G 1319 : 2000
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は、工業標準化法に基づいて、日本工業標準調査会の審議を経て、通商産業大臣が改正した日
本工業規格である。これによって,JIS G 1319 : 1982は改正され,この規格に置き換えられる。
今回の改正では,国際規格との整合化を図るために,ISO規格の翻訳を附属書2として規定している。
JIS G 1319には,次に示す附属書がある。
附属書1(規定) チタン定量方法−アルミニウム還元硫酸アンモニウム鉄 (III) 滴定法(1)
附属書2(規定) チタン定量方法−アルミニウム還元硫酸アンモニウム鉄 (III) 滴定法(2) (ISO 7692)
附属書3(規定) 炭素定量方法−燃焼−赤外線吸収法
附属書4(規定) けい素定量方法−二酸化けい素重量法
附属書5(規定) けい素定量方法−チタン分離モリブドけい酸青吸光光度法
附属書6(規定) マンガン定量方法−過マンガン酸吸光光度法
附属書7(規定) りん定量方法−チタン分離モリブドりん酸青吸光光度法
附属書8(規定) 硫黄定量方法−燃焼−赤外線吸収法
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
G 1319 : 2000
フェロチタン分析方法
Methods for chemical analysis of ferrotitanium
序文 この規格は,附属書2(規定)に1983年に発行されたISO 7692, Ferrotitanium−Determination of titanium
content−Titrimetric methodを翻訳し,技術的内容及び規格票の様式を変更することなく規定し,さらに,
対応国際規格には規定されていない定量方法を附属書1,3,4,5,6,7及び8として追加して作成した
日本工業規格である。
1. 適用範囲 この規格は,フェロチタン中のチタン,炭素,けい素,マンガン,りん及び硫黄の定量方
法について規定する。
備考 この規格の対応国際規格を,次に示す。
ISO 7692 : 1983 Ferrotitanium−Determination of titanium content−Titrimetric method
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
JIS G 1301 フェロアロイ分析方法の通則
JIS G 2309 フェロチタン
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS Z 2615 金属材料の炭素定量方法通則
JIS Z 2616 金属材料の硫黄定量方法通則
3. 一般事項 定量方法に共通な一般事項は,JIS G 1301による。ただし,JIS G 1301は,附属書2(規
定)には適用しない。
4. 定量方法の区分 定量方法の区分は,表1による。

2
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1 定量方法の区分
成分
定量方法
適用含有率範囲
% (m/m)
附属書(規定)
番号
チタン
アルミニウム還元硫酸アンモニウム鉄 (III) 滴定法(1)
20以上75以下
1
アルミニウム還元硫酸アンモニウム鉄 (III) 滴定法(2)
(ISO 7692)
20以上75以下
2
炭素
燃焼−赤外線吸収法
0.001以上0.40以下
3
けい素
二酸化けい素重量法
0.10以上10以下
4
チタン分離モリブドけい酸青吸光光度法
0.01以上0.10以下
5
マンガン 過マンガン酸吸光光度法
0.05以上2.0以下
6
りん
チタン分離モリブドりん酸青吸光光度法
0.005以上0.05以下
7
硫黄
燃焼−赤外線吸収法
0.001以上0.10以下
8
3
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(規定) チタン定量方法−
アルミニウム還元硫酸アンモニウム鉄 (III) 滴定法(1)
1. 要旨 試料を硫酸と塩酸とで分解し,硝酸で酸化した後,加熱して硫酸白煙を発生させる。アルミニ
ウムでチタンを還元した後,チオシアン酸アンモニウムを指示薬として硫酸アンモニウム鉄 (III) 標準溶
液で滴定する。
2. 試薬 試薬は,次による。
a) 塩酸
b) 塩酸 (1+1, 1+4)
c) 硝酸
d) ふっ化水素酸
e) 硫酸
f)
硫酸 (1+1)
g) 水酸化ナトリウム溶液 (200g/λ)
h) チオシアン酸アンモニウム溶液 (100g/λ)
i)
炭酸水素ナトリウム溶液(飽和,約100g/λ)
j)
炭酸ナトリウム溶液 (20g/λ)
k) 二硫酸カリウム
l)
アルミニウム 99% (m/m) 以上でチタン及び銅の含有率が0.01% (m/m) 以下のもの。1個が約1gの長
方形,棒又は線状のものを使用前に塩酸 (1+5) で洗浄する。
m) 1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液 硫酸アンモニウム鉄 (III) ・12水16gを塩酸 (1+1)
10mlを含む水約300mlに溶解し,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線ま
で薄める。この溶液のファクターは,次の手順によって求める。
1) 1/30mol/λチオ硫酸ナトリウム標準溶液の調製及び標定
1.1)
調製 チオ硫酸ナトリウム五水和物8.3gと炭酸ナトリウム0.2gをはかり取り,溶存酸素を含まない
水[JIS K 8001の3.6(4)による。]1000mlに溶解し,気密容器に入れて2日間放置する。
1.2)
標定方法
1.2.1) JIS K 8005の2.(11)で規定するよう素酸カリウムを,JIS K 8005の4.表1の乾燥条件に従って乾燥
し,デシケーター中で放冷した後,1.0〜1.3gを0.1mgのけたまではかり取って,1 000mlの全量フ
ラスコに移し入れ,水を加えて溶解し,水で標線まで薄める。この溶液25mlを正確に共通すり合
わせ三角フラスコ (200ml) に取る。
1.2.2) よう化カリウム2g及び硫酸 (1+1) 2mlを加え,直ちに栓をして穏やかに振り混ぜ,暗所に5分間
放置した後,1.1)で調製した1/30mol/λチオ硫酸ナトリウム溶液で滴定し,溶液が薄い黄色になって
から指示薬としてでんぷん溶液(調製は,JIS K 8001の4.4表8による。)0.5mlを加え,引き続き
滴定して溶液の色の青が消える点を終点とし,1/30mol/λチオ硫酸ナトリウム溶液の使用量を求める。
1.2.3) 別の共通すり合わせ三角フラスコ (200ml) に水25mlを取り,以下,1.2.2)の操作を行う。
1.2.4) 次の式によって1/30mol/λチオ硫酸ナトリウム標準溶液のファクターを算出する。
4
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
P
)
V
V
(
185
001
.
