K 2601:1998
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目次
ページ
1. 適用範囲 ························································································································ 1
2. 試験方法の種類 ··············································································································· 1
3. 密度試験方法 ·················································································································· 2
4. 蒸気圧試験方法 ··············································································································· 2
5. 動粘度試験方法 ··············································································································· 2
6. 流動点試験方法 ··············································································································· 2
7. 残留炭素分試験方法 ········································································································· 2
8. 硫黄分試験方法 ··············································································································· 3
9. 窒素分試験方法 ··············································································································· 3
10. 灰分試験方法 ················································································································ 3
11. 発熱量試験方法·············································································································· 3
12. 水分試験方法 ················································································································ 3
13. 引火点試験方法 ············································································································· 3
14. 水でい分試験方法 ·········································································································· 8
15. 塩分試験方法(滴定法)································································································· 14
16. 塩分試験方法(導電率法)······························································································ 22
17. 原油常圧法蒸留試験方法································································································· 29
18. ワックス分試験方法 ······································································································ 39
19. 硫化水素分試験方法 ······································································································ 44
付表1 引用規格 ················································································································ 51
附属書1 水でい分試験用水飽和トルエン調製方法 ···································································· 54
附属書2 水でい分試験用試料の均一化方法 ············································································· 56
参考 理論段数15段の精留塔を使用した蒸留試験方法 ······························································· 58
0. 序文 ····························································································································· 58
1. 適用範囲 ······················································································································· 60
2. 引用規格 ······················································································································· 60
3. 用語の定義 ···················································································································· 61
4. 試験方法の概要 ·············································································································· 63
5. 試料採取 ······················································································································· 63
6. 装置 ····························································································································· 63
7. 装置の準備 ···················································································································· 66
8. 試験の手順 ···················································································································· 66
K 2601:1998 目次
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ページ
9. 計算 ····························································································································· 70
10. 試験報告書 ·················································································································· 71
参考附属書A 蒸留カラムの効率測定方法················································································ 83
参考附属書B カラムの動的ホールドアップの測定方法 B1.1 原理 B1.1.1 ···································· 92
参考附属書C 蒸留カラムの熱損失の測定方法 C1.1 原理 C1.1.1 ················································ 94
参考附属書D 温度検出器の位置の確認方法 D1.1 目的 ···························································· 95
参考附属書E 温度応答時間の確認方法 ··················································································· 97
参考附属書F 検出器の校正方法 ···························································································· 98
参考附属書G 還流分割弁の性能検査方法··············································································· 103
参考附属書H 含水原油試料の脱水方法 ················································································· 105
参考付属書I 蒸留温度の計算方法 ························································································· 107

2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K 2601:1998
原油試験方法
Testing methods for crude petroleum
序文 この規格は,1983年に第3版として発行されたISO 3680,Paints,varnishes,petroleum and related
products−Flash/no flash test−Rapid equilibrium methodと,1990年に第1版として発行されたISO 9030,Crude
petroleum−Determination of water and sediment−Centrifuge methodを基に,対応する部分については技術的
内容を変更することなく作成した日本工業規格であるが、対応国際規格には規定されていない規定項目[密
度試験方法,蒸気圧試験方法,動粘度試験方法,流動点試験方法,残留炭素分試験方法,硫黄分試験方法,
窒素分試験方法,灰分試験方法,発熱量試験方法,水分試験方法,塩分試験方法(滴定法),塩分試験方法
(導電率法),原油常圧法蒸留試験方法,ワックス分試験方法,硫化水素分試験方法]を日本工業規格とし
て追加している。
1. 適用範囲 この規格は,原油の試験方法について規定する。
なお,理論段数15段の精留塔を使用した蒸留試験方法を参考に示す。
備考1. この規格は,危険な試薬,操作及び装置を使うことがあるが,安全な使用方法をすべてにわ
たって規定しているわけではないので,この試験方法の使用者は使用に先立って,適切な安
全上及び健康上の禁止事項を決めておかなければならない。
2. この規格の引用規格を,付表1に示す。
3. この規格の対応国際規格を,表1に示す。
4. この規格の中で{ }を付けて示してある単位及び数値は,従来単位によるものであって,
参考として併記したものである。
表1 対応国際規格
試験方法
対応国際規格
引火点
ISO 3680:1983
Paints,varnishes,petroleum
and
related
products-Flash/no flash test−Rapid equilibrium method
水でい分
ISO 9030:1990
Crude petroleum−Determination of water and sediment
−Centrifuge method
2. 試験方法の種類 試験方法の種類は,表2のとおりとする。

2
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表2 試験方法の種類
試験方法
箇条番号
密度試験方法
3.
