T 3217:2016
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 2
4 寸法及び形状 ··················································································································· 2
5 設計······························································································································· 3
5.1 一般的要求事項 ············································································································· 3
5.2 空気含有量 ··················································································································· 4
5.3 加圧取出し ··················································································································· 4
5.4 パイロットサンプル ······································································································· 4
5.5 採血能 ························································································································· 4
5.6 チューブ ······················································································································ 4
5.7 採血針 ························································································································· 5
5.8 アウトレットポート(取出口) ························································································ 5
5.9 懸垂用孔 ······················································································································ 5
6 要求事項························································································································· 6
6.1 一般 ···························································································································· 6
6.2 物理的要求事項 ············································································································· 6
6.3 化学的要求事項 ············································································································· 7
6.4 生物学的要求事項 ·········································································································· 8
7 包装······························································································································· 8
8 ラベル···························································································································· 9
8.1 一般 ···························································································································· 9
8.2 バッグのラベル ············································································································· 9
8.3 包装のラベル ················································································································ 9
8.4 出荷用箱に付けるラベル ································································································· 9
8.5 ラベルの要求事項 ·········································································································· 9
附属書A(規定)化学的試験 ································································································· 11
附属書B(規定)物理的試験 ································································································· 15
附属書C(規定)生物学的試験 ······························································································ 17
参考文献 ···························································································································· 20
