G 1216-1997
(1)
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。これによってJIS G 1216-1981は改正され,この規格に置き換えられる。
今回の改正では,国際規格との整合化を図るために,ISO規格の翻訳を附属書3及び附属書6として規
定している。
JIS G 1216には,次に示す附属書がある。
附属書1 ジメチルグリオキシムニッケル重量法
附属書2 ジメチルグリオキシム沈殿分離エチレンジアミン四酢酸二水素二ナトリウム・亜鉛逆滴定
法
附属書3 ジメチルグリオキシム分離定量法 (ISO 4938)
附属書4 ジメチルグリオキシム吸光光度法(1)
附属書5 ジメチルグリオキシム吸光光度法(2)
附属書6 ジメチルグリオキシム吸光光度法(3) (ISO 4939)
日本工業規格 JIS
G 1216-1997
鉄及び鋼−ニッケル定量方法
Iron and steel−Methods for determination of nickel content
序文 この規格は,附属書3に1988年に発行されたISO 4938, Steel and iron−Determination of nickel content
−Gravimetric or titrimetric methodを翻訳し,また,附属書6には,1984年に発行されたISO 4939, Steel and
cast iron−Determination of nickel content−Dimethylglyoxime spectrophotometric methodを翻訳し,技術的内容
及び規格票の様式を変更することなく作成した日本工業規格であるが,対応国際規格には規定されていな
い規定事項を日本工業規格として追加している。
なお,この規格で点線の下線を施してある“参考”は,原国際規格にはない事項である。
1. 適用範囲 この規格は,鉄及び鋼中のニッケルの定量方法について規定する。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
JIS G 1201 鉄及び鋼の分析方法通則
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS Z 8402 分析・試験の許容差通則
ISO 4938 Steel and iron−Determination of nickel content−Gravimetric or titrimetric method
ISO 4939 Steel and cast iron−Determination of nickel content−Dimethylglyoxime spectrophotometric
method
3. 一般事項 定量方法に共通な一般事項は,JIS G 1201による。ただし,JIS G 1201は,附属書3及び
附属書6には適用しない。
4. 定量方法の区分 ニッケルの定量方法は,次のいずれかによる。
(1) ジメチルグリオキシムニッケル重量法 この方法は,ニッケル含有率0.1% (m/m) 以上30% (m/m) 以
下の試料に適用し,その定量方法は,附属書1による。
(2) ジメチルグリオキシム沈殿分離エチレンジアミン四酢酸二水素二ナトリウム・亜鉛逆滴定法 この方
法は,ニッケル含有率0.1% (m/m) 以上30% (m/m) 以下の試料に適用し,その定量方法は,附属書2
による。
(3) ジメチルグリオキシム分離定量法 (ISO 4938) この方法は,ニッケル含有率0.5% (m/m) 以上30%
(m/m) 以下の試料に適用し,その定量方法は,附属書3による。
(4) ジメチルグリオキシム吸光光度法(1) この方法は,ニッケル含有率0.01% (m/m) 以上1.0% (m/m) 以
2
G 1216-1997
下の試料に適用し,その定量方法は,附属書4による。
(5) ジメチルグリオキシム吸光光度法(2) この方法は,ニッケル含有率1.0% (m/m) 以上5.0% (m/m) 以下
の試料に適用し,その定量方法は,附属書5による。
(6) ジメチルグリオキシム吸光光度法(3) (ISO 4939) この方法は,ニッケル含有率0.10% (m/m) 以上4%
(m/m) 以下の試料に適用し,その定量方法は,附属書6による。

3
G 1216-1997
附属書1 ジメチルグリオキシムニッケル重量法
1. 要旨 試料を適切な酸で分解し,くえん酸又はL (+) -酒石酸を加え,アンモニア水でアルカリ性とし
た後,ジメチルグリオキシムを加え,生成するジメチルグリオキシムニッケルの沈殿をこし分け,その質
量をはかる。
2. 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1, 2+100)
(3) 硝酸
(4) 硝酸 (1+2)
(5) 過塩素酸
(6) 過塩素酸 (2+100)
(7) ふっ化水素酸
(8) アンモニア水
(9) アルミニウム片 約1.7〜3mmの薄片状に切削したもの。
(10) 臭素酸カリウム
(11) 二硫酸カリウム
(12) アンモニア性洗浄水 温水約500mlにアンモニア水1,2滴を加え,かき混ぜる。この洗浄水は,使
用の都度調製する。
(13) 酢酸
(14) くえん酸溶液 くえん酸−水和物500gを水に溶解し,水で液量を1 000mlとする。
(15) L (+)-酒石酸溶液 (500g/l)
(16) L (+)-アスコルビン酸溶液 (10g/l) この溶液は,使用の都度調製する。
(17) くえん酸水素二アンモニウム溶液 (100g/l)
(18) ジメチルグリオキシム溶液 ジメチルグリオキシム [(CH3)2C2 (NOH)2] 10gをエタノール (95) 1 000ml
に溶解する。
又は,ジメチルグリオキシム5gを水酸化ナトリウム溶液 (20g/l) 250mlに溶解し,水で液量を500ml
とする。
3. 試料はかり採り量 試料はかり採り量は,附属書1表1による。
附属書1表1 試料はかり採り量
ニッケル含有率
% (m/m)
試料はかり採り量
g
0.1以上 0.5未満
3.0
0.5以上 2 未満
1.0
2 以上 30 以下
0.50
4. 操作
4
G 1216-1997
参考 警告 過塩素酸の蒸気は,アンモニア,亜硝酸蒸気又は有機物が存在すると爆発する危険があ
る。過塩素酸の蒸発処理は,過塩素酸を使用しても安全な排気設備を備えた場所で行わなけれ
ばならない。
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
(1) 酸で分解容易な試料 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆い,塩酸20m1
及び硝酸5mlを加え,加熱して分解する。時計皿の下面を水で洗って時計皿を取り除き,引き続き穏
やかに加熱して蒸発乾固する。放冷した後,塩酸 (1+1) 20mlを加え,加熱して塩類を溶解し,温水
約50mlを加える。溶液をろ紙(5種B)を用いてろ過し,ろ紙と残さを温めた塩酸 (2+100) と温水
で交互に数回洗浄する。ろ液と洗液をビーカー (500ml) に集める(1)。残さは捨てる。
(2) 酸で分解困難な試料
(a) 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆い,塩酸20ml及び硝酸5mlを加え,
加熱して分解する。時計皿の下面を水で洗って時計皿を取り除き,引き続き穏やかに加熱して蒸発
乾固する。放冷した後,塩酸 (1+1) 20mlを加え,加熱して塩類を溶解し,温水約50mlを加える。
溶液をろ紙(5種B)を用いてろ過し,ろ紙と残さを温めた塩酸 (2+100) と温水で交互に数回洗浄
する。ろ液と洗液をビーカー (500ml) に集め,主溶液として保存する。
(b) 残さをろ紙と共に白金るつぼ(30番)に移し入れ,乾燥した後,強熱してろ紙を灰化する。放冷し
た後,二硫酸カリウム約2gを加え,加熱して残さを融解する。放冷した後,融成物を少量の塩酸 (1
+1) 及び温水で溶解し,溶液をろ紙(5種B)でろ過する。ろ紙を温水で洗浄し,ろ液及び洗液を
先に保存しておいた主溶液を合わせる(1)。残さは捨てる。
(3) 多量のけい素,クロム,銅,コバルトなどを含む試料 試料をはかり採ってビーカー (300ml) に移し
入れ,時計皿で覆い,塩酸20ml及び硝酸5mlを加え,加熱して分解する。時計皿の下面を水で洗っ
て時計皿を取り除き,ふっ化水素酸数滴を加え,さらに過塩素酸を試料はかり採り量が3.0gのときに
は30ml,1.0gのときには20ml,0.50gのときには15ml加え,加熱して過塩素酸の白煙が発生するま
で蒸発する。乾いた時計皿で覆い,クロムが酸化されて二クロム酸になるまで加熱する(2)(3)。放冷し
た後,水を加えて液量を約100mlとし,塩類が溶解するまで加熱して煮沸する。溶液をろ紙(5種B)
を用いてろ過し,ろ紙及び残さを過塩素酸 (2+100) と温水で交互に数回,最後に温水で十分に洗浄
する。ろ液と洗液をビーカー (500ml) に集め,主溶液として保存する。以下,(2)(b)の操作を行う。
注(1) 試料中のニッケル含有率が5% (m/m) 以上の場合には,溶液を常温まで冷却した後,250mlの全
量フラスコに水を用いて移し入れ,水で標線まで薄める。この溶液をニッケル量が10〜25mgに
なるように分取し,ビーカー (500ml) に移し入れる。
(2) 多量の銅とコバルトを含む試料で,4.2(4)の操作に従ってジメチルグリオキシムニッケルの沈殿
を生成させる場合には,過塩素酸の残量が10m1以下になるまで蒸発させる。
(3) 多量のクロムを含む場合には,過塩素酸の白煙を発生させながら,塩酸を滴加して大部分のク
ロムを二酸化二塩化クロムとして揮散させてもよい。
4.2
沈殿の生成 沈殿の生成は,次のいずれかの手順によって行う。
(1) 調製した試料溶液中にコバルト及び銅を含まない場合
(a) 4.1の(1),(2)(b)又は(3)で得た溶液に,くえん酸溶液 [2.(14)] 又はL (+) -酒石酸溶液を試料はかり
採り量が3.0gのときには30ml,試料はかり採り量が1.0g及び0.50gのときには10ml加えた後,さ
らに溶液中のクロム量0.1gにつき20mlを追加し,水で液量を200mlとする。アンモニア水を加え
て中和し,さらに過剰に約2ml加えた後,温水で液量を300mlとする。
5
G 1216-1997
(b) この溶液を煮沸近くまで加熱した後,溶液中のニッケル予想量10mgにつき10mlの割合でジメチル
グリオキシム溶液 [2.(18)] をかき混ぜながら少量ずつ加える。アンモニア水を加えて中和し,さら
に過剰に約2mlを加え,3〜5分間溶液をかき混ぜ,30分間から数時間静置する(4)(5)。
(2) 調製した試料溶液中にコバルト5mg以上を含む場合
(a) 4.1の(1),(2)(b)又は(3)で得た溶液に,くえん酸溶液 [2.(14)] 又はL (+) -酒石酸溶液を試料はかり
採り量が3.0gのときには30ml,試料はかり採り量が1.0g及び0.50gのときには10ml加えた後,さ
らに溶液中のクロム量0.1gにつき20mlを追加し,水で液量を200mlとする。
(b) アンモニア水を加えて中和し,さらに過剰に約2ml加えた後,酢酸を加えて弱酸性とする。臭素酸
カリウム1〜3gを加え,加熱して液温を80℃に約10分間保持してコバルトを酸化した後,温水で
液量を300mlとする。以下,(1)(b)の操作を行う。ただし,ジメチルグリオキシム溶液 [2.(18)] は,
溶液中のニッケルとコバルトとの予想合量10mgに対して10mlの割合で加える。
(3) 調製した試料溶液中に銅5mg以上を含む場合 4.1の(1),(2)(b)又は(3)で得た溶液に,L (+) -アスコ
ルビン酸溶液 [2.(16)] 20mlを加えてかき混ぜ,5〜10分間静置する。くえん酸溶液 [2.(14)] 又はL (+)
-酒石酸溶液を試料はかり採り量が3.0gのときには30ml,試料はかり採り量が1.0g及び0.50gのとき
には10ml加えた後,さらに溶液中のクロム量0.1gにつき20mlを追加し,さらに10ml過剰に加え,
水で液量を200mlとする。アンモニア水を加えて中和し,さらに過剰に約2ml加えた後,温水で液量
を300mlとする。以下,(1)(b)の操作を行う。ただし,ジメチルグリオキシム溶液 [2.(18)] は,溶液
中のニッケルと銅との予想合量10mgに対して10mlの割合で加える。
(4) 調製した試料溶液中にコバルト2mg以上及び銅5mg以上を同時に含む場合
(a) 4.1の(1),(2)(b)又は(3)で得た溶液に,くえん酸水素二アンモニウム溶液5mlを加え,水で液量を約
150ml(6)とする。アルミニウム片 [2.(9)] 約1gを加え,穏やかに加熱して二クロム酸を還元した後,
さらに3〜5分間加熱を続ける。析出した金属銅を残ったアルミニウム片と共に,ろ紙(5種B)を
用いてろ別し,ろ紙及び残さを温水で7,8回洗浄する。ろ液と洗液をビーカー (500ml) に集め,
硝酸10mlを加え,加熱して5,6分間煮沸し,鉄などを酸化する。
(b) くえん酸溶液 [2.(14)] 又はL (+) -酒石酸溶液を試料はかり採り量が3.0gのときには30ml,試料は
かり採り量が1.0g及び0.50gのときには10ml加えた後,さらに溶液中のクロム量0.1gにつき20ml
を追加し,水で液量を200mlとする。以下,(2)(b)の操作を行う(7)。
