K 4101-1993
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目次
ページ
1. 適用範囲 ························································································································ 1
2. 一般事項 ························································································································ 1
3. 試料採取方法 ·················································································································· 2
3.1 要旨 ···························································································································· 2
3.2 試料の採取 ··················································································································· 2
3.3 製品の種別と容器の種別·································································································· 2
3.3.1 製品の種別 ················································································································· 2
3.3.2 容器の種別 ················································································································· 2
3.4 ロット及び代表試料 ······································································································· 2
3.5 試料採取の時期及び場所·································································································· 2
3.6 容器の抜取個数 ············································································································· 2
3.7 液体試料採取方法 ·········································································································· 2
3.7.1 器具 ·························································································································· 2
3.7.2 操作 ·························································································································· 5
3.7.2.1 小形容器の場合 ········································································································· 5
3.7.2.2 大形容器の場合 ········································································································· 5
3.8 固体試料採取方法 ·········································································································· 6
3.8.1 器具 ·························································································································· 6
3.8.2 操作 ·························································································································· 7
4. 試料乾燥方法 ·················································································································· 7
4.1 要旨 ···························································································································· 7
4.2 液体の場合 ··················································································································· 7
4.3 固体の場合 ··················································································································· 7
4.3.1 デシケーターによる方法 ······························································································· 7
4.3.2 恒温乾燥器による方法 ·································································································· 8
4.3.3 乾燥ガスによる方法 ····································································································· 8
5. 融点測定方法 ·················································································································· 9
5.1 目視による方法 ············································································································· 9
5.1.1 要旨 ·························································································································· 9
5.1.2 装置及び器具 ·············································································································· 9
5.1.3 試料の前処理 ············································································································· 11
5.1.4 操作 ························································································································· 11
5.2 光透過量の測定による方法······························································································ 12
5.2.1 要旨 ························································································································· 12
2
K 4101-1993 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.2.2 装置及び器具 ············································································································· 12
5.2.3 試料の前処理 ············································································································· 13
5.2.4 操作 ························································································································· 13
5.3 “とけ始め”及び“とけ終わり”の判定方法 ······································································ 13
6. 凝固点測定方法 ·············································································································· 15
6.1 要旨 ··························································································································· 15
6.2 装置及び器具 ··············································································································· 15
6.3 試料の前処理 ··············································································································· 16
6.4 操作 ··························································································································· 16
7. 蒸留試験方法 ················································································································· 17
7.1 要旨 ··························································································································· 17
7.2 装置及び器具 ··············································································································· 17
7.3 操作 ··························································································································· 20
7.4 蒸留温度の計算 ············································································································ 20
8. 密度及び比重測定方法 ····································································································· 21
8.1 浮ひょう法 ·················································································································· 21
8.1.1 要旨 ························································································································· 21
8.1.2 装置及び器具 ············································································································· 21
8.1.3 操作 ························································································································· 22
8.1.4 計算 ························································································································· 23
8.2 比重瓶法 ····················································································································· 24
8.2.1 要旨 ························································································································· 24
8.2.2 装置及び器具 ············································································································· 24
8.2.3 校正 ························································································································· 27
8.2.4 操作 ························································································································· 28
8.2.5 計算 ························································································································· 29
9. 水分試験方法 ················································································································· 29
9.1 カールフィッシャー滴定法······························································································ 29
9.1.1 要旨 ························································································································· 29
9.1.2 力価 ························································································································· 30
9.1.3 試料の採取 ················································································································ 30
9.1.4 容量滴定法 ················································································································ 30
9.1.4.1 要旨 ······················································································································· 30
9.1.4.2 装置及び器具 ··········································································································· 30
9.1.4.3 試薬 ······················································································································· 33
9.1.4.4 試料採取操作 ··········································································································· 35
9.1.4.5 操作 ······················································································································· 36
9.1.4.6 計算 ······················································································································· 37
9.1.5 電量滴定法 ················································································································ 38
9.1.5.1 要旨 ······················································································································· 38
3
K 4101-1993 目次
(3)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.1.5.2 装置及び器具 ··········································································································· 38
9.1.5.3 試薬 ······················································································································· 40
9.1.5.4 操作 ······················································································································· 40
9.1.5.5 計算 ······················································································································· 40
9.1.6 水分気化法 ················································································································ 40
9.1.6.1 要旨 ······················································································································· 40
9.1.6.2 装置及び器具 ··········································································································· 40
9.1.6.3 試薬 ······················································································································· 42
9.1.6.4 操作 ······················································································································· 42
9.1.6.5 計算 ······················································································································· 43
9.2 乾燥減量法 ·················································································································· 43
9.2.1 要旨 ························································································································· 43
9.2.2 装置及び器具 ············································································································· 43
9.2.3 試料 ························································································································· 43
9.2.4 操作 ························································································································· 43
9.2.5 計算 ························································································································· 44
10. 灰分試験方法 ··············································································································· 44
10.1 要旨 ·························································································································· 44
10.2 装置及び器具 ·············································································································· 44
10.3 操作 ·························································································································· 44
10.4 計算 ·························································································································· 44
11. 滴定方法 ····················································································································· 45
11.1 滴定方法の種類 ··········································································································· 45
11.2 試薬 ·························································································································· 45
