T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 2
4 一般要求事項 ··················································································································· 5
4.1 一般 ···························································································································· 5
4.2 品質システム ················································································································ 5
4.3 サンプリング ················································································································ 5
4.4 試験方法 ······················································································································ 5
4.5 文書化 ························································································································· 6
5 材料及び成形前無菌バリアシステム ····················································································· 6
5.1 一般要求事項 ················································································································ 6
5.2 微生物バリア特性 ·········································································································· 8
5.3 滅菌プロセスとの適合性 ································································································· 9
5.4 ラベル表示システムとの適合性 ························································································ 9
5.5 保管及び輸送 ··············································································································· 10
6 包装システムの設計及び開発に関する要求事項 ····································································· 10
6.1 一般 ··························································································································· 10
6.2 設計 ··························································································································· 10
6.3 包装システム性能試験 ··································································································· 11
6.4 安定性試験 ·················································································································· 11
7 情報提供························································································································ 12
附属書A(参考)医療用包装に関する指針 ··············································································· 13
附属書B(参考)要求事項への適合を実証するために使用できる標準試験方法及び手順 ···················· 16
附属書C(規定)非透気性材料の透気抵抗の試験方法 ································································ 32
参考文献 ···························································································································· 33
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,一般社団法人日本
医療機器学会(JSMI)から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,日本工業
標準調査会の審議を経て,厚生労働大臣及び経済産業大臣が改正した日本工業規格である。
これによって,JIS T 0841-1:2009は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣,経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の
特許出願及び実用新案権に関わる確認について,責任はもたない。
JIS T 0841の規格群には,次に示す部編成がある。
JIS T 0841-1 第1部:材料,無菌バリアシステム及び包装システムに関する要求事項
JIS T 0841-2 第2部:成形,シール及び組立プロセスのバリデーション
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
T 0841-1:2019
(ISO 11607-1:2006,Amd.1:2014)
最終段階で滅菌される医療機器の包装−
第1部:材料,無菌バリアシステム及び包装
システムに関する要求事項
Packaging for terminally sterilized medical devices-Part 1: Requirements
for materials, sterile barrier systems and packaging systems
序文
この規格は,2006年に第1版として発行されたISO 11607-1及びAmendment 1(2014)を基に,技術的
内容及び構成を変更することなく作成した日本工業規格である。ただし,追補(amendment)については,
編集し,一体とした。無菌バリアシステムについては,附属書Aに示す。
なお,この規格で点線の下線を施してある箇所は,対応国際規格にはない事項である。
1
適用範囲
この規格は,最終段階で滅菌される医療機器の無菌性を使用時点まで維持できるように意図した材料,
成形前無菌バリアシステム,無菌バリアシステム及び包装システムについて規定する。
この規格は,製造業(例えば,包装材料製造業),ヘルスケア施設及び医療機器を無菌バリアシステムの
中に置いて滅菌するあらゆるところに適用できる。
この規格は,無菌的に製造する医療機器の無菌バリアシステム及び包装システムについての全ての要求
事項を対象とはしていない。医薬品と医療機器との組合せには,追加要求事項が必要なこともある。
この規格は,製造段階の管理のための品質保証システムについては規定しない。また,この規格は,汚
染された医療機器を再処理場又は廃棄処分場へ運搬するときに使用する包装材料及び/又は包装システム
には適用しない。
注記 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 11607-1:2006,Packaging for terminally sterilized medical devices−Part 1: Requirements for
materials, sterile barrier systems and packaging systems及びAmendment 1:2014(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”こ
とを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。この引用
規格は,その最新版(追補を含む。)を適用する。
JIS P 8117 紙及び板紙−透気度及び透気抵抗度試験方法(中間領域)−ガーレー法
注記 対応国際規格:ISO 5636-5,Paper and board−Determination of air permeance (medium range)−
2
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
Part 5: Gurley method
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
無菌提供(aseptic presentation)
微生物汚染を除去する条件及び手順を使用した無菌製品の提供。
3.2
バイオバーデン(bioburden)
製品又は無菌バリアシステムの上又は内部に存在する生育可能な微生物群(ISO/TS 11139:2006参照)。
3.3
クロージャ(closure)
無菌バリアシステムを閉鎖するために用いる手段。
注記 例えば,無菌バリアシステムでは,再使用可能なコンテナのガスケットによるか,又は曲線経
路を作るための繰返しの折畳みによって閉鎖することができる。
3.4
クロージャの完全性(closure integrity)
クロージャが滅菌プロセス,取扱い,流通,輸送及び保管を考慮した試験条件下で微生物の侵入を防止
することを保証するクロージャの特性。
3.5
使用期限(expiry date)
その日までに製品を使用しなければならない日付の表示で,少なくとも年及び月で表したもの。
3.6
ラベル表示(labelling)
医療機器若しくはその包装システムに貼付されるか又は医療機器に添付される手書き,印刷,電子式又
は図式のもの。
注記 ラベル表示は,医療機器の識別,技術的な記載及び使用と関連するが,出荷に関する文書は除
外する。
3.7
医療機器(medical device)
あらゆる計器,器械,用具,機械,器具,埋込み用具,体外診断薬又は検定物質,ソフトウェア,材料
又は他の同類の若しくは関連する物質であって,単独使用か又は組合せ使用かを問わず,製造業者が人体
への使用を意図し,その使用目的が次の一つ以上であるもの。
a) 疾病の診断,予防,監視,治療又は緩和
b) 負傷の診断,監視,治療,緩和又は補助
c) 解剖学的又は生理学的なプロセスの検査,代替,又は修復
d) 生命支援又は維持
e) 受胎調節
f)
医療機器の消毒
g) 人体から採取される標本の体外試験法による医療目的のための情報提供
3
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
薬学,免疫学,又は新陳代謝の手段によって体内又は体表において意図したその主機能を達成すること
はないが,それらの手段によって機能を補助するものである(JIS Q 13485:2005参照)。
3.8
微生物バリア(microbial barrier)
滅菌プロセス,取扱い,流通,輸送及び保管を考慮した試験条件下で微生物の侵入を防止することを保
証する無菌バリアシステムの特性。
3.9
包装材料(packaging material)
包装システムの組立又はシールに使用する材料。
3.10
包装システム(packaging system)
無菌バリアシステムと保護的包装との組合せ(ISO/TS 11139:2006参照)。
3.11
成形前無菌バリアシステム(preformed sterile barrier system)
充塡及び最終クロージャ又はシールのために部分的に組み立てて提供される,3.22に定義する無菌バリ
アシステム。
例 パウチ,バッグ及び開放形再使用可能コンテナ(ISO/TS 11139:2006参照)。
3.12
製品(product)
プロセスの結果(JIS Q 9000:2006参照)。
注記 滅菌規格では,製品は有形のものであり,原料,中間品,半組立品及びヘルスケア製品をいう
(ISO/TS 11139:2006参照)。
3.13
保護的包装(protective packaging)
組立時から使用時点まで,無菌バリアシステム及びその内容物に対する損傷を防止するために設計した
