2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
Z 3906-1988
パラジウムろう分析方法
Methods for Chemical Analysis of Palladium Brazing Filler Metals
1. 適用範囲 この規格は,JIS Z 3267(パラジウムろう)に規定されたパラジウム,銀,銅,マンガン,
ニッケル及び鉛の定量方法について規定する。
引用規格:
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析のための通則
JIS K 0121 原子吸光分析のための通則
JIS Z 3267 パラジウムろう
JIS Z 3900 貴金属ろうのサンプリング方法
JIS Z 8401 数値の丸め方
2. 一般事項 分析方法に共通な一般事項は,JIS K 0050(化学分析方法通則),JIS K 0115(吸光光度分
析のための通則)及びJIS K 0121(原子吸光分析のための通則)による。
3. 分析試料の採り方及び取扱い方
3.1
試料の採り方
3.1.1
試料の採り方は,JIS Z 3900(貴金属ろうのサンプリング方法)による。
3.1.2
試料は,平均品質を代表するようにし,特に偏析,汚染などに注意しなければならない。
3.2
試料のはかり方
3.2.1
試料をはかり取る際には,よくかき混ぜて平均組成を表すように注意し,また,異物が混入してい
ないことを確かめなければならない。
3.2.2
試料のはかり取りには,化学はかりを用い,0.1mgまで読み取る。
4. 分析値のまとめ方
4.1
分析回数 分析は,同一試料について原則として2回以上行わなければならない。
4.2
空試験 分析に当たっては,全操作を通じて空試験を行い,含有率を補正しなければならない。
4.3
分析値の表示 分析値は質量百分率で表し,二つ以上の分析値を平均して,JIS Z 8401(数値の丸め
方)によってパラジウム,銀,銅,マンガン及びニッケルは小数点以下1けたに,鉛は小数点以下2けた
に丸める。
5. 分析試料による分類及び分析順序 パラジウムろう分析方法は,系統分析方法によるため,便宜上パ
ラジウムろうの分析試料を4種別に分類し,その分析順序は次の表による。
2
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表 分析試料による分類及び分析順序
分析試料による分類
パラジウム
ろうの種類
分析順序
種別
成分系
(1)
Pd−Ag−Cu系
B Pd-1, 2, 4, 6
銀定量−ろ液 (A) −パラジウム定量−ろ液 (B) −銅定量
(6.2.4)
(7.2.4.1)
(8.2.4)
Pd−Ag系
B Pd-7
銀定量−ろ液 (A) −パラジウム定量
(6.2.4)
(7.2.4.1)
(2)
Pd−Ag−Mn系
B Pd-9, 10
銀定量−ろ液 (A) −パラジウム定量−ろ液 (B) −マンガン
定量
(6.2.4)
(7.2.4.1)
(9.2.3.1
又は9.3.3)
(3)
Pd−Cu−Mn−Ni系 B Pd-12
パラジウム定量−ろ液 (B) −銅定量−電解残液−マンガン
(7.2.4.2)
(8.2.4)
(9.2.3.2)
定量−試料溶液 (D) −ニッケル定量
(10.2.3.2)
(4)
Pd−Mn−Ni系
B Pd-11
パラジウム定量−ろ液 (B) −マンガン定量−試料溶液 (C)
(7.2.4.2)
(9.2.3.1)
−ニッケル定量
(10.2.3.1)
Pd−Ni系
B Pd-14
パラジウム定量−ろ液 (B) −ニッケル定量
(7.2.4.2)
(10.2.3.1)
6. 銀定量方法
6.1
定量方法 銀の定量方法は,塩化銀重量法による。この方法は,種別(1)及び(2)に適用する。
6.2
塩化銀重量法
6.2.1
要旨 試料を硝酸で分解し,アンモニア水を過剰に加えてパラジウムをアンミン錯体とした後,酢
酸を用いて弱酸性とし,塩酸を加えて塩化銀を沈殿させる。生成した塩化銀はガラスろ過器を用いてこし
