G 2404:2015
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 一般事項························································································································· 1
4 金属アルミニウム定量方法 ································································································· 2
4.1 定量方法の区分 ············································································································· 2
4.2 金属アルミニウム分解分離ICP発光分光分析方法 ································································ 2
4.3 金属アルミニウム分解分離亜鉛逆滴定法············································································· 4
4.4 塩酸溶解ガス容量法 ······································································································· 5
4.5 反応液温度測定法 ·········································································································· 8
5 窒素定量方法 ·················································································································· 10
5.1 定量方法 ····················································································································· 10
5.2 要旨 ··························································································································· 10
5.3 試薬 ··························································································································· 10
5.4 水蒸気蒸留装置 ············································································································ 11
5.5 分析用試料 ·················································································································· 11
5.6 試料はかりとり量 ········································································································· 11
5.7 操作 ··························································································································· 11
5.8 空試験 ························································································································ 13
5.9 計算 ··························································································································· 13
6 塩化物イオン定量方法 ······································································································ 13
6.1 定量方法の区分 ············································································································ 13
6.2 イオンクロマトグラフィー ····························································································· 13
6.3 硝酸銀滴定法 ··············································································································· 16
7 全けい素定量方法 ············································································································ 18
7.1 定量方法の区分 ············································································································ 18
7.2 モリブドけい酸吸光光度法 ····························································································· 18
7.3 二酸化けい素重量法 ······································································································ 21
8 酸化アルミニウム定量方法 ································································································ 22
8.1 定量方法の区分 ············································································································ 22
8.2 金属アルミニウム分解分離ICP発光分光分析方法 ······························································· 22
8.3 金属アルミニウム分解分離亜鉛逆滴定法············································································ 25
9 炭素定量方法 ·················································································································· 26
9.1 定量方法 ····················································································································· 26
9.2 要旨 ··························································································································· 26
9.3 試薬 ··························································································································· 26
G 2404:2015 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
9.4 器具及び材料 ··············································································································· 27
9.5 装置 ··························································································································· 28
9.6 分析用試料 ·················································································································· 29
9.7 試料はかりとり量 ········································································································· 29
9.8 操作 ··························································································································· 29
9.9 空試験 ························································································································ 31
9.10 検量線の作成 ·············································································································· 31
9.11 検量線の校正 ·············································································································· 33
9.12 計算 ·························································································································· 34
10 水分定量方法 ················································································································ 34
10.1 定量方法 ···················································································································· 34
10.2 要旨 ·························································································································· 34
10.3 試料 ·························································································································· 34
10.4 装置及び器具 ·············································································································· 34
10.5 操作 ·························································································································· 34
10.6 計算 ·························································································································· 34
10.7 許容差 ······················································································································· 35
10.8 分析値 ······················································································································· 35
11 安全衛生及び環境に関する注意 ························································································ 35
G 2404:2015
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第12条第1項の規定に基づき,一般社団法人日本アルミニウム協会(JAA)
及び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を制定すべきとの申出
があり,日本工業標準調査会の審議を経て,経済産業大臣が制定した日本工業規格である。
これによって,JIS G 2402:2009及びJIS G 2403:2003は改正され,その一部を分割して制定したこの規
格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
G 2404:2015
鉄鋼用アルミニウムドロス分析方法
Methods for chemical analysis of aluminium dross for iron and steel making
1
適用範囲
この規格は,鉄鋼製造時のフラックスとして使用する鉄鋼用アルミニウムドロス中の金属アルミニウム,
窒素,塩化物イオン,全けい素,酸化アルミニウム及び炭素並びに水分の定量方法について規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS G 2403 鉄鋼用アルミニウムドロス−サンプリング及び試料調製方法
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0127 イオンクロマトグラフィー通則
JIS K 0970 ピストン式ピペット
JIS K 1101 酸素
JIS K 8005 容量分析用標準物質
JIS R 1306 化学分析用磁器燃焼ボート
JIS Z 2615 金属材料の炭素定量方法通則
3
一般事項
分析方法に共通な一般事項は,JIS K 0050によるほか,次による。
a) サンプリング及び試料調製は,JIS G 2403による。
b) 水は,JIS K 0050の附属書D(化学分析に用いる水)に規定する種別A2又はA3の水を用いる。
c) 試薬及びガスは,該当する日本工業規格がある場合,その種類の最上級又は適切な用途のものを用い,
該当する日本工業規格がない場合,分析に支障がない品質のものを用いる。