11.3 装置及び器具 ·············································································································· 46
11.4 滴定用溶液(規定液),指示薬溶液及び試験紙 ··································································· 47
11.4.1 滴定用溶液(規定液) ································································································ 47
11.4.1.1 調製,標定及び保存 ································································································· 47
11.4.1.2 ファクターの温度補正 ······························································································ 55
11.4.2 指示薬溶液及び試験紙 ································································································ 56
11.5 ニトロソ化滴定法 ········································································································ 57
11.5.1 要旨 ························································································································ 57
11.5.2 操作 ························································································································ 57
11.5.3 計算 ························································································································ 57
11.6 ジアゾ化滴定法 ··········································································································· 58
11.6.1 要旨 ························································································································ 58
11.6.2 直接法(一般法) ······································································································ 58
11.6.3 直接法(別法) ········································································································· 58
11.6.4 逆滴定法 ·················································································································· 59
11.6.5 還元法 ····················································································································· 59
4
K 4101-1993 目次
(4)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
11.6.6 零電流電位差滴定法 ··································································································· 60
11.6.7 定電圧分極電流滴定法 ································································································ 60
11.6.8 定電流分極電位差滴定法 ····························································································· 61
11.7 カップリング滴定法 ····································································································· 61
11.7.1 要旨 ························································································································ 61
11.7.2 操作 ························································································································ 61
11.7.3 計算 ························································································································ 62
11.8 臭素化滴定法(逆滴定法) ···························································································· 62
11.8.1 要旨 ························································································································ 62
11.8.2 操作 ························································································································ 62
11.8.3 計算 ························································································································ 62
11.9 中和滴定法 ················································································································· 63
11.9.1 要旨 ························································································································ 63
11.9.2 操作 ························································································································ 63
11.9.3 計算 ························································································································ 63
12. 濁度測定方法 ··············································································································· 63
12.1 要旨 ·························································································································· 63
12.2 試薬 ·························································································································· 63
12.3 器具 ·························································································································· 63
12.4 塩化物標準液の調製 ····································································································· 63
12.5 試料溶液の調製 ··········································································································· 63
12.6 濁度標準液の調製 ········································································································ 63
12.7 操作 ·························································································································· 64
13. 色数試験方法 ··············································································································· 64
13.1 要旨 ·························································································································· 64
13.2 装置及び器具 ·············································································································· 64
13.3 試薬 ·························································································································· 64
13.4 ハーゼン標準比色液の調製 ···························································································· 66
13.5 操作 ·························································································································· 67
13.6 結果の記録 ················································································································· 67
14. 酸分試験方法 ··············································································································· 67
14.1 定性法 ······················································································································· 67
14.1.1 要旨 ························································································································ 67
14.1.2 指示薬 ····················································································································· 67
14.1.3 器具 ························································································································ 67
14.1.4 操作 ························································································································ 67
14.1.5 評価 ························································································································ 67
14.2 定量法(H2SO4として) ······························································································ 67
14.2.1 要旨 ························································································································ 67
14.2.2 滴定用溶液(規定液)及び指示薬 ················································································· 67
5
K 4101-1993 目次K 4101-1993
(5)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
14.2.3 器具 ························································································································ 68
14.2.4 操作 ························································································································ 68
14.2.5 計算 ························································································································ 68
15. 塩酸不溶分試験方法 ······································································································ 68
15.1 要旨 ·························································································································· 68
15.2 試薬 ·························································································································· 68
15.3 器具 ·························································································································· 68
15.4 操作 ·························································································································· 68
15.5 計算 ·························································································································· 69
16. ガスクロマトグラフ分析方法 ··························································································· 69
17. 不揮発分試験方法 ········································································································· 69
17.1 要旨 ·························································································································· 69
17.2 装置及び器具 ·············································································································· 69
17.3 操作 ·························································································································· 70
17.4 計算 ·························································································································· 70
18. 高速液体クロマトグラフ分析方法 ····················································································· 70
19. 有機中間物を取り扱うときの注意事項 ··············································································· 71
付表1 引用規格 ················································································································· 71
付表2 関連規格 ················································································································· 73
参考 安全衛生,防災,環境保全及び公害対策上の措置······························································ 74
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 4101-1993
有機中間物一般試験方法
General testing methods for organic intermediates
1. 適用範囲 この規格は,有機中間物(主として染顔料中間物)の試験に共通する一般事項及び一般試
験方法について規定する。
備考1. この規格の引用規格を,付表1に示す。
2. この規格の中で,{ }を付けて示してある単位及び数値は従来単位によるものであって,参
考として併記したものである。
2. 一般事項 一般事項は,次のとおりとする。
(1) 試験において共通する事項は,JIS K 0050による。
(2) 原子量は最新の国際原子量表による。式量はこれによって計算し,JIS Z 8401によって小数点以下2
けたに丸める。
(3) 分析に用いる器具などは,特に規定するもののほかはJIS K 0050のものを用いる。
(4) 単位記号は,JIS Z 8202及びJIS Z 8203による。
(5) 全量ピペット,全量フラスコ及びビュレットの補正は,JIS K 0050の9.3(全量ピペット・全量フラス
コ及びビュレットの校正方法)による。
(6) 操作上の注意は,次のとおりとする。
(6.1) 試験を行うときの温度 試験は,特に規定するもののほかは,JIS Z 8703に規定する室温で行う。
なお,操作直後に試験室温度を記録するのが望ましい。
(6.2) 計量値の示し方 体積,質量,温度,時間などを数値で示す場合は,特に規定するもののほかは,
次のとおりとする。
(a) “約”と付記してあるのは,示した数値の±10%を許容するものとする。
(b) 必要のある場合は,範囲で示す。
例1. 100〜150ml
例2. 100±5g
(c) 単に数値だけを書いてあるのは,記載されている数値の最下位までを有効数字とする。すなわち,
最下位の次のけたまで計測又は計量し,最下位に丸める。
(d) 数値に対し特に正確さを要求する場合は,要求する最下位を指定する。ただし,体積の場合は最下
位を指定せず,次の例のとおりとする。
例1. 25mlを正しく取る…全量ピペット又はビュレットを用いて量る。
例2. 正しく250mlにする…全量フラスコを用いて標線まで満たす。
(6.3) 目盛の読み方は,次のとおりとする。
(a) 体積計の目盛は,特に規定するもののほかは,水平面(下縁)で読む。

2
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(b) 浮ひょう(浮きばかり)の目盛は,特に規定するもののほかは,上縁で読む。
(c) 目盛は,特に規定するもののほかは,最小目盛の次のけたまで読まなければならない。
(6.4) 滴数で溶液を加えるときは,水20滴を滴下するとき,その質量が0.9〜1.1gとなるような器具を用
いる。
3. 試料採取方法
3.1
要旨 試験に用いる試料を得るために,試料の採取,製品の種別と容器の種別,ロット及び代表試
料,試料採取の時期及び場所,容器の抜取個数,液体試料採取方法,固体試料採取方法について規定する。
3.2
試料の採取 試料の採取は製品1ロットごとに,製品の種別に従い,3.7及び3.8によって行う。
3.3
製品の種別と容器の種別
3.3.1
製品の種別 その状態によって液体製品及び固体製品に区別する。
3.3.2
容器の種別 製品の種別と貯蔵及び輸送に用いる容器によって次のとおり区分する。
(1) 液体製品の場合 液体製品の場合は,次のとおりとする。
(a) 小形容器(18l缶,ドラム缶,プラスチック製容器など)
(b) 大形容器(タンク,タンク車,タンクローリー,タンカーなど)
(2) 固体製品の場合 固体製品の場合は,次のとおりとする。
(a) 小形容器(紙袋,18l缶,ドラム缶,プラスチック製容器など)
(b) 大形容器(フレキシブルコンテナなど)
3.4
ロット及び代表試料
(1) ロット 同一の管理条件下に生産及び貯蔵され,同一の品質とみなして,同じ取扱いをする製品の集
まり。
(2) 代表試料 1ロットの製品の平均品質を代表するように3.7.2及び3.8.2によって採取した試料で,少
なくとも,分析に必要な量の2倍以上の量がなければならない。
3.5
試料採取の時期及び場所 試料採取の時期及び場所は,当事者間の協議による。
3.6
容器の抜取個数 複数の容器からなるロットの代表試料を採取するときは,表1に示す個数の容器
をランダムに抜き取る。ただし,容器の抜取個数は,当事者間の合意によって,別に定めることができる。
表1 抜取個数
容器数
抜取個数
(最低)
容器数
抜取個数
(最低)
1〜 3
全数
126〜216
6
4〜64
4
217〜343
7
65〜125
5
344〜512
8
備考 容器数65以上の抜取個数は,容器数の立方根
(整数にならない場合は,切り上げる。)に基
づいたものである。
3.7
液体試料採取方法
3.7.1
器具 器具は,次のとおりとする。
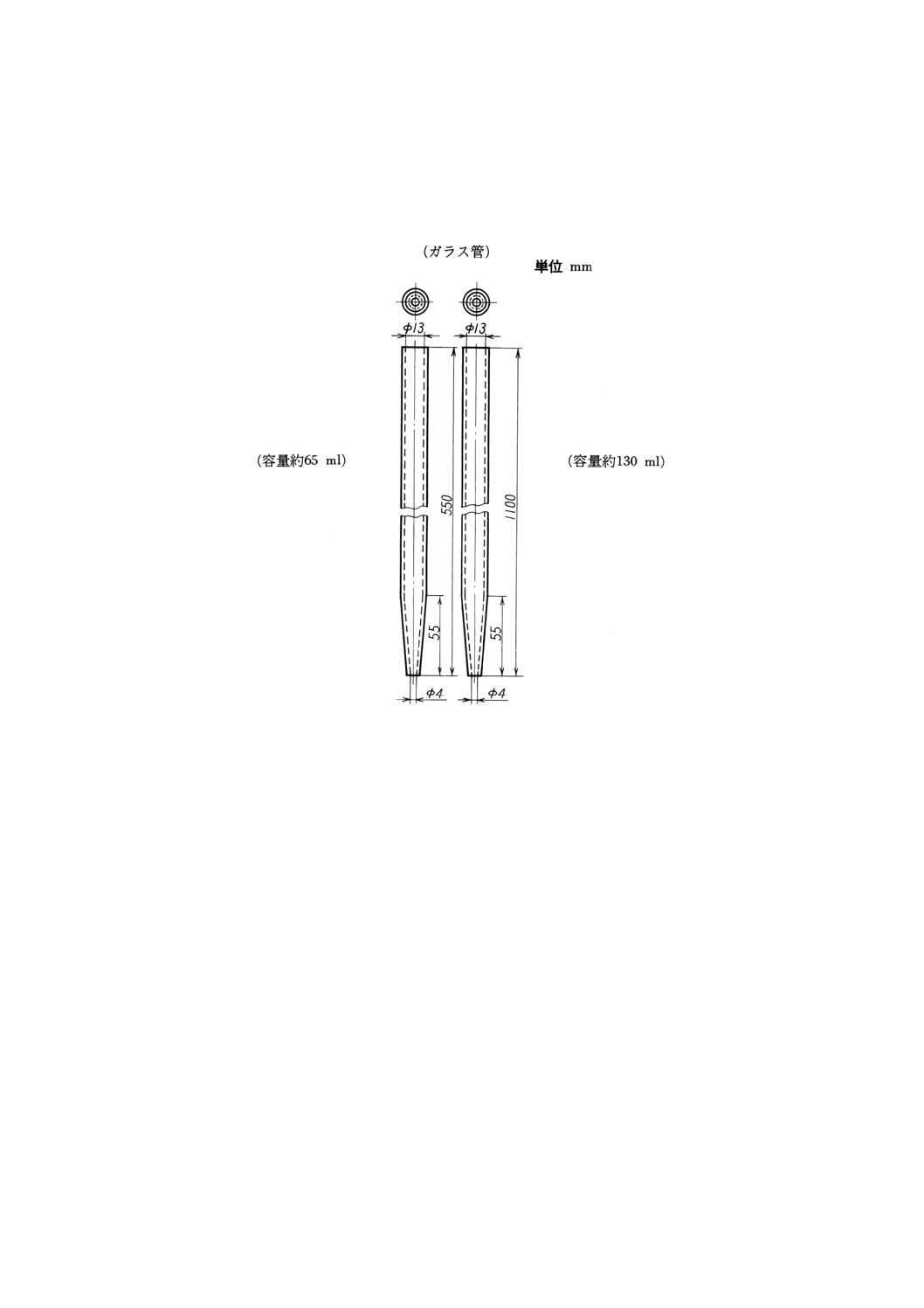
(1) 小形液体試料採取器 一例を図1に示す。
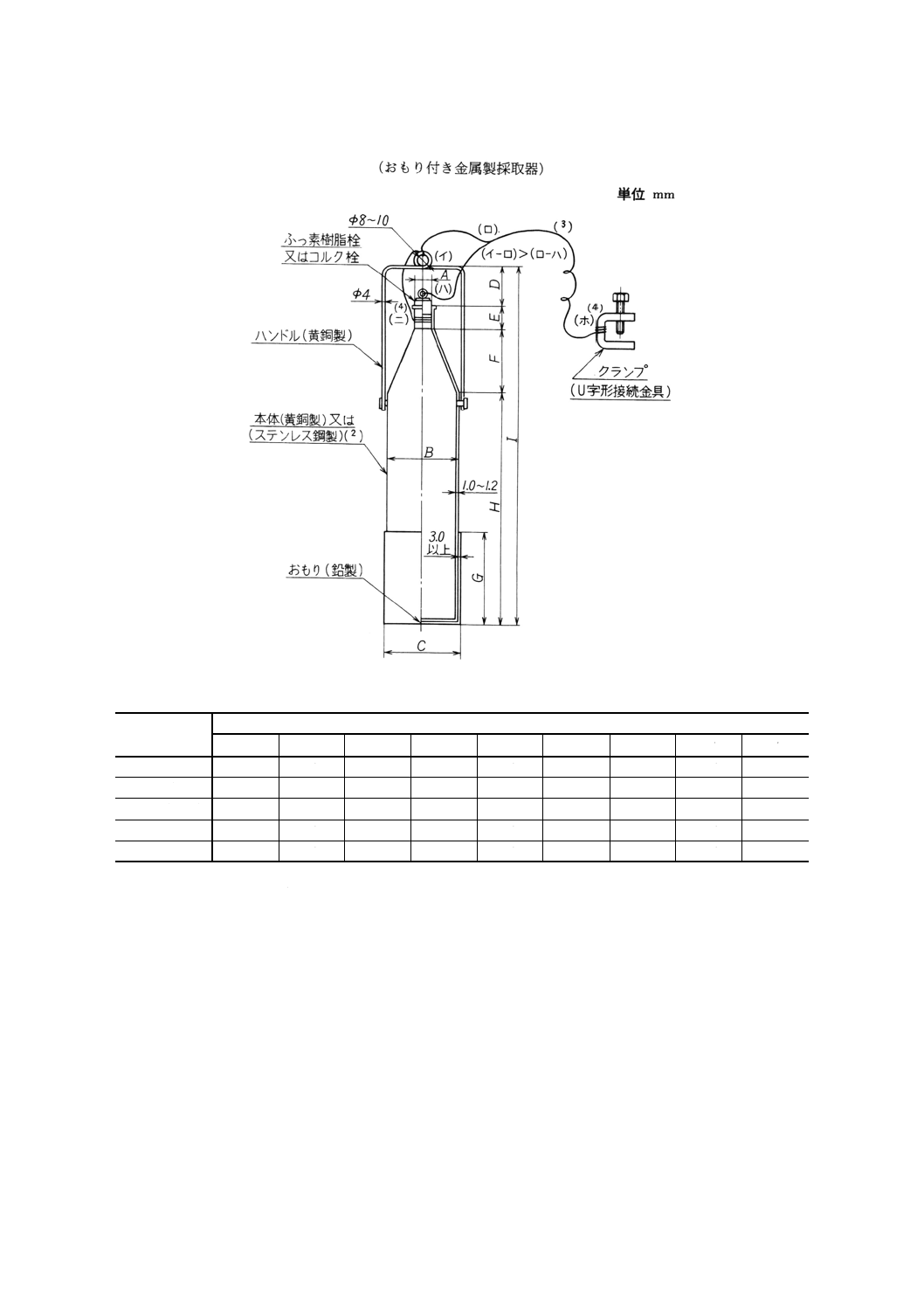
(2) 大形液体試料採取器 一例を図2に示す。
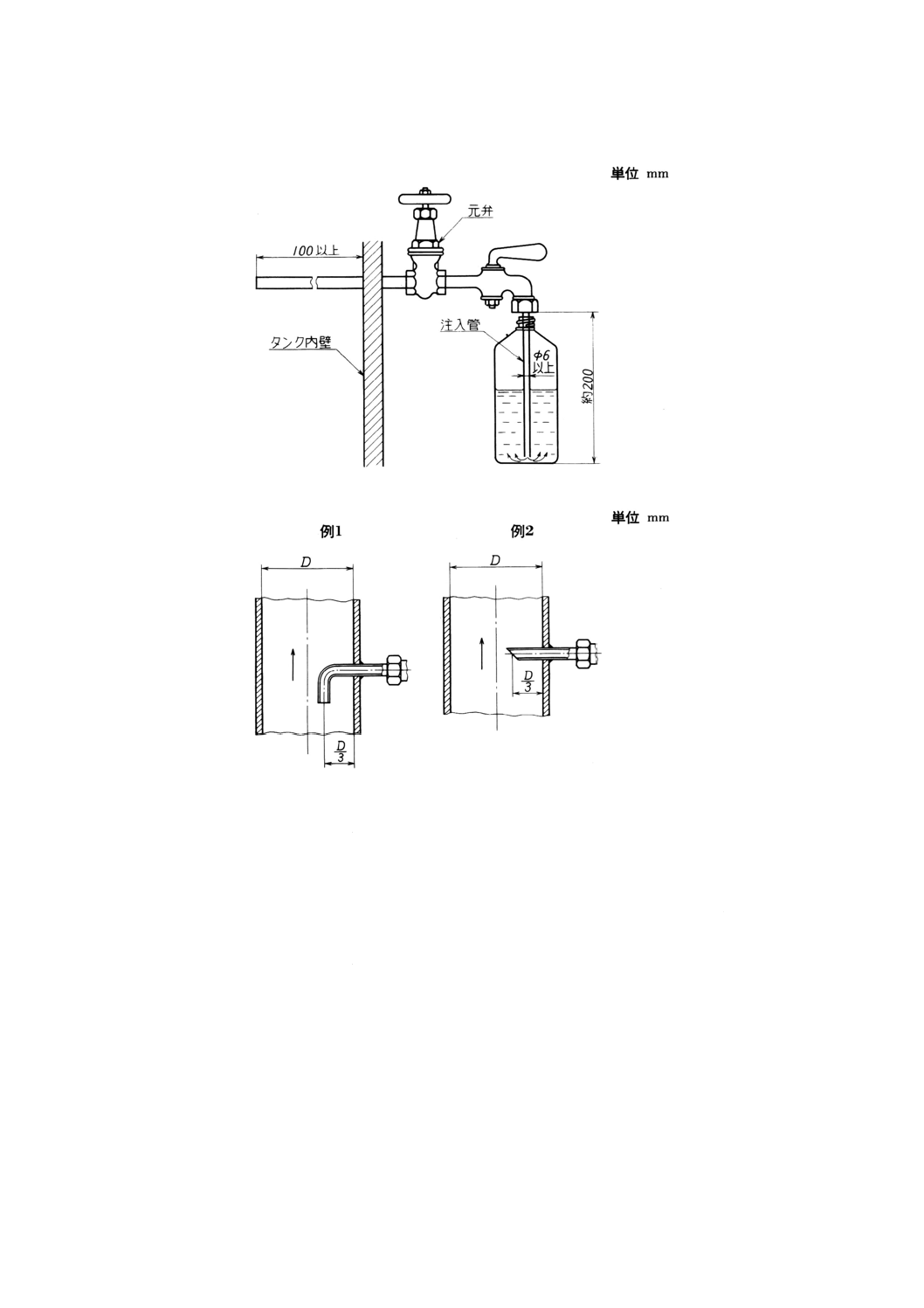
(3) タンク試料採取用ノズル(1) 一例を図3に示す。
タンク試料採取用ノズルは,タンクの高さに沿って等間隔に少なくとも3個設ける。
3
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(1) ノズルのタンク内部の先端は,液面との静電気放電の危険を防止するための十分な対策が必要
である。
(4) パイプライン試料採取用ノズル その例を図4に示す。
図1 小形液体試料採取器の一例
4
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図2 大形液体試料採取器の一例
おもり付き金属製採取器の各部の寸法
単位mm
品名
各部の寸法記号
A
B
C
D
E
F
G
H
I
260ml(細口)
20±1
45±1
51±2
50±3
15±2
40±3
60±3
155±3
260±10
500ml(細口)
20±1
55±1
61±2
45±3
20±2
55±3
80±3
220±3
340±10
500ml(広口)
40±1
55±1
61±2
45±3
20±2
55±3
80±3
220±3
340±10
1l (細口)
20±1
76±1
82±2
45±3
25±2
70±3
100±3
255±3
395±10
