Q 13485:2018 (ISO 13485:2016)
(1)
目 次
ページ
0 序文······························································································································· 1
0.1 一般 ···························································································································· 1
0.2 概念の明確化 ················································································································ 2
0.3 プロセスアプローチ ······································································································· 3
0.4 JIS Q 9001との関係········································································································ 3
0.5 他のマネジメントシステムとの両立性················································································ 3
1 適用範囲························································································································· 3
2 引用規格························································································································· 4
3 用語及び定義 ··················································································································· 4
4 品質マネジメントシステム ································································································· 8
4.1 一般要求事項 ················································································································ 8
4.2 文書化に関する要求事項 ································································································· 9
5 経営者の責任 ·················································································································· 11
5.1 経営者のコミットメント ································································································ 11
5.2 顧客重視 ····················································································································· 11
5.3 品質方針 ····················································································································· 11
5.4 計画 ··························································································································· 11
5.5 責任,権限及びコミュニケーション·················································································· 11
5.6 マネジメントレビュー ··································································································· 12
6 資源の運用管理 ··············································································································· 13
6.1 資源の提供 ·················································································································· 13
6.2 人的資源 ····················································································································· 13
6.3 インフラストラクチャ ··································································································· 13
6.4 作業環境及び汚染管理 ··································································································· 13
7 製品実現························································································································ 14
7.1 製品実現の計画 ············································································································ 14
7.2 顧客関連のプロセス ······································································································ 14
7.3 設計・開発 ·················································································································· 15
7.4 購買 ··························································································································· 17
7.5 製造及びサービスの提供 ································································································ 18
7.6 監視機器及び測定機器の管理 ·························································································· 21
8 測定,分析及び改善 ········································································································· 21
8.1 一般 ··························································································································· 21
8.2 監視及び測定 ··············································································································· 21
8.3 不適合製品の管理 ········································································································· 23
Q 13485:2018 (ISO 13485:2016) 目次
(2)
ページ
8.4 データの分析 ··············································································································· 24
8.5 改善 ··························································································································· 24
附属書A(参考)JIS Q 13485:2005とJIS Q 13485:2018との内容の比較 ········································· 26
附属書B(参考)JIS Q 13485:2018とJIS Q 9001:2015との関係 ···················································· 30
参考文献 ···························································································································· 36
Q 13485:2018 (ISO 13485:2016)
(3)
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,一般社団法人日本
医療機器産業連合会(JFMDA)から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,
日本工業標準調査会の審議を経て,厚生労働大臣及び経済産業大臣が改正した日本工業規格である。これ
によって,JIS Q 13485:2005は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。厚生労働大臣,経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の
特許出願及び実用新案権に関わる確認について,責任はもたない。
日本工業規格 JIS
Q 13485:2018
(ISO 13485:2016)
医療機器−品質マネジメントシステム−
規制目的のための要求事項
Medical devices-Quality management systems-
Requirements for regulatory purposes
0
序文
0.1
一般
この規格は,2016年に第3版として発行されたISO 13485を基に,技術的内容及び構成を変更すること
なく作成した日本工業規格である。
なお,この規格で点線の下線を施してある参考事項は,対応国際規格にはない事項である。
附属書AにJIS Q 13485:2005とJIS Q 13485:2018との内容の比較を示す。
この規格は,医療機器の設計・開発,製造,保管及び流通,据付け,附帯サービス,最終的な廃棄・処
分,並びに関連する活動(例 技術支援)の設計・開発及び提供を含む医療機器のライフサイクルの一つ
以上の段階に関係する組織が使うことができる品質マネジメントシステムの要求事項を規定する。製品
(例 原料,部品,組立品,医療機器,滅菌サービス,校正サービス,流通サービス,保守サービス)を
そのような組織に提供する供給者又は外部パーティも,この規格の要求事項を使用することができる。供
給者又は外部パーティは,この規格の要求事項を満たすことを自主的に選択できるし,又は契約によって
満たすことを要求される場合がある。
幾つかの法的管轄においては,医療機器のサプライチェーンの中の様々な役割の組織に対して,品質マ
ネジメントシステムの適用に対する規制要求事項がある。このため,この規格は,組織に対して次を期待
している。
