G 1212-1997
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。これによってJIS G 1212-1981は改正され,この規格に置き換えられる。
今回の改正では,国際規格との整合化を図るために,ISO規格の翻訳を附属書2,4及び附属書5として
規定している。
JIS G 1212には,次に示す附属書がある。
附属書1 二酸化けい素重量法(1)
附属書2 二酸化けい素重量法(2) (ISO 439)
附属書3 モリブドけい酸青吸光光度法(1)
附属書4 モリブドけい酸青吸光光度法(2) (ISO 4829-1)
附属書5 モリブドけい酸青吸光光度法(3) (ISO 4829-2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
G 1212-1997
鉄及び鋼−けい素定量方法
Iron and steel−Methods for
determination of silicon content
序文 この規格は,附属書2に1994年に第2版として発行されたISO 439, Steel and iron−Determination of
total silicon content−Gravimetric methodを翻訳し,また,附属書4には,1986年に発行されたISO 4829-1, Steel
and cast iron−Determination of total silicon content−Reduced molybdosilicate spectrophotometric method−Part
1 : Silicon contents between 0.05 and 1.0%を,附属書5には,1988年に発行されたISO 4829-2, Steel and iron
−Determination of total silicon content−Reduced molybdosilicate spectrophotometric method−Part 2 : Silicon
contents between 0.01 and 0.05%を翻訳し,技術的内容及び規格票の様式を変更することなく作成した日本工
業規格であるが,対応国際規格には規定されていない規定事項を日本工業規格として追加している。
なお,この規格で点線の下線を施してある“参考”は,原国際規格にはない事項である。
1. 適用範囲 この規格は,鉄及び鋼中のけい素の定量方法について規定する。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
JIS G 1201 鉄及び鋼の分析方法通則
JIS Z 8402 分析・試験の許容差通則
ISO 439 Steel and iron−Determination of total silicon content−Gravimetric method
ISO 4829-1 Steel and cast iron−Determination of total silicon content−Reduced molybdosilicate
spectro-photometric method−Part 1 : Silicon contents between 0.05 and 1.0%
ISO 4829-2 Steel and iron−Determination of total silicon content−Reduced molybdosilicate
spectrophotometric method−Part 2 : Silicon contents between 0.01 and 0.05%
3. 一般事項 定量方法に共通な一般事項は,JIS G 1201による。ただし,JIS G 1201は,附属書2,附
属書4及び附属書5には適用しない。
4. 定量方法の区分 けい素の定量方法は,次のいずれかによる。
(1) 二酸化けい素重量法(1) この方法は,全けい素含有率0.1% (m/m) 以上8.0% (m/m) 以下の試料に適
用し,その定量方法は附属書1による。
(2) 二酸化けい素重量法(2)(ISO 439) この方法は,全けい素含有率0.10% (m/m) 以上5.0% (m/m) 以下の
試料に適用し,その定量方法は附属書2による。
2
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) モリブドけい酸青吸光光度法(1) この方法は,けい素含有率0.01% (m/m) 以上1.0% (m/m) 以下の試
料に適用し,その定量方法は附属書3による。
(4) モリブドけい酸青吸光光度法(2) (ISO 4829-1) この方法は,全けい素含有率0.05% (m/m) 以上1.0%
(m/m) 以下の試料に適用し,その定量方法は附属書4による。
(5) モリブドけい酸青吸光光度法(3) (ISO 4829-2) この方法は,全けい素含有率0.01% (m/m) 以上0.05%
(m/m) 以下の試料に適用し,その定量方法は附属書5による。
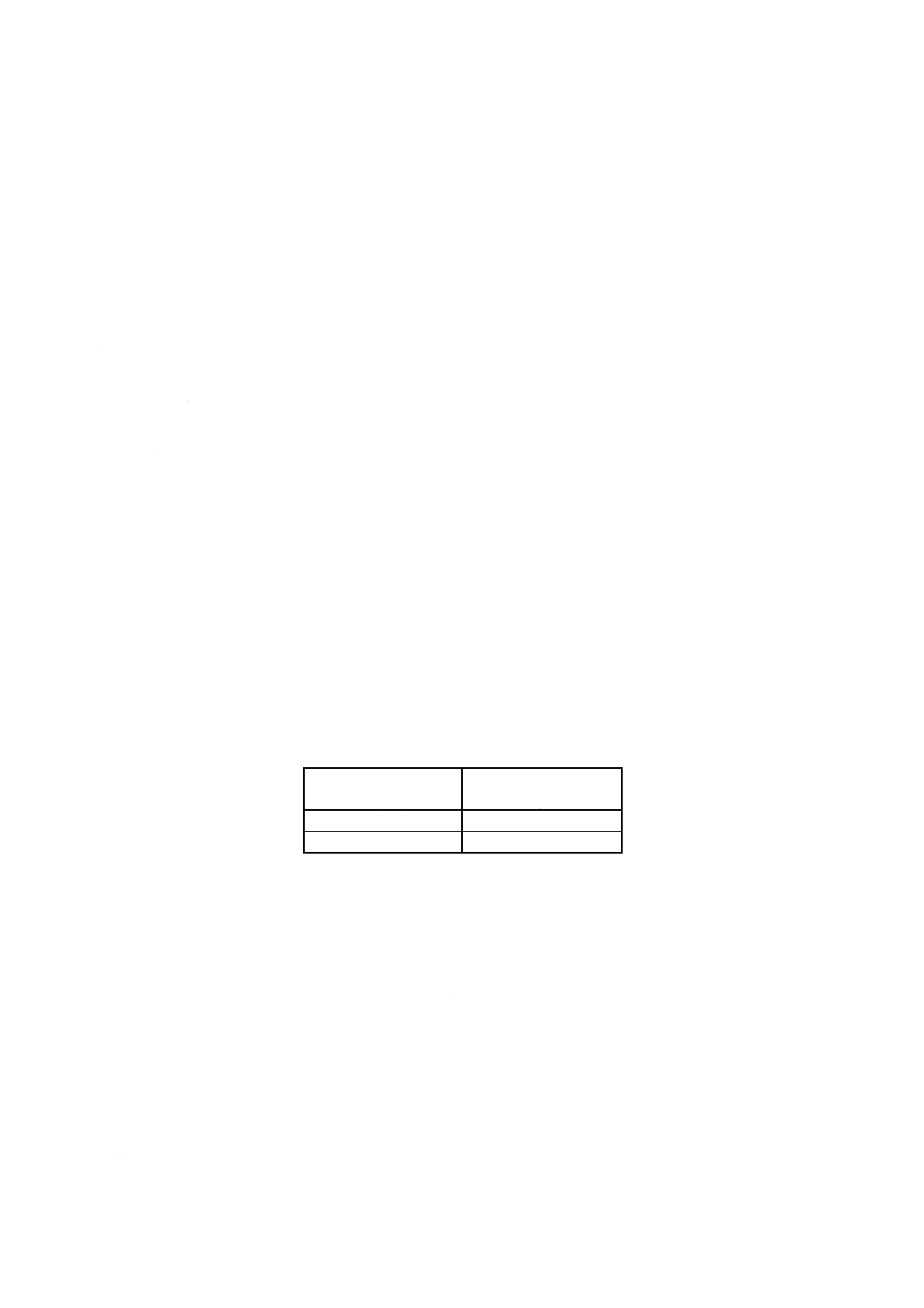
3
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1 二酸化けい素重量法(1)
1. 要旨 試料を適切な酸で分解し,過塩素酸又は硫酸を加えて加熱蒸発し,けい素を不溶性けい酸とす
る。沈殿をこし分けた後,強熱して二酸化けい素とし,その質量をはかる。さらに硫酸とふっ化水素酸を
加え,加熱して二酸化けい素を揮散させた後,その質量をはかる。
2. 試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (2+1, 1+4, 1+10)
(3) 硝酸
(4) 硝酸 (1+1)
(5) 過塩素酸
(6) ふっ化水素酸
(7) 硫酸
(8) 硫酸 (1+1, 1+3)
(9) 王水(塩酸3,硝酸1)
(10) 過酸化水素
(11) 炭酸ナトリウム
(12) チオシアン酸アンモニウム溶液(飽和)
3. 試料はかり採り量 試料はかり採り量は,附属書1表1による。
附属書1表1 試料はかり採り量
けい素含有率
% (m/m)
試料はかり採り量
g
0.10以上1.0未満
3.0
1.0以上8.0以下
1.0
4. 操作
参考
警告
過塩素酸の蒸気は,アンモニア,亜硝酸蒸気又は有機物が存在すると爆発する危険
がある。過塩素酸の蒸発処理は,過塩素酸を使用しても安全な排気設備を備えた場
所で行わなければならない。
4.1
試料の分解及びけい酸の脱水処理 試料の分解及びけい酸の脱水処理は,次のいずれかの手順によ
って行う。
(1) 過塩素酸と硝酸で分解容易な試料
(a) 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆う。
(b) 過塩素酸を試料はかり採り量が1.0gのときには20ml,3.0gのときには40ml加え,更に硝酸 (1+1)
を試料はかり採り量1.0gについて15〜20ml加え,穏やかに加熱して分解する。
(c) 引き続き強く加熱し,蒸発した過塩素酸の蒸気がビーカーの内壁を伝わって逆流する状態になった
4
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
後,15〜20分間加熱を続ける。
(2) 過塩素酸と硝酸で分解困難な試料又はニオブ若しくはタンタルを含む試料
(a) 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆う。
(b) 王水20〜30mlを加え,穏やかに加熱して分解する。
(c) 過塩素酸を試料はかり採り量が1.0gのときには20ml,3.0gのときには40ml加える。
(d) (1)(c)の操作を行う。
(3) クロム又はタングステンを含む試料
(a) 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆う。
(b) 塩酸 (2+1) を試料はかり採り量が1.0gのときには20ml,3.0gのときには40ml少量ずつ加え,穏
やかに加熱して分解する。
(c) 過塩素酸を試料はかり採り量が1.0gのときには20ml,3.0gのときには40ml加える。
(d) 引き続き強く加熱してクロムが酸化されて赤いニクロム酸となり,過塩素酸の蒸気がビーカーの内
壁を伝わって逆流する状態になった後,15〜20分間加熱を続ける。
(4) ニッケル,クロム,タングステン,コバルトなどを多量に含む試料
(a) 試料(1)をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆う。
(b) 水50ml及び塩酸10mlを加え,更に過酸化水素30mlを少量ずつ加え,激しい反応が収まった後,
穏やかに加熱して分解する。
(c) 過塩素酸20mlを加える。
(d) (3)(d)の操作を行う。
注(1) 試料がニッケル,クロム,タングステン,コバルトなどを多量に含有する場合には,試料はか
り採り量はけい素含有率にかかわらず1.0gとする。
(5) モリブデン又はチタンを含む試料
(a) 試料(1)をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆う。
(b) 硫酸 (1+3) を試料はかり採り量が1.0gのときには40ml,3.0gのときには60ml加え,加熱して分
解する。
(c) 硝酸を試料はかり採り量1.0gについて5ml加え,煮沸する。
(d) 塩酸5〜10mlを加え,時計皿の下面を水で洗って時計皿を取り除き,引き続き砂浴上で加熱して濃
厚な硫酸白煙を15〜20分間発生させる。
(6) 硝酸と塩酸で分解容易な試料でけい酸の脱水に硫酸を用いる場合
(a) 試料(1)をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆う。
(b) 硝酸 (1+1) を試料はかり採り量1.0gについて15〜20ml少量ずつ加え,穏やかに加熱して分解する。
(c) 塩酸5〜10mlを加える。
(d) 硫酸 (1+1) を試料はかり採り量が1.0gのときには20ml,3.0gのときには30ml加える。
(e) 時計皿の下面を水で洗って時計皿を取り除き,引き続き砂浴上で加熱して濃厚な硫酸白煙を15〜20
分間発生させる。
4.2
ろ過洗浄 ろ過洗浄は,次のいずれかの手順によって行う。
(1) 過塩素酸で脱水処理をした場合
(a) 4.1の(1)(c),(2)(d),(3)(d)又は(4)(d)で得た塩類を室温近くまで放冷する。
(b) 時計皿の下面を水で洗って時計皿を取り除き,温水約120mlを加え,かき混ぜて塩類を溶解する。
(c) 直ちに,ろ紙(5種B)で沈殿をこし分け,ビーカーの内壁に付着したけい酸を,ゴム付きガラス
5
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
棒でこすり落としてろ紙上に移す。
(d) 始めは温水と40〜60℃に加熱した塩酸 (1+10) とで交互に約5回ずつ,最後に温水でろ液に鉄 (III)
イオンがなくなるまで洗浄する(2)(3)(4)。沈殿は保存し,ろ液及び洗液は捨てる(5)。
注(2) 洗液の少量を取り,チオシアン酸アンモニウム溶液(飽和)を加えたとき,とう(橙)赤色を
示さなくなるまで十分に洗浄する。
(3) 洗浄が不十分であると4.3(1)(b)において灰化した際に,小爆発が起こって危険であるばかりで
なく,残さの損失を招くおそれがある。
(4) ろ過洗浄を行った沈殿中に試料の未分解残さが認められた場合には,沈殿と残さをろ紙と共に
白金るつぼ(30番)に移し入れ,乾燥し,灰化した後,約6倍量の炭酸ナトリウムを加え,加
熱して融解する。放冷した後,融成物を白金るつぼと共にビーカー (300ml) に移し入れ,温水
100mlを加えて融成物を抽出し,白金るつぼを水で洗って取り出す。時計皿で覆い,酸性とな
るまで少量ずつ塩酸を加えて塩類を溶解する。次に過塩素酸30mlを加えた後,4.1(1)(c)及び
4.2(1)の操作を行う。
(5) けい素含有率が1% (m/m) 以上の試料では,次のように操作してろ液及び洗液からけい素を回
収する必要がある。ろ液及び洗液をビーカー (500ml) に集めて時計皿で覆い,過塩素酸20ml
を加えて加熱し,蒸発した過塩素酸の蒸気がビーカーの内壁を伝わって逆流する状態になって
から更に15〜20分間加熱を続けた後,室温まで放冷する。以下,(b)〜(d)の手順に従って操作
してけい素を回収し,保存しておいた沈殿に合わせる。ろ液及び洗液は捨てる。
(2) 硫酸で脱水処理をした場合
(a) 4.1の(5)(d)又は(6)(e)で得た塩類を室温近くまで放冷する。
(b) 塩酸 (1+4) 50mlを少量ずつ加え,加熱して塩類を溶解する。
(c) (1)(c)の操作を行う。
(d) 始めは温水と40〜60℃に加熱した塩酸 (1+10) とで交互に約5回ずつ,最後に温水でろ液に鉄 (III)
イオンがなくなるまで洗浄する(2)(6)(7)。沈殿は主沈殿として保存する。
(e) ろ液及び洗液をビーカー (500ml) に集めて時計皿で覆い,加熱して蒸発させる。時計皿の下面を水
で洗って時計皿を取り除き,再び砂浴上で加熱して濃厚な硫酸白煙を15〜20分間発生させた後,放
冷する。
(f) (b)及び(c)の操作を行う。
(g) 始めは温水と40〜60℃に加熱した塩酸 (1+10) とで交互に約5回ずつ,最後に温水でろ液に鉄 (III)
イオンがなくなるまで洗浄する(2)。ろ液及び洗液は捨てる。沈殿は(d)で保存しておいた主沈殿に合
わせる。
注(6) 試料にモリブデン又はチタンが含まれる場合には,始めは40〜60℃に加熱した塩酸 (1+10) で5,
6回洗浄し,次に温水でろ液に鉄 (III) イオンがなくなるまで洗浄する。
(7) ろ過洗浄を行った沈殿中に試料の未分解残さが認められた場合には,沈殿と残さをろ紙と共に
白金るつぼ(30番)に移し入れ,乾燥し,灰化した後,約6倍量の炭酸ナトリウムを加え,加
