H 1151 : 1999
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。これによってJIS H 1151 : 1993は改正され,この規格に置き換えられる。
今回の改正では,日本工業規格と国際規格との対比,国際規格に一致した日本工業規格の作成,及び日
本工業規格を基礎にした国際規格原案の提案を容易にするために,ISO 6351 : 1985,ISO 7523 : 1985,ISO
7524 : 1985,ISO 7525 : 1985,ISO 7526 : 1985及びISO 7527 : 1985を基礎として用いた。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
H 1151 : 1999
ニッケル地金分析方法
Methods for chemical analysis of nickel metal
序文 この規格は,ISO 6351 : 1985, Nickel−Determination of silver, bismuth, cadmium, cobalt, copper, iron,
manganese, lead and zinc contents−Flame atomic absorption spectrometric method, ISO 7523 : 1985, Nickel−
Determination of silver, arsenic, bismuth, cadmium, lead, antimony, selenium, tin, tellurium and thallium contents−
Electrothermal atomic absorption spectrometric method, ISO 7524 : 1985, Nickel, ferronickel and nickel alloys−
Determination of carbon content−Infra-red absorption method after induction furnace combustion, ISO 7525 : 1985,
Nickel−Determination of sulfur content−Metylene blue molecular absorption spectrometric method after
generation of hydrogen sulfide, ISO 7526 : 1985, Nickel, ferronickel and nickel alloys−Determination of sulfur
content−Infra-red absorption method after induction furnace combustion, 及びISO 7527 : 1985, Nickel,
ferronickel and nickel alloys−Determination of sulfur content−Iodimetric titration method after induction furnace
combustionの対応する部分 (ISO 6351) を元に,対応する部分については対応国際規格を翻訳し,技術的
内容を変更することなく,作成した日本工業規格であるが,鉛の定量方法のうち,電気加熱原子吸光法に
ついては対応する国際規格で規定されている四塩化炭素を削除した。
1. 適用範囲 この規格は,JIS H 2104に規定するニッケル地金中のコバルト,鉄,銅,鉛,マンガン,
炭素,硫黄及びけい素の定量方法について規定する。
備考 この規格の対応国際規格を,次に示す。
ISO 6351 : 1985 Nickel−Determination of silver, bismuth, cadmium, cobalt, copper, iron, manganese,
lead and zinc contents−Flame atomic absorption spectrometric method
ISO 7523 : 1985 Nickel−Determination of silver, arsenic, bismuth, cadmium, lead, antimony,
selenium, tin, tellurium and thallium contents−Electrothermal atomic absorption spectrometric
method
ISO 7524 : 1985 Nickel, ferronickel and nickel alloys−Determination of carbon content−Infra-red
absorption method after induction furnace combustion
ISO 7525 : 1985 Nickel−Determination of sulfur content−Metylene blue molecular absorption
spectrometric method after generation of hydrogen sulfide
ISO 7526 : 1985 Nickel, ferronickel and nickel alloys−Determination of sulfur content−Infra-red
absorption method after induction furnace combustion
ISO 7527 : 1985 Nickel, ferronickel and nickel alloys−Determination of sulfur content−Iodimetric
titration method after induction furnace combustion
3
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
2
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS H 0301 非鉄金属地金のサンプリング,試料調製及び分析検査通則
JIS H 2104 ニッケル地金
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS Z 2615 金属材料の炭素定量方法通則
JIS Z 2616 金属材料の硫黄定量方法通則
JIS Z 8401 数値の丸め方
3. 一般事項 分析方法に共通な一般事項は,JIS H 0301,JIS K 0050,JIS K 0115,JIS K 0116,JIS K 0121,
JIS Z 2615及びJIS Z 2616の規定による。
4. 分析試料の採り方及び取扱い方
4.1
試料の採り方 試料の採り方は,次による。
a) 地金から切粉を取るときは,削りとった試料がその地金の品質を代表するように,採取する箇所は地
金の中央部,周辺に近い部分などとする。また,地金面に直角にボーリングして貫通させるか,又は
地金から切断した試料片からフライス盤などで切削して採取する。
b) ボーリングなどによって切粉試料を取るときは,あらかじめドリルその他の工具をエタノールなどを
用いて清浄にする。試料採取箇所を清浄にし,次に油類その他の減磨剤を用いないで,切粉が酸化し
ない程度の力を与えてボーリングを行う。この際,ドリルの圧力,回転数などを加減して,極端に発
熱しないようにしなければならないが,冷却のため水などを注加してはならない。
c) 切粉試料はその全部を集め,切粉試料がひも状の場合は,清浄なはさみなどを用いて約5mm以下に
切断した後,よく混ぜ合わせて分析用試料とする。ただし,燃焼法による炭素及び硫黄の定量に用い
る試料は,約2mm以下の細片とする。
d) 分析用試料の採取と調製が,a)〜c)の規定によることができない場合には,受渡当事者間の協議によ
って定める。
4.2
試料の取扱い方 試料の取扱い方は,次による。
a) 分析用試料は,異物などによる汚染を防止するため,適宜なふた付きガラス容器などに入れ,密封し
て保存する。
b) 分析用試料は,その表面に油などが付着しているおそれのあるときは,あらかじめエタノール,アセ
トンなどで洗浄して乾燥する。
c) 分析用試料を鉄の定量に用いる場合で,鉄の汚染のおそれがあるときには,あらかじめ次の処理を行
う。
分析用試料の必要量をビーカーに取り,塩酸 (1+10) を試料片が沈む程度に加え,加熱して5分間
煮沸するか,又は約80℃で約30分間加熱して,表面に付着又は混入した鉄分を溶解する。水で洗浄
した後,エタノール,アセトンで順次洗浄して乾燥する。
4.3
試料のはかり方 試料のはかり方は,次による。
a) 分析試料のはかり取りに際しては,平均組成を代表するように注意しなければならない。
b) 分析試料のはかり取りには,化学はかり,電子はかりなどを用いる。
3
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5. 分析値のまとめ方
5.1
分析回数 通常同一分析所において,2回の繰返し分析を行う。
5.2
空試験 分析に当たっては,空試験を行い,測定値を補正する。
5.3
分析値の表示 分析値は質量百分率で表し,JIS H 2104に規定された数値の次の2けたまで算出し,
JIS Z 8401によってJIS H 2104に規定された数値の次の位に丸める。
6. コバルト定量方法
6.1
定量方法の区分 コバルトの定量方法は,次のいずれかによる。
a) チオシアン酸・トリオクチルアミン抽出吸光光度法 この方法は,コバルト含有率0.001% (m/m) 以
上0.4% (m/m) 以下の試料に適用する。
b) フレーム原子吸光法 この方法は,コバルト含有率0.001% (m/m) 以上1.0% (m/m) 以下の試料に適用
する。
c) ICP発光分光分析法 この方法は,コバルト含有率0.001% (m/m) 以上0.5% (m/m) 以下の試料に適用
する。
d) 陰イオン交換分離ICP発光分光分析法 この方法は,コバルト含有率0.000 1% (m/m) 以上0.05%
(m/m) 以下の試料に適用する。
6.2
チオシアン酸・トリオクチルアミン抽出吸光光度法
6.2.1
要旨 試料を硝酸で分解した後,塩酸,チオ尿素及びチオシアン酸カリウムを加える。トリオクチ
ルアミン−キシレンを加え,生成するトリオクチルアミンチオシアン酸コバルト錯体をキシレンに抽出す
る。同時に抽出された鉄などをチオ硫酸ナトリウムで洗浄して除去した後,分光光度計を用いて有機相の
吸光度を測定する。
6.2.2
試薬 試薬は,次による。
a) 塩酸
b) 硝酸 (1+1)
c) 硫酸ナトリウム(無水)
d) チオ硫酸ナトリウム溶液 チオ硫酸ナトリウム五水和物25gを水に溶解し,水で液量を1 000mlとす
る。
e) チオシアン酸カリウム溶液 (300g/l)
f)
チオ尿素溶液 (100g/l)
g) トリオクチルアミン−キシレン溶液 トリオクチルアミン10mlをキシレンに溶解し,キシレンで液
量を200mlとする。
h) キシレン
i)
標準コバルト溶液 (20μgCo/ml) コバルト[99.9% (m/m) 以上]1.000gを硝酸 (1+1) 20mlで分解し,
常温まで冷却した後,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原
液 (1mgCo/ml) とする。この原液を使用の都度,必要量だけ水で正確に50倍に薄めて標準コバルト溶
液とする。
6.2.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
6.2.4
操作
6.2.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取って,ビーカー (300ml) に移し入れる。
4
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 時計皿で覆い,硝酸 (1+1) 40mlを加えて穏やかに加熱して分解した後,煮沸して酸化窒素などを追
い出す。常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除き,溶液を100mlの全量フ
ラスコに水を用いて移し入れ,水で標線まで薄める。
6.2.4.2
コバルトの分離 コバルトの分離は,次の手順によって行う。
a) 6.2.4.1 b)で得た溶液から正確に10ml(1)を分液漏斗 (100ml) に分取する。
b) 塩酸2ml,チオ尿素溶液10ml及びチオシアン酸カリウム溶液3mlを加える(2)。
c) トリオクチルアミン−キシレン溶液 [6.2.2 g)] を正確に10ml加え,約2分間激しく振り混ぜ,静置し
て2層に分離した後,水相を取り除く。
d) 有機相にチオ硫酸ナトリウム溶液 [6.2.2 d)] 5mlを加え,約1分間振り混ぜ,静置して2層に分離した
後,水相を取り除く。さらに,この操作をもう1回繰り返す。
注(1) 分取した溶液10ml中のコバルト量が200μgを超える場合には,正確に20mlを100mlの全量フラ
スコに分取し,水で標線まで薄めた後,コバルト量が50〜200μgとなるように分液漏斗に分取
する。
(2) 注(1)を適用したときは,水を加えて液量を約25mlとする。
6.2.4.3
吸光度の測定 6.2.4.2 d)で得た有機相の水分を取り除き(3),その一部を分光光度計の吸収セル
(10mm) に取り,キシレンを対照液として,波長630nm付近の吸光度を測定する。
注(3) 乾いたろ紙又は無水硫酸ナトリウムなどを用いて脱水する。
6.2.5
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
6.2.6
検量線の作成 標準コバルト溶液 [6.2.2 i)] 0〜10.0ml(コバルトとして0〜200μg)を段階的に数個
の分液漏斗 (100ml) に取り,以下6.2.4.2 b)〜6.2.4.3の手順に従って試料と同じ操作を試料と並行して行い,
得た吸光度とコバルト量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
6.2.7
計算 6.2.4.3及び6.2.5で得た吸光度と6.2.6で作成した検量線とからコバルト量を求め,試料中の
コバルト含有率を,次の式によって算出する。
100
100
)
(
2
1
×
×B
m
A
A
Co
−
=
ここに, Co: コバルト含有率 [% (m/m)]
A1: 分取した試料溶液中のコバルト検出量 (g)
A2: 分取した空試験液中のコバルト検出量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
6.3
フレーム原子吸光法
6.3.1
要旨 試料を硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,
その吸光度を測定する。
6.3.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] コバルト含有率が既知(4)で,かつ,そのコバルト含有率が試料中の
コバルト含有率より低いもの。
c) 標準コバルト溶液A (50μgCo/ml) 6.2.2 h)の原液 (1mgCo/ml) を使用の都度,必要量だけ水で正しく
20倍に薄めて標準コバルト溶液Aとする。
d) 標準コバルト溶液B (10μgCo/ml) 6.2.2 h)の原液 (1mgCo/ml) を使用の都度,必要量だけ水で正しく
5
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100倍に薄めて標準コバルト溶液Bとする。
注(4) コバルト含有率は,6.2のチオシアン酸・トリオクチルアミン抽出吸光光度法,又は6.5の陰イオ
ン交換分離ICP発光分光分析法によって求める。
6.3.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
6.3.4
操作