− 適用される規制要求事項におけるその役割を明確にする。
− この役割における当該活動に適用される規制要求事項を明確にする。
− 適用される規制要求事項を,その品質マネジメントシステム内に含める。
適用される規制要求事項における定義は国によって,また,地域によって異なる。組織は,医療機器が
使用可能とされている法的管轄における規制の定義に照らして,この規格の用語がどのように訳されてい
るか理解する必要がある。
さらに,この規格は,品質マネジメントシステム及び組織の要求事項に適用される顧客要求事項及び適
用される規制要求事項を満たす組織の能力を,組織自身が内部で評価するためにも,また,審査登録機関
を含む外部パーティが評価するためにも使用することができる。この規格が規定する品質マネジメントシ
ステムの要求事項は,安全及び性能に対する顧客要求事項並びに適用される規制要求事項への合致に必要
な製品に対する技術的要求事項を補完するものであることを強調しておく。
品質マネジメントシステムを採用することは,組織の戦略的な決定である。組織の品質マネジメントシ
2
Q 13485:2018 (ISO 13485:2016)
ステムの設計及び実施は,次によって影響を受ける。
a) 組織的環境,環境の変化,及び組織的環境が医療機器の適合性に与える影響
b) 組織のニーズの変化
c) 組織固有の目的
d) 組織が提供する製品
e) 組織が採用するプロセス
f)
組織の大きさ及び構造
g) 組織の活動に適用される規制要求事項
この規格は,品質マネジメントシステムの構造の均一化,文書の画一化又はこの規格の箇条の構造に文
書化の構造を合わせることを意図していない。
医療機器には,様々な種類があり,この規格の特定の一部の要求事項は,特定の医療機器のグループに
だけ適用される。これらの医療機器のグループは,箇条3に定義している。
0.2
概念の明確化
この規格の次の用語及びフレーズは,次に記載する意味で用いられる。
− 要求事項が“適切な場合”という用語で特定された場合,組織が他の方法によることの正当性を示す
ことができなければ,その要求事項の適用は“適切”であるとみなされる。次のために必要であるな
らば,その要求事項は“適切”であると考えられる。
− 製品が規定要求事項を満たす。
− 適用される規制要求事項に適合する。
− 組織が是正処置を実行する。
− 組織がリスクを管理する。
− “リスク”という用語が用いられた場合,この規格の適用範囲内におけるこの用語の利用は,医療機
器の安全上及び性能上の要求事項,又は適用される規制要求事項への適合に関連する。
− 要求事項が,“文書化する”という用語で要求された場合,確立し,実施し,維持することが要求され
る。
− “製品”という用語が用いられた場合,それは“サービス”も意味する。“製品”は,意図するアウト
プット,顧客から要求されたアウトプット,又は製品実現プロセスから得られた意図するアウトプッ
トへ適用する。
− “規制要求事項”という用語が用いられた場合,それは,この規格の利用者に適用される全ての法律
上の要求事項を包含している(例えば,法律,規則,条例,指令)。“規制要求事項”という用語の利
用は,品質マネジメントシステム及び医療機器の安全又は性能に関する要求事項に限定される。
この規格では,次のような表現形式を用いている。
− “〜する”(shall)は,要求事項を示す。
− “〜するとよい”(should)は,推奨を示す。
− “〜してもよい”(may)は,許容を示す。
− “〜することができる,〜できる”(can)は,可能性又は実現能力を示す。
“注記”と記載されている情報は,その関連する要求事項を理解するための,又は明確にするための手
引である。
3
Q 13485:2018 (ISO 13485:2016)
0.3
プロセスアプローチ
この規格は,品質マネジメントに対するプロセスアプローチに基づいている。インプットを受け,それ
らをアウトプットに変換する活動は,プロセスとみなすことができる。一つのプロセスからのアウトプッ
トは,多くの場合,次のプロセスの直接のインプットとなる。
効果的に機能するために,組織は,数多くの関連し合うプロセスを明確にし,管理する必要がある。組
織内において,所望の結果をもたらすために,プロセスを明確にし,その相互関係を把握し,運営管理す
ることと併せて,一連のプロセスをシステムとして適用することを,“プロセスアプローチ”という。
品質マネジメントシステム内において,このようなアプローチを用いる場合,そのアプローチは次の重
要性を強調する。
a) 要求事項の理解及び適合
b) 付加価値の観点から,プロセスを考慮すること。
c) プロセスパフォーマンス及び有効性の結果を得ること。
d) 客観的測定に基づく,プロセスの改善
0.4
JIS Q 9001との関係
この規格は独立した規格であるが,既にJIS Q 9001:2015に置き換わっているJIS Q 9001:2008に基づい
ている。ユーザの便宜のため,附属書Bに,この規格とJIS Q 9001:2015との対応を示す。
この規格は,医療機器のライフサイクルの一つ以上の段階に関与する組織に適用される品質マネジメン
トシステムのための適切な規制要求事項の世界的な整合を容易にすることを意図している。この規格は,
医療機器のライフサイクルに関与する組織のための幾つかの特別な要求事項を含んでおり,規制要件とし
て適切でない,JIS Q 9001の要求事項の一部を除外している。このような除外があるため,組織の品質マ
ネジメントシステムがこの規格に適合していても,除外したJIS Q 9001の要求事項を満たしていなければ,
組織はJIS Q 9001への適合を主張することはできない。
0.5
他のマネジメントシステムとの両立性
この規格には,環境マネジメントシステム,労働安全衛生マネジメントシステム,財務マネジメントシ
ステムなど他のマネジンメントシステムに固有な要求事項は含まれていない。しかし,この規格は,組織
が品質マネジメントシステムを,関連するマネジメントシステム要求事項に合わせたり,統合したりでき
るようになっている。組織がこの規格の要求事項に適合した品質マネジメントシステムを構築するに当た
って,既存のマネジメントシステムを適応させることも可能である。
1
適用範囲
この規格は,組織が顧客要求事項及び適用される規制要求事項を一貫して満たす医療機器及び関連する
サービスを提供する能力を実証する必要がある場合の品質マネジメントシステムの要求事項について規定
する。そのような組織は,医療機器の設計・開発,製造,保管及び流通,据付け,附帯サービス,並びに
関連する活動(例 技術支援)の設計・開発及び提供を含む医療機器のライフサイクルの一つ以上の段階
の活動に関わることができる。
さらに,この規格は,品質マネジメントシステムに関連するサービスを含み,製品をそのような医療機
器組織に提供する供給者又は外部パーティも使用することができる。
この規格の要求事項は,その組織の規模を問わず適用でき,また,明確に規定している場合を除き,そ
の組織の形態を問わず適用できる。要求事項が医療機器に適用するとしている場合でも,その要求事項は,
組織が提供する関連するサービスに対して同様に適用される。
4
Q 13485:2018 (ISO 13485:2016)
この規格が要求するプロセスで,その組織に適用できるが,組織が実行していないプロセスについては,
その組織に責任があり,それらのプロセスは,監視,維持及びプロセス管理によって組織の品質マネジメ
ントシステム内で明らかにする必要がある。
適用される規制要求事項が設計・開発の管理を除外してよいとしている場合には,品質マネジメントシ
ステムからそれらを除外することを正当化するために使用することができる。そのような規制要求事項は,
品質マネジメントシステムで対応する別のアプローチを規定していることもある。設計・開発の管理を除
外している場合,この規格への適合宣言の中にそのことを確実に反映させることは,組織の責任である。
組織で実行する活動又はその品質マネジメントシステムを適用する医療機器の性質のため,この規格の
箇条6〜箇条8の要求事項のいずれかが適用できない場合,組織は自己の品質マネジメントシステムに,
それらの要求事項を含める必要はない。適用できないと判断する全ての箇条について,組織は4.2.2に規
定しているようにその正当化の理由を記録する。
注記1 2019年2月28日までJIS Q 13485:2005を適用することができる。
注記2 この規格の対応国際規格及びその対応の程度を表す記号を,次に示す。
ISO 13485:2016,Medical devices−Quality management systems−Requirements for regulatory
purposes(IDT)
なお,対応の程度を表す記号“IDT”は,ISO/IEC Guide 21-1に基づき,“一致している”
ことを示す。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。この引用
規格は,記載の年の版を適用し,その後の改正版(追補を含む。)は適用しない。
JIS Q 9000:2015 品質マネジメントシステム−基本及び用語
注記 対応国際規格:ISO 9000:2015,Quality management systems−Fundamentals and vocabulary
3
用語及び定義
この規格で用いる主な用語及び定義は,JIS Q 9000:2015によるほか,次による。
3.1
通知書(advisory notice)
医療機器を引き渡した後に,組織によって発行される通知であって,補足的情報を提供し,又は次に対
し,とるべき処置を助言するもの。
− 医療機器の使用
− 医療機器の改造
− その医療機器を供給した組織への返却
− 医療機器の破壊
注記 通知書の発行は,国又は地域の規制要求事項に適合させるために要求される場合がある。
3.2
指定代理人(authorized representative)
ある国又は法的管轄の法規制の下で製造業者の義務に関して規定された業務を製造業者の代わりに行う
よう製造業者から文書で委任を受けた,その国又は法的管轄に設置されたあらゆる自然人又は法人
(GHTF/SG1/N055:2009の5.2参照)。
5
Q 13485:2018 (ISO 13485:2016)
3.3
臨床評価(clinical evaluation)
製造業者の意図に従って使用したとき,医療機器の臨床的安全性及び性能を検証するための,医療機器
に関する臨床データの評価及び分析(GHTF/SG5/N4:2010の箇条4参照)。
3.4
苦情(complaint)
組織の管理下からリリースされた医療機器の同一性,品質,耐久性,信頼性,ユーザビリティ,安全性
若しくは性能,又は医療機器の性能に影響を及ぼすサービスに関連した不具合を申し立てるための文書,
電子媒体又は口頭によるコミュニケーション。
注記 この定義は,JIS Q 9000:2015の定義とは異なる。
3.5
ディストリビュータ(distributor)
製造業者に代わって,最終使用者に医療機器を利用できるようにするサプライチェーン内の自然人又は
法人(GHTF/SG1/N055:2009の5.3参照)。
注記1 一つ以上のディストリビュータがサプライチェーンに関与することがある。
注記2 サプライチェーンの活動で,製造業者,輸入業者及びディストリビュータに代わって,保管
及び輸送のような活動を行う者は,この定義の下ではディストリビュータではない。
3.6
埋込み医療機器(implantable medical device)
内科的又は外科的介入によってだけ除去可能な医療機器であり,次のいずれかを意図する医療機器。
− 医療機器の全体又は一部分を人体内又は体表開口部に挿入し,処置後少なくとも30日間留置させる機
器
− 皮膚表面又は眼の表面を代替させ,処置後少なくとも30日間留置させる機器
注記 この定義は,能動埋込み医療機器を含んでいる。
3.7
輸入業者(importer)
別の国又は法的管轄で製造された医療機器を,その医療機器が上市される国又は法的管轄で利用できる
医療機器とするサプライチェーン内の一番初めの自然人又は法人(GHTF/SG1/N055:2009の5.4参照)。
3.8
ラベリング(labelling)
出荷書類を除く,医療機器の識別子,技術情報,使用目的及び適正使用に関わるラベル,取扱説明書又
はそれ以外の情報(GHTF/SG1/N70:2011の箇条4参照)。
3.9
ライフサイクル(life-cycle)
医療機器の寿命の全ての段階であって,最初の構想から最後の使用停止及び廃棄までの段階(JIS T
14971:2012の2.7参照)。
3.10
製造業者(manufacturer)
医療機器の設計及び/又は製造が自分自身によるか,又は他の人による行為かにかかわらず,その名の
下に,使用に供するために医療機器を作ることを意図し,医療機器の設計及び/又は製造に責任をもつ自
6
Q 13485:2018 (ISO 13485:2016)
然人又は法人(GHTF/SG1/N055:2009の5.1参照)。
注記1 法的管轄で規制当局によって他の人に特別に責任を負わす場合を除き,利用可能とする又は
販売することを意図した国又は法的管轄において,適用される全ての医療機器の規制要求事
項に適合させる最終的な法的責任をもつ自然人又は法人。
注記2 製造業者の責任は,この定義の出典とは異なる他のGHTF指針文書に記載されている。これ
らの責任には,市販前要求事項並びに有害事象報告及び是正措置の通知のような市販後要求
事項の両方に適合することを含んでいる。
注記3 上記の定義が示すとおり,“設計及び/又は製造”は,仕様開発,生産,成型加工,組立,加
工,包装,再包装,ラベリング,ラベル変更,滅菌,据付け又は医療機器の再製造,及び医
療目的のために利用可能な他の製品及び医療機器を一緒に収集してまとめることを含む。
