G1237-1997
(1)
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。これによってJIS G 1237−1981は改正され,この規格に置き換えられる。
今回の改正では,誘導結合プラズマ発光分光分析方法を新たに規定し,使用されていない五酸化ニオブ
重量法及びピロガロール吸光光度法を廃止している。
JIS G 1237には,次に示す附属書がある。
附属書1 スルホクロロフェノールS吸光光度法
附属書2 スルホクロロフェノールS抽出吸光光度法
附属書3 誘導結合プラズマ発光分光分析方法
日本工業規格 JIS
G1237-1997
鉄及び鋼−ニオブ定量方法
Iron and steel−Methods for
determination of niobium content
1. 適用範囲 この規格は,鉄及び鋼中のニオブの定量方法について規定する。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
JIS G 1201 鉄及び鋼の分析方法通則
JIS K 0116 発光分光分析通則
JIS Z 8402 分析・試験の許容差通則
3. 一般事項 定量方法に共通な一般事項は,JIS G 1201による。
4. 定量方法の区分 ニオブの定量方法は,次のいずれかによる。
(1) スルホクロロフェノールS吸光光度法 この方法は,ニオブ含有率0.5% (m/m) 以上2.5% (m/m) 以下
の試料に適用し,その定量方法は附属書1による。ただし,この方法は,呈色のために分取した試料
溶液中に,タングステンが1mg,チタンが0.2mg,モリブデンが0.1mg,銅が0.05mg,ジルコニウム
が0.01mg又はタンタルが0.005mg以上を,単独ででも共存する試料には適用できない。
(2) スルホクロロフェノールS抽出吸光光度法 この方法は,ニオブ含有率0.01% (m/m) 以上0.5% (m/m)
以下の試料に適用し,その定量方法は附属書2による。ただし,この方法は,呈色のために分取した
試料溶液中に,マンガン,ニッケル,銅,アルミニウム,コバルト又はバナジウムが,それぞれ5mg
以上,すず,ひ素,タングステン又はイットリウムが,それぞれ1mg以上,チタンが0.2mg以上,モ
リブデンが0.1mg以上,ジルコニウムが0.02mg以上又はタンタルが0.005mg以上を,単独ででも共
存する試料には適用できない。
(3) 誘導結合プラズマ発光分光分析方法 この方法は,ニオブ含有率0.001% (m/m) 以上2.5% (m/m) 以下
の試料に適用し,その定量方法は附属書3による

2
G1237-1997
附属書1 スルホクロロフェノールS吸光光度法
1. 要旨 試料を王水で分解し,硫酸及びりん酸を加え,加熱して三酸化硫黄の白煙を発生させて硝酸を
除去する。L (+) -酒石酸及び塩酸を加えて塩類を溶解し,L (+) -アスコルビン酸及びエチレンジアミン四
酢酸二水素二ナトリウム(以下,EDTA2Naという。)で鉄などをマスキングした後,スルホクロロフェノ
ールSを加えてスルホクロロフェノールSニオブ錯体を生成させ,光度計を用いて,その吸光度を測定す
る。
2. 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1)
(3) 硝酸
(4) 王水(塩酸3,硝酸1)
(5) 混酸(硫酸2,りん酸1,水7)
(6) 鉄 できるだけ純度の高い鉄で,ニオブ含有率が0.001% (m/m) 以下であるもの。
(7) L (+) -酒石酸溶液 (200g/l)
(8) L (+) -アスコルビン酸溶液 (10g/l) この溶液は,使用の都度調製する。
(9) EDTA2Na溶液 エチレンジアミン四酢酸二水素二ナトリウム二水和物1gを水に溶解し,水で液量を
100mlとする。
(10)スルホクロロフェノールS溶液 スルホクロロフェノールS (C22H10N4O16S4Na4Cl2) 0.05gを水に溶解し,
水で液量を100mlとする。褐色瓶に保存し,使用の都度,乾いたろ紙(5種A)でろ過する。
(11) 標準ニオブ溶液 (100μgNb/ml) ニオブ[99.9% (m/m) 以上]0.100gをはかり採って白金皿(100番)