d) ICP発光分光分析方法に共通な一般事項は,JIS K 0116による。
e) イオンクロマトグラフィーに共通な一般事項は,JIS K 0127による。
f)
吸光光度分析に共通な一般事項は,JIS K 0115による。
g) 炭素定量方法に共通な一般事項は,JIS Z 2615による。
h) 分析値は,はかりとった試料の質量に対する百分率(質量分率)で表し,小数点以下2桁に丸める。
2
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4
金属アルミニウム定量方法
4.1
定量方法の区分
金属アルミニウムの定量は,次のいずれかによる。
a) 金属アルミニウム分解分離ICP発光分光分析方法 この方法は,金属アルミニウム含有率1 %(質量
分率)以上の試料に適用する。
b) 金属アルミニウム分解分離亜鉛逆滴定法 この方法は,金属アルミニウム含有率5 %(質量分率)以
上の試料に適用する。
c) 塩酸溶解ガス容量法 この方法は,金属アルミニウムの日常の管理分析に用いる簡易定量方法として
規定するもので,金属アルミニウム含有率5 %(質量分率)以上の試料に適用する。
d) 反応液温度測定法 この方法は,金属アルミニウムの日常の管理分析に用いる簡易定量方法として規
定するもので,金属アルミニウム含有率15 %(質量分率)以上の試料に適用する。
4.2
金属アルミニウム分解分離ICP発光分光分析方法
4.2.1
要旨
試料をメタノール中臭素で分解し,金属アルミニウムを溶解する。不溶解残さをろ過した後,一定量と
し,ICP発光分光分析方法によってアルミニウムを定量する。
4.2.2
試薬
試薬は,次による。
4.2.2.1
塩酸(1+1)
4.2.2.2
過酸化水素
4.2.2.3
臭素
4.2.2.4
アルゴン
4.2.2.5
メタノール
4.2.2.6
アルミニウム標準液(Al:1 mg/mL) アルミニウム[99.9 %(質量分率)以上]1.000 gをはか
りとり,コニカルビーカー(500 mL)に移す。時計皿で覆い,塩酸(1+1)20 mL及び硝酸(1+1)20 mL
を加えて穏やかに加熱して分解する。完全に分解した後,2〜3分間穏やかに煮沸して窒素酸化物を追い出
す。流水中で常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除く。洗液は,コニカルビー
カーに入れる。溶液を1 000 mL全量フラスコに水を用いて移し入れ,水で標線までうすめる。
4.2.3
ICP発光分光分析装置
分析装置の構成は,JIS K 0116による。
4.2.4
分析用試料
分析用試料は,JIS G 2403によって,0.5 mm以下に粉砕する。この粉砕した試料は,ポリエチレン製袋
などに入れて密封して保存する。
4.2.5
試料はかりとり量
試料はかりとり量は,2.0 gとし,1 mgの桁まではかる。
4.2.6
操作
4.2.6.1
試料溶液の調製
試料溶液の調製は,次による。
a) 試料をはかりとって乾燥したコニカルビーカー(500 mL)に移し入れる。
b) メタノール200 mLを加え,棒状温度計(0〜100 ℃)を入れ,乾燥した時計皿で覆う。乾燥したピペ
ットを用いて臭素10 mLを初めは1 mLずつ,反応が穏やかになれば2 mLずつ加える。反応時の溶液
3
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の温度は40 ℃以下1)に保つように,必要ならば,流水中で冷却するなどして調整する。
c) 臭素の添加が終了し,金属アルミニウムなどの分解反応がほぼ終了した後,約1時間放置して完全に
分解する。
d) 時計皿の下面を少量のメタノールで洗って時計皿を取り除く。洗液は,コニカルビーカーに入れる。
ろ紙(5種C)又は四ふっ化エチレン樹脂製若しくはポリカーボネート樹脂製の孔径1 μmのメンブレ
ンフィルターで溶液をろ過し,ろ紙(又はメンブレンフィルター)及び不溶解残さを少量のメタノー
ルで4,5回洗浄する。ろ液及び洗液は,あらかじめ塩酸(1+1)40 mLを入れた500 mL全量フラス
コに受ける。
e) 水で約450 mLにうすめ,流水中で常温まで冷却した後,水で標線までうすめる。
f)
この溶液50 mLをコニカルビーカー(300 mL)に分取する。塩酸(1+1)5 mLを加えた後,熱板上
で加熱し,約5 mLになるまで蒸発して濃縮する。熱板から下ろして放冷した後,過酸化水素1 mLを
加え加熱して臭素を除去する。さらに,加熱を続けて溶液がシロップ状となるまで濃縮する。
g) 塩酸(1+1)10 mLを加えて,析出した塩類を溶かし1〜2分間穏やかに煮沸する。流水中で常温まで
冷却した後,100 mL全量フラスコに水を用いて移し入れ,水で標線までうすめる。
h) この溶液10 mLを100 mL全量フラスコに分取し,水で標線までうすめる。
注1) 分解時の溶液温度が40 ℃を超えると,金属アルミニウム以外のアルミニウム化合物が溶け
始める。
4.2.6.2
測定
測定は,次による。
a) ICP発光分光分析装置を所定の測定条件にセットしてアルゴンプラズマを点灯し,安定した測定がで
きる状態としておく。
b) ICP発光分光分析装置のアルゴンプラズマ中に4.2.6.1 h)で調製した溶液の一部を噴霧し,309.271 nm,
309.284 nm,396.152 nmなどのアルミニウムの発光線波長における発光強度を測定する。バックグラ
ウンドが同時測定可能な装置では,選択した測定波長付近でアルミニウム及び他の共存元素が影響し
ない波長でバックグラウンドを測定して差し引き,アルミニウムの正味の発光強度を求める。
c) 4.2.8によって,同時に作成した検量線からアルミニウム量を求める。
4.2.7
空試験
空試験は,行わない。
4.2.8
検量線の作成
8個の100 mL全量フラスコのそれぞれに,アルミニウム標準液(4.2.2.6)をアルミニウムとして0,0.5,
1,3,5,10,15及び20 mgを加える。塩酸(1+1)1 mLを加えた後,水で標線までうすめる。この溶液
について4.2.6.2によって測定を行い,発光強度と添加アルミニウム量との関係線を作成して検量線とする。
4.2.9
計算
次の式によって試料中の金属アルミニウム含有率を算出する。
100
10
100
50
500
×
×
×
=
W
a
Al
ここに, Al: 金属アルミニウム含有率[%(質量分率)]
a: 検量線から求めたアルミニウム量(g)
W: 試料はかりとり量(g)
4
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.3
金属アルミニウム分解分離亜鉛逆滴定法
4.3.1
要旨
試料をメタノール中臭素で分解し,金属アルミニウムを溶解する。不溶解残さをろ過する。ろ液に過剰
のエチレンジアミン四酢酸二水素二ナトリウム(EDTA2Na)を加え,その過剰をキシレノールオレンジ指
示薬を用いて亜鉛で滴定する。ふっ化ナトリウムを加えてアルミニウムと結合しているEDTA2Naを遊離
させ,亜鉛を用いて前と同様に滴定する。
4.3.2
試薬
試薬は,次による。
4.3.2.1
塩酸(1+1)
4.3.2.2
アンモニア水(1+2)
4.3.2.3
過酸化水素
4.3.2.4
臭素
4.3.2.5
ふっ化ナトリウム飽和溶液 ポリエチレン製ビーカーを用いて,ふっ化ナトリウムを水に溶解し
た飽和溶液。この溶液は,ポリエチレン製瓶に保存する。
4.3.2.6
メタノール
4.3.2.7
エチレンジアミン四酢酸二水素二ナトリウム溶液(EDTA2Na溶液) エチレンジアミン四酢酸
二水素二ナトリウム二水和物37.2 gをはかりとり,水1 Lを加えて溶かした後,ポリエチレンなどの樹脂
製気密容器に入れて保存する。
4.3.2.8
0.1 mol/L亜鉛標準液 亜鉛[99.99 %(質量分率)以上]6.538 gを1 mgの桁まではかりとり,
コニカルビーカー(1 L)に移す。塩酸(1+1)100 mLを加え,加熱して分解する。完全に分解した後,
流水中で室温まで冷却する。液量を約500 mLとし,アンモニア水を加えてアルカリ性とした後,酢酸で
溶液のpHを5〜6に調整する。常温まで流水中で冷却後,1 000 mL全量フラスコに水を用いて移し入れ,
水で標線までうすめる。密栓して保存する。
4.3.2.9
キシレノールオレンジ溶液 キシレノールオレンジ0.1 gを100 mLの水に溶かす。冷暗所に保存
する。
4.3.2.10 緩衝液(pH5.8) 酢酸アンモニウム100 gを700 mLの水に溶かし,酢酸を加えてpH5.8とした
後,水で1 Lにうすめる。
4.3.3
分析用試料
4.2.4による。
4.3.4
試料はかりとり量
試料はかりとり量は,2.0 gとし,1 mgの桁まではかる。
4.3.5
操作
操作は,次による。
a) 試料溶液の調製 試料溶液の調製は,4.2.6.1のa)〜f)の手順に従って行った後,塩酸(1+1)5 mL及
び少量の水で析出した塩類を溶解する。加熱して1〜2分間煮沸した後,流水中で常温まで冷却し,100
mL全量フラスコに水を用いて移し入れ,水で標線までうすめる。
b) 共存イオンのマスキング 共存イオンのマスキングは,次による。
1) a)で調製した溶液の20 mLをコニカルビーカー(300 mL)に分取し,EDTA2Na溶液(4.3.2.7)20 mL
を加え,よく振り混ぜる。アンモニア水(1+2)を用いて溶液をpHメーターでpH5〜pH6に調整
した後,緩衝液(pH5.8)(4.3.2.10)15 mLを加える。
5
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) 加熱して約3分間煮沸した後,流水中で常温まで冷却する。キシレノールオレンジ溶液(4.3.2.9)4,
5滴を加え,0.1 mol/L亜鉛標準液(4.3.2.8)で滴定して溶液の色が黄から赤とう(橙)となったと
ころで止める。
c) 滴定 滴定は,次による。
1) 直ちにふっ化ナトリウム飽和溶液(4.3.2.5)50 mLを加え,再び加熱して約3分間煮沸した後,流
水中で常温まで冷却する。
2) キシレノールオレンジ溶液(4.3.2.9)2,3滴を追加し,アルミニウム量に相当する遊離したエチレ
ンジアミン四酢酸を0.1 mol/L亜鉛標準液(4.3.2.8)で滴定する。溶液の色が黄から赤とうに変化し
たところを終点とし,そのときの滴定量a(mL)を記録する。
4.3.6
空試験
空試験は,行わない。
4.3.7
計算
次の式によって試料中の金属アルミニウム含有率を求める。
100
20
100
50
500
698
002
.0
×
×
×
×
=
W
a
Al
ここに,
Al: 金属アルミニウム含有率[%(質量分率)]
a: 4.3.5 c) 2) で記録した0.1 mol/L亜鉛標準液の滴定量
(mL)
W: 試料はかりとり量(g)
0.002 698: 0.1 mol/L亜鉛標準液1 mLに相当するアルミニウムの質
量を示す換算係数(g/mL)
4.4
塩酸溶解ガス容量法
4.4.1
要旨
試料を塩酸で溶かし,水素ガスを発生させ,ガスメーターで水素ガス量を測定して金属アルミニウム含
有率に換算する。
注記 本法は金属アルミニウム粉末の塩酸溶解時の発生水素ガス量と比較することで,間接的に金属
アルミニウム含有率を算出する簡易法であり,広範な種類の試料について実用範囲内での十分
な分析精度を有するが,種々の誤差要因も内在する。このため4.4.6の操作手順に正しく従うこ
とで誤差を予防し,4.4.7及び4.4.8に述べる補正計算によって誤差を縮小する必要がある。特
に定期的に4.3 金属アルミニウム分解分離亜鉛逆滴定法との比較検証及び補正を行うことが
重要である。
4.4.2
試薬
試薬は,次による。
4.4.2.1
塩酸(1+1)
4.4.2.2
アルミニウム粉末 純度が99.5 %(質量分率)以上で粒度が200 μm以下のもの。
4.4.2.3
吸収液 硫酸(1+11)
4.4.3
器具及び装置
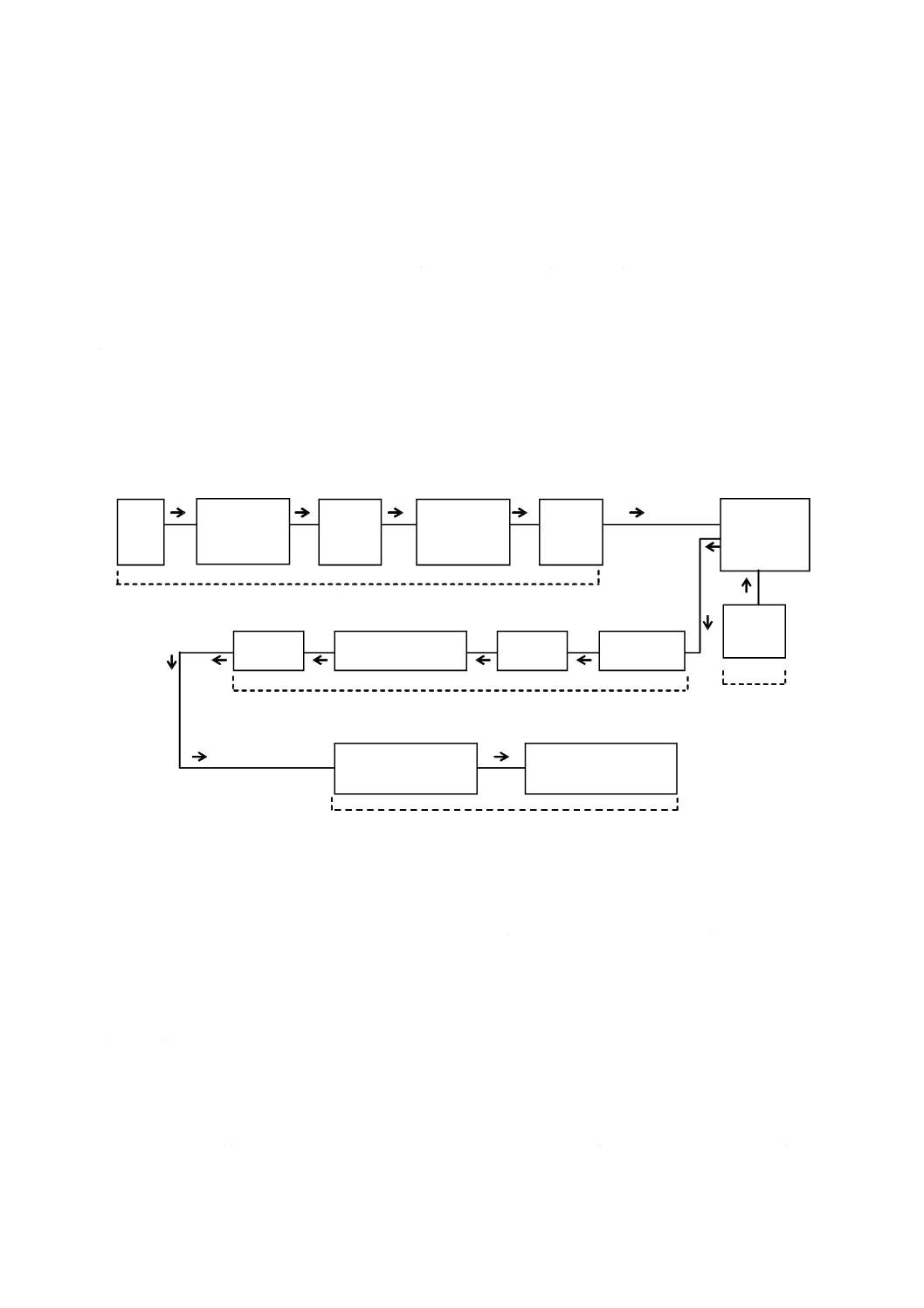
器具及び装置は,次のものを用意し,図1の例のように組み立てる。
4.4.3.1
反応用三角フラスコ ほうけい酸ガラス製の三角フラスコ(500 mL)を用いる。
4.4.3.2
随伴ガス吸収フラスコ ほうけい酸ガラス製の三角フラスコ(500 mL)を用いる。
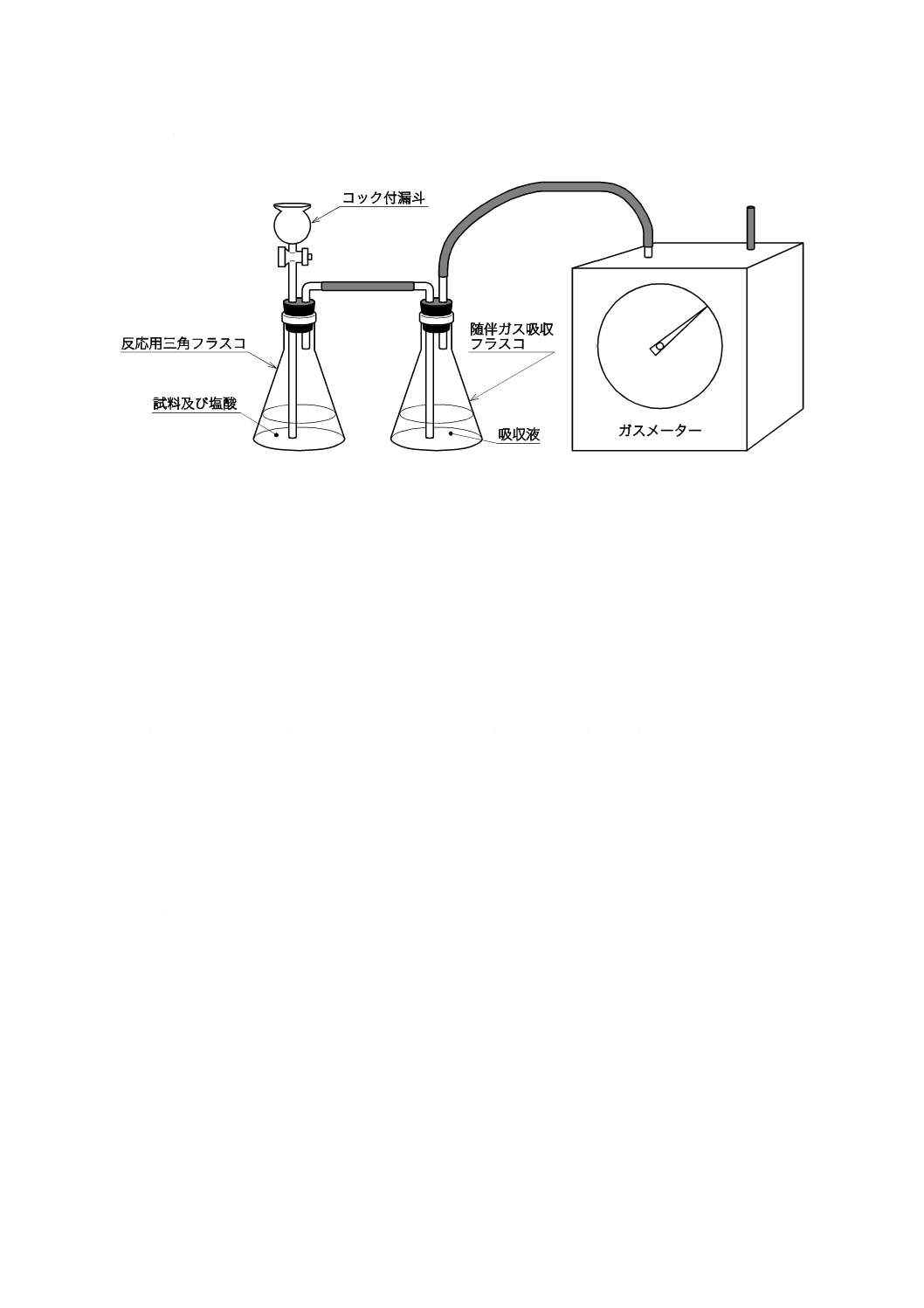
6
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.4.3.3
ガスメーター 湿式実験用ガスメーター(5〜300 L/h,最小読取量 0.005 L)。
図1−塩酸溶解ガス容量法の装置構成の例
4.4.4
分析用試料
試料は,JIS G 2403によって,0.1 mm以下に粉砕する。この粉砕した試料は,ポリエチレン製袋などに
入れて密封して保存する。
4.4.5
試料はかりとり量
試料はかりとり量は,5.0 gとし,1 mgの桁まではかる。
4.4.6
操作
操作は,次による。
a) 試料及び純水75 mLを反応用三角フラスコに入れてゴム栓をする。
b) 反応用三角フラスコに塩酸(1+1)100 mLをコック付漏斗から添加し,直ちにコックを閉める。
c) 溶解は,室温で10分間行う。
d) 水素以外の発生ガス(随伴ガス)を除くこと及び発生水素ガスの冷却を目的として,溶解によって発
生したガスを随伴ガス吸収フラスコ内の吸収液(4.4.2.3)に通じて随伴ガスを除去し,水素ガスの温
度を安定化した後にガスメーターに送る2)。
e) ガスメーターで水素ガス量を測定すると同時に,ガスメーター内に設置した温度計によって水素ガス
温度を測定する。
注2) 吸収液の温度管理及びpH管理が重要であり,酸性を維持し,温度は室温+10 ℃以内に常時
管理することが望ましい。また,随伴ガス吸収フラスコに満たす吸収液の量は,250 mL以上
350 mL以下を目安とする。
4.4.7
アルミニウム粉末測定時の発生水素ガス量とアルミニウム質量との換算係数算出方法
アルミニウム粉末(4.4.2.2)1.000 g程度を0.1 mgの桁まで2個以上はかりとり,それぞれを別々の反応
用三角フラスコに入れ,4.4.6の操作と同様に操作して水素ガス量及び温度を測定する。測定した水素ガス
温度から式(1)によって温度補正係数F0を算出する。アルミニウム粉末溶解時と鉄鋼用アルミニウムドロ
ス溶解時の水素ガス温度とが異なる場合は,それぞれt1(℃)及びt2(℃)とし,温度補正係数もそれぞ
れF01,F02とする。
7
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
273
1
0
t
F
+
=
·············································································· (1)
273
1
273
1
2
02
1
01
t
F
t
F
+
=
+
=
ここに,
F0: 水素ガス温度による温度補正係数
F01: アルミニウム粉末測定時の水素ガス温度による温度補正係
数
F02: 鉄鋼用アルミニウムドロス測定時の水素ガス温度による温
度補正係数
t: 測定時のガスメーター内の水素ガス温度(℃)
t1: アルミニウム粉末溶解時の水素ガス温度の平均値(℃)
t2: 鉄鋼用アルミニウムドロス溶解時の水素ガス温度の平均値
(℃)
アルミニウム1 molから発生する標準状態での理論水素ガス量V01(L)とアルミニウム粉末の塩酸溶解
時に発生する水素ガス量実測値V1(L)との比率を換算係数F1とする。
1
01
1
V
V
F=
ここで,純度P0 %(質量分率)のアルミニウム粉末W1(g)の場合には標準状態で当てはめると,
100
98
26
62
33
0
1
01
P
.
W
.
V
×
×
=
したがって,標準状態では,
100
98
26
62
33
0
1
1
1
01
1
P
V
.
W
.
V
V
F
×
×
×
=
=
上の式を温度補正係数F01によって補正して式(2)を得る。
100
98
26
62
33
01
0
1
1
1
F
P
V
.
W
.
F
×
×
×
×
=
······························································· (2)
ここに,
F1:
P0:
1 atmにおけるアルミニウム粉末による水素ガス量の理論値
との換算係数
アルミニウム粉末の純度[%(質量分率)]
V1: アルミニウム粉末の溶解時に発生する水素ガス量実測値(L)
F01: アルミニウム粉末の溶解時の水素ガス温度による温度補正
係数
W1:
26.98:
33.62:
アルミニウム粉末はかりとり量(g)
アルミニウム原子のモル質量(g/mol)
標準状態(0 ℃,1 atm)におけるアルミニウム1 molに相当
する発生する水素ガス量(L/mol)
4.4.8
計算
計算は,次による。
a) 式(2)を,アルミニウム量を求める式に変形するため,W1を鉄鋼用アルミニウムドロス試料のはかりと
り量W2に,V1を4.4.6の操作によって得られた試料の溶解水素ガス量V2にそれぞれ置き換え,式(3)
によって鉄鋼用アルミニウムドロス試料中の金属アルミニウム含有率P1を得る。
100
62
33
98
26
02
2
1
2
1
×
×
×
×
×
=
F
W
.
F
V
.
