2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
R 7223-1997
黒鉛素材の化学分析方法
Chemical analysis of graphite materials
1. 適用範囲 この規格は,黒鉛素材(以下,素材という。)の化学分析方法について規定する。
備考 この規格の引用規格を,付表1に示す。
2. 分析項目 この規格で規定する分析項目は,次のとおりとする。
(1) ほう素
(2) 灰分
3. 共通な装置
3.1
はかり 使用するはかりのひょう量は100g以上とし,感量は0.1mgとする。
3.2
化学分析用ガラス器具及び陶磁器類 この試験に用いる化学分析用ガラス器具及び陶磁器類は,特
に指定のない限り,次の規格に適合するものとする。
(1) JIS R 1301
(2) JIS R 1302
(3) JIS R 1303
(4) JIS R 1305
(5) JIS R 1306
(6) JIS R 1307
(7) JIS R 3503
(8) JIS R 3505
3.3
温度計 温度計は,0〜150℃の範囲をもつ棒状水銀温度計を用いること。
3.4
乾燥装置 乾燥装置は,温度105〜110℃に保つことのできる自動温度調節器付電気恒温器を用いる
こと。
3.5
蒸留水の容器 蒸留水の容器は,ポリエチレン製容器を用いること。
4. 数値の丸め方 測定値・計算値を丸める場合の数値の丸め方は,JIS Z 8401の規定による。
5. ほう素の分析方法
5.1
要旨 ほう素含有量0.01〜2ppm程度の素材中のほう素の分析方法について規定する。ほう素の定量
方法は,(1)比色分析方法,(2)灰化担体蒸留発光分析方法,(3)固結直接発光分析方法の3種とし,そのいず
れを使用してもよい。
5.2
分析試料の採り方 分析試料の採り方は,次のとおりとする。
2
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 素材から試料片を採取する。その採取方法は,受渡当事者間の協定による。
(2) 原則的には試料片から,次の事項に注意しながら必要量の粉末試料を削り取る。
なお,試料は清浄な容器に入れて保存する。
(a) 試料片の各所から必要量の粉末試料を得る。
(b) きりを用いて粉末試料を採取する場合には,きりはJIS H 5501に規定するG2又はこれと同等以上
の硬度をもつドリルを用いること。使用に先立ちアルコールなどで清浄にし,さらにあらかじめ素
材にきりもみして十分に清浄にする。
(c) 粉末試料の採取場所は,じんあい(塵挨)などによる汚染のおそれのない清浄な場所でなければな
らない。
5.3
分析結果の表し方 分析結果は百万分率 (ppm) をもって表し,百万分率は2けたの有効数字に丸め
る。ただし,2けたの有効数字が小数点以下3けたにわたるときは,小数点以下2けたに丸める。
5.4
比色分析方法
5.4.1
要旨 試料を水酸化カルシウムとともに灰化し,試料中のほう素を酸化カルシウムに捕そくし,こ
の灰化物を硫酸酸性でエタノール蒸留を行い,ほう素をほう酸メチルとして留出分離させて,石灰水中に
吸収させ,クルクミン試薬とともに蒸発乾固し,生成した赤色化合物をエタノールで抽出し,その赤色溶
液の吸光度を測定する。
5.4.2
灰化処理 あらかじめ空試験を実施して,内容物がほう素による汚染のないことを確認した白金容
器(1)に,粉末試料をほう素が約0.5〜2μgになるように採取(2)し,3%水酸化カルシウム(3)懸濁液(4)を試料
1gにつき1mlの割合で加え,よく混合浸潤させて乾燥し,白金棒で乾燥試料を容器からはく離(5)した後,
電気炉(6)に入れて880±20℃で清浄な酸素(7)を通じ,完全に灰化させる。
注(1) 白金るつぼ又は白金ボートを用いること。それらの容器は清浄でなければならない。
多量のほう素とともに強熱された白金容器は,ほう素による汚染があり,通常行われる濃塩
酸による煮沸洗浄や,ピロ硫酸カリウムによる溶融洗浄によっても清浄にならない場合がある。
ほう素に汚染された白金容器は,例えば,精製水酸化カルシウムとともに強熱(約1 200℃)す
ると,強熱後の酸化カルシウム中に強熱前に比較して,ほう素の増加が認められる。この強熱
によるほう素増加は,新しい酸化カルシウムを入れ替えて強熱を繰り返すことによって次第に
きん(僅)少となり,ついには認められなくなる。この状態になった白金容器を,ここでは清
浄であるという。
(2) ほう素含有量0.1ppm以下の試料については10g,0.1〜1ppmの試料については2〜5g,1ppm以
上の試料については1gを採取する。
(3) 水酸化カルシウムは,精製して使用する。
精製方法の一例 無水塩化カルシウム11gをJIS K 8891に規定するメタノール約60mlに溶
解し,塩化水素ガスを吹き込み十分に酸性とする。これをほう素蒸留器によってメタノール蒸
留を行って脱ほう素を行い,A液とする。別にJIS K 8519に規定するしゅう酸13gをJIS K 8891
に規定するメタノール約60mlに溶解し,塩化水素ガスを吹き込み十分に酸性として,A液の場
合と同様に,脱ほう素を行い,B液とする。A液とB液とを混合してアンモニア蒸気を吹き込