0
1000
/
25
m
F
2
1
1
×
−
×
×
=
ここに, F1: 1/30mol/λチオ硫酸ナトリウム標準溶液のファクター
m: 1.2.1)はかり取ったよう素酸カリウムの質量 (g)
P: よう素酸カリウムの純度 [% (m/m)]
V1: 1.2.2)で得た1/30mo1/λチオ硫酸ナトリウム溶液の使用量 (ml)
V2: 1.2.3)で得た1/30mo1/λチオ硫酸ナトリウム溶液の使用量 (ml)
2) 1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液の標定
2.1)
共通すり合わせ三角フラスコ (300ml) に1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液を正確に
25ml取り,水35ml,塩酸 (1+1) 3ml及びよう化カリウム3gを加え,直ちに栓をして穏やかに振り
混ぜ,暗所に10分間放置する。
2.2)
遊離したよう素を1)で調製及び標定した1/30mol/λチオ硫酸ナトリウム標準溶液で滴定し,溶液が薄
い黄色になってから酢酸ナトリウム溶液 (250g/λ) 10ml及び指示薬としてでんぷん溶液(調製は,JIS
K 8001の4.4表8による。)3mlを加え,引き続き滴定して溶液の色の青が消える点を終点とし,
1/30mol/λチオ硫酸ナトリウム標準溶液の使用量を求める。
2.3)
別の共通すり合わせ三角フラスコ (300ml) に水60mlを取り,塩酸 (1+1) 3ml及びよう化カリウム
3gを加え,直ちに栓をして穏やかに振り混ぜ,暗所に10分間放置し,以下,2.2)の操作を行う。
2.4)
次の式によって1/30mo1/λ硫酸アンモニウム鉄 (III) 標準溶液のファクターを算出する。
1
4
3
2
25
)
(
F
V
V
F
×
−
=
ここに, F2: 1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液のファクター
V3: 2.2)で得た1/30mol/λチオ硫酸ナトリウム標準溶液の使用量
(ml)
V4: 2.3)で得た1/30mol/λチオ硫酸ナトリウム標準溶液の使用量
(ml)
F1: 1/30mol/λチオ硫酸ナトリウム標準溶液のファクター
3. 還元用器具 還元用器具は,次のいずれかを用いる。
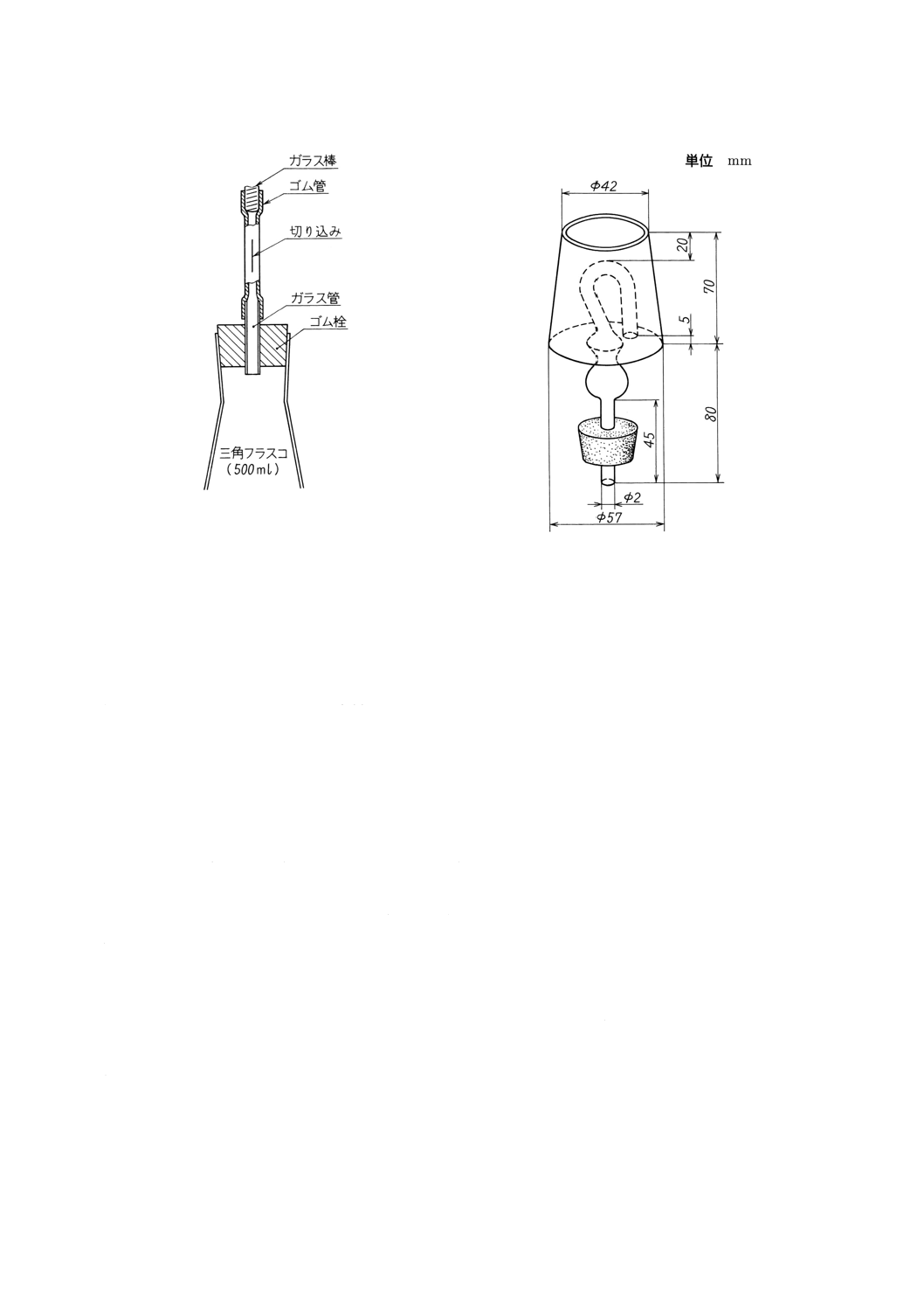
a) 弁付きゴム栓(附属書1図1参照) ゴム栓にガラス管を通し,これにゴム管を付ける。ゴム管は先
端をガラス棒などで閉じ,先端とガラス管の中間部にかみそりなどを用いて10〜15mmの切り込みを
付ける。
b) キャップ付きゴム栓(附属書1図2参照) キャップに炭酸水素ナトリウム溶液(飽和,約100g/λ)
を入れて用いる。
5
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1図1 弁付きゴム栓
附属書1図2 キャップ付きゴム栓
4. 試料はかり取り量 試料はかり取り量は,1.0gとし,0.1mgのけたまで読み取る。
5. 操作
5.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってビーカー (500ml) に移し入れ,水50mlを加え,時計皿で覆い,硫酸30mlをかき
混ぜながら少量ずつ加え,次の塩酸50mlをかき混ぜながら少量ずつ加える。ビーカーを穏やかに加
熱して試料を分解し,硝酸10mlを加えた後,時計皿の下面を水で洗って時計皿を取り除き,引き続
き加熱して硫酸の白煙を発生させる。放冷した後,ビーカーの内壁を少量の水で洗い,再び加熱して
濃厚な硫酸の白煙を約5分間発生させる。放冷した後,水200mlを加えてよくかき混ぜ,穏やかに加
熱して可溶性塩類を溶解する(1)。
注(1) この溶液に未分解残さが認められない場合には,次のb)及びc)の操作は行わない。
b) 溶液をろ紙(5種C)を用いてろ過し,ろ紙及び未分解残さを温めた塩酸 (1+4) 及び温水で洗浄して
ろ液及び洗液をビーカー (500ml) に集めて保存する。
c) 残さをろ紙とともに白金るつぼ(30番)に移し入れ,乾燥した後,徐々に加熱してろ紙を灰化する。
放冷した後,硫酸 (1+1) 2,3滴で残さを湿し,ふっ化水素酸1〜2mlを加え,加熱して二酸化けい素
及び硫酸を揮散させる。放冷した後,白金るつぼに二硫酸カリウム3gを加え,加熱して徐々に温度を
上げ,約700℃で加熱して残さを融解する。放冷した後,白金るつぼをb)で保存しておいたろ液及び
洗液中に入れ,加熱して融成物を溶解し,るつぼを水で洗って取り出す。
d) 常温まで冷却した後,溶液を500mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
5.2
チタンの還元 チタンの還元は,次のいずれかの手順によって行う。
a) 試料中にパナジウム,モリブデン,又はすずを含有していない場合
1) 5.1d)で得た溶液から50mlを分取して三角フラスコ (500ml) に移し入れ,塩酸60mlを加え,水で液
6
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
量を約200mlとする。
2) アルミニウム[2.1)]約3gを加え,還元用器具(3.)のゴム栓をはめ込んで加熱し,アルミニウムが分解
し始めたら熱源から降ろして放置し,アルミニウムの分解が終了したら再び加熱して1〜2分間沸騰
させた後,流水中で常温まで冷却する。
b) 試料中にバナジウム,モリブデン又はすずを含有する場合
1) 5.1d)で得た溶液から50mlを分取してビーカー (300ml) に移し入れ,水酸化ナトリウム溶液を滴加
して中和し,さらに,その10mlを過剰に加え,加熱して1〜2分間沸騰させた後,常温まで冷却す
る。
2) 沈殿をろ紙(5種B又は6種)を用いてこし分け,炭酸ナトリウム溶液でビーカー内を1回,ろ紙
及び沈殿を2,3回洗浄する。ろ液及び洗液は捨てる。
3) 沈殿に温水を吹き付けて元のビーカーに洗い落とし,ろ紙に付着している沈殿は,温めた塩酸 (1
+1) 20mlを注いで溶解し,溶液を元のビーカーに受ける。ろ紙を温めた塩酸 (1+4) で洗浄して洗
液を元のビーカーに合わせる。ビーカーを穏やかに加熱して沈殿を完全に溶解した後,三角フラス
コ (500ml) に移し入れ,塩酸50mlを加え,水で液量を約200mlとする。
4) a)2)の操作を行う。
5.3
滴定5.2のa)2)又はb)4)で得た溶液が入っている三角フラスコから還元用器具を取り外し,直ちに指
示薬としてチオシアン酸アンモニウム溶液10mlを加え,1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液
[2.m)]で滴定し,約1分間赤褐色が残る点を終点とし,1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液の使用
量を求める。
6. 空試験 試薬だけを用いて5.1〜5.3の手順に従って試料と同じ操作を試料と併行して行う。
7. 計算 試料中のチタン含有率を,次の式によって算出する。
100
10
/
1
m
597
001
.