熱して融解する。放冷した後,融成物を白金るつぼと共にビーカー (300ml) に移し入れ,温水
100mlを加えて融成物を抽出し,白金るつぼを水で洗って取り出す。時計皿で覆い,酸性とな
るまで少量ずつ塩酸を加えて塩類を溶解する。次に硫酸 (1+1) 20mlを加えた後,4.1(6)(e)及び
4.2(2)の操作を行う。
4.3
灰化及びひょう量 灰化及びひょう量は,次のいずれかの手順によって行う。
6
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) ニオブ,タンタル,チタン,ジルコニウム,タングステン及びモリブデンを含まない試料
(a) 4.2の(1)(d)又は(2)(g)で得たけい酸の沈殿を,ろ紙と共に白金るつぼ(30番)に移し入れ,乾燥した
後,低温でろ紙を灰化する。
(b) 1100℃以上で30〜45分間強熱し,デシケーター中で室温まで放冷した後,白金るつぼの質量をはか
る。恒量となるまでこの操作を繰り返す。
(2) ニオブ,タンタル,チタン,ジルコニウム,タングステン又はモリブデンを含む試料
(a) (1)の(a)及び(b)の操作を行う。
(b) 硫酸 (1+1) 約2mlを加えて加熱し,硫酸白煙が発生しなくなるまで注意して加熱を続ける。
(c) 800℃で強熱し,デシケーター中で室温まで放冷した後,白金るつぼの質量をはかる。恒量となるま
でこの操作を繰り返す。
4.4
ふっ化水素酸処理及びひょう量 ふっ化水素酸処理及びひょう量は,次のいずれかの手順によって
行う。
(1) ニオブ,タンタル,チタン,ジルコニウム,タングステン及びモリブデンを含まない試料
(a) 4.3(1)(b)で得た白金るつぼ中の二酸化けい素を硫酸 (1+1) で湿し,ふっ化水素酸約5mlを加え,注
意して加熱して二酸化けい素を揮散させる。
(b) 硫酸白煙が発生しなくなるまで加熱を続ける。
(c) 1100℃以上で強熱し,デシケーター中で室温まで放冷した後,白金るつぼの質量をはかる。恒量と
なるまでこの操作を繰り返す。
(2) ニオブ,タンタル,チタン,ジルコニウム,タングステン又はモリブデンを含む試料
(a) 4.3(2)(c)で得た白金るつぼ中の二酸化けい素に硫酸 (1+1) 約2ml及びふっ化水素酸約5mlを加え,
注意して加熱して二酸化けい素を揮散させる。
(b) 硫酸白煙が発生しなくなるまで加熱を続ける。
(c) 800℃で10分間強熱してデシケーター中で室温まで放冷した後,白金るつぼの質量をはかる。恒量
となるまでこの操作を繰り返す。
5. 空試験 試料を用いないで,試料と同じ操作を試料と併行して行う。
6. 計算 試料中のけい素含有率を,次の式によって算出する。
100
4
467
.0
)]
(
)
[(
4
3
2
1
×
×
−
−
−
=
m
m
m
m
m
Si
ここに,
Si: 試料中のけい素含有率 [% (m/m)]
m1: 試料において4.3の(1)(b)又は(2)(c)で得た白金るつぼの質量
(g)
m2: 試料において4.4の(1)(c)又は(2)(c)で得た白金るつぼの質量
(g)
m3: 空試験において4.3の(1)(b)又は(2)(c)で得た白金るつぼの質
量 (g)
m4: 空試験において4.4の(1)(c)又は(2)(c)で得た白金るつぼの質
量 (g)
m: 試料はかり採り量 (g)
7. 許容差 許容差(8)は,附属書1表2による。

7
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1表2 許容差
単位 % (m/m)
けい素含有率
室内再現許容差
室間再現許容差
0.1以上8.0以下
D [0.002 7× (Si) +0.002 9]
D [0.006 7× (Si) +0.001 6]
注(8) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一分析室内における分析回数,室間再現許容差の場合は分析に関与
した分析室数である。
また, (Si) は,許容差を求めるけい素含有率 [% (m/m)] である。
参考 この許容差は,けい素含有率0.1% (m/m) 以上3.3% (m/m) 以下の試料を用い,共同実験した結
果から求めたものである。
この方法の英文名は,Method for determination of silicon content−Gravimetric method as silicon
dioxide である。
8
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2 二酸化けい素重量法(2)
附属書2としてのまえがき
この附属書2は,1994年第2版として発行されたISO 439(Steel and iron−Determination of total silicon content
−Gravimetric method) を翻訳し,技術的内容及び規格票の様式を変更することなく作成した日本工業規格
である。
なお,この規格で下線(点線)を施してある箇所は,原国際規格にはない事項である。
1. 適用範囲 この附属書2は,鉄及び鋼中に含有する全けい素を重量法によって定量する方法について
規定する。
この方法は,けい素含有率0.10% (m/m) 以上5.0% (m/m) 以下の試料に適用する(1)。
注(1) モリブデン,ニオブ,タンタル,チタン,タングステン,ジルコニウム又はクロムを含む鋼で
は,普通鋼の場合より精確さの劣る結果が得られる。
2. 引用規格 次に記載する規格は,この国際規格の本文中で引用するので,国際規格の規定の一部を構
成する。この規格の発行の時点では,それぞれ規格の発行版表示は正しいものであるが,国際規格はすべ
て改訂されるものであるので,この国際規格を使用することに合意した当事者は,常に最新版の規格を参
照するように努力されたい。IEC及びISOのメンバーには最新の国際規格のリストが配布されている。
ISO 377-2 : 1989 Selection and preparation of samples and test pieces of wrought steels−Part 2 : Samples
for the determination of the chemical composition
ISO 385-1 : 1984 Laboratory glassware−Burettes−Part 1 : General requirements
ISO 648 : 1977 Laboratory glassware−One-mark pipettes
ISO 1042 : 1983 Laboratory glassware−One-mark volumetric flasks
ISO 3696 : 1987 Water for analytical laboratory use−Specification and test methods
ISO 5725 : 1986 Precision of the test methods−Determination of repeatability and reproducibility for a
standard test method by inter-laboratory tests
参考 ISO 377-2は,次の規格で置き換えられている。
ISO 14284 : 1996 Steel and iron−Sampling and preparation of samples for the determination of
chemical composition
3. 原理 試料を塩酸と硝酸で分解する。過塩素酸白煙処理をして酸可溶性けい素化合物を含水二酸化け
い素とする。含水二酸化けい素及び酸不溶性けい素化合物をこし分け,強熱して不純二酸化けい素として
ひょう量する。強熱残さをふっ化水素酸と硫酸で処理し,強熱してひょう量する。
4. 試薬 分析の際は,特に記述しない限り,分析用保証試薬及びISO 3696の第2級の水を使用する。
4.1
塩酸 (密度 約1.19g/ml)
4.2
塩酸 (密度 約1.19g/ml)の希釈液1+1
4.3
塩酸 (密度 約1.19g/ml)の希釈液1+19
4.4
硝酸 (密度 約1.42g/ml)の希釈液3+1

9
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.5
ふっ化水素酸 (密度 約1.14g/ml)
4.6
過塩素酸 (密度 約1.67g/ml)(2)
4.7
硫酸 (密度 約1.83g/ml)の希釈液1+1
注(2) 過塩素酸は,密度約1.54g/mlのものも使用できる。
5. 装置 体積計用ガラス器具は,ISO 385-1,ISO 648又はISO 1042に従った適切なものでA級のもの
を使用しなければならない。
通常の分析室用器具のほか,次のものを用いる。
5.1
白金るつぼ 容量約30mlのもの
5.2
マッフル炉 800〜1 100℃に調節可能なもの
6. サンプリング サンプリングは,ISO 377-2か又は鉄に関する適切な国内規格に従って実施する。
7. 操作
警告
過塩素酸の蒸気は,一般にアンモニア,亜硝酸蒸気又は有機物の存在で爆発する危険がある。
過塩素酸の蒸発処理は,過塩素酸を使用しても安全な排気設備を備えた場所で行わなければな
らない。
7.1
試料はかり採り量 試料には,最大厚さ約0.2mmに平削又はドリルで切削したものを使用する。
試料は,けい素の予想含有率に従って,次に示す質量 (m0) を0.001gのけたまではかり採る。
a) けい素含有率が,0.10% (m/m) 以上0.50% (m/m) 未満の場合のはかり採り量 (m0) は,約5g
b) けい素含有率が,0.50% (m/m) 以上2.5% (m/m) 未満の場合のはかり採り量 (m0) は,約2.5g
c) けい素含有率が,2.5% (m/m) 以上5.0% (m/m) 以下の場合のはかり採り量 (m0) は,約1g
7.2
空試験 空試験は,試料の分析と併行して同一操作に従って全試薬の同量を使用して行う。
7.3
定量
7.3.1
試料の分解と含水二酸化けい素の生成 はかり採った試料(7.1)を適切な体積の耐酸ガラス製ビー
カーに移し入れる。
塩酸(4.1)30mlを加え,ビーカーを時計皿で覆い,反応が終了するまで穏やかに加熱する。硝酸(4.4)15ml
を少量ずつ加えて酸化する。激しい反応が終了した後,時計皿を少量の熱水で洗浄し,洗液をビーカーに
入れる。過塩素酸(4.6)を附属書2表1に従って加える。
附属書2表1 過塩素酸添加量
はかり採った試料
(7.1)の質量
g
過塩素酸(4.6)の量
ml
密度=1.67g/ml
密度=1.54g/ml
5
2.5
1
60
40
25
75
50
35
警告
過塩素酸(4.6)は,少量ずつ注意深く加えなければならない。特に試料はかり採り量が5gの場
合は反応が非常に激しくなって煮沸こぼれを避けるためにも必要なことである。
参考 過塩素酸(4.6)の添加は,試料を塩酸及び硝酸で分解した後に加えているので煮沸こぼれを引き
起こす原因とはならない。少量ずつ注意深く加えなければならないのは硝酸(4.4)を加えるとき
10
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
である。
分解が完了するまで時計皿を外したままわずかに加熱し,次第に温度を上げる。過塩素酸の白煙が発生
し始めたなら,直ちにビーカーを時計皿で覆い,約20分間白煙を発生させる。放冷した後,塩酸(4.1)5ml
で注意深く湿らせ,わずかに加熱し,70〜80℃の水100mlを加えて薄め,塩類が溶解するまで再び加熱す
る(ここで沸騰させてはならない)。
7.3.2
ろ過と洗浄 ビーカーに付着した二酸化けい素をゴム付きガラス棒でこすり落とし,直ちに,使用
するろ紙と同質のろ紙パルプを少量入れた灰分が少なく,既知の中程度の粗さのろ紙(5種B)を用いて
ろ過する。
ビーカーとろ紙を80℃以上に加熱した塩酸(4.3)で洗浄し,二酸化けい素をろ紙上に移し,最初に80℃以
上に加熱した塩酸(4.2)で,次に冷水で鉄塩が完全になくなるまでよく洗浄する(3)。
注(3) 強熱の際,過塩素酸による小爆発や残さの損失を避けるため,ろ紙は十分に洗浄しておく必要
がある。
7.3.3
ろ液中のけい素化合物の回収 ろ液と洗液を分解の際使用した元のビーカーに移し入れ,過塩素酸
の濃厚な白煙が発生するまで加熱して蒸発し,ビーカーの内壁に沿って逆流する状態で約20分間保持する。
以下,7.3.1に従って塩酸(4.1)で湿らせ,水で薄める操作を行い,次に7.3.2に記述したようにして2枚目
のろ紙(5種B)を用いてろ過する。
7.3.4
強熱,二酸化けい素の揮散及びひょう量 2枚のろ紙とその内容物を合わせて白金るつぼ(5.1)に移
し入れる。ろ紙が完全に灰化するまで500〜600℃で加熱し,るつぼを白金ふたで部分的にふたをして二酸
化けい素の量に応じて30〜45分間,1 100℃のマッフル炉中で強熱する。モリブデンを含む試料では恒量
となるまで強熱する。
放冷した後,るつぼへ硫酸(4.7)約2mlを加え,注意深く加熱し,引き続き硫酸の蒸気が完全になくなる
まで加熱する。次に恒量になるまで800℃のマッフル炉中で強熱する。
デシケーター中で放冷しるつぼと内容物をひょう量する (m1)。
強熱した二酸化けい素を硫酸(4.7)数滴(4)で湿らせ,ふっ化水素酸(4.5)約5mlを加え,乾固するまで蒸発
し,引き続き硫酸が完全になくなるまで加熱する。
注(4) モリブデン,ニオブ,タンタル,チタン又はジルコニウムが存在する場合は,硫酸(4.7)2mlを
加えてそれらの元素のふっ化物が部分的に揮散することを避ける。
800℃のマッフル炉中で10分間十分に強熱した後,デシケーター中で放冷し,るつぼと内容物をひょう
量する (m2)。
8. 結果の表示
8.1
計算方法 けい素含有率 [% (m/m)] を次の式で計算する。
100
)
(
)
(
4
467
.0
0
4
3
2
1
×
−
−
−
×
=
m
m
m
m
m
Si
0
4
3
2
1
)
(
)
(
74
.
