G 1222 : 1999
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。これによってJIS G 1222 : 1981は改正され,この規格によって置き換えられる。
今回の改正では,国際規格との整合化を図るため,ISO規格の翻訳を附属書4として規定している。
この規格の一部が,技術的性質をもつ特許権,出願公開後の特許出願,実用新案権又は出願公開後の実
用新案登録出願に抵触する可能性があることに注意を喚起する。
通商産業大臣及び日本工業標準調査会は,このような技術的性質をもつ特許権,出願公開後の特許出願,
実用新案権又は出願公開後の実用新案登録出願にかかわる確認について,責任はもたない。
JIS G 1222には,次に示す附属書がある。
附属書1(規定) 1−ニトロソ−2−ナフトール沈殿分離四酸化三コバルト重量法
附属書2(規定) 1−ニトロソ−2−ナフトール−3,6−ジスルホン酸二ナトリウム吸光光度法
附属書3(規定) 2−ニトロソ−1−ナフトール抽出吸光光度法
附属書4(規定) イオン交換分離電位差滴定法
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
G 1222 : 1999
鉄及び鋼−コバルト定量方法
Iron and steel−Methods for determination of cobalt content
序文 この規格は,JIS G 1222 : 1981の様式を変更して附属書1(規定)〜3(規定)とし,附属書4(規
定)に1997年に第1版として発行されたISO 11653, Steel−Determination of high cobalt content−
Potentiometric titration method after separation by ion exchangeを翻訳し,技術的内容及び規格票の様式を変更
することなく作成した日本工業規格である。
1. 適用範囲 この規格は,鉄及び鋼中のコバルト定量方法について規定する。
備考 この規格の対応国際規格を,次に示す。
ISO 11653 Steel−Determination of high cobalt content−Potentiometric titration method after
separa-tion by ion exchange
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
JIS G 1201 鉄及び鋼の分析方法通則
JIS G 1257 鉄及び鋼−原子吸光分析方法
JIS Z 8402 分析・試験の許容差通則
3. 一般事項 定量方法に共通な一般事項は,JIS G 1201による。ただし,JIS G 1201は,附属書4(規
定)には適用しない。
4. 定量方法の区分 コバルトの定量方法は,次のいずれかによる。
a) 1−ニトロソ−2−ナフトール沈殿分離四酸化三コバルト重量法 この方法は,コバルト含有率0.5%
(m/m) 以上20% (m/m) 以下の試料に適用するもので附属書1(規定)による。
b) 1−ニトロソ−2−ナフトール−3,6−ジスルホン酸二ナトリウム吸光光度法 この方法は,コバルト
含有率0.1% (m/m) 以上20% (m/m) 以下の試料に適用するもので附属書2(規定)による。ただし,
この方法は,呈色のために分取した試料溶液中に,ニッケルが60mg以上,クロムが20mg以上,モ
リブデン及びバナジウムが1mg以上,チタンが0.2mg以上,タングステンが2mg以上,及びニオブ
が0.5mg以上を単独ででも共存する試料には適用できない。
c) 2−ニトロソ−1−ナフトール抽出吸光光度法 この方法は,コバルト含有率0.001% (m/m) 以上0.1%
(m/m) 以下の試料に適用するもので附属書3(規定)による。
d) イオン交換分離電位差滴定法 (ISO 11653) この方法は,コバルト含有率5.0% (m/m) 以上17.0% (m/m)
以下の試料に適用するもので附属書4(規定)による。

2
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(規定) 1−ニトロソ−2−ナフトール沈殿分離
四酸化三コバルト重量法
1. 要旨 試料を塩酸で分解し,硝酸で鉄などを酸化した後,酸化亜鉛で鉄,クロムなどを沈殿させてろ
別し,ろ液に塩酸及び1−ニトロソ−2−ナフトールを加え,生成する1−ニトロソ−2−ナフトールコバル
トの沈殿をこし分ける。この沈殿を強熱して四酸化三コバルトとし,その質量をはかる。さらに四酸化三
コバルトを塩酸で溶解し,四酸化三コバルト中の酸化モリブデン (VI) 及び酸化銅 (II) の量を求め,四酸
化三コバルトの質量から差し引く。
2. 試薬 試薬は,次による。
a) 塩酸
b) 塩酸 (1+1, 2+100)
c) 硝酸
d) 融解合剤(炭酸ナトリウム1,炭酸カリウム1)
e) 酸化亜鉛乳状液 微細粉にした酸化亜鉛50gに水300mlを加えて十分にかき混ぜ,乳状にする。
f)
1−ニトロソ−2−ナフトール溶液 1−ニトロソ−2−ナフトール (NOC10H6OH) 1.0gを酢酸15mlに溶
解し,ろ過する。この溶液は,使用の都度,調製する。
3. 試料はかり採り量 試料はかり採り量は,附属書1表1による。
附属書1表1 試料はかり採り量
コバルト含有率
% (m/m)
試料はかり採り量
g
0.5 以上
5未満
1.0
5 以上
10未満
0.50
10 以上
20以下
0.25
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 酸で分解容易な試料
1) 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆い,塩酸 (1+1) 30mlを加え,加熱
して分解する。硝酸2mlを加えて鉄などを酸化し,引き続き加熱する(1)。時計皿の下面を少量の水
で洗って時計皿を取り除き,穏やかに加熱して液面に皮膜が生じるようになるまで蒸発する。
注(1) タングステンを含む試料の場合は,タングステンが完全に黄色のタングステン酸になるまで十
分に煮沸する。
2) 放冷した後,温水約200mlを加えて塩類を溶解する。
b) 酸で分解困難な試料
1) a)1)の操作を行う。
2) 放冷した後,温水約100mlを加えて塩類を溶解する。
3) 溶液をろ紙(5種B)を用いてビーカー (300ml) にろ過し,ろ紙及び不溶解残さを塩酸 (2+100) で
3
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
数回洗浄し,ろ液と洗液を合わせて主液として保存する。
4) 不溶解残さは,ろ紙と共に白金るつぼ(30番)に移し入れ,乾燥した後,強熱してろ紙を灰化する。
放冷した後,残さの約10倍量の融解合剤 [2.d)] を加えて混合し,強熱して残さを融解する。放冷
した後,融成物を少量の塩酸 (1+1) 及び水に溶解し,溶液を3)で保存した主液に合わせる。この
溶液を穏やかに加熱して蒸発し,乾固する。
5) 放冷した後,塩酸 (1+1) 30mlを加えて塩類を溶解し,再び穏やかに加熱して液面に皮膜が生じる
ようになるまで蒸発する。
6) a)2)の操作を行う。
4.2
鉄,クロムなどの分離 鉄,クロムなどの分離は,次の手順によって行う。
a) 4.1a)2)又はb)6)で得た溶液を室温まで冷却し,かき混ぜながら酸化亜鉛乳状液 [2.e)] を少量ずつ加え,
鉄,クロムなどを完全に沈殿させた後,更に上澄み液が白濁するまで少過剰を加える。しばらく放置
して沈殿を沈降させた後,ろ紙(2種,15cm)を用いてビーカー (500ml) にろ過する。元のビーカー
と沈殿は,冷水で各3回洗浄して洗液をろ液に合わせる。ろ液及び洗液に塩酸10mlを加えた後,液
量が約100mlになるまで溶液を加熱して蒸発し,主液として保存する。
b) 沈殿とろ紙は,元のビーカーに移し入れ,塩酸 (1+1) 20mlを加えて振り混ぜ,沈殿を溶解(2)した後,
ろ紙を破砕し,水で液量を約200mlとする。
注(2) 沈殿が完全に溶解しない場合は,できるだけ少量の塩酸 (1+1) を追加して溶解する。
c) 溶液に再び酸化亜鉛乳状液 [2.e)] を少量ずつ加えて鉄,クロムなどを完全に沈殿させた後,さらに上
澄み液が白濁するまでその少過剰を加える。しばらく放置して沈殿を沈降させた後,ろ紙(2種,15cm)
を用いて1)で保存した主液の入っているビーカー (500ml) にろ過する。元のビーカーと沈殿は,冷水
で各3回洗浄し,洗液をろ液に合わせる。
d) 水で液量を約400mlとする。沈殿は,捨てる。