注(4) ニッケル含有率が多い試料の場合には,沈殿生成後30分間以上静置して,またニッケル含有率
が少ない試料又は沈殿の生成が不完全と認めた場合には,数時間静置して,沈殿を熟成させる。
(5) 生成した沈殿にジメチルグリオキシムニッケル以外の不純物を含有するおそれのある場合には,
沈殿をろ紙(5種A)を用いてこし分けた後,沈殿を硝酸 (1+2) 30mlで溶解し,ろ紙を温水で
洗浄し,ろ液と洗液を元のビーカーに集め,温水約50ml及びくえん酸溶液 [2.(14)] 又はL (+)
-酒石酸溶液10mlを加え,水で液量を200mlとする。アンモニア水を加えて中和し,さらに過
剰に約2ml加え,温水で液量を300mlとした後,(1)(b)の操作を行う。ただし,沈殿を静置する
時間は,30〜60分間でよい。
(6) 注(2)を適用した場合には,過塩素酸濃度が0.5〜1.0mol/lになるように適宜液量を減らす。
(7) 溶液中のコバルト量が2〜5mgの場合には,(2)(b)の操作を行うかわりに,アンモニア水を加え
て中和し,さらに過剰に約2ml加え,温水で液量を300mlとした後,(1)(b)の操作を行ってもよ
い。

6
G 1216-1997
4.3
沈殿のひょう量 4.2の(1)(b),(2)(b),(3)又は(4)(b)で得た沈殿を,あらかじめ115±5℃で乾燥して
恒量としたガラスろ過器 (3G) でこし分け,アンモニア性洗浄水 [2.(12)] で十分に洗浄する。沈殿をガラ
スろ過器とともに115±5℃で約1時間乾燥し,デシケーター中で常温まで放冷した後,質量をはかる。恒
量となるまでこの操作を繰り返し,得た質量からガラスろ過器の質量を差し引いてジメチルグリオキシム
ニッケルの質量とする。
5. 空試験 試料を用いないで,試料と同じ操作を試料と併行して行う。
6. 計算 計算は,次のいずれかによる。
(1) 試料中のニッケル含有率が5% (m/m) 未満の場合
(
)
100
2
203
.0
2
1
×
×
−
=
m
m
m
Ni
ここに, Ni: 試料中のニッケル含有率 [% (m/m)]
m1: 4.3で得たジメチルグリオキシムニッケルの質量 (g)
m2: 5.で得たジメチルグリオキシムニッケルの質量 (g)
m: 試料はかり採り量 (g)
(2) 試料中のニッケル含有率が5% (m/m) 以上の場合
(
)
100
250
2
203
.0
2
1
×
×
×
−
=
B
m
m
m
Ni
ここに,
Ni: 試料中のニッケル含有率 [% (m/m)]
m1: 4.3で得たジメチルグリオキシムニッケルの質量 (g)
m2: 5.で得たジメチルグリオキシムニッケルの質量 (g)
m: 試料はかり採り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
7. 許容差 許容差(8)は,附属書1表2による。
附属書1表2 許容差
単位% (m/m)
ニッケル含有率
室内再現許容差
室間再現許容差
0.1以上 19.6以下 D [0.002 7× (Ni) +0.004 9] D [0.008 5× (Ni) −0.010 5]
注(8) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一室内における分析回数,室間再現許容差の場合は分析に関与した
分析室数である(n=2のとき,D=2.8である)。
また, (Ni) は,許容差を求める試料中のニッケル含有率 [% (m/m)] である。
参考 この許容差は,日本鉄鋼認証標準物質の認証値決定の集計値から,ジメチルグリオキシムニッ
ケル重量法による定量値を集計して解析して求めたものである。
7
G 1216-1997
附属書2 ジメチルグリオキシム沈殿分離エニチレンジアミン
四酢酸二水素二ナトリウム・亜鉛逆滴定法
1. 要旨 試料を適切な酸で分解し,くえん酸又はL (+) -酒石酸を加え,アンモニア水でアルカリ性とし
た後,ジメチルグリオキシムを加え,生成するジメチルグリオキシムニッケルの沈殿をこし分ける。沈殿
を硝酸で溶解し,一定量のエチレンジアミン四酢酸二水素二ナトリウム(以下,EDTA2Naという。)を加
えてニッケルとの錯体を生成させ,pHを調節した後,亜鉛標準溶液で過剰のEDTA2Naを滴定する。
2. 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1, 2+100)
(3) 硝酸
(4) 硝酸 (1+2)
(5) 過塩素酸
(6) 過塩素酸 (2+100)
(7) ふっ化水素酸
(8) アンモニア水
(9) アンモニア水 (1+1)
(10) アルミニウム片 約1.7〜3mmの薄片状に切削したもの。
(11) 臭素酸カリウム
(12) 二硫酸カリウム
(13) アンモニア性洗浄水 温水約500mlにアンモニア水1,2滴を加え,かき混ぜる。この洗浄水は,使
用の都度調製する。
(14) 酢酸
(15) くえん酸溶液 くえん酸一水和物500gを水に溶解し,水で液量を1 000mlとする。
(16) L (+) -酒石酸溶液 (500g/l)
(17) L (+) -アスコルビン酸溶液 (10g/l) この溶液は,使用の都度調製する。
(18) 酢酸アンモニウム溶液 (200g/l)
(19) くえん酸水素二アンモニウム溶液 (100g/l)
(20) 酒石酸ナトリウム溶液 酒石酸ナトリウム二水和物10gを水に溶解し,水で液量を1 000mlとする。
(21) ジメチルグリオキシム溶液 ジメチルグリオキシム [(CH3)2C2 (NOH)2] 10gをエタノール (95) 1 000ml
に溶解する。
又は,ジメチルグリオキシム5gを水酸化ナトリウム溶液 (20g/l) 250mlに溶解し,水で液量を500ml
とする。
(22) 0.02mol/l EDTA2Na標準溶液 エチレンジアミン四酢酸二水素二ナトリウム二水和物
(C10H14N2Na2O8・2H2O) 7.5gをはかり採り,水約100mlに溶解し,溶液を1 000mlの全量フラスコに水
を用いて移し入れ,水で標線まで薄める。この溶液のファクターは,次のようにして決める。

8
G 1216-1997
コニカルビーカー (200ml) に0.02mol/l亜鉛標準溶液 [2.(23)] を正確に25ml取り,水で液量を100ml
とした後,ビュレットを用いて0.02mol/l EDTA2Na標準溶液20.0mlを加える。水酸化ナトリウム溶液
(100g/l) でpHを6〜8に調節し,アンモニア性塩化アンモニウム緩衝液[調製は,JIS K 8001の4.2
表4(試薬溶液)による。]2ml及びエリオクロムブラックT溶液[調製は,JIS K 8001の4.4表8(沈
殿滴定,酸化還元滴定,錯滴定用など)による。]数滴を指示薬として加え,引き続き先のビュレット
を用いて0.02mol/l EDTA2Na標準溶液で滴定し,溶液の色が赤から青に変わった点を終点として
EDTA2Na標準溶液の使用量を求め,次の式によってファクターを算出する。
V
F
F
25
2
1
×
=
ここに, F1: 0.02mol/l EDTA2Na標準溶液のファクター
F2: 0.02mol/l 鉛標準溶液のファクター
V: 0.02mol/l EDTA2Na標準溶液の使用量 (ml)
(23) 0.02mol/l亜鉛標準溶液 JIS K 8005で規定する亜鉛をJIS K 8005の4.表1(乾燥条件)に従って処理
した後,その1.31gを0.1mgのけたまではかり採ってビーカー (300ml) に移し入れる。時計皿で覆い,
水約50ml及び塩酸 (1+1) 20mlを加え,更に臭素水5滴を加え,加熱して分解する。引き続き加熱し,
煮沸して臭素を追い出す。常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除き,酢酸
20mlを加え,アンモニア水でpHを6.0±0.2に調節する。溶液を1 000mlの全量フラスコに水を用い
て移し入れ,水で標線まで薄める。この標準溶液のファクターは,次の式によって算出する。
100
8
307
.1
2
A
m
F
×
=
ここに, F2: 0.02mol/l亜鉛標準溶液のファクター
m: はかり採った亜鉛の質量 (g)
A: 亜鉛の純度 [% (m/m)]
(24) キシレノールオレンジ溶液 調製は,JIS K 8001の4.4表8(沈殿滴定,酸化還元滴定,錯滴定用な
ど)による。
3. 試料はかり採り量 試料はかり採り量は,附属書2表1による。
附属書2表1 試料はかり採り量
ニッケル含有率
% (m/m)
試料はかり採り量
g
0.1以上 0.5未満
3.0
0.5以上 2 未満
1.0
2 以上 30 以下
0.50
4. 操作
参考 警告 過塩素酸の蒸気は,アンモニア,亜硝酸蒸気又は有機物が存在すると爆発する危険があ
る。過塩素酸の蒸発処理は,過塩素酸を使用しても安全な排気設備を備えた場所で行わなけれ
ばならない。
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
(1) 酸で分解容易な試料 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆い,塩酸20ml
及び硝酸5mlを加え,加熱して分解する。時計皿の下面を水で洗って時計皿を取り除き,引き続き穏
やかに加熱して蒸発乾固(1)する。放冷した後,塩酸 (1+1) 20mlを加え,加熱して塩類を溶解し,温
9
G 1216-1997
水約50mlを加える。溶液をろ紙(5種B)を用いてろ過し,ろ紙と残さを温めた塩酸 (2+100) と温
水で交互に数回洗浄する。ろ液と洗液をビーカー (500ml) に集める(2)。残さは捨てる。
(2) 酸で分解困難な試料
(a) 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆い,塩酸20ml及び硝酸5mlを加え,
加熱して分解する。時計皿の下面を水で洗って時計皿を取り除き,引き続き穏やかに加熱して蒸発
乾固する。放冷した後,塩酸 (1+1) 20mlを加え,加熱して塩類を溶解し,温水約50mlを加える。
溶液をろ紙(5種B)を用いてろ過し,ろ紙と残さを温めた塩酸 (2+100) と温水で交互に数回洗浄
する。ろ液と洗液をビーカー (500ml) に集め,主溶液として保存する。
(b) 残さをろ紙と共に白金るつぼ(30番)に移し入れ,乾燥した後,強熱してろ紙を灰化する。放冷し
た後,二硫酸カリウム約2gを加え,加熱して残さを融解する。放冷した後,融成物を少量の塩酸 (1
+1) 及び温水で溶解し,溶液をろ紙(5種B)でろ過する。ろ紙は,温水で洗浄し,ろ液及び洗液
を先に保存しておいた主溶液に合わせる(2)。残さは捨てる。
(3) 多量のけい素,クロム,銅,コバルトなどを含む試料 試料をはかり採ってビーカー (300ml) に移し
入れ,時計皿で覆い,塩酸20ml及び硝酸5mlを加え,加熱して分解する。時計皿の下面を水で洗っ
て時計皿を取り除き,ふっ化水素酸数滴を加え,さらに過塩素酸を試料はかり採り量が3.0gのときに
は30ml,1.0gのときには20ml,0.50gのときには15ml加え,加熱して過塩素酸の白煙が発生するま
で蒸発する。乾いた時計皿で覆い,クロムが酸化されて二クロム酸になるまで加熱する(3)(4)。放冷し
た後,水を加えて液量を約100mlとし,塩類が溶解するまで加熱して煮沸する。溶液をろ紙(5種B)
を用いてろ過し,ろ紙及び残さを過塩素酸 (2+100) と温水で交互に数回,最後に温水で十分に洗浄
する。ろ液と洗液をビーカー (500ml) に集め,主溶液として保存する。以下,(2)(b)の操作を行う。
注(1) 試料を分解した溶液中に二酸化けい素,その他の残さが認められない場合には,蒸発乾固及び
ろ過操作を省略してもよい。
(2) 試料中のニッケル含有率が5% (m/m) 以上の場合には,溶液を常温まで冷却した後,250mlの全
量フラスコに水を用いて移し入れ,水で標線まで薄める。この溶液をニッケル量が10〜25mg
になるように分取し,ビーカー (500ml) に移し入れる。
(3) 多量の銅とコバルトを含む試料で,ニッケルの沈殿の生成を4.2(4)の操作に従ってジメチルグリ
オキシムニッケルの沈殿を生成させる場合には,過塩素酸の残量が10ml以下になるまで蒸発さ
せる。
(4) 多量のクロムを含む場合には,過塩素酸の白煙を発生させながら,塩酸を滴加して大部分のク
ロムを二酸化二塩化クロムとして揮散させてもよい。
4.2
ニッケルの沈殿分離 ニッケルの沈殿分離は,次のいずれかの手順によって行う。
(1) 調製した試料溶液中にコバルト及び銅を含まない場合
(a) 4.1の(1),(2)(b)又は(3)で得た溶液に,くえん酸溶液 [2.(15)] 又はL (+) -酒石酸溶液を試料はかり
採り量が3.0gのときには30ml,試料はかり採り量が1.0g及び0.50gのときには10ml加えた後,さ
らに溶液中のクロム量0.1gにつき20mlを追加し,水で液量を200mlとする。アンモニア水を加え
て中和し,さらに過剰に約2ml加えた後,温水で液量を300mlとする。
(b) この溶液を煮沸近くまで加熱した後,溶液中のニッケル予想量10mgにつき10mlの割合でジメチル
グリオキシム溶液 [2.(21)] をかき混ぜながら少量ずつ加える。アンモニア水を加えて中和し,さら
に過剰に約2mlを加え,3〜5分間溶液をかき混ぜ,30分間から数時間静置する(5)(6)。
(2) 調製した試料溶液中にコバルト5mg以上を含む場合
