2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
G 1220-1994
鉄及び鋼−タングステン定量方法
Iron and steel−Methods for
determination of tungsten content
1. 適用範囲 この規格は,鉄及び鋼中のタングステン定量方法について規定する。
備考 この規格の引用規格を,次に示す。
JIS G 1201 鉄及び鋼の分析方法通則
JIS G 1217 鉄及び鋼中のクロム定量方法
JIS G 1218 鉄及び鋼−モリブデン定量方法
JIS G 1221 鉄及び鋼中のバナジウム定量方法
JIS K 8001 試薬試験方法通則
JIS Z 8402 分析・試験の許容差通則
2. 一般事項 定量方法に共通な一般事項は,JIS G 1201による。
3. 定量方法の区分 タングステンの定量方法は,次のいずれかによる。
(1) シンコニン沈殿分離酸化タングステン (VI) 重量法 この方法は,タングステン含有率0.5% (m/m) 以
上20% (m/m) 以下の試料に適用するもので,附属書1による。
(2) タンニン酸ニオブ共沈分離チオシアン酸塩吸光光度法 この方法は,タングステン含有率0.05% (m/m)
以上7.0% (m/m) 以下の試料に適用するもので,附属書2による。ただし,チタン含有率0.25% (m/m)
以上,ニオブ含有率1.5% (m/m) 以上及び/又はタンタル含有率1.5% (m/m) 以上の試料には適用でき
ない。
(3) モリブデン分離テトラフェニルアルソニウムクロリド・チオシアン酸塩抽出吸光光度法 この方法は,
タングステン含有率0.01% (m/m) 以上7.0% (m/m) の試料に適用するもので,附属書3による。
2
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1 シンコニン沈殿分離酸化タングステン (VI) 重量法
1. 要旨 試料を適切な酸で分解し,タングステンをタングステン酸とし,シンコニンを加えてタングス
テンを完全に沈殿させる。沈殿をこし分け,強熱した後,硫酸とふっ化水素酸とで処理して二酸化けい素
を除去し,再び,強熱して不純酸化タングステン (VI) の質量をはかる。次に,不純酸化タングステンを
炭酸ナトリウムで融解し,温水に溶かしてこし分け,不溶解残さを強熱して質量をはかり,不純酸化タン
グステン (VI) の質量から差し引く。
2. 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1)
(3) 硝酸
(4) 過塩素酸
(5) ふっ化水素酸
(6) ふっ化水素酸 (1+10)
(7) 硫酸 (1+1)
(8) りん酸
(9) アンモニア水
(10) 水酸化ナトリウム溶液 (500g/l)
(11) 炭酸ナトリウム(無水)
(12) 炭酸カリウム(無水)
(13) 鉄溶液 (10mgFe/ml) できるだけ純度が高く,モリブデンを含有しないか,又はモリブデン含有率が
できるだけ低く,既知である鉄1.00gを過塩素酸10mlで加熱して分解し,さらに,加熱を続けて過塩
素酸の白煙を発生させる。室温まで放冷した後,水約50mlを加えて塩類を溶解する。常温まで冷却
した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
(14) 硫酸マグネシウム・塩化アンモニウム溶液 硫酸マグネシウム七水和物20g及び塩化アンモニウム20g
をはかり採ってビーカー (300ml) に移し入れ,アンモニア水2ml及び水を加えて溶解し,水で液量を
250mlとした後,ときどきかき混ぜながら穏やかに加熱して溶解し,室温まで冷却する。この溶液は,
使用の都度調製する。
(15) 硫酸マグネシウム洗浄液 硫酸マグネシウム七水和物20g及び塩化アンモニウム20gをはかり採って
ビーカー (1l) に移し入れ,水約300mlとアンモニア水4mlを加え,穏やかに加熱して溶解する。さ
らに,炭酸カリウム(無水)10gを加え,かき混ぜながら穏やかに加熱して溶解し,室温まで冷却し
た後,水で液量を1 000mlとする。
(16) 酒石酸溶液 (500g/ml)
(17) シンコニン溶液 シンコニン (C19H22N2O) 12.5gを塩酸 (1+1) 100mlに溶解する。
(18) シンコニン洗浄液 シンコニン溶液 [(17)] 30mlを水で1 000mlに薄める。
(19) ローダミンB溶液 ローダミンB (C28H31O3N2Cl) 2gを温水200mlに溶解する。
この溶液は,使用の都度調製する。

3
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(20) ローダミンB洗浄液 ローダミンB溶液 [(19)] 10mlを水で1 000mlに薄め,これに塩酸3mlを加え
る。この溶液は,使用の都度調整する。
(21) メチルオレンジ溶液 調製方法は,JIS K 8001の4.4(指示薬)表7(中和滴定用)による。
3. 試料はかり採り量 試料はかり採り量は,附属書1表1による。
附属書1表1 試料はかり採り量及び塩酸 (1+1) の添加量
タングステン含有率
% (m/m)
試料はかり採り量
g
塩酸 (1+1) の添加量
ml
0.5以上 1.0未満
5.0
80
1.0以上 10.0未満
2.0
60
10.0以上 20.0以下
1.0
50
4. 操作
参考 警告 過塩素酸の蒸気は,アンモニア,亜硝酸蒸気又は有機物が存在すると爆発する危険があ
る。
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかによる。
(1) 塩酸・硝酸で分解容易な試料 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆い,塩