材料構成(ISO/TS 11139:2006参照)。
3.14
リサイクル材料(recycled material)
本来又はその他の目的で発生した廃棄物を製造プロセスを経て再生処理した材料。
3.15
繰返し性(repeatability)
同一の測定条件下で行われた,同一の測定量の繰返し測定結果の間の一致の度合い。
注記1 これらの測定条件を繰返し性条件という。
注記2 繰返し性の測定条件には,次のものを含むことができる。
a) 同一の測定手順
b) 同一の測定者
c) 同一の条件下で使用する同一の測定機器
d) 同一の場所
e) 短期間での繰返し
注記3 繰返し性は,結果の分散特性を定量的に表してもよい。
4
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注記4 “国際計量基本用語集”による。
3.16
再現性(reproducibility)
測定条件を変更して行われた,同一の測定量の測定結果の間の一致の度合い。
注記1 再現性の報告が有効であるためには,変更した条件の明示が必要である。
注記2 変更した測定条件には,次のものを含むことができる。
a) 測定の原理
b) 測定の方法
c) 測定者
d) 測定機器
e) 参照標準
f)
場所
g) 使用条件
h) 時間
注記3 再現性は,結果の分散特性を定量的に表してもよい。
注記4 “国際計量基本用語集”参照。
3.17
再使用可能なコンテナ(reusable container)
繰返し使用するように設計された硬質の無菌バリアシステム。
3.18
シール(seal)
接着面同士を一つに結合した結果。
注記 例えば,接着面同士は,接着剤又は熱溶融によって一つに結合することができる。
3.19
シールの完全性(seal integrity)
シールが,滅菌プロセス,取扱い,流通,輸送及び保管を考慮した試験条件下で微生物の侵入を防止す
ることを保証するシールの特性。
3.20
シールの強さ(seal strength)
シールの機械的強度。
3.21
無菌(sterile)
生育可能な微生物が存在しないこと(ISO/TS 11139:2006参照)。
3.22
無菌バリアシステム(sterile barrier system)
微生物の侵入を防止し,かつ,使用時点での製品の無菌提供を可能にする最低限の包装(ISO/TS
11139:2006参照)。
3.23
無菌流体経路包装(sterile fluid-path packaging)
流体との接触を意図した医療機器の一部分の無菌性を確実にするように設計した,保護ポートカバー及
5
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
び/又は包装システム。
注記 無菌流体経路包装の例には,静脈注射液の投与のためのチューブの内部がある。
3.24
滅菌適合性(sterilization compatibility)
包装材料及び/又は包装システムが滅菌プロセスに耐え,また,包装システム内での必要な滅菌条件に
達することができるようにする,包装材料及び/又は包装システムの属性。
3.25
滅菌剤(sterilizing agent)
あらかじめ定めた条件下で無菌性を達成するために十分な殺菌作用をもつ,物理的若しくは化学的媒体
又はその組合せ(ISO/TS 11139:2006参照)。
3.26
最終滅菌(terminal sterilization)
製品が,無菌バリアシステム内で滅菌されるプロセス。
3.27
耐用寿命(useful life)
全ての性能要求事項が満たされる期間。
3.28
バリデーション(validation)
(一般)意図された特定の仕様に関する特定の要求事項を一貫して満たすことができることの,検査に
よる確認及び客観的証拠の提出。この定義は,試験方法及び設計のバリデーションに適用する。
(プロセスの場合)プロセスが恒常的に,あらかじめ定めた仕様に適合する製品を得ることができるこ
とを確立するために,要求される結果を得て,記録し,解釈するための文書化した手順(ISO/TS 11139:2006
参照)。
4
一般要求事項
4.1
一般
個別の製品規格がある場合には,それによってもよい(附属書B及び参考文献参照)。
4.2
品質システム
4.2.1
この規格で規定する要求事項は,品質システムの一環として実施しなければならない。
注記 JIS Q 9001及びJIS Q 13485には,適切な品質システムに関する要求事項が含まれている。
4.2.2
この規格の要件を満たすために品質システムの第三者認証を取得する必要はない。
注記 医療機器によっては,個別製品で第三者認証を要求される場合がある。
4.2.3
ヘルスケア施設は,国又は都道府県が要求する品質システムを用いることを検討しなければならな
い。
4.3
サンプリング
サンプリング計画は,包装システムの選定及び試験に用いられなければならない。そのサンプリング計
画は,統計的に有効な根拠に基づかなければならない。
注記 適切なサンプリング計画の例は,JIS Z 9015-1又はJIS P 8110がある。
4.4
試験方法
4.4.1
この規格への適合を示すために用いる全ての試験方法は,バリデーションを実施し,文書化しなけ
6
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ればならない。
注記 附属書Bに適切な試験方法の例を示している。
4.4.2
試験方法のバリデーションには,使用した方法の妥当性を実証しなければならない。このとき,次
の項目を含めなければならない。
a) 包装システムに適した試験の選定根拠の確立
b) 合否判定基準の確立
注記 合格又は不合格が,判定基準の一つである。
c) 試験方法の繰返し性の決定
d) 試験方法の再現性の決定
e) 完全性試験での,試験方法の検出限界の確立
4.4.3
試験方法に特に規定がない限り,試験試料は,温度23±1 ℃及び相対湿度 (50±2) %の雰囲気中で
24時間以上前処理しなければならない。
4.5
文書化
4.5.1
この規格の要求事項に適合している実証を,文書化しなければならない。
4.5.2
全ての文書は,あらかじめ定めた期間保管しなければならない。保管期間は,医療機器又は無菌バ
リアシステムの要求事項,使用期限,トレーサビリティなどの要素を考慮しなければならない。
4.5.3
要求事項への適合の文書は,性能データ,仕様及びバリデーションされた試験方法による試験結果
を含んでもよいが,これらに限らない。
4.5.4
バリデーション,プロセス管理又はその他の品質の意思決定プロセスに関係する電子記録,電子署
名及び電子記録に署名された手書きの署名は,信頼できるものでなければならない。
5
材料及び成形前無菌バリアシステム
5.1
一般要求事項
5.1.1
材料に関する要求事項は,成形前無菌バリアシステム及び無菌バリアシステムで使用する材料に適
用する。
5.1.2
5.1に記載した要求事項は,全てを含んだものではない。材料がもつこの細分箇条に記載していな
い特性も,箇条6の性能判定基準に従って評価を行う場合がある。
5.1.3
材料及び/又は成形前無菌バリアシステムの製造及び取扱い条件を確立,管理及び記録し,必要が
あれば次の条件を確実にしなければならない。
a) 条件が,材料及び/又は無菌バリアシステムの設計された用途に適合している。
b) 材料及び/又は無菌バリアシステムの性能特性が維持されている。
5.1.4
次の事項は,少なくとも考慮していなければならない。
a) 温度範囲
b) 圧力範囲
c) 湿度範囲
d) 必要に応じて,上記事項の最大変化率
e) 直射日光又は紫外線に対する露出
f)
清浄度
g) バイオバーデン
h) 静電気特性
7
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.1.5
最終製品が,この規格の要求事項に適合することを確実にするために,全ての材料,特にリサイク
ル材料は,原産地,履歴及びトレーサビリティを明らかにし,かつ,管理しなければならない。
注記 現行技術では,リサイクル材料の安全な管理ができないため,未使用製造廃棄物以外のものを
リサイクル材料の中で使用する可能性は少ない。
5.1.6
次の特性を評価しなければならない。
a) 微生物バリア(5.2参照)
b) 生体適合性及び毒物学的属性
注記 これは通常,機器と接触する材料に限定する。生体適合性に関する評価指針は,JIS T 0993-1
に示されている。
生体適合性に関する滅菌の影響を評価することが望ましい。
c) 物理的及び化学的特性
d) 成形及びシールプロセスに関する適合性
e) 意図された滅菌プロセスに関する適合性(5.3参照)
f)
滅菌前及び滅菌後の保管についての品質保持期間
5.1.7
包装材料(例えば,紙,プラスチックフィルム,不織布,再使用布など)は,次の要求事項に適合
しなければならない。
a) 材料は,製造業者が定めた使用条件で,性能及び安全性を損なわず,また,材料が接触する医療機器
に悪い影響を与えない範囲で非浸出性かつ無臭でなければならない。
注記 臭気は容易に分かるため,臭気判定には,標準化された試験方法を必要としない。
b) 材料は,機能を損なうような穴(孔),亀裂,破れ,しわ又は局部的な厚薄がないものでなければなら
ない。
c) 材料は,坪量(目付)があらかじめ定めた値に適合又は合致したものでなければならない。
d) 材料は,清浄度,粒子状物質及び毛羽立ちの許容レベルを示さなければならない。
e) 材料は,引張強さ,厚さのばらつき,引裂強さ,透気度,破裂強さなどの,あらかじめ定めた値又は
最低限の物理的特性に適合しなければならない。
f)
材料は,医療機器,包装システム又は滅菌プロセスの要求事項に適合するために,定められた化学的
特性(pH値,塩化物及び硫酸塩含有量など)に適合しなければならない。
g) 材料は,使用条件下の滅菌前,途中又は後のいずれにおいても,健康に害を及ぼすほどの量の有害物
質を含有又は放出してはならない。
5.1.8
接着剤を塗布した材料は,5.1.1〜5.1.7に加えて,次の要求事項に適合しなければならない。
a) 塗布の形状は,シールに不連続箇所を生じるほど,形状の中に飛越し又は破断がなく,連続していな
ければならない。
b) 塗布量は,あらかじめ定めた量どおりでなければならない。
c) 別の材料とシールを行う場合,あらかじめ定めた条件下で最小限のシールの強さを実証しなければな
らない。
5.1.9
無菌バリアシステム及び成形前無菌バリアシステムは,5.1.1〜5.1.7の要求事項,及び5.1.8に該当
する場合はその要求事項に適合することに加え,次の要求事項に適合しなければならない。
a) 例えば,塗料,インキ又はケミカルインジケータなどの材料及び構成要素は,あらかじめ定めた滅菌
プロセス前,プロセス中又はプロセス後に,反応,汚染及び/又は移送によって医療機器に悪影響を
与えてはならない。
8
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) シールする場合には,シール幅及びシールの強さ(引張及び/又は破裂)に関して,製造業者があら
かじめ定めた要求事項に適合しなければならない。
c) 引剝がし開封特性は,無菌開封及び提供に影響することのある材料の層剝離又は破れがなく,連続し,
かつ,均一でなければならない。
注記1 ペーパーバッグ,ヒートシール式パウチ及びロールには,構造上及び設計上の要求事項並
びに性能上の要求事項がある。
注記2 シールが無菌提供のために開封するように意図されている場合,最大のシールの強さが必
要なことがある。
d) シール及び/又はクロージャは,微生物バリア性をもたなければならない。
5.1.10 再使用可能なコンテナは,5.1.1〜5.1.7に加えて,次の要求事項に適合しなければならない。
a) コンテナには,クロージャの完全性が損なわれた場合に,明確な表示を提供するための改ざん防止シ
ステムを備えなければならない。
b) 滅菌剤の入口は,滅菌装置から取り出すとき並びに輸送及び保管のとき,微生物バリア性をもたなけ
ればならない(5.2参照)。
c) 無菌バリアシステムの成形後,クロージャは,微生物バリア性をもたなければならない。
d) コンテナは,全ての重要部分の検査が容易になるような構造でなければならない。
e) それぞれの再使用に先立つ検査のための,合否判定基準を確立しなければならない。
注記1 目視検査が最も一般的な手順であるが,ほかにも容認できる方法はある。
f)
互換性のない場合は,それが明らかになるような設計でなければならない。
注記2 適切なコード化及び/又はラベル表示で,この設計要求事項に対処することができる。
g) 構成要素の整備及び浄化手順並びに点検,維持及び交換方法をあらかじめ定めなければならない。
注記3 再使用可能なコンテナの追加の評価指針は,EN 868-8を参照。
5.1.11 再使用布は,5.1.1〜5.1.7及び該当する場合は5.1.8の要求事項に加えて,次の要求事項に適合しな
ければならない。
a) 材料の修理後及び滅菌サイクル後に,性能要求事項に適合しなければならない。
b) 洗浄及び再生の処理手順を確立し,文書化しなければならない。
注記 これには,再使用のための目視検査,その他の試験方法及び合否判定基準を含めてもよい。
c) 処理手順は,製品ラベル表示内容に適合しなければならない。
5.1.12 再使用可能なコンテナ及び布を含む無菌バリアシステムの場合,製造業者が指示した方法にて使用
し,耐用寿命の期間内において,製品機能が維持されていることを確認しなければならない。劣化が予測
される場合は,許容される再処理サイクル数を製品ラベルに記載するか,又は耐用寿命の終了が検出可能
でなければならない。
5.2
微生物バリア特性
5.2.1
材料の非透気性を,附属書Cに従って試験しなければならない。
注記 無菌バリアシステムの製造に使用する材料の微生物バリア特性は,完全性及び製品の安全性を