10
G 1216-1997
(a) 4.1の(1),(2)(b)又は(3)で得た溶液に,くえん酸溶液 [2.(15)] 又はL (+) -酒石酸溶液を試料はかり
採り量が3.0gのときには30ml,試料はかり採り量が1.0g及び0.50gのときには10ml加えた後,さ
らに溶液中のクロム量0.1gにつき20mlを追加し,水で液量を200mlとする。
(b) アンモニア水を加えて中和し,さらに過剰に約2ml加えた後,酢酸を加えて弱酸性とする。臭素酸
カリウム1〜3gを加え,加熱して液温を80℃に約10分間保持してコバルトを酸化した後,温水で
液量を300mlとする。以下,(1)(b)の操作を行う。ただし,ジメチルグリオキシム溶液 [2.(21)] は,
溶液中のニッケルとコバルトとの予想合量10mgに対して10mlの割合で加える。
(3) 調製した試料溶液中に銅5mg以上を含む場合 4.1の(1),(2)(b)又は(3)で得た溶液に,L (+) -アスコ
ルビン酸溶液 [2.(17)] 20mlを加えてかき混ぜ,5〜10分間静置する。くえん酸溶液 [2.(15)] 又はL (+)
-酒石酸溶液を試料はかり採り量が3.0gのときには30ml,試料はかり採り量が1.0g及び0.50gのとき
には10ml加えた後,さらに溶液中のクロム量0.1gにつき20mlを追加し,さらに10m1過剰に加え,
水で液量を200mlとする。アンモニア水を加えて中和し,さらに過剰に約2m1加えた後,温水で液量
を300mlとする。以下,(1)(b)の操作を行う。ただし,ジメチルグリオキシム溶液 [2.(21)] は,溶液
中のニッケルと銅との予想合量10mgに対して10mlの割合で加える。
(4) 調製した試料溶液中にコバルト2mg以上及び銅5mg以上を同時に含む場合
(a) 4.1の(1),(2)(b)又は(3)で得た溶液に,くえん酸水素二アンモニウム溶液5mlを加え,水で液量を約
150ml(7)とする。アルミニウム片 [2.(l0)] 約1gを加え,穏やかに加熱して二クロム酸を還元した後,
さらに3〜5分間加熱を続ける。析出した金属銅を残ったアルミニウム片と共に,ろ紙(5種B)を
用いてろ別し,ろ紙及び残さを温水で7,8回洗浄する。ろ液と洗液をビーカー (500ml) に集め,
硝酸10mlを加え,加熱して5,6分間煮沸し,鉄などを酸化する。
(b) くえん酸溶液 [2.(15)] 又はL (+) -酒石酸溶液を試料はかり採り量が3.0gのときには30ml,試料は
かり採り量が1.0g及び0.50gのときには10ml加えた後,さらに溶液中のクロム量0.1gにつき20ml
を追加し,水で液量を200mlとする。以下,(2)(b)の操作を行う(8)。
注(5) ニッケル含有率が多い試料の場合には,沈殿生成後30分間以上静置して,またニッケル含有率
が少ない試料又は沈殿の生成が不完全と認めた場合には,数時間静置して,沈殿を熟成させる。
(6) 生成した沈殿にジメチルグリオキシムニッケル以外の不純物を含有するおそれのある場合には,
沈殿をろ紙(5種A)を用いてこし分けた後,沈殿を硝酸 (1+2) 30mlで溶解し,ろ紙を温水で
洗浄し,ろ液と洗液を元のビーカーに集め,温水約50ml及びくえん酸溶液 [2.(15)] 又はL (+)
-酒石酸溶液10mlを加え,水で液量を200mlとする。アンモニア水を加えて中和し,さらに過
剰に約2ml加え,温水で液量を300mlとした後,(1)(b)の操作を行う。ただし,沈殿を静置する
時間は,30〜60分間でよい。
(7) 注(3)を適用した場合には,過塩素酸濃度が0.5〜1.0mol/lになるように適宜液量を減らす。
(8) 溶液中のコバルト量が2〜5mgの場合には,(2)(b)の操作を行う代わりに,アンモニア水を加え
て中和し,さらに過剰に約2ml加え,温水で液量を300mlとした後,(1)(b)の操作を行ってもよ
い。
4.3
滴定 滴定は,次の手順によって行う。
(1) 4.2の(1)(b),(2)(b),(3)2 又は(4)(b)で得た沈殿を,ろ紙(5種A)を用いてこし分け,アンモニア性
洗浄水 [2.(13)] 又は酒石酸ナトリウム溶液 [2.(20)] で約10回洗浄する。漏斗にろ紙をはったまま水
でろ紙上の沈殿を元のビーカーに洗い落とし,硝酸 (1+2) 30mlを加え,時計皿で覆い,穏やかに加
熱して溶解する。溶液を元のろ紙に注いでろ紙上に残っている沈殿を溶解しながらビーカー (300ml)
11
G 1216-1997
にろ過する。温水でろ紙を十分に洗浄した後,ろ液と洗液を数分間煮沸してジメチルグリオキシムニ
ッケルを分解する。
(2) 溶液を常温まで冷却した後,ビュレットを用いて溶液中のニッケル予想量10mgにつき0.02mol/l
EDTA2Na標準溶液 [2.(22)] 10mlの割合で加え,さらに3mlを過剰に加えた後,0.02mol/l EDTA2Na標
準溶液 [2.(22)] の使用量を正確に読み取る。酢酸アンモニウム溶液15mlを加え,水で液量を約150ml
とした後,アンモニア水 (1+1) 及び塩酸 (2+100) を用いて溶液のpHを6.0±0.2に調節し,約10
分間静置する。キシレノールオレンジ溶液 [2.(24)] 3〜5滴を指示薬として加え,よくかき混ぜながら
0.02mol/l亜鉛標準溶液 [2.(23)] で滴定し,最後の1滴で溶液の色が赤紫を呈する点を終点とし,
0.02mol/l亜鉛標準溶液 [2.(23)] の使用量を求める。
5. 空試験 試料を用いないで,試料と同じ操作を試料と併行して行う。ただし,0.02mol/l EDTA2Na標
準溶液 [2.(22)] の使用量は,3mlとする。
6. 計算 計算は,次のいずれかによる。
(1) 試料中のニッケル含有率が5% (m/m) 未満の場合
(
)(
)
[
]
100
174
001
.0
4
2
3
1
2
2
1
1
×
×
×
−
×
−
×
−
×
=
m
V
F
V
F
V
F
V
F
Ni
ここに, Ni: 試料中のニッケル含有率 [% (m/m)]
F1: 0.02mol/l EDTA2Na標準溶液 [2.(22)] のファクター
F2: 0.02mol/l亜鉛標準溶液 [2.(23)] のファクター
V1: 試料溶液の滴定における0.02mol/l EDTA2Na標準溶液
[2.(22)] の使用量 (ml)
V2: 試料溶液の滴定における0.02mol/l亜鉛標準溶液 [2.(23)] の
使用量 (ml)
V3: 空試験液の滴定における0.02mol/l EDTA2Na標準溶液
[2.(22)] の使用量 (ml)
V4: 空試験液の滴定における0.02mol/l亜鉛標準溶液 [2.(23)] の
使用量 (ml)
m: 試料はかり採り量 (g)
(2) 試料中のニッケル含有率が5% (m/m) 以上の場合
(
)(
)
[
]
100
250
174
001
.0
4
2
3
1
2
2
1
1
×
×
×
×
−
×
−
×
−
×
=
B
m
V
F
V
F
V
F
V
F
Ni
ここに, Ni: 試料中のニッケル含有率 [% (m/m)]
F1: 0.02mol/l EDTA2Na標準溶液 [2.(22)] のファクター
F2: 0.02mol/l亜鉛標準溶液 [2.(23)] のファクター
V1: 試料溶液の滴定における0.02mol/l EDTA2Na標準溶液
[2.(22)] の使用量 (ml)
V2: 試料溶液の滴定における0.02mol/l亜鉛標準溶液 [2.(23)] の
使用量 (ml)
V3: 空試験液の滴定における0.02mol/l EDTA2Na標準溶液
[2.(22)] の使用量 (ml)
V4: 空試験液の滴定における0.02mol/l亜鉛標準溶液 [2.(23)] の
使用量 (ml)
m: 試料はかり採り量 (g)

12
G 1216-1997
B: 試料溶液及び空試験液の分取量 (ml)
7. 許容差 許容差(9)は,附属書2表2による。
附属書2表2 許容差
単位% (m/m)
ニッケル含有率
室内再現許容差
室間再現許容差
0.1以上 5.0未満 D [0.002 9× (Ni) +0.002 9] D [0.005 2× (Ni) +0.004 0]
5.0以上 19.6以下 D [0.011 1× (Ni) −0.056 6] D [0.016 0× (Ni) −0.066 0]
注(9) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一室内における分析回数,室間再現許容差の場合は分析に関与した
分析室数である(n=2のとき,D=2.8である)。
また, (Ni) は,許容差を求める試料中のニッケル含有率 [% (m/m)] である。
参考 この許容差は,ニッケル含有率0.30% (m/m) 以上19.6% (m/m) 以下の試料を用い,共同実験し
た結果から求めたものである。
13
G 1216-1997
附属書3 ジメチルグリオキシム分離定量法
附属書3としてのまえがき
この附属書3は,1988年第1版として発行されたISO 4938 (Steel and iron−Determination of nickel content
−Gravimetric or titrimetric method) を翻訳し,技術的内容及び規格票の様式を変更することなく作成した日
本工業規格である。
なお,この規格で下線(点線)を施してある箇所は,原国際規格にはない事項である。
1. 適用範囲 この附属書3は,鋼及び鉄中のニッケルを重量法か又は滴定法のいずれかによって定量す
る方法について規定する。
この方法は,ニッケル含有率0.5% (m/m) 以上30% (m/m) 以下の試料に適用する。
2. 引用規格 次に記載する規格は,この国際規格の本文中で引用するので,国際規格の規定の一部を構
成する。この規格の発行の時点では,それぞれの規格の発行版表示は正しいものであるが,国際規格はす
べて改訂されるものであるので,この規格を使用することに合意した当事者は,常に最新版の規格を参照
するように努力されたい。IEC及びISOのメンバーには最新の国際規格のリストが配布されている。
ISO 377 : 1985 Wrought steel−Selection and preparation of samples and test pieces
ISO 385-1 : 1984 Laboratory glassware−Burettes−Part 1 : General requirements
ISO 648 : 1977 Laboratory glassware−One-mark pipettes
ISO 1042 : 1983 Laboratory glassware−One-mark volumetric flasks
ISO 4793 : 1980 Laboratory sintered (fritted) filters−Porosity grading, classification and designation
ISO 5725 : 1986 Precision of test methods−Determination of repeatability and reproducibility for a standard
test method by interlaboratory tests
参考 ISO 377は,次の規格で置き換えられている。
ISO 14284 : 1996 Steel and iron−Sampling and preparation of samples for the determination of chemical
composition
3. 原理 試料を適切な酸で分解する。ニッケルをニッケルジメチルグリオキシムとして沈殿させる。
− コバルトが共存するときは,ヘキサシアノ鉄 (III) 酸カリウムで酸化する。
− 銅とコバルトが共存するときは,定電位電解で分離するのがよい。
沈殿を酸に溶解してろ過し,引き続いてニッケルを再度ニッケルジメチルグリオキシムとして沈殿させ
る。重量法による場合は,ニッケルジメチルグリオキシムの沈殿を乾燥してひょう量する。滴定法による
場合は,沈殿を酸に溶解して過剰のEDTA. Na2溶液を加え,その過剰をキシレノールオレンジを指示薬と
して亜鉛標準溶液で逆滴定する。
4. 試薬 分析の際は,特に記述しない限り,分析用保証試薬及び蒸留水又はこれと同等の純度をもつ水
を使用する。
4.1
硫酸水素ナトリウム
14
G 1216-1997
4.2
エタノール 95% (V/V)
4.3
酢酸 密度約1.05g/ml
4.4
ふっ化水素酸 密度約1.15g/ml
4.5
硝酸 密度約1.40g/ml
4.6
過塩素酸 密度約1.54g/ml
4.7
硫酸 密度約1.84g/ml
4.8
アンモニア水 密度約0.90g/ml
4.9
塩酸 密度約1.19g/ml,希釈液1+1
4.10 塩酸 密度約1.19g/ml,希釈液1+99
4.11 硝酸 密度約1.40g/ml,希釈液2+3
4.12 過塩素酸 密度約1.54g/ml,希釈液1+49
4.13 アンモニア水 密度約0.90g/ml,希釈液1+1
4.14 アンモニア水 密度約0.90g/ml,希釈液1+3
4.15 塩酸/硝酸の混液 塩酸(密度約1.19g/ml)3容に,硝酸 (4.5) 1容を混合する。
この溶液は,使用の直前に調製する。
4.16 酢酸アンモニウム溶液2 00g/l溶液
4.17 くえん酸アンモニウム緩衝溶液 くえん酸一水和物 (C6H8O7.H2O) 500gをアンモニア水 (4.8) 675ml