分け,乾燥した後その質量を量る[ろ液及び洗液はろ液 (A) とし,パラジウムの定量に用いる。]。
6.2.2
試薬 試薬は,次による。
(1) 塩酸 (1+9)
(2) 硝酸 (1+1, 1+100)
(3) アンモニア水 (1+1)
(4) 酢酸 (1+1)
6.2.3
試料はかり取り量 試料はかり取り量は0.5gとする。
6.2.4
操作 定量操作は,次の手順によって行う。
(1) 試料の分解 試料をはかり取ってコニカルビーカー (300ml) に移し入れ,硝酸 (1+1) 20mlを加え,
時計皿で覆い加熱して分解する。
(2) 塩化銀沈殿の生成 時計皿の下面及びビーカーの内壁を水で洗い,水を加えて液量を約150mlとし,
アンモニア水 (1+1) を加えて微アルカリ性とした後,酢酸 (1+1) を弱酸性となるまで加える。溶液
をかき混ぜながら塩酸 (1+9) を少量ずつ加えて塩化銀の沈殿を生成させる。沈殿が生じなくなった
後,更にその1mlを過剰に加えて十分にかき混ぜ,時計皿の下面及びビーカーの内壁を水で洗い,加
熱して約5分間静かに煮沸する。
(3) 沈殿のこし分け及び洗浄 暗所に1夜間放置した後,あらかじめ恒量としたガラスろ過器 (1G4) を用
3
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
いてこし分け,初めは硝酸 (1+100) で,次に冷水で十分に洗浄する[ろ液及び洗液は,ろ液 (A) と
し,種別(1)及び(2)のパラジウムの定量に用いる。]。
(4) 沈殿の乾燥及びひょう量 沈殿をガラスろ過器と共に約130℃の空気浴中で約1時間乾燥し,デシケ
ーター中で約1時間放冷した後その質量を量り,恒量となるまでこの操作を繰り返す。
6.2.5
空試験 試薬だけを用いて6.2.4(1)以降の手順に従って試料と並行して操作する。ろ液及び洗液は
7.2.5の空試験に用いる。
6.2.6
計算 試料中の銀含有率は,次の式によって算出する。
(
)
00
1
6
752
.0
%
0
2
1
×
×
m
m
m
wt
−
=
銀
ここに,
m1: 6.2.4で得られた塩化銀の質量 (g)
m2: 6.2.5で得られた質量 (g)
m0: 試料はかり取り量 (g)
7. パラジウム定量方法
7.1
定量方法 パラジウムの定量方法は,ジメチルグリオキシムパラジウム重量法による。この方法は,
種別(1)及び(2)並びに種別(3)及び(4)に適用する。
7.2
ジメチルゲリオキシムパラジウム重量法
7.2.1
要旨 種別(1)及び(2)の場合は,6.2.1で得られたろ液 (A) に塩酸を加え,煮沸した後,水で薄める。
種別(3)及び(4)の場合は,試料を王水で分解し,加熱蒸発してシロップ状とし,塩酸を加えてこの操作を繰
り返し硝酸を除去する。次いで塩酸を加え,水で薄める。
このいずれかの操作によって得られた溶液にジメチルグリオキシムを加えてパラジウム錯体を沈殿させ,
ガラスろ過器を用いてこし分け,乾燥した後その質量を量る[ろ液及び洗液は,ろ液 (B) とし,銅又はマ
ンガン,ニッケルの定量に用いる。]。
7.2.2
試薬 試薬は,次による。
(1) 塩酸
(2) 塩酸 (1+1)
(3) 王水(塩酸3,硝酸1)
(4) ジメチルグリオキシム溶液 ジメチルグリオキシム1.0gをエタノール (95vol%) 100mlに溶解する。
(5) エタノール (95vol%)
7.2.3
試料はかり取り量 種別(1)及び(2)の場合はろ液 (A) の全量を用い,種別(3)及び(4)の場合は試料は
かり取り量は0.5gとする。
7.2.4
操作
7.2.4.1
種別(1)及び(2)の場合 種別(1)及び(2)の場合の定量操作は,次の手順によって行う。
(1) 塩酸処理 6.2.4(3)で得られたろ液 (A) に,塩酸10mlを加え,加熱して静かに5〜10分間煮沸した後,
水で約700mlに薄める。
(2) ジメチルグリオキシムパラジウム沈殿の生成 溶液をかき混ぜながらジメチルグリオキシム溶液
