H 1306 : 1999
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。これによってJIS H 1306 : 1992は改正され,この規格に置き換えられる。
今回の改正では,原子吸光法による定量9元素のうち,マグネシウム及び亜鉛について国際規格ISO
3256 : 1977, Aluminium and aluminium alloys−Determination of magnesium−Atomic absorption
spectrophotometric method(アルミニウム及びアルミニウム合金−マグネシウムの定量−原子吸光法)及び
ISO 5194 : 1981, Aluminium and aluminium alloys−Determination of zinc content−Flame atomic absorption
spectrophotometric methodと整合させた。また,国際規格に規定されていない鉄,マンガン及びビスマスの
定量方法を追加規定している。
なお,ISO 3980(銅の定量),ISO 3981(ニッケルの定量),ISO 4192(鉛の定量)及びISO 4193(クロ
ムの定量)の原子吸光分析方法の4元素の定量方法については,試料の分解に水銀を使用しているために
採択せず,別方法を規定している。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
H 1306 : 1999
アルミニウム及びアルミニウム合金の
原子吸光分析方法
Methods for atomic absorption spectrometric
analysis of aluminium and aluminium alloys
序文 この規格は,1977年に第1版として発行されたISO 3256, Aluminium and aluminium alloys−
Determination of magnesium−Atomic absorption spectrophotometric method, ISO 3980, Aluminium and
aluminium alloys−Determination of copper−Atomic absorption spectrophotometric method, ISO 3981,
Aluminium and aluminium alloys−Determination of nickel−Atomic absorption spectrophotometric method,及び
1981年に第1版として発行されたISO 4192, Aluminium and aluminium alloys−Determination of lead content
−Flame atomic absorption spectrophotometric method, ISO 4193, Aluminium and aluminium alloys−
Determination of chromium content−Flame atomic absorption spectrophotometric method, ISO 5194, Aluminium
and aluminium alloys−Determination of zinc content−Flame atomic absorption spectrophotometric methodが対応
国際規格としてあるが,ISO 3980,ISO 3981,ISO 4192及びISO 4193の4規格は,試料分解時に水銀を
使用しているため採択せず,ISO 3256及びISO 5194の2規格の対応する部分については技術的内容を変
更することなく作成した日本工業規格である。
なお,国際規格が制定されていない鉄,マンガン,ビスマス及び対応する国際規格が制定されているが,
前記の理由で採択しなかった銅,ニッケル,鉛,クロムの原子吸光法の七つの定量方法を日本工業規格と
して追加規定している。
1. 適用範囲 この規格は,アルミニウム及びアルミニウム合金の原子吸光分析方法について規定する。
備考 この規格の対応国際規格を,次に示す。
ISO 3256 : 1977 Aluminium and aluminium alloys−Determination of magnesium−Atomic
absorption spectrophotometric method
ISO 3980 : 1977 Aluminium and aluminium alloys−Determination of copper−Atomic absorption
spectrophotometric method
ISO 3981 : 1977 Aluminium and aluminium alloys−Determination of nickel−Atomic absorption
spectrophotometric method
ISO 4192 : 1981 Aluminium and aluminium alloys−Determination of lead content−Flame atomic
absorption spectrophotometric method
ISO 4193 : 1981 Aluminium and aluminium alloys−Determination of chromium content−Flame
atomic absorption spectrophotometric method
ISO 5194 : 1981 Aluminium and aluminium alloys−Determination of zinc content−Flame atomic

2
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
absorption spectrophotometric method
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
JIS H 1351 アルミニウム及びアルミニウム合金の分析方法通則
JIS K 0121 原子吸光分析通則
3. 一般事項 分析方法に共通な一般事項は,JIS H 1351及びJIS K 0121の規定による。
4. 定量元素及び定量範囲 定量元素及び定量範囲は,表1による。
表1 定量元素及び定量範囲
定量元素
定量範囲
% (m/m)
鉄
0.005以上 1.5以下
銅
0.005以上 5.0以下
マンガン
0.005以上 1.5以下
亜鉛
0.005以上 6.0以下
マグネシウム
0.005以上 5.0以下
クロム
0.01 以上 0.5以下
ニッケル
0.005以上 3.0以下
ビスマス