み,酸を中和してしゅう酸カルシウムの沈殿をつくり,さらに湯浴上に一昼夜放置して沈殿を
成長させる。この沈殿を,デカンテーションによってブフナー漏斗でろ過洗浄し,乾燥させる。
石英ガラス製さらに沈殿を移し,電気炉で強熱(約1 000℃)して酸化カルシウムとし,水を
作用させて精製水酸化カルシウムを得る。
3
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(4) ほう素による汚染のない容器,例えば,ポリエチレン容器に保存する。
(5) 乾燥後の白金容器の内容物を清浄な薬包紙上にあけ,白金容器内に付着している試料を白金棒
を用いてできるだけ完全に落とし,希塩酸で白金容器を洗い,水洗乾燥させ,先に薬包紙上に
移した乾燥試料を再び白金容器内へ移し入れる。
(6) 石英ガラス製管状炉又はマッフル炉を用いる。マッフル炉の場合,炉の内壁に石英ガラス板の
覆いをし,底部に石英ガラス板を敷く。
(7) クロム酸硫酸混液で洗い,次にアルカリ溶液で洗って中和し,乾燥剤の中を通し,最後にガラ
スウールなどの脱じん槽を通して清浄にする。
5.4.3
蒸留 蒸留装置(8)の分解フラスコ内へ灰化試料を少量のメタノールを洗浄剤として完全に洗い落
とし(9),装置に連結して,分解フラスコ上部の漏斗管から,硫酸メタノール(10)混液(11)20mlを注加して,
分解フラスコとメタノール蒸気発生フラスコの両者を湯浴で加熱し,あらかじめ水酸化カルシウム飽和液
10mlを入れた受器に留分10mlを採取する。
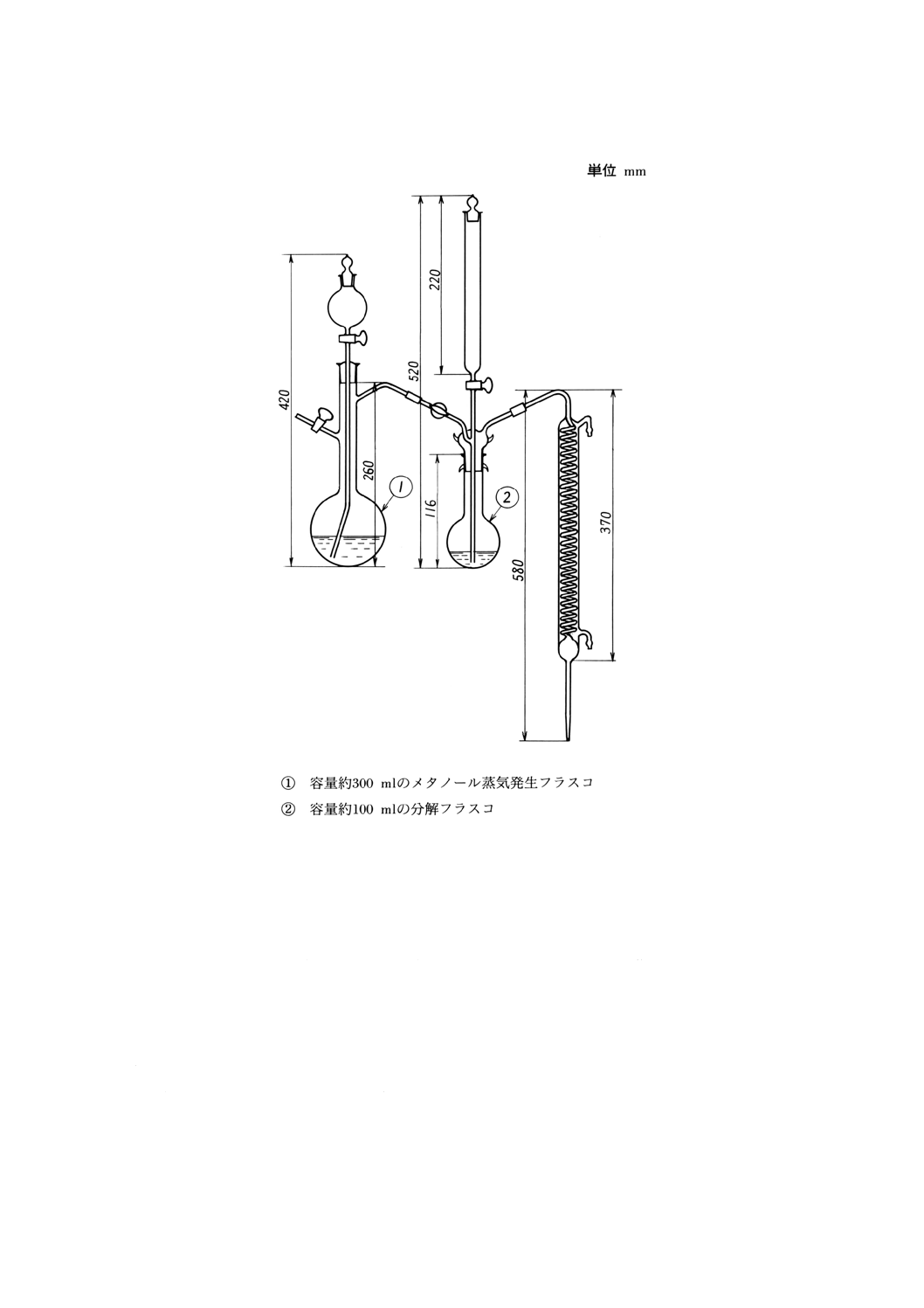
注(8) 蒸留装置は,石英ガラス又は無ほう酸ガラス製とする。その一例を図1に示す。
メタノール蒸気発生フラスコ内には,あらかじめJIS K 8882に規定するマンニトール約1g
及びJIS K 8799に規定するフェノールフタレインを指示薬として,その紅色を呈するようにJIS
K 8574に規定する水酸化カリウム約1gを加えたメタノール溶液を入れておく。
4
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図1 ほう素蒸留装置
(9) 灰化処理後の空試験の場合は,酸化カルシウムが固まっていて蒸留時に分解フラスコ内で硫酸
とメタノールの混液によってよく分散しないことがある。それを防ぐには,白金容器内に水を
数滴加えて粉末状の水酸化カルシウムにしてから,分解フラスコに移すとよい。
(10) ポリエチレン容器内でJIS K 8891に規定するメタノール2.5容にJIS K 8951に規定する硫酸1
容を冷却しながら加え,混合する。
(11) 混液は,使用する前に約60mlを分解フラスコに入れてメタノール蒸留を行い,留分約30mlを
捨てて残りの精製された約30mlを使用する。
5.4.4
蒸発発色 受器に集めた留分を蒸発容器(12)に移し,クルクミン試薬(13)4mlを加えて55〜60℃に調
節した恒温水槽に入れて直射日光の当たらない場所で蒸発乾固し,乾固後2時間,同温度に保持して発色
を完全にする。
蒸発発色の操作は,次の吸光度測定も含めて中途で中止放置してはならない。連続操作して定量を完了
させる。
注(12) 操作中にほう素が溶出するおそれのない器具(例えば,白金,石英ガラス又は磁製)を用いる
5
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
が,同一形状,同一寸法でなければならない。
(13) JIS K 8297に規定するクルクミン0.04gとJIS K 8519に規定するしゅう酸(特級)5gとをJIS K
8101に規定するエタノール(特級)100mlに溶解し,1時間以上還流煮沸した後冷却してポリ
エチレン容器に保存する。
5.4.5
吸光度測定 発色乾固物にJIS K 8101に規定するエタノール(14)25mlを加えて着色物を完全に溶解
抽出し,この抽出液をJIS P 3801に規定する6種のろ紙(直径11cm)でろ過し,ろ液を吸収槽に入れて,
波長550nm付近で対比液にエタノールを用い吸光度を測定して,別に作成した検量線(15)によってほう素
量を求める。これと空試験値(16)から,次の式によって計算する。
[]
s
c
a−
=
B
ここに,
[B]: ほう素 (ppm)
s: 試料の質量 (g)
a: ほう素の検出量 (μg)
c: 空試験値 (μg)
少なくとも2回以上定量し,平均値をもって示す。
注(14) 1級品程度でよい。
(15) 蒸留装置の反応フラスコ内へ0〜2μgの範囲で含有量既知のほう素標準液を数種作り,これらを
蒸留する。以下,本文操作によって吸光度を測定し,方眼紙上に吸光度とほう素量との関係を
求めて検量線とする。ほう素標準液は,JIS K 8863に規定するほう酸(特級)0.572gを蒸留水
に溶解して1 000mlとする。この溶液100mlを分取し,蒸留水で1 000mlに薄めて原液とする。
この原液をさらに蒸留水で10倍に薄め,1μg/mlの標準液とする。
(16) 空試験は,試料を用いずに3%水酸化カルシウム懸濁液添加から以後全操作にわたり,試料分析
の場合と同一操作でその都度行う。
5.5
灰化担体蒸留発光分光分析方法
5.5.1
要旨 試料を酸化ランタンとともに灰化して,試料中のほう素を酸化ランタンの中に補そくし,こ
