T 9010 : 1999
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣及び厚生大臣
が制定した日本工業規格である。
JIS T 9010には,次に示す附属書がある。
附属書1(規定) 溶出方法及び条件
附属書2(参考) 分析対象とする老化防止剤及び加硫促進剤
附属書3(参考) DTC系加硫促進剤の定量法
附属書4(参考) 参考文献
T 9010 : 1999
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目次
ページ
1. 適用範囲 ························································································································ 1
2. 引用規格 ························································································································ 1
3. 試験 ······························································································································ 2
3.1 カドミウム及び鉛 ·········································································································· 2
3.1.1 試験の概要 ················································································································· 2
3.1.2 試料の採取方法 ··········································································································· 2
3.1.3 試験溶液の調製 ··········································································································· 2
3.1.4 カドミウム ················································································································· 4
3.1.5 鉛 ····························································································································· 5
3.2 2-メルカプトイミダゾリン ······························································································· 5
3.2.1 試験の概要 ················································································································· 5
3.2.2 試料の採取方法 ··········································································································· 5
3.2.3 試験溶液の調製 ··········································································································· 5
3.2.4 試験 ·························································································································· 5
3.3 溶出物 ························································································································· 6
3.3.1 試験の概要 ················································································································· 6
3.3.2 溶出方法 ···················································································································· 6
3.3.3 フェノール ················································································································· 6
3.3.4 ホルムアルデヒド ········································································································ 7
3.3.5 重金属 ······················································································································· 8
3.3.6 亜鉛 ·························································································································· 8
3.3.7 ひ素 ·························································································································· 8
3.3.8 蒸発残留物 ················································································································ 10
3.3.9 過マンガン酸カリウム消費量 ························································································ 11
3.3.10 紫外吸収スペクトル ··································································································· 12
3.4 N-ニトロソ化合物 ········································································································· 12
3.4.1 試験の概要 ················································································································ 12
3.4.2 総N-ニトロソアミン量とN-ニトロソアミン類の分別定量 ··················································· 12
3.4.3 N-ニトロソアミン及びN-ニトロソ化可能物質の溶出量······················································· 16
3.5 細胞毒性及び感作性物質································································································· 17
3.5.1 試験の概要 ················································································································ 17
3.5.2 細胞毒性試験 ············································································································· 17
3.5.3 感作性試験 ················································································································ 20
3.5.4 老化防止剤及び加硫促進剤の分析 ·················································································· 23
3.6 水溶性タンパク質 ········································································································· 27
2
T 9010 : 1999 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.6.1 試験の概要 ················································································································ 27
3.6.2 試薬 ························································································································· 27
3.6.3 器具及び装置 ············································································································· 27
3.6.4 操作 ························································································································· 28
3.7 エンドトキシン ············································································································ 28
3.7.1 試験の概要 ················································································································ 28
3.7.2 試薬及び試液 ············································································································· 29
3.7.3 器具 ························································································································· 29
3.7.4 試料溶液の調製 ·········································································································· 29
3.7.5 試験及び判定方法 ······································································································· 30
3.8 エチレンオキシド ········································································································· 30
3.8.1 試験の概要 ················································································································ 30
3.8.2 試薬及び試液 ············································································································· 30
3.8.3 装置及び器具 ············································································································· 30
3.8.4 操作 ························································································································· 31
附属書1(規定) 溶出方法及び条件 ······················································································ 33
1. 適用範囲 ······················································································································· 33
2. 溶媒 ····························································································································· 33
2.1 食品用器具・容器包装の場合 ··························································································· 33
2.2 医療用具及び医薬品容器の場合 ························································································ 33
3. 溶出方法 ······················································································································· 33
3.1 充てん溶出法 ··············································································································· 33
3.2 浸せき溶出法 ··············································································································· 33
3.3 片面溶出法 ·················································································································· 34