46
m
m
m
m
m
−
−
−
×
=
ここに,
Si: 試料中のけい素含有率 [% (m/m)]
m0: はかり採った試料の質量 (g)
m1: るつぼと不純二酸化けい素の質量 (g)
m2: 二酸化けい素を揮散した後のるつぼと残さの質量 (g)
m3: 空試験のるつぼと不純二酸化けい素の質量 (g)
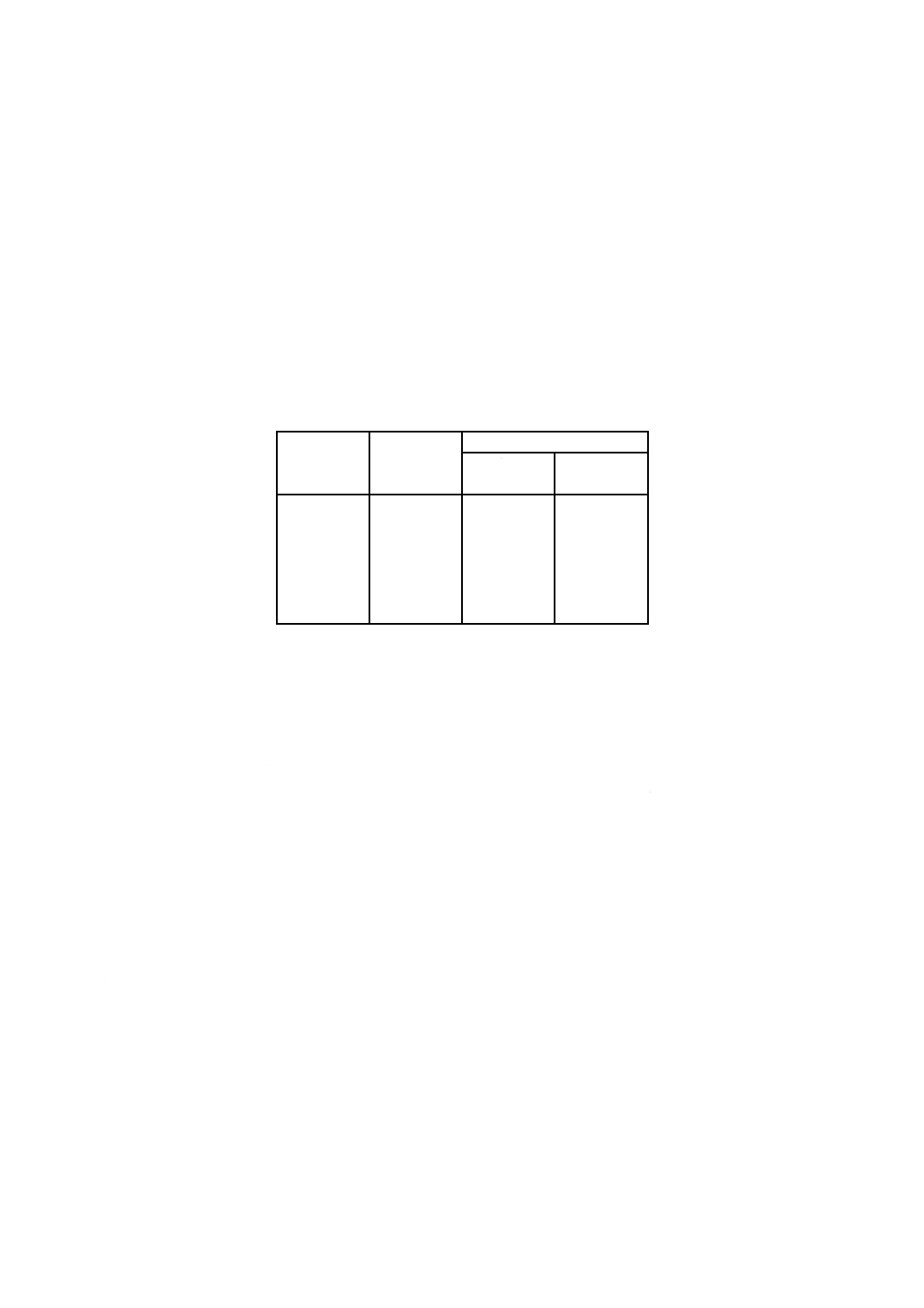
11
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m4: 空試験の二酸化けい素を揮散した後のるつぼと残さの
質量 (g)
0.467 4: Si/SiO2の係数
結果は,小数点第2位まで求める。
8.2
許容差 この方法の共同実験は,9水準の全けい素含有率試料を用いて26分析室で行い,各分析室
はそれぞれ3回ずつ(5)(6)全けい素を定量した。
実験に供した試料と得られた結果を,参考A表A.1に示した。
得られた結果は,ISO 5725に従って統計的に処理した。
得られたデータは,附属書2表2に要約したように,けい素含有率と併行許容差 (r) 及び再現許容差(R
及びRw)(7)との間に対数的比例関係があった。これを参考Bに図示した。
附属書2表2 許容差
全けい素
含有率
% (m/m)
併行許容差
r
再現許容差
室間
R
室内
Rw
0.10
0.20
0.50
1.00
2.00
5.00
0.010 5
0.013 8
0.019 8
0.025 9
0.034 0
0.048 7
0.013 9
0.019 3
0.030 0
0.041 9
0.058 5
0.090 9
0.010 5
0.014 8
0.023 3
0.032 9
0.046 5
0.073 4
注(5) 3回の定量の内の2回は,ISO 5725に規定されている併行測定条件,すなわち,一人の分析者が
同一装置,同一条件,同一検量線で最小の時間内に実験した。
参考 “同一検量線で”と記載されているが,附属書2の方法は,重量法であるので検量線を作成す
ることはない。
注(6) 3回目の定量は,注(5)と同じ分析者が同じ装置を用いて新しく作成した検量線で異なった時間
(異なった日)に実験した。
(7) 第1日に得られた結果から併行許容差 (r) 及び室間再現許容差 (R) をISO 5725に規定された
手順で計算した。第1日の最初の結果と第2日の結果から室内再現許容差 (Rw) を計算した。
9. 分析報告書 分析報告書には,次の事項を記載しなければならない。
a) 試料,分析室及び分析月日を識別させるために必要なすべての情報
b) この附属書2の引用
c) 結果及び表示した形態
d) 定量の際に気づいた非定常的なすべての特筆すべき点
e) この附属書2の中に規定されていないすべての操作,又は結果に影響を与えそうなすべての任意操作

12
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考A 国際共同実験に関する追加情報
表A.1は,8個の鋼試料及び1個の鋳鉄試料を用い,11か国,26分析室で1991年に国際共同実験
した分析結果から求めたものである。
この結果は,1992年4月に発行されたISO/TC17/SC1資料N 934に記載されている。その許容差に
関するデータは,参考Bに示した。
実験に供された試料及び得られた結果は,表A.1に示した。
表A.1
試料
全けい素含有率
% (m/m)
許容差
認証値
分析値
併行
r
室間
R
室内
Rw
wsi.1
wsi.2
BCS 460/1低合金鋼
[0.066Nb]
0.09 8
0.097 3
0.097 2
0.007 6
0.010 3
0.007 8
BCS 220/2工具鋼
[5Cr, 0.3Co, 5Mo, 2V, 7W]
0.19
0.194
0.192
0.024 9
0.032 4
0.022 6
JSS 030-6普通鋼
0.26
0.255
0.255
0.013 4
0.016 3
0.015 7
ECRM 292-1ステンレス鋼
[18Cr, 10Ni, 0.6Nb]
0.402
0.404
0.403
0.021 8
0.043 4
0.026 4
BCS 410/2低合金鋼
[1.7Cr, 2Ni, 0.4Mo, 0.4V]
1.10
1.090
1.091
0.015 0
0.024 6
0.020 6
ECRM 481-1鋳鉄
[1Ni, 0.05Mg]
2.29
2.284
2.289
0.028 0
0.052 0
0.042 0
BCS 206/3 高Si, P鋼
[3Si, 1.6P]
3.17
3.155
3.152
0.055 9
0.082 9
0.069 5
ECRM F 114-1高Si鋼
4.01)
4.012
4.014
0.068 2
0.077 6
0.083 9
CE 034 Si鋼
5.18
5.152
5.149
0.037 4
0.115
0.068 7
wsi.1 日内総平均
wsi.2 日間総平均
1) 非認証値
13
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
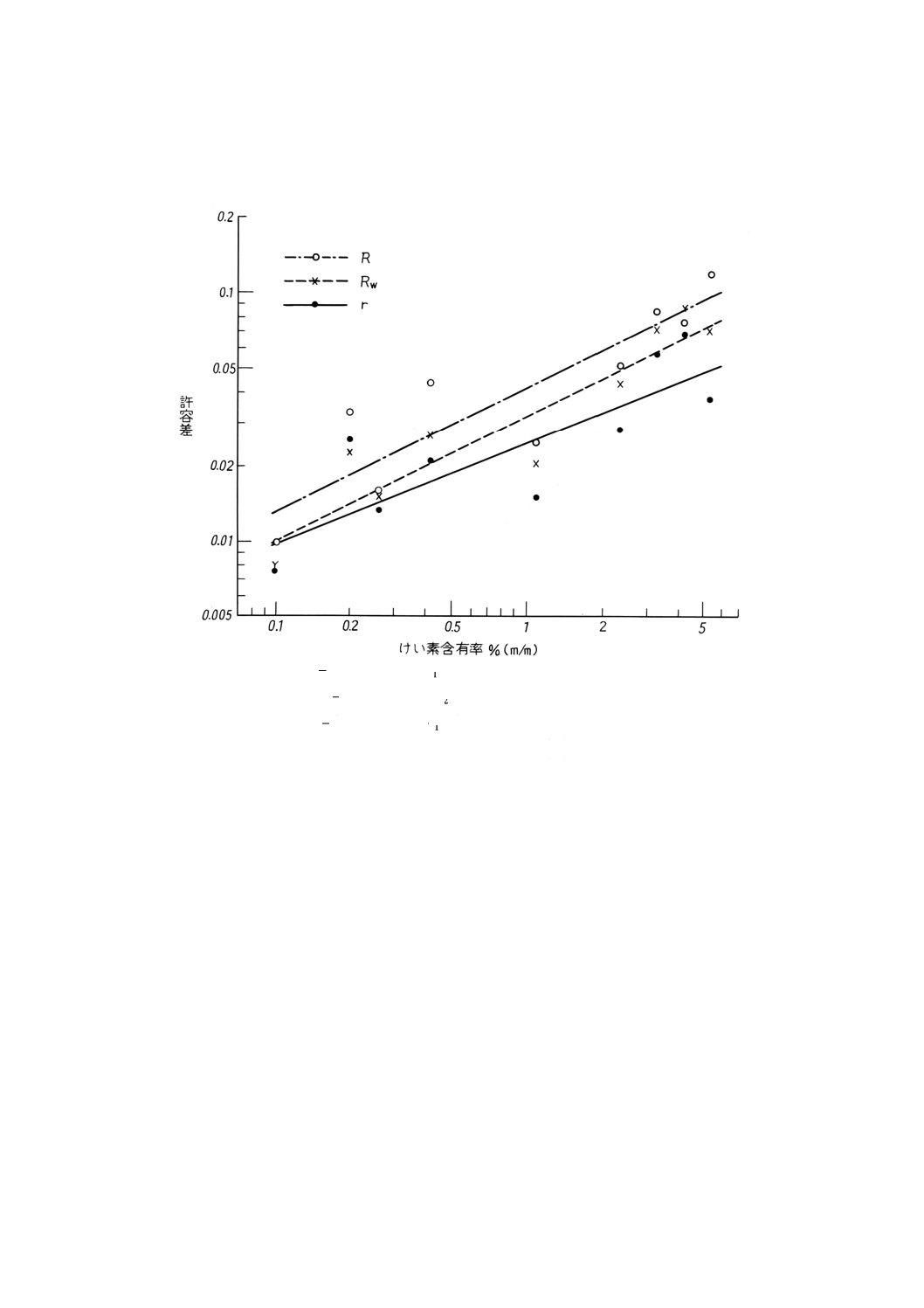
参考B 許容差データの図示
586
.1
log
3918
.0
log
1.−
=
Si
w
r
482
.1
log
4981
.0
log
2.−
=
Si
w
Rw
377
.1
log
4805
.0
log
1.−
=
Si
w
R
ここに,
wsi.1 日内総平均,けい素含有率 % (m/m)
wsi.2 日間総平均,けい素含有率 % (m/m)
図B.1 全けい素含有率 (wsi) と併行許容差 (r) 又は再現許容差(R及びRW)との対数的比例関係
14
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3 モリブドけい酸青吸光光度法(1)
1. 要旨 試料を塩酸と過酸化水素又は塩酸と硝酸との混酸で分解し,七モリブデン酸六アンモニウムを
加えてモリブドけい酸とし,しゅう酸を加えてりん,ひ素,鉄などの影響を除いた後,硫酸アンモニウム
鉄 (II) で還元してモリブドけい酸青を生成させ,光度計を用いてその吸光度を測定する。
2. 試薬 試薬は,次による(1)。
(1) 塩酸
(2) 混酸[塩酸 (1+1) 1, 硝酸 (1+1) 2] この溶液は,使用の都度調製する。
(3) 鉄 できるだけ純度の高い鉄で,けい素を含有しないか又はけい素含有率ができるだけ低く,既知で
あるもの。
(4) 過酸化水素
(5) 七モリブデン酸六アンモニウム溶液 七モリブデン酸六アンモニウム四水和物100gに温水を加えて
溶解し,室温まで冷却した後,水で液量を1 000mlとする。
この溶液は,使用の都度,ろ過して用いる。
(6) 硫酸アンモニウム鉄 (II) 溶液 硫酸アンモニウム鉄 (II) 六水和物150gに温水約500ml及び硫酸 (1
+1) 200mlを加えて溶解し,室温まで冷却した後,溶液をろ紙(5種A)を用いてろ過する。ろ紙を
水で数回洗浄し,ろ液と洗液を合わせ,水で液量を1 000mlとする。
(7) しゅう酸溶液 しゅう酸二水和物100gに温水を加えて溶解し,室温まで冷却した後,水で液量を1
000mlとする。
(8) 標準けい素容液(50μgSi/ml) あらかじめ1 000℃で1時間強熱し,デシケーター中で常温まで放冷
した二酸化けい素[99.95% (m/m) 以上]0.428 0gをはかり採って白金るつぼ(30番)に移し入れ,炭
酸ナトリウム2.5gを加えて混ぜ合わせ,加熱して融解する。放冷した後,白金るつぼを温水100ml
を入れた白金皿(150番)又はポリエチレンビーカー (200ml) 中に浸して融成物を完全に溶解し,白
金るつぼを水で洗って取り出す。常温まで冷却した後,溶液を1 000mlの全量フラスコに水を用いて
移し入れ,水で標線まで薄めて原液 (200μgSi/ml) とする。この原液は,ポリエチレン製容器に入れて
保存する。
使用の都度,この原液50.0mlをビーカー (200ml) に取り,塩酸で中和(pH7付近)した後,200ml
の全量フラスコに水を用いて移し入れ,水で標線まで薄めて標準けい素溶液とする。
注(1) この方法では,使用する試薬中に含まれるけい素が誤差の原因となるので,できるだけ純度が
高く,けい素含有率が低い試薬を使用する。
また,調製した試薬溶液は,ポリエチレン製容器に保存する必要がある。
3. 試料はかり採り量 試料はかり採り量は,附属書3表1による。
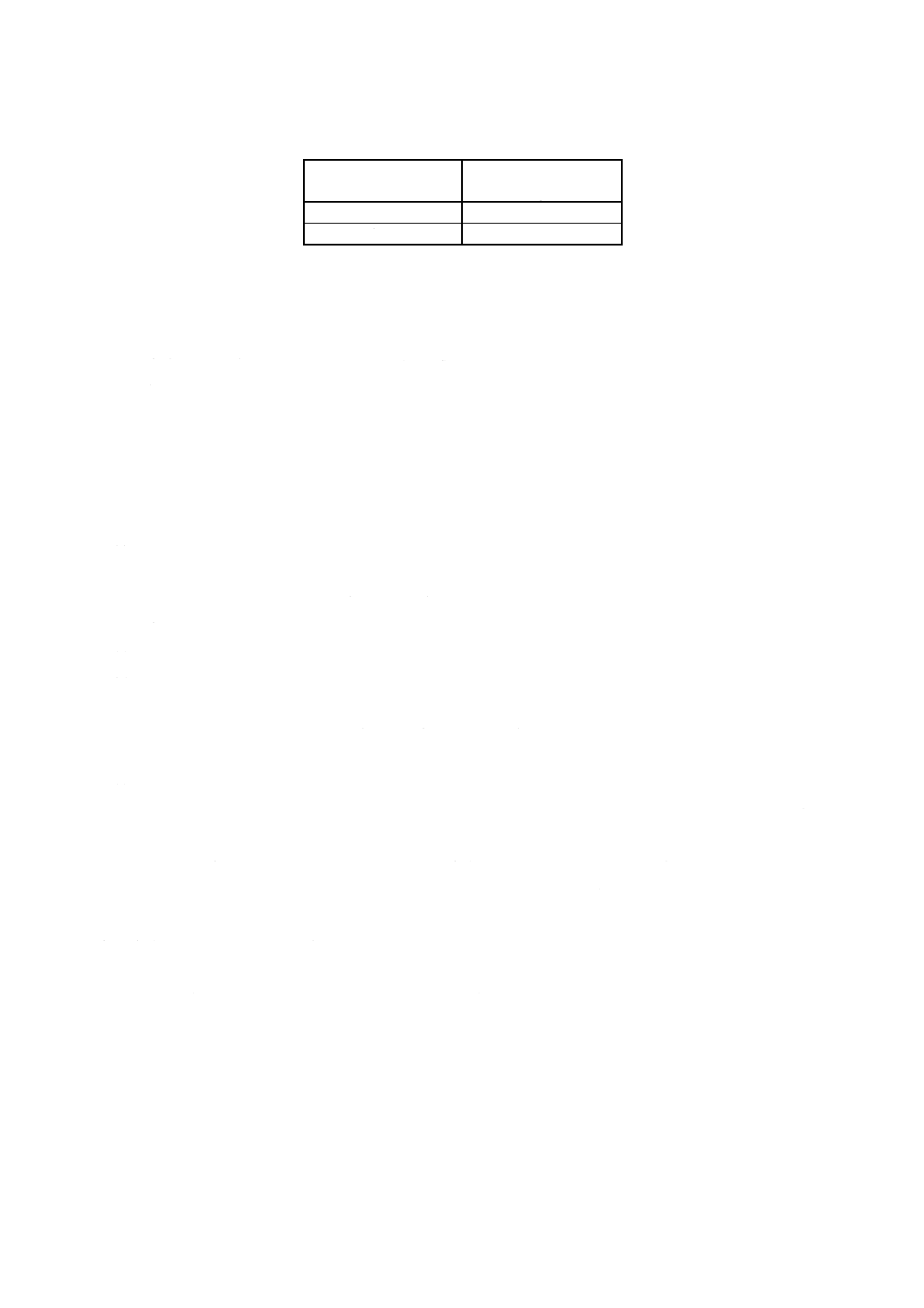
15
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3表1 試料はかり採り量
けい素含有率
% (m/m)
試料はかり採り量
g
0.01以上0.10 未満
0.50
0.10以上1.0 以下
0.10
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次のいずれかの手順によって行う。
(1) 塩酸と過酸化水素で分解容易な試料
(a) 試料をはかり採ってトールビーカー (200ml) に移し入れ,時計皿で覆う。
(b) 塩酸を試料はかり採り量が0.50gのときには20ml,0.10gのときには15ml加え,過酸化水素10ml
を少量ずつ注意しながら加え,激しい反応がおさまった後,穏やかに加熱して分解する。
(c) 温水20mlを加え,沸騰が始まるまで加熱した後,引き続き沸騰しない程度に約3分間加熱して過