6.3.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取って,ビーカー (300ml) に移し入れる。
b) 時計皿で覆い,硝酸 (1+1) 40mlを加えて穏やかに加熱して分解した後,煮沸して酸化窒素などを追
い出す。常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除く。
c) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(5)。
注(5) この溶液中のコバルト量が1 000μgを超える場合には,コバルト量が100〜1 000μgとなるように
100mlの全量フラスコに分取し,水で標線まで薄める。
6.3.4.2
吸光度の測定 6.3.4.1 c)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長240.7nmにおける吸光度を測定する。
6.3.5
空試験 空試験は,次のいずれかによる。
a) 6.3.4.1 c)で分取しない場合 6.3.6 a)の検量線の作成操作において得られる,標準コバルト溶液を添加
しない溶液の吸光度を,空試験の吸光度とする。
b) 6.3.4.1 c)で分取する場合 6.3.6 b)の検量線の作成操作において得られる,標準コバルト溶液を添加し
ない溶液の吸光度を,空試験の吸光度とする。
6.3.6
検量線の作成 検量線の作成は,次のいずれかの手順によって行う。
a) 6.3.4.1 c)で分取しない場合
1) ニッケル [6.3.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
2) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
3) これらの溶液に,標準コバルト溶液A [6.3.2 c)] 及び標準コバルト溶液B [6.3.2 d)] の各種液量(コ
バルトとして0〜1 000μg)を段階的に加え,水で標線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長240.7nmにおける吸光度を試料と並行して測定し,得た吸光度とコバルト量との
関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 6.3.4.1 c)で分取する場合
1) ニッケル [6.3.2 b)] を5.0gはかり取り,ビーカー (300ml) に移し入れる。
2) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄
める。
3) この溶液から6.3.4.1 c)で分取した試料溶液と同じ量を数個の100mlの全量フラスコに分取し,標準
コバルト溶液A [6.3.2 c)] 及び標準コバルト溶液B [6.3.2 d)] の各種液量(コバルトとして0〜1
000μg)を段階的に加え,水で標線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長240.7nmにおける吸光度を試料と並行して測定し,得た吸光度とコバルト量との
関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
6.3.7
計算 計算は,次のいずれかによる。
a) 6.3.4.1 c)で分取しなかった場合 6.3.4.2及び6.3.5 a)で得た吸光度と6.3.6 a)で作成した検量線とから
6
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
コバルト量を求め,試料中のコバルト含有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Co
−
−
=
ここに, Co: コバルト含有率 [% (m/m)]
A1: 試料溶液中のコバルト検出量 (g)
A2: 空試験液中のコバルト検出量 (g)
A3: ニッケル [6.3.2 b)] 5.0g中に含まれるコバルト量 (g)
m: 試料はかり取り量 (g)
b) 6.3.4.1 c)で分取した場合 6.3.4.2及び6.3.5 b)で得た吸光度と6.3.6 b)で作成した検量線とからコバル
ト量を求め,試料中のコバルト含有率を,次の式によって算出する。
100
100
100)
(
6
5
4
×
×
×
B
m
B
A
A
A
Co
−
−
=
ここに, Co: コバルト含有率 [% (m/m)]
A4: 分取した試料溶液中のコバルト検出量 (g)
A5: 分取した空試験液中のコバルト検出量 (g)
A6: ニッケル [6.3.2 b)] 5.0g中に含まれるコバルト量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
6.4
ICP発光分光分析法
6.4.1
要旨 試料を硝酸で分解した後,溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,そ
の発光強度を測定する。
6.4.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 6.3.2 b)による。
c) 標準コバルト溶液A (1mgCo/ml) 6.2.2 h)の原液 (1mgCo/ml) を使用する。
d) 標準コバルト溶液B (100μgCo/ml) 標準コバルト溶液A [c)] を使用の都度,必要量だけ水で正しく
10倍に薄めて標準コバルト溶液Bとする。
e) 標準コバルト溶液C (10μgCo/ml) 6.3.2 d)による。
6.4.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
6.4.4
操作
6.4.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
6.4.4.2
発光強度の測定 6.4.4.1 b)で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に
噴霧し,波長238.892nmにおける発光強度を測定する(6)。
注(6) 精度及び真度を確認してあれば,他の波長を用いて測定してもよい。また,高次のスペクトル
線が使用可能な装置では高次のスペクトル線を用いてもよい。バックグラウンド補正機構が付
いている装置では,バックグラウンド補正機構を用いてもよい。
6.4.5
空試験 6.4.6の検量線の作成操作において得られる,標準コバルト溶液を添加しない溶液の発光
強度を,空試験の発光強度とする。
7
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.4.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) ニッケル [6.4.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
b) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
c) これらの溶液に,標準コバルト溶液A [6.4.2 c)],標準コバルト溶液B [6.4.2 d)] 及び標準コバルト溶液
C [6.4.2 e)] の各種液量(コバルトとして0〜25mg)を段階的に加え,水で標線まで薄める。
d) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長238.892nmにお
ける発光強度を試料と並行して測定し(6),得た発光強度とコバルト量との関係線を作成し,その関係
線を原点を通るように平行移動して検量線とする。
6.4.7
計算 6.4.4.2及び6.4.5で得た発光強度と6.4.6で作成した検量線とからコバルト量を求め,試料中
のコバルト含有率を,次の式によって算出する。
100
)
(
3
2
1
×
=
m
A
A
A
Co
−
−
ここに, Co: コバルト含有率 [% (m/m)]
A1: 試料溶液中のコバルト検出量 (g)
A2: 空試験液中のコバルト検出量 (g)
A3: ニッケル [6.4.2 b)] 5.0g中に含まれるコバルト量 (g)
m: 試料はかり取り量 (g)
6.5
陰イオン交換分離ICP発光分光分析法
6.5.1
要旨 試料を硝酸で分解した後,シロップ状とし,塩酸を加えて濃縮を繰り返して硝酸を除去する。
水を加えて可溶性塩類を溶解した後,塩類を加えて塩類濃度を調節する。陰イオン交換カラムに通してコ
バルトを吸着させた後,塩酸とアスコルビン酸を通してコバルトを溶出させる。溶出液をICP発光分光分
析装置のアルゴンプラズマ中に噴霧し,その発光強度を測定する。
6.5.2
試薬 試薬は,次による。
a) 塩酸
b) 塩酸 (3+1,1+100)
c) 硝酸 (1+1)
d) 水酸化ナトリウム溶液 (40g/l)
e) L (+) −アスコルビン酸溶液 (10g/l) 使用の都度,調製する。
f)
アスコルビン酸・塩酸溶液 L (+) −アスコルビン酸1gを塩酸 (1+9) 1 000mlに溶解する。この溶液
は,使用の都度,調製する。
g) 標準コバルト溶液A (50μgCo/ml) 6.3.2 c)による。
h) 標準コバルト溶液B (10μgCo/ml) 6.3.2 d)による。
6.5.3
器具 器具は,次による。
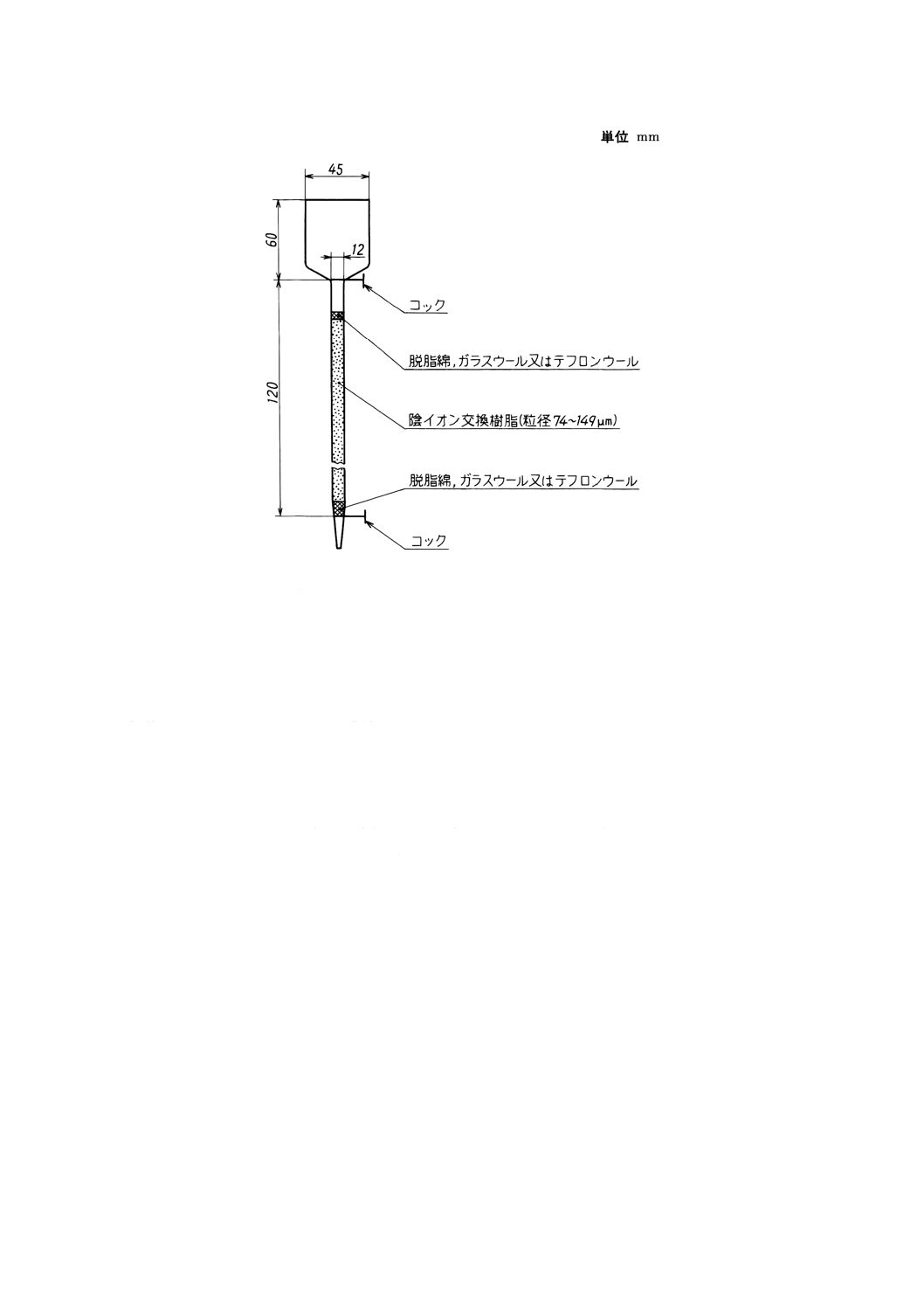
陰イオン交換カラム ガラス管(内径約12mm)に水でほぐした脱脂綿,ガラスウール又はテフロンウ
ールを約5mmの厚さに緩く詰め,水で膨潤させた強塩基性陰イオン交換樹脂(Cl型,粒径74〜149μm)
約20mlをスラリー状にして流し入れる。樹脂が沈降した後,その上に水でほぐした脱脂綿,ガラスウー
ル又はテフロンウールを約5mmの厚さに緩く詰める。脱脂綿,ガラスウール又はテフロンウールの詰め
方を調節するなどして流出液の流量を毎分0.5〜1mlになるようにしておく。陰イオン交換カラムの例を図
1に示す。
8
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図1 陰イオン交換カラムの例
6.5.4
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
6.5.5
操作
6.5.5.1
準備操作 陰イオン交換カラム (6.5.3) に塩酸 (3+1) 50mlを通す。
6.5.5.2
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 加熱してシロップ状になるまで濃縮する。
c) 放冷した後,時計皿で覆い,塩酸20mlを加えて加熱し,数分間煮沸する。放冷した後,時計皿の下
面を水で洗って時計皿を取り除き,再び加熱してシロップ状になるまで濃縮する。
d) c)の操作をもう一度繰り返す。
e) 水20mlを加え,加熱して塩類を溶解した後,塩酸60mlを加え,放冷する。
6.5.5.3
コバルトの分離 6.5.5.2 e)で得た溶液を毎分0.5〜1mlの流量で6.5.5.1の準備操作を終わった陰イ
オン交換カラムに通す。樹脂上に溶液がなくなってから塩酸 (3+1) 50mlを数回に分けてビーカー内壁を
洗浄し,その都度カラムに通す。これまでの流出液は,すべて捨てる。次にアスコルビン酸・塩酸溶液 [6.5.2
f)] 90mlを毎分0.5〜1mlの流量でカラムに通し,溶出液を100mlの全量フラスコに受けた後,アスコルビ
ン酸・塩酸溶液 [6.5.2 f)] で標線まで薄める(7)。
注(7) 使用したカラムを再使用する場合には,塩酸 (1+100),水,水酸化ナトリウム溶液 (40g/l),
水のそれぞれ約50mlずつを順次カラムに通す。
6.5.5.4
発光強度の測定 6.5.5.3で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴
霧し,波長238.892nmにおける発光強度を測定する。
6.5.6
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
6.5.7
検量線の作成 検量線の作成は,次の手順によって行う。
a) 標準コバルト溶液A [6.5.2 g)] 及び標準コバルト溶液B [6.5.2 h)] の各種液量(コバルトとして0〜2
9
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
500μg)を段階的に数個の100mlの全量フラスコに取る。
b) これらの溶液に,L (+) −アスコルビン酸溶液 [6.5.2 e)] 10ml及び塩酸20mlを加え,水で標線まで薄
める。
c) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長238.892nmにお
ける発光強度を試料と並行して測定し(6),得た発光強度とコバルト量との関係線を作成し,その関係
線を原点を通るように平行移動して検量線とする。
6.5.8
計算 6.5.5.4及び6.5.6で得た発光強度と6.5.7で作成した検量線とからコバルト量を求め,試料中
のコバルト含有率を,次の式によって算出する。
100
2
1
×
m
A
A
Co
−
=
ここに, Co: コバルト含有率 [% (m/m)]
A1: 試料溶液中のコバルト検出量 (g)
A2:
空試験液中のコバルト検出量 (g)
m: 試料はかり取り量 (g)
7. 鉄定量方法
7.1
定量方法の区分 鉄の定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,鉄含有率0.000 5% (m/m) 以上1.0% (m/m) 以下の試料に適用する。
b) ICP発光分光分析法 この方法は,鉄含有率0.000 5% (m/m) 以上1.0% (m/m) 以下の試料に適用する。
c) 陰イオン交換分離ICP発光分光分析法 この方法は,鉄含有率0.000 1% (m/m) 以上0.05% (m/m) 以
下の試料に適用する。
7.2
フレーム原子吸光法
7.2.1
要旨 試料を硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,
その吸光度を測定する。
7.2.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 鉄含有率が既知(8)で,かつ,その鉄含有率が試料中の鉄含有率より
低いもの。
c) 標準鉄溶液A (50μgFe/ml) 鉄[99.9% (m/m) 以上]1.000gを硝酸 (1+1) 20mlで分解し,常温まで冷
却した後,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原液 (1mgFe/ml)
とする。この原液を使用の都度,必要量だけ水で正しく20倍に薄めて標準鉄溶液Aとする。
d) 標準鉄溶液B (10μgFe/ml) c)の原液 (1mgFe/ml) を使用の都度,必要量だけ水で正しく100倍に薄め
て標準鉄溶液Bとする。
注(8) 鉄含有率は,7.4の陰イオン交換分離ICP発光分光分析法によって求める。
7.2.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
7.2.4
操作
7.2.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(9)。
注(9) この溶液中の鉄量が1 000μgを超える場合には,鉄量が100〜1 000μgとなるように100mlの全量
10
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
フラスコに分取し,水で標線まで薄める。
7.2.4.2
吸光度の測定 7.2.4.1 b)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長248.3nmにおける吸光度を測定する。
7.2.5
空試験 空試験は,次のいずれかによる。