P
····························································· (3)
ここに,
P1: 金属アルミニウム含有率[%(質量分率)]
8
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V2: 試料の溶解水素ガス量(L)
F02: 式(1)を用いた鉄鋼用アルミニウムドロス試料測定時の水素
ガス温度の温度補正係数
F1: 式(2)で得たアルミニウム粉末による換算係数
W2: 試料はかりとり量(g)
b) 式(3)によって得られた金属アルミニウム含有率P1の誤差を,補正係数F2を使用して最小化する。補
正係数F2は,過去の同一試料を用いて,塩酸溶解ガス容量法によって求めた金属アルミニウム含有率
P1ʼ及び金属アルミニウム分解亜鉛逆滴定法によって求めた金属アルミニウム含有率P2ʼの測定値を
集積してデータベース化した平均値を使用して両者の比率として求める。補正係数F2は,式(4)によっ
て算出しておく。
注記 補正係数F2は,試料のアルミニウム量によっても影響を受けるので,アルミニウム含有率
30 %以下,40 %,50 %,60 %及び70 %の各水準について求めておくことが望ましい。
,
n
n
n
P
P
F
1
2
2
,
=
∑
=
n
n
F
n
F
1
2
2
1
············································································ (4)
ここに, P2nʼ: 金属アルミニウム分解分離亜鉛逆滴定法によって過去にn
個の試料によって得た金属アルミニウム含有率[%(質量分
率)]
P1nʼ: 塩酸溶解ガス容量法によって過去にn個の試料によって得
た金属アルミニウム含有率[%(質量分率)]
F2n : 過去のデータによるP2nʼとP1nʼとの比率
F2 : 金属アルミニウム分解分離亜鉛逆滴定法による補正係数
c) 式(5)によって試料中の補正した金属アルミニウム含有率Al[%(質量分率)]を確定する。
Al=P1×F2 ··············································································· (5)
ここに,
Al: 補正した金属アルミニウム含有率[%(質量分率)]
P1: 金属アルミニウム含有率[%(質量分率)]
F2: 金属アルミニウム分解分離亜鉛逆滴定法による補正係数
d) 補正係数F2は,同一試料を用いたP1ʼとP2ʼとの差分ΔPʼとし,これを用いて式(6)によって試料中
の補正した金属アルミニウム含有率Al[%(質量分率)]を確定してもよい。
Al=P1+ΔPʼ ·········································································· (6)
4.5
反応液温度測定法
4.5.1
要旨
試料をジュワー瓶内の塩酸反応液中に投入し,試料中の金属アルミニウムと塩酸とを反応させ,その反
応によって生じた反応熱で反応液を加熱する。反応熱総量が,反応したアルミニウム量に比例すること及
びジュワー瓶を使用することで反応液に蓄積された熱量に等しいことから,反応前後の反応液温度の差に
も比例することとなる。ここで,反応前後の反応液温度の差から金属アルミニウム含有率を算出する簡易
測定法である。この分析方法は簡便であることから多用されているが,不純物元素の影響を受けやすいの
でこの点の注意が必要である。
4.5.2
試薬 反応液は,塩酸(1+2)とする。
4.5.3
器具
器具は,次による。
4.5.3.1
ジュワー瓶 反応中,反応液があふれ出ない内容量のもの。
9
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.5.3.2
温度計 目量0.1 ℃のもの。
注記 温度計は,校正してある温度計を使用するのが望ましい。
4.5.4
分析用試料
4.2.4による。
4.5.5
試料はかりとり量
試料はかりとり量は,2.0 gとし,1 mgの桁まではかる。
4.5.6
操作
操作は,次による。
a) 反応液300 mLを,塩酸が極力揮発しない方法で(30±1)℃に調節する。
b) a) の反応液をジュワー瓶に移し,反応液温度(T1 ℃)を0.1 ℃の桁まで測定する。
c) 試料をジュワー瓶に入れ,直ちにゴム栓をする。
d) ジュワー瓶を緩やかに揺すって反応液をかくはんし,反応液温度を1分ごとに測定し,反応液の温度
が均一化され,安定したときの温度(T2 ℃)を0.1 ℃の桁まで測定する。
4.5.7
検量線の作成
検量線の作成は,次による。
a) 検量線作成用試料 検量線作成用試料は,4.3の方法で測定された金属アルミニウム含有率が既知のも
のを段階的に3試料以上準備する。
b) 検量線作成用試料はかりとり量 検量線作成用試料はかりとり量は,2.0 gとし,1 mgの桁まではか
る。
c) 操作 4.5.6の手順に従って操作する。
なお,温度測定には,試料の場合と同一の温度計を使用する。
d) 検量線の換算係数の算出 c) で測定された3試料以上の反応前後の反応液温度差の測定結果(ΔT)及
び4.3で得られた金属アルミニウム含有率(Alʼ)に基づき,次の式のf及びa(検量線の換算係数)
を最小二乗法で算出する。
ʼ
ʼ
W
a
f
T
Al
0.2
]
)
[(
×
+
×
∆
=
ここに,
Alʼ: 金属アルミニウム含有率[%(質量分率)]
ΔT : 反応前後の反応液温度差(℃)
Wʼ: 検量線作成用試料はかりとり量(g)
f,a : 検量線の換算係数
この測定法は,不純物含有量の影響を受けるので,鉄鋼用アルミニウムドロスの供給元が変わるなどし
て内容物が変動すると予測される場合には,検量線を別途作成することが必要である。
4.5.8
計算
4.5.6の操作によって得られた反応前後の反応液温度差を用いて,次の式によって試料中の金属アルミニ
ウム含有率を算出する。
W
a
f
T
T
Al
0.2
]
)
[(
1
2
×
+
×
−
=
ここに,
Al: 金属アルミニウム含有率[%(質量分率)]
T1: 反応前の反応液温度(℃)
T2: 反応後の定常になった反応液温度(℃)
W: 試料はかりとり量(g)
f,a: 検量線の換算係数
10
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5
窒素定量方法
5.1
定量方法
窒素の定量方法は,アンモニア蒸留分離硫酸・水酸化ナトリウム逆滴定法による。この方法は,窒素含
有率0.2 %(質量分率)以上7 %(質量分率)以下の試料に適用する。
5.2
要旨
試料を塩酸及び過酸化水素で分解する。未分解残さ及び溶液を蒸留フラスコに移し,水酸化ナトリウム
でアルカリ性とする。水蒸気蒸留によってアンモニアを,あらかじめ一定量の硫酸標準液を入れた受け器
に留出する。残存する硫酸を水酸化ナトリウム標準液で逆滴定してアンモニウムイオンを定量し,窒素含
有率を算出する。
5.3
試薬
試薬は,次による。
5.3.1
塩酸(1+1)
5.3.2
水酸化ナトリウム溶液(500 g/L) ポリエチレン製瓶に栓をして保存する。
5.3.3
過酸化水素(1+9)
5.3.4
0.5 mol/L硫酸標準液 約900 mLの水に硫酸28 mLを少量ずつ加えた後,水で1 Lにうすめ,気密
容器に保存する。標定は,次による。
この溶液10 mLを三角フラスコ(300 mL)に分取し,約100 mLの水を加える。0.2 mol/L水酸化ナトリ
ウム標準液(5.3.5)でブロモチモールブルーを指示薬として滴定し,溶液の色が緑に変化した点を終点と
する。次の式によって0.5 mol/L硫酸標準液のファクターを算出し,小数点以下3桁に丸める。
10
2.0
NaOH
SO
H
4
2
f
a
f
×
×
=
ここに,
4
2SO
Hf
: 0.5 mol/L硫酸標準液のファクター
10: 0.5 mol/L硫酸標準液分取量(mL)
a: 0.2 mol/L水酸化ナトリウム標準液の滴定量(mL)
fNaOH: 0.2 mol/L水酸化ナトリウム標準液のファクター
容量分析用として標定済みの0.5 mol/L硫酸標準液市販品を使用してもよい。
5.3.5
0.2 mol/L水酸化ナトリウム標準液 0.2 mol/L水酸化ナトリウム標準液の調製,標定及び計算は,
次による。
a) 調製 調製後4,5日経過した水酸化ナトリウム溶液(500 g/L)の上澄み液16 mLをポリエチレンな
どの樹脂製気密容器1 Lに入れ,二酸化炭素を除いた水を加えて1 Lとし,混合した後,ソーダ石灰
管を付けて保存する。
b) 標定及び計算 標定及び計算は,次のいずれかによる。
1) アミド硫酸(HOSO2NH2,JIS K 8005に規定する容量分析用標準物質)を粉砕することなく50 ℃
で2時間乾燥した後,シリカゲルデシケーター中で30分間放冷する。その約4 gを1 mgの桁まで
はかりとり,水に溶かして200 mL全量フラスコに移し入れ,水を標線まで加える。その20 mLを
三角フラスコ(300 mL)に分取し,水で約100 mLとする。指示薬としてブロモチモールブルー溶
液2,3滴を加え,この0.2 mol/L水酸化ナトリウム標準液で滴定し,溶液の色が緑に変化した点を
終点とする。次の式によって0.2 mol/L水酸化ナトリウム標準液のファクターを算出し,小数点以下
3桁に丸める。
11
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
42
019
.0
1
200
20
100
NaOH
×
×
×
×
=
c
b
a
f
ここに,
fNaOH: 0.2 mol/L水酸化ナトリウム標準液のファクター
a: アミド硫酸のはかりとり量(g)
b: アミド硫酸の純度[%(質量分率)]
c: 0.2 mol/L水酸化ナトリウム標準液の滴定量(mL)
0.019 42: 0.2 mol/L水酸化ナトリウム標準液1 mLに相当するアミド
硫酸の質量を示す換算係数(g/mL)
2) 標定済みの0.5 mol/L硫酸標準液市販品を正しく5倍にうすめた溶液を20 mL分取して,ブロモチ
モールブルー溶液を用い,0.2 mol/L水酸化ナトリウム標準液で滴定して標定する。その場合は,次
の式によって0.2 mol/L水酸化ナトリウム標準液のファクターを算出する。
b
a
f
f
×
×
×
=
2
1.0
4
2SO
H
NaOH
ここに,
fNaOH: 0.2 mol/L水酸化ナトリウム標準液のファクター
4
2SO
Hf
: 0.5 mol/L硫酸標準液のファクター
a: 0.5 mol/L硫酸標準液を1/5にうすめた溶液の分取量
(mL)
b: 0.2 mol/L水酸化ナトリウム標準液の滴定量(mL)
5.3.6
ブロモチモールブルー溶液 ブロモチモールブルー0.1 gを20 mLのエタノールに加温して溶かし
た後,水を加えて100 mLとし,褐色ガラス製瓶に保存する。
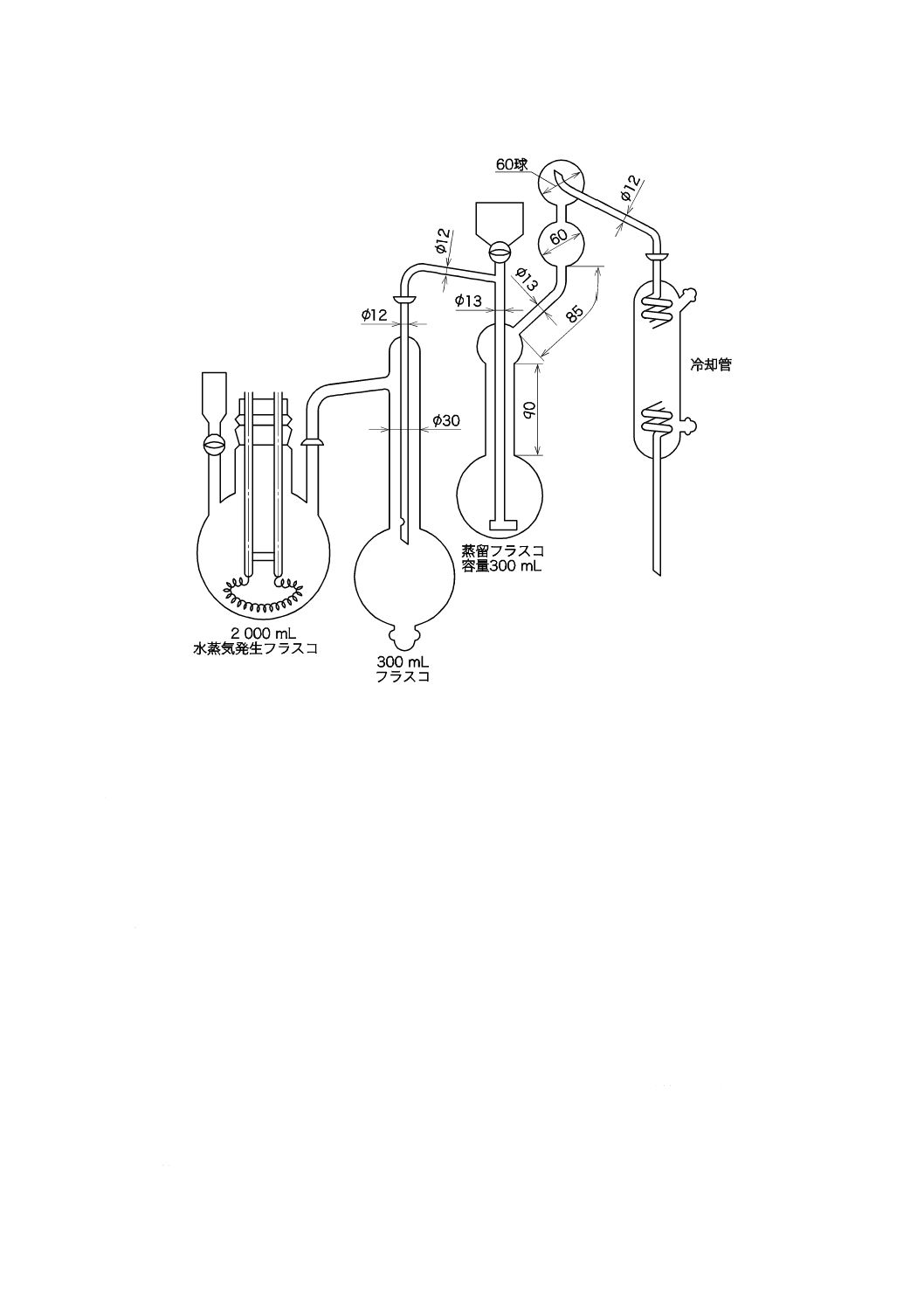
5.4
水蒸気蒸留装置
水蒸気蒸留装置の例を図2に示す。ガラス器具類は,使用前に水でよく洗う。
5.5
分析用試料
4.2.4による。
5.6
試料はかりとり量
試料はかりとり量は,2.0 gとし,1 mgの桁まではかる。
5.7
操作
5.7.1
試料溶液の調製
試料溶液の調製は,次による。
a) 試料をはかりとってビーカー(500 mL)に移し入れる。
b) 時計皿で覆い,塩酸(1+1)30 mLを少量ずつ加える。必要ならば,加熱して金属状アルミニウムな
どを分解する。反応が穏やかになった後,過酸化水素(1+9)10 mLを数回に分けて加え,加熱して
可溶分を完全に分解する。流水中で室温まで冷却した後,時計皿の下面を水で洗って,洗液はビーカ
ーに入れる。
12
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図2−水蒸気蒸留装置の例
5.7.2
アンモニウムイオンの蒸留分離
アンモニウムイオンの蒸留分離は,次による。
a) 水蒸気蒸留装置を組み立て,蒸留開始に先立って水蒸気発生フラスコの水を加熱して15分間以上水蒸
気を発生させておく。また,冷却管には冷却水を流しておく。
b) 5.7.1で調製した試料溶液を未分解残さとともに水蒸気蒸留装置の蒸留フラスコへ少量の水で洗い移
す。
c) 水蒸気蒸留装置へ蒸留フラスコをセットする。次に,あらかじめ0.5 mol/L硫酸標準液(5.3.4)10.00 mL,
水10 mL及びブロモチモールブルー溶液(5.3.6)3,4滴を加えてある三角フラスコ(300 mL)を,
冷却管の留出口がフラスコ中の溶液に浸るようにセットする。
d) 蒸留フラスコ上部に接続されている漏斗から,水酸化ナトリウム溶液(5.3.2)を50 mL加えた後,少
量の水で漏斗部を洗う。漏斗部のコックを閉じ,水蒸気発生フラスコで発生させた水蒸気を蒸留フラ
スコに導入する。
e) 留出液の量が約150 mLになるまで蒸留を続ける。
なお,蒸留の途中で溶液の色が緑に変化した場合は,窒素含有率が7 %(質量分率)以上であるか
ら,試料はかりとり量を減らして再分析する。
f)
留出液の量が約150 mLとなった後,冷却管の留出口を留出液から離し,蒸留フラスコ上部のコック
を開放する。
13
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) 冷却管を外し,その内管の内壁を少量の水で洗浄して留出液に合わせる。
5.7.3
滴定
0.2 mol/L水酸化ナトリウム標準液(5.3.5)で,留出液中に残存する硫酸を逆滴定する。溶液の色が緑に
変化した点を終点とする。
5.8
空試験
空試験は,試料を用いずに5.7の手順に従って操作する。
5.9
計算
5.7.3及び5.8で得た滴定量から,次の式によって試料中の窒素含有率を算出する。
100
000
1
01
.
