M 8123:2006
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,日本鉱業協会(JMIA)
/財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を改正すべきとの申出があり,日
本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格である。
これによって,JIS M 8123:1999は改正され,この規格に置き換えられる。
改正に当たっては,日本工業規格と国際規格との対比,国際規格に一致した日本工業規格の作成及び日
本工業規格を基礎にした国際規格原案の提案を容易にするために,ISO 11441:1995,Lead sulfide concentrates
−Determination of lead content−Back titration of EDTA after precipitation of lead sulfate及びISO 13545:2000,
Lead sulfide concentrates−Determination of lead content−EDTA titration method after acid digestionを基礎とし
て用いた。
この規格の一部が,技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の
実用新案登録出願に抵触する可能性があることに注意を喚起する。経済産業大臣及び日本工業標準調査会
は,このような技術的性質をもつ特許権,出願公開後の特許出願,実用新案権,又は出願公開後の実用新
案登録出願にかかわる確認について,責任をもたない。
JIS M 8123には,次に示す附属書がある。
附属書1(規定)分析用試料の吸着水分の測定方法
附属書2(規定)酸化するおそれがある分析用試料の吸着水分の測定方法
附属書3(参考)JISと対応する国際規格との対比表
M 8123:2006
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1. 適用範囲 ························································································································ 1
2. 引用規格 ························································································································ 1
3. 一般事項 ························································································································ 1
4. 分析試料のとり方及び取扱い方法 ······················································································· 1
4.1 試料の採取及び調製 ······································································································· 2
4.2 試料のはかり方 ············································································································· 2
5. 分析値の表し方及び操作上の注意 ······················································································· 2
5.1 分析値の表し方 ············································································································· 2
5.2 分析操作上の注意 ·········································································································· 3
6. 定量方法 ························································································································ 3
6.1 定量方法の区分 ············································································································· 3
6.2 酸分解・硫酸鉛沈殿分離・EDTA滴定法 ············································································· 3
6.3 アルカリ融解・硫酸鉛沈殿分離・EDTA−亜鉛逆滴定法 ························································ 10
6.4 フレーム原子吸光法 ······································································································ 13
附属書1(規定)分析用試料の吸着水分の測定方法 ···································································· 17
附属書2(規定)酸化するおそれがある分析用試料の 吸着水分の測定方法 ····································· 19
附属書3(参考)JISと対応する国際規格との対比表 ·································································· 21
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
M 8123:2006
鉱石中の鉛定量方法
Ores-Methods for determination of lead
序文 この規格は,1995年に第1版として発行されたISO 11441,Lead sulfide concentrates−Determination of
lead content−Back titration of EDTA after precipitation of lead sulfate及び2000年に第1版として発行された
ISO 13545,Lead sulfide concentrates−Determination of lead content−EDTA titration method after acid digestion
を翻訳し,技術的内容の一部を変更して作成した日本工業規格である。
なお,この規格で側線又は点線の下線を施してある箇所は,原国際規格を変更している,又は原国際規
格にない事項である。変更の一覧表をその説明を付けて,附属書3に示す。
1. 適用範囲 この規格は,鉱石中の鉛の定量方法について規定する。ただし,他の日本工業規格で鉛定
量方法が規定されている鉱石には適用しない。
備考 この規格の対応国際規格を,次に示す。
なお,対応の程度を表す記号は,ISO/IEC Guide 21に基づき,IDT(一致している),MOD
(修正している),NEQ(同等でない)とする。
ISO 11441:1995,Lead sulfide concentrates−Determination of lead content−Back titration of EDTA
after precipitation of lead sulfate (MOD)
ISO 13545:2000,Lead sulfide concentrates−Determination of lead content−EDTA titration method
after acid digestion (MOD)
2. 引用規格 次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成す
る。これらの引用規格は,その最新版(追補を含む。)を適用する。
JIS K 0050 化学分析方法通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS M 8083 銅,鉛及び亜鉛硫化精鉱−サンプリング及び水分決定方法
JIS M 8101 非鉄金属鉱石のサンプリング,試料調製及び水分決定方法
JIS Z 8401 数値の丸め方
ISO 9599,Copper,lead and zinc sulfide concentrates−Determination of hygroscopic moisture in the analysis
sample−Gravimetric method
3. 一般事項 定量方法に共通な一般事項は,JIS K 0050,JIS K 0116及びJIS K 0121による。
4. 分析試料のとり方及び取扱い方法
2
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4.1
