T 0816-1:2010 (ISO 17665-1:2006)
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲 ························································································································· 1
1.1 適用 ···························································································································· 1
1.2 適用除外 ······················································································································ 2
2 引用規格 ························································································································· 2
3 用語及び定義 ··················································································································· 3
4 品質マネジメントシステムの要素 ······················································································· 10
4.1 文書化 ························································································································ 10
4.2 経営者の責任 ··············································································································· 10
4.3 製品実現 ····················································································································· 10
4.4 測定,分析及び改善−不適合製品の管理 ············································································ 10
5 滅菌剤の特性 ·················································································································· 10
5.1 滅菌剤 ························································································································ 10
5.2 微生物殺滅効果の有効性································································································· 11
5.3 材料への影響 ··············································································································· 11
5.4 環境への配慮 ··············································································································· 11
6 プロセス及び装置の特性 ··································································································· 11
6.1 プロセスの特性 ············································································································ 11
6.2 装置 ··························································································································· 12
7 製品の定義 ····················································································································· 14
8 プロセスの定義 ··············································································································· 14
9 バリデーション ··············································································································· 16
9.1 一般 ··························································································································· 16
9.2 据付適格性の確認 (IQ) ·································································································· 16
9.3 運転適格性の確認 (OQ) ································································································· 17
9.4 稼働性能適格性の確認 (PQ) ···························································································· 17
9.5 バリデーションのレビュー及び承認 ·················································································· 18
10 日常監視及び管理 ·········································································································· 18
11 滅菌からの製品のリリース ······························································································ 19
12 プロセス有効性の維持 ···································································································· 19
12.1 継続的な有効性の立証 ·································································································· 19
12.2 再校正 ······················································································································· 20
12.3 装置のメンテナンス ····································································································· 20
12.4 適格性の再確認 ··········································································································· 20
12.5 変更の評価 ················································································································· 20
T 0816-1:2010 (ISO 17665-1:2006)
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
附属書A(参考)この規格の使用上の指針 ··············································································· 21
附属書B(参考)自然状態にある微生物群の不活化に基づくプロセスの定義(バイオバーデン法) ····· 25
附属書C(参考)標準菌の不活化及び滅菌する製品上のバイオバーデンの知見に基づく
プロセスの定義(バイオバーデン/バイオロジカルインジケータ併用法) ············· 26
附属書D(参考)標準菌の不活化に基づく安全率を見込んだプロセスの定義(オーバーキル法) ········ 27
附属書E(参考)運転サイクル······························································································· 29
参考文献 ···························································································································· 33
T 0816-1:2010 (ISO 17665-1:2006)
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第12条第1項の規定に基づき,日本医療機器学会(JSMI)及び財団法人日本規
格協会(JSA)から,工業標準原案を具して日本工業規格を制定すべきとの申出があり,日本工業標準調査会
の審議を経て,厚生労働大臣が制定した日本工業規格である。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願,実用新案権又は出願公開後の実用新案登録出願に
抵触する可能性があることに注意を喚起する。厚生労働大臣及び日本工業標準調査会は,このような特許
権,出願公開後の特許出願,実用新案権及び出願公開後の実用新案登録出願にかかわる確認について,責
任はもたない。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格
JIS
T 0816-1:2010
(ISO 17665-1:2006)
ヘルスケア製品の滅菌−湿熱−
第1部:医療機器の滅菌プロセスの開発,
バリデーション及び日常管理の要求事項
Sterilization of health care products-moist heat-
Part 1: Requirements for the development, validation and
routine control of a sterilization process for medical devices
序文
この規格は,2006年に第1版として発行されたISO 17665-1を基に,技術的内容及び対応国際規格の構
成を変更することなく作成した日本工業規格である。
なお,この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
医療機器を滅菌するための湿熱滅菌プロセスにおいて,この規格に適合することで適切な微生物の殺滅
が実施できる。さらに,この要求事項に適合することで,この作業自体の信頼性及び再現性を保証し,そ
れによって一定の信頼度をもって滅菌処理後の製品上に生育可能な微生物の存在する確率が低いことを,
あらかじめ知ることができる。
附属書A〜附属書Eに参考として示す事項は,要求事項ではなく,また,監査員のためのチェックリス
トとして作成されたものでもない。参考として示した事項は,要求事項に適合するための適切な方法を示
すものであり,この規格の要求事項への適合性を達成することに有効な場合は,これらに示す以外の方法
を使用してもよい。
1
適用範囲
1.1
適用
1.1.1
この規格は,医療機器の湿熱滅菌プロセスの開発,バリデーション及び日常管理の要求事項につい
て規定する。
注記 この規格の適用範囲は医療機器に限定しているが,他のヘルスケア製品にも適用できる。
1.1.2
この規格において,湿熱滅菌プロセスには次を含むが,これに限定しない。
a) 重力置換式飽和蒸気システム
b) 真空脱気式飽和蒸気システム
c) 空気蒸気混合
d) 水散布
e) 水浸せき(漬)
注記 附属書Eも参照。
2
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1.2
適用除外
1.2.1
