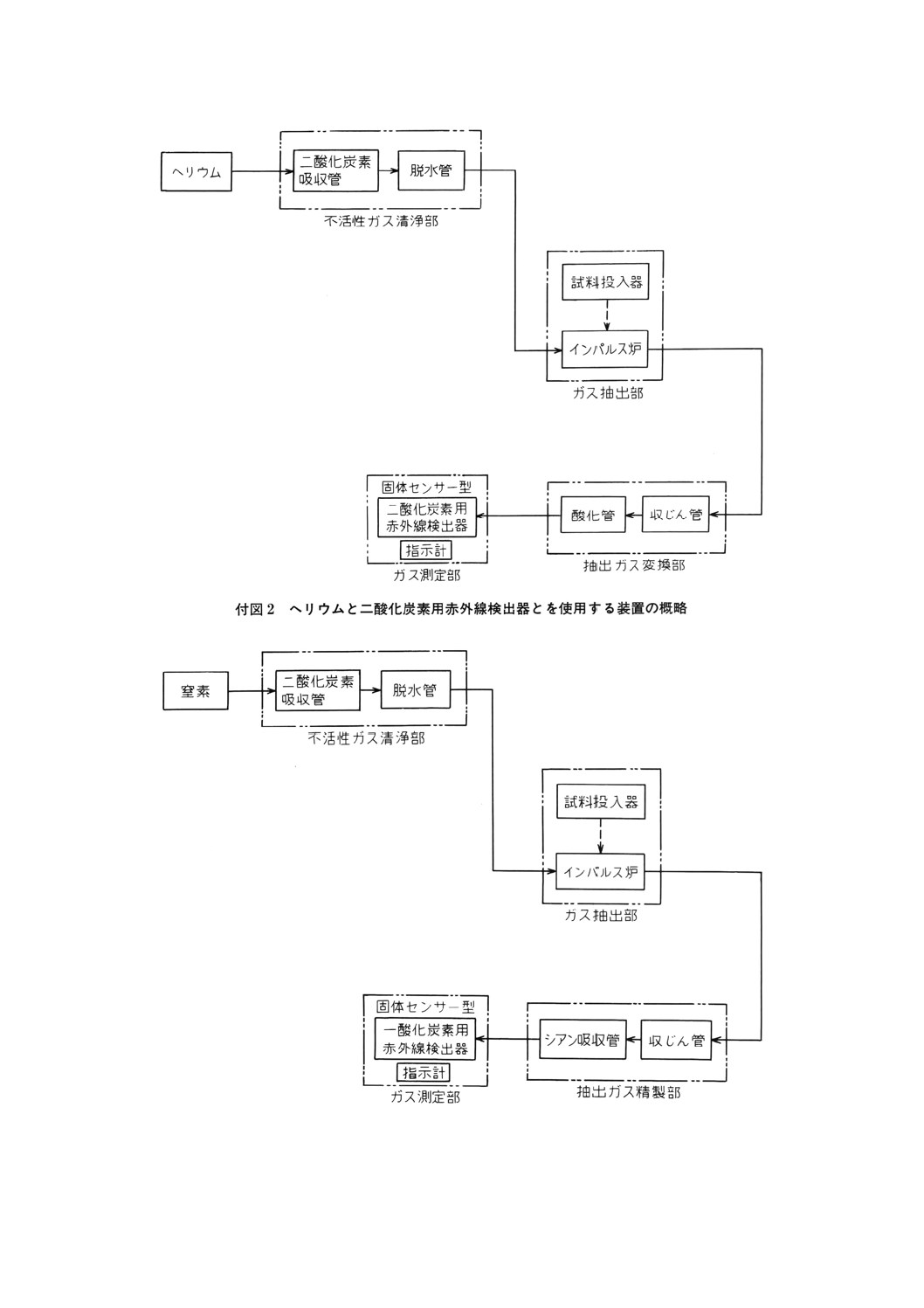
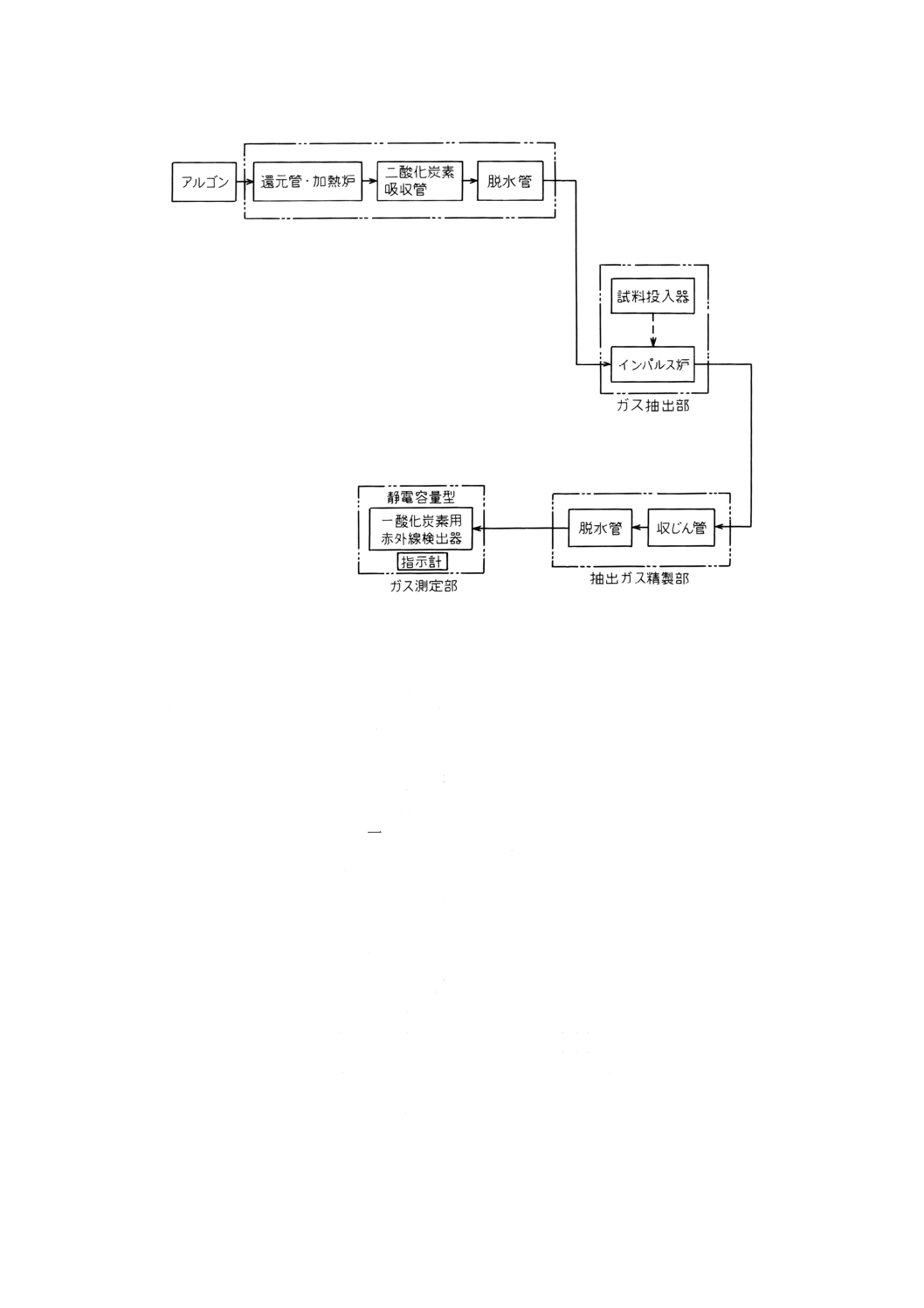
13.4.3 装置(23) 装置は,不活性ガス清浄部,ガス抽出部,抽出ガス変換部又は抽出ガス精製部,ガス測
定部などで構成する。ガス抽出部以外の各部の構成は,装置に用いられている赤外線検出器及び使用する
不活性ガスの種類によって異なる。装置の概略を付図2〜4に示す。
注(23) 装置の構成,構造及び使用条件は,使用する装置によって異なる。
13.4.3.1 ヘリウムと二酸化炭素用赤外線検出器とを使用する装置(付図2参照)
a) 不活性ガス清浄部 不活性ガス清浄部は,二酸化炭素吸収管,脱水管などで構成する
1) 二酸化炭素吸収管 ガラス管にソーダ石灰又は水酸化ナトリウムを詰めたもの。
2) 脱水管 ガラス管に過塩素酸マグネシウムを詰めたもの。
b) ガス抽出部 ガス抽出部は,試料投入器,インパルス炉などで構成する。
1) 試料投入器 不活性ガス雰囲気中で試料をインパルス炉に投入できるもの。
2) インパルス炉 銅製の固定された上部水冷電極及び上下に移動できる下部水冷電極で構成し,両電
極の間に挟んだ黒鉛るつぼ [13.4.2 c)] を通電によって数秒間で2 000〜2 500℃に昇温できるもの。
c) 抽出ガス変換部 抽出ガス変換部は,収じん管,酸化管などで構成する。
1) 収じん管 ガラス管にガラスウールを詰めたもの。
2) 酸化管 ステンレス管又はガラス管に酸化銅(II)を詰めたもの。電気抵抗加熱炉によって加熱して使
用する。
d) ガス測定部 ガス測定部は,赤外線検出器,指示計などで構成する。
1) 赤外線検出器 固体センサー形の二酸化炭素用赤外線検出器で,二酸化炭素による赤外線吸収量の
変化を測定できるもの。
2) 指示計 赤外線検出器で検出された二酸化炭素に基づく信号を読み取ることのできるもの。
30
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.4.3.2 窒素と一酸化炭素用赤外線検出器とを使用する装置(付図3参照)
a) 不活性ガス清浄部 13.4.3.1 a)による。
b) ガス抽出部 13.4.3.1 b)による。
c) 抽出ガス精製部 抽出ガス精製部は収じん管,ジシアン吸収管などで構成する。
1) 収じん管 13.4.3.1 c)1)による。
2) ジシアン吸収管 ガラス管に水酸化ナトリウムを詰めたもの。
d) ガス測定部 ガス測定部は,赤外線検出器,指示計などで構成する。
1) 赤外線検出器 固体センサー形の一酸化炭素用赤外線検出器で,一酸化炭素による赤外線吸収量の
変化を測定できるもの。
2) 指示計 赤外線検出器で検出された一酸化炭素に基づく信号を読み取ることのできるもの。
13.4.3.3 アルゴンと一酸化炭素用赤外線検出器とを使用する装置(付図4参照)
a) 不活性ガス清浄部 不活性ガス清浄部は,還元管,二酸化炭素吸収管,脱水管などで構成する。
1) 還元管 ステンレス鋼管に金属銅(粒状)又は活性炭を詰めたもの。電気抵抗加熱炉で加熱して使
用する。
2) 二酸化炭素吸収管 13.4.3.1 a)1)による。
3) 脱水管 13.4.3.1 a)2)による。
b) ガス抽出部 13.4.3.1 b)による。
c) 抽出ガス精製部 抽出ガス精製部は,収じん管,脱水管などで構成する。
1) 収じん管 13.4.3.1 c)1)による。
2) 脱水管 13.4.3.1 a)2)による。
d) ガス測定部 ガス測定部は,赤外線検出器,指示計などで構成する。
1) 赤外線検出器 内部に一酸化炭素を封入した静電容量形の一酸化炭素用赤外線検出器で,一酸化炭
素による赤外線吸収量の変化を測定できるもの。
2) 指示計 13.4.3.2 d)2)による。
13.4.4 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,0.1mgのけたまでは
かる。
13.4.5 操作(24)
注(24) 操作の細かい手順は,装置によって異なるので,その装置の指定する手順に従う。
13.4.5.1 準備操作 準備操作は,次の手順によって行う。
a) 装置に使用する不活性ガス [13.4.2 b)] 及び冷却水を供給した後,電源を入れ,装置各部を指定の条件
に設定し,装置の各部を安定させる。
b) 新しい黒鉛るつぼ [13.4.2 c)] を所定の位置に設置し,インパルス炉に通電して黒鉛るつぼを脱ガス温
度に加熱する(25)。
注(25) 黒鉛るつぼの温度のパラメータとして,黒鉛るつぼに流れる電流値又は電力値を読み,ガス抽