の灰分物に担体(酸化インジウム)を2%になるように加えてよく混合する。この100mgをはかりとり,
電極に詰めて直流弧光法によって励起発光させ,分光写真器によってそのスペクトルを撮影し,スペクト
ル線の強度を測定して定量する。
5.5.2
灰化処理 ほう素が酸化ランタン(17)中で約0.4〜5ppmになるように粉末試料を採取(18)して,白金
容器(1)に入れ,これに酸化ランタンを加えて白金棒でよく混合し,蒸留水で浸潤させた後熱板上で乾燥す
る。乾燥した試料を白金棒で容器からはく離(5)した後,電気炉(6)に入れて880±20℃でJIS K 1101に規定
する清浄な酸素を通しながら完全に灰化させる。灰化後,電気炉から取り出し,直ちにデシケーター(乾
燥剤:五酸化りん)に入れ,冷却後乳鉢でよく混合する。
注(17) 酸化ランタンは,精製して使用する。その精製方法の一例として,次の方法がある。
再結晶による方法 市販の酸化ランタンを最小既知量のJIS K 8541に規定する硝酸に溶解し
た後,使用した硝酸の32量をJIS K 8085に規定するアンモニア水で中和する。この際に,もし
沈殿ができればこれをろ別除去する。得た溶液を10%硝酸水溶液となるように蒸留水で希釈し
た後,溶液の表面に小結晶が生じるまで蒸発濃縮し,その表面に少量の水を吹きかけ,一昼夜
放置する。沈殿した結晶を母液から分離し,蒸留水で洗浄する。得られたランタンアンモニウ
ム複塩 [La (NO3)3・2NH4NO3・4H2O] に少量の蒸留水を加えて加熱し,溶解後急冷して再結晶を
6
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
行う。再結晶を4〜5回行った後,その一部を取って酸化物とし,酸化インジウムを酸化ランタ
ンに対して2%になるように加えて発光させ,撮影する。ほう素のスペクトル線が強く検出され
れば,さらに再結晶を繰り返す。精製された複塩は白金容器に入れ,ブンゼンバーナーの直火
で加熱し,酸化窒素ガスを追い出した後電気炉に入れ,880±20℃で約3時間加熱する。加熱後
取り出してデシケーターに入れ,冷却後乳鉢でよくすりつぶし,混合する。精製した酸化ラン
タンは,ポリエチレン製のふた付瓶に入れ,デシケーター中に保存する。
蒸留による方法 市販の酸化ランタンを石英ガラス製容器又は無ほう酸ガラス製容器中で
JIS K 8180に規定する12mol/l特級塩酸に溶解し,これを蒸留濃縮して含水塩酸ランタン
(LaCl3・7H2O) とする。含水塩化ランタンを粉砕し,これに昇華精製したJIS K 8116に規定する
塩化アンモニウムを同量加えてよく混合し,石英ガラス製ボードに入れ,約200℃で加熱脱水
を行う。途中から350℃にする。以上の操作で無水塩化ランタンを得る。これをデシケーター
中に保存する。
無水塩化ランタン約16.6gをメタノール約100mlに溶解し,塩化水素ガスを吹き込み,十分
に酸性とする。これを比色分析に用いるほう素蒸留装置によって蒸留を行って脱ほう素してA
液とする。初期留出量は約30mlとして,これを破棄する。
別にしゅう酸20gをメタノール約100mlに溶解し,塩化水素ガスを吹き込み,十分に酸性と
する。これをA液と同様に蒸留を行って脱ほう素してB液とする。
A液とB液を混合してアンモニア蒸気を吹き込み,酸を中和してしゅう酸ランタンの沈殿を
成長させる。これをブフナー漏斗でろ過洗浄し,乾燥させる。次に,これを石英ガラス製さら
に入れ,電気炉で約1 000℃に加熱し,酸化ランタンとする。これをデシケーター中に保存する。
(18) 酸化ランタン1gに対してほう素含有量0.05ppm以下の試料は40g,0.05〜0.1ppmでは20g,0.1
〜1ppmでは2〜10g,1ppm以上では1gを採取する。ただし,10g以上の場合には,累積灰化が
望ましい。
5.5.3
灰化試料の処理 灰化した試料(19)をはかりとり,担体として酸化インジウム(20)を灰化試料に対し
て2%になるように加えてよく混合し,これを100mgずつに分ける。分光分析用黒鉛棒を加工して(21)試料
を詰める電極とし,これを灰化試料の100mgを充てんし,なるべく密に押し付け,中心にピンホールをあ
けて(22)おく。これを下極とする。1試料に付き,このような下極3個を準備する。
分光分析用黒鉛棒の先端を削って,これを上極(21)とする。
注(19) 灰化試料の処理は手早く行わなければならない。酸化ランタンは吸湿しやすく,吸湿した灰化
試料を電極に充てんして発光させると,5〜10秒の後に試料が電極からとび出してしまう。した
がって,酸化ランタンや灰化試料は,電気炉から取り出した後,発光までの操作は手早く行わ
なければならない。放置する場合には必ずデシケーターの中へ入れる。できれば相対湿度50%
以下の恒湿室で操作することが望ましい。
夏期では,電気炉から取り出した後3〜4時間以内,冬期では,7〜8時間以内に発光を完了
することが望ましい。恒湿室のない場合及び翌日に発光以降の操作をする場合には,担体混合
の後,発光の前に電気炉で500℃程度に1〜2時間加熱してから取り出し,手早く操作を行うと
よい。
(20) 酸化インジウムは,ほう素のスペクトル線が検出されないもの又はなるべくほう素の少ないも
