R 2014 : 1998
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が制定した日
本工業規格である。
JIS R 2014には,次に示す附属書がある。
附属書1(規定) アルミナ−マグネシア質耐火物中の酸化マグネシウムの陽イオン交換分離−EDTA
滴定法
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
R 2014 : 1998
アルミナ−マグネシア質耐火物
の化学分析方法
Methods for chemical analysis of
refractories containing alumina and magnesia
序文 耐火物の化学分析では,耐火物の各々の材質に合わせた分析方法を用いないと,正しい定量値を得
ることができない。このため,日本工業規格では,各材質についての分析方法を制定,整備しているので
材質に合わせて適用しなければならない。
この規格は,このような一連の耐火物の化学分析方法に関する規格整備の一環として,アルミナ−マグ
ネシア質耐火物を対象に制定されたもので,ほかの材質へは適用できない。
1. 適用範囲 この規格は,アルミナ−マグネシア質耐火物の化学分析方法について規定する。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版を適用する。
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS K 8001 試薬試験方法通則
JIS R 2001 耐火物用語
JIS R 2551 キャスタブル耐火物の試験試料採取方法
JIS Z 8401 数値の丸め方
JIS Z 8801 試験用ふるい
3. 一般事項 分析方法に共通な一般事項は,JIS K 0050,JIS K 0115,JIS K 0116及びJIS K 0121の規
定による。
4. 定義 この規格で用いる主な用語の定義は,JIS R 2001によるほか,次による。
a) アルミナ−マグネシア質耐火物 化学成分として酸化アルミニウム10〜95mass%及び酸化マグネシウ
ム3〜80mass%を含有する耐火物。
b) 乾状不定形耐火物 粒及び粉末で構成される耐火物。
c) 湿状不定形耐火物 粒及び粉末に液状物質を加えて構成される耐火物。
2
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5. 分析項目 この規格で規定する分析項目は,次による。
a) 強熱減量 (LOI)
b) 酸化けい素(IV) (SiO2)
c) 酸化アルミニウム (Al2O3)
d) 酸化鉄(III)(Fe2O3として全鉄を表す。)
e) 酸化チタン(IV) (TiO2)
f)
酸化カルシウム (CaO)
g) 酸化マグネシウム (MgO)
h) 酸化ナトリウム (Na2O)
i)
酸化カリウム (K2O)
j)
酸化りん(V) (P2O5)
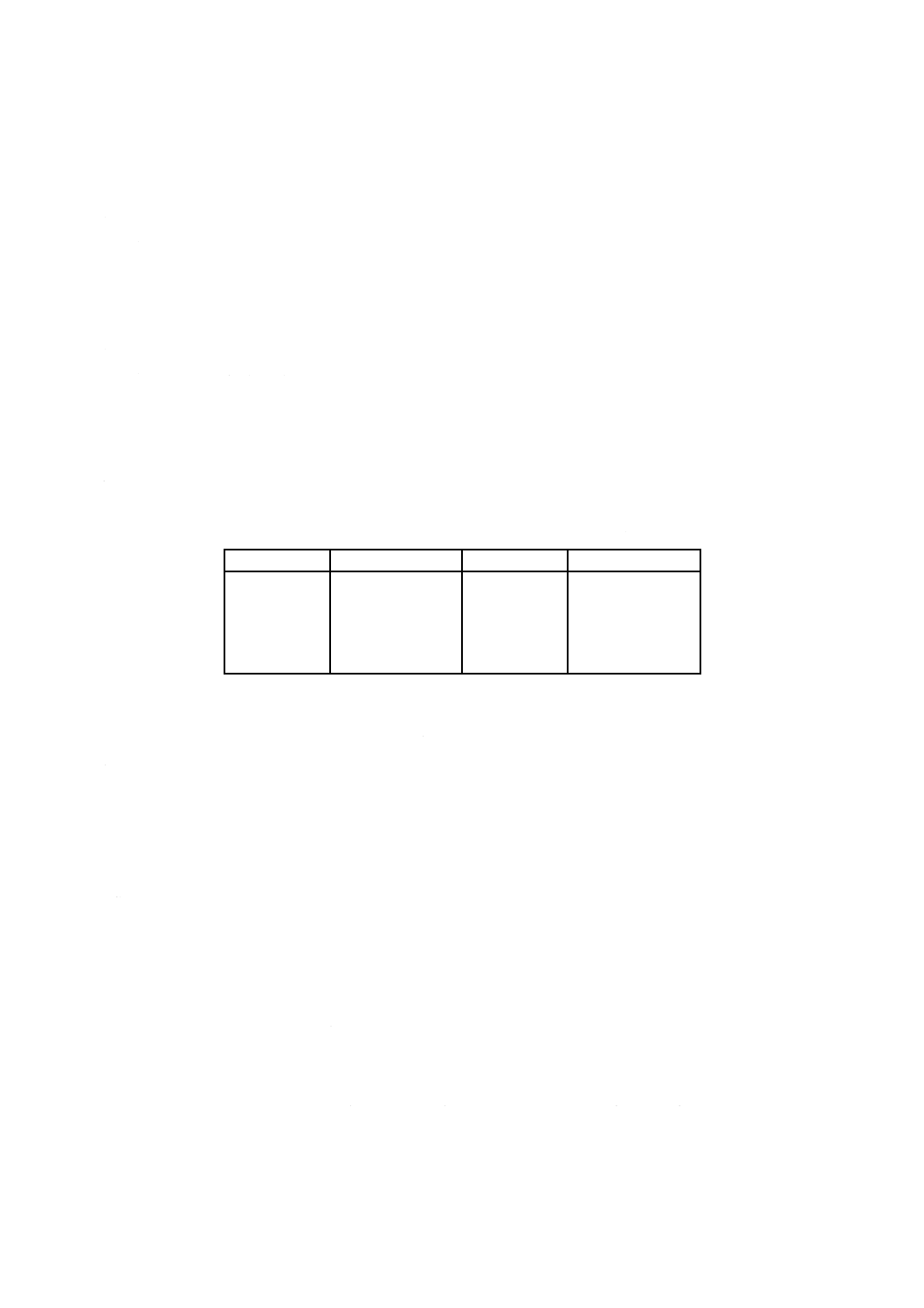
6. 定量範囲 この規格で規定する定量範囲は,表1による。
表1 定量範囲
単位mass%
成分
定量範囲
成分
定量範囲
LOI
−1〜40
CaO
0.01〜 5
SiO2
0.1〜 5
MgO
3〜80
Al2O3
10〜95
Na2O
0.01〜 3
Fe2O3
0.01〜 5
K2O
0.01〜 1
TiO2
0.01〜 3
P2O5
0.01〜 1
7. 試料
7.1
試料の採取及び調製 試料の採取及び調製は,次による。
a) 耐火れんがは,ロットから受渡当事者間の協定に基づく数量の試料をランダムに採取する。採取した
試料は,全量を粉砕してJIS Z 8801に規定する網ふるい6.7mmを通過させ,二分器を用いるか,又は
四分法によって約100gになるまで縮分する。次に,この縮分した全量が網ふるい300μmを通過する
まで粉砕する。
b) 不定形耐火物は,その性状によって乾状と湿状に区分し,次によって試料約100gを調製する。
1) 乾状不定形耐火物は,ロットからランダムに1袋又は50kgを採取し,二分器を用いるか,又は四分
法によって約100gになるまで縮分する。次に,この縮分した全量が網ふるい300μmを通過するま
で粉砕する。
2) 湿状不定形耐火物は,JIS R 2551に規定された一定量を採取し(1),湿状耐火物と反応しない耐熱性
板(例えば,四ふっ化エチレン樹脂板)上に厚みが10mm以下の薄い円盤状になるように広げ,110
±5℃の空気浴中で2時間(2)乾燥させ,全量を粉砕して網ふるい6.7mmを通過させ,二分器を用い
るか,又は四分法によって約100gになるまで縮分する。次に,この縮分した全量が網ふるい300μm
を通過するまで粉砕する。
注(1) 湿状の耐火モルタルの場合は,一容器全量を採取し,その容器内又は不定形耐火物と反応しな
い清浄な容器に移し,清浄なかくはん機などを用いて均一になるまで十分混合し,このうちの
1kgを採取する。
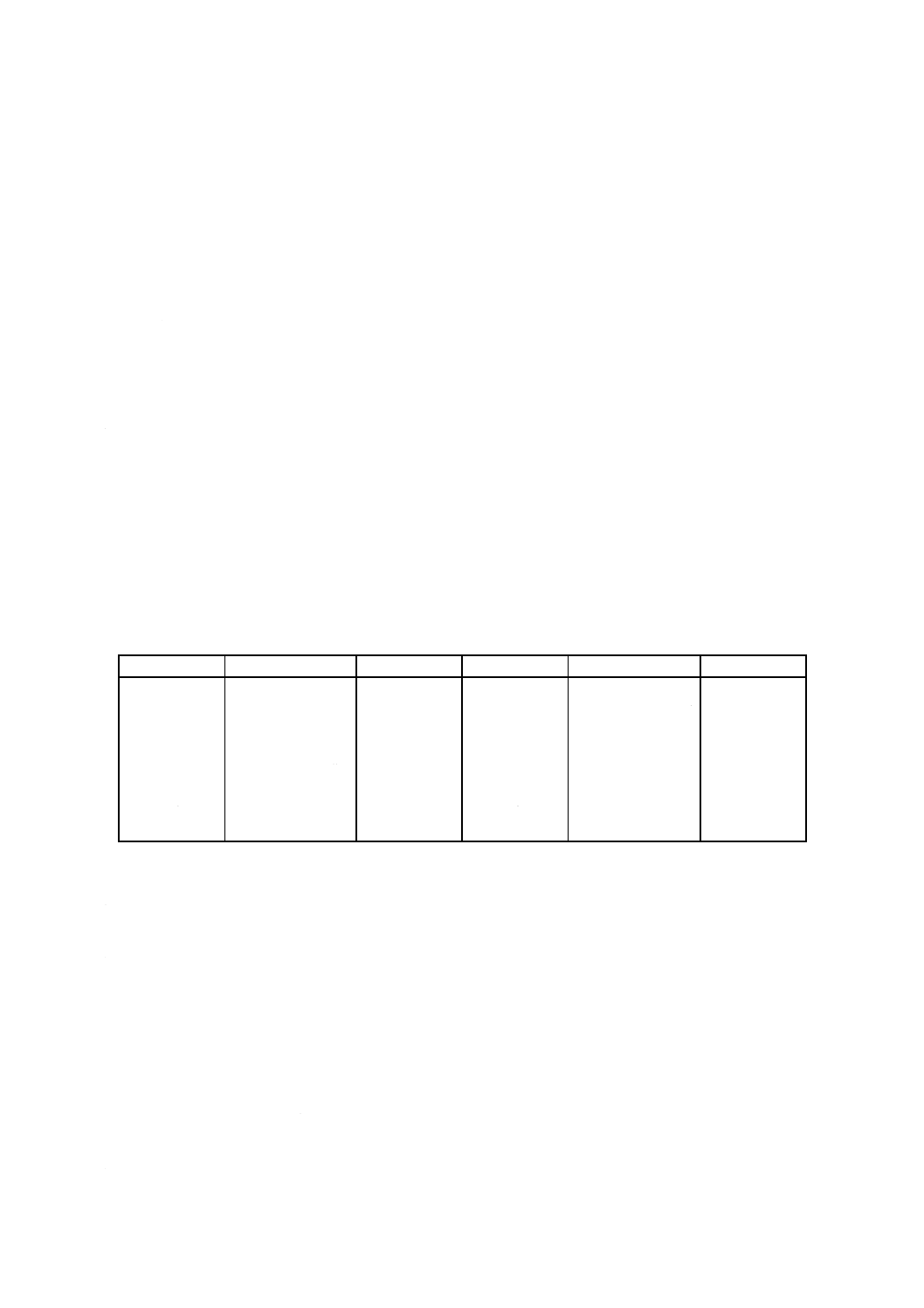
3
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 湿状の耐火モルタルの場合は,10時間以上乾燥する。
c) a)又はb)によって得られた試験室試料を,四分法によって縮分して約10gとする。これを網ふるい
106μmを通過する程度まで微粉砕し,平形はかり瓶 (50×30mm) に薄く広げ,110±5℃の空気浴中で
2時間以上乾燥した後,デシケーター中で放冷し保存する。これを分析用試料とする。
7.2
試料のはかり方 分析試料は,分析用試料から化学はかりを用いて規定された量を,0.1mgのけたま
ではかり取る。
8. 分析値のまとめ方
8.1
分析回数 分析は,日を変えて2回繰り返す。
8.2
空試験 分析に当たっては空試験を行い,測定値を補正する。
8.3
分析値の表示 分析値は乾燥ベースの質量百分率で表し,JIS Z 8401によって次のように丸める。
a) 含有率の整数部が2けたの場合,小数点以下1けた。
b) 含有率の整数部が1けた以下の場合,小数点以下2けた。
8.4
分析値の検討・採択
8.4.1
2個の分析値の差が,表2の許容差を超えないときは,その平均を報告値とする。
8.4.2
2個の分析値の差が許容差を超えるときは,更に2回の分析を繰り返し,その差が許容差を超えな
いときは,その平均を報告値とする。これも許容差を超えるときは,4個の分析値のメジアンを報告値と
する。
表2 分析値の許容差
単位mass%
成分
含有率
許容差
成分
含有率
許容差
LOI
5未満
0.10
CaO
0.08
5以上
0.20
MgO
7未満
0.15
SiO2
2未満
0.10
7以上
20未満
0.25
2以上
5未満
0.20
20以上
50未満
0.40
Al2O3
40未満
0.25
50以上
0.50
40以上
0.40
Na2O
0.08
Fe2O3
0.05
K2O
0.08
TiO2
0.10
P2O5
0.10
8.5
分析報告 分析報告には,次の事項を記録する。
a) 分析所名
b) 分析年月日
c) 試料名及び試料に関する情報
d) 分析成分名及び分析値(報告値)
9. 強熱減量の定量方法
9.1
定量方法 強熱減量の定量方法は,重量法による。
9.2
重量法
9.2.1
要旨 試料を1 050±25℃で強熱し,質量の増減を測定する。
9.2.2
試料はかり取り量 試料はかり取り量は,1.0gとする。
9.2.3
操作 定量操作は,次の手順によって行う。
a) 白金るつぼ(例えば,20番)又は磁器るつぼ(例えば,B形15ml)を1 050±25℃で一定時間(3)強熱
4
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
し,デシケーター中で放冷した後,その質量をはかる。
注(3) 白金るつぼの場合は,約15分間,磁器るつぼの場合は,約60分間強熱する。
b) 試料をるつぼの底に薄く広げるように移し入れ,その質量をはかる。
c) るつぼにふたをしないで最初は低温で加熱し,次第に温度を上げ,最後は電気炉中で1 050±25℃で約
60分間強熱する。るつぼにふたをしてデシケーター中で放冷した後,ふたを取ってその質量をはかる。
9.2.4
計算 試料中の強熱減量は,次の式によって算出する(4)。
100
0
1
2
1
×
m
m
m
m
LOI
−
−
=
ここに, LOI: 強熱減量 (mass%)
m0: 9.2.3 a)で得た質量 (g)
m1: 9.2.3 b)で得た質量 (g)
m2: 9.2.3 c)で得た質量 (g)
注(4) 質量が増加した場合は,“−”(負符号)をつけて表示する。
10. 酸化けい素(IV)の定量方法
10.1 定量方法の区分 酸化けい素(IV)の定量方法は,次のいずれかによる。
a) モリブデン青吸光光度法
b) ICP発光分光法
10.2 モリブデン青吸光光度法
10.2.1 要旨 試料を炭酸ナトリウムとほう酸で融解し,塩酸で溶解した後,酸濃度を調節し,七モリブデ
ン酸六アンモニウム,L (+) −酒石酸及びL (+) −アスコルビン酸を加えてモリブデン青を呈色させ,吸
光度を測定する。
10.2.2 試薬 試薬は,次による。プラスチック瓶に保存する。
a) ふっ化水素酸(1+9)
b) 塩酸(1+2)
c) 硫酸(1+15)
d) ほう酸
e) ほう酸溶液 (40g/l)
f)
炭酸ナトリウム(無水)(5)
注(5) 試薬によっては,カルシウムを微量含むものがある。高純度の試薬を用いる。
g) 七モリブデン酸六アンモニウム溶液 七モリブデン酸六アンモニウム四水和物20gを水200mlに溶か
し,必要ならろ過する。保存中にモリブデン酸が析出したときは,新しく調製する。
h) L (+) −酒石酸溶液 (200g/l)
i)
L (+) −アスコルビン酸溶液 (100g/l) 冷暗所に保存する。調製後2週間以上経過したものは,使用
しないほうがよい。
