2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
G 1328-1982
フェロニオブ分析方法
Methods for Chemical Analysis of Ferroniobium
1. 適用範囲 この規格は,フェロニオブ中のニオブ,タンタル,炭素,けい素,りん,硫黄,すず及び
アルミニウムの定量方法について規定する。
引用規格:
JIS G 1301 フェロアロイ分析方法の通則
JIS K 8006 試薬の含量試験中滴定に関する基本事項
JIS R 1306 化学分析用磁器燃焼ボート
JIS R 1307 化学分析用磁器燃焼管
JIS R 1308 化学分析用高周波燃焼るつぼ
JIS Z 2615 金属材料の炭素定量方法通則
2. 一般事項 定量方法に共通な一般事項は,JIS G 1301(フェロアロイ分析方法の通則)による。
3. ニオブ定量方法
3.1
方法の区分 ニオブの定量方法は,次のいずれかによる。
(1) 加水分解分離五酸化ニオブ重量法 この方法は,フェロニオブの全範囲に適用する。
(2) イオン交換分離五酸化ニオブ重量法 この方法は,フェロニオブの全範囲に適用する。
3.2
加水分解分離五酸化ニオブ重量法
3.2.1
要旨 試料をふっ化水素酸と硝酸で分解し,硫酸で白煙処理をする。これを塩酸と過酸化水素水に
溶解し,亜硫酸とタンニン酸でニオブ及びタンタル(チタンの一部を含む)を加水分解させる。沈殿をこ
し分けて灰化し,ニオブ及びタンタルなどの混合酸化物をふっ化水素酸と硫酸で溶解し,再び亜硫酸とタ
ンニン酸でニオブ及びタンタルなどを沈殿させる。沈殿をこし分けて強熱し,恒量として混合酸化物の質
量をはかる。この混合酸化物をピロ硫酸カリウムで融解し,しゅう酸アンモニウムに溶解してジアンチピ
リルメタン吸光光度法又はアリザリンS吸光光度法で二酸化チタン量を求める。別にこの溶液から4.3ピ
ロガロール吸光光度法又は4.4マラカイトゲリーン吸光光度法でタンタル量を求める。先に求めた混合酸
化物の質量からチタンとタンタルの酸化物の質量を差し引く。
3.2.2
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1, 1+50)
(3) 硝酸 (1+1)
(4) ふっ化水素酸
(5) 硫酸 (1+1)
2
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(6) 亜硫酸水(飽和,約6%)
(7) 過酸化水素水
(8) ピロ硫酸カリウム
(9) 塩化第一すず溶液 塩化第一すず (SnCl2・2H2O) 100gを塩酸250mlに溶解して水で500mlにうすめる。
(10) ニオブ・タンタル混合溶液 五酸化ニオブ(純度99.5%以上)0.186gと五酸化タンタル(純度99.5%
以上)0.017gをはかり取って白金又は石英るつぼ (30ml) に移し,ピロ硫酸カリウム5gを混合して加
熱融解する。放冷後,るつぼをビーカー (300ml) に移し,しゅう酸アンモニウム溶液〔3.2.2(12)〕約
150mlを加えて加熱溶解し,るつぼをしゅう酸アンモニウム溶液〔3.2.2(12)〕で洗浄して取り出す。常
温まで冷却後,この溶液を250mlのメスフラスコに移し,しゅう酸アンモニウム溶液〔3.2.2(12)〕で
標線までうすめる。この溶液25mlは,ニオブ含有率65%,タンタル含有率7%のフェロニオブ0.200g
を処理してその101を分取した量に相当するニオブ及びタンタルを含有する。この溶液の調製は,
4.3.3(2)及び(6)に示してあるニオブ溶液及びタンタル溶液を別々に調製して必要量を混合してもよい。
(11) タンニン酸 (C14H10O9)
(12) しゅう酸アンモニウム溶液 しゅう酸アンモニウム〔(NH4)2C2O4・H2O〕40gを水に溶解して1000ml
とする。
(13) ジアンチピリルメタン溶液 ジアンチピリルメタン〔CH2〔C3N2O(CH3)2C6H5〕2・H2O〕15gを水300ml
と硫酸 (1+1) 30mlに加熱して溶解し,常温まで冷却して水で1000mlにうすめ,褐色瓶に入れて保存
する。
(14) アリザリンS溶液 アリザリンスルホン酸ナトリウム (C14H7O7S・Na・H2O) 0.2gを水に溶解して100ml
にうすめる。
(15) 標準チタン溶液 (20μgTiO2/ml) あらかじめ900〜950℃で約30分間強熱して冷却した二酸化チタン
(純度99.0%以上)0.2000gをはかり取って白金又は石英るつぼに移し,ピロ硫酸カリウム5gを混合
して加熱融解する。放冷後,るつぼをビーカー (500ml) に移してしゅう酸アンモニウム溶液〔3.2.2(12)〕
約300mlを加え,加熱溶解してるつぼをしゅう酸アンモニウム溶液で洗浄して取り出し,常温まで冷
却して500mlのメスフラスコに移し,しゅう酸アンモニウム溶液で標線までうすめ,標準原液
(400μgTiO2/ml) とする。使用の都度,この標準溶液の一部をしゅう酸アンモニウム溶液で正しく20
倍にうすめて標準チタン溶液とする。
3.2.3
試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
3.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料の分解 はかり取った試料〔3.2.3〕を白金皿(100番)に移してふたをし,ふっ化水素酸10ml
を加え,硝酸 (1+1) 5mlを滴加して分解した後,硫酸 (1+1) 5mlを加え,加熱して硫酸白煙を7〜8
分間発生させ(1),冷却する。
注(1) 激しく白煙を発生させると飛散などによって誤差を生ずることがあるので,白煙処理は注意す
る。
(2) 混合酸化物の分離 3.2.4(1)で得た白金皿の内容物を,あらかじめ過酸化水素水2mlを入れてあるビー
カー (500ml) に水で洗い移し,更に白金皿に過酸化水素水1mlを加えて付着物を完全に溶解し,水で
洗浄して先のビーカーに合わせる。これを温水で約250mlにうすめ,塩酸15mlを加えた後,かき混
ぜながら亜硫酸水(飽和,約6%)80mlを加えて沈殿が生成し始めたとき,タンニン酸約1gと少量の
ろ紙パルプを加えて加熱し(2),約10分間煮沸した後,流水中で1時間以上静置する。
沈殿は,ろ紙(6種)を用いてこし分け(3),塩酸 (1+50) で5〜6回洗浄する。沈殿をろ紙と共に乾
3
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
燥して磁器るつぼ (30ml) に移し,暗赤色に加熱して灰化する。この灰化残さを白金皿(100番)に移
して(4)ふっ化水素酸10mlと硫酸 (1+1) 5mlを加え,加熱して硫酸白煙を7〜8分間発生させ(1),冷却
する。
この白金皿の内容物を,あらかじめ過酸化水素水2mlを入れてあるビーカー (500ml) に水で洗い移
し,更に白金皿に過酸化水素水1mlを加えて付着物を完全に溶解し,水で洗浄して先のビーカーに合
わせる。これを温水で約250mlにうすめ,塩酸15mlを加えた後,かき混ぜながら亜硫酸水(飽和,
約6%)80mlを加えて沈殿が生成し始めたとき,タンニン酸約1gと少量のろ紙パルプを加えて加熱し,
約10分間煮沸する。これを流水中で常温以下に冷却した後,ろ紙(6種)を用いて沈殿をこし分け(3),
塩酸 (1+50) で5回以上洗浄する。ろ液及び洗液は捨てる。
注(2) 沈殿生成後にろ紙パルプを加えてもよい。この場合は十分にかき混ぜてから,ろ過する必要が
ある。
(3) 沈殿量が多い場合は,複数個の漏斗を用いる。
(4) るつぼに酸化物が残ったときは,ふっ化水素酸の少量で溶解して白金皿に移す。
(3) 混合酸化物のひょう量 3.2.4(2)で得た沈殿をろ紙と共に白金るつぼ(30番)に移し,徐々に加熱温度
を強め,灰化した後,900〜1000℃で強熱して恒量とし,デシケーター中で常温まで放冷し,その質量
をはかって混合酸化物量 (w1) とする。
(4) 補正溶液の調製 3.2.4(3)で得た混合酸化物に,ピロ硫酸カリウム5gを混合して加熱融解する。放冷
後,白金るつぼをビーカー (200ml) に移し,しゅう酸アンモニウム溶液〔3.2.2(12)〕約50mlを加え,
加熱して融解物を溶解し,白金るつぼをしゅう酸アンモニウム溶液で洗浄して取り出し,常温まで冷
却して100mlのメスフラスコに移し,しゅう酸アンモニウム溶液で標線までうすめる。
(5) 補正用二酸化チタンの定量
(a) 3.2.4(4)で得た補正溶液から10mlを分取して50mlのメスフラスコに移し,塩酸 (1+1) 14mlを加え
て振り混ぜる。
(b) これにジアンチピリルメタン溶液〔3.2.2(13)〕20mlを加えて水で標線までうすめ,20分間以上放置
後,その一部を光度計の吸収セルに移して波長385nm付近の吸光度を測定する。
(c) この吸光度を検量線(5)に挿入して二酸化チタン量 (w2) を求める。
注(5) 二酸化チタン定量用検量線の作成 数個の50mlのメスフラスコを準備し,標準チタン溶液0〜
15ml(二酸化チタンとして0〜300μg)を段階的に正確に加え,しゅう酸アンモニウム溶液
〔3.2.2(12)〕でそれぞれの液量を15mlとし,塩酸 (1+1) 14mlを加えて振り混ぜる。以下
3.2.4(5)(b)の手順に従って操作して吸光度を測定する。吸光度と呈色溶液中の二酸化チタン量と
の関係を求めて検量線とする。
(6) 五酸化タンタルの定量 4.3又は4.4のタンタル定量方法に従ってタンタル含有率 (w3) を求める。
3.2.5
計算 試料中のニオブ含有率を次の式によって算出する。
()
(
)
[
]
100
6991
.0
%
3
2
1
×
×
+
−
=
W
W
W
w
ニオブ
ここに,
w1: 3.2.4(3)で求めた混合酸化物の質量 (g)
W2: 同上中に含有される二酸化チタンの質量 (g)
W2=10w2
w2:分取した補正溶液中の二酸化チタンの質量 (g)
W3: 混合酸化物中に含有される五酸化タンタルの質量 (g)
W3=w3×0.01221×W
4
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
w3:試料中のタンタル含有率 (%)
W: 試料はかり取り量 (g)
備考 補正用二酸化チタンの定量は,次の手順に従ってもよい。
3.2.4(4)で得た補正溶液から10mlを分取してビーカー (100ml) に移す。これを加熱濃縮して
液量を約5mlとし,常温まで冷却して50mlのメスフラスコに塩酸20mlとしゅう酸アンモニウ
ム溶液〔3.2.2(12)〕10mlで洗い移し,15℃以下に冷却した後,アリザリンS溶液〔3.2.2(14)〕
3.5mlと塩化第一すず溶液〔3.2.2(9)〕10mlを順次加え,塩酸 (1+1) で標線までうすめる(6)。
この溶液を約15℃に保ち,30分間静置した後,その一部を光度計の吸収セルに移し,空試験溶
液(7)を対照液として波長760nm付近の吸光度を測定する。この吸光度を検量線(8)に挿入して二
酸化チタン量 (w2) を求める。
注(6) 最終試料溶液の塩酸濃度が6Nになるようにする。
(7) 空試験溶液は,試料のみを除いて全操作を試料と同様に処理した溶液である。
(8) この場合の検量線は,次のようにして作成する。
数個のビーカー (100ml) を準備し,それぞれにニオブ・タンタル混合溶液〔3.2.2(10)〕を正
確に25mlずつ加える。これらに標準チタン溶液0〜10ml(二酸化チタンとして0〜200μg)を
段階的に正確に加える。これらの溶液を加熱濃縮して液量を5mlとし,常温まで冷却して50ml
のメスフラスコに塩酸20mlとしゅう酸アンモニウム溶液〔3.2.2(12)〕10mlで洗い移し,15℃以
下に冷却する。これらにアリザリンS溶液〔3.2.2(14)〕3.5mlと塩化第一すず溶液〔3.2.2(9)〕10ml
を順次正確に加え,塩酸 (1+1) で標線までうすめる(6)。これらの溶液を約15℃に保ち,30分
間静置した後,それぞれ溶液の一部を光度計の吸収セルに移し,標準チタン溶液添加量0の溶
液を対照液として波長760nm付近の吸光度を測定する。これら吸光度と呈色溶液中の二酸化チ
タン量との関係を求めて検量線とする。
3.3
イオン交換分離五酸化ニオブ重量法
3.3.1
要旨 試料を適切な酸で分解してふっ化水素酸と硝酸の溶液とする。この溶液をイオン交換樹脂カ
ラムに通してニオブ及びタンタルなどを樹脂に吸着させ,溶離液 (A) で鉄などを溶出させた後,溶離液
(B) でニオブのみを分離溶出させる。この溶出液の酸度をアンモニア水と酢酸アンモニウムで調節し,タ
ンニン酸でニオブを沈殿させてこし分け,灰化強熱してその質量をはかる。
3.3.2
適用分野 試料がふっ化水素酸と硝酸で容易に分解する場合と分解が不完全な場合は,試料溶液の
調製をそれぞれ別操作による。
3.3.3
試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) ふっ化水素酸
(3) 硫酸 (1+1)
(4) アンモニア水 (1+1)
(5) 溶離液 (A) 硝酸308mlとふっ化水素酸296mlを1000mlのポリエチレン製メスフラスコに入れて水
で標線までうすめ,ポリエチレン瓶に移して保存する。
(6) 溶離液 (B) 硝酸308mlとふっ化水素酸37mlを1000mlのポリエチレン製メスフラスコに入れて水で
標線までうすめ,ポリエチレン瓶に移して保存する。
(7) 硝酸アンモニウム溶液 硝酸アンモニウム20gを水約900mlに溶解し,アンモニア水 (1+1) でpHを
6.5±0.5に調節して水で1000mlにうすめる。
5
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(8) タンニン酸溶液 タンニン酸 (C14H10O9) 10gをエチルアルコール (30v/v%) 90mlに溶解してアンモニ
ア水でpHを6.5±0.5に調節する。
(9) 酢酸アンモニウム (CH3COONH4) 溶液 (10w/v%)
(10) メチルレッド溶液 メチルレッド〔(CH3)2NC6H4N : NC6H4・COOH〕0.1gをエチルアルコール (95v/v%)
50mlに溶解して水で100mlにうすめる。
3.3.4
装置及び器具 イオン交換樹脂カラム(付図1参照) 一端を細くした長さ約250mm,内径約11mm
のポリエチレン管に,水でほぐしたポリプロピレンウールを約5mmの厚さにゆるく詰め,水で膨潤させ
た強塩基性陰イオン交換樹脂 (74〜149μm) 10mlを流し入れて沈降させた後,上部にポリプロピレンウー
ルを約5mmの厚さに詰める。このイオン交換樹脂カラムからの水の流出速度が1.0±0.2ml/minになるよう
にポリプロピレンウールの詰め方を調節する。なお,新しいイオン交換樹脂カラムは,溶離液 (A)〔3.3.3(5)〕
約60mlを通してから使用する。
3.3.5
試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
3.3.6
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の言同製
(a) ふっ化水素酸と硝酸で分解容易な試料 はかり取った試料〔3.3.5〕をポリエチレンビーカー (100ml)
又は白金皿(100番)に移してふたをし,ふっ化水素酸8mlを加えた後,硝酸 (1+1) 2mlを滴加し
て分解する。
(b) ふっ化水素酸と硝酸で分解不完全な試料 はかり取った試料〔3.3.5〕をテフロンビーカー (100ml)
又は白金皿(100番)に移してふたをし,ふっ化水素酸8ml,硝酸 (1+1) 2ml及び硫酸 (1+1) 1ml
を加えて分解し,加熱蒸発して乾固する。放冷後,ふっ化水素酸8mlと硝酸 (1+1) 2mlを加えて低
温で加熱して溶解する(9)。
注(9) 加熱があまり長過ぎると酸濃度が変化するので,できるだけ短時間で溶解するように注意する。
(2) イオン交換分離 溶離液 (A)〔3.3.3(5)〕30mlを通したイオン交換樹脂カラム〔3.3.4〕に,3.3.6(1)で
得た試料溶液を通す。
次に溶離液 (A) 10mlでふたとビーカーを洗浄して同じイオン交換樹脂カラムを通す。この操作を2
回繰り返した後,最後に溶離液 (A) を10mlずつ3回通し,溶出液はポリエチレンビーカー (300ml) に
受けて保存する(10)。次にイオン交換樹脂カラムに溶離液 (B)〔3.3.3(6)〕50mlを数回に分けて通し,
溶出液をテフロンビーカー (300ml) に受ける(11)。
注(10) この溶出液は,アルミニウム及びすずなどの定量に使用することができる。これらの定量をし
ない場合は捨てる。
(11) タンタルの定量を行う場合は,このときのイオン交換樹脂カラムを保存する。
(3) 沈殿分離 3.3.6(2)で得た溶出液にアンモニア水 (1+1) を徐々に加え,中和点近くでメチルレッド溶
液2〜3滴を指示薬として加え,更に溶液が黄色に変わるまでアンモニア水 (1+1) を滴加する。これ
に酢酸アンモニウム溶液 (10w/v%) 15mlを加えた後,熱水で約200mlにうすめ,約80℃に加熱してか
き混ぜながらタンニン酸溶液〔3.3.3(8)〕50mlを加え,約80℃の水浴中で約1時間加熱した後,流水
中で常温以下に冷却し,ろ紙 (5種B) を用いて沈殿をこし分け(3),硝酸アンモニウム溶液〔3.3.3(7)〕
で十分に洗浄する。ろ洗液は捨てる。
(4) ひょう量 3.3.6(3)で得た沈殿をろ紙と共に質量既知の白金るつぼ(30番)に移して灰化した後,1000℃
で強熱して恒量とし,デシケーター中で常温まで冷却してその質量をはかる。
3.3.7
計算 試料中のニオブ含有率を次の式によって算出する。
6
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
()(
)
100
6991
.0
%
2
1
×
×
−
=
W
w
w
ニオブ
ここに, w1: 五酸化ニオブの入った白金るつぼの質量 (g)
