T 14971:2020 (ISO 14971:2019)
(1)
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 2
3 用語及び定義··················································································································· 2
4 リスクマネジメントシステムの一般要求事項 ········································································· 7
4.1 リスクマネジメントプロセス ··························································································· 7
4.2 経営者の責任 ················································································································ 9
4.3 要員の力量 ··················································································································· 9
4.4 リスクマネジメント計画 ································································································· 9
4.5 リスクマネジメントファイル ·························································································· 10
5 リスク分析····················································································································· 11
5.1 リスク分析プロセス ······································································································ 11
5.2 意図する使用及び合理的に予見可能な誤使用 ····································································· 11
5.3 安全に関する特質の明確化 ····························································································· 12
5.4 ハザード及び危険状態の特定 ·························································································· 12
5.5 リスク推定 ·················································································································· 12
6 リスク評価····················································································································· 13
7 リスクコントロール········································································································· 13
7.1 リスクコントロール手段の選択 ······················································································· 13
7.2 リスクコントロール手段の実施 ······················································································· 14
7.3 残留リスクの評価 ········································································································· 14
7.4 ベネフィット・リスク分析 ····························································································· 15
7.5 リスクコントロール手段によって発生したリスク ······························································· 15
7.6 リスクコントロールの完了 ····························································································· 15
8 全体的な残留リスクの評価 ································································································ 15
9 リスクマネジメントのレビュー ·························································································· 16
10 製造及び製造後の活動 ···································································································· 16
10.1 一般 ·························································································································· 16
10.2 情報の収集 ················································································································· 16
10.3 情報のレビュー ··········································································································· 17
10.4 処置 ·························································································································· 17
附属書A(参考)要求事項の根拠 ··························································································· 18
附属書B(参考)医療機器のリスクマネジメントプロセス ·························································· 28
附属書C(参考)リスクの基礎的な概念 ·················································································· 32
参考文献 ···························································································································· 37
T 14971:2020 (ISO 14971:2019)
(2)
まえがき
この規格は,産業標準化法第16条において準用する同法第12条第1項の規定に基づき,一般社団法人
日本医療機器産業連合会(JFMDA)から,産業標準原案を添えて日本産業規格を改正すべきとの申出が
あり,日本産業標準調査会の審議を経て,厚生労働大臣及び経済産業大臣が改正した日本産業規格であ
る。これによって,JIS T 14971:2012は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣,経済産業大臣及び日本産業標準調査会は,このような特許権,出願公開後の
特許出願及び実用新案権に関わる確認について,責任はもたない。
日本産業規格 JIS
T 14971:2020
(ISO 14971:2019)
医療機器−リスクマネジメントの医療機器への適用
Medical devices-Application of risk management to medical devices
序文
この規格は,2019年に第3版として発行されたISO 14971を基に,技術的内容及び構成を変更すること
なく作成した日本産業規格である。
なお,この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
本文中の太字で示した用語は,この規格で定義している用語である。
1
適用範囲
この規格は,医療機器としてのソフトウェア及び体外診断用医療機器を含む医療機器のリスクマネジメ
ントの用語,原則及びプロセスについて規定する。この規格で規定するプロセスは,医療機器に関連する
ハザードを特定し,関連するリスクの推定及び評価を行い,これらのリスクをコントロールし,そのコン
トロールの有効性を監視するために,医療機器の製造業者を支援することを意図している。
この規格の要求事項は,医療機器のライフサイクルのいずれの段階にも適用可能である。この規格に規
定するプロセスは,医療機器に関連する,生体適合性,データ及びシステムのセキュリティ,電気,動く
部分,放射線,ユーザビリティなどに関するリスクに適用する。
この規格に規定するプロセスは,法的管轄によっては必ずしも医療機器とはならない製品に対しても適
用可能であり,また,医療機器のライフサイクルに関与する他の人々が用いることも可能である。
この規格は,次のいずれにも適用しない。
− 個別の臨床上の手順における医療機器の使用判断
− ビジネス上のリスクマネジメント
この規格は,製造業者がリスクの受容可能性の客観的な判断基準を確立することを要求するが,受容可
能なリスクレベルは規定しない。
リスクマネジメントは,品質マネジメントシステムの不可欠な部分である。しかし,この規格は,製造
業者が品質マネジメントシステムを構築することは要求しない。
注記1 この規格の適用の指針は,TR T 24971[9]にある。
注記2 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 14971:2019,Medical devices−Application of risk management to medical devices(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”こ
とを示す。
2
T 14971:2020 (ISO 14971:2019)
2
引用規格
この規格には,引用規格はない。
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
附属資料(accompanying documentation)
医療機器(3.10)に附属し,医療機器(3.10)の据付け,使用,保守,使用停止及び廃棄に責任をもつ
者又はユーザーに対する情報で,特に安全な使用に関する情報を記載した資料
注釈1 附属資料は,取扱説明書,技術解説,据付手順書,簡易参照ガイドなどで構成可能である。
注釈2 附属資料は,必ずしも,書面又は印刷した文書である必要はなく,視覚的,聴覚的又は触覚的
な資料及び複数の媒体を含むことがある。
3.2
ベネフィット(benefit)
医療機器(3.10)の使用が,個人の健康に与える良い影響若しくは望ましい結果,又は患者管理若しく
は公衆衛生に与える有益な影響
注釈1 ベネフィットには,臨床結果,患者の生活の質[QOL(quality of life)],診断に関係する結果に
与える有益な影響,臨床結果に与える診断機器の有益な影響又は公衆衛生への有益な影響が含
まれる。
3.3
危害(harm)
人の受ける傷害若しくは健康障害,又は財産若しくは環境の受ける害
(出典:JIS T 0063:2020の3.1)
3.4
ハザード(hazard)
危害(3.3)の潜在的な源
(出典:JIS T 0063:2020の3.2)
3.5
危険状態(hazardous situation)