酸 (1+1) を試料はかり採り量に応じて表1によって加え,加熱して分解する。硝酸5〜10mlを加え,
煮沸してタングステンをタングステン酸とする。時計皿の下面を水で洗って時計皿を取り除き,液量
が約15mlになるまで加熱して蒸発し,再び,時計皿で覆い,さらに,加熱して酸化窒素などを発生
させ,約10mlになるまで濃縮する(1)。室温まで放冷した後,時計皿の下面を水で洗って時計皿を取
り除き,塩酸 (1+1) 10ml及び温水を加えて液量を約100mlとする。
注(1) 工具鋼,高速度鋼など,タングステン含有率が5% (m/m) 以上の試料の場合は,ふっ化水素酸 (1
+10) 5mlを加え,引き続き加熱して液量が約5mlになるまで濃縮する。
(2) 塩酸・硝酸で分解困難な試料 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆い,塩
酸及び硝酸をそれぞれ30mlずつ加えて加熱して分解する。過塩素酸を15ml,更に試料1gにつき7ml
ずつ加えた後,ふっ化水素酸1〜3mlを加えて加熱を続け,過塩素酸の白煙を発生させてクロムを酸
化し,引き続き約5分間白煙を発生させる。室温まで放冷した後,時計皿の下面を水で洗って時計皿
を取り除き,温水を加えて液量を約200mlとする。
4.2
酸化タングステン (VI) の分離とひょう量 酸化タングステン(VI)の分離とひょう量は,次のいずれ
かによる。
(1) ニオブ及びタンタルを含まない試料
(a) 4.1の(1)又は(2)で得た溶液に,シンコニン溶液 [2.(17)] 5mlを加えて加熱し,ときどき振り混ぜなが
ら,30〜60分間約90℃に保持する。室温まで冷却した後(2),沈殿を少量のろ紙パルプを加えたろ紙
(6種)を用いてこし分け,温シンコニン洗浄液 [2.(18)] で洗液に鉄イオンが認められなくなるま
で洗浄する。ろ液と洗液は捨てる。沈殿は,ろ紙と共に,質量既知の白金るつぼ(30番)に移し入
れる。ビーカーの内壁に沈殿が付着している場合は,アンモニア水で湿したろ紙(6種)の小片で
ぬぐい取って,沈殿を入れた白金るつぼに合わせる。るつぼを穏やかに加熱して乾燥し,次第に温
度を高めて,ろ紙などを灰化した後,750〜800℃の範囲で強熱し,室温まで放冷する。硫酸 (1±1)
2,3滴及びふっ化水素酸3〜5mlを加え,加熱して蒸発乾固する。
4
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(b) 沈殿の入った白金るつぼを750〜800℃の範囲で強熱し,デシケーター中で常温まで放冷して質量を
はかる。
(c) (b)の操作を繰り返して恒量とし,その質量から(a)で用いた質量既知の白金るつぼの質量を差し引く。
注(2) タングステン含有率が1% (m/m) 未満の場合は,室温で更に一夜間放置する。
(2) ニオブ及びタンタルを含む試料
(a) (1)(a)の操作を行う。
(b) 沈殿の入った白金るつぼを750〜800℃の範囲で強熱し,室温まで放冷する。炭酸カリウム(無水)
4gを加え,加熱して融解する。室温まで放冷した後,温水約20mlを加え,穏やかに加熱して融成
物を溶解する。
(c) 溶液をビーカー (500ml) に水を用いて移し入れ,水を加えて液量を約200mlとし,硫酸マグネシウ
ム・塩化アンモニウム溶液 [2.(14)] 25mlを加えてよく振り混ぜ,1〜4時間放置する。沈殿は,ろ紙
(6種)を用いてろ過する。硫酸マグネシウム洗浄液 [2.(15)] で5,6回洗浄し,ろ紙と残さは捨て
る。ろ液及び洗液をビーカー (500ml) に集め,メチルオレンジ溶液 [2.(21)] を指示薬として1,2
滴加え,溶液の色が黄色から赤に変わるまで塩酸を滴加した後,溶液100mlにつき,塩酸1mlを加
える。この溶液を煮沸するまで加熱した後,かき混ぜながらローダミンB溶液 [2.(19)] 20mlを加え,
1〜2分間煮沸して沈殿を凝集させる。しばらく静置し沈殿を沈降させた後,沈殿を少量のろ紙パル
プを加えたろ紙(6種)を用いてこし分け,ローダミンB洗浄液 [2.(20)] で5,6回洗浄する。沈殿
を,ろ紙と共に,質量既知の白金るつぼ(30番)に移し入れ,ビーカーの内壁に付着した沈殿は,
アンモニア水で湿したろ紙(6種)の小片でぬぐい取って沈殿を入れたるつぼ内の沈殿に合わせる。
ろ液と洗液は捨てる。るつぼを穏やかに加熱して乾燥し,次第に温度を高め,ろ紙を灰化する。
(d) るつぼを750〜800℃の範囲で強熱し,デシケーター中で常温まで放冷した後,質量をはかる。
(e) (d)の操作を繰り返して恒量とし,その質量から(c)で用いた質量既知の白金るつぼの質量を差し引く。
4.3
不純酸化タングステン (VI) の分解及び炭酸ナトリウム不溶性不純物の定量 4.2の(1)(c)又は(2)(e)
で得た質量の約10倍量(約1〜3g)の炭酸ナトリウム(無水)を4.2の(1)(c)又は(2)(e)で恒量とした白金
るつぼに加え,加熱して融解する。室温まで放冷した後,白金るつぼ中に温水約10mlを加え,穏やかに
加熱して融成物を溶解する。溶液をろ紙(5種A)を用いてろ過し,白金るつぼ及びろ紙を温水でよく洗
浄し,ろ液及び洗液をビーカー (200ml) に集めて保存する。ろ紙上の残さは,ろ紙と共に,元の白金るつ
ぼに移し,穏やかに加熱して乾燥する。次第に温度を高めて,ろ紙を灰化し,以下,4.2(2)の(d)及び(e)の
手順に従って操作する。
4.4
不純酸化タングステン (VI) 中のクロム,モリブデン及びバナジウムの定量 不純酸化タングステン
(VI) 中のクロム,モリブデン及びバナジウムの定量は,次の手順によって行う。
(1) 定量用溶液の調製 4.3で保存しておいた溶液を加熱して蒸発し,液量が約70mlになるまで濃縮する。
