M 8228 : 1997
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法に基づいて,日本工業標準調査会の審議を経て,通商産業大臣が改正した日
本工業規格である。これによってJIS M 8228-1983は改正され,この規格によって置き換えられる。
今回の改正では,国際規格との整合化を図るため,ISO規格の翻訳を附属書2として規定している。
JIS M 8228には,次に示す附属書がある。
附属書1(規定) 共存元素分離エチレンジアミン四酢酸二水素二ナトリウム(EDTA二ナトリウム)
滴定法
附属書2(規定) 原子吸光法
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
M 8228 : 1997
鉄鉱石−亜鉛定量方法
Iron ores−Methods for determination of zinc content
序文 この規格の,附属書1は,JIS M 8228-1983のEDTA滴定法を改正し規定した日本工業規格である。
附属書2は1987年に発行されたISO 8753, Iron ores−Determination of lead and/or zinc content−Flame atomic
absorption spectrometric methodの亜鉛部分を翻訳し,技術的内容及び規格票の様式を変更することなく作成
した日本工業規格である。
1. 適用範囲 この規格は,鉄鉱石中の亜鉛定量方法について規定する。
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。
これらの引用規格は,その最新版を適用する。
JIS K 8001 試薬試験方法通則
JIS M 8202 鉄鉱石−分析方法通則
3. 一般事項 定量方法に共通な一般事項は,JIS M 8202の規定による。
4. 定量方法 亜鉛の定量方法は,次のいずれかによる。
a) 共存元素分離エチレンジアミン四酢酸二水素二ナトリウム(EDTA二ナトリウム)滴定法 この方法
は,亜鉛含有率0.2% (m/m) 以上30% (m/m) 以下の試料に適用するもので,附属書1による。
b) 原子吸光法[国際一致規格 (ISO 8753)] この方法は,亜鉛含有率0.001% (m/m) 以上0.5% (m/m) 以
下の試料に適用するもので,附属書2による。
2
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(規定) 共存元素分離EDTA二ナトリウム滴定法
1. 要旨 試料を塩酸及び硝酸で分解し,乾固した後塩酸に溶解し,ろ過する。残さは,ふっ化水素酸で
処理した後,二硫酸カリウムで融解し,ろ液に合わせる。硫酸で白煙処理を行い,硫酸鉛などの沈殿をろ
過し,更にアンモニア水及びペルオキソ二硫酸アンモニウムで鉄,マンガンなどを沈殿分離した後,pHを
調節し,チオ硫酸ナトリウム及びふっ化アンモニウムで銅及びアルミニウムをマスキングし,キシレノー
ルオレンジを指示薬として,EDTA二ナトリウム標準溶液で滴定する。
2. 試薬 試薬は,次による。
a) 塩酸
b) 塩酸(1+1,1+100)
c) 硝酸
d) ふっ化水素酸
e) 硫酸(1+1,1+10)
f)
アンモニア水(1+1)
g) 塩化アンモニウム
h) 二硫酸カリウム
i)
ペルオキソ二硫酸アンモニウム
j)
ふっ化アンモニウム溶液 (50g/l)
k) 緩衝溶液 酢酸アンモニウム250gを水に溶解して,水で液量を1 000mlとし,これに酢酸約25mlを
加えて,pHを5.5に調節する。
l)
チオ硫酸ナトリウム溶液 チオ硫酸ナトリウム五水和物100gを水に溶解して,水で液量を1 000ml
とする。
m) ジメチルグリオキシムエタノール溶液 ジメチルグリオキシム1gをエタノール (95) 100mlに溶解す
る。不溶解分があればろ過する。
n) 0.01mol/lEDTA二ナトリウム標準溶液 この溶液の調製,保存及び標定方法は,JIS K 8001の4.5(3.3)
[0.01mol/lエチレンジアミン四酢酸二水素二ナトリウム溶液(0.01mol/lEDTA2Na溶液)]による。
o) p−ニトロフェノール溶液 (2g/l)
p) キシレノールオレンジ溶液 キシレノールオレンジ0.1gを水100mlに溶解する。不溶解分があればろ
過する。
3. 試料はかり採り量 試料はかり採り量は,附属書1表1による。

3
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1表1 試料はかり採り量
亜鉛含有率
% (m/m)
試料はかり採り量
g
0.2以上2未満
2.0
2以上 5未満
1.0
5以上 10未満
0.50
10以上 30以下
0.20
4. 操作
4.1
試料溶液の調製 試料溶液の調製は,次の手順によって行う。