1l (広口)
40±1
76±1
82±2
45±3
25±2
70±3
100±3
255±3
395±10
注(2) 黄銅製採取器を使用すると試料が汚染するおそれがある場合は,ステンレス鋼製採取器を用いてもよい。
ただし,ステンレス鋼は,SUS 304相当品で,底板の厚さ15mmのものとする。この場合,下部の鉛製の
おもりは要らない。
(3) JIS C 3105に規定する銅製より線で,末端にアース用クランプを取り付ける。
(4) (ニ)及び(ホ)は,容易に離れないように接続した後,それぞれろう付けする。
5
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図3 タンク試料採取用ノズルの一例
図4 パイプライン試料採取用ノズルの例
3.7.2
操作 操作は,次のとおり行う。
3.7.2.1
小形容器の場合 小形液体試料採取器の上部開口部を開いたまま容器内に垂直に入れ,器底に達
した後開口部を閉じ,取り出して中身を適当な試料容器に移し,栓をする。採取した試料は,試験室で等
量ずつ混合して代表試料とする。
3.7.2.2
大形容器の場合 大形容器の場合は,次のとおりとする。
(1) 大形液体試料採取器を用いて採取する場合 タンクのふたを開き,アースした大形液体試料採取器を
口を閉じたままタンク内に垂直に入れ,採取器の口が所定の採取位置に達したならば,これを開いて
試料を流し込み,取り出して中身を適当な試料容器に移し,栓をする。採取した試料は,試験室で所
定の混合割合で混合して代表試料とする。
試料の採取位置及び試料の混合割合は,横置円筒形タンクの場合は表2による。その他のタンクの
場合は,内容物をほぼ3等分した各層の中心から採取し,それぞれ等量ずつ混合する。
6
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表2 横置円筒形タンクの試料の採取位置及び試料の混合割合
内容物の深さ
(直径に対する%)
採取位置(底からの高さ)
(直径に対する%)
混合割合(体積)
上部
中部
下部
上部
中部
下部
100
80
50
20
3
4
3
90
75
50
20
3
4
3
80
70
50
20
2
5
3
70
−
50
20
−
6
4
60
−
50
20
−
5
5
50
−
40
20
−
4
6
40
−
−
20
−
−
10
30
−
−
15
−
−
10
20
−
−
10
−
−
10
10
−
−
5
−
−
10
(2) タンク試料採取用ノズルから採取する場合 試料の採取に先立ち,ノズルからタンクの内容物を流出
させ,ノズル及び試料の通路を十分に洗い,次に,清浄な注入管(5)をノズルに連結する。注入管の例
を図3中に示す。
次に,中部,下部及び上部の各ノズルからそれぞれ試料を採取する。試料容器に試料を満たした後,
試験室で試料容器から等量ずつ試料を取って混合し,代表試料とする。
注(5) 試料容器内に差し込んで,試料を注入するための管である。試料を採取するとき,製品を汚染
するなど悪影響を与えないもので,底まで届く長さがなければならない。
(3) 移送中のパイプラインから採取する場合 試料採取に先立ち,コックを開いて内容物を流出させ,コ
ック及び試料の通路を十分に洗っておく。
次に,全移送時間を等分する中間時点においてコックを少し開き,試料を3回以上試料容器に採取
した後,等量の各試料を混合して代表試料とする。ただし,試料採取回数は,当事者間の協議による。
3.8
固体試料採取方法
3.8.1
器具 器具は,次のとおりとする。
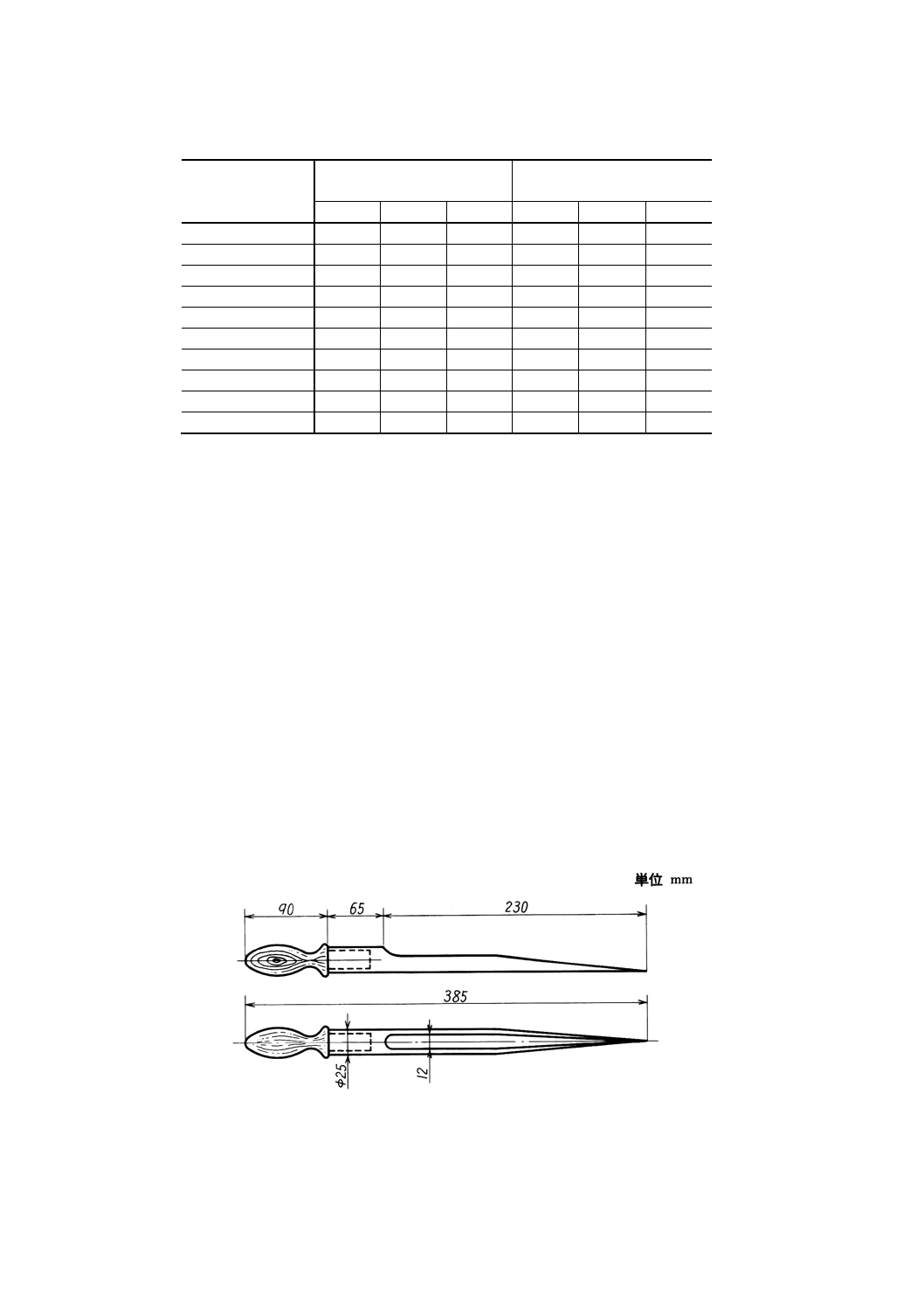
(1) 細粒及び粒末試料採取器 一例を図5に示す。
(2) サンプリングスコップ 一例を図6及び図7に示す。
図5 細粒及び粉末試料採取器の一例
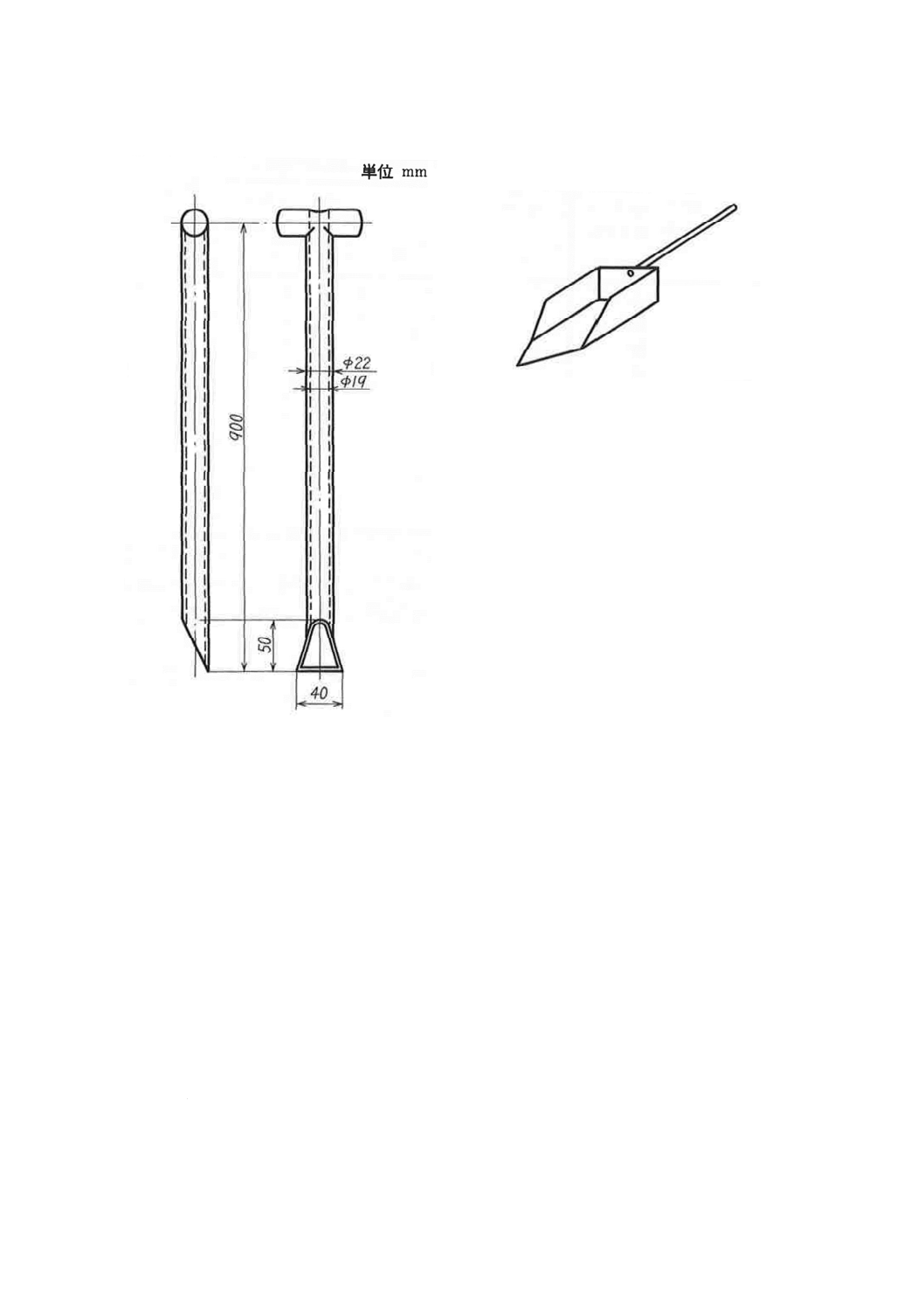
7
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図6 サンプリングスコップ(A)の一例 図7 サンプリングスコップ(B)の一例
3.8.2 操作 操作は,次のとおり行う。ただし,縮分が必要な場合は,JIS M 8100の6.5(試料の縮分)
によって縮分し,代表試料とする。
(1) 小形容器の場合 容器の口を開き,できるだけ中心部から試料を採取し,適当な試料容器に移して栓
をする。採取した試料は,実験室で等量ずつ混合して代表試料とする。
(2) 大形容器の場合 容器の口を開き,内容物の上・中・下から試料を採取し,適当な試料容器に移して
栓をする。採取した試料は,実験室で等量ずつ混合して代表試料とする。
4. 試料乾燥方法
4.1
要旨 試験に用いる試料の乾燥方法として,液体の場合及び固体の場合について規定する。
4.2
液体の場合 試料に個々の製品規格に規定する乾燥剤を入れ,よく振り混ぜた後 静置し,乾燥ろ紙
でろ過して,その ろ液を用いる。
4.3 固体の場合
4.3.1 デシケーターによる方法 常圧で行う場合には図8(1)のデシケーターを用いて,減圧で行う場合に
は図8(2)のデシケーターを用いて,試料を入れたはかり瓶及び個々の製品規格に規定する乾燥剤を入れ,
20〜24時間放置する。
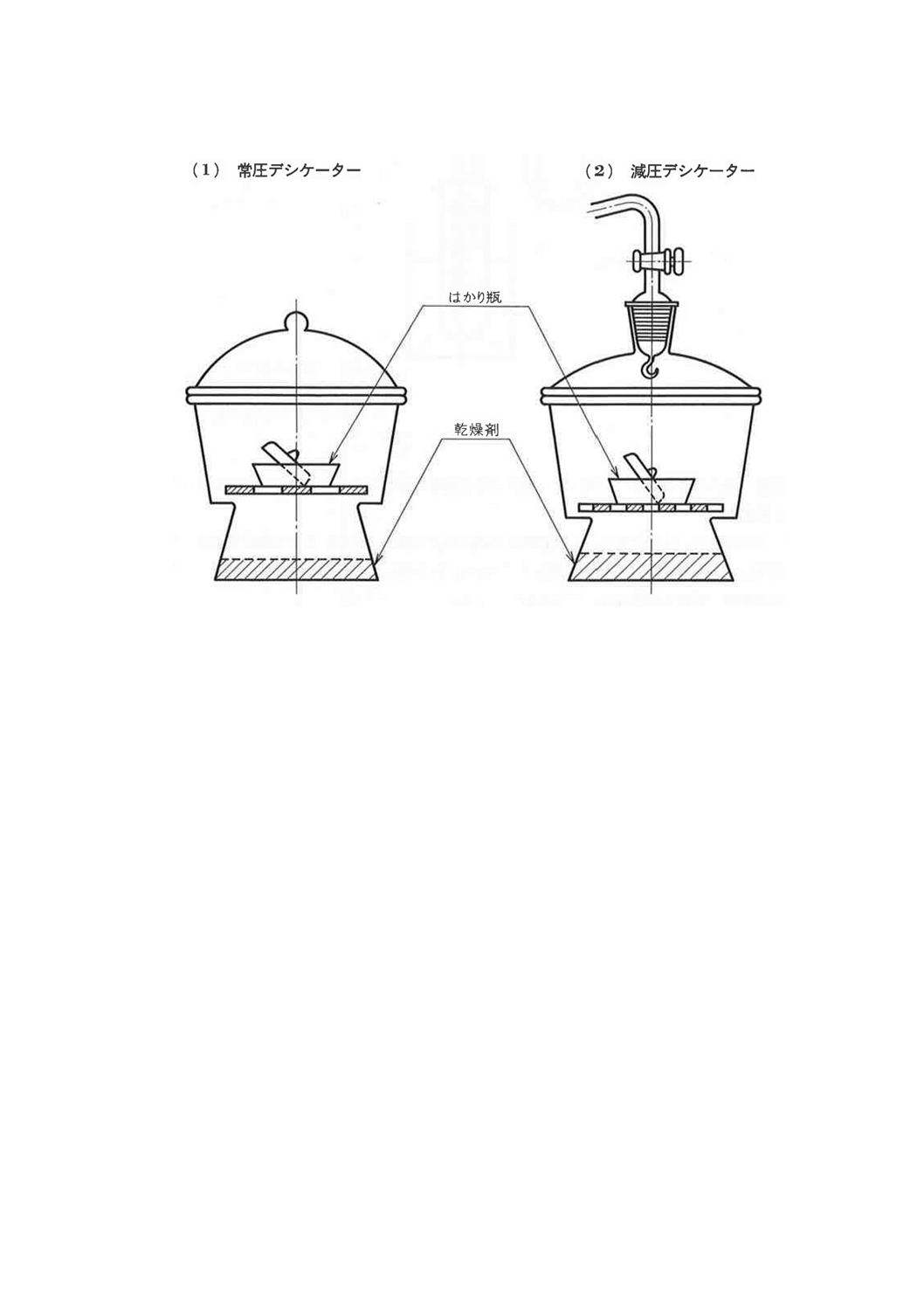
8
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図8 デシケーターによる乾燥剤
4.3.2 恒温乾燥器による方法 常圧で行う場合は,個々の製品規格に規定するもののほかは105〜110℃に
設定した恒温乾燥機に試料を入れ,約2時間乾燥した後,デシケーター中で放冷する。減圧で行う場合は,
個々の製品規格に規定する温度に設定した減圧恒温乾燥器に試料を入れ,個々の製品規格に規定する時間
乾燥した後,デシケーター中で放冷する。
4.3.3 乾燥ガスによる方法 図9に示す装置に試料を入れ,個々の製品規格に規定するもののほかは,空
気(6),窒素又は二酸化炭素のいずれかを約1 ℓ/minの割合で約2時間通じる。
なお,水浴使用の場合の温度は,60℃以下とするのが望ましい。
注(6) 濃硫酸及び塩化カルシウムで乾燥したものを用いる。
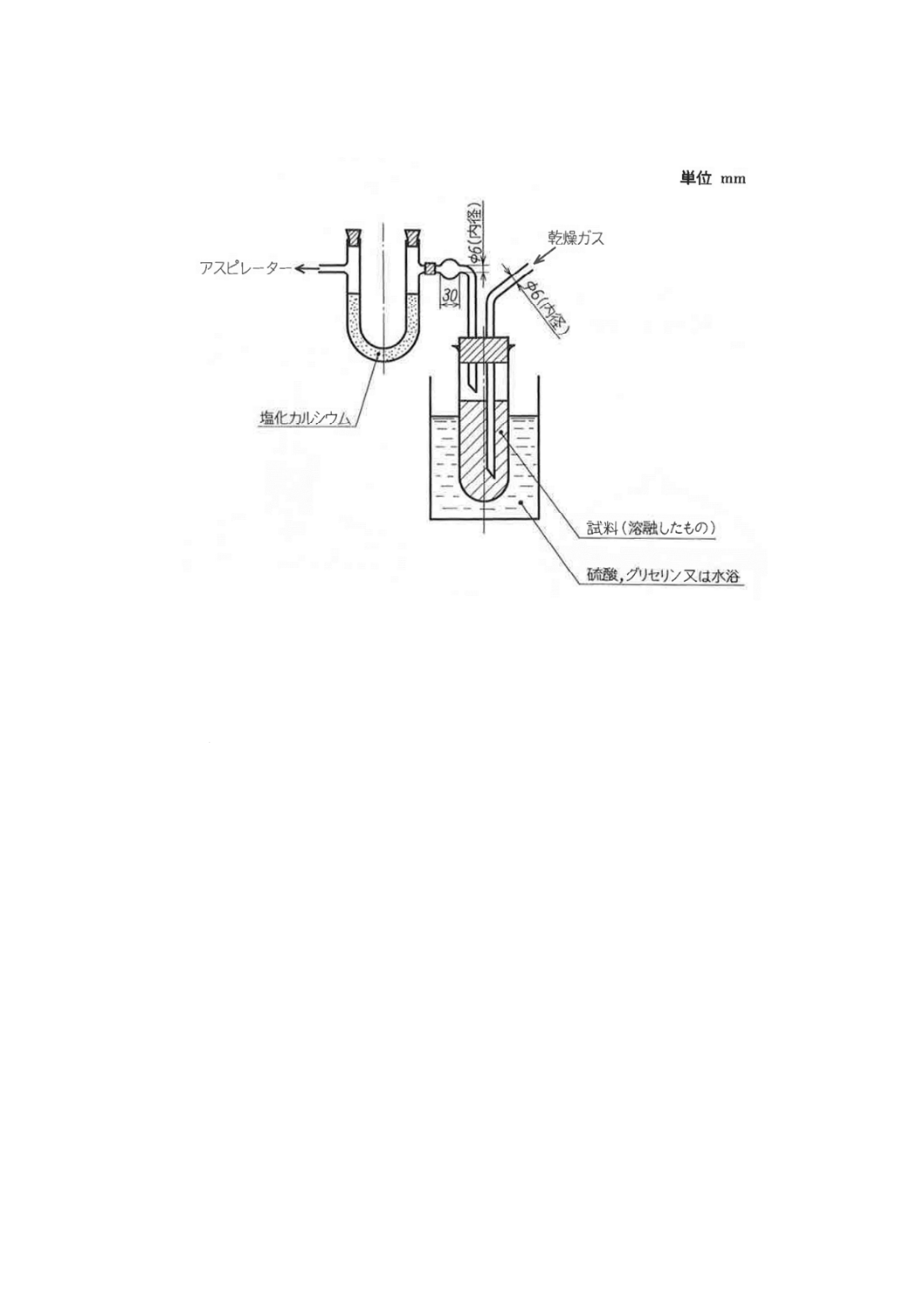
9
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図9 固体試料乾燥器(溶融法)の一例
5. 融点測定方法 融点測定方法は、目視による方法又は光透過量の測定による方法のいずれかによる。
5.1
目視による方法
5.1.1
要旨 この方法は,毛管に充てんした試料を加熱液中で加熱し,目視によって融点を求める方法で
ある。目視による融点測定は,JIS K 0064の3.1(目視による方法)によるほか,次のとおりとする。
5.1.2
装置及び器具 装置及び器具は,次のとおりとする。
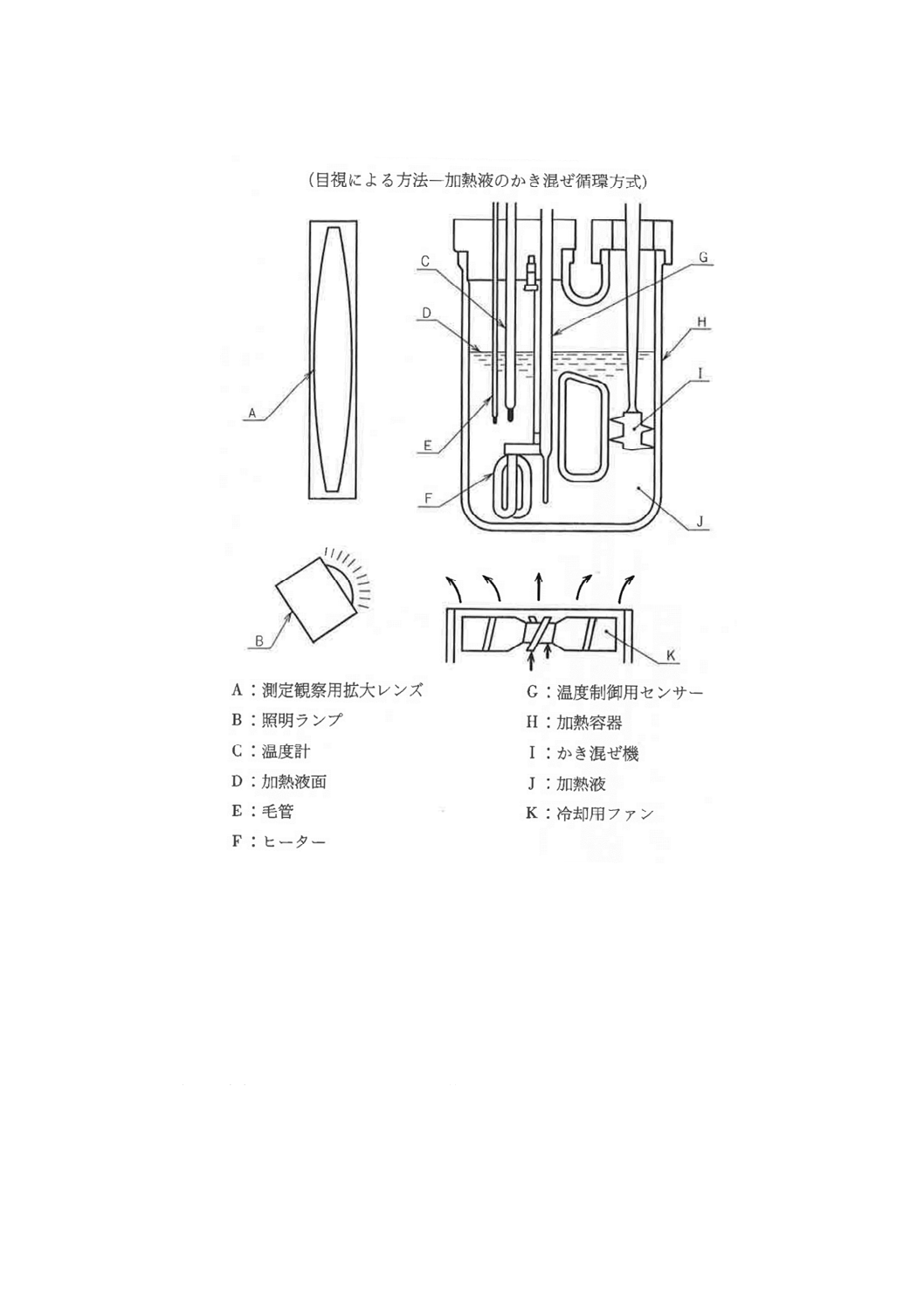
(1) 融点測定装置 (2)以下の器具を用いて組み立てたもので,一例を図10に示す。温度計は,その水銀
球に毛管の試料充てん部が密接するように固定し,加熱容器中の加熱液のほぼ中央,加熱容器の底か
ら温度計の先端まで約20㎜のところに取り付ける。
また,加熱液のかき混ぜ循環方式による融点測定装置を用いてもよい。一例を図11に示す。このと
きは,試料を充てんした毛管の下端が温度計の水銀球に近接するように固定する。
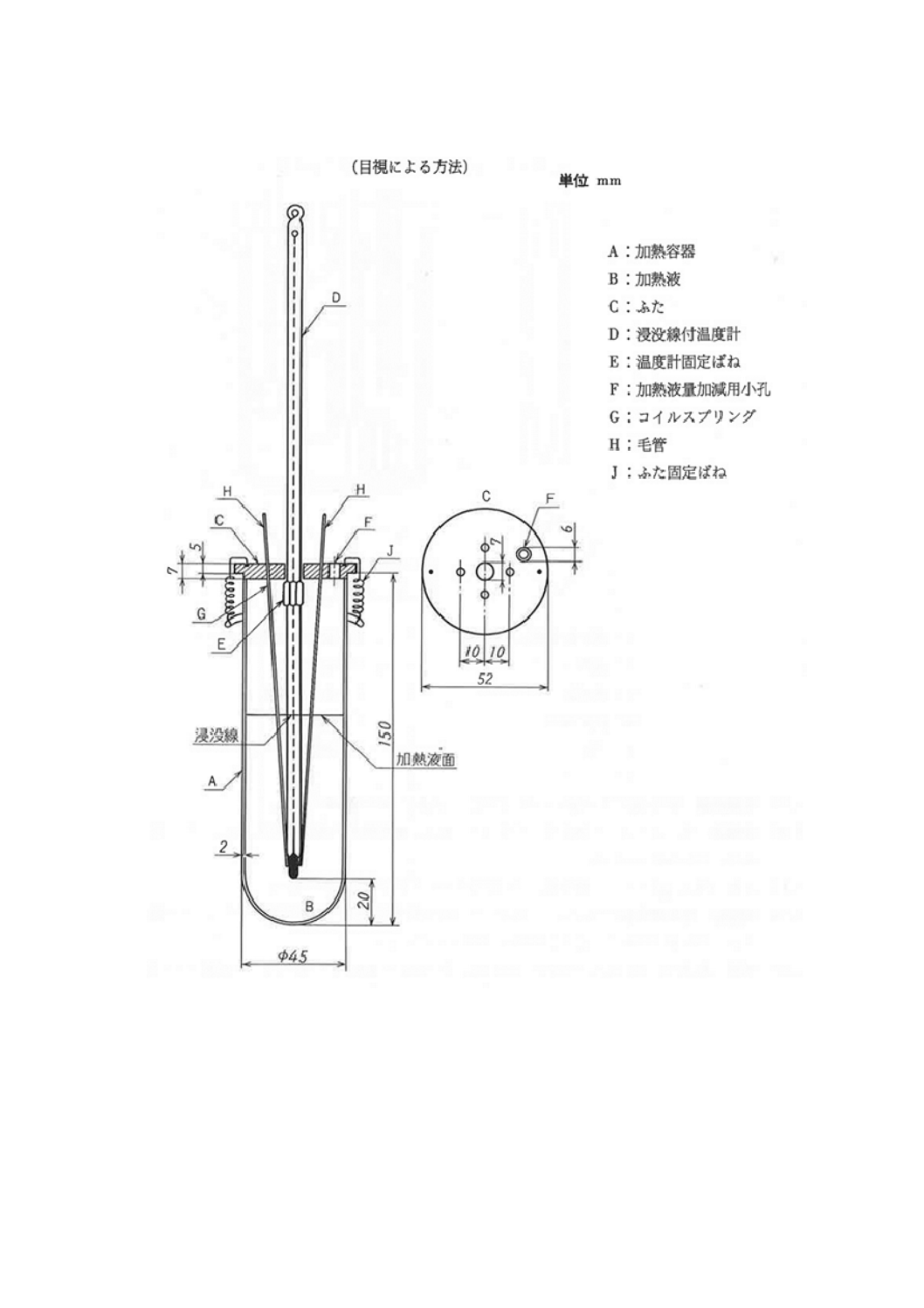
10
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図10 融点測定装置の一例
11
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図11 融点測定装置の一例
(2) 加熱容器 硬質1級ガラス製のもの。形状及び寸法の一例を図10に示す。
(3) 加熱液 水,シリコーン油など。シリコーン油は,耐熱温度が測定温度以上のもので,25℃における
動粘度が50〜100mm2/sのもの。
(4) ふた 四ふっ化エチレン樹脂製,ゴム製又はコルク製のいずれかのもの。
(5) 温度計 日本薬局方(第12改正)に規定する浸線付温度計(棒状)1号〜6号。ただし,予想融点が
70℃以下のときは,JIS B 7410に規定するSOP57〜59を用いてもよい。
(6) 毛管 内径0.8〜1.2mm,肉厚0.2〜0.3mm,長さ約150mmで,一端を閉じた硬質ガラス製のもの。
(7) 加熱器 加熱液を予想融点より15℃低い温度から5℃高い温度まで加熱することができ,かつ,加熱
液の温度上昇速度を毎分約3℃及び約1℃に調節可能なもの。
5.1.3
試料の前処理 あらかじめ,個々の製品規格の試験方法中に記載する方法によって,試料を乾燥す
る。
5.1.4
操作 操作は,次のとおり行う。
12
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 乾燥した試料を毛管に充てんし,閉じた一端を下にして,ガラス板又は陶板上に立てた長さ約70cm
のガラス管の内部に落とし,はずませて固く詰め,試料の層を約3mmとする。
(2) 加熱液を加熱し,予想した融点より約10℃低い温度まで徐々に上昇させる。
(3) 温度計の浸没線を加熱液の液面に合わせる。
(4) 試料を入れた毛管を図10又は図11に示すとおり固定する。
(5) 加熱液の温度が1分間に約3℃上昇するように加熱し,予想した融点より約5℃低い温度となった後,
1分間に約1℃上昇するように加熱を続ける。
(6) 試料が毛管内で溶融して固体を認めなくなったとき,温度計の最小目盛の101まで読み取り,融点の測
定値とする。
(7) (1)〜(6)の操作を3回以上行い,測定値の平均値を小数点以下第1位に丸めて融点とする。
5.2
光透過量の測定による方法
5.2.1
要旨 この方法は,試料の温度による状態変化を光の透過量によって電気的に検出し,同時にその
温度を読み取って融点を求める方法である。光透過量の測定による融点測定は,JIS K 0064の3.2(光透過
量の測定による方法)によるほか,次のとおりとする。
備考 溶融状態において,光を透過しない製品には適用できない。
5.2.2
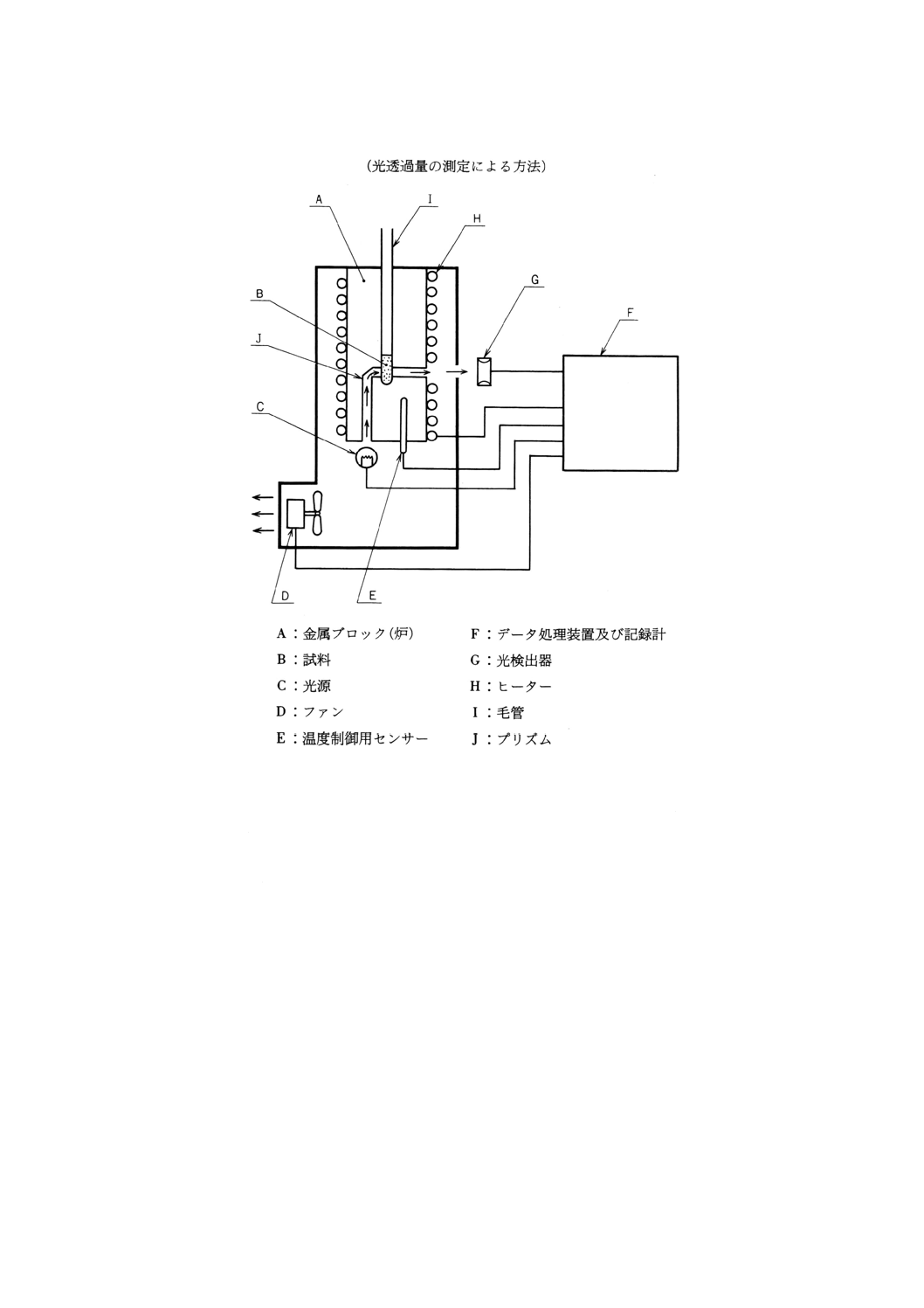
装置及び器具 装置及び器具は,次のとおりとする。
(1) 融点自動検出測定装置 装置の一例を図12に示す。装置は,必要に応じて融点既知の物質を用いて予
備試験を行い,その測定結果が5.1と差がないことを確認する。
13
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図12 融点自動検出測定装置の一例
参考 測定原理 金属ブロック(炉)に差し込まれた毛管は,あらかじめセットさ
れた昇温速度で加熱される。光源からの光は,光路を通って試料に達する。
試料の溶融過程で光透過率は増加し,光検出器で検出され,更に電位に変換
出力される。出力はデータ処理された後,記録計に出力され融点曲線が記録
される。
(2) 毛管 内径0.8〜1.2mm,肉厚0.2〜0.3mmで,一端を閉じた硬質ガラス製のもの。長さは,装置によ
って異なるが,装着したときの露出部分の長さが10〜20mmのもの。
5.2.3
試料の前処理 試料の前処理は,5.1.3による。
5.2.4
操作 操作は,次のとおり行う。
(1) 5.1.4(1)によって,試料の充てんを行う。
(2) 試料の昇温速度が1分間に約1℃となるように操作条件を設定して融点を測定する。
(3) (1)及び(2)の操作を3回以上行い,測定値の平均値を小数点以下第1位に丸めて融点とする。
5.3
“とけ始め”及び“とけ終わり”の判定方法 個々の製品規格に,融点として,とけ始めの温度及
びとけ終わりの温度を規定するとき,“とけ始め”及び“とけ終わり”は,次によって判定する。
(1) 目視による方法の場合 図13の湿潤点 [(a)] をとけ始めとし,溶融終点 [(e)] をとけ終わりとする。

14
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図13 融点付近における試料の変化の一例
(a) 湿潤点 試料が毛管内壁に接する面に細かい液滴が一様に生じるとき(とけ始め)。
(b) 収縮点 試料が収縮し,毛管内壁との間に明らかにすきまが生じるとき。
(c) 崩壊点 収縮した試料が下方に崩壊して,液化が始まるとき。
(d) 液化点 崩壊した試料がいくらか固体のままで液中に残っているが,液面の上部が完全なメニスカ
スを形成するとき。
(e) 溶融終点 液中に残っている固体の試料が完全に液化するとき(とけ終わり)。
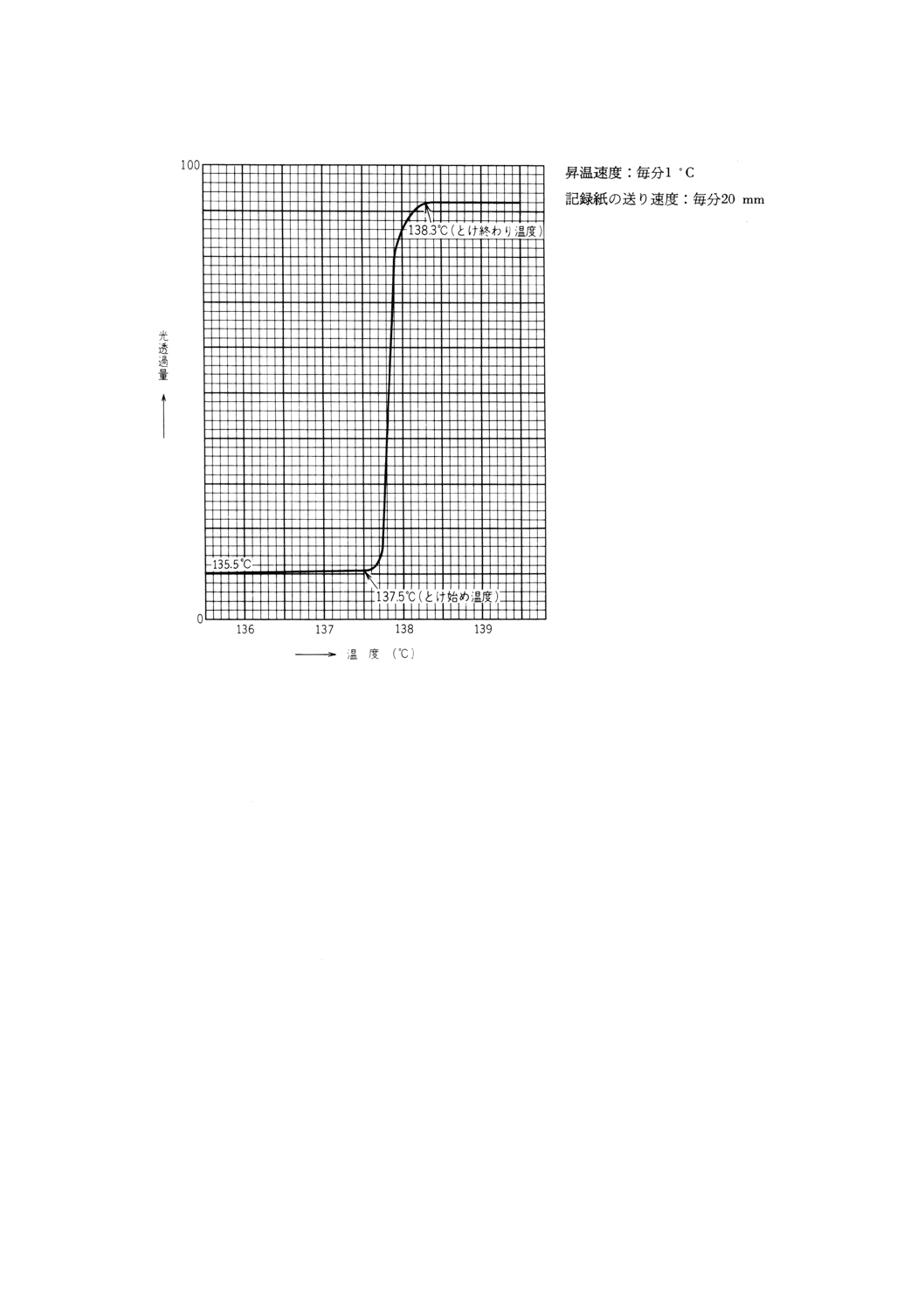
(2) 光透過量の測定による方法の場合 光透過量測定装置によって記録された溶融曲線の光透過量が増加
し始める点をとけ始めとし,光透過量が一定になる点をとけ終わりとする。
試料の溶融過程の記録の一例を図14に示す。
15
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図14 溶融過程の記録の一例
6. 凝固点測定方法
6.1
要旨 この方法は,試料容器に入れた試料を冷却水,寒剤又はシリコーン油などの熱媒体によって
間接的に冷却し,目視によって凝固点を求める方法である。目視による凝固点測定は,JIS K 0065による
ほか,次のとおりとする。
6.2
装置及び器具 装置及び器具は,次のとおりとする。
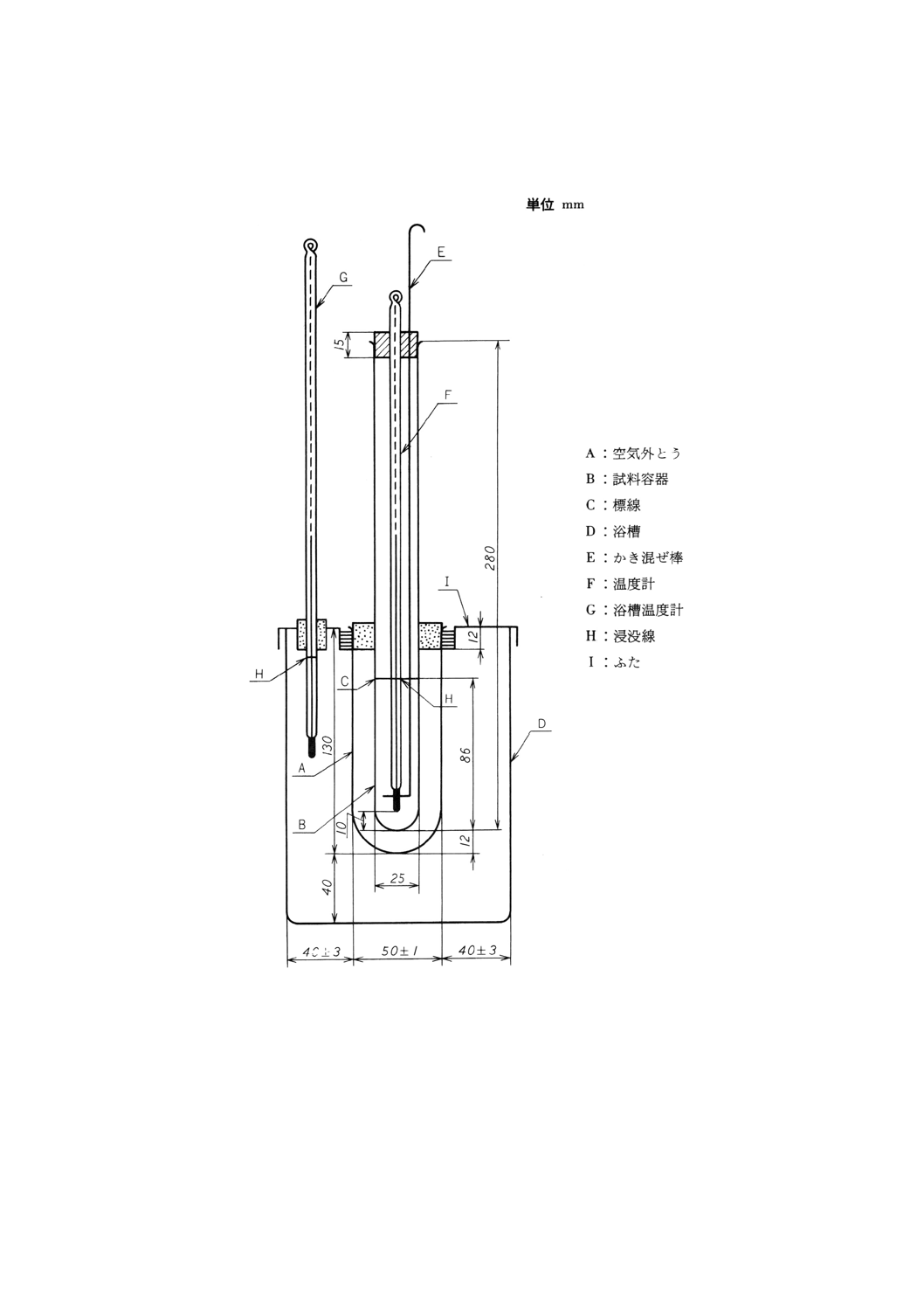
(1) 凝固点測定装置 (2)以下の器具を用いて組み立てたもの。一例を図15に示す。
(2) 空気外とう ガラス製で,図15に示す形状及び寸法のもの。肉厚は2〜3mmとし,内外面にシリコ
ーン油を塗る。
(3) 試料容器 硬質ガラス製で,図15に示す形状及び寸法のもの。肉厚は1.2〜1.5mmとし,図15に示
す位置に標線を入れる。空気外とう中に差し込み,コルク栓で固定する。
備考 試料容器が曇って温度計の目盛が読み取れないときは,容器壁の必要な部分にシリコーン油な
どを薄く塗ってもよい。ただし,シリコーン油などが試料中に混入しないように注意する。
(4) 浴槽 ガラス製又は透明なプラスチック製の容器で,図15に示す形状及び寸法のもの。
(5) かき混ぜ棒 ガラス製又はステンレス鋼製で,下端を外径約18mmの輪状にしたもの。
(6) 温度計 JIS B 7410に規定するSOP56〜SOP59。ただし,予想凝固点が70℃以上のものを測定すると
きは,JIS B 7410に規定するAP40を用いてもよい。
また,予想凝固点が100℃以上のものを測定するときは,日本薬局方(第12改正)に規定する浸線
付温度計(棒状)3〜6号を用いる。
16
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(7) 浴槽温度計 JIS B 7413に規定する浸没線付温度計で,必要な測定温度範囲のもの。
図15 凝固点測定装置の一例
6.3
試料の前処理 あらかじめ,個々の製品規格に規定する方法によって試料を乾燥する。
6.4
操作 操作は,次のとおり行う。
(1) 試料を試料容器の標線まで入れる。試料が固体のときは,予想する凝固点よりも10℃以上高い温度ま
で注意しながら加熱して溶かし,試料容器に入れる。
(2) 浴槽に,予想する凝固点よりも5〜10℃低い温度の水,寒剤又はシリコーン油などの熱媒体を入れる。
(3) 試料容器を空気外とう中に入れ,浸没線付温度計の浸没線を試料の液面に合わせる。
(4) 試料の温度が予想する凝固点よりも5℃高い温度まで冷却した後,かき混ぜ棒を毎分60〜80回の割合

17
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
で上下に動かし,30秒ごとに温度を読み取る。
(5) 温度が徐々に下がり,結晶が析出し始めたとき,かき混ぜを止める。
(6) このとき,温度上昇がない場合には,静止したときの温度を読み取り,温度上昇がある場合には,温
度上昇後の最高温度を読み取る。
温度は温度計の最小目盛の101まで読み取る。
(7) (1)〜(6)の操作を3回以上行い,連続3回の測定値の差が0.2℃以内となったとき,その平均値を小数
点以下第1位に丸めて凝固点とする。
7. 蒸留試験方法
7.1
要旨 この方法は,揮発性液体試料を蒸留し,蒸留特性を試験する方法である。蒸留試験は,JIS K
0066の附属書(B法による方法)によるほか,次のとおりとする。
7.2
装置及び器具 装置及び器具は,次のとおりとする。
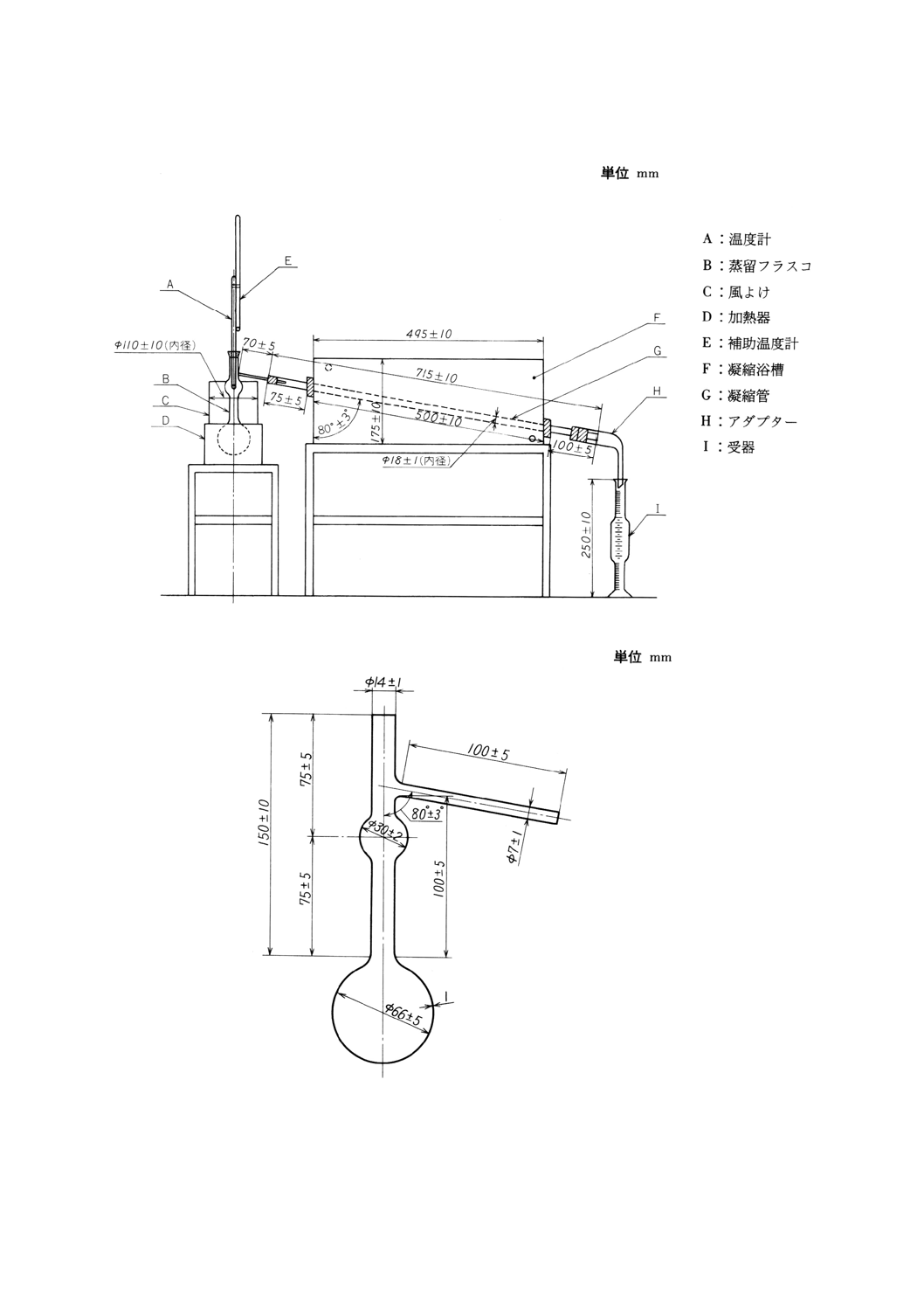
(1) 蒸留試験装置 (2)以下に規定する器具を用いて,図16のように組み立てたもの。
(2) 蒸留フラスコ 硬質ガラス製で,一例を図17(7)に示す。
注(7) 図18と図19を連結してもよい。
(3) 風よけ 金属製で,温度計の水銀球部に風があたらない高さにしたもの。
(4) アダプター 硬質ガラス製で,一例を図20(8)に示す。
注(8) 図21を用いてもよい。
(5) 受器 硬質ガラス製のメスシリンダーで,一例を図22に示す。
(6) 凝縮器 凝縮浴槽及び凝縮管(9)からなり,一例を図16に示す。
なお,凝縮浴槽は幅120±10mmの金属製で,凝縮管は肉厚1mmの硬質ガラス製のもの。
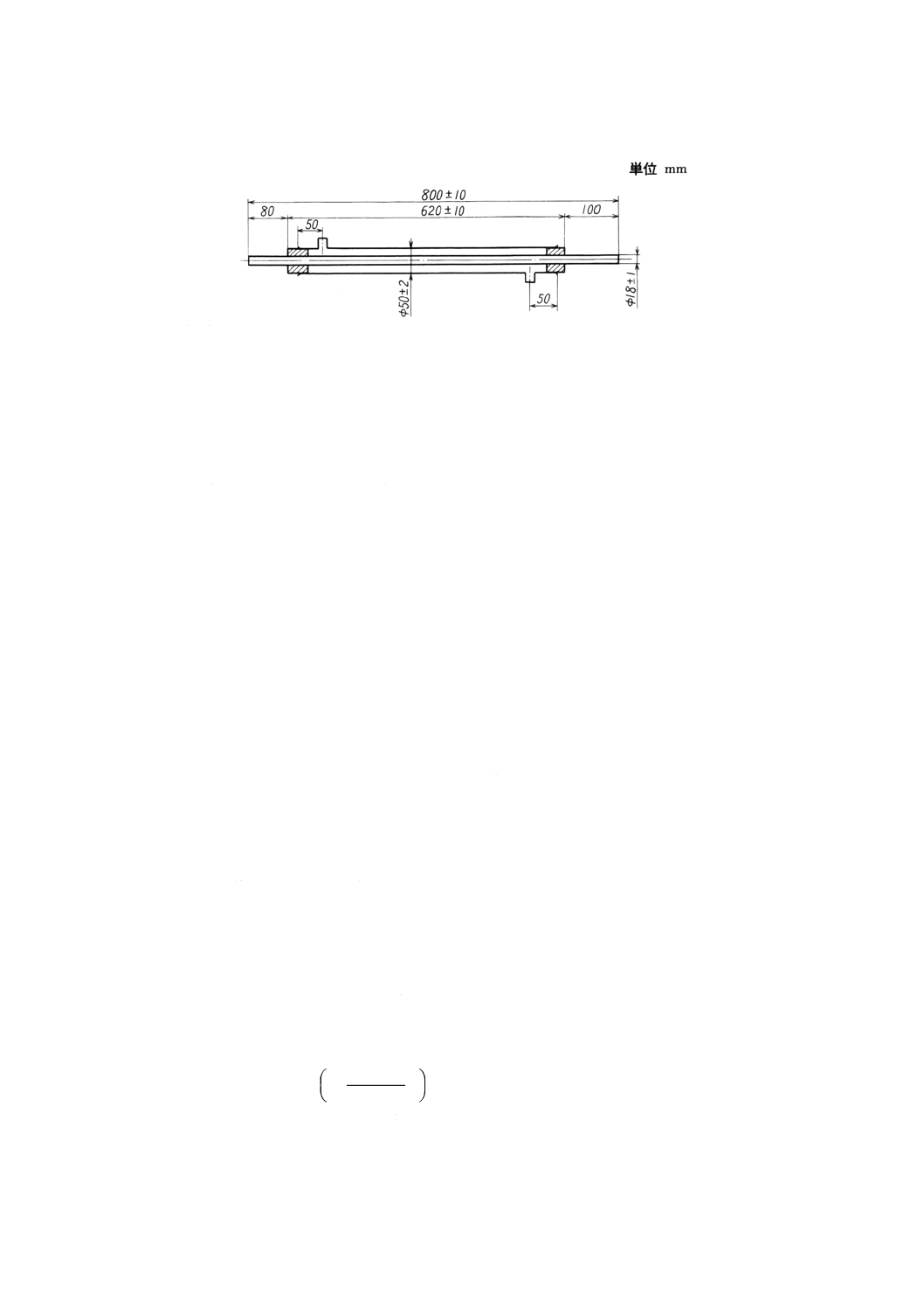
注(9) 図23を用いてもよい。
(7) 温度計 JIS B 7411に規定するガラス製棒状温度計(全浸没)又は表3に規定する全浸没水銀棒状温
度計で,校正をしたもの。
表3 全浸没水銀棒状温度計
温度範囲
−10〜120℃
90〜220℃
190〜320℃
最小目盛
0.5℃
全長
330〜350mm
(8) 補助温度計 水銀温度計で,温度範囲0〜100℃,細分目盛1℃以下のもの。
18
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図16 蒸留試験装置の一例
図17 蒸留フラスコの一例(容量100ml)
19
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
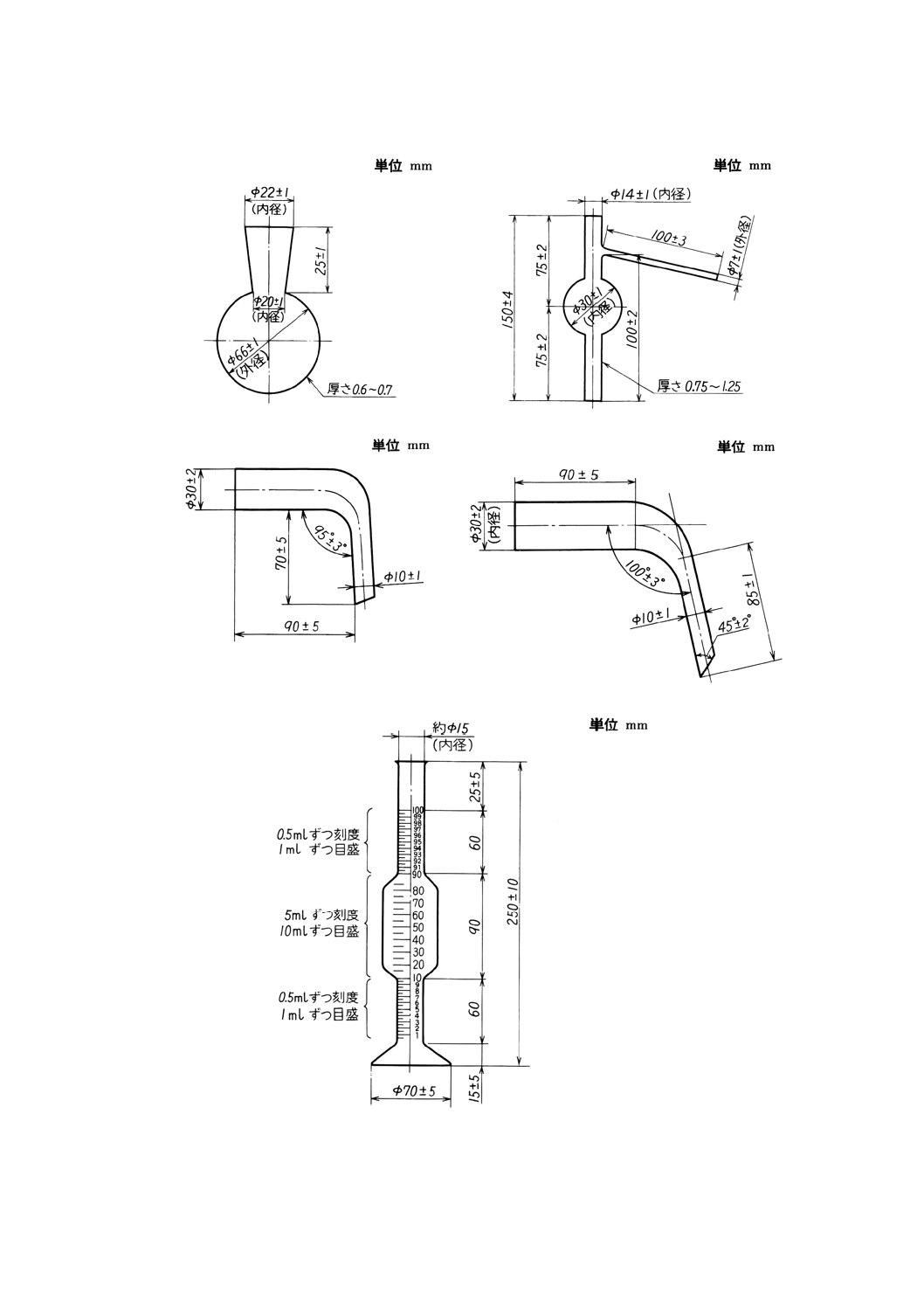
図18 蒸留フラスコの一例
図19 分留管の一例
図20 アダプターの一例
図21 アダプターの一例
図22 受器(メスシリンダー)の一例(容量100ml)
20
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図23 硬質ガラス製凝縮管の一例
7.3
操作 操作は,次のとおり行う。
(1) 試料100mlをメスシリンダーに取り(10),磁器片又はガラス製毛管数個を入れた蒸留フラスコに注入し,
コルク栓,シリコーン栓などに取り付けた温度計を蒸留フラスコに差し込む。試料を量り取ったメス
シリンダーは,洗浄しないでそのまま受器として用いる。
注(10) 試料の予想初留点が80℃以下のときは,試料をあらかじめ10〜15℃に冷却してその体積を量る。
蒸留中は,アダプターとメスシリンダーのすき間は脱脂綿でふさぎ,メスシリンダーの100ml
の標線まで10〜15℃に保った透明の浴に浸す。
2) 試料の予想初留点が80℃以下のときは,風よけを用いない。試料の予想初留点が150℃以上のときは,
蒸留フラスコのフラスコ支え板より上の部分に,あらかじめ耐熱性保温材を巻いて保温する。ただし,
枝部は保温しない。
(3) アダプターの先端は,受器の内壁に触れるようにする。
(4) 温度計の位置は,その水銀部の中央が蒸留フラスコの上部球部の中心にくるようにし,補助温度計の
位置は,その水銀部が温度計の露出水銀線の中央部にくるようにする。
(5) 凝縮浴に冷却水(11)を満たす。
注(11) 蒸留温度60℃以下のときは氷水を用い,蒸留温度150℃以上のときは,温水冷却又は空気冷却と
する。
(6) 蒸留フラスコを加熱し,10分間で留出を始め,その後毎分4〜5mlの留出速度で蒸留する。試験終了
後,温度計露出部の補正及び大気圧による沸点補正(12)を行う。
注(12) 温度計露出部及び大気圧による沸点補正は,試験の都度行う。
(7) 留出量とそれに対応する温度計の示度,補助温度計の示度及び試験時の大気圧(13)を記録する。
注(13) 蒸留の初めと終わりの平均値を求める。
7.4
蒸留温度の計算 蒸留温度の計算は,次のとおりとする。
(1) 水銀露出部の温度補正を行った蒸留温度は,次の式によって算出する。
T1=t+0.000 16 (t−t1) n
ここに, T1: 水銀露出部の温度補正を行った蒸留温度 (℃)
t: 水銀露出部の温度補正を行った温度計の示度 (℃)
t1: 補助温度計の示度 (℃)
n: 露出部にある水銀柱の度数範囲
(2) 補正大気圧は,次の式によって算出する。
′
−
t
P
p
133322
.0
000163
.0
1
=
ここに, p: 補正大気圧 (kPa)
P: 試験時の大気圧 (kPa)
t': 気圧計附属温度計の示度 (℃)
21
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 大気圧補正を行った蒸留温度は,次の式によって算出する。
(
)(
)
1
1
273
32
.
101
133322
.0
00012
.0
T
p
T
T
+
−
+
=
ここに,
T: 大気圧補正を行った蒸留温度 (℃)
T1: 水銀露出部の温度補正を行った蒸留温度 (℃)
p: 補正大気圧 (kPa)
8. 密度及び比重測定方法 密度及び比重測定方法は,浮ひょう法又は比重瓶法のいずれかによる。
備考 比重 (15/4℃) を測定するときは,測定温度を15.0±0.1℃とする。
8.1
浮ひょう法
8.1.1
要旨 この方法は,液体中に浮かべた浮ひょうの目盛を読み取り,その液体の密度又は比重を求め
る方法である。浮ひょうを用いる密度及び比重測定は,JIS K 0061の4.1(浮ひょう法)によるほか,次の
とおりとする。
8.1.2
装置及び器具 装置及び器具は,次のとおりとする。
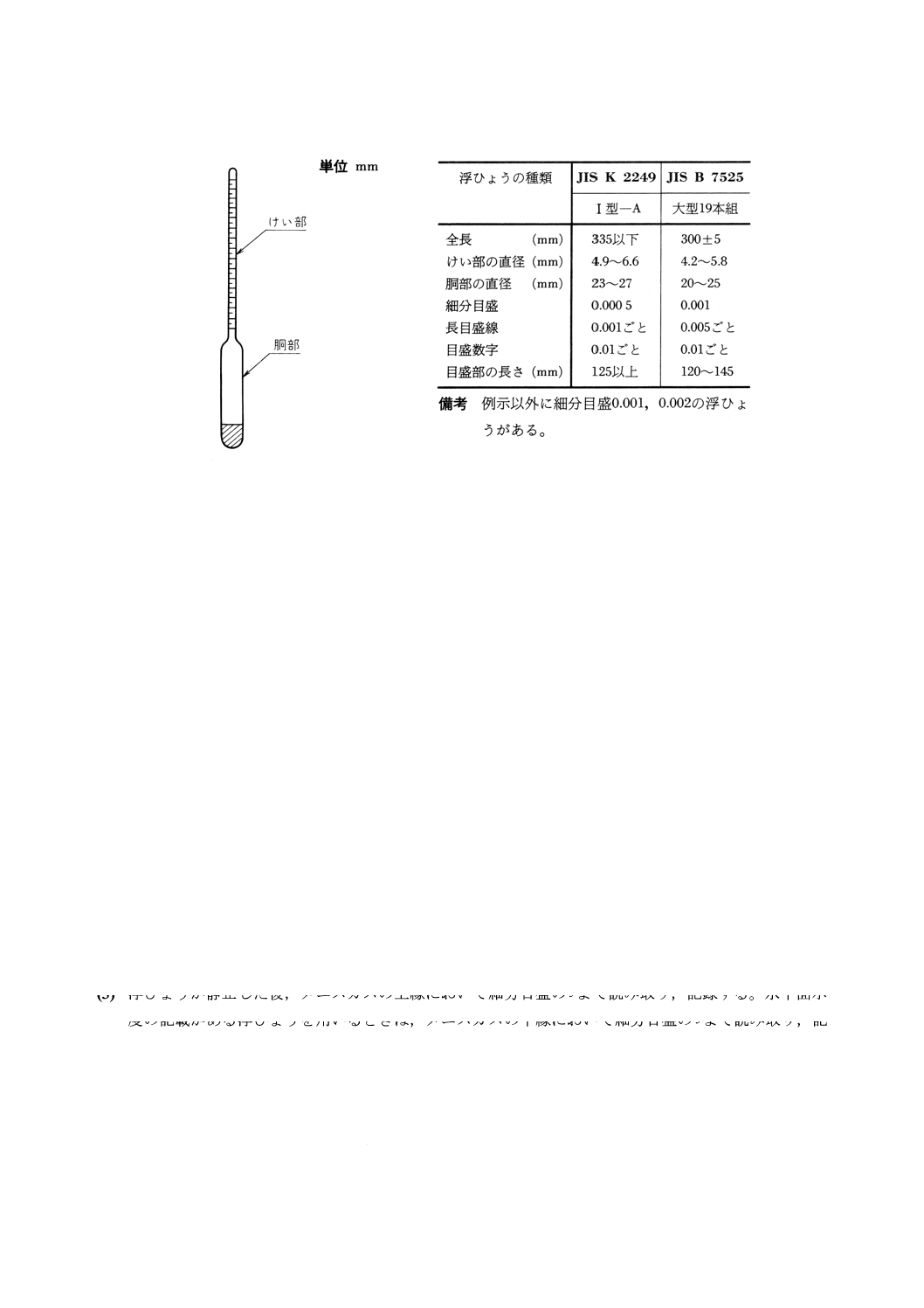
(1) 浮ひょう JIS K 2249に規定するI形浮きばかり(1形−A),若しくはソーダ石灰ガラス製浮ひょう
で,密度 (15℃) で目盛られ,密度を0.600 0g/cm3から1.100 0g/cm3の範囲において測定できるもの又
はJIS B 7525に規定する大型19本組若しくは軟質ガラス製のもので,比重 (15/4℃) で目盛られ,比
重を0.700から1.850の範囲において測定できるもので,いずれも器差(14)が既知のものを用いる。
浮ひょうの一例を図24に,浮ひょうの規格例を表4に示す。
注(14) 器差とは,計量器固有の誤差をいう。
備考1. 密度 (20℃) 又は比重 (20/20℃) で目盛られた浮ひょうで,器差が既知のものを用いてもよ
い。
2. 器差の求め方は,校正された密度浮ひょう又は比重浮ひょうとの比較で,次の式による。
E=R− (Rs−e)
ここに, E: 器差
R: 用いる浮ひょうの示度
Rs: 校正した浮ひょうの示度
e: 校正した浮ひょうの器差
3. 測定精度を0.001のけたまで必要としないときは,器差を求めなくてもよい。
22
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図24 浮ひょうの一例
表4 浮ひょうの規格例
(2) 温度計 JIS B 7410に規定するSG42又はSG44。
(3) シリンダー 流し出し口付ガラス製で,内径は,浮ひょうの最大直径より25mm以上大きく,高さは,
浮ひょうをシリンダーに入れたとき,浮ひょうの下端がシリンダーの底から25mm以上の位置にくる
もの。
備考 JIS K 2839の図35に規定するI形用ガラス製シリンダー又は透明プラスチック製シリンダー。
透明プラスチック製シリンダーは,試料の性状に影響を及ぼさないものでなければならない。
(4) 恒温水槽 シリンダーに入れた試料を20.0±0.1℃に保持できるもの。
(5) かき混ぜ棒 試料に侵されず,その性状に影響を及ぼさない材質を用い,シリンダー中の試料の密度
及び比重を一様にするため,十分にかき混ぜることができるもの。
8.1.3
操作 操作は,次のとおり行う。
(1) 気泡が入らないように試料をシリンダーに取り,恒温水槽中に保持してかき混ぜ棒で試料を上下にか
き混ぜた後,温度計を全浸没(15)にして試料の温度を測る。
注(15) 水銀球部の下端から水銀柱頂部(温度指示部)までの水銀部全体を試料に浸す。
(2) 試料の温度が20.0±0.1℃になった後,あらかじめ20℃近くに保った清浄な(16)浮ひょうを静かに試料
中に入れて静止させ,約2目盛だけ液中に沈めて手を離す(17)。
注(16) 浮ひょうは中性洗剤で洗い,ジエチルエーテル,エタノールなどを含ませた布や紙でけい部を
ぬぐったものを用いる。けい部が汚れていると,表面張力の影響で示度が変わる。
また,けい部は上端を手でつまみ,目盛部分には手を触れてはならない。
(17) 手を離すときに,浮ひょうを少し回転させると,シリンダー内壁に触れずに静止させることが
できる。
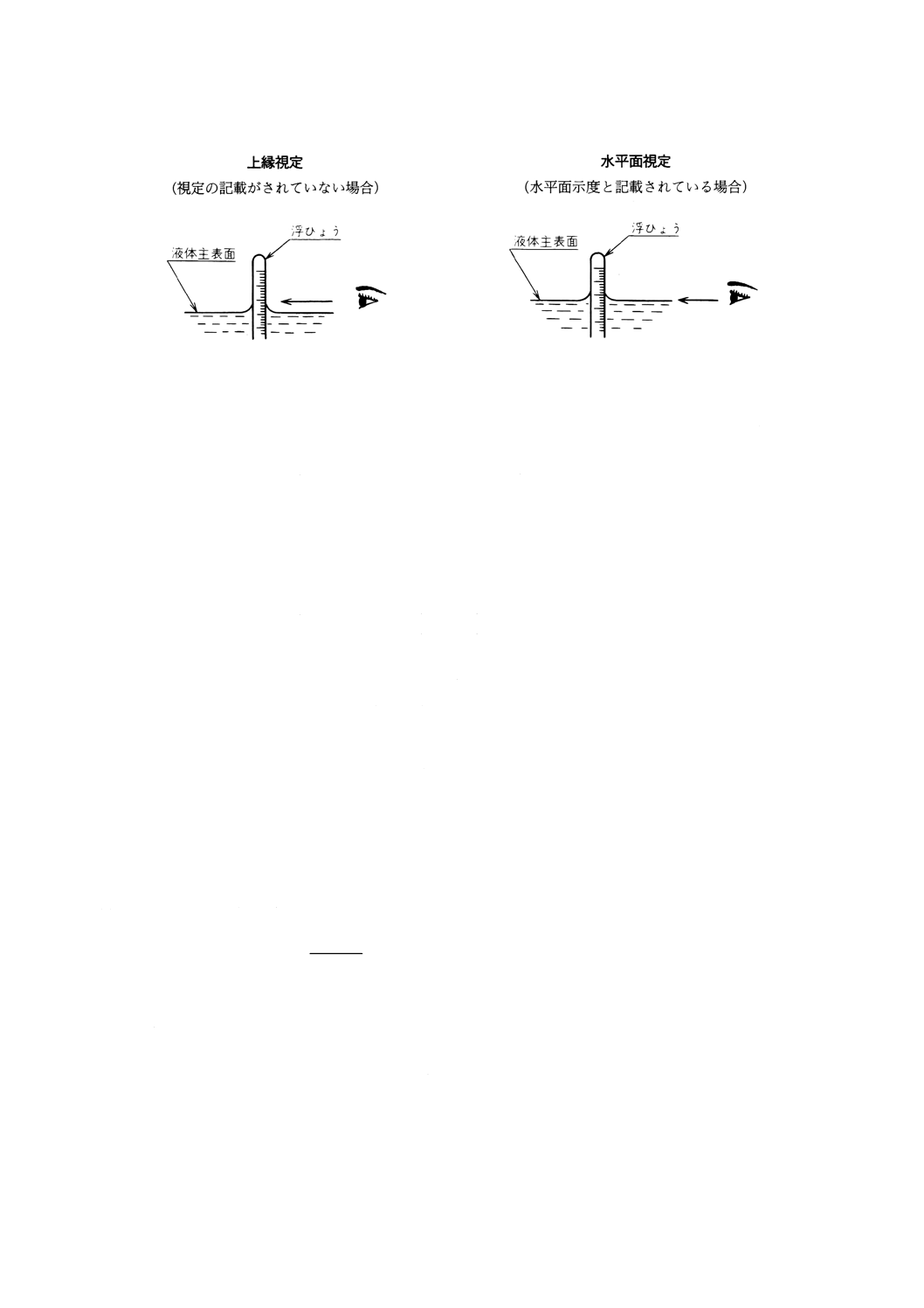
(3) 浮ひょうが静止した後,メニスカスの上縁において細分目盛の51まで読み取り,記録する。水平面示
度の記載がある浮ひょうを用いるときは,メニスカスの下縁において細分目盛の51まで読み取り,記
録する。浮ひょうの目盛の読み方を図25に示す。
備考1. 半透明試料は,眼を試料面のわずか下方から静かに上げていく際に,最初長円形に見えた試
料面が直線になったとき読み取る。
2. 不透明試料は,試料面メニスカスの上縁において目盛を読み,これにあらかじめ求めた補正
値を用いて下縁相当値を求める。
23
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図25 浮ひょうの目盛の読み方
8.1.4
計算 密度 (20℃),比重 (20/20℃) は,次によって小数点以下第4位まで求める。ただし,測定
精度を0.001のけたまで必要としないときは,器差の補正を省略してよい。
器差の補正は,浮ひょうの示度から器差を差し引く。
(1) 密度 (20℃) で目盛られた浮ひょうを用いて密度 (20℃) を求める場合は,次の式によって算出する。
D=D20−E
ここに,
D: 密度 (20℃) (g/cm3)
D20: 密度 (20℃) で目盛られた浮ひょうの示度 (g/cm3)
E: 器差
(2) 比重 (20/20℃) で目盛られた浮ひょうを用いて比重 (20/20℃) を求める場合は,次の式によって算出
する。
S=S20−E
ここに,
S: 比重 (20/20℃)
S20: 比重 (20/20℃) で目盛られた浮ひょうの示度
E: 器差
(3) 密度 (15℃) で目盛られた浮ひょうを用いて密度 (20℃) を求める場合は,次の式によって算出する。
D=0.999875 (D15−E)
ここに,
D: 密度 (20℃) (g/cm3)
D15: 密度 (15℃) で目盛られた浮ひょうの示度 (g/cm3)
E: 器差
(4) 比重 (15/4℃) で目盛られた浮ひょうを用いて密度 (20℃) を求める場合は,次の式によって算出する。
D=0.999845 (S15−E)
ここに,
D: 密度 (20℃) (g/cm3)
S15: 比重 (15/4℃) で目盛られた浮ひょうの示度
E: 器差
(5) 密度(20。C) から比重 (20/20℃) を求める場合は,次の式によって算出する。
99820
.0
D
S=
ここに,
S: 比重 (20/20℃)
D: 密度 (20℃) (g/cm3)
(6) 比重 (20/20℃) から密度 (20℃) を求める場合は,次の式によって算出する。
D=0.99820S
ここに,
D:密度 (20℃) (g/cm3)
S:比重 (20/20℃)
備考 浮ひょう法によって得たS又はDの値に対する空気の浮力の影響は,液面上に出ているけい部
の体積と同体積の空気の質量だけであるので,通常無視することができる。
24
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.2
比重瓶法
8.2.1
要旨 この方法は,比重瓶を用いて試料と水の質量を量ることによって,その液体の密度又は比重
を求める方法である。比重瓶を用いる密度及び比重測定は,JIS K 0061の4.2(比重瓶法)によるほか,次
のとおりとする。
8.2.2
装置及び器具 装置及び器具は,次のとおりとする。
(1) 比重瓶
(a) ワードン形比重瓶 JIS R 3503の図15に規定するもの。図26に示す。
図26 ワードン形比重瓶
備考1. 容量は,約50mlとする.