常温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
(2) クロムの定量(3)
(a) (1)で得た溶液から20ml(4)を分取してビーカー (200ml) に移し入れる。りん酸で中和し,更に2ml
を過剰に加え,水で液量を約60mlとした後,硫酸 (1+1) 1mlを加える。
(b) (a)で得た溶液を用いて,JIS G 1217の7.(ジフェニルカルバジド吸光光度法)の操作の項の7.5.3
(呈色)の“ジフェニルカルバジド溶液 [7.3(10)] 3mlを正確に加えて振り混ぜ,約1分間静置して
から,ふっ化水素酸 (1+11) 5mlを加え,水で標線まで薄める。”以降,7.5.4(吸光度の測定)及び
7.6(検量線の作成)の手順に従って処理し,7.5.4で得た吸光度と7.6で作成した検量線(5)とからク
5
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ロムの量 (Cr) を求める。
(c) 次の式によって酸化クロム (III) の量 (Cr2O3) を求める。
Cr2O3=Cr×1.462
注(3) 試料中にクロムが含まれる場合に行う。
(4) (1)で得た溶液中に含まれるクロムの量によっては,20mlの分取液量を増減する必要がある。
(5) クロム定量用の検量線は,鉄を添加せずに作成する。
(3) モリブデンの定量(6) (1)で得た溶液から20ml(7)を分取してビーカー (100ml) に移し入れる。塩酸を
滴加してわずかに酸性とし,鉄溶液 [2.(13)] 5ml及び過塩素酸2mlをそれぞれ正確に加え,時計皿で
覆い,加熱してわずかに過塩素酸の白煙が発生するまで濃縮した後,室温まで放冷し,酒石酸溶液5ml
を加える。溶液を水酸化ナトリウム溶液で中和して,わずかにアルカリ性として酸化タングステン
(VI) を溶解する。室温まで冷却した後,時計皿の下面を洗って時計皿を取り除く。過塩素酸を滴加し
て中和した後,更に8mlを過剰に加え,常温まで冷却した後,溶液を50mlの全量フラスコに水を用
いて移し入れる。以下,JIS G 1218の附属書2(チオシアン酸塩吸光光度法)の4.3(呈色)〜6.(検
量線の作成)の手順に従って操作し,4.4で得た吸光度と6.で作成した検量線とからモリブデンの量
(Mo) を求め,次の式によって酸化モリブデン (VI) の量 (MoO3) を求める。
MoO3=Mo×1.500
注(6) 試料中にモりブデンが含まれる場合に行う。
(7) (1)で得た溶液中に含まれるモリブデンの量によっては,20mlの分取液量を増減する必要がある。
(4) バナジウムの定量(8)
(1)で得た溶液から20ml(9)を分取してビーカー (200ml) に移し入れる。りん酸
5ml及び過塩素酸20mlを加え,時計皿で覆い,加熱して過塩素酸の白煙を発生させ,ビーカーの内部
が透明になってから,更に2〜3分間加熱を続ける。以下,JIS G 1221の5.(N-BPHA抽出吸光光度法)
における5.5.2(二クロム酸の還元)〜5.6(検量線の作成)の手順に従って操作し,5.5.5(吸光度の測
定)で測定した吸光度と5.6で作成した検量線とからバナジウムの量 (V) を求め,次の式によって酸
化バナジウム (V) の量 (V2O5) を求める。
V2O5=V×1.785
注(8) 試料中にバナジウムが含まれる場合に行う。
(9) (1)で得た溶液中に含まれるバナジウムの量によっては,20mlの分取液量を増減する必要がある。
5. 空試験 試薬だけを用いて,試料と同じ操作を試料と併行して行う。
6. 計算 試料中のタングステン含有率 [W% (m/m)] を,次の式によって算出する。
100
0
793
.0
)
100
100
100
(
m/m
%
W
0
3
5
2
4
1
3
2
1
×
×
+
×
+
×
+
×
+
−
=
m
m
B
m
B
m
B
m
m
m
)
(
ここに, m1: 4.2の(1)(c)又は(2)(e)で得た質量 (g)
m2: 4.3で得た質量 (g)
m3: 4.4.(2)(c)で得た酸化クロム (III) の量 (g)
B1: 4.4(2)(a)で分取した溶液の量 (ml)
m4: 4.4(3)で得た酸化モリブデン (VI) の量 (g)
B2: 4.4(3)で分取した溶液の量 (ml)
m5: 4.4(4)で得た酸化バナジウム (V) の量 (g)

6
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
B3: 4.4(4)で分取した溶液の量 (ml)
m0: 5.で得た質量 (g)
m: 試料はかり採り量 (g)
7. 許容差 許容差(10)は,附属書1表2による。
附属書1表2 許容差(10)
単位% (m/m)
タングステン含有率
室内再現許容差
室間再現許容差
0.5以上 6.0未満
D [0.004 7× (W) +0.004 4]
D [0.008 3× (W) +0.009 8]
6.0以上 20.0以下
D [0.001 2× (W) +0.026 3]
D [0.000 1× (W) +0.074 5]
注(10) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4[D (n,
0.95) の値]による。nの値は,室内再現許容差の場合は同一分析室内における
分析回数,室間再現許容差の場合は分析に関与した分析室数である。
また, (W) は,許容差を求めるタングステン含有率 [% (m/m)] である。
参考 この許容差は,タングステン含有率1.1% (m/m) 以上17.1% (m/m) 以下の試料を用い,共同実
験した結果から求めたものである。
7