剰の過酸化水素を完全に分解する。わずかに冷却した後,時計皿の下面を温水で洗浄して時計皿を
取り除く。
(d) 溶液(2)(3)をろ紙(5種B)を用いてろ過し,少量の温水で洗浄し,ろ液と洗液を合わせる。
(e) 常温まで冷却した後,溶液を100mlの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
注(2) 溶液中に炭化物又は残さが認められない場合には,この(d)の操作を省略してもよい。
(3) 試料中にタングステンが含まれる場合は,この(d)の操作は行わない。
(2) 混酸に分解容易な試料
(a) 試料をはかり採ってビーカー (300ml) に移し入れ,時計皿で覆う。
(b) 混酸 [2.(2)] を試料はかり採り量が0.50gのときには25ml, 0.10gのときには15ml加え,穏やかに加
熱して分解する。
(c) 温水20mlを加えて加熱し,沸騰させて窒素酸化物を追い出す。
(d) (1)(d)の操作を行う。
(e) (1)(e)の操作を行う。
4.2
呈色 4.1の(1)(e)又は(2)(e)で得た溶液(4)を,試料中の予想けい素含有率が0.1% (m/m) 未満の場合に
は20ml,0.1% (m/m) 以上の場合には10ml分取して100mlの全量フラスコに移し入れる。七モリブデン酸
六アンモニウム溶液 [2.(5)] を試料中の予想けい素含有率が0.1% (m/m) 未満の場合には15ml,0.1% (m/m)
以上の場合には10ml加えて振り混ぜ,10分間放置した後,しゅう酸溶液 [2.(7)] を試料中の予想けい素含
有率が0.1% (m/m) 未満の場合には25ml,0.1% (m/m) 以上の場合には15ml加えて振り混ぜる。30秒間以
内に硫酸アンモニウム鉄 (II) 溶液 [2.(6)] 5mlを加え,振り混ぜた後,水で標線まで薄める(5)。
注(4) 注(3)を適用した場合には,溶液を乾いたろ紙(5種C)でろ過し,そのろ液を用いる。
(5) 呈色溶液は,液温が20〜30℃のときは,呈色した後,60分間は安定であるが,液温が15℃以
下又は35℃以上のときは,定量値が低値を示す原因となるので,操作中の液温に注意する必要
がある。
4.3
吸光度の測定 4.2で得た呈色溶液を,呈色1分間後にその一部を光度計の吸収セル (1cm) に取り,
水を対照液として波長810nm付近における吸光度を測定する(6)(7)。
注(6) 波長810nm付近で測定した吸光度が,0.7を超える場合には,波長660nm付近の吸光度を測定す
ればよい。ただし,その場合には,検量線も波長660nm付近で測定したものを用いる。
16
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(7) 試料中のけい素含有率が0.1% (m/m) 未満の場合で,波長660nm付近の吸光度を測定するとき
は,ニッケル20% (m/m) ,クロム10% (m/m) ,コバルト25% (m/m) 又はバナジウム2.4% (m/m)
以上を含有する試料では,その有色イオンの影響が無視できないので,次のように操作して補
正する。
試料溶液及び空試験液の調製において4.1の(1)(e)又は(2)(e)で得た溶液のそれぞれをもう1個
20mlずつ分取し,別の100ml全量フラスコに移し入れる。次に4.2の呈色操作を七モリブデン
酸六アンモニウム溶液 [2.(5)] の添加だけを省略して行い,得た溶液の吸光度を呈色溶液及び空
試験液と同じ条件で測定し,その値をそれぞれ試料溶液及び空試験液の吸光度から差し引く。
5. 空試験 試料の代わりに試料と同量の鉄 [2.(3)] をはかり採り,4.1〜4.3の手順に従って試料と同じ操
作を試料と併行して行う。
6. 検量線の作成 附属書3表2のけい素含有率の範囲ごとに数個のトールビーカー (200ml) を準備し,
それぞれに附属書3表2に従ってはかり採った鉄 [2.(3)] を移し入れ,時計皿で覆い,4.1(1)の(b)及び(c)
又は4.1(2)の(b)及び(c)の手順に従って操作した後,附属書3表2に従って標準けい素溶液 [2.(8)] を段階
的に添加する。以下,4.1(1)(e)〜4.3又は4.1(2)(e)〜4.3の手順に従って試料と同じ操作を試料と併行して行
い(8),得た吸光度と標準けい素溶液として加えたけい素量との関係線を作成し,その関係線を原点を通る
ように平行移動して検量線とする。
注(8) 注(4)及び注(7)は適用しない。
附属書3表2 検量線溶液の調製
けい素含有率
% (m/m)
鉄 [2.(3)] はかり採り量
g
標準けい素溶液 [2.(8)] 添加量
ml
0.01以上0.10未満
0.500
0, 2, 4, 6, 8, 10
0.10以上1.0以下
0.100
0, 4, 8, 12, 16, 20
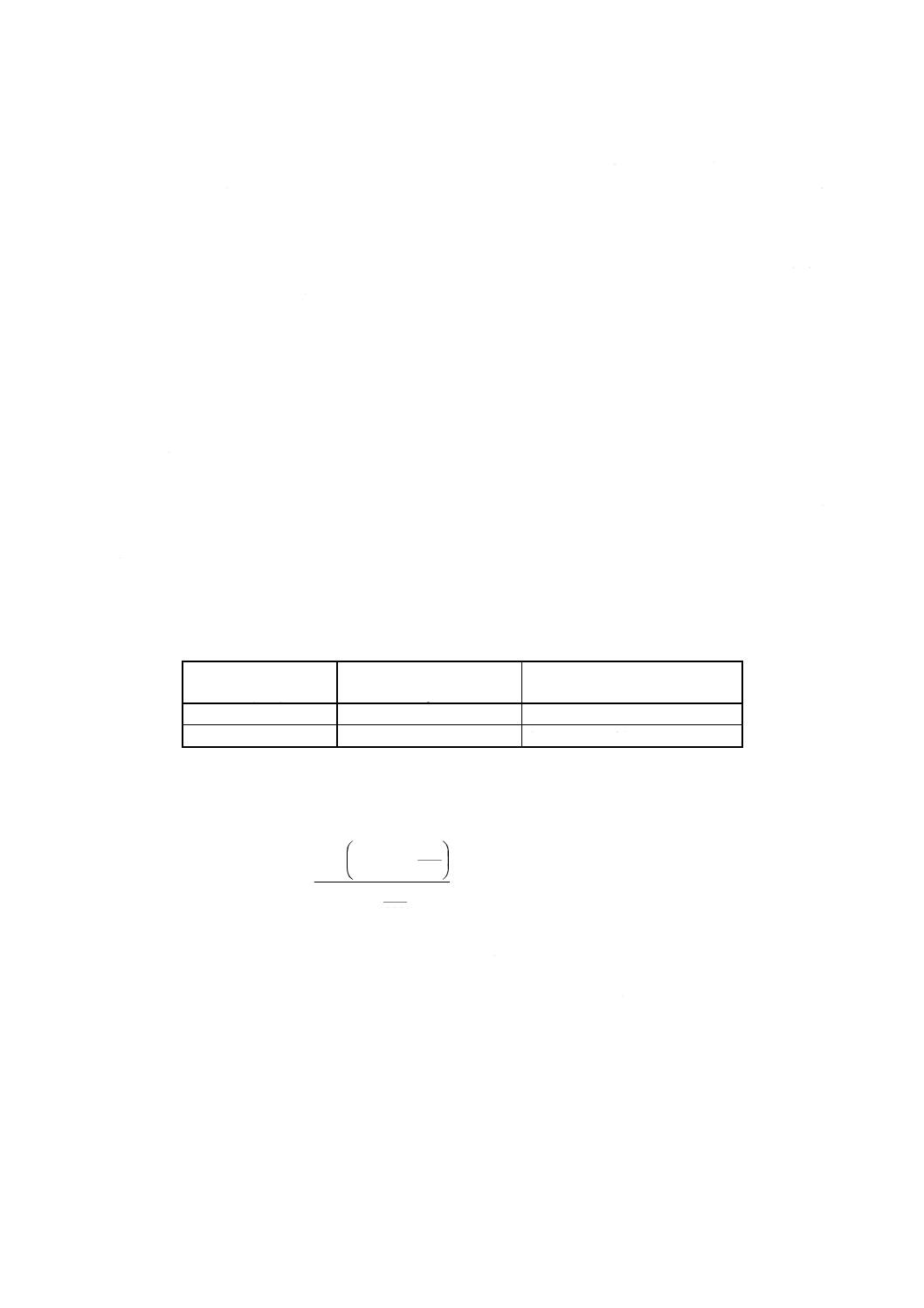
7. 計算 4.3及び5.で得た吸光度と6.で作成した検量線とからけい素量を求め,試料中のけい素含有率を,
次の式によって算出する。
100
100
100
3
2
1
×
×
×
−
−
=
B
m
B
A
A
A
Si
ここに, Si: けい素含有率 % (m/m)
A1: 分取した試料溶液中のけい素検出量 (g)
A2: 分取した空試験液中のけい素検出量 (g)
A3: 5.ではかり採った鉄 [2.(3)] 中に含まれるけい素の量 (g)
m: 試料はかり採り量 (g)
B: 試料溶液及び空試験液の分取量 (ml)
8. 許容差 許容差(9)は,附属書3表3による。

17
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3表3 許容差
単位 % (m/m)
けい素含有率
室内再現許容差
室間再現許容差
0.01以上0.10 未満
D [0.0094× (Si) +0.000 8]
D [0.0306× (Si) +0.000 9]
0.10以上1.0 以下
D [0.0052× (Si) +0.001 8]
D [0.0076× (Si) +0.004 4]
注(9) 許容差計算式中のDは,D (n, 0.95) を意味し,その値は,JIS Z 8402の表4による。nの値は,
室内再現許容差の場合は同一分析室内における分析回数,室間再現許容差の場合は分析に関与
した分析室数である。
また, (Si) は,許容差を求めるけい素含有率 [% (m/m) ] である。
参考 この許容差は,けい素含有率0.01% (m/m) 以上1.0% (m/m) 以下の試料を用い,共同実験した
結果から求めたものである。
この方法の英文名は,Method for determination of silicon content−Reduced molybdosilicate
spectrophotometric methodである。
18
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4 モリブドけい酸青吸光光度法(2)
附属書4としてのまえがき
この附属書4は,1986年第1版として発行されたISO 4829-1 (Steel and cast iron−Determination of total silicon
content−Reduced molybdosilicate spectrophotometric method−Part 1 : Silicon contents between 0.05 and 1.0%)
を翻訳し,技術的内容及び規格票の様式を変更することなく作成した日本工業規格である。
なお,この規格で下線(点線)を施してある箇所は,原国際規格にはない事項である。
1. 目的及び適用範囲 この附属書4は,鋼及び鋳鉄中に含有する全けい素をモリブドけい酸青吸光光度
法によって定量する方法について規定する。
この方法は,規定する酸に可溶な鋼及び鋳鉄中に含有するけい素(参考参照)で0.05% (m/m) 以上1.0%
(m/m) 以下の試料に適用する。
参考 この方法は,酸分解後に不溶解残さの処理を行い,主液に合わせているので,表題どおり“全
けい素の定量方法”であるのが正しい。
2. 引用規格 ISO/R 377 Selection and preparation of samples and test pieces of wrough steels(参考参照)
参考 ISO/R 377は,次の規格で置き換えられている。
ISO 14284 : 1996 Steel and iron−Sampling and preparation of samples for the determination of
chemical composition
3. 原理 試料を合金成分に応じて適切な混酸に溶解し,酸不溶解残さを過酸化ナトリウムで融解する。
弱酸性溶液中で酸化モリブドけい酸塩(黄色)を生成させる。硫酸濃度を増加させ,さらにりん,ひ素及
びバナジウムの妨害を除去するためにしゅう酸を加え,L (+) −アスコルビン酸でモリブドけい酸塩を青
色錯体に還元する。還元した青色錯体の吸光度を約810nmの波長で測定する。
4. 試薬 分析に際しては,特に記述しない限り,分析用保証試薬及び蒸留水又はそれと同等の純度をも
つ水を使用する。溶液は,すべて新たに調製してポリテトラフルオロエチレン(PTFEと略)製容器に保
存しなければならない。
4.1
純鉄 けい素含有量5μg/g未満のもの。
4.2
過酸化ナトリウム 粒度500μm以下のもの。
4.3
