R 9101:2018
(1)
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 用語及び定義 ··················································································································· 1
4 分析項目························································································································· 2
5 試料の調製及び保存 ·········································································································· 2
6 一般事項························································································································· 3
7 分析結果のまとめ方 ·········································································································· 3
8 水分の定量方法 ················································································································ 3
8.1 方法の区分 ··················································································································· 3
8.2 要旨 ···························································································································· 3
8.3 天然せっこう及び化学せっこう ························································································ 3
8.4 焼せっこう ··················································································································· 4
9 化合水の定量方法 ············································································································· 4
9.1 方法の区分 ··················································································································· 4
9.2 要旨 ···························································································································· 4
9.3 試料はかりとり量 ·········································································································· 4
9.4 操作 ···························································································································· 4
9.5 計算 ···························································································································· 4
10 二酸化けい素+不溶残分の定量方法 ··················································································· 5
10.1 方法の区分 ·················································································································· 5
10.2 要旨 ··························································································································· 5
10.3 試薬 ··························································································································· 5
10.4 試料はかりとり量 ········································································································· 5
10.5 操作 ··························································································································· 5
10.6 計算 ··························································································································· 5
11 酸化アルミニウム+酸化鉄(III)の定量方法 ······································································· 6
11.1 方法の区分 ·················································································································· 6
11.2 要旨 ··························································································································· 6
11.3 試薬 ··························································································································· 6
11.4 操作 ··························································································································· 6
11.5 計算 ··························································································································· 6
12 酸化鉄(III)の定量方法 ·································································································· 7
12.1 方法の区分 ·················································································································· 7
12.2 EDTA滴定法 ··············································································································· 7
R 9101:2018
(2)
ページ
12.3 1,10-フェナントロリン吸光光度法 ···················································································· 8
12.4 原子吸光分析法 ············································································································ 9
12.5 ICP発光分光分析法 ····································································································· 10
13 酸化カルシウムの定量方法 ······························································································ 11
13.1 方法の区分 ················································································································· 11
13.2 EDTA滴定法 ·············································································································· 11
13.3 過マンガン酸カリウム滴定法 ························································································· 12
14 酸化マグネシウムの定量方法 ··························································································· 13
14.1 方法の区分 ················································································································· 13
14.2 原子吸光分析法 ··········································································································· 13
14.3 ICP発光分光分析法 ····································································································· 14
14.4 EDTA滴定法 ·············································································································· 15
15 三酸化硫黄の定量方法 ···································································································· 16
15.1 方法の区分 ················································································································· 16
15.2 要旨 ·························································································································· 16
15.3 試薬 ·························································································································· 16
15.4 試料はかりとり量 ········································································································ 16
15.5 操作 ·························································································································· 16
15.6 計算 ·························································································································· 17
16 二酸化硫黄の定量方法 ···································································································· 17
16.1 方法の区分 ················································································································· 17
16.2 要旨 ·························································································································· 17
16.3 試薬 ·························································································································· 17
16.4 試料はかりとり量 ········································································································ 18
16.5 操作 ·························································································································· 18
16.6 計算 ·························································································································· 18
17 二酸化炭素の定量方法 ···································································································· 19
17.1 方法の区分 ················································································································· 19
17.2 要旨 ·························································································································· 19
17.3 試薬 ·························································································································· 19
17.4 装置 ·························································································································· 19
17.5 試料はかりとり量 ········································································································ 21
17.6 操作 ·························································································································· 21
17.7 空試験 ······················································································································· 22
17.8 計算 ·························································································································· 22
18 酸化ナトリウム及び酸化カリウムの定量方法 ······································································ 22
18.1 方法の区分 ················································································································· 22
18.2 要旨 ·························································································································· 22
18.3 試薬 ·························································································································· 22
目次
R 9101:2018
(3)
ページ
18.4 装置 ·························································································································· 22
18.5 操作 ·························································································································· 22
18.6 検量線の作成 ·············································································································· 22
18.7 計算 ·························································································································· 23
19 ふっ素の定量方法 ·········································································································· 23
19.1 方法の区分 ················································································································· 23
19.2 ランタン−アリザリンコンプレキソン吸光光度法 ······························································ 23
19.3 イオン電極法 ·············································································································· 27
20 全りん酸の定量方法 ······································································································· 28
20.1 方法の区分 ················································································································· 28
20.2 モリブデン青吸光光度法 ······························································································· 28
20.3 りんバナドモリブデン酸吸光光度法 ················································································ 30
21 水溶性りん酸の定量方法 ································································································· 31
21.1 方法の区分 ················································································································· 31
21.2 モリブデン青吸光光度法 ······························································································· 31
21.3 りんバナドモリブデン酸吸光光度法 ················································································ 32
22 塩素の定量方法 ············································································································· 33
22.1 方法の区分 ················································································································· 33
22.2 硝酸銀滴定法 ·············································································································· 33
22.3 イオン電極法 ·············································································································· 34
23 遊離酸の定量方法 ·········································································································· 35
23.1 方法の区分 ················································································································· 35
