T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
(
1
)
目 次
ページ
序文
···································································································································
1
1
目的及び適用範囲
·············································································································
3
1.1
*
目的
···························································································································
3
1.2
*
適用範囲
·····················································································································
3
1.3
他の規格との関係
··········································································································
4
1.4
適合性
·························································································································
4
2
*
引用規格
·······················································································································
4
3
*
用語及び定義
·················································································································
4
4
*
一般要求事項
················································································································
10
4.1
*
品質マネジメントシステム
····························································································
10
4.2
*
リスクマネジメント
·····································································································
10
4.3
*
ソフトウェア安全クラス分類
·························································································
10
4.4
*
レガシーソフトウェア
··································································································
12
5
ソフトウェア開発プロセス
································································································
13
5.1
*
ソフトウェア開発計画
··································································································
13
5.2
*
ソフトウェア要求事項分析
····························································································
16
5.3
*
ソフトウェアアーキテクチャの設計
················································································
18
5.4
*
ソフトウェア詳細設計
··································································································
18
5.5
*
ソフトウェアユニットの実装
·························································································
19
5.6
*
ソフトウェア結合及び結合試験
······················································································
20
5.7
*
ソフトウェアシステム試験
····························································································
21
5.8
*
システムレベルで使用するためのソフトウェアリリース
·····················································
21
6
ソフトウェア保守プロセス
································································································
22
6.1
*
ソフトウェア保守計画の確立
·························································································
22
6.2
*
問題及び修正の分析
·····································································································
23
6.3
*
修正の実装
·················································································································
24
7
*
ソフトウェアリスクマネジメントプロセス
·········································································
24
7.1
*
危険状態を引き起こすソフトウェアの分析
·······································································
24
7.2
リスクコントロール手段
································································································
25
7.3
リスクコントロール手段の検証
·······················································································
25
7.4
ソフトウェア変更のリスクマネジメント
············································································
25
8
*
ソフトウェア構成管理プロセス
························································································
26
8.1
*
構成識別
····················································································································
26
8.2
*
変更管理
····················································································································
26
8.3
*
構成状態の記録
···········································································································
27
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
目次
(
2
)
ページ
9
*
ソフトウェア問題解決プロセス
························································································
27
9.1
問題報告の作成
············································································································
27
9.2
問題の調査
··················································································································
27
9.3
関係者への通知
············································································································
27
9.4
変更管理プロセスの使用
································································································
27
9.5
記録の保持
··················································································································
27
9.6
問題の傾向分析
············································································································
27
9.7
ソフトウェア問題解決の検証
··························································································
28
9.8
試験文書の内容
············································································································
28
附属書
A
(参考)この規格の要求事項の根拠
············································································
29
附属書
B
(参考)この規格の適用についての指針
·······································································
32
附属書
C
(参考)他の規格との関係
························································································
49
附属書
D
(参考)実装
··········································································································
66
附属書
JA
(参考)定義した用語の索引
····················································································
68
参考文献
····························································································································
69
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
(
3
)
まえがき
この規格は,工業標準化法第
14
条によって準用する第
12
条第
1
項の規定に基づき,一般社団法人電子
情報技術産業協会(
JEITA
)から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,日
本工業標準調査会の審議を経て,厚生労働大臣及び経済産業大臣が改正した日本工業規格である。
これによって,
JIS T 2304
:2012
は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣,経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の
特許出願及び実用新案権に関わる確認について,責任はもたない。
日本工業規格
JIS
T
2304
:
2017
(IEC 62304
:
2006
,
Amd.1
:
2015
)
医療機器ソフトウェア-
ソフトウェアライフサイクルプロセス
Medical device software-Software life cycle processes
序文
この規格は,
2006
年に第
1
版として発行された
IEC 62304
及び
Amendment 1
(
2015
)を基に,技術的内
容及び構成を変更することなく作成した日本工業規格である。ただし,追補(
amendment
)については,
編集し,一体とした。
なお,この規格で点線の下線を施してある参考事項及び
附属書
JA
は,対応国際規格にはない事項であ
る。
この規格でアスタリスク(
*
)がある箇所は,指針又は根拠についての説明を,
附属書
B
に記載してい
る。また,本文中の太字は,この規格で定義した用語である。この規格で定義した用語が,太字で表記し
ていない場合,定義は適用せず意味は文脈に沿って解釈する。
この規格は,
医療機器ソフトウェア
の
安全
設計及び保守に必要な
アクティビティ
及び
タスク
から成るラ
イフサイクル
プロセス
のフレームワーク並びに各ライフサイクル
プロセス
に対する要求事項を規定する。
各ライフサイクル
プロセス
は,一連の
アクティビティ
で構成する。さらに,大部分の
アクティビティ
は,
それぞれ一連の
タスク
で構成する。
通常,
医療機器ソフトウェア
は,品質マネジメントシステム(
4.1
参照)及び
リスクマネジメントプロ
セス
(
4.2
参照)の範囲内で開発し,維持することを前提とする。
リスクマネジメントプロセス
は,
JIS T 14971
に規定しているので,
JIS T 14971
を引用規格として参照する。また,ソフトウェアが
危険状態
1)
を引き起
こす要因であるかどうか特定する場合など,ソフトウェアの
リスクマネジメント
要求事項について若干の
追加が必要になる。これらの要求事項は,ソフトウェア
リスクマネジメントプロセス
として箇条
7
に規定
する。
注
1)
対応国際規格の
ハザード
を
危険状態
とした。
ソフトウェアが
危険状態
を引き起こす要因であるかは,
リスクマネジメントプロセス
の
ハザード
を特定
する
アクティビティ
で判断できる。判断する場合は,間接的にソフトウェアに起因する
危険状態
(例えば,
不適切な治療行為につながる可能性のある,誤解を招く情報の提供に起因する
危険状態
)も考慮する必要
がある。
リスクコントロール
手段としてソフトウェアを使用するかは,
リスクマネジメントプロセス
の
リ
スクコントロールアクティビティ
において決定する。この規格が要求する
リスクマネジメントプロセス
は,
JIS T 14971
に基づいて,機器の
リスクマネジメントプロセス
に組み込むことを規定している。
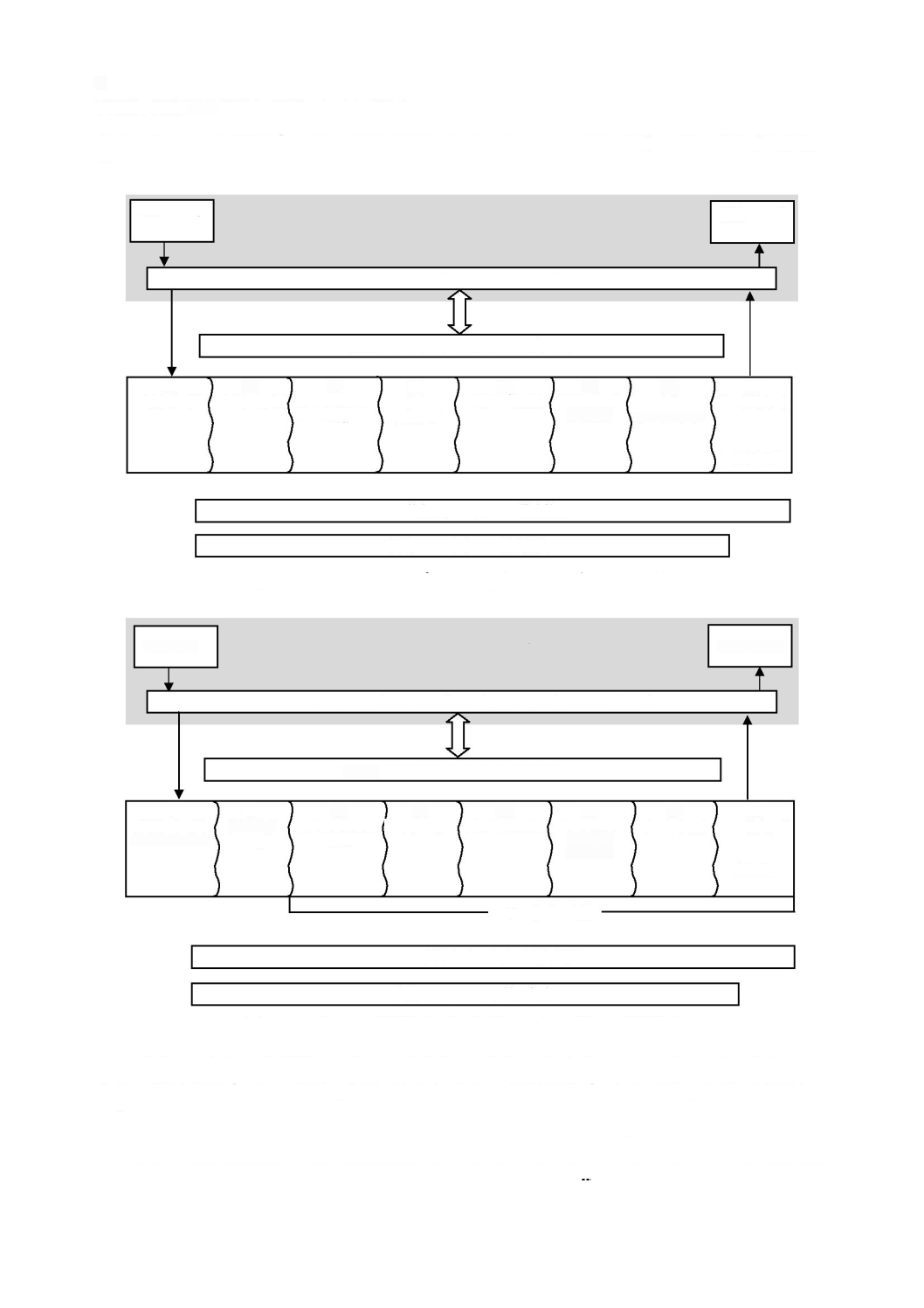
ソフトウェア開発
プロセス
は,多くの
アクティビティ
によって構成する。これらの
アクティビティ
につ
いては,
図
1
に示し,箇条
5
に記載する。現場で発生する多くの事故が,ソフトウェアの不適切なアップ
デート及びアップグレードを含む
医療機器システム
のサービス又は保守に関連するため,ソフトウェア保
守
プロセス
は,ソフトウェア開発
プロセス
と同様に重要とみなすことができる。ソフトウェア保守
プロセ
2
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
ス
は,ソフトウェア開発
プロセス
と非常に類似している。これについては,
図
2
に示し,箇条
6
に記載す
る。
図
1
-
ソフトウェア開発プロセス及びアクティビティの関連図
図
2
-
ソフトウェア保守プロセス及びアクティビティの関連図
この規格は,安全な
医療機器ソフトウェア
を開発するために不可欠な二つの
プロセス
,すなわち,ソフ
トウェア構成管理
プロセス
(箇条
8
参照)及びソフトウェア問題解決
プロセス
(箇条
9
参照)を規定して
いる。
欧州指令への規格適合性を示さなければならない
製造業者
への一助とするため,
レガシーソフトウェア
[この規格の対応国際規格の,初版発行以前に設計したソフトウェア
2)
]の取扱いについての要求事項を
保守要求
この規格の範囲外の
アクティビティ
要求の満足
システム保守
アクティビティ
(
リスクマネジメント
を含む。)
箇条
7
ソフトウェア
リスクマネジメント
6.1
ソフトウェア
保守計画の確立
6.2
問題及び
修正の分析
5.3
ソフトウェア
アーキテクチャ
の設計
5.4
ソフトウェア
詳細設計
5.5
ソフトウェア
ユニット
の実装
5.6
ソフトウェア
結合及び
結合試験
5.7
ソフトウェア
システム
試験
5.8
システムレベ
ルで使用する
ための
ソフトウェア
リリース
箇条
8
ソフトウェア構成管理
箇条
9
ソフトウェア問題解決
6.3
修正の実装
顧客ニーズ
顧客ニーズ
の満足
この規格の範囲外の
アクティビティ
箇条
7
ソフトウェア
リスクマネジメント
箇条
8
ソフトウェア構成管理
5.3
ソフトウェア
アーキテクチャ
の設計
5.2
ソフトウェア
要求事項分析
5.1
ソフトウェア
開発計画
5.4
ソフトウェア
詳細設計
5.5
ソフトウェア
ユニット
の実装
5.6
ソフトウェア
結合及び
結合試験
5.7
ソフトウェア
システム
試験
5.8
システムレベ
ルで使用する
ための
ソフトウェア
リリース
システム開発
アクティビティ
(
リスクマネジメント
を含む。)
箇条
9
ソフトウェア問題解決
3
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
規定する。また,ソフトウェア安全クラス分類を変更して,要求事項を明確化し,
リスク
に基づくアプロ
ーチを規定している。
注
2)
対応国際規格では“この規格の現行版発行以前に設計したソフトウェア”と記載しているが,
ここでは欧州指令への規格適合性に言及しているので,適用時期を明確化するため変更した。
この規格は,
製造業者
の組織の構成,及び
プロセス
,
アクティビティ
又は
タスク
を実行する組織の部門
は規定していない。この規格が要求するのは,この規格への適合性を確立するために
プロセス
,
アクティ
ビティ
又は
タスク
を完備しなければならないということだけである。
この規格は,作成する文書の名称,書式及び記載すべき内容のいずれも規定しない。この規格は,
タス
ク
の文書化を要求しているが,文書をどのようにまとめるかは規格の利用者に任せている。
この規格は,特定のライフサイクルモデルを規定していない。この規格の利用者は,ソフトウェアプロ
ジェクトのライフサイクルモデルを選択し,そのモデルにこの規格の
プロセス
,
アクティビティ
及び
タス
ク
を割り付けることが必要である。
附属書
A
は,この規格の要求事項の根拠を,
附属書
B
には,この規格の適用についての指針を記載する。
1
目的及び適用範囲
1.1
*
目的
この規格は,
医療機器ソフトウェア
のライフサイクルの要求事項について規定する。この規格に規定す
る一連の
プロセス
,
アクティビティ
及び
タスク
は,
医療機器ソフトウェア
ライフサイクル
プロセス
に共通
のフレームワークを確立する。
1.2
*
適用範囲
この規格は,ソフトウェアそれ自体が
医療機器
である場合,又はソフトウェアが完成品である
医療機器
に組み込まれているか若しくは不可欠な部分となっている場合の,
医療機器ソフトウェア
の開発及び保守
について規定する。
注記
1
この規格は,それ自体が
医療機器
であるソフトウェアの開発及び保守において使用できる。
ただし,この種のソフトウェアの使用開始前には,追加の開発
アクティビティ
が
システム
レ
ベルで必要となる。こうした
システムアクティビティ
について,この規格では規定していな
いが,
IEC 82304-1
[18]
に規定している。
この規格は,プロセッサ上で実行するソフトウェア,又はプロセッサ上で実行する他のソフトウェア(例
えば,インタプリタ)によって実行されるソフトウェアに適用するための
プロセス
について規定する。
この規格は,ソフトウェアの格納に使用する恒久的な記憶装置(例えば,ハードディスク,光ディスク
又はフラッシュメモリ)の種類にかかわらず適用する。
この規格は,ソフトウェアの提供方法(例えば,ネットワーク若しくは電子メールでの転送,光ディス
ク,フラッシュメモリ又は
EEPROM
)にかかわらず適用する。ソフトウェアの提供方法自体は,
医療機器
ソフトウェア
とはみなさない。
この規格は,その
医療機器
が全てソフトウェアで構成されている場合でも,
医療機器
の妥当性確認及び
最終的なリリースは,その対象としない。
注記
2
プロセッサ上で実行することを意図したソフトウェアが
医療機器
に組み込まれている場合
は,この規格の要求事項は,
開発過程が不明なソフトウェア
(
SOUP
)に関する要求事項も
含め,そのソフトウェアに適用する(
8.1.2
参照)。
注記
3
ソフトウェア及び
医療機器
の使用開始前には,妥当性確認などの開発
アクティビティ
が
シス
4
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
テム
レベルで必要となる。こうした
システムアクティビティ
について,この規格では規定し
ていないが,関連する製品規格(
JIS T 0601-1
,
IEC 82304-1
など)に規定している。
注記
4
この規格の対応国際規格及びその対応の程度を示す記号を,次に示す。
IEC 62304
:2006
,
Medical device software
-
Software life cycle processes
及び
Amendment 1:2015
(
IDT
)
なお,対応の程度を表す記号“
IDT
”は,
ISO/IEC Guide 21-1
に基づき,“一致している”
ことを示す。
1.3
他の規格との関係
この規格は,
医療機器
を開発する場合に,適切な他の規格と併せて使用するよう意図している。
附属書
C
に,この規格と他の関連規格との関係を示す。
1.4
適合性
この規格に適合するとは,ソフトウェア安全クラス(
4.3
参照)に従って,この規格で特定した全ての
プ
ロセス
,
アクティビティ
及び
タスク
を実行することをいう。
注記
1
各要求事項で指定するソフトウェア安全クラスは,その要求事項の末尾に記載している。
適合性の判断は,
リスクマネジメントファイル
を含むこの規格が要求する全ての文書の調査,並びにソ
フトウェア安全クラスに要求する
プロセス
,
アクティビティ
及び
タスク
を総合評価して行う。
注記
2
この総合評価は,内部監査又は外部監査によって行うこともある。
注記
3
規定した
プロセス
,
アクティビティ
及び
タスク
は実行するが,これらの
プロセス
を実装する
方法並びにこれらの
アクティビティ
及び
タスク
を実行する方法には,柔軟性がある。
注記
4
“適宜”という表記を含む要求事項を実施しなかった場合は,総合評価には実施しなかった
ことの正当性を説明するための文書が必要である。
注記
5
レガシーソフトウェア
の適合性は,
4.4
を参照する。
2
*
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。この引用
規格は,その最新版(追補を含む。)を適用する。
JIS T 14971
医療機器-リスクマネジメントの医療機器への適用
注記
対応国際規格:
ISO 14971
,
Medical devices
-
Application of risk management to medical devices
(
IDT
)
3
*
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
アクティビティ
(
ACTIVITY
)
一組以上の相互関係又は相互作用のある
タスク
。
3.2
異常
(
ANOMALY
)
要求仕様書,設計文書,規格など,又は既存の認識若しくは経験に基づいて予想した結果を逸脱する状
態。
異常
は,
医療機器ソフトウェア
又は該当する文書のレビュー,試験,分析,コンパイル又は使用中に
発見されることがあるが,これには限定しない。
5
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
(
IEEE 1044
:1993
の
3.1
参照)
3.3
アーキテクチャ
(
ARCHITECTURE
)
システム
又はコンポーネントの構造。
(
IEEE 610.12
:1990
参照)
3.4
変更要求
(
CHANGE REQUEST
)
医療機器ソフトウェア
に対する変更内容を文書化した仕様。
3.5
構成アイテム
(
CONFIGURATION ITEM
)
決められた時点で一意に特定できる“もの(
entity
)”。
(
JIS X 0160
:2012
の
4.7
参照)
3.6
成果物
(
DELIVERABLE
)
アクティビティ
又は
タスク
で要求される結果又はアウトプット(文書を含む。
)。
3.7
評価
(
EVALUATION
)
対象とする“もの(
entity
)”が,特定の基準に達していることを系統的に決定すること。
(
JIS X 0160
:2012
の
4.12
参照)
3.8
危害
(
HARM
)
人の受ける身体的傷害若しくは健康障害,又は財産若しくは環境の受ける害。
(
JIS T 14971
:2012
の
2.2
参照)
3.9
ハザード
(
HAZARD
)
危害
の潜在的な源。
(
JIS T 14971
:2012
の
2.3
参照)
3.10
製造業者
(
MANUFACTURER
)
医療機器
の市場出荷又は使用開始の前に,
医療機器
の設計,製造,こん(梱)包若しくはラベリング又
は
システム
の組合せ若しくは変更に責任を負う個人又は法人。その業務をその個人若しくは法人又は代理
を受けた第三者が行うか否かを問わない。
(
JIS T 14971
:2012
の
2.8
参照)
注記
1
国又は地域の法規制で
製造業者
を定義している場合があることに注意する。
注記
2
ラベリングの定義は,
JIS Q 13485
:2005
の
3.6
を参照する。