蒸気圧試験方法
4.
動粘度試験方法
5.
流動点試験方法
6.
残留炭素分試験方法
7.
硫黄分試験方法
8.
窒素分試験方法
9.
灰分試験方法
10.
発熱量試験方法
11.
水分試験方法
12.
引火点試験方法
13.
水でい分試験方法
14.
塩分試験方法(滴定法)
15.
塩分試験方法(導電率法)
16.
原油常圧法蒸留試験方法
17.
ワックス分試験方法
18.
硫化水素分試験方法
19.
備考1. 水でい分試験方法に用いる水飽和トルエンの調整方法を附属
書1(水でい分試験用水飽和トルエン調整方法)に示す。
2. かくはん機を用いて水でい分試験用試料を均一化する方法を,
附属書2(水でい分試験用試料の均一化方法)に示す。
3. 密度試験方法 JIS K 2249の規定による。ただし,浮ひょう法,振動式密度計法以外で測定した場合
は,その試験方法の種類を明記する。
また,15℃以外の温度で測定した測定密度を,15℃の密度に換算する場合は,JIS K 2249に規定する付
表IA(原油の温度に対する密度換算表)から求める。
4. 蒸気圧試験方法 JIS K 2258の規定による。ただし,精度の規定は適用しない。
なお,流動しにくい試料のときは,流動可能なできるだけ低い温度で試料を採取する。
5. 動粘度試験方法 JIS K 2283に規定する動粘度試験方法によって30℃,50℃又は75℃における動粘度
を測定する(1)。ただし,精度の規定は適用しない。
注(1) 動粘度の試験温度は,試料が完全に流動する温度以上とする。
また,動粘度を測定する際には,“揮発性成分を含む場合”の操作に従い,試験操作に要する
時間はできるだけ短くする。
6. 流動点試験方法 JIS K 2269に規定する流動点試験方法による。ただし,精度の規定は適用しない。
7. 残留炭素分試験方法 JIS K 2270の規定による。ただし,精度の規定は適用しない。
なお,水でい分又は軽質分の多い原油については,JIS K 2270の附属書の規定によって残油を回収し,
得られた残油中の残留炭素分を測定し,次の式によって算出してもよい。
M
m
B
A
×
=
3
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ここに,
A: 原油の残留炭素分(質量%)
B: 10%残油の調製方法で得られた残油の残留炭素分(質量%)
m: 10%残油の調製方法で得られた残油の質量(g)
M: 10%残油の調製方法で蒸留フラスコにはかり採った試料の質
量(g)
8. 硫黄分試験方法 JIS K 2541に規定する燃焼管式(空気法)又は放射線式(励起法)による。ただし,
室温で軽質分が蒸発するような試料に対しては,精度の規定は適用しない。
9. 窒素分試験方法 JIS K 2609の規定による。ただし,精度の規定は適用しない。
10. 灰分試験方法 JIS K 2272に規定する灰分試験方法による。
なお,水でい分又は軽質分の多い原油については17.5などの常圧法蒸留試験によって残油を回収し,得
られた残油中の灰分を測定し,次の式によって算出してもよい。
M
m
D
C
×
=
ここに,
C: 原油の灰分(質量%)
D: 常圧法蒸留試験で得られた残油の灰分(質量%)
m: 常圧法蒸留試験で得られた残油の質量(g)
M: 常圧法蒸留試験で蒸留フラスコにはかり採った試料の質量
(g)
11. 発熱量試験方法 JIS K 2279の規定による。ただし,精度の規定は適用しない。
12. 水分試験方法 JIS K 2275の規定による。