0.1
以上 1.0以下
鉛
0.1
以上 1.0以下
備考 表1の亜鉛の定量範囲は,精度及び正確度に
十分注意をすることによって,定量下限を
0.002% (m/m) まで拡大してもよい。
5. 鉄定量方法
5.1
要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
5.2
試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,鉄を含有しないもの又は鉄含有率が既知で,
できるだけ低いもの。
e) 過酸化水素
f)
すず溶液 すず[99.9% (m/m) 以上]0.1gをはかり取ってビーカー (200ml) に移し入れ,時計皿で覆
い,塩酸 (1+1) 30mlを加え,白金を接触させながら50〜80℃に加熱して分解する。常温まで冷却し
た後,時計皿の下面及びビーカーの内壁を水で洗って時計皿を取り除き,塩酸 (1+5) で液量を100ml
とする。
g) ニッケル溶液 ニッケル[99.9% (m/m) 以上]0.1gをはかり取ってビーカー (200ml) に移し入れ,時
計皿で覆い,硝酸 (1+1) 10mlを加え,穏やかに加熱して分解する。常温まで冷却した後,時計皿の

3
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
下面及びビーカーの内壁を水で洗って時計皿を取り除き,水で液量を100mlとする。
h) 標準鉄溶液A (1 000μgFe/ml) 鉄[99.9% (m/m) 以上]1.000gをはかり取ってビーカー (200ml) に移
し入れ,時計皿で覆い,硝酸 (1+1) 30mlを加え,穏やかに加熱して分解する。常温まで冷却した後,
時計皿の下面及びビーカーの内壁を水で洗浄し,時計皿を取り除く。溶液を1 000mlの全量フラスコ
に水を用いて移し入れ,水で標線まで薄めて標準鉄溶液Aとする。
i)
標準鉄溶液B (100μgFe/ml) 標準鉄溶液A [h)] を使用の都度,必要量だけ水で正確に10倍に薄めて
標準鉄溶液Bとする。
5.3
試料はかり取り量 試料はかり取り量は,1.00gとし,1mgのけたまではかる。
5.4
操作
5.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する(1)。過酸化水素1mlを加え(2),
加熱して試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水を
用いて洗浄し,時計皿を取り除く。溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線ま
で薄める(5)。
c) 溶液を試料中の鉄含有率に応じて表2に従って100mlの全量フラスコに分取し,水で標線まで薄める。
表2 分取量
試料中の鉄含有率
% (m/m)
分取量
ml
0.1以上 0.5未満
50.0
0.5以上 1.5以下
20.0
注(1) 試料が分解しにくい場合には,すず溶液 [5.2f)] 2m1又はニッケル溶液 [5.2g)] 2mlを加える。
(2) 銅含有率が高い場合には,金属銅が残ることがあるので,更に過酸化水素を加える。
(3) 不溶解物を認めた場合には,ろ紙(5種B)でこし分け,温水で洗浄した後,ろ液及び洗液を合
わせて保存する。ろ紙及び不溶解物を白金るつぼ(20番)に移し入れ,徐々に温度を上げ,
700~800℃で加熱し,ろ紙を完全に灰化した後,放冷する。硝酸 (1+1) 5mlを加え,ふっ化水
素酸約3mlを少量ずつ加えて不溶解物を完全に分解し,更に加熱して乾固した後,塩酸 (1+1)
数滴を加えて塩類を溶解し,溶液を少量の水を用いて保存しておいたろ液及び洗液に合わせる。
(4) 過酸化水素が残存すると低値を示すことがあるので,十分に加熱しなければならない。
(5) 試料中の鉄含有率が0.005% (m/m) 以上0.1% (m/m) 未満の場合には,次のc)の操作は行わない。
5.4.2
吸光度の測定 5.4.1のb)又はc)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度
計の空気・アセチレンフレーム中に噴霧し,波長248.3nm又は372.0nmにおける吸光度を測定する。
5.5
空試験 5.6の操作において得られる標準鉄溶液を添加しない溶液の吸光度を,空試験の吸光度とす
る。
5.6
検量線の作成 アルミニウム [5.2d)] を1.00gずつはかり取って数個のビーカー (300ml) に移し入
れ,試料中の鉄含有率に応じて,表3に従って標準鉄溶液を段階的に加える。以下,5.4.1b)〜5.4.2の手順
に従って試料と同じ操作を試料と並行して行い,得た吸光度と標準鉄溶液として加えた鉄量(6)との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
注(6) 5.4.1c)の操作を行った場合には,分取した溶液中の標準鉄溶液として加えた鉄量 (g)。

4
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3 標準鉄溶液添加量
試料中の鉄含有率
% (m/m)
使用する標準鉄溶液
標準鉄溶液添加量
ml
0.005以上 0.1未満
B [5.2i)]
0〜10.0
0.1
以上 0.5未満
A [5.2h)]
0〜 5.0
0.5
以上 1.5以下
A [5.2h)]
0〜15.0
5.7
計算 計算は,次のいずれかによる。
a) 5.4.1c)の操作を行わなかった場合 5.4.2及び5.5で得た吸光度と5.6で作成した検量線とから鉄量を
求め,試料中の鉄含有率を,次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Fe
ここに, Fe: 試料中の鉄含有率 [% (m/m)]
A1: 試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
A3: アルミニウム [5.2d)] 1.00g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
b) 5.4.1c)の操作を行った場合 5.4.2及び5.5で得た吸光度と5.6で作成した検量線とから鉄量を求め,
試料中の鉄含有率を,次の式によって算出する。
100
100
)
(
3
2
1
×
+
×
−
=
m
A
B
A
A
Fe
ここに, Fe: 試料中の鉄含有率 [% (m/m)]
A1: 分取した試料溶液中の鉄検出量 (g)
A2: 分取した空試験液中の鉄検出量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
A3: アルミニウム [5.2d)] 1.00g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