のとし,これを灰化試料980mgに対して20mg加える。
(21) 分光分析用黒鉛棒は,分析操作と同じ条件でそのスペクトルを撮影して,ほう素のスペクトル
7
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
線が検出されないか,又はほう素がなるべく少ないもので,直径6mm程度のものを使用する。
電極の加工は,適当な形状のバイト又はドリルを使用する。バイト又はドリルの材質は,電極
を汚染するものであってはならない。
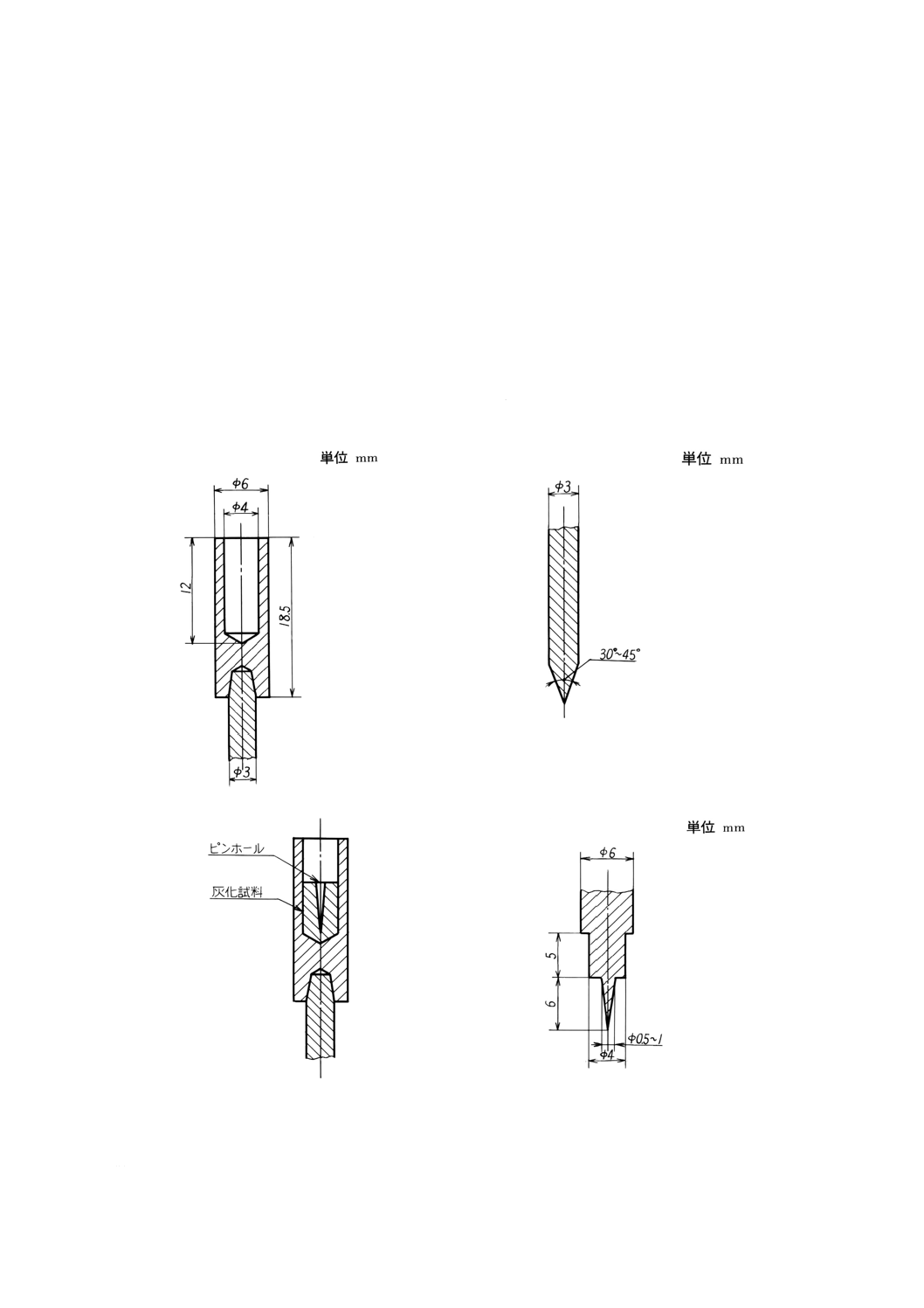
下極は,図2のように加工し,その下部に直径3mm程度の黒鉛棒を差し込む。
寸法は,図2に記載した程度が望ましく,適当に規正して一定の大きさのものを試験に用い
ること。
上極は,図3のように直径3mm程度で先端をとがらせる。
(22) 試料を図4のように充てんするため,例えば,図5のようなステンレス鋼製の充てん用ピンを
使用する。
試料充てんの際に電極の内壁に試料がなるべく付着しないようにする。
充てん用ピンの大きさは,図4に記載した程度で,電極に合わせて作る。
図2 下極
図3 上極
図4 試料の充てん
図5 充てん用ピン
5.5.4
分光分析操作 乾板(23)を分光写真器(24)に装てんし,スリット幅及びスリットの高さを調整し,電
極を電極支持器に保持する。電極投影装置によって分析間げきを正確に調整した後,発光装置(25)の諸元を
選択調整して試料及び標準試料を励起発光させ(26)撮影する。したがって,1試料につき3個のスペクトル
が得られる。

8
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
以上の操作は,表1に示す条件に近い範囲で,それぞれの分光写真器に適当な一定条件で行う。乾板校
正を行うため,鉄スペクトルを階段フィルタ(27)又は階段セクタを用いて乾板の中央近辺に撮影し,乾板の
現像・定着・水洗・乾燥を行う。
表1 分光分析操作条件の一例
分光写真器
大形(リトロ形)水晶プリズム分光写真器
光源
直流連続弧光
弧光電源電圧
100〜300V
弧光電流
12〜15A
露光
30〜40秒
予備放電
0秒
中間絞り(28)
3〜5mm
分析間げき(29)
4mm
スリット高さ
5mm以下
スリット幅
10/1 000〜20/1 000mm
波長領域(30)
220〜282nm
乾板
この波長範囲で強コントラスト,かつ,微粒子の乾板
現像(31)
強コントラスト現像液
定着
水洗
乾燥
ミクロホトメータ(32)
自動記録式ミクロホトメータ又は連続式ミクロホトメータ
分析線波長(33)
ほう素BI249.678 8nm
乾板校正
ザイデル黒化度を用いる2段セクタ法又は2段フィルタ法
バックグランド補正
ザイデル黒化度差引き
注(23) 分光分析用乾板としては,全面的に均質で波長による特性の差異が少なく,特性曲線の直
線部分が長く,コントラストが大で,相反則不規が少なく,かつ,微粒子乾板が望ましい。
(24) 分光写真器は,中形水晶プリズム分光写真器が使用可能であるが,できれば線分散が
0.5nm/mmよりよいことが望ましい。
また,大形(リトロ形)水晶プリズム分光写真器及び回析格子式のものを使用すること
ができる。
(25) 発光装置は,直流弧光発生装置又は直流弧光発生可能の発生装置を使用する。
容器はできるだけ大きいもの(200V,20A程度)が望ましい。
(26) 発光中に試料が電極から飛び出してしまったスペクトルは,実験失敗として結果から除
く,発光中,弧光の安定時間が15秒以上あったものを実験値としてとることが望ましい。
(27) 階段フィルタを使用する場合,前もって分析線波長における階段フィルタ各段の透過率を
調べ,その結果の値を用いなければならない。
(28) 電極赤熱部からの連続スペクトルを避けるために,適当な絞りを光学系中に挿入する。分
光器の外部集光系に中間結像法が用いられているときは,中間絞りを適当に規正すること
によって容易に行うことができる。
(29) 分析間げきは,発光中も4mmに保つように調節する。
(30) 波長領域は,分析線波長を乾板の中央部分にするように固定したほうがよい。
(31) 現像液は強コントラスト現像液を用い,ロッキング現像を行う。
(32) 測定に際しては光源を安定させるため,自動電圧調整器又は蓄電池を使用する。
自動電圧調整器の場合は,作動後30〜90分間,蓄電池の場合には,少なくとも5分間点
灯させてから使用する。
測定する乾板を支持台に固定し,スリットを測定するスペクトル線が平行になるように
調節する。スリット幅は,受光面のスリット上に撮影されたスペクトル線像の幅の31〜43程
度の一定値にする。
9
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
光源の光度は,測定中は完全に一定でなければならない。
自記記録式の場合には,スペクトル線像をスリットの左右いずれか一方に低速度で移動
させながら記録させる。読取式の場合には,線像をスリットの左右にわずかずつ移動しなが
ら検流計の揺れの最小値(バックグランドは最大値)を読み取る。測定室は暗室でなくても
よいが,光電池に他から光が入らないように注意する。測定する乾板及びミクロホトメータ