保証するために重要である。微生物バリア特性の評価に使用する方法は,次の二つのカテゴリ
に分類できる。
− 非透気性の材料にとって適切な方法
− 多孔質材料にとって適切な方法
5.2.2
材料が非透気性であることを実証することは,微生物バリア要求事項を満足する。
9
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.2.3
多孔質材料は,無菌バリアシステムの完全性及び製品の安全性を提供するために,微生物に対する
適切な微生物バリア性をもたなければならない。
注記 微生物バリア特性を実証するための普遍的に容認された方法はない。多孔質材料の微生物バリ
ア特性の評価は,代表的には,試験時の吸引流量,微生物の投与量及び試験時間をあらかじめ
定めた一連の試験条件の下で,細菌の胞子又は微粒子のエアロゾルで試料の微生物透気特性を
求めることで実施する。これらのあらかじめ定めた試験条件における材料の微生物バリア特性
は,細菌又は微粒子の透気量と投与量とを比較して判定する。バリデーションされた微生物バ
リア特性と相関する,バリデーションされた物理学的試験方法からのデータは,微生物バリア
特性を判定するのに容認できるものとみなす。材料及び無菌バリアシステムのバリデーション
された微生物バリア特性は,使用可能になっており,この規格の将来の版への包含が検討され
ることになる(詳細については,Sinclair・Tallentire,2002年[39],Tallentire・Sinclair,1996年[38],
Schollaほか,1995年[37]及びSchollaほか,2000年[36]参照)。
5.3
滅菌プロセスとの適合性
5.3.1
材料及び成形後無菌バリアシステムは,あらかじめ定めた滅菌プロセス及びサイクルパラメータで
の使用に適していることを実証しなければならない。
5.3.2
滅菌適合性は,関連JIS及び国際規格の要求事項に従って設計,製造及び使用する滅菌装置を用い
て判定することが望ましい。
注記 例えば,JIS T 7322,JIS T 7323,JIS T 7324,JIS T 7325,JIS T 0816-1,JIS T 0801,JIS T 0806-1,
JIS T 0806-2,JIS T 0806-3及びISO 14937の最新版を参照。
5.3.3
あらかじめ定めた全ての滅菌プロセスへのばく(曝)露後,材料性能があらかじめ定めた範囲内に
あることを評価しなければならない。
5.3.4
あらかじめ定めた滅菌プロセスには,同一の又は異なった滅菌プロセスの複数のばく(曝)露を含
めてもよい。
5.3.5
意図した目的に対する適合性の判定には,通常の定期供給中に発生する材料のばらつきを考慮する
ことを含めなければならない。
5.3.6
製品が複数のラッピング材料又は多層材料で封入される場合は,内層及び外層に関して,材料特性
に関する異なる限度値を設定してもよい。
5.3.7
適合性の判定は,使用する滅菌プロセスのバリデーションと同時に実施してもよい。
5.4
ラベル表示システムとの適合性
ラベル表示システムは,次による。
a) 使用時点までむきずで,また,容易に判読できる状態でなければならない。
b) あらかじめ定めた滅菌プロセス以降,材料,無菌バリアシステム及び医療機器に適合し,かつ,滅菌
プロセスに悪影響を与えてはならない。
c) 医療機器に移行するようなインキで印刷又は表示せず,包装材料及び/又はシステムと反応してその
有用性を損なってはならない。また,ラベルが読みにくくなるほど色が変化してはならない。
注記 ラベル表示システムは,包装材料及び/又は無菌バリアシステムへの直接印刷又は書込み,
また,接着,溶着,又はその他の手段によって材料及び/又はシステムの表面に付着させた
別の材料の層によるラベルを含め,多様な形態をとることができる。
10
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.5
保管及び輸送
5.5.1
材料及び成形前無菌バリアシステムは,輸送及び保管中に性能特性を維持するために必要な保護を
するように包装しなければならない。
5.5.2
材料及び成形前無菌バリアシステムは,性能特性が5.1の限度値内になることを保証する条件下で
輸送し,保管しなければならない。
これは,次によって達成できる。
a) これらの特性が,あらかじめ定めた保管条件下で保持されていることを実証する。
b) 保管条件が,あらかじめ定めた範囲内に維持されていることを確実にする。
6
包装システムの設計及び開発に関する要求事項
6.1
一般
6.1.1
包装システムは,意図したあらかじめ定めた使用条件において,使用者及び患者に対する危害を最
小限にする設計でなければならない。
6.1.2
包装システムは,物理的保護を提供し,かつ,無菌バリアシステムの完全さを維持しなければなら
ない。
6.1.3
無菌バリアシステムは,滅菌を可能にし,かつ,選択されたプロセスに適合しなければならない。
6.1.4
無菌バリアシステムは,使用時点又は使用期限まで,無菌性を維持しなければならない(6.4.1参
照)。
6.1.5
無菌バリアの完全性の維持は,無菌性の維持を実証するために使用してもよい。
注記 ANSI/AAMI ST65:2008及びHansenほか,1995年[34]参照。無菌性の喪失は,時間関連ではなく
事象関連とみなす。
6.1.6
類似の医療機器が同一の包装システムを使用する場合は,類似性(プロダクト・ファミリ)を確立
し,かつ,限界条件を特定する根拠を文書化しなければならない。少なくとも,限界条件によって製造し
たワーストケースの製品を使用して,この規格への適合を判定しなければならない。
注記 例えば,類似性は,異なるサイズの同一製品で確立できる。
6.2
設計
6.2.1
包装システムの設計及び開発の手順を文書化しなければならない。
6.2.2
無菌バリアシステムは,製品が無菌操作法によって提供されることを許容しなければならない。
6.2.3
包装システムの設計及び開発は,次の事項を考慮しなければならないが,これらに限らない。
a) 顧客要求事項
b) 製品の質量及び構成
c) 鋭いエッジ又は突起の存在
d) 物理的及びその他の保護の必要性
e) 特定のリスク(例えば,放射線,湿気,機械的衝撃,静電放電)に対する製品の感度
f)
包装システム当たりの製品数
g) 包装ラベル表示要求事項
h) 環境上の制限事項
i)
製品の使用期限
j)
流通,取扱い及び保管の環境
k) 滅菌適合性及び残留物
11
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2.4
無菌流体経路を構成する,製品の構成要素及び構造を明確にし,あらかじめ定めなければならない。
これらには,次のことを含めることが望ましいが,これらに限らない。
a) 材料
b) 仕上げ
c) 構成要素の寸法
d) アセンブリ寸法(例えば,締まりばめの許容差)
6.2.5
設計及び開発プロセス(6.2.1,6.2.3及び6.2.4)の結果を記録し,検証し,製品の出荷前に承認し
なければならない。
6.3
包装システム性能試験
6.3.1
滅菌及びその後の性能試験後に,無菌バリアシステムの完全性を実証しなければならない。
6.3.2
多孔質包装材料については,微生物バリア試験以外に,物理的な試験をすることによって,無菌性
の維持のための能力を確立することができる。
注記 この問題のレビューについては,ANSI/AAMI ST65:2008及びHansenほか,1995年[34]を参照。
6.3.3
無菌バリアシステムの完全性を評価するための標準化試験方法を優先する。ただし,該当するバリ
デーション済みの無菌バリアシステム完全性試験を行わない場合,微生物バリア性能特性は,材料の無菌
バリア特性並びにシール及びクロージャの完全性を試験して確認することができる。
6.3.4
性能試験は,成形及びシールの定められたプロセスの限界条件で製造したワーストケースの無菌バ
リアシステムを使用して,製造業者が定めた全ての滅菌プロセスへのばく(曝)露後に実施しなければな
らない。
注記 製造業者が定めた滅菌プロセスには,同一の又は異なった滅菌プロセスの複数のばく(曝)露
を含めてもよい。
6.3.5
包装システムは,取扱い,配送及び保管に関連する危険全体を通して,製品の適切な保護をしなけ
ればならない。
6.4
安定性試験
6.4.1
安定性試験は,無菌バリアシステムが,所定の期間にわたって完全性を維持することを実証しなけ
ればならない。
6.4.2
安定性試験は,実時間劣化試験を用いて実施しなければならない。
6.4.3 加速劣化試験プロトコルを使用する安定性試験は,実時間劣化試験からのデータを入手するまでは,
申告された使用期限に対する十分な証拠とみなす。
6.4.4
実時間劣化試験及び加速劣化試験は,同時に開始するのがよい。
注記 安定性試験と性能試験とは目的が異なる。性能試験は,包装システムと,製造及び滅菌プロセ
ス,並びに取扱い,保管及び出荷環境によって負荷された応力に対応する製品との間の相互作
用を評価するものである。
6.4.5
使用期限が製品性能に基づく場合は,使用期限を決定するための安定性試験を,包装安定性試験と
同時に実施するのがよい。
6.4.6
加速劣化試験を実施する場合は,加速劣化条件及び選択する試験期間の根拠を確立し,文書化しな
ければならない。
6.4.7
製品が所定の期間にわたってあらかじめ定めた無菌バリアシステムと相互作用しないことを実証
した場合,安定性試験に関して以前に文書化されたデータは,6.4.1に従わなければならない。
12
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7
情報提供
7.1
製造販売業者又は製造業者は,材料,成形前無菌バリアシステム又は無菌バリアシステムとともに,
次の情報を提供しなければならない。
− 製造販売業者若しくは製造業者の氏名又は名称及び住所
− タイプ,サイズ又はグレード
− 製品のバッチ番号又は製造履歴の追跡のためのその他の手段
− 意図する滅菌プロセス
− 使用期限(必要な場合)
− 保管条件(必要な場合)
− 取扱い又は使用に関する既知の制限事項。例えば,環境条件など(必要な場合)。
− 材料及び/又は成形前無菌バリアシステムの再使用可能可否
− 再使用可能な材料及び/又は成形前無菌バリアシステムの場合,保守の種類及び頻度
− 取扱説明書を添付する場合,発行日又は最新版の改訂日
7.2
国又は都道府県が,ヘルスケア市場に出荷する材料,成形前無菌バリアシステム又は無菌バリアシ
ステムに関する追加情報を要求した場合は,その追加情報を提供しなければならない。
13
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
医療用包装に関する指針
A.1 材料の選択及び包装システムの設計に影響を与える要素
特定の種類の医療機器,意図した滅菌方法,意図した用途,使用期限,輸送及び保管の全てが,包装シ
ステム設計及び材料の選択に影響する。最終段階で滅菌する医療機器包装システムのための適切な材料の
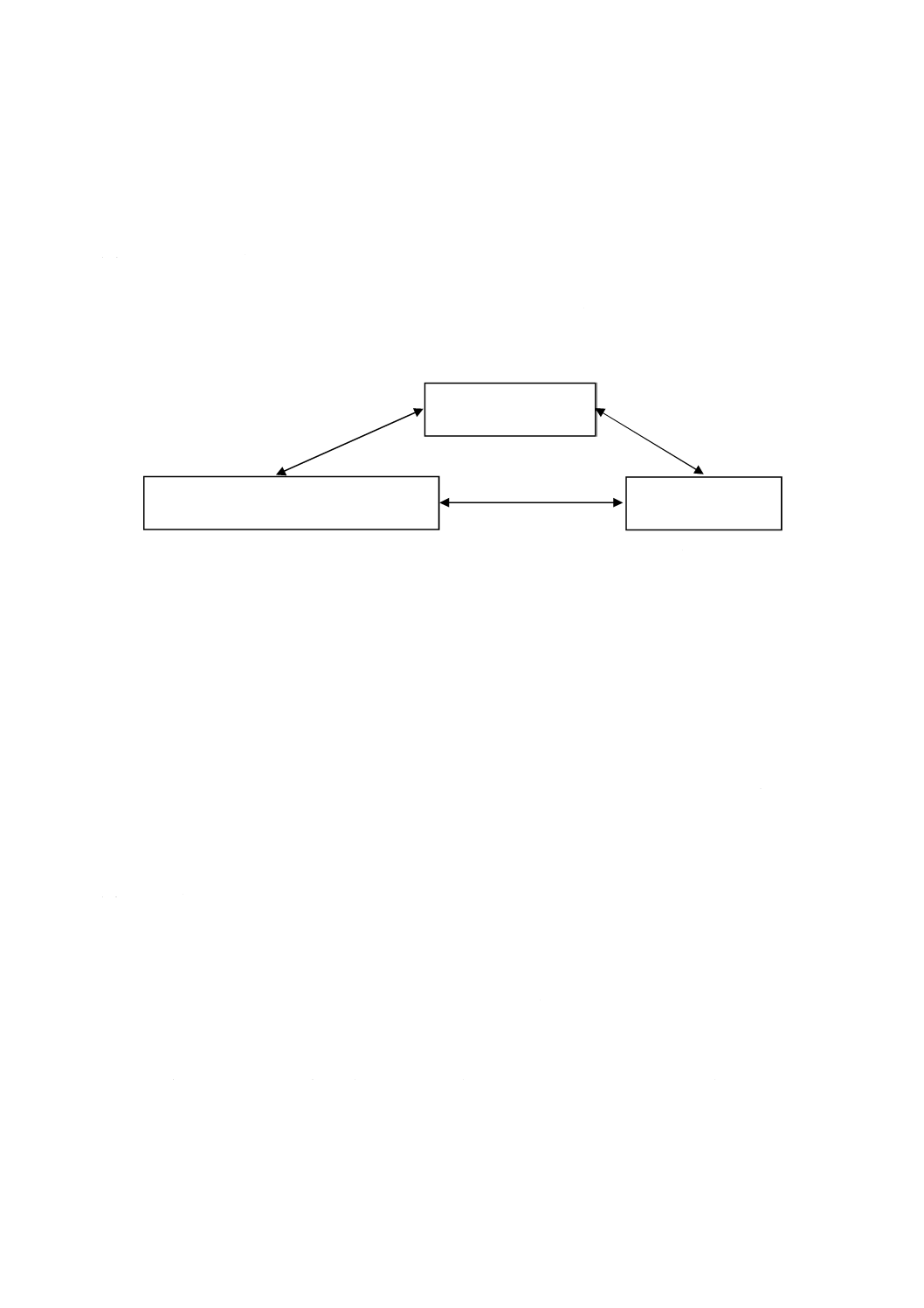
選択は,図A.1に示す相互関係によって影響を受ける。
図A.1−最終段階で滅菌する医療機器包装システムのための適切な材料の選択に影響を与える相互関係
A.2 滅菌プロセス及び考慮事項
A.2.1 滅菌プロセスの選択には,エチレンオキサイド(EO),ガンマ線,電子線,湿熱及び低温酸化的滅
菌プロセスが含まれるが,これらに限らない。医療機器が,EO,湿熱又は低温酸化的滅菌プロセスによっ
て滅菌するように意図されている場合,無菌バリアシステムは,滅菌ガスが入り,微生物を殺し,滅菌ガ
スが著しく残留しない透気性材料を使用する。
A.2.2 医療機器を放射線(ガンマ線又は電子線)で滅菌する場合は,透気性材料を使用しなくてもよい。
また,無菌バリアシステムは,完全に非透気性材料で製造することができる。医療機器の製造業者は,各
機器に適切な滅菌プロセスを選択し,その選択は,幾つかの要素に依存する。医療機器を放射線安定でな
い材料で製造する場合,一般的には,EO,湿熱及び酸化剤が使用される。一方,医療機器が,高いEOの
残留濃度を保持する傾向がある場合には,医療機器の製造業者は放射線滅菌を選択してもよい。
A.3 無菌バリアシステム
A.3.1 医療機器の無菌バリアシステムは,多くの共通的な特性をもつ。大半は,蓋材及びボトム材並びに
それらの材料を一つに結合する手段をもつ。剝離可能なシールが要求される場合は,二つの層を一つにヒ
ートシールすることを可能にするためのシーリング材を使用する。透気性材料には,一般に塗布材として
知られるシーリング材が伝統的に使用されてきた。今日では,多くのフィルムが,シーリング材層をフィ
ルムの構造内に層として組み入れている。剝離可能なシールが要求されない場合は,熱又はその他の方法,
例えば,超音波シールによる結合を可能にするための材料の親和性が要求される。
A.3.2 医療機器を滅菌する場合に,包装に使用する無菌バリアシステムは,タイプが数多く,また,種類
も多様である。第1のタイプは,打ち抜いた蓋材をもつ成形前硬質トレイである。このトレイは,通常,
熱成形又は圧力成形プロセスによって事前成形される。打ち抜いた蓋材は,透気性でも又は非透気性でも
よく,一般的には,蓋材をトレイにヒートシールするために使用するシーリング材層をもつ。打ち抜いた
医療機器
包装システム設計及びプロセス
滅菌プロセス
14
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
蓋材をもつ硬質トレイは,通常,整形インプラント,ペースメーカ,外科キットなど,大きな形状の質量
の大きい機器に使用される。
A.3.3 第2のタイプは,パウチである。パウチは,一般的には,一方の側がフィルムで他方の側がフィル