3.11
医療機器
(
MEDICAL DEVICE
)
あらゆる計器,器械,用具,機械,器具,埋め込み用具,体外診断薬,検定物質,ソフトウェア,材料
又はその他の同類のもの若しくは関連する物質(
article
)であって,単独使用か組合せ使用かを問わず,
製造業者
が人体への使用を意図し,その使用目的が次の一つ以上であり,
6
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
-
疾病の診断,予防,監視,治療又は緩和
-
負傷の診断,監視,治療,緩和又は補助
-
解剖学的又は生理学的な
プロセス
の検査,代替,又は修復
-
生命支援又は維持
-
受胎調整
-
医療機器
の殺菌
-
人体から採取される標本の体外試験法による医療目的のための情報提供
薬学,免疫学,又は新陳代謝の手段によって体内又は体表において意図したその主機能を達成すること
はないが,それらの手段によって機能の実現を補助するものである。
(
JIS Q 13485
:2005
の
3.7
参照)
注記
1
この定義は,
GHTF
(
Global Harmonization Task Force
)によって作成されたものである[参考
文献
[13]
参照]。
注記
2
(
JIS
に直接関係がない記載なので,削除した。)
注記
3
JIS T 0601-1
:2012
及び追補
1:2014
との関連においては,用語“
医療機器
”は,用語“
ME
機
器
”又は“
ME
システム
”と同じ意味であるとみなす。
3.12
医療機器ソフトウェア
(
MEDICAL DEVICE SOFTWARE
)
医療機器
に組み込むことを目的として開発した,又は
医療機器
として使用することを意図した
ソフトウ
ェアシステム
。
注記
それ自体が
医療機器
である
医療機器ソフトウェア
製品を含む。
3.13
問題報告
(
PROBLEM REPORT
)
ユーザ又はその他の関係者が,安全でない,意図する使用に対して不適切である又は仕様に反すると判
断した,
医療機器ソフトウェア
の実際の又は潜在的な動作の記録。
注記
1
この規格は,全ての
問題報告
に対して
医療機器ソフトウェア
の変更を要求するものではない。
製造業者
は,誤解,故障又は軽微な事象について,
問題報告
を処置の対象としなくてもよい。
注記
2
問題報告
は,リリースした
医療機器ソフトウェア
又は開発中の
医療機器ソフトウェア
に適用
する。
注記
3
この規格は,リリースした製品についての
問題報告
の法的な対応処置を,確実に特定及び実
行できるようにするため,
製造業者
に別途方針決定を行うことを要求している(箇条
6
参照)
。
3.14
プロセス
(
PROCESS
)
インプットをアウトプットに変換する,相互に関連する又は相互に作用する一連の
アクティビティ
。
(
JIS Q 9000
:2006
の
3.4.1
参照)
注記
用語“
アクティビティ
”は,資源を利用することも含む。
3.15
回帰テスト
(
REGRESSION TESTING
)
システム
コンポーネントの変更が,機能性,信頼性又は性能に悪影響を与えないこと,及び更なる欠陥
を招かないことを判定するために要求される試験。
(
ISO/IEC 90003
:2004
の
3.11
参照)
7
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
3.16
リスク
(
RISK
)
危害
の発生確率とその
危害
の重大さとの組合せ。
(
JIS T 14971
:2012
の
2.16
参照)
3.17
リスク分析
(
RISK ANALYSIS
)
利用可能な情報を体系的に用いて
ハザード
を特定し,
リスク
を推定すること。
(
JIS T 14971
:2012
の
2.17
参照)
3.18
リスクコントロール
(
RISK CONTROL
)
規定したレベルまで
リスク
を低減するか又はそのレベルで
リスク
を維持するという決定に到達し,かつ,
そのための手段を実施する
プロセス
。
(
JIS T 14971
:2012
の
2.19
参照)
3.19
リスクマネジメント
(
RISK MANAGEMENT
)
リスク
の分析,
評価
及びコントロールに対して,管理方針,手順及び実施を体系的に適用すること。
(
JIS T 14971
:2012
の
2.22
参照)
3.20
リスクマネジメントファイル
(
RISK MANAGEMENT FILE
)
リスクマネジメント
によって作成した記録及び他の文書のまとまり。
(
JIS T 14971
:2012
の
2.23
参照)
3.21
安全
(
SAFETY
)
受容できない
リスク
がないこと。
(
JIS T 14971
:2012
の
2.24
参照)
3.22
セキュリティ
(
SECURITY
)
権限を与えられていない者又は
システム
が読み込んだり変更できないように,かつ,権限を与えられて
いる者又は
システム
がアクセスを拒否されないように,情報及びデータを保護すること。
(
JIS X 0160
:2012
の
4.39
参照)
3.23
重傷
(
SERIOUS INJURY
)
次のいずれかの結果を引き起こすけが又は病気。
a)
生命の危険
b)
身体機能又は身体構造の永久的障害
c)
身体機能又は身体構造の永久的障害を防止するために,内科的又は外科的処置を必要とする障害
注記
永久的障害とは,軽微な障害又は損害を除く,身体構造又は機能の不可逆性の障害若しくは損
害を意味する。
3.24
ソフトウェア開発ライフサイクルモデル
(
SOFTWARE DEVELOPMENT LIFE CYCLE MODEL
)
8
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
ソフトウェア要求事項の定義からリリースまでの,ソフトウェアのライフサイクルに関わる次のような
概念上の構造。
-
医療機器ソフトウェア
の開発に関与している
プロセス
,
アクティビティ
及び
タスク
を明確にする。
-
アクティビティ
と
タスク
との間のシーケンス及び依存性を表す。
-
規定した
成果物
の完全性を検証するマイルストーンを明確にする。
3.25
ソフトウェアアイテム
(
SOFTWARE ITEM
)
コンピュータプログラムの識別可能な部分(例えば,ソースコード,オブジェクトコード,制御コード,
制御データ又はこれらのアイテムの集まり)。
注記
1
ソフトウェアの構造は,三つの用語によって識別できる。最上位のレベルは,
ソフトウェア
システム
である。最下位のレベルは,それ以上分解できない
ソフトウェアユニット
である。
最上位及び最下位レベルを含む構成の全てのレベルを,
ソフトウェアアイテム
ということが
できる。
ソフトウェアシステム
は,一つ以上の
ソフトウェアアイテム
で構成され,各
ソフト
ウェアアイテム
は,一つ以上の
ソフトウェアユニット
又は分割可能な
ソフトウェアアイテム
で構成される。
製造業者
は,
ソフトウェアアイテム
及び
ソフトウェアユニット
の粒度
(
granularity
)を提示する責任がある。
注記
2
ISO/IEC 90003
:2014
の
3.14
,
JIS X 0160
:2012
の
4.41
に基づく。
3.26
(対応国際規格で削除されている。)
3.27
ソフトウェアシステム
(
SOFTWARE SYSTEM
)
特定の機能又は特定の機能群を達成するために組む,複数の
ソフトウェアアイテム
を結合した集合体。
3.28
ソフトウェアユニット
(
SOFTWARE UNIT
)
他のアイテムに分割できない
ソフトウェアアイテム
。
注記
ソフトウェアユニット
の粒度は,
製造業者
が定義する(
B.3
参照)
。
3.29
開発過程が不明なソフトウェア
,
SOUP
(
software of unknown provenance
,
SOUP
)
既に開発されていて一般に利用できるが,
医療機器
に組み込むことを目的に開発したものではない
ソフ
トウェアアイテム
[
“
OTS
ソフトウェア(
off-the-shelf
:既製品)
”として知られているソフトウェア]又は
以前開発された
ソフトウェアアイテム
でその開発
プロセス
についての十分な記録が利用できないもの。
注記
医療機器ソフトウェアシステム
全体を
SOUP
であると主張することはできない。
3.30
システム
(
SYSTEM
)
一つ以上の
プロセス
,ハードウェア,ソフトウェア,設備及び人を統合化して,規定のニーズ又は目的
を満たす能力を提供するまとまり。
3.31
タスク
(
TASK
)
行う必要がある一つの作業。
9
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
3.32
トレーサビリティ
(
TRACEABILITY
)
開発
プロセス
の二つ以上の
成果物
間の関係を明らかにできる程度。
(
IEEE 610.12
:1990
参照)
注記
要求事項,
アーキテクチャ
,
リスクコントロール
手段などは,開発
プロセス
の
成果物
の例であ
る。
3.33
検証
(
VERIFICATION
)
客観的証拠を提示することによって,規定要求事項が満たされていることを確認すること。
注記
1
“検証済み”という用語は,
検証
が済んでいる状態を示すために用いられる。
(
JIS Q 9000
:2006
の
3.8.4
参照)
注記
2
設計及び開発における
検証
は,ある
アクティビティ
に対する要求事項に適合しているかを確
定するために,その
アクティビティ
の結果に対して吟味を行う
プロセス
である。
3.34
バージョン
(
VERSION
)
ある
構成アイテム
の識別された実例。
注記
医療機器ソフトウェア
の
バージョン
の変更を行って新しい
バージョン
とする場合は,ソフトウ
ェア構成管理を実施する必要がある。
(
JIS X 0160
:2012
の
4.56
参照)
3.35
危険状態
(
HAZARDOUS SITUATION
)
人,財産又は環境が,一つ又は複数の
ハザード
にさらされる状況。
(
JIS T 14971
:2012
の
2.4
参照)
3.36
レガシーソフトウェア
(
LEGACY SOFTWARE
)
法規制に適合して市場に出荷され,現在も市販されているが,この規格の現行版に適合して開発された
という客観的な証拠が不十分な
医療機器ソフトウェア
。
3.37
リリース
(
RELEASE
)
特定の目的のために用意された
構成アイテム
の特定の
バージョン
。
(
JIS X 0160
:2012
の
4.35
参照)
3.38
残留リスク
(
RESIDUAL RISK
)
リスクコントロール
手段を講じた後にも残る
リスク
。
注記
対応国際規格の
注記
1
及び
注記
2
は,
ISO/IEC Guide 51
:2014 [14]
で,
“
residual risk
”の定義が変
更されたことによって記載不要となり,削除した。
(
JIS T 14971
:2012
の
2.15
参照)
3.39
リスク推定
(
RISK ESTIMATION
)
危害
の発生確率とその
危害
の重大さとに対して重み付けをするために用いる
プロセス
。
10
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
(
JIS T 14971
:2012
の
2.20
参照)
3.40
リスク評価
(
RISK EVALUATION
)
判断基準に照らして推定した
リスク
が受容できるかを判断する
プロセス
。
(
JIS T 14971
:2012
の
2.21
参照)
4
*
一般要求事項
4.1
*
品質マネジメントシステム
医療機器ソフトウェア
の
製造業者
は,顧客要求事項及び該当する規制要求事項に適合する
医療機器ソフ
トウェア
を提供する能力があることを実証する。
注記
1
この能力は,次のいずれかに適合する品質マネジメントシステムを使用して実証できる。
-
JIS Q 13485
[5]
-
医療機器
及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令
注記
2
品質マネジメントシステム要求事項をソフトウェアに適用するための指針は,
ISO/IEC 90003
[13]
に規定している。
4.2
*
リスクマネジメント
製造業者
は,
JIS T 14971
に規定した
リスクマネジメントプロセス
を適用する。
4.3
*
ソフトウェア安全クラス分類
a)
製造業者
は,
ソフトウェアシステム
に起因する
危険状態
が,最悪の場合に患者,操作者,又はその他
の人にもたらす
危害
の
リスク
に応じて,
図
3
に示すように,各
ソフトウェアシステム
をソフトウェア
安全クラス(
A
,
B
又は
C
)に分類する。
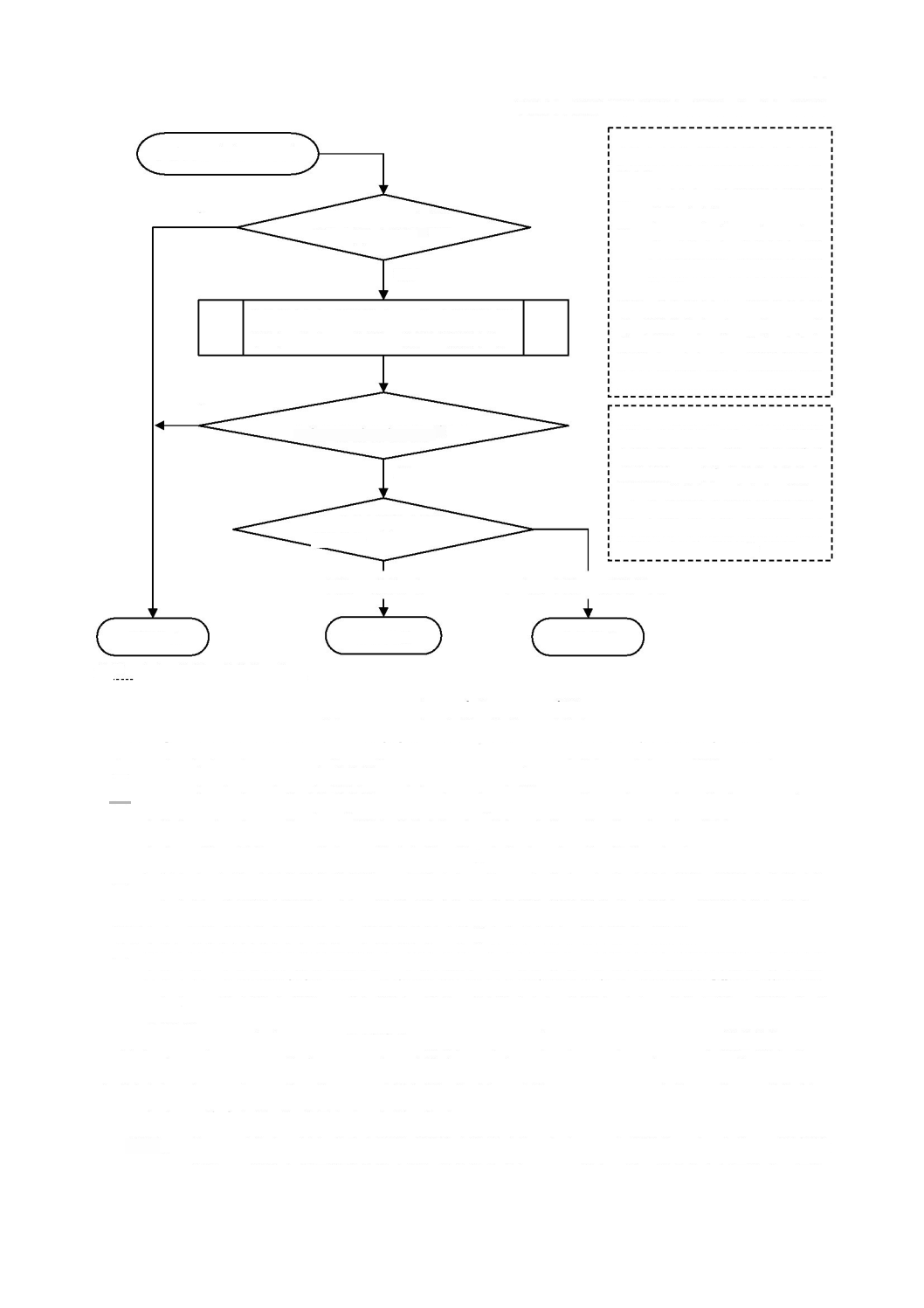
11
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
注
a)
本文の記載に整合させた。
図
3
-
ソフトウェア安全クラスの割当て
ソフトウェアシステム
のソフトウェア安全クラスが
A
となるのは,次のいずれかの場合である。
-
ソフトウェアシステム
が
危険状態
の一因とならない場合。
-
ソフトウェアシステム
が
危険状態
の一因となるが,その
ソフトウェアシステム
以外(
external to
)で
実施する
リスクコントロール
手段を考慮すれば,受容できない
リスク
は生じない場合。
ソフトウェアシステム
のソフトウェア安全クラスが
B
となるのは,次の場合である。
-
ソフトウェアシステム
が
危険状態
の一因となり,その
ソフトウェアシステム
以外で実施する
リスク
コントロール
手段を考慮しても,受容できない
リスク
が生じる場合で,
重傷
の可能性はない場合。
ソフトウェアシステム
のソフトウェア安全クラスが
C
となるのは,次の場合である。
-
ソフトウェアシステム
が
危険状態
の一因となり,その
ソフトウェアシステム
以外で実施する
リスク
コントロール
手段を考慮しても,受容できない
リスク
が生じる場合で,死亡又は
重傷
の可能性があ
る場合。
当初,ソフトウェア安全クラスを
B
又は
C
に分類した
ソフトウェアシステム
について,
製造業者
は,そ
の
ソフトウェアシステム
以外(
external to
)で実施する
リスクコントロール
手段(その
ソフトウェアシステ
ム
が含まれる
システムアーキテクチャ
の改善など)を追加で実施して,その
ソフトウェアシステム
を新し
いソフトウェア安全クラスに分類することができる。
注記
1
その
ソフトウェアシステム
以外で実施する
リスクコントロール
手段は,ソフトウェアが
危険
状態
の一因となる可能性を最小限に抑えるために,ハードウェア,独立した
ソフトウェアシ
ソフトウェアは,
危険状
態
の一因になるか
a)
そのソフトウェア以外(
external to
)で実施する
リスクコントロール
手段の有効性を評価する。
クラス
C
(デフォルト)
クラス
C
クラス
A
クラス
B
ソフトウェアによって,受容で
きない
リスク
を生じるか
a)
どのような障害の可能性
があるか
はい
いいえ
いいえ
ソフトウェアシステム
のソフトウェ
ア安全クラスを決める場合には,次
による。
ソフトウェアの故障の発生確
率は
1
とする。
その
ソフトウェアシステム
以
外(
external to
)で実施する
リス
クコントロール
手段だけを考
慮する。
注記
その
ソフトウェアシステム
以
外で実施する
リスクコントロール
手
段は,ソフトウェアの故障が
危害
を
起こす発生確率及び/又は
危害
の重
大さを減少させることができる。
注記
リスクコントロール
手段を実
装した
ソフトウェアシステム
が故障
することもあり,また,この故障が
危 険 状 態
の 一因 に な る か もし れ な
い。その結果として発生する
危害
に
は,
リスクコントロール
手段が
リス
ク
低減をすることを意図していた
危
害
のこともある[
7.2.2 b)
参照]。
重傷
の可能性はない
死亡又は
重傷
の可能性がある
はい
12
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
ステム
,医療処置又は他の手段とすることができる。
注記
2
リスク
の受容可能性の定義は,
JIS T 14971
:2012
の
3.2
(経営者の責任)を参照する。
b)
(対応国際規格で削除されている。)
c)
製造業者
は,分類した各
ソフトウェアシステム
の安全クラスを,
リスクマネジメントファイル
に文書
化する。
d)
製造業者
は,
ソフトウェアシステム
を
ソフトウェアアイテム
に分割する場合,及び
ソフトウェアアイ
テム
を更に幾つかの
ソフトウェアアイテム
に分割する場合,それらの
ソフトウェアアイテム
は,元の
ソフトウェアアイテム
(又は
ソフトウェアシステム
)のソフトウェア安全クラスを継承する。ただし,
別のソフトウェア安全クラスに分類することの正当な根拠を文書で示せば,その分類を変更してもよ
い。根拠の文書化に当たっては,新しい
ソフトウェアアイテム
をどのように分離するのかを説明して,
別分類としてもよいことを示す[ソフトウェア安全クラスは,
4.3 a)
の“
ソフトウェアシステム
”を“
ソ
フトウェアアイテム
”に読み替えて分類する。]。
e)
分割によって作成した
ソフトウェアアイテム
のソフトウェア安全クラスが,元の
ソフトウェアアイテ
ム
のクラスと異なる場合,
製造業者
は,各
ソフトウェアアイテム
のソフトウェア安全クラスを文書化
する。
f)
この規格をある
ソフトウェアアイテム
のグループに適用する場合,この規格に適合するためには,
製
造業者
は,そのグループの中で最も高い安全クラスに分類している
ソフトウェアアイテム
が必要とす
る
プロセス
及び
タスク
を使用する。ただし,
リスクマネジメントファイル
の中で根拠を示すことによ
って,より低いクラスの
プロセス
及び
タスク
を使用できる。
g)
ソフトウェア安全クラスを分類するまで,各
ソフトウェアシステム
には,クラス
C
の要求事項を適用
する。
注記
これ以降の箇条及び細分箇条において,ある特定の要求事項を適用するソフトウェア安全ク
ラスは,要求事項の後に(クラス
...
)の形式で記載している。
4.4
*
レガシーソフトウェア
4.4.1
一般
レガシーソフトウェア
の適合性は,この規格の箇条
5
~箇条
9
を適用する代わりに,
4.4.2
~
4.4.5
に示す
方法で実証してもよい。
4.4.2
リスクマネジメントアクティビティ
この規格の
4.2
に従って,
製造業者
は,次を実施する。
a)
レガシーソフトウェア
に関連する事故事例及び/又はヒヤリ・ハット事例について,社内及び/又は
使用者からの,製造後情報を含むあらゆるフィードバック情報を評価する。
b)
レガシーソフトウェア
の継続使用に伴う
リスクマネジメントアクティビティ
を,次の点を考慮して実
施する。
-
レガシーソフトウェア
の
医療機器アーキテクチャ
全体への統合
-
レガシーソフトウェア
の一部として実装した
リスクコントロール
手段の継続的有効性
-
レガシーソフトウェア
の継続使用に伴う
危険状態
の特定
-
レガシーソフトウェア
が
危険状態
の一因となる場合の潜在的原因の特定
-
レガシーソフトウェア
が
危険状態
の一因となる場合の潜在的原因のそれぞれに対する
リスクコント
ロール
手段の定義
4.4.3
ギャップ分析
13
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
製造業者
は,
レガシーソフトウェア
のソフトウェア安全クラス(
4.3
参照)に基づいて,
5.2
,
5.3
,
5.7
及び箇条
7
の要求事項に対し,使用可能な
成果物
とのギャップ分析を行う。
a)
製造業者
は,使用可能な
成果物
の継続的有効性を評価する。
b)
ギャップが特定された場合,
製造業者
は,不足する
成果物
を作成し関連
アクティビティ
を実施するこ
とによって,
リスク
をどの程度低減できるか
評価
する。
c)
製造業者
は,この
評価
に基づいて,作成する
成果物
と実施する関連
アクティビティ
とを決定する。
成
果物
は,少なくとも
ソフトウェアシステム
試験記録(
5.7.5
参照)を含むものとする。
注記
こうしたギャップ分析によって,
レガシーソフトウェア
に実装した
リスクコントロール
手段
をソフトウェア要求事項に確実に含めることが望ましい。
4.4.4
ギャップ解消アクティビティ
a)
製造業者
は,特定した
成果物
を作成するための計画を確立し実行する。客観的な証拠を利用できる場
合は,
5.2
,
5.3
,
5.7
及び箇条
7
で要求する
アクティビティ
を行わずに,その証拠を用いて必要な
成果
物
を作成してもよい。
注記
特定したギャップに対応するための計画は,ソフトウェア保守計画(
6.1
参照)に含めること
ができる。
b)
この計画では,箇条
9
に従って発見した
レガシーソフトウェア
及び
成果物
の問題に対処するため,問
題解決
プロセス
を使用する。
c)
レガシーソフトウェア
に対する変更は,箇条
6
に従って実施する。
4.4.5
レガシーソフトウェアを使用する根拠
製造業者
は,
レガシーソフトウェア
の
バージョン
とともに,そのソフトウェアを継続使用する根拠を,
4.4
のアウトプットに基づいて文書化する。
注記
4.4
を満たしていれば,この規格に従って,
レガシーソフトウェア
を今後も使用できる。
5
ソフトウェア開発プロセス
5.1
*
ソフトウェア開発計画
5.1.1
ソフトウェア開発計画
製造業者
は,開発する
ソフトウェアシステム
の適用範囲,規模及びソフトウェア安全クラス分類に適し
た,ソフトウェア開発
プロセス
の
アクティビティ
を実施するために,(一つ又は複数の)ソフトウェア開
発計画を確立する。
ソフトウェア開発ライフサイクルモデル
は,その計画の中に全てを定義するか,又は
引用するかのいずれかとする。計画は,次の事項を扱った内容とする(クラス
A
,
B
,
C
)。
a)
ソフトウェアシステム
の開発に使用する
プロセス
(
注記
4
参照)
b)
アクティビティ
及び
タスク
の
成果物
(文書化を含む。
)
c)
システム
要求事項,ソフトウェア要求事項,
ソフトウェアシステム
試験及びソフトウェアに実装する
リスクコントロール
手段の間の
トレーサビリティ
d)
SOUP
構成アイテム
及び開発支援用ソフトウェアを含む,ソフトウェア構成管理及び変更管理
e)
ライフサイクルの各段階で発見される
医療機器ソフトウェア
,
成果物
及び
アクティビティ
の問題に対
処するためのソフトウェア問題解決
注記
1
ソフトウェア開発ライフサイクルモデル
では,
ソフトウェアシステム
の各
ソフトウェアア
イテム
の安全クラス分類に従って,異なる
ソフトウェアアイテム
に対し,それぞれ別の要
素(
プロセス
,
アクティビティ
,
タスク
及び
成果物
)を割り付けることができる。
14
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
注記
2
これらの
アクティビティ
及び
タスク
は,重複するものであっても相互に影響を与え合うも
のであってもよく,反復的又は循環的に実行してもよい。特定のライフサイクルモデルを
使用することが望ましいということを意図したものではない。
注記
3
この規格には,開発
プロセス
と区別して説明している他の
プロセス
があるが,それらを別
の
アクティビティ
及び
タスク
として実装しなければならないということを意味するもので
はない。それらの
プロセス
の
アクティビティ
及び
タスク
は,開発
プロセス
に統合できる。
注記
4
ソフトウェア開発計画では,既存の
プロセス
を引用しても新たに
プロセス
を定義してもよ
い。
注記
5
ソフトウェア開発計画は,全体の
システム
開発計画に統合してもよい。
5.1.2
ソフトウェア開発計画の継続更新
製造業者
は,開発の進捗に応じて,計画を適宜更新する(クラス
A
,
B
,
C
)。
5.1.3
ソフトウェア開発計画におけるシステム設計及びシステム開発の引用
(クラス
A
,
B
,
C
)
a)
ソフトウェア開発のためのインプットとなる
システム
要求事項は,
製造業者
がソフトウェア開発計画
の中で引用する。
b)
製造業者
は,ソフトウェア開発計画に,
4.1
に適合するために必要な,ソフトウェア開発と
システム
開
発との整合性をとるための手順(
システム
結合,
検証
,妥当性確認など)を示すか又は引用する。
注記
ソフトウェアシステム
がスタンドアロン
システム
(ソフトウェア単独)の場合は,
ソフトウ
ェアシステム
要求事項と
システム
要求事項とに差異がない場合もある。
5.1.4
ソフトウェア開発規格,方法及びツールの計画
製造業者
は,ソフトウェア開発計画書に,クラス
C
の
ソフトウェアアイテム
の開発に関連する次の項目
を示すか又は引用する(クラス
C
)。
a)
規格
b)
方法
c)
ツール
5.1.5
ソフトウェア結合及び結合試験計画
製造業者
は,
ソフトウェアアイテム
(
SOUP
を含む。
)を結合して,結合時に試験を実施するための計画
を,ソフトウェア開発計画書に示すか又は引用する(クラス
B
,
C
)
。
注記
1
結合試験及び
ソフトウェアシステム
試験は,一つの計画及び一連の
アクティビティ
に統合し
てもよい。
注記
2
5.6
参照。
5.1.6
ソフトウェア検証計画
製造業者
は,ソフトウェア開発計画書に,次の
検証
情報を示すか又は引用する(クラス
A
,
B
,
C
)。
a)
検証
が必要な
成果物
b)
各ライフサイクル
アクティビティ
に必要な
検証タスク
c)
成果物
を
検証
するマイルストーン
d)
成果物検証
の合否判定基準
5.1.7
ソフトウェアリスクマネジメント計画
製造業者
は,ソフトウェア
リスクマネジメントプロセス
の
アクティビティ
及び
タスク
の実行計画(
SOUP
に関連した
リスクマネジメント
を含む。
)を,ソフトウェア開発計画に示すか又は引用する(クラス
A
,
B
,
15
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
C
)。
注記
箇条
7
参照。
5.1.8
文書化計画
製造業者
は,ソフトウェア開発ライフサイクルにおいて作成する文書についての情報を,ソフトウェア
開発計画書に示すか又は引用する。記載した文書又は文書のタイプそれぞれについて,次の情報を示すか
又は引用する(クラス
A
,
B
,
C
)。
a)
題名,名称又は命名規則(
naming convention
)
b)
目的
c)
(対応国際規格で削除されている。)
d)
開発,レビュー,承認及び修正のための手順並びに責任
注記
文書の構成管理については,箇条
8
を参照する。
5.1.9
ソフトウェア構成管理計画
製造業者
は,ソフトウェア開発計画書に,ソフトウェア構成管理情報を示すか又は引用する。記載又は
引用するソフトウェア構成管理情報とは,次をいう(クラス
A
,
B
,
C
)。
a)
管理対象アイテムのクラス,タイプ,カテゴリ又はリスト
b)
ソフトウェア構成管理
アクティビティ
及び
タスク
c)
ソフトウェア構成管理
アクティビティ
の実行に責任を負う組織
d)
それらの組織と他の組織(例えば,ソフトウェア開発又は保守など)との関係
e)
アイテムを構成管理下に置く時期
f)
問題解決
プロセス
を使用する時期
注記
箇条
8
参照。
5.1.10
管理が必要な支援アイテム
管理対象のアイテムには,
医療機器ソフトウェア
に影響を及ぼす可能性のある,
医療機器ソフトウェア
の開発に使用するツール,アイテム又は設定を含める(クラス
B
,
C
)。
注記
1
このようなアイテムの例には,コンパイラ又はアセンブラの
バージョン
,メイクファイル,
バッチファイル及び特定の環境設定を含む。
注記
2
箇条
8
参照。
5.1.11
検証前のソフトウェア構成アイテムのコントロール
製造業者
は,ソフトウェア
構成アイテム
を,その
検証
前に,構成管理の管理下に置くように計画する(ク
ラス
B
,
C
)。
5.1.12
既知のソフトウェア欠陥の特定及び回避
製造業者
は,ソフトウェア開発計画書に,次の手順を示すか又は引用する(クラス
B
,
C
)。
a)
ソフトウェアシステム
に対して選択したプログラミング技術によって生じる可能性のある欠陥を分類
するための手順。
b)
これらの欠陥が受容できない
リスク
の一因にならないことを示す証拠を文書化する手順。
注記
欠陥の分類又は
危険状態
の一因となる原因の例については,
IEC TR 80002-1
:2009
の
附属書
B
を参照する。
16