硝酸 水600ml中に硝酸(密度約1.40g/ml)150mlを少量ずつ加える。放冷した後,水で液量を1 000ml
とする。
4.4
硫酸 水600mlを振り混ぜながら,硫酸(密度約1.84g/ml)250mlを少量ずつ加える。放冷した後,
水で液量を1 000mlとする。
4.5
硫酸 水800mlを振り混ぜながら,硫酸(密度約1.84g/ml)50mlを少量ずつ加える。放冷した後,
水で液量を1 000mlとする。
4.6
塩酸・硝酸の混酸 水500mlに塩酸(密度約1.19g/ml)180mlと硝酸(密度約1.40g/ml)65mlを加
え,放冷した後,水で液量を1 000mlとする。
19
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.7
硫酸・硝酸の混酸 水500mlを振り混ぜながら,硫酸(密度約1.84g/ml)35mlを少量ずつ加え,さ
らに硝酸(密度約1.40g/ml)45mlを加える。放冷した後,水で液量を1 000mlとする。
4.8
L (+) −アスコルビン酸溶液 (20g/l) この溶液は,使用の直前に調製する(参考参照)。
参考 原文には,“アスコルビン酸”と記載されているが.IUPAC命名法に従って修正した。
4.9
しゅう酸溶液 しゅう酸二水和物 (C2H2O4・2H2O) 5gを水に溶解し,水で液量を100mlとする。
4.10 過酸化水素 過酸化水素 (300g/l) 200mlを水で液量を1000mlとする。
4.11 過マンガン酸カリウム溶液 (22.5g/l) 使用する前にろ過する。
4.12 モリブデン酸ナトリウム溶液 モリブデン酸ナトリウム二水和物 (Na2MoO4・2H2O) 2.5gを水50ml
に溶解し,中程度の粗さのろ紙(5種B)でろ過する。使用直前に,硫酸(4.5)15mlを加え,水で液量を100ml
とする。
4.13 標準けい素溶液
4.13.1 標準けい素溶液原液 (1gSi/l) 高純度の二酸化けい素(99.9%SiO2以上)(1)2.139 3gを0.1mgのけ
たまではかり採って白金るつぼへ移し入れる。炭酸ナトリウム16gを加えてよく混合し,1 050℃で30分
間融解する。融成物をポリプロピレン又はPTFE製のビーカーに入れて水100mlに溶解する(2)。溶け残り
がないように溶解し,抽出液を1 000mlの全量フラスコに移し入れ,水で標線まで薄める。直ちに栓付き
PTFE製瓶に保存する。この原液1mlは,けい素1mgを含有する。
4.13.2 標準けい素溶液 (200mgSi/l) 標準けい素溶液原液(4.13.1)50mlを分取して250mlの全量フラスコ
に移し入れる。水で標線まで薄める。直ちに栓付きPTFE製瓶に保存する。この標準溶液1mlは,けい素
200μgを含有する。
注(1) 高純度二酸化けい素は,使用直前に1 100℃で1時間焼成し,デシケーター中で放冷しておかな
ければならない。
(2) 融成物の抽出は,徐々に加熱しながら長時間かけて水で溶解する必要がある。
5. 装置 通常の分析室器具と次のものを使用する。
5.1
ビーカー類 容量250mlのポリプロピレン製又はPTFE製のもの
5.2
るつぼ類 容量50mlのジルコニウム製のもの(3)
注(3) ジルコニウムるつぼの代わりにアルミナるつぼも使用できる。
5.3
分光光度計 分光光度計は,波長810nmにおいて分光バンド幅が10nm以下で吸光度を測定できる
装置でなければならない。測定波長は,803nmにおけるジジミウム (didy-mium) フィルターの最大吸収で
測定するか又は他の適当な検量方法で測定して±2nmの正確さで測定できなければならない。最大吸収の
溶液に対する吸光度測定は,相対偏差 (relative deviation) で表現した併行精度 (repeatability) で±0.3%より
よくなければならない。
6. サンプリング サンプリングは,ISO/R 377か又は鉄に関する適切な国内規格に従って実施する。
7. 操作
7.1
試料はかり採り量 旋盤,フライス盤又はやすり (filings) で調製した細粒のチップ試料0.50g±
0.01g (m) を0.001gのけたまではかり採る。
7.2
空試験 空試験は,試料と併行して同じ操作で,全試薬の同じ量を使用して行う。ただし,試料の
代わりに純鉄(4.1)(4)0.50g±0.01gを使用する。
20
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(4) 純鉄は,硫酸・硝酸の混酸(4.7)による溶解が非常に遅いので,硫酸(4.5)85mlで溶解した後,硝
酸(4.3)35mlを加えて溶解するのがよい。
7.3
定量
7.3.1
試料の分解 はかり採った試料(7.1)を250mlのポリプロピレン製又はPTFE製のビーカー(5.1)に移
し入れ,硫酸・硝酸の混酸(4.7)120mlを加え,穏やかに加熱して溶解する(5)(6)。
溶解反応が終了したら,灰分が低く,既知の硬質ち密ろ紙(5種C)で溶液をろ過し,ろ液を500mlの
ビーカーに集める。ビーカーを熱水20mlで洗浄し,ビーカーに付着した粒子をゴム付きガラス棒でこす
り落とし,同じろ紙で洗液をろ過する。ろ紙を毎回20mlの熱水で数回洗浄する。ろ液を保存して7.3.3の
操作で使用する(7)。
注(5) 試料の分解が遅い場合は,試料を硫酸(4.5)85mlに溶解した後,硝酸(4.3)35mlを加えて分解して
もよい。
(6) 試料が硫酸・硝酸の混酸(4.7)で分解しない場合は,その代わりに塩酸・硝酸の混酸(4.6)85mlを
使用してもよい。
(7) 酸可溶性けい素だけが必要な場合は直接7.3.3の操作に続けるが,二,三の品種の試料について
は溶解時間によって異なった結果が得られるので満足できる方法ではない。
7.3.2
不溶解残さの処理 ろ紙と残さをジルコニウムるつぼ(5.2)中に移し入れ,低温で炭素分がなくなる
まで灰化し,引き続き炉の中で600℃で強熱する。放冷して過酸化ナトリウム(4.2)0.25gを加えて残さと混
合する。さらに過酸化ナトリウム(4.2)0.25gを加えて覆い,炉の中で600℃で10分間加熱する。放冷し,
水15mlを加え,るつぼにふたをかぶせて反応が終了するまで放置する。硫酸(4.5)15mlを加え,るつぼを
動かして融成物を完全に溶解し,7.3.1で得られたろ液に合わせる。水でるつぼとふたを洗浄し,ろ液に合
わせる。
7.3.3
試料溶液の調製 7.3.1(酸可溶性のけい素だけを定量する場合)又は7.3.2の溶液を水で約300ml
に薄めて冷却する。過マンガン酸カリウム溶液(4.11)5mlを加え,さらに必要ならばその数滴を加えて少な
くとも1分間退色しない赤桃色を生じさせる。加熱して沸騰させ,2分間穏やかに沸騰を続ける。必要な
らば,過酸化水素(4.10)を二酸化マンガンの沈殿が溶解するまで滴加し,5分間穏やかに沸騰させる。常温
まで冷却した後,1 000mlの全量フラスコに移し入れ,水で標線まで薄める。
7.3.4
呈色 ピペットを用いて各試料溶液(7.3.3)(8)と空試験溶液(7.2)からそれぞれ2個ずつ20mlを分取
してそれぞれ2個の50mlのほうけい酸ガラス製全量フラスコの中へ移し入れる。それぞれの場合,一つ
は試料溶液用であり,他の一つは補償溶液用である。
各試料溶液と補償溶液の双方の液温を,15℃から25℃の範囲に調整し,次に示すように処理する。この
とき,全試薬は,ピペットを用いて添加する。
a) 試料溶液 次の順序で試薬を加える。
モリブデン酸ナトリウム溶液(4.12)10.0mlを加え,振り混ぜて20分間静置する。
硫酸(4.4)5.0mlを加えて振り混ぜる。
しゅう酸溶液(4.9)5.0mlを加えて振り混ぜる。
直ちに,L (+) −アスコルビン酸溶液(4.8)5.0mlを加えて振り混ぜる。
b) 補償溶液 次の順序で試薬を加える。
硫酸(4.4)5.0mlを加えて振り混ぜる。
しゅう酸溶液(4.9)5.0mlを加えて振り混ぜる。
モリブデン酸ナトリウム溶液(4.12)10.0mlを加え,振り混ぜて20分間静置する。
21
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
直ちに,L (+) −アスコルビン酸溶液(4.8)5.0mlを加えて振り混ぜる。
a)及びb)を,標線まで水で薄める。各溶液(試料溶液と空試験溶液)及びそれぞれの補償溶液を30分間
静置する。
注(8) ニオブ又はタンタルを含む試料溶液には,希釈したときに微細な沈殿を生じる。沈殿は沈降さ
せ,分取する前にち密なろ紙(5種C)を用いて上澄液を乾いた容器中にろ過する。最初のろ液
数mlは捨てる。
7.3.5
吸光度の測定 各溶液(7.3.4)の吸光度を,波長約810nm(注参照)で水を対照液として適切な光路
長の吸収セル(附属書4表1参照)を用いて測定する。
各溶液(試料及び空試験)の吸光度から各々に相当する補償溶液の吸光度を差し引いて補正する。
注 使用する分光光度計によって検量線シリーズの吸光度の範囲が適切であれば,810nm以外の吸光
度 (760〜860nm) も使用できる。810nmにおける特性質量吸収係数 (specific mass absorbance
coeffcient) は,780 (gSi/l) −1cm−1である。
7.4
検量線の作成
7.4.1
検量線溶液の調製 純鉄(4.1)0.50g±0.01gずつをはかり採って250mlのポリプロピレン又はPTFE
製のビーカー(5.1)の各々に移し入れ,7.3.1及び7.3.2に従って溶解する[7.2に示す注(4)参照]。