4.3
沈殿の生成 4.2 d)で得た溶液を時計皿で覆い,加熱して煮沸し,ビーカーを熱源から降ろして溶液
をかき混ぜながらコバルト予想含有量0.01gにつき1−ニトロソ−2−ナフトール溶液 [2.f)] を6mlの割合
で加える。ときどきかき混ぜながら30分間以上放置した後,ろ紙(5種A)を用いてこし分ける。ろ紙及
び沈殿は,初め冷水で,次に塩酸 (1+1) と水で交互に数回洗浄し,最後に温水で数回洗浄する。ろ液及
び洗液は,捨てる。
4.4
沈殿のひょう量 沈殿のひょう量は,次の手順によって行う。
a) 4.3で得た沈殿を,あらかじめ750〜850℃で強熱して恒量とした磁器るつぼ(PC1B形30ml)にろ紙
と共に移し入れる。乾燥した後,低温で加熱してろ紙を灰化する。
b) 750〜850℃に強熱し,デシケーター中で室温まで放冷した後,磁器るつぼの質量をはかる。恒量とな
るまでこの操作を繰り返す。
c) b)で得た質量から磁器るつぼの質量を差し引いて不純四酸化三コバルトの質量とする。
4.5
不純四酸化三コバルト中のモリブデン及び銅の定量 不純四酸化三コバルト中のモリブデン及び銅
の定量は,次の手順によって行う。
a) 定量用溶液の調製 4.4 c)で得た不純四酸化三コバルトを塩酸10mlで分解し,溶液を100mlの全量フ
ラスコに水を用いて移し入れ,水で標線まで薄める。
b) モリブデンの定量(3)
注(3) 試料中にモリブデンが含まれる場合に行う。
1) a)で得た溶液から20ml(4)を分取して100mlの全量フラスコに移し入れ,硝酸5ml及び塩酸 (1+1)

4
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
30mlを加える。
注(4) a)で得た溶液中に含まれるモリブデンの量によっては,20mlの分取液量を増減する必要がある。
2) 1)の溶液を用いて,JIS G 1257の附属書7(モリブデン定量方法−酸分解直接法)の操作の項の4.1
(1)(d)の“アルミニウム溶液 [2.(8)] を正確に10ml加え”から4.2(吸光度の測定)及び6.(検量線
の作成)の手順に従って操作し,4.2で得た吸光度と6.で作成した検量線(5)とからモリブデンの量を
求める。
注(5) モリブデン定量用の検量線は,鉄を添加しないで作成する。
c) 銅の定量(6)
注(6) 試料中に銅が含まれる場合に行う。
1) a)で得た溶液から20ml(7)を分取して100mlの全量フラスコに移し入れ,硝酸5ml及び塩酸 (1+1)
10mlを加え,水で標線まで薄める。
注(7) a)で得た溶液中に含まれる銅の量によっては,20mlの分取液量を増減する必要がある。
2) 1)の溶液を用いて,JIS G 1257の附属書8(銅定量方法−酸分解直接法)の操作の項の4.2(吸光度
の測定)及び6.(検量線の作成)の手順に従って操作し,4.2で得た吸光度と6.で作成した検量線(8)
とから銅の量を求める。
注(8) 銅定量用の検量線は,鉄を添加しないで作成する。
5. 空試験 空試験は,行わない。
6. 計算 試料中のコバルト含有率を,次の式によって算出する。
100
1
734
.0
}]
8
251
.1
100
(
3
500
.1
100
{
[
2
3
1
2
1
×
×
×
×
+
×
×
−
=
m
B
m
B
m
m
Co
ここに,
Co: 試料中のコバルト含有率 [% (m/m)]
m1: 不純四酸化三コバルトの質量 (g)
m2: 4.5 b)2)で得たモリブデンの量 (g)
m3: 4.5 c)2)で得た銅の量 (g)
B1: 4.5 b)1)で分取した溶液の量 (ml)
B2: 4.5 c)1)で分取した溶液の量 (ml)
m: 試料はかり採り量 (g)
参考
1.500 3: モリブデンの量を酸化モリブデン (VI) に変換する係数
1.251 8: 銅の量を酸化銅 (II) に変換する係数
7. 許容差 許容差(9)は,附属書1表2による。
附属書1表2 許容差
単位% (m/m)
コバルト含有率
室内再現許容差
室間再現許容差
0.5以上 12以下
D [0.005 6× (Co) +0.002 3]
D [0.015 4× (Co) −0.007 5]
注(9) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4[D (n, 0.95) の値]
による。nの値は,室内再現許容差の場合は同一室内における分析回数,室間再現許容差の場
合は分析に関与した分析室数である(n=2のとき,D=2.8である)。
また, (Co) は,許容差を求めるコバルト含有率 [% (m/m)] である。
5
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考 この許容差は,コバルト含有率0.23% (m/m) 以上11.3% (m/m) 以下の試料を共同実験した結果
から求めたものである。
6
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2(規定) 1−ニトロソ−2−ナフトール−3,6−ジスルホン酸
二ナトリウム吸光光度法
1. 要旨 試料を適切な酸で分解し,硝酸で鉄などを酸化する。ニッケル,酢酸ナトリウム及び1−ニト
ロソ−2−ナフトール−3,6−ジスルホン酸二ナトリウム(以下,ニトロソR塩という。)を加えてニトロ
ソR塩コバルト錯体を生成させ,光度計を用いて,その吸光度を測定する。
2. 試薬 試薬は,次による。
a) 硝酸
b) 硫酸 (1+100)
c) りん酸 (1+1)
d) 王水(塩酸3,硝酸1)
e) 混酸(硫酸3,りん酸3,水14)
f)
鉄 できるだけ純度が高い鉄で,コバルトを含有しないか,又はコバルト含有率ができるだけ低く既
知であるもの。
g) ニッケル溶液 できるだけ純度が高く,コバルト含有率が0.001% (m/m) 以下のニッケル2.000gをは
かり採ってビーカー (300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 20mlを加え,加熱して分解す
る。常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除き,溶液を500mlの全量フラス
コに水を用いて移し入れ,水で標線まで薄める。
h) 鉄溶液 できるだけ純度が高く,コバルト含有率が0.001% (m/m) 以下の鉄1.000gをはかり採ってビ
ーカー (300ml) に移し入れ,時計皿で覆い,硫酸 (1+3) 10ml及び過酸化水素5mlを加えて加熱して
分解する。引き続き加熱して過酸化水素を完全に分解し,常温まで冷却した後,時計皿の下面を水で
洗って時計皿を取り除き,溶液を500mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
この溶液1mlは,鉄2mgを含む。
i)
酢酸ナトリウム溶液 酢酸ナトリウム三水和物500gを水約800mlに溶解し,水で液量を1 000mlとす
る。
j)
ニトロソR塩溶液 ニトロソR塩 [NOC10H4OH (SO3Na) 2] 2gを温水約80mlに溶解し,室温まで冷却
した後,水で液量を100mlとする。
k) 標準コバルト溶液A (1mgCo/ml) コバルト[99.9% (m/m) 以上]1.000gをはかり採ってビーカー
(300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 20mlを加え,加熱して分解する。常温まで冷却した
後,時計皿の下面を水で洗って時計皿を取り除き,溶液を1 000mlの全量フラスコに水を用いて移し
入れ,水で標線まで薄めて標準コバルト溶液Aとする。
l)
標準コバルト溶液B (500μgCo/ml) 標準コバルト溶液A [k)] を使用の都度,必要量だけ水で正しく2
倍に薄めて標準コバルト溶液Bとする。
3. 試料はかり採り量 試料はかり採り量は,附属書2表1による。

7
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2表1 試料はかり採り量
コバルト含有率
% (m/m)
試料はかり採り量
g
0.10 以上
2.0 未満
0.50
2.0 以上
20 以下
0.20
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) 混酸に分解容易な試料
1) 試料をはかり採ってビーカー (200ml) に移し入れる。
2) 時計皿で覆い,混酸20mlを加え,加熱して分解する。硝酸3mlを加えて鉄などを酸化し,引き続
き加熱して窒素酸化物などを追い出す。時計皿の下面を少量の水で洗って時計皿を取り除き,再び