に溶解し,水で1 000mlに薄める。使用前にろ過する。
4.18 くえん酸溶液 くえん酸一水和物 (C6H8O7.H2O) 500gを水に溶解し,水で1 000mlに薄める。使用前
にろ過する。
4.19 ジメチルグリオキシム溶液 30g/lアルカリ性溶液 水400mlに水酸化カリウム20gを溶解し,ジメ
チルグリオキシム (C4H8N2O2) 30gを加えて完全に溶解するまで振り混ぜる。水で1 000mlに薄めて混合す
る。使用前にろ過する。
4.20 ジメチルグリオキシム溶液 10g/lエタノール溶液 ジメチルグリオキシム (C4H8N2O2) 10gをエタ
ノール (4.2) 1000mlに溶解する。使用前にろ過する。
4.21 硫酸ヒドラジニウム (2+) 溶液 N2H6SO4, 100g/l溶液
4.22 ヘキサシアノ鉄 (III) 酸カリウム溶液 K3 [Fe (CN) 6], 100g/l溶液 この溶液は,約30日間安定であ
る。この溶液1mlは,コバルト及びマンガンのそれぞれの約0.02gに相当する。
4.23 洗浄水 アンモニア水 (4.13) の数滴を加えてpHを8に調節した水。
4.24 エチレンジアミン四酢酸二水素二ナトリウム (EDTA. Na2) 滴定用標準溶液
4.24.1 溶液の調製 エチレンジアミン四酢酸二水素二ナトリウム二水和物 (C10H14O8N2Na2.2H2O) 6.33gを
水に溶解し,1 000mlの全量フラスコに移し入れ,水で標線まで薄めて混合する。この標準溶液1mlは,
ニッケルの約1mgに相当する。
4.24.2 溶液の標定 ニッケル基準溶液 (4.24.3) 25.0mlを250mlのビーカーに分取し,EDTA. Na2溶液
(4.24.1) 33mlを加える。これに酢酸アンモニウム溶液 (4.16) 15mlを加えて水で約150mlに薄める。以下,
7.2.5の“溶液のpHを”以降の第3分節の操作に続けて処理する。EDTA. Na2溶液 (4.24.1) のニッケル相
当濃度 (mg/ml) cは,次の式から求める。
(
)(
)
2
1
2
125
V
V
m
m
c
×
+
×
=
ここに, m1: ニッケル基準溶液 (4.24.3) 1mlに含有するニッケルの質量 (mg)
15
G 1216-1997
m2: 亜鉛標準溶液 (4.25) 1mlに相当するニッケルの質量 (mg)
V1: 滴定に使用した亜鉛標準溶液 (4.25) の容積 (ml)
V2: 標定に使用したEDTA. Na2溶液 (4.24.1) の容積 (ml)
4.24.3 ニッケル基準溶液の調製 高純度ニッケル[純度99.95% (m/m) 以上]1.000 0gを0.1mgのけたま
ではかり採る。硝酸 (4.11) 20mlに溶解する。煮沸して亜硝酸ガスを追い出し,冷却した後,全量を1 000ml
の全量フラスコに移し入れ,水で標線まで薄めて混合する。この基準溶液1mlは,ニッケル1.0mgを含有
する。
4.25 亜鉛標準溶液 金属亜鉛[純度99.9% (m/m) 以上]1.114 0gを0.1mgのけたまではかり採り,300ml
のビーカーに移し入れる。もし,金属亜鉛が酸化しているときは,塩酸 (4.9),水及びアセトンで洗浄し,
110℃で5分間乾燥するのがよい。水約50ml,塩酸 (4.9) 20ml及び臭素飽和水の5滴を加える。時計皿で
覆い,加熱して溶解する。臭素の色がなくなるまで引き続き加熱し,室温まで冷却し,酢酸 (4.3) 20mlを
加える。アンモニア水 (4.14) で溶液のpHを6.0±0.2に調節する。全量を1 000mlの全量フラスコに移し
入れ,水で標線まで薄めて混合する。この標準溶液1mlは,ニッケル1.0mgに,またEDTA. Na2滴定用標
準溶液 (4.24) の約1mlに相当する。
4.26 キシレノールオレンジ溶液 1g/l溶液 キシレノールオレンジ (C31H28N2O13SNa4) 0.1gを粉状にし,
少量の水でのり(糊)状にする。水で100mlに薄める。ろ過して共栓付き褐色ガラス瓶中に保存する。こ
の溶液は,1週間は安定である。
5. 装置 体積測定用ガラス器具は,すべてISO 385-1,ISO 648又はISO 1042に従う適切なものでA級
のものを使用する。
通常の分析室用の器具のほか,次のものを使用する。
5.1
ガラスろ過器 ISO 4793の細孔等級 (porosity) P16に従うもの。
5.2
pHメータ
5.3
定電位電解装置 飽和カロメル参照電極と白金電極をもったもの。
6. サンプリング サンプリングは,ISO 377又は鉄に関する適切な国家規格に従って実施する。
7. 操作
警告 過塩素酸の蒸気は,一般にアンモニア,亜硝酸の蒸気又は有機物の存在で爆発の危険がある。
7.1
試料のはかり採り試料は,沈殿するニッケルの量が,重量法にあっては25〜70mgの範囲,滴定法に
あっては25〜40mgの範囲になるように選択しなければならない。例えば,ニッケルの予想含有率が3.5%
(m/m) の場合は,試料はかり採り量は,約1gである。いずれの場合も0.1mgのけたまではかり採る。
7.2
定量
7.2.1
試料溶液の調製 はかり採った試料 (7.1) を適切な容積のビーカー(例えば,試料はかり採り量が
2.5gまでの場合は400ml,2.5gを超える場合は600ml)に移す。塩酸/硝酸の混酸 (4.15) を試料はかり採
り量が2.5gまでの場合は30ml,2.5gを超える場合は50mlを加える。ビーカーを時計皿で覆い,反応が完
了するまで50℃〜60℃の温度で加熱し,さらにふっ化水素酸 (4.4) 0.5ml〜1mlを加える。過塩素酸 (4.6) を,
試料はかり採り量が2.5gの場合は30ml,その他の場合は50mlを加える。
温度を約180℃に上げて過塩素酸の白煙が発生するまで蒸発する。乾燥した時計皿でビーカーを覆い,
引き続きクロムが完全に酸化するまで白煙を発生させる。ビーカーを熱源から降ろして放冷する。水100ml
16
G 1216-1997
を加え,加熱して塩類を溶解する。約5分間煮沸して塩素化合物を追い出す。
ろ紙(5種B)(参考参照)を用いて黒鉛並びにけい素,タングステン,ニオブ及びタンタルの酸化物を
ろ別しろ液を800mlのビーカーに集め,温めた過塩素酸 (4.12) で8〜10回洗浄し,引き続き水で2回洗浄
する(これを主液とする。)。
ろ紙と残さを白金るつぼに移し入れる。乾燥し,灰化して900℃で強熱する。ふっ化水素酸 (4.4) で残
さを処理し,蒸発した後,少量の硫酸水素ナトリウム (4.1) を加えて白金るつぼを注意深く加熱して残さ
を融解する。融成物を放冷し,熱水に溶解して主液に合わせる。
参考 原文は,迅速ろ紙 (rapid filter) である。
7.2.2
初回のニッケル沈殿生成
7.2.2.1
はかり採った試料中に銅が5mg未満及びコバルトが5mg未満含有の場合 7.2.1で調製した試料
溶液を水で約400mlに薄めてくえん酸溶液 (4.18) を50ml加える。アンモニア水 (4.13) で中和して塩酸
(4.9) で微酸性に戻す。90℃に加熱してこれにジメチルグリオキシム溶液 (4.20) をニッケル量10mgごと
に10mlの割合で直接注ぎ込む。
溶液をアンモニア水 (4.13) で中和し,過剰に2ml加えてよくかき混ぜる。溶液を約65℃で約2時間静
置する。速やかに室温まで冷却する。直径12.5cmの迅速硬質ろ紙でろ過して冷洗浄水 (4.23) で6〜8回洗
浄する。
7.2.2.2
はかり採った試料中に銅を5mg以上含有する場合 7.2.2.1のように操作するが,しかし,沈殿生
成の際にジメチルグリオキシム溶液 (4.20) の量を増加する。すなわち,ニッケル量10mgごとに10mlを
加えてさらにその過剰30mlを加える。
7.2.2.3
はかり採った試料中にコバルトを5mg以上含有する場合 7.2.1又は7.2.2.4で調製した試料溶液
を約100mlに蒸発する。この溶液をくえん酸アンモニウム緩衝溶液 (4.17) 100mlとアンモニア水 (4.8) 65ml
を入れた600mlのビーカーに移し入れる。元のビーカーを水ですすいだ後,アンモニア水 (4.13) 15mlで
洗浄して洗液を試料溶液に加える。
これにヘキサシアノ鉄 (III) 酸カリウム溶液 (4.22) を共存するコバルトとマンガンの量を酸化するの
に十分な量(コバルトとマンガンの0.1gごとに6ml)にその10%を追加して加える。よくかき混ぜ(この
とき溶液は,赤色でなければならない)pHメータ (5.2) を使用してアンモニア水 (4.8) 又は酢酸 (4.3) で
溶液のpHを8.0±0.2に調節する。これにエタノール (4.2) 50mlとジメチルグリオキシム溶液 (4.19) 100ml
を加えてよくかき混ぜる。この溶液を室温に4時間,溶液のpHが8になっているかどうかを確認しなが
ら静置する。直径12.5cmの迅速硬質ろ紙でろ過して冷洗浄水 (4.23) で6〜8回洗浄する。
7.2.2.4
はかり採った試料中にコバルト及び高濃度の銅を含有する場合 7.2.1で得た試料溶液に硫酸ヒ
ドラジニウム (2+) 溶液 (4.21) を滴加してクロムを完全に還元する。
溶液を定電位電解にかけ,銅が析出し始める陰極電位−0.15V(対飽和カロメル電極)から,電位を−0.30V
まで徐々に下げて銅を除去する。銅の析出は,約40分後に完全に終了し,そのときの電流は非常に低い一
定値を示すようになる。析出が完全であるかどうかは,水20mlを加えて引き続いて電解することによっ
て確認することができる。溶液に浸したばかりの陰極部に5分たっても銅が析出しなければ,析出は完了
である。カロメル電極回路への電流を切る。カロメル電極を取り出し,さらに白金電極を取り出して水で
すすぐ。
硝酸 (4.5) 約5mlを加えて過塩素酸の濃厚な白煙が発生するまで溶液を蒸発する。ビーカーに乾燥した
時計皿をかぶせて引き続きクロムが完全に酸化するまで加熱する。ビーカーを熱源から降ろして放冷する。
溶液を水100mlで薄め,加熱して塩類を溶解し,約5分間煮沸して塩素化合物を追い出す。
17
G 1216-1997
以下,7.2.2.3に示す手順に従って操作する。
7.2.3
2回目のニッケル沈殿生成(1) 7.2.2で得たろ紙と沈殿は,沈殿を生成させたビーカーに入れ,時計
皿でふたをする。これに硝酸 (4.5) 15ml,硝酸 (4.7) 10ml及び過塩素酸 (4.6) 4mlを加えて加熱する。次に
温度を上げて濃厚な白煙が発生するまで溶液を蒸発する(2)。冷却した後,水で約400mlに薄める。
くえん酸溶液 (4.18) 10mlを加え,アンモニア水 (4.13) で中和して塩酸 (4.9) で再度酸性に戻す。溶液
を90℃に加熱してニッケルの存在量10mgごとにジメチルグリオキシム溶液 (4.20) を10mlの割合で加え
る。アンモニア水 (4.13) で中和してさらにその過剰2mlを加える。よくかき混ぜて80℃で2時間静置す
る。溶液を徐々に50℃まで冷却し,エタノール (4.2) 50ml(3) を加え,よく混合して35℃まで冷却する。
注(1) 2回目のニッケル沈殿生成は,滴定法による場合,はかり採った試料中に銅が20mg以上及びコ
バルトが25mg以上含有しないとき省略してもよく,そのとき7.2.5の操作に続けて処理する。
(2) 沈殿とろ紙に含有する有機物の除去は,通常20分〜30分間必要とする。
(3) エタノールは,蒸発による損失を補い,低温においてジメチルグリオキシムが分離するのを避
けるために添加する。
7.2.4
重量法 あらかじめ110℃で乾燥し,デシケーター中で放冷し,10分間隔でひょう量して一定質量
になったガラスろ過器 (5.1) で7.2.3で得た沈殿を回収する。ビーカーとガラスろ過器を45℃に加温した
洗浄水 (4.23) で15回洗浄する。洗浄操作中は,いずれのときでも沈殿を乾燥状態にしてはならない。ガ
ラスろ過器と沈殿は,110℃で2時間乾燥し,デシケーター中で放冷した後10分間隔で一定質量になるま
でひょう量する。
7.2.5
滴定法 7.2.2又は7.2.3(4)で得た沈殿を硝酸 (4.11) と温水の少量ずつを交互に使用してろ紙から
溶解し,その溶液を沈殿を生成した元のビーカーへ集める。
注(4) 7.2.3から継続するときは,7.2.5の操作に移る前に次のようにしてろ過しなければならない。
直径12.5cmの迅速硬質ろ紙でろ過し,冷洗浄水 (4.23) で十分に洗浄する。
250mlのビーカーに移し入れて数分間煮沸する。冷却した後,ニッケル存在量の10mgごとにEDTA. Na2
滴定用標準溶液 (4.25) 12mlを添加し,さらにその3mlを過剰に加える。添加したEDTA. Na2滴定用標準
溶液の合計量を記録する。これに酢酸アンモニウム溶液 (4.16) 15mlを加えて水で約150mlに薄める。
溶液のpHをアンモニア水 (4.14) 又は塩酸 (4.10) で6.0±0.2に調節して約10分間保持する。指示薬と
してキシレノールオレンジ溶液 (4.26) の数滴を加え,かき混ぜて亜鉛標準溶液 (4.25) で赤紫色の終点が
得られるまで滴定する。
8. 結果の表示
8.1
計算方法
8.1.1
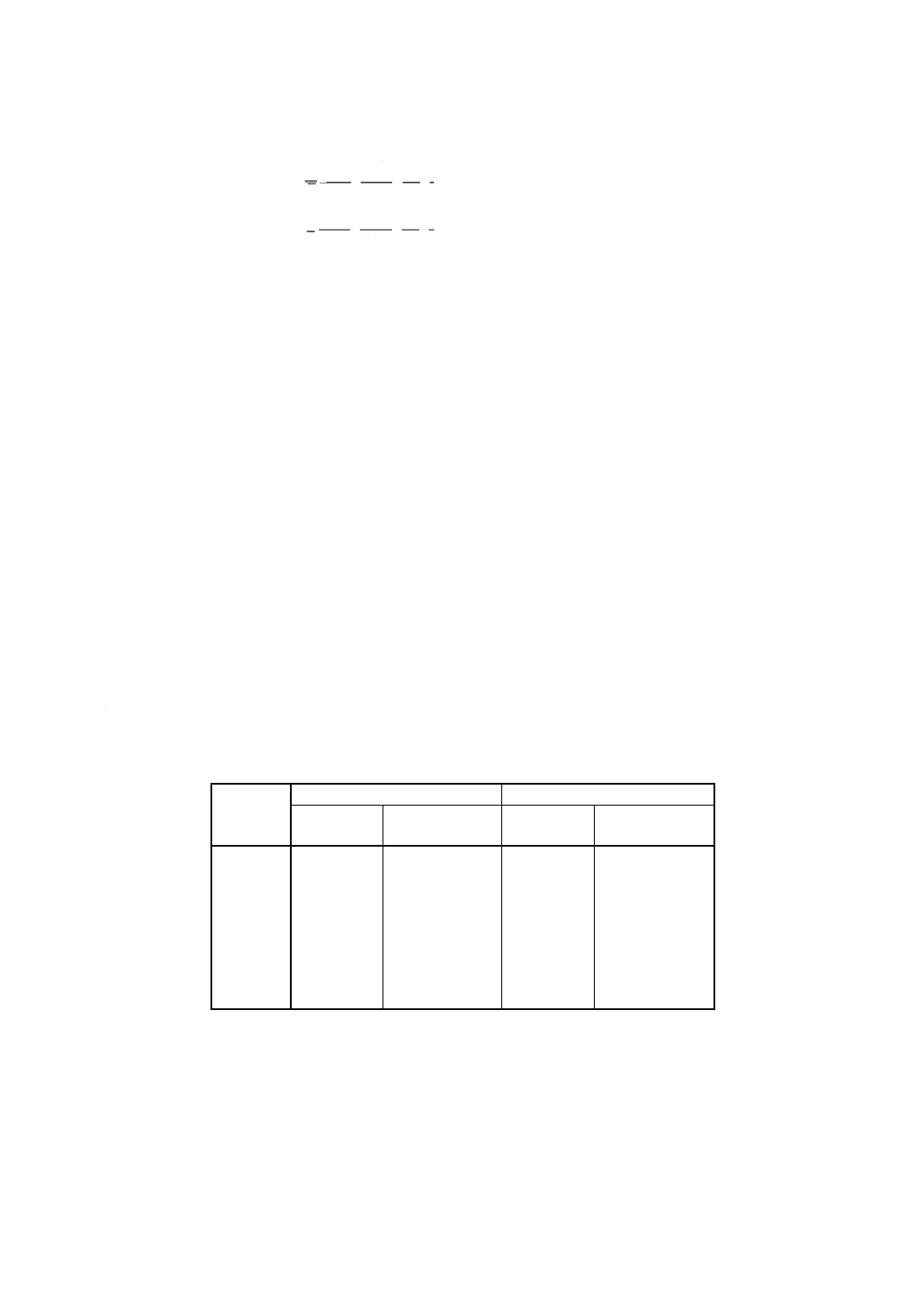
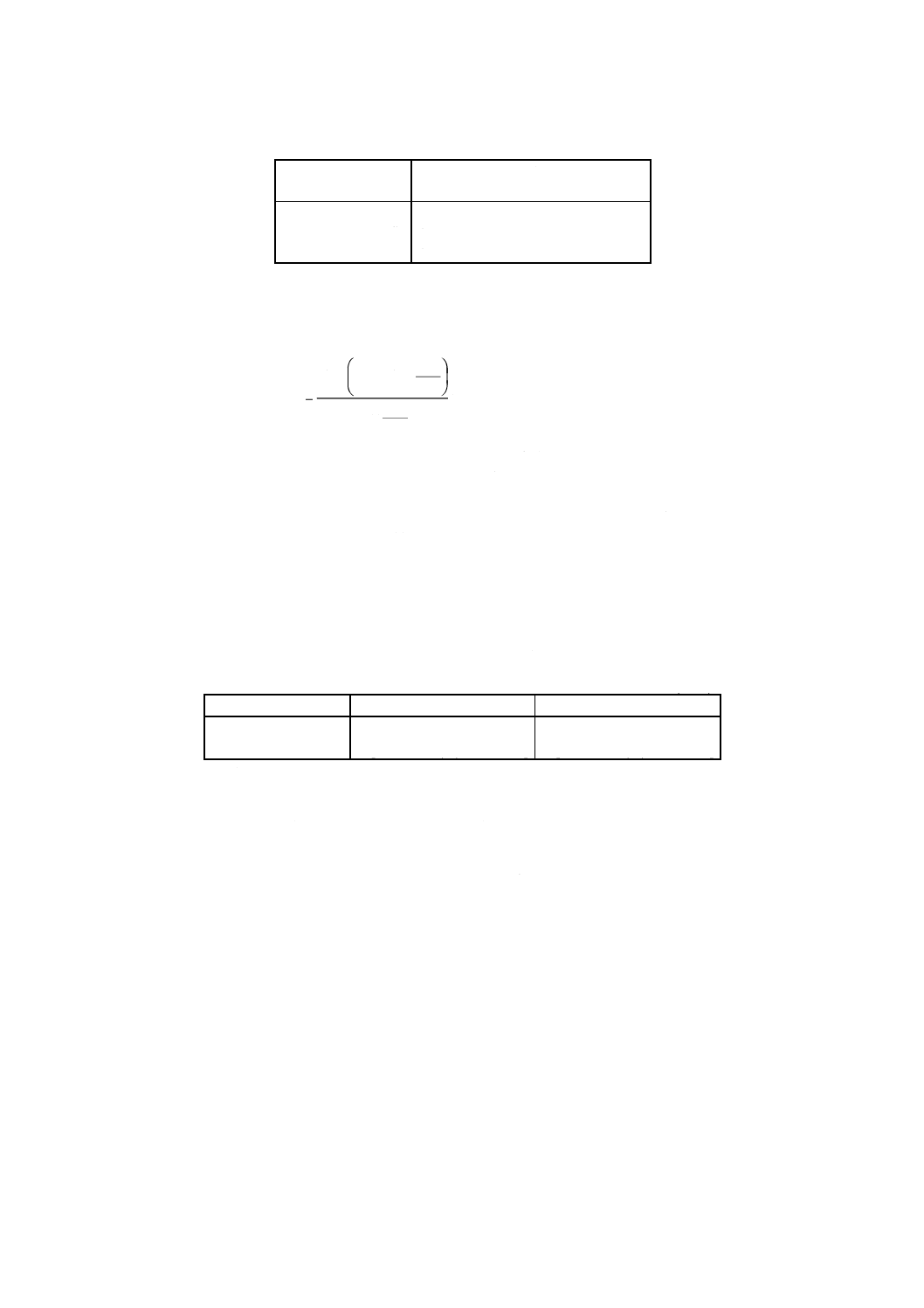
重量法の場合 ニッケル含有率 [% (m/m)] を次の式で計算する。
(
)
0
3
4
32
.