23.2 要旨 ·························································································································· 35
23.3 試薬 ·························································································································· 35
23.4 操作 ·························································································································· 36
23.5 計算 ·························································································································· 36
24 pHの測定方法 ·············································································································· 36
24.1 方法の区分 ················································································································· 36
24.2 要旨 ·························································································································· 36
24.3 試薬 ·························································································································· 36
24.4 装置 ·························································································································· 36
24.5 試料はかりとり量 ········································································································ 36
24.6 操作 ·························································································································· 36
附属書A(参考)分析操作図 ································································································· 37
R 9101:2018 目次
R 9101:2018
(4)
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,無機マテリアル学
会(SIMJ)及び一般財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を改正すべ
きとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格である。
これによって,JIS R 9101:1995は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
日本工業規格 JIS
R 9101:2018
せっこうの化学分析方法
Methods for chemical analysis of gypsum
序文
この規格は,1953年に制定され,その後7回の改正を経て今日に至っている。前回の改正は1995年に
行われたが,その後JIS R 9111の改正に伴い,酸化鉄(III)及び酸化マグネシウムの定量方法にICP発光
分光分析法が追加され,これに対応するために改正した。
今回の改正に当たっては,原子吸光分析法とICP発光分光分析法との比較を行い,原子吸光分析法も見
直すこととした。また,試験の利便性を考慮し,一部の分析項目において市販の標準溶液等を使用するこ
ととした。
なお,対応国際規格は現時点で制定されていない。
1
適用範囲
この規格は,天然せっこう,化学せっこう及び焼せっこうの化学分析方法について規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS K 0122 イオン電極測定方法通則
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS M 8100 粉塊混合物−サンプリング方法通則
JIS R 9112 陶磁器型材用せっこうの物理試験方法
JIS Z 8401 数値の丸め方
JIS Z 8801-1 試験用ふるい−第1部:金属製網ふるい
JIS Z 8802 pH測定方法
3
用語及び定義
この規格で用いる主な用語及び定義は,JIS K 0050,JIS K 0115,JIS K 0116,JIS K 0121,JIS K 0122,
JIS K 8005,JIS M 8100,JIS R 9112,JIS Z 8401,JIS Z 8801-1及びJIS Z 8802によるほか,次による。
2
R 9101:2018
3.1
水分
せっこう中の化合水を除く付着水。
3.2
ICP発光分光分析
誘導結合プラズマによって測定対象元素を気化励起し,得られる原子スペクトル線の発光強度を測定す
ることによって定量する分析方法。
3.3
標準原液
試薬を用いて直接,調製した標準溶液又は化学分析用に用いるファクターが明記されている市販の標準
溶液。
3.4
標準溶液
標準原液を希釈した溶液。
4
分析項目
この規格で規定する分析項目は,次とする。
a) 水分,Wf
b) 化合水,Wc
c) 二酸化けい素+不溶残分,GS,i
d) 酸化アルミニウム+酸化鉄(III),GAF
e) 酸化鉄(III),GF
f)
酸化カルシウム,GC
g) 酸化マグネシウム,GM
h) 三酸化硫黄,Gs3
i)
二酸化硫黄,Gs2
j)
二酸化炭素,Gc2
k) 酸化ナトリウム及び酸化カリウム,GN,GK
l)
ふっ素,Gf
m) 全りん酸,GtP
n) 水溶性りん酸,GsP
o) 塩素,Gc
p) 遊離酸,GH
q) pH
5
試料の調製及び保存
試料は,次によって採取調製し,外気の影響を受けない気密な容器に保存する。
a) 天然せっこう 試料は,JIS M 8100によって採取し,水分定量用試料は約4 mm以下の細かさとし,
水分以外の分析用試料は,JIS Z 8801-1に規定する標準網ふるい250 μmを全量通過したものとする。
採取した試料が著しく湿っていて粉砕・縮分に適さない場合には,粉砕・縮分できるまで自然乾燥
3
R 9101:2018
によるか,又は40±2 ℃の空気浴で24時間以上乾燥を行う。
なお,250 μmでは,試料の均一性を保証できない場合は,150 μmを全量通過したものとする。
b) 化学せっこう a)と同様に採取・調製する。
c) 焼せっこう JIS R 9112の箇条3(試料の採取方法)に規定する方法によって調製した試料を水分定
量用試料とする。また,これを40±2 ℃に調節した空気浴で恒量になるまで乾燥し,水分以外の分析
用試料とする。
6
一般事項
分析方法に共通な一般事項は,通常,JIS K 0050,JIS K 0115,JIS K 0116,JIS K 0121,JIS K 0122及
びJIS Z 8802による。
警告 各自の責任において安全及び健康に対する適切な措置を取らなければならない。
7
分析結果のまとめ方
分析値のまとめ方は,次による。分析を行うに当たっては,全操作を通じて空試験(blank test)を行い,
測定値を補正する。分析操作を参考として附属書Aに示す。
a) 水分は,受け入れたままの試料に対する百分率で表し,JIS Z 8401によって小数点以下1桁に丸める。
b) 化合水,酸化カルシウム及び三酸化硫黄は,乾燥した試料に対する百分率で表し,JIS Z 8401によっ
て小数点以下1桁に丸める。
c) 二酸化けい素+不溶残分,酸化アルミニウム+酸化鉄(III),酸化鉄(III),酸化マグネシウム,二酸
化硫黄,二酸化炭素,酸化ナトリウム,酸化カリウム,ふっ素,全りん酸,水溶性りん酸,塩素及び
遊離酸は,乾燥した試料に対する百分率で表し,JIS Z 8401によって小数点以下2桁に丸める。
d) pHは,JIS Z 8401によって小数点以下1桁に丸める。
8
水分の定量方法
8.1
方法の区分
水分の定量は,質量法による。ただし,天然せっこう及び化学せっこうの場合と焼せっこうの場合とで,
試料のはかりとり量が異なる。
8.2
要旨
試料を40±2 ℃で恒量となるまで乾燥したときの減量をはかり,水分の付着率を求める。
8.3
天然せっこう及び化学せっこう
8.3.1
試料はかりとり量
試料は,約200 gを0.1 gまで正しくはかりとる。
8.3.2
操作
定量操作は,次の手順による。
a) 試料を質量既知の適切な乾燥皿にはかりとり,乾燥しやすいように10 mm程度の厚さの薄層に広げ
る。これを40±2 ℃に調節した空気浴で2時間乾燥し,デシケーター中で冷却した後,質量をはかる。
b) 以降1時間乾燥し,質量をはかり,前後の質量差が0.5 g以下になるまで繰り返し,減量を求める。
8.3.3
計算
試料中の水分の付着率は,次の式によって算出する。
4
R 9101:2018
Wf
100
×
s
m
=
ここに,
Wf: 水分の付着率(%)
m: 減量(g)
s: 8.3.1ではかりとった試料の質量(g)
8.4
焼せっこう
8.4.1
試料はかりとり量
試料は,約10 gを0.01 gまで正しくはかりとる。
8.4.2
操作
定量操作は,次の手順による。
a) 試料を質量既知の平形ひょう(秤)量瓶(60 mm×30 mm)にはかりとり,これを40±2 ℃に調節し
た空気浴で2時間乾燥し,デシケーター中で冷却した後,質量をはかる。
b) 以降1時間乾燥し,質量をはかり,前後の質量差が0.02 g以下になるまで繰り返し,減量を求める。
8.4.3
計算
試料中の水分の付着率は,次の式によって算出する。
Wf
100
×
s
m
=
ここに,
Wf: 水分の付着率(%)
m: 減量(g)
s: 8.4.1ではかりとった試料の質量(g)
9
化合水の定量方法
9.1
方法の区分
化合水の定量は,質量法による。
9.2
要旨
試料を240〜260 ℃で恒量になるまで加熱したときの減量をはかり,化合水の含有率を求める。
9.3
試料はかりとり量
試料は,約1.0 gを0.1 mgまで正しくはかりとる。
9.4
操作
定量操作は,次の手順による。
a) 試料を質量既知の筒形ひょう(秤)量瓶(30 mm×45 mm)にはかりとり,緩く蓋をして240〜260 ℃
に調節した電気炉で1時間加熱し,デシケーター中で冷却した後,質量をはかる。
b) 以降30分間乾燥し,質量をはかり,前後の質量差が0.2 mg以下になるまで繰り返し,減量を求める。
9.5
計算
試料中の化合水の含有率は,次の式によって算出する。
Wc
100
×
s
m
=
ここに,
Wc: 化合水の含有率(%)
m: 減量(g)
s: 9.3ではかりとった試料の質量(g)
5
R 9101:2018
10 二酸化けい素+不溶残分の定量方法
10.1 方法の区分
二酸化けい素+不溶残分の定量は,質量法による。
10.2 要旨
試料を塩酸及び過塩素酸で溶かした後,加熱して二酸化けい素を脱水し,不溶性とした後,可溶分をろ
過する。ろ紙上の二酸化けい素+不溶残分は,加熱して質量をはかる。
10.3 試薬
試薬は,次のものを用いる。
a) 塩酸 (1+1)
b) 過塩素酸(60 %)
10.4 試料はかりとり量
試料は,約1.0 gを0.1 mgまで正しくはかりとる。
10.5 操作
定量操作は,次の手順による。
a) 試料をビーカー(200 mL)にはかりとり,少量の水を加えてスラリー状とし,時計皿で覆って塩酸 (1
+1) 5 mLをビーカーの縁から少しずつ加える。
なお,ふっ素を含有している試料については,四ふっ化けい素によるふっ素の揮散を防ぐため,ほ
う酸約1 g及び水約20 mLを加えてスラリー状とし,加熱してほう酸を溶かし,四ふっ化けい素の気
散を防いだ後,塩酸を加える操作に移る。
b) 時計皿を除き,過塩素酸(60 %)5 mLを加えてかき混ぜ,砂浴の温度が砂の中程に温度計を差し込ん
だときに180〜200 ℃を示すように調節した砂浴上で加熱し,過塩素酸(60 %)の濃い白煙が出始め
たら,再び時計皿で覆い,引き続き10分間加熱する。
c) ビーカーを砂浴から降ろして放冷した後,塩酸 (1+1) 5 mL及び温水約50 mLを加えてかき混ぜ,水
浴上で約15分間加熱する。この間,時々かき混ぜ,ガラス棒の先で固まりを潰して可溶分を完全に溶
かす。
d) ろ紙(5種B)を用いてろ過し,温水で十分に洗浄する。
なお,洗浄が不十分であると,ろ紙の灰化時に飛散するおそれがある。特に,不溶解物が多い場合
は注意する必要がある。ろ液及び洗液はビーカー(300 mL)に受け,室温まで冷却した後,250 mL
の全量フラスコに洗い移し,水で標線までうすめる。
この溶液を試料溶液(A)とし,酸化アルミニウム+酸化鉄(III),酸化鉄(III),酸化マグネシウ
ム,酸化ナトリウム及び酸化カリウム,全りん酸などの定量に用いる。
e) 不溶解物はろ紙とともに磁器るつぼに入れて乾燥し,緩く蓋をして加熱して,炎の出ないように注意
しながらろ紙を灰化した後,1 000±50 ℃に調節した電気炉で1時間均熱保持し,デシケーター中で
放冷した後,質量を0.1 mgまで正しくはかる。
10.6 計算
試料中の二酸化けい素+不溶残分の含有率は,次の式によって算出する。
GS,i
100
×
s
m
=
ここに,
GS,i: 二酸化けい素+不溶残分の含有率(%)
m: 不溶解物の質量(g)
6
R 9101:2018
s: 10.4ではかりとった試料の質量(g)
11 酸化アルミニウム+酸化鉄(III)の定量方法
11.1 方法の区分
酸化アルミニウム+酸化鉄(III)の定量は,質量法による。
11.2 要旨
10.5 d)で保存した試料溶液(A)を用い,塩化アンモニウム及びアンモニア水を加えて,アルミニウム
を鉄などとともに沈殿させて,ろ過する。
沈殿物は加熱して質量をはかり,酸化アルミニウム+酸化鉄(III)の含有率を求める。
注記 試料中にりん酸,チタンなどを含有する場合は,これらが共沈する。
11.3 試薬
試薬は,次のものを用いる。
a) 硝酸
b) 塩化アンモニウム飽和溶液
c) アンモニア水 (1+1)
d) 硝酸アンモニウム洗浄液(20 g/L) 硝酸アンモニウム洗浄液は,メチルレッド溶液2,3滴を加え,
溶液の色が赤から黄に変わるまでアンモニア水 (1+1) を滴加して用いる。この溶液を加熱したとき
色が赤に戻ったら,更にアンモニア水 (1+1) を滴加し黄色にしたものを用いる。
e) メチルレッド溶液 調製方法は,JIS K 8001のJA.5(指示薬)による。
11.4 操作
定量操作は,次の手順による。
a) 10.5 d)で保存した試料溶液(A)から100 mLをビーカー(300 mL)に分取し,硝酸1,2滴を加えて
数分間煮沸する。
b) 塩化アンモニウム飽和溶液10 mL及びメチルレッド溶液1,2滴を加え,かき混ぜながら溶液の色が
赤から黄に変わるまでアンモニア水 (1+1) を滴加し,更に1,2滴過剰に加える。
c) 加熱して1〜2分間煮沸した後,熱源から降ろし,沈殿が沈降したら直ちにろ紙(5種A)でろ過し,
硝酸アンモニウム温洗浄液(20 g/L)で沈殿を約8回洗浄する。
d) 沈殿はろ紙とともに磁器るつぼに入れて乾燥し,緩く蓋をして緩やかに加熱して炎の出ないように注
意しながら,ろ紙を灰化した後,1 000±50 ℃に調節した電気炉で1時間均熱保持し,デシケーター
中で放冷した後,質量を0.1 mgまで正しくはかる。
11.5 計算
試料中の酸化アルミニウム+酸化鉄(III)の含有率は,次の式によって算出する。
GAF
100
250×
×v
s
m
=
ここに,
GAF: 酸化アルミニウム+酸化鉄(III)の含有率(%)
m: 沈殿の質量(g)
s: 10.4ではかりとった試料の質量(g)
v: 試料溶液(A)からの分取量(mL)
7
R 9101:2018
12 酸化鉄(III)の定量方法
12.1 方法の区分
酸化鉄(III)の定量は,次のいずれかの方法による。
a) EDTA滴定法 この方法は,酸化鉄(III)の含有率0.1 %以上の試料に適用する。
b) 1,10-フェナントロリン吸光光度法
c) 原子吸光分析法 この方法は,酸化鉄(III)の含有率1.0 %以下の試料に適用する。
d) ICP発光分光分析法 この方法は,酸化鉄(III)の含有率1.0 %以下の試料に適用する。
12.2 EDTA滴定法
12.2.1 要旨
10.5 d)で保存した試料溶液(A)を用い,pHを2.2〜2.5とした後,サリチル酸溶液を指示薬としてEDTA
標準溶液で滴定する。
12.2.2 試薬
試薬は,次のものを用いる。
a) 酢酸アンモニウム溶液(250 g/L)
b) 塩酸 (1+1)
c) 水酸化ナトリウム溶液(100 g/L)
d) サリチル酸溶液(20 g/L) サリチル酸2 gをメタノール(95 vol%)100 mLに溶かす。
e) エリオクロムブラックT溶液 調製方法は,JIS K 8001のJA.5による。
f)
緩衝溶液 (pH10) 塩化アンモニウム70 gを適量の水に溶かし,アンモニア水570 mLを加え,水
を加えて1 Lにうすめる。
g) 0.01 mol/L亜鉛標準溶液 JIS K 8005に規定する亜鉛を塩化カルシウムデシケーター又は硫酸デシケ
ーター中に,24時間以上保った後使用する。亜鉛の表面が酸化しているおそれのある場合は,塩酸 (1
+1),水,アセトンで順次に洗い,110 ℃で5分間乾燥して用いる。この亜鉛約0.65 gを0.1 mgまで
正しくはかりとって,ビーカー(200 mL)に入れ,塩酸 (1+1) 20 mLを加え,少し温めて完全に溶か
した後1 Lの全量フラスコに洗い移し,冷却した後水で標線までうすめる。
この標準溶液のファクターは,次の式によって算出し,小数点以下3桁に丸める。
8
653
.0
1
m
f=
ここに,
f1: 0.01 mol/L亜鉛標準溶液のファクター
m: はかりとった亜鉛の質量(g)
h) 0.01 mol/L EDTA標準溶液 エチレンジアミン四酢酸二ナトリウム二水和物3.72 gを適量の水に溶か
して1 Lの全量フラスコに入れ,水で標線までうすめ,ポリエチレン瓶に保存する。この標準溶液は,
次のようにして標定する。
なお,市販のEDTA標準溶液を用いることができる。
0.01 mol/L亜鉛標準溶液25 mLを分取してビーカー(200 mL)に移し,水を加えて約100 mLとし,
水酸化ナトリウム溶液(100 g/L)を加えて中和した後,pHを9.5〜10.0に調節し,緩衝溶液(pH10)
2 mL及びエリオクロムブラックT溶液2,3滴を加えて0.01 mol/L EDTA標準溶液で滴定し,溶液の
色が赤紫から赤みが全く消えて鮮明な青となった点を終点とする。この標準溶液のファクターは,次
の式によって算出し,小数点以下6桁に丸める。
8
R 9101:2018
v
f
f
1
2
25×
=
ここに,
f2: 0.01 mol/L EDTA標準溶液のファクター
v: 0.01 mol/L EDTA標準溶液の使用量(mL)
f1: 0.01 mol/L亜鉛標準溶液のファクター
12.2.3 操作
定量操作は,次の手順による。
a) 10.5 d)で保存した試料溶液(A)から100 mLを分取し,ビーカー(300 mL)に移す。これに酢酸アン
モニウム溶液(250 g/L)10 mLを加え,塩酸 (1+1) を用いてpHを2.2〜2.5に調節する。
b) 指示薬としてサリチル酸溶液(20 g/L)1 mLを加え,0.01 mol/L EDTA標準溶液で滴定し,溶液の色
が赤紫から無色又は淡黄色になったときを終点とする。
12.2.4 計算
試料中の酸化鉄(III)の含有率は,次の式によって算出する。
GF
100
250
5
798
000
.0
2
2
1
×
×
×
×
v
s
f
v
=
ここに,
GF: 酸化鉄(III)の含有率(%)
v1: 0.01 mol/L EDTA標準溶液の使用量(mL)
f2: 0.01 mol/L EDTA標準溶液のファクター
s: 10.4ではかりとった試料の質量(g)
v2: 試料溶液(A)からの分取量(mL)
12.3 1,10-フェナントロリン吸光光度法
12.3.1 要旨
10.5 d)で保存した試料溶液(A)を用い,アスコルビン酸で鉄を還元した後,1,10-フェナントロリンを
加え,酢酸アンモニウムでpHを調節して呈色させ,その吸光度を測定して酸化鉄(III)の含有率を求め
る。
12.3.2 試薬
試薬は,次のものを用いる。
a) アスコルビン酸溶液(50 g/L) 冷暗所に保存し,保存中に着色したときは,新しく調製する。
b) 1,10-フェナントロリン溶液 1,10-フェナントロリン塩酸塩−水和物1 gを適量の水に溶かした後,1 L
にうすめる。冷暗所に保存し,保存中に着色したときは新しく調製する。
c) 酢酸アンモニウム溶液(200 g/L)
d) 塩酸 (1+1)
e) 鉄標準原液(1 000 mg/L)
12.3.3 装置
装置は,吸光光度分析装置を用いる。
12.3.4 操作
定量操作は,次の手順による。
a) 10.5 d)で保存した試料溶液(A)から,適量を分取して,100 mLの全量フラスコに移し,水を加えて
約50 mLとする。
なお,試料溶液(A)からの分取量は,酸化鉄(III)の含有率に応じて表1による。
b) これに,アスコルビン酸溶液(50 g/L)2 mLを加えて振り混ぜ,1,10-フェナントロリン溶液10 mL及

9
R 9101:2018
び酢酸アンモニウム溶液(200 g/L)10 mLを加え,標線まで水を加えて振り混ぜ,30分間放置する。
c) この溶液の一部を吸光光度分析装置の吸収セルにとり,波長510 nm付近の吸光度を測定する。
表1−試料溶液の分取量
酸化鉄(III)の含有率 %
分取量 mL
0.15未満
50
0.15以上
0.30未満
25
0.30以上
0.75未満
10
0.75以上
1.50未満
5
12.3.5 検量線の作成
12.3.2 e)の鉄標準原液(1 000 mg/L)を水で正しく100倍にうすめ,その0〜20 mL(鉄として0〜0.2 mg)
を100 mLの全量フラスコに段階的にとり,水を加えて約50 mLとする。12.3.4のb)及びc)の手順に従っ
て操作して吸光度を測定し,その吸光度と鉄の濃度との関係線を作成して検量線とする。
12.3.6 計算
12.3.5で作成した検量線を用い,12.3.4で測定した吸光度から鉄の濃度を求め,次の式によって試料中の
酸化鉄(III)の含有率を算出する。
GF
7
429
.1
100
250
103
×
×
×
×
−
v
s
C
=
ここに,
GF: 酸化鉄(III)の含有率(%)
C: 吸光度から求めた鉄の濃度(mg/100 mL)
s: 10.4ではかりとった試料の質量(g)
v: 試料溶液(A)からの分取量(mL)
1.429 7: 鉄から酸化鉄(III)への換算値
12.4 原子吸光分析法
12.4.1 要旨
10.5 d)で保存した試料溶液(A)を用いて,原子吸光分析装置によって鉄の吸光度を測定し,酸化鉄(III)
の含有率を求める。
12.4.2 試薬
試薬は,次のものを用いる。
a) 塩酸 (1+1)
b) 過塩素酸(60 %)
c) 硫酸
d) 炭酸カルシウム(アルカリ分析用)
e) マトリックス溶液-I 炭酸カルシウム(アルカリ分析用)11.6 gをはかりとってビーカー(500 mL)
に入れ,適量の水を加えて分散させ,時計皿で覆い,塩酸 (1+1) 200 mLをビーカーの縁から少しず
つ加えて溶かす。これに過塩素酸(60 %)100 mLを加え,放冷した後,1 Lの全量フラスコに移し,
水で標線までうすめる。この溶液は,ポリエチレン瓶に移して保存する。
f)
マトリックス溶液-II 適量の水に硫酸6.5 mLを加え,放冷した後,1 Lの全量フラスコに移し,水で
標線までうすめる。この溶液は,ポリエチレン瓶に移して保存する。
g) 鉄標準原液(1 000 mg/L)
10
R 9101:2018
12.4.3 装置
装置は,原子吸光分析装置を用いる。
12.4.4 操作
定量操作は,次の手順による。
10.5 d)で保存した試料溶液(A)の一部を分取し,水を加えて2倍にうすめ,その一部を原子吸光分析
装置のアセチレン・空気フレーム中に噴霧し,鉄用光源ランプを用いて,波長248.3 nmにおける吸光度を
測定する。
12.4.5 検量線の作成
12.4.2 g)の鉄標準原液(1 000 mg/L)を水で正しく10倍にうすめ,その0〜15 mL(鉄として0〜1.5 mg)
を100 mLの全量フラスコに段階的にとり,水を加えて約50 mLとする。これに12.4.2 e)のマトリックス
溶液-I 10 mL1)及び12.4.2 f)のマトリックス溶液-II 10 mL2)を加え,水で標線までうすめる。これらの標
準液を原子吸光分析装置のアセチレン・空気フレーム中に噴霧し,波長248.3 nmにおける吸光度を測定し,
吸光度と鉄の濃度との関係線を作成して検量線とする。
注1) 試料中の酸化カルシウム(CaO)の含有率GC(%)が既知の場合は,マトリックス溶液-Iの添
加量νは次の式によって算出する。
10
6.
32
C×
=G
ν
(mL)
注2) 試料中の三酸化硫黄(SO3)の含有率Gs3(%)が既知の場合は,マトリックス溶液-IIの添加量
νは次の式によって算出する。
10
5.