に移し入れ,白金製のふたで覆う。ふっ化水素酸10ml及び硝酸数滴を加え,加熱して分解する。白
金製のふたの下面を水で洗ってふたを取り除き,硫酸(1+1)5mlを加え,加熱して三酸化硫黄の白
煙を発生させる。放冷した後,水で冷却しながらL (+) -酒石酸溶液 (200g/l) 20mlを加えてかき混ぜ
る。さらに,流水中で常温まで冷却し,溶液を1 000mlの全量フラスコに酒石酸溶液 (8g/l) を用いて
移し入れ,L (+) -酒石酸溶液 (8g/l) で標線まで薄める。
3. 試料はかり採り量 試料はかり採り量は,附属書1表1による。
附属書1表1 試料はかり採り量
ニオブ含有率
% (m/m)
試料はかり採り量
g
0.5以上1.2未満
0.20
1.2以上2.5以下
0.10
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採ってビーカー (200ml) に移し入れ,時計皿で覆う。
3
G1237-1997
(2) 王水15mlを加え,加熱して分解した後,混酸25.0mlを加える。
(3) 引き続き加熱して濃縮し(1),三酸化硫黄の白煙が発生する直前に時計皿の下面を水で洗って時計皿を
取り除き,引き続き加熱して三酸化硫黄の白煙を4,5分間発生させた後,乾いた時計皿で覆って放冷
する。
(4) L (+) -酒石酸溶液 (200g/l) 40ml及び塩酸15mlを加え,穏やかに加熱して塩類を溶解する。
(5) 常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除き,溶液を250mlの全量フラスコに
水を用いて移し入れ,水で標線まで薄める(2)。
注(1) 試料中にニオブの炭化物などが存在する場合には,熱源温度を500℃以上として完全に分解する。
(2) この溶液を長時間放置すると,ニオブが加水分解して定量値が低値となる場合があるので,直
ちに4.2の操作を行う。
4.2
呈色 4.1(5)で得た溶液から5mlを分取し,50mlの全量フラスコに移し入れ,塩酸(1+1)15ml及
びL (+) -アスコルビン酸溶液 [2.(8)] 5mlを加えて振り混ぜる。3分間放置した後,EDTA2Na溶液 [2.(9)]
5mlを加えて振り混ぜ,スルホクロロフェノールS溶液 [2.(10)] を3.0ml又は4.0ml(3)加えて振り混ぜ,水
で標線まで薄める。次に,あらかじめ60〜65℃に調節してある水浴中に全量フラスコを5分間浸して加熱
した後,常温まで冷却する。
注(3) スルホクロロフェノールS溶液 [2.(10)] は,使用する試薬のロットによってその使用量が3.0ml
では不足することがあるので,必要によっては4.0mlを添加する。
4.3
吸光度の測定 4.2で得た溶液の一部を,呈色1分間後に光度計の吸収セル (1cm) に取り,5.で得た
空試験溶液を対照液として波長655nm付近における吸光度を測定する。
5. 空試験 試料の代わりに同量の鉄 [2.(6)] をはかり採り,4.1〜4.3の手順に従って試料と同じ操作を試
料と併行して行う。
6. 検量線の作成 ニオブ含有率の範囲ごとに6個のビーカー (200ml) を準備し,それぞれにはかり採っ
た試料と同量の鉄 [2.(6)] をはかり採って移し入れ,標準ニオブ溶液 [2.(11)] を0,5.0,10.0,15.0,20.0
及び25.0ml(ニオブとして0,500,1 000,1 500,2 000及び2 500μg)加え,時計皿で覆う。以下,4.1(2)
〜4.3の手順に従って試料と同じ操作を試料と併行して行い(4),得た吸光度と標準ニオブ溶液として加えた
ニオブ量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
注(4) 標準ニオブ溶液を添加しない溶液を対照液として吸光度を測定する。
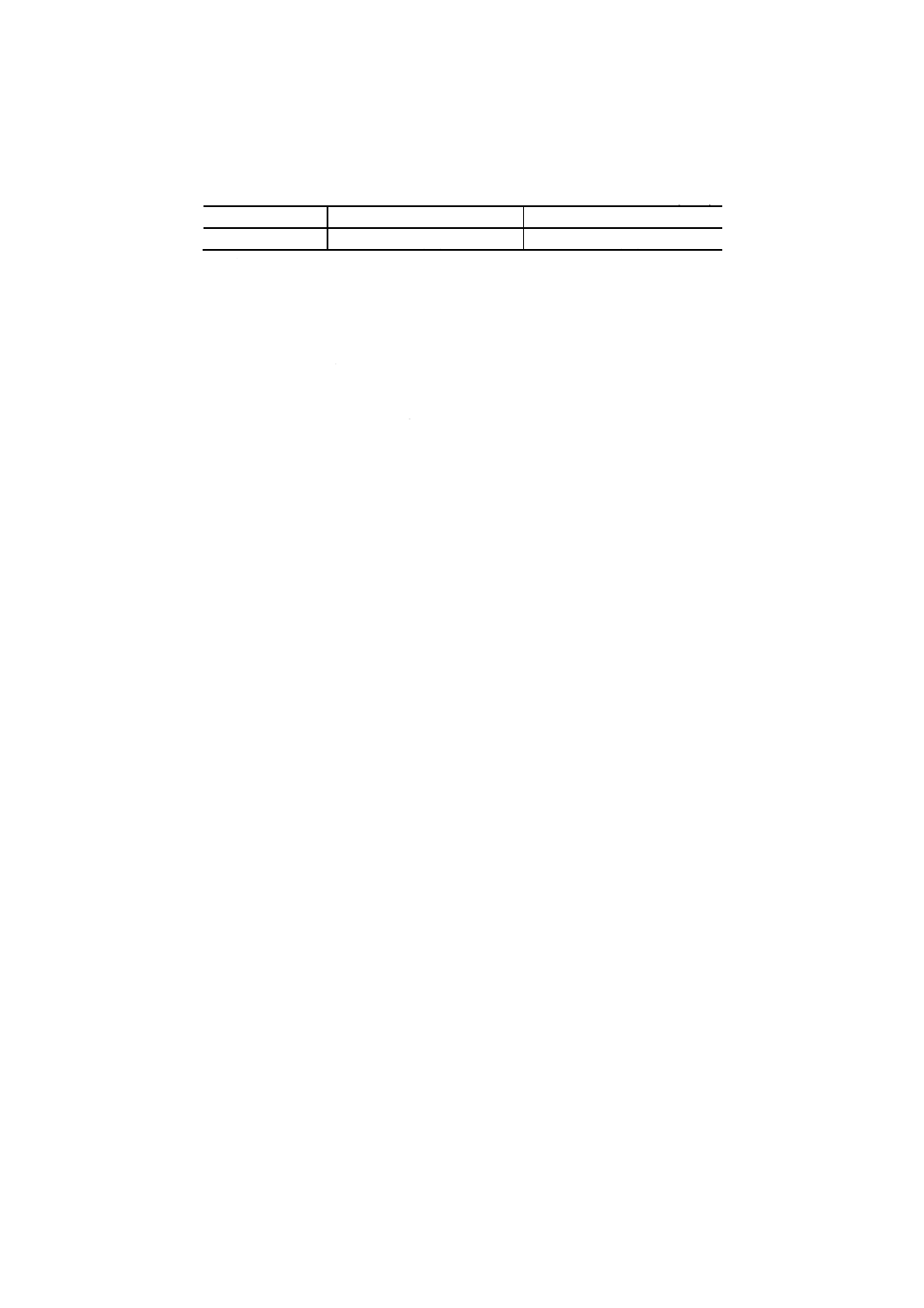
7. 計算 4.3で得た吸光度と6.で作成した検量線とからニオブ量を求め,試料中のニオブ含有率を,次の
式によって算出する。
100
250
5
Nb
×
×
m
A
=
ここに, Nb: 試料中のニオブ含有率 [% (m/m) ]
A: 分取した試料溶液中のニオブ検出量 (g)
m: 試料はかり採り量 (g)
4