注記4 取扱説明書に従って,個々の患者に対して他の人が既に供給した医療機器を組み立てる人又
は適応する人は,製造業者ではない。ただし,指定された組立及び適応は,医療機器の意図
する用途を変更しないことが前提である。
注記5 医療機器の元々の製造業者の代理としてではなく,医療機器の意図する用途を変更する人,
医療機器を改造する人,又は自身の名の下に利用できるようにする人は,変更した医療機器
の製造業者とみなされる。
注記6 既存のラベルを覆ったり,変更することなく,医療機器又はその包装に自身の所在地及び連
絡先だけを表示する指定代理人,ディストリビュータ及び輸入業者は,製造業者とはみなさ
ない。
注記7 附属品は医療機器の規制要求事項の範囲になるため,その附属品の設計及び/又は製造に関
して責任をもつ者は,製造業者とみなす。
3.11
医療機器(medical device)
計器,器械,用具,機械,器具,埋込み用具,体外診断薬,ソフトウェア,材料又はその他の同類のも
の若しくは関連する物質であって,単独使用又は組合せ使用かを問わず,製造業者が人体への使用を意図
し,その使用目的が次の一つ以上であるもの。
− 疾病の診断,予防,監視,治療又は緩和
− 負傷の診断,監視,治療,緩和又は補助
− 解剖学的又は生理学的なプロセスの検査,代替,修復又は支援
− 生命支援又は維持
− 受胎調整
− 医療機器の消毒
− 人体から採取される標本の体外試験法による情報提供
さらに,薬学,免疫学又は新陳代謝の手段によって,体内又は体表において意図するその主機能を達成
することはないものである。しかし,それらの手段によって意図する機能の実現が補助されてもよい
(GHTF/SG1/N071:2012の5.1参照)。
注記 法的管轄によって医療機器に該当するか否かが分かれる製品には,次のものがある。
− 消毒剤
− 身体障害者用の補助器具
− 動物及び/又はヒト組織を伴う機器
7
Q 13485:2018 (ISO 13485:2016)
− 体外受精又は生殖補助技術用の機器
3.12
医療機器ファミリ(medical device family)
同一の組織によって又は同一の組織のために製造され,安全,意図する用途及び機能に関して同一の基
本的設計及び性能的特徴をもつ一群の医療機器。
3.13
性能評価(performance evaluation)
体外診断用医療機器を,その意図する用途を達成する能力を確立又は検証するためにデータを分析し,
評価すること。
3.14
市販後監視(post-market surveillance)
上市後の段階で,医療機器から得られた経験を収集し,分析する体系的なプロセス。
3.15
製品(product)
プロセスの結果(JIS Q 9000:2006の3.4.2を一部変更)。
注記1 製品には,次の4種の包括的なカテゴリがある。
− サービス(例 輸送)
− ソフトウェア(例 コンピュータプログラム,辞書)
− ハードウェア(例 エンジンの機械部品)
− 加工した材料(例 潤滑剤)
多くの製品は異なる包括的な製品カテゴリと関連した要素で構成される。製品がサービス,
ソフトウェア,ハードウェア又は加工した材料のいずれで呼ばれるかは主要な構成要素によ
る。例えば,提供された製品“自動車”は,ハードウェア(例 タイヤ),加工した材料(例
燃料,冷却剤),ソフトウェア(例 エンジンコントロールソフトウェア,運転者マニュアル)
及びサービス(例 セールスマンが提供する説明)から成る。
注記2 サービスとは,少なくとも供給者と顧客とを結び付けるために不可欠な活動の結果であり,
一般的には無形物である。サービスの提供には,例えば次が含まれる。
− 顧客に提供した有形の製品に対して行われる活動(例 自動車修理)
− 顧客に提供した無形の製品に対して行われる活動(例 税金の返還のために準備する損
益計算書)
− 無形の製品の配送(例 知識を伝達することを目的とした情報の配送)
− 顧客のための雰囲気作り(例 ホテル,レストラン)
ソフトウェアは,情報で構成され,一般に無形であり,アプローチ,処理又は手順の形を
取る場合がある。
ハードウェアは,一般に有形で,その量は数えることができる特性である。加工した材料
は,一般に有形で,その量は連続的な特性である。ハードウェア及び加工した材料は,品物
ということが多い。
3.16
購買製品(purchased product)
組織の品質マネジメントシステムの外のパーティによって提供される製品。
8
Q 13485:2018 (ISO 13485:2016)
注記 製品の提供は,必ずしも商業的又は財政的な処理を必要としない。
3.17
リスク(risk)
危害の発生確率とその危害の重大さとの組合せ(JIS T 14971:2012の2.16参照)。
注記 この定義はJIS Q 9000:2015の定義とは異なる。
3.18
リスクマネジメント(risk management)
リスクの分析,評価,コントロール及び監視に対して,管理方針,手順及び実施を体系的に適用するこ
と(JIS T 14971:2012の2.22参照)。
3.19
無菌バリアシステム(sterile barrier system)
微生物の侵入を防止し,かつ,使用時点での製品の無菌提供を可能にする最低限の包装(JIS T
0841-1:2009の3.22参照)。
3.20
滅菌医療機器(sterile medical device)
滅菌に対する要求事項を満たすことを意図した医療機器。
注記 医療機器の滅菌に対する要求事項が,適用される規制要求事項又は規格に規定されている場合
がある。
4
品質マネジメントシステム
4.1
一般要求事項
4.1.1
組織は,この規格の要求事項及び適用される規制要求事項に従って,品質マネジメントシステムを
文書化する。また,品質マネジメントシステムの有効性を維持する。
組織は,この規格又は適用される規制要求事項で文書化することを要求している全ての要求事項,手順,
活動又は取決めを,確立し,実施し,維持する。
組織は,適用される規制要求事項に従って,組織が引き受けている役割を文書化する。
注記 組織が引き受けている役割の例には,製造業者,指定代理人,輸入業者,ディストリビュータ
がある。
4.1.2
組織は,次を実施する。
a) 組織の役割を考慮し,品質マネジメントシステムに必要なプロセス及びそれらの組織への適用を明確
にする。
b) 品質マネジメントシステムのために必要とする適切なプロセスの管理においては,リスクに基づくア
プローチを適用する。
c) これらのプロセスの順序及び相互関係を明確にする。
4.1.3
それぞれの品質マネジメントシステムプロセスにおいて,組織は次を行う。
a) これらのプロセスの運用及び管理のいずれもが効果的であることを確実にするために必要な判断基準
及び方法を定める。
b) これらのプロセスの運用及び監視の支援をするために必要な資源及び情報を利用できることを確実に
する。
c) これらのプロセスについて,計画どおりの結果が得られるように,かつ,それらのプロセスの有効性
9
Q 13485:2018 (ISO 13485:2016)
を維持するために必要な処置をとる。
d) これらのプロセスを監視し,適切な場合,測定し,分析する。
e) この規格への適合及び適用される規制要求事項への適合を立証するために,必要な記録を確立し,維
持する(4.2.5参照)。
4.1.4
組織は,これらのプロセスを,この規格の要求事項及び適用される規制要求事項に従って運営管理
する。これらのプロセスを変更する場合は,次による。
a) 品質マネジメントシステムへの影響度を評価する。
b) この品質マネジメントシステムで製造する医療機器への影響度を評価する。
c) この規格及び適用される規制要求事項に従って管理する。
4.1.5
要求事項に対する製品の適合性に影響を与えるプロセスをアウトソースすることを組織が決めた
場合には,組織はアウトソースしたプロセスを監視し,その管理を確実にする。組織は,アウトソースし
たプロセスに関して,この規格並びに顧客の要求事項及び適用される規制要求事項への適合に対する責任
をもつ。管理の程度は,7.4による要求事項を満たすために,関連するリスク及び外部パーティの能力に見
合ったものとする。管理する事項には,文書化した品質上の合意を含む。
4.1.6
組織は,品質マネジメントシステムで使用するコンピュータソフトウェアの適用のバリデーション
の手順を文書化する。このようなソフトウェアの適用は,初回の使用前にバリデーションを行う。また,
適切な場合,そのソフトウェア又は適用への変更後に,バリデーションを行う。
ソフトウェアのバリデーション及び再バリデーションに関する固有のアプローチ及び活動は,ソフトウ
ェアの使用に伴うリスクに見合ったものとする。
この活動の記録(4.2.5参照)は,維持する。
4.2
文書化に関する要求事項
4.2.1
一般
品質マネジメントシステムの文書(4.2.4参照)には,次を含める。
a) 文書化した,品質方針及び品質目標の表明
b) 品質マニュアル
c) この規格が要求する文書化した手順及び記録
d) プロセスの効果的な計画,運用及び管理を確実にするために必要であると組織が決めた記録を含む文
書
e) 適用される規制要求事項によって規定された他の文書
4.2.2
品質マニュアル
組織は,次を含む品質マニュアルを文書化する。
a) 品質マネジメントシステムの適用範囲。除外及び/又は不適用がある場合には,その詳細及びそれが
正当であるとする理由
b) 品質マネジメントシステムについて文書化された手順又はそれらを参照できる情報
c) 品質マネジメントシステムのプロセス間の相互関係に関する記述
品質マニュアルには,品質マネジメントシステムで使用している文書体系の概要を記載する。
4.2.3
医療機器ファイル
組織は,それぞれの医療機器の型式又は医療機器ファミリに対して,この規格及び適用される規制要求
事項への適合を立証するために作成した文書を含むか又は参照する一つ以上のファイルを確立し,維持す
る。
10
Q 13485:2018 (ISO 13485:2016)
このファイルの内容には次を含むが,これに限らない。
a) 医療機器の一般的記述,意図する用途・目的及び全ての使用説明を含むラベリング
b) 製品仕様
c) 製造,保管,取扱い及び配送の仕様又は手順
d) 測定及び監視手順
e) 適切な場合,据付けに対する要求事項
f)
適切な場合,サービス手順に対する要求事項
4.2.4
文書管理
品質マネジメントシステムで必要とする文書は管理する。ただし,記録は文書の一種ではあるが,4.2.5
に規定する要求事項に従って管理する。
文書化した手順は,次の活動に必要な管理を規定する。
a) 発行前に,適切かどうかの観点から文書をレビューし,承認する。
b) 文書をレビューする。また,必要に応じて更新し,再承認する。
c) 文書の現在の改訂版の識別及び変更の識別を確実にする。
d) 該当する文書の適切な版が,必要なときに,必要なところで使用可能な状態にあることを確実にする。
e) 文書が読みやすく,容易に識別可能な状態であることを確実にする。
f)
外部で作成され,組織が品質マネジメントシステムの計画及び運営に必要と判断した文書を明確にし,
その配付の管理を確実にする。
g) 文書の劣化又は紛失を防ぐ。
h) 廃止文書が誤って使用されないようにする。また,廃止文書に適切な識別をする。
組織は,その決定の基礎となる関連する背景情報を入手できる立場にある,最初に承認した部署又はそ
の他の指名した部署が,文書の変更をレビューし,承認することを確実にする。
組織は,廃止した管理文書の少なくとも一部を保管しておく期間を定める。この期間は,その医療機器
の製造及び検査に使用した文書が,少なくとも組織が定めたその医療機器の寿命の期間は入手できること
を確実にする。ただし,この期間は,結果として得られる全ての記録(4.2.5参照)の保管期間又は適用さ
れる規制要求事項によって定められた期間より短くしない。
4.2.5
記録の管理
要求事項への適合及び品質マネジメントシステムの効果的運用の証拠を示すために,記録を作成し,維
持する。
組織は,記録の識別,保管,セキュリティ及び完全性の維持,検索,並びに保管期間及び廃棄に関して
必要な管理を規定するために,手順を文書化する。
組織は,適用される規制要求事項に従い,記録に含まれる機密健康情報を保護するための方法を規定し,
実施する。
記録は,読みやすく,容易に識別可能で,検索可能とする。記録の変更は,識別可能とする。
組織は,少なくとも自ら定めたその医療機器の寿命に相当する期間,又は適用される規制要求事項で規
定された期間,記録を保管する。ただし,この期間は,組織が医療機器をリリースしてから2年間より短
くしない。
11
Q 13485:2018 (ISO 13485:2016)
5
経営者の責任
5.1
経営者のコミットメント
トップマネジメントは,品質マネジメントシステムの構築及び実施,並びにその有効性の維持に対する
コミットメントの証拠を次によって示す。
a) 適用される規制要求事項を満たすことは当然のこととして,顧客要求事項を満たすことの重要性を組
織内に周知する。
b) 品質方針を設定する。
c) 品質目標が設定されることを確実にする。
d) マネジメントレビューを実施する。
e) 資源が使用できることを確実にする。
5.2
顧客重視
トップマネジメントは,顧客要求事項及び適用される規制要求事項を決定し,満たしていることを確実
にする。
5.3
品質方針
トップマネジメントは,品質方針について次を確実にする。
a) 組織の目的に適用できる。
b) 品質マネジメントシステムの要求事項への適合及び品質マネジメントシステムの有効性の維持に対す
るコミットメントを含む。
c) 品質目標の設定及びレビューのための枠組みを与える。
d) 組織全体に伝達され,理解される。
e) 適切性の維持のためにレビューする。
5.4
計画
5.4.1
品質目標
トップマネジメントは,組織内のそれぞれの部門及び階層で,適用される規制要求事項及び製品要求事