出に相当する電流値又は電力値より高いことを確認する。
c) b)の黒鉛るつぼをガス抽出温度に加熱し(26),指示計の値を読み取る。
注(26) ガス抽出温度のパラメータとして,あらかじめ試料を用いて,酸素を十分に抽出できる温度の
最適な電流値又は電力値を求めておく。
d) 安定した値が得られるまでc)の操作を繰り返す(27)。
注(27) b)で脱ガスした黒鉛るつぼを繰り返して用いる。
31
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.4.5.2 定量操作 定量操作は,準備操作,空試験及び検量線の作成に引き続き,次の手順によって行う。
a) 新しい黒鉛るつぼ [13.4.2 c)] を所定の位置に設置する。
b) 試料及び浴金属 [13.4.2 a)] をはかり取る(28)。
注(28) 浴金属の使用量は,使用する装置によって異なるので,あらかじめ試料を用いて,その装置に
適した使用量を求めておく。
c) はかり取った試料を浴金属で包んで,試料投入器に入れる。
d) インパルス炉に通電して黒鉛るつぼを脱ガス温度に加熱し(25),黒鉛るつぼの脱ガスを行う。
e) c) の浴金属及び試料を黒鉛るつぼに投入し,インパルス炉に通電して黒鉛るつぼを抽出温度(26)に加
熱し,指示計の値を読み取る。
13.4.6 空試験 空試験は,準備操作に引き続き,次の手順によって行う。
a) 新しい黒鉛るつぼ [13.4.2 c)] を所定の位置に設置する。
b) 13.4.5.2 d)と同じ条件でインパルス炉に通電して黒鉛るつぼを脱ガス温度に加熱し,黒鉛るつぼの脱ガ
スを行う。
c) 13.4.5.2 b)で用いるのと同じ浴金属の同じ量を試料投入器に入れ,13.4.5.2 e)と同じ条件でインパルス
炉に通電して黒鉛るつぼをガス抽出温度に加熱し,指示計の値を読み取る。
d) a)〜c)の操作を数回繰り返し,読み取った値の平均を求める。
13.4.7 検量線の作成 検量線の作成は,準備操作及び空試験に引き続き,次の手順(24)によって行う。
a) 新しい黒鉛るつぼ [13.4.2 c)] を所定の位置に設置する。
b) 検量線用試料 [13.4.2 d)] を0.1mgのけたまではかり取る。
c) はかり取った検量線用試料 [13.4.2 d)] を13.4.5.2 b)で用いるのと同じ浴金属の同じ量(29)で包んで試
料投入器に入れる。
注(29) 検量線用試料の種類に応じて浴金属の種類や量を変更してもよい。この場合,浴金属の使用量
は,使用する装置によって異なるので,あらかじめ検量線用試料を用いて,その装置に適した
使用量を求めておく。ただし,あらかじめ酸素が十分に抽出されることがわかっている場合は,
浴金属を使用しなくてもよい。
d) 13.4.5.2 d)と同じ条件でインパルス炉に通電して黒鉛るつぼを脱ガス温度に加熱し,黒鉛るつぼの脱ガ
スを行う。
e) c) の浴金属及び検量線用試料を黒鉛るつぼに投入し,13.4.5.2 e)と同じ条件でインパルス炉に通電し
て黒鉛るつぼをガス出抽温度に加熱し,指示計の値を読み取り,13.4.6 d)で得た平均値(30)を差し引く。
注(30) c)で注(29)を適用し13.4.5.2 c)と異なる浴金属を用いる場合は,c)で用いるのと同じ浴金属の同じ
量を試料投入器に入れ,d)と同じ条件でインパルス炉に通電して黒鉛るつぼを脱ガス温度に加
熱し,黒鉛るつぼの脱ガスを行う。次に,浴金属を黒鉛るつぼに投入し,13.4.5.2 e)と同じ条件
でインパルス炉に通電して黒鉛るつぼをガス抽出温度に加熱し,指示計の値を読み取る。この
操作を数回繰り返し,読み取った値の平均を求める。
なお,浴金属を使用しない場合でもd)〜f)の操作を行う。
f)
a)〜e)の操作を数回繰り返し,読み取った値の平均を求める。
g) 検量線用試料の酸素含有率とはかり取った質量から酸素量(31)を求め,酸素量とf)で得た値との関係を
プロットする。プロットした点と原点とを通る直線を作成し,その直線(酸素と指示値との関係線)
を検量線とする。
注(31) 酸素の量は,次の式によって算出する。
32
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
P
G
M
×
=
ここに, M: はかり取った検量線用試料中の酸素量 (g)
G: はかり取った検量線用試料の質量 (g)
P: 検量線用試料中の酸素含有率 [% (m/m)]
13.4.8 計算 13.4.5.2 e)で読み取った値及び13.4.6 d)で得た平均値と13.4.7で作成した検量線とから酸素