附属書JA(参考)JISと対応国際規格との対比表 ······································································ 21
T 3217:2016
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,一般社団法人日本
医療機器テクノロジー協会(MTJAPAN)及び一般財団法人日本規格協会(JSA)から,工業標準原案を具
して日本工業規格を改正すべきとの申出があり,日本工業標準調査会の審議を経て,厚生労働大臣が改正
した日本工業規格である。
これによって,JIS T 3217:2011は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
T 3217:2016
血液成分分離バッグ
Plastic collapsible containers for human blood and blood components
序文
この規格は,2013年に第2版として発行されたISO 3826-1を基とし,我が国の実情に合わせるため,
技術的内容を変更して作成した日本工業規格である。
なお,この規格で側線又は点線の下線を施してある箇所は,対応国際規格を変更している事項である。
変更の一覧表にその説明を付けて,附属書JAに示す。
1
適用範囲
この規格は,血液及び血液成分の採取,保存,処理,輸送,分離及び投与を行うための,プラスチック
製の折り畳み可能な血液成分分離バッグ(以下,バッグという。)について規定する。
ただし,抗凝固剤及び保存液を含む製品は除く。
注記1 平成31年9月30日までJIS T 3217:2011を適用することができる。
注記2 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 3826-1:2013,Plastics collapsible containers for human blood and blood components−Part 1:
Conventional containers(MOD)
なお,対応の程度を表す記号“MOD”は,ISO/IEC Guide 21-1に基づき,“修正している”
ことを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS T 0307 医療機器−医療機器のラベル,ラベリング及び供給される情報に用いる図記号
JIS T 0993-1 医療機器の生物学的評価−第1部:リスクマネジメントプロセスにおける評価及び試験
注記 対応国際規格:ISO 10993-1,Biological evaluation of medical devices−Part 1: Evaluation and
testing within a risk management process(MOD)
JIS T 3212 滅菌済み輸血セット
注記 対応国際規格:ISO 1135-4,Transfusion equipment for medical use−Part 4: Transfusion sets for
single use(MOD)
ISO 10993-4,Biological evaluation of medical devices−Part 4: Selection of tests for interactions with blood
ISO 10993-5,Biological evaluation of medical devices−Part 5: Tests for in vitro cytotoxicity
ISO 10993-10,Biological evaluation of medical devices−Part 10: Tests for irritation and skin sensitization
ISO 10993-11,Biological evaluation of medical devices−Part 11: Tests for systemic toxicity
2
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ISO 10993-12,Biological evaluation of medical devices−Part 12: Sample preparation and reference materials
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
バッグ(plastics container)
血液及び血液成分を採取,保存,処理,輸送,分離又は投与するための,採血用口,輸血用口又は血し
ょう(漿)などの取出し口が付いた袋。直接採血するものを採血バッグという。
3.2
使用期限(shelf life)
滅菌年月から有効期限までの期間。有効期限後はバッグを使用できない。
4
寸法及び形状
バッグは主にバッグ本体とチューブとからなる。また,バッグを複数もつものもあり,分岐管,びん針,
採血針,雄雌コネクタ(ロック付きを含む。),ベント,逆止弁,補助バッグ,サンプリングポートなどが
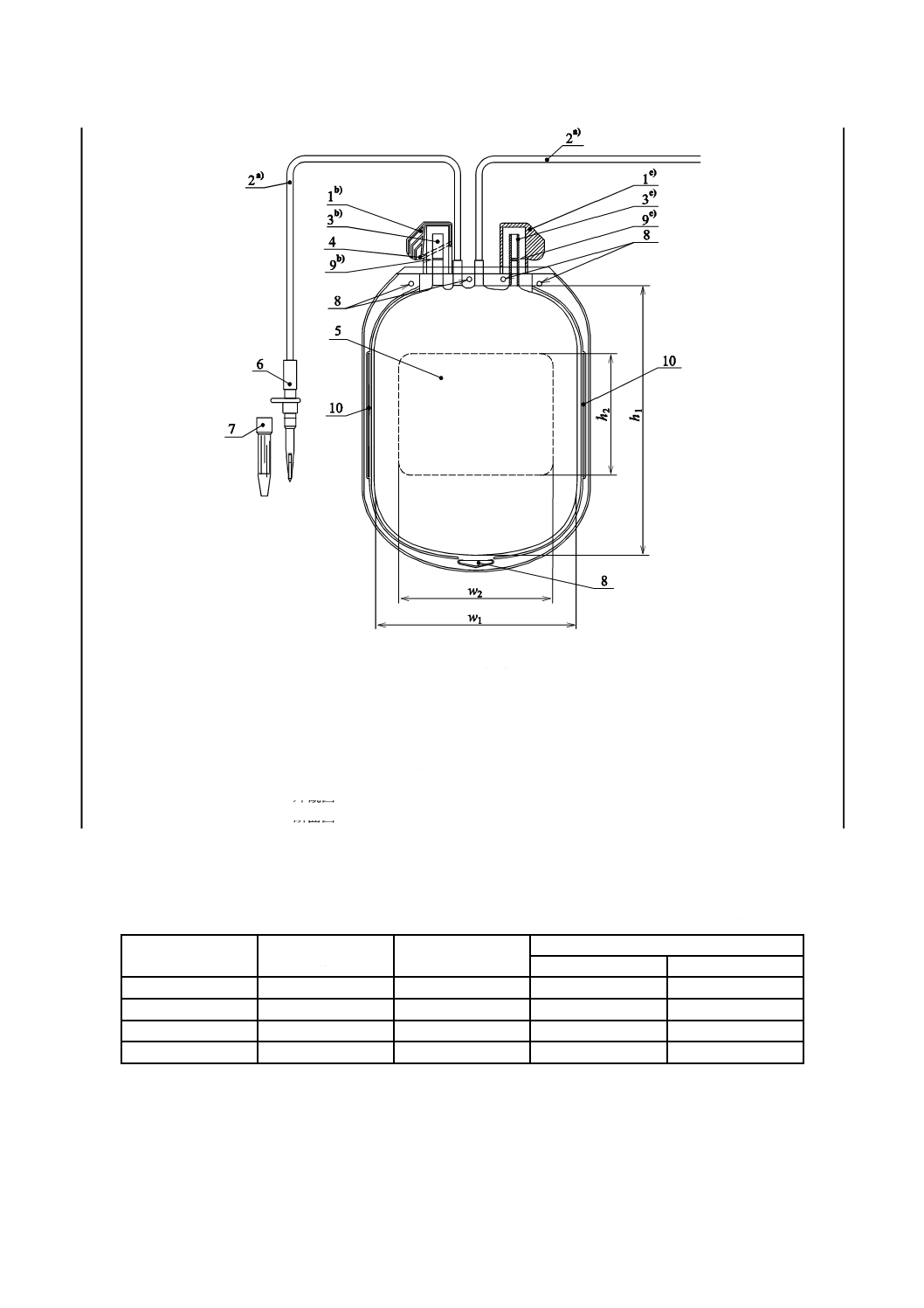
附属するものもある。バッグの一般的な構成を図1に示す。表1の寸法は参考に示す。
3
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1 取出口カバー
7 保護キャップ
2 チューブ
8 懸垂用孔
3 取出口
9 膜
4 破断線
10 サイドスリット
5 ラベル領域
6 びん針
注a) 内径2.7 mm以上,肉厚0.5 mm以上
b) 外観図
c) 断面図
図1−バッグの概略図(参考)
表1−バッグ,ラベル領域及び公称容量の寸法(参考)
単位 mm
公称容量
mL
内側の幅
w1
内側の高さ
h1
ラベル領域の大きさ
w2±5
h2±5
100
75
120
60
85
250
120
130
90
85
400
120
170
105
105
500/600
120
185
105
105
5
設計
5.1
一般的要求事項
バッグは,血液及び血液成分の採取,保存,処理,輸送,分離及び投与が安全かつ効率よく行えるよう
4
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
に設計及び製造しなければならない。バッグは,微生物による汚染の危険を最小限に抑えて,血液を採取
し,かつ,血しょう(漿)を調製,又は遠心分離若しくは再懸濁によって細胞成分を調製できるものでな
ければならない。バッグは,JIS T 3212に規定する輸血セットと機能適合性のあるものでなければならな
い。
さらに,遠心分離に用いられるバッグは,遠心分離機のカップと機能適合性のあるものでなければなら
ない。
5.2
空気含有量
5.2.1
バッグ内に含まれる空気の総量は,1袋に対して15 mLを超えてはならない。
注記 エチレンオキサイドガス滅菌品など,その滅菌の原理上,バッグ内外の空気の流通ができる構
造になっているバッグは除く。
5.2.2
使用者が製造販売業者の指示に従って使用するとき,バッグは空気を混入させず血液を充塡できる
ものでなければならない。
5.3
加圧取出し
バッグに公称容量と同量の温度23±5 ℃の水を入れ,JIS T 3212に規定する輸血セット(静脈針を除き,
流量調節器を閉じたもの)をアウトレットポート(取出口)(5.8参照)に接続し,バッグを2枚のプレー
トで挟み,内圧が大気圧より50 kPa高くなるように圧縮したとき,接続した輸血セットを通して水が漏れ
ずに平均流速250 mL/min以上で取り出せるものでなければならない。