項を満たすために必要なものを含む品質目標を設定していることを確実にする。品質目標は,その達成度
が判定可能で,品質方針との整合がとれている。
5.4.2
品質マネジメントシステムの計画
トップマネジメントは,次を確実にする。
a) 品質目標に加えて4.1に規定する要求事項を満たすために,品質マネジメントシステムの計画を実施
する。
b) 品質マネジメントシステムの変更を計画し,実施する場合には,品質マネジメントシステムが“完全
に整っている状態(integrity)”を維持する。
5.5
責任,権限及びコミュニケーション
5.5.1
責任及び権限
トップマネジメントは,責任及び権限を定め,文書化し,組織全体に周知していることを確実にする。
トップマネジメントは,品質に影響を与える業務を運営管理し,実施し,検証する全ての要員の相互関
係を文書化し,それらの任務の遂行に必要な独立性及び権限を確実にする。
5.5.2
管理責任者
トップマネジメントは,管理層の中から管理責任者を任命する。管理責任者は,与えられている他の責
任と関わりなく次の責任及び権限をもつ。
12
Q 13485:2018 (ISO 13485:2016)
a) 品質マネジメントシステムに必要なプロセスを文書化していることを確実にする。
b) 品質マネジメントシステムの有効性及び改善の必要性の有無について,トップマネジメントに報告す
る。
c) 組織全体にわたって,適用される規制要求事項及び品質マネジメントシステム要求事項に対する認識
を高めることを確実にする。
5.5.3
内部コミュニケーション
トップマネジメントは,組織内にコミュニケーションのための適切なプロセスが確立されることを確実
にする。また,品質マネジメントシステムの有効性に関しての情報交換が行われることを確実にする。
5.6
マネジメントレビュー
5.6.1
一般
組織は,マネジメントレビューの手順を文書化する。トップマネジメントは,組織の品質マネジメント
システムが,引き続き適切で,妥当で,かつ,有効であることを確実にするために,あらかじめ文書化し
て定めた間隔で品質マネジメントシステムをレビューする。このレビューでは,品質マネジメントシステ
ムの改善の機会の評価,品質方針及び品質目標を含む品質マネジメントシステムの変更の必要性の評価も
行う。
マネジメントレビューの結果の記録は,維持する(4.2.5参照)。
5.6.2
マネジメントレビューへのインプット
マネジメントレビューへのインプットには,次からの情報を含むが,これに限らない。
a) フィードバック
b) 苦情処理
c) 規制当局への報告
d) 監査
e) プロセスの監視及び測定
f)
製品の監視及び測定
g) 是正処置
h) 予防処置
i)
前回までのマネジメントレビューの結果に対するフォローアップ
j)
品質マネジメントシステムに影響を及ぼす可能性のある変更
k) 改善のための提案
l)
適用される新しい又は改正された規制要求事項
5.6.3
マネジメントレビューからのアウトプット
マネジメントレビューからのアウトプットは,記録し(4.2.5参照),レビューしたインプット及び次に
関する決定及び処置を含める。
a) 品質マネジメントシステム及びそのプロセスの適切性,妥当性及び有効性の維持に必要な改善
b) 顧客要求事項に関連した製品の改善
c) 適用される新しい又は改正された規制要求事項への対応に必要な変更
d) 資源の必要性
13
Q 13485:2018 (ISO 13485:2016)
6
資源の運用管理
6.1
資源の提供
組織は,次のために必要な資源を明確にし,提供する。
a) 品質マネジメントシステムを実施し,その有効性を維持する。
b) 適用される規制要求事項及び顧客要求事項を満たす。
6.2
人的資源
製品の品質に影響を与える業務を行う要員は,適切な教育,訓練,技能及び経験に基づいた力量をもつ。
組織は,要員の力量の確立,必要な教育訓練の提供及び認識を確実にするためのプロセスを文書化する。
組織は,次を実施する。
a) 製品の品質に影響を及ぼす業務を行う要員に必要な力量を明確にする。
b) 必要な力量を達成又は維持できるよう訓練し,又は他の処置をとる。
c) とった処置の有効性を評価する。
d) 組織の要員が自らの活動のもつ意味及び重要性を認識し,品質目標の達成に向けて自らどのように貢
献できるかを認識することを確実にする。
e) 教育,訓練,技能及び経験について,該当する記録を維持する(4.2.5参照)。
注記 有効性を確認するために用いる方法は,訓練をした,又は他の処置をとった業務に伴うリスク
に見合ったものとする。
6.3
インフラストラクチャ
組織は,製品要求事項への適合を達成し,製品の混同を防止し,秩序だった取扱いを保証するために必
要なインフラストラクチャの要求事項を文書化する。インフラストラクチャには,適切な場合,次のよう
なものが含まれる。
a) 建物,作業場所及び関連するユーティリティ
b) 設備(ハードウェア及びソフトウェアを含む。)
c) 支援業務(例えば,輸送,通信,情報システム)
組織は,保守活動又はその欠如が製品の品質に影響を与える場合,保守活動の実施の間隔を含む保守活
動の要求事項を文書化する。適切な場合,製造,作業環境の管理並びに監視及び測定に用いる設備にその
要求事項を適用する。
そのような保守の記録は,維持する(4.2.5参照)。
6.4
作業環境及び汚染管理
6.4.1
作業環境
組織は,製品要求事項への適合を達成するために必要な作業環境の要求事項を文書化する。
作業環境の状態が製品の品質に対して悪影響を与える可能性がある場合,組織は作業環境に関する要求
事項及び作業環境を監視し,管理するための手順を文書化する。
組織は,次を実施する。
a) 要員の製品又は作業環境との接触が医療機器の安全性又は性能に悪影響を与えるおそれがある場合,
要員の健康,清潔さ及び衣服に対する要求事項を文書化する。
b) 作業環境内の特殊な環境条件下で一時的に作業するように要求した全ての要員に対して,本人に力量
があるか,又は力量がある者によって監督することを確実にする。
注記 更なる情報は,ISO 14644規格群及びJIS B 9918規格群を参照。
14
Q 13485:2018 (ISO 13485:2016)
6.4.2
汚染管理
適切な場合,組織は,汚染された又は汚染されている可能性がある製品の管理に対して,作業環境,要
員又は製品の汚染防止のための取決めを計画し,文書化する。
滅菌医療機器について,組織は,微生物又は微粒子による製品の汚染を管理するための要求事項を文書
化し,製品の組立又は包装プロセスにおいて要求する清浄性を維持する。
7
製品実現
7.1
製品実現の計画
組織は,製品実現のために必要なプロセスを計画して,構築する。製品実現の計画は,品質マネジメン
トシステムのその他のプロセスの要求事項と整合性をとる。
組織は,製品実現におけるリスクマネジメントの一つ以上のプロセスを文書化する。リスクマネジメン
ト活動による記録は,維持する(4.2.5参照)
製品実現の計画に当たって,適切な場合,組織は次を明確にする。
a) 製品に対する品質目標及び要求事項
b) インフラストラクチャ及び作業環境を含む,製品に特有なプロセス及び文書(4.2.4参照)の確立の必
要性,並びに資源の提供の必要性
c) 製品合否判定基準とともに,要求する検証,バリデーション,監視,測定,検査及び試験,取扱い,
保管,流通並びに製品特有のトレーサビリティ活動
d) 製品実現のプロセス及びその結果としての製品が要求事項を満たしていることを実証するために必要
な記録(4.2.5参照)
この計画のアウトプットは,組織の計画の実行に適した形式で文書化する。
注記 更なる情報は,JIS T 14971を参照。
7.2
顧客関連のプロセス
7.2.1
製品に関連する要求事項の明確化
組織は,次を明確にする。
a) 顧客が規定した要求事項。これには,引渡し時及び引渡し後の活動に関する要求事項を含む。
b) 顧客が明示してはいないが,指定する用途又は意図する用途が既知である場合,それらの用途に応じ
た要求事項
c) 製品に関連し,適用される規制要求事項
d) 医療機器の指定された性能及び安全で有効な使用を保証するために必要となる全てのユーザトレーニ
ング
e) 組織が必要と判断する追加要求事項
7.2.2
製品に関連する要求事項のレビュー
組織は,製品に関連する要求事項をレビューする。このレビューは,組織が顧客に製品を提供すること
についてのコミットメント(例 提案書の提出,契約又は注文の受諾,契約又は注文への変更の受諾)を
する前に実施する。レビューでは,次を確実にする。
a) 製品要求事項を定め,文書化している。
b) 契約又は注文の要求事項が以前に提示されたものと異なる場合には,それについて解決している。
c) 適用される規制要求事項を満たしている。
d) 7.2.1の要求事項によって明確にした全てのユーザトレーニングが利用できるか,利用できるように計
15
Q 13485:2018 (ISO 13485:2016)
画する。
e) 組織が,定められた要求事項を満たす能力をもっている。
このレビューの結果及びレビューの結果に基づいて取った処置の記録を維持する(4.2.5参照)。
顧客がその要求事項を書面で示さない場合には,組織は,受諾する前に顧客要求事項を確認する。
製品要求事項が変更された場合には,組織は,関連する文書を修正し,変更後の要求事項を関連する要
員に周知することを確実にする。
7.2.3
コミュニケーション
組織は,次に関して顧客とのコミュニケーションを図るための方法を計画し,文書化する。
a) 製品情報
b) 引き合い,契約又は注文,及びそれらの変更
c) 苦情を含む顧客からのフィードバック
d) 通知書
組織は,適用される規制要求事項に従い,規制当局とコミュニケーションを図る。
7.3
設計・開発
7.3.1
一般
組織は,設計・開発の手順を文書化する。
7.3.2
設計・開発の計画
組織は,製品の設計・開発の計画を策定し,管理する。適切な場合,策定した計画文書は維持し,設計・
開発の進行に応じて,更新する。
設計・開発の計画において,組織は次について文書化する。
a) 設計・開発の段階
b) 設計・開発の各段階で必要なレビュー
c) 設計・開発の各段階に適した検証,バリデーション及び設計移管の活動
d) 設計・開発に関する責任及び権限
e) 設計・開発へのインプットに対する設計・開発アウトップットのトレーサビリティを確実にする方法
f)
要員の力量を含む必要な資源
7.3.3
設計・開発へのインプット
製品要求事項に関連するインプットを明確にし,記録を維持する(4.2.5参照)。インプットには次を含
める。
a) 意図する用途に対応する機能,性能,ユーザビリティ及び安全上の要求事項
b) 適用される規制要求事項及び規格
c) リスクマネジメントからの適用できるアウトプット
d) 適切な場合,以前の類似した設計から得られた情報
e) 製品及びプロセスの設計・開発に不可欠なその他の要求事項
これらのインプットについては,その適切性をレビューし,承認する。
要求事項は,漏れがなく,曖昧ではなく,検証又はバリデーションが可能で,かつ,相反しない。
注記 更なる情報は,IEC 62366-1を参照。
7.3.4
設計・開発からのアウトプット
設計・開発からのアウトプットは,次に適合させる。
a) 設計・開発へのインプットで与えられた要求事項を満たす。
16
Q 13485:2018 (ISO 13485:2016)
b) 購買,製造及びサービス提供に対して適切な情報を提供する。
c) 製品の合否判定基準を含むか又はそれを参照する。
d) 安全な使用及び適正な使用に不可欠な製品の特性を明確にする。
設計・開発からのアウトプットは,設計・開発へのインプットと対比した検証に適した形式とする。ま
た,次の段階に進める前に,承認を受ける。
設計・開発からのアウトプットの記録は,維持する(4.2.5参照)。
7.3.5
設計・開発のレビュー
設計・開発の適切な段階において,次を目的として,計画し,文書化した取決めに従い,体系的なレビ
ューを行う。
a) 設計・開発の結果が要求事項に適合する能力があるかどうかを評価する。
b) 必要な処置を明確にし,提案する。
レビューへの参加者として,レビューの対象となっている設計・開発段階に関連する部門の代表及びそ
の他の専門家を含める。
対象とした設計,参加者の識別及び日付を含め,このレビューの結果及び全ての必要な処置の記録を維
持する(4.2.5参照)。
7.3.6
設計・開発の検証
設計・開発からのアウトプットが,設計・開発へのインプットの要求事項を満たしていることを確実に
するために,計画し,文書化した取決めに従って設計・開発の検証を実施する。
組織は,方法及び許容基準を含む検証計画を文書化する。また,適切な場合,サンプルサイズの根拠と
なる統計的手法を検証計画に含める。
注記 他の方法が正当であることを示せる場合は,統計的手法ではない方法でサンプルサイズを設定
することもできる(例 規制・規格要求事項,文献,類似の医療機器の経験,評価対象の性質
など)。
意図する用途が,医療機器の他の機器への接続又はインタフェースを要求している場合,検証には,接
続又はインタフェースした状態で,設計からのアウトプットが,設計へのインプットを満たしていること
の確認を含む。
検証の結果及び結論並びに必要な処置の記録を維持する(4.2.4及び4.2.5参照)。
7.3.7
設計・開発のバリデーション
結果として得られる製品が,規定した適用又は意図する用途への要求事項を満たす能力があることを確
実にするために,計画し,文書化した取決めに従って設計・開発のバリデーションを実施する。
組織は,方法及び許容基準を含むバリデーション計画を文書化する。また,適切な場合,サンプルサイ
ズの根拠となる統計的手法をバリデーション計画に含める。
注記 他の方法が正当であることを示せる場合は,統計的手法ではない方法でサンプルサイズを設定
することもできる(例 規制・規格要求事項,文献,類似の医療機器の経験,評価対象の性質