j)
標準酸化けい素(IV)溶液 (0.05mgSiO2/ml) 二酸化けい素(99.9mass%以上)を強熱し,デシケーター
中で放冷後,0.050 0gを白金るつぼにはかり取り,炭酸ナトリウム(無水)1gと混合した後,加熱融
解する。放冷後,白金るつぼごと水100mlの入ったプラスチックビーカー (200ml) に移し,加熱する
ことなく融成物を溶解して1 000mlの全量フラスコに移し入れ,水で標線まで薄める。使用の都度調
製する。

5
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.2.3 試料はかり取り量 試料のはかり取り量は,0.50gとする。
10.2.4 操作 定量操作は,次の手順によって行う。
a) 試料を白金皿(例えば,75番)にはかり取り,炭酸ナトリウム(無水)3.0g及びほう酸2.0gを加えて,
初めは低温で加熱し(6),次第に温度を上げ,最後は電気炉中で1 100±25℃で加熱し,未分解物が認め
られなくなるまで強熱して融解する(7)。時計皿でふたをして放冷後,水15ml,塩酸(1+2)30ml及び硫
酸(1+15)10mlを加え,ときどきかき混ぜながら水浴上で加熱溶解する。放冷後,少量の水で時計皿を
水洗して取り除き,得られた溶液を250mlの全量フラスコに移し入れ,水で標線まで薄める。この溶
液を試料溶液(A)とし,酸化けい素(IV),酸化アルミニウム,酸化鉄(III),酸化チタン(IV),酸化カルシ
ウム,酸化マグネシウム及び酸化りん(V)の定量に用いる。
注(6) 急激に加熱すると,ほう酸の脱水のために試料が飛散するおそれがある。
(7) 融解時間が長すぎると融成物が塩酸に溶けにくくなる。
b) この試料溶液(A)から正しく定量(8)を2個のプラスチックビーカー (100ml) に分取し,10.2.5の空試験
液(A)の一定量(8)を加える。ふっ化水素酸(1+9)2mlを加え,プラスチック棒でかき混ぜて約10分間放
置した後,ほう酸溶液50mlを加え,水で約90mlに薄めて液温を25℃付近にする。七モリブデン酸六
アンモニウム溶液5mlを加えてかき混ぜ,10分間放置する。L (+) −酒石酸溶液20mlを加えかき混
ぜ,1分間後にL (+) −アスコルビン酸溶液10mlを加え,200mlの全量フラスコに移し入れ,水で標
線まで薄めて60分間放置する。この溶液の一部を吸光光度計の吸収セル (10mm) に取り,波長650nm
付近で水を対照液にして吸光度を測定し,2個の測定値(9)を平均する。
注(8) 試料溶液(A)の分取量及び空試験液(A)の添加量は,試料中の酸化けい素(IV)含有率に応じて表3
による。
表3 試料溶液(A)の分取量及び空試験液(A)の添加量
酸化けい素(IV)含有率
mass%
試料溶液(A)の分取量
ml
空試験液(A)の添加量
ml
2未満
20
0
2以上 5未満
10
10
(9) 吸光度の差が,0.005を超えるときは,10.2.4 b)以降の操作を再び行う。
吸光光度計は,吸光度1.00付近の溶液を繰り返し測定したとき,吸光度の差が0.002以内で
あるものが望ましい。
10.2.5 空試験 試料を用いないで10.2.4の操作を行う。ただし,融解操作は,省略する。ここで得た試料
溶液(A)に対応する溶液を空試験液(A)とする。
10.2.6 検量線の作成 標準酸化けい素(IV)溶液0〜20.0ml[酸化けい素(IV)として0〜1mg]を数個のプラ
スチックビーカー (100ml) に段階的に取り,それぞれに10.2.5の空試験液(A)20.0mlを加え,10.2.4 b)のふ
っ化水素酸(1+9)添加以降の操作を行い,吸光度と酸化けい素(IV)量との関係線を作成し,原点を通るよう
に平行移動して検量線とする。
10.2.7 計算 試料中の酸化けい素(IV)含有率は,10.2.4 b)及び10.2.5で得た吸光度と10.2.6で作成した検
量線とから酸化けい素(IV)量を求め,次の式によって算出する。
100
250
2
1
2
×
×V
m
A
A
SiO
−
=
ここに, SiO2: 酸化けい素(IV)の含有率 (mass%)
A1: 分取した試料溶液(A)中の酸化けい素(IV)量 (g)
A2: 分取した空試験液(A)中の酸化けい素(IV)量 (g)
6
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V: 試料溶液(A)の分取量 (ml)
m: 試料はかり取り量 (g)
10.3 ICP発光分光法
10.3.1 要旨 試料を炭酸ナトリウムとほう酸で融解し,塩酸で溶解した後,定容とする。この溶液の一部
を取り,ICP発光分光装置を用いてけい素の分析線の発光強度を測定する。
10.3.2 試薬は,次による。
a) 塩酸(1+2)
b) 硫酸(1+15)
c) ほう酸
d) 炭酸ナトリウム(無水)(5)
e) 酸化アルミニウム溶液 (5mgAl2O3/ml) アルミニウム(99.5mass%以上,Si0.01mass%以下)2.6gを白
金皿(例えば,100番)にはかり取り,白金皿を時計皿で覆い,塩酸(1+1)100mlを加えて水浴上で加
熱融解し,冷却後1 000mlの全量フラスコに移し入れ,水で標線まで薄める。
f)
酸化マグネシウム溶液 (5mgMgO/ml) マグネシウム(99.5mass%以上,Si0.01mass%以下)3.0gをは
かり取り,ビーカー (200ml) に移し入れ,ビーカーを時計皿で覆い,塩酸(1+1)50mlを徐々に加えて
溶解し,冷却後1 000mlの全量フラスコに移し入れ,水で標線まで薄める。
g) 混合添加液 250mlの全量フラスコに酸化アルミニウム溶液及び酸化マグネシウム溶液の一定量(10)を
取り,水で標線まで薄める。この溶液は,使用の都度調製する。
注(10) 酸化アルミニウム溶液及び酸化マグネシウム溶液の分取量は,試料中の酸化アルミニウム及び
酸化マグネシウム含有率によって決定する。例えば,酸化アルミニウム含有率52mass%及び酸
化マグネシウム含有率38mass%の場合,それぞれ50ml及び40mlとする。
なお,分取量は,含有率に対し±5ml程度の精度でよい。
h) 標準酸化けい素(IV)溶液 10.2.2 j)と同じ。
10.3.3 試料はかり取り量 試料のはかり取り量は,0.50gとする。
10.3.4 操作 定量操作は,次の手順によって行う。
a) 10.2.4 a)に準じて操作する。得られた溶液を試料溶液(A')とし,酸化けい素(IV),酸化アルミニウム,
酸化鉄(III),酸化チタン(IV),酸化カルシウム,酸化マグネシウム及び酸化りん(V)の定量に用いる。
b) この試料溶液(A')から正しく10mlを100mlの全量フラスコに分取し,水で標線まで薄める。
c) この溶液の一部をICP発光分析装置のアルゴンプラズマ中に噴霧し,例えば,波長251.61nmにおけ
る発光強度を測定する。
10.3.5 空試験 試料を用いないで10.3.4の操作を行う。ただし,融解操作は,省略する。ここで得た試料
溶液(A')に対応する溶液を空試験液(A')とする。
10.3.6 検量線の作成 標準酸化けい素(IV)溶液0〜20.0ml[酸化けい素(IV)として0〜1mg]を数個の100ml
の全量フラスコに段階的に取り,それぞれに混合添加液10.0ml及び空試験液(A')10.0mlを加え,標線まで
水で薄める。以下,これら溶液を用いて10.3.4 c)の操作を行い(11),発光強度と酸化けい素(IV)量との関係
線を作成し,原点を通るように平行移動して検量線とする。
注(11) 検量線用溶液系列の測定は,試料溶液及び空試験液の測定との一連の操作として行い,検量線
は,測定ごとに新しいものを作成する。
10.3.7 計算 試料中の酸化けい素(IV)含有率は,10.3.4 c)及び10.3.5で得た発光強度と10.3.6で作成した
検量線とから酸化けい素(IV)量を求め,次の式によって算出する。

7
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
10
250
2
1
2
×
×
−
m
A
A
SiO=
ここに, SiO2: 酸化けい素(IV)の含有率 (mass%)
A1: 10.3.4の希釈試料溶液中の酸化けい素(IV)量 (g)
A2: 10.3.5の希釈空試験液中の酸化けい素(IV)量 (g)
m: 10.3.4 a)の試料はかり取り量 (g)
11. 酸化アルミニウムの定量方法
11.1 定量方法の区分 酸化アルミニウムの定量方法は,CyDTA−亜鉛逆滴定法による。
11.2 CyDTA−亜鉛逆滴定法
11.2.1 要旨 10.2.4又は10.3.4の試料溶液(A又はA')を分取し,過剰のCyDTAを加え,アンモニア水
でpHを調節してアルミニウム−CyDTAキレートを生成させ,ヘキサメチレンテトラミンを加えてpHを
再調整した後,キシレノールオレンジを指示薬として過剰のCyDTAを亜鉛溶液で逆滴定する。
11.2.2 試薬 試薬は,次による。
a) 塩酸(1+1)
b) アンモニア水(1+1,1+9)
c) ヘキサメチレンテトラミン(ヘキサミン)
d) 0.02mol/l CyDTA溶液 シクロヘキサンジアミン四酢酸−水和物7.30gに水酸化ナトリウム溶液
(100g/l) 16ml及び水約150mlを加え,加熱して溶解する。冷却後,水で1 000mlに薄める。
e) 0.02mol/l亜鉛溶液 調製方法及びファクターの計算方法は,JIS K 8001の4.5(滴定用溶液) (1.3) に
準じる。ただし,亜鉛0.66g及び硝酸(1+1)10mlを用い,ファクターの計算の分母は,0.653 9とする。
f)
キシレノールオレンジ溶液 調製方法及び保存方法は,JIS K 8001の4.4(表8)による。
11.2.3 操作 操作は,次の手順によって行う。
a) 10.2.4 a)又は10.3.4 a)で得た試料溶液(A又はA')から正しく一定量(12)をビーカー (300ml) に分取し,