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2 タンニン酸ニオブ共沈分離チオシアン酸塩吸光光度法
1. 要旨 試料に共沈剤としてニオブを加え,王水,過塩素酸及びふっ化水素酸で分解し,過塩素酸の白
煙を発生させてクロムを酸化した後,塩酸,亜硫酸ナトリウム及びタンニン酸を加えてタングステンをニ
オブと共に完全に沈殿させる。沈殿をこし分け,硝酸,過塩素酸,硫酸及びりん酸の混酸で分解した後,
塩化すず (II) でタングステンを還元し,チオシアン酸アンモニウムでタングステンのチオシアン酸錯体を
生成させ,光度計を用いて,その吸光度を測定する。
2. 試薬 試薬は,次による。
(1) 塩酸 (1+1)
(2) 硝酸
(3) 過塩素酸
(4) ふっ化水素酸 (1+3)
(5) 硫酸 (3+7)
(6) りん酸 (1+1)
(7) 王水(塩酸3,硝酸1)
(8) 混酸(過塩素酸1,硫酸1,りん酸1,水1)
(9) 銅溶液 銅1.0gを硝酸20mlで分解し,過塩素酸20mlを加え,加熱して白煙を発生させる。室温まで
放冷した後,水約50mlで塩類を溶解し,水で液量を100mlとする。
(10) ニオブ溶液 (5mgNb/ml) ニオブ5.00gをはかり採って白金皿 (100ml) に移し入れ,硝酸20mlを加
え,ふっ化水素酸を滴加して分解し,室温まで冷却した後,ふっ化水素酸 (1±10) で液量を1 000ml
とする。この溶液は,ポリエチレン製容器に保存する。
(11) 塩化すず (II) 溶液 塩化すず (II) 二水和物35gを塩酸100mlに溶解し,塩酸で液量を500mlとする。
この溶液は,使用の都度調製する。
(12) 亜硫酸ナトリウム溶液 亜硫酸ナトリウム七水和物100gを水に溶解し,水で液量を100mlとする。
この溶液は,使用の都度調製する。
(13) よう化カリウム溶液 (300g/l) この溶液は,使用の都度調製する。
(14) 過マンガン酸カリウム溶液 (3g/l)
(15) チオシアン酸アンモニウム溶液 (200g/l)
(16) L (+) −アスコルビン酸溶液 (10g/l) この溶液は,使用の都度調製する。
(17) タンニン酸溶液 (10g/l) この溶液は,使用の都度調製する。
(18) タンニン酸洗浄液 塩酸 (1+100) 1 000mlに,タンニン酸溶液 [(17)] 10mlを加える。この溶液は,使
用の都度調製する。
(19) しゅう酸アンモニウム溶液(飽和,約50g/l)
(20) 標準モリブデン溶液 (1.0mgMo/ml) モリブデン[99.9% (m/m) 以上]1.00gをはかり採ってビーカー
(300ml) に移し入れ,時計皿で覆い,混酸(硝酸1,硫酸1,水2)10mlを加え,加熱して分解する。
時計皿の下面を水で洗って時計皿を取り除き,再び加熱して硫酸の白煙を発生させる。室温まで放冷

8
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
した後,水約30mlを加えて塩類を溶解し,常温まで冷却する。溶液を1 000mlの全量フラスコに水を
用いて移し入れ,水で標線まで薄める。
(21) 標準バナジウム溶液 (1.0mgV/ml) バナジン酸アンモニウム (NH4VO3) 0.229 6gをはかり採ってビー
カー (100ml) に移し入れ,時計皿で覆い,水約50mlを加えて加熱して溶解する。常温まで冷却した
後,時計皿の下面を水で洗って時計皿を取り除き,溶液を100mlの全量フラスコに水を用いて移し入
れ,水で標線まで薄める。
(22) 標準タングステン溶液 (1.0mgW/ml) タングステン酸ナトリウム二水和物 (Na2WO4・2H2O) 1.794g
を水約300mlに溶解し,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
3. 試料はかり採り量 試料はかり採り量は,附属書2表1による。
附属書2表1 試料はかり採り量
タングステン含有率
% (m/m)
試料はかり採り量
g
0.05以上2.0未満
1.0
2.0 以上 4.0未満
0.50
4.0 以上 7.0以下
0.20
4. 操作
参考 警告 過塩素酸の蒸気は,アンモニア,亜硝酸蒸気又は有機物が存在すると爆発する危険があ
る。
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採ってビーカー (300ml) に移し入れ,はかり採った試料中のニオブ量が5mg以下の場合
には,ニオブ溶液 [2.(10)] 1.0mlを加える。時計皿で覆い,王水30mlを加え,加熱して分解する。過
塩素酸20〜30ml及びふつ化水素酸1mlを加え,引き続き加熱して過塩素酸の白煙を発生させ,クロ
ムを酸化した後,さらに,5〜10分間加熱する。室温まで放冷した後,水約50mlを加えて塩類を溶解
し,塩酸 (1+1) 10mlを加え,水で液量を約100mlとする。亜硫酸ナトリウム溶液 [2.(12)] 20ml,タン
ニン酸溶液 [2.(17)] 10ml及びろ紙(6種)の小片を加え,加熱して約5分間煮沸する。約30分間放置
した後,時計皿の下面を水で洗って時計皿を取り除き,沈殿をろ紙(6種)を水中で細かく砕いたろ
紙パルプを用いてこし分け,タンニン酸洗浄液 [2.(18)] で5,6回洗浄する。ろ液と洗液は捨てる。
(2) 沈殿をろ紙パルプと共に元のビーカーに移し入れ,時計皿で覆い,混酸20ml及び硝酸30mlを加え,
加熱してろ紙などを分解する(1)。時計皿の下面を水で洗って時計皿を取り除き,引き続き加熱して硫
酸白煙を発生させ,さらに,1〜2分間加熱する。室温まで放冷した後,水約50mlを加えて振り混ぜ,
常温まで冷却する。溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める(2)。