[7.2.2(4)]をパラジウム含有量0.01gにつき3.0mlの割合で加える(1)。70〜80℃に約20分間加熱した後,
温所に約2時間放置する。
(3) 沈殿のこし分け及び洗浄 沈殿をあらかじめ恒量としたガラスろ過器 (2G4) を用いてこし分け,温水
で十分に洗浄し[ろ液及び洗液は,ろ液 (B) とし,種別(1)の銅又は種別(2)のマンガンの定量に用い
4
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。],更にエタノールで1回洗浄する。
(4) 沈殿の乾燥及びひょう量 沈殿は,ガラスろ過器と共に約110℃の空気浴中で約1時間乾燥し,デシ
ケーター中で約1時間放冷した後,その質量を量り,恒量となるまでこの操作を繰り返す(2)。
注(1) ジメチルグリオキシム溶液の必要計算量は,パラジウム0.01gにつき2.2mlであるが,完全沈殿
には過剰が必要である。
(2) ジメチルグリオキシムパラジウムは,水にわずかに溶解するので,精度を上げる必要がある場
合には,既知合成試料を用いて並行試験を行い,パラジウム測定値を補正する。
7.2.4.2
種別(3)及び(4)の場合 種別(3)及び(4)の場合の定量操作は,次の手順によって行う。
(1) 試料の分解 試料をはかり取ってビーカー (200ml) に移し入れ,王水20mlを加えて時計皿で覆い加
熱して分解した後,静かに加熱蒸発してシロップ状とする。
(2) 脱硝酸 塩酸10mlを加え,加熱蒸発して再びシロップ状とする。この操作を3,4回繰り返す。
(3) 塩酸溶解 塩酸 (1+1) 35mlを加え(3),析出した塩類を溶解した後,ビーカー (1 000ml) に水を用い
て移し入れ,水で約700mlに薄める。
(4) ジメチルグリオキシムパラジウム沈殿の生成及び分離 以下7.2.4.1(2)〜(4)の手順に従って操作する。
ただし,7.2.4.1(3)の操作で得られたろ液 (B) は,種別(3)の銅又は種別(4)のマンガン,ニッケルの定
量に用いる。
注(3) 沈殿生成の酸度は,ニッケルが共存しているので塩酸0.3mol/l程度がよい。
7.2.5
空試験 試薬だけを用いて7.2.4.1(1)又は7.2.4.2(1)以降の手順に従って試料と並行して操作する。
ろ液及び洗液は8.2.5の空試験に用いる。
7.2.6
計算 試料中のパラジウム含有率は,次の式によって算出する。
(
)
100
1
316
.0
%
0
2
1
×
×
m
m
m
wt
−
=
パラジウム
ここに, m1: 7.2.4.1又は7.2.4.2で得られたジメチルグリオキシムパラジウ
ムの質量 (g)
m2: 7.2.5で得られた質量 (g)
m0: 試料はかり取り量 (g)
8. 銅定量方法
8.1
定量方法 銅の定量方法は,銅電解重量法による。この方法は,種別(1)及び(3)に適用する。
8.2
銅電解重量法
8.2.1
要旨 7.2.1で得られたろ液 (B) に硝酸を加えて濃縮した後,硫酸を加えて加熱蒸発して白煙を発
生させる。放冷後,水及び硝酸を加えて電解し,陰極に析出した銅の質量を量る(電解残液は,マンガン,
ニッケルの定量に用いる。)。
8.2.2
試薬 試薬は,次による。
(1) 硝酸
(2) 硝酸 (1+1)
(3) 硫酸 (1+1)
(4) エタノール (95vol%)
8.2.3
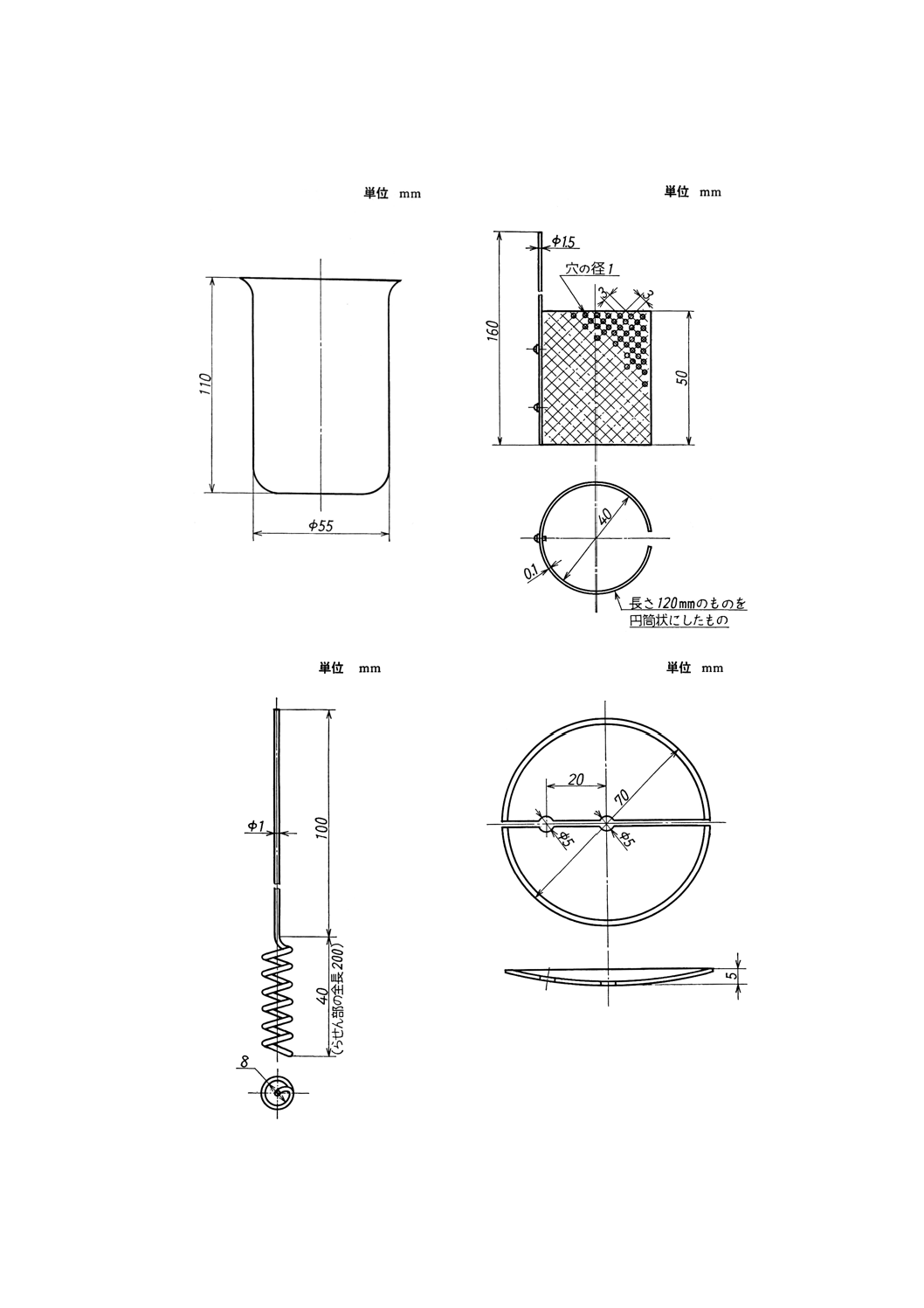
装置及び器具 装置及び器具は,原則として次のものを用いる。
(1) 電解用ビーカー(付図1参照)
5
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 白金電極A(付図2参照)
(3) 白金電極B(付図3参照)
(4) 半円形時計皿(付図4参照)
8.2.4
操作 定量操作は,次の手順によって行う。
(1) ろ液の酸処理 種別(1)の場合は7.2.4.1(3)で得られたろ液 (B) を,種別(3)の場合は7.2.4.2(4)で得られ