試料の採取及び調製 試料の採取及び調製は,JIS M 8083又はJIS M 8101による。
4.2
試料のはかり方 試料のはかり方は,次による。
a) 試料のはかりとりにおいては,試料をよくかき混ぜて平均組成を表すように注意し,また,異物が混
入していないことを確かめなければならない。
b) 試料のはかりとりは,次のいずれかによる。ただし,事前乾燥法Aは,酸化性が大きな試料には適用
しない。事前乾燥法Bは,吸湿性及び/又は酸化性が大きな試料には適用しない。
1) 吸着水分補正法 附属書1の4.2又は附属書2の5.2によって調製した試料から,吸着水分測定試料
をはかりとった後,5分以内に6.定量方法に各々規定する量の試料を0.1 mgのけたまではかりとる。
2) 事前乾燥法A 事前乾燥法Aは,次の手順によって行う。
2.1) 105±5 ℃で1時間乾燥し,デシケーター中で室温まで放冷したはかり瓶(20 g以下のもの)に,
各定量方法に規定する量とほぼ同じ量の分析試料をはかりとり,はかり瓶のふたとともに,あら
かじめ105±5 ℃に調節してある空気浴に入れて2時間乾燥する。空気浴から取り出し,はかり瓶
にふたをして,デシケーター中で室温まで放冷する。
2.2) デシケーターからはかり瓶を取り出し,はかり瓶のふたを少し持ち上げ,再度ふたをした後,は
かり瓶の質量を0.1 mgのけたまではかる。規定の時間の乾燥で十分であることが確認されていな
い試料の場合には,試料,はかり瓶及びそのふたの合計質量をはかった後,更に2時間乾燥し,
デシケーター中で放冷し,その合計質量をはかる。この操作を2時間の乾燥減量が0.5 mg以下な
るまで繰り返す。
2.3) 分析試料を,各定量方法に規定した容器に移し入れた後,空のはかり瓶の質量を0.1 mgのけたま
ではかり,得た質量を2.2)で得た質量から差し引いて得られる質量を,試料のはかりとり量とす
る。
3) 事前乾燥法B 事前乾燥法Bは,次の手順によって行う。
3.1) 分析試料5〜10 gを平形はかり瓶(直径約50 mmのもの。)に移し入れ,平らに広げた後,平形は
かり瓶のふたとともにあらかじめ105±5 ℃に調節してある空気浴に入れて2時間乾燥する。空気
浴から取り出し,平形はかり瓶にふたをして,デシケーター中で室温まで放冷する。規定の時間
の乾燥で十分であることが確認されていない試料の場合には,試料,はかり瓶及びそのふたの合
計質量をはかった後,更に2時間乾燥し,デシケーター中で放冷し,その合計質量をはかる。こ
の操作を2時間の乾燥減量が1 mg以下となるまで繰り返す。
3.2) デシケーターから平形はかり瓶を取り出し,直ちに各定量方法に規定する量の試料を0.1 mgのけ
たまではかりとる(1)。
注(1) ひょう量の都度,平形はかり瓶をデシケーターから取り出し,平形はかり瓶のふたを取り,速
やかに試料をはかりとる。
5. 分析値の表し方及び操作上の注意
5.1
分析値の表し方 分析値の表し方は,次による。
a) 分析値は,質量百分率で表し,JIS Z 8401によって小数点以下2けたに丸める。
b) 分析回数は,通常,併行条件において2回分析(併行2個掛け)とする。2個の分析値の差が併行許
容差(以下,許容差という。)以下のとき,その平均値を求め,JIS Z 8401によって小数点以下2けた
に丸めて報告値とする。
c) 併行2個掛けを行った分析値の差が許容差を超えるときは,改めて2個掛けの分析をやり直す。
3
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d) 許容差は,表1による。2個の分析値が二つの区分にまたがるときは,2個の分析値の平均値が属する
区分の許容差を適用する。
表 1 許容差
定量方法
鉛含有率の区分
%(質量分率)
許容差(併行)
%(質量分率)
酸分解・硫酸鉛沈殿分離・EDTA滴定法
5.0以上 10未満
0.100
10以上 40未満
0.150
40以上 80以下
0.250
アルカリ溶融・硫酸鉛沈殿分離・EDTA−亜鉛逆滴定法
10以上 40未満
0.250
40以上 80以下
0.430
フレーム原子吸光法
0.01以上 0.05未満
0.005
0.05以上 0.2未満
0.015
0.2以上 0.5未満
0.040
0.5以上 2.0未満
0.055
2.0以上 5.0以下
0.150
5.2
分析操作上の注意 分析に当たっては,全操作を通じて空試験を行い,測定値を補正する。
6. 定量方法
6.1
定量方法の区分 鉱石中の鉛定量方法は,次のいずれかによる。
a) 酸分解・硫酸鉛沈殿分離・EDTA滴定法 この方法は,鉛含有率5.0 %(質量分率)以上80 %(質
量分率)以下の試料に適用する。
b) アルカリ溶融・硫酸鉛沈殿分離・EDTA−亜鉛逆滴定法 この方法は,鉛含有率10 %(質量分率)以
上80 %(質量分率)以下の硫化鉛精鉱に適用する。
c) フレーム原子吸光法 この方法は,鉛含有率0.01 %(質量分率)以上5.0 %(質量分率)以下の試料
に適用する。
6.2
酸分解・硫酸鉛沈殿分離・EDTA滴定法
6.2.1
要旨 試料を硝酸,硫酸及び臭素で分解した後,臭化水素酸を用いてひ素,アンチモン及びすずを
揮散分離する。硫酸を加え,加熱して硫酸の白煙を発生させ,硫酸鉛の沈殿を生成させる。水で可溶性塩
を溶解した後,エタノール又はメタノールを加え,硫酸鉛の沈殿をこし分ける。沈殿を酢酸アンモニウム
と酢酸の混液とで溶解し,ろ過して不溶解残物を除いた後,溶液のpHを5.5〜5.7とし,キシレノールオ
レンジを指示薬として,EDTA(エチレンジアミン四酢酸二水素二ナトリウム)標準溶液で滴定する。
6.2.2
試薬 試薬は,次による。
a) 塩酸
b) 塩酸(1+1)
c) 硝酸
d) 過塩素酸
e) ふっ化水素酸
f)
臭化水素酸
g) 硫酸(1+1)
4
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
h) 硫酸洗浄溶液 硫酸(1+99) 500 mLにエタノール50 mL又はメタノール50 mLを加え,冷却して用い
る。
i)
酢酸
j)
硝硫酸混液 冷却しながら硝酸250 mLに硫酸250 mLをゆっくり加える。
k) アンモニア水
l)
アンモニア水(1+1)
m) 臭素
n) 酢酸アンモニウム混液 酢酸アンモニウム250 gを水に溶解し,水で液量を1 000 mLとした後,酢酸
25 mLを加える。
o) 酢酸アンモニウム洗浄液 酢酸アンモニウム混液[6.2.2 n)]を水で20倍に薄める。
p) 塩化アンモニウム
q) 炭酸ナトリウム(無水)
r) 炭酸アンモニウム
s)
炭酸アンモニウム溶液(20 g/L)
t)
塩化カルシウム溶液 塩化カルシウム二水和物18.4 gを水に溶解し,水で液量を500 mLとする。こ
の溶液1 mLは,カルシウム約10 mgを含む。
u) 塩化ナトリウム
v) 塩化カリウム
w) 混合融剤 炭酸ナトリウムと炭酸カリウムとを1:1の割合で混合する。
x) 過酸化ナトリウム
y) 0.01 mol/L EDTA溶液 エチレンジアミン四酢酸二水素二ナトリウム二水和物0.38 gを水に溶解し,
水で液量を100 mLとする。この溶液1 mLは,ビスマス約0.002 1 gに相当する。
z) 0.025 mol/L EDTA標準溶液A エチレンジアミン四酢酸二水素二ナトリウム二水和物9.4 gを水に溶
解し,水で液量を1 000 mLとする。この溶液1 mLは,鉛約0.005 2 gに相当するが,標定は,次のよ
うにして行う。
鉛[99.99 %(質量分率)以上]0.20 gを0.1 mgのけたまで3個はかりとり,それぞれ別のビーカ
ー(500 mL)に移し入れ,硝酸(1+1) 20 mLを加え,時計皿で覆い,穏やかに加熱して分解する。時計
皿の底面を少量の水で洗浄して時計皿を取り除き,加熱して穏やかに沸騰させ,窒素酸化物のガスを
逃がした後,室温まで放冷する。酢酸アンモニウム混液[6.2.2 n)] 30 mLを加え,水で液量を約150 mL
とする。pH計を用いてアンモニア水(1+1)でpH 5.5〜5.7に調節する。3個の溶液にそれぞれキシレ
ノールオレンジ溶液(1 g/L) 0.5 mLを指示薬として加え,直ちに0.025 mol/L EDTA標準溶液A [6.2.2
z)]で滴定し,溶液の色が赤から黄に変わった点を終点とし,0.025 mol/L EDTA標準溶液Aの使用量を
それぞれ求め,個々の滴定で得た0.025 mol/L EDTA標準溶液A 1 mLに相当する鉛量を,式(1)によっ
て算出する。
3 ,2 ,1
x
x
x
x
=
=
V
m
f
································································· (1)
ここに, fx: 個々の滴定で得た0.025 mol/L EDTA標準溶液1 mLに相当す
る鉛量 (g/mL)
mx: 個々の滴定ではかりとった鉛の質量 (g)
Vx: 個々の滴定で得た0.025 mol/L EDTA標準溶液の使用量 (mL)
5
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
f1,f2及びf3の各算出値の範囲が0.000 02 g/mLを超える場合には,標定をやり直す。各算出値の範
囲が0.000 02 g/mL以下の場合には,式(2)によって0.025 mol/L EDTA標準溶液A 1 mLに相当する鉛
量を,求める。
3
3
2
1
f
f
f
f
+
+
=
········································································· (2)
ここに,
f: 0.025 mol/L EDTA標準溶液1 mLに相当する鉛量 (g/mL)