5.4
パイロットサンプル
バッグは,適切な適合検査ができるよう,パイロットサンプルをバッグに孔をあけず採取できる設計と
する。例えば,チューブにバッグ固有のナンバリングをする。
5.5
採血能
採血針をもつバッグは,B.2 a) 又はB.2 b) に基づいて試験したとき,8分以内でバッグの公称容量を充
塡できる設計になっていなければならない。
5.6
チューブ
5.6.1
バッグには,血液及び血液成分の採取又は分離を可能にする1本又は複数本のチューブがあり,分
岐しているものもある。意図しないバッグ間の流通を回避する必要がある場合,初めは密封として機能し,
密封を解いた後は,両方向いずれの方向にでも血液成分が流れるようにする器具を取り付けなければなら
ない。
5.6.2
チューブは密封することができ,通常の使用状態の下では潰れないものでなければならない。
5.6.3
公称容量まで水を充塡し,密封したバッグ及びそれに接続したチューブは,室温(23±5 ℃)で接
合部端面に20 Nの引張力を15秒間加えたとき,接合部分から水漏れがあってはならない。室温(23±5 ℃)
の温度において,バッグの平面の縦軸に沿って,接合部端面と直角方向に張力を加えるものとする。また,
バッグは,6.2.7に適合したものでなければならない。
5.6.4
目視で検査したとき,チューブには裂け目,膨れ,よじれ,又はその他の欠陥があってはならない。
5.6.5
チューブを無菌接合する場合の要件を,次に示す。
− チューブ形状は,容器間の血液及び血液成分の効率的な移動を可能にする。
− 設計上,単一の製造販売業者によって,又は無菌接合装置を使用して,異なる製造販売業者から供給
されたチューブの接合を可能とする。
− 一般的に,二次的処理によって,血液成分を調製する際に個別の容器の接続を可能にする。
− 無菌接合装置は,無菌状態を維持しながらチューブの二つの端部を接ぎ合わせる。
5
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
− 無菌接合装置の製造販売業者は,一般的には,その機器の使用に許容されるチューブの寸法(外径及
び/又は内径並びに肉厚)を指定する。バッグの製造販売業者は,輸血業務におけるチューブ接合の
ための適合性を評価できるようにするために,製品資料などで,原材料,内径,外径及び肉厚を指定
する必要がある。
− 輸血業務従事者は,異なる仕様のチューブを接合することを希望する場合,実施する前に検証を行う
べきである。このためのプロトコルを,それらの検証のための最低限の基準(B.5参照)として設け
る(参考文献[9]参照)。
5.7
採血針
採血針をもつバッグの場合には,針はチューブと一体のもので,保護キャップで覆われていなければな
らない。保護キャップは,保存期間中,バッグから液が漏れるのを防ぎ,かつ,液の通路を無菌状態に保
ち,容易に取外し可能なものでなければならない。
保護キャップはキャップを交換できないような機構,又はキャップに不正操作したことが一目瞭然に分
かるような機構を備えなければならない。
採血針の内外面は,清潔で,仕上げが滑らかでなければならない。また,刃面は鋭利で,凹凸,ばり,
切り粉があってはならない。
採血針は,チューブの軸方向に20 Nの引張力及び押し込み力を15秒間加えたとき,組立部品から緩む
ことなく同じ引張力及び押し込み力に耐えなければならない。
採血針には,針刺し防止形のものも含む。
5.8
アウトレットポート(取出口)
5.8.1
バッグには,血液及び血液成分を投与するときに使用する1か所又は数箇所の取出口を設けなけれ
ばならない。取出口の再密閉不可能な閉鎖部(単数又は複数)は,JIS T 3212に規定するせん(穿)刺器
具を備えた輸血セットの接続時又は使用時に,加圧取出し(5.3参照)で漏れてはならない。
閉鎖部は,せん(穿)刺器具の先端によってせん(穿)刺される前に,取出口がしっかりとせん(穿)
刺器具によって閉塞されなければならない。使用者が製造販売業者の定める操作手順に従って使用すると
き,せん(穿)刺器具は,バッグをきずつけるものであってはならない。
注記1 せん(穿)刺器具は,JIS T 3212を参照する。
注記2 せん(穿)刺器具との良好なかん(嵌)合を確保できる取出口を設計する場合,製造販売業
者は,過度に硬いチューブの使用は避けるべきである。ねじれ及び座屈を起こしやすい肉厚
が薄い(1 mm未満)チューブの使用も避けるべきである。
5.8.2
取出口は完全に密封し,不正開封が一目瞭然であり,内側表面の無菌状態を維持するための機構を
もつものとする。
5.8.3
JIS T 3212に適合したせん(穿)刺器具がバッグの取出口に挿入されたとき,15秒間15 Nの引張
力に耐えなければならない。
5.8.4
5.3に従って試験したとき,せん(穿)刺器具及びバッグの取出口との間の接続から,漏れがあっ
てはならない。
5.9
懸垂用孔
バッグには,採取,保存,処理,輸送及び投与中に,バッグの使用に支障を来さない懸垂用孔又は位置
決め用の孔がなければならない(例えば,図1の8参照)。懸垂用孔又は位置決め用の孔は,23±5 ℃の
温度で,取出口の軸方向に20 Nの引張力を60分間加えたとき,破損があってはならない。
6
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6
要求事項
6.1
一般
バッグは,透明で,実質的に無色(6.2.4参照)であり,柔軟性があって,無菌,非発熱性で,毒性(6.4
参照)がなく,使用条件の下ではもろ(脆)くないもの(6.2.5参照)とする。バッグは,通常の使用条件
の下では,内容物に適合するものとする。バッグは,製造の最終段階で滅菌し,滅菌中又はその後に,使
用期限中において40 ℃を超えない温度で保管した場合,粘着するものであってはならない。
バッグは,その使用期限中,内容物に関して生物学的,化学的及び物理的に安定しているものであり,
微生物の侵入を許すものであってはならない。
抗凝固剤及び/又は保存液,血液及び血液成分との化学的相互作用又は物理的溶解のいずれかによって
バッグから浸出する物質は,規定された限度内でなければならない。
6.2
物理的要求事項
6.2.1
製造条件
バッグの製造,組立及び保存に関する全プロセスは,清浄で衛生的な条件下で行う。微生物又は異物に
よる偶発的汚染のリスクを低減するため,実行可能なあらゆる予防措置が全ての段階で取られなければな
らない。
6.2.2
滅菌
6.2.2.1
バッグは,高圧蒸気滅菌又はその他の実証された方法によって滅菌されなければならない。
6.2.2.2
使用する滅菌方法は,材料又は内容物に悪影響を与えず,接続部の緩み及びプラスチック材料の
溶接の劣化,又はバッグの形状に大きな変化を引き起こしてはならない。
6.2.2.3
製造販売業者は,実際に使用する滅菌プロセスの有効性に関し,行政機関などが承諾できる証拠
を作成しなければならない。行政機関などから要求された場合は,滅菌の有効性を確認するための陽性コ
ントロールを各滅菌ロットに含める。
6.2.3
透明性
B.1に従って試験したとき,精製水を注入した同様のバッグと比較して,バッグを通して見たときに,
懸濁液の乳白色を確認できるものとする。
6.2.4
変色
滅菌されたバッグは,血液の色の評価に悪影響を及ぼすような程度まで変色してはならない。
6.2.5
熱安定性
この要求事項は,主に血しょう(漿)凍結バッグに適用する。
バッグは,公称容量の半分まで水を充塡して,−80 ℃で24時間保管し,さらに,37±2 ℃の水中に60
分間浸し,室温に戻したとき,5.6.3,5.9,6.2.7及び6.2.8の要求事項に適合しなければならない。急速冷
凍又は照射を行うことを意図したバッグは,それらの適用に関し検証するものとする。冷凍剤溶液を使用