の光学系に,ほこりやきずがつかないように注意しなければならない。
(33) 分析線としてBI249.678 8nmを選定したのは,BI249.773 3nmの妨害線としてランタンのス
ペクトル線が存在することが確かめられたからである。ランタンのこのスペクトル線は,MIT
の波長表には記載されていない。
5.5.5
検量方法
(1) 標準試料の調製 精製した酸化ランタン8gをはかりとり,よく混合する。これを正確に1gずつに分
けて白金容器に入れ,これにほう素標準液(15)を添加して,ほう素含有量(34)が0〜5ppmぐらいの間で
5〜6種類の適当量のほう素を含有した酸化ランタンを作る。これを熱板上で乾燥した後,電気炉に入
れて880±20℃で30〜60分間加熱する。取り出したならば直ちにデシケーターに入れ,冷却後めのう
乳鉢でよく混合する。
注(34) 添加するほう素標準液の量は,正確でなければならない。したがって,繰り返し添加する回数
はなるべく少なくする。添加するための器具として,例えば,マイクロピペットを使用すると
よい。
(2) 検量線(35)の作成 調製した標準試料は,5.5.3の処理に従って操作し,撮影・現像・定着・水洗・乾
燥を行う。乾燥した乾板は,ほこりその他の汚れをよくふき取り,ミクロホトメータによって
BI249.678 8nm及びバックグランドの透過率を測定し,得た透過率の値からザイデル黒化度を求める。
測定操作については,試料の場合と同様の注意を要する。検量線は,両対数方眼紙を用いて,縦軸に
ザイデル黒化度をとり,横軸に酸化ランタンに対するほう素含有量 (ppm) をとり,これを結んで検量
線(36)を作成する。
なお,この検量線は,濃度補正(37)をしなければならない。
注(35) 検量線を作る場合には,1個の標準試料について撮影するスペクトルの数は3本以上,できれば5
〜7本程度が望ましい。
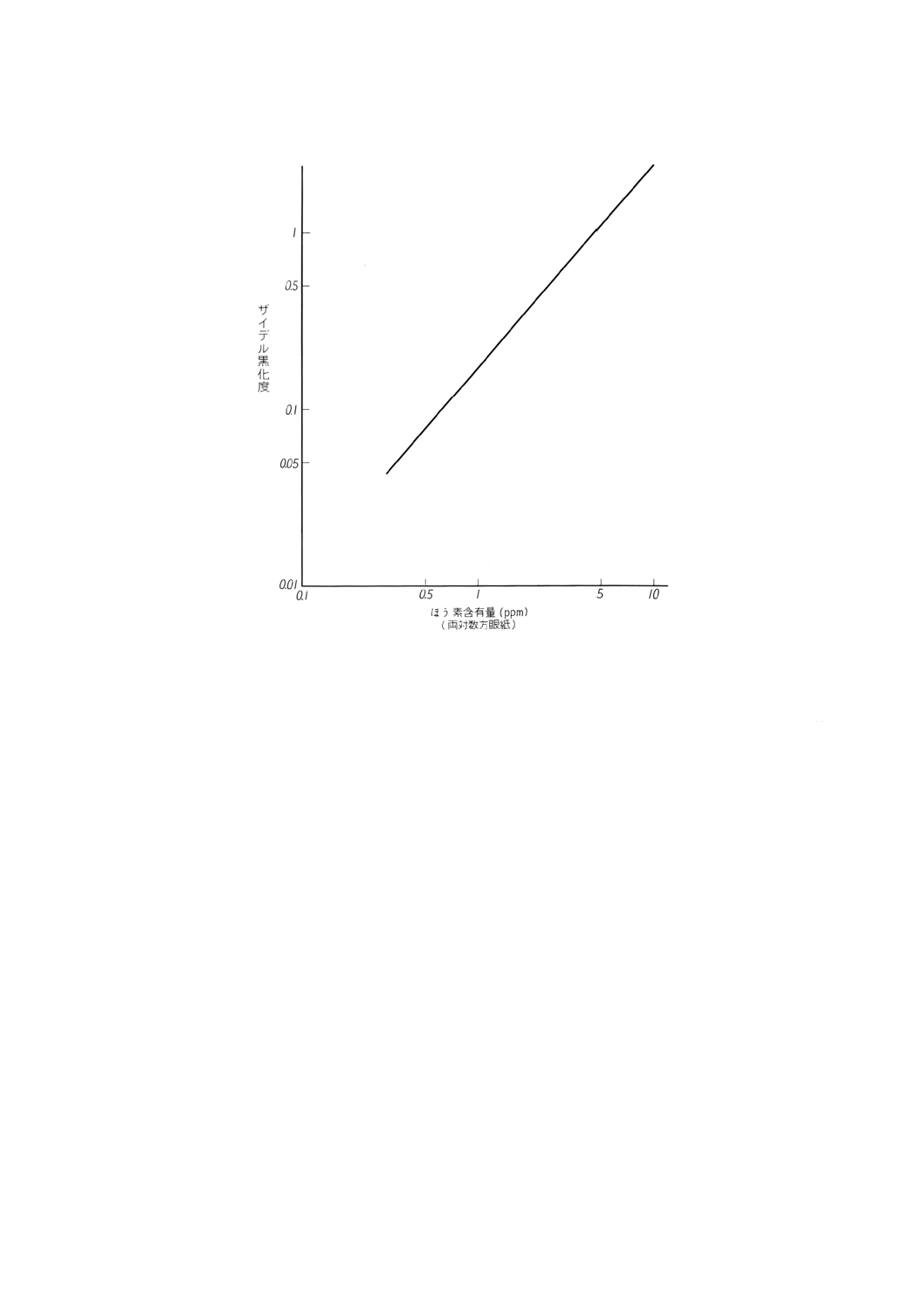
(36) 得られた検量線の一例を,図6に示す。
10
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図6 検量線の一例
(37) 精製した酸化ランタンに,まだ残留しているほう素量を補正しなければならない。
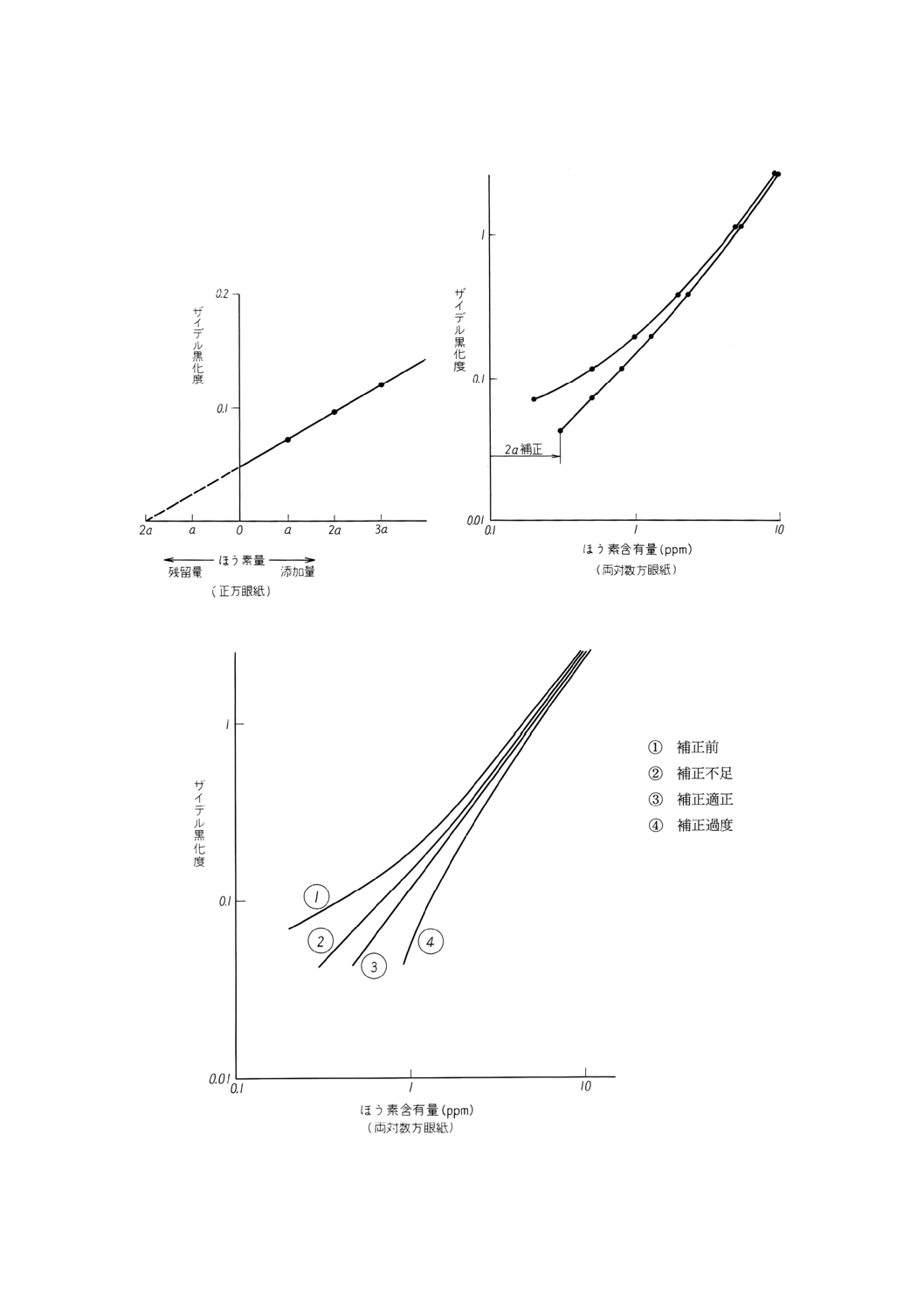
残留不純物の補正方法には,添加方法(図7)及び検量線が直線になるまで高含有量側へ移
行する方法(図8)がある。どちらを用いても分析値は,ばらつきの範囲内で一致する。添加
方法は,補正前の検量線から残留ほう素の量を想定し,これと同程度の量 (a) 及び2a,3aと整
数倍量のほう素を酸化ランタンに添加し,外挿して酸化ランタン中の残留ほう素量を求める。
補正した値が得られたならば,検量線の各点を補正値だけ高濃度側へ移して,これを検量線
として用いること。
11
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図7 残留不純物の補正方法の一例
図8 残留不純物の補正方法の一例
12
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) ほう素含有量の算出 試料及び空試験(38)の両者のほう素の分析線のザイデル黒化度からバックグラ
ンドのザイデル黒化度を差し引いた値を検量線と照合して酸化ランタンに対するほう素の含有量を求
め,次の式によって計算する。この場合,3個のスペクトルから得られた値を平均して分析結果とす
る。
[]
d
c
a−
=
B
ここに,
[B]: ほう素 (ppm)
a: 灰化試料の酸化ランタン中のほう素含有量 (ppm)
c: 空試験値(酸化ランタン中のほう素含有量) (ppm)
d: 濃縮率[黒鉛粉末試料の量 (g) /酸化ランタンの量 (g)]
注(38) 空試験は,試料を用いずに,酸化ランタンだけを全操作にわたって試料分析の場合と同一に処
理する。
空試験は,試料灰化の都度行うこととする。空試験に用いる酸化ランタンは,灰化試料中の
酸化ランタンと同時に精製したものとする。
5.6
固結直接発光分光分析方法(39)
5.6.1
要旨 試料に粘結剤としてフェノール樹脂を加え,これを加熱成形して円筒とする。この円筒の一
端に穴をあけ,これに細い黒鉛棒を差し込んだものを上下の電極として,細い黒鉛棒を電極支持具に挟み,
直流連続弧光法によって励起発光させ,分光写真器によって撮影する。得られたほう素BI249.773 3nmと
NO (γ) 249.717nmのスペクトル線の強度比を測定して試料中のほう素を定量する。
注(39) この方法の定量下限は,標準試料調製の際に[注(34)参照]用いる試料のほう素含有量によって
変わる。試料のほう素含有量が0.3ppmを超える場合には,ほう素の少ない含有量既知の黒鉛粉
末で適当に希釈して上記濃度範囲とする。
5.6.2
試料電極の成形 黒鉛粉末試料(40)の約5gをめのう乳鉢にはかりとり,粘結剤(41)として適当量の
フェノール樹脂(42)アルコール溶液(43)を粉末試料に加えて赤外線ランプの下で穏やかに加熱しつつ混練し,
乾燥(44)させる。この乾燥物の0.5gをとり,金型(45)に入れて加熱しつつ加圧成形(46)する。成形は,適当な