46
3s×
=G
ν
(mL)
12.4.6 計算
12.4.5で作成した検量線を用い,12.4.4で測定した吸光度から鉄の濃度を求め,次の式によって試料中の
酸化鉄(III)の含有率を算出する。
GF
7
429
.1
100
100
2
250
103
×
×
×
×
×
−
s
C
=
ここに,
GF: 酸化鉄(III)の含有率(%)
C: 吸光度から求めた鉄の濃度(mg/100 mL)
s: 10.4ではかりとった試料の質量(g)
1.429 7: 鉄から酸化鉄(III)への換算値
12.5 ICP発光分光分析法
12.5.1 要旨
10.5 d)で保存した試料溶液(A)を用いて,ICP発光分光分析装置によって発光強度を測定し,酸化鉄(III)
の含有率を求める。
12.5.2 試薬
試薬は,次のものを用いる。
a) 塩酸 (1+1)
b) 過塩素酸(60 %)
c) 硫酸
d) 炭酸カルシウム(アルカリ分析用)
11
R 9101:2018
e) マトリックス溶液-I 12.4.2 e)で調製した溶液。
f)
マトリックス溶液-II 12.4.2 f)で調製した溶液。
g) 鉄標準原液(1 000 mg/L)
12.5.3 装置
装置は,JIS K 0116によるICP発光分光分析装置を用いる。
12.5.4 操作
定量操作は,次の手順による。
10.5 d)で保存した試料溶液(A)の一部を分取し,水を加えて2倍にうすめ,その一部をICP発光分光
分析装置の発光部に導入し,波長238.204 nmにおける発光強度を測定する。
12.5.5 検量線の作成
12.5.2 g)の鉄標準原液(1 000 mg/L)を水で正しく10倍にうすめ,その0〜15 mL(鉄として0〜1.5 mg)
を100 mLの全量フラスコに段階的にとり,水を加えて約50 mLとする。これに12.5.2 e)のマトリックス
溶液-I 10 mL及び12.5.2 f)のマトリックス溶液-II 10 mLを加え,水で標線までうすめる。これらの標準
液をICP発光分光分析装置を用いて,波長238.204 nmで各々の溶液の発光強度を測定し,発光強度と鉄濃
度との関係線を作成して検量線とする。
12.5.6 計算
12.5.5で作成した検量線を用い,12.5.4で測定した発光強度から鉄の濃度を求め,次の式によって試料中
の酸化鉄(III)の含有率を算出する。
GF
7
429
.1
100
100
2
250
103
×
×
×
×
×
−
s
C
=
ここに,
GF: 酸化鉄(III)の含有率(%)
C: 発光強度から求めた鉄の濃度(mg/100 mL)
s: 10.4ではかりとった試料の質量(g)
1.429 7: 鉄から酸化鉄(III)への換算値
13 酸化カルシウムの定量方法
13.1 方法の区分
酸化カルシウムの定量は,次のいずれかの方法による。
a) EDTA滴定法
b) 過マンガン酸カリウム滴定法
13.2 EDTA滴定法
13.2.1 要旨
試料を塩酸で溶かした後,ろ過する。この溶液を用い,pHを12.7〜13.2とした後,HSNN希釈粉末を用
いてEDTA標準溶液で滴定する。
13.2.2 試薬
試薬は,次のものを用いる。
a) 塩酸 (1+1)
b) トリエタノールアミン (1+1)
c) 水酸化カリウム溶液(200 g/L)
d) HSNN希釈粉末2-ヒドロキシ-1-(2-ヒドロキシ-4-スルホ-1-ナフチルアゾ)-3-ナフトエ酸 調製方法
12
R 9101:2018
は,JIS K 8001のJA.5による。
e) 0.01 mol/L EDTA標準溶液 12.2.2 h)で調製したもの。
13.2.3 試料はかりとり量
試料は,約1.0 gを0.1 mgまで正しくはかりとる。
13.2.4 操作
定量操作は,次の手順による。
a) 試料をビーカー(200 mL)にはかりとり,塩酸 (1+1) 20 mL及び水約100 mLを加え,数分間煮沸し
て可溶分を溶かした後,ろ紙(5種B)でろ過し3),温水で8回洗浄する。
b) 冷却した後,ろ液及び洗液を500 mLの全量フラスコに移し,水で標線までうすめる。この溶液を試
料溶液(B)とし,酸化カルシウム,酸化マグネシウム及び三酸化硫黄の定量に用いる。
c) 試料溶液(B)から25 mLを分取して,ビーカー(300 mL)に入れ,水を加えて約100 mLとする。
これに,トリエタノールアミン (1+1) 2 mLを加え,適量の水酸化カリウム溶液(200 g/L)を加えて
よくかき混ぜ,pHを12.7〜13.2に調節し,2〜3分間静置する。この溶液に,HSNN希釈粉末約0.1 g
を加え,0.01 mol/L EDTA標準溶液で滴定し,溶液の色が赤紫から赤みが全く消えて鮮明な青となった
点を終点とする。
なお,滴定の終点は,タングステンランプの光を透して見ると分かりやすい。また,マグネシウム
が共存するときは,一度青となっても,放置すると色が戻ることがある。このような場合は,最初に
青になったときを終点とする。
注3) 微粒子がろ紙から漏れるおそれがある場合には,少量の粉末ろ紙を加えて少し煮沸した後,
ろ過する。
13.2.5 計算
試料中の酸化カルシウムの含有率は,次の式によって算出する。
GC
100
25
500
8
560
000
.0
×
×
×
×
s
f
v
=
ここに,
GC: 酸化カルシウムの含有率(%)
v: 0.01 mol/L EDTA標準溶液の使用量(mL)
f: 12.2.2 h)の0.01 mol/L EDTA標準溶液のファクター
s: 13.2.3ではかりとった試料の質量(g)
13.3 過マンガン酸カリウム滴定法
13.3.1 要旨
13.2.4 b)で保存した試料溶液(B)を用い,しゅう酸アンモニウムを加えてしゅう酸カルシウムを沈殿さ
せる。この沈殿をろ過した後,硫酸で溶かし,分離したしゅう酸を過マンガン酸カリウムで滴定する。
13.3.2 試薬
試薬は,次のものを用いる。
a) しゅう酸アンモニウム溶液(40 g/L)
b) 酢酸アンモニウム溶液(500 g/L)
c) 硫酸 (1+4)
d) 過マンガン酸カリウム標準溶液 過マンガン酸カリウム2.26 gを水に溶かして1 Lとし,フラスコに
入れて静かに一度煮沸した後,一夜暗所に放置し,ガラスろ過器G4(漏斗形又はブフナー漏斗形)で
ろ過して,褐色瓶に保存する。この溶液1 mLは,約0.002 gの酸化カルシウムに相当する。
13
R 9101:2018
この溶液は,次のようにして標定する。
JIS K 8005に規定するしゅう酸ナトリウムを150〜200 ℃に1〜1.5時間保ち,硫酸デシケーター中
で放冷したものを用いる。このしゅう酸ナトリウム0.2 gを0.1 mgまで正しくはかりとり,ビーカー
(500 mL)に入れ,温水150 mLを加えて溶かし,硫酸 (1+4) 50 mLを加えて約70 ℃に加熱し,熱
いうちに過マンガン酸カリウム標準溶液で滴定し,溶液の微紅色が約10秒間消えなくなったときを終
点とする。次の式によって標準溶液1 mLの酸化カルシウム相当量を算出し,小数点以下5桁に丸め
る。
v
m
E
5
418
.0
×
=
ここに,
E: 過マンガン酸カリウム標準溶液1 mLの酸化カルシウム
相当量(g)
v: 過マンガン酸カリウム標準溶液の使用量(mL)
m: しゅう酸ナトリウムのはかりとった量(g)
13.3.3 操作
定量操作は,次の手順による。
a) 13.2.4 b)で保存した試料溶液(B)から100 mLを分取してビーカー(500 mL)に入れ,加熱後,しゅ
う酸アンモニウム温溶液(40 g/L)30 mLをかき混ぜながら少しずつ加える。続いて酢酸アンモニウ
ム温溶液(500 g/L)20 mLをかき混ぜながら加え,2分間煮沸した後,沈殿するのを待って直ちにろ
紙(6種)でろ過し,温水で8回洗浄する。
b) 沈殿をろ紙とともにビーカー(500 mL)に入れ,ろ紙を開いてビーカーの内側に密着させ,沈殿を洗
い落とす。これに温水約150 mL及び硫酸 (1+4) 50 mLを加え,約70 ℃に加熱して沈殿を溶かし,
熱いうちに過マンガン酸カリウム標準溶液で紅色になるまで滴定する。さらに,ビーカーに付着して
いるろ紙を落として滴定を続け,微紅色が10秒間消えなくなったときを終点とする。
13.3.4 計算
試料中の酸化カルシウムの含有率は,次の式によって算出する。
GC
100
100
500×
×
×
s
E
v
=
ここに,
GC: 酸化カルシウムの含有率(%)
v: 過マンガン酸カリウム標準溶液の使用量(mL)
E: 過マンガン酸カリウム標準溶液1mLの酸化カルシウム
相当量(g)
s: 13.2.3ではかりとった試料の質量(g)
14 酸化マグネシウムの定量方法
14.1 方法の区分
酸化マグネシウムの定量は,次のいずれかの方法による。
a) 原子吸光分析法
b) ICP発光分光分析法
c) EDTA滴定法
14.2 原子吸光分析法
14.2.1 要旨
10.5 d)で保存した試料溶液(A)を用い,原子吸光分析装置によってマグネシウムの吸光度を測定し,
14
R 9101:2018
酸化マグネシウムの含有率を求める。
14.2.2 試薬
試薬は,次のものを用いる。
a) マトリックス溶液-III 12.4.2 e)で調製したマトリックス溶液-I及び12.4.2 f)で調製したマトリックス
溶液-IIを同量分取し,水で25倍にうすめる。
b) マグネシウム標準原液(1 000 mg/L)
14.2.3 装置
装置は,原子吸光分析装置を用いる。
14.2.4 操作
定量操作は,次の手順による。
10.5 d)で保存した試料溶液(A)の一部を分取し,水を加えて50倍にうすめ,その一部を原子吸光分析
装置のアセチレン・空気フレーム中に噴霧し,マグネシウム用光源ランプを用いて,波長285.2 nmにおけ
る吸光度を測定する。
14.2.5 検量線の作成
14.2.2 b)のマグネシウム標準原液(1 000 mg/L)を水で正しく1 000倍にうすめ,その0〜50 mL(マグネ
シウムとして0〜0.050 mg)を100 mLの全量フラスコに段階的にとり,水を加えて約50 mLとする。これ
に14.2.2 a)のマトリックス溶液-III 10 mLを加え,水で標線までうすめる。これらの標準溶液を原子吸光
分析装置のアセチレン・空気フレーム中に噴霧し,波長285.2 nmにおける吸光度を測定し,吸光度とマグ
ネシウム濃度との関係線を作成して検量線とする。
14.2.6 計算
14.2.5で作成した検量線を用い,14.2.4で測定した吸光度からマグネシウムの濃度を求め,次の式によっ
て試料中の酸化マグネシウムの含有率を算出する。
GM
4
658
.1
100
100
50
250
103
×
×
×
×
×
−
s
C
=
ここに,
GM: 酸化マグネシウムの含有率(%)
C: 吸光度から求めたマグネシウムの濃度(mg/100 mL)
s: 10.4ではかりとった試料の質量(g)
1.658 4: マグネシウムから酸化マグネシウムへの換算値
14.3 ICP発光分光分析法
14.3.1 要旨
10.5 d)で保存した試料溶液(A)を用い,ICP発光分光分析装置によってマグネシウムの発光強度を測定
し,酸化マグネシウムの含有率を求める。
14.3.2 試薬
試薬は,次のものを用いる。
a) マトリックス溶液-III 14.2.2 a)で調製した溶液。
b) マグネシウム標準原液(1 000 mg/L)
14.3.3 装置
装置は,JIS K 0116によるICP発光分光分析装置を用いる。
14.3.4 操作
定量操作は,次の手順による。
15
R 9101:2018
10.5 d)で保存した試料溶液(A)の一部を分取し,水を加えて50倍にうすめ,その一部をICP発光分光
分析装置の発光部に導入し,波長279.553 nmにおける発光強度を測定する。
14.3.5 検量線の作成
14.3.2 b)のマグネシウム標準原液(1 000 mg/L)を水で正しく1 000倍にうすめ,その0〜50 mL(マグネ