w2: 白金るつぼの質量 (g)
W: 試料はかり取り量 (g)
4. タンタル定量方法
4.1
方法の区分 タンタルの定量方法は,次のいずれかによる。
(1) 五酸化タンタル重量法 この方法は,タンタル含有率1%以上の試料に適用する。
(2) ピロガロール吸光光度法 この方法は,タンタル含有率10%未満の試料に適用する。
(3) マラカイトゲリーン吸光光度法 この方法は,タンタル含有率1%未満の試料に適用する。
4.2
五酸化タンタル重量法
4.2.1
要旨 試料を適切な酸で分解してふっ化水素酸と硝酸の溶液とする。この溶液をイオン交換樹脂カ
ラムを通してニオブ,タンタルなどを樹脂に吸着させ,ニオブなどを溶出分離させた後,溶離液 (C) でタ
ンタルを分離溶出させる。この溶出液の酸度をアンモニア水と酢酸アンモニウムで調節し,タンニン酸で
タンタルを沈殿させてこし分け,灰化強熱してその質量をはかる。
4.2.2
試薬 試薬は,次による。
(1) アンモニア水 (1+1)
(2) 溶離液 (C) 硝酸アンモニウム240g,ふっ化水素アンモニウム14g及びふっ化アンモニウム18gを
ポリエチレンビーカーに取り,水約900mlに溶解し,1000ml近くまで水でうすめる。ブロムクレゾー
ルグリーン試験紙を用い,硝酸 (1+1) 又はアンモニア水でpHを3.5に調節して水で1000mlにうす
める。この溶液は,ポリエチレン瓶に入れて保存する。
(3) 硝酸アンモニウム溶液 硝酸アンモニウム20gを水約900mlに溶解し,アンモニア水 (1+1) でpHを
6.5±0.5に調節して水で1000mlにうすめる。
(4) タンニン酸溶液 タンニン酸 (C14H10O9) 10gをエチルアルコール (30v/v%) 90mlに溶解してアンモニ
ア水でpHを6.5±0.5に調節する。
(5) 酢酸アンモニウム溶液 (10w/v%)
(6) メチルレッド溶液 メチルレッド〔(CH3)2NC6H4N : NC6H4COOH〕0.1gをエチルアルコール (95v/v%)
50mlに溶解して水で100mlにうすめる。
4.2.3
装置及び器具 3.3.4による。
4.2.4
試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
4.2.5
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製 3.3.6(1)の手順に従って操作する。
(2) イオン交換分離 3.3.6(2)の手順に従って操作し,溶離液 (B) を通した後のイオン交換樹脂カラムに溶
離液 (C)〔4.2.2(2)〕10mlずつを5回通し,溶出液をポリエチレンビーカー (300ml) に集める。
(3) 沈殿分離 4.2.5(2)で得た溶出液にアンモニア水 (1+1) を徐々に加え,中和点近くでメチルレッド溶
液2〜3滴を指示薬として加え,更に溶液が黄色に変わるまでアンモニア水 (1+1) を滴加する。これ
に酢酸アンモニウム溶液 (10w/v%) 15mlを加えた後,熱水で約200mlにうすめ,約80℃に加熱してか
き混ぜながらタンニン酸溶液〔4.2.2(4)〕50mlを加え,約80℃の水浴中で約1時間加熱した後,流水
中で常温以下に冷却し,ろ紙(5種B)を用いて沈殿をこし分け,硝酸アンモニウム溶液〔4.2.2(3)〕
7
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
で十分に洗浄する。ろ洗液は捨てる。
(4) ひょう量 4.2.5(3)で得た沈殿は,ろ紙と共に質量既知の白金るつぼ(30番)に移して灰化した後,
1000℃で強熱して恒量とし,デシケーター中で常温まで冷却してその質量をはかる。
4.2.6
計算 試料中のタンタル含有率を次の式によって算出する。
()(
)
100
8190
.0
2
1
×
×
−
=
W
w
w
%
タンタル
ここに,
w1: 五酸化タンタルの入った白金るつぼの質量 (g)
w2: 白金るつぼの質量 (g)
W: 試料はかり取り量 (g)
4.3
ピロガロール吸光光度法
4.3.1
要旨 (加水分解分離の場合) 試料をふっ化水素酸と硝酸で分解し,硫酸で白煙処理して塩酸と
過酸化水素水に溶解し,亜硫酸とタンニン酸でタンタルをニオブなどと共に加水分解させてこし分ける。
この沈殿を再溶解して加水分解操作を繰り返す。沈殿をこし分けて灰化し,ピロ硫酸カリウムで融解して
しゅう酸アンモニウムに溶解する。
(イオン交換分離の場合) 試料を適切な酸で分解してふっ化水素酸と硝酸の溶液とし,イオン交換樹
脂にタンタルをニオブなどと共に吸着させ,ニオブなどを溶出させた後,タンタルを溶離液 (C) で溶出さ
せる。この溶出液を硫酸で白煙処理してしゅう酸アンモニウムに溶解する。
いずれかの方法で妨害成分を分離した溶液からタンタルをピロガロール錯体として呈色させ,その吸光
度を測定する。
4.3.2 適用分野 加水分解分離の場合とイオン交換分離の場合は,試料溶液の調製と検量線の作成をそれ
ぞれ別操作による。
4.3.3
試薬 試薬は,次による。
(1) 硫酸 (1+1)
(2) ニオブ溶液 五酸化ニオブ(純度99.5%以上)0.372gをはかり取って白金又は石英るつぼ (30ml) に
移し,ピロ硫酸カリウム5gを混合して加熱融解する。放冷後,るつぼをビーカー (500ml) に移し,
しゅう酸アンモニウム溶液〔4.3.3(3)〕300mlを加え,加熱して融成物を溶解する。るつぼはしゅう酸
アンモニウム溶液で洗浄して取り出し,常温まで冷却して500mlのメスフラスコに移し,しゅう酸ア
ンモニウム溶液で標線までうすめる。この溶液25ml中にはニオブ13mgを含み,ニオブ含有率65%の
フェロニオブ0.2gを処理し,その101を分取した溶液中のニオブ量に相当する。
(3) しゅう酸アンモニウム溶液 3.2.2(12)による。
(4) ピロガロール溶液 ピロガロール (C6H6O3) 30gをりん酸6.7mlを含む水に溶解して水で100mlとする。
この溶液は使用の都度調製する。
(5) 標準チタン溶液 3.2.2(15)による。
(6) 標準タンタル溶液 (100μgTa/ml) 五酸化タンタル(純度99.5%以上)0.1527gをはかり取って白金る
つぼ(30番)に移し,ピロ硫酸カリウム5gを混合して加熱融解する。放冷後,るつぼをビーカー (300ml)
に移してしゅう酸アンモニウム溶液〔4.3.3(3)〕150mlを加え,加熱して融成物を溶解し,白金るつぼ
はしゅう酸アンモニウム溶液で洗浄して取り出す。常温まで冷却して溶液を250mlのメスフラスコに
移し,しゅう酸アンモニウム溶液で標線までうすめて標準原液とする。使用の都度,この標準溶液の
一部をしゅう酸アンモニウム溶液で正しく5倍にうすめて標準タンタル溶液とする。
4.3.4
試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
8
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.3.5
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製
(a) 加水分解分離の場合 3.2.4(4)で得た溶液を用いる。
(b) イオン交換分離の場合 4.2.5(2)で得た溶液を白金皿(100番)に移し,硫酸 (1+1) 1mlを加え,加
熱してアンモニウム塩を揮散させ,硫酸の白煙が発生し始めたならば放冷する。硫酸 (1+1) 2mlを
加え,白金皿の内壁を水で洗浄した後,硫酸の白煙が発生し始めるまで加熱蒸発する。放冷後,し
ゅう酸アンモニウム溶液〔4.3.3(3)〕30mlを加え,低温で加熱して溶解し,常温まで冷却して100ml
のメスフラスコに移し,しゅう酸アンモニウム溶液で標線までうすめる。
(2) 呈色
(a) 4.3.5(1)で得た試料溶液10mlを分取して100mlのメスフラスコに移し,しゅう酸アンモニウム溶液
〔4.3.3(3)〕40mlを加えて一定液温にする(12)。
(b) この溶液と同温度のピロガロール溶液〔4.3.3(4)〕30mlを加えて振り混ぜ,約15分間液温を先の液
温と同じに保ち,同温度のしゅう酸アンモニウム溶液で標線までうすめる。
注(12) タンタルのピロガロール錯体の吸光度は,液温の影響が大きいので検量線作成時の温度と同じ
条件にする。
(3) 吸光度の測定 4.3.5(2)で得た溶液の一部を直ちに光度計の吸収セルに取り,空試験溶液を対照液とし
て波長430nm付近の吸光度(13)を測定する。
注(13) 4.3.5(1)(a)の溶液(加水分解分離の場合)を呈色させた場合は,3.2.4(5)又は備考で求めた混合酸
化物中の二酸化チタン量 (w2) をチタン補正用検量線(14)に挿入して相当する吸光度を求めて差
し引く必要がある。
(14) チタン補正用検量線は,次のようにして作成する。
数個の100mlのメスフラスコを準備し,それぞれにニオブ溶液〔4.3.3(2)〕25mlを正確に加え,
これらに標準チタン溶液を0〜15ml(二酸化チタンとして0〜300μg)を段階的に加え,しゅう
酸アンモニウム溶液〔4.3.3(3)〕で50mlにうすめて一定温度にする。以下4.3.5(2)(b)以降の手順
に従って操作し,吸光度を測定する。吸光度と呈色溶液中の二酸化チタン量との関係を求めて
検量線とする。
4.3.6
検量線の作成 検量線は,次のようにして作成する。
(1) 加水分解分離の場合の検量線 数個の100mlのメスフラスコを準備し,それぞれにニオブ溶液
〔4.3.3(2)〕25mlを正確に加え,これらに標準タンタル溶液0〜20ml(タンタルとして0〜2.00mg)を
段階的に正確に加え,しゅう酸アンモニウム溶液〔4.3.3(3)〕で50mlにうすめる。以下4.3.5(2)(b)以降
の手順に従って操作し,吸光度を測定する。吸光度と呈色溶液中のタンタル量との関係を求めて検量
線とする。
(2) イオン交換分離の場合の検量線 数個の100mlのメスフラスコを準備し,標準タンタル溶液0〜20ml
(タンタルとして0〜2.00mg)を段階的に正確に加え,しゅう酸アンモニウム溶液〔4.3.3(3)〕で50ml
にうすめて一定温度にする。以下4.3.5(2)(b)以降の手順に従って操作し,吸光度を測定する。吸光度
と呈色溶液中のタンタル量との関係を求めて検量線とする。
4.3.7
計算 4.3.5(3)で得た吸光度を,4.3.5(1)(a)の溶液(加水分解分離の場合)を呈色させたときは,
4.3.6(1)で作成した検量線に,4.3.5(1)(b)の溶液(イオン交換分離の場合)を呈色させたときは,4.3.6(2)で
作成した検量線に挿入してタンタル量を求めて試料中のタンタル含有率を次の式によって算出する。
9
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
()
100
%
×
×
=
B
W
A
タンタル
ここに,
A: 分取した試料溶液中のタンタル量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比。ここでは101
4.4
マラカイトグリーン吸光光度法
4.4.1
要旨 (加水分解分離の場合) 試料をふっ化水素酸と硝酸で分解し,硫酸で白煙処理して塩酸と
過酸化水素水に溶解し,亜硫酸とタンニン酸でタンタルをニオブなどと共に加水分解させてこし分ける。
この沈殿を再溶解して加水分解操作を繰り返す。沈殿をこし分けて灰化し,ピロ硫酸カリウムで融解して
しゅう酸アンモニウム溶液に溶解する。
(イオン交換分離の場合) 試料を適切な酸に分解してふっ化水素酸と硝酸の溶液とし,イオン交換樹
脂にタンタルをニオブなどと共に吸着させ,ニオブなどを溶出させた後,タンタルを溶離液 (C) で溶出さ
せる。この溶出液を硫酸で白煙処理してしゅう酸アンモニウム溶液に溶解する。
いずれかの方法で妨害成分を分離した溶液からタンタルをマラカイトグリーン錯体としてその吸光度を
測定する。
4.4.2
適用分野 加水分解分離の場合とイオン交換分離の場合は,試料溶液の調製方法をそれぞれ別操作
による。
4.4.3
試薬 試薬は,次による。
(1) ふっ化水素酸 (1+17)
(2) 硫酸 (1+9)
(3) しゅう酸アンモニウム溶液 3.2.2(12)による。
(4) マラカイトグリーン (C23H25N2Cl) 溶液 (0.2w/v%)
(5) ベンゼン (C6H6)
(6) 標準タンタル溶液 (0.100mgTa/ml) 4.3.3(6)による。
4.4.4
試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
4.4.5
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製
(a) 加水分解分離の暢合 3.2.4(4)で得た溶液を用いる。
(b) イオン交換分離の場合 4.3.5(1)(b)で得た溶液を用いる。
(2) 呈色 4.4.5(1)で得た試料溶液からタンタル含有率に応じ,0.5%未満の場合は5mlを,0.5%以上の場合
は2mlを分取してポリエチレン分液漏斗 (100ml) に移し,硫酸 (1+9) 2mlとふっ化水素酸 (1+17)
4mlを正しく加えて振り混ぜ,次にマラカイトグリーン溶液 (0.2w/v%) 1.5mlを正しく加えて振り混ぜ,
直ちにベンゼン20mlを正確に加えて20秒間激しく振り混ぜた後,静置して下層の水溶液を捨てる。
(3) 吸光度の測定 4.4.5(2)で得た有機相を乾燥ろ紙(5種A)でろ過し,最初のろ液は捨て,次のろ液か
らろ液の一部を光度計の吸収セルに取り,ベンゼンを対照液として波長635nm付近の吸光度を測定す
る。
4.4.6
検量線の作成 検量線は,次のようにして作成する。
数個の100mlのメスフラスコを準備し,標準タンタル溶液0〜20ml(タンタルとして0〜2.00mg)を段
階的に正確に加え,しゅう酸アンモニウム溶液 (4.4.3(3)) で標線までうすめる。この溶液から5mlを分取
してポリエチレン分液漏斗 (100ml) に移し,硫酸 (1+9) 2mlとふっ化水素酸 (1+17) 4mlを正しく加えて

10
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
振り混ぜ,次にマラカイトグリーン溶液 (0.2w/v%) 1.5mlを正しく加えて振り混ぜ,直ちにベンゼン20ml
を正確に加えて30秒間激しく振り混ぜた後,静置して下層の水溶液を捨てる。以下4.4.5(3)の手順に従っ
て吸光度を測定する。吸光度と呈色溶液中のタンタル量との関係を求めて検量線とする。
4.4.7
計算 4.4.6で作成した検量線に4.4.5(3)で得た吸光度を挿入してタンタル量を求め,試料中のタン
タル含有率を次の式によって算出する。
()
100
%
×
×
=
B
W
A
タンタル
ここに,
A: 分取した試料溶液中のタンタル量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
5. 炭素定量方法
5.1
方法の区分 炭素の定量方法は,次のいずれかによる。
(1) ガス容量法 この方法は,炭素含有率0.05%以上の試料に適用する。
(2) 導電率法 この方法は,炭素含有率0.001%以上の試料に適用する。
(3) 電量法 この方法は,炭素含有率0.001%以上の試料に適用する。
(4) 赤外線吸収法 この方法は,炭素含有率0.001%以上の試料に適用する。
5.2
ガス容量法
5.2.1
要旨 試料を酸素気流中で加熱し,炭素を十分に酸化して二酸化炭素とし,これを酸素と共にビュ
レットに捕集してガス容積を測定し,次に二酸化炭素をアルカリ溶液に吸収させて除き,残りのガス容積
を測定してその容積減を求める。
5.2.2
装置及び器具 装置及び器具は,原則としてJIS Z 2615(金属材料の炭素定量方法通則)の5.及び
6.3.2による。
5.2.3
試料はかり取り量及び助燃剤添加量 試料は炭素含有率に応じ,原則として表1に従ってはかり取
り,助燃剤を添加する。助燃剤は,JIS Z 2615の5.(13)に示したものから最も適したものを選び,試料の2
〜4倍量を添加してよく混合するか,試料の上を覆うようにする。
表1 試料はかり取り量
炭素含有率 %
試料はかり取り量
0.05以上0.10未満 2.0gを1mgのけたまで
0.10以上0.20以下 1.0gを1mgのけたまで
5.2.4
予備操作 予備操作は,JIS Z 2615の6.3.3による。
なお,管状電気抵抗加熱炉を用いる場合は,燃焼管内温度を1300〜1400℃に保つ(15)。
また,高周波誘導加熱炉を用いる場合及び高周波誘導加熱に関する条件を設定する(16)。
注(15) 高温計の温度指示と燃焼管内温度との差に注意して補正する。
(16) 例えば,高周波発振機の陽極電流及び格子電流などを使用する装置の仕様に応じて決められた
条件のことである。
5.2.5
定量操作 定量操作は,次の手順によって行う。
(1) 管状電気抵抗加熱炉を用いる場合 JIS Z 2615の6.3.4による。
(2) 高周波誘導加熱炉を用いる場合 JIS Z 2615の6.3の備考による。
5.2.6
空試験 空試験は,次の手順によって行う。
(1) 管状電気抵抗加熱炉を用いる場合 JIS Z 2615の6.3.5による。
11
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 高周波誘導加熱炉を用いる場 試料を除き,試料に添加した量と同量の助燃剤を入れたるつぼを用い
て5.2.5.(2)の操作を行う。
なお,高周波を誘導しない助燃剤を用いた場合は,炭素含有率既知(できるだけ低いもの)の鉄な
ど0.5〜1.0gを追加して行い,追加した鉄などの中の炭素含有量を差し引いて空試験値とする。
5.2.7
計算 JIS Z 2615の6.2.6による。
5.3
導電率法
5.3.1
要旨 試料を酸素気流中で加熱し,炭素を十分に酸化して二酸化炭素とし,一定量のアルカリ溶液
に吸収させ,吸収前後のアルカリ溶液の導電率の変化を測定する。
5.3.2
試薬 試薬は,JIS Z 2615の6.6.2による。
5.3.3