一定条件のもとで行う。表2に成形条件の一例を示す。
表2 成形条件の一例
金型中心温度 ℃ 150〜170
成形圧力 MPa
450〜950
2分間加熱加圧後加圧を止め,1分間金型内に放置してから取り出す。取り出した円筒(47)の一端に直径
約2.8mm,深さ3〜4mmの穴(48)をあけ,この穴に直径3mm程度の黒鉛棒(49)を差し込む。
同じ試料を成形して得た2個の試料を1組にして試料電極とする。1試料につき3組のこのような電極
を調整する。
注(40) 試料は成形を容易にするため,JIS Z 8801に規定する網ふるい75μmを通過するように微粉砕す
ることが望ましい。
なお,微粉砕中の汚染を避けるため,自動らいかい機を使用するのがよい。
(41) 粘結剤としてのフェノール樹脂の添加量は,10〜20%の範囲で一定にする。
(42) 粘結剤に用いるフェノール樹脂は,使用に先立って無ほう素であることを確かめなければなら
ない。樹脂を炭化して粉砕し,試料と同一操作を行ってほう素含有量を求める。ほう素含有量
は0.02ppm以下であることが望ましい。
(43) フェノール樹脂アルコール溶液のアルコールには,メタノールを使用する。低沸点のアルコー
13
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ルを用いるほうが操作上便利である。使用するメタノールは,石英ガラス製又は無ほう酸ガラ
ス製の蒸留器又はほう素の比色分析に使用する蒸留装置を用いて精製する。
フェノール樹脂の濃度は,試料に添加する百分率と同率のものが便利である。作ったフェノ
ール樹脂アルコール溶液は,ふた付ポリエチレン製瓶に入れ,冷暗所に保存する。
(44) 混練物の乾燥は,アルコール分が蒸発した後,全体がばさばさになるまで行う。
(45) 金型及び押棒の材質は,なるべく硬質のものが望ましく,少なくとも硬鋼以上の硬度をもつ材
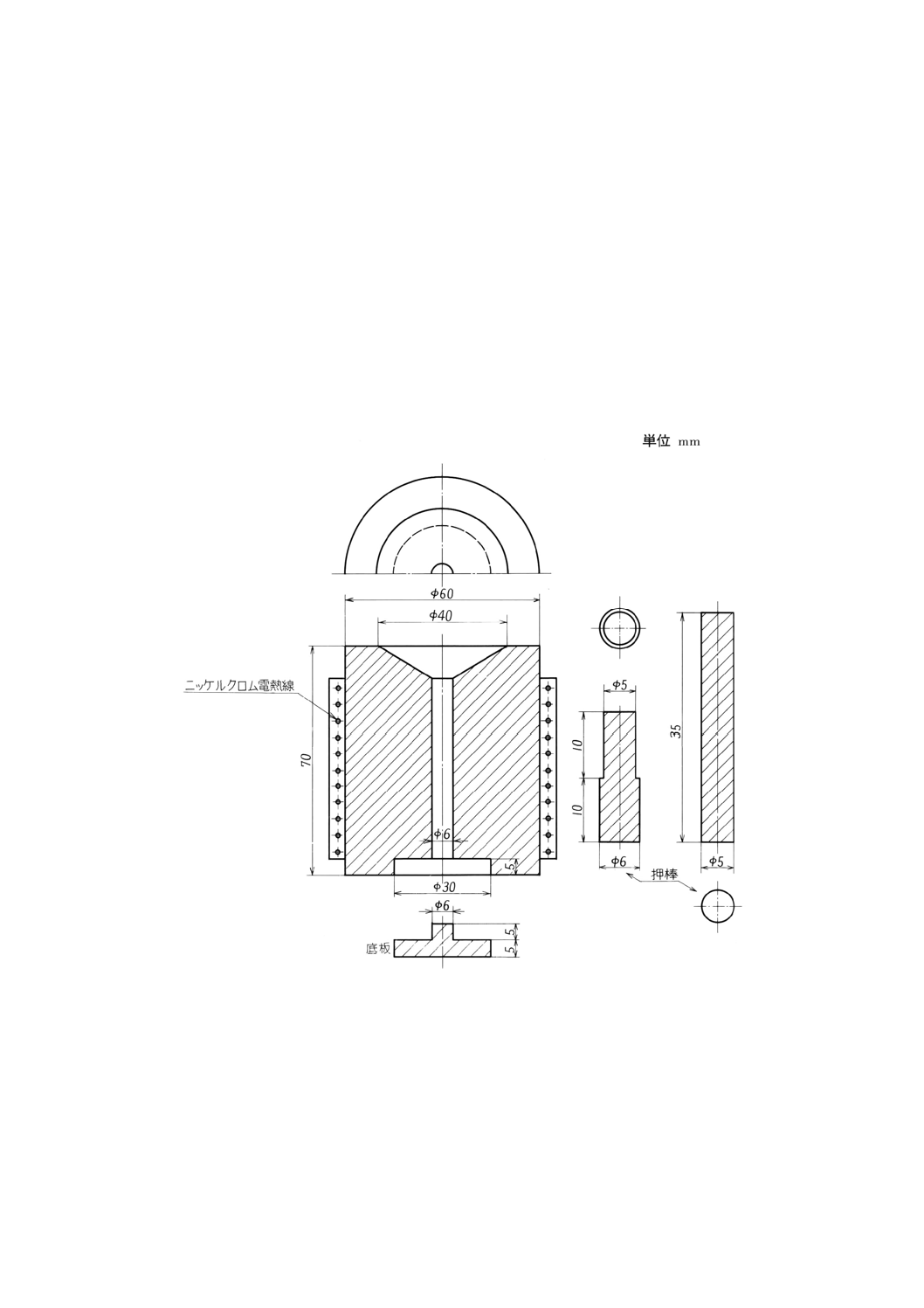
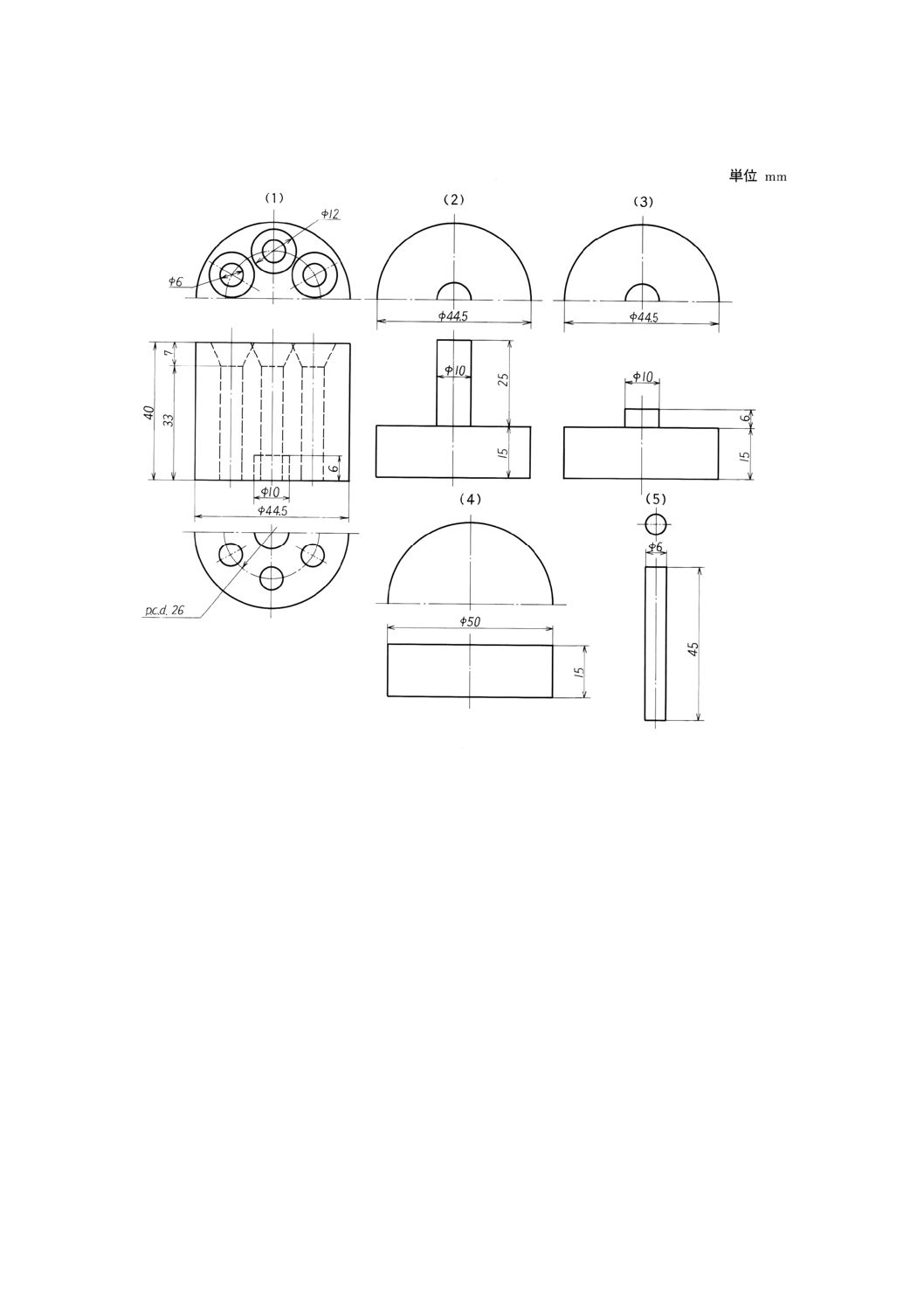
質とし,押棒はさらに焼入れするのがよい。金型の一例を,図9及び図10に示す。
寸法はおよその値であって,直径6mmの円筒を成形できるような寸法であればよい。金型の
外筒をニッケルクロム電熱線で巻き加熱する。試料と接する部分には,例えば,クロムめっき
などを施したほうがよい。
図9 金型の一例
14
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図10 6個押しの金型の例
(3)に(1)を載せて混練物を成形穴に入れ,押棒(5)を6本,各穴に入れる。
(4)を6本の押棒の上に載せて加圧する。
(2)は成形後(3)と置き換えて成形物を押し出す。
(46) 成形する際には,金型に粘結剤と混練した試料0.5gを入れ,300MPa程度の圧力で一度加圧し
た後,圧力を0にしてガス抜きを行い,再び測定圧力で加圧成形するのがよい。
(47) 成形された円筒は,およそ直径6mm,長さ10mmとなる。円筒の長さが7mm以下になると結
果がばらつく傾向があるので望ましくない。
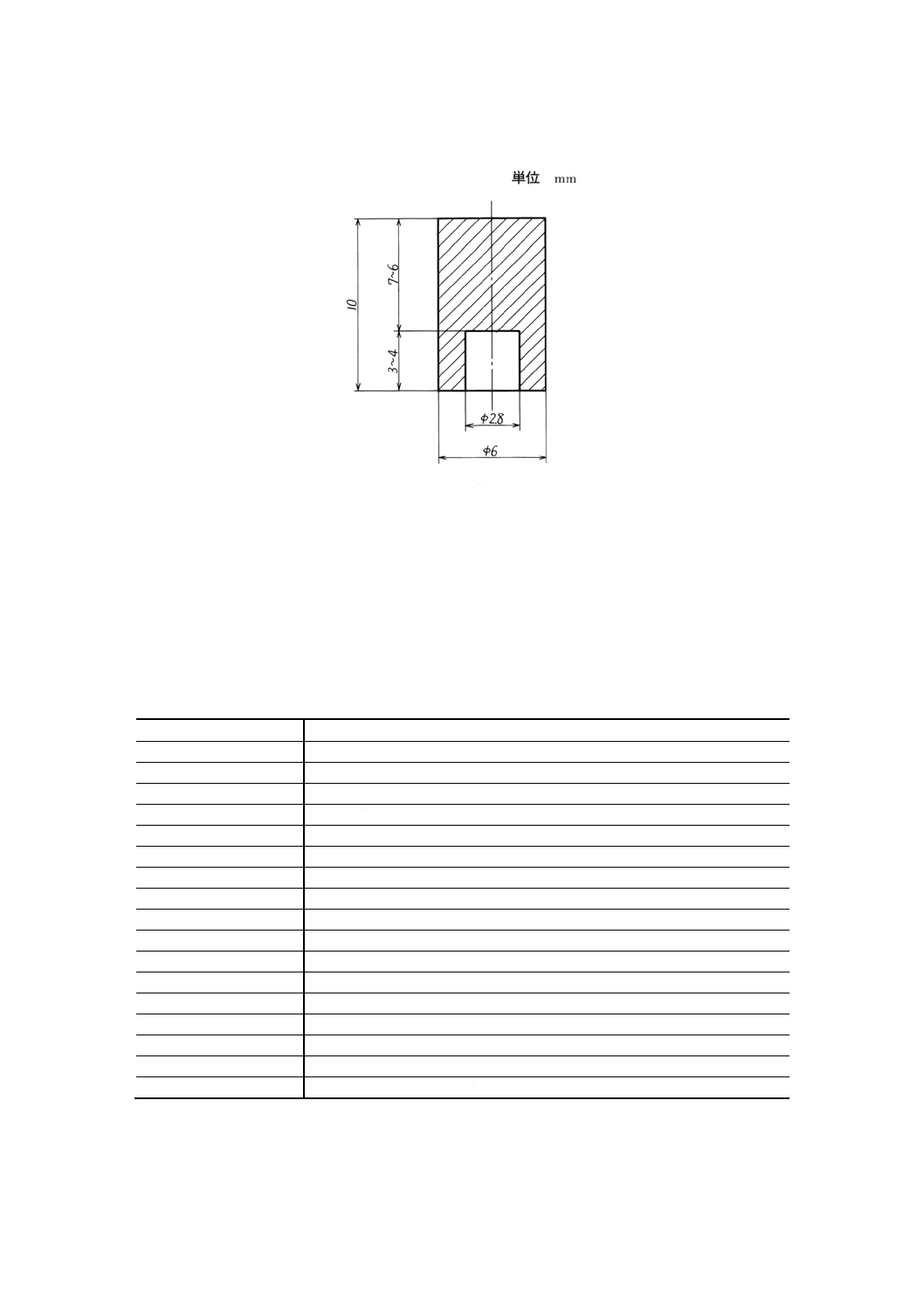
(48) 円筒の一端にあける穴の一例を,図11に示す。
穴には,緩いテーパをつけてもよい。穴をあけるための刃物の材質はなるべく硬いもので,
試料を汚染しないものを使用する。ボール盤を使用する場合には,試料が動かないように押さ
えなければならない。無ほう素黒鉛の板を適当に加工して,試料を固定する台を使用すること
が望ましい。
15
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図11 円筒の一端にあける穴の一例
(49) 試料電極の一端に差し込む直径3mmの黒鉛棒には,緩いテーパを付け,試料が崩れないように
しっかり差し込む。
5.6.3
分光分析操作 乾板(23)を分光写真器(50)に装てんし,スリット幅及びスリット高さを調節し,電極
を電極支持台に保持する。電極投影装置によって試料間げきを正確に調節した後,発光装置の諸元を選択
調節して試料を励起発光させ,撮影する。
以上の操作は,表3に示す条件に近い範囲内で,それぞれの分光写真器に適当な一定条件で,1試料に
付き3回繰り返して行う。したがって,1試料当たり3個のスペクトルを得ることとなる。撮影操作終了
後,乾板の現像・定着・水洗・乾燥を行う。
表3 分光分析操作条件の一例
分光写真器
大形(リトロ形)水晶プリズム分光写真器
光源
直流連続弧光
弧光電源電圧
100〜300V
弧光電流
7A
露光(51)
60〜90秒
予備放電(52)
60〜90秒
中間絞り(28)
3〜5mm
分析間げき(29)
4mm
スリット高さ
5mm以下
スリット幅
10/1 000〜20/1 000mm
波長領域(30)
220〜282nm
乾板
この波長範囲で強コントラスト,かつ,微粒子の乾板
現像(31)
強コントラスト現像液
定着
水洗
乾燥
ミクロホトメータ(53)
低速走査自記記録式ミクロホトメータ又はブラウン管表示式ミクロホトメータ
分析線波長
ほう素BI249.773 3nm及びNO (γ) 249.717nm
16
