M 8222-1:2018
(1)
目 次
ページ
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 一般事項························································································································· 1
4 要旨······························································································································· 1
5 試薬······························································································································· 2
6 試料のはかりとり ············································································································· 3
7 操作······························································································································· 3
7.1 試料の分解及び不溶解残さの処理······················································································ 3
7.2 水酸化物分離 ················································································································ 3
7.3 妨害元素の分離 ············································································································· 4
7.4 カルシウムとマグネシウムとの合量の滴定 ·········································································· 4
7.5 カルシウムの滴定 ·········································································································· 4
8 空試験···························································································································· 5
9 計算······························································································································· 5
10 許容差 ·························································································································· 5
M 8222-1:2018
(2)
まえがき
この規格は,工業標準化法第12条第1項の規定に基づき,一般社団法人日本鉄鋼連盟(JISF)から,工
業標準原案を具して日本工業規格を制定すべきとの申出があり,日本工業標準調査会の審議を経て,経済
産業大臣が制定した日本工業規格である。
これによって,JIS M 8222:1997は廃止され,その一部を分割して制定したこの規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格の一部が,特許権,出願公開後の特許出願又は実用新案権に抵触する可能性があることに注意
を喚起する。経済産業大臣及び日本工業標準調査会は,このような特許権,出願公開後の特許出願及び実
用新案権に関わる確認について,責任はもたない。
JIS M 8222の規格群には,次に示す部編成がある。
JIS M 8222-1 第1部:共存元素分離エチレンジアミン四酢酸二水素二ナトリウム滴定法
JIS M 8222-2 第2部:原子吸光分析法
日本工業規格 JIS
M 8222-1:2018
鉄鉱石−マグネシウム定量方法−
第1部:共存元素分離
エチレンジアミン四酢酸二水素二ナトリウム滴定法
Iron ores-Determination of magnesium-
Part 1: Disodium dihydrogen ethylenediamine tetraacetate titrimetric
determination after separation of co-existed elements
1
適用範囲
この規格は,鉄鉱石中のマグネシウム定量方法のうち,共存元素分離エチレンジアミン四酢酸二水素二
ナトリウム滴定法について規定する。