人,財産又は環境が,一つ以上のハザード(3.4)にさらされる状況
注釈1 ハザード及び危険状態の関係については,附属書Cを参照。
(出典:JIS T 0063:2020の3.3に注釈1を追加)
3.6
意図する使用,意図する目的(intended use,intended purpose)
製造業者(3.9)が提供する仕様,取扱説明及び情報で意図している,製品,プロセス(3.14)又はサ
ービスの使用
注釈1 意図する医学的適応,患者集団,相互に作用し合う対象の体の部分又は生体組織の種類,ユー
ザープロファイル,使用環境及び動作原理が,意図する使用の典型的要素である。
3
T 14971:2020 (ISO 14971:2019)
(出典:JIS T 0063:2020の3.4)
3.7
体外診断用医療機器(in vitro diagnostic medical device,IVD medical device)
試薬,キャリブレーター(標準物質),管理物質,検体容器,ソフトウェア,及び関連する器具若しく
は器械又はその他の品目を含む,単独使用か又は組合せ使用かを問わず,診断,監視又は適合性の情報を
提供することを唯一の又は主な目的とし,人体から採取した検体の体外(in vitro)検査に使用することを
製造業者(3.9)が意図した機器
(出典:ISO 18113-1:2009の3.27の注記1を削除)
3.8
ライフサイクル(life cycle)
医療機器(3.10)の初期構想から最終的な使用停止及び廃棄に至るまでの一連の全ての段階
(出典:JIS T 0063:2020の3.5)
3.9
製造業者(manufacturer)
医療機器(3.10)の設計及び/又は製造が自分自身によるか,又は他の人による行為かにかかわらず,
その名の下に,使用に供するために医療機器(3.10)を作ることを意図し,医療機器(3.10)の設計及び
/又は製造に責任をもつ自然人又は法人
注釈1 この“自然人又は法人”は,利用可能とする又は販売することを意図した国又は法的管轄にお
いて,法的管轄で規制当局によって他の人に特別に責任を負わす場合を除き,適用される全て
の医療機器の規制要求事項に適合させる最終的な法的責任をもっている。
注釈2 製造業者の責任は,他のGHTF指針文書に記載されている。これらの責任には,市販前要求事
項並びに有害事象報告及び是正措置の通知のような市販後要求事項の両方に適合することを含
んでいる。
注釈3 “設計及び/又は製造”は,仕様開発,生産,成型加工,組立,加工,包装,再包装,ラベリ
ング,再ラベリング,滅菌,据付け若しくは医療機器の再製造,又は医療目的のために利用可
能な他の製品及び機器を一緒に収集してまとめることを含む場合がある。
注釈4 個々の患者に対して他の人が既に供給した医療機器を,取扱説明書に従って組立又は調整する
人は,製造業者ではない。ただし,指定された組立及び調整は,医療機器の意図する使用を変
更しないことが前提である。
注釈5 医療機器の元々の製造業者の代理としてではなく,医療機器の意図する使用を変更する人,医
療機器を改造する人,又は自身の名の下にそのような医療機器を利用可能にする人は,変更し
た医療機器の製造業者とみなされる。
注釈6 既存のラベルを覆ったり,変更したりすることなく,医療機器又はその包装に自身の所在地及
び連絡先だけを表示する指定代理人,ディストリビューター及び輸入業者は,製造業者とはみ
なさない。
注釈7 附属品は医療機器の規制要求事項の対象となるため,その附属品の設計及び/又は製造に関し
て責任をもつ者は,製造業者とみなす。
(出典:JIS T 0063:2020の3.6)
3.10
医療機器(medical device)
4
T 14971:2020 (ISO 14971:2019)
計器,器械,用具,機械,器具,植込み用具,体外診断薬,ソフトウェア,材料又はその他の同類のも
の若しくは関連する物質であって,単独使用か又は組合せ使用かを問わず,製造業者(3.9)が人体への
使用を意図し,その使用目的が次の一つ以上であり,
− 疾病の診断,予防,監視,治療又は緩和,
− 負傷の診断,監視,治療,緩和又は補助,
− 解剖学的又は生理学的なプロセス(3.14)の検査,代替,修復又は支援,
− 生命支援又は維持,
− 受胎調整,
− 医療機器(3.10)の消毒,
− 人体から採取される検体の体外試験法による情報提供,
さらに,薬学,免疫学又は新陳代謝の手段によって,体内又は体表において意図するその主機能を達成す
ることはないが,それらの手段によって意図する機能の実現が補助される場合があるもの
注釈1 法的管轄によって医療機器に該当するか否かが分かれる場合がある製品には,次のものがある。
− 消毒剤
− 身体障害者用の補助器具
− 動物及び/又はヒト組織に由来する機器
− 体外受精又は生殖補助技術用の機器
(出典:JIS T 0063:2020の3.7)
3.11
客観的証拠(objective evidence)
あるものの存在又は真実を裏付けるデータ
注釈1 客観的証拠は,観察,測定,試験又はその他の手段によって得ることが可能である。
(出典:JIS Q 9000:2015の3.8.3の注記2を削除)
3.12
製造後(post-production)
医療機器(3.10)のライフサイクル(3.8)のうち,設計を完了し,製造した後の段階
例 輸送,保管,据付け,製品使用,保守,修理,製品変更,使用停止及び廃棄
3.13
手順(procedure)
活動又はプロセス(3.14)を実行するために規定された方法
注釈1 手順は,文書にすることもあれば,しないこともある。
(出典:JIS Q 9000:2015の3.4.5)
3.14
プロセス(process)
インプットを使用して意図した結果を生み出す,相互に関連する又は相互に作用する一連の活動
注釈1 プロセスの“意図した結果”を,アウトプット,製品又はサービスのいずれと呼ぶかは,それ
が用いられている文脈による。
注釈2 プロセスへのインプットは,通常,他のプロセスからのアウトプットであり,また,プロセス
からのアウトプットは,通常,他のプロセスへのインプットである。
5
T 14971:2020 (ISO 14971:2019)
注釈3 連続した二つ以上の相互に関連する及び相互に作用するプロセスを,一つのプロセスと呼ぶこ
ともあり得る。
(出典:JIS Q 9000:2015の3.4.1の注記4〜注記6を削除)
3.15
合理的に予見可能な誤使用(reasonably foreseeable misuse)
容易に予測可能な人間の行動によって引き起こされる使用であるが,製造業者(3.9)が意図しない方
法による製品又はシステムの使用
注釈1 容易に予測可能な人間の行動は,様々なタイプのユーザー(例えば,一般の人及び専門家)の
行動を含む。
注釈2 合理的に予見可能な誤使用は,意図的である場合も意図的でない場合もある。
(出典:JIS T 0063:2020の3.8)
3.16
記録(record)
達成した結果を記述した,又は実施した活動の証拠を提供する文書
注釈1 記録は,例えば,トレーサビリティを正式なものにするため,又は検証,予防処置及び是正処
置の証拠を提供するために使用されることがある。
注釈2 通常,記録の改訂管理を行う必要はない。
(出典:JIS Q 9000:2015の3.8.10)
3.17
残留リスク(residual risk)
リスクコントロール(3.21)手段を実施した後にも残るリスク(3.18)
(出典:JIS T 0063:2020の3.9)
3.18
リスク(risk)
危害(3.3)の発生確率とその危害(3.3)の重大さ(3.27)との組合せ
(出典:JIS T 0063:2020の3.10の注釈1を削除)
3.19
リスク分析(risk analysis)
ハザード(3.4)を特定するための及びリスク(3.18)を推定するための利用可能な情報の体系的な使
用
(出典:JIS T 0063:2020の3.11)
3.20
リスクアセスメント(risk assessment)
リスク分析(3.19)及びリスク評価(3.23)からなる全てのプロセス(3.14)
(出典:JIS Z 8051:2015の3.11)
3.21
リスクコントロール(risk control)
規定したレベルまでリスク(3.18)を低減するか又はそのレベルでリスク(3.18)を維持するという決
定に到達し,かつ,そのための手段を実施するプロセス(3.14)
6
T 14971:2020 (ISO 14971:2019)
(出典:JIS T 0063:2020の3.12)
3.22
リスク推定(risk estimation)
危害(3.3)の発生確率とその危害(3.3)の重大さ(3.27)に対して重み付けをするために用いるプロ
セス(3.14)
(出典:JIS T 0063:2020の3.13)
3.23
リスク評価(risk evaluation)
判断基準に照らして推定したリスク(3.18)の受容可能性を判断するプロセス(3.14)
(出典:JIS T 0063:2020の3.14)
3.24
リスクマネジメント(risk management)
リスク(3.18)の分析,評価,コントロール及び監視に対する,マネジメント方針,手順(3.13)及び
実施の体系的な適用
(出典:JIS T 0063:2020の3.15)
3.25
リスクマネジメントファイル(risk management file)
リスクマネジメント(3.24)によって作成した記録(3.16)及びその他の文書のまとまり
3.26
安全(safety)
受容できないリスク(3.18)がないこと
(出典:JIS T 0063:2020の3.16)
3.27
重大さ(severity)
ハザード(3.4)から生じる可能性がある結果の尺度
(出典:JIS T 0063:2020の3.17)
3.28
最新の技術水準(state of the art)
ある時点での,科学,技術及び経験を結集した知見に基づいた,製品,プロセス(3.14)及びサービス
に関する技術力の到達段階
注釈1 最新の技術水準は,技術及び医学の優れた実践として現在一般に受け入れられているものを具
体化したものである。最新の技術水準は,必ずしも技術的に最も進んだ解決策を意味しない。
最新の技術水準は,この規格では“一般に認められた最新の技術水準”として規定する場合が
ある。
(出典:JIS T 0063:2020の3.18)
3.29
トップマネジメント(top management)
最高位で製造業者(3.9)を指揮し,管理する個人又はグループ
(出典:JIS Q 9000:2015の3.1.1の“組織”を“製造業者”に変更し,注記を削除)
7
T 14971:2020 (ISO 14971:2019)
3.30
使用エラー(use error)
製造業者(3.9)が意図するものとは異なる,又はユーザーが期待するものとは異なる結果を引き起こ
す,医療機器(3.10)を使用する際のユーザーの行為又はユーザーの行為の欠如
注釈1 使用エラーには,ユーザーがタスクを完遂できないことが含まれる。
注釈2 使用エラーは,ユーザー,ユーザーインターフェイス,タスク又は使用環境の間の特性の不一
致によって発生することもある。
注釈3 ユーザーが,使用エラーとなっていることに気付くことも気付かないこともある。
注釈4 患者の想定外の生理学的反応は,それ自体を使用エラーとはみなさない。
注釈5 想定外の結果を引き起こす医療機器の誤作動は,使用エラーとはみなさない。
(出典:JIS T 62366-1:2019の3.21の注記6を削除)
3.31
検証(verification)
客観的証拠(3.11)を提示することよって,規定要求事項が満たされていることを確認すること
注釈1 検証のために必要な客観的証拠は,検査の結果,又は別の方法での計算の実施若しくは文書の
レビューのような他の形の確定の結果であることがある。
注釈2 検証のために行われる活動は,適格性確認プロセスと呼ばれることがある。
注釈3 “検証済み”という言葉は,検証が済んでいる状態を示すために用いられる。
(出典:JIS T 0063:2020の3.19)
4
リスクマネジメントシステムの一般要求事項
4.1
リスクマネジメントプロセス
製造業者は,次に対する一連のプロセスを確立し,実施し,文書化し,維持する。
a) 医療機器に関連するハザード及び危険状態の特定
b) 関連するリスクの推定及び評価
c) リスクのコントロール
d) リスクコントロール手段の有効性の監視
このプロセスは,医療機器のライフサイクルを通して適用する。
このプロセスには,次の要素を含める。
− リスク分析
− リスク評価
− リスクコントロール
− 製造及び製造後の活動
文書化した製品実現のプロセスがある場合には,リスクマネジメントプロセスの該当する部分を取り入
れる。
注記1 製品実現のプロセスは,例えば,JIS Q 13485:2018[5]の箇条7に規定している。
注記2 品質マネジメントシステムの文書化したプロセスは,安全を確保するために体系的に使用可能
8
T 14971:2020 (ISO 14971:2019)
である。特に複雑な医療機器において,早い段階でのハザード及び危険状態の特定を可能にす
る。
注記3 リスクマネジメントプロセスの概略図を図1に示す。特定のライフサイクル段階によってリス
クマネジメントの各要素は,様々な重点をもつことがある。リスクマネジメント活動は,医療
機器によって必要な場合,繰り返し又は分割した複数段階で実施することも可能である。附属
書Bに,リスクマネジメントプロセスの各段階の更に詳細な概要を示す。
適合性は,適切な文書の調査によって確認する。