この規格は,スクレイピー,牛海綿状脳症,クロイツフェルト・ヤコブ病のような海綿状脳症を引
き起こす病原物質の不活化のプロセスに対する開発,バリデーション及び日常管理についての要求事項は
規定しない。これらの病原物質による汚染の可能性のあるものの処理に対する勧告が,厚生労働省によっ
て通知されている。
注記1 ISO 22442-1,ISO 22442-2及び ISO 22442-3参照。
注記2 平成9年4月24日薬機第71号厚生省薬務局医療機器開発課長通知“クロイツフェルト・ヤ
コブ病感染防止のための医療用具の消毒について”参照。
1.2.2
この規格は,滅菌剤として湿熱と他の殺菌剤(例えば,ホルムアルデヒド)との組合せに基づく滅
菌プロセスには適用しない。
1.2.3
この規格は,医療機器を“無菌”又は“滅菌済み”と表示するための要求事項の詳細についても規
定しない。
注記1 医療機器に“無菌”又は“滅菌済み”と表示するための国又は地域の規制要求事項に注意を
払う必要がある。例えば,欧米ではEN 556-1又はANSI/AAMI ST67を参照。
注記2 日本では“無菌”又は“滅菌済み”である旨を医療機器に表示する場合,低リスク品を除き
無菌性保証水準(SAL)は10−6以下であることが規制上求められている。低リスク品について
はSALは10−3以下の場合がある。
1.2.4
この規格は,医療機器の製造のすべての段階を管理するための品質マネジメントシステムを規定す
るものではない。
注記 製造における完全な品質マネジメントシステムは,この規格の要求事項ではないが,滅菌プロ
セスの管理に最低限必要な品質マネジメントシステムの要素は,この規格の該当する箇所(特
に箇条4参照)で要求事項として引用する。医療機器の滅菌を含む製造のすべての段階を管理
する品質マネジメントシステム規格(JIS Q 13485参照)に注意を払う必要がある。薬事法令は,
医療機器の製造について完全な品質マネジメントシステムの適用並びに規制当局又は第三者機
関によるシステムの評価を要求している場合がある。
1.2.5 この規格は,湿熱滅菌装置の設計及び運転に伴う従業員の安全の要求事項を規定するものではない。
注記1 操作上の安全についての要求事項は,IEC 61010-2-040に規定されている。さらに,安全につ
いての労働安全衛生法などの法的規制が存在する。
注記2 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 17665-1:2006,Sterilization of health care products−Moist heat−Part 1: Requirements for the
development, validation and routine control of a sterilization process for medical devices (IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”
ことを表す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格のうちで,西暦年を付記してあるものは,記載の年の版を適用し,その後の改正版(追補を含む。)
は適用しない。西暦年の付記がない引用規格は,その最新版(追補を含む。)を適用する。
JIS Q 13485:2005 医療機器−品質マネジメントシステム−規制目的のための要求事項
注記 対応国際規格:ISO 13485:2003,Medical devices−Quality management systems−Requirements
3
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
for regulatory purposes (IDT)
ISO 10012,Measurement management systems−Requirements for measurement processes and measuring
equipment
ISO 11138-1,Sterilization of health care products−Biological indicators−Part 1: General requirements
ISO 11138-3,Sterilization of health care products−Biological indicators−Part 3: Biological indicators for
moist heat sterilization processes
ISO 11140-1,Sterilization of health care products−Chemical indicators−Part 1: General requirements
ISO 11140-3,Sterilization of health care products−Chemical indicators−Part 3: Class 2 indicator systems for
use in the Bowie and Dick-type steam penetration test
ISO 11140-4,Sterilization of health care products−Chemical indicators−Part 4: Class 2 indicators as an
alternative to Bowie and Dick-type test for detection of steam penetration
ISO 11140-5,Sterilization of health care products−Chemical indicators−Part 5: Class 2 indicators for Bowie
and Dick-type air removal tests
ISO 11607-1,Packaging for terminally sterilized medical devices−Part 1: Requirements for materials, sterile
barrier systems and packaging systems
ISO 11607-2,Packaging for terminally sterilized medical devices−Part 2: Validation requirements for forming,
sealing and assembly processes
ISO 11737-1,Sterilization of medical devices−Microbiological methods−Part 1: Determination of a
population of microorganisms on products
ISO 11737-2,Sterilization of medical devices−Microbiological methods−Part 2: Tests of sterility performed
in the validation of a sterilization process
ISO 17664,Sterilization of medical devices−Information to be provided by the manufacturer for the
processing of resterilizable medical devices
3
用語及び定義
この規格で用いる主な用語及び定義は,次による。
3.1
空気検知器 (air detector)
蒸気及びその凝縮物の流れの中又は滅菌チャンバ内の空気などの非凝縮性ガスの存在を検出するように
設計された機器。
3.2
自動制御器 (automatic controller)
あらかじめ規定された運転サイクル(3.29)の変数によって運転サイクルの要求された工程の間,滅菌器を
連続的に操作する装置。
3.3
バイオバーデン (bioburden)
製品及び/又は無菌バリアシステムの上又は内部に存在する生育可能な微生物群(ISO/TS 11139定義2.2
参照)。
4
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.4
バイオロジカルインジケータ (biological indicator)
ある特定の滅菌プロセスに対して,あらかじめ定めた抵抗性を示す生育可能な微生物を含む試験システ
ム(ISO/TS 11139定義2.3参照)。
3.5
校正 (calibration)
計器若しくは測定系の示す値,又は実量器若しくは標準物質の表す値と,標準によって実現される値と
の間の関係を確定する一連の作業(JIS Z 8103参照)。
注記 校正には,計器を調整して誤差を修正することは含まない。
3.6
ケミカルインジケータ (chemical indicator)
非バイオロジカルインジケータ (non-biological indicator)
プロセスにばく(曝)露することで生じる化学的又は物理的な変化に基づき,あらかじめ定義した一つ
又は複数の滅菌プロセス変数の変化を表すシステム(ISO/TS 11139定義2.6参照)。
3.7
容器封入製品 (contained product)
滅菌プロセスのすべての段階において,滅菌器の中の環境が,直接その製品に接触しない製品。
注記 滅菌器の中の環境は加熱及び冷却のためにだけ用いられ,滅菌効果の達成には用いられない。
例えば,封止された瓶の中の液体。
3.8
修正 (correction)
検出された不適合を除去するための処置。
注記 是正処置と併せて,修正が行われることもある(JIS Q 9000定義3.6.6参照)。
3.9
是正処置 (corrective action)
検出された不適合又はその他の検出された望ましくない状況の原因を除去するための処置(JIS Q 9000
定義3.6.5参照)。
注記1 不適合の原因は,一つ以上のことがあり得る。
注記2 予防処置は発生を未然に防止するためにとるのに対し,是正処置は再発を防止するためにと
る。
注記3 修正と是正処置とは異なる。
3.10
D値 (D value)
D10値 (D10 value)
定められた条件下で,試験に用いる微生物数の90 %を不活化するのに要する時間又はばく露量
(ISO/TS 11139定義2.11参照)。
注記 この規格では,D値は90 %減少を達成するために必要とされるばく露時間のことをいう。
3.11
開発 (development)
仕様を作り上げる行為(ISO/TS 11139定義2.13参照)。
5
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.12
環境管理 (environmental control)
定義した領域の条件を,あらかじめ定めた限度値内に収めるために実施する工学的及び/又は手続き上
のシステム(ISO/TS 11139定義2.16参照)。
注記 そのようなシステムには,空気及び流体用のフィルタ,表面消毒操作,作業員服装及び管理上
の諸手順を含むことがある。
3.13
平衡時間 (equilibration time)
参照測定点(3.41)が滅菌温度(3.54)に達した後,滅菌負荷内のすべての点が滅菌温度に達するまでに要す
る時間。
3.14
確立 (establish)
理論的評価によって決定し,実験によって確認すること(ISO/TS 11139定義2.17参照)。
3.15
ばく(曝)露時間 (exposure time)
プロセスパラメータを,それぞれにあらかじめ定めた許容範囲内に維持した時間(ISO/TS 11139定義2.18
参照)。
3.16
許容外 (fault)
あらかじめ定めた許容幅から一つ又は複数のプロセスパラメータが外れること(ISO/TS 11139定義2.19
参照)。
3.17
F0値 (F0 value)
滅菌プロセスの微生物致死量であって,10 ℃のz値をもつ微生物について,121.1 ℃の温度に等価な時
間(分)で表される値。
3.18
ヘルスケア製品[health care product(s)]
体外診断用医療機器を含む医療機器,又は生物製剤を含む医薬品(ISO/TS 11139定義2.20参照)。
3.19
保持時間 (holding time)
参照測定点及び滅菌負荷ですべての点の温度が継続的に滅菌温度幅に保たれている時間。
3.20
据付適格性の確認 (IQ) [installation qualification (IQ)]
装置がその仕様を満たして提供され,かつ,据え付けられたことの証拠を取得して文書化するプロセス
(ISO/TS 11139定義2.22参照)。
3.21
載荷形態 (load configuration)
滅菌する製品の滅菌チャンバ内での,チャンバ固定の架台,数,型,配置及び方向についてあらかじめ
定めた組合せ。
6
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.22
メンテナンス (maintenance)
目的とするものが,その要求された機能を果たせる状態に維持し,また,修理することを意図したすべ
ての技術的及びそれに付随する管理的活動の組合せ。
3.23
医療機器 (medical device)
あらゆる計器,器械,用具,機械,器具,埋込み用具,体外診断薬,検定物質,ソフトウェア,材料又
はその他の同類のもの若しくは関連する物質であって,単独使用か組合せ使用かを問わず,製造業者が人
体への使用を意図し,その使用目的が次の一つ以上であり,
− 疾病の診断,予防,監視,治療,又は緩和
− 負傷の診断,監視,治療,緩和,又は補助
− 解剖学的又は生理学的なプロセスの検査,代替,又は修復
− 生命支援又は維持
− 受胎調整
− 医療機器の殺菌
− 人体から採取される標本の体外試験法による医療目的のための情報提供
薬学,免疫学,又は新陳代謝の手段によって体内又は体表において意図したその主機能を達成すること
はないが,それらの手段によって機能の実現を補助するものである(JIS Q 13485定義3.7参照)。
3.24
測定系 (measuring chain)
インプット(測定対象の量)からアウトプット(測定結果)に至る測定信号の経路を構成する測定装置
及び測定システムの一連の要素。
3.25
微生物 (microorganism)