0
F
)
V
V
(
Ti
2
6
5
×
×
×
×
−
=
ここに, Ti: 試料中のチタン含有率 [% (m/m) ]
V5: 5.3で得た1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液の使
用量 (ml)
V6: 6.で得た1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液の使用
量 (ml)
F2: 1/30mol/λ硫酸アンモニウム鉄 (III) 標準溶液のファクター
m: 試料はかり取り量 (g)
7
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2(規定) チタン定量方法−
アルミニウム還元硫酸アンモニウム鉄 (III) 滴定法(2)
序文 この附属書は,1983年に第1版として発行されたISO 7692, Ferrotitanium−Determinaton of titanium
content−Titrimetric methodを翻訳し,技術的内容及び規格票の様式を変更することなく作成した日本工業
規格である。
なお,この附属書で点線の下線を施してある“参考”は,原国際規格にはない事項である。
1. 目的及び適用範囲 この附属書2(規定)は,フェロチタン中のチタンを滴定法で定量する方法につ
いて規定する。
この方法は,チタン含有率20〜75% (m/m) のフェロチタンに適用する。
参考 ISOの原文では,80% (m/m) であるが,ISO 5454及びJIS G 2309の製品規格上限値である75%
(m/m) とした。
2. 引用規格
ISO 3713 : 1987 Ferroalloys−Sampling and preparation of samples−General rules
参考 ISOの原文では,注(1)現在案文作成中になっている。
ISO 5454 : 1980 Ferrotitanium−Specification and conditions of delivery
JIS G 2309 フェロチタン
3. 原理 試料を硫酸,ふっ化水素酸,硝酸及び塩酸に溶解する。妨害元素(クロム,バナジウム,モリ
ブデン及びすず)が存在する場合には,過酸化水素を共存させてチタンを水酸化チタンとして分離する。
チタンを二酸化炭素又は窒素の雰囲気中でアルミニウムによってチタン (III) に還元する。チタン (III)
を,チオシアン酸塩を指示薬として硫酸アンモニウム鉄 (III) 標準溶液で滴定する。
4. 試薬 分析に際しては,特に規定しない限り,保証された分析級の試薬及び蒸留水又はそれに相当す
る水だけを使用する。
4.1
アルミニウム純度99.5% (m/m) 以上,厚さ0.05mmのはく(箔)状のものでチタンを含有しないも
の。
4.2
炭酸水素ナトリウム (NaHC03)
4.3
硝酸 密度1.42g/ml
4.4
硫酸 密度1.84g/ml
4.5
塩酸 密度1.19g/ml
4.6
ふっ化水素酸 密度1.14g/ml
4.7
硫酸 (1+1) 水1容に硫酸(4.4)1容を少量ずつかき混ぜなから加え,かき混ぜなから冷却する。
4.8
硫酸 (1+4) 水4容に硫酸(4.4)1容を少量ずつかき混ぜながら加え,かき混ぜなから冷却する。
4.9
水酸化ナトリウム溶液 100g/λ
4.10 水酸化ナトリウム溶液 20g/λ
8
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.11 チオシアン酸アンモニウム溶液 100g/λ
4.12 窒素 酸素を含まず(10ppm/V以下),純度99.998% (m/m) のもの。又は同様な純度をもつ二酸化炭
素。
4.13 過酸化水素 30%又は原液
4.14 硫酸アンモニウム鉄 (III) 標準溶液 硫酸アンモニウム鉄 (III) [Fe2 (SO4)3 (NH4)2SO4・24H2O] 10.2g
をビーカー (400ml) にはかり取って,冷水100ml及び硫酸(4.7)50mlを加えて溶解し,水を用いて1 000ml
の全量フラスコに移し入れ,水で標線まで薄める。
この溶液1mlは,チタン約1mgに相当する。
4.15 硫酸鉄 (III) 溶液 純鉄2gに塩酸(4.5)50mlを加えて溶解し,硝酸(4.3)10mlを加えて酸化し,硫酸
(4.7)40mlを加え,硫酸の白煙が発生するまで加熱する。放冷して水で200mlに薄め,15分間煮沸する。
冷却した後,水を用いて500mlの全量フラスコに移し入れ,水で標線まで薄める。
4.16 スポンジチタン又は金属チタン 純度99.9% (m/m) 以上のもの。
4.17 チタン標準溶液 (0.500gTi/λ) この溶液は,次に示すいずれかの方法によって調製する。
4.17.1 酸化チタン (IV) を用いる場合 あらかじめ800℃で2時間焼成してある酸化チタン (IV) 0.8340±
0.0002gをはかり取って,同質のふた付き白金るつぼ又は板状のふたがついた体積170mlの石英るつぼ若
しくは石英コニカルフラスコに移し入れる。これに二硫酸カリウム10gを加え,穏やかに加熱して融解す
る。
放冷した後,石英るつぼ及び石英コニカルフラスコの場合には,直接温水100mlを加えて融成物を溶解
し,ビーカー (400ml) に静かに移し入れる。石英るつぼ及び石英コニカルフラスコを水で数回洗う。
白金るつぼを使用した場合には,放冷した後,温水100mlの入ったビーカー (400ml) に入れて融成物を
溶解する。融成物が溶解してからるつぼを水で数回洗浄して取り出す。
硫酸(4.8)50mlを融成物の溶液に加え,熱板上で穏やかに加熱して透明な溶液とする。冷却した後,水を
用いて1 000mlの全量フラスコに移し入れ,水で標線まで薄める。
4.17.2 ヘキサフルオロチタン酸カリウムを用いる場合 あらかじめ105℃で2時間乾燥しておいたヘキサ
フルオロチタン酸カリウム [K2TiF6] 2.5060±0.0002gをはかり取って白金るつぼに移し入れる。白金るつぼ
をビーカー (100ml) に入れ,硫酸(4.8)50mlを加え,ヘキサフルオロチタン酸カリウムが溶解するまで(15
分間)加熱する。冷却した後,るつぼを取り出して水で数回洗浄する。この溶液を水を用いて1 000mlの
全量フラスコに移し入れ,水で標線まで薄める。
5. 装置 通常,分析室で使用する器具及び次の器具を使用する。
5.1
ポリテトラフルオロエチレン (PTFE) ビーカー 体積250mlのもの。
5.2
ガラスビーカー 体積600mlのもの。
5.3
フラスコ 平底でコニカル又は円筒状で体積500mlのもの。
5.4
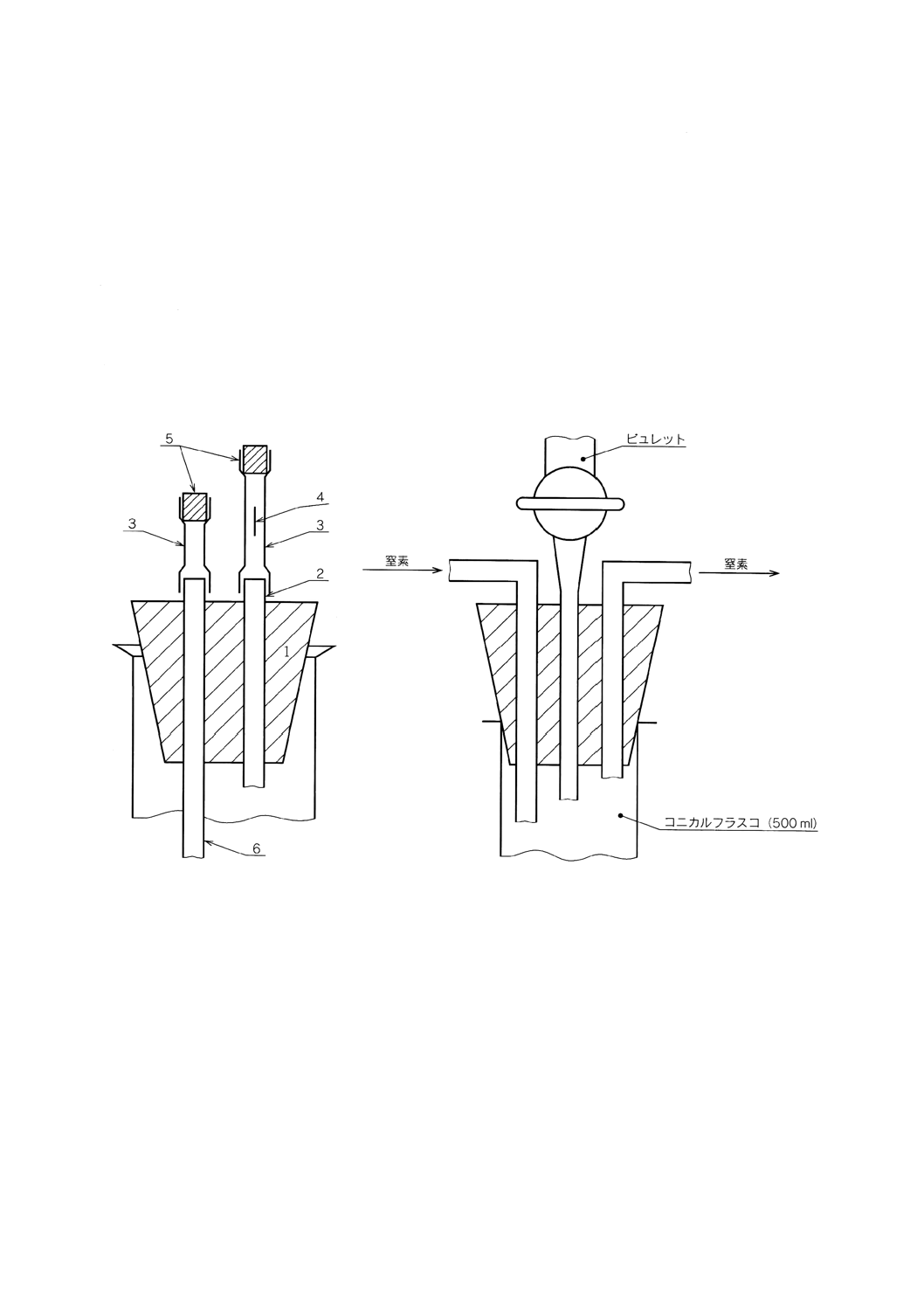
Bunsen弁又はGoeckel球管冷却器 Bunsen弁(附属書2図1参照)は,2本のガラス管(2及び6)
が通ったゴム栓(1)から成っている。ゴム管(3)がそれらのガラス管に固定してあり,それらの上端はガラス
栓(5)で閉じてある。ガラス管(2)に固定してあるゴム管(3)は,長さ方向の中央部に安全かみそりで長さ10
〜15mmのスリット(4)が切り込まれている。管(6)は,滴定溶液の表面上5cmのところに端末があり,ガス
をゴム管(3)の方向へ流すとき,ゴム管(3)を取り替えることによって窒素又は二酸化炭素を送り込むことが
できる。
5.5
磁気かき混ぜ器 PTFEでカバーした回転子付き
9