加熱して硫酸の白煙を発生させる(1)。
注(1) クロム,タングステンなどの炭化物が存在しない場合には,硫酸白煙の発生を省略できる。
3) 放冷した後,温水約50mlを加え,穏やかに加熱して塩類を溶解する。溶液をろ紙(5種A)でろ過
し,ろ紙及び不溶解残さを温硫酸 (1+100) で十分に洗浄する。ろ液及び洗液は,ビーカー (300ml)
に集め,常温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで
薄める。
b) 混酸に分解困難な試料
1) 試料をはかり採ってビーカー (200ml) に移し入れる。
2) 時計皿で覆い,王水20mlを加え,加熱して分解する。混酸20mlを加え,引き続き加熱して窒素酸
化物などを追い出す。時計皿の下面を少量の水で洗って時計皿を取り除き,再び加熱して硫酸の白
煙を発生させる。
3) a)3)の操作を行う。
4.2
呈色 呈色は,次の手順によって行う。
a) 4.1 a)3)又はb)3)で得た溶液を附属書2表2に従って分取して,ビーカー (300ml) に移し入れる。ニッ
ケル溶液 [2.g)] 10mlを加え,水で液量を約25mlとし,溶液を十分振り混ぜながら酢酸ナトリウム溶
液 [2.i)] 20ml及びニトロソR塩溶液 [2.j)] 10mlを加える。
b) 溶液を75〜80℃の水浴中に約10分間浸した後,流水中で常温まで冷却する。
c) 溶液を十分振り混ぜながらりん酸 (1+1) 5ml及び硝酸25mlを加え,再び75〜80℃の水浴中に約10
分間浸した後,流水中で常温まで冷却する。
d) コバルト含有率範囲に応じて附属書2表2に示す希釈量に従って,溶液を100ml又は200mlの全量フ
ラスコに水を用いて移し入れ(2),水で標線まで薄める。
注(2) 分取した溶液中の鉄量がクロム量の2倍以下である場合は,鉄量がクロム量の2倍以上になるよ
うに鉄溶液 [2.h)] を加える。ただし,鉄量が50mgを超えてはならない。
附属書2表2 分取量と希釈量
コバルト含有率
% (m/m)
分取量
ml
希釈量
ml
0.10 以上
2.0 未満
10
100
2.0 以上
20 以下
5
200
8
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.3
対照液の調製 対照液の調製は,次の手順によって行う。
a) 4.1 a)3)又はb)3)で得た溶液を4.2 a)で分取した量と同じ量を分取して,ビーカー (300ml) に移し入れ
る。ニッケル溶液 [2.g)] 10mlを加え,水で液量を約25mlとし,溶液を十分振り混ぜながら酢酸ナト
リウム溶液 [2.i)] 20mlを加える。次にりん酸 (1+1) 5ml及び硝酸25mlを加えて沈殿を溶解する。
b) 溶液を75〜80℃の水浴中に約10分間浸した後,流水中で常温まで冷却し,溶液を十分振り混ぜなが
らニトロソR塩溶液 [2.j)] 10mlを加える。再び75〜80℃の水浴中に約10分間浸した後,流水中で常
温まで冷却する。
c) 4.2 d)の操作を行う。
4.4
吸光度の測定 4.2 4)で得た溶液の一部を,光度計の吸収セル (1cm) に取り,4.3 c)で得た溶液を対
照液として,コバルト含有率範囲に従って附属書2表3の波長における吸光度を測定する。
附属書2表3 吸光度の測定波長
コバルト含有率
% (m/m)
波長
mm
0.10 以上
0.40 未満
530
0.40 以上
20
以下
570
5. 空試験 試料の代わりに試料と同量の鉄 [2.f)] をはかり採り,4.1〜4.4の手順に従って試料と同じ操
作を試料と併行して行う。
6. 検量線の作成 検量線の作成は,次の手順によって行う。
a) 附属書2表4のコバルト含有率の範囲ごとに数個のビーカー (200ml) を準備し,それぞれに附属書2
表4に従って鉄 [2.f)] をはかり採って移し入れ,標準コバルト溶液を附属書2表4に従って正確に加
える。
b) 4.1 a)2)及び3)の操作を行う。
c) 4.2〜4.4の操作を試料と併行して行う。
d) 得た吸光度と標準コバルト溶液として加えた呈色液中のコバルト量との関係線を作成し,その関係線
を原点を通るように平行移動して検量線とする。
附属書2表4 検量線溶液の調製
コバルト含有率
% (m/m)
鉄 [2.f)] はかり採り量
g
使用する標準コバルト溶液
標準コバルト溶液添加量
ml
0.10 以上
0.40 未満
0.500
B [2.1)]
0, 1, 2, 3, 4
0.40 以上
2.0 未満
0.500
A [2.k)]
0, 2, 4, 6, 8, 10
2.0 以上
20
以下
0.200
A [2.k)]
0, 5, 10, 20, 30, 40
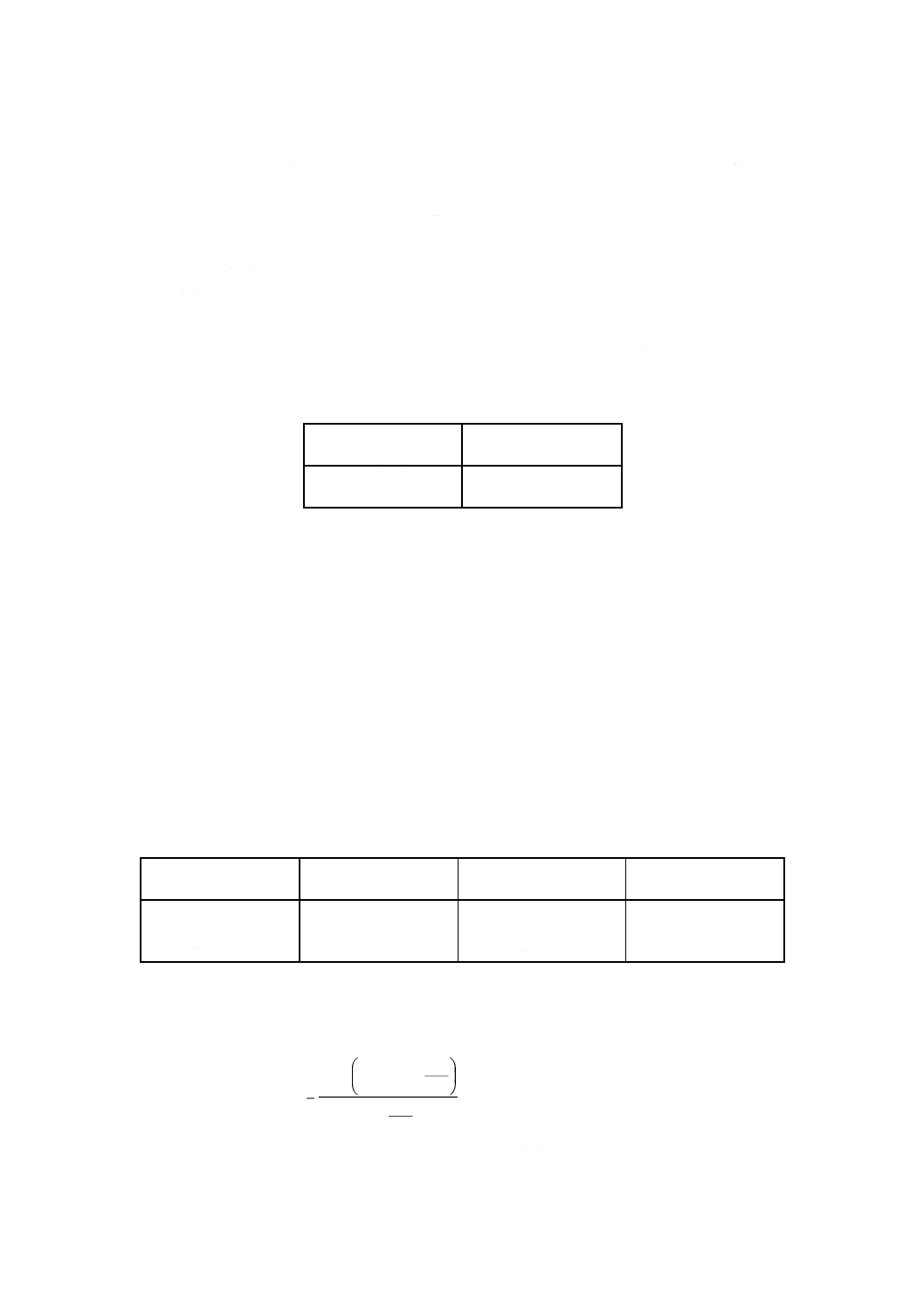
7. 計算 4.4及び5.で得た吸光度と6.で作成した検量線とからコバルト量を求め,試料中のコバルト含有
率を,次の式によって算出する。
100
100
100
3
2
1
×
×
×
−
−
=
B
m
B
m
m
m
Co
ここに, Co: 試料中のコバルト含有率 [% (m/m)]
9
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m1: 分取した試料溶液中のコバルト検出量 (g)
m2: 分取した空試験液中のコバルト検出量 (g)
m3: 5.ではかり採った鉄 [2.f)] 中に含まれるコバルトの量 (g)
m: 試料はかり採り量 (g)
B: 4.2のa)で分取した試料溶液及び空試験液の分取量 (ml)
8. 許容差 許容差(3)は,附属書2表5による。
附属書2表5 許容差
単位% (m/m)
コバルト含有率
室内再現許容差
室間再現許容差
0.10 以上
2.0 未満
D [0.019 6× (Co) +0.000 6]
D [0.036 4× (Co) −0.001 6]
2.0 以上
20 以下
D [0.005 1× (Co) +0.036 4]
D [0.009 3× (Co) +0.045 1]
注(3) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4[D (n, 0.95) の値]
による。nの値は,室内再現許容差の場合は同一室内における分析回数,室間再現許容差の場
合は分析に関与した分析室数である(n=2のとき,D=2.8である。)。
また, (Co) は,許容差を求めるコバルト含有率 [% (m/m)] である。
参考 この許容差は,コバルト含有率0.11% (m/m) 以上17.91% (m/m) 以下の試料を共同実験した結
果から求めたものである。

10
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3(規定) 2−ニトロソ−1−ナフトール抽出吸光光度法
1. 要旨 試料を王水で分解し,過塩素酸を加え,加熱して過塩素酸の白煙を発生させる。くえん酸で鉄
などをマスキングした後,溶液のpHをアンモニア水で調節し,2−ニトロソ−1−ナフトールを加え,生
成する2−ニトロソ−1−ナフトールコバルト錯体をベンゼンで抽出し,光度計を用いて有機相の吸光度を
測定する。
2. 試薬 試薬は,次による。
a) 塩酸
b) 塩酸 (1+10)
c) 過塩素酸
d) 過塩素酸 (1+100)
e) 王水(塩酸3,硝酸1)
f)
アンモニア水 (1+1, 1+2)
g) 水酸化ナトリウム溶液 (40g/l)
h) 鉄 できるだけ純度が高い鉄で,コバルト含有率が0.001% (m/m)以下で既知であるもの。
i)
過酸化水素
j)
マンガン溶液 マンガン(99%以上)0.5gをはかり採ってビーカー (300ml) に移し入れ,時計皿で覆
い,硝酸10ml及び過塩素酸15mlを加え,加熱して分解する。引き続き加熱して過塩素酸の濃厚な白
煙を発生させる。放冷した後,水50mlを加えて加熱し,過酸化水素を酸化マンガン (IV) の沈殿が溶
解するまで滴加し,煮沸して過剰の過酸化水素を分解する。室温まで冷却した後,時計皿の下面を水
で洗って時計皿を取り除き,水で液量を500mlとする。
k) くえん酸溶液 (500g/l)
l)
2−ニトロソ−1−ナフトール溶液 2−ニトロソ−1−ナフトール (NOC10H6OH) 0.1gに水酸化ナトリ
ウム溶液 (40g/l) 15滴及び水1mlを加えて溶解した後,水で液量を100mlとする。
m) ベンゼン
n) 標準コバルト溶液 (10μFgCo/ml) コバルト(99.9%以上)0.100gをはかり採ってビーカー (200ml) に