シウムとして0〜0.050 mg)を100 mLの全量フラスコに段階的にとり,水を加えて約50 mLとする。これ
に14.3.2 a)のマトリックス溶液-III 10 mLを加え,水で標線までうすめる。これらの標準溶液をICP発光
分光分析装置を用いて,波長279.553 nmで各々の溶液の発光強度を測定し,発光強度とマグネシウム濃度
との関係線を作成して検量線とする。
14.3.6 計算
14.3.5で作成した検量線を用い,14.3.4で測定した発光強度からマグネシウムの濃度を求め,次の式によ
って試料中の酸化マグネシウムの含有率を算出する。
GM
4
658
.1
100
100
50
250
103
×
×
×
×
×
−
s
C
=
ここに,
GM: 酸化マグネシウムの含有率(%)
C: 発光強度から求めたマグネシウムの濃度(mg/100 mL)
s: 10.4ではかりとった試料の質量(g)
1.658 4: マグネシウムから酸化マグネシウムへの換算値
14.4 EDTA滴定法
14.4.1 要旨
13.2.4 b)で保存した試料溶液(B)を用い,塩酸ヒドロキシルアミンを加えて鉄を還元し,トリエタノー
ルアミン及び硫化ナトリウムを加えて妨害イオンをマスキングした後,緩衝溶液を加えてpHを約10に調
節する。エリオクロムブラックT溶液を指示薬として,試料溶液中のカルシウムとマグネシウムとをEDTA
標準溶液で滴定する。この滴定量からカルシウム分として13.2.4 c)によるEDTA標準溶液の滴定量を差し
引いて,酸化マグネシウムの含有率を求める。
14.4.2 試薬
試薬は,次のものを用いる。
a) 緩衝溶液(pH10) 12.2.2 f)で調製したもの。
b) 塩酸ヒドロキシルアミン溶液(50 g/L)
c) エリオクロムブラックT溶液 12.2.2 e)で調製したもの。
d) トリエタノールアミン (1+1)
e) 硫化ナトリウム溶液 硫化ナトリウム九水和物10 gを水に溶かして100 mLとする。使用の都度調製
するのが望ましい。
f)
0.01 mol/L EDTA標準溶液 12.2.2 h)で調製したもの。
14.4.3 操作
定量操作は,次の手順による。
a) 13.2.4 b)で保存した試料溶液(B)から25 mLを分取してビーカー(300 mL)に入れ,水を加えて,
約100 mLとする。
塩酸ヒドロキシルアミン溶液(50 g/L)5 mL,トリエタノールアミン (1+1) 2 mL及び硫化ナトリ
ウム溶液1 mLを加え,次に緩衝溶液(pH10)を加えてpHを9.8〜10.2に調節する。
b) エリオクロムブラックT溶液2,3滴を加え,0.01 mol/L EDTA標準溶液で滴定し,溶液の色が赤紫か
16
R 9101:2018
ら赤みが全く消えて鮮明な青となった点を終点とする。
14.4.4 計算
試料中の酸化マグネシウムの含有率は,次の式によって算出する。
GM(
)
100
25
500
1
403
000
.0
1
2
×
×
×
×
−
s
f
v
v
=
ここに,
GM: 酸化マグネシウムの含有率(%)
v1: 13.2.4 c)の0.01 mol/L EDTA標準溶液の使用量(mL)
v2: 14.4.3 b)の0.01 mol/L EDTA標準溶液の使用量(mL)
f: 12.2.2 h)の0.01 mol/L EDTA標準溶液のファクター
s: 13.2.3ではかりとった試料の質量(g)
15 三酸化硫黄の定量方法
15.1 方法の区分
三酸化硫黄の定量は,質量法による。
15.2 要旨
13.2.4 b)で保存した試料溶液(B)を用い,塩化バリウム溶液を加えて,硫酸バリウムを沈殿させる。沈
殿はろ過し,加熱して質量をはかる。
試料に亜硫酸カルシウムを含有する場合には,試料を別途にはかりとり,亜硫酸カルシウム分を過酸化
水素で酸化した後,これを試料溶液として,全三酸化硫黄を求め,16.6で得られる二酸化硫黄の量を差し
引いて求める。
15.3 試薬
試薬は,次のものを用いる。
a) 塩化バリウム溶液(50 g/L) 塩化バリウム二水和物60 gを水に溶かして1 Lとする。
b) 過酸化水素水 (1+9) 過酸化水素水(30 %)を水でうすめる。
c) 緩衝溶液(pH4) 酢酸ナトリウム75 gを酢酸 (1+2) 500 mLに溶かす。
d) 塩酸 (1+1)
15.4 試料はかりとり量
試料に亜硫酸カルシウムを含有する場合には,試料0.2 gを0.1 mgまで正しくはかりとる。
15.5 操作
15.5.1 試料に亜硫酸カルシウムを含有しない場合
操作は,次の手順による。
a) 13.2.4 b)で保存した試料溶液(B)から200 mLをビーカー(500 mL)に分取し,水を加えて全量を約
300 mLとする。
b) 加熱して煮沸しながら塩化バリウム温溶液(50 g/L)25 mLを少しずつ約10分間かけて滴下し,沈殿
物が成長して溶液が清澄するまで煮沸を続ける。
c) これを煮沸に近い温度で約3時間静置する。この間,溶液の量がほぼ300 mLに保たれるように注意
し,必要ならば適宜温水を加える。
d) 沈殿をろ紙(6種,11 cm)でろ過し,温水で8〜10回洗浄する。
e) 沈殿をろ紙とともに磁器るつぼに入れて乾燥し,緩やかに加熱して炎の出ないように注意しながらろ
紙を灰化し,800±50 ℃に調節した電気炉で30分間均熱保持し,冷却した後沈殿の質量をはかる。
17
R 9101:2018
15.5.2 試料に亜硫酸カルシウムを含有する場合
操作は,次の手順による。
a) 試料をビーカー(300 mL)に正しくはかりとり,過酸化水素水 (1+9) 10 mL及び緩衝溶液(pH4)10
mLを加えて,水で全量を約150 mLとする。かき混ぜながら亜硫酸カルシウムを酸化し,溶かした後,
ろ紙(6種,11 cm)でろ過し,温水で数回洗浄する。ろ液及び洗液はビーカー(500 mL)に受ける。
b) ろ液に塩酸 (1+1) 2 mL及び水を加えて全量を約300 mLとする。
c) 以後は,15.5.1 b)からの操作による。
15.6 計算
試料中の三酸化硫黄の含有率は,次のa)又はb)の式によって算出する。
a) 試料に亜硫酸カルシウムを含有しない場合
Gs3
100
200
500
343
.0
1
1
×
×
×
s
m
=
ここに,
Gs3: 三酸化硫黄の含有率(%)
m1: 沈殿の質量(g)
s1: 13.2.3ではかりとった試料の質量(g)
b) 試料に亜硫酸カルシウムを含有する場合
Cs3
100
5
012
.0
343
.0
2
3
2
×
×
−
×
s
m
m
=
ここに,
Gs3: 三酸化硫黄の含有率(%)
m2: 沈殿の質量(g)
m3: 16.6で求めた二酸化硫黄の含有率(%)×s2
s2: 15.4ではかりとった試料の質量(g)
16 二酸化硫黄の定量方法
16.1 方法の区分
二酸化硫黄の定量は,チオ硫酸ナトリウム滴定法による。
16.2 要旨
試料によう素標準溶液を加えて酸化した後,塩酸を加えて溶かし,チオ硫酸ナトリウム標準溶液で滴定
する。
16.3 試薬
試薬は,次のものを用いる。
a) 0.1 mol/Lチオ硫酸ナトリウム標準溶液 チオ硫酸ナトリウム五水和物26 g及び炭酸ナトリウム(無
水)0.2 gをはかりとり,水を加えて溶かし1 Lになるようにうすめる。これに3-メチル-1-ブタノール
(イソアミルアルコール)約10 mLを加え,よく振り混ぜて2日間放置する。この溶液は,次のよう
にして標定する。
よう素酸カリウム(標準試薬)を120〜140 ℃で2時間乾燥し,デシケーター中で放冷した後,約
0.71 gを0.1 mgまで正しくはかりとり,水で溶かした後,200 mLの全量フラスコに移して水で標線ま
でうすめる。この溶液20 mLを分取して共通すり合わせよう素フラスコA形(300 mL)に入れ,よう
化カリウム2 g及び硫酸 (1+5) 5 mLを加え速やかに栓をして静かに振り混ぜた後,水100 mLを加え,
直ちに遊離したよう素をチオ硫酸ナトリウム標準溶液で滴定する。滴定の終点近くで溶液の色が微黄
色になったとき,でん(澱)粉溶液数滴を加えて滴定を続け,青が消えたときを終点とする。次の式
18
R 9101:2018
によってファクターを算出する。
7
566
003
.0
1
200
20
100
1
×
×
×
×
v
a
m
f=
ここに,
f1: 0.1 mol/Lチオ硫酸ナトリウム標準溶液のファクター
m: よう素酸カリウム(標準試薬)の質量(g)
a: よう素酸カリウム(標準試薬)の純度(%)
v: 0.1 mol/Lチオ硫酸ナトリウム標準溶液の使用量(mL)
b) 塩酸 (1+1)
c) 0.05 mol/Lよう素標準溶液 よう化カリウム40 gを少量の水に溶かし,これによう素12.7 gを加えて
完全に溶かした後,塩酸 (1+1) 1 mL及び水を加えて1 Lとする。この溶液は,次のようにして標定
する。
よう素標準溶液20 mLを分取し,水100 mLを加え,0.1 mol/Lチオ硫酸ナトリウム標準溶液で滴定
する。滴定の終点近くで溶液の色が微黄色になったとき,でん(澱)粉溶液数滴を加えて滴定を続け,
青が消えたときを終点とする。この標準溶液のファクターは,次の式によって算出する。
20
1
2
f
v
f
×
=
ここに,
f2: 0.05 mol/Lよう素標準溶液のファクター
v: 0.1 mol/Lチオ硫酸ナトリウム標準溶液の使用量(mL)
f1: 0.1 mol/Lチオ硫酸ナトリウム標準溶液のファクター
d) でん(澱)粉溶液 でん(澱)粉1 gをビーカー(100 mL)にはかりとり,冷水10 mLを加えてよく
かき混ぜた後,熱湯200 mLの中へかき混ぜながら少しずつ加える。液が透明になるまで煮沸し,放
冷した後,上澄液を使用する。この溶液を保存するときは,少量のトルエン又は二硫化炭素を添加す
る。
16.4 試料はかりとり量
試料は,約2 gを0.1 mgまで正しくはかりとる。
16.5 操作
定量操作は,次の手順による。
a) 試料を共通すり合わせよう素フラスコA形(300 mL)に正しくはかりとり,水約50 mLを加えた後,
0.05 mol/Lよう素標準溶液を一定量(25 mL)加えて振り混ぜ,塩酸 (1+1) 20 mLを加えた後,速や
かに栓をして10〜15分間振り混ぜて試料を溶かす。
b) フラスコの栓を除き,未反応のよう素を0.1 mol/Lチオ硫酸ナトリウム標準溶液で滴定する。滴定の終
点近くで溶液の色が微黄色になったとき,でん(澱)粉溶液数滴を加えて滴定を続け,青が消えたと
きを終点とする。
16.6 計算
試料中の二酸化硫黄の含有率は,次の式によって算出する。
Gs2(
)
100
203
003
.0
1
1
2
2
×
×
×
−
×
s
f
v
f
v
=
ここに,
Gs2: 二酸化硫黄の含有率(%)
v1: 0.1 mol/Lチオ硫酸ナトリウム標準溶液の使用量(mL)
f1: 0.1 mol/Lチオ硫酸ナトリウム標準溶液のファクター
v2: 0.05 mol/Lよう素標準溶液の使用量(mL)
f2: 0.05 mol/Lよう素標準溶液のファクター
s: 16.4ではかりとった試料の質量(g)
19
R 9101:2018
17 二酸化炭素の定量方法
17.1 方法の区分
二酸化炭素の定量は,水酸化ナトリウム溶液吸収−塩酸滴定法による。
17.2 要旨
試料を塩酸で分解し,生成した二酸化炭素を塩化バリウム及び水酸化ナトリウム溶液中に導入して吸収
させ,塩酸標準溶液で滴定する。
17.3 試薬
試薬は,次のものを用いる。
a) メチルオレンジ溶液(1 g/L) 調製方法は,JIS K 8001のJA.5による。
b) フェノールフタレイン溶液(10g/Lエタノール溶液) 調製方法は,JIS K 8001のJA.5による。
c) ブロモフェノールブルー溶液(1 g/L) 調製方法は,JIS K 8001のJA.5による。