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(50) 分光写真器は,大形水晶プリズム分光写真器又はこれと同等以上の性能をもつ回析格子式分光写
真器を使用する。250nm付近で線分散0.2nm/mm程度をもつことが望ましい。
(51) 露光時間は,分光器の明るさによって異なるので,スリット幅を考え合わせてNO (γ) のパター
ンが検出される程度まで露光する必要がある。
(52) 予備放電は,粘結剤として混合した樹脂が炭化し,すすが出終わるまで行う必要があり,加える
樹脂の量によって一定時間を定めるべきである。
(53) この方法では,従来広く用いられている読取式ミクロホトメータを利用することはできない。
249.7nm付近のスペクトル構造を拡大表示して,図12のようなパターンを得ることができるよう
なミクロホトメータを使用しなければならない。これには,試料乾板を一定方向にゆっくり送り,
試料スペクトルの各点の黒度を自動記録するか(低速走査表示式ミクロホトメータ)又は試料乾
板とミクロホトメータの光源からの光束を相対的に繰り返し往復振動させ,それをブラウン管に
表示する(ブラウン管表示式ミクロホトメータ)方法とがある。
5.6.4
検量方法
(1) 標準試料の調製 灰化担体蒸留発光分光分析方法又は比色分析方法で分析されたほう素含有量既知の
黒鉛粉末を使用する。黒鉛粉末は,ほう素含有量の多い試料(A) (0.3〜1ppm) 及び少ない試料(B)
(0.02ppm以下)の2種類を用意する(54)。黒鉛粉末(A)・(B)のそれぞれ計算量をはかりとり,よく混
合して(55)ほう素含有量0.02〜0.3ppmぐらいの間で5〜6種類の適当量のほう素を含有した標準試料を
作る。
注(54) 標準試料に使用する黒鉛粉末は,灰化担体蒸留発光分光分析方法又は比色分析方法で3回以上分
析され,試料(A) (0.3〜1ppm) の分析値の標準偏差が±15%以内であることが望ましい。試料(B)
は0.02ppm以下で,なるべく低含有量であることが望ましい。
(55) (A)・(B)2種類の黒鉛粉末は,めのう乳鉢又は不純物混入のおそれのない器具で十分に混合しな
ければならない。
(2) 検量線の作成 調整した標準試料は,5.6.2及び5.6.3に従って順次行う。
乾燥した乾板は,ほこりその他の汚れをよくふき取り,ミクロホトメータによって測定し,分析線
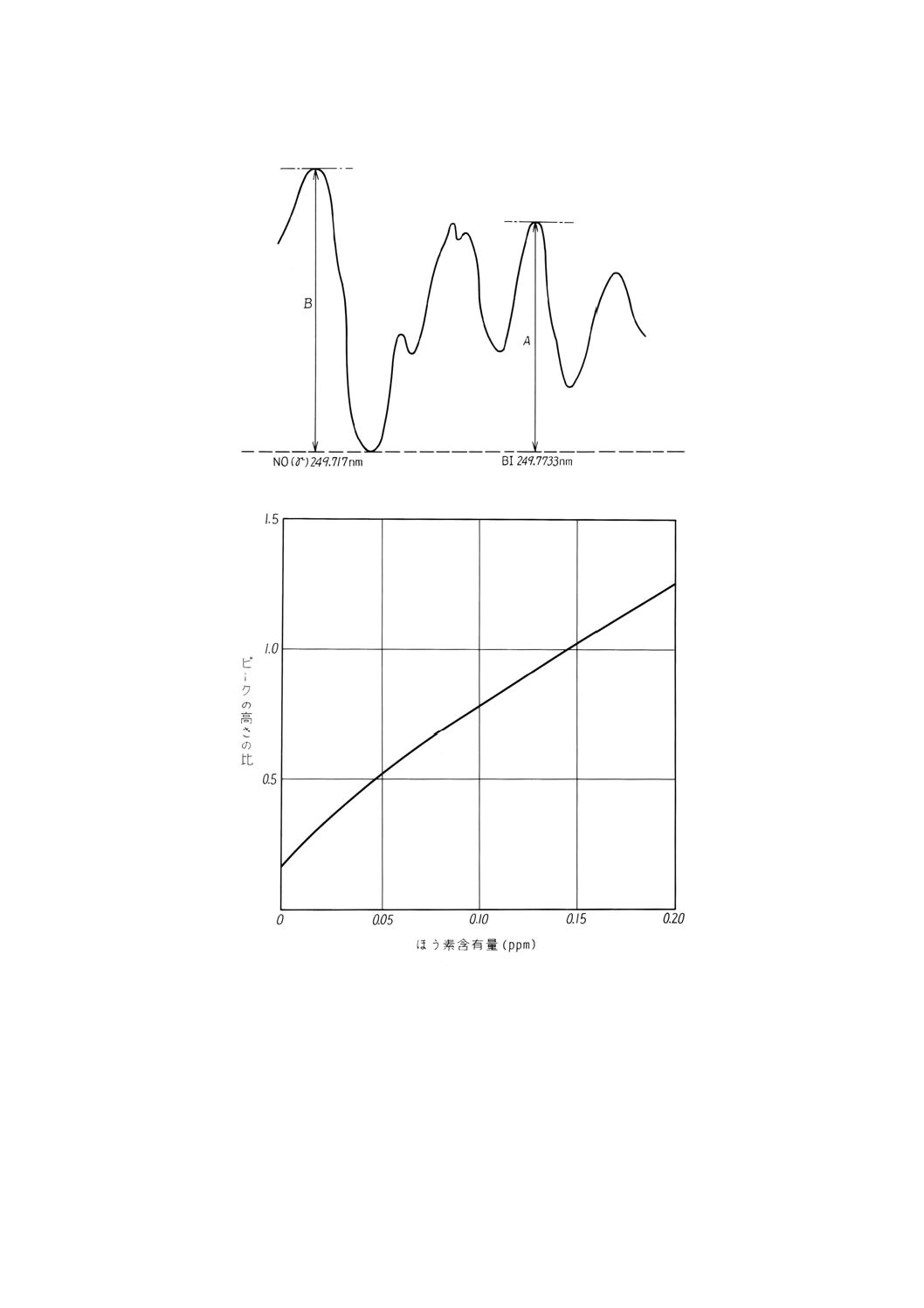
BI249.773 3nm及びNO (γ) 249.717nmを含む範囲のパターンを求める(56)。
パターンからそれぞれのピークの高さの比(56)[BI249.773 3nm/NO (γ) 249.717nm]を求めて,ほう素
含有量に対してプロットして検量線を作成する(57)。
注(56) BI249.773 3nm及びNO (γ) 249.717nm付近のパターンは,分光写真器,乾板,ミクロホトメータ
によって異なるが,パターンの一例は図12のようであって,それぞれのピークの高さA,Bか
らその比A/Bを求める。BI249.773 3nm/NO (γ) 249.717nm=A/B
(57) 検量線は,図13のようにプロットして作成する。
17
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図12 パターンの一例
図13 検量線の一例
(3) ほう素含有量の算出 試料のBI249.773 3nm及びNO (γ) 249.717nmのパターンから,それぞれのピー
クの高さの比を求めて,検量線と照合して,試料中のほう素含有量を求める(58)。
三つのスペクトルから得られた値を平均して分析結果とする。
注(58) 試料のほう素含有量が0.3ppm以上のため,ほう素の少ない粉末で希釈して分析を行った場合に
は,その希釈率で割って試料のほう素含有量を求める。
18
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6. 灰分の分析方法
6.1
要旨 素材中の灰分含有量0.01%未満の場合及び灰分含有量0.01%以上の場合の二つに付き灰分の
分析方法を規定する。
6.2
灰分含有量0.01%未満の場合
6.2.1
分析試料の採り方 5.2による。
6.2.2
定量方法 定量方法は,次のとおりとする。
(1) 操作 必要な量の試料(59)を清浄な白金容器にとり,これを電気炉(60)の中で800±20℃で灰化する。必
要があれば清浄な空気又は酸素を通じて(61)灰化を促す。灰化後,灰の入った容器を脱水したけい酸ゲ
ルを入れたデシケーター中に1時間,次にはかり箱内に10分間置いた後,質量をはかる(62)。再び電
気炉の中に移し,1時間ごとに質量をはかる。これを質量が変わらなくなるまで繰り返す。白金容器
中の灰を捨て,なお,残っているかもしれない灰を除くために,合成洗剤溶液(63)を含ませた柔らかい
布で数回軽く容器の内壁をこする。次に,水で洗浄して乾かした後,灰化温度で,20〜30分間焼き,
白金容器の質量をはかる。
(2) 計算及び表示 試料の灰分は,次の式によって計算する。
100
2
1
×
−
=
s
m
m
a
ここに,
a: 灰分 (%)
s: 試料の質量 (g)
