T 3250:2013
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲 ························································································································· 1
2 引用規格 ························································································································· 1
3 用語及び定義 ··················································································································· 2
4 要求事項 ························································································································· 4
4.1 生物学的安全性 ············································································································· 4
4.2 無菌性 ························································································································· 4
4.3 非発熱性 ······················································································································ 4
4.4 機械的特性 ··················································································································· 4
4.5 性能特性 ······················································································································ 6
4.6 使用期限 ······················································································································ 6
5 試験方法 ························································································································· 6
5.1 一般 ···························································································································· 6
5.2 生物学的安全性 ············································································································· 7
5.3 無菌性 ························································································································· 7
5.4 非発熱性 ······················································································································ 7
5.5 機械的特性 ··················································································································· 7
5.6 性能特性 ······················································································································ 8
5.7 使用期限 ····················································································································· 11
6 表示······························································································································ 11
6.1 本体の表示 ·················································································································· 11
6.2 一次包装(該当機器の個包装)························································································ 12
6.3 二次包装(外箱) ········································································································· 12
6.4 添付する文書 ··············································································································· 12
6.5 図記号の使用 ··············································································································· 13
附属書JA(参考)JISと対応国際規格との対比表 ······································································ 15
T 3250:2013
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,日本医療器材工業
会(JMED)及び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を改正すべ
きとの申出があり,日本工業標準調査会の審議を経て,厚生労働大臣が改正した日本工業規格である。
これによって,JIS T 3250:2011は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案登録に抵触する可能性があることに注
意を喚起する。厚生労働大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び
実用新案権に関わる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
T 3250:2013
血液透析器,血液透析ろ(濾)過器,
血液ろ(濾)過器及び血液濃縮器
Haemodialysers, haemodiafilters, haemofilters and haemoconcentrators
序文
この規格は,2010年に第3版として発行されたISO 8637及び2011年に発行されたAmendment 1を基と
し,我が国の実情に合わせるため,技術的内容を変更して作成した日本工業規格である。
なお,この規格で点線の下線を施してある箇所は,対応国際規格を変更している事項である。変更の一
覧表にその説明を付けて,附属書JAに示す。
1
適用範囲
この規格は,人に用いる血液透析器,血液透析ろ(濾)過器,血液ろ(濾)過器及び血液濃縮器(以下,
該当機器という。)について規定する。ただし,次の機器には適用しない。
− 体外循環用血液回路
− 血しょう(漿)分離器
− 血液吸着器
− 血管アクセス機器
− 血液ポンプ
− 体外循環用血液回路の圧力モニタ装置
− 気泡検知器
− 透析液を調製し維持管理する血液透析装置
− 血液透析,血液透析ろ過,血液ろ過又は血液濃縮を行うために使用する装置(該当機器を用いるため
の装置)
− 再生手順及び装置
注記1 血液透析器,血液透析ろ過器及び血液ろ過器の体外循環血液回路の要求事項については,JIS
T 3248に規定されている。
注記2 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 8637:2010,Cardiovascular implants and extracorporeal systems−Haemodialysers,