ム,紙又は不織布の構造である。パウチは,一般的には1か所(底部)を除き,全てのシールが,成形さ
れた成形前無菌バリアシステムとして供給される。これは,機器を内部に収納し,次に滅菌前に最終シー
ルすることができるように開いた状態のままである。パウチは,様々なサイズを広範に入手できるので,
多様な医療機器が,これを無菌バリアシステムとして使用している。これらの機器は,一般的には,形状
が小さく,かつ,軽量である。パウチは,多様な設計が可能である(例えば,より大きな形状の医療機器
を収納するために,ガセット袋などがある。)。
A.3.4 第3のタイプは,滅菌紙を使用した連続のロール状(又はリール状)のものである。これは,サイ
ドガセットをもつ,又はサイドガセットをもたない長いロール状の管を成形するために折り畳まれた滅菌
紙の単一ウェブ構造である。そのロール状の管は,長さ方向に,2本の接着剤によってシールされている。
次に,必要なサイズに切断し,その一端をシールする。開いている端には,通常,開封を容易にするため
に切り口又はサムカットが付いている。この必要なサイズにカットされた袋の最終クロージャは,滅菌前
に適用する。
A.3.5 第4のタイプは,ヘッダバッグである。ヘッダバッグは,主として,2枚の非透気性でシール性の
あるフィルム層から製造したヒートシールバッグである。材料の1枚は,通常,数cmずらす。このずれ
の部分に,接着剤が付いた透気性材料を熱接着する。この透気性材料は,内容品を取り出すときに剝がす
ことができる。ヘッダバッグは,キットなどのかさばる品目には一般的である。
A.3.6 第5のタイプは,成形・充塡・シール(FFS:Form Fill Seal)として知られるプロセスである。FFS
によって製造される無菌バリアシステムは,パウチ及び蓋付きの硬質トレイであることも,又は引っ張ら
れた若しくは成形された軟質フィルムボトム材をもつこともある。FFSでは,蓋材及びボトム材はFFSの
機械に設置する。機械は,ボトム材を成形し,その成形物に機器を充塡し,蓋材をシールして,無菌バリ
アシステムを製造する。
A.3.7 第6のタイプは,四方シール(4SS:Four Side Sealing)プロセスである。4SSは,ピロー包装のよ
うな連続的包装プロセスである。最も一般的には,シールを成形するための回転シール装置を使用してい
る。4SSプロセスでは,ボトム材及び蓋材を4SSの機械に設置する。製品はボトム材の上に置く。蓋材を
その上に貼付し,最後に四つの面をシールする。4SSは,例えば,手袋及び創傷治療用製品の包装に使用
する。
A.3.8 A.3.2〜A.3.7に記載した無菌バリアシステムのリストは,全てを網羅したものではない。このほか
の構造も,無菌バリアシステムとして使用できる。
A.3.9 無菌流体経路付き医療機器は,医療機器の流体経路入口点に直接固定した,固有の無菌流体経路包
装システムを使用してもよい。これらは,キャップ,プラグ,カバー又はその他の機器特定クロージャ設
計で構成してもよい。これらの場合,製品包装の第一層は,上記の四つのスタイルの一つによって表して
もよいが,医療機器に微生物バリアをもつことが要求されないこともある。
A.3.10 ヘルスケア施設は,一般的にはパウチ,ロール,ペーパーバッグ,CSR(中央材料室)ラップ又は
再使用可能なコンテナの形態の無菌バリアシステムを使用する。
A.3.11 CSRラップは,ヘルスケア施設で滅菌する多くの機器に無菌バリアシステムを提供するために使
用する。熱又は接着剤によるシールをする代わりのラッピング及び折畳みプロセスで,無菌性を維持する
曲線経路を提供する。医療機器は,一般的には,ラッピング及びその後の滅菌の前にセット用トレイに収
15
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
納する。
A.3.12 再使用可能なコンテナは,病院の滅菌サイクルへの繰返しのばく(曝)露に耐えることができる金
属又は合成樹脂で製造される。これらのコンテナは,一般的には蓋と底との二つの間を,非透気性のシー
ルを提供するガスケットで合わせている。透気システム(フィルタ)は,滅菌剤がコンテナに入り,そこ
から抜け出ることができるようにしている。微生物バリアをもっている透気の設計及び材料は,多様で広
範である。コンテナ内で滅菌される機器は,滅菌プロセスが完全であることを保証するために,特定の前
処理又はより長いばく(曝)露時間を必要とすることがある。
A.3.13 最終滅菌及び無菌性の維持は,これらのプロセスを実施する施設に関わりなく,患者の安全にとっ
て不可欠である。この規格は,適切な無菌バリアシステムを提供する包装システムの使用に関する最低限
の要求事項について規定する。
16
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(参考)
要求事項への適合を実証するために使用できる
標準試験方法及び手順
B.1
一般
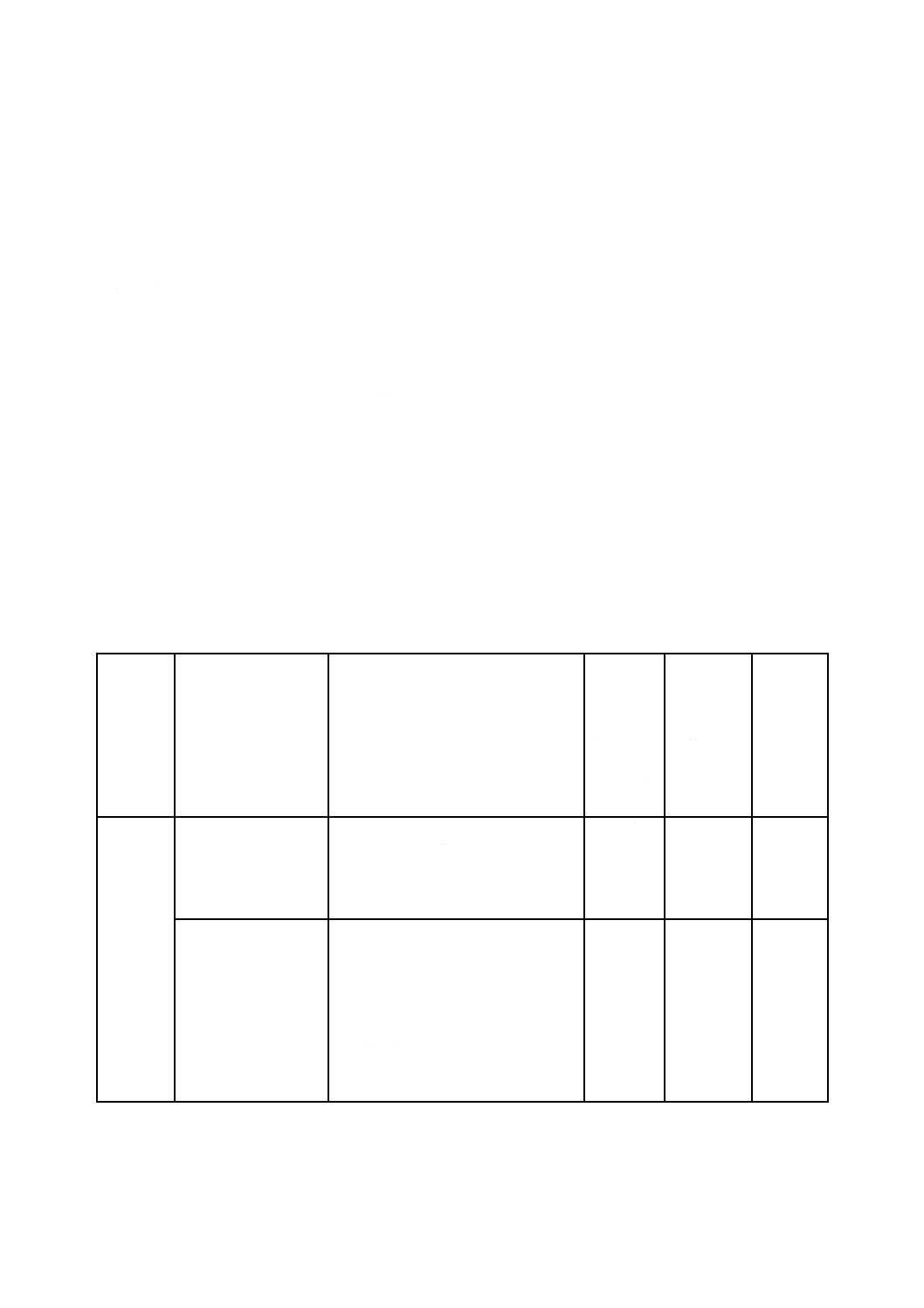
表B.1には,この規格の規定への適合を実証するために使用できる試験方法が含まれている。表B.1に
記載されている試験方法及び試験手順を用いる場合,当該文書の発行年に注意することが重要である。試
験方法の使用に関する特定要求事項は,4.4に示されている。
表B.1に示す試験方法及び試験手順を含めた基準は,各国の標準機関規格開発組織又は業者団体から商
業的に入手できるものである。参考文献には,資料として公開されている追加の試験方法が記載されてい
る。この附属書は,全てを網羅しようと意図されているものではなく,また,発行の時点で新しい試験方
法の開発が進行中のものもある。
注記 JISの試験方法には,最新のISO規格の試験方法に対応していない場合がある。B.2にはISO
規格の最新版でない試験方法も記載されているので変更点を把握することが必要である。
B.2
包装材料及び成形前無菌バリアシステム
表B.1−試験方法及びその状況
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
加速劣化
ASTM F1980
Standard Guide for Accelerated Aging of
Sterile Barrier Systems for Medical
Devices
(滅菌医療機器包装の加速劣化の標準
ガイド)
NAa)
NA
YES
EN 868-8
Packaging
for
terminally
sterilized
medical devices−Part 8: Re-usable
sterilization
containers
for
steam
sterilizers conforming to EN 285−
Requirements and test methods
(最終段階で滅菌される医療機器の包
装−第8部:EN 285に準拠した蒸気滅
菌器の再使用滅菌コンテナ−要求事項
及び試験方法)
NA
NA
YES
17
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
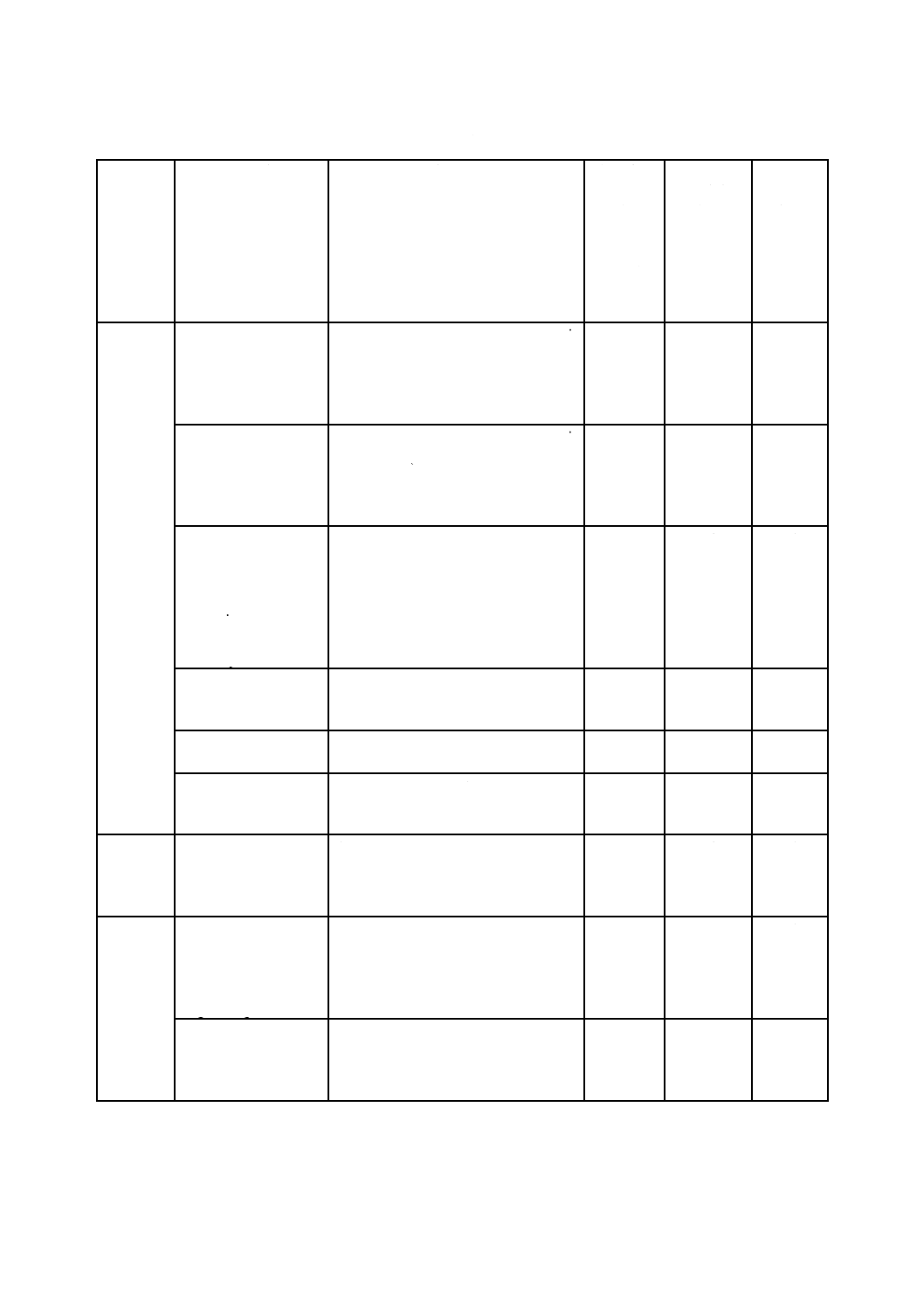
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
透気度
ISO 5636-2
Paper and board−Determination of air
permeance (medium range)−Part 2:
Schopper method
[紙及び板紙−透気度試験方法(中間
領域)−第2部:ショッパー法]
NO
NO
NA
ISO 5636-3
Paper and board−Determination of air
permeance (medium range)−Part 3:
Bendtsen method
[紙及び板紙−透気度試験方法(中間
領域)−第3部:ベントセン法]
NO
NO
NA
JIS P 8117
注記 対応国際規格:
ISO 5636-5,Paper and
board−Determination
of
air
permeance
(medium range)−Part
5: Gurley method
紙及び板紙−透気度及び透気抵抗度試
験方法(中間領域)−ガーレー法
NO
NO
NA
ASTM D737
Standard Test Method for Air Permeability
of Textile Fabrics
(織物の透気度の標準試験方法)
YES
−
NA
TAPPI T460
Air Resistance of Paper (Gurley Method)
[紙の透気度(ガーレー法)]
YES
−
NA
TAPPI T536
Resistance of Paper to Passage of Air
(High-Pressure Gurley Method)
[紙の透気度(高圧ガーレー法)]
YES
−
NA
はつ(撥)
アルコー
ル性
AATCC-193
Aqueous Liquid Repellency: Water/
Alcohol Solution Resistance Test
[はつ(撥)アルコール性:耐水/ア
ルコール溶液試験]
NO
NO
NA
坪量(目
付)
JIS P 8124
注記 対応国際規格:
ISO 536,Paper and
board−Determination
of grammage
紙及び板紙−坪量の測定方法
NO
NO
NA
ASTM D4321
Standard Test Method for Package Yield
of Plastic Film
(プラスチックフィルムのパッケー
ジ・イールドの標準試験方法)
YES
−
NA

18
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
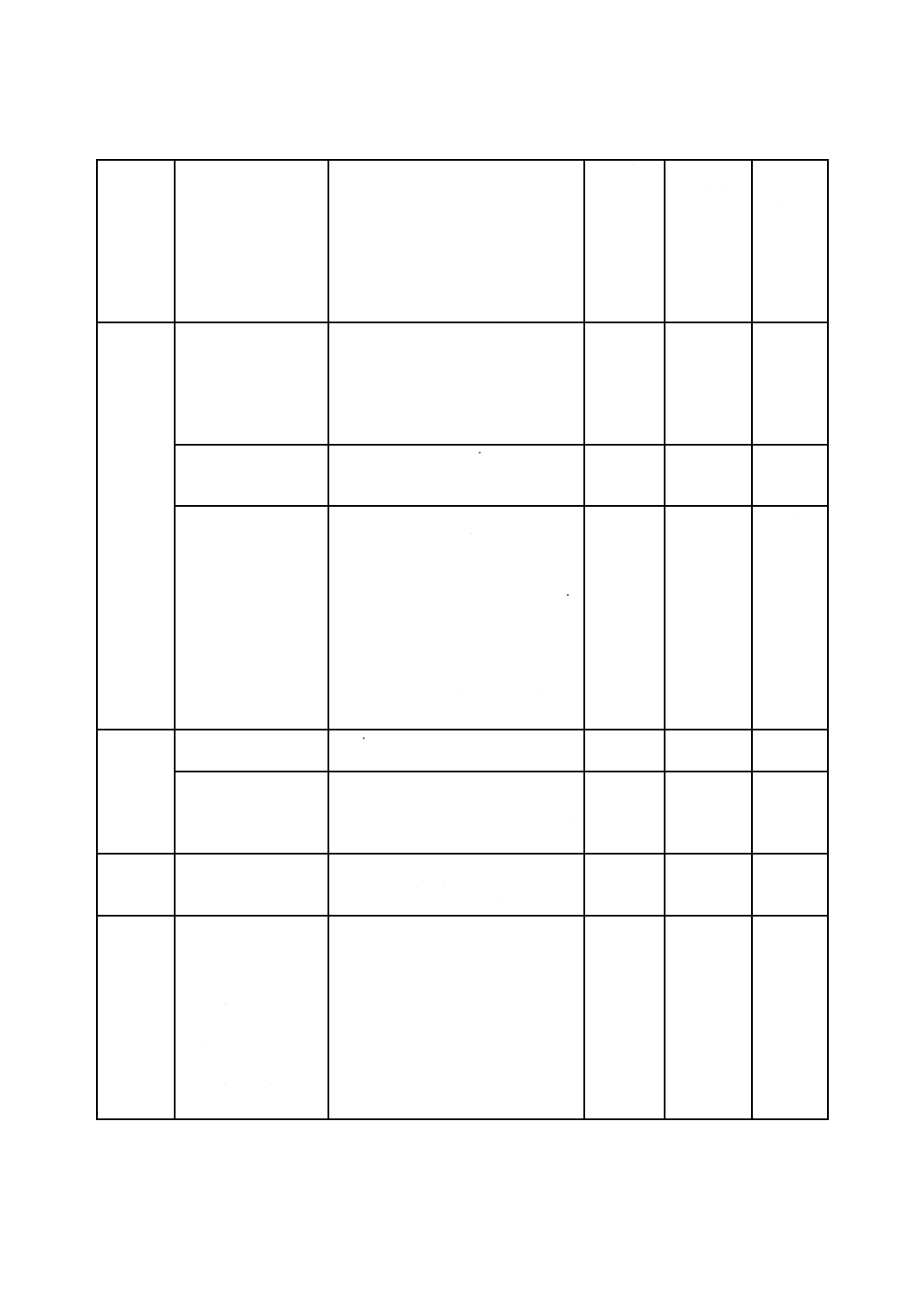
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
坪量(目
付)(続き)
ASTM D3776/D3776M Standard Test Methods for Mass Per Unit
Area (Weight) of Fabric
[織物の単位面積当たりの質量(重量)
の標準試験方法]
YES
−
NA
TAPPI T410
Grammage of Paper and Paperboard
(Weight per Unit Area)
[紙及び板紙の坪量(単位面積当たり
の重量)]
YES
−
NA
生体適合
性
JIS T 0993-1
注記 対応国際規格:
ISO 10993-1,
Biological evaluation of
medical devices−Part
1:
Evaluation
and
testing within a risk
management process
医療機器の生物学的評価−第1部:リ
スクマネジメントプロセスにおける評
価及び試験
NA
NA
YES
ASTM F2475
Standard Guide for Biocompatibility
Evaluation of Medical Device Packaging
Materials
(医療機器包装材料生体適合性評価標
準ガイド)
NA
NA
YES
破裂強さ
JIS P 8112
注記 対応国際規格:
ISO 2758,Paper−
Determination of
bursting strength
紙−破裂強さ試験方法
YES
−
NA
TAPPI T403
Bursting Strength of Paper
(紙の破裂強さ)
YES
−
NA
ASTM F1140/F1140M
Standard Test Methods for Internal
Pressurization Failure Resistance of
Unrestrained Packages
(医療のための非拘束及び非硬質包装
の故障耐性の標準試験方法)
YES
−
NA
ASTM F2054/F2054M
Standard Test Method for Burst Testing of
Flexible Package Seals Using Internal Air
Pressurization Within Restraining Plates
(軟質包装シールの破裂試験のための
拘束板内の内部空気圧を使用する標準
試験方法)
YES
−
NA
19
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
塩化物
JIS P 8144
注記 対応国際規格:
ISO 9197,Paper, board
and pulps−
Determination of
water-soluble chlorides
紙,板紙及びパルプ−水溶性塩化物の
測定方法
YES
−
NA
TAPPI T256
Water-Soluble Chlorides in Pulp and Paper
(パルプ及び水に含まれる水溶性塩化
物)
−
YES
NA
EN 868-4
Packaging
for
terminally
sterilized
medical devices−Part 4: Paper bags−
Requirements and test methods
(Annex B: Method for the determination
of pH value, chloride and sulphate in
paper bags)
[最終段階で滅菌される医療機器の包
装−第4部:ペーパーバッグ−要求事
項及び試験方法(附属書B:ペーパー
バッグのpH値,塩化物,及び硫化物
の試験方法)]
NO
NOb)
NA
清浄度
TAPPI T437
Dirt in Paper and Paperboard
(板紙及び板紙中のきょう雑物)
YES
−
NA
TAPPI T564