m1: 白金容器と灰化後の試料の質量 (g)
m2: 白金容器の質量 (g)
少なくとも2回以上定量し,その平均値をもって示す。ただし,最大値と最小値との差が平均値の
10%以内でなければならない。この平均値は,2けたの有効数字に丸める。試料が粉末であるとき以
外は,得られた結果に試料の採取方法を付記する。
注(59) 灰の質量が,使うはかりの感量の少なくとも2倍以上になるように試料をとる。
(60) 石英ガラス板又は白金板などで内壁を囲み,白金容器を載せるのに適した引き出すことのでき
る石英ガラス板を敷いたものとする。酸素を導き入れる管を炉壁の適当なところに取り付ける
ときは,この管の先端は炉の内壁から少なくとも2cm程度離れなくてはならず,また,白金容
器の内容物が吹き飛ばされるほど,灰化容器に近くしてはならない。
(61) クロム酸−硫酸混液で洗浄し,次に蒸留水で2回洗浄した酸素を使う。白金容器の内容物を吹
き飛ばさないように注意しながら酸素を送らなければならない。すなわち,酸素を導き入れる
管の単位断面積 (cm2) 当たり2 000ml/min以下の速さで酸素を送るのが適当である。
(62) 感量が1mg以下のはかりを使い,注意して質量をはかる。特に白金容器とはかり箱内の温度差
による空気の対流などによる調整を避けなければならない。微量はかりを使う場合は,相対湿
度40〜60%程度,温度20〜25℃程度の部屋に設備して用いる。
(63) その洗剤に適した濃度に溶かしたものを用いること。
6.3
灰分含有量0.01%以上の場合
6.3.1
分析試料の採り方 試料を粗砕した後十分に混合し,縮分した約30gとし,めのう乳鉢で全量が
JIS Z 8801に規定する網ふるい250μmを通過するまで砕いて用いること。
6.3.2
定量方法 定量方法は,次のとおりとする。
(1) 操作 試料約1〜3gを灰化容器(64)にはかりとり電気炉(65)に入れ,空気を通しながら通電し,800℃ま
19
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
で昇温して800±20℃に保持する。黒点が認められなくなり恒量となるまで灰化した後,デシケータ
ー中で室温まで冷却し,直ちに質量をはかる。
(2) 計算及び表示 試料の灰分は,次の式によって計算し,小数点以下2けたに丸める。
100
×
′
=m
m
a
ここに,
a: 灰分 (%)
m': 灰化後の試料の質量 (g)
m: 試料の質量 (g)
供試素材の灰分は,2回の測定値の平均を小数点以下1けたに丸めて示す。
注(64) 灰化容器は,JIS R 1301又はJIS R 1306に規定されたものを使用する。
(65) 電気炉は,次の条件を備えたものを使用する。
(a) 炉内通気量が十分に大きいこと。
(b) 均熱帯 (800±20℃) が広いこと。
付表1 引用規格
JIS H 5501 超硬合金
JIS K 1101 酸素
JIS K 8085 アンモニア水(試薬)
JIS K 8101 エタノール(99.5)(試薬)
JIS K 8116 塩化アンモニウム(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8297 クルクミン(試薬)
JIS K 8519 しゅう酸二水和物(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8574 水酸化カリウム(試薬)
JIS K 8799 フェノールフタレイン(試薬)
JIS K 8863 ほう酸(試薬)
JIS K 8882 D (−) -マンニトール(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8951 硫酸(試薬)
JIS P 3801 ろ紙(化学分析用)
JIS R 1301 化学分析用磁器るつぼ
JIS R 1302 化学分析用磁器蒸発ざら
JIS R 1303 化学分析用磁器ビーカー
JIS R 1305 化学分析用磁器カッセロール
JIS R 1306 化学分析用磁器燃焼ボート
JIS R 1307 化学分析用磁器燃焼管
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS Z 8401 数値の丸め方
JIS Z 8801 試験用ふるい
20
R 7223-1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
窯業部会 炭素・黒鉛製品専門委員会
氏名
所属
(委員会長)
稲 垣 道 夫
北海道大学
宮 川 義 正
大同特殊鋼株式会社
本 庄 昭 郎
合同製鉄株式会社船橋製造所
山 地 厳
東京製鉄株式会社高松工場
篠 原 泰 明
新日本製鐵株式会社
宮 原 忍
日本鋼管株式会社
野 竹 毅
日本カーボン株式会社
富 安 稔
東海カーボン株式会社
岡 村 政 彦
昭和電工株式会社
藤 本 博 明
株式会社エスイーシー
相 沢 淳 一
日立化成工業株式会社
馬 場 清 治
炭素協会
富 田 育 男
通商産業省生活産業局
岡 林 哲 夫
工業技術院標準部
(関係者)
羽 鳥 暢 淑
炭素協会
宮 内 正 行
昭和電工株式会社
檪 原 龍 也
東海カーボン株式会社
(事務局)
池 川 澄 夫
工業技術院標準部繊維化学規格課