塩酸(1+1)2mlを加えた後,0.02mol/l CyDTA溶液の正しく一定量(12)を加え,水で約100mlに薄める。
注(12) 試料溶液(A又はA')の分取量及びCyDTAの添加量は,試料中の酸化アルミニウム,酸化鉄(III)
及び酸化チタン(IV)の含有率に応じて表4による。
表4 試料溶液(A又はA')の分取量及び0.02mol/l CyDTA溶液の添加量
酸化アルミニウム,酸化鉄(III)及び
酸化チタン(IV)含有率の合量
mass%
試料溶液(A又はA')の分取量
ml
0.02mol/l CyDTA溶液の添加量
ml
20未満
50
30
20以上 30未満
50
40
30以上 50未満
40
50
50以上 75未満
30
50
75以上
25
50
b) pH計を用いてpHが2.8〜3.3になるまでアンモニア水(1+1),次いでアンモニア水(1+9)を加える。
もし,アンモニア水を加え過ぎたときは,塩酸(1+1)を加えてpH3以下に戻してから同様の調節を行
う。pH計を用いてpHが5.5〜5.8になるまでヘキサミンを加え,キシレノールオレンジ溶液4, 5滴を
指示薬として加えて0.02mol/l亜鉛溶液で滴定する。終点付近になったら,よくかき混ぜながらゆっく
りと滴定し,黄色がわずかに赤みを帯びた点を終点とする。
8
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.2.4 空試験 10.2.5又は10.3.5で得た空試験液(A又はA')を用いて11.2.3の操作を行う。
11.2.5 計算 試料中の酸化アルミニウム含有率は,次の式によって算出する。
(
)
(
)
2
3
2
3
1
2
3
2
638
.0
100
250
0010196
.0
TiO
O
Fe
V
m
F
V
V
O
Al
+
−
−
=
×
×
×
×
×
ここに, Al2O3: 酸化アルミニウムの含有率 (mass%)
V1: 11.2.3 b)の0.02mol/l亜鉛溶液使用量 (ml)
V2: 11.2.4の0.02mol/l亜鉛溶液使用量 (ml)
V3: 11.2.3 a)の試料溶液(A又はA')の分取量 (ml)
F: 0.02mol/l亜鉛溶液のファクター
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
Fe2O3: 12.で求めた酸化鉄(III)の含有率 (mass%)
TiO2: 13.で求めた酸化チタン(IV)の含有率 (mass%)
12. 酸化鉄(III)の定量方法
12.1 定量方法の区分 酸化鉄(III)の定量方法は,次のいずれかによる。
a) 1, 10−フェナントロリニウム吸光光度法
b) ICP発光分光法
12.2 1, 10−フェナントロリニウム吸光光度法
12.2.1 要旨 10.2.4又は10.3.4の試料溶液(A又はA')を分取し,L (+) −アスコルビン酸で鉄を還元し,
塩化1, 10−フェナントロリニウムを加え,酢酸アンモニウムでpHを調節して鉄を呈色させ,吸光度を測
定する。
12.2.2 試薬 試薬は,次による。
a) L (+) −酒石酸溶液 (100g/l) 10.2.2 h)と同じ。
b) 酢酸アンモニウム溶液 (200g/l)
c) L (+) −アスコルビン酸溶液 (100g/l) 10.2.2 i)と同じ。
d) 塩化1, 10−フェナントロリニウム溶液 塩化1, 10−フェナントロリニウム−水和物1gを水に溶かし
て1 000mlに薄め,冷暗所に保存する。ただし,保存中に着色したときは,新しく調製する。
e) 標準酸化鉄(III)溶液 (1.0mgFe2O3/ml) 鉄(99.9mass%以上)(13)0.699 4gをはかり取り,ビーカー
(200ml) に移し,ビーカーを時計皿で覆い,塩酸(1+1)30mlを加えて水浴上で加熱溶解し,冷却後1
000mlの全量フラスコに移し入れ,水で標線まで薄める。
注(13) 表面が酸化している場合には,表面酸化層を塩酸(1+3)で溶解し,水,エタノール,ジエチルエ
ーテルで順次洗浄した後,デシケーターに入れ乾燥させて用いる。
f)
希釈標準酸化鉄(III)溶液 (0.05mgFe2O3/ml) e)の標準酸化鉄(III)溶液を水で正しく20倍に薄める。使
用の都度調製する。
12.2.3 操作 定量操作は,次の手順によって行う。
a) 10.2.4 a)又は10.3.4 a)で得た試料溶液(A又はA')から正しく一定量(14)を100mlの全量フラスコに分
取する。
注(14) 試料溶液(A又はA')の分取量は,試料中の酸化鉄 (III) 含有率に応じて表5による。

9
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表5 試料溶液(A又はA')の分取量
酸化鉄 (III) 含有率
mass%
分取量
ml
1未満
25
1以上 3未満
10
3以上
5
b) 水で約60mlに薄め,L (+) −酒石酸溶液5ml及びL (+) −アスコルビン酸溶液2mlを加えて振り混
ぜ,塩化1, 10−フェナントロリニウム溶液10ml及び酢酸アンモニウム溶液10mlを加え,水で標線ま
で薄め,30分間放置する。この溶液の一部を吸光光度計の吸収セル (10mm) に取り,波長510nm付
近で水を対照液にして吸光度を測定する。
12.2.4 空試験 10.2.5又は10.3.5で得た空試験液(A又はA')を用いて12.2.3の操作を行う。ただし,空
試験液の分取量は,試料溶液の場合と同量とする。
12.2.5 検量線の作成 希釈標準酸化鉄(III)溶液0〜15.0ml[酸化鉄(III)として0〜0.75mg]を数個の100ml
の全量フラスコに段階的に取り,12.2.3 b)の操作を行い,吸光度と酸化鉄(III)量との関係線を作成し,原点
を通るように平行移動して検量線とする。
12.2.6 計算 試料中の酸化鉄(III)含有率は,12.2.3 b)及び12.2.4で得た吸光度と12.2.5で作成した検量線
とから酸化鉄(III)量を求め,次の式によって算出する。
100
250
2
1
3
2
×
×V
m
A
A
O
Fe
−
=
ここに, Fe2O3: 酸化鉄(III)の含有率 (mass%)
A1: 分取した試料溶液(A又はA')中の酸化鉄(III)量 (g)
A2: 分取した空試験液(A又はA')中の酸化鉄(III)量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
V: 12.2.3 a)の試料溶液(A又はA')の分取量 (ml)
12.3 ICP発光分光法
12.3.1 要旨 10.2.4又は10.3.4の試料溶液(A又はA')を分取し,ICP発光分光装置を用いて鉄の発光強
度を測定する。
12.3.2 試薬 試薬は,次による。
a) 標準酸化鉄(III)溶液 (1.0mgFe2O3/ml) 12.2.2 e)と同じ。
b) 標準酸化チタン(IV)溶液 (1.0mgTiO2/ml) チタン(99.9mass%以上)0.599 4gを白金皿(例えば,100
番)に取り,白金皿を四ふっ化エチレン樹脂製時計皿で覆い,ふっ化水素酸20ml,硫酸(1+1)15ml,
及び硝酸0.5mlを加え,水浴上で加熱溶解する。時計皿を水で洗って取り除き,砂浴上で硫酸の濃い
白煙が出るまで加熱する。冷却後,白金皿の内壁を少量の水で洗い,再び加熱して白煙を発生させる。
冷却後,水を加え,1 000mlの全量フラスコに移し入れ,水で標線まで薄める。
c) 標準酸化カルシウム溶液 (1.0mgCaO/ml) 炭酸カルシウム(99.9mass%以上)2〜2.5gを白金るつぼ(例
えば,20番)又は磁器るつぼ(例えば,B形15ml)に取り,600±25℃で約60分間加熱した後,デシ
ケーターに入れ放冷する。この中から1.784 8gをはかり取り,ビーカー (200ml) に移し入れ(15),ビ
ーカーを時計皿で覆い,塩酸(1+1)10mlを徐々に加えて溶解し,冷却後1 000mlの全量フラスコに移
し入れ,水で標線まで薄める。
注(15) 例えば,金属製(例えば,白金製)はかり取り皿に正しくはかり取り,飛散しないように注意
してビーカーに移し,少量の水で金属製はかり取り皿の付着残留物を洗い移す。
d) 標準酸化マグネシウム溶液 (1.0mgMgO/ml) マグネシウム(99.9mass%以上)(13)0.603 0gをはかり取
10
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
り,ビーカー (200ml) に移し入れ,ビーカーを時計皿で覆い,塩酸(1+1)10mlを徐々に加えて溶解し,
冷却後1 000mlの全量フラスコに移し入れ,水で標線まで薄める。
e) 混合標準液I(16) (0.05mgFe2O3/ml, 0.03mgTiO2/ml, 0.05mgCaO/ml, 0.08mgMgO/ml) 1 000mlの全量フラ
スコに標準酸化鉄(III)溶液,標準酸化チタン(IV)溶液,標準酸化カルシウム溶液及び標準酸化マグネシ
ウム溶液のそれぞれを正しく50ml,30ml,50ml及び80ml(16)取り,水で標線まで薄める。この溶液は,
使用の都度調製する。
注(16) 各成分の濃度及び分取量は,試料中の分析成分の含有率によって変えてよい。
なお,試料中の酸化マグネシウム含有率が7mass%を超える場合には,酸化マグネシウムの定
量は,EDTA滴定法を用いるので標準酸化マグネシウム溶液の添加は不要である。
f)
添加液 白金皿(例えば,75番)に酸化アルミニウム及び酸化マグネシウムの一定量(17)をはかり取
り,以下10.2.4又は10.3.4に準じて試料溶液(A又はA')に相当する溶液を調製する(18)。
注(17) 試料中の酸化アルミニウム及び酸化マグネシウム含有率に相当する量(例えば,酸化アルミニ
ウム含有率が50mass%及び酸化マグネシウムの含有率が30mass%の場合,酸化アルミニウムを
0.25g及び酸化マグネシウムを0.15g)をはかり取る。