d) 0.2 mol/L水酸化ナトリウム標準溶液 水酸化ナトリウム9 gを水1 Lに溶かし,ポリエチレン瓶に保
存する。
なお,市販の水酸化ナトリウム標準溶液を用いることができる。
この溶液は調製のときに炭酸塩を除く必要はない。また,あらかじめ標定しておく必要もない。
e) 塩化バリウム溶液 塩化バリウム二水和物10 gを水1 Lに溶かし,これにフェノールフタレイン溶液
(10 g/Lエタノール溶液)約1 mLを加える。
f)
塩酸 (1+5)
g) 0.2 mol/L塩酸標準溶液 塩酸20 mLを適量の水でうすめた後,1 Lとする。
なお,市販の塩酸標準溶液を用いることができる。この溶液は,次のようにして標定する。
JIS K 8005の炭酸ナトリウムを白金るつぼ中で500〜650 ℃に40〜50分間保ち,硫酸デシケーター
中で放冷したものを用いる。この炭酸ナトリウム(標準試薬)2.5〜3.0 gを0.1 mgまで正しくはかり
とり,適量の水に溶かして250 mLの全量フラスコに移し,水で標線までうすめる。この溶液25 mL
を分取し,ブロモフェノールブルー溶液を用いて塩酸標準溶液で滴定し,溶液が青紫から黄色になっ
たときを終点とする。次の式によってファクターを算出し,小数点以下3桁に丸める。
6
010
.0
1.0
×
×
v
m
f=
ここに,
f: 0.2 mol/L塩酸標準溶液のファクター
m: はかりとった炭酸ナトリウムの質量(g)
v: 0.2 mol/L塩酸標準溶液の使用量(mL)
17.4 装置
装置は,図1の装置を用いる。
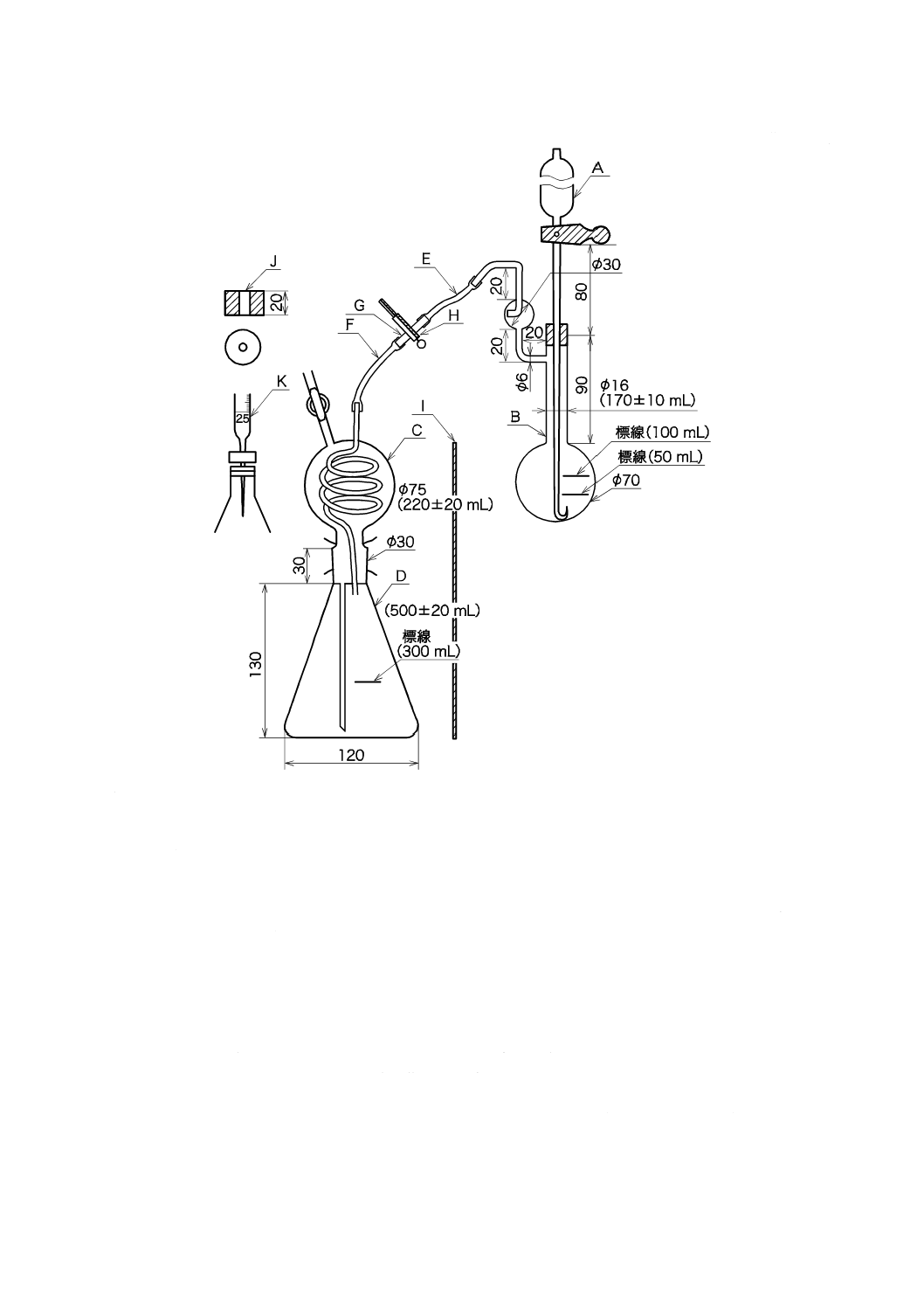
20
R 9101:2018
単位 mm
A
:活栓付漏斗 容量約60 mL,足の下方はやや細く,先端は上向きに曲げる。
B
:分解フラスコ 首までの容積170±10 mLのもので,50 mL及び100 mLの所にそれぞれ標線を付ける。側
管には小形の蒸留管を備える。
C
:ガス吸収器球状部 下部三角フラスコから押し上げられた吸収液をこの中にためるとともに,分解フラス
コから導入される熱水蒸気の冷却器としての役目をする。容積220±20 mL,内部に封入された曲管及び下
部の足管は,内径2 mm,外径5〜6 mmとする。
D
:ガス吸収器下部三角フラスコ 上部球状部とすり合わせによって一体をなす。容積500±20 mLのもので,
300 mLの所に標線を付ける。
なお,球状部と連結するときに,ゴムバンドを架けるように球状部及びフラスコのすり合わせの上下に
はバンド止めの角を備える。
E・F :ゴム管 Fは,これをGから外して球状部の活栓口に連結できるような長さにしておく。
G
:ガラス管
H
:ピンチコック
I
:遮蔽板 分解フラスコを加熱するときに吸収器への放射熱を防ぐために用いる。
J
:ゴム栓 吸収器の三角フラスコに合う薄形のゴム栓で,ビュレットの先を深く差し込めるように小孔が開
けてあるもので,滴定のときに用いる。
K
:滴定用ビュレット 容量25 mL,コックから先の部分を約7 cmに引き伸ばし,先端の孔を細くして1滴が
0.02〜0.03 mLになるようにする。
図1−二酸化炭素定量装置

21
R 9101:2018
17.5 試料はかりとり量
試料は,二酸化炭素の含有率に応じて表2に示す量を0.1 mgまで正しくはかりとる。
表2−試料はかりとり量
二酸化炭素の含有率 % 試料の質量 g
2未満
2
2以上
5未満
1
5以上
0.5
17.6 操作
定量操作は,次の手順による。
a) 試料をはかりとって分解フラスコBに入れ,50 mLの標線まで水を加え,メチルオレンジ溶液(1 g/L)
1,2滴を加える。煮沸を円滑にするために,一端を封じた細いガラス毛細管(長さ2〜3 cm)数本を
入れ,活栓付漏斗Aを差し込んで密栓する。
b) 0.2 mol/L水酸化ナトリウム標準溶液25 mLを分取してガス吸収器下部三角フラスコDに入れ,直ち
に塩化バリウム溶液を300 mLの標線まで加え,ガス吸収器球状部Cをはめてバンドをかけた後,図
1のようにガス吸収器と分解フラスコとを連結する。
なお,定量操作の前に,分解フラスコ及び吸収器は一度水を満たした後,水を流出して内部を実験
室内の空気で置換しておく。特に数個の試料について連続して定量を行う場合には,前回の操作で装
置内の空気が実験室内の空気と異なった組成になっているから,新たに試験を行うたびにこれを実行
する必要がある。
警告 換気の悪い室内で多数のバーナーを使用し,室内の空気中の炭酸ガス量が甚だしく変動する
おそれのある場合には,装置内の空気の置換を室外で行えばよい。
c) 連結管のピンチコックHでガラス管Gを挟み,ガスの通路を開き,ガス吸収器球状部の活栓を開く。
d) 活栓付漏斗Aに塩酸 (1+5) を満たし,活栓を開いて分解フラスコ中に塩酸を少しずつ流下させ,試
料が溶けて溶液が赤変してから,更に1〜2 mL過剰に加える。ただし,分解フラスコ内の溶液は最後
まで赤を保っていなければならない。
なお,塩酸 (1+5) を大過剰に加えることは避けなければならない。
e) 分解フラスコを緩やかに加熱し,溶液が煮沸してガス吸収器球状部の曲管の露出部が指頭で長く触れ
られない程度に熱せられてから,更に10分間煮沸を継続する。加熱の強さは,凝縮水が曲管から数秒
間に1,2滴の割合で滴下する程度とする。
f)
加熱をやめ,手早くピンチコックHでゴム管Fを閉じ,ガス吸収器球状部の活栓も閉じる。ゴム管F
をガラス管Gから離し活栓の先端を連結させ,吸収器を約5分間激しく振り混ぜた後,ピンチコック
H及び活栓を開けて球状部内に残留している吸収液を三角フラスコ内に流下させる。
なお,振り混ぜたとき,三角フラスコ内の溶液の赤が消えた場合は,ゴム管Fの先をガラスコック
の方に連結して閉回路を作り,外気と遮断した状態でピンチコック及び活栓を少し開いて球状部内の
液の一部を三角フラスコに流下させた後,赤の消えない状態で,更に約5分間振り混ぜる。もし球状
部内の液の半分近くを流下させてもなお赤が消えたならば,試料を少なくしてもう一度操作をやり直
す。
g) 三角フラスコから球状部を取り外してゴム栓Jをはめ,滴定用ビュレットKの先端を深く差し込み,
22
R 9101:2018
0.2 mol/L塩酸標準溶液で滴定し,赤が消えたときを終点とする。
17.7 空試験
定量操作と同一の条件で空試験を行い,0.2 mol/L塩酸標準溶液の使用量を求める。
17.8 計算
試料中の二酸化炭素の含有率は,次の式によって算出する。
Gc2(
)
100
4
004
.0
2
1
×
×
×
−
s
f
v
v
=
ここに,
Gc2: 二酸化炭素の含有率(%)
v1: 空試験の0.2 mol/L塩酸標準溶液の使用量(mL)
v2: 試料分析時の0.2 mol/L塩酸標準溶液の使用量(mL)
f: 0.2 mol/L塩酸標準溶液のファクター
s: 17.5ではかりとった試料の質量(g)
18 酸化ナトリウム及び酸化カリウムの定量方法
18.1 方法の区分
酸化ナトリウム及び酸化カリウムの定量は,原子吸光分析法による。
18.2 要旨
10.5 d)で保存した試料溶液(A)を用いて,原子吸光分析装置によって,ナトリウム及びカリウムの吸
光度を測定し,酸化ナトリウムの含有率及び酸化カリウムの含有率を求める。
18.3 試薬
試薬は,次のものを用いる。
a) マトリックス溶液-I 12.4.2 e)で調製したもの。
b) マトリックス溶液-II 12.4.2 f)で調製したもの。
c) 酸化ナトリウム及び酸化カリウム標準原液(1.0 mgNa2O/mL, 1.0 mgK2O/mL) 500〜650 ℃で40〜
50分間乾燥した後,放冷した塩化ナトリウム1.886 g及び110 ℃で2〜3日間乾燥した後,放冷した塩
化カリウム1.583 gをはかりとって,1 Lの全量フラスコに入れ,水を加えて溶かした後,水で標線ま
でうすめる。この溶液は,ポリエチレン瓶に移して保存する。
18.4 装置
装置は,原子吸光分析装置を用いる。
18.5 操作
定量操作は,次の手順による。
a) 10.5 d)で保存した試料溶液(A)の一部を分取し,水を加えて10倍にうすめる。その一部を原子吸光
分析装置のアセチレン・空気フレーム中に噴霧し,ナトリウム用光源ランプを用いて波長589.0 nmに
おけるナトリウムの吸光度を測定する。
b) 次に,同じ操作で,カリウム用光源ランプを用いて,波長766.5 nmにおけるカリウムの吸光度を測定
する。
18.6 検量線の作成
検量線の作成は,次の手順による。
a) 18.3 c)の酸化ナトリウム及び酸化カリウム標準原液(1.0 mgNa2O/mL,1.0 mgK2O/mL)を水で50倍に
うすめ,その0〜20 mL(酸化ナトリウム及び酸化カリウムとして,それぞれ0〜0.4 mg)を100 mL
の全量フラスコに段階的にとり,水を加えて約50 mLとする。これに18.3 a)のマトリックス溶液-I及
23
R 9101:2018
び18.3 b)のマトリックス溶液-IIをそれぞれ水で5倍にうすめた希釈溶液10 mLずつを分取して加え,
水で標線までうすめる。
b) これらの標準溶液を原子吸光分析装置のアセチレン・空気フレーム中に噴霧し,波長589.0 nmにおけ
る吸光度を測定し,吸光度と酸化ナトリウムの濃度との関係線を作成して,酸化ナトリウム用検量線
とする。
c) 次に,同じ操作で波長766.5 nmにおける吸光度を測定し,吸光度と酸化カリウムの濃度との関係線を
作成して,酸化カリウム用検量線とする。
18.7 計算
18.6のa),b)及びc)で作成した検量線を用い,18.5のa)及びb)で測定した吸光度から,酸化ナトリウム
の濃度及び酸化カリウムの濃度を求め,次の式によって試料中の酸化ナトリウムの含有率及び酸化カリウ
ムの含有率を算出する。
GN
100
10
100
250
103
1
×
×
×
×
−
s
C
=
GK
100
10
100
250
103
2
×
×
×
×
−
s
C
=
ここに,
GN: 酸化ナトリウムの含有率(%)
GK: 酸化カリウムの含有率(%)
C1: 吸光度から求めた酸化ナトリウムの濃度(mg/100 mL)
C2: 吸光度から求めた酸化カリウムの濃度(mg/100 mL)
s: 10.4ではかりとった試料の質量(g)
19 ふっ素の定量方法
19.1 方法の区分
ふっ素の定量は,次のいずれかの方法による。
a) ランタン−アリザリンコンプレキソン吸光光度法
b) イオン電極法
19.2 ランタン−アリザリンコンプレキソン吸光光度法
19.2.1 要旨
試料に過塩素酸及び二酸化けい素を加えて,水蒸気蒸留によってふっ素をけいふっ化水素酸として留出
させ,その留出液を用いて,ランタン−アリザリンコンプレキソン溶液で呈色させ,その吸光度を測定す
る。
19.2.2 試薬
試薬は,次のものを用いる。
a) 二酸化けい素 粒度100〜150 μmのもの。又はメタけい酸ナトリウムを用いてもよい。
b) りん酸
c) 過塩素酸 加熱して白煙を発生させた後,放冷したもの。
d) ランタン−アリザリンコンプレキソン溶液 アリザリンコンプレキソン(1,2-ジヒドロキシアントラ
キノニル-3-メチルアミン-N,N二酢酸)0.192 gをアンモニア水 (1+10) 4 mL及び酢酸アンモニウム
溶液(200 g/L)4 mLに溶かし,これを酢酸ナトリウム溶液(酢酸ナトリウム三水和物41 gを水400 mL
に溶かし,酢酸24 mLを加えもの)中にかき混ぜながら加える。この溶液にアセトン400 mLをかき
混ぜながら少しずつ加え,更にランタン溶液[酸化ランタン0.163 gを塩酸 (1+5) 10 mLに加熱し溶
24
R 9101:2018
かしたもの]を加えてかき混ぜる。冷却した後,酢酸又はアンモニア水でpH計を用いてpHを約4.7
に調節した後,水を加えて1 Lとする。
なお,ランタン−アリザリンコンプレキソン溶液として,市販のアルフッソン2.5 gを水に溶かして