装置及び器具 装置及び器具は,原則としてJIS Z 2615の5.及び6.6.3による。
5.3.4
試料はかり取り量及び助燃剤添加量 試料は,使用する装置に最も適した量(通常は0.5〜1.0g)
をはかり取り助燃剤を添加する。助燃剤はJIS Z 2615の5.(13)に示したものから最も適したものを選び,
試料の2〜4倍量を添加してよく混合するか,試料の上を覆うようにする。
5.3.5
予備操作 予備操作は,JIS Z 2615の6.6.4による。
なお,管状電気抵抗加熱炉を用いる場合は,燃焼管内温度を1300〜1400℃(15)に保つ。また,高周波誘
導加熱炉を用いる場合は,高周波誘導加熱に関する条件を設定する(16)。
5.3.6
定量操作 定量操作は,次の手順によって行う。
(1) 管状電気抵抗加熱炉を用いる場合 JIS Z 2615の6.5.5による。
(2) 高周波誘導加熱炉を用いる場合
(a) 試料と助燃剤を入れたるつぼを受台に置き,助燃管を閉じる。指定された流量で酸素を送入して管
内の空気を置換した後,高周波誘導加熱炉を作動させる(試料が燃焼し,燃焼ガス中の二酸化炭素
は吸収液に吸収され,指示値が次第に増加する。)。
(b) 記録計又は指示計が一定値を示したとき,指示値を読み取り,高周波スイッチを切ってるつぼを取
り出す。
5.3.7
空試験 空試験は,次の手順によって行う。
(1) 管状電気抵抗加熱炉を用いる場合 JIS Z 2615の6.6.6による。
(2) 高周波誘導加熱炉を用いる場合 試料を除き,試料に添加した量と同量の助燃剤を入れたるつぼを用
いて5.3.6(2)の操作を行う。
なお,高周波を誘導しない助燃剤を用いた場合は,炭素含有率既知(できるだけ低いもの)の鉄な
ど0.5〜1.0gを追加して行い,追加した鉄などの中の炭素含有量を差し引いて空試験値とする。
5.3.8
計算 JIS Z 2615の6.6.7による。
5.4
電量法
5.4.1
要旨 試料を酸素気流中で加熱し,炭素を十分に酸化して二酸化炭素とし,一定のpHにした弱ア
ルカリ性のバリウム塩溶液に吸収させ,吸収によって減少したpHをバリウム塩溶液の電解によって元の
pHに戻すために要した電気量を測定する。
5.4.2
試薬 試薬は,JIS Z 2615の6.7.2による。
5.4.3
装置及び器具 装置及び器具は,原則としてJIS Z 2615の5.及び6.7.3による。
5.4.4
試料はかり取り量及び助燃剤添加量 試料は,使用する装置に最も適した量(通常は0.5〜1.0g)
をはかり取り,助燃剤を添加する。助燃剤は,JIS Z 2615の5.(13)に示したものから最も適したものを選
び,試料の2〜4倍量を添加してよく混合するか,試料の上を覆うようにする。
12
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.4.5
予備操作 予備操作は,JIS Z 2615の6.7.4による。
なお,管状電気抵抗加熱炉を用いる場合は,燃焼管内温度を1300〜1400℃(15)に保つ。また,高周波誘
導加熱炉を用いる場合は,高周波誘導加熱に関する条件を設定する(16)。
5.4.6
定量操作 定量操作は,次の手順によって行う。
(1) 管状電気抵抗加熱炉を用いる場合 JIS Z 2615の6.7.5による。
(2) 高周波誘導加熱炉を用いる場合
(a) 試料と助燃剤を入れたるつぼを受台に置き,燃焼管を閉じる。指定された流量で酸素を送入して管
内の空気を置換した後,高周波誘導加熱炉を作動させ,同時に指示値を零に戻す(試料が燃焼し,
二酸化炭素の吸収が始まると指示値が次第に増加する。)。
(b) 指示計が一定値を示したとき,指示値を読み取り,高周波スイッチを切ってるつぼを取り出す。
5.4.7
空試験 空試験は,次の手順によって行う。
(1) 管状電気抵抗加熱炉を用いる場合 JIS Z 2615の6.7.6による。
(2) 高周波誘導加熱炉を用いる場合 試料を除き,試料に添加した量と同量の助燃剤を入れたるつぼを用
いて5.4.6(2)の操作を行う。
なお,高周波を誘導しない助燃剤を用いた場合は,炭素含有率既知(できるだけ低いもの)の鉄な
ど0.5〜1.0gを追加して行い,追加した鉄などの中の炭素含有量を差し引いて空試験値とする。
5.4.8
計算 JIS Z 2615の6.7.7による。
5.5
赤外線吸収法
5.5.1
要旨 試料を酸素気流中で加熱し,炭素を十分に酸化して二酸化炭素とし,これを酸素と共に赤外
線吸収セルに送り,二酸化炭素による赤外線吸収量を測定する(積分法)。又は,試料を一定容積内の一定
圧力下の循環酸素気流中で加熱し,炭素を二酸化炭素及び一酸化炭素に酸化し,過剰の酸素と共に循環ル
ープの赤外線吸収検出器に送り,二酸化炭素及び一酸化炭素の赤外線吸収量をそれぞれ測定する(循環法)。
5.5.2
材料 材料は,JIS Z 2615の5.及び6.9.2による。
5.5.3
装置 装置は,JIS Z 2615の6.9.3(積分法)又は6.10.2(循環法)による。
5.5.4
試料はかり取り量及び助燃剤添加量 試料は,使用する装置に最も適した量(通常は0.5〜1.0g)
をはかり取り,助燃剤を添加する。助燃剤は,JIS Z 2615の5.(13)に示したものから最も適したものを選
び,試料の2〜4倍量を添加してよく混合するか,試料の上を覆うようにする。
5.5.5
予備操作 JIS Z 2615の6.9.4(積分法)又は6.10.3(循環法)による。
5.5.6
定量操作 JIS Z 2615の6.9.5(積分法)又は6.10.4(循環法)による。
5.5.7
空試験 JIS Z 2615の6.9.6(積分法)又は6.10.5(循環法)による。
5.5.8
計算 JIS Z 2615の6.9.7(積分法)又は6.10.6(循環法)による。
6. けい素定量方法
6.1
方法の区分 けい素の定量方法は,次のいずれかによる。
(1) 二酸化けい素重量法 この方法は,けい素含有率0.1%以上の試料に適用する。
(2) 中和滴定法 この方法は,けい素含有率0.8%以上の試料に適用する。
(3) 原子吸光法 この方法は,けい素含有率3.0%以下の試料に適用する。
6.2
二酸化けい素重量法
13
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2.1
要旨 試料をピロ硫酸カリウム又は過酸化ナトリウムで融解し,硫酸で白煙処理してけい素を不溶
性二酸化けい素とする。可溶性塩類をしゅう酸アンモニウムに溶解して残さをこし分け,強熱して恒量と
する。次に硫酸とふっ化水素酸で処理して二酸化けい素をふっ化物として揮散させ,その減量をはかる。
6.2.2
適用分野 ピロ硫酸カリウムで融解できる試料と過酸化ナトリウムを融解剤として用いる場合は,
試料溶液の調製をそれぞれ別操作による。
6.2.3
試薬 試薬は,次による。
(1) ふっ化水素酸
(2) 硫酸 (1+1, 1+4)
(3) 過酸化ナトリウム
(4) ピロ硫酸カリウム
(5) しゅう酸アンモニウム溶液3.2.2(12)による。
6.2.4
試料はかり取り量 試料は,1.0gを1mgのけたまではかり取る。
6.2.5
操作 定量操作は,次の手順によって行う。
(1) 試料の分解
(a) ピロ硫酸カリウムで融解できる試料 はかり取った試料〔6.2.4〕を白金るつぼ(30番)に移し,ピ
ロ硫酸カリウム10gを加えてよくかき混ぜて徐々に加熱し,次第に温度を高めて試料を融解する。
放冷後,白金るつぼをビーカー (500ml) に移して硫酸 (1+4) 約100mlを加え,加熱して融成物を
溶解し,白金るつぼを水で洗浄して取り出す。
(b) 過酸化ナトリウムで融解を必要とする試料 はかり取った試料〔6.2.4〕をニッケルるつぼ (30ml) に
移し,過酸化ナトリウム7gを加えてよくかき混ぜ,更にその上を過酸化ナトリウム3gで覆い,徐々
に加熱し,次第に温度を高めて試料を融解する。放冷後,るつぼを硫酸 (1+1) 100mlを入れてある
ビーカー (500ml) に移して融成物を溶解し,るつぼを水で洗浄して取り出す。
(2) 二酸化けい素の脱水処理及びろ過洗浄 6.2.5(1)で得た試料溶液を加熱蒸発し,硫酸白煙を5〜7分間
発生させた後,放冷する。これにしゅう酸アンモニウム溶液〔6.2.3(5)〕約200mlを加え,加温して可
溶性塩類を溶解し,直ちに少量のろ紙パルプを加え,ろ紙(5種A)を用いてこし分け,残さとろ紙
を温しゅう酸アンモニウム溶液で十分洗浄する。
(3) 灰化及びひょう量 6.2.5(2)で得た残さをろ紙と共に湿ったまま白金るつぼ(30番)に移し,徐々に加
熱してろ紙を灰化した後,約1100℃で強熱して恒量とし,デシケーター中で放冷し,強熱残さの入っ
ている白金るつぼの質量 (w1) をはかる。
(4) ふっ化水素酸処理及びひょう量 6.2.5(3)で得た白金るつぼ内の残さを硫酸 (1+1) 2〜3滴で湿し,ふ
っ化水素酸約5mlを加え,徐々に加熱して二酸化けい素と硫酸を揮散させる。更に加熱を強め,約
1100℃で強熱して恒量とし,デシケーター中で放冷して不純物の残っている白金るつぼの質量 (w2)
をはかる。
6.2.6
計算 試料中のけい素含有率を次の式によって算出する。
()(
)
100
4674
.0
%
2
1
×
×
−
=
W
w
w
けい素
ここに, w1: 第1回目に恒量とした不純二酸化けい素の入っている白金るつ
ぼの質量 (g)
w2: 第2回目に恒量とした不純物の入っている白金るつぼの質量
(g)
W: 試料はかり取り量 (g)
14
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.3
中和滴定法
6.3.1
要旨 試料を硝酸とふっ化カリウム共存のふっ化水素酸で分解し,けいふっ化カリウム塩を沈殿さ
せてこし分け,温水に溶解してフェノールフタレインを指示薬として水酸化ナトリウム標準溶液で滴定す
る。
6.3.2
試薬 試薬は,次による。
(1) 硝酸
(2) 硝酸カリウム溶液 (5w/v%)
(3) ふっ化カリウム溶液 ふっ化水素酸400mlを冷却し,これにふっ化カリウム(無水)60gを少量ずつ
加えて溶解し,一夜間静置してろ紙(5種A)でろ過し,ろ液を使用する。
(4) ふっ化カリウム・硝酸カリウム混液 ふっ化カリウム溶液〔6.3.2(3)〕1容に硝酸カリウム溶液 (5w/v%)
2容を混合する。
(5) N/10水酸化ナトリウム標準溶液 (4.000gNaOH/l) 調製及び標定方法は,JIS K 8006の2.(14)による。
(6) フェノールフタレイン溶液 フェノールフタレイン (C20H14O4) 0.5gをエチルアルコール (95v/v%)
100mlに溶解する。
6.3.3
試料はかり取り量 試料は,0.50gを0.1mgのけたまではかり取る。
6.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製 はかり取った試料〔6.3.3〕を白金皿(100番)又はポリエチレンビーカー (100ml) に
移してふたをし,硝酸20mlを加え,次にふっ化カリウム溶液〔6.3.2(3)〕10mlを滴加してときどき揺
り動かし,必要な場合は水浴中で70℃以下に加熱して分解する。
(2) 沈殿の分離 6.3.4(1)で得た溶液に少量のろ紙パルプを加え,ポリエチレン製の棒又は磁気かくはん器
を用いて約3分間かき混ぜた後,10〜15℃に冷却して約30分間静置する。生じた沈殿は,ポリエチレ
ン漏斗でろ紙(5種A)を用いてこし分け,15℃以下に冷却したふっ化カリウム・硝酸カリウム混液
〔6.3.2(4)〕で5回,次に硝酸カリウム溶液 (5w/v%) で洗液が青色リトマス試験紙を赤変しなくなるま
で洗浄する。
(3) 滴定 6.3.4(2)で得た沈殿をろ紙と共に三角フラスコ (200ml) に移し,温水約50mlを加えてよく振り
混ぜ,ろ紙を破壊する。沸騰するまで加熱し,直ちにフェノールフタレイン溶液2〜3滴を指示薬とし
て加え,N/10水酸化ナトリウム標準溶液で滴定して溶液が微紅色に変わる点を終点とする(17)。
注(17) 終点が不明りょうな場合は,滴定の終点近くで指示薬を追加するとよい。
6.3.5
計算 試料中のけい素含有率を次の式によって算出する。
()
100
0007022
.0
%
×
×
=
W
V
けい素
ここに,
V: N/10水酸化ナトリウム標準溶液の使用量 (ml)
W: 試料はかり取り量 (g)
6.4
原子吸光法
6.4.1
要旨 試料をふっ化水素酸と硝酸で分解して一定量にうすめ,原子吸光光度計の亜酸化窒素−アセ
チレンフレーム中に噴霧してけい素の吸光度を測定する。
6.4.2
試薬試薬は,次による。
(1) 硝酸
(2) ふっ化水素酸 (1+1)
(3) ニオブ溶液 (10mgNb/ml) ニオブ(純度99.7%以上)1.000gをはかり取ってポリエチレンビーカー
15
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(100ml) に移してふたをし,水10mlとふっ化水素酸5mlを加えた後,硝酸1mlを徐々に滴加して分解
する。これを100mlのポリエチレン製のメスフラスコに移して水で標線までうすめる。
(4) 鉄溶液 (10mgFe/ml) 鉄(純度99.9%以上)2.500gをはかり取ってポリエチレンビーカー (100ml) に
移してふたをし,水10mlとふっ化水素酸5mlを加えた後,硝酸約5mlを徐々に加えて分解する。こ
れを250mlのポリエチレン製のメスフラスコに移して水で標線までうすめる。
(5) 標準けい素溶液 (1.0mgSi/ml) 二酸化けい素(純度99.9%以上)0.5349gをはかり取ってポリエチレ
ンビーカー (100ml) に移し,ふたをして硝酸 (1+2) 15mlとふっ化水素酸7mlを加え,室温で一夜間
放置して分解する。次にこれを250mlのポリエチレン製のメスフラスコに移して水で標線までうすめ,
標準けい素溶液とする。
6.4.3
試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
6.4.4
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製 はかり取った試料 (6.4.3) をポリエチレンビーカー (100ml) に移してふたをし,ふ
っ化水素酸 (1+1) 10mlを加えて室温で約1〜2時間放置して試料の大部分を分解した後,硝酸数滴
(1ml以内) を加えて試料を完全に溶解する(18)。この溶液を100mlのポリエチレン製のメスフラスコに
移して水で標線までうすめる。
注(18) 難溶性試料の場合は,70℃以下で加熱してもよい。
(2) 吸光度の測定 原子吸光光度計を用い,6.4.4(1)で得た試料溶液の一部を亜酸化窒素−アセチレンフレ
ーム中に噴霧し,分析線251.6nmにおける吸光度を測定する。
6.4.5
検量線の作成 検量線は,次のようにして作成する(19)。
数個の100mlのポリエチレン製メスフラスコを準備し,それぞれにニオブ溶液〔6.4.2(3)〕13mlと鉄溶
液〔6.4.2(4)〕5.5mlを正確に加え,これらに標準けい素溶液0〜6.0ml(けい素として0〜6.0mg)を段階的
に正確に加えて水で標線までうすめる。以下6.4.4(2)の手順に従って操作して吸光度を測定する。吸光度と
溶液中のけい素量との関係を求めて検量線とする。
注(19) 検量線は,試料の分析と併行して作成する。
6.4.6
計算 6.4.5で作成した検量線に6.4.4(2)で得た吸光度を挿入して試料溶液中のけい素量を求め,試
料中のけい素含有率を次の式によって算出する。
()
100
%
×
=WA
けい素
ここに,
A: 試料溶液中のけい素量 (g)
W: 試料はかり取り量 (g)
7. りん定量方法
7.1
方法の区分 りんの定量方法は,次のいずれかによる。
(1) 中和滴定法 この方法は,りん含有率0.01%以上の試料に適用する。
(2) モリブデン青吸光光度法 この方法は,りん含有率0.2%未満の試料に適用する。
7.2
中和滴定法
7.2.1
要旨 (酸分解の場合) 試料を硝酸とふっ化水素酸で分解し,過塩素酸で白煙処理をしてふっ化
水素酸で溶解し,りんを過マンガン酸カリウムで酸化し,過剰の過マンガン酸を硫酸第一鉄で還元する。
(アルカリ融解の場合) 試料を過酸化ナトリウムで融解し,水で抽出してニオブなどを分離した後,
りんをりん酸鉄としてこし分け,過塩素酸で白煙処理して可溶性塩類を水に溶解する。
16
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
上記いずれかの試料溶液をアンモニア水と硝酸で酸濃度を調節し,適量の硝酸アンモニウムを共存させ,
りんをモリブデン酸アンモニウムでりんモリブデン酸アンモニウムとして分離する。これを過剰の水酸化
ナトリウム標準溶液に溶解し,硝酸標準溶液でフェノールフタレインを指示薬として滴定する。
7.2.2
適用分野 酸で分解する場合と過酸化ナトリウムで融解する場合は,試料溶液の調製とりんモリブ
デン酸アンモニウムの分離をそれぞれ別操作による。
7.2.3
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1)
(3) 硝酸
(4) 硝酸 (1+50, 1+5000)
(5) 過塩素酸
(6) ふっ化水素酸
(7) ふっ化水素酸 (1+4)
(8) 臭化水素酸 (40v/v%)
(9) 酸洗浄液(ふっ化水素酸1, 硫酸2, 水10)
(10) アンモニア水
(11) アンモニア水 (1+50)
(12) 過酸化ナトリウム
(13) 硝酸アンモニウム
(14) 硫酸第一鉄アンモニウム
(15) 融解合剤〔炭酸ナトリウム(無水)5+硝酸ナトリウム〕
(16) 塩化第二鉄溶液 塩化第二鉄 (FeCl3・6H2O) 17gを塩酸 (1+19) 100mlに溶解する。
(17) 硝酸カリウム溶液 (1w/v%) アルカリ性を呈するものは,あらかじめ硝酸で中和しておく。
(18) 過マンガン酸カリウム溶液 (3w/v%)
(19) モリブデン酸アンモニウム溶液 モリブデン酸アンモニウム〔(NH4)6Mo7O24・4H2O〕40gを水300ml