なお,試料中の酸化マグネシウム含有率が7mass%未満の場合,酸化マグネシウムは,e)の混
合標準液Iに加えられるので,添加液には添加する必要はない。
(18) 試料と同じ操作法を行って調製する。
g) 検量線用溶液系列I(19) 混合標準液Iを段階的に正しく数個の100mlの全量フラスコに取り,それぞ
れに添加液10mlを加え,水で標線まで薄める。
注(19) 表6に調製例を示す。分析試料の組成及び使用する分析装置の種類・性能に応じて最適な検量線
用溶液系列を調製する。
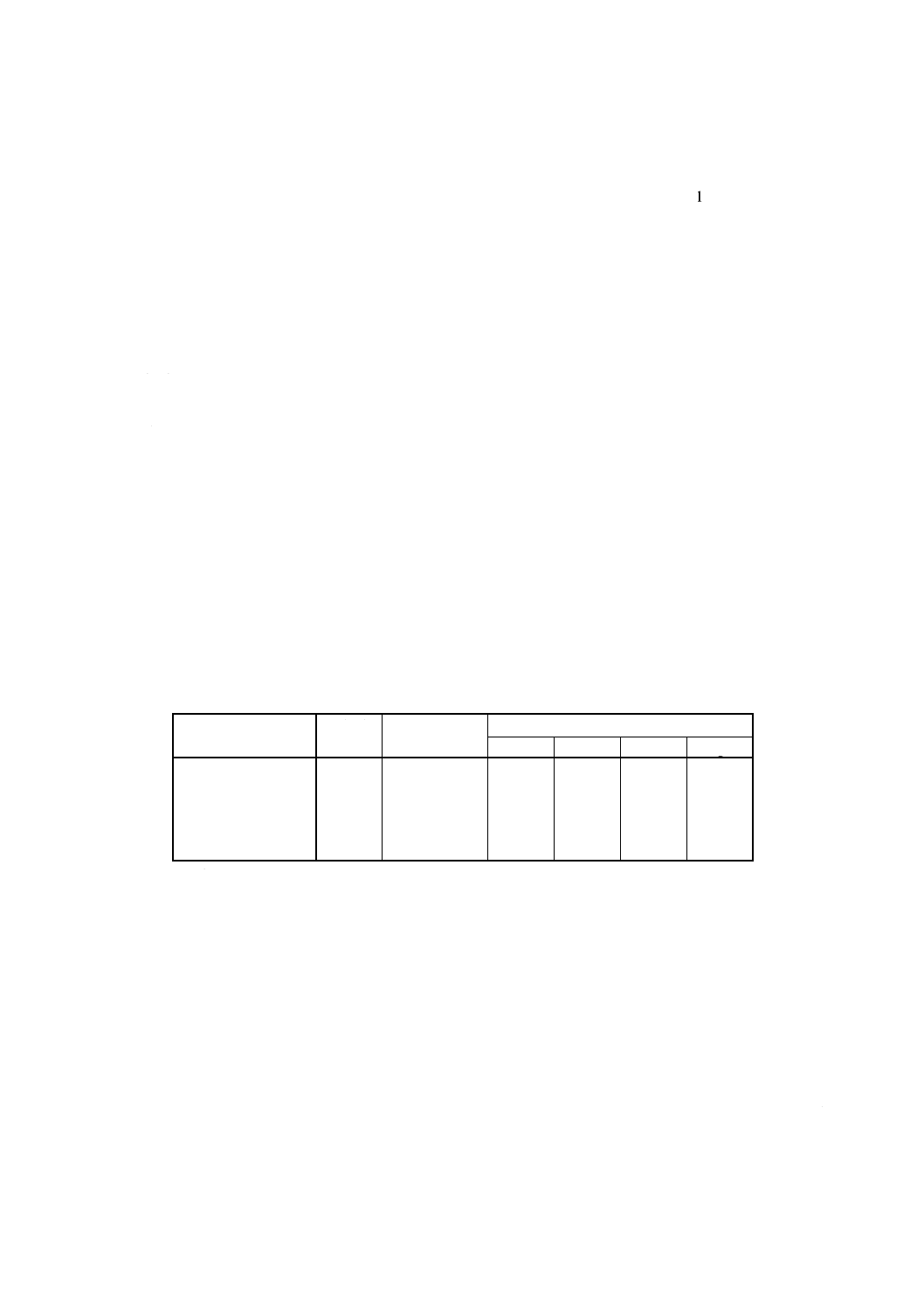
表6 検量線用溶液系列Iの調製例
検量線用溶液系列I
添加液
ml
混合標準液I
ml
溶液濃度 (mg/100ml)
Fe2O3
TiO2
CaO
MgO
No.1
10
0
0
0
0
0
No.2
10
5
0.25
0.15
0.25
0.40
No.3
10
10
0.50
0.30
0.50
0.80
No.4
10
15
0.75
0.45
0.75
1.20
No.5
10
20
1.00
0.60
1.00
1.60
12.3.3 操作 定量操作は,次の手順によって行う。
a) 10.2.4 a)又は10.3.4 a)で得た試料溶液(A又はA')の正しく10mlを100mlの全量フラスコに分取し,
水で標線まで薄める。この溶液を試料溶液(B)とする。
b) 試料溶液(B)の一部をICP発光分析装置のアルゴンプラズマ中に噴霧し,例えば,波長259.94nmにお
ける発光強度を測定する。
12.3.4 空試験 10.2.5又は10.3.5で得た空試験液(A又はA')を用いて12.3.3の操作を行う。試料溶液(B)
に対応する溶液を,空試験液(B)とする。
12.3.5 検量線の作成 検量線用溶液系列Iを用いて12.3.3 b)の操作を行い(11),発光強度と酸化アルミニウ
ム量との関係線を作成し,原点を通るように平行移動して検量線とする。
12.3.6 計算 試料中の酸化鉄(III)含有率は,12.3.3及び12.3.4で得た発光強度と12.3.5で作成した検量線
とから酸化鉄(III)量を求め,次の式によって算出する。

11
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
10
250
2
1
3
2
×
×
m
A
A
O
Fe
−
=
ここに, Fe2O3: 酸化鉄(III)の含有率 (mass%)
A1: 12.3.3の試料溶液(B)中の酸化鉄(III)量 (g)
A2: 12.3.4の空試験液(B)中の酸化鉄(III)量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
13. 酸化チタン(IV)の定員方法
13.1 定量方法の区分 酸化チタン(IV)の定量方法は,次のいずれかによる。
a) ジアンチピリルメタン吸光光度法
b) ICP発光分光法
13.2 ジアンチピリルメタン吸光光度法
13.2.1 要旨 10.2.4又は10.3.4の試料溶液(A又はA')を分取し,酸濃度を調節した後,L (+) −アスコ
ルビン酸を加えて鉄を還元し,ジアンチピリルメタンで呈色させ,吸光度を測定する。
13.2.2 試薬 試薬は,次による。
a) 塩酸(1+1)
b) L (+) −アスコルビン酸溶液 (100g/l) 10.2.2 i)と同じ。
c) ジアンチピリルメタン溶液 (15g/l) ジアンチピリルメタン1.5gを塩酸(1+5)45mlに溶かし,水で
100mlに薄める。
d) 希釈標準酸化チタン(IV)溶液 (0.01mgTiO2/ml) 12.3.2 b)の標準酸化チタン(IV)溶液を水で正しく100
倍に薄める。使用の都度調製する。
13.2.3 操作 定量操作は,次の手順によって行う。
a) 10.2.4 a)又は10.3.4 a)で得た試料溶液(A又はA')の正しく一定量(20)を50mlの全量フラスコに分取す
る。
注(20) 試料溶液(A又はA')の分取量は,試料中の酸化チタン(IV)含有率に応じて表7による。
表7 試料溶液(A又はA')の分取量
酸化チタン(IV)含有率
mass%
分取量
ml
0.5未満
25
0.5以上 1.5未満
10
1.5以上
5
b) 塩酸(1+1)5ml及びL (+) −アスコルビン酸溶液2mlを加え,1分間放置した後,ジアンチピリルメタ
ン溶液10mlを加え,水で標線まで薄め,90分間放置する。この溶液の一部を吸光光度計の吸収セル
(10mm) に取り,波長390nm付近で水を対照液にして吸光度を測定する。
13.2.4 空試験 10.2.5又は10.3.5で得た空試験液(A又はA')を用いて13.2.3の操作を行う。ただし,空
試験液の分取量は,試料溶液の場合と同量とする。
13.2.5 検量線の作成 13.2.2 d)の希釈標準酸化チタン(IV)溶液0〜30.0ml[酸化チタン(IV)として0〜
0.30mg]を数個の50mlの全量フラスコに段階的に取り,13.2.3 a)の塩酸(1+1)5ml添加以降の操作を行い,
吸光度と酸化チタン(IV)量との関係線を作成し,原点を通るように平行移動して検量線とする。
12
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
13.2.6 計算 試料中の酸化チタン(IV)含有率は,13.2.3 b)及び13.2.4で得た吸光度と13.2.5で作成した検
量線とから酸化チタン(IV)量を求め,次の式によって算出する。
100
250
2
1
2
×
×V
m
A
A
TiO
−
=
ここに, TiO2: 酸化チタン(IV)の含有率 (mass%)
A1: 分取した試料溶液(A又はA')中の酸化チタン(IV)量 (g)
A2: 分取した空試験液(A又はA')中の酸化チタン(IV)量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
V: 13.2.3 a)の試料溶液(A又はA')の分取量 (ml)
13.3 ICP発光分光法
13.3.1 要旨 12.3.3の試料溶液(B)を取り,ICP発光分光装置を用いてチタンの発光強度を測定する。
13.3.2 操作 12.3.3 a)で得た試料溶液(B)の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,例え
ば,波長334.94mmにおける発光強度を測定する。
13.3.3 空試験 12.3.4で得た空試験液(B)を用いて,13.3.2の操作を行う。
13.3.4 検量線の作成 12.3.2 g)の検量線用溶液系列Iを用いて,13.3.2の操作を行い(11),得た発光強度と
酸化チタン(IV)量との関係線を作成し,原点を通るように平行移動して検量線とする。
13.3.5 計算 試料中の酸化チタン(IV)含有率は,13.3.2及び13.3.3で得た発光強度と13.3.4で作成した検
量線とから酸化チタン(IV)量を求め,次の式によって算出する。
100
10
250
2
1
2
×
×
m
A
A
TiO
−
=
ここに, TiO2: 酸化チタン(IV)の含有率 (mass%)
A1: 13.3.2の試料溶液(B)中の酸化チタン(IV)量 (g)
A2: 13.3.3の空試験液(B)中の酸化チタン(IV)量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
14. 酸化カルシウム定量方法
14.1 定量方法の区分 酸化カルシウムの定量方法は,次のいずれかによる。
a) 原子吸光法
b) ICP発光分光法
14.2 原子吸光法
14.2.1 要旨 10.2.4又は10.3.4の試料溶液(A又はA')を分取し,原子吸光光度計を用いてカルシウムの
吸光度を測定する。
14.2.2 試薬 試薬は,次による。
a) 標準酸化カルシウム溶液 (1.0mgCaO/ml) 12.3.2 c)と同じ。
b) 標準酸化マグネシウム溶液 (1.0mgMgO/ml) 12.3.2 d)と同じ。
c) 混合標準液Ⅱ(21) (0.1mgCaO/ml, 0.1mgMgO/ml) 1 000mlの全量フラスコに標準酸化カルシウム溶液
及び標準酸化マグネシウム溶液をそれぞれ正しく100ml取り,水で標線まで薄める。
注(21) 各成分の濃度及び分取量は,試料中の分析成分の含有率によって変えてよい。
なお,試料中の酸化マグネシウム含有率が7mass%を超える場合には,酸化マグネシウムの定
量は,EDTA滴定法を用いるので,標準酸化マグネシウム溶液の添加は不要である。
d) 添加液 12.3.2 f)と同じ。
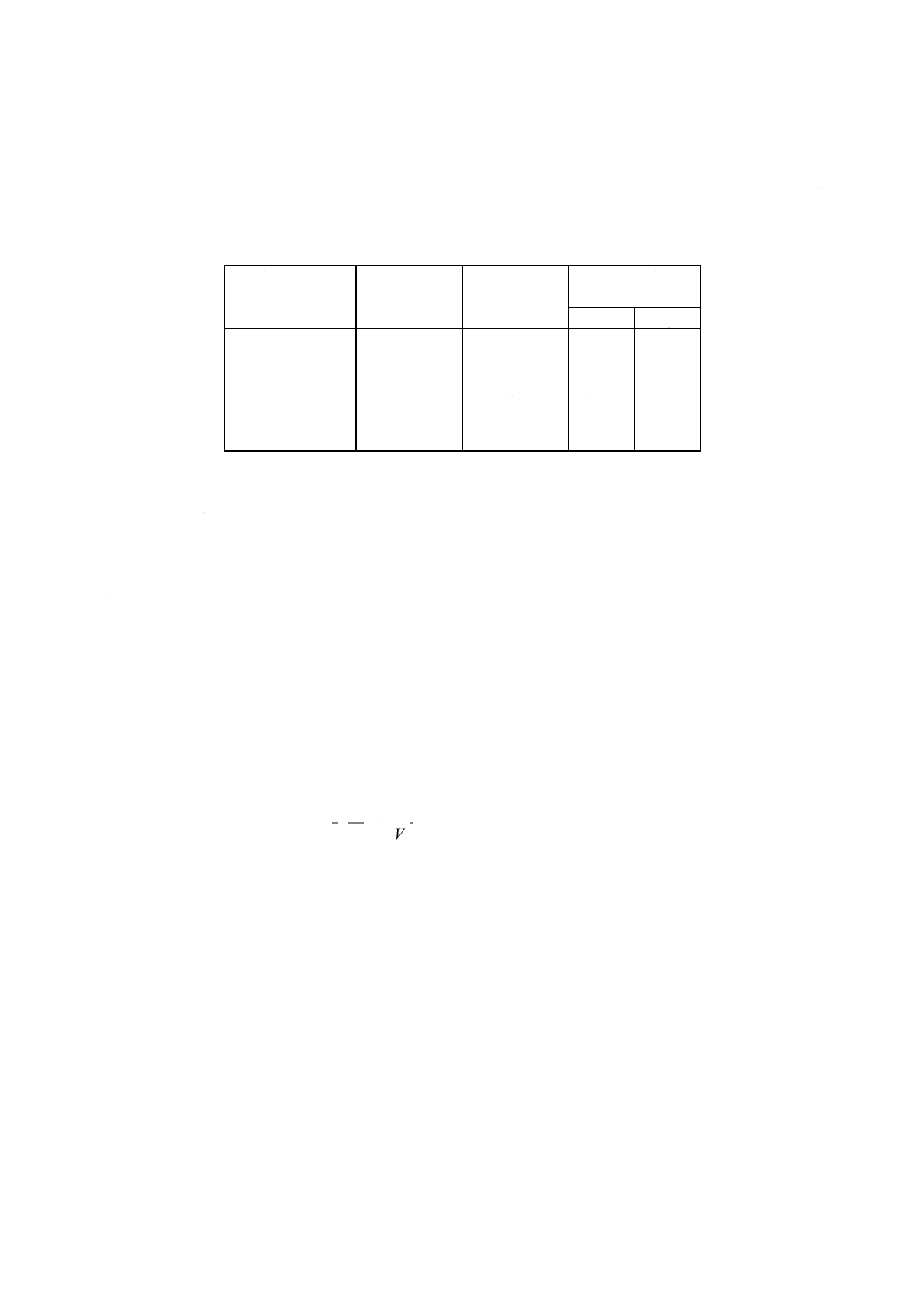
13
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
e) 検量線用溶液系列Ⅱ(22) 混合標準液ⅡIを段階的に正しく数個の100mlの全量フラスコに取り,それ
ぞれに添加液25ml(23)を加え,水で標線まで薄める。
注(22) 表8に調製例を示す。分析試料の組成及び使用する分析装置の種類・性能に応じて最適な検量線
用溶液系列を調製する。
表8 検量線用溶液系列IIの調製例
検量線用溶液系列Ⅱ
添加液(23)
ml
混合標準液Ⅱ
ml
溶液濃度
(mg/100ml)
CaO
MgO
No.1
25
0
0
0
No.2
25
5
0.5
0.5
No.3
25
10
1.0
1.0
No.4
25
15
1.5
1.5
No.5
25
20
2.0
2.0
No.6
25
25
2.5
2.5
(23) 分析試料中の酸化カルシウム及び酸化マグネシウムの含有率に応じ,また,使用する分析装置
の種類・性能に応じ,10mlとしてもよい。
14.2.3 操作 定量操作は,次の手順によって行う。
a) 10.2.4 a)又は10.3.4 a)で得た試料溶液(A又はA')の正しく25ml(23)を100mlの全量フラスコに分取し,
水で標線まで薄める。この溶液を試料溶液(C)とする。