G1237-1997
8. 許容差 許容差(5)は,附属書1表2による。
附属書1表2 許容差
単位% (m/m)
ニオブ含有率
室内再現許容差
室間再現許容差
0.5以上2.5以下
D [0.007 2× (Nb) +0.000 9]
D [0.010 0× (Nb) +0.004 7]
注(5) 許容差計算式中のDは,D (n,0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一分析室内における分析回数,室間再現許容差の場合は分析に関与
した分析室数である。
また, (Nb) は,許容差を求めるニオブ含有率 [% (m/m)] である。
参考 この許容差は,ニオブ含有率0.6% (m/m) 以上2.5% (m/m) 以下の試料を用い,共同実験した結
果から求めたものである。
この方法の英文名は,Method for determination of niobium content−Sulfochlorophenol S
spectro-photometric method である。

5
G1237-1997
附属書2 スルホクロロフェノールS抽出吸光光度法
1. 要旨 試料を王水で分解し,硫酸及びりん酸を加え,加熱して三酸化硫黄の白煙を発生させて硝酸を
除去する。L (+) -酒石酸及び塩酸を加えて塩類を溶解し,L (+) -アスコルビン酸及びエチレンジアミン四
酢酸二水素二ナトリウム(以下,EDTA2Naという。)で鉄などをマスキングした後,スルホクロロフェノ
ールSを加え,生成するスルホクロロフェノールSニオブ錯体を1-ブタノールで抽出し,光度計を用いて
有機相の吸光度を測定する。
2. 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1)
(3) 硝酸
(4) 王水(塩酸3,硝酸1)
(5) 混酸(硫酸2,りん酸1,水7)
(6) 鉄 できるだけ純度の高い鉄で,ニオブ含有率0.001% (m/m) 以下であるもの。
(7) L (+) -酒石酸溶液 (200g/l)
(8) L (+) -アスコルビン酸溶液 (10g/l) この溶液は,使用の都度調製する。
(9) EDTA2Na溶液 エチレンジアミン四酢酸二水素二ナトリウム二水和物1gを水に溶解し,水で液量を
100mlとする。
(10) エタノール (99.5)
(11) 1-ブタノール(別名:n-ブチルアルコール)
(12) 3-メチル-1-ブタノール(別名:イソアミルアルコール)
(13)スルホクロロフェノールS溶液 スルホクロロフェノールS (C22H10N4O16S4Na4C12) 0.05gを水に溶解し,
水で液量を100mlとする。褐色瓶に保存し,使用の都度,乾いたろ紙(5種A)でろ過する。
(14) 標準ニオブ溶液 (100μgNb/ml) ニオブ[99.9% (m/m) 以上]0.100gをはかり採って白金皿(100番)
に移し入れ,白金製のふたで覆う。ふっ化水素酸10ml及び硝酸数滴を加え,加熱して分解する。白
金製のふたの下面を水で洗ってふたを取り除き,硫酸(1+1)5mlを加え,加熱して三酸化硫黄の白
煙を発生させる。放冷した後,水で冷却しながらL (+) -酒石酸溶液 (200g/l) 20mlを加えてかき混ぜ
る。さらに,流水中で常温まで冷却し,溶液を1 000mlの全量フラスコに酒石酸溶液 (8g/l) を用いて
移し入れ,L (+) -酒石酸溶液 (8g/l) で標線まで薄める。
3. 試料はかり採り量 試料はかり採り量は,附属書2表1による。
附属書2表1 試料はかり採り量
ニオブ含有率
% (m/m)
試料はかり採り量
g
0.01以上0.10未満
0.50
0.10以上0.25未満
0.20
0.25以上0.50以下
0.10
6
G1237-1997
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採ってビーカー (200ml) に移し入れ,時計皿で覆う。
(2) 王水15mlを加え,加熱して分解した後,混酸25.0mlを加える。
(3) 時計皿の下面を水で洗って時計皿を取り除き,引き続き加熱して濃縮し(1),三酸化硫黄の白煙を4,5
分間発生させた後,乾いた時計皿で覆って放冷する。