これに標準けい素溶液(4.13.2)を正確に加えて附属書4表1に示すけい素含有率の各々の範囲の検量線シ
リーズを作成する。
検量線シリーズは,さらに7.3.3から7.3.4までの操作に続けて処理する。各検量線シリーズの範囲では
補償溶液は1個でよい。
各検量線溶液と補償溶液は,30分間静置する。
7.4.2
吸光度の測定 各溶液(7.4.1)の吸光度は,約810nm(7.3.5における注参照)で水を対照液として適
切な光路長の吸収セル(附属書4表1参照)を用いて測定する。
各検量線溶液の吸光度から補償溶液の吸光度を差し引く。次に,シリーズの各検量線溶液の補正吸光度
から検量線ゼロ溶液の吸光度を差し引いて正味の吸光度を求める。
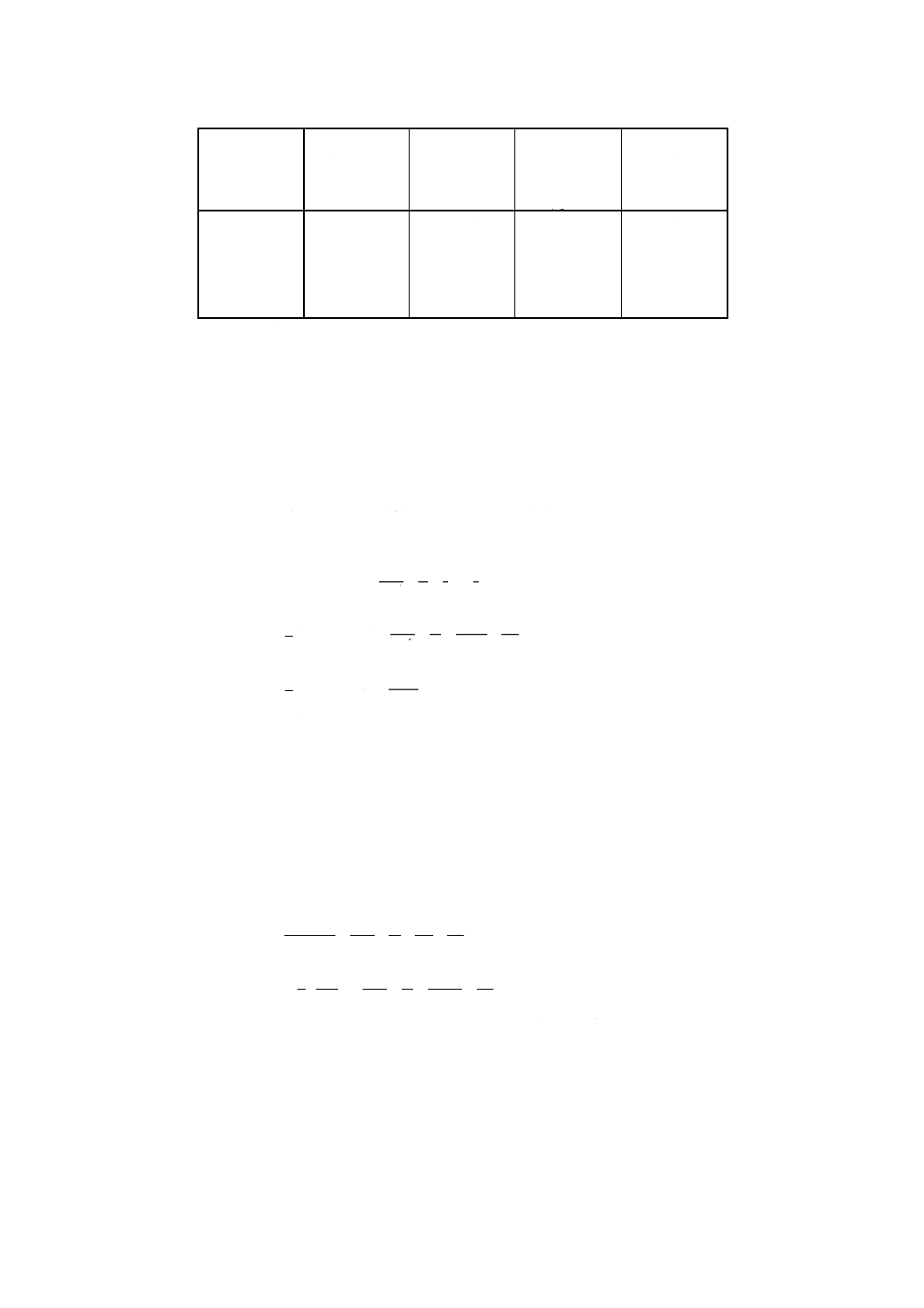
附属書4表1 検量線溶液の調製と吸収セル光路長
けい素含有
率範囲
% (m/m)
標準けい素
溶液(4.13.2)
ml
相当する
けい素量
μg
呈色後の分取
液中のけい素
濃度
μg/ml
セルの
光路長
cm
0.05〜0.10
0.00
0.50
1.00
1.50
2.00
2.50
0
100
200
300
400
500
0.00
0.04
0.06
0.12
0.16
0.20
2又は4
0.10〜0.50
0.00
2.50
5.00
7.50
10.00
12.50
0
500
1 000
1 500
2 000
2 500
0.00
0.20
0.40
0.60
0.80
1.00
2
22
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
けい素含有
率範囲
% (m/m)
標準けい素
溶液(4.13.2)
ml
相当する
けい素量
μg
呈色後の分取
液中のけい素
濃度
μg/ml
セルの
光路長
cm
0.50〜1.00
0.00
10.00
15.00
20.00
25.00
0
2 000
3 000
4 000
5 000
0.00
0.80
1.20
1.60
2.00
1
7.4.3
検量線の作成及び角度係数aの計算 測定溶液中のけい素濃度 (μg/ml) に対して正味の吸光度を
光路長1cmの吸収セルで測定した値に換算してプロットし,検量線を作成する。
検量線が直線であれば,その傾斜から角度係数aを計算する。
8. 結果の表示
8.1
計算方法
8.1.1
検量線が直線でない場合 各溶液(試料溶液及び空試験溶液)の補正吸光度(7.3.5)を,検量線(7.4.3)
を用いて相当するけい素濃度 (μg/ml) に換算し,けい素 (Si) 含有率 [% (m/m)] を,次の式によって算出
する。
100
m
V
V
V
d
1
10
1
)
(
t
1
0
6
0
Si
1
i
S
×
×
×
×
×
−ρ
ρ
100
50
20
1000
1
10
1
)
(
6
0
1
×
×
×
×
×
−
=
m
d
Si
Si
ρ
ρ
dm
Si
Si
4
1
)
(
0
1
×
−
=
ρ
ρ
ここに,
ρsi0: 空試験溶液中のけい素濃度(その補償溶液で補正) (μg/ml)
ρsi1: 試料溶液中のけい素濃度(その補償溶液で補正) (μg/ml)
d: 測定に使用したセルの光路長 (cm)
V0: 試料溶液(7.3.3)の体積 (ml)
V1: 分取液(7.3.4)の体積 (ml)
Vt: 呈色試料溶液(7.3.4)の体積 (ml)
m: 試料はかり採り量(7.1) (g)
8.1.2
検量線が直線の場合
100
1
10
1
1
0
6
0
1
×
×
×
×
×
−
m
V
V
V
d
a
A
A
t
100
50
20
000
1
1
10
1
6
0
1
×
×
×
×
×
−
=
m
d
a
A
A
ここに, A0: 空試験溶液(7.3.5)の補正吸光度
A1: 試料溶液(7.3.5)の補正吸光度
a: 角度係数又は光路長1cmで測定した溶液中のけい素μg/ml当
たりの吸光度
d: 吸光度測定に使用した吸収セルの光路長 (cm)
V0: 試料溶液(7.3.3)の体積 (ml)
V1: 分取液(7.3.4)の体積 (ml)
Vt: 呈色試料溶液(7.3.4)の体積 (ml)

23
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
m: 試料はかり採り量(7.1) (g)
8.2
許容差 この方法の国際共同実験は,10水準のけい素含有率試料を用いて18分析室で行い,各分析
室は,それぞれ4回ずつけい素を定量した。
実験に供した試料を,参考Aに示した。
得られた結果は,ISO 5725 (Precision of test methods−Determination of repeatability and reproducibility for a
standard test method by inter-laboratory tests) に従って統計的に処理した。
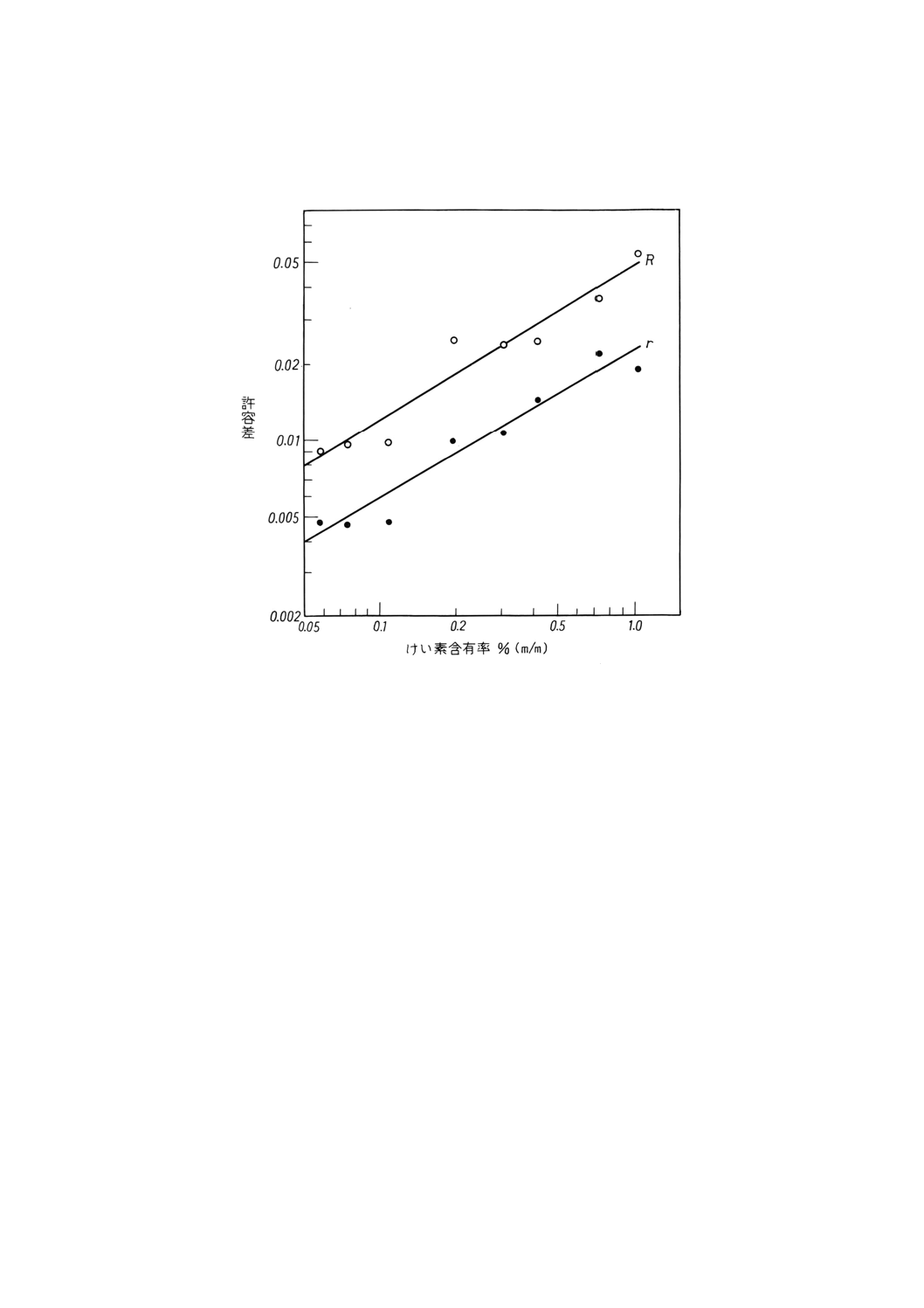
得られたデータは,附属書4表2に要約したようにけい素含有率と併行許容差 (r) 及び再現許容差 (R)
との間に対数的比例関係があった。これを参考Bに図示した。
附属書4表2 許容差
けい素含有率
% (m/m)
併行許容差
r
再現許容差
R
0.05
0.1
0.2
0.5
1.0
0.004
0.006
0.009
0.015
0.023
0.008
0.012
0.018
0.032
0.049
9. 分析報告書 分析報告書には,次の情報を記載しなければならない。
a) 試料,分析室及び分析月日を識別させるために必要なすべての情報
b) この附属書4の引用
c) 結果,及び表示した形態
d) 定量の際に気づいた非定常的なすべての特筆すべき点
e) この附属書4の中に規定されていないすべての操作,又は結果に影響を与えそうなすべての任意操作

24
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考A 国際共同実験に関する追加情報
8.2の附属書4表2の結果は,1個の鋳鉄試料及び9個の鋼試料を用い,7か国,18分析室で1983年に
国際共同実験した分析結果から求められたものである。
この結果は,1984年4月に発行されたISO/TC 17/SC 1資料に記載されている。その許容差に関するデ
ータは,参考Bに示す。
実験に供された試料は,表A.1に表示する。
表A.1
試料
けい素含有率
% (m/m)
ECRM 085-1
0.3%硫黄快削鋼
JSS 023-5
炭素鋼
BCS 452/1
1.30%Mn鋼
ECRM 020-1
炭素鋼
ECRM 081-1
炭素鋼
ECRM 254-1
7%W, 5%Mo, 2%V, 5%Cr鋼
ECRM 077-2
1.25%Mn鋼
ECRM 277-1
18%Cr, 10%Ni鋼
ECRM 484-1
可鍛白鋳鉄
ECRM 276-1
5%Cr, 1.5%Mo, 0.5%V鋼
0.008
0.024
0.055
0.072
0.105
0.19
0.293
0.417
0.717
0.985
備考1. 統計的処理は,ISO 5725に従った。
2. 共同実験には,10試料を使用した。しかし,この方法のけい素含有率適用範囲が0.05% (m/m)
以上1.0% (m/m) 以下であったので,試料ECRM 085-1及びJSS 023-5のデータを削除した8
点だけを参考Bに図示した。
25
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考B 許容差データの図示
図B.1 けい素含有率と併行許容差 (r) ,又は再現許容差 (R) との関係
26
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書5 モリブドけい酸青吸光光度法(3)
附属書5としてのまえがき
この附属書5は,1988年第1版として発行されたISO 4829-2 (Steel and iron−Determination of total silicon
content−Reduced molybdosilicate spectrophotometric method−Part2 : Silicon contents between 0.01 and 0.05%)
を翻訳し,内容及び規格票の様式を変更することなく作成した日本工業規格である。
なお,この規格で下線(点線)を施してある箇所は,原国際規格にはない事項である。
1. 目的及び適用範囲 この附属書5は,鉄及び鋼中に含有する全けい素をモリブドけい酸青吸光光度法
によって定量する方法について規定する。
この方法は,鉄及び鋼中に0.01% (m/m) 以上0.05% (m/m) 以下を含有するけい素の定量に適用する。
2. 引用規格
ISO 377 : Selection and preparation of samples and test pieces of wrought steels(参考参照)
参考 ISO 377は,次の規格で置き換えられている。
ISO 14284 : 1996 Steel and iron−Sampling and preparation of samples for the determination of
chemical composition
ISO 385-1 : 1984 Laboratory glassware−Burettes−Part 1 : General requirements
ISO 648 : 1977 Laboratory glassware−One-mark pipettes