2. 容量表示,メモ用スペースは,砂目又は焼付けを施す.
3. 質量は,栓,キャップを含めて35g以下とする.
4. ワードン形比重瓶の呼び方は,名称による.
例 ワードン形比重瓶
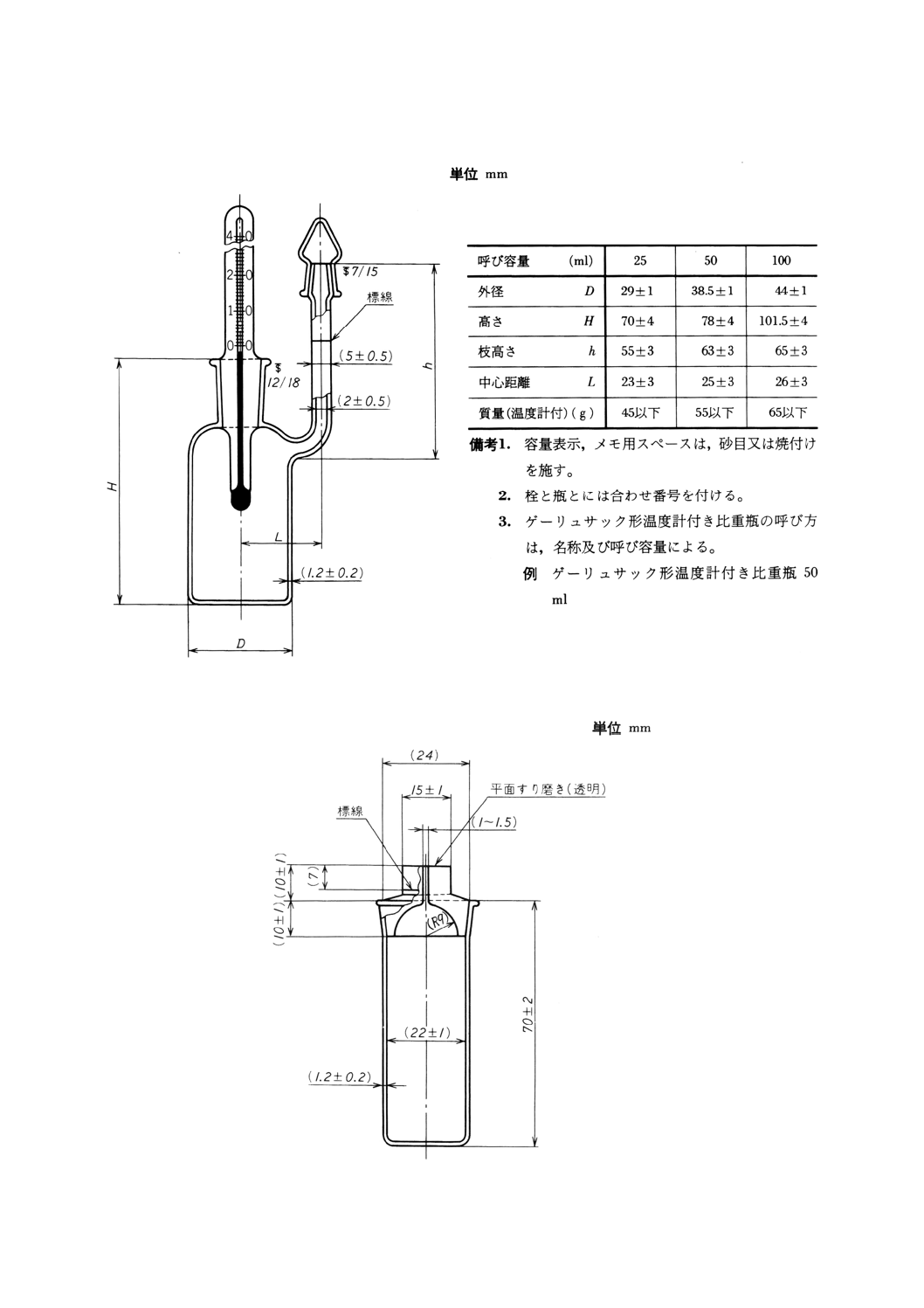
(b) ゲーリュサック形温度計付比重瓶 JIS R 3503の図54に規定するもの。図27に示す。
25
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図27 ゲーリュサック形温度計付き比重瓶
(c) ハーバート形比重瓶 JIS R 3503の図56に規定するもの。図28に示す。
図28 ハーバート形比重瓶
26
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
備考1. 質量は,栓を含めて31g以下とする.
2. 口形状は,リップ付きとする.
3. 栓と瓶とには合わせ番号を付ける.
4. 容量表示,メモ用スペースは,砂目又は焼付けを施す.
5. 栓のすり合わせは,水密でなければならない.
6. ハーバート形比重瓶の呼び方は,名称による.
例 ハーバート形比重瓶
(d) 目盛ピクノメーター JIS K 2839の図37に規定する目盛ピクノメーター(I型)。図29に示す。
図29 目盛ピクノメーター
備考 質量は,30g以下とする。
(e) オストワルドピクノメーター JIS K 2839の図39に規定するもの。図30に示す。
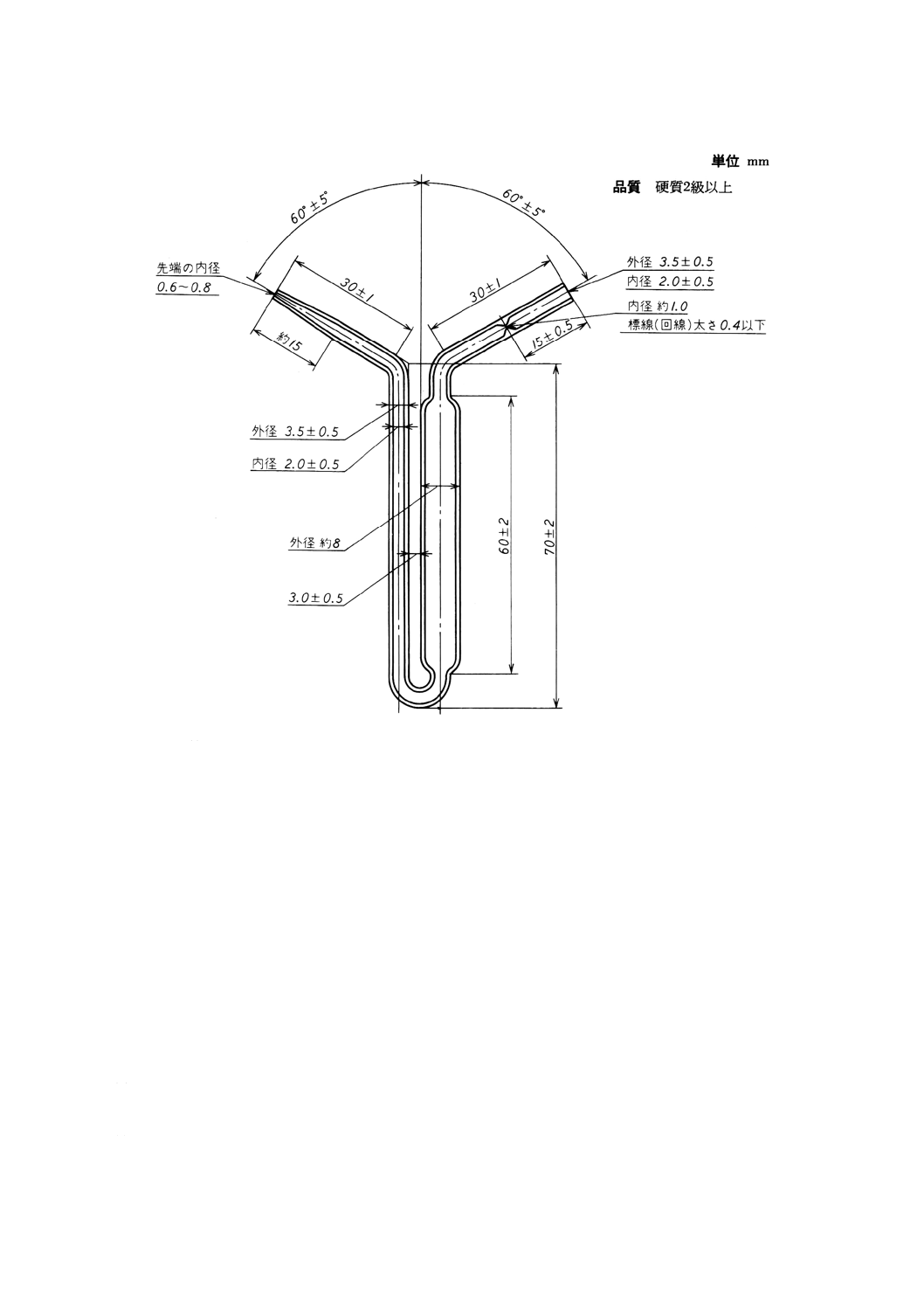
27
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図30 オストワルドピクノメーター
備考1. 標線(回線)までの容量2±0.2ml。
2. 質量は,6g以下とする。
(2) 恒温水槽 比重瓶のけい部以下を水面下に保持できる構造で,温度を20.0±0.1℃(18)に調節できるも
の。
注(18) 目盛ピクノメーターの場合,温度を20.00±0.05℃に調節できるものを用いる。
(3) はかり(天びん) 化学はかり又は電子はかり。
備考 化学はかりとは,測定できる最大の質量が100〜200gで,0.1mgの差を読み取れる等比式化学
はかり又は定感量直示式はかり。
(4) 温度計 JIS B 7410に規定するSG44。
なお,これと同程度の性能をもつ温度計であって,正しく校正したものであれば,このほかのガラ
ス製温度計,抵抗温度計,熱電温度計などを用いてもよい。
8.2.3
校正 器具及び装置の校正は,水当量の測定によって,次のとおり行う。
(1) ワードン形,ゲーリュサック形温度計付及びハーバート形比重瓶の校正
(a) 洗浄乾燥した比重瓶の質量を1mgのけたまで量り,これをW0とする。
(b) あらかじめ,測定温度より1〜3℃低い温度に調節した蒸留水を気泡が残らないように注意深く比重
瓶に満たし,20.0±0.1℃に保った恒温水槽にそのけい部まで入れる。
(c) 30分間以上放置して温度が一定になった後,ほぼ同温に保った栓(19)を差し込み,過剰の水をピペ
ット又は細く切ったろ紙で吸い取り,液面を標線又はふたの細孔上端に合わせる。
28
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(19) ゲーリュサック形温度計付比重瓶の場合は,温度計付きの栓を差し込む。
(d) 恒温水槽から比重瓶を取り出し,清浄な乾燥している布で外面をぬぐって水分を除き,ふたのある
比重瓶の場合は,ふたを付けて室温近くになるまで放置する。
(e) 測定温度が室温より低い場合,比重瓶の表面に結露することがあるので,その表面をよくぬぐって
全質量を1mgのけたまで量り,これをW1とする。
(f) (a)及び(e)から比重瓶の水当量 (W1−W0) を求める。水当量は,必要に応じて測定する。
(2) 目盛ピクノメーターの校正
(a) 洗浄乾燥したピクノメーターの質量を0.1 mgのけたまで量り,これをW0とする。
(b) ピクノメーターを立てたまま,かぎ状の一端を水中に入れ,長管の端から軽く吸引した後,サイホ
ン作用によって球上方の細管を超える位置まで満たす。
(c) 指先で軽くはじいてピクノメーター内の気泡を除き,細管の端の内外に付着する水は,細く切った
ろ紙などでぬぐい取る。
(d) 20.00±0.05℃に保った恒温水槽にその細管目盛の上まで入れ,温度が一定になって細管中の水面が
静止するまで静置する。
(e) 両細管の細分目盛をその51まで読み取って記録し,水槽からピクノメーターを取り出して外面の水
をぬぐい取り,室温になるまで放置する。
(f) 測定温度が室温より低いときは,ピクノメーターの表面に結露することがあるので,その表面をよ
くぬぐい,全質量を0.1mgのけたまで量り,これをW1とする。
(g) 次に細管の水を徐々に増加して,細管目盛の3点以上について,(a)〜(f)の操作を行って,両細管の
目盛の読みの合計値とそれに対応する水当量の関係線図を作成する。この関係線図から任意の読み
に選んだ点における読みの合計値に対する水当量 (W1−W0) を求める。この関係線図は,必要に応
じて調整する。
(3) オストワルドピクノメーターの校正
(a) 洗浄乾燥したピクノメーターの質量を0.1mgのけたまで量り,これをW0とする。
(b) 標線の付いている細管の端にゴム管を付け,標線を超える位置まで水を吸い上げる。
(c) 指先で軽くはじいて管内の気泡を完全に除いた後,ピクノメーターを20.0±0.1℃に保った恒温水槽
中にその標線付近まで入れ,ピクノメーター内の水の温度が一定になって,細管中の液面が静止す
るまで保持する。
(d) 液面が静止した後,ピクノメーターを水槽中に保持したまま,標線の付いていない細管の端から細
く切ったろ紙で過剰の水を吸い出して液面を標線に合わせ,細管に付着する水はぬぐい取る。
(e) 恒温水槽からピクノメーターを取り出し,清浄な乾燥している布で外面をぬぐって水分を除き,室
温になるまで放置する。
(f) 測定温度が室温より低いときは,ピクノメーターの表面に結露することがあるので,その表面をよ
くぬぐい,質量を0.1mgのけたまで量り,これをW1とする。
(g) (a)及び(f)からオストワルドピクノメーターの水当量 (W1−W0) を求める。水当量は,必要に応じて
測定する。
8.2.4
操作 操作は,次のとおり行う。
(1) ワードン形,ゲーリュサック形温度計付及びハーバート形比重瓶の場合
(a) 洗浄乾燥した比重瓶の質量を1mgのけたまで量り,これをW0とする。
(b) これにあらかじめ測定温度より1〜3℃低い温度に調節した試料を8.2.3(1)(b)〜(e)によって満たし,
29
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
その質量をW2とする。
(2) 目盛ピクノメーターの場合
(a) 洗浄乾燥したピクノメーターの質量を0.1mgのけたまで量り,これをW0とする。
(b) 8.2.3(2)(b)〜(f)によって任意の目盛まで試料を取った後,目盛の読みを記録して質量を量り,これを
W2とする。
(c) 8.2.3(2)(g)の関係線から,試料を測定したときのピクノメーターの目盛の読みの合計に対応する水当
量 (W1−W0) を求める。
(3) オストワルドピクノメーターの場合
(a) 洗浄乾燥したピクノメーターの質量を0.1mgのけたまで量り,これをW0とする。
(b) 8.2.3(3)(b)〜(f)によって試料をピクノメーターに満たし,その全質量を0.1mgのけたまで量り,これ
をW2とする。
8.2.5
計算
(1) 比重 (20/20℃) は,次の式によって算出し,小数点以下第3位に丸める。
0
1
0
2W
W
W
W
S
−
−
=
ここに,
S: 試料の比重 (20/20℃)
W0: 比重瓶の質量 (g)
W1: 比重瓶を水で満たしたときの質量 (g)
W2: 比重瓶を試料で満たしたときの質量 (g)
W2−W0: 比重瓶中の試料の質量 (g)
W1−W0: 水当量 (g)
(2) 密度 (20℃) は,次の式によって算出し,小数点以下第3位に丸める。
D=S×0.998 2
ここに,
D: 試料の密度 (20℃) (g/cm3)
0.998 2: 水の密度 (20℃) (g/cm3)
参考 W0,W1及びW2を量るとき,空気の浮力による補正を行わない場合でも,試料のS又はDが0.600
〜1.400のときに生じる誤差は0.000 5未満である。
9. 水分試験方法 水分試験方法は,カールフィッシャー滴定法又は乾燥減量法のいずれかによる。
9.1
カールフィッシャー滴定法
9.1.1
要旨 この方法は,カールフィッシャー試薬を用いて水を滴定し,その滴定量から水分量を求める
方法であり,試料の性質及び水分量に応じて,容量滴定法,電量滴定法又は水分気化法のいずれかを用い
る。カールフィッシャー滴定による水分試験は,JIS K 0068の4.(カールフィッシャー滴定法)によるほ
か,次のとおりとする。
備考1. 水以外でカールフィッシャー試薬と反応する物質(以下,妨害物質という。)を含む試料には
適用できない。ただし,妨害を除去できるように調製された滴定溶剤や電解液,又は水分気
化法を用いるなどによって適用できる場合がある。代表的な妨害物質には,次のようなもの
がある。
(1) よう素と反応するもの:強塩基性物質,酸化性物質,還元性物質など。
(2) その他の成分と反応するもの:メタノールと反応して水を生成するケトン類など。
2. 電量滴定法は,通常,低水分の場合に適用し,2%以上の水分を含む試料には,容量滴定法を
30
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
用いる方がよい。
9.1.2
力価 力価とは,カールフィッシャー試薬を用いて水を滴定するときのカールフィッシャー試薬の
単位体積当たりの水の当量で,mg/mlで表す。
9.1.3
試料の採取 試料の採取は,次のとおり行う。
(1) 試料容器は,密栓できる構造のもので,使用前に約105℃で乾燥し,デシケーター中で放冷する。デ
シケーターから取り出したときは,直ちに密栓する。
(2) 試料の採取は,試料容器の容量の80%以上となる量を速やかに採取し,直ちに密栓する。
(3) 試料容器に採取した試料は,よく振り混ぜて均一にする。
(4) 固体の試料が塊状又は粒状のときは,適切な粒度に粉砕して粉状とする。この場合,水分の揮散,吸
湿などが生じないように注意する。
(5) 試料の温度が室温と異なるときの試料採取は,試料を室内に放置して試料の温度が室温になった後行
う。
(6) 試料容器を開栓した後,直ちに試料を採取し,速やかに密栓する。
(7) 試料量は,水分の量に応じて,表5及び表6によって決める。
表5 容量滴定法の場合の試料量
表6 電量滴定法の場合の試料量
予想水分 (%)
試料量(20)
(ml又はg)
予想水分 (%)
試料量
(ml又はg)
0.1以下
10〜20
5
2
0.5
0.3
0.1
0 〜0.05
5〜10
2
1
0.5
0.1
0.1〜0.5
0.05〜0.1
0.5〜1
0.1 〜0.2
1 〜5
0.2 〜0.5
5 〜10
0.5 〜2.0
10 〜50
50以上
0.03
注(20) 表5の試料量は,カールフィッシャー試薬の力価を3mgH2O/mlとして計算したも
のである。力価がこの値と大きく異なるときは,試料量を調節する必要がある。
9.1.4
容量滴定法
9.1.4.1
要旨 この方法は,滴定槽に滴定溶剤を入れ,カールフィッシャー試薬を滴加して無水状態とし,
これに試料を加えて溶解又は水分を抽出し,次に,カールフィッシャー試薬を用いて滴定し,その滴定量
から水分量を求める方法である。
9.1.4.2
装置及び器具 装置及び器具は,次のとおりとする。
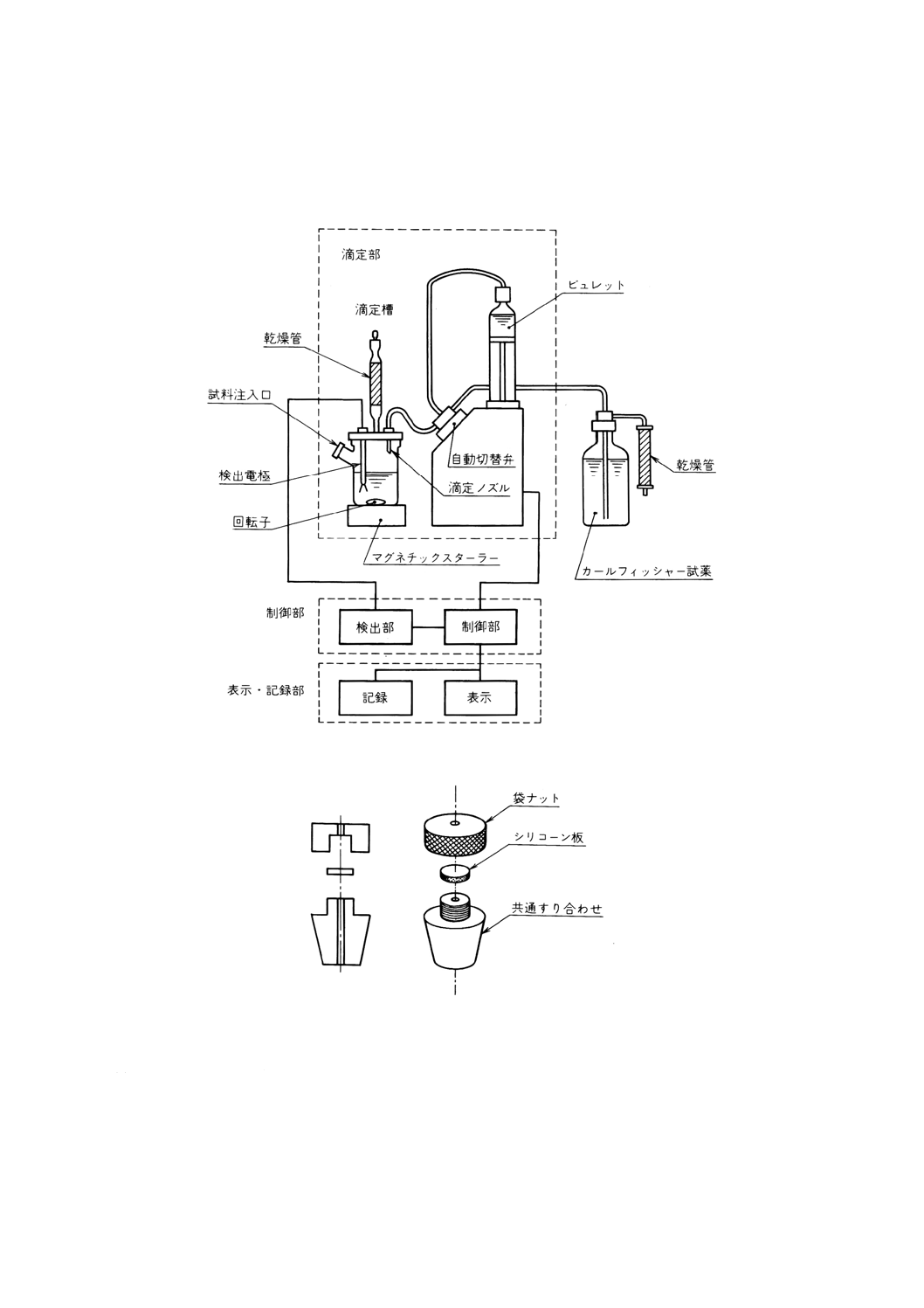
(1) 容量滴定装置(21) 滴定部,制御部及び表示・記録部で構成する自動滴定装置。構成の一例を図31に
示す。滴定装置は,JIS K 0113によるほか,次による。
注(21) ガラス器具の連結部は,すべてすり合わせとし,カールフィッシャー試薬と反応又は溶解しな
いグリースを塗って大気からの吸湿を防ぐ。
備考 容量滴定装置は,終点検出にマジックアイ又は電流計を用いた電圧制御電流検出方式を用いて
もよい。この場合は,手動によって滴定を行う。
(a) 滴定槽 試料注入口,検出器(一対の白金電極),滴定ノズル及びシリカゲルなどの乾燥剤を入れた
乾燥管を備えた容量100〜250mlのガラス製平底フラスコとし,回転子を入れた後,かき混ぜ速度
を調節できるマグネチックスターラーの上に置く。試料注入口はガラス栓で密栓するが,試料採取
にマイクロシリンジ又は注射器を用いるときは,この代わりにパッキン付ステンレス鋼製又は四ふ
っ化エチレン樹脂製のストッパーを装着する。ストッパーの一例を図32に示す。
31
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(b) ビュレット 容量10ml又は20mlのパルスモーター駆動によるピストンビュレットとし,最小排出
量が0.01〜0.02mlのもの。
図31 容量滴定法自動滴定装置の構成の一例
図32 ストッパーの一例
(2) 試料採取器 試料採取器は,次のいずれかを用いる。
(2.1) 液体試料採取器
(a) マイクロシリンジ 容量10μl,25μl,50μl又は100μlで,針の長さは50mm以上のもの。
(b) 注射器 容量1〜20ml,注射針は,外径1mmで長さ50〜100mmのもの。シール用として注射針の
先端にゴム栓を付けるが,ゴム栓には,直径13〜15mm,厚さ5〜7mmの丸型のシリコーンゴム製
のものなどがある。
32
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(c) 全量ピペット 5〜20ml
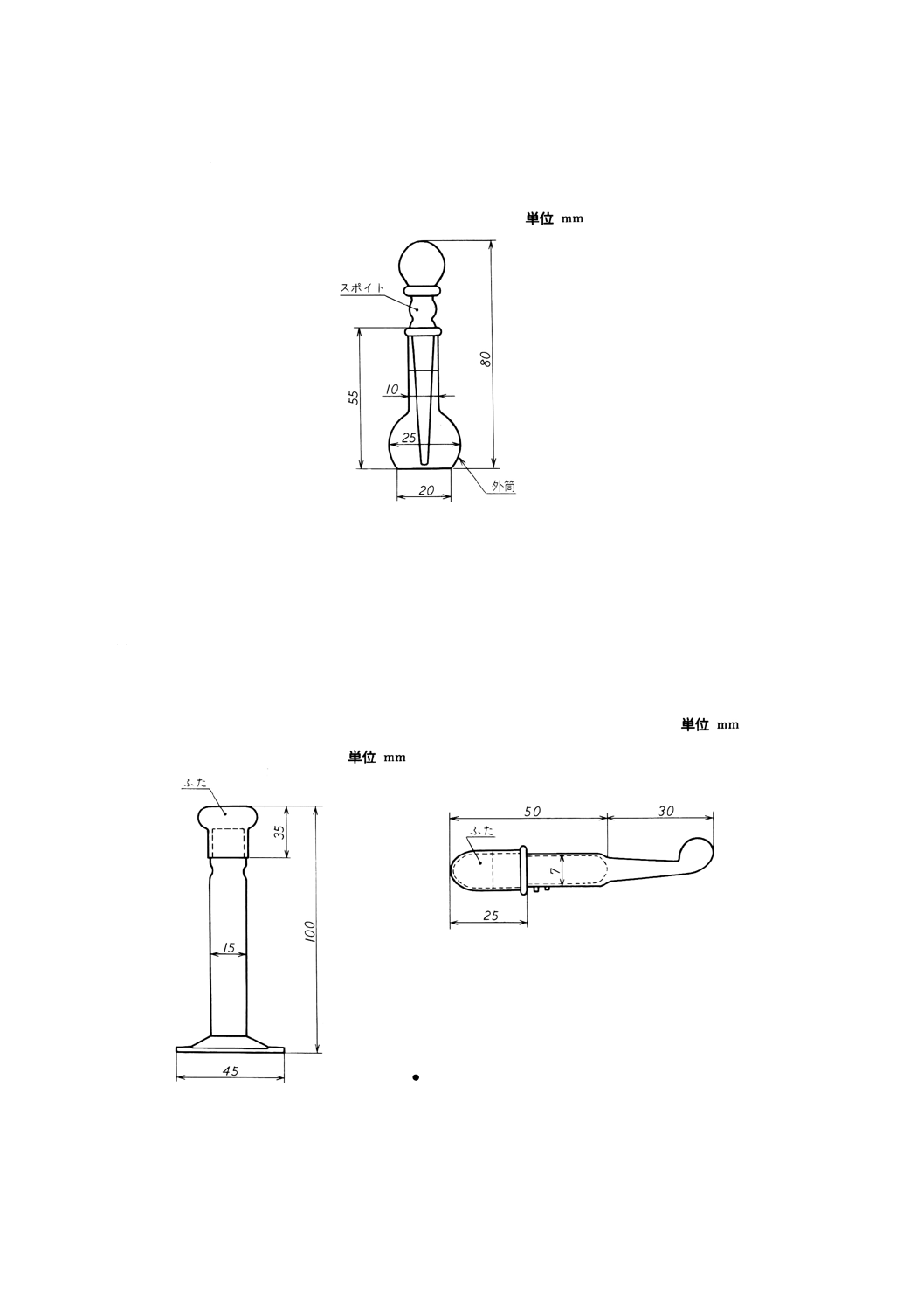
(d) 滴瓶 1滴が約0.03mlのスポイト付。一例を図33に示す。
図33 滴瓶の一例
(2.2) 固体試料採取器
(a) 直管型固体試料採取器 一例を図34に示す。この採取器は,流動性のある粉末,粒状及び塊状の試
料に用いる。
(b) 小量用固体試料採取器 一例を図35に示す。この採取器は,0.4g以下の試料の採取に用いる。
(c) 高粘度半固体試料採取器 一例を図36に示す。この採取器は,高粘度の半固体試料に用いる。
(d) 注射筒型固体試料採取器 一例を図37に示す。この採取器は,固結性粉末又は粘性のある試料に用
いる。
図34 直管型固体試料採取器の一
例
図35 小量用固体試料採取器の一例
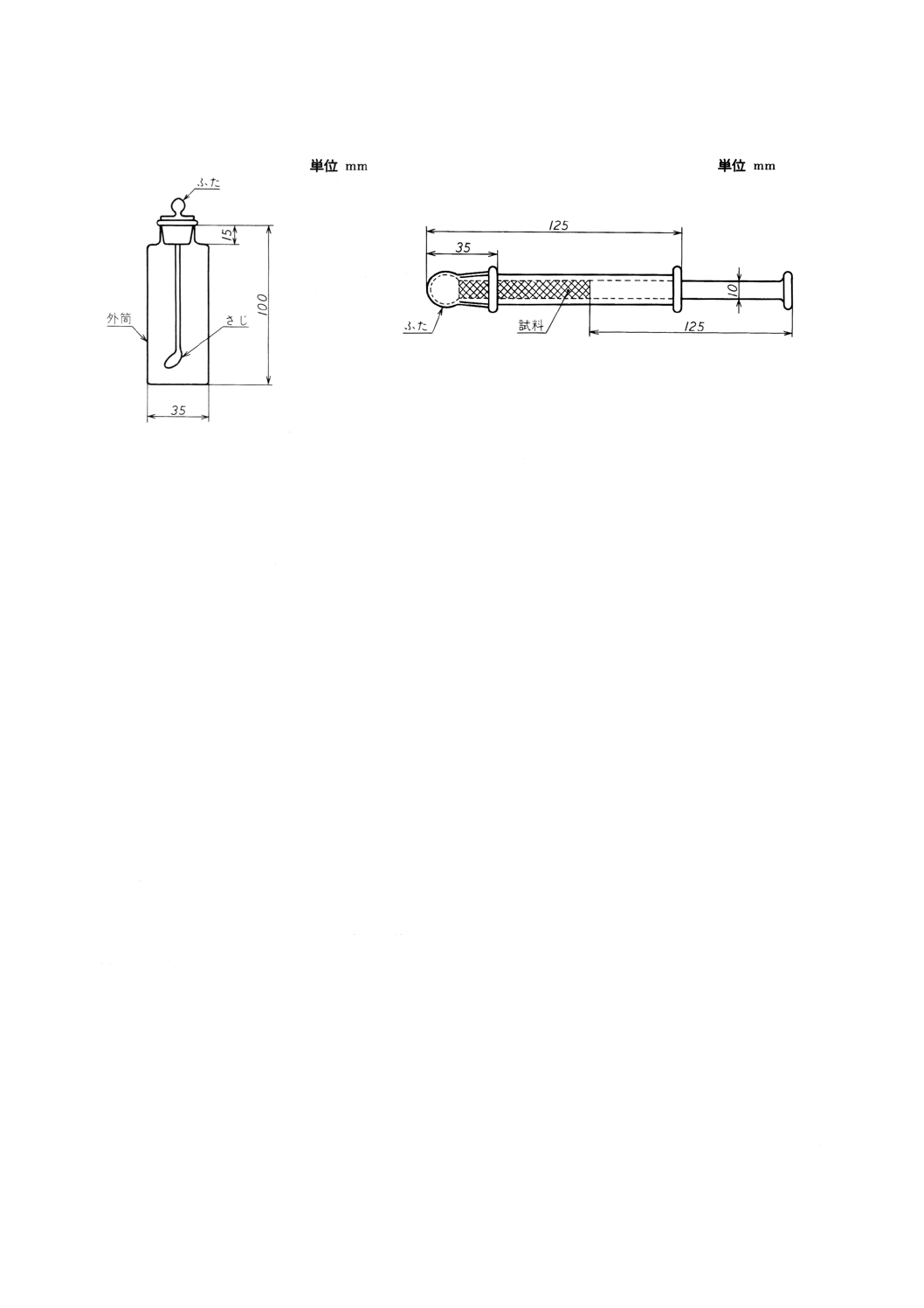
33
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図36 高粘度半固体試料採取器の一例
図37 注射筒型固体試料採取器の一例
(3) はかり(天びん) 化学はかり又は電子はかり。
備考 電子はかりは,適切なインターフェイスによって自動滴定装置と接続できるものを用いると便
利である。
(4) デシケーター JIS R 3503に規定するもので,乾燥剤としてシリカゲル(22)を入れたもの。
注(22) シリカゲルは,約150℃で加熱したものを用い,再生する場合も同じ処理を行う。
(5) フラスコ 共通すり合わせ三角フラスコ2 000ml
9.1.4.3
試薬 試薬は,次のとおりとする。
(1) メタノール JIS K 8891に規定するメタノール1 000mlをフラスコに取り,JIS K 8876に規定するマ
グネシウム末5gを加え,必要に応じて,反応促進用にJIS K 8920に規定する よう素0.5gを加える。
ガスの発生が終わった後,湿気を遮ってメタノールを蒸留し,密栓して保存する。水分は,0.05%以
下とする。
(2) クロロホルム JIS K 8322に規定するクロロホルム1 000mlを共通すり合わせ三角フラスコ2lに取り,
乾燥用合成ゼオライト30gを加えて密栓し,約8時間放置する。その間,ときどき軽く振り混ぜる。
さらに,約16時間放置した後,クロロホルムを分取し,密栓して保存する。水分は,0.01%以下とす
る。
(3) 2−メトキシエタノール JIS K 8895に規定する2−メトキシエタノールを湿気を遮って蒸留し,初留
分を除き,中間留出物を集め,密栓して保存する。水分は,0.05%以下とする。
(4) ピリジン JIS K 8777に規定するピリジン1 000mlを共通すり合わせ三角フラスコ2 000mlに取り,
JIS K 8574に規定する水酸化カリウム約10g又はJIS K 8428に規定する酸化バリウム約10gを加え,
数日間密栓して放置後,湿気を遮って蒸留し,密栓して保存する。水分は,0.05%以下とする。
(5) 酒石酸ナトリウム二水和物 JIS K 8540に規定する酒石酸ナトリウム二水和物を水溶液から再結晶さ
せ,十分に付着水分を除いてから,室温,相対湿度約50%で含水量を一定としたものを用いる(23)。
注(23) 水分調整用デシケーターに入れ,10〜30℃において48時間上保ったもの。
(6) 滴定溶剤 滴定溶剤は,試料の性質及び試験の目的に応じ,次のいずれかを用いる。そのいずれも,
水分は,0.05%以下とする。
参考 滴定溶剤は,市販品を用いてもよい。
(a) メタノール (1)による。
(b) メタノール−クロロホルム混合溶剤 メタノールとクロロホルムとを体積比1 : 3又は1 : 4に混合
34
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
したもの。この溶剤は石油,油脂類などに用いる。
備考 メタノールとクロロホルムとの混合比は,試料の溶解性に応じていずれかを選ぶ。微量水分の
ため試料量を多く採取するときは,クロロホルムの混合比が大きいものを用いる。
(c) メタノール−ホルムアミド混合溶剤 メタノールとJIS K 8873に規定するホルムアミドとを体積比
1 : 2に混合したもの。この溶剤は糖類,たん白質類などに用いる。
(d) クロロホルム−プロピレンカーボネート混合溶剤 クロロホルムとプロピレンカーボネートとを混
合したもの。この溶剤はケトン類,有機酸,アミノベンゼン類などに用いる。
(e) ピリジン−エチレングリコール混合溶剤 ピリジンとJIS K 8105に規定するエチレングリコールと
を体積比5 : 1に混合したもの。この溶剤はカルボニル化合物などに用いる。
(f) ピリジン−プロピレングリコール混合溶剤 ピリジンとJIS K 8837に規定するプロピレングリコー
ルとを体積比3 : 1に混合したもの。この溶剤はアルデヒド類などに用いる。
(g) サリチル酸−メタノール溶液 JIS K 8392に規定するサリチル酸150gをメタノール1 000mlに溶か
す。この溶剤は,電離指数pKa値9以上のアミン類に用いる。
(7) カールフィッシャー試薬
参考 カールフィッシャー試薬は,市販品を用いてもよい。
(7.1) 調製方法 JIS K 8920に規定する よう素85gをメタノール670mlに溶かし,ピリジン270mlを加
え,よく混合してから氷冷する。これに20℃を超えないように注意しながら乾燥二酸化硫黄を通じ,
その増量が65gに達するまで行う(24)。この場合,メタノールの代わりに2−メトキシエタノール又
はクロロホルムを用いてもよい。
カールフィッシャー試薬の力価を希釈によって調整するときは,溶液の調製に使用した溶剤を用
いて行う。この試薬は,光を遮り,湿気を避けて冷所に密栓して保存し,標定は24時間以後に行う。
力価は,日時の経過とともに低下するので,使用の都度,標定しなければならない。
注(24) この調製方法によって調製されたカールフィッシャー試薬の体積は,約1 000mlとなり,その初
期力価は,3.5〜4.5 H2Omg/mlである。
(7.2) 標定 標定は,次のとおり行う。
(a) 滴定槽にメタノール25〜50mlを入れる。
(b) カールフィッシャー試薬で終点まで滴定し,滴定槽内を無水状態にする。このときの滴定量は,読
み取る必要はない。
(c) この溶液をかき混ぜながら,0.1 mgのけたまで量り取った水30〜50mg又は酒石酸ナトリウム二水
和物約150 mgを直ちに加える(25)。
注(25) 水の場合は,滴瓶を用い,酒石酸ナトリウム二水和物の場合は,少量用固体試料採取器を用い
る。
(d) この溶液をかき混ぜながら,カールフィッシャー試薬で終点まで滴定する。
(7.3) 計算 力価は,次の式によって算出し,小数点以下第2位に丸める。
(a) 水を用いた場合
a
V
m
F
1
=
(b) 酒石酸ナトリウム二水和物を用いた場合
a
V
m
F
1566
.0
2×
=
35
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
F: 力価 (mg/ml)
m1: 量り取った水の質量 (mg)
m2: 量り取った酒石酸ナトリウム二水和物の質量 (mg)
Va: 滴定に用いたカールフィッシャー試薬の量 (ml)
0.156 6: 酒石酸ナトリウム二水和物中の水分の含有率
備考 標定には,水の濃度が既知である水−メタノール溶液を用いてもよい。
(8) 水−メタノール溶液
参考 水−メタノール溶液は,市販品を用いてもよい。
(8.1) 調製方法 メタノール約500mlを乾燥した全量フラスコ1 000mlに取り,水約2gを0.1 mgのけた
まで量って加え,更にメタノールを標線まで加える。この溶液は,湿気及び光を遮り,温度変化の
小さい場所に保存し,標定は測定の前に行う。
(8.2) 標定 標定は,次のとおり行う。
(a) 滴定槽にメタノールを25〜50ml入れる。
(b) メタノールをかき混ぜながら,カールフィッシャー試薬で終点まで滴定し,滴定槽内を無水状態に
する。このときの滴定量は,読み取る必要はない。
(c) この溶液をかき混ぜながら,全量ピペット又はビュレットを用いて量り取った水−メタノール溶液
10.0mlを直ちに加える。
(d) この溶液をかき混ぜながら,カールフィッシャー試薬で終点まで滴定する。
(8.3) 計算 水−メタノール溶液の水の濃度は,次の式によって算出し,小数点以下第2位に丸める。
10
F
V
f
b×
=
ここに,
f: 水−メタノール溶液中の水の濃度 (mg/ml)
Vb: 滴定に用いたカールフィッシャー試薬の量 (ml)
F: カールフィッシャー試薬の力価 (mg/ml)
9.1.4.4
試料採取操作 試料採取操作は,用いる採取器具によって次のとおり行う。
(1) マイクロシリンジを用いる場合
(a) マイクロシリンジは,あらかじめ洗浄し,空気又は窒素で乾燥する。
(b) 試料で共洗いを2回以上行う。
(c) 全容量の21以下になるように試料を採取する。
(d) 針を上にして空気を抜き,目盛に合わせる。
(c) 試料を採取したマイクロシリンジの質量を0.1mgのけたまで量る。
(f) 針をストッパーに刺し,プランジャーを押して滴定槽内に試料を注入する。
(g) マイクロシリンジの質量を0.1 mgのけたまで量る。
(2) 注射器を用いる場合
(a) 注射器は,あらかじめ洗浄し,乾燥した後,デシケーター中に放冷する。
(b) デシケーターから注射器を取り出し,ゴム栓を注射針の先端に付ける。
(c) ゴム栓を外し,試料容器中の試料の液面からできる限り深く注射針を入れて,注射器の内筒を静か
に引き,注射器の容量の約101まで試料を採取する。
(d) 注射器を試料容器から引き出し,針を上向きにして空気を除いた後,注射針の先端にゴム栓を付け,
軽く圧力を加えて注射器のすり合わせ部分に試料を行きわたらせた後,試料を排出する。この共洗
いを2,3回行う。
36
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(e) 試料を採取量の目盛以上まで徐々に取り,注射針を上にして目盛まで静かに試料を排出し,注射器
の内部の空気を追い出す。直ちに注射針の先端にゴム栓を付ける。
(f) 試料を採取した注射器の質量を0.1mgのけたまで量る。
(g) 注射針に装着したゴム栓を外してストッパーに刺し,内筒を押して滴定槽内に試料を注入する。
(h) 注射針の先端に再び同じゴム栓を付け,その質量を0.1mgのけたまで量る。
(3) 全量ピペットを用いる場合
(a) 全量ピペットはあらかじめ洗浄し,乾燥する。
(b) 試料で共洗い(26)を2回以上行う。
注(26) 試料の吸引は,安全スポイト又はゴムスポイトを用いて行い,口で吸引してはならない。
(c) 試料を素早く採取し(26),標線に合わせる。
(d) 滴定槽の試料注入口の栓を開けて試料を槽内に注入し,直ちに栓をする。このとき,試料注入のた
め栓が開いていた時間を測定し,記録しておく。
(e) 体積から,試料の質量を求めるときは,次の式によって算出する。
S=D×Vs
ここに,
S: 試料の質量 (g)
D: 試料採取時の温度で測定した密度 (g/ml)
Vs: 試料の体積 (ml)
(4) 滴瓶を用いる場合
(a) スポイトと滴瓶は,あらかじめ洗浄し,約105℃で1時間以上乾燥した後,デシケーター中に放冷
する。
(b) スポイトに試料を吸い上げ,2回以上共洗いする。
(c) スポイトに内容量の約60%の試料を取る。
(d) 外筒に装着し,その質量を0.1mgのけたまで量る。
(e) 滴定槽の試料注入口の栓を開けて,スポイトから試料を滴加し(27),直ちに栓をする。このとき,試
料注入のため栓が開いていた時間を測定し,記録しておく。
注(27) 滴加する滴数は,あらかじめ予想水分から決めておく。
(f) スポイトを再び外筒に装着し,質量を0.1mgのけたまで量る。
(5) 固体試料採取器を用いる場合
(a) 固体試料採取器は,あらかじめ洗浄し,約105℃で1時間以上乾燥して,デシケーター中に放冷す
る。
(b) 試料を採取器に採取し,質量を0.1mgのけたまで量る。高粘度半固体試料採取器を用いるときは,
さじに試料を取り,外筒に装着後,質量を0.1mgのけたまで量る。
(c) 滴定槽の試料注入口の栓を開けて試料を投入し,直ちに栓をする。このとき,試料投入のため栓が
開いていた時間を測定し,記録しておく。
なお,高粘度半固体試料採取器を用いるときは,採取した試料が付着した状態のさじを滴定溶剤
に浸し,試料を溶かす。
(d) 試料投入後の固体試料採取器の質量を0.1mgのけたまで量る。
9.1.4.5
操作 操作は,直接滴定又は逆滴定によって行う。
(1) 直接滴定
(1.1) 試料の滴定
37
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(a) 滴定溶剤20〜50mlを滴定槽に入れ,かき混ぜながらカールフィッシャー試薬で終点まで滴定し,
滴定槽内を無水状態にする。このときの滴定量は,読み取る必要はない。
(b) この滴定溶剤をかき混ぜながら,9.1.4.4によって滴定槽内に試料を注入又は投入する。
(c) 滴定槽内の溶液をかき混ぜて,試料を溶解する。試料が溶解しないときは,一定時間かき混ぜて水
分を滴定溶剤に抽出する。
(d) この溶液をよくかき混ぜながら,カールフィッシャー試薬で終点まで滴定する。
(1.2) 空試験 試料採取に全量ピペット,滴瓶,固体試料採取器を用いたときは,次によって空試験を行
う。