4. 溶出温度と時間 ·············································································································· 34
附属書2(参考) 分析対象とする老化防止剤及び加硫促進剤 ····················································· 36
1. 老化防止剤及び加硫促進剤の化合物名と略称 ······································································· 36
附属書3(参考) DTC系加硫促進剤の定量法 ········································································· 37
1. DTC系加硫促進剤のコバルト錯体化 ·················································································· 37
2. 複数のDTC系加硫促進剤が共存する場合のコバルト錯体の生成 ·············································· 37
3. 未知試料中のDTC系加硫促進剤の定性・定量分析 ································································ 38
附属書4(参考) 参考文献 ·································································································· 41
1. N-ニトロソアミン関連 ····································································································· 41
2. 細胞毒性・感作性関連 ····································································································· 41
3. 水溶性タンパク質関連 ····································································································· 42
4. エチレンオキシド関連 ····································································································· 42
3
T 9010 : 1999 目次
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5. 附属書1関連 ················································································································· 42
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
T 9010 : 1999
ゴム製品の生物学的安全性に関する
試験方法
Test methods relevant to biological safety of rubber products
1. 適用範囲 この規格は,ゴム製品の生物学的安全性を評価するための試験方法について規定する。
備考 この規格は,すべてのゴム(1)製品に適用することが必要なわけではなく,また,適用する場合
にも,この規格に規定したすべての項目の試験を実施することが必要なわけではないことに留
意しなければならない。必要な試験項目は,個々のゴム製品の規格の中に定められる。
試験項目の中には,すべての種類のゴムに適用可能なもの(又は,適用するもの)と,一部
の種類のゴムだけにしか適用できないもの(又は,一部の種類のゴム以外に適用する意味のな
いもの)がある。また,ある種の試験は特殊な用途の製品に適用するために作られたものであ
る。その他の用途の製品に適用する場合には,当初の目的や意図を理解して適用しなければな
らない。これらについては各試験項目中の「試験の概要」に記述する。
規格値は,この規格においては定めない。それらは個々のゴム製品の規格の中で定められる。
注(1) ここでいうゴムとは,天然ゴム,合成ゴム(シリコーンゴム,ウレタンゴムなどを含む)及び
それらのラテックス並びに熱可塑性ゴムを含み,プラスチックとの複合材で,ゴムの含有量が
50%以上のものを含む。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格のうちで発行年を付記してあるものは,記載の年の版だけがこの規格の規定を構成
するものであって,その後の改訂版・追補には適用しない。発行年を付記しないものは,最新の版を適用
する。
JIS K 0115 吸光光度分析通則
JIS K 0121 原子吸光分析通則
JIS K 8012 亜鉛(試薬)
JIS K 8085 アンモニア水(試薬)
JIS K 8872 ホルムアルデヒド液(試薬)
JIS P 8140 紙及び板紙−吸水度試験方法−コッブ法
ISO 10993-7 : 1995 Biological evaluation of medical devices−Part 7 : Ethylene oxide sterilization residuals
第13改正日本薬局方・一般試験法 エンドトキシン試験法
第13改正日本薬局方・一般試験法 発熱性物質試験法
第13改正日本薬局方・一般試験法 注射剤用ガラス容器試験法
第13改正日本薬局方第II部“注射用水”
2
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3. 試験
参考 この規格に規定する試験の多くには何らかの抽出操作又は溶出過程が含まれている。温度及び
かき混ぜ条件が一定であれば,ある物質が材料から溶出する量及び溶出する速度は次のパラメ
ータによって規定される。すなわち,(1)その物質のゴム中の含有量,(2)その物質のゴムと外部
液(溶出溶媒)間の平衡定数,(3)その物質のゴム中での拡散定数,(4)ゴムの外部液による膨潤
度,(5)外部液量とゴム表面積の比である。各試験項目には一般的に適用可能な溶出(抽出)条
件が記述されているが,例えば,試料ゴム製品の細切方法などを厳密には記述していない。そ
れはこの試験を適用するゴム製品が多岐にわたると推定されるためである。したがって,厳密
にいえば,各試験において規定していない程度の溶出(抽出)条件の細部の違いによっても試
験結果が微妙に異なることは否定できない。どの程度厳密に溶出(抽出)条件を規定するかは,
試験結果にどの程度の精確さを要求するかにも関わる問題である。この試験を個別のゴム製品
に適用する場合には,その点を留意して,適切な溶出(抽出)方法を選択する。
3.1
カドミウム及び鉛
3.1.1
試験の概要 この試験は,ゴム製品中のカドミウム及び鉛(2)の含有量を定量する試験である。この
試験は,すべての種類のゴムに適用できる。
注(2) 配合剤として用いる酸化亜鉛などの不純物として混入する可能性がある。
3.1.2
試料の採取方法 試験に用いるゴム試料は,2mm角程度に細かく切断し,水で洗い,風乾したも
のから1〜2g(3)を0.1mgまで量り採り,これを試料採取量とする。
注(3) カドミウム及び鉛の含有量が多い試料については約1gを,含有量の少ない試料については約2g
を0.1mgまで量り採る。
3.1.3
試験溶液の調製 試験溶液は,次に示すJISに規定する試薬,器具,装置及び操作によって調製す
る。
a) 試薬
1) 硫酸
2) 塩酸
3) くえん酸溶液 (500g/l)
4) 酢酸アンモニウム溶液 (400g/l)
5) 硝酸
6) 王水(塩酸3+硝酸1)
7) ふっ化水素酸
8) 過塩素酸 (70%)
b) 器具及び装置
1) 白金皿又は石英皿
2) ガラスろ過器 ブフナー漏斗形ガラスろ過器
3) 全量フラスコ 100ml
4) 電気炉 温度を±30℃以下に自動的に調節できるものが望ましい。
5) ケルダールフラスコ 200〜300ml
c) 操作
1) 乾式分解 試料を白金皿又は石英皿にとり,硫酸2mlを滴下して潤し,徐々に加熱し,大部分の硫
酸分を蒸発させた後,直火上で乾固する(4)。これを電気炉に入れ,470±30℃で加熱し,灰化する。
3
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
24時間後なお灰化が十分でない場合(5)には,蒸発皿の内容物を硫酸で潤して再び加熱し,ほとんど
白色の灰分が得られるまでこの操作を繰り返す。この残留物に0.1mol/l硝酸10mlを加えて加温溶解
する。不溶解物が残存するときはガラスろ過器でろ過する。得られた溶液を全量フラスコ100mlに
移し,0.1mol/l硝酸を標線まで加えて試験溶液とする。別に試料を含まないものについて,試験溶
液と同様に操作し,空試験溶液とする。
注(4) 硫酸を添加して灰化する際に,急激に加熱すると試料が飛散しやすいので,ホットプレート,
砂浴などを用いて徐々に加熱し,ゴムと硫酸を反応させた後,加熱温度を上げ硫酸分を蒸発さ
せる。蒸発後,さらに温度を上げ乾固する。ただし,500℃以上に加熱しないようにする。
(5) 470±30℃で24時間灰化した後,なお灰化が十分でない場合には,低温灰化装置を用いると灰
化時間を早めることができる。
備考1. カーボンブラック配合の試料には,灰化が非常に遅いものがある。
2. けい酸塩などの不溶性残留物が多量にある場合には,ガラスろ過器でろ過し,水で洗浄した
後,不溶物につき次のいずれかの方法によって処理する。
(a) 不溶物を白金皿に移し,ふっ化水素酸5mlを加え水浴上で蒸発乾固する。これに水及び塩
酸を数滴ずつ加えて溶かし,これを初めのろ液及び洗液に合わせて試験溶液とする。
(b) 不溶物に王水を加え,2時間煮沸し抽出する。不溶物をガラスろ過器でろ過し,ろ液を水
浴上で蒸発乾固する。これに水及び塩酸を加えて溶かし,得られた溶液を初めのろ液及び
洗液に合わせて試験溶液とする。
(c) 試料がシリコーンゴムの場合は,次の操作によって行う。
試料を白金皿にとり,硫酸約2mlで潤し,硫酸の白煙がやや出る程度の温度で,徐々に
加熱して炭化する。これにふっ化水素酸約5mlを加え,砂浴上で蒸発乾固した後,電気炉
に入れ470±30℃で3時間灰化し,以下本文操作における灰化後の手順に従って塩酸及び
塩酸・くえん酸混合液などによる溶解処理を行い,試験溶液とする。
なお,分解時に炭化物が生成する場合は,ふっ化水素酸添加の前後に硝酸1〜2mlを加え
る。
2) 湿式分解 試料をケルダールフラスコ (200〜300ml) にとり,硝酸(6)10mlを加え穏やかに熱する。
この液が減少したら加熱をやめ,硝酸少量を追加して再び加熱を続け,この液が減少したら同じ操
作を繰り返す。20〜30分間加熱を続け,原形が崩れたら加熱をやめ,硫酸5mlを注意しながら加え
再び加熱する。この液が黒色泥状となったら硝酸少量を滴下し,二酸化窒素の発生が弱くなるまで
加熱し,液が減少し黒色となったら硝酸を滴下する。この操作を繰り返し,液が茶褐色になったら
加熱をやめ,過塩素酸(7)1ml,硝酸2mlを注意しながら加え加熱する。反応の強弱の度合によって
加熱を加減し,液が乾固しないように注意して濃縮する。加熱をやめ,硝酸1mlを加え加熱を続け
る。この液が濃縮されたら再び硝酸少量を滴下し加熱を続ける。この操作を液が茶褐色から黄色を
経て,ほとんど無色になるまで繰り返す。硫酸の白煙が発生したら加熱を止め冷却する。ケルダー
ルフラスコを水で洗いながら100mlの全量フラスコに移し,水を標線まで加えて試験溶液とする。
別に試料を含まないものについて試験溶液と同様に操作し,空試験溶液とする。
注(6) ゴム質によっては,分解の初期段階で試料が黒く固まることがある。このような試料は,分解
しにくいから初めに硫酸10mlを加えて加熱し,原形を崩してから硝酸を加える。以後,硫酸は
加えない。
(7) 有機物が多量に残存しているときに過塩素酸を加え乾固すると爆発を起こすから十分に注意す
4
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。
3.1.4
カドミウム
a) 試薬
1) ジエチルジチオカルバミン酸ナトリウム溶液 (50g/l) (NaEDC溶液)
2) カドミウム標準溶液 (0.01mgCd/ml) 金属カドミウム(純度99.9%以上)0.100gをとり,1mol/l硝
酸100mlに溶かし煮沸して窒素酸化物を除去し,冷却後1mol/l硝酸を加えて1 000mlとし,保存溶
液とする。使用時にこの保存溶液を0.5mol/l硝酸で10倍に希釈し,標準溶液とする。
3) ブロムチモールブルー溶液(BTB溶液) ブロムチモールブルー0.1gをエタノール (50v/v%) 100ml
に溶かす。
4) アンモニア水 (1+2)
5) メチルイソブチルケトン (MIBK)
6) くえん酸二アンモニウム溶液 (250g/l) くえん酸二アンモニウム25gを水約80mlに溶かす。BTB
溶液2滴を加え,液の色が黄から緑になるまでアンモニア水 (1+2) を滴下した後,水で100mlと
する。これを分液漏斗に移し入れ,NaEDC溶液 (50g/l) 2mlとMIBK10mlを加え,激しく振り混ぜ
静置する。くえん酸二アンモニウム溶液層を分離し,乾いたろ紙でろ過し,MIBKの細かい泡を取
り除く。
7) 硫酸アンモニウム溶液 (400g/l) 硫酸アンモニウム40gを水約80mlに溶かし,以下6)と同様に操
作する。
b) 使用ガス
1) 可燃性ガス アセチレン又は水素
2) 支燃性ガス 空気
c) 器具及び装置
1) 分液漏斗 300ml
2) 原子吸光分析装置
3) カドミウム中空陰極ランプ
d) 操作 試験溶液及び空試験溶液のそれぞれ一定量(8)を分液漏斗にとり,それぞれにくえん酸二アンモ
ニウム溶液 (250g/l) 10ml及び指示薬としてBTB溶液2滴を加え,液の色が黄から緑になるまでアン
モニア水 (1+2) を加えて中和し(9),これに硫酸アンモニウム溶液 (400g/l) 10ml及び水を加えて100ml
とする。これにNaEDC溶液 (50g/l) 20mlを加えて混和し,数分間放置した後,MIBK20.0mlを加え,
激しく振り混ぜる。これを静置してMIBK層を分取する。カドミウム中空陰極ランプを点灯し安定し
た後228.8nmの波長でJIS K 0121の6.の分析操作に従って行い,次の検量線からカドミウムの量を求
める。検量線は,カドミウム標準溶液の濃度を段階的にかえて,試験溶液と同様に操作し,JIS K 0121
の6.4(検量線の作成と定量法)によってカドミウム量と吸光度との関係線から作成する。
注(8) 試験溶液の一定量は,規格値に応じて適正な量を採取する。
(9) 指示薬BTB溶液を加え,液の色が黄から緑になるまでアンモニア水で中和するときpHは6.6
になる。
備考1. 測定時に妨害となる金属が抽出される場合は,pHの調節,マスキング剤の使用などによって,
金属イオンの相互分離を行う。
2. ジエチルジチオカルバミン酸ナトリウムと鉛,カドミウムのキレート化合物が1回のMIBK
抽出で完全に抽出されないときは,抽出を2回繰り返す。
5
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3. 測定時に共存物質などによる光の吸収,散乱で空試験に大きな吸収があるときは,重水素ラ
ンプで補正し,試験溶液の吸光度が空試験溶液の2倍以上あった場合に定量を行う。
3.1.5
鉛
a) 試薬
1) ジエチルジチオカルバミン酸ナトリウム溶液 (50g/l) (NaEDC溶液)
2) 鉛標準溶液 (0.01mgPb/ml) 硝酸鉛1.598gを1mol/l硝酸10mlに溶かし,水を加えて1 000mlとし
保存溶液とする。保存溶液の調製及び保存には可溶性鉛塩を含まないガラス器具を用いる。保存溶
液を水で100倍に希釈し,標準溶液とする。この液は使用時に調製する。
3) くえん酸二アンモニウム溶液 (250g/l) 3.1.4 a)6)による。
4) ブロムチモールブルー溶液(BTB溶液) 3.1.4 a)3)による。
5) アンモニア水 (1+2)
6) 硫酸アンモニウム溶液 (400g/l) 3.1.4 a)7)による。
7) メチルイソブチルケトン (MIBK)
b) 使用ガス
1) 可燃性ガス アセチレン又は水素
2) 支燃性ガス 空気
c) 器具及び装置
1) 分液漏斗 300ml
2) 原子吸光分析装置
3) 鉛中空陰極ランプ
d) 操作 3.1.4 d)に規定する方法によって行う。
なお,鉛中空陰極ランプを用い,測定波長は217.0nm又は283.3nmとする。
3.2
2-メルカプトイミダゾリン
3.2.1試験の概要 この試験は塩素を含むゴム材料中の2-メルカプトイミダゾリンを検出する試験である。