細菌,真菌,原虫及びウィルスを包含する微小体(ISO/TS 11139定義2.26参照)。
注記 ある種の規格によっては,滅菌プロセスのバリデーション及び/又は日常管理において,上記
で定義したすべてのタイプの微生物の不活化の滅菌プロセスの有効性を立証することを要求し
ないこともある。
3.26
湿熱 (moist heat)
微生物の死滅を達成する目的のため,蒸気又は液体の水によって供給される,湿り気のある状態での熱
エネルギー。
3.27
非凝縮性ガス (non-condensable gas)
飽和蒸気プロセスの状態下で凝縮しない空気及び/又は他の気体。
3.28
運転適格性の確認 (OQ) [operational qualification (OQ)]
据え付けられた装置をその操作手順によって用いたとき,あらかじめ定めた限度内で作動する証拠を取
得し,文書化するプロセス(ISO/TS 11139定義2.27参照)。
7
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.29
運転サイクル (operating cycle)
あらかじめ定めた順序によって実行する段階的なプロセスの一式(IEC 61010-2-040参照)。
3.30
包装システム (packaging system)
無菌バリアシステムと保護包装との組合せ(ISO/TS 11139定義2.28参照)。
3.31
稼働性能適格性の確認 (PQ) [performance qualification (PQ)]
操作手順によって据え付けられ,運転されている装置が,あらかじめ定めた判断基準に恒常的に適合し
て稼働し,その結果仕様に適合する製品を生産することができるという証拠を取得し,文書化するプロセ
ス(ISO/TS 11139定義2.30参照)。
3.32
予防処置 (preventive action)
起こり得る不適合,又はその他の望ましくない起こり得る状況の原因を除去するための処置(JIS Q 9000
定義3.6.4参照)。
注記1 起こり得る不適合の原因は,一つ以上のことがある。
注記2 是正処置は再発を防止するためにとるのに対し,予防処置は発生を未然に防止するためにと
る。
3.33
平たん(坦)期間 (plateau period)
平衡時間及び保持時間の合計。
3.34
プロセスチャレンジデバイス (PCD) [process challenge device (PCD)]
滅菌プロセスに対する定義された抵抗性を示すように設計された,滅菌プロセスの性能を評価するため
に用いられる物(ISO/TS 11139定義2.33参照)。
3.35
プロセスパラメータ (process parameter)
あらかじめ定めたプロセス変数の値(ISO/TS 11139定義2.34参照)。
注記 滅菌プロセスの仕様には,プロセスパラメータ及びその許容範囲が含まれる。
3.36
プロセス変数 (process variable)
滅菌プロセスの条件で,その変化が微生物の殺滅効果に変動を与えるような条件。
例えば,時間,温度,圧力,濃度,湿度,波長(ISO/TS 11139定義2.35参照)。
3.37
製品 (product)
プロセスの結果(ISO/TS 11139定義2.36参照)。
注記 この規格では,製品は有形のものであり,原料,中間品,半組立品及びヘルスケア製品でもあ
り得る。
8
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.38
製品ファミリー (product family)
質量,材質,構造,形状,内腔,包装システム,そして滅菌プロセス(3.53参照)の同等の困難性の存
在のような属性によって特徴づけられる製品のグループ及びサブグループ。
3.39
参照チャレンジデバイス (reference challenge device)
容器封入製品又は滅菌負荷と既知の熱的な関係をもつデバイス。
3.40
参照負荷 (reference load)
滅菌が困難な対象の組合せを代表するものとしてあらかじめ定めた滅菌負荷。
3.41
参照測定点 (reference measuring point)
滅菌サイクルを制御するための温度センサの位置。
3.42
標準菌 (reference microorganism)
公的微生物保存機関から得られる菌株(ISO/TS 11139定義2.39参照)。
3.43
適格性の再確認 (requalification)
規定された滅菌プロセスが引き続き許容できるものであることを確認するために,バリデーションの一
部分を反復実施すること(ISO/TS 11139定義2.40参照)。
3.44
飽和蒸気 (saturated steam)
凝結と蒸発との間で平衡状態にある気化した水。
3.45
サービス (services)
外部から供給を受けるもので,装置が機能を発揮するのに必要なもの。
例えば,電気,水,圧縮空気,排水(ISO/TS 11139定義2.41参照)。
3.46
仕様書 (specification)
要求事項を記述した文書(JIS Q 9000 定義3.7.3参照)。
3.47
あらかじめ定める (specify)
承認を受けた文書の中で詳細を明記すること(ISO/TS 11139定義2.42参照)。
3.48
無菌 (sterile)
生育可能な微生物が存在しないこと(ISO/TS 11139定義2.43参照)。
3.49
無菌性 (sterility)
生育可能な微生物が存在しない状態(ISO/TS 11139定義2.45参照)。
注記 実際には,そのような微生物が存在しない絶対的な状態を証明することはできない[滅菌(3.51)
9
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参照]。
3.50
無菌性保証水準 (SAL) [sterility assurance level (SAL)]
滅菌後に,生育可能な1個の微生物が製品上に存在する確率(ISO/TS 11139定義2.46参照)。
注記 SALは定量値として一般的に,10−3又は10−6と表す。この定量値を無菌性保証に適用すると
きは,10−6のSALは10−3のSALよりも小さい値であるが,より高い無菌性保証を与える。
3.51
滅菌 (sterilization)
製品(3.37)を生育可能な微生物が存在しない状態にするために用いる,バリデートされたプロセス
(ISO/TS 11139定義2.47参照)。
注記 滅菌プロセスでは,微生物の不活化は指数関数で表現される。したがって,個々の製品に存在
する生育可能な微生物は,確率論の観点から表現が可能である。この確率は,非常に低い数に
減らすことはできるが,決してゼロに低減することはできない。
3.52
滅菌負荷 (sterilization load)
滅菌プロセスを用いて一緒に滅菌される,又はされた製品(ISO/TS 11139定義2.48参照)。
3.53
滅菌プロセス (sterilization process)
あらかじめ定めた無菌性についての要求事項を達成するための一連の活動又は操作(ISO/TS 11139定義
2.49参照)。
注記 この一連の活動又は操作には,(必要ならば)前処理,規定された条件下での滅菌剤へのばく露
及び必要な後処理のすべてを含む。滅菌プロセスに先立つすべての洗浄,消毒又は包装操作は
含まない。
3.54
滅菌温度 (sterilization temperature)
滅菌温度幅の最低温度。
3.55
滅菌温度幅 (sterilization temperature band)
保持時間の間,滅菌負荷において実現されていると考えられる,滅菌温度及び最高許容温度で表現され
る温度範囲。
3.56
滅菌チャンバ (sterilizer chamber)
滅菌器の滅菌負荷を受け入れる部分。
3.57
滅菌剤 (sterilizing agent)
あらかじめ定めた条件下で,無菌性を達成するために十分な微生物の殺滅効果をもつ物理的若しくは化
学的媒体又はその組合せ(ISO/TS 11139定義2.50参照)。
3.58
熱エネルギー (thermal energy)
熱として与えられるエネルギー。
10
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3.59
無菌性の試験 (test of sterility)
開発,バリデーション又は適格性の再確認の一部として実施する技術的操作で,製品又はその一部に生
育可能な微生物の存在の有無を判定するために行う試験(ISO/TS 11139定義2.54参照)。
3.60
バリデーション (validation)
プロセスが,恒常的にあらかじめ定めた仕様に適合する製品を得ることができることを確立するために,
要求される結果を得て,記録し,解釈するための文書化した手順(ISO/TS 11139定義2.55参照)。
3.61
z値 (z value)
D値が10倍変化するのに要する温度変化。
4
品質マネジメントシステムの要素
4.1
文書化
4.1.1
開発,バリデーション,日常管理及び滅菌からの製品リリースの手順をあらかじめ定めなければ
ならない。
4.1.2
この規格が要求する文書及び記録は,あらかじめ指名した職員(4.2.1参照)によってレビュー及
び承認されなければならない。文書及び記録は,JIS Q 13485の該当する箇条によって管理しなければなら
ない。
4.2
経営者の責任
4.2.1
この規格の要求事項を実施し,これに適合するための責任及び権限をあらかじめ定めなければな
らない。責任は,JIS Q 13485の該当する箇条によって,力量のある職員に割り当てなければならない。
4.2.2
この規格の要求事項を,他の品質マネジメントシステムの組織によって実行する場合は,それぞ
れの組織の責任及び権限をあらかじめ定めなければならない。
4.3
製品実現
4.3.1
購買の手順をあらかじめ定めなければならない。これらの手順は,JIS Q 13485の該当する箇条に
適合しなければならない。
4.3.2
製品の識別及びトレーサビリティの手順をあらかじめ定めなければならない。この手順は,JIS Q
13485の該当する箇条に適合しなければならない。
4.3.3
この規格の要求事項に適合するために,試験用の計器を含むすべての機器の校正について,JIS Q
13485又はISO 10012の該当する箇条に適合したシステムを構築しなければならない。
4.4
測定,分析及び改善−不適合製品の管理
不適合と認定した製品の管理,修正,是正処置及び予防処置の手順をあらかじめ定めなければならない。
これらの手順は,JIS Q 13485の該当する箇条に適合しなければならない。
5
滅菌剤の特性
5.1
滅菌剤
5.1.1
この規格では,滅菌剤は湿熱でなければならない。
5.1.2
滅菌剤に含まれる汚染物質は,意図する使用方法に対して,製品の安全性に悪影響を及ぼしては
ならない。
11
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.2
微生物殺滅効果の有効性
湿熱を広く理解されている条件幅の外で採用する場合は,微生物殺滅効果の有効性を確立し,文書化し
なければならない。
注記 湿熱の微生物殺滅効果の有効性及び滅菌プロセスへの適用については,広く文書化されており,
刊行物によって入手が可能である。
5.3
材料への影響
滅菌剤が,材料に及ぼす影響を箇条6及び箇条7の要求事項によって評価しなければならない。
5.4
環境への配慮
湿熱は,環境への影響が大きいとは通常考えられていないが,滅菌プロセスの運用による環境への影響
の可能性を評価し,すべての環境の保護に必要な手段を明確にしなければならない。この評価は,(ある場
合はすべての)環境への影響及び管理する手段(定めた場合)を含んで文書化しなければならない。
6
プロセス及び装置の特性
6.1
プロセスの特性
6.1.1
一般
すべての滅菌プロセスをあらかじめ定めなければならない。この仕様には,次の事項を含まなければな
らない。
a) 運転サイクルの記載。
b) プロセスパラメータ及びその許容範囲。
c) 滅菌できる製品ファミリー。
d) 滅菌プロセスの効果を確実にするために滅菌前の製品の状態調整(プレコンディショニング)が必要
な場合,状態調整についての要求事項。
e) 参照測定点の位置。
f)
空の滅菌チャンバ(固定された滅菌器内の部品を含む。)での最低及び最高圧力。
g) プロセスの各段階での圧力降下及び上昇速度並びにその許容幅。
h) 滅菌チャンバに供給する液体,空気,ガス又は蒸気の中に製品に対して悪影響を及ぼす汚染物質が存
在する可能性がある場合は,各汚染物質の最大許容量。
i)
滅菌プロセスが運用されていることを検証するために測定し,使用するプロセス変数。
j)
載荷形態。
k) 滅菌負荷のサイズ及び/又は質量についてのあらゆる制限事項。
l)
滅菌プロセスが,再現性を保持していることを検証するために使用する,定期的な試験及び許容限界
(該当する場合)。
m) バイオロジカルインジケータ(8.5又は8.6)の位置及び判定基準(用いる場合)。
n) ケミカルインジケータ(8.8)の位置及び判定基準(用いる場合)。
o) 微生物学的方法を滅菌プロセスの有効性の確立に用いる場合には,割り当てた滅菌プロセスで滅菌負
荷全体で達成される最低のサイクル致死率,及び滅菌負荷においてその致死率を測定した方法。
p) 運転サイクル後の処理を滅菌プロセスに含む場合は,その処理。
6.1.2
飽和蒸気プロセス
6.1.1の要求事項に加え,滅菌器に注入する蒸気を滅菌剤として用いる飽和蒸気滅菌プロセスの仕様には,
次の事項を含まなければならない。
12
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
a) 保持時間の間,空の滅菌チャンバ(固定されたチャンバ内の部品を含む。)の中で測定される保持時間,
最低及び最高温度(並びにその場所)。
b) 保持時間の間,参照測定点で測定された温度と,滅菌チャンバ内で測定した圧力から蒸気表によって
求められる理論的蒸気温度との最大許容差。
注記 飽和蒸気圧から求められる理論的蒸気温度は,ISO/TS 17665-2では蒸気表とともに,次の式
によって求められることが示されている。
T=42.677 6+[−389 2.7/(ln P−9.486 54)]−273.27
ここに,
T: 理論的蒸気温度 (℃)
P: 圧力 (MPa)
c) 6.1.1 c) で定めた製品ファミリーが,その構造,材質又は載荷形態によって蒸気浸透が困難であるこ