とアンモニア水80mlに溶解して冷却し,流水中で冷却した硝酸 (1+1) 600mlをかき混ぜながら少量
ずつ注加する。この溶液は使用の都度,沈殿物をろ紙(5種A)でろ過して使用する。
(20) N/10硝酸標準溶液 硝酸7.5mlを水でうすめて1000mlとする。この溶液の標定は,N/10水酸化ナト
リウム標準溶液25mlを分取して三角フラスコ (300ml) に移し,フェノールフタレイン溶液2〜3滴を
指示薬として加え,N/10硝酸標準溶液で滴定し,次の式によってN/10に対するファクターを求める。
2
1
25F
V
F
×
=
ここに, F1: N/10硝酸標準溶液のファクター
V: N/10硝酸標準溶液の使用量 (ml)
F2: N/10水酸化ナトリウム溶液のファクター
(21) N/10水酸化ナトリウム標準溶液 (4.000gNaOH/l) 調製及び標定方法は,JIS K 8006(試薬の含量試
験中滴定に関する基本事項)の2.(14)による。
(22) フェノールフタレイン溶液 (0.1w/v%) 調製方法は,JIS K 8006の3.による。
7.2.4
試料はかり取り量 試料は,1.0gを1mgのけたまではかり取る。
7.2.5
操作 定量操作は,次の手順によって行う。
17
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 試料溶液の調製
(a) 酸分解の場合
(i) はかり取った試料 (7.2.4) を白金皿(100番)に移してふたをし,硝酸20mlと過塩素酸10mlを加え,
ふっ化水素酸を徐々に滴加して分解する。激しい反応が終わった後,更にふっ化水素酸5mlを加え
て加熱蒸発し,わずかに過塩素酸の白煙を発生させて(20)放冷する(21)。これにふっ化水素酸 (1+4)
15mlを加え,加熱して(22)可溶性塩類を溶解する(23)。
(ii) これに水15mlと過マンガン酸カリウム溶液 (3w/v%) 数滴を滴加して過マンガン酸の紅色を保ちな
がら2〜3分間煮沸し,硫酸第一鉄アンモニウムを少量ずつ加えて過マンガン酸を分解した後,三角
フラスコ (300ml) に移す。
注(20) 過塩素酸の白煙を激しく長時間発生させると,析出した塩類の再溶解が困難となる。
(21) 試料溶液中にひ素が0.5mg以上含有する場合は,次のように操作してから,次の操作に移る。
塩酸20mlと臭化水素酸 (40v/v%) 5mlを加え,加熱蒸発して濃厚な白煙を発生させ,放冷する。
(22) ふっ化水素酸の揮散を少なくするため,低温で短時間行う。
(23) 不溶解物がある場合は,ろ紙(5種C)を用いてろ過し,硝酸 (1+50) で洗浄する。ろ洗液は
ポリエチレンビーカー (300ml) に集め,主液として保存する。残さは,ろ紙と共に白金るつぼ
(30番)に移して灰化し,融解合剤〔7.2.3(15)〕1gを加えて融解し,冷却後,融成物を主液で
溶出する。以下7.2.5(1)(a)(ii)以降の手順に従って操作する。
(b) アルカリ融解の場合 はかり取った試料 (7.2.4) をニッケル又はアルミナるつぼ (30ml) (24)に移し,
過酸化ナトリウム(25)7gを加えてよくかき混ぜ,更にその上を過酸化ナトリウム(25)3gで覆い,徐徐
に加熱して次第に加熱を強め,ときどき揺り動かしながら試料を融解する。放冷後,るつぼをビー
カー (500ml) に移し,熱水約100mlを加えて融成物を抽出し,るつぼを水で洗浄して取り出し,約
5分間煮沸して常温まで冷却する。これを250mlのメスフラスコに移して水で標線までうすめる。
上澄み液を乾いたろ紙(6種)を用いて乾いたビーカー (500ml) にろ過し,初めのろ液少量を捨て,
次のろ液から100mlを分取してビーカー (500ml) に移し,塩酸 (1+1) 30mlを加えて振り混ぜる。
これに塩化第二鉄溶液〔7.2.3(16)〕5mlを加え,約70℃に加熱する。次にアンモニア水を加えて中
和し,更にその過剰約10mlを加えて約1分間静かに煮沸し,沈殿はろ紙(5種A)を用いてこし分
け,アンモニア水 (1+50) で洗浄する。大部分の沈殿を水で元のビーカーに洗い移し,このビーカ
ーを漏斗下に置き,ろ紙上から温塩酸 (1+1) を滴加して溶解し,温水で十分洗浄する。この溶液
に過塩素酸10mlと硝酸5m1を加えて加熱蒸発し,過塩素酸の白煙を発生させて放冷する(21)。これ
に温水約50mlを加え,加熱して可溶性塩類を溶解し,ろ紙(5種A)を用いてろ過し,温水で十分
に洗浄して残さは捨て,ろ洗液を三角フラスコ (300ml) に集める。
注(24) ここで使用するニッケル又はアルミナるつぼは,りん含有率の低いものがよい。
(25) 融剤は,融解合剤〔7.2.3(15)〕を用いてもよい。
(2) りんモリブデン酸アンモニウムの分離
(a) 酸分解の場合 7.2.5(1)(a)で得た試料溶液に硝酸アンモニウム3gを加え,振り混ぜて溶解し,アン
モニア水を加えてわずかに水酸化鉄の沈殿を生成させ,直ちに硝酸で中和して更にその過剰5mlを
加え,水で約100mlにうすめる。これにモリブデン酸アンモニウム溶液〔7.2.3(19)〕150mlを加え,
50℃の水浴中で液温が50℃になるまで加熱し,フラスコにゴム栓をして約3分間振り混ぜ,常温で
30〜60分間放置する。この沈殿をろ紙(6種)に少量のろ紙パルプを加え(26)てこし分け,酸洗浄液
〔7.2.3(9)〕でフラスコ内部を2回,沈殿を2〜3回洗浄する。次に硝酸 (1+5000) でフラスコ内部
18
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を3回,沈殿を5回洗浄する。更に硝酸カリウム溶液〔7.2.3(17)〕でフラスコ内部を2回,沈殿を1
回洗浄する(27)。
注(26) ろ紙の代わりにろ紙パルプを用いてもよい。ろ紙パルプは,ろ紙を小さく切って三角フラスコ
に入れ,適量の水を加えた後,激しく振り混ぜて破砕して作る。ろ紙パルプは口径6cmの漏斗
を用い,磁器ふるい板は脱脂綿の上にろ紙パルプを漏斗円すい部の高さの約21まで入れる。
なお,漏斗脚部は水を満たして用いる。本文操作に従ってろ過と洗浄が終了した後,沈殿は
ろ紙パルプと共に元のフラスコに移し,漏斗壁上部に付着した少量の沈殿は,ろ紙の小片でふ
き取って元のフラスコに加える。
(27) 洗浄完了の確認方法は,硝酸カリウム溶液〔7.2.3(17)〕による3回目以降の洗液約5mlを試験
管に取り,フェノールフタレイン溶液2滴を指示薬として加え,これにN/10水酸化ナトリウム
標準溶液0.1mlをビュレットから加えて振り混ぜ,溶液が赤色になれば洗浄完了とする。
(b) アルカリ融解の場合 7.2.5(1)(b)で得た試料溶液に硝酸アンモニウム3gを加え,振り混ぜて溶解し,
アンモニア水を加えてわずかに水酸化鉄の沈殿を生成させ,直ちに硝酸で中和して更にその過剰
5mlを加え,水で約100mlにうすめる。これにモリブデン酸アンモニウム溶液〔7.2.3(19)〕100ml
を加え,50℃の水浴中で溶液が約50℃になるまで加熱し,フラスコにゴム栓をして約3分間振り混
ぜ,常温で30〜60分間放置する。この沈殿をろ紙 (6種, 9cm) (26)を用いてこし分け(28),硝酸 (1+
50) でフラスコ内部を2〜3回洗浄して沈殿を完全にろ紙上に移した後,硝酸 (1+50) での洗浄を続
け,洗液に鉄イオンの反応がなくなるまで洗浄する。次に硝酸 (1+5000) でフラスコ内部を3回,
沈殿を5回洗浄し,次に硝酸カリウム溶液〔7.2.3(17)〕でフラスコ内部を2回,沈殿を1回洗浄す
る(27)。
注(28) こし分けるのに,ろ紙パルプを加えてもよい。
(3) 滴定 7.2.5(2)で得た沈殿をろ紙と共に元のフラスコに移し,水約50mlを加え,フラスコを激しく振
り混ぜてろ紙を十分に破砕し,N/10水酸化ナトリウム標準溶液を加えて沈殿を溶解し,更にその過剰
約5mlを正確に加える。次にフェノールフタレイン溶液3〜4滴を指示薬として加え,N/10硝酸標準
溶液で滴定し,溶液の赤色が消失する点を終点とする。
7.2.6
計算 試料中のりん含有率を次の式によって算出する。
()(
)
100
000135
.0
%
2
1
×
×
×
−
=
B
W
V
V
りん
ここに, V1: N/10水酸化ナトリウム標準溶液の使用量 (ml)
V2: N/10硝酸標準溶液の使用量 (ml)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
7.3
モリブデン青吸光光度法
7.3.1
要旨 試料を過酸化ナトリウムで融解し,温水で抽出してニオブなどを分離した後,塩酸溶液とし
て鉄を添加し,アンモニア水でりんをりん酸鉄として水酸化鉄と共にこし分ける。この沈殿を過塩素酸で
溶解して白煙処理して亜硫酸水素ナトリウムで鉄を還元し,モリブデン酸アンモニウムと硫酸ヒドラジン
を反応させて生じたモリブデン青の吸光度を測定する。
7.3.2
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1)

19
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 硝酸
(4) 過塩素酸
(5) 臭化水素酸 (40w/v%)
(6) アンモニア水
(7) アンモニア水 (1+50)
(8) 過酸化ナトリウム
(9) 塩化第二鉄溶液 塩化第二鉄 (FeCl3・6H2O) 17gを塩酸 (1+9) 100mlに溶解する。
(10) 亜硫酸水素ナトリウム溶液 (10w/v%)
(11) 呈色試薬溶液
A液 モリブデン酸アンモニウム〔(NH4)6Mo7O24・4H2O〕20gを温水100mlに溶解し,これに硫酸 (1
+1) 700mlを加え,常温まで冷却して水で1000mlにうすめる。
B液 硫酸ヒドラジン〔(NH2)2・H2SO4〕溶液 (0.15w/v%)
使用の都度,A液25ml, B液10ml及び水65mlを混合し,その25mlを使用する。
(12) 標準りん溶液 (100μgP/ml) りん酸一カリウム (KH2PO4) を110℃で乾燥して恒量とした後,デシケ
ーター中に保存したものから0.4394gをはかり取って水に溶解し,1000mlのメスフラスコに移して水
で標線までうすめる。
7.3.3
試料はかり取り量 試料はりん含有率に応じ,表2に従って1mgのけたまではかり取る。
表2
りん含有率 %
試料はかり取り量 g
0.10未満
1.00
0.10以上0.20以下
0.50
7.3.4
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製 はかり取った試料〔7.3.3〕をニッケル又はアルミナるつぼ (30ml) (29)に移し,過酸
化ナトリウム7gを加えてかき混ぜ,更にその上を過酸化ナトリウム3gで覆い,徐々に加熱してとき
どき揺り動かしながら融解する。放冷後,るつぼをビーカー (500ml) に移し,熱水約100mlを加えて
融成物を抽出し,るつぼは水で洗浄して取り出し,約5分間煮沸して常温まで冷却し,250mlのメス
フラスコに移して水で標線までうすめる。上澄み液を乾いたろ紙(6種)を用いて乾いたビーカー
(500ml) にろ過し,初めのろ液の少量を捨て,次のろ液を集める。
注(29) 使用するるつぼは,りん含有率の少ないものがよい。
(2) りんの分離 7.3.4(1)で得たろ液から100mlを分取してビーカー (300ml) に移し,塩酸 (1+1) 20mlを
加え,塩化第二鉄溶液〔7.3.2(9)〕5mlを正確に加えて約70℃に加熱する。これをアンモニア水で中和
し,更にその過剰10mlを加え,約1分間静かに煮沸し,生成した沈殿をろ紙(5種A)を用いてこし
分け,アンモニア水 (1+50) で洗浄する。この沈殿の大部分を元のビーカーに水で洗い移した後,こ
のビーカーを漏斗下に置き,ろ紙上から温塩酸 (1+1) を滴加して沈殿を溶解し,温水で十分に洗浄
する。過塩素酸10mlと硝酸5mlを加えて加熱蒸発し,過塩素酸の白煙を発生させて(30)放冷する。
これに水約50mlを加えて溶解し,ろ紙(5種A)を用いてろ過し,温水で十分洗浄して残さを捨て
る。ろ洗液を250mlのメスフラスコに集めて常温まで冷却し,水で標線までうすめる。
注(30) 試料溶液中にひ素を15mg以上含む場合は,注(21)のように塩酸と臭化水素酸を用いてひ素を除
去した後,次の操作に移る。
(3) 呈色 7.3.4(2)で得た溶液から25mlを分取して100mlのメスフラスコに移し,亜硫酸水素ナトリウム
20
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
溶液 (10w/v%) 10mlを加えて溶液が無色になるまで沸騰水浴中で加熱し,直ちに呈色試薬溶液
〔7.3.2(11)〕25mlを加え,再び沸騰水浴中で約15分間加熱する。この溶液の入ったフラスコを流水中
で常温まで冷却し,水で標線までうすめる。
(4) 吸光度の測定 7.3.4(3)で得た呈色溶液の一部を光度計の吸収セルに移して波長825nm付近(31)の吸光
度を測定する。
注(31) 定量範囲を広げるために,700nm付近としてもよい。
7.3.5
検量継の作成 検量線は,次のようにして作成する。
数個のビーカー (100ml) を準備し,それぞれに過塩素酸5mlを加え,これらに標準りん溶液0〜5ml(り
んとして0〜500μg)を段階的に正確に加え,加熱して過塩素酸の白煙を発生させ,水30mlに溶解し,常
温まで冷却して250mlのメスフラスコに移し,以下7.3.4(3)以降の手順に従って操作し,吸光度を測定する。
吸光度と呈色溶液中のりん量との関係を求めて検量線とする。
7.3.6
計算 7.3.5で作成した検量線に7.3.4(4)で得た吸光度を挿入してりん量を求め,試料中のりん含有
率を次の式によって算出する。
()
100
%
×
×
=
B
W
A
りん
ここに,
A: 分取した試料溶液中のりん量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比。ここでは251
8. 硫黄定量方法
8.1
方法の区分 硫黄の定量方法は,次のいずれかによる。
(1) 中和滴定法 この方法は,硫黄含有率0.005%以上の試料に適用する。
(2) 導電率法 この方法は,原則として硫黄含有率0.001%以上0.10%未満の試料に適用する。
(3) 電量法 この方法は,原則として硫黄含有率0.001%以上の試料に適用する。
(4) 赤外線吸収法 この方法は,原則として硫黄含有率0.001%以上の試料に適用する。
8.2
中和滴定法
8.2.1
要旨 試料を酸素気流中で高温に加熱し,硫黄を酸化して二酸化硫黄などとし,これを過酸化水素
水に吸収させて硫酸とし,水酸化ナトリウム標準溶液で滴定する。
8.2.2
試薬 試薬は,次による。
(1) 吸収液 過酸化水素水3.5mlを取り,あらかじめ煮沸して二酸化炭素を追い出し冷却した水を加えて
約1000mlにうすめる。この溶液の一定量を分取し,メチルレッド・メチレンブルー混合指示薬3〜5
滴を加えてN/100水酸化ナトリウム標準溶液で滴定し,その結果によって必要量のN/100水酸化ナト
リウム標準溶液を母液に加えて中和し,褐色瓶に入れて貯蔵する。この溶液は,使用の都度更に正確
に中和して使用する。
(2) N/100水酸化ナトリウム標準溶液 (0.400gNaOH/l) N/10水酸化ナトリウム標準溶液を調製し,使用
の都度,あらかじめ煮沸して二酸化炭素を追い出し冷却した水で正しく10倍にうすめてN/100水酸化
ナトリウム標準溶液とする。N/10水酸化ナトリウム標準溶液の調製及び保存方法は,JIS K 8006の
2.(14)による。
また,標定については,予備操作 (8.2.5) による。
(3) メチルレッド・メチレンブルー混合指示薬 メチルレッド0.125gとメチレンブルー0.083gをエチルア
21
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ルコール (95v/v%) に溶解して,エチルアルコール (95v/v%) で100mlにうすめる。
8.2.3
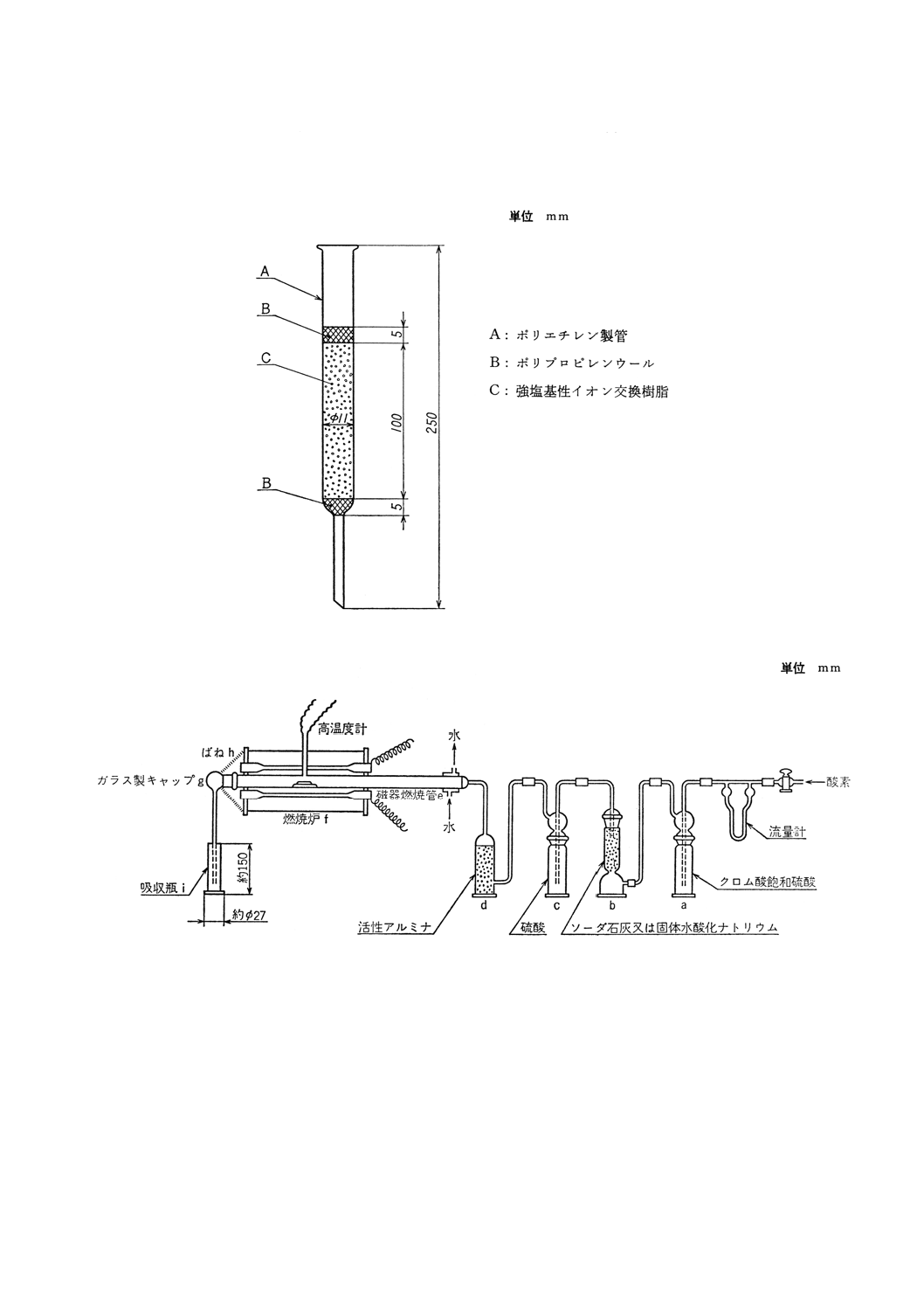
装置,器具及び材料 装置,器具及び材料は,原則として次のものを用いる(付図2参照)。
(1) 酸素精製部 酸素ボンベから供給される酸素の圧力及び流量を調節し,酸素中に含まれる硫黄酸化物,
有機硫黄化合物,水などの硫黄定量の妨害となる成分を除去するための部分で,酸素ボンベ,減圧弁,
流量計,酸化管,硫黄酸化物吸収管,脱水管などから成り,この順序に連結して使用する。
この酸化管には,クロム酸飽和硫酸を入れた洗瓶 (a) (32),硫黄酸化物吸収管には,粒状水酸化ナト
リウム又はソーダ石灰を詰めた塔 (b),脱水管には,硫酸を入れた洗瓶 (c) 及び活性アルミナを詰め
た塔 (d) を用いる。
なお,使用する酸素に硫黄定量を妨害する不純物を含まない場合は,この一部又は全部を省略する
ことができる。
(2) 試料燃焼部 試料を酸素気流中で加熱燃焼させるための部分で,燃焼管及び加熱炉から成る。その入
口は酸素精製部,出口は二酸化硫黄吸収部に連結して使用する。加熱炉には,電気抵抗加熱方式と高
周波誘導加熱方式とがある。
(a) 管状電気抵抗加熱炉 管状電気抵抗加熱炉は,原則として次のものを用いる。
管状電気抵抗加熱炉は,内径約30mm,長さ約300mmで,電気抵抗加熱体を用いて加熱し,電流を
調節して温度を加減し,炉の中央部において長さ約150mmを1400〜1450℃の一定温度に保つことが
できるようにする。
炉内には,長さ約600mm,内径約24mmで1400〜1450℃耐える磁器燃焼管〔JIS R 1307(化学分析
用磁器燃焼管)のCT0又はCT1〕(e) を挿入し(33)(34)燃焼管の出口部は炉壁から40〜60mm突き出さ
せる。
また,出口部にはテーパーを付けてすり合わせたガラス製キャップ (g) をはめ,ばね (h) で炉壁
に締めつける。
炉の中央部の燃焼管の真上の温度を熱電高温度計で測定する。熱電高温度計の指示値は,一般に