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.6
窒素雰囲気中における滴定用装置(附属書2図2参照)
6. 試料 ISO 3713に従って調製した160μmのふるい目を通過した粉末を使用する。又は,JIS G 2309
による。
7. 操作
7.1
試料のはかり取り 試料のはかり取り量は,1±0.000 2gとする。
7.2
空試験 空試験は,試料だけを用いないで,試料と同じ操作を同じ試薬の同量を用いて,試料の分
析と併行して行う。
7.3
確認試験 チタン含有率既知の同じ種類の1個又は数個の試料を用い,チタンの定量を,試料の分
析と併行して同じ操作で実施することによって,作業する操作の確認試験を行う。
7.4
硫酸アンモニウム鉄 (III) 標準溶液の標定
7.4.1
スポンジチタンを使用する方法 スポンジチタン(4.16)を0.75〜0.85gはかり取って定量する。
7.4.2
標準チタン溶液を使用する方法 ピペットを用いてチタン標準溶液(4.17)50mlをコニカルフラス
コ (500ml) に取り,硫酸(4.7)5ml及び塩酸(4.5)80mlを加える。次に7.5.3.2の手順に従って操作する。
標準溶液を調製する際に使用した試薬中にチタンが含有しなかったことを確認するために同じ条件で空
試験を行う。
7.5
定量
7.5.1
試料の分解 はかり取った試料を250mlのPTFEビーカーに移し入れ,水約20ml及び硫酸(4.7)35ml
を加える。反応が緩慢な場合には,塩酸(4.5)20ml及びふっ化水素酸(4.6)1mlを加える。ビーカーをPTFE
製のふたで覆う。溶液を冷却しながら硝酸(4.3)5mlを滴加して酸化する。
反応が終了したら,ふたを取り去って硫酸の白煙が発生するまで穏やかに加熱する。引き続き約5分間
加熱する。放冷し,塩酸(4.5)30mlを加え,穏やかに振り混ぜ,ふたをして熱板上で加熱して溶液を透明に
する。塩酸(4.5)5mlでふたを洗浄する。冷却した後,水を用いて溶液を500mlの全量フラスコに移し入れ,
水で標線まで薄める(溶液Aとする。)。
試料中のチタンの予想含有率が,20〜45% (m/m) の範囲であるならば,溶液Aから正確に50mlを分取
する。チタンの予想含有率が,45〜75% (m/m) の範囲であるならば,溶液Aから正確に25mlを分取する。
この分取した溶液は,硫酸鉄 (III) 溶液(4.15)10mlの入っているビーカー (250ml) に移し入れ,妨害元
素を含む場合には,7.5.2の手順に従って分離するか,又は分離操作が必要ない場合にはコニカルフラスコ
(500ml) に移し入れて7.5.3.1の手順に従って操作する。
7.5.2
妨害元素の分離 ビーカー (250ml) に分取した溶液Aに水酸化ナトリウム溶液(4.9)50ml及び過酸
化水素(4.13)2mlを加え,加熱して5分間沸騰を続ける。
沈殿を沈降させ,2枚の迅速ろ紙又はろ紙パルプを加えたろ紙を用いてろ過する。水酸化物の沈殿をす
べてろ紙上に移してから,ろ紙と沈殿を水酸化ナトリウム溶液(4.10)で洗浄し,さらにビーカーと沈殿を水
酸化ナトリウム溶液(4.10)約10mlで6回洗浄する。
漏斗の脚部を水で洗浄し,漏斗をコニカルフラスコ (500ml) の上に置く。
ビーカー (250ml) 中に塩酸(4.5)45ml及び硫酸(4.8)15mlを加えて60〜70℃に加熱する。この混酸を用い
てろ紙上の水酸化物沈殿を溶解する。その際,混酸は,10mlずつに分けて加え,各回の溶液が完全にろ紙
を通過してから次の添加を行うようにする。ビーカーが空になってから塩酸(4.5)35mlを加え,加熱し,そ
の約10mlずつを用いてろ紙を洗浄する。最後にビーカー,ろ紙及び漏斗の脚部を熱水 (70〜80℃) で洗浄
10
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
する。この際,洗浄する熱水の量は,40mlを超えてはならない。
7.5.3.2の手順に従って操作する。
7.5.3
滴定
7.5.3.1
妨害成分が共存しない場合 溶液Aから分取した溶液をコニカルフラスコ (500ml) に移し入れ,
塩酸(4.5)85ml及び水25mlを加える。
7.5.3.2の手順に従って操作する。
7.5.3.2
チタンの還元 試料溶液(7.5.2又は7.5.3.1で調製された)に炭酸水素ナトリウム(4.2)2±0.2g及
び数片に切断したアルミニウム (4.1) 4±0.2gを加える。直ちに,フラスコにBunsen弁又は飽和炭酸水素
ナトリウム溶液を充満したGoeckel球管冷却器(5.4参照)を取り付ける(1)。還元操作中かき混ぜを続ける。
注(1) 還元の終了時点で,Goeckel球管冷却器のバルブに吸引が起こるかもしれないので,そのときは,
飽和炭酸水素ナトリウム溶液を迅速に添加する必要がある。
7.5.3.3
窒素雰囲気中における滴定 アルミニウムが完全に溶解する前に,Bunsen弁の下向き管(6)を,
ゴム管(3)を取り除いて窒素源に接続する。窒素の流速を0.7±0.1λ/minに調節する。アルミニウムが溶解し
たら(泡立ちが止んだら),フラスコを完全に冷水に浸し,窒素をゴム管(3)のスリット(4)から逃避させ,
溶液を室温まで下げる(約7分間必要とする。)。
窒素の流れを中断することなく,Bunsen弁を取り外し,チオシアン酸アンモニウム溶液(4.11)10mlを加
え,磁気かき混ぜ器(5.5参照)の回転子を挿入する。下向き管(6)を冷水で洗浄し,窒素源に接続したBunsen
弁を再び滴定装置に接続する(附属書2図2参照)。
磁気かき混ぜ器(5.5)の上に置き,硫酸アンモニウム鉄 (III) 標準溶液(4.14)で,永続的な桃色が生じるま
で滴定する。
8. 結果の表示
8.1
硫酸アンモニウム鉄 (III) 標準溶液が,スポンジチタンで標定されている場合 硫酸アンモニウム
鉄 (III) 標準溶液が,スポンジチタン(7.4.1参照)で標定されている場合には,次の式によってチタン含
有率 [% (m/m)] を計算する。
[m1/ (V2−V1)] × [(V3−V1) /m2] ×100
ここに, V1: 空試験で消費した硫酸アンモニウム鉄 (III) 標準溶液(4.14)の
量 (ml)
V2: スポンジチタンを使用して標定した際に消費した硫酸アンモ
ニウム鉄 (III) 標準溶液(4.14)の量 (ml)
V3: 試料溶液で消費した硫酸アンモニウム鉄 (III) 標準溶液(4.14)
の量 (ml)
m1: 7.5.1で分取した溶液中に含まれるスポンジチタン(4.16)の量
(g)
m2: 7.5.1で分取した溶液中に含まれるフェロチタンの量 (g)
8.2
硫酸アンモニウム鉄 (III) 標準溶液が,チタン標準溶液で標定されている場合 硫酸アンモニウム
鉄 (III) 標準溶液が,チタン標準溶液(7.4.2参照)で標定されている場合には,次の式によってチタン含
有率 [% (m/m) ] を計算する。
[m1/ (V2−V1)] × [(V3−V4) /m2] ×100
ここに, V1: 標定の際の試薬の空試験で消費した硫酸アンモニウム鉄
(III) 標準溶液(4.14)の量 (ml)
V2: チタン標準溶液(4.17)50mlを滴定した際に消費した硫酸アン
モニウム鉄 (III) 標準溶液(4.14)の量 (ml)
11
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V3: 試料溶液で消費した硫酸アンモニウム鉄 (III) 標準溶液