など)。
設計・開発のバリデーションは,製品を代表するもので実施する。製品を代表するものは,初回生産品
のユニット,バッチ,又はこれらと同等なものを含む。バリデーションに用いる製品の選択の根拠を記録
する(4.2.5参照)。
設計・開発のバリデーションの一部として,組織は,適用される規制要求事項に従い,医療機器の臨床
評価又は性能評価を実施する。臨床評価又は性能評価に用いる医療機器は,顧客の使用のためのリリース
17
Q 13485:2018 (ISO 13485:2016)
とはみなさない。
意図する用途が,医療機器の他の機器への接続又はインタフェースを要求している場合,バリデーショ
ンには,接続又はインタフェースした状態で,その製品が規定した適用又は意図する用途に合致すること
の確認を含める。
バリデーションは,顧客の製品使用のためのリリースに先立ち完了する。
バリデーションの結果及び結論並びに必要な処置の記録を維持する(4.2.4及び4.2.5参照)。
7.3.8
設計・開発の移管
組織は,設計・開発のアウトプットを製造に移管するための手順を文書化する。この手順は,設計・開
発のアウトプットが最終製造仕様になる前に,製造に適していることが検証されていることを確実にし,
その製造能力が製品要求事項を満たすことができることを確実にする。
移管の結果及び結論は記録する(4.2.5参照)。
7.3.9
設計・開発の変更管理
組織は,設計・開発の変更を管理する手順を文書化する。組織は,医療機器の機能,性能,ユーザビリ
ティ,安全性,適用される規制要求事項,及び意図する用途に対する変更の重要性を決定する。
設計・開発の変更は,識別する。変更を実施する前に次を行う。
a) レビューする。
b) 検証する。
c) 適切な場合,バリデーションを行う。
d) 承認する。
設計・開発の変更のレビューには,その変更が,構成部品及びプロセス内又は引渡し済みの製品,並び
にリスクマネジメントのインプット又はアウトプット及び製品実現プロセスに与える影響の評価を含める。
変更内容,変更のレビュー及び全ての必要な処置の記録(4.2.5参照)を維持する。
7.3.10 設計・開発ファイル
組織は,個々の医療機器の型式又は医療機器ファミリについて,設計・開発のファイルを維持する。こ
のファイルは,設計・開発の要求事項への適合性を示すために作成した記録,及び設計・開発の変更の記
録を含むか又は参照する。
7.4
購買
7.4.1
購買プロセス
組織は,購買製品が規定した購買情報を満たすことを確実にするための手順を文書化する(4.2.4参照)。
組織は,供給者の評価及び選定の基準を確立する。この基準は,次による。
a) 組織の要求事項に合致する製品を提供する供給者の能力に基づく。
b) 供給者のパフォーマンスに基づく。
c) 購買製品が医療機器の品質に与える影響に基づく。
d) 医療機器に付随するリスクに見合う。
組織は,供給者の監視及び再評価を計画する。購買製品が要求事項に合致する上での供給者のパフォー
マンスを監視する。その監視の結果を,再評価のプロセスに入力する。
購買要求事項を満たさないときは,購買製品及び適用される規制要求事項への適合に伴うリスクに見合
うように,供給者とともに解決する。
供給者の能力又はパフォーマンスの評価,選定,監視及び再評価の結果並びにそれらから必要とした処
置の記録は維持する(4.2.5参照)。
18
Q 13485:2018 (ISO 13485:2016)
7.4.2
購買情報
購買情報には,購買する製品を記述又は参照し,適切な場合,次を含める。
a) 製品仕様
b) 製品受入条件,手順,プロセス及び設備に対する要求事項
c) 供給者の要員の資格認定に関する要求事項
d) 品質マネジメントシステムに関する要求事項
組織は,供給者に伝達する前に,規定した購買要求事項が妥当であることを確実にする。
購買情報には,適用できる場合,購買製品が規定した購買要求事項を満たす能力に影響がある全ての変
更について,供給者が購買製品への変更を実施する前に組織に通知することへの書面の合意を含む。
7.5.9で規定したトレーサビリティに対して要求される範囲で,組織は,関連する購買情報を,文書(4.2.4
参照)及び記録(4.2.5参照)として維持する。
7.4.3
購買製品の検証
組織は,購買製品が,規定した購買要求事項を満たしていることを確実にするために,必要な検査又は
その他の活動を確立し,実施する。検証活動の範囲は,供給者の評価の結果に基づき,購買製品に伴うリ
スクに見合ったものとする。
組織は,購買製品の変更に気付いた場合,その変更が製品実現プロセス又は医療機器に影響を与えるか
どうか判断する。
組織又はその顧客が,供給者先で検証を実施することにした場合には,組織は,その検証活動及び購買
製品のリリースの方法を購買情報の中で明確にする。
検証の記録は維持する(4.2.5参照)。
7.5
製造及びサービスの提供
7.5.1
製造及びサービス提供の管理
製造及びサービス提供は,製品がその仕様を満たすことを確実にするように計画し,実施し,監視し,
管理する。適切な場合,製造管理は,次を含むが,これに限らない。
a) 製造管理の文書化した手順及び方法(4.2.4参照)
b) インフラストラクチャの認定
c) プロセスパラメータ及び製品の特性の監視及び測定の実施
d) 監視機器及び測定機器が利用でき,使用している。
e) 定められたラベリング及び包装作業の実施
f)
製品リリース,引渡し時及び引渡し後の活動の実施
組織は,個々の医療機器又はバッチに対し,7.5.9で規定した範囲のトレーサビリティを提供し,製造し
た数量及び出荷承認された数量を明確にした記録(4.2.5参照)を確立し維持する。この記録は検証し,承
認する。
7.5.2
製品の清浄性
組織は,次のいずれかに該当する場合,製品の清浄性又は汚染の管理に対する要求事項を文書化する。
a) 製品を,滅菌又はその使用に先立ち,組織によって清浄する場合
b) 製品を,非滅菌で供給し,滅菌又はその使用に先立ち清浄する場合
c) 製品は滅菌又は使用に先立ち清浄できないが,使用時の清浄性が重要である場合
d) 製品は滅菌されずに使用されるが,使用時の清浄性が重要である場合
e) 製造工程内で製品から副資材を除去することになっている場合
19
Q 13485:2018 (ISO 13485:2016)
上記のa)又はb)に従って製品を清浄する場合,6.4.1に含まれている要求事項は,清浄化プロセスの前の
段階には適用しない。
7.5.3
据付け活動
適切な場合,組織は,医療機器の据付け及び据付けの検証の合否判定基準のための要求事項を文書化す
る。
合意した顧客要求事項が,組織又はその供給者以外の外部パーティによる医療機器の据付けを許容して
いる場合,組織は,据付け及び据付けの検証に対する文書化した要求事項を提供する。
組織又はその供給者による医療機器の据付け及び据付けの検証の記録は,維持する(4.2.5参照)。
7.5.4
附帯サービス活動
医療機器の附帯サービスが規定要求事項である場合,組織は,附帯サービス活動を実施し,製品要求事
項を満たしていることを検証するために,必要な附帯サービス活動の手順,参照物質及び参照測定手順を
文書化する。
組織は,組織又は供給者が実施する附帯サービス活動の記録を次のために分析する。
a) その情報を苦情として扱うかどうかを判断する。
b) 適切な場合,改善プロセスへのインプットとする。
組織又はその供給者が実施した附帯サービス活動の記録は維持する(4.2.5参照)。
7.5.5
滅菌医療機器に対する特別要求事項
組織は,各滅菌バッチに対して使用した滅菌プロセスパラメータの記録を維持する(4.2.5参照)。滅菌
の記録は,医療機器の各製造バッチに対してトレースできるようにする。
7.5.6
製造及びサービス提供に関するプロセスのバリデーション
製造及びサービス提供の過程で結果として生じるアウトプットが,それ以降の監視又は測定で検証する
ことが不可能であるか検証を実施しない場合は,製品が使用され又はサービスを提供した後でだけしか不
具合が顕在化しないため,組織は,その製造及びサービス提供の該当するプロセスのバリデーションを行
う。
バリデーションによって,これらのプロセスが計画どおりの結果を一貫して出せることを実証する。
組織は,次を含むプロセスのバリデーションの手順を文書化する。
a) プロセスのレビュー及び承認のために定めた判断基準
b) 設備の認定及び要員の資格認定
c) 特定の方法,手順及び判断基準の使用
d) 適切な場合,サンプルサイズの根拠となる統計的手法
e) 記録に関する要求事項(4.2.5参照)
f)
再バリデーションの判断基準を含む,再バリデーション
g) プロセスに対する変更の承認
組織は,製造及びサービス提供のために使用するコンピュータソフトウェアの適用のバリデーションの
手順を文書化する。このようなソフトウェアの適用は,初回の使用前にバリデーションを行う。また,適
切な場合,そのソフトウェア又は適用の変更後に,バリデーションを行う。ソフトウェアのバリデーショ
ン及び再バリデーションに関する固有のアプローチ及び活動は,製品がその仕様に適合する能力への影響
を含むソフトウェアの使用に伴うリスクに見合ったものとする。
バリデーションの結果及び結論並びに必要な処置の記録は,維持する(4.2.4及び4.2.5参照)。
20
Q 13485:2018 (ISO 13485:2016)
7.5.7
滅菌及び無菌バリアシステムのプロセスのバリデーションに対する特別要求事項
組織は,滅菌及び無菌バリアシステムのプロセスのバリデーションに対して手順(4.2.4参照)を文書化
する。
滅菌及び無菌バリアシステムのプロセスは,最初の使用及び適切な場合には,その後の製品又はプロセ
スの変更に先立ってバリデーションを行う。
バリデーションの結果及び結論並びに必要な処置の記録は,維持する(4.2.4及び4.2.5参照)。
注記 更なる情報は,JIS T 0841-1及びJIS T 0841-2を参照。
7.5.8
識別
組織は,製品を識別するための手順を文書化し,製品実現の全過程において製品を適切な手段で識別す
る。
組織は,製品実現の全過程において,監視及び測定の要求事項に関連して製品の状態を識別する。製品
の状態の識別は,要求する検査及び試験に合格したか,又は正式な特別採用手続の下でリリースした製品
だけを出荷し,使用し,又は据え付けることを確実にするために,製造,保管,据付け及び附帯サービス
の全過程において維持する。
適用される規制要求事項によって要求される場合,医療機器に機器固有識別(UDI)を割り当てるシス
テムについて文書化する。
組織は,組織に返却された医療機器を明確にし,適合製品から識別することを確実にするための手順を
文書化する。
7.5.9
トレーサビリティ
7.5.9.1
一般
組織は,トレーサビリティに対して手順を文書化する。この手順には,適用される規制要求事項に基づ
いてトレーサビリティの範囲及び維持する記録を規定する(4.2.5参照)。
7.5.9.2
埋込み医療機器に対する特別要求事項
構成部品,材料及び用いられた作業環境条件が,医療機器の規定した安全性及び性能の要求事項を満た
さない原因となり得る場合,トレーサビリティのために要求する記録として,構成部品,材料及び用いた
作業環境条件の記録を含める。
組織は,流通サービスの供給者又はディストリビュータに対し,トレーサビリティを可能にする医療機
器の流通の記録を維持し,そのような記録を監査のときに提示できることを要求する。
出荷こん(梱)包荷受人の氏名及び住所の記録を維持する(4.2.5参照)。
7.5.10 顧客の所有物
組織は,顧客の所有物が組織の管理下にある又は組織がそれを使用している間,使用するため又は製品
に組み込むために提供された顧客の所有物の識別,検証,保護及び防護を実施する。顧客の所有物を紛失,
損傷した場合又は使用に適さないと分かった場合には,組織は,これを顧客に報告し,記録を維持する(4.2.5
参照)。
7.5.11 製品の保存
組織は,処理,保管,取扱い及び流通の間,製品を要求事項に適合した状態のまま保存するための手順
を文書化する。保存は,医療機器の構成部品にも適用する。
処理,保管,取扱い及び流通の間,想定される状態及びハザードにさら(晒)される場合,組織は,変
質,汚染又は損傷から,次のいずれかの方法によって,製品を保護する。
a) 適切な医療機器の包装及び出荷コンテナを設計し,構築する。
21
Q 13485:2018 (ISO 13485:2016)
b) 包装だけでは保存状態を確保できない場合,必要な特別条件の要求事項を文書化する。
特別な条件が要求される場合は,それを管理し,記録する(4.2.5参照)。
7.6
監視機器及び測定機器の管理
規定した要求事項に対する製品の適合性を実証するために,組織は,実施する監視及び測定を明確にす
る。また,そのために必要な監視機器及び測定機器を明確にする。
組織は,監視及び測定の要求事項との整合性を確保できる方法で監視及び測定が実施できること,及び
実施することを確実にする手順を文書化する。
測定値の妥当性を保証するために,必要がある場合は,測定機器に関し,次の事項を満たす。
a) 定めた間隔又は使用前に,国際計量標準又は国家計量標準にトレース可能な計量標準に照らして校正
若しくは検証,又はその双方を実施する。そのような標準が存在しない場合には,校正又は検証に用
いた基準について記録する(4.2.5参照)。
b) 測定機器の調整をする,又は必要に応じて再調整をする。そのような調整又は再調整は記録する(4.2.5
参照)。
c) 校正の状態が明確になるような識別がある。
d) 測定した結果が無効になるような操作ができないようにする。
e) 取扱い,保守及び保管において,損傷及び劣化しないように保護する。
組織は,文書化した手順に従い,校正又は検証を実施する。
さらに,測定機器が要求事項に適合していないことが判明した場合には,組織は,その測定機器でそれ