14
)
(
2.0
NaOH
×
×
×
−
×
×
=
W
b
BT
f
N
ここに,
N: 窒素含有率[%(質量分率)]
fNaOH: 0.2 mol/L水酸化ナトリウム標準液のファクター
b: 0.2 mol/L水酸化ナトリウム標準液の滴定量(mL)
BT: 空試験での0.2 mol/L水酸化ナトリウム標準液の滴定量(mL)
W: 試料はかりとり量(g)
14.01: 窒素原子のモル質量(g/mol)
6
塩化物イオン定量方法
6.1
定量方法の区分
塩化物イオンの定量は,次のいずれかによる。
a) イオンクロマトグラフィー この方法は,塩化物イオン含有率0.05 %(質量分率)以上3.0 %(質量
分率)以下の試料に適用する。
b) 硝酸銀滴定法 この方法は,塩化物イオン含有率0.2 %(質量分率)以上3.0 %(質量分率)以下の試
料に適用する。
6.2
イオンクロマトグラフィー
6.2.1
要旨
試料に水を加えて約1時間煮沸して塩化物イオンを溶出する。残さをろ過した後,一定量とし,イオン
クロマトグラフィーによって塩化物イオンを定量する。
6.2.2
試薬
試薬は,次による。
6.2.2.1
溶離液 溶離液の調製方法の例を次に示す。
a) サプレッサーを用いる場合の例
1) 炭酸水素ナトリウム溶液(1.7 mmol/L)・炭酸ナトリウム溶液(1.8 mmol/L) 炭酸水素ナトリウム
0.143 g及び炭酸ナトリウム0.191 gをはかりとり,水に溶かして1 Lとする。
2) 炭酸水素ナトリウム溶液(0.3 mmol/L)・炭酸ナトリウム溶液(2.7 mmol/L) 炭酸水素ナトリウム
0.025 g及び炭酸ナトリウム0.286 gをはかりとり,水に溶かして1 Lとする。
3) 炭酸ナトリウム溶液(3 mmol/L) 炭酸ナトリウム0.318 gをはかりとり,水に溶かして1 Lとする。
b) サプレッサーを用いない場合の例
1) グルコン酸カリウム溶液(1.3 mmol/L)・四ほう酸ナトリウム溶液(1.3 mmol/L)・ほう酸溶液(30
mmol/L)・アセトニトリル溶液(100 g/L)・グリセリン溶液(5 g/L) グルコン酸カリウム0.31 g,
四ほう酸ナトリウム十水和物0.50 g,ほう酸1.86 g,アセトニトリル100 g(128 mL)及びグリセリ
14
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ン5 g(4 mL)をはかりとり,水に溶かして1 Lとする。
2) フタル酸溶液(2.5 mmol/L)・2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール溶液(2.4 mmol/L)
フタル酸0.415 g及び2-アミノ-2-ヒドロキシメチル-1,3-プロパンジオール[トリス(ヒドロキシメ
チル)アミノメタン]0.291 gをはかりとり,水に溶かして1 Lとする。
注記 溶離液は,装置の種類及び分離カラムに充塡した陰イオン交換体の種類によって異なるの
で,あらかじめ塩化物イオン及び亜硝酸イオンの分離を,6.2.3 a)の操作を行って確認する。
6.2.2.2
再生液 再生液の調製方法の例を次に示す。
硫酸(12.5 mmol/L) 0.5 mol/L硫酸25 mLを水で1 Lにうすめる。
注記 再生液は,サプレッサーを用いる場合に使用するが,装置の種類及びサプレッサーの種類によ
って異なる。あらかじめ分離カラムと組み合わせて,6.2.3 a)の操作を行って再生液の性能を確
認する。
6.2.2.3
塩化物イオン標準液A(Cl−:1 mg/mL) 塩化ナトリウム(JIS K 8005に規定する容量分析用
標準物質)を600 ℃で2時間乾燥した後,シリカゲルデシケーターに入れて放冷する。これの1.648 gを
はかりとり,少量の水に溶かして1 000 mL全量フラスコに水を用いて移し入れ,水で標線までうすめる。
6.2.2.4
塩化物イオン標準液B(Cl−:0.1 mg/mL) 調製した塩化物イオン標準液A 10 mLを100 mL全
量フラスコに分取し,水で標線までうすめる。
6.2.3
イオンクロマトグラフ
イオンクロマトグラフは,分離カラムとサプレッサーとを組み合わせた方式のもの又は分離カラム単独
の方式のものいずれでもよいが,次の条件を満たすもので,塩化物イオン,亜硝酸イオン,臭化物イオン,
硝酸イオン,硫酸イオンなどが分離定量できなければならない。
なお,サプレッサーは,溶離液中の陽イオンを水素イオンに変換するもので,溶離液中の陽イオンに対
して十分なイオン交換容量をもつ陽イオン交換膜(膜形及び電気透析形がある。)又は同様な性能をもった
陽イオン交換体を充塡したものとし,再生液と組み合わせて用いる。ただし,電気透析形の場合は,再生
液として検出器からの流出液(検出器から排出される溶液)を用いる。
a) 分離カラム ステンレス鋼製又は合成樹脂製3) のものに,強塩基性陰イオン交換体(表層被覆形,全
多孔性シリカ形など)を充塡したものとする。
なお,分離カラムには,溶離液を一定の流量(例えば,1〜2 mL/min)で流し,陰イオン混合希釈標
準液[JIS K 0127の箇条7 p)(混合希釈標準液)]の一定量をイオンクロマトグラフに注入し,クロマ
トグラムを求め,それぞれの陰イオンが分離(分離度1.3以上)できるものを用いる。また,定期的
に分離カラムの性能4)を確認するとよい。
陰イオン混合希釈標準液(Cl−:5 μg/mL,NO2−:10 μg/mL,Br−,NO3−及びSO42−:それぞれ20 μg/mL)
は,次のように調製する。塩化物イオン標準液A(Cl−:1 mg/mL)5 mL,亜硝酸イオン標準液(NO2 −:
1 mg/mL)10 mL,臭化物イオン標準液(Br−:1 mg/mL)20 mL,硝酸イオン標準液(NO3−:1 mg/mL)
20 mL,及び硫酸イオン標準液(SO42−:1 mg/mL)20 mLをそれぞれ分取して,1 000 mL全量フラス
コに移し入れ,水で標線までうすめる。
注3) 例えば,四ふっ化エチレン樹脂製,ポリエーテルケトン樹脂製などがある。
4) 例えば,確認する項目は,JIS K 0127の11.3 c)(分離度の確認)のほか,メーカーが推奨す
るピークの対称性,保持時間及び圧力などがある。
b) 検出器 電気伝導度検出器
c) 記録部 記録部は,データ処理装置又は記録計とする。

15
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2.4
分析用試料
4.2.4による。
6.2.5
試料はかりとり量
試料はかりとり量は,1.0 gとし,1 mgの桁まではかる。
6.2.6
操作
操作は,次による。
a) 試料溶液の調製 試料溶液の調製は,次による。
1) 試料をはかりとってビーカー(300 mL)に移し入れる。
2) 時計皿で覆い,水約150 mLを加える。
3) 熱板上で加熱して,約1時間穏やかに煮沸する。この間,溶液量を150 mLに保つよう必要に応じ
て水を適宜,追加する。
4) 流水中で室温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除く。洗液は,ビーカーに
入れる。
5) ろ紙(5種C)又は孔径1 μmのメンブレンフィルターで溶液をろ過し,ろ紙(又はメンブレンフィ
ルター)及び残さを少量の水で4,5回洗浄する。ろ液は,洗液とともに200 mL全量フラスコに受
け,水で標線までうすめる。
6) 試料中の塩化物イオン含有率に応じて,表1に従い,5) で得た溶液を200 mL全量フラスコに分取
し,水で標線までうすめて試料溶液とする。
表1−分取量
試料中の塩化物イオン含有率
%(質量分率)
分取量
mL
0.05以上
0.2未満
全量
0.2以上
2.0未満
20
2.0以上
3.0以下
10
b) 測定 測定は,次による。
1) イオンクロマトグラフを所定の測定条件にセットして作動できる状態にし,分離カラムなどに溶離
液(6.2.2.1)を一定の流量(例えば,1〜2 mL/min)で流しておく。サプレッサーを必要とする装置
では,再生液(6.2.2.2)を一定の流量で流しておく。
2) 孔径0.2 μmのカートリッジフィルターを装着したシリンジを用い,a) 6) で得た試料溶液の一定量
を装置のサンプリング部に注入する。
3) 装置の試料導入部を作動させて,サンプリング部の試料溶液(50〜200 μLの一定量)をカラム部に
注入し,クロマトグラムを記録する。
4) クロマトグラム上の塩化物イオンに相当するピークについて,指示値を読み取る。
なお,指示値は,ピーク高さ又はピーク面積とする。
6.2.7
空試験
ビーカー(300 mL)に水150 mLを加え,時計皿で覆う。以下,6.2.6 a) の3)〜6) 及び6.2.6 b) の手順
に従って操作し,空試験の指示値を読み取る。ただし,分取量及び装置への注入量は,試料溶液と同じ量
とする。
16
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2.8
検量線の作成
塩化物イオン標準液A又は塩化物イオン標準液Bから塩化物イオンとして0〜2 mgを段階的に数個の
200 mL全量フラスコに分取し,水で標線までうすめる。この溶液を6.2.6 b) の手順に従って操作し,指示
値と塩化物イオン量との関係線を作成し,原点を通るように平行移動して検量線とする。
6.2.9
計算
6.2.6及び6.2.7によって得た指示値と6.2.8によって作成した検量線とから塩化物イオン量を求め,次の
式によって試料中の塩化物イオン含有率を算出する。
100
200
×
×
−
=
c
W
b
a
Cl
ここに,
Cl: 塩化物イオン含有率[%(質量分率)]
a: 検量線から求めた試料溶液200 mL中の塩化物イオン量(g)
b: 空試験値(g)
c: 分取量(mL)
(分取をしない場合は,cは200とする。)
W: 試料はかりとり量(g)
6.3
硝酸銀滴定法
6.3.1
要旨
試料に水を加えて煮沸し,塩化物イオンを溶出する。水酸化ナトリウムでアルカリ性とし,過酸化水素
を加えて還元性物質を酸化する。硝酸で中和し,析出した水酸化物などをろ過する。ろ液についてウラニ
ンを指示薬とし,硝酸銀で滴定して塩化物イオンを定量する。
6.3.2
試薬
試薬は,次による。
6.3.2.1
硝酸(1+1)
6.3.2.2
硫酸
6.3.2.3
水酸化ナトリウム溶液(20 g/100 mL)
6.3.2.4
過酸化水素(1+9)
6.3.2.5
塩化ヒドロキシルアンモニウム
6.3.2.6
硫酸ヒドロキシルアンモニウム
6.3.2.7
デキストリン溶液 デキストリン水和物2 gに水を加え,煮沸して溶かし,100 mLにうすめる。
使用の都度,調製する。
6.3.2.8
40 mmol/L硝酸銀標準液 硝酸銀6.8 gを水に溶かし1 Lにうすめて,着色ガラス瓶に保存する。
標定は,次のように行う。
塩化ナトリウム(JIS K 8005に規定する容量分析用標準物質)を600 ℃で2時間乾燥した後,シリカゲ
ルデシケーターに入れて放冷する。0.467 5 gをはかりとり,少量の水に溶かして200 mL全量フラスコに
水を用いて移し入れ,水で標線までうすめる。この溶液20 mLをビーカー(200 mL)に分取し,水で液量
を約50 mLとする。これにデキストリン溶液5 mL及びウラニン溶液1,2滴を加え,静かに振り混ぜなが
ら40 mmol/L硝酸銀標準液で滴定する。黄緑の蛍光が消失して僅かに赤くなったときを終点とする。次の
式によって,40 mmol/L硝酸銀標準液のファクターを算出し,小数点以下3桁に丸める。
7
337
002
.0
1
200
20
100
3
AgNO
×
×
×
×
c
b
a
f
=
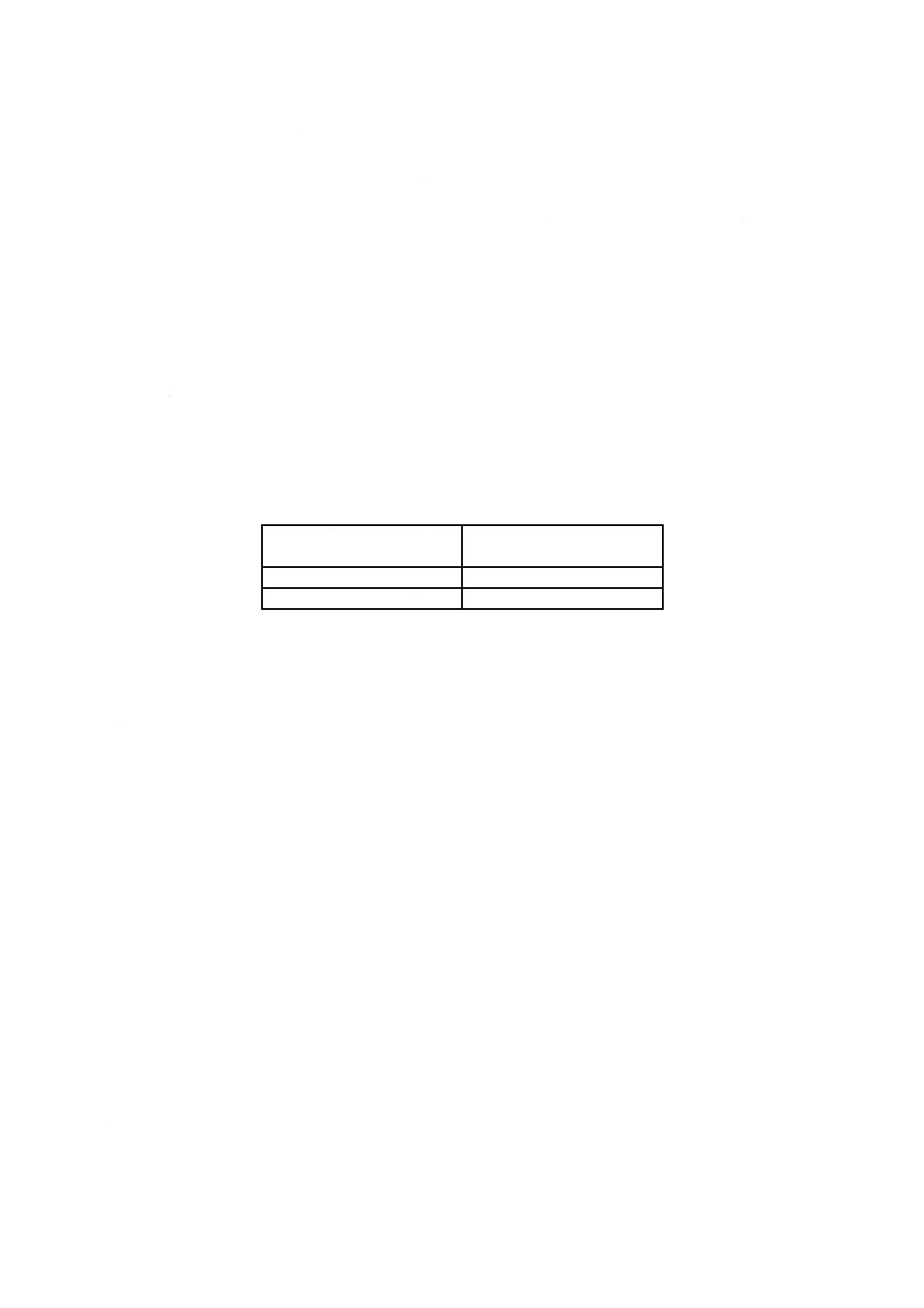
17
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
3
AgNO
f
: 40 mmol/L硝酸銀標準液のファクター
a: 塩化ナトリウムはかりとり量(g)
b: 塩化ナトリウムの純度[%(質量分率)]
c: 40 mmol/L硝酸銀標準液の滴定量(mL)
0.002 337 7: 40 mmol/L硝酸銀標準液1 mLに相当する塩化ナトリウ
ムの質量を示す換算係数(g/mL)
6.3.2.9
クロム酸カリウム溶液(5 g/100 mL)
6.3.2.10 ウラニン溶液 ウラニン(フルオレセインナトリウム)0.2 gを水に溶かして100 mLにうすめる。
6.3.2.11
pH試験紙 pH2〜pH10の範囲で赤から黄,緑を経て紫に変色し,pHが7付近のとき緑を示す
もの。
6.3.3
分析用試料
4.2.4による。
6.3.4
試料はかりとり量
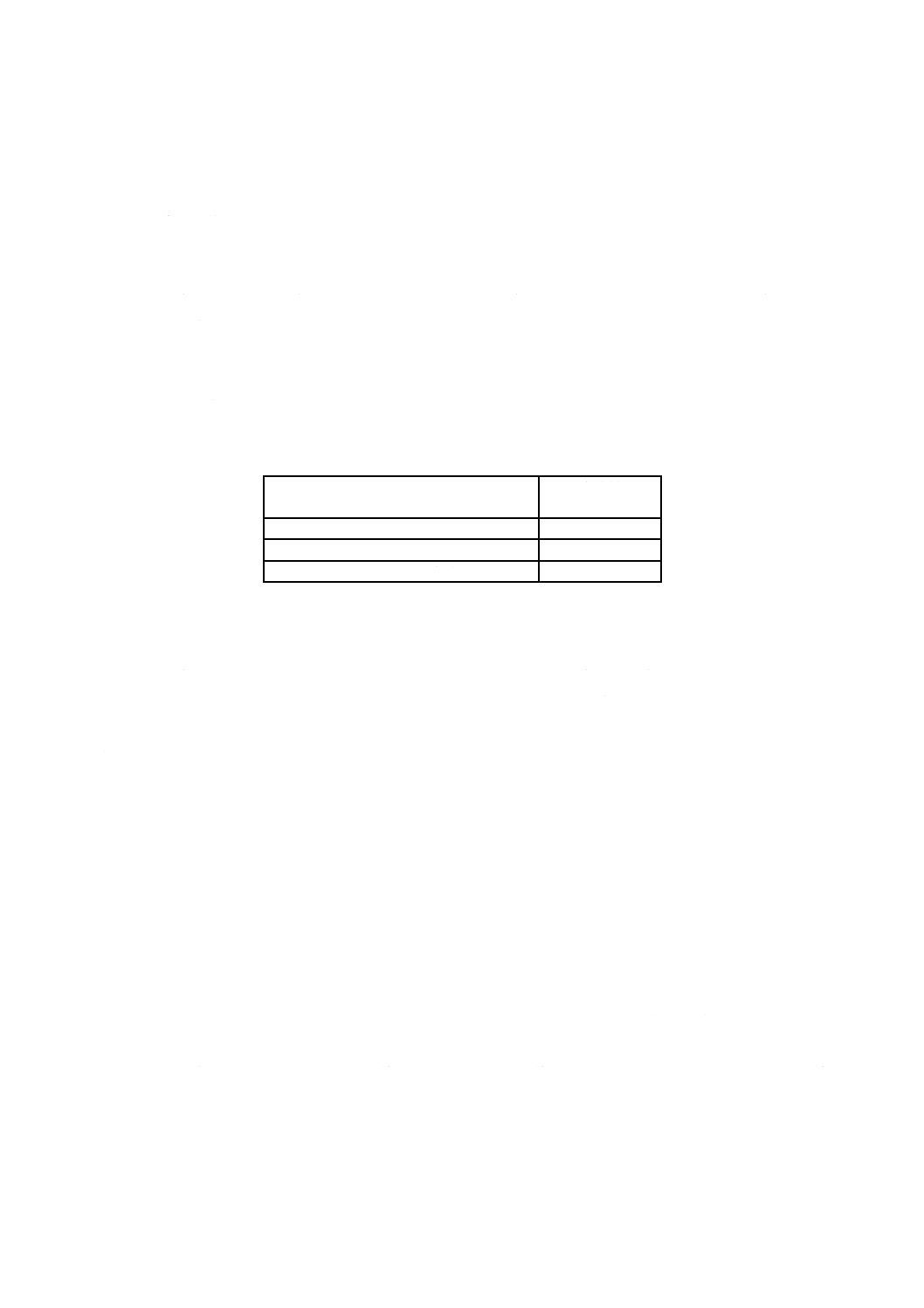
試料は,塩化物イオン含有率に応じ,表2によって1 mgの桁まではかる。
表2−試料はかりとり量
試料中の塩化物イオン含有率
%(質量分率)
試料はかりとり量
g
0.2以上 1.0未満
2.0
1.0以上 3.0以下
1.0
6.3.5
操作
操作は,次による。
a) 試料溶液の調製 試料溶液の調製は,次による。