6. 銅定量方法
6.1
要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
6.2
試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,銅を含有しないもの又は銅含有率が既知で,
できるだけ低いもの。
e) 過酸化水素
f)
すず溶液 5.2f)による。
g) ニッケル溶液 5.2g)による。
h) 標準銅溶液A (1 000μgCu/ml) 銅[99.9% (m/m) 以上]1.000gをはかり取ってビーカー (200ml) に移
し入れ,時計皿で覆い,硝酸 (1+1) 30mlを加え,穏やかに加熱して分解する常温まで冷却した後,
時計皿の下面及びビーカーの内壁を水で洗浄し,時計皿を取り除く。溶液を1 000mlの全量フラスコ
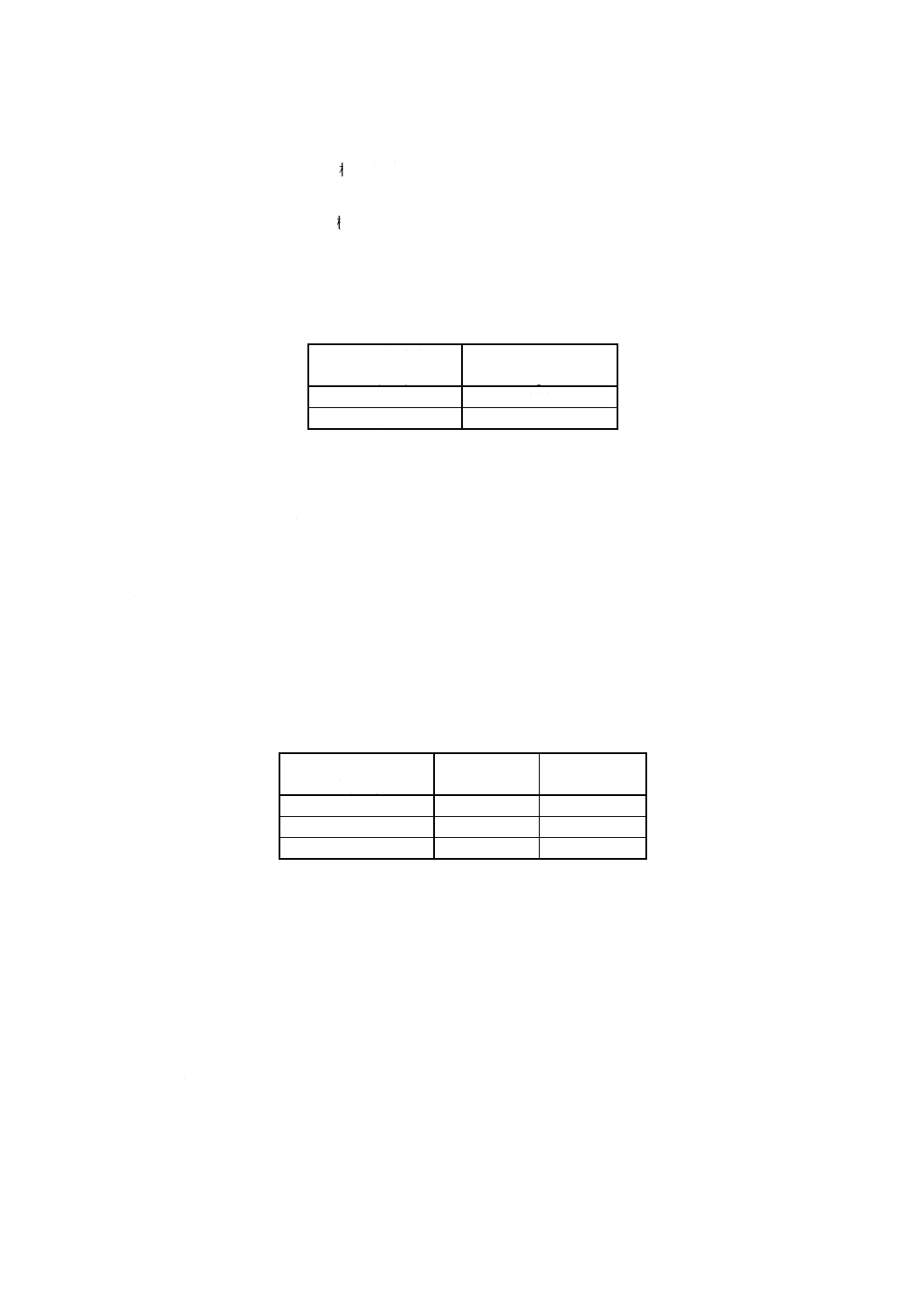
5
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に水を用いて移し入れ,水で標線まで薄めて標準銅溶液Aとする。
i)
標準銅溶液B (500μgCu/ml) 標準銅溶液A [h)] を使用の都度,必要量だけ水で正確に2倍に薄めて
標準銅溶液Bとする。
j)
標準銅溶液C (100μgCu/ml) 標準銅溶液A [h)] を使用の都度,必要量だけ水で正確に10倍に薄めて
標準銅溶液Cとする。
6.3
試料はかり取り量 試料はかり取り量は,試料中の銅含有率に応じて表4に従い,1mgのけたまで
はかる。
表4 試料はかり取り量
試料中の銅含有率
% (m/m)
試料はかり取り量
g
0.005以上 1.5未満
1.00
1.5 以上 5.0未満
0.20
6.4
操作
6.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する(1)。過酸化水素1mlを加え(2),
加熱して試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水を
用いて洗浄し,時計皿を取り除く。溶液を試料中の銅含有率が1.5% (m/m) 未満の場合には100mlの
全量フラスコに,1.5% (m/m) 以上の場合には200mlの全量フラスコに水を用いて移し入れ,水で標線
まで薄める(7)。
c) 溶液を試料中の銅含有率に応じて表5に従って分取し,表5に示した全量フラスコに移し入れ,水で
標線まで薄める。
注(7) 試料中の銅含有率が0.005% (m/m) 以上0.1% (m/m) 未満の場合には,次のc)の操作は行わない。
表5 分取量
銅含有率
% (m/m)
分取量
ml
全量フラスコ
ml
0.1以上 0.5未満
50.0
100
0.5以上 1.5未満
20.0
100
1.5以上 5.0以下
20.0
200
6.4.2
吸光度の測定 6.4.1のb)又はc)で得た溶液の一部を水を用いてゼロ点を調整した原子吸光光度計
の空気・アセチレンフレーム中に噴霧し,波長324.8nm又は327.4nmにおける吸光度を測定する。
6.5
空試験 6.6の操作において得られる標準銅溶液を添加しない溶液の吸光度を,空試験の吸光度とす
る。
6.6
検量線の作成 アルミニウム [6.2d)] を試料中の銅含有率に応じて表4の試料はかり取り量と同じ
になるようにはかり取って,数個のビーカー (300ml) に移し入れ,試料中の銅含有率に応じて表6に従い,
標準銅溶液を段階的に加える。以下,6.4.1b)〜6.4.2の手順に従って試料と同じ操作を試料と並行して行い,
得た吸光度と標準銅溶液として加えた銅量(8)との関係線を作成し,その関係線を原点を通るように平行移
動して検量線とする。
注(8) 6.4.1c)の操作を行った場合には,分取した溶液中の標準銅溶液として加えた銅量 (g)。

6
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表6 標準銅溶液添加量
試料中の銅含有率
% (m/m)
使用する標準銅溶液
標準銅溶液添加量
ml
0.005以上 0.1未満
C [6.2j)]
0〜10.0
0.1
以上 0.5未満
A [6.2h)]
0〜 5.0
0.5
以上 1.5未満
A [6.2h)]
0〜15.0
1.5
以上 5.0以下
B [6.2i)]
0〜20.0
6.7
計算 計算は,次のいずれかによる。
a) 6.4.1c)の操作を行わなかった場合 6.4.2及び6.5で得た吸光度と6.6で作成した検量線とから銅量を
求め,試料中の銅含有率を,次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Cu
ここに, Cu: 試料中の銅含有率 [% (m/m)]
A1: 試料溶液中の銅検出量 (g)
A2: 空試験液中の銅検出量 (g)
A3: 6.6ではかり取ったアルミニウム [6.2d)] 中に含まれる銅量
(g)
m: 試料はかり取り量 (g)
b) 6.4.1c)の操作を行った場合 6.4.2及び6.5で得た吸光度と6.6で作成した検量線とから銅量を求め,
試料中の銅含有率を,次の式によって算出する。
100
)
(
3
2
1
×
+
×
−
=
m
A
B
C
A
A
Cu
ここに, Cu: 試料中の銅含有率 [% (m/m)]
A1: 分取した試料溶液中の銅検出量 (g)
A2: 分取した空試験液中の銅検出量 (g)
C: 6.4.1b)で使用した全量フラスコの容量 (ml)
B: 試料溶液及び空試験液の分取量 (ml)
A3: 6.6ではかり取ったアルミニウム [6.2d)] 中に含まれる銅量
(g)
m: 試料はかり取り量 (g)
7. マンガン定量方法
7.1
要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
7.2
試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,マンガンを含有しないもの又はマンガン含有
率が既知で,できるだけ低いもの。
e) 過酸化水素
f)
すず溶液 5.2f)による。
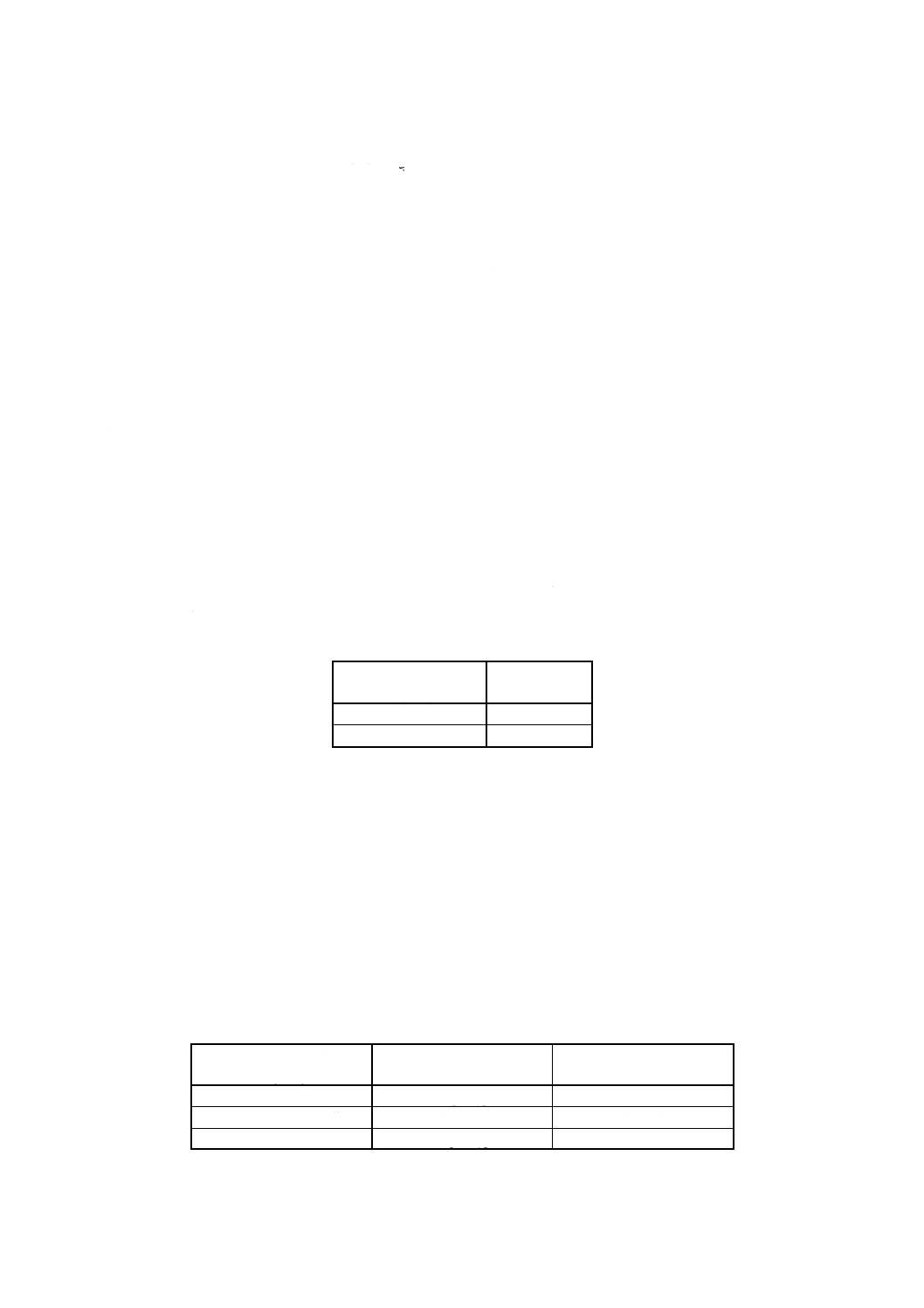
7
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) ニッケル溶液 5.2g)による。
h) 標準マンガン溶液A (1 000μgMn/ml) マンガン[99.9% (m/m) 以上]1.000gをはかり取ってビーカー
(200ml) に移し入れ,時計皿で覆い,塩酸 (1+1) 30mlを加えて穏やかに加熱して分解する。常温まで
冷却した後,時計皿の下面及びビーカーの内壁を水で洗浄し,時計皿を取り除く。溶液を1 000mlの