この方法は,鉄鉱石中のマグネシウム含有率(質量分率)0.05 %以上5.0 %以下の定量に適用する。
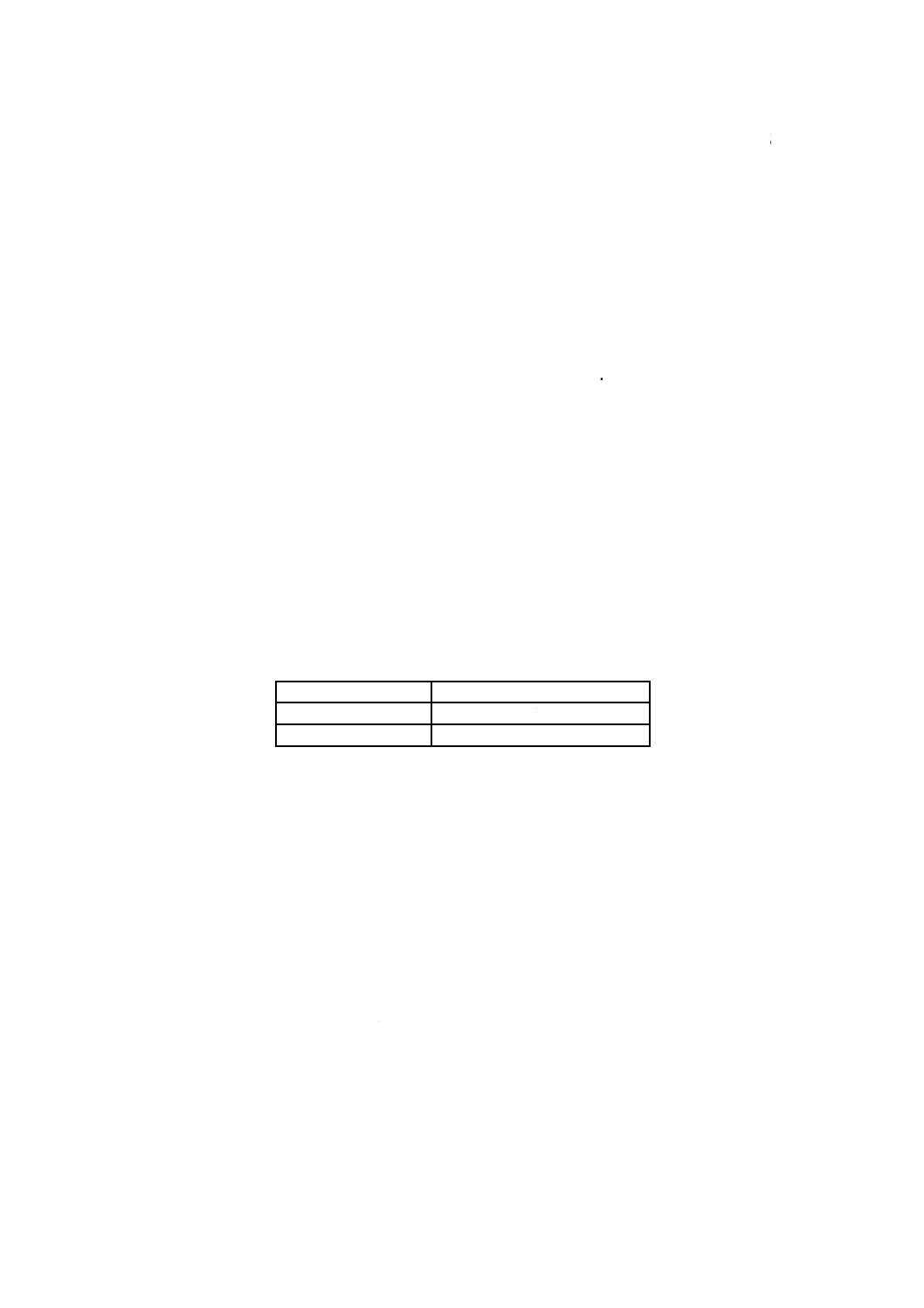
注記 JIS M 8222の規格群の定量範囲を表1に示す。
表1−JIS M 8222規格群の定量範囲
規格番号
定量範囲[質量分率(%)]
JIS M 8222-1
0.05 以上 5.0 以下
JIS M 8222-2
0.010 以上 2.00 以下
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS K 8001 試薬試験方法通則
JIS M 8202 鉄鉱石−分析方法通則
JIS Z 8402-6 測定方法及び測定結果の精確さ(真度及び精度)−第6部:精確さに関する値の実用的
な使い方
3
一般事項
定量方法に共通な一般事項は,JIS M 8202による。
4
要旨
試料を塩酸及び硝酸で分解した後,ろ過する。ろ液中の鉄を4-メチル-2-ペンタノンを用いて抽出して除
去する。残さは,ふっ化水素酸で処理した後,二硫酸カリウムで融解し,鉄を抽出分離した溶液に融成物
を合わせる。得た溶液を水酸化カリウムで中和し,塩酸を滴加してpHを調節した後,ヘキサメチレンテ
2
M 8222-1:2018
トラミンで水酸化物の沈殿を生成させてろ過する。ろ液の一部を取り,N,N-ジエチルジチオカルバミド酸
ナトリウム三水和物及びクロロホルムで共存元素を分離した後,アンモニア緩衝液で溶液のpHを調節し,
エリオクロムブラックT(EBT)溶液を指示薬としてエチレンジアミン四酢酸二水素二ナトリウム溶液で
カルシウムとマグネシウムとの合量を滴定する。別にカルシウムをカルセイン・チモールフタレインを指
示薬としてエチレンジアミン四酢酸二水素二ナトリウム溶液で滴定し,合量から差し引く。
5
試薬
試薬は,次による。
5.1
塩酸
5.2
塩酸(10+6,1+3,2+100)
5.3
硝酸
5.4
ふっ化水素酸
5.5
硫酸(1+1)
5.6
水酸化カリウム溶液(280 g/L)
5.7
鉄 純度の高い鉄で,カルシウム含有率(質量分率)及びマグネシウム含有率(質量分率)が,0.001 %
以下のもの。
5.8
アンモニア緩衝液
塩化アンモニウム54 gを水に溶解し,アンモニア水350 mLを加え,水で液量を1 000 mLにする。
5.9
鉄溶液
硫酸アンモニウム鉄(III)・12水8.6 gを水800 mL及び硫酸(1+1)10 mLに溶解し,1 000 mLの全量
フラスコに水を用いて移し入れ,水で標線までうすめる。
5.10 二硫酸カリウム
5.11 ヘキサメチレンテトラミン溶液(250 g/L)
5.12 ヘキサメチレンテトラミン洗浄溶液(5 g/L)
5.13 N,N-ジエチルジチオカルバミド酸ナトリウム三水和物(NaDDTC)