b) 試料溶液(C)の一部を,原子吸光光度計の空気−アセチレンフレーム中に噴霧し,波長422.7nmにおけ
る吸光度を測定する。
14.2.4 空試験 10.2.5又は10.3.5で得た空試験液(A又はA')を用いて14.2.3の操作を行う。試料溶液(C)
に対応する溶液を,空試験液(C)とする。
14.2.5 検量線の作成 検量線用溶液系列Ⅱを用いて14.2.3 b)の操作を行い(11),吸光度と酸化カルシウム
量との関係線を作成し,原点を通るように平行移動して検量線とする。
14.2.6 計算 試料中の酸化カルシウム含有率は,14.2.3 b)及び14.2.4で得た吸光度と14.2.5で作成した検
量線とから酸化カルシウム量を求め,次の式によって算出する。
100
250
2
1
×
×V
m
A
A
CaO
−
=
ここに, CaO: 酸化カルシウムの含有率 (mass%)
A1: 試料溶液(C)中の酸化カルシウム量 (g)
A2: 空試験液(C)中の酸化カルシウム量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
V: 14.2.3 a)の試料溶液(A又はA')の分取量 (ml)
14.3 ICP発光分光法
14.3.1 要旨 12.3.3の試料溶液(B)を取り,ICP発光分光装置を用いてカルシウムの発光強度を測定する。
14.3.2 操作 12.3.3a)で得た試料溶液(B)の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,例え
ば,波長393.37nmにおける発光強度を測定する。
14.3.3 空試験 12.3.4で得た空試験液(B)を用いて,14.3.2の操作を行う。
14.3.4 検量線の作成 12.3.2 g)の検量線用溶液系列Iを用いて14.3.2の操作を行い(11),得た発光強度と酸
化カルシウム量との関係線を作成し,原点を通るように平行移動して検量線とする。
14
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.3.5 計算 試料中の酸化カルシウム含有率は,14.3.2及び14.3.3で得た発光強度と14.3.4で作成した検
量線とから酸化カルシウム量を求め,次の式によって算出する。
100
10
250
2
1
×
×
m
A
A
CaO
−
=
ここに, CaO: 酸化カルシウムの含有率 (mass%)
A1: 14.3.2の試料溶液(B)中の酸化カルシウム量 (g)
A2: 14.3.3の空試験液(B)中の酸化カルシウム量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
15. 酸化マグネシウムの定量方法
15.1 定量方法の区分 酸化マグネシウムの定量方法は,次のいずれかによる。
a) 原子吸光法 酸化マグネシウム含有率7mass%以下の試料に適用する。
b) ICP発光分光法 酸化マグネシウム含有率7mass%以下の試料に適用する。
c) EDTA滴定−ICP発光分光併用法
15.2 原子吸光法
15.2.1 要旨 14.2.3の試料溶液(C)を取り,原子吸光光度計を用いてマグネシウムの吸光度を測定する。
15.2.2 操作 14.2.3 a)で得た試料溶液(C)の一部を原子吸光光度計の酸化二窒素−アセチレンフレーム又
は空気−アセチレンフレーム中に噴霧し,波長285.2nmにおける吸光度を測定する。
15.2.3 空試験 14.2.4で得た空試験液を用いて,15.2.2の操作を行う。
15.2.4 検量線の作成 14.2.2 e)の検量線用溶液系列Ⅱを用いて15.2.2の操作を行い(11),吸光度と酸化マグ
ネシウム量との関係線を作成し,原点を通るように平行移動して検量線とする。
15.2.5 計算 試料中の酸化マグネシウム含有率は,15.2.2及び15.2.3で得た吸光度と15.2.4で作成した検
量線とから酸化マグネシウム量を求め,次の式によって算出する。
100
100
2
1
×
×V
m
A
A
MgO
−
=
ここに, MgO: 酸化マグネシウムの含有率 (mass%)
A1: 試料溶液(C)中の酸化マグネシウム量 (g)
A2: 空試験液(C)中の酸化マグネシウム量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
V: 14.2.3 a)の試料溶液(A又はA')の分取量 (ml)
15.3 ICP発光分光法
15.3.1 要旨 12.3.3の試料溶液(B)を取り,ICP発光分光装置を用いてマグネシウムの発光強度を測定する。
15.3.2 操作 12.3.3 a)で得た試料溶液(B)の一部をICP発光分光装置のアルゴンプラズマ中に噴霧し,例え
ば,波長279.55nmにおける発光強度を測定する。
15.3.3 空試験 12.3.4で得た空試験液(B)を用いて,15.3.2の操作を行う。
15.3.4 検量線の作成 12.3.2 g)の検量線用溶液系列Iを用いて15.3.2の操作を行い(11),得た発光強度と酸
化マグネシウム量との関係線を作成し,原点を通るように平行移動して検量線とする。
15.3.5 計算 試料中の酸化マグネシウム含有率は,15.3.2及び15.3.3で得た発光強度と15.3.4で作成した
検量線とから酸化マグネシウム量を求め,次の式によって算出する。
100
10
250
2
1
×
×
m
A
A
MgO
−
=
ここに, MgO: 酸化マグネシウムの含有率 (mass%)

15
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A1: 15.3.2の試料溶液(B)中の酸化マグネシウム量 (g)
A2: 15.3.3の空試験液(B)中の酸化マグネシウム量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
15.4 EDTA滴定−ICP発光分光併用法
15.4.1 要旨 10.2.4又は10.3.4の試料溶液(A又はA')を分取し,アンモニア水でアルミニウム,鉄及び
チタンを分離した後,ろ液に2, 2', 2"−ニトリロトリエタノール及び緩衝液を加えてpHを調節し,エリオ
クロムブラックTを指示薬としてEDTA溶液で酸化カルシウムと酸化マグネシウムの合量を滴定し,14.
で求めた酸化カルシウム量を補正する。沈殿は,塩酸に溶解しICP発光分光法で沈殿中の残存酸化マグネ
シウム量を求め,EDTA滴定法とICP発光分光法の和から酸化マグネシウム含有率を算出する。
15.4.2 試薬 試薬は,次による。ただし,a),c),e),f)及びg)は,プラスチック瓶に保存する。
a) 塩酸(1+3)
b) 塩化アンモニウム
c) アンモニア水(1+1,1+9)
d) 塩化アンモニウム溶液 (20g/l) 塩化アンモニウム10gを水500mlに溶かし,メチルレッド溶液1滴
を加え,黄色を呈するまでアンモニア水(1+9)を滴下する。加熱して赤色に戻ったら,アンモニア水(1
+9)を追加する。
e) シアン化カリウム溶液 (50g/l)
f)
塩化ヒドロキシルアンモニウム溶液 (100g/l)
g) 緩衝液 (pH10) 塩化アンモニウム70gにアンモニア水570mlを加え,水で1 000mlにする。
h) 2, 2', 2"−ニトリロトリエタノール(1+1)
i)
0.02mol/l EDTA溶液 調製方法及び標定方法は,JIS K 8001の4.5(3.3)(0.01mol/lエチレンジアミン四
酢酸二水素二ナトリウム溶液)に準じる。ただし,エチレンジアミン四酢酸二水素二ナトリウム二水
和物7.5gを用いる。
j)
メチルレッド溶液 調製方法及び保存方法は,JIS K 8001の4.4(表7)による。
k) エリオクロムブラックT溶液 調製方法及び保存方法は,JIS K 8001の4.4(表8)による。
15.4.3 操作 操作は,次の手順によって行う。
a) 10.2.4 a)又は10.3.4 a)で得た試料溶液(A又はA')から正しく一定量(24)をビーカー (200ml) に分取し,
分取量によって水浴上で加熱濃縮するか水で薄めるかして,液量を約50mlにする。塩化アンモニウ
ム3g及びメチルレッド溶液1滴を加え,ビーカーを時計皿で覆って穏やかに加熱し,煮沸し出したな
ら熱源から降ろし時計皿を取り,かき混ぜながら最初はアンモニア水(1+1)を滴下し,溶液の変色間
際になったならアンモニア水(1+9)に替えて滴下を続け,かすかにアンモニア臭を呈してから更に過
剰に10滴を加え,時計皿で覆って約1分間煮沸を続けた後,水浴上に移し15分間温浸する。
注(24) 試料溶液(A又はA')の分取量は,試料中の酸化マグネシウムと酸化カルシウムの含有率の合
量に応じて表9による。
表9 試料溶液(A又はA')の分取量
酸化マグネシウムと酸化カルシウ
ムの含有率の合量
分取量
mass%
ml
15未満
100
15以上 30未満
50
30以上 45未満
30
45以上 60未満
25

16
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
酸化マグネシウムと酸化カルシウ
ムの含有率の合量
分取量
mass%
ml
60以上 75未満
20
75以上
15
b) ろ紙(5種A)を用いてろ過し,熱塩化アンモニウム溶液で6回洗浄する。ろ液及び洗液は,ビーカ
ー (300ml) に受ける。
c) 水浴上で液量が200mlになるまで濃縮し,放冷後塩化ヒドロキシルアンモニウム溶液5ml,2, 2', 2"−
ニトリロトリエタノール(1+1)2ml,緩衝液 (pH10) 10ml,シアン化カリウム溶液2ml及び指示薬とし
てエリオクロムブラックT溶液3, 4滴を加え,かき混ぜながら0.02mol/l EDTA溶液で滴定する。終点
付近になったら,よくかき混ぜながらゆっくり滴定し,溶液が赤紫色から青色に変わる点を終点とす
る(25)(26)。
注(25) タングステンランプの光を透過させた乳白色のガラス又はプラスチック板の上で滴定を行うと