a) 7.2.4.1 b)で分取しない場合 7.2.6 a)の検量線の作成操作において得られる,標準鉄溶液を添加しない
溶液の吸光度を,空試験の吸光度とする。
b) 7.2.4.1 b)で分取する場合 7.2.6 b)の検量線の作成操作において得られる,標準鉄溶液を添加しない溶
液の吸光度を,空試験の吸光度とする。
7.2.6
検量線の作成 検量線の作成は,次のいずれかの手順によって行う。
a) 7.2.4.1 b)で分取しない場合
1) ニッケル [7.2.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
2) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
3) これらの溶液に,標準鉄溶液A [7.2.2 c)] 及び標準鉄溶液B [7.2.2 d)] の各種液量(鉄として0〜1
000μg)を段階的に加え,水で標線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長248.3nmにおける吸光度を試料と並行して測定し,得た吸光度と鉄量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 7.2.4.1 b)で分取する場合
1) ニッケル [7.2.2 b)] を5.0gはかり取り,ビーカー (300ml) に移し入れる。
2) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄
める。
3) この溶液から7.2.4.1 b)で分取した試料溶液と同じ量を数個の100mlの全量フラスコに分取し,標準
鉄溶液A [7.2.2 c)] 及び標準鉄溶液B [7.2.2 d)] の各種液量(鉄として0〜1 000μg)を段階的に加え,
水で標線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長248.3nmにおける吸光度を試料と並行して測定し,得た吸光度と鉄量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
7.2.7
計算 計算は,次のいずれかによる。
a) 7.2.4.1 b)で分取しなかった場合 7.2.4.2及び7.2.5 a)で得た吸光度と7.2.6 a)で作成した検量線とから
鉄量を求め,試料中の鉄含有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Fe
−
−
=
ここに, Fe: 鉄含有率 [% (m/m)]
A1: 試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
A3: ニッケル [7.2.2 b)] 5.0g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
b) 7.2.4.1 b)で分取した場合 7.2.4.2及び7.2.5 b)で得た吸光度と7.2.6 b)で作成した検量線とから鉄量を
求め,試料中の鉄含有率を,次の式によって算出する。
11
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
100
100)
(
6
5
4
×
×
×
B
m
B
A
A
A
Fe
−
−
=
ここに, Fe: 鉄含有率 [% (m/m)]
A4: 分取した試料溶液中の鉄検出量 (g)
A5: 分取した空試験液中の鉄検出量 (g)
A6: ニッケル [7.2.2 b)] 5.0g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
7.3
ICP発光分光分析法
7.3.1
要旨 試料を硝酸で分解した後,溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,そ
の発光強度を測定する。
7.3.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 7.2.2 b)による。
c) 標準鉄溶液A (1mgFe/ml) 7.2.2 c)の原液 (1mgFe/ml) を使用する。
d) 標準鉄溶液B (100μgFe/ml) 標準鉄溶液A [c)] を使用の都度,必要量だけ水で正しく10倍に薄めて
標準鉄溶液Bとする。
e) 標準鉄溶液C (10μgFe/ml) 7.2.2 d)による。
7.3.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
7.3.4
操作
7.3.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
7.3.4.2
発光強度の測定 7.3.4.1 b)で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に
噴霧し,波長259.940nmにおける発光強度を測定する(6)。
7.3.5
空試験 7.3.6の検量線の作成操作において得られる,標準鉄溶液を添加しない溶液の発光強度を,
空試験の発光強度とする。
7.3.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) ニッケル [7.3.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
b) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
c) これらの溶液に,標準鉄溶液A [7.3.2 c)],標準鉄溶液B [7.3.2 d)] 及び標準鉄溶液C [7.3.2 e)] の各種
液量(鉄として0〜50mg)を段階的に加え,水で標線まで薄める。
d) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長259.940nmにお
ける発光強度を試料と並行して測定し(6),得た発光強度と鉄量との関係線を作成し,その関係線を原
点を通るように平行移動して検量線とする。
7.3.7
計算 7.3.4.2及び7.3.5で得た発光強度と7.3.6で作成した検量線とから鉄量を求め,試料中の鉄含
有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Fe
−
−
=
12
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに, Fe: 鉄含有率 [% (m/m)]
A1: 試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
A3: ニッケル [7.3.2 b)] 5.0g中に含まれる鉄量 (g)
m: 試料はかり取り量 (g)
7.4
陰イオン交換分離ICP発光分光分析法
7.4.1
要旨 試料を硝酸で分解した後,シロップ状とし,塩酸を加えて濃縮を繰り返して硝酸を除去する。
水を加えて可溶性塩類を溶解した後,塩酸を加えて塩酸濃度を調節する。陰イオン交換カラムに通して鉄
を吸着させた後,塩酸とアスコルビン酸を通して鉄を溶出させる。溶出液をICP発光分光分析装置のアル
ゴンプラズマ中に噴霧し,その発光強度を測定する。
7.4.2
試薬 試薬は,次による。
a) 塩酸
b) 塩酸 (3+1, 1+100)
c) 硝酸 (1+1)
d) 水酸化ナトリウム溶液 (40g/l)
e) L (+) −アスコルビン酸溶液 (10g/l) 使用の都度,調製する。
f)
アスコルビン酸・塩酸溶液 6.5.2 f)による。
g) 標準鉄溶液A (50μgFe/ml) 7.2.2 c)による。
h) 標準鉄溶液B (10μgFe/ml) 7.2.2 d)による。
7.4.3
器具 器具は,次による。
陰イオン交換カラム 6.5.3の陰イオン交換カラムによる。
7.4.4
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
7.4.5
操作
7.4.5.1
準備操作 陰イオン交換カラム (7.4.3) に塩酸 (3+1) 50mlを通す。
7.4.5.2
試料溶液の調製 試料溶液の調製は,6.5.5.2による。
7.4.5.3
鉄の分離 7.4.5.2で得た溶液を毎分0.5〜1mlの流量で7.4.5.1の準備操作を終わった陰イオン交
換カラムに通す。樹脂上に溶液がなくなってから塩酸 (3+1) 50mlを数回に分けてビーカー内壁を洗浄し,
その都度,カラムに通す。これまでの流出液は,すべて捨てる。次にアスコルビン酸・塩酸溶液 [7.4.2 f)]
90mlを毎分0.5〜1mlの流量でカラムに通し,溶出液を100mlの全量フラスコに受けた後,アスコルビン
酸・塩酸溶液 [7.4.2 f)] で標線まで薄める(7)。
7.4.5.4
発光強度の測定 7.4.5.3で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴
霧し,波長259.940nmにおける発光強度を測定する(6)。
7.4.6
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
7.4.7
検量線の作成 検量線の作成は,次の手順によって行う。
a) 標準鉄溶液A [7.4.2 g)] 及び標準鉄溶液B [7.4.2 h)] の各種液量(鉄として0〜2 500μg)を段階的に数
個の100mlの全量フラスコに取る。
b) これらの溶液に,L (+) −アスコルビン酸溶液 [7.4.2 e)] 10ml及び塩酸20mlを加え,水で標線まで薄
める。
c) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長259.940nmにお
ける発光強度を試料と並行して測定し(6),得た発光強度と鉄量との関係線を作成し,その関係線を原
点を通るように平行移動して検量線とする。
13
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.4.8
計算 7.4.5.4及び7.4.6で得た発光強度と7.4.7で作成した検量線とから鉄量を求め,試料中の鉄含
有率を,次の式によって算出する。
100
2
1
×
m
A
A
Fe
−
=
ここに, Fe: 鉄含有率 [% (m/m)]
A1: 試料溶液中の鉄検出量 (g)
A2: 空試験液中の鉄検出量 (g)
m: 試料はかり取り量 (g)
8. 銅定量方法
8.1
定量方法の区分 銅の定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,銅含有率0.000 5% (m/m) 以上1.0% (m/m) 以下の試料に適用する。
b) ICP発光分光分析法 この方法は,銅含有率0.000 5% (m/m) 以上0.5% (m/m) 以下の試料に適用する。
c) 陰イオン交換分離ICP発光分光分析法 この方法は,銅含有率0.000 1% (m/m) 以上0.05% (m/m) 以
下の試料に適用する。
8.2
フレーム原子吸光法
8.2.1
要旨 試料を硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,
その吸光度を測定する。
8.2.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 銅含有率が既知(10)で,かつ,その銅含有率が試料中の銅含有率より
低いもの。
c) 標準銅溶液A (50μgCu/ml) 銅[99.9% (m/m) 以上]1.000gを硝酸 (1+1) 20mlで分解し,常温まで
冷却した後,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原液
(1mgCu/ml) とする。この原液を使用の都度,必要量だけ水で正しく20倍に薄めて標準銅溶液Aとす
る。
d) 標準銅溶液B (10μgCu/ml) c)の原液 (1mgCu/ml) を使用の都度,必要量だけ水で正しく100倍に薄
めて標準銅溶液Bとする。
注(10) 銅含有率は,8.4の陰イオン交換分離ICP発光分光分析法によって求める。
8.2.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
8.2.4
操作
8.2.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(11)。
注(11) この溶液中の銅含有率が1 000μgを超える場合には,銅量が100〜1 000μgとなるように100ml
の全量フラスコに分取し,水で標線まで薄める。
8.2.4.2
吸光度の測定 8.2.4.1 b)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長324.8nmにおける吸光度を測定する。
8.2.5
空試験 空試験は,次のいずれかによる。
a) 8.2.4.1 b)で分取しない場合 8.2.6 a)の検量線の作成操作において得られる,標準銅溶液を添加しない
14
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
溶液の吸光度を,空試験の吸光度とする。
b) 8.2.4.1 b)で分取する場合 8.2.6 b)の検量線の作成操作において得られる,標準銅溶液を添加しない溶
液の吸光度を,空試験の吸光度とする。
8.2.6
検量線の作成 検量線の作成は,次のいずれかの手順によって行う。
a) 8.2.4.1 b)で分取しない場合
1) ニッケル [8.2.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
2) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
3) これらの溶液に,標準銅溶液A [8.2.2 c)] 及び標準銅溶液B [8.2.2 d)] の各種液量(銅として0〜1
000μg)を段階的に加え,水で標線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長324.8nmにおける吸光度を試料と並行して測定し,得た吸光度と銅量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 8.2.4.1 b)で分取する場合
1) ニッケル [8.2.2 b)] を5.0gはかり取り,ビーカー (300ml) に移し入れる。
2) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄
める。
3) この溶液から8.2.4.1 b)で分取した試料溶液と同じ量を数個の100mlの全量フラスコに分取し,標準
銅溶液A [8.2.2 c)] 及び標準銅溶液B [8.2.2 d)] の各種液量(銅として0〜1 000μg)を段階的に加え,
水で標線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長324.8nmにおける吸光度を試料と並行して測定し,得た吸光度と銅量との関係線
を作成し,その関係線を原点を通るように平行移動して検量線とする。
8.2.7
計算 計算は,次のいずれかによる。
a) 8.2.4.1 b)で分取しなかった場合 8.2.4.2及び8.2.5 a)で得た吸光度と8.2.6 a)で作成した検量線とから
銅量を求め,試料中の銅含有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Cu
−
−
=
ここに, Cu: 銅含有率 [% (m/m)]
A1:
試料溶液中の銅検出量 (g)
A2: 空試験液中の銅検出量 (g)
A3: ニッケル [8.2.2 b)] 5.0g中に含まれる銅量 (g)
m: 試料はかり取り量 (g)
b) 8.2.4.1 b)で分取した場合 8.2.4.2及び8.2.5 b)で得た吸光度と8.2.6 b)で作成した検量線とから銅量を
求め,試料中の銅含有率を,次の式によって算出する。
100
100