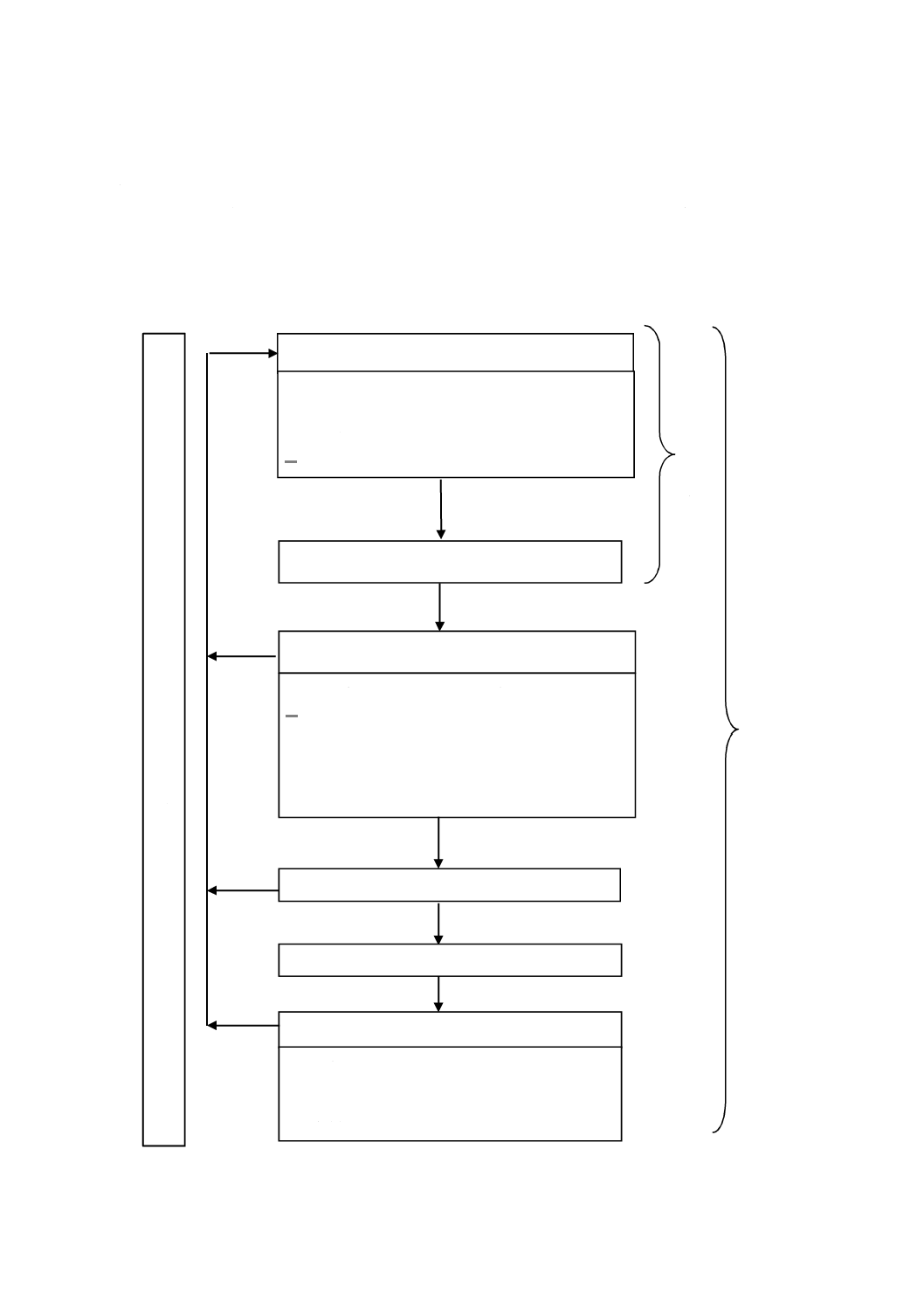
図1−リスクマネジメントプロセスの概略図
リスク分析
‒
意図する使用及び合理的に予見可能な誤使用
‒
安全に関する特質の明確化
‒
ハザード及び危険状態の特定
‒
リスク推定
リスク評価
リスクコントロール
‒
リスクコントロール手段の選択
‒
リスクコントロール手段の実施
‒
残留リスクの評価
‒
ベネフィット・リスク分析
‒
リスクコントロール手段によって発生したリスク
‒
リスクコントロールの完了
全体的な残留リスクの評価
製造及び製造後の活動
リ
ス
ク
ア
セ
ス
メ
ン
ト
リ
ス
ク
マ
ネ
ジ
メ
ン
ト
‒
一般
‒
情報の収集
‒
情報のレビュー
‒
処置
リ
ス
ク
マ
ネ
ジ
メ
ン
ト
計
画
リスクマネジメントのレビュー
9
T 14971:2020 (ISO 14971:2019)
4.2
経営者の責任
トップマネジメントは,リスクマネジメントプロセスに対するコミットメントの証拠を次によって示す。
− 十分な資源の提供
− リスクマネジメントの力量がある要員(4.3参照)の割当て
トップマネジメントは,リスクの受容可能性についての判断基準を確立するための方針を定義し,文書
化する。この方針は,判断基準が適用される国又は地域の規制及び関連のあるJIS又は国際規格に基づく
ことを確実にするための枠組みを提供し,並びに一般に認められた最新の技術水準及び既知である利害関
係者の懸念などの利用可能な情報を考慮する。
注記1 リスクの受容可能性についての判断基準を確立する製造業者の方針によってリスクコントロ
ールのアプローチ(リスクを合理的に実施可能な範囲で低減する,リスクを合理的に達成可能
な限り低減する,リスクをベネフィット・リスク比に悪影響を与えることなしに可能な限り低
減する)は,定義可能である。そのような方針を定義するための指針は,TR T 24971[9]を参照。
トップマネジメントは,リスクマネジメントプロセスの継続的な有効性を保証するため,あらかじめ計
画した間隔で,リスクマネジメントプロセスの適切性をレビューし,あらゆる決定及び講じた処置を文書
化する。製造業者が品質マネジメントシステムを構築している場合には,このレビューを品質マネジメン
トシステムのレビューの一環としてもよい。
注記2 製造及び製造後の情報をレビューした結果は,リスクマネジメントプロセスの適切性のレビュ
ーのインプットとすることが可能である。
注記3 この細分箇条で規定する文書は,製造業者の品質マネジメントシステムによって作成した文書
の中に含めることが可能である。また,これらの文書は,リスクマネジメントファイルにおい
て参照可能である。
適合性は,適切な文書の調査によって確認する。
4.3
要員の力量
リスクマネジメント作業を実施する人は,割り当てられた作業に対して適切な,教育,訓練,技能及び
経験に基づいた力量がなければならない。その要員は,適切な場合,特定の医療機器(又は類似の医療機
器)及びその使用,関連技術又は採用したリスクマネジメント技法についての知識及び経験をもたなけれ
ばならない。適切な記録を維持する。
注記 リスクマネジメント作業は,幾つかの部門の代表がそれぞれ専門知識を提供することによって実
施可能である。
適合性は,適切な記録の調査によって確認する。
4.4
リスクマネジメント計画
リスクマネジメント活動を計画する。検討対象となる特定の医療機器について,製造業者は,リスクマ
ネジメントプロセスに従ってリスクマネジメント計画を確立し,文書化する。リスクマネジメント計画は,
リスクマネジメントファイルの一部とする。
この計画には,少なくとも次を含める。
a) 計画したリスクマネジメント活動の適用範囲,医療機器の特定及び説明,並びに計画の各要素が適用
されるライフサイクルの段階
10
T 14971:2020 (ISO 14971:2019)
b) 責任及び権限の割当て
c) リスクマネジメント活動のレビューについての要求事項
d) リスクの受容可能性についての判断基準(受容可能なリスクを決定するための製造業者の方針に基づ
くもので,危害の発生確率が推定不可能な場合にはリスクを受容するための判断基準も含む。)
注記1 リスクの受容可能性についての判断基準は,リスクマネジメントプロセスを本質的に有効
にするために極めて重要である。製造業者は,各々のリスクマネジメント計画に対して,
個々の医療機器に適切なリスクの受容可能性の判断基準を確立する必要がある。
e) 全体的な残留リスクを評価する方法,及びリスクが受容可能かどうかを判断するための製造業者の方
針に基づく全体的な残留リスクの受容可能性の判断基準
注記2 全体的な残留リスクを評価する方法には,対象とする医療機器及び既に市販されている類
似の医療機器についてのデータ及び文献を収集し,レビューすることが含まれる。また,
適用分野及び臨床の専門知識をもつ専門家の職域を横断したチームによる判断を伴う場合
がある。
f)
リスクコントロール手段の実施及びその有効性についての検証の活動
g) 関連のある製造及び製造後情報の収集並びにレビューに関する活動
必ずしも全ての部分の計画を同時に作成する必要はない。計画又はその一部を継続して策定可能である。
注記3 リスクマネジメント計画の策定及びリスクの受容可能性の判断基準の確立の指針については,
TR T 24971[9]を参照。
注記4 対応国際規格のNOTE 4は許容事項のため,本文に移動した。
個別の医療機器のライフサイクルにおいて計画を変更した場合は,リスクマネジメントファイルにその
変更を記録する。
適合性は,リスクマネジメントファイルの調査によって確認する。
4.5
リスクマネジメントファイル
対象とする特定の医療機器について,製造業者はリスクマネジメントファイルを作成し,維持する。こ
の規格に含む他の箇条の要求事項に加え,リスクマネジメントファイルは,特定した各ハザードについて,
次の活動に対してのトレーサビリティをもたなければならない。
− リスク分析
− リスク評価
− リスクコントロール手段の実施及び検証
− 残留リスクの評価結果
注記1 リスクマネジメントファイルを構成する記録及びその他の文書は,例えば,製造業者の品質マ
ネジメントシステムが要求する他の文書及びファイルの一部とすることが可能である。リスク
マネジメントファイルには,物理的に全ての記録及びその他の文書を含める必要はない。ただ
し,少なくとも,製造業者がリスクマネジメントファイルで参照した情報を適時集めて整理可
能なように,要求する全ての文書に対する参照又は指定先を含める必要がある。
注記2 リスクマネジメントファイルは,あらゆる形式又はあらゆる種類の媒体で作成することが可能
である。
注記3 この規格を用いずに設計した部品及び機器に対するリスクマネジメントファイルを作成する
11
T 14971:2020 (ISO 14971:2019)
ための指針は,TR T 24971[9]を参照。
5
リスク分析
5.1
リスク分析プロセス
製造業者は,5.2〜5.5に規定したリスク分析を個別の医療機器に対し実施する。計画したリスク分析活
動の実施及びそのリスク分析結果は,リスクマネジメントファイルに記録する。
注記1 類似の医療機器についてのリスク分析又はその他の関連する情報を用いることが可能な場合は,
その分析又は情報を新規のリスク分析の出発点として使用可能である。関連の程度は,医療機
器間の差異及びそれらが新しいハザードを生じるか,又は出力,特性,性能若しくは結果に対
して大きな違いをもたらすかどうかに依存する。既存のリスク分析をどの程度使用するかは,
その違いが危険状態の発生に与え得る影響についての体系的な評価に基づく。
注記2 選択したリスク分析技法に対する指針,及び体外診断用医療機器に対するリスク分析技法につ
いての指針は,TR T 24971[9]を参照。
リスク分析の実施及びその結果の文書には,5.2〜5.5で要求した記録に加え,少なくとも次を含めなけ
ればならない。
a) 分析を行った医療機器の特定及び説明
b) リスク分析を行った人及び組織の特定
c) リスク分析の適用範囲及び行った日付
注記3 リスク分析の適用範囲は,(例えば,製造業者が全く又はほとんど経験がない新しい医療機器の
開発においては)非常に広くなる。製造業者が,(製造業者のファイルに多くの情報をもってい
る既存の医療機器について,変更の影響の分析をする場合は,)適用範囲を限定することが可能
である。
適合性は,リスクマネジメントファイルの調査によって確認する。
5.2
意図する使用及び合理的に予見可能な誤使用
製造業者は,分析対象とする医療機器の意図する使用を文書化する。
意図する使用は,例えば,意図する医学的適応,患者集団,相互に作用し合う対象の体の部分又は生体
の組織,ユーザープロファイル,使用環境,動作原理などの情報を考慮することが望ましい。
製造業者は,合理的に予見可能な誤使用についても文書化する。
この文書は,リスクマネジメントファイルに維持する。
注記1 使用関連仕様(JIS T 62366-1:2019[13]の3.23参照)は,意図する使用を決定するためのインプ
ットにすることが可能である。
注記2 意図する使用を決定する際に検討する要因及び合理的に予見可能な誤使用の説明については,
TR T 24971[9]を参照。
適合性は,リスクマネジメントファイルの調査によって確認する。
12
T 14971:2020 (ISO 14971:2019)
5.3
安全に関する特質の明確化
分析対象とする医療機器について,製造業者は,医療機器の安全に影響する定性的及び定量的特質を特
定し,文書化する。該当する場合,製造業者は,それらの特質の限度値を決定する。この文書は,リスク
マネジメントファイルに維持する。
注記1 医療機器の安全に影響する特質を特定するときの指針にできる質問事項の一覧については,TR
T 24971[9]を参照。
注記2 医療機器の臨床性能の喪失又は低下に関する特質で,受容できないリスクにつながるものは,
基本性能と呼ばれることがある(例は,JIS T 0601-1[12]を参照)。
適合性は,リスクマネジメントファイルの調査によって確認する。
5.4
ハザード及び危険状態の特定
製造業者は,医療機器についての既知及び予見可能なハザードを,意図する使用,合理的に予見可能な
誤使用及び安全に関する特質に基づいて,正常状態及び故障状態の両方において特定し,文書化する。
特定した各ハザードに対して,製造業者は,危険状態を起こすような合理的に予見可能な一連の事象又
は事象の組合せを検討し,その結果起こる危険状態を特定し,文書化する。
注記1 一連の事象は,例えば,輸送,保管,据付け,保守,日常点検,使用停止及び廃棄のライフサ
イクルの全ての段階で発生する可能性がある。
注記2 ハザード,危険状態及び危害の関係の説明並びに例を,附属書Cに示す。
注記3 リスク分析には,様々な危険状態につながる可能性のある一つのハザードに関して,様々な一
連の事象又は様々な事象の組合せを検討することを含める。その各々の危険状態は,異なるタ
イプの危害につながることがある。
注記4 過去に認識されなかった危険状態を特定する際,その状況を扱うリスク分析の体系的な技法を
用いることが可能である。利用可能な技法の幾つかについての指針をTR T 24971[9]に示す。
この文書は,リスクマネジメントファイルに維持する。
適合性は,リスクマネジメントファイルの調査によって確認する。
5.5
リスク推定
特定した各危険状態に対して,製造業者は,利用可能な情報又はデータを用いて関連するリスクを推定
する。危害の発生確率が推定できない危険状態に対しては,リスク評価及びリスクコントロールに用いる
ために,起こり得る結果をリストする。これらの活動の結果は,リスクマネジメントファイルに記録する。
危害の発生確率及び危害の重大さの定性的又は定量的な分類に用いた方法は,リスクマネジメントファ
イルに記録する。
注記1 リスク推定には,危害の発生確率及び危害の重大さの分析を含んでいる。適用分野によっては,
リスク推定のプロセスのある特定の要素だけを詳細に検討する必要があるかもしれない。例え
ば,危害が極めて小さい場合には,初期のハザード及びその結果(危害)の分析だけで十分か
もしれない。十分な情報又はデータが利用できない場合には,発生確率の慎重な推定(例えば,
発生確率=1)によってリスクの度合いが示されるかもしれない。TR T 24971[9]も参照。
注記2 リスク推定は,定性的又は定量的に行うことが可能である。系統的な故障の結果生じるリスク
も含め,リスク推定の方法は,体外診断用医療機器のためのリスク推定に有益な情報とともに,
13
T 14971:2020 (ISO 14971:2019)
TR T 24971[9]に示している。
注記3 リスク推定のための情報又はデータは,例えば,次によって得ることが可能である。
− 発行済み規格
− 科学的又は技術的調査
− 一般に利用可能な事故報告を含め,既に使用している類似の医療機器の市場データ
− 標準的なユーザーによるユーザビリティの評価
− 臨床エビデンス
− 関連する調査又はシミュレーションの結果
− 専門家の意見
− 体外診断用医療機器の外部品質評価スキーム
適合性は,リスクマネジメントファイルの調査によって確認する。
6
リスク評価
製造業者は,特定した各危険状態について,リスクマネジメント計画で定義したリスクの受容可能性の
判断基準を用いて,推定したリスクを評価し,リスクが受容可能かどうかを決定する。
リスクが受容可能な場合は,この危険状態には,7.1〜7.5の要求事項を適用する必要はなく(すなわち,
7.6に進み),その推定したリスクを残留リスクとして扱う。