haemodiafilters, haemofilters and haemoconcentrators及びAmendment 1:2011(MOD)
なお,対応の程度を表す記号“MOD”は,ISO/IEC Guide 21-1に基づき,“修正している”
ことを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
2
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
引用規格は,記載の西暦年の版を適用し,その後の改正版(追補を含む。)は適用しない。
JIS T 0307:2004 医療機器−医療機器のラベル,ラベリング及び供給される情報に用いる図記号
JIS T 0993-1:2012 医療機器の生物学的評価−第1部:リスクマネジメントプロセスにおける評価及
び試験
注記 対応国際規格:ISO 10993-1:2009,Biological evaluation of medical devices−Part 1: Evaluation and
testing within a risk management process(MOD)
ISO 594-2:1998,Conical fittings with 6 % (Luer) taper for syringes, needles and certain other medical
equipment−Part 2: Lock fittings
ISO 10993-11:2006,Biological evaluation of medical devices−Part 11: Tests for systemic toxicity
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
血液側(blood compartment)
該当機器の血液を流す部分。中空糸型機器においては,中空糸及びヘッダー部の容量を含む。
3.2
クリアランス(Clearance)
単位時間当たりに溶質が完全に除去された溶液の量。
3.3
ろ過(convection)
ろ液とともに生じる,圧力勾配又は膜間圧力差による半透膜を介した溶質の移動。
3.4
透析液(dialysis fluid)
血液透析又は血液透析ろ過したとき,血液中の溶質及び/又は水と交換するための溶液。
3.5
透析液側(dialysis fluid compartment)
血液透析器又は血液透析ろ過器の透析液を流す部分。
3.6
拡散(diffusion)
濃度勾配による半透膜を介した溶質の移動。
3.7
ろ液(filtrate)
半透膜間の圧力勾配によって血液から半透膜を介して該当の透析液側又はろ液側に移動する流体。
3.8
血液濃縮(haemoconcentration)
半透膜を通して希釈された血液から余分な液体を除去するプロセス。
3.9
血液濃縮器(haemoconcentrator)
血液濃縮を目的とする機器。
3
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.10
血液透析ろ過器(haemodiafilter)
血液透析ろ過を目的とする機器。
3.11
血液透析ろ過(haemodiafiltration)
半透膜を介し拡散とろ過とを同時に行い,また,適切な生理的溶液との置換によって患者の血液中の溶
質不均衡を是正するプロセス。
注記 通常,このプロセスは除水を伴う。
3.12
血液透析器(haemodialyser)
血液透析を目的とする機器。
3.13
血液透析(haemodialysis)
主に半透膜を介し拡散によって患者の血液中の溶質不均衡を是正するプロセス。
注記 通常,このプロセスは除水を伴う。
3.14
血液ろ過器(haemofilter)
血液ろ過を目的とする機器。
3.15
血液ろ過(haemofiltration)
主に半透膜を介したろ過と適切な生理的溶液との置換によって患者の血液中の溶質不均衡を是正するプ
ロセス。
注記 通常,このプロセスは除水を伴う。
3.16
表示(labelling)
記載,印刷,図表化又は電子化された次のもの。
− 医療機器の容器及び包装に貼付又は印刷されたもの。
− 医療機器に同封されているもので,製品識別に関係するもの。添付文書,技術的説明書及び取扱説明
書。ただし,出荷案内書は含まない。
3.17
ふるい係数(sieving coefficient)
同時点での血しょう(漿)とろ液との同一溶質の濃度比。
3.18
膜間圧力差(transmembrane pressure)(以下,TMPという。)
半透膜を介して生じる圧力差。
注記 実用的には,平均TMPは一般に次のいずれかである。
− 血液透析器又は血液透析ろ過器の,血液側の入口・出口における圧力の算術平均と透析液