注(1) ろ紙の分解が不完全なときは,硝酸を適宜添加して分解する。
なお,ろ紙が未分解の間は,必ず硝酸が溶液中に残存するようにして,過塩素酸と有機物と
の反応による爆発を防止しなければならない。
(2) この溶液は,2時間以上放置してはならない。
4.2
呈色 呈色は,次のいずれかによる。
(1) タングステン含有率が0.25% (m/m) 未満の試料
(a) 4.1(2)で得た溶液を20ml分取してビーカー (100ml) に移し入れ,時計皿で覆い,加熱して液量が5ml
9
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
以下になるまで濃縮し,常温まで冷却する。
(b) 溶液をあらかじめ乾燥してある100mlの全量フラスコに塩化すず (II) 溶液 [2.(11)] 40mlを用いて
移し入れ,60℃の水浴中で10分間加熱する。
(c) しゅう酸アンモニウム溶液(飽和)10mlを加え,流水中で常温まで冷却した後,チオシアン酸アン
モニウム溶液10mlを加え,水で標線まで薄め,15〜20℃の水浴中に10分間放置する。
(2) タングステン含有率が0.25% (m/m) 以上の試料 4.1(2)で得た溶液5mlを分取して,あらかじめ乾燥
してある100mlの全量フラスコに移し入れ,塩化すず (II) 溶液 [2.(11)] 40mlを加え,60℃の水浴中で
10分間加熱する。以下,(1)(c)の操作を行う。
4.3
吸光度の測定 4.2の(1)(c)又は(2)で得た呈色溶液の一部を,呈色後20分以内に光度計の吸収セル (
10mm) に移し入れ,水を対照液として波長400nm付近の吸光度を測定する(3)。
注(3) 4.2の(1)(a)又は(2)で分取した溶液中に,モリブデンが0.25mg以上及び/又はバナジウムが
0.02mg以上共存する場合には,ここで得た吸光度を7.によって得た補正値で補正する。
5. 空試験 試薬だけを用いて,試料と同じ操作を試料と併行して行う。
6. 検量線の作成 7個のビーカー (300ml) を準備し,それぞれに標準タングステン溶液 [2.(22)] を正確
に,0,2,4,8,12,16,20ml(タングステンとして0〜20mg)取り,混酸20mlを加え,加熱して硫酸
の白煙を発生させ,さらに,1〜2分間加熱する。室温まで放冷した後,水約50mlを加えて振り混ぜる。
常温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。これら
の溶液を20mlずつ分取してビーカー (100ml) に移し入れ,時計皿で覆い,加熱して液量が5mlになるま
で濃縮して常温まで冷却する。以下,4.2(1)の(b)及び(c)並びに4.3の手順に従って試料と併行して操作し,
得た吸光度と呈色溶液中のタングステン量との関係線を作成し,その関係線を原点を通るように平行移動
して検量線とする。
7. 妨害元素吸光度補正値の測定
7.1
モリブデン吸光度補正値の測定 モリブデン吸光度補正値の測定は,次の手順によって行う。
(1) モリブデンの定量
(a) 4.1(2)で得た溶液5mlを分取して100mlの全量プラスコに移し入れ,硫酸 (3+7) 25mlを加えて振り
混ぜ,さらに,チオシアン酸アンモニウム溶液10mlを加えて振り混ぜる。次に,よう化カリウム
溶液 [2.(13)] 10mlを加えて振り混ぜ,20〜25℃で5分間放置した後,L (+) −アスコルビン酸溶液
[2.(16)] 0.5mlを加えて振り混ぜ,20〜25℃の水で標線まで薄めて約10分間放置する。この呈色溶液
の一部を,光度計の吸収セル (10mm) に取り,水を対照液として波長460nm付近における吸光度を
測定する。
(b) 7個のビーカー (300ml) を準備し,それぞれに標準モリブデン溶液 [2.(20)] を正確に,0,1,2,4,
6,8,10ml(モリブデンとして0〜10mg)取り,混酸20mlを加え,加熱して硫酸の白煙を発生さ
せ,さらに,2〜5分間加熱を続ける。室温まで放冷した後,水50mlを加えて塩類を溶解する。常
温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。こ
れらの溶液から5mlずつ分取してそれぞれの100mlの全量フラスコに移し入れ,硫酸 (3+7) 25ml
を加えて振り混ぜ,さらに,チオシアン酸アンモニウム溶液10mlを加えて振り混ぜる。次に,よ
う化カリウム溶液 [2.(13)] 10mlを加えて振り混ぜ,20〜25℃で5分間放置した後,L (+) −アスコ
10
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ルビン酸溶液 [2.(16)] 0.5mlを加えて振り混ぜ,20〜25℃の水で標線まで薄めて約10分間放置する。
この呈色溶液の一部を,光度計の吸収セル (10mm) に取り,水を対照液として波長460nm付近にお
ける吸光度を測定し,得た吸光度と呈色溶液中のモリブデン量との関係線を作成して検量線とする。
(c) (a)で得た吸光度と(b)で作成した検量線とからモリブデン量を求める。
(2) 吸光度補正値の読取り
(a) 7個のビーカー (300ml) を準備し,それぞれに標準モリブデン溶液 [2.(20)] を正確に,0,1,2,4,
6,8,10ml(モリブデンとして0〜10mg)取り,混酸20mlを加え,加熱して硫酸の白煙を発生さ
せ,更に1〜2分間加熱する。室温まで放冷した後,水約50mlを加えて振り混ぜる。常温まで冷却
した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。これらの溶液
を5mlずつ分取して,あらかじめ乾燥してある100mlの全量フラスコに移し入れ,塩化すず (II) 溶