リスクが受容可能でない場合は,製造業者は7.1〜7.6に規定するリスクコントロール活動を実施する。
このリスク評価の結果は,リスクマネジメントファイルに記録する。
適合性は,リスクマネジメントファイルの調査によって確認する。
7
リスクコントロール
7.1
リスクコントロール手段の選択
製造業者は,リスクを受容可能なレベルまで低減するための適切なリスクコントロール手段を決定する。
製造業者は,次の優先順位に従って,一つ以上のリスクコントロール手段を用いる。
a) 本質的に安全な設計及び製造
b) 医療機器自体又は製造プロセスにおける保護手段
c) 安全に関する情報,及び適切な場合,ユーザートレーニング
注記1 リスクコントロール手段を選択する際の優先順位の根拠は,A.2.7.1に示している。
注記2 リスクコントロール手段は,危害の重大さ若しくは危害の発生確率又はその両者を減少させる
ことが可能である。
注記3 安全に関する情報を提示するための指針については,TR T 24971[9]を参照。
リスクコントロール手段の選択の一部として,関連する規格を適用することが望ましい。
注記4 多くの規格で,医療機器の本質的な安全,保護手段及び安全に関する情報を規定している。そ
14
T 14971:2020 (ISO 14971:2019)
れに加えて,リスクマネジメントプロセスの要素を取り込んだ医療機器の規格(例えば,電磁
両立性,ユーザビリティ,生物学的評価など)もある。リスクマネジメントにおけるJIS及び
国際規格の役割についての情報は,TR T 24971[9]を参照。
選択したリスクコントロール手段は,リスクマネジメントファイルに記録する。
リスクコントロール手段を選択するときに,製造業者は,リスク低減が現実的でないと判断した場合は,
残留リスクについてベネフィット・リスク分析を実施する(7.4に進む。)。
適合性は,リスクマネジメントファイルの調査によって確認する。
7.2
リスクコントロール手段の実施
製造業者は,7.1で選択したリスクコントロール手段を実施する。
各リスクコントロール手段の実施を検証し,その検証をリスクマネジメントファイルに記録する。
注記1 リスクコントロール手段の実施の検証は,品質マネジメントシステムの設計・開発の検証又は
プロセスの適格性確認の一部として実施することが可能である。
リスクコントロール手段の有効性を検証する。この検証の結果をリスクマネジメントファイルに記録す
る。
注記2 有効性の検証は,品質マネジメントシステムの設計・開発のバリデーションの一部として実施
することが可能であり,ユーザーを伴う試験を含めることが可能である。A.2.7.2参照。
注記3 有効性の検証は,リスク低減の有効性と設計・開発の検証又はプロセスの適格性確認の結果と
の関係が分かっている場合には,設計・開発の検証又はプロセスの適格性確認の一部としても
実施可能である。
例1 薬剤注入器の投与量の精度などの,特定の性能特性の設計検証は,薬剤の安全な投与
を保証するリスクコントロール手段の有効性の検証に活用可能である。
例2 プロセスの適格性確認は,製造のアウトプットのばらつきに起因するリスクに関する
リスクコントロール手段の有効性の検証に活用可能である。
注記4 設計・開発の検証及びバリデーションに関する詳細情報は,JIS Q 13485[5]を参照。更なる指針
については,TR T 24971[9]も参照。
適合性は,リスクマネジメントファイルの調査によって確認する。
7.3
残留リスクの評価
製造業者は,リスクコントロール手段の実施後の残留リスクを,リスクマネジメント計画で定義したリ
スクの受容可能性についての判断基準を用いて評価する。この評価の結果は,リスクマネジメントファイ
ルに記録する。
残留リスクが,この判断基準を用いて受容可能と判断されない場合は,更にリスクコントロール手段を
検討する(7.1に戻る。)。
適合性は,リスクマネジメントファイルの調査によって確認する。
15
T 14971:2020 (ISO 14971:2019)
7.4
ベネフィット・リスク分析
リスクマネジメント計画で確立した判断基準に照らし残留リスクが受容できないと判断し,かつ,それ
以上のリスクコントロールも現実的ではない場合,製造業者は,意図する使用のベネフィットが残留リス
クを上回るか否かを判断するために,データ及び文献を収集しレビューしてもよい。
この証拠から,ベネフィットがこの残留リスクを上回るという結論が裏付けられない場合は,製造業者
は,医療機器又はその意図する使用を変更することを検討してもよい(5.2に戻る。)。そうでない場合は,
このリスクは受容できないものとして残る。
ベネフィットが残留リスクを上回る場合は,7.5に進む。
ベネフィット・リスク分析の結果は,リスクマネジメントファイルに記録する。
注記 ベネフィット・リスク分析を行うための指針については,TR T 24971[9]を参照。
適合性は,リスクマネジメントファイルの調査によって確認する。
7.5
リスクコントロール手段によって発生したリスク
製造業者は,リスクコントロール手段の影響を次の点に関してレビューする。
− 新たなハザード又は危険状態が発生しないかどうか
− 既に特定した危険状態について推定したリスクがリスクコントロール手段の導入によって変わらな
いかどうか
新たに発生,又は増加した全てのリスクには,5.5〜7.4を適用する。
このレビューの結果は,リスクマネジメントファイルに記録する。
適合性は,リスクマネジメントファイルの調査によって確認する。
7.6
リスクコントロールの完了
製造業者は,特定した全ての危険状態から発生するリスクを検討しており,全てのリスクコントロール
活動が完了されることを確実にするために,リスクコントロール活動をレビューする。
このレビューの結果は,リスクマネジメントファイルに記録する。
適合性は,リスクマネジメントファイルの調査によって確認する。
8
全体的な残留リスクの評価
全てのリスクコントロール手段が実施及び検証された後,製造業者は,全ての残留リスクの寄与を考慮
し,意図する使用のベネフィットとの関連において,リスクマネジメント計画で確立した全体的な残留リ
スクの受容可能性についての評価方法及び判断基準を用いて,医療機器の全体的な残留リスクを評価する
[4.4 e)参照]。
全体的な残留リスクを受容可能と判断した場合,製造業者は,重大な残留リスクをユーザーに通知し,
残留リスクを開示するために必要な情報を附属資料に記載する。
注記1 重大な残留リスクを開示する根拠をA.2.8に示す。
16
T 14971:2020 (ISO 14971:2019)
注記2 全体的な残留リスクの評価及び残留リスクの開示についての指針は,TR T 24971[9]を参照。
意図する使用のベネフィットに関連して,全体的な残留リスクを受容できないと判断した場合は,製造
業者は,追加のリスクコントロール手段を実施すること(7.1に戻る。),又は医療機器若しくはその意図す
る使用を変更すること(5.2に戻る。)を検討してもよい。そうでない場合は,全体的な残留リスクは受容
できないものとして残る。
全体的な残留リスクの評価結果は,リスクマネジメントファイルに記録する。
適合性は,リスクマネジメントファイル及び附属資料の調査によって確認する。
9
リスクマネジメントのレビュー
製造業者は,医療機器の市場出荷に先立ってリスクマネジメント計画の実行についてレビューする。こ
のレビューでは,少なくとも次を確認する。
− リスクマネジメント計画が適切に実施されている。
− 全体的な残留リスクが受容可能である。
− 製造及び製造後の段階において,情報を収集しレビューする適切な方法が定められている。
このレビューの結果はリスクマネジメント報告書に記録して維持し,リスクマネジメントファイルに含
める。
そのレビューの責任者には,リスクマネジメント計画で指定した適切な権限をもつ者を選ばなければな
らない[4.4 b)参照]。
適合性は,リスクマネジメントファイルの調査によって確認する。
10 製造及び製造後の活動
10.1 一般
製造業者は,製造及び製造後の段階において,その医療機器に関連する情報を積極的に収集及びレビュ
ーするシステムを確立し,文書化し,維持する。このシステムを確立する場合には,製造業者は,情報を
収集し処理する適切な方法を検討する。
注記1 JIS Q 13485:2018[5]の7.3.3,8.2.1,8.4及び8.5も参照。
注記2 製造及び製造後の活動についての指針は,TR T 24971[9]参照。
適合性は,適切な文書の調査によって確認する。
10.2 情報の収集
製造業者は,該当する場合,次の情報を収集する。
a) 製造中及び製造プロセスの監視から得られる情報
b) ユーザーからの情報
c) 医療機器の据付け,使用及び保守の責任者からの情報
d) サプライチェーンからの情報
17
T 14971:2020 (ISO 14971:2019)
e) 一般的に入手可能な情報
f)
一般に認められた最新の技術水準に関する情報
注記 一般に認められた最新の技術水準に関する情報には,新規又は改正された規格,対象とする医療
機器の適用に特有な公表データ,代替えの医療機器及び/又は治療方法の利用可能性その他の情
報が含まれる場合がある(TR T 24971[9]も参照)。
製造業者は,市販されている類似の医療機器及びその他の類似の製品についての一般に利用可能な情報
についても,積極的に収集し,レビューする必要があるかを検討する。
適合性は,適切な文書の調査によって確認する。
10.3 情報のレビュー
製造業者は,収集した情報を,安全との関連の有無に対して,特に次についてレビューする。
− 以前に認識されていなかったハザード又は危険状態が存在するかどうか
− 危険状態によって発生したリスクが,もはや受容できないかどうか
− 全体的な残留リスクが,意図する使用のベネフィットに関連して,もはや受容できないかどうか
− 一般に認められた最新の技術水準に変更があるかどうか
レビューの結果は,リスクマネジメントファイルに記録する。
適合性は,リスクマネジメントファイルの調査によって確認する。
10.4 処置
収集した情報が安全に関連すると判断した場合には,次による。
1) 個別の医療機器に関して
− 製造業者は,リスクマネジメントファイルをレビューして,リスクの再アセスメント及び/又は新し
いリスクのアセスメントが必要かどうかを決定する。
− 残留リスクがもはや受容可能でなくなった場合には,既に実施したリスクコントロール手段への影響
を評価する。さらに,医療機器の変更のためのインプットとして検討することが望ましい。
− 製造業者は,市場にある医療機器に関する処置の必要性を検討することが望ましい。
− 全ての決定及び処置をリスクマネジメントファイルに記録する。
2) リスクマネジメントプロセスに関して
− 製造業者は,既に実施したリスクマネジメント活動への影響を評価する。
− この評価の結果は,トップマネジメントによるリスクマネジメントプロセスの適切性のレビューへの
インプットとして検討する(4.2参照)。
注記 製造後監視の幾つかの側面は,一部の国の規制対象となっている。そのような場合,追加対策が
要求されることがある(例えば,前向き製造後評価)。
適合性は,リスクマネジメントファイル及び他の適切な文書の調査によって確認する。
18
T 14971:2020 (ISO 14971:2019)
附属書A
(参考)
要求事項の根拠
A.1 一般
このJIS T 14971の基となったISO 14971の作成では,次のような経緯があった。
ISO/TC 210-IEC/SC 62A合同作業グループ1(JWG1)(リスクマネジメントの医療機器への適用)は,
この規格に盛り込んだ要求事項を解説するために,この附属書を作成した。この規格の使用経験を基に今
後改正するに当たって,製造業者,規制当局及び医療従事者にとってより有用な規格とするためにこの附
属書を使用することが可能である。
ISO/TC 210及びIEC/SC 62Aは,リスクマネジメントについての取組を統合し,リスクマネジメントを
医療機器へ適用するための規格を開発するためにJWG1を編成した。リスクマネジメントの国際規格の作
成開始に当たって,医療機器のリスクとベネフィットとのバランスをとることはもちろん,リスク評価の
プロセスなどをリスクマネジメントの不可欠な特徴と位置付けることにした。製造業者,規制当局及び医
療従事者は,医療機器の“絶対安全”の達成は不可能であるということを認識してきた。さらに製品の安
全規格において,医療機器の多様性の増加及びその適用に起因するリスクを全て明らかにすることは不可
能である。これら事実の認識,及び医療機器のライフサイクルにおけるリスクをマネジメントする必要か
ら,医療機器の安全を積極的に改善するためのツールとしてISO 14971を作成するに至った。この規格の
第1版は,2000年に発行された。
ISO 14971の第2版は,リスクマネジメントの適用及びハザードと危険状態との関係についての指針を
追加する必要性も考慮して,2007年に作成し発行された。本文での変更は,製造後の監視をリスクマネジ
メント計画に含める要求事項の追加,トレーサビリティの要求事項をリスクマネジメント報告書から削除
するなど,軽微なものであった。
2010年の定期見直しによって,幾つかのトピックスについて追加の指針が必要であることが明らかにな
った。指針に対する軽微な変更であっても,規格の改訂が必要になるので,国際標準報告書ISO/TR 24971[9]
を開発することが決定された。この国際標準報告書の第1版は,2013年に発行された。
ISO 14971の第3版は,規定要求事項を明確にして,特に全体的な残留リスクの評価,リスクマネジメ