終点の判定が容易である。
(26) 測定後の排液は,シアンを分解した上で廃棄する。
d) b)の沈殿を少量の水で元のビーカー (200ml) に移し,塩酸(1+3)20mlを加え,加熱して沈殿を完全に
溶解する。この溶液を元のろ紙に少量ずつ注ぎ,ろ紙に残留する沈殿を完全に溶解させる。ビーカー
及びろ紙を熱水で十分に洗浄後,ろ液及び洗液を200mlの全量フラスコに移し,冷却後水で標線まで
薄める。
e) d)の溶液の一部を,ICP発光分光装置のアルゴンプラズマ中に噴霧し,波長393.37nm及び279.55nm
におけるカルシウム及びマグネシウムの発光強度を測定する(27)。
注(27) 原子吸光法を用いてもよい。このときは,d)の溶液は100mlの全量フラスコに受け,14.2.2 e)の
ランタン溶液10mlを加えて標線まで薄める。この溶液の一部を原子吸光光度計のフレーム中に
噴霧し,波長259.94nm及び285.2nmにおける吸光度を測定する。
15.4.4 空試験 10.2.5又は10.3.5で得た空試験液(A又はA')を用いて,15.4.3の操作を行う。
15.4.5 検量線の作成 12.3.2 c)及びd)の標準酸化カルシウム溶液及び標準酸化マグネシウム溶液をそれぞ
れ水で正しく10倍に薄める。これら溶液各0〜10.0ml(酸化カルシウム及び酸化マグネシウムとしてそれ
ぞれ0〜1mg)を数個の200mlの全量フラスコに段階的に取り,それぞれに塩酸(1+3)20mlを加え,水で
標線まで薄める。この検量線用溶液を用いて15.4.3 e)の操作を行い(11),発光強度と酸化カルシウム量及び
酸化マグネシウム量との関係線を作成し,原点を通るように平行移動して検量線とする(28)。
注(28) 15.4.3 e)において原子吸光法を用いた場合には,100mlの全量フラスコにランタン溶液10mlを加
え,検量線用溶液を作製する。検量線用溶液には,12.3.2 f)に準じて調製した試料中の酸化アル
ミニウム含有率に相当する添加液を加えるのが望ましい。
この検量線用溶液について注(27)の操作を行い,吸光度と酸化カルシウム量及び酸化マグネシ
ウム量との関係線を作成し,原点を通るように平行移動して検量線とする。
15.4.6 計算 試料中の酸化マグネシウム含有率は,15.4.3 e)及び15.4.4で得た発光強度と15.4.5で作成し
た検量線とから残留酸化マグネシウム量を求め,次の式によって算出する。
(
)
(
)(
)
719
.0
100
250
719
.0
1
806
000
.0
3
4
3
2
1
2
1
×
×
×
+
×
×
×
CaO
V
m
A
A
A
A
F
V
V
MgO
−
−
−
+
−
=
ここに, MgO: 酸化マグネシウムの含有率 (mass%)
V1: 15.4.3 c)の0.02mol/l EDTA溶液使用量 (ml)
17
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V2: 15.4.4の0.02mol/l EDTA溶液使用量 (ml)
V3: 15.4.3 a)の試料溶液(A又はA')の分取量 (ml)
F: 0.02mol/l EDTA溶液のファクター
A1: 15.4.3 e)の残留酸化カルシウム量 (g)
A2: 15.4.4の残留酸化カルシウム量 (g)
A3: 15.4.3 e)の残留酸化マグネシウム量 (g)
A4: 15.4.4の残留酸化マグネシウム量 (g)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
CaO: 14.で求めた酸化カルシウム含有率 (mass%)
16. 酸化ナトリウム定量方法
16.1 定量方法の区分 酸化ナトリウムの定量方法は,次のいずれかによる。
a) 炎光光度法
b) 原子吸光法
16.2 炎光光度法
16.2.1 要旨 試料をふっ化水素酸,過塩素酸及び硝酸を用いて,加熱分解する。蒸発乾固した後,塩酸に
溶解して定容とする。この溶液の一部を取り,炎光光度計のフレーム中に噴霧し,カルシウムの発光強度
を測定する。
16.2.2 試薬 試薬は,次による。
a) 塩酸(1+1)
b) 硝酸
c) 過塩素酸
d) ふっ化水素酸
e) ランタン溶液 酸化ランタン(III)50gをビーカー (1 000ml) にはかり取り,塩酸(1+1)200mlを加えて
加熱分解し,水で1 000mlに薄める。
f)
標準酸化ナトリウム溶液 (0.5mgNa2O/ml) 塩化ナトリウム1〜1.5gを白金るつぼ(例えば,30番)
に取り,600±25℃で約60分間加熱した後,デシケーターに入れ放冷する。この中から0.942 9gをは
かり取り,ビーカー (200ml) に移し入れ(15),水約100mlを加えて溶解し,1 000mlの全量フラスコに
移し入れ,水で標線まで薄める。
g) 標準酸化カリウム溶液 (0.5mgK2O/ml) 塩化カリウム1〜1.5gを白金るつぼ(例えば,30番)に取り,
600±25℃で約60分間加熱した後,デシケーターに入れ放冷する。この中から0.791 4gをはかり取り,
ビーカー (200ml) に移し入れ(15),水約100mlを加えて溶解し,1 000mlの全量フラスコに移し入れ,
水で標線まで薄める。
h) 混合標準液III (0.02mgNa2O/ml, 0.02mgK2O/ml) 1 000mlの全量フラスコに標準酸化ナトリウム溶液
及び標準酸化カリウム溶液のそれぞれ正しく40mlを取り,水で標線まで薄める。
i)
検量線用溶液系列III(29) 混合標準液IIIを段階的に正しく100mlの全量フラスコに取り,ランタン溶
液10.0ml及び塩酸(1+1)5.0mlを加えた後,水で標線まで薄める。
注(29) 表10に調製例を示す。分析試料の組成及び使用する分析装置の種類・性能に応じて最適な検量
線用溶液系列を調製する。
18
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
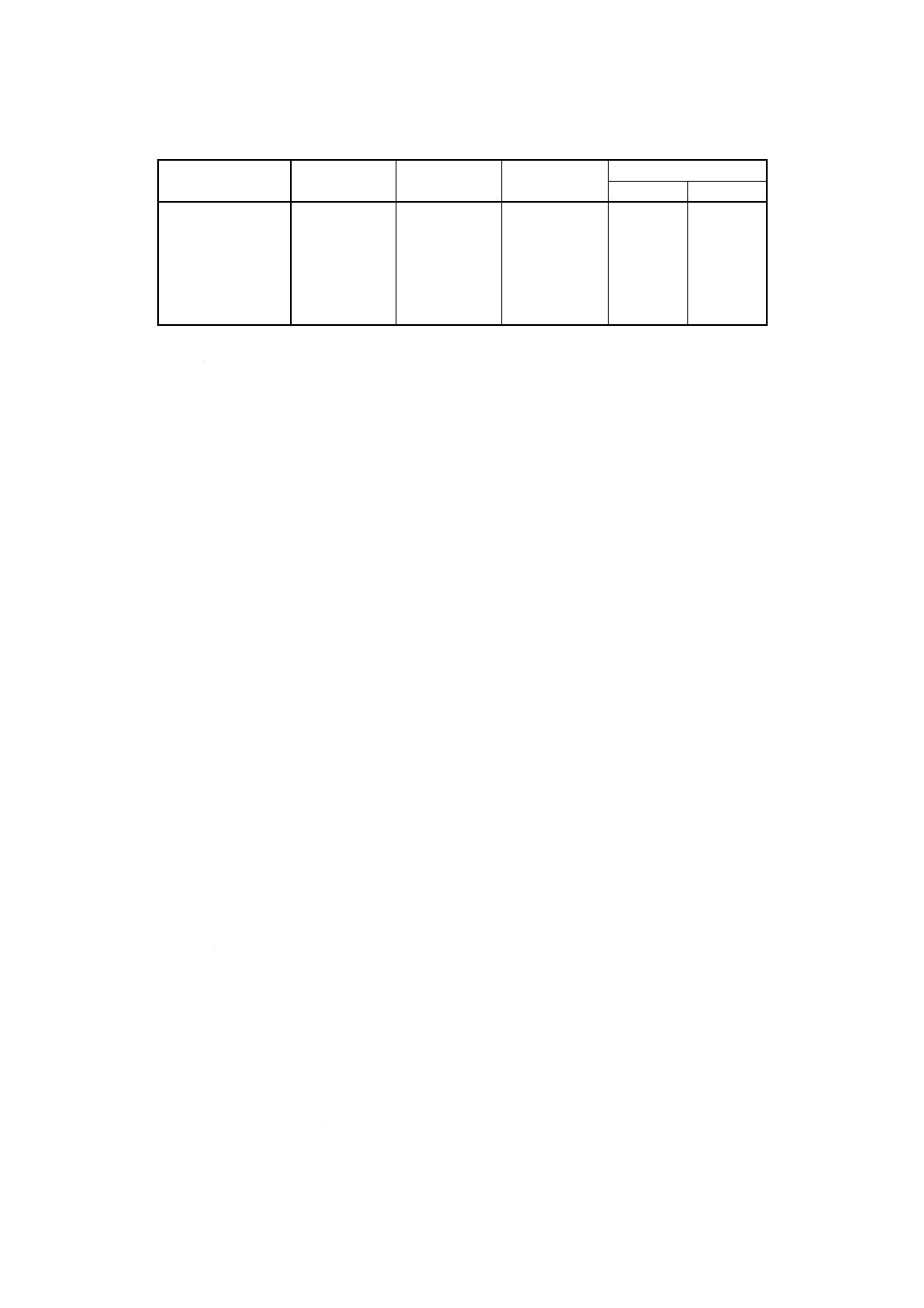
表10 検量線用溶液系列IIIの調製例
検量線用溶液系列
III
塩酸
ml
ランタン溶液
ml
混合標準液III
ml
溶液濃度 (mg/100ml)
Na2O
K2O
No.1
5
10
0
0
0
No.2
5
10
5
0.1
0.1
No.3
5
10
10
0.2
0.2
No.4
5
10
15
0.3
0.3
No.5
5
10
20
0.4
0.4
No.6
5
10
25
0.5
0.5
16.2.3 試料はかり取り量 試料はかり取り量は,0.20gとする。
16.2.4 操作 定量操作は,次の手順によって行う。
a) 試料を白金皿(例えば,150番)にはかり取り,水で潤し,過塩素酸5ml,硝酸2ml及びふっ化水素
酸10mlを加え,よくかき混ぜ,砂浴上で注意して加熱分解し(30),過塩素酸の白煙を激しく発生させ
て蒸発乾固する。放冷後,白金皿の内壁を少量の水で洗い,再び過塩素酸3ml,硝酸2ml及びふっ化
水素酸5mlを加え,砂浴上で蒸発乾固する。放冷後,白金皿の内壁を少量の水で洗い,過塩素酸3ml
を加え,砂浴上で加熱し蒸発乾固して残留するふっ化物を揮散させる。放冷後,塩酸(1+1)5.0ml及び
水約20mlを加え時計皿で覆い,水浴上で加熱溶解し(31),プラスチックビーカー (200ml) に受け,プ
ラスチック漏斗及びろ紙(5種B)を用いてろ過し,熱水で十分洗浄する(32)。
注(30) 白金皿内容物のかき混ぜには,太めの白金合金(例えば,白金−ロジウム)線の先端を折り曲
げたもの,白金製さじ,四ふっ化エチレン樹脂製棒又はさじなどが利用できる。加熱していく
と試料が白金皿の底に固化して試薬と反応しにくくなるので,砂浴から降ろし放冷後,固化物
を白金皿の底からはがし,よくつぶすとよい。加熱分解を続け,液量が少なくなり,過塩素酸
の白煙が発生する直前になると試料によっては激しく反応し,飛散することがあるので注意す
る。
(31) 塩酸が揮発するので,できるだけ短時間で溶解する。
(32) 溶液中に微粒子が漏れることがあるが,測定上差し支えない。
b) 放冷後,ランタン溶液10mlを加え,100mlの全量フラスコに移し入れ,水で標線まで薄めた後,直ち
にプラスチック瓶に移す。この溶液を試料溶液(D)とし,炎光光度法による酸化ナトリウム及び酸化カ
リウムの定量に用いる。
c) この試料溶液(D)(33)の一部を炎光光度計のフレーム中に噴霧し,波長589.0nm(34)における発光強度を
測定する。
注(33) 試料溶液(D)中の濃度が原子吸光光度計の定量上限を超えるときは,試料溶液(D)から正しく一
定量 (x ml) を100mlの全量フラスコに取り,塩酸(1+1) (5.0−5.0×x/100) ml及びランタン溶液
(10−10×x/100) mlを加え,水で標線まで薄め,この溶液について測定する。
(34) ナトリウム用フィルターを使用してもよい。
16.2.5 空試験 試料を用いないで16.2.4の操作を行う(35)。ここで得た試料溶液(D)に対応する溶液を空試
験液(D)とする。
注(35) 注(33)によるときは,空試験液(D)も試料溶液と同様にして調製する。
16.2.6 検量線の作成 検量線用溶液系列IIIを用いて16.2.4 c)の操作を行い(11),発光強度と酸化ナトリウ
ム量との関係線を作成し,検量線とする。
19
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
16.2.7 計算 試料中の酸化ナトリウム含有率は,16.2.4 c)及び16.2.5で得た発光強度と16.2.6で作成した
検量線とから酸化ナトリウム量を求め,次の式によって算出する。
100
100
2
1
2
×
×V
m
A
A
O
Na
−
=
ここに, Na2O: 酸化ナトリウムの含有率 (mass%)
A1: 試料溶液(D)又は希釈試料溶液中の酸化ナトリウム量 (g)
A2: 空試験液(D)又は希釈空試験液中の酸化ナトリウム量 (g)