全量フラスコに水を用いて移し入れ,水で標線まで薄めて標準マンガン溶液Aとする。
i)
標準マンガン溶液B (100μgMn/ml) 標準マンガン溶液A [h)] を使用の都度,必要量だけ水で正確に
10倍に薄めて標準マンガン溶液Bとする。
7.3
試料はかり取り量 試料はかり取り量は,1.00gとし,1mgのけたまではかる。
7.4
操作
7.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する(1)。過酸化水素1mlを加え(2),
加熱して試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水を
用いて洗浄し,時計皿を取り除く。溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線ま
で薄める(9)。
c) 溶液を試料中のマンガン含有率に応じて表7に従って100mlの全量フラスコに分取し,水で標線まで
薄める。
注(9) 試料中のマンガン含有率が0.005% (m/m) 以上0.1% (m/m) 未満の場合には,次のc)の操作は行
わない。
表7 分取量
試料中のマンガン含有率
% (m/m)
分取量
ml
0.1以上 0.5未満
50.0
0.5以上 1.5以下
20.0
7.4.2
吸光度の測定 7.4.1のb)又はc)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度
計の空気・アセチレンフレーム中に噴霧し,波長279.5nmにおける吸光度を測定する。
7.5
空試験 7.6の操作において得られる標準マンガン溶液を添加しない溶液の吸光度を,空試験の吸光
度とする。
7.6
検量線の作成 アルミニウム [7.2d)] を1.00gずつはかり取って数個のビーカー (300ml) に移し入
れ,試料中のマンガン含有率に応じて表8に従い,標準マンガン溶液を段階的に加える。以下,7.4.1b)〜
7.4.2の手順に従って試料と同じ操作を試料と並行して行い,得た吸光度と標準マンガン溶液として加えた
マンガン量(10)との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
注(10) 7.4.1c)の操作を行った場合には,分取した溶液中の標準マンガン溶液として加えたマンガン量
(g)。
表8 標準マンガン溶液添加量
試料中のマンガン含有率
% (m/m)
使用する標準マンガン溶液
標準マンガン溶液添加量
ml
0.005以上 0.1未満
B [7.2i)]
0〜10.0
0.1
以上 0.5未満
A [7.2h)]
0〜 5.0
0.5
以上 1.5以下
A [7.2h)]
0〜15.0
7.7. 計算 計算は,次のいずれかによる。
8
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 7.4.1c)の操作を行わなかった場合 7.4.2及び7.5で得た吸光度と7.6で作成した検量線とからマンガ
ン量を求め,試料中のマンガン含有率を,次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Mn
ここに, Mn: 試料中のマンガン含有率 [% (m/m)]
A1: 試料溶液中のマンガン検出量 (g)
A2: 空試験液中のマンガン検出量 (g)
A3: アルミニウム [7.2d)] 1.00g中に含まれるマンガン量 (g)
m: 試料はかり取り量 (g)
b) 7.4.1c)の操作を行った場合 7.4.2及び7.5で得た吸光度と7.6で作成した検量線とからマンガン量を
求め,試料中のマンガン含有率を,次の式によって算出する。
100
100
)
(
3
2
1
×
+
×
−
=
m
A
B
A
A
Mn
ここに, Mn: 試料中のマンガン含有率 [% (m/m)]
A1: 分取した試料溶液中のマンガン検出量 (g)
A2: 分取した空試験液中のマンガン検出量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
A3: アルミニウム [7.2d)] 1.00g中に含まれるマンガン量 (g)
m: 試料はかり取り量 (g)
8. 亜鉛定量方法
8.1
要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
8.2
試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,亜鉛を含有しないもの又は亜鉛含有率が既知
で,できるだけ低いもの。
e) 過酸化水素
f)
すず溶液 5.2f)による。
g) ニッケル溶液 5.2g)による。
h) 標準亜鉛溶液A (1 000μgZn/ml) 亜鉛[99.9% (m/m) 以上]1.000gをはかり取ってビーカー (200ml) に
移し入れ,時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解するまで冷却した後,時
計皿の下面及びビーカーの内壁を水で洗浄し,時計皿を取り除く。溶液を1 000mlの全量フラスコに
水を用いて移し入れ,水で標線まで薄めて標準亜鉛溶液Aとする。
i)
標準亜鉛溶液B (100μgZn/ml) 標準亜鉛溶液A [h)] を使用の都度,必要量だけ水で正確に10倍に薄
めて標準亜鉛溶液Bとする。
8.3
試料はかり取り量 試料はかり取り量は,試料中の亜鉛含有率に応じて表9に従い,1mgのけたま
ではかる。
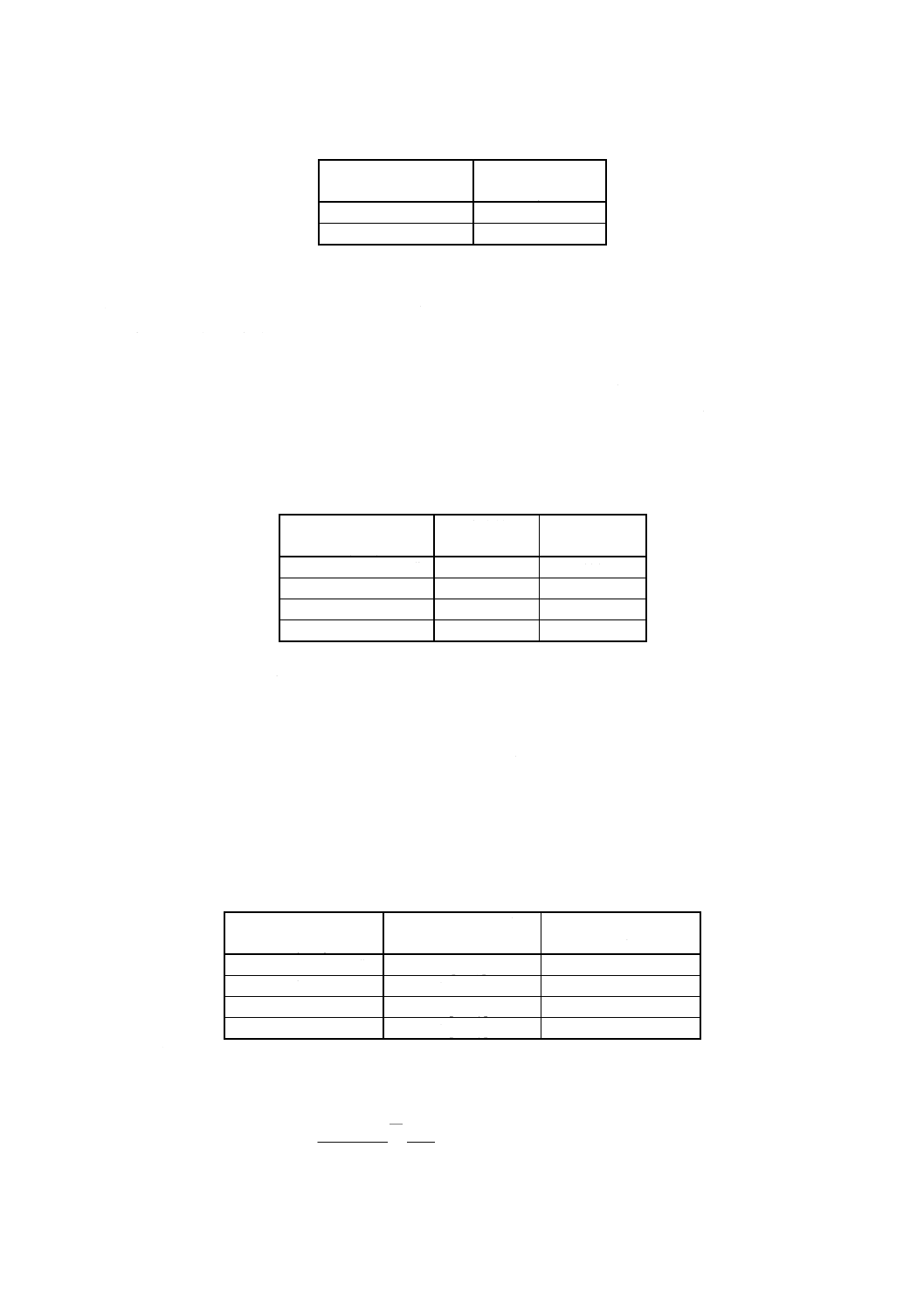
9
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表9 試料はかり取り量
試料中の亜鉛含有率
% (m/m)
試料はかり取り量
g
0.005以上 1.5未満
1.00
1.5
以上 6.0以下
0.20
8.4
操作
8.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,加熱して分解する(1)。過酸化水素1mlを加え(2),加熱して
試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水で洗浄し,
時計皿を取り除く。溶液を試料中の亜鉛含有率が1.5% (m/m) 未満の場合は100mlの全量フラスコに,
1.5% (m/m) 以上の場合には200mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
c) 溶液を試料中の亜鉛含有率に応じて表10に従って分取し,表10に示した全量フラスコに移し入れ,
水で標線まで薄める。
表10 分取量
試料中の亜鉛含有率
% (m/m)
分取量
ml
全量フラスコ
ml
0.005以上 0.1未満
50.0
100
0.1
以上 0.5未満
20.0
100
0.5