1) 試料をはかりとってビーカー(300 mL)に移し入れる。
2) 時計皿で覆い,水80 mLを加える。加熱して溶液を約1時間穏やかに煮沸した後,流水中で室温ま
で冷却する。
3) 時計皿の下面を水で洗って時計皿を取り除く。洗液は,ビーカーに入れる。ろ紙(5種B)を用い
て溶液をろ過し,ろ紙及び残さを少量の水で4,5回洗浄する。ろ液及び洗液は,合わせて別のビー
カー(300 mL)に受ける。
4) 水酸化ナトリウム溶液(6.3.2.3)1 mLを加え,煮沸し始めるまで加熱する。過酸化水素(1+9)5 mL
を少量ずつ加えた後,約10分間煮沸する。
5) 流水中で室温まで冷却後,硝酸(1+1)を加え,pH7付近まで中和する。pH試験紙で溶液のpHが
7付近にあることを確認する。
6) 水酸化物などの沈殿物が認められれば,ろ紙(5種A)を用いて溶液をろ過し,ろ紙及び沈殿物を
少量の水で数回洗浄する。ろ液及び洗液は,合わせて新しいビーカー(300 mL)に受け試料溶液と
する。溶液量が100 mLを超えていた場合は,加熱蒸発して約100 mLとする。
なお,ろ紙の代わりに,孔径0.4〜1 μmのメンブレンフィルターを使用して吸引ろ過を行っても
よい。
b) 塩化物イオンの滴定 塩化物イオンの滴定は,次による。
1) a) 6) で得た試料溶液にデキストリン溶液(6.3.2.7)5 mL及びウラニン溶液(6.3.2.10)2,3滴を加
18
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
える。
2) 溶液を静かに振り混ぜながら,40 mmol/L硝酸銀標準液(6.3.2.8)で滴定する。溶液の黄緑の蛍光が
消失して僅かに赤くなった点を終点とする。
なお,この滴定操作は,次のいずれかによる。
2.1) 試料中の塩化物イオン含有率が1 %(質量分率)未満の場合は,ミクロビュレット(10 mL)を用
いて滴定を行う。
2.2) 滴定時の塩化物イオン量が10 mgより少ない場合,終点の判定には高度の熟練を要する。終点の
判定ができない場合は,ウラニン溶液に替えてクロム酸カリウム溶液(6.3.2.9)1 mLを使用して
滴定を行ってもよい(保護コロイドのデキストリン溶液は添加しない。)。その場合,溶液が黄色
から僅かに赤褐色に着色した点を終点とする。
警告 滴定終了後の溶液は6価クロムを含むので,少量の硫酸及び硫酸ヒドロキシルアンモニ
ウム又は塩化ヒドロキシルアンモニウムを加えて,クロムを3価に還元して廃棄処理を
行う。
6.3.6
空試験
水80 mLをビーカー(300 mL)に取り,6.3.5 a) 2) の煮沸操作以降,試料の場合と同様に操作して空試
験の滴定量を求める。
6.3.7
計算
次の式によって試料中の塩化物イオン含有率を算出する。
100
000
1
418
.1
)
(
3
AgNO
×
×
×
×
=
W
b
a
f
Cl
−
ここに,
Cl: 塩化物イオン含有率[%(質量分率)]
3
AgNO
f
: 40 mmol/L硝酸銀標準液のファクター
a: 試料の滴定量(mL)
b: 空試験の滴定量(mL)
W: 試料はかりとり量(g)
1.418: 40 mmol/L硝酸銀標準液1 mLに相当する塩化物イオンの
質量を示す換算係数(mg/mL)
7
全けい素定量方法
7.1
定量方法の区分
全けい素の定量は,次のいずれかによる。
a) モリブドけい酸吸光光度法 この方法は,全けい素含有率(SiO2表示)0.5 %(質量分率)以上20 %
(質量分率)以下の試料に適用する。
b) 二酸化けい素重量法 この方法は,全けい素含有率(SiO2表示)0.5 %(質量分率)以上20 %(質量
分率)以下の試料に適用する。
7.2
モリブドけい酸吸光光度法
7.2.1
要旨
試料を塩酸と硝酸との混酸で分解した後,未分解残さをこし分け,白金るつぼ中で強熱して灰化する。
炭酸ナトリウムとほう酸とで残さを融解し,水に溶解した後,ろ液と合わせる。一部を分取し,溶液のpH
を0.8付近に調節する。モリブデン酸アンモニウムを加えてモリブドけい酸を生成させ,分光光度計を用
いて吸光度を測定する。分析結果は,SiO2として表示する。
19
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.2.2
試薬
試薬は,次による。
7.2.2.1
硝酸(1+1)
7.2.2.2
混酸(塩酸1,硝酸1,水2:容量比)
7.2.2.3
亜硝酸ナトリウム溶液(1 g/100 mL)
7.2.2.4
融解合剤[ほう酸3,炭酸ナトリウム5:質量比]
7.2.2.5
モリブデン酸アンモニウム溶液 七モリブデン酸六アンモニウム四水和物10 gを水に溶かした
後,100 mLとする。ポリエチレン製瓶に保存する。
7.2.2.6
けい素標準液(Si:500 μg/mL) けい素標準液の調製は,次による。
a) あらかじめ1 000 ℃で強熱し,デシケーター中で常温まで放冷した二酸化けい素[99.95 %(質量分率)
以上]0.535 0 gを白金るつぼ(30番又は40番)にはかりとり,炭酸ナトリウム4.0 gを混和し,加熱
して溶融する。
b) 放冷した後,白金るつぼを温水100 mLを入れた四ふっ化エチレン樹脂製ビーカー(300 mL)中に浸
して融成物を溶かし,白金るつぼを水洗して取り出す。室温まで冷却した後,溶液を少量のろ紙パル
プを入れたろ紙(5種B)を用いてろ過し,ろ紙を炭酸ナトリウム溶液(10 g/L)で洗浄する。ろ紙及
び洗液を500 mL全量フラスコに水で洗い移し,水で標線までうすめる。ポリエチレン製瓶に保存す
る。
c) この溶液50 mLを,あらかじめ過塩素酸30 mL,硝酸(1+1)5 mL及び水20 mLを入れてあるビー
カー(500 mL)に分取し,7.3.6 a) 3) の過塩素酸白煙発生操作から7.3.6 c) 4) の手順に従って操作し,
けい素濃度を求める。二酸化けい素質量をけい素質量に変換する係数は,0.467 4である。
7.2.3
装置及び器具
7.2.3.1
分光光度計 分析装置の構成は,JIS K 0115による。
7.2.3.2
白金るつぼ 30番又は40番
7.2.4
分析用試料
4.2.4による。
7.2.5
試料はかりとり量
試料はかりとり量は,1.0 gとし,1 mgの桁まではかる。
7.2.6
操作
操作は,次による。
a) 試料溶液の調製 試料溶液の調製は,次による。
1) 試料をはかりとって,ビーカー(500 mL)に移し入れる。
2) 時計皿で覆い,混酸(7.2.2.2)30 mLを少量ずつ加え,加熱して金属アルミニウムなどを分解する。
分解反応が終了したら必要以上に加熱せずに,流水中で室温まで冷却する。
3) 時計皿の下面を水で洗って時計皿を取り除く。洗液は,ビーカーに入れる。ろ紙(5種B)を用い
て溶液をこし分け,ろ紙及び未分解残さを少量の水で数回洗浄する。ろ液及び洗液は,合わせて新
しいビーカー(500 mL)に受け,保存する。
4) ろ紙及び未分解残さを白金るつぼ(30番又は40番)に移し,加熱してろ紙を乾燥した後,強熱し
て灰化する。金属状けい素などが完全に酸化されて残さがほとんど白色になるまで強熱を続ける。
5) 放冷した後,融解合剤(7.2.2.4)6 gを白金るつぼに加え,白金棒で混合する。その混合物の上に融
解合剤2 gをかぶせるようにして加える。
20
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6) 穏やかに加熱して水分を完全に除去した後,徐々に加熱を強くして残さを融解する。融解合剤が溶
け落ちて,白金るつぼ内壁に付着物がない状態になってから約20分間強熱を続ける。
7) 加熱を止め,放冷した後白金るつぼを融成物とともに四ふっ化エチレン樹脂製ビーカー(300 mL)
に移し,約100 mLの水を加え,加熱して融成物を溶解する。
8) 融成物が溶け,白金るつぼ内に固着物がなくなった後,硝酸(1+1)30 mLを加え,加熱して水酸
化物などの沈殿物を溶解する。白金るつぼを水洗して取り出した後,流水中で常温まで冷却する。
この溶液及び3) で保存したろ液及び洗液を,500 mL全量フラスコに水を用いて移し入れ合わせる。
水で標線までうすめて試料溶液とする。
なお,マンガンの酸化物が認められる場合には,亜硝酸ナトリウム溶液(7.2.2.3)数滴を加える。
b) 呈色 呈色は,次による。
1) 全けい素含有率に応じて表3に従い,a) 8) で得た試料溶液をビーカー(300 mL)に分取する。
表3−分取量
試料中の全けい素含有率(SiO2表示)
%(質量分率)
分取量
mL
0.5 以上 2未満
100
2 以上 10未満
50
10 以上 20以下
25
2) 水で液量を150〜200 mLとし,溶液のpHをpHメーターを用いて硝酸(1+1)でpH0.7〜pH0.9に
調整する。
3) この溶液を250 mL全量フラスコに少量の水を用いて移し入れ,溶液の温度を20〜30 ℃に調整する。
モリブデン酸アンモニウム溶液(7.2.2.5)15 mLを加え,水で標線までうすめ,よく混合して,約
10分間放置する。
c) 吸光度の測定 b) 3) で得た呈色溶液の一部を分光光度計の吸収セルにとり,モリブデン酸アンモニウ
ム溶液の添加から15分間以内に,水を対照液として,波長420 nm付近の吸光度を測定する。a) 8) で
得た試料溶液が着色している場合は,その溶液を呈色用と同量ビーカーに分取し,モリブデン酸アン
モニウム溶液を加えずに試料と同様に操作して得た溶液を対照液に使用する。
7.2.7
空試験
試料の場合と同量の試薬を用い,同様に操作して吸光度を求める。
7.2.8
検量線の作成
数個の四ふっ化エチレン樹脂製ビーカーに融解合剤(7.2.2.4)8 gをとり,約200 mLの水を加え,加熱
して溶解する。けい素標準液(7.2.2.6)を,けい素として0〜25 mgを段階的に加えた後,溶液を加熱して
3〜4分間煮沸する。室温まで冷却後,硝酸(1+1)30 mLを加えて酸性溶液とし,500 mL全量フラスコ
に水を用いて移し入れ,水で標線までうすめる。この溶液100 mLをビーカー(300 mL)に分取する。次
いで,7.2.6 b) の2),3) 及び7.2.6 c) の手順に従って操作し,吸光度を測定する。得た吸光度と分取した
溶液50 mLに含まれるけい素量との関係線を作成し,その関係線を原点を通るように平行移動して検量線
とする。
7.2.9
計算
7.2.6 c) で得た吸光度から7.2.7で得た吸光度を差し引いて得られる吸光度と7.2.8で作成した検量線と
21
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
からけい素量を求め,次の式によって試料中の全けい素含有率(SiO2表示)を算出する。
100
500
/
3
139
.2
2
×
×
×
=
b
W
a
SiO
ここに,
SiO2: 全けい素含有率(SiO2表示)[%(質量分率)]
a: 検量線から求めたけい素検出量(g)
b: 分取量(mL)
W: 試料はかりとり量(g)
2.139 3: SiからSiO2への換算係数
7.3
二酸化けい素重量法
7.3.1
要旨
試料を塩酸と硝酸との混酸で分解した後,未分解残さをこし分け,白金るつぼ中で強熱して灰化する。
炭酸ナトリウムとほう酸とで残さを融解し,水に溶解した後,ろ液と合わせる。過塩素酸を加えて加熱し,
白煙を発生させて,けい素を不溶性の二酸化けい素とする。温水で可溶性塩類を溶解後,沈殿をこし分け,
強熱した後,その質量をはかる。次に,硫酸及びふっ化水素酸を加え,加熱して二酸化けい素をふっ化け
い素として揮散させ,強熱した後,その質量をはかる。質量の差を二酸化けい素として全けい素含有率(SiO2
表示)を求める。
7.3.2
試薬
試薬は,次による。
7.3.2.1
塩酸(1+100)
7.3.2.2
硝酸(1+1)
7.3.2.3
過塩素酸
7.3.2.4
ふっ化水素酸
7.3.2.5
硫酸(1+1)
7.3.2.6
混酸(塩酸1,硝酸1,水2:容量比)
7.3.2.7
亜硝酸ナトリウム溶液(1 g/100 mL)
7.3.2.8
融解合剤[ほう酸3,炭酸ナトリウム5:質量比]
7.3.2.9
硝酸銀溶液(1 g/100 mL)
7.3.3
器具 白金るつぼは,30番又は40番とする。
7.3.4
分析用試料
4.2.4による。
7.3.5
試料はかりとり量
試料はかりとり量は,1.0 gを,1 mgの桁まではかる。
7.3.6
操作
操作は,次による。
a) 試料の分解及び二酸化けい素の脱水処理 試料の分解及び二酸化けい素の脱水処理は,次による。
1) 7.2.6 a)の 1)〜7) の手順に従って操作する。
2) 融成物が溶け,白金るつぼ内に固着物がなくなった後,硝酸(1+1)30 mLを加え,加熱して水酸
化物などの沈殿物を溶解する。白金るつぼを水洗して取り出した後,7.2.6 a) 3) で保存したろ液及
び洗液の入ったビーカー(500 mL)に溶液を少量の水を用いて移し入れ,合わせる。
なお,マンガンの酸化物が認められる場合には,亜硝酸ナトリウム溶液(7.3.2.7)数滴を加える。
3) 過塩素酸50 mLを加え,加熱して過塩素酸白煙を発生させる。時計皿で覆い,熱板上で強熱して過