とが既知の場合,非凝縮性ガスが,滅菌する製品の表面における飽和蒸気の存在を妨害しないことを
検証するための蒸気浸透試験にかかわる事項。
なお,蒸気浸透試験を実施しなくてもよい根拠があれば試験を実施する必要はない。
d) 滅菌する製品及びその包装システムの完全性に対して,水が悪影響を与える可能性がある場合,滅菌
チャンバに注入する飽和蒸気内に浮遊してもよい水の最大量。
e) 6.1.1 c),6.1.1 j),6.1.1 k)で定めた滅菌負荷に対する滅菌プロセスの効果を確認又は判定するために用
いる参照負荷。
f)
滅菌プロセスの運用をチェックするために監視機器を用いる場合,その監視機器,配置(設置場所)
及び結果を判断する方法。
g) 質量変化又は感覚的な湿り気で測る参照負荷の乾燥度。
6.1.3
容器封入製品プロセス
滅菌プロセスの仕様には6.1.1の要求事項に加えて,6.1.1 c)で識別した製品ファミリーに割り当てた製
品の中で最も滅菌が困難である滅菌負荷及び載荷形態について,次の情報を含まなければならない。
a) 製品及びその容器,又は該当する場合は,参照製品の詳細。
b) 滅菌負荷のサイズ,並びに滅菌チャンバ内での配置場所,方向及び支持システム。
c) 設定した致死率に対応した運転サイクル時間を決定するために,空のチャンバ及び滅菌負荷周辺の自
由空間の両方において測定した温度プロファイル及びその測定場所。
d) 設定した致死率に対応した運転サイクル時間についての,最高及び最低温度並びに変化速度。
e) 最高及び最低温度を測定できる場所を確立するための方法。
6.2
装置
6.2.1
滅菌プロセスを運用するために使用する装置を,あらかじめ定めなければならない。この仕様には,
次の事項を含まなければならない。
a) 装置及び必要な附属品。
b) 滅菌チャンバ内へ蒸気,ほかのすべてのガス又は液体を貯蔵し運搬するために用いる装置及び附属品
のあらゆる部分の構造材質[6.1.1 h)参照]。
c) 滅菌プロセスの制御,指示,監視及び電子的な記録又は恒久的な記録を提供するそれぞれの測定系に
ついての次の事項。
1) 測定系についての記載。
2) センサの特性及び位置。
3) 国家計量標準へトレースできる測定系の校正の検証方法。
13
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 圧力の最大変化速度。
e) すべての目視,音,記録などの警報によって滅菌装置で認識される許容外(3.16参照)事項。
f)
職員及び環境保護を含む安全側面。
g) 装置から環境への排出についての国又は自治体の規制への適合についての表明。
h) 運転サイクルで真空を用いる場合,滅菌チャンバへの空気漏れの水準を測定するために使う試験及び
合否判定基準。
i)
非凝縮性ガス(空気を含む。)を検出するための機器(例えば,空気検知器)を装着している場合,設
定を含むこの機器についての情報。
6.2.2
装置及び附属品の操作手順をあらかじめ定めなければならない。この仕様には,次の事項を含ま
なければならない。
a) 自動制御器にプログラムした運転サイクル及びプログラムを変更する方法。
b) ステップごとの運転方法。
c) プロセスパラメータに影響を与える失敗の識別を可能にする方法及びそのような場合に取る処置。
d) 校正及びメンテナンスの方法。
e) 制御,表示又は記録のための測定の間違いを識別する手段。
f)
技術支援への連絡方法。
6.2.3
装置を据え付ける場所をあらかじめ定めなければならない。この仕様には,次の事項を含まなけ
ればならない。
a) 装置が据え付けられる場所,空間及び環境。
b) 据付け方法の説明書。
c) 装置が正常な機能を発揮するために必要なそれぞれのサービスの詳細。これには(該当する場合)次
を含む。
1) 断熱方法
2) 最高及び最低圧力
3) 最高温度
4) 最小流量
5) ろ過
6) 最高及び最低電圧並びに最大電圧電流
7) 飽和蒸気中の非凝縮性ガス及び液体の水の最大水準
8) 各汚染物質の最大量
d) 装置の基本的な重量部分を支えることのできる耐荷重構造。
e) 蒸気,ガス,空気及び水を供給するための配管などを構成する材質。
f)
装置から環境への排出物質に関連する国及び自治体の規制への適合性を示す文書。
6.2.4
滅菌チャンバの中で滅菌負荷を保持するシステムは,滅菌条件の均一な達成を妨害するか,又は
製品及びその包装に損傷を与える原因となってはならない。
6.2.5
あらかじめ定めたプロセスパラメータの達成に失敗したときに,無効な滅菌プロセスを有効であ
ると誤判断しないことを確実にするための手段を備えなければならない。
6.2.6
装置の製造業者は,ソフトウェア及び/又はその製造への適用が,その装置仕様への適合性に影
響を与える可能性がある場合について,ソフトウェアの適用及び変更についてのバリデーションの文書化
した手順を確立しなければならない。
14
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7
製品の定義
7.1
滅菌する製品を,あらかじめ定めなければならない。
7.2
ISO 11607-1及びISO 11607-2に適合した製品の包装システム(用いる場合)を,あらかじめ定めな
ければならない。
7.3
製品ファミリーに割り当てる製品及びその包装システムを,あらかじめ定めなければならない。
7.4
製品ファミリーに割り当てるための判断基準を,あらかじめ定めなければならない。
7.5
プロセスチャレンジデバイス(PCD)を,製品及びその包装システムを代表するチャレンジとして使用
する場合は,それを定義しなければならない。
7.6
製品及びその包装システム(用いる場合)を,ばく露する各プロセス変数について,その限界値を
あらかじめ定めなければならない。限界値は,使用するすべての材質及びその組合せに対して明確にしな
ければならない。すべてのプロセスパラメータの組合せの下での製品の物理的,化学的特性及び生体適合
性に対する滅菌剤へのばく露,又は反復のばく露(該当する場合)のワーストケースの影響を明確にしな
ければならない。
プロセス変数の例を,次に示す。
− 温度
− 限界値での滞留時間
− 圧力
− 圧力変化速度
− 温度変化速度
注記 これらのプロセスパラメータの限界値を定めておくのは,定めた値を超えることによって製品
及び/又はその包装の性能に悪影響を与える場合があるからである。
7.7
滅菌前の製品及び/又はその包装システム内に存在する湿り気が滅菌プロセスの効果に影響を与え
る場合は,湿り気の水準の限界値をあらかじめ定めなければならない。
7.8
予定している滅菌温度及びばく露時間で容器封入製品を処理するとき,製品の安定性及び有効性に
悪影響があってはならない。
7.9
滅菌後に製品上に残っている汚染物質が,製品の品質に影響を与える可能性がある場合,汚染物質
及びその最大許容限界をあらかじめ定めなければならない。
7.10 滅菌する製品及び/又は包装システムの状態が,滅菌プロセスの有効性を損なわないことを保証す
るためのシステムをあらかじめ定めなければならない。このシステムには少なくとも次の要素を含まなけ
ればならない。
a) 洗浄又は消毒(再処理を意図する場合)を実施する場合,再使用包装システム(例えば,滅菌コンテ
ナ)を含む有効な洗浄及び消毒。
b) 滅菌プロセスばく露前後の包装システムの完全性。
c) 製品のバイオバーデンに影響を与える可能性のある環境管理。
d) プロセスパラメータをバイオバーデン手法によって決定する場合は,ISO 11737-1によるバイオバーデ
ンの見積り。
8
プロセスの定義
8.1
プロセスパラメータ及びその限界値を含む滅菌プロセスをあらかじめ定めなければならない。この
プロセスの確立に当たっては,物理的プロセスパラメータを測定し,該当する場合,再現性の確認に利用
15
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
しなければならない。
8.2
滅菌プロセスによって達成する製品上及び/又は製品内の最低の無菌性保証水準(SAL)を,あらかじ
め定めなければならない。
8.3
滅菌プロセスは7.6で定めたプロセスパラメータの範囲を超え,また,製品及びその包装システム(用
いる場合)を7.9で定めた汚染物質にばく露してはならない。
8.4
製品を飽和蒸気によって滅菌する場合,保持時間の開始時の残存空気及び非凝縮性ガスは,内孔,
中空及びチューブを含む製品の全表面の飽和蒸気との接触を妨げるものであってはならない。
8.5
バイオロジカルインジケータシステムを8.11 a)で規定した滅菌プロセスの確立の一部として用いる
場合,これらはISO 11138-1及びISO 11138-3に適合しなければならない。その微生物,微生物数,抵抗性
及び配置の方法は,使用する滅菌プロセスの性質及び予想できるか又は確立したバイオバーデンを考慮し
て定めなければならない。
配置の方法は,製品に直接接種しても,接種した担体を製品の中に入れてもよい。
8.6
バイオロジカルインジケータシステムを8.11 b),c)及び/又はd)で規定した飽和蒸気滅菌プロセス
を確立する方法の一部として用いる場合,これらはISO 11138-1及びISO 11138-3に適合しなければならな
い。
8.7
容器封入製品について,あらかじめ定めた滅菌プロセスにばく露したときの製品及びその包装シス
テムが試験微生物致死率に与える影響は,既知でなければならない。
8.8
ケミカルインジケータを滅菌プロセスの確立の一部として用いる場合,これらはISO 11140シリー
ズの該当する部分に適合したものを用い,滅菌プロセスの実施前,実施中又は実施後における医療機器と
の反応,汚染及び/又は移動による悪影響がないようにしなければならない。
8.9
箇条7で定めた製品の処理のための滅菌プロセスの有効性の評価に,プロセスチャレンジデバイス
を用いる場合,プロセスチャレンジデバイスの正当性,試験方法及び許容限界を確立し文書化しなければ
ならない。
8.10 滅菌プロセスは,少なくとも次の一つから確立しなければならない。
− 医療機器の製造業者及び/又は包装材料の製造業者及び/又は滅菌器の製造業者から提供されるデー
タ(ISO 17664参照)。
− 既に製品ファミリーに割り当てた製品との類似性。
− 必要とするSALを得ることができる運転サイクルの開発。
8.11
滅菌プロセスによって製品上及び/又は製品内で得られるSALは,次のいずれかによるものでなけ
ればならない。
a) バイオバーデンの知見(附属書B及び附属書C参照)によって確立するもの。
b) オーバーキル法(附属書D参照)で決定するもの。
c) 保持時間の間,製品のすべての部分が,薬局方から選択したプロセスパラメータにばく露されている
ことを立証することによって定めるもの。
注記 薬局方から選ばれたプロセスパラメータの例として,ISO/TS 17665-2では次の値が示されて
いる。
温度 (℃)
時間(分)
121
15
126
10
134
3
16
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 製品を滅菌プロセスに対応する製品ファミリーに割り当て,その製品ファミリーに属する製品の平衡
時間の最大値を超えないことを示すことによって,c)で定めた要求事項以上であるとみなされるもの。
8.12 微生物学的方法で滅菌プロセスを確立する場合は,次を適用する。
− バイオバーデンの測定は,ISO 11737-1に従って実施する。
− 無菌性の試験は,ISO 11737-2に従って実施する。
− プロセスの確立で用いる製品は,日常的に製造するものを代表するもの。
− プロセス確立に使用する装置は,実際の滅菌プロセスで目標とする致死率と比べて低い致死率(微生
物不活化の程度が小さい)のプロセスパラメータの組合せを再現性よく運用できる能力があるもの。
8.13 滅菌プロセスのばく露の後で,無菌性の維持のために製品及びその包装システムの処理が必要な場
合は,この処理についてあらかじめ定めなければならない。
9
バリデーション
9.1
一般
9.1.1
バリデーションは,文書化した手順に従って実施しなければならない。
9.1.2
バリデーションの間に使用する固定及び可搬式の装置及び機器は,その仕様に適合していることを
検証しなければならない。
9.1.3
バリデーションの間に製品,装置及び滅菌プロセスに対して行ったすべての変更は,記録し,根拠
を明確にし,関連する仕様書を変更しなければならない(箇条12参照)。
9.1.4
バリデーションで使う目的の試験計器のそれぞれの測定系は,次の状態でなければならない。
− 国家計量標準にトレース可能な校正状態にある。
− 該当する場合,妥当なメンテナンスの証明をもつ。
− 技術及び該当するマネジメントシステムの要求事項によって検証した校正状態にある。
− 校正の検証は,試験結果の判定又は滅菌プロセスを制御するために使用する値で実施されている。
9.1.5
滅菌器に固定した計器の指示及び記録は,近傍の独立した試験計器のセンサによって得られた値と
の関係を検証しなければならない。
9.1.6
該当する場合,据付適格性の確認(IQ),運転適格性の確認(OQ)又は稼働性能適格性の確認(PQ)の間
に,警報などの許容外を認識するシステムの機能及びその性能仕様への適合を検証しなければならない。
9.1.7
既存の滅菌装置及び滅菌プロセスを新しい製品の処理に用いる場合,既存の滅菌プロセスの運用に
影響を及ぼすような装置又は既存の滅菌負荷への変更がないことを示すことによって,バリデーションの
IQ及びOQの段階を省略してもよい。
9.1.8
予定している定期的な試験の妥当性を検証しなければならない[6.1.1 l)及び10.3参照]。
9.2
据付適格性の確認 (IQ)
9.2.1
装置
装置及び文書化が,6.2.1,6.2.2及び6.2.3に適合し,サービスが6.2.3に適合していることを検証しなけ
ればならない。
9.2.2
据付け
据付けが,6.2.3に適合していることを検証しなければならない。
9.2.3
機能
6.2.1で定めた装置及び安全システムが,その仕様によって機能し,運転サイクルが,6.1.1 a)で定めたも
のに従っており,サービス及び装置からの漏れを示すような状況がないことを検証しなければならない。
17
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.3
運転適格性の確認 (OQ)
9.3.1