以上 1.5未満
10.0
100
1.5
以上 6.0以下
20.0
200
8.4.2
吸光度の測定 8.4.1c)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・
アセチレンフレーム中に噴霧し,波長213.9nmにおける吸光度を測定する。
8.5
空試験 8.6の操作において得られる標準亜鉛溶液を添加しない溶液の吸光度を,空試験の吸光度と
する。
8.6
検量線の作成 アルミニウム [8.2d)] を試料中の亜鉛含有率に応じて表9の試料はかり取り量と同
じになるようにはかり取って,数個のビーカー (300ml) に移し入れ,試料中の亜鉛含有率に応じて表11
に従って標準亜鉛溶液を段階的に加える。以下,8.4.1b)〜8.4.2の手順に従って試料と同じ操作を試料と並
行して行い,得た吸光度と分取した溶液中の標準亜鉛溶液として加えた亜鉛量との関係線を作成し,その
関係線を原点を通るように平行移動して検量線とする。
表11 標準亜鉛溶液添加量
試料中の亜鉛含有率
% (m/m)
使用する標準亜鉛溶液
標準亜鉛溶液添加量
ml
0.005以上 0.1未満
B [8.2i)]
0〜10.0
0.1 以上 0.5未満
A [8.2h)]
0〜 5.0
0.5 以上 1.5未満
A [8.2h)]
0〜15.0
1.5 以上 6.0以下
A [8.2h)]
0〜12.0
8.7
計算 8.4.2及び8.5で得た吸光度と8.6で作成した検量線とから亜鉛量を求め,試料中の亜鉛含有率
を,次の式によって算出する。
100
)
(
3
2
1
×
+
×
−
=
m
A
B
C
A
A
Zn
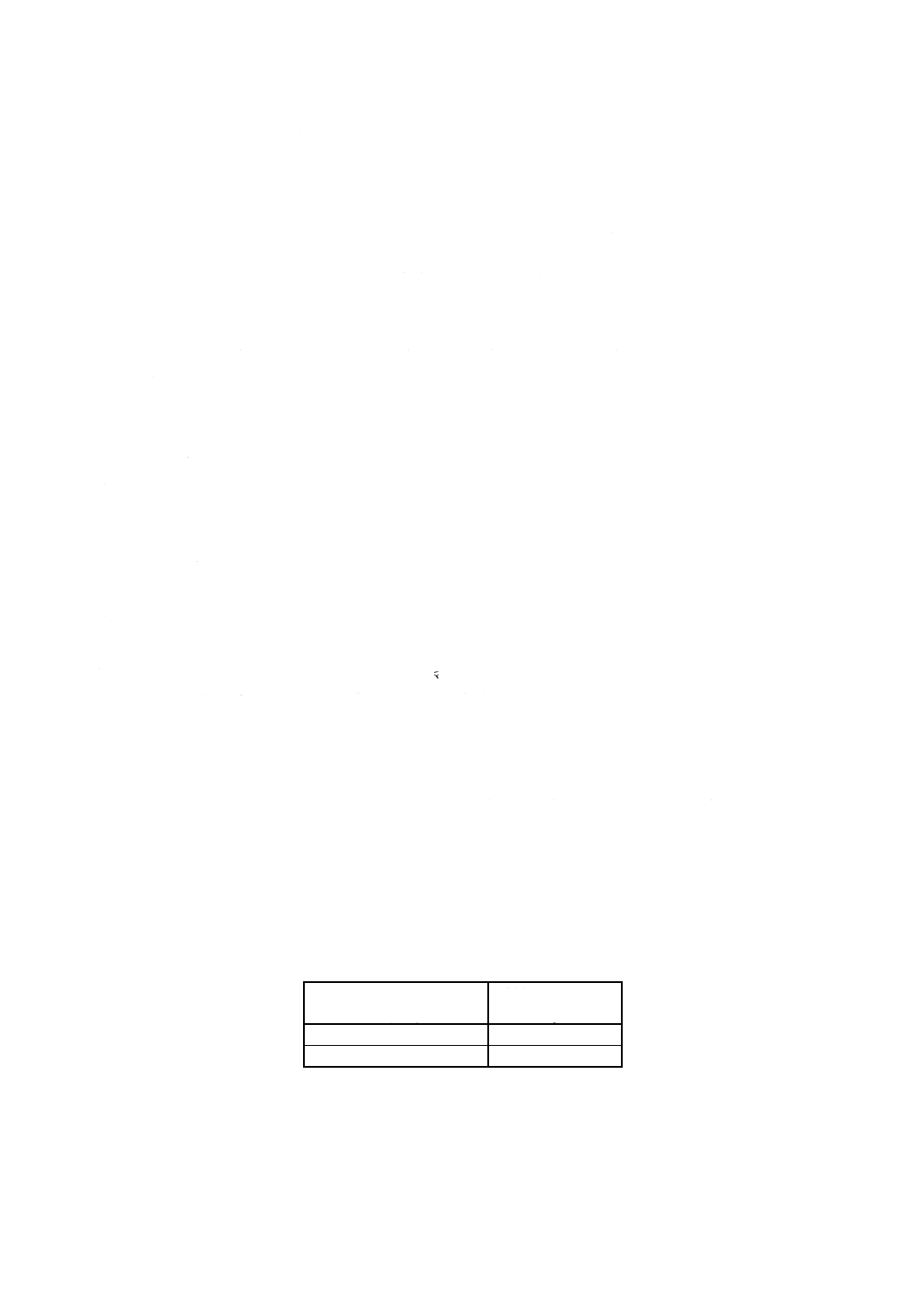
10
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに, Zn: 試料中の亜鉛含有率 [% (m/m)]
A1: 分取した試料溶液中の亜鉛検出量 (g)
A2: 分取した空試験液中の亜鉛検出量 (g)
C: 8.4.1b)で使用した全量フラスコの容量 (ml)
B: 試料溶液及び空試験液の分取量 (ml)
A3: 8.6ではかり取ったアルミニウム [8.2d)] 中に含まれる亜鉛
量 (g)
m: 試料はかり取り量 (g)
9. マグネシウム定量方法
9.1
要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
9.2
試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,マグネシウムを含有しないもの又はマグネシ
ウム含有率が既知で,できるだけ低いもの。
e) 過酸化水素
f)
ストロンチウム溶液 二塩化ストロンチウム六水和物30.0gを水に溶解し,水で液量を100mlとする。
g) すず溶液 5.2f)による。
h) ニッケル溶液 5.2g)による。
i)
標準マグネシウム溶液A (1 000μgMg/ml) マグネシウム[99.9% (m/m) 以上]1.000gをはかり取って
ビーカー (200ml) に移し入れ,時計皿で覆い,塩酸 (1+1) 30mlを少量ずつ加え,穏やかに加熱して
分解する。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水で洗浄し,時計皿を取り除く。
溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて標準マグネシウム溶液A
とする。
j)
標準マグネシウム溶液B (500μgMg/ml) 標準マグネシウム溶液A [i)] を使用の都度,必要量だけ水
で正確に2倍に薄めて標準マグネシウム溶液Bとする。
k) 標準マグネシウム溶液C (100μgMg/ml) 標準マグネシウム溶液A [i)] を使用の都度,必要量だけ水
で正確に10倍に薄めて標準マグネシウム溶液Cとする。
9.3
試料はかり取り量 試料はかり取り量は,試料中のマグネシウム含有率に応じて表12に従い,1mg
のけたまではかる。
表12 試料はかり取り量
試料中のマグネシウム含有率
% (m/m)
試料はかり取り量
g
0.005以上 1.5未満
1.00
1.5
以上 5.0以下
0.20
9.4
操作
9.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する(1)。過酸化水素1mlを加え(2),
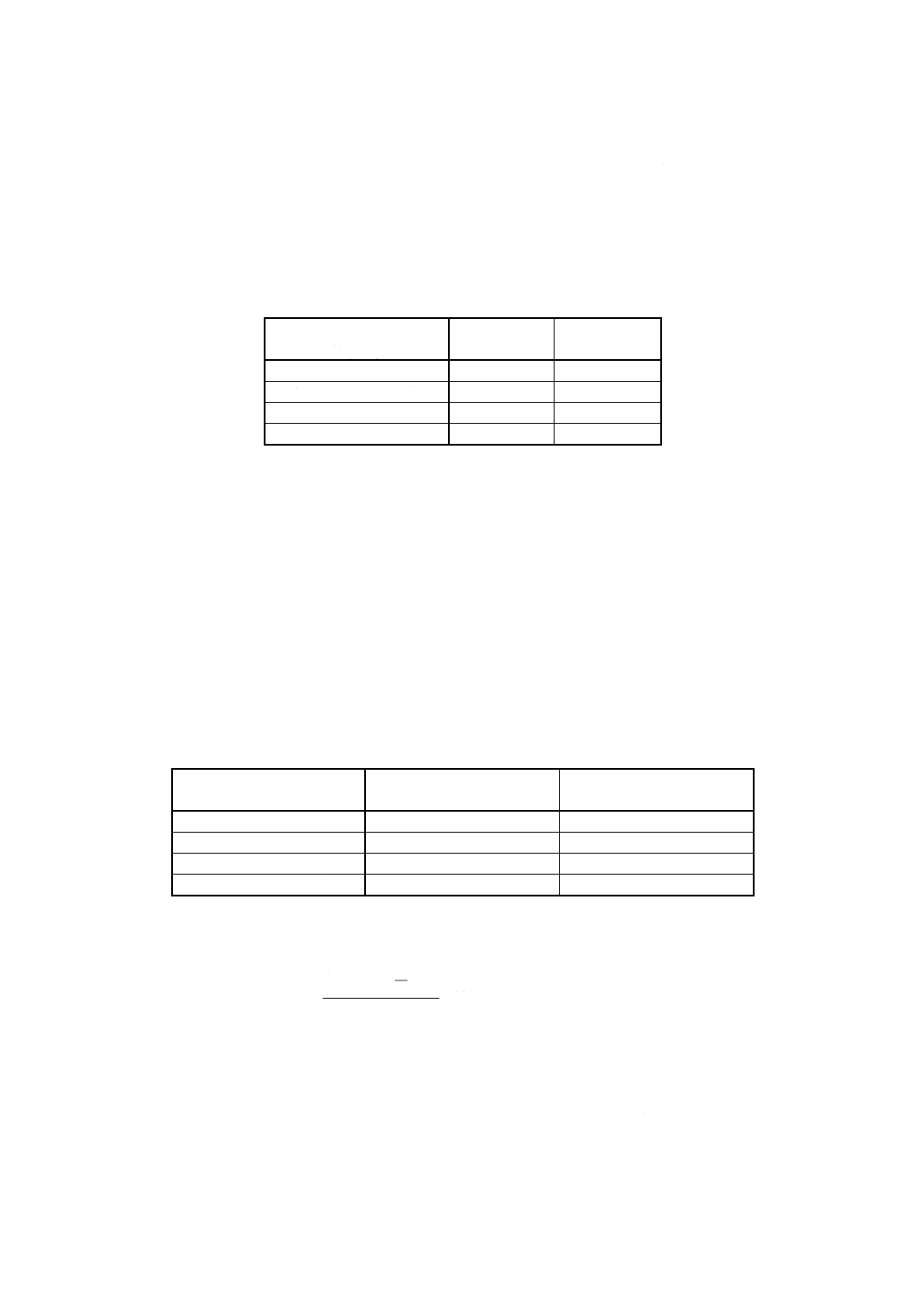
11
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
加熱して試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水で
洗浄し,時計皿を取り除く。溶液を試料中のマグネシウム含有率が1.5% (m/m) 未満の場合は100ml
の全量フラスコに,1.5% (m/m) 以上の場合には200mlの全量フラスコに水を用いて移し入れ,水で標
線まで薄める。
c) 溶液を試料中のマグネシウム含有率に応じて表13に従って分取し,表13に示した全量フラスコに移
し入れ,ストロンチウム溶液 [9.2f)] 2mlを加え(11),水で標線まで薄める。
表13 分取量
試料中のマグネシウム含有率
% (m/m)
分取量
ml
全量フラスコ
ml
0.005以上 0.1未満
20.0
100
0.1 以上 0.5未満
10.0
100
0.5 以上 1.5未満
5.0
100
1.5 以上 5.0以下
10.0
200
注(11) 分取した溶液中のアルミニウム量が0.1g以上の場合には,ストロンチウム溶液を加える必要は
ない。
9.4.2
吸光度の測定 9.4.1c)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・
アセチレンフレーム中に噴霧し,波長285.2nmにおける吸光度を測定する。
9.5
空試験 9.6の操作において得られる標準マグネシウム溶液を添加しない溶液の吸光度を,空試験の
吸光度とする。
9.6
検量線の作成 アルミニウム [9.2d)] を試料中のマグネシウム含有率に応じて表12の試料はかり取
り量と同じになるようにはかり取って,数個のビーカー (300ml) に移し入れ,試料中のマグネシウム含有
率に応じて表14に従い,標準マグネシウム溶液を段階的に加える。以下,9.4.1b)〜9.4.2の手順に従って
試料と同じ操作を試料と並行して行い,得た吸光度と分取した溶液中の標準マグネシウム溶液として加え
たマグネシウム量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
表14 標準マグネシウム溶液添加量
試料中のマグネシウム含有率
% (m/m)
使用する標準マグネシウム溶液 標準溶液マグネシウム添加量