100)
(
6
5
4
×
×
×
B
m
B
A
A
A
Cu
−
−
=
ここに, Cu: 銅含有率 [% (m/m)]
A4: 分取した試料溶液中の銅検出量 (g)
A5: 分取した空試験液中の銅検出量 (g)
A6: ニッケル [8.2.2 b)] 5.0g中に含まれる銅量 (g)
15
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
8.3
ICP発光分光分析法
8.3.1
要旨 試料を硝酸で分解した後,溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,そ
の発光強度を測定する。
8.3.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 8.2.2 b)による。
c) 標準銅溶液A (1mgCu/ml) 8.2.2 c)の原液 (1mgCu/ml) を使用する。
d) 標準銅溶液B (100μgCu/ml) 標準銅溶液A [c)] を使用の都度,必要量だけ水で正しく10倍に薄めて
標準銅溶液Bとする。
e) 標準銅溶液C (10μgCu/ml) 8.2.2 d)による。
8.3.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
8.3.4
操作
8.3.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
8.3.4.2
発光強度の測定 8.3.4.1 b)で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に
噴霧し,波長324.754nmにおける発光強度を測定する(6)。
8.3.5
空試験 8.3.6の検量線の作成操作において得られる,標準銅溶液を添加しない溶液の発光強度を,
空試験の発光強度とする。
8.3.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) ニッケル [8.3.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
b) 5.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
c) これらの溶液に,標準銅溶液A [8.3.2 c)] 及び標準銅溶液B [8.3.2 d)] 及び標準銅溶液C [8.3.2 e)] の各
種液量(銅として0〜25mg)を段階的に加え,水で標線まで薄める。
d) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長324.754nmにお
ける発光強度を試料と並行して測定し,得た発光強度と銅量との関係線を作成し,その関係線を原点
を通るように平行移動して検量線とする。
8.3.7
計算 8.3.4.2及び8.3.5で得た発光強度と8.3.6で作成した検量線とから銅量を求め,試料中の銅含
有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Cu
−
−
=
ここに, Cu: 銅含有率 [% (m/m)]
A1: 試料溶液中の銅検出量 (g)
A2: 空試験液中の銅検出量 (g)
A3: ニッケル [8.3.2 b)] 5.0g中に含まれる銅量 (g)
m: 試料はかり取り量 (g)
8.4
陰イオン交換分離ICP発光分光分析法
16
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.4.1
要旨 試料を硝酸で分解した後,シロップ状とし,塩酸を加えて濃縮を繰り返して硝酸を除去する。
水を加えて可溶性塩類を溶解した後,塩酸を加えて塩酸濃度を調節する。陰イオン交換カラムに通して銅
を吸着させた後,塩酸とアスコルビン酸を通して銅を溶出させる。溶出液をICP発光分光分析装置のアル
ゴンプラズマ中に噴霧し,その発光強度を測定する。
8.4.2
試薬 試薬は,次による。
a) 塩酸
b) 塩酸 (3+1, 1+100)
c) 硝酸 (1+1)
d) 水酸化ナトリウム溶液 (40g/l)
e) L (+) -アスコルビン酸溶液 (10g/l) 使用の都度,調製する。
f)
アスコルビン酸・塩酸溶液 6.5.2 f)による。
g) 標準銅溶液A (50μgCu/ml) 8.2.2 c)による。
h) 標準銅溶液B (10μgCu/ml) 8.2.2 d)による。
8.4.3
器具 器具は,次による。
陰イオン交換カラム 6.5.3の陰イオン交換カラムによる。
8.4.4
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
8.4.5
操作
8.4.5.1
準備操作 陰イオン交換カラム (8.4.3) に塩酸 (3+1) 50mlを通す。
8.4.5.2
試料溶液の調製 試料溶液の調製は,6.5.5.2による。
8.4.5.3
銅の分離 8.4.5.2で得た溶液を毎分0.5〜1mlの流量で8.4.5.1の準備操作で終わった陰イオン交
換カラムに通す。樹脂上に溶液がなくなってから塩酸 (3+1) 50mlを数回に分けてビーカー内壁を洗浄し,
その都度,カラムに通す。これまでの流出液は,すべて捨てる。次にアスコルビン酸・塩酸溶液 [8.4.2 f)]
90mlを毎分0.5〜1mlの流量でカラムに通し,溶出液を100mlの全量フラスコに受けた後,アスコルビン
酸・塩酸溶液 [8.4.2 f)] で標線まで薄める(7)。
8.4.5.4
発光強度の測定 8.4.5.3で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴
霧し,波長324.754nmにおける発光強度を測定する(6)。
8.4.6
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
8.4.7
検量線の作成 検量線の作成は,次の手順によって行う。
a) 標準銅溶液A [8.4.2 g)] 及び標準銅溶液B [8.4.2 h)] の各種液量(銅として0〜2 500μg)を段階的に数
個の100mlの全量フラスコに取る。
b) これらの溶液に,L (+) −アスコルビン酸溶液 [8.4.2 e)] 10ml及び塩酸20mlを加え,水で標線まで薄
める。
c) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長324.754nmにお
ける発光強度を試料と並行して測定し(6),得た発光強度と銅量との関係線を作成し,その関係線を原
点を通るように平行移動して検量線とする。
8.4.8
計算 8.4.5.4及び8.4.6で得た発光強度と8.4.7で作成した検量線とから銅量を求め,試料中の銅含
有率を,次の式によって算出する。
100
2
1
×
m
A
A
Cu
−
=
ここに, Cu: 銅含有率 [% (m/m)]
17
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A1: 試料溶液中の銅検出量 (g)
A2: 空試験液中の銅検出量 (g)
m: 試料はかり取り量 (g)
9. 鉛定量方法
9.1
定量方法の区分 鉛の定量方法は,次のいずれかによる。
a) 水酸化鉄共沈分離フレーム原子吸光法 この方法は,鉛含有率0.000 2% (m/m) 以上0.01% (m/m) 以下
の試料に適用する。
b) 水酸化鉄共沈分離ICP発光分光分析法 この方法は,鉛含有率0.000 1% (m/m) 以上0.01% (m/m) 以
下の試料に適用する。
c) フレーム原子吸光法 この方法は,鉛含有率0.000 5% (m/m) 以上0.01% (m/m) 以下の試料に適用する。
d) 電気加熱原子吸光法 この方法は,鉛含有率0.000 1% (m/m) 以上0.001% (m/m) 以下の試料に適用す
る。
9.2
水酸化鉄共沈分離フレーム原子吸光法
9.2.1
要旨 試料を硝酸で分解した後,鉄 (III),アンモニア水及び炭酸アンモニウムを加え,鉛を水酸
化鉄 (III) と共沈させて,こし分ける。沈殿を塩酸に溶解した後,溶液を原子吸光光度計の空気・アセチ
レンフレーム中に噴霧し,その吸光度を測定する。
9.2.2
試薬 試薬は,次による。
a) 塩酸 (1+1,1+50)
b) 硝酸 (1+1)
c) アンモニア水
d) アンモニア洗浄液 アンモニア水 (2+50) 500mlに炭酸アンモニウム15gを溶解する。
e) 炭酸アンモニウム
f)
鉄 (III) 溶液 鉄[99.9% (m/m) 以上]0.50gを塩酸 (1+1) 20mlと過酸化水素2mlとで分解した後,
加熱して過剰の過酸化水素を追い出す。常温まで冷却した後,水で液量を100mlとする。この溶液1ml
は鉄 (III) 5mgを含む。
g) 標準鉛溶液A (50μgPb/ml) 鉛[99.9% (m/m) 以上]0.100gを硝酸 (1+1) 20mlで分解し,常温まで
冷却した後,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原液
(100μgPb/ml) とする。この原液を使用の都度,必要量だけ水で正しく2倍に薄めて標準鉛溶液Aとす
る。
h) 標準鉛溶液B (10μgPb/ml) g)の原液を使用の都度,必要量だけ水で正しく5倍に薄めて標準鉛溶液
Bとする。
9.2.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
9.2.4
操作
9.2.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取って,ビーカー (500ml) に移し入れる。
b) 時計皿で覆い,硝酸 (1+1) 40mlを加えて穏やかに加熱して分解した後,煮沸して酸化窒素などを追
い出す。
c) 放冷した後,時計皿の下面を水で洗って時計皿を取り除き,水を加えて液量を約150mlとする。
9.2.4.2
鉛の分離 鉛の分離は,次の手順によって行う。
18
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 9.2.4.1 c)で得た溶液に鉄 (III) 溶液 [9.2.2 f)] 5mlを加え,この溶液をかき混ぜながらアンモニア水
20mlを少量ずつ加え,さらに50mlを加える。次に炭酸アンモニウム約15gを加えて溶解し,時計皿
で覆い,加熱して約5分間煮沸した後,60〜80℃で1〜2時間放置し,時計皿の下面を水で洗って時計
皿を取り除く。
b) 沈殿をろ紙(5種A)を用いてこし分け,温アンモニア洗浄液 [9.2.2 d)] で数回洗浄する。ろ液及び
洗液は捨てる。
c) ろ紙上の沈殿を温水で元のビーカーに洗い移し,漏斗の下に元のビーカーを置き,ろ紙上に塩酸 (1
+1) 約10mlを滴加して,ろ紙上及びビーカー中に残存する沈殿を溶解し,ろ紙を温塩酸 (1+50) で
十分に洗浄する。
d) 溶液を加熱して乾固直前まで濃縮する。塩酸 (1+1) 5ml及び水5mlを加えて加熱して塩類を溶解し,
常温まで冷却した後,溶液を25mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
9.2.4.3
吸光度の測定 9.2.4.2 d)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長217.0nm又は波長283.3nmにおける吸光度を測定する。
9.2.5
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
9.2.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) 標準鉛溶液A [9.2.2 g)] 及び標準鉛溶液B [9.2.2 h)] の各種液量(鉛として0〜500μg)を段階的に数個
の25mlの全量フラスコに取る。
b) 鉄 (III) 溶液 [9.2.2 f)] 5ml及び塩酸 (1+1) 5mlを加え,水で標線まで薄める。
c) 溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム中に噴霧し,
波長217.0nm又は波長283.3nmにおける吸光度を試料と並行して測定し,得た吸光度と鉛量との関係
線を作成し,その関係線を原点を通るように平行移動して検量線とする。
9.2.7
計算 9.2.4.3及び9.2.5で得た発光強度と9.2.6で作成した検量線とから鉛量を求め,試料中の鉛含
有率を,次の式によって算出する。
100
2
1
×
m
A
A
Pb
−
=
ここに, Pb: 鉛含有率 [% (m/m)]
A1: 試料溶液中の鉛検出量 (g)
A2: 空試験液中の鉛検出量 (g)
m: 試料はかり取り量 (g)
9.3
水酸化鉄共沈分離ICP発光分光分析法
9.3.1
要旨 試料を硝酸で分解した後,鉄 (III),アンモニア水及び炭酸アンモニウムを加え,鉛を水酸
化鉄 (III) と共沈させて,こし分ける。沈殿を塩酸に溶解した後,溶液をICP発光分光分析装置のアルゴ
ンプラズマ中に噴霧し,その発光強度を測定する。
9.3.2
試薬 試薬は,次による。
a) 塩酸 (1+1,1+50)
b) 硝酸 (1+1)
c) アンモニア水
d) アンモニア洗浄液 9.2.2 d)による。
e) 炭酸アンモニウム
f)
鉄 (III) 溶液 9.2.2 f)による。
19
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) 標準鉛溶液A (50μgPb/ml) 9.2.2 g)による。
h) 標準鉛溶液B (10μgPb/ml) 9.2.2 h)による。
9.3.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
9.3.4
操作
9.3.4.1
試料溶液の調製 試料溶液の調製は,9.2.4.1による。
9.3.4.2
鉛の分離 鉛の分離は,次の手順によって行う。
a) 9.3.4.1で得た溶液に鉄 (III) 溶液 [9.3.2 f)] 5mlを加え,この溶液をかき混ぜながらアンモニア水20ml
を少量ずつ加え,さらに50mlを加える。次に炭酸アンモニウム約15gを加えて溶解し,時計皿で覆
い,加熱して約5分間煮沸した後,60〜80℃で1〜2時間放置し,時計皿の下面を水で洗って時計皿を
取り除く。
b) 沈殿をろ紙(5種A)を用いてこし分け,温アンモニア洗浄液 [9.3.2 d)] で数回洗浄する。ろ液及び
洗浄液は捨てる。
c) 9.2.4.2のc)及びd)の手順に従って操作する。
9.3.4.3
発光強度の測定 9.3.4.2 c)で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に
噴霧し,波長220.353nmにおける発光強度を測定する(6)。
9.3.5
空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
9.3.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) 標準鉛溶液A [9.3.2 g)] 及び標準鉛溶液B [9.3.2 h)] の各種液量(鉛として0〜500μg)を段階的に数個
の25mlの全量フラスコに取る。
b) 鉄 (III) 溶液 [9.3.2 f)] 5ml及び塩酸 (1+1) 5mlを加え,水で標線まで薄める。
c) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長220.353nmにお
ける発光強度を試料と並行して測定し(6),得た発光強度と鉛量との関係線を作成し,その関係線を原
点を通るように平行移動して検量線とする。
9.3.7
計算 9.3.4.3及び9.3.5で得た発光強度と9.3.6で作成した検量線とから鉛量を求め,試料中の鉛含
有率を,次の式によって算出する。
100
2
1
×
m
A
A
Pb
−
=
ここに, Pb: 鉛含有率 [% (m/m)]