運転適格性の確認は,据え付けた装置が,箇条8で定めた滅菌プロセスを運用できることを立証し,
該当する場合,6.1で定めた要求事項それぞれに対するデータを確立しなければならない。
9.3.2
空の滅菌チャンバ(チャンバ内に固定した部品を含む。)及び試験負荷(用いる場合)の中の温度
分布が,要求事項に合致していることを立証するために用いる温度センサの数及び場所の根拠を文書化し
なければならない。
9.4
稼働性能適格性の確認 (PQ)
9.4.1
稼働性能適格性の確認は,日常の滅菌に用いる装置によって,製品があらかじめ定めた滅菌プロセ
スにばく露できることを立証しなければならない。
9.4.2
滅菌負荷の中が,要求事項に合致していることを立証するために用いる温度センサの数及び場所の
根拠を文書化しなければならない。
9.4.3
次の事項を確認し,検証しなければならない。
a) IQ及びOQが成功したことを示す文書。
b) 試験滅菌負荷は,日常処理され,その滅菌プロセスに割り当てた製品ファミリーに属する製品又は製
品ファミリーを代表する最も大きいチャレンジがある製品で構成している。
c) 包装システムは,日常の製造又は再処理を意図しているものと同じである。
d) プレコンディショニングは,6.1.1 d)に適合している。
e) 載荷形態は,6.1.1 j)に従っており,最も滅菌が困難なことが既知である。
f)
滅菌負荷のサイズ及び/又は質量は,6.1.1 k)に適合している。
9.4.4
次の事項のそれぞれについての検討を確立しなければならない。
a) 箇条8で定めた滅菌プロセス,及び7.6で定めたプロセス値の限界値への適合性。
b) 6.1.3で要求するデータ(該当する場合)。
c) 6.1.1 e),6.1.1 m)及び6.1.1 n)で定めた位置における製品上及び製品全体のばく露プロファイル。
d) 6.1.2で定めたプロセスにおいて,保持時間の長さ,その時間中に滅菌負荷の中で測定した最高及び最
低温度並びにその位置。
e) 6.1.2 a)で定めた滅菌プロセスの平たん期間内の,次に示す温度プロファイル。
− 参照測定点で測定した値
− 滅菌負荷の上又は中で測定した値
− 滅菌チャンバ圧力から求めた値
注記 測定された温度と計算された温度との間に最大許容差を定める場合,国又は地域の要求に注
意を払う必要がある。例えば,EN 285を参照。
f) ケミカルインジケータが示す結果(用いる場合)(8.8参照)。
g) プロセスチャレンジデバイスが示す結果(用いる場合)。
h) 包装システムの完全性(用いる場合)。
9.4.5
物理パラメータの測定に加えて,滅菌プロセスをバイオバーデンに基づいて決定する場合,又は微
生物学的手法によって検証する場合は,9.4.4で識別した位置における製品の中及び/又は上にバイオロジ
カルインジケータ(8.5又は8.6参照)を配置し,次の一つによってばく露しなければならない。
− 設定した滅菌プロセスに対して減少させた処理。設定した滅菌プロセスが,最低微生物殺滅効果の有
効性への要求事項に合致することを立証するために,この処理の結果を設定した処理条件に外挿する。
− 滅菌プロセスパラメータの低い側の限界ですべてを実施した処理。この結果は,滅菌プロセスを適用
18
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
したときに,微生物の殺滅効果の有効性への最低限の要求事項を満足していることを確認するのに用
いる。
− “オーバーキル”プロセス
注記 附属書B,附属書C及び附属書Dを参照。
9.4.6 PQには,滅菌プロセス仕様の適合及び滅菌プロセスの再現性を立証するため,少なくとも3回の
連続した滅菌負荷の滅菌プロセスへのばく露を含まなければならない。
9.4.7 PQの間に発生した滅菌プロセス仕様への不適合は,レビューし,修正しなければならない。
バリデートしている滅菌プロセスの有効性に関係しない要因(例参照)によって失敗が発生した場合,
追加の3回連続したばく露なしで,その試験は滅菌プロセスの有効性に関連ないとして文書化してもよい。
例 停電及びサービスの停止,外部監視装置の故障など。
9.5
バリデーションのレビュー及び承認
9.5.1 IQ,OQ及びPQの間に収集し,作成した情報は,バリデーションプロセスの各段階であらかじめ
定めた合否判定基準に適合しているかをレビューしなければならない。レビューの結果は文書化し,権限
のある者が承認しなければならない(4.1.2参照)。
9.5.2 プロセスパラメータ及びその許容幅を含めた滅菌プロセスの仕様を確定しなければならない。この
仕様には,個別の滅菌負荷について適用した滅菌プロセスが適合していると認定するための基準を含め,
少なくとも次の事項を文書化しなければならない。
a) 処理可能な製品ファミリー
b) 載荷形態
c) 滅菌負荷のサイズ及び/又は質量
d) 製品のあらゆるプレコンディショニングの手順
e) 包装システム及びその方法
f)
複数の医療機器を含む包装内での医療機器の配置(該当する場合)
g) 定期的な試験(10.3参照)
h) プロセスチャレンジデバイスと関連する製品ファミリー
i)
バイオバーデン(該当する場合)
10 日常監視及び管理
10.1 滅菌サイクルのそれぞれについて,日常監視及び管理を実行しなければならない。
10.2 メンテナンス及び(該当する場合)適格性の再確認が成功した証拠を検証しなければならない。
10.3 装置の運転状態は,該当する場合,次に挙げるような定期的な試験の結果によって,検証しなけれ
ばならない。ただし,これに限定しない。
a) 滅菌チャンバへの空気漏れ
b) 滅菌チャンバに供給する飽和蒸気又は熱媒体の品質(これには非凝縮性ガス,供給水の導電率,汚染
源及び湿り度のチェックを含む場合がある)
c) 自動制御(例えば,運転サイクルが正しく機能して継続することを検証するための試験)
d) 蒸気浸透
e) 滅菌プロセス(例えば,滅菌プロセスが再現性を維持していることを検証するための試験)
10.4 滅菌プロセスの運用は,日常の監視で記録したデータが,あらかじめ定めた許容範囲内であり,バ
リデーションで得たデータと一致していることを検証しなければならない。また,ケミカルインジケータ
19
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(8.8参照)又はバイオロジカルインジケータシステム(8.5又は8.6参照)を用いる場合は,その結果を
含めて検証しなければならない。
10.5 飽和蒸気プロセスについては,該当する場合,次のデータを含まなければならない。
a) 平たん期間での滅菌温度及びチャンバ内圧力,理論的蒸気温度。
b) 平たん期間の持続時間。
c) 少なくとも運転サイクルの各段階のチャンバ内温度及びチャンバ内圧力。
d) プロセスチャレンジデバイスから得られた結果。
e) プロセス制御の一部として用いる場合,プロセス監視システム内での温度及び/又は圧力。
10.6 容器封入製品の場合,次のデータを含まなければならない(該当する場合)。
a) プロセス制御の一部として用いる場合,参照チャレンジデバイス内の温度。
b) 加熱,ばく露及び冷却のチャンバ内温度並びにチャンバ内圧力のプロファイル。
c) プロセス制御の一部として用いる場合,9.4.4 c)で定めた場所での製品内の加熱,ばく露及び冷却の温
度プロファイル。
d) 平たん期間又は保持時間。
e) 滅菌チャンバ内の加熱媒体の均一性を示すプロセスパラメータの値。
f) 包装システムの乾燥度及び完全性を確認するための滅菌負荷の検査結果。
10.7 すべての記録は,4.1.2によって保管しなければならない。
11 滅菌からの製品のリリース
11.1 記録のレビュー及び滅菌プロセスからの製品リリースの手順をあらかじめ定めなければならない。
この手順には,滅菌プロセスが適合したと判定するための要求事項(9.5.2及び該当する場合10.3参照)を
含めなければならない。要求事項に合致しない場合は,製品は不適合とし,4.4によって取り扱わなければ
ならない。
11.2 処理済みと未処理の製品とを明確に区別することを確実にするシステムを,あらかじめ定めなけれ
ばならない。
12 プロセス有効性の維持
12.1 継続的な有効性の立証
12.1.1 滅菌する製品は,次の事項に適合しなければならない。
a) 箇条7で定めた製品
b) 6.1.1 j)で定めた載荷形態
c) 6.1.1 k)で定めたサイズ及び質量の制限事項
12.1.2 あらかじめ定めた間隔で実行した定期的な試験,校正,メンテナンス業務及び適格性の再確認が,
成功したことを検証しなければならない。
12.1.3 製品を製造及び/又は包装を行う区域の環境の質をあらかじめ定め,定期的に検証しなければなら
ない。
12.1.4 製造及び/又は包装を行う区域での職員の健康,清潔及び衣服についての要求事項をあらかじめ定
め,遵守しなければならない。
12.1.5
滅菌プロセスで真空を用いる場合は,空気漏れ試験をあらかじめ定めた間隔で実行しなければな
らない。
20
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.1.6
迅速で均等な滅菌負荷への蒸気浸透のために,滅菌プロセスにおいて滅菌チャンバからの空気除
去を実施する場合は,滅菌器を使用する前に蒸気浸透試験を毎日実施しなければならない。
蒸気浸透試験は,通常,プロセスに対して空気除去及び蒸気浸透の定義したチャレンジをもつ機器を使
用して実施する。工業的な滅菌において定常的に飽和蒸気プロセスを用いる場合は,物理的な他の方法に
よる確認及びプロセスの失敗の可能性についてのリスク評価に基づき,蒸気浸透を妨げないことが既知で
ある滅菌負荷について蒸気浸透試験を省略してもよい。
12.1.7
該当する場合,製品はバイオバーデン要求事項に適合しなければならない。
12.2 再校正
滅菌プロセスの制御,指示又は記録に用いる各測定系の正確さ及び信頼性は,4.3.3によって定期的に検
証しなければならない。
12.3 装置のメンテナンス
12.3.1
予防メンテナンスを,あらかじめ定めた手順によって計画し,実行しなければならない。
12.3.2
あらかじめ定めたすべてのメンテナンス活動が,完了し,記録するまでは,装置を製品の処理に
使用してはならない。
12.3.3
メンテナンス計画,メンテナンス手順及びメンテナンス記録を保管し(4.1.2参照),指名した職
員によってあらかじめ定めた間隔でレビューしなければならない。レビューの結果は,文書化しなければ
ならない。
12.4 適格性の再確認
12.4.1
あらかじめ定めた間隔及びすべての変更の評価(12.5参照)を行った後に,指定した製品及び装
置について,滅菌プロセスの適格性の再確認を実施しなければならない。適格性の再確認の実施範囲は,
正当な理由付けをしなければならない。
12.4.2
適格性の再確認の手順をあらかじめ定め,適格性の再確認の記録を保管しなければならない(4.1.2
参照)。
12.4.3
文書化した手順に従って,適格性の再確認のデータをあらかじめ定めた判定基準に対してレビュ
ーしなければならない。適格性の再確認のレビューの記録は,実施した修正及び実施した是正処置の記録
と共に保管しなければならない(4.1.2参照)。
12.5 変更の評価
すべての変更は,滅菌プロセスの有効性に与える影響の大きさについて評価しなければならない。考慮
すべき変更には,(該当する場合)次の事項を含まなければならない。
a) プロセスパラメータに変化を起こす可能性のある部品の交換。
b) 滅菌チャンバへの漏れの増加を引き起こす可能性のある部品の交換。
c) 滅菌チャンバの中の均一性の変化。
d) 新しい又は変更したソフトウェア及び/又はハードウェア。
e) すべてのプロセスパラメータの変更。
f)
すべてのサービス及びサービスに対するメンテナンスの結果の変更。
g) すべての包装及び/又は包装手順の変更。
h) すべての載荷形態の変更。
i)
すべての製品材質,原料の起源又は設計の変更。
この評価の結果は,決定の根拠,及び該当する場合,滅菌プロセス,製品又は適格性の再確認に対する
要求事項に対して行った変更の範囲を含めて文書化しなければならない。
21
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書A
(参考)
この規格の使用上の指針
注記1 この附属書の中で与えられる指針は,この規格への適合性を評価するチェックリストとして
意図したものではない。この指針はこの規格の一様な理解及び適用を助け,規定要求事項へ
の適合のための許容できる方法及び説明を提供することを意図している。この指針で示した
以外の方法を使用することができるが,他の方法を用いる場合,この規格への適合に有効で
あることを立証する必要がある。
注記2 参照を容易にするため,この附属書の箇条番号は,この規格の本体と対応している。
A.1 適用範囲
A.1.1 適用
指針はない。
A.1.2 適用除外
A.1.2.1 指針はない。
A.1.2.2 指針はない。
A.1.2.3 指針はない。
A.1.2.4 医療機器の滅菌プロセスの開発,バリデーション,日常管理に当たっては,定義し文書化した手
順の効果的な実行が必要である。このような手順は,通常,品質マネジメントシステムの要素として考慮
される。この規格では,JIS Q 13485で示される医療機器の品質マネジメントシステムを引用規格とするこ
とによって,滅菌の有効な管理のための基本的な品質マネジメントシステムの要素を明確にし,規定して
いる。この規格は,JIS Q 13485に適合する完全な品質マネジメントシステムの適用を要求しているのでは
なく,第三者機関による品質マネジメントシステム要素の評価を要求しているのでもない。医療機器製造
業者には,薬事法令によって品質マネジメントシステム及び規制当局又は第三者機関によるシステム評価
に対する要求事項が存在することに注意を払う必要がある。
A.1.2.5 指針はない。
A.2 引用規格
引用規格の要求事項は,それがこの規格の要求事項の部分において引用した範囲においてだけ,この規
格の要求事項であり,その引用は規格の完全な引用である場合及び特定の箇条に限定された場合がある。
A.3 用語及び定義
指針はない。
A.4 品質マネジメントシステムの要素
注記 A.1.2.4を参照。
A.4.1 文書化
文書化及び記録の管理についての要求事項は,JIS Q 13485の4.2.3及び4.2.4にそれぞれ規定している。
22