たろ液 (B) をそれぞれビーカー (1 000ml) に移し入れ,硝酸10mlを加えて時計皿で覆い,静かに加
熱蒸発さぜ約20mlとし,時計皿の下面とビーカーの内壁を水で洗い,電解用ビーカーに移し入れる。
硫酸 (1+1) 10mlを加え,加熱蒸発して硫酸の白煙を発生させる(4)。
(2) 硝酸溶解 放冷後,水50mlと硝酸 (1+1) 5mlを加え,加熱して可溶性塩類を溶解し,1〜2分間静か
に煮沸して酸化窒素などを追い出し,室温まで冷却する。
(3) 銅の電解分離 水を加えて液量を約150mlとし,あらかじめ質量を量った白金電極Aを陰極に,白金
電極Bを陽極に用い,2個の半円形時計皿で覆い,室温で0.3〜0.5Aの電流を通じ,5〜6時間電解す
る(5)。少量の水で時計皿の下面,ビーカーの内壁及び両極の柄の液面上に露出した部分を洗い,電解
液面を約5mm上昇させて更に約30分間電解を続ける。新しく電解液中に浸った電極Aの柄に銅が析
出しなくなったならば電流を通じたまま水洗いしながら両極を徐々に引き上げる(6)[種別(3)の場合に
は,電解残液をマンガン,ニッケルの定量に用いる。]。
(4) 電極の洗浄,乾燥及びひょう量 次に,手早く新たな水中に浸して電極Aを離し,静かに数回上下し
て水洗した後,エタノールに浸してよく洗い直ちに約80℃の空気浴中で速やかに乾燥し,デシケータ
ー中で約30分間放冷した後その質量を量る。
注(4) この際,有機物の分解が不十分で褐色を呈している場合には,更に硝酸10mlを加え,白煙発生
を繰り返す。
(5) この電解条件は,昼間操作する場合の一応の目標を示したもので,0.2〜0.3Aで1夜間電解して
もよい。
(6) 電解残液からマンガンを定量する場合は,[9.2.3.2.(1)]を参照する。
8.2.5
空試験 試薬だけを用いて8.2.4(1)以降の手順に従って,試料と並行して操作する。電解残液は9.3.4
の空試験に用いる。
8.2.6
計算 試料中の銅含有率は,次の式によって算出する。
100
%
0
2
1
×
m
m
m
wt
−
=
銅
ここに, m1: 8.2.4で得られた質量 (g)
m2: 8.2.5で得られた質量 (g)
m0: 試料はかり取り量 (g)
9. マンガン定量方法
9.1
定量方法の区分 マンガンの定量方法は,次のいずれかによる。
(1) ペルオキソ二硫酸アンモニウム酸化亜ひ酸ナトリウム滴定法 この方法は,種別(2),(3)及び(4)に適用
する。
(2) 原子吸光法 この方法は,種別(2)の場合だけに適用する。
9.2
ペルオキソ二硫酸アンモニウム酸化亜ひ酸ナトリウム滴定法
6
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.2.1
要旨 種別(2),(4)の場合は,7.2.1で得られたろ液 (B) を定容とし,その一定垂を分取し[残液は,
試料溶液 (C) とし,ニッケルの定量に用いる。],硝酸及び硫酸を加えて加熱蒸発し,硫酸の白煙を発生さ
せ,放冷した後水を加えて塩類を溶解する。種別(3)の場合は,8.2.1で得られた電解残液,電極B及び電
解用ビーカーなどに析出しているマンガンの酸化物を過酸化水素処理をして分解した後,定容とし,その
一定量を分取し[残液は,試料溶液 (D) とし,ニッケルの定量に用いる。],硫酸を加える。
このいずれかの操作で得られた溶液に混酸及びペルオキソ二硫酸アンモニウムを加えて煮沸し,マンガ
ンを過マンガン酸に酸化するとともに過剰のペルオキソ二硫酸アンモニウムを分解する。冷却後,亜ひ酸
ナトリウム標準溶液で滴定する。
9.2.2
試薬 試薬は,次による。
(1) 硝酸
(2) 硫酸 (1+1)
(3) 混酸 水435mlに硫酸150mlを加え,冷却した後硝酸250ml,りん酸150ml及び硝酸銀溶液 (200g/l)
15mlを混和する。
(4) 過酸化水素
(5) ペルオキソ二硫酸アンモニウム溶液 (200g/l) この溶液は,使用の都度調製する。
(6) 亜ひ酸ナトリウム標準溶液 三酸化ひ素[0.500gをはかり取って,ビーカー (200ml) に入れ,水酸化