側圧力の算術平均との差。
− 血液ろ過器又は血液濃縮器の,血液側の入口・出口における圧力の算術平均とろ液圧力と
の差。
4
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.19
限外ろ過率(ultrafiltration coefficient)
膜の透水性。一般的には,時間当たりの圧力(水銀柱)当たりの流量(mL/mmHg/hr)で表現する。
4
要求事項
4.1
生物学的安全性
該当機器の血液と直接又は間接的に接触する機器の部分は,5.2によって,生物学的な危険性がないこと
を評価しなければならない。
注記 我が国においては,機器に用いられる材料で,血液と直接又は間接的に接触する新規材料は,
JIS T 0993-1によって,生物学的安全性を確認する。
4.2
無菌性
該当機器の血液が通過する部分は,滅菌バリデーション基準又はこれと同等以上の基準に基づき,無菌
性の担保を行う。試験は,5.3によって行う。
注記 滅菌バリデーション基準には,厚生労働省の定めた滅菌バリデーション基準がある。
4.3
非発熱性
該当機器の血液が通過する部分に発熱性物質があってはならない。試験は5.4に従って行う。
4.4
機械的特性
4.4.1
全体的な構造
5.5.1によって試験を行ったとき,該当機器に,漏れがあってはならない。該当機器は,次の条件下で試
験する。
a) 規定の最大圧力の1.5倍
b) 製造販売業者が規定する最大陰圧の1.5倍[ただし,−700 mmHg(−93.3 kPa)より低くてはならな
い。]又は実施可能な最大陰圧
なお,この要求事項は該当機器の外部容器に対するものである。
4.4.2
血液側の構造
製造販売業者が指定した最大推奨TMPの1.5倍で血液側を試験したとき,血液側は漏れてはならない。
要求事項は,5.5.2によって検証する。
4.4.3
血液透析器,血液透析ろ過器及び血液ろ過器の血液側接続部分
血液透析器,血液透析ろ過器及び血液ろ過器と血液回路とが一体化されているものを除き,血液側接続
部分の寸法は,図1のとおりとする。この要求事項は,5.5.3によって検証する。
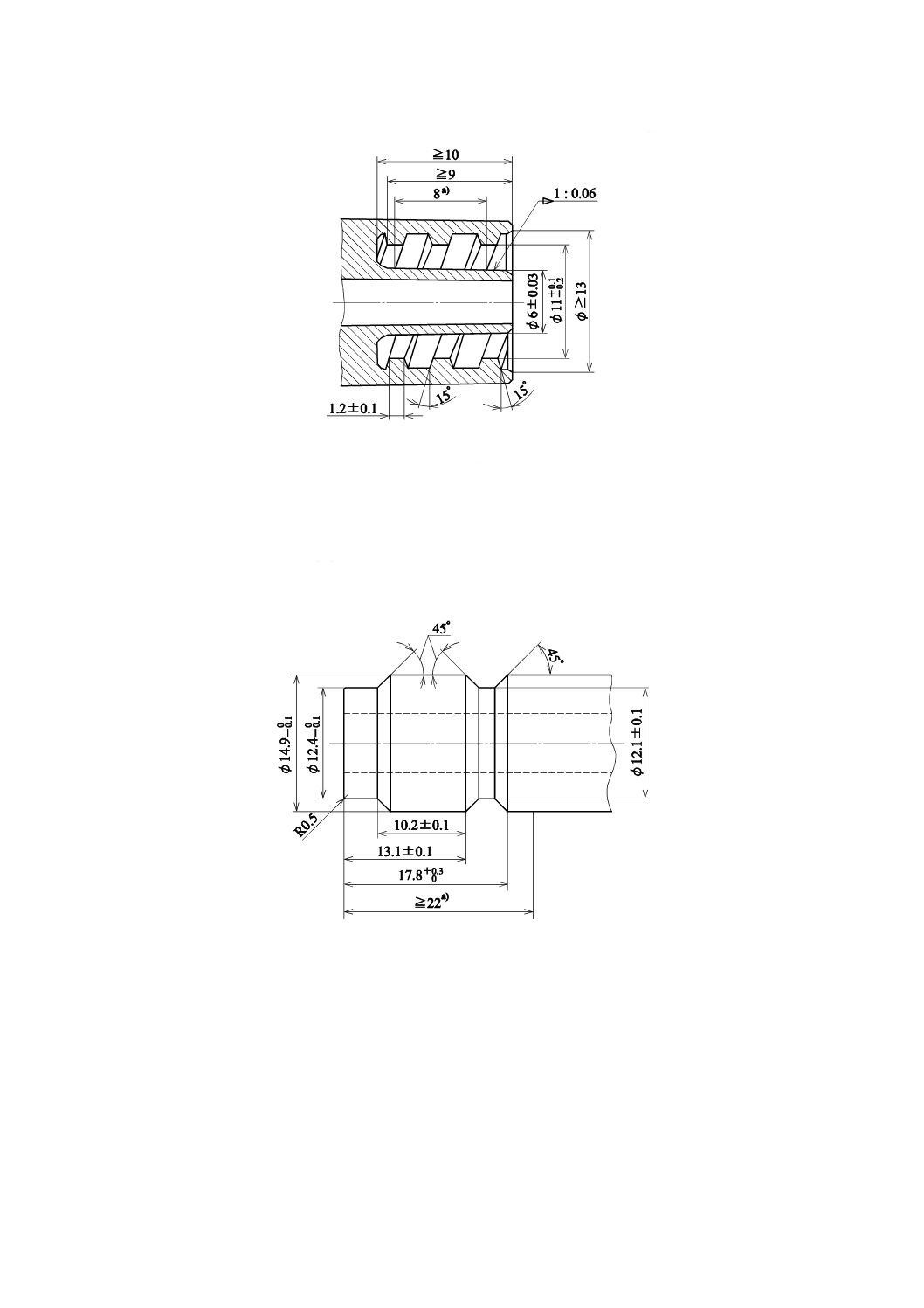
5
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
注a):二条ねじ
図1−血液の出入口接続部分の主要寸法
4.4.4
血液透析器及び血液透析ろ過器の透析液側接続部分
血液透析器及び血液透析ろ過器と透析液回路とが一体化されているものを除き,透析液側接続部分は,
図2に示す形状・寸法とする。試験は,5.5.4によって行う。
単位 mm
注a) 透析液回路コネクタとのかん(嵌)合のために必要な長さ及び直径
図2−透析液の出入口接続部分の主要寸法
4.4.5
血液ろ過器のろ液側接続部分
血液ろ過器とろ液回路とが一体化されているものを除き,血液ろ過器のろ液側接続部分は,図2又はISO
594-2に規定するルアー固定フィッティングの要求事項のいずれかに適合しなければならない。試験は
5.5.5に従って行う。
4.4.6
血液濃縮器の血液側及びろ液側接続部分
血液濃縮器の血液側及びろ液側接続部分は,この該当機器に用いられるチューブと安全に接続できなけ
6
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ればならない。この要求事項は,5.5.6によって試験する。
4.5
性能特性
4.5.1
血液透析器及び血液透析ろ過器のクリアランス
尿素,クレアチニン,りん酸及びビタミンB12のクリアランスは,5.6.1に従って決定する。血液と透析
液との流量は,製造販売業者が定めた範囲内でなければならない。
注記 補足として,KoA(総括物質移動面積係数)を含めることができる。
4.5.2
血液透析ろ過器,血液ろ過器及び血液濃縮器のふるい係数
アルブミン,イヌリン及びβ2ミクログロブリン又はミオグロビンのふるい係数は,5.6.2に従って決定す
る。試験条件は,製造販売業者によるものとする。
4.5.3
限外ろ過率(UFR)
限外ろ過率は,5.6.3に従って確認する。試験は,製造販売業者が指定したTMP及び血液流量の範囲内
で行う。
4.5.4
血液側容量(充塡量)
血液側の充塡量は,製造販売業者が指定するTMPの範囲で,5.6.4の試験方法に従って確認する。
注記 血液側の充塡量がTMPによって変化しない場合,一つのTMPの値に従って決定するとよい。
4.5.5
血液側の圧力損失
血液側の圧力損失は,5.6.5に従って決定する。
4.6
使用期限
機器の生物学的安全性,無菌性及び機械的特性は,使用期限に相当する期間について証明しなければな
らない。試験は,5.7によって行う。
5
試験方法
5.1
一般
4.5で要求する性能については,新製品を市場に出す前に測定しなければならない。また,製品仕様変更
後に性能が変わっていることが予想される場合は,再評価しなければならない。