(a) 試料滴定後の滴定槽内の溶液をかき混ぜながら,カールフィッシャー試薬で終点まで滴定し,滴定
槽内の溶液を無水状態にする。このときの滴定量は,読み取る必要はない。
(b) この溶液をかき混ぜながら,滴定槽の試料注入口の栓を試料採取のために開いた時間だけ開き,再
び閉じる。
(c) これをかき混ぜながら,カールフィッシャー試薬で終点まで滴定する。
(2) 逆滴定
(2.1) 試料の滴定
(a) 滴定溶剤20〜50mlを滴定槽に入れ,かき混ぜながらカールフィッシャー試薬で終点まで滴定し,
滴定槽内を無水状態にする。このときの滴定量は,読み取る必要はない。
(b) この滴定溶剤をかき混ぜながら,9.1.4.4によって滴定槽内に試料を注入又は投入する。
(c) 滴定槽内の溶液をよくかき混ぜて,試料を溶解する。試料が溶解しないときは,一定時間かき混ぜ
て水分を滴定溶剤に抽出する。
(d) この溶液をかき混ぜながら,これにカールフィッシャー試薬を必要な体積より更に数ml過剰に加
え,加えたカールフィッシャー試薬の量を読み取る。
(e) この溶液をかき混ぜながら,水−メタノール溶液で過剰のカールフィッシャー試薬を終点まで逆滴
定する。
(2.2) 空試験 試料採取に全量ピペット,滴瓶,固体試料採取器を用いたときは,次によって空試験を行
う。
(a) 試料滴定後の滴定槽内の溶液をかき混ぜながら,カールフィッシャー試薬で終点まで滴定し,滴定
槽内の溶液を無水状態にする。このときの滴定量は,読み取る必要はない。
(b) この溶液をかき混ぜながら,滴定槽の試料注入口の栓を試料採取のために開いた時間だけ開き,再
び閉じる。
(c) これをかき混ぜながら,カールフィッシャー試薬で終点まで滴定する。
9.1.4.6
計算 水分は,次の式のいずれかによって算出する。
(1) 直接滴定
(a) 空試験を行わない場合
100
103×
×
×
=S
V
F
W
ここに, W: 水分 (%)
F: カールフィッシャー試薬の力価 (mg/ml)
V: 滴定に用いたカールフィッシャー試薬の量 (ml)
S: 試料の質量 (g)
38
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(b) 空試験を行った場合
(
)100
103×
×
−
=S
B
V
F
W
ここに, W: 水分 (%)
F: カールフィッシャー試薬の力価 (mg/ml)
V: 滴定に用いたカールフィッシャー試薬の量 (ml)
B: 空試験に用いたカールフィッシャー試薬の量 (ml)
S: 試料の質量 (g)
(2) 逆滴定
(a) 空試験を行わない場合
(
)(
)100
103
×
×
×
−
×
=
S
V
f
V
F
W
m
a
ここに,
W: 水分 (%)
F: カールフィッシャー試薬の力価 (mg/ml)
Va: 加えたカールフィッシャー試薬の量 (ml)
Vm: 滴定に用いた水−メタノール溶液の量 (ml)
f: 水−メタノール溶液の水の濃度 (mg/ml)
S: 試料の質量 (g)
(b) 空試験を行った場合
(
)
[
](
)100
103
×
×
×
−
−
×
=
S
V
f
B
V
F
W
m
a
ここに, W: 水分 (%)
F: カールフィッシャー試薬の力価 (mg/ml)
Va: 加えたカールフィッシャー試薬の量 (ml)
B: 空試験に用いたカールフィッシャー試薬の量 (ml)
Vm: 滴定に用いた水−メタノール溶液の量 (ml)
f: 水−メタノール溶液の水の濃度 (mg/ml)
S: 試料の質量 (g)
9.1.5
電量滴定法
9.1.5.1
要旨 この方法は,電解セルに電解液と試料を入れ,電解電流を流して電解を行い,生成したよ
う素と試料中の水をカールフィッシャー反応させ,滴定の終点までに要した電気量から水分量を求める方
法である。終点は,電流制御電圧検出方式によって検出する。
備考 電量滴定法は,それ自身電極反応をするアニリン,フェノールなどを含む試料,不溶物などに
よって隔膜を閉そく(塞)し,又は電流効率に影響を与える試料には適用できない。特に,固
体試料の場合は,完全に溶解していることを確認しなければならない。
9.1.5.2
装置及び器具 装置及び器具は,次のとおりとする。
(1) 電量滴定装置 滴定部,制御部及び表示・記録部で構成する自動滴定装置。構成の一例を図38に示す。
滴定装置は,JIS K 0113によるほか,次による。
(a) 電解セル 容量約200mlのガラス製容器で,それぞれ白金網電極を備えた陽極室,陰極室の2室か
ら構成し,両室は,セラミック又はイオン交換膜などの隔膜で仕切る。試料採取にマイクロシリン
ジ又は注射器を用いるときは,試料注入口にはパッキン付ステンレス鋼製又は四ふっ化エチレン樹
脂製のストッパーを装着する。電解セルには,検出器及びシリカゲルなどの乾燥剤を入れた乾燥管
を取り付ける。電解セルの一例を図39に示す。回転子は,滴定時に電解セルに入れる。
備考 滴定装置の校正 電解セル内の陽極液をかき混ぜながら,終点まで電量滴定して電解セル内を
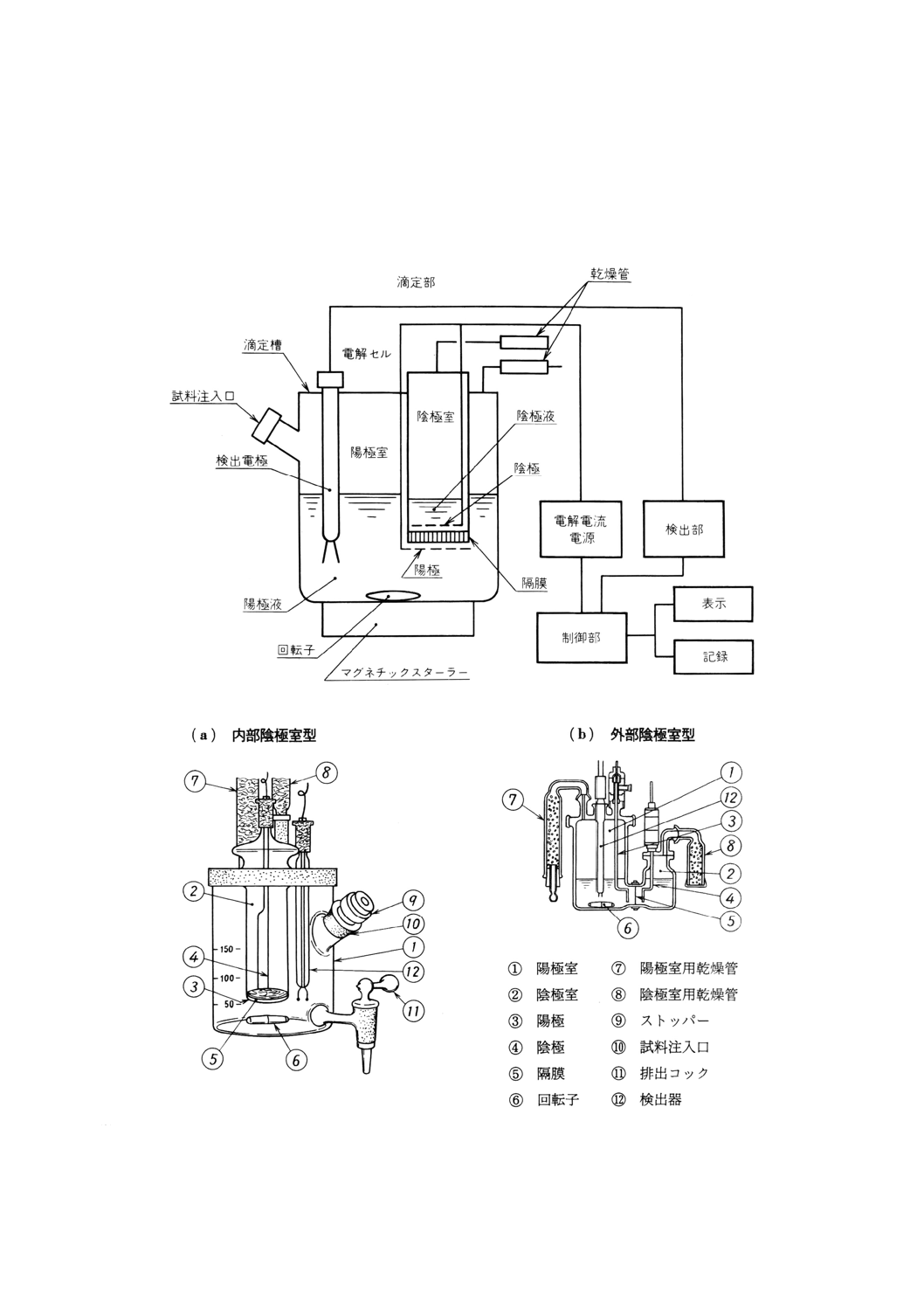
39
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
無水状態とした後,マイクロシリンジを用いて水5μlを取り,陽極液に注入し,終点まで電量
滴定を行って水分を求める。この操作を2回以上繰り返して行い,求めた水分量が5 000±750μg
の範囲内にあることを確認する。この範囲内にないときは,電解液の入れ替え又は電極及び隔
膜の洗浄などを行った後,校正を行う。
図38 電量滴定装置の構成の一例
図39 電解セルの一例
(2) 試料採取器 9.1.4.2に規定するマイクロシリンジ,注射器又は固体試料採取器。
(3) はかり(天びん) 9.1.4.2(3)による。
40
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.1.5.3
試薬 試薬は,次のとおりとする。
参考 電解液及び水−アルコール溶液は,市販品を用いてもよい。
(1) 電解液 電解液は,次の陽極液と陰極液を組み合わせて用いる。
(a) 陽極液 よう化物イオン,二酸化硫黄,ピリジン又はそれに代わる塩基などとメタノールなどの有
機溶剤との混合溶液(28)。
注(28) アルデヒド類,ケトン類などメタノールと反応する化合物を含む試料に対しては,メタノール
の代わりにクロロホルム,2−メトキシエタノール又はプロピレンカーボネートなどを用いる。
(b) 陰極液 二酸化硫黄,ピリジン又はそれに代わる塩基などとメタノールなどの有機溶剤との混合溶
液(28)。
(2) 水−アルコール溶液 2−メトキシエタノールに水を加え,その1mlが水約4mgを含むように調製し
たもの。微量の水の添加に用いる。
9.1.5.4
操作 操作は,次のとおり行う。
(a) 電解セルの陽極室には陽極液を約100ml,陰極室には陰極液を約5ml入れる。電解セルを密封し,
組み立てた後,滴定装置に取り付ける。
(b) 電解セル内の陽極液をかき混ぜながら終点まで電量滴定し(29),電解セル内を無水状態にする。この
ときの滴定量は,読み取る必要はない。
注(29) 陽極液に遊離よう素が存在している場合には,電解電流が流れないので,水又は水−アルコー
ル溶液を加え,水が2 000〜3 000μg程度過剰になるようにするとよい。
(c) 9.1.4.4によって採取した試料をストッパーを通して又は注入口の栓を開けて,電解セル中の陽極液
に注入又は投入する。
(d) 陽極液をよくかき混ぜて,試料を溶解する。
(e) 陽極液をかき混ぜながら終点まで電量滴定し,水分の表示値を読み取る。
9.1.5.5
計算 水分は,次の式によって算出する。
100
106×
×
S
G
W=
ここに, W: 水分 (%)
G: 水分の表示値 (μg)
S: 試料の質量 (g)
9.1.6
水分気化法
9.1.6.1
要旨 この方法は,乾燥した窒素又は空気の気流中で試料を加熱して水分を気化させ,滴定溶剤
又は電解液に捕集してカールフィッシャー滴定を行い,その滴定量から水分量を求める方法である。カー
ルフィッシャー滴定法は,水分量に応じて容量滴定法又は電量滴定法のいずれかを用いる。
備考1. 加熱によって妨害物質が揮散する試料には適用しない。ただし,加熱温度,滴定溶剤,電解
液などの試験条件を選ぶことによって,適用可能となる場合がある。
2. 適切な加熱温度を設定することによって,付着水(遊離水)と化合水(結晶水)の分別定量
が可能である。
9.1.6.2
装置及び器具 装置及び器具は,次のとおりとする。
(1) 水分気化装置(21) 気化部,乾燥部及びキャリヤーガス供給部から構成する装置。構成の一例を図40
に示す。
(2) 気化部 加熱管,加熱炉及び温度制御装置などで構成する。
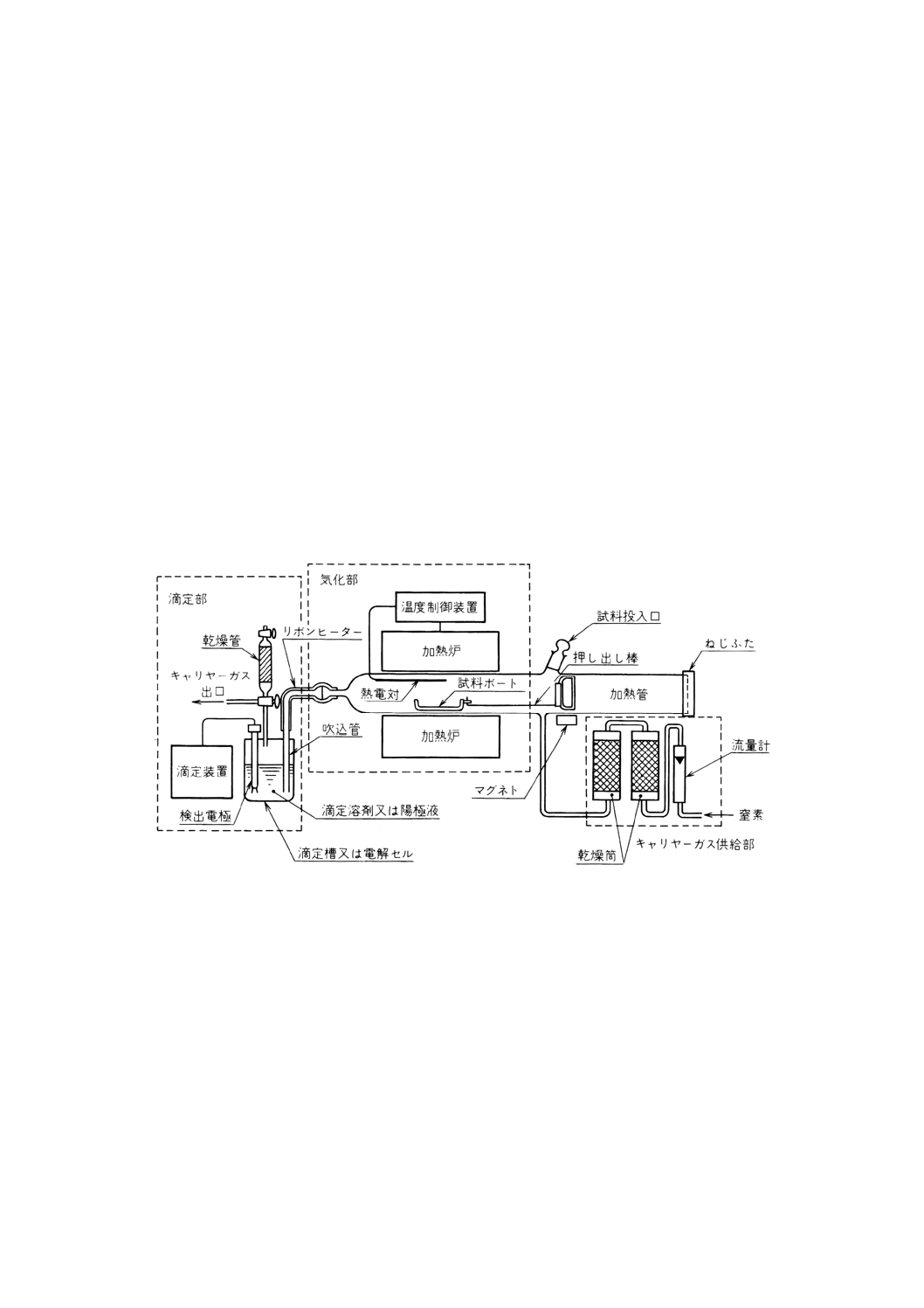
41
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(a) 加熱炉 試験温度に保持できるもの。加熱炉内の温度は,試料ボートに近接した場所の温度分布を
熱電対などを用いて測り,加熱帯の温度が均一になっていることをあらかじめ確認しておく。
参考 市販の水分気化用加熱炉には,最高使用温度が300℃までのものと1 000℃までのものがある。
また,両方式を一つの炉に直列に組み合わせたものがあり,300℃までの加熱炉で付着水の測
定又は除去を行い,1 000℃までの加熱炉で化合水の測定を行うなどの使い方ができる。
(b) 加熱管 両端が開放され,内径約20〜30mm,長さ約400〜1 000mmのガラス(30)又は石英ガラス製
の直管で,試料投入口,キャリヤーガス供給口,吹込管との接続口及び試料ボートの出入口を備え
たもので,石英ガラス製などの押し出し棒(31)を内部に挿入できるようにする。
注(30) 300℃までの加熱炉に用いる。
(31) 押し出し棒の先は,かぎ型にして試料ボートを引っかけ,接続できるようにしておき,更に,
マグネットを用いて駆動し,押し出し棒全体が加熱管の中に入るようにしておくと,押し出し
棒に吸着した水分による誤差を小さくすることができる。
(c) 試料ボート ガラス又は石英ガラス製の平底ボートで,試料を入れたとき,約半量の容積を占める
大きさのもの。試料ボートの一端には,押し出し棒とつなぐことのできる取っ手を付ける。一例を
図41に示す。
図40 水分気化装置の構成の一例
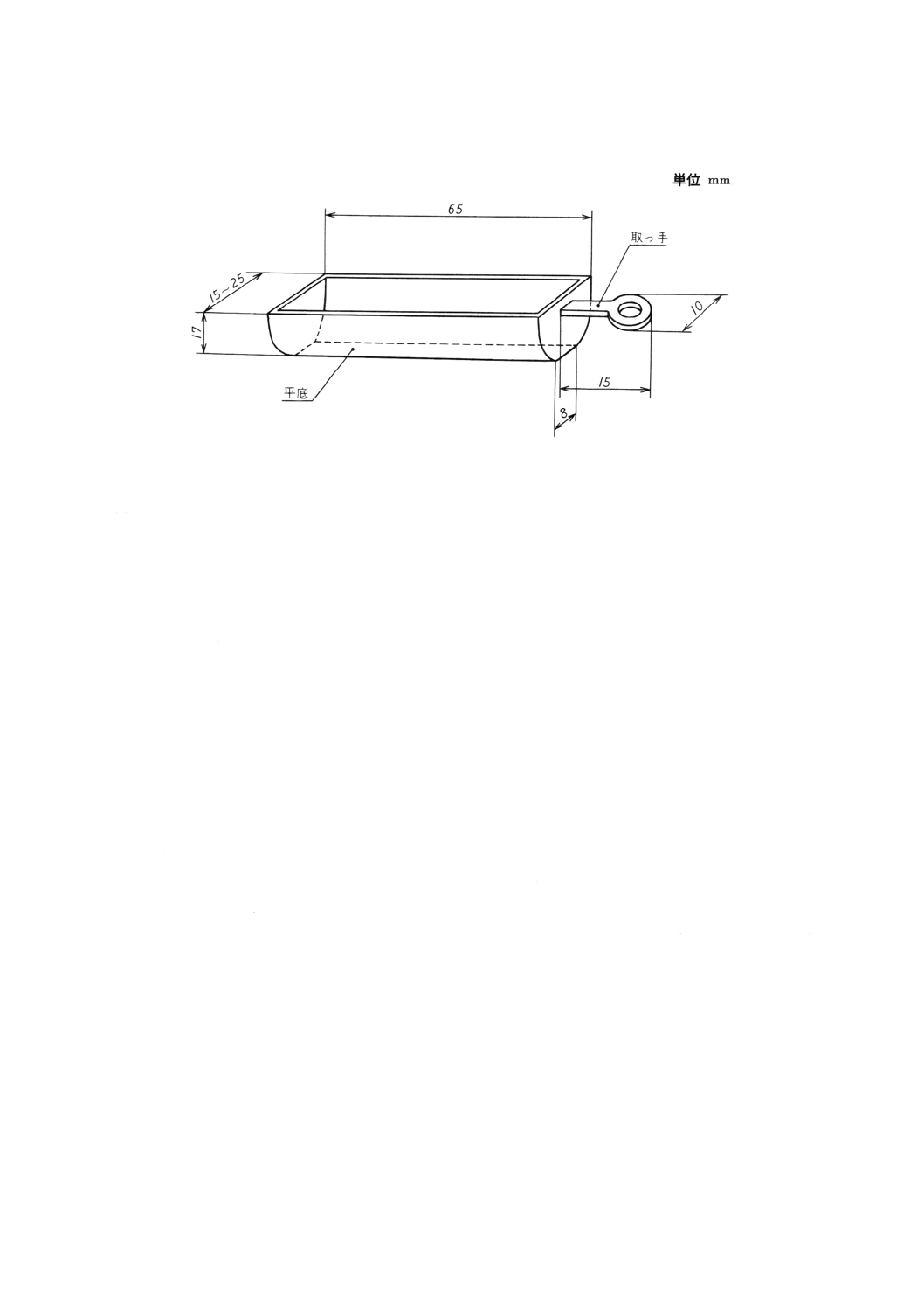
42
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図41 試料ボートの一例
(3) キャリヤーガス供給部 キャリヤーガス源,乾燥筒及び流量計などで構成する。
(a) キャリヤーガス源 ボンベ又はコンプレッサー。
(b) 乾燥筒 乾燥剤を入れ,1個又は2個を直列に接続したもの。
(c) 流量計 500ml/minまで測定できるもの。
(4) 滴定装置 9.1.4.2(1)又は9.1.5.2(1)による。ただし,滴定槽又は電解セルに吹込管を付け,加熱管と接
続する。吹込管の露出部分にはリボンヒーターなどを巻き,加熱できるようにする。
(5) 試料採取器 9.1.4.2(2)による。
(6) はかり(天びん) 9.1.4.2(3)による。
9.1.6.3
試薬 試薬は,次のとおりとする。
(1) キャリヤーガス 窒素又は空気(32)。
注(32) 空気によって変質しない試料の場合に用いる。
(2) 乾燥剤 キャリヤーガスを十分に乾燥し,気化装置内の無水状態を維持できる性能をもつもので,シ
リカゲル,合成ゼオライト,五酸化りんなどを用いる。
(3) 滴定溶剤 滴定溶剤は,次のいずれかを用い,いずれも水分は0.05%以下とする。
参考 滴定溶剤は,市販品を用いてもよい。
(a) メタノール 9.1.4.3(1)による。
(b) メタノール−エチレングリコール混合溶剤 メタノールとJIS K 8105に規定するエチレングリコー
ルとを体積比1 : 1に混合したもの。
(c) メタノール−プロピレングリコール混合溶剤 メタノールとJIS K 8837に規定するプロピレングリ
コールとを体積比3 : 1に混合したもの。
(4) カールフィッシャー試薬 9.1.4.3(7)による。
(5) 電解液 9.1.5.3(1)による。
9.1.6.4
操作 操作は,次のとおり行う。
(1) 装置の準備
(a) 容量滴定装置を用いるときは,滴定槽へ滴定溶剤を約150ml入れる。電量滴定装置を用いるときは,
電解セルの陽極室に陽極液を約150ml,陰極室に陰極液を約10ml入れる。
(b) 滴定装置に吹込管を付けて,加熱管と接続する。
43
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(c) キャリヤーガスの流量を100〜300ml/minに設定して系内に流す。
(d) 温度制御装置を試験温度に設定し,加熱炉を昇温する。
(e) 試料ボートを加熱管の中に入れて加熱炉の中央部にくるように置き,規定の温度で空焼きをする。
(f) 加熱管内及び滴定系内を無水状態(33)にする。
注(33) 容量滴定法のときは9.1.4.4,電量滴定法のときは9.1.5.4による。
(g) 空焼きした試料ボートを試料投入口の真下に置き,室温まで放冷する。
(2) 試料の投入
(a) 試料を固体試料採取器に採取し,0.1mgのけたまで量る。
(b) 試料投入口を開けて,試料を試料ボートに投入する。
(c) 直ちに試料投入口を閉じる。
(d) 固体試料採取器の質量を0.1mgのけたまで量る。
(3) 滴定
(a) 試料を入れた試料ボートを加熱炉の均一加熱帯のほぼ中央部に置く。
(b) 3〜5分後(34)に,9.1.4.5又は9.1.5.4によって滴定を始め,試料の加熱及びキャリヤーガスの送入を
続けながら終点まで滴定する。
注(34) 滴定は,水分の一部又は全部が気化し,滴定溶剤に捕集された後開始する。通常,この程度の
時間でよいが,あらかじめ最適時間を測定しておくのがよい。
9.1.6.5
計算 計算は,容量滴定法を用いるときは9.1.4.6によって,電量滴定法を用いるときは9.1.5.5
による。
9.2
乾燥減量法
9.2.1
要旨 この方法は,試料を約105℃で恒量になるまで加熱乾燥して乾燥後の減量を量り,その量を
水分とする方法である。この方法は,加熱に安定な固体試料に適用する。乾燥減量による水分試験は,JIS
K 0068の5.(乾燥減量法)によるほか,次のとおりとする。
備考 乾燥減量法による水分試験は,水以外の揮発性物質又は加熱による化学変化で生成した揮散す
る物質も水分として定量されるので,注意が必要である。
9.2.2
装置及び器具 装置及び器具は,次のとおりとする。
(1) はかり瓶 JIS R 3503に規定する平形はかり瓶。容量は,試料を入れたとき,その厚さが5mm以下に
なるもの。
(2) 乾燥器 105±2℃に保持できるもの。
(3) デシケーター JIS R 3503に規定するもので,乾燥剤としてシリカゲルを入れたもの。
(4) はかり(天びん) 化学はかり又は電子はかり。
9.2.3
試料 大きな結晶又は塊のときは,粉砕し,粒径2mm以下にして試料とする。このとき,大気中
から吸湿し又は水分の損失がないように注意する。
9.2.4
操作 操作は,次のとおり行う。
(1) はかり瓶を105±2℃に調節した乾燥器で乾燥し,デシケーター中で放冷後,質量を0.1mgのけたまで
量る。恒量(35)になるまでこの操作を繰り返す。
注(35) 試料を乾燥し,放冷後,質量を測定する操作を2回繰り返したとき,1回目の質量に対して1回目
と2回目の質量の差が
000
10
10以下のとき,恒量とする。ただし,試料の質量が0.3g以下のときは,
1回目と2回目の質量の差が0.3mg以下のとき,恒量とする。
(2) 試料約10gをはかり瓶に取り,試料の表面を平らにならしてから,質量を0.1mgのけたまで量る。
44
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 試料を入れたはかり瓶を105±2℃に調節した乾燥器に入れる(36)。はかり瓶のふたは,少しずらして
おくか又は外して同時に乾燥する。
注(36) 試料が約105℃より低い温度で融解するときは,その温度より約10℃低い温度で1〜2時間加熱後,
105℃で加熱する。
(4) 2〜4時間乾燥後,はかり瓶とふたとを速やかにデシケーターに移し,放冷する。放冷後,はかり瓶に
ふたをして質量を0.1mgのけたまで量る。
(5) 恒量(35)になるまで(3)及び(4)を繰り返す。この場合,乾燥時間は約1時間とする。
9.2.5
計算 水分は,次の式によって算出する。
100
3
1
2
1
×
−
−
=
S
S
S
S
W
ここに, W: 水分 (%)
S1: 乾燥前の試料とはかり瓶の質量 (g)
S2: 乾燥後の試料とはかり瓶の質量 (g)
S3: はかり瓶の質量 (g)
10. 灰分試験方法
10.1 要旨 この方法は,試料を徐々に加熱して灰化した後強熱し,強熱後の残分の質量を量ることによ
って灰分を求める方法である。この方法は,容易に灰化ができる有機物試料に適用する。灰分試験は,JIS
K 0067の4.4.1(1)(第1法 灰化後強熱する方法)によるほか,次のとおりとする。
10.2 装置及び器具 装置及び器具は,次のとおりとする。
(1) 電気炉 600〜700℃に保持できるもの。
(2) 熱板 電気加熱式のもの。
(3) デシケーター JIS R 3503に規定するもので,乾燥剤としてシリカゲルを入れたもの。
(4) はかり(天びん) 化学はかり又は電子はかり。
10.3 操作 操作は,次のとおり行う。
(1) 恒量(37)にした るつぼ又は蒸発皿に,試料を0.1mgのけたまで量り取る。
注(37) 試料を乾燥し,放冷後,質量を測定する操作を2回繰り返したとき,1回目の質量に対して1回目
と2回目の質量の差が
000
10
10以下のとき,恒量とする。ただし,試料の質量が0.3g以下のときは,
1回目と2回目の質量の差が0.3mg以下のとき,恒量とする。
(2) 試料を入れた るつぼ又は蒸発皿を熱板上で徐々に加熱し,試料の大部分が揮散,分解又は炭化した後,
電気炉に入れ,徐々に温度を上げて灰化する。灰化温度は,特に規定しない限り600〜700℃とする。
炭化物の灰化が困難な試料については,放冷後水で潤した後,再び徐々に温度を上げて加熱する。
(3) 電気炉で1時間強熱する。
(4) 電気炉から取り出した るつぼ又は蒸発皿を速やかにデシケーターに移し,放冷後,その質量を0.1mg
のけたまで量る。
(5) 恒量(37)になるまで(3),(4)を繰り返す。
10.4 計算 灰分は,次の式によって算出する。
100
3
1
3
2
×
−
−
=
W
W
W
W
D
ここに,
D: 灰分 (%)
W1: 強熱前の試料とるつぼ又は蒸発皿の質量 (g)
45
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
W2: 強熱後の試料とるつぼ又は蒸発皿の質量 (g)
W3: るつぼ又は蒸発皿の質量 (g)
11. 滴定方法
11.1 滴定方法の種類 滴定方法には,次の種類がある。
(1) ニトロン化滴定法
(2) ジアゾ化滴定法
(a) 直接法(一般法)
(b) 直接法(別法)
(c) 逆滴定法
(d) 還元法
(e) 零電流電位差滴定法
(f) 定電圧分極電流滴定法
(g) 定電流分極電位差滴定法
(3) カップリング滴定法
(4) 臭素化滴定法(逆滴定法)
(5) 中和滴定法
参考 滴定方法は,上記のほかにJIS K 0113に規定する方法もある。
11.2 試薬 試薬は,次のとおりとする。
(1) 塩酸 JIS K 8180に規定する特級。
(2) 炭酸ナトリウム(無水) JIS K 8005に規定する容量分析用標準物質。
(3) 硫酸 JIS K 8951に規定する特級。
(4) 飽和水酸化バリウム溶液 JIS K 8577に規定する特級約5gを水100mlに溶解したもの。
(5) アミド硫酸 JIS K 8005に規定する容量分析用標準物質。
(6) 水酸化ナトリウム JIS K 8576に規定する特級。
(7) 亜硝酸ナトリウム JIS K 8019に規定する特級。
(8) 臭素酸カリウム JIS K 8530に規定する特級。
(9) チオ硫酸ナトリウム五水和物 JIS K 8637に規定する特級。
(10) 炭酸ナトリウム(無水) JIS K 8625に規定する特級。
(11) 3−メチル−1−ブタノール JIS K 8051に規定する特級。
(12) よう素酸カリウム JIS K 8005に規定する容量分析用標準物質。
(13) よう化カリウム JIS K 8913に規定する特級。
(14) エタノール (95) [エチルアルコール (95)] JIS K 8102に規定する特級。
(15) 1 mol/l水酸化カリウムエタノール溶液 JIS K 8001に規定するもの。
(16) 過塩素酸マグネシウム JIS K 8228に規定する乾燥用。
(17) スルファニル酸 JIS K 8586に規定する特級。
(18) アンモニア水 JIS K 8085に規定する特級。
(19) p-トルイジン(4−アミノトルエン) 市販の試薬を用いる。
(20) 臭化カリウム JIS K 8506に規定する特級。
(21) アミド硫酸アンモニウム溶液 (100g/l) JIS K 8588に規定する特級を用いて調製したもの。
46
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(22) p-ニトロアニリン JIS K 8708に規定するもの。
(23) m-トルイレンジアミン 市販のもの。
(24) エタノール (99.5) [エチルアルコール (99.5)] JIS K 8101に規定する特級。
(25) 活性炭 市販のもの。
(26) 五酸化二りん(無水りん酸) JIS K 8342に規定する特級。
(27) メタノール JIS K 8891に規定する特級。
(28) 酢酸 JIS K 8355に規定する特級。
(29) ブロモフェノールブルー JIS K 8844に規定する特級。
(30) コンゴーレッド JIS K 8352に規定する特級。
(31) メチルオレンジ JIS K 8893に規定する特級。
(32) ブロモチモールブルー JIS K 8842に規定する特級。
(33) クレゾールレッド JIS K 8308に規定する特級。
(34) フェノールフタレイン JIS K 8799に規定する特級。
(35) でんぷん(溶性) JIS K 8659に規定する特級。
(36) 4−アミノ−5−ヒドロキシ−2,7−ナフタレンジスルホン酸モノナトリウム塩(H酸モノナトリウム
塩) JIS K 4134に規定するもの。
(37) 炭酸ナトリウム溶液 (100g/l) JIS K 8625に規定する特級を用いて調製したもの。
(38) 亜鉛粉末 JIS K 8013に規定する特級。
(39) 塩化ナトリウム JIS K 8150に規定する特級。
(40) よう化カリウム溶液 (100g/l) JIS K 8913に規定する特級を用いて調製したもの。
(41) ソーダ石灰 JIS K 8603に規定する1号。
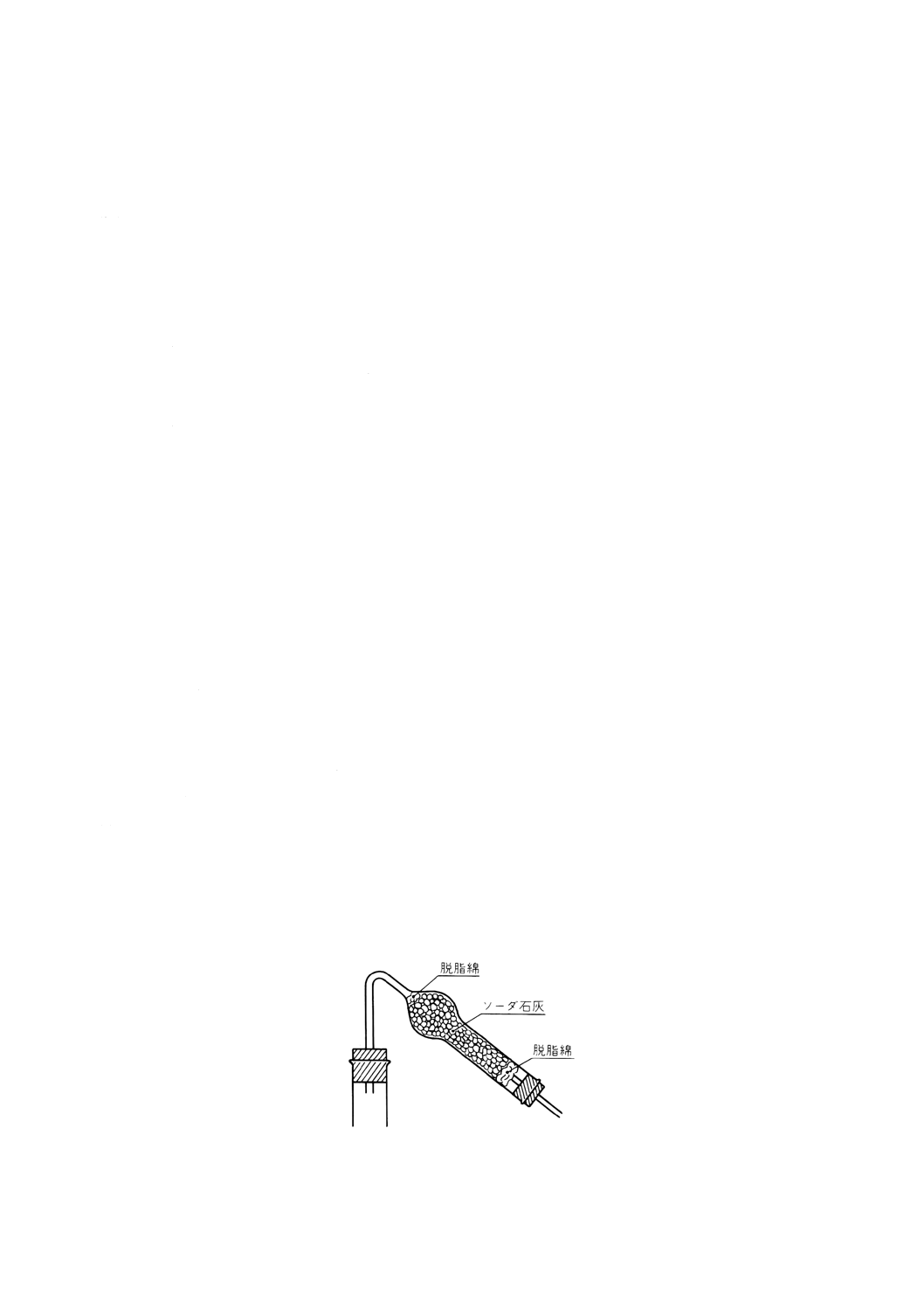
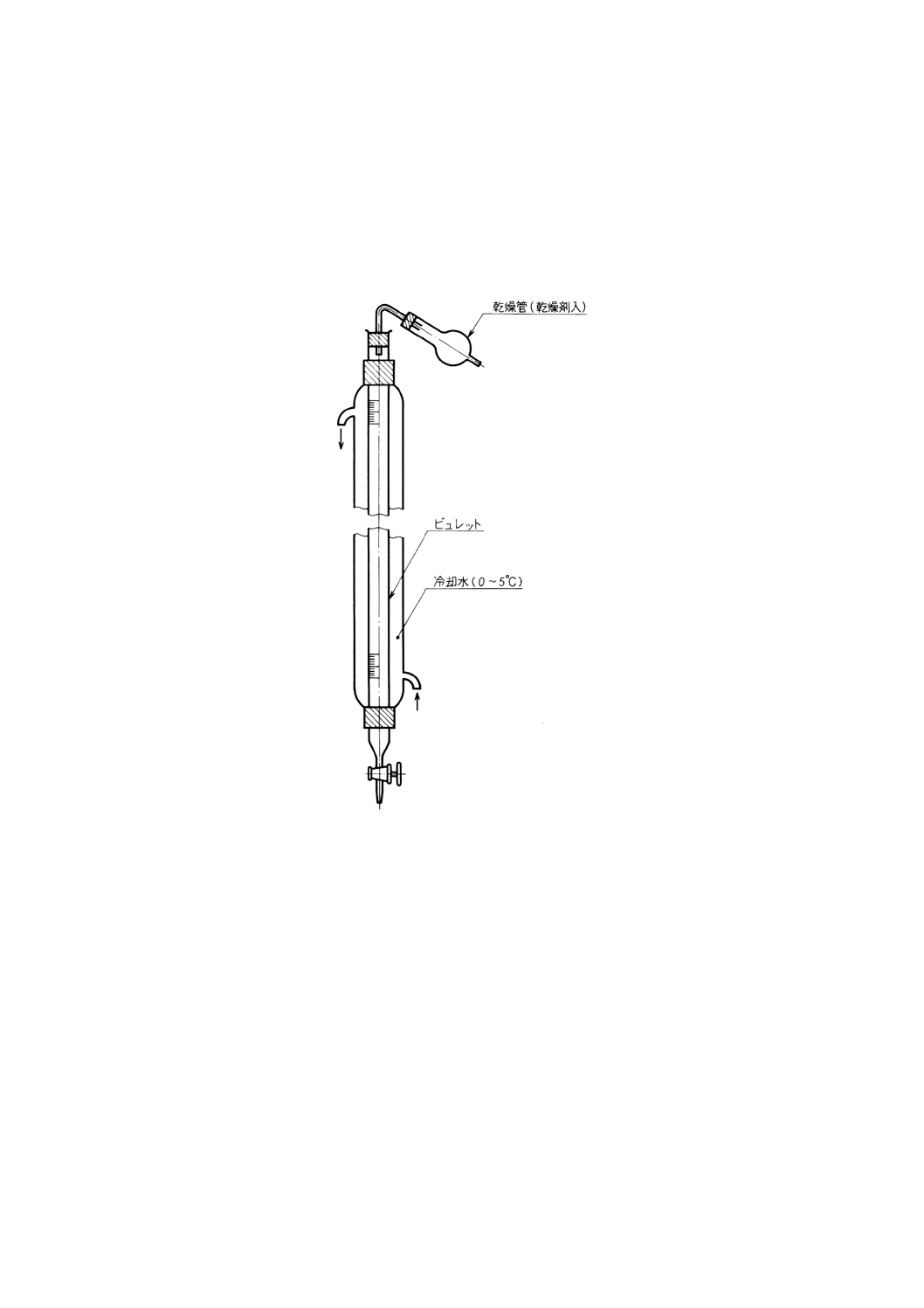
11.3 装置及び器具 装置及び器具は,次のとおりとする。
(1) 電位差滴定装置 JIS K 0113に規定するもの。
(2) 電流滴定装置 JIS K 0113に規定するもの。
(3) 化学はかり JIS K 0050に規定するもの。
(4) 微量化学はかり JIS K 0050に規定するもの。
(5) 気密容器 液体の浸入,内容物の変質,揮散,損失などを防ぐために密閉することができる容器で,
JIS R 3503に規定する広口共栓瓶又は細口共栓瓶,若しくは,JIS Z 1703に規定するポリエチレン瓶
又はこれらと同等の品質のものを用いる。
(6) ソーダ石灰管 図42に示すもので,吸収剤として11.2(41)のソーダ石灰を入れたもの。
図42 ソーダ石灰管の一例
(7) よう素フラスコ JIS R 3503に規定するもの。
47
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(8) 全量フラスコ JIS R 3503に規定するもの。
(9) 着色メスフラスコ 市販のもの。
(10) 減圧デシケーター 図8(2)に示すもので,乾燥剤として過塩素酸マグネシウムを入れたもの。
(11) ろ紙 JIS P 3801に規定する定性分析用2種。
(12) 冷却式ビュレット 図43に一例を示す。
図43 冷却式ビュレットの一例
11.4 滴定用溶液(規定液),指示薬溶液及び試験紙
11.4.1 滴定用溶液(規定液)
11.4.1.1 調製,標定及び保存 滴定用溶液(規定液)は,次に示す方法によって調製,標定及び保存を行
う。ただし,0.1mol/lよりもうすい溶液で個々の製品規格に規定するものは,必要量の0.1mol/l溶液を水で
薄めて作る。この滴定用溶液(規定液)は標定を行わないで,ファクターは0.1mol/lのファクターと薄め
た割合から求める。
(1) 1mol/l {1N} 塩酸 (36.46gHCl/l)
(1.1) 調製 塩酸90mlを量り取り,水を加えて1 000mlとする。
(1.2) 標定
(a) 炭酸ナトリウム(無水)(容量分析用標準物質)をJIS K 8005の4.(乾燥条件)によって乾燥した
後,その1.3〜1.4gを0.1mgのけたまで量り取り,水20mlに溶解する。
(b) ブロモフェノールブルー溶液を指示薬として用い,(1.1)の塩酸で滴定する。この場合,終点付近で
煮沸して二酸化炭素を追い出し,常温まで冷却した後,引き続き滴定を行う。
48
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(c) 滴定の終点は,(1.1)の塩酸を1滴加えて青紫から黄色に変わる点とする。
(1.3) 計算 1mol/l {1 N} 塩酸のファクターは,次の式によって算出し,小数点以下4けたに丸める。
100
052994
.0
P
V
S
F
×
×
=
ここに, F: 1 mol/l {1N} 塩酸のファクター
V: 滴定に要した(1.1)の塩酸の量 (ml)
S: 炭酸ナトリウム(無水)(容量分析用標準物質)の質量 (g)
P: 炭酸ナトリウム(無水)(容量分析用標準物質)の純度 (%)
(1.4) 保存 気密容器に入れて保存する。
(2) 0.1mol/l {0.1N} 塩酸 (3.646gHCl/l)
(2.1) 調製 塩酸9mlを量り取り,水を加えて1 000mlとする。
(2.2) 標定 (1)の1mol/l {1N} 塩酸による。ただし,炭酸ナトリウム(無水)(容量分析用標準物質)は,
0.13〜0.14gを0.1mgのけたまで量り取る。
(2.3) 計算 0.1 mol/l {0.1N} 塩酸のファクターは,次の式によって算出し,小数点以下4けたに丸める。
100
0052994
.0
P
V
S
F
×
×
=
ここに, F: 0.1 mol/l {0.1 N} 塩酸のファクター
V: 滴定に要した(2.1)の塩酸の量 (ml)