20
m
m
m
Ni
−
×
=
Λ
Λ
ここに,
Ni: 試料中のニッケル含有率 [% (m/m)]
m0: 試料はかり採り量 (g)
m3: 空のガラスろ過器の質量 (g)
m4: ガラスろ過器とニッケルジメチルグリオキシム沈殿の質
量 (g)
20.32: ニッケルジメチルグリオキシムのニッケルヘの変換係数
の100倍
18
G 1216-1997
8.1.2
滴定法の場合 ニッケル含有率 [% (m/m)] を次の式で計算する。
(
)(
)
(
)(
)
0
3
2
4
3
0
3
2
4
10
100
10
m
V
m
V
c
m
V
m
V
c
Ni
×
−
×
=
×
×
×
−
×
=
Λ
Λ
ここに, Ni: 試料中のニッケル含有率 [% (m/m)]
c: EDTA. Na2滴定用標準溶液のニッケル当量 (mg/ml)
V3: 消費した亜鉛標準溶液の容積 (ml)
V5: 添加したEDTA. Na2滴定用標準溶液の全容積 (ml)
m0: 試料はかり採り量 (g)
m2: 使用した亜鉛標準溶液1mlに相当するニッケルの質量 (mg)
8.2
許容差 この方法は,重量法については12分析室,滴定法については10分析室で共同実験を行っ
た。すなわち,ニッケルを含む7水準の試料を用い,各試料について各分析室はニッケルを4回ずつ定量
した。得られた結果は,ISO 5725に従って統計計算を行った。
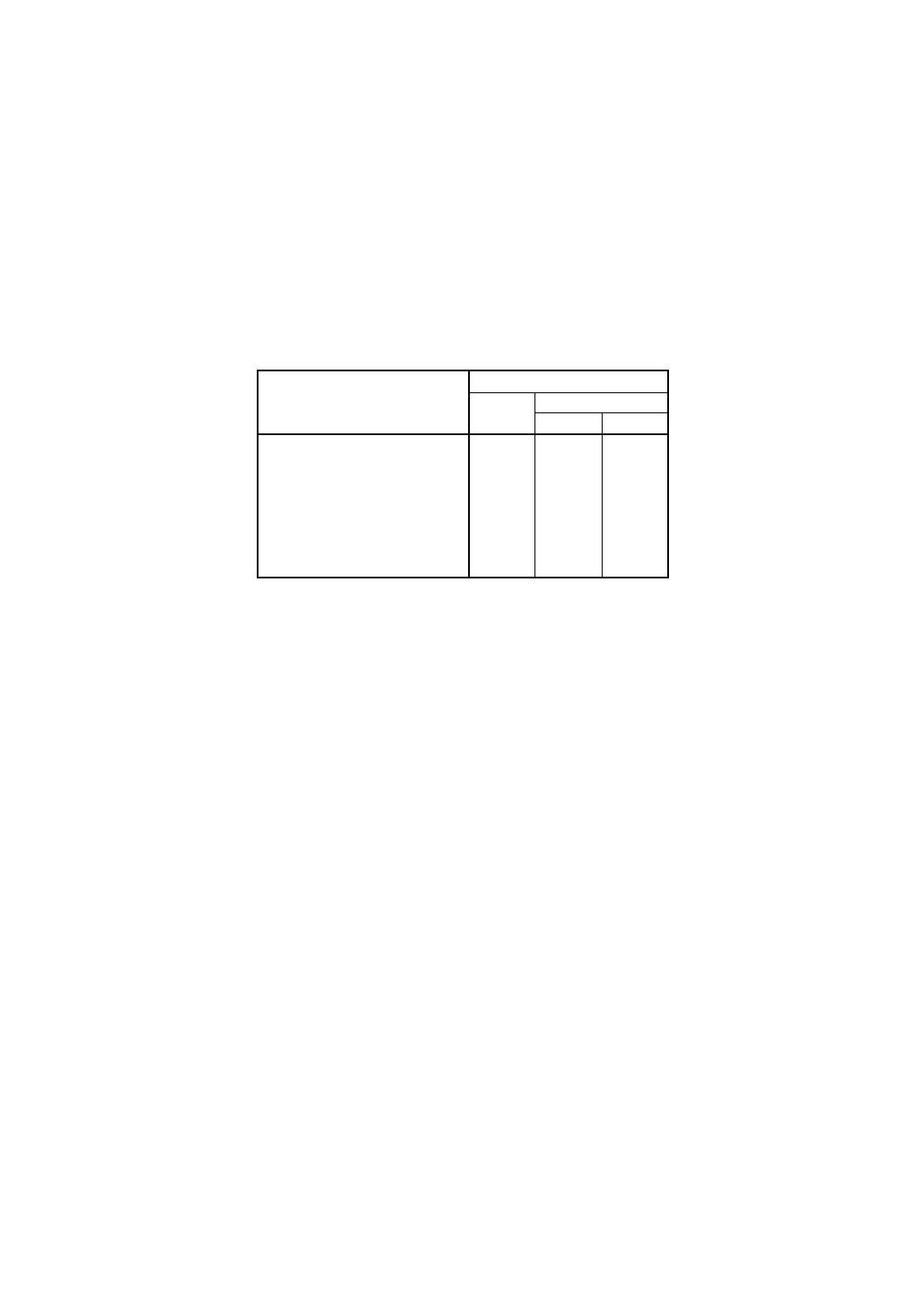
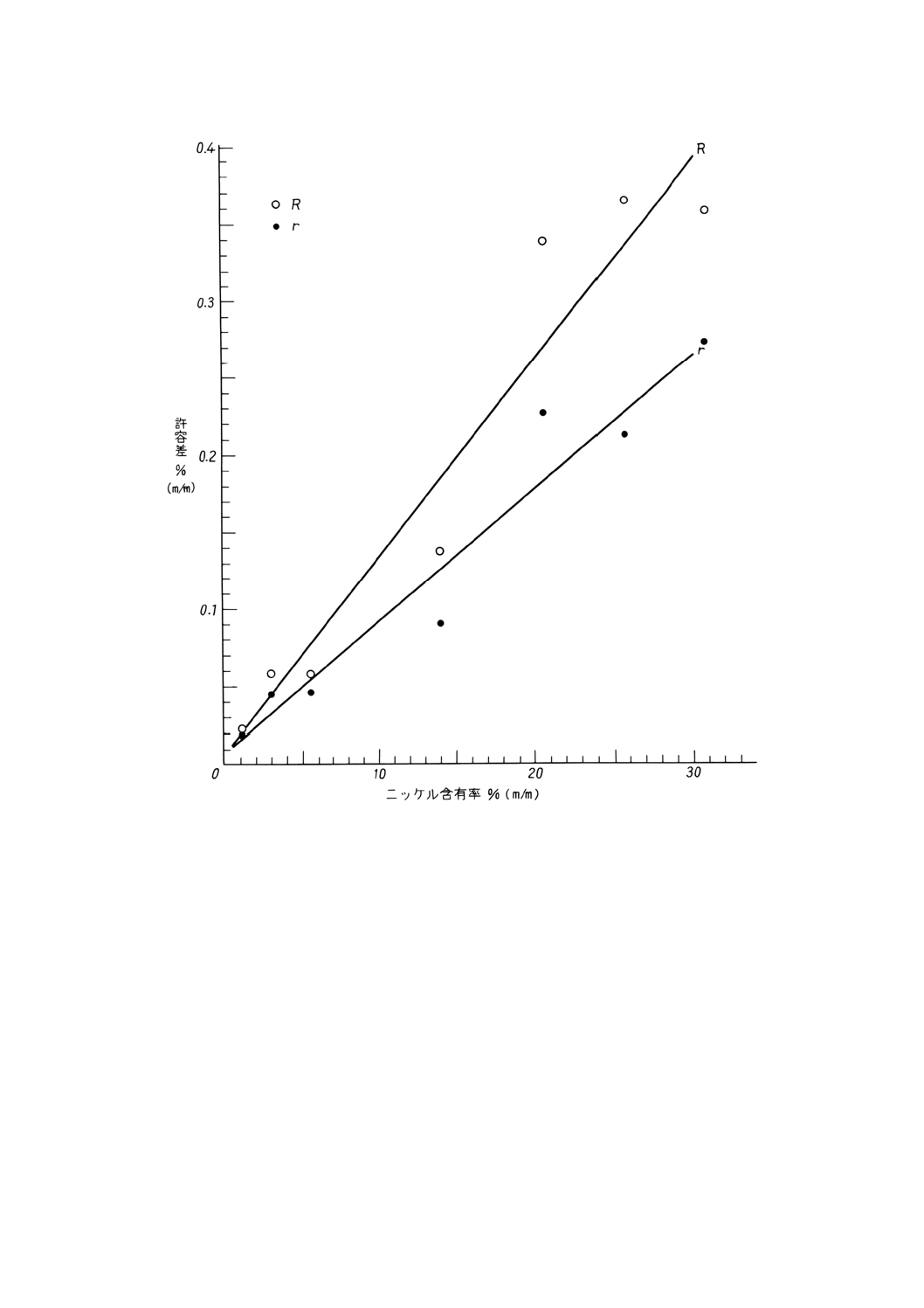
得られたデータは,附属書3表1にとりまとめたように,ニッケル含有率と分析結果の併行許容差及び
室間再現許容差は直線的関係を示した。使用した試料に関する情報は,参考Aに,データの図示を参考B
に示した。
9. 分析報告書 分析報告書には,次の事項を記載しなければならない。
a) 試料,分析室及び分析日時など証明に必要なすべての情報
b) この国際規格の引用
c) 使用した定量方法の種類(重量法か滴定法か)
d) 結果,及び表示した形態
e) 分析時に注目された非定常的なすべての特筆すべき点
f)
この国際規格に規定されてないすべての操作,又は結果に影響を与えそうなすべての任意操作
附属書3表1 許容差
ニッケル
含有率
% (m/m)
重量法
滴定法
併行許容差
r
室間再現許容差
R
併行許容差
r
室間再現許容差
R
0.5
0.032
0.025
0.011
0.012
1.0
0.035
0.030
0.015
0.018
5.0
0.063
0.071
0.049
0.070
10.0
0.098
0.132
0.092
0.134
15.0
0.132
0.174
0.135
0.198
20.0
0.167
0.226
0.178
0.262
25.0
0.202
0.278
0.220
0.326
30.0
0.236
0.329
0.263
0.390
19
G 1216-1997
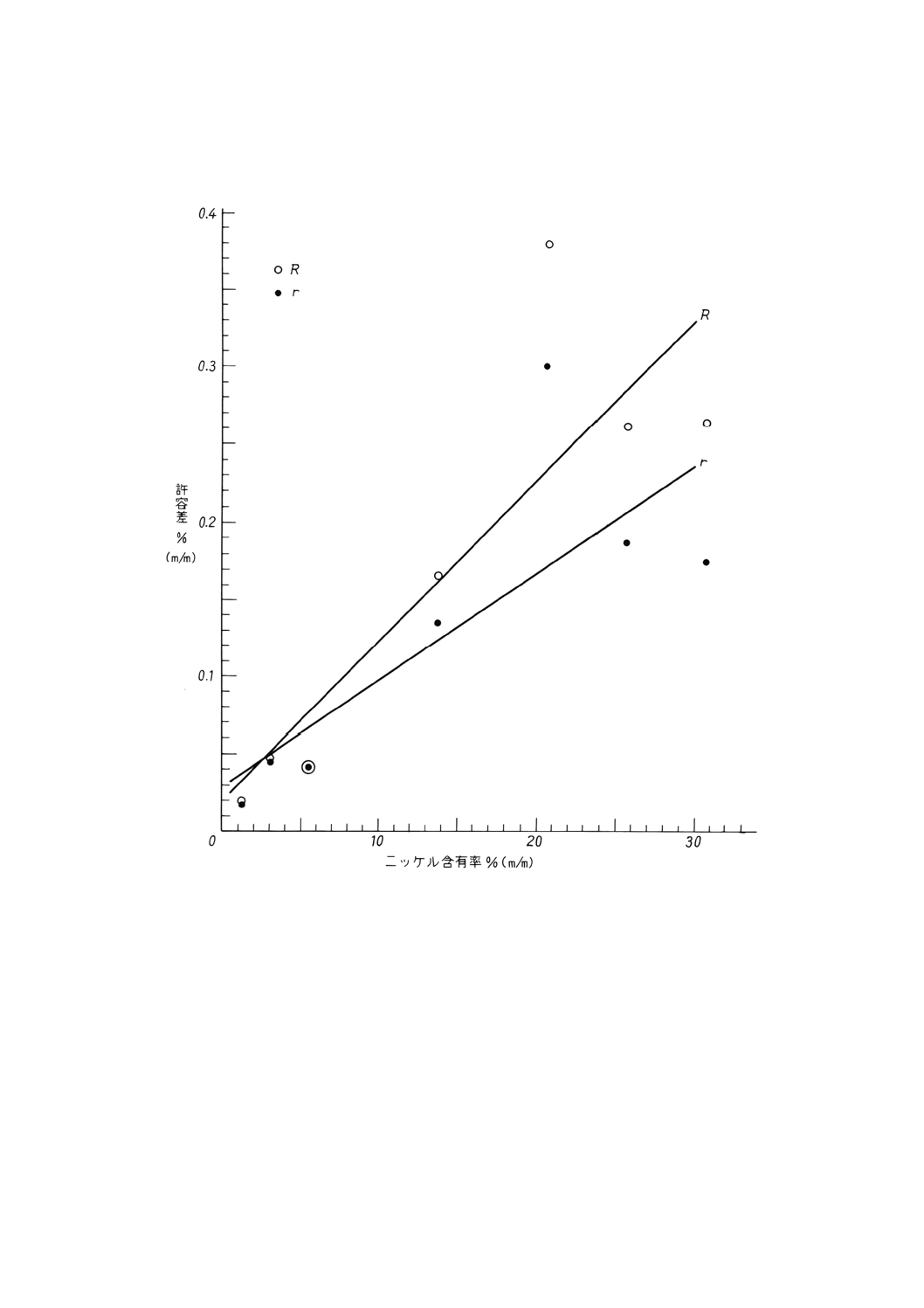
参考A 国際共同実験に関する追加情報
表1は,1983年に国際共同実験を実施した結果,すなわち,5か国,重量法は12分析室,滴定法は10
分析室で,銑鉄試料1個と鋼試料6個を実験した結果から求めたものである。
実験結果は,1984年3月に発行された文書17/1N598に記載されている。許容差データの図示を参考B
に示してある。
使用した試料を表A.1に示した。
表A.1
試料
ニッケル含有率% (m/m)
認証値
定量値
重量法
滴定法
ECRM 481-1(銑鉄)
1.19
1.183
1.190
JSS 151-7(低合金鋼)
2.99
2.959
2.985
BAM 230-1(ステンレス鋼)
5.55
5.555
5.576
JSS 653-1(ステンレス鋼)
13.91
13.897
13.935
BCS 334(ステンレス鋼)
20.60
20.478
20.560
NBS 348(高合金鋼)
25.8
25.719
25.751
JK 37(ステンレス鋼)
30.82
30.743
30.802
20
G 1216-1997
参考B 許容差データの図示
図B.1 ニッケル含有率と併行許容差 (r) 及びニッケル含有率と室間再現許容差 (R) との関係(重量法)
21
G 1216-1997
図B.2 ニッケル含有率と併行許容差 (r) 及びニッケル含有率と室間再現許容差 (R) との関係(滴定法)
22
G 1216-1997
附属書4 ジメチルグリオキシム吸光光度法 (1)
1. 要旨 試料を適切な酸で分解し,よう素を加えてアンモニアアルカリ性でニッケルを酸化した後,ジ
メチルグリオキシムを加えてジメチルグリオキシムニッケル錯体を生成させ,水酸化鉄などをろ過した後,
光度計を用いてその吸光度を測定する。
2. 試薬 試薬は,次による。
(1) 塩酸
(2) 硝酸
(3) 過塩素酸
(4) アンモニア水
(5) 鉄 できるだけ純度の高い鉄で,ニッケルを含有しないか,又はニッケル含有率ができるだけ低く既
知であるもの。
(6) よう素溶液 よう素2.6gによう化カリウム10gを加え,水約100mlを加えて完全に溶解した後,水で
液量を1 000mlとする。この溶液は,褐色瓶に保存する。
(7) 塩化ナトリウム
(8) エチレンジアミン四酢酸二水素二ナトリウム二水和物(以下,EDTA2Naという。)
(9) ジメチルグリオキシム溶液 ジメチルグリオキシム [(CH3)2C2 (NOH)2] 1gを水酸化ナトリウム溶液
(10g/l) 100mlに溶解する。
(10) 標準ニッケル溶液(250μgNi/ml) ニッケル[99.9% (m/m) 以上]0.250gをはかり採って,ビーカー
(300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 10mlを加えて穏やかに加熱して分解する。過塩素酸
20mlを加え,加熱して過塩素酸の濃厚な白煙が発生してビーカーの内部が透明になってからさらに2,
3分間加熱を続ける。放冷した後,時計皿の下面を水で洗って時計皿を取り除き,水約100mlを加え
て塩類を溶解する。常温まで冷却した後,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水
で標線まで薄める。
3. 試料はかり採り量 試料はかり採り量は,0.50gとする。
4. 操作
参考 警告 過塩素酸の蒸気は,アンモニア,亜硝酸蒸気又は有機物が存在すると爆発する危険があ
る。過塩素酸の蒸発処理は,過塩素酸を使用しても安全な排気設備を備えた場所で行わなけれ
ばならない。
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
(1) 硝酸と過塩素酸で分解容易な試料
(a) 試料をはかり採って三角フラスコ (300ml) に移し入れる。
(b) 過塩素酸15ml及び硝酸5mlを加え,加熱して分解し,さらに加熱して過塩素酸の濃厚な白煙を発
生させる。放冷した後,水約100mlを加えて振り混ぜて塩類を溶解する。常温まで冷却した後,溶
液を250mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。

23
G 1216-1997
(2) クロムなどの合金成分を含む試料
(a) 試料をはかり採って三角フラスコ (300ml) に移し入れる。
(b) 塩酸10ml及び硝酸5mlを加え,加熱して分解した後,過塩素酸15〜20mlを加え,引き続き加熱し
て過塩素酸の濃厚な白煙を発生させ,クロムを二クロム酸に酸化する。さらに加熱を続けながら塩
化ナトリウム0.1〜0.5gを少量ずつ数回に分けて加え,大部分のクロムを二酸化二塩化クロムとして
揮散させる(1)。放冷した後,水約100mlを加え,振り混ぜて塩類を溶解する。常温まで冷却した後,
溶液を250mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
注(1) 附属書4表1に示す分取量中のクロム量が1mg以下になる場合には,塩化ナトリウムによるクロ
ムの揮散処理を省略してもよい。
4.2
呈色 4.1の(1)(b)又は(2)(b)で得た溶液を附属書4表1に従って100mlの全量フラスコに分取し,水
で液量を約50mlとする。よう素溶液 [2.(6)] 10mlを加えて振り混ぜ,5〜10分間静置する。アンモニア水
20mlを加え,流水中で常温(2)まで冷却した後,ジメチルグリオキシム溶液 [2.(9)] 3mlを加え,水で標線ま
で薄める。
附属書4表1 分取量
ニッケル含有率
% (m/m)
分取量
ml
0.01以上 0.20未満
50
0.20以上 0.50未満
20
0.50以上 1.0 以下
10
注(2) 分取した試料溶液中に銅0.3mg以上を含み,吸光度の測定を4.3(2)によって行う場合には,15℃
以下に冷却する。
4.3
吸光度の測定 吸光度の測定は,次のいずれかの手順によって行う。
(1) 分取溶液中の銅含有量が0.3mg未満の場合 4.2で得た呈色液の一部を乾いたろ紙(5種A)でろ過す
る。最初のろ液の数mlは捨て,その後のろ液の一部を光度計の吸収セル (1cm) に取り,呈色後30
分以内に,水を対照液として波長440nm付近の吸光度を測定する。