の量を求め,試料中の酸素含有率を次の式によって算出する。
100
2
1
×
−
=
m
A
A
O
ここに,
O: 試料中の酸素含有率 [% (m/m)]
A1: 13.4.5.2 e)で読み取った値から求めた酸素量 (g)
A2: 13.4.6 d)の平均値から求めた酸素の量 (g)
m: 試料はかり取り量 (g)
14. 全炭素定量方法
14.1 定量方法の区分 全炭素の定量方法は,次のいずれかによる。
a) 燃焼−導電率法 この方法は,炭素含有率0.001% (m/m) 以上の試料に適用する。
b) 燃焼−電量法 この方法は,炭素含有率0.001% (m/m) 以上の試料に適用する。
c) 燃焼−熱伝導度法 この方法は,炭素含有率0.001% (m/m) 以上の試料に適用する。
d) 燃焼−赤外線吸収法(積分法) この方法は,炭素含有率0.001% (m/m) 以上の試料に適用する。
e) 燃焼−赤外線吸収法(循環法) この方法は,炭素含有率0.001% (m/m) 以上の試料に適用する。
14.2 焼燃−導電率法
14.2.1 要旨 試料を酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,一定量の水酸化ナトリウム溶
液に吸収させ,吸収前後の溶液の導電率の変化を測定する。
14.2.2 試薬 試薬は,JIS Z 2615の6.6.2(試薬)による。ただし,水酸化ナトリウム溶液の濃度は0.01
〜0.1mol/Lとする。
14.2.3 装置,器具及び材料 装置は,JIS Z 2615の6.6.3(装置)による。器具及び材料は,JIS Z 2615
の5. (器具及び材料)による。ただし,JIS Z 2615の6.6.3(4)(二酸化炭素定量部)の水酸化ナトリウム
溶液濃度は0.01〜0.1mol/Lとする。
14.2.4 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,0.1mgのけたまでは
かる。
14.2.5 操作
14.2.5.1 準備操作 準備操作は,JIS Z 2615の6.6.4(予備操作)による。ただし,管状電気抵抗加熱炉中
央部での燃焼管内温度を1 200〜1 300℃に保持する。
14.2.5.2 定量操作 定量操作は,JIS Z 2615の6.6.5(定量操作)による。
14.2.6 空試験 空試験は,JIS Z 2615の6.6.6(空試験)による。
14.2.7 計算 計算は,JIS Z 2615の6.6.7(計算)による。
14.3 燃焼−電量法
33
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.3.1 要旨 試料を酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,一定のpHに調整した弱アル
カリ性のバリウム塩溶液に吸収させ,吸収によって低下したpHをバリウム塩溶液の電解によって元のpH
に戻すために要した電気量を測定する。
14.3.2 試薬 試薬は,JIS Z 2615の6.7.2(試薬)による。
14.3.3 装置,器具及び材料 装置は,JIS Z 2615の6.7.3(装置)による。器具及び材料は,JIS Z 2615
の5.(器具及び材料)による。
14.3.4 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,0.1mgのけたまでは
かる。
14.3.5 操作
14.3.5.1 準備操作 準備操作は,JIS Z 2615の6.7.4(予備操作)による。ただし,管状電気抵抗加熱炉中
央部での燃焼管内温度を1 200〜1 300℃に保持する。
14.3.5.2 定量操作 定量操作は,JIS Z 2615の6.7.5(定量操作)による。
14.3.6 空試験 空試験は,JIS Z 2615の6.7.6(空試験)による。
14.3.7 計算 計算は,JIS Z 2615の6.7.7(計算)による。
14.4 燃焼−熱伝導度法
14.4.1 要旨 試料を酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,合成ゼオライトを入れた捕集
管に吸着させた後,この捕集管を加熱して吸着した二酸化炭素を放出させ,酸素とともに熱伝導度検出器
に送り,二酸化炭素による熱伝導度の変化を測定する。
14.4.2 試薬 試薬は,JIS Z 2615の6.8.2(試薬)による。
14.4.3 装置,器具及び材料 装置は,JIS Z 2615の6.8.3(装置)による。器具及び材料は,JIS Z 2615
の5.(器具及び材料)による。