する場合,バッグは,冷凍剤溶液とバッグとの直接の接触を避けるため,保護バッグ内に封入してもよい。
6.2.6
水蒸気透過
バッグは,外部包装のない状態で,公称容量まで精製水を注入し,密封し,いつでも使用できるように
ラベルを貼る。バッグはその後,温度4±2 ℃,42日間,溶液から質量分率で2 %を超える水を失うこと
なく保存できなければならない。
注記 血小板濃厚液などのある種の血液成分の保存には,酸素及び二酸化炭素について特定のガス交
換率が要求されることがある。
6.2.7
漏出抵抗性

7
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
公称容量まで水を注入して密封したとき,バッグは,37 ℃で10分間,5 000 gの遠心分離条件下で漏出
を生じてはならない。バッグは,その後,23±5 ℃で10分間,内部圧力が大気圧を50 kPa超えるまで2
枚のプレートで圧搾する。目視検査で漏出があってはならない。ポリ塩化ビニル(PVC)のバッグについ
ては,さらに,4 ℃で両方の試験を繰り返す。溶液なしで通常に遠心分離されるバッグは,溶液のない状
態で上記と同じ遠心分離条件を適用する。この後バッグは,公称容量まで溶液を注入した後,大気圧を50
kPa超える内部圧力に耐えなければならない。
注記 バッグは,ACD溶液などの抗凝固剤溶液又は同様のpHの溶液で満たされた場合,漏出は,バ
ッグを青色リトマス紙のシートに押しつけ,赤色の発生が観察されることで検出できる。
他のpHの溶液についても,適切な指標で同じ方法を用いることができる。同程度の感度を
もつ代替的な方法を用いてもよい。
6.2.8
微粒子汚染
バッグは,粒子による汚染を最小限にするように製造する。
B.4に規定した方法で試験したとき,バッグ内の液の通路は,目に見える粒子がない状態でなければな
らない。
注記 当面の間,例えば,非経口溶液について,欧州薬局方で指定されるものなど,薬局方が定める
限度及び試験手順を使用してもよい。
6.3
化学的要求事項
6.3.1
バッグ又はシートの要求事項
バッグ又はシートは,各国の薬局方の要求事項を満たさなければならない。また,表2に規定する強熱
残分試験を行うことでもよい。
表2−ポリオレフィン及びポリ塩化ビニル(PVC)の強熱残分
試験
バッグの材料
規格
規定する試験方法
強熱残分
ポリオレフィン
0.5 mg/g以下
A.2
可塑剤を含むPVC
1 mg/g以下
6.3.2
試験液の要求事項
A.3に従って得られた抽出物について適切に試験をしたとき,表3に適合しなければならない。

8
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3−バッグからの抽出液に対する化学的限度
試験
規格
試験方法
還元性物質
1.5 mL以下
A.4.1
アンモニア
比較液より濃くない
A.4.2
塩化物イオン(Cl−)
標準溶液より混濁しない
A.4.3
金属類b)
Ba,Cr,Cu,Pb
各金属につき1 mg/L以下
A.4.4.1
Sn,Cd
各金属につき0.1 mg/L以下
Al
0.05 mg/L以下
重金属類
2 mg/L以下
A.4.4.2
酸性度又はアルカリ度
溶液はだいだい(橙)色−赤色
A.4.5
蒸発残留物
5 mg又は50 mg/L以下
A.4.6
乳光度
僅かに乳光を呈するが,対照懸濁液よりも顕著でない。
A.4.7
着色度
無色である。
A.4.8
UV吸収
230 nm〜360 nmの範囲で
公称容量≦100 mLのバッグについては,0.25以下
公称容量>100 mLのバッグについては,0.2以下
A.4.9
抽出可塑剤 例 ジ(2-エチルヘ
キシル)フタレート(DEHP)a)
15 mg/100 mL以下
A.4.10
注a) DEHPを含むポリ塩化ビニル(PVC)にだけ適用。
b) バリウム(Ba),クロム(Cr),銅(Cu),鉛(Pb),すず(Sn),カドミウム(Cd),アルミニウム
(Al)を指す(以下同じ)。
バッグの製造に使用される材料は,製品に化学成分が浸出することから生じるリスクを最小限にするよ
う注意深く選ばなければならない。使用する材料の毒性及びバッグの生物学的安全性に対して,特別な注
意を払わなければならない。
注記 各国の薬局方には,様々な成分の組成及び限度だけでなく,Ba,Pb,Cd,Sn,Crなどの重金
属類,及び該当する場合は,例えば,塩化ビニルモノマーなどの限度を指定するプラスチック
材料に関するモノグラフが存在する。
6.4
生物学的要求事項
6.4.1
一般
バッグは,血液及び血液成分の治療上の効果に悪影響を与えず,また,過度の毒性,細胞毒性,静菌性,
殺菌性,発熱性又は溶血性反応を示す可能性のある物質が遊離してはならない。
注記 代表的毒性試験は,JIS T 0993-1及びISO 10993シリーズに規定されている(表C.1参照)。
6.4.2
微生物の不透過性
バッグは,C.3で規定する試験を行ったとき,微生物に対し不透過性とする。
6.4.3
適合性
C.4,C.5及びC.6で規定する試験を行ったとき,バッグは,血液若しくは血液成分に対し,発熱性,毒
性又は溶血性効果を示す量の物質を一切放出してはならない。
7
包装
7.1
7.2〜7.6の要求事項は,密封された一次包装1) 内のバッグに関するものである。
7.2
バッグの使用期限(3.2参照)は,安定性データを基準として製造販売業者が指定する。
7.3
一次包装1) 又はその内表面の処理をする物質は,バッグ及びその内容物のいずれにも相互作用があ
9
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ってはならない。また,かび(黴)の生長を助長してはならない。化学的防ばい(黴)剤を使用する場合
には,バッグ及びその内容物に有害な浸透又は影響を生じていないことを示す証拠を提示する。
7.4
一次包装1) は,不正操作があったか判別可能で,封が破られた証拠を残さずに包装を開閉すること
ができない方法で密封しなければならない。
7.5
一次包装1) は,通常の取扱い及び使用条件下で損傷に耐える十分な強度がなければならない。
7.6
バッグ及び構成部品は,チューブのねじれ及び永久的ひずみを最小限とする方法で配置・包装しな
ければならない。
注1) バッグを直接に覆う包装で,バッグの無菌性を保持するためのもの。
8
ラベル
8.1
一般
ラベルには,8.2〜8.5で規定する事項を含まなければならない。JIS T 0307で示す図記号を使用しても
よい。
8.2
バッグのラベル
バッグのラベルには,可能な場合,また,該当する場合,a) 〜g) に規定する情報を記載する。
a) 製造販売業者の名称及び住所
b) 採取される血液及び血液成分の容量(mL)又は質量(g)
c) 滅菌の状態を明示する記載
d) ロット表示2)
e) 再使用禁止であることの指示
f)
外観で異常が認められた場合は使用しないことの指示
g) 使用説明書等を参照する旨
注2) ロット表示は,製造番号,製造記号又は滅菌年月を使用してもよい。
8.3
包装のラベル
包装には,次の情報を記載する。
a) 製造販売業者の名称及び住所
b) ロット表示2)
c) 使用期限
透明な一次包装1) を使用する場合,8.2及び8.3で要求する事項は,全てバッグのラベルに記載する。
8.4
出荷用箱に付けるラベル
ラベルは,パレットに載せた状態で確認でき,次の情報を記載する。
a) 製造販売業者の名称及び住所
b) ロット表示2)
c) 使用期限
d) 保管条件
8.5
ラベルの要求事項
バッグのラベルは,次による。
a) バッグの製造販売業者及び使用者に関する情報のための適切な表示面積を確保する。
注記 通常,表示面積の30 %は製造販売業者の記入を意図し,残り70 %はバッグに血液を採取する
者の記入又は表示の追加貼付けを意図するのがよい。