aa) 0.025 mol/L EDTA標準溶液B エチレンジアミン四酢酸二水素二ナトリウム二水和物9.4 gを水に溶
解し,水で液量を1 000 mLとする。この溶液1 mLは,鉛約0.005 2 gに相当するが,標定は,次のよ
うにして行う。
鉛[99.99 %(質量分率)以上]0.10〜0.20 gを,6.2.3ではかりとった試料中に含まれる鉛量とそれ
ぞれほぼ同じになるように0.1 mgのけたまで3個はかりとり,それぞれ別のビーカー(500 mL)に移し
入れ,硝酸(1+1) 20 mLを加え,時計皿で覆い,穏やかに加熱して分解した後,6.2.4のa) 2)〜b) 4)
の手順に従って操作し,得た3個の溶液にそれぞれキシレノールオレンジ溶液(1 g/L) 0.5 mLを指示
薬として加え,直ちに0.025 mol/L EDTA標準溶液B [6.2.2 aa)]で滴定して溶液の色が赤から黄に変わ
った点を終点とし,0.025 mol/L EDTA標準溶液Bの使用量をそれぞれ求め,個々の滴定で得た0.025
mol/L EDTA標準溶液B 1 mLに相当する鉛量を,式(1)によって算出する。
f1,f2及びf3各算出値の範囲が0.000 02 g/mLを超える場合には,標定をやり直す。各算出値の範囲
が0.000 02 g/mL以下の場合には,式(2)によって0.025 mol/L EDTA標準溶液B 1 mLに相当する鉛量
を求める。
備考 この標定方法は,硫酸鉛をろ過したときにろ液に損失する鉛量を6.2.4 d)の操作によって補正す
る代わりに,0.025 mol/L EDTA標準溶液Bの標定操作を試料と同じ条件で行い,標準溶液の鉛
相当量をろ液中に損失する鉛量を考慮して求める方法である。硫酸鉛をろ過したときのろ液に
損失する鉛量は,操作条件によって異なるので,この標定操作は,操作中の液量,液温,試薬
量,洗浄操作及び鉛量を試料の場合と同一にしなければならない。
ab) 標準鉛溶液(Pb:0.1 mg/mL) 鉛[99.99 %(質量分率)以上]0.10 gを0.1 mgのけたまではかりと
り,ビーカー(300 mL)に移し入れ,硝酸(1+1) 20 mLを加え,時計皿で覆い,穏やかに加熱して分解
する。時計皿の底面を少量の水で洗浄して時計皿を取り除き,加熱して穏やかに沸騰させ,窒素酸化
物のガスを逃がした後,室温まで放冷する。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,
水で標線まで薄める。
ac) 鉄溶液(Fe:10 mg/mL) 鉄[99.99 %(質量分率)以上]1.0 gを塩酸(1+1) 30 mLで加熱分解し,
硝酸(1+1) 5 mLを加え,加熱する。放冷した後,水で液量を100 mLとする。
ad) 亜鉛溶液(Zn:10 mg/mL) 亜鉛[99.99 %(質量分率)以上で,鉛含有率0.001 %(質量分率)以下]
1.0 gを塩酸(1+1) 20 mLで加熱分解する。放冷した後,水で液量を100 mLとする。
ae) エタノール(95)
af) メタノール(95)
ag) L-アスコルビン酸
ah) キシレノールオレンジ溶液(1 g/L)
6.2.3
試料はかりとり量 試料のはかりとり量は,鉛の含有率に応じて表2によって,0.1 mgのけたまで
はかる(2)。
注(2) 試料は,なるべく鉛量100〜200 mgとなるようにはかりとる。ただし,鉛含有率50 %(質量

6
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
分率)以上の場合には,鉛量が200 mgに近くなるようにはかりとる。
表 2 試料はかりとり量
試料中の鉛含有率
%(質量分率)
試料はかりとり量
g
5.0以上 10未満
1.0 〜2.0
10 以上 40未満
0.5 〜1.0
40 以上 80以下
0.25〜0.5
6.2.4
操作 操作は,次による。
a) 試料の分解 試料の分解は,次の手順によって行う。
1) 試料をはかりとり,ビーカー(300〜500 mL)に移し入れ,少量の水で試料を湿らせ,硝酸10〜30 mL
及び臭素1〜2 mLをそれぞれ少量ずつ加え(3),時計皿で覆い,穏やかに加熱して分解する(4)。
注(3) 酸化鉱,焼鉱などの場合には,試料をはかりとった後,塩酸20〜40 mLを加えて加熱し,
次いで硝酸10〜30 mL及び臭素1〜2 mLを加える。
(4) 硫化鉱など析出した硫黄の分解が不十分な場合には,更に少量の臭素を加え,加熱して完
全に分解する。
2) 時計皿の底面を少量の水で洗浄して時計皿を取り除き,更にビーカーの壁面を少量の水で洗浄する。
硫酸(1+1) 10 mLを加えて加熱し,硫酸の白煙を十分に発生させた後(5),室温まで放冷する(6)(7)。
注(5) 分解残物中に炭素が残存し,黒色がかって見える場合は,硝硫酸混液[6.2.2 j)]を試料溶液が
熱いうちに黒色が消えるまで少量ずつ滴下し,その後,硫酸の白煙を十分に発生させる。
(6) 多量[約0.5 %(質量分率)以上]のひ素,アンチモン,すず及び/又はセレンを含む試
料の場合には,放冷した後,水約5 mL及び臭化水素酸10 mLを加え,加熱蒸発して硫酸
の白煙を十分に発生させる。放冷した後,硫酸(1+1) 5 mL及び臭化水素酸10 mLを加え,
再び加熱蒸発して硫酸の白煙を十分に発生させ,ひ素,アンチモンなどを揮散除去する。
(7) ビスマスを含む試料の場合には,硫酸白煙発生後,長時間放置するとビスマスが不溶性と
なるので,できるだけ速やかに次の操作を行う。また,ビスマスが多量に存在する場合及
び/又は硫酸白煙を発生させた後,やむを得ず長時間放置する場合には,ビスマスが不溶
性となり,鉛と共沈して鉛の滴定に影響するおそれがある。このときは,注(12)の操作を行
って,その影響を除く必要がある。
b) 硫酸鉛の分離 硫酸鉛の分離は,次の手順によって行う。
1) a) 2)で得た溶液に,水をビーカーの内壁を洗いながら加え,液量を約100 mLとする。時計皿で覆
い,加熱して穏やかに煮沸して,可溶性塩を溶解した後,室温まで放冷する。エタノール(95),又
はメタノール(95) 10 mLを加えて振り混ぜ,1時間以上放置する(8)。
注(8) 沈殿の沈降を確保するのに十分な放置時間(1夜間程度)がとれる場合は,次の2)の操作
を行う代わりに,次の操作を行ってもよい。
時計皿を外し,沈殿をビーカーに残し,上澄み液をろ紙(5種C)を通して別のビーカー
に移し入れる。ビーカー中の沈殿に硫酸洗浄溶液[6.2.2 h)]をビーカーの内壁を洗浄しなが
ら静かに加え,デカンテーションによって数回洗浄し,次に水を用いてデカンテーション
によって洗浄する。このとき,洗液は,上澄み液をろ過したろ紙(5種C)を通して上澄み
液を入れたビーカーに移し入れる。ろ紙を硫酸洗浄溶液[6.2.2 h)]及び水を用いて数回洗浄
し,この洗液も上澄み液を入れたビーカーに移し入れる。外した時計皿の底面を少量の水
7
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
で洗浄し,この洗液も上澄み液を入れたビーカーに移し入れる。上澄み液及び洗液を合わ
せ,保管しておく。
2) 時計皿の底面を少量の水で洗浄して時計皿を取り除き,ろ紙(5種C)を用いて沈殿をこし分けた
後,硫酸洗浄溶液[6.2.2 h)]を用いてビーカーの内壁及び沈殿を数回洗い,更に,少量の水を用いて
洗浄する。温水を用いて沈殿を元のビーカーに洗い移す(9)。ろ液及び洗液を合わせ,別のビーカー
(300〜500 mL)に保管しておく(10)。
注(9) 比較的多量のバリウムを含む試料の場合は,次の3)の操作で酢酸アンモニウム混液[6.2.2
n)]だけでは,硫酸鉛の沈殿を完全に溶解できない。この場合には,ここで塩酸10 mLを加
え,加熱して乾固近くまで濃縮する。
(10) 滴定をc) 2)によって行う場合は,ろ液及び洗液は保管しなくてよい。
3) 酢酸アンモニウム混液[6.2.2 n)] 30 mLをろ紙上から加え,更に小量の温水でろ紙を洗浄し,ろ液及
び洗液は沈殿の入っている元のビーカーに受ける。小量の酢酸アンモニウム混液[6.2.2 n)]を用いて
ビーカーの内側を洗浄する。時計皿で覆い,加熱して約5分間煮沸し,硫酸鉛の沈殿を溶解する。
4) 時計皿の底面を少量の水で洗浄して時計皿を取り除き,元のろ紙を用いてビーカー(300〜500 mL)
にろ過し,温めた酢酸アンモニウム洗浄液[6.2.2 o)]を用いて十分に洗浄する。ろ液及び洗液を合わ
せ,水で液量を約150 mLとし,室温まで冷却する(11)。不溶解残物及びろ紙は,保管しておく。
注(11) この溶液のpHは,約5.5である。滴定にはpH 5.5〜5.7の範囲がよいので,pHがこの範囲
外になっているおそれがあるときは,pH計を用い,酢酸又はアンモニア水を滴加してpH
を調節する。
c) 滴定 滴定は,次のいずれかによる(12)。
注(12) 試料中にビスマスが0.1 %(質量分率)以上共存する場合は,b) 4)で得た溶液に硝酸を滴加し
てpH計を用いてpHを1.5に調節した後,キシレノールオレンジ溶液(1 g/L) 0.5 mLを指示薬
として加え,0.01 mol/L EDTA溶液[6.2.2 y)]で溶液の色が赤から黄に変わるまで滴定し,ビスマ
スEDTA錯体を完全に生成させる。次いでアンモニア水(1+1)を滴下してpHを5.5〜5.7に調
節した後,次のいずれかによって滴定を行う。
1) 0.025 mol/L EDTA標準溶液Aを用いる場合 b) 4)で得た溶液に(13),キシレノールオレンジ溶液(1
g/L) 0.5 mLを指示薬として加え,直ちに0.025mol/L EDTA標準溶液A [6.2.2 z)]で滴定し,溶液の色
が赤から黄に変わった点を終点とし,0.025 mol/L EDTA標準溶液Aの使用量を求める。
注(13) この溶液中に小量でも鉄(Ⅲ)のイオンが含まれるときは,終点が不明りょうになる。そこ