特に,食品用器具・容器包装などに適用する。
3.2.2
試料の採取方法 3.1.2による。ただし,2-メルカプトイミダゾリンは,水で洗うと溶出してしまう
ので,水で洗わない。
3.2.3
試験溶液の調製 試料2.0gを円筒ろ紙に入れ,ソックスレー抽出器を用いてメタノール約90mlで
8時間抽出する。抽出液を水浴上で約2mlまで濃縮し,試験溶液とする。
3.2.4
試験
a) 試薬
1) 薄層板 乳鉢又はビーカーにシリカゲル(10)(薄層クロマトグラフ用)30gを量り採り,水60mlを
加えてよく混ぜ合わせて均一なぺースト状にする。これを約200×200mmのガラス板上に厚さ
250μmに均一に塗布し,5〜10分間放置後,120℃で1時間乾燥して活性化する。デシケーター中で
放冷保存する。
2) 展開溶媒 展開溶媒は次のものを用いる。
酢酸エチル・ベンゼン (5 : 1)
酢酸エチル・メタノール・アンモニア水 (1+1) (15 : 1 : 1)
3) 発色液 2, 6-ジクロロキノン-4-クロルイミドエタノール溶液 (10g/l)。2, 6-ジクロロキノン-4-クロル
イミド0.1gをエタノールに溶かして10mlとする。この液は,使用時に調製する。
6
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4) 2-メルカプトイミダゾリン標準液 2-メルカプトイミダゾリン0.20gをメタノールに溶かして100ml
とする。この液をメタノールで100倍に希釈して標準液とする。
b) 操作 薄層板の左右両端から約20mmを残し,下端から約20mmの位置に幅20mm以上の間隔で試験
溶液及び2-メルカプトイミダゾリン標準液各10μlを,それぞれ別のマイクロシリンジを用いてスポッ
トする。スポットの大きさは,できるだけ小さくなるようにする。あらかじめガラス製の密閉できる
展開槽に深さ約10mmになるように展開溶媒(11)を入れ,槽内を溶媒蒸気で十分に飽和させた後薄層板
を入れ,上昇法によって展開する。溶媒の先端が原点から約100mmに達したとき,展開槽から薄層
板を取り出し風乾する。発色液を噴霧し,120℃で10分間加熱する。試験溶液に2-メルカプトイミダ
ゾリンを含むときは,2-メルカプトイミダゾリン標準液と同じRf値の位置に褐色のスポットが認めら
れる。
注(10) 固着剤として硫酸カルシウムを5%含有したもの。
(11) a)2)に示した2種類の展開溶媒を別々に用いる。
3.3
溶出物
3.3.1
試験の概要 この試験は,食品衛生上又は医薬品容器の品質上その他で一般的に検査が必要とされ
る各種の物質が実際の使用条件下でどれほど溶出するかを,模擬的な溶出条件を用いて評価するためのも
のである。どのような種類のゴム材料についても適用できる。
3.3.2
溶出方法 附属書1に示した溶媒,溶出方法,溶出温度及び時間から最も適切な条件を選択し,そ
の条件で溶出して得た液を試料溶液とする。別に試料を含まないものについて同様に操作して得た液を空
試験液とする。
備考 完全に使用条件を再現することは困難であるが,食品用器具・容器包装や医療用具,医薬品容
器などにおいて従来から用いられてきた模擬溶出溶媒があるので,それらの中から最も近いと
思われる溶媒を選択する(附属書1参照)。
なお,溶出温度などのその他の条件の選択に当たっては,想定される使用条件のうちの最高
温度条件,最大の試料/溶媒比を使用する。
3.3.3
フェノール
a) 試薬
1) 4-アミノアンチピリン溶液 4-アミノアンチピリン1.36gを水に溶かして全量を1 000mlとする。
2) ほう酸緩衝液 1mol/l水酸化ナトリウム溶液9容量と1mol/lほう酸10容量の混合溶液。
3) フェリシアン化カリウム溶液 フェリシアン化カリウム8.6gを水に溶かし,アンモニア水 (28%)
1.8ml及び水を加えて全量を1 000mlとする。
4) フェノール標準溶液 フェノールを定量し,1ml中に10mgのフェノールを含有するように溶かし
原液とする。次に原液1.0mlを正確にとり,これを水で100mlとした液を標準溶液とする。
フェノール標準溶液1ml=100μgフェノール
b) 操作 附属書1の操作によって得た試験溶液30.0mlを50mlの全量フラスコにとり(12),ほう酸緩衝液
3mlを加えてよく振り混ぜた後,4-アミノアンチピリン溶液5ml及びフェリシアン化カリウム溶液
2.5mlを加える。さらに水を加えて全量50mlとし,よく振り混ぜ,室温に10分間放置した後,空試
験溶液(12)30.0mlを試験溶液と同様に操作したものを対照とし,波長510nm付近における吸光度を測
定する。検量線からフェノールの量を求め,試験溶液中のフェノールの濃度 (μg/ml) を算出する。
c) 検量線の作成 別にフェノール標準溶液0.5,1.0,1.5,2.0及び2.5mlを50mlの全量フラスコにとり,
それぞれに水を加えて30mlとし,これにほう酸緩衝液3mlを加え,以下,試験溶液と同様に操作し
7
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
てフェノールの量と吸光度との検量線を作成する。対照液は空試験で得た液を用いる。
注(12) 試験溶液及び空試験溶液が4%酢酸を用いて作られた場合は,4mol/l水酸化ナトリウム溶液で中
和し,BTB試験紙を用いてpHを約7に調整する。
3.3.4
ホルムアルデヒド
a) 試薬
1) アセチルアセトン溶液 酢酸アンモニウム150gを水に溶かし,酢酸3ml及びアセチルアセトン2ml
を加え,さらに水を加えて1 000mlとする。この液は使用時に調整する。
2) りん酸 (200g/l)
3) よう素溶液
4) 水酸化カリウム溶液
5) チオ硫酸ナトリウム溶液
6) 硫酸
7) ホルムアルデヒド液 (HCHO) JIS K 8872
8) ホルムアルデヒド標準溶液 ホルムアルデヒド液約1gを水5mlを入れたはかり瓶に精密に量り,
水を加えて正確に100mlとする。この溶液10mlをとり,0.1mol/lよう素溶液50mlを加え,さらに
1mol/l水酸化カリウム溶液20mlを加える。15分間室温で放置した後,10%硫酸15mlを加え,0.1mol/l
チオ硫酸ナトリウム溶液 (Na2S2O3) で滴定する(指示薬:でんぷん溶液1ml)。別に水10mlについ
て同様にして空試験を行う。
ホルムアルデヒド液中のホルムアルデヒド含量 (%) を次式によって算出する。
ホルムアルデヒド含量C (%) =
W
F
V
V
)
(
51
.1
0−
×
ここに,
V: 本試験における0.1mol/l Na2S2O3溶液の滴定量 (ml)
V0: 空試験における0.1mol/l Na2S2O3溶液の滴定量 (ml)
F: 0.1mol/l Na2S2O3溶液のファクター
W: ホルムアルデヒド液の採取量 (g)
ホルムアルデヒド液200/C (g) を正確に量り,水に溶かして100mlとする (HCHO20 000mg/l)。こ
れを水で10倍に希釈する操作を4回繰り返して,ホルムアルデヒド標準溶液とする。
ホルムアルデヒド標準溶液1ml=2μgHCHO
b) 操作 附属書1の操作によって得た試験溶液10mlを蒸留フラスコにとり,20%りん酸1mlを加えた後,
200mlのメスシリンダーに水5〜10mlを入れて受器とし,冷却器のアダプターが水に浸るようにして
水蒸気蒸留を行う(13)。留出液が約190mlになったとき蒸留をやめ,水を加えて200mlとする。この液
5.0mlを共栓試験管にとり,アセチルアセトン溶液5mlを加えて混和し,沸騰水浴中で10分間加熱す
る。冷後,溶液の一部を吸収セルに移し,空試験の液を対照として波長415nm付近における吸光度を
測定する。別に,空試験液として水5.0mlをとり,以下,試験溶液の場合と同様に操作を行った空試
験の液を対照とする。検量線からホルムアルデヒドの濃度を求め,試験溶液中の濃度 (μg/ml) を算出
する。
c) 検量線の作成 別にホルムアルデヒド標準溶液 (0.5,1.0,2.0,4.0,6.0及び8.0μg/ml) 5.0mlについて
試験溶液と同様の操作を行い,検量線を作成する。
注(13) 妨害物質が試験溶液内に存在するのが一般的なので,水蒸気蒸留によってこれを除去するが,
特に妨害物質の存在しない場合には,以上の水蒸気蒸留の操作は省略できる。
8
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.3.5
重金属
a) 試薬
1) シアン化カリウム溶液 (100g/l) シアン化カリウム10gを水に溶かし100mlとする。この液は使用
時に調製する。
2) 鉛標準溶液 (0.01mgPb/ml) 3.1.5 a)2)による。
3) 硫化ナトリウム溶液 硫化ナトリウム5gを水10ml及びグリセリン30mlの混液に溶かして調製す
る。又は水酸化ナトリウム5gを水30ml及びグリセリン90mlの混液に溶かし,その半容量をとり,
冷時硫化水素を飽和し,これに残りの半容量を混和して調製する。遮光した小瓶になるべく空気が
入らないように満たし,密栓して保存する。この液は調製後3か月以内に使用する。
b) 器具 ネスラー管 無色,厚さ1.0〜1.5mmのほうけい酸ガラス製,共栓付き円筒で,管径20〜22mm
で栓の下端から目盛線上部の空間が長さ35〜45mmのものを用いる。ただし,それぞれの管の50ml
目盛線の高さの差が2mm以下のものを用いる。
c) 操作 附属書1の操作によって得た試験溶液をネスラー管にとり(14),水を加えて50mlとし,検液と
する。比較液は規定する量の鉛標準溶液をネスラー管にとり,検液の調製と同様に操作する。両液に
硫化ナトリウム溶液2滴ずつを加えてよく混和し5分間放置した後,両管を白色を背景として上方及
び側方から観察し,鉛標準溶液の呈する色と比較する。
注(14) 硫化亜鉛の生成による影響を考慮する必要がある場合,アンモニア水で中和してpH7以上とし,
シアン化カリウム溶液を加える操作を行えば,亜鉛溶出量が200μgくらいまでは影響はない。
しかし,亜鉛の溶出量が50μg以下であれば,前記処理はしなくても濁りはなく,1μgの鉛を十
分検出できる。白濁を生じ測定困難なときは,前記のようにあらかじめ中和しておけば,シア
ン化カリウム溶液の使用量は2mlで十分である。
3.3.6
亜鉛
a) 試薬
1) 亜鉛(試薬)
2) 亜鉛標準液 (0.01mgZn/ml) 亜鉛1.000gに塩酸 (1+1) 10mlを加え加温して溶かす。水浴上で乾固
した後,1mol/l塩酸に溶かして1 000mlとし保存溶液とする。使用時に保存溶液を0.1mol/l塩酸で
100倍に希釈し,標準溶液とする。
b) 使用ガス
1) 可燃性ガス アセチレン
2) 支燃性ガス 空気
c) 装置
1) 原子吸光分析装置
2) 亜鉛中空陰極ランプ
d) 操作 附属書1の操作によって得た試験溶液及び空試験溶液をそれぞれ20mlとり,塩酸数滴を加え
て振り混ぜ,亜鉛中空陰極ランプを点灯し安定した後,213.9nmの波長でJIS K 0121の6.の分析操作
によって行い,次の検量線から亜鉛の量を求める。検量線は,亜鉛標準溶液の濃度を段階的にかえて,
JIS K 0121の6.4(検量線の作成と定量法)によって亜鉛量と吸光度との関係線から作成する。
3.3.7
ひ素
a) 試薬
1) ブロムフェノールブルー試液 ブロムフェノールブルー0.1gを量り,50%エタノール溶液100mlを
9
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
加えて溶かし,必要があればろ過する。
2) アンモニア試液 (10%) アンモニア水 (JIS K 8085) 400mlに水を加えて1 000mlとする。
3) よう化カリウム試液 (1mol/l) よう化カリウム16.5gを水に溶かし100mlとする。遮光して保存す
る。
4) 酸性塩化第一すず試液 塩化第一すず4gを量り,無ひ素塩酸125mlを加えて溶かし,水を加えて
250mlとし,共栓瓶に入れ,密栓して保存する。この液は,調製後1か月以内に用いる。
5) 無ひ素亜鉛 Zn(JIS K 8012,ひ素分析用)粒径約800μmのものを用いる。ただし,多孔性のもの
は,一般に溶解が速すぎるので使用しない。操作終了後なお少量が溶けきれずに残り,水素の発生
が持続しているものがよい。
6) ひ化水素吸収液 ジエチルジチオカルバミン酸銀0.50gをピリジンに溶かし100mlとする。この液
は遮光した共栓瓶に入れ,冷所に保存する。
7) ひ素標準原液 三酸化二ひ素を微細な粉末とし,105℃で4時間乾燥し,その0.10gを正確に量り,
水酸化ナトリウム溶液 (200g/l) 5mlを加えて溶かす。この液を硫酸 (1+19) で中和し,さらに硫酸
(1+19) 10mlを追加し,新たに煮沸し冷却した水を加えて正確に1 000mlとする。この液1mlは,
三酸化二ひ素 (As2O3) 0.1mgを含む。
8) ひ素標準液 ひ素標準原液10mlを正確に量り,硫酸 (1+19) 10mlを加え,新たに煮沸し冷却した
水を加えて正確に1 000mlとする。この液1mlは三酸化二ひ素 (As2O3) 1μgを含む。使用時に調製
し,共栓瓶に保存する。
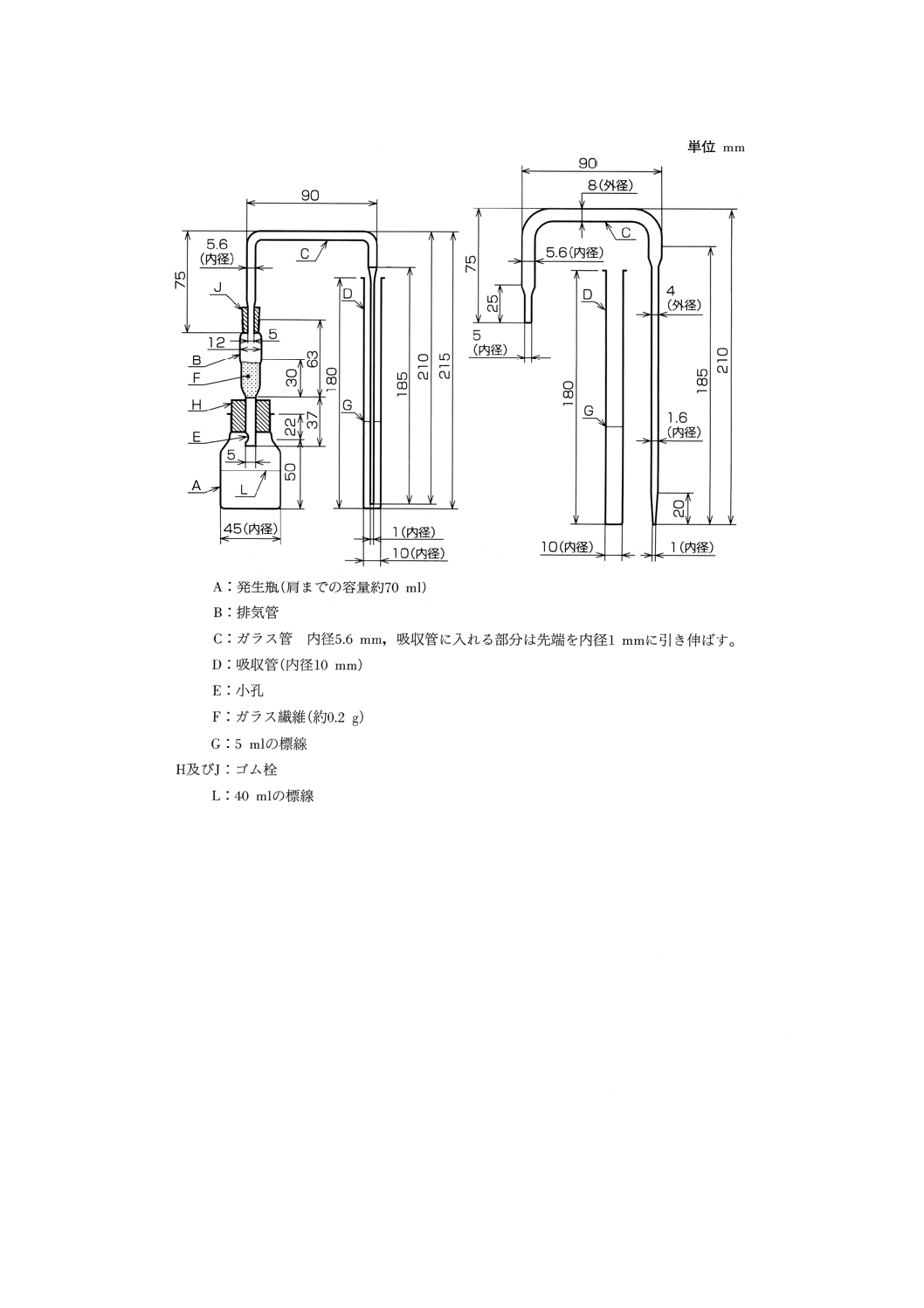
b) 装置 概略は図1による。排気管Bに約30mmの高さにガラス繊維Fを詰め,酢酸鉛試液及び水のな
ど等容量混液で均等に潤した後,下端から弱く吸引して,過量の液を除く。これをゴム栓Hの中心に
垂直に差し込み,Bの下部の小孔Eは下にわずかに突き出るようにして発生瓶Aにつける。Bの上端
にはガラス管Cを垂直に固定したゴム栓Jをつける。Cの排気管側の下端はゴム栓Jの下端と同一平
面とする。
10
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図1 ひ素試験装置の例
c) 操作 附属書1の操作によって得た試験溶液を発生瓶に入れ,ブロムフェノールブルー試液1滴を加
え,アンモニア水,アンモニア試液又は塩酸 (1+3) で中和し,塩酸 (1+1) 5ml及びよう化カリウム
試液5mlを加え,2〜3分間放置した後,酸性塩化第一すず試液5mlを加えて室温で10分間放置した
後,次に水を加えて40mlとし,無ひ素亜鉛2gを加え,直ちにB及びCを連結したゴム栓Hを発生
瓶につける。Cの細管部の端はあらかじめひ化水素吸収液5mlを入れた吸収管Dの底に達するように
入れておく。次に発生瓶は25℃の水中に肩まで浸し,1時間放置する。吸収管をはずし,必要があれ
ばピリジンを加えて5mlとし,検液の吸収液とする。この色を次の標準液と比較する。標準液の調製
は,検液の試験と同時に行い,ひ素標準液2.0mlを量り,発生瓶に入れ塩酸 (1+1) 5ml及びよう化カ
リウム試液5mlを加えて2〜3分間放置した後,酸性塩化第一すず試液5mlを加え,室温で10分間放
置する。以下検液の場合と同様に操作して得た吸収液の呈色を標準色とする。この液は三酸化ひ素
(As2O3) 2μgに対応する。
3.3.8
蒸発残留物
11
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 器具 白金製又は石英製蒸発皿
b) 操作 附属書1の操作によって得た試験溶液200〜300mlをあらかじめ105℃で乾燥した質量既知の白
金製又は石英製蒸発皿にとり,水浴上で蒸発乾固する(15)。次いで105℃で2時間乾燥した後,デシケ
ーター中で放冷する。冷却後,質量を量り,蒸発皿の試験前後の質量の差amgを求める。同時に試験
溶液と同量の空試験溶液を用いて同様に操作し,試験前後の蒸発皿の質量の差bmgを求める。蒸発残
留物の量は次の式によって算出する。
S
b
a
R
000
1
)
(
×
−
=
ここに, R: 蒸発残留物 (μg/ml)
S: 試験溶液の採取量 (ml)
注(15) なす形フラスコ (1 000ml) に試験溶液200〜300mlをとり,ロータリーエバポレーターによって
20〜30mlに濃縮した後,蒸発皿に移してもよい。この場合,新しい溶出溶媒10mlずつで,加温
しながら3回洗浄を行い,洗液もまとめて蒸発皿に移す。
3.3.9
過マンガン酸カリウム消費量
a) 試薬
1) 硫酸 (1+2) 水2容に硫酸1容をかき混ぜながら徐々に加えた後,微紅色を呈するまで過マンガ
ン酸カリウム溶液を加える。
2) しゅう酸ナトリウム溶液 (0.01mol/l) 150〜200℃に1〜1.5時間保った後,硫酸デシケーター中で
放冷したしゅう酸ナトリウム(分析用標準試薬)0.670gを水に溶かして1 000mlとする。遮光した
共栓瓶に保存し,調製後,1か月以内に使用する。
3) 過マンガン酸カリウム溶液 (0.002mol/l) 過マンガン酸カリウム約0.33gを水に溶かして1 000ml
とする。遮光した共栓瓶に保存し,使用に際し,しゅう酸ナトリウム溶液を用いて標定する。標定
は次の方法による。水100mlをフラスコ (300ml) にとり,硫酸 (1+2) 5ml及びここで調製した過マ
ンガン酸カリウム溶液5mlを加えて5分間煮沸する。次いで加熱をやめ,直ちに,しゅう酸ナトリ
ウム溶液10mlを加えて脱色した後,過マンガン酸カリウム溶液を微紅色が消えずに残るまで滴下
する。この液に硫酸 (1+2) 5ml及び過マンガン酸カリウム溶液5mlを加え,5分間煮沸した後,し
ゅう酸ナトリウム溶液10mlを加え,直ちに過マンガン酸カリウム溶液で滴定し,次式によって過