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
5.2
*
ソフトウェア要求事項分析
5.2.1
システム要求事項からのソフトウェア要求事項の定義及び文書化
製造業者
は,
医療機器
の
ソフトウェアシステム
ごとに,
システム
レベルの要求事項から
ソフトウェアシ
ステム
要求事項を定義して文書化する(クラス
A
,
B
,
C
)。
注記
ソフトウェアシステム
がスタンドアロン
システム
(ソフトウェア単独の機器)の場合は,
ソフ
トウェアシステム
要求事項と
システム
要求事項とに差異がない場合もある。
5.2.2
ソフトウェア要求事項の内容
製造業者
は,
医療機器ソフトウェア
の要求事項に必要な場合は,次の事項を適宜含める(クラス
A
,
B
,
C
)。
a)
機能及び能力についての要求事項
注記
1
要求事項の例を,次に示す。
-
性能(例えば,ソフトウェアの目的,タイミング要求事項)
-
物理的特性(例えば,コード言語,プラットフォーム,オペレーティングシステム)
-
ソフトウェアの実行環境(例えば,ハードウェア,メモリサイズ,処理ユニット,時
間帯の設定,ネットワークインフラストラクチャ)
-
アップグレード,複数の
SOUP
又は他機器との互換性の必要性
b)
ソフトウェアシステム
のインプット及びアウトプット
注記
2
要求事項の例を,次に示す。
-
データの型(例えば,数字,英数字,フォーマット)
-
範囲
-
制限
-
既定値
c)
ソフトウェアシステム
と他の
システム
との間のインタフェース
d)
ソフトウェアによる警報,警告及び操作者へのメッセージ
e)
セキュリティ
要求事項
注記
3
要求事項の例を,次に示す。
-
機密情報の漏えい(洩)に関連する事項
-
認証
-
認可
-
監査証跡(
audit trail
)
-
通信の完全性
-
システム
セキュリティ
・マルウェアからの保護
f)
ソフトウェアで実装するユーザインタフェースの要求事項
注記
4
要求事項に関連する例を,次に示す。
-
手動操作の支援
-
人間と機器との相互作用
-
人員についての制約
-
人間の注意を集中する必要がある領域
注記
5
ユーザビリティエンジニアリング要求事項についての情報は,
IEC 62366-1
[17]
他[例えば,
IEC 60601-1-6
[16]
]に規定している。
17
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
g)
データ定義及びデータベース要求事項
注記
6
要求事項の例を,次に示す。
-
形式
-
整合性
-
機能
h)
納入した
医療機器ソフトウェア
の,操作現場及び保守現場におけるインストール及び受入れの要求事
項
i)
操作及び保守の方法に関わる要求事項
j)
IT
ネットワークに関連する要求事項
注記
7
要求事項の例を,次に示す。
-
ネットワーク通信する,アラーム,警告及びオペレータメッセージ
-
ネットワークプロトコル
-
ネットワークサービスが利用できない場合の扱い
k)
ユーザ保守要求事項
l)
規制要求事項
注記
8
a)
~
l)
の要求事項は,重複することもある。
注記
9
ソフトウェア開発の初期には,必ずしも,これらの要求事項の全てが利用できるとは限らな
い。
注記
10
JIS X 25010
[9]
は,ソフトウェア要求事項の定義付けに有用な品質特性についての情報を規
定している。
5.2.3
リスクコントロール手段のソフトウェア要求事項への包含
製造業者
は,ソフトウェアに実装する
リスクコントロール
手段を,
医療機器ソフトウェア
の要求事項に
含める(クラス
B
,
C
)。
注記
これらの要求事項は,ソフトウェア開発の初期には利用できないこともあり,ソフトウェアの
設計及び
リスクコントロール
手段の追加定義を行うことで変更できる。
5.2.4
医療機器のリスク分析の再評価
製造業者
は,ソフトウェア要求事項が確定した時点で
医療機器
の
リスク分析
を再
評価
し,適宜,更新す
る(クラス
A
,
B
,
C
)。
5.2.5
要求事項の更新
製造業者
は,ソフトウェア要求事項分析
アクティビティ
の結果を受けて,適宜,
システム
要求事項を含
む既存の要求事項の再
評価
及び更新を確実に実施する(クラス
A
,
B
,
C
)。
5.2.6
ソフトウェア要求事項の検証
製造業者
は,ソフトウェア要求事項について次の点を
検証
し文書化する(クラス
A
,
B
,
C
)。
a)
システム
要求事項(
リスクコントロール
に関わるものを含む。)を実装している。
b)
相互に矛盾しない。
c)
曖昧さを回避した用語で表現している。
d)
試験基準を確立して,試験が実施できる表現で記載している。
e)
一意に識別できる。
f)
システム
要求事項又は他の要求事項を追跡できる。
18
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
注記
この規格は,形式仕様記述言語の使用を要求するものではない。
5.3
*
ソフトウェアアーキテクチャの設計
5.3.1
ソフトウェア要求事項のアーキテクチャへの変換
製造業者
は,
医療機器ソフトウェア
の要求事項を,文書化した
アーキテクチャ
(ソフトウェアの構造の
説明及び
ソフトウェアアイテム
の特定をしているもの)に変換する(クラス
B
,
C
)。
5.3.2
ソフトウェアアイテムのインタフェース用アーキテクチャの開発
製造業者
は,
ソフトウェアアイテム
と
ソフトウェアアイテム
外部のコンポーネント(ソフトウェア及び
ハードウェア)との間,及び
ソフトウェアアイテム
間のインタフェースについて,
アーキテクチャ
を開発
し,文書化する(クラス
B
,
C
)。
5.3.3 SOUP
アイテムの機能及び性能要求事項の指定
ソフトウェアアイテム
を
SOUP
と特定している場合,
製造業者
は,その
SOUP
アイテムについて,その
意図する使用に必要な機能性能要求事項を明確にする(クラス
B
,
C
)。
5.3.4 SOUP
アイテムが要求するシステムハードウェア及びシステムソフトウェアの指定
ソフトウェアアイテム
を
SOUP
と特定している場合,
製造業者
は,
SOUP
アイテムの正常な動作に必要
な
システム
ハードウェア及び
システム
ソフトウェアを明確にする(クラス
B
,
C
)
。
注記
例としては,プロセッサのタイプ及び速度,メモリのタイプ及びサイズ,
システム
ソフトウェ
アのタイプ,通信及び表示のソフトウェア要求事項などがある。
5.3.5
リスクコントロールに必要な分離の特定
製造業者
は,
リスクコントロール
に必要な
ソフトウェアアイテム
間の分離を特定し,分離が有効である
ことを確実にする方法について明示する(クラス
C
)
。
注記
分離の例としては,異なるプロセッサ上で
ソフトウェアアイテム
を実行させることがある。分
離の効果は,プロセッサ間に共有リソースをもたないことによって保証できる。ソフトウェア
アーキテクチャ
の設計(
B.4.3
参照)によって効果を保証できる場合は,他の分離手段を適用で
きる。
5.3.6
ソフトウェアアーキテクチャの検証
製造業者
は,次の事項を検証し,文書化する(クラス
B
,
C
)。
a)
ソフトウェアの
アーキテクチャ
が,
リスクコントロール
に関わる要求事項を含む,
システム
及びソフ
トウェアの要求事項を実装している。
b)
ソフトウェア
アーキテクチャ
が,
ソフトウェアアイテム
間及び
ソフトウェアアイテム
とハードウェア
との間のインタフェースを支援できる。
c)
医療機器アーキテクチャ
が,全ての
SOUP
アイテムの正常な動作を支援している。
注記
要求事項
a)
を満たすために,ソフトウェア要求事項に対する
アーキテクチャ
の
トレーサビリテ
ィ
分析を行ってもよい。
5.4
*
ソフトウェア詳細設計
5.4.1
ソフトウェアのソフトウェアユニットへの分解
製造業者
は,ソフトウェアを
ソフトウェアユニット
に分解する(クラス
B
,
C
)。
注記
ソフトウェアシステム
によっては,それ以上分解できないこともある。
5.4.2
ソフトウェアユニットごとの詳細設計の開発
製造業者
は,それぞれの
ソフトウェアユニット
を正しく実装するために,十分な詳細設計をして,文書
化する(クラス
C
)。
19
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
5.4.3
インタフェース用詳細設計の開発
製造業者
は,
ソフトウェアユニット
と外部コンポーネント(ハードウェア又はソフトウェア)との間,
及び
ソフトウェアユニット
間の全てのインタフェースについての設計を文書化する。この文書化は,各
ソ
フトウェアユニット
及びそのインタフェースを正しく実装するために十分詳細なものとする(クラス
C
)。
5.4.4
詳細設計の検証
製造業者
は,ソフトウェアの詳細設計が次によることを検証し,文書化する(クラス
C
)。
a)
ソフトウェア
アーキテクチャ
を実装している。
b)
ソフトウェア
アーキテクチャ
との矛盾がない。
注記
要求事項
a)
を満たすために,ソフトウェア詳細設計に対する
アーキテクチャ
の
トレーサビリテ
ィ
分析を行ってもよい。
5.5
*
ソフトウェアユニットの実装
5.5.1
各ソフトウェアユニットの実装
製造業者
は,各
ソフトウェアユニット
を実装する(クラス
A
,
B
,
C
)。
5.5.2
ソフトウェアユニット検証プロセスの確立
製造業者
は,
ソフトウェアユニット
を検証するための方針,方法及び手順を確立する。
検証
を試験によ
って実施する場合は,その試験手順の適切性について
評価
する(クラス
B
,
C
)。
注記
結合試験及び
ソフトウェアシステム
試験は,一つの計画及び一連の
アクティビティ
に統合して
もよい。
5.5.3
ソフトウェアユニットの合否判定基準
製造業者
は,より大きな
ソフトウェアアイテム
に結合する前に,必要に応じて
ソフトウェアユニット
の
合否判定基準を確立し,
ソフトウェアユニット
が合否判定基準を確実に適合するようにする(クラス
B
,
C
)。
注記
合否判定基準の例を,次に示す。
-
ソフトウェアコードが,
リスクコントロール
手段を含む要求事項を実装しているか。
-
ソフトウェアコードが,
ソフトウェアユニット
のインタフェース設計と矛盾しないか。
-
ソフトウェアコードが,プログラミング手順又はコーディング標準に従っているか。
5.5.4
追加のソフトウェアユニット合否判定基準
設計に当たって,
製造業者
は,必要に応じて次の事項についての追加の合否判定基準を含める(クラス
C
)。
a)
適正なイベントシーケンス
b)
データ及び制御フロー
c)
計画したリソース配分
d)
異常処理(エラーの定義,特定及び復帰)
e)
変数の初期化
f)
自己診断
g)
メモリ管理及びメモリオーバーフロー
h)
境界条件
5.5.5
ソフトウェアユニットの検証
製造業者
は,
ソフトウェアユニット
の
検証
を実行し,結果を文書化する(クラス
B
,
C
)。
20
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
5.6
*
ソフトウェア結合及び結合試験
5.6.1
ソフトウェアユニットの結合
製造業者
は,結合計画に従って
ソフトウェアユニット
を結合する(
5.1.5
参照)(クラス
B
,
C
)
。
5.6.2
ソフトウェア結合の検証
製造業者
は,
ソフトウェアユニット
が
ソフトウェアアイテム
及び/又は
ソフトウェアシステム
に結合さ
れていることを結合計画(
5.1.5
参照)に従って検証し,
検証
を行った証拠を記録として保存する(クラス
B
,
C
)。
注記
ここでは,結合が計画に従って実施されているかについてだけ
検証
する。この
検証
は,何らか
の調査で行うのが望ましい。
5.6.3
ソフトウェア結合試験
製造業者
は,結合計画(
5.1.5
参照)に従って,結合した
ソフトウェアアイテム
を試験し,結果を文書化
する(クラス
B
,
C
)。
5.6.4
ソフトウェア結合試験の内容
ソフトウェア結合試験では,
製造業者
は,結合した
ソフトウェアアイテム
が意図したとおりに機能する
かを明確にする(クラス
B
,
C
)。
注記
1
考慮することが望ましい事項の例を次に示す。
-
ソフトウェアに要求している機能
-
リスクコントロール
手段の実装
-
指定したタイミング及びその他の動作
-
内部及び外部インタフェースの指定した機能
-
予見可能な誤使用を含む異常な条件下での試験
注記
2
結合試験及び
ソフトウェアシステム
試験は,一つの計画及び一連の
アクティビティ
に統合し
てもよい。
5.6.5
ソフトウェア結合試験手順の評価
製造業者
は,結合試験手順の適切性を
評価
する(クラス
B
,
C
)。
5.6.6
回帰テストの実施
製造業者
は,
ソフトウェアアイテム
を既存の結合済みソフトウェアに結合する場合,
回帰テスト
を適宜
実施して,結合前にはなかった欠陥が新たに生じていないことを実証する(クラス
B
,
C
)。
5.6.7
結合試験記録の内容
製造業者
は,次の事項を実施する(クラス
B
,
C
)。
a)
試験結果(合否及び
異常
箇所のリスト)を文書化する。
b)
試験を再現できるように,十分な記録を保存する。
c)
試験者を明示する。
注記
要求事項
b)
は,例えば,次のものを保存することによって行うこともある。
-
要求される処置及び期待される結果を示すテストケース仕様
-
装置の記録
-
試験に使用した,(ソフトウェアツールを含む。)試験環境の記録
5.6.8
ソフトウェア問題解決プロセスの使用
製造業者
は,ソフトウェア結合及び結合試験中に発見した
異常
を,ソフトウェア問題解決
プロセス
で処
理する(クラス
B
,
C
)。
21
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
注記
箇条
9
参照。
5.7
*
ソフトウェアシステム試験
5.7.1
ソフトウェア要求事項についての試験の確立
a)
製造業者
は,
ソフトウェアシステム
試験の実施のために,個々のソフトウェア要求事項を対象として,
インプット内容,予想する結果,合否判定基準及び手順を定めた一連の試験を確立し,実施する。
b)
製造業者
は,
検証
方針及び試験手順の適切性を
評価
する。
(クラス
A
,
B
,
C
)
注記
1
結合試験及び
ソフトウェアシステム
試験は,一つの計画及び一連の
アクティビティ
に統合し
てもよい。また,ソフトウェア要求事項は,より早い段階で試験してもよい。
注記
2
特に要求事項の間に依存性が存在する場合は,要求事項ごとの個別試験だけでなく,要求事
項を組み合わせた試験も実施可能である。
5.7.2
ソフトウェア問題解決プロセスの使用
製造業者
は,
ソフトウェアシステム
試験中に発見した
異常
を,ソフトウェア問題解決
プロセス
で処理す
る(クラス
A
,
B
,
C
)。
5.7.3
変更後の再試験
製造業者
は,
ソフトウェアシステム
試験の実施中に変更があった場合,次の処理をする(クラス
A
,
B
,
C
)。
a)
必要に応じた試験のやり直し,試験の修正及び実施,又は追加試験の実施によって,変更が問題の訂
正にどの程度有効かを検証する。
b)
副作用が発生しなかったことを示すための適切な試験を実施する。
c)
7.4
で規定する,関連する
リスクマネジメントアクティビティ
を実行する。
5.7.4
ソフトウェアシステム試験の評価
製造業者
は,検証方針及び試験手順の適切性を
評価
する。
製造業者
は,次の事項を検証する(クラス
A
,
B
,
C
)。
a)
全てのソフトウェア要求事項を対象に,試験又は
検証
を実施している。
b)
ソフトウェア要求事項と試験又は
検証
との間の
トレーサビリティ
が記録されている。
c)
試験結果が,要求する合否判定基準に適合する。
5.7.5
ソフトウェアシステム試験記録の内容
試験の再現性を図るために,
製造業者
は,次の事項を文書化する(クラス
A
,
B
,
C
)。
a)
要求される処置及び期待される結果を示すテストケース手順書への参照表記
b)
試験結果(合否及び
異常
箇所のリスト)
c)
試験したソフトウェアの
バージョン
d)
関連するハードウェア及びソフトウェアテスト構成
e)
関連試験ツール
f)
試験実施日
g)
試験の実施及び試験結果の記録に関わる責任者の識別
5.8
*
システムレベルで使用するためのソフトウェアリリース
5.8.1
ソフトウェア検証の完了確認
製造業者
は,全てのソフトウェア
検証アクティビティ
が完了し,結果を
評価
したことを,ソフトウェア
のリリース前に確認する(クラス
A
,
B
,
C
)。
22
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
5.8.2
既知の残留異常の文書化
製造業者
は,残留している既知の
異常
を全て文書化する(クラス
A
,
B
,
C
)。
5.8.3
既知の残留異常の評価
製造業者
は,残留している既知の
異常
を全て
評価
したことを確認し,受容できない
リスク
の原因になら
ないことを確実にする(クラス
B
,
C
)。
5.8.4
リリースするバージョンの文書化
製造業者
は,リリースする
医療機器ソフトウェア
の
バージョン
を文書化する(クラス
A
,
B
,
C
)。
5.8.5
リリースするソフトウェアの作成方法の文書化
製造業者
は,リリースするソフトウェアの作成手順及び作成環境を文書化する(クラス
B
,
C
)
。
5.8.6
アクティビティ及びタスクの完了確認
製造業者
は,ソフトウェア開発計画(又は保守計画)の全ての
アクティビティ
及び
タスク
が,関連する
文書化とともに完了していることを確認する(クラス
B
,
C
)。
注記
5.1.3 b)
参照。
5.8.7
ソフトウェアのアーカイブ
製造業者
は,次について,
製造業者
自身が決定した
医療機器ソフトウェア
の耐用期間,又は関連する規
制要求事項が規定する期間の,いずれか長い方を最低保管期間として保管する(クラス
A
,
B
,
C
)。
a)
医療機器ソフトウェア
及び
構成アイテム
b)
文書
5.8.8
ソフトウェアリリースの信頼性の確保
製造業者
は,リリースする
医療機器ソフトウェア
が,変造又は無断で変更されることなく使用する場所
に確実に納品されるようにするための手順を確立する。具体的には
医療機器ソフトウェア
を格納した媒体
の製造及び取扱いについての手順であり,必要に応じて次のものを含める(クラス
A
,
B
,
C
)。
-
複製
-
媒体のラベリング
-
こん(梱)包
-
保護
-
保管
-
納品
6
ソフトウェア保守プロセス
6.1
*
ソフトウェア保守計画の確立
製造業者
は,保守
プロセス
の
アクティビティ
及び
タスク
を実行するためのソフトウェア保守計画を確立
する。計画の内容は,次による(クラス
A
,
B
,
C
)。
a)
医療機器ソフトウェア
のリリース後に発生する情報をフィードバックするための,次の手順
-
取得
-
文書化
-
評価
-
解決
-
追跡
b)
フィードバックした情報に問題があるかを判断するための基準
23
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
c)
ソフトウェア
リスクマネジメントプロセス
の使用
d)
医療機器ソフトウェア
のリリース後に発生した問題を分析及び解決するためのソフトウェア問題解決
プロセス
の使用
e)
既存
ソフトウェアシステム
の修正を管理するための,ソフトウェア構成管理
プロセス
(箇条
8
参照)
の使用
f)
SOUP
について,次の事項を
評価
し実行する手順
-
アップグレード
-
バグ修正
-
パッチ
-
陳腐化の確認
6.2
*
問題及び修正の分析
6.2.1
フィードバックの文書化及び評価
6.2.1.1
フィードバックの監視
製造業者
は,意図する使用のためにリリースした
医療機器ソフトウェア
についてのフィードバックを監
視する(クラス
A
,
B
,
C
)。
6.2.1.2
フィードバックの文書化及び評価
フィードバックを文書化するとともにそれを
評価
し,リリースした
医療機器ソフトウェア
に問題がない
かを判断する。問題があった場合は,
問題報告
として記録する(箇条
9
参照)。
問題報告
には,実際に悪
影響を及ぼす又はその可能性のある事象,及び仕様から逸脱した事象を含める(クラス
A
,
B
,
C
)。
6.2.1.3
安全性に影響する問題報告の評価
問題報告
は,個々に
評価
を実施し,意図する使用のためにリリースした
医療機器ソフトウェア
の
安全
性
にどのような影響があるかを判断するとともに(
9.2
参照),問題に対処するためにそのソフトウェアに変
更を加える必要があるかを判断する(クラス
A
,
B
,
C
)。
6.2.2
ソフトウェア問題解決プロセスの使用
製造業者
は,
問題報告
の対処に当たり,ソフトウェア問題解決
プロセス
(箇条
9
参照)を使用する(ク
ラス
A
,
B
,
C
)。
注記
問題を調べると,
ソフトウェアシステム
又は
ソフトウェアアイテム
が,正しいソフトウェア安
全クラスに分類されていないことが分かることがある。問題解決
プロセス
では,ソフトウェア
安全クラスの変更が示唆されることがある。その場合は,
プロセス
が完了した時点で,
ソフト
ウェアシステム
又はその
ソフトウェアアイテム
の安全クラスの変更を明らかにし,文書化して
いることが望ましい。
6.2.3
変更要求の分析
製造業者
は,箇条
9
で要求している分析を実施するほか,各
変更要求
が,組織,意図する使用のために
リリースした
医療機器ソフトウェア
及び連携する
システム
に及ぼす影響について分析を行う(クラス
A
,
B
,
C
)。
6.2.4
変更要求の承認
製造業者
は,リリースした
医療機器ソフトウェア
に修正が生じる
変更要求
を
評価
し,承認する(クラス
A
,
B
,
C
)。
6.2.5
ユーザ及び規制当局への通知
製造業者
は,リリースした
医療機器ソフトウェア
に影響がある,承認済みの
変更要求
を明らかにする。
24
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
地域の法令の要求に応じて,
製造業者
は,ユーザ及び規制当局に対して次の事項を通知する(クラス
A
,
B
,
C
)。
a)
リリースした
医療機器ソフトウェア
についての全ての問題及び変更せずに継続使用した場合の結果。
b)
リリースした
医療機器ソフトウェア
に対して利用可能な変更の本質(
nature
),並びにそれらの変更の
入手及びインストールの方法。
6.3
*
修正の実装
6.3.1
確立したプロセスを使用した修正の実装
製造業者
は,修正を行った結果,再度実施する必要性がある箇条
5
の
アクティビティ
を特定し,実施す
る(クラス
A
,
B
,
C
)。
注記
ソフトウェア変更の
リスクマネジメント
に関わる要求事項については,
7.4
参照。
6.3.2
修正ソフトウェアシステムの再リリース
製造業者
は,
5.8
に従って,作成した修正版をリリースする(クラス
A
,
B
,
C
)。
注記
修正版は,
ソフトウェアシステム
の完全再リリース版の一部としてリリースすることもできる
し,変更した
ソフトウェアアイテム
及び修正版の変更内容を既存の
ソフトウェアシステム
にイ
ンストールするために必要なツールから成る修正キットとしてリリースすることもできる。
7
*
ソフトウェアリスクマネジメントプロセス
7.1
*
危険状態を引き起こすソフトウェアの分析
7.1.1
危険状態の一因となるソフトウェアアイテムの特定
製造業者
は,
JIS T 14971
に規定した
医療機器
の
リスク分析
を行い,
危険状態
及び
危険状態
を引き起こす
可能性のある
ソフトウェアアイテム
を特定する(
4.2
参照)(クラス
B
,
C
)。
注記
危険状態
は,ソフトウェアの故障が直接の原因となる場合,又はソフトウェアに実装した
リス
クコントロール
手段の故障が原因となる場合が考えられる。
7.1.2
危険状態の一因となるソフトウェアアイテムの潜在的原因の特定
製造業者
は,
7.1.1
で特定した
危険状態
の一因となる
ソフトウェアアイテム
の,潜在的原因を特定する。
製造業者
は,必要に応じて,次に挙げるような潜在的原因を検討する(クラス
B
,
C
)。
a)
誤った又は不完全な機能仕様
b)
特定した
ソフトウェアアイテム
の,機能におけるソフトウェア不具合
c)
SOUP
に起因する,故障又は予期せぬ結果
d)
予測できないソフトウェア動作を引き起こす可能性のある,ハードウェアの故障又は他のソフトウェ
アの欠陥
e)
合理的に予見可能な誤使用
7.1.3
公開された
SOUP
異常リストの評価
SOUP
に起因する故障又は予期せぬ結果が,
危険状態
の一因となる
ソフトウェアアイテム
の潜在的原因
になっている場合,
製造業者
は,当該
医療機器
に使用している
SOUP
アイテムの
バージョン
に関係する,
SOUP
アイテムの供給者が公開している
異常
リストを最低限度として
評価
し,既知の
異常
のいずれかによ
って,
危険状態
の原因となる可能性がある一連の事象が生じるかを判断する(クラス
B
,
C
)。
7.1.4
潜在的原因の文書化
製造業者
は,
危険状態
の一因となる
ソフトウェアアイテム
の潜在的原因を,
リスクマネジメントファイ
ル
に文書化する(
JIS T 14971
参照)(クラス
B
,
C
)。
25
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
7.1.5
(対応国際規格で削除されている。
)
7.2
リスクコントロール手段
7.2.1
リスクコントロール手段の選択
製造業者
は,
リスクマネジメントファイル
に文書化した,
ソフトウェアアイテム
が
危険状態
の一因とな
るケースのそれぞれについて,
JIS T 14971
に従って
リスクコントロール
手段を選択し,文書化する(クラ
ス
B
,
C
)。
注記
リスクコントロール
手段は,ハードウェア,ソフトウェア若しくは動作環境において実施する
か,又は取扱説明書への記載による。
7.2.2
ソフトウェアに実装するリスクコントロール手段
リスクコントロール
手段を
ソフトウェアアイテム
の機能の一部として実装する場合,
製造業者
は,次の
事項を実施する(クラス
B
,
C
)。
a)
リスクコントロール
手段をソフトウェア要求事項に含める。
b)
リスクコントロール
手段の実施に寄与する各
ソフトウェアアイテム
に対して,その
リスクコントロー
ル
手段によってコントロールしている
リスク
に基づいて,ソフトウェア安全クラスの分類を行う[
4.3
a)
参照]。
c)
箇条
5
に従って
ソフトウェアアイテム
を開発する。
注記
この要求事項は,
JIS T 14971
の
リスクコントロール
の要求事項を詳細にしたものである。
7.3
リスクコントロール手段の検証
7.3.1
リスクコントロール手段の実施の検証
7.2
で文書化した
リスクコントロール
手段を全て実施していることを
検証
し,その
検証
結果を文書化す
る。
製造業者
は,
リスクコントロール
手段をレビューし,それによって新たな
危険状態
に至ることがない
か判断する(クラス
B
,
C
)
。
7.3.2
(対応国際規格で削除されている。
)
7.3.3
トレーサビリティの文書化
製造業者
は,次のソフトウェアに関連する事項の
トレーサビリティ
について,適宜文書化する(クラス
B
,
C
)。
a)
危険状態
から
ソフトウェアアイテム
まで
b)
ソフトウェアアイテム
から特定のソフトウェアの原因まで
c)
ソフトウェアの原因から
リスクコントロール
手段まで
d)
リスクコントロール
手段から
リスクコントロール
手段の
検証
まで
注記
JIS T 14971
参照。
7.4
ソフトウェア変更のリスクマネジメント
7.4.1
医療機器ソフトウェアの安全性に関わる変更の分析
製造業者
は,
医療機器ソフトウェア
(
SOUP
を含む。)の変更内容を分析して,次を確認する(クラス
A
,
B
,
C
)。
a)
危険状態
の一因となる潜在的原因が新たに生じていないか。
b)
新たなソフトウェア
リスクコントロール
手段が必要でないか。
7.4.2
ソフトウェア変更が既存のリスクコントロール手段に与える影響の分析
製造業者
は,ソフトウェアの変更(
SOUP
の変更を含む。)を分析して,ソフトウェアの修正が既存の
リ
スクコントロール
手段の妨げとなる危険性がないかを確定する(クラス
B
,
C
)。
26
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
7.4.3
分析に基づくリスクマネジメントアクティビティの実行
製造業者
は,これらの分析に基づき,
7.1
~
7.3
で規定した当該