ISO 1042 : 1983 Laboratory glassware−One-mark volumetric flasks
ISO 5725 : 1986 Precision of the test methods−Determination of repeatability and reproducibility for a
standard test method by interlaboratory tests
3. 原理 試料を塩酸と硝酸の混酸に溶解し,酸不溶解残さを過酸化ナトリウムで融解する。弱酸性溶液
中で酸化モリブドけい酸塩(黄色)を生成させる。硫酸濃度を増加させ,しゅう酸を加えてりん,ひ素及
びバナジウムの妨害を除いた後,L (+) −アスコルビン酸でモリブドけい酸錯塩を選択的に青色錯体に還
元する。その吸光度を約810nmの波長で測定する。
4. 試薬 分析の際は,特に記述しない限り,分析用保証試薬を使用する。ガラス瓶に入っている試薬は,
一たん(旦)開封すると吸湿したり,ガラスと反応しやすくなる。特に,アルカリ性の試薬,例えば,炭
酸ナトリウム及び過酸化ナトリウムは,その影響を受けやすい。この原因から起こる汚染の危険を避ける
ため,試薬溶液を調製するのに使用する試薬は,すべて開封直後の新鮮なものだけを使用することをすす
める。
さらに,水も試薬溶液調製の際から操作全体を通じて蒸留した直後のものを使用しなければならない。
イオン交換水は,コロイド性のけい酸を含む可能性があるので使用してはならない。
偶発的な汚染を避けるため,水は,必要ならばその目的のために調製し,使用直前までポリプロピレン
製の容器に保存しなければならない。
全試薬は,使用直前に調製し,ポリプロピレン又はポリテトラフルオロエチレン(PTFEと略)製の容
器に保存しなければならない。
27
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.1
純鉄 けい素含有量2μg/g未満のもの
4.2
過酸化ナトリウム 粒度500μm以下のもの
4.3
硫酸 (1+3) 水600mlをかき混ぜながら,硫酸(密度約1.84g/ml)250mlを注意深く加える。放冷
した後,水で液量を1 000mlとする。
4.4
硫酸 (1+19) 水800mlをかき混ぜながら,硫酸(密度約1.84g/ml)50mlを注意深く加える。放冷
した後,水で液量を1 000mlとする。
4.5
塩酸・硝酸の混酸 水500mlに塩酸(密度約1.19g/ml)180mlと硝酸(密度約1.40g/ml)65mlを加
える。放冷した後,水で液量を1 000mlとする。
4.6
L (+) −アスコルビン酸溶液 (20g/l) この溶液は,使用の直前に調製する(参考参照)。
参考 原文には,“アスコルビン酸”と記載されているが,IUPACの命名法に従って修正した。
4.7
しゅう酸溶液 しゅう酸二水和物 (C2H2O4・2H2O) 5gを水に溶解し,水で液量を100mlとする。
4.8
過酸化水素 過酸化水素 (300g/l) 200mlを水で液量を1000mlとする。
4.9
過マンガン酸カリウム溶液 (22.5g/l) 使用する前にろ過する。
4.10 モリブデン酸ナトリウム溶液 モリブデン酸ナトリウム二水和物 (Na2MoO4・2H2O) 2.5gを水50ml
に溶解し,中程度の粗さのろ紙(5種B)でろ過する。使用直前に,硫酸(4.4)15mlを加え,水で液量を100ml
とする。
4.11 標準けい素溶液
4.11.1 標準けい素溶液原液 (1gSi/l) 焼成したばかりの高純度の二酸化けい素(99.9%SiO2以上)2.139 3g
を0.1mgのけたまではかり採って白金るつぼに移し入れる(高純度の二酸化けい素は,使用直前に1 100℃
で1時間焼成してデシケーターの中で放冷する。)。これに炭酸ナトリウム16gを加えて十分に混合し,1
050℃で30分間融解する。融成物をポリプロピレン又はPTFE製のビーカー(1)の中で水100mlに抽出する。
抽出液を1 000mlの全量フラスコに移し入れ,水で標線まで薄める。直ちに,共栓付きPTFE製の瓶に移
して保存する。
この原液1mlには,けい素1mgを含有する。
注(1) 融成物の抽出は,穏やかに加熱する必要がある。
4.11.2 標準けい素溶液 (20mgSi/l) 標準けい素溶液原液(4.11.1)10.0mlを分取して500mlの全量フラスコ
に移し入れる。水で標線まで薄める。直ちに,共栓付きPTFE製の瓶に移し入れる。この溶液は,使用直
前に調製する。
この標準溶液1mlは,けい素20μgを含有する。
5. 装置 通常の分析室器具及び次のものを使用する。
5.1
ビーカー及びふた ポリプロピレン又はPTFE製のもの
5.2
るつぼ(2) ジルコニウム製で容積50mlのもの
注(2) ジルコニウムるつぼの代わりに,ガラス質の黒鉛るつぼも使用することができる。
5.3
体積用ガラス器具 体積用ガラス器具はすべてA級のもので,ISO 385-1,ISO 648又はISO 1042
に従った適切なものを使用する。
ガラス器具を使用する際は接触時間を最小に制限し,可能な限り,ほうけい酸ガラス製のものを使用す
べきである。
28
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.4
分光光度計 分光光度計は,波長810nmにおいて分光バンド幅が10nm以下で吸光度を測定できる
装置でなければならない。測定波長は,803nmにおけるジジミウム (didy-mium) フィルターの最大吸収で
測定するか又は他の適当な検量方法で測定して±2nmの正確さで測定できなければならない。最大吸収の
溶液に対する吸光度測定は,相対偏差 (relative deviation) で表現した併行精度 (repeatability) で±0.3%より
よくなければならない。
6. サンプリング サンプリングは,ISO 377か又は鉄に関する適切な国内規格に従って実施する。
7. 操作
7.1
試料はかり採り量 旋盤,フライス盤又はやすり (filings) で調製した細粒のチップ試料0.50g±
0.01g (m) を0.001gのけたまではかり採る。
7.2
空試験 空試験は2個,試料と併行して同じ操作で全試薬の同じ量を使用して行う。ただし,試料
の代わりに純鉄(4.1)0.50g±0.01gを使用する。
空試験値は,常に低値で,しかも再現性のよい値で管理されなければならない。空試験は,分析ごとに
併行して2回行い,その平均値を計算の際に使用するのがよい。空試験値が高い場合やばらついた場合に
は,採用してはならず,次の操作を行う前に水や個々の試薬について品質をチェックして汚染の根源をつ
きとめる必要がある。特に,過酸化ナトリウムと過マンガン酸カリウムは高い空試験値を与える試薬とし
て二,三の等級のものを注意深く選択する必要がある。空試験の読みは,吸光度として0.050[4cmの光路
長の吸収セルを使用したとき,けい素の0.008% (m/m) に相当する。]を超えないようにするのがよい。
7.3
定量
7.3.1
試料の分解 はかり採った試料(7.1)を250mlのポリプロピレン又はPTFE製のビーカー(5.1参照)
に移し入れる。塩酸・硝酸の混酸(4.5)85mlを加え,ふた(5.1参照)をかぶせ,液量が減少しないように
注意しながら穏やかに加熱して試料を分解する。
分解反応が終了したら,溶液を灰分が低く,既知の硬質でち密なろ紙(5種C)を用いてろ過し,ろ液
を500mlのビーカーに集める。ビーカーを熱水20mlで洗浄し,ビーカーに付着した粒子をゴム付きガラ
ス棒でこすり落とし,同じろ紙で洗液をろ過する。ろ紙を毎回20mlの熱水で数回洗浄する。ろ液を保存
して7.3.2の操作で使用する。
7.3.2
不溶解残さの処理 ろ紙と残さをジルコニウムるつぼ(5.2)に移し入れ,低温で炭素分がなくなるま
で灰化し,さらに炉の中で600℃で強熱する。放冷して過酸化ナトリウム(4.2)0.25gを加えて残さとよく混
合する。さらに過酸化ナトリウム(4.2)0.25gでそれらの上を覆い,炉の中に入れて600℃で10分間加熱す
る。放冷し,水15mlを加え,るつぼにふたをして反応が終わるまで放置する。硫酸(4.4)15mlを加え,ゆ
り動かして沈殿を完全に溶解し7.3.1で得られたろ液に合わせる。水でるつぼとふたを洗浄し,その洗液も
ろ液に合わせる。
7.3.3
試料溶液の調製 7.3.2で得た溶液を水で約300mlに薄めて冷却する。過マンガン酸カリウム溶液
(4.9)5mlを加え,さらに必要ならば,その数滴を加えて少なくとも1分間退色しない赤桃色を生じさせる
[試料溶液に赤桃色を生じさせるのに必要であった同じ量の過マンガン酸カリウム溶液(4.9)を空試験溶液
(7.2参照)にも添加する。]。加熱して沸騰させ,2分間穏やかに沸騰を続ける。二酸化マンガンの沈殿が
生じたならば,過酸化水素(4.8)を少量ずつ注意して滴加して沈殿を溶解する(二酸化マンガンの沈殿が生
じなくとも空試験溶液は,試料溶液と同じように正確に処理する。)。冷却した後,1 000mlの全量フラス
コに移し入れ,水で標線まで薄める。直ちに,ポリプロピレン又はPTFE製の容器に移し入れる。
29
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7.3.4
呈色 ピペットで,試料溶液(7.3.3参照)から20.0mlの分取液2個と,空試験溶液(7.2参照)か
らも同じように20mlの分取液2個を取り,別々のほうけい酸ガラス製の50ml全量フラスコに入れる。そ