マンガン酸カリウム溶液のファクターを算出する。
a
f
+
=510
ここに,
f: 過マンガン酸カリウム溶液のファクター
a: 過マンガン酸カリウム溶液滴定量 (ml)
b) 器具
1) ビュレット
2) 全量ピペット 5ml及び10ml
3) 三角フラスコ 300ml
c) 操作 三角フラスコ (300ml) に水100ml,硫酸 (1+2) 5ml及び過マンガン酸カリウム溶液5mlを入れ,
5分間煮沸した後,液を捨て,水でよく洗浄する。この三角フラスコに,水を用いて附属書1に従い,
調製した試験溶液100mlを量り採り,硫酸 (1+2) 5ml及び過マンガン酸カリウム溶液10mlを加えて
5分間煮沸する。次いで加熱をやめ,直ちにしゅう酸ナトリウム溶液10mlを加えて脱色した後,過マ
12
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ンガン酸カリウム溶液で微紅色を呈するまで滴定する。別に試験溶液の代わりに空試験溶液100mlを
用いて同様の方法で空試験を行い,次式によって過マンガン酸カリウム消費量を算出する。
f
b
a
M
×
×
×
316
.0
100
000
1
)
(−
=
ここに, M: 過マンガン酸カリウム消費量 (μg/ml)
a: 試験溶液の過マンガン酸カリウム溶液滴定量 (ml)
b: 空試験溶液の過マンガン酸カリウム溶液滴定量 (ml)
f: 過マンガン酸カリウム溶液のファクター
3.3.10 紫外吸収スペクトル
a) 器具及び装置
1) 吸収セル 石英製,層長10mm
2) 光電分光光度計
b) 操作 附属書1に従い,水を用いて調製した試験溶液を吸収セルに入れ,空試験溶液を対照液として
波長220〜350nmにおける吸光度をJIS K 0115によって測定する。
3.4
N-ニトロソ化合物
3.4.1
試験の概要 ゴム製品の製造時に添加される特定の加硫促進剤などが分解すると,各種のアミン化
合物が生成する。これらのアミン化合物がニトロソ化されるとN-ニトロソアミンが生成する。このN-ニ
トロソアミンの一部は発がん性である。この試験は,そのようなゴム製品中の発がん性N-ニトロソアミン
の存在量 (3.4.2) 及び人工だ液による溶出量 (3.4.3) を測定するものである。この試験は,食品用器具及び
容器包装,乳幼児用おもちゃ及び医薬品容器などに用いられるゴム製品に適用できる。この試験で分析対
象とするN-ニトロソアミンは次のとおりとする。
a) N-ニトロソジメチルアミン (NDMA)
b) N-ニトロソジエチルアミン (NDEA)
c) N-ニトロソジブチルアミン (NDBA)
d) N-ニトロソピペリジン (NPIP)
e) N-ニトロソモルホリン (NMOR)
備考 上記の試験項目のほかに,ジアルキル型と環状アルキル型のN-ニトロソアミンもこの試験法で
定量可能である。ただし,N-ニトロソメチルフェニルアミンとN-ニトロソエチルフェニルアミ
ンは熱に不安定なため定量困難である。
また,上記のN-ニトロソ化合物は強い発がん性をもつので,その取り扱いには十分な注意を
払わなければならない。
3.4.2
総N-ニトロソアミン量とN-ニトロソアミン類の分別定量
備考 総N-ニトロソアミン量の測定は,N-ニトロソアミンを臭化水素酸 (HBr) で分解し,遊離する
総一酸化窒素 (NO) 量を熱エネルギー検出器 (Thermal Energy Analyzer:TEA) で測定する。ま
た,個々のN-ニトロソアミンの分別定量は,TEA付きガスクロマトグラフによって行う。
a) 試薬
1) ジクロロメタン(16)
2) 酢酸エチル(16)
3) アセトン(16)
4) 臭化水素酸・酢酸溶液 (15g/l) 臭化水素酸1.5gに酢酸を加えて100mlとする。
13
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5) L-アスコルビン酸パルミチン酸エステル(16)
6) N-ニトロソアミン
N-ニトロソジメチルアミン (NDMA)
N-ニトロソジエチルアミン (NDEA)
N-ニトロソジブチルアミン (NDBA)
N-ニトロソピペリジン (NPIP)
N-ニトロソモルホリン (NMOR)
7) N-ニトロソアミン標準溶液 各N-ニトロソアミン0.050gを,それぞれ100mlの全量フラスコに正
確に量り採り,エタノールに溶かしてそれぞれ100mlとする。これらをN-ニトロソアミンの標準原
液とする。この標準原液からそれぞれ1mlをl00mlの全量フラスコにとり,エタノールで100mlと
し,さらにこの溶液から4mlをとり,エタノールで100mlとしたものをN-ニトロソアミン標準溶液
とする(この溶液はN-ニトロソアミンそれぞれを1ml当たりに0.2μg含む)。
8) 総N-ニトロソアミン量測定用標準溶液 亜硝酸ナトリウム(17)0.69gを水に溶かして100mlとする。
さらにこの溶液から1mlをとり,水で100mlにしたものを総N-ニトロソアミン量測定用標準溶液と
する(この溶液はNOとして1ml当たり100nmolを含む)。
注(16) 使用する前にあらかじめN-ニトロソアミンが含まれていないことを確かめておく。
(17) 乾燥剤を入れたデシケーター中で一昼夜乾燥する。
b) 器具及び装置
1) 共栓付き円筒形クデルナ・ダニシュ (K-D) 濃縮受器 100ml
2) ソックスレー抽出器
3) ジュワー瓶
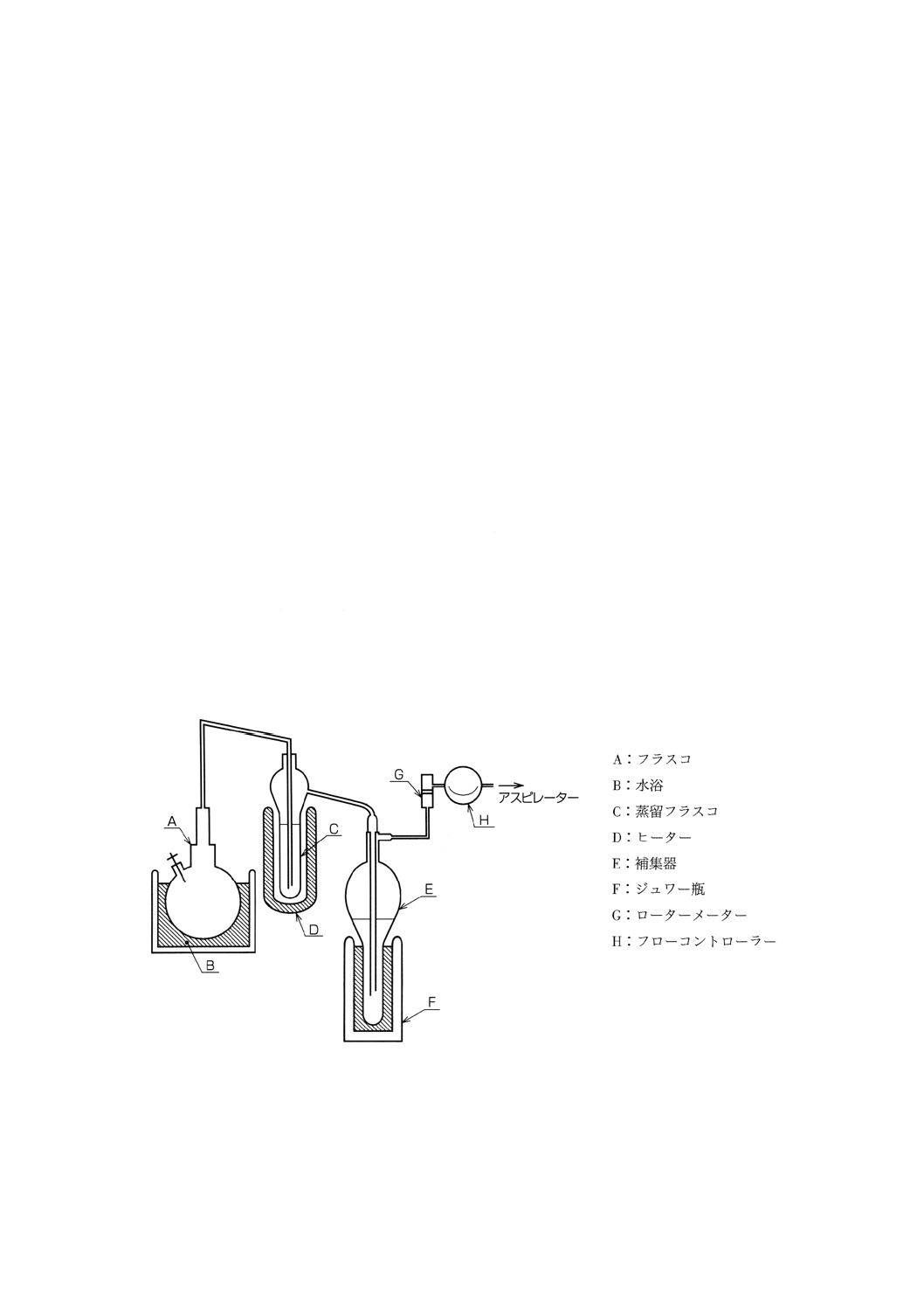
4) 減圧蒸留装置(図2)
図2 減圧蒸留装置
14
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
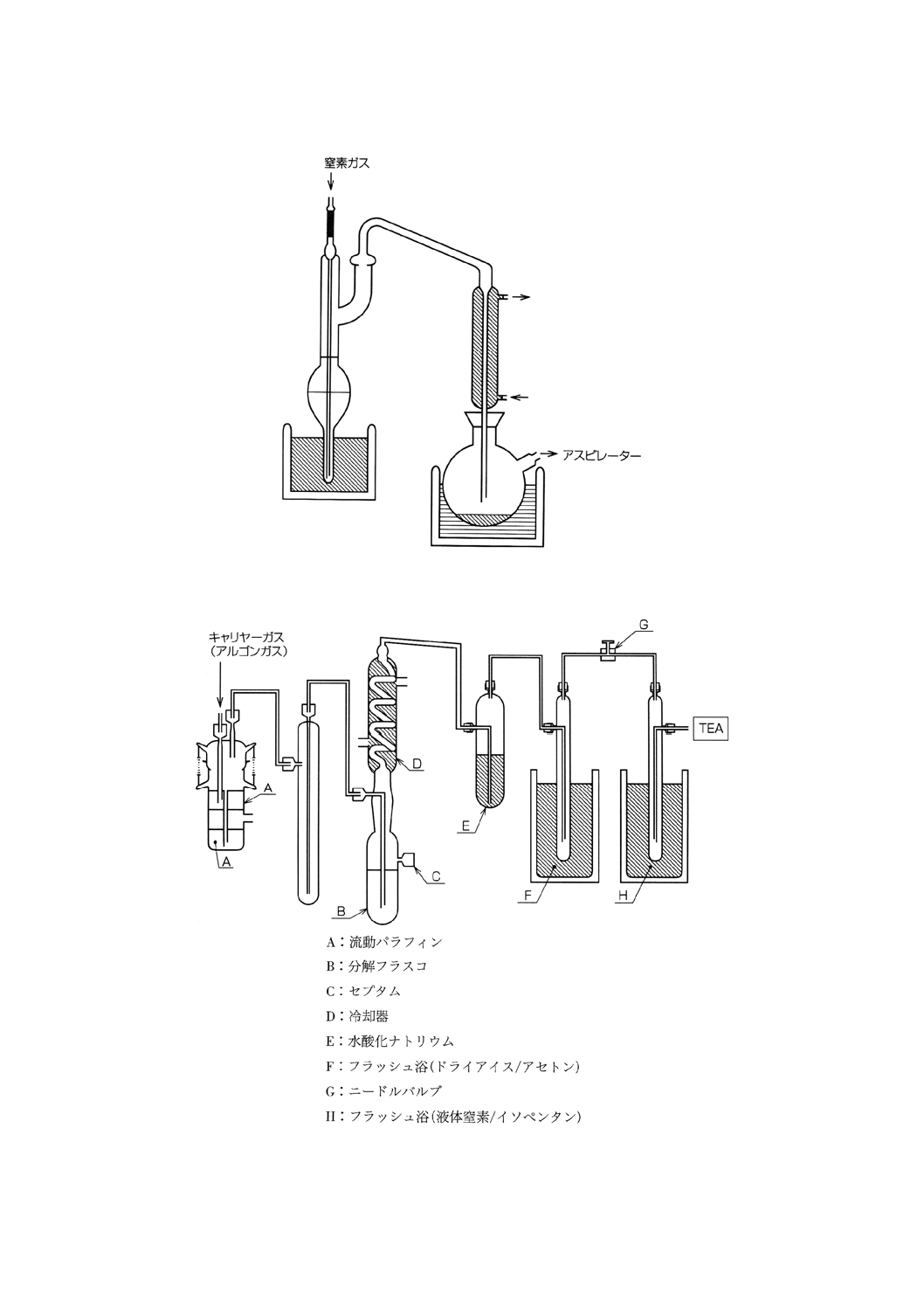
5) K-D濃縮装置(図3)(18)
図3 K-D濃縮装置
6) NO蒸留装置(図4)
図4 NO蒸留装置
15
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) 投込式小形冷却機(19)
8) ハンディー型アスピレーター
9) 振とう機付き恒温水槽
10) TEA付きガスクロマトグラフ (GC-TEA)
注(18) スニーダ分留管を取り外した装置。
(19) この装置の代わりにドライアイス/アセトンのフラッシュ浴でもよい。
c) 試料の調製 この試験に用いるゴム試料は,1〜2mm角程度に細かく切断したものを用いる。
d) 試験溶液の調製
1) 浸せき抽出法 3.4.2 c)で調製した試料5gを共栓付き円筒形K-D濃縮受器にとり,ジクロロメタン
50ml及びL-アスコルビン酸パルミチン酸エステル(20)200mgを加え,密栓した後,40℃の振とう器
付き恒温水槽で,12時間振とう抽出する。抽出後,この円筒形K-D濃縮受器を図2に示した減圧
蒸留装置のCに装着し,減圧蒸留を行う。
2) ソックスレー抽出法 3.4.2 c)で調製した試料5gをソックスレー抽出器用円筒ろ紙を入れた抽出部
に入れ,抽出フラスコにはジクロロメタン50ml及びL-アスコルビン酸パルミチン酸エステル
(20)200mgをそれぞれ加え,抽出フラスコの加熱温度を40℃で12時間還流抽出を行う。フラスコ内
のジクロロメタン抽出液を,共栓付き円筒形K-D濃縮受器に移し,これを図2に示した減圧蒸留装
置のCに装着し,減圧蒸留を行う。
3) 減圧蒸留 図2に示したAのフラスコにジクロロメタン150mlを加え,Bの水浴温度を約10℃,C
の蒸留フラスコの加熱温度を約80℃,Eの補集器(K-D濃縮フラスコ付きK-D濃縮受器)の冷却温
度を−50℃(21),Hの減圧度を約10mmHgにそれぞれ調整し減圧蒸留を1時間行う。
4) 濃縮 蒸留液を捕集した捕集器をK-D濃縮装置に装着し,水浴温度40℃以下で,約5mlまで緩や
かに濃縮する。濃縮終了後,濃縮装置系内を少量のジクロロメタンでK-D濃縮受器内へ洗い込み,
濃縮受器を濃縮フラスコから取り外し,濃縮受器を氷水中で冷却しながら緩やかに窒素ガスを通じ
て1mlまで濃縮(22)し,これを総N-ニトロソアミン量及びその分別定量それぞれの測定用試験溶液
とする。
注(20) N-ニトロソアミンの操作中の生成抑制剤で,この物質の代わりに没食子酸プロピルでもよい。
(21) 冷却用溶媒にメタノールを用いた場合の冷却温度。
(22) N-ニトロソアミンは極めて揮発性が高く濃縮操作中に損失することがあるので,濃縮操作に伴
う回収率を確認する。
e) 総N-ニトロソアミン量の測定 図4に示したNO蒸留装置をTEAに結合させ,Bの分解フラスコに
酢酸エチル5mlを入れ,NO蒸留装置に装着しキャリヤーガス(アルゴンガス)を流し,TEAの減圧
計の目盛が1〜2になるようにFのニードルバルブで調整する。調整後,Bの分解フラスコをNO蒸留
装置から外し,新たに酢酸エチル5mlを分解フラスコに加え,次に総N-ニトロソアミン量測定用標準
溶液100μlを入れる。この分解フラスコをNO蒸留装置に装着し,Cのセプタムから臭化水素酸・酢
酸溶液5mlを注入し,遊離するNOをTEAに導き,NOのTEAクロマトグラムをとり,NOのピーク
高を測定する。これを検量用とする。次に別の分解フラスコに3.4.2 d)で得られた試験溶液0.5mlをと
り,さらに酢酸エチル5mlを加える。この分解フラスコをNO蒸留装置に装着し,Cのセプタムから
酢酸(23)5mlを注入し,発生するNOをTEAに導き,NOのTEAクロマトグラムをとった後,Cのセ
プタムから臭化水素酸・酢酸溶液5mlを注入する。N-ニトロソアミンから遊離するNOをTEAに導き,
NOのTEAクロマトグラムをとり,上記同様NOのピーク高を測定し検量用のピーク高と比較するこ
16
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
とによって総N-ニトロソアミン量をNO量として算出する。
注(23) 試験溶液に溶存する亜硝酸又はNOxなどを酢酸でNOを発生させ,除去する。
f)
N-ニトロソアミン類の分別定量 N-ニトロソアミン標準溶液5μlをGC-TEAに注入し,標準N-ニトロ
ソアミンNDMA,NDEA,NDBA,NPIP及びNMORのガスクロマトグラムをとり,それぞれのN-ニ
トロソアミンのピーク高を測定し検量用とする。次に,3.4.2 d)で得られた試験溶液5μlを注入し,N-
ニトロソアミンのガスクロマトグラムをとり,上記同様N-ニトロソアミンのピーク高を測定し,標準
N-ニトロソアミンの検量用のピーク高と比較してN-ニトロソアミン量を測定する。
参考 次のGC-TEAの条件が推奨される。
GC部
カラム:5%PEG20M/ChromosorbW(粒径177〜250μm)を内径3mm,長さ3mのガラス管に充
てんしたもの
注入口及びカラム温度:170℃
キャリヤーガス:アルゴン
流量:50ml/min
TEA部
インターフェース温度:200℃
パイロライザー温度:550℃
コールドトラップ温度:−160℃(液体窒素/イソペンタン)
3.4.3
N-ニトロソアミン及びN-ニトロソ化可能物質の溶出量
備考 ゴム製品から亜硝酸を含有する人工だ液へ溶出するN-ニトロソアミン及びN-ニトロソ化可能
物質(アミン化合物)を定量する方法である。得られた溶出液の半量を用いて分析を行って得
られたN-ニトロソアミン量を“溶出N-ニトロソアミン量”と定義し,残りの半量を酸性下で
N-ニトロソ化後に分析を行って得られたN-ニトロソアミン量を“N-ニトロソ化可能物質”と定
義する。すなわち,この定量法は,だ液に溶出するN-ニトロソ化合物,及びだ液に溶出した2
級アミンとだ液中に含まれる亜硝酸イオンが胃酸酸性下で反応して生成するN-ニトロソ化合
物の総量を定量するものである。
a) 試薬
1) ジクロロメタン(16) 3.4.2 a)1)による。
2) 人工だ液 炭酸水素ナトリウム4.2g,塩化ナトリウム0.5g,炭酸カリウム0.2g,硝酸ナトリウム0.005g
及び亜硝酸ナトリウム0.02gを1 000mlの水に溶かしたもの。
3) N-ニトロンアミン 3.4.2 a)6)による。
4) N-ニトロソアミン標準溶液 3.4.2 a)7)による。
b) 器具及び装置
1) 共栓付き三角フラスコ 100ml
2) 分液漏斗 100ml
3) K-D濃縮装置(図3参照)
4) 振とう機付き恒温水槽
5) GC-TEA
c) 試料の調製方法 この試験に用いる試料は3.4.2 c)による。
d) 試験溶液の調製 試料10gを共栓付き三角フラスコにとり,人工だ液25mlを加え,密栓した後,40℃
17
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の振とう器付き恒温水槽で24時間振とうし,溶出する。溶出後,ろ紙でろ過し,さらに少量の人工だ
液で三角フラスコ,ろ紙及び残さを洗い,洗液を合わせ人工だ液で50mlに定容する。この溶出液の
半量25mlを分液漏斗にとり,1mol/lの塩酸でpH7.0に調整した後,ジクロロメタン25mlで2回振と
う抽出する。ジクロロメタン層を合わせ,無水硫酸ナトリウムで脱水した後,K-D濃縮装置(図3)
で約5mlに濃縮し,さらにK-D濃縮装置内を少量のジクロロメタンでK-D濃縮受器に洗い込み,氷
水中で緩やかに窒素ガスを通じて1mlまで濃縮する(22)。これを溶出N-ニトロソアミン測定用試験溶
液とする。溶出液の残り25mlを共栓付き三角フラスコに取り,1mol/lの塩酸でpH1.0に調整し,室温
で30分間N-ニトロソ化反応を行う。反応終了後1mol/lの水酸化ナトリウムでpH7.0に調整した後,
分液漏斗に移し,ジクロロメタン25mlで2回振とう抽出する。ジクロロメタン層を合わせ,無水硫
酸ナトリウムで脱水した後,K-D濃縮装置(図3)で約5mlまで濃縮し,さらにK-D濃縮装置系内を
少量のジクロロメタンでK-D濃縮受器中に洗い込み,氷水中で窒素ガスを通じて1mlまで濃縮する(22)。
これをN-ニトロソ化可能物質(N-ニトロソアミンとして)の測定用試験溶液とする。
e) 溶出N-ニトロソアミンの測定 N-ニトロソアミン標準溶液5μlをGC-TEAに注入し,標準N-ニトロ
ソアミンNDMA,NDEA,NDBA,NPIP及びNMORのガスクロマトグラムをとり,それぞれのN-ニ
トロソアミンのピーク高を測定し検量用とする。次に溶出N-ニトロソアミン測定用試験溶液5μlを注
入し,N-ニトロソアミンのガスクロマトグラムをとり,上記と同様にピーク高を測定し,標準N-ニト
ロソアミンの検量用のピーク高からN-ニトロソアミン量を算出する。
参考 次のGC-TEA条件が推奨される。
GC部 3.4.2 f)による。
TEA部 3.4.2 f)による。
f)
溶出N-ニトロソ化可能物質の測定 溶出N-ニトロソ化可能物質測定用試験溶液を3.4.3 e)によってN-
ニトロソアミンを測定し,得られたN-ニトロソアミンを溶出N-ニトロソ化可能物質とする。
3.5
細胞毒性及び感作性物質
3.5.1
試験の概要 ゴム製品で細胞毒性や接触感作性が問題となることがある。これらの作用はゴム製品
中に残留する加硫促進剤や老化防止剤が原因であることが多い。この試験法中の3.5.2及び3.5.3はゴム製
品の細胞毒性及び接触感作性の強さを評価するためのものであり,すべての種類のゴムに適用できる。3.5.4
は現時点でその原因となると考えられているゴム製品中に残留する加硫促進剤及び老化防止剤の分析方法
である。
3.5.2
細胞毒性試験
a) 細胞 L929細胞又はV79細胞を用いる(24)。
注(24) 上記細胞のほか,ヒトを含むほ乳類の細胞株を用いてもよい。その際は明らかなコロニーを形
成し,再現性のある結果が出ることをあらかじめ確認しておくこと。細胞の形態や増殖速度な
どを常時よく観察し,できるだけ同一性質の細胞を用いるよう心がける。
b) 培地及び試薬
1) 培地(25) 高圧蒸気滅菌用イーグルMEM粉末培地9.4gを水に溶かして1 000mlとした後,121℃で
15〜20分間高圧蒸気滅菌する。室温まで冷却した後,炭酸水素ナトリウム溶液 (75g/l) 16.6〜29.3ml
を加えて冷蔵庫で保存する。使用時,グルタミン溶液 (29.2g/l) を培地1 000mlに対して10ml,さ
らにウシ胎児血清(26)を培地の10v/v%になるように加える。
2) 炭酸水素ナトリウム溶液 (75g/l) 炭酸水素ナトリウム7.5gを水に溶かして100mlとし,密栓して
121℃で15〜20分間高圧蒸気滅菌する(27)。
18
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) グルタミン溶液 (29.2g/l) L-グルタミン2.92gを水に溶かして100mlとし,孔径0.22μm以下のメ
ンブランフィルターを用いてろ過して滅菌する(27)。
4) りん酸塩緩衝生理食塩水 塩化カリウム0.2g,りん酸二水素カリウム0.2g,塩化ナトリウム8.0g及
びりん酸水素二ナトリウム(無水)1.15gに徐々に水を加えて溶解し全量を1 000mlとし,121℃で
15〜20分間高圧蒸気滅菌する(27)。
5) トリプシン溶液 トリプシン0.5g,エチレンジアミン四酢酸四ナトリウム02g及び塩化ナトリウム
0.85gを水に溶かし1 000mlとし,ろ過滅菌する(27)。
6) ホルムアルデヒド溶液 ホルムアルデヒド液を水で10倍に希釈する。
7) 希釈液 りん酸二水素カリウム4.54gとりん酸水素二ナトリウム(無水)4.75gを別々に水に溶解し,