(4.14)の量 (ml)
V4: 空試験で消費した硫酸アンモニウム鉄 (III) 標準溶液 (4.14)
の量 (ml)
m1: チタン標準溶液(4.17)50ml中に含まれるチタンの量 (g)
m2: 7.5.1で分取した溶液中に含まれるフェロチタンの量 (g)
9. 結果の報告 試験結果の報告は,次の情報を記載する。
a) この規格に準拠したこと。
b) 試料の識別に関すること。
c) 分析結果。
d) 定量時に注目された非定常的な事項。
e) この規格に規定されていない操作又は任意的な操作の詳細な事項。
附属書2図1 Bunsen弁
附属書2図2 窒素雰囲気中における滴定用装置
12
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3(規定) 炭素定量方法−燃焼−赤外線吸収法
1. 要旨 試料を酸素気流中で高温に加熱し,炭素を二酸化炭素及び一酸化炭素とし,これを酸素ととも
に赤外線吸収セルに送り,二酸化炭素及び一酸化炭素による赤外線吸収量を測定する。
2. 装置 装置は,JIS Z 2615の6.9[赤外線吸収法(積分法)]の6.9.3(装置)又は6.10[赤外線吸収法
(循環法)]の6.10.2(装置)による。
3. 器具及び材料 器具及び材料は,JIS Z 2615の5.(器具及び材料)及び6.9.2(材料)による。
4. 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,0.1mgのけたまで読み
取る。
参考 試料はかり取り量は,一般的には,0.5g又は1.0gである。
5. 操作
安全上での警告 燃焼操作においては,高温に加熱された磁器燃焼ボート又は磁器燃焼るつぼの取扱いに
は,必ずるつぼ挟みなどを使用し,やけどをしないように注意しなければならない。また,過剰の酸素排
気の取扱いに留意して火災発生の防止に努めなければならない。
5.1
準備操作 準備操作は,JIS Z 2615の6.9.4(予備操作)又は6.10.3(予備操作)による。ただし,
管状電気抵抗加熱炉を用いる場合には,燃焼管内温度を1 200〜1 300℃とする。
5.2
定量操作 定量操作は,次のいずれかの手順によって行う(1)。
注(1) 日常作業にあっては,作業時間の初期,中期及び後期に,必ず炭素含有率既知の試料を用いて
分析試料と同じ操作を行い,装置及びその他が正常に作動しているかどうかを試験しなければ
ならない。
a) 管状電気抵抗加熱炉を使用する積分法の場合
1) 試料をはかり取って磁器燃焼ボートに移し入れる。
2) 助燃剤(2)を正確に1〜2gをはかり取って1)の磁器燃焼ボート中の試料の上に置く。
注(2) 助燃剤は,JIS Z 2615の5.(器具及び材料)の (13) (助燃剤)に示されたものから最も適した
もの(一般に,銅,すずなど)を選び,その使用量は,使用する装置に最適な量をあらかじめ
調査しておく。
3) 燃焼管の入口部を開いて試料及び助燃剤の入った磁器燃焼ボートを燃焼管内の適切な部位に挿入し,
直ちに気密に栓をする。
4) 適切な量の酸素を流し,生成した燃焼生成ガスを酸素とともに赤外線吸収セルに送り込む。
5) 燃焼生成ガス中の二酸化炭素及び一酸化炭素含有量に相当する赤外線吸収量の積分値を指示値とし
て読み取る。
b) 高周波誘導加熱炉を使用する積分法の場合
1) 試料をはかり取って磁器燃焼るつぼに移し入れる。
2) 助燃剤(3)を正確に1〜2gをはかり取って1)の磁器燃焼るつぼ中の試料の上に置く。
注(3) 助燃剤は,JIS Z 2615の5.(器具及び材料)の(13)(助燃剤)に示されたものから最も適したも
13
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の(一般に,タングステンとすずとの混合物,銅など。)を選び,その使用量は,使用する装置
に最適な量をあらかじめ調査しておく。
3) 試料及び助燃剤の入った磁器燃焼るつぼを高周波誘導加熱炉の受台上に置き,操作ハンドルを操作
して加熱コイルの中心部に挿入し,燃焼管を閉じる。
4) JIS Z 2615の6.9の6.9.5(定量操作)の(2)及び(3)の手順に従って操作する。
c) 高周波誘導加熱炉を使用する循環法の場合 JIS Z 2615の6.10.4(定量操作)に従って操作する。
6. 空試験 試料に添加するのと同量の助燃剤だけを用いて試料と同じ操作を試料と併行して行う。
7. 計算 計算は,JIS Z 2615の6.9.7(計算)又は6.10.6(計算)による。
14
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4(規定) けい素定量方法−二酸化けい素重量法
1. 要旨 試料を硫酸と塩酸とで分解し,加熱して硫酸白煙を発生させ,けい素を不溶性のけい酸とする。
沈殿をこし分けた後,強熱して二酸化けい素とし,その質量をはかる。
次にふっ化水素酸を加え,加熱して二酸化けい素を四ふっ化けい素として蒸発揮散させ,強熱した後,
その質量をはかる。
2. 試薬 試薬は,次による。
a) 塩酸
b) ふっ化水素酸
c) 硫酸 (1+1, 1+4)
d) 過酸化水素
e) 洗浄溶液 塩酸2,過酸化水素2及び水96の割合で混合する。
3. 試料はかり取り量 試料はかり取り量は,3.0gとし,1mgのけたまで読み取る。
4. 操作
4.1
試料の分解 試料をはかり取ってビーカー (500ml) に移し入れ,時計皿で覆い,硫酸 (1+4) 100ml
を加え,かき混ぜながら塩酸30mlを少量ずつ加えた後,加熱して分解する。室温まで冷却した後,溶液
をかき混ぜながら過酸化水素10mlを少量ずつ加えてチタンなどを酸化し,再び加熱して過酸化水素を分
解する。時計皿の下面及びビーカーの内壁を少量の水で洗って時計皿を取り除き,再び加熱して蒸発し,
硫酸の濃厚な白煙を約5分間発生させた後,室温まで放冷する。
4.2
けい酸の分離 4.1で得た溶液に塩酸20ml及び水約200mlを加えてかき混ぜ,時計皿で覆い,穏や
かに加熱して可溶性塩類を溶解し,更に約1分間煮沸する(1)。直ちに沈殿をろ紙(5種B)でこし分け,
ビーカー内壁に付着した沈殿をゴム付きガラス棒でこすってろ紙上に移し,ろ紙及び沈殿を初めは温めた
洗浄溶液[2.e)]で洗液に過チタン酸の黄色が認められなくなるまで洗浄し,次に温水で十分に洗浄する。
注(1) 長時間の加熱は,不溶性けい酸が再び可溶性になるおそれがあるので,加熱溶解はできるだけ
短時間で行い,直ちにろ過する。
4.3
ひょう量 ひょう量は,次の手順によって行う。
a) 4.2で得た沈殿をろ紙とともに白金るつぼ(30番)に移し入れ,加熱してろ紙を乾燥した後,低温で
灰化する。
b) 1100℃以上で30〜45分間強熱し,デシケーター中で常温まで放冷した後,その質量をはかる。この操
作を恒量となるまで繰り返す。
c) 白金るつぼ中の沈殿を硫酸 (1+1) 2,3滴で湿し,ふっ化水素酸5mlを加え,穏やかに加熱して二酸
化けい素及び硫酸を揮散させる。
d) 1 100℃以上で強熱し,デシケーター中で常温まで放冷した後,その質量をはかる。この操作を恒量と
なるまで繰り返す。
c) b)で得た質量からd)で得た質量を差し引く。
15
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5. 空試験 試料を用いないで,試料と同じ操作を試料と併行して行う。
6. 計算 試料中のけい素含有率を,次の式によって算出する。
100
m
4
467
.