(2) 分取溶液中の銅含有量が0.3mg以上の場合 4.2で得た呈色液の一部を乾いたろ紙(5種A)でろ過す
る。最初のろ液の数mlは捨て,その後のろ液の約30mlを乾いたビーカー (100ml) に取る。
EDTA2Na0.1gを加え,振り混ぜて溶解し,直ちに溶液の一部を光度計の吸収セル (1cm) に取り,
EDTA2Na添加後5分以内に,水を対照液として波長440nm付近における吸光度を測定する。
5. 空試験 鉄 [2.(5)] 0.500gを三角フラスコ (300ml) にはかり採り,以下,4.1(1)(b),4.2及び4.3の手順
又は4.1(2)(b),4.2及び4.3の手順に従って試料と同じ操作を試料と併行して行う。
6. 検量線の作成 鉄 [2.(5)] 0.500gずつを6個はかり採り,それぞれを三角フラスコ (300ml) に移し入
れ,さらに標準ニッケル溶液 [2.(10)] を附属書4表2に従って正確に加える。以下,4.1(1)(b),4.2及び4.3
の手順に従って操作して,得た吸光度と標準ニッケル溶液として加えたニッケル量との関係線を作成し,
その関係線を原点を通るように平行移動して検量線とする。
24
G 1216-1997
附属書4表2 標準ニッケル溶液添加量
ニッケル含有率
% (m/m)
標準ニッケル溶液 [2.(10)] 添加量
ml
0.01以上0.20未満
0, 0.50, 1.0,
2.0,
3.0,
4.0
0.20以上0.50未満
0, 2.0,
4.0,
6.0,
8.0, 10.0
0.50以上1.0 以下
0, 4.0,
8.0,
12.0, 16.0, 20.0
7. 計算 4.3及び5.で得た吸光度と6.で作成した検量線とからニッケル量を求め,試料中のニッケル含有
率を,次の式によって算出する。
100
250
250
3
2
1
×
×
×
−
−
=
B
m
B
A
A
A
Ni
ここに, Ni: 試料中のニッケル含有率 [% (m/m)]
A1: 分取した試料溶液中のニッケル検出量 (g)
A2: 分取した空試験液中のニッケル検出量 (g)
A3: 5.ではかり採った鉄 [2.(5)] 0.500g中に含まれるニッケルの
量 (g)
m: 試料はかり採り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
8. 許容差 許容差(3)は,附属書4表3による。
附属書4表3 許容差
単位% (m/m)
ニッケル含有率
室内再現許容差
室間再現許容差
0.018以上0.20未満 D [0.002 7× (Ni) +0.000 5] D [0.006 0× (Ni) +0.001 3]
0.20 以上1.0 以下
D [0.003 2× (Ni) +0.001 0] D [0.014 1× (Ni) −0.001 3]
注(3) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一室内における分析回数,室間再現許容差の場合は分析に関与した
分析室数である(n=2のとき,D=2.8である)。
また, (Ni) は,許容差を求める試料中のニッケル含有率 [% (m/m)] である。
参考 この許容差は,ニッケル含有率0.018% (m/m) 以上0.86% (m/m) 以下の試料を用い,共同実験
した結果から求めたものである。
25
G 1216-1997
附属書5 ジメチルグリオキシム吸光光度法 (2)
1. 要旨 試料を適切な酸で分解し,くえん酸で鉄などをマスキングした後,アンモニアアルカリ性でよ
う素を加えてニッケルを酸化し,ジメチルグリオキシムを加えてジメチルグリオキシムニッケル錯体を生
成させ,光度計を用いてその吸光度を測定する。
2. 試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) 過塩素酸
(3) 王水(塩酸3,硝酸1)
(4) アンモニア水
(5) アルミニウム片 約1.7mm以上の切削薄片にしたもの。
(6) 鉄 できるだけ純度の高い鉄で,ニッケル含有率が0.01% (m/m) 以下であるもの。
(7) よう素溶液 よう素2.6gによう化カリウム10gを加え,水約100mlを加えて完全に溶解した後,水で
液量を1 000mlとする。この溶液は,褐色瓶に保存する。
(8) 塩化ナトリウム
(9) くえん酸溶液 (500g/l)
(10) ジメチルグリオキシム溶液 ジメチルグリオキシム [(CH3)2C2 (NOH)2] 1gを水酸化ナトリウム溶液
(10g/l) 100mlに溶解する。
(11) 標準ニッケル溶液 (500μgNi/ml) ニッケル[99.9% (m/m) 以上]0.500gをはかり採って,ビーカー
(300ml) に移し入れ,時計皿で覆い,硝酸 (1+1) 20mlを加えて穏やかに加熱して分解する。過塩素酸
20mlを加え,加熱して過塩素酸の濃厚な白煙が発生してビーカーの内部が透明になってから更に2,
3分間加熱を続ける。放冷した後,時計皿の下面を水で洗って時計皿を取り除き,水約100mlを加え
て塩類を溶解する。常温まで冷却した後,溶液を1 000mlの全量フラスコに水を用いて移し入れ,水
で標線まで薄める。
3. 試料はかり採り量 試料はかり採り量は,0.20gとする。
4. 操作
参考 警告 過塩素酸の蒸気は,アンモニア,亜硝酸蒸気又は有機物が存在すると爆発する危険があ
る。過塩素酸の蒸発処理は,過塩素酸を使用しても安全な排気設備を備えた場所で行わ
なければならない。
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
(1) 王水と過塩素酸で分解容易な試料
(a) 試料をはかり採って三角フラスコ (300ml) に移し入れる。
(b) 王水20mlを加え,加熱して分解した後,過塩素酸15mlを加え,引き続き加熱して過塩素酸の濃厚
な白煙を発生させ,クロムを二クロム酸に酸化する。さらに加熱を続けながら塩化ナトリウム0.1
〜0.5gを少量ずつ数回に分けて加え,大部分のクロムを二酸化二塩化クロムとして揮散させる(1)。
26
G 1216-1997
放冷した後,水約100mlを加え,振り混ぜて塩類を溶解する。常温まで冷却した後,溶液を250ml
の全量フラスコに水を用いて移し入れ,水で標線まで薄める。
(2) 銅含有率3% (m/m) 以上の試料
(a) 試料をはかり採って三角フラスコ (300ml) に移し入れる。
(b) 王水20mlを加え,加熱して分解した後,過塩素酸15mlを加え,引き続き加熱して過塩素酸の濃厚
な白煙を発生させ,クロムを二クロム酸に酸化し,過塩素酸の残量が7〜10mlになるまで蒸発する。
放冷した後,水約100mlを加えて塩類を溶解し,アルミニウム片 [2.(5)] 約1gを加えて穏やかに加
熱し,二クロム酸を還元してからさらに3〜5分間加熱を続ける。析出した銅を残ったアルミニウム
片と共にろ紙(5種B)でろ過する。ろ紙及び残さを温水で7,8回洗浄し,ろ液と洗液はビーカー
(300ml) に集める。この溶液に硝酸 (1+1) 5mlと過塩素酸5mlを加え,時計皿で覆い,加熱して過
塩素酸の濃厚な白煙を発生させる。放冷した後,時計皿の下面を水で洗って時計皿を取り除き,水
約100mlを加え,振り混ぜて塩類を溶解する。常温まで冷却した後,溶液を250mlの全量フラスコ
に水を用いて移し入れ,水で標線まで薄める。
注(1) 試料中のクロム含有率が10% (m/m) 未満の場合には,塩化ナトリウムによるクロムの揮散操作
を省略してもよい。
4.2
呈色 呈色は,次のいずれかの手順によって行う。
(1) コバルト含有率3% (m/m) 未満の試料の場合 4.1の(1)(b)又は (2)(b)で得た溶液10mlを100mlの全量
フラスコに分取し,くえん酸溶液10mlを加え,水で液量を約50mlとする。アンモニア水10mlを加
えて振り混ぜた後,全量フラスコを流水に浸して常温まで冷却する。よう素溶液 [2.(7)] 10ml及びジ
メチルグリオキシム溶液 [2.(10)] 3mlを加え,水で標線まで薄める。
(2) コバルト含有率3% (m/m) 以上の試料の場合 2個の100mlの全量フラスコに4.1の(1)(b)又は(2)(b)
で得た溶液を10mlずつを分取し,それぞれにくえん酸溶液10mlを加え,水で液量を約50mlとする。
アンモニア水10mlを加えて振り混ぜた後,全量フラスコを流水に浸して常温まで冷却する。一方の
全量フラスコには,よう素溶液 [2.(7)] 10ml及びジメチルグリオキシム溶液 [2.(10)] 3mlを加え,水で
標線まで薄めて呈色液とする。もう一方の全量フラスコには,よう素溶液 [2.(7)] だけを加え,水で
標線まで薄めて補償用対照液とする。
4.3
吸光度の測定 吸光度の測定は,次のいずれかの手順によって行う。
(1) コバルト含有率3% (m/m) 未満の試料の場合 4.2(1)で得た呈色溶液の一部を,光度計の吸収セル
(1cm) に取り,水を対照として,波長530nm付近の吸光度を呈色後30分間以内に測定する。
(2) コバルト含有率3% (m/m) 以上の試料の場合 4.2(2)で得た呈色液及び補償用対照液の一部を,光度
計の吸収セル (1cm) に取り,水を対照として,波長530nm付近の吸光度を呈色後30分間以内に測定
する。