移し入れ,時計皿で覆い,硝酸 (1+1) 10ml及び過塩素酸5mlを加え,加熱して分解する。引き続き
加熱して過塩素酸の濃厚な白煙を発生させる。放冷した後,時計皿の下面を水で洗って時計皿を取り
除き,水約50mlを加える。常温まで冷却した後,溶液を1 000mlの全量フラスコに水を用いて移し入
れ,水で標線まで薄めて原液 (100μgCo/ml) とする。この原液を使用の都度,必要量だけ水で正確に
10倍に薄めて標準コバルト溶液とする。
3. 試料はかり採り量 試料はかり採り量は,附属書3表1による。
附属書3表1 試料はかり採り量
コバルト含有率
% (m/m)
試料はかり採り量
g
0.001 以上
0.025 未満
1.0
0.025 以上
0.10 以下
0.25

11
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
a) クロム含有率4% (m/m) 未満の試料の場合
1) 試料をはかり採ってビーカー (200ml) に移し入れる。
2) 時計皿で覆い,王水20mlを加え,加熱して分解する。過塩素酸15mlを加え,加熱して過塩素酸の
白煙を発生させ,さらに4〜5分間濃厚な白煙を発生させる。
3) 放冷した後,時計皿の下面を水で洗って時計皿を取り除き,水約50mlを加えて塩類を溶解する。
常温まで冷却した後,溶液をろ紙(5種A)でろ過し,ろ紙及び不溶解残さを水(1)で洗浄する。ろ
液及び洗液は,100mlの全量フラスコに集め,水で標線まで薄める。不溶解残さは,捨てる。
注(1) タングステンを含有する試料の場合は,過塩素酸 (1+100) で洗浄する。
b) クロム含有率4% (m/m) 以上の試料の場合
1) a)1)及び2)の操作を行う。
2) 加熱しながら塩酸10mlを少量ずつ添加し,大部分のクロムを二塩化二酸化クロムとして揮散させ,
引き続き加熱して残ったクロムが二クロム酸に酸化されるまで過塩素酸の白煙を発生させる。放冷
した後,水約50mlを加えて塩類を溶解し,ニクロム酸が還元されるまで過酸化水素を滴加する。
加熱して煮沸し,過剰の過酸化水素を分解する。
3) a)3)の操作を行う。
4.2
呈色 呈色は,次の手順によって行う。
a) 4.1 a)3)又はb)3)で得た溶液を附属書3表2に従って分取し,ビーカー (200ml) に移し入れる。
b) くえん酸溶液4mlを加え,pH計を用いて分取量が25mlの場合にはアンモニア水 (1+1) で,分取量
が10mlの場合にはアンモニア水 (1+2) で,溶液のpHを7.0±0.2に調節する。室温まで冷却した後,
溶液を分液漏斗に水を用いて移し入れ,水で50mlとする。
附属書3表2 試料溶液の分取量
コバルト含有率
% (m/m)
試料溶液の分取量
ml
0.001 以上
0.010 未満
25
0.010 以上
0.10 以下
10
c) 2−ニトロソ−1−ナフトール溶液 [2.1)] 5.0mlを加えて振り混ぜ,5分間以上放置した後,ベンゼン
20.0mlを加え,約30秒間激しく振り混ぜる。静置して二層に分離した後,下層の水相を捨てる。有
機相に塩酸 (1+10) 5mlを加えて再び約10秒間激しく振り混ぜ,静置して二層に分離した後,下層の
水相を捨てる。
d) 有機相に水酸化ナトリウム溶液5mlを加え,約10秒間激しく振り混ぜて有機相を洗浄し,静置して
二層に分離した後,下層の水相を捨てる。
e) d)の操作を水相に黄色の着色が認められなくなるまで繰り返す。
f)
有機相に塩酸 (1+10) 5mlを加え,かるく振り混ぜ,静置して二層に分離した後,下層の水相を捨て
る。
4.3
吸光度の測定 4.2 f)で得た有機相を乾いたろ紙(5種A)でろ過し,最初のろ液は捨て,次のろ液
からその一部を光度計の吸収セル (1cm) に取り,ベンゼンを対照液として波長370nm付近の吸光度を測
定する。
12
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5. 空試験 試料の代わりに試料と同量の鉄 [2.h)] をはかり採り,4.1〜4.3の手順に従って試料と同じ操
作を試料と併行して行う。
6. 検量線の作成 検量線の作成は,次の手順によって行う。
a) 附属書3表3のコバルト含有率範囲ごとに6個のビーカー (200ml) を準備し,それぞれに附属書3表
3に従って鉄 [2.h)] をはかり採って移し入れる。
附属書3表3 検量線溶液の調製
コバルト含有率
% (m/m)
鉄 [2.h)] はかり採り量
g
分取量
ml
標準コバルト溶液 [2.n)] 添加量
ml
0.001以上 0.010 未満
1.00
25
0, 0.5, 1, 1.5, 2, 2.5
0.010以上 0.025 未満
1.00
10
0.025以上 0.10 以下
0.25
10
b) 4.1a)2)の操作を行う。
c) 放冷した後,時計皿の下面を水で洗って時計皿を取り除き,水約50mlを加えて塩類を溶解する。常
温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。それ
ぞれの溶液を附属書3表3のコバルト含有率範囲に従ってビーカー (200ml) に分取し,附属書3表3
に従って標準コバルト溶液 [2.n)] を正確に添加した後,マンガン溶液 [2.j)] 1mlを加える。
d) 4.2 b)〜4.3の操作を試料と併行して行う。
e) 得た吸光度と標準コバルト溶液として加えたコバルト量との関係線を作成し,その関係線を原点を通
るように平行移動して検量線とする。
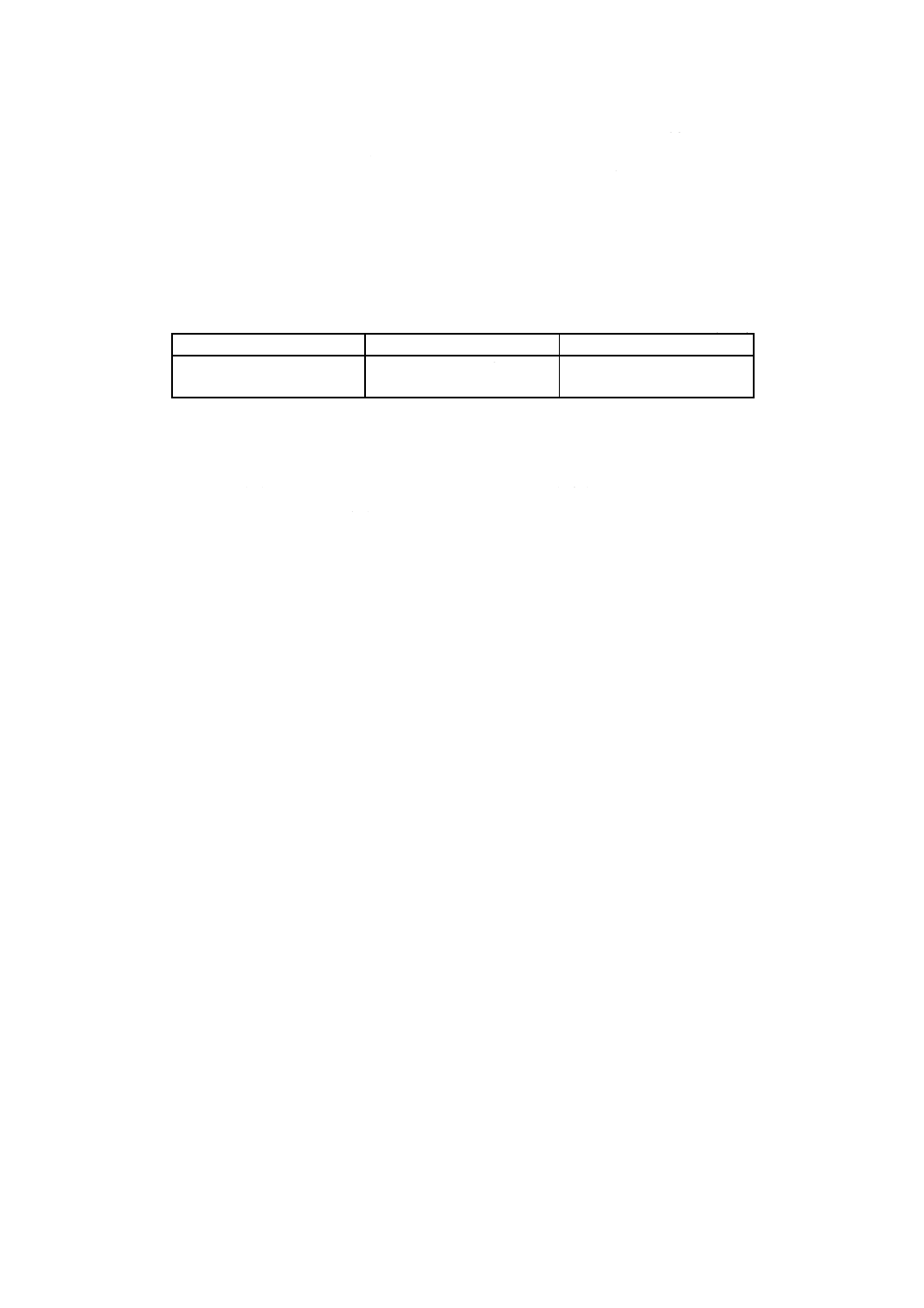
7. 計算 4.3及び5.で得た吸光度と6.で作成した検量線とからコバルト量を求め,試料中のコバルト含有
率を次の式によって算出する。
100
100
100
3
2
1
×
×
×
−
−
=
B
m
B
A
A
A
Co
ここに, Co: 試料中のコバルト含有率 [% (m/m)]
A1: 分取した試料溶液中のコバルト検出量 (g)
A2: 分取した空試験液中のコバルト検出量 (g)
A3: 5.ではかり採った鉄 [2.h)] 中に含まれるコバルトの量 (g)
B: 試料溶液の分取量 (ml)
m: 試料はかり採り量 (g)
8. 許容差 許容差(2)は,附属書3表4による。
附属書3表4 許容差
単位% (m/m)
コバルト含有率
室内再現許容差
室間再現許容差
0.010以上 0.020以下
D×0.000 5
D×0.001 2
注(2) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4[D (n, 0.95) の値]
による。nの値は,室内再現許容差の場合は,同一室内における分析回数,室間再現許容差の
場合は分析に関与した分析室数である(n=2のとき,D=2.8である。)。
13
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考 この許容差は,コバルト含有率0.012% (m/m) 及び0.018% (m/m) の試料を共同実験した結果か
ら求めたものである。
14
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4(規定) イオン交換分離電位差滴定法
序文 この附属書4(規定)は,1997年に第1版として発行されたISO 11653 (Steel−Determination of high
cobalt content−Potentiometric titration method after separation by ion exchange) を翻訳し,技術的内容及び規格
票の様式を変更することなく作成した日本工業規格である。
なお,この規格で点線の下線を施してある“参考”は,原国際規格にはない事項である。
1. 適用範囲 この附属書4(規定)は,鋼中のコバルトをイオン交換で分離した後,電位差滴定法によ
って定量する方法について規定する。
この方法は,コバルト含有率5.0% (m/m) 以上17.0% (m/m) 以下の試料に適用する。
2. 引用規格 次に記載する規格は,この附属書4(規定)の本体中に引用されることによって,この規
格の規定の一部を構成する。この規格の発行の時点では,それぞれの規格の発行版は正しいものであるが,
国際規格はすべて改訂されるものであるので,この規格を使用することに合意した当事者は,常に最新版
の規格を参照するように努力されたい。IEC及びISOメンバーには最新の国際規格のリストが配布されて
いる。
ISO 385-1 : 1984, Laboratory glassware−Burettes−Part 1 : General rquirements.