リスクマネジメントアクティビティ
を実
行する(クラス
B
,
C
)。
8
*
ソフトウェア構成管理プロセス
8.1
*
構成識別
8.1.1
構成アイテム識別手段の確立
製造業者
は,
5.1
に規定した開発及び構成管理計画に従って管理することが望ましい
構成アイテム
及び
その
バージョン
を,一意に識別するための仕組みを確立する(クラス
A
,
B
,
C
)。
8.1.2 SOUP
の特定
現在使用中の
SOUP
構成アイテム
(標準ライブラリを含む。)のそれぞれについて,
製造業者
は,次を
文書化する(クラス
A
,
B
,
C
)。
a)
名称
b)
製造業者
c)
SOUP
を特定する識別子
注記
SOUP
を特定する識別子の例として,
バージョン
,リリース年月日,パッチ番号,アップグレ
ードの識別子などが考えられる。
8.1.3
システム構成文書の特定
製造業者
は,
ソフトウェアシステム
の構成要素である
構成アイテム
及びその
バージョン
一式を文書化す
る(クラス
A
,
B
,
C
)。
8.2
*
変更管理
8.2.1
変更要求の承認
製造業者
は,承認した
変更要求
に応じる場合に限り,
8.1
に従って識別する
構成アイテム
を変更する(ク
ラス
A
,
B
,
C
)。
注記
1
変更要求
承認の決定が,変更管理
プロセス
又は他の
プロセス
の一部に不可欠となる場合があ
る。この細分箇条が要求するのは,変更の実装の前に変更の承認が必要ということだけであ
る。
注記
2
ライフサイクルの様々な段階で受け取る
変更要求
に対して,様々な受入れ
プロセス
を使用で
きる[
5.1.1 d)
及び
6.1 e)
参照]。
8.2.2
変更の実装
製造業者
は,
変更要求
で指定されているとおりに変更を実装する。
製造業者
は,変更の結果やり直しが
必要な全ての
アクティビティ
(
ソフトウェアシステム
及び
ソフトウェアアイテム
のソフトウェア安全クラ
ス分類の変更を含む。)を特定し,実装する(クラス
A
,
B
,
C
)。
注記
この細分箇条では,十分な変更管理を達成するために,どのように変更を実装するのが望まし
いかということを明記している。これは,変更の実装が,変更管理
プロセス
に不可欠であると
いうことを意味するものではない。実装には,計画した
プロセス
を使用することが望ましい
[
5.1.1 e)
及び
6.1 e)
参照]
。
8.2.3
変更の検証
製造業者
は,
5.7.3
及び
9.7
を考慮しながら,変更によって無効になった
検証
のやり直しも含めて,変更
を検証する(クラス
A
,
B
,
C
)。
27
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
注記
この細分箇条が要求するのは,変更を
検証
することだけであり,
検証
が変更管理
プロセス
に不
可欠であるということを意味するものではない。
検証
には,計画した
プロセス
を使用すること
が望ましい[
5.1.1 e)
及び
6.1 e)
参照]。
8.2.4
変更のトレーサビリティを実現する手段の提示
製造業者
は,次の事項の間の関連性及び依存関係の記録を維持する(クラス
A
,
B
,
C
)。
a)
変更要求
b)
当該
問題報告
c)
変更要求
の承認
8.3
*
構成状態の記録
製造業者
は,
システム
構成を含む,管理している
構成アイテム
について,検索可能な履歴記録を保存す
る(クラス
A
,
B
,
C
)。
9
*
ソフトウェア問題解決プロセス
9.1
問題報告の作成
製造業者
は,発見した
医療機器ソフトウェア
の問題ごとに
問題報告
を作成する。
問題報告
には,重大性
に関する記載(例えば,性能,
安全
又は
セキュリティ
への影響)のほか,問題解決に役立ちそうな他の情
報(例えば,影響を受ける機器,影響を受けるサポート対象附属品)を含める(クラス
A
,
B
,
C
)。
注記
問題は,リリースの前後及び
製造業者
の組織内外を問わず発見される可能性がある。
9.2
問題の調査
製造業者
は,次を実施する(クラス
A
,
B
,
C
)。
a)
問題を調査し,可能であれば原因を特定する。
b)
ソフトウェア
リスクマネジメントプロセス
(箇条
7
参照)を用いて,その問題の
安全
性への関わりを
評価
する。
c)
調査及び評価の結果を文書化する。
d)
問題の是正に必要な処置のための
変更要求
を作成する,又は処置を行わない場合の正当な根拠を文書
化する。
注記
問題が
安全
性に関連がない場合,
製造業者
は,ソフトウェア問題解決
プロセス
に従って問題を
解決する必要はない。
9.3
関係者への通知
製造業者
は,問題の存在を関係者に適宜通知する(クラス
A
,
B
,
C
)。
注記
問題は,リリースの前後及び
製造業者
の組織内外を問わず発見される可能性がある。
製造業者
は,状況に応じて通知先関係者を決定する。
9.4
変更管理プロセスの使用
製造業者
は,変更管理
プロセス
の要求事項を遵守した上で,全ての
変更要求
を承認し,実行する(
8.2
参照)(クラス
A
,
B
,
C
)。
9.5
記録の保持
製造業者
は,
問題報告
の記録及びその
検証
も含めた解決についての記録を保持する。
製造業者
は,
リスクマネジメントファイル
を適宜更新する(クラス
A
,
B
,
C
)。
9.6
問題の傾向分析
製造業者
は,
問題報告
を分析してその傾向を把握する(クラス
A
,
B
,
C
)。
28
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
9.7
ソフトウェア問題解決の検証
製造業者
は,問題の解決を検証し,次の事項を判断する(クラス
A
,
B
,
C
)。
a)
問題を解決し,
問題報告
を完了した。
b)
好ましくない傾向を改善した。
c)
変更要求
を,適切な
医療機器ソフトウェア
及び
アクティビティ
に実装した。
d)
新たな問題が発生していない。
9.8
試験文書の内容
製造業者
は,変更の後に実施する,
ソフトウェアアイテム
及び
システム
の試験,再試験又は
回帰テスト
に当たって,試験文書の中に次を含める(クラス
A
,
B
,
C
)。
a)
試験結果
b)
発見した
異常
c)
試験したソフトウェアの
バージョン
d)
関連するハードウェア及びソフトウェアテスト構成
e)
関連試験ツール
f)
試験実施日
g)
試験者の識別
29
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
附属書
A
(参考)
この規格の要求事項の根拠
この規格の各箇条の根拠を,この
附属書
に記載する。
A.1
根拠
この規格が第一に求めているのは,
医療機器ソフトウェア
の開発及び保守が,一連の
プロセス
に従うと
ともに,患者及びその他の人への
リスク
に対して適切な
プロセス
を選択することである。これは,ソフト
ウェアの試験を実施しただけでは,その使用が安全であると判断するには十分ではないという信念に基づ
いている。
この規格が要求する
プロセス
は,次の
2
種類のカテゴリに分類できる。
-
ソフトウェアの各
ソフトウェアアイテム
の動作に起因する
リスク
を評価するために必要となる
プロセ
ス
-
評価した
リスク
に基づいて選択される,各
ソフトウェアアイテム
にソフトウェアの故障が発生する確
率を低い水準に抑えるために必要となる
プロセス
この規格は,最初のカテゴリは全ての
医療機器ソフトウェア
に対して実施し,
2
番目のカテゴリは選択
した
ソフトウェアアイテム
に対して実施することを要求している。
したがって,この規格への適合性を示す場合は,ソフトウェアを含む
危険状態
を引き起こす可能性のあ
る,予見可能な一連の事象の特定を行う
リスク分析
を文書化して盛り込むことが望ましい(
JIS T 14971
参
照)。ソフトウェアに起因する
危険状態
(例えば,不適切な治療行為につながる可能性のある,誤解を招
く情報の提供に起因する
危険状態
)も,この
リスク分析
に記載するのがよい。
プロセス
の最初のカテゴリの一部として要求する全ての
アクティビティ
は,本文では“(クラス
A
,
B
,
C
)”と記載し,ソフトウェアの分類に関係なく必要であることを示している。
次の場合,
アクティビティ
をクラス
A
,
B
,
C
のいずれにも要求している。
-
アクティビティ
が,
リスクマネジメント
又はソフトウェア安全クラス分類に関係する計画を策定する。
-
アクティビティ
のアウトプットが,
リスクマネジメント
又はソフトウェア安全クラス分類のインプッ
トとなる。
-
アクティビティ
が,
リスクマネジメント
又はソフトウェア安全クラス分類の一部である。
-
アクティビティ
が,管理システム,文書化システム又は記録管理システムを確立して,
リスクマネジ
メント
又はソフトウェア安全クラス分類を支援する。
-
アクティビティ
は,関係するソフトウェアのクラス分類が不明なときに通常実施する。
-
アクティビティ
が,関連するソフトウェアの現在のソフトウェア安全クラス分類を無効にするような
変更をもたらす。これには,リリース後の安全性に関わる問題の検出及び分析が含まれる。
その他の
プロセス
は,ソフトウェア安全クラス
B
又は
C
に分類される
ソフトウェアシステム
又は
ソフト
ウェアアイテム
だけに要求している。これらの
プロセス
の一部として要求する
アクティビティ
は,本文で
は“(クラス
B
,
C
)”又は“(クラス
C
)”と記載し,適用されるソフトウェアのクラス分類に応じて選択的
に要求することを示している。
次の場合,
アクティビティ
は,クラス
B
及び
C
のソフトウェアに選択的に要求される。
30
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
-
アクティビティ
が,設計,試験又はその他の
検証
を,より詳細に又はより厳密にすることを要求し,
ソフトウェアの信頼性を高める。
-
アクティビティ
が,クラス
B
又は
C
に要求される別の
アクティビティ
を支援する管理
アクティビティ
である。
-
アクティビティ
が,安全性に関わる問題の是正を支援する。
-
アクティビティ
が,安全性に関わるソフトウェアの設計,実装,
検証
及びリリースの記録を作成する。
次の場合,
アクティビティ
は,クラス
C
のソフトウェアに選択的に要求される。
-
アクティビティ
が,設計,試験又はその他の
検証
を,より詳細に又はより厳密にすること若しくは特
定の問題点に注意を払うことを要求し,ソフトウェアの信頼性を更に高める。
なお,この規格で定義している全ての
プロセス
及び
アクティビティ
は,高品質ソフトウェアの開発及び
保守を確実に行うために有益なものとみなされていることに留意する。クラス
A
のソフトウェアの要求事
項に,これらの
プロセス
及び
アクティビティ
の多くが含まれていないが,それらに価値がない,又はそれ
らを推奨しないということを意味するものではない。
危険状態
1)
の発生の可能性がないソフトウェアの場
合,主には
医療機器
の設計中に行う総合的な妥当性確認
アクティビティ
によって,又はソフトウェアライ
フサイクルの管理項目の幾つかを単純に実施することで,その
安全
性及び有効性が保証できるということ
を考慮した結果,これらの
アクティビティ
を省略している(
医療機器
全体の妥当性確認は,この規格の適
用範囲外である。)。
注記
注
1)
は,序文の
注
1
)
を参照。
A.2
ソフトウェア安全クラス別要求事項のまとめ
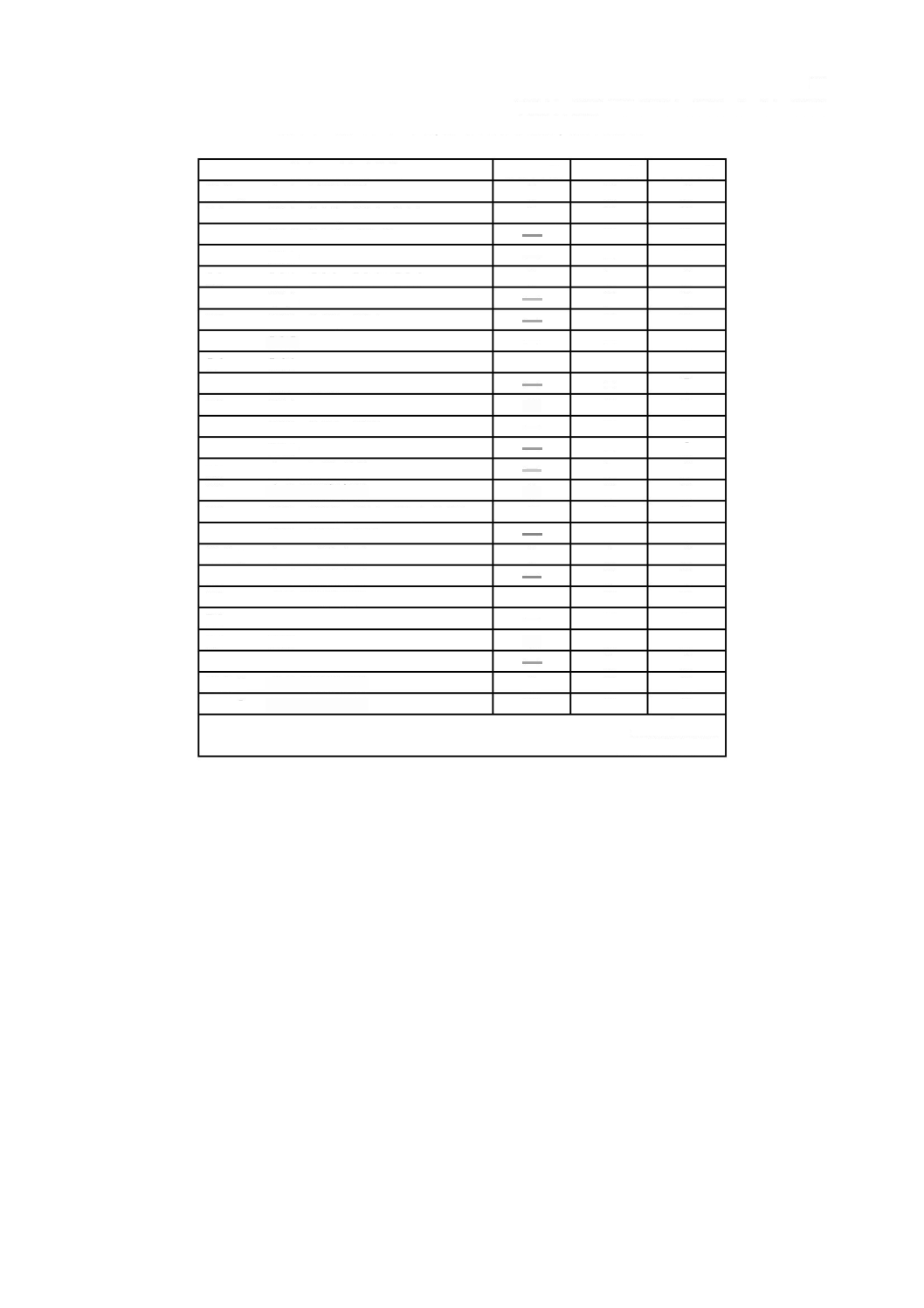
表
A.1
は,個々の要求事項がどのソフトウェア安全クラスを指定しているかをまとめている。
この表は,参考であり,便宜上示すにとどめる。個々の要求事項のためのソフトウェア安全クラスは,
本文で規定している。
31
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
A.1
-
ソフトウェア安全クラス別要求事項のまとめ
箇条及び細分箇条
クラス
A
クラス
B
クラス
C
箇条
4
全ての要求事項
○
○
○
5.1
5.1.1
~
5.1.3
,
5.1.6
~
5.1.9
○
○
○
5.1.5
,
5.1.10
~
5.1.12
-
○
○
5.1.4
-
-
○
5.2
5.2.1
,
5.2.2
,
5.2.4
~
5.2.6
○
○
○
5.2.3
-
○
○
5.3
5.3.1
~
5.3.4
,
5.3.6
-
○
○
5.3.5
-
-
○
5.4
5.4.1
-
○
○
5.4.2
~
5.4.4
-
-
○
5.5
5.5.1
○
○
○
5.5.2
,
5.5.3
,
5.5.5
-
○
○
5.5.4
-
-
○
5.6
全ての要求事項
-
○
○
5.7
全ての要求事項
○
○
○
5.8
5.8.1
,
5.8.2
,
5.8.4
,
5.8.7
,
5.8.8
○
○
○
5.8.3
,
5.8.5
,
5.8.6
-
○
○
箇条
6
全ての要求事項
○
○
○
7.1
全ての要求事項
-
○
○
7.2
全ての要求事項
-
○
○
7.3
全ての要求事項
-
○
○
7.4
7.4.1
○
○
○
7.4.2
,
7.4.3
-
○
○
箇条
8
全ての要求事項
○
○
○
箇条
9
全ての要求事項
○
○
○
○:該当する
-:該当しない
32
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
附属書
B
(参考)
この規格の適用についての指針
B.1
適用範囲
B.1.1
目的
この規格は,高品質で安全な
医療機器ソフトウェア
を,常に製造する開発
プロセス
を示すことを目的と
している。この目的を達成するために,この規格では,信頼性の高い安全な
ソフトウェア
を生産できる方
法でソフトウェア開発が行われたということを確実にするために,最低限実施すべき
アクティビティ
及び
タスク
を特定している。
この附属書は,この規格の要求事項の適用についての指針を記載したものであり,この規格の要求事項
に追加又は変更を加えるものではない。この附属書は,規格の要求事項をより明確に理解する目的で使用
できる。
ここで注意することは,この規格において,
アクティビティ
は,
プロセス
の中で定義している細分箇条
であり,
タスク
は,
アクティビティ
の範囲内で定義している。例えば,ソフトウェア開発
プロセス
で定義
している
アクティビティ
は,ソフトウェア開発計画,ソフトウェア要求事項分析,ソフトウェア
アーキテ
クチャ
設計,ソフトウェア詳細設計,
ソフトウェアユニット
の実装及び
検証
,ソフトウェア結合及び結合
試験,
ソフトウェアシステム
試験並びにソフトウェアリリースである。これらの
アクティビティ
に含まれ
る
タスク
は,個別の要求事項である。
この規格は,特定の
ソフトウェア開発ライフサイクルモデル
を要求するものではない。しかし,ある
プ
ロセス
へのインプットは,別の
プロセス
によって生じるため,この規格に適合することは,
プロセス
間の
依存性があるということを意味する。例えば,
ソフトウェアシステム
の故障からどんな
危害
が生じるかを
リスク分析プロセス
で確立した後に,
ソフトウェアシステム
のソフトウェア安全クラス分類を完了するこ
とが望ましい。
このように
プロセス
間に論理的依存性があるため,この規格の
プロセス
は,“ウォータフォール”又は
“ワンススルー(
once-through
)”ライフサイクルモデルを示唆するシーケンスで説明するのが最も容易で
ある。しかし,他のライフサイクルモデルも使用可能である。
JIS X 0160
[7]
で定義している開発モデルに
は,次のようなものがある(
表
B.1
も参照)。
-
ウォータフォール:“ウォータフォール”ともいわれる“ワンススルー(
once-through
)”モデルでは,
開発
プロセス
を一度実施する。簡単にいえば,顧客ニーズの決定,要求事項の定義,
システム
の設計,
システム
の実装,試験,修理及び出荷から成るモデルである。
-
繰返し(
Incremental
):“繰返し”モデルでは,顧客ニーズの決定及び
システム
要求事項の定義付けを
行い,その後,残りの開発を一連の版(
build
)ごとに行う。計画した仕様の一部を最初の版(
build
)
で組み込み,次の版(
build
)で更に仕様を追加し,
システム
完成に至るまでこれを続ける。
-
進展的(
Evolutionary
)
:
“進展的”モデルもまた,版(
build
)での
システム
開発であるが,ユーザの要
求が完全には理解されておらず,全ての要求事項を前もって定義することが不可能であるとの認識が
ある点で,繰返しモデルとは異なる。このモデルでは,顧客ニーズ及び
システム
要求事項の定義を前
もって部分的に行い,次の版(
build
)で改良(
refine
)する。
33
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
B.1
-
JIS X 0160
の定義に従った開発ライフサイクルモデル
開発モデル
最初に全ての要求事項
を定義するか
複数の開発サイクルか
暫定版ソフトウェアを
配布するか
ウォータフォール
(ワンススルー)
する
該当せず
しない
繰返し(
Incremental
)
(事前計画された改良)
する
該当する
可能性あり
進展的(
Evolutionary
)
しない
該当する
する
どのライフサイクルモデルを選択する場合でも,仕様書,設計書,ソフトウェアなどの
プロセス
アウト
プット間の論理的依存性を維持する必要がある。これは,ウォータフォールライフサイクルモデルの場合,
その
プロセス
のためのインプットが完了し承認されるまで,
プロセス
の開始を延期することで達成する。
他のライフサイクルモデル,特に進展的(
Evolutionary
)ライフサイクルモデルでは,
プロセス
アウトプ
ットを,当該
プロセス
の全てのインプットが利用可能になる前に作成できる。例えば,新しい
ソフトウェ
アアイテム
の仕様,分類,実装及び
検証
は,ソフトウェア
アーキテクチャ
全体が完成する前でも可能であ
る。このようなライフサイクルモデルには,一つの
プロセス
アウトプットの変更又は開発によって,別の
プロセス
アウトプットが無効になるという
リスク
が伴う。このため,全ての
プロセス
アウトプットを整合
させ依存性を維持するために,どのライフサイクルモデルにおいても包括的な構成管理システムを使用す
る。
次の原則は,使用する
ソフトウェア開発ライフサイクルモデル
にかかわらず重要である。
-
全ての
プロセス
アウトプットの整合性を維持することが望ましい。
プロセス
アウトプットを作成する
又は変更するときは,関連する全ての
プロセス
アウトプットを直ちに更新して,相互の整合性を維持
し,この規格が明示的又は暗示的に要求している全ての依存性を維持することが望ましい。
-
プロセス
アウトプットは,ソフトウェアについて更に作業するためのインプットとして必要なとき,
全てが利用可能になっていることが望ましい。
-
医療機器ソフトウェア
をリリースする前の段階で,全ての
プロセス
アウトプットに相互に整合性があ
り,この規格が明示的又は暗示的に要求している
プロセス
アウトプット間の依存性が,全てあること
が望ましい。
B.1.2
適用範囲
この規格は,
医療機器ソフトウェア
の開発及び保守,並びに
SOUP
を含む
医療機器
の開発及び保守に適
用する。
この規格を使用する場合,
製造業者
が
医療機器
について
JIS T 14971
に適合する
リスクマネジメント
を
実施することを要求している。したがって,
医療機器
の
システムアーキテクチャ
が,入手したコンポーネ
ント(購入したコンポーネント又は出所が不明なコンポーネント,例えば
SOUP
を含むプリンタ,プロッ
タなど)を含む場合,
製造業者
は,入手したコンポーネントに対する責任者となり,これを
医療機器
の
リ
スクマネジメント
対象に含めなければならない。
製造業者
は,
医療機器
の
リスクマネジメント
を適切に実
施することで,コンポーネントについて理解し,それに
SOUP
が含まれていることを認識しているとみな
される。この規格を利用する
製造業者
は,ソフトウェア
リスクマネジメントプロセス
を
医療機器
の
リスク
マネジメントプロセス
の一環として実施する。
リリースした
医療機器ソフトウェア
の保守は,その生産後の技術的行為に適用する。ソフトウェア保守
は,監視行為を含めたあらゆる技術的及び管理的手段の組合せを含む。その目的は,アイテムを維持又は
34
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
回復させるための行為を
問題報告
に基づいて実施し,リリースした
医療機器ソフトウェア
に関連する修正
要求だけでなく,
医療機器ソフトウェア
に要求されている機能を正しく実行できる状態にすることである。
例えば,問題是正,規制当局への報告,妥当性再確認,予防措置などが含まれる[
JIS X 0161
[8]
参照]。
B.2
引用規格
ISO/IEC 90003
[13]
は,品質マネジメントシステムをソフトウェア開発に適用するための指針を示してい
る。この規格が要求する指針ではないが,推奨する。
B.3
用語及び定義
用語の定義には,可能な範囲で国際規格の定義を使用している。
この規格は,
ソフトウェアシステム
(最上位レベル)の構造を説明する場合に,三つの用語を用いてい
る。
ソフトウェアシステム
は,
医療機器
のサブシステム(
IEC 60601-1-4
[15]
参照)又はそれ自体が
医療機
器
であり,結果としてソフトウェア
医療機器
になる。試験又はソフトウェア構成管理を目的にしたとき,
それ以上分割することができない最下位レベルは,
ソフトウェアユニット
である。最上位から最下位の構
成レベルまで,全て含めて
ソフトウェアアイテム
といえる。このように
ソフトウェアシステム
は,一つ以
上の
ソフトウェアアイテム
で構成し,各
ソフトウェアアイテム
は,一つ以上の
ソフトウェアユニット
又は
分割可能な
ソフトウェアアイテム
で構成する。
ソフトウェアアイテム
及び
ソフトウェアユニット
の定義並
びに粒度を示す責任は,
製造業者
にある。これらの用語は,明確に定義していないため,
医療機器
に使用
されるソフトウェアには多種多様な開発方法及びタイプが適用できる。
B.4
一般要求事項
いずれのソフトウェアについても,
100 %
の
安全
を保証する既知の方法はない。
医療機器ソフトウェア
の
安全
性を向上させる次の三つの大原則が存在する。
-
リスクマネジメント
-
品質マネジメント
-
ソフトウェアエンジニアリング
安全な
医療機器ソフトウェア
の開発及び保守には,適切なソフトウェアエンジニアリングの方法及び技
術を適用する総合的フレームワークとして,品質マネジメントシステムに不可欠な
リスクマネジメント
を
確立する必要がある。上記三つのコンセプトを組み合わせれば,
医療機器
の
製造業者
の意思決定
プロセス
が明確な体系をとり,首尾一貫した再現性のあるものとなり,
医療機器ソフトウェア
の
安全
性が促進され
る。
B.4.1
品質マネジメントシステム
統制がとれ,かつ,効果的な一連のソフトウェア
プロセス
には,マネジメント,インフラストラクチャ,
改善,訓練などの系統的な
プロセス
が含まれる。重複を避け,また,この規格の焦点をソフトウェアエン
ジニアリングに絞るため,これらの
プロセス
は,この規格から外している。これらの
プロセス
は,品質マ
ネジメントシステムが対象とするところである。
JIS Q 13485
[5]
は,品質マネジメントのコンセプトを特
に
医療機器
に応用するための規格である。ただし,
JIS Q 13485
の品質マネジメントシステム要求事項に適
合しても,それが自動的に法の要求事項に適合することにはならない。該当する規制要求事項に適合して
いるかを確認し,適合性を確立するのは,
製造業者
の責任である。
35
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
B.4.2
リスクマネジメント
ソフトウェア開発では,
リスクマネジメントアクティビティ
に十分に関与し,
医療機器ソフトウェア
関
連の合理的に予見可能な
リスク
を確実に考慮する。
このソフトウェア規格は,適切な
リスクマネジメントプロセス
を定義するものではなく,
医療機器
のた
めの
リスクマネジメント
を明確に扱っている規格,
JIS T 14971
に適合する
リスクマネジメントプロセス
の
適用を
製造業者
に要求する。ソフトウェアを一因とする
危険状態
に対して発生する,具体的なソフトウェ
ア
リスクマネジメントアクティビティ
については,箇条
7
の対象
プロセス
に示す。
B.4.3
ソフトウェア安全クラス分類
医療機器
の一部,
医療機器
の附属品又は一つの
医療機器
としてのソフトウェアに関連する
リスク
は,ソ
フトウェア安全クラス分類の仕組みに対するインプットとして使い,この仕組みによってソフトウェアの
開発及び保守を行うときに使用する
プロセス
を決定する。
リスク
は,
危害
の発生確率とその
危害
の重大さとの組合せと定義される。しかし,ソフトウェアの故障
が発生する確率を定量的に推定する広く認められた方法はない。
危険状態
の原因となる一連の事象又は事
象の組合せの中にソフトウェアが存在する場合,
危険状態
に対する
リスク
の推定においてソフトウェアの
故障の発生確率を考慮することはできない。このような場合,最悪の場合の確率を考えるのが適切であり,
ソフトウェアの故障の発生確率は,
1
とするのがよい。一連の事象のうち,ソフトウェア以外の事象の確
率を推定できる場合は,その確率を
危険状態
の発生確率として使用できる(
図
B.2
の
P
1
)。
しかし,ソフトウェア以外の事象の確率を推定できないことも多く,その場合は
危害
の性質だけに基づ
いて
リスク
を
評価
するのがよい(
危険状態
の発生確率は,
1
とするのがよい。
)。この場合の
リスク
の推定
は,
危険状態
から生じる
危害
の重大性に焦点を合わせるのがよい。医療従事者によって発見される可能性
が高い故障と,発見されずに
危害
の原因になる可能性がより高い故障を見分けるため,臨床知識に基づい
て主観的な確率ランキングを定めることもできる。
一般的に,
危険状態
が
危害
に至る確率(
図
B.2
の
P
2
)を推定するには,医療現場で
危害
を防げる可能性
が高い
危険状態
と,
危害
の原因となる可能性がより高い
危険状態
とを見分けるための臨床知識が必要とな
る。
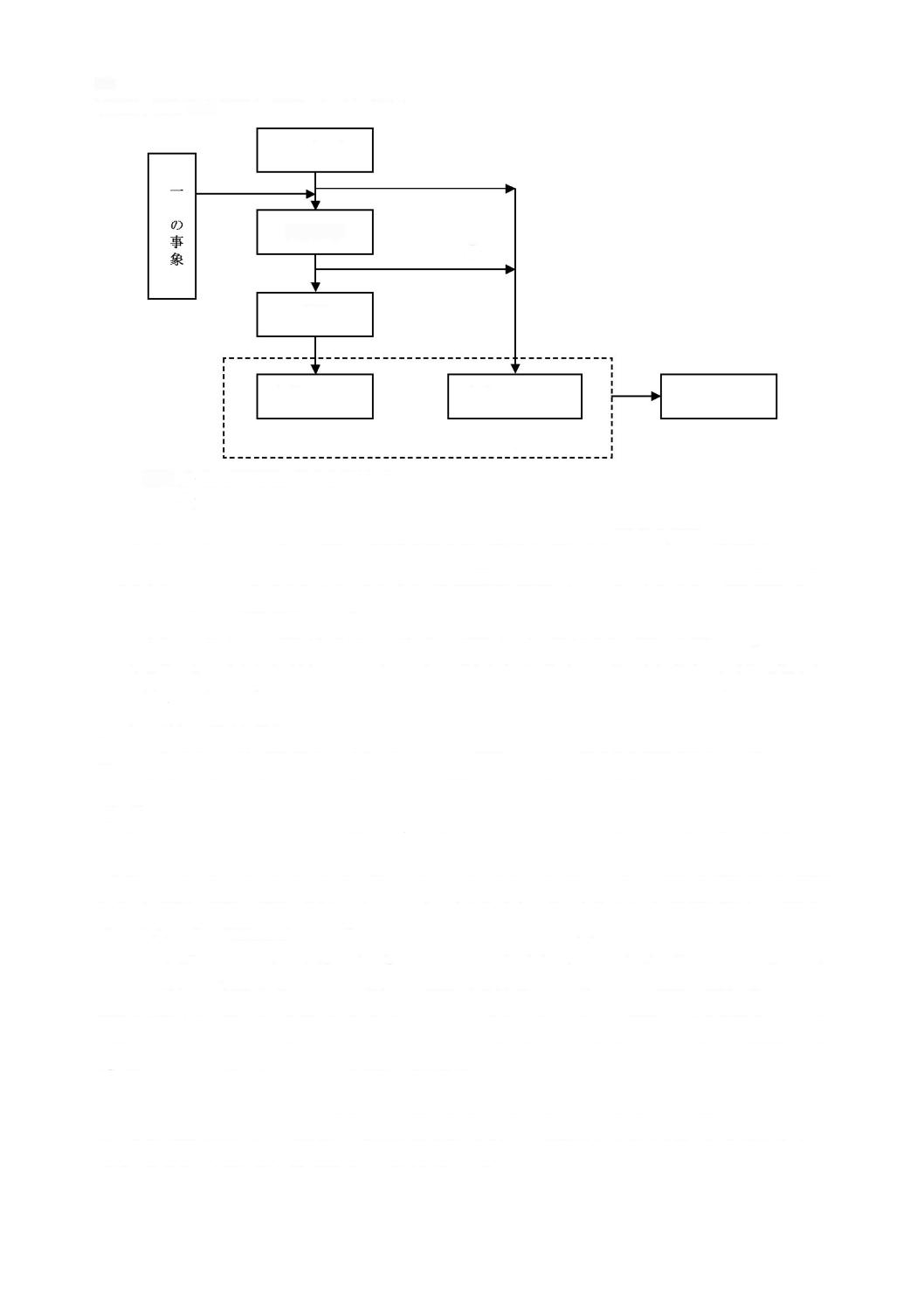
36
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
注記
P
1
は,
危険状態
の発生確率を示す。
P
2
は,
危険状態
が
危害
に至る確率を示す。
図
B.2
-
ハザード,一連の事象,危険状態及び危害の関係の図式(
JIS T 14971:2012
の附属書
E
)
ソフトウェアシステム
を
ソフトウェアアイテム
に分割した場合,
ソフトウェアアイテム
それぞれにソフ
トウェア安全クラス分類を割り当てる。
ソフトウェアアイテム
の故障に関連した
リスク
の判断は,次の場合に限り可能である。
-
システムアーキテクチャ
及びソフトウェア
アーキテクチャ
が,
ソフトウェアアイテム
の役割を,それ
自体の目的,及び他のソフトウェア及びハードウェアアイテムとのインタフェースの観点で定義付け
ている。
-
システム
の変更を管理している。
-
アーキテクチャ
及び指定した
リスクコントロール
手段についての
リスク分析
が完了している。
この規格は,全てのソフトウェアクラスについて前述の条件が整う,最低数の
アクティビティ
を要求し
ている。
ソフトウェアの
アーキテクチャアクティビティ
が終了するのは,
ソフトウェアアイテム
群全体を定義し,
リスクマネジメントアクティビティ
によって
ソフトウェアアイテム
がどのように
安全
に関わるかが明確
になる,開発の最初の時点である。したがって,これは,
ソフトウェアアイテム
の分類を
安全
上の役割に
応じて決定できる最初の時点である。
この時点は,
JIS T 14971
において
リスクコントロール
を開始する時点に対応する。
これに先立って,
リスクマネジメントプロセス
によって
アーキテクチャリスクコントロール
手段が明確
になる。例えば,保護サブシステムの追加又は
危害
の原因となるソフトウェアの故障の低減である。この
段階を過ぎると,
リスクマネジメントプロセス
は,
ソフトウェアアイテム
の故障の確率を低減させること
を狙いとした
プロセス
を使用する。つまり,
ソフトウェアアイテム
の分類が,そのアイテムに適用される
プロセス
ベースの
リスクコントロール