ントのレビュー及び報告書並びに製造及び製造後の情報に関する箇条について,更に細かく規定するため
に開発された。この明確化は,ISO 14971の2016年の定期見直しにおいて説明が要求されたこと,規制当
局からより厳密な要求事項を求められていることから,必要があると考えられた。医療機器を使用するこ
とによって期待されるベネフィット及び(全体的な)残留リスクとそれらのベネフィットとのバランスに
さらに力点が置かれてた。ISO 14971に規定するプロセスは,例えば,生体適合性,データ及びシステム
のセキュリティ,電気,動く部分,放射線,ユーザビリティなどの,医療機器に関連する全てのタイプの
ハザード及びリスクに適用可能であると説明された。参考情報を記載した附属書の幾つかは,この規格か
ら,同時に改訂している指針文書のISO/TR 24971[9]に移動された。これによって,指針文書を,規格の改
訂とは独立して,より頻繁に改正することが可能となる。
19
T 14971:2020 (ISO 14971:2019)
A.2 個別の箇条及び細分箇条の要求事項の根拠
A.2.1 適用範囲
このJISの基であるISO 14971:2019の序文に記載しているように,医療機器のライフサイクルに適用す
るリスクマネジメント規格が必要とされている。医療機器としてのソフトウェア及び体外診断用医療機器
については異なる規制があるため,これらの機器がこの規格から除外されるという誤解を生じないように
適用範囲において言及している。
医療機器のライフサイクルのあらゆる場面でリスクは存在する可能性がある。ライフサイクルのある時
点で明らかになるリスクは,それとは異なるライフサイクルの時点での処置によってリスクの対策が可能
である。この理由から,この規格は全てのライフサイクルをカバーする必要がある。つまり,この規格は,
リスクマネジメントの原則を,初期構想から最終的な使用停止及び廃棄に至るまで医療機器に適用するよ
う製造業者に指示している。
この規格に規定するプロセスは,医療機器に関連するハザード及びリスクに対して適用可能である。医
療機器に関するセキュリティリスクをマネジメントするためには別のプロセスが必要になるという誤解
を避けるために,データ及びシステムのセキュリティに関するリスクについては,適用範囲で特に言及し
ている。これは,セキュリティリスクのアセスメント及びコントロールのための特別な方法及び要求事項
を規定する,特定の規格の開発を妨げるものではない。このような規格は,ユーザビリティに対するJIS
T 62366-1 [13],生物学的評価に対するJIS T 0993-1[4]又は電気的及び機械的リスクに対するJIS T 0601-
1[12]と同様のやり方で,この規格と併せて用いることが可能である。
この規格の適用範囲には,臨床的な意思決定,すなわち個別の臨床上の手順における医療機器の使用に
関する判断は含まない。そうした決定は,残留リスクが,手順に期待されるベネフィット,又は代替手順
に伴うリスク及び期待されるベネフィットに対してバランスがとられている必要がある。このような決定
は,臨床上の手順又は使用状況に伴うリスク及びベネフィットと同様に,意図する使用,性能及び医療機
器に伴うリスクを考慮している。そうした判断には,個々の患者の健康状態及び患者自身の意見を知って
いる医師だけが下せるものもある。
この規格の適用範囲には,ビジネス上の意思決定についても含まれていない。組織のリスクマネジメン
ト及び関連する課題については,JIS Q 31000[10]などの他の規格がある。
リスクの受容可能なレベルをどう決めるかについて重要な議論がなされたが,この規格は受容レベルを
規定していない。受容可能なリスクの普遍的なレベルを規定することは適切でない。この判断は,次に基
づいている。
− この規格の対象となる医療機器及び状況は広範囲にわたっており,受容可能なリスクについて,画一
的なレベルは意味をもたない。
− 地域の法律,慣習,価値観及びリスクの認識は,世界各国の独自の文化又は地域性に適したリスクの
受容可能性を決定するのにより適切である。
医療機器の製造業者に対する品質マネジメントシステムは,全ての国において要求されているわけでは
ないので,この規格は品質マネジメントシステムを要求していない。しかし,品質マネジメントシステム
はリスクマネジメントを適切に行うのに極めて有益である。そのため,ほとんどの医療機器の製造業者が
品質マネジメントシステムを導入しているので,この規格は,製造業者が採用している品質マネジメント
システムに容易に組み込むことが可能なように構成している。
20
T 14971:2020 (ISO 14971:2019)
A.2.2 引用規格
この規格に従うリスクマネジメントプロセスを確立し,維持するためには,他の規格を必要としない。
JIS Z 8301:2019の箇条15は,この記載を規格に含めることを要求している。
A.2.3 用語及び定義
この規格の定義のほとんどは,JIS Q 9000:2015[3]及びJIS T 0063:2020[2]から用いられている。JIS T
0063:2020[2]は,JIS Z 8051:2015[1]の定義及びGHTF(Global Harmonization Task Force)によって開発され
た定義の多くを採用し,一部を変更している。JIS T 0063:2020[2]及びこの規格の定義の幾つかは,他の規
格と若干意味が異なるものがある。
例えば,JWG1は,危害(harm)(3.3)の定義を幅広いものとしており,過度の心理的ストレス又は予
想外の妊娠を“人の受ける健康障害”の一部として含めることを意図している。そうしたストレスは,疾
病の偽陽性の診断の後に起こることがある。“財産及び環境の受ける害”は望ましいものではないので,関
連するリスクは同様に考える必要がある。例えば,医療機器の使用又は廃棄に伴って生成される危険な廃
棄物に関するリスクなどである。“身体的な”という言葉は,JIS Z 8051:2015[1]において危害の定義から除
外され,JIS T 0063:2020[2]及びこの規格においても同様であるが,これは傷害が元々身体的な障害を含ん
でいるからである。データ及びシステムのセキュリティ侵害は,例えば,データの喪失,データの管理さ
れないアクセス,診断情報の破損又は喪失,医療機器の機能不全につながるソフトウェアの破損などによ
って危害につながることがある。
意図する使用(intended use)(3.6)という用語の定義は,米国で使用されている意図する使用(intended
use)の定義と欧州連合の用語である意図する目的(intended purpose)とを組み合わせた定義にしている。
これらの用語の定義は,本質的に同一である。この定義では,製造業者が医療機器の意図する使用を決定
する上で,意図する医学的適応,患者集団,相互に作用し合う対象の体の部分又は生体の組織,ユーザー
プロファイル,使用環境及び動作原理を考慮に入れることが意図されている。この規格において使用する
ライフサイクル(life cycle)(3.8)の用語は,医療機器のあらゆる側面を含んでいることを明らかにするよ
うに定義した。リスクマネジメント(risk management)(3.24)の用語は,体系的なアプローチの使用及び
その運営の監視の必要性を強調している。トップマネジメント(top management)(3.29)の定義は,JIS Q
9000:2015 [3]から引用した。この定義は,製造業者の組織において最高レベルに位置する個人又はグルー
プに対して適用する。
ベネフィット(benefit)(3.2),製造後(post-production)(3.12),及びリスクマネジメントファイル(risk
management file)(3.25)という用語は,JIS T 0063:2020[2]又は他の規格の定義に基づくものではない。ベ
ネフィットという用語は,規制当局が(残留)リスクと医療機器のベネフィットとのバランスをますます
強調しているので定義した。同じ理由でベネフィット・リスク分析という語句が使用されている。製造後
(post-production)の定義は,医療機器のライフサイクル全体にわたってリスクマネジメントが重要である
ことを強調するために追加した。リスクマネジメントファイルの概念は,今ではよく理解されている。
A.2.4 リスクマネジメントシステムの一般要求事項
A.2.4.1 リスクマネジメントプロセス
リスクマネジメントシステムは,4.1〜4.5の要素からなる。
製造業者は,医療機器の設計・開発の一部としてリスクマネジメントプロセスを確立する必要がある。
これは,製造業者がプロセスに必要な要素を取り入れることを体系的に保証するために要求されている。
21
T 14971:2020 (ISO 14971:2019)
リスク分析,リスク評価及びリスクコントロールは,リスクマネジメントの重要な要素であることは広く
認識されている。これらの要素に加えてこの規格は,リスクマネジメントプロセスが医療機器の設計及び
製造(関連する滅菌,包装及びラベリングを含む。)で終わりではなく,製造後の段階まで継続することを
強調している。したがって,製造及び製造後の情報の収集及びレビューは,リスクマネジメントプロセス
の一部として要求されている。さらに製造業者が品質マネジメントシステムを採用する場合は,リスクマ
ネジメントプロセスは,品質マネジメントシステムに完全に統合されることが望ましい。
リスクマネジメント活動は,対象とする医療機器ごとに大きく異なるが,リスクマネジメントプロセス
に含める必要のある基本的な要素があり,4.1では,必要な要素について言及している。また,医療機器に
リスクマネジメントを適用するための規制上のアプローチには,様々な差異があることが分かっている。
4.2及び4.3は,品質マネジメントシステム規格のリスクに関連する要求事項に密接に関連している。医
療機器を上市する場合に,(その医療機器が特に除外されていない限り)品質マネジメントシステムが要求
される国もある。製造業者が,品質マネジメントシステムを適用するか否かを選択することができる国も
ある。しかし,4.2及び4.3の要求事項は,製造業者が品質マネジメントシステムの他の要素を適用するか
否かにかかわらず,効果的なリスクマネジメントプロセスにおいて常に必要とされる。
A.2.4.2 経営者の責任
トップマネジメントによるコミットメントが,効果的なリスクマネジメントプロセスには重要である。
各トップマネジメントは,リスクマネジメントプロセスの全体的指針に対して責任があり,この細分箇条
では,彼らの役割,特に次について強調している。
− 十分な資源を確保しなければ,この規格の他の要求事項に適合してもリスクマネジメント活動の効果
は低い。
− リスクマネジメントは,専門的な分野であるので,リスクマネジメント技法の訓練を受けた力量のあ
る人員の参加が必要である(A.2.4.3参照)。
− この規格は,受容可能なリスクレベルを規定していないため,トップマネジメントは,受容可能なリ
スクをどのように決定するかの方針を確立することが要求されている。
− リスクマネジメントは絶えず進化していくプロセスであるため,それが適切に実施されているかどう
かを確認し,あらゆる課題点を修正し,改善を行い,さらに,変化に適応するためには,リスクマネ
ジメント活動についての定期的なレビューを行う必要がある。
A.2.4.3 要員の力量
リスクマネジメント活動の実施に必要な知識及び経験をもつ力量のある人を確保することは最も重要
である。リスクマネジメントプロセスには,次のような領域の知識及び経験をもつ人が要求されている。
− 医療機器は,どう構成されているか。
− 医療機器は,どのように作動するか。
− 医療機器は,どのように製造されるか。
− 医療機器は,実際どのように使用されるか。
− どのようにリスクマネジメントプロセスを適用するか。
一般には,様々な職務,又は専門分野の多くの代表者が,それぞれの専門知識を生かしてこれにあたる
ことが要求される。その代表者間のバランス及び関係を考慮することが望ましい。
力量に対する客観的証拠を示すためには,記録が要求される。重複を避けるため,また,機密性及びデ
22
T 14971:2020 (ISO 14971:2019)
ータ保護を理由として,この規格は,その記録をリスクマネジメントファイルで維持することを要求して
いない。
A.2.4.4 リスクマネジメント計画
リスクマネジメント計画は,次の理由から要求されている。
− 適切なリスクマネジメントには,組織的なアプローチが不可欠であるため
− 計画では,リスクマネジメントのロードマップ(工程表)を規定するため
− 計画は,客観性を促進し,不可欠な要素の見落としを防ぐのに役立つため
4.4のa)〜g)は,次の理由から必要である。
a) 計画の適用範囲には,二つの異なる要素がある。第1に医療機器を特定することであり,第2に計画
の各要素がライフサイクルのどの段階で適用可能かを特定することである。適用範囲を明確にするこ
とで,製造業者は,リスクマネジメント活動を具現化していく上での基礎ができるようになる。
b) 責任及び権限の割当ては,責任の所在が不明確になるのを防ぐために必要である。
c) リスクマネジメント活動のレビューは,一般的にはマネジメントの責任と考えられている。
d) リスクの受容可能性についての判断基準は,リスクマネジメントの基本となるので,リスク分析を開
始する前に決定することが望ましい。これは,箇条6のリスク評価を客観的に行うのに役立つ。
e) 全てのリスクコントロール手段を実施した後,製造業者は全ての残留リスクを合わせた全体的影響を