ISO 648 : 1977, Laboratory glassware−One-mark pipettes.
ISO 1042 : 1983, Laboratory glassware−One-mark volumetric flasks.
ISO 3696 : 1987, Water for analytical laboratory use−Specification and test methods.
ISO 5725-1 : 1994, Accuracy (trueness and precision) of measurement methods and results−Part 1 : General
principles and definitions.
ISO 5725-2 : 1994, Accuracy (trueness and precision) of measurement methods and results−Part 2 : Basic
method for the determination of repeatability and reproducibility of a standard measurement method.
ISO 5725-3 : 1994, Accuracy (trueness and precision) of measurement methods and results−Part 3 :
Intermediate measures of the precision of a standard measurement method.
ISO 14284 : 1996, Steel and iron−Sampling and preparation of samples for the determination of chemical
composition.
3. 原理 はかり採った試料を硝酸と塩酸との混酸で溶解する。
陰イオン交換樹脂にコバルトなどを吸着させた後,塩酸を用いて陰イオン交換カラムからコバルトを選
択的に溶出させ,妨害元素から分離する。
過塩素酸で酸化する。窒素を通して溶存する塩素と酸素を追い出す。
くえん酸アンモニウム,アンモニア水及び過剰のヘキサシアノ鉄 (III) 酸カリウム標準溶液を含む溶液
に試料溶液を加え,電位差終点決定方法を用いてヘキサシアノ鉄 (III) 酸カリウムの過剰分を標準コバル
ト溶液で滴定する。
4. 試薬 分析に際しては,特に記述しない限り,分析用保証試薬及びISO 3696に規定された等級2の水
だけを使用する。
15
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.1
塩酸 密度 約1.19g/ml
4.2
塩酸 密度 約1.19g/ml,希釈7+5
4.3
塩酸 密度 約1.19g/ml,希釈2+3
4.4
塩酸 密度 約1.19g/ml,希釈1+2
4.5
塩酸 密度 約1.19g/ml,希釈1+19
4.6
塩酸 密度 約1.40g/ml
4.7
過塩素酸 密度 約1.67g/ml(1)
注(1) 過塩素酸(密度約1.54g/ml)を使用してもよい。過塩素酸(密度約1.67g/ml)100mlは,125ml
の過塩素酸(密度約1.54g/ml)と同等である。
4.8
アンモニア水 密度 約0.89g/ml
4.9
くえん酸アンモニウム溶液 (200g/l) くえん酸一水和物 (C6H8O7・H2O) 100gを水約250mlに溶解す
る。注意して常にかき混ぜながらアンモニア水 (4.8) 170mlを加える。冷却した後,水で500mlに薄めて
かき混ぜる。
4.10 標準コバルト溶液 (2.00gCo/l) 金属コバルト[99.95% (m/m) 以上](2)2.000gを0.001gのけたまでは
かり採ってビーカー (600ml) に移し入れ,硝酸(密度約1.40,希釈1+1)40mlで溶解する。溶解が完了
するまで加熱し,静かに煮沸して窒素酸化物を追い出す。常温まで冷却した後,水を用いて溶液を1 000ml
の全量フラスコに移し入れ,水で標線まで薄めてかき混ぜる。ポリエチレン製の瓶に保存する。
この溶液1mlはコバルト2.00mgを含有する。
注(2) 高純度コバルト粉末は,表面が酸化されているので使用してはならない。例えば,塊,粒又は
線を使用する。
4.11 ヘキサシアノ鉄 (III) 酸カリウム標準溶液
4.11.1 標準溶液の調製 ヘキサシアノ鉄 (III) 酸カリウム [K3Fe (CN) 6] 5.6gを水250mlに溶解する。溶液
をろ紙でろ過し,水で十分に洗浄する。ろ液及び洗液を500mlの全量フラスコに移し入れ,水で標線まで
薄めてかき混ぜる。この溶液は,使用直前に再度乾いたろ紙でろ過し,4.11.2に従って標定する。
4.11.2 標準溶液の標定 標準コバルト溶液 (4.10) 20.0mlを2回取り,2個のビーカーに移し入れる。以下,
7.2.3及び7.2.4の手順に従って操作する。
次の式によって各溶液のファクターTを算出する。
3
2
1
V
V
V
T
+
=
ここに, V1: 2で取った標準コバルト溶液 (4.10) の量 (ml) (=20.0ml)
V2: 滴定に使用した標準コバルト溶液 (4.10) の量 (ml)
V3: 滴定に使用したヘキサシアノ鉄 (III) 酸カリウム標準溶液の
量 (ml)
T: 求めるファクター。溶液が理論値以下なら係数は1未満とな
る。算出した2個のT値の平均を計算する。
4.12 イオン交換樹脂 架橋度が公称8%,径が公称200〜400メッシュの球状粒子で構成される第四級ア
ルキルアンモニウム形(塩化物形)の陰イオン交換樹脂を用いる。極端に細かい粒子や直径約180μm以上
の粒子を除去するために樹脂を次のように処理する。
十分な量の樹脂をビーカーに移し入れ,水で覆い,粒子を最大限に膨潤させるため十分な時間(30分以
上)静置する。ビーカー (2l) の上に直径150mmの180μmふるいを置く。樹脂を薄いスラリー状にして,
ふるいの上から注ぐ。流水を注ぎ,細かい粒子をふるい下のビーカーに洗い落とす。ふるいの目の過度の
16
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
詰まりを避けるため,必要があれば周期的にふるい上に残っている粒子を捨てる。ビーカーを静置して集
めた樹脂の大部分が沈んだら,ビーカーを傾むけて水を捨て,樹脂約100mlを400mlのビーカーに移し入
れる。塩酸 (4.5) 200mlを加えて激しくかき混ぜ,4〜6分間樹脂を安定させる。傾けて懸濁物のうち150
〜175mlを捨てる。塩酸 (4.5) による処理を2回以上繰り返して,カラム作成用の粗い樹脂を確保する。