手段を特定する。
この時点以前にソフトウェアの分類を行うと,例えば,調査を実施する領域に焦点を絞ることができる
という点で
製造業者
にとって好都合であると予想されるが,この分類は,予備的なものとして捉え,
プロ
セス
の省略を正当化する目的に使わないことが望ましい。
一
連
の
事
象
ハザード
危険状態
危害
危害
の重大さ
危害
の発生確率
リスク
P
1
×
P
2
(
ハザード
の顕在)
P
1
P
2
37
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
ソフトウェア安全クラス分類の仕組みは,
JIS T 14971
の
リスク
に合わせることを意図するものではない。
JIS T 14971
の仕組みでは,重大さ及び発生確率に応じて
リスク
を分類しているが,ソフトウェア安全クラ
ス分類の仕組みでは,
ソフトウェアシステム
及び
ソフトウェアアイテム
を,開発及び保守で適用する
プロ
セス
に応じて分類している。
設計が進展するにつれ,新たな
リスク
が判明してくる可能性がある。そのため,
リスクマネジメント
を,
開発
プロセス
に不可欠な部分として適用することが望ましい。それによって,確実で安全に動作するため
に正確な機能が要求される
ソフトウェアアイテム
,及び
危害
を引き起こす故障を防止する
ソフトウェアア
イテム
を含む,全ての
ソフトウェアアイテム
を特定する
アーキテクチャ
設計の開発が可能になる。
ソフトウェア
アーキテクチャ
が,安全な動作に要求される
ソフトウェアアイテム
の分離を促すとともに,
その
ソフトウェアアイテム
を確実に効果的に分離するために使用する方法を説明付けることが望ましい。
分離は,物理的な分離(プロセッサ又はメモリの分割)に限定するものではなく,一つの
ソフトウェアア
イテム
が他の
ソフトウェアアイテム
に悪影響を及ぼさないようにするための,あらゆるメカニズムを含む。
分離の適切性は,関係する
リスク
と分離の根拠とに基づいて判断し,根拠については文書化する必要があ
る。
B.3
に記載したとおり,この規格では,
ソフトウェアシステム
(最上位レベル)の構成を説明する場
合に,三つの用語を用いる。
図
B.1
は,
ソフトウェアシステム
の中での
ソフトウェアアイテム
の分割の例,及び構成内の
ソフトウェ
アアイテム
群にどのようにソフトウェア安全クラスが割り当てられるかを示す。
図
B.1
-
ソフトウェアアイテムの分割例
この例では,
製造業者
は,開発中の
医療機器ソフトウェア
のタイプから,
ソフトウェアシステム
の予備
的なソフトウェア安全クラス分類が,ソフトウェア安全クラス
C
であるということを理解している。
製造
ソフトウェアシステム
/
ソフトウェアアイテム
(クラス
C
)
ソフトウェアアイテム
X
(クラス
A
)
ソフトウェアアイテム
Y
(クラス
C
)
ソフトウェアアイテム
W
(クラス
B
)
ソフトウェアアイテム
Z
(クラス
C
)
38
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
業者
は,ソフトウェア
アーキテクチャ
の設計時に,
システム
を
図
B.1
のように三つの
ソフトウェアアイテ
ム
,
X
,
W
及び
Z
に分割することを決定した。
製造業者
は,死亡又は
重傷
を引き起こす可能性のある
危険
状態
の要因となる全ての
ソフトウェアシステム
を,
ソフトウェアアイテム
Z
に分離し,
重傷
の可能性のな
い
危険状態
の要因となる,残り全ての
ソフトウェアシステム
を
ソフトウェアアイテム
W
に分離できる。
ソ
フトウェアアイテム
W
は,ソフトウェア安全クラス
B
に分類され,
ソフトウェアアイテム
Z
は,ソフト
ウェア安全クラス
C
に分類される。したがって,
ソフトウェアアイテム
Y
は,
4.3 d)
に従い,クラス
C
と
して分類する。
ソフトウェアシステム
も,また,この要求事項に従ってソフトウェア安全クラス
C
にする。
ソフトウェアアイテム
X
は,ソフトウェア安全クラス
A
に分類された。
製造業者
は,分離の完全性を徹底
するために,
ソフトウェアアイテム
X
及び
Y
並びに
W
及び
Z
について,分離の根拠を文書化できる。
ソフトウェアアイテム
X
及び
Y
の分離が不可能な場合は,
ソフトウェアアイテム
X
を,ソフトウェア
安全クラス
C
に分類しなければならない。
B.4.4
レガシーソフトウェア
4.4
は,この規格を
レガシーソフトウェア
に適用する
プロセス
を確立する。地域によっては,この規格
の現行版ができる前に設計されたもの(
レガシーソフトウェア
)であっても,
医療機器ソフトウェア
の規
制当局の承認を得るために,
製造業者
が規格への適合性を示さなければならないことがある。この場合,
製造業者
は,
4.4
に規定する方法によって,
レガシーソフトウェア
が規格に適合していることを示すこと
ができる。
製造業者
は,既に終了した開発ライフサイクルを過去に遡って単独の
アクティビティ
として文書化を行
っても,製品の使用に伴う
リスク
の低減にはつながらない,と判断するかもしれない。この
プロセス
では,
この規格で定義する
アクティビティ
のどれを実施すれば
リスク
の低減につながるかを特定する。この
プロ
セス
では,次を前提事項としている。
-
必要な
アクティビティ
及びそれに伴う文書化は,可能な限り既存の文書を信頼して,これを活用する。
-
製造業者
は,
リスク
を低減するために,リソースをできるだけ有効活用する。
この
プロセス
では,実行する必要のある
アクティビティ
を特定し,
レガシーソフトウェア
を安全に継続
使用するための客観的な証拠を集め,継続使用する根拠をまとめる。
レガシーソフトウェア
を計画的に継続使用することに伴う
リスク
は,そのソフトウェアを
ソフトウェア
システム
の作成に使用する状況によって変わる。
製造業者
は,
レガシーソフトウェア
に関連する
医療機器
の
ハザード
を特定し,全て文書化する。
4.4
では,
レガシーソフトウェア
が製造及び使用されていた期間に得られた,製造後の市場データの総
合的評価が必要となる。製造後データの一般的な情報源としては,次がある。
-
機器に起因する有害事象
-
機器の使用者からのフィードバック
-
製造業者
が発見した
異常
ソフトウェアの故障の発生確率を事前に定量的に推定する方法については,意見の一致が得られていな
いが,
レガシーソフトウェア
については,ソフトウェアの使用状況と製造後データの
評価
とに基づいてそ
うした情報が得られるかもしれない。そのような場合に,一連の事象それぞれの確率の定量的推定が可能
であれば,一連の事象全体の発生確率を定量的な値として使用してもよい。定量的に推定できない場合は,
最悪の場合の確率を考えるのが適切であり,ソフトウェアの故障の発生確率は
1
とするのがよい。
製造業者
は,
医療機器システムアーキテクチャ
全体において
レガシーソフトウェア
をどのように使用す
るかを決定し,
リスク
アセスメントのインプットとする。考慮すべき
リスク
は,それによって変わる。
39
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
-
レガシーソフトウェア
が安全,かつ,確実に使用されていて,その継続使用を
製造業者
が希望する場
合,継続使用の根拠は,主に製造後の記録に基づく
リスク
アセスメントによる。
-
レガシーソフトウェア
を再利用して新しい
ソフトウェアシステム
を作成する場合,
レガシーソフトウ
ェア
の意図する使用が当初のものと異なることがある。この場合,
レガシーソフトウェア
の故障に起
因する
危険状態
を見直して,
リスク
アセスメントで考慮しなければならない。
-
再利用する
レガシーソフトウェア
が新しい
ソフトウェアシステム
に結合されている場合でも,同様の
意図する使用に用いていることがあるかもしれない。この場合は,
5.3
に従って,
アーキテクチャ
上の
リスクコントロール
手段を修正して,
リスク
アセスメントで考慮するのがよい。
レガシーソフトウェア
を変更して新しい
ソフトウェアシステム
で使用する場合,
製造業者
は,これまで
安全で確実に使用されていたことの記録が,変更によってどの程度無効になるか検討するのが望ましい。
レガシーソフトウェア
の変更は,
リスクコントロール
手段への影響の評価(
7.4
)を含め,この規格の箇
条
4
~箇条
9
に従って実施するのがよい。
レガシーソフトウェア
の場合は,既存の
リスクコントロール
手
段が完全に文書化されていない可能性があり,使用可能な設計記録文書に加えて,
システム
について知識
をもつ個人の専門知識を活用して,変更がもたらす潜在的影響を慎重に
評価
するのがよい。
製造業者
は,
4.4
に従ってギャップ分析を実施する。この分析は,使用可能な文書を決定するために行い,
対象文書には,
レガシーソフトウェア
の開発中に行った
タスク
による客観的証拠を含み,
5.2
,
5.3
,
5.7
及
び箇条
7
の要求と比較して分析する。ギャップ分析を遂行するための一般的な手順には,次がある。
a)
レガシーソフトウェア
を
バージョン
,リビジョン及びその他の手段によって,明確に特定する。
b)
5.2
,
5.3
,
5.7
及び箇条
7
で要求する
成果物
に相当する,既存の
成果物
を
評価
する。
c)
使用可能な客観的証拠を
評価
する。必要な場合,以前適用した
ソフトウェア開発ライフサイクルモデ
ル
を文書化する。
d)
JIS T 14971
を考慮して,既存の
リスクマネジメント
文書が適切であるか
評価
する。
製造業者
は,実施したギャップ分析を考慮し,不足している
成果物
を作成して関連
アクティビティ
を実
施することで
リスク
をどの程度低減できるか
評価
し,ギャップを解消するための
アクティビティ
の実施及
び
成果物
の作成についての計画を立案する。
リスク
の低減においては,箇条
5
のソフトウェア開発
プロセス
を適用する利益と,開発履歴を十分知ら
ずに
レガシーソフトウェア
を修正することで新しい欠陥が生じ,
リスク
が上昇する可能性とのバランスを
とるのがよい。箇条
5
の要素の中には,事後に行っても,ほとんど又は全く
リスク
が低減されないと思わ
れるものもある。例えば,詳細設計及びユニット
検証
は,主に新しいソフトウェアの開発
プロセス
,又は
既存のソフトウェアのリファクタリング
プロセス
において
リスク
を低減する。目的が計画に明示されず,
ただ
アクティビティ
を行うだけでは,文書は作成されても
リスク
の低減にはつながらない可能性がある。
ギャップ解消計画では,少なくとも,不足している
ソフトウェアシステム
試験の記録に対処する。試験
記録がない又は
レガシーソフトウェア
を継続使用する根拠の裏付けとして適切でない場合は,
ソフトウェ
アシステム
要求事項の機能レベルでの作成(
5.2
)と試験(
5.7
)とを,ギャップ解消計画に含めるのがよ
い。
レガシーソフトウェア
を継続使用する根拠を示す文書は,
リスク
アセスメントの過程及びそのソフトウ
ェアを再利用する状況に適したギャップ解消計画を立案する過程で得られた,使用可能な客観的証拠と分
析とに基づいて作成する。
この根拠は,
レガシーソフトウェア
について使用可能な製造後の記録と,
プロセス
ギャップの解消によ
って達成される
リスクコントロール
手段とを共に考慮しており,計画した再利用状況における
レガシーソ
40
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
フトウェア
の安全で確実な性能を肯定的に論証するものとなる。
レガシーソフトウェア
を
4.4
に従って再利用した後は,そのソフトウェアのうち
成果物
のギャップが残
っている部分が,引き続き
レガシーソフトウェア
となる。この部分については,
4.4
に従って,今後,ま
た,再利用を検討してもよい。
レガシーソフトウェア
を変更して
成果物
のギャップを解消する場合は,こ
の規格の箇条
4
~箇条
9
に従って変更を実施するのがよい。
B.5
ソフトウェア開発プロセス
B.5.1
ソフトウェア開発計画
この
アクティビティ
の目的は,ソフトウェアに起因する
リスク
を低減するためのソフトウェア開発
タス
ク
を計画し,開発チームのメンバに手順及び目標を周知し,
医療機器ソフトウェア
の
システム
品質要求事
項に確実に適合することである。
ソフトウェア開発計画
アクティビティ
では,
タスク
を単一の計画書又は複数の計画書で文書化すること
が可能である。
製造業者
によっては,
医療機器ソフトウェア
全ての開発に適用する方針及び手順を既に確
立している可能性がある。その場合,計画は,単純に既存の方針及び手順を引用可能である。
製造業者
に
よっては,各
医療機器ソフトウェア
の開発に限定した,単一又は複数の計画書を作成する可能性もある。
この場合,固有の
アクティビティ
の内容を詳細に明記し,他は一般手順書を引用する。このほか,単一又
は複数の計画書を,
医療機器ソフトウェア
それぞれの開発に合わせて調整する可能性もある。計画は,開
発
プロセス
を実行するために必要なレベルまで詳細に定義し,
リスク
に比例したものにすることが望まし
い。例えば,
リスク
が高い
システム
又はアイテムは,開発
プロセス
をより厳密にする必要があり,
タスク
は,より詳細に規定することが望ましい。
計画とは,繰り返し行うことが望ましい
アクティビティ
であり,開発が進むにつれて,再検討及び更新
することが望ましい。
システム
及び
システム
開発に必要なレベルの取組みについての理解が深まるにつれ
て,計画は,より多くの,より十分な情報を包含したものへと発展できる。例えば,
リスクマネジメント
プロセス
の実行及びソフトウェア
アーキテクチャ
の開発の結果,
システム
の当初のソフトウェア安全クラ
ス分類が変わることがある。又は
システム
に
SOUP
を組み入れる決定がなされる可能性もある。開発
プロ
セス
を適切に管理できるよう,
システム
についての最新知識,及び
システム
又は
システム
におけるアイテ
ムに必要なレベルの厳密さについての最新知識を反映させ,計画を更新することが重要である。
B.5.2
ソフトウェア要求事項分析
この
アクティビティ
は,
製造業者
が
医療機器ソフトウェア
のソフトウェア要求事項を確立,検証するこ
とを要求する。何を構築するべきかの判断,
医療機器ソフトウェア
が受容できる動作を示しているかの判
断,及び完成した
医療機器ソフトウェア
が使用可能状態にあることを実証するためには,検証可能な要求
事項の確立が不可欠である。要求事項が要求どおりに実装されていることを実証するためには,要求事項
が正確に実装されているか否かを判断する客観的基準が確立できるような方法で,各要求事項を提示する
ことが望ましい。
医療機器
(
device
)の
リスクマネジメントプロセス
において,特定した
リスク
を管理す
るためにソフトウェアに要求事項が課されている場合,それらの要求事項をソフトウェア要求事項におい
て明確にすることが望ましい。このとき,
リスクコントロール
手段を追跡すると,ソフトウェア要求事項
に帰着できるような方法で明確にする。全てのソフトウェア要求事項は,要求事項と
ソフトウェアシステ
ム
試験との間の
トレーサビリティ
が実証できるように明確にすることが望ましい。規制当局の承認に特定
の規制又は規格への適合が求められる場合は,この適合要求事項をソフトウェア要求事項において文書化
することが望ましい。ソフトウェア要求事項が何をソフトウェアに実装するかを定義するため,要求事項
41
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
分析
アクティビティ
が完成する前に,要求事項を
評価
することが要求される。
顧客ニーズ,設計インプット,ソフトウェア要求事項,ソフトウェア機能仕様書及びソフトウェア設計
仕様書の区別には,しばしば混乱が見られる。設計インプットとは,顧客ニーズを解釈して正式に文書化
した
医療機器
要求事項である。ソフトウェア要求事項とは,顧客ニーズ及び設計インプットに適合するた
めにソフトウェアが果たすべき役割を正式に文書化した仕様書である。ソフトウェア機能仕様書は,ソフ
トウェア要求事項に付随することが多く,要求事項に適合する代替ソフトウェアが数多く存在する可能性
があっても,要求事項に適合するためにソフトウェアが果たすべき役割を詳細に定義する。ソフトウェア
設計仕様書は,ソフトウェアの要求事項及び機能仕様を実装するために,ソフトウェアをどのように設計,
分割するかを定義する。
従来,ソフトウェア要求事項,機能仕様書及び設計仕様書は,一つ以上の文書のまとまりとして書かれ
てきた。現在はこの情報を共通データベース内のデータアイテムとして捉えることが可能である。各アイ
テムには,その目的とデータベースの他のアイテムとの関連が定義される属性が一つ以上ある。この手法
によって,各対象ユーザ(
製造業者
,マーケティング担当者,試験者,監査員など)に最適な,様々な情
報の提示及び印刷が可能となり,実装の適切性及びテストケースによる要求事項の試験実施範囲を実証す
るための
トレーサビリティ
が確保されている。この手法の支援ツールには,リンク機能を用いた
HTML
文
書のように単純なものから,
CASE
ツール及び要求事項分析ツールのような複雑で高度なものまである。
システム
要求事項
プロセス
は,この規格の適用範囲外である。しかし,
医療機器
の機能をソフトウェア
を用いて実装するかは,通常
システム
設計時に決定する。一部又は全ての
システム
要求事項は,ソフトウ
ェアで実装するように割り当てられる。ソフトウェア要求事項分析
アクティビティ
は,
システム
要求事項
プロセス
がソフトウェアに割り当てた要求事項を分析する作業,及び割り当てた要求事項を反映した広範
囲にわたるソフトウェア要求事項を抽出する作業から成る。
システム
の完全性を確実にするために,
製造業者
は,親
システム
の要求事項及びソフトウェア要求事項
における非実用性,不整合性又は曖昧性を是正するために,
システム
要求事項に対する変更及び明確化の
実現を協議できる仕組みを提供することが望ましい。
システム
要求事項及びソフトウェア要求事項の収集及び分析の
プロセス
は,繰り返し行うことが可能で
ある。この規格は,その
プロセス
を厳密に二層に分離させることを要求するものではない。実際には,
シ
ステムアーキテクチャ
及びソフトウェア
アーキテクチャ
を同時に設計し,それに続いて
システム
要求事項
及びソフトウェア要求事項を階層化された形で文書化することが多い。
B.5.3
ソフトウェアアーキテクチャ設計
この
アクティビティ
は,
製造業者
がソフトウェアの主要構造コンポーネントを定義し,コンポーネント
の重要な役割,外部に表れるコンポーネントの特性及びその間の関係について特定することを要求してい
る。あるコンポーネントの動作が他のコンポーネントに影響を与える可能性がある場合は,その動作をソ
フトウェア
アーキテクチャ
で説明することが望ましい(
5.3.5
及び
B.4.3
参照)。この説明は,ソフトウェア
外部の
医療機器
のコンポーネントに影響を与え得る動作について特に重要である。
アーキテクチャ
の決定
は,
リスクコントロール
手段の実装に非常に重要である。他のコンポーネントに影響を与える可能性があ
るコンポーネントの動作を理解(及び文書化)しなければ,
システム
が安全であることを証明するのは不
可能に近い。ソフトウェア要求事項の正確な実装を確実にするためには,ソフトウェア
アーキテクチャ
が
必要である。ソフトウェア
アーキテクチャ
は,指定した
ソフトウェアアイテム
がソフトウェア要求事項の
全てを実装しない限り,完全とはいえない。ソフトウェアの設計及び実装は,
アーキテクチャ
に依存する
ため,この
アクティビティ
を完了するには
アーキテクチャ
を
検証
する。
アーキテクチャ
の
検証
は通常,技
42
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
術
評価
によって実施する。
ソフトウェア
アーキテクチャアクティビティ
での
ソフトウェアアイテム
のソフトウェア安全クラス分
類は,それに続くソフトウェア
プロセス
を選択する基準となる。分類の記録は,
リスクマネジメントファ
イル
の一部として変更管理する。
引き続いて起こるイベントによっては,分類が無効になる可能性があることも多い。例えば,次のよう
なイベントが該当する。
-
システム
仕様,ソフトウェア仕様又は
アーキテクチャ
の変更
-
リスク分析
のエラー,特に予見できない
ハザード
の発見
-
要求事項,特に
リスクコントロール
手段の実行不可能性の発見
したがって,ソフトウェア
アーキテクチャ
の設計に続く全ての
アクティビティ
で,
ソフトウェアシステ
ム
及び
ソフトウェアアイテム
の分類は,再
評価
することが望ましく,改訂が必要となる可能性もある。結
果,
ソフトウェアアイテム
の分類が上位クラスに引き上げられることによって見直しが発生し,
ソフトウ
ェアアイテム
に追加
プロセス
を適用するに至る可能性がある。ソフトウェア構成管理
プロセス
(箇条
8
)
は,全ての必要な見直しを確定し完了させることを確認するために用いる。
B.5.4
ソフトウェア詳細設計
この
アクティビティ
は,
製造業者
が
アーキテクチャ
で定義した
ソフトウェアアイテム
及びインタフェー
スをリファインし,
ソフトウェアユニット
及びそのインタフェースを作成することを要求する。
ソフトウ
ェアユニット
は,単一の関数又はモジュールとして考えることが多いが,この考え方は,常に適切である
わけではない。この規格では,
ソフトウェアユニット
を,それ以上小さなアイテムに細分化できない
ソフ
トウェアアイテム
と定義している。
ソフトウェアユニット
の試験は,ユニット単独で実行できる。
製造業
者
は,
ソフトウェアユニット
の詳細レベルを定義することが望ましい。詳細設計によって,アルゴリズム,
データ表記,異なった
ソフトウェアユニット
間のインタフェース及び
ソフトウェアユニット
とデータ構造
との間のインタフェースを定義する。詳細設計は,
ソフトウェア
のこん(梱)包も考慮する。
ソフトウェ
アユニット
及びインタフェースの設計については,十分な詳細を定義し,その
安全
性及び有効性を客観的
に
検証
できるようにする必要がある。他の要求事項又は設計文書を使用することで客観的な
検証
を担保で
きる場合もある。詳細設計は,プログラマがその場しのぎの設計を行う必要がないよう,完全であること
が望ましい。詳細設計は,
医療機器ソフトウェア
の
アーキテクチャ
も考慮しなければならない。
ソフトウェアアイテム
は,少数の新しい
ソフトウェアアイテム
だけが元の
ソフトウェアアイテム
の
安全
関連要求事項を実装するように分割可能である。残りの
ソフトウェアアイテム
は,
安全
関連機能を実装す
ることがなく,下位のソフトウェア安全クラスに再分類できる。しかし,これを実行する決定そのものが
リスクマネジメントプロセス
の一環であり,これは,
リスクマネジメントファイル
で文書化する。
実装は,詳細設計に依存するため,
アクティビティ
の完了前に詳細設計を
検証
する必要がある。詳細設
計の
検証
は,通常,技術
評価
によって実施する。
5.4.4
は,
製造業者
が詳細設計
アクティビティ
のアウトプ
ットを検証することを要求している。設計が,要求事項をどのように実装するかを定義する。設計を
検証
することで,設計がソフトウェア
アーキテクチャ
を実装していること,及び設計がソフトウェア
アーキテ
クチャ
と矛盾しないことが保証される。設計に欠陥があれば,コードは要求事項を正確に実装できない。
製造業者
は,安全性にとって重要であると思われる設計特性が設計中に存在する場合は,設計特性を検
証することが望ましい。こうした設計特性の例としては,次のようなものがある。
-
意図したイベント,インプット,アウトプット,インタフェース,論理フロー,
CPU
負荷の配分,メ
モリの配分,エラー及び例外の定義並びに分離,エラーからの復帰処理の実装
43
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
-
危険状態
を引き起こし得る全ての故障について,イベント及びその移行を伴う既定状態の定義
-
変数の初期化,メモリ管理
-
リスクコントロール
手段に影響し得るコールド及びウォームリセット,待機並びに他の状態変化。
B.5.5
ソフトウェアユニットの実装
この
アクティビティ
は,
製造業者
が
ソフトウェアユニット
のコードを作成し,検証することを要求する。
詳細設計は,ソースコードに変換される。コーディングは,仕様の分解が終了し,実行可能なソフトウェ
アの組立を開始する時点を表す。望ましいコード特性を一貫して実現するために,コーディング規約にお
いて,適切なコーディングスタイルを規定することが望ましい。コーディング規約の例としては,分かり
やすさ,言語使用法又は制限,複雑性管理などに対する要求事項がある。各ユニットのコードは,詳細設
計の定義どおりに機能すること,及び規定したコーディング規約に準拠することを確実にするために
検証
される。
5.5.5
は,
製造業者
がコードを検証することを要求している。コードが正確に設計を実装していない場合,
医療機器ソフトウェア
は,意図したとおりに機能しない。
B.5.6
ソフトウェア結合及び結合試験
この
アクティビティ
は,
製造業者
が,
ソフトウェアアイテム
を
ソフトウェアアイテム
の上位集合体に結
合することに加え,
ソフトウェアユニット
を
ソフトウェアアイテム
の集合体に結合することを計画及び実
施し,結合後の
ソフトウェアアイテム
が意図したとおりに作動するかを検証することを要求している。
この結合方法は,非繰返し結合から,様々な繰返し結合に及ぶ。アセンブルする
ソフトウェアアイテム
の特性によって,結合方法が決定される。
ソフトウェア結合試験では,
ソフトウェアアイテム
内外のインタフェースを介したデータ転送及び制御
が重点的な対象となっている。外部インタフェースとは,オペレーティングシステムのソフトウェアを含
む他のソフトウェア及び
医療機器
のハードウェアとのインタフェースである。
結合試験の厳密さ及び結合試験に関連する文書の詳細レベルは,次のものに見合うことが望ましい。す
なわち,機器に関連する
リスク
,潜在的に
危害
を発生させる可能性がある機能に対する機器のソフトウェ
アへの依存度,及び高
リスク
機器の機能における特定の
ソフトウェアアイテム
の役割である。例えば,全
ての
ソフトウェアアイテム
について試験を実施することが望ましいが,
安全
に影響を及ぼすアイテムは,
より直接的,徹底的,かつ,詳細な試験を実施することが望ましい。
規定どおりに,結合試験によって,インプット及びアウトプット領域の境界におけるプログラムの動作
を実証し,無効なインプット,予期せぬインプット及び特殊なインプットにプログラムが反応することを
確認する。複数のインプットの組合せ又は予期せぬインプットシーケンスが与えられたとき,若しくは規
定したタイミング要求事項の違反が生じたときに,プログラムの実際の動作が明らかになる。計画書の試
験要求事項には,適宜結合試験の一環として実施するホワイトボックス試験を含めることが望ましい。
ホワイトボックス試験は,グラスボックス,構造,クリアボックス及びオープンボックス試験としても
知られ,試験対象の
ソフトウェアアイテム
の内部動作についての十分な知識を使用して試験データを選択
する試験方法である。ホワイトボックス試験は,
ソフトウェアアイテム
についての特定の知識を用いてア
ウトプットを調べる試験である。試験者が
ソフトウェアアイテム
の使用目的を知っている場合にだけ,正
確な試験となる。試験者は,
ソフトウェアアイテム
が意図した目的から逸脱していないかを確認できる。
ホワイトボックス試験は,
ソフトウェアアイテム
の実装の試験が中心であるため,完全な仕様が実装され
ていることを保証するとは限らない。ブラックボックス試験は,動作,機能,不透明ボックス及びクロー
ズドボックス試験としても知られ,機能仕様の試験が中心であるため,実装のあらゆる部分について試験
44
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
済みであることを保証するとは限らない。このようにブラックボックス試験は,仕様を確認するための試
験であり,仕様の一部が適合していないことを示すことで,仕様漏れによる不具合を検出する。ホワイト
ボックス試験は,実装を確認するための試験であり,実装の一部に誤りがあることを示すことで,動作の
不具合を検出する。
医療機器ソフトウェア
に対して完全な試験を実施するためには,ブラックボックス試
験及びホワイトボックス試験の両方が要求される可能性がある。