14.4.4 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,0.1mgのけたまでは
かる。
14.4.5 助燃剤 助燃剤は,酸素気流中で試料を加熱燃焼させる際に酸化反応を円滑に進め,試料中の炭素
を二酸化炭素とするため,その適量で試料を覆い使用するもので,次の材料を単独又は組み合わせて用い
る(32)。いずれも炭素含有率ができるだけ低いものが望ましい。
注(32) 試料を用いて炭素を十分に酸化できることを確かめた後使用する。
a) 鉄 例えば,粒状[粒度250〜1 000mm(60〜16メッシュ相当)]
b) 銅 例えば,粒状[粒度850mm(20メッシュ相当)]
c) タングステン(33) 例えば,粒状[粒度850〜2 000mm(20〜9メッシュ相当)]
注(33) タングステンを用いる場合は,他の助燃剤と組み合わせて用いなければならない。
14.4.6 操作
14.4.6.1 準備操作 準備操作は,JIS Z 2615の6.8.4(予備操作)による。
14.4.6.2 定量操作 定量操作は,JIS Z 2615の6.8.5(定量操作)による。
14.4.7 空試験 空試験は,JIS Z 2615の6.8.6(空試験)による。
14.4.8 計算 計算は,JIS Z 2615の6.8.7(計算)による。
14.5 燃焼−赤外線吸収法(積分法)
14.5.1 要旨 試料を酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,これを酸素とともに赤外線吸
収検出器に送り,その赤外線吸収量を積分法で測定する。
34
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.5.2 装置,器具及び材料 装置は,JIS Z 2615の6.9.3(装置)による。ただし,高周波誘導加熱炉の
代わりに,JIS Z 2615の5.(6)(6.1)(管状電気抵抗加熱炉)を用いることができる。器具及び材料は,JIS Z
2615の5.(器具及び材料)及びJIS Z 2615の6.9.2(材料)による。
14.5.3 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,0.1mgのけたまでは
かる。
14.5.4 助燃剤 助燃剤は,14.4.5による。
14.5.5 操作
14.5.5.1 準備操作 準備操作は,JIS Z 2615の6.9.4(予備操作)による。ただし,試料燃焼部に管状電気
抵抗加熱炉を用いる場合は,管状電気抵抗加熱炉中央部での燃焼管内温度を1 200〜1 300℃に保持する。
14.5.5.2 定量操作 定量操作は,JIS Z 2615の6.9.5(定量操作)による。ただし,試料燃焼部に管状電気
抵抗加熱炉を用いる場合は,次の手順による。
a) 試料をはかり取って磁器燃焼ボートに移し入れ,燃焼管の中央部に挿入し,直ちに気密に栓をし,指
定された割合で酸素を送入する(試料が燃焼し,燃焼ガスは,精製部を経て赤外線吸収検出器の試料
セルに送られ,指示値が次第に増加する。)。
b) 指示値が一定になったときの指示値を読みとる。
14.5.6 空試験 空試験は,JIS Z 2615の6.9.6(空試験)による。
14.5.7 計算 計算は,JIS Z 2615の6.9.7(計算)による。
14.6 燃焼−赤外線吸収法(循環法)
14.6.1 要旨 試料を一定容積内の一定圧力下の循環酸素気流中で加熱し,炭素を二酸化炭素及び一酸化炭
素に酸化し,過剰の酸素とともに循環ループの赤外線吸収検出器に送り,その赤外線吸収量をそれぞれ測
定する。
14.6.2 装置,器具及び材料 装置は,JIS Z 2615の6.10.2(装置)による。器具及び材料は,JIS Z 2615
の5.(器具及び材料)による。
14.6.3 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,0.1mgのけたまでは
かる。
14.6.4 助燃剤 助燃剤は,14.4.5による。
14.6.5 操作
14.6.5.1 準備操作 準備操作は,JIS Z 2615の6.10.3(予備操作)による。
14.6.5.2 定量操作 定量操作は,JIS Z 2615の6.10.4(定量操作)による。
14.6.6 空試験 空試験は,JIS Z 2615の6.10.5(空試験)による。
14.6.7 計算 計算は,JIS Z 2615の6.10.6(計算)による。
15. 遊離炭素定量方法
15.1 定量方法の区分 遊離炭素の定量方法は,次のいずれかによる。