それぞれ全量を500mlとする。これらの溶液を1 : 1の割合で混合し,塩酸又は水酸化ナトリウム溶
液でpH6.8に調整する。
8) ギムザ液 ギムザ粉末(メチレンブルー,アズールB,エオシンYの混合体)1gを乳鉢にとり,少
量のグリセリンを加え,よく練る。全量66mlとなるようにグリセリンを加え,ビーカー又は三角
フラスコに移し,55〜60℃で2時間加温して溶かす。冷後,メタノール66mlを加え,ろ紙でろ過
する(28)。使用時,10倍に希釈した希釈液を用いて2.5v/v%に希釈し,ろ紙でろ過して使用する。
注(25) 培地はその処方に従って実験室で調製してもよいが,調製された粉末培地又は滅菌済の液体培
地が市販されているのでこれを用いることができる。粉末培地は,使用説明書に従って指定量
をとり,指定の方法で滅菌して用いる。炭酸水素ナトリウム溶液の添加量は,5%炭酸ガス下,
37℃で培地のpHが7.1〜7.4になるように加減する。
(26) 56℃で30分間加温し氷冷したものを加える。血清の中には細胞増殖促進性が極めて低いものも
あるので,対象細胞の培養に適したロットを選ぶことが重要である。
(27) 調製された市販品を用いてもよい。
(28) 市販品を用いてもよい。希釈は希釈液にギムザ液を加える。逆にすると沈殿物を生じる。いっ
たん希釈した後は,時間の経過にともないアズールBとエオシンYが結合し染色性が悪くなる。
希釈後は5〜6時間が使用の限度である。
c) 器具及び装置
1) ピペット パスツールピペット,メスピペット,チップ式微量ピペット(29)
2) スクリューキャップ式ガラス瓶 50〜1 000ml(30)
3) プラスチック製遠心管 15ml及び50ml(30)
4) プラスチック製培養フラスコ 25cm2(30)
5) プラスチック製培養プレート (24穴)(30)
6) セラムチューブ(30)
7) ろ過滅菌装置 孔径0.22μm以下の大きさのメンブランフィルター及びフィルターホルダー(29)
8) 血球計算盤
9) 炭酸ガス培養器 器内を37℃,炭酸ガス濃度5%に設定できるもの。
10) 顕微鏡 倒立顕微鏡及び実体顕微鏡
11) クリーンベンチ
12) 高圧蒸気滅菌器
13) 乾熱滅菌器
14) 超純水製造装置
19
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
15) 遠心機
16) 細胞凍結保存機器 超低温槽 (−70〜−80℃),液体窒素タンク
注(29) 器具類は滅菌しようとするものの性質を損なわないように,それぞれに合った滅菌法を選ぶ。
高圧蒸気滅菌は121℃で15〜20分間,乾熱滅菌は160〜170℃で2〜4時間加熱する。
(30) 市販の細胞培養用で滅菌済のものを用いる。
d) 操作
1) 試料の採取方法 試料を2×15mm角程度に細かく切断し,水で洗い,風乾したものから1.0gを正
しく量り採る。
2) 試験溶液の調製 試料をスクリューキャップ式ガラス瓶又はプラスチック製遠心管に入れ,軽く栓
をした後清浄なアルミニウムはくで覆い,121℃で20分間高圧蒸気滅菌する。室温まで冷却後,試
料1.0gに対して培地10mlを加えて,軽く栓をした後,炭酸ガス培養器中に24時間静置して抽出す
る。抽出液はスクリューキャップ式ガラス瓶又はプラスチック製遠心管に移し,これを100%試料
溶液とする。さらに培地を用いて,公比2の割合で希釈し,種々の濃度の試験溶液を調製する。
3) 細胞浮遊液の調製 凍結細胞が入ったセラムチューブを37℃の温水につけ急速に融解させ,直ちに
培地5mlを入れた15mlプラスチック製遠心管に内容物を移し,毎分1 000回転で5分間遠心し,上
清を捨てる。これに新しい培地10mlを加え,細胞を分散させ,プラスチック製培養フラスコに分
注して,炭酸ガス培養器内で2〜3日間培養する。細胞が十分に増殖したら(対数増殖期にあるうち
に),培地をパスツールピペットで吸引して除き,りん酸塩緩衝生理食塩液2mlを入れ,プラスチッ
ク製培養フラスコを2〜3回傾けて細胞を軽く洗った後,りん酸塩緩衝生理食塩液を捨てる。トリプ
シン溶液1mlを加え,炭酸ガス培養器中で1〜2分間静置する(31)。培地0.5〜1mlを加え,パスツー
ルピペットで静かにピペッティングして(32),細胞をばらばらにし,単細胞の浮遊液とする。この浮
遊液を15mlプラスチック製遠心管に移し,りん酸塩緩衝生理食塩液5mlを加え,毎分1 000回転で
2〜5分間遠心して上清を捨てる。新しいりん酸塩緩衝生理食塩液5〜10mlを加え,ピペッティング
した後,再度遠心し上清を捨てる。培地を適当量 (1〜5ml) 加え,ピペッティングして均一な細胞
浮遊液をつくる。細胞浮遊液の一部 (10μl) をとり,血球計算盤で細胞数を計数した後,培地で希釈
して100個/mlの細胞浮遊液を調製する。
細胞の保存用には2×106個/mlの細胞浮遊液を調製し,これをあらかじめ調製,保冷したジメ
チルスルホキシドを20%含有する培地と1 : 1の割合で混合し,セラムチューブに1mlずつ分注する。
冷凍庫に2〜3時間放置後,超低温槽又は液体窒素容器に移し保存する。
4) コロニー数の算定 100個/mlの濃度の細胞浮遊液0.5mlを,プラスチック製培養プレートの各穴
に分注し,炭酸ガス培養器中で4〜6時間静置して,細胞をプラスチック製培養プレートの底面に接
着させる。各穴から培地を除いた後,種々の濃度の試験溶液0.5mlを加える。各試験溶液について
4穴を使用する。6〜9日間培養後(33),各穴から上清を捨て,ホルムアルデヒド溶液約0.5mlを加え,
約30分間放置して細胞を固定する。上清を除いて,ギムザ液約0.5mlを加えて約30分間染色する
(34)。コロニーがよく染色されたのを確認した後,染色液を捨て,水道水で余分なギムザ液を洗い落
として乾燥する。各穴について,実体顕微鏡で観察して50個以上の細胞集団からなるものをコロニ
ーとして数える。培地のみで培養した穴のコロニー数を対照として,試験溶液でのコロニー数と比
較する(35)。
注(31) 細胞が底面からはく(剥)離してくる時間は,細胞の種類によって異なる。あまり長い時間の
トリプシン処理は細胞障害をもたらすので,倒立顕微鏡ではく離状態を観察しながら,なるべ
20
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
く短時間にとどめるようにする。
(32) ピペットによって溶液を吸入噴出させる操作のことで,この操作を繰り返すことによって細胞
を十分にばらばらにして単細胞浮遊液とする。
(33) L929細胞は7〜9日間,V79細胞は6〜7日間培養する。
(34) ギムザ染色の時間はあくまで目安であり,ギムザ液の濃度が高いときは短く,低いときは長く
なければならない。
(35) 各濃度の試験溶液処理群のコロニー数を培地処理群と比べたときの百分率(コロニー形成率)
を縦軸に,横軸を試験溶液の濃度 (%) の対数とするグラフに表す。コロニー形成率が50%を示
す試験溶液の濃度を,試料の細胞毒性の強度として表すこともできる。
備考 標準となる物質(例えばジエチルジチオカルバミン酸亜鉛など)又は対照材料の抽出液につい
て,同時に試験して相対的な細胞毒性強度を確認することが望ましい。対照材料としては,“医
療用具及び医用材料の基礎的な生物学的試験のガイドライン”に規定した標準材料がある。
3.5.3
感作性試験
a) 動物 雌性モルモット(36)
注(36) 動物は実験開始時に体重が300〜400gで,妊娠していない未経産の健康な若齢白色モルモット
(通常1〜2か月齢)を使用する。
b) 試薬
1) フロイント完全アジュバントW/O型乳化物(FCA-W/O型乳化物) フロイント完全アジュバント
(FCA) に等量の注射用蒸留水を加えて混合し乳化させたもの(37)。
2) ラウリル硫酸ナトリウム軟こう (10%) (SLS軟こう) 白色ワセリン90gを加温して溶かし,そ
の中にラウリル硫酸ナトリウム10gを加えて均質になるまで練り合わせたもの。
3) 陽性対照物質(38) 2, 4-ジニトロクロロベンゼン (DNCB)
注(37) 最終的にW/O型とするためには,アジュバント側に注射用蒸留水を少量ずつ加えていくとよい。
(38) 陽性対照物質は,実験手技の感度及び信頼性をチェックするために必要である。DNCB以外に
も,α-ヘキシルシンナムアルデヒド,2-メルカプトベンゾチアゾール,4-アミノ安息香酸エチル
などが用いられている。
c) 器具
1) 電気バリカン
2) 電気かみそり
3) ろ紙
4) 不浸透性ばん(絆)創こう(膏)
5) 伸縮性包帯
6) 粘着性テープ
d) 操作
1) 試料の採取方法 試験に用いるゴム製品を2×15mm角程度に細かく切断し,水で洗い,風乾した
ものから2.0gを量り採り,これを試料採取量とする。
2) 試験試料の調製法 試料を50mlの遠心管にとり,アセトン・クロロホルム (1 : 1) 20mlを加え,振
とう機で室温下,30分間振とうする。さらに,同様の操作を4回繰り返し,全抽出液を合わせ,ろ
紙でろ過する。50℃の水浴上でロータリーエバポレーターを用いて溶媒を留去して残留物を得る
(3.5.4を参照)。
21
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なお,得られた残留物が試験に必要な量に足りない場合は,新たな試料について同様の操作を繰
り返して必要な量の残留物を得る。試料量及び残留物量を精密に量り記録する。この残留物を試験
試料とする。
3) 試験試料溶液の調製法 試験試料はオリーブ油に溶解,又は均一に分散させる。これらに溶解せず,
アセトンに溶解する場合は,アセトンに溶解させた後,その溶液をオリーブ油に混ぜながらアセト
ンを揮散させる方法が推奨される。濃度は10%とする。ただし,10%濃度で刺激性が認められる場
合には,5%以下の濃度でもよいがその理由を記録に残すこと。
4) 感作性試験(マキシミゼーションテスト法) 実験群は試験試料群,溶媒対照群及び陽性対照群の
3群を設ける。動物は,試験試料群には1群10匹,その他の群には1群5匹のモルモットを用いる。
4.1)
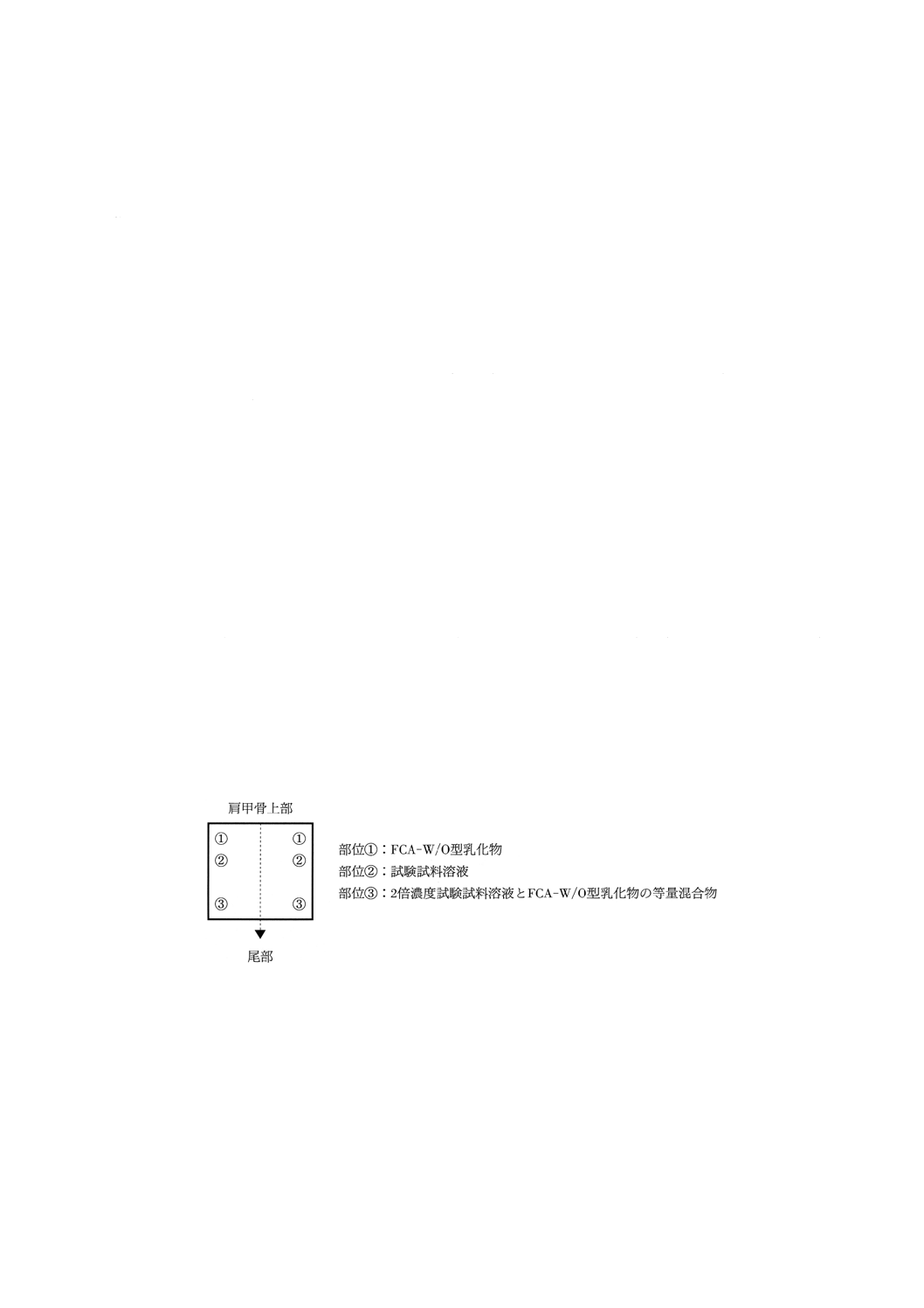
感作 モルモットの肩甲骨上,4×6cmの面積を電気バリカンで毛を苅り,さらに電気かみそりで毛
をそる。皮内注射による第1回目の感作は図5に示すように,2×4cmの広さのところに,FCA-W/O
型乳化物,試験試料溶液,2倍濃度試験試料溶液とFCA-W/O型乳化物の等量混合物を左右対称に
0.1mlずつ皮内注射する。
溶媒対照群(39)及び陽性対照群(40)についても同様の操作を行う。
第1回目感作(皮内注射)終了後1週間目に,塗布による第2回目感作を行う。肩甲骨上の毛を
再びそり,試験試料を塗布する(41)。試験試料は,ワセリンと混合するか,適切な溶媒に溶かし,2
×4cmのろ紙上に塗る。不浸透性絆創膏の上に試験試料を塗ったろ紙を置き,肩甲骨上に48時間し
っかりと閉そくパッチする。溶媒対照群及び陽性対照群についても同様の操作を行う。
4.2)
じゃっ(惹)起 第2回目感作終了後[48時間閉そく(塞)パッチ除去後]2週間目に,背部又は
側腹部を電気バリカンで毛を苅り,さらに電気カミソリで毛をそる。試験試料は,ワセリンと混合
するか,アセトンなどの適当な溶媒に溶かし(42),段階希釈した試験試料をろ紙上に塗り(約0.1g
又は0.1ml),不浸透性絆創膏の上に試験試料を塗ったろ紙を置き,背部又は側腹部に24時間しっ
かりと閉塞パッチする。
なお,アセトンに溶解する場合は開放で塗布する。溶媒対照群及び陽性対照群についても同様の
操作を行う。
図5 皮内注射部位
4.3)
評価 閉そくパッチの場合は,パッチ除去後24及び48時間目に,開放塗布の場合には24,48及び
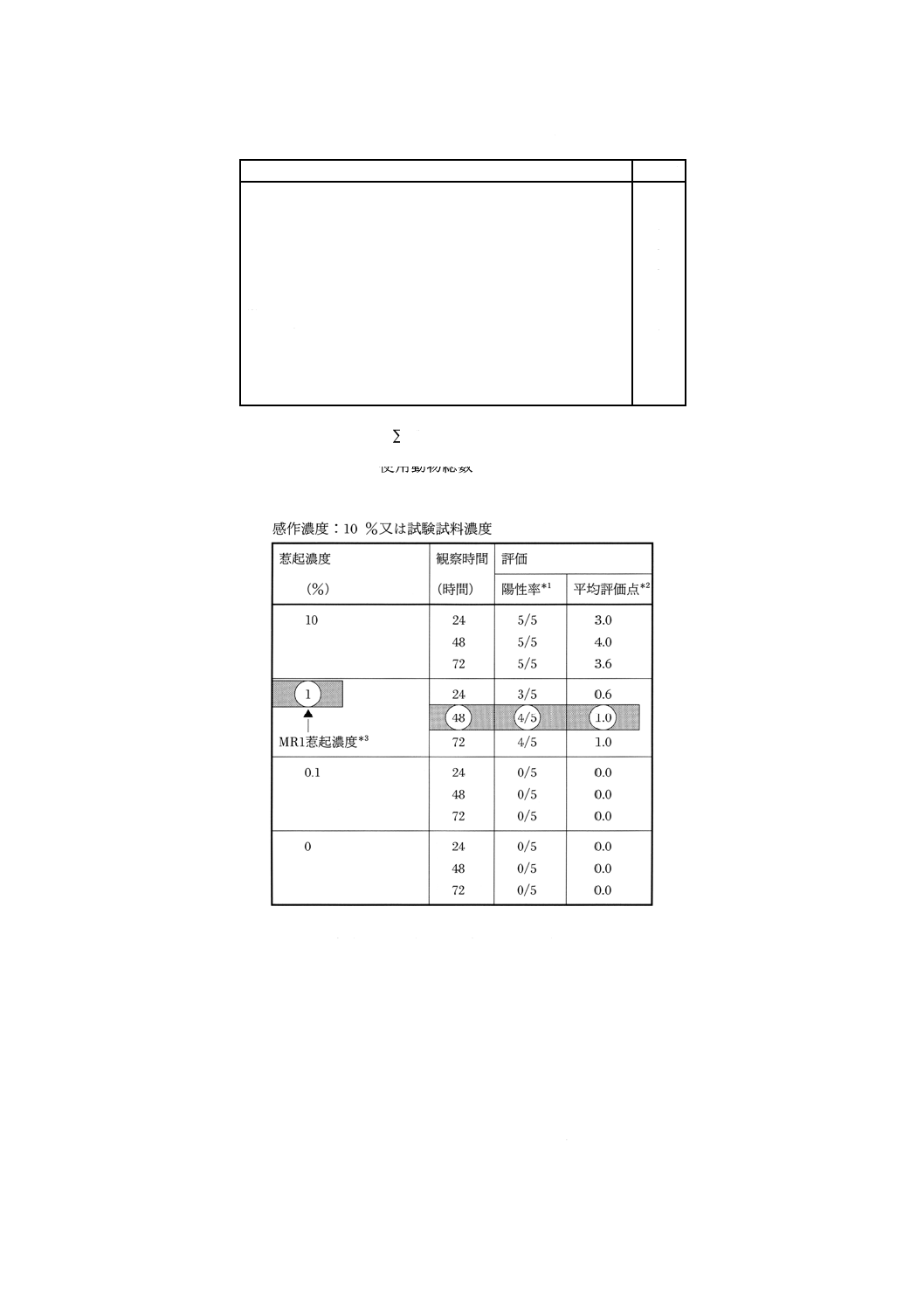
72時間目に,表1の基準に従って皮膚反応を判定し,陽性率及び平均評価点を算出する。さらに,
48時間目における総括表(表2参照)を作成し,平均評価点がおおよそ1.0を示す最も低いじゃっ
起濃度(MR1じゃっ起濃度)を求める。
22
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1 皮膚反応の評価[紅はん(斑)及び浮しゅ(腫)]
皮膚反応
スコア
(1) 紅斑形成
紅はんなし
0
ごく軽度の紅はん
1
明らかな紅はん
2
中〜強度の紅はん
3
反応が深く,わずかにか(痂)皮をつけた深紅色紅はん
4
(2) 浮しゅ形成
浮しゅなし
0
軽度の浮しゅ
1
中程度の浮しゅ
2
投与域を出る反応で,1mm以上にもなる強い浮しゅ
3
平均評価点=使用動物総数
+)]
2(
)1
[(
1
n∑
表2 試験結果の総括表(例)
*1
陽性動物数/当該群の使用動物総数
*2
当該群における個々の評価点の総点/使用動物総数
*3
平均評価点がほぼ1.0を示す最も低い惹起濃度
注(39) 部位①にFCA-W/O型乳化物を,部位②に試験試料溶液の調製に用いた溶媒を,③には溶媒と
FCA-W/O型乳化物の等量混合物をそれぞれ皮内注射する。溶媒としては,植物油(オリーブ油,
コーン油),注射用蒸留水,生理食塩液,流動パラフィン,プロピレングリコール,アセトンな
どが用いられる。
(40) 部位①にFCA-W/O型乳化物を,部位②に0.05〜0.1%のDNCB溶液を,部位③には2倍濃度DNCB
溶液とFCA-W/O型乳化物の等量混合物をそれぞれ皮内注射する。
(41) 試験試料が無刺激性の場合には,塗布24時間前にSLS軟こうを塗り,軽度の炎症を生じさせ
る。この操作によって,感作が増強される。
23
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(42) 植物油(オリーブ油,綿実油,ゴマ油など)は,皮膚刺激性があるのでじゃっ起塗布時の溶媒
として不適当である。じゃっ起濃度は,対照群の動物で刺激性を示さない最高濃度から段階的
に希釈して,例えば,10,1,0.1,0.01及び0.001%のような希釈系列を用いる。
備考 感作濃度を5%として試験を行う場合,部位②には,5%の試験試料溶液を皮内注射する。部位
③には,試験試料が水溶性のときは,注射用蒸留水で最終濃度の倍の10%の試験試料溶液を作
り,等量のFCAと乳化して皮内注射する。また,油溶性のときは,FCAに10%になるよう溶
解し,等量の注射用蒸留水と乳化して皮内注射する。
e) 結果の報告 成績は,各観察時期における個々の動物の皮膚反応がはっきりわかるように,表などに
まとめることが望ましい。試験結果を報告する際には次の事項を含めるようにする。
1) 試料採取量及び抽出残留物の質量
2) 用いたモルモットの系統
3) 使用動物数/週齢/性
4) 実験開始時及び終了時の個別体重
5) 実験期間中の動物の一般状態
6) 結果の評価及び考察
参考 例えば,次の器具や材料がある。
1) 電気バリカン:小動物用電気バリカン(スライブモデル900,刃先#40,夏目製作所)
2) 電気カミソリ:ブラウンシェーバー(Braun Dual 500 AC/RC,一枚刃,ブラウンジャパン
社)
3) ろ紙:No.2
4) 不浸透性ばん創こう:パッチテスト用ばん創こう(鳥居薬品)
5) 伸縮性包帯:シルキーテックス(東京衛材研究所)
6) 粘着性テープ:サージカルテープ(3M社)
3.5.4
老化防止剤及び加硫促進剤の分析
a) 試薬
1) ゴム添加剤 分析対象とする老化防止剤,チウラム系,チオウレア系,ジチオカルバメート (DTC)
系,メルカプトベンゾチアゾール (MBT) 系加硫促進剤並びにアミン類について,化合物名及び略
称をグループごとに附属書2に示す。
2) ろ紙
3) 分液ろ紙 強はっ水性を有し,水相を保持し,有機溶媒相を通過させるろ紙で,分液漏斗に代わり,
液相を分離できるもの。
4) 液体クロマトグラフィー用フィルター 保留粒子径0.45μmのもの。
5) カラムクロマトグラフィー用シリカゲル 粒径62〜210μmのもの。
b) 器具及び装置
1) 振とう機
2) ロータリーエバポレーター
3) 活栓付きクロマトグラフィー用ガラス管 (内径10mm)
4) ガスクロマトグラフ (GC) 水素炎イオン化検出器 (FID) ,窒素−りん検出器 (NPD) 又は熱イオ
ン化検出器 (FTD) 付き。
5) ガスクロマトグラフ/質量分析計 (GC-MS) データ処理システム付き。
24
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6) 高速液体クロマトグラフ (HPLC) 波長可変型紫外部可視部吸収検出器又はフォトダイオードア
レイ紫外部可視部吸収検出器付き。
c) 試料溶液の調製 細切したゴム製品2.0gを50mlの遠心管に正確に量り採り,アセトン・クロロホル
ム (1 : 1) (A-C混液)20mlを加えて,振とう機で室温下,30分間振とうし,抽出液を傾斜して分取
する。さらに,4回同様の操作を繰り返し,全抽出液を合わせ,ろ紙でろ過する。ろ液を合わせた後,
全量にA-C混液を加えて正確に100mlとする。その抽出液25mlずつを①老化防止剤,②チウラム系,
MBT系及びチオウレア系加硫促進剤,③DTC系加硫促進剤及び④アミン類の分析用試料溶液とする。
d) 操作
1) 老化防止剤 試料溶液25mlについて,50℃の水浴上でロータリーエバポレーターを用いて濃縮し,
さらに窒素を吹きつけて溶媒を完全に留去する。カラムクロマトグラフィー用シリカゲル5gをヘキ