22
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
塩素酸の蒸気がビーカー内壁を伝わって逆流する状態になってから,更に15〜20分間加熱を続ける。
放冷した後,時計皿の下面を少量の水で洗って時計皿を取り除く。洗液は,ビーカーに入れる。
b) ろ過及び洗浄 ろ過及び洗浄は,次による。
1) a) 3) で得た,塩類の入っているビーカー(500 mL)中に温水を加えて液量を約300 mLとし,ガラ
ス棒でかき混ぜ,塩類を溶解する。
2) 静置して,沈殿物が沈降した後,速やかに沈殿をろ紙(5種B)を用いてこし分け,ビーカーの内
壁に付着した二酸化けい素は,ゴム管付きガラス棒を用いてこすり落とし,少量の温塩酸(1+100)
でろ紙上に洗い移す。
3) ろ紙及び沈殿物を,温塩酸(1+100)で5,6回,次いで温水で洗液に塩化物イオンがなくなるまで,
十分に洗浄する。この場合,洗液の少量を取り,硝酸銀溶液(7.3.2.9)4,5滴を加えて溶液が白濁
しなくなればよい。
c) ひょう量 ひょう量は,次による。
1) b) 3) で得た二酸化けい素の沈殿を,ろ紙とともに白金るつぼに移し入れ,加熱してろ紙を乾燥した
後,強熱して灰化する。
2) 1 100〜1 150 ℃で約1時間強熱して,デシケーター中で常温まで放冷した後,その質量をはかる。
0.3 mg以下の恒量となるまでこの操作を繰り返す。
3) 白金るつぼ中に硫酸(1+1)0.5 mL及びふっ化水素酸約5 mLを加え,穏やかに加熱して二酸化け
い素をふっ化けい素として揮散させ,引き続き硫酸白煙が発生しなくなるまで加熱する。
4) 1 100〜1 150 ℃で約30分間強熱して,デシケーター中で常温まで放冷した後,その質量をはかる。
0.3 mg以下の恒量となるまでこの操作を繰り返す。
7.3.7
空試験
空試験は,行わない。
7.3.8
計算
次の式によって試料中の全けい素含有率(SiO2表示)を算出する。
100
2
1
2
×
−
=
W
m
m
SiO
ここに, SiO2: 全けい素含有率(SiO2表示)[%(質量分率)]
m1: 7.3.6 c) 2)で得た質量(g)
m2: 7.3.6 c) 4)で得た質量(g)
W: 試料はかりとり量(g)
8
酸化アルミニウム定量方法
8.1
定量方法の区分
酸化アルミニウムの定量は,次のいずれかによる。
a) 金属アルミニウム分解分離ICP発光分光分析方法 この方法は,酸化アルミニウム含有率2.0 %(質
量分率)以上の試料に適用する。
b) 金属アルミニウム分解分離亜鉛逆滴定法 この方法は,酸化アルミニウム含有率10 %(質量分率)以
上の試料に適用する。
8.2
金属アルミニウム分解分離ICP発光分光分析方法
8.2.1
要旨
23
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試料をメタノール中臭素で分解した後,不溶解残さをこし分けて回収する。回収した不溶解残さを炭酸
ナトリウムとほう酸とで融解した後,融成物を水に溶解する。塩酸酸性とした後,一定量とし,ICP発光
分光分析方法によってアルミニウムを定量する。得られたアルミニウム量から,別途定量した窒化アルミ
ニウムとしてのアルミニウム量を差し引き,その残部を酸化アルミニウム量に換算して含有率を求める。
8.2.2
試薬
試薬は,次による。
8.2.2.1
塩酸(1+1)
8.2.2.2
臭素
8.2.2.3
融解合剤[ほう酸3,炭酸ナトリウム5:質量比]
8.2.2.4
アルゴン
8.2.2.5
メタノール
8.2.2.6
アルミニウム標準液(Al:1 mg/mL) 4.2.2.5による。
8.2.3
装置及び器具
8.2.3.1
ICP発光分光分析装置 分析装置の構成は,JIS K 0116による。
8.2.3.2
白金るつぼ 25番又は30番
8.2.4
分析用試料
4.2.4による。
8.2.5
試料はかりとり量
試料はかりとり量は,1.0 gとし,1 mgの桁まではかる。
8.2.6
操作
操作は,次による。
a) 試料溶液の調製 試料溶液の調製は,次による。
1) 試料をはかりとって乾燥したコニカルビーカー(500 mL)に移し入れる。
2) メタノール100 mLを加え,乾燥した時計皿で覆う。乾燥したピペットを用いて臭素を1 mLずつに
分けて合計5 mL加える。試料分解時の溶液温度が40 ℃以下5)となるよう臭素を加える時間間隔を
調整し,必要ならば,流水中で冷却しながら試料を分解する。分解反応による細かな気泡が生じな
くなったらそのまま一夜放置する。
注5) 分解時の溶液温度が40 ℃を超えると,金属アルミニウム以外のアルミニウム化合物が溶
け始める。
3) 時計皿の下面を少量のメタノールで洗って時計皿を取り除く。洗液は,ビーカーに入れる。ろ紙(5
種B)又はポリカーボネート樹脂製の孔径1 μmのメンブレンフィルターで溶液をこし分け,ろ紙(又
はメンブレンフィルター)及び残さを少量のメタノールで4,5回洗浄する。ろ液及び洗液は捨て,
残さは,ろ紙(又はメンブレンフィルター)とともに白金るつぼに移す。白金るつぼをセラミック
付き金網に載せ,バーナーの弱火で加熱して乾燥した後,バーナーを少し強くしてろ紙(又はメン
ブレンフィルター)を炭化する。さらに,マッフル炉の三角架に白金るつぼを置き,バーナー直火
で強熱してろ紙(又はメンブレンフィルター)を灰化した後,バーナーから下ろして室温まで放冷
する。炭素,金属状けい素などが共存していると,融解時に白金るつぼを損傷する原因となるので,
強熱してこれらを完全に酸化させておく。
4) 融解合剤(8.2.2.3)8.0 gをとり,その約半分を回収した残さが入っている白金るつぼに加え,ガラ
ス棒又は白金棒でよく混合する。残りの融解合剤をその上にかぶせるように加え,白金るつぼをセ
24
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ラミック付き金網に載せ,バーナーの弱火でしばらく加熱する。マッフル炉の三角架に白金るつぼ
を置き,徐々に加熱を強め,融解が始まったら,白金トングスを用いて白金るつぼをゆっくり回し
ながら加熱して残さを融解する。融解がほぼ終了したら,白金るつぼを三角架に置き,マッフル炉
中で加熱を続けて残さを完全に融解する。白金るつぼをセラミック付き金網上で室温まで放冷する。
5) ビーカー(300 mL)中にるつぼを横倒しに置き,水100 mLを加える。時々振り混ぜながら穏やか
に加熱して白金るつぼ内の融成物を溶かす。完全に溶けた後,るつぼを水洗して取り出す。溶液を
ガラス棒でかき混ぜながら塩酸(1+1)30 mLを少しずつ加える。加熱して溶液が透明になったら
流水中で常温まで冷却した後,250 mL全量フラスコに少量の水を用いて移し入れ,水で標線までう
すめる。
6) この溶液10 mLを100 mL全量フラスコに分取し,水で標線までうすめて,ICP発光強度測定用試
料溶液とする。
b) 測定 測定は,次による。
1) ICP発光分光分析装置を所定の測定条件にセットしてアルゴンプラズマを点灯し,安定した測定が
できる状態としておく。
2) ICP発光分光分析装置のアルゴンプラズマ中にa) 6)で調製したICP発光強度測定用試料溶液の一部
を噴霧し,309.271 nm,309.284 nm,396.152 nmなどのアルミニウムの発光線波長における発光強
度を測定する。バックグラウンドが同時測定可能な装置では,選択した測定波長に対して最も適正
と判断される方法にてバックグラウンド補正を行い,アルミニウムの真の発光強度を求める。
3) 8.2.8によって,同時に作成した検量線からアルミニウム量を求める。
8.2.7
空試験
空試験は,行わない。
8.2.8
検量線の作成
融解合剤8.0 gをビーカー(300 mL)にはかりとり,水100 mLを加える。加熱して溶かした後,溶液を
振り混ぜながら塩酸(1+1)30 mLを少量ずつ加える。加熱して1〜2分間煮沸した後,流水中で常温まで
冷却し,250 mL全量フラスコに水を用いて移し入れ,水で標線までうすめる。この溶液を10 mLずつ7
個の100 mL全量フラスコに分取する。8.2.6 a) 6)で調製したICP発光強度測定用試料溶液中のアルミニウ
ム量が内挿されるように,アルミニウム標準液(8.2.2.6)でアルミニウムとして0,1,3,5,10,15及び
20 mgをそれぞれ加え,水で標線までうすめる。この溶液について8.2.6 b)によって測定を行い,発光強度
と添加アルミニウム量との関係線を作成して検量線とする。
8.2.9
計算
計算は,次による。
a) 次の式によって回収した不溶解残さ中のアルミニウム量(A)を算出する。
10
/
250
×
=a
A
ここに,
A: 回収した不溶解残さ中のアルミニウム量(g)
a: 検量線によって求めたICP発光強度測定用試料溶液中のア
ルミニウム量(g)
b) 箇条5に従って別途分析した窒素量によって算出した窒化アルミニウム含有量から,はかりとった試
料中に含有される窒化アルミニウムとしてのアルミニウム量(B)を算出し,これをa)で求めたアル
ミニウム量(A)から差し引いて,酸化アルミニウムとしてのアルミニウム量を求める。これを“補
正Al量”とする。
25
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 次の式によって試料中の酸化アルミニウム含有率を算出する。
100
4
1.889
3
2
×
×
=
W
Al
O
Al
量
補正
ここに,
Al2O3: 酸化アルミニウム含有率[%(質量分率)]
補正Al量: A−B(g)
A:
回収した不溶解残さ中のアルミニウム量(g)
B:
窒化アルミニウムとしてのアルミニウム量(g)
W: 試料はかりとり量(g)
1.889 4: AlからAl2O3への換算係数
8.3
金属アルミニウム分解分離亜鉛逆滴定法
8.3.1
要旨
試料をメタノール中臭素で分解した後,不溶解残さをこし分けて回収する。回収した不溶解残さを炭酸
ナトリウムとほう酸とで融解した後,融成物を水に溶解する。塩酸酸性とした後,溶液中のアルミニウム
をエチレンジアミン四酢酸と亜鉛とによる逆滴定法によって定量する。得られたアルミニウム量から,別
途定量した窒化アルミニウムとしてのアルミニウム量を差し引き,その残部を酸化アルミニウム量に換算
して含有率を求める。
8.3.2
試薬
試薬は,次による。
8.3.2.1
塩酸(1+1)
8.3.2.2
アンモニア水(1+2)
8.3.2.3
臭素
8.3.2.4
ふっ化ナトリウム飽和溶液 4.3.2.5による。
8.3.2.5
融解合剤[ほう酸3,炭酸ナトリウム5:質量比]
8.3.2.6
メタノール
8.3.2.7
エチレンジアミン四酢酸二水素二ナトリウム溶液(EDTA2Na溶液) 4.3.2.7による。
8.3.2.8
0.1 mol/L亜鉛標準液 4.3.2.8による。
8.3.2.9
キシレノールオレンジ溶液 4.3.2.9による。
8.3.2.10 緩衝液(pH5.8) 4.3.2.10による。
8.3.3
分析用試料
4.2.4による。
8.3.4
試料はかりとり量
試料はかりとり量は,1.0 gとし,1 mgの桁まではかる。
8.3.5
操作
操作は,次による。
a) 試料溶液の調製 8.2.6 a)の 1)〜5)の手順に従って調製した溶液を試料溶液とする。
b) 共存イオンのマスキング 共存イオンのマスキングは,次による。
1) a)で調製した試料溶液の20 mLをコニカルビーカー(300 mL)に分取し,EDTA2Na溶液(8.3.2.7)
20 mLを加え,よく振り混ぜる。アンモニア水(1+2)を用いて溶液をpHメーターでpH5〜pH6
に調整した後,緩衝液(pH5.8)(8.3.2.10)15 mLを加える。
2) 加熱して約3分間煮沸した後,流水中で常温まで冷却する。キシレノールオレンジ溶液(8.3.2.9)4,
26
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5滴を加え,0.1 mol/L亜鉛標準液(8.3.2.8)で滴定して溶液の色が黄から赤とうとなったところで
止める。
c) 滴定 適定は,次による。
1) 直ちにふっ化ナトリウム飽和溶液(8.3.2.4)50 mLを加え,再び加熱して約3分間煮沸した後,流
水中で常温まで冷却する。
2) キシレノールオレンジ溶液(8.3.2.9)2,3滴を追加し,アルミニウム量に相当する遊離したエチレ
ンジアミン四酢酸を0.1 mol/L亜鉛標準液(8.3.2.8)で滴定する。溶液の色が黄から赤とうに変化し
たところを終点とし,そのときの滴定量a(mL)を記録する。
8.3.6
空試験
空試験は,行わない。
8.3.7
計算
計算は,次による。
a) 次の式によって回収した不溶解残さ中のアルミニウム量(A)を算出する。
20
/
250
698
002
.0
×
×
=a
A
ここに,
A: 回収した不溶解残さ中のアルミニウム量(g)
a: 8.3.5 c) 2)で記録した0.1 mol/L亜鉛標準液の滴定量(mL)
0.002 698: 0.1 mol/L亜鉛標準液1 mLに相当するアルミニウムの質
量を示す換算係数(g/mL)