Transparent Chart for the Estimation of
Defect Size
(不具合の大体の大きさを測定する透
明チャート)
NO
NO
NA
コート量
ASTM F2217/F2217M
Standard Practice for Coating/Adhesive
Weight Determination
(コート量測定の標準実施要領)
NA
NA
YES
前処理
JIS P 8111
注記 対応国際規格:
ISO 187,Paper, board
and pulps−Standard
atmosphere for
conditioning and testing
and procedure for
monitoring the
atmosphere and
conditioning of samples
紙,板紙及びパルプ−調湿及び試験の
ための標準状態
NA
NA
YES
20
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
前処理
(続き)
ASTM D4332
Standard
Practice
for
Conditioning
Containers, Packages, or Packaging
Components for Testing
(試験のためのコンテナ,包装又は包
装構成要素の前処理の標準実施要領)
NA
NA
YES
ISO 2233
Packaging−Complete, filled transport
packages and unit loads−Conditioning
for testing
(包装−包装貨物試験方法−試験のた
めの前処理)
NA
NA
YES
寸法
ASTM F2203
Standard
Test
Method
for
Linear
Measurement Using Precision Steel Rule
(鋼尺を使用する長さ測定の標準試験
方法)
YES
−
NA
ドレープ
性
ISO 9073-9
Textiles−Test methods for nonwovens−
Part 9: Determination of drapability
including drape coefficient
(織物−不織布の試験−第9部:ドレ
ープ係数の測定)
NO
NO
NA
JIS P 8125-1
注記 対応国際規格:
ISO 2493-1,Paper and
board−Determination
of bending resistance
Part 1: Constant rate of
deflection
紙及び板紙−曲げ抵抗試験方法−第1
部:定速曲げ法
NO
NO
NA
JIS P 8125-2
注記 対応国際規格:
ISO 2493-2,Paper and
board−Determination
of bending resistance−
Part 2: Taber-type tester
紙及び板紙−曲げ抵抗試験方法−第2
部:テーバー型試験機法
YES
−
NA
DIN 53121
Testing of paper and board−
Determination of the bending stiffness by
the beam method
(紙及び板紙の試験−ビーム法による
曲げ鋼度の測定)
NO
NO
NA
TAPPI T489
Bending Resistance (Stiffness) of Paper
and Paperboard (Taber-Type Stiffness
Tester in Basic Configuration)
[紙及び板紙のこわさ(剛度)(基本構
成のテーバー型こわさ試験機)]
YES
−
NA

21
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
ドレープ
性(続き)
TAPPI T566
Bending Resistance (Stiffness) of Paper
(Taber-Type Tester 0 to 10 Taber Stiffness
Unit Configuration)
[紙のこわさ(剛度)(テーバー剛度単
位0〜10のテーバーこわさ試験機)]
YES
−
NA
曲げ耐久
性
ASTM F392/F392M
Standard
Practice
for
Conditioning
Flexible Barrier Materials for Flex
Durability
(軟質バリア材料の屈曲耐久性の標準
試験方法)
YES
−
NA
微生物バ
リア性
ASTM F1608
Standard Test Method for Microbial
Ranking of Porous Packaging Materials
(Exposure Chamber Method)
[多孔質包装材料の微生物順位法の標
準試験方法(露出室法)]
YES
−
NA
ASTM F2638
Standard Test Method for Using Aerosol
Filtration for Measuring the Performance
of Porous Packaging Materials as a
Surrogate Microbial Barrier
(多孔質包装材料の微生物バリア性を
測定するエアロゾルろ過を使用した標
準試験方法)
YES
−
NA
DIN 58953-6
Sterilization
−
Sterile
Supply
−
Sterilization Paper for bags and Tube
packaging−Test; Subclause 2.14: Testing
for Germ Proofness in Moisture and
Clause 15: Testing for Germ Proofness
with Passage of Air
(滅菌−滅菌供給−バッグ及び管包装
の滅菌紙−試験:2.14:湿気による防
菌性の試験及び第15節:透気による防
菌性の試験)
YESf), g)
NAf)
NA
BS 6256
Specification for Paper for Steam
Sterilization Paper Bags, Pouches, and
Reels for Medical Use Appendix C:
Methods for Determination of Methylene
Blue Particulate Penetration
[医療用蒸気滅菌ペーパーバッグ,パ
ウチ及びロール用の仕様(附属書C:
メチレンブルー粒子浸透の測定方法)]
NO
NO
NA
22
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
微生物バ
リア性
(続き)
ASTM F2101
Standard Test Method for Evaluating the
Bacterial Filtration Efficiency (BFE) of
Medical Face Mask Materials, Using a
Biological Aerosol of Staphylococcus
aureus
[生物学的ぶどう状球菌エアロゾルを
使用する医療用顔マスクの微生物ろ過
効率 (BFE)を評価する試験方法]
−
YES
NA
SS 876 0019
Health
Care
Textiles−Bacterial
Penetration−Wet
(ヘルスケア織物−微生物透過−湿
潤)
NO
NO
NA
酸素透過
性
ASTM D3985
Standard Test Method for Oxygen Gas
Transmission Rate Through Plastic Film
and Sheeting Using a Coulometric Sensor
(クーロメトリックセンサーを用い,
酸素ガスがプラスチックフィルム及び
シートを透過する速度を測る標準試験
方法)
YES
−
NA
ASTM F1307
Standard Test Method for Oxygen
Transmission Rate Through Dry Packages
Using a Coulometric Sensor
(クーロメトリックセンサーを用い,
酸素ガスがドライパックを透過する速
度を測る標準試験方法)
YES
−
NA
ASTM F1927
Standard Test Method for Determination
of Oxygen Gas Transmission Rate,
Permeability and Permeance at Controlled
Relative
Humidity
Through
Barrier
Materials Using a Coulometric Detector
(クーロメトリック検出器を用い,相
対湿度管理下で,酸素ガスがバリア材
料を通る透過率,透過性,透過を測定
する標準試験方法)
YES
−
NA
ASTM F2622
Standard Test Method for Oxygen Gas
Transmission Rate Through Plastic Film
and Sheeting Using Various Sensors
(各種センサーを用い,酸素ガスがプ
ラスチックフィルム及びシートを透過
する速度を測る標準試験方法)
YES
−
NA
23
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
引剝がし
開封特性
EN 868-5
Packaging
for
terminally
sterilized
medical devices Part 5: Sealable pouches
and reels of porous materials and plastic
film construction−Requirements and test
methods
(Annex E determination of peel
characteristics of paper/plastic laminate
products)
[最終段階で滅菌される医療機器の包
装−第5部:多孔質材料及びプラスチ
ックフィルム構造のシール可能なパウ
チ及びロール−要求事項及び試験方法
(附属書E:紙/プラスチック積層製
品のピール特性の測定方法)]
NO
NO
NA
性能試験
ASTM D4169
Standard Practice for Performance Testing
of Shipping Containers and Systems
(出荷コンテナ及びシステムの性能試
験の実施要領)
NA
NA
YES
ISTA 1,2 and 3 Series
International Safe Transit Association
Preshipment Test Procedures
(国際安全輸送協会出荷前試験手順)
NA
NA
YES
ISTA 4AB
Packaged−Product for Shipment in
Known Distribution Channels
(既知の流通経路による出荷用の包装
済み製品)
NA
NA
YES
ISTA 7D
Thermal Controlled Transport Packaging
for Parcel Delivery System Shipment
(小荷物配達システム出荷用温度制御
運送包装)
NA
NA
YES
JIS Z 0200
注記 対応国際規格:
ISO 4180,Packaging
−
Complete,
filled
transport packages−
General rules for the
compilation
of
performance
test
schedules
包装貨物−性能試験方法一般通則
NA
NA
YES
24
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
性能試験
(続き)
EN 868-8
Packaging
for
terminally
sterilized
medical devices−Part 8: Re-usable
sterilization
containers
for
steam
sterilizers conforming to EN 285−
Requirements and test methods
(最終段階で滅菌される医療機器の包
装−第8部:EN 285に準拠した蒸気滅
菌器の再使用滅菌コンテナ−要求事項
及び試験方法)
NO
NO
NA
ASTM F2825
Standard Practice for Climatic Stressing of
Packaging Systems for Single Parcel
Delivery
(単一小荷物配達用包装システムの気
候ストレス対応標準実施要領)
NA
NA
YES
ASTM D7386
Standard Practice for Performance Testing
of Packages for Single Parcel Delivery
Systems
(単一小荷物配達用包装の性能試験の
標準実施要領)
NA
NA
YES
pH値
JIS P 8133-1
注記 対応国際規格:
ISO 6588-1,Paper,
board and pulps−
Determination of pH of
aqueous extracts−Part
1: Cold extraction
紙,板紙及びパルプ−水抽出液pHの
試験方法−第1部:冷水抽出
YES
−
NA
JIS P 8133-2
注記 対応国際規格:
ISO 6588-2,Paper,
board and pulps−
Determination of pH of
aqueous extracts−Part
2: Hot extraction
紙,板紙及びパルプ−水抽出液pHの
試験方法−第2部:熱水抽出
YES
−
NA