5.14 4-メチル-2-ペンタノン
5.15 クロロホルム
5.16 0.01 mol/L エチレンジアミン四酢酸二水素二ナトリウム溶液(0.01 mol/L EDTA2Na溶液)
(C10H14O8N2Na2・2H2O:3.722 g/L)
調製,保存,標定及びファクターの計算は,JIS K 8001のJA.6.4 c) 4)[0.01 mol/Lエチレンジアミン四
酢酸二水素二ナトリウム溶液(0.01 mol/L EDTA2Na溶液)]による。
5.17 0.01 mol/Lカルシウム溶液
あらかじめ105 ℃で約2時間乾燥した後,デシケーター中で常温まで放冷した炭酸カルシウム1.000 9 g
をはかりとってビーカー(300 mL)に移し入れ,時計皿で覆い,塩酸(1+3)100 mLを加える。完全に
溶解させて常温まで冷却した後,時計皿の下面を水で洗って時計皿を取り除き,溶液を1 000 mLの全量フ
ラスコに水を用いて移し入れ,水で標線までうすめて0.01 mol/ Lカルシウム溶液とする。
5.18 エリオクロムブラックT(EBT)溶液
この溶液の調製及び保存方法は,JIS K 8001の表JA.7[指示薬(沈殿滴定用,酸化還元滴定用,錯滴定
用など)の調製]による。
3
M 8222-1:2018
5.19 カルセイン・チモールフタレイン混合指示薬
カルセイン(C30H26N2O13)1,チモールフタレイン1,塩化カリウム100の割合で混合し,粉砕する。
6
試料のはかりとり
試料はかりとり量は,1.0 gとする。
7
操作
7.1
試料の分解及び不溶解残さの処理
試料の分解及び不溶解残さの処理は,次の手順によって行う。
a) 試料をはかりとってビーカー(300 mL)に移し入れ,時計皿で覆い,塩酸(5.1)25 mLを加えて初め
は熱板周辺の低温部(60 ℃〜100 ℃)にビーカーを置き,約1時間保持した後,更に熱板の高温部に
移して約10分間沸騰直前まで加熱して分解する。次に,硝酸(5.3)5 mLを加えて鉄などを酸化し,
引き続き加熱して蒸発させる。放冷した後,これに塩酸(5.1)5 mL〜10 mLを加えて加熱し,温水約
100 mLを加えて振り混ぜ,可溶性塩類を溶解する。時計皿の下面を温水で洗って時計皿を取り除き,
ろ紙(5種B)を用いて不溶解残さをろ過する。ビーカーの内壁をポリスマンを用いてこすり,付着
物をできるだけ少量の温塩酸(2+100)を用いてろ紙上に移す。ろ紙は,温塩酸(2+100)で,ろ紙
に塩化鉄(III)の黄色が認められなくなるまで洗浄し,次に温水で,3,4回洗浄する。不溶解残さは,
ろ紙とともに白金るつぼ(30 mL)に移し入れ,保存する。ろ液及び洗液はビーカー(300 mL)に受
ける。
b) ろ液及び洗液を液面に皮膜が生じるまで加熱蒸発させ,これに塩酸(10+6)20 mLを加えて可溶性塩