サンを用いて湿式法によって充てんして調製したカラムに得られた抽出物を吸着させ,ヘキサン,
次いでエーテル・ヘキサン (5 : 95) ,エーテル・ヘキサン (10 : 90) ,エーテル・ヘキサン (50 : 50)
及びメタノール各50mlずつで溶離し,五つの分画を得る。各分画について,50℃の水浴上でロー
タリーエバポレーターを用いて溶媒を留去後,残留物をジクロロメタン0.5mlに溶かし,GC-FID,
GC-MS用の試験溶液とする。老化防止剤の定性分析は次に示すGC-FID及びGC-MS条件によって,
老化防止剤の標品と比較して行う。また,定量分析はGC-FIDによって,絶対検量線法を用いて行
う。
GC-FID条件
カラム:溶融シリカキャピラリーカラム(液相50%メチルシリコーン+50%フェニルシリコーン,
内径0.53mm,長さ15m,膜厚1.0μm)
カラム温度:150〜275℃(16℃/min,昇温)
注入口及び検出器温度:280℃
キャリヤーガス及び流量:窒素,10ml/min
検出器:FID
GC-MS条件
カラム:溶融シリカキャピラリーカラム(液相50%メチルシリコーン+50%フェニルシリコーン,
内径0.53mm,長さ15m,膜厚1.0μm)
カラム温度:150〜275℃(16℃/min,昇温)
注入口温度:250℃
キャリヤーガス及び流量:ヘリウム,20ml/min
電子衝撃イオン化 (EI) 法
イオン化電圧:70eV
イオン化電流:300μA
イオン加速電圧:3kV
イオン源温度:250℃
参考 老化防止剤分析用のGCカラムとしては,DB-17(J&W社),HP-50(Hewlett Packard社),CPB-5
(島津製作所),Thikotes MPS-50(Quadrex社)などがある。
2) チウラム系加硫促進剤 試料溶液25mlを,50℃の水浴上でロータリーエバポレーターを用いて濃
縮し,さらに空気を吹きつけて溶媒を完全に留去する。残留物をジクロロメタンに溶かし,液体ク
ロマトグラフィー用フィルターを通過させた後,ジクロロメタンを加えて全量を正確に1mlにする。
25
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定量分析は,その2.5μlを用い,次に示すHPLC条件を用い,ピーク高による絶対検量線法によっ
て行う。
HPLC条件
カラム:オクタデシルシリル化シリカゲルカラム(粒径5μm,内径4.6mm,長さ15cm)
移動相:メタノール・水 (95 : 5)
流速1.0ml/min
検出器:波長可変型UV-VIS検出器又はフォトダイオードアレイUV-VIS検出器 (254nm)
参考 チウラム系加硫促進剤分析用のHPLCカラムとしては,SenshupakODS-1151-N(センシュー科
学),Capcell Pak C18(資生堂),Nucleosil 5C18(Nagel社),LiChrosorb RP-18(Merck社)など
がある。
3) チオウレア系加硫促進剤 2)で調製した試験溶液2.5μlを用いて,次に示すHPLC条件によって分析
する。定量はピーク高による絶対検量線法によって行う。
HPLC条件
カラム:シリカゲルカラム(粒径5μm,内径4.6mm,長さ15cm)
移動相:クロロホルム
流速:1.0ml/min
検出器:波長可変型UV-VIS検出器又はフォトダイオードアレイUV-VIS検出器 (254nm)
参考 チオウレア系加硫促進剤分析用のHPLCカラムとしては,Senshupak Silica-1151-N(センシュー
科学),Capcell Pakシリカ(資生堂),Nucleosil Si(Nagel社),LiChrosorb Si-60(Merck社)な
どがある。
4) MBT系加硫促進剤
4.1) MBT系加硫促進剤の種類の推定 CBS及びMMBTは加硫工程において分解してアミンとしてCHA,
MORを生成することから,CHA又はMORを確認することによって,抽出液中にCBS又はMMBT
が存在していることを推定できる。すなわち,2)で調製した試験溶液25mlに,塩酸 (1+6) 5mlを
加え,5分間振とうしてアミン類を水層に抽出し,傾斜して分取する。この操作を3回繰り返す。
水層を合わせ,炭酸水素ナトリウムの粉末を加えて中和後,水を加えて全量を正確に20mlとする。
アミン類の定性分析は,次に示すGC-NPD又はGC-FTD条件によって,アミン類の標品と比較して
行う。また,定量分析はピーク高さによる絶対検量線法によって行う。
GC-NPD,GC-FTD条件
カラム:アミン分析用カラム(粒径149〜177μm,内径2mm,長さ1.2m,ガラスカラム)
カラム温度:80℃ (4min),80〜210℃(32℃/min,昇温)
注入口及び検出器温度:220℃
キャリヤーガス及び流量:窒素,10ml/min
検出器:NPD又はFTD
参考 アミン類分析用のGCカラムとしては,Amipack 124,Amipack 141,Unisole 10T+KOH(いず
れもジーエルサイエンス)などがある。
4.2)
定性及び定量分析 2)で調製した試験溶液2.5μlを用いて,次に示すHPLC条件1又は2によって分
析する。定量はピーク高による絶対検量線法によって行う。
HPLC条件1
カラム:オクタデシルシリル化シリカゲルカラム(粒径5μm,内径4.6mm,長さ15cm)
26
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
移動相:メタノール・水 (95 : 5)
流速:1.0ml/min
検出器:波長可変型UV-VIS検出器又はフォトダイオードアレイUV-VIS検出器 (275nm)
HPLC条件2
カラム:シリカゲルカラム(粒径5μm,内径4.6mm,長さ15cm)
移動相:クロロホルム・ヘキサン (3 : 1)
流速:1.0ml/min
検出器:波長可変型UV-VIS検出器又はフォトダイオードアレイUV-VIS検出器 (275nm)
参考 MBT系加硫促進剤分析用のオクタデシルシリル化シリカゲルカラムとしては,2)のチウラム系
加硫促進剤分析用のHPLCカラム,シリカゲルカラムとしては,3)のチオウレア系加硫促進剤
分析用のHPLCカラムを参照。
5) DTC系加硫促進剤
5.1)
DTC系加硫促進剤の種類の推定 DTC系加硫促進剤は加硫工程において分解してアミンを生成す
ることから,4.1)の操作によってDMA,DEA,DBA,PIP又はEANを確認することで,抽出液中
にDTC系加硫促進剤のZnMDC,ZnEDC,ZnBDC,ZnPDC又はZnEPDCが存在していることを推
定できる。
5.2)
DTC系加硫促進剤の定性及び定量分析 分析手順の概要を,次に示す。
なお,詳細な手順は附属書3を参照。
5.2.1) 抽出液中のDTC系加硫促進剤のコバルトしゃく(錯)体化 抽出液25mlを,50℃の水浴上でロー
タリーエバポレーターを用いて濃縮し,さらに空気を吹きつけて溶媒を完全に留去する。残留物を
ジクロロメタン約1mlに溶かし,1mol/l塩化コバルト溶液1mlを加え,室温下30分間振とうして,
抽出液中のDTC系加硫促進剤(亜鉛しゃく体として使用されたもの)をコバルトしゃく体化する。
分液ろ紙を用いてジクロロメタン層を分取し,溶媒を留去後,ジクロロメタンに溶かして全量を1ml
にし,その2.5μlを次に示すHPLC条件によって分析する。
HPLC条件
カラム:オクタデシルシリル化シリカゲルカラム(粒径5μm,内径4.6mm,長さ15cm),
移動相:メタノール・水 (95 : 5), (90 : 10) 又は (85 : 15)
流速:1.0ml/min
検出器:波長可変型UV-VIS検出器又はフォトダイオードアレイUV-VIS検出器 (320nm)
参考 DTC系加硫促進剤分析用のオクタデシルシリル化シリカゲルカラムは,2)のチウラム系加硫促
進剤分析用のHPLCカラムを参照。
5.2.2) 標準混合系の調製 操作2)によって抽出液中に複数のDTC系加硫促進剤が存在すると推定された
場合,おのおののDTC系加硫促進剤の標準溶液(約1μmol/mlのジクロロメタン溶液)を調製し,
さらにそれらを混合して,混合比の異なる標準混合系を調製する。その標準混合系について5.2.1)
と同様に処理してコバルトしゃく体化し,5.2.1)に示すHPLC条件によって分析する。
5.2.3) 試料中のDTC系化合物の定量 試料及び標準混合系について,HPLCクロマトグラムにおける出現
ピーク間のピーク高比を算出する。標準混合系の分析結果をもとに作成したDTC系加硫促進剤の混
合比とピーク高比の関係図から,試料中のDTC系化合物のおおまかな混合比を求める。次いで,そ
の混合比に近い標準混合系数種を用いた内挿法によって,試料中のDTC系化合物の混合比を近似的
に求める。さらに,試料に最も近似する混合比の標準混合系を用いて,ピーク高の総和及び混合比
27
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
から,試料中のDTC系化合物の含有量を求める。
3.6
水溶性タンパク質
3.6.1
試験の概要 天然ゴムラテックス中の水溶性タンパク質が抗原となって起こる即時型アレルギー
が知られている。この試験法は,天然ゴムラテックス製品から水に溶出するタンパク質の総量を定量する
方法である。粘膜及び皮膚に接触する製品,又は呼吸器や循環器の近傍で用いられる製品などに適用でき
る。
備考 総水溶性タンパク質量が総アレルゲン量や即時型アレルギーのリスクを直接に反映するとは必
ずしもいえない。しかし,製品中の総水溶性タンパク質量が減少すれば,全体としては即時型
アレルギーのリスクは減少するといえる。
3.6.2
試薬
a) りん酸緩衝生理食塩水 (PBS) 塩化ナトリウム8gをpH7.4の10mmol/lりん酸緩衝液に溶かして1
000mlとする。
b) トリクロロ酢酸 (TCA) 溶液 (300g/l)
c) りんタングステン酸 (PTA) 溶液 (14g/l)
d) 水酸化ナトリウム溶液 (0.1mol/l)
e) 試薬A(43) 炭酸ナトリウム2gを水酸化ナトリウム溶液に溶かして100mlとする。
f)
試薬B(43) くえん酸三ナトリウム二水和物1g,酒石酸ナトリウム二水和物1gを硫酸銅五水和物溶液
(5g/l) に溶かして100mlとする。
g) フェノール試液(43)(44) タングステン酸ナトリウム二水和物100gとモリブデン酸ナトリウム二水和物
25gを700mlの水に溶解し,りん酸50mlと塩酸100mlを加え,10時間還流加熱する。冷却後,硫酸
リチウム一水和物150gと水50mlを加え溶解させる。さらに臭素を数滴加えた後,黄色透明な溶液が
得られるまで溶液を加熱する。冷却後,水を加えて1 000mlとし,不溶物をろ過して除いた後,褐色
瓶中で冷暗所に保存する。使用時にフェノールフタレインを指示薬として酸度を求め,水で1規定に
なるように希釈する。
h) 試薬C(45) 4, 4ʼ-ジカルボキシ-2, 2ʼ-ビキノリン二ナトリウム塩(ビシンコニン酸二ナトリウム)1g,
炭酸ナトリウム一水和物2g,酒石酸ナトリウム二水和物0.16g,水酸化ナトリウム0.4g,炭酸水素ナ
トリウム0.95gを水95.5mlに溶解し,炭酸水素ナトリウムを添加して溶液のpHを11.25に調整する。
i)
試薬D(45) 硫酸銅五水和物水溶液 (40g/l)
j)
ウシ血清アルブミン (BSA) 希釈列(43)(45) 結晶BSAを水酸化ナトリウム溶液に溶解し,BSA希釈列
(20〜300μg/mlで10種程度)を調製する。
注(43) 試薬A,試薬B,フェノール試液及び標準BSA溶液からなる市販試薬キット(Lowry法用)が
各社から入手できる。
(44) フェノール試液だけの市販品も各社から入手できる。
(45) 試薬C,試薬D及び標準BSA溶液からなる市販試薬キット(BCA法用)がある。
参考 Pierce社製BCA Protein Assay Reagentキットなどがある。
3.6.3
器具及び装置
a) ふた付きプラスチック製チューブ 50ml
b) 振とう機
c) 遠心機
d) 恒温水槽
28
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
e) 可視分光光度計
3.6.4
操作
a) タンパク質の抽出 試料を約1cm角の薄片に切断し(46),その2.00gをふた付きプラスチック製チュー
ブに入れ,PBS10.0mlを加える。振とう機を用いて激しく振とうしながら室温で2時間抽出する。ゴ
ム片をろ過して除いた後,ろ液を1 500Gで10分間遠心し,上清を抽出液とする。
注(46) 試料は素手で触れないようにする。
b) 試験液の調製 5.00mlの抽出液にTCA溶液1.0mlを加え,かくはん後5分間静置する。さらに,PTA
溶液1.0mlを加えかくはん後20分間静置する。次に,1 500Gで30分間遠心して上清を完全に除く。
このようにして得られたタンパク質の沈殿を水酸化ナトリウム溶液500μlに溶解し,試験液とする。
沈殿したタンパク質の量が非常に多いか,又は少ない場合には,比色定量における吸光度が検量線の
範囲内に収まるように,加える水酸化ナトリウム溶液の量を増減させて試験液を調製する。
備考 測定試料液中に混在するチオール類,フェノール類,糖類,キレート剤,界面活性剤,カリウ
ムイオンなどはLowry法の呈色反応を妨害する。また,BCA法においてはチオール類,還元糖,
チメロサール,脂質などが呈色反応を妨害する。これらの妨害物質を除去するため,また,測
定試料液のタンパク質濃度を上昇させるため,抽出液中のタンパク質を酸を用いて沈殿させて
から測定する。標準タンパク質としてはウシ血清アルブミン (BSA) を用いる。
c) Lowry法による比色定量 試験液及びBSA希釈列それぞれ100μlを試験管にとる。別に空試験として
水酸化ナトリウム溶液100μlを試験管にとる。試薬Aと試薬Bを50 : 1の容量比で混合し試験試薬と
する。試験試薬2.00mlを各試験管に加えてよくかくはんした後,室温で20分間静置する。次に,フ
ェノール試液200μlを素早く加え,1〜2秒のうちに完全に混和する。室温で30分間静置した後,750nm
での吸光度を測定する。試験液及びBSA希釈列の測定吸光度から,空試験の測定吸光度を差し引いた
値をそれぞれの試料についての吸光度とする。
d) BCA法による比色定量 試験液及びBSA希釈列それぞれ100μlを試験管にとる。別に空試験として
水酸化ナトリウム溶液100μlを試験管にとる。試薬Cと試薬Dを50 : 1の容量比で混合し試験試薬と
する。試験試薬2.00mlを各試験管に加えてよくかくはんした後,60℃で30分間保つ。室温まで冷却
した後,直ちに562nmでの吸光度を測定する。試験液及びBSA希釈列の測定吸光度から,空試験の
測定吸光度を差し引いた値をそれぞれの試料についての吸光度とする。
e) 溶出タンパク質量の算出 BSA希釈列のタンパク質濃度に対して吸光度をプロットし,検量線を作成
する。試験液の吸光度を検量線に当てはめ,タンパク質濃度を求める。求めた試験液のタンパク質濃
度から溶出タンパク質量を算出する。
3.7
エンドトキシン
3.7.1
試験の概要 血流中に移行したエンドトキシンは,発熱やショックを起こすことがある。この試験
は,グラム陰性菌に由来するエンドトキシンを定量する方法である。血液,粘膜,組織などに直接又は間
接的に接触するゴム製品に適用できる。
備考1. エンドトキシンの検出には,従来ウサギを用いた発熱性物質試験法が適用されてきたが,カ
ブトガニの血球抽出成分LAL (Limulus Amebocyte Lysate) がエンドトキシンによって活性化
されてゲル化する反応を利用する定量的なエンドトキシン試験法が動物実験代替法として用
いられている。この試験法は,LALのゲル形成を指標とするゲル化法とゲル形成過程におけ
る濁度変化を指標とする比濁法並びに発色合成基質の加水分解による発色を指標とする比色
法からなる。
29
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2. この試験は,できるだけ速やかに,微生物による汚染を避けて行う。この試験で再試験が必
要な場合,又は反応阻害又は促進の影響が除けない試料の場合は,第13改正日本薬局方・一
般試験法の発熱性物質試験法を適用してもよい
3.7.2
試薬及び試液
a) エンドトキシン試験用水 第13改正日本薬局方第二部「注射用水」又はその他の方法によって製造し
た水で,エンドトキシン試験に用いるLAL試薬で反応を示さないもの。
b) エンドトキシン試験用0.1mol/l塩酸試液 塩酸9.0mlにエンドトキシン試験用水を加えて1 000mlと
する。
c) エンドトキシン試験用0.1mol/l水酸化ナトリウム試液 水酸化ナトリウム4.3gをエンドトキシン試験
用水に溶かし,1 000mlとする。
d) LAL試薬 本品はカブトガニ(Limulus polyphemus又はTachypleus tridentatus)のamebocyte lysateを原
料として調製された凍結乾燥品である。
備考 LALはエンドトキシンのほかに, (1→3) -β-D-グルカンに対しても反応するので注意が必要で
ある。β-グルカンと反応するG因子を除去,又はG因子系の反応を抑制したLAL試薬もある
ので,検体中にβ-グルカンの存在が考えられる場合にはこれらのエンドトキシンに特異的な
LAL試薬を用いる。