10
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) バッグの一部に目に見えるようマーキングのない部分を残し,内容物を目視で十分に検査できるよう
にする。
c) ラベルから印字が,バッグの材料に伝ぱ(播)することがないようにする。
d) ラベル上の印字が使用時まで判読できなければならない。
e) ラベルに使用する接着剤は,かび(黴)の生長を助長しないこととし,バッグ及びその内容物に有害
な影響がないことを示す証拠を提供する。
f)
ラベルを剝がそうとしたときには,ラベルが破壊されなければならない。
g) B.3によって試験したとき,ラベルは水から取り出した後バッグから剝がれない。ラベル上又はバッ
グ上の印字が判読可能とする。
11
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(規定)
化学的試験
A.1 一般
滅菌された,完成品で空のバッグ,すなわち,輸血,採血,分離及び投与に使用される状態にある容器
の,血液及び血液派生物と接触する材料(バッグに使用するシート,チューブ並びにその他血液及び血液
成分と接触するあらゆる部品を含む。)から,試験用の材料を採取する。
A.2 強熱残分の測定方法
あらかじめ強熱,冷却,ひょう(秤)量し,恒量化した適切なるつぼに1.00 g〜2.00 g(小片)の材料を
量る。1時間,100 ℃〜105 ℃に加熱する。その後,550±25 ℃に強熱する。デシケーター内で冷却して
量る。一定の質量が得られるまでの強熱を繰り返す。初めの物質のグラム当たりの強熱残分の質量を計算
する。各国の薬局方で規定された同等な方法を使用してもよい。
A.3 試験液の準備
空のバッグに,注射用水を公称容量まで2回注入し,約1分間振とうし,その後空にする。すすぎ水を
排出した後,空のバッグに,注射用水を公称容量まで注入する。その後,残っている空気を除くためバッ
グを圧縮し,密封する。121±2 ℃で,加圧した飽和蒸気内で少なくとも30分間バッグを抽出する。比較
液(ブランクサンプル)として,250 mLの注射用水を使用する。加熱及び冷却の時間は,30分サイクル
タイムに含まれない。
適切な場合,抽出は,シート片又は未処理バッグの一片で行ってもよい。プラスチックシートの両面を
含め合計1 500 cm2の表面積をもつ小片を使用する。この材料を100 mLの水で2回洗い,使用後の水を捨
てる。小片から水を拭き取り,それらに250 mLの注射用水を注ぎ,121±2 ℃で,加圧した飽和蒸気内で
30分間バッグを抽出する。空試験液(ブランクサンプル)として,注射用水を同じ方法で処理する。
シート片に対する試験は,プラスチック材料が均質である場合にだけ可能である。ラミネートシートは
内表面だけを選択的に試験するため,上記のものと同等のシートに置き換える必要がある。
バッグが少なくとも121 ℃の温度で滅菌することが意図されていない場合,抽出は,代わりに,100±
2 ℃で2時間,又は70±2 ℃で24±2時間行ってもよい。この場合,選択される温度は,バッグが使用さ
れる温度よりも低くしてはならない。
1個のバッグ又は1個のシートサンプルの抽出の結果得られる溶液が,規定した試験の全部を行うため
の量として不十分である場合は,2回又はそれ以上の抽出から得られた溶液を組み合わせ,混合試験液を
作ってもよい。例えば,γ線照射,エチレン酸化物,電子線など,加熱滅菌以外の代替の滅菌方法をバッ
グに使用する場合,試験液の調製には滅菌されたバッグを使用する。
A.4 試験
A.4.1 還元性物質の測定
試験液20 mLを20 mLの過マンガン酸カリウム溶液[c(KMnO4)=0.002 mol/L]及び1 mLの硫酸[c(H2SO4)
=1 mol/L]とともに3分間沸騰させる。1.0 gのよう化カリウムを加え,明るい茶色になるまで溶液をチオ
12
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
硫酸ナトリウム溶液[c(Na2S2O3)=0.01 mol/L]で滴定する。
その後,でんぷん溶液を5滴加え,無色になるまで滴定する。
試験液及び比較液として使用する水について,過マンガン酸カリウム溶液[c(KMnO4)=0.01 mol/L]の
消費量を計算する。二つの値の差は,1.5 mLを超えてはならない。
A.4.2 アンモニアの測定
2 mLの苛性ソーダ[c(NaOH)=1 mol/L]を添加することによって10 mLの試験液をアルカリ性にし,蒸
留水で15 mLまで希釈し,その後0.3 mLのネスラー試薬3)を加える。
同時に,2 mLの苛性ソーダを加えて8 mLのアンモニア標準液[ρ (NH+4)=1 mg/L]をアルカリ性にし,
15 mLまで蒸留水で希釈し,その後0.3 mLのネスラー試薬3) を添加することによって,比較液を調製す
る。30秒後,溶液は比較液よりも黄色が強くなってはならない。
注3) 各国の薬局方を参照する。
A.4.3 塩化物イオンの測定
硝酸銀溶液[c(AgNO3)=0.1 mol/L]0.3 mLを0.15 mLの希硝酸に加える。その結果できる溶液を15 mL
の抽出液に加える。12 mLの塩化物標準液(1 L当たり5 mgのCl−)と3 mLの水を使い,同じ方法で標準
溶液を調製する。混合物を振とうする。2分後,抽出液を使用して調製された溶液は,標準溶液よりも混
濁していてはならない。直射日光は避ける。
A.4.4 金属類の測定
A.4.4.1 Pb2+に関連する重金属類
金属類Ba,Cd,Cr,Cu,Pb,Sn及びAlは,原子吸光光度法によって判定される。AAS(原子吸光光
度法)を用いた検出限界は,A.2に従い蒸発によって試験液を濃縮することで引き上げることができる。
この場合,2.5 mLの塩酸溶液[ρ(HCl)=10 g/L]を250 mLの試験液に加える。
A.4.4.2 重金属類の代替試験法
A.3に従った試験液中の金属類の原子吸光分光分析の判定の代わりに,重金属類の合計量の化学的判定
を使用することができる。1.2 mLのチオアセトアミド試薬を12 mLの試験液と2 mLのアンモニアアセテ
ート緩衝液(pH=3.5)に加え,直ちに混合する。
10 mLの鉛溶液[ρ(Pb2+)=2 mg/L]を使い,2 mLの試験液を加え,同じ方法で比較溶液を調製する。2
分後,溶液は比較溶液よりも深い暗茶色であってはならない。
A.4.5 酸性度又はアルカリ度の測定
フェノールフタレイン溶液2滴を加えた後,10 mLの試験液は,赤く着色してはならない。しかし,0.4
mL未満の苛性ソーダ[c(NaOH)=0.01 mol/L]を加えると同時に,赤色を呈するものとする。この色は,
0.8 mLの塩酸[c(HCl)=0.01 mol/L]を加えた後,再び消えなければならない。5滴のメチルレッド溶液を
加えると,溶液は,だいだい(橙)色−赤色にならなければならない。
A.4.6 蒸発残留物の測定
水浴で100 mLの試験液を蒸発させ,105 ℃で一定の重量まで乾燥させる。
A.4.7 混濁度及び乳光度の測定
A.4.7.1 一般
無色透明,中性ガラスの底が平らで,内径15 mm〜25 mmの同一の試験管を使い,次に規定するとおり
に調製し,層の深さが40 mmの新鮮な標準懸濁液と,試験する液体とを比較する。標準懸濁液の調製5分
後,拡散する昼光の中で,背景を黒にして垂直に見て,溶液を比較する。光の拡散は,標準懸濁液1を水
と容易に見分けられ,標準懸濁液2を標準懸濁液1と容易に見分けられるようにする。

13
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A.4.7.2 試薬
A.4.7.2.1 ヒドラジン硫酸塩溶液 1 gのヒドラジン硫酸塩を水に溶かし,100 mLに希釈する。4時間〜6