ナトリウム溶液 (40g/l) 20mlと水約100mlを加えて加熱して溶解し,冷却した後1 000mlの全量フラ
スコに移し入れる。これにフェノールフタレインエタノール溶液 (10g/l) を指示薬として2,3滴加え,
硫酸 (1+35) を加えて微酸性とし,炭酸水素ナトリウム溶液 (5g/l) 20mlを加えて水で標線まで薄める。
この標準溶液1mlに相当するマンガン量は,次によって標定する。
標準マンガン溶液(7)の一定量を正しく取り,三角フラスコ (1 000ml) に入れ,硫酸 (1+1) 10mlと
混酸30mlを加え,水で400mlに薄める[ニッケルを含む試料溶液の場合は分取した試料溶液中の含
有量に相当する電解ニッケルをはかり取り,三角フラスコ (1 000ml) に入れ,硫酸 (1+1) 10mlと混
酸30mlで分解した後,標準マンガン溶液の一定量を加え,水で400mlに薄める。]。
以下,9.2.3.1(3)の加熱操作以降の手順に従って操作し,次の式によって相当するマンガン量を算出
する。
2
1
2
000
.0
V
V
f
×
=
ここに,
f: 亜ひ酸ナトリウム標準溶液1mlに相当するマンガン量 (g)
V1: 標準マンガン溶液の使用量 (ml)
V2: 亜ひ酸ナトリウム標準溶液の使用量 (ml)
注(7) 標準マンガン溶液 (0.2mg/ml) の調製 マンガン(99.9wt%以上)0.100gをはかり取って,ビー
カー (200ml) に移し入れ,硫酸 (1+4) 50mlを加え加熱して分解し,常温まで冷却した後500ml
の全量フラスコに水を用いて移し入れ,水で標線まで薄める。
9.2.3
操作
9.2.3.1
種別(2)又は(4)の場合 種別(2)又は(4)の場合の定量操作は,次の手順によって行う。
(1) ろ液 (B) の分取 種別(2)の場合は7.2.4.1(3)で得られたろ液 (B) を,種別(4)の場合は7.2.4.2(4)で得ら
れたろ液 (B) をそれぞれ1 000mlの全量フラスコに移し入れ,水で標線まで薄める。その一定量を分
取し(8)[種別(4)の場合は残液を試料溶液 (C) とし,ニッケルの定量に用いる。]ビーカー (300ml) に
入れる。
7
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 硝酸及び硫酸による処理 硝酸10ml及び硫酸 (1+1) 10mlを加えた後,時計皿で覆い加熱蒸発して硫
酸の白煙を発生させ(9)放冷する。水約100mlを加え,加熱して析出した塩類を溶解した後,水を用い
て三角フラスコ (1 000ml) に移し入れる。
(3) マンガンの酸化 溶液に混酸 [9.2.2(3)] 30mlを加えた後,水で約400mlに薄め加熱する。煮沸し始め
たときに,ペルオキソ二硫酸アンモニウム溶液10mlを注意して加え,小気泡が大気泡となるまで2
〜3分間煮沸する。
(4) 滴定 溶液を流水中で25℃以下に冷却し,速やかに亜ひ酸ナトリウム標準溶液で滴定する。
注(8) マンガン量が約3mgとなるように分取する。
(9) 有機物の分解が不十分で褐色を呈している場合は,更に硝酸10mlを加え白煙発生を繰り返す。
9.2.3.2
種別(3)の場合 種別(3)の場合の定量操作は,次の手順によって行う。
(1) マンガンの酸化物の分解 8.2.4(3)で得られた電解残液の約半分及び白金電極Bをビーカー (500ml)
に移し入れ,残りの電解残液に過酸化水素を少量ずつ加えて,電解用ビーカーの内壁に付着したマン
ガンの酸化物を分解した後,水を用いて白金電極Bを入れたビーカーに移し入れ,加熱してマンガン
の酸化物を分解した後,白金電極Bを水で洗浄して取り去り常温まで冷却する。
(2) 試料溶液の分取,マンガンの酸化及び滴定 溶液を1 000mlの全量フラスコに移し入れ,水で標線ま
で薄める。その一定量を分取し(8)[残液は試料溶液 (D) とし,種別(3)のニッケルの定量に用いる。],
三角フラスコ (1 000ml) に移し入れ,硫酸 (1+1) 10mlを加える。