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS Q 13485では,文書化に対する要求事項は,文書(仕様及び手順を含む。)の作成,管理及び記録に
ついてである。
A.4.2 経営者の責任
責任及び権限についての要求事項は,JIS Q 13485の5.5に規定しており,人的資源についての要求事項
は,JIS Q 13485の6.2に規定している。
JIS Q 13485では,経営者の責任に対する要求事項は,経営者のコミットメント,顧客重視,品質方針,
計画,責任,権限,コミュニケーション及びマネジメントレビューに関連している。
滅菌プロセスの開発,バリデーション及び日常管理は,幾つかの別々の組織を包含することがあり,そ
れぞれがある要素について責任をもつことがある。この規格では,組織が受け入れたそれぞれの責任を定
め,その定められた責任を文書化することを要求している。この定めた権限及び責任は,それぞれの組織
の品質マネジメントシステムの中で文書化する。各要素に責任をもつ組織は,これらの要素について,適
切な訓練及び資格認定によってその能力が立証された力量のある職員に割り当てることを要求している。
A.4.3 製品実現
注記 JIS Q 13485では,製品実現に対する要求事項は,顧客要求事項の決定,設計・開発,購買,製
造管理,監視及び測定機器の校正からなる製品ライフサイクルが製品実現に関連している。
A.4.3.1 購買についての要求事項は,JIS Q 13485の7.4に規定している。特にJIS Q 13485の7.4.3の購
買製品の検証についての要求事項が,外部の組織から受け取るすべての製品及びサービスに適用されるこ
とに注意するとよい。
A.4.3.2 識別及びトレーサビリティについての要求事項は,JIS Q 13485の7.5.3に規定している。
A.4.3.3 監視及び測定装置の校正についての要求事項は,JIS Q 13485の7.6に規定している。
A.4.4 測定,分析及び改善−不適合製品の管理
不適合製品の管理及び是正処置の手順は,JIS Q 13485の8.3及び8.5.2にそれぞれ規定している。
JIS Q 13485では,測定,分析及び改善の要求事項は,プロセス監視,不適合製品の管理,データの分析
及び改善(是正処置及び予防処置を含む。)に関連している。
A.5 滅菌剤の特性
箇条5の目的は,滅菌剤の特性を明確にし,その微生物殺滅効果の有効性を立証し,滅菌剤へのばく露
が材質に与える影響を評価し,職員の安全及び環境保護についての要求事項を明確にすることである。
この活動は,試験又は試作システムで行ってもよいが,最終的な装置仕様は,このようなすべての試験
又は試作システムを用いて実施した実験的な検討と関連づけるとよい。
A.6 プロセス及び装置の特性
箇条6の目的は,滅菌プロセスの全体及び滅菌プロセスを安全に,かつ,再現性よく運用するために必
要な装置の特性を明確にすることである。
滅菌プロセスは,特定の製品ファミリー及び載荷形態に対して確立する。確立した滅菌プロセスの仕様
には,運転サイクル中のばく露プロファイルを定義するプロセスパラメータ,及び再現性を検証するのに
使用するプロセスパラメータを含むのがよい。致死率を確立するのに使用した部分的なばく露を明確にし
ておき,致死率及び製品の性能に影響を与える可能性のあるプロセスのパラメータの上限及び下限を定め
る。滅菌プロセス及びプロセスを運用する装置の仕様には,オプションとして新製品又は載荷形態を提案
するときのために,プロセスの定義(箇条8参照)で議論する場合に考慮すべき詳細な事項を十分に含む
23
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
のがよい。
A.7 製品の定義
箇条7の目的は,滅菌前の製品の微生物学的品質(バイオバーデン),製品の包装方法及び製品を滅菌器
に搬入する形態(載荷形態)を含み,滅菌する製品を定義することである。
製品の構成及び包装に使われる材質の組合せは,湿熱滅菌プロセスにおいて代表的なプロセスパラメー
タに耐える必要がある。製品設計及び使用材料によるすべての制限事項を明確にするとよい。
A.8 プロセスの定義
箇条8の目的は,製品の安全及び品質及び有効性を損なうことなしに定義した製品に適用する滅菌プロ
セスの詳細な仕様を定義することである。
装置がバリデートされており,既存の滅菌プロセスが製品が属する製品ファミリーの製品を処理できる
ことが既知であれば,その滅菌プロセスを使ってもよい。この滅菌プロセスは製品の製造業者によって示
されたり,対象とする製品が属する製品ファミリーのために開発されたものである場合がある。すべての
ケースについて製品の定義で定めたプロセスパラメータの限界値及びばく露への制限事項を監視するのが
よい。
運転サイクルの例を,附属書Eに示す。
A.9 バリデーション
バリデーションの目的は,プロセスの定義で開発した滅菌プロセスが滅菌負荷に対して有効で再現性よ
く運用できることを確立することである。バリデーションは,据付適格性の確認,運転適格性の確認,及
び稼働性能適格性の確認の段階から構成する。装置,サービス及び据付けの仕様への適合は据付適格性の
確認で確立する。滅菌プロセスの運用は,運転適格性の確認において確立し,製品内部及び/又は製品上
で要求されるSALの達成は,稼働性能適格性の確認の間に確立する。
A.10 日常監視及び管理
日常監視及び管理の目的は,バリデートした滅菌プロセスが製品に対して運用されることを確実にする
ことである。これは,滅菌プロセスの間に得たデータ,及びあらかじめ定めた滅菌プロセスが運用できる
ことを検証するために使用する定期的な試験によって立証する。
A.11 滅菌からの製品のリリース
滅菌からの製品のリリースの目的は,製品が,あらかじめ定めた滅菌プロセスに問題なくばく露されて
おり,使用のためにリリース可能であることを確証することである。
A.12 プロセス有効性の維持
箇条12の目的は,日常の処理において,製品に対してあらかじめ定めた滅菌プロセスを継続的に運用す
るために必要な定期的なチェック及びテストを明確にし,実施することである。
滅菌負荷に対する致死率に疑問を抱かせるような変更があった場合は,レビューを開始するのがよい。
バリデーション及び日常監視の各要素に関係する責任部門を,表A.1に示す。

24
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表A.1−滅菌剤の特性,滅菌プロセスの開発,バリデーション及び日常管理の要素
要素
目的
構成要素
責任部門
品質システム
滅菌プロセスのすべての段階
を管理する構造を提供する。
経営者の責任,設計管理,製品
実現,測定・分析及び改善。
それぞれの要素に関連する
すべての組織。
滅菌剤の特性
滅菌剤を定義し,その微生物殺
滅効果の有効性を定義する。
滅菌剤の定義,微生物殺滅効果
の有効性及び材質への影響。
滅菌プロセスの開発者。
滅菌プロセス
及び装置の特
性
滅菌プロセス全体を定義し,そ
れを実行するために必要な装
置を定義する。
滅菌プロセスの記載,装置の仕
様,附属装置,サービスの定義,
安全及び環境。
滅菌器の製造業者(該当する
場合)滅菌プロセスの開発者
の協力を得て。
製品定義
滅菌する製品を定義する。
製品仕様,包装材料及び滅菌前
の製品品質。
滅菌する製品の製造業者(及
び滅菌装置の契約によって
は滅菌器の製造業者)。
滅菌プロセス
の定義
識別した製品について製品の
安全性及び機能を維持しなが
ら無菌性を達成するために滅
菌プロセスを定義する。
開発,生物学的安全性,プロセ
ス残さ(渣),製品適合性及び
再滅菌における制限。
滅菌する製品の製造業者,滅
菌器の製造業者の協力及び
該当する場合,ヘルスケア施
設の協力を得て。
バリデーショ
ン
定義した滅菌プロセスが,滅菌
負荷に対して,効果的に再現性
よく運用できることを立証す
る。
据付適格性の確認,運転適格性
の確認,稼働性能適格性の確認
並びにバリデーションのレビ
ュー及び承認。
製品の滅菌に責任をもつ組
織(製品製造業者又は再処理
施設のいずれか),(該当す
る場合,滅菌器の製造業者の
協力を得て。)製造業者又は
再処理施設(該当する場合,
製品の滅菌を実施する組織
の協力を得て)。
日常監視及び
管理
バリデートした滅菌プロセス
が,あらかじめ定めた許容範囲
内で滅菌負荷のすべての製品
について運用されていること
を立証する。
滅菌負荷,載荷形態,滅菌プロ
セスの監視,記録の作成,定期
的試験及び記録の保持。
製品製造業者又は再処理施
設。
滅菌からの製
品のリリース
日常の管理手順の記録をレビ
ューし,各滅菌負荷の処置を決
める。
記録のレビュー,(用いる場合
はすべての)インジケータの試
験,製品の処置及び(あればす
べての)是正処置。
製品製造業者又は再処理施
設。
プロセスの有
効性の維持
バリデートした滅菌プロセス
が継続して許容できることを
確実にする。
滅菌器製造業者,製品製造業
者又は再処理施設(該当する
場合,製品を滅菌する組織と
共同して)。
25
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書B
(参考)
自然状態にある微生物群の不活化に基づくプロセスの定義
(バイオバーデン法)
B.1
一般
この方法についての指針及び議論は,例えば,Halverson and Zeigler 1932[37],Pflug and Holcomb 1983 [44],
PDA 1978[42],Pflug 1999[43]の文献で示されている。
この方法は,自然に存在するバイオバーデンについての広範な知見を必要とする。代表的な製品のバイ
オバーデンは,ISO 11737-1によって測定するとよい。熱抵抗性による分離株のスクリーニングが必要とな
る場合がある。
B.2
サンプリング
プロセスの定義の検討に使用する製品には,日常生産品の代表的な製品を選ぶのがよい。
B.3
手順
次のようにするのがよい。
B.3.1 予定している滅菌プロセスに対して一定の増分で,製品を滅菌剤にばく露する。
B.3.2 その増加部分に要求される正確さ及び精度を確立し,その限界値に合致するよう滅菌剤の導入を制
御し,監視する。
B.3.3 滅菌剤へのばく露の後,個々の製品について無菌性の試験を実施する(ISO 11737-2参照)。
B.3.4 無菌性の試験で生育を示さない製品の比率と滅菌剤へのばく露量との関係から得られる知見を使
って,滅菌プロセスを決定する。
B.3.5 再現性を立証するために,決定した水準の処理を3回繰り返す。
B.4
フォローアップ
滅菌プロセスの継続的な適切性は,日常生産品を代表する製品を用いて,定期的に確認するのがよい。
この方法ではバイオバーデンについての継続的な監視及び管理を必要とする。
26
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書C
(参考)
標準菌の不活化及び滅菌する製品上のバイオバーデンの
知見に基づくプロセスの定義
(バイオバーデン/バイオロジカルインジケータ併用法)
C.1 一般
この方法への指針及び議論は,例えば,ISO 14161[10],Halverson and Zeigler 1932[37],Pflug and Holcomb
1983[44],PDA 1978[42],Pflug 1999[43]の文献で示されている。
C.2 手順
次のようにするのがよい。
C.2.1 滅菌が一番困難な製品内の場所を明確にする。
C.2.2 滅菌剤に対する既知の抵抗性をもつ微生物の既知の数からなる滅菌プロセスへのチャレンジを,次
のいずれかによって作成する。
a) 製品内の滅菌条件の達成が最も困難な部位,又はそれらを代表する部位にバイオロジカルインジケー
タを配置する。
b) 製品内の滅菌条件の達成が最も困難な部位に標準菌を接種する。
注記 菌を接種した製品は,バイオロジカルインジケータとみなされる(8.5及びISO 11138-1参照)。
C.2.3 この方法で作られたチャレンジを日常製造する製品と同じ方法で包装し,滅菌条件の達成が最も困
難な場所に配置する滅菌負荷に含める。
C.2.4 すべての標準菌が不活化しないような,日常の滅菌条件より低い致死率を与える条件を選択し,そ
の条件で滅菌負荷を滅菌剤にばく露する。
C.2.5 再現性を立証するために,前項の条件で処理を3回繰り返す。
C.2.6 微生物の生残数を,直接の計数又は最確数法(MPN法)で測定する。
C.2.7 標準菌の不活化速度を計算する。
C.2.8 バイオバーデン[8.11 a)によって確立した]の知見及び標準菌の不活化速度から,必要とする無菌
性に対する要求事項を満たす処理の程度を求める。
27
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書D
(参考)
標準菌の不活化に基づく安全率を見込んだプロセスの定義
(オーバーキル法)
D.1 一般
D.1.1 この附属書は,標準菌の不活化に基づくオーバーキルのプロセスについて記載する。このプロセス
は広範に採用され,再使用可能な製品の滅菌によく使われる方法である。このような製品の場合,滅菌へ
のチャレンジを定義するのが難しく,また,洗浄のような滅菌前の処理をバリデートし,管理するのが困
難であることから,滅菌プロセスの適格性確認において,未使用の製品のときとは異なるアプローチを採
用することが必要である。
この状況において,滅菌プロセスはしばしば安全側であり,無菌に対して定めた要求事項を達成するの
に必要とするものよりも過剰な処理を行うように設計する。“オーバーキル”と呼ばれるこのタイプの処
理は,経験的な微生物に基づく数学的処理(フルサイクルアプローチ),及び定義した微生物に対する減少
させた処理(部分サイクルアプローチ)で決定することができる。
D.1.2 オーバーキル法は,不活化が直線的に起こることが立証される場合,滅菌プロセスに最も適したも
のである。
D.1.3 この方法への指針及び議論は,例えば,ISO 14161[10],Halverson and Zeigler 1932[37],Pflug and
Holcomb 1983[44],PDA 1978[42],Pflug 1999[43]の文献に示されている。
D.2 手順
次のようにするのがよい。
D.2.1 製品内の無菌性の達成が最も困難な場所を確定する。
D.2.2 滅菌プロセスへのチャレンジを,次の一つの方法によって作成する。
a) 製品内の滅菌条件の達成が最も困難な部位又はそれらを代表する部位にバイオロジカルインジケータ
を入れる。