A1: 試料溶液中の鉛検出量 (g)
A2: 空試験液中の鉛検出量 (g)
m: 試料はかり取り量 (g)
9.4
フレーム原子吸光法
9.4.1
要旨 試料を硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,
その吸光度を測定する。
9.4.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 鉛含有率が既知(12)で,かつ,その鉛含有率が試料中の鉛含有率より
低いもの。
c) 標準鉛溶液A (50μgPb/ml) 9.2.2 g)による。
d) 標準鉛溶液B (10μgPb/ml) 9.2.2 h)による。
20
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(12) 鉛含有率は,9.2の水酸化鉄共沈分離原子吸光法又は9.3の水酸化鉄共沈分離ICP発光分光分析法
によって求める。
9.4.3
試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
9.4.4
操作
9.4.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
9.4.4.2
吸光度の測定 9.4.4.1 b)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長217.0nm又は283.3nmにおける吸光度を測定する。
9.4.5
空試験 9.4.6の検量線の作成操作において得られる,標準鉛溶液を添加しない溶液の吸光度を,
空試験の吸光度とする。
9.4.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) ニッケル [9.4.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
b) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
c) これらの溶液に,標準鉛溶液A [9.4.2 c)] 及び標準鉛溶液B [9.4.2 d)] の各種液量(鉛として0〜500μg)
を段階的に加え,水で標線まで薄める。
d) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム中
に噴霧し,波長217.0nm又は283.3nmにおける吸光度を試料と並行して測定し,得た吸光度と鉛量と
の関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
9.4.7
計算 計算は,9.4.4.2及び9.4.5で得た吸光度と9.4.6で作成した検量線とから鉛量を求め,試料中
の鉛含有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Pb
−
−
=
ここに, Pb: 鉛含有率 [% (m/m)]
A1: 試料溶液中の鉛検出量 (g)
A2: 空試験液中の鉛検出量 (g)
A3: ニッケル [9.4.2 b)] 5.0g中に含まれる鉛量 (g)
m: 試料はかり取り量 (g)
9.5
電気加熱原子吸光法
9.5.1
要旨 試料を硝酸で分解した後,溶液を原子吸光光度計の電気加熱炉に導入し,加熱原子化してそ
の吸光度を測定する。
9.5.2
試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 9.4.2 b)による。
c) 標準鉛溶液 (1μgPb/ml) 9.2.2 g)の原液 (100μgPb/ml) を使用の都度,必要量だけ水で正しく100倍に
薄めて標準鉛溶液 (1μgPb/ml) とする。
9.5.3
試料はかり取り量 試料はかり取り量は,1.0gとし,10mgのけたまではかる。
9.5.4
操作
9.5.4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
21
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.5.4.2
吸光度の測定 9.5.4.1 b)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の電
気加熱炉に導入し,加熱原子化して波長283.3nmにおける吸光度を測定する。
9.5.5
空試験 9.5.6の検量線の作成操作において得られる,標準鉛溶液を添加しない溶液の吸光度を,
空試験の吸光度とする。
9.5.6
検量線の作成 検量線の作成は,次の手順によって行う。
a) ニッケル [9.5.2 b)] を1.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
b) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
c) これらの溶液に,標準鉛溶液 [9.5.2 c)] の各種液量(鉛として0〜10μg)を段階的に加え,水で標線ま
で薄める。
d) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の電気加熱炉に導入し,加熱原
子化して波長283.3nmにおける吸光度を試料と並行して測定し,得た吸光度と鉛量との関係線を作成
し,その関係線を原点を通るように平行移動して検量線とする。
9.5.7
計算 計算は,9.5.4.2及び9.5.5で得た吸光度と9.5.6で作成した検量線とから鉛量を求め,試料中
の鉛含有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Pb
−
−
=
ここに, Pb: 鉛含有率 [% (m/m)]
A1: 試料溶液中の鉛検出量 (g)
A2: 空試験液中の鉛検出量 (g)
A3: ニッケル [9.5.2 b)] 5.0g中に含まれる鉛量 (g)
m: 試料はかり取り量 (g)
10. マンガン定量方法
10.1 定量方法の区分 マンガンの定量方法は,次のいずれかによる。
a) フレーム原子吸光法 この方法は,マンガン含有率0.000 1% (m/m) 以上0.2% (m/m) 以下の試料に適
用する。
b) ICP発光分光分析法 この方法は,マンガン含有率0.000 1% (m/m) 以上0.004% (m/m) 以下の試料に
適用する。
c) 水酸化鉄共沈分離ICP発光分光分析法 この方法は,マンガン含有率0.000 05% (m/m) 以上0.004%
(m/m) 以下の試料に適用する。
10.2 フレーム原子吸光法
10.2.1 要旨 試料を硝酸で分解した後,溶液を原子吸光光度計の空気・アセチレンフレーム中に噴霧し,
その吸光度を測定する。
10.2.2 試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] マンガン含有率が既知(13)で,かつ,そのマンガン含有率が試料中の
マンガン含有率より低いもの。
c) 標準マンガン溶液 (10μgMn/ml) マンガン[99.9% (m/m) 以上]1.000gを硝酸 (1+1) 20mlで分解し,
常温まで冷却した後,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原
液 (1mgMn/ml) とする。この原液を使用の都度,必要量だけ水で正しく100倍に薄めて標準マンガン
溶液とする。
22
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(13) マンガン含有率は,10.4の水酸化鉄共沈分離ICP発光分光分析法によって求める。
10.2.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
10.2.4 操作
10.2.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(14)。
注(14) この溶液中のマンガン含有率が200μgを超える場合には,マンガン量が20〜200μgとなるように
100mlの全量フラスコに分取し,水で標線まで薄める。
10.2.4.2 吸光度の測定 10.2.4.1 b)で得た溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長279.5nmにおける吸光度を測定する。
10.2.5 空試験 空試験は,次のいずれかによる。
a) 10.2.4.1 b)で分取しない場合 10.2.6 a)の検量線の作成操作において得られる,標準マンガン溶液を添
加しない溶液の吸光度を,空試験の吸光度とする。
b) 10.2.4.1 b)で分取する場合 10.2.6 b)の検量線の作成操作において得られる,標準マンガン溶液を添加
しない溶液の吸光度を,空試験の吸光度とする。
10.2.6 検量線の作成 検量線の作成は,次のいずれかの手順によって行う。
a) 10.2.4.1 b)で分取しない場合
1) ニッケル [10.2.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
2) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
3) これらの溶液に,標準マンガン溶液 [10.2.2 c)] の各種液量(マンガンとして0〜200μg)を段階的に
加え,水で標線まで薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長279.5nmにおける吸光度を試料と並行して測定し,得た吸光度とマンガン量との
関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
b) 10.2.4.1 b)で分取する場合
1) ニッケル [10.2.2 b)] を5.0gはかり取り,ビーカー (300ml) に移し入れる。
2) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄
める。
3) この溶液から10.2.4.1 b)で分取した試料溶液と同じ量を数個の100mlの全量フラスコに分取し,標
準マンガン溶液 [10.2.2 c)] の各種液量(マンガンとして0〜200μg)を段階的に加え,水で標線まで
薄める。
4) これらの溶液の一部を,水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム
中に噴霧し,波長279.5nmにおける吸光度を試料と並行して測定し,得た吸光度とマンガン量との
関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
10.2.7 計算 計算は,次のいずれかによる。
a) 10.2.4.1 b)で分取しなかった場合 10.2.4.2及び10.2.5 a)で得た吸光度と10.2.6 a)で作成した検量線と
からマンガン量を求め,試料中のマンガン含有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Mn
−
−
=
ここに, Mn: マンガン含有率 [% (m/m)]
23
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A1: 試料溶液中のマンガン検出量 (g)
A2: 空試験液中のマンガン検出量 (g)
A3: ニッケル [10.2.2 b)] 5.0g中に含まれるマンガン量 (g)
m: 試料はかり取り量 (g)
b) 10.2.4.1 b)で分取した場合 10.2.4.2及び10.2.5 b)で得た吸光度と10.2.6 b)で作成した検量線とからマ
ンガン量を求め,試料中のマンガン含有率を,次の式によって算出する。
100
100
)
100
6
5
4(
×
×
×
B
m
B
A
A
A
Mn
−
−
=
ここに, Mn: マンガン含有率 [% (m/m)]
A4: 分取した試料溶液中のマンガン検出量 (g)
A5: 分取した空試験液中のマンガン検出量 (g)
A6: ニッケル [10.2.2 b)] 5.0g中に含まれるマンガン量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
10.3 ICP発光分光分析法
10.3.1 要旨 試料を硝酸で分解した後,溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,そ
の発光強度を測定する。
10.3.2 試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 10.2.2 b)による。
c) 標準マンガン溶液 (10μgMn/ml) 10.2.2 c)による。
10.3.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
10.3.4 操作
10.3.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
10.3.4.2 発光強度の測定 10.3.4.1 b)で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に
噴霧し,波長257.610nmにおける発光強度を測定する(6)。
10.3.5 空試験 10.3.6の検量線の作成操作において得られる,標準マンガン溶液を添加しない溶液の発光
強度を,空試験の発光強度とする。
10.3.6 検量線の作成 検量線の作成は,次の手順によって行う。
a) ニッケル [10.3.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
b) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
c) これらの溶液に,標準マンガン溶液 [10.3.2 c)] の各種液量(マンガンとして0〜200μg)を段階的に加
え,水で標線まで薄める。
d) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長257.610nmにお
ける発光強度を試料と並行して測定し(6),得た発光強度とマンガン量との関係線を作成し,その関係
線を原点を通るように平行移動して検量線とする。
10.3.7 計算 10.3.4.2及び10.3.5で得た発光強度と10.3.6で作成した検量線とからマンガン量を求め,試
料中のマンガン含有率を,次の式によって算出する。
24
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
)
(
3
2
1
×
m
A
A
A
Mn
−
−
=
ここに, Mn: マンガン含有率 [% (m/m)]
A1: 試料溶液中のマンガン検出量 (g)
A2: 空試験液中のマンガン検出量 (g)
A3: ニッケル [10.3.2 b)] 5.0g中に含まれるマンガン量 (g)
m: 試料はかり取り量 (g)
10.4 水酸化鉄共沈分離ICP発光分光分析法
10.4.1 要旨 試料を硝酸で分解した後,鉄 (III),アンモニア水及びペルオキソ二硫酸アンモニウムを加
え,マンガンを水酸化鉄 (III) と共沈させて,こし分ける。沈殿を塩酸及び過酸化水素に溶解した後,溶
液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,その発光強度を測定する。
10.4.2 試薬 試薬は,次による。
a) 塩酸 (1+1, 1+50)
b) 硝酸 (1+1)
c) 塩酸混液 塩酸 (1+3) 200mlに過酸化水素2mlを加える。この溶液は,使用の都度,調製する。
d) アンモニア水
e) ペルオキソ二硫酸アンモニウム
f)
ペルオキソ二硫酸アンモニウム洗浄液 ペルオキソ二硫酸アンモニウム10gをアンモニア水 (1+4)
500mlに溶解する。
g) 鉄 (III) 溶液 9.2.2 f)による。
h) 標準マンガン溶液 (10μgMn/ml) 10.2.2 c)による。