5.6
及び
5.7
に示す計画書及び試験文書は,特定の開発フェーズ又は進展的(
Evolutionary
)プロトタイプ
に関連する個別の文書になる可能性がある。これらの文書を結合させて,単独の文書又はひとまとまりの
文書で複数の細分箇条の要求事項を対象とすることも可能である。また,上位のプロジェクト文書に,文
書の全て又は一部を組み入れることも可能である。上位のプロジェクト文書とは,ソフトウェア又はプロ
ジェクトの品質保証計画書,若しくはハードウェア及びソフトウェア試験のあらゆる点に対応した包括的
な試験計画書などである。この場合,様々なプロジェクト文書がどのように各ソフトウェア結合
タスク
に
関連するかを記載した相互参照表を作成することが望ましい。
ソフトウェア結合試験は,模擬環境,実際のターゲットハードウェア又は
医療機器
全体のいずれにおい
ても実施可能である。
5.6.2
は,
製造業者
が,ソフトウェア結合
アクティビティ
のアウトプットを検証することを要求している。
ソフトウェア結合
アクティビティ
のアウトプットとは,結合した
ソフトウェアアイテム
である。結合した
ソフトウェアアイテム
は,
医療機器ソフトウェア
全体が正確かつ安全に機能するよう適切に機能しなけれ
ばならない。
B.5.7
ソフトウェアシステム試験
この
アクティビティ
は,
製造業者
が,ソフトウェアの要求事項を正しく実装していることを検証するこ
とによって,ソフトウェアの機能を検証するよう要求している。
ソフトウェアシステム
試験は,定義した機能が存在することを実証する。この試験によって,ソフトウ
ェアの要求事項に従って構築したプログラムの,機能性及び性能を
検証
することができる。
ホワイトボックス手法(
B.5.6
参照)を用いて,特定の試験の遂行,負荷条件又は故障の誘発,コードの
品質試験範囲の拡大を実現することが望ましいが,
ソフトウェアシステム
試験では機能試験(ブラックボ
ックス試験)が集中的に行われる。タイプ及び試験段階ごとの試験の体系には柔軟性があるが,要求事項
の網羅性,
リスクコントロール
,有用性及び試験タイプ(故障,インストール,負荷など)を実証及び文
書化することが望ましい。
ソフトウェアシステム
試験は,結合したソフトウェアを試験するもので,模擬環境,実際のターゲット
ハードウェア又は
医療機器
全体のいずれにおいても実施可能である。
ソフトウェアシステム
を変更する場合(小さな変更であっても)
,(個々の変更の試験だけではない)
回
帰テスト
の実施の程度を決めて,意図しない副作用が発生していないことを確実にすることが望ましい。
この
回帰テスト
(及び
ソフトウェアシステム
試験全体を繰り返して行わない根拠)は,計画し,文書化す
ることが望ましい(
B.6.3
参照)。
ソフトウェアシステム
試験に対する責任は,分散させてもよく,異なる場所及び組織で実施可能である。
しかし,
タスク
の実行組織,実行場所,契約関係,コンポーネントの出所又は開発環境にかかわらず,機
器
製造業者
には,ソフトウェアが意図する使用に対して適切に機能することを徹底する最終責任がある。
試験中に発見した
異常
が繰り返し発生する可能性があっても,それを修正しないことが決定されている
場合,
リスク分析
と関連付けてこの
異常
を
評価
し,それが機器の
安全
性に影響しないことを検証する必要
がある。
異常
の根本的原因及び兆候を理解し,修正しない根拠を文書化することが望ましい。
45
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
5.7.4
は,期待した結果が得られたことを確認するために,
ソフトウェアシステム
試験の結果を
評価
する
ことを要求している。
B.5.8
システムレベルで使用するためのソフトウェアリリース
この
アクティビティ
は,
製造業者
が,リリースする
医療機器ソフトウェア
の
バージョン
を文書化し,ど
のように製造したかを明確にするとともに,そのソフトウェアのリリースに当たり適切な手順を踏むこと
を要求している。
製造業者
は,開発
プロセス
を用いて開発したソフトウェアが,リリースするソフトウェアであることを
証明できることが望ましい。また,
製造業者
は,ソフトウェア及びその生成に使用したツールを,今後必
要とされる場合に備え,回収可能にしておくことが望ましく,ソフトウェアの損傷又は誤使用が最小限に
抑えられるよう,ソフトウェアを保管,こん(梱)包及び出荷することが望ましい。これらの
タスク
を適
切に一貫した結果を伴って実施することを確実にするために,定義付けした手順を確立することが望まし
い。
B.6
ソフトウェア保守プロセス
B.6.1
ソフトウェア保守計画の確立
ソフトウェア保守
プロセス
は,次の
2
点でソフトウェア開発
プロセス
と異なる。
-
製造業者
は,緊急の問題に対処して即急に変更を実装するため,完全な形のソフトウェア開発
プロセ
ス
より小規模の
プロセス
を用いることが許されている。
-
製造業者
は,リリースした製品に関連するソフトウェア
問題報告
を受けて問題に対処するだけでなく,
(主に,現場から問題データを収集するために市販後監視の仕組みを施行し,問題についてユーザ及
び規制当局と連絡をとることによって)法規制に適合させる。
6.1
は,保守計画の中でこれらの
プロセス
を確立することを要求している。
この
アクティビティ
は,
製造業者
が保守
アクティビティ
及び
タスク
を実行するための手順を,考案又は
明確にすることを要求している。
製造業者
は,是正措置を実行し,保守時に変更を管理し,改訂されたソ
フトウェアの
リリース
を管理することを目的に,ユーザから報告された問題及び要請を文書化して解決す
るとともに,
医療機器ソフトウェア
の修正を管理することが望ましい。この
プロセス
は,ある問題を理由
として,又は改善,適合の必要性があるという理由から,
医療機器ソフトウェア
のコード及び関連文書が
修正される場合に有効となる。その目的は,リリースした
医療機器ソフトウェア
の完全性を維持しながら
これを修正することにある。この
プロセス
は,
医療機器ソフトウェア
のリリース時には想定していなかっ
た環境又はプラットフォームへの移行を含む。この細分箇条に示した
アクティビティ
は,保守
プロセス
固
有のものではあるが,この規格の他の
プロセス
を保守
プロセス
で使用してもよい。
製造業者
は,保守
プロセス
の
アクティビティ
及び
タスク
をどのように実行するかを計画する必要がある。
B.6.2
問題及び修正の分析
この
アクティビティ
は,
製造業者
が得たフィードバックの影響について分析し,報告された問題を検証
することを要求し,修正の選択肢の実装について検討,選択及び承認を得ることを要求している。問題及
びその他の変更要請は,
医療機器
の性能,
安全
性又は法律上の認可に影響する可能性がある。
問題報告
に
よる何らかの影響が存在するか,又は問題を是正したり要請内容を実装したりするための修正によって影
響が生じるかを判断するため,分析が必要である。ソフトウェア保守
アクティビティ
の一環として実装す
るソフトウェア変更によって,機器に組み込まれた
リスクコントロール
手段が誤った形で変更又は修正さ
れないことを,トレース分析又は回帰的分析を通して検証することが非常に重要である。また,修正前に
46
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
は
危険状態
を引き起こす又は
リスク
を低減させることのなかったソフトウェアについて,修正が原因で
危
険状態
が生じる又は
リスク
が低減されることがないことを
検証
することも重要である。ソフトウェア修正
で
危険状態
が生じる又は
リスク
が低減される可能性がある場合は,
ソフトウェアアイテム
のソフトウェア
安全クラス分類が変わっている可能性がある。
ソフトウェア保守(箇条
6
)とソフトウェア問題解決(箇条
9
)とを区別することは,重要である。
ソフトウェア保守
プロセス
において焦点となるのは,
医療機器ソフトウェア
リリース後のフィードバッ
クに適切に対処することである。ソフトウェア保守
プロセス
では,
医療機器
の一部として次の事項を徹底
する必要がある。
-
安全
に関連する
問題報告
について対応し,担当の規制当局及び影響を受けるユーザに対して報告をす
る。
-
医療機器ソフトウェア
は,問題の是正及び更なる問題の回避を確実にするために,正式な管理の下で
修正した後,再確認して再リリースされる。
-
製造業者
は,ほかのどの
医療機器ソフトウェア
が影響を受け得るかを考慮し,適切な処置をする。
ソフトウェア問題解決において焦点となるのは,次のような包括的マネジメントシステムの運用である。
-
問題報告
を分析し,問題が示唆する事柄全てを明確にする。
-
変更の数を決定し,変更による全ての副作用を明確にする。
-
リスクマネジメントファイル
を含むソフトウェア
構成アイテム
の一貫性を維持した上で,変更を実装
する。
-
変更の実装を
検証
する。
ソフトウェア保守
プロセス
では,ソフトウェア問題解決
プロセス
を使用する。ソフトウェア保守
プロセ
ス
では,
問題報告
についての上層部での決定(問題が存在するか,問題が
安全
性に重大な影響を与えるか,
どのような変更が必要か,及び変更をいつ実装するか)を扱うとともに,ソフトウェア問題解決
プロセス
を用いて
問題報告
分析を行い,引き起こされると思われる内容を全て検出し,変更を要する全ての
構成ア
イテム
,及び必要な全ての
検証
工程を明確にする,実行可能な
変更要求
を作成する。
B.6.3
修正の実装
この
アクティビティ
では,
製造業者
が修正を行う場合に,確立した
プロセス
を使用することを要求して
いる。保守
プロセス
を定義していない場合は,適切な開発
プロセスタスク
を使用して修正できる。さらに,
製造業者
は,修正によって当該
医療機器ソフトウェア
の他の部分に副作用が生じないことを確実にするこ
とが望ましい。
医療機器ソフトウェア
が新規の開発品として扱われていない場合は,修正が
医療機器ソフ
トウェア
全体に与える影響を分析する必要がある。回帰的分析及び
回帰テスト
を使用すると,変更によっ
て
医療機器ソフトウェア
の他の部分に問題が生じていないことを確認できる。回帰的分析では,関連文書
(ソフトウェア要求仕様書,ソフトウェア設計仕様書,ソースコード,試験計画書,テストケース,テス
ト手順など)のレビューに基づいて,変更のもたらす影響を判定し,実施すべき
回帰テスト
を特定する。
回帰テスト
では,プログラムがこれまで正しく実行してきたテストケースを再実行し,現在の結果を以前
の結果と比較して,ソフトウェアの変更がもたらす意図しない影響を見出す。
医療機器ソフトウェア
の修
正対象外の部分は,修正前と同じように機能するかを
回帰テスト
で確認するが,そのテストの量について
根拠付けをする。
B.7
ソフトウェアリスクマネジメントプロセス
ソフトウェア
リスクマネジメント
は,
医療機器リスクマネジメント
全体の一部であり,分離した場合,
47
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
十分に扱うことはできない。この規格は,
JIS T 14971
に適合する
リスクマネジメントプロセス
を使用する
ことを要求している。
JIS T 14971
で定義している
リスクマネジメント
は,特に
医療機器
の使用に関連する
リスク
を効果的に管理するためのフレームワークを扱う。
JIS T 14971
の一部は,
リスク分析
で確定した各
ハザード
に関連する特定の
リスク
の管理に関係する。この規格のソフトウェア
リスクマネジメントプロセ
ス
は,
リスク分析
で
危険状態
の潜在的な一因とされたソフトウェア又は
医療機器
の
リスクマネジメント
に
使用するソフトウェアなどの
リスクコントロール
に要求事項を追加する。次の二つの理由から,ソフトウ
ェア
リスクマネジメントプロセス
がこの規格に含まれている。
a)
この規格の利用者は,ソフトウェアという責任領域における
リスクコントロール
手段の最低限の要求
事項を理解する必要がある。
b)
この規格に引用規格として示している,一般的な
リスクマネジメント
を扱う規格
JIS T 14971
は,ソ
フトウェアの
リスクコントロール
及びソフトウェア開発ライフサイクルへの
リスクコントロール
導入
について特に扱うものではない。
ソフトウェア
リスクマネジメント
は,
医療機器リスクマネジメント
全体の一部である。ソフトウェア
リ
スクマネジメントアクティビティ
に要求する計画書,手順書及び資料は,一連の個別の文書でも単独の文
書でもよく,この規格の要求事項全てに適合する限りにおいては,
医療機器リスクマネジメント
の
アクテ
ィビティ
及び資料と統合させてもよい。
B.7.1
危険状態を引き起こすソフトウェアの分析
医療機器
(
device
)の
リスク分析
3)
によって,
危険状態
及び
危険状態
に対応する
リスクコントロール
手
段が明確になり,
危険状態
の確率及び/又は重大さが受容可能なレベルにまで低減することを期待する。
また,
リスクコントロール
手段を実装することを期待するソフトウェア機能に,
リスクコントロール
手段
が割り当てられることを期待する。
注
3)
対応国際規格の
ハザード
分析を,
リスク分析
とした。
しかし,ソフトウェア
アーキテクチャ
の設計完了までに,全ての機器の
危険状態
を確定することは期待
できない。
アーキテクチャ
の設計完了時点では,ソフトウェア機能をどのようにソフトウェアコンポーネ
ントの中で実装するかが明らかになり,ソフトウェア機能に割り当てた
リスクコントロール
手段の実用性
を
評価
する。その時点で,
医療機器
の
リスク分析
3)
を改訂して,次の事項を含めることが望ましい。
-
改訂された
危険状態
-
改訂された
リスクコントロール
手段及びソフトウェア要求事項
-
人的要因に関係した
危険状態
など,ソフトウェアから生じる新たな
危険状態
ソフトウェア
アーキテクチャ
には,ソフトウェアコンポーネント間で危険な相互作用が起きないように
ソフトウェアコンポーネントを分離する確実な方針を含むのが望ましい。
B.8
ソフトウェア構成管理プロセス
ソフトウェア構成管理
プロセス
は,ソフトウェアライフサイクル全般にわたって管理手順及び技術的手
順を適用する
プロセス
であり,文書を含む
システム
における
ソフトウェアアイテム
の識別及び定義,アイ
テムの修正及び
リリース
の管理,並びにアイテム及び
変更要求
の状況の文書化及び報告を行う。ソフトウ
ェア構成管理は,
ソフトウェアアイテム
の再製作,その構成部分の確定及び実施した変更履歴の提供に必
要である。
B.8.1
構成の識別
この
アクティビティ
は,
製造業者
がソフトウェア
構成アイテム
及びその
バージョン
を一意に識別するこ
48
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
とを要求する。この識別は,
医療機器ソフトウェア
に含まれるソフトウェア
構成アイテム
及び
バージョン
の識別に必要である。
B.8.2
変更管理
この
アクティビティ
は,
製造業者
がソフトウェア
構成アイテム
の変更を管理するとともに,
変更要求
を
明確にし,その性質を記載するための情報を文書化するよう要求している。この
アクティビティ
は,ソフ
トウェア
構成アイテム
に許可していない又は意図しない変更を決して加えることがなく,承認した
変更要
求
を完全に実装し
検証
することを確実にするために必要である。
変更要求
は,変更管理委員会,又はマネージャ若しくは技術リーダーのいずれかが,ソフトウェア構成
管理計画に基づいて承認できる。承認した
変更要求
は,ソフトウェアの実際の修正及び
検証
に対して追跡
可能なものにする。ここでの要求事項は,実際の変更それぞれが
変更要求
と関連付けられ,
変更要求
が承
認されたことを示す文書が存在することである。この文書は,変更管理会議議事録,承認署名又はデータ
ベース上の記録であってもよい。
B.8.3
構成状況の記録
この
アクティビティ
は,
製造業者
がソフトウェア
構成アイテム
の履歴を保持することを要求している。
この
アクティビティ
は,変更がいつ,なぜ行われたかを判断するために必要である。この情報の入手は,
ソフトウェア
構成アイテム
が認可された修正だけを含むことを確実にするために,必要である。
B.9
ソフトウェア問題解決プロセス
ソフトウェア問題解決
プロセス
は,問題の性質及び原因にかかわらず,問題(不適合を含む。
)を分析し
解決する
プロセス
であり,問題には,開発,保守又はその他の
プロセス
の実行時に発見したものも含まれ
る。その目的は,適時の,責任を伴う文書化した手段によって,発見した問題を分析,解決し,その傾向
を認識するようにすることである。この
プロセス
は,ソフトウェアエンジニアリングの文献で“欠陥追跡”
といわれることもある。
JIS X 0160
[7]
及び
IEC 60601-1-4
[15]
の
Amendment 1
では,
“問題解決”と規定し
ている。この規格では“ソフトウェア問題解決”と規定している。
この
アクティビティ
は,問題又は不適合が特定されたときに,
製造業者
がソフトウェア問題解決
プロセ
ス
を使用することを要求している。この
アクティビティ
は,発見した問題を,
安全
との関連性について分
析及び
評価
することを確実にするために必要である(
JIS T 14971
の規定)。
ソフトウェア開発計画又は手順は,
5.1
で要求しているように,問題又は不適合の扱い方について検討す
る。これには,ライフサイクルの各段階で,文書化する正式なソフトウェア問題解決
プロセス
の各側面を
明確にするとともに,問題及び不適合点をどの時点でソフトウェア問題解決
プロセス
に取り入れるかを指
定することが含まれる。
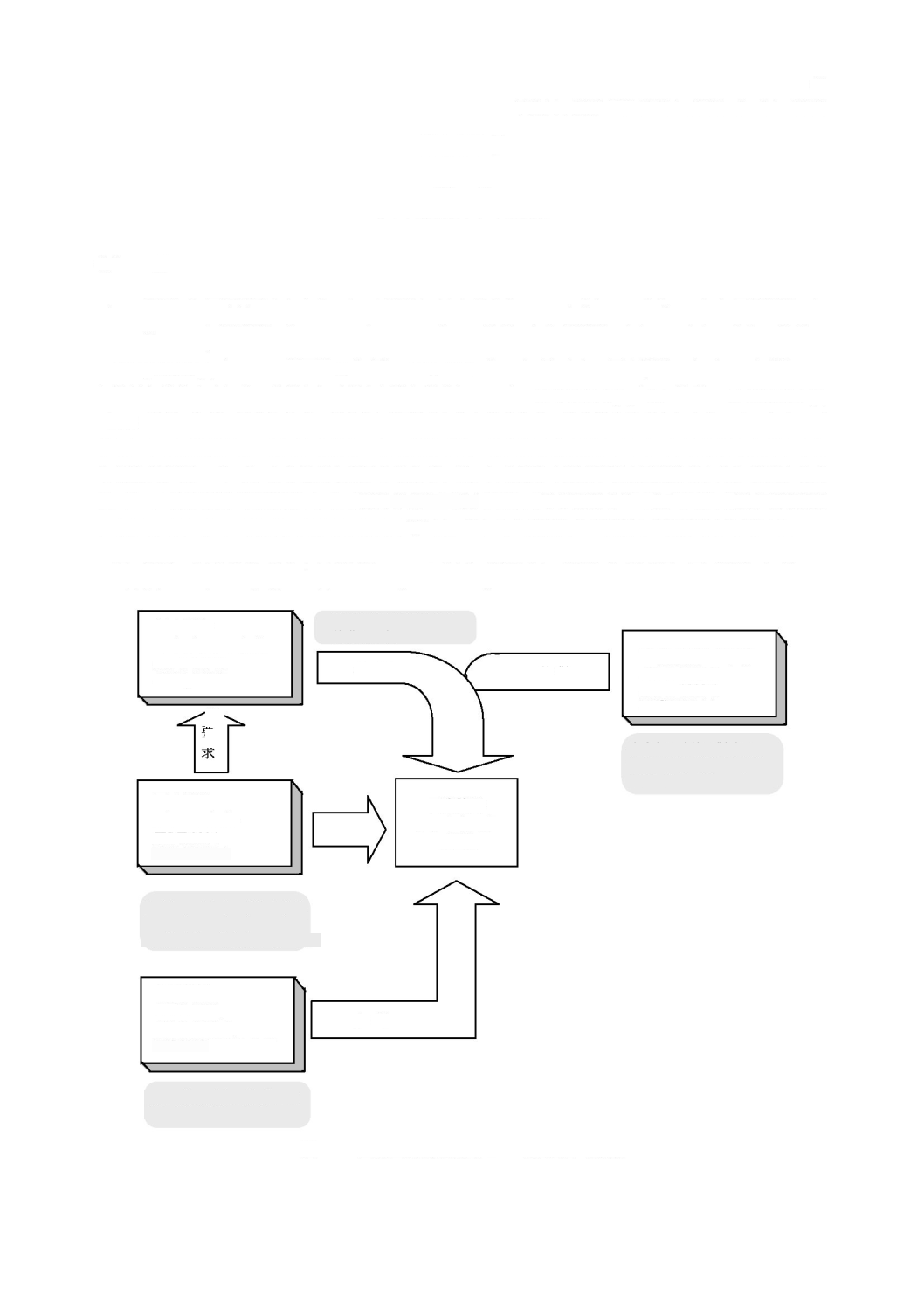
49
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
附属書
C
(参考)
他の規格との関係
C.1
一般
この規格は,
医療機器ソフトウェア
の開発及び保守を対象としている。ソフトウェアは,
医療機器
のサ
ブシステム又は
医療機器
自体であるとみなされる。この規格は,
医療機器
の開発時に,他の適切な規格と
ともに使用する。
JIS Q 13485
[5]
(
C.2
及び
附属書
D
参照),
JIS T 14971
(
C.3
参照)などの
医療機器
マネジメント規格は,
組織がを開発するための基盤となる管理環境を規定している。
JIS T 0601-1
[6]
(
C.4
参照),
JIS C 1010-1
[2]
(
C.5
参照)などの安全規格は,安全な
医療機器
を作り出すための具体的な指示を与える。ソフトウェア
がこれらの
医療機器
の一部である場合,この規格では,安全な
医療機器ソフトウェア
を開発し維持するた
めの要求事項について,より詳細な指示を与えている。この規格の要求事項を実装するために使用する方
法,ツール及び技術の情報源として,
JIS X 0160
[7]
(
C.6
参照),
JIS C 0508-3
[1]
(
C.7
参照),
ISO/IEC 90003
[13]
など,他の多くの規格を参照できる。
図
C.1
は,これらの規格とこの規格との関係を示している。
他の規格から箇条又は要求事項を引用している場合,引用文内の用語は,引用元の他の規格で定義して
いる用語であり,この規格で定義しているものではない。
図
C.1
-
主要医療機器規格とこの規格との関係
医療機器
マネジメント規格
JIS T 14971
JIS Q 13485
医療機器
プロセス規格
JIS T 2304
IEC 62366-1
他の規格
JIS X 0160
,
JIS C 0508-3
,
ISO/IEC 90003
など
医療機器
製品規格
JIS T 0601-1
JIS C 1010-1
IEC 82304-1
利用可能な追加の指針,技
術などを規定している。
安全な
ソフトウェアシステム
の開発及び保守の仕方に関す
る詳細な指示を規定している。
医療機器開発のための
基礎を示す。
安全な医療機器製造の
ための具体的指示を規定
している。
医療機器
ソフトウェア
の実装
参
考
影
響
影
響
影
響
要
求

50
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
C.2
JIS Q 13485
との関係
この規格は,
製造業者
が品質マネジメントシステムを採用することを要求している。
製造業者
が
JIS Q
13485
[5]
を使用する場合,
表
C.1
に示すように,この規格の要求事項が
JIS Q 13485
の要求事項の一部と
直接関係する。
表
C.1
-
JIS Q 13485:2005
との関係
この規格の箇条及び細分箇条
JIS Q 13485
:2005
の関連箇条及び細分箇条
5.1
ソフトウェア開発計画
7.3.1
設計・開発の計画
5.2
ソフトウェア要求事項分析
7.3.2
設計・開発へのインプット
5.3
ソフトウェアアーキテクチャの設計
5.4
ソフトウェア詳細設計
5.5
ソフトウェアユニットの実装及び検証
5.6
ソフトウェア結合及び結合試験
5.7
ソフトウェアシステム試験
7.3.3
設計・開発からのアウトプット
7.3.4
設計・開発のレビュー
5.8
システムレベルで使用するためのソフトウェアリリ
ース
7.3.5
設計・開発の検証
7.3.6
設計・開発の妥当性確認
6.1
ソフトウェア保守計画の確立
7.3.7
設計・開発の変更管理
6.2
問題及び修正の分析
6.3
修正の実装
7.3.5
設計・開発の検証
7.3.6
設計・開発の妥当性確認
7.1
危険状態を引き起こすソフトウェアの分析
7.2
リスクコントロール手段
7.3
リスクコントロール手段の検証
7.4
ソフトウェア変更のリスクマネジメント
8.1
構成識別
7.5.3
識別及びトレーサビリティ
8.2
変更管理
7.5.3
識別及びトレーサビリティ
8.3
構成状態の記録
9
ソフトウェア問題解決プロセス
C.3 JIS
T 14971
との関係
表
C.2
は,
JIS T 14971
で要求する
リスクマネジメントプロセス
の要求事項が,この規格で規定している
分野を示している。

51
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.2
-
JIS T 14971:2012
との関係
JIS T 14971
:2012
の箇条及び細分箇条
この規格の関連箇条及び細分箇条
4.1
リスク分析プロセス
4.2
意図する使用及び医療機器の安全に関する特質の明
確化
4.3
ハザードの特定
7.1
危険状態を引き起こすソフトウェアの分析
4.4
個々の危険状態に対するリスクの推定
4.3
ソフトウェア安全クラス分類
5
リスク評価
6.1
リスクの低減
6.2
リスクコントロール手段の選択
7.2.1
リスクコントロール手段の選択
6.3
リスクコントロール手段の実施
7.2.2
ソフトウェアに実装するリスクコントロール手段
7.3.1
リスクコントロール手段の実装の検証
6.4
残留リスクの評価
6.5
リスク/効用 分析
6.6
リスクコントロール手段によって発生したリスク
6.7
リスクコントロールの完了
7
残留リスクの全体的な受容可能性の評価
8
リスクマネジメント報告書
7.3.3
トレーサビリティの文書化
9
製造及び製造後情報
7.4
ソフトウェア変更のリスクマネジメント
C.4 JIS
T 0601-1:2012
及び追補
1:2014
の
PEMS
要求事項との関係
C.4.1
一般
ソフトウェア要求事項は,プログラマブル電気医用システム(
PEMS
)の要求事項のサブセットである。
この規格では,
PEMS
についての
JIS T 0601-1
:2012
及び追補
1:2014 [6]
の要求事項に矛盾しない追加のソフ
トウェア要求事項を規定している。
PEMS
にはソフトウェア以外の要素も含まれるため,
PEMS
について
の
JIS T 0601-1
:2012
及び追補
1:2014
の要求事項の全てをこの規格で扱っているわけではない。
JIS T
0601-1
:2012
及び追補
1:2014
の発行とともに,この規格は
JIS T 0601-1
の引用規格となり,
JIS T 0601-1
:2012
及び追補
1:2014
の箇条
14
に適合する(ひいては,規格に適合する)ためには,この規格の一部に適合し
なければならなくなった(
JIS T 0601-1
:2012
及び追補
1:2014
は,この規格の市販後及び保守に関する要求
事項への適合を求めていないため,この規格全体ではない。)。また,
JIS T 0601-1
:2012
及び追補
1:2014
が
使用されるのは,ソフトウェアが
PEMS
の一部となっている場合だけであり,ソフトウェアがそれ自体で
医療機器
となっている場合には適用されないことを覚えておくことが重要である。
C.4.2
ソフトウェアと
PEMS
開発との関係
PEMS
開発の内容を説明した
図
C.2
の
V
字形モデルから,このソフトウェア規格の要求事項が,ソフト
ウェア要求事項の指定から,結合による
ソフトウェアアイテム
の
ソフトウェアシステム
化までの,
PEMS
コンポーネントレベルで適用されるということが分かる。この
ソフトウェアシステム
は,プログラマブル
電子サブシステム(
PESS
)の一部であり,このサブシステムは,
PEMS
の一部である。
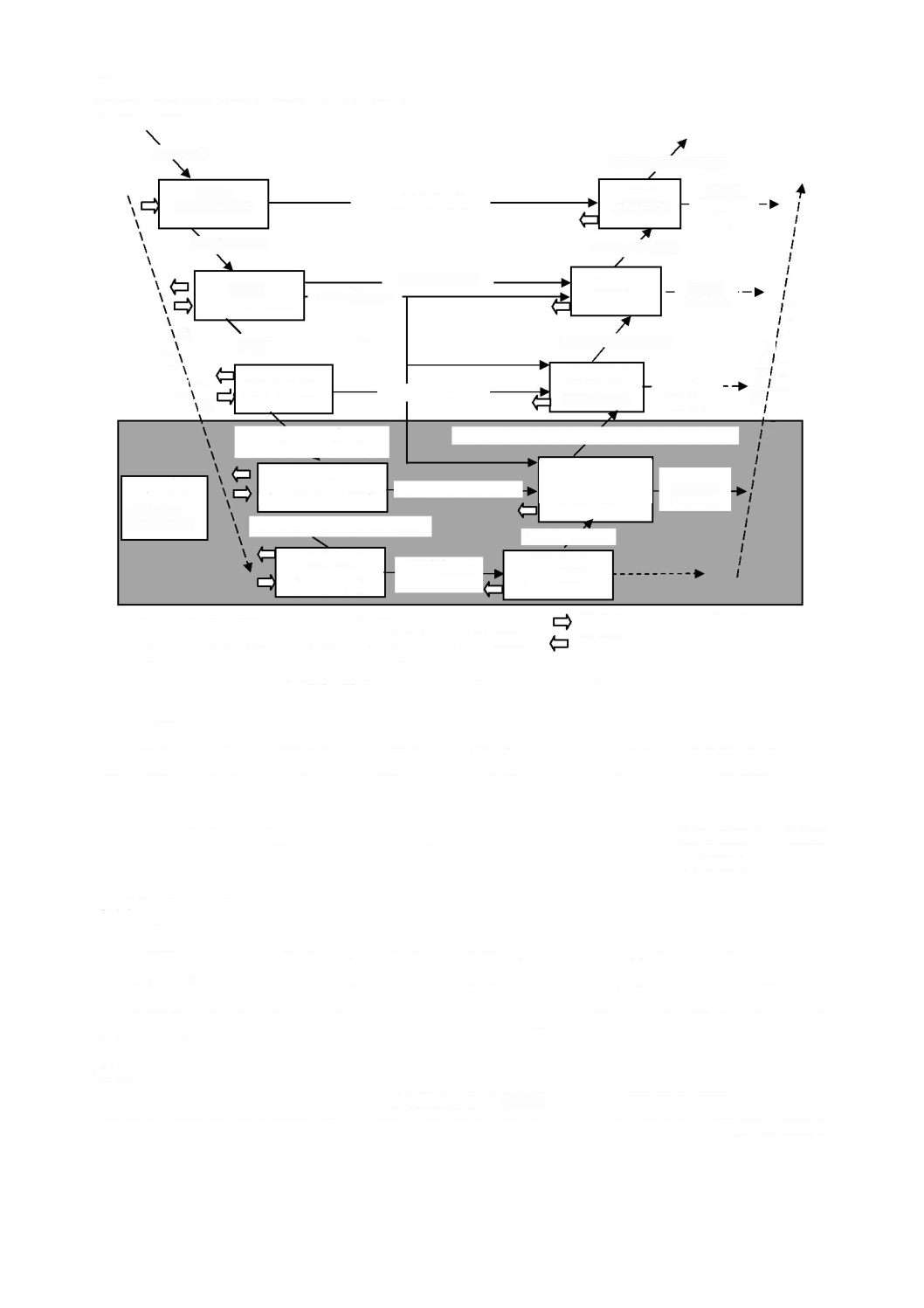
52
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
図
C.2
-
V
字形モデルの一部としてのソフトウェア
C.4.3
開発プロセス
この規格のソフトウェア開発
プロセス
(箇条
5
)に適合するために,ソフトウェア開発計画を定めて,
それに従うことを要求している。これに当たって,ある特定のライフサイクルモデルの使用が必要ではな
いが,計画に特定の
アクティビティ
及び属性を含めることが必要である。これらの要求事項は,開発ライ
フサイクル,要求事項の指定,
アーキテクチャ
,設計及び実装,並びに
検証
についての
JIS T 0601-1
の
PEMS
要求事項に関係している。ソフトウェア開発についてのこの規格の要求事項は,
JIS T 0601-1
よりも詳し
い内容になっている。
C.4.4
保守プロセス
この規格のソフトウェア保守
プロセス
(箇条
6
)に適合するためには,ソフトウェアを変更するときの
手順を確立し,それに従うことが要求される。これらの要求事項は,
PEMS
の修正についての
JIS T 0601-1
の要求事項に対応している。この規格のソフトウェア保守についての要求事項は,ソフトウェア保守のた
めに必ず行われなければならない事項について,
JIS T 0601-1
の
PEMS
修正の要求事項よりも詳しい内容
になっている。
C.4.5
その他のプロセス
この規格のその他の
プロセス
では,
JIS T 0601-1
にある
PEMS
についての類似要求事項以外の,追加の
ソフトウェア要求事項を規定している。ほとんどの場合,
PEMS
についての一般要求事項が
JIS T 0601-1
にあり,それを発展させたのがこの規格の
プロセス
である。
ボックスは代表的な開発ライフサイクル
アクティビティ
実線矢印は
アクティビティ
から又は
アクティビティ
へ転送される代表的な
成果物
破線矢印は
リスクマネジメントファイル
だけへの
成果物
問題解決
プロセス
へのインプット
問題解決
プロセス
からのアウトプット
PEMS
要求事項の把握
顧客の要求
リ
ス
ク
分
析
PEMS
要求仕様
PEMS
アーキテクチャ仕様
PEMS
アーキテクチャ設計
サブシステム
(
PESS
など)アー
キテクチャ設計
ソフトウェア要求仕様
(コンポーネント要求事項)
PEMS
V
字形
モデルで
この規格に
含まれる部分
ソフトウェア
アーキテクチャ設計
(コンポーネント設計)
ソフトウェア
アーキテクチャ
仕様
ソフトウェア
詳細設計
(ユニット設計)
ソフトウェア
ユニット実装
ソフトウェア
ユニット
検証
(ユニット
検証
)
結果
ユニット
検証
ソフトウェア結合及び
ソフトウェアシステム
検証
(コンポーネント
結合及び
検証
)
ソフトウェア試験仕様
ソフトウェア
結合及び
検証
結果
サブシステム
(
PESS
など)
結合及び
検証
サブシステム
検証
結果
検証済みコード
検証済みソフトウェアサブシステム(コンポーネント)
検証済みサブシステム
PEMS
検証
結果
検証
済みの
PEMS
PEMS
妥当性確認
PEMS
結合・