燃焼管内の温度と異なるのでその差を求めておき,必要があれば指示値から燃焼管内の温度を補正
する。
燃焼管と酸素精製部との連結は,すり合わせ又は耐熱性のシリコーンゴム栓を用いる。
(b) 高周波誘導加熱炉 高周波誘導加熱炉は,原則として次のものを用いる。
高周波誘導加熱炉は,石英管(外径30〜44mm,内径26〜37mm,長さ200〜220mm),その外側
に巻いた加熱コイル(高さ35〜55mm,巻数4〜5回),これに高周波電流を供給する高周波発振機
などから成り,高周波燃焼用磁器るつぼに試料を入れ,このるつぼを加熱コイルのほぼ中央に保持
して電流を通じたとき,試料が燃焼して硫黄が十分に酸化される温度が得られようにする。
酸素の供給にはバイパス回路を設けて,るつぼ受台の動きと連動させるものなどがある。燃焼ガ
ス出口は,燃焼管と一体として硫黄酸化物の凝縮を防止するか,凝縮した場合に除去できるものが
必要である。いずれも試料を完全に燃焼させて試料中の硫黄を酸化し,燃焼管から送り出させる能
力が必要である。
(3) 二酸化硫黄定量部 吸収瓶 (i) には,指定量の吸収液〔8.2.2(1)〕を入れ,管状電気抵抗炉の場合はガ
ラス製キャップ (g),高周波誘導加熱炉の場合は耐熱ゴム管で接続したガラス管を通じて燃焼管と連
結する。
(4) 酸素ボンベ及び減圧弁 試料を燃焼させるための酸素(純度99.5%以上)を供給するボンベには,酸
素の圧力及び流量を調節するために減圧弁を付ける。減圧弁は二段式のものが望ましい。
22
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(5) 流量計 供給する酸素の流量を監視するためのもので,一般には毎分0〜4000ml程度のロータメータ
ーを用いる。あまり高い精度を必要としないので,ほかの適当なものを用いてもよい。
(6) 磁器燃焼ボート及び磁器燃焼ボートカバー(以下,ボート及びカバーという。) 管状電気抵抗加熱炉
に使用するボートは,JIS R 1306(化学分析用磁器燃焼ボート)のCB1又はCB2とする。ボートには,
必要に応じてカバーを使用する。カバーは,JIS R 1306のCBC1とする(33)(34)。
(7) 高周波磁器燃焼るつほ(以下,るつぼという。) 高周波誘導加熱炉に使用するるつぼは,JIS R 1308
(化学分析用高周波燃焼るつぼ)のFC1又はFC2とし(33)(34),受台は,JIS R 1308のFCB1とする(33)(34)。
るつぼには必要に応じてふたをする。ふたは,JIS R 1308のFCC1又はFCC2とする(33)(34)。
(8) 助燃剤 酸素気流中で試料を加熱燃焼させる際に,酸化反応を円滑に進めるため,あらかじめ試料と
混和するか又は試料の上に載せて使用するもので,次の材料を単独又は2〜3種を組み合わせて用いる。
いずれも硫黄含有率ができるだけ低いものが望ましい。高温に加熱することによって硫黄含有率(空
試験値)を下げることができる材料は,あらかじめ空焼きを行う。
(i) 鉄
例えば粒状,149〜1000μm
(ii) すず
例えば粒状,250〜1000μm
(iii) タングステン 例えば粒状,250〜1000μm
注(32) この中の溶液が緑色を帯びてきたときは,新しい溶液と交換する。
(33) 新しい燃焼管を使用する場合は,定量操作の温度で酸素を送入しながら空試験値が安定するま
で空焼きを行う。燃焼管の内壁が酸化鉄などで汚染された場合は,使用前に洗う。汚染が著し
く,その除去が十分でないときは更新する必要がある。
ボート,カバー,るつぼ,ふた及び受台は,あらかじめ空気中又は酸素中で空焼きしたもの
を使用する。この際の空焼き温度は,試料の燃焼温度と同一であることが望ましいが,一般に
不可能である場合が多いので通常は1100〜1200℃で空焼きし,空試験 (8.2.7) を行って補正す
る。一度に多数を空焼きした場合は,冷却した後,グリースなどを塗らないデシケーター中に
保存する。ピンセットなどで扱い,直接手を触れてはならない。長時間保存したものは,空試
験値が高くなっているおそれがあるから使用を避けて再度空焼きを行う。
(34) 燃焼管,ボート,るつぼなどは,JIS R 1306〜R 1308などに示された耐火度以上のものを用い
てもよい。なお燃焼管,ボート,カバー,るつぼ,ふた及び受台は,使用する装置の指定する
形状及び寸法のものを用いてもよい。
8.2.4
試料はかり取り量及び助燃剤添加量 試料は,1.0gを1mgのけたまではかり取り,助燃剤〔8.2.3(8)〕
を試料の1〜5倍量(35)添加する。ただし,高周波誘導加熱炉を用いる場合は,試料0.50gを1mgのけたま
ではかり取ってもよい。
注(35) 助燃剤の種類及び量は,使用する装置に最も適したものを選ぶ。
8.2.5
予備操作 予備操作は,次の手順によって行う。
(1) 装置 (8.2.3) を気密に連結した後,電源を入れて各部を安定させ,管状電気抵抗加熱炉を使用する場
合は,燃焼管内温度を1400〜1450℃に保ち,高周波誘導加熱炉を使用する場合は,高周波誘導加熱に
関する条件(36)を設定する。
(2) 空試験 (8.2.7) を行って空試験値を安定化させる。
(3) 試料と組成の類似する硫黄含有率既知の試料(37)を用い,8.2.6の手順に従って操作して滴定量を求め,
8.2.7で求めた空試験値を差し引き,次の計算式によって標準溶液1mlの硫黄相当量を求める。
23
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
0
×
×
=V
S
W
f
ここに,
f: N/100水酸化ナトリウム標準溶液1mlの硫黄相当量 (g)
W0: 既知試料のはかり取り量 (g)
S: 既知試料中の硫黄含有率 (%)
V: N/100水酸化ナトリウム標準溶液の使用量 (ml)
注(36) 例えば,高周波発振機の陽極電流及び格子電流などを使用する装置の仕様に応じて決められた
条件のことである。
(37) 分析試料と硫黄含有率既知試料の硫黄含有率は,可及的に類似するのがよい。
8.2.6
定量操作 定量操作は,次の手順によって行う。
(1) 管状電気抵抗加熱炉を用いる場合
(a) 吸収液〔8.2.2(1)〕40mlを吸収瓶 (i) に移し,メチルレッド・メチレンブルー混合指示薬3〜5滴を
加えて酸素を毎分700〜900mlの割合で約5分間送入し,溶液が赤紫色になった場合は,N/100水酸
化ナトリウム標準溶液で正確に中和する。
(b) 乾燥したガラス製キャップ (g) を燃焼管の出口部にはめ,8.2.6(1)(a)で正確に中和した吸収液の入っ
ている吸収瓶 (i) を連結する。
(c) はかり取った試料及び助燃剤 (8.2.4) をボートに移し,ボートの中央部にカバーをかぶせ,燃焼管
(e) の加熱部の中央に挿入して直ちに気密に酸素精製部と連結する。
(d) 吸収液が逆流しない程度に酸素をわずかに流しながら,3〜5分間予熱する。次に毎分200mlの割合
で酸素を送入して試料を燃焼させた後,引き続き酸素を毎分700〜900mlの割合で送入して燃焼し
たガスを吸収液に導いて吸収させる。燃焼が終了してから約10分後に酸素の送入をやめる。
(e) 挿入棒でボートを管外に引き出し,カバーを外して試料の燃焼状態が完全であるかどうかを調べる。
(f) 吸収瓶 (i) とガラス製キャップ (g) を取り外し(38),キャップを放冷しておき,吸収瓶中の吸収液を
三角フラスコ (300ml) に少量の水で洗い移す。これにメチルレッド・メチレンブルー混合指示薬3
滴を加え,N/10水酸化ナトリウム標準溶液で滴定し,溶液の赤紫色が緑色に変わったならば,先に
保存したキャップの内部をこの溶液の一部で洗い,更に少量の水で洗って両者を先の三角フラスコ
内の溶液に合わせ,引き続いてN/100水酸化ナトリウム標準溶液で滴定し,溶液の赤紫色が緑色に
変わる点を終点とする (V1ml) 。
注(38) キャップ部に酸化鉄などが飛来して付着した場合は,再分析する。
(2) 高周波誘導加熱炉を用いる場合
(a) 吸収液〔8.2.2(1)〕40mlを取って吸収瓶 (i) に移し,メチルレッド・メチレンブルー混合指示薬3〜
5滴を加えて酸素を毎分700〜900mlの割合で約5分間送入し,溶液が赤紫色になった場合は,N/100
水酸化ナトリウム標準溶液で正確に中和する。
(b) 燃焼管の出口部に取り付けられたガラス管を吸収瓶の吸収液中に挿入する。
(c) はかり取った試料及び助燃剤 (8.2.4) をるつぼに移し,そのるつぼにふたをして燃焼管の加熱コイ
ルの中心部に挿入して気密に酸素精製部と連結する。
(d) 酸素を毎分1500mlの割合で通じ,高周波スイッチを入れて試料を燃焼させ,発生した二酸化硫黄
などを吸収液に導いて吸収させる。4〜5分後に高周波スイッチを切り,引き続き燃焼管内の二酸化
硫黄などを送り終わるまで約10分間酸素を送入する。
(e) 吸収瓶を取り外した後に酸素の送入をやめ,るつぼを管外に取り出してふたを外し,試料の燃焼状
24
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
態が完全であるかどうかを調べる。
(f) 吸収瓶中の吸収液を三角フラスコ (300ml) に少量の水で洗い移す。これにメチルレッド・メチレン
ブルー混合指示薬3滴を加え,N/100水酸化ナトリウム標準溶液で滴定し,溶液の赤紫色が緑色に
変わる点を終点とする (V1ml) 。
8.2.7
空試験 試料のみを入れないボート又はるつぼ(39)を用いて8.2.6の手順に従って操作し,N/100水
酸化ナトリウム標準溶液の滴定量 (V2ml) を求める。
注(39) 高周波誘導加熱炉を用いる場合は,硫黄含有率の低い既知試料(適当な試料がない場合は,純
度の高い鉄を用いてもよい。)を添加し,既知量に相当するN/100水酸化ナトリウム標準溶液量
を差し引く。
8.2.8
計算 試料中の硫黄含有率を次の式によって算出する。
()(
)
100
%
2
1
×
×
−
=
W
f
V
V
硫黄
ここに, V1: N/100水酸化ナトリウム標準溶液の全滴定量 (ml)
V2: 空試験におけるN/100水酸化ナトリウム標準溶液の滴定量 (ml)
f: 使用したN/100水酸化ナトリウム標準溶液1ml中の硫黄相当量
(g)
W: 試料はかり取り量 (g)
8.3
導電率法
8.3.1
要旨 試料を酸素気流中で高温に加熱し,硫黄を酸化して二酸化硫黄などとし,これを一定量の硫
酸酸性の過酸化水素水に吸収させて硫酸とし,吸収前後の酸性溶液の導電率の変化を測定する。
8.3.2
試薬 試薬は,次による。
(1) 吸収液N/10 硫酸100mlに過酸化水素水2.5mlを加えて水で5000mlにうすめる。N/10硫酸は,JIS K
8006の2.(6)に準じて調製する。
8.3.3
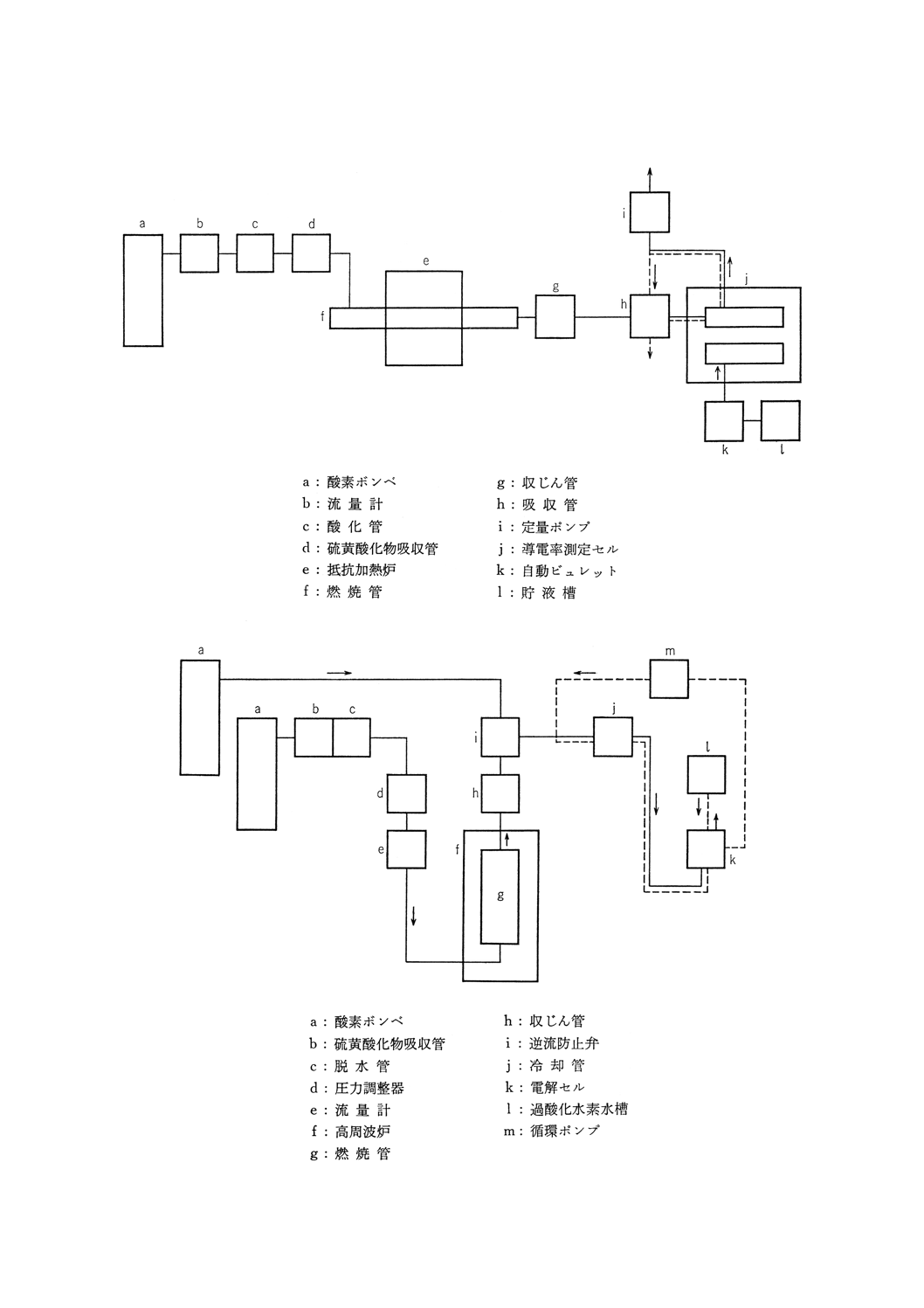
装置,器具及び材料 装置,器具及び材料は,原則として次のものを用いる。装置の概念図を付図
3に示す。
(1) 醸素精製部 8.2.3(1)に準ずる。ただし,硫黄酸化物の吸収剤にはソーダ石灰又はソーダ石綿を,脱水
剤には過塩素酸マグネシウムを用いる。
(2) 試料燃焼部 8.2.3(2)に準ずる。ただし,管状電気抵抗加熱炉を用いる(40)場合は,燃焼管の入口は,こ
こから過剰の酸素を放出して空気が管内に侵入するのを防ぐようにすれば開放してもよい。この場合
には,一定の割合で燃焼ガスを吸収管内に送り込むための定量ポンプを用いる必要がある。
(3) 燃焼ガス精製部 燃焼ガス中の酸化物ダストを除去するためのもので,石英綿を詰めた収じん管を用
いる。
(4) 二酸化硫黄定量部(図1参照) 測定セルは試料セル,参照セル及び吸収管から成り,それぞれのセ
ルには導電率測定用の電極を封入し,吸収管及びそれぞれのセルに一定量の吸収液〔8.3.2(1)〕を入れ
て恒温槽に浸す。測定セルの吸収管は,燃焼ガス回路に連結する測定回路は,ホーイトストンブリッ
ジ回路,直線化回路などから成り,二酸化硫黄などの吸収によって生じた導電率の変化を,硫黄量に
対応した値として指示計又は記録計に指示又は記録させる。指示計又は記録計は,指定された試料は
かり取り量の場合に,硫黄含有率を直示するものが望ましい。
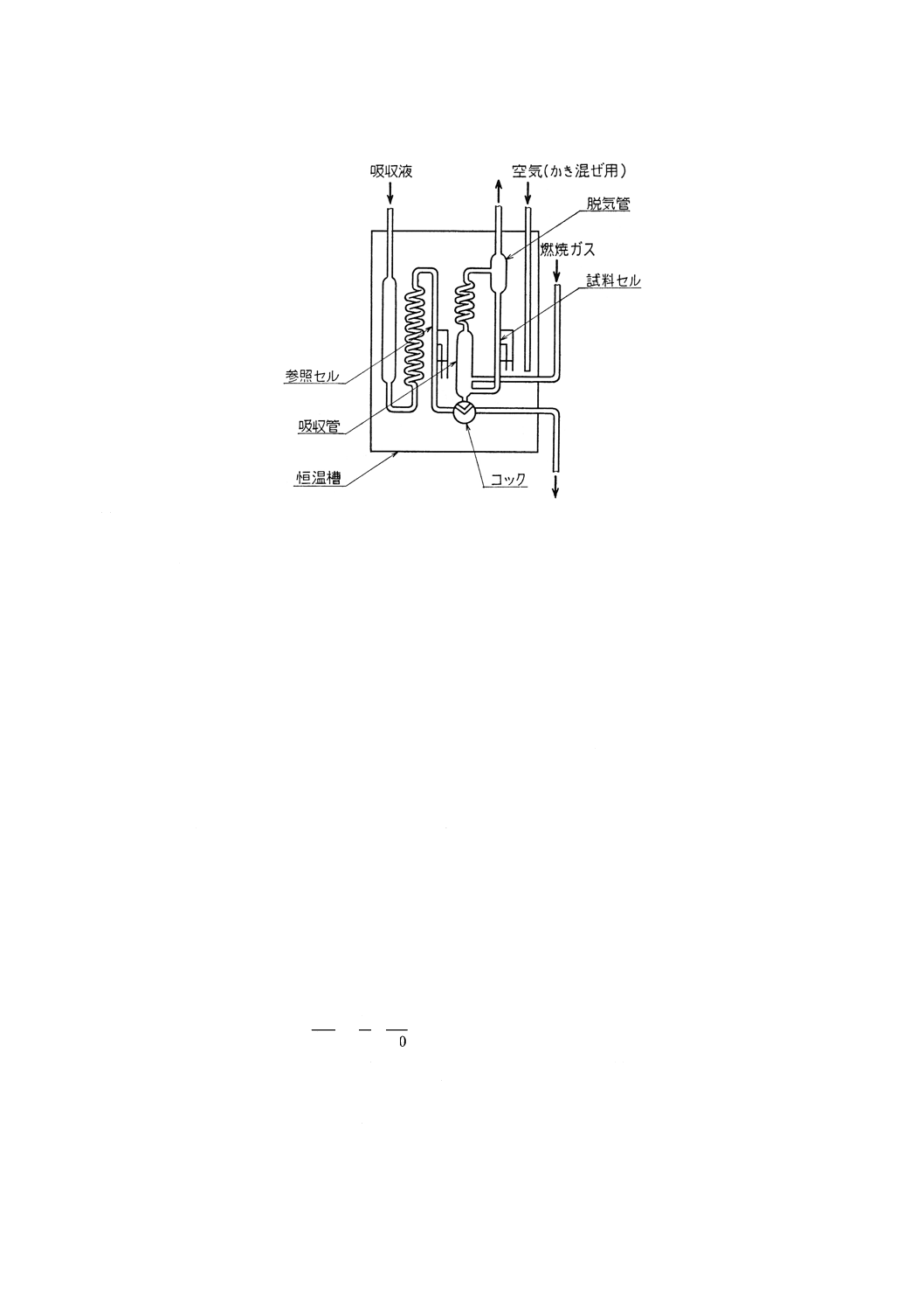
25
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図1 導電率測定セルの例
(5) 酸素ボンベ及び減圧弁,流量計,磁器燃煙ボート及び磁器燃焼ボートカバー,磁器燃焼るつぼ,助燃
剤
これらについては8.2.3による。
注(40) 燃焼管の位置及び連結は,使用する装置の取扱説明書の指示に従う。
8.3.4
試料はかり取り量及び助燃剤添加量 試料は,使用する装置に最も適した量(通常は0.5〜1.0g)
をはかり取り,その1〜4倍量の助燃剤を添加する(41)。
注(41) 試料はかり取り量,助燃剤の種類,量及び添加方法は,硫黄含有率既知の試料を分析して確認
する。
8.3.5
予備操作 予備操作は,次の手順によって行う。
(1) 装置 (8.3.3) を気密に連結した後,電源を入れて各部を安定させ,管状電気抵抗加熱炉を用いる場合
は,燃焼管内温度を1400〜1450℃に保ち,高周波誘導加熱炉を用いる場合は,高周波誘導加熱に関す
る条件(36)を設定する。
また,導電率測定セルの恒温槽を指定の温度に保つ。
(2) 酸素を使用する装置によって指定された流量(例えば毎分2l)で送りながら試料セル及び参照セルに
一定量の吸収液〔8.3.2(1)〕を入れ,約10分間酸素を送った後,導電率を測定し,3分間その変化がな
いことを確かめる。もし変化があれば,酸素を更に約10分間送入した後に導電率を測定するか,又は
吸収液を新しいものと取り替える。次に指示計又は記録計の零点を調節する。
(3) 試料と組成の類似する硫黄含有率既知の試料(37)を用い,8.3.6の手順に従って操作して指示値を求め,
空試験 (8.3.7) を行って補正し,次の計算式によって指示値1単位当たりの硫黄量を求める(42)。
(
)100
0
0
0
×
−
×
=
B
A
S
W
f
ここに,
f: 指示値1単位当たりの硫黄量 (g)
W0: 既知試料のはかり取り量 (g)
S: 既知試料の硫黄含有率 (%)
A0: 本試験における指示値
B0: 空試験における指示値
注(42) 市販の装置には,試料はかり取り量及び空試験値を補正し,硫黄含有率を直示するものがある。
26
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
この場合には,指示値が既知の硫黄含有率と一致するように調節する。
8.3.6
定量操作 定量操作は,次の手順によって行う。
(1) 管状電気抵抗加熱炉による場合
(a) はかり取った試料及び助燃剤〔8.3.4〕をボートに移し,ボートの中央部にふたをかぶせ,燃焼管 (e)
の加熱部の中央に挿入し,必要があれば直ちに酸素精製部と連結し,使用する装置によって指定さ
れた酸素を送入する。
(b) 試料が燃焼し,燃焼ガスが吸収管に送られ,指示計又は記録計の指示値が次第に増加する。指示又
は記録が安定したときの値を読み取る。
(2) 高周波誘導加熱炉を用いる場合
(a) はかり取った試料及び助燃剤〔8.3.4〕をるつぼに移し,そのるつぼにふたをして燃焼管の加熱コイ
ルの中心部に挿入し,気密に酸素精製部と連結する。
(b) 酸素を使用する装置によって指定された流量で送り,高周波スイッチを入れて試料を燃焼させ,3
〜5分後に高周波スイッチを切る。発生した燃焼ガスは吸収管に送られ,指示計又は記録計の指示
値が次第に増加する。指示又は記録が安定したときの値を読み取る。
8.3.7
空試験 試料のみを入れないボート又はるつぼ(43)を用いて8.3.6の手順に従って操作し,指示計又
は記録計の指示値を読み取る。
注(43) 高周波誘導加熱炉を用いる場合は,硫黄含有率の低い既知試料(適当な試料がない場合は,純
度の高い鉄を用いてもよい)を添加し,既知量に相当する指示値を差し引く。
8.3.8
計算 試料中の硫黄含有率を次の式によって算出する(44)。
()(
)
100
%
×
×
−
=
W
f
B
A
硫黄
ここに,
A: 本試験における指示値
B: 空試験における指示値
f: 指示値1単位当たりの硫黄量 (g)
W: 試料はかり取り量 (g)
注(44) 注(42)の場合には,指示値は試料中の硫黄含有率を示す。
8.4
電量法
8.4.1
要旨 試料を酸素気流中で高温に加熱し,硫黄を酸化して二酸化硫黄などとし,あらかじめ一定の
pH値に設定した過酸化水素水・硫酸ナトリウム吸収液に吸収させる。このとき増加した水素イオンを中和
するのに必要なアルカリを電気分解によって発生させるために消費された電気量を測定する。
8.4.2
試薬 試薬は,次による。
(1) 過酸化水素水
(2) 硫酸ナトリウム吸収液 使用する装置に指定されたものを調製する。一般に硫酸ナトリウム20gを水
に溶解して1000mlとしたものを使用する。
(3) 陽極セル液 使用する装置に指定されたものを調製する。一般に陽極セルに水100mlを入れ,硫酸ナ
トリウム約30gを加えて素早くかき混ぜ,飽和溶液として使用する。
(4) 参照セル液 使用する装置に指定されたものを調製する。一般に硫酸ナトリウム吸収液〔8.4.2(2)〕
100mlに塩化ナトリウム2〜4gを加え,十分にかき混ぜて溶解したものを使用する。
8.4.3
装置,器具及び材料 装置,器具及び材料は,原則として次のものを用いる。装置の概念図を付図
4に示す。
27
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 酸素精製部 8.2.3(1)に準ずる。ただし,脱水管には,過塩素酸マグネシウムを詰める。逆流防止用の
酸素は精製しなくてもよい。
(2) 試料燃焼部 8.2.3(2)に準ずる。ただし,高周波誘導加熱炉のみを用いる(図2参照)。
(3) 燃焼ガス精製部 8.3.3(3)による(図2参照)。
(4) 二酸化硫黄定量部 収じん管の上部に逆流防止用酸素入口及び燃焼ガス出口を取り付け,ヒーターで