50 mLとした溶液を用いてもよい。使用時に調製する。
e) ふっ化物イオン標準原液(1 000 mg/L)
f)
ふっ化物イオン標準溶液(2 mg/L) ふっ化物イオン標準原液(1 000 mg/L)を水で正しく10倍にう
すめ,それを10 mLとり,全量フラスコ500 mLに入れ,水で標線までうすめる。
19.2.3 装置
装置は,次による。
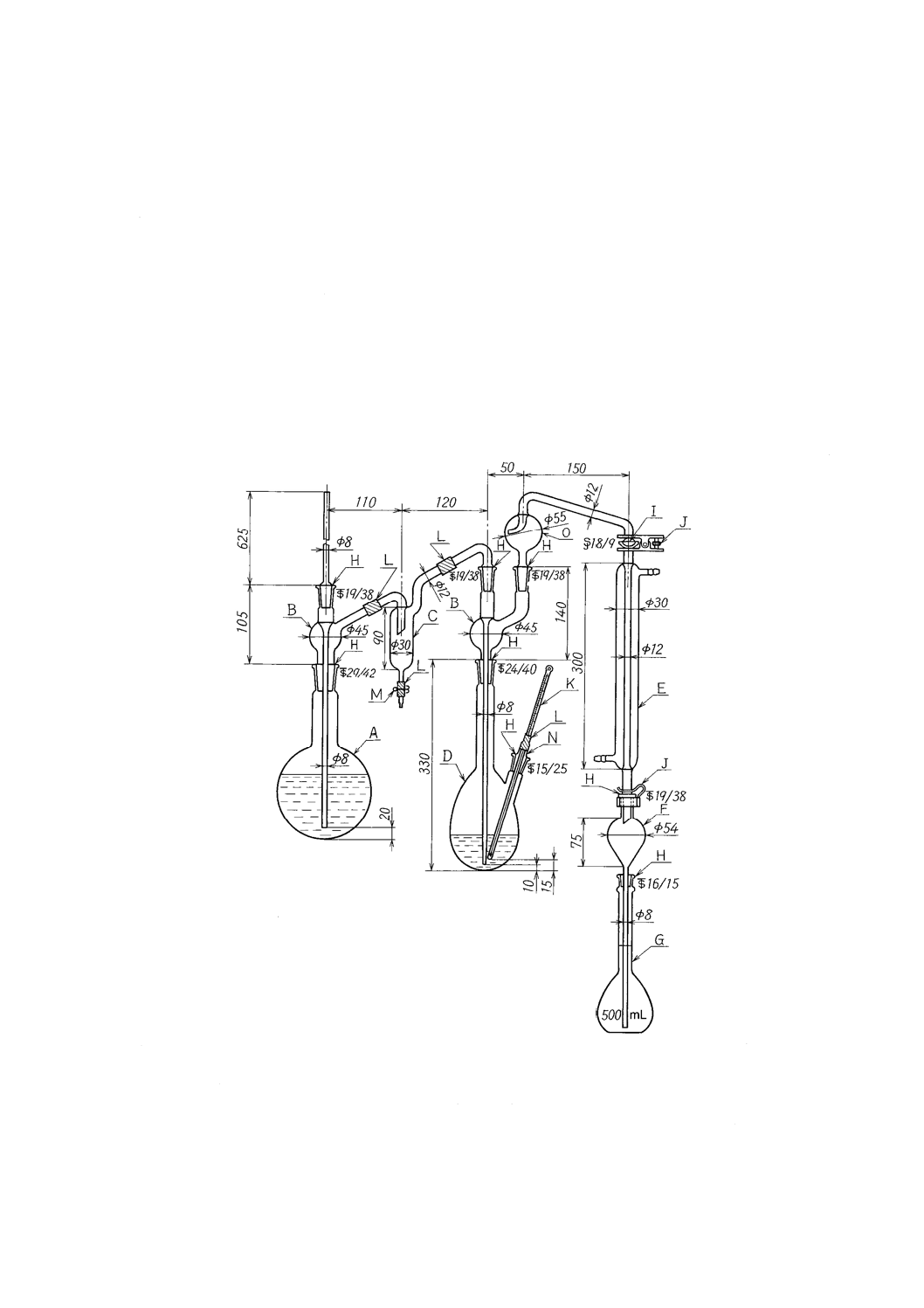
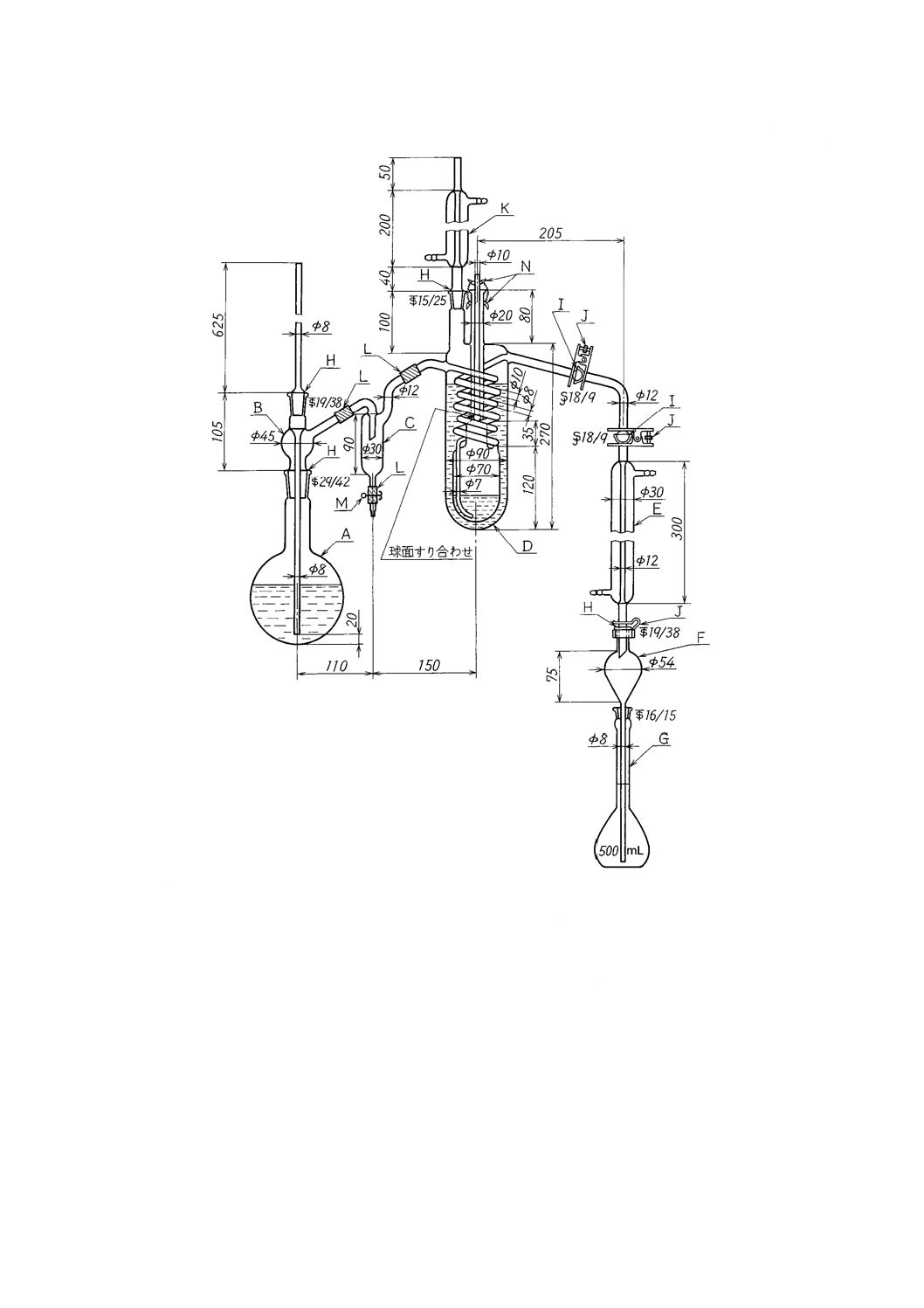
a) 蒸留装置 図2及び図3に装置の一例を示す。
b) 吸光光度分析装置
単位 mm
A :水蒸気発生フラスコ1 000 mL
F :逆流止め(約50 mL)
K :温度計200 ℃
B :連結導入管
G :受器(全量フラスコ500 mL)
L :ゴム管
C :トラップ
H :共通テーパーすり合わせ
M :ピンチコック
D :ケルダールフラスコ500 mL
I :共通球面すり合わせ
N :温度計差込み栓
E :リービッヒ冷却器300 mm
J :押さえばね
O :トラップ球
図2−蒸留装置の一例(A)
25
R 9101:2018
単位 mm
A :水蒸気発生フラスコ1 000 mL
H :共通テーパーすり合わせ
B :連結導入管
I :共通球面すり合わせ
C :トラップ
J :押さえばね
D :蒸留フラスコ300 mL
K :リービッヒ冷却器200 mm
(外筒に1,1,2,2-テトラクロロエタンを入れる)
L :ゴム管
E :リービッヒ冷却器300 mm
M :ピンチコック
F :逆流止め(約50 mL)
N :スプリングフック
G :受器(全量フラスコ500 mL)
図3−蒸留装置の一例(B)

26
R 9101:2018
19.2.4 試料はかりとり量
試料は,ふっ素の含有率に応じて表3に示す量を0.1 mgまで正しくはかりとる。
表3−試料はかりとり量
ふっ素の含有率 %
試料の質量 g
0.5未満
3
0.5以上
1.0未満
2
1.0以上
1
19.2.5 操作
定量操作は,次の手順による。
a) 試料をはかりとって蒸留フラスコDに入れ,二酸化けい素約0.5 g,りん酸1 mL及び過塩素酸40 mL
を加える。留出液の受器G(全量フラスコ500 mL)には水20 mLを加え,逆流止めFの先端は,水
面下に保つ。
なお,試料中にふっ化物イオン以外のハロゲン化物イオンが多量に含まれる場合には,水酸化ナト
リウム溶液(40 g/L)数滴及びフェノールフタレイン溶液1滴を加えておく。受器中の溶液は蒸留が
終わるまで微紅色を保つように必要に応じ水酸化ナトリウム溶液(40 g/L)を滴加する。さらに,こ
の場合は,蒸留が終わった後,留出液に硫酸 (1+35) を微紅色が消えるまで滴加し,e)以下の操作を
行う。
b) 図2のようにケルダールフラスコを使用する場合には,フラスコを直接加熱し,図3のように蒸留フ
ラスコを使用する場合には,外筒を加熱する。
なお,加熱には油浴,グリセリン浴などを用いてもよい。必要な場合,径2〜3 mmの沸騰石を約
10個入れる。
c) 図2の場合には,フラスコ内の液温が約140 ℃に達してから,また,図3の場合には外筒中の1,1,2,2-
テトラクロロエタンが煮沸し始めてから,水蒸気を通す。
d) 蒸留温度を145±5 ℃,留出速度を3〜5 mL/minに調節し,受器の液量が約400 mLになるまで蒸留を
続ける。
e) 冷却器及び逆流止めを取り外し,冷却器の内管及び逆流止めの内外を少量の水で洗い,洗液も受器に
加え,更に水で標線までうすめる。
f)
留出液から30 mL以下の適量(F−として0.004〜0.05 mgを含む。)を分取して,全量フラスコ50 mL
にとる。
なお,ふっ素の含有率が高い場合は,必要に応じて希釈操作を行う。
g) ランタン−アリザリンコンプレキソン溶液20 mLを加え,更に水で標線までうすめて振り混ぜ,1時
間放置する。
h) この溶液の一部を吸光光度分析装置の吸収セルにとり,波長620 nm付近の吸光度を測定する。
19.2.6 検量線の作成
19.2.2 f)のふっ化物イオン標準溶液(2 mg/L)2〜25 mLを段階的にとり,19.2.5のf),g)及びh)の操作に
よって吸光度を測定し,その吸光度とふっ化物イオン濃度との関係線を作成して検量線とする。
19.2.7 計算
19.2.6で作成した検量線を用い,19.2.5 h)で測定した吸光度からふっ化物イオン濃度を求め,次の式によ
って試料中のふっ素の含有率を算出する。
27
R 9101:2018
Gf
100
500
103
×
×
×
−
v
s
C
=
ここに,
Gf: ふっ素の含有率(%)
C: 吸光度から求めたふっ化物イオンの濃度(mg/50 mL)
s: 19.2.4ではかりとった試料の質量(g)
v: 19.2.5 f)の試料溶液の分取量(mL)
19.3 イオン電極法
19.3.1 要旨
19.2.5と同じ操作で水蒸気蒸留を行い,その留出液を用い,緩衝溶液(全イオン強度調節液)を加えて,
pH5.0〜5.5に調節した後,ふっ化物イオン電極を用いて電位を測定する。
19.3.2 試薬
試薬は,次のものを用いる。
a) 緩衝溶液(pH5.2)4) 塩化ナトリウム58 g及びクエン酸水素アンモニウム二水和物1 gに水500 mL
を加えて溶かし,酢酸50 mLを加え,水酸化ナトリウム溶液(200 g/L)を滴下して,pH計を用いて
pH5.2に調節した後,水を加えて1 Lとする。
b) ふっ化物イオン標準原液(1 000 mg/L)
注4) 緩衝溶液として,次の組成のものを使用してもよい。
1) 水500 mLに酢酸57 mL,塩化ナトリウム58 g,1,2-シクロヘキサンジアミン四酢酸
(CyDTA)4 gを加えて溶かし,水酸化ナトリウム溶液(200 g/L)を滴下し,pH計を用
いてpH5.0〜5.5に調節した後,水を加えて1 Lにする。
2) 水500 mLに酢酸57 mL,塩化ナトリウム58 g,クエン酸ナトリウム二水和物0.3 gを加
えて溶かし,水酸化ナトリウム溶液(200 g/L)を加え,pH計を用いてpH5.0〜5.5に調
節した後,水を加えて1 Lにする。
19.3.3 装置
装置は,次による。
a) 電位差計 最小目盛1 mVの高入力抵抗電位差計(例えば,デジタル式pH-mV計,拡大スパン付pH-mV
計,イオン電極用電位差計など)。
b) ふっ化物イオン電極
c) 参照電極 二重液絡型(又は塩橋)参照電極(ダブルジャンクションのスリーブ型参照電極又はセラ
ミック型参照電極で抵抗の小さいもの)。内筒液には塩化カリウム溶液(3.3 mol/L又は飽和溶液)を
入れる。外筒液には塩化カリウム溶液(3.3 mol/L又は飽和溶液)又は硝酸カリウム溶液(100 g/L)を
入れる。
d) マグネチックスターラー 回転による発熱で液温に変化を与えないもの。
19.3.4 操作
定量操作は,次の手順による。
a) 19.2.5のa)〜e)と同じ操作で水蒸気蒸留を行い,留出液を500 mLの全量フラスコに受けた後,水を標
線まで加える。この試料溶液から100 mLをビーカー(200 mL)に分取し,緩衝溶液(pH5.2)10 mL
を加える。
b) これにふっ化物イオン電極と参照電極とを浸し,マグネチックスターラーを用いて,泡が電極に触れ
ない程度に強くかき混ぜる。
28
R 9101:2018
なお,ふっ化物イオン電極の感応膜に傷がつくと,検量線の勾配(電位勾配)が小さくなり,応答
速度も遅くなるので注意する。また,イオン電極の感応膜が汚れると,反応速度が遅くなるので,脱
脂綿にアルコールを含ませて汚れを拭き取るか,又は柔らかい紙(ティッシュペーパーなど)で汚れ
を拭き取り,水で洗浄する。
c) 液温の測定を行い,電位差計で電位を測定する。ふっ化物イオン電極の応答時間は,ふっ化物イオン
濃度が0.1 mg/Lで約1分間,1 mg/L以上では約30秒間である。
19.3.5 検量線の作成
検量線の作成は,次の手順による。
a) ふっ化物イオン標準原液(1 000 mg/L)を水で正しく10倍にうすめ,ふっ化物イオン標準溶液(100
mg/L)を調製する。
b) ふっ化物イオン標準溶液(100 mg/L)20 mLを200 mLの全量フラスコに分取し,水を標線まで加えて,
ふっ化物イオン標準溶液(10 mg/L)を調製する。
c) ふっ化物イオン標準溶液(10 mg/L)20 mLを200 mLの全量フラスコに分取して,ふっ化物イオン標
準溶液(1 mg/L)を調製し,更にこれを10倍にうすめて,ふっ化物イオン標準溶液(0.1 mg/L)を調
製する。
d) 段階的に調製したふっ化物イオン標準溶液(0.1〜100 mg/L)のそれぞれ100 mLをビーカー(200 mL)
にとり,緩衝溶液(pH5.2)10 mLを加える。
e) それぞれのふっ化物イオン標準溶液の液温を試料溶液の液温の±1 ℃以内になるように調節し,19.3.4
のb)及び c)と同じ操作で電位を測定する。
f)
片対数方眼紙の対数軸にふっ化物イオンの濃度をとり,均等軸に電位をとって,ふっ化物イオン濃度
(mg/L)と電位との関係線を作成して検量線とする。ふっ化物イオン標準溶液(1 mg/L)と同溶液(100
mg/L)との電位の差は,110〜120 mVの範囲に入り,ふっ化物イオンの濃度0.1 mg/Lから100 mg/L
の間の検量線は直線になる。
19.3.6 計算
19.3.5で作成した検量線を用い,19.3.4で測定した電位からふっ化物イオン濃度を求め,次の式によって
試料中のふっ素の含有率を算出する。
Gf
100
100
500
103
×
×
×
−
s
C
=
ここに,
Gf: ふっ素の含有率(%)
C: 測定電位から求めたふっ化物イオン濃度(mg/100 mL)
s: 19.2.4ではかりとった試料の質量(g)
20 全りん酸の定量方法
20.1 方法の区分
全りん酸の定量は,次のいずれかの方法による。
a) モリブデン青吸光光度法 この方法は,五酸化りんの含有率1.0 %未満の試料に適用する。
b) りんバナドモリブデン酸吸光光度法 この方法は,五酸化りんの含有率0.0l %以上の試料に適用する。
20.2 モリブデン青吸光光度法
20.2.1 要旨
10.5 d)で保存した試料溶液(A)を用い,水酸化ナトリウム及び硫酸で酸濃度を調節した後,モリブデ
29
R 9101:2018
ン酸アンモニウム及びアスコルビン酸を加え,加熱して呈色させ,その吸光度を測定する。
20.2.2 試薬
試薬は,次のものを用いる。
a) p-ニトロフェノール指示薬(2 g/L)
b) 水酸化ナトリウム溶液(100 g/L) ポリエチレン瓶に保存する。
c) 硫酸 (1+1)
d) モリブデン酸アンモニウム溶液 モリブデン酸アンモニウム溶液四水和物2 gを温水約20 mLに溶か
し,必要ならばろ過し,硫酸 (1+1) 60 mLを加えた後,水で100 mLにうすめる。
e) アスコルビン酸溶液(50 g/L) 12.3.2 a)で調製したもの。
f)
五酸化りん標準原液(100 mgP2O5/L) りん酸二水素カリウムを105〜110 ℃で3時間乾燥し,デシ
ケーター中で放冷した後,0.192 gをはかりとって水に溶かし,1 Lの全量フラスコに移して水で標線
までうすめる。
20.2.3 装置
装置は,吸光光度分析装置を用いる。
20.2.4 操作
定量操作は,次の手順による。
a) 10.5 d)で保存した試料溶液(A)から全りん酸の含有率に応じて表4に示す量を分取して,100 mLの
全量フラスコに移し,p-ニトロフェノール指示薬(2 g/L)1滴を加え,溶液の色が黄色になるまで水
酸化ナトリウム溶液(100 g/L)を滴加し,次に硫酸 (1+1) を滴加して無色とした後,2,3滴過剰に
加える。
b) これにモリブデン酸アンモニウム溶液10 mL及びアスコルビン酸溶液(50 g/L)2 mLを加え,水で標