で,L-アスコルビン酸0.2 gを添加し,鉄(Ⅱ)イオンに還元しておく。
備考 この方法によって滴定を行った場合は,d)の操作を行う。
2) 0.025 mol/L EDTA標準溶液Bを用いる場合 b) 4)で得た溶液に(13),キシレノールオレンジ溶液(1
g/L) 0.5 mLを指示薬として加え,直ちに0.025 mol/L EDTA標準溶液B [6.2.2 aa)]で滴定し,溶液の
色が赤から黄に変わった点を終点とし,0.025 mol/L EDTA標準溶液Bの使用量を求める。
備考 この方法によって滴定を行った場合は,ろ液中に溶解する鉛量を標準溶液Bの標定時に考
慮しているので,d)の操作は行わない。
d) ろ液中の鉛の定量 c) 1)によって滴定を行った場合には,次の手順によって,ろ液中の鉛の定量を行
う。
1) 測定溶液の調製 b) 2)又は注(8)で保管しておいたろ液及び洗液を加熱して,約100 mLになるまで
濃縮し,塩酸(1+1) 10 mLを加える。室温まで冷却した後,溶液を200 mLの全量フラスコに水を
8
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
用いて移し入れ,水で標線まで薄める。
2) 原子吸光光度法による吸光度の測定 1)で得た溶液の一部を,水を用いて零点を調整した原子吸光
光度計の空気・アセチレンフレーム中に噴霧し,波長217.0 nm又は283.3 nmにおける吸光度を測
定し(14)(15),得た吸光度と3)で作成した検量線とから溶液中の鉛量を求める。
注(14) バックグラウンド補正機能がある原子吸光光度計を用いて測定する場合は,バックグラウ
ンド補正を行う。
(15) 原子吸光光度計の代わりに,ICP発光分光装置を用いて鉛量を求めてもよい。このとき用
いる測定波長の一例としては,220.35 nmがある。
なお,ICP発光分光装置は,装置の種類によって波長分解能が異なっており,事前に妨
害の有無を確認し,適切な波長を選択する。高次のスペクトル線が使用可能な装置では,
高次のスペクトル線を用いてもよい。また,バックグラウンド補正機構が付いている装置
では,バックグラウンド補正機構を用いてもよい。
3) 検量線の作成 標準鉛溶液[6.2.2 ab)]の各種液量(鉛として0〜0.5 mg)を段階的に数個の200 mL
の全量フラスコに取り,塩酸(1+1) 10 mL及び硫酸(1+1) 10 mLを加え,更に,試料溶液に含まれ
る鉄及び亜鉛の量と同じになるように鉄溶液[6.2.2 ac)]及び亜鉛溶液[6.2.2 ad)]を加え,水で標線ま
で薄める。これらの溶液の吸光度を,2)の手順に従って測定溶液と併行して測定して,得た吸光度
と鉛量との関係線を作成し,その関係線を原点を通るように平行移動して検量線とする。
e) 不溶解残物中の鉛の定量(16) 不溶解残物中の鉛の定量は,次の手順によって行う。
注(16) 残物中に鉛が含まれないことを確認していれば,省略することができる。
1) 試料溶液の調製 不溶解残物中の鉛の定量は,次のいずれかによる。
1.1) 融解による試料溶液の調製
1.1.1) b) 4)で保管しておいた残物及びろ紙をジルコニウムるつぼへ入れ,乾燥した後,600 ℃で灰化す
る。
1.1.2) 放冷した後,混合融剤[6.2.2 w)] 4 g及び過酸化ナトリウム1 gを加え,加熱して融解する。放冷
した後,融解物をジルコニウムるつぼとともにビーカー(300〜500 mL)に入れ,水約50 mLを加
え,時計皿で覆い,静置して可溶性塩を溶解する。さらに,穏やかに煮沸するまで加熱する。塩
酸(1+1) 25 mLを加え,反応が終了した後,時計皿の底面を少量の水で洗浄して時計皿を取り
除き,ジルコニウムるつぼは,温水で十分に洗浄して取り除く。
1.1.3) 溶液を250 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
1.2) ふっ化水素酸処理による試料溶液の調製
1.2.1) b) 4)で保管しておいた残物及びろ紙を磁性るつぼ(20番)へ入れ,乾燥した後,700〜750 ℃で
灰化する。放冷した後,灰化物を四ふっ化エチレン樹脂ビーカー(50〜100 mL)へ少量の水を用
い移し入れる。
1.2.2) 硝酸3 mL及びふっ化水素酸2〜5 mLを加え,更に過塩素酸5 mLを加えた後,穏やかに加熱し
て分解し,引き続き加熱して二酸化けい素を揮散させ,ほとんど乾固する。放冷した後,ビーカ
ーの内壁を少量の水で洗い,塩酸(1+1) 10 mLを加え,加熱して可溶性塩を溶解する。
1.2.3) 溶液を100 mLの全量フラスコに水を用いて移し入れ,水で標線まで薄める。
1.3) 試料中に比較的多量のバリウムを含むときの試料溶液の調製
1.3.1) b) 4)で保管しておいた残物及びろ紙を磁性るつぼ(20番)へ入れ,乾燥した後,できるだけ低
温でろ紙を灰化する。放冷した後,灰化物を白金るつぼ(20番)に移し入れる。
9
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1.3.2) 炭酸ナトリウム(無水)5 gを加え,加熱して融解する。放冷した後,融解物を白金るつぼとと
もにビーカー(300 mL)に入れ,温水約150 mLを加え穏やかに加熱して可溶性塩を溶解し,白金
るつぼは温水で十分に洗浄して取り除く。
1.3.3) 塩化アンモニウム5 g,アンモニア水10 mL及び炭酸アンモニウム5 gを順次加えてかき混ぜ,
引き続き溶液をかき混ぜながら,塩化カルシウム溶液[6.2.2 t)] 10 mLを加える。時計皿で覆い,
80〜90 ℃で約1時間加熱するか,又は約10分間煮沸した後,10〜15分間温所に静置して沈殿
を熟成させる。時計皿の底面を少量の水で洗浄して時計皿を取り除き,沈殿をろ紙(5種C)を
用いてこし分け,温炭酸アンモニウム溶液(20 g/L)で十分に洗浄した後,温水で元のビーカーに
洗い移し,温塩酸(1+1) 10 mLをろ紙上から滴下し,更にろ紙を温水で十分に洗浄し,ろ液及
び洗液を沈殿の入っているビーカーに受ける。溶液を再び加熱し,残物があるときは,ろ紙(5
種C)を用いてろ過し,温水で十分に洗浄する。
1.3.4) この溶液を,必要に応じて加熱して濃縮し,放冷した後100 mLの全量フラスコに水を用いて移
し入れ,水で標線まで薄める。
2) 原子吸光光度法による吸光度の測定 1.1.3),1.2.3)又は1.3.4)で得た溶液の一部を,水を用いて零点
を調整した原子吸光光度計の空気・アセチレンフレーム中に噴霧し,波長217.0 nm又は283.3 nm
における吸光度を測定し(14)(15),得た吸光度と3)で作成した検量線とから溶液中の鉛量を求める。
3) 検量線の作成 検量線の作成は,次のいずれかによる。
3.1) 1.1)によって試料溶液を調製した場合 標準鉛溶液[6.2.2 ab)]の各種液量(鉛として0〜0.5 mg)を
段階的に数個の250 mLの全量フラスコにとり,塩酸(1+1) 10 mL,塩化ナトリウム2.1 g及び塩
化カリウム3.7 gを加え,水で標線まで薄める。この溶液の吸光度を,2)の手順に従って試料溶液
と併行して測定し得た吸光度と鉛量との関係線を作成し,この関係線を,原点を通るように平行
移動して検量線とする。
3.2) 試料溶液の調製を行った場合 1.2)又は1.3)によって,標準鉛溶液[6.2.2 ab)]の各種液量(鉛とし
て0〜0.5 mg)を段階的に数個の100 mLの全量フラスコに取り,塩酸(1+1) 10 mLを加え,水で
標線まで薄める。これらの溶液の吸光度を,2)の手順に従って,試料溶液と併行して測定し,得
た吸光度と鉛量との関係線を作成し,この関係線を原点を通るように平行移動して検量線とする。
6.2.5
空試験 試料を用いないで,試料と同じ操作を試料と併行して行う。
6.2.6
計算 試料中の鉛含有率を,式(3)によって算出する。
(
)
H
m
m
m
f
V
V
Pb
−
×
×
+
+
×
−
=
100
100
100
b
a
b
t
······································· (3)
ここに, Pb: 試料中の鉛含有率[%(質量分率)]
Vt: 6.2.4 c)で得た,0.025 mol/L EDTA標準溶液の使用量 (mL)
Vb: 6.2.5で得た,0.025 mol/L EDTA標準溶液の使用量 (mL)
f: 0.025 mol/L EDTA標準溶液1 mLに相当する鉛量 (g/mL)
ma: 6.2.4 d) 2)で得たろ液中の鉛量 (g)
mb: 6.2.4 e) 2)で得た不溶解残物中の鉛量 (g)
m: 試料はかりとり量 (g)
H: 附属書1又は附属書2によって求めた分析試料中の吸着水分含
有率[%(質量分率)]{試料のはかりとりを事前乾燥法[4.2 b)
の2)又は3)]で行った場合には,H=0とする。}
10
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.3
アルカリ融解・硫酸鉛沈殿分離・EDTA−亜鉛逆滴定法
6.3.1 要旨 試料を過酸化ナトリウムを用いて融解し,融解物を水で溶解する。ふっ化水素酸及び過塩素
酸で処理した後,硫酸を加え,加熱して硫酸の白煙を発生させる。水で可溶性塩を溶解した後,エタノー
ルを加え,硫酸鉛として鉛を沈殿分離する。沈殿を水酸化カリウム及び一定量のEDTA(エチレンジアミ
ン四酢酸二水素ナトリウム)標準溶液で溶解し,キシレノールオレンジを指示薬として,亜鉛標準溶液で
過剰のエチレンジアミン四酢酸を滴定する。
6.3.2
試薬 試薬は,次による。
a) 硝酸
b) 硝酸(1+1)
c) 過塩素酸
d) ふっ化水素酸
e) 硫酸
f)
硫酸(1+1)
g) 酸洗浄混液 過塩素酸50 mLと温水100 mLとを混合し,過酸化水素水2 mLを加える。この溶液は使