a) 試料をはかり採ってビーカー (300ml) に移し入れて時計皿で覆い,塩酸30mlを加え,初めは熱板周
辺の低温部 (60〜100℃) にビーカーを置き,約1時間保持した後,更に高温部に移して約10分間煮
沸直前まで加熱して分解する。次に硝酸5mlを加えて鉄などを酸化し,更に加熱を続けてほとんど乾
固する。放冷した後,塩酸 (1+1) 30mlを加え,加熱して塩類を溶解し,ろ紙(5種B)と少量のろ紙
パルプを用いてビーカー (300ml) にろ過し,ろ紙及び残さを約40〜60℃に加熱した温塩酸(1+100)で
3〜4回,次に温水で3〜4回洗浄する。ろ液及び洗液は主液として保存する。また,試料溶解に用い
たビーカーも保存する。
b) 不溶解残さ(1)はろ紙とともに白金るつぼ(30番)に移し入れ,乾燥した後,強熱灰化して放冷する。
この強熱残さを硫酸 (1+1) で湿し,ふっ化水素酸約5mlを加えて穏やかに加熱し,二酸化けい素及
び硫酸を揮散させる放冷した後,これに二硫酸カリウム約2gを加え,ふたをして初めは徐々に加熱し,
次第に温度を高めて暗赤熱状に加熱して融解する。放冷した後,白金るつぼをa)で保存したビーカー
に入れ,温水約30mlを加え,加熱して融成物を溶解する。白金るつぼは水で洗って取り出し,溶液
はa)で保存した主液に合わせる。
注(1) 炭素が多量に含まれる場合は,磁器るつぼ (30ml) で乾燥した後,強熱して灰化する。強熱残
さを白金るつぼ(30番)に移し入れ,ふっ化水素酸処理以降の操作を行う。
4.2
鉛の分離 4.1で得た試料溶液に硫酸 (1+1) 20ml(2)を加え,加熱して濃厚な硫酸白煙を約10分間発
生させる。放冷した後,水約100ml(2)を加え,加熱して塩類を溶解し,常温まで冷却する。ろ紙(5種B)
を用いて250mlの全量フラスコにろ過し,硫酸 (1+10) で2〜3回,水で3〜4回洗浄した後,水で標線ま
で薄める。沈殿は捨てる。
注(2) 試料はかり採り量が2.0gの場合は,硫酸(1+1)40ml及び水約200mlとする。
4.3
マンガンなどの分離 マンガンなどの分離は,次の手順によって行う。
a) 4.2で得た試料溶液から50mlを分取して,ビーカー (300ml) に移し入れ,塩化アンモニウム5g及び
水約50mlを加え,溶液をかき混ぜながらアンモニア水 (1+1) を加えて中和し,更に過剰に20mlを
加える。次にペルオキソ二硫酸アンモニウム0.5gを加え,加熱して2分間煮沸する。生じた沈殿をろ
紙(5種A)を用いて三角フラスコ (500ml) にろ過し,温水で2〜3回洗浄する。ろ液及び洗液は主液
として保存する。
b) 沈殿を元のビーカーへ射水して洗い移し,ろ紙の上から約40〜60℃に加熱した温塩酸(1+1)10mlを加
え,ろ紙に付着している沈殿及びビーカー内の沈殿を溶解する。ろ紙は温水で3〜4回洗浄して,保存
する。この溶液に温水を加えて液量を約100mlとして,a)の塩化アンモニウム添加以降の手順に従っ
て操作して再び沈殿を生じさせ,保存したろ紙を用いてろ過し,温水で3〜4回洗浄する。ろ液及び洗
4
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
液はa)で保存した主液に合わせる。
4.4
pHの調整 4.3で得た溶液を穏やかに加熱して煮沸し,過剰のペルオキソ二硫酸アンモニウムを分
解し,引き続き煮沸を継続して大部分のアンモニアを除去する。常温まで冷却した後,p−ニトロフェノー
ル溶液2〜3滴を指示薬として加え(3),塩酸 (1+1) を滴加して溶液が黄色から無色に変化するまで中和す
る。これに緩衝溶液[2.k)]15mlを加え,pHを約5.5に調整する。
注(3) p−ニトロフェノール溶液を滴加しても溶液が黄色に変化しない場合は,アンモニア水(1+1)を
滴加して黄色にする。
4.5
滴定 滴定は,次のいずれかによる。
a) ニッケルを含まない試料
1) 4.4で得た溶液にチオ硫酸ナトリウム溶液[2.l)]5m1及びふっ化アンモニウム溶液5mlを加えた後,
水で液量を約350mlにする。
2) キシレノールオレンジ溶液[2.p)]4〜5滴を指示薬として加え,0.01mo1/lEDTA二ナトリウム標準溶液
[2.n)]で滴定し,溶液の色の赤が最後の1滴で黄色に変化する点を終点とする
b) ニッケルを含む試料
1) 4.4で得た溶液にチオ硫酸ナトリウム溶液[2.l)]5mlを加えた後,ジメチルグリオキシムエタノール溶
液[2.m)]10mlを加えて十分に振り混ぜ,ニッケルジメチルグリオキシムを沈殿させる。これをろ紙
(5種A)を用いて三角フラスコ (500ml) にろ過し,水で3〜4回洗浄する。次にふっ化アンモニウ
ム溶液5mlを加え,水で液量を約350mlにする。
2) a)2)に従って操作する。
5. 空試験 試料を用いないで,4.1〜4.5の手順に従って,試料と同じ操作を試料と併行して行う。
6. 計算 計算は,次による。
a) 亜鉛含有率の計算 4.5及び5.で得た滴定量から試料中の亜鉛含有率を,次の式によって算出する。