線までうすめる。沸騰水中に浸し15分間加熱した後,流水中で室温まで冷却する。
c) この溶液の一部を吸光光度分析装置の吸収セルにとり,波長830 nm付近で吸光度を測定する。
表4−試料溶液の分取量
全りん酸の含有率 %
分取量 mL
0.10未満
25
0.10以上
0.50未満
10
0.50以上
5
20.2.5 検量線の作成
20.2.2 f)の五酸化りん標準原液(100 mgP2O5/L)を水で正しく10倍にうすめ,その0〜20 mL(五酸化り
んとして0〜0.20 mg)を100 mLの全量フラスコに段階的にとり,20.2.4のa),b)及びc)の手順に従って操
作して吸光度を測定し,その吸光度と五酸化りんの濃度との関係線を作成して検量線とする。
20.2.6 計算
20.2.5で作成した検量線を用い,20.2.4 c)で測定した吸光度から五酸化りんの濃度を求め,次の式によっ
て試料中の全りん酸の含有率を算出する。
GtP
100
250
103
×
×
×
−
v
s
C
=
ここに,
GtP: 全りん酸の含有率(%)
C: 吸光度から求めた五酸化りんの濃度(mg/100 mL)

30
R 9101:2018
s: 10.4ではかりとった試料の質量(g)
v: 試料溶液(A)からの分取量(mL)
20.3 りんバナドモリブデン酸吸光光度法
20.3.1 要旨
試料に塩酸及び硝酸を加え,加熱し溶かした後,ろ過し,ろ液にメタバナジン酸アンモニウム及びモリ
ブデン酸アンモニウムを加えて,りんバナドモリブデン酸として呈色させ,その吸光度を測定する。
20.3.2 試薬
試薬は,次のものを用いる。
a) 塩酸
b) 硝酸
c) フェノールフタレイン溶液(10 g/Lエタノール溶液) 17.3 b)で調製したもの。
d) アンモニア水 (1+1)
e) 硝酸 (1+1)
f)
発色試薬溶液 メタバナジン酸アンモニウム1.12 gを適量の水に溶かし,硝酸250 mLを加える。こ
の溶液に,モリブデン酸アンモニウム27 gを水に溶かして加え,更に水を加えて1 Lとする。この溶
液は褐色瓶に入れて保存する。
g) 五酸化りん標準原液(100 mgP2O5/L) 20.2.2 f)で調製したもの。
20.3.3 装置
装置は,吸光光度分析装置を用いる。
20.3.4 試料はかりとり量
試料は,約2.5gを0.1 mgまで正しくはかりとる。
20.3.5 操作
定量操作は,次の手順による。
a) 試料をビーカー(300 mL)に正しくはかりとり,塩酸30 mL及び硝酸10 mLを加えて加熱し,約20
分間煮沸する。
なお,煮沸中,試料溶液の蒸発が進んだ場合は,せっこうの結晶が析出することがある。そのとき
は,水又は塩酸を加え,ろ過前に加熱して溶かす必要がある。
b) 温水約100 mLを加えて可溶分を溶かした後,直ちにろ紙(5種B)でろ過し,水で数回洗浄する。冷
却した後,ろ液及び洗液を250 mL全量フラスコに移し,水で標線までうすめる。
c) 試料溶液の全りん酸の含有率に応じて表5に示す量を分取して100 mL全量フラスコに移し,フェノ
ールフタレイン溶液(10 g/Lエタノール溶液)を1,2滴加え,アンモニア水 (1+1) を加えて中和し,
硝酸 (1+1) を滴下して微酸性とする。水を加えて全量を約70 mLとした後,発色試薬溶液20 mLを
加え,水で標線までうすめ,約30分間放置する。
d) この溶液の一部を吸光光度分析装置の吸収セルにとり,波長400〜420 nmで吸光度を測定する。
表5−試料溶液の分取量
全りん酸の含有率 %
分取量 mL
0.1未満
25
0.1以上
1.0未満
10
1.0以上
5
31
R 9101:2018
20.3.6 検量線の作成
20.3.2 g)の五酸化りん標準原液(100 mgP2O5/L)0〜20 mL(五酸化りんとして0〜2.0 mg)を100 mLの
全量フラスコに段階的にとり,20.3.5のc)及びd)の手順に従って操作して吸光度を測定し,その吸光度と
五酸化りん濃度との関係線を作成して検量線とする。
20.3.7 計算
20.3.6で作成した検量線を用い,20.3.5 d)で測定した吸光度から五酸化りんの濃度を求め,次の式によっ
て試料中の全りん酸の含有率を求める。
GtP
100
250
103
×
×
×
−
v
s
C
=
ここに,
GtP: 全りん酸の含有率(%)
C: 吸光度から求めた五酸化りんの濃度(mg/100 mL)
s: 20.3.4ではかりとった試料の質量(g)
v: 20.3.5 c)の試料溶液の分取量(mL)
21 水溶性りん酸の定量方法
21.1 方法の区分
水溶性りん酸の定量は,次のいずれかの方法による。
a) モリブデン青吸光光度法 この方法は,五酸化りんの含有率1.0 %未満の試料に適用する。
b) りんバナドモリブデン酸吸光光度法 この方法は,五酸化りんの含有率0.01 %以上の試料に適用する。
21.2 モリブデン青吸光光度法
21.2.1 要旨
試料に20倍量の水を加えて水溶性物質を溶出させ,不溶解物をろ過する。そのろ液を用い,水酸化ナト
リウム及び硫酸で酸濃度を調節し,モリブデン酸アンモニウム及びアスコルビン酸を加え加熱して呈色さ
せ,その吸光度を測定する。
21.2.2 試薬
試薬は,次のものを用いる。
a) p-ニトロフェノール指示薬(2 g/L)
b) 水酸化ナトリウム溶液 (100 g/L)
c) 硫酸 (1+1)
d) モリブデン酸アンモニウム溶液 20.2.2 d)で調製したもの。
e) アスコルビン酸溶液(50 g/L) 12.3.2 a)で調製したもの。
f)
五酸化りん標準原液(100 mgP2O5/L) 20.2.2 f)で調製したもの。
21.2.3 装置
装置は,吸光光度分析装置を用いる。
21.2.4 試料はかりとり量
試料は,約15.0 gを0.1 gまで正しくはかりとる。
21.2.5 操作
定量操作は,次の手順による。
a) 試料をはかりとり500 mLの振とうフラスコに入れ,正しく水300 mLを加え,30分間よく振り混ぜ
た後,乾いたろ紙(5種B)でろ過する。

32
R 9101:2018
このろ液を試料溶液(C)とし,水溶性りん酸,塩素及び遊離酸の定量に用いる。
b) 試料溶液(C)から20 mLを分取して250 mLの全量フラスコに移し,水で標線までうすめる。その溶
液の水溶性りん酸の含有率に応じて表6に示す量を分取して100 mLの全量フラスコに移し,20.2.4 a)
と同様の操作によって酸濃度を調節する。
c) 20.2.4 b)と同様の操作で呈色させ,20.2.4 c)の操作で吸光度を測定する。
表6−試料溶液の分取量
水溶性りん酸の含有率%
分取量mL
0.10未満
25
0.10以上
0.50未満
10
0.50以上
5
21.2.6 検量線の作成
20.2.5と同様の方法で検量線を作成する。
21.2.7 計算
21.2.6で作成した検量線を用い,21.2.5 c)で測定した吸光度から五酸化りんの濃度を求め,次の式によっ
て試料中の水溶性りん酸の含有率を算出する。
GsP
100
20
250
300
103
×
×
×
×
×
−
v
s
C
=
ここに,
GsP: 水溶性りん酸の含有率(%)
C: 吸光度から求めた五酸化りんの濃度(mg/100 mL)
s: 21.2.4ではかりとった試料の質量(g)
v: 21.2.5 b)の試料溶液の分取量(mL)
21.3 りんバナドモリブデン酸吸光光度法
21.3.1 要旨
21.2.5 a)で保存した試料溶液(C)を用い,メタバナジン酸アンモニウム及びモリブデン酸アンモニウム
を加えて,りんバナドモリブデン酸として呈色させ,その吸光度を測定する。
21.3.2 試薬
試薬は,次のものを用いる。
a) フェノールフタレイン溶液(10 g/Lエタノール溶液) 17.3 b)で調製したもの。
b) アンモニア水 (1+1)
c) 硝酸 (1+1)
d) 発色試薬溶液 20.3.2 f)で調製したもの。
e) 五酸化りん標準原液(100 mgP2O5/L) 20.2.2 f)で調製したもの。
21.3.3 装置
装置は,吸光光度分析装置を用いる。
21.3.4 操作
定量操作は,次の手順による。
21.2.5 a)で保存した試料溶液(C)の水溶性りん酸の含有率に応じて表6に示す量を分取して100 mLの
全量フラスコに移し,20.3.5のc)及びd)の操作によって,吸光度を測定する。
21.3.5 検量線の作成
20.3.6と同様の方法で検量線を作成する。
33
R 9101:2018
21.3.6 計算
21.3.5で作成した検量線を用い,21.3.4で測定した吸光度から五酸化りんの濃度を求め,次の式によって
試料中の水溶性りん酸の含有率を算出する。
GsP
100
300
103
×
×
×
−
v
s
C
=
ここに,
GsP: 水溶性りん酸の含有率(%)
C: 吸光度から求めた五酸化りんの濃度(mg/100 mL)
s: 21.2.4ではかりとった試料の質量(g)
v: 試料溶液(C)からの分取量(mL)
22 塩素の定量方法
22.1 方法の区分
塩素の定量は,次のいずれかの方法による。
a) 硝酸銀滴定法
b) イオン電極法
22.2 硝酸銀滴定法
22.2.1 要旨
21.2.5 a)で保存した試料溶液(C)を用い,クロム酸カリウムを指示薬として硝酸銀標準溶液で滴定する。
22.2.2 試薬
試薬は,次のものを用いる。
a) 炭酸カルシウム
b) 硝酸 (1+10)
c) クロム酸カリウム溶液(50 g/L)
d) 0.05 mol/L硝酸銀標準溶液 硝酸銀8.5 gを水1 Lに溶かし,褐色瓶に保存する。
なお,市販の硝酸銀標準溶液を用いることができる。この溶液は,次のようにして標定する。
500〜650 ℃で40〜50分間乾燥した後放冷した塩化ナトリウム約2.9 gを0.1 mgまで正しくはかり
とり,適量の水に溶かして1 Lの全量フラスコに移し,水で標線までうすめる。この溶液25 mLを分
取し,クロム酸カリウム溶液(50 g/L)0.5 mLを加えて硝酸銀標準溶液で滴定し,溶液の色が黄色か
ら赤になったときを終点とする。次の式によって0.05 mol/L硝酸銀標準溶液1 mLの塩素相当量を算
出し,小数点以下5桁に丸める。
v
m
E
025
.0
6
606
.0
×
×
=
ここに,
E: 0.05 mol/L硝酸銀標準溶液1 mLの塩素相当量(g)
m: はかりとった塩化ナトリウム質量(g)
v: 0.05 mol/L硝酸銀標準溶液の使用量(mL)
22.2.3 操作
定量操作は,次の手順による。
a) 21.2.5 a)で保存した試料溶液(C)から20〜50 mLの適量を分取してビーカー(200 mL)に入れ,水
を加えて約100 mLとする。
b) 試料溶液のpHが7〜10であることを確認した後,クロム酸カリウム溶液(50 g/L)1 mLを加え,0.05
mol/L硝酸銀標準溶液で滴定し,溶液の色が黄色から赤になったときを終点とする。
34
R 9101:2018
なお,試料溶液のpHが7以下であるときは,溶液に少量の炭酸カルシウムを加えて酸を中和し,
煮沸して炭酸を除き,冷却した後滴定する。pHが10以上であるときは,硝酸 (1+10) を滴下して僅
かに酸性とした後,上記のように炭酸カルシウムで中和する。
22.2.4 計算
試料中の塩素の含有率は,次の式によって算出する。
Gc
100
300
2
1
×
×
×
v
s
E
v
=
ここに,
Gc: 塩素の含有率(%)
v1: 0.05 mol/L硝酸銀標準溶液の使用量(mL)
E: 0.05 mol/L硝酸銀標準溶液1 mLの塩素相当量(g)
s: 21.2.4ではかりとった試料の質量(g)
v2: 試料溶液(C)からの分取量(mL)
22.3 イオン電極法
22.3.1 要旨
21.2.5 a)で保存した試料溶液(C)を用い,緩衝溶液を加えてpHを5に調節し,塩化物イオン電極を用
いて電位を測定する。