0
)
m
m
(
Si
2
1
×
×
−
=
ここに,
Si: 試料中のけい素含有率 [% (m/m)]
m1: 4.3.e)で得た質量 (g)
m2: 5.で得た質量 (g)
m: 試料はかり取り量 (g)
16
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書5(規定) けい素定量方法−
チタン分離モリブドけい酸青吸光光度法
1. 要旨 試料をふっ化水素酸と硝酸とで分解し,過マンガン酸カリウムを加えて加熱し,チタンを加水
分解させてろ別する。ろ液に七モリブデン酸六アンモニウムを加え,けい素をモリブドけい酸とした後,
L (+) −酒石酸及び還元試薬を加えてモリブドけい酸青を生成させ,光度計を用いてその吸光度を測定す
る。
2. 試薬 試薬は,次による(1)。
注(1) 試薬は,けい素含有率の低いものを使用し,水は蒸留水を用いる。また調製した試薬溶液は,
ポリエチレン容器に保存する。
a) 塩酸 (1+1)
b) 硝酸 (1+1)
c) ふっ化水素酸 (1+2)
d) ほう酸
e) アンモニア水 (1+1)
f)
過マンガン酸カリウム溶液 (30g/λ)
g) 七モリブデン酸六アンモニウム溶液 七モリブデン酸六アンモニウム四水和物106gを約800mlの温
水に溶解し,室温まで冷却した後,水で液量を1 000mlとする。
この溶液は,使用の都度,ろ紙(5種B)でろ過して用いる。
h) 還元試薬溶液 亜硫酸水素ナトリウム30g,亜硫酸ナトリウム1g及び1−アミノ−2−ナフト−ル−4
−スルホン酸0.50gを,水約180ml中に加え,約50℃に加熱して溶解する。室温まで冷却した後,水
で液量を200mlとし,ろ過する。
この溶液は,調製した後,10日以上経過したものは使用してはならない。
i)
L (+) −酒石酸溶液 (200g/λ)
j)
標準けい素溶液A(100μgSi/ml) あらかじめ1 000℃で強熱し,デシケーター中で常温まで放冷した
二酸化けい素0.2140gをはかり取って白金るつぼ(30番)に移し入れ,炭酸ナトリウム1gを加えて混
合し,加熱して融解する。放冷した後,温水約100mlを入れたポリエチレンビーカー (200ml) 中に浸
し,水浴上で温めて融成物を溶解した後,白金るつぼを水洗して取り出す。常温まで冷却した後,溶
液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
k) 標準けい素溶液B(10μSi/ml) 標準けい素溶液A[j)]を使用の都度,必要量だけ水で正確に10倍に薄
めて標準けい素溶液Bとする。
3. 試料はかり取り量 試料はかり取り量は,0.50gとし,0.1mgのけたまで読み取る。
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり取ってポリエチレンビーカー (200ml) に移し入れ,ポリエチレン時計皿で覆い,水40ml,
17
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ふっ化水素酸 (1+2) 5ml及び硝酸 (1+1) 1mlを加え,沸騰水浴中で加熱して分解した後,水約100ml
及びほう酸5gを加えて振り混ぜる。
b) 沸騰水浴中で加熱してほう酸を溶解する。
4.2
チタンの分離 4.1で得た溶液に過マンガン酸カリウム溶液を滴加し,溶液が微紅色を呈してからさ
らに過剰に5,6滴加えた後,ときどき振り混ぜながら沸騰水浴中で約90分間加熱してチタンを完全に加
水分解させる。室温まで冷却した後,時計皿の下面を少量の水で洗って時計皿を取り除く。溶液をろ紙パ
ルプを入れたろ紙(5種B)とポリエチレン漏斗とを用いて別のポリエチレンビーカー (500ml) にろ過し,
ろ紙と沈殿を液量が約200mlになるまで水で洗浄する。
4.3
呈色 4.2で得た溶液のpHが0.8〜1.5の範囲にあることを確認した後(2), 七モリブデン酸六アンモ
ニウム溶液[2.g)]5mlを正確に加えてよく振り混ぜ,20〜30℃で15〜20分間放置する。L (+) −酒石酸溶
液5mlを加えて振り混ぜ,次に還元試薬溶液[2.h)]3mlを正確に加えてよく振り混ぜた後,溶液を250mlの
全量フラスコに水を用いて移し入れ,水で標線まで薄め,20〜30℃で約10分間放置する。
注(2) このときpHが0.8以下の場合には,アンモニア水 (1+1) を用いて0.8〜1.5に調節する。また,
pHが1.5以上の場合には,塩酸 (1+1) を用いて0.8〜1.5に調節する。
4.4
吸光度の測定 4.3で得た溶液の一部を光度計の吸収セル (10mm) に取り,水を対照液として,けい
素含有率が0.05% (m/m) 未満の場合には,波長が810nm付近の,けい素含有率が0.05% (m/m) 以上の場合
には,波長650nm付近の吸光度を測定する。
5. 空試験 6.の検量線の作成操作において得られる標準けい素溶液を添加しない溶液の吸光度を,空試
験の吸光度とする。
6. 検量線の作成 検量線の作成は,次のいずれかによる。
6.1
試料中のけい素含有率が0.05% (m/m) 未満の場合 数個のポリエチレンビーカー (200ml) を準備
し,標準けい素溶液B[2.k)]0〜25.0ml(けい素として0〜250μg)を段階的に加え,水で液量を約40mlとし
た後,ポリエチレン時計皿で覆い,ふっ化水素酸 (1+2) 5ml及びほう酸5gを加える。以下,4.1.b)〜4.4
の手順に従って試料と同じ操作を試料と併行して行い,得た吸光度とけい素標準溶液として加えたけい素
量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
6.2
試料中のけい素含有率が0.05% (m/m) 以上の場合 数個のポリエチレンビーカー (200ml) を準備
し,標準けい素溶液A[2.j)]0〜5.0ml(けい素として0〜500μg)を段階的に加え,水で液量を約40mlとし
た後,ポリエチレン時計皿で覆い,ふっ化水素酸 (1+2) 5ml及びほう酸5gを加える。以下,4.1.b)〜4.4
の手順に従って試料と同じ操作を試料と併行して行い,得た吸光度とけい素標準溶液として加えたけい素
量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
7. 計算 4.4及び5.で得た吸光度と6.で作成した検量線とからけい素量を求め,試料中のけい素含有率を,
次の式によって算出する。
100
2
1
×
−
=
m
A
A
Si
ここに, Si: 試料中のけい素含有率 [% (m/m)]
A1: 試料溶液中のけい素検出量 (g)
A2: 空試験液中のけい素検出量 (g)
18
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m: 試料はかり取り量 (g)

19
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書6(規定)
マンガン定量方法−過マンガン酸吸光光度法
1. 要旨 試料を硫酸と塩酸とで分解し,過酸化水素でチタンなどを酸化した後,加熱して硫酸白煙を発
生させる。過よう素酸ナトリウムを加えて煮沸してマンガンを過マンガン酸に酸化し,光度計を用いてそ
の吸光度を測定する。
2. 試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硫酸 (1+1)
c) 過酸化水素
d) 過よう素酸ナトリウム溶液 (50g/λ)