までに測定した結果の妥当性を評価し,記録する。組織は,その装置及び影響を受けた製品に関して,適
切な処置をとる。
校正及び検証の結果の記録を維持する(4.2.5参照)。
組織は,監視及び測定の要求事項のために使用するコンピュータソフトウェアの適用のバリデーション
の手順を文書化する。このようなソフトウェアの適用は,初回の使用前にバリデーションを行う。また,
適切な場合,そのソフトウェア又は適用への変更後にバリデーションを行う。ソフトウェアのバリデーシ
ョン及び再バリデーションに関する固有のアプローチ及び活動は,製品がその仕様に適合する能力への影
響を含むソフトウェアの使用に伴うリスクに見合ったものとする。
バリデーションの結果及び結論並びに必要な処置の記録は,維持する(4.2.4及び4.2.5参照)。
注記 更なる情報は,JIS Q 10012を参照。
8
測定,分析及び改善
8.1
一般
組織は,次のために必要となる監視,測定,分析及び改善のプロセスを計画し,実施する。
a) 製品の適合性を実証する。
b) 品質マネジメントシステムの適合性を確実にする。
c) 品質マネジメントシステムの有効性を維持する。
これには,統計的手法を含む,適切な方法及びその使用の程度を決定することを含める。
8.2
監視及び測定
8.2.1
フィードバック
組織は,品質マネジメントシステムの有効性の測定の一つとして,顧客要求事項を満たしているかどう
かに関する情報を収集し,監視する。この情報の入手及び利用の方法を文書化する。
22
Q 13485:2018 (ISO 13485:2016)
組織は,フィードバックプロセスの手順を文書化する。フィードバックプロセスには,製造後の活動と
ともに,製造からのデータ収集の規定を含める。
フィードバックプロセスで収集した情報は,製品実現又は改善プロセスへの潜在的なインプットである
と同様に,製品要求事項の監視及び維持のために,リスクマネジメントへの潜在的なインプットとして働
く。
適用される規制要求事項が組織に製造後の活動からの特定の経験を得ることを要求している場合,この
経験のレビューは,フィードバックプロセスの一部とする。
8.2.2
苦情処理
組織は,タイムリな苦情処理のための手順を,適用される規制要求事項に従って文書化する。
この手順には,少なくとも,次の活動に対する要求事項及び責任を含む。
a) 情報の受領及び記録
b) フィードバックが苦情を含んでいるかを決めるための情報の評価
c) 苦情の調査
d) 適切な規制当局への報告の必要性の決定
e) 苦情に関連する製品の取扱い
f)
修正又は是正処置の開始の必要性の決定
苦情を調査しない場合には,その理由を文書化する。苦情処理プロセスからの結果として実施する全て
の修正及び是正処置は文書化する。
調査の結果,組織外の活動が苦情の一因である場合,関連する情報を組織と関与している外部パーティ
との間で交換する。
苦情処理の記録は維持する(4.2.5参照)。
8.2.3
規制当局への報告
適用される規制要求事項が,規定された有害事象の報告基準に該当する苦情又は通知書の発行の通知を
要求している場合,組織は,適切な規制当局への通知を提供するための手順を文書化する。
規制当局への報告の記録は維持する(4.2.5参照)。
8.2.4
内部監査
組織は,品質マネジメントシステムが次の事項を満たしているか否かを明確にするために,計画した間
隔で内部監査を実施する。
a) 品質マネジメントシステムが,計画し,文書化した取決めに適合しているか,この規格の要求事項に
適合しているか,組織が決めた品質マネジメントシステム要求事項に適合しているか,及び適用され
る規制要求事項に適合しているか。
b) 品質マネジメントシステムを効果的に実施し,維持しているか。
組織は,監査の計画及び実施,並びに記録及び監査結果の報告に関する責任及び要求事項を規定した手
順を文書化する。
組織は,監査の対象となるプロセス及び領域の状態及び重要性,並びにこれまでの監査結果を考慮して,
監査プログラムを策定する。監査の基準,範囲,頻度及び方法は,定義し,記録する(4.2.5参照)。監査
員の選定及び監査の実施においては,監査プロセスの客観性及び公平性を確保する。監査員は,自らの仕
事は監査しない。
監査したプロセス及び領域の識別,並びに結論を含む,監査及びその結果の記録を維持する(4.2.5参照)。
監査された領域に責任をもつ管理者は,発見された不適合及びその原因を除去するために遅滞なく全て
23
Q 13485:2018 (ISO 13485:2016)
の必要な修正及び是正処置がとられることを確実にする。フォローアップには,とられた処置の検証及び
検証結果の報告を含める。
注記 更なる情報は,JIS Q 19011を参照。
8.2.5
プロセスの監視及び測定
組織は,品質マネジメントシステムのプロセスを適切な方法で監視し,適切な場合,測定をする。これ
らの方法は,プロセスが計画どおりの結果を達成する能力があることを実証するものである。計画どおり
の結果が達成できない場合は,適切な場合,修正及び是正処置をとる。
8.2.6
製品の監視及び測定
組織は,製品要求事項を満たしていることを検証するために,製品の特性を監視し,測定する。監視及
び測定は,計画し,文書化した取決め及び文書化した手順に従って,製品実現プロセスの適切な段階で実
施する。
合否判定基準への適合の証拠を維持する。製品のリリースを正式に許可した要員の識別を記録する(4.2.5
参照)。適切な場合,記録には,測定活動の実施のために使用した試験機器を識別する。
計画し,文書化した取決めが問題なく完了するまでは,製品のリリース及びサービス提供を行わない。
埋込み医療機器に関して,組織は,全ての検査又は試験を実施した要員の識別を記録する。
8.3
不適合製品の管理
8.3.1
一般
組織は,製品要求事項に適合しない製品が誤って使用されたり,又は引き渡されることを防ぐために,
それらを識別し,管理することを確実にする。組織は,不適合製品の識別,文書化,隔離,評価,及び処
分の管理並びに関連する責任及び権限を定めるための手順を文書化する。
不適合製品の評価には,調査の必要性の決定及び不適合に対する責任をもつ全ての外部パーティへの通
知の必要性の決定を含める。
不適合の性質及び不適合の評価,全ての調査及び決定の理由を含む全てのその後にとった処置の記録は,
維持する(4.2.5参照)。
8.3.2
引渡し前に発見した不適合製品における処置
組織は,次の一つ以上の方法で不適合製品を処理する。
a) 発見した不適合を除去するための処置をとる。
b) 本来の意図する用途又は適用のために用いることができないような処置をとる。
c) 特別採用によって,その使用,リリース又は合格と判定することを正式に許可する。
組織は,正当性を提供し,承認が得られ,適用される規制要求事項が満たされる場合に限って,特別採
用によって不適合製品を受け入れることを確実にする。特別採用による受入れ及び特別採用を許可した人
の識別の記録は,維持する(4.2.5参照)。
8.3.3
引渡し後に発見された不適合製品における処置
引渡し後又は使用開始後に不適合製品が発見された場合には,組織は,不適合の影響,又は潜在的影響
に対して適切な処置をとる。とった処置の記録は維持する(4.2.5参照)。
組織は,適用される規制要求事項に基づいて,通知書を発行するための手順を文書化する。この手順は,
いつでも実施できるものとする。通知書の発行に関する処置の記録は,維持する(4.2.5参照)。
8.3.4
手直し
組織は,その手直しが製品に与える潜在的悪影響を考慮の上,文書化した手順に従い,手直しを実施す
る。この手順は,元の手順と同様のレビュー及び承認に基づいて発行する。
24
Q 13485:2018 (ISO 13485:2016)
手直しの完了後,製品が,適用される合否判定基準及び規制要求事項に適合していることを確実にする
ために,製品を検証する。
手直しの記録は,維持する(4.2.5参照)。
8.4
データの分析
組織は,品質マネジメントシステムの適切性,妥当性及び有効性を実証するため,適切なデータを明確
にし,それらのデータを収集し,分析するための手順を文書化する。この手順には,統計的手法及びその
使用の範囲を含む,適切な方法を決定することを含める。
データの分析には,監視及び測定の結果から得られたデータ及びそれ以外の該当する情報源からのデー
タを含める。それには,少なくとも次からのインプットを含める。
a) フィードバック
b) 製品要求事項への適合性
c) 改善の機会を得ることを含む,プロセス及び製品の特性及び傾向
d) 供給者
e) 監査
f)
適切な場合,附帯サービスの報告書
もし,このデータ分析によって,品質マネジメントシステムの適切性,妥当性又は有効性がないことが
示された場合は,8.5で要求しているように,組織はこの分析を改善のためにインプットとして用いる。
データの分析結果の記録は,維持する(4.2.5参照)。
8.5
改善
8.5.1
一般
組織は,品質方針,品質目標,監査結果,市販後監視,データ分析,是正処置,予防処置及びマネジメ
ントレビューを通じて,医療機器の安全性及び性能,並びに品質マネジメントシステムの継続的な適切性,
妥当性及び有効性を確実にし,維持するために必要な全ての変更を明確にし,実施する。
8.5.2
是正処置
組織は,再発防止のため,不適合の原因を除去する処置をとる。全ての必要な是正処置は,遅滞なく実
施する。是正処置は,発見した不適合の影響に見合ったものとする。
組織は,次に関する要求事項を規定するための手順を文書化する。
a) 不適合(苦情を含む。)のレビュー
b) 不適合の原因の特定
c) 不適合の再発防止を確実にするための処置の必要性の評価
d) 必要な処置の計画及び文書化,並びにその処置の実施。適切な場合,文書の更新を含む。
e) 是正処置が,適用される規制要求事項へ適合するための能力又は医療機器の安全性及び性能への悪影
響を与えていないことの検証
f)
とった是正処置の有効性のレビュー
全ての調査及びとった処置の結果の記録は,維持する(4.2.5参照)。
8.5.3
予防処置
組織は,起こり得る不適合が発生することを防止するために,その原因を除去する処置を決める。予防
処置は,起こり得る問題の影響に見合ったものとする。
組織は,次に関する要求事項を規定するための手順を文書化する。
a) 起こり得る不適合及びその原因の特定
25
Q 13485:2018 (ISO 13485:2016)
b) 不適合の発生を予防するための処置の必要性の評価
c) 必要な処置の計画及び文書化,並びにその処置の実施。適切な場合,文書の更新を含む。
d) 処置が,適用される規制要求事項へ適合するための能力又は医療機器の安全性及び性能への悪影響を
与えていないことの検証
e) 適切な場合,とった予防処置の有効性のレビュー
全ての調査及びとった処置の結果の記録は,維持する(4.2.5参照)。

26
Q 13485:2018 (ISO 13485:2016)
附属書A
(参考)
JIS Q 13485:2005とJIS Q 13485:2018との内容の比較
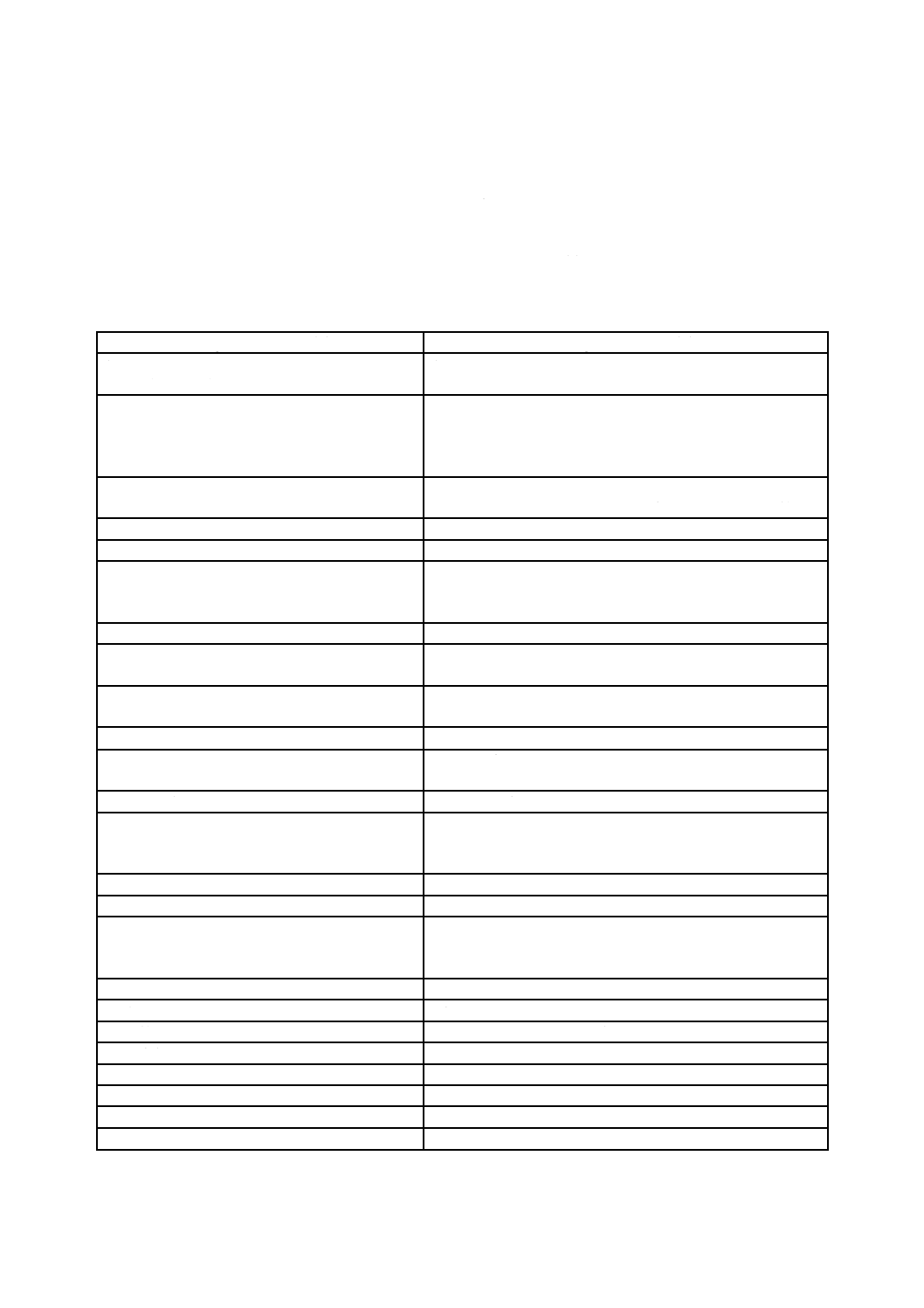
表A.1に,旧規格(JIS Q 13485:2005)とこの規格(JIS Q 13485:2018)との変更の概要を示す。
表A.1−JIS Q 13485:2005とJIS Q 13485:2018との内容の比較
JIS Q 13485:2018の箇条
JIS Q 13485:2005と比較した変更点に関する解説
序文
0.1 一般
− この規格の要求事項及び規格でカバーされるライフサイクルステージに
おける組織の性質に関連した実質的なより多くの詳細が含まれている。
− 自発的に又は契約の取決めの結果,供給者又は他の外部パーティによっ
て,この要求事項を利用することができることを説明している。
− 品質マネジメントシステムにフォーカスされた規制要求事項に関連した
組織の義務に関して組織に警告している。
− 地域規制の定義における相違及び組織の品質マネジメントシステムにこ
れらの定義がどのように影響するかを理解する組織の義務に関して組織
に警告している。
− 組織自身の品質マネジメントシステム要求事項を満たす義務を追加して
いる。
− 特に,“顧客要求事項,安全性及び性能に適用される規制要求事項を満た
す”必要性に焦点を当てている。
− 重要な製品要求事項が,安全性及び性能に関連するものであることを強
調している。
− 元のリストにはなかった,品質マネジメントシステムの特質上の二つの
影響を追加する(組織環境及び規制要求事項)。
− 組織が,この規格の箇条の構造に,そのマニュアルの構造をそろ(揃)
える必要がないことを明確にしている。
0.2 概念の明確化
− 適切な要求事項の記載に関連する次の二つの基準を追加した。
− 規制要求事項の遵守
− 組織がリスクを管理するために必要な要求事項
− リスクの適用を医療機器の安全性若しくは性能の要求事項,又は適用さ
れる規制要求事項を満たすために制限した。
− 用語“文書化”は,確立し,実施し,維持する必要が含まれていること
を明確にした。
− 用語“製品”は顧客へ意図するアウトプット,顧客から要求されたアウ
トプット,又は製品実現プロセスから得られた意図したアウトプットへ
適用することを明確にした。
0.3 プロセスアプローチ
− プロセスアプローチの説明を拡張した。
0.4 JIS Q 9001との関係
− JIS Q 13485:2018とJIS Q 9001との関係を記載した。
− JIS Q 13485:2018とJIS Q 9001:2015との構造的関係を附属書Bに概説し
ていることを記載した。
− この規格ではJIS Q 9001:2008からの変更を示すためのイタリック体の
表記を利用することを取りやめた。

27
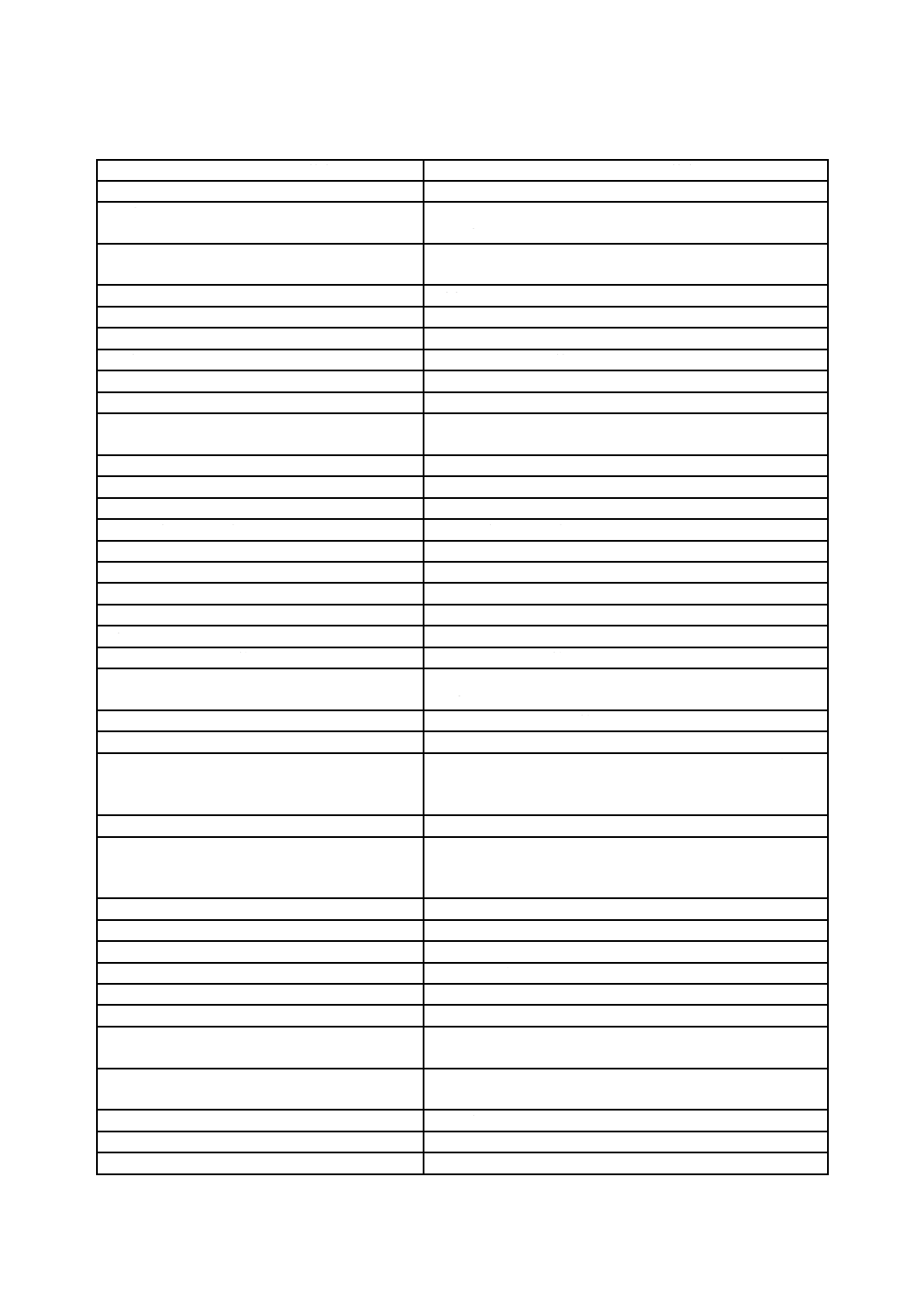
Q 13485:2018 (ISO 13485:2016)
表A.1−JIS Q 13485:2005とJIS Q 13485:2018との内容の比較(続き)