TAPPI T509
Hydrogen Ion Concentration (pH) of
Paper Extracts (Cold Extraction Method)
[ペーパー抽出物の水素イオン濃度
(pH)(冷感抽出)]
YES
−
NA
TAPPI T435
Hydrogen Ion Concentration (pH) of
Paper Extracts (Hot Extraction Method)
[ペーパー抽出物の水素イオン濃度
(pH)(熱感抽出)]
YES
−
NA

25
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
気孔サイ
ズ
EN 868-2
Packaging
for
terminally
sterilized
medical devices−Part 2: Sterilization
wrap−Requirements and test methods
(Annex C: Method for the determination
of pore size)
[最終段階で滅菌される医療機器の包
装−第2部:滅菌ラップ−要求事項及
び試験方法(附属書C:気孔サイズの
測定方法)c)]
NO
NOb)
NA
印刷及び
コート
ASTM F2250
Standard Practice for Evaluation of
Chemical Resistance of Printed Inks and
Coatings on Flexible Packaging Materials
(軟質包装材料上の印刷インキ及びコ
ート材の耐化学性の評価の標準実施要
領)
NA
NA
YES
ASTM F2252/F2252M
Standard Practice for Evaluating Ink or
Coating Adhesion to Flexible Packaging
Materials Using Tape
(軟質包装材料上へのインキ又はコー
ト材の接着を評価するためのテープを
使用する標準実施要領)
NA
NA
YES
導通
ASTM D1709
Standard Test Methods for Impact
Resistance of Plastic Film by the
Free-Falling Dart Method
(プラスチックフィルムの耐衝撃性の
ための自由落下ダーツによる標準試験
方法)
YES
−
NA
ASTM F1306
Standard Test Method for Slow Rate
Penetration Resistance of Flexible Barrier
Films and Laminates
(軟質バリアフィルム及び積層板の耐
低速貫通性の標準試験方法)
YES
−
NA
ASTM D3420
Standard Test Method for Pendulum
Impact Resistance of Plastic Film
(プラスチックフィルムの耐振り子衝
撃性のための標準試験方法)
YESd)
−
NA
シールの
強さh)
ASTM F88/F88M
Standard Test Method for Seal Strength of
Flexible Barrier Materials
(軟質バリア材料のシール強さの標準
試験方法)
YES
−
NA
26
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
シールの
強さh)
(続き)
EN 868-5
Packaging
for
terminally
sterilized
medical devices−Part 5: Sealable
pouches and reels of porous materials and
plastic film construction−Requirements
and test methods
(最終段階で滅菌される医療機器の包
装−第5部:多孔質材料及びプラスチ
ックフィルム構造のシール可能なパウ
チ及びロール−要求事項及び試験方
法)
NO
NO
NA
仕様作成
ASTM F2559/F2559M
Standard Guide for Writing a Specification
for Sterilizable Peel Pouches
(滅菌可能引剝がし型パウチの仕様を
書くための標準ガイド)
NA
NA
YES
ASTM F99
Standard Guide for Writing a Specification
for Flexible Barrier Rollstock Materials
(軟質バリアのロールストック材料の
仕様を書くための標準ガイド)
NA
NA
YES
ASTM F17
Standard Terminology Relating to Primary
Barrier Packaging
(軟質バリア包装関連標準用語集)
NA
NA
YES
ASTM F2097
Standard Guide for Design and Evaluation
of Primary Flexible Packaging for Medical
Products
(医療用品一次軟質包装の設計及び評
価標準ガイド)
NA
NA
YES
静電気
BS 6524
Method for determination of the surface
resistivity of a textile fabric
(織物の表面低効率の測定方法)
NO
NO
NA
ASTM D257
Standard Test Methods for DC Resistance
or Conductance of Insulating Materials
(絶縁体のDC抵抗又は伝導性の標準
試験方法)
−
YES
NA
無菌バリ
アシステ
ムの完全
性
ASTM F2228
Standard Test Method for Non-Destructive
Detection of Leaks in Packaging Which
Incorporates Porous Barrier Material by
CO2 Tracer Gas Method
(多孔質バリア材料を組み込む医療包
装の漏れの非破壊検出のためのCO2ト
レーサガス法による標準試験方法)
YES
−
NA
27
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
無菌バリ
アシステ
ムの完全
性(続き)
ASTM F1929
Standard Test Method for Detecting Seal
Leaks in Porous Medical Packaging by
Dye Penetration
(医療用多孔質包装のシール漏れを検
出するための染料浸透による標準試験
方法)
YES
−
NA
ASTM F2227
Standard Test Method for Non-Destructive
Detection of Leaks in Non-sealed and
Empty Packaging Trays by CO2 Tracer
Gas Method
(非密封及び空の医療包装トレイの漏
れを非破壊検出するためのCO2トレー
サガス法による標準試験方法)
YES
−
NA
ASTM F2391
Standard Test Method for Measuring
Package and Seal Integrity Using Helium
as the Tracer Gas
(トレーサガスとしてヘリウムを用い
て包装及びシールの完全性を調べる標
準試験方法)
YES
−
NA
ASTM F2096
Standard Test Method for Detecting Gross
Leaks
in
Packaging
by
Internal
Pressurization (Bubble Test)
(医療用多孔質包装の大きな漏れを検
出するための内圧による標準試験方
法)
YES
−
NA
ASTM F1886/F1886M
Standard Test Method for Determining
Integrity of Seals for Flexible Packaging
by Visual Inspection
(医療包装のためのシールの完全性を
判定するための目視検査による標準試
験方法)
YES
−
NA
ASTM F2338
Standard Test Method for Nondestructive
Detection of Leaks in Packages by
Vacuum Decay Method
(包装の漏れの非破壊検出の真空崩壊
による標準試験方法)
YES
−
NA
ASTM D3078
Standard Test Method for Determination
of Leaks in Flexible Packaging by Bubble
Emission
(軟質包装における漏れを測定するた
めの気泡放出による標準試験方法)
YES
−
NA
28
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
無菌バリ
アシステ
ムの完全
性(続き)
ASTM F2095
Standard Test Methods for Pressure Decay
Leak Test for Flexible Packages With and
Without Restraining Plates
(拘束板がある場合とない場合の軟質
包装の圧減衰漏れ試験の標準試験方
法)
YES
−
NA
硫化物
ISO 9198
Paper, board and pulp−Determination of
water-soluble sulfates
(紙,板紙及びパルプ−水可溶性硫酸
塩試験方法)
−
YES
NA
TAPPI T255
Water-Soluble Sulfates in Pulp and Paper,
Test Method
(パルプ及び水に含まれる水可溶性硫
化物−試験方法)
−
YES
NA
EN 868-4
Packaging
for
terminally
sterilized
medical devices−Part 4: Paper bags−
Requirements and test methods
(Annex B: Method for the determination
of pH value, chloride and sulphate in
paper bags)
[最終段階で滅菌される医療機器の包
装−第4部:ペーパーバッグ−要求事
項及び試験方法(附属書B:ペーパー
バッグのpH値,塩化物,及び硫化物
の試験方法)]
NO
NOb)
NA
引裂抵抗
ASTM D1922
Standard Test Method for Propagation
Tear Resistance of Plastic Film and Thin
Sheeting by Pendulum Method
(プラスチックフィルム及び薄シート
の引裂伝ぱ抵抗の振り子方法による標
準試験方法)
YES
−
NA
ASTM D1938
Standard
Test
Method
for
Tear-
Propagation Resistance (Trouser Tear) of
Plastic Film and Thin Sheeting by a
Single-Tear Method
[プラスチックフィルム及び薄シート
の引裂伝ぱ抵抗(トラウザー引裂き)
の単一引裂き法による標準試験方法]
YES
−
NA
29
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
引裂抵抗
(続き)
JIS P 8116
注記 対応国際規格:
ISO 1974,Paper−
Determination
of
tearing
resistance
(Elmendorf method)
紙−引裂強さ試験方法−エルメンドル
フ形引裂試験機法
NO
NO
NA
引張特性
JIS P 8113
注記 対応国際規格:
ISO 1924-2,Paper and
board−Determination
of tensile Properties−
Part 2: Constant rate of
elongation method
紙及び板紙−引張特性の試験方法−第
2部:定速伸張法
YES
−
NA
ISO 1924-3
Paper and board−Determination of
tensile properties−Part 3: Constant rate of
elongation method (100 mm/min)
[紙及び板紙−引張特性の試験方法−
第3部:定速伸張法(100 mm/分)]
YES
−
NA
ASTM D882
Standard Test Method for Tensile
Properties of Thin Plastic Sheeting
(薄プラスチックシートの引張特性の
標準試験方法)
YES
−
NA
TAPPI T494
Tesile Properties of Paper and Paperboard
(Using Constant Rate of Elongation
Apparatus)
[紙及び板紙の引張特性(定速伸張装
置を使用)]
YES
−
NA
厚さ・密度 JIS P 8118
注記 対応国際規格:
ISO 534,Paper and
board−Determination
of thickness, density
and specific volume
紙及び板紙−厚さ,密度及び比容積の
試験方法
YES
−
NA
ASTM F2251
Standard Test Method for Thickness
Measurement of Flexible Packaging
Material
(軟質包装材料の厚さ測定の標準試験
方法)
YES
−
NA
TAPPI T551