(
)
100
0006539
.0
2
1
×
×
×
−
=
B
m
V
V
Zn
ここに, Zn: 試料中の亜鉛含有率 [% (m/m)]
V1: 分取した試料溶液の滴定における0.01mol/lEDTA二ナトリウ
ム標準溶液の使用量 (ml)
V2: 分取した空試験液の滴定における0.01mol/lEDTA二ナトリウ
ム標準溶液の使用量 (ml)
m: 試料はかり採り量 (g)
B: 試料溶液及び空試験液の分取比
b) 酸化亜鉛含有率の計算 試料中の酸化亜鉛含有率は,亜鉛含有率から次の式によって算出する。
ZnO=1.2447×Zn
ここに, ZnO: 試料中の酸化亜鉛含有率 [% (m/m)]
Zn: a)に同じ
7. 許容差 許容差は,附属書1表2による。

5
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1表2 許容差
単位 % (m/m)
室内許容差
室間許容差
D (n) × [0.008 0×(亜鉛含有率)+0.014 4]
2.8× [0.018 5×(亜鉛含有率)+0.030 5]
n=2のとき,D (n) =2.8
n=3のとき,D (n) =3.3
n=4のとき,D (n) =3.6
参考 この許容差は,亜鉛含有率0.2% (m/m) 以上21% (m/m) 以下の試料を用いて求めたものである。
6
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2(規定) 原子吸光法
序文 この附属書は,1987年第一版として発行されたISO 8753 (Iron ores−Determination of lead and/or zinc
content−Flame atomic absorption spectrometric method) の亜鉛部分を翻訳し,技術的内容及び規格票の様式
を変更することなく作成した日本工業規格である。
なお,この附属書で下線(点線)を施してある“参考”は原国際規格にはない事項である。
1. 適用範囲
この附属書は,鉄鉱石中の亜鉛をフレーム原子吸光法によって定量する方法について規定する。
この方法は,天然鉄鉱石,精鉱及び焼結鉱を含む塊成鉱で,亜鉛の含有率0.001% (m/m) 以上0.5% (m/m)
以下の範囲のものに適用する。
参考 この方法は,硫化鉄焼鉱,スケール及びダスト又はこれらの粉粒状のものを加工した団鉱など
の鉄原料にも適用できる。
2. 引用規格
ISO 648 : 1977 Laboratory glassware−One-mark pipettes
ISO 1042 : 1983 Laboratory glassware−One-mark volumetric flasks
ISO 3081 : 1986 Iron ores−Increment sampling−Manual method
ISO 3082: 1987 Iron ores−Increment sampling and sample preparation−Mechanical method
ISO 3083 : 1986 Iron ores−Preparation of samples−Manual method
ISO 7764 : 1985 Iron ores−Preparation of predried test samples for chemical analysis
3. 原理
試料を分解し,塩酸とふっ化水素酸による処理で二酸化けい素を除去する。硝酸で酸化する。
蒸発乾固した後,希釈してろ過する。残さを灰化して炭酸ナトリウムで融解する。冷却した融成物を塩
酸で溶解する。溶液を保存する。
ろ液中の鉄を4−メチル−2−ペンタノンで抽出する。抽出された亜鉛を回収する。4−メチル−2−ペン
タノンを硝酸で分解する。蒸発乾固した後,保存溶液と塩酸で塩類を溶解する。
原子吸光光度計の空気・アセチレンバーナーのフレームに溶液を噴霧する。
亜鉛の吸光度を検量線溶液の吸光度と比較する。
4. 試薬
分析の際は,分析用保証試薬 (recognized analytical grade),蒸留水又はこれと同等の純度の水を使用する。
4.1
無水炭酸ナトリウム
4.2
4−メチル−2−ペンタノン 高純度のもの。
4.3
塩酸(密度1.16〜1.19g/ml)
4.4
塩酸(密度1.16〜1.19g/ml)の希釈液10+6
4.5
塩酸(密度1.16〜1.19g/ml)の希釈液1+1
4.6
塩酸(密度1.16〜1.19g/ml)の希釈液2+98
7
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.7
硝酸(密度1.4g/ml)
4.8
ふっ化水素酸,40% (m/m),(密度1.13g/ml)
4.10 標準亜鉛溶液
4.10.1 標準亜鉛原液
高純度金属亜鉛(鉄を含まない。)1.000gを塩酸(4.5)40mlに溶解する。冷却した後,1 000mlの全量フラ