検証
妥当性確認済みの
PEMS
PEMS
妥当性確認
結果
PEMS
妥当性確認計画
PEMS
試験仕様
PEMS
検証
計画
サブシステム試験仕様
P
E
M
S
結
合
要
求
事
項
分
割
リ
ス
ク
コ
ン
ト
ロ
|
ル
検
証

53
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
この規格のソフトウェア
リスクマネジメントプロセス
は,
JIS T 0601-1
で特定している
PEMS
について
の補足的
リスクマネジメント
要求事項に対応している。
この規格のソフトウェア問題解決
プロセス
は,
JIS T 0601-1
の
PEMS
についての問題解決要求事項に対
応している。
この規格のソフトウェア構成管理
プロセス
は,
JIS T 0601-1
の
PEMS
についての要求事項にはない,追
加の要求事項(文書化は除く。)を規定している。
C.4.6 JIS
T 0601-1:2012
及び追補
1:2014
の
PEMS
要求事項の範囲
表
C.3
は,
JIS T 0601-1
の
PEMS
要求事項及びそれに対応するこの規格の要求事項である。
注記
JIS T 0601-1
から引用した箇所では,
JIS T 0601-1
で定義した用語を太字で表記した。
表
C.3
-
JIS T 0601-1
との関係
JIS T 0601-1
:2012
及び追補
1:2014
の
PEMS
要求事項
PEMS
のソフトウェアサブシステムに関係する,
この規格の要求事項
14.1
一般
14.2
~
14.12
の要求事項は,次に該当する場合は,
PEMS
に適用する。
-
プログラマブル電子サブシステム
(
PESS
)が,
基礎安
全
又は
基本性能
に必要な機能を提供する場合。
-
4.2
に従った
リスクマネジメント
の適用によって,
PESS
の故障が受容できない
リスク
を生じないことを立証で
きない場合。
14.2
~
14.12
の要求事項を適用するかどうかによらず,
14.13
の要求事項は,
IT
ネットワーク
に組み込むことを意図
した
PEMS
に適用できる。
14.2
~
14.13
の要求事項を適用する場合は,各
PESS
のソフ
トウェアの開発又は変更管理に対して,
JIS T 2304
の
4.3
,
箇条
5
,箇条
7
,箇条
8
及び箇条
9
の要求事項も適用する。
4.3
ソフトウェア安全クラス分類
JIS T 0601-1
の
PEMS
要求事項は,ソフトウェア安全
クラス
B
及び
C
にだけ適用される。この規格には,ソ
フトウェア安全クラス
A
の要求事項が幾つか含まれる。
JIS T 0601-1
への適合に必要なソフトウェア開発
プロ
セス
には,この規格の箇条
6
で要求される市販後の監視
及び保守は含まれない。
14.2
文書化
箇条
14
で要求する文書は,正式な文書管理
手順
に従って,
検討,承認,発行及び変更する。
5.1
ソフトウェア開発計画
ソフトウェア開発計画
アクティビティ
の特定の要求
事項に加え,
リスクマネジメント
ファイルの一部となる
文書を保持することも
JIS T 14971
によって要求され
る。さらに,品質システムが要求する文書は,
JIS Q
13485
[5]
によって,その管理が要求される。
14.3
リスクマネジメント計画
4.2.2
で要求する
リスクマネジメント
計画は,
PEMS
妥当
性確認
計画を参照することも含める(
14.11
参照)
。
特定の要求事項はない。
ソフトウェア妥当性確認計画は含まない。
PEMS
妥当
性確認計画は,
システム
レベルであり,このソフトウェ
ア規格の適用範囲外である。この規格では,
ハザード
か
ら,
リスクコントロール
手段のための特定のソフトウェ
ア及び
リスクコントロール
手段の
検証
までを追跡する
トレーサビリティ
が要求される(
7.3
参照)
。
14.4
PEMS
開発ライフサイクル
PEMS
開発ライフサイクル
を,文書化する。
5.1
ソフトウェア開発計画
5.1.1
ソフトウェア開発計画
ソフトウェア開発計画で扱う項目は,ソフトウェア開
発ライフサイクルの一部とみなす。
PEMS
開発ライフサイクル
には,一連のマイルストーンを
決定する。
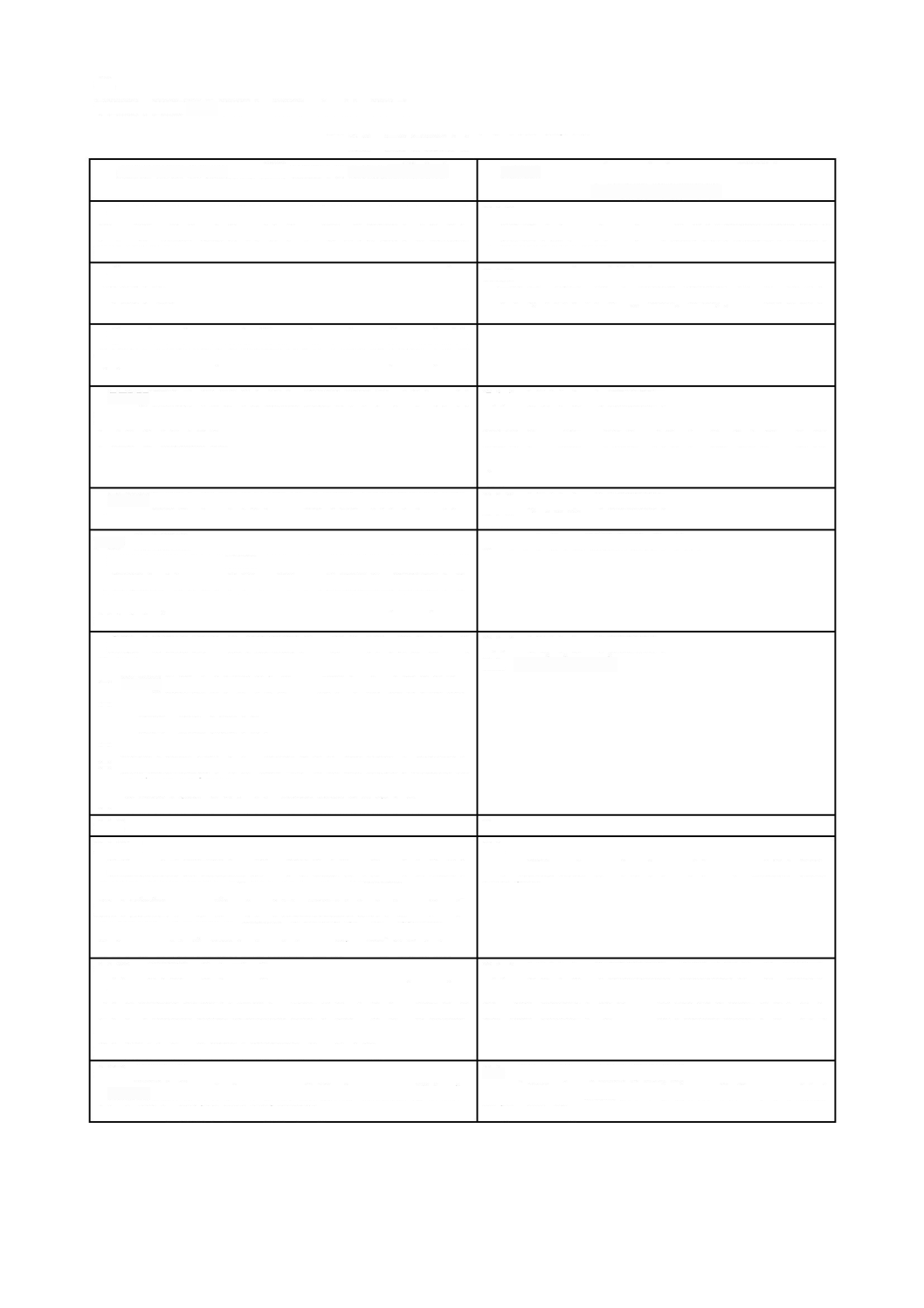
54
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.3
-
JIS T 0601-1
との関係(続き)
JIS T 0601-1
:2012
及び追補
1:2014
の
PEMS
要求事項
PEMS
のソフトウェアサブシステムに関係する,
この規格の要求事項
各マイルストーンにおいて,完了させることが必要なアク
ティビティ及びこれらのアクティビティに適用する
検証
方
法を決定する。
5.1.6
ソフトウェア検証計画
検証タスク
,マイルストーン及び合否判定基準を計画
しなければならない。
各アクティビティは,インプット及びアウトプットを含め
て決定する。
5.1.1
ソフトウェア開発計画
アクティビティ
は,この規格で定義している。作成す
べき文書は,
アクティビティ
ごとに定義している。
各マイルストーンには,そのマイルストーンまでに完了さ
せることが必要な
リスクマネジメント
アクティビティを特
定する。
PEMS
開発ライフサイクル
は,詳細なアクティビティ,日
程及びマイルストーンを含めた計画を作成して,特定の開発
に合わせて作成する。
5.1.1
ソフトウェア開発計画
この規格では,開発ライフサイクルの文書化は,開発
計画において行ってもよいとしている。したがって,開
発計画は,その開発に応じた開発ライフサイクルを含
む。
PEMS
開発ライフサイクル
には,文書化の要求事項を含め
る。
5.1.1
ソフトウェア開発計画
5.1.8
文書化計画
14.5
問題解決
該当する場合は,
PEMS
開発ライフサイクル
における全て
の段階及び全てのアクティビティの期間内及び期間外にお
ける問題解決のための文書化システムを開発し,かつ,その
保守をする。
9
ソフトウェア問題解決プロセス
製品の種類に応じて,問題解決システムでは,次を行って
もよい。
-
PEMS
開発ライフサイクル
の一部として,文書化する。
-
基礎安全
又は
基本性能
に影響する潜在的若しくは既知
の問題の報告を認める。
-
関連する
リスク
に対する各問題の評価を含める。
-
問題を解決済みとして処理するために満たす必要があ
る基準を特定する。
-
各問題を解決するために必要な活動を特定する。
5.1.1
ソフトウェア開発計画
9.1
問題報告の作成
14.6
リスクマネジメントプロセス
7
ソフトウェアリスクマネジメントプロセス
14.6.1
既知及び予測可能なハザードの特定
既知又は予測可能な
ハザード
を特定してリストを作成す
る場合,
製造業者
は,
IT
ネットワーク
への
PEMS
の組込み,
第三者製造のコンポーネント及び継承したサブシステムに
関係するものを含んだ
PEMS
のソフトウェア,並びにハー
ドウェアの側面に関連したこれらの
ハザード
を考慮する。
7.1
危険状態を引き起こすソフトウェアの分析
この規格では,
IT
ネットワークについては特に触れ
ていない。
14.6.2
リスクコントロール
各
リスクコントロール
手段を実行するために,適切に
検証
された手法及び
手順
を選択し,特定する。これらの手法及び
手順は,各
リスクコントロール
手段が,特定した
リスク
を十
分に低減するのを保証する適切なものとする。
5.1.4
ソフトウェア開発規格,方法及びツールの計画
この規格は,
リスクコントロール
手段ごとではなく開
発一般に使用されるツール及び方法を特定することを
要求している。
14.7
要求仕様
PEMS
及びそのサブシステム(例えば,
PESS
に対する)
については,要求仕様を文書化する。
5.2
ソフトウェア要求事項分析
この規格は,
PEMS
のソフトウェアサブシステムだけ
を扱っている。
55
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.3
-
JIS T 0601-1
との関係(続き)
JIS T 0601-1
:2012
及び追補
1:2014
の
PEMS
要求事項
PEMS
のソフトウェアサブシステムに関係する,
この規格の要求事項
システム又はサブシステムに対する要求仕様は,そのシス
テム又はサブシステムによって実施されるいかなる
基本性
能
及びいかなる
リスクコントロール
手段をも含める。
5.2.1
システム要求事項からのソフトウェア要求事項
の定義及び文書化
5.2.2
ソフトウェア要求事項の内容
5.2.3
リスクコントロール手段のソフトウェア要求事
項への包含
この規格は,重要な性能及び
リスクコントロール
手段
に関わる要求事項を他の要求事項と区別することは要
求していないが,全ての要求事項が一意に識別できるこ
とは要求している。
14.8
アーキテクチャ
アーキテクチャは,
PEMS
及びそのサブシステムのそれぞ
れについて,要求仕様を満たすように規定する。
5.3
ソフトウェアアーキテクチャの設計
該当する場合,アーキテクチャの仕様は,
リスク
を受容可
能なレベルに低減するため,次の一つ以上を使用する。
a)
高信頼性部品
b)
フェールセーフ機能
c)
冗長性
d)
多様性
e)
機能の分割
f)
防御設計
例えば,利用可能な出力を制限することによ
って,又はアクチュエータの動きを制限する手段の導入
によって,潜在的に危険な影響を抑制する。
5.3.5
リスクコントロールに必要な分離の特定
分割が唯一特定されている技術である(分割の完全性
をどのように保証するかについて説明するという要求
事項があることが,唯一の特定理由である。
)
。
アーキテクチャの仕様には,次の
g)
~
n)
を考慮する。
g)
PEMS
のサブシステム及びコンポーネントに対する
リ
スクコントロール
手段の配分
h)
コンポーネントの故障モード及びそれらへの影響
i)
共通原因による故障
j)
系統的な故障
k)
試験間隔及び診断範囲
l)
保守性
m)
合理的に予見できる誤使用に対する保護
n)
該当する場合は,
IT
ネットワーク
の仕様
この規格には含まれない。
14.9
設計及び実装
該当する場合,設計をサブシステムに分割し,それぞれは,
設計仕様及び試験仕様をもつ。
5.4
ソフトウェア詳細設計
5.4.2
ソフトウェアユニットごとの詳細設計の開発
この規格では,詳細設計に試験仕様書は要求していな
い。
設計環境に対する説明データは,文書化する。
5.4.2
ソフトウェアユニットごとの詳細設計の開発
14.10
検証
検証
は,
基礎安全
,
基本性能
又は
リスクコントロール
手段
を実施する全ての機能に対して要求する。
5.1.6
ソフトウェア検証計画
アクティビティ
ごとに
検証
を要求する。
検証
計画は,どのようにこれらの機能を
検証
するかを示す
ために作成する。計画には,次の事項を含める。
-
各機能に対してどのマイルストーンで
検証
の実施が必
要か。
-
検証
方針,アクティビティ,技法,並びに
検証
を実行す
る要員の適切な独立性のレベルの選択及び文書化
-
検証
手段の選択及び活用
-
検証
に対する適用範囲の基準
5.1.6
ソフトウェア検証計画
要員の独立性は,この規格に含まれていない。
JIS Q
13485
で網羅されているとみなしている。
56
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.3
-
JIS T 0601-1
との関係(続き)
JIS T 0601-1
:2012
及び追補
1:2014
の
PEMS
要求事項
PEMS
のソフトウェアサブシステムに関係する,
この規格の要求事項
検証
は,
検証
計画に従って実施する。
検証
活動の結果は,
文書化する。
検証
の要求事項は,大部分の
アクティビティ
に入って
いる。
14.11
PEMS
妥当性確認
PEMS
妥当性確認
計画は,
基礎安全
及び
基本性能
の妥当性
確認を含む。
この規格は,ソフトウェア妥当性確認を扱っていな
い。
PEMS
妥当性確認は,
システム
レベルの
アクティビ
ティ
であり,この規格の適用範囲外である。
PEMS
妥当性確認
で用いた方法は,文書化する。
この規格は,ソフトウェア妥当性確認を扱っていな
い。
PEMS
妥当性確認は,
システム
レベルの
アクティビ
ティ
であり,この規格の適用範囲外である。
PEMS
妥当性確認
は,
PEMS
妥当性確認
計画に従って実施
する。
PEMS
妥当性確認
アクティビティの結果は,文書化す
る。
この規格は,ソフトウェア妥当性確認を扱っていな
い。
PEMS
妥当性確認は,
システム
レベルの
アクティビ
ティ
であり,この規格の適用範囲外である。
PEMS
妥当性確認
に対して全体的な責任をもつ者は,設計
チームから独立したものとする。
製造業者
は,独立性のレベ
ルに対する根拠を文書化する。
この規格は,ソフトウェア妥当性確認を扱っていな
い。
PEMS
妥当性確認は,
システム
レベルの
アクティビ
ティ
であり,この規格の適用範囲外である。
設計チームのいかなる要員も,自らの設計した結果に対し
て
PEMS
妥当性確認
の最終判定責任を負ってはならない。
この規格は,ソフトウェア妥当性確認を扱っていな
い。
PEMS
妥当性確認は,
システム
レベルの
アクティビ
ティ
であり,この規格の適用範囲外である。
PEMS
妥当性確認
チームの要員と設計チームの要員との
全ての職務上の関係を,
リスクマネジメントファイル
に文書
化する。
この規格は,ソフトウェア妥当性確認を扱っていな
い。
PEMS
妥当性確認は,
システム
レベルの
アクティビ
ティ
であり,この規格の適用範囲外である。
14.12
変更管理
初期段階の設計に起因する一部又は全ての設計変更の場
合は,新規設計と同じものとしてこの箇条の全てを適用する
か,又は変更前の設計文書に記載してあった変更の前後で共
通な妥当性については,文書化した修正又は変更
手順
に従っ
て評価する。
6
ソフトウェア保守プロセス
この規格では,ソフトウェア保守を計画し,修正の実
装にはソフトウェア開発
プロセス
又は確立されたソフ
トウェア保守
プロセス
を使うのが望ましいという方針
をとっている。
ソフトウェアを変更管理する場合には,
JIS T 2304
:2012
の
4.3
,箇条
5
,箇条
7
,箇条
8
及び箇条
9
の要求事項も適用
する。

57
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.3
-
JIS T 0601-1
との関係(続き)
JIS T 0601-1
:2012
及び追補
1:2014
の
PEMS
要求事項
PEMS
のソフトウェアサブシステムに関係する,
この規格の要求事項
14.13
IT
ネットワークに組み込むことを意図する
PEMS
PEMS
を
PEMS
製造業者
による
妥当性確認
がされていな
い
IT
ネットワーク
に組み込むことを意図した場合には,
製
造業者
は,そのような接続を実行するための,次の事項を含
む取扱い指示を提供する。
a)
IT
ネットワーク
への
PEMS
接続の目的。
b)
PEMS
を組み込む
IT
ネットワーク
に必要な特性。
c)
PEMS
を組み込む
IT
ネットワーク
に必要な構成。
d)
セキュリティ仕様を含む,
PEMS
のネットワーク接続の
技術仕様。
e)
PEMS
,
IT
ネットワーク
,及び
IT
ネットワーク
に接続
されている他の装置との間の意図する情報の流れ,及び
IT
ネットワーク
を通じる意図する経路。
注記
1
これには,
基礎安全
及び
基本性能
に関連する
有効性並びにデータの側面及びシステムセキ
ュリティの側面を含めてもよい。
H.6
及び
IEC 80001-1
:2010
も参照する。
f)
IT
ネットワーク
への
PEMS
接続の目的に合わせるため
に必要な特性を提供する
IT
ネットワーク
が,故障した
場合に生じる
危険状態
のリスト。
注記
2
データを転送する目的で
PEMS
を他の装置に
接続すると,二つのノードの
IT
ネットワー
ク
を形成する。例えば,
PEMS
をプリンタに
接続すると,
IT
ネットワーク
を形成する。
製
造業者
が,そのプリンタとともに
PEMS
の妥
当性を確認していれば,
製造業者
が,その形
成されたネットワーク全体の妥当性を確認し
ているとみなしてもよい
a)
。
製造業者
は,
附属文書
の中で,
責任部門
に次を指示する。
-
その他の機器を含む
IT
ネットワーク
への
PEMS
の接続
が,
患者
,
操作者
又は第三者に対して事前に特定してい
なかった受容できない
リスク
を生じる可能性がある。
-
責任部門
は,これらの
リスク
を特定,分析,評価及び管
理をすることが望ましい。
注記
3
IEC 80001-1
:2010
は,これらの
リスク
を扱う
ためのガイダンスを,
責任部門
に提供してい
る。
-
IT
ネットワーク
を後から変更した場合には,新たな
リ
スク
を招き,追加の分析が必要となる。
-
IT
ネットワーク
の変更には,次を含める。
-
IT
ネットワーク
の構成における変更。
-
IT
ネットワーク
への追加アイテムの接続。
-
IT
ネットワーク
からのアイテムの取外し。
-
IT
ネットワーク
に接続された機器のアップデート。
-
IT
ネットワーク
に接続された機器のアップグレード。
IT
ネットワークの要求事項は,この規格に含まれて
いない。
注
a)
対応国際規格の記載漏れを補った。
58
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
C.4.7 IEC
60601-1-4
の要求事項との関係
IEC 60601-1-4
は,廃止となっている。
C.5 JIS
C
1010-1
との関係
JIS C 1010-1
[2]
の適用範囲は,測定,制御及び研究室用電気機器である。研究室用機器は,医療環境に
おいて又は体外診断(
IVD: In Vitro Diagnostic
)機器として,一部が使用されているにとどまる。
法規制又は引用規格があるため,
IVD
機器は,
医療機器
に分類するが,
JIS T 0601-1
[6]
の適用範囲外で
ある。これは,
IVD
機器が厳密には直接患者に接触する
医療機器
ではないという理由のほか,そのような
製品が,各種の研究室での多種多様な用途のために製造されているという理由もある。
IVD
機器又は
IVD
機器用附属品としての使用は,まれである。
研究室用機器を
IVD
機器として使用する場合,医療基準に従って測定結果を
評価
する。
リスクマネジメ
ント
については,
JIS T 14971
の適用が要求される。ソフトウェアに起因する医療データ(測定結果)の不
要な変更を引き起こす故障など,
危険状態
の原因になる可能性のあるソフトウェアがそのような製品に含
まれている場合は,この規格を考慮する。
JIS C 1010-1
:2014
の箇条
17
には
リスクマネジメント
の一般要求事項があるが,これは
JIS T 14971
の詳
細な
リスクマネジメント
要求事項を簡素化したものである。この規格の
リスクマネジメント
要求基準は,
JIS T 14971
の
リスクマネジメント
要求事項に基づいているため,
JIS C 1010-1
の箇条
17
を適用しただけ
では,この規格の
リスクマネジメント
要求基準に適合しない。このことを念頭において,この規格では,
IVD
医療機器
にソフトウェア関連の
リスク
がある場合には,
JIS C 1010-1
の箇条
17
だけではなく
JIS T
14971
に従って
リスクマネジメントプロセス
を実施することを期待している。
JIS C 1010-1
の箇条
17
への
注記で詳述するように,
JIS C 1010-1
の箇条
17
への適合性も達成される。
注記
リスク
アセスメント手順の一つは,
JIS C 1010-1
の
附属書
J
に概説している。他の
リスク
アセ
スメント手順は,
JIS T 14971
,
SEMI S10-1296
,
JIS C 0508
規格群,
ISO 14121-1
及び
ANSI
B11.TR3
にある。類似のステップを踏む他の確立した手順も用いることができる。
図
C.3
のフローチャートは,
JIS C 1010-1
の箇条
17
及びこの規格の適用について示している。
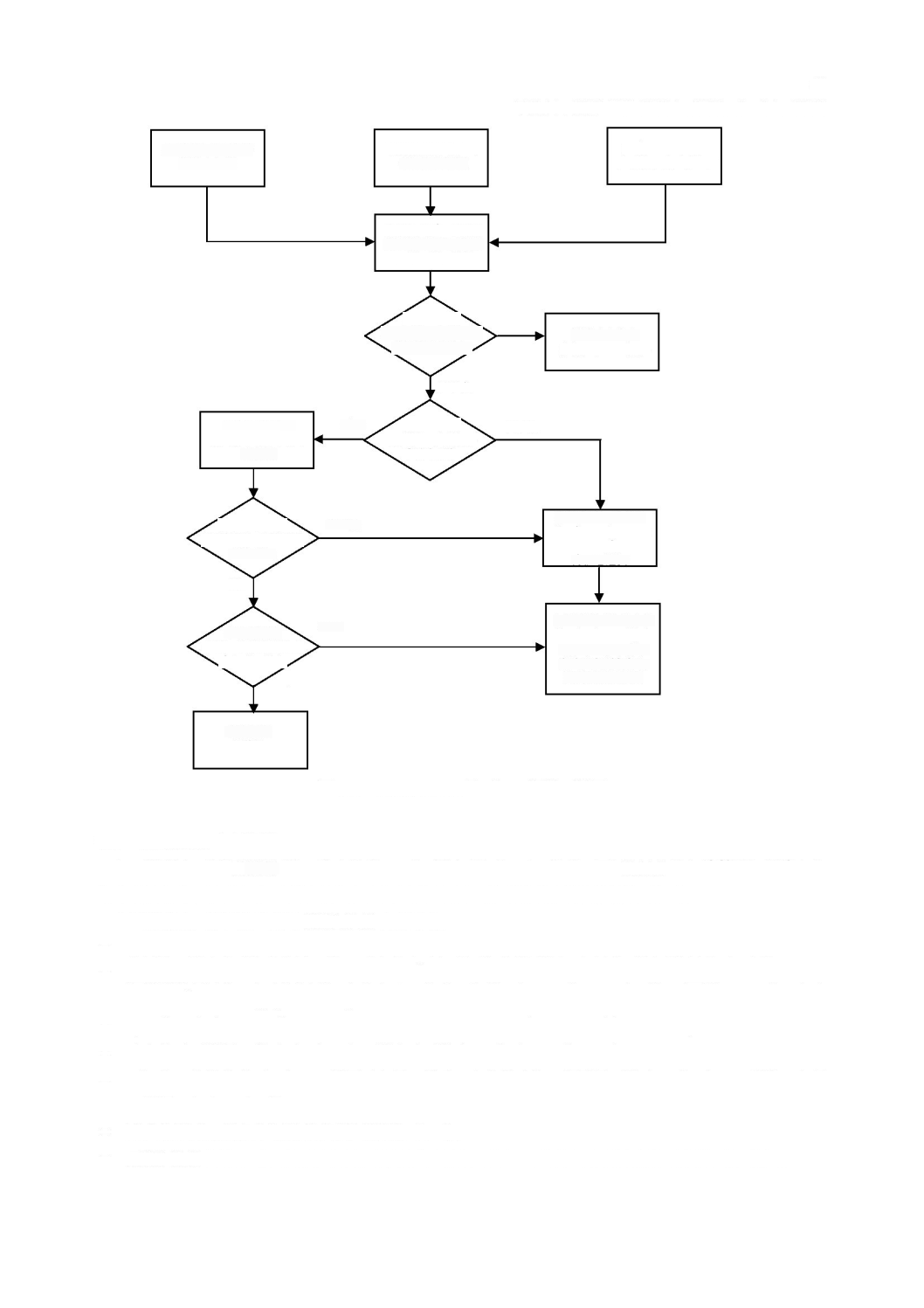
59
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
図
C.3
-
JIS C 1010-1
及びこの規格の適用
C.6 JIS
X
0160
との関係
この規格は,
JIS X 0160
[7]
の様式及びコンセプトに基づいているが,
JIS X 0160
は
医療機器
に限定した
ものではなく,ソフトウェアライフサイクル
プロセス
一般の要求事項を定義している。
この規格は,特に次の点で
JIS X 0160
と異なる。
-
システム
要求事項,
システムアーキテクチャ
,妥当性確認などの
システム
面を除外している。
-
医療機器
を対象とする別文書に
アクティビティ
が記載されているとして,それと重複しているとみな
される
プロセス
を省略している。
-
リスクマネジメントプロセス
及びソフトウェアリリース
プロセス
を追加している。
-
文書化及び
検証
の補助
プロセス
を,開発及び保守の
プロセス
に組み入れている。
-
プロセス
実装と各
プロセス
の計画
アクティビティ
とを統合し,開発及び保守の
プロセス
の単独
アクテ
ィビティ
になっている。
-
安全
性要求の観点から要求事項を分類している。
-
JIS X 0160
と違って,主
プロセス
,補助
プロセス
,グループ
プロセス
を明確に分類していない。
意図する目的及び
用途を定義
考えられる
ハザード
の原因の特定
医療データ取扱い
に関連する
ハザード
既知の
ハザード
及び
合理的に予知可能な
ハザード
の特定
関連する安全
規格に従って検証
ハザード
は
関連する安全規格の
適用を受けるか
医療目的で使用する
前に誤データが検出
されるようにする
ために必要となる
追加の要求事項
JIS T 14971
を
リスクマネジメント
に使用
この規格
を使用
機器は医療
関連データを提供
するものか
ソフトウェア
は医療データに影響
があるか
手順の使用
がデータ検証に要求
されているか
はい
はい
はい
いいえ
安全規格に基づき
リスクコントロール
のための適切な
方法を選択
はい
いいえ
いいえ
いいえ
60
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
これらの変更点は,当該規格を次のような方法で
医療機器
分野の要求に合わせようとする意図に基づい
ている。
-
安全
面及び
医療機器リスクマネジメント
規格
JIS T 14971
に重点を置く。
-
規制環境における有用かつ適切な
プロセス
を選択する。
-
ソフトウェア開発が,
(
JIS X 0160
の
プロセス
及び要求事項の一部を扱う)品質システムに組み込まれ
ることを考慮する。
-
曖昧さを抑えて,使いやすくする。
この規格は,
JIS X 0160
と矛盾するものではない。
JIS X 0160
は,この規格の要求事項を盛り込んだ,
うまく構造化された
ソフトウェア開発ライフサイクルモデル
を設定するのに有用である。
表
C.5
は,
ISO/IEC JTC1/SC7
が作成したもので,この規格と
JIS X 0160
との関係を示している。
表
C.5
-
JIS X 0160
との関係
この規格の
プロセス
JIS X 0160
の
プロセス
アクティビティ
タスク
プロセス
アクティビティ・タスク
5
ソフトウェア開発プロセス
5.1
ソフ トウ ェア 開
発計画
5.1.1
ソフトウェア開発計
画
7.1.1
ソフトウェア実装プ
ロセス
7.1.1.3.1
ソ フ ト ウ ェ ア 実 装
戦略
7.1.1.3.1.1
7.1.1.3.1.3
7.1.1.3.1.4
6.3.1.3.2
プロジェクト計画
6.3.1.3.2.1
5.1.2
ソフトウェア開発計
画の継続更新
6.3.2
プロジェクトアセス
メント及び制御プロセス
6.3.2.3.2
プ ロ ジ ェ ク ト の 制
御
6.3.2.3.2.1
5.1.3
ソフトウェア開発計
画におけるシステム設計及
びシステム開発の引用
6.4.3
システム方式設計プ
ロセス
6.4.5
システム結合プロセ
ス
7.2.5
ソフトウェア妥当性
確認プロセス
6.4.3.3.1
システム方式の確立
6.4.3.3.1.1
6.4.5.3.1
結合
6.4.5.3.1.1
7.2.5.3.1
プロセスの実施
7.2.5.3.1.4
5.1.4
ソフトウェア開発規
格,方法及びツールの計画
7.1.1
ソフトウェア実装プ
ロセス
7.1.1.3.1
ソ フ ト ウ ェ ア 実 装
戦略
7.1.1.3.1.3
5.1.5
ソフトウェア結合及
び結合試験計画
7.1.6
ソフトウェア結合プ
ロセス
7.1.6.3.1
ソフトウェア結合
7.1.6.3.1.1
5.1.6
ソフトウェア検証計
画
7.2.4
ソフトウェア検証プ
ロセス
7.1.5
ソフトウェア構築プ
ロセス
7.1.6
ソフトウェア結合プ
ロセス
7.1.7
ソフトウェア適格性
確認テストプロセス
7.2.4.3.1
プロセスの実施
7.2.4.3.1.4
7.2.4.3.1.5
7.1.5.3.1
ソフトウェア構築
7.1.5.3.1.5
7.1.6.3.1
ソフトウェア結合
7.1.6.3.1.5
7.1.7.3.1
ソ フ ト ウ ェ ア 適 格
性確認テスト
7.1.7.3.1.3
5.1.7
ソフトウェアリスク
マネジメント計画
6.3.4
リスク管理プロセス
61
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.5
-
JIS X 0160
との関係(続き)
この規格の
プロセス
JIS X 0160
の
プロセス
アクティビティ
タスク
プロセス
アクティビティ・タスク
5.1
ソフ トウ ェア 開
発計画
5.1.8
文書化計画
7.2.1
ソフトウェア文書化
管理プロセス
7.2.1.3.1
プロセスの実施
7.2.1.3.1.1
5.1.9
ソフトウェア構成管
理計画
7.2.2
ソフトウェア構成管
理プロセス
7.2.8
ソフトウェア問題解
決プロセス
7.2.2.3.1
プロセスの実施
7.2.2.3.1.1
7.2.8.3.1
プロセス開始の準備
7.2.8.3.1.1
5.1.10
管理が必要な支援ア
イテム
6.2.2
インフラストラクチ
ャ管理プロセス
6.2.2.3.2
イ ン フ ラ ス ト ラ ク
チャの確立
6.2.2.3.2.1
6.2.2.3.3
イ ン フ ラ ス ト ラ ク
チャの保守
6.2.2.3.3.1
5.1.11
検証前のソフトウェ
ア構成アイテムのコントロ
ール
7.2.2
ソフトウェア構成管
理プロセス
7.2.2.3.2
構成識別
7.2.2.3.2.1
5.2
ソフ トウ ェア 要
求事項分析
5.2.1
システム要求事項か
らのソフトウェア要求事項
の定義及び文書化
6.4.3
システム方式設計プ
ロセス
6.4.3.3.1
シ ス テ ム 方 式 の 確
立
6.4.3.3.1.1
5.2.2
ソフトウェア要求事
項の内容
7.1.2
ソフトウェア要求事
項分析プロセス
7.1.2.3.1
ソ フ ト ウ ェ ア 要 求
事項分析
7.1.2.3.1.1
5.2.3
リスクコントロール
手段のソフトウェア要求事
項への包含
5.2.4
医療機器のリスク分
析の再評価
なし
なし
5.2.5
要求事項の更新
7.1.2
ソフトウェア要求事
項分析プロセス
7.1.2.3.1
ソ フ ト ウ ェ ア 要 求
事項分析
7.1.2.3.1.1
の
a)
及び
b)
5.2.6
ソフトウェア要求事
項の検証
7.2.4
ソフトウェア検証プ
ロセス
7.2.4.3.2
検証
7.2.4.3.2.1
5.3
ソフ トウ ェア ア
ーキテクチャの設計
5.3.1
ソフトウェア要求事
項のアーキテクチャへの変
換
7.1.3
ソフトウェア方式設
計プロセス
7.1.3.3.1
ソ フ ト ウ ェ ア 方 式
設計
7.1.3.3.1.1
5.3.2
ソフトウェアアイテ
ムのインタフェース用アー
キテクチャの開発
7.1.3.3.1
ソ フ ト ウ ェ ア 方 式
設計
7.1.3.3.1.2
5.3.3
SOUP
アイテムの機能
及び性能要求事項の指定
なし
なし
5.3.4
SOUP
アイテムが要求
するシステムハードウェア
及びシステムソフトウェア
の指定
なし
なし
5.3.5
リスクコントロール
に必要な分離の特定
なし
なし
5.3.6