類を溶解する。放冷した後,分液漏斗(200 mL)に移し入れ,更にビーカー内壁を塩酸(10+6)を
用いて洗浄し,分液漏斗に移す。これに試料溶液量より少過剰の4-メチル-2-ペンタノン(5.14)を加
え,約1分間振り混ぜ,静置して二層に分離した後,下層の水相を元のビーカーに移し入れる。有機
相に塩酸(10+6)5 mLを加え,約30秒間振り混ぜ,静置して二層に分離した後,下層の水相を元の
ビーカーに加え,上層の有機相は捨てる。元のビーカー上部を,適正な開放部を作るように時計皿で
覆い,溶液を加熱して約5分間煮沸し,大部分の有機溶媒を揮散させた後,硝酸5 mLを加え,再び
時計皿で覆って引き続き液面に被膜を生じるまで加熱蒸発する。塩酸(5.1)10 mLを加えて可溶性塩
類を溶解し,これを主液として保存する。
c) a)で保存した不溶解残さを乾燥した後,ろ紙を強熱し,灰化する。放冷した後,強熱残さを硫酸(5.5)
2,3滴で湿し,ふっ化水素酸(5.4)約5 mLを加えて穏やかに加熱して二酸化けい素を揮散させ,更
に乾固するまで加熱して硫酸を揮散させる。放冷した後,二硫酸カリウム(5.10)約3 gを加え,白金
製の蓋をして初めは徐々に加熱し,次第に温度を高めて暗赤熱状に加熱して,残さを融解する。放冷
した後,白金るつぼの蓋を外して,蓋とともにビーカー(300 mL)に入れ,温水60 mLを加え穏やか
に加熱して融成物を溶解する。白金るつぼ及び蓋を温水で洗って取り出し,この溶液をb)で保存した
主液に合わせる。
7.2
水酸化物分離
7.1 c)で得た溶液に鉄溶液(5.9)10 mL〜20 mLを加え,水で液量を約100 mLとした後,50 ℃に加熱し
て水酸化カリウム溶液(5.6)を最初の沈殿が生じるまで加えて中和する。pH計を用いて塩酸(1+3)を
滴加してpHを1.5〜3.0に調節する。次に,ヘキサメチレンテトラミン溶液(5.11)20 mLを加え,温浴上
で水酸化物の沈殿を生成させる。この場合,温浴上の代わりに熱板上で数分間煮沸してもよい。沈殿は,
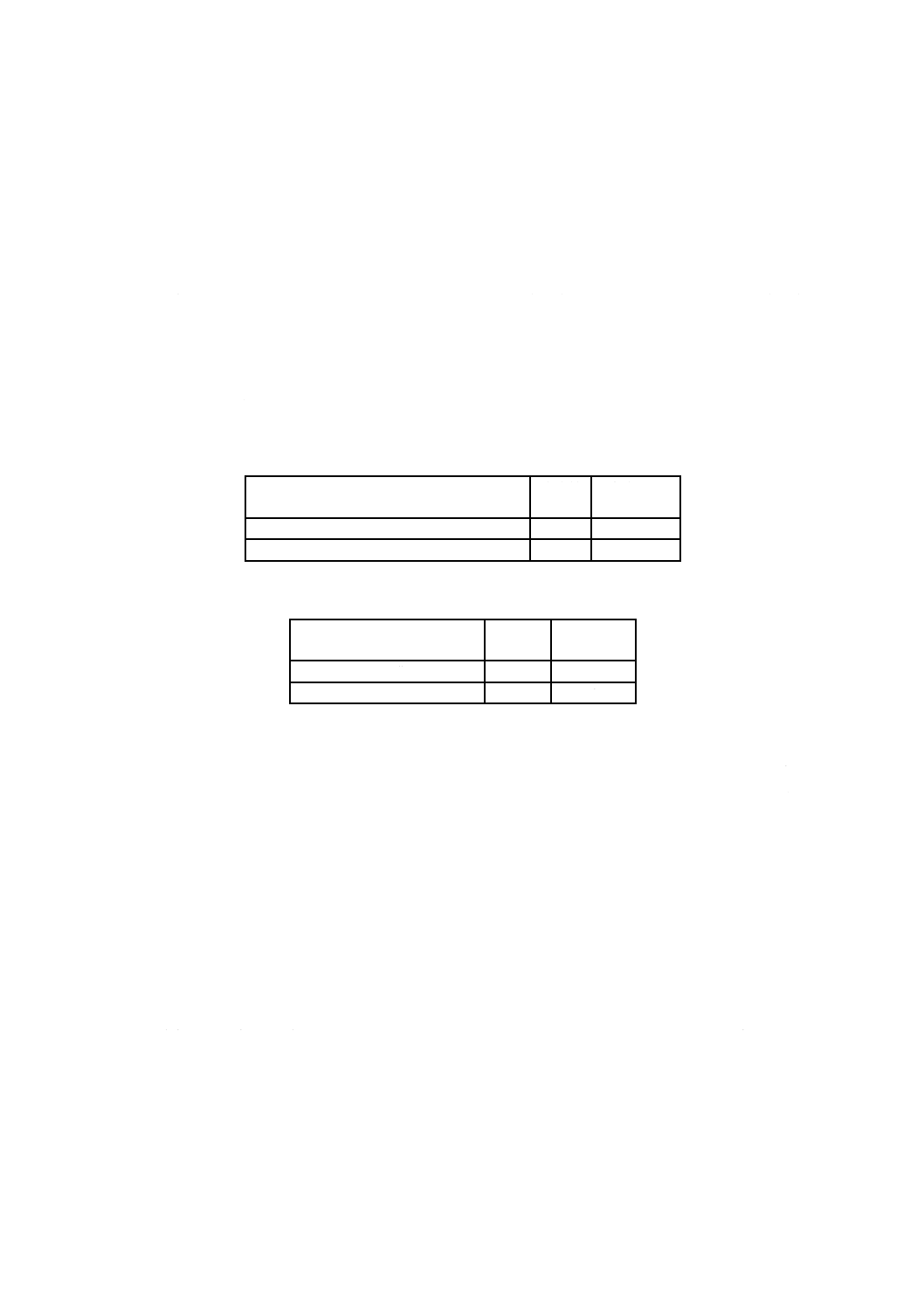
4
M 8222-1:2018
ろ紙(5種A)でろ過し,ヘキサメチレンテトラミン洗浄溶液(5.12)で数回洗浄する。ろ液及び洗液は
250 mLの全量フラスコに受け,保存する。沈殿は捨てる。
7.3
妨害元素の分離
7.2で得た溶液を常温まで冷却した後,水で標線までうすめて試料溶液とする。2個の分液漏斗(500 mL)
を用意し,試料溶液からカルシウムとマグネシウム含有率との合量に応じて一定量を表2に従って分取し
て,1個の分液漏斗に移し入れる。次に同じ試料溶液からカルシウム含有率に応じて表3に従って分取し
て,もう1個の分液漏斗に移し入れる。それぞれにNaDDTC(5.13)約0.1 g及びクロロホルム(5.15)50