スコに移し入れ,水で希釈し振り混ぜる。
この原液1mlは亜鉛1 000μgを含有する。
参考 高純度金属亜鉛は,純度が99.9% (m/m) 以上のものを使用する。
4.10.2 標準溶液
標準亜鉛原液(4.10.1)10.0mlを1 000mlの容量フラスコに移し入れ,水で標線まで薄めて振り混ぜる。
この標準溶液1mlは亜鉛10μgを含有する。
参考 試薬番号に4.9がないのはISO 8753を二分割したことによるものであり,鉛に関する番号を削
除したためである。
5. 装置
注 ピペット及びフラスコは,ISO 648及びISO 1042に規定されている1標線付きピペット及び1標
線付き容量フラスコでなければならない。
通常の実験器具及び次のものを使用する。
5.1
ポリテトラフロロエチレン (PTFE) 製ビーカー PTFE製ふた付きの容量250mlのもの。
5.2
原子吸光光度計 空気・アセチレンバーナーを備えているもの。この方法で使用する原子吸光光度
計は,次の装置基準を満足することが望ましい。
a) 最小感度−検量線最高濃度(7.4.4参照)の溶液の吸光度は,少なくとも0.25。
b) 検量線の直進性−検量線の上部20%範囲のこう配(吸光度の変化で表す。)と,同じやり方で算出し
た下部20%範囲のこう配の比が,0.7以上。
c) 最小安定性 (minimum stability) −検量線最高濃度溶液とゼロ検量線溶液をそれぞれ十分な回数の繰
り返し測定をして,得た標準偏差がそれぞれ最高濃度溶液の平均吸光度の1.5%,0.5%以下。
参考 最小安定性の求め方
最高濃度の検量線溶液をn回噴霧し,個々の吸光度の読みAAiを求めて,平均値AAを計算す
る。
最低濃度の検量線溶液(ゼロ検量線溶液を除く。)をn回噴霧し,個々の吸光度の読みABiを
求めて,平均値ABを計算する(nは10回以上)。
最高及び最低濃度の検量線溶液の各々の標準偏差sA及びsBを次の式で計算する。
(
)
(
)
1
1
2
2
2
−
Σ
−
Σ
=
−
−
Σ
=
n
n
Ai
Ai
n
A
A
s
A
Ai
A
(
)
(
)
1
1
2
2
2
−
Σ
−
Σ
=
−
−
Σ
=
n
n
Bi
Bi
n
A
A
s
B
Bi
B
最高濃度及び最低濃度の検量線溶液の各々の最小安定性はsA×100/AA及びsB×100/AAの式
で求める。
8
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注1. 基準a),b)及びc)の評価及び引き続き行われるすべての測定に対しては,チャート式記録装置
及び/又はデジタル表示装置の使用を推奨する。
2. 測定条件は装置ごとに変わる可能性がある。次に示す測定条件は,数箇所の分析室で支障なく
用いられた条件であり,操作の指針として用いることができる。
亜鉛中空陰極ランプの電流,mA
8
波長,nm
213.9
空気の流量,l/min
14
アセチレンのガス流量,l/min
3
上に示したガス流量が適当でない装置においても,ガス流量の比率は操作の指針となる。
参考 装置基準については,JIS M 8202(鉄鉱石−分析方法通則)の解説に記載されている。
6. サンプリング及び試料
6.1
分析用試料 (laboratory sample)
分析には,ISO 3081又はISO 3082に従って採取され,ISO 3082又はISO 3083に従って調製された粒度
−100μmの分析用試料を用いる。化合水又は酸化しやすい化合物の含有率が著しく高い鉱石の場合には,
粒度−160μmの試料を用いる。
注 化合水及び酸化しやすい化合物の著しく高い含有率についてのガイドラインは,ISO 7764に記載
されている。
参考 化合水及び酸化しやすい化合物の含有率については,JIS M 8202に記載されている。
6.2
事前乾燥試験試料 (predried test samples) の調製
分析用試料を十分に混合し,容器の全量を代表するように数インクリメントで分析試料を採取する。分
析試料をISO 7764に従って105±2℃で乾燥する(これを事前乾燥試料という。)。
7. 操作
7.1
分析回数
分析は,事前乾燥試料1個について,附属書2Aに従って少なくとも独立に2回の分析を行う。
注 “独立に”という表現は,2度目又は続いて行った分析結果が以前の結果によって影響を受けな
いことを意味する。特にこの分析方法では,この条件は操作の繰り返しが同一人が異なった時間
に,又は異なった人によって,いずれの場合も適切な再校正を含めて行われなければならないこ
とを意味する。
7.2
空試験及びチェック試験
一連の定量ごとに,1回の空試験と,同一種類の鉄鉱石認証標準物質の1個を,1分析試料(1個又は数
個)と併行して同一条件で分析しなければならない。認証標準物質の事前乾燥試料は6.2に従って調製し
なければならない。
注 認証標準物質は分析試料と同一種類で,両者の性質が分析操作の重大な変更を必要としない程度