(4) L (+) -酒石酸溶液 (200g/l) 40ml及び塩酸15mlを加え,緩やかに加熱して塩類を溶解する。
(5) 常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除き,溶液を100mlの全量フラスコに
水を用いて移し入れ,水で標線まで薄める(2)。
注(1) 試料中にニオブの炭化物などが存在する場合には,熱源温度を500℃以上として完全に分解する。
(2) この溶液を長時間放置すると,ニオブが加水分解して定量値が低値となる場合があるので,直
ちに4.2の操作を行う。
4.2
呈色 4.1(5)で得た溶液から5mlを分取し,50mlの全量フラスコに移し入れ,塩酸(1+1)15ml及
びL (+) -アスコルビン酸溶液 [2.(8)] 5mlを加えて振り混ぜる。3分間放置した後,EDTA2Na溶液 [2.(9)]
5mlを加えて振り混ぜ,スルホクロロフェノールS溶液 [2.(13)] を3.0ml又は4.0ml(3)加えて振り混ぜ,水
で標線まで薄める。次に,あらかじめ60〜65℃に調節してある水浴中に全量フラスコを5分間浸して加熱
した後,常温まで冷却する。
注(3) スルホクロロフェノールS溶液 [2.(13)] は,使用する試薬のロットによってその使用量が3.0ml
では不足することがあるので,必要によっては4.0mlを添加する。
4.3
抽出 4.2で得た溶液を,分液漏斗 (100ml) に移し入れ,全量フラスコ内を水5mlで洗って合わせる。
1-ブタノール(4)を正確に20ml加え,30秒間激しく振り混ぜて静置する。二層に分離した後,下層の水相
を捨て,有機相にエタノールを正確に5ml加え,振り混ぜる(5)。
注(4) 1-ブタノールの代わりに,3-メチル-1-ブタノールを使用することができる。
(5) エタノールを加えないで,有機相を乾いたろ紙(5種A)でろ過して,水滴を除去してもよい。
4.4
吸光度の測定 4.3で得た有機相の一部を,光度計の吸収セル (1cm) に取り,5.で得た空試験液を対
照液として波長655nm付近における吸光度を測定する。
5. 空試験 試料の代わりに同量の鉄 [2.(6)] をはかり採り,4.1〜4.4の手順に従って試料と同じ操作を試
料と併行して行う。
6. 検量線の作成 ニオブ含有率の範囲ごとに6個のビーカー (200ml) を準備し,それぞれにはかり採っ
た試料と同量の鉄 [2.(6)] をはかり採って移し入れ,標準ニオブ溶液 [2.(14)] を0,1.0,2.0,3.0,4.0及
び5.0ml(ニオブとして0,100,200,300,400及び500μg)加え,時計皿で覆う。以下,4.1(b) 〜4.4の
手順に従って試料と同じ操作を試料と併行して行い(6),得た吸光度と標準ニオブ溶液として加えたニオブ
量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
注(6) 標準ニオブ溶液を添加しない溶液を対照液として吸光度を測定する。
7. 計算 4.4で得た吸光度と6.で作成した検量線とからニオブ量を求め,試料中のニオブ含有率を,次の
式によって算出する。

7
G1237-1997
100
100
5
Nb
×
×
m
A
=
ここに, Nb: 試料中のニオブ含有率 [% (m/m)]
A: 分取した試料溶液中のニオブ検出量 (g)
m: 試料はかり採り量 (g)
8. 許容差 許容差(7)は,附属書2表2による。
附属書2表2 許容差
単位% (m/m)
ニオブ含有率
室内再現許容差
室間再現許容差
0.01以上0.5以下
D [0.007 2× (Nb) +0.001 0]
D [0.024 8× (Nb) +0.000 2]
注(7) 許容差計算式中のDは,D (n,0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一分析室内における分析回数,室間再現許容差の場合は分析に関与
した分析室数である。
また,(Nb) は,許容差を求めるニオブ含有率 [% (m/m)] である。
参考 この許容差は,ニオブ含有率0.04% (m/m) 以上0.4% (m/m) 以下の試料を用い,共同実験した
結果から求めたものである。
この方法の英文名は,Method for determination of niobium content−Sulfochlorophenol S 1-butanol