ソフトウェアアーキ
テクチャの検証
7.1.3
ソフトウェア方式設
計プロセス
7.1.3.3.1
ソ フ ト ウ ェ ア 方 式
設計
7.1.3.3.1.6
62
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.5
-
JIS X 0160
との関係(続き)
この規格の
プロセス
JIS X 0160
の
プロセス
アクティビティ
タスク
プロセス
アクティビティ・タスク
5.4
ソフ トウ ェア 詳
細設計
5.4.1
ソフトウェアのソフ
トウェアユニットへの分解
7.1.4
ソフトウェア詳細設
計プロセス
7.1.4.3.1
ソ フ ト ウ ェ ア 詳 細
設計
7.1.4.3.1.1
5.4.2
ソフトウェアユニッ
トごとの詳細設計の開発
5.4.3
インタフェース用詳
細設計の開発
7.1.4.3.1
ソ フ ト ウ ェ ア 詳 細
設計
7.1.4.3.1.2
5.4.4
詳細設計の検証
7.1.4
ソフトウェア詳細設
計プロセス
7.1.4.3.1
ソ フ ト ウ ェ ア 詳 細
設計
7.1.4.3.1.7
5.5
ソフ トウ ェア ユ
ニットの実装
5.5.1
各ソフトウェアユニ
ットの実装
7.1.5
ソフトウェア構築プ
ロセス
7.1.5.3.1
ソフトウェア構築
7.1.5.3.1.1
5.5.2
ソフトウェアユニッ
ト検証プロセスの確立
7.1.4
ソフトウェア詳細設
計プロセス
7.1.5
ソフトウェア構築プ
ロセス
7.1.4.3.1
ソ フ ト ウ ェ ア 詳 細
設計
7.1.4.3.1.5
7.1.5.3.1
ソフトウェア構築
7.1.5.3.1.5
5.5.3
ソフトウェアユニッ
トの合否判定基準
7.1.5
ソフトウェア構築プ
ロセス
7.1.5.3.1
ソフトウェア構築
7.1.5.3.1.5
5.5.4
追加のソフトウェア
ユニット合否判定基準
7.1.5
ソフトウェア構築プ
ロセス
7.2.4
ソフトウェア検証プ
ロセス
7.1.5.3.1
ソフトウェア構築
7.1.5.3.1.2
5.5.5
ソフトウェアユニッ
トの検証
7.1.5
ソフトウェア構築プ
ロセス
7.1.5.3.1
ソフトウェア構築
7.1.5.3.1.2
5.6
ソフ トウ ェア 結
合及び結合試験
5.6.1
ソフトウェアユニッ
トの結合
7.1.6
ソフトウェア結合プ
ロセス
7.1.6.3.1
ソフトウェア結合
7.1.6.3.1.2
5.6.2
ソフトウェア結合の
検証
7.1.6
ソフトウェア結合プ
ロセス
6.4.5
システム結合プロセ
ス
7.1.6.3.1
ソフトウェア結合
7.1.6.3.1.2
6.4.5.3.1
結合
5.6.3
ソフトウェア結合試
験
7.1.7
ソフトウェア適格性
確認テストプロセス
7.1.7.3.1
ソ フ ト ウ ェ ア 適 格
性確認テスト
7.1.7.3.1.1
5.6.4
ソフトウェア結合試
験の内容
7.1.7
ソフトウェア適格性
確認テストプロセス
7.1.7.3.1
ソ フ ト ウ ェ ア 適 格
性確認テスト
7.1.7.3.1.3
5.6.5
ソフトウェア結合試
験手順の評価
なし
なし
5.6.6
回帰テストの実施
7.1.6
ソフトウェア結合プ
ロセス
7.1.6.3.1
ソフトウェア結合
7.1.6.3.1.2
5.6.7
結合試験記録の内容
7.1.6
ソフトウェア結合プ
ロセス
7.1.6.3.1
ソフトウェア結合
7.1.6.3.1.2
5.6.8
ソフトウェア問題解
決プロセスの使用
7.2.4
ソフトウェア検証プ
ロセス
7.2.4.3.1
プ ロ セ ス の 実 施
7.2.4.3.1.6
63
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.5
-
JIS X 0160
との関係(続き)
この規格の
プロセス
JIS X 0160
の
プロセス
アクティビティ
タスク
プロセス
アクティビティ・タスク
5.7
ソフ トウ ェア シ
ステム試験
5.7.1
ソフトウェア要求事
項についての試験の確立
7.1.6
ソフトウェア結合プ
ロセス
7.1.7
ソフトウェア適格性
確認テストプロセス
7.1.6.3.1
ソフトウェア結合
7.1.6.3.1.4
7.1.7.3.1
ソ フ ト ウ ェ ア 適 格
性確認テスト
7.1.7.3.1.1
5.7.2
ソフトウェア問題解
決プロセスの使用
7.2.4
ソフトウェア検証プ
ロセス
7.2.4.3.1
プロセスの実施
7.2.4.3.1.6
5.7.3
変更後の再試験
7.2.8
ソフトウェア問題解
決プロセス
7.2.8.3.1
プロセス開始の準備
7.2.8.3.1.1
5.7.4
ソフトウェアシステ
ム試験の評価
7.1.7
ソフトウェア適格性
確認テストプロセス
7.1.7.3.1
ソ フ ト ウ ェ ア 適 格
性確認テスト
7.1.7.3.1.3
5.7.5
ソフトウェアシステ
ム試験記録の内容
7.1.7
ソフトウェア適格性
確認テストプロセス
7.1.7.3.1
ソ フ ト ウ ェ ア 適 格
性確認テスト
7.1.7.3.1.1
5.8
シス テム レベ ル
で 使 用 す る た め の ソ
フトウェアリリース
5.8.1
ソフトウェア検証の
完了確認
6.4.9
ソフトウェア運用プ
ロセス
7.2.2
ソフトウェア構成管
理プロセス
6.4.9.3.2
運 用 の 開 始 及 び 終
了
6.4.9.3.2.1
6.4.9.3.2.2
7.2.2.3.6
リ リ ー ス 管 理 及 び
納入
7.2.2.3.6.1
5.8.2
既知の残留異常の文
書化
7.2.2
ソフトウェア構成管
理プロセス
7.1.7
ソフトウェア適格性
確認テストプロセス
7.2.2.3.5
構成評価
7.2.2.3.5.1
7.1.7.3.1
ソ フ ト ウ ェ ア 適 格
性確認テスト
7.1.7.3.1.3
5.8.3
既知の残留異常の評
価
5.8.4
リリースするバージ
ョンの文書化
7.2.2
ソフトウェア構成管
理プロセス
7.2.2.3.6
リ リ ー ス 管 理 及 び
納入
7.2.2.3.6.1
5.8.5
リリースするソフト
ウェアの作成方法の文書化
5.8.6
アクティビティ及び
タスクの完了確認
5.8.7
ソフトウェアのアー
カイブ
5.8.8
ソフトウェアリリー
スの信頼性の確保
6
ソフトウェア保守プロセス
6.4.10
ソフトウェア保守プロセス
6.1
ソフ トウ ェア 保
守計画の確立
6.4.10
ソフトウェア保守プ
ロセス
なし
6.2
問題 及び 修正 の
分析
6.2.1
フィードバックの文
書化及び評価
なし
なし
6.2.1.1
フィードバックの監
視
6.4.10
ソフトウェア保守プ
ロセス
なし
6.2.1.2
フィードバックの文
書化及び評価
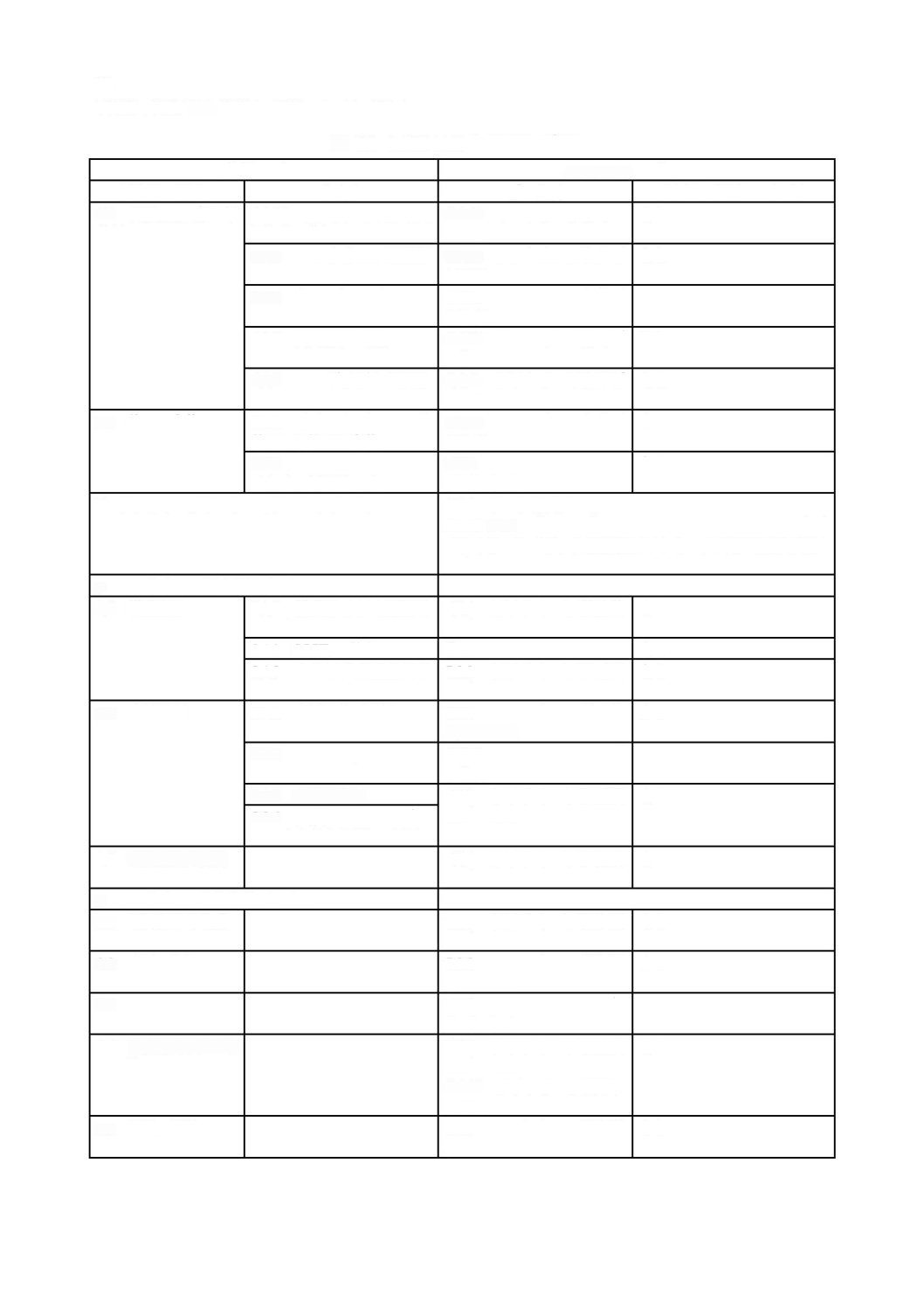
64
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.5
-
JIS X 0160
との関係(続き)
この規格の
プロセス
JIS X 0160
の
プロセス
アクティビティ
タスク
プロセス
アクティビティ・タスク
6.2
問題 及び 修正 の
分析
6.2.1.3
安全性に影響する問
題報告の評価
6.4.10
ソフトウェア保守プ
ロセス
なし
6.2.2
ソフトウェア問題解
決プロセスの使用
6.4.10
ソフトウェア保守プ
ロセス
なし
6.2.3
変更要求の分析
6.4.10
ソフトウェア保守プ
ロセス
なし
6.2.4
変更要求の承認
6.4.10
ソフトウェア保守プ
ロセス
なし
6.2.5
ユーザ及び規制当局
への通知
6.4.10
ソフトウェア保守プ
ロセス
なし
6.3
修正の実装
6.3.1
確立したプロセスを
使用した修正の実装
6.4.10
ソフトウェア保守プ
ロセス
なし
6.3.2
修正ソフトウェアシ
ステムの再リリース
7.2.2
ソフトウェア構成管
理プロセス
なし
7
ソフトウェアリスクマネジメントプロセス
6.3.4
リスク管理プロセス
JIS X 0162
に基づく。若干の共通性はあるが,
リスクマネジ
メント
については,
医療機器ソフトウェア
特有の要求事項に
対応したものではない。
8
ソフトウェア構成管理プロセス
8.1
構成識別
8.1.1
構成アイテム識別手
段の確立
7.2.2
ソフトウェア構成管
理プロセス
なし
8.1.2
SOUP
の特定
なし
なし
8.1.3
システム構成文書の
特定
7.2.2
ソフトウェア構成管
理プロセス
なし
8.2
変更管理
8.2.1
変更要求の承認
7.2.2
ソフトウェア構成管
理プロセス
なし
8.2.2
変更の実装
6.4.10
ソフトウェア保守プ
ロセス
なし
8.2.3
変更の検証
7.2.2
ソフトウェア構成管
理プロセス
なし
8.2.4
変更のトレーサビリ
ティを実現する手段の提示
8.3
構成状態の記録
7.2.2
ソフトウェア構成管
理プロセス
なし
9
ソフトウェア問題解決プロセス
9.1
問題報告の作成
7.2.8
ソフトウェア問題解
決プロセス
なし
9.2
問題の調査
7.2.8
ソフトウェア問題解
決プロセス
なし
9.3
関係者への通知
7.2.8
ソフトウェア問題解
決プロセス
なし
9.4
変更 管理 プロ セ
スの使用
7.2.2
ソフトウェア構成管
理プロセス
6.4.10
ソフトウェア保守プ
ロセス
なし
9.5
記録の保持
7.2.8
ソフトウェア問題解
決プロセス
なし
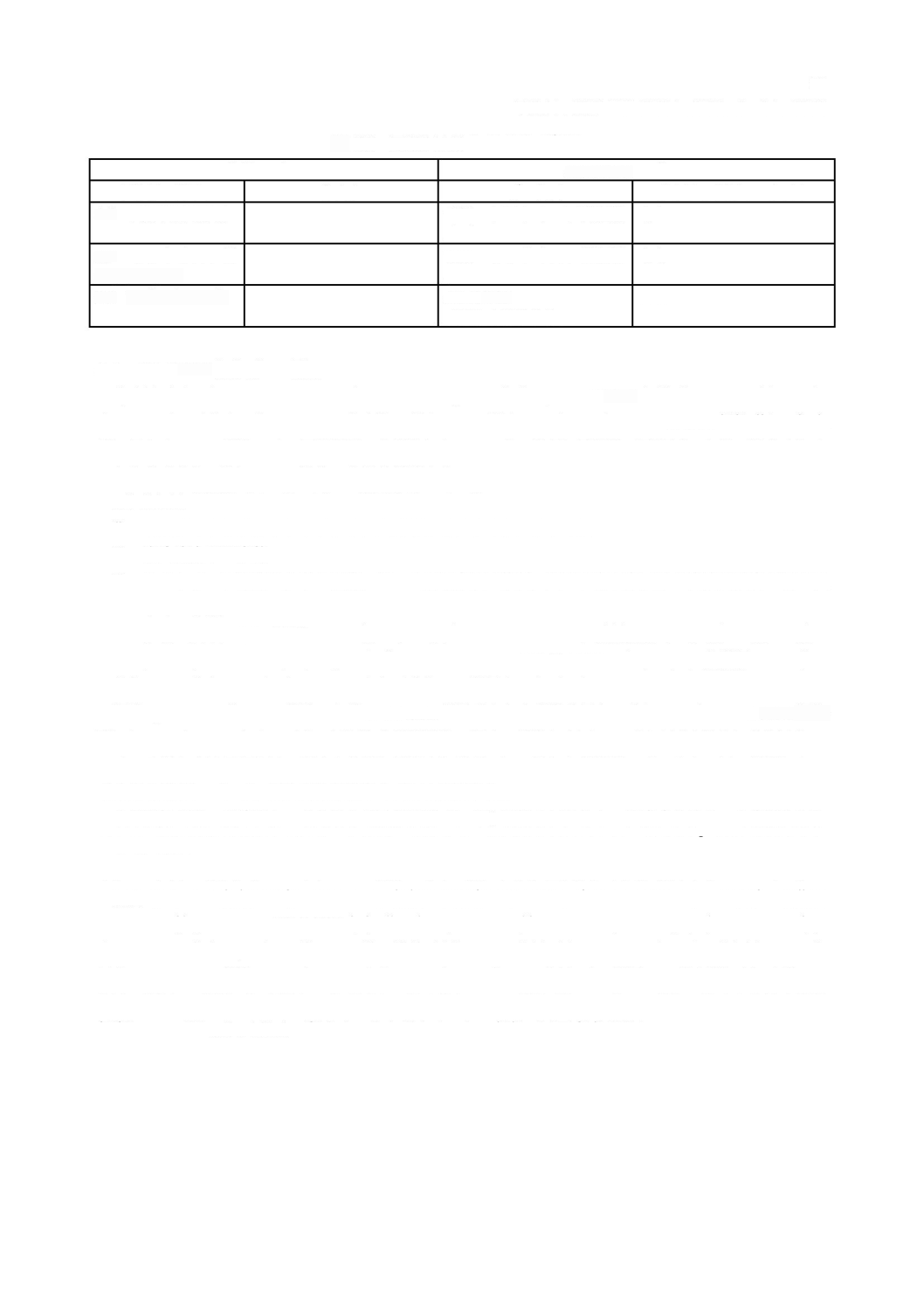
65
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
C.5
-
JIS X 0160
との関係(続き)
この規格の
プロセス
JIS X 0160
の
プロセス
アクティビティ
タスク
プロセス
アクティビティ・タスク
9.6
問題の傾向分析
7.2.8
ソフトウェア問題解
決プロセス
なし
9.7
ソフ トウ ェア 問
題解決の検証
7.2.8
ソフトウェア問題解
決プロセス
なし
9.8
試験文書の内容
JIS X 0160
の全ての試験は,
文書化を要求する。
なし
C.7 JIS
C
0508
規格群との関係
安全
性が重要なソフトウェアの設計に関わるため,この規格が,
JIS C 0508
規格群の原則に従うのが望
ましいか否かが論点になっている。
安全
性に対するこの規格のアプローチは,
JIS C 0508
規格群とは根本
的に異なる。この規格では,
医療機器
の有効性によって,その使用に関連する
残留リスク
が正当化される
ことを考慮する。次に,この規格の立場を説明する。
JIS C 0508
規格群では,次の三つの課題を扱っている。
1)
リスクマネジメント
ライフサイクル及びライフサイクル
プロセス
2)
安全度水準の定義
3)
ソフトウェア開発のための技術,ツール及び方法の推奨,並びに各種
タスク
の実施責任者の独立性
水準の推奨
この規格は,課題
1)
を
JIS T 14971
(
リスクマネジメント
についての
医療機器
分野の規格)の引用で補っ
ている。この引用によって,
リスクマネジメント
についての
JIS T 14971
の趣旨が,
医療機器ソフトウェ
ア
対象のソフトウェア
プロセス
の不可欠な要素として採用された形になっている。
課題
2)
については,この規格の方が
JIS C 0508
規格群よりも単純な取組みをしている。
JIS C 0508
規格
群においては,ソフトウェアは,目標とする信頼性の観点で定義した四つの“安全度水準”に分類されて
いる。目標とする信頼性は,
リスク分析
に特定されるが,この
リスク分析
は,ソフトウェアの故障によっ
て生じる
危害
について,重大性及び確率の両方を推定する。
この規格では,故障によって生じる
リスク
に基づいて
3
種類のソフトウェア安全クラスへの分類を行う
ことで,課題
2)
を簡略化している。分類後,ソフトウェア安全クラスごとに,異なる
プロセス
が要求され
る。この要求の意図は,ソフトウェアの故障の確率(及び/又は重大性)を更に低くすることである。
課題
3)
は,この規格では扱っていない。規格の利用者に対しては,望ましいソフトウェア技法,技術及
びツールの情報源として
JIS C 0508
規格群を使用することを奨励している。ただし,現存するものと今後
出てくるものを含め,別の手法でも同等の結果が得られる可能性があることも承知の上で使用することを
勧めている。この規格では,あるソフトウェア
アクティビティ
(例えば,
検証
)の責任者とそれ以外の
ア
クティビティ
(例えば,設計)の責任者との独立性について推奨はしていない。特に,独立した安全性評
価者については
JIS T 14971
で扱うものとなるため,この規格では要求事項がない。
66
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
附属書
D
(参考)
実装
D.1
序文
この
附属書
は,この規格を
製造業者
の
プロセス
にどのように実装できるかについて概説している。また,
JIS Q 13485
[5]
のような他の規格が,この規格と同等の適切な
プロセス
を要求している点も,この附属書
では考慮している。
D.2
品質マネジメントシステム
この規格に関連する
医療機器ソフトウェア
も含めて,
医療機器
の
製造業者
は,
4.1
で品質マネジメント
システム(
QMS
)の確立が必要である。この規格は,
QMS
の認証が必ず必要であるということを要求す
るものではない。
D.3
品質管理プロセスの評価
確立し,文書化した
QMS
の
プロセス
が,ソフトウェアライフサイクルの
プロセス
に既にどの程度対応
できているかについて,
製造業者
の責任の下に監査,調査又は分析することによって,
評価
するよう推奨
している。不足部分を確認した場合は,品質管理
プロセス
を拡大して対応する,又は個別に説明して対応
できる。
製造業者
が,ソフトウェアの開発,
検証
及び妥当性確認の統制をはかる
プロセス
記述を既にもち
合わせている場合,どの程度この規格と一致するかを
評価
することが望ましい。
D.4
この規格の要求事項と製造業者の品質管理プロセスとの統合
QMS
に,既に導入している
プロセス
を適応若しくは拡大させるか,又は新しい
プロセス
を統合させるこ
とで,この規格を実施できる。それをどのように行うべきかについては,この規格では規定していない。
したがって,
製造業者
は,適切な方法で自由に行うことができる。
製造業者
は,文書化した独自の
QMS
をもたない相手先商標製品製造業者(
OEM
)又は外部委託契約者
にその
医療機器ソフトウェア
の開発を任せた場合,規格で説明している
プロセス
を適切に実行に移したこ
とを確実にする責任がある。
D.5
認証
QMS
がない小規模製造業者のためのチェックリスト
製造業者
が,ソフトウェアのソフトウェア安全クラス(
A
,
B
又は
C
)を分類する場合,最も
リスク
の
高いものに決定するのが望ましい。
表
D.1
は,この規格で説明している
アクティビティ
を全て列挙してい
る。
JIS Q 13485
を参照すれば,
QMS
における位置付けを明らかにするのに役立つ。
製造業者
は,要求さ
れるソフトウェア安全クラスに基づき,各要求
アクティビティ
を既存
プロセス
と突き合わせて総合評価す
るのがよい。要求事項が既に盛り込まれている場合は,該当する
プロセス
記述の参照先を示すのが望まし
い。
矛盾がある場合は,
プロセス
改善のための措置が必要である。
この一覧表は,改善措置実施後の
プロセス
を対象とした
評価
にも使うことができる。
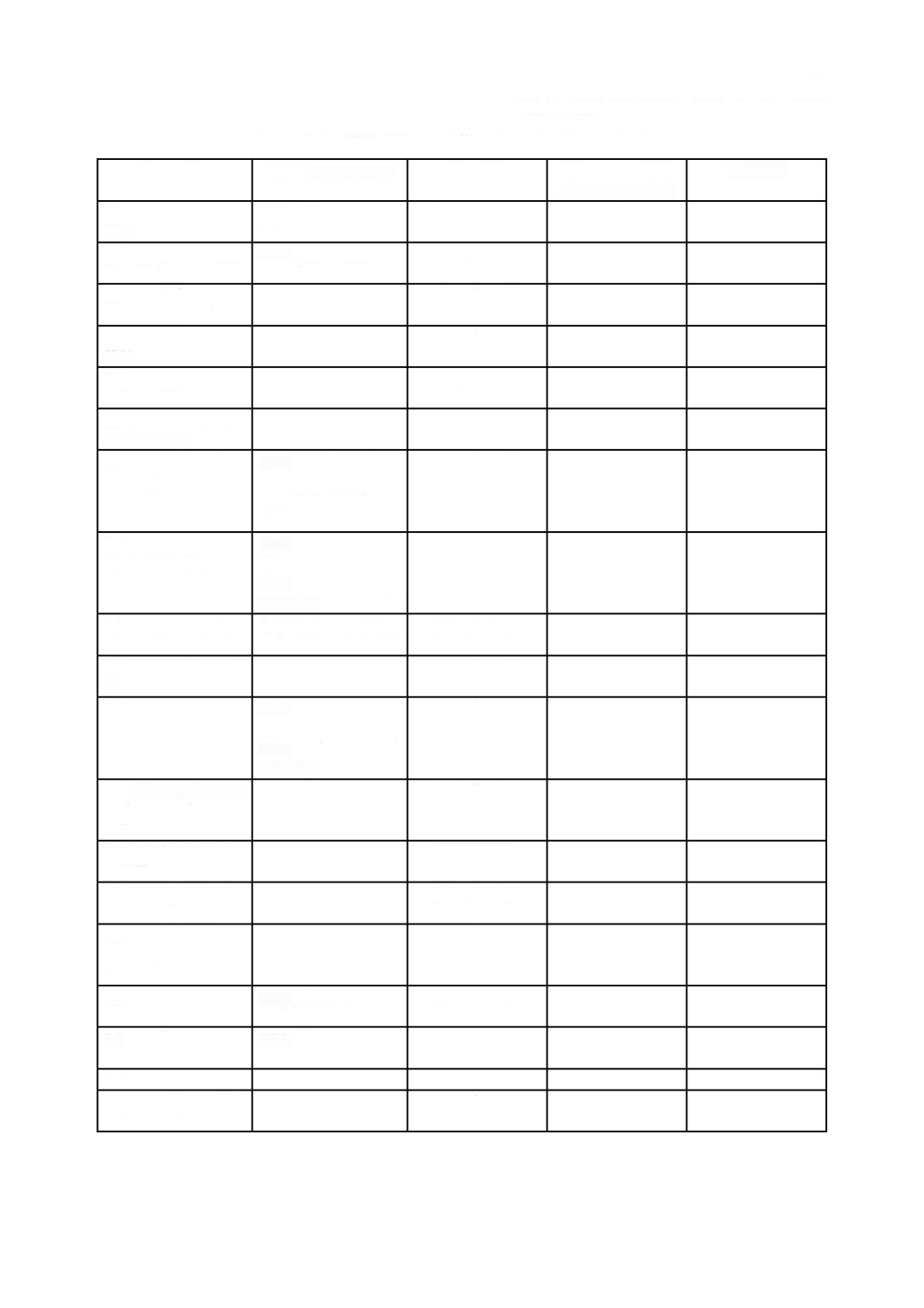
67
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
表
D.1
-
認証
QMS
がない小規模会社のためのチェックリスト
アクティビティ
JIS Q 13485
:2005
の
関連箇条
既存手順で
補われているか
補われている場合
はその引用を記入
実施措置
5.1
ソフトウェア開発
計画
7.3.1
設計・開発の計
画
はい/いいえ
5.2
ソフトウェア要求
事項分析
7.3.2
設計・開発への
インプット
はい/いいえ
5.3
ソフトウェアアー
キテクチャの設計
はい/いいえ
5.4
ソフトウェア詳細
設計
はい/いいえ
5.5
ソフトウェアユニ
ットの実装
はい/いいえ
5.6
ソフトウェア結合
及び結合試験
はい/いいえ
5.7
ソフトウェアシス
テム試験
7.3.3
設計・開発から
のアウトプット
7.3.4
設計・開発のレ
ビュー
はい/いいえ
5.8
システムレベルで
使 用 す る た め の ソ フ
トウェアリリース
7.3.5
設計・開発の検
証
7.3.6
設計・開発の妥
当性確認
はい/いいえ
6.1
ソフトウェア保守
計画の確立
7.3.7
設計・開発の変
更管理
はい/いいえ
6.2
問題及び修正の分
析
はい/いいえ
6.3
修正の実装
7.3.5
設計・開発の検
証
7.3.6
設計・開発の妥
当性確認
はい/いいえ
7.1
危険状態を引き起
こ す ソ フ ト ウ ェ ア の
分析
はい/いいえ
7.2
リスクコントロー
ル手段
はい/いいえ
7.3
リスクコントロー
ル手段の検証
はい/いいえ
7.4
ソフトウェア変更
の リ ス ク マ ネ ジ メ ン
ト
はい/いいえ
8.1
構成識別
7.5.3
識別及びトレー
サビリティ
はい/いいえ
8.2
変更管理
7.5.3
識別及びトレー
サビリティ
はい/いいえ
8.3
構成状態の記録
はい/いいえ
9
ソ フ ト ウ ェ ア 問 題
解決プロセス
はい/いいえ

68
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
附属書
JA
(参考)
定義した用語の索引
この規格で定義した用語を五十音順に,次に示す。
定義した用語
細分箇条
あ
アーキテクチャ
(
ARCHITECTURE
)
アクティビティ
(
ACTIVITY
)
安全
(
SAFETY
)
3.3
3.1
3.21
い
異常
(
ANOMALY
)
医療機器
(
MEDICAL DEVICE
)
医療機器ソフトウェア
(
MEDICAL DEVICE SOFTWARE
)
3.2
3.11
3.12
か
回帰テスト
(
REGRESSION TESTING
)
開発過程が不明なソフトウェア,
SOUP
(
software of unknown provenance
,
SOUP
)
3.15
3.29
き
危害
(
HARM
)
危険状態
(
HAZARDOUS SITUATION
)
3.8
3.35
け
検証
(
VERIFICATION
)
3.33
こ
構成アイテム
(
CONFIGURATION ITEM
)
3.5
さ
残留リスク
(
RESIDUAL RISK
)
3.38
し
システム
(
SYSTEM
)
重傷
(
SERIOUS INJURY
)
3.30
3.23
せ
成果物
(
DELIVERABLE
)
製造業者
(
MANUFACTURER
)
セキュリティ
(
SECURITY
)
3.6
3.10
3.22
そ
ソフトウェアアイテム
(
SOFTWARE ITEM
)
ソフトウェア開発ライフサイクルモデル
(
SOFTWARE DEVELOPMENT LIFE CYCLE MODEL
)
ソフトウェアシステム
(
SOFTWARE SYSTEM
)
ソフトウェアユニット
(
SOFTWARE UNIT
)
3.25
3.24
3.27
3.28
た
タスク
(
TASK
)
3.31
と
トレーサビリティ
(
TRACEABILITY
)
3.32
は
ハザード
(
HAZARD
)
バージョン
(
VERSION
)
3.9
3.34
ひ
評価
(
EVALUATION
)
3.7
ふ
プロセス
(
PROCESS
)
3.14
へ
変更要求
(
CHANGE REQUEST
)
3.4
も
問題報告
(
PROBLEM REPORT
)
3.13
り
リスク
(
RISK
)
リスクコントロール
(
RISK CONTROL
)
リスク推定
(
RISK ESTIMATION
)
リスク評価
(
RISK EVALUATION
)
リスク分析
(
RISK ANALYSIS
)
リスクマネジメント
(
RISK MANAGEMENT
)
リスクマネジメントファイル
(
RISK MANAGEMENT FILE
)
リリース
(
RELEASE
)
3.16
3.18
3.39
3.40
3.17
3.19
3.20
3.37
れ
レガシーソフトウェア
(
LEGACY SOFTWARE
)
3.36
69
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
参考文献
[1]
JIS C 0508
(規格群)
電気・電子・プログラマブル電子安全関連系の機能安全
注記
対応国際規格:
IEC 61508
(all parts)
,
Functional safety of electrical/electronic/programmable
electronic safety-related systems
(
IDT
)
[2]
JIS C 1010-1
:2014
測定用,制御用及び試験室用電気機器の安全性-第
1
部:一般要求事項
注記
対応国際規格:
IEC 61010-1
:2010
,
Safety requirements for electrical equipment for measurement
,
control, and laboratory use
-
Part 1: General requirements
(
MOD
)
[3]
JIS Q 9000
:2006
品質マネジメントシステム-基本及び用語
注記
対応国際規格:
ISO 9000
:2005
,
Quality management systems
-
Fundamentals and vocabulary
(
IDT
)
[4]
JIS Q 9001
:2008
品質マネジメントシステム-要求事項
注記
対応国際規格:
ISO 9001
:2008
,
Quality management systems
-
Requirements
(
IDT
)
[5]
JIS Q 13485
:2005
医療機器-品質マネジメントシステム-規制目的のための要求事項
注記
対応国際規格:
ISO 13485
:2003
,
Medical devices
-
Quality management systems
-
Requirements for
regulatory purposes
(
IDT
)
[6]
JIS T 0601-1
:2012
医用電気機器-第
1
部:基礎安全及び基本性能に関する一般要求事項及び追補
1:2014
注記
対応国際規格:
IEC 60601-1
:2005
,
Medical electrical equipment
-
Part 1: General requirements for
basic safety and essential performance
及び
Amendment 1:2012
(
MOD
)
[7]
JIS X 0160
:2012
ソフトウェアライフサイクルプロセス
注記
対応国際規格:
ISO/IEC 12207
:2008
,
Systems and software engineering
-
Software life cycle
processes
(
IDT
)
[8]
JIS X 0161
:2008
ソフトウェア技術-ソフトウェアライフサイクルプロセス-保守
注記
1
対応国際規格:
ISO/IEC 14764
:2006
,
Software Engineering
-
Software Life Cycle Processes
-
Maintenance
(
IDT
)
注記
2
この規格の対応国際規格(
IEC 62304
)では,
ISO/IEC 14764
:1999
が記載されていたが,
規格名称が,
2006
年の変更後の名称となっており,西暦年の記載を変更した。
[9]
JIS X 25010
:2013
システム及びソフトウェア製品の品質要求及び評価(
SQuaRE
)-システム及びソ
フトウェア品質モデル
注記
対応国際規格:
ISO/IEC 25010
:2011
,
Systems and software engineering
-
Systems and software
Quality Requirements and Evaluation (SQuaRE)
-
System and software quality models
(
IDT
)
[10]
ISO/IEC 15504-5
:2012
,
Information technology
-
Process assessment
-
Part 5: An exemplar software life
cycle process assessment model
[11]
ISO/IEC 33001
:2015
,
Information technology
-
Process assessment
-
Concepts and terminology
[12]
ISO/IEC 33004
:2015
,
Information technology
-
Process assessment
-
Requirements for process reference,
process assessment and maturity models
[13]
ISO/IEC 90003
:2014
,
Software engineering
-
Guidelines for the application of ISO 9001:2008 to computer
software
[14]
ISO/IEC Guide 51
:2014
,
Safety aspects
-
Guidelines for their inclusion in standards
70
T 2304
:
2017 (IEC 62304
:
2006
,
Amd.1
:
2015)
[15]
IEC 60601-1-4
:1996
,
Medical electrical equipment
-
Part 1: General requirements for safety
-
4. Collateral
standard: Programmable electrical medical systems
及び
Amendment 1 (1999)
(廃止)
[16]
IEC 60601-1-6
,
Medical electrical equipment
-
Part 1-6: General requirements for basic safety and essential
performance
-
Collateral standard: Usability
[17]
IEC 62366-1
:2015
,
Medical devices
-
Part 1: Application of usability engineering to medical devices
[18]
IEC 82304-1
:2016
,
Health software
-
Part 1: General requirements for product safety
[19]
IEEE 610.12
:1990
,
IEEE standard glossary of software engineering terminology
[20]
IEEE 1044
:1993
,
IEEE standard classification for software anomalies
[21] U.S. Department Of Health and Human Services, Food and Drug Administration, Guidance for the Content of
Premarket Submissions for Software Contained in Medical Devices, May 11, 2005,
<http://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm089543.htm>
[22] U.S. Department Of Health and Human Services, Food and Drug Administration, General Principles of
Software Validation; Final Guidance for Industry and FDA Staff, January 11, 2002,
<http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm126955.pdf>