によく類似したものでなければならない。
参考 空試験は,はかり採り試料と同量の純酸化鉄[亜鉛含有率0.000 1% (m/m) 以下のもの]をはか
り採って加えなければならない。
同時に数試料を分析する場合,操作が同じで同一の試薬瓶からの試薬を使うのであれば,1個の空試験
値で代表することができる。
9
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
同時に同一種類の鉱石の数試料を分析する場合は,1個の認証標準物質の分析値を使用することができ
る。
7.3
はかり採り試料 (test portion)
6.2に従って得られた事前乾燥試料から数インクリメントを採って,約2gを0.000 2gのけたまではかる。
注 はかり採り試料は,水分の再吸収を防ぐために迅速にはかり採るべきである。
7.4
定量
7.4.1
試料の分解
はかり採り試料(7.3)を250mlのPTFE製ビーカー(5.1)に移す。水数mlで湿し,塩酸(4.3)40mlとふっ化水
素酸(4.8)10mlを加え,PTFE製のふたで覆う。100℃の熱板で加熱し,次に200℃で加熱する。蒸発乾固す
る。硝酸(4.7)5mlを加え,溶液が約1mlになるまで加熱蒸発する。塩酸(4.3)10mlを加え塩類を溶解した後,
再び蒸発乾固する。
塩類を塩酸(4.3)5mlで溶解する。水10mlを加え,ち密なろ紙を用いて250mlのビーカーにろ過する。ビ
ーカーに付着している粒子はすべてゴム帽付きガラス棒でこすりおとし,ろ紙に鉄イオンの色が認められ
なくなるまで塩酸(4.6)で洗い,さらに熱水でろ紙を3回洗浄する。残さ,ろ液及び洗液を保存する。
参考 “ち密なろ紙”には,5種Cが相当する。
7.4.2
残さ処理
残さとろ紙を白金るつぼに入れ,低温でろ紙を乾燥して炭化し,次に550℃のマッフル炉中で強熱する。
無水炭酸ナトリウム(4.1)0.5gを加え,ブンゼンバーナー(約900〜1 000℃)上で透明な融成物ができるま
で融解する。冷却した融成物を塩酸(4.5)5mlで溶解した後,加熱して二酸化炭素を追い出し,溶液を保存
する。
7.4.3
ろ液及び洗液の処理
ろ液及び洗液(7.4.1)をほとんど乾固するまで蒸発する。塩類を塩酸(4.4)20mlで溶解し,200mlの分液漏
斗に移す。塩酸(4.4)20mlでビーカーを洗い,この洗液を主液に合わせる。4−メチル−2−ペンタノン
(4.2)50mlを加え,1分間激しく振り混ぜる。放置して二層に分離したら,下層の水溶液を250mlのビーカ
ーに流出させる。有機相を塩酸(4.4)10mlで抽出することによって洗浄し,洗液は先のビーカーに移す。
この溶液を穏やかに加熱し,溶液中のほとんど全部の4−メチル−2−ペンタノンを揮散させた後,硝酸
(4.7)5mlを加え,蒸発乾固する。塩類は塩酸(4.5)15mlで溶解した後,7.4.2で保存した溶液に合わせる。
溶液を50mlの全量フラスコに移し,水で標線まで薄めて振り混ぜる。吸光度測定のため,この溶液中
の亜鉛の含有率に応じて希釈しないか又は規定されたように希釈する(附属書2表1参照)。希釈する場合
は適当量を分液して250mlのビーカーに移し,附属書2表1に示す量の無水炭酸ナトリウム(4.1)と塩酸(4.5)
を加えた後,加熱して二酸化炭素を追い出す。冷却した後,100mlの全量フラスコに移し,水で標線まで
薄めて振り混ぜる(附属書2表2の注を参照)(この溶液を,最終試料溶液という。)。
参考 “附属書2表2の注”は,附属書2表1の注*の誤り。
空試験溶液の相当量を250mlのビーカーに移し,試料溶液に用いたのと同じ量の無水炭酸ナトリウム
(4.1)と塩酸(4.5)を加えた後,加熱して二酸化炭素を追い出す。冷却した後,100mlの全量フラスコに移し,
水で標線まで薄めて振り混ぜる(この溶液を,希釈空試験溶液という。)。
参考 この節は,試料溶液を希釈する(附属書2表1参照)場合にだけ適用する。
10
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
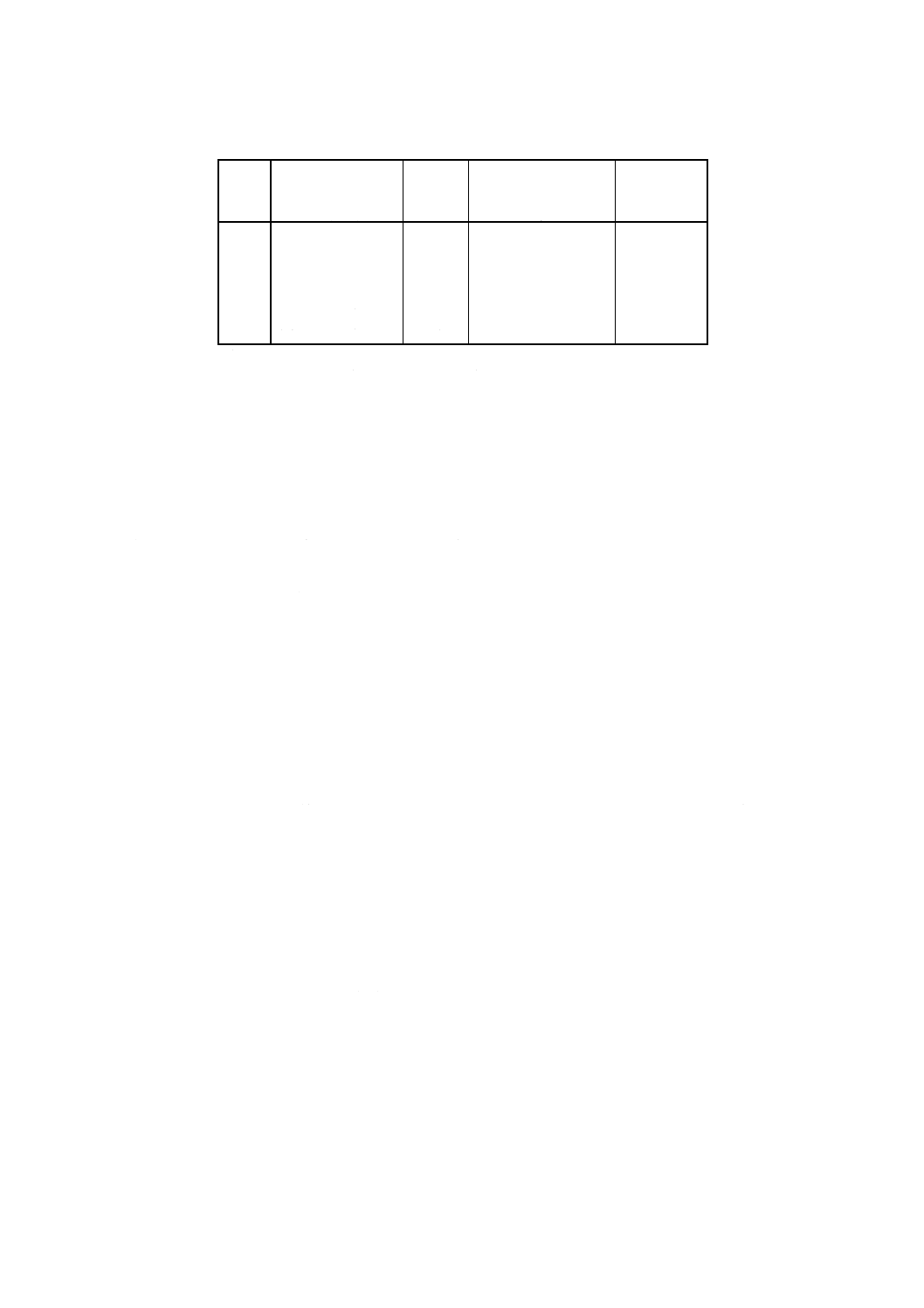
附属書2表1 試料溶液の希釈指針*
元素
試料中予想含有率
% (m/m)
分取量
ml
無水炭酸ナトリウム
(4.1)添加量
g
塩酸(4.5)
添加量
ml
亜鉛
0.001<wZn≦0.006
−
−
1
0.006<wZn≦0.02
25
0.75
30
0.02 <wZn≦0.06
10
0.9
36
0.06 <wZn≦0.12
5
0.95
38
0.12 <wZn≦0.3
2
0.98
39
0.3 <wZn≦0.5
1
0.99
40
注*
表に示した希釈に従えば,亜鉛の濃度が検量線溶液(7.4.4)の濃度範囲
に入る。高感度の装置では,試料溶液を少量分取することが好ましい。
1ml又は2mlの分取については,希釈誤差を避けるためにあらかじめ