13. 引火点試験方法
13.1 室温で流動する試料の場合 JIS K 2265に規定するタグ密閉式又はペンスキーマルテンス密閉式に
よる。
ただし,精度の規定は適用しない。
13.2 室温で流動しない試料の場合 迅速平衡式引火点試験方法による。迅速平衡式引火点試験方法を次
に示す。
(1) 試験の原理 引火するか否かを確認しようとする温度(以下,この温度を“判定温度”とする。)を,
0〜110℃の範囲内で決定する。試料2〜3gを判定温度に保った試料カップ中に入れ,60秒間(2)放置す
る。試験炎をのぞかせて引火するか否かを観察し,判定温度において引火,又は未引火とする。
注(2) 判定温度が室温以下の場合には,5〜10分間放置する。
参考1
迅速平衡式引火点試験方法は,セタ密閉式引火点試験方法と呼ぶこともある。
2. 判定温度は,消防法による第一石油類,第二石油類又は第三石油類を区分する温度,すなわ
ち21℃又は70℃とするか,軽質留分の含有量から推定した温度とする。
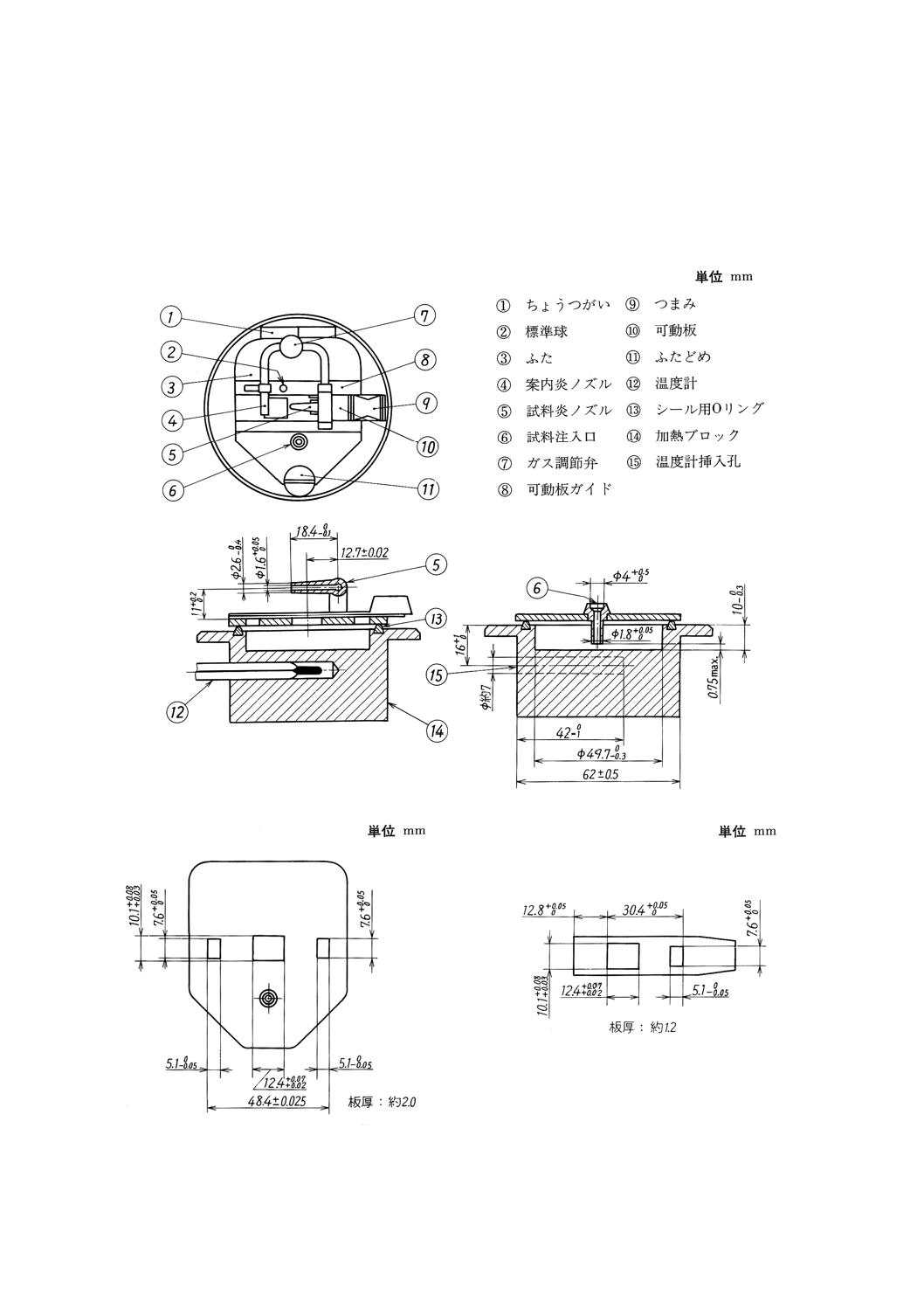
(2) 迅速平衡式引火点試験器 迅速平衡式引火点試験器は,(2.1)〜(2.4)からなり,その一例を図1に示す。
(2.1) 加熱ブロック アルミニウム合金などの熱伝導性のよい耐腐食性金属製のブロックで,試料を保持
するための深さ約10mm,直径約50mmの円筒状のくぼみ(試料カップ),ふたと密着して気密を保

4
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
つためのOリング及び温度計挿入孔を設けたもの。その詳細は,図1に示す。
(2.2) ふた 黄銅などの金属製のもので試料カップ上面に取り付けて,ちょうつがい及びふたどめによっ
て開閉する機構とし,閉の状態でOリングに密着して試料上の空間を密閉する。ふたには3個の窓,
試料注入管,試験炎のぞき機構及び可動板を設ける。その詳細は,図2に示す。
(a) 可動板 黄銅などの金属製のもので,可動板ガイドに沿ってふたに密着してしゅう動して,ふたに
設けた3個の窓を開閉し,試験炎をのぞかせる機構とする。開の状態とした際,可動板の2個の窓
は対応するふたの2個の窓と正確に合致しなければならない。
また,ふたの上面と可動板との間の滑り面はすり合わせて,試料蒸気が漏れないようにしなけれ
ばならない。その詳細は,図3に示す。
(b) 試験炎のぞき機構 黄銅又はステンレス鋼製のもので,試験炎の大きさを3.5±0.5mmに調節する
ことができ,可動板を開としたときふた中央の窓へ試験炎をのぞかせる構造とする。
なお,試験炎をのぞかせたとき,試験炎ノズルの先端の中心がふたの下側の平面と±0.1mmの範
囲内で一致しなければならない。その詳細は,図1に示す。
(2.3) 温度計 表3に規定するもの。
なお,温度計の水銀球と加熱ブロックの温度計挿入孔との透き間には熱伝導性のよいペーストを
詰める。
参考 熱伝導性のよいペーストとしては,シリコン系のグリースに金属酸化物を混入したものが適切
である。
市販品には,Dow Corning 340 Heat Sink Compound,KS 609などがある。
表3 迅速平衡式引火点試験用ガラス温度計
目盛範囲 ℃
0〜110
浸没 mm
44.5±1
目盛 ℃
目量
1
長目盛線
5
目盛数字
10
目盛の誤差
0.5
膨張室
温度計許容加熱温度 ℃
120
全長 mm
20024
−
+
直径 mm
6〜7
球 mm
長さ
10〜14