加熱して二酸化硫黄などの凝縮を防ぐ。燃焼ガス出口には吸収液還流管を取り付け(図2参照),陰極
セルの吸収液〔8.4.2(2)〕を循環させる。燃焼ガス出口と陰極セルとの間には冷却管を,陰極セルと燃
焼ガス出口との間には循環ポンプ(45)を取り付ける。
電解セル(図3参照)は,陰極セル,陽極セル及び参照セルから成り,各セルは多孔板隔膜で連結
する。陰極セルには電解用白金電極(46),pH測定用ガラス電極及びかき混ぜ機を,陽極セルには電解
用白金電極(46)を,参照セルには参照電極(47)を,それぞれ取り付ける。陰極セルには硫酸ナトリウム
吸収液〔8.4.2(2)〕(48)を,陽極セルには陽極セル液〔8.4.2(3)〕(49)を,参照セルには参照セル液〔8.4.2(4)〕
をそれぞれ入れる。また,過酸化水素水槽を設げ,陰極セルに一定の割合(50)で過酸化水素水を滴加で
きるようにする。
測定回路は,電解電流パルス発生回路,pH測定回路,パルス数制御回路などから成る。電解電流を
一定の大きさの電気量(51)のパルスとして供給し,陰極セルの吸収液のpHが設定値からずれるとパル
スの発生を始め,ずれが大きい間は,単位時間当たりのパルス数を多くし,ずれが小さくなると単位
時間当たりのパルス数を少なくし,設定値と一致するとパルスの発生を停止する。この間に発生した
パルスの全数を,硫黄量に対応した値として指示計に指示させる。指示計は,指定された試料はかり
取り量の場合に,硫黄含有率を直示するものが望ましい。
(5) 酸素ボンベ及び減圧弁,流量計,磁器燃焼るつほ及びふた,助燃剤 これらについては8.2.3による。
注(45) 通常,吸収液の循環速度が毎分80〜100mlの割合となる循環ポンプを用いる。
(46) 白金電極の大きさは,少なくとも2cm2以上とすることが望ましい。
(47) 参照電極には,一般に銀・塩化銀電極を用いる。
(48) 硫酸ナトリウム吸収液は,汚染した場合は交換する。
(49) 陽極セル液は,白金電極の10mm上まで加える。硫酸ナトリウムの結晶が底部に十分に残って
いなければならない。
(50) 通常約3秒間に過酸化水素水2滴が滴加するように調節する。
(51) 市販の装置では,1パルス当たりの電気量が3×10−3クーロン,すなわち,硫黄0.5×10−6gに
対応するように設計されている。
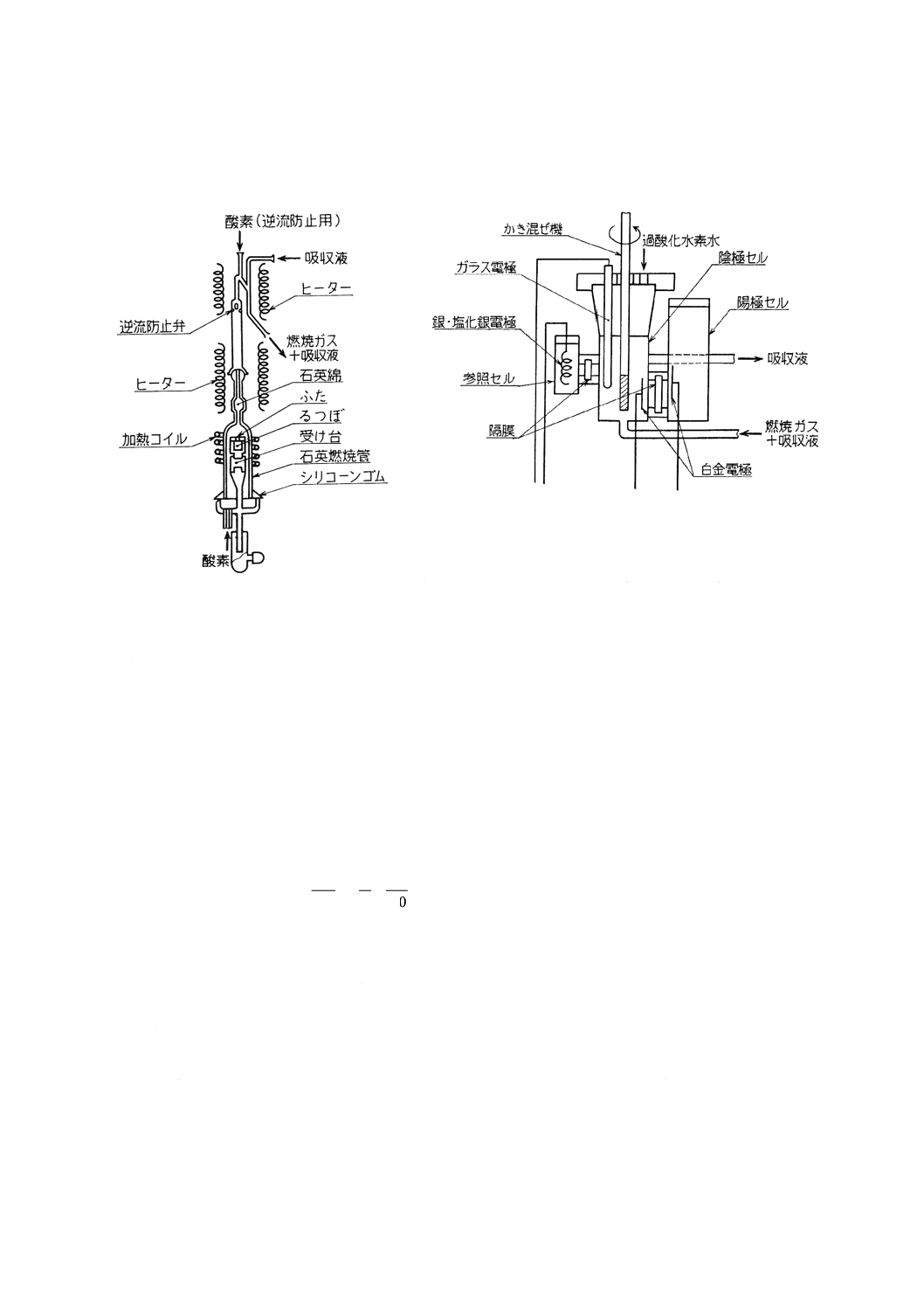
28
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図2 高周波誘導加熱炉の燃焼部,燃焼ガス精製部
及び吸収液還流管の例
図3 電解セルの例
8.4.4
試料はかり取り量及び助燃剤添加量 試料は,使用する装置に最も適した量(通常は0.5〜1.0g)
をはかり取り,その1〜4倍量の助燃剤を添加する(41)。
8.4.5
予備操作 予備操作は,次の手順によって行う。
(1) 装置 (8.4.3) を気密に連結した後,電源を入れて各部を安定させ,高周波誘導加熱に関する条件(36)を
設定する。
(2) 各セルに指定の高さまでそれぞれの溶液が入っていることを確かめ,酸素を使用する装置によって指
定された流量(52)で供給し,陰極セル吸収液のかき混ぜ,吸収液の循環及び冷却水の供給(53)を始める。
過酸化水素水20滴を加え,タイマーを作動させて過酸化水素水を自動的に補給する。
(3) 吸収液を電解し,そのpHを4〜5の一定値に設定し(54),指針を零に調節する。
(4) 試料と組成の類似する硫黄含有率既知の試料(37)を用い,8.4.6の手順に従って操作して指示値を求め
(55),空試験 (8.4.7) を行って補正し,次の計算式によって指示値1単位当たりの硫黄量を求める(42)。
(
)100
0
0
0
×
−
×
=
B
A
S
W
f
ここに,
f: 指示値1単位当たりの硫黄量 (g)
W0: 既知試料のはかり取り量 (g)
S: 既知試料の硫黄含有率 (%)
A0: 本試験における指示値
B0: 空試験における指示値
注(52) 通常,燃焼用酸素は毎分1500ml,逆流防止用酸素は毎分300〜400mlとする。
(53) 通常,冷却水は毎分80mlを流す。
(54) 市販の装置には,pHの設定値が直接読み取れないものがある。この場合には,所定の操作に従
ってpHを設定する。
(55) 初めの2〜3試料の分析値は採用しない。
8.4.6
定量操作 定量操作は,次の手順によって行う。
29
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) はかり取った試料及び助燃剤 (8.4.4) をるつぼに移し,そのるつぼにふたをして燃焼管の加熱コイル
の中心部に挿入して気密に酸素精製部と連結する。
(2) 酸素を使用する装置によって指定された流量で通じ,高周波スイッチを入れて試料を燃焼させる。発
生した燃焼ガスは,陰極セルに送られて一定時間後(56)に電解が始まり,指示値が次第に増加する。
(3) 指示値が一定となったとき,高周波誘導加熱をやめて(57)指示値を読み取る。
注(56) 吸収液中に吸収された二酸化炭素を追い出すのに必要な時間で,通常は5〜6分間とし,タイマ
ーを用いて設定する。
(57) 燃焼タイマー付きの場合には,自動的に停止する。
8.4.7
空試験 試料の代わりに硫黄含有率の低い既知試料(58)を用いて8.4.6の手順に従って操作し,指示
値を読み取って既知量に相当する指示値を差し引く。
注(58) 適当な試料がない場合は,高純度の鉄を用いてもよい。
8.4.8
計算 試料の硫黄含有率を次の式によって算出する(44)。
()(
)
100
%
×
×
−
=
W
f
B
A
硫黄
ここに,
A: 本試験における指示値
B: 空試験における指示値
f: 指示値1単位当たりの硫黄量 (g)
W: 試料はかり取り量 (g)
8.5
赤外線吸収法(積分法)
8.5.1
要旨 試料を酸素気流中で高温に加熱し,硫黄を酸化して二酸化硫黄などとし,過剰の酸素と共に
赤外線吸収検出器に送って二酸化硫黄の赤外線吸収量を測定する。
8.5.2
装置,器具及び材料 装置,器具及び材料は,原則として次のものを用いる。装置の概念図の例を
付図5に示す。
(1) 酸素精製部 8.2.3(1)に準ずる。ただし,脱水管には過塩素酸マグネシウムを詰める。酸素は二分して
一定の圧力で,一方は燃焼管に,他方は赤外線吸収検出器の対照セルに送る。
(2) 試料燃焼部 8.2.3(2)に準ずる。ただし,高周波誘導加熱炉のみを用いる(図4参照)。
(3) 燃焼ガス精製部 8.3.3(3)に準ずる。ただし,収じん管にはガラス綿を,脱水管には過塩素酸マグネシ
ウムをそれぞれ詰める(図4参照)。
(4) 二酸化硫黄定量部 赤外線吸収検出器(図5参照)は,赤外線発生源,フィルターセル,試料セル,
対照セル,赤外線検出部などから成り,二酸化硫黄の赤外線吸収量を測定できるものを用いる。
測定回路は,直線化回路,積分回路,演算回路などから成り,検出器から取り出された電気信号を,
二酸化硫黄濃度と直線関係に変換・積分し,硫黄量に比例した値として指示計に指示させる。指示計
は,指定された試料はかり取り量の場合に硫黄含有率を直示するものが望ましい。
(5) 酸素ボンベ及び減圧弁,流量計,磁器燃焼るつぼ及びふた,助燃剤 これらについては8.2.3による。
8.5.3
試料はかり取り量及び助燃剤添加量 試料は,使用する装置に最も適した量(通常は0.5〜1.0g)
をはかり取り,その1〜4倍量の助燃剤を添加する(41)。
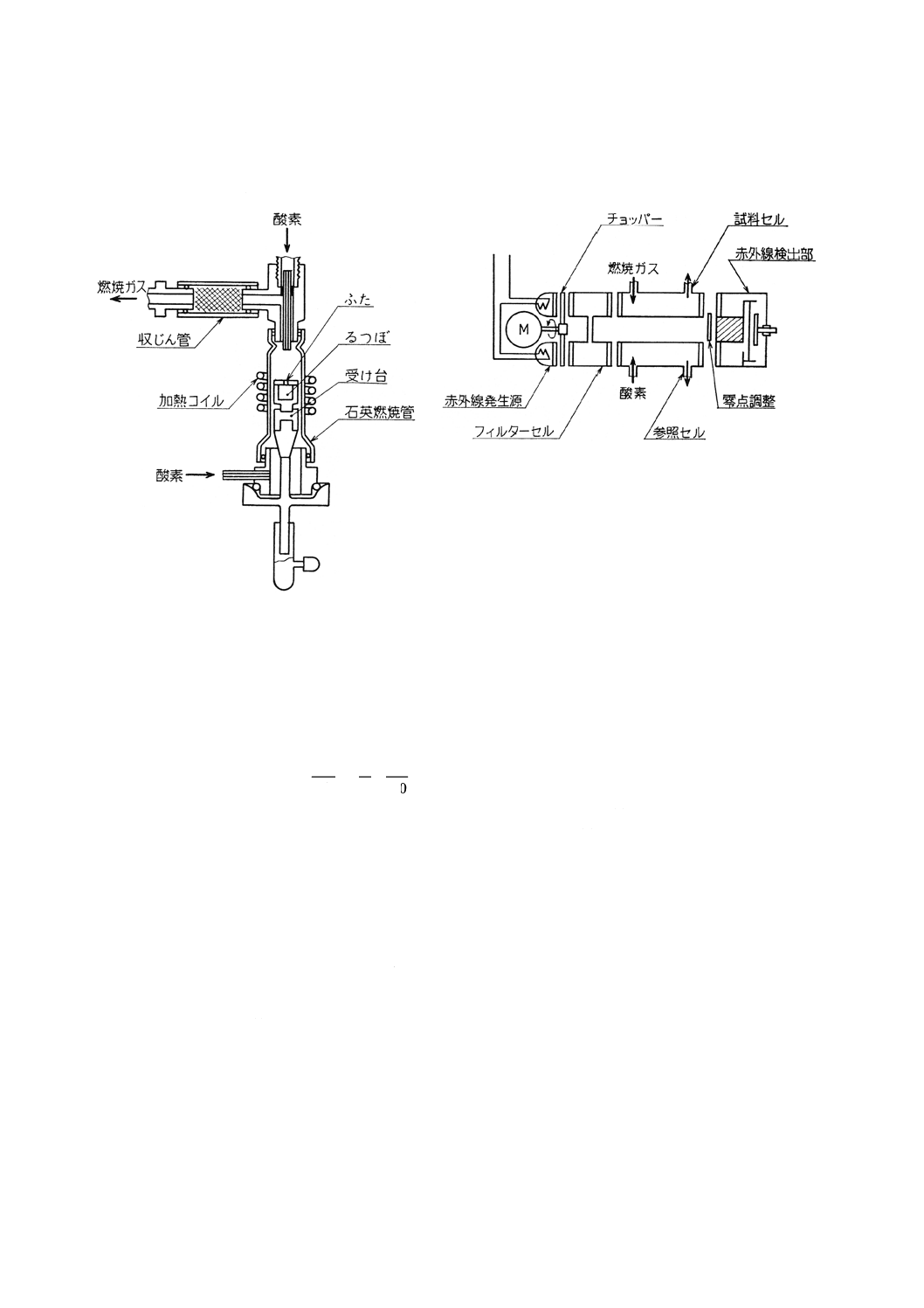
30
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図4 高周波誘導加熱炉における試料燃焼
部と燃焼ガス精製部の例
図5 赤外線吸収検出器の例
8.5.4
予備操作 予備操作は,次の手順によって行う。
(1) 装置 (8.5.2) を気密に連結した後,電源を入れて各部を安定させ,高周波誘導加熱に関する条件(36)を
設定する。
(2) 酸素を使用する装置によって指定された流量で送り,指示計の零点調節などを行う。
(3) 試料と組成の類似する硫黄含有率既知の試料(37)を用い,8.5.5の手順に従って操作して指示値を求め
(59),空試験 (8.5.6) を行って補正し,次の計算式によって指示値1単位当たりの硫黄量を求める(42)。
(
)100
0
0
0
×
−
×
=
B
A
S
W
f
ここに,
f: 指示値1単位当たりの硫黄量 (g)
W0: 既知試料のはかり取り量 (g)
S: 既知試料の硫黄含有率 (%)
A0: 本試験における指示値
B0: 空試験における指示値
注(59) 初めの3〜5試料の分析値は採用しない。
8.5.5
定量操作 定量操作は,次の手順によって行う。
(1) はかり取った試料及び助燃剤 (8.5.3) をるつぼに移し,そのるつぼにふたをして燃焼管の加熱コイル
の中心部に挿入し,気密に酸素精製部と連結する。
(2) 酸素を通じて燃焼管内を所定の圧力にし,高周波スイッチを入れて試料を燃焼させる。発生した燃焼
ガスは精製部を経て赤外線吸収検出器の試料セルに送られ,指示値が次第に増加する。
(3) 指示値が一定になったとき,高周波誘導加熱をやめて(57)指示値を読み取る。
8.5.6
空試験 試料の代わりに硫黄含有率の低い既知試料(58)を用いて8.5.5の手順に従って操作し,指示
値を読み取って既知量に相当する指示値を差し引く。
8.5.7
計算 試料中の硫黄含有率を次の式によって算出する(44)。
31
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
()(
)
100
%
×
×
−
=
W
f
B
A
硫黄
ここに,
A: 本試験における指示値
B: 空試験における指示値
f: 指示値1単位当たりの硫黄量 (g)
W: 試料はかり取り量 (g)
8.6
赤外線吸収法(循環法)
8.6.1
要旨 試料を一定容積内の一定圧力下の循環酸素気流中で高温に加熱し,硫黄を酸化して二酸化硫
黄などとし,過剰の酸素と共に循環ループ中の赤外線吸収検出器に送り,二酸化硫黄の赤外線吸収量を測
定する。
8.6.2
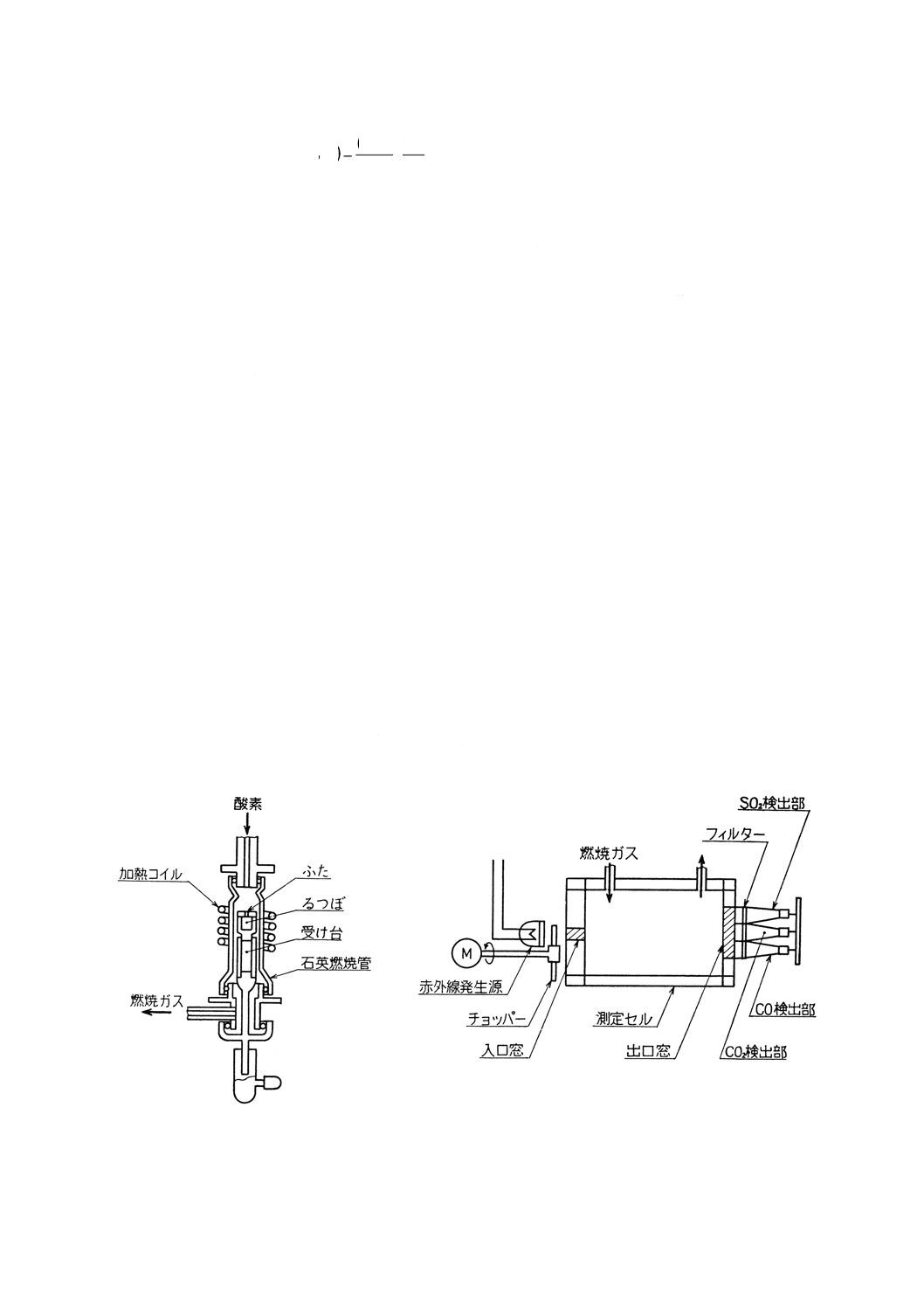
装置,器具及び材料 装置,器具及び材料は,原則として次のものを用いる。装置の概念図の例を
付図6に示す。
(1) 酸素精製部 8.2.3(1)に準ずる。ただし,硫黄酸化物吸収管にはソーダ石綿を,脱水管には過塩素酸マ
グネシウムをそれぞれ詰める。酸素は,圧力調整器によって一定圧力で循環ループへ供給する。
(2) 試料燃焼部 8.2.3(2)に準ずる。ただし,高周波誘導加熱炉のみを用いる(図6参照)。
(3) 燃焼ガス精製部 8.5.2(3)による。
(4) 二酸化硫黄定量部 赤外線吸収検出器(図7参照)は,赤外線発生源,試料セル,赤外線検出部など
から成り,二酸化硫黄の赤外線吸収量を測定できるものを用いる。
測定回路は,直線化回路,演算回路などから成る。赤外線吸収検出器から取り出された電気信号を,
二酸化硫黄と直線関係に変換し,硫黄量に比例した電圧を指示計に供給する。指示計は,指定された
試料はかり取り量の場合に硫黄含有率を直示するものが望ましい。
(5) 循環ループ 試料の燃焼を迅速にし,燃焼ガスの均一化を促進するために循環ポンプを用いて酸素及
び燃焼ガスを一定の圧力及び流量で,試料燃焼部,燃焼ガス精製部及び二酸化硫黄定量部を循環させ
る。燃焼によって消費された酸素は,圧力調整器から循環ループに補給する。
(6) 酸素ボンベ及び減圧弁,流量計,磁器燃焼るつぼ及びふた,助燃剤 これらについては8.2.3による。
図6 高周波誘導加熱炉における試料燃焼部の例
図7 赤外線吸収検出器の例
8.6.3
試料はかり取り量及び助燃剤添加量 試料は,使用する装置に最も適した量(通常は0.5〜1.0g)
をはかり取り,その1〜4倍量の助燃剤を添加する(41)。
8.6.4
予備操作 予備操作は,次の手順によって行う。
32
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 装置 (8.6.2) を気密に連結した後,電源を入れて各部を安定させ,高周波誘導加熱に関する条件(36)を
設定する。
(2) 酸素を使用する装置によって指定された流量で送り,指示計の零点調節などを行う。
(3) 試料と組成の類似する硫黄含有率既知の試料(37)を用い,8.6.5の手順に従って操作して指示値を求め
(59),空試験 (8.6.6) を行って補正し,次の計算式によって指示値1単位当たりの硫黄量を求める(42)。
(
)100
0
0
0
×
−
×
=
B
A
S
W
f
ここに,
f: 指示値1単位当たりの硫黄量 (g)
W0: 既知試料のはかり取り量 (g)
S: 既知試料の硫黄含有率 (%)
A0: 本試験における指示値
B0: 空試験における指示値
8.6.5
定量操作 定量操作は,次の手順によって行う。
(1) はかり取った試料及び助燃剤 (8.6.3) をるつぼに移し,そのるつぼにふたをして燃焼管の加熱コイル
の中心部に挿入し,気密に酸素精製部と連結する。
(2) 排出弁を開き,酸素を送入して空気と置換した後,排出弁を閉じる。
(3) 循環ポンプを作動させ,燃焼管内が所定の圧力及び流量になるのを待って高周波スイッチを入れて試
料を燃焼させる。燃焼ガスは精製部,赤外線吸収検出器及び燃焼管を循環し,指示値が次第に増加す
る。
(4) 指示値が一定となったとき,高周波誘導加熱をやめて(57)指示値を読み取る。
8.6.6
空試験 試料の代わりに硫黄含有率の低い既知試料(58)を用いて8.6.5の手順に従って操作し,指示
値を読み取って既知量に相当する指示値を差し引く。
8.6.7
計算 試料の硫黄含有率を,次の式によって算出する(44)。
()(
)
100
%
×
×
−
=
W
f
B
A
硫黄
ここに,
A: 本試験における指示値
B: 空試験における指示値
f: 指示値1単位当たりの硫黄量 (g)
W: 試料はかり取り量 (g)
9. すず定量方法
9.1
方法の区分 すずの定量方法は,次のいずれかによる。
(1) よう素酸カリウム滴定法 この方法は,すず含有率0.1%以上の試料に適用する。
(2) ピロカテコールバイオレット吸光光度法 この方法は,すず含有率3.0%以下の試料に適用する。
(3) 原子吸光法 この方法は,すず含有率3.0%以下の試料に適用する。
9.2
よう素酸カリウム滴定法
9.2.1
要旨 (加水分解分離の場合) 試料を過酸化ナトリウムで融解し,塩酸で大部分のニオブ及びタ
ンタルを加水分解させて分離し,ろ洗液を塩酸溶液とする。
(イオン交換分離の場合) 試料を適切な酸で分解してふっ化水素酸と硝酸に溶解し,イオン交換樹脂
にニオブなどを吸着させ,すずを溶離液 (A) で溶出し,硫酸白煙処理をして塩酸溶液とする。
上記いずれかの試料溶液中のすずをアルミニウムなどの還元剤で還元し,よう素酸カリウム標準溶液で
でんぷんを指示薬として滴定する。
33
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.2
適用分野 ニオブなどを塩酸で加水分解分離する場合と,イオン交換分離する場合とは,試料溶液
の調製をそれぞれ別操作による。
9.2.3
試薬 試薬は,次による。
(1) 塩酸
(2) 硫酸 (1+1)
(3) 過酸化ナトリウム
(4) 還元剤 アルミニウムはく,すずを含まない粒状鉛又は図8に示すような円筒状にしたニッケル環
図8 ニッケル環の寸法例
(5) 炭酸水素ナトリウム溶液(飽和,約10w/v%)
(6) よう化カリウム
(7) よう素酸カリウム標準溶液 よう素酸カリウム1.1gをはかり取って水に溶解し,1000mlのメスフラ
スコに移して水で標線までうすめる。この溶液のすず相当量は,次のようにして求める。
すず(純度99.5%以上)0.5000gをはかり取って白金皿(100番)に移し,硫酸 (1+1) 20mlを加え
て加熱分解し,常温まで冷却して1000mlのメスフラスコに移し,硫酸 (1+6) で標線までうすめる。
この溶液50mlを分取して三角フラスコ (500ml) に移し,塩酸70mlを加えて水で約250mlにうすめる。
以下9.2.5(2)及び9.2.5(3)の手順に従って操作し,よう素酸カリウム標準溶液の滴定量を求める。
よう素酸カリウム標準溶液1mlのすず相当量を,次の式によって算出する。
1000
1
25×
=V
f
ここに,
f: よう素酸カリウム標準溶液1mlのすず相当量 (g)
V: よう素酸カリウム標準溶液の滴定量 (ml)
(8) でんぷん溶液 調製方法はJIS K 8006の3.による。
9.2.4
試料はかり取り量 試料は,沈殿分離の場合1.0gを,イオン交換分離の場合0.20gを0.1mgのけた
まではかり取る。
9.2.5
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製
(a) 加水分解分離の場合 はかり取った試料 (9.2.4) をアルミナるつぼ (30ml) に移し,過酸化ナトリウ
ム7gを加えてよくかき混ぜ,更にその上を過酸化ナトリウム3gで覆い,徐々に加熱して次第に加
熱を強め,ときどき揺り動かしながら試料を融解する。放冷後,るつぼをビーカー (500ml) に移し,
熱水約150mlを加えて融成物を抽出し,塩酸で中和して更にその過剰30mlを加え,るつぼを水で
洗浄して取り出し,約10分間煮沸して常温まで冷却する。この溶液を250mlのメスフラスコに移
して水で標線までうすめる。これを乾いたろ紙(6種)を用いて乾いたビーカー (300ml) にろ過し,
34
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
初めのろ液の少量を捨て,次のろ液から50mlを分取して三角フラスコ (500ml) に移し,塩酸50ml
を加えて水で約250mlにうすめる。
(b) イオン交換分離の場合 3.3.6(2)で溶離液 (A) で溶出してポリエチレンビーカー (300ml) に保存し