希釈しておく。空試験溶液についても同様に取り扱う。
7.4.4
亜鉛検量線溶液の調製
6個の250mlのビーカーの各々に無水炭酸ナトリウム(4.1)1.0gを入れる。塩酸(4.3)20mlと標準亜鉛溶液
を表2に従って加える。加熱して二酸化炭素を追い出す。冷却した後,100mlの全量フラスコに移し,水
で標線まで薄めて振り混ぜる。
注 適用できる亜鉛の濃度範囲は,原子吸光光度計によって異なる。5.2で示す装置基準に注意すべき
である。高感度の装置では,標準溶液の分取量を減じて使用することができる。
7.4.5
原子吸光光度計の調整
亜鉛の波長 (213.9nm) を合わせ,吸光度が最小になるようにセットする。バーナーを合わせ,装置の取
扱書に従って,適正なフレームを点火する。バーナーを2分間予備加熱した後,最高濃度(7.4.4参照)検
量線溶液を噴霧しながら吸光度が最大になるように燃料とバーナーを調節し,5.2に定める装置基準を確認
する。
水と検量線溶液を噴霧して,吸光度の読取り値が変動していないことを確認した後,水に対する読み値
を吸光度ゼロに合わせる。
7.4.6
吸光度の測定
希釈空試験溶液とゼロ検量線溶液から始めて,最終試験溶液が一連の測定の適切な時点で噴霧できるよ
うに,検量線溶液及び最終試料溶液を吸光度が増加していくような順序で噴霧する。安定した応答が得ら
れたら,その読取り値を記録し,各溶液の噴霧の間には水を噴霧する。
測定は,少なくとも2回以上繰返す。必要ならば,各溶液の読取り値の平均を吸光度に変換する。ゼロ
検量線溶液の吸光度を差し引いて,各検量線溶液の真 (net) の吸光度を求める。同様に希釈空試験溶液の
吸光度を差し引いて,最終試料溶液の真 (net) の吸光度を求める。
亜鉛の濃度μg/mlに対して,検量線溶液の真 (net) の吸光度をプロットして,検量線を作成する。
検量線を用いて最終試料溶液の真 (net) の吸光度をml当たりの亜鉛のμgに変換する

11
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2表2 検量線溶液
溶液番号
亜鉛
標準溶液 (4.10.2)
ml
濃度
μg/ml
0
0
0
1
5
0.5
2
10
1
3
15
1.5
4
20
2
5
25
2.5
8. 結果の表示
8.1
亜鉛含有率の計算
亜鉛含有率(質量百分率)wZnは次の式を用いて小数点以下5けたまで計算する。
10000
1×
=m
V
wZn
ρ
········································································ (1)
ここに,
ρ: 最終試料溶液中の亜鉛の濃度 (μg/ml)
V: 最終試料溶液の量 (ml)
m1: 希釈率を計算に入れた最終試料溶液中の試料の質量 (g)
50
1
1
V
m
m
×
=
ここに,
m: はかり採り試料の質量 (g)
V1: 7.4.3に従って分取した量 (ml) (表1参照)
希釈なしの場合は,V1=50
8.2
結果の一般的処理
8.2.1
精度及び許容差
この分析方法の精度は,次の回帰式で表される。1)
r=0.041 5X+0.001 1 ··································································· (2)
P=0.104 8X+0.003 0 ·································································· (3)
σr=0.014 7X+0.000 4 ································································· (4)
σL=0.035 5X+0.001 0 ································································· (5)
ここに,
X: 分析試料の亜鉛を質量百分率で表したもの
− 室内計算式[式(2)及び式(4)の場合]:2回の分析値の算術平均
− 室間計算式[式(3)及び式(5)の場合]:2か所の分析室の最終結
果 (8.2.3) の算術平均
r: 室内許容差
P: 室間許容差
σr: 室内標準偏差
σL: 室間標準偏差
参考 PはRに,σLはσRに改正されている (ISO 5725 : 1994)。
8.2.2
分析値の採択
認証標準物質で求めた結果において,この分析結果と標準物質の認証値との間に統計的に有意差が認め
られてはならない。真度 (accuracy) 及び精度 (precision) ともにこの方法に相当する分析方法を用いて,
注1) 追加の情報は附属書2B及び附属書2Cに記載されている。
12
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
少なくとも10か所の分析室で分析した標準物質に対しては,有意差の検定には次の式を用いる。