時間静置する。
A.4.7.2.2 ヘキサメチレンテトラミン溶液 ガラス栓付100 mLフラスコ内で,2.5 gのヘキサメチレンテ
トラミンを25 mLの水に溶かす。
A.4.7.2.3 一次乳光懸濁液 ヘキサメチレンテトラミン溶液(A.4.7.2.2参照)に25 mLのヒドラジン硫酸
塩溶液(A.4.7.2.1参照)を加える。混合し,24時間静置する。この懸濁液は,表面に欠陥のないガラス容
器で保存する場合,2か月間安定である。懸濁液は,ガラスに付着してはならず,使用前に十分にかくは
ん(攪拌)する。
A.4.7.2.4 乳光の標準 15 mLの一次乳光懸濁液(A.4.7.2.3参照)を水で1 000 mLに希釈する。
この懸濁液は,新たに調製する。最大で24時間保存することができる。
A.4.7.2.5 標準懸濁液 表A.1によって標準懸濁液を調製する。使用前に混合し,振とうする。
表A.1−標準懸濁液
単位 mL
標準懸濁液
1
2
3
4
乳光の標準
5
10
30
50
水
95
90
70
50
A.4.7.3 結果の表現
A.4.7.3.1 液体は,A.4.7.1及びA.4.7.2で説明した条件下で試験したときに透明度が水若しくは使用した
溶液と同じである場合,又は乳光度が標準懸濁液1と見分けがつかない場合に,透明であるとみなす。
A.4.7.3.2 液体は,乳光度がA.4.7.3.1で説明したよりもはっきりしているが,標準懸濁液2とは見分けが
つかない場合に,僅かに乳光度を呈しているとみなす。
A.4.7.3.3 液体は,乳光度がA.4.7.3.2で説明したよりもはっきりしているが,標準懸濁液3とは見分けが
つかない場合に,乳光度を呈しているとみなす。
A.4.7.3.4 液体は,乳光度がA.4.7.3.3で説明したよりもはっきりしているが,標準懸濁液4とは見分けが
つかない場合に,強い乳光度を呈しているとみなす。
A.4.8 着色度の測定
A.4.8.1 一般
茶−黄−赤の範囲における液体の着色度の試験は,次のA.4.8.2及びA.4.8.3で規定する二つの方法のう
ちの一つで実施しなければならない。
A.4.8.2 方法1 内径12 mmで無色透明な中性ガラス製の同形管を用い,試験する液体2 mLを水2 mL
と比較する。拡散光の中で背景を白にして水平に見て,色を比較する。
A.4.8.3 方法2 内径16 mmで無色透明の中性ガラス製の同形管を用い,試験する液体10 mLを水10 mL
と比較する。拡散光の中で背景を白にして,管の軸方向に見下ろす方向で液を吟味する。
A.4.8.4 結果の表現
液体は,方法1又は方法2に規定した条件下で試験したとき,水の外観をもっている場合に,無色であ
るとみなす。
A.4.9 UV吸収の測定
14
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ブランク状態と対比し,1 cmの試料容器内の抽出液のUV吸収度を測定する。吸収度は,230 nm〜360 nm
の範囲で測定する。
A.4.10 抽出フタル酸ジ-2-エチルヘキシル(DEHP)可塑剤の測定
注記 この測定は,DEHPを含むポリ塩化ビニル(PVC)に対してだけ行う。
A.4.10.1 試薬
A.4.10.1.1 エタノール 体積分率(φ)95.1 %〜96.6 %,密度(ρ)0.805 0 g/mL〜0.812 3 g/mLの範囲
A.4.10.1.2 抽出溶媒 密度(ρ)0.937 3 g/mL〜0.937 8 g/mLのエタノールと水との混合液
A.4.10.1.3 フタル酸ジ-2-エチルヘキシル (C24H38O4),無色,水に不溶の油性の液体,有機溶剤に可溶で
密度(ρ)0.982 g/mL〜0.986 g/mL,屈折率nD20 1.486〜1.487
A.4.10.2 DEHP測定用の標準溶液の準備
A.4.10.2.1 溶液1 1 gのDEHP(A.4.10.1.3参照)をエタノール(A.4.10.1.1参照)に溶かし,100 mLの
エタノールで希釈する。
A.4.10.2.2 溶液2 10 mLの溶液1(A.4.10.2.1参照)を100 mLのエタノールで希釈する。
A.4.10.2.3 標準溶液A〜標準溶液E
a) 溶液A:20 mLの溶液2(A.4.10.2.2参照)を抽出溶媒(A.4.10.1.2参照)で100 mLに希釈する
(DEHP含有量:20 mg/100 mL)。
b) 溶液B:10 mLの溶液2を抽出溶媒で100 mLに希釈する(DEHP含有量:10 mg/100 mL)。
c) 溶液C:5 mLの溶液2を抽出溶媒で100 mLに希釈する(DEHP含有量:5 mg/100 mL)。
d) 溶液D:2 mLの溶液2を抽出溶媒で100 mLに希釈する(DEHP含有量:2 mg/100 mL)。
e) 溶液E:1 mLの溶液2を抽出溶媒で100 mLに希釈する(DEHP含有量:1 mg/100 mL)。
A.4.10.3 検量線
標準溶液として抽出溶媒を使い,272 nmで標準溶液(A.4.10.2.3参照)の最大吸収度を測定し,DEHP
濃度に対する吸光曲線をプロットする。
A.4.10.4 抽出手順
37 ℃まで加熱した抽出溶媒を,採血管を通して空のバッグに公称容量の半分まで注入する。バッグから
空気を完全に排出し,採血管を密封する。振とうすることなく,充塡したバッグを水平に保ち,37±1 ℃
に維持した湯浴に60±1分間浸す。湯浴からバッグを取り出し,ゆっくりと10回回転させてから,内容液
をガラス製フラスコに移す。標準溶液として抽出溶媒を使用し,272 nmで最大吸収度を測定する。
A.4.10.5 結果の表現
バッグについて得られた結果(A.4.10.4参照)を標準溶液の吸光検量線(A.4.10.3参照)と比較すること
によって,抽出可能なDEHPの量を決定する。
15
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(規定)
物理的試験
B.1
透明性試験
空のバッグに,1 cm光路長をもつキュベットで測定した場合に640 nmで0.37〜0.43の吸収度まで希釈
した(希釈係数約1:16)所定体積の一次乳光懸濁液(A.4.7.2.3参照)を公称容量まで注入する。
B.2
採血速度の試験
採血速度の試験は,次のa) 又はb) のいずれかに適合しなければならない。
a) 公称容量の約2倍量の,次に掲げる保存血液又は表B.1のPVP・ソルビット混合液が入っている瓶に
採血針を刺し込み,液面からバッグの頂点までの高さ50 cmの落差によって,公称容量に相当する液
量をバッグに満たすとき,空気の混入がなく,また,その所要時間は,8分以内とする。
保存血液:比重が1.052以上の血液を生物学的製剤基準の保存血液の条に規定する割合で血液保存
液に混入したものとする。ただし,既に調製されている保存血液にあっては,生物学的製剤基準の保
存血液の条の規定に従って貯蔵されたものに限る。
表B.1−PVP・ソルビット混合液
溶質及び溶液
用量
ポリビニルピロリドン(PVP)(平均分子量35 000)
30.0 g
D−ソルビット
50.0 g
注射用水
適量
全量
1 000 mL
b) 37 ℃で3.4×10−6 m2/sの粘度をもつ十分な試験液の容器から,同じ液面から5.7に適合した採血針を
通じ,圧力9.3 kPa,温度23±5 ℃で公称容量に相当する液量をバッグに満たす。
注記 この試験で使用に適した試験液は,ぶどう糖水溶液(400 g/L)である。
B.3
ラベルの耐久性試験
容量まで注入して密封したバッグを,温度4±2 ℃で24時間保存する。この最初の保存期間の後,温度
−30±5 ℃で24時間保存する。その後,温度37±2 ℃に1時間保たれた水中にバッグを沈める。
B.4
微粒子汚染の試験
B.4.1 クリーンルームの条件下で,空のバッグに,孔径0.2 μmの膜フィルタを通してあらかじめろ過さ