評価することが要求される。全体的な残留リスクの評価方法及び受容可能性の判断基準は,この評価
を実施する前に決定していることが望ましい。これは,箇条8の全体的な残留リスクの評価を客観的
に行うのに役立つ。
f)
検証は重要な活動であり,7.2で要求されている。この活動を計画することは,必要に応じて不可欠な
資源を利用可能にすることを確実にするために役立つ。検証が計画されない場合は,検証の重要部分
が欠落する可能性がある。
g) 製造及び製造後情報をリスクマネジメントプロセスにフィードバックするための正式かつ適切な方
法を確保するために,製造及び製造後情報を収集し,レビューする方法を確立する必要がある。
変更の記録を維持するための要求事項は,医療機器についてのリスクマネジメントプロセスの監査及び
レビューを容易にする。
A.2.4.5 リスクマネジメントファイル
この規格では,リスクマネジメントに適用する全ての記録及び他の文書を製造業者がファイルする,又
はこれらがどこにあるかの場所を特定するために,リスクマネジメントファイルという用語を用いた。こ
れは,リスクマネジメントプロセスの実践を容易にし,また,この規格に対してより効率的な監査を可能
にする。トレーサビリティは,特定した各ハザードにリスクマネジメントプロセスが適用されたことを明
らかにするために必要である。
確実に完了することは,リスクマネジメントにおいて非常に重要である。作業が不十分だと,特定した
ハザードがコントロールされず,その結果として危害が発生する可能性がある。このような問題は,リス
クマネジメントのあらゆる段階における不十分な活動の結果として生じる可能性がある。不十分な活動と
して,次がある。ハザードを特定しきれていない,正しくアセスメントされていないリスクがある,リス
クコントロール手段を特定しきれていない,リスクコントロール手段が未実施又はリスクコントロール手
段の効果があることを確認していない。トレーサビリティは,リスクマネジメントプロセスを確実に完了
23
T 14971:2020 (ISO 14971:2019)
するために必要とされる。
A.2.5 リスク分析
A.2.5.1 リスク分析プロセス
5.1の注記1は,類似の医療機器についてのリスク分析が利用可能な場合に,どのように扱うかを示し
ている。既に十分な情報が存在する場合は,時間,取組及び資源を節約するために,その情報を適用する
ことが可能である。しかし,この規格の利用者は,過去のリスク分析の結果が現在のリスク分析に適用で
きるかを体系的に評価するよう留意する必要がある。
5.1のa)〜c)で要求する事項は,トレーサビリティを保証するための基本的な最小限のデータであり,マ
ネジメントレビュー及び後の監査に重要である。c)の要求事項は,分析の適用範囲の内容を明確にし,確
実に完了したことを検証するためにも役立つ。
A.2.5.2 意図する使用及び合理的に予見可能な誤使用
医療機器の意図する使用は,リスク分析の重要な側面であり,開始点である。意図する使用には,適切
な場合,3.6の注釈1に列挙した要素を含めることが望ましい。製造業者は,医療機器の意図したユーザー
を考慮に入れることが望ましい。例えば,専門家でない人が医療機器を使うのか,トレーニングを受けた
医療従事者が使用することになるのかなどである。この分析は,医療機器の製造業者が意図した状況以外
での使用,及び初期構想時に予見した状況以外での使用も考慮することが望ましい。その医療機器の潜在
的な使用に起因するハザード及び合理的に予見可能な誤使用が将来起こり得ることを,製造業者が予見す
ることが重要である。
A.2.5.3 安全に関する特質の明確化
この段階において,製造業者は医療機器の安全に影響する可能性のある全ての特質を考慮する必要があ
る。この特質は,定性的でも定量的でもよく,医療機器の動作原理,意図する使用及び/又は合理的に予
見可能な誤使用に関連する。この特質は,医療機器の性能又は動作原理,医療機器の測定機能又は無菌性,
患者に接触する部分に使われる材料,診断又は治療の目的で放射線を使用すること,その他にも関係する
可能性がある。該当する場合,これらの特質の限度値を同様に検討する必要がある。なぜならば,医療機
器の操作及び/又は安全は,限度値を超えたときに影響を受けるからである。
A.2.5.4 ハザード及び危険状態の特定
この段階において,製造業者は正常状態及び故障状態において予見されるハザードを体系的に特定する
必要がある。この特定は,5.2で明確化した意図する使用及び合理的に予見可能な誤使用並びに5.3で明確
化した安全に関する特質に基づいて行うことが望ましい。
リスクのアセスメント及びマネジメントは,危険状態を特定して初めて可能となる。ハザードを危険状
態に変え得る事象について,合理的に予見可能な一連の事象を文書化することによって,リスクの評価及
びマネジメントを体系的に実施可能である。附属書Cは,製造業者がハザード及び危険状態を特定する際
の手助けとなることを目的としている。典型的なハザードを列挙し,ハザード,予見可能な一連の事象,
危険状態,及びそれに伴う可能性がある危害の間の関連性を説明している。
A.2.5.5 リスク推定
これは,リスク分析の最終段階である。個々の医療機器についてと同様に,調査対象の個々の危険状態
24
T 14971:2020 (ISO 14971:2019)
に対するリスク推定は異なるという難しさのため,この細分箇条は,一般的に記載している。医療機器が
正常に機能している場合にも,また,不具合を生じている場合にも,危険状態が発生し得るため,いずれ
の状況も厳密に調査することが望ましい。実際には,リスクの構成要素である危害の発生確率及び重大さ
の両方について個別に分析することが望ましい。製造業者が危害の重大さレベル又は発生確率を体系的な
方法で分類する場合は,その分類表を定義し,リスクマネジメントファイルに記録することが望ましい。
これは製造業者が,同じレベルをもつリスクを整合的に扱うことを可能とし,製造業者がそのように実施
したという証拠になる。
系統的な故障又は一連の事象が原因となって発生する危険状態もある。系統的な故障の確率を計算する
ための広く認められた方法はない。危害の発生確率が計算可能でない場合でも,ハザードには対処する必
要があり,製造業者は,危険状態のリストを個別に作成することによって,これら危険状態によるリスク
の低減が図れることになる。
特に全く新規の医療機器を開発する際又はセキュリティリスクに対しては,十分な定量的データが利用
可能でない場合も多い。したがって,リスク推定は必ず定量的に行うことが望ましいという表現は避けた。
A.2.6 リスク評価
リスクの受容可能性について決定することが必要である。製造業者は,リスクマネジメント計画で定義
したリスクの受容可能性についての判断基準によって,推定したリスクを評価する。製造業者は,どのリ
スクをコントロールする必要があるかを判断するためにリスクを調査することが可能である。箇条6は,
この規格の利用者が不必要な作業を行わなくてよいよう規定した。
A.2.7 リスクコントロール
A.2.7.1 リスクコントロール手段の選択
リスクを低減する方法は,多くの場合,複数ある。三つの手段を列挙しているが,これらは,いずれも
標準的なリスク低減手段であり,JIS T 0063:2020[2]から抜粋した。列挙した優先順位は重要である。この
原則は,IEC TR 60513 [11]でも採用されている。設計及び製造による本質的な安全は,リスクコントロー
ル手段の選択においては,最初に検討すべき最も重要な選択肢である。なぜならば,医療機器の特性に本
来備わるような設計による解決策は,継続して有効である可能性があるが,それに対して,ガード及び保
護手段はうまく設計しても故障する又は破壊される可能性があり,安全に関する情報に従わないことがあ
ることが経験上示されているからである。実施可能な場合,医療機器は,本質的に安全であるように設計
及び製造されることが望ましい。これが実施可能でない場合には,防壁(バリア)又はアラームのような
保護手段が適切である。第三の選択肢は,文書による警告又は禁忌などの安全に関する情報を提供するこ
とである。ユーザートレーニングは,安全に関する情報を提供する上で重要な意味をもつ可能性がある。
製造業者は,意図するユーザーに対して必須のトレーニングを提供することが検討可能である。
製造プロセスは,例えば,部品の汚染,プロセスで使用する有害物質の残留又は部品の取り違えなどに
起因するリスクにつながることがある。そうしたリスクは,製造プロセスを本質的に安全に設計すること
(例えば,有害物質を使うのをやめる又は別の製造ラインを用いる)又は保護手段をとること(例えば,
プロセス内の目視検査)によってコントロール可能である。
リスクコントロール手段の選択の結果の一つとして,事前に決めたリスク受容可能性についての判断基
準に従ってリスクを受容可能なレベルまで低減するための実施可能な方法がない場合がある。例えば,全
ての残留リスクが受容可能であるような生命維持医療機器の設計は,現実的ではないかもしれない。この
場合は,患者に対する医療機器のベネフィットが残留リスクを上回るかどうかを判断するため,7.4に記
25
T 14971:2020 (ISO 14971:2019)
載するベネフィット・リスク分析が実施可能である。事前に決めた受容可能なレベルまでリスク低減する
ため最初にあらゆる手段を施すことを確実にするため,この選択に関して7.1に含めている。
A.2.7.2 リスクコントロール手段の実施
二つの異なる検証がある。第1の検証は,医療機器の最終設計又は製造プロセスにおいてリスクコント
ロール手段が実施されたことを確認するために要求される。第2の検証は,実施したリスクコントロール
手段(安全に関する情報を含む)が実際にリスクを低減していることを確実にするためである。場合によ
っては,バリデーションを,リスクコントロール手段の効果を検証するために用いることが可能である。
リスク推定の十分なデータ及び情報を得ることは難しく,結果として残留リスクの評価の不確かさにつ
ながる。そのため,残留リスクの評価が納得できるものになるように,製造業者は,リスクコントロール
手段の有効性の検証に労力を集中することが実際的である。労力のレベルはリスクのレベルに見合ったも
のとすることが望ましい。ユーザーに対する試験が,リスクコントロールの有効性を検証するために必要
になるかもしれない。例えば,ユーザビリティ試験(JIS T 62366-1 [13]参照),医療機器の臨床試験(ISO
14155[6]参照),体外診断用医療機器の臨床性能試験(ISO 20916[8]参照)などである。ユーザビリティ試
験によって安全に関する情報の有効性が検証可能である。試験規格に従って試験をすることで,例えば,
機械的強度などに関する,設計したリスクコントロール手段の有効性が検証可能である。
A.2.7.3 残留リスクの評価
残留リスクの評価は,実施したリスクコントロール手段によってリスクが受容可能になったかどうかを
判断するために導入された。リスクがリスクマネジメント計画で確立した受容可能性の判断基準を超える
場合には,製造業者は,追加のリスクコントロール手段を調査するように指示される。それ以上のリスク
コントロールが現実的でなく,残留リスクがリスクマネジメント計画で確立した受容可能性の判断基準を
超えることがなくなるまで,残留リスクの評価を繰り返し行うことが望ましい。
A.2.7.4 ベネフィット・リスク分析
リスクが製造業者のリスク受容可能性の判断基準を超えるような,特定の危険状態が発生する場合もあ
る。この細分箇条は,製造業者が慎重な評価を実施し,かつ,医療機器のベネフィットが残留リスクを上
回ることが示せた場合には,判断基準を上回る高い残留リスクがあっても医療機器を提供可能にするもの
である。しかし,この細分箇条は,経済的利点又はビジネス上の利点が残留リスクを上回るかの判断(す
なわち,ビジネス上の意思決定)には使用可能ではない。
A.2.7.5 リスクコントロール手段によって発生したリスク
リスクコントロール手段を単独で用いるか,又は組み合わせることによって,新たな全く異なるハザー
ド又は危険状態が発生する可能性があること,さらに,あるリスクを低減するために導入したリスクコン
トロール手段が別のリスクを増加させる可能性があるため,この細分箇条を設けた。
A.2.7.6 リスクコントロールの完了
この時点では,特定した全ての危険状態についてのリスク評価が終了していることが望ましい。複雑な
リスク分析において,全ての危険状態に関係したリスクを検討したかを確認するようにするためである。
A.2.8 全体的な残留リスクの評価
箇条5〜箇条7で規定したプロセスに従い,製造業者は,ハザード及び危険状態を特定し,リスクを評
26
T 14971:2020 (ISO 14971:2019)
価し,医療機器の設計において個々のリスクコントロール手段を実施する。製造業者は,この時点で振り
返り,残留リスク全ての組合せによる影響を検討し,医療機器の開発を続けるか否かの決定を行う。個々
の残留リスクは受容可能であっても,全体的な残留リスクが製造業者のリスク受容可能性の判断基準を上
回る可能性がある。これは,多数のリスクを伴う,複雑なシステム及び医療機器の場合に特に当てはまる。
リスクマネジメント計画で定めた,全体的な残留リスクを評価する方法には,全体的な残留リスクと医療
機器のベネフィットとのバランスをとることが含まれる。これは,リスクは高いが極めて有益な医療機器
を商品化してもよいかどうかを判断する場合に特に関係する。
製造業者は,重要な残留リスクに関連のある情報をユーザーに提供する責任があり,これによってユー