e) 亜硝酸ナトリウム溶液 (100g/λ)
f)
尿素溶液 (100g/λ)
g) 標準マンガン溶液 (100μgMn/ml) マンガン[99.5% (m/m) 以上]0.100gをはかり取ってビーカー
(300ml) に移し入れ,時計皿で覆い,硫酸 (1+1) 20mlを加え,穏やかに加熱して分解する。常温まで
冷却した後,時計皿の下面及びビーカーの内壁を水で洗浄し,時計皿を取り除く。溶液を100mlの全
量フラスコに水を用いて移し入れ,水で標線まで薄めて原液 (1000μg/ml) とする。この原液を使用の
都度,必要量だけ水で正確に10倍に薄めて標準マンガン溶液とする。
3. 試料はかり取り量 試料はかり取り量は,試料中のマンガン含有率に応じて附属書6表1によって,
0.1mgのけたまで読み取る。
附属書6表1 試料はかり取り量
マンガン含有率% (m/m)
試料はかり取り量g
0.05以上1.0未満
1.0
1.0 以上2.0以下
0.50
4. 操作
4.1
試料溶液の調製 試料をはかり取ってビーカー (300ml) に移し入れ,時計皿で覆い,水約20ml,硫
酸 (1+1) 20ml及び塩酸 (1+1) 10mlを加え,穏やかに加熱して分解する。少し放冷した後,過酸化水素
5mlを少量ずつ加えてチタンなどを酸化し,再び加熱して過酸化水素を分解する。時計皿の下面及びビー
カーの内壁を少量の水で洗浄し,時計皿を取り除く。引き続き加熱して硫酸の白煙を2〜3分間発生させる。
放冷した後,塩酸 (1+1) 20ml及び水約50mlを加えて振り混ぜ,穏やかに加熱して可溶性の塩類を溶解す
る。常温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
4.2
呈色 呈色は,次の手順によって行う。
a) 4.1で得た溶液を乾いたろ紙(5種B)でろ過し,初めのろ液は捨て,次のろ液から正確に20mlをビ
ーカー (200ml) に取り,硫酸 (1+1) 10mlを加える。
b) 加熱して硫酸の白煙を2〜3分間発生させる。放冷した後,水約60mlを加え,時計皿で覆い,加熱し
て2〜3分間沸騰させる。過よう素酸ナトリウム溶液10mlを加え,引き続き沸騰する程度に加熱し,
20
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
溶液が過マンガン酸の赤紫を呈してから5分間以上(1)加熱してマンガンを完全に過マンガン酸に酸化
する。この溶液を流水中で常温まで冷却し,尿素溶液を加えた後,100mlの全量フラスコに水を用い
て移し入れ,水で標線まで薄める。
注(1) 試料中のマンガン含有率が0.1% (m/m) 未満の場合は,10〜20分間加熱する。
4.3
吸光度の測定 吸光度の測定は,次の手順によって行う。
a) 4.2b)で得た溶液の一部を光度計の吸収セル (10ml) に取り,水を対照液として,波長530nm付近の吸
光度を測定する。
b) 全量フラスコに残った溶液を振り混ぜながら,亜硝酸ナトリウム溶液を1滴ずつ加え,過マンガン酸
の色を完全に消した後,溶液を約1分間振り混ぜて亜硝酸ナトリウムの分解による泡立ちを消失させ
る。溶液の一部を光度計の吸収セル (10mm) に取り,水を対照液として,波長530nm付近の吸光度を
測定する。
c) a)で得た吸光度からb)で得た吸光度を差し引く。
5. 空試験 6.の検量線の作成操作において得られる標準マンガン溶液を添加しない溶液の吸光度を,空
試験の吸光度とする。
6. 検量線の作成 数個のビーカー (200ml) を用意し,標準マンガン溶液 [2.g)] 0〜20.0ml(マンガン量
として0〜2000μg)を段階的に取り,硫酸 (1+1) 10mlを加える。以下,4.2b)〜4.3c)の手順に従って試料
と同じ操作を試料と併行して行い,得た吸光度と標準マンガン溶液として加えたマンガン量との関係線を
作成し,その関係線を原点を通るように平行移動して検量線とする。
7. 計算 4.3c)及び5.で得た吸光度と5.で作成した検量線とからマンガン量を求め,試料中のマンガン含
有率を,次の式によって算出する。
100
5
1
2
1
×
×
−
=
m
A
A
Mn
ここに, Mn: 試料中のマンガン含有率 [% (m/m)]
A1: 分取した試料溶液中のマンガン検出量 (g)
A2: 分取した空試験液中のマンガン検出量 (g)
m: 試料はかり取り量 (g)
21
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書7(規定)
りん定量方法−チタン分離モリブドりん酸青吸光光度法
1. 要旨 試料をふっ化水素酸と硝酸とで分解し,硫酸を加え,加熱して硫酸の白煙を発生させた後,水
酸化ナトリウムを加えてチタンなどを沈殿させ,ろ別する。ろ液を塩酸で酸性とし,塩化鉄(III)加え,ア
ンモニア水でアルカリ性とし,りんを水酸化鉄と共沈させてこし分ける。沈殿を硝酸で溶解し,過塩素酸
を加え,加熱して過塩素酸の白煙を発生させた後,亜硫酸水素ナトリウムで鉄 (III) を還元する。七モリ
ブデン酸六アンモニウムを加えてりんをモリブドりん酸とした後,硫酸ヒドラジニウムで還元してモリブ
ドりん酸青を生成させ,光度計を用いてその吸光度を測定する。
2. 試薬 試薬は,次による。
a) 塩酸 (1+1)
b) 硝酸
c) 硝酸 (1+2, 1+50)
d) ふっ化水素酸
e) 過塩素酸
f)
硫酸 (1+1)
g) アンモニア水 (1+1)
h) 水酸化ナトリウム溶液 (200g/λ)
i)
塩化鉄 (III) 溶液 塩化鉄 (III) 六水和物10gを塩酸 (1+1) 10mlに溶解し,水で液量を100mlとする。
j)
亜硫酸水素ナトリウム溶液 (100g/λ)
k) 呈色試薬溶液 あらかじめ次の二つの溶液を調製しておき,使用の都度,七モリブデン酸六アンモニ
ウム溶液25ml,硫酸ヒドラジニウム溶液10ml及び水65mlの割合で混合する。
1) 七モリブデン酸六アンモニウム溶液 七モリブデン酸六アンモニウム四水和物20gを温水約100ml
に溶解し,硫酸 (1+1) 700mlを加え,室温まで冷却した後,水で液量を1 000mlとする。
2) 硫酸ヒドラジニウム溶液 (1.5g/λ)
l)
標準りん溶液 (100μgP/ml) あらかじめ110℃で恒量となるまで乾燥してデシケーター中で常温ま
で放冷したりん酸二水素カリウム0.4394gをはかり取り,ビーカー (200ml) に移し入れ,水約100ml
に溶解し,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
3. 試料はかり取り量 試料はかり取り量は,1.0gとし,1mgのけたまで読み取る。
4. 操作
22
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.1
試料溶液の調製 試料をはかり取って白金皿(100番)又はポリテトラフルオロエチレンビーカー
(100ml) に移し入れ,ポリエチレン時計皿で覆い,硝酸20mlを加えた後,ふっ化水素酸を少量ずつ滴加し
て試料を分解する。ポリエチレン時計皿の下面を水で洗って時計皿を取り除き,硫酸 (1+1) 10mlを加え,
再び加熱して硫酸の白煙を発生させ,引き続き加熱して硫酸の白煙がほとんど出なくなるまで乾固する。
放冷した後,塩酸 (1+1) 20mlを加え,穏やかに加熱して可溶性塩類を溶解し,溶液をビーカー (500ml) に
移し入れ,温水で液量を約200mlとする。
4.2