以下,9.2.3.1 3),(4)の手順に従って操作する。
9.2.4
空試験 試薬だけを用いて9.2.3.1(1)又は9.2.3.2(1)以降の手順に従って試料と並行して操作する。
残液は10.2.4の空試験に用いる。
9.2.5
計算 試料中のマンガン含有率は,次の式によって算出する。
(
)
100
%
1
0
4
3
×
×
×
B
m
f
V
V
wt
−
=
マンガン
ここに, V3: 9.2.3.1(4)又は9.2.3.2(2)で得た亜ひ酸ナトリウム標準溶液の使
用量 (ml)
V4: 9.2.4で得た亜ひ酸ナトリウム標準溶液の使用量 (ml)
f: 亜ひ酸ナトリウム標準溶液1mlに相当するマンガン量 (g)
m0: 試料はかり取り量 (g)
B1: 試料溶液及び空試験液の分取比
9.3
原子吸光法
9.3.1
要旨 7.2.1で得られたろ液 (B) に,硝酸を加えた後加熱してシロップ状とする。硝酸を加えた後
溶液を空気・アセチレンフレーム中に噴霧し,原子吸光光度計を用いてその吸光度を測定する。
9.3.2
試薬 試薬は,次による。
(1) 硝酸
(2) 硝酸 (1+1)
(3) 標準マンガン溶液 (0.2mgMn/ml) マンガン(99.9wt%以上)0.100gをはかり取って,ビーカー (200ml)
に移し入れ,硝酸 (1+1) 10mlを加えて時計皿で覆い,加熱して分解し,時計皿の下面及び内壁を
水を用いて洗浄し,常温まで冷却した後,500mlの全量フラスコに水を用いて移し入れ,水で標線ま
で薄める。
9.3.3
操作 定量操作は,次の手順によって行う。
(1) 試料溶液の調製 7.2.4.1(3)で得られたろ液 (B) を1 000mlの全量フラスコに移し入れ,水で標線まで
8
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
薄める。その一定量を分取し(10),ビーカー (200ml) に入れ,硝酸20mlを加え時計皿で覆い加熱蒸発
してシロップ状とする。硝酸 (1+1) 10mlを加え,加熱して可溶性塩を溶解した後,時計皿の下面及
びビーカーの内壁を水を用いて洗浄し,常温まで冷却し,水を用いて100mlの全量フラスコに移し入
れ,水で標線まで薄める。
(2) 測定 溶液の一部を水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム中に噴
霧し,波長280.1nmの吸光度を測定する。
注(10) マンガン量が1〜3mgとなるように分取する。
9.3.4
空試験 試薬だけを用いて9.3.3(1)以降の手順に従って試料と並行して操作する。
9.3.5
検量線の作成 標準マンガン溶液 [9.3.2(3)] の0〜15.0ml(マンガンとして0〜3.0mg)を段階的に
数個の100mlの全量フラスコに取り,硝酸 (1+1) 10mlを加え,水で標線まで薄める。以下,9.3.3(2)の手
順に従って操作し,試料と並行して測定して得た吸光度とマンガン量との関係線を作成し,その関係線を
原点を通るように平行移動して検量線とする。
9.3.6
計算 9.3.3(2)及び9.3.4で得た吸光度と9.3.5で作成した検量線とからマンガン量を求め,試料中
のマンガン含有率を,次の式によって算出する。
100
%
2
0
2
1
×
×B
m
A
A
wt
−
=
マンガン
ここに, A1: 分取した試料溶液中のマンガン検出量 (g)
A2: 分取した空試験液中のマンガン検出量 (g)
m0: 試料はかり取り量 (g)
B2: 試料溶液及び空試験液の分取比
10. ニッケル定量方法
10.1 定量方法 ニッケルの定量方法は,ジメチルグリオキシムニッケル重量法による。この方法は,種
別(4)及び(3)に適用する。
10.2 ジメチルグリオキシムニッケル重量法
10.2.1 要旨 種別(4)の場合は9.2.1で得られた試料溶液 (C) を用い,その定量を分取し,硝酸及び硫酸を