カラムは次のように作製する。
イオン交換カラム (5.2) の底にグラスウール又はポリ(塩化ビニル)プラスチック製繊維を10〜30mm
の厚さに敷き,準備した樹脂をカラムの高さ約140mmまで満たすに十分な量だけ入れる。
溶液を入れる際に,懸濁液になるのを防止するため樹脂層最上部に,ガラスウール又はポリ(塩化ビニ
ル)プラスチック製繊維を20mmの厚さに敷く。樹脂層最上部の上で,静水圧が100mmになるよう塩酸
(4.2) 35mlを少しずつカラムに流しながら,流量が毎分3.0ml以上にならないよう調節する。樹脂層最上部
の上で液量10〜20mlになるまで流し出してから,下側の栓を閉める。
5. 装置 分析に際しては,特に記載しない限り,通常の実験室器具を用いる。
ガラス製体積計はすべて,ISO 385-1,ISO 648又はISO 1042に規定された等級Aの適切なものでなけ
ればならない。
5.1
電位差滴定装置
5.1.1
指示電極 曇りのない白金製で,清浄かつ高度に研磨した状態に保たれたもの。電極は硝酸(4.6)
に浸して清浄にし,使用の前に水で洗う。
5.1.2
参照電極 銀/塩化銀,塩化水銀 (I) 又は硫酸水銀 (I) 製で,それぞれの電極の取扱いと保守につ
いては,製造業者の指示書に従わなければならない。
5.1.3
滴定部 ビーカー (400ml) 1個,ISO 385-1に規定された等級Aのビュレット2本及び磁気かき混
ぜ機で構成する。
5.1.4
高インピーダンス電圧計 通常,電圧計としてpH計が使用できる。市販の滴定装置又は電位差計
は滴定曲線が作図でき,終点を一次又は二次の導関数(9.参照)から計算するよりも,むしろ曲線への内
挿法で判定できるので手動のシステムより有利である。
5.2
イオン交換カラム 直径約25mm,長さ300mmで一方の端部で先細になっていて,流量を制御する
ための活栓と,流れを停止させるためにさらに下側に活栓の付いているもの。カラムの先端に溶離液の貯
留部を付けてもよい。
6. サンプリング サンプリングは,ISO 14284に従って行う。
7. 操作
警告 白煙を発生している過塩素酸は強力な酸化剤で,アンモニア,亜硝酸ガス又は有機物質と接触すれ
ば爆発性の混合物となる。蒸発操作はすべて過塩素酸の使用に適したドラフト内で実施しなければならな
い。
7.1
試料はかり採り量 試料はかり採り量は,0.5gを0.000 1gのけたまではかり採る。
7.2
定量
17
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.2.1
試料溶液の調製 はかり採った試料 (7.1) をビーカー (150ml) に移し入れる。塩酸 (4.1) と硝酸
(4.6) を体積比で5:1に混合した混酸(3)20mlを加える。時計皿で覆い,試料の分解が完了するまで60〜70℃
で加熱する。時計皿の下面を水で洗って時計皿を外し,ビーカーに溝付きのガラスふたを置き,焼き付か
ないように注意しながら溶液を乾固近くまで蒸発させる。放冷した後,塩酸 (4.2) 20mlを加え,塩類が溶
解するまで(約10分)(4)60〜70℃で加熱する。
注(3) 特殊な合金鋼の分解に対しては,ふっ化水素酸を添加したり又は添加しなかったり,異なった
酸の比率及び濃度を用いる。
(4) タングステン酸化物及び/又はモリブデン酸化物は少し溶け残ることがある。これらは,後の
ろ過で取り除く。
7.2.2
イオン交換樹脂による分離 溶液を室温まで冷却して緻密なろ紙(5種C)でろ過し,水で十分に
洗浄する。ろ液及び洗液を集めてイオン交換カラムに移し入れる。カラムの下にビーカーを置いて下側の
活栓を開ける。溶液が樹脂層の上10〜20mmの液面に達したとき,元のビーカーを塩酸 (4.2) 5〜6mlで洗
い,洗液をカラムに移し入れる。このビーカー洗浄操作を2分間隔で4回繰り返す。カラムの上部を塩酸
(4.2) で2,3回洗浄し,その度に液面を樹脂層の上10〜20mmまで降下させる。流量を毎分3.0ml以下に
保ち,主にクロム,マンガン及びニッケルを含有する溶液(試料溶液及び洗液)が合計で175〜185ml集
まるまで塩酸 (4.2) をカラムに入れる。カラム内の溶液の液面が樹脂層の上10〜20mmに達したときに流
出液を捨て,それからビーカー (400ml) を用いてコバルトの溶出液を集める。
コバルトの損失を少しでも防止するためにコバルト帯の先端が樹脂の底部から25mmを少しでも超えさ
せないことが重要である。通常,コバルトがカラム内のこの点に到達したとき,クロム,マンガン及びニ
ッケルは除去されている。集めた総量が175ml以下であってもこの点で溶出を停止させる。
カラムに塩酸 (4.4) を入れ,毎分3.0mlの流量を維持しながら165〜175mlの溶液を集める。この溶液は
保存しておく。試料溶液中の鉄が0.200g以上含有していなければ,次の試料溶液用にカラムを次のように
再生する。カラム内に残っている溶液を樹脂層の上10〜20mlになるまで流し出し,塩酸 (4.2) 35〜50ml
をカラムに流して溶液が樹脂層の上に10〜20ml残るまで続けてから下側の栓を閉める。試料溶液中の鉄
が0.200g以上含有していれば,各回の溶出後には鉄 (II) を洗い流すために塩酸 (4.3) 約500mlで,その
後,鉄 (III) を洗い流すために塩酸 (4.5) 約200mlでカラムを再生しなければならない。
7.2.3
溶出液の処理 7.2.2で保存した溶液に硝酸 (4.6) 30ml及び過塩素酸 (4.7) 15mlを加え,加熱して
過塩素酸の白煙が発生して乾固近く(1〜2mlが残る)(5)になるまで蒸発させる。放冷した後,水25〜35ml
を加えて1〜2分間煮沸する。冷却した後,くえん酸アンモニウム溶液 (4.9) 10mlを加える。
注(5) 乾固が不十分だとヘキサシアノ鉄 (III) 酸カリウム標準溶液 (4.11) を添加した後に沈殿が生じ
る。
溶液を冷却し,溶存している塩素及び酸素を除去するために溶液中に窒素を10〜15分間激しく通す。室
温まで冷却する。
7.2.4
滴定 50mlビュレットを用いて,コバルトを酸化するのに十分な量のヘキサシアノ鉄 (III) 酸カリ
ウム標準溶液 (4.11) を滴定部のビーカー (400ml) に移し入れ,更に過剰に約5mlを加える。添加したヘ
キサシアノ鉄 (III) 酸カリウム標準溶液 (4.11) の使用量 (V4) を記録しておく。ヘキサシアノ鉄 (III) 酸カ
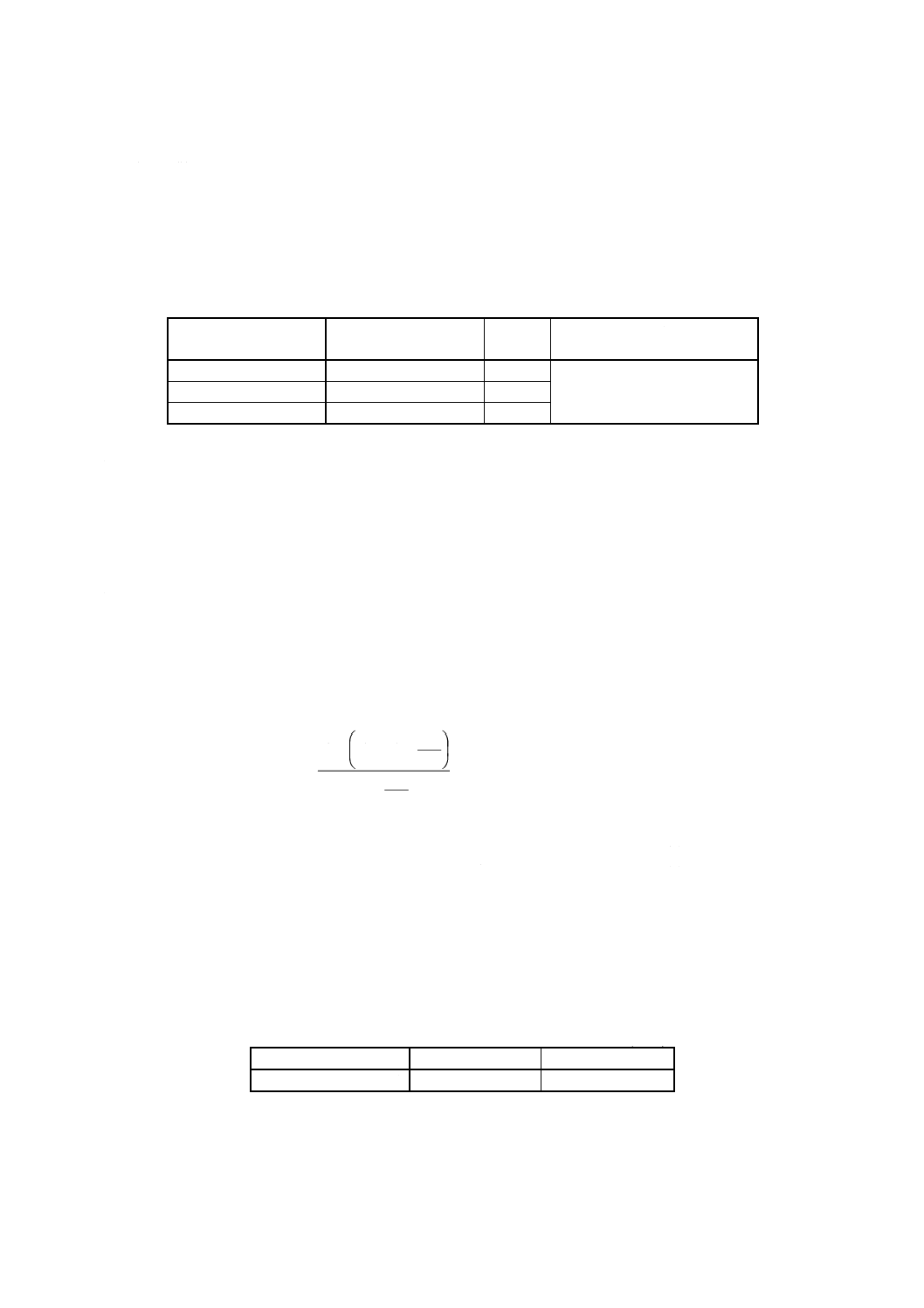
リウム標準溶液 (4.11) の使用量巧は,次の式で推定する。
過剰分
+
×
×
=
000
1
2.