ml
0.005以上 0.1未満
C [9.2k)]
0〜10.0
0.1
以上 0.5未満
A [9.2i)]
0〜 5.0
0.5
以上 1.5未満
A [9.2i)]
0〜15.0
1.5
以上 5.0以下
B [9.2j)]
0〜20.0
9.7
計算 9.4.2及び9.5で得た吸光度と9.6で作成した検量線とからマグネシウム量を求め,試料中のマ
グネシウム含有率を,次の式によって算出する。
100
)
(
3
2
1
×
+
×
−
=
m
A
B
C
A
A
Mg
ここに, Mg: 試料中のマグネシウム含有率 [% (m/m)]
A1: 分取した試料溶液中のマグネシウム検出量 (g)
A2: 分取した空試験液中のマグネシウム検出量 (g)
C: 9.4.1b)で使用した全量フラスコの容量 (ml)
B: 試料溶液及び空試験液の分取量 (ml)
A3: 9.6ではかり取ったアルミニウム [9.2.d)] 中に含まれるマグ
ネシウム量 (g)
12
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m: 試料はかり取り量 (g)
10. クロム定量方法
10.1 要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中又は酸化二窒素・アセチレンフレーム中に噴霧し,その吸光度を測定する。
10.2 試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,クロムを含有しないもの又はクロム含有率が
既知で,できるだけ低いもの。
e) 過酸化水素
f)
塩化アンモニウム溶液 (270g/1 000ml)
g) すず溶液 5.2f)による。
h) ニッケル溶液 5.2g)による。
i)
標準クロム溶液A (1 000μgCr/ml) クロム[99.9% (m/m) 以上]1.000gをはかり取ってビーカー
(200ml) に移し入れ,時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する。常温まで
冷却した後,時計皿の下面及びビーカーの内壁を水で洗浄し,時計皿を取り除く。溶液を1 000mlの
全量フラスコに水を用いて移し入れ,水で標線まで薄めて標準クロム溶液Aとする。
j)
標準クロム溶液B (100μgCr/ml) 標準クロム溶液A [i)] を使用の都度,必要量だけ水で正確に10倍
に薄めて標準クロム溶液Bとする。
10.3 試料はかり取り量 試料はかり取り量は,1.00gとし,1mgのけたまではかる。
10.4 操作
10.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,加熱して分解する(1)。過酸化水素1mlを加え(2),加熱して
試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水を用いて洗
浄し,時計皿を取り除く。溶液を100mlの全量フラスコに水を用いて移し入れ,塩化アンモニウム溶
液1.5mlを加え(12),水で標線まで薄める(13)。
c) この溶液50.0mlを100mlの全量フラスコに分取し,水で標線まで薄める。
注(12) 10.4.2で酸化二窒素・アセチレンフレームを用いて吸光度を測定する場合には,塩化アンモニウ
ムを加える必要はない。
(13) 試料中のクロム含有率が0.01% (m/m) 以上0.1% (m/m) 未満の場合には,次のc)の操作は行わ
ない。
10.4.2 吸光度の測定 10.4.1のb)又はc)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度
計の空気・アセチレンフレーム中又は酸化二窒素・アセチレンフレーム中に噴霧し,波長357.9nmにおけ
る吸光度を測定する。
10.5 空試験 10.6の操作において得られる標準クロム溶液を添加しない溶液の吸光度を,空試験の吸光
度とする。
13
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.6 検量線の作成 アルミニウム [10.2d)] を1.00gずつはかり取って数個のビーカー (300ml) に移し入
れ,試料中のクロム含有率に応じて表15に従って標準クロム溶液を段階的に加える。以下,10.4.1b)〜10.4.2
の手順に従って試料と同じ操作を試料と並行して行い,得た吸光度と標準クロム溶液として加えたクロム
量(14)との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
注(14) 10.4.1c)の操作を行った場合には,分取した溶液中の標準クロム溶液として加えたクロム量 (g)。
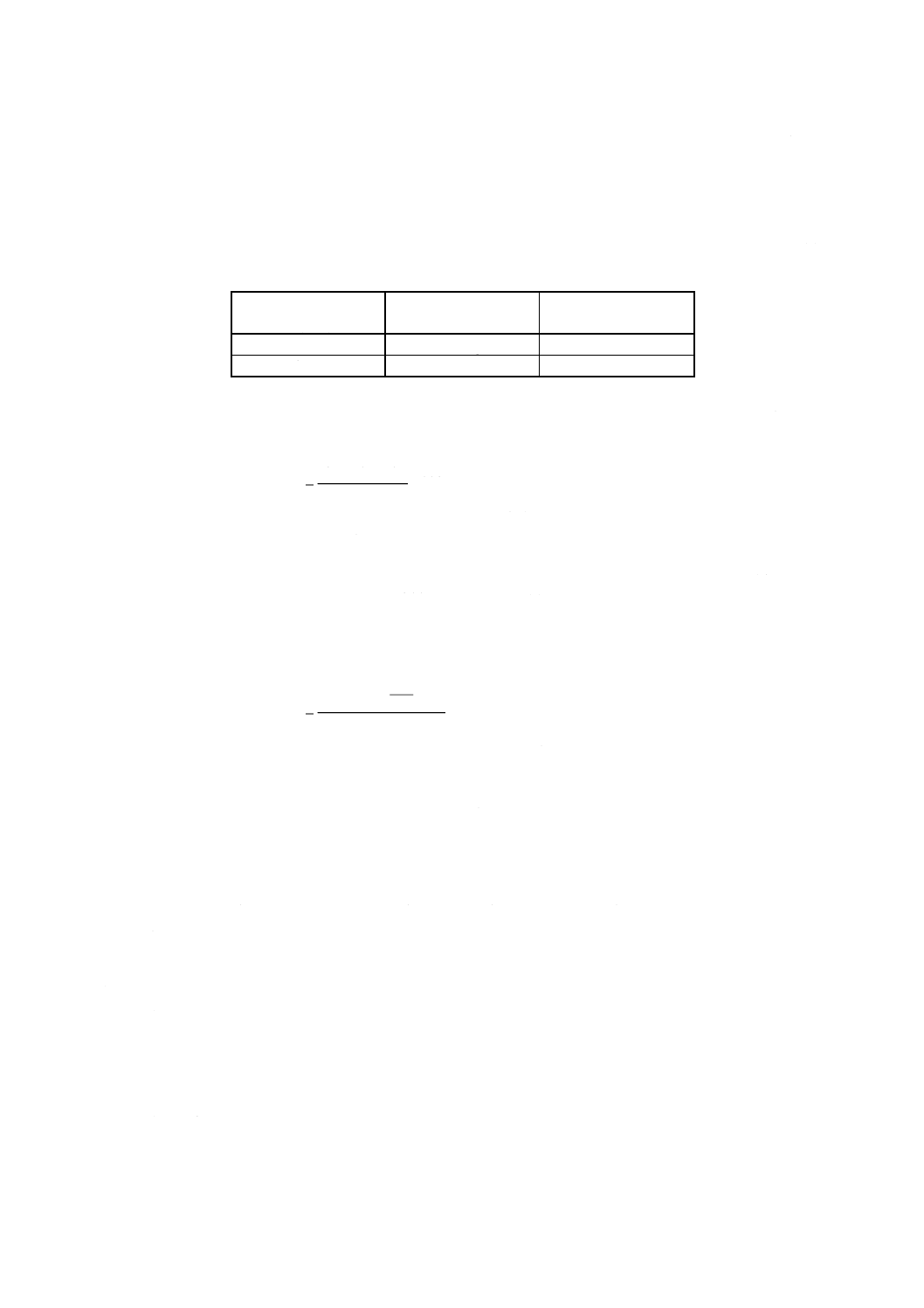
表15 標準クロム溶液添加量
試料中のクロム含有率
% (m/m)
使用する標準クロム溶液 標準クロム溶液添加量
ml
0.01以上 0.1未満
B [10.2j)]
0〜10.0
0.1 以上 0.5以下
A [10.2i)]
0〜 5.0
10.7 計算 計算は,次のいずれかによる。
a) 10.4.1でc)の操作を行わなかった場合 10.4.2及び10.5で得た吸光度と10.6で作成した検量線とから
クロム量を求め,試料中のクロム含有率を,次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Cr
ここに, Cr: 試料中のクロム含有率 [% (m/m)]
A1: 試料溶液中のクロム検出量 (g)
A2: 空試験液中のクロム検出量 (g)
A3: アルミニウム [10.2d)] 1.00g中に含まれるクロム量 (g)
m: 試料はかり取り量 (g)
b) 10.4.1でc)の操作を行った場合 10.4.2及び10.5で得た吸光度と10.6で作成した検量線とからクロム
量を求め,試料中のクロム含有率を,次の式によって算出する。
100
50
100
)
(
3
2
1
×
+
×
−
=
m
A
A
A
Cr
ここに, Cr: 試料中のクロム含有率 [% (m/m)]
A1: 分取した試料溶液中のクロム検出量 (g)
A2: 分取した空試験液中のクロム検出量 (g)
A3: アルミニウム [10.2d)] 1.00g中に含まれるクロム量 (g)
m: 試料はかり取り量 (g)
11 ニッケル定量方法
11.1 要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
11.2 試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,ニッケルを含有しないもの又はニッケル含有
率が既知で,できるだけ低いもの。
e) 過酸化水素
f)
すず溶液 5.2f)による。
14