extraction spectrophotometric methodである。
8
G1237-1997
附属書3 誘導結合プラズマ発光分光分析方法
1. 要旨 試料を王水で分解し,硫酸及びりん酸を加え,加熱して三酸化硫黄の白煙を発生させて塩酸及
び硝酸を除去し,L (+) -酒石酸及び塩酸を加えて塩類を溶解した後,又は試料を塩酸と硝酸の混酸で分解
してL (+) -酒石酸を添加し,溶液をろ過し,残さを二硫酸カリウムで融解してろ液に合わせた後,溶液に
イットリウムを添加し,溶液をICP発光分光分析装置のアルゴンプラズマ中に噴霧してニオブとイットリ
ウムの発光強度を測定する。
2. 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (2+100)
(3) ふっ化水素酸
(4) 硫酸 (1+1)
(5) 王水(塩酸3,硝酸1)
(6) 混酸A(硫酸2,りん酸2,水6)
(7) 混酸B(塩酸1,硝酸1,水2)
(8) 過酸化水素
(9) 過酸化水素 (1+100)
(10) 鉄 できるだけ純度の高い鉄で,ニオブ含有率0.000 1% (m/m) 以下のもの。
(11) 二硫酸カリウム
(12) L (+) -酒石酸溶液 (200g/l)
(13) しゅう酸溶液 しゅう酸二水和物35gを水に溶解し,水で液量を1lとする。
(14) イットリウム溶液 (1mgY/ml) 酸化イットリウム[99.9% (m/m) 以上]1.269 9gを塩酸(1+1)50ml
で加熱して分解し,常温まで冷却した後,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水
で標線まで薄める。
(15) 標準ニオブ溶液A (1mgNb/ml) ニオブ[99.9% (m/m) 以上]0.500 0gをはかり採って白金皿(100番)
に移し入れ,白金製のふたで覆う。ふっ化水素酸10ml及び硝酸数滴を加え,加熱して分解する。白
金製のふたの下面を水で洗ってふたを取り除き,硫酸(1+1)10mlを加え,穏やかに加熱して三酸化
硫黄の白煙を3〜5分間発生させる(1)。放冷した後,水で冷却しながら過酸化水素(1+9)100mlを加
えて塩類を溶解する。常温まで冷却した後,溶液を500mlの全量フラスコに過酸化水素(1+100)を
用いて移し入れ,過酸化水素(1+100)で標線まで薄める。
(16) 標準ニオブ溶液B (100ugNb/ml) 標準ニオブ溶液A [ (15) ] を必要量だけ,過酸化水素(1+100)で
正確に10倍に薄めて標準ニオブ溶液Bとする。この溶液は,使用の都度調製する。
注(1) 強く加熱したり,長時間白煙を発生させすぎると塩類が溶解しなくなるので,注意しなければ
ならない。
9
G1237-1997
3. 装置の性能基準 装置の性能基準は,次による。
3.1
性能基準 この規格で用いるICP発光分光分析装置は,次の性能基準のいずれをも満足するよう分
析線,励起条件,測光条件などを選定しなければならない。
(1) 定量下限域短期間安定性 鉄 [2.(10)] 0.500gをはかり採ってビーカー (200ml) に移し入れ,標準ニオ
ブ溶液B [2.(16)] を過酸化水素(1+100)で正確に20倍に薄めた溶液(5μgNb/ml)1.0ml[鋼中のニ
オブ含有率として0.001% (m/m) に相当]を添加した後,時計皿で覆う。以下,適用する試料溶液の
調製方法[5.1(1)による場合には,5.1(1) (b)〜(e),5.1(2)による場合には,5.1(2) (b)〜(g)]の手順に従
って操作した後,5.2の操作を,連続10回行い,得た10個の発光強度比と7.で作成した検量線とから
ニオブ量を求め,試料中のニオブ含有率に換算した値を小数第4位のけたまで求める。得た定量値の
標準偏差を計算し,得た標準偏差が0.000 5% (m/m) (定量下限域における標準偏差許容上限値)未満
でなければならない。
(2) 定量上限域短期間安定性 鉄 [2.(10)] 0.500gをはかり採ってビーカー (200ml) に移し入れ,標準ニオ
ブ溶液A [2.(15)] 12.5ml[鋼中のニオブ含有率として2.50% (m/m) に相当]を添加した後,時計皿で覆