用直前に調製する。
h) 水酸化カリウム溶液(200 g/L)
i)
アルカリ洗浄溶液 水酸化カリウム溶液(200 g/L) 50 mLを水で1 Lに希釈する。
j)
酢酸アンモニウム溶液(500 g/L)
k) ふっ化ナトリウム
l)
過酸化ナトリウム
m) 硝酸ジルコニウム溶液 オキシ塩化ジルコニウム(ZrOCl2・8H2O) 3.53 gを硝酸20 mLに加える。約10
分間煮沸し,室温まで冷却した後,水で1 Lに希釈する。
n) エタノール(95)
o) エタノール・硫酸洗浄混液 水200 mLにエタノール100 mL及び硫酸50 mLを加える。
p) ヘキサメチレンテトラミン水溶液(飽和溶液)
q) 0.1 mol/L亜鉛標準溶液 亜鉛[99.99 %(質量分率)以上]6.537 gをビーカー(1 000 mL)にはかりと
り,水25 mLと硝酸20 mLとの混合液を少量ずつ添加する。時計皿で覆い,穏やかに加熱して完全に
分解し,引き続き窒素酸化物がなくなるまで煮沸する。放冷した後,時計皿の下面を水で洗って時計
皿を取り除き,水を加えて液量を約700 mLとする。ヘキサメチレンテトラミン溶液(飽和)を滴加
して,pH計を用いてpH 5.5に調節する。溶液を1 000 mLの全量フラスコに水を用いて移し入れ,水
で標線まで薄める。この溶液の標定は,次のようにして行う。
0.1 mol/L EDTA標準溶液[6.3.2 r)]を正確に25 mLずつ,3個の別々のビーカー(500 mL)にとる。水
酸化カリウム溶液(200 g/L) 25 mLを加え,水で液量を約350 mLとし,更に,キシレノールオレンジ
溶液(5 g/L)を指示薬として0.3 mL及び酢酸アンモニウム溶液(500 g/L) 5 mLを加える。pH計を用い,
硝酸(1+1)又はヘキサメチレンテトラミン水溶液(飽和)を加えて,溶液のpHを5.5±0.1に調節す
る。3個の溶液をそれぞれ0.1 mol/L亜鉛標準溶液で滴定し,溶液の色が明りょうに赤に変わった点を
終点として,0.1 mol/L亜鉛標準溶液の使用量を求め,式(4)によって亜鉛標準溶液使用量のEDTA標
準溶液使用量への換算係数を算出する。
11
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3 ,2 ,1
x
25
x
x
=
=
V
t
·································································· (4)
ここに, tx: 個々の滴定で得た0.1 mol/L亜鉛標準溶液使用量の0.1 mol/L
EDTA標準溶液使用量への換算係数
Vx: 個々の滴定で得た0.1 mol/L亜鉛標準溶液の使用量 (mL)
t1,t2及びt3各算出値の範囲が0.001を超える場合には,標定をやり直す。算出値の範囲が0.001以
下の場合には,亜鉛標準溶液使用量の0.1 mol/L EDTA標準溶液使用量への換算係数を,式(5)によっ
て求める。
3
3
2
1
t
t
t
t
+
+
=
··········································································· (5)
ここに,
t: 0.1 mol/L亜鉛標準溶液使用量の0.1 mol/L EDTA標準溶液使用
量への換算係数
r) 0.1 mol/L EDTA標準溶液 エチレンジアミン四酢酸二水素二ナトリウム二水和物37.22 gを水に溶解
し,水で液量を1 Lとする。この溶液1 mLは,鉛約0.020 7 gに相当するが,標定は,次のようにし
て行う。
鉛[99.99 %(質量分率)以上]の0.1〜0.8 gを0.1 mgのけたまで3個はかりとり(17),それぞれ別々
のニッケルるつぼに移し入れ,別に空試験として3個のニッケルるつぼを用意する。以下6個のるつ
ぼについて6.3.4のa) 2)〜c)の手順に従って操作し,0.1 mol/L EDTA標準溶液1 mLに相当する鉛量を,
式(6)によって算出する。
(
)
3
,2
,1
x
Zx
Bx
x
x
V
V
t
m
f
=
−
=
······················································ (6)
ここに,
fx: 個々の滴定で得た0.1 mol/L EDTA標準溶液1 mLに相当する
鉛量 (g/mL)
mx: 個々の滴定に用いた鉛の質量 (g)
t: 6.3.2 q)で求めた0.1 mol/L亜鉛標準溶液使用量の0.1 mol/L
EDTA標準溶液使用量への換算係数
VBx: 個々の空試験で得た,0.1 mol/L亜鉛標準溶液の使用量 (mL)
VZx: 個々の滴定で得た0.1 mol/L亜鉛標準溶液の使用量 (mL)
f1,f2及びf3各算出値の範囲が0.000 02 g/mLを超える場合には,標定をやり直す。算出値の範囲が
0.000 02 g/mL以下の場合には,式(7)によってEDTA標準溶液1 mLに相当する鉛量を求める。
3
3
2
1
f
f
f
f
+
+
=
······································································· (7)
ここに,
f: 0.1 mol/L EDTA標準溶液1 mLに相当する鉛量 (g/mL)
注(17) 6.3.3ではかりとった試料中に含まれる鉛量とほぼ同じ量の鉛をはかりとる。
s)
キシレノールオレンジ溶液(5 g/L)
6.3.3
試料のはかりとり量 試料のはかりとり量は,1 gとし,0.1 mgのけたまではかりとる。
6.3.4
操作 操作は,次による。
a) 試料の分解 試料の分解は,次の手順によって行う。
1) 試料をはかりとり,ニッケルるつぼ又は,ジルコニウムるつぼへ移し入れる。
2) るつぼに過酸化ナトリウム2 gを加え,混合する。過酸化ナトリウム3 gで混合物を覆い,融解が始
まるまで穏やかに加熱した後,暗赤色になるまで温度を上昇させ,振り混ぜながら2〜3分間保持す
る。放冷した後,るつぼ及び融解物を四ふっ化エチレン樹脂ビーカー(500 mL,トール型)に入れ,
水約125 mLを加える。

12
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) 融解物が溶解するまで放置した後,水で,続いて酸洗浄混液[6.3.2 g)]を用いてるつぼを洗浄しなが
らビーカーから取り出す。
4) ふっ化水素酸3 mLを加え,時計皿で覆い,10分間煮沸する。時計皿の底面を少量の水で洗浄して
時計皿を取り除き,過塩素酸20 mLを加え,加熱して最終液量が約10 mLになるまで過塩素酸の白
煙を発生させる。冷却した後,溶液を少量の水を用いてビーカー(500 mL)に移し入れる。
b) 硫酸鉛の分離 硫酸鉛の分離は,次の手順によって行う。
1) a) 4)で得た溶液に硫酸(1+1) 50 mLを加え,大量の硫酸白煙が発生するまで加熱する。放冷した後,
注意して水250 mLを加え,室温まで冷却し,エタノール(95) 150 mLを加える。
2) 1〜2時間静置した後,硫酸鉛の沈殿をメンブランフィルタ(ニトロセルロース製,孔径:0.45〜0.65
µm)(18)を用いて吸引してこし分け,エタノール・硫酸洗浄混液[6.3.2 o)]で2〜3回ビーカーを洗
浄し,この洗液もろ過する。メンブランフィルタ及びろ過装置をエタノール・硫酸洗浄混液[6.3.2 o)]
で2〜3回洗浄する。
注(18) セルロース製メンブランフィルタは,以後の操作で溶解し,硫酸鉛の沈殿生成及びろ過操
作に支障を来さない。したがって,この操作におけるメンブランフィルタは,硫酸及び硝
酸の混合液によって容易に溶解するものであれば,他の代用品を用いてもよい。
3) 沈殿をメンブランフィルタとともに元のビーカーに移し入れ,湿ったろ紙片でろ過装置をぬぐい,
そのろ紙片もビーカーに入れる。
4) ビーカーに硫酸50 mL及び硝酸5 mLを加え,時計皿で覆い,加熱して十分に硫酸白煙を発生させ
る。放冷した後,時計皿を取り除き,水20 mLを注意して加え,再び加熱して,十分に硫酸白煙を
発生させる。
5) 放冷した後,十分にかき混ぜながら,水100 mL,更に温水50 mLを加える。時計皿でビーカーを覆
い,加熱して5分間穏やかに煮沸する。室温まで冷却し,エタノール(95) 100 mLを加えた後,再び
冷却し,1時間静置する。
6) 時計皿の底面を少量の水で洗浄して時計皿を取り除き,あらかじめエタノール(95)で湿らしたメン
ブランフィルタ(塩化ビニル製,孔径:0.45〜0.65 µm)(19)を用いて硫酸鉛の沈殿を吸引してこし
分ける。エタノール・硫酸洗浄混液[6.3.2 o)]でビーカー,メンブランフィルタ及びろ過装置を2〜3
回洗浄した後,沈殿がほとんど乾燥するまで吸引を続ける。
注(19) 塩化ビニル(PVC)製メンブランフィルタは,以後の操作によっても溶解しない。一方,セ
ルロース製メンブランフィルタは,以後の操作で溶解し,滴定で呈色妨害を引き起こす。
そこで,この操作におけるメンブランフィルタは,以後の操作において溶解しなければ代
用品を用いてもよい。
7) 沈殿をメンブランフィルタとともに元のビーカーに移し入れる。ろ過装置に付着している微量の沈
殿をゴム管付きガラス棒を用いてすべて元のビーカーに移し入れる。
8) かき混ぜながら,水酸化カリウム溶液(200 g/L) 25 mL及び0.1 mol/L EDTA標準溶液[6.3.2 r)]とを
表3に従って加える。
表 3 0.1 mol/L EDTA標準溶液の添加量
試料中の鉛含有率
%(質量分率)
0.1 mol/L EDTA標準溶液の添加量
mL
10〜60
50.00
60〜80
70.00
13
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9) 時計皿で覆い,加熱して沈殿が溶けるまで穏やかに煮沸した後,時計皿の底面を少量の水で洗浄し