b) 製品内の滅菌条件の達成が最も困難な部位に標準菌を接種する。
注記 この方法で接種した製品は,バイオロジカルインジケータとみなされる(8.5又は8.6及び
ISO 11138-1参照)。
D.2.3 この方法で作ったチャレンジを,日常製造する製品と同じ方法で包装し,滅菌条件の達成が最も困
難な場所に配置する滅菌負荷に含める。
D.3 部分サイクルアプローチ
次のようにするのがよい。
D.3.1 低減させた処理となるように設定した条件で滅菌負荷を滅菌剤にばく露する。
D.3.2 処理の程度は,ISO 11138-3に適合するバイオロジカルインジケータ上の106の微生物を不活化す
るのに必要なものとする。
D.3.3 再現性を立証するために,決定した水準の処理を3回繰り返す。
28
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D.3.4
106の微生物の不活化が確認された場合,滅菌剤の不活性化速度の性質及びバイオロジカルインジ
ケータの微生物の抵抗性及び数を考慮に入れた上で,外挿によって,10−6又はそれより低い生残率に外挿
することによって,滅菌プロセスの処理条件を決定する。
D.3.5
処理条件は安全度を見込んで,ここで行った減少処理条件の2倍で定義できる。
D.4 フルサイクルアプローチ
次のようにするのがよい。
D.4.1 ISO 11138-3に適合するバイオロジカルインジケータを不活化する処理となるよう設定した条件で,
滅菌負荷を滅菌剤にばく露する。
D.4.2
バイオロジカルインジケータの菌の公称菌数は,Fbio12及びバイオロジカルインジケータの成績書
記載のD121から求められた菌数に対して,少なくとも常用対数で0.5過剰なものである必要がある。これ
には微生物学的操作における変動及び製品と汚染物質との接触によって起こり得る微生物のD値の変化を
考慮に入れている。
Fbio は,次の式で求められる。
Fbio=D121(log N0−log N) ························································· (D.1)
ここに,
D121: 121 ℃でばく露したときのバイオロジカルインジケー
タのD値
N0: ばく露前のバイオロジカルインジケータの生育可能な
菌数
N: ばく露後のバイオロジカルインジケータの生育可能な
菌数
D.4.3
湿熱に高い抵抗性を示し,このアプローチでの使用に適した菌の例として,G. stearothermophilus,
B. coagulans,C. sporogenes,及びB. atrophaeusがある。
D.4.4
目標とするF0の計算には,滅菌チャンバ内で生じる可能性のある致死率の変動及びバイオロジカ
ルインジケータが陽性となる確率を考慮するのがよい。
D.4.5
目標とするF0分間,滅菌負荷を滅菌剤にばく露し,生残がないことを確認する。テストの結果,
この処理の水準が受け入れられることを示す場合,更に2回の繰返しを行い,再現性を立証し,滅菌プロ
セスの処理の定義を確実なものとするのがよい。
29
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書E
(参考)
運転サイクル
注記 この附属書は,湿熱滅菌プロセスで用いられる代表的な運転サイクルについて記載する。図は,
概念を示す例である。
E.1
重力置換式飽和蒸気サイクル
E.1.1
この運転サイクルでは,繊維及び中空孔からの空気除去が不確かなので,表面接触の滅菌を基本的
に意図している。また,容器に入った流体の滅菌のように蒸気が加熱媒体として作用する滅菌プロセスに
も使われる。
E.1.2
飽和蒸気−排気のチャンバ内温度及び圧力のプロファイルの例を,図E.1に示す。
E.1.3
滅菌プロセスは,次の三つの主要な段階で構成する。
a) 加熱段階 排気口が開き,所定の条件(通常,温度測定によって決定される。)になるまで飽和蒸気を
供給するか又はチャンバ内で発生させる。排気口が閉じ滅菌温度及びこれに対応する飽和蒸気圧に到
達するまで,飽和蒸気を引き続き供給するか滅菌チャンバ内で生成させる。
b) 平たん(坦)期間段階 所定の時間,滅菌チャンバ内を飽和蒸気で滅菌温度を維持する。
c) 冷却段階 この段階は,製品のタイプでいろいろ異なる。滅菌チャンバに空気を供給し大気圧に戻す。
密閉容器内の溶液の場合は,突沸を避けるため,滅菌チャンバにフィルタを通した圧縮空気を入れる。
この段階は,滅菌チャンバの圧力が大気圧になったときに終了し,密閉容器の場合は容器内が安全な
温度になったときに終了する。
E.2
真空脱気式飽和蒸気サイクル
E.2.1
この運転サイクルは,主に空気を除去するのが困難な多孔質材料,管こう(腔)及び/又は中空並
びに包装した対象物に用いられる。また,これは表面接触の滅菌にも使用できる。
E.2.2
チャンバ内温度及び圧力のプロファイルの例を,図E.2に示す。これは,最新の滅菌器で適用され
る数多い中の一例に過ぎない。
E2.3
この滅菌プロセスは,次の六つの主要な段階で構成する。
a) 脱気段階 滅菌チャンバ及び滅菌負荷から高真空,又は真空及び大気圧を超える及び/又は大気圧以
下の蒸気パルスの組合せで空気を除去する。
b) 蒸気注入段階 滅菌チャンバ内の滅菌温度及び圧力が達成されるまで飽和蒸気を滅菌チャンバに入れ
る。
c) 平たん期間段階 所定の時間,滅菌チャンバ内を飽和蒸気によって滅菌温度及び圧力を維持する。
d) 排気段階 滅菌チャンバから蒸気を排出し,所定の水準まで真空にする。
e) 乾燥段階 乾燥が必要な製品については,滅菌チャンバのジャケットの温度及び滅菌チャンバの真空
を所定の時間維持する。
f)
真空解除段階 大気圧に戻るまで,微生物除去フィルタを通した空気を滅菌チャンバに供給する。
30
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
E.3
空気加圧運転サイクル
E.3.1 一般
この運転サイクルは,主として容器に入った製品を滅菌するときに,サイクルのある段階で容器の中の
圧力がチャンバ内の圧力を超過するような場合に使用する。このような場合,滅菌時に容器の破裂及びシ
ールの完全性を損なうことになるので,対策のためにチャンバ内への空気加圧を使用する。
このような容器封入製品については,製品の外側と内側の圧力の均衡を確実にとるための幾つかの方法
を使用する。
E.3.2 空気蒸気混合
E.3.2.1 チャンバ内温度及び圧力のプロファイルの例を,図E.3に示す。
E.3.2.2 この滅菌プロセスは,次の三つの主要な段階で構成する。
a) 加熱段階 製品の完全性が蒸気圧の上昇に影響を受け排気ができないような場合を除いて,この段階
の最初の部分は,排気システムと同じである。滅菌温度に到達するまで,蒸気を滅菌チャンバに継続
して導入する。この段階で製品が,加圧を必要とする場合及び残った空気の分圧が,製品の保護に不
十分な場合は,加圧した空気を導入する。均一な環境を維持するため,通常,循環を必要とする。
b) 平たん期間段階 所定の時間,循環及び滅菌温度を維持する。
c) 冷却段階 製品冷却は,加圧した空気又は冷却した水を散布して行う。この段階では,急速な滅菌チ
ャンバの圧力降下による製品の完全性への損傷を加圧空気によって避ける。製品が十分冷却するまで
滅菌チャンバ内の必要な圧力を維持し,その後大気圧に戻す。
E3.3
水散布(スプレー)
E.3.3.1 チャンバ内温度及び圧力のプロファイルの例を,図E.4に示す。
E.3.3.2 この滅菌プロセスは,次の四つの主要な段階で構成する。
a) 水充てん(填)段階 滅菌サイクルの始めに,滅菌器システムに一定量の水を導入するか.又は蒸気
を凝縮して作る。その後,製品全体に散布する。
b) 加熱段階 空気及び蒸気を循環システムへ導入するか,又は水を熱交換器によって加熱し,加圧した
空気を滅菌チャンバへ導入して,滅菌温度に加熱する。
c) 平たん期間段階 循環システムを運転し,所定の時間,水を滅菌温度に保つ。
d) 冷却段階 滅菌チャンバの圧力を加圧した空気で保ち,循環水の温度を管理した速度で冷やしながら,
製品を冷やす。製品が安全な温度になったとき,滅菌チャンバの圧力を落とす。
E3.4
水浸せき(漬)
E.3.4.1 チャンバ内温度及び圧力のプロファイルの例を,図E.5に示す。
E.3.4.2 この運転サイクルは,製品の形を保つために製品全体が水につ(漬)けられることを除いて,水
散布システムと類似している。
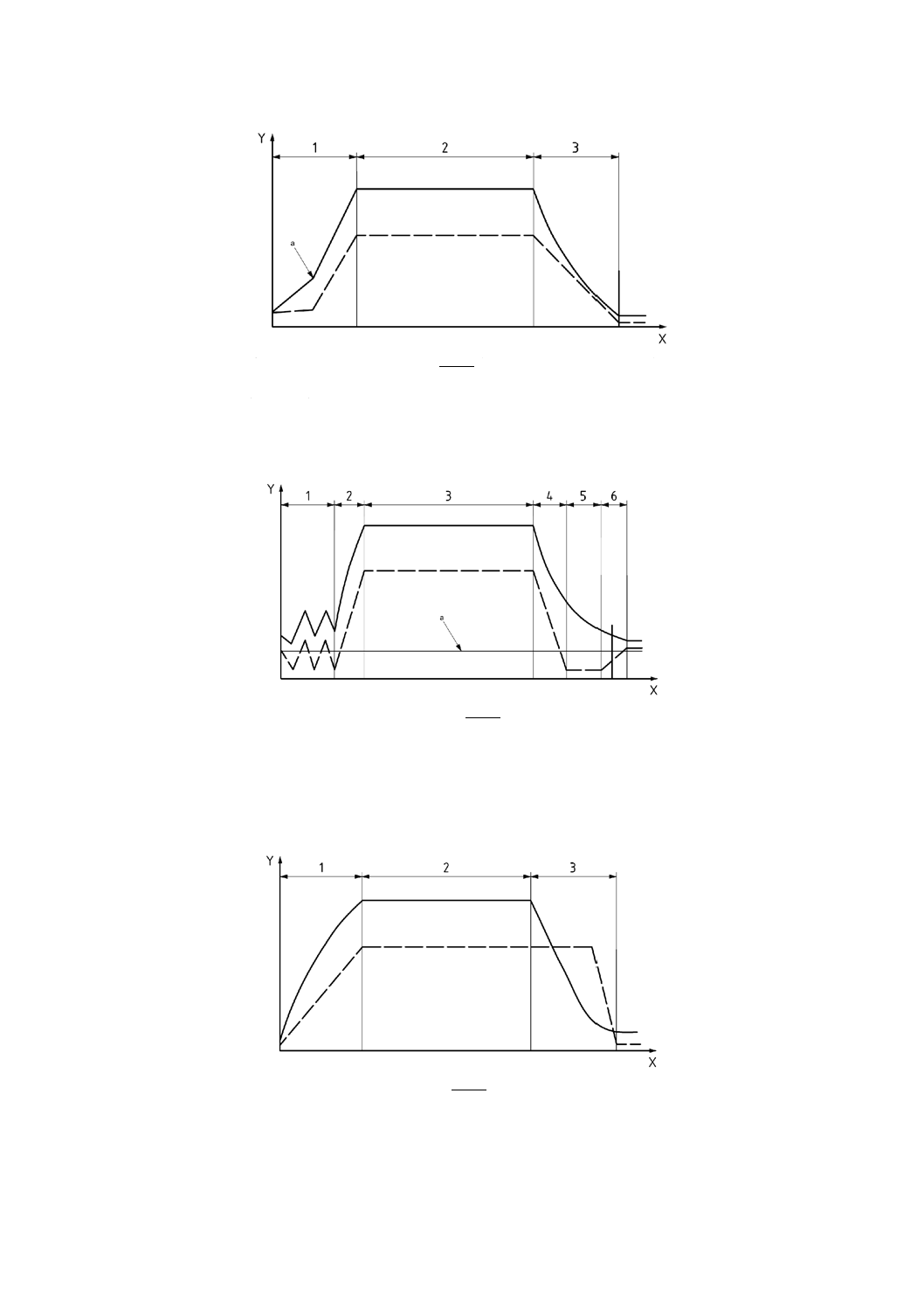
31
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
X 時間
1 加熱段階
a 換気口閉鎖
Y 温度( )
2 平たん期間段階
飽和蒸気圧(− − −)
3 冷却段階
図E.1−重力置換式飽和蒸気サイクルのチャンバ温度及び圧力のプロファイルの例
X 時間
1 脱気段階
4 排気段階
a 大気圧
Y 温度( )
2 蒸気注入段階
5 乾燥段階
飽和蒸気圧(− − −)
3 平たん期間段階
6 真空解除段階
図E.2−真空脱気式飽和蒸気サイクルのチャンバ温度及び圧力のプロファイルの例
X 時間
1 加熱段階
Y 温度( )
2 平たん期間段階
空気加圧(− − −)
3 冷却段階
図E.3−空気蒸気混合サイクルのチャンバ温度及び圧力のプロファイルの例
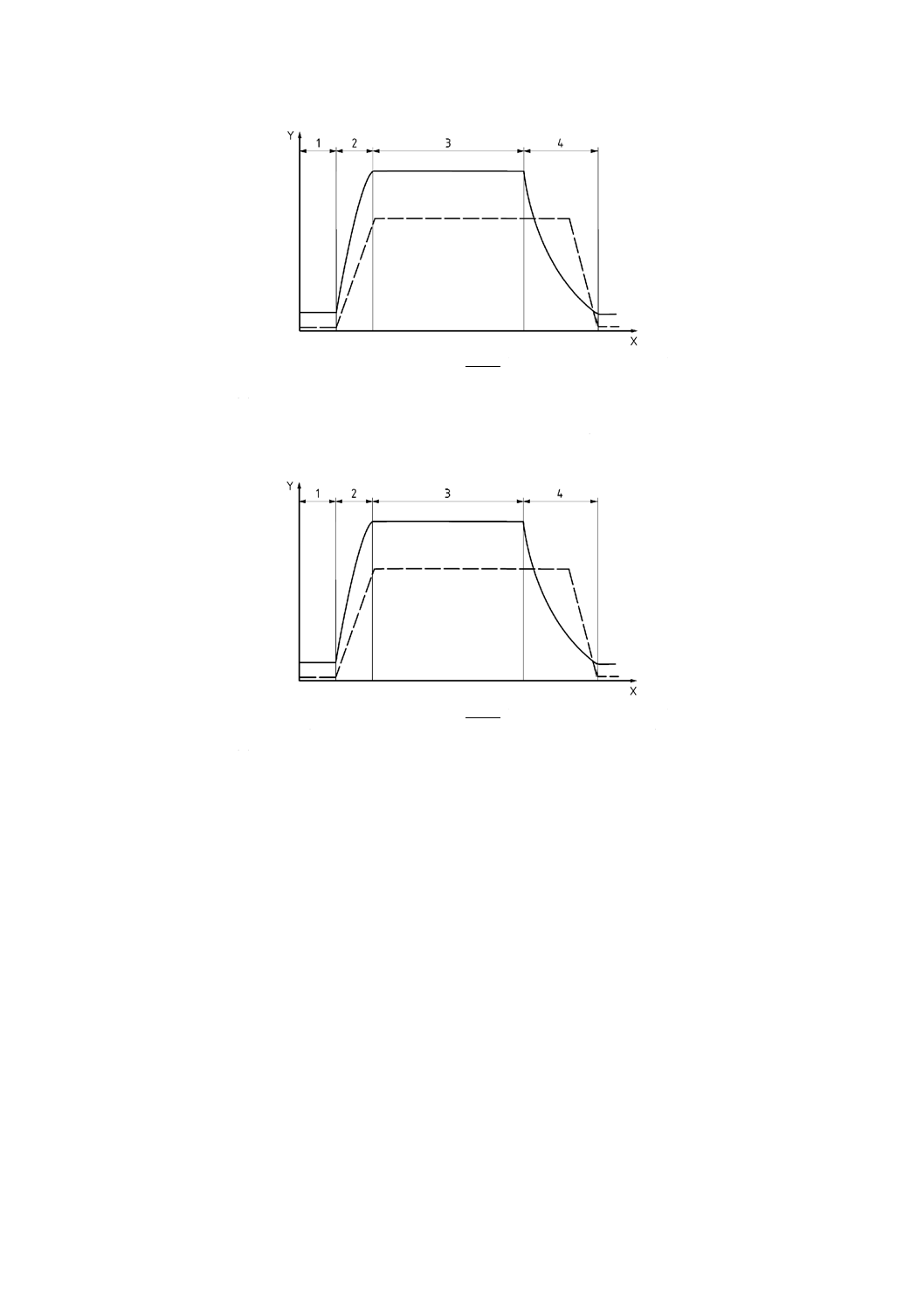
32
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
X 時間
1 水充てん段階
4 冷却段階
Y 温度( )
2 加熱段階
空気加圧(− − −)
3 平たん期間段階
図E.4−水散布サイクルのチャンバ温度及び圧力のプロファイルの例
X 時間
1 水充てん段階
4 冷却段階
Y 温度( )
2 加熱段階
空気加圧(− − −)
3 平たん期間段階
図E.5−水浸せきサイクルのチャンバ温度及び圧力のプロファイルの例
33
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考文献
[1] JIS Q 9000:2006 品質マネジメントシステム−基本及び用語
注記 対応国際規格:ISO 9000:2005,Quality management systems−Fundamentals and vocabulary
(IDT)
[2] ISO 9000-3:1991,Quality management and quality assurance standards−Part 3: Guidelines for the
application of ISO 9001 to the development, supply and maintenance of computer software
[3] JIS Q 9001:2000 品質マネジメントシステム−要求事項
注記 対応国際規格:ISO 9001:2000,Quality management systems−Requirements (IDT)
[4] JIS Q 9004:2000 品質マネジメントシステム−パフォーマンス改善の指針
注記 対応国際規格:ISO 9004:2000,Quality management systems−Guidelines for performance
improvements (IDT)
[5] JIS T 0993-1 医療機器の生物学的評価−第1部:評価及び試験
注記 対応国際規格:ISO 10993-1,Biological evaluation of medical devices−Part 1: Evaluation and
testing (IDT)
[6] ISO 10993-17,Biological evaluation of medical devices−Part 17: Establishment of allowable limits for
leachable substances
[7] ISO/TS 11139,Sterilization of health care products−Vocabulary
[8] ISO 14001,Environmental management systems−Requirements with guidance for use
[9] ISO 14040,Environmental management−Life cycle assessment−Principles and framework
[10] ISO 14161:2000,Sterilization of health care products−Biological indicators−Guidance for the selection,
use and interpretation of results
[11] ISO 14937:2000,Sterilization of health care products−General requirements for characterization of a
sterilizing agent and the development, validation and routine control of a sterilization process for medical
devices
[12] JIS T 14971 医療機器−リスクマネジメントの医療機器への適用
注記 対応国際規格:ISO 14971,Medical devices−Application of risk management to medical devices
(IDT)
[13] ISO 15882:2003,Sterilization of health care products−Chemical indicators−Guidance for selection, use
and interpretation of results
[14] ISO 15883-1,Washer-disinfectors−Part 1: General requirements, terms and definitions and tests
[15] ISO 15883-2,Washer-disinfectors−Part 2: Requirements and tests for washer-disinfectors employing