S: 炭酸ナトリウム(無水)(容量分析用標準物質)の質量 (g)
P: 炭酸ナトリウム(無水)(容量分析用標準物質)の純度 (%)
(2.4) 保存 (1)の1mol/l {1N} 塩酸と同じ。
(3) 0.05mol/l {0.1N} 硫酸 (4.904gH2S04/l)
(3.1) 調製 硫酸3mlを量り取り,かき混ぜながら,水1 000mlに徐々に加えて放冷する。
(3.2) 標定
(a) 炭酸ナトリウム(無水)(容量分析用標準物質)をJIS K 8005の4.(乾燥条件)によって乾燥した
後,その0.13〜0.16gを0.1mgのけたまで量り取り,水20mlに溶解する。
(b) ブロモフェノールブルー溶液を指示薬として用い,(3.1)の硫酸で滴定する。この場合,終点付近で
煮沸して二酸化炭素を追い出し,常温まで冷却した後,引き続き滴定を行う。
(c) 滴定の終点は,(3.1)の硫酸を1滴加えて青紫から黄色に変わる点とする。
(3.3) 計算 0.05mol/l {0.1N} 硫酸のファクターは,次の式によって算出し,小数点以下4けたに丸める。
100
0052994
.0
P
V
S
F
×
×
=
ここに, F: 0.05mol/l {0.1N} 硫酸のファクター
V: 滴定に要した(3.1)の硫酸の量 (ml)
S: 炭酸ナトリウム(無水)(容量分析用標準物質)の質量 (g)
P: 炭酸ナトリウム(無水)(容量分析用標準物質)の純度 (%)
(3.4) 保存 気密容器に入れて保存する。
(4) 1mol/l {1N} 水酸化カリウム溶液 (56.11 gKOH/l)
(4.1) 調製
(a) 水酸化カリウム70gを量り取り,JIS K 8001の3.6(3)(二酸化炭素を含まない水)に規定する二酸
化炭素を含まない水1 000mlに溶解する。
(b) 軽く振り混ぜながら,調製した直後の飽和水酸化バリウム溶液を沈殿が生じなくなるまで少量ずつ
49
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
加える。
(c) 更に強く振り混ぜた後,二酸化炭素を遮るためにソーダ石灰管を取り付けたポリエチレン瓶に入れ,
2〜3日間放置し,その上澄み液を標定に用いる。
(4.2) 標定
(a) アミド硫酸(容量分析用標準物質)をJIS K 8005の4.(乾燥条件)によって乾燥した後,その2.4
〜2.9gを0.1mgのけたまで量り取り,水25mlに溶解する。
(b) ブロモチモールブルー溶液を指示薬として用い,(4.1)の水酸化カリウム溶液で滴定する。
(c) 滴定の終点は,(4.1)の水酸化カリウム溶液を1滴加えて黄色から青に変わる点とする。
(4.3) 計算 1mol/l {1N} 水酸化カリウム溶液のファクターは,次の式によって算出し,小数点以下4け
たに丸める。
100
097095
.0
P
V
S
F
×
×
=
ここに, F: 1 mol/l {1N} 水酸化カリウム溶液のファクター
V: 滴定に要した(4.1)の水酸化カリウム溶液の量 (ml)
S: アミド硫酸(容量分析用標準物質)の質量 (g)
P: アミド硫酸(容量分析用標準物質)の純度 (%)
(4.4) 保存 気密容器(ポリエチレン瓶)に入れ,ソーダ石灰管を取り付けて保存する。
(5) 0.1mol/l {0.1N} 水酸化カリウム溶液 (5.611gKOH/l)
(5.1) 調製 (4)の1mol/l {1N} 水酸化カリウム溶液100mlをJIS K 8001の3.6(3)に規定する二酸化炭素を
含まない水で薄めて1 000mlとする。
(5.2) 標定 (4)の1mol/l {1N} 水酸化カリウム溶液による。ただし,アミド硫酸(容量分析用標準物質)
は,0.24〜0.29gを0.1mgのけたまで量り取る。
(5.3) 計算 0.1mol/l {0.1 N} 水酸化カリウム溶液のファクターは,次の式によって算出し,小数点以下4
けたに丸める。
100
0097095
.0
P
V
S
F
×
×
=
ここに, F: 0.1mol/l {0.1 N} 水酸化カリウム溶液のファクター
V: 滴定に要した(5.1)の水酸化カリウム溶液の量 (ml)
S: アミド硫酸(容量分析用標準物質)の質量 (g)
P: アミド硫酸(容量分析用標準物質)の純度 (%)
(5.4) 保存 (4)の1mol/l {1N} 水酸化カリウム溶液と同じ。
(6) 1mol/l {1N} 水酸化ナトリウム溶液 (40.00gNaOH/l)
(6.1) 調製
(a) 水酸化ナトリウム42gを量り取り,JIS K 8001の3.6(3)に規定する二酸化炭素を含まない水1 000ml
に溶解する。
(b) 軽く振り混ぜながら,調製した直後の飽和水酸化バリウム溶液を沈殿が生じなくなるまで少量ずつ
加える。
(c) さらに,強く振り混ぜた後,二酸化炭素を遮るためにソーダ石灰管を取り付けたポリエチレン瓶に
入れ,2〜3日間放置し,その上澄み液を標定に用いる。
(6.2) 標定
(a) アミド硫酸(容量分析用標準物質)をJIS K 8005の4.(乾燥条件)によって乾燥した後,その2.4
50
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
〜2.6gを0.1mgのけたまで量り取り,水25mlに溶解する。
(b) ブロモチモールブルー溶液を指示薬として用い,(6.1)の水酸化ナトリウム溶液で滴定する。
(c) 滴定の終点は,(6.1)の水酸化ナトリウム溶液を1滴加えて黄色から青に変わる点とする。
(6.3) 計算 1mol/l {1N} 水酸化ナトリウム溶液のファクターは,次の式によって算出し,小数点以下4
けたに丸める。
100
097095
.0
P
V
S
F
×
×
=
ここに, F: 1 mol/l {1N} 水酸化ナトリウム溶液のファクター
V: 滴定に要した(6.1)の水酸化ナトリウム溶液の量 (ml)
S: アミド硫酸(容量分析用標準物質)の質量 (g)
P: アミド硫酸(容量分析用標準物質)の純度 (%)
(6.4) 保存 気密容器(ポリエチレン瓶)に入れ,ソーダ石灰管を取り付けて保存する。
(7) 0.1mol/l {0.1N} 水酸化ナトリウム溶液 (4.000gNaOH/l)
(7.1) 調製 (6)の1mol/l {1N} 水酸化ナトリウム溶液100mlをJIS K 8001の3.6(3)に規定する二酸化炭素
を含まない水で薄めて1 000mlとする。
(7.2) 標定 (6)の1mol/l {1N} 水酸化ナトリウム溶液による。ただし,アミド硫酸(容量分析用標準物質)
は,0.24〜0.29gを0.1mgのけたまで量り取る。
(7.3) 計算 0.1mol/l {0.1N} 水酸化ナトリウム溶液のファクターは,次の式によって算出し,小数点以下
4けたに丸める。
100
0097095
.0
P
V
S
F
×
×
=
ここに, F: 0.1 mol/l {0.1N} 水酸化ナトリウム溶液のファクター
V: 滴定に要した(7.1)の水酸化ナトリウム溶液の量 (ml)
S: アミド硫酸(容量分析用標準物質)の質量 (g)
P: アミド硫酸(容量分析用標準物質)の純度 (%)
(7.4) 保存 (6)の1mol/l {1N} 水酸化ナトリウム溶液と同じ。
(8) 0.5mol/l {0.5N} 亜硝酸ナトリウム溶液 (34.50gNaN02/l)
(8.1) 調製 亜硝酸ナトリウム35gを量り取り,水に溶解して1 000mlとする。
(8.2) 標定
(a) アミド硫酸(容量分析用標準物質)をJIS K 8005の4.(乾燥条件)によって乾燥した後,その1.0
〜1.2gを0.1mgのけたまで量り取り,水25mlに溶解する。
(b) 塩酸5mlを加えて冷却し,15℃以下に保ちながら(8.1)の亜硝酸ナトリウム溶液で滴定する。
(c) 滴定の終点は,(8.1)の亜硝酸ナトリウム溶液を1滴加えて5分後によう化カリウムでんぷん紙で試
験して,わずかに青が現れる点とする。
(8.3) 計算 0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクターは,次の式によって算出し,小数点以下
4けたに丸める。
100
048548
.0
P
V
S
F
×
×
=
ここに, F: 0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクター
V: 滴定に要した(8.1)の亜硝酸ナトリウム溶液の量 (ml)
S: アミド硫酸(容量分析用標準物質)の質量 (g)
P: アミド硫酸(容量分析用標準物質)の純度 (%)

51
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(8.4) 保存 気密容器に入れて保存する。
(9) 0.1mol/l {0.1N} 亜硝酸ナトリウム溶液 (6.900gNaNO2/l)
(9.1) 調製 (8)の0.5mol/l {0.5N} 亜硝酸ナトリウム溶液200mlを水で薄めて1 000mlとする。
(9.2) 標定 (8)の0.5mol/l {0.5N} 亜硝酸ナトリウム溶液による。ただし,アミド硫酸(容量分析用標準物
質)は,0.20〜0.25gを0.1 mgのけたまで量り取る。
(9.3) 計算 0.1mol/l {0.1N} 亜硝酸ナトリウム溶液のファクターは,次の式によって算出し,小数点以下
4けたに丸める。
100
0097095
.0
P
V
S
F
×
×
=
ここに, F: 0.1 mol/l {0.1N} 亜硝酸ナトリウム溶液のファクター
V: 滴定に要した(9.1)の亜硝酸ナトリウム溶液の量 (ml)
S: アミド硫酸(容量分析用標準物質)の質量 (g)
P: アミド硫酸(容量分析用標準物質)の純度 (%)
(9.4) 保存 (8)の0.5mol/l {0.5N} 亜硝酸ナトリウム溶液と同じ。
(10) 601mol/l {0.1N} 臭素酸カリウム溶液 (2.783gKBrO3/l)
(10.1) 調製 臭素酸カリウム2.8g及び臭化カリウム50gを水に溶解して1 000mlとする。
(10.2) 標定
(a) (10.1)の臭素酸カリウム溶液25mlを共通すり合わせよう素フラスコA型300ml又はB型300ml(以
下,よう素フラスコという。)に正しく取り,よう化カリウム3g及び塩酸 (2+1) 5mlを速やかに加
えて,手早く栓をする。
(b) よう素フラスコのリップの周りに水を入れ,緩やかに振り混ぜ,暗所に2〜3分間放置した後,(11)
の0.1mol/l {0.1N} チオ硫酸ナトリウム溶液で滴定する。この場合,溶液が極くうすい黄色になった
後,指示薬のでんぷん溶液を加える。
(c) 滴定の終点は,0.1mol/l {0.1N} チオ硫酸ナトリウム溶液を1滴加えて青から無色に変わる点とする。
(d) 同一条件で空試験を行い,0.1mol/l {0.1N} チオ硫酸ナトリウム溶液の滴定量を補正する。
(10.3) 計算
60
1mol/l {0.1N} 臭素酸カリウム溶液のファクターは,次の式によって算出し,小数点以下4
けたに丸める。
25
V
f
F
×
=
ここに, F:
60
1mol/l {0.1N} 臭素酸カリウム溶液のファクター
V: 滴定に要した0.1mol/l {0.1N} チオ硫酸ナトリウム溶液の量 (ml)
f: 滴定に要した0.1mol/l {0.1N} チオ硫酸ナトリウム溶液のファク
ター
(10.4) 保存 気密容器(褐色ガラス瓶)に入れて暗所に保存する。
(11) 0.1mol/l {0.1N} チオ硫酸ナトリウム溶液 (15.811gNa2S203/l)
(11.1) 調製
(a) チオ硫酸ナトリウム五水和物26g及び炭酸ナトリウム(無水)0.2gを量り取り,JIS K 8001の3.6(4)
に規定する溶存酸素を含まない水1 000mlに溶解する。
(b) 3−メチル−1−ブタノール10mlを加えて2日間放置した後,標定に用いる。
(11.2) 標定
(a) よう素酸カリウム(容量分析用標準物質)をJIS K 8005の4.(乾燥条件)によって乾燥した後,そ
52
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
の0.10〜0.15gを0.1mgのけたまで量り取り,よう素フラスコに入れ,水25mlを加えて溶解する。
(b) よう化カリウム2g及び硫酸 (1+5) 5mlを速やかに加えて,手早く栓をする。
(c) よう素フラスコのリップの周りに水を入れ,緩やかに振り混ぜ,暗所に5分間放置した後,(11.1)
のチオ硫酸ナトリウム溶液で滴定する。この場合,溶液がごくうすい黄色になってから,指示薬の
でんぷん溶液を加える。
(d) 滴定の終点は,(11.1)のチオ硫酸ナトリウム溶液を1滴加えて青から無色に変わる点とする。
(e) 同一条件で空試験を行い,チオ硫酸ナトリウム溶液の滴定量を補正する。
(11.3) 計算 0.1mol/l {0.1N} チオ硫酸ナトリウム溶液のファクターは,次の式によって算出し,小数点以
下4けたに丸める。
100
0035667
.0
P
V
S
F
×
×
=
ここに, F: 0.1mol/l {0.1N} チオ硫酸ナトリウム溶液のファクター
V: 滴定に要した(11.1)のチオ硫酸ナトリウム溶液の量 (ml)
S: よう素酸カリウム(容量分析用標準物質)の質量 (g)
P: よう素酸カリウム(容量分析用標準物質)の純度 (%)
(11.4) 保存 気密容器に入れて保存する。
(12) 0.1mol/l {0.1N} フタル酸カリウム溶液 [24.231gC5H4 (COOK) 2/l]
(12.1) 調製 フタル酸(38)16.613g(100%として)及びエタノール (95) [エチルアルコール (95)]690ml
を全量フラスコ1 000mlに入れ,これに1mol/l水酸化カリウムエタノール溶液200mlを正しく取っ
て加え(ファクター1.000 0として),緩やかに振り混ぜた後,水を加えて正しく1 000mlとする。
この溶液は,ブロモフェノールブルー溶液を加えたとき青紫,フェノールフタレイン溶液を加え
たとき無色を示し,中性であることが必要である。
注(38) フタル酸の精製及び純度試験方法 無水フタル酸を約100倍の水と煮沸して溶解し,冷却してフ
タル酸を析出させる。この結晶を水から1回再結晶して減圧デシケーター中で恒量になるまで乾
燥し,次によって純度を求める。
フタル酸0.1 gを0.1 mgのけたまで量り取り,JIS K 8001の3.6(3)に規定する二酸化炭素を含
まない水100mlに溶解し,(5)の0.1mol/l {0.1N} 水酸化カリウム溶液40mlを正しく取って加え,
フェノールフタレイン溶液を指示薬として(2)の0.1mol/l {0.1N} 塩酸で滴定し,約1mlを過剰に
加えた後,その合量を正しく量る。次に徐々に煮沸し,冷却して,再び0.1mol/l {0.1N} 水酸化
カリウム溶液で逆滴定を行い,次の式によって純度を算出し,小数点以下2けたに丸める。
(
)
100
008306
.0
40
2
1
×
×
+
−
S
V
V
P=
ここに,
P: 純度 (%)
V1: 添加した0.1mol/l {0.1N} 塩酸の量 (ml)
V2: 逆滴定に要した0.1mol/l {0.1N} 水酸化カリウム溶液の量 (ml)
S: フタル酸の質量 (g)
(12.2) 標定 ファクターは計算値を用い,標定は行わない。
(12.3) 保存 気密容器に入れて保存する。
(13) 0.1mol/l {0.1N} スルファニル酸溶液 (17.319gp-C6H4・SO3H・NH2/l)
(13.1) 調製 スルファニル酸17.4gをアンモニア水約8mlと共に水に溶解して1 000mlとする。
(13.2) 標定
53
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(a) (13.1)のスルファニル酸溶液25mlを正しく取り,水175mlを加えて冷却し,0〜5℃に保ちながら以
下の操作を行う。
(b) (9)の0.1mol/l {0.1N} 亜硝酸ナトリウム溶液滴定の所要量の95〜98% (23.7〜24.5ml) を加え,かき混
ぜながら塩酸4mlを一時に加えて酸性とし,0.1mol/l {0.1N} 亜硝酸ナトリウム溶液で滴定する。
(c) 滴定の終点は,0.1mol/l {0.1N} 亜硝酸ナトリウム溶液を1滴加えて2分後によう化カリウムでんぷ
ん紙で試験して,わずかに青が現れる点とする。
(13.3) 計算 0.1mol/l {0.1N} スルファニル酸溶液のファクターは,次の式によって算出し,小数点以下4
けたに丸める。
25
V
f
F
×
=
ここに, F: 0.1mol/l {0.1N} スルファニル酸溶液のファクター
f: 滴定に要した0.1mol/l {0.1N} 亜硝酸ナトリウム溶液のファクタ
ー
V: 滴定に要した0.1mol/l {0.1N} 亜硝酸ナトリウム溶液の量 (ml)
(13.4) 保存 気密容器に入れて暗所に保存する。
(14) 0.1mol/l {0.1N} p−トルイジンジアゾ溶液
(14.1) 0.5mol/l {0.5N} p−トルイジン原液 (53.58 gp-C6H4・CH3・NH2/l)
(14.1.1) 調製 p−トルイジン(4-アミノトルエン)54gを塩酸104mlと共に水に溶解して1000mlとする。
(14.1.2) 標定
(a) (14.1.1) のp−トルイジン溶液25mlを正しく取り,塩酸20ml及び水175mlを加えて冷却し,0〜5℃
に保ちながら以下の操作を行う。
(b) 臭化カリウム5gを加えて溶解し,かき混ぜながら(8)の0.5mol/l {0.5N} 亜硝酸ナトリウム溶液で滴
定する。
(c) 滴定の終点は,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を1滴加えて10分後によう化カリウムでん
ぷん紙で試験して,わずかに青が現れる点とする。
(14.1.3) 計算 0.5mol/l {0.5N} p−トルイジン原液のファクターは,次の式によって算出し,小数点以下4
けたに丸める。
25
V
f
F
×
=
ここに, F: 0.5mol/l {0.5N} p−トルイジン原液のファクター
f: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクタ
ー
V: 滴定に要した0.5mol/l {0.5 N} 亜硝酸ナトリウム溶液の量 (ml)
(14.1.4) 保存 気密容器に入れて暗所に保存する。
(14.2) 0.1mol/l {0.1N} p−トルイジンジアゾ溶液
(14.2.1) 調整
(a) (14.1)の0.5mol/l {0.5N} p−トルイジン原液50mlを着色メスフラスコ250ml(以下,着色メスフラス
コという。)に正しく取り,臭化カリウム5gを加えて溶解し,これを冷却して0〜5℃に保ちながら
以下の操作を行う。
(b) 別に,(8)の0.5mol/l {0.5N} 亜硝酸ナトリウム溶液55mlを0〜5℃に冷却し,着色メスフラスコを振
り動かしながら,その中に一時に加え,ときどき振り混ぜながら10分間放置する。

54
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(c) 次に,アミド硫酸アンモニウム溶液 (100g/l) 5mlを加え,よく振り混ぜた後,0〜5℃の水を加えて
正しく250mlとする。
(14.2.2) 標定 0.1mol/l {0.1N} p−トルイジンジアゾ溶液のファクターは,(14.1)の0.5mol/l {0.5N} p−トル
イジン原液のファクターを用いる。
標定の必要がある場合は,カップリング法によって行う(備考参照)。
備考 各種ジアゾ溶液のカップリング法による標定は,次のとおり行う。
1. m−トルイレンジアミンの精製 m−トルイレンジアミンをエタノール (99.5) [エチルアル
コール (99.5)]から4回再結晶を行う。この場合,3回目までは活性炭を用い,4回目は熱ろ
過だけとする。結晶は,約60℃で減圧の状態に約6時間保ってアルコール分を追い出した後,
デシケーター中で48時間以上乾燥し,デシケーター中に保存する。この場合,乾燥剤には五
酸化二りんを用いる。
凝固点97.6℃以上,水分(39)0.1%以下のものを精製品とする。
注(39) 水分は,精製品2gを0.1mgのけたまで量り取り,9.1.4.5(1)によって測定する。この場合,
溶解にはメタノール25ml及び酢酸5mlを用いる。
2. 0.102mol/l {0.102N} m−トルイレンジアミン溶液[12.461 gC6H3・CH3・ (NH2) 2/l]
(1) 調製 1.のm−トルイレンジアミン12.461gを1mgのけたまで量り取り,塩酸40mlと共に水
に溶解して正しく1 000 mlとする。
(2) 標定 (1)のm−トルイレンジアミン採取量から水分を差し引いたものを純分として,ファク
ターを算出する。
(3) 保存 この溶液は,使用時に調製する。
3. カップリング滴定法 2.の0.102mol/l {0.102N} m−トルイレンジアミン溶液25mlを正しく取
り,次のカップリング条件によって,11.5.3に準じて滴定する。
カップリング条件
ジアゾ溶液の種類 水添加量
ml
酢酸ナトリウム
溶液 (164g/l)
添加量 ml
酢酸
添加量
ml
温度
℃
終点判定
時間
min
P−トルイジン
100
50
−
15〜20
2
p−ニトロアニリン
50
50
2.5
15
2
(14.2.3) 保存 気密容器に入れ,0〜5℃で光を遮って保存する。この場合,滴定には調製後10時間以内の
ものを用いる。
(15) 0.1mol/l {0.1N} p−ニトロアニリンジアゾ溶液
(15.1) 0.25mol/l {0.25N} p−ニトロアニリン原液 (34.532gp-C6H4・NO2・NH2/l)
(15.1.1) 精製
(a) p-ニトロアニリンをエタノール (99.5) [エチルアルコール (99.5)]で4回再結晶して用いる。この
場合,3回目までは活性炭を用い,4回目は熱ろ過だけとする。
(b) 結晶は,50〜60℃で減圧とし,アルコール分を追い出した後,減圧デシケーター中で24時間以上乾
燥する。
(c) 凝固点147.4℃以上,水分(39)0.1%以下のものを精製品とする。
(15.1.2) 調製 (15.1.1)のp−ニトロアニリン34.5gを塩酸200mlと共に水に溶解して1 000mlとする。
(15.1.3) 標定
55
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(a) (15.1.2)のp−ニトロアニリン溶液50mlを正しく取り,塩酸10mlを加えてかき混ぜた後,水150ml
を加え,これを冷却して0〜5℃に保ちながら以下の操作を行う。
(b) かき混ぜながら,(8)の0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の滴定所要量の95〜98% (23.7〜
24.5ml) をビュレットの先端を液中に浸したまま一度に加え,かき混ぜながら0.5mol/l {0.5N} 亜硝
酸ナトリウム溶液で滴定する。
(c) 滴定の終点は,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を1滴加えて2分後によう化カリウムでんぷ
ん紙で試験して,わずかに青が現れる点とする。
(15.1.4) 計算 0.25mol/l {0.25N} p−ニトロアニリン原液のファクターは,次の式によって算出し,小数点
以下4けたに丸める。
25
V
f
F
×
=
ここに, F: 0.25mol/l {0.25N} p−ニトロアニリン原液のファクター
f: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクタ
ー
V: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
(15.1.5) 保存 気密容器に入れて暗所に保存する。
(15.2) 0.1mol/l {0.1N} p−ニトロアニリンジアゾ溶液
(15.2.1) 調製
(a) (15.1)の0.25mol/l {0.25N} p−ニトロアニリン原液100m1を着色メスフラスコに正しく取り,これを
冷却して0〜5℃に保ちながら以下の操作を行う。
(b) 別に、(8)の0.5mol/l {0.5N} 亜硝酸ナトリウム溶液55mlを0〜5℃に冷却し,着色メスフラスコを振
り動かしながら,その中に一時に加え,ときどき振り混ぜながら3分間放置する。
(c) アミド硫酸アンモニウム溶液 (100g/l) 5mlを加え,よく振り混ぜた後,0〜5℃の水を加えて正しく
250mlとする。
(15.2.2) 標定 0.1 mol/l {0.1N} p−ニトロアニリンジアゾ溶液のファクターは,(15.1)の0.25mol/l {0.25N} p
−ニトロアニリン原液のファクターを用いる。
標定の必要がある場合は,カップリング法によって行う[(14.2)の備考参照]。
(15.2.3) 保存 気密容器に入れ,0〜5℃で光を遮って保存する。この場合,滴定には調製後8時間以内の
ものを用いる。
11.4.1.2 ファクターの温度補正 滴定用溶液(規定液)のファクターは,標定したときの温度 (t1) と使用
するときの温度 (t2) が異なるときは,次の式によって補正しなければならない。
1
2
1
2
1000
1000
t
t
t
t
F
b
a
b
a
F
×
×
−
×
−
=
ここに, Ft2: 使用するときの温度 (t2) におけるファクター
Ft1: 標定したときの温度 (t1) におけるファクター
at1: 標定したときの温度 (t1) における水の温度補正値 (ml) (表7
から求める。)
at2: 使用するときの温度 (t2) における水の温度補正値 (ml) (表7
から求める。)
b: 各種滴定用溶液(規定液)の水に対する補正係数(40)(温度20℃)
(表8から求める。)
56
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(40) 表8に示す係数は,11.4.1.1によって調製したものだけに適用する。
また,この係数は,20℃の値であって,特に低温部においてはあまり正しくない。
したがって,10℃以下の場合は,滴定用溶液(規定液)標定時の温度と,滴定時の
温度差が5℃以内であることが望ましい。しかし,15〜30℃の温度範囲では,表8を
適用しても誤差は無視できるほど小さい。
表7 水の温度補正値
温度
℃
補正値
ml
温度
℃
補正値
ml
温度
℃
補正値
ml
5
+1.4
16
+0.6
27
−1.5
6
+1.4
17
+0.5
28
−1.8
7
+1.4
18
+0.3
29
−2.0
8
+1.4
19
+0.2
30
−2.3
9
+1.3
20
±0.0
31
−2.6
10
+1.2
21
−0.2
32
−2.9
11
+1.2
22
−0.4
33
−3.2
12
+1.1
23
−0.6
34
−3.5
13
+1.0
24
−0.8
35
−3.8
14
+0.9
25
−1.0
15
+0.8
26
−1.3
表8 各種滴定用溶液(規定液)の水に対する補正係数
(温度20℃)
滴定用溶液(規定液)
モル濃度 mol/l
1
0.5
0.25