a) 炭素沈殿分離燃焼−導電率法 この方法は,遊離炭素含有率0.01% (m/m) 以上の試料に適用する。
b) 炭素沈殿分離燃焼−電量法 この方法は,遊離炭素含有率0.01% (m/m) 以上の試料に適用する。
c) 炭素沈殿分離燃焼−熱伝導度法 この方法は,遊離炭素含有率0.01% (m/m) 以上の試料に適用する。
d) 炭素沈殿分離燃焼−赤外線吸収法(積分法) この方法は,遊離炭素含有率0.01% (m/m) 以上の試料
に適用する。
e) 炭素沈殿分離燃焼−赤外線吸収法(循環法) この方法は,遊離炭素含有率0.01% (m/m) 以上の試料
35
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に適用する。
15.2 炭素沈殿分離燃焼−導電率法
15.2.1 要旨 試料を適切な酸で分解し,石綿でこし分け,遊離炭素を石綿とともに磁器燃焼ボートに移し
て乾燥し,酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,一定量の水酸化ナトリウム溶液に吸収
させ,吸収前後の溶液の導電率の変化を測定する。
15.2.2 試薬 試薬は,JIS Z 2615の6.6.2(試薬)及び次による。ただし,水酸化ナトリウム溶液の濃度
は0.01〜0.1mol/Lとする。
a) 硝酸
b) ふっ化水素酸
c) 混酸D(硝酸4,りん酸1,水10)
d) アンモニア水
e) 石綿 あらかじめ酸素気流中で約1 000℃で約1時間強熱したもの。
15.2.3 装置,器具及び材料 装置,器具及び材料は,14.2.3による。
15.2.4 試料はかり取り量 試料はかり取り量は,2.0gとし,10mgのけたまではかる。
15.2.5 操作
15.2.5.1 準備操作 準備操作は,14.2.5.1による。
15.2.5.2 試料の分解 試料の分解は,次のいずれかによる。
a) 硝酸・ふっ化水素酸による分解
1) 試料をはかり取って,白金皿(75番又は90番)に移し入れ,白金ふたで覆い,ふっ化水素酸約10ml
を加え,硝酸を1滴ずつ加え,放置又は加熱(34)しながら穏やかに分解する。分解した後,白金ふた
の下面を水で洗浄して白金ふたを取り除く。更に硝酸を5m1加えた後,約10分間加熱を続ける。
注(34) 分解反応は,粒度の小さいものほど激しく加熱なしに分解する場合が多い。
2) 室温まで冷却した後,水で液量を約50mlとし,アンモニア水を加えて弱アルカリ性とする。
b) 混酸Dによる分解 試料をはかり取って,ビーカー (200〜300ml) に移し入れ時計皿で覆い,混酸
D75mlを加え加熱して分解する。時計皿の下面及びビーカーの内壁を少量の水で洗浄し,更に数分間
加熱する。室温まで冷却した後,時計皿の下面を水で洗浄して時計皿を取り除く。
15.2.5.3 遊離炭素の分離 15.2.5.2のa)2)又はb)で得た溶液を,石綿 [15.2.2 e)](35)(36)を使って上澄み液を
吸引ろ過する。次に残りを温水を用いてその上に移し,温水で十分に洗浄する。ろ液及び洗液は捨てる。
注(35) 石綿は,ろ過漏れを起こさない程度に水に湿らせた後,圧縮する。
(36) 混酸分解による遊離炭素の分離には,石綿の代わりにバインダーを含まないガラス繊維ろ紙を
使用してもよい。
15.2.5.4 定量操作 定量操作は,15.2.5.3で得た残さを,石綿とともに磁器燃焼ボートに移し入れ,約130℃
で1時間以上乾燥した後,14.2.5.2に準じて操作する。ただし,管状電気抵抗加熱炉中央部での燃焼管内温
度を約1 000℃とする。
15.2.6 空試験 試料を用いないで試料と同じ操作を試料と並行して行い,得られた指示値を空試験の指示
値とする。
15.2.7 計算 計算は,14.2.7による。
15.3 炭素沈殿分離燃焼−電量法
36
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
15.3.1 要旨 試料を適切な酸で分解し,石綿でこし分け,遊離炭素を石綿とともに磁器燃焼ボートに移し
て乾燥し,酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,一定のpHに調整した弱アルカリ性の
バリウム塩溶液に吸収させ,吸収によって低下したpHをバリウム塩溶液の電解によって元のpHに戻すた
めに要した電気量を測定する。
15.3.2 試薬 試薬は,15.2.2のa)〜e)及びJIS Z 2615の6.7.2(試薬)による。
15.3.3 装置,器具及び材料 装置,器具及び材料は,14.3.3による。
15.3.4 試料はかり取り量 試料はかり取り量は,2.0gとし,10mgのけたまではかる。