22.3.2 試薬
試薬は,次のものを用いる。
a) 緩衝溶液(pH5) 硝酸カリウム100 g及び酢酸50 mLを水500 mLに加えて溶かし,これに水酸化ナ
トリウム溶液(40 g/L)を加え,pH計を用いてpH5に調節し,水を加えて1 Lとする。
b) 塩化物イオン標準原液(1 000 mg/L)
22.3.3 装置
装置は,次による。
a) 電位差計 19.3.3 a)と同じもの。
b) 塩化物イオン電極
c) 参照電極 19.3.3 c)と同じもの。ただし,外筒液には硝酸カリウム溶液(100 g/L)を用いる。
d) マグネチックスターラー 19.3.3 d)と同じもの。
22.3.4 操作
定量操作は,次の手順による。
a) 21.2.5 a)で保存した試料溶液(C)から100 mLを分取して,ビーカー(200 mL)に入れる。試料溶液
が酸性の場合には,水酸化ナトリウム溶液(40 g/L)を用い,アルカリ性の場合には,酢酸 (1+10) を
用いて,あらかじめpHを約5に調節する。また,硫化物イオンが含まれている場合には,酢酸亜鉛
溶液(100 g/L)を加え,硫化物を固定してろ過した後,ろ液をpH5に調節する。これに,緩衝溶液(pH5)
10 mLを加えた後,塩化物イオン電極と参照電極とを浸し,マグネチックスターラーを用いて,泡が
電極に触れない程度に強くかき混ぜる。
b) 液温の測定を行い,電位差計で電位を測定する。塩化物イオン電極の応答時間は,塩化物イオンの濃
度が5 mg/L以上ならば1分間以内である。
22.3.5 検量線の作成
a) 塩化物イオン標準原液(1 000 mg/L)20 mLを200 mLの全量フラスコに分取し,水で標線までうすめ
て,塩化物イオン標準溶液(100 mg/L)を調製する。
b) 塩化物イオン標準溶液(100 mg/L)20 mLを200 mLの全量フラスコに分取して,塩化物イオン標準溶
35
R 9101:2018
液(10 mg/mL)を調製し,更にこれを10倍にうすめて塩化物イオン標準溶液(1 mg/mL)を調製する。
c) 段階的に調製した塩化物イオン標準溶液(1〜1 000 mg/L)のそれぞれ100 mLをビーカー(200 mL)
にとり,緩衝溶液(pH5)10 mLを加える。
d) それぞれの塩化物イオン標準溶液の液温を試料溶液の液温の±1 ℃以内になるように調節し,22.3.4
のa)及びb)と同じ操作で電位を測定する。
e) 片対数方眼紙の対数軸に塩化物イオンの濃度を取り,均等軸に電位を取って,塩化物イオン濃度
(mg/L)と電位との関係線を作成して検量線とする5)。
注5) 塩化物イオン標準溶液(10 mg/L)及び塩化物イオン標準溶液(1 000 mg/L)との電位の差は,
110〜120 mVの範囲に入り,塩化物イオンの濃度1〜1 000 mg/Lの間の検量線は直線になる。
22.3.6 計算
22.3.5で作成した検量線を用い,22.3.4で測定した電位から塩化物イオン濃度を求め,次の式によって試
料中の塩素の含有率を算出する。
Gc
100
100
300
103
×
×
×
−
s
C
=
ここに,
Gc: 塩素の含有率(%)
C: 測定電位から求めた塩化物イオン濃度(mg/100 mL)
s: 21.2.4ではかりとった試料の質量(g)
23 遊離酸の定量方法
23.1 方法の区分
遊離酸の定量は,水酸化ナトリウム滴定法による。
23.2 要旨
21.2.5 a)で保存した試料溶液(C)を用い,フェノールフタレインを指示薬として,水酸化ナトリウム標
準溶液で滴定する。
23.3 試薬
試薬は,次のものを用いる。
a) フェノールフタレイン溶液(10 g/Lエタノール溶液) 17.3 b)で調製したもの。
b) ブロモチモールブルー溶液(1 g/L)
c) 0.1 mol/L水酸化ナトリウム標準溶液 水酸化ナトリウム約4.5 gを水1 Lに溶かし,これに新しく作
った水酸化バリウム飽和溶液を沈殿が生じなくなるまで振り混ぜながら加え,密栓をして2〜3日間放
置した後,上澄み液をとり,ポリエチレン瓶に保存する。
なお,市販の水酸化ナトリウム標準溶液を用いることができる。この溶液は,スルファミン酸又は
標定済みの0.1 mol/L塩酸標準溶液を用いて標定する。
スルファミン酸を用いる場合は,次のようにして標定する。あらかじめデシケーター中に約48時間
放置して乾燥したスルファミン酸(標準試薬)2.0〜2.5 gを0.l mgまで正しくはかりとり,適量の水
に溶かして250 mLの全量フラスコに入れ,水で標線までうすめる。この溶液25 mLを分取し,ブロ
モチモールブルー溶液(1 g/L)3,4滴を加え,0.1 mo1/L水酸化ナトリウム標準溶液で滴定し,次の
式によってファクターを算出し,小数点以下3桁に丸める。
709
009
.0
1.0
1
×
×
v
m
f=
36
R 9101:2018
ここに,
f1: 0.1 mol/L水酸化ナトリウム標準溶液のファクター
m: はかりとったスルファミン酸の質量(g)
v: 0.1 mol/L水酸化ナトリウム標準溶液の使用量(mL)
23.4 操作
定量操作は,次の手順による。
a) 21.2.5 a)で保存した試料溶液(C)から,100 mLを分取してビーカー(300 mL)に入れる。
b) フェノールフタレイン溶液(10 g/Lエタノール溶液)2,3滴を加え,0.1 mol/L水酸化ナトリウム標準
溶液で滴定し,溶液の色が無色から微紅色になったときを終点とする。
なお,終点が分かりにくいときは,試料溶液(C)からの分取量を少なくし,水でうすめた後,滴
定する。
23.5 計算
試料中の遊離酸の含有率は,これを全て硫酸(H2SO4)とみなし,次の式によって算出する。
GH
100
300
90
004
.0
2
1
1
×
×
×
×
v
s
f
v
=
ここに,
GH: 硫酸とみなして表示した遊離酸の含有率(%)
v1: 0.1 mol/L水酸化ナトリウム標準溶液の使用量(mL)
f1: 0.1 mol/L水酸化ナトリウム標準溶液のファクター
s: 21.2.4ではかりとった試料の質量(g)
v2: 試料溶液(C)からの分取量(mL)
24 pHの測定方法
24.1 方法の区分
pHの測定は,ガラス電極pH計を用いて行う。
24.2 要旨
試料を水中でかき混ぜ,その試料懸濁液のpHをガラス電極pH計を用いて測定する。
24.3 試薬
試薬は,JIS Z 8802の箇条7(pH標準液)に規定するpH標準液を用いる。
24.4 装置
装置は,次による。
a) ガラス電極pH計 JIS Z 8802の8.1(pH計の試験)b)(直線性試験)に規定するpH計のうちで,標
準液のpHの値を測定したとき,繰返性が±0.1のもの。
b) マグネチックスターラー 19.3.3 d)と同じもの。
24.5 試料はかりとり量
試料は,5.0 gをはかりとる。
24.6 操作
測定操作は,次の手順による。
a) 試料5.0 gをはかりとり,あらかじめ水100 mLを入れたビーカー(200 mL)中に投入し,直ちにマグ
ネチックスターラーを用いて4分間かき混ぜる。
b) pH計の電極を試料懸濁液に浸し,引き続きかき混ぜ,試料投入時から5分間経過したときのpHの値
を読み取る。
37
R 9101:2018
附属書A
(参考)
分析操作図
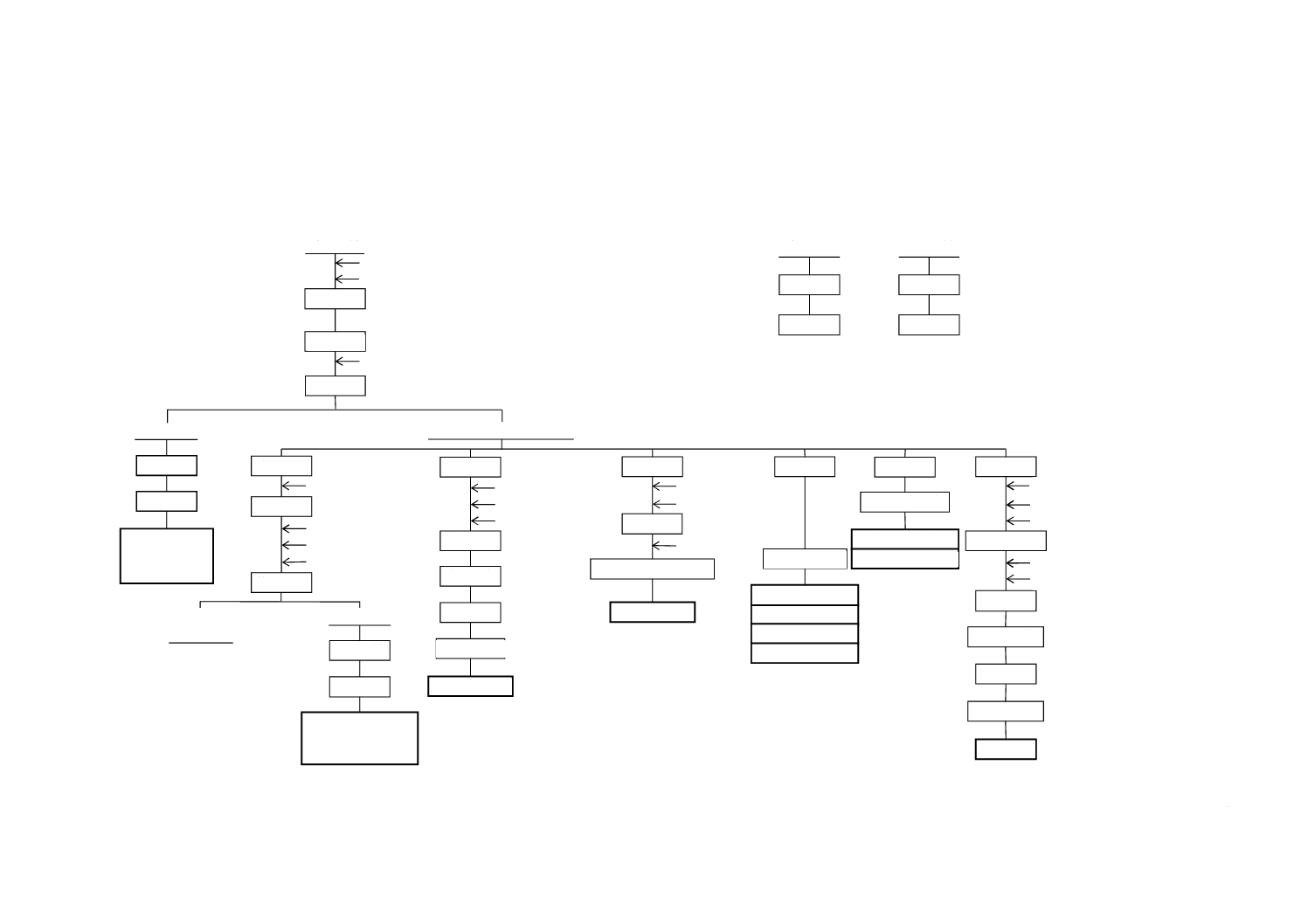
図A.1−二酸化けい素+不溶残分・酸化アルミニウム+酸化鉄(III)・酸化鉄(III)・酸化マグネシウム・酸化ナトリウム・酸化カリウム・全りん酸
試 料
水 分
恒量乾燥
試 料
化合水
加 熱
定 容
放 置
吸光度測定
酸化鉄(III)
分 取
アスコルビン酸
1,10-フェナントロリン
酢酸アンモニウム
呈 色
不溶解物
強 熱
ひょう量
二酸化けい素
+
不溶残分
分 取
原子吸光測定
酸化鉄(III)
酸化マグネシウム
酸化ナトリウム
酸化カリウム
分 取
発光強度測定
酸化鉄(III)
酸化マグネシウム
酸濃度調節
吸光度測定
全りん酸
分 取
p-ニトロフェノール
水酸化ナトリウム
硫酸
定 容
加熱・呈色
冷 却
モリブデン酸アンモニウム
アスコルビン酸
分 取
酢酸アンモニウム
塩酸
pH調節
酸化鉄(III)
サリチル酸
EDTA標準溶液滴定
ろ 過
白煙発生
分 解
試 料
塩酸
過塩素酸
塩酸・水
ろ 液(試料溶液A)
分 取
硝酸
煮 沸
ろ 過
塩化アンモニウム
アンモニア水
メチルレッド溶液
ろ 液
(不要)
沈 殿
酸化アルミニウム
+
酸化鉄(III)
強 熱
ひょう量
2
0
R
9
1
0
1
:
2
0
1
8
38
R 9101:2018
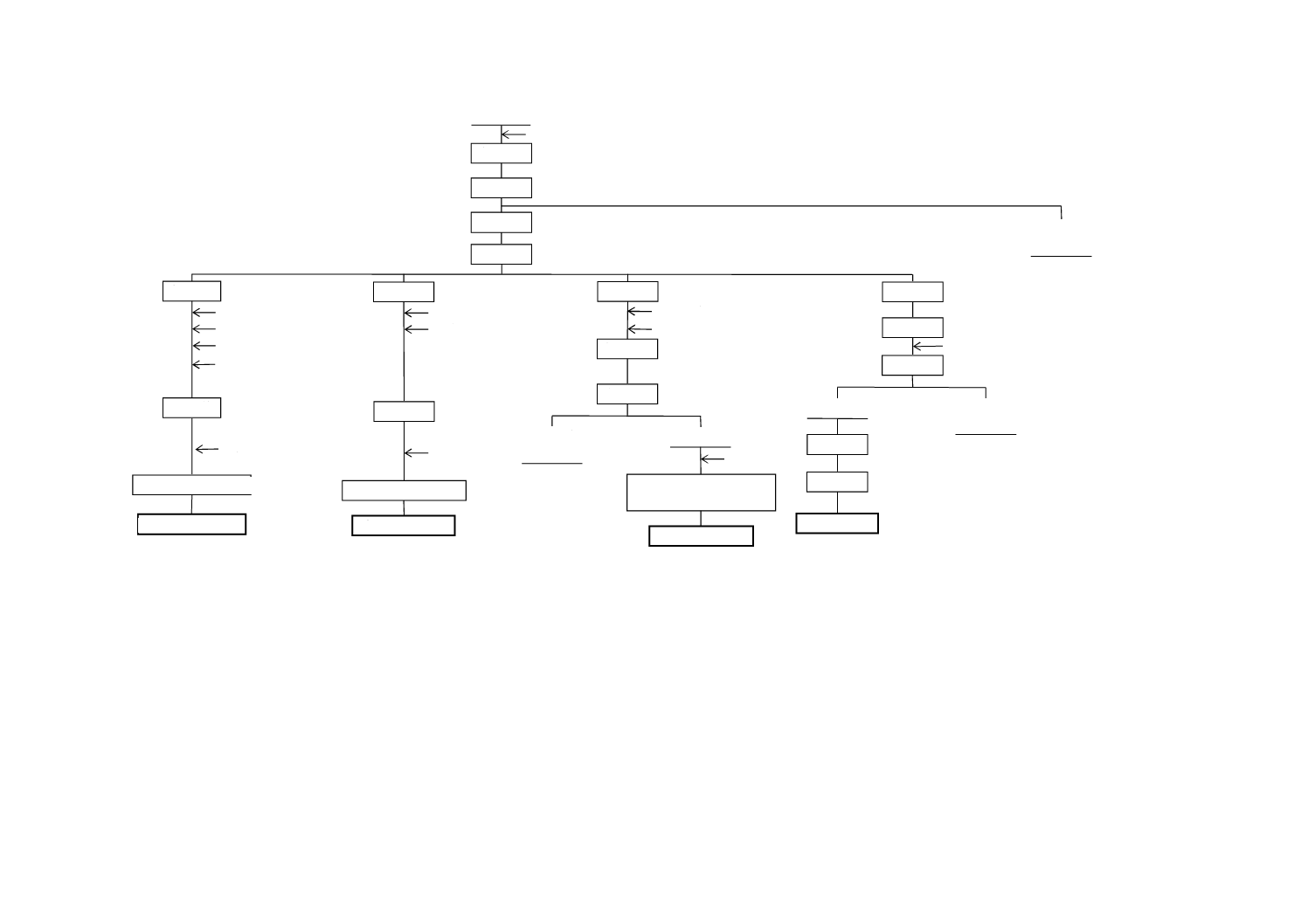
図A.2−酸化マグネシウム・酸化カルシウム・三酸化硫黄
ろ 液
ろ 過
分 解
試 料
塩酸
(試験溶液B)
定 容
分 取
トリエタノールアミン
水酸化カリウム
分 取
しゅう酸アンモニウム
酢酸アンモニウム
煮 沸
pH調節
EDTA標準溶液滴定
酸化カルシウム
HSNN希釈粉末
塩酸ヒドロキシルアミン
トリエタノールアミン
硫化ナトリウム
緩衝溶液
(pH10)
分 取
EDTA標準溶液滴定
酸化マグネシウム
pH調節
エリオクロムブラック
T溶液
ろ 過
ろ 液
(不要)
沈 殿
硫酸・水
過マンガン酸カリウム
標準溶液滴定
酸化カルシウム
分 取
ろ 過
加 熱
沈 殿
ろ 液
(不要)
ひょう量
強 熱
三酸化硫黄
(亜硫酸カルシウムを含有しない場合)
塩化バリウム
不溶解物
(不要)
2
0
R
9
1
0
1
:
2
0
1
8
39
R 9101:2018
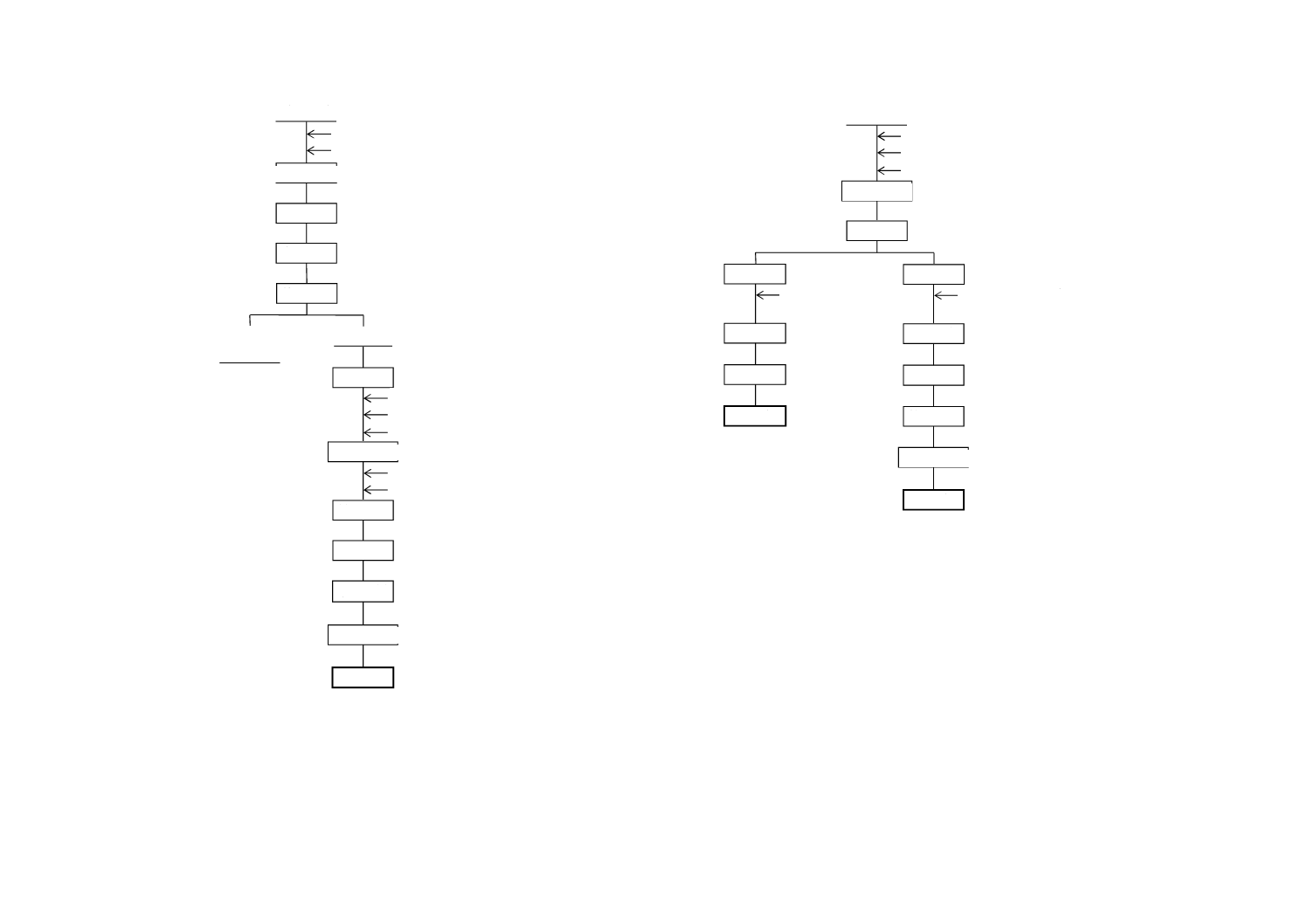
図A.3−全りん酸
図A.4−ふっ素
試 料
分 取
水蒸気蒸留
pH調節
定 容
分 取
呈 色
放 置
定 容
吸光度測定
ふっ素
ランタン−アリザリン
コンプレキソン溶液
二酸化けい素
りん酸
過塩素酸
緩衝溶液
(pH5.2)
電位測定
ふっ素
試 料
放 冷
煮沸・溶解
塩酸
硝酸
定 容
ろ 過
不溶解物
(不要)
ろ 液
分 取
酸濃度調節
呈 色
定 容
放 置
吸光度測定
全りん酸
バナジン酸アンモニウム
モリブデン酸アンモニウム
フェノールフタレイン
アンモニア水
硝酸
2
0
R
9
1
0
1
:
2
0
1
8
40
R 9101:2018
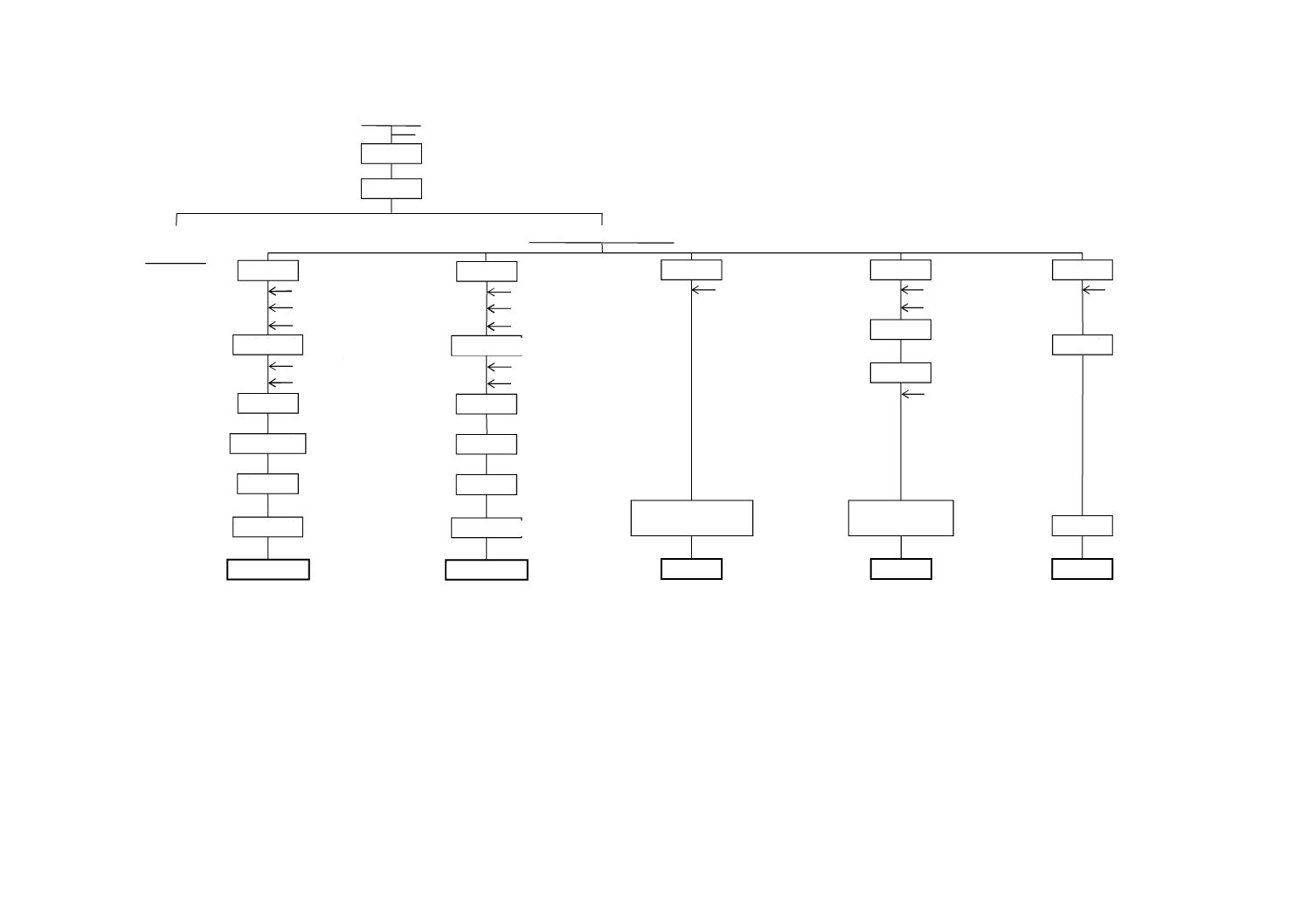
図A.5−水溶性りん酸・遊離酸・塩素
pH調節
定 容
不溶解物
(不要)
ろ 過
振とう
試 料
水
分 取
フェノールフタレイン
水酸化ナトリウム
標準溶液滴定
遊離酸
ろ 液(試料溶液C)
分 取
酸濃度調節
呈 色
定 容
放 置
吸光度測定
バナジン酸アンモニウム
モリブデン酸アンモニウム
フェノールフタレイン
アンモニア水
硝酸
分 取
酸濃度調節
定 容
加熱・呈色
冷 却
吸光度測定
水溶性りん酸
モリブデン酸アンモニウム
アスコルビン酸
p-ニトロフェノール
水酸化ナトリウム
硫酸
水溶性りん酸
分 取
炭酸カルシウム
硝酸
硝酸銀
標準溶液滴定
塩 素
分 取
緩衝溶液
(pH5)
塩 素
クロム酸カリウム
電位測定
pH調節
2
0
R
9
1
0
1
:
2
0
1
8
41
R 9101:2018
図A.6−三酸化硫黄
図A.7−二酸化硫黄
図A.8−二酸化炭素
図A.9−pH
試 料
かくはん
水
pH
試 料
吸 収
HCI ガス発生
水酸化ナトリウム
塩化バリウム
塩酸標準溶液滴定
二酸化炭素
試 料
二酸化硫黄
チオ硫酸ナトリウム
標準溶液
よう素標準溶液
塩酸
でん粉
試 料
強 熱
沈 殿
過酸化水素
緩衝溶液
(pH4)
ひょう量
ろ 過
ろ 液
(不要)
ろ 過
ろ 液
不溶解物
(不要)
塩酸
塩化バリウム
三酸化硫黄
(亜硫酸カルシウムを含有する場合)
2
0
R
9
1
0
1
:
2
0
1
8