う。以下,適用する試料溶液の調製方法[5.1(1)による場合には,5.1(1) (b)〜(e),5.1(2)による場合に
は,5.1 (2) (b)〜(g)]の手順に従って操作した後,5.2の操作を,連続10回行い,得た10個の発光強
度比と7. で作成した検量線とからニオブ量を求め,試料中のニオブ含有率に換算した値を小数第4
位のけたまで求める。得た定量値の標準偏差を計算し,得た標準偏差が0.015% (m/m) (定量上限域
における標準偏差許容上限値)未満でなければならない。
3.2
性能基準の調査頻度 性能基準の調査は,期間を定めて定期的に行う。分析条件を変更したときや
オーバーホールを行ったときなど,装置の状態が変わる可能性がある場合には,必ず行われなければなら
ない。
4. 試料はかり採り量 試料はかり採り量は,0.50gとする。
5. 操作
5.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
(1) 王水分解法を用いる場合
(a) 試料をはかり採ってビーカー (200ml) に移し入れ,時計皿で覆う。
(b) 王水15mlを加え,加熱して分解した後,混酸A20.0mlを加える。
(c) 時計皿の下面を水で洗って時計皿を取り除き,引き続き加熱して濃縮し(2),三酸化硫黄の白煙を4,
5分間発生させた後(3),放冷する。
(d) L (+) -酒石酸溶液10ml及び塩酸10mlを加え,穏やかに加熱して塩類を溶解する。
(e) 常温まで冷却した後,イットリウム溶液 [2.(14)] 10.0mlを加え,溶液を100mlの全量フラスコに塩
酸 (2+100) を用いて移し入れ,塩酸 (2+100) で標線まで薄める。
注(2) けい素含有率が0.5% (m/m) 以上の試料の場合には,三酸化硫黄の白煙発生の手前でふっ化水素
酸3mlを添加する。
(3) 未分解の炭化物が認められる場合には,わずかに放冷した後,硝酸5mlを加え,再び三酸化硫
黄白煙を2,3分間発生させる。
10
G1237-1997
(2) 混酸分解法を用いる場合
(a) 試料をはかり採ってビーカー (200ml) に移し入れ,時計皿で覆う。
(b) 混酸B25mlを加え,穏やかに加熱して分解し,引き続き加熱して窒素酸化物などを追い出した後,
放冷する。
(c) L (+) -酒石酸溶液10mlを加え,時計皿の下面を塩酸(2+100)で洗って時計皿を取り除く。
(d) 少量のろ紙パルプを加えてかき混ぜ,未分解物をろ紙(5種C)を用いてこし分け,ビーカー内壁
に付着した残さをゴム付きガラス棒を用いてこすり落としてろ紙上に移し,40〜60℃に加熱した塩
酸(2+100)及び温水を用いてろ紙に塩化鉄 (III) による黄色が認められなくなるまで洗浄する。
ろ液及び洗液はビーカー (200ml) に集める。残さは保存する。
(e) ろ液及び洗液を穏やかに加熱して液量が約40mlになるまで濃縮し,約40℃になるまで放冷する (4) 。
過酸化水素2mlを添加して生成したニオブの沈殿を溶解し(5),主液として保存する。
(f) (d) で保存しておいた残さを,ろ紙と共に白金るつぼ(30番)に移し入れ,加熱して乾燥し,低温
でろ紙を灰化した後,強熱する。放冷した後,残さを硫酸(1+1)2,3滴で湿し,ふっ化水素酸5
〜7mlを加え,穏やかに加熱して二酸化けい素を揮散させ,更に400℃(6)以下に加熱して硫酸を揮
散させる。放冷した後,二硫酸カリウム1.0gを加え,白金製のふたをして初めは徐々に加熱し,次
第に温度を高めて暗赤熱状に加熱して残さを融解する。放冷した後,白金るつぼにしゅう酸溶液
[2.(13)] 15mlを加え,穏やかに加熱して融成物を溶解し,溶液を(e)で保存しておいた主液に合わせ
る。
(g) 常温まで冷却した後,イットリウム溶液 [2.(14)] 10.0mlを加え,溶液を100mlの全量フラスコに塩
酸(2+100)を用いて移し入れ,塩酸(2+100)で標線まで薄める。
注(4) 過酸化水素を添加するときの液温が高すぎると過酸化水素が分解してニオブ沈殿の溶解が不完
全になるおそれがある。
(5) ニオブ沈殿の溶解が不完全な場合には,過酸化水素を追加して完全に溶解する。
(6) 400℃を超えて加熱するとニオブが飛散するので加熱温度に注意しなければならない。