mLを加えて振り混ぜ,妨害元素をクロロホルム中に抽出する。静置後有機相を捨て,再びNaDDTC約0.1
g及びクロロホルム50 mLを加えて振り混ぜる操作を行い,有機相が透明になるまで繰り返し,完全に妨
害元素を除去する。それぞれの水相を別々の三角フラスコ(500 mL)に移し入れ,分液漏斗の内壁を水で
洗浄して合わせる。有機相は捨てる。分取量が50 mLの場合は,水を加えて液量を100 mLにする。
表2−試料溶液分取量及び分取比
カルシウムとマグネシウム含有率との合量
[質量分率(%)]
分取量
mL
分取比B1
2未満
100
100/250
2以上
50
50/250
表3−試料溶液分取量及び分取比
カルシウム含有率
[質量分率(%)]
分取量
mL
分取比B2
2未満
100
100/250
2以上
50
50/250
7.4
カルシウムとマグネシウムとの合量の滴定
7.3において,試料溶液から表2に従って分取して得た溶液にアンモニア緩衝液(5.8)10 mLを加えて
pHを調節し1),EBT溶液(5.18)数滴を加え,0.01 mol/L EDTA2Na溶液(5.16)で滴定し,溶液の色の赤
が消失して鮮明な青に変わる点を終点とし,0.01 mol/L EDTA2Na溶液の使用量を求める。
注1) このときpHは約10となる。
7.5
カルシウムの滴定
7.3において,試料溶液から表3に従って分取して得た溶液に水酸化カリウム溶液(5.6)20 mL及びカ
ルセイン・チモールフタレイン混合指示薬(5.19)約0.05 gを加えて振り混ぜ,水を加えて液量を約200 mL
にした後,0.01 mol/L EDTA2Na溶液(5.16)で滴定する。溶液の色が黄緑から最後の1滴で紫に変わる点
を終点とし,0.01 mol/L EDTA2Na溶液の使用量を求める。
カルセイン・チモールフタレイン混合指示薬による呈色が不鮮明な場合は,カルセイン・チモールフタ
レイン混合指示薬を加えた溶液に0.01 mol/Lカルシウム溶液(5.17)を1 mL〜3 mLの間の一定量を正確に
加え,0.01 mol/L EDTA2Na溶液で滴定してもよい。
注記 0.01 mol/Lカルシウム溶液1 mLは,0.01 mol/L EDTA2Na溶液1 mLに相当する。また,この溶
液は,空試験においても同量添加されるので結果的に相殺される。
5
M 8222-1:2018
8
空試験
鉄(5.7)0.5 gをはかりとってビーカー(300 mL)に移し入れ,時計皿で覆い,7.1〜7.5の手順に従って,
試料と同じ操作を試料と併行して行う。
9
計算
計算は,次による。
a) マグネシウム含有率の計算 7.4,7.5及び箇条8の滴定に要した0.01 mol/L EDTA2Na溶液(5.16)の
使用量から試料中のマグネシウム含有率を,次の式によって算出する。
[
]