b) 箇条5に従って別途分析した窒素量によって算出した窒化アルミニウム含有量から,はかりとった試
料中に含有される窒化アルミニウムとしてのアルミニウム量(B)を算出し,これをa)で求めたアル
ミニウム量(A)から差し引いて,酸化アルミニウムとしてのアルミニウム量を求める。これを“補
正Al量”とする。
c) 次の式によって試料中の酸化アルミニウム含有率を算出する。
100
4
1.889
3
2
×
×
=
W
Al
O
Al
量
補正
ここに,
Al2O3: 試料中の酸化アルミニウム含有率[%(質量分率)]
補正Al量: A−B(g)
A:
回収した不溶解残さ中のアルミニウム量(g)
B:
別途分析した窒素量によって算出した窒化ア
ルミニウムとしてのアルミニウム量(g)
W: 試料はかりとり量(g)
1.889 4: AlからAl2O3への換算係数
9
炭素定量方法
9.1
定量方法
炭素の定量方法は,燃焼−赤外線吸収法による。この方法は,炭素含有率0.10 %(質量分率)以上7.0 %
(質量分率)以下の試料に適用する。
9.2
要旨
試料を助燃剤とともに酸素気流中で燃焼させ,炭素を酸化して炭素酸化物とし,これを酸素とともに赤
外線吸収セルに導き,二酸化炭素又は二酸化炭素と一酸化炭素とによる赤外線吸収量を連続測定して積分
することによって炭素量を求める。
9.3
試薬
27
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試薬は,次による。
9.3.1
二酸化炭素を除いた水 JIS K 0050のE.2(二酸化炭素を除いた水の場合)による。
9.3.2
酸素 酸素は,JIS K 1101に規定するもの。
9.3.3
鉄 純度が高く,炭素含有率が0.001 0 %(質量分率)以下で既知のもの。
9.3.4
過塩素酸マグネシウム 元素分析用で粒径0.7〜1.7 mmのもの。
警告 過塩素酸マグネシウムは,強力な酸化剤であり,有機物との接触は避けなければならない。廃
棄するときは,そのまま廃棄箱に捨てず,水に溶かして処理する。
9.3.5
炭酸ナトリウム 炭酸ナトリウム[99.9 %(質量分率)以上]を使用前に約300 ℃で約2時間乾燥
した後,グリースなどを塗らないデシケーター中で放冷する。
9.3.6
しゅう酸ナトリウム しゅう酸ナトリウム[99.95 %(質量分率)以上]を使用前に約200 ℃で約
1時間乾燥した後,グリースなどを塗らないデシケーター中で放冷する。
9.3.7
助燃剤 炭素含有率が0.001 0 %(質量分率)以下の鉄,すず又はタングステンを単独又は合わせ
たもの。高周波誘導加熱燃焼に用いる助燃剤は,タングステン・すず・鉄の複合使用が望ましく,管状電
気抵抗加熱燃焼に用いる助燃剤は,すず又はタングステンが望ましい。それらの使用量は,使用する装置
に最適な量をあらかじめ調査しておく。
なお,助燃剤は,JIS Z 2615の8.13(助燃剤)に示されている形状のものを用いることが望ましい。
9.3.8
炭素標準液A(C:25 mg/mL) スクロース(C12H22O11)をあらかじめ100〜105 ℃で約2.5時間
乾燥してグリースなどを塗らないデシケーター中で放冷しておく。これの14.843 gをはかりとり,二酸化
炭素を除いた水(9.3.1)約100 mLを加えて溶解し,250 mLの全量フラスコに二酸化炭素を除いた水を用
いて移し入れ,二酸化炭素を除いた水で標線までうすめる。
9.3.9
炭素標準液B(C:25 mg/mL) 炭酸ナトリウム(9.3.5)55.152 gをはかりとり,二酸化炭素を除
いた水(9.3.1)約200 mLを加えて溶解し,250 mLの全量フラスコに二酸化炭素を除いた水を用いて移し
入れ,二酸化炭素を除いた水で標線までうすめる。
9.3.10
水酸化ナトリウムを含浸させた不活性磁器粒子(繊推質粘土鉱物) 粒径0.7〜1.2 mmのもの。
9.3.11
鉄鋼認証標準物質 炭素含有率の認証値が得られている,鉄鋼認証標準物質。
9.3.12
検量線校正用試料 炭素含有率が検量線の上限付近で,均質な試料。鉄鋼認証標準物質(9.3.11)
を用いてもよい。
9.4
器具及び材料
器具及び材料は,JIS Z 2615の箇条8(器具及び材料)によるほか,次による。
9.4.1
ピストン式ピペット JIS K 0970に規定された容量100 μLで,誤差が1 μL以内のもの。
9.4.2
すずカプセル 直径約6 mm,高さ約18 mm,質量約0.3 g及び体積約0.4 mLで,炭素含有率が
0.001 0 %(質量分率)以下のもの。
9.4.3
高周波磁器燃焼るつぼ及び蓋
JIS Z 2615の8.11(高周波磁器燃焼るつぼ)による。
高周波磁器燃焼るつぼ及び蓋6)は,あらかじめ空気中又は酸素(9.3.2)中約1 100 ℃で約2時間空焼き
を行う。一度に多数空焼きした場合は,放冷した後,グリースなどを塗らないデシケーター中で保管する。
取扱いはピンセットなどを用い,直接手を触れてはならない。長時間保管したものは,空試験値が高くな
っているおそれがあるので使用を避け,再度空焼きを行う。
注6) 蓋は燃焼時の燃焼ダスト類の飛散防止に効果があり,分析精度の改善,燃焼ガス系内の汚染防
止につながる。
28
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.4.4
磁器燃焼ボート及び磁器燃焼ボートカバー
JIS Z 2615の8.10(磁器燃焼ボート及び磁器燃焼ボートカバー)による。
管状電気抵抗加熱炉に使用する磁器燃焼ボート及び磁器燃焼ボートカバーは,JIS R 1306に規定する
CB1及びCBC1とする。磁器燃焼ボート及び磁器燃焼ボートカバーは,あらかじめ空気中又は酸素(9.3.2)
中約1 250 ℃で約5分間空焼きを行う。一度に多数空焼きした場合は,放冷した後,若干の余熱をもつ状
態から使用直前までグリースなどを塗らないデシケーター中で保管する。取扱いはピンセットなどを用い,
直接手を触れてはならない。長時間保管したものは,空試験値が高くなっているおそれがあるので使用を
避け,再度空焼きを行う。
9.5
装置
使用する装置は,市販の高周波誘導加熱炉燃焼−赤外線吸収炭素分析装置若しくは管状電気抵抗加熱炉
燃焼−赤外線吸収炭素分析装置又はそれらと同等の装置で,酸素精製部,試料燃焼部,燃焼ガス精製部及
び炭素酸化物定量部からなる。装置の組立ては,JIS Z 2615の9.7.2(装置の組立て)による。装置構成の
例を,図3に示す。
炭素酸化物定量部
図3−装置構成の例
9.5.1
酸素精製部
二酸化炭素吸収管にはシリカゲル,雲母などの無機質の支持体に水酸化ナトリウムを含浸させたもの,
粒状の水酸化ナトリウム,ソーダ石灰などを,脱水管には過塩素酸マグネシウムを詰める。圧力調整器及
びバルブによって一定の圧力に保ち,燃焼管を通り,赤外線吸収検出器の試料セルに送る。燃焼管の酸素
は,流路切換器によって加熱前には流路外にパージし,測定開始(加熱)時には赤外線吸収検出器側に流
路を切り換える。
注記 市販の赤外線吸収検出器の装置には,対照セルを用いるものがある。
9.5.2
試料燃焼部
試料を酸素気流中で加熱燃焼する部分で,試料挿入部,燃焼管及び加熱炉からなる。加熱炉には,高周
波誘導加熱炉又は管状電気抵抗加熱炉が使用される。その入口は酸素精製部に,出口は燃焼ガス精製部に
燃焼ガス精製部
試料燃焼部
赤外線吸収セル
赤外線吸収検出器
酸素
圧力及び
酸化管
二酸化炭素
吸収管
脱水管
酸素精製部
試料+
助燃剤
脱水管
酸化管及び加熱炉
脱硫管
集じん管
流量調整器
燃焼管
及び加熱炉
29
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
連結して使用する。
a) 高周波誘導加熱炉 高周波誘導加熱炉は,JIS Z 2615の8.6 b)(高周波誘導加熱炉)による。
b) 管状電気抵抗加熱炉 管状電気抵抗加熱炉は,約1 250 ℃以上の温度に加熱できるもので,JIS Z 2615
の8.6 a)(管状電気抵抗加熱炉)による。
9.5.3
燃焼ガス精製部
燃焼ガス精製部は,JIS Z 2615の7.1.4(燃焼ガス精製部)による。
集じん管には石英ガラスウールを,脱水管には過塩素酸マグネシウムを詰める。酸化管には白金触媒を
詰め,約350 ℃に加熱する。
9.5.4
炭素酸化物定量部(赤外線吸収部)
赤外線吸収検出器は,赤外線発生源(IRソース),試料セル,検出部などから構成され,炭素酸化物の
赤外線吸収量を測定できるものを用いる。その測定回路は,直線化回路,演算回路などから構成する。赤
外線吸収検出器から取り出した電気信号を,炭素酸化物に変換・加算して,炭素の量に比例した電圧を指
示計に供給する。指示計は,試料はかりとり量を指定する場合には,炭素の含有率を直読できることが望
ましい。
注記 市販の装置には,二酸化硫黄の検出器及び硫黄用の指示器をもち,硫黄との同時定量ができる
ものがある。
9.6
分析用試料
4.2.4による。
9.7
試料はかりとり量
試料はかりとり量は,表4に従い,1 mgの桁まではかる。
表4−試料はかりとり量
使用装置
炭素含有率
%(質量分率)
試料はかりとり量
g
高周波誘導加熱炉
0.10以上 7.0以下
0.1
管状電気抵抗加熱炉
0.10以上 0.5未満
0.2
0.5 以上 7.0以下
0.1
9.8
操作
9.8.1
予備操作
予備操作は,次の手順によって行う。
a) 電源を入れ,各部が十分に安定した後,酸素(9.3.2)を送入して装置の気密性を確認する。酸化管を
指定の温度に保つ。また,管状電気抵抗加熱炉の場合は,燃焼管の温度を約1 250 ℃に保つ。
b) 装置で指定する圧力及び流量で酸素(9.3.2)を通気し,指示計のゼロ点などを調節する。
9.8.2
定量操作
定量操作は,次のいずれかの手順によって行う。
a) 高周波誘導加熱炉による場合 予備操作を行った後,次の手順によって行う。
1) はかりとった試料(9.7)を高周波磁器燃焼るつぼ(9.4.3)に入れる。
2) 助燃剤(9.3.7)を高周波磁器燃焼るつぼ(9.4.3)中の試料にかぶせるように入れる。事前に,用い
る分析装置で試料が完全燃焼できる最適な助燃剤の組合せ条件を求めておく。助燃剤の一般的な添
30
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
加量の例を表5に示す。
表5−高周波誘導加熱炉における助燃剤の例
種類
添加量
g
添加順
添加方法
タングステン(粒状250〜1 000 μm)
1.5
1
試料の上に添加順に従っ
てかぶせて置く。偏りがあ
れば,平らにする。
すず(粒状250〜1 000 μm)
0.3
2
鉄(粒状150〜1 000 μm)
0.6
3
3) 試料及び助燃剤を入れた高周波磁器燃焼るつぼに蓋(9.4.3)をかぶせて受台に置き,加熱コイルの
中心部に挿入し,燃焼管を気密になるように閉じる。
4) 燃焼管内が所定の圧力になるのを待ち,高周波誘導加熱炉を作動させ,試料を燃焼する。
5) 発生した燃焼ガスは,燃焼ガス精製部を経て赤外線吸収セルに送る。
6) 指示値が次第に増加し,指示計が一定値を示したとき高周波誘導加熱を止め,指示値を読み取る。
7) 読み取った指示値は,9.10.1による検量線を用いて炭素量に変換する。
注記 市販の装置では,燃焼管を閉じると,酸素は燃焼管,燃焼ガス精製部及び赤外線吸収検出
器の試料セルを経て大気中に放出するパージ時間が設定できる。高周波磁器燃焼るつぼ
(9.4.3)挿入のため燃焼管を開けるときは,ランス(試料の燃焼を促進するために,酸素
を試料の直上へ効率よく供給する搬送管)から酸素が噴出しているので,空気は燃焼管内
に侵入しない。
市販の装置では,燃焼時間タイマーの設定によって,加熱開始,加熱停止などの動作を
自動的に行うものがある。
指示値は,赤外線吸収量の積分値又は炭素含有率に換算された値で示される。
b) 管状電気抵抗加熱炉による場合 予備操作を行った後,次の手順によって行う。
1) 磁器燃焼ボート(9.4.4)に助燃剤(9.3.7)を表6に従い入れる。この上にはかりとった試料(9.7)
を加え入れ,更に残りの助燃剤をかぶせるようにして加えて3層状にし,磁器燃焼ボートカバー
(9.4.4)をかぶせる。事前に,用いる分析装置で試料が完全燃焼できる最適な助燃剤又はその組合
せ条件を求めておく。助燃剤の一般的な添加量の例を表6に示す。
表6−管状電気抵抗加熱炉における助燃剤の例
種類
添加量
g
添加方法
すず(粒状140〜250 μm)又は
タングステン(粒状200〜900 μm)
1.0
又は
2.0
試料の下に助燃剤1.0 g又は
2.0 gを層状に置く。
2.0
試料の上に助燃剤2.0 gを層状
に置く。
2) 1 250 ℃に加熱された燃焼管の挿入口を開いて,試料及び助燃剤を入れた磁器燃焼ボート及び磁器
燃焼ボートカバー(9.4.4)を燃焼管内の適切な部位に挿入し,燃焼管を気密になるように直ちに閉
じる。
3) 適切な流量(例えば,5 L/min程度)で酸素(9.3.2)を流し,試料を燃焼する。
31
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) 発生した燃焼ガスを,酸素(9.3.2)とともに燃焼ガス精製部を経て赤外線吸収セルに送る。