n
N
n
s
s
s
A
A
r
L
c
Wc
Wc
Lc
c
2
2
2
2
σ
σ+
+
+
≤
−
···················································· (6)
ここに,
AC: 認証値
A: 認証標準物質を分析して得られた結果又はその平均値
SLc: 認証値を決定した分析室の室間標準偏差
SWc: 認証値を決定した分析室の室内標準偏差
nWc: 認証値を決定した分析室の分析回数の平均
Nc: 認証値を決定した分析室の数
n: 認証標準物質の分析回数(ほとんどの場合n=1)
σL及びσr: 8.2.1に定義してあるとおり。
もし,式(6)の左辺が右辺より小さいか又は等しければ,差|Ac−A|は統計的に有意ではなく,逆の場合は
統計的に有意である。
差が有意であるときは,試料の分析と同時に認証標準試料の分析を繰り返す。もし,差が再び有意であ
るならば,同じ種類で別の認証標準物質を用いて同じ操作を繰り返さなければならない。
分析試料の二つの分析値の範囲が8.2.1の式(2)に従って計算されたrの限界を超えるときは,附属書2A
のフローシートに従って,更にもう一度同じ種類の認証標準物質とともに分析試料の分析を行わなければ
ならない。
分析試料の結果の採択の可否は,いつの場合も認証標準物質の結果の採択の可否に従わなければならな
い。
注 認証標準物質の情報が不十分なときには,次の手順を用いる。
a) 室間標準偏差を推定するのに十分なデータがあれば,s2Wc/nWcを削除して,sLcを室平均値の標
準偏差とみなす。
b) もし,認証標準物質の認証が1分析室だけで行われている場合,又は室間の分析結果がない
場合には,この認証標準物質はこの規格には適用しないのがよい。その使用が避けられない
場合は,次の式を用いる。
n
A
A
r
L
c
2
2
2
2
σ
σ+
≤
−
································································· (7)
8.2.3
最終結果の計算
最終結果は,分析試料の採択し得る値の算術平均か,又は附属書2Aに規定した手順によって求めた値
で,小数点以下5けたまで計算し,次のようにして小数点以下3けたに丸めなければならない。
a) 小数点以下4けた目の数値が5より小さいときには,それを切り捨て,小数点以下3けた目の数値は
そのままとする。
b) 小数点以下4けた目の数値が5で,小数点以下5けた目に0以外の数値があるとき,又は小数点以下
4けた目の数値が5より大きいときには,小数点以下3けた目の数値を一つだけ増加させる。
c) 小数点以下4けた目の数値が5で,小数点以下5けた目が0のときは,小数点以下4けた目の5を切
り捨て,小数点以下3けた目の数値が0,2,4,6又は8であれば,小数点以下3けた目の数値はその
ままとし,1,3,5,7又は9であれば小数点以下3けた目の数値は切り上げて数を一つだけ増加させ
る。
8.3
酸化物換算係数
wZnO (%) =1.2447×wZn (%)
13
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9. 試験結果の報告
試験結果の報告には,次の情報を記載する。
a) この附属書の引用
b) 試料の識別に必要な事項
c) 分析結果
d) 分析結果の参照番号
e) 定量時に気がついた特定事項及びこの附属書に規定がない操作で,分析試料又は認証標準物質の分析
結果に影響を与えているおそれのある操作。
14
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
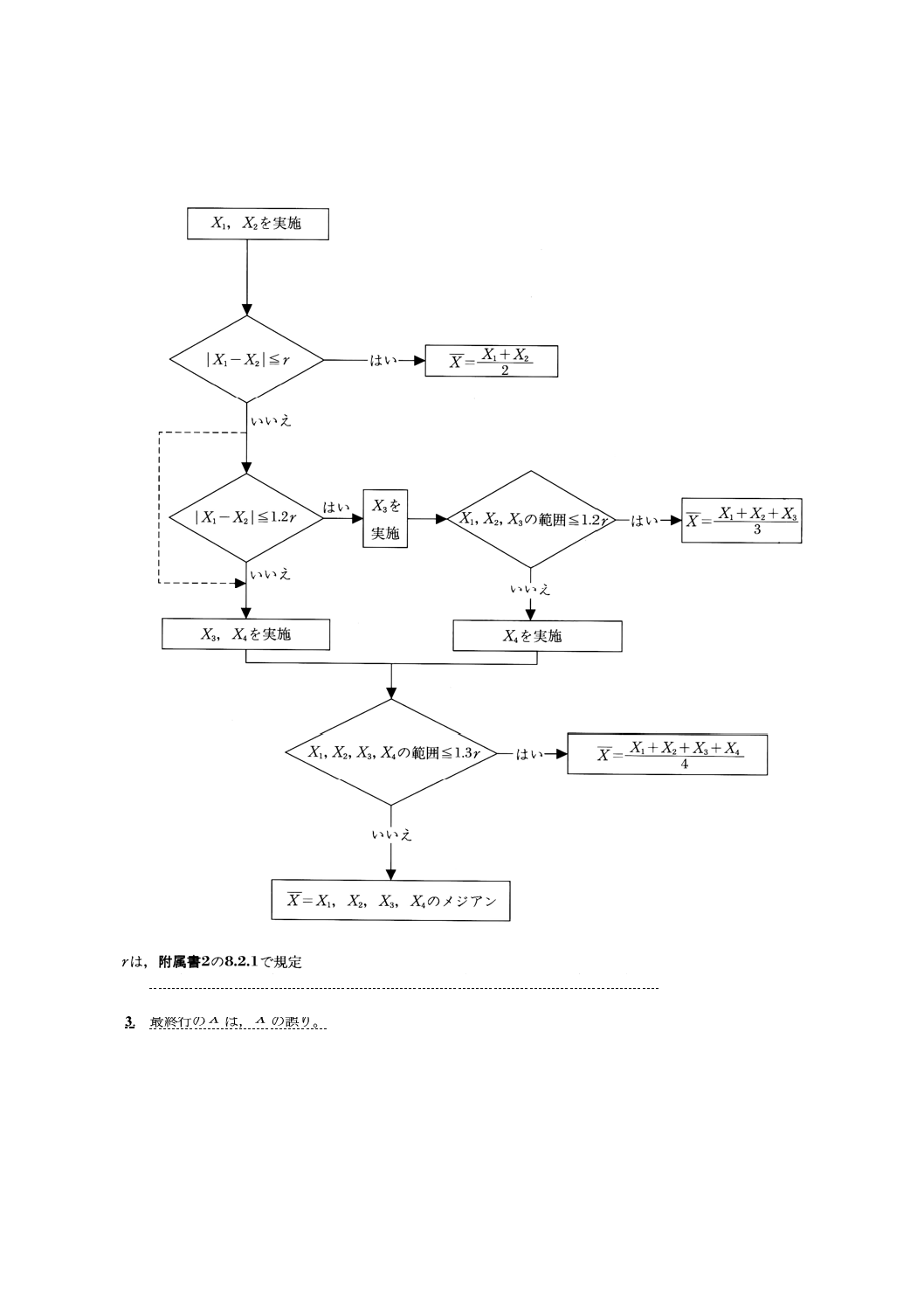
附属書2A(規定) 分析値の採択手順のフローシート
参考1. 偶数個に対するメジアンは,数値を大きさの順に並べたときの中央2個の平均値。
2. 1.2rで検定せずに,直ちにX3,X4を実施してもよい(破線部)。
3. 最終行のXは,X~の誤り。
15
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2B(参考) 精度及び許容差回帰式の典拠
附属書2の8.2.1の回帰式は,9か国の22分析室で5種類の鉄鉱石試料を用い,1981年/1982年に実施
した国際共同分析実験の結果から求められている。
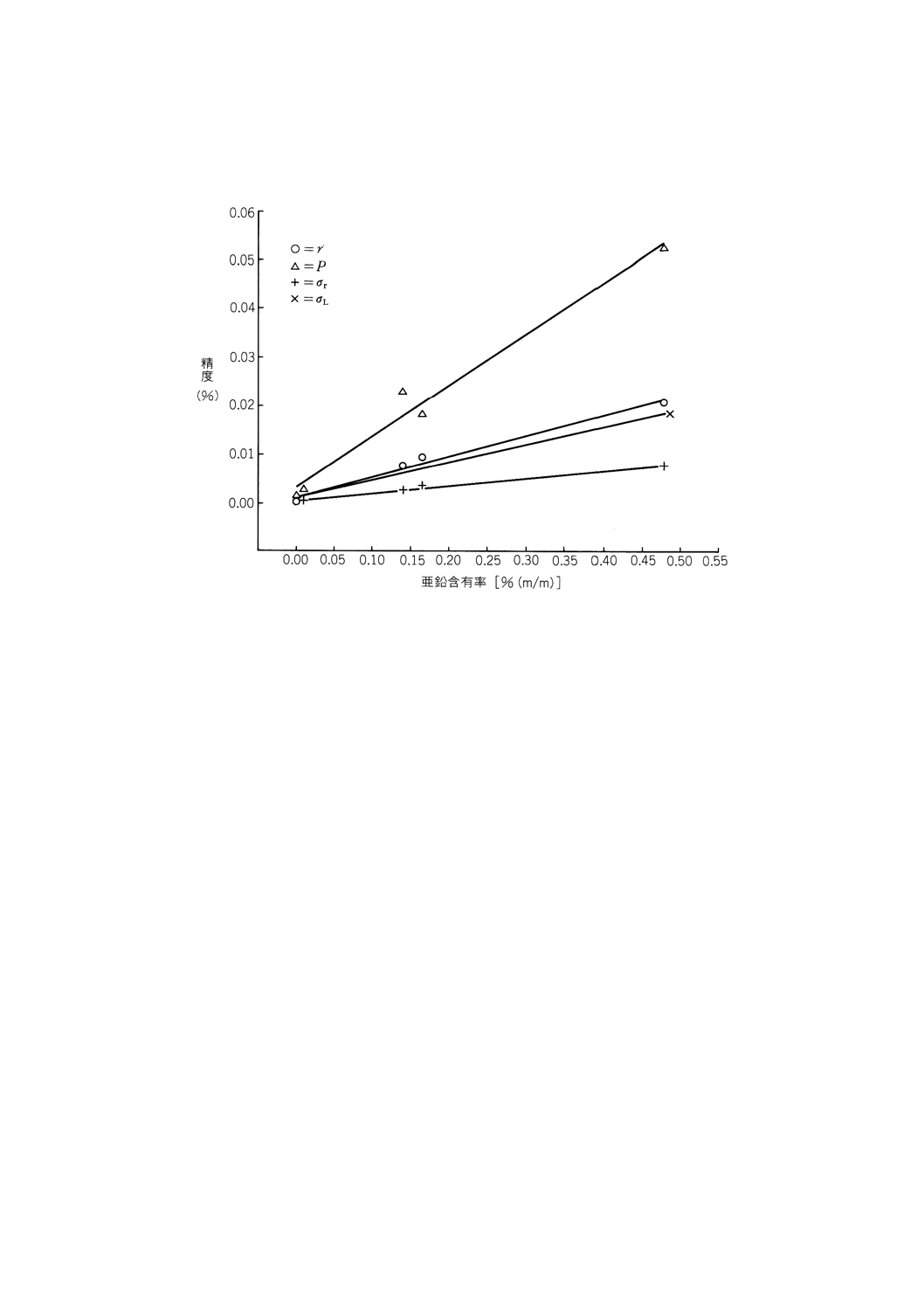
精度データは,附属書2C図1に図示してある。
分析試料には,次のものを使用した。
試料
亜鉛含有率
[% (m/m)]
Sishen iron ore (76−16)
0.001 6
Robe River (76−21)
0.009 9
Stollberg (79−1)
0.139 8
Whyalla Ore (79−13)
0.477 8
Purpur Ore (80−1)
0.164 9