5.2
発光強度の測定 5.1の(1) (e)又は(2) (g)で得た溶液(7)の一部を,ICP発光分光分析装置 [3.2] のアル
ゴンプラズマ中へ噴霧し,309.42nm,313.08nm又は319.50nmにおけるニオブの発光強度及び371.03nmに
おけるイットリウムの発光強度を測定し,イットリウムの発光強度に対するニオブの発光強度の比を求め
る。基本的な測定操作は,JIS K 0116の5.8による。
注(7) 二酸化けい素の沈殿が認められる場合には,乾いたろ紙(5種A)でこし分けた後,ろ液を用い
る。
6. 空試験 7. の検量線の作成操作において得られる標準ニオブ溶液を添加しない溶液の発光強度比を
空試験の発光強度比とする。
7. 検量線の作成 附属書3表1に示すニオブ含有率の範囲ごとに7個のビーカー (200ml) を準備し,そ
れぞれに鉄 [2.(10)] 0.500gをはかり採って移し入れ,附属書3表1に従って標準ニオブ溶液を加え,時計
皿で覆う。以下,5.1(1)の(b)〜(e)及び5.2の手順,又は5.1(2) (b)〜(g)及び5.2の手順に従って試料と同じ
操作を試料と併行して行い,得た発光強度比と標準ニオブ溶液として加えたニオブ量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
検量線は,一定時間ごと又は一定試料数ごとに検量線校正溶液(検量線用ニオブゼロ溶液及び検量線用
11
G1237-1997
最高濃度溶液)を用いて校正を行う。
附属書3表1 検量線溶液
ニオブ含有率
% (m/m)
使用する標準
ニオブ溶液
標準ニオブ溶液添加量
ml
0.001以上0.20未満
B [2.16]
0,0.5,2.0,4.0,6.0,8.0,10.0
0.20 以上2.50以下
A [2.15]
0,1.0,2.0,4.0,7.0,10.0,12.5
8. 元素間干渉補正 あらかじめ各共存元素のニオブ分析線へのスペクトル線の重なりについて調べ,ス
ペクトル線の重なりがある場合には,スペクトル線の重なり係数lj(8)を次の手順によって求める。
(1) 7. の手順に従ってニオブ含有率0.001% (m/m) 以上0.20% (m/m) 未満の検量線を作成する。
(2) 鉄と共存元素jの二元系溶液を,5.1の(1)又は(2)の手順に従って試料と同じ操作を行って,鉄含有率
と共存元素含有率の合計で100% (m/m) になるように共存元素jを段階的(3〜4水準)に加えた溶液
を調製した後,5.2の操作を行う。
(3) (2)で得た発光強度比から空試験の発光強度比を差し引いて得られる発光強度比と(1)で作成した検量
線とから見掛けのニオブ量を求め,試料中のニオブ含有率換算値⊿Nb [% (m/m)] を求める。
(4) ⊿Nb [% (m/m) ] をY軸,二元系溶液の共存元素jの試料中含有率換算値Mj [% (m/m)] をX軸として,
両者の関係を最小二乗法によって一次回帰計算を行う。
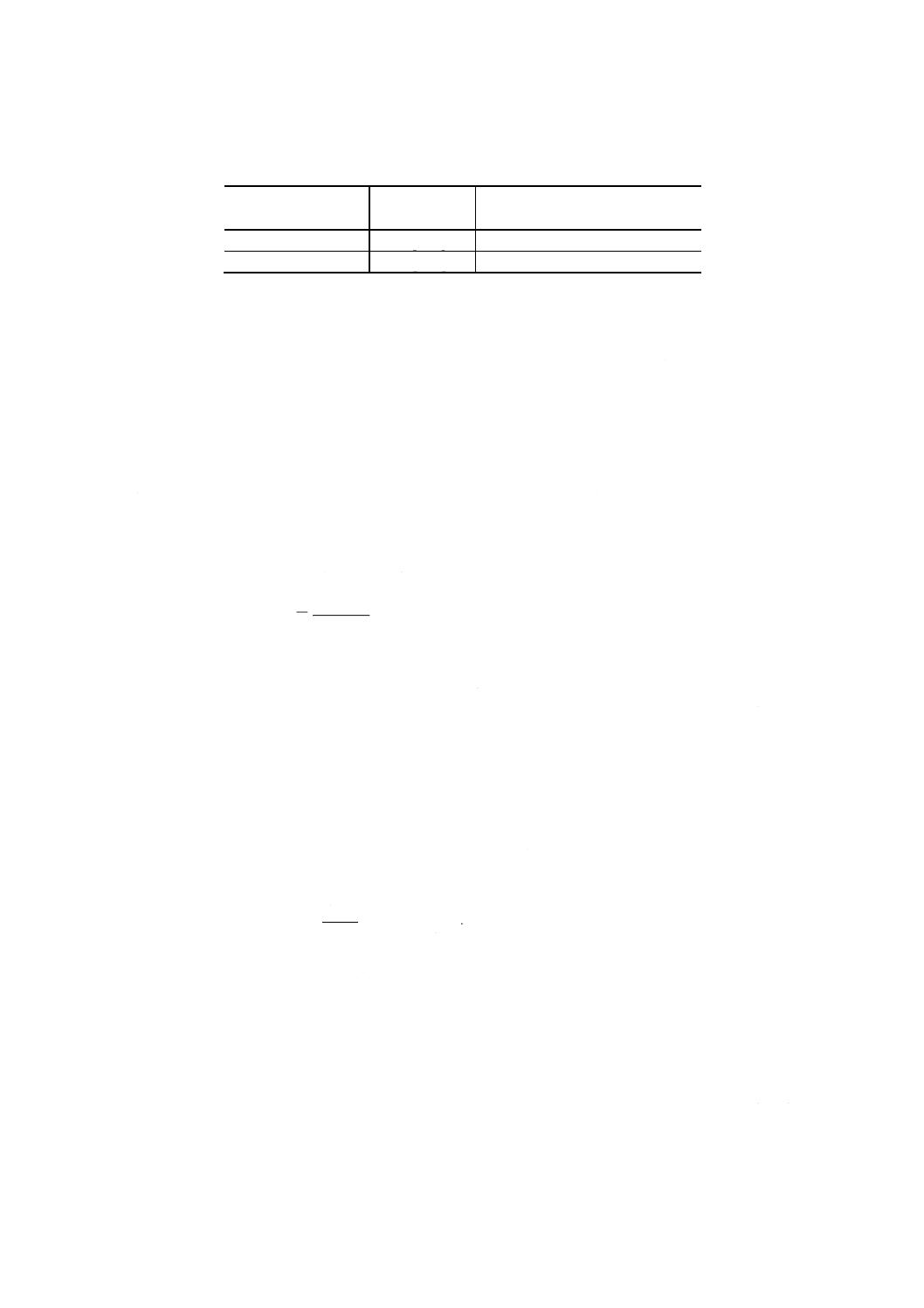
(5) 次の式によってスペクトル線の重なり補正係数ljを求める。
j
j
M
b
Nb
l
−
⊿
=
ここに,
lj: Nbに対する共存元素jのスペクトル線の重なり補正係数
⊿Nb: Fe-j二元系溶液中のニオブの見掛けの試料中含有率換算
値 [% (m/m)]
Mj: Fe-j二元系溶液中の共存元素jの試料中含有率換算値 [%
(m/m)]
b: 一次回帰によって得られた定数
注(8) スペクトル線の重なり補正係数は,装置の分解能などによって異なるので,装置ごとに実測し
て求める必要がある。
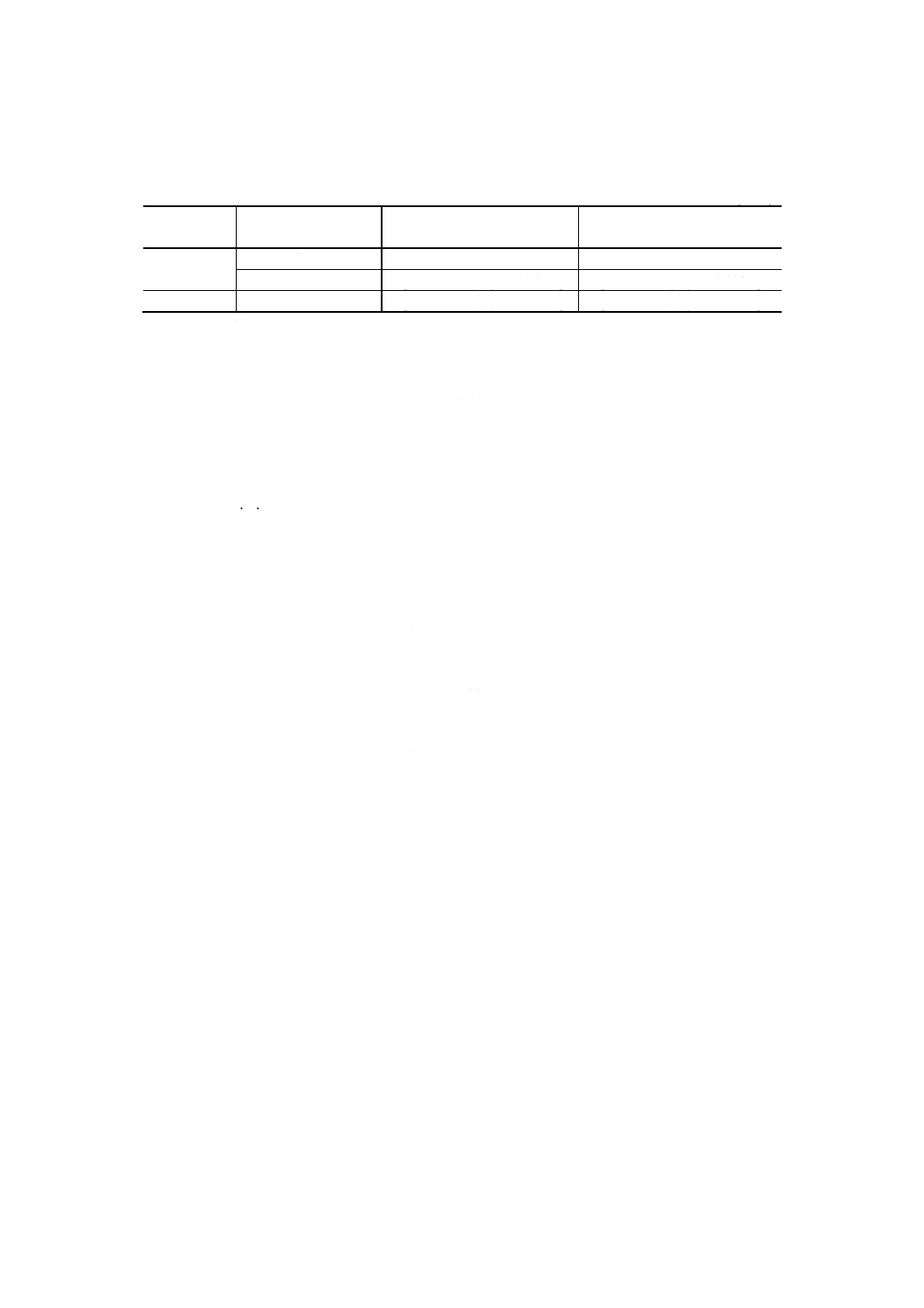
9. 計算 5.2及び6.で得た発光強度比と,7.で作成した検量線とからニオブ量を求め,試料中のニオブ含
有率を,次の式によって算出する。
∑
−
×
−
j
jM
l
m
B
A
100
Nb=
ここに, Nb: 試料中のニオブ含有率 [% (m/m)]
A: 試料溶液中のニオブ検出量 (g)
B: 空試験液中のニオブ検出量 (g)
m: 試料はかり採り量 (g)
lj: ニオブに対する共存元素jのスペクトル線の重なり補正係数
Mj: 試料中の共存元素jの含有率 [% (m/m)] (9)