液 [2.(11)] 40mlを加え,60℃の水浴中で10分間加熱する。以下,4.2(1)(c)及び4.3の手順に従って
操作し,得た吸光度と呈色溶液中のモリブデン量との関係線を作成し,その関係線を原点を通るよ
うに平行移動してモリブデン吸光度補正線とする。
(b) (a)で得た補正線と(c)で求めたモリブデンの量とから(1)(a)で分取した溶液中のモリブデン量に相当
する吸光度を求め,モリブデンによる吸光度補正値とする。
7.2
バナジウム吸光度補正値の測定 バナジウム吸光度補正値の測定は,次の手順によって行う。
(1) バナジウムの定量
(a) 4.1(2)で得た溶液10mlを分取して分液漏斗 (200ml) に移し入れ,りん酸 (1+1) 7m1及び銅溶液
[2.(9)] 1mlを加える。溶液を振り混ぜながら過マンガン酸カリウム溶液を滴加してわずかに赤紫を
呈してから,更に0.05mlを過剰に加えて5分間静置する。
(b) 以下,JIS G 1221の5.の5.5.4(呈色)〜5.6の手順に従って操作し,5.5.5で得た吸光度と5.6で作
成した検量線とからバナジウム量を求める。
(2) 吸光度補正値の読取り
(a) 7個のビーカー (300ml) を準備し,それぞれに標準バナジウム溶液 [2.(21)] を正確に,0,1,2,4,
6,8,10ml(バナジウムとして0〜10mg)取り,混酸20mlを加え,加熱して硫酸の白煙を発生さ
せ,更に1〜2分間加熱する。室温まで放冷した後,水約50mlを加えて振り混ぜる。常温まで冷却
した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。これらの溶液
から10mlずつ分取してビーカー (100ml) に移し入れ,時計皿で覆い,加熱して液量が5mlになる
まで濃縮し,常温まで冷却する。以下,4.2(1)の(b)及び(c)並びに4.3の手順に従って操作し,得た吸
光度と呈色溶液中のバナジウム量との関係線を作成し,その関係線を原点を通るように平行移動し
てバナジウム吸光度補正線とする。
(b) (a)で得た補正線と(1)(b)で求めたバナジウム量とから7.2(1)(a)で分取した溶液中のバナジウム量に
相当する吸光度を求め,バナジウムによる吸光度補正値とする。
7.3
吸光度の補正 吸光度の補正は,次のいずれかによる。
(1) タングステン含有率が0.25% (m/m) 未満の試料
A=A1− [(A2×4) + (A3×2)]
ここに,
A: 4.2(1)(a)で試料溶液を20ml分取した場合のタングステンだけ
の吸光度
A1: 4.2(1)(a)で試料溶液を20ml分取した場合の4.3で得た吸光度
A2: 7.1(1)(a)で試料溶液を5ml分取した場合の7.1(2)(b)で得たモ
リブデン吸光度補正値
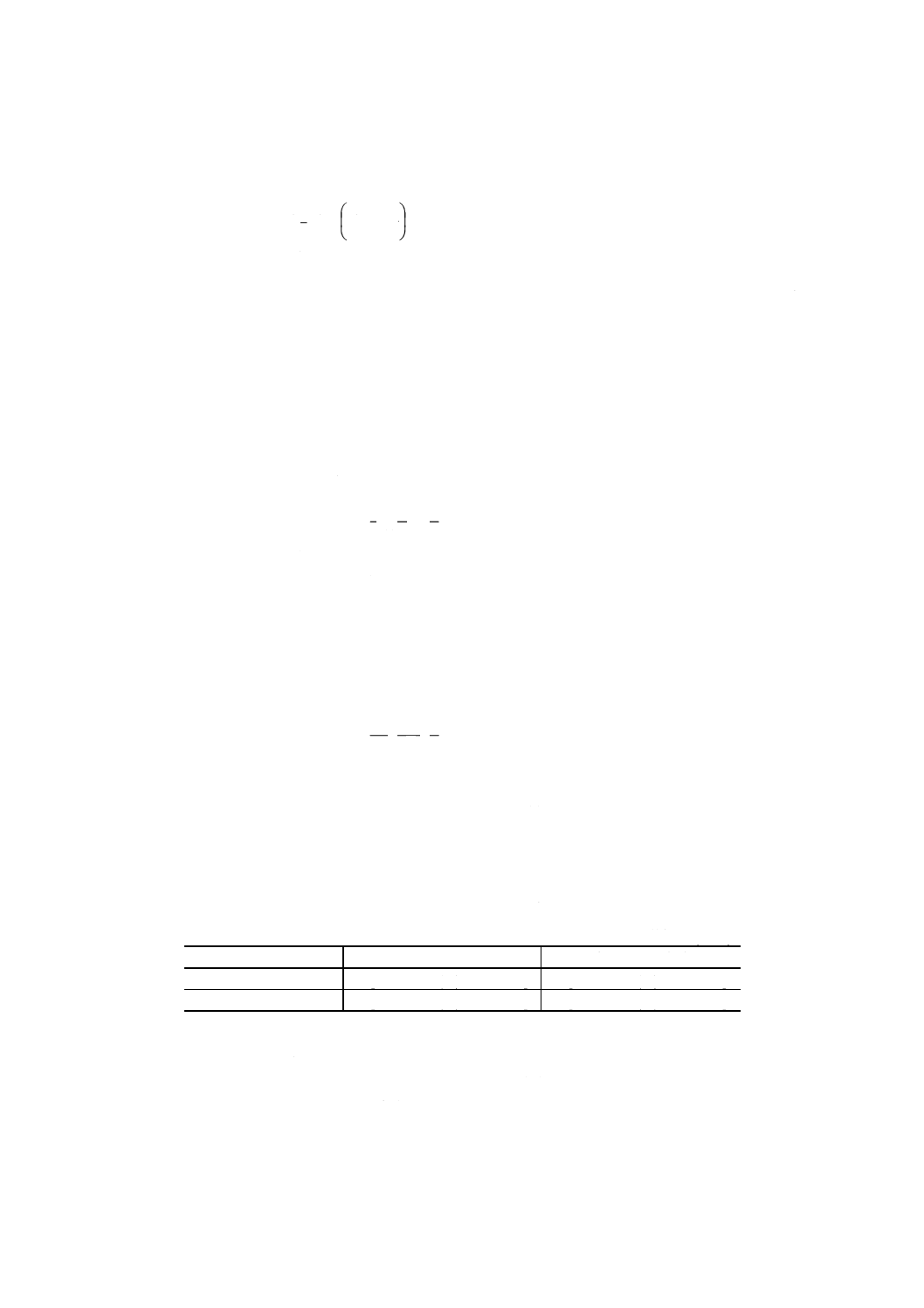
11
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A3: 7.2(1)(a)で試料溶液を10ml分取した場合の7.2(2)(b)で得たバ
ナジウム吸光度補正値
(2) タングステン含有率が0.25% (m/m) 以上の試料
+
−
=
2
3
2
1
A
A
A
A
ここに,
A: 4.2(2)(a)で試料溶液を5ml分取した場合のタングステンだけ
の吸光度
A1: 4.2(2)(a)で試料溶液を5ml分取した場合の4.3で得た吸光度
A2: 7.1(1)(a)で試料溶液を5ml分取した場合の7.1(2)(b)で得たモ
リブデン吸光度補正値
A3: 7.2(1)(a)で試料溶液を10ml分取した場合の7.2(2)(b)で得たバ
ナジウム吸光度補正値
8. 計算 計算は,次のいずれかによる。
(1) 注(3)の補正を行わない場合 4.3及び5.で得た吸光度と6.で作成した検量線とからタングステン量を
求め,試料中のタングステン含有率 [W% (m/m)] を次の式によって算出する。
100
/
m/m
W%
0
1
B
m
m
m
×
−
=
)
(
ここに, m1: 分取した試料溶液中のタングステン検出量 (g)
m0: 分取した空試験液中のタングステン検出量 (g)
m: 試料はかり採り量 (g)
B: 試料溶液及び空試験溶液の分取量 (ml)
(2) 注(3)の補正を行った場合 7.3の(1)又は(2)で得たタングステンだけの吸光度及び5.で得た吸光度と6.
で作成した検量線とからタングステン量を求め,試料中のタングステン含有率 [W% (m/m)] を次の式
によって算出する。
100
/
m/m
W%
0
1
B
m
m
m
×
−
=
)
(
ここに, m1: 分取した試料溶液中のタングステン検出量 (g)
m0: 分取した空試験液中のタングステン検出量 (g)
m: 試料はかり採り量 (g)
B: 試料溶液及び空試験溶液の分取量 (ml)
9. 許容差 許容差(4)は,附属書2表2による。
附属書2表2 許容差(4)
単位% (m/m)
タングステン含有率
室内再現許容差
室間再現許容差
0.05以上1.0未満
D [0.009 3× (W) +0.001 9]
D [0.013 5× (W) +0.003 9]
1.0 以上7.0以下
D [0.008 7× (W) +0.004 6]
D [0.014 2× (W) −0.003 4]
注(4) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4による。
nの値は,室内再現許容差の場合は同一分析室内における分析回数,室間再現許
容差の場合は分析に関与した分析室数である。
また, (W) は,許容差を求めるタングステン含有率 [% (m/m)] である。