れた精製水を注入する。バッグの公称容量に対応する所定の体積の精製水を使用する。
B.4.2 目視できる粒子を容易に検出可能な適切な方法によって,バッグ内の液を検査する。
B.5
チューブの無菌接合試験
無菌接合装置を設置及び調整し,開始する前に,使用者を訓練する。
チューブの口径が,無菌接合装置の製造販売業者の指定された許容範囲内にあることを確認する。

16
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
無菌接合装置の製造販売業者の指示に厳密に従い,表B.2に示すチューブの組合せごとに12 cm片の間
の無菌接合を行う。個別に各接合部を特定する。
完了後,目視で欠陥がないか,全ての接合部を検査する。
密閉し,次に水面下に沈め,接合チューブの開口端部に10秒間150 kPaの圧力を加え,チューブの一端
を封止することによる圧力試験をしたとき,全ての接合部から漏れてはならない。接合部から気泡の発生
を認めない。欠陥は許容されない。
万能引張試験機を用いて500 mm/minの速度でチューブ片を延伸して各接合部の破断ひずみを測定する。
各接合部は,23±5 ℃で最小40 Nに耐えなければならない(参考文献[9]参照)。
表B.2−要求される無菌接合と試験
チューブ内容
Dry/dry
Wet/wet
Dry/wet
Wet/dry
チューブX/チューブY
5 サンプル
5 サンプル
10 サンプル
10 サンプル
湿潤状態は,体液[例えば,血しょう(漿)]又は抗凝固剤/保存液のいずれかで達成することができる。
17
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C
(規定)
生物学的試験
C.1 一般
一般的な生物学的試験には,JIS T 0993-1又はISO 10993シリーズを参照する。ISO 10993-12は試料の
調製及び標準物質を定義している。製造販売業者は,生体適合性を保証するために適切な評価を実施しな
ければならない。
C.2 試験液の調製
C.2.1 試験液I(極性溶媒)
空のバッグに,注射用水を2回公称容量まで注入し,約1分間振とうし,その後空にする。すすぎ水を
排出した後,空のバッグにエンドトキシンがなく十分に滅菌した塩化ナトリウム溶液4)[ρ(NaCl)=9 g/L]
を注入し,cm2単位で表した空のバッグの内部表面の比率が,mL単位で表した塩化ナトリウム溶液の体積
に対し,少なくとも6:1になるようにする。その後,残っている空気をバッグから出すためにバッグを圧
縮し,その後閉じる。バッグが外部包装される場合には,121±2 ℃で加圧された飽和蒸気内で少なくとも
60±12分間それを抽出する。少なくとも約250 mLの抽出物が利用可能になるよう,十分な数のバッグに
抽出を行う。それらを冷却した後,個別のバッグから得られた抽出液を混合する。比較液(ブランクサン
プル)として250 mLの滅菌したエンドトキシンのない等浸透圧の塩化ナトリウム溶液を,フラスコ内で
同じ方法で処理する。
C.2.2 試験液II(非極性溶媒)
C.2.1による試験液Iと同じ方法で試験液IIを調製する。ただし,次によって行わなければならない。
− 注射用水ですすいだ後50 ℃で1時間か,又は目視検査で水分が検出できなくなるまで空のバッグを
乾燥させる。
− 抽出剤として非経口用ごま油4)か又は綿実油4)を使用する。
− 比較液として,使用する抽出剤に従い,非経口用ごま油4)か又は綿実油4)を使用する。
− 対象の生物学的試験で記載された非極性溶媒を使用する。
注4) 各国の薬局方を参照する。
C.3 微生物の不透過性に関する試験
空のバッグに,滅菌条件下で,例えば,カゼインペプトン大豆粉ペプトンブイヨン(CaSo)などの培地
を注入し,密封する。バッグ又はバッグの適切な部分を試験に供する微生物(例えば,枯草菌:Bacillus
atropheus,NCTC 10073)を含む懸濁液(およそ106 CFU/mL)に少なくとも30分間漬ける。バッグを当該
微生物を含む懸濁液から取り出し,無菌水ですすぐ。バッグを,試験に供する微生物にとって適切な温度
(例えば,枯草菌:Bacillus atropheusでは37 ℃)で少なくとも7日間培養する。
陽性コントロールとして,同じ方法で準備されたバッグ,及び負荷する微生物の培養菌1 mLを植え付
けた内容物が使用される。代替方法として,培地を注入したユニットをきずつけることによって陽性コン
トロールを作成する。これは,試験に供するバッグの特定の部分に孔をあけることによって達成すること
ができる。微生物の生長に関し内容物を試験する。陽性コントロールは,混濁を表さなければならない。
18
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
試験物は,混濁してはならない。
C.4 エンドトキシンに関する試験
関連する各国の薬局方に従い,エンドトキシンの試験を行う。
C.5 細胞毒性に関する試験
ISO 10993-5に従い,細胞毒性試験を行う。
C.6 溶血性に関する試験
C.6.1 一般
ISO 10993-4を参照する。
C.6.2 赤血球懸濁液の調製
各国の薬局方に従い抗凝固化した,採取したばかりの新鮮なヒトの血液の体積1に対し滅菌した塩化ナ
トリウム溶液[ρ(NaCl)=9 g/L]の体積5の割合で希釈し,1 500 g〜2 000 gで遠心分離機で5分間遠心分
離を行う。上澄み液を吸引し,同じ条件,同じ体積の塩化ナトリウム溶液で赤血球の処理を繰り返す。
この方法で得られた赤血球を,滅菌した塩化ナトリウム溶液[ρ(NaCl)=9 g/L]を用いて1:9の割合に
希釈する。この懸濁液は,室温で保存した場合,最長で6時間使用することができる。
C.6.3 手順
A.3によって調製した試験液125 mLを温度100 ℃で蒸発させる。蒸発残留物を5 mLの塩化ナトリウム
溶液[ρ(NaCl)=9 g/L]に溶かし,1 mLの赤血球懸濁液と混合し,20分間37±1 ℃に保つ。その後,1 500
g〜2 000 gで5分間混合液を遠心分離する。
同時に,同じ条件で空試験液を調製する。ただし,試験液の蒸発残留物は加えない。
光路長が1 cmのキュベット内で,540 nmで空試験液と比較して,試験液の吸収度を測定する。試験液
と空試験液との吸収度の差は,10 %を超えてはならない。
注記 試験液中の揮発成分はこの試験では検出できない。しかし,試験液を濃縮することによって,
より高い試験感度が得られる。
C.7 生物学的試験方法
表C.1に生物学的試験方法を示す。
生物学的安全性の評価に当たっては,JIS T 0993-1又はISO 10993-1がガイダンスとして考慮されなけれ
ばならない。

19
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表C.1−生物学的試験方法
箇条
生物学的試験
ガイドラインが存在しない場合に推奨される試験方法
C.7.1
血液との相互作用
ISO 10993-4a)
C.7.2
細胞培養の細胞毒性
ISO 10993-5
合衆国薬局方,生物学的反応性試験,In-vitro<87>
C.7.3
溶血性
ISO 10993-4
欧州薬局方(3.2.3)
C.7.4
全身性投与(急性毒性)
ISO 10993-11
欧州薬局方
合衆国薬局方,生物学的反応性試験,In-vivo<88>
C.7.5
感作
ISO 10993-10
C.7.6
皮内注射(刺激)
ISO 10993-10
合衆国薬局方,生物学的反応性試験,In-vivo<88>
C.7.7
発熱性物質に関する試験
欧州薬局方(2.6.8)
合衆国薬局方(General Tests and Analysis,<151>)
日本薬局方(4.04)