15.3.5 操作
15.3.5.1 準備操作 準備操作は,14.3.5.1による。
15.3.5.2 試料の分解 試料の分解は,15.2.5.2による。
15.3.5.3 遊離炭素の分離 遊離炭素の分離は,15.2.5.3による。
15.3.5.4 定量操作 定量操作は,15.3.5.3で得た残さを,石綿とともに磁器燃焼ボートに移し入れ,約130℃
で1時間以上乾燥した後,14.3.5.2に準じて操作する。ただし,管状電気抵抗加熱炉中央部での燃焼管内温
度を約1 000℃とする。
15.3.6 空試験 試料を用いないで,試料と同じ操作を試料と並行して行い,得られた指示値を空試験の指
示値とする。
15.3.7 計算 計算は,14.3.7による。
15.4 炭素沈殿分離燃焼−熱伝導度法
15.4.1 要旨 試料を適切な酸で分解し,石綿でこし分け,遊離炭素を石綿とともに高周波燃焼るつぼに移
して乾燥し,酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,合成ゼオライトを入れた捕集管に吸
着させた後,この捕集管を加熱して吸着した二酸化炭素を放出させ,酸素とともに熱伝導度検出器に送り,
二酸化炭素による熱伝導度の変化を測定する。
15.4.2 試薬 試薬は,15.2.2のa)〜e)及びJIS Z 2615の6.8.2(試薬)による。
15.4.3 装置,器具及び材料 装置,器具及び材料は,14.4.3による。
15.4.4 試料はかり取り量 試料はかり取り量は,2.0gとし,10mgのけたまではかる。
15.4.5 助燃剤 助燃剤は,14.4.5による。
15.4.6 操作
15.4.6.1 準備操作 準備操作は,14.4.6.1による。
15.4.6.2 試料の分解 試料の分解は,15.2.5.2による。
15.4.6.3 遊離炭素の分離 遊離炭素の分離は,15.2.5.3による。
15.4.6.4 定量操作 定量操作は,15.4.6.3で得た残さを,石綿とともに高周波燃焼るつぼに移し入れ,約
130℃で1時間以上乾燥した後,14.4.6.2に準じて操作する。
15.4.7 空試験 試料を用いないで,試料と同じ操作を試料と並行して行い,得られた指示値を空試験の指
示値とする。
15.4.8 計算 計算は,14.4.8による。
15.5 炭素沈殿分離燃焼−赤外線吸収法(積分法)
15.5.1 要旨 試料を適切な酸で分解し,石綿でこし分け,遊離炭素を石綿とともに磁器燃焼ボート又は高
周波燃焼るつぼに移して乾燥し,酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,これを酸素とと
もに赤外線吸収検出器に送り,その赤外線吸収量を積分法で測定する。
15.5.2 試薬 試薬は,15.2.2のa)〜e)による。
37
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
15.5.3 装置,器具及び材料 装置,器具及び材料は,14.5.2による。
15.5.4 試料はかり取り量 試料はかり取り量は,2.0gとし,10mgのけたまではかる。
15.5.5 助燃剤 助燃剤は,14.4.5による。
15.5.6 操作
15.5.6.1 準備操作 準備操作は,14.5.5.1による。
15.5.6.2 試料の分解 試料の分解は,15.2.5.2による。
15.5.6.3 遊離炭素の分離 遊離炭素の分離は,15.2.5.3による。
15.5.6.4 定量操作 定量操作は,15.5.6.3で得た残さを,石綿とともに磁器燃焼ボート又は高周波燃焼る
つぼに移し入れ,約130℃で1時間以上乾燥した後,14.5.5.2に準じて操作する。ただし,試料燃焼部に管
状電気抵抗加熱炉を用いる場合は,管状電気抵抗加熱炉中央部での燃焼管内温度を約1 000℃とする。
15.5.7 空試験 試料を用いないで,試料と同じ操作を試料と並行して行い,得られた指示値を空試験の指
示値とする。
15.5.8 計算 計算は,14.5.7による。
15.6 炭素沈殿分離燃焼−赤外線吸収法(循環法)
15.6.1 要旨 試料を適切な酸で分解し,石綿でこし分け,遊離炭素を石綿とともに高周波燃焼るつぼに移
して乾燥し,一定容積内の一定圧力下の循環酸素気流中で加熱し,炭素を二酸化炭素及び一酸化炭素に酸
化し,過剰の酸素とともに循環ループの赤外線吸収検出器に送り,その赤外線吸収量をそれぞれ測定する。
15.6.2 試薬 試薬は,15.2.2のa)〜e)による。
15.6.3 装置,器具及び材料 装置,器具及び材料は,14.6.2による。