5) 燃焼ガス中の炭素酸化物含有量に相当する指示値を読み取る。炭素含有率換算値を指示値とする形
式の場合は,試料はかりとり量の入力を0.100 g又は0.200 gとする。試料中の炭素の化学形態によ
って所要燃焼時間が異なるので,あらかじめ確認した積分時間で測定する。通常120秒間とし,一
連の測定の途中で変更しない。
6) 読み取った指示値は,9.10.2による検量線を用いて炭素量に変換する。
9.9
空試験
a) 高周波誘導加熱炉による場合 試料に加えるのと同量の助燃剤だけをはかりとって高周波磁器燃焼る
つぼに入れた後,9.8.2 a) の3)〜6)の手順に従って試料と同じ操作を,試料と併行して行い,空試験指
示値を読み取る。
b) 管状電気抵抗加熱炉による場合 試料に加えるのと同量の助燃剤だけをはかりとって磁器燃焼ボート
に入れた後,9.8.2 b) の2)〜5)の手順に従って試料と同じ操作を,試料と併行して行い,空試験指示
値を読み取る。
空試験は,2回行う。空試験値は,2個の空試験指示値から平均空試験指示値を計算し,9.10による検量
線を用いて空試験値を炭素量に変換する。
9.10
検量線の作成
9.10.1
高周波誘導加熱炉による場合
注記 市販の装置には,試料はかりとり量及び空試験値を補正し,炭素含有率を直読できるものがあ
る。この場合には,指示値が既知の炭素含有率と一致するように調節できる。
9.10.1.1 試薬を用いる検量線の作成
a) 炭素含有率0.10 %(質量分率)以上1.0 %(質量分率)未満の試料の場合
1) 検量線シリーズの調製 検量線溶液の調製は,5個の50 mL全量フラスコのそれぞれに,表7に示
す炭素標準液A(9.3.8)又は炭素標準液B(9.3.9)の体積を5 段階はかりとり,二酸化炭素を除い
た水(9.3.1)を標線まで加えて混合して希釈溶液とする。各希釈溶液の100 μLずつを,ピストン式
ピペット(9.4.1)を用いて分取して5個のすずカプセル(9.4.2)のそれぞれに入れ,90〜95 ℃で約
2時間乾燥する。グリースなどを塗らないデシケーター中で室温まで放冷する。
表7−炭素含有率0.10 %(質量分率)以上1.0 %(質量分率)未満の場合の検量線シリーズ
炭素標準液A(9.3.8)又は
炭素標準液B(9.3.9)の量
mL
希釈溶液中の
炭素含有量
mg/mL
すずカプセル(9.4.2)に
入れた炭素量
mg
はかりとり試料中の
炭素含有率
%(質量分率)
0a)
0a)
0
0
2
1.0
0.10
0.10
4
2.0
0.20
0.20
10
5.0
0.50
0.50
20
10.0
1.00
1.00
注a) ゼロメンバー(分析対象成分の標準液を添加していない溶液)
2) 測定 1)で調製した,炭素標準液の入ったすずカプセル(9.4.2)を高周波磁器燃焼るつぼ(9.4.3)
に入れ,カプセルを高周波磁器燃焼るつぼの底の方へ軽く押さえ付けて鉄(9.3.3)0.100 gを加え,
はかりとり試料に加えるのと同じ種類及び量の助燃剤(9.3.7)で覆う。高周波磁器燃焼るつぼと内

32
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
容物を9.8.2 a)の3)〜6)の手順に従って処理する。
3) 検量線の作成 検量線シリーズの各溶液について,2)で得た指示値からゼロメンバーの指示値を差
し引いて正味の指示値とする。検量線シリーズの各溶液の炭素量(mg)に対する正味の指示値をプ
ロットした検量線を作成する。
b) 炭素含有率1.0 %(質量分率)以上7.0 %(質量分率)以下の試料の場合
1) 検量線シリーズの調製 炭酸ナトリウム(9.3.5)又はしゅう酸ナトリウム(9.3.6)を,表8の各行
に示す質量をはかりとり,5個のすずカプセル(9.4.2)のそれぞれに移し入れる。はかりとった試
薬をすずカプセルの中に入れることができない場合は,それを高周波磁器燃焼るつぼ(9.4.3)の底
に直接置いてもよい。
表8−炭素含有率1.0 %(質量分率)以上7.0 %(質量分率)以下の場合の検量線シリーズ
試薬のはかりとり質量
すずカプセル(9.4.2)に
入れた炭素量
mg
はかりとり試料中の
炭素含有率
%(質量分率)
炭酸ナトリウム
(9.3.5)の場合
mg
しゅう酸ナトリウム
(9.3.6)の場合
mg
0a)
0a)
0
0
12.9
5.6
1.0
1.0
37.7
16.8
3.0
3.0
62.9
27.9
5.0
5.0
88.1
39.1
7.0
7.0
注a) ゼロメンバー
2) 測定 1)で調製した,炭酸ナトリウム(9.3.5)又はしゅう酸ナトリウム(9.3.6)の入ったすずカプ
セル(9.4.2)を高周波磁器燃焼るつぼ(9.4.3)に入れ,カプセルを高周波磁器燃焼るつぼの底の方
へ軽く押さえ付けて鉄(9.3.3)0.100 gを加え,はかりとり試料に加えるのと同量の助燃剤(9.3.7)
で覆う。高周波磁器燃焼るつぼと内容物を9.8.2 a)の3)〜6)の手順に従って処理する。
3) 検量線の作成 検量線シリーズの各溶液について,2)で得た指示値からゼロメンバーの指示値を差
し引いて正味の指示値とする。検量線シリーズの各溶液の炭素量(mg)に対する正味の指示値をプ
ロットした検量線を作成する。
9.10.1.2 鉄鋼認証標準物質を用いる検量線の作成
a) 検量線作成用試料の選定 検量線を作成したい炭素含有率範囲に対して,その上下限近傍の含有率を
含み,かつ,認証された炭素含有率が適用範囲を均等に満足するように,できるだけ4種類以上の鉄
鋼認証標準物質(9.3.11)を選定する。鉄鋼認証標準物質を2種類準備する場合[例えば,JSS 150-15
炭素含有率0.469 %(質量分率)及びJSS 110-7炭素含有率4.12 %(質量分率)],これら標準物質の採
取量を0.1〜0.5 gの範囲で変化させて,はかりとったときの標準物質中の炭素量が0.5〜10 mgの間で
5〜6段階になるようにはかりとり量を決める。
注記 鉄鋼認証標準物質(9.3.11)の使用によってトレーサビリティを確保する。一般的な鉄鋼認証
標準物質のはかりとり量は,0.5 g又は1 gとする場合が多い。含有率の高い定量域を検量で
きない場合(例えば,5 %以上)は,鉄鋼認証標準物質(9.3.11)のはかりとり量を変動させ
て補完することができる。
b) 測定 測定は,次による。
33
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 9.8.2 a)の1)及び2)と同様にして鉄鋼認証標準物質(9.3.11)及び助燃剤をはかりとり,高周波磁器
燃焼るつぼ(9.4.3)に移し入れる。
2) 9.8.2 a)の3)〜6)の手順に従って操作し,指示値を読み取る。
3) 読み取った指示値から9.9 a)で測定した平均空試験指示値を差し引き,正味の指示値とする。
c) 検量線の作成 正味の指示値を縦軸,使用した鉄鋼認証標準物質(9.3.11)の炭素量を横軸として,指
示値と炭素量との関係線を作成し,検量線とする。
注記 検量線は一次回帰直線とすることから,広範囲で認証標準物質と正味の指示値との直線関係
が重要となる。使用する検量線は,一次回帰計算で得られる相関係数が0.999以上となる範
囲内で適用する。
9.10.2
管状電気抵抗加熱炉による場合
a) 検量線作成用試料の選定 9.10.1.2 a)と同様に,検量線を作成したい炭素含有率範囲に対して,その上
下限近傍の含有率を含み,かつ,認証された炭素含有率が適用範囲を均等に満足するように,できる
だけ4種類以上の鉄鋼認証標準物質(9.3.11)を選定する。
注記 炭素含有率0.10〜4.5 %(質量分率)の鉄鋼認証標準物質(9.3.11)として,日本鉄鋼認証標
準物質(JSS),アメリカ国立標準技術研究所認証標準物質(NIST SRM)などが適用できる。
b) 測定 測定は,次による。
1) 9.8.2 b) 1)と同様にして鉄鋼認証標準物質(9.3.11)及び助燃剤をはかりとり,磁器燃焼ボート(9.4.4)
に移し入れる。
2) 9.8.2 b)の2)〜5)の手順に従って操作し,指示値を読み取る。
3) 読み取った指示値から9.9 b)で測定した平均空試験指示値を差し引き,正味の指示値とする。
c) 検量線の作成 正味の指示値を縦軸,使用した鉄鋼認証標準物質(9.3.11)の炭素量を横軸として,指
示値と炭素量との関係線を作成し,検量線とする。
9.11
検量線の校正
検量線の校正は,次による。
a) 検量線校正用試料(9.3.12)を定期的に定量し,あらかじめ実験的に求めた室内許容差を満足すること
を確認する。これを満足できない場合,又は装置条件の変動があり作成した検量線に経時変化がある
ときは,検量線の校正を行う。
b) 含有率が検量線の範囲の上限及び下限付近にある2個の均質な試料を経時変化補正試料(以下,補正
試料という。)として用いる。
c) 補正試料を,9.10.1.2 b)又は9.10.2 b)によって測定する。
d) 検量線作成時の指示値からの変化を,2個の補正試料の指示値を用いて,式(7)によって補正する。
I=α×Iʼ+β ································································································· (7)
ʼ
ʼL
H
L
H
I
I
I
I
−
−
=
α
β=IH−α×IHʼ
ここに,
I : 分析試料分析時の補正後の指示値
Iʼ : 分析試料分析時の未補正の指示値
IH : 高濃度側検量線校正試料の検量線作成時の指示値
IL : 低濃度側検量線校正試料の検量線作成時の指示値
IHʼ: 高濃度側検量線校正試料の指示値
34
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ILʼ: 低濃度側検量線校正試料の指示値
注記 検量線の校正の計算は,通常は装置に組み込まれていて自動計算される。
9.12
計算
次の式によって試料中の炭素の含有率を算出する。
100
000
1
×
×
−
=W
B
A
C
ここに,
C: 炭素含有率[%(質量分率)]
A: 検量線によって求めた炭素量(mg)
B: 空試験で得た炭素量(mg)
W: 試料はかりとり量(g)
注記 市販の測定装置では,赤外線吸収量の積分値を炭素含有率に直接換算する計算処理ソフトウェ
アを内蔵し,試料はかりとり量が入力されていれば炭素含有率が測定指示値として示されるも
のが多い。検量線法を用いずに装置の測定指示値をそのまま炭素含有率分析結果とする場合は,
測定に先立って,適用範囲内の鉄鋼認証標準物質(9.3.11)を用いて真度を十分確認する必要が
あり,異常があれば,空試験値設定を含めた計算処理ソフトウェアの校正を必ず実施する。
10 水分定量方法
10.1 定量方法
水分の定量方法は,乾燥減量法による。
10.2 要旨
水分試験試料を規定温度で一定時間乾燥し,乾燥減量を求めて水分量を算出する。
10.3 試料
JIS G 2403の7.5(水分試験試料)によって調製した水分試験試料を,3個以上採取する。
10.4 装置及び器具
装置及び器具は,次による。
10.4.1
乾燥器 乾燥器は,保持温度の許容差が±5 ℃の性能をもつものを用いる。
10.4.2
乾燥容器 乾燥容器は,耐食耐熱性のもので,広げた試料の厚さが10 mm以下となる底面積をも
つ乾燥皿を用いる。また,乾燥容器の質量は,測定する試料の質量より小さくしなければならない。
10.4.3
はかり はかりは,最小読取値が試料量の1/2 000以下のものを用いる。
10.5 操作
操作は,10.3で採取した各試料について次の手順によって行う。
a) あらかじめ乾燥した質量既知の乾燥容器(W1)に試料を移し,試料の厚さがほぼ一定になるように平
らに広げ,それぞれの質量(W2)をはかる。
b) あらかじめ(105±5)℃に調節されている乾燥器に1試料1乾燥容器とし,試料を入れたそれぞれの
乾燥容器を同時に入れて乾燥する。
注記 変質しやすいものは,受渡当事者間において定めた乾燥方法による。
c) 約6時間乾燥した後,試料を入れたそれぞれの乾燥容器を取り出して直ちにそれぞれの質量(W3)を
はかる。
10.6 計算
次の式によって乾燥減量率M[%(質量分率)]を算出する。
35
G 2404:2015
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
1
2
3
2
×
−
−
=
W
W
W
W
M
ここに,
M: 乾燥減量率[%(質量分率)]
W1: 乾燥容器の質量(g)
W2: 10.5 a)で得た質量(g)
W3: 10.5 c)で得た質量(g)
10.7 許容差
各試料の乾燥減量率Mの最大値と最小値との差が0.1 %(質量分率)を超える場合は,再試験を行う。
10.8 分析値
乾燥減量率の各測定値を算術平均し,小数点以下2桁とした値を水分の分析値とする。
11 安全衛生及び環境に関する注意
ICP発光分光分析及び燃焼−赤外線吸収法における高圧ガスの取扱い,危険薬品(臭素,ふっ化水素酸,
有機溶媒など)の使用・廃棄処理などには十分注意し,災害の防止及び環境の保全に努めなければならな
い。