加えて加熱蒸発し,硫酸の白煙を発生させ,冷却した後水で薄める。種別(3)の場合は9.2.1で得られた試
料溶液 (D) の一定量を分取する。
この溶液に酒石酸及び塩化アンモニウムを加えた後,アンモニウム水を加えてアルカリ性とし,ジメチ
ルグリオキシムを加えてニッケル錯体を沈殿さぜる。ガラスろ過器でこし分け,乾燥した後その質量を量
る。
10.2.2 試薬 試薬は,次による。
(1) 硝酸
(2) 硫酸 (1+1)
(3) アンモニア水 (1+1)
(4) 塩化アンモニウム溶液 (250g/l)
(5) 酒石酸溶液 (250g/l)
(6) ジメチルグリオキシム溶液 ジメチルグリオキシム1.0gをエタノール (95vol%) 100mlに溶解する。
10.2.3 操作
10.2.3.1 種別(4)の場合 種別(4)の場合の定量操作は,次の手順によって行う。
9
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 硫酸処理 9.2.3.1(1)で得られた試料溶液 (C) から一定量を分取し(11)ビーカー (500ml) に入れ,硝酸
10ml及び硫酸 (1+1) 10mlを加えた後,加熱蒸発して硫酸の白煙を発生させる(12)。放冷した後水約
100mlを加え,加熱して析出した塩類を溶解した後,ビーカー (1 000ml) に移し入れる。
(2) ジメチルグリオキシムニッケル沈殿の生成 溶液に酒石酸溶液10ml及び塩化アンモニウム溶液20ml
を加え,アンモニア水 (1+1) で中和した後,その3mlを過剰に加え,液量を約500mlに薄める。
溶液を約90℃に加熱し,かき混ぜながらジメチルグリオキシム溶液 [10.2.2(6)] をニッケル予想含
有量0.01gにつき7mlの割合で加え,更にその5mlを過剰に加えて十分にかき混ぜた後,室温まで冷
却する。
(3) 沈殿のこし分け及び洗浄 沈殿をあらかじめ恒量としたガラスろ過器 (1G4) を用いてこし分け,温水
で十分に洗浄する。
(4) 沈殿の乾燥及びひょう量 沈殿は,ガラスろ過器と共に110〜120℃の空気浴中で約1時間乾燥し,デ
シケーター中で約1時間放冷した後,その質量を量り,恒量となるまでこの操作を繰り返す。
注(11) ニッケル量として30〜100mgとなるように分取する。
(12) 有機物の分解が不十分で褐色を呈している場合には,更に硝酸10mlを加えて白煙操作を繰り返
す。
10.2.3.2 種別(3)の場合 種別(3)の場合の定量操作は,次の手順によって行う。
(1) 試料溶液の調製 9.2.3.2(2)で得られた試料溶液 (D) の一定量を正しく分取し(11),ビーカー (1 000ml)
に入れる。以下,10.2.3.1(2)〜(4)の手順に従って操作する。
10.2.4 空試験 試薬だけを用いて10.2.3.1(1)又は10.2.3.2(1)以降の手順に従って試料と並行して操作する。
10.2.5 計算 試料中のニッケル含有率は,次の式によって算出する。
(
)
100
32
.2.0
%
0
2
1
×
×
×
B
m
m
m
wt
−
=
ニッケル
ここに,
m1: 10.2.2.1又は10.2.2.2で得られたジメチルグリオキシムニッ
ケルの質量 (g)
m2: 10.2.4で得られた質量 (g)
m0: 試料はかり取り量 (g)
B: 試料溶液及び空試験液の分取比
11. 鉛定量方法
11.1 定量方法 鉛の定量方法は,原子吸光法による。この方法は,鉛含有率0.01wt%以上0.1wt%未満の
試料に適用する。
11.2 原子吸光法
11.2.1 要旨 種別(1),(2)の場合は試料を硝酸で分解し,種別(3),(4)の場合は試料を王水で分解して定容
とする。その定量を数個分取し,各溶液に鉛の標準溶液を段階的に添加した後,定容とし,溶液を空気・
アセチレンフレーム中に噴霧し原子吸光光度計を用いてその吸光度を測定する。
11.2.2 試薬 試薬は,次による。
(1) 硝酸 (1+1)
(2) 王水(塩酸3,硝酸1)
(3) 標準鉛溶液(25μgPb/ml)鉛(99.99wt%以上)0.100gをはかり取ってビーカー (200ml) に移し入れ,