た溶液(10)を白金皿(100番)に移し,硫酸 (1+1) 15mlを加えて加熱蒸発し,硫酸白煙を約5分間
発生させて冷却する。水約15mlと塩酸約30mlを加え,加熱して可溶性塩類を溶解し,水で三角フ
ラスコ (500ml) に移して塩酸20mlを加え,水で約250mlにうすめる。
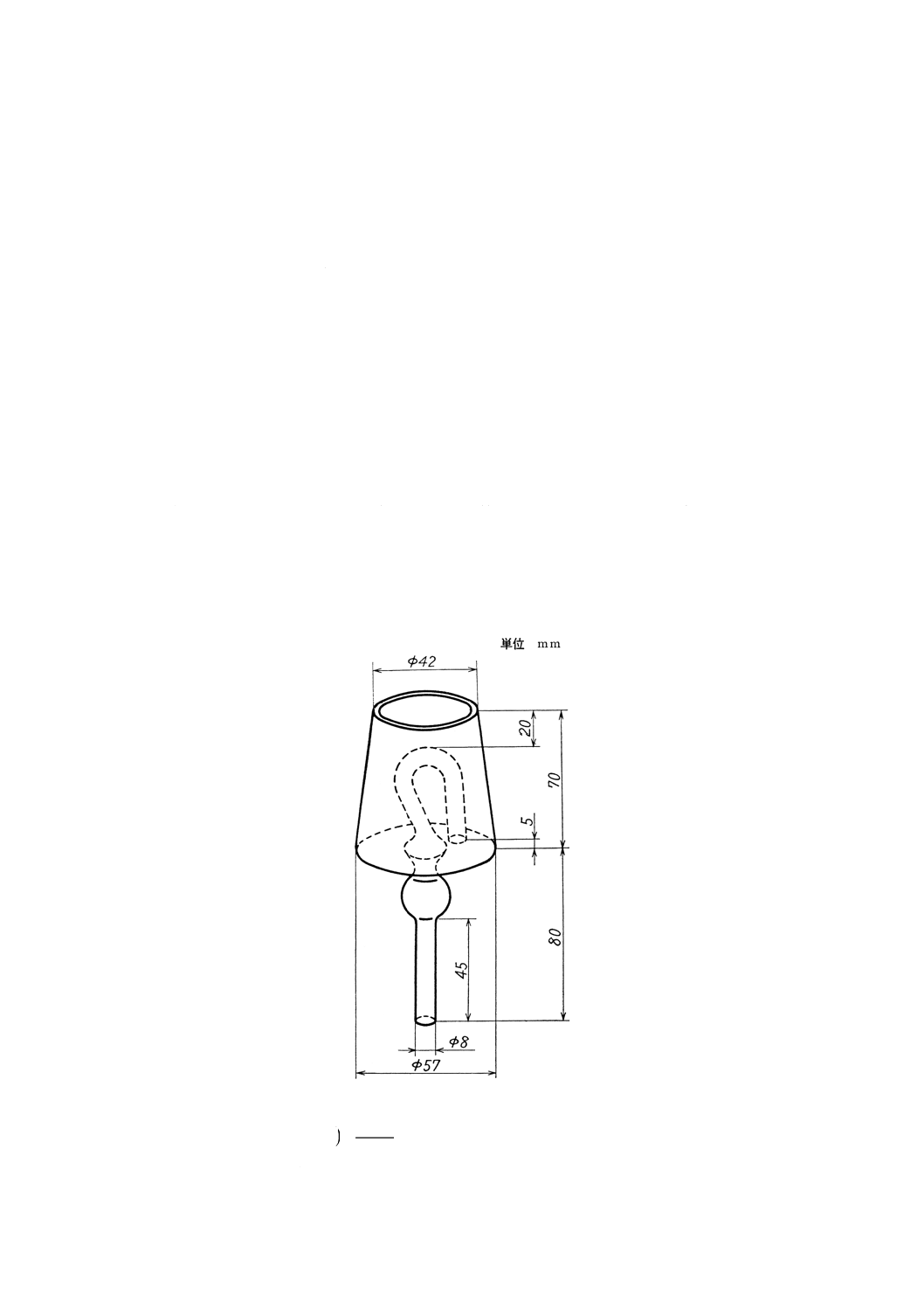
(2) 還元 9.2.5(1)で得た試料溶液に還元剤〔9.2.3(4)〕(60)を加え,その三角フラスコに図9に示すような
還元用キャップを付けたゴム栓をし,キャップ内には水約50mlを入れる。試料溶液を加熱煮沸(61)(62)
してすずを第一すずイオンに還元する。還元が完了したらフラスコ内に空気が入らないようにキャッ
プ内に炭酸水素ナトリウム溶液(飽和約10w/v%)を補充しながら,水又は氷水で15℃以下に冷却する。
注(60) 還元剤としてアルミニウムはくを使用する場合は約1.5gを,鉛の場合は約10gを,またニッケ
ル環の場合は3個程度を加える。
(61) アルミニウムはくを使用した場合,急激に反応し始めたときに三角フラスコを水又は氷水で急
冷する。
(62) 鉛又はニッケル環を用いた場合,約300℃の熱板上で約40分間煮沸すればよい。
(3) 滴定 9.2.5(2)で還元冷却した三角フラスコのキャップを外してキャップの内壁及び脚部を水で洗い,
その洗液を9.2.5(2)で得た溶液に加える。直ちによう化カリウム0.5gとでんぷん溶液〔9.2.3(8)〕5ml
を指示薬として加え,よう素酸カリウム標準溶液で滴定して溶液が紫色に変わる点を終点とする。
図9 還元キャップの寸法例
9.2.6
計算 試料中のすず含有率を次の式によって算出する。
()
100
%
×
×
×
=
B
W
f
V
すず
ここに,
V: よう素酸カリウム標準溶液の使用量 (ml)
35
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
f: よう素酸カリウム標準溶液1mlのすず相当量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
9.3
ピロカテコールバイオレット吸光光度法
9.3.1
要旨 (単独定量の場合) 試料をふっ化水素酸と硝酸で分解して硫酸で白煙処理する。
(イオン交換分離の場合) 試料を適切な酸で分解してふっ化水素酸と硝酸に溶解し,イオン交換樹脂
にニオブなどを吸着させ,すずを溶離液 (A) で溶出し,硫酸白煙処理をする。
上記いずれかの試料溶液によう化カリウムを反応させてすずをよう化物とし,ベンゼンで抽出した後,
塩酸に逆抽出し,ピロカテコールバイオレットを反応させてpHを調節し,吸光度を測定する。
9.3.2
適用分野 単独に定量する場合とイオン交換分離して他の成分と系統的に定量する場合とでは,試
料溶液の調製をそれぞれ別操作による。
9.3.3
試薬 試薬は,次による。
(1) 塩酸 (1+9)
(2) 硝酸 (1+1)
(3) ふっ化水素酸
(4) 硫酸 (1+1)
(5) アンモニア水
(6) よう化カリウム溶液 (50w/v%)
(7) よう化カリウム洗浄液〔よう化カリウム溶液 (50w/v%) 1,硫酸 (1+1) 1,水1〕
(8) 酢酸 (1+3)
(9) 硫酸ヒドラジン〔NH2)2H2SO4〕溶液 (1w/v%)
(10) ピロカテコールバイオレット〔C19H14O7S〕溶液 (0.1w/v%)
(11) ベンゼン
(12) 標準すず溶液 (5μgSn/ml) すず(純度99.5%以上)0.500gをはかり取って白金皿(100番)に移し,
硫酸 (1+1 )20mlを加え,加熱して分解する。常温まで冷却して1000mlのメスフラスコに移し,硫酸
(1+6) で標線までうすめて標準原液とする。使用の都度,この標準原液の一部を水で正しく100倍に
うすめて標準すず溶液とする。
9.3.4
試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
9.3.5
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製
(a) 単独定量の場合 はかり取った試料〔9.3.4〕を白金皿(100番)に移し,ふっ化水素酸10mlと硝酸
(1+1) 5mlを加えて試料を分解し,硫酸 (1+1) 20mlを加えて加熱し,硫酸の白煙を5〜7分間発生
させて放冷する。
(b) イオン交換分離の場合 3.3.6(2)で溶離液 (A) で溶出してポリエチレンビーカー (300ml) に保存し
た溶液(10)を白金皿(100番)に移し,硫酸 (1+1) 20mlを加えて硫酸の白煙が発生し始めるまで加
熱蒸発する。放冷後,白金皿の内壁を少量の水で洗浄し,再び加熱して硫酸の白煙を5〜10分間発
生させて放冷する。
(2) 抽出 9.3.5(1)で得た試料溶液に水10mlを加え,加熱して可溶性塩類を溶解した後,冷却する。内容
物(63)を硫酸 (1+1) 10mlで分液漏斗 (100ml) に移してよう化カリウム溶液 (50w/v%) 5mlを加えて振
り混ぜ,これにベンゼン10mlを加えて約2分間激しく振り混ぜる。2層に分離するまで静置して水相

36
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を捨て,有機相によう化カリウム洗浄液〔9.3.3(7)〕10mlを加えて約30秒間激しく振り混ぜる。2層
に分離するまで静置して水相を捨てる。
有機相に塩酸 (1+9) 10mlを加えて約1分間激しく振り混ぜ,2層に分離するまで静置した後,水相
をビーカー (100ml) に移して硫酸ヒドラジン溶液 (1w/v%) 5mlを加え,加熱してよう素の色を脱色さ
せ,常温まで冷却する。この溶液をすず含有率に応じて表3に従ってうすめた後,同表に従って一定
量を分取してビーカー (100ml) に移し,水で約50mlにうすめる。
注(63) ニオブなどの不溶解分が認められてもそのまま分液漏斗に移す。
表3
すず含有率
%
用いるメスフ
ラスコの容積
ml
うすめる溶液
分取量
ml
0.2未満
100
水
50
0.2以上0.5未満
100
水
20
0.5以上1.0未満
100
水
10
1.0以上2.5未満
250
塩酸 (1+9) 15ml+水
10
2.5以上3.0以下
250
塩酸 (1+9) 15ml+水
5
(3) 呈色 9.3.5(2)で分取した溶液にピロカテコールバイオレット溶液 (0.1w/v%) 1.0mlと酢酸 (1+3) 5ml
を正確に加え,pHメーターを用いてアンモニア水でpHを4.0±0.2に調節し,100mlのメスフラスコ
に移して水で標線までうすめ,10〜20分間放置する。
(4) 吸光度の測定 9.3.5(3)で得た呈色溶液の一部を光度計の吸収セルに取り,空試験溶液を対照液として
波長500nm付近における吸光度を測定する。
9.3.6
検量線の作成 検量線は,次のようにして作成する。
数個のビーカー (100ml) を準備し,標準すず溶液0〜40ml(すずとして0〜200μg)を段階的に正確に加
え,それぞれに塩酸 (1+9) 10mlを加える。以下9.3.5(3)及び9.3.5(4)の手順に従って操作し,吸光度を測
定する。吸光度と呈色溶液中のすず量との関係を求めて検量線とする。
9.3.7
計算 9.3.6で作成した検量線に9.3.5(4)で得た吸光度を挿入してすず量を求め,試料中のすず含有
率を次の式によって算出する。
()
100
%
×
×
=
B
W
A
すず
ここに,
A: 分取した試料溶液中のすず量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
9.4
原子吸光法
9.4.1
要旨 (ニオブ共存の場合) 試料をふっ化水素酸と硝酸で分解して一定量にうすめる。
(加水分解の場合) 試料をふっ化水素酸と硝酸で分解した後,硫酸で白煙処理する。可溶性塩類を水
と過酸化水素水に溶解し,ニオブなどを加水分解させて一定量とし,乾いたろ紙でろ過する。
(イオン交換分解の場合) 試料を適切な酸で分解してふっ化水素酸と硝酸に溶解し,イオン交換樹脂
にニオブなどを吸着させ,すずを溶離液 (A) で溶出し,硫酸白煙処理をした後,塩酸溶液として一定量に
うすめる。
上記のいずれかの方法で得た試料溶液を原子吸光光度計の亜酸化窒素−アセチレンフレーム中に噴霧し
てすずの吸光度を測定する。
37
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.4.2
適用分野 ニオブ共存のままの場合と分離する場合とでは,試料溶液の調製及び検量線の作成をそ
れぞれ別操作による。
9.4.3
試薬
(1) 塩酸
(2) 硝酸
(3) 硝酸 (1+1)
(4) ふっ化水素酸
(5) ふっ化水素酸 (1+1)
(6) 硫酸 (1+1)
(7) 過酸化水素水
(8) 鉄溶液A (10mgFe/ml) 鉄(純度99.9%以上)2.50gをはかり取ってビーカー (300ml) に移し,時計皿
で覆って塩酸20mlを加え,静かに加熱して分解する。過酸化水素水5mlを徐々に加えて鉄などを酸
化し,2〜3分間煮沸して過剰の過酸化水素を分解する。常温まで冷却し,250mlのメスフラスコに移
して水で標線までうすめる。
(9) 鉄溶液B (10mgFe/ml) 鉄(純度99.9%以上)2.50gをはかり取ってポリエチレンビーカー (300ml) に
移してふたをし,水10mlとふっ化水素酸5mlを加えた後,硝酸約5mlを加えて徐々に分解する。こ
れを250mlのポリエチレン製のメスフラスコに移して水で標線までうすめる。
(10) ニオブ溶液 (10mgNb/ml) ニオブ(純度99.7%以上)1.00gをはかり取ってポリエチレンビーカー
(300ml) に移してふたをし,水10mlとふっ化水素酸5mlを加えた後,硝酸1mlを徐々に滴加して分解
する。これを100mlのポリエチレン製のメスフラスコに移して水で標線までうすめる。
(11) 標準すず溶液A (500FgSn/ml) すず(純度99.9%以上)0.5000gをはかり取って白金皿(100番)に移
し,時計皿で覆い,硫酸,(1+1),20mlを加えて加熱分解し,常温まで冷却して1000mlのメスフラ
スコに移し,塩酸 (1+1) 300mlを加えて水で標線までうすめる。
(12) 標準すず溶液B (1.00mgSn/ml) すず(純度99.9%以上)0.2500gをはかり取ってポリエチレンビーカ
ー (300ml) に移してふたをし,ふっ化水素酸10mlを加えた後,硝酸1mlを加えて徐々に分解する。
これを250mlのポリエチレン製のメスフラスコに移して水で標線までうすめる。
9.4.4
試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
9.4.5
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製
(a) ニオブ共存の場合 はかり取った試料 (9.4.4) をポリエチレンビーカー (300ml) に移してふたをし,
ふっ化水素酸 (1+1) 10mlを加えて常温で約1〜2時間放置して試料の大部分を分解した後,硝酸数
滴(1ml以内)を加えて試料を完全に溶解する(18)。この溶液を100mlのポリエチレン製のメスフラ
スコに移して水で標線までうすめる。
(b) 加水分解分量の場合 はかり取った試料 (9.4.4) を白金皿(100番)に移してふたをし,ふっ化水素
酸10mlを加え,硝酸 (1+1) 5mlを滴加して分解し,硫酸 (1+1) 10mlを加えて加熱し,硫酸の白煙
を5〜7分間発生させる。放冷後,水約10ml,塩酸15ml及び過酸化水素水2mlを滴加して可溶性塩
類を溶解し,ビーカー (200ml) に水で洗い移す。白金皿は,再び水10mlと過酸化水素水1mlを加
えて加熱し,先のビーカーに合わせる。塩酸15mlを加え,水で80mlにうすめた後,加熱して10
〜15分間煮沸し(64),常温まで冷却して100mlのメスフラスコに移して水で標線までうすめる。こ
れを乾いたろ紙(5種C)を用いて乾いたビーカー (300ml) にろ過し,最初のろ液を捨て,次のろ
38
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
液を集める。
注(64) 静かに沸騰させて過酸化水素を分解し,すずの加水分解を防ぐ。
(c) イオン交換分離の場合 3.3.6(2)で溶離液 (A) で溶出してポリエチレンビーカー (300ml) に保存し
た溶液(10)を白金皿(100番)に移し,硫酸 (1+1) 10mlを加えて硫酸の白煙が発生するまで加熱す
る。放冷後,水約10mlと塩酸15mlを加えて可溶性塩類を溶解し,常温まで冷却して100mlのメス
フラスコに移し,水で標線までうすめる。
(2) 吸光度の測定 原子吸光光度計を用い,9.4.5(1)で得た試料溶液の一部を亜酸化窒素−アセチレンフレ
ーム中に噴霧して分析線224.6nm又は286.3nmにおける吸光度を測定する。
9.4.6
検量線の作成 検量線は,次のようにして作成する(19)。
(1) ニオブ共存の場合 数個の100mlのポリニチレン製のメスフラスコを準備する。それぞれにニオブ溶
液〔9.4.3(10)〕13mlと鉄溶液B〔9.4.3(9)〕5.5mlを正確に加え,これらに標準すず溶液B〔9.4.3(12)〕
0〜6ml(すずとして0〜6.00mg)を段階的に正確に加えて水で標線までうすめる。以下9.4.5(2)の手順
に従って操作して吸光度を測定する。吸光度と溶液中のすず量との関係を求めて検量線とする。
(2) ニオブを分離する場合 数個のビーカー (100ml) を準備する。それぞれに鉄溶液A〔9.4.3(8)〕6mlと
硫酸 (1+1) 5mlずつを加え,これらに標準すず溶液A〔9.4.3(11)〕0〜12ml(すずとして0〜6.00mg)
を段階的に正確に加えて硫酸の白煙が発生するまで加熱する。冷却後,塩酸15mlを加えて溶解し,
常温まで冷却して100mlのメスフラスコに移し,水で標線までうすめる。以下9.4.5(2)の手順に従って
操作して吸光度を測定する。吸光度と溶液中のすず量との関係を求めて検量線とする。
9.4.7
計算 9.4.5(1)(a)の試料溶液を用いた場合は,9.4.6(1)の検量線に,9.4.5(1)(b)及び9.4.5(1)(c)の試料
溶液を用いた場合は9.4.6(2)の検量線に,それぞれ9.4.5(2)で得た吸光度を挿入して試料溶液中のすず量を
求め,試料中のすず含有率を次の式によって算出する。
()
100
%
×
=WA
すず
ここに,
A: 試料溶液中のすず量 (g)
W: 試料はかり取り量 (g)
10. アルミニウム定量方法
10.1 方法の区分 アルミニウムの定量方法は,次のいずれかによる。
(1) EDTA滴定法 この方法は,アルミニウム含有率0.1%以上の試料に適用する。
(2) クロムアズロールS吸光光度法 この方法は,アルミニウム含有率3.0%未満の試料に適用する。
(3) 原子吸光法 この方法は,アルミニウム含有率2.5%未満の試料に適用する。
10.2 EDTA滴定法
10.2.1 要旨 (加水分解分離の場合) 試料を硝酸とふっ化水素酸で分解し,硫酸で白煙処理し,過酸化
水素水と硫酸に溶解し,ニオブ,タンタルなどを加水分解させて分離する。
(イオン交換分離の場合) 試料を適切な酸で分解してふっ化水素酸と硝酸に溶解し,イオン交換樹脂
にニオブなどを吸着させ,アルミニウムを溶離液 (A) で溶出し,硫酸白煙処理して硫酸溶液とする。
これらの溶液からクペロン−クロロホルムで鉄などの妨害元素を抽出除去し 加熱してクロロホルムを
揮散させる。
(バリウム共存分離の場合) 試料を融解合剤で融解し,塩化バリウム溶液で融成物を抽出し,妨害元
素を分離し,塩酸溶液とする。
39
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
上記のいずれかの溶液をアンモニア水と酢酸アンモニウムでpHを調節して煮沸させ,Cu−PANを指示
薬として,EDTA標準溶液の少過剰と反応させ,硫酸銅標準溶液で過剰のEDTAを滴定する。
10.2.2 適用分野 ニオブ,タンタルを加水分解又はイオン交換樹脂で分離し,クペロン−クロロホルムで
鉄などを分離する場合と試料をアルカリ融解する場合は,試料溶液の調製及び共存元素の分離をそれぞれ
別操作による。
10.2.3 試薬 試薬は,次による。
(1) 塩酸 (1+1)
(2) 硝酸
(3) 硝酸 (1+1)
(4) ふっ化水素酸
(5) 硫酸 (1+1)
(6) アンモニア水 (1+1)
(7) 過酸化水素水
(8) 融解合剤〔過酸化ナトリウム7,炭酸ナトリウム(無水)1〕
(9) 塩化バリウム溶液 (25w/v%)
(10) 酢酸アンモニウム〔CH3CO2NH4〕溶液 (10w/v%)
(11) クペロン溶液 (6w/v%) クペロン(ニトロソフェニルヒドロキシルアミンアンモニウム塩)
(C6H5N2O2・NH4) 6gと炭酸アンモニウム約1gを水100mlに溶解し,分解生成物などをろ紙(5種A)
を用いてろ過し,ろ液を使用する。この溶液は使用の都度調製する。
(12) クロロホルム
(13) M/100エチレンジアミン四酢酸二ナトリウム (EDTA) 標準溶液〔3.722gC10H14O8N2Na2・2H2O/l〕 調製
及び標定方法は,JIS K 8006の2.(20)による。
(14) M/100硫酸銅溶液 硫酸銅(5水塩)2.5gをはかり取って水約100mlに溶解し,1000mlのメスフラス
コに移して水で標線までうすめる。この溶液の標定は,M/100EDTA標準溶液10mlを分取してビーカ
ー (300ml) に移し,水で約100mlにうすめる。塩酸 (1+1) 5mlを加えた後,pHメーターを用いて酢
酸アンモニウム溶液 (10w/v%) でpHを3.0±0.2に調節して沸騰するまで加熱する。Cu−PAN溶液4
〜5滴を指示薬として加え,M/100硫酸銅標準溶液で滴定し,微紅色を呈する点を終点とする。次の
式によってM/100に対するファクターを算出する。
2
00
.