チタンなどの分離 4.1で得た溶液に水酸化ナトリウム溶液を加えて中和し,更に10mlを過剰に加
え,時計皿で覆い,穏やかに加熱して3〜5分間沸騰させる。常温まで冷却した後,時計皿の下面を水で洗
って時計皿を取り除き,沈殿及び溶液を500mlの全量フラスコに水を用いて移し入れ,水で標線まで薄め
る。この溶液を乾いたろ紙(5種B)でろ過し,初めのろ液は捨て,その後のろ液を乾いた250mlの全量
フラスコに受けて標線まで入れる。
4.3
りんの共沈分離 りんの共沈分離は,次の手順によって行う。
a) 4.2で得た溶液の全量をビーカー (500ml) に移し入れ,全量フラスコ内を少量の水で洗ってビーカー
内の溶液に合わせ,溶液を塩酸 (1+1) で中和し,更に10mlを過剰に加えた後,時計皿で覆い,穏や
かに加熱して1〜2分間沸騰させる。
b) 塩化鉄 (III) 溶液 [2.i)] 5mlを加え,アンモニア水 (1+1) で中和し,更に5mlを過剰に加え,穏やか
に加熱して約1分間沸騰させる。
c) 静置して沈殿が沈降した後,時計皿の下面を少量の温水で洗って時計皿を取り除き,沈殿をろ紙(5
種A)を用いてこし分け,温水で2, 3回洗浄する,洗液は捨てる。元のビーカーを漏斗の下に置き,温
めた硝酸 (1+2) をろ紙上に注いで沈殿を溶解し,溶液を元のビーカーに受ける。ろ紙を温めた硝酸 (1
+50) で十分に洗浄し,洗液をろ液に合わせる。過塩素酸10mlを加え,時計皿で覆い,加熱して過塩
素酸の蒸気がビーカー内壁に逆流する状態を約10分間持続させる。
d) 放冷した後,温水約30mlを加えて塩類を溶解し,加熱して1〜2分間沸騰させる。時計皿の下面を水
で洗って時計皿を取り除き,溶液をろ紙(5種A)でろ過し,温水で十分に洗浄する。ろ液及び洗液
を100mlの全量フラスコに水を用いて移し入れ,常温まで冷却した後,水で標線まで薄める。
4.4
呈色 呈色は,次の手順によって行う。
a) 4.3d)で得た溶液から10mlを分取して100mlの全量フラスコに移し入れる。
b) 亜硫酸水素ナトリウム溶液10mlを加え,沸騰水浴中で溶液が無色になるまで加熱した後,呈色試薬
溶液[2.k)]25mlを加えて再び沸騰水浴中で約15分間加熱する。この溶液を流水で常温まで冷却した後,
水で標線まで薄める。
4.5
吸光度の測定 4.4b)で得た溶液の一部を光度計の吸収セル (20mm) に取り,水を対照液として波長
825nm付近の吸光度を測定する。
5. 空試験 試薬だけを用いて,試料と同じ操作を試料と併行して行う。
6. 検量線の作成 検量線の作成は,次の手順によって行う。
a) 数個のビーカー (100ml) を準備し,標準りん溶液[2.1)]0〜5.0ml(りんとして0〜500μg)を段階的に
加える。これらに塩化鉄 (III) 溶液[2.i)]5ml及び過塩素酸5mlを加え,時計皿で覆い,加熱して過塩
素酸の白煙を発生させる。放冷した後,時計皿の下面を水で洗って時計皿を取り除き,温水約30ml
を加えて塩類を溶解する。常温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,
23
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
水で標線まで薄める。
b) この溶液を10ml分取して100mlの全量フラスコに移し入れ,以下,4.4b)及び4.5の手順に従って試料
と同じ操作を試料と併行して行い,得た吸光度と標準りん溶液として加えたりん量との関係線を作成
し,その関係線を原点を通るように平行移動して検量線とする。
7. 計算 4.5及び5.で得た吸光度と6.で作成した検量線とからりん量を求め,試料中のりん含有率を,次
の式によって算出する。
100
20
1
2
1
×
×
−
=
m
A
A
P
ここに,
P: 試料中のりん含有率 [% (m/m)]
A1: 分取した試料溶液中のりん検出量 (g)
A2: 分取した空試験液中のりん検出量 (g)
m: 試料はかり取り量 (g)
24
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書8(規定) 硫黄定量方法−燃焼−赤外線吸収法
1. 要旨 試料を酸素気流中で高温に加熱し,硫黄を二酸化硫黄とし,これを酸素とともに赤外線吸収セ
ルに送り,二酸化硫黄による赤外線吸収量を測定する。
2. 装置 装置は,JIS Z 2616の7.7[赤外線吸収法(積分法)]の(2)(装置)又は7.8[赤外線吸収法(循
環法)]の(2)(装置)による。
3. 試料はかり取り量 試料はかり取り量は,使用する装置に最も適した量とし,0.1mgのけたまで読み
取る。
参考 試料はかり取り量は,一般的には,0.5g又は1.0gである。
4. 操作
安全上での警告 燃焼操作においては,高温に加熱された磁器燃焼るつぼの取扱いには,必ずるつぼ挟 み
などを使用し,やけどをしないように注意しなければならない。また,過剰の酸素排気の取扱いに留意し
て火災発生の防止に努めなければならない。
4.1
準備操作 準備操作は,JIS Z 2616の7.7(3)(予備操作)又は7.8(3)(予備操作)による。
4.2
定量操作 定量操作は,JIS Z 2616の7.7(4)(定量操作)又は7.8(4)(定量操作)による(1)(2)。
注(1) 日常作業にあっては,作業時間の初期,中期及び後期に,必ず硫黄含有率既知の試料を用いて
分析試料と同じ操作を行い,装置及びその他が正常に作動しているかどうかを試験しなければ
ならない。
(2) 助燃剤は,JIS Z 2616の6.12(助燃剤)に示されたものから最も適したもの(一般に,タング
ステン,すずなど。)を選び,その使用量は,使用する装置に最適な量をあらかじめ調査してお
く。
5. 空試験 試料に添加するのと同量の助燃剤だけを用いて試料と同じ操作を試料と併行して行う。
計算 計算は,JIS Z 2616の7.7(6)(計算)又は7.8(6)(計算)による。
25
G 1319 : 2000
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
原案作成委員会 構成表
社団法人 日本チタン協会技術委員会分析分科会 構成表
氏名
所属
(主査)
稲 本 勇
新日本製鐵株式会社・株式会社日鐵テクノリサーチ
岡 圭 男
住友金属工業株式会社
福 田 建一郎
株式会社住友シチックス尼崎
乾 道 春
株式会社神戸製鋼所・株式会社コベルコ科研
久留須 一 彦
古河電気工業株式会社
豊 嶋 雅 康
住友軽金属工業株式会社
服 部 兆 隆
東邦チタニウム株式会社・株式会社テスコ
前 田 繁 則
株式会社ジャパンエナジー分析センター
横 溝 耿
三菱マテリアル株式会社
吉 川 裕 泰
日本鋼管株式会社
(事務局)
伊 藤 均
社団法人日本チタン協会
日本フェロアロイ協会分析専門委員会 構成表
氏名
所属
(主査)
鈴 木 邦 輝
日本重化学工業株式会社
大 槻 孝
前社団法人日本鉄鋼連盟
稲 本 勇
社団法人日本鉄鋼連盟
戸 舘 一
社団法人日本海事検定協会
金 築 宏 治
株式会社神戸製鋼所
杉 山 鉄 男
大平洋金属株式会社
佐 藤 正 文
昭和電工株式会社
松 本 誠
中央電気工業株式会社
河 野 政 治
日本電工株式会社
紅 谷 紀 生
日本鋼管株式会社
見 持 洋 司
日本重化学工業株式会社
佐 藤 哲 哉
通商産業省基礎産業局
大 嶋 清 治
通商産業省工業技術院標準部
橋 本 繁 晴
財団法人日本規格協会
(関係者)
増 田 正 純
通商産業省工業技術院標準部
(事務局)
奥 山 満 之
日本フェロアロイ協会