製品の供試品は,製造工程からランダムに抽出されたもので,適用されている全ての該当機器は,品質
管理工程において適合したものでなければならない。また,臨床使用されるものと同様に,製造販売業者
が推奨する方法に従って,準備する。
測定は,液温37±1 ℃で,インビトロで行わなければならない。変数間の関係が非線形である場合には,
測定点間の内挿を行うことが可能となるように,十分な測定を行わなければならない。この規格の試験方
法は参考方法である。十分な精度及び再現性が得られる場合は,他の試験方法を採用してもよい。
次に示す試験方法は,実際の試験装置において必要な詳細事項全てを示しているわけではない。実際の
試験方法の設計,構成及びその構築は,測定誤差の原因となる多くの要因に注意しなければならない。要
因とは,落差と動的圧力損失とによる圧力測定誤差,パラメータの安定化に要する時間,不定流量におけ
る制御できない温度変化,pH,熱・光・時間による試験物質の劣化,試験液の脱気,貯留空気及び未知物
質・藻類・バクテリアによるシステム汚染(ここに示したことに限定されない。)である。
注記 箇条5において,5.5.1,5.5.3,5.5.4,5.6.1,5.6.2,5.6.3及び5.6.4に規定するものは,新しい
該当機器を市場に出す前,又は該当機器若しくは製造過程を変更するときの形式検査を含む。
また,5.3,5.4及び5.5.2に規定するものは,品質管理システムの必要条件に従って定期的に実
施する品質管理項目も含まれる。
7
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.2
生物学的安全性
患者の血液と直接又は間接的に接触する該当機器における生物学的安全性については,製品を市場に出
す前に,また,その製品で用いる材料の変更後及び滅菌方法の変更後は,該当製品の供試品を用いて評価
を実施する。
試験は,この規格と関連するJIS T 0993-1によって実施する。
5.3
無菌性
4.2に示す要求事項との整合性は,該当機器が検証された滅菌工程を経たことを示す製品の検査記録によ
って確認する。
5.4
非発熱性
4.3の試験は,ISO 10993-11に適合しなければならない。
5.5
機械的特性
5.5.1
全体的な構造
5.5.1.1
一般
4.4.1の確認は,次の試験に従って行う。
5.5.1.2
陽圧試験
脱気した37±1 ℃の水で該当機器を完全に満たす。次に圧力を加える部分を除き全ての接続部分を閉め
る。製造販売業者が規定する1.5倍の陽圧を加え,該当機器一式を密封する。10分後,圧力を記録し,目
視で該当機器の水漏れの有無を確認する。
5.5.1.3
陰圧試験
脱気した37±1 ℃の水で該当機器を完全に満たす。次に圧力を加える部分を除き全ての接続部分を閉め
る。製造販売業者が規定する1.5倍の負圧を加える。ただし,その圧力値が−700 mmHg(−93.3 kPa)を
超える場合は,−700 mmHg(−93.3 kPa)を加える。該当機器一式を密封し,10分後,圧力を記録し,目
視で機器の漏れを確認する。
5.5.2
血液側の構造
血液側の構造は,製造業者(又は製造販売業者)の試験手順に従った試験結果の確認によって行う。
5.5.3
血液透析器,血液透析ろ過器及び血液ろ過器の血液側接続部分
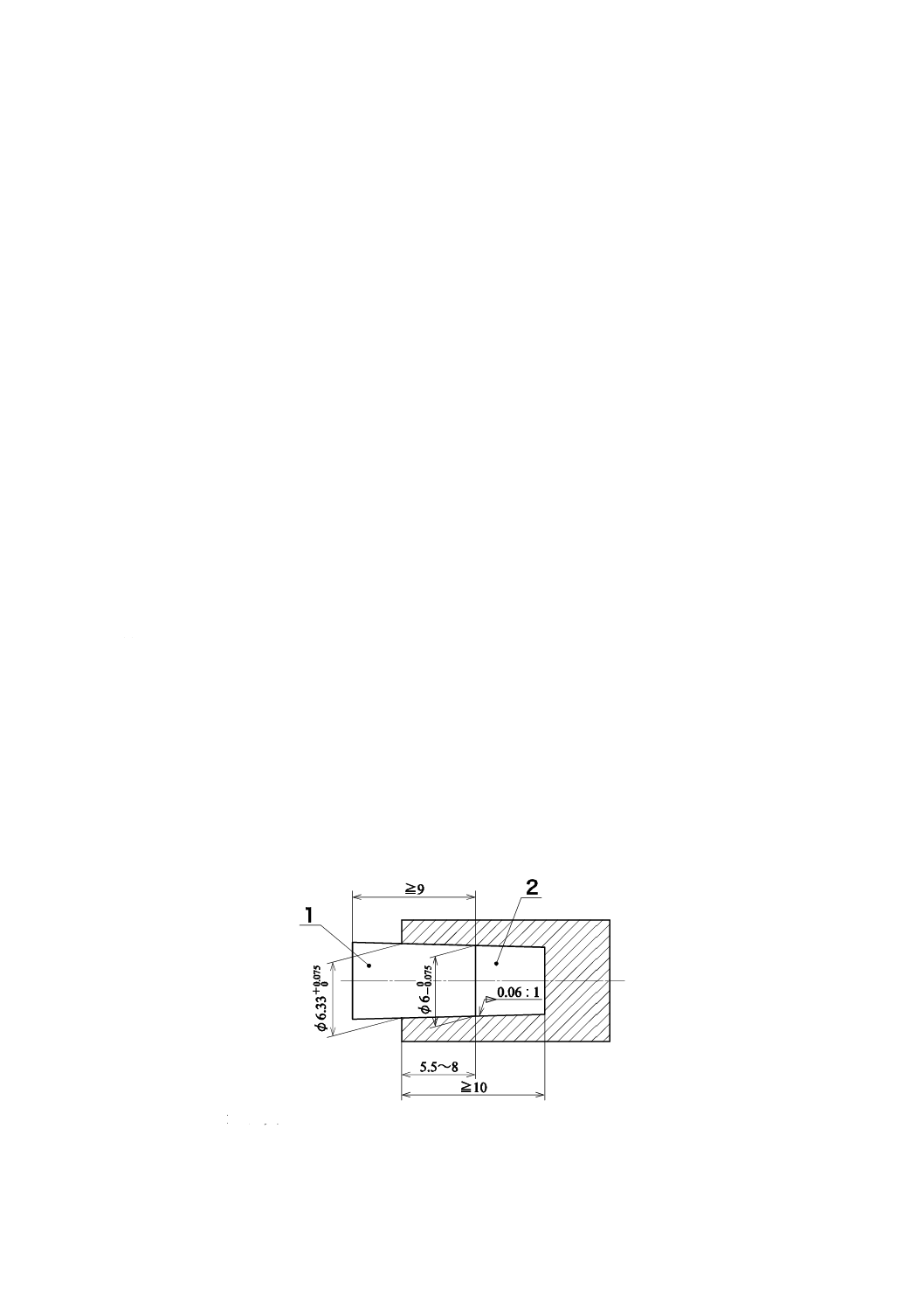
4.4.3の要求事項への適合性は,検査によって確認する(図1及び図3参照)。
単位 mm
1:アウタコーン
2:インナコーン
図3−血液出入口接続部のかん(嵌)合長測定に用いるゲージ
8
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.5.4
血液透析器及び血液透析ろ過器の透析液側接続部分
4.4.4の要求事項への適合性は,検査によって確認する(図2参照)。
5.5.5
血液ろ過器のろ液側接続部分
4.4.5の要求事項への適合性は,検査によって確認する。図2又はISO 594-2の必要条件に適合しなけれ
ばならない。
5.5.6
血液濃縮器の血液側及びろ液側接続部分
4.4.6の要求事項への適合性は,検査によって確認し,15 N以下の引張り力で外れてはならない。
5.6
性能特性
5.6.1
クリアランス
5.6.1.1
一般
4.5.1の試験は,次のとおり行う。
5.6.1.2
試験液
血液側には,一つ又は二つ以上の次の試験物質を含む透析液,生理食塩液,りん酸緩衝液又は水をかん
(灌)流させる。
血液透析器及び血液透析ろ過器の透析液側には,透析液,生理食塩液,りん酸緩衝液又は水をかん(灌)
流させる。
注記 血液側及び透析液側に当初かん(灌)流させる試験液は,イオン強度を一致させる。
尿素
15〜35 mmol/L
クレアチニン
500〜1 000 μmol/L
りん酸
1〜5 mmol/L,pH 7.4±0.1に調整する。
ビタミンB12
15〜40 μmol/L