Thickness of Paper and Paperboard (Soft
Platen Method)
[紙及び板紙の厚さ(ソフトプラテン
法)]
YES
−
NA
30
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
厚さ・密度
(続き)
TAPPI T411
Thickness (Caliper) of Paper, Paperboard
and Combined Paper
[紙,板紙及び結合ボードの厚さ(キ
ャリパー)]
YES
−
NA
真空漏れ
EN 868-8
Packaging
for
terminally
sterilized
medical devices−Part 8: Re-usable
sterilization
containers
for
steam
sterilizers conforming to EN 285−
Requirements and test methods
(最終段階で滅菌される医療機器の包
装−第8部:EN 285に準拠した蒸気滅
菌器の再使用滅菌コンテナ−要求事項
及び試験方法)
NO
NO
NA
目視検査
EN 868-8
Packaging
for
terminally
sterilized
medical devices−Part 8: Re-usable
sterilization
containers
for
steam
sterilizers conforming to EN 285−
Requirements and test methods
(最終段階で滅菌される医療機器の包
装−第8部:EN 285に準拠した蒸気滅
菌器の再使用滅菌コンテナ−要求事項
及び試験方法)
NO
NO
NA
耐水性
JIS L 1092
注記 対応国際規格:
ISO 811,Textiles−
Determination of
resistance to water
penetration−
Hydrostatic pressure
test
繊維製品の防水性試験方法
(JIS L 1096参照)
NO
NO
NA
EDANA 170-1
Wet Barrier−Mason Jar
(水分バリア−メーソンジャー)
NO
NO
NA
EN 20535
Paper and Board−Determination of Water
Absorptiveness−Cobb Method
(紙及び板紙−吸水性の測定−コッブ
法)
NO
NO
NA
AATCC-127
Water Resistance: Hydrostatic Pressure
Test
(耐水性:静水圧試験)
NO
NO
NA

31
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表B.1−試験方法及びその状況(続き)
属性・特性
引用規格
引用規格のタイトル
試験方法
に,精度及
び/又は
バイアス,
繰返し性
及び再現
性の記載
がある。
試験方法
に,精度及
び/又はバ
イアスだけ
記載されて
いる。
ガイダン
ス,標準
実施要領
耐水性
(続き)
TAPPI T441
Water
Absorptiveness
of
Sized
(Non-Bibulous) Paper, Paperboard and
Corrugated Fiberboard (Cobb Test)
[サイズ(非吸水性)紙,板紙及び段
ボールの吸水性(コッブ試験)]
YES
−
NA
EN 868-2
Packaging
for
terminally
sterilized
medical devices−Part 2: Sterilization
wrap−Requirements and test methods
(Annex B: Method for the determination
of water repellency)
[最終段階で滅菌される医療機器の包
装−第2部:滅菌ラップ−要求事項及
び試験方法(附属書B:はっ(撥)水
性の測定方法)e)]
NO
NOb)
NA
湿潤条件
での湿潤
破裂
ISO 3689
Paper and board−Determination of
bursting strength after immersion in water
(紙及び板紙−浸水後の破裂強さ試験
方法)
NO
NO
NA
湿潤引張
特性
JIS P 8135
注記 対応国際規格:
ISO 3781,Paper and
board−Determination
of tensile strength after
immersion in water
紙及び板紙−湿潤引張強さ試験方法
NO
NO
NA
TAPPI T456
Tensile Breaking Strength of Water-
saturated Paper and Paperboard (“Wet
Tensile Strength”)
[水分飽和状態の紙及び板紙の引張破
断強さ(湿潤引張強さ)]
YES
−
NA
注a) NA:適用不可
b) 精度及び/又はバイアスについては,Berry,C.W.,Harding,L. 共著(2012),“滅菌包装の構造に用いられ
る材料の特性決定及び特定のための試験方法のバリデーション”(Packag.Technol.Sci.)を参照。
c) 気孔サイズを測る試験方法は,EN 868-3:2009,EN 868-6:2009及びEN 868-7:2009の附属書Dにも記載されて
いる。
d) ASTMの基準にあるのは精度の記載だけである。
e) はっ(撥)水性の試験方法は,EN 868-3:2009,EN 868-6:2009及びEN 868-7:2009の附属書Dにも記載されて
いる。
f) 試験は合否試験。バイアスは,一般にはもう使われていない。
g) 繰返し性及び再現性については,D.Zahn,ISEGA Forschungs- und Untersuchungsgesellschaft mbH.“平行試験−
DIN 58953-6:2010に従った滅菌される医療機器の包装材料の微生物バリア試験”の記載を参照。
h) 包装の規格としてJIS Z 0238(ヒートシール軟包装袋及び半剛性容器の試験方法)がある。
32
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C
(規定)
非透気性材料の透気抵抗の試験方法
C.1 無菌バリアシステムの非透気性材料は,JIS P 8117に規定する装置及び温度条件(JIS P 8111を参照)
を用い,非透気性を確認しなければならない。
判定基準は,1時間以上経った後に,±1 mmの許容差で,シリンダに目に見える動きがあってはならな
い。
C.2 日常管理及び製品検査には,上記以外の試験方法を用いてもよいが,これらの方法は,使用する材
料に関して,標準試験方法(C.1参照)と比較確認しバリデーションを行わなければならない。
注記 上記以外の試験方法の例を,附属書Bに示す。
33
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] JIS P 8110 紙及び板紙−平均品質を測定するためのサンプリング方法
注記 対応国際規格:ISO 186:2002,Paper and board−Sampling to determine average quality
[2] JIS Q 9000:2006 品質マネジメントシステム−基本及び用語
注記 対応国際規格:ISO 9000:2005,Quality management systems−Fundamentals and vocabulary
なお,ISO 9000は,ISO 9000:2015が最新である。
[3] JIS Q 9001 品質マネジメントシステム−要求事項
注記 対応国際規格:ISO 9001:2008,Quality management systems−Requirements
なお,ISO 9001は,ISO 9001:2015が最新である。
[4] JIS Q 13485:2005 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 対応国際規格:ISO 13485:2003,Medical devices−Quality management systems−Requirements for
regulatory purposes
なお,ISO 13485は,ISO 13485:2016が最新である。
[5] JIS T 0801 ヘルスケア製品の滅菌−エチレンオキサイド−医療機器の滅菌プロセスの開発,バリデ
ーション及び日常管理の要求事項
注記 対応国際規格:ISO 11135:2014,Sterilization of health-care products−Ethylene oxide−
Requirements for the development, validation and routine control of a sterilization process for
medical devices
[6] JIS T 0806-1 ヘルスケア製品の滅菌−放射線−第1部:医療機器の滅菌プロセスの開発,バリデーシ
ョン及び日常管理の要求事項
注記 対応国際規格:ISO 11137-1:2006,Sterilization of health care products−Radiation−Part 1:
Requirements for development, validation and routine control of a sterilization process for medical
devices
なお,ISO 11137-1:2006には,Amendment 1:2013が発行されている。
[7] JIS T 0806-2 ヘルスケア製品の滅菌−放射線−第2部:滅菌線量の確立
注記 対応国際規格:ISO 11137-2:2006,Sterilization of health care products−Radiation−Part 2:
Establishing the sterilization dose
なお,ISO 11137-2は,ISO 11137-2:2013が最新である。
[8] JIS T 0806-3 ヘルスケア製品の滅菌−放射線−第3部:線量測定にかかわる指針
注記 対応国際規格:ISO 11137-3:2006,Sterilization of health care products−Radiation−Part 3:
Guidance on dosimetric aspects
なお,ISO 11137-3は,ISO 11137-3:2017が最新である。
[9] JIS T 0816-1 ヘルスケア製品の滅菌−湿熱−第1部:医療機器の滅菌プロセスの開発,バリデーショ
ン及び日常管理の要求事項
注記 対応国際規格:ISO 17665-1:2006,Sterilization of health care products−Moist heat−Part 1:
Requirements for the development, validation and routine control of a sterilization process for
medical devices
[10] JIS T 0841-2 最終段階で滅菌される医療機器の包装−第2部:成形,シール及び組立プロセスのバ
34
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
リデーション
注記 対応国際規格:ISO 11607-2:2006,Packaging for terminally sterilized medical devices−Part 2:
Validation requirements for forming, sealing and assembly processes
なお,ISO 11607-2:2006には,Amendment 1:2014が発行されている。
[11] JIS T 0993-1 医療機器の生物学的評価−第1部:リスクマネジメントプロセスにおける評価及び試験
注記 対応国際規格:ISO 10993-1:2009,Biological evaluation of medical devices−Part 1: Evaluation and
testing within a risk management process
[12] JIS T 7322 医療用高圧蒸気滅菌器
[13] JIS T 7323 医療用酸化エチレンガス滅菌器
[14] JIS T 7324 医療用小型高圧蒸気滅菌器
[15] JIS T 7325 医療用小型酸化エチレンガス滅菌器
[16] JIS Z 9015-1 計数値検査に対する抜取検査手順−第1部:ロットごとの検査に対するAQL指標型
抜取検査方式
注記 対応国際規格:ISO 2859-1:1999,Sampling procedures for inspection by attributes−Part 1:
Sampling schemes indexed by acceptance quality limit (AQL) for lot-by-lot inspection
なお,ISO 2859-1:1999には,Amendment 1:2011が発行されている。
[17] ISO 5636-2:1984,Paper and board−Determination of air permeance (medium range)−Part 2: Schopper
method
[18] ISO/TS 11139:2006,Sterilization of health care products−Vocabulary
[19] ISO 14937:2000,Sterilization of health care products−General requirements for characterization of a