注(9) 補正計算 (∑ljMj) は,スペクトル線の重なり補正係数と共存元素の含有率との積を用いて行う
ので,共存元素jの試料中含有率 [% (m/m)] を知る必要があるが,その値は,他の定量方法で
得られる値を用いる。
12
G1237-1997
10. 許容差 許容差(10)は,附属書3表2による。
附属書3表2 許容差
単位% (m/m)
試料溶液
調製方法
ニオブ含有率範囲
室内再現許容差
室間再現許容差
王水分解法
0.001以上 0.40未満 D [0.004 6× (Nb) +0.000 7]
D [0.013 4× (Nb) +0.000 3]
0.40 以上 2.5以下 D [0.008 6× (Nb) −0.000 3]
D [0.027 1× (Nb) −0.003 3]
混酸分解法
0.001以上 2.5以下
D [0.010 0× (Nb) +0.000 9]
D [0.025 9× (Nb) +0.000 1]
注(10) 許容差計算式中のDは,D (n,0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一分析室内における分析回数,室間再現許容差の場合は分析に関与
した分析室数である。
また, (Nb) は,許容差を求めるニオブ含有率 [% (m/m)] である。
参考 この許容差は,ニオブ含有率0.004% (m/m) 以上2.3% (m/m) 以下の試料を用い,共同実験した
結果から求めたものである。
この方法の英文名は,Method for determination of niobium content−Inductively coupled plasma
atomic emission spectrometric methodである。
原案作成委員会構成表
社団法人日本鉄鋼協会共同研究会鉄鋼分析部会化学分析分科会ニオブICP発光分光分析検討WG 構成表
氏名
所属
(化学分析分科会主査)
岩 田 英 夫
日本鋼管株式会社
(WGリーダー)
茂 木 文 吉
大同特殊鋼株式会社
(WGメンバー)
山 本 佳 博
新日本製鐵株式会社
小 倉 正 之
日本鋼管株式会社
岡 野 輝 雄
川崎製鉄株式会社
蔵 保 浩 文
住友金属工業株式会社
河 村 恒 夫
株式会社コベルコ科研
大 濱 熙 久
日新製鋼株式会社
余 語 英 俊
愛知製鋼株式会社
13
G1237-1997
社団法人日本鉄鋼連盟鋼材標準委員会JE4分科会 構成表
氏名
所属
(主査)
佐 伯 正 夫
新日本製鐵株式会社
(委員)
大 磯 義 和
工業技術院標準部
大 野 義 信
新日本製鐵株式会社
土 屋 武 久
新日本製鐵株式会社
船 曳 佳 弘
日本鋼管株式会社
磯 部 健
日本鋼管株式会社
岡 野 輝 雄
川崎製鉄株式会社
滝 沢 佳 郎
川崎製鉄株式会社
蔵 保 浩 文
住友金属工業株式会社
山 下 良 一
住友金属工業株式会社
金 子 晃 司
株式会社神戸製鋼所
河 村 恒 夫
株式会社コベルコ科研
伊 藤 清 孝
大同特殊鋼株式会社
藤 田 昇 平
日新製鋼株式会社
余 語 英 俊
愛知製鋼株式会社
永 井 宣太郎
日本冶金工業株式会社
(編集WG)
稲 本 勇
新日本製鐵株式会社
吉 川 裕 泰
日本鋼管株式会社
(幹事)
小 野 昭 紘
新日本製鐵株式会社(編集WG兼務)
柿 田 和 俊
社団法人日本鉄鋼連盟(編集WG兼務)
大 槻 孝
社団法人日本鉄鋼連盟(編集WG兼務)
社団法人日本鉄鋼連盟JIS三者委員会 構成表
氏名
所属
(委員長)
大河内 春 乃
科学技術庁金属材料技術研究所
(委員)
青 柳 桂 一
通商産業省基礎産業局
服 部 幹 雄
工業技術院標準部
加 山 英 男
財団法人日本規格協会
藤 貫 正
社団法人日本分析化学会
広 川 吉之助
東北大学金属材料研究所
永 山 宏
日立マテリアルエンジニアリング株式会社
束 原 巌
古河電気工業株式会社
橋 本 勝
株式会社日産アーク
岩 田 英 夫
日本鋼管株式会社
岡 野 輝 雄
川崎製鉄株式会社
蔵 保 浩 文
住友金属工業株式会社
河 村 恒 夫
株式会社コベルコ科研
成 田 正 尚
大同特殊鋼株式会社
藤 田 昇 平
日新製鋼株式会社
(幹事)
佐 伯 正 夫
新日本製鐵株式会社
(事務局)
柿 田 和 俊
社団法人日本鉄鋼連盟
大 槻 孝
社団法人日本鉄鋼連盟