e) LAL試液 LAL試薬にエンドトキシン試験用水,又はエンドトキシンが検出されないことを確認した
緩衝液を加えて,静かにかき混ぜて溶かす。
備考 トリス(ヒドロキシメチル)アミノメタン−塩酸緩衝液などを使うことができる。
3.7.3
器具 ガラス容器 第13改正日本薬局方・一般試験法注射剤ガラス容器試験法のアルカリ溶出試
験に適合するもので,250℃で1時間加熱処理したもの。
3.7.4
試料溶液の調製 ガラス容器に試料をとり,附属書1の3.2 c),d),e)及びf)のうちのいずれかの
割合でエンドトキシン試験用水を加え,適切に栓をした後,附属書1の4c)又はd)のいずれかの時間で浸
せきし,さらに室温になるまで放置し,この内容液を試料溶液とする。別に同様の方法で空試験液を調製
する。試料溶液は必要ならば,エンドトキシン試験用0.1mol/l塩酸試液又はエンドトキシン試験用0.1mol/l
水酸化ナトリウム試液を用いてpH6.0〜8.0に調整する。
備考1. エンドトキシン試験用水の代わりにエンドトキシンが検出されないことを確認した緩衝液を
用いることができる。
2. 試料溶液がこの方法において反応促進又は反応阻害を示す場合は,限外ろ過その他適当な方
法によって反応の促進又は阻害要因を除去するか,又はエンドトキシン試験用水,若しくは,
エンドトキシンが検出されないことを確認した緩衝液を用いて,最大有効希釈倍数を超えな
い範囲で希釈した希釈試料溶液によって試験することができる。
3. 最大有効希釈倍数とは,反応促進又は反応阻害などの作用によって,試料溶液での試験がで
きない場合,許容範囲内で希釈した希釈試料溶液を調製することによって,エンドトキシン
の測定を可能とするための最大希釈倍数である。最大有効希釈倍数は,次の式によって求め
る。
最大有効希釈倍数=
)
(
1λ
λ又は
エンドトキシン規格値
30
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
エンドトキシン規格値: 各試料溶液毎に規定されたエンドトキ
シン規格値 (EU/ml)
λ: ゲル化法の場合のLAL試薬の表示感
度 (EU/ml)
λ1: 比濁法又は比色法の場合の検量線の最
小エンドトキシン濃度 (EU/ml)
3.7.5
試験及び判定方法 この方法によって試験を行うことが規定されている場合は,別に規定するもの
のほか,ゲル化法,比濁法及び比色法のいずれかの方法によって試験を行う。試験及び判定は,第13改正
日本薬局方・一般試験法のエンドトキシン試験法による。
3.8
エチレンオキシド
3.8.1
試験の概要 エチレンオキシド (EO) ガスを用いて滅菌したゴム製品中に残留するEOの総量を測
定する方法である。
備考 この方法はISO 10993-7の附属書B5.4(エタノール抽出・ヘッドスペース法)を基本としてい
るが,B5.3(気相抽出法),B5.5(溶媒抽出法),B5.6(ブロモヒドリン・ECD法)も適用でき
る。
3.8.2
試薬及び試液
a) EO標準原液 テフロンセプタム付きバイアル瓶を寒剤(47)で十分に冷やす。EOガスボンベの調節弁
口の先に適切なチューブを接続し,その先端に注射針をつける。注射針の先端をセプタムを通してバ
イアル瓶に接続する。別の同様の注射針をセプタムに突き刺しておく。ボンベのバルブをゆっくり開
いてEOガスをバイアル瓶内に導入し,適当量の液化したEOをバイアル瓶に捕集する。別にテフロ
ンセプタム付き100mlの全量フラスコの質量を0.1mgの単位まで正確に量り,寒剤(47)で十分に冷却す
る。捕集した液体EOの5滴を全量フラスコに加え,再度ひょう量してEO量を算出する。エタノー
ルを標線まで加える。
警告 EOガスによる障害を防ぐために,これらの操作はドラフトチャンバー中で,よく注意して
行う必要がある。
注(47) ドライアイス/2-プロパノールやドライアイス/アセトンなどがある。
備考 適切に調製された品質保証済の市販アンプル入り標準原液を使用してもよい。
b) EO標準液 EO標準原液をエタノールで薄めて,0.4,0.8,1.2,1.6,2.0μg/mlなどの濃度のEO標準
液を調製する。
c) プロピレンオキシド (PO) 標準液 プロピレンオキシド (PO) をエタノールを薄めて,0.5μg/mlの濃
度のPO標準液を調製する。
3.8.3
装置及び器具
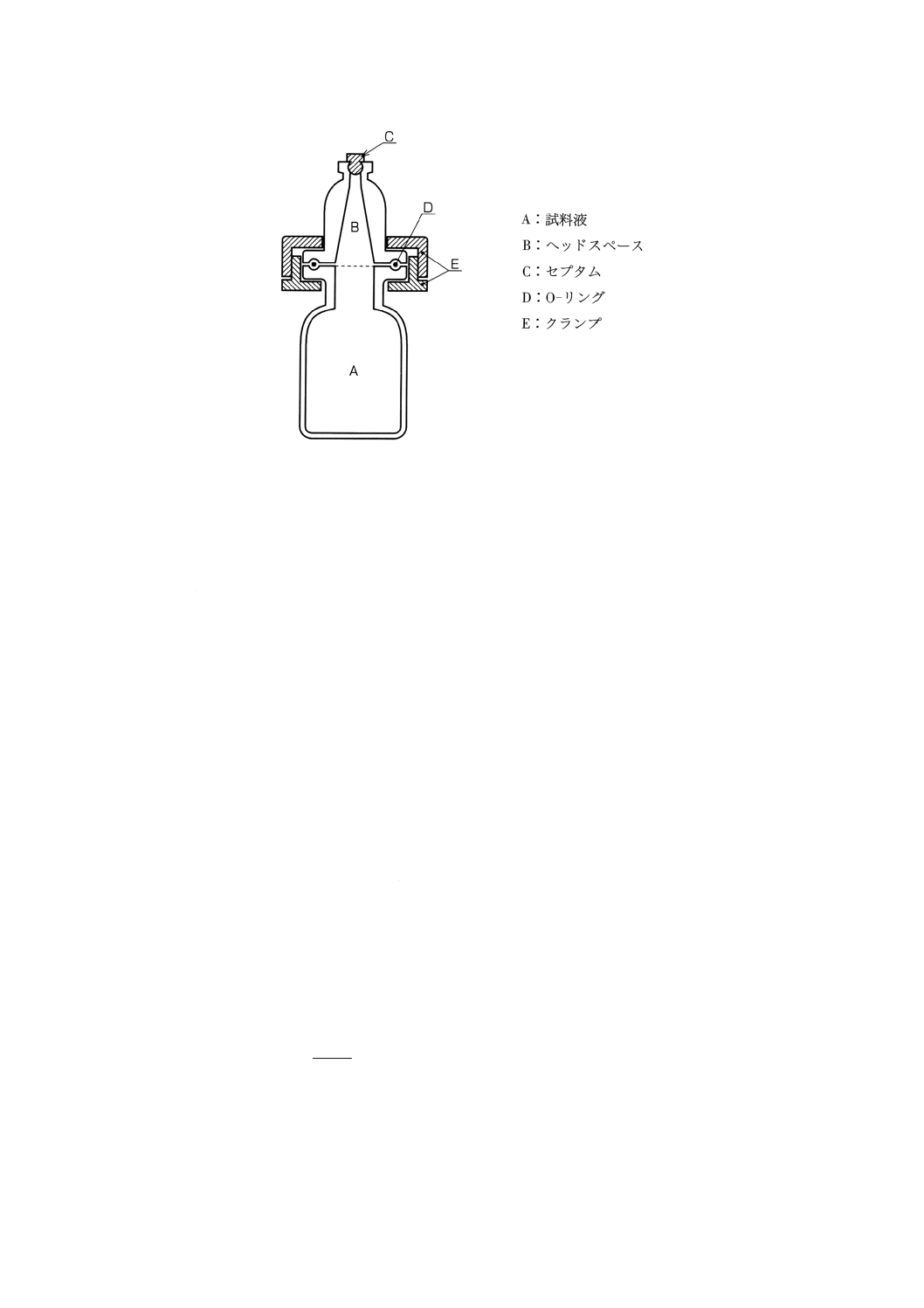
a) ヘッドスペースボトル 図6に示したような内容積50mlのヘッドスペースボトルを用いる。
備考 70℃に加温しても安全でヘッドスペース・ガスの漏れがなければ,適切なバイアル瓶を用いて
もよい。
31
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図6 ヘッドスペースボトルの例
b) ガスクロマトグラフ 水素炎検出器付き。EO,PO,ホルムアルデヒド,アセトアルデヒド,エタノ
ール,メタノール,エチレンクロロヒドリン,エチレングリコールなどが良好に分離するようにカラ
ムと分析条件を整える。
参考 次に示す条件がある。
カラム:25%Flexol 8N8/Chromosorb W AW(粒径149〜177μm,内径3mm,長さ2m,ガラス
カラム)
カラム温度:50℃
検出器及び注入口温度:120℃
キャリヤーガス及び流量:窒素,40ml/min
c) 恒温振とう器
3.8.4
操作
a) 検量線の作成 5個のヘッドスペースボトルおのおのに,0.4,0.8,1.2,1.6及び2.0μg/mlのEO標準
液10mlを速やかに加え,さらにPO標準液を10mlずつ加えて密栓する。これらのボトルを恒温振と
う器中で70℃で30分間緩やかに振とうした後,セプタムを通してヘッドスペースガス100μlをガス
タイトシリンジで採取し,ガスクロマトグラフに注入する。EOとPOのピークの高比を求め,その比
と標準混液中のEO濃度との間の検量線を求める。
b) 分析 試料約2gをとり,約10mm角に切断し,0.1mg単位まで正確にはかった後,ヘッドスペースボ
トルに入れ,20mlのPO標準液を加えて素早く密栓し,70℃で3時間恒温振とう器中で緩やかに振り
混ぜながら加温する。ガスタイトシリンジで100μlのヘッドスペースガスをとり,ガスクロマトグラ
フに注入し,EOとPOのピーク高比を測定する。検量線から溶液中のEO濃度 (Yμg/ml) を求める。
ゴム製品中の残留EO濃度 (C) を,次の式によって算出する。
S
Y
C
×
20
=
ここに, C: ゴム製品中の残留EO濃度 (μg/g)
Y: 溶液中のEO濃度 (μg/ml)
S: 試料採取量 (g)
32
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考 ここに示した操作は,製品中の残留EO濃度として1〜10μg/gまでの分析に適したものである。
さらに高濃度の残留EOを分析する場合は,試料採取量,標準液濃度や注入ガス量などを適切
に変化させて対応しなければならない。

33
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(規定) 溶出方法及び条件
1. 適用範囲 この附属書は,本体3.3に適用する溶出方法及び条件の詳細事項について規定する。
備考 材料からの物質の溶出には,時間,温度,試料/溶媒比,溶媒の種類,相平衡などが影響する。
次に示すように,実際の使用条件に似せた標準的な模擬溶出条件がいろいろな分野で用いられ
ているので,その中から適切と考えられる条件を選ぶこともできる。その場合には,当該条件
を選択した理由を明らかにしなければならない。
2. 溶媒
2.1
食品用器具・容器包装の場合 試験(本体3.3)の項目と器具などの種類の違いによって,附属書1
表1の中から選択する。
附属書1表1 試験項目及び試験に用いる溶媒
試験項目
溶媒
ほ乳用器具 ほ乳用器具以外の器具・容器包装
フェノール
蒸留水
蒸留水
ホルムアルデヒド 蒸留水
蒸留水
亜鉛
蒸留水
4%酢酸
重金属
4%酢酸
4%酢酸
蒸発残留物
蒸留水
蒸留水:pH5を超える食品用
4%酢酸:pH5以下の食品用
20%エタノール:アルコール性食品,油脂及び脂肪性食品用
2.2
医療用具及び医薬品容器の場合 溶媒は,蒸留水を用いる。
3. 溶出方法
3.1
充てん溶出法 容器又は王冠ディスク,シーリング材のようなものについて行う方法である。容器
の場合には口元部から6〜7mm以内のところまで,又は規定容量まで,あらかじめ規定温度にした水又は
その他の浸出用液を満たし,試料の口元を時計皿又は清浄なアルミニウムはくで覆う(1)。王冠ディスク,
シーリング材などの栓類の場合は,同様に浸出用液を満たし,液が漏れないように栓をして倒立させ,規
定温度の水浴中に入れ規定時間放置する。この間水浴の温度が規定温度を保持するよう監視する。規定時
間放置後,直ちに浸出液を清浄なビーカー中に移し室温まで放冷し,試験溶液とする。容器の場合には,
別に等量の浸出用液をビーカーに量りとり,時計皿又は清浄なアルミニウムはくで覆い(1),同様に操作し
て空試験溶液とし,栓類の場合には容器に充てん後,倒立させずに口元を時計皿又は清浄なアルミニウム
はくで覆い(1),同様に操作して,空試験溶液とする。
3.2
浸せき溶出法 医療用具などで一般的に用いられる方法である。試料を硬質ガラス製容器に入れ,
次の試料/溶媒比の中から適切な比を選択し,その比率で規定の溶媒を加え,適切な溶出温度及び時間(4.
に規定)で溶出する。放冷後,試料を取り除いた液を試料溶液とする。別に試料を含まないものについて
同様に操作し,空試験溶液とする。
a) 試料1gに対し,溶媒20ml
b) 試料1cm2に対し,溶媒2ml
c) 試料(厚さ≦1.0mm)3cm2に対し,溶媒1ml
34
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 試料(厚さ>1.0mm)1.25cm2に対し,溶媒1ml
e) 試料(不規則な形状の場合)0.1〜0.2gに対し,溶媒1ml
f)
試料(不規則な形状の場合)6cm2に対し,溶媒1ml
3.3
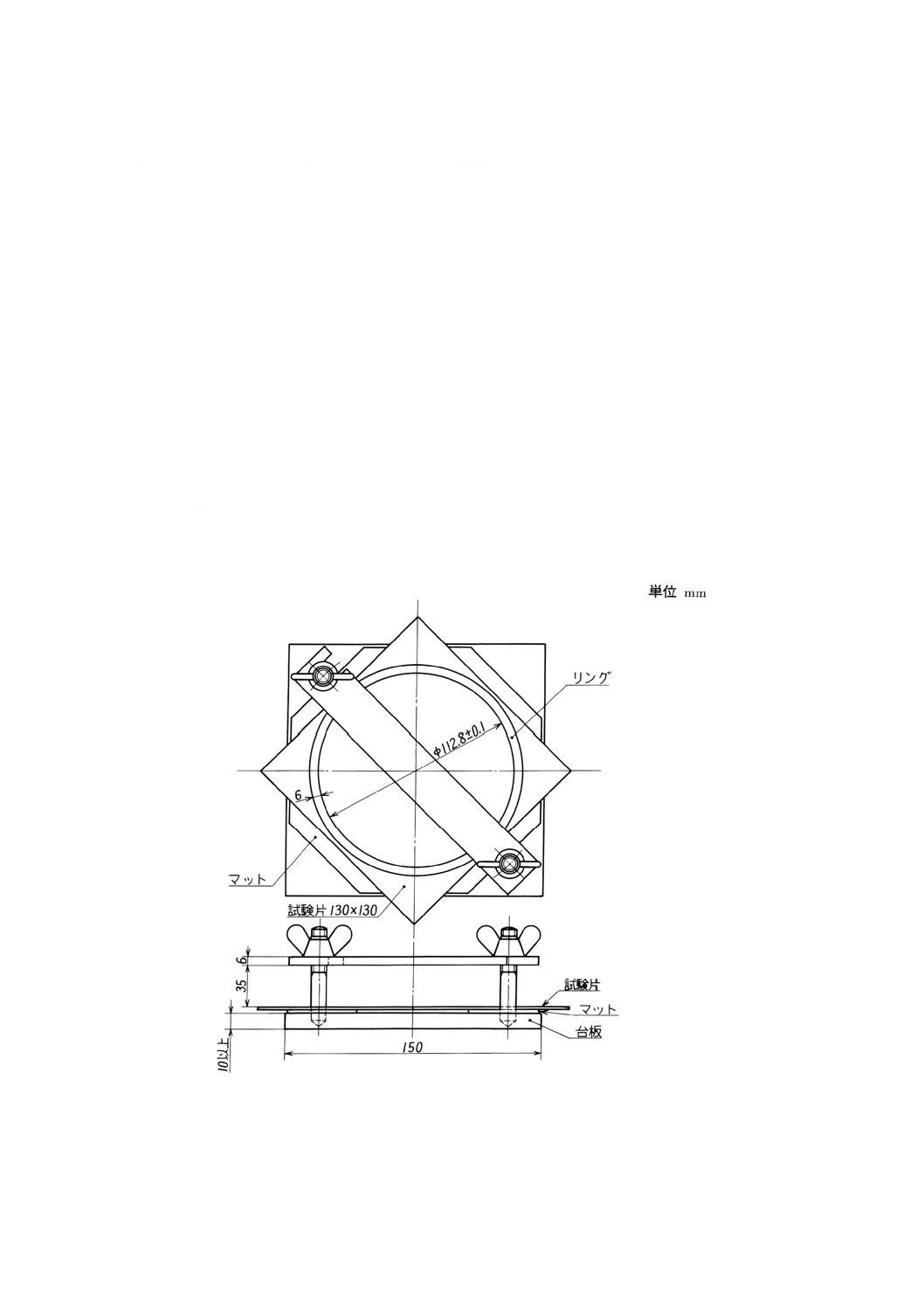
片面溶出法 食品などに接する面が片面である包装材,手袋などについて行う方法である。附属書1
図1の溶出用器具の台板上のマットの上にそれより大きめのアルミニウムはくを1枚載せる。その上に食
品に接する面が浸出用液と接するように試料を置き,リングを載せ,締付金具を浸出用液が漏れないよう
に平均に締めつける。この器具を規定温度にした水浴中にセットし,器具が規定温度になるようにする。
この器具は,リングの高さの2/3以上水浴中に没するように置く。この加温された器具の中に,あらかじ
め規定温度にした水又はその他の浸出用液を規定の割合で満たし,清浄なアルミニウムはくで覆い(1)規定
時間放置する。この間,水浴の温度が規定温度を保持するよう監視する。規定時間後,器具を水浴から取
り出し,直ちに浸出用液を清浄なビーカーに移し,室温になるまで放冷し,これを試験溶液とする。別に
等量の浸出用液をビーカーに量り採り,時計皿又は清浄なアルミニウムはくで覆い(1),同様に操作して空
試験溶液とする。浸出用器具は,附属書1図1に示すように,底面が平滑に仕上げられたリングとシリコ
ーンゴムのマットを張りつけた台板及び締付金具からなっている。図面の寸法は,溶出表面積を100cm2
にしたときの基準寸法である(JIS P 8140参照)。
注(1) 浸出用液が酢酸の場合には,金属リング及びアルミニウムはくは使用できないので,ガラスリ
ングを用い,締付金具に浸出用液及びその蒸気が接触しないようにガラス板でふたをする。
附属書1図1 片面溶出法に用いる溶出用器具
4. 溶出温度と時間
35
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 60℃,0.5h
b) 40℃,24h
c) 37±1℃,24±2h
d) 37±1℃,72±2h
e) 50±2℃,72±2h
f)
70±2℃,24±2h
g) 121±2℃,1±0.2h

36
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2(参考) 分析対象とする老化防止剤及び加硫促進剤
序文 この附属書(参考)は,本体3.5.4の分析対象とする老化防止剤及び加硫促進剤について記述するも
のであり,規定の一部ではない。
1. 老化防止剤及び加硫促進剤の化合物名と略称 対象となる老化防止剤及び加硫促進剤とその略称を
附属書2表1
附属書2表1
化合物名
略称
老化防止剤
6-エトキシ-2, 2, 4-トリメチル-1, 2-ジヒドロキノリン
ETMDQ
フェニル-1-ナフチルアミン
PAN
フェニル-2-ナフチルアミン
PBN
N-イソプロピル-Nʼ-フェニル-p-フェニレンジアミン
IPPD
N-1, 3-ジメチルブチル-Nʼ-フェニル-p-フェニレンジアミン
DMBPPD
N, Nʼ-ジフェニル-p-フェニレンジアミン
DPPD
2, 6-ジ-tert-ブチル4-メチルフェノール
DTBMP
スチレン化フェノール
SP
2, 5-ジ-tert-ブチルヒドロキノン
DBHQ
チウラム系加硫促進剤
テトラメチルチウラムジスルフィド
TMTD
テトラエチルチウラムジスルフィド
TETD
テトラブチルチウラムジスルフィド
TBTD
ジペンタメチレンチウラムテトラスルフィド
DPTT
チオウレア系加硫促進剤
ジエチルチオウレア
DEU
ジブチルチオウレア
DBU
ジラウリルチオウレア
DLU
ジフェニルチオウレア
DPU
メルカプトベンゾチアゾール (MBT) 系加硫促進剤
2-メルカプトベンゾチアゾール
MBT
ジベンゾチアジルジスルフィド
MBTS
N-シクロヘキシル-2-ベンゾチアジルスルフェンアミド
CBS
2-モルホリノチオベンゾチアゾール
MMBT
ジチオカルバメート (DTC) 系加硫促進剤
ジメチルジチオカルバミン酸亜鉛
ZnMDC
ジエチルジチオカルバミン酸亜鉛
ZnEDC
ジブチルジチオカルバミン酸亜鉛
ZnBDC
エチルフェニルジチオカルバミン酸亜鉛
ZnEPDC
ペンタメチレンジチオカルバミン酸亜鉛
ZnPDC
アミン類
ジメチルアミン
DMA
ジエチルアミン
DEA
ジブチルアミン
DBA
N-エチルアニリン
EAN
ピペリジン
PIP
シクロヘキシルアミン
CHA
モルホリン
MOR
37
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3(参考) DTC系加硫促進剤の定量法
序文 この附属書(参考)は,本体3.5.4d) 5)DTC系加硫促進剤の定量手順について記述するものであり,
規定の一部ではない。
1. DTC系加硫促進剤のコバルト錯体化 DTC系加硫促進剤のZnMDC,ZnEDC,ZnBDC,ZnEPDC及び
ZnPDCは,亜鉛 (Zn) (II) 錯体のままでは,通常のHPLC用ステンレスカラム中に含まれているニッケル
と反応してしまい,ピークとして確認できない。そこで,Zn (II) 錯体を安定なコバルト (Co) (III) 錯体に
変換して分析する。すなわち,Co (MDC)3(M3と略す。),Co (EDC)3(E3と略す。),Co (BDC)3(B3と略す。),
Co (EPDC)3(EP3と略す。),Co (PDC)3(P3と略す。)などに変換後,移動相としてメタノール及び水の混
液(メタノール・水)(95 : 5) を用いて逆相系カラムによって分析する。その場合の保持時間はM3<E3<
Ep3<P3<B3の順である。
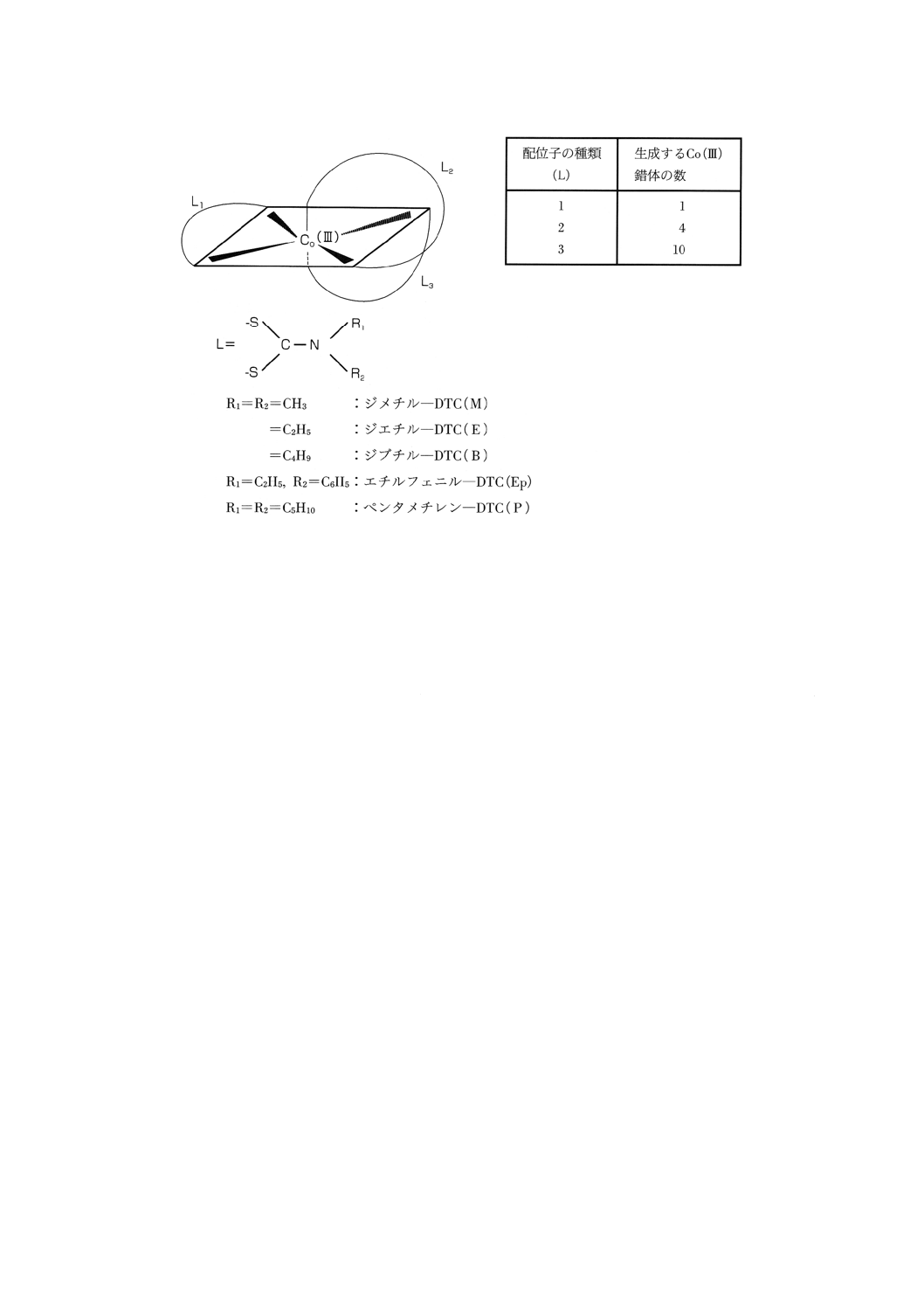
2. 複数のDTC系加硫促進剤が共存する場合のコバルト錯体の生成 複数のDTC系加硫促進剤が共存す
る場合には,Zn (II) 錯体をCo (III) 錯体に変換する際に配位子(リガンド:この場合はジアルキルジチオ
カルバモイル基)の交換が起こり,単一リガンド錯体と混合リガンド錯体の混合物が生成する(附属書3
図1)。すなわち,2種のDTC系加硫促進剤を含む標準混合系では,単一リガンド錯体2種及び混合リガン
ド錯体2種の計4種のCo錯体が生成する。オクタデシルシリル化シリカゲルカラムで移動相にメタノー
ル・水 (95 : 5) を用いてHPLC分析すると,ZnEPDC-ZnPDC混合系を除いて,いずれの混合系も四つのピ
ークを分離でき,混合系間の識別は容易である。
なお,ZnEPDC-ZnPDC混合系はメタノール・水 (85 : 15) を用いることによって四つのピークを分離で
きる。
一方,3種のDTC系加硫促進剤が共存する場合には,単一リガンド錯体3種及び混合リガンド錯体7種
の計10種のCo錯体が生成する。この場合,HPLCクロマトグラムにおけるピークの分離は不十分な場合
が多く,HPLC分析の結果だけでは混合系間の識別は容易ではない。
38
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3図1 共存するDTC系加硫促進剤から生成するCo (III) 混合錯体の構造
3. 未知試料中のDTC系加硫促進剤の定性・定量分析 未知試料については,複数のDTC系加硫促進剤
が共存する場合を想定して,GC-NPD又はGC-FTDによるアミン類の分析を行い,共存するDTC系加硫
促進剤の種類をまず推定してから,DTC系加硫促進剤の定性及び定量分析を行うことが必要である。参考