10.4.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
10.4.4 操作
10.4.4.1 試料溶液の調製 試料溶液の調製は,10.2.4.1による。
10.4.4.2 マンガンの分離 マンガンの分離は,次の手順によって行う。
a) 10.4.4.1で得た溶液に鉄 (III) 溶液 [10.4.2 g)] 5mlを加え,この溶液をかき混ぜながらアンモニア水
20mlを少量ずつ加え,さらに50mlを加える。次にペルオキソ二硫酸アンモニウム約5gを加えて溶解
し,時計皿で覆い,加熱して約5分間煮沸した後,60〜80℃で約15分間放置し,時計皿の下面を水で
洗って時計皿を取り除く。
b) 沈殿をろ紙を用いてこし分け,温ペルオキソ二硫酸アンモニウム洗浄液 [10.4.2 f)] で数回洗浄する。
ろ液及び洗液は捨てる。
c) ろ紙上の沈殿を温水で元のビーカーに洗い移し,漏斗の下に元のビーカーを置き,ろ紙上に塩酸混液
[10.4.2 c)] 約20mlを滴加して,ろ紙上及びビーカー中に残存する沈殿を溶解し,ろ紙を温塩酸 (1+
50) で十分に洗浄する。
d) 溶液を加熱して乾固直前まで濃縮する。塩酸 (1+1) 5ml及び水5mlを加えて加熱して塩類を溶解し,
常温まで冷却した後,溶液を50mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
10.4.4.3 発光強度の測定 10.4.4.2 d)で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に
噴霧し,波長257.610nmにおける発光強度を測定する(6)。
10.4.5 空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
10.4.6 検量線の作成 検量線の作成は,次の手順によって行う。
a) 標準マンガン溶液 [10.4.2 b)] の各種液量(マンガンとして0〜200μg)を段階的に数個の50mlの全量
25
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
フラスコに取る。
b) 鉄 (III) 溶液 [10.4.2 g)] 5ml及び塩酸 (1+1) 5mlを加え,水で標線まで薄める。
c) 溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長257.610nmにおける発光
強度を試料と並行して測定し(6),得た発光強度とマンガン量との関係線を作成し,その関係線を原点
を通るように平行移動して検量線とする。
10.4.7 計算 10.4.4.3及び10.4.5で得た発光強度と10.4.6で作成した検量線とからマンガン量を求め,試
料中のマンガン含有率を,次の式によって算出する。
100
2
1
×
m
A
A
Mn
−
=
ここに, Mn: マンガン含有率 [% (m/m)]
A1: 試料溶液中のマンガン検出量 (g)
A2: 空試験液中のマンガン検出量 (g)
m: 試料はかり取り量 (g)
11. 炭素定量方法
11.1 定量方法の区分 炭素の定量方法は,次のいずれかによる。
a) 燃焼−電量法 この方法は,炭素含有率0.001% (m/m) 以上0.1% (m/m) 以下の試料に適用する。
b) 燃焼−赤外線吸収法(積分法) この方法は,炭素含有率0.001% (m/m) 以上0.1% (m/m) 以下の試料
に適用する。
c) 燃焼−赤外線吸収法(循環法) この方法は,炭素含有率0.001% (m/m) 以上0.1% (m/m) 以下の試料
に適用する。
11.2 燃焼−電量法
11.2.1 要旨 試料を酸素気流中で加熱し,炭素を酸化して二酸化炭素とし,あらかじめ一定のpHに設定
した弱アルカリ性の過塩素酸バリウム溶液に吸収させる。このとき増加した水素イオンを,電気分解によ
って発生させた水酸化物イオンで中和し,そのときに要した電気量を測定する。
11.2.2 試薬 試薬は,JIS Z 2615の6.7.2(試薬)による。
11.2.3 装置 装置は,JIS Z 2615の6.7.3(装置)による。
11.2.4 試料はかり取り量 試料はかり取り量は,0.5〜10gとし,1mgのけたまではかる。
11.2.5 操作 操作は,次による。
a) 準備操作 準備操作は,JIS Z 2615の6.7.4(予備操作)による(15)。ただし,管状電気抵抗炉中央部で
の燃焼管内温度を1 300〜1 400℃に保持し,酸素流量は,毎分300mlとする。
注(15) 試料と同程度の炭素含有率をもつ鉄鋼標準試料を用いてもよい。
b) 定量操作 定量操作は,JIS Z 2615の6.7.5(定量操作)による(16)(17)。
注(16) 磁器燃焼ボート及び磁器燃焼ボートカバーは,あらかじめ1 400〜1 450℃で2時間以上空焼きし
たものを用いる。
(17) 助燃剤は,JIS Z 2615の5.(13)(助燃剤)に規定されたものを単独又は2,3種類組み合わせて
用いる。あらかじめ,最適な助燃剤の種類とその添加量及び加え方を炭素含有率既知の試料を
用いて調べておく。
11.2.6 空試験 試料に添加したのと同量の助燃剤だけを用いて11.2.5 b)の手順に従って操作する。
11.2.7 計算 計算は,JIS Z 2615の6.7.7(計算)による。
26
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.3 燃焼−赤外線吸収法(積分法)
11.3.1 要旨 試料を酸素気流中で加熱し,炭素を酸化して二酸化炭素及び一酸化炭素とし,酸素とともに
赤外線吸収検出器に送り,その赤外線吸収量を測定する。
11.3.2 材料 材料は,JIS Z 2615の6.9.2(材料)による。
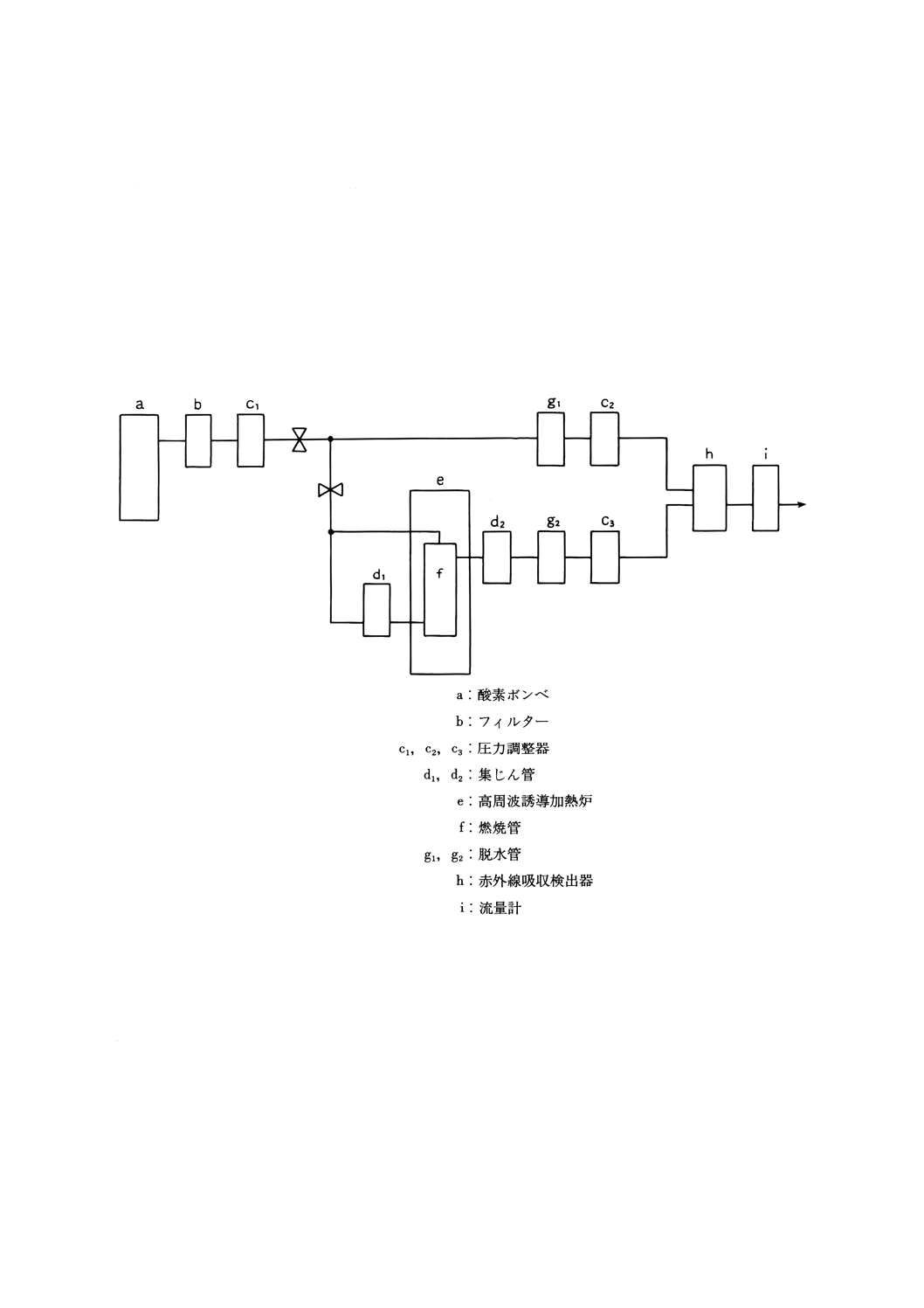
11.3.3 装置 装置は,JIS Z 2615の5.(6)の(a)の管状電気抵抗加熱炉又は(b)の高周波誘導加熱炉とJIS Z
2615の6.9.3の(1)の酸素精製部,(3)の燃焼ガス精製部及び(4)の二酸化炭素定量部とを接続して用いる。た
だし,JIS Z 2615の6.9.3(4)の二酸化炭素定量部の代わりに二酸化炭素及び一酸化炭素の合量の赤外線吸収
量を測定する方式のものを用いてもよい。この方式を用いる場合には,JIS Z 2615の6.9.3(3)の燃焼ガス精
製部の代わりに,JIS Z 2615の6.10.2(3)の燃焼ガス精製部を用いる。この場合の装置の概略図の例を図2
に示す。
図2 赤外線吸収法(二酸化炭素及び一酸化炭素積分法)の概略図
11.3.4 試料はかり取り量 試料はかり取り量は,0.5〜1.0gとし,1mgのけたまではかる。
11.3.5 操作 操作は,次のいずれかの手順によって行う。
a) 管状電気抵抗加熱炉を用いる場合
1) 準備操作
1.1)
電源を入れ,各部を十分に安定させ,酸素を340kPaで供給して装置の気密を確認する。燃焼管の中
央部の温度を1 250〜1 450℃に,酸化管の温度を700℃に保つ(18)。
1.2)
指示計のゼロ点を調整する。
1.3)
分析試料と同程度の炭素含有率の標準試料(15)を用い,2)の手順に従って操作し,次の式によって換
27
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
算係数を求める。
100
0
0
0
0
C
B
A
W
K
×
−
=
ここに,
K: 測定値 (A0−B0) を炭素量 (g) に換算するための係数
A0: 標準試料を用いて2) 2.2)で得た指示値
B0: 11.3.6 a)で得た指示値
C0: 標準試料の炭素含有率 [% (m/m)]
W0: 標準試料はかり取り量 (g)
2) 定量操作
2.1)
試料及び助燃剤(17)を磁器燃焼ボート(16)にはかり取り,磁器燃焼ボートカバー(16)で覆い,燃焼管の
中央部に挿入し,直ちに気密に栓をする(試料が燃焼し,燃焼ガスは精製部を経て赤外線吸収検出
器の試料セルに送られ,指示値が次第に増加する。)。
2.2)
指示計が一定値を示したとき指示値を読み取る。
注(18) 各部の器具及び材料の充てん,保守,機密試験などは,使用する装置の手引書に従って行う。
b) 高周波誘導加熱炉を用いる場合
1) 準備操作 準備操作は,JIS Z 2615の6.9.4(予備操作)による(15)。ただし,二酸化炭素定量部に二
酸化炭素及び一酸化炭素の合量の赤外線吸収量を測定する方式の装置を使用する場合は,JIS Z
2615の6.9.4(2)の窒素,アルゴン又は圧縮空気の代わりに酸素を用いてもよい。
2) 定量操作 定量操作は,JIS Z 2615の6.9.5(定量操作)による(17)(19)。
注(19) 高周波磁器燃焼るつぼ及びるつぼのふたは,1 400〜1 450℃で2時間以上空焼きしたものを用い
る。
11.3.6 空試験 空試験は,次のいずれかによる。
a) 管状電気抵抗加熱炉を用いる場合 試料に添加するのと同量の助燃剤をはかり取った磁器燃焼ボート
を用い,11.3.5 a) 2)の2.1)及び2.2)の手順に従って操作する。
b) 高周波誘導加熱炉を用いる場合 試料に添加するのと同量の助燃剤をはかり取ったるつぼを用い,
11.3.5 b) 2)の手順に従って操作する。
11.3.7 計算 計算は,JIS Z 2615の6.9.7(計算)による。
11.4 燃焼−赤外線吸収法(循環法)
11.4.1 要旨 試料を一定体積内,一定圧力下の循環酸素気流中で加熱し,炭素を酸化して二酸化炭素及び
一酸化炭素とし,これを過剰の酸素とともに循環ループの赤外線吸収検出器に送ってその赤外線吸収量を
それぞれ測定する。
11.4.2 装置 装置は,JIS Z 2615の6.10.2(装置)による。
11.4.3 試料はかり取り量 試料はかり取り量は,0.5〜10gとし,1mgのけたまではかる。
11.4.4 操作 操作は,次による。
a) 準備操作 準備操作は,JIS Z 2615の6.10.3(予備操作)による(15)。
b) 定量操作 定量操作は,JIS Z 2615の6.10.4(定量操作)による(17)(19)。
11.4.5 空試験 空試験は,JIS Z 2615の6.10.5(空試験)による。
11.4.6 計算 計算は,JIS Z 2615の6.10.6(計算)による。
12. 硫黄定量方法
12.1 定量方法の区分 硫黄の定量方法は,次のいずれかによる。
28
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 硫化水素気化分離メチレンブルー吸光光度法 この方法は,硫黄含有率0.000 1% (m/m) 以上0.002%
(m/m) 以下の試料に適用する(20)。
b) 燃焼−電量法 この方法は,硫黄含有率0.001% (m/m) 以上0.1% (m/m) 以下の試料に適用する。
c) 燃焼−赤外線吸収法(積分法) この方法は,硫黄含有率0.000 1% (m/m) 以上0.1% (m/m) 以下の試
料に適用する。
d) 燃焼−赤外線吸収法(循環法) この方法は,硫黄含有率0.001% (m/m) 以上0.1% (m/m) 以下の試料
に適用する。
e) 燃焼−よう素酸カリウム滴定法 この方法は,硫黄含有率0.001% (m/m) 以上0.3% (m/m) 以下の試料
に適用する。
注(20) この方法は他の方法よりも複雑であり,他の方法では適用できない試料,又は他の方法で要求
される装置がない場合に勧められる。
12.2 硫化水素気化分離メチレンブルー吸光光度法
12.2.1 要旨 試料を硝酸と塩素酸カリウムとで分解し,硫黄を硫酸に酸化する。塩酸とぎ酸を加え,蒸発
させて硝酸を除去する。アルゴン又は窒素雰囲気中でよう化水素酸と次亜りん酸とによって,硫酸を硫化
水素に還元して気化させ,亜鉛アンミン錯体溶液に吸収させた後,N, N−ジメチル−p−フェニレンジアミ
ン及びFe (III) を加えてメチレンブルーを生成させ,光度計を用いてその吸光度を測定する。
12.2.2 試薬 試薬は,次による。
a) 塩酸
b) 塩酸 (3+2, 1+4)
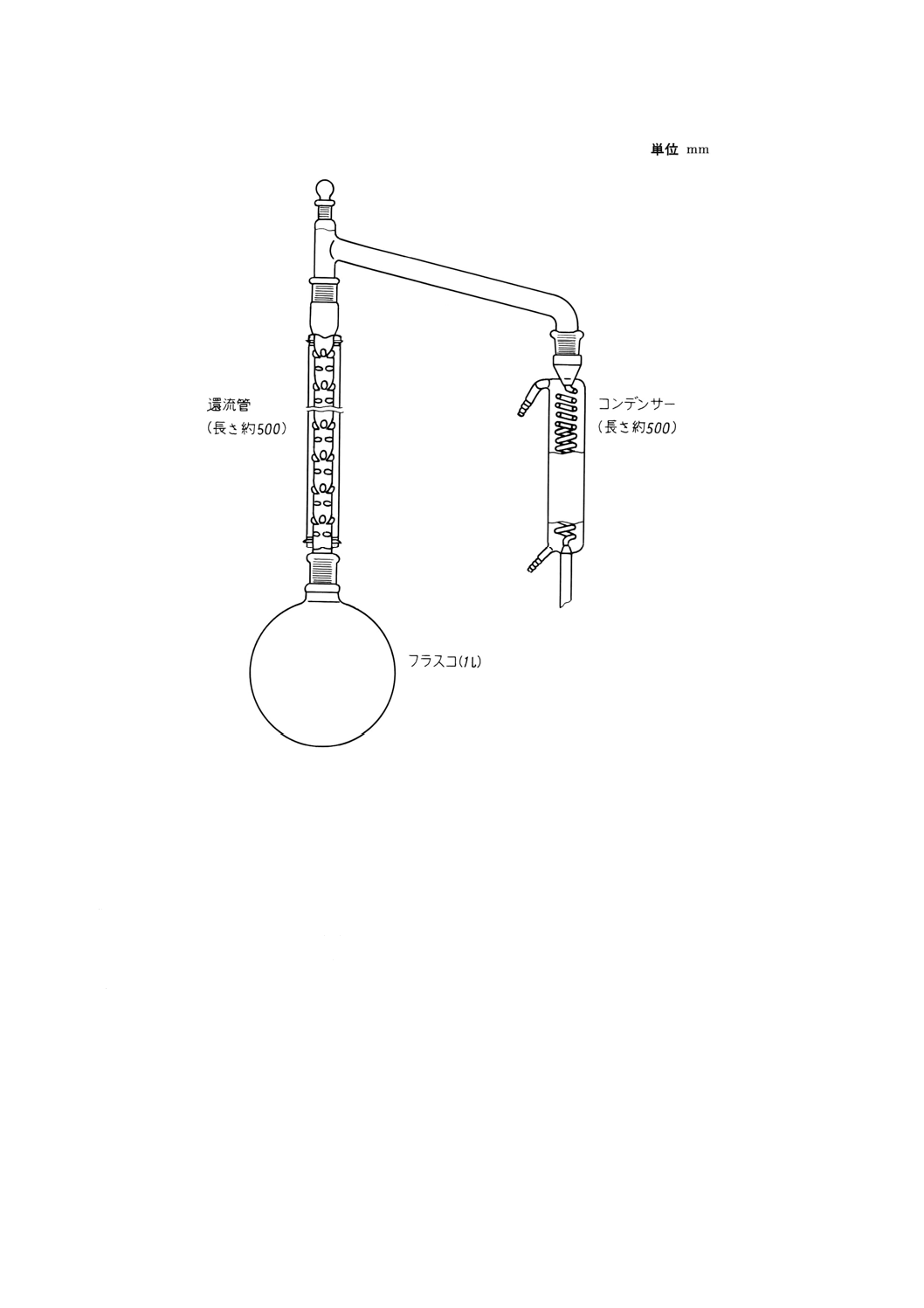
c) 精製塩酸 塩酸 (3+2) を図3に示す装置を使用して蒸留する。この際,留出液全量の101に相当する
最初の留出液及び最後の数mlの留出液は捨てる。留出液を清浄なガラス瓶の中に集める。この精製
塩酸に含まれる硫黄は0.005mg/l以下でなければならない。
d) 硝酸
e) 精製硝酸 硝酸を図3に示す装置を使用して蒸留する。この際,留出液全量の101に相当する最初の留
出液及び最後の数mlの留出液は捨てる。留出液を清浄なガラス瓶の中に集める。
29
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図3 酸精製装置の例
f)
硝酸・塩素酸混酸 塩素酸カリウム3gを水30mlに溶解し,精製硝酸 [e)] を100ml加える。使用の
都度,調製する。
g) アルゴン[99.99% (v/v) 以上](21)
h) 窒素[99.5% (v/v) 以上](21)
i)
塩化カリウム溶液 (10g/l)
j)
塩化鉄 (III) 溶液 塩化鉄 (III) 六水和物1.0gに塩酸10ml及び水40mlを加えて溶解し,水で液量を
100mlとする。ガラス瓶に保存する。
k) 還元混合液 よう化水素酸420ml,次亜りん酸溶液 [ρ20=1.2g/ml, 50% (m/m)] 80ml及びよう化ナトリ
ウム70gを図4に示す精製装置のフラスコに入れる。酢酸亜鉛吸収液 [n)] 50mlを入れた硫化水素ト
ラップを装着する。アルゴン又は窒素を流量200〜300ml/minで10分間送入し,装置から空気を追い
出す。マントルヒーターのスイッチを入れ,混合液を113〜115℃で4時間アルゴン又は窒素を流しな
がら加熱した後,アルゴン又は窒素を流したまま混合液を冷却する。室温まで冷却した後直ちに混合
液を褐色ガラス瓶に移し,栓をして冷暗所に保存する。
30
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図4 還元混合液精製装置の例
l)
ぎ酸
m) 精製ぎ酸 ぎ酸を図3に示す装置を使用して蒸留する。この際留出液全量の101に相当する最初の留出
液及び最後の数mlの留出液は捨てる。留出液を清浄なガラス瓶の中に集める。
n) 酢酸亜鉛吸収液 酢酸亜鉛二水和物5g及び塩化アンモニウム70gを約350mlの水で溶解する。水酸
化ナトリウム7.5gを加え,かき混ぜて溶解し,水で液量を500mlとする。ガラス瓶の中に保存する。
o) ジアミン塩溶液 N, N−ジメチル−p−フェニレンジアミン塩酸塩又はN, N−ジメチル−p−フェニレ
ンジアミン硫酸塩0.1gを塩酸26mlに溶解し,水で液量を100mlとする。冷暗所に保存し,毎週新し
いものを調製する。
p) 標準硫黄溶液 (100mgS/l) あらかじめ105℃で1時間乾燥させた硫酸カリウムを正確に0.543 5gは
かり取り,水を加えて溶解し,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで
薄める。
注(21) アルゴン又は窒素に硫黄化合物が含まれている場合は,過マンガン酸カリウム溶液(過マンガ
31
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ン酸カリウム1g及び水酸化ナトリウム10gを水100mlに溶解する。)を入れたガス洗浄瓶を通し
て,硫黄化合物を吸収し除去する。
12.2.3 装置及び器具 装置及び器具は,次による。
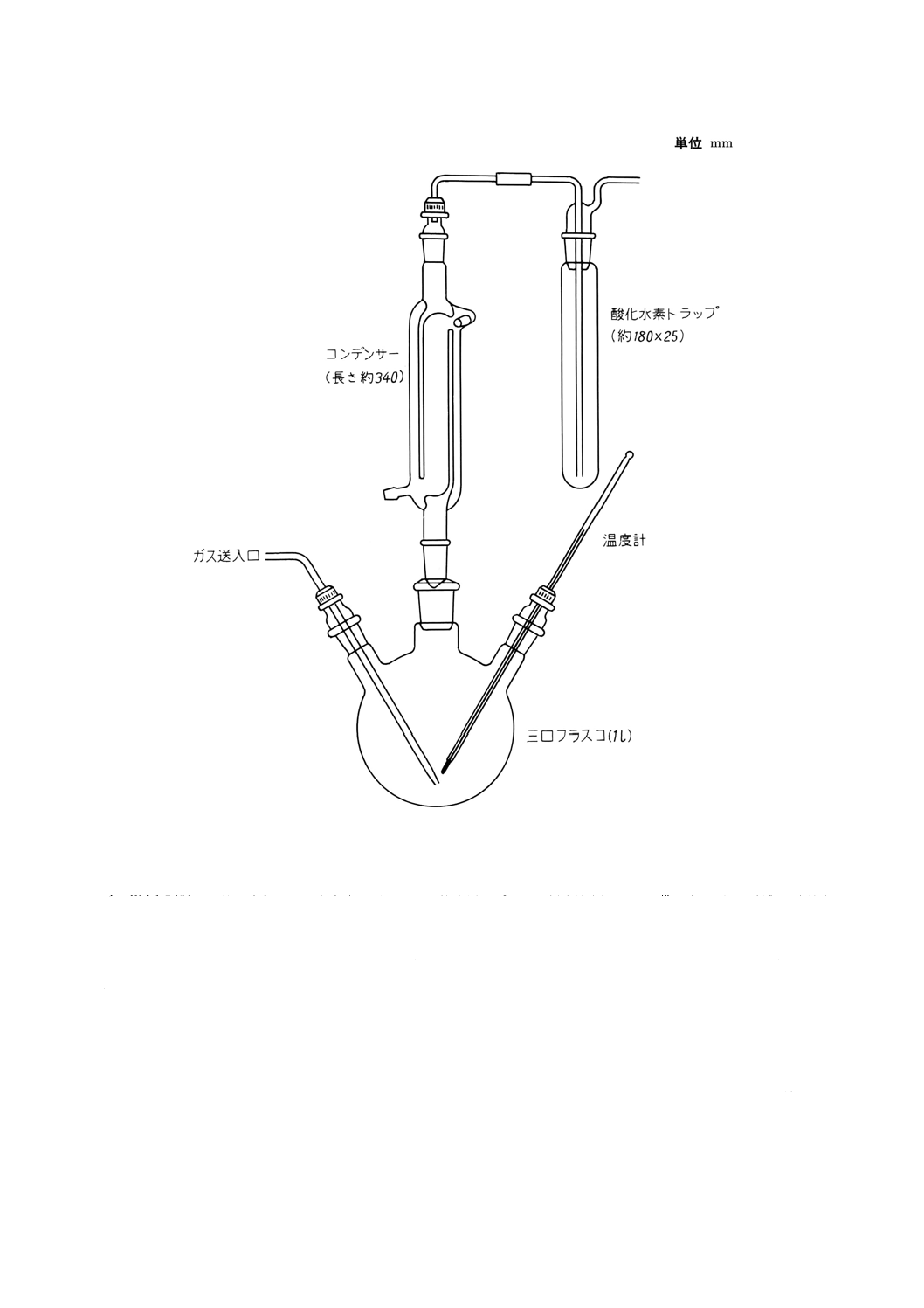
a) 硫化水素気化装置 通常図5に示すものを用いる。
図5 硫化水素気化装置の例
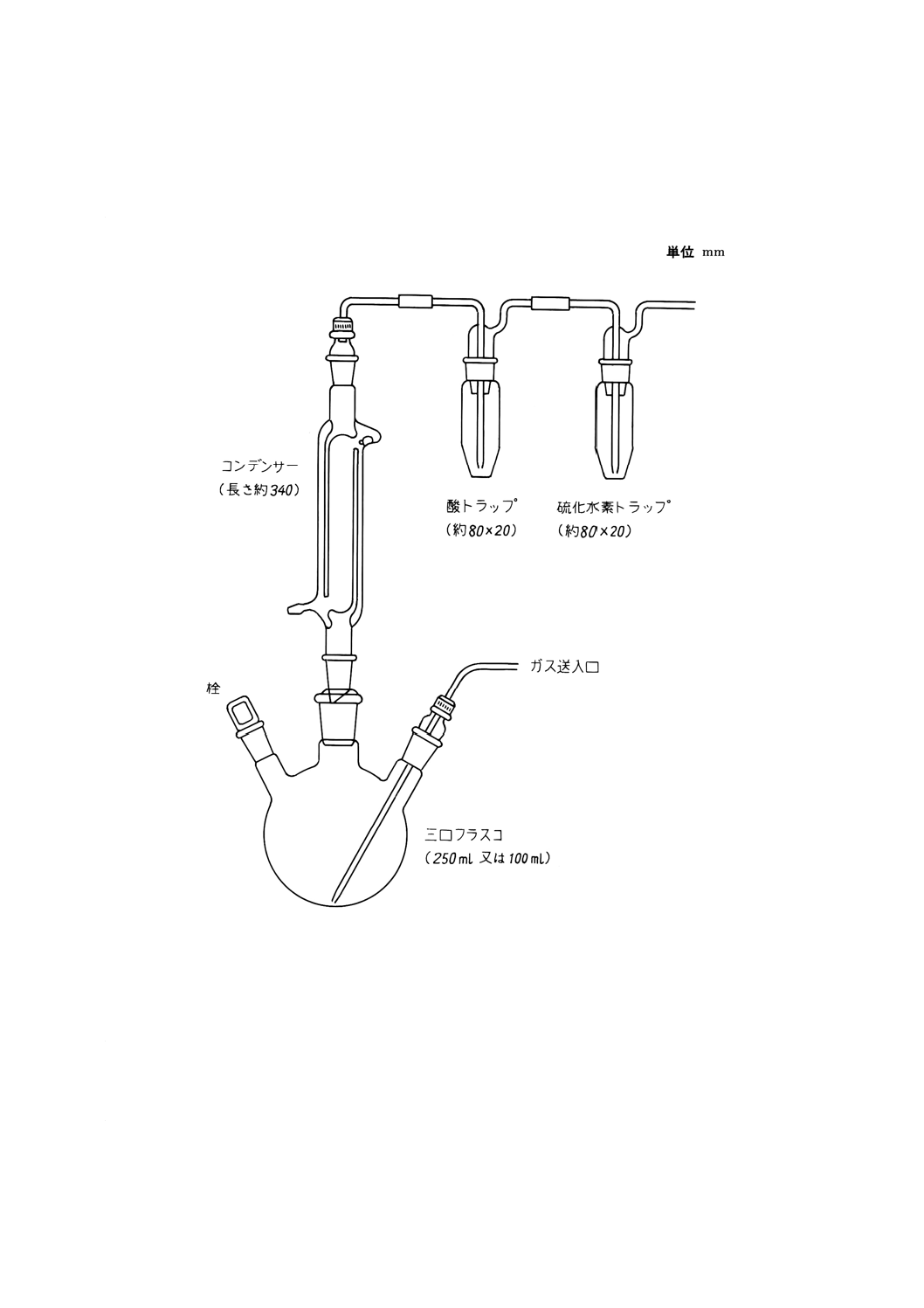
12.2.4 試料はかり取り量 試料はかり取り量は,1.0gとし,10mgのけたまではかる。
12.2.5 操作(22)
12.2.5.1 試料の分解(23) 試料の分解は,次の手順によって行う。