注1. この国際共同分析実験及び統計的解析の結果報告書(文書ISO/TC 102/SC 2N702E及び2N740E)
は,ISO/TC 102/SC 2事務局又はISO/TC 102事務局で入手できる。
2. 統計的解析はISO 5725で規定している原則に従って行った。
参考 ISO 5725 : 1986は,次の改訂版が発行済みである。
ISO 5725-1 : 1994 Accuracy (trueness and precision) of measurement methods and results−Part
1 : General principles and definitions
ISO 5725-2 : 1994 Accuracy (trueness and precision) of measurement methods and results−Part
2 : Basic method for the determination of repeatability and reproducibility of a standard
measure-ment method
ISO 5725-3 : 1994 Accuracy (trueness and precision) of measurement methods and results−Part
3 : Intermediate measures of the precision of a standard measurement method
ISO 5725-4 : 1994 Accuracy (trueness and precision) of measurement methods and results−Part
4 : Basic methods for the determination of the trueness of a standard measurement method
ISO 5725-6 : 1994 Accuracy (trueness and precision) of measurement methods and results−Part
6 : Use in practice of accuracy values
16
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2C(参考) 国際共同分析実験で得られた精度データ
注 この図は,附属書2の8.2.1の回帰式をグラフ表示したものである。
附属書2C図1 亜鉛含有率X [% (m/m)] に対する精度の最小二乗法による回帰線
17
M 8228 : 1997
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
原案作成委員会
氏名
所属
(鉄鋼分析部会部会長)
佐 伯 正 夫
新日本製鐵株式会社
(化学分析分科会主査)
岩 田 英 夫
日本鋼管株式会社
(鉄鉱石JIS改正WGリーダー)
岩 田 英 夫
日本鋼管株式会社
(直属幹事)
石 橋 耀 一
日本鋼管株式会社
(委員)
岡 野 輝 雄
川崎製鉄株式会社
杉 原 孝 志
川崎テクノリサーチ株式会社
中 川 孝
川崎テクノリサーチ株式会社
秋 窪 英 敏
合同製鐵株式会社
金 築 宏 治
株式会社神戸製鋼所
河 村 恒 夫
株式会社コベルコ科研
稲 本 勇
新日本製鐵株式会社
大 水 勝
新日本製鐵株式会社
笠 井 茂 夫
新日本製鐵株式会社
鈴 木 興 三
新日本製鐵株式会社
鈴 木 節 雄
新日本製鐵株式会社
土 屋 武 久
新日本製鐵株式会社
蔵 保 浩 文
住友金属工業株式会社
中 里 福 和
住友金属工業株式会社
西 野 和 美
住友金属工業株式会社
平 松 茂 人
住友金属工業株式会社
菅 野 清
株式会社中山製綱所
西 田 宏
日新製鋼株式会社
小 倉 正 之
日本鋼管株式会社
船 曵 佳 弘
日本鋼管株式会社
大 槻 孝
社団法人日本鉄鋼協会
増 喜 浩 二
社団法人日本鉄鋼協会
(原料標準委員会委員長)
安 達 良 英
新日本製鐵株式会社
(JM2分科会主査)
松 村 泰 治
川崎テクノリサーチ株式会社
(委員)
中 林 賢 司
通商産業省工業技術院
藤 本 京 子
川崎製鉄株式会社
滝 沢 佳 郎
川崎テクノリサーチ株式会社
岡 山 和 生
合同製鐵株式会社
金 築 宏 治
株式会社神戸製鋼所
今 北 毅
株式会社コベルコ科研
西 埜 誠
株式会社島津製作所
笠 井 茂 夫
新日本製鐵株式会社
菊 池 統 一
新日本製鐵株式会社
鈴 木 節 雄
新日本製鐵株式会社
松 本 義 朗
住友金属工業株式会社
西 野 和 美
住友金属テクノロジー株式会社
原 田 幹 雄
株式会社中山製綱所
槌 尾 武 久
日新製鋼株式会社
林 三 男
社団法人日本海事検定協会
石 橋 耀 一
日本鋼管株式会社
吉 岡 豊
日本鋼管株式会社
河 野 久 征
理学電機工業株式会社
大 槻 孝
社団法人日本鉄鋼連盟
脊 戸 雄 功
社団法人日本鉄鋼連盟