5.6.1.3
クリアランス試験手順
図4に示す試験回路を組み立てる。血液の流れと透析液の流れとを安定させる。温度,圧力及び限外ろ
過量が安定していることを確認する。血液透析器又は血液透析ろ過器から,エアが除去されていることを
確認する。指定された範囲の血液側流量と透析液側流量とで,定常状態に達した後,試験液を採取する。
限外ろ過流量はそれぞれの条件で記載しなければならない。試験液を分析し,5.6.1.4の計算式に基づいて
クリアランスを計算する。
注記1 図4では,試験液を血液透析器又は血液透析ろ過器の下側から血液側に流入させているが,
血液側と透析液側とが向流になっていれば,上側から流入させても問題ない。また,血液透
析器又は血液透析ろ過器の配置は,垂直方向と水平方向との同等性が確認されていれば,水
平方向で試験を行ってもよい。
注記2 測定の信頼性を確認するための実際的な方法は,マスバランス・エラーを評価することであ
る。
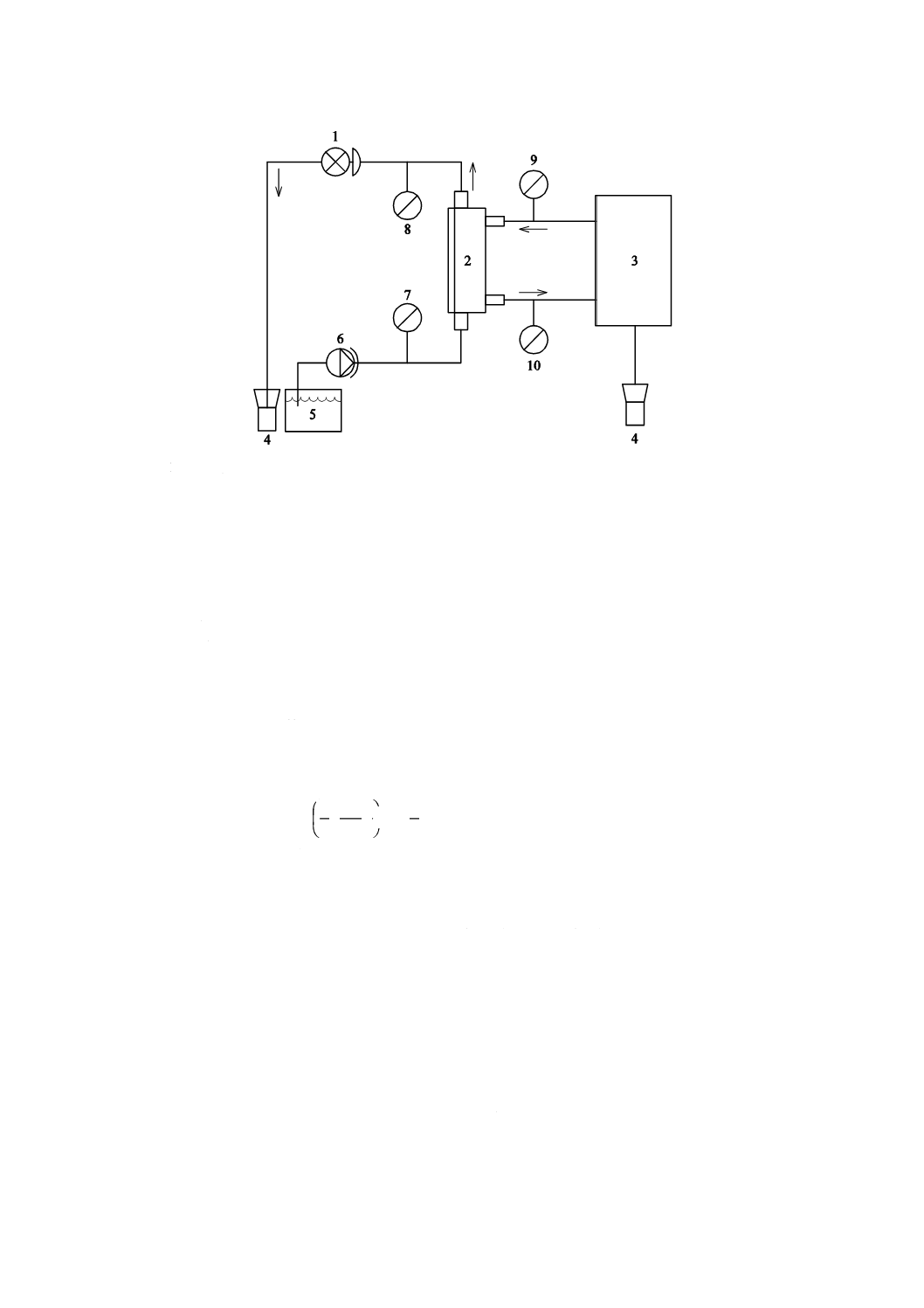
9
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1 圧力調整器
2 血液透析器又は血液透析ろ過器
3 除水調整機能付き透析液供給装置
4 廃液
5 試験液リザーバ
6 血液ポンプ
7 血液入口側圧力計
8 血液出口側圧力計
9 透析液入口側圧力計
10 透析液出口側圧力計
図4−血液透析器又は血液透析ろ過器のクリアランス測定のための試験回路図
5.6.1.4
クリアランスの計算式
クリアランスの計算式は,次による。式において,CA及びCVの測定は,同じ単位を用いなければなら
ない。
F
A
V
B
A
V
A
q
C
C
q
C
C
C
K
+
−
=
ここに,
K: クリアランス
CA: 血液透析器又は血液透析ろ過器の入口側の溶質濃度
CV: 血液透析器又は血液透析ろ過器の出口側の溶質濃度
qB: 該当機器の入口の血液流量
qF: ろ過流量(限外ろ過流量)
5.6.2
血液透析ろ過器,血液ろ過器及び血液濃縮器のふるい係数
5.6.2.1
一般
4.5.2への適合性の確認は,5.6.2.2〜5.6.2.4の試験によって実施する。
5.6.2.2
試験液
試験液として,総たん(蛋)白濃度6.0±0.5 g/dLの抗凝固化した牛血しょう(漿),又はヘマトクリッ
ト値(32±3)%及びたん(蛋)白濃度6.0±0.5 g/dLの抗凝固化した血液を用いる。
血液側は,4.5.2に示す一つ又は二つ以上の試験物質を含む試験液を,かん(灌)流させる。
5.6.2.3
試験手順
図5に示す試験回路を組み立てる。血液及びろ液の流れを安定させる(温度,流速及び圧力)。血液透析
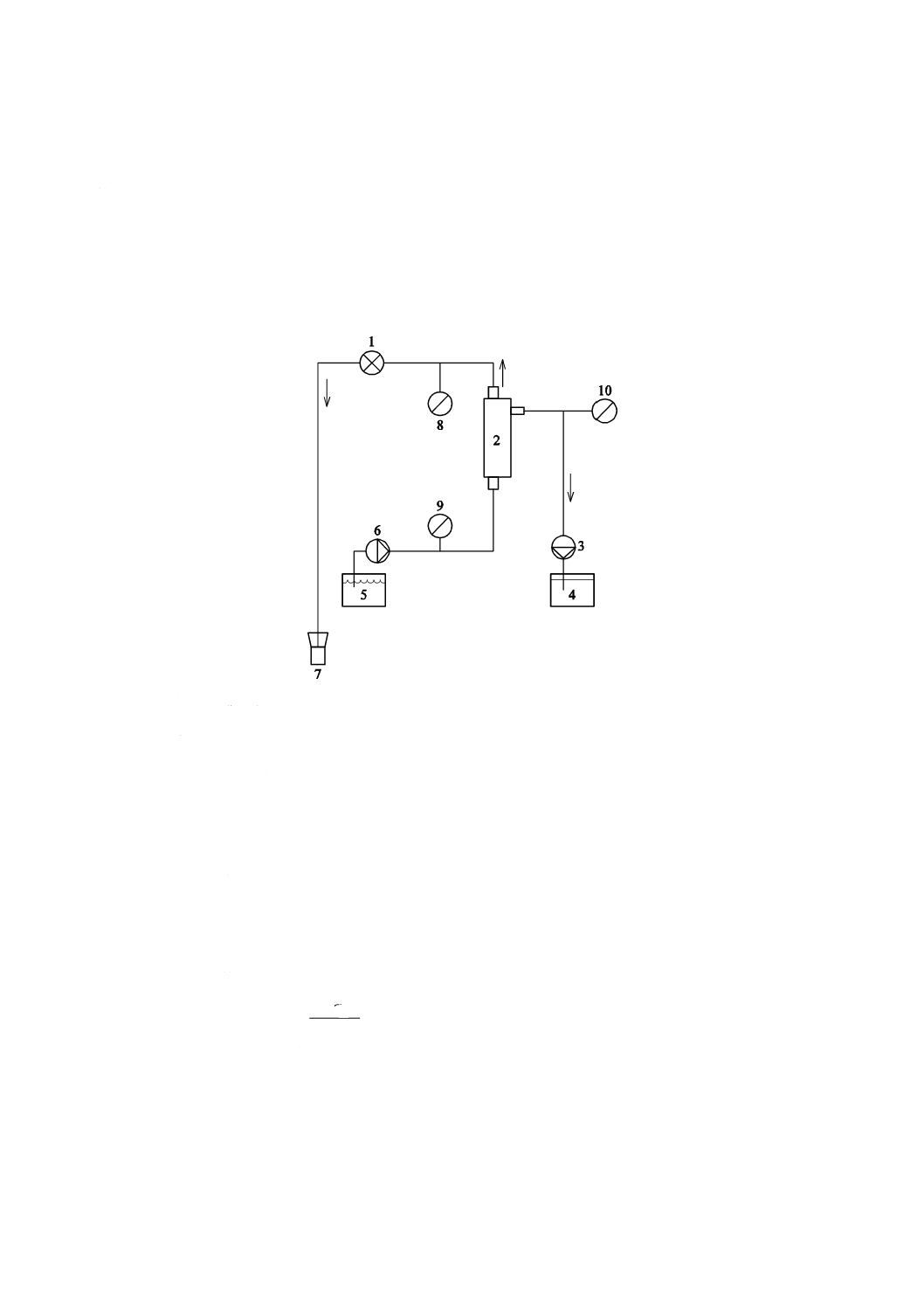
10
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ろ過器,血液ろ過器又は血液濃縮器から,エアが除去されていることを確認する。製造販売業者の定めた
UFR範囲で調節する。血液及びろ液の一対の試験液を収集する。5.6.2.4の計算式によってふるい係数を計
算する。
注記 図5では,試験液を血液透析ろ過器,血液ろ過器又は血液濃縮器の下側から血液側に流入させ
ているが,上側から流入させても問題ない。また,血液透析ろ過器,血液ろ過器又は血液濃縮
器の配置は,垂直方向と水平方向との同等性が確認されていれば,水平方向で試験を行っても
よい。
1 圧力調整器
2 該当機器
3 ろ過ポンプ
4 ろ液
5 試験液リザーバ
6 血液ポンプ
7 廃液
8 血液出口側圧力計
9 血液入口側圧力計
10 ろ液側圧力計
図5−該当機器のふるい係数,限外ろ過率測定のための回路図
5.6.2.4
ふるい係数の計算式
ふるい係数の計算式は,次による。
V
A
F
2
C
C
C
S
+
=
ここに,
S: ふるい係数
CA: 血液透析ろ過器,血液ろ過器又は血液濃縮器の入口側溶
質濃度
CV: 血液透析ろ過器,血液ろ過器又は血液濃縮器の出口側溶
質濃度
CF: 血液透析ろ過器,血液ろ過器又は血液濃縮器のろ液側溶
質濃度
11
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.6.3
限外ろ過率
5.6.3.1
試験液
血液透析器,血液透析ろ過器又は血液ろ過器の場合,試験液はヘマトクリット値(32±3)%及びたん(蛋)
白濃度6.0±0.5 g/dLの抗凝固化した牛血又は人血を使用する。血液濃縮器の場合,試験液はヘマトクリッ
ト値(25±3)%及びたん(蛋)白濃度5.0±0.5 g/dLの抗凝固化した牛血又は人血を使用してもよい。透析
液側又はろ液側は液体をかん(灌)流させない。
5.6.3.2
試験手順
図5に示すように試験回路を組み立てる。血液及びろ液の流れを安定させる(温度,流速及び圧力)。該
当機器から,エアが除去されていることを確認する。製造業者(又は製造販売業者)が定めた範囲でろ過