5. 空試験 鉄 [2.(6)] 0.200gを三角フラスコ (300ml) にはかり採り,以下,4.1(1)(b),4.2及び4.3の手順
又は4.1(2)(b),4.2及び4.3の手順に従って試料と同じ操作を試料と併行して行う。
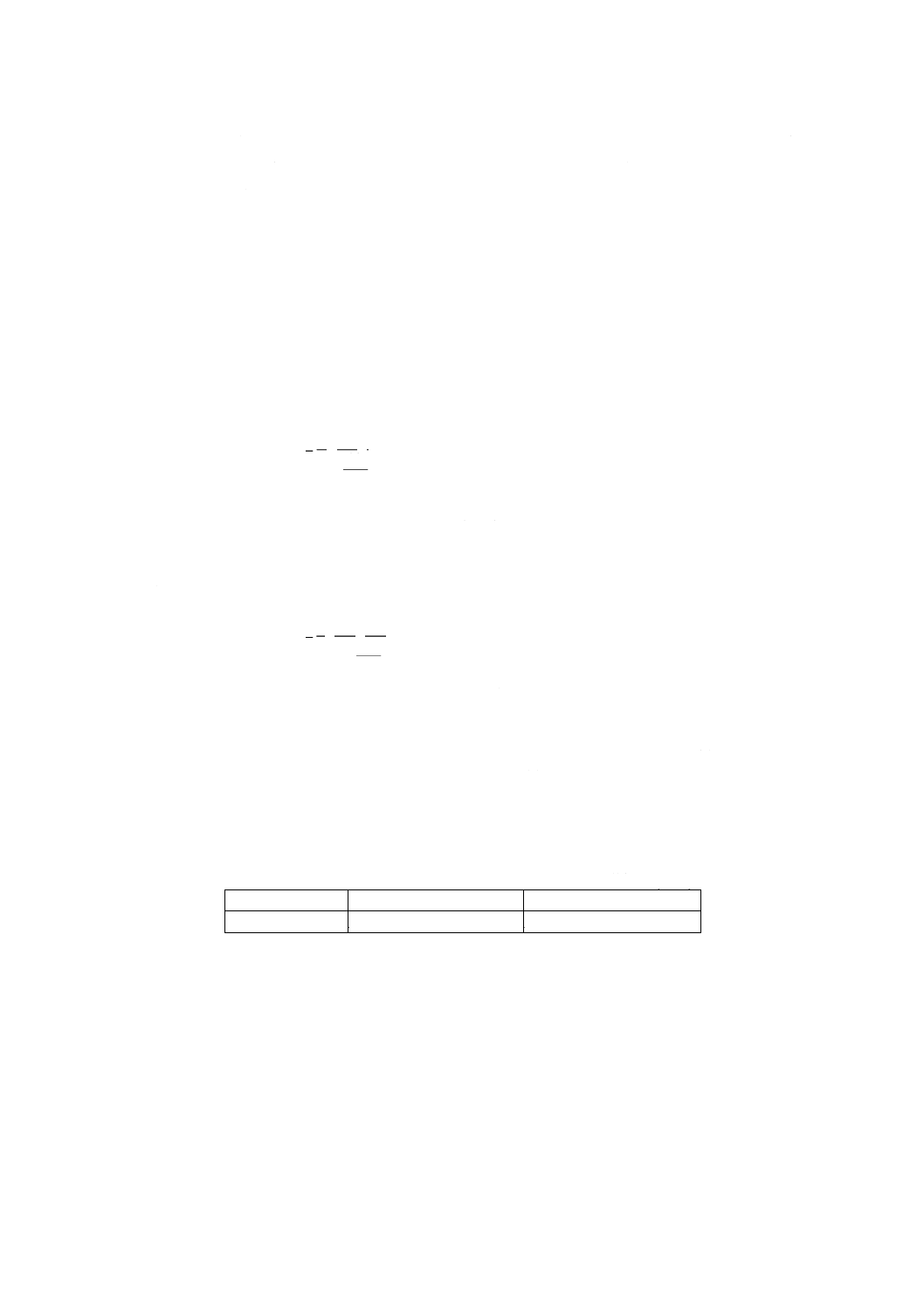
27
G 1216-1997
6. 検量線の作成 鉄 [2.(6)] 0.200gずつを6個はかり採り,それぞれを三角フラスコ (300ml) に移し入
れ,さらに標準ニッケル溶液 [2.(11)] を0,4.0,8.0,12.0,16.0及び20.0ml(ニッケル量として0,2 000,
4 000,6 000,8 000及び10 000μg)加える。王水20ml及び過塩素酸15mlを加え,加熱して分解し,引き
続き加熱して過塩素酸の濃厚な白煙を発生させる。放冷した後,水約100mlを加え,振り混ぜて塩類を溶
解する。常温まで冷却した後,溶液を250mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
以下,4.2(1)及び4.3(1)の手順に従って操作して,得た吸光度と標準ニッケル溶液として加えたニッケル量
との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
7. 計算 4.3及び5.で得た吸光度と6.で作成した検量線とからニッケル量を求め,試料中のニッケル含有
率を,次のいずれかの式によって算出する。
(1) コバルト含有率3% (m/m) 未満の試料の場合
100
250
10
2
1
×
×
−
=
m
A
A
Ni
ここに, Ni: 試料中のニッケル含有率% (m/m)
A1: 分取した試料溶液中のニッケル検出量 (g)
A2: 分取した空試験液中のニッケル検出量 (g)
m: 試料はかり採り量 (g)
(2) コバルト含有率3% (m/m) 以上の試料の場合
100
250
10
3
2
1
×
×
−
−
=
m
A
A
A
Ni
ここに, Ni: ニッケル含有率% (m/m)
A1: 分取した試料溶液中のニッケル検出量 (g)
A2: 分取した空試験液中のニッケル検出量 (g)
A3: 分取した補償用対照液中のニッケル検出量 (g)
m: 試料はかり採り量 (g)
8. 許容差 許容差(2)は,附属書5表1による。
附属書5表1 許容差
単位% (m/m)
ニッケル含有率
室内再現許容差
室間再現許容差
1.0以上5.0以下 D [0.002 7×(Ni) +0.002 4] D [0.006 2×(Ni) +0.006 2]
注(2) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一室内における分析回数,室間再現許容差の場合は分析に関与した
分析室数である(n=2のとき,D=2.8である)。
また, (Ni) は,許容差を求める試料中のニッケル含有率 [% (m/m)] である。
参考 この許容差は,日本鉄鋼認証標準物質の認証値決定の集計値から,ジメチルグリオキシム吸光
光度法(2)による定量値を集計して解析したものである。
28
G 1216-1997
附属書6 ジメチルグリオキシム吸光光度法 (3)
附属書6としてのまえがき
この附属書6は,1984年第1版として発行されたISO 4939 (Steel and cast iron−Determination of nickel content
−Dimethylglyoxime spectrophotometric method) を翻訳し,技術的内容及び規格票の様式を変更することな
く作成した日本工業規格である。
なお,この規格で下線(点線)を施してある箇所は,原国際規格にはない事項である。
1. 目的及び適用範囲 この附属書6は,鋼及び鋳鉄中のニッケルをジメチルグリオキシム吸光光度法に
よって定量する方法について規定する。
この方法は,ニッケル含有率0.10% (m/m) 以上4% (m/m) 以下の範囲の試料に適用する。コバルト,銅
及びマンガンが存在すると妨害の原因となる[7.3.2の注(2)参照]。
2. 引用規格
ISO/R 377 Selection and preparation of samples and test pieces for wrought steel
参考 ISO 377は,次の規格で置き換えられている。
ISO 14284 : 1996 Steel and iron−Sampling and preparation of samples for the determination of chemical
composition
3. 原理 試料を塩酸,硝酸及び過塩素酸で分解する。よう素とよう化カリウムを含むアンモニア性溶液
中でニッケル (III) とジメチルグリオキシムとの錯体を生成させる。この錯体の吸光度を波長約535nmで
測定する。
4. 試薬 分析に際しては,特に記述しない限り,分析保証試薬及び蒸留水又はそれに同等の純度をもつ
水を使用する。
4.1
混酸 塩酸(密度約1.19g/ml)2容,硝酸(密度約1.40g/ml)1容及び水2容を混合する。
4.2
過塩素酸 密度約1.54g/ml(1)
注(1) 過塩素酸(密度約1.67g/ml)も使用できる。過塩素酸(密度約1.54g/ml)100mlは,過塩素酸(密
度約1.67g/ml)79mlに相当する。
4.3
くえん酸アンモニウム溶液 くえん酸一水和物 (C6H8O7.H2O) 250gをアンモニア水(密度約
0.91g/ml)250mlに溶解し,冷却した後,水で1lに薄めて混合する。
4.4
よう素溶液 よう化カリウム25gとよう素12.7gを最少量の水に溶解する。水で1lに薄めて混合す
る。
4.5
ジメチルグリオキシム溶液 ジメチルグリオキシム1gをアンモニア水(密度約0.91g/ml)500mlに
溶解し,水で1lに薄めて混合する。
4.6
アンモニア水 密度約0.91g/ml,希釈液1+1
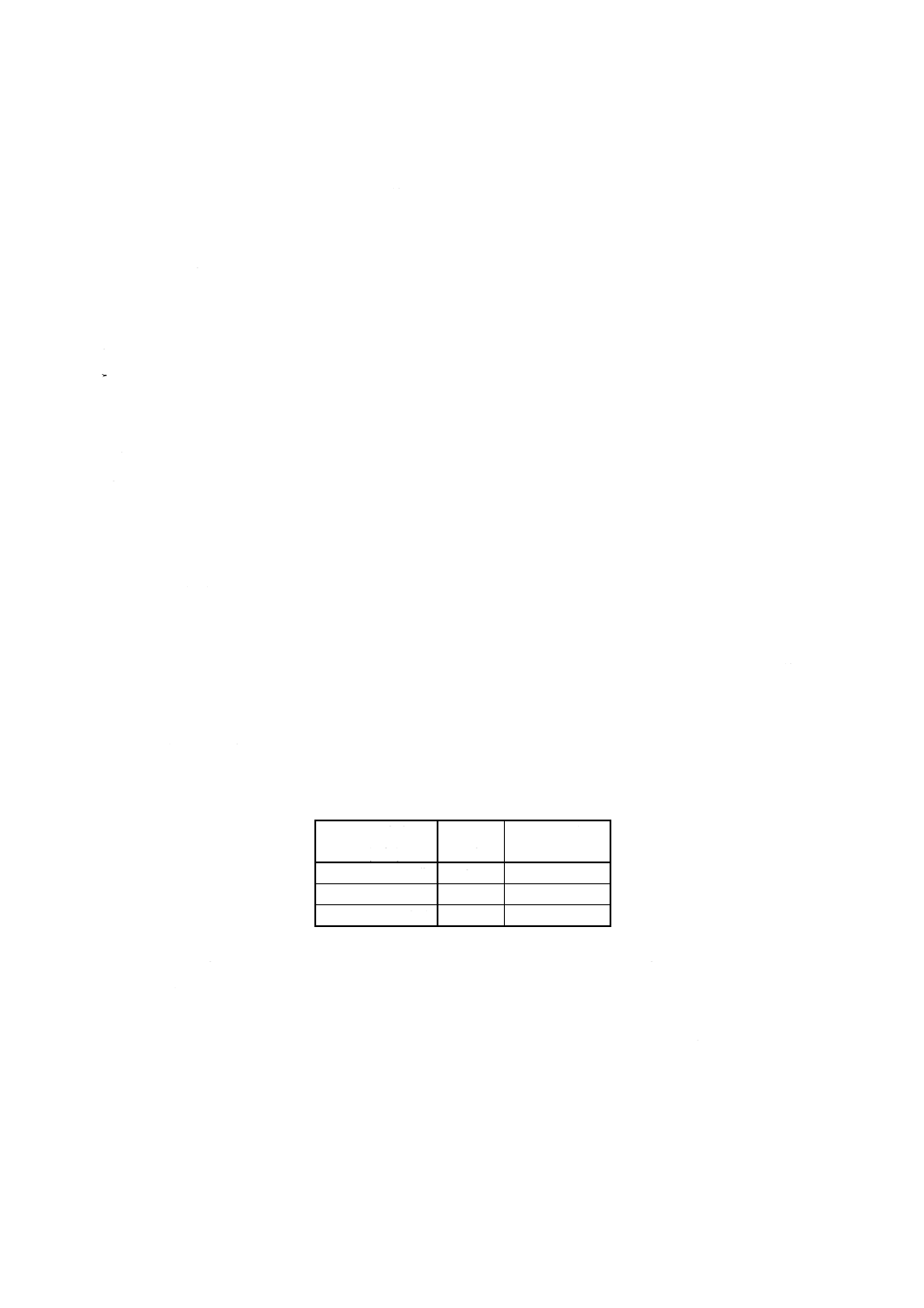
29
G 1216-1997
4.7
ニッケル標準溶液 0.5gNi/l相当溶液高純度ニッケル0.500 0gを0.000 1gのけたまではかり採り,硝
酸(密度約1.40g/ml,希釈液2+3)20mlで分解する。煮沸して窒素酸化物を除去し,冷却する。これを1
000mlの全量フラスコに移し入れ,水で標線まで薄めて混合する。この標準溶液1ml中には,Niを0.5mg
含有する。
5. 装置 通常の分析室器具と次のものを使用する。
分光光度計
6. サンプリング サンプリングは,ISO/R 377によるか,又は鋼若しくは鋳鉄用の適切な国家規格に従
う。
7. 操作
警告 過塩素酸の蒸気は,一般にアンモニア,亜硝酸の蒸気,又は有機物の存在で爆発の危険がある。
7.1
試料のはかり採り 試料は,約0.5gを0.001gのけたまではかり採る。
7.2
空試験 試料と併行して同一操作に従って全試薬の同一量を用いて空試験を行う。
7.3
定量
7.3.1
試料溶液の調製 はかり採った試料 (7.1) を250mlのビーカーに移し入れる。混酸 (4.1) 10m1及
び過塩素酸 (4.2) 10mlを加え,ビーカーを時計皿で覆い,反応が終了するまで加熱する。過塩素酸の白煙
が発生するまで蒸発し,過塩素酸の白煙がビーカーの側壁を伝わって逆流するような状態が維持できる温
度で少なくとも3分間を継続させ,すべてのクロムを二クロム酸に酸化する。熱源から降ろして放冷する。
水50mlを加え,加熱して塩類を溶解し,冷却した後,250mlの全量フラスコに移し入れる。水で標線まで
薄めて混合する。乾いたろ紙(5種A)を用いて傾斜法によってろ過し,ろ液の最初の部分を捨てた後,
残さや沈殿物を全部除去し,ろ液を乾燥ビーカーに集める。
7.3.2
呈色 試料溶液 (7.3.1) をニッケルの含有率に応じて附属書6表1に従って,25ml又は10mlを分
取して100mlの全量フラスコに移し入れる(2)。
附属書6表1 分取量及びセルの光路長
ニッケル含有率
% (m/m)
分取量
ml
セルの光路長
cm
0.1以上 0.5未満
25
4
0.5以上 1.6未満
25
1
1.6以上 4.0以下
10
1
注(2) 試料溶液 (7.3.1) 中にコバルト,銅,又はマンガンが含まれる場合は,分取した溶液中にそれら
の元素のそれぞれの量が次の制限量を超えないように分取しなければならない。コバルト5mg,
銅0.5mg,マンガン1mg。
くえん酸アンモニウム溶液 (4.3) 20mlを加え,振り混ぜ,次によう素溶液 (4.4) 3mlを加えて再度振り混
ぜる。5分間静置する。これにジメチルグリオキシム溶液 (4.5) 20mlを加え,水で標線まで薄めて混合す
る。これを20℃の温度に10分間以上20分間を超えないように静置する。

30
G 1216-1997
7.3.3
対照溶液の調製 試料溶液の分取量 (7.3.2) と同量の試料溶液を分取して別の100mlの全量フラス
コに移し入れる。これにくえん酸アンモニウム溶液 (4.3) 20mlを加え,振り混ぜ,次によう素溶液 (4.4) 3ml
を加えて再度振り混ぜる。5分間静置する。これにアンモニア水 (4.6) 20mlを加え,水で標線まで薄めて
混合する。これを20℃の温度に10分間以上20分間を超えないように静置する。