注a) 血液との相互作用に関する試験について提案される選択:レベル1−間接的血液通路,レベル2−循環血液
20
T 3217:2016
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] ISO 3826-2,Plastics collapsible containers for human blood and blood components−Part 2: Graphical symbols
for use on labels and instruction leaflets
[2] ISO 3826-3,Plastics collapsible containers for human blood and blood components−Part 3: Blood bag systems
with integrated features
[3] ISO 9626:1991/Amd.1:2001,Stainless steel needle tubing for the manufacture of medical devices/Amendment
1
[4] ISO 15223-1,Medical devices−Symbols to be used with medical device labels, labelling and information to be
supplied−Part 1: General requirements
[5] EN 15986,Symbol for use in the labelling of medical devices−Requirements for labelling of medical devices
containing phthalates
[6] European Pharmacopoeia
[7] United States Pharmacopeia
[8] EC GMP Good Manufacturing Practice
[9] Nightingale M.J., Lees B., Biset R., Mertens W. Use of a (proposed) standard protocol to validate Terumo
TSCD-II connections between dissimilar blood bag tubing. Vox Sang. 2006, 91 pp. 241-269
[10] Nightingale M.J. Improving compatibility between blood packs and transfusion sets. Transfus. Med. 2006, 16
pp. 11-15
[11] Nightingale M.J., & Leimbach R. An evaluation of proposed changes to International Standards for blood bags
and transfusion sets to improve their compatibility. Transfus. Med. 2008, 18 pp. 281-286
附属書JA
(参考)
JISと対応国際規格との対比表
JIS T 3217:2016 血液成分分離バッグ
ISO 3826-1:2013,Plastics collapsible containers for human blood and blood components
−Part 1: Conventional containers
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇条ごと
の評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
箇条番号
及び題名
内容
箇条
番号
内容
箇条ごと
の評価
技術的差異の内容
1 適用範囲
1
JISとほぼ同じ
変更
JISでは抗凝固剤及び保存液を含む
製品を含めていない。
JISの適用範囲は抗凝固剤及び保
存液を含まない血液成分バッグで
あるため。
3 用語及び
定義
3.1
JISとほぼ同じ
変更
JISでは単に“バッグ”とした。
実質的技術的差異なし。
4 寸法及び
形状
4.1
JISとほぼ同じ
変更
JISでは文章を追記した。
JISでは不要な構成部品などを削除
した。
実質的技術的差異なし。
−
4.2
JISとほぼ同じ
削除
設計時の事例説明のため削除した。 実質的技術的差異なし。
5 設計
5.3 加圧取出し
圧縮したとき,接続
した輸血セットを
通して水がバッグ
から漏れずに平均
流速250 mL/min以
上で取り出せる。
5.3
JISとほぼ同じ
変更
水の取出し時間を変更した。
ISO規格とJISとではバッグの容
量が異なるため。
5.5 採血能
試験方法B.2 a)及び
試験方法B.2 b)
5.5
JISとほぼ同じ
追加
試験方法にB.2 a) を追加した。
ISO規格とJISとではバッグの容
量が異なるため。
5.7 採血針
採血針には,針刺し
防止形のものも含
む。
5.7
JISとほぼ同じ
変更
ISO 3826-3の文言を削除した。
対象品の補足説明であり針の規格
まで規定する必要はないため。
2
T
3
2
1
7
:
2
0
1
6
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(I)JISの規定
(II)国際
規格番号
(III)国際規格の規定
(IV)JISと国際規格との技術的差異の箇条ごと
の評価及びその内容
(V)JISと国際規格との技術的差
異の理由及び今後の対策
箇条番号
及び題名
内容
箇条
番号
内容
箇条ごと
の評価
技術的差異の内容
7 包装
7.2
7.2
JISとほぼ同じ
変更
JISでは本項の抗凝固剤及び保存液
を含む製品に関わる要求事項を含
めない。
JISの適用範囲は抗凝固剤及び保
存液を含まない血液成分分離バッ
グであるため。
8 ラベル
8
JISとほぼ同じ
変更
JISはManufacturerを“製造販売業
者”とした。その他JISに不要な項
目は含めない。
医薬品,医療機器等の品質,有効
性及び安全性の確保等に関する法
律における法定表示を考慮したた
め。
−
−
9
Anticoagulant
and/or
preservative solution
削除
JISでは本項を規定しない。
JISの適用範囲は抗凝固剤及び保
存液を含まない血液成分分離バッ
グであるため削除した。
附属書B
(規定)
B.2 採血速度の試験
a)
B.2
−
追加
試験方法にB.2 a) を追加した。
ISO規格とJISとではバッグの容
量が異なるため。
B.4 微粒子汚染の試
験
B.4
B.4.1
Test for particulate
contamination
削除
−
JISの適用は抗凝固剤及び保存液
を含まない血液成分分離バッグで
あるため削除した。
JISと国際規格との対応の程度の全体評価:ISO 3826-1:2013,MOD
注記1 箇条ごとの評価欄の用語の意味は,次による。
− 削除 ················ 国際規格の規定項目又は規定内容を削除している。
− 追加 ················ 国際規格にない規定項目又は規定内容を追加している。
− 変更 ················ 国際規格の規定内容を変更している。
注記2 JISと国際規格との対応の程度の全体評価欄の記号の意味は,次による。
− MOD ··············· 国際規格を修正している。
2
T
3
2
1
7
:
2
0
1
6
2019年7月1日の法改正により名称が変わりました。まえがきを除き、本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。