11
8
055
.0
4
w
x
V
ここに,
x: コバルト予想量 [% (m/m)]
18
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ω: 試料はかり採り量 (g)
0.055 8: ヘキサシアノ鉄 (III) 酸カリウムに換算するファクター
11.2: ヘキサシアノ鉄 (III) 酸カリウム濃度 (g/l) (4.11.1参照)
1 000: mlに換算するファクター
アンモニア水 (4.8) 50mlを加えて冷却する。ビーカーを滴定部 (5.1.3) の磁気かき混ぜ機に載せ,かき
混ぜ機を始動させる。
絶えずかき混ぜながら試料溶液を全量,上述のビーカー (400ml) に移し入れる。溶液中に指示電極及び
参照電極(5.1.1及び5.1.2)を挿入する。終点に達するまで標準コバルト溶液 (4.10) で静かに滴定する。
0.1ml又は一滴ずつ増して滴定を続け,増加分を添加した後,平衡に達したときのビュレットと電位差計
の読み取り値を記録する。終点を過ぎるまで滴定を続ける。滴定曲線の内挿法によって終点を決定し,終
点に対応する使用量 (V5) を決定する(9.参照)。
8. 結果の表示
8.1
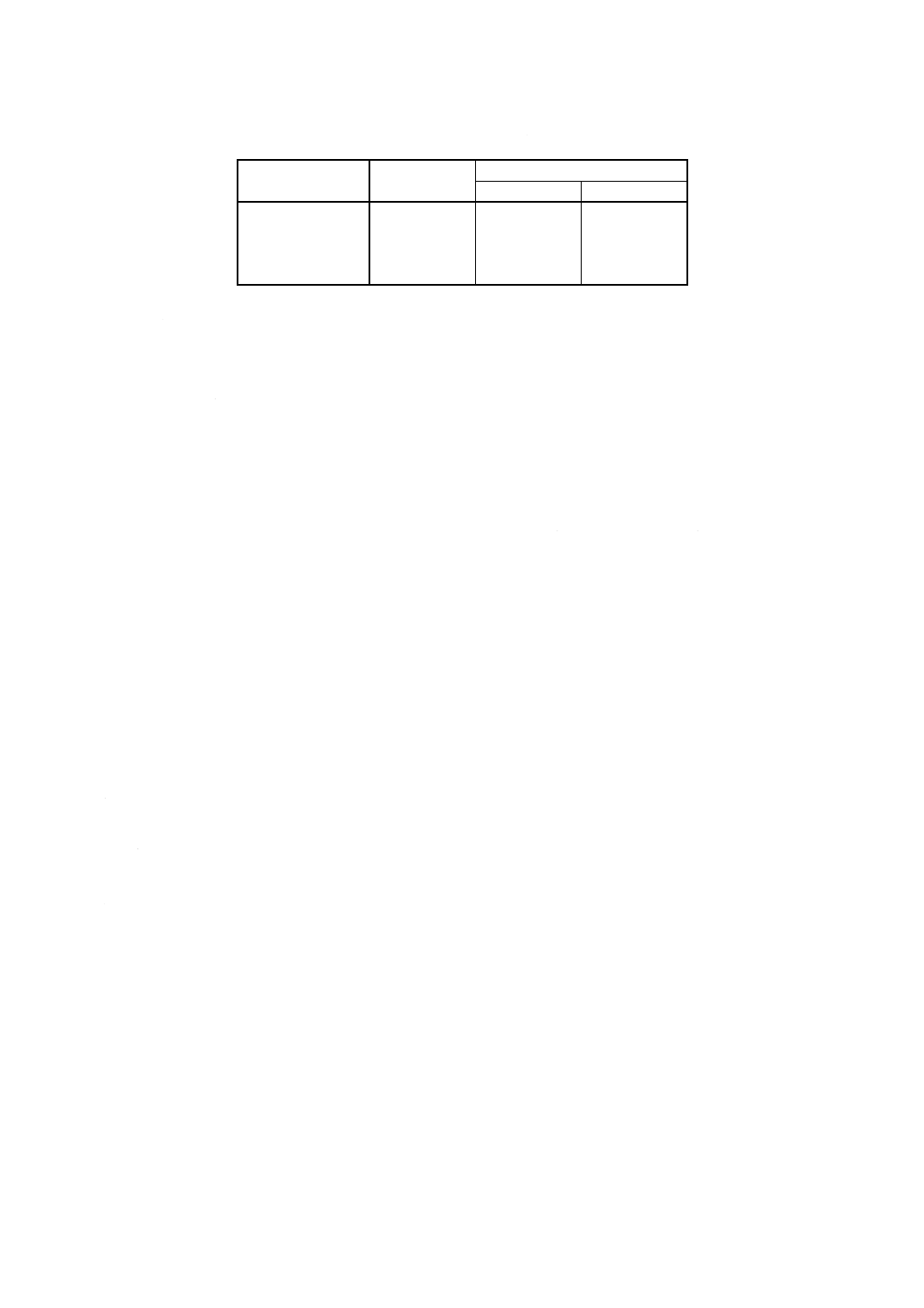
計算方法 コバルト含有率wco [% (m/m)]を次の式によって算出する。
1.0
)
(
100
10
)
(
5
4
3
5
4
×
×
−
×
=
×
×
×
−
×
=
m
V
T
V
m
V
T
V
w
co
co
co
ρ
ρ
ここに,
V4: 滴定に用いたヘキサシアノ鉄 (III) 酸カリウム標準溶液
(4.11) の使用量 (ml)
V5: 計算した終点 (7.2.4) に示された標準コバルト溶液 (4.10)
の使用量 (ml)
T: 4.11.2で計算したヘキサシアノ鉄 (III) 酸カリウム標準溶液
のファクター
ρco: 標準コバルト溶液 (4.10) の濃度 (mg/ml)
m: 試料はかり採り量 (mg)
8.2
許容差 この方法の共同実験は,8水準のコバルト含有率試料を用いて,8分析室が参加して行った
(6)(7)。
使用した試料及び得た平均値の結果を,附属書4参考A表A.1に示した。
得た結果は,ISO 5725-1,-2及び-3によって統計処理した。
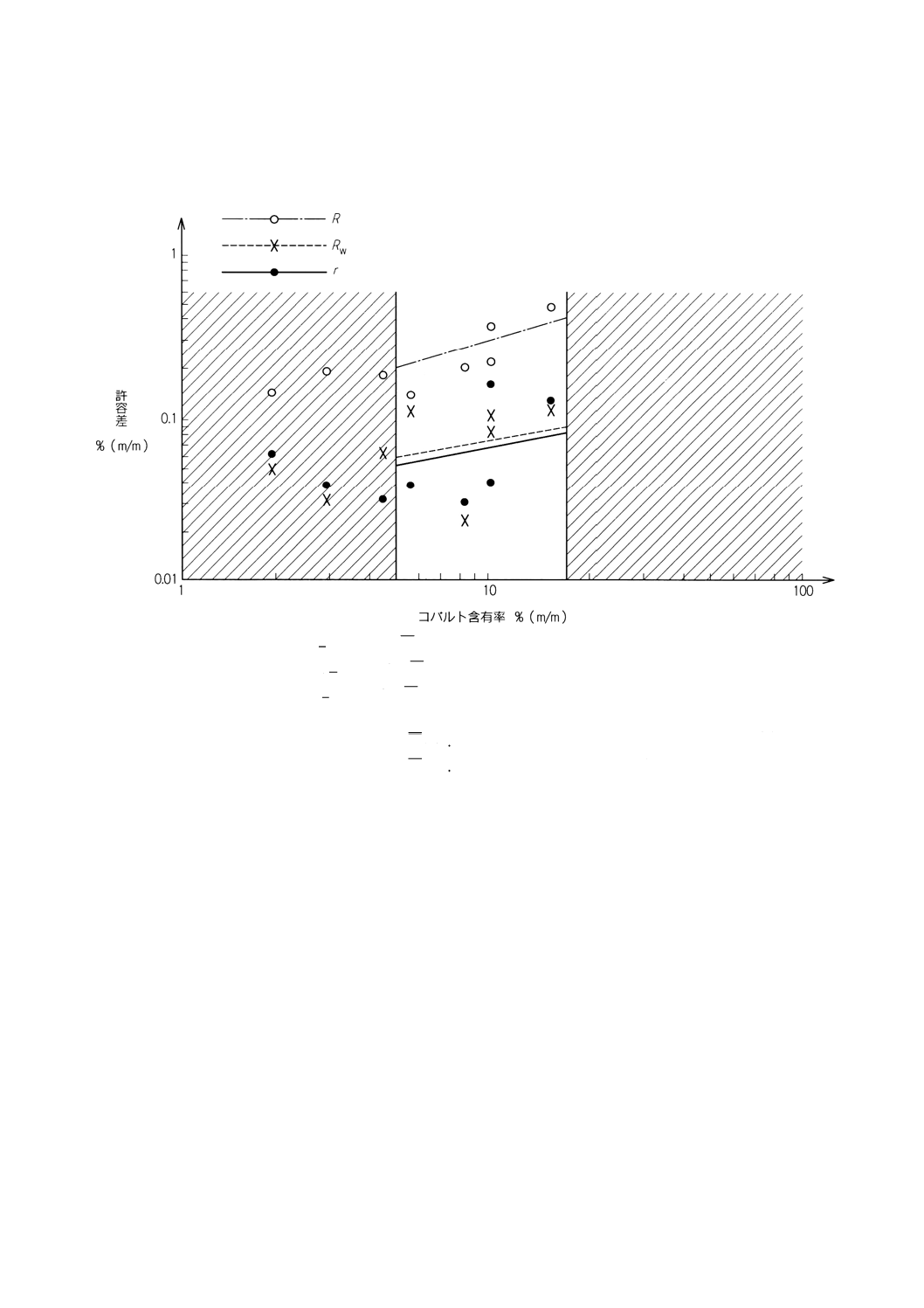
得たデータは,コバルト含有率と分析結果の併行許容差 (r) 及び再現許容差(R及びRw)(8)との間に対
数的比例関係があり,附属書4表1にその結果を要約した。許容差データを附属書4参考B図B.1に図示
した。
注(6) 3回の定量のうち2回はISO 5725-1に定義している併行測定条件のもとで実験した。すなわち,
一人の分析者が同じ装置で同一操作条件で同一標準溶液ファクターで最小の短時間内で行った。
(7) 3回目の定量は,同じ分析者によって同じ装置で新しい標準溶液ファクターを用い,異なった
時間(異なった日)に行った。
(8) 第1日目に得た結果から,ISO 5725-2の定義に従い併行許容差 (r) 及び再現許容差 (R) を計算
した。1日目の最初に得た結果と2日目に得た結果から室内再現許容差 (Rw) を計算した。
参考 原文では検量線になっているが,滴定法では検量線を使用しないので標準溶液のファクターと
した。
19
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4表1 併行許容差及び再現許容差
コバルト含有率
% (m/m)
併行許容差
r
再現許容差
R
Rw
5.0
0.051
0.202
0.061
10.0
0.067
0.285
0.079
15.0
0.079
0.348
0.093
17.0
0.084
0.365
0.098
9. 滴定終点決定の留意点 当量点を十分に過ぎるまで普通に滴定を実施した際の,電位差計による終点
の正確かつ再現性のある数値は,普通の目視的な終点検出とは異なる。古典的なS字型滴定曲線は,当量
点付近で急激な電位の立ち上がりを示す。曲線の急激な立ち上がり部分の中間点は,通常,変曲点であり
対称的な滴定曲線については,中間点は当量点と一致する。真の当量点と,中間点が一致しない非対称の
滴定曲線については,電位の変化は通常,滴定誤差を無視するに十分な大きさである。
滴定試薬を添加する度に,平衡電位を安定させて記録しなければならないので,手動の電位差滴定は遅
い。終点付近では,滴定試薬を少量ずつ添加しなければならず,電位差の大きな変化を観察した後,少な
くとも3回測定しなければならない。終点の鋭敏さを改善し手動滴定を速くするために,約6滴のエチレ
ンジアミンを滴定時に加えてもよい。記録したデータから滴定曲線を作図し,その曲線の急こう(勾)配
の部分から終点を内挿して,当量点を決定する。しかし,当量点で極大となる一次導関数 (dE/dV) を計算
するのが,より望ましい。当量点の体積の正確な値は,体積に対する電位差の二次導関数が数的にゼロに
等しくなる値を計算して,決定する。もし,滴定試薬の等量が大きな電位変化の直前と直後に添加されて
いれば,2回の添加の間で二次導関数の記号が変わるのが容易に分かる。つまり,内挿法で決定したある
点で,二次導関数がゼロを通過するはずである。
滴定曲線を直接記録し,データをデジタル形で処理する自動滴定装置を使用する際には,大きな利点が
ある。この附属書4(規定)ではそのような機器を推奨する。
10. 分析報告書 この分析報告書には,次の情報を記載しなければならない。
a) 試料,分析室及び分析日時を証明するのに必要なすべての情報。
b) この附属書4(規定)の引用。
c) 結果及び表示様式。
d) 定量に際して注目された異常な特徴。
e) この附属書4(規定)に規定されていない操作,又は結果に影響を与えるようなすべて任意の操作。
20