流量を測定する。
ろ過流量とこう(膠)質浸透圧を考慮した膜間圧力差との回帰直線の傾きから限外ろ過率を計算する。
注記 膜間圧力差が一定の値以上になると,ろ過流量と膜間圧力差とに直線性がなくなる。この値以
上の膜間圧力差では,それぞれの該当機器に応じて最大ろ過流量と表される一定のろ過流量と
なる傾向がある。
5.6.4
血液側容量(充塡量)
中空糸型の該当機器にあっては,容量は該当機器の寸法と中空糸の本数とによって計算することが可能
である。膜がぬ(濡)れることによって明らかに容量が変化することが分かっている場合には,別の方法
を用いる。
別の方法は,膜は透過しないが容易に除去できる溶液で血液側を満たす。血液側を満たすために必要な
溶液の量を測定する。製造販売業者が指定したTMPの範囲で測定を実行する。血液側の容量が変化しな
い場合は,単圧での測定でもよい。
5.6.5
血液側の圧力損失
5.6.5.1
一般
4.5.5への適合性の確認は,次の試験方法を用いて確認する。
5.6.5.2
試験液
血液側は,ヘマトクリット値(32±3)%及びたん(蛋)白濃度6.0±0.5 g/dLの抗凝固化した牛血又は同
様の粘度の液体(例えば,グリセリン水溶液又はキサンタンガム/グリセリン溶液)で作られた試験液で
満たす。透析液側又はろ液側は,通常の透析液又は生理食塩液で満たす。
5.6.5.3
試験手順
血液流量を設定し,血液側の入口及び出口圧力を測定し,圧力損失を決定する。製造販売業者の定めた
血液流量の範囲で測定を繰り返す。
積層型透析器では,透析液流量の設定並びに圧力及び血液流量の測定が必要である。
5.7
使用期限
4.6の要求事項への適合は,加速試験又は実時間試験によって,使用期限に相当する期間の保管の後,該
当機器の生物学的安全性,無菌性及び機械的特性について確認する。
6
表示
6.1
本体の表示
該当機器本体には,次の事項を表示する。
a) 製造販売業者の氏名又は名称
12
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 販売名
c) 製造販売業者の規定する該当機器の識別コード
d) ロット番号
e) 血液及び透析液の流れる方向(必要な場合)
f)
最大TMP
g) 使用期限(例えば,YYYY-MM)
h) 滅菌方法
i)
再使用禁止の旨(“ディスポーザブル”の表現は,使用しない。)
6.2
一次包装(該当機器の個包装)
次の事項を該当機器の個包装上に直接,又は個包装を通して見えるように表示する。
a) 製造販売業者の氏名又は名称,及び住所
b) 販売名
c) 製造販売業者の規定する該当機器の識別コード
d) ロット番号
e) 無菌性及び非発熱性の表示。
注記 次の三つの可能性がある。
1) 包装の中全体が滅菌されている。
2) 液体の流路(血液側及び透析液側)が滅菌されている。
3) 血液の流路だけが滅菌されている。
f)
滅菌方法
g) 使用期限(例えば,YYYY-MM)
h) 再使用禁止の旨(“ディスポーザブル”の表現は,使用しない。)
i)
“使用前に添付文書を読む”旨の記載,又は同等の内容の記載
j)
UFコントローラ装置が必要である旨の記載(該当する場合)
6.3
二次包装(外箱)
外箱上には,次の事項を表示する。
a) 製造販売業者の氏名又は名称,及び住所
b) 販売名,内容物の説明及び外箱の中に納められている該当機器の数量
c) 製造販売業者の規定する該当機器の識別コード
d) ロット番号
e) 滅菌済み及び非発熱性である旨の表示
f)
取扱い及び貯蔵についての注意・警告
g) 使用期限(例えば,YYYY-MM)
6.4
添付する文書
該当機器の外箱ごとに,次の情報を提供する。
a) 製造販売業者の氏名又は名称,及び住所
b) 販売名
c) 用法
1) 装置の製造販売業者の取扱説明書に従う旨の記載。必要な場合,補助的な該当機器の取付方法
2) 体外回路連結部の位置決め及び透析液チューブ連結部の位置決め(該当する場合)
13
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) 該当機器の推奨されるプライミング,リンス及び終了の手順
4) 血液の流れの方向(必要な場合)
5) 回路図
6) 抗凝固の必要性及び医師の処方に従う旨の表示
7) 必要な関連機器の詳細
d) 注意及び警告事項
1) 圧力の制限
2) 透析液流量の制限(血液透析器及び血液透析ろ過器だけに適用。)
3) 血液流量の制限
4) 使用前に該当機器を推奨する方法で洗浄するという指示
5) 必要性のある特別な装置
6) 既知の副作用のリスト
7) 小児には推奨しない,透析液の脱気機能をもたない装置では使用しないなどの一般的又は特別な禁
忌のリスト
8) 該当機器を一定の流量以下,一定の圧力以下,又は特定の位置(水平,垂直など)で使用した場合
に性能が低下する,という適切な警告及び禁忌
e) 製造販売業者の規定する該当機器の識別コード(カタログ番号)