参考 この許容差は,タングステン含有率0.05% (m/m) 以上7.0% (m/m) 未満の試料を用いて共同実
験した結果から求めたものである。
12
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3 モリブデン分離テトラフェニルアルソ
ニウムクロリド・チオシアン酸塩抽出
吸光光度法
1. 要旨 試料を王水で分解し,硫酸及びりん酸を加え,硫酸の白煙を発生させた後,塩酸の濃度を調節
し,モリブデンをイソプロピルエーテルで抽出して分離する。塩化すず (II) を加えてタングステンを還元
した後,しゅう酸,テトラアルソニウムクロリド(以下,TPACという。)及びチオシアン酸カリウムを加
え,生成するTPAC・チオシアン酸・タングステン錯体をクロロホルムに抽出し,光度計を用いて,その
吸光度を測定する。
2. 試薬 試薬は,次による。
(1) 塩酸 (2+1)
(2) 王水(塩酸3,硝酸1)
(3) 混酸(硫酸1,りん酸1,水1)
(4) 鉄 できるだけ純度の高い鉄で,タングステンを含有しないか,又はタングステン含有率ができるだ
け低く,既知であるもの。
(5) 鉄溶液 (1mgFe/ml) 鉄 [(4)] 0.100gを王水10mlで加熱して分解し,混酸15mlを加え,加熱して2〜
3分間硫酸の白煙を発生させる。室温まで放冷した後,塩酸 (2+1) 約50mlを加えて塩類を溶解する。
常温まで冷却した後,溶液を100mlの全量フラスコに塩酸 (2+1) を用いて移し入れ,塩酸 (2+1) で
標線まで薄める。
(6) 塩化すず (II) 溶液 塩化すず (II) 二水和物20gを塩酸 (2+1) 約30mlに溶解し,塩酸 (2+1) で液
量を100mlとする。この溶液は,使用の都度調製する。
(7) チオシアン酸カリウム溶液 (150g/l)
(8) しゅう酸溶液 しゅう酸15gを塩酸 (2+1) 500mlに溶解する。
(9) TPAC溶液 TPAC [(C6H5) 4AsCl] 1gを水100mlに溶解する。
(10) イソプロピルエーテル
(11) クロロホルム
(12) 標準タングステン溶液 (1.0mgW/ml) タングステン酸ナトリウム二水和物 (Na2WO4・2H2O) 1.794g
をはかり採り,温水約100mlに溶解した後,常温まで冷却し,溶液を1 000mlの全量フラスコに水を
用いて移し入れ,水で標線まで薄める。
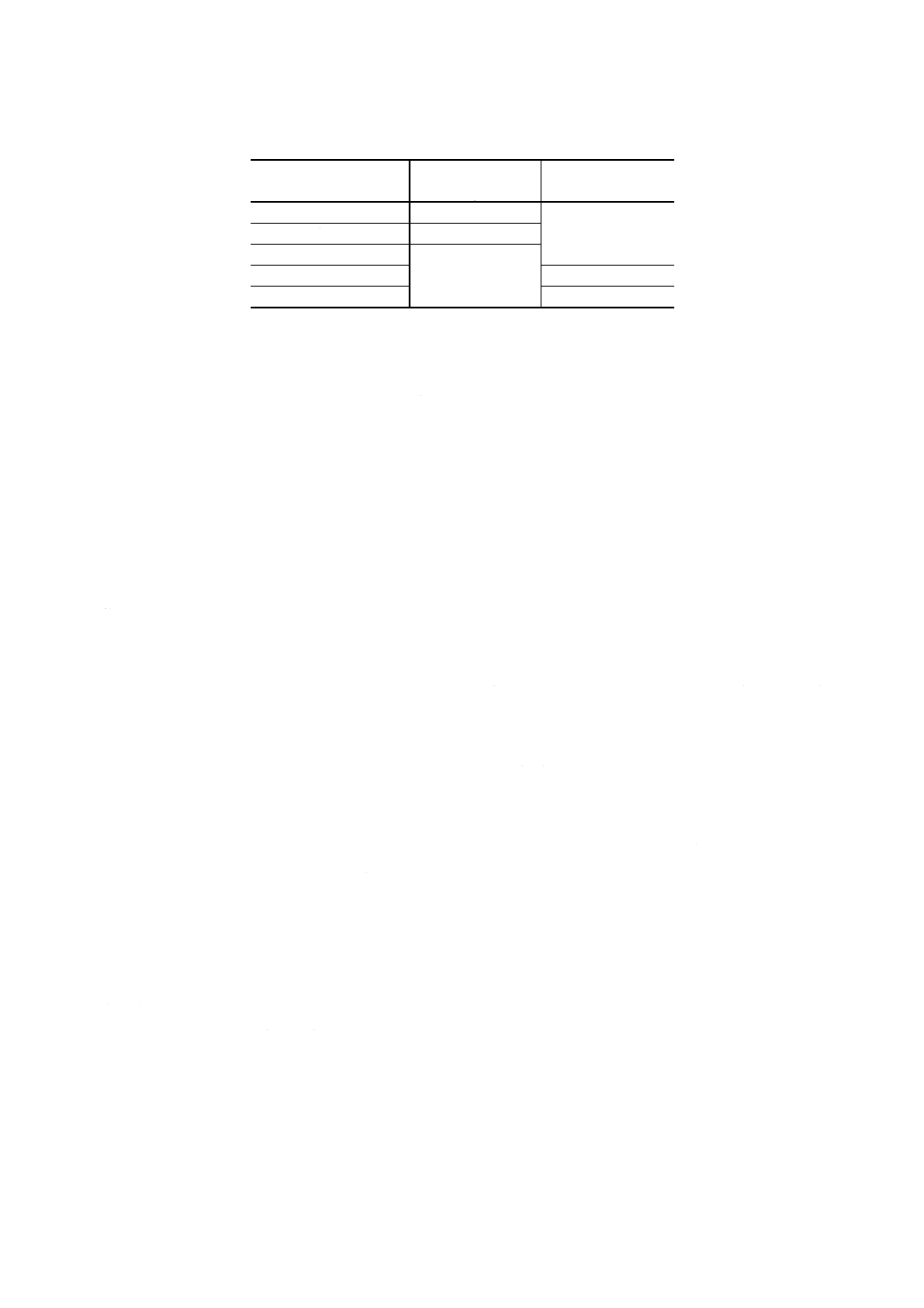
3. 試料はかり採り量 試料はかり採り量は,附属書3表1による。
13
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3表1 試料はかり採り量及び試料溶液の分取量
タングステン含有率
% (m/m)
試料はかり採り量
g
試料溶液の分取量
ml
0.01 以上 0.40 未満
0.50
10
0.40 以上 1.0 未満
0.20
1.0 以上 2.0 未満
0.10
2.0 以上 4.0 未満
5
4.0 以上 7.0 以下
2
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
(1) 試料をはかり採ってビーカー (100ml) に移し入れる。
(2) 時計皿で覆い,王水10mlを加え,加熱して分解する。混酸15mlを加え,加熱して濃縮する。時計皿
の下面を水で洗って取り除き,再び加熱して2〜3分間硫酸の白煙を発生させた後,室温まで放冷する。
塩酸 (2+1) 約30mlを加えてよく振り混ぜ,再び時計皿で覆い,穏やかに加熱して塩類を溶解する。
常温まで冷却した後,時計皿の下面を塩酸 (2+1) で洗って時計皿を取り除き,溶液を100mlの全量
フラスコに塩酸 (2+1) を用いて移し入れ,塩酸 (2+1) で標線まで薄める。
4.2
モリブデンの分離 モリブデンの分離は,次の手順によって行う。
(1) 4.1 (2) で得た溶液を表1に従って分取する(1)。
(2) 分取した溶液を分液漏斗 (100ml) に移し入れる。分取量が5mlの場合は,鉄溶液 [2.(5)] を正確に5ml,
分取量が2mlの場合は,鉄溶液 [2.(5)] を正確に8ml加える。イソプロピルエーテル20mlを加え,10
〜15秒間激しく振り混ぜる。静置して二層に分離した後,下層の水相をビーカー (100ml) に移し入れ,
分液漏斗内を塩酸 (2+1) 5mlで2回洗浄し,その都度,水相を先のビーカー (100ml) に合わせる。有
機相は捨てる。
注(1) 分取した溶液中のモリブデン量が0.4mg未満の場合には,附属書3表1に従って分取した試料溶
液を直接ビーカー (100ml) に移し入れ,次の(2)の操作を省略し,4.3の手順に移る。
4.3
呈色及び抽出 呈色及び抽出は,次の手順によって行う。
(1) 4.2(2)で得た溶液に塩化すず (II) 溶液 [2.(6)] 10mlを加えて振り混ぜる。
(2) 時計皿で覆い,熱板上で加熱し,煮沸による気泡が出始めたら,直ちに熱板から降ろし,常温まで冷
却する。溶液を分液漏斗 (200ml) に移し入れ,ビーカー内をしゅう酸溶液 [2.(8)] 30mlで洗って洗液
を分液漏斗に合わせた後,TPAC溶液 [2.(9)] を正確に1.6ml加え,1分間激しく振り混ぜる。チオシ
アン酸カリウム溶液3mlを加えて振り混ぜた後,クロロホルムを正確に20ml加え,1分間激しく振り
混ぜた後,静置する。
4.4
吸光度の測定 4.3(2)で得た下層の有機相を乾いたろ紙(5種A)を用いてろ過し,その一部を光度
計の吸収セル (10mm) に移し入れ,クロロホルムを対照液として波長404nm付近における吸光度を4.3(2)
の操作終了後10分間以内に測定する。
5. 空試験 4.1(1)ではかり採った試料と同量の鉄 [2.(4)] をはかり採ってビーカー (100ml) に移し入れ,