a) 試料をはかり取って三口フラスコ(100ml又は250ml)に移し入れ,硝酸・塩素酸混酸 [12.2.2 f)] 10ml
を加えて分解する。反応が穏やかになったら加熱し,溶液が粘りけのあるシロップ状になるまで注意
して蒸発させる(24)。
b) 精製塩酸 [12.2.2 c)] 10mlを加え,残留物が溶解するまで加熱する。精製ぎ酸 [12.2.2 l)] 2mlを加え,
加熱して乾固するまで蒸発させる。
32
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 精製塩酸 [12.2.2 c)] 10ml及び精製ぎ酸 [12.2.2 l)] 0.5mlを加え,数分間加熱して塩類を完全に溶解(25)
した後,室温まで冷却する。
注(22) すべての試料の取扱いは,硫黄を含む蒸気,ダストなどのない実験室で行う。
(23) 空試験値の変動が大きい場合は,試料の分解は,次のように行ってもよい。試料10.0gを10mg
のけたまではかり取ってビーカー (300ml) に移し入れ,時計皿で覆い,精製硝酸 [12.2.2 e)]
100mlを加え,穏やかに加熱して分解する。時計皿の下面を水で洗って時計皿を取り除き,加
熱して液量が約80mlになるまで濃縮する。常温まで冷却した後,溶液を100mlの全量フラスコ
に水を用いて移し入れ,水で標線まで薄める。この溶液から正確に10mlをビーカー (100ml) に
分取し,塩化カリウム溶液5mlを加え,加熱して乾固する。放冷した後,精製塩酸 [12.2.2 c)] 20ml
をビーカーの内壁を洗浄しながら加えて塩類を溶解し,再び加熱して乾固する。さらに,この
操作を2回繰り返した後,精製塩酸 [12.2.2 c)] 10mlと精製ぎ酸 [12.2.2 l)] 0.5mlを加え,加熱し
て塩類を溶解する。この溶液を水を用いて三口フラスコに移し入れる。
(24) 加熱するとき,三口フラスコは,ホットプレート上に円筒状の金属ホルダーで保持するとよい。
また,ホットプレート上の砂浴を使用するか,又は適切な大きさの低い形状のビーカーの上に
かけてもよい。
(25) もしc)の溶解時に褐色の煙が出るならば,再び乾固するまで蒸発させ,c)の操作を繰り返す。
12.2.5.2 硫化水素の気化分離
a) 硫化水素気化装置に,12.2.5.1 c)又は注(23)で得た溶液が入っている三口フラスコを取り付ける。
b) 酸トラップの中に塩酸 (1+4) 3mlを入れ,硫化水素トラップの中に酢酸亜鉛吸収液 [12.2.2 n)] 5.0ml
を入れる。三口フラスコの横の口から還元混合液 [12.2.2 k)] 30mlを加え,栓をする。
c) すべてのジョイントがしっかりと閉まっていることを確かめ,アルゴン又は窒素を流量30ml/minで装
置に通じ,約2分後にマントルヒーターのスイッチを入れ,114〜116℃で30分間加熱する(26)。硫化
物トラップを取り外し,マントルヒーターのスイッチを切る。
注(26) 硫酸塩から硫化水素への迅速な還元の最適温度は約114℃である。溶液はゆっくりと還流しなけ
ればならない。このような加熱条件を得るためのマントルヒーターの出力設定値は,あらかじ
め予備試験(加熱しながら三口フラスコ中の溶液に温度計を浸し,その温度を測定する。)を行
って求めておく。
12.2.5.3 呈色 呈色は,次の手順によって行う。
a) 12.2.5.2 b)で得た硫化水素トラップ中の溶液(27)に,ジアミン塩溶液 [12.2.2 o)] 3.0ml(27)を硫化物トラッ
プのガス導入管を通して加え,引き続き塩化鉄 (III) 溶液(27)0.5mlを素早く加え,穏やかに混合する。
b) ガス導入管の内側と外側を少量の水で洗う。この洗液を硫化物トラップ内の溶液と混合し,25mlの全
量フラスコ(27)に移し入れる。硫化物トラップを水で洗い,その洗液も全量フラスコに移し入れる。水
で標線まで薄めて混合し,30分間放置する(28)。
注(27) この溶液中の硫黄量が10μgを超える場合には,添加するジアミン塩溶液 [12.2.2 o)] を6.0ml及
び塩化鉄 (III) 溶液を1.0mlとして,25mlの全量フラスコの代わりに50mlの全量フラスコを用
いる。
(28) 一度生成したメチレンブルーの呈色は,少なくとも24時間安定している。
12.2.5.4 吸光度の測定 12.2.5.3 c)で得た溶液の一部を光度計の吸収セル(10mm又は20mm)に取り,水
を対照液として波長665nm付近の吸光度を測定する。
12.2.6 空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
33
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.2.7 検量線 三口フラスコを数個準備し,試料を用いないで,12.2.5.1 a)〜12.2.5.2 c)の手順に従って操
作した後,三口フラスコをアルゴン又は窒素を通じながら50℃以下に冷却する。三口フラスコ内の溶液に
マイクロピペットで標準硫黄溶液 [12.2.2 p)] の各種液量(硫黄として0〜10μg)(29)を段階的に加えた後,
三口フラスコを硫化水素気化装置に取り付ける。以下,12.2.5.2 b)〜12.2.5.4の手順に従って試料と同じ操
作を試料と並行して行い,得た吸光度と硫黄量との関係線を作成し,その関係線を原点を通るように平行
移動して検量線とする。
注(29) 注(27)を適用した場合には,添加するジアミン塩溶液 [12.2.2 o)] を6.0ml及び塩化鉄 (III) 溶液
を1.0mlとして,25mlの全量フラスコの代わりに50mlの全量フラスコを用いる。さらに,標準
硫黄溶液 [12.2.2 p)] の各種液量は硫黄として0〜25μgとなる量とする。
12.2.8 計算 12.2.5.4及び12.2.6で得た吸光度と12.2.7で作成した検量線とから硫黄量を求め,試料中の
硫黄含有率を次のいずれかの式によって算出する。
a) 試料の分解を12.2.5.1のa)〜c)によって行った場合
100
2
1
×
m
A
A
S
−
=
ここに,
S: 硫黄含有率 [% (m/m)]
A1: 試料溶液中の硫黄検出量 (g)
A2: 空試験液中の硫黄検出量 (g)
m: 試料はかり取り量 (g)
b) 試料の分解を注(23)によって行った場合
100
100
10
2
1
×
×
m
A
A
S
−
=
ここに,
S: 硫黄含有率 [% (m/m)]
A1: 分取した試料溶液中の硫黄検出量 (g)
A2: 分取した空試験液中の硫黄検出量 (g)
m: 試料はかり取り量 (g)
12.3 燃焼−電量法
12.3.1 要旨 試料を酸素気流中で加熱し,硫黄を酸化して二酸化硫黄とし,あらかじめ一定のpHに設定
した過酸化水素・硫酸ナトリウム吸収液に吸収させる。このとき増加した水素イオンを,電気分解によっ
て発生させた水酸化物イオンで中和し,そのときに要した電気量を測定する。
12.3.2 試薬 試薬は,JIS Z 2616の7.6(2)(試薬)による。
12.3.3 装置 装置は,JIS Z 2616の7.6(3)(装置)による。
12.3.4 試料はかり取り量 試料はかり取り量は,0.5〜1.0gとし,1mgのけたまではかる。
12.3.5 操作 操作は,次による。
a) 準備操作 準備操作は,JIS Z 2616の7.6(4)(予備操作)による(30)。
注(30) 硫黄含有率既知の試料として,鉄鋼標準試料を用いてもよい。
b) 定量操作 定量操作は,JIS Z 2616の7.6 (5)(定量操作)による(31)。ただし適切な助燃剤(32)を用いる。
注(31) 高周波磁器燃焼るつぼ及びるつぼのふたは,1 400〜1 450℃で2時間以上空焼きしたものを用い
る。
(32) 助燃剤は,JIS Z 2616の6.12(助燃剤)に規定されたものを単独又は2,3種類組み合わせて用
いる。あらかじめ,最適な助燃剤の種類とその添加量を,硫黄含有率既知の試料を用いて調べ
34
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ておく。
12.3.6 空試験 試料に添加したのと同量の助燃剤だけを用いて12.3.5.2の操作を行う。
12.3.7 計算 計算は,JIS Z 2616の7.6(7)(計算)による。
12.4 燃焼−赤外線吸収法(積分法)
12.4.1 要旨 試料を酸素気流中で加熱し,硫黄を酸化して二酸化硫黄とし,酸素とともに赤外線吸収検出
器に送って,その赤外線吸収量を測定する。
12.4.2 装置 装置は,JIS Z 2616の6.6の(1)の管状電気抵抗加熱炉又は(2)の高周波誘導加熱炉とJIS Z
2616の7.7(2)の(a)の酸素精製部,(c)の燃焼ガス精製部及び(d)の二酸化硫黄定量部の装置とを接続して用い
る。
12.4.3 試料はかり取り量 試料はかり取り量は,0.5〜10gとし,1mgのけたまではかる。
12.4.4 操作 操作は,次のいずれかの手順によって行う。
a) 管状電気抵抗加熱炉を用いる場合
1) 準備操作
1.1)
電源を入れ,各部を十分に安定させ,酸素を340kPaで供給して装置の気密を確認する。燃焼管の中
央部の温度を1 250〜1 450℃に,酸化管の温度を700℃に保つ(33)。
1.2)
指示計のゼロ点を調整する。
1.3)
分析試料と同程度の硫黄含有率の標準試料(30)を用い,2)の手順に従って操作し,次の式によって換
算係数を求める。
100
0
0
0
0
C
B
A
W
K
×
−
=
ここに,
K: 測定値 (A0−B0) を硫黄量 (g) に換算するための係数
A0: 標準試料を用いて2) 2.2)で得た指示値
B0: 12.4.5 a)で得た指示値
C0: 標準試料の硫黄含有率 [% (m/m)]
W0: 標準試料はかり取り量 (g)
2) 定量操作
2.1)
試料及び助燃剤(32)を磁器燃焼ボート(33)にはかり取り,磁器燃焼ボートカバー(34)で覆い,燃焼管の
中央部に挿入し,直ちに気密に栓をする(試料が燃焼し,燃焼ガスは精製部を経て赤外線吸収検出
器の試料セルに送られ,指示値が次第に増加する。)。
2.2)
指示計が一定値を示したとき指示値を読み取る。
b) 高周波誘導加熱炉を用いる場合
1) 準備操作 準備操作は,JIS Z 2616の7.7(3)(予備操作)による(30)。
2) 定量操作 定量操作は,JIS Z 2615の7.7(4)(定量操作)による(31)(32)。
注(33) 各部の器具及び材料の充てん(填),保守,気密試験などは,使用する装置の手引書に従って行
う。
(34) 磁器燃焼ボート及びボートカバーは,1 400〜1 450℃で2時間以上空焼きしたものを用いる。
12.4.5 空試験 空試験は,次のいずれかによる。
a) 管状電気抵抗加熱炉を用いる場合 試料に添加するのと同量の助燃剤をはかり取った磁器燃焼ボート
を用い,12.4.4 a) 2)の手順に従って操作する。
b) 高周波誘導加熱炉を用いる場合 試料に手順に従って添加するのと同量の助燃剤をはかり取ったるつ
ぼを用い,12.4.4 b) 2)の手順に従って操作する。
35
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.4.6 計算 計算は,JIS Z 2616の7.7(6)(計算)による。
12.5 燃焼−赤外線吸収法(循環法)
12.5.1 要旨 試料を一定体積,一定圧力下の循環酸素気流中で加熱し,硫黄を酸化して二酸化硫黄とし,
過剰の酸素とともに循環ループの赤外線吸収検出器に送ってその赤外線吸収量を測定する。
12.5.2 装置 装置は,JIS Z 2616の7.8(2)(装置)による。
12.5.3 試料はかり取り量 試料はかり取り量は,0.5〜10gとし,1mgのけたまではかる。
12.5.4 操作
a) 準備操作 準備操作は,JIS Z 2616の7.8(3)(予備操作)による(30)。
b) 定量操作 定量操作は,JIS Z 2616の7.8(4)(定量操作)による(31)(32)。
12.5.5 空試験 試料に添加したのと同量の助燃剤だけを用いて,12.5.4 b)の手順に従って操作する。
12.5.6 計算 計算は,JIS Z 2616の7.8(6)(計算)による。
12.6 燃焼−よう素酸カリウム滴定法
12.6.1 要旨 試料を酸素気流中で高温に加熱し,硫黄を酸化して二酸化硫黄とし,これを吸収液に吸収さ
せ,でんぷんを指示薬として,よう素酸カリウム標準溶液で滴定する。
12.6.2 試薬 試薬は,JIS Z 2616の7.3(2)(試薬)による。
12.6.3 装置 装置は,JIS Z 2616の7.3(3)(装置)による。ただし,試料燃焼部には,JIS Z 2616の6.6(2)
の高周波誘導加熱炉を用いる。
12.6.4 試料はかり取り量 試料はかり取り量は,0.5〜10gとし,1mgのけたまではかる。
12.6.5 操作 操作は,次の手順によって行う。
a) 準備操作
1) 電源を入れ,各部を十分に安定させ,酸素を供給して装置の気密を確認する。
2) 試料と同種の金属で,硫黄含有率既知の試料(30)を用い,b)及び12.6.6の手順に従って操作し,その
滴定量から次の計算式によって標準溶液1mlに相当する硫黄量を求める。
V
S
f=
ここに,
f: よう素酸カリウム標準溶液1mlに相当する硫黄量 (g)
S: 硫黄含有率既知の試料中の硫黄量 (g)
V: よう素酸カリウム溶液の使用量 (ml)
b) 定量操作
1) 試料及び助燃剤(32)を高周波磁器燃焼るつぼ(33)にはかり取り,磁器燃焼るつぼふた(31)で覆う。
2) 炉の受け台に試料の入ったるつぼを置いて酸素を流しながら燃焼位置まで上げる。
3) 塩酸50〜70ml及びでんぷん溶液2mlを吸収容器へ入れる。
4) 滴定終点として設定した青色の濃さを示すまでよう素酸カリウム標準溶液をビュレットから加える。
よう素酸カリウム標準溶液をビュレットに加え,ビュレットのゼロマークに合わす。
5) 炉のスイッチを入れて酸素を流し続けながら試料を燃焼させる。
6) 色調対照液と比較しながら,燃焼中に絶えずよう素酸カリウム標準溶液を滴下して,最初の色調を
保持させ(35),退色しなくなった点を終点とする。
注(35) 二酸化硫黄が損失するので,滴定の間は常に溶液が無色にならないようにする。
12.6.6 空試験 試料に添加した量と同量の助燃剤をはかり取ったるつぼを用い,12.6.5 b)の手順に従って
操作する。
36
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.6.7 計算 計算は,JIS Z 2616の7.3(7)(計算)による。
13. けい素定量方法
13.1 定量方法の区分 けい素の定量方法は,次のいずれかによる。
a) モリブドけい酸抽出吸光光度法 この方法は,けい素含有率0.000 2% (m/m) 以上0.01% (m/m) 以下の
試料に適用する。ただし,試料中にりん又はひ素が含まれる場合には適用しない。
b) モリブドけい酸抽出分離モリブドけい酸吸光光度法 この方法は,けい素含有率0.000 8% (m/m) 以上
0.01% (m/m) 以下の試料に適用する。
c) ICP発光分光分析法 この方法は,けい素含有率0.000 2% (m/m) 以上0.01% (m/m) 以下の試料に適用
する。
13.2 モリブドけい酸抽出吸光光度法
13.2.1 要旨 試料を硝酸で分解した後,硝酸濃度を調節し,七モリブデン酸六アンモニウムを加え,加熱
してモリブドけい酸を生成させる。硝酸濃度を調節した後,3−メチル−1−ブタノールでモリブドけい酸
を抽出し,分光光度計を用いて有機相の吸光度を測定する。
13.2.2 試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] けい素含有率が0.001% (m/m) 以下で,かつ,試料中のけい素含有
率と同じかそれより低いもの(36)。
c) 硫酸ナトリウム(無水)
d) 七モリブデン酸六アンモニウム溶液 七モリブデン酸六アンモニウム四水和物10gを水に溶解し,水