硝酸 (1+1) 10mlを加え時計皿で覆い加熱して分解し,時計皿の下面及びビーカーの内壁を水を用い
て洗浄し,常温まで冷却する。水を用いて100mlの全量フラスコに移し入れ,水で標線まで薄め原液
10
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1 000μgPb/ml)とする。使用の都度この原液の一定量を水で40倍に薄め標準鉛溶液とする。
11.2.3 試料はかり取り量 試料はかり取り量は,5.0gとする。
11.2.4 操作
11.2.4.1 種別(1)及び(2)の場合 種別(1)及び(2)の場合の定量操作は,次の手順によって行う。
(1) 試料の分解 試料をはかり取ってコニカルビーカー (300ml) に移し入れ,硝酸 (1+1) 50mlを加え,
時計皿で覆い加熱して分解する。時計皿の下面及びビーカーの内壁を水を用いて洗浄し,常温まで冷
却する。水を用いて100mlの全量フラスコに移し入れ,水で標線まで薄める。
(2) 試料溶液の調製 溶液10.0mlずつを4個の100mlの全量フラスコに分取し,標準鉛溶液 [11.2.2(3)] の
0〜20.0ml(鉛として0〜500μg)を段階的に加えた後,水で標線まで薄める。
(3) 測定 4個の試料溶液を水を用いてゼロ点を調整した原子吸光光度計の空気・アセチレンフレーム中
に噴霧し,波長283.3nm(13)の吸光度を測定する。
注(13) 波長217.0mmを用いてもよい。
11.2.4.2 種別(3)及び(4)の場合 種別(3)及び(4)の場合の定量操作は,次の手順によって行う。
(1) 試料の分解 試料をはかり取ってコニカルビーカー (300ml) に移し入れ,王水50mlを加えて,時計
皿で覆い加熱して分解する。時計皿の下面及びビーカーの内壁を水を用いて洗浄し,常温まで冷却す
る。水を用いて100mlの全量フラスコに移し入れ,水で標線まで薄める。
(2) 試料溶液の調製及び測定 1.2.4.1(2),(3)の手順に従って操作する。
11.2.5 検量線の作成 11.2.4.1(3)及び11.2.4.2(2)で得た鉛の吸光度を縦軸とし,11.2.4.1(2)及び11.2.4.2(2)
で添加した鉛の添加量を横軸として,関係線を作成し検量線とする。
11.2.6 計算 11.2.5で作成した検量線が横軸と交わる点から鉛添加量0の点までの距離に対応する鉛の量
を求め,試料中の鉛の含有率を,次の式によって算出する。
100
10
1
%
×
×
m
A
wt=
鉛
ここに,
A: 試料溶液中の鉛検出量 (g)
m: 試料はかり取り量 (g)
11
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
付図1 電解用ビーカー
付図2 白金電極A
付図3 白金電極B
付図4 半円形時計皿
12
Z 3906-1988
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
パラジウムろう分析方法工業標準改正原案調査作成委員会 構成表
氏名
所属
(委員長)
俣 野 宣 久
川崎製線株式会社
山 本 博 信
株式会社徳力本店
有 賀 正
東海大学
大河内 春 乃
科学技術庁金属材料技術研究所
荒 木 誠 司
大蔵省造幣局
足 立 芳 寛
通商産業省基礎産業局
加 藤 康 宏
工業技術院標準部
池 田 純 一
財団法人日本規格協会
前 田 修
田中貴金属工業株式会社
吉光寺 博
石福金属興業株式会社
橋 本 謙 三
橋本貴金属工業株式会社
乾 昌 弘
乾庄貴金属化工株式会社
堀 仁
水野ハンディ・ハーマン株式会社
鈴 木 健 史
合資会社徳力商店
有 井 満
株式会社東芝
古 矢 元 佑
清峰金属工業株式会社
松 井 文 夫
日本アイ・ティー・エス株式会社
岡 島 義 昭
株式会社日立製作所
今 村 孝
三菱電機株式会社
堀 泰 治
東京ラヂエーター製造株式会社
(事務局)
池 原 平 晋
社団法人日本溶接協会