以下,4.1(2)〜4.4の手順に従って試料と同じ操作を試料と併行して行う。
6. 検量線の作成 検量線の作成は,次の手順によって行う。

14
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 7個のビーカー (100ml) を準備し,それぞれに鉄 [2.(4)] を0.200gずつはかり採って移し入れ,標準
タングステン溶液 [2.(12)] を正確に,0,0.2,0.4,0.8,1.2,1.6,2.0ml(タングステンとして0〜2.0mg)
加える。時計皿で覆い,王水10mlを加え,加熱して分解する。さらに,混酸15mlを加え,加熱して
濃縮する。時計皿の下面を水で洗って時計皿を取り除き,再び加熱して2〜3分間硫酸の白煙を発生さ
せた後,室温まで放冷する。塩酸 (2+1) 30mlを加えてよく振り混ぜ,再び時計皿で覆い,穏やかに
加熱して塩類を溶解し,常温まで冷却した後,時計皿の下面を塩酸 (2+1) で洗って時計皿を取り除
き,100mlの全量フラスコに塩酸 (2+1) を用いて移し入れ,塩酸 (2+1) で標線まで薄める。
(2) (1)で得た溶液から10mlを分取して(2)分液漏斗 (100ml) に移し入れる。イソプロピルエーテル20ml
を加え,10〜15秒間激しく振り混ぜる。静置して二層に分離した後,下層の水相をビーカー (100ml) に
移し入れ,分液漏斗内を塩酸 (2+1) 5mlで2回洗浄し,その都度,水相を先のビーカー (100ml) に合
わせる。有機相は捨てる。
(3) 塩化すず (II) 溶液 [2.(6)] 10mlを加えて振り混ぜた後,4.3(2)及び4.4の手順に従って試料と併行して
操作し,得た吸光度と呈色溶液中の標準タングステン溶液として加えたタングステン量との関係線を
作成し,その関係線を原点を通るように平行移動して検量線とする。
注(2) 注(1)を適用した場合は,直接,ビーカー (100ml) に移し入れて,次の(3)の操作に移る。
7. 計算 4.4及び5.で得た吸光度と6.で作成した検量線とからタングステン量を求め,試料中のタングス
テン含有率 [W% (m/m)] を次の式によって算出する。
2
0
1
100
100
/
m/m
W%
m
B
m
m
m
+
×
×
−
=
)
(
ここに, m1: 分取した試料溶液中のタングステン検出量 (g)
m0: 分取した空試験液中のタングステン検出量 (g)
m: 試料はかり採り量 (g)
B: 試料溶液及び空試験溶液の分取量 (ml)
m2: 鉄 [2.(4)] 中のタングステン含有率 [% (m/m)]
8. 許容差 許容差(3)は,附属書3表2による。
附属書3表2 許容差(3)
単位% (m/m)
タングステン含有率
室内再現許容差
室間再現許容差
0.01 以上 1.0未満
D [0.006 4× (W) +0.000 9]
D [0.020 9× (W) −0.000 1]
1.0 以上 7.0以下
D [0.008 1× (W) −0.000 9]
D [0.008 2× (W) +0.024 7]
注(3) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4による。
nの値は,室内再現許容差の場合は同一分析室内における分析回数,室間再現許
容差の場合は分析に関与した分析室数である。
また, (W) は,許容差を求めるタングステン含有率 [% (m/m)] である。
参考 この許容差は,タングステン含有率0.05% (m/m) 以上7.0% (m/m) 以下の試料を用いて共同実
験した結果から求めたものである。
15
G 1220-1994
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
原案作成委員会 構成表
(1) 社団法人日本鉄鋼協会標準化委員会JE4分科会
氏名
所属
(主査)
佐 伯 正 夫
新日本製鐵株式会社
(委員)
大 磯 義 和
工業技術院標準部
大 野 義 信
新日本製鐵株式会社
土 屋 武 久
新日本製鐵株式会社
船 曳 佳 弘
日本鋼管株式会社
磯 部 健
日本鋼管株式会社
岡 野 輝 雄
川崎製鉄株式会社(編集WG兼務)
滝 沢 佳 郎
川鉄テクノリサーチ株式会社
蔵 保 浩 文
住友金属工業株式会社
山 下 良 一
住友金属工業株式会社
金 子 晃 司
株式会社神戸製鋼所
河 村 恒 夫
株式会社コベルコ科研
伊 藤 清 孝
大同特殊鋼株式会社
藤 田 昇 平
日新製鋼株式会社
余 語 英 俊
愛知製鋼株式会社
永 井 宣太郎
日本冶金工業株式会社
(編集WG)
稲 本 勇
新日本製鐵株式会社
吉 川 裕 泰
日本鋼管株式会社
(幹事)
小 野 昭 紘
新日本製鐵株式会社(編集WG兼務)
柿 田 和 俊
社団法人日本鉄鋼協会(編集WG兼務)
大 槻 孝
社団法人日本鉄鋼協会(編集WG兼務)
(2) 社団法人日本鉄鋼協会鉄鋼JIS三者委員会鉄鋼分析JIS三者小員会
氏名
所属
(委員長)
大河内 春 乃
科学技術庁金属材料技術研究所
(委員)
青 柳 挂 一
通商産業省基礎産業局
服 部 幹 雄
工業技術院標準部
加 山 英 男
財団法人日本規格協会
藤 貫 正
社団法人日本分析化学会
広 川 吉之助
東北大学金属材料研究所
永 山 宏
日立マテリアルエンジニア株式会社
束 原 巌
古河電気工業株式会社
橋 本 勝
株式会社日産アーク
岩 田 英 夫
日本鋼管株式会社
岡 野 輝 雄
川崎製鉄株式会社
蔵 保 浩 文
住友金属工業株式会社
河 村 恒 夫
株式会社コベルコ科研
成 田 正 尚
大同特殊鋼株式会社
藤 田 昇 平
日新製鋼株式会社
(幹事)
佐 伯 正 夫
新日本製鐵株式会社
(事務局)
柿 田 和 俊
社団法人日本鉄鋼協会
大 槻 孝
社団法人日本鉄鋼協会