15.6.4 試料はかり取り量 試料はかり取り量は,2.0gとし,10mgのけたまではかる。
15.6.5 助燃剤 助燃剤は,14.4.5による。
15.6.6 操作
15.6.6.1 準備操作 準備操作は,14.6.5.1による。
15.6.6.2 試料の分解 試料の分解は,15.2.5.2による。
15.6.6.3 遊離炭素の分離 遊離炭素の分離は,15.2.5.3による。
15.6.6.4 定量操作 定量操作は,15.6.6.3で得た残さを,石綿とともに高周波燃焼るつぼに移し入れ,約
130℃で1時間以上乾燥した後,14.6.5.2に準じて操作する。
15.6.7 空試験 試料を用いないで,試料と同じ操作を試料と並行して行い,得られた指示値を空試験の指
示値とする。
15.6.8 計算 計算は,14.6.7による。
16. 硫黄定量方法
16.1 定量方法 硫黄の定量方法は,燃焼−赤外線吸収法(積分法)による。この方法は,硫黄含有率0.000
5% (m/m) 以上の試料に適用する。
16.2 要旨 試料を酸素気流中で加熱し,硫黄を酸化して二酸化硫黄とし,これを酸素とともに赤外線吸
収検出器に送り,二酸化硫黄による赤外線吸収量を積分法で測定する。
16.3 装置,器具及び材料 装置は,JIS Z 2616の7.7(2)(装置)による。ただし,高周波誘導加熱炉の代
わりに,JIS Z 2616の6.6(1)(管状電気抵抗加熱炉)(37)を用いることができる。器具及び材料は,JIS Z 2616
の6.(器具及び材料)(38)による。
注(37) 燃焼管の入口は,ここから過剰の酸素を放出し,空気が管内に侵入するのを防ぐようにすれば
38
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
開放してもよい。この場合には,一定の割合で燃焼ガスを吸収管内に送り込むための定量ポン
プを用いる必要がある。
(38) 磁器燃焼ボート,磁器燃焼ボートカバー及び高周波磁器燃焼るつぼは,1 200℃以上で30分間
以上空焼きしたものを用いる。
16.4 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,1mgのけたまではか
る。
16.5 助燃剤 助燃剤は,JIS Z 2616の6.12(助燃剤)に示すものの中から単独又は2,3種類組み合わせ
て用いる。あらかじめ最適な種類とその添加量を,試料を用いて調べておく。
16.6 操作
16.6.1 準備操作 準備操作は,JIS Z 2616の7.7(3)(予備操作)による。ただし,管状電気抵抗加熱炉を
用いる場合は,管状電気抵抗加熱炉中央部での燃焼管内温度を1 200〜1 300℃に保持する。
16.6.2 定量操作 定量操作は,JIS Z 2616の7.7(4)(定量操作)による。ただし,試料燃焼部に管状電気
抵抗加熱炉を用いる場合は,次の手順による。
a) はかり取った試料を入れたボートを燃焼管の中央部に挿入し,直ちに気密に栓をし,指定された割合
で酸素を送入する(試料が燃焼し,燃焼ガスは精製部を経て赤外線検出器の試料セルに送られ,指示
値が次第に増加する。)。
b) 指示値が一定になったときの,指示値を読み取る。
16.7 空試験 空試験は,JIS Z 2616の7.2(6)(空試験)に準じる。
16.8 計算 計算は,JIS Z 2616の7.7(6)(計算)による。
17. 不揮発分定量方法
17.1 定量方法 不揮発分の定量方法は,揮発分分離重量法による。この方法は,不揮発分含有率0.002%
(m/m) 以上の試料に適用する。
17.2 要旨 試料を加熱して酸化物にし,塩化水素を通じて揮発分を揮発させ,残分の質量をはかる。
17.3 試薬 試薬は,次による。
a) 塩酸
b) 硫酸
c) 液化塩化水素
17.4 装置 不揮発分定量装置は,通常図4に示すものを用いる。
不揮発分定量装置は,塩化水素発生部と反応部とからなる。
a) 塩化水素発生部(39) 塩化水素発生部は,塩酸を入れた塩化水素発生用フラスコ (a),塩化水素を発生
させるために滴下する硫酸を入れた硫酸滴下漏斗 (b),発生した塩化水素を清浄にするための硫酸を
入れた塩化水素洗浄瓶 (e) 及び塩化水素流量計 (f) からなる。
注(39) 液化塩化水素ボンベを用いてもよい。
b) 反応部 反応部は,電気抵抗加熱炉 (h),反応管 (i) 及び熱電温度計 (j) からなり,炉の中央部にお
いて約800℃の温度を保つことのできるもので,反応管 (i) の加熱中央部の温度を熱電温度計 (j) を
用いて測定できるもの。
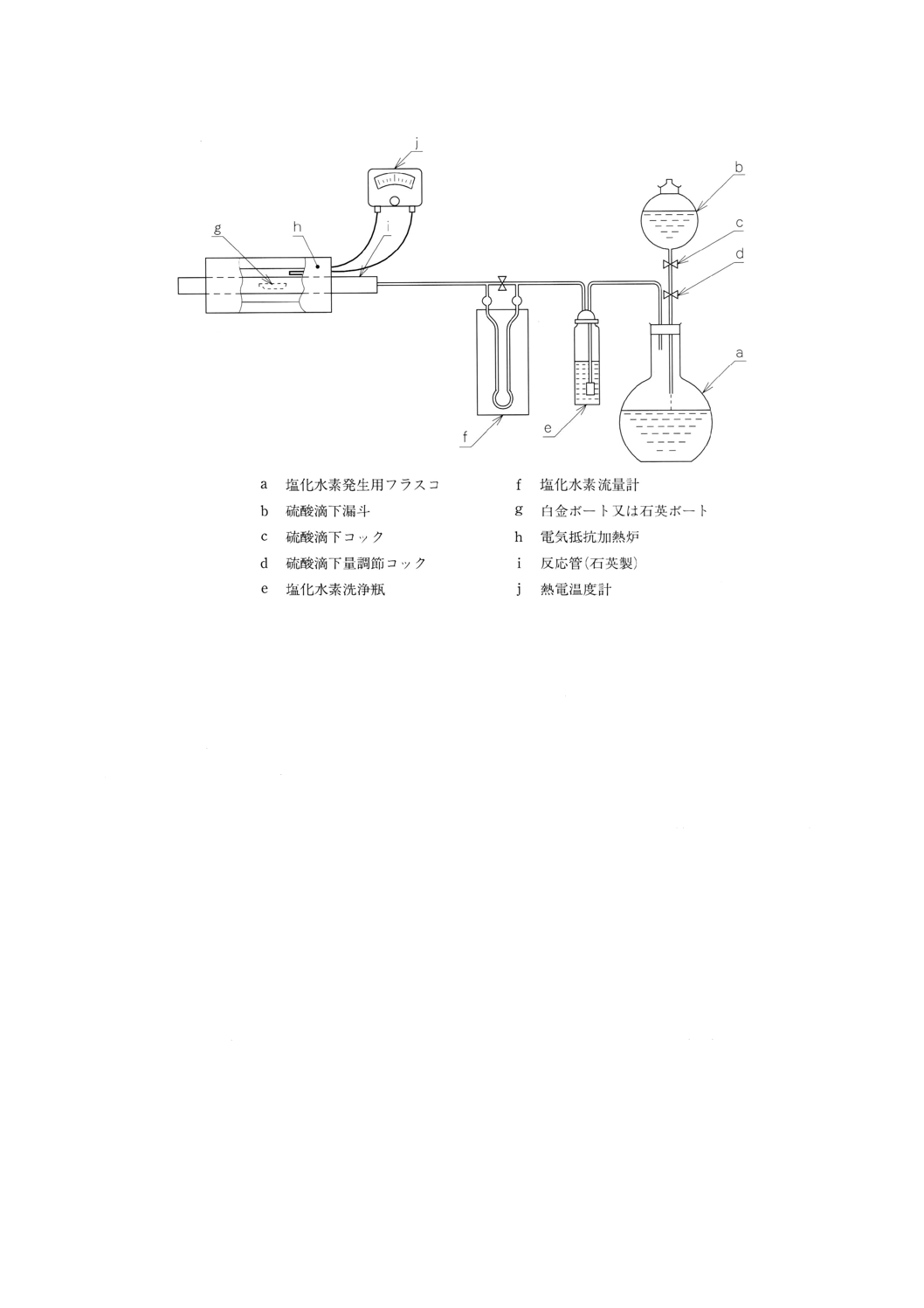