thermal disinfection for surgical instruments, anaesthetic equipment, bowls, dishes, receivers, utensils,
glassware, etc.
[16] ISO 15883-4,Washer-disinfectors−Part 4: Requirements and tests for washer-disinfectors employing
chemical disinfection for thermolabile endoscopes
[17] ANSI/AAMI ST67:2003,Sterilization of health care products−Requirements for products labeled
“STERILE”
34
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[18] BLOCK, S.S., ed., Disinfection, sterilization, and preservation, Lippincott, Williams and Wilkins,
Philadelphia, PA; 5th Edition, 2001
[19] BOWIE, J.H., KELSEY, J.C. and THOMSON, G.R. The Bowie and Dick autoclave tape test, Lancet 16, pp
586 - 587 (1963)
[20] CHEN, J.H.S., Methods of testing virucides, Disinfection, Sterilization and Preservation, Block, S.S., ed., Lea
and Febinger, Philadelphia, PA, 1983
[21] EN 285,Sterilization−Steam sterilizers−Large sterilizers
[22] EN 556-1,Sterilization of medical devices−Requirements for medical devices to be designated “STERILE”
−Part 1: Requirements for terminally-sterilized medical devices
[23] EN 868-1:1997,Packaging materials and systems for medical devices which are to be sterilized−Part 1:
General requirements and test methods
[24] EN 868-2:1999,Packaging materials and systems for medical devices which are to be sterilized−Part 2:
Sterilization wrap−Requirements and test methods
[25] EN 868-3:1999,Packaging materials and systems for medical devices which are to be sterilized−Part 3:
Paper for use in the manufacture of paper bags (specified in EN 868-4) and in the manufacture of pouches
and reels (specified in EN 868-5)−Requirements and test methods
[26] EN 868-4:1999,Packaging materials and systems for medical devices which are to be sterilized−Part 4:
Paper bags−Requirements and test methods
[27] EN 868-5:1999,Packaging materials and systems for medical devices which are to be sterilized−Part 5:
Heat and self-sealable pouches and reels of paper and plastic film construction−Requirements and test
methods
[28] EN 868-8:1999,Packaging materials and systems for medical devices which are to be sterilized−Part 8:
Re-usable sterilization containers for steam sterilizers conforming to EN 285−Requirements and test
methods
[29] EN 868-9:2000,Packaging materials and systems for medical devices which are to be sterilized−Part 9:
Uncoated nonwoven materials of polyolefines for use in the manufacture of heat sealable pouches, reels and
lids−Requirements and test methods
[30] EN 868-10:2000,Packaging materials and systems for medical devices which are to be sterilized−Part 10:
Adhesive coated nonwoven materials of polyolefines for use in the manufacture of heat sealable pouches,
reels and lids−Requirements and test methods
[31] EN 12442-1:2000,Animal tissues and their derivatives utilized in the manufacture of medical devices−Part
1: Analysis and management of risk
[32] EN 12442-2:2000,Animal tissues and their derivatives utilized in the manufacture of medical devices−Part
2: Controls on sourcing, collection and handling
[33] EN 12442-3:2000,Animal tissues and their derivatives utilized in the manufacture of medical devices−Part
3: Validation of the elimination and/or inactivation of viruses and transmissible agents
[34] EN 13060,Small steam sterilizers
[35] GAMP 4, The Good Automated Manufacturing Practice (GAMP) Guide for Validation of Automated Systems
in Pharmaceutical Manufacture, December 2001, ISPE(International Society for Pharmaceutical Engineering),
3109 W. Dr. Martin Luther King, Jr. Blvd., Suite 250, Tampa, FL 33607
35
T 0816-1:2010 (ISO 17665-1:2006)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
[36] Global Harmonization Task Force (GHTF)−Study Group 1 (SG1), Document N029R15, dated 3 December,
2004
[37] HALVORSON, H. O. and ZIEGLER, N.R. Applications of statistics to problems in bacteriology, Journal of
Bacteriology, 25(2): pp. 101-118, 1932
[38] IEC 61010-2-040, Safety requirements for electrical equipment for measurement, control, and laboratory use
−Part 2-040: Particular requirements for sterilizers and washer-disinfectors used to treat medical materials
[39] IRVINE, Th. F. and LILEY, P.E. Steam and Gas tables with computer equations, Academic Press, 1984
[40] MORRISSEY, R.F. and PHILLIPS, G.B., eds. Sterilization technology: A practical guide for manufacturers
and users of health care products, New York, Van Nostrand Reinhold, 1995
[41] National Canners Association, Manual for food Canners and Processors, Vol. 1, AVI Publishing Co., Westport,
CT, 1968
[42] PDA, Validation of steam sterilization cycles, Technical monograph No. 1, Parenteral Drug Association 1978
[43] PFLUG, I.J. Microbiology and Engineering of Sterilization Processes, 1999 Tenth Edition, Environmental
Sterilization Laboratory, 1920 South First St, Minneapolis, Philadelphia, PA, 1978
[44] PFLUG, I.J. and HOLCOMB, R.G., Principles of thermal destruction of microorganisms, Disinfection,
Sterilization and Preservation, Block, S.S., ed., Lea and Febinger, Philadelphia, PA, 1983
[45] PFLUG, I.J. and KRISTEN, D.E., Carrying out Biological Qualification, the Control Operation of Moist-Heat
(Steam Sterilization) Processes for Producing Sterile Pharmaceuticals and Medical Devices. PDA, Journal of
Pharmaceutical Science and Technology, 54 (2) 2000
[46] SATTAR, S.A. and SPRINGTHORPE, V.S. Methods under development for evaluating the antimicrobial
activity of germicides, Proceedings of the International Symposium on Chemical Germicides in Health Care,
Cincinnati, Ohio, May 1994, Polyscience Publications Inc, pp 237-254, 1995
[47] ISO 18472,Sterilization of health care products−Biological and chemical indicators−Test equipment
[48] ISO 22442-1,Medical devices utilizing animal tissues and their derivatives−Part 1: Application of risk
management
[49] ISO 22442-2,Medical devices utilizing animal tissues and their derivatives−Part 2: Controls on sourcing,
collection and handling
[50] ISO 22442-3,Medical devices utilizing animal tissues and their derivatives−Part 3: Validation of the
elimination and/or inactivation of viruses and transmissible spongiform encephalopathy (TSE) agents
[51] International Vocabulary of Basic and General Terms in Metrology (VIM), BIPM, IEC, IFCC, ISO, IUPAC,
IUPAP, OIML, 2nd ed, Geneva, 1993
[52] ISO/TS 17665-2,Sterlization of health care products−Moist heat−Part 2: Guidance on the application of
ISO 17665-1