れぞれの場合,1個の分取液は試料溶液用であり,他の1個は補償溶液用である。
試料中にニオブ又はタンタルを含む溶液にあっては希釈の際に細かい沈殿を生じる。このような場合は,
沈殿が沈降するまで静置して分取する前に,その上澄液を乾いたち密なろ紙(5種C)を用いて乾いた容
器の中にろ過する。最初の数mlは捨てる。
15〜25℃の温度で各試料溶液と補償溶液を以下に述べるように処理する。ただし,すべての試薬溶液は,
ピペットを用いて添加する。
a) 試料溶液 試薬を,次に述べる順序で加える。
モリブデン酸ナトリウム溶液(4.10)10.0mlを加え,振り混ぜて20分間静置する。
硫酸(4.3)5.0mlを加えて振り混ぜる。
しゅう酸溶液(4.7)5.0mlを加えて振り混ぜる。
直ちに,L (+) −アスコルビン酸溶液(4.6)5.0mlを加えて振り混ぜる。
b) 補償溶液 試薬を,次に述べる順序で加える。
硫酸(4.3)5.0mlを加えて振り混ぜる。
しゅう酸溶液(4.7)5.0mlを加えて振り混ぜる。
モリブデン酸ナトリウム溶液(4.10)10.0mlを加えて振り混ぜる。
直ちに,L (+) −アスコルビン酸溶液(4.6)5.0mlを加えて振り混ぜる。
各試薬を加えた試料溶液及び補償溶液を,水で標線まで薄める。各溶液(試料溶液と空試験溶液)
とそれに対応する補償溶液を30分間静置する。
7.3.5
吸光度の測定 7.3.4で得られた溶液の吸光度を,水を対照液として用い,光路長4cmの吸収セル
で,波長約810nm(3)で測定する。
各溶液(試料溶液及び空試験溶液)の吸光度から各々に相当する補償溶液の吸光度を差し引いて補正す
る。
注(3) 使用する分光光度計によって検量線シリーズの吸光度の範囲が適切であれば,810nm以外の吸
光度 (760〜860nm) も使用できる。810nmにおける特性質量吸収係数 (specific mass absorbance
coefficient) は,780 (gSi/l) −1cm−1である。
7.4
検量線の作成
7.4.1
検量線溶液の調製 純鉄(4.1)0.50±0.01gずつをはかり採って別々の250mlのポリプロピレン又は
PTFE製のビーカー(5.1参照)に移し入れ,7.3.1及び7.3.2に従って分解する。
これに標準けい素溶液(4.11.2)の正確な量を加え,附属書5表1に示すけい素含有率範囲のシリーズを作
成する。
30
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書5表1 検量線溶液の調製
標準けい素溶液
(4.11.2) の量
ml
相当するけい素の
質量
μg
呈色後の分取液中の
けい素の濃度
μg/ml
0.0
0
0.000
2.5
50
0.020
5.0
100
0.040
7.5
150
0.060
10.0
200
0.080
12.5
250
0.100
7.3.3に示す試料溶液の調製の処理操作に続ける。しかし,空試験の操作を省略する。
さらに7.3.4に示す操作に続けるが,その際,補償溶液の操作を省略する(4)。
注(4) 空試験溶液と補償溶液は,検量線シリーズの検量線ゼロ溶液で代用できるので,両方とも調製
の必要はない。
7.4.2
吸光度の測定 7.4.1で調製した各検量線溶液の吸光度を,水を対照液として用い,光路長4cmの
吸収セルで波長約810nm(7.3.5の注参照)で測定する。
検量線ゼロ溶液の吸光度を各検量線溶液の吸光度から差し引く。
7.4.3
検量線の作成 正味の吸光度値と測定溶液中のけい素含有量 (μg/ml) との関係線を作成する。
8. 結果の表示
8.1
計算方法 各溶液(試料溶液及び空試験溶液)の補正吸光度(7.3.5参照)を,7.4.3で作成した検量
線に挿入して相当するけい素含有量 (μg/ml) に変換する。
けい素含有率 [% (m/m)] を,次の式によって算出する。
100
10
1
)
(
1
0
6
0
1
×
×
×
×
−
m
V
V
V
t
Si
Si
ρ
ρ
100
50
20
000
1
10
1
)
(
6
0
1
×
×
×
×
−
=
m
Si
Si
ρ
ρ
m
Si
Si
4
1
)
(
0
1
ρ
ρ
−
=
ここに,
ρsi0: 空試験溶液(その補償溶液で補正した)中のけい素含
有量 (μg/ml)
ρsi1: 試料溶液(その補償溶液で補正した)中のけい素含有
量 (μg/ml)
V0: 試料溶液の体積 (ml) (7.3.3参照)
V1: 分取液の体積 (ml) (7.3.4参照)
Vt: 発色試料溶液の体積 (ml) (7.3.4参照)
m: はかり採った試料の量 (g) (7.1)
8.2
許容差 この方法の国際共同実験は,5水準のけい素含有率試料を用いて15分析室で行い,各分析
室は,それぞれ3回ずつけい素を定量した。
得られた結果は,ISO 5725に従って統計的に処理をした(5)(6)(7)。
得られたデータは,けい素含有率と併行許容差 (r) 及び再現許容差(Rw及びR)との間に系統的な関係
はなかった。代表的な値として,併行許容差 (r) は0.004% (m/m) Si, 室内再現許容差 (Rw) は0.005% (m/m)
Si, 室間再現許容差 (R) は0.006% (m/m) Siである。
31
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
実験に供した試料とその試料について得られた結果は,参考に示した。
注(5) 3回の定量の内の2回は,ISO 5725に規定されている併行測定条件,すなわち,一人の分析者が
同一装置,同一条件(同じ検量線)で最小の時間内に実験した。
(6) 3回目の定量は,注(5)と同じ分析者が同じ装置を使用して新しく作成した検量線で,異なった
時間(異なった日)に実験した。
(7) 第1日に得られた結果から併行許容差 (r) と室間再現許容差 (R) をISO 5725に規定された手
順で計算した。第1日の最初の結果と第2日の結果から,室内再現許容差 (Rw) を計算した。
9. 分析報告書 分析報告書には,次の情報を記載しなければならない。
a) 試料,分析室及び分析月日を識別させるために必要なすべての情報
b) この附属書5の引用
c) 結果及び表示した形態
d) 定量の際に気づいた非定常的なすべての特筆すべき点
e) この附属書5に規定されていないすべての操作,又は結果に影響を与えそうなすべての任意操作

32
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考 国際共同実験に関する追加情報
表1は,5個の鋼試料を用い,7か国,15分析室で1985年に国際共同実験した分析結果から求められた
ものである。この結果は,1986年3月に発行されたISO/TC 17/SC1資料N 655に記載されている。
表1
試料
けい素
% (m/m)
含有率
併行
許容差
r
室内再現
許容差
RW
室間再現
許容差
R
ECRM 085-1
(0.3%S快削鋼)
0.008
0.002 0
0.002 8
0.005 1
ECRM 285-1
(9%Co, 5%Mo, 18%Ni, 0.7%Ti鋼)
0.015
0.002 2
0.005 7
0.007 2
JSS 023-5
(炭素鋼)
0.024
0.004 5
0.005 3
0.005 2
BCS 432/1
(炭素鋼)
0.043
0.006 0
0.007 0
0.009 8
BCS 452/1
(1.3%Mn鋼)
0.053
0.003 8
0.003 2
0.003 8
原案作成委員会 構成表
(1) 社団法人日本鉄鋼連盟鋼材標準委員会JE4分科会
氏名
所属
(主査)
佐 伯 正 夫
新日本製鐵株式会社
(委員)
大 磯 義 和
工業技術院標準部
大 野 義 信
新日本製鐵株式会社
土 屋 武 久
新日本製鐵株式会社
船 曳 佳 弘
日本鋼管株式会社
磯 部 健
日本鋼管株式会社
岡 野 輝 雄
川崎製鉄株式会社
滝 沢 佳 郎
川崎製鉄株式会社
蔵 保 浩 文
住友金属工業株式会社
山 下 良 一
住友金属工業株式会社
金 子 晃 司
株式会社神戸製鋼所
河 村 恒 夫
株式会社コベルコ科研
伊 藤 清 孝
大同特殊鋼株式会社
藤 田 昇 平
日新製鋼株式会社
余 語 英 俊
愛知製鋼株式会社
永 井 宣太郎
日本冶金工業株式会社
(編集WG)
稲 本 勇
新日本製鐵株式会社
吉 川 裕 泰
日本鋼管株式会社
(幹事)
小 野 昭 紘
新日本製鐵株式会社
柿 田 和 俊
社団法人日本鉄鋼連盟(編集WG兼務)
大 槻 孝
社団法人日本鉄鋼連盟(編集WG兼務)
33
G 1212-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 社団法人日本鉄鋼連盟鉄鋼JIS三者委員会
氏名
所属
(委員会)
大河内 春 乃
科学技術庁金属材料技術研究所
(委員)
青 柳 佳 一
通商産業省基礎産業局
服 部 幹 雄
工業技術院標準部
加 山 英 男
財団法人日本規格協会
藤 貫 正
社団法人日本分析化学会
広 川 吉之助
東北大学金属材料研究所
永 山 宏
日立マテリアルエンジニアリング株式会社
束 原 巌
古河電気工業株式会社
橋 本 勝
株式会社日産アーク
岩 田 英 夫
日本鋼管株式会社
岡 野 輝 雄
川崎製鉄株式会社
蔵 保 浩 文
住友金属工業株式会社
河 村 恒 夫
株式会社コベルコ科研
成 田 正 尚
大同特殊鋼株式会社
藤 田 昇 平
日新製鋼株式会社
(幹事)
佐 伯 正 夫
新日本製鐵株式会社
(事務局)
柿 田 和 俊
社団法人日本鉄鋼連盟
大 槻 孝
社団法人日本鉄鋼連盟