39
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図4 不揮発分定量装置の例
17.5 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
17.6 操作
17.6.1 準備操作 不揮発分定量装置 [17.4] の電気抵抗加熱炉 (h) に通電して反応管 (i) を加熱し,管内
温度を750〜800℃(21)に保持する。
17.6.2 試料の酸化 試料の酸化は,次のいずれかによる。
a) 白金皿又は磁器るつぼ中で行う場合 試料をはかり取って,白金皿(50番又は75番)又は磁器るつ
ぼ(A形50ml)に移し入れ750〜800℃の温度で約1時間加熱し完全に酸化する。室温まで放冷した
後,酸化物を白金ボート又は石英ボート (g) に移し入れ17.6.1で昇温した反応管 (i) の中央部に挿入
する。
b) 白金ボート又は石英ボート中で行う場合 試料をはかり取って,白金ボート又は石英ボート (g) に移
し入れ17.6.1で昇温した反応管 (i) の中央部に挿入し,約1時間加熱して酸化する(40)。
注(40) 試料によっては酸化の過程でボートからあふれ出るものがあるので,あふれ出ていないか確認
する。あふれ出る場合の試料の酸化は,a)による。
17.6.3 揮発分の揮発 揮発分の揮発は,次の手順によって行う。
a) 塩化水素発生部 [17.4 a)](39)と反応管 (i) とを接続し,塩化水素発生用フラスコ (a) の塩酸中に滴下漏
斗 (b) のコック (c) 及び (d) を開いて硫酸を滴下し,反応管 (i) の断面積 (cm2) 当たり20〜40ml/
分になるようにコック (d) を調節して,塩化水素を発生させる(41)。
注(41) 塩化水素発生部に塩化水素ボンベを用いる場合には,ボンベと塩化水素流量計 (f) とを接続し,
反応管 (i) の断面積 (cm2) 当たり20〜40ml/分になるように塩化水素流量を調整する。
b) 塩化水素を流してから1.5〜2時間経過して揮発が完結したことを確かめた後(42),ボートを取り出し,
40
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
デシケーターに移し入れ室温まで放冷する。
注(42) 三酸化タングステンによる黄色が認められないことを確かめる。
17.6.4 ひょう量 ひょう量は,次の手順によって行う。
a) 17.6.3 b)で室温まで放冷したボートの質量を0.1mgのけたまではかる。
b) ボートの内容物を羽毛で払い出した後,ボートの質量を0.1mgのけたまではかる。
17.7 計算 試料中の不揮発分含有率を,次の式によって算出する。
100
0
2
1
×
−
=
m
m
m
NVR
ここに,
NVR: 試料中の不揮発分含有率 [% (m/m)]
m1: 17.6.4 a)で得た質量 (g)
m2: 17.6.4 b)で得た質量 (g)
m0: 試料はかり取り量 (g)
付図1 黒鉛るつぼの例
41
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図3 窒素と一酸化炭素用赤外線検出器とを使用する装置の概略
42
H 1402 : 2001
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図4 アルゴンと一酸化炭素用赤外線検出器とを使用する装置の概略
タングステン・モリブデン関係JIS原案策定委員会 構成表
氏名
所属
(委員長)
辻 川 正 弘
日本工業標準調査会臨時委員
(委員)
橋 本 進
財団法人日本規格協会技術部
藤 井 忠 行
科学技術庁金属材料研究所
菊 地 正
山口東京理科大学基礎工学部
杉 浦 稔
岩崎電気株式会社
斉 藤 武 志
東芝タンガロイ株式会社
石 塚 昌 泰
東芝ライテック株式会社
瀬 戸 啓 之
東京タングテスン株式会社
鮫 島 進 一
日本新金属株式会社
荒 木 敏 春
株式会社東芝
鮎 川 昇
日本タングステン株式会社
(分科会)
仙 場 謙 次
日本タングステン株式会社
秋 吉 直 義
東邦金属株式会社
堀 田 幸 男
松下電子工業株式会社
山 口 悟
株式会社東芝
国 本 宏
東京タングステン株式会社
福 田 政 則
日本新金属株式会社
仲 田 公 夫
東邦金属株式会社
幸 本 京 一
東邦金属株式会社
安 宅 とも子
松下電子工業株式会社
児 玉 吉 弘
松下電子工業株式会社
内 藤 光 博
東芝電子エンジニアリング株式会社
(事務局)
小 泉 英 雄
タングステン・モリブデン工業会