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4参考A 国際共同実験に関する追加情報
附属書4表A.1は,1993年に4か国,8分析室で,8個の鋼試料を用いて実施した国際共同実験の結果
から求めた。
共同実験の結果は,1994年4月に発行されたISO/TC 17/SC 1 N 1015の文書に報告されている。許容差
データを附属書4参考Bに図示する。
使用した試料及び得た結果の平均値を,附属書4表A.1に示した。
附属書4表A.1 使用した試料及び実験結果
試料
コバルト含有率
% (m/m)
許容差データ
認証値
分析値
併行許容差
r
再現許容差
WCo.1
WCo.2
R
Rw
BCS 483(高速度鋼)
[3Cr, 11W, 0.5V, 0.2Mo]
1.94
1.94
1.95
0.061
0.143
0.052
DAIDO(高合金鋼)
[14Cr, 6Mo]
3.051)
2.96
2.96
0.039
0.195
0.032
JSS 607-8(高速度鋼)
[4Cr, 0.8V, 17W, 0.5Mo]
4.59
4.56
4.58
0.032
0.186
0.064
ECRM 251-1(高速度鋼)
[5Cr, 1.6V, 20W, 0.5Mo]
5.70
5.64
5.64
0.039
0.138
0.116
NBS 153a(高速度鋼)
[3.7Cr, 2V, 1.8W, 8.8Mo]
8.47
8.42
8.44
0.031
0.211
0.024
ECRM 283-1(高速度鋼)
[4Cr, 3V, 9.7W, 3.4Mo]
10.27
10.17
10.19
0.041
0.225
0.086
BCS 484(高速度鋼)
[5Cr, 1V, 22W, 1.1Mo]
10.2
10.15
10.15
0.169
0.380
0.107
NBS 868(耐熱鋼)
[38Ni]
16.1
15.84
15.82
0.133
0.490
0.122
WCO.1:日内総平均
WCO.2:日間総平均
1) 非認証値
21
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4参考B 許容差データの図
309
.1
log
4
493
.0
log
500
.1
log
1
399
.0
log
577
.1
log
0
403
.0
log
1.
2.
1.
−
=
−
=
−
=
Co
Co
w
Co
W
R
W
R
W
r
ここに,
1.
Co
W
: 日内で得たコバルト含有率 [% (m/m)]の平均値
2.
Co
W
: 日間で得たコバルト含有率 [% (m/m)]の平均値
附属書4図B.1 コバルト含有率 (WCo) と併行許容差 (r) 又は再現許容差(R及びRw)の間の対数的比例
関係
22
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
原案作成委員会 構成表
(1) 社団法人 日本鉄鋼連盟鋼材規格検討会F02.03分野
氏名
所属
(委員長)
前 原 郷 治
社団法人日本鉄鋼連盟
(幹事)
森 下 昇
社団法人日本鉄鋼連盟
余 語 英 俊
愛知製鋼株式会社
安 原 久 雄
川鉄テクノリサーチ株式会社
滝 沢 佳 郎
川鉄テクノリサーチ株式会社
岡 山 和 生
合同製鐵株式会社
金 築 宏 治
株式会社神戸製鋼所
河 村 恒 夫
株式会社コベルコ科研
大 石 隆 司
山陽特殊鋼株式会社
岡 圭 男
住友金属工業株式会社
伊 藤 清 孝
大同特殊鋼株式会社
猪 股 重 宏
大平洋金属株式会社
中 嶋 康 博
株式会社中山製鋼所
向 奥 巌
日新製鋼株式会社
山 本 満 治
株式会社日鐵テクノリサーチ
田 中 耕 一
日本金属工業株式会社
堀 籠 秀 和
日本鋼管株式会社
宮 川 利 弘
日本高周波鋼業株式会社
増 田 正 純
通商産業省工業技術院
(関係者)
稲 本 勇
社団法人日本鉄鋼連盟
23
G 1222 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 社団法人 日本鉄鋼連盟鋼材規格三者委員会
氏名
所属
(委員長)
佐久間 健 人
東京大学工学部
(副委員長)
大河内 春 乃
東京理科大学
二 瓶 正 俊
科学技術庁金属材料技術研究所
土 門 斉
東京工科大学
脇 本 眞 也
通商産業省基礎産業局
大 嶋 清 治
通商産業省工業技術院
林 央
理化学研究所
馬 木 秀 雄
社団法人火力原子力発電技術協会(石川島播磨重工業株式会社)
金 沢 孝
社団法人日本自動車工業会(いすゞ自動車株式会社)
井 上 一 朗
日本建築学会(大阪大学)
松 田 邦 男
川崎製鉄株式会社
岡 井 遼 二
社団法人高圧ガス保安協会
石 田 安 正
株式会社神戸製鋼所
小 峰 武 夫
日本工具工業会(コベルコツールエンジニアリング株式会社)
大 橋 守
新日本製鋼株式会社
福 永 規
住友金属工業株式会社
富 沢 精 治
線材製品協会(鈴木金属工業株式会社)
白 谷 勝 典
大同特殊鋼株式会社
大 山 康 郎
鉄管継手協会
上津原 政 則
トーア・スチール株式会社
山 田 健太郎
社団法人土木学会(名古屋大学)
三 浦 恒 幸
エンジニアリング振興協会(日揮株式会社)
北 田 博 重
社団法人日本海事検定協会
橋 本 繁 晴
財団法人日本規格協会
柴 田 正 宣
日本鋼管株式会社
本 野 光 彦
社団法人日本水道協会
川 原 雄 三
日本機械工業連合会(三菱重工業株式会社)
金 子 純 一
日本大学
井 波 隆 夫
社団法人軽金属協会
菅 野 久 勝
日本試験機工業会
藤 沢 裕
日本伸銅協会
束 原 巌
株式会社第一原子力グループ放射線研究所
橋 本 勝
株式会社日産アーク
嶋 貫 孝
社団法人日本分析化学会
永 山 宏
日立協和エンジニアリング株式会社
(事務局)
桃 木 明 和
社団法人日本鉄鋼連盟
(関係者)
森 下 昇
社団法人日本鉄鋼連盟
稲 本 勇
社団法人日本鉄鋼連盟