で液量を100mlとする。
e) 3−メチル−1−ブタノール
f)
標準けい素溶液A (2μgSi/ml) あらかじめ1 000℃で強熱し,デシケーター中で室温まで放冷した二
酸化けい素[99.95% (m/m) 以上]0.214gを白金るつぼにはかり取り,炭酸ナトリウム(無水)4gを
加えて十分に混合し,約30分間強熱して融解する。放冷した後,温水100mlを入れたポリエチレン
ビーカー中に浸して融成物を溶解した後,白金るつぼを水で洗って取り出す。常温まで冷却した後,
溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄めて原液 (100μgSi/ml) とする。
この原液はポリエチレン瓶に入れて保存する。この原液を使用の都度,必要量だけ水で正しく50倍に
薄めて標準けい素溶液Aとする。
g) 標準けい素溶液B (1μgSi/ml) f)の原液 (100μgSi/ml) を使用の都度,必要量だけ水で正しく100倍に
薄めて標準けい素溶液Bとする。
注(36) けい素含有率は,13.2のモリブドけい酸抽出吸光光度法,又は13.3のモリブドけい酸抽出分離モ
リブドけい酸吸光光度法によって求める。
13.2.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
13.2.4 操作
13.2.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1のa)及びb)の手順に従って操作する。
b) 溶液を,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
13.2.4.2 呈色 呈色は,次の手順によって行う。
a) 13.2.4.1 b)で得た溶液から正確に10ml(37)をビーカー (100ml) に分取し,水を加えて液量を約40ml(38)
37
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
とする。
b) 溶液に七モリブデン酸六アンモニウム溶液 [13.2.2 d)] 2mlを加え,よく振り混ぜた後約50℃に20分
間放置する。硝酸 (1+1) 5mlを加え,分液漏斗 (150ml) に水を用いて移し入れ,水で液量を約60ml(39)
とした後,室温まで放冷する。
注(37) 分取した溶液中のけい素量が25μgを超える場合には,けい素が5〜20μgとなるように分取量を
少なくする。
(38) このときの硝酸濃度は,0.05〜0.3mol/lの範囲でなくてはならない。10mlを分取したときは,特
に調節を行わなくてもこの濃度となるが,分取量を変えたいときには別に分取した溶液を用い
て,中和滴定法によって酸濃度を求め,硝酸 (1+1) を用いて硝酸濃度を0.05〜0.3mol/lに調節
する。
(39) このときの硝酸濃度は,0.9〜1.3mol/lの範囲でなくてはならない。
13.2.4.3 抽出 抽出は,次の手順によって行う。
a) 13.2.4.2 b)で得た溶液に3−メチル−1−ブタノールを正確に10ml加え,約1分間激しく振り混ぜ,静
置して2層に分離した後,水相を取り除く。
b) 硝酸 (1+1) 10mlを加え,約1分間振り混ぜ,静置して2層に分離した後,水相を取り除く。
13.2.4.4 吸光度の測定 13.2.4.3 b)で得た有機相の水分を取り除き(3),その一部を,分光光度計の吸収セル
(10mm) に取り,3−メチル−1−ブタノールを対照液として,波長370nm付近の吸光度を測定する。
13.2.5 空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
13.2.6 検量線の作成
13.2.6.1 試料用検量線の作成 試料用検量線の作成は,次の手順によって行う。
a) ニッケル [13.2.2 b)] を5.0gはかり取り,ビーカー (300ml) に移し入れる。
b) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄め
る。
c) この溶液を正確に10ml(40)ずつを数個のビーカー (100ml) に分取し,標準けい素溶液A [13.2.2 f)] 及
び標準けい素溶液B [13.2.2 g)] の各種液量(けい素として0〜20μg)を段階的に加え,水を加えて液
量を約40mlとする。
d) 13.2.4.2 b)〜13.2.4.4の手順に従って試料と同じ操作を試料と並行して行い,得た吸光度とけい素量と
の関係線を作成し,その関係線を原点を通るように平行移動して試料用検量線とする。
注(40) 注(37)を適用した場合には,試料溶液と同量分取する。
13.2.6.2 空試験用検量線の作成 空試験用検量線の作成は,次の手順によって行う。
a) 標準けい素溶液B [13.2.2 g)] の各種液量(けい素として0〜5μg)を段階的に数個のビーカー (100ml)
に取り,硝酸 (1+1) 1mlを加えた後,水を加えて液量を約40mlとする。
b) 13.2.4.2 b)〜13.2.4.4の手順に従って空試験と同じ操作を空試験と並行して行い,得た吸光度とけい素
量との関係線を作成し,その関係線を原点を通るように平行移動して空試験用検量線とする。
13.2.7 計算 13.2.4.4及び13.2.5で得た吸光度と13.2.6.1及び13.2.6.2で作成した検量線とからそれぞれけ
い素量を求め,試料中のけい素含有率を,次の式によって算出する。
100
100
2
1
×
×B
m
A
A
Si
−
=
ここに, Si: けい素含有率 [% (m/m)]
38
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A1: 分取した試料溶液中のけい素検出量 (g)
A2: 分取した空試験液中のけい素検出量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
13.3 モリブドけい酸抽出分離モリブドけい酸吸光光度法
13.3.1 要旨 試料を硝酸で分解した後,硝酸濃度を調節し,七モリブデン酸六アンモニウムを加え,加熱
してモリブドけい酸を生成させる。硝酸濃度を調節し,3−メチル−1−ブタノールでモリブドけい酸を抽
出した後,水相に逆抽出し,分光光度計を用いてその吸光度を測定する。
13.3.2 試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 13.2.2 b)による。
c) 水酸化ナトリウム・ほう酸緩衝溶液 水酸化ナトリウム溶液 (4g/l) 400mlに,ほう酸溶液 (4g/l) を
滴加して,pHを11.0〜11.5に調節した後,水で2倍に薄める。
d) 七モリブデン酸六アンモニウム溶液 13.2.2 d)による。
e) 3−メチル−1−ブタノール
f)
標準けい素溶液A (2μgSi/ml) 13.2.2 f)による。
g) 標準けい素溶液B (1μgSi/ml) 13.2.2 g)による。
13.3.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
13.3.4 操作
13.3.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1 a)及びb)の手順に従って操作する。
b) 溶液を,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
13.3.4.2 けい素の分離 けい素の分離は,次の手順によって行う。
a) 13.3.4.1 b)で得た溶液から正確に10ml(41)をビーカー (100ml) に分取し,水を加えて液量を約40ml(38)
とする。
b) この溶液に七モリブデン酸六アンモニウム溶液 [13.3.2 d)] 2mlを加え,よく振り混ぜた後約50℃に20
分間放置する。硝酸 (1+1) 5mlを加え,分液漏斗 (150ml) に水を用いて移し入れ,水で液量を約
60ml(39)とした後,室温まで放冷する。
c) 3−メチル−1−ブタノールを正確に10ml加え,約1分間激しく振り混ぜ,静置して2層に分離した
後,水相を取り除く。
d) 硝酸 (1+1) 10mlを加え,約1分間振り混ぜ,静置して2層に分離した後,水相を取り除く。
e) 水酸化ナトリウム・ほう酸緩衝溶液 [13.3.2 c)] を正確に40ml加え,約1分間激しく振り混ぜ,静置
して2層に分離する。
注(41) 分取した溶液中のけい素量が40μgを超える場合には,けい素量が10〜40μgとなるように分取量
を少なくする。
13.3.4.3 吸光度の測定 13.3.4.2 e)で得た水相の一部を,分光光度計の吸収セル (10mm) に取り,水を対
照液として,波長340nm付近の吸光度を測定する。
13.3.5 空試験 試料を用いないで,試料と同じ操作を試料と並行して行う。
13.3.6 検量線の作成
13.3.6.1 試料用検量線の作成 試料用検量線の作成は,次の手順によって行う。
39
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) ニッケル [13.3.2 b)] を5.0gはかり取り,ビーカー (300ml) に移し入れる。
b) 5.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄め
る。
c) この溶液を正確に10ml(42)ずつ数個のビーカー (100ml) に分取し,標準けい素溶液A [13.3.2 f)] 及び
標準けい素溶液B [13.3.2 g)] の各種液量(けい素として0〜40μg)を段階的に加え,水を加えて液量
を約40mlとする。
d) 13.3.4.2 b)〜13.3.4.3の手順に従って試料と同じ操作を試料と並行して行い,得た吸光度とけい素量と
の関係線を作成し,その関係線を原点を通るように平行移動して試料用検量線とする。
注(42) 注(41)を適用した場合には,試料溶液と同量分取する。
13.3.6.2 空試験用検量線の作成 空試験用検量線の作成は,次の手順によって行う。
a) 標準けい素溶液B [13.3.2 g)] の各種液量(けい素として0〜5μg)を段階的に数個のビーカー (100ml)
に取り,硝酸 (1+1) 1mlを加えた後,水を加えて液量を約40mlとする。
b) 13.3.4.2 b)〜13.3.4.3の手順に従って空試験と同じ操作を空試験と並行して行い,得た吸光度とけい素
量との関係線を作成し,その関係線を原点を通るように平行移動して空試験用検量線とする。
13.3.7 計算 13.3.4.3及び13.3.5で得た吸光度と13.3.6.1及び13.3.6.2で作成した検量線とからそれぞれけ
い素量を求め,試料中のけい素含有率を,次の式によって算出する。
100
100
2
1
×
×B
m
A
A
Si
−
=
ここに, Si: けい素含有率 [% (m/m)]
A1: 分取した試料溶液中のけい素検出量 (g)
A2: 分取した空試験液中のけい素検出量 (g)
m: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
13.4 ICP発光分光分析法
13.4.1 要旨 試料を硝酸で分解した後,溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧し,そ
の発光強度を測定する。
13.4.2 試薬 試薬は,次による。
a) 硝酸 (1+1)
b) ニッケル[99.9% (m/m) 以上] 13.2.2 b)による。
c) 標準けい素溶液A (10μgSi/ml) 13.2.2 f)の原液 (100μgSi/ml) を使用の都度,必要量だけ水で正しく
10倍に薄めて標準けい素溶液Aとする。
d) 標準けい素溶液B (2μgSi/ml) 13.2.2 f)による。
13.4.3 試料はかり取り量 試料はかり取り量は,5.0gとし,10mgのけたまではかる。
13.4.4 操作
13.4.4.1 試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 6.3.4.1 a)及びb)の手順に従って操作する。
b) 溶液を,100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
13.4.4.2 発光強度の測定 13.4.4.1 b)で得た溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に
噴霧し,波長251.611nmにおける発光強度を測定する(6)。
40
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.4.5 空試験 13.4.6の検量線の作成操作において得られる,標準けい素溶液を添加しない溶液の発光強
度を,空試験の発光強度とする。
13.4.6 検量線の作成 検量線の作成は,次の手順によって行う。
a) ニッケル [13.4.2 b)] を5.0gずつ数個はかり取り,それぞれをビーカー (300ml) に移し入れる。
b) 6.3.4.1 b)の操作を行った後,溶液を100mlの全量フラスコに水を用いて移し入れる。
c) これらの溶液に,標準けい素溶液A [13.4.2 c)] 及び標準けい素溶液B [13.4.2 d)] の各種液量(けい素
として0〜500μg)を段階的に加え,水で標線まで薄める。
d) これらの溶液の一部を,ICP発光分光分析装置のアルゴンプラズマ中に噴霧し,波長251.611nmにお
ける発光強度を試料と並行して測定し,得た発光強度とけい素量との関係線を作成し,その関係線を
原点を通るように平行移動して検量線とする。
13.4.7 計算 13.4.4.2及び13.4.5で得た発光強度と13.4.6で作成した検量線とからけい素量を求め,試料
中のけい素含有率を,次の式によって算出する。
100
)
(
3
2
1
×
m
A
A
A
Si
−
−
=
ここに, Si: けい素含有率 [% (m/m)]
A1: 試料溶液中のけい素検出量 (g)
A2: 空試験液中のけい素検出量 (g)
A3: ニッケル [13.4.2 b)] 5.0g中に含まれるけい素量 (g)
m: 試料はかり取り量 (g)
41
H 1151 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS H 1151原案作成委員会 構成表
氏名
所属
(委員長)
奥 谷 忠 雄
日本大学理工学部
(委員)
長 尾 捷 彦
大蔵省造幣局東京支局試験課
長谷川 良 佑
科学技術庁金属材料研究所
揖 斐 敏 夫
通商産業省資源エネルギー庁鉱業課
◎ 大 嶋 清 治
工業技術院標準部材料規格課
◎ 山 村 修 蔵
財団法人日本規格協会技術部
大 屋 武 夫
ステンレス協会
長谷部 守 邦
日本電線工業会技術部
相 馬 南海雄
日本伸銅協会技術部
小 林 芳 夫
日新製鋼株式会社商品技術部
野 坂 洋 一
三井金属鉱業株式会社ダイカスト事業部製造部
(主査)
◎ 町 田 克 己
住友金属鉱山株式会社中央研究所分析センター
◎ 丹 野 一 雄
東邦亜鉛株式会社安中製錬所品質保証部
◎ 尾 上 喬
同和鉱業株式会社中央研究所分析室
◎ 天 川 義 勝
株式会社ジャパンエナジー分析センター
◎ 井 出 光 良
三井金属鉱業株式会社上尾分析センター
◎ 佐 山 恭 正
三菱マテリアル株式会社分析・材料評価センター
○ 塚 原 涼 一
住友金属鉱山株式会社中央研究所分析センター
○ 清 水 透
同和鉱業株式会社中央研究所分析室
○ 樫 村 寛
株式会社ジャパンエナジー分析センター
○ 志 村 和 俊
三菱マテリアル株式会社分析・材料評価センター
(関係者)
中 村 靖
株式会社ジャパンエナジー分析センター
渡 辺 義 治
三井金属鉱業株式会社上尾分析センター
端 洋 志
三井金属鉱業株式会社上尾分析センター
(事務局)
渡 辺 昌 弘
日本鉱業協会技術部
稲 垣 勝 彦
日本鉱業協会技術部
備考:◎印は本委員会及び分科会委員を示す。
○印は分科会委員を示す。