て時計皿を取り除き,メンブランフィルタを水で洗浄しながら取り除く。水で液量を約170 mLと
し,指示薬としてキシレノールオレンジ溶液(5 g/L) 3〜4滴を加え,更に硝酸(1+1)を溶液の色が
黄に変わるまで加える(20)(21)。
注(20) バリウムが含まれている場合には,硫酸バリウムが沈殿することがある。
(21) ビスマス含有率が0.05 %(質量分率)以下の場合には,次の10)の操作は省略してもよい。
10) 硝酸ジルコニウム溶液[6.3.2 m)] 5 mLを加え,更に水酸化カリウム溶液(200 g/L) 5 mLとを1滴ず
つ加えた後,時計皿で覆い,加熱して約5分間溶液を煮沸し,温度を保ちながら30分間静置する(22)。
溶液を室温まで冷却し,時計皿の底面を少量の水で洗浄して時計皿を取り除き,エタノール(95)で
湿らせたメンブランフィルタ(塩化ビニル製,孔径:0.45〜0.65 µm)(19)で吸引ろ過し,ろ液をビ
ーカー(500 mL)に受ける。アルカリ洗浄溶液[6.3.2 i)]を用いて元のビーカーを少なくとも5回洗浄
し,その都度洗液を吸引ろ過してろ液に合わせる。再びアルカリ洗浄液[6.3.2 i)]を用いてメンブラ
ンフィルタ及びろ過装置を3回洗浄し,洗液をろ液に合わせ,溶液の色が黄となるまで硝酸(1+1)
を滴加する(23)。
注(22) 9)の操作で生成した硫酸バリウム沈殿は,この操作によって再び溶解する。
(23) 硫酸バリウムが沈殿するが,この操作では沈殿に硫酸鉛が包含されて残存することはない。
c) 滴定 6.3.4のb) 9)又は,b) 10)で得た溶液にふっ化ナトリウム2〜3 gを加え,ビーカーを時計皿で覆
い,加熱して3分間煮沸した後,水で液量を約350 mLとする。室温まで冷却し,時計皿の底面を少
量の水で洗浄して時計皿を取り除き,酢酸アンモニウム溶液(50 g/L) 5 mL加える。pH計を用い,硝酸
(1+1)又はヘキサメチレンテトラミン水溶液(飽和)を加え,溶液のpHを5.5±0.1に調節する。0.1 mol/L
亜鉛標準溶液[6.3.2 q)]で滴定し,溶液の色が明りょうに赤に変わった点を終点とし,0.1 mol/L亜鉛標
準溶液の使用量を求める。
6.3.5
空試験 試料を用いないで,試料と同じ操作を試料と併行して行う。
6.3.6
計算 試料中の鉛含有率を,式(8)によって算出する。
(
)
H
m
f
V
V
t
Pb
−
×
×
×
−
=
100
100
100
Z
B
·················································· (8)
ここに, Pb: 試料中の鉛含有率[%(質量分率)]
t: 6.3.2 q)で求めた0.1 mol/L亜鉛標準溶液使用量の0.1 mol/L
EDTA標準溶液使用量への換算係数
VB: 6.3.5で得た,0.1 mol/L亜鉛標準溶液の使用量 (mL)
VZ: 6.3.4 c)で得た,0.1 mol/L亜鉛標準溶液の使用量 (mL)
f: 6.3.2 r)で求めた0.1 mol/L EDTA標準溶液1 mLに相当する鉛量
(g/mL)
m: 試料はかりとり量 (g)
H: 附属書1又は附属書2によって求めた分析試料の吸着水分含有
率{試料のはかりとり方を事前乾燥法[4.2 b)の2)又は3)]で
行った場合には,H=0とする。}
6.4
フレーム原子吸光法
6.4.1
要旨 試料を塩酸と硝酸とで分解し,蒸発乾固する。塩酸を加えて乾固物を溶解し,溶液をろ過し
た後,原子吸光光度計を用いて,その吸光度を測定する。
6.4.2
試薬 試薬は,次による。
a) 塩酸

14
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
b) 塩酸(1+1,1+9,1+50)
c) 硝酸
d) 過塩素酸
e) ふっ化水素酸
f)
臭化水素酸
g) アンモニア水
h) アンモニア洗浄液 アンモニア水(2+50) 500 mLに炭酸アンモニウム15 gを加え溶解する。
i)
炭酸アンモニウム
j)
炭酸アンモニウム溶液(20 g/L)
k) 炭酸ナトリウム(無水)
l)
塩化鉄(Ⅲ)溶液 鉄[99.9 %(質量分率)以上]5.0 gを塩酸(1+1) 50 mLと硝酸10 mLとで分解し,
穏やかに加熱して蒸発乾固する。放冷した後,塩酸5 mLを加えて乾固物を溶解し,水で液量を500 mL
とする。
m) 標準鉛溶液(Pb:0.2 mg/mL又は0.1 mg/mL) 鉛[99.9 %(質量分率)以上]1.0 gを1 mgのけた
まではかりとり,ビーカー(300 mL)に移し入れ,硝酸(1+1) 20 mLを加え,加熱して時計皿で覆い,
穏やかに加熱して分解する。時計皿の底面を少量の水で洗浄して時計皿を取り除き,加熱して穏やか
に沸騰させ,窒素酸化物のガスを逃がした後,室温まで放冷する。溶液を1 000 mLの全量フラスコに
水を用いて移し入れ,水で標線まで薄める。使用の都度,必要量だけ水で正確に5倍又は10倍に薄め
て標準鉛溶液とする。
6.4.3
試料はかりとり量 試料のはかりとり量は,鉛の含有率に応じて表4によるものとし,0.1 mgのけ
たまではかる。
表 4 試料はかり取り量
試料中の鉛含有率
%(質量分率)
試料はかりとり量
g
0.01以上 0.1未満
1.0〜2.0
0.1 以上 0.5未満
0.5
0.5 以上 5.0以下
0.2
6.4.4
操作 操作は,次による。
a) 試料の分解及び測定溶液の調製 試料の分解及び測定溶液の調製は,次の手順によって行う。
1) 試料をはかりとり,ビーカー(300〜500 mL)に移し入れ,時計皿で覆い,塩酸20〜40 mLを加えて
加熱し,更に硝酸10〜30 mLを加え,穏やかに加熱して分解する(24)(25)。時計皿の底面を少量の水
で洗浄して時計皿を取り除いた後,引き続き加熱して蒸発乾固する。
注(24) 比較的多量の二酸化けい素などを含む試料の場合には,試料をはかりとった後,四ふっ化
エチレン樹脂ビーカー(100〜200 mL)に移し入れ,硝酸15〜20 mL及びふっ化水素酸10〜
20 mLを加え,更に必要に応じて過塩素酸約10 mLを加え,穏やかに加熱して分解し,引
き続き加熱して二酸化けい素を揮散させ,蒸発乾固する。少し放冷した後,ビーカーの内
壁を少量の水で洗い,硝酸又は過塩素酸5 mLを加え,再び加熱して蒸発乾固する。以下,
2)以降の手順に従って操作する。
(25) 多量のひ素,アンチモン,すず又はセレンを含む試料の場合には,過塩素酸10 mLを加え,
加熱蒸発して過塩素酸の白煙を十分に発生させる。放冷した後,水約5mL及び臭化水素酸

15
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5〜10 mLを加え,加熱蒸発して過塩素酸の白煙を十分に発生させる。以下,2)以降の手順
に従って操作する。
2) 放冷した後,塩酸(1+1) 20 mL及び水15〜20 mLを加え,時計皿で覆い,加熱して可溶性塩を溶解
する。時計皿の底面を少量の水で洗浄して時計皿を取り除き,溶液をろ紙(5種B)を用いてろ過
し,温水で十分に洗浄する(26)(27)。冷却した後,ろ液及び洗液(28)を100 mLの全量フラスコに水
を用いて移し入れ,水で標線まで薄める(29)。
注(26) 残物中に鉛が含まれる場合には,残物を少量の水で四ふっ化エチレン樹脂ビーカー(100〜
200 mL)に洗い移し,硝酸5〜10 mL,ふっ化水素酸5〜10 mL及び過塩素酸5 mLを加え,
加熱して蒸発乾固する。少し放冷した後,ビーカーの内壁を少量の水で洗い,過塩素酸約
5 mLを加え,再び加熱して蒸発乾固する。放冷した後,塩酸(1+1) 10 mL及び少量の水を
加え,時計皿で覆い,加熱して可溶性塩を溶解し,時計皿の底面を少量の水で洗浄して時
計皿を取り除き,溶液をろ紙(5種B)を用いてビーカー(200 mL)にろ過し,温水で十分
に洗浄する。必要に応じてこの残物処理操作を更に一回繰り返す。ろ紙及び洗液は合わせ,
加熱して1〜2 mLに濃縮した後,主液に合わせる。
なお,残物をろ紙とともに磁器るつぼに移し入れ,乾燥した後,できるだけ低温でろ紙
を灰化してから,四ふっ化エチレン樹脂ビーカーに移し入れて操作してもよい。
(27) 試料中に比較的多量のバリウムを含み,注(26)の操作では残物中の鉛が補正できない場合に
は,6.2.4 e)の1.3)〜3)の手順に従って残物中の鉛を定量し,補正する。また,カルシウムの
影響が無視できない場合には,検量線の作成において注(31)を準用する。
(28) 試料中に多量の銅,亜鉛などを含み,特に試料はかりとり量が多い場合など,その影響が
無視できないときは,次の操作によって銅,亜鉛などを分離する。
この溶液に,必要に応じ塩化鉄(Ⅲ)溶液[6.4.2 l)]の所要量を加え,溶液をかき混ぜなが
らアンモニア水を,水酸化鉄の沈殿が生成するまで滴加した後,更に過剰に約50 mLを加
える。
次いで炭酸アンモニウム約15 gを加えて加熱し,時計皿で覆い,穏やかに約5分間煮沸
した後,時計皿の底面を少量の水で洗浄して時計皿を取り除き,沈殿をろ紙(5種A)を
用いてこし分け,温めたアンモニア洗浄液[6.4.2 h)]で数回洗浄する。沈殿を温水で元のビ
ーカーに洗い移し,塩酸(1+1) 20 mLをろ紙上から滴下して,ろ紙及びビーカー中の沈殿
を溶解する。ろ紙は温塩酸(1+50)で十分に洗浄し,洗液は元のビーカーに合わせる。
なお,塩化鉄(Ⅲ)溶液[6.4.2 l)]の添加量は,6.4.3ではかりとった試料中の鉄含有量を考
慮して,この溶液中の全鉄量が約60 mg以上約250 mg以下となるように決定する。また,
試料中に多量の鉄を含み,鉄量がこの範囲を超える場合には,試料はかりとり量を少なく
する。