sterilizing agent and the development, validation and routine control of a sterilization process for medical
devices
[20] EN 285:1996+Amd.2:2009,Steam sterilizers−Large sterilizers
[21] EN 868-2:1999,Packaging materials and systems for medical devices which are to be sterilized−Part 2:
Sterilization wrap−Requirements and test methods
[22] EN 868-3:2009,Packaging for terminally sterilized medical devices−Part 3: Paper for use in the manufacture
of paper bags (specified in EN 868-4) and in the manufacture of pouches and reels (specified in EN 868-5)
−Requirements and test methods
[23] EN 868-4:1999,Packaging materials and systems for medical devices which are to be sterilized−Part 4:
Paper bags−Requirements and test methods
[24] EN 868-5:2009,Packaging for terminally sterilized medical devices−Part 5: Sealable pouches and reels of
porous materials and plastic film construction−Requirements and test methods
[25] EN 868-6:2009,packaging for terminally sterilized medical devices−Part 6: Paper for low temperature
sterilization processes−Requirements and test methods
[26] EN 868-7:2009,Packaging for terminally sterilized medical devices−Part 7: Adhesive coated paper for low
temperature sterilization processes−Requirements and test methods
[27] EN 868-8:1999,Packaging materials and systems for medical devices which are to be sterilized−Part 8:
Re-usable sterilization containers for steam sterilizers conforming to EN 285−Requirements and test
methods
[28] EN 868-9:2000,Packaging materials and systems for medical devices which are to be sterilized−Part 9:
35
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
Uncoated nonwoven materials of polyolefines for use in the manufacture of heat sealable pouches, reels
and lids−Requirements and test methods
[29] EN 868-10:2000,Packaging materials and systems for medical devices which are to be sterilized−Part 10:
Adhesive coated nonwoven materials of polyolefines for use in the manufacture of heat sealable pouches,
reels and lids−Requirements and test methods
[30] EN 1422:1997,Sterilizers for medical purposes−Ethylene oxide sterilizers−Requirements and test methods
[31] EN 13795-1:2002+Amd.1:2009,Surgical drapes, gowns and clean air suits, used as medical devices for
patients, clinical staff and equipment−Part 1: General requirements for manufacturers, processors and
products
[32] EN 14180:2003+Amd.2:2009,Sterilizers for medical purposes−Low temperature steam and formaldehyde
sterilizers−Requirements and testing
[33] ANSI/AAMI ST65:2008,Processing of reusable surgical textiles for use in health care facilities
[34] HANSEN, J., JONES, L., ANDERSON, H., LARSEN, C., SCHOLLA, M., SPITZLEY, J., and BALDWIN, A.
1995. In quest of sterile packaging: Part 1: Approaches to package testing. Med. Dev. & Diag. Ind. 17 (8): pp.
56-61
[35] JONES, L., HANSEN, J., ANDERSON, H., LARSEN, C., SCHOLLA, M., SPITZLEY, J., and BALDWIN, A.
1995. In quest of sterile packaging: Part 2: Approaches to package testing. Med. Dev. & Diag. Ind. 17 (9): pp.
72-79
[36] SCHOLLA, M., HACKETT, S., RUDYS, S., MICHELS, C. and BLETSOS, J. 2000. A potential method for
the specification of microbial barrier properties. Med. Dev. Technol. 11 (3): pp. 12-16
[37] SCHOLLA, M., SINCLAIR, C.S., and TALLENTIRE, A. (1995). A European Consortium Effort to Develop a
Physical Test for Assessing the Microbial Barrier Properties of Porous Medical Packaging Materials. In:
Pharm. Med. Packaging 95, Copenhagen, Denmark
[38] TALLENTIRE, A. and SINCLAIR, C.S. (1996). A Discriminating Method for Measuring the Microbial
Barrier Performance of Medical Packaging Papers. Med. Dev. Diag. Ind., 18 (5), pp. 228-241
[39] SINCLAIR, C.S. and TALLENTIRE, A. (2002) Definition of a correlation between microbiological and
physical articulate barrier performances for porous medical packaging materials. PDA J. Pharm. Sci. Technol.
56 (1): pp. 11-9
[40] JUNGHANNß, U., WINTERFELD, S., GABELE, L. and KULOW: U. Hygienic-Microbiological and
Technical Testing of Sterilizer Container Systems, Zentr. Steril. 1999: 7 (3) pp. 154-162 under Sterile barrier
systems, Package Integrity
[41] GABELE, L. and JUNGHANNß, U. Untersuchung zur Lagerdauer von Sterilgut unter Einbezug des
Sterilcontainers: Aseptica 6, 2000, pp. 5-7
[42] Merkblatt 45, Verpackungs-Rundschau 5/1982: Prüfung von Heißsiegelnähten auf Dichtigkeit, Herausgegeben
von den Arbeitsgruppen der Industrievereinigung für Lebensmitteltechnologie und Verpackung e. V. am
Fraunhofer-Institut für Lebensmitteltechnologie und Verpackung, Institut an der Technischen Universität
München
[43] DUNKELBERG, H. and WEDEKIND, S. A New Method for Testing the Effectiveness of the Microbial
Barrier Properties of Packaging Materials for Sterile Products: Biomed. Technik, 47 (2002), pp. 290-293
[44] Test method for the microbial barrier properties of wrapping materials, new approach: Report No. 319 011.
36
T 0841-1:2019 (ISO 11607-1:2006,Amd.1:2014)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
007 RIVM (Rijksinstituut voor volksgezondheid en milieuhygiene), Netherlands
[45] Test method for the microbial barrier properties of packaging for medical devices: Report No. 31900, RIVM
(Rijksinstituut voor volksgezondheid en milieuhygiene), Netherlands
[46] International Vocabulary of Basic and General Terms in Metrology: 1993, BIPM, IEC, IFCC, ISO, IUPAC,
IUPAP, OIML
[47] AORN Journal 26 (21:334-350) Microbiology of Sterilization. Litsky, Bertha, Y. 1977
[48] USP 27<1031> The biocompatibility of materials used in drug containers, medical devices and implants
[49] Berry,C.W. and Harding,L.(2012),Validation of Test Methods for Characterizing and Specifying Materials
Used in the Construction of Sterilization Packaging. Packag. Technol.Sci.
[50] D. Zahn, ISEGA Forschungs- und Untersuchungsgesellschaft mbH. Interlaboratory Test-“Microbial barrier
testing of packaging materials for medical devices which are to be sterilized” according to DIN 58953-6:2010.
Available on line from the Sterile Barrier Association (SBA)
http://www.sterilebarrier.org/media/43501/Final-validation-report-germproofness-plus-author.pdf