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) 標準ニッケル溶液A (1 000μgNi/ml) ニッケル[99.9% (m/m) 以上]1.000gをはかり取ってビーカー
(200ml) に移し入れ,時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する。常温まで
冷却した後,時計皿の下面及びビーカーの内壁を水で洗浄し,時計皿を取り除く。溶液を1 000mlの
全量フラスコに水を用いて移し入れ,水で標線まで薄めて標準ニッケル溶液Aとする。
h) 標準ニッケル溶液B (500μgNi/ml) 標準ニッケル溶液A [g)] を使用の都度,必要量だけ水で正確に2
倍に薄めて標準ニッケル溶液Bとする。
i)
標準ニッケル溶液C (100μgNi/ml) 標準ニッケル溶液A [g)] を使用の都度,必要量だけ水で正確に
10倍に薄めて標準ニッケル溶液Cとする。
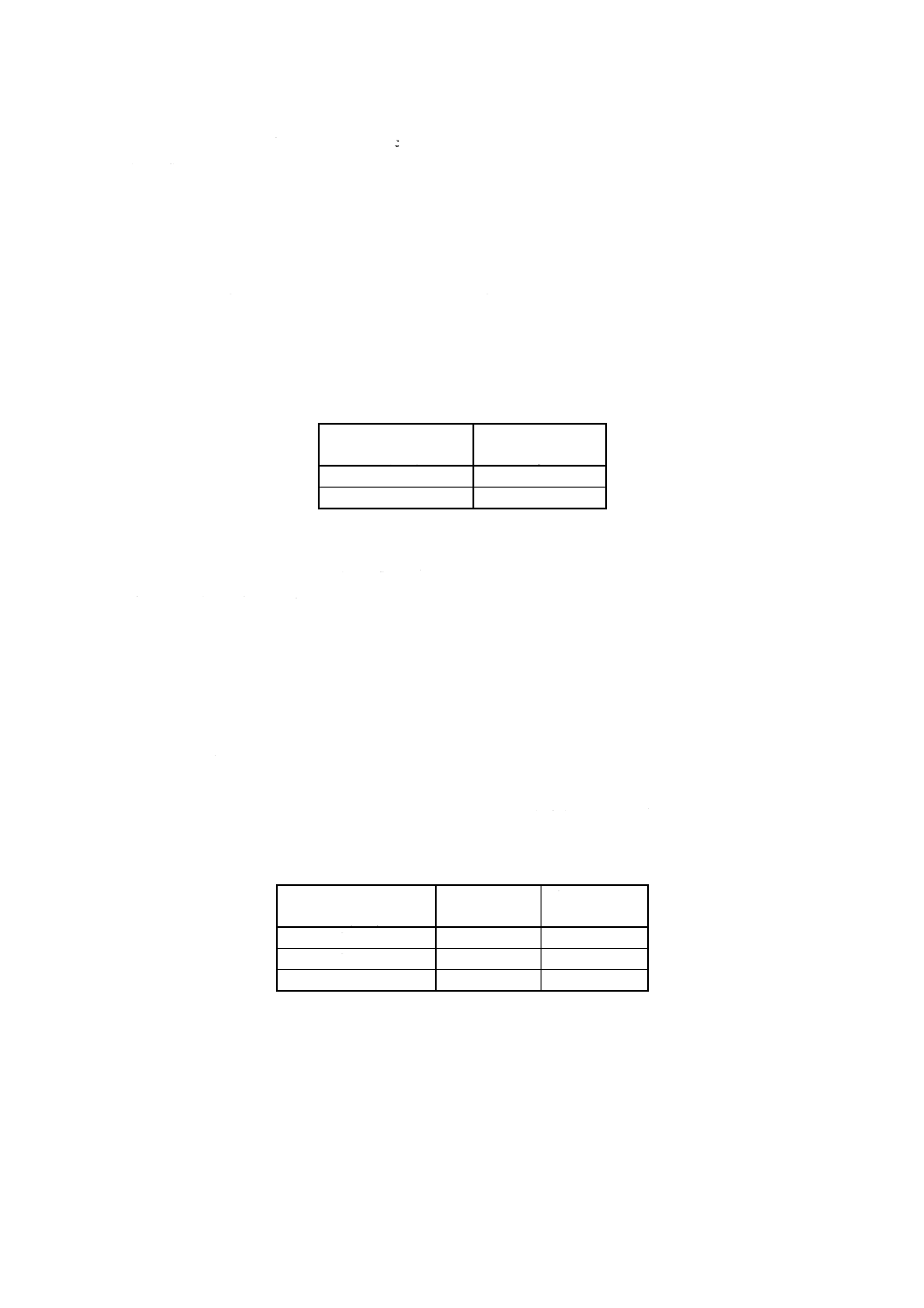
11.3 試料はかり取り量 試料はかり取り量は,試料中のマグネシウム含有率に応じて表16に従い,1mg
のけたまではかる。
表16 試料はかり取り量
試料中のニッケル含有率
% (m/m)
試料はかり取り量
g
0.005以上 1.5未満
1.00
1.5
以上 3.0以下
0.20
11.4 操作
11.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する(15)。この溶液に過酸化水素1ml
を加え(2),加熱して試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの
内壁を水で洗浄し,時計皿を取り除く。溶液を試料中のニッケル含有率が1.5% (m/m) 未満の場合は
100mlの全量フラスコに,1.5% (m/m) 以上の場合には200mlの全量フラスコに水を用いて移し入れ,
水で標線まで薄める(16)。
c) 溶液を試料中のニッケル含有率に応じて表17に従って分取し,表17に示した全量フラスコに移し入
れ,水で標線まで薄める。
注(15) 試料が分解しにくい場合は,すず溶液 [11.2f)] 2mlを加える。
(16) 試料中のニッケル含有率が0.005% (m/m) 以上0.1% (m/m) 未満の場合には,次のc)の操作は行
わない。
表17 分取量
ニッケル含有率
% (m/m)
分取量
ml
全量フラスコ
ml
0.1以上 0.5未満
50.0
100
0.5以上 1.5未満
20.0
100
1.5以上 3.0以下
20.0
200
11.4.2 吸光度の測定 11.4.1のb)又はc)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度
計の空気・アセチレンフレーム中に噴霧し,波長232.0nm又は341.5nmにおける吸光度を測定する。
11.5 空試験 11.6の操作において得られる標準ニッケル溶液を添加しない溶液の吸光度を,空試験の吸
光度とする。
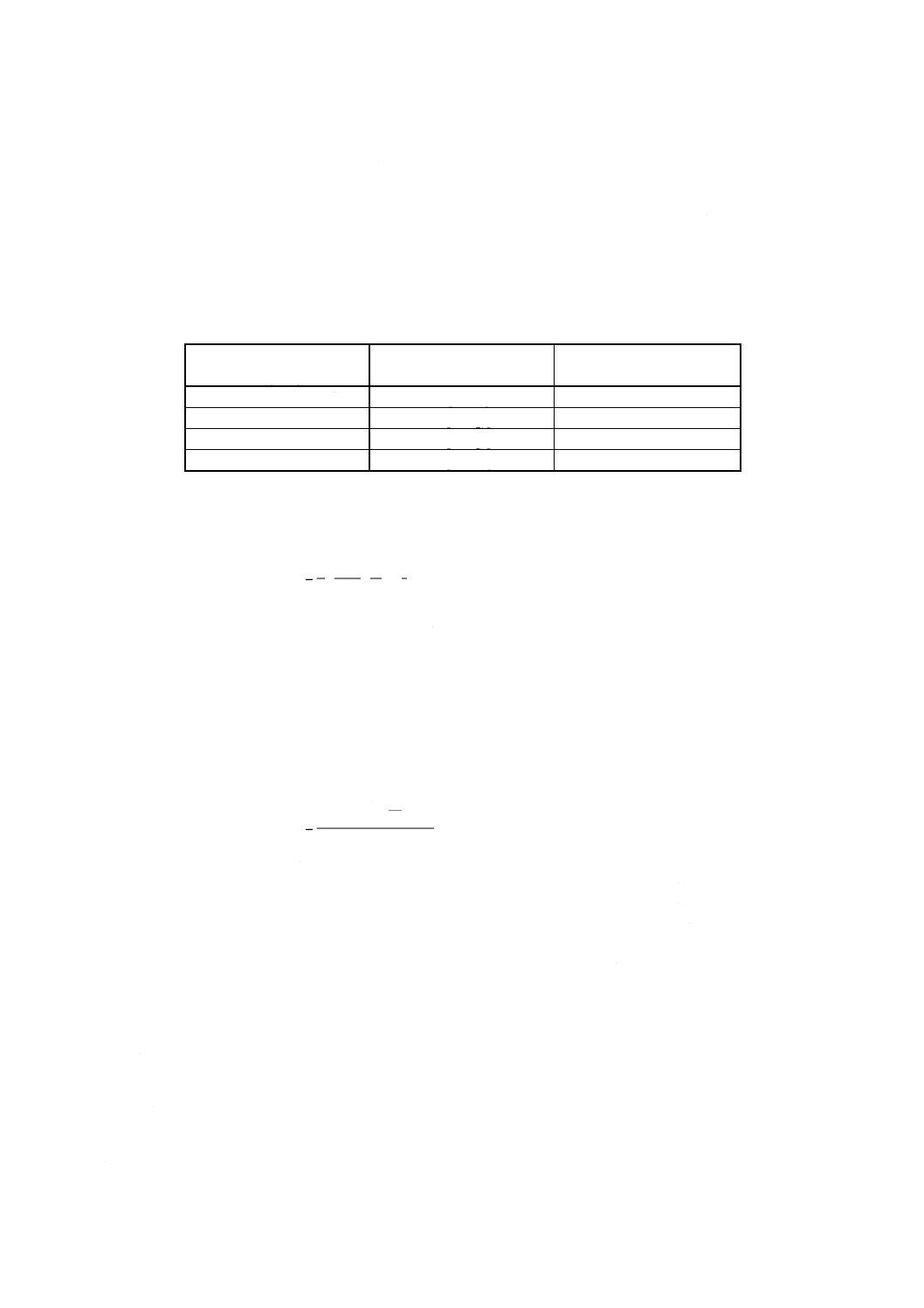
15
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.6 検量線の作成 アルミニウム [11.2d)] を試料中のニッケル含有率に応じて表16の試料はかり取り
量と同じになるようにはかり取って,数個のビーカー (300ml) に移し入れ,試料中のニッケル含有率に応
じて表18に従って標準ニッケル溶液を段階的に加える。以下,11.4.1b)〜11.4.2の手順に従って試料と同
じ操作を試料と並行して行い,得た吸光度と標準ニッケル溶液として加えたニッケル量(17)との関係線を作
成し,その関係線を原点を通るように平行移動して検量線とする。
注(17) 11.4.1c)の操作を行った場合には,分取した溶液中の標準ニッケル溶液として加えたニッケル量
(g)。
表18 標準ニッケル溶液添加量
試料中のニッケル含有率
% (m/m)
使用する標準ニッケル溶液
標準溶液ニッケル添加量
ml
0.005以上 0.1未満
C [11.2i)]
0〜10.0
0.1 以上 0.5未満
A [11.2g)]
0〜 5.0
0.5 以上 1.5未満
A [11.2g)]
0〜15.0
1.5 以上 3.0以下
B [11.2h)]
0〜12.0
11.7 計算 計算は,次のいずれかによる。
a) 11.4.1c)の操作を行わなかった場合11.4.2及び11.5で得た吸光度と11.6で作成した検量線とからニッ
ケル量を求め,試料中のニッケル含有率を,次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Ni
ここに, Ni: 試料中のニッケル含有率 [% (m/m)]
A1: 試料溶液中のニッケル検出量 (g)
A2: 空試験液中のニッケル検出量 (g)
A3: 11.6ではかり取ったアルミニウム [11.2d)] 中に含まれるニ
ッケル量 (g)
m: 試料はかり取り量 (g)
b) 11.4.1c)の操作を行った場合 11.4.2及び11.5で得た吸光度と11.6で作成した検量線とからニッケル量
を求め,試料中のニッケル含有率を,次の式によって算出する。
100
)
(
3
2
1
×
+
×
−
=
m
A
B
C
A
A
Ni
ここに, Ni: 試料中のニッケル含有率 [% (m/m)]
A1: 分取した試料溶液中のニッケル検出量 (g)
A2: 分取した空試験液中のニッケル検出量 (g)
C: 11.4.1b)で使用した全量フラスコの容量 (ml)
B: 試料溶液及び空試験液の分取量 (ml)
A3: 11.6ではかり取ったアルミニウム [11.2d)] 中に含まれるニ
ッケル含有量 (g)
m: 試料はかり取り量 (g)
12. ビスマス定量方法
12.1 要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
12.2 試薬 試薬は,次による。
a) 塩酸 (1+1)
16
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,ビスマスを含有しないもの又はビスマス含有
率が既知で,できるだけ低いもの。
e) 過酸化水素
f)
すず溶液 5.2f)による。
g) ニッケル溶液 5.2g)による。
h) 標準ビスマス溶液 (1 000μgBi/ml) ビスマス[99.9% (m/m) 以上]1.000gをはかり取ってビーカー
(200ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 30mlを加え,穏やかに加熱して分解する。常温まで
冷却した後,時計皿の下面及びビーカーの内壁を硝酸 (1+5) で洗浄し,時計皿を取り除く。溶液を1