(29) 鉛量が多い場合には,検量線の直線領域で,測定精度の良い濃度範囲に入るように,この
溶液の適量を100 mLの全量フラスコに正確に分取し,塩酸(1+9)で標線まで薄める。
b) 吸光度の測定 a) 2)又は注(29)で得た溶液の一部を,水を用いて零点を調整した原子吸光光度計の空
気・アセチレンフレーム中に噴霧し,波長217.0 nm又は283.3 nmにおける吸光度を測定する(30)。
注(30) 原子吸光光度計の代わりに,ICP発光分光装置を用いて鉛量を求めてもよい。このとき用いる
測定波長の一例としては,220.35 nmがある。
なお,ICP発光分光装置は,装置の種類によって波長分解能が異なっており,事前に妨害の

16
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
有無を確認し,適切な波長を選択する。高次のスペクトル線が使用可能な装置では,高次のス
ペクトル線を用いてもよい。また,バックグラウンド補正機構が付いている装置では,バック
グラウンド補正機構を用いてもよい。
6.4.5
空試験値 試料を用いないで,試料と同じ操作を試料と併行して行う。
6.4.6
検量線の作成 標準鉛溶液[6.4.2 m)]の各種液量(鉛として0〜5 mg)を段階的に数個の100 mLの
全量フラスコに取り,塩酸(1+1) 20 mLを加え(31),水で標線まで薄める。この溶液の一部を水を用いて零
点を調整した原子吸光光度計の空気・アセチレンフレーム中に噴霧し,波長217.0 nm又は283.3 nmにお
ける吸光度を試料と併行して測定し,得た吸光度と鉛量との関係線を作成し,その関係線を原点を通るよ
うに平行移動して検量線とする。
注(31) 装置及び/又は測定条件によっては,試料に含まれる鉄,カルシウムなどが鉛の測定に影響す
ることがある。特に試料中に多量の鉄を含むとき及び注(28)で塩化鉄(Ⅲ)溶液[6.4.2 l)]を添加し
たときは注意する。また,ICP発光分光装置を用いて測定するときは,原子吸光光度計を用い
たときよりもそれらの影響が大きい。このように試料に含まれる共存元素の影響を受けるとき
は,検量線用溶液を調製するとき,これら共存元素の影響を相殺できる濃度範囲となるように
塩化鉄(Ⅲ)溶液[6.4.2 l)]などを添加する。
なお,カルシウムなど鉄以外の元素の影響がある場合にも,この操作に準じて,その影響を
相殺する。
6.4.7
計算 6.4.4 b)で得た吸光度から6.4.5で得た吸光度を差し引いて得られた吸光度と6.4.6で作成し
た検量線とから鉛量を求め,試料中の鉛含有率を,式(9)又は式(10)のいずれかによって算出する。
a) 注(29)を適用しなかった場合
H
m
A
Pb
−
×
×
=
100
100
100
································································ (9)
ここに, Pb: 試料中の鉛含有率[%(質量分率)]
A: 試料溶液中の鉛検出量 (g)
m: 試料はかりとり量 (g)
H: 附属書1又は附属書2によって求めた分析試料中の吸着水分含
有率[%(質量分率)]{試料のはかりとり方を事前乾燥法[4.2
b)の2)又は3)]で行った場合には,H=0とする。}
b) 注(29)を適用した場合
H
B
m
A
Pb
−
×
×
×
=
100
100
100
100
······················································· (10)
ここに, Pb: 試料中の鉛含有率[%(質量分率)]
A: 分取した試料溶液中の鉛検出量 (g)
m: 試料はかりとり量 (g)
B: 注(33)で分取した試料溶液の量 (mL)
H: 附属書1又は附属書2によって求めた分析試料中の吸着水分含
有率[%(質量分率)]{試料のはかりとり方を事前乾燥法[4.2
b)の2)又は3)]で行った場合には,H=0とする。}
17
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1(規定)分析用試料の吸着水分の測定方法
序文 この附属書は,ISO 9599を翻訳(要約)している。
1. 適用範囲 この附属書は,分析用試料の吸着水分の測定方法について規定する。この方法は,例えば,
ケロシンなどの揮発性成分を含まない吸着水分含有率が0.05 %(質量分率)以上2 %(質量分率)以下
の試料に適用する。この方法は,酸化するおそれのある硫化精鉱に用いることはできない。この場合は,
附属書2による。
2. 要旨 試料を規定された温度で恒量となるまで乾燥し,熱乾燥減量を求める。
3. 装置 装置は,次による。
a) 平形はかり瓶 ガラス製,石英製又は耐食性金属製の直径約50 mmのふた付きのもの。
b) 平皿 耐食性のもので,試料の厚さが3〜5 mmとなる底面積をもつもの。
4. 試料
4.1
分析用試料 粒径150 µm以下の試料を用いる。
4.2
試験試料の調製 化学分析及び吸着水分含有率の測定に十分な量の分析用試料を平皿[3.b)]に移し
入れ,3〜5 mmの厚さになるように平らに広げる。粉じん(塵)による試料の汚染を防ぐため,試料の上
部を空気が自由に流れるようにしてふたなどで覆う。試験試料を実験室大気と平衡にするため,2時間又
は平衡に達するために十分な時間放置する。試験試料の質量の変化が,放置2時間当たり0.1 %(質量分
率)よりも小さくなるまで放置する。
5. 操作
5.1
平形はかり瓶の準備 平形はかり瓶[3. a)]及びそのふたを空気浴中で105±5 ℃で1時間乾燥する。
平形はかり瓶にふたをしてデシケーター中で室温まで放冷した後,デシケーターから取り出してふたをわ
ずかにずらし,直ちに元に戻し,その質量を0.1 mgのけたまではかる。
5.2
試料のはかりとり 4.2で大気と平衡にした試験試料から約10 gを5.1で質量をはかった平形はかり
瓶にはかりとり,約3〜5 mmの厚さになるように平らに広げる。平形はかり瓶,ふた及び試料の合計質量
を0.1 mgのけたまではかる(1)。
注(1) 5分以内に本体の各定量方法に規定する量の試料をはかりとる。
5.3
乾燥及びひょう量 5.2で質量をはかった試料が入っている平形はかり瓶及びふたを空気浴に入れ,
105±5 ℃で2時間乾燥する。試料が入っている平形はかり瓶とそのふたを取り出し,ふたをしてデシケー
ター中で室温まで放冷した後,デシケーターから取り出してふたをわずかにずらし,直ちに元に戻し,そ
の質量を0.1 mgのけたまではかる。再び空気浴中で105±5 ℃で2時間乾燥し,デシケーター中で室温ま
で放冷した後,デシケーターから取り出してふたをわずかにずらし,直ちに元に戻し,その質量を0.1 mg
のけたまではかる(2)。
注(2) 乾燥前後の質量の差が±1 mgを超えたときは,再び乾燥及びひょう量を繰り返す。2時間の乾
燥を3回繰り返しても乾燥前後の質量の差が±1 mg以下とならないときは,附属書2に規定す
18
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る方法で吸着水分を求める。
6. 計算 試料中の吸着水分含有率を,次の式によって小数点以下2位まで算出する。
100
1
2
3
2
×
−
−
=
m
m
m
m
H
······································································ (1)
ここに,
H: 試料中の吸着水分含有率[%(質量分率)]
m2: 5.2で得た乾燥前の試料,平形はかり瓶及びふたの合計質量 (g)
m3: 5.3で得た乾燥後の試料,平形はかり瓶及びふたの合計質量 (g)
m1: 5.1で得た乾燥した平形はかり瓶及びふたとの合計質量 (g)
19
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2(規定)酸化するおそれがある分析用試料の
吸着水分の測定方法
序文 この附属書は,ISO 9599を翻訳(要約)している。
1. 適用範囲 この附属書は,酸化するおそれがある分析用試料の吸着水分の測定方法について規定する。
この方法は,例えば,ケロシンなどの揮発性成分を含まない吸着水分含有率が0.05 %(質量分率)以上2 %
(質量分率)以下で,空気中で乾燥するとき酸化するおそれがある試料に適用する。
2. 要旨 試料を,乾燥した窒素中で規定された温度で恒量となるまで乾燥し,熱乾燥減量を求める。
3. 試薬 試薬は,次による。
a) 窒素 酸素含有率が 30 µL/L以下の乾燥ガス。
4. 装置 装置は,次による。
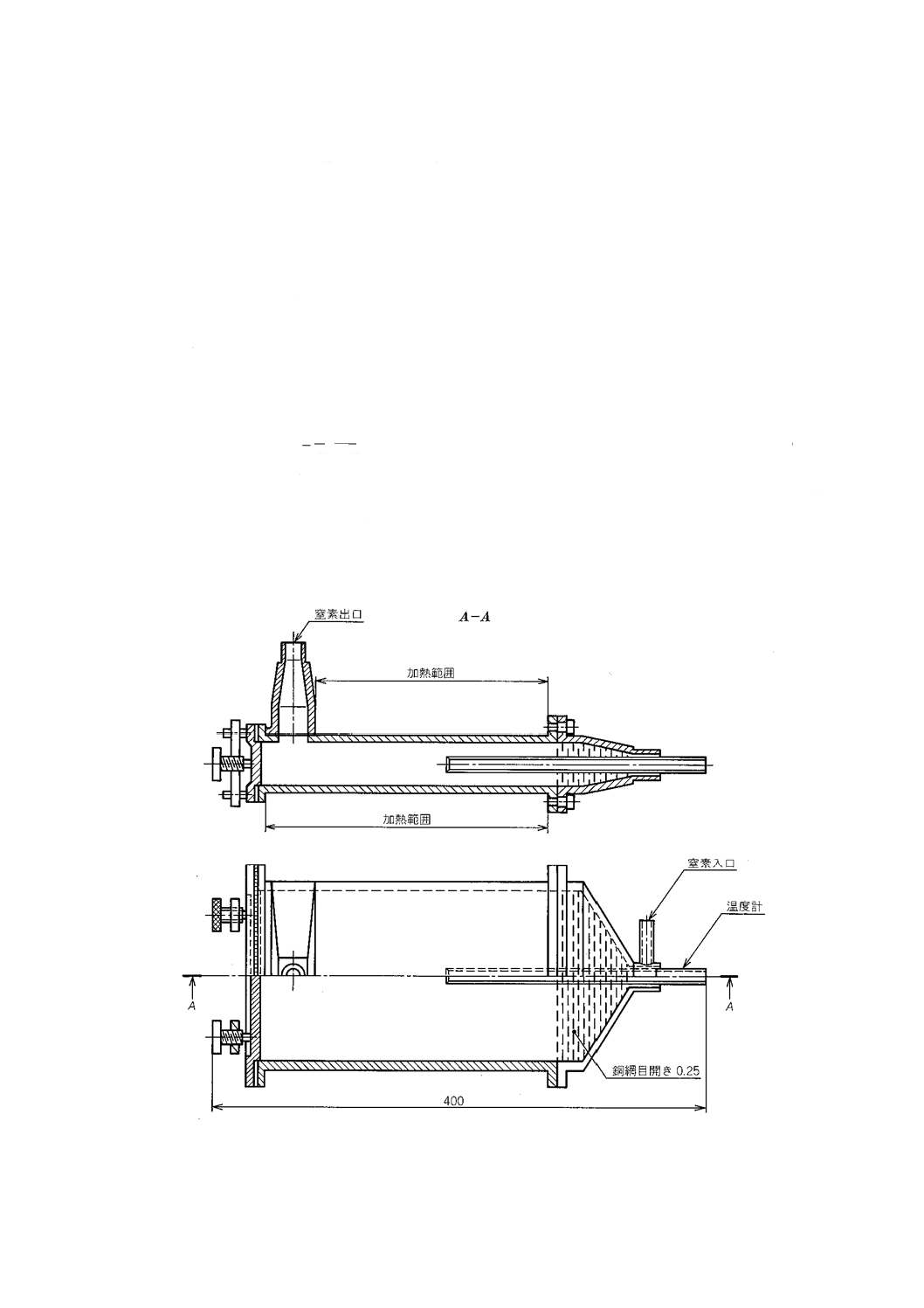
a) 乾燥器 1時間当たり乾燥器の容量の15〜20倍の予熱した窒素を供給できる装置が付いた,空間容量
が小さく,105±5 ℃に保持できるもの。一例を附属書2図1に示す。
b) 平形はかり瓶 ガラス製,石英製又は耐食性金属製の直径約50 mmのふた付きのもの。
c) 平皿 耐食性のもので,試料の厚さが3〜5 mmとなる底面積をもつもの。
d) 乾燥塔 容量約250 mLで,窒素を乾燥するための無水過塩素酸マグネシウムを詰めたもの。
5. 試料
5.1
分析用試料 粒径150 µm以下の試料を用いる。
5.2
試験試料の調製 化学分析及び吸着水分含有率の測定に十分な量の分析用試料を平皿[4. c)]に移し
入れ,3〜5 mmの厚さになるように平らに広げる。粉じん(塵)による試料の汚染を防ぐため,試料の上
部を空気が自由に流れるようにしてふたなどで覆う。試験試料を実験室大気と平衡にするため,2時間又
は平衡(1)に達するために十分な時間放置する。
注(1) 試験試料の質量の変化が,放置2時間当たり0.1 %(質量分率)よりも小さくなったときに平
衡に達したものとする。
6. 操作
6.1
平形はかり瓶の準備 平形はかり瓶[4. b)]及びそのふたを乾燥器[4. a)]中で1時間当たり乾燥器の