例として,家庭用ゴム手袋中のDTC系加硫促進剤の定性・定量分析を行った手順及び結果を次に示す。
a) アミン類の分析(共存するチウラム系及びDTC系加硫促進剤の推定) 抽出液を本体3.5.4 d)5.1)に
従って処理し,GC-NPD又はGC-FTDによってアミン類の分析を行う。すなわち,参考例の家庭用ゴ
ム手袋の抽出液を分析したところ,アミンとしてDEA108μg/g及びPIP502μg/gが確認できた[附属書
3図2]。したがって,この家庭用ゴム手袋の抽出液中には,チウラム系加硫促進剤のTETD,DPTT
又はDTC系加硫促進剤のZnEDC,ZnPDCが含まれていると推定された。
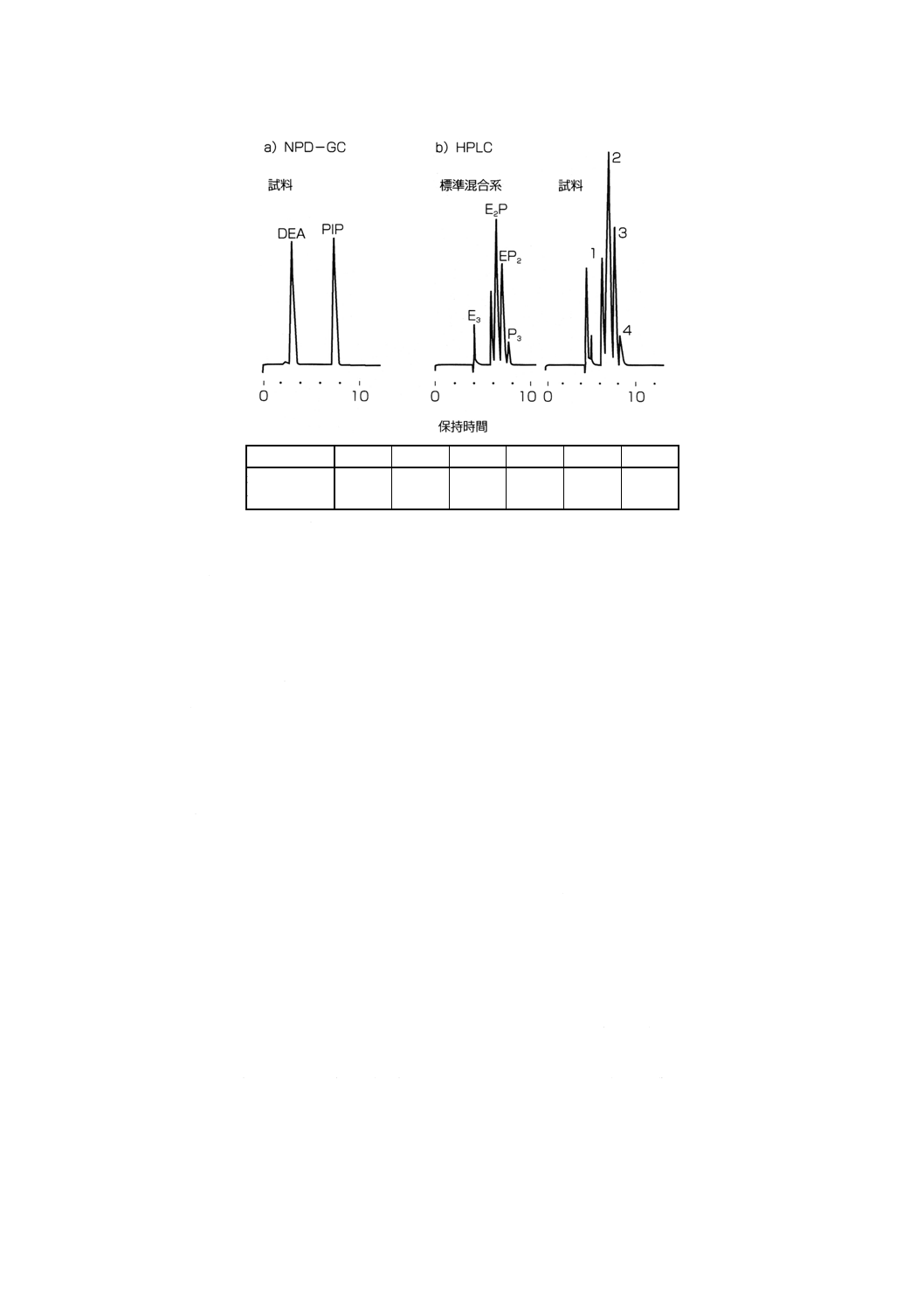
39
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ピーク高さ比 E2P/E3
EP2/E3
P3/E3
EP2/E2P P3/E2P
P3/EP2
標準混合系
試料
1.99
1.96
1.37
1.28
0.32
0.28
0.69
0.65
0.16
0.14
0.23
0.22
附属書3図2 家庭用ゴム手袋中の2種のDTC系加硫促進剤 (ZnEDC,ZnPDC) の確認例
GC-NPD条件
カラム:Amipack 124カラム(粒径149〜177μm,内径2mm,長さ1.2m)
カラム温度:80℃(4分間保持)〜210℃(32℃/min,昇温)
注入口及び検出器温度:220℃
キャリヤーガス及び流量:窒素,10ml/min
検出器:NPD
HPLC条件
カラム:Senshupak ODS-1151-Nカラム(内径4.6mm,長さ15cm)
移動相:メタノール・水 (90 : 10)
流速:1.0ml/min
検出波長:UV320nm
標準混合系
ZnEDC:ZnPDC=1.13
定量値
アミン:DEA108μg/g,PIP502μg/g
DTC系加硫促進剤:ZnEDC935μg/g,ZnPDC830μg/g
b) チウラム系加硫促進剤の定性分析 抽出液を本体3.5.4 d)2)に従って処理し,HPLCによってチウラム
系加硫促進剤を分析する。すなわち,この家庭用ゴム手袋の抽出液中には,TETD,DPTTいずれも確
認できなかった。
c) DTC系加硫促進剤の定性分析 抽出液を本体3.5.4 d)5.2)に従って,Co錯体化後,HPLCによってDTC
系加硫促進剤について分析する。すなわち,この家庭用ゴム手袋の抽出液中には,四つの成分ピーク
が認められ,そのピーク1及び4はZnEDC及びZnPDCのCo錯体 (E3,P3) と一致した。
d) 標準混合系の調製 抽出液中に存在が認められたDTC系加硫促進剤おのおのの標準溶液を調製し,さ
らにそれらを混合して標準混合系(混合比として0.1〜10相当のもの)を調製する。すなわち,この
家庭用ゴム手袋については,ZnEDC及びZnPDCの標準溶液(おのおの約1μmol/mlのジクロロメタン
溶液)を調製後,それらを混合して,標準混合系(ZnEDC/ZnPDC=0.56〜4.50のもの)を調製した。
e) 標準混合系におけるピーク高さ比と混合比との関係図の作成 これらの標準混合系を本体3.5.4 d)5.2)
40
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に従ってCo錯体化後,HPLC分析し,Co錯体のクロマトグラムパターンの変化,並びに成分ピーク
高さ比と混合比との関係を求める。すなわち,この家庭用ゴム手袋に関しては,四つのCo錯体 (E3,
E2P,EP2,P3) のピーク高さ比 (E2P/E3,EP2/E3,P3/E3,EP2/E2P,P3/E2P,P3/EP2) と混合比との関係を
求めた。
f)
試料におけるピーク高さ比と混合比との算出 抽出液を本体3.5.4 d)5.2)に従ってCo錯体化後,HPLC
分析し,Co錯体のクロマトグラムパターンの変化,並びにピーク高さ比と混合比との関係を求める。
すなわち,試料におけるCo錯体のピーク高さ比を算出し,e)で作成したピーク高さ比と混合比との関
係図と照合したところ,混合比ZnEDC/ZnPDC=1.13のピーク高さ比に最も近似していた(附属書3
図2)。
g) 試料中のDTC系加硫促進剤の定量分析 最も近似していた標準混合系を用い,総ピーク高さ法によ
ってDTC系加硫促進剤の定量を行う。すなわち,混合比ZnEDC/ZnPDC=1.13の標準混合系を用いて
定量したところ,家庭用ゴム手袋中のZnEDCは935μg/g,ZnPDCは830μg/gという値が得られた。
41
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書4(参考) 参考文献
序文 この附属書(参考)は,参考文献について記述するものであり,規定の一部ではない。
1. N-ニトロソアミン関連
a) Druckrey, H., Preussmann, R., Ivankovic, S. and Schmahl, D. : Organotrope carcinogene wirkungen bei 65
verschiedenen N-nitroso-verbindungen an BD-ratten, Z. Krebsforsch., 69, 103-201 (1967)
b) Fine, D. H., Lieb, D. and Rufeh, F. : Principle of operation of the thermal energy analyzer for the trace analysis
of volatile N-nitroso compounds, J. Chromatogr. , 107, 351-357 (1975)
c) Spiegelhalder, B. and Preussmann R. : Nitrosamines and rubber. “N-Nitroso compounds : Occurrence and
biological effects”edited by Bartsch, H., O'Neil, I. K., Castegnaro, M. and Okada, M., IARC Scientific
Publications No.41, Lyon, International Agency for Research on Cancer, p.231-243 (1982)
d) Kim, Y.-K., Tannenbaum, S. R. and Wishnok, J. S. : Effects of ascorbic acid on the nitrosation of dialkylamines.
“Ascorbic acid : Chemistry, metabolism and uses” edited by Seib, P. A. and Tolbert, B. M., American chemical
society,Washington DC, p.571-585 (1982)
e) Havery, D. C. and Fazio, T. : Estimation of volatile N-nitosamines in rubber nipples for babie's bottles, Food
Chem. Toxicol., 20, 939-944 (1982)
f)
Walters, C. L., Hart, R. J. and Smith, P. L. R. : Analysis of total N-nitroso compounds as a group by
denitrosation to nitrite oxide with detection using a chemiluminescence analyzer. “Environmental carcinogens
selected methods of analysis” edited by Preussmann, R., O'Neill, I. K., Eisenbrand, G., Spiegelhalder, B. and
Bartsch, H., IARC Scientific Publications No.45, Lyon, International Agency for Research on Cancer,
p.295-308 (1983)
g) Castegnaro, M., Pollock, J. R. A. and Iriesen, M. : Possible underestimation of nitrosatable amine levels
artificial saliva extracts of children rubber pacifiers and baby-bottle teats. “The relevance of N-nitroso
compounds to human cancer : Exposures and mechanisms” edited by Bartsch, H., O'Neill, I. K. and
Schulte-Hermann, R., IARC Scientific Publications No.84, Lyon, International Agency for Research on Cancer,
p.377-379 (1987)
h) Sen, N. P. and Seman, S. W. : Improved methods for determination of volatile nitrosamines in baby bottle
rubber nipples and pacifiers, J. A. O. A. C., 70, 434-438 (1987)
i)
石橋 亨,松居正己:環境中のN-ニトロソ化合物の分析法に関する研究:N-ニトロソジエタノールア
ミン分析の精製濃縮法,環境化学,1,571-574 (1991)
2. 細胞毒性・感作性関連
a) Tsuchiya, T. Studies on the standardization of cytotoxicity tests and new standard reference materials useful for
evaluating the safety of biomaterials, J. Biomaterials Appl., 9, 138-157 (1994)
b) ISO 10993-10 : Biological evaluation of medical devices. Part 10 : Tests for irritation and sensitization
c) 厚生省薬務局医療機器開発課監修:医療用具及び医用材料の基礎的な生物学的試験のガイドライン
1995解説,薬事日報社 (1996)
d) 厚生省薬務局審査第一課:医薬品毒性試験法ガイドライン・皮膚感作性試験(平成元年9月11日薬審
42
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1第24号):医薬品非臨床試験ガイドライン解説1991,厚生省薬務局新医薬品課監修,薬事日報社,
p.60-64 (1991)
e) OECD GUIDELINES FOR TESTING OF CHEMICALS. No.406・Skin Sensitization OECD毒性試験ガイド
ライン,厚生省生活衛生局企画課生活化学安全対策室監修,薬事日報社,p.37-43 (1991)
f)
Magnusson,B. and Kligman, A. M. : The identification of contact allergens by animal assay. The guinea pig
maximization test, J. Invest. Dermatol., 52, 268-276 (1969)
g) Sato, Y., Katsumura, Y., Ichikawa, H., Kobayashi, T., Kozuka, T., Morikawa, F. and Ohta, S. : A modified
technique of guinea pig testing to identify delayed hypersensitivity allergens, Contact Dermatitis, 7, 225-237
(1981)
h) Kaniwa, M.-A., Momma, J., Ikarashi, Y., Kojima, S., Nakamura, A., Nakaji, Y., Kurokawa, Y., Kantoh, H. and
Itoh, M. : A method for identifying causative chemicals of allergic contact dermatitis using a combination of
chemical analysis and patch testing in patients and animal groups : Application to a case of rubber boot
dermatitis, Contact Dermatitis, 27, 166-173 (1992)
i)
Nakamura, A., Momma, J., Sekiguchi, H., Noda, T., Yamano, T., Kaniwa, M.-A., Kojima, S., Tsuda, M. and
Kurokawa, Y. : A new protocol and criteria for quantitative determination of sensitization potencies of
chemicals by guinea pig maximization test, Contact Dermatitis, 31, 72-85 (1994)
j)
Kaniwa, M.-A., Isama, K., Nakamura, A., Kantoh, H., Itoh, M., Miyoshi, K., Saito, S. and Shono, M. :
Identification of causative chemicals of allergic contact dermatitis using a combination of patch testing in
patients and chemical analysis, Contact Dermatitis, 30, 26-34 (1994)
3. 水溶性タンパク質関連
a) 中村晃忠:ゴムラテックスによる即時型アレルギー,生体材料,9, 303-310 (1991)
b) 矢上 健,佐藤道夫,中村晃忠:ラテックス製ゴム手袋から溶出する蛋白質の比色定量,衛生試験所
報告,111, 84-87 (1993)
c) Yeang, H. Y., Yusof, F. and Abdullah, L. : Precipitation of Hevea brasiliensis latex proteins with trichloroacetic
acid and phosphotungstic acid in preparation for the Lowry protein assay, Anal. Biochem., 226, 35-43 (1995)
4. エチレンオキシド関連
a) 大場琢磨,辻楠雄,水町彰吾,菊池寛,新谷英晴,飯田和子,目黒賢二:医療用具に残留するエチレ
ンオキサイドに関する研究 (I) ガスクロマトグラフ法によるエチレンオキサイドの定量について,医
科器械学,52, 134-139 (1982)
b) 大場琢磨,辻楠雄,菊池寛,水町彰吾,新谷英晴,三沢明:医療用具に残留するエチレンオキサイド
に関する研究 (II) 手術用ゴム手袋及び各種ゴム類の残留と蒸散について,医科器械学,52, 140-144
(1982)
5. 附属書1関連
a) 日本薬学会編:衛生試験法・注解1990付.追補 (1995) ,金原出版,p.729
43
T 9010 : 1999
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
原案作成委員会 構成表
(原案作成当時の氏名,所属)
氏名
所属
(委員長)
中 村 晃 忠
国立衛生試験所療品部(現 国立医薬品食品衛生研究所
療品部)
増 田 優
通商産業省基礎産業局
西 出 徹 雄
工業技術院
山 村 修 蔵
財団法人日本規格協会
五十嵐 良 明
国立衛生試験所療品部(現 国立医薬品食品衛生研究所
療品部)
石 橋 亨
財団法人食品等分析調査研究所(現 財団法人東京顕微鏡
院)
大 武 義 人
財団法人化学品検査協会(現 財団法人化学物質評価研究
機構)
小 川 義 之
国立衛生試験所大阪支所(現 国立医薬品食品衛生研究
所)
風 間 成 孔
東京薬科大学大学院(現 東京薬科大学薬学物)
鹿 庭 正 昭
国立衛生試験所療品部第二室(現 国立医薬品食品衛生研
究所 療品部)
幕 内 恵 三
日本原子力研究所
門 馬 純 子
国立衛生試験所毒性部(現 医薬品副作用被害救済・研究
振興調査機構)
石 井 一 弥
住友化学工業株式会社
河 野 政 美
昭和ゴム株式会社
澤 谷 保
澤谷ゴム工業株式会社
芹 澤 俊 夫
オカモト株式会社
龍 久 則
大内新興化学工業株式会社
鈴 木 守
社団法人日本ゴム協会
(事務局)
中 川 剛 章
社団法人日本ゴム協会