JIS Q 13485:2018の箇条
JIS Q 13485:2005と比較した変更点に関する解説
1 適用範囲
− 医療機器のライフサイクルの一つ以上の段階に関与している組織に,こ
の規格の適用の可能性を記載している。
− この規格は,医療機器の組織に品質マネジメントシステム関連サービス
を含む製品を提供する供給者及び外部パーティが使用できることを記載
している。
− 特にアウトソースしたプロセスを監視し,維持し,管理する責任を喚起
した。
− 非適用可能な要求事項の範囲を箇条6〜箇条8の要求事項に拡大した。
− 用語“規制要求事項”は,法令,規則,条例又は指令が含まれているこ
とを明確にし,“適用される規制要求事項”の適用範囲を品質マネジメン
トシステム及び医療機器の安全性及び性能のための要求事項に制限する
ことを明確にした。
3 用語及び定義
− 定義を追加した。また,旧規格から一部の定義を修正した。
4 品質マネジメントシステム
4.1 一般要求事項
− 組織の役割の文書化の要求事項を追加した。
− “組織が取る役割を考慮に入れる”プロセスを定めることを要求した。
− “品質マネジメントシステムに必要な適切なプロセスの管理にリスクに
基づくアプローチ”を適用することを要求した。
− プロセスの変更に関する要求事項を追加した。
− 品質マネジメントシステムで用いるコンピュータソフトウェアの適用の
バリデーションに関する要求事項を追加した。
4.2 文書化の要求事項
− 文書管理の要求事項に記録の管理を含めた。
− 医療機器ファイルに含まれる文書をリスト化した。
− 機密健康情報の保護に関する新たな要求事項を追加した。
− 文書の劣化及び紛失に関する新たな要求事項を追加した。
5.6 マネジメントレビュー
− マネジメントレビューの一つ以上の手順の文書化の要求事項及び“あら
かじめ定め,文書化した間隔で”マネジメントレビューを実施する要求
事項を追加した。
− マネジメントレビューのインプット及びアウトプットのリストを拡大し
た。
6.2 人的資源
− 要員の力量,要員に必要な訓練を提供する,要員の認識を確実にする文
書化のプロセスに関する新しい要求事項を追加した。
6.3 インフラストラクチャ
− 製品の混同を防止し,製品の秩序だった取扱いを確実にする要求事項を
追加した。
− 支援体制のリストに情報システムを追加した。
6.4 作業環境及び汚染管理
− 作業環境に関する文書化の要求事項を追加した。
− 滅菌医療機器に関し,微粒子又は微生物による製品の汚染を管理する要
求事項を追加した。
7.1 製品実現の計画
− リストに要求事項を追加した。
7.2 顧客関連のプロセス
− リストに要求事項を追加した。
− 規制当局とのコミュニケーションに関する新しい要求事項を追加した。
7.3.2 設計・開発の計画
− リストに要求事項を追加した。
− 設計・開発に関与する異なったグループ間のインタフェースの管理に関
する要求事項を削除した。
7.3.3 設計・開発へのインプット
− リストに要求事項を追加した。
− 要求事項が検証又はバリデーション可能であるという要求事項を追加し
た。
7.3.5 設計・開発のレビュー
− 記録の内容の詳細を追加した。

28
Q 13485:2018 (ISO 13485:2016)
表A.1−JIS Q 13485:2005とJIS Q 13485:2018との内容の比較(続き)
JIS Q 13485:2018の箇条
JIS Q 13485:2005と比較した変更点に関する解説
7.3.6 設計・開発の検証
− 検証の計画の文書化及びインタフェースの考慮に関する要求事項を追加
した。
− 検証の記録に関する要求事項を追加した。
7.3.7 設計・開発のバリデーション
− バリデーションの計画の文書化,バリデーションに使用する製品及びイ
ンタフェースの考慮に関する要求事項を追加した。
− バリデーションの記録に関する要求事項を追加した。
7.3.8 設計・開発の移管
− 新しい細分箇条を追加した。
7.3.9 設計・開発の変更管理
− 設計・開発の変更の効果の評価は,プロセス内の製品及びリスクマネジ
メントのアウトプット並びに製品実現のプロセスに対してなされるとい
う要求事項を追加した。
− 設計・開発の変更の重要性の決定に際し,検討する詳細内容を追加した。
7.3.10 設計・開発ファイル
− 新しい細分箇条を追加した。
7.4.1 購買プロセス
− 医療機器の品質,医療機器に関連するリスク,及び規制要求事項に適合
する製品への供給者の能力の影響に関する供給者の選択基準にフォーカ
スした。
− 供給者の監視及び再評価に関連する新しい要求事項及び購買要求事項に
合致しなかったときにとるべき処置に関する新しい要求事項を追加し
た。
− 記録の内容に関する追加の詳細内容を提供した。
7.4.2 購買情報
− 購買製品の変更の通知を含める新たな要求事項を追加した。
7.4.3 購買製品の検証
− 検証活動の範囲及び購買製品に関する全ての変更に組織が気付いたとき
にとるべき処置に関する新規要求事項を追加した。
7.5.1 製造及びサービス提供の管理
− 製造及びサービス提供の実行の管理に関連する詳細内容を追加した。
7.5.2 製品の清浄性
− リストに要求事項を追加した。
7.5.4 附帯サービス活動
− 附帯サービス活動の記録の分析について,新しい要求事項を追加した。
7.5.6 製造及びサービス提供に関す
るプロセスバリデーション
− リストに要求事項を追加した。
− 手順を要求する状況に関して詳細内容を追加した。
− ソフトウェアの利用に関するリスクに対するソフトウェアバリデーショ
ンの特有のアプローチを関連付けた。
− バリデーションの記録に関する要求事項を追加した。
7.5.7 滅菌及び無菌バリアシステム
のプロセスバリデーションに対す
る特別要求事項
− 無菌バリアシステムの要求事項を追加した。
7.5.8 識別
− 機器固有識別(UDI)に関する要求事項を追加した。
− 製品の識別及び製造途中の識別並びに製品の状態に関わる文書化した手
順に関する新たな要求事項を追加した。
7.5.11 製品の保存
− 保存を行う方法についての詳細を追加した。
8.2.1 フィードバック
− フィードバックが製造中の活動及び生産製造後の活動から得られること
を明確にした。
− 製品要求事項を監視,維持するためにリスクマネジメントプロセスにお
いてフィードバックを活用する要求事項を追加した。
8.2.2 苦情処理
− 新規細分箇条
8.2.3 規制当局への報告
− 新規細分箇条
8.2.6 製品の監視及び測定
− 測定活動に用いた検査機器を特定する要求事項を追加した。

29
Q 13485:2018 (ISO 13485:2016)
表A.1−JIS Q 13485:2005とJIS Q 13485:2018との内容の比較(続き)
JIS Q 13485:2018の箇条
JIS Q 13485:2005と比較した変更点に関する解説
8.3 不適合製品の管理
− 文書化しなければならない管理の種類に関する詳細を追加した。
− 全ての調査及び決定の理由を含む要求事項を一般化した。
− 特別採用に関する要求事項を追加した。
− 引渡し前に発見した不適合製品の処置及び引渡し後に発見された不適合
製品の処置に対する要求事項及び手直しに関する要求事項を個別の要求
事項とした。
− 通知書の発行に関する記録の要求事項を追加した。
8.4 データの分析
− 統計的手法及びその使用の範囲を含む,適切な可能な方法を決定するこ
とを含むという要求事項を追加した。
− インプットのリストに内容を追加した。
8.5.2 是正処置
− 是正処置が悪影響を与えていないことを検証する要求事項を追加した。
− 是正処置が過度な遅延なくとられるようにする要求事項を追加した。
8.5.3 予防処置
− 予防処置が悪影響を与えていないことを検証する要求事項を追加した。
30
Q 13485:2018 (ISO 13485:2016)
附属書B
(参考)
JIS Q 13485:2018とJIS Q 9001:2015との関係
表B.1及び表B.2は,JIS Q 13485:2018とJIS Q 9001:2015との関係を示している。
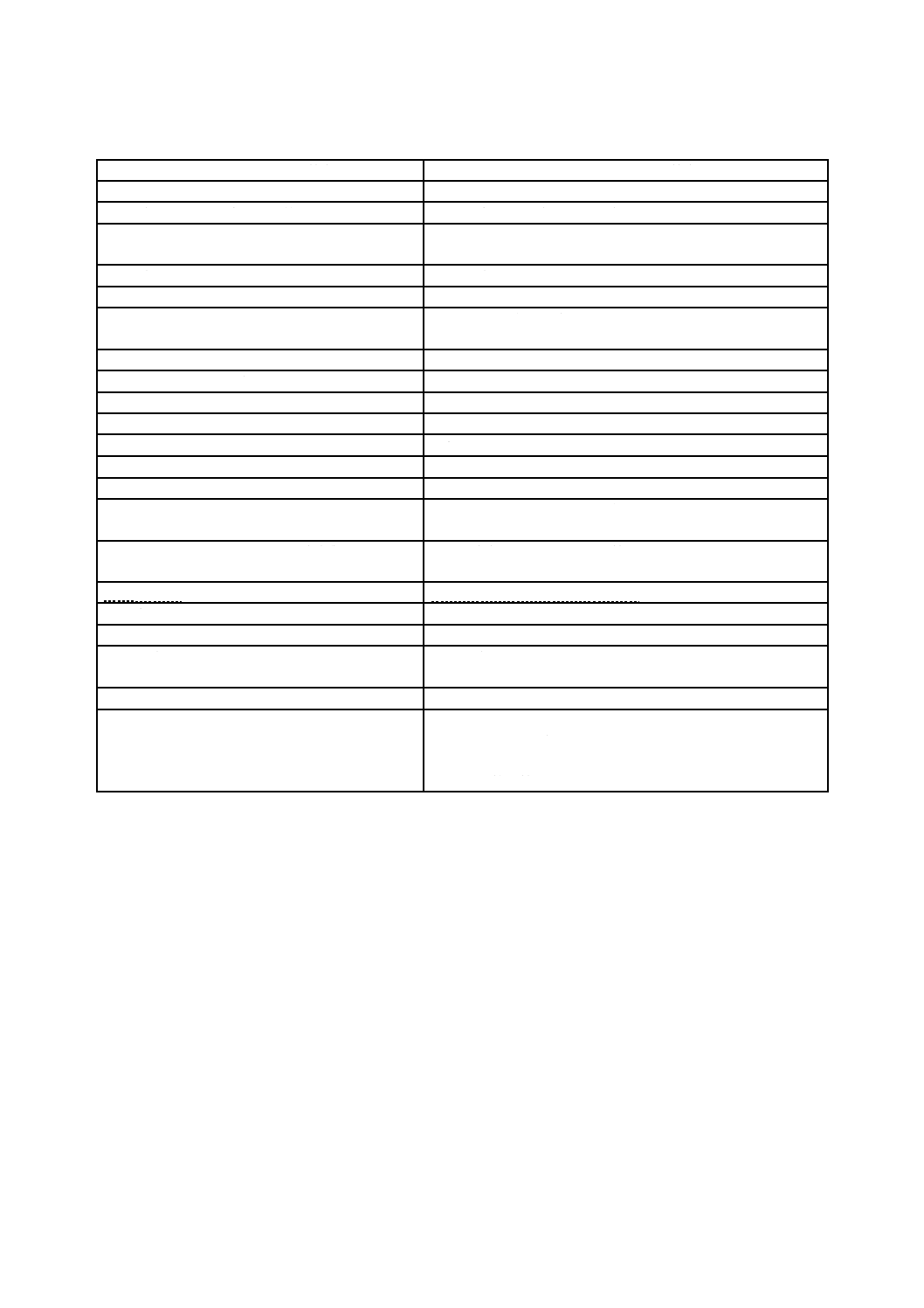
表B.1−JIS Q 13485:2018とJIS Q 9001:2015との関係
JIS Q 13485:2018の箇条
JIS Q 9001:2015の箇条
1 適用範囲
4.1.1 (表題なし)
1 適用範囲
4.3 品質マネジメントシステムの適用範囲の決定
4 品質マネジメントシステム
4 組織の状況
4.1 組織及びその状況の理解
4.2 利害関係者のニーズ及び期待の理解
4.3 品質マネジメントシステムの適用範囲の決定
4.1 一般要求事項
4.4 品質マネジメントシステム及びそのプロセス
8.4 外部から提供されるプロセス,製品及びサービスの管理
4.2 文書化に関する要求事項
7.5 文書化した情報
4.2.1 一般
7.5.1 一般
4.2.2 品質マニュアル
4.3 品質マネジメントシステムの適用範囲の決定
4.4 品質マネジメントシステム及びそのプロセス
7.5.1 一般
4.2.3 医療機器ファイル
対応する箇条なし
4.2.4 文書管理
7.5.2 作成及び更新
7.5.3 文書化した情報の管理
4.2.5 記録の管理
7.5.2 作成及び更新
7.5.3 文書化した情報の管理
5 経営者の責任
5 リーダーシップ
5.1 経営者のコミットメント
5.1 リーダーシップ及びコミットメント
5.1.1 一般
5.2 顧客重視
5.1.2 顧客重視
5.3 品質方針
5.2 方針
5.2.1 品質方針の確立
5.2.2 品質方針の伝達
5.4 計画
6 計画
5.4.1 品質目標
6.2 品質目標及びそれを達成するための計画策定
5.4.2 品質マネジメントシステムの計画
6 計画
6.1 リスク及び機会への取組み
6.3 変更の計画
5.5 責任,権限及びコミュニケーション
5 リーダーシップ
5.5.1 責任及び権限
5.3 組織の役割,責任及び権限
5.5.2 管理責任者
5.3 組織の役割,責任及び権限
5.5.3 内部コミュニケーション
7.4 コミュニケーション
5.6 マネジメントレビュー
9.3 マネジメントレビュー
5.6.1 一般
9.3.1 一般
5.6.2 マネジメントレビューへのインプット
9.3.2 マネジメントレビューへのインプット
5.6.3 マネジメントレビューからのアウトプット
9.3.3 マネジメントレビューからのアウトプット
31
Q 13485:2018 (ISO 13485:2016)
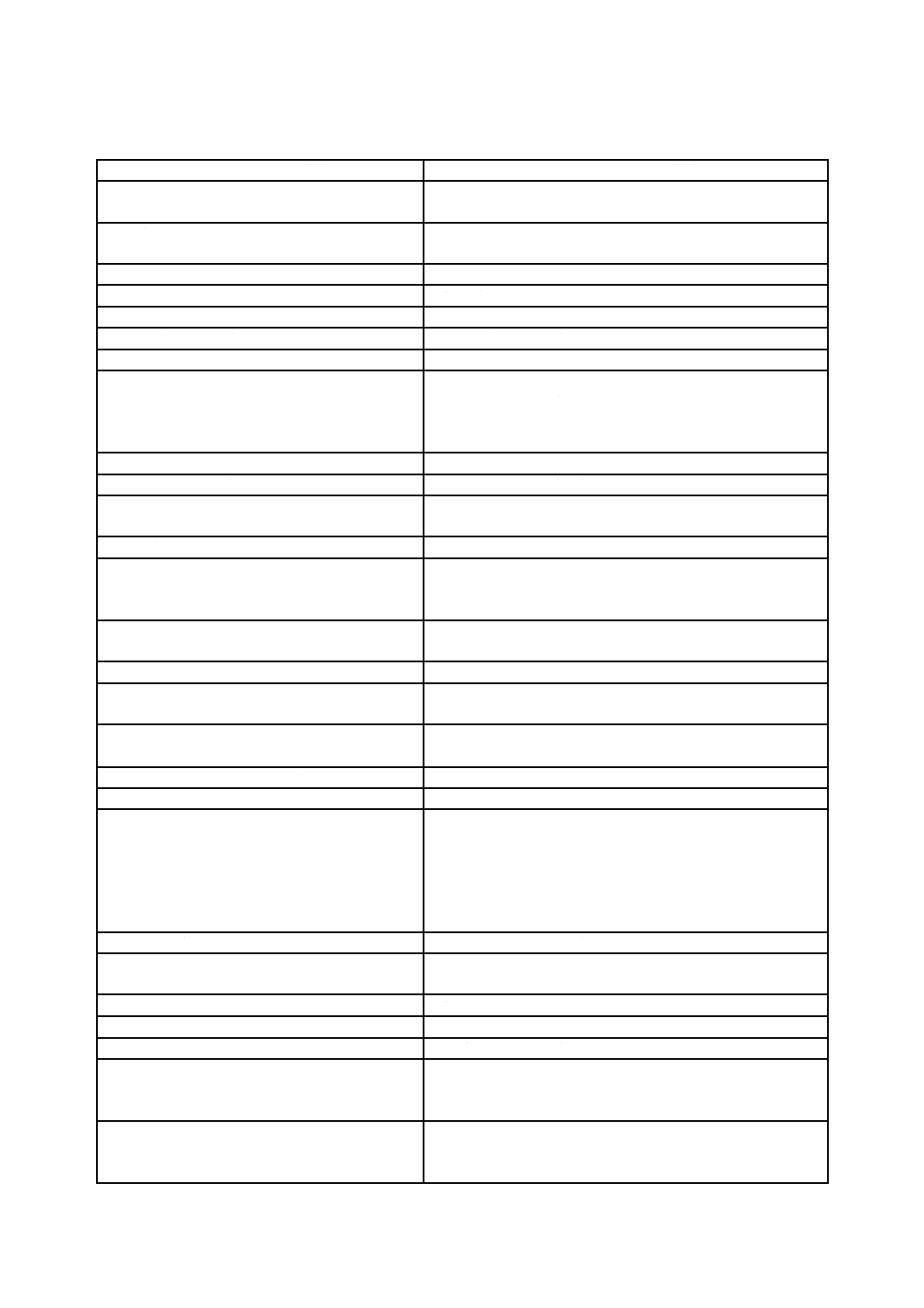
表B.1−JIS Q 13485:2018とJIS Q 9001:2015との関係(続き)
JIS Q 13485:2018の箇条
JIS Q 9001:2015の箇条
6 資源の運用管理
7.1 資源
6.1 資源の提供
7.1.1 一般
7.1.2 人々
6.2 人的資源
7.2 力量
7.3 認識
6.3 インフラストラクチャ
7.1.3 インフラストラクチャ
6.4 作業環境及び汚染管理
7.1.4 プロセスの運用に関する環境
7 製品実現
8 運用
7.1 製品実現の計画
8.1 運用の計画及び管理
7.2 顧客関連のプロセス
8.2 製品及びサービスに関する要求事項
7.2.1 製品に関連する要求事項の明確化
8.2.2 製品及びサービスに関する要求事項の明確化
7.2.2 製品に関連する要求事項のレビュー
8.2.3 製品及びサービスに関する要求事項のレビュー
8.2.4 製品及びサービスに関する要求事項の変更
7.2.3 コミュニケーション
8.2.1 顧客とのコミュニケーション
7.3 設計・開発
8.3 製品及びサービスの設計・開発
7.3.1 一般
8.3.1 一般
7.3.2 設計・開発の計画
8.3.2 設計・開発の計画
7.3.3 設計・開発へのインプット
8.3.3 設計・開発へのインプット
7.3.4 設計・開発からのアウトプット
8.3.5 設計・開発からのアウトプット
7.3.5 設計・開発のレビュー
8.3.4 設計・開発の管理
7.3.6 設計・開発の検証
8.3.4 設計・開発の管理
7.3.7 設計・開発のバリデーション
8.3.4 設計・開発の管理
7.3.8 設計・開発の移管
8.3.4 設計・開発の管理
7.3.9 設計・開発の変更管理
8.3.6 設計・開発の変更
8.5.6 変更の管理
7.3.10 設計・開発ファイル
7.5.3 文書化した情報の管理
7.4 購買
8.4 外部から提供されるプロセス,製品及びサービスの管理
7.4.1 購買プロセス
8.4 外部から提供されるプロセス,製品及びサービスの管理
8.4.1 一般
8.4.2 管理の方式及び程度
7.4.2 購買情報
8.4.3 外部提供者に対する情報
7.4.3 購買製品の検証
8.4.2 管理の方式及び程度
8.4.3 外部提供者に対する情報
8.6 製品及びサービスのリリース
7.5 製造及びサービスの提供
8.5 製造及びサービス提供
7.5.1 製造及びサービス提供の管理
8.5.1 製造及びサービス提供の管理
7.5.2 製品の清浄性
対応する箇条なし
7.5.3 据付け活動
対応する箇条なし
7.5.4 附帯サービス活動
対応する箇条なし
7.5.5 滅菌医療機器に対する特別要求事項
対応する箇条なし
7.5.6 製造及びサービス提供に関するプロセスバ
リデーション
8.5.1 製造及びサービス提供の管理
7.5.7 滅菌及び無菌バリアシステムのプロセスの
バリデーションに対する特別要求事項
対応する箇条なし
7.5.8 識別
8.5.2 識別及びトレーサビリティ
7.5.9 トレーサビリティ
8.5.2 識別及びトレーサビリティ
7.5.10 顧客の所有物
8.5.3 顧客又は外部提供者の所有物
32