ザーは医療機器の使用について,情報に基づいて判断することが可能になる。したがって,製造業者は,
附属資料に残留リスクに関連する情報を含めることを指示されている。ただし,どの残留リスクに関する
情報を,どれくらい提供すべきかについては,製造業者が決定する。この要求事項は,多くの国及び地域
で採用されているアプローチと一致している。
A.2.9 リスクマネジメントのレビュー
リスクマネジメントのレビューは,医療機器の市場出荷前の重要なステップである。リスクマネジメン
ト計画を実行することによって得られる,リスクマネジメントプロセスの最終的な結果をレビューする。
リスクマネジメント報告書は,このレビューの結果を含み,リスクマネジメントファイルの重要な部分で
ある。この報告書は,リスクマネジメント計画が適切に実行され,製造業者が,要求された目的を達成し
たことを確認した証拠となる重要な文書である。リスクマネジメント計画を実行した後に行うレビュー及
びリスクマネジメント報告書の更新が,医療機器のライフサイクルを通して,製造及び製造後の活動の実
行結果として必要となることがある。
A.2.10 製造及び製造後の活動
医療機器が製造段階に入ってもリスクマネジメントを継続することが必要である。リスクマネジメント
は,医療機器の試作品がない,構想の段階で開始することがしばしばある。製造業者は,類似の医療機器
及び類似の技術に関する経験を含む,多くの情報源からの情報を収集する。設計プロセスを実施していく
中でリスク推定の精度が高まり,実際に機能する試作品ができると,更に正確になっていく。しかし,実
際のユーザーがどう医療機器を使うかは推定しきれない。
したがって,製造業者は,製造及び製造後情報を収集し,レビューして,その安全との関係を評価する
必要がある。その情報は,新たなハザード又は危険状態に関連することがあり,リスク推定又はベネフィ
ットと全体的な残留リスクとのバランスに影響することがある。いずれの情報も製造業者のリスクマネジ
メントにおける決定に影響することがある。製造業者は,新規又は改正された規格を含む,一般に認めら
れた最新の技術水準についても考慮することが望ましい。情報が安全に関連すると判断した場合は,リス
クマネジメントプロセスでは,その情報を医療機器の変更のインプットとして,及びプロセス自体を改善
するためのインプットとしても検討する必要がある。効果的な製造及び製造後の活動によって,リスクマ
ネジメントプロセスは,医療機器が継続的に安全であることを確実にするための,真に反復的なクローズ
ドループのプロセスとなる。
フィードバック,追加指針を求める要求及び規制要求事項の変化に応え,JIS T 14971:2020では製造及
び製造後の活動に関する要求事項を詳細に規定している。この箇条は,複数の細分箇条に分割した。一般
に認められた最新の技術水準についての情報及びサプライチェーンからのフィードバックを含む,情報源
を追加して列挙している。サプライチェーンには,部品又はサブシステムのほかにサードパーティ製ソフ
トウェアの供給者も含む。既に市場にある医療機器に関する処置の必要性の有無がより明確にされている。
27
T 14971:2020 (ISO 14971:2019)
フォローアップの処置を検討する必要がある状況は,安全に関係する可能性のある最新の技術水準の変化
に伴って拡張される。例えば,新しい医療機器及び/又は治療方法が市場で利用可能になる,リスクの認
知又はリスクの受容可能性に変化がある,などである。

28
T 14971:2020 (ISO 14971:2019)
附属書B
(参考)
医療機器のリスクマネジメントプロセス
B.1
第2版と第3版との対応
JIS T 14971:2020では,箇条及び細分箇条の番号付けを変更した。表B.1にJIS T 14971:2012(第2版)
とJIS T 14971:2020(第3版)との箇条及び細分箇条の対応を示す。この表は,この規格の利用者が第2版
から第3版へ移行することを支援し,この規格を参照している他の規格の更新を容易にするために用意し
た。
表B.1−JIS T 14971:2012及びJIS T 14971:2020の要素の対応
JIS T 14971:2012
JIS T 14971:2020
序文
序文
1
適用範囲
1
適用範囲
(新しい箇条)
2
引用規格
2
用語及び定義
3
用語及び定義
2.1
附属文書
3.1
附属資料
(新しい用語)
3.2
ベネフィット
2.2
危害
3.3
危害
2.3
ハザード
3.4
ハザード
2.4
危険状態
3.5
危険状態
2.5
意図する使用(意図する目的)
3.6
意図する使用,意図する目的
2.6
体外診断用医療機器
3.7
体外診断用医療機器
2.7
ライフサイクル
3.8
ライフサイクル
2.8
製造業者
3.9
製造業者
2.9
医療機器
3.10
医療機器
2.10
客観的証拠
3.11
客観的証拠
2.11
製造後
3.12
製造後
2.12
手順
3.13
手順
2.13
プロセス
3.14
プロセス
(新しい用語)
3.15
合理的に予見可能な誤使用
2.14
記録
3.16
記録
2.15
残留リスク
3.17
残留リスク
2.16
リスク
3.18
リスク
2.17
リスク分析
3.19
リスク分析
2.18
リスクアセスメント
3.20
リスクアセスメント
2.19
リスクコントロール
3.21
リスクコントロール
2.20
リスク推定
3.22
リスク推定
2.21
リスク評価
3.23
リスク評価
2.22
リスクマネジメント
3.24
リスクマネジメント
2.23
リスクマネジメントファイル
3.25
リスクマネジメントファイル
2.24
安全
3.26
安全
2.25
重大さ
3.27
重大さ
(新しい用語)
3.28
最新の技術水準
2.26
トップマネジメント
3.29
トップマネジメント
2.27
誤使用
3.30
使用エラー

29
T 14971:2020 (ISO 14971:2019)
表B.1−JIS T 14971:2012及びJIS T 14971:2020の要素の対応(続き)
JIS T 14971:2012
JIS T 14971:2020
2.28
検証
3.31
検証
3
リスクマネジメントの一般要求事項
4
リスクマネジメントシステムの一般要求事項
3.1
リスクマネジメントプロセス
4.1
リスクマネジメントプロセス
3.2
経営者の責任
4.2
経営者の責任
3.3
要員の資格認定
4.3
要員の力量
3.4
リスクマネジメント計画
4.4
リスクマネジメント計画
3.5
リスクマネジメントファイル
4.5
リスクマネジメントファイル
4
リスク分析
5
リスク分析
4.1
リスク分析プロセス
5.1
リスク分析プロセス
4.2
意図する使用及び医療機器の安全に関する
特質の明確化
5.2
意図する使用及び合理的に予見可能な誤使用
5.3
安全に関する特質の明確化
4.3
ハザードの特定
5.4
ハザード及び危険状態の特定
4.4
個々の危険状態に対するリスクの推定
5.5
リスク推定
5
リスク評価
6
リスク評価
6
リスクコントロール
7
リスクコントロール
6.1
リスクの低減
(削除)
6.2
リスクコントロール手段の選択
7.1
リスクコントロール手段の選択
6.3
リスクコントロール手段の実施
7.2
リスクコントロール手段の実施
6.4
残留リスクの評価
7.3
残留リスクの評価
6.5
リスク/効用 分析
7.4
ベネフィット・リスク分析
6.6
リスクコントロール手段によって発生した
リスク
7.5
リスクコントロール手段によって発生した
リスク
6.7
リスクコントロールの完了
7.6
リスクコントロールの完了
7
残留リスクの全体的な受容可能性の評価
8
全体的な残留リスクの評価
8
リスクマネジメント報告書
9
リスクマネジメントのレビュー
9
製造及び製造後情報
10
製造及び製造後の活動
10.1
一般
10.2
情報の収集
10.3
情報のレビュー
10.4
処置
附属書A 指針及び根拠
附属書A 要求事項の根拠
附属書B 医療機器についてのリスクマネジメントプ
ロセスの概要
附属書B 医療機器のリスクマネジメントプロセス
附属書C 安全に影響する医療機器の特質を明確化す
るために使用できる質問事項
TR T 24971に移動
附属書D 医療機器に適用するリスクの概念
附属書E ハザード,予見可能な一連の事象及び危険状
態の例
附属書C リスクの基礎的な概念
附属書F リスクマネジメント計画
TR T 24971に移動
附属書G リスクマネジメント手法に関する情報
附属書H 体外診断用医療機器に関するリスクマネジ
メントの指針
附属書I
生物学的なハザードに関するリスク分析プ
ロセスの指針
(削除)
附属書J 安全に関する情報及び残留リスクについて
の情報
TR T 24971に移動
参考文献
参考文献
30
T 14971:2020 (ISO 14971:2019)
B.2
リスクマネジメントプロセスの概要
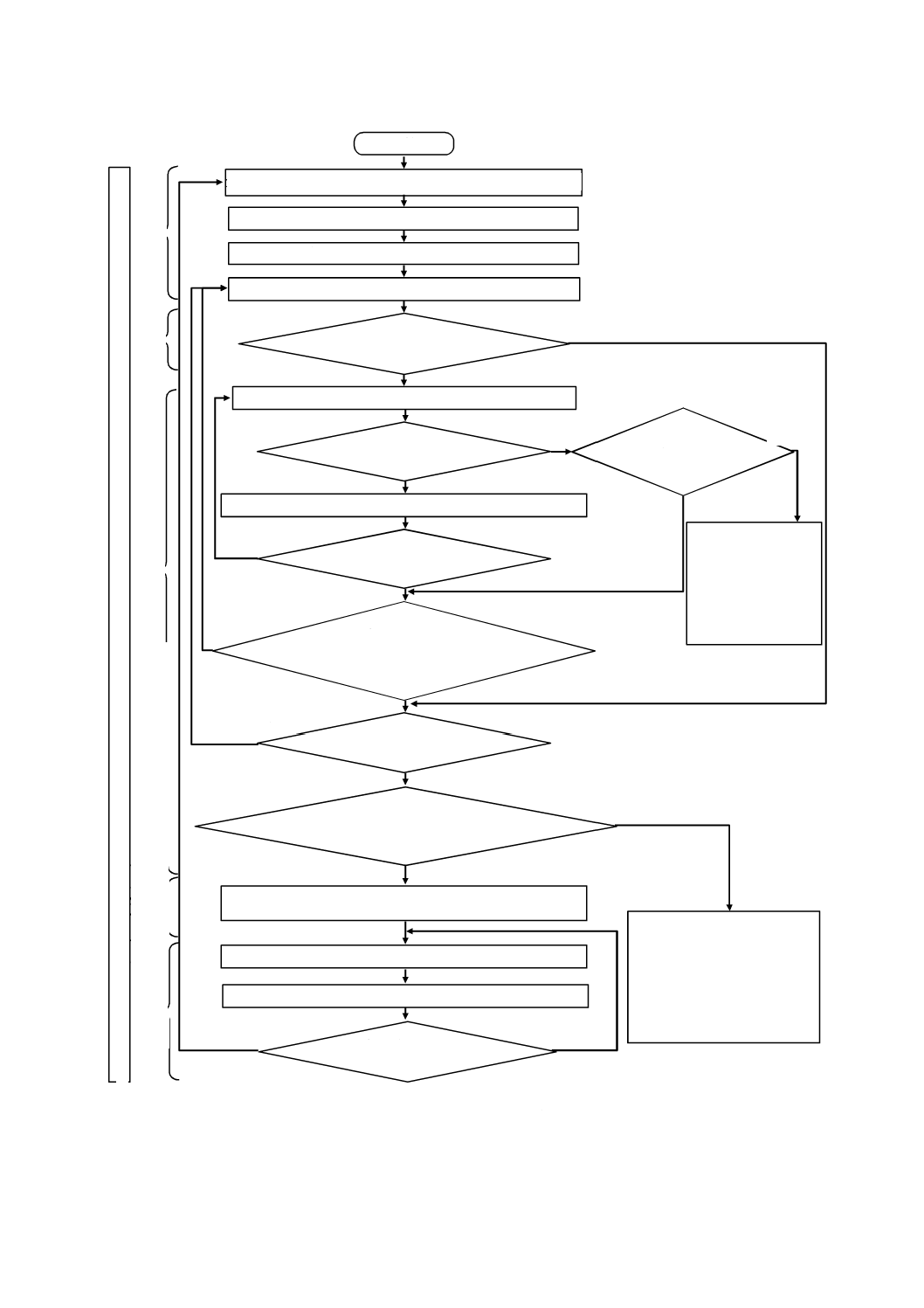
図B.1は,この規格の利用者に対し,リスクマネジメントプロセスの概要を例示するための図である。
この図は,解説目的のためだけのものである。図B.1に示すとおり,個々のリスクについてプロセスは反
復的であり,かつ,リスクコントロール手段が新たなハザード又は危険状態を発生した場合,又は新たな
情報が得られた場合に初期のステップに戻ることを示している。
31
T 14971:2020 (ISO 14971:2019)
図B.1−医療機器に適用するリスクマネジメント活動の概要
開始
意図する使用及び合理的に予見可能な誤使用を特定する(5.2)
安全に関する特質を明確化する(5.3)
ハザード及び危険状態を特定する(5.4)
製造及び製造後情報をレビューする(10.3)
リ
ス
ク
マ
ネ
ジ
メ
ン
ト
計
画
(
4
.4
)
リスクマネジメント計画の実行をレビューし,
リスクマネジメント報告書を作成する(箇条9)
製造及び製造後情報を収集する(10.2)
各危険状態に対するリスクを推定する(5.5)
リスクコントロールは必要か
(箇条6)
適切なリスクコントロール手段を選択する(7.1)
選択したリスクコントロール手段を実施して検証する(7.2)
リスクコントロールは
実施可能か(7.1)
ベネフィットが
リスクを上回るか
(7.4)
残留リスクは受容可能か
(7.3)
新たなハザード又は危険状態が発
生するか,又は既に特定したリス
クの評価に影響するか(7.5)
全ての特定した危険状態
が検討されたか(7.6)
ベネフィットに関連して,全体的な
残留リスクは,受容可能か(箇条8)
リスクの再アセスメント
が必要か(10.4)
製造業者は,医療機器
又は意図する使用を変
更すること(5.2に戻
る)を検討してもよい。
そうでない場合は,リ
スクは受容できないも
のとして残る。
製造業者は,追加のリスクコント
ロール手段を実施すること(7.1
に戻る),又は医療機器若しくは
その意図する使用を変更するこ
と(5.2に戻る)を検討してもよ
い。そうでない場合は,全体的な
残留リスクは受容できないもの
として残る。
リ
ス
ク
分
析
リ
ス
ク
評
価
リ
ス
ク
コ
ン
ト
ロ
ー
ル
全
体
的
な
残
留
リ
ス
ク
の
評
価
製
造
及
び
製
造
後
の
活
動
はい
いいえ
いいえ
いいえ
いいえ
いいえ
いいえ
いいえ
いいえ
はい
はい
はい
はい
はい
はい
リ
ス
ク
マ
ネ
ジ
メ
ン
ト
の
レ
ビ
ュ
ー
はい
32
T 14971:2020 (ISO 14971:2019)
附属書C
(参考)
リスクの基礎的な概念
C.1 一般
この規格は,製造業者に対して,医療機器の正常状態及び故障状態の両方について,医療機器に関連す
る既知及び予見可能なハザードのリストを作成し,危険状態及び危害の原因になると予見可能な一連の事
象の検討を要求している。定義によれば,一連の事象又はその他の周囲の状況(通常使用を含む。)によっ
て危険状態が生じない限り,ハザードが危害に至ることはない。危害に至る時点では,発生する可能性の
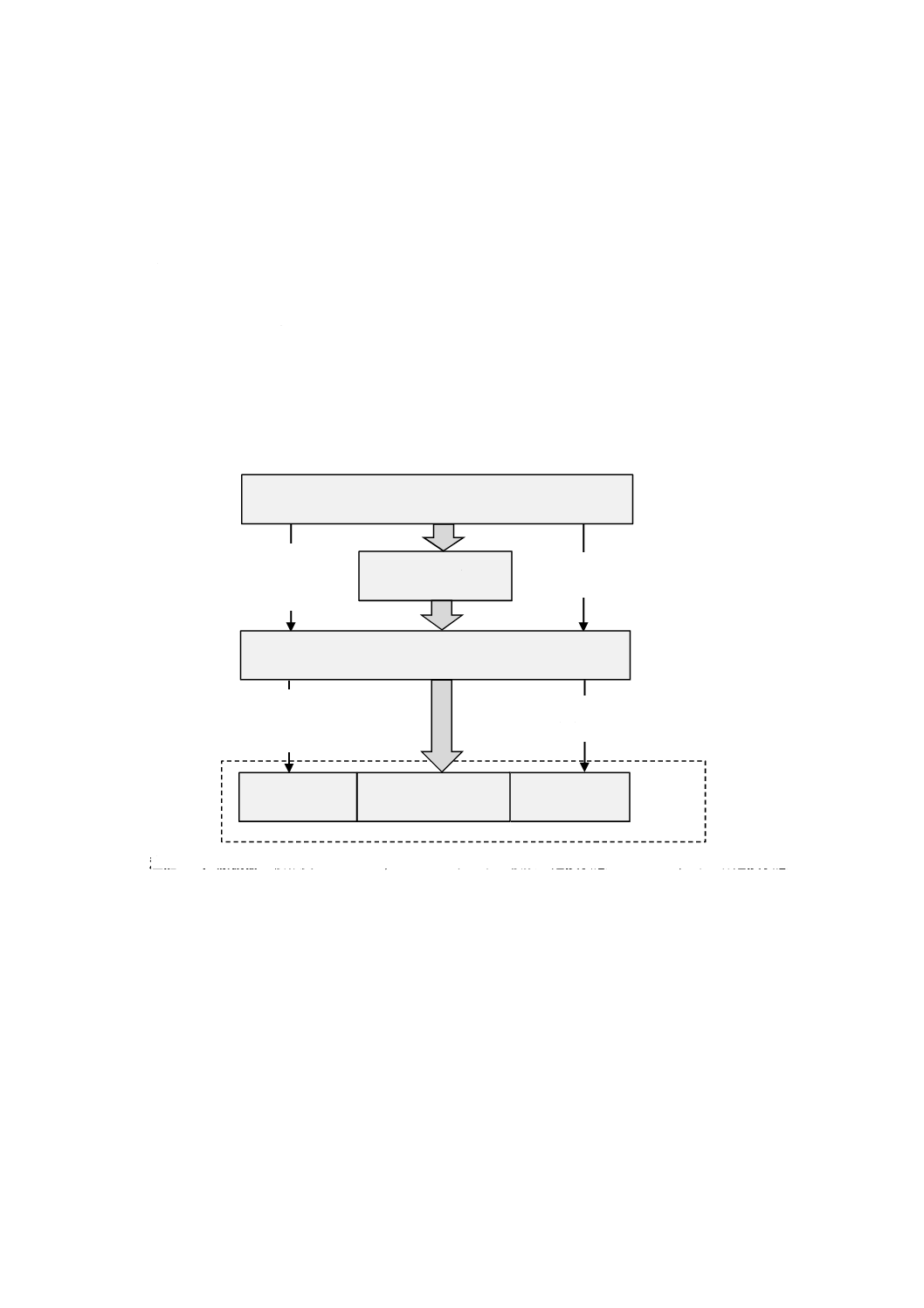
ある危害の重大さと発生確率を推定することによってリスクがアセスメント可能である(図C.1を参照)。
危害の発生確率は,二つの確率(P1,P2)の組合せとして表すことも可能であり,一つの確率(P)で表す
ことも可能である。P1及びP2に分解することは必須ではない。
注記1 医療機器の複雑度によっては,一つのハザードが複数の危険状態に至ったり,その各危険状態
が複数の危害に至ったりすることがある。
注記2 危害の発生確率(P)は,独立した確率P1及びP2の組合せとすることが可能である。
注記3 細い矢印はリスク分析の要素を表し,太い矢印はハザードがどのように危害に至るかを示す。
図C.1−ハザード,一連の事象,危険状態及び危害の関係の図解例(JIS T 0063:2020[2]による)
ハザードのリスト作成は,同じか又は類似の医療機器に関連する経験をレビューすることから始めるの
が適切である。このレビューでは,自社の経験,適切な場合は,有害事象データベースで報告されている
他社の経験,刊行物,科学文献,及びその他利用可能な情報源も考慮することが望ましい。医療機器に典
型的なハザード,危険状態及びそれに起因して発生し得る危害を特定しリストを作成する上で,このレビ
ューは特に有用である。次に,作成したリスト及び表C.1の事例リストなどの補助ツールによって,初期
のハザードリストを作成可能である。
ハザードとともに危険状態及び危害の発生に至る一連の事象の幾つかは,この時点で特定が開始可能で
リスク
ハザード
危険状態