V: 試料溶液(D)の分取量 (ml) (分取しない場合は100)
m: 試料はかり取り量 (g)
16.3 原子吸光法
16.3.1 要旨 試料をふっ化水素酸,過塩素酸及び硝酸を用いて加熱分解する。蒸発乾固した後,塩酸に溶
解して定容とする。この溶液の一部を取り,原子吸光光度計を用いてナトリウムの吸光度を測定する。
16.3.2 試薬 16.2.2に同じ。
16.3.3 操作
a) 16.2.4 a)及びb)に準じて操作する。得られた溶液を試料溶液(D')とし,原子吸光法による酸化ナトリウ
ム及び酸化カリウムの定量に用いる。
b) この試料溶液(D')(33)の一部を原子吸光光度計の空気−アセチレンフレーム中に噴霧し,波長589.0nm
における吸光度を測定する。
16.3.4 空試験 試料を用いないで16.3.3の操作を行う(35)。ここで得た試料溶液(D')に対応する溶液を空試
験液(D')とする。
16.3.5 検量線の作成 16.2.2 i)の検量線用溶液系列IIIを用いて16.3.3の操作を行い(11),吸光度と酸化ナ
トリウム量との関係線を作成し,原点を通るように平行移動して検量線とする。
16.3.6 計算 試料中の酸化ナトリウム含有率は,16.3.3及び16.3.4で得た吸光度と16.3.5で作成した検量
線とから酸化ナトリウム量を求め,次の式によって算出する。
100
100
2
1
2
×
×V
m
A
A
O
Na
−
=
ここに,
Na2O: 酸化ナトリウムの含有率 (mass%)
A1: 試料溶液(D')又は希釈試料溶液中の酸化ナトリウム量 (g)
A2: 空試験液(D')又は希釈空試験液中の酸化ナトリウム量 (g)
V: 試料溶液(D')の分取量 (ml) (分取しない場合は100)
m: 試料はかり取り量 (g)
17. 酸化カリウム定量方法
17.1 定量方法の区分 酸化カリウムの定量方法は,次のいずれかによる。
a) 炎光光度法
b) 原子吸光法
17.2 炎光光度法
17.2.1 要旨 16.2.4の試料溶液(D)を取り,炎光光度計のフレーム中に噴霧し,カリウムの発光強度を測
定する。
17.2.2 操作 16.2.4 b)で得た試料溶液(D)(33)の一部を炎光光度計のフレーム中に噴霧し,波長766.5nm(36)
における発光強度を測定する。
注(36) カリウム用フィルターを使用してもよい。
20
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
17.2.3 空試験 16.2.5で得た空試験液(D)(35)を用いて,17.2.2の操作を行う。
17.2.4 検量線の作成 16.2.2 i)の検量線用溶液系列IIIを用いて17.2.2の操作を行い(11),発光強度と酸化
カリウム量との関係線を作成して検量線とする。
17.2.5 計算 試料中の酸化カリウム含有率は,17.2.2及び17.2.3で得た発光強度と17.2.4で作成した検量
線とから酸化カリウム量を求め,次の式によって算出する。
100
100
2
1
2
×
×V
m
A
A
O
K
−
=
ここに, K2O: 酸化カリウムの含有率 (mass%)
A1: 試料溶液(D)又は希釈試料溶液中の酸化カリウム量 (g)
A2: 空試験液(D)又は希釈空試験液中の酸化カリウム量 (g)
V: 試料溶液(D)の分取量 (ml) (分取しない場合は100)
m: 16.2.4 a)の試料はかり取り量 (g)
17.3 原子吸光法
17.3.1 要旨 16.3.3の試料溶液(D')を取り,原子吸光光度計を用いてカリウムの吸光度を測定する。
17.3.2 操作 16.3.3 a)で得た試料溶液(D')(33)の一部を原子吸光光度計の空気−アセチレンフレーム中に噴
霧し,波長766.5nm(37)における吸光度を測定する。
注(37) 試料溶液中の酸化カリウム濃度が高い場合は,波長769.9nm又は404.4nmを用いてもよい。
17.3.3 空試験 16.3.4で得た空試験液(D')(35)を用いて17.3.2の操作を行う。
17.3.4 検量線の作成 16.2.2 i)の検量線用溶液系列III(38)を用いて17.3.2の操作を行い(11),吸光度と酸化
カリウム量との関係線を作成し,原点を通るように平行移動して検量線とする。
注(38) 注(37)による場合は,その濃度に合わせた検量線用溶液系列IIIを作成し,試料溶液と同じ波長
を用いて検量線を作成する。
17.3.5 計算 試料中の酸化カリウム含有率は,17.3.2及び17.3.3で得た吸光度と17.3.4で作成した検量線
とから酸化カリウム量を求め,次の式によって算出する。
100
100
2
1
2
×
×V
m
A
A
O
K
−
=
ここに, K2O: 酸化カリウムの含有率 (mass%)
A1: 試料溶液(D')又は希釈試料溶液中の酸化カリウム量 (g)
A2: 空試験液(D')又は希釈空試験液中の酸化カリウム量 (g)
V: 試料溶液(D')の分取量 (ml) (分取しない場合は100)
m: 16.3.3 a)の試料はかり取り量 (g)
21
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
18. 酸化りん(V)の定量方法
18.1 定量方法 酸化りん(V)の定量方法は,モリブデン青吸光光度法による。
18.2 モリブデン青吸光光度法
18.2.1 要旨 10.2.4又は10.3.4の試料溶液(A又はA')を分取し,酸濃度を調節した後,七モリブデン酸
六アンモニウム及びL (+) −アスコルビン酸を加え,加熱してモリブデン青を呈色させ,吸光度を測定す
る。
18.2.2 試薬 試薬は,次による。
a) 硫酸(1+1)
b) 水酸化ナトリウム溶液 (100g/l)
c) 七モリブデン酸六アンモニウム溶液 (20g/l) 七モリブデン酸六アンモニウム四水和物2gを温水
20mlに溶かし,必要ならばろ過し,硫酸(1+1)60mlを加えて水で100mlに薄める。
d) L (+) −アスコルビン酸溶液 (100g/l) 10.2.2 i)と同じ。
e) 標準酸化りん(V)溶液 (0.1mgP2O5/ml) りん酸二水素カリウム約0.5gを110±5℃で3時間乾燥し,
デシケーター中で放冷する。この中から0.191 7gをはかり取り,ビーカー (200ml) に入れ(15),水約
100mlを加えて溶解し,水で正しく1 000mlに薄める。
f)
p−ニトロフェノール溶液 (2g/l)
18.2.3 操作 操作は,次の手順によって行う。
a) 10.2.4 a)又は10.3.4 a)で得た試料溶液(A又はA')から正しく一定量(39)を100mlの全量フラスコに分
取する。
注(39) 試料溶液(A又はA')の分取量は,試料中の酸化りん(V)の含有率に応じて表11による。
表11 試料溶液(A又はA')の分取量
酸化りん(V)の含有率
mass%
試料溶液(A)の分取量
ml
0.4未満
25
0.4以上
10
b) p−ニトロフェノール溶液2,3滴を指示薬として加え,溶液が黄色になるまで水酸化ナトリウム溶液
を滴下した後,直ちに硫酸(1+1)を滴下し,無色になった後更に2,3滴過剰に加える。七モリブデン
酸六アンモニウム溶液10ml及びL (+) −アスコルビン酸溶液2mlを加え,水で標線まで薄める。沸
騰水浴中で15分間加熱した後,流水中で冷却する。この溶液の一部を吸光光度計の吸収セルに取り,
波長830nm付近で水を対照液として吸光度を測定する。
18.2.4 空試験 10.2.5又は10.3.5で得た空試験液(A又はA')を用いて18.2.3の操作を行う。
18.2.5 検量線の作成 標準酸化りん溶液を水で正しく10倍に薄める。この溶液0〜25.0ml[酸化りん(V)
として0〜0.25mg]を数個の100mlの全量フラスコに段階的に取り,18.2.3 b)の操作を行い,吸光度と酸化
りん(V)量との関係線を作成し,原点を通るように平行移動して検量線とする。
18.2.6 計算 試料中の酸化りん(V)含有率は,18.2.3 b)及び18.2.4で得た吸光度と18.2.5で作成した検量線
とから酸化りん(V)量を求め,次の式によって算出する。
100
250
2
1
5
2
×
×V
m
A
A
O
P
−
=
ここに, P2O5: 酸化りん(V)の含有率 (mass%)
A1: 分取した試料溶液(A又はA')中の酸化りん (V) 量 (g)
A2: 分取した空試験液(A又はA')中の酸化りん (V) 量 (g)
22
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V: 18.2.3 a)の試料溶液(A又はA')の分取量 (ml)
m: 10.2.4 a)又は10.3.4 a)の試料はかり取り量 (g)
23
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(規定) アルミナ−マグネシア質耐火物中の酸化マグネシウム
の陽イオン交換分離−EDTA滴定法
序文 この附属書(規定)は,アルミナ−マグネシア質耐火物中の酸化マグネシウムの陽イオン交換分離
−キレート滴定法について記述するものであり,規定の一部である。アルミナ−マグネシア質耐火物は,
酸化アルミニウムと酸化マグネシウムの二元系をなすため,当然ながら,本体6.(定量範囲)に規定する
ように,酸化マグネシウムの定量範囲も3〜80mass%と広い。そのため,本体15.(酸化マグネシウムの定
量方法)では,酸化マグネシウム含有率7mass%以下を対象に原子吸光法とICP発光分光分析法を,また,
全領域を対象にEDTA滴定−ICP発光分光併用法を規定している。しかし,後者のEDTA滴定法で採用し
ている酸化アルミニウム及び酸化鉄(III)の沈殿分離法では,沈殿中に酸化カルシウム及び酸化マグネシウ
ムの一部が残留しやすく,ICP発光分光分析法によってそれら残留量を補足しなければならない。
この陽イオン交換分離−EDTA滴定法は,EDTA滴定−ICP発光分光併用法のこれら問題点を解消する目