容量の15〜20倍の予熱した窒素[3. a)]を乾燥塔[4. d)]を通して供給しながら,105±5 ℃で1時間乾燥す
る。平形はかり瓶にふたをしてデシケーター中で室温まで放冷した後,デシケーターから取り出してふた
をわずかにずらし,直ちに元に戻し,その質量を0.1 mg のけたまではかる。
6.2
試料のはかりとり 5.2で大気と平衡にした試験試料から約10 gを,6.1で質量をはかった平形はか
り瓶にはかりとり,約3〜5 mmの厚さになるように平らに広げる。平形はかり瓶,ふた及び試料の合計質
量を0.1 mgのけたまではかる(2)。
20
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(2) 5分以内に本体の各定量方法に規定する量の試料をはかりとる。
6.3
乾燥及びひょう量 5.2で質量をはかった試料が入っている平形はかり瓶及びふたを乾燥器[4. a)]に
入れ,1時間当り乾燥器の容量の15〜20倍の予熱した窒素[3. a)]を乾燥塔[4. d)]を通じて供給しながら,
105±5 ℃で恒量(3)となるまで乾燥する(4)。恒量となったら,試料が入っている平形はかり瓶及びそのふ
たを取り出し,ふたをしてデシケーター中で室温まで放冷した後,デシケーターから取り出して,ふたを
わずかにずらし,直ちに元に戻し,その質量を0.1 mgのけたまではかる。
注(3) 30分間乾燥した前後の質量の差が±1 mg以下のとき恒量となったものとする。
注(4) この方法では,1.5〜3時間で乾燥は完了する。その後105±5 ℃で更に30分間乾燥すれば恒量
となる。
7. 計算 試料中の吸着水分含有率を,式(1)によって小数点以下2位まで算出する。
100
1
2
3
2
×
−
−
=
m
m
m
m
H
······································································ (1)
ここに, H: 試料中の吸着水分含有率[%(質量分率)]
m2: 6.2で得た乾燥前の試料,平形はかり瓶及びふたの合計質量 (g)
m3: 6.3で得た乾燥後の試料,平形はかり瓶及びふたの合計質量 (g)
m1: 6.1で得た乾燥した平形はかり瓶及びふたの合計質量 (g)
単位 mm
附属書2図 1 窒素を用いる乾燥器の例
21
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書3(参考)JISと対応する国際規格との対比表
JIS M 8123:2005 鉱石中の鉛定量方法
ISO 11441:1995,硫化鉛精鉱−鉛の定量方法−沈殿分離EDTA逆滴定法
ISO 13545:2000,硫化鉛精鉱−鉛の定量方法−酸分解EDTA滴定法
(Ⅰ) JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ) 国際規格の規定
(Ⅳ) JISと国際規格との技術的差異の項目ごとの評価
及びその内容
表示箇所:本体,附属書
表示方法:点線の下線又は側線
(Ⅴ) JISと国際規格との技
術的差異の理由及び今後の
対策
項目
番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
1. 適用範囲
鉱石
Pb 0.01〜80 %(質量分率)
ISO
11441
ISO
13545
1
硫化鉛精鉱
Pb 10〜80 %(質量分率)
硫化鉛精鉱
Pb 50〜80 %(質量分率)
MOD/追加 JISは,全鉱石について適用,ISO規
格は,硫化鉛精鉱だけ適用。
規格の適用対象の差によ
るもので,技術的差異は
ない。
2. 引用規格
JIS K 0050
JIS K 0116
JIS K 0121
JIS M 8083
JIS M 8101
JIS Z 8401
2
ISO 385-1:1984
ISO 648:1977
ISO 1042:1998
ISO 3696:1987
ISO 4787:1984
ISO 9599:1991
ISO Guide 35:1989
MOD/追加 ・発光分光分析通則及び原子吸光分析
通則が必要なため,JIS K 0116及び
JIS K 0121を追加した。
・サンプリング,試料調製及び水分決
定方法が必要なため,JIS M 8083及
びJIS M 8101を追加した。
・数値の丸め方JIS Z 8401を追加し
た。
規格の構成上の差
3. 一般事項
JIS K 0050,JIS K 0116及
びJIS K 0121を引用
−
−
ISO規格には,規定されていない。
規格の構成上の差
4. 分析試料
のとり方及
び取扱い方
法
4.1 試料の採取及び調製:
JIS M 8083及びJIS M
8101による。
−
MOD/追加 a) ISO規格には,規定されていない。 a) 規格の構成上の差
4.2 試料のはかり方:附属
書1,2(吸着水分測定)
による。又は,105±
5 ℃で恒量となるまで
(事前乾燥法A,B)。
6
b) 試料の乾燥:ISO 9599
に従い吸着水分を測定
する。又は各ISO規格
の附属書の事前乾燥法
(105±5 ℃)による。
MOD/追加 b) JISには簡便な事前乾燥法Bを追
加した。
b) 軽微な差
技術的な骨格に差はない
ため,JISの独自規定と
する。
2
1
M
8
1
2
3
:
2
0
0
6
22
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(Ⅰ) JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ) 国際規格の規定
(Ⅳ) JISと国際規格との技術的差異の項目ごとの評価及
びその内容
表示箇所:本体,附属書
表示方法:点線の下線又は側線
(Ⅴ) JISと国際規格との技
術的差異の理由及び今後
の対策
項目
番号
内容
項目
番号
内容
項目ごとの
評価
技術的差異の内容
5. 分析値の
表し方及び操
作上の注意
5.1 分析値の表し方
a) JIS Z 8401に従い小数
点以下2位に丸める。
ISO
11441
ISO
13545
8
a) 結果の算出:小数点
以下2位まで算出
する。
IDT
a) 技術的に同等である。
d) 併行許容差(表1)
9
b) 共同実験結果から
の許容差。
MOD/追加
b) 一致している。
ただし,ISO規格には規定されて
いない濃度範囲及び分析方法の許
容差を追加している。
b) 規格の適用対象の
差によるもので,技
術的差異はない。
5.2 全操作を通じて空試
験を行い,測定値を補
正する。
7
c) 空試験:全操作を通
じて実施する。
IDT
6. 定量方法
6.2 酸分解・硫酸鉛沈殿分
離・EDTA滴定法[Pb
5〜80 %(質量分率)]
ISO
13545
3
4
5
7
a) 酸分解・硫酸鉛沈殿
分離・EDTA滴定法
[Pb 50〜80 %(質
量分率)]
MOD/追加
a) ISO規格と技術的に一致している。
ただし,JISは全鉱石種対象のた
め,定量範囲を拡大。
滴定用標準溶液の標定法につい
て,方法間の整合を計るため,別法
を追加。
酸化鉱,焼鉱,高ビスマス含有鉱
及び高バリウム含有鉱の場合の操
作を追加。
a) 規格の適用対象の
差
ISO13545は,JIS M
8123を基に日本が提
案し,作成されたISO
である。審議を経て一
部JISとの差異が発生
したが,技術的には,
JISが基盤となってい
おり,差がない。
6.3 アルカリ溶融・硫酸鉛
沈殿分離・EDTA−亜
鉛逆滴定法[Pb 10〜
80 %(質量分率)]
ISO
11441
3
4
5
7
b) アルカリ溶融・硫酸
鉛沈殿分離・EDTA
−亜鉛逆滴定法[Pb
10〜80 %(質量分
率)]
MOD/追加,
削除
b) ISO規格と技術的に一致している。
硫酸鉛の分離操作時に危険を伴
う過塩素酸の使用を削除した。
メンブランフィルタの選定条件
を追加した。
b) 作業安全上の修正
各国で流通してい
る商品名に差がある
ため,規定を明確化し
た(技術上の変更はな
い)。
2
2
M
8
1
2
3
:
2
0
0
6

23
M 8123:2006
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(Ⅰ) JISの規定
(Ⅱ)
国際
規格
番号
(Ⅲ) 国際規格の規定
(Ⅳ) JISと国際規格との技術的差異の項目ごとの評
価及びその内容
表示箇所:本体,附属書
表示方法:点線の下線又は側線
(Ⅴ) JISと国際規格との技術
的差異の理由及び今後の対策
項目
番号
内容
項目
番号
内容
項目ごと
の評価
技術的差異の内容
6.4 フレーム原子吸光法[Pb
0.01〜5 %(質量分率)]
−
MOD/追加 c) ISO規格には規定されていない。 c) 規格の適用対象の差
附属書1(規定) 試料を規定された温度で恒量
となるまで乾燥し,熱乾燥減
量率を求める(ISO 9599本体
と同内容)。
ISO
11441
ISO
13545
6
ISO 9599を引用
IDT
附属書2(規定) 試料を乾燥した窒素中で,規
定された温度で恒量となるま
で乾燥し,熱乾燥減量率を求
める(ISO 9599附属書Aと同
内容)。
ISO
11441
ISO
13545
6
ISO 9599を引用
IDT
JISと国際規格との対応の程度の全体評価:MOD
備考1. 項目ごとの評価欄の記号の意味は,次のとおりである。
― IDT……………… 技術的差異がない。
― MOD/削除……… 国際規格の規定項目又は規定内容を削除している。
― MOD/追加……… 国際規格にない規定項目又は規定内容を追加している。
2. JISと国際規格との対応の程度の全体評価欄の記号の意味は,次のとおりである。
― MOD…………… 国際規格を修正している。
2
3
M
8
1
2
3
:
2
0
0
6