直径
4.50〜4.65
目盛の位置 mm
球下端から0℃目盛線までの距離
50±2
球下端から110℃目盛線までの距離
165〜185
膨らみ
なし
備考 温度計は,JIS B 7410の附属書に規定する補正試験を行って,あらかじ
め目盛の誤差を求めておく。
(2.4) 加熱器 設定温度が常温以上の場合に,試料カップを設定温度±0.2℃に保つことができる温度調節
機構をもち,加熱ブロックが設定温度に達したことを表示する機能を備えた電熱式のもの。
(2.5) 試料管 ステンレス鋼などの金属製で,内径は約10mmで長さは適切なもの。
(2.6) 冷却袋 設定温度が室温以下の場合に,加熱ブロックを冷却するために使用するもので,プラスチ
ック製の袋に凍結温度の低い液体を封入した市販の保冷剤を冷凍庫などで冷却したものか,又は試
験温度に応じて,プラスチック製の袋などに砕氷若しくは砕氷と塩化ナトリウムの混合物を入れた
5
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
もの。
(2.7) 試料ならし具 試料カップに採取した試料を押し付けて,試料カップ底部に均一な厚さとなるよう
に広げるために使用するもので,試料カップの内径よりやや小さい直径の適切な金属製円板に柄を
取り付けたもの,又は適切な大きさのガラス製若しくは金属製のビーカーを用いてもよい。
図1 迅速平衡式引火点試験器
図2 ふた
図3 可動板
(3) 試料の採取及び調製 試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法,
又はそれに準じた方法によって採取及び調製し,試験を実施するまで密閉した容器内に保存する。
6
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
また,容器内の内容物上の空間は容器全容量の10%を超えないように注意する。
備考 プラスチック容器(ポリプロピレン製,ポリエチレン製など)は,試料中の揮発性物質が壁面
を透過して拡散する可能性があるので使用してはならない。
(4) 試験器の準備 試験器は,できるだけ通風の少ない場所に置く。直射日光や強い照明は避ける。
(5) 温度の設定 判定温度を整数単位で設定し,これを設定温度とする。
備考 気圧が99.4〜103.2kPa{746〜774mmHg}の範囲を超えている場合には,次の式によって判定温度
から設定温度を計算し,整数位に丸める。
TC=T−0.25 (101.3−P1)
{TC=T−0.033 (760−P2) }
ここに,
TC: 設定温度(℃)
T: 判定温度(℃)
P1: 気圧(kPa)
P2: 気圧(mmHg)
(6) 試験の手順 (5)で求めた設定温度に応じて,(6.1)設定温度が室温を超える場合,又は(6.2)設定温度が
室温以下の場合に従って試験を行う。
(6.1) 設定温度が室温を超える場合
(a) 試料,試料ならし具及び清浄な試料管を設定温度より約10℃低い温度に保つ。ただし,設定温度が
室温より10℃以上高い場合は,試料,試料ならし具及び試料管を室温に保つ。
(b) 試料カップの内面,ふた及び可動板をティッシュペーパーなどでふき,清浄にする。試料注入口を
耐熱性のゴム栓などで密閉した後,ふたを閉じる。
(c) 加熱器のスイッチを入れ,温度調節器を調整して温度計の読みを設定温度に正確に合わせて,温度
を安定させる。
(d) 試料中に試料管を突き刺した後,試料管上端を指でふさいで引き抜くことによって,試料を採る。