的で検討された方法であり,酸化マグネシウムを酸化アルミニウム及び酸化鉄から分離して滴定できる優
れた方法である。しかし,特殊なカラムを要し,必ずしも普及した方法とはいえないため,本体から分離
して附属書として規定することになった。
1. 適用範囲 この附属書(規定)は,アルミナ−マグネシア質耐火物中の酸化マグネシウム (3〜80mass%)
の陽イオン交換分離−EDTA滴定法について規定する。
2. 要旨 試料を炭酸ナトリウムとほう酸で融解し,硝酸と硫酸で溶解後,その一部を分取して陽イオン
交換樹脂カラムに流し,イオン交換樹脂にマグネシウムなどの陽イオンを吸着させる。カラムにふっ化水
素酸(1+150)を流してアルミニウム及びチタンを溶離し,次にふっ化水素酸(1+19)を流して鉄を溶離し,
カラムに水を流してふっ化水素酸を除去する。ここまでの溶出液は不要である。次に塩酸(1+1)を流して
マグネシウム及びカルシウムを溶離する。溶離液を蒸発乾固させた後塩酸に溶解し,緩衝液を加えてpH
を調節し,エリオクロムブラックTを指示薬としてEDTA溶液で酸化マグネシウムと酸化カルシウムの合
量を滴定し,本体14.で求めた酸化カルシウム量を補正する。
3. 試薬 試薬は,次による。プラスチック瓶に保存する。
a) ふっ化水素酸(1+150,1+19)
b) 塩酸(1+1)
c) 硫酸(1+9)
d) 硝酸 (1+4,1+20)
e) ほう酸
f)
炭酸ナトリウム(無水)(1)
注(1) 試薬によっては,カルシウムを微量含むものがある。高純度の試薬を用いる。
g) 塩化ヒドロキシルアンモニウム溶液 (100g/l)
h) 緩衝液 (pH 10) 塩化アンモニウム70gにアンモニア水570mlを加え,水で1 000mlにする。
24
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
i)
0.02mol/l EDTA溶液 調製方法及び標定方法は,JIS K 8001の4.5(3.3)(0.01mol/lエチレンジアミン四
酢酸二水素二ナトリウム溶液)に準じる。ただし,エチレンジアミン四酢酸二水素二ナトリウム二水
和物7.5gを用いる。
j)
エリオクロムブラックT溶液 調製方法及び保存方法は,JIS K 8001の4.4(表8)による。
4. 器具 器具は,次による。
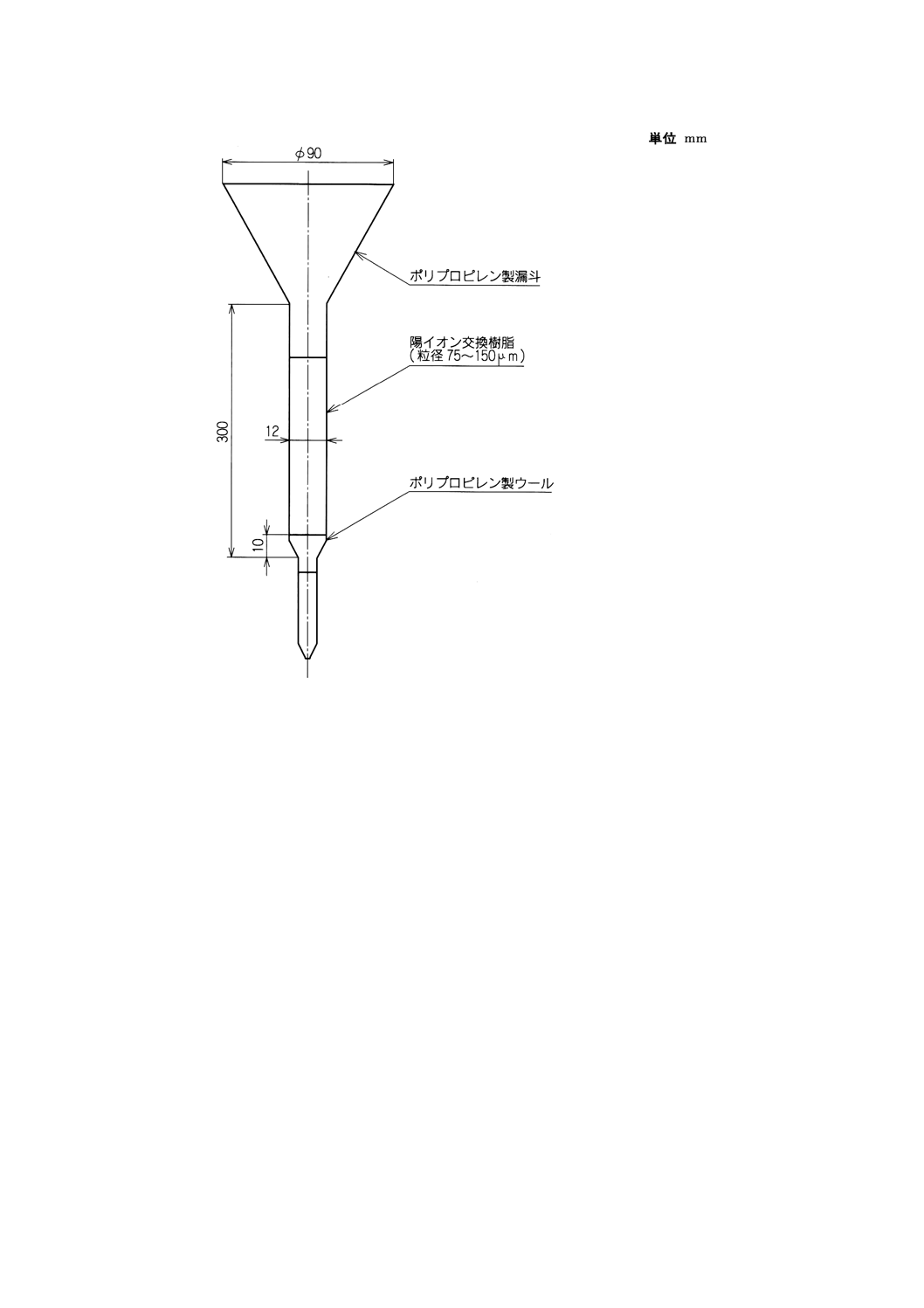
a) 陽イオン交換樹脂カラム 附属書付図1に示すものを標準とする。
上端にプラスチック漏斗を取り付け,下端を細く引き伸ばしたプラスチック管(長さ200mm,内径
12mm)に水でほぐしたプラスチックウールを厚さ約10mmに詰め,水で膨潤させた強酸性陽イオン
交換樹脂(8%DVB, 75〜150μm)約18mlをスラリー状にして流し入れ,沈殿させる。ウールの詰め方
を加減するなどして流量を毎分1.0〜1.5mlになるように調節した後,塩酸(1+2)120ml,水70mlを順
次流しておく。
5. 試料はかり取り量 試料のはかり取り量は,0.50gとする。
6. 操作 定量操作は,次の手順によって行う。
a) 試料を白金皿(例えば,75番)にはかり取り,炭酸ナトリウム(無水)3.0g及びほう酸2.0gを加えて,
始めは低温で加熱し(2),次第に温度を上げ,最後は電気炉中で1 100±25℃で加熱し,未分解物が認め
られなくなるまで強熱して融解する(3)。時計皿でふたをして放冷後,硝酸(1+4)45ml及び硫酸(1+
9)10mlを加え,ときどきかき混ぜながら水浴上で加熱溶解する。放冷後,少量の水で時計皿を水洗し
て取り除き,得られた溶液を500mlの全量フラスコに移し入れ,水で標線まで薄め試料溶液とする。
注(2) 急激に加熱すると,ほう酸の脱水のために試料が飛散するおそれがある。
(3) 融解時間が長すぎると,融成物が硫酸及び硝酸に溶けにくくなる。
b) 陽イオン交換樹脂カラムの下にプラスチック製ビーカー (1 000ml) を受け,試料溶液から100mlを正
確に分取し,陽イオン交換樹脂カラムに流す(4)。硝酸(1+20)10mlずつを2回漏斗の内壁を洗浄しなが
らカラムに流し,更に硝酸(1+20)60mlを流す。ふっ化水素酸(1+150)10mlずつを2回漏斗の内壁を洗
浄しながらカラムに流し,更にふっ化水素酸(1+150)80mlを流す。続いて,ふっ化水素酸(1+19)10ml
ずつを2回漏斗の内壁を洗浄しながらカラムに流し,更にふっ化水素酸(1+19)80mlを流す。次に,水
10mlずつを2回漏斗の内壁を洗浄しながらカラムに流し,更に水80mlを流す。ここまでの流出液は,
不要である。
注(4) この溶液がカラムの先端から滴下しなくなってから,次の溶液を流す。以下同様。
c) 白金皿(例えば,150番)(5)を受け,塩酸(1+1)10mlずつを2回漏斗の内壁を洗浄しながらカラムに
流し,更に80mlを流してカルシウム及びマグネシウムを溶出させる。
備考 使用後のカラムに水70mlを流すと,カラムは再生する。
注(5) 四ふっ化エチレン樹脂製ビーカー(例えば,200ml)を用いてもよい。
d) c)の溶出液に硫酸(1+9)10mlを加え,熱板上で注意しながら加熱蒸発し,硫酸白煙を発生させる。冷
却後,塩酸(1+1)5ml及び水30mlを加えて加熱溶解する。放冷後,100mlの全量フラスコに移し,水
で標線まで薄める(6)。
注(6) 酸化マグネシウム含有率と酸化カルシウム含有率の合量が30mass%以下の場合,100mlの全量フ
ラスコに移さないで,直接e)のビーカー (300ml) に移す。

25
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
e) d)で得た試料溶液から正しく一定量(7)をビーカー (300ml) に取り,水で約100mlに薄め,塩化ヒドロ
キシルアンモニウム溶液5ml,緩衝液 (pH 10) 20ml及び指示薬としてエリオクロムブラックT溶液3,
4滴を加え,かき混ぜながら0.02mol/l EDTA溶液で滴定する。終点付近になったら,よくかき混ぜな
がらゆっくり滴定し,溶液が赤紫色から青色に変わる点を終点とする(8)。
注(7) d)で得た試料溶液の分取量は,試料中の酸化マグネシウムと酸化カルシウムの含有率の合量に
応じて附属書表1による。
附属書表1 試料溶液の分取量
酸化マグネシウムと酸化カルシウ
ムの含有率の合量
mass%
分取量
ml
45未満
60
45以上 60未満
50
60以上 75未満
40
75以上
30
(8) タングステンランプの光を透過させた乳白色のガラス又はプラスチック板の上で滴定を行うと,
終点の判定が容易である。
7. 空試験 試料を用いないで6.の操作を行う。ただし,融解操作は,省略する。
8. 計算 試料中の酸化マグネシウムの含有率は,5.から7.の測定値と本体14.で求めた酸化カルシウムの
含有率とから次の式によって算出する。
(
)
719
.0
100
500
1
806
000
.0
3
2
1
×
×
×
×
×
CaO
V
m
F
V
V
MgO
−
−
=
ここに,
MgO: 酸化マグネシウムの含有率 (mass%)
V1: 6.e)の0.02mol/l EDTA溶液の使用量 (ml)
V2: 7.の0.02mol/l EDTA溶液の使用量 (ml)
V3: 6.e)の試料溶液の分取量 (ml)
[ただし,6.d)で試料溶液を全量用いた場合には,100mlと
する。]
F: 0.02mol/l EDTA溶液のファクター
m: 6.a)の試料のはかり取り量 (g)
CaO: 本体14.で求めた酸化カルシウムの含有率 (mass%)
26
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書付図1 陽イオン交換樹脂カラム
27
R 2014 : 1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS R 2014制定原案作成委員会 構成表
氏名
所属
(委員長)
中 川 善兵衛
秋田大学鉱山学部
(委員)
○ 村 田 守
鳴門教育大学自然系
(分科会主査)
○ 三 橋 久
岡山セラミックス技術振興財団
(委員)
福 水 健 文
通商産業省生活産業局
大 嶋 清 治
工業技術院標準部材料規格課
黒 木 勝 也
財団法人日本規格協会技術部
○ 藤 貫 正
日本磁気共鳴医学会
○ 多 田 格 三
(元)株式会社東芝
荒 木 慎 介
耐火物協会
沢 田 正 志
秩父小野田株式会社生産部
小 松 英 雄
旭硝子株式会社セラミックス事業部
石 井 章 生
新日本製鐵株式会社設備技術センター機械技術部
磯 村 敬一郎
川崎製鉄株式会社技術研究所
長 岡 博
NKK京浜製鉄所製鋼部炉材室
(分科会幹事)
○ 朝 倉 秀 夫
品川白煉瓦株式会社技術研究所
(委員)
宮 川 三 郎
川崎炉材株式会社管理部
○ 久保田 裕
黒崎窯業株式会社技術研究所
沓 掛 行 徳
旭硝子株式会社高砂工場
○ 鹿 野 弘
黒崎窯業株式会社東京支社
○ 下 司 誠
ハリマセラミック株式会社研究開発部
渡 辺 高
東芝セラミックス株式会社
● 森 邦 夫
旭硝子株式会社高砂工場
● 福 井 洋 一
ハリマセラミック株式会社技術開発部
● 鬼 塚 浩 次
大光炉材株式会社技術研究所
● 松 岡 文 子
黒崎窯業株式会社測定評価センター
● 池 上 克 重
品川白煉瓦株式会社技術研究所
● 吉 田 清 志
川崎炉材株式会社技術研究所
● 藤 原 昇
株式会社ヨータイ技術研究所
● 宮 脇 正 夫
日本特殊炉材株式会社技術部
● 鈴 木 俊 宏
東芝セラミックス株式会社刈谷製造所
● 戸 松 一 郎
株式会社TYK多治見工場研究所
● 河 野 久 征
理学電気工業株式会社
● 中 山 信 司
東芝モノフラックス株式会社神崎工場
(事務局)
細 川 周 明
耐火物技術協会
備考 ○印は分科会委員併任。●印は分科会委員専任。
文責:JIS R 2014(アルミナ−マグネシア質耐火物の化学分析方法)
制定原案作成委員会分析分科会