f)
滅菌済み,非発熱性である旨,及び滅菌方法の表示
g) 再使用禁止の旨(“ディスポーザブル”の表現は,使用しない。)
h) 該当機器の性能データは,含まれるか又は参照できるようにする。透析器の性能データは,新しい機
器の場合は,膜面積,クリアランス,ふるい係数,限外ろ過率,透析液側及び血液側の圧力損失,並
びに血液容量を含む。
なお,性能データには,次を含むか又は参照可能とする。
1) 妥当な場合,インビトロの結果がインビボの結果と異なる可能性があることが分かれば,その違い
の大きさの推定についての記載
2) 観察の期間において性能が変化する場合がある旨の記載(該当する場合)
3) 性能特性の決定に用いた試験方法
i)
膜の一般名,及び該当する場合,その商標名
注記 膜の一般的な名称には,膜素材の完全な化学名を含む。
j)
機器の一般的説明
注記 この情報には,特別な制御装置を必要とする限外ろ過性能,又は透析液側の気泡による弊害
などの特徴を記載する。
k) 透析液側接続部分,又はろ液側接続部分に推奨するコネクタ
l)
血液側接続部分が図1及び図2でない場合,該当機器と適合する血液回路コネクタのタイプを明記す
る。
m) 血液と直接又は間接的に接触する該当機器を構成する材料の一般名
6.5
図記号の使用
6.1〜6.4の事項は,JIS T 0307に規定する適切な図記号を使用することによってこれに替えてもよい。
注記 JIS T 0307に規定する主な図記号の例を,表1に示す。

14
T 3250:2013
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表1−JIS T 0307に規定する主な図記号の例
参考文献 JIS T 3248 透析用血液回路
注記 対応国際規格:ISO 8638,Cardiovascular implants and extracorporeal systems−
Extracorporeal blood circuit for haemodialysers, haemodiafilters and haemofilters(MOD)
附属書JA
(参考)
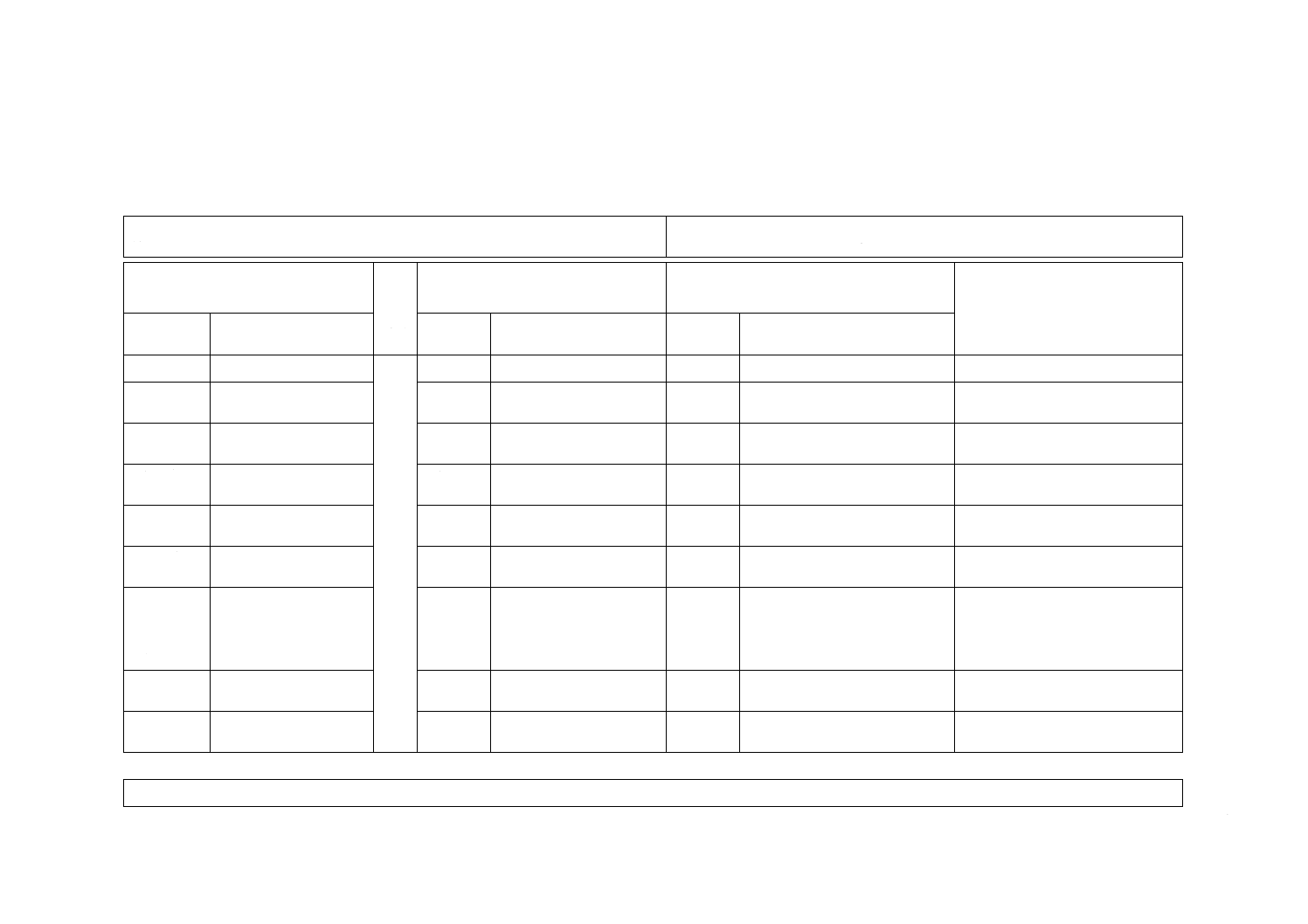
JISと対応国際規格との対比表
JIS T 3250:2013 血液透析器,血液透析ろ(濾)過器,血液ろ(濾)過器及び血液濃
縮器
ISO 8637:2010 Cardiovascular implants and extracorporeal systems−Haemodialysers,
haemodiafilters, haemofilters and haemoconcentrators及びAmendment 1:2011
(I)JISの規定
(II)
国際
規格
番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇条
ごとの評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
箇条番号及
び題名
内容
箇条番号
内容
箇条ごと
の評価
技術的差異の内容
2 引用規格
2
4.1 生物学
的安全性
4.1
JISとほぼ同じ
削除
再使用可能な機器についての記
述を削除
再使用可能な機器は我が国では販
売されていない。
4.2 無菌性
4.2
追加
他のJISとの整合。
実質的な差異はない。
5.1 一般
5.1
JISとほぼ同じ
削除
再使用可能な機器についての記
述を削除
再使用可能な機器は我が国では販
売されていない。
5.2 生物学
的安全性
5.2
JISとほぼ同じ
削除
再使用可能な機器についての記
述を削除
再使用可能な機器は我が国では販
売されていない。
6.1 本体の
表示
6.1
JISとほぼ同じ
追加
再使用禁止の表示を追加
6.2 一次包
装(該当機
器の個包
装)
6.2
JISとほぼ同じ
追加
再使用禁止の表示を追加
6.4 添付す
る文書
6.4
JISとほぼ同じ
追加
再使用禁止の表示を追加
6.5 図記号
の使用
追加
他のJISとの整合。
実質的な差異はない。
JISと国際規格との対応の程度の全体評価:(ISO 8637:2010,Amd.1:2011,MOD)
2
T
3
2
5
0
:
2
0
1
3
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。

注記1 箇条ごとの評価欄の用語の意味は,次による。
− 削除……………… 国際規格の規定項目又は規定内容を削除している。
− 追加……………… 国際規格にない規定項目又は規定内容を追加している。
注記2 JISと国際規格との対応の程度の全体評価欄の記号の意味は,次による。
− MOD…………… 国際規格を修正している。
2
T
3
2
5
0
:
2
0
1
3
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。