この際,試料を採る位置は試料管の先端が試料容器の側壁及び試料容器の中心からほぼ等距離で,
深さは試料上面及び試料容器底面からほぼ等距離の点に位置することが望ましい。
備考 試料の固化状態によっては,試料管で試料を採るのが困難なことがある。この場合には,スパ
チュラなどを用いて試料表面を避けて試料採取を行う。
(e) 試験器のふたを開き,試料管の上端にゴム球などで圧力を加えて,試料2〜3g(3)を試料カップ中に
押し出す。直ちに試料カップの上に薄いプラスチックフィルム(4)を載せ,その上から試料ならし具
を押し付けて,できるだけ試料が試料カップ底部全面に均一な厚さになるように広げる。試料なら
し具及びプラスチックフィルムを取り除き,ふたを閉じてふたどめで固定し,秒時計を始動する。
(e)の操作は,できるだけ迅速に行う。
注(3) 試料管の内径及び試料の密度から,押し出す長さを計算しておく。
(4) プラスチックフィルムは,JIS K 2541に規定する放射線励起法で,セル窓材として使用するポ
リエステル製フィルム,又は家庭で電気冷蔵庫や電子レンジに食品を入れる際,包装に使用す
るもの。
(f) ガス調節弁を開き,案内炎と試験炎に点火して,試験炎の大きさを直径3.5±0.5mmに調節する。
(g) 試料カップに試料を入れてから60秒経過したら,2.5±0.5秒間可動板を開いて試験炎をのぞかせ,
引火(5)の有無を観察する。
注(5) 引火点に近い温度では,試験炎の周りに青白い輪が現れることがあるが,これは引火とみなさ
7
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ず,試料の表面に炎が広がった場合に引火したとみなす。
(h) 引火した場合には(7.1)によって“引火”と表し,試験を終了する。未引火の場合には(a)〜(g)の操作
を繰り返し,引火した場合には(7.1)によって“引火”と表す。2回の試験結果がいずれも未引火の
場合には,(7.1)によって“未引火”と表す。
(6.2) 設定温度が室温以下の場合
(a) 試料,試料ならし具及び清浄な試料管を設定温度より3〜5℃低い温度に保つ。
(b) 試料カップの内面,ふた及び可動板をティッシュペーパーなどでふき,清浄にする。試料注入管を
耐熱性のゴム栓などで密閉する。
(c) ふたを開けて試料カップ中に冷却モジュール(6)を置くか,又はふたを閉じてふたの上に冷却袋をか
ぶせて,温度計が設定温度より3〜5℃低い温度を示すまで加熱ブロックを冷却する。
注(6) 冷却モジュールは,ステンレス鋼などの金属製のシリンダ状の容器で,外径は試料カップ内に
密着する大きさとし,高さは200mm以上とする。
なお,試料カップに密着する部分以外の外壁を適切な保温材で覆う。この中に砕氷又は砕氷
と塩化ナトリウムの混合物を入れて,試料カップ中に置き加熱ブロックを冷却する。
(d) 試料中に試料管を突き刺した後,試料管上端を指でふさいで引き抜くことによって,試料を採取す
る。この際,試料を採取する位置は試料管の先端が試料容器の側壁及び試料容器の中心からほぼ等
距離で深さは試料上面及び試料容器底面からほぼ等距離の点に位置することが望ましい。
備考 試料の固化状態によって,試料管による試料採取が困難なことがある。この場合には,スパチ
ュラなどを用いて試料表面を避けて試料採取を行う。
(e) 試験器のふたを開き,凝縮水が認められる場合は,ティッシュペーパーなどでふき取る。試料管の
上端にゴム球などで圧力を加えて,試料2〜3g(3)を試料カップ中に押し