一連の事象
危険状態が
起こる確率
(P1)
危険状態が危害に
至る確率
(P2)
重大さに影響を
与える状況
危害の発生確率
(P=P1×P2)
危害の重大さ
危害
重大さに影響を
与える状況
33
T 14971:2020 (ISO 14971:2019)
ある。ハザードの多くは危害には至らない可能性があるため,更なる検討対象から外せる場合がある。し
たがって,医療機器に関連して発生し得る危害の分析から始めて,そこから危険状態,ハザード,きっか
けとなる原因へと遡る方法が効果的である。このアプローチは上記理由で有効であるが,完全な分析では
ないことを認識するのが望ましい。(TR T 24971[9]に記載されているような)リスク分析技法などを系統
立てて使用することによってだけ,一連の事象の多くは特定可能となる。表C.2に示すように,考慮すべ
き多くの事象及び周囲の状況によって,分析及び特定は更に複雑となる。したがって,包括的な分析を実
施するには,二つ以上のリスク分析技法,特に補完的な技法がしばしば使用される。表C.3にハザード,
一連の事象,危険状態及び危害の関係の例を示す。
リスクコントロールを容易にするため,設計・開発プロセスのできるだけ早い段階でハザード,危険状
態,及び一連の事象のリストの作成を完了しておくことが望ましいが,特定及びリストの作成は,実際の
運用では製造後から廃棄に至る医療機器のライフサイクルにおける継続的な活動である。
この附属書では,種々の医療機器に関連するハザードのリスト(表C.1)を示すが,これは全てを網羅
しているわけではない。また,事象及び周囲の状況で,危険状態を生じ結果的に危害に至る事例のリスト
(表C.2)を示す。表C.3は,一連の事象又は周囲の状況によってハザードがどのようにして危険状態と
なり,危害に至るかの過程の例を示す。
ハザードが危険状態に至る過程を認識するのは,結果的に生じる危害の発生確率及び重大さを推定する
上で不可欠である。このプロセスは,包括的に危険状態を網羅することが目的である。ハザード及び一連
の事象を認識するのはこのための足掛かりとなる。この附属書の各表のリストは,危険状態を特定する手
助けとして使用可能である。何をハザードとするかについては,個別の分析目的に合うように製造業者が
決定する必要がある。
C.2 ハザードの例
表C.1のリストは,個別の医療機器に関連して,最終的に危害を生じさせるハザードを特定する手助け
として使用可能である。

34
T 14971:2020 (ISO 14971:2019)
表C.1−ハザードの例
エネルギーに関連するハザード
生物学的及び化学的なハザード
性能に関連するハザード
音響エネルギー
− 低周波音
− 音圧
− 超音波
電気エネルギー
電界
漏れ電流
− 接地漏れ電流
− 接触電流
磁界
静電気
電圧
機械的エネルギー
運動エネルギー
− 物体の落下
− 高圧液体流入
− 動く部分
− 振動する部分
位置(蓄積)エネルギー
− 曲げ
− 圧縮
− 切断,せん断
− 引力
− 懸垂物体
− 張力
− ねじれ
放射線エネルギー
電離放射線
− 加速粒子(アルファ粒子,電
子,陽子,中性子)
− ガンマ線
− X線
非電離放射線
− 赤外線
− レーザー
− マイクロ波
− 紫外線熱エネルギー
低温効果
温熱効果
生物学的因子
細菌
菌類
寄生生物
プリオン
毒素
ウイルス
化学的因子
発がん性,変異原性,生殖性
腐食性(caustic),腐食性(corrosive)
− 酸
− アルカリ
− 酸化剤
引火性,可燃性,爆発物
煙,蒸気
浸透性
粒子(微小粒子,ナノ粒子を含む)
発熱性
溶剤
中毒性
− アスベスト
− 重金属
− 無機毒性物質
− 有機毒性物質
− シリカ
免疫学的因子
アレルギー性
− 防腐剤
− ラテックス
免疫抑制剤
刺激物
− 洗浄時の残留物
感作物
データ
− アクセス
− 可用性
− 機密性
− 転送
− 完全性
デリバリー
− 量
− 速度
診断情報
− 検査結果
− 画像のアーチファクト
− 画像の向き
− 画像の解像度
− 患者の識別・患者情報
− 機能性
− アラーム
− 重要な性能
− 測定
C.3 事象及び周囲の状況の例
予見可能な一連の事象を特定するためには,事象及び周囲の状況についての検討が有効である場合が多
い。表C.2は,事象及び周囲の状況の例を一般的な分類にまとめたものである。このリストは網羅的なも
のではないが,医療機器に関連して予見可能な一連の事象を特定する場合に,考慮する必要がある事象及
び周囲の状況を多岐にわたって示すことを意図している。
35
T 14971:2020 (ISO 14971:2019)
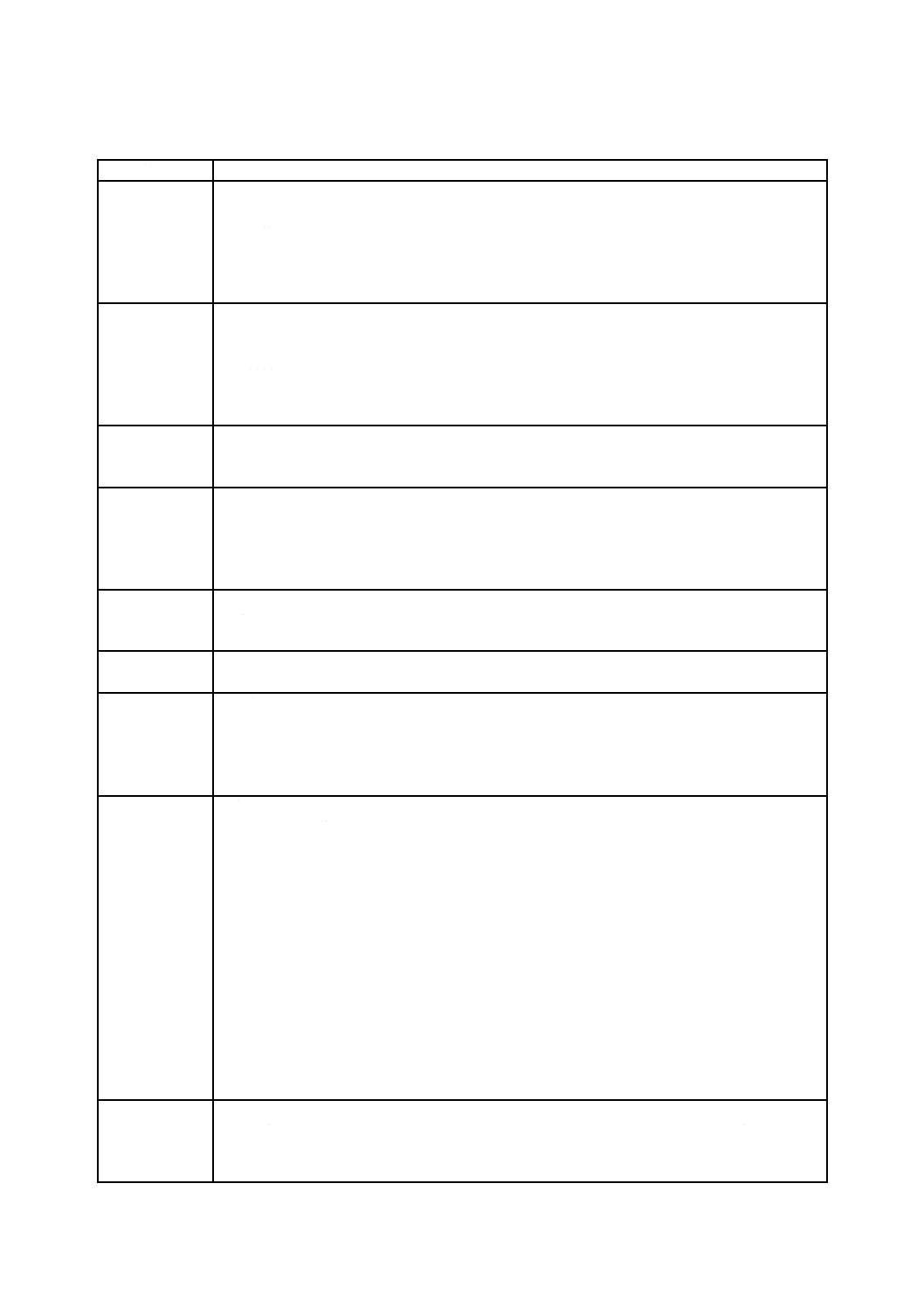
表C.2−事象及び周囲の状況の例
一般的な分類
事象及び周囲の状況の例
要求事項
次に関する不完全な仕様
− 設計のパラメーター
− 操作のパラメーター
− 性能の要求事項
− サービスに関する要求事項(例えば,保守,再処理)
− 製品寿命
製造プロセス
次に関する管理が不十分
− 製造プロセス
− 製造プロセスに対する変更
− 材料
− 材料の適合性に関する情報
− 下請負業者
輸送及び保管
不適切な包装
汚染又は劣化
不適切な環境条件
環境要因
物理的要因(例えば,熱,圧力,時間)
化学的要因(例えば,腐食,分解,汚染)
電磁場(例えば,電磁妨害による影響)
電力の不適切な供給
冷却材の不適切な供給
洗浄,消毒及び
滅菌
バリデーションされた手順が存在しない
不適切な仕様
洗浄,消毒及び滅菌の不適切な実施
廃棄及び解体
情報が提供されない,又は不適切な情報提供
使用エラー
組成
生分解性
生体適合性
情報が提供されない,又は提供された仕様が不適切
不正確な組成
使用エラー
ユーザビリティ
取扱説明書が分かりにくい,又はない。
制御システムが複雑又は分かりにくい。
医療機器の状態が紛らわしい,又は明確でない。
設定,計測値又はその他の情報の表示が紛らわしい,又は明確でない。
結果の誤表示
視認性,可聴性又は感触性が不十分
動作に対する制御の割当て,又は実際の状態に対する表示情報の割当てが不適切
既存の装置と比べ問題を引き起こしやすいモード又は配置
熟練していない者,又は訓練を受けていない者が使用
副作用に関する警告が不十分
単回使用医療機器を再使用した場合のハザードに関する警告が不適切
計測又はその他の計量が不正確
消耗品,附属品,その他の医療機器との不適合
患者の取り違え
うっかりミス,過失及び誤り
機能性
電気的又は機械的な完全性の喪失
老化,摩耗及び反復使用による性能の劣化(例えば,液体又はガス流路が徐々に閉塞する,流動
抵抗及び電気伝導度の変化)
経年変化,摩耗又は疲労故障による部品故障
36
T 14971:2020 (ISO 14971:2019)
表C.2−事象及び周囲の状況の例(続き)
一般的な分類
事象及び周囲の状況の例
セキュリティ
外部からアクセス可能な,セキュアでないデータポート(例えば,ネットワーク,シリアルポー
ト,USBポート)
暗号化されていないデータ
悪用可能なソフトウェアぜい(脆)弱性
真正性が確認されていないソフトウェアの更新
C.4 ハザード,予見可能な一連の事象,危険状態及び起こり得る危害の関係の事例
表C.3にハザード,予見可能な一連の事象,危険状態と危害との関係について簡略化した例を示す。一
つのハザードから複数の危害が発生することもあり,また,複数の一連の事象が併発することで,危険状
態が引き起こされる場合もあるので留意する。
実施する個別の分析に合わせて,何が危険状態であるかを決定する必要がある。場合によっては高電圧
端子のカバーが開いたままになっているのを危険状態とすることがよい場合もあるし,人が高電圧端子に
接触しているのを危険状態とすることがよい場合もある。
表C.3−ハザード,予見可能な一連の事象,危険状態と起こり得る危害との関連
ハザード
予見可能な一連の事象
危険状態
危害
電磁エネルギー
(高電圧)
(1) 電極ケーブルを意図せずに商用電源ソケットに接
続する。
商用電圧が電極上に生
じる。
重篤な熱傷
心臓の細動
化学物質
(揮発性溶剤,塞
栓)
(1) 製造中に使用した揮発性溶剤を完全に除去しきれ
ていない。
(2) 溶剤残留物が体温で気化する。
透析中に梗塞(血流中で
気泡が発生)が起こる。
ガス塞栓症
脳障害
生物学的
(微生物による
汚染)
(1) 再使用麻酔チューブの浄化に関する指示が不適切
である。
(2) 麻酔時に汚染したチューブを使用する。
麻酔時に患者の気道内
に細菌が付着する。
細菌感染
機能性
(投与されない)
(1) 静電気に帯電した患者が注入ポンプに触れる。
(2) 静電気放電(ESD)が原因となってポンプ及びポ
ンプアラームが故障する。
血糖値が上昇している
患者にインスリンが投
与されず,警報も作動し
ない。
軽微な臓器
障害
意識低下
機能性
(出力停止)
(1) 植込み型除細動器のバッテリーが寿命に達する。
(2) 臨床的な経過観察受診間隔が不適切に長い。
不整脈の発生時に除細
動器が作動しない。
死亡
測定
(不正確な情報)
(1) 測定エラー
(2) 測定エラーがユーザーによって検出されない。
医師に不正確な情報が
報告され,誤診する,及
び/又は適切な治療が
施されない。
疾病の進行
重傷
37
T 14971:2020 (ISO 14971:2019)
参考文献
[1] JIS Z 8051:2015 安全側面−規格への導入指針
注記 対応国際規格では,ISO/IEC Guide 51:2014,Safety aspects−Guidelines for their inclusion in
standardsを記載している。
[2] JIS T 0063:2020 医療機器規格における安全側面の開発及び導入の指針
注記 対応国際規格では,ISO/IEC Guide 63:2019,Guide to the development and inclusion of aspects of
safety in International Standards for medical devicesを記載している。
[3] JIS Q 9000:2015 品質マネジメントシステム−基本及び用語
注記 対応国際規格では,ISO 9000:2015,Quality management systems−Fundamentals and vocabulary
を記載している。
[4] JIS T 0993-1 医療機器の生物学的評価−第1部:リスクマネジメントプロセスにおける評価及び試
験
注記 対応国際規格では,ISO 10993-1,Biological evaluation of medical devices−Part 1: Evaluation and
testing within a risk management processを記載している。
[5] JIS Q 13485:2018 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 対応国際規格では,ISO 13485:2016,Medical devices−Quality management systems−
Requirements for regulatory purposesを記載している。
[6] ISO 14155,Clinical investigation of medical devices for human subjects−Good clinical practice
[7] ISO 18113-1:2009,In vitro diagnostic medical devices −Information supplied by the manufacturer (labelling)
−Part 1: Terms, definitions and general requirements
[8] ISO 20916,In vitro diagnostic medical devices−Clinical performance studies using specimens from human
subjects−Good study practice
[9] TR T 24971 医療機器−JIS T 14971適用の指針
注記 対応国際規格では,ISO/TR 24971,Medical devices−Guidance on the application of ISO 14971
を記載している。
[10] JIS Q 31000 リスクマネジメント−指針
注記 対応国際規格では,ISO 31000,Risk management−Guidelinesを記載している。
[11] IEC TR 60513,Fundamental aspects of safety standards for medical electrical equipment
[12] JIS T 0601-1 医用電気機器−第1部:基礎安全及び基本性能に関する一般要求事項
注記 対応国際規格では,IEC 60601-1,Medical electrical equipment−Part 1: General requirements for
basic safety and essential performanceを記載している。
[13] JIS T 62366-1:2019 医療機器−第1部:ユーザビリティエンジニアリングの医療機器への適用
注記 対応国際規格では,IEC 62366-1:2015,Medical devices−Part 1: Application of usability
engineering to medical devicesを記載している。