7.3.4
吸光度の測定 適切な光路長のセル(附属書6表1参照)を用い,対照溶液 (7.3.3) でゼロ点を調
節した分光光度計で波長約535nmにおける試料溶液の吸光度を測定する。
7.4
検量線の作成
7.4.1
検量線溶液の調製 混酸 (4.1) 10mlと過塩素酸 (4.2) 10mlをあらかじめ添加した250mlのビーカー
のシリーズを用意して,それぞれにニッケルの予想含有率に適するように附属書6表2に示す量のニッケ
ル標準溶液 (4.7) を加える。
7.3.1及び7.3.2の手順に従って処理し,対照溶液には検量線溶液“0”のものを使用する。
附属書6表2 検量線溶液の調製
ニッケル含有率
(7.3.2)
% (m/m)
ニッケル標準溶液
(4.7)
ml
ニッケル相当濃度
μg/ml
セルの
光路長
cm
0.1以上0.5未満
0
0
4
1.0
5
2.0
10
3.0
15
4.0
20
5.0
25
0.5以上1.6未満
0
0
1
2.0
10
4.0
20
8.0
40
10.0
50
14.0
70
16.0
80
1.6以上4.0以下
0
0
1
5.0
25
10.0
50
15.0
75
20.0
100
25.0
125
30.0
150
35.0
175
40.0
200
7.4.2
吸光度の測定 波長約535nmで,対照溶液として検量線溶液“0”のものを使用してゼロ点を調節
した分光光度計を用いて各溶液の吸光度を測定する。
7.4.3
検量線の作図 7.4.2で得た吸光度とニッケル標準溶液として加えたニッケル量との関係線を作成
し,その関係線を原点を通るように平行移動して検量線とする。
参考 原文では,検量線の傾斜から角度係数aを計算して8.結果の表示に用いているが,作図するJIS
の一般的な記載様式に変更した。

31
G 1216-1997
8. 結果の表示 7.3.4で得た吸光度と7.4.3で作図した検量線とからニッケル量を求め,試料中のニッケ
ル含有率を,次の式によって算出する。
100
250
2
1
×
×
−
=
V
m
A
A
Ni
ここに, Ni: 試料中のニッケル含有率% (m/m)
A1: 分取した試料溶液中のニッケル検出量 (g)
A2: 分取した空試験液中のニッケル検出量 (g)
V: 分取液量 (ml) (附属書6表1)
m: 試料はかり採り量 (g)
参考 原文では,8.結果の表示を8.1検量線が直線でない場合及び8.2検量線が直線の場合にわけて記
載しているが,8.1検量線が直線でない場合では,4cmセルで測定した吸光度を1cmで測定し
た値に補正することによる計算式に誤りがあるので採用していない。
9. 許容差 この方法の共同実験は,5分析室で6水準のニッケル含有率の試料を用いて行い,各分析室
とも各試料を6回ずつ定量した。
得られた結果は,ISO 5725に従って統計計算を行った。得られたデータは,附属書6表3に要約したよ
うにニッケル含有率と試験結果の併行許容差及び室間再現許容差との間に対数的比例関係を示した。参考
Bに,その結果を図示した。
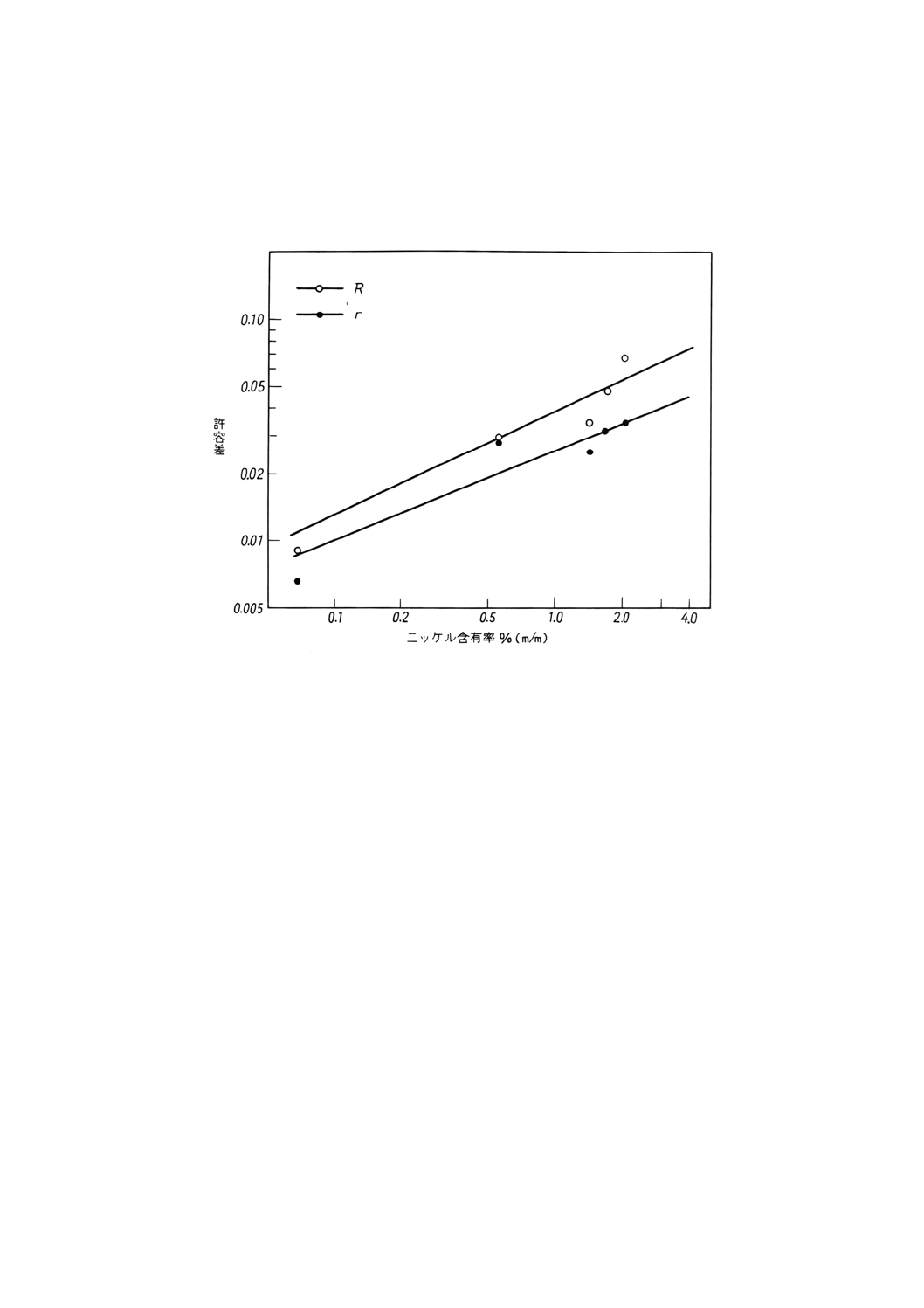
附属書6表3 許容差
ニッケル含有率
% (m/m)
併行許容差
r
室間再現許容差
R
0.1
0.010
0.013
0.2
0.013
0.018
0.5
0.019
0.027
1.0
0.025
0.038
2.0
0.034
0.053
4.0
0.046
0.075
短期間内で同一装置を使用して一人の分析者が同一試料について分析した2個の分析値の差は,方法を
標準的に正確に操作した場合,併行許容差 (r) を超えることは20回の分析のうちで1回より多くはない。
同一試料について異分析所間で2人の分析者が分析した独立の2個の分析値の差は,方法を標準的に正
確に操作した場合,室間再現許容差 (R) を超えることは20回の分析のうち1回より多くはない。
10. 分析報告書 分析報告書には,次の事項を記載しなければならない。
a) この国際規格の引用
b) 結果と表示した形態
c) 分析時に注目された非定常的な特筆すべき点
d) この国際規格に規定されていないすべての操作,又は結果に影響を与えそうなすべての任意操作

32
G 1216-1997
参考A 国際共同実験に関する追加情報
(この国際規格の必要な部分ではない)
附属書6表3は,1979年に,2か国,5分析所で6個の鋼試料を用いて実施した国際共同実験の結果か
ら求めたものである。
実験結果は,1980年11月に発行された文書17/1 N 433に報告されている。許容差データを参考Bに図
示した。分析試料を表A.1に示した。
表A.1
試料
ニッケル含有率%
(m/m)
認証値
分析値
BCS 260/4(高純度鉄)
0.003
0.005 5
BCS 431 (炭素鋼)
0.069
0.066 4
BCS 341 (24%クロムステンレス鋼)
0.56
0.558
BCS 225/2(Ni, Cr, Mo鋼)
1.43
1.434
BCS 406 (低合金鋼)
1.69
1.690
BCS 410 (低合金鋼)
2.04
2.034
備考1. 統計解析は,ISO 5725に従って行った。
2. この国際共同実験には,6個の試料が使用された
が,この方法の適用範囲がニッケル含有率0.1%
(m/m) 以上4% (m/m) 以下となったので,参考B
には,5点だけが図示され,BCS 260/4 [Ni : 0.003%
(m/m)] のデータを省略した。
33
G 1216-1997
参考B 許容差データの図示
(この国際規格の必要な部分ではない)
図B.1 ニッケル含有率と併行許容差r又は室間再現許容差Rとの関係
34
G 1216-1997
原案作成委員会 構成表
(1) 社団法人日本鉄鋼連盟鋼材標準委員会JE6分科会
氏名
所属
(主査)
松 村 泰 治
川鉄テクノリサーチ株式会社
(ISO TC17/SC1議長)
佐 伯 正 夫
富士物産株式会社
(主幹事)
小 野 昭 紘
新日本製鐵株式会社
(幹事)
天 野 徹
通商産業省工業技術院標準部
広 川 吉之助
東北大学金属材料研究所
大河内 春 乃
科学技術庁金属材料技術研究所
(幹事)
余 語 英 俊
愛知製鋼株式会社
(幹事)
安 原 久 雄
川崎製鉄株式会社
杉 原 孝 志
川崎製鉄株式会社
合 田 明 弘
川鉄テクノリサーチ株式会社
(幹事)
滝 沢 佳 郎
川鉄テクノリサーチ株式会社
瀬 野 英 夫
鋼管計測株式会社
岡 山 和 生
合同製鐵株式会社
吉 野 信一郎
株式会社神戸製鋼所
(幹事)
金 築 宏 治
株式会社神戸製鋼所
(幹事)
河 村 恒 夫
株式会社コベルコ科研
大 石 隆 司
山陽特殊鋼株式会社
(幹事)
鈴 木 眞
新日本製鐵株式会社
八 塚 隆
新日本製鐵株式会社
橋 本 光 生
新日本製鐵株式会社
大 塚 祐 二
新日本製鐵株式会社
大 野 義 信
新日本製鐵株式会社
佐 藤 明 久
新日本製鐵株式会社
梶 間 透
新日本製鐵株式会社
兼 松 勤 治
新日本製鐵株式会社
高 橋 譲
新日本製鐵株式会社
黒 岩 猛
新日本製鐵株式会社
藤 生 卓
住友金属工業株式会社
(幹事)
岡 沢 亨
住友金属工業株式会社
(幹事)
蔵 保 浩 文
住友金属工業株式会社
藤 城 泰 文
住友金属工業株式会社
西 野 和 美
住友金属工業株式会社
遠 藤 丈
住友金属テクノロジー株式会社
(幹事)
伊 藤 清 孝
大同特殊鋼株式会社
山 村 英 二
株式会社中山製鋼所
小 澤 幸 男
日鋼検査サービス株式会社
(幹事)
槌 尾 武 久
日新製鋼株式会社
永 本 弘 信
ニッテクリサーチ株式会社
桝 井 為 則
株式会社日鐵テクノリサーチ
山 本 満 治
株式会社日鐵テクノリサーチ
遠 山 直 人
日本金属工業株式会社
(幹事)
吉 岡 豊
日本鋼管株式会社
(幹事)
石 橋 耀 一
日本鋼管株式会社
吉 川 裕 泰
日本鋼管株式会社
藤 原 民 雄
株式会社日本製鋼所
(幹事)
永 井 宣太郎
日本冶金工業株式会社
野 原 努
日立金属株式会社
羽 毛 和 記
三菱製鋼株式会社
35
G 1216-1997
氏名
所属
竹 田 秀 俊
株式会社室蘭試験分析センター
(事務局)
柿 田 和 俊
社団法人日本鉄鋼連盟
稲 本 勇
社団法人日本鉄鋼連盟
脊 戸 雄 功
社団法人日本鉄鋼連盟
(2) 社団法人日本鉄鋼連盟鉄鋼分析JIS三者委員会
氏名
所属
(委員長)
大河内 春 乃
科学技術庁金属材料技術研究所
(幹事)
松 村 泰 治
川鉄テクノリサーチ株式会社
近 藤 隆 明
日本鋼管株式会社
(委員)
小 島 彰
通商産業省基礎産業局
天 野 徹
通商産業省工業技術院標準部
因 幸二郎
財団法人日本規格協会
倉 橋 正 保
通商産業省工業技術院物質工学工業技術研究所
島 貫 孝
社団法人日本分析化学会
広 川 吉之助
東北大学金属材料研究所
浦 谷 文 博
大阪府立産業技術総合研究所
鈴 木 勝
社団法人日本海事検定協会
永 山 宏
日立マテリアルエンジニアリング株式会社
束 原 巌
古河電気工業株式会社
橋 本 勝
株式会社日産アーク
蔵 保 浩 文
住友金属工業株式会社
河 村 恒 夫
株式会社コベルコ科研
伊 藤 清 孝
大同特殊鋼株式会社
槌 尾 武 久
日新製鋼株式会社
(事務局)
柿 田 和 俊
社団法人日本鉄鋼連盟
稲 本 勇
社団法人日本鉄鋼連盟