000mlの全量フラスコに硝酸 (1+5) を用いて移し入れ,硝酸 (1+5) で標線まで薄めて標準ビスマス
溶液とする。
12.3 試料はかり取り量 試料はかり取り量は,1.00gとし,1mgのけたまではかる。
12.4 操作
12.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する(1)。過酸化水素1mlを加え(2),
加熱して試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水を
用いて洗浄し,時計皿を取り除く。溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線ま
で薄める。
12.4.2 吸光度の測定 12.4.1b)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長223.1nm又は306.8nmにおける吸光度を測定する。
12.5 空試験 12.6の操作において得られる標準ビスマス溶液を添加しない溶液の吸光度を,空試験の吸
光度とする。
12.6 検量線の作成 アルミニウム [12.2d)] を1.00gずつはかり取って数個のビーカー (300ml) に移し入
れ,標準ビスマス溶液 [12.2h)] 0〜10.0ml(ビスマスとして0〜10.0mg)を段階的に加える。以下,12.4.1b)
〜12.4.2の手順に従って試料と同じ操作を試料と並行して行い,得た吸光度と標準ビスマス溶液として加
えたビスマス量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
12.7 計算 12.4.2及び12.5で得た吸光度と12.6で作成した検量線とからビスマス量を求め,試料中のビ
スマス含有率を,次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Bi
ここに, Bi: 試料中のビスマス含有率 [% (m/m)]
A1: 試料溶液中のビスマス検出量 (g)
A2: 空試験液中のビスマス検出量 (g)
A3: アルミニウム [12.2d)] 1.00g中に含まれるビスマス量 (g)
m: 試料はかり取り量 (g)
13. 鉛定量方法
13.1 要旨 試料を塩酸と過酸化水素とで分解した後,溶液を原子吸光光度計の空気・アセチレンフレー
ム中に噴霧し,その吸光度を測定する。
17
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.2 試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸
d) アルミニウム できるだけ純度の高いアルミニウムで,鉛を含有しないもの又は鉛含有率が既知で,
できるだけ低いもの。
e) 過酸化水素
f)
すず溶液 5.2.f)による。
g) ニッケル溶液 5.2g)による。
h) 標準鉛溶液 (1 000μgPb/ml) 鉛[99.9% (m/m) 以上]1.000gをはかり取ってビーカー (200ml) に移し
入れ,時計皿で覆い,硝酸 (1+1) 30mlを加え,穏やかに加熱して分解する。常温まで冷却した後,
時計皿の下面及びビーカーの内壁を水を用いて洗浄し,時計皿を取り除く。溶液を1 000mlの全量フ
ラスコに水を用いて移し入れ,水で標線まで薄めて標準鉛溶液とする。
13.3 試料はかり取り量 試料はかり取り量は,1.00gとし,1mgのけたまではかる。
13.4 操作
13.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (300ml) に移し入れる。
b) 時計皿で覆い,塩酸 (1+1) 30mlを加え,穏やかに加熱して分解する(1)。過酸化水素1mlを加え(2),
加熱して試料を完全に分解する(3)(4)。常温まで冷却した後,時計皿の下面及びビーカーの内壁を水を
用いて洗浄し,時計皿を取り除く。溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線ま
で薄める。
13.4.2 吸光度の測定 13.4.1b)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長217.0nm又は283.3nmにおける吸光度を測定する。
13.5 空試験 13.6の操作において得られる標準鉛溶液を添加しない溶液の吸光度を,空試験の吸光度と
する。
13.6 検量線の作成 アルミニウム [13.2d)] を1.00gずつはかり取って数個のビーカー (300ml) に移し入
れ,標準鉛溶液 [13.2h] 0〜10.0ml(鉛として0〜10.0mg)を段階的に加える。以下,13.4.1b)〜13.4.2の手順
に従って試料と同じ操作を試料と並行して行い,得た吸光度と標準鉛溶液として加えた鉛量との関係線を
作成し,その関係線を原点を通るように平行移動して検量線とする。
13.7 計算 13.4.2及び13.5で得た吸光度と13.6で作成した検量線とから鉛量を求め,試料中の鉛含有率
を,次の式によって算出する。
100
)
(
3
2
1
×
−
−
=
m
A
A
A
Pb
ここに, Pb: 試料中の鉛含有率 [% (m/m)]
A1: 試料溶液中の鉛検出量 (g)
A2: 空試験液中の鉛検出量 (g)
A3: アルミニウム [13.2d)] 1.00g中に含まれる鉛量 (g)
m: 試料はかり取り量 (g)
18
H 1306 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS改正原案作成委員会 構成表
氏名
所属
(委員長)
畦 上 尚
株式会社日軽分析センター
藤 沼 弘
東洋大学工学部
村 上 徹 朗
工学院大学
大河内 春 乃
東京理科大学
俣 野 宣 久
川崎製線株式会社
村 山 拓 己
通商産業省基礎産業局
大 嶋 清 治
工業技術院標準部
橋 本 繁 晴
財団法人日本規格協会
井 川 洋 志
昭和電工株式会社千葉事業所
泉 巌
日本軽金属株式会社蒲原製造所
坂 巻 博
新日軽株式会社管理部
勝間田 一 宏
三菱アルミニウム株式会社富士製作所
川 口 修
スカイアルミニウム株式会社技術研究所
北 村 照 夫
昭和アルミニウム株式会社研究開発部
豊 嶋 雅 康
住友軽金属工業株式会社研究開発センター
中 田 滋
古河電気工業株式会社福井事業所
坂 本 敏 正
株式会社神戸製鋼所アルミ・銅事業本部
安 部 正 明
東洋アルミニウム株式会社研究開発本部
久留須 一 彦
古河電気工業株式会社横浜研究所分析技術センター
水 砂 博 文
住友電気工業株式会社研究開発部
船 渡 好 人
株式会社島津製作所分析機器事業部
本 多 和 人
株式会社パーキンエルマージャパン応用技術部
(事務局)
井 波 隆 夫
社団法人軽金属協会