Q 13485:2018 (ISO 13485:2016)
表B.1−JIS Q 13485:2018とJIS Q 9001:2015との関係(続き)
JIS Q 13485:2018の箇条
JIS Q 9001:2015の箇条
7.5.11 製品の保存
8.5.4 保存
7.6 監視機器及び測定機器の管理
7.1.5 監視及び測定のための資源
8 測定,分析及び改善
9 パフォーマンス評価
9.1 監視,測定,分析及び評価
8.1 一般
9.1.1 一般
8.2 監視及び測定
9.1 監視,測定,分析及び評価
8.2.1 フィードバック
8.5.5 引渡し後の活動
9.1.2 顧客満足
8.2.2 苦情処理
9.1.2 顧客満足
8.2.3 規制当局への報告
8.5.5 引渡し後の活動
8.2.4 内部監査
9.2 内部監査
8.2.5 プロセスの監視及び測定
9.1.1 一般
8.2.6 製品の監視及び測定
8.6 製品及びサービスのリリース
8.3 不適合製品の管理
8.7 不適合なアウトプットの管理
8.3.1 一般
10.2 不適合及び是正処置
8.3.2 引渡し前に発見した不適合製品における処
置
8.7 不適合なアウトプットの管理
8.3.3 引渡し後に発見された不適合製品における
処置
8.7 不適合なアウトプットの管理
8.3.4 手直し
8.7 不適合なアウトプットの管理
8.4 データの分析
9.1.3 分析及び評価
8.5 改善
10 改善
8.5.1 一般
10.1 一般
10.3 継続的改善
8.5.2 是正処置
10.2 不適合及び是正処置
8.5.3 予防処置
0.3.3 リスクに基づく考え方
6.1 リスク及び機会への取組み
10.1 一般
10.3 継続的改善

33
Q 13485:2018 (ISO 13485:2016)
表B.2−JIS Q 9001:2015とJIS Q 13485:2018との関係
JIS Q 9001:2015の箇条
JIS Q 13485:2018の箇条
1 適用範囲
1 適用範囲
4 組織の状況
4 品質マネジメントシステム
4.1 組織及びその状況の理解
4.1 一般要求事項
4.2 利害関係者のニーズ及び期待の理解
4.1 一般要求事項
4.3 品質マネジメントシステムの適用範囲の決定
4.1 一般要求事項
4.2.2 品質マニュアル
4.4 品質マネジメントシステム及びそのプロセス
4.1 一般要求事項
5 リーダーシップ
5 経営者の責任
5.1 リーダーシップ及びコミットメント
5.1 経営者のコミットメント
5.1.1 一般
5.1 経営者のコミットメント
5.1.2 顧客重視
5.2 顧客重視
5.2 方針
5.3 品質方針
5.2.1 品質方針の確立
5.3 品質方針
5.2.2 品質方針の伝達
5.3 品質方針
5.3 組織の役割,責任及び権限
5.4.2 品質マネジメントシステムの計画
5.5.1 責任及び権限
5.5.2 管理責任者
6 計画
5.4.2 品質マネジメントシステムの計画
6.1 リスク及び機会への取組み
5.4.2 品質マネジメントシステムの計画
8.5.3 予防処置
6.2 品質目標及びそれを達成するための計画策定
5.4.1 品質目標
6.3 変更の計画
5.4.2 品質マネジメントシステムの計画
7 支援
6 資源の運用管理
7.1 資源
6 資源の運用管理
7.1.1 一般
6.1 資源の提供
7.1.2 人々
6.2 人的資源
7.1.3 インフラストラクチャ
6.3 インフラストラクチャ
7.1.4 プロセスの運用に関する環境
6.4.1 作業環境
7.1.5 監視及び測定のための資源
7.6 監視機器及び測定機器の管理
7.1.5.1 一般
7.6 監視機器及び測定機器の管理
7.1.5.2 測定のトレーサビリティ
7.6 監視機器及び測定機器の管理
7.1.6 組織の知識
6.2 人的資源
7.2 力量
6.2 人的資源
7.3 認識
6.2 人的資源
7.4 コミュニケーション
5.5.3 内部コミュニケーション
7.5 文書化した情報
4.2 文書化に関する要求事項
7.5.1 一般
4.2.1 一般
7.5.2 作成及び更新
4.2.4 文書管理
4.2.5 記録の管理
7.5.3 文書化した情報の管理
4.2.3 医療機器ファイル
4.2.4 文書管理
4.2.5 記録の管理
7.3.10 設計・開発ファイル
8 運用
7 製品実現
8.1 運用の計画及び管理
7.1 製品実現の計画
8.2 製品及びサービスに関する要求事項
7.2 顧客関連のプロセス
8.2.1 顧客とのコミュニケーション
7.2.3 コミュニケーション
34
Q 13485:2018 (ISO 13485:2016)
表B.2−JIS Q 9001:2015とJIS Q 13485:2018との関係(続き)
JIS Q 9001:2015の箇条
JIS Q 13485:2018の箇条
8.2.2 製品及びサービスに関する要求事項の明確
化
7.2.1 製品に関連する要求事項の明確化
8.2.3 製品及びサービスに関する要求事項のレビ
ュー
7.2.2 製品に関連する要求事項のレビュー
8.2.4 製品及びサービスに関する要求事項の変更
7.2.2 製品に関連する要求事項のレビュー
8.3 製品及びサービスの設計・開発
7.3 設計・開発
8.3.1 一般
7.3.1 一般
8.3.2 設計・開発の計画
7.3.2 設計・開発の計画
8.3.3 設計・開発へのインプット
7.3.3 設計・開発へのインプット
8.3.4 設計・開発の管理
7.3.5 設計・開発のレビュー
7.3.6 設計・開発の検証
7.3.7 設計・開発のバリデーション
7.3.8 設計・開発の移管
8.3.5 設計・開発からのアウトプット
7.3.4 設計・開発からのアウトプット
8.3.6 設計・開発の変更
7.3.9 設計・開発の変更管理
8.4 外部から提供されるプロセス,製品及びサービ
スの管理
4.1 一般要求事項(4.1.5参照)
7.4.1 購買プロセス
8.4.1 一般
7.4.1 購買プロセス
8.4.2 管理の方式及び程度
4.1 一般要求事項(4.1.5参照)
7.4.1 購買プロセス
7.4.3 購買製品の検証
8.4.3 外部提供者に対する情報
7.4.2 購買情報
7.4.3 購買製品の検証
8.5 製造及びサービス提供
7.5 製造及びサービスの提供
8.5.1 製造及びサービス提供の管理
7.5.1 製造及びサービス提供の管理
7.5.6 製造及びサービス提供に関するプロセスバリデーション
8.5.2 識別及びトレーサビリティ
7.5.8 識別
7.5.9 トレーサビリティ
8.5.3 顧客又は外部提供者の所有物
7.5.10 顧客の所有物
8.5.4 保存
7.5.11 製品の保存
8.5.5 引渡し後の活動
7.5.1 製造及びサービス提供の管理
7.5.3 据付け活動
7.5.4 附帯サービス活動
8.2.2 苦情処理
8.2.3 規制当局への報告
8.3.3 引渡し後に発見された不適合製品における処置
8.5.6 変更の管理
7.3.9 設計・開発の変更管理
8.6 製品及びサービスのリリース
7.4.3 購買製品の検証
8.2.6 製品の監視及び測定
8.7 不適合なアウトプットの管理
8.3 不適合製品の管理
9 パフォーマンス評価
8 測定,分析及び改善
9.1 監視,測定,分析及び評価
8 測定,分析及び改善
9.1.1 一般
8.1 一般
8.2.5 プロセスの監視及び測定
8.2.6 製品の監視及び測定
9.1.2 顧客満足
7.2.3 コミュニケーション
8.2.1 フィードバック
8.2.2 苦情処理
35
Q 13485:2018 (ISO 13485:2016)
表B.2−JIS Q 9001:2015とJIS Q 13485:2018との関係(続き)
JIS Q 9001:2015の箇条
JIS Q 13485:2018の箇条
9.1.3 分析及び評価
8.4 データの分析
9.2 内部監査
8.2.4 内部監査
9.3 マネジメントレビュー
5.6 マネジメントレビュー
9.3.1 一般
5.6.1 一般
9.3.2 マネジメントレビューへのインプット
5.6.2 マネジメントレビューへのインプット
9.3.3 マネジメントレビューからのアウトプット
5.6.3 マネジメントレビューからのアウトプット
10 改善
8.5 改善
10.1 一般
8.5.1 一般
10.2 不適合及び是正処置
8.3 不適合製品の管理
8.5.2 是正処置
10.3 継続的改善
5.6.1 一般
8.5.3 予防処置
36
Q 13485:2018 (ISO 13485:2016)
参考文献
[1] JIS Q 9001:2015 1) 品質マネジメントシステム−要求事項
注記 対応国際規格:ISO 9001:2015,Quality management systems−Requirements
[2] JIS Q 10012 計測マネジメントシステム−測定プロセス及び測定機器に関する要求事項
注記 対応国際規格:ISO 10012,Measurement management systems−Requirements for measurement
processes and measuring equipment
[3] JIS T 0841-1 最終段階で滅菌される医療機器の包装−第1部:材料,無菌バリアシステム及び包装シ
ステムに関する要求事項
注記 対応国際規格:ISO 11607-1,Packaging for terminally sterilized medical devices−Part 1:
Requirements for materials, sterile barrier systems and packaging systems
[4] JIS T 0841-2 最終段階で滅菌される医療機器の包装−第2部:成形,シール及び組立プロセスのバリ
デーション
注記 対応国際規格:ISO 11607-2,Packaging for terminally sterilized medical devices−Part 2: Validation
requirements for forming, sealing and assembly processes
[5] ISO 14644 (all parts),Cleanrooms and associated controlled environments
注記1 ISO 14644-1については,JIS B 9920 クリーンルームの空気清浄度の評価方法(ISO
14644-1,MOD)がある。
注記2 ISO 14644-3については,JIS B 9917-3 クリーンルーム及び付属清浄環境−第3部:試験
方法(ISO 14644-3,MOD)がある。
注記3 ISO 14644-5 については,JIS B 9917-5 クリーンルーム運転における管理及び清浄化(ISO
14644-5,MOD)がある。
注記4 ISO 14644-7 については,JIS B 9917-7 クリーンルーム及び関連制御環境−第7部:隔離
装置(ISO 14644-7,MOD)がある。
注記5 ISO 14644-8 については,JIS B 9917-8 クリーンルーム及び関連制御環境−第8部:浮遊
分子状汚染物質に関する空気清浄度(ISO 14644-8,MOD)がある。
[6] JIS B 9918(全ての部) クリーンルーム及び関連制御環境−微生物汚染制御
注記 対応国際規格:ISO 14698 (all parts),Cleanrooms and associated controlled environments−
Biocontamination control
[7] JIS T 14971:2012 医療機器−リスクマネジメントの医療機器への適用
注記 対応国際規格:ISO 14971:2007,Medical devices−Application of risk management to medical
devices
[8] JIS Q 19011 マネジメントシステム監査のための指針
注記 対応国際規格:ISO 19011,Guidelines for auditing management systems
[9] IEC 62366-1,Medical devices−Part 1: Application of usability engineering to medical devices
[10] GHTF/SG1/N055:2009,Definition of the Terms “Manufacturer”, “Authorised Representative”, “Distributor”
and “Importer”2)
[11] GHTF/SG5/N4:2010,Post-Market Clinical Follow-Up Studies 3)
[12] GHTF/SG1/N70:2011,Label and Instructions for Use for Medical Devices 4)

37
Q 13485:2018 (ISO 13485:2016)
[13] GHTF/SG1/N071:2012,Definition of the Terms “Medical Device” and “In Vitro Diagnostic (IVD) Medical
Device”5)
注1) JIS Q 9001:2008から改正された。
2) http://www.imdrf.org/documents/doc-ghtf-sg1.asp から入手できる。
3) http://www.imdrf.org/documents/doc-ghtf-sg5.asp から入手できる。
4) http://www.imdrf.org/documents/doc-ghtf-sg1.asp から入手できる。
5) http://www.imdrf.org/documents/doc-ghtf-sg1.asp から入手できる。