0.1
0.05
60
1
塩酸
1.25
1.15
−
1.03
−
−
硫酸
−
1.45
1.25
−
1.05
−
水酸化カリウム
1.40
1.20
−
1.04
−
−
水酸化ナトリウム
1.40
1.25
−
1.05
−
−
亜硝酸ナトリウム
−
1.30
−
1.06
−
−
臭素酸カリウム
−
−
−
−
−
1.35
チオ硫酸ナトリウム
−
−
−
1.10
−
−
フタル酸カリウム
−
−
−
5.40
−
−
スルファニル酸
−
1.20
−
1.04
−
−
p-トルイジン
−
1.51
−
−
−
−
p-ニトロアニリン
−
−
1.63
−
−
−
参考表1
滴定用溶液(規定液)
規定度 N
1
0.15
0.25
0.1
塩酸
1.25
1.15
−
1.03
硫酸
1.45
1.25
−
1.05
水酸化カリウム
1.40
1.20
−
1.04
水酸化ナトリウム
1.40
1.25
−
1.05
亜硝酸ナトリウム
−
1.30
−
1.06
臭素酸カリウム
−
−
−
1.35
チオ硫酸ナトリウム
−
−
−
1.10
フタル酸カリウム
−
−
−
5.40
スルファニル酸
−
1.20
−
1.04
p-トルイジン
−
1.51
−
−
p-ニトロアニリン
−
−
1.63
−
備考1. 表7は,t℃の水1lを20℃にしたときに示す体積の変化量である。したがって,t℃の水1lを20℃のときの体積
に換算するには,表7の補正値を加える。
この補正値は,1lについての値であるから,500mlについてはその21となる。
表8は,表7の補正値に対する各種滴定用溶液(規定液)の膨張補正係数である。したがって,各種滴定
用溶液(規定液)の場合は,表7の補正値に表8の各欄に示す係数を乗じて,各々該当する滴定用溶液(規
定液)の補正値とする。
2. 通常の場合,滴定用溶液(規定液)を標定してから使用し終わるまでの温度変化は,気温の激変がない限り,
あまり大きいものではないから,標定時の温度から数度範囲のファクターの変化をあらかじめ計算しておく
と便利である。
3. ジアゾ溶接については,調製時とできるだけ同一温度で全滴定を完了して,補正は省略する。
11.4.2 指示薬溶液及び試験紙 指示薬溶液及び試験紙は,表9に規定するものを用いる。
57
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表9 指示薬溶液及び試験紙の調製方法
名称
変色範囲
溶液の調製方法
備考
ブロモフェノール
ブルー溶液
黄色3.0〜4.6青紫
ブロモフェノールブルー0.10gをエタノール
(95) [エチルアルコール (95)]50mlに溶解し,
水を加えて100mlとする。
褐色ガラス瓶
に保存する。
コンゴーレッド溶
液
青紫3.0〜5.0赤みのだいだい色
コンゴーレッド0.10gを水に溶解して100mlと
する。
−
メチルオレンジ溶
液
赤3.1〜4.4赤みの黄
メチルオレンジ0.10gを水に溶解して100mlと
する。
褐色ガラス瓶
に保存する。
ブロモチモールブ
ルー溶液
黄色6.0〜7.6青
ブロモチモールブルー0.10gをエタノール (95)
[エチルアルコール (95)]50mlに溶解し,水を
加えて100mlとする。
褐色ガラス瓶
に保存する。
クレゾールレッド
溶液
黄色7.2〜8.8赤
クレゾールレッド0.10gをエタノール (95) [エ
チルアルコール (95)]20mlに溶解し,水を加え
て100mlとする。
−
フェノールフタレ
イン溶液
無色7.8〜10.0紅色
フェノールフタレイン1.0gをエタノール (95)
[エチルアルコール (95)]90mlに溶解し,水を
加えて100mlとする。
−
でんぷん溶液
−
でんぷん(溶性)1.0gを水10mlと混ぜ合わせ,
熱水200mlにかき混ぜながら加え,約1分間煮
沸した後冷却し,その上澄み液を用いる。
使用時に調製
する。
H酸溶液
−
4−アミノ−5−ヒドロキシ−2,7−ナフタレン
スルホン酸モノナトリウム塩(H酸モノナトリ
ウム塩)(41)0.5gを炭酸ナトリウム溶液 (100g/l)
5mlに溶解する。
使用時に調製
する。
よう化カリウムで
んぷん紙
−
でんぷん(溶性)5.0gを水25mlと混ぜ合わせ,
熱水500mlにかき混ぜながら加え,約1分間煮
沸した後冷却し,よう化カリウム1.0 gを5ml
の水に溶解したものを加える。この溶液にろ紙
を浸し,空気の清浄な室内で,直射日光を避け
て自然乾燥する。
褐色ガラス瓶
に保存する。
注(41) 市販の4−アミノ−5−ヒドロキシ−2,7−ナフタレンスルホン酸モノナトリウム塩(H酸モノナトリウ
ム塩)を活性炭を用いて水で1回再結晶し,メタノールで洗浄した後,約70℃で2時間以上乾燥する。
11.5 ニトロソ化滴定法
11.5.1 要旨 この方法は,フェノール類又は芳香族第2アミン類と亜硝酸塩が定量的に結合してニトロソ
化物を生成する反応を用い,亜硝酸塩の消費量から試料の純度を求める方法である。
11.5.2 操作 操作は,次のとおり行う。
(a) 個々の製品規格に規定する量(42)の試料を0.1mgのけたまで量り取り,その規定によって溶解し,水
を加えて全量を約300mlとする。
注(42) 試料の純度を100%とした場合の量。
(b) 規定の温度に保ち,かき混ぜながら0.5mol/l {0.5N} 亜硝酸ナトリウム溶液で滴定する。
(c) 滴定の終点は,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を1滴加えて5分後によう化カリウムでんぷ
ん紙で試験して,わずかに青が現れる点とする。
11.5.3 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
2
×
×
×
×
×
=S
M
F
V
P
ここに,
P: 純度 (%)
58
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
V: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
F: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファク
ター
M: 試料の分子量
S: 試料の質量 (g)
11.6 ジアゾ化滴定法 ジアゾ化滴定法は,直接法(一般法),直接法(別法),逆滴定法又は還元法のい
ずれかによる。
備考 本法によって滴定を行うときの終点判定には,11.6.2〜11.6.5に示す試験紙を用いる方法のほか,
JIS K 0113の5.(電位差滴定方法)及び6.(電流滴定方法)に規定する方法(以下,電示終点
判定方法という。)を用いることができる。試験紙を用いる方法では終点判定が困難な場合,電
示終点判定方法を用いることによって,簡便,かつ,高精度に測定を行うことができる。
11.6.6〜11.6.8に電示終点判定方法を用いるときの操作法を示すが,本法の直接法によって滴
定するときは11.6.6に規定する零電流電位差滴定法を用いて終点判定を行い,逆滴定法によっ
て滴定するときは11.6.8に規定する定電流分極電位差滴定法を用いて終点判定を行う。
また,11.6.6に規定する方法で明確な終点が得られないときは,11.6.7に規定する定電圧分極
電流滴定法を用いる。
11.6.1 要旨 この方法は,芳香族第1アミン類と亜硝酸塩が定量的に結合してジアゾ化物を生成する反応
を用い,亜硝酸塩の消費量から試料の純度を求める方法である。
11.6.2 直接法(一般法)
(1) 操作 操作は,次のとおり行う。
(a) 試料1000.1〜1001.1mol(42)を0.1mgのけたまで量り取り,水100ml及び塩酸20mlに溶解し,水を加えて全
量を200〜250mlとする。
(b) 臭化カリウム5gを加えて溶解し,これを冷却して0〜5℃に保ち,かき混ぜながら0.5mol/l {0.5N} 亜
硝酸ナトリウム溶液で滴定する。
(c) 滴定の終点は,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を1滴加えて2分後によう化カリウムでんぷ
ん紙で試験して,わずかに青が現れる点とする。
(2) 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
2
×
×
×
×
×
=S
M
F
V
P
ここに,
P: 純度 (%)
V: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
F: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクタ
ー
M: 試料の分子量
S: 試料の質量 (g)
11.6.3 直接法(別法)
(1) 操作 操作は,次のとおり行う。
(a) 試料1000.1〜1001.1mol(42)を0.1mgのけたまで量り取り,水100ml及び個々の製品規格に規定するアルカリ
に溶解し,水を加えて全量を約300mlとする。
(b) 臭化カリウム5gを加えて溶解し,これを冷却して0〜5℃に保ちながら以下の操作を行う。
(c) かき混ぜながら,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の滴定所要量の90%以上を加え,次に塩酸
25mlを一時に加えて酸性とし,かき混ぜながら0.5mol/l {0.5N} 亜硝酸ナトリウム溶液で滴定する。
59
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(d) 滴定の終点は,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を1滴加えて2分後によう化カリウムでんぷ
ん紙で試験して,わずかに青が現れる点とする。
(2) 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
2
×
×
×
×
×
=S
M
F
V
P
ここに,
P: 純度 (%)
V: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
F: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクタ
ー
M: 試料の分子量
S: 試料の質量 (g)
11.6.4 逆滴定法
(1) 操作 操作は,次のとおり行う。
(a) 試料1000.1〜1001.1mol(42)を0.1 mgのけたまで量り取り,水100ml及び個々の製品規格に規定するアルカ
リに溶解し,水を加えて全量を約300mlとする。
(b) 臭化カリウム5gを加えて溶解し,これを冷却して0〜5℃に保ちながら以下の操作を行う。
(c) かき混ぜながら,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を計算量より約1ml過剰になるように加え,
塩酸25mlを一時に加えて2〜5分間この温度で緩やかにかき混ぜる。
(d) 0.1mol/l {0.1N} スルファニル酸溶液で過剰の亜硝酸を滴定する。
(e) 滴定の終点は,0.1 mol/l {0.1 N} スルファニル酸溶液を1滴加えて2分後によう化カリウムでんぷ
ん紙で試験して,色が現れなくなった点とする。
(2) 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
2
5
2
2
1
1
×
×
×
×
×
−
×
=
S
M
F
V
F
V
P
ここに, P: 純度 (%)
V1: 添加した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
F1: 添加した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクター
V2: 逆滴定に要した0.1mol/l {0.1N} スルファニル酸溶液の量 (ml)
F2: 逆滴定に要した0.1mol/l {0.1N} スルファニル酸溶液のファクター
M: 試料の分子量
S: 試料の質量 (g)
11.6.5 還元法
(1) 操作 操作は,次のとおり行う。
(a) 試料1000.1〜1001.1mol(42)を0.1mgのけたまで量り取り,酢酸40mlに溶解する。
(b) 塩酸30mlを加えてかき混ぜ,これを冷却して0〜5℃に保ちながら以下の操作を行う。
(c) かき混ぜながら,亜鉛粉末6gを少量ずつ約10分間かけて加え,更に10分間かき混ぜた後,ろ過す
る。
(d) 残さは約150mlの水で洗い,ろ液と洗液とを合わせ,水を加えて全量を200〜250mlとする。
(e) 塩酸15ml及び臭化カリウム5gを加えて溶解し,かき混ぜながら0.5mol/l {0.5N} 亜硝酸ナトリウム
溶液で滴定する。
(f) 滴定の終点は,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を1滴加えて2分後によう化カリウムでんぷ
ん紙で試験して,わずかに青が現れる点とする。
60
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
2
×
×
×
×
×
=S
M
F
V
P
ここに,
P:純度 (%)
V:滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
F:滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクター
M:試料の分子量
S:試料の質量 (g)
11.6.6 零電流電位差滴定法
(1) 要旨 この方法は,指示電極1個を用いる電位差滴定法で,滴定途中の加電圧電流曲線の電流値がゼ
ロのときの電位差変化から終点を決定する方法である。
(2) 操作 操作は,次のとおり行う。
(a) 試料1000.1〜1001.1mol(42)を0.1mgのけたまで量り取り,水100ml及び塩酸20mlに溶解し,水を加えて全
量を150〜200mlとする。
(b) 臭化カリウム5gを加えて溶解し,これを冷却して0〜5℃に保ちながら以下の操作を行う。
(c) 白金電極と飽和カロメル電極を挿入し,両電極間の電位差を電位差滴定装置を用いて測定する。
(d) 0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を一定量滴下し,よくかき混ぜて被滴定溶液と反応させた後,
電極間の電位差を測定する。
(e) (d)の操作を繰り返し,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の添加量に対応する電極間の電位差を
記録し,JIS K 0113の5.2.1(滴定曲線又は示差滴定曲線の作成方法)によって滴定曲線を作成する。
(f) 滴定の終点は,JIS K 0113の5.2.2(終点決定方法)による。
(3) 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
2
×
×
×
×
×
=S
M
F
V
P
ここに, P: 純度 (%)
V: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
F: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクター
M: 試料の分子量
S: 試料の質量 (g)
11.6.7 定電圧分極電流滴定法
(1) 要旨 この方法は,指示電極2個を用いる電流滴定法で,電極間に一定の微小電圧を加え,当量点付
近の電流変化から終点を決定する方法である。
(2) 操作 操作は,次のとおり行う。
(a) 試料1000.1〜1001.1mol(42)を0.1mgのけたまで量り取り,水100ml及び塩酸20mlに溶解し,水を加えて全
量を150〜200mlとする。これを冷却して0〜5℃に保ちながら以下の操作を行う。
(b) 2本の白金電極を挿入し,両電極間に微小直流電圧を加え,電極間の電流を電流滴定装置を用いて
測定する。
(c) 0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を一定量滴下し,よくかき混ぜて被滴定溶液と反応させた後,
電極間の電流を測定する。
(d) (c)の操作を繰り返し,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の添加量に対応する電極間の電流を記
録し,JIS K 0113の6.2.1(滴定曲線の作成方法)によって滴定曲線を作成する。
(e) 滴定の終点は,JIS K 0113の6.2.2(終点決定方法)による。
61
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
2
×
×
×
×
×
=S
M
F
V
P
ここに,
P: 純度 (%)
V: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
F: 滴定に要した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクタ
ー
M: 試料の分子量
S: 試料の質量 (g)
11.6.8 定電流分極電位差滴定法
(1) 要旨 この方法は,指示電極2個を用いる電位差滴定法で,電極間に一定の微小電流を流し,当量点
付近の電位差変化から終点を決定する方法である。
(2) 操作 操作は,次のとおり行う。
(a) 試料1000.1〜1001.1mol(42)を0.1mgのけたまで量り取り,水100ml及び個々の製品規格に規定するアルカリ
に溶解し,水を加えて全量を約150mlとする。
(b) 臭化カリウム5gを加えて溶解し,これを冷却して0〜5℃に保ちながら以下の操作を行う。
(c) かき混ぜながら,0.5mol/l {0.5N} 亜硝酸ナトリウム溶液を計算量より約1ml過剰になるように加え,
次に,塩酸20mlを一時に加えて2〜5分間この温度で緩やかにかき混ぜた後,2本の白金電極を挿
入し,電極間に微小直流電流を流し,両電極間の電位差を電位差滴定装置を用いて測定する。
(d) 次に,0.1 mol/l {0.1N} スルファニル酸溶液を一定量滴下し,よくかき混ぜて被滴定溶液と反応させ
た後,電極間の電位差を測定する。
(e) (d)の操作を繰り返し,0.1mol/l {0.1N} スルファニル酸溶液の添加量に対応する電極間の電位差を記
録し,JIS K 0113の5.2.1(滴定曲線又は示差滴定曲線の作成方法)によって滴定曲線を作成する。
(f) 滴定の終点は,JIS K 0113の5.2.2(終点決定方法)による。
(3) 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
2
5
2
2
1
1
×
×
×
×
×
−
×
=
S
M
F
V
F
V
P
ここに,
P: 純度 (%)
V1: 添加した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液の量 (ml)
F1: 添加した0.5mol/l {0.5N} 亜硝酸ナトリウム溶液のファクター
V2: 逆滴定に要した0.1 mol/l {0.1N} スルファニル酸溶液の量 (ml)
F2: 逆滴定に要した0.1 mol/l {0.1N} スルファニル酸溶液のファクタ
ー
M: 試料の分子量
S: 試料の質量 (g)
11.7 カップリング滴定法
11.7.1 要旨 この方法は,フェノール類又は芳香族アミン類とジアゾニウム塩が定量的に結合してジアゾ
化合物を生成する反応を用い,ジアゾニウム塩の消費量から試料の純度を求める方法である。
11.7.2 操作 操作は,次のとおり行う。
(a) 試料400
01
.1〜400
03
.1mol(42)を0.1mgのけたまで量り取り,個々の製品規格の規定によって溶解し,水を加え
て全量を約50mlとする。これを冷却して0〜5℃に保ちながら以下の操作を行う。
(b) かき混ぜながら,冷却式ビュレット又は冷却したピペットを用いて0.1mol/l {0.1N} ジアゾ溶液の滴
62
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
定所要量の98%を一時に加え,2〜5分間かき混ぜる。塩析の規定がある場合は,規定量の塩化ナト
リウムを加える。
(c) 試料溶液の1滴をろ紙上に取り,その無色湿潤部(色素の周りにできる無色の部分)の一部分にジ
アゾ溶液を,また,他の部分にH酸溶液(指示薬)をそれぞれ接触させる。
(d) ジアゾ溶液との接触部に着色帯が生じることを確かめた後,冷却式ビュレット又は冷却したメスピ
ペット1mlを用いてジアゾ溶液を少量ずつ滴加し,その都度,(c)のはん点試験を行う。
(e) 滴定の終点は,ジアゾ溶液を1滴加えて2分後に(c)のはん点試験を行い,ジアゾ溶液との接触部に
着色帯を認めなくなり,かつ,H酸溶液(指示薬)との接触部に紫から赤の着色帯がわずかに生じ
る点とする。
11.7.3 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000
10
×
×
×
×
×
=S
M
F
V
P
ここに,
P: 純度 (%)
V: 滴定に要した0.1 mol/l {0.1N} ジアゾ溶液の量 (ml)
F: 滴定に要した0.1 mol/l {0.1N} ジアゾ溶液のファクター
M: 試料の分子量
S: 試料の質量 (g)
11.8 臭素化滴定法(逆滴定法)
11.8.1 要旨 この方法は,フェノール類又は芳香族アミン類と臭素酸塩が定量的に結合して臭素化物を生
成する反応を用い,臭素酸塩の消費量から試料の純度を求める方法である。
11.8.2 操作 操作は,次のとおり行う。
(a) 試料2009.0〜2000.1mol(42)を0.1mgのけたまで量り取り,各品目の規定によって溶解し,水を加えて全量を
正しく250mlとする。これを20〜25℃に保ちながら以下の操作を行う。
(b) 共通すり合わせよう素フラスコA型500ml又はB型500ml(以下,よう素フラスコという。)に水
約150mlを入れ,これに601mol/l {0.1N} 臭素酸カリウム溶液35ml及び試料溶液25mlを正しく取っ
て加え,振り混ぜた後,よう素フラスコの栓を少し開けて塩酸5mlを速やかに加え,手早く栓をす
る。
(c) よう素フラスコのリップの周りに水を入れ,約10分間暗所に放置する。
(d) 緩やかに振り混ぜて臭素蒸気を溶液に吸収させた後,よう化カリウム溶液 (100g/l) 10mlを速やかに
加えて手早く栓をし,よく振り混ぜる。
(e) 遊離したよう素を0.1mol/l {0.1N} チオ硫酸ナトリウム溶液で滴定する。この場合,溶液がごくうす
い黄色になった後,指示薬のでんぷん溶液8〜10mlを加える。
(f) 滴定の終点は,0.1mol/l {0.1N} チオ硫酸ナトリウム溶液を1滴加えて青から無色に変わる点とする。
11.8.3 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
(
)
100
250
25
1000
10
35
×
×
×
×
×
×
×
−
=
n
S
M
F
V
P
ここに,
P: 純度 (%)
V: 滴定に要した0.1mol/l {0.1N} チオ硫酸ナトリウム溶液の量 (ml)
F: 滴定に要した0.1mol/l {0.1N} チオ硫酸ナトリウム溶液のファクタ
ー
M: 試料の分子量
63
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S: 試料の質量 (g)
n: 臭素原子置換数×2
11.9 中和滴定法
11.9.1 要旨 この方法は,酸がアルカリと定量的に結合して塩及び水を生成する反応を用い,アルカリの
消費量から試料の純度を求める方法である。
11.9.2 操作 操作は,次のとおり行う。
(a) 試料500.1〜501.1mol(42)を0.1mgのけたまで量り取り,エタノール (99.5) [エチルアルコール (99.5)]50ml
に溶解する。
(b) フェノールフタレイン溶液を指示薬として,1mol/l {1N} 水酸化ナトリウム溶液で滴定する。
(c) 滴定の終点は,1mol/l {1N} 水酸化ナトリウム溶液を1滴加えてうすい紅色が現れる点とする。
11.9.3 計算 純度は,次の式によって算出し,小数点以下2けたに丸める。
100
1000×
×
×
×
=S
M
F
V
P
ここに,
P: 純度 (%)
V: 滴定に要した1mol/l {1N} 水酸化ナトリウム溶液の量 (ml)
F: 滴定に要した1mol/l {1N} 水酸化ナトリウム溶液のファクター
M: 試料の分子量
S: 試料の質量 (g)
12. 濁度測定方法
12.1 要旨 この方法は,試料の濁度を濁度標準液と目視によって比較して求める方法である。
12.2 試薬 試薬は,次のとおりとする。
(1) 塩化ナトリウム JIS K 8150に規定する特級。
(2) 硝酸 (1+3) JIS K 8541に規定する特級を用いて調製したもの。
(3) 0.1mol/l {0.1N} 硝酸銀溶液 JIS K 8550に規定する特級を用いて調製したもの。
12.3 器具 器具は,次のとおりとする。
(1) 比色管 内径23mm,全長約180mm,容量50mlの共通すり合わせ平底試験管で,それぞれ同質,同
形で肉厚の等しいもの5本。
12.4 塩化物標準液の調製 塩化物標準液は,次のとおり調製する。
(1) 塩化物標準液1号 塩化ナトリウム0.164gを水1 000mlに溶解したもので,1ml中に塩素分として0.1
mgを含む。
(2) 塩化物標準液2号 塩化物標準液1号を水で10倍に希釈したもので,1ml中に塩素分として0.01mg
を含む。
(3) 塩化物標準液3号 塩化物標準液1号を水で100倍に希釈したもので,1ml中に塩素分として0.001mg
を含む。
12.5 試料溶液の調製 試料溶液の調製は,個々の製品規格に規定する方法による。
12.6 濁度標準液の調製 5本の比色管を用意し,表10に示した塩化物標準液をそれぞれの比色管に取り,
水を加えて20mlとし,これに硝酸 (1+3) 1mlと0.1mol/l {0.1N} 硝酸銀溶液5滴を加えた後,栓をしてよ
く振り混ぜる。
なお,この溶液は,経時変化によって沈殿するため,調製して15分間経過した後,12.7の操作を行うこ
と。
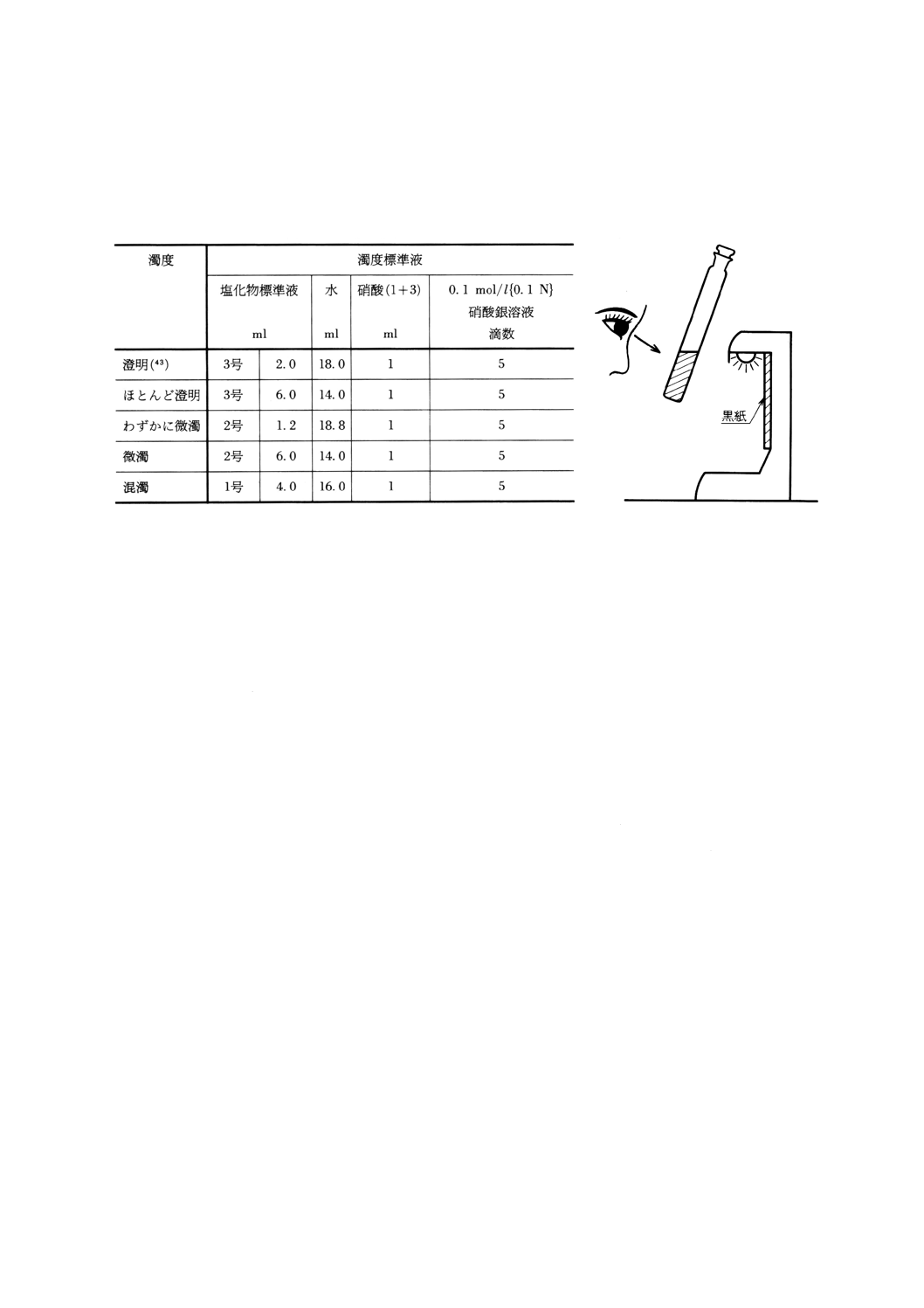
64
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
12.7 操作 試料溶液20mlを比色管に取り,調製後15分経過した濁度標準液と試料溶液とを並べて静置
し,図44のように黒紙を背に後方から光線を透過させ,光源を直視しないように斜め上方から目視して,
試料溶液の濁りを標準液と比較して濁度を求める。
表10 濁度標準液
図44 濁度の見方
注(43) 浮遊物などの異物の混入をほとんど認めないもの。
13. 色数試験方法
13.1 要旨 この方法は,液体化学製品及び加熱して溶融状態になる化学製品について,試料の透過色を
ハーゼン標準比色液と目視で比較してハーゼン単位色数を求める方法である。標準白金コバルトに近い色
の透明で淡黄褐色の着色液体についてだけ適用できる。ハーゼン単位による色数試験は,JIS K 0071の5.