10
F
V
F
×
=
ここに,
F: M/100硫酸銅標準溶液のファクター
V: M/100硫酸銅標準溶液の使用量 (ml)
F2: M/100EDTA標準溶液のファクター
(15) Cu−PAN溶液 Cu−PAN(Cu−EDTAと1−ピリジルアゾ−2−ナフトールの混合物)1gをイソプロ
ピルアルコール (50v/v%) 100mlに溶解する。Cu−PAN溶液の空試験値は負になることがあるので注意
する。
10.2.4 試料はかり取り量 試料は,0.50gを0.1mgのけたまではかり取る。ただし,イオン交換分離の場
合は0.20gとする。
10.2.5 操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製
40
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(a) 酸分解の場合 はかり取った試料 (10.2.4) を白金皿(100番)に移し,ふっ化水素酸10mlを加え,
硝酸 (1+1) 5mlを滴加して分解し,硫酸 (1+1) 10mlを加えて加熱し,硫酸の白煙を5〜7分間発生
させる。放冷後,硫酸 (1+1) 15mlと水10mlを加え,加熱して可溶性塩類を溶解し,水でビーカー
(300ml) に洗い移す。白金皿に水10mlと過酸化水素水2mlを加え,加熱して付着物を溶解し,先の
ビーカーに合わせる。この溶液を水で約150mlにうすめ,約15分間煮沸し,常温に冷却して250ml
のメスフラスコに移し,水で標線までうすめる。
(b) 融解合剤で融解する場合 はかり取った試料 (10.2.4) を鉄るつぼ (30ml) に移し,融解合剤
〔10.2.3(8)〕18gを加えてよく混合し,徐々に加熱温度を上げ,ときどき揺り動かしながら融解する。
放冷後,るつぼをビーカー (500ml) に移し,塩化バリウム溶液 (25w/v%) 20mlと水約130mlを加え
て融成物を抽出し,るつぼを水で洗って取り出す。この液を約5分間静かに煮沸した後,常温に冷
却して250mlのメスフラスコに移し,水で標線までうすめる。
(c) イオン交換分量の場合 3.3.6(2)で溶離液 (A) で溶出してポリエチレンビーカー (300ml) に保存し
た溶液(10)を白金皿(100番)に移し,硫酸 (1+1) 10mlを加え,硫酸の白煙が出始めるまで加熱す
る。放冷後,少量の水で白金皿の内壁を洗い,再び加熱して硫酸の白煙が発生し始めたならば放冷
する。水約20mlを加え,加熱して可溶性塩類を溶解し,常温まで冷却して100mlのメスフラスコ
に移し,水で標線までうすめる。
(2) 共存元素の分離
(a) クペロン−クロロホルム抽出分離の場合 10.2.5(1)(a)で得た溶液を乾いたろ紙(5種C)で乾いたビ
ーカー (300ml) にろ過し,最初のろ液は捨て,次のろ液から50mlを分取して分液漏斗 (200ml) に
移す。又は10.2.5(1)(c)で得た溶液から50mlを分取して分液漏斗 (200ml) に移す。クペロン溶液
〔10.2.3(11)〕10mlとクロロホルム15mlを加え,約30秒間激しく振り混ぜ,静置して2層に分離し
た後,下層の有機相を捨てる。更にクロロホルム10mlを用いて有機相に着色が認められなくなる
まで抽出操作を繰り返す。水相をビーカー (300ml) に移し,加熱して硫酸白煙が発生し始めるまで
蒸発し,クロロホルムを除去する(65)。
注(65) 溶液が淡黄色に着色することがあるが,これは残存したクペロンが分解したためで,アルミニ
ウムの定量値には影響しない。着色が著しいときは,硝酸を滴加して有機物を分解し,引き続
き加熱して硫酸白煙を発生させて硝酸を除去する。
(b) バリウム共存分離の場合 10.2.5(1)(b)で得た溶液を乾いたビーカー (300ml) に移し,静置して上澄
み液を乾いたろ紙(6種)でろ過する。初めのろ液は捨て,次のろ液から50mlを分取してビーカー
(300ml) に移し,塩酸 (1+1) を加えて微酸性にする。
(3) 滴定 10.2.5(2)で得た溶液を水で約200mlにうすめ,酢酸アンモニウム溶液 (10w/v%) 10mlを加え,pH
メーターを用いてアンモニア水 (1+1) 又は塩酸 (1+1) でpH3.0±0.2に調節する。この溶液を加熱
して煮沸し始めたならばCu−PAN溶液4〜5滴を指示薬として加え,引き続き沸騰させながら
M/100EDTA標準溶液を溶液の赤紫色が黄色に変わった後,更にその過剰2〜3mlを正確に加え,約10
分間煮沸し,M/100硫酸銅標準溶液で滴定(66)して溶液が黄色から赤紫色に変わる点を終点とする。
注(66) 過剰のバリウムにより白濁することがあるが,終点が判別できれば支障はない。
10.2.6 計算 試料中のアルミニウム含有率を次の式によって算出する。
()(
)
100
0002698
.0
%
2
1
×
×
×
−
=
B
W
V
V
アルミニウム
ここに, V1: M/100EDTA標準溶液の使用量 (ml)
41
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V2: M/100硫酸銅標準溶液の使用量 (ml)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
10.3 クロムアズロールS吸光光度法
10.3.1 要旨 (加水分解分離の場合) 試料をふっ化水素酸と硝酸で分解し,硫酸白煙処理して過酸化水
素水と硫酸に溶解し,ニオブなどを加水分解させて分離する。
(イオン交換分離の場合) 試料を適切な酸で分解し,ふっ化水素酸と硝酸に溶解し,イオン交換樹脂
にニオブなどを吸着させ,アルミニウムを溶離液 (A) で溶出し,硫酸白煙処理して硫酸溶液とする。
これらの溶液からクペロンとクロロホルムで鉄などを抽出し,クロムアズロールSと塩酸ヒドロキシル
アミンでアルミニウムを呈色させ,アンモニア水でpHを調節してその吸光度を測定する。
10.3.2 適用分野 ニオブ,タンタルを加水分解させて分離する場合とイオン交換樹脂で分離する場合は,
試料溶液の調製をそれぞれ別操作による。
10.3.3 試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) ふっ化水素酸
(3) 硫酸 (1+1)
(4) アンモニア水 (1+1)
(5) 過酸化水素水
(6) 塩酸ヒドロキシルアミン (NH2OH・HCl〕 溶液 (10v/v%)
(7) クペロン溶液 (6w/v%) 10.2.3(11)による。
(8) クロムアズロールS (C23H13O9SCl2Na3) 溶液 (0.1v/v%)
(9) クロロホルム
(10) 標準アルミニウム溶液 (10μgAl/ml) アルミニウム(純度99.9%以上)0.100gをはかり取ってビーカ
ー (300ml) に移し,塩酸 (1+1) 50mlに加熱して溶解し,常温まで冷却して1000mlのメスフラスコに
移し,水で標線までうすめてアルミニウム標準原液とする。使用の都度,この標準原液の一部を水で
正しく10倍にうすめて標準アルミニウム溶液とする。
10.3.4 試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
10.3.5 操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製
(a) 加水分解分離の場合 はかり取った試料 (10.3.4) を白金皿(100番)に移し,ふっ化水素酸10ml
を加え,硝酸 (1+1) を滴加して分解した後,硫酸 (1+1) 10mlを加え,加熱して硫酸の白煙を発生
させて放冷する。硫酸 (1+1) 15ml,水10ml及び過酸化水素水2mlを加えて加熱し,可溶性塩類を
溶解してビーカー (300ml) に移す。白金皿に水10mlと過酸化水素水2mlを加え,付着物を溶解し
て先のビーカーに合わせる。熱水を加えて約200mlにうすめ,約15分間煮沸して常温まで冷却し
た後,250mlのメスフラスコに移し,水で標線までうすめて乾いたビーカー (300ml) に移す。上澄
み液を乾いたろ紙(5種C)を用いてろ過し,初めのろ液は捨て,次のろ液から25mlを分取する。
(b) イオン交換分離の場合 3.3.6(2)で保存した溶液(10)を白金皿(100番)に移し,硫酸 (1+1) 10mlを
加え,硫酸の白煙が発生し始めるまで加熱蒸発して放冷する。白金皿の内壁を少量の水で洗って再
び加熱し,硫酸の白煙を5〜10分間発生させて放冷する。硫酸 (1+1) 15mlと水10mlを加えて加熱
し,可溶性塩類を溶解して常温に冷却した後,250mlのメスフラスコに移して水で標線までうすめ,

42
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
その25mlを分取する。
(2) 妨害元素の分離 10.3.5(1)で分取した溶液を分液漏斗 (100ml) に移し,クペロン溶液〔10.3.3(7)〕10ml
とクロロホルム15mlを加え,約30秒間激しく振り混ぜ,静置して2層に分離後,下層の有機相を捨
てる。クロロホルム10mlずつを用いて有機相に着色が認められなくなるまで抽出を繰り返し,更に
クロロホルム10mlで抽出した後,水相をビーカー (200ml) に移し,硫酸の白煙が発生し始めるまで
加熱してクロロホルムを除去する(65)。冷却後,水約50mlを加えて可溶性塩類を溶解し,100mlのメ
スフラスコに移して水で標線までうすめる。
(3) 呈色 10.3.5(2)で得た溶液からアルミニウム含有率に応じ,原則として表4に従って分取してビーカ
ー (100ml) に移し,水で約60mlにうすめる。クロムアズロールS溶液 (0.1w/v%) 2mlを正確に加えて
振り混ぜた後,塩酸ヒドロキシルアミン溶液 (10w/v%) 5mlを加え,pHメーターを用いてアンモニア
水 (1+1) でpH6.0±0.1に調節し,直ちに100mlのメスフラスコに移して水で標線までうすめる。
表4
アルミニウム含有率
%
分取量
ml
0.2未満
50
0.2以上 0.5未満
25
0.5以上 1.5未満
10
1.5以上 3.0未満
5
(4) 吸光度の測定 10.3.5(3)で得た溶液を10分間静置後(67),溶液の一部を光度計の吸収セルに移し,空試
験溶液を対照液として波長550nm付近の吸光度を測定する。
注(67) 吸光度は約5分間で最大となり,約20分間は安定である。
10.3.6 検量線の作成 検量線は,次のようにして作成する。
数個のビーカー (100ml) を準備し,それぞれに標準アルミニウム溶液0〜5ml(アルミニウムとして0
〜50μg)を段階的に正確に加え,それぞれに硫酸 (1+1) 10滴を加えて水で約60mlにうすめる。以下
10.3.5(3)クロムアズロール溶液添加以降の手順に従って操作し,吸光度とアルミニウム量との関係を求め
て検量線とする。
10.3.7 計算 10.3.6で作成した検量線に10.3.5(4)で得た吸光度を挿入して試料溶液中のアルミニウム量を
求め,試料中のアルミニウム含有率を次の式によって算出する。
()
100
%
×
×
=
B
W
A
アルミニウム
ここに,
A: 分取した試料溶液中のアルミニウム量 (g)
W: 試料はかり取り量 (g)
B: 試料溶液の分取比
10.4 原子吸光法
10.4.1 要旨 (ニオブ共存の場合) 試料をふっ化水素酸と硝酸で分解して一定量にうすめる。
(加水分解分離の場合) 試料をふっ化水素酸と硝酸で分解し,硫酸で白煙処理をした後,可溶性塩類
を水と過酸化水素水で溶解し,ニオブなどを加水分解させて一定量とし,乾いたろ紙でろ過する。
(イオン交換分離の場合) 試料を適切な酸で処理してふっ化水素酸と硝酸の溶液とし,ニオブなどを
イオン交換樹脂に吸着させ,アルミニウムを溶離液 (A) で溶出させ,硫酸で白煙処理をして一定量にうす
める。
上記のいずれかの試料溶液を,原子吸光光度計の亜酸化窒素−アセチレンフレーム中に噴霧してアルミ
43
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ニウムの吸光度を測定する。
10.4.2 適用分野 ニオブ共存の場合とニオブを分離する場合とは,試料溶液の調製と検量線の作成を,そ
れぞれ別操作による。
10.4.3 試薬 試薬は,次による。
(1) 塩酸
(2) 硫酸 (1+1)
(3) 鉄溶液A (10mgFe/ml) 9.4.3(8)による。
(4) 鉄溶液B (10mgFe/ml) 9.4.3(9)による。
(5) ニオブ溶液 (10mgNb/ml) 9.4.3(10)による。
(6) 標準アルミニウム溶液A (500μgAl/ml) アルミニウム(純度99.9%以上)0.500gをはかり取ってビー
カー (300ml) に移して時計皿で覆い,硫酸 (1+1) 20mlを加えて加熱分解し,常温まで冷却して水で
約200mlにうすめる。これを1000mlのメスフラスコに移して水で標線までうすめる。
(7) 標準アルミニウム溶液B (1.00mgAl/ml) アルミニウム(純度99.9%以上)0.250gをはかり取ってポ
リエチレンビーカー (300ml) に移してふたをし,水20mlとふっ化水素酸5mlを加えて常温で徐々に
分解する。これを250mlのポリエチレン製のメスフラスコに移して水で標線までうすめる。
10.4.4 試料はかり取り量 試料は,0.20gを0.1mgのけたまではかり取る。
10.4.5 操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製
(a) ニオブ共存の場合 9.4.5(1)(a)の手順に従って操作する。
(b) 加水分解分離の場合 9.4.5(1)(b)の手順に従って操作する。
(c) イオン交換分離の場合 9.4.5(1)(c)の手順に従って操作する。
(2) 吸光度の測定 原子吸光光度計を用い,10.4.5(1)で得た試料溶液の一部を亜酸化窒素−アセチレンフ
レーム中に噴霧し 分析線309.3nmにおける吸光度を測定する。
10.4.6 検量線の作成 検量線は 次のようにして作成する(19)。
(1) ニオブ共存の場合 数個の100mlのポリエチレン製メスフラスコを準備してそれぞれにニオブ溶液
〔10.4.3(5)〕13mlと鉄溶液B〔10.4.3(4)〕5.5mlを正確に加え,これらに標準アルミニウム溶液B
〔10.4.3(7)〕0〜8ml(アルミニウムとして0〜8.00mg)を段階的に正確に加えて水で標線までうすめる。
以下10.4.5(2)の手順に従って操作し,吸光度を測定する。吸光度と溶液中のアルミニウム量との関係
を求めて検量線とする。
(2) ニオブを分離する場合 数個のビーカー (100ml) を準備してそれぞれに鉄溶液A〔10.4.3(3)〕6mlと
硫酸 (1+1) 5mlずつを加え,これらに標準アルミニウム溶液A〔10.4.3(6)〕を0〜16ml(アルミニウ
ムとして0〜8.00mg)を段階的に正確に加え,硫酸白煙が発生するまで加熱する。冷却後,塩酸15ml
を加えて溶解し,常温まで冷却して100mlのメスフラスコに移し,水で標線までうすめる。以下
10.4.5(2)の手順に従って操作し,吸光度を測定する。吸光度と溶液中のアルミニウム量との関係を求
めて検量線とする。
10.4.7 計算 10.4.5(1)(a)の試料溶液を用いた場合は10.4.6(1)の検量線に,10.4.5(1)(b)及び10.4.5(1)(c)の試
料溶液を用いた場合は10.4.6(2)の検量線に,それぞれ10.4.5(2)で得た吸光度を挿入して試料溶液中のアル
ミニウム量を求め,試料中のアルミニウム含有率を次の式によって算出する。
()
100
%
×
=WA
アルミニウム
44
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
A: 試料溶液中のアルミニウム量 (g)
W: 試料はかり取り量 (g)
付図1 イオン交換樹脂カラムの例
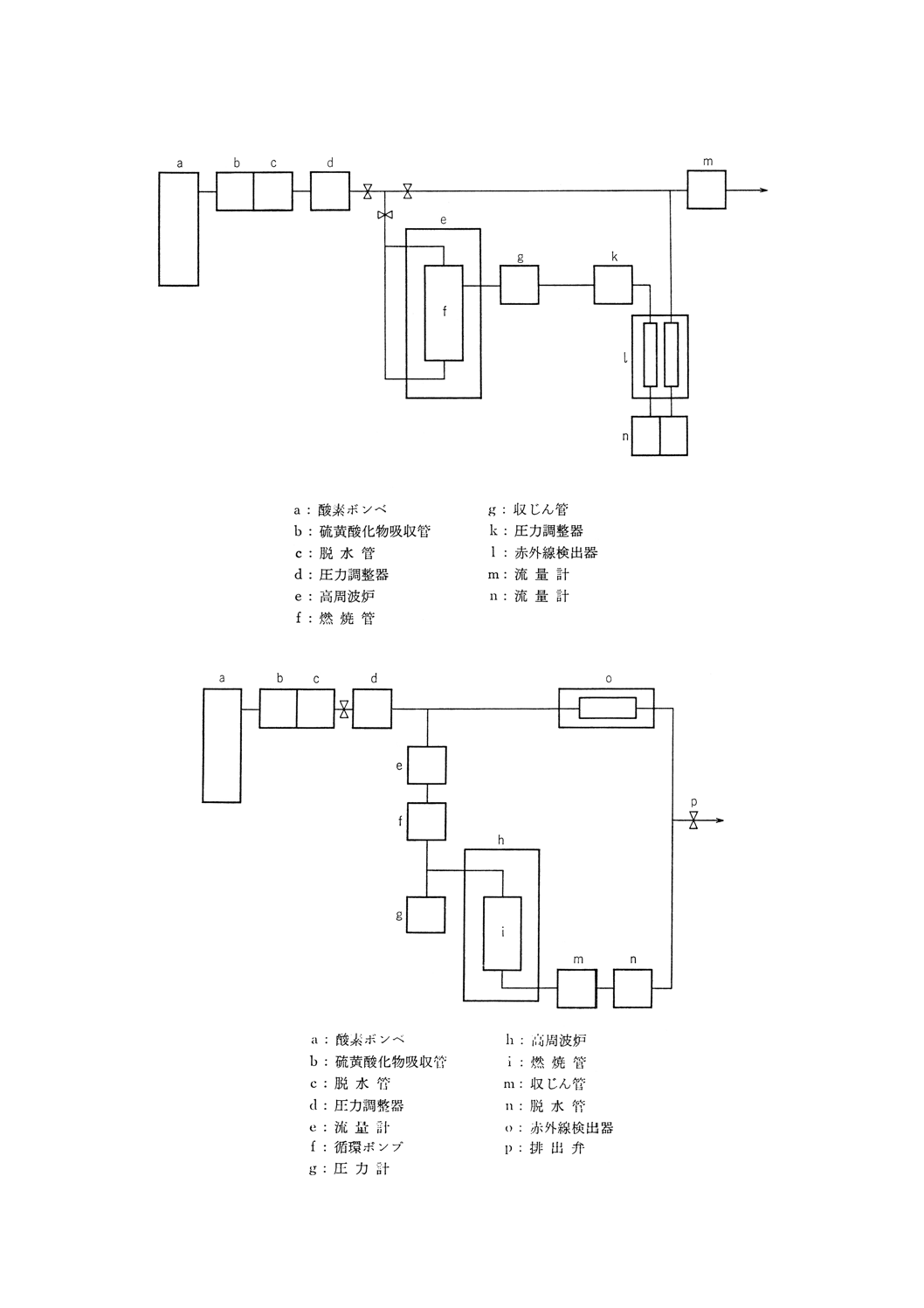
付図2 フェロニオブ中の硫黄定量装置(8.2中和滴定法)の例
45
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図3 フェロニオブ中の硫黄定量装置(8.3導電率法)の概念図
付図4 フェロニオブ中の硫黄定量装置(8.4電量法)の概念図
46
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図5 フェロニオブ中の硫黄定量装置〔8.5赤外線吸収法(積分法)〕の概念図
付図6 フェロニオブ中の硫黄定量装置〔8.6赤外線吸収法(循環法)〕の概念図
47
G 1328-1982
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本フエロアロイ協会分析専門委員会
氏名
所属
(主査)
吉 山 鍈 一
日本重化学工業株式会社
(委員)
大 田 一 身
日本鋼管株式会社
大 槻 孝
新日本製鐵株式会社
岡 田 実
粟村金属工業株式会社
小埜木 昭 三
日本重化学工業株式会社
鈴 木 好 道
社団法人日本海事検定協会
世 良 昇
電気化学工業株式会社
高 橋 利 雄
日本電工株式会社
棚 田 實
日本重化学工業株式会社
中 島 武 次
日本鋼管株式会社
長谷川 宗 治
昭和電工株式会社
針間矢 宣 一
川崎製鐵株式会社
星 野 友三郎
太平洋金属株式会社
三 矢 進 彦
昭和電工株式会社
森 本 勲
日本電工株式会社
吉 田 信 之
工業技術院標準部
渡 辺 千 之
太陽鉱工株式会社
(幹事)
神 谷 圭 三
日本フエロアロイ協会
鉄鋼部会 フェロアロイ分析方法専門委員会 構成表
氏名
所属
(委員会長)
神 森 大 彦
社団法人化学情報協会
飯 田 芳 男
成蹊大学
大河内 春 乃
科学技術庁金属材料技術研究所
林 俊 太
工業技術院標準部
吉 山 鍈 一
日本重化学工業株式会社
松 村 正
日本電工株式会社
三 矢 進 彦
昭和電工株式会社
三 馬 興 治
粟村金属工業株式会社
神 谷 圭 三
日本フエロアロイ協会
岩 田 晶 夫
住友金属鉱山株式会社
世 良 昇
電気化学工業株式会社
大 槻 孝
新日本製鐵株式会社
針間矢 宣 一
川崎製鐵株式会社
井樋田 睦
日本鋼管株式会社
藤 野 允 克
住友金属工業株式会社
伊 藤 六 仁
大同特殊鋼株式会社
栗 原 国 勝
日本冶金工業株式会社
谷 口 政 行
株式会社神戸製鋼所
水 野 幸四郎
社団法人日本鉄鋼協会
今 井 康 夫
社団法人日本海事検定協会
加 藤 智 也
愛知製鋼株式会社
鈴 木 孝 範
株式会社日本製鋼所
(事務局)
吉 田 信 之
工業技術院標準部材料規格課
宮 崎 正 治
工業技術院標準部材料規格課