100
243
000
.0
/)
(
/)
(
2
4
2
1
3
1
1
×
×
−
−
−
=
m
B
V
V
B
V
V
F
Mg
ここに,
Mg: 試料中のマグネシウム含有率[質量分率(%)]
F1: 0.01 mol/L EDTA2Na溶液のファクター[JIS K 8001の
JA.6.4 c) 4)で算出した値]
V1: 分取した試料溶液のカルシウムとマグネシウム含有率
との合量の滴定における0.01 mol/L EDTA2Na溶液の使
用量(mL)
V2: 分取した試料溶液のカルシウム含有率の滴定における
0.01 mol/L EDTA2Na溶液の使用量(mL)
V3: カルシウムとマグネシウム含有率との合量の滴定にお
ける空試験で得た0.01 mol/L EDTA2Na溶液の使用量
(mL)
V4: カルシウム含有率の滴定における空試験で得た0.01
mol/L EDTA2Na溶液の使用量(mL)
m: 試料はかりとり量(g)
B1: カルシウムとマグネシウム含有率との合量滴定時の分
取比
B2: カルシウム含有率滴定時の分取比
0.000 243: 0.01 mol/L EDTA2Na溶液1 mLに相当するマグネシウム
の質量を示す換算係数(g/mL);0.01/1 000×24.305 0で
求められる。
b) 酸化マグネシウム含有率の計算 試料中の酸化マグネシウム含有率は,マグネシウム含有率から次の
式によって算出する。
MgO=1.658×Mg
ここに,
MgO: 試料中の酸化マグネシウム含有率[質量分率(%)]
Mg: 試料中のマグネシウム含有率[質量分率(%)]
なお,酸化マグネシウム含有率を報告値とする場合,丸めを行っていないマグネシウム含有率から酸化
マグネシウムの含有率を求め,最終報告値とする。
10 許容差
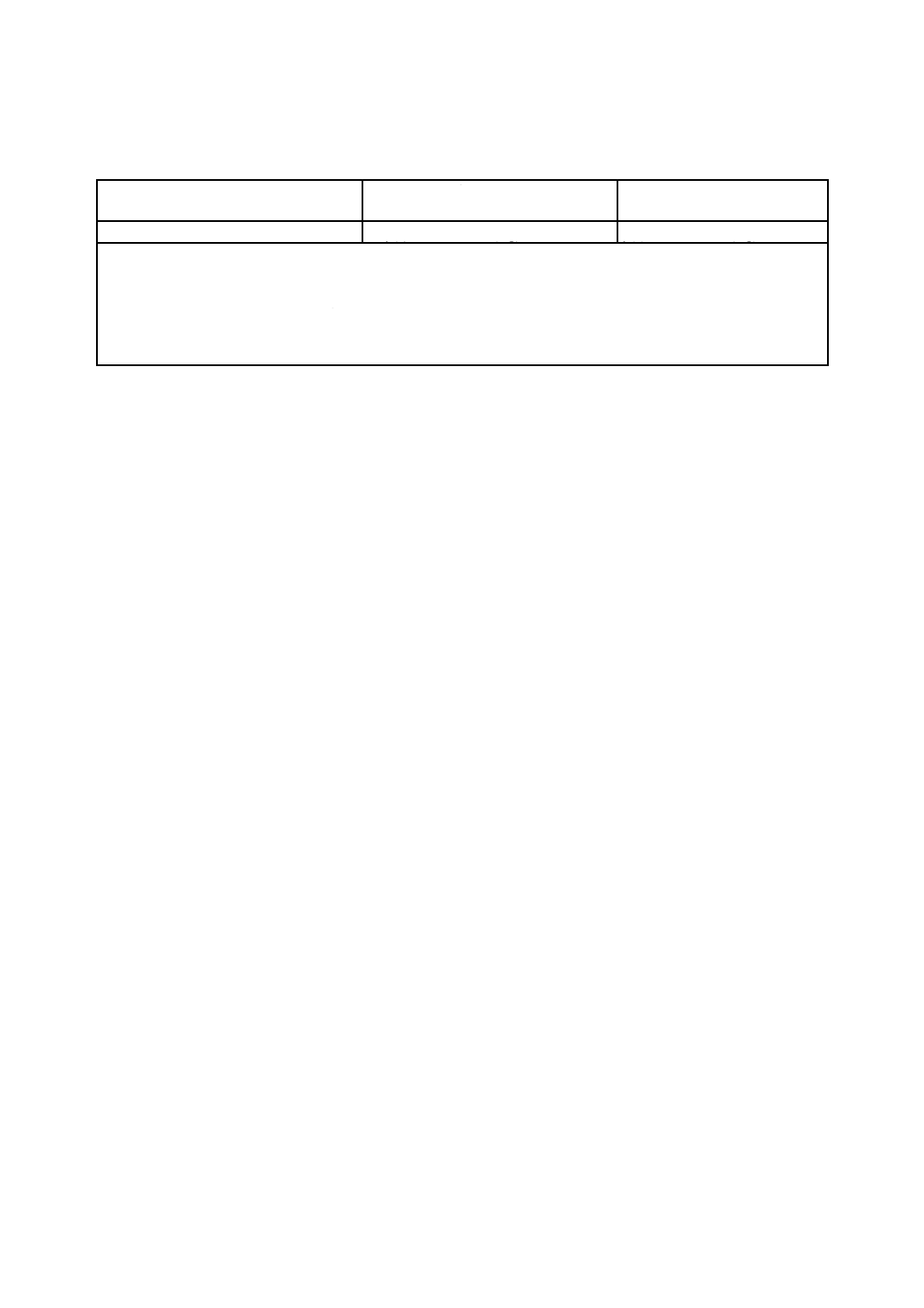
許容差は,表4による。
6
M 8222-1:2018
表4−許容差
単位 質量分率(%)
マグネシウム含有率
室内再現許容差
Rd
室間許容差a)
P
0.05以上5.0以下
f(n)×[0.006 2×(Mg)+0.003 6]
f(n)×[0.028 4×(Mg)+0.000 5]
許容差計算式中のf(n)の値は,JIS Z 8402-6の表1(許容範囲の係数)による。nの値は,室内再現許容差の場合
は同一分析室内における分析回数,室間許容差の場合は分析に関与した分析室数である。また,(Mg)は,許容差を
求めるマグネシウム定量値の平均値[質量分率(%)]である。
注記 この許容差は,マグネシウム含有率(質量分率)0.06 %以上2.42 %以下の試料を用いて求めたものである。
注a) この規格における室間許容差は,各分析室においてJIS M 8202の6.5(分析値の採択)によって求めた分析
値を用いて判定する。