(ハーゼン単位色数試験方法)によるほか,次のとおりとする。
13.2 装置及び器具 装置及び器具は,次のとおりとする。
(1) 分光光度計 JIS K 0115に規定するもの。
(2) 吸収セル 光路長10mmの角形セル
(3) 比色管
(a) 液体試料用 直径20mm以上の共通すり合わせ平底ガラス管で,同質,同型で肉厚の等しいもの,
かつ,液量が約100mlになるように底面から同じ高さのところに標線を刻んだもの。
(b) 固体試料用 JIS R 3503に規定する外径25mm(硬質1級:呼び寸法25×200)の肉厚の等しい試験
管。
(4) 白色板 JIS P 3801に規定する定性分析用角形ろ紙又はこれと同等以上の品質のもの。
(5) 比色計 2本以上の比色管を立てる指示枠,白色平滑なガラス底板,側面からの散乱光を防ぐための
黒色遮光板及び底板を透して光線を導入する反射鏡とからなり,拡散昼光(44)のもとで,管の上から見
透して管の液の色を比較することができる装置。一例を図45に示す。
注(44) 日の出3時間後から日没3時間前までの間の日光の直射を避けた北窓からの光をいう。ただし,
JIS Z 8720に規定する標準の光D65又は標準の光C,若しくは,JIS Z 8716に規定する蛍光ランプ
の光を拡散昼光として用いることができる。
(6) 溶融装置 試料を短時間に溶融でき,試験温度±2〜3℃に調節できるもの。装置の一例を図46に示す。
13.3 試薬 試薬は,次のとおりとする。
(1) 塩化コバルト (II) 六水和物(試薬) JIS K 8129に規定する特級。
65
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) ヘキサクロロ白金 (IV) 酸カリウム(塩化白金酸カリウム)(試薬) JIS K 8163に規定する特級
(3) 塩酸(試薬) JIS K 8180に規定する特級。
図45 比色計の一例
66
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図46 溶融装置の一例
13.4 ハーゼン標準比色液の調製 ヘキサクロロ白金 (IV) 酸カリウム1.245gと塩化コバルト (II) 六水和
物1.000gを水に溶かし,塩酸100mlを加え,水で薄めて1 000mlとする。これをハーゼン標準比色液500
番とする。比色液の吸光度は,分光光度計(吸収セル10mm)を用いて,水を対照液として測定したとき,
表11の許容範囲内に入らなければならない。
500番未満のハーゼン標準比色液は,ハーゼン標準比色液500番を表12によって比色管に取り,水を加
えて100mlとする。
標準比色液は,密栓した着色ガラス瓶に入れ,暗所に保存する。保存期間は,原則として調製後,ハー
ゼン500の標準比色液については1年以内とし,ハーゼン500未満の標準比色液は1か月以内とする。
表11 ハーゼン標準比色液500番の吸光度許容範囲
波長nm
吸光度
430
0.110〜0.120
455
0.130〜0.145
480
0.105〜0.120
510
0.055〜0.065
67
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表12 ハーゼン標準比色液
標準比色液
の番号
標準比色液
500番 ml
標準比色液
の番号
標準比色液
500番 ml
5
1
70
14
10
2
100
20
15
3
150
30
20
4
200
40
25
5
250
50
30
6
300
60
35
7
350
70
40
8
400
80
50
10
450
90
60
12
500
100
13.5 操作 操作は,次のとおり行う。
(1) 試料とハーゼン標準比色液の色調が近いことを確認する。
(2) 液体試料のときは,試料とハーゼン標準比色液をそれぞれ比色管の標線まで入れて白色板上に置き,
拡散昼光(44)のもとで比色管の上方から下方に透かして色を比較するか又は両管を比色計の中に置い
て色を比較する。試料に最も近似したハーゼン標準比色液の番号を読み取る。
(3) 固体試料の場合は,必要量を比色管に入れふたをして,試験温度に調節した溶融装置で溶融させ,こ
れを直ちにハーゼン標準比色液と並べて直立させ,その背面に白色板を置き,拡散昼光(44)のもとで比
色管の側面から透かして色を比較する。試料に最も近似したハーゼン標準比色液の番号を読み取る。
13.6 結果の記録 試料と最も近似したハーゼン標準比色液の番号を記録する。二つの中間にあるときは,
二つのうち濃い方の番号を選ぶ。
記録例 ハーゼン色数50
14. 酸分試験方法 酸分試験方法は,定性法又は定量法(H2SO4として)のいずれかによる。
14.1 定性法
14.1.1 要旨 この方法は,指示薬の色の変化によって酸分の有無を定性的に求める方法である。
14.1.2 指示薬 指示薬は,次のとおりとする。
(1) コンゴーレッド溶液 表9によって調製したもの。
14.1.3 器具 器具は,次のとおりとする。
(1) 試験管 直径20mm以上,容量約50mlの共通すり合わせ試験管
14.1.4 操作 操作は,次のとおり行う。
(a) 個々の製品規格に規定する質量の試料を試験管に取り,水10mlを加えてよく振り混ぜる。ただし,
固体試料の場合は,水10mlを加えて加熱溶融させ,よく振り混ぜた後,常温まで冷却する。
(b) 指示薬としてコンゴーレッド溶液2〜3滴を加え,緩やかに振り混ぜた後,目視によって色の変化を
調べる。
14.1.5 評価 評価は,個々の製品規格に規定する。
14.2 定量法(H2SO4として)
14.2.1 要旨 この方法は,中和滴定によって酸分(H2SO4として)を求める方法である。
14.2.2 滴定用溶液(規定液)及び指示薬 滴定用溶液(規定液)及び指示薬は,次のとおりとする。
(1) 0.1mol/l {0.1N} 水酸化ナトリウム溶液 11.4.1.1(7)によって調製したもの。
68
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) メチルオレンジ溶液 表9によって調製したもの。
14.2.3 器具 器具は,次のとおりとする。
(1) 化学はかり JIS K 0050に規定するもの。
14.2.4 操作 操作は,次のとおり行う。
(a) 試料10gを1mgのけたまで量り取り,水50mlを加え,ガラス棒でかき混ぜながら,10分間緩やか
に煮沸する。
(b) 常温まで冷却した後,ろ過し,残さを約30mlの水で洗浄する。
(c) ろ液と洗液とを合わせ,指示薬としてメチルオレンジ溶液1〜2滴を加え,0.1mol/l {0.1N} 水酸化
ナトリウム溶液で滴定する。
(d) 滴定の終点は,0.1mol/l {0.1N} 水酸化ナトリウム溶液を1滴加えてうすい赤みの黄色が現れる点と
する。
14.2.5 計算 酸分(H2SO4として)は,次の式によって算出し,小数点以下2けたに丸める。
100
00490
.0
×
×
×
=
S
F
V
X
ここに,
X: 酸分(H2SO4として) (%)
V: 滴定に要した0.1mol/l {0.1N} 水酸化ナトリウム溶液の量 (ml)
F: 滴定に要した0.1mol/l {0.1N} 水酸化ナトリウム溶液のファクター
S: 試料の質量 (g)
15. 塩酸不溶分試験方法
15.1 要旨 この方法は,試料を塩酸に溶解し,残さをろ過洗浄後,乾燥し,乾燥後の残分の質量を量る
ことによって塩酸不溶分を求める方法である。
15.2 試薬 試薬は,次のとおりとする。
(1) 塩酸 JIS K 8180に規定する特級。
(2) 硝酸 JIS K 8541に規定する特級。
(3) 硝酸銀溶液 (20g/l) JIS K 8550に規定する特級を用いて調製したもの。
15.3 器具 器具は,次のとおりとする。
(1) るつぼ形ガラスろ過器 JIS R 3503に規定する1G3又は1G4。
(2) 化学はかり JIS K 0050に規定するもの。
(3) デシケーター JIS R 3503に規定するもので,乾燥剤としてシリカゲルを入れたもの。
15.4 操作 操作は,次のとおり行う。
(a) 試料5gを1mgのけたまで量り取り,塩酸25ml及び水250mlを加え,ガラス棒でかき混ぜながら,
40〜50℃で5分間加熱する。
(b) あらかじめ,100〜105℃で恒量(45)にしたるつぼ形ガラスろ過器を用いて吸引ろ過し,残さを塩酸 (1
+13) 50mlを用いて洗浄した後,塩化物イオンを認めなくなるまで(46)水で洗浄する。
注(45) 質量を測定する操作を2回繰り返したとき,1回目の質量に対して1回目と2回目の質量の差が
000
10
10以下のとき,恒量とする。ただし,試料の質量が0.3g以下のときは,1回目と2回目の質量
の差が0.3mg以下のとき,恒量とする。
(46) 洗液20mlに硝酸 (1+2) 5mlを加えてかき混ぜ,硝酸銀溶液 (20g/l) 1mlを加える。溶液が白濁
しなくなるまでこの操作を繰り返す。
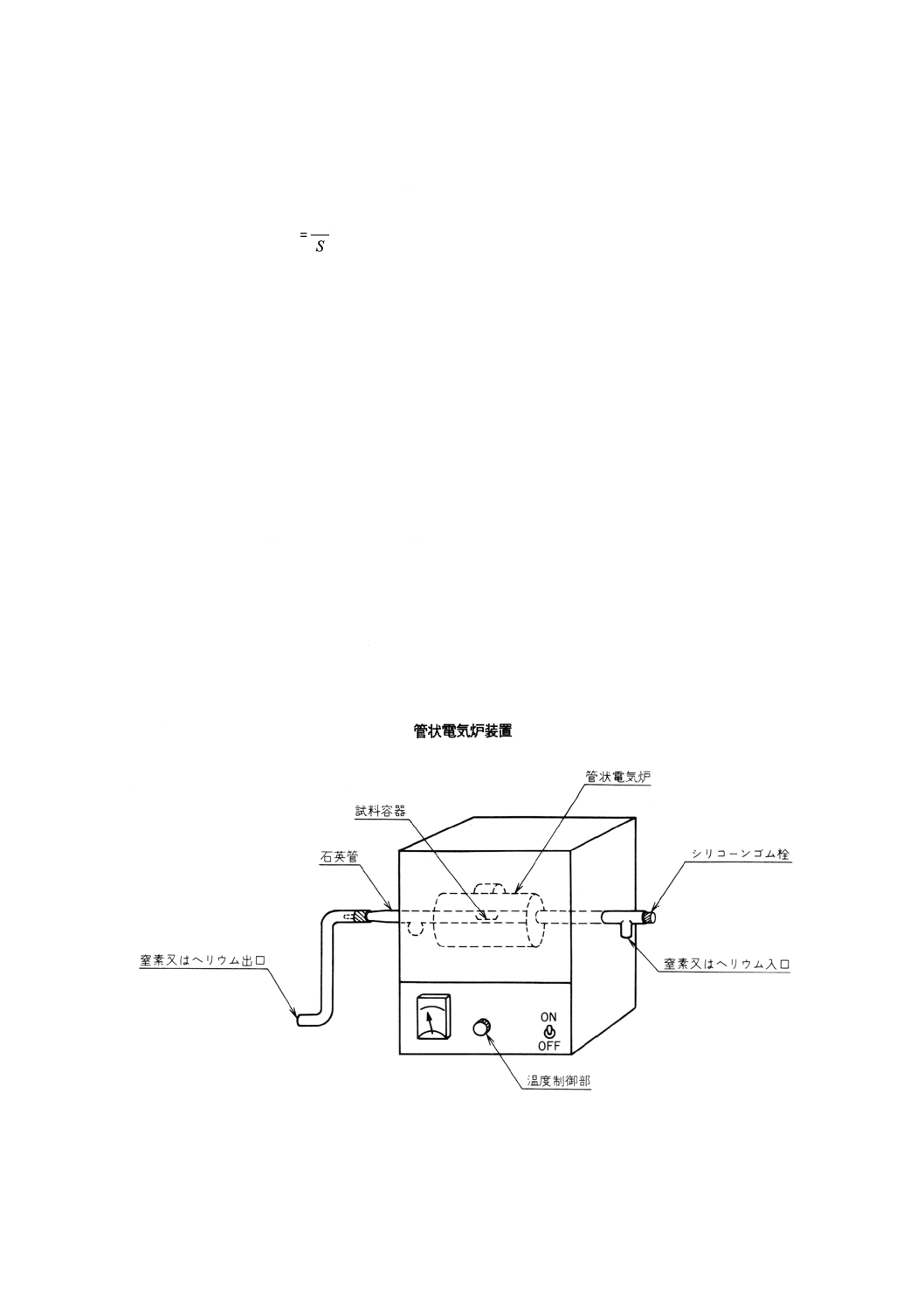
69
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(c) るつぼ形ガラスろ過器を100〜105℃で恒量(46)になるまで乾燥し,速やかにデシケーターに移し,
常温まで放冷した後,その質量を1mgのけたまで量る。
15.5 計算 塩酸不溶分は,次の式によって算出し,小数点以下2けたに丸める。
100
×
=SM
R
ここに,
R: 塩酸不溶分 (%)
M: 残さの質量 (g)
S: 試料の質量 (g)
16. ガスクロマトグラフ分析方法 ガスクロマトグラフ分析方法は,JIS K 0114による。この場合,検出
器の種類,分析条件及び定量法などは,個々の製品規格の試験方法中に規定する。
17. 不揮発分試験方法
17.1 要旨 この方法は,管状電気炉を用いて,試料中の揮発分をガスクロマトグラフ分析条件の試料気
化室温度で揮発させ,揮発後の残分の質量を量ることによって不揮発分を求める方法である。
17.2 装置及び器具 装置及び器具は,次のとおりとする。
(1) 不揮発分試験装置 管状電気炉及び石英管で構成されるもので,一例を図47に示す。
(1.1) 管状電気炉 100〜400℃の温度範囲に設定できるもので,±10℃以内の精度をもつもの。
(1.2) 石英管 石英ガラス製のもので,一例を図48に示す。
(1.3) 試料容器 容量約1mlの白金製,硬質ガラス製又はアルミニウム製のもの。
(2) 微量化学はかり JIS K 0050に規定するもの。
(3) デシケーター JIS R 3503に規定するもので,乾燥剤としてシリカゲルを入れたもの。
図47 不揮発分試験装置の一例

70
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図48 石英管の一例
17.3 操作 操作は,次のとおり行う。
(a) 100〜200mgの試料をあらかじめ恒量(47)にした試料容器に入れ,その質量を0.01 mgのけたまで量
る。
注(47) 質量を測定する操作を2回繰り返したとき,1回目の質量に対して1回目と2回目の質量の差が
000
10
10以下のとき,恒量とする。ただし,試料の質量が0.3g以下のときは,1回目と2回目の質量
の差が0.3mg以下のとき,恒量とする。
(b) あらかじめ,個々の製品規格で規定する試料気化室温度に設定した不揮発分試験装置の石英管内に
試料容器を挿入し,シリコーンゴム栓をする。
(c) 流量約200ml/minの窒素又はヘリウム気流中で,個々の製品規格で規定する時間加熱する。
(d) 石英管内から取り出した試料容器を速やかにデシケーターに移し,常温まで放冷した後,その質量
を0.01mgのけたまで量る。
17.4 計算 不揮発分は,次の式によって算出し,その結果を小数点以下2けたに丸める。
100
×
S
m
R=
ここに, R: 不揮発分 (%)
S: 試料の質量 (mg)
m: 揮発後の残分の質量 (mg)
18. 高速液体クロマトグラフ分析方法 高速液体クロマトグラフ分析方法は,JIS K 0124による。この場
合,検出器の種類,分析条件及び定量法などは,個々の製品規格の試験方法中に規定する。ただし,定量
法については,JIS K 0124に規定されている定量法のほか,次の方法によってもよい。
(1) 面積百分率法(48) クロマトグラムから得た試料各成分のピーク面積を測定して,それらの総和を100
とし,それに対するそれぞれのピーク面積の比率を各成分の含有率として,次の式によって算出する。
100
1
×
∑
=
n
i
i
i
i
A
A
C=
ここに, Ci: 各成分の含有率 (%)
Ai: i成分のピーク面積
n: 全ピーク数
注(48) 本法の場合は,導入した試料の全成分が溶出することが必要である。
また,使用した検出器によって,各成分の相対感度が大幅に異なることがあるので,十分注
意する必要がある。
71
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) 補正面積百分率法 導入した試料の全成分が溶出し,溶出全成分の相対感度(49)が求められ,かつ,そ
れぞれの相対感度が測定濃度域にわたって一定とみなされる場合,次の式によって,各成分の含有率
を算出する。
100
1
×
∑
=
n
i
i
i
i
i
i
f
A
f
A
C=
ここに, Ci: 各成分の含有率 (%)
Ai: i成分のピーク面積
fi: i成分の相対感度
n: 全ピーク数
注(49) 相対感度の求め方 成分量既知の混合試料のクロマトグラムから各成分のピーク面積を測定し,
単位成分量当たりの面積(感度)を算出する。適当な成分を基準にとり,その感度に対する比
をとれば相対感度となる。相対感度の逆数を相対補正係数という。
19. 有機中間物を取り扱うときの注意事項 有機中間物を取り扱うときには,まずその物質の名称を確認
し,その安全性について確認する。その物質の物性などの情報が不十分で安全性の確認ができないときは,
事前に調査を行い,十分な安全性の対策を施した上で取り扱わなければならない。
危険性,有害性などに関して法規上の規制があるものについては,十分な準備と対策を施した後,関連
する法令・規則に従って取り扱わなければならない。
付表1 引用規格
JIS B 7410 石油類試験用ガラス製温度計
JIS B 7411 ガラス製棒状温度計(全浸没)
JIS B 7413 浸没線付ガラス製水銀棒状温度計
JIS B 7525 比重浮ひょう
JIS C 3105 硬銅より線
JIS K 0050 化学分析方法通則
JIS K 0061 化学製品の密度及び比重測定方法
JIS K 0064 化学製品の融点及び溶融範囲測定方法
JIS K 0065 化学製品の凝固点測定方法
JIS K 0066 化学製品の蒸留試験方法
JIS K 0067 化学製品の減量及び残分試験方法
JIS K 0068 化学製品の水分測定方法
JIS K 0071 化学製品の色及び硫酸着色試験方法
JIS K 0113 電位差・電流・電量・カールフィッシャー滴定方法通則
JIS K 0114 ガスクロマトグラフ分析のための通則
JIS K 0115 吸光光度分析のための通則
JIS K 0124 高速液体クロマトグラフ分析通則
JIS K 2249 原油及び石油製品の密度試験方法並びに密度・質量・容量換算表
JIS K 2839 石油類試験用ガラス器具
72
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 4134 4−アミノ−5−ヒドロキシ−2,7−ナフタレンジスルホン酸モノナトリウム塩(H酸モ
ノナトリウム塩)
JIS K 8001 試薬試験方法通則
JIS K 8005 容量分析用標準物質
JIS K 8013 亜鉛粉末(試薬)
JIS K 8019 亜硝酸ナトリウム(試薬)
JIS K 8051 3−メチル−1−ブタノール(試薬)
JIS K 8085 アンモニア水(試薬)
JIS K 8101 エタノール (99.5) [エチルアルコール (99.5)](試薬)
JIS K 8102 エタノール (95) [エチルアルコール (95)](試薬)
JIS K 8105 エチレングリコール(グリコール)(試薬)
JIS K 8129 塩化コバルト (II) 六水和物(試薬)
JIS K 8150 塩化ナトリウム(試薬)
JIS K 8163 ヘキサクロロ白金 (IV) 酸カリウム(塩化白金酸カリウム)(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8228 過塩素酸マグネシウム(試薬)
JIS K 8308 クレゾールレッド(試薬)
JIS K 8322 クロロホルム(試薬)
JIS K 8342 五酸化二りん(無水りん酸)(試薬)
JIS K 8352 コンゴーレッド(試薬)
JIS K 8355 酢酸(試薬)
JIS K 8392 サリチル酸(試薬)
JIS K 8428 酸化バリウム(試薬)
JIS K 8506 臭化カリウム(試薬)
JIS K 8530 臭素酸カリウム(試薬)
JIS K 8540 酒石酸ナトリウム二水和物(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8550 硝酸銀(試薬)
JIS K 8574 水酸化カリウム(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8577 水酸化バリウム八水和物(試薬)
JIS K 8586 スルファニル酸(試薬)
JIS K 8588 アミド硫酸アンモニウム(試薬)
JIS K 8603 ソーダ石灰(試薬)
JIS K 8625 炭酸ナトリウム(試薬)
JIS K 8637 チオ硫酸ナトリウム五水和物(試薬)
JIS K 8659 でんぷん(溶性)(試薬)
JIS K 8708 p−ニトロアニリン(試薬)
JIS K 8777 ピリジン(試薬)
JIS K 8799 フェノールフタレイン(試薬)
73
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 8837 プロピレングリコール(試薬)
JIS K 8842 ブロモチモールブルー(試薬)
JIS K 8844 ブロモフェノールブルー(試薬)
JIS K 8873 ホルムアミド(試薬)
JIS K 8876 マグネシウム末(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8893 メチルオレンジ(試薬)
JIS K 8895 2−メトキシエタノール(試薬)
JIS K 8913 よう化カリウム(試薬)
JIS K 8920 よう素(試薬)
JIS K 8951 硫酸(試薬)
JIS M 8100 粉塊混合物−サンプリング方法通則
JIS P 3801 ろ紙(化学分析用)
JIS R 3503 化学分析用ガラス器具
JIS Z 1703 ポリエチレンびん
JIS Z 8202 量記号,単位記号及び化学記号
JIS Z 8203 国際単位系 (SI) 及びその使い方
JIS Z 8401 数値の丸め方
JIS Z 8703 試験場所の標準状態
JIS Z 8716 表面色の比較に用いる常用光源蛍光ランプD65−形式及び性能
JIS Z 8720 測色用の標準の光及び標準光源
付表2 関連規格
ISO 760 水分の測定−カールフィッシャー法(一般方法)
ISO 758 工業用液体化学製品の20℃における比重の測定
ISO 759 工業用揮発性有機液状物−水浴上での蒸発残分測定−一般法
ISO 918 工業用揮発性有機液状物−蒸留特性の測定
ISO 1393 工業用液体ハロゲン化炭化水素−酸性度の測定−滴定法
ISO 1995 芳香族炭化水素−試料採取方法
ISO 2209 工業用ハロゲン化炭化水素−試料採取方法
ISO 2211 液体化学製品−ハーゼン単位による色の測定(プラチナ−コバルトスケール)
ISO 2718 ガスクロマトグラフィーによる化学分析方法の一般的方法
ISO 3165 工業用化学製品の試料採取−試料採取の際の安全
ISO 5297 芳香族炭化水素−沸点150℃以下の製品についての蒸発残分定量方法
日本薬局方
74
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考 安全衛生,防災,環境保全及び公害対策上の措置
この参考は,本体の規定に関連する事柄を補足するもので,規定の一部ではない。
この規格によって試験を行う場合は,試料の採取,試料,材料,装置及び器具の取扱い並びに試験操作
については,必要に応じ,該当する法令に準拠し,適切な予防措置をとるものとする。
主な関連法令としては,次のものがある。
(1) 労働安全衛生法に関係するもの
・ 労働安全衛生法(昭和47年6月8日法律第57号)
・ 有機溶剤中毒予防規則(昭和47年9月30日労働省令第36号)
・ 特定化学物質等障害予防規則(昭和47年9月30日労働省令第39号)
(2) 毒物及び劇物取締法に関係するもの
・ 毒物及び劇物取締法(昭和25年12月28日法律第303号)
(3) 作業環境測定法に関係するもの
・ 作業環境測定法(昭和50年5月1日法律第28号)
(4) 消防法に関係するもの
・ 消防法(昭和23年7月24日法律第186号)
・ 危険物の規制に関する政令(昭和34年9月26日政令第306号)
(5) 環境保全・公害対策に関係するもの
・ 公害対策基本法(昭和42年8月3日法律第132号)
・ 大気汚染防止法(昭和43年6月10日法律第97号)
・ 水質汚濁防止法(昭和45年12月25日法律第138号)
・ 廃棄物の処理及び清掃に関する法律(昭和45年12月25日法律第137号)
・ 地方自治体の定める環境保全・公害防止条例
75
K 4101-1993
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
解説表7 原案作成委員会 構成表
氏名
所属
委員会
分科会
第1
第2
第3
(委員長)
小 川 昭二郎 お茶の水女子大学
○
横 手 正 夫 日本大学
○
地 崎 修 工業技術院標準部
○
○
○
○
(細川 幹夫) (工業技術院標準部)
○
○
○
○
中 島 邦 雄 通商産業省基礎産業局
○
渡 辺 義 生 通商産業省通商産業検査所
○
(石毛 和之) (通商産業省通商産業検査所)
○
黒 木 勝 也 財団法人日本規格協会
○
(第1分科会主査) 広 明 修 住友化学工業株式会社
○
○
(第2分科会主査) 熊 本 和 夫 三菱化成株式会社
○
○
(第3分科会主査) 佐 野 和 明 日本化薬株式会社
○
○
渡 辺 哲 男 三井東圧化学株式会社
○
○
岩 野 三 夫 保土谷化学工業株式会社
○
○
松 本 良 暉 スガイ化学工業株式会社
○
○
(矢野 公孝) (スガイ化学工業株式会社)
○
○
堀 田 善 和 三星化学工業株式会社
○
○
金 子 正 弥 株式会社三星化学研究所
○
○
井 上 省 三 三和化学工業株式会社
○
○
今 元 建 三 大日本インキ化学工業株式会社
○
○
彦 坂 道 邇 東洋インキ製造株式会社
○
○
大 山 松 二 精工化学株式会社
○
○
小薗井 勝 新日鐵化学株式会社
○
清 水 正 裕 川口化学工業株式会社
○
(事務局)
柳 瀬 齋 彦 化成品工業協会
○
○
○
○
(関係者)
高 橋 潔 工業技術院標準部
平 塚 智 章 工業技術院標準部
御 須 孝 通商産業省基礎産業局
住 吉 和 夫 住友化学工業株式会社
吉 岡 武 司 三井東圧化学株式会社
境 清 スガイ化学工業株式会社
藤 川 昇 一 三和化学工業株式会社
正 田 邦 之 大日本インキ化学工業株式会社
川 島 義 範 東洋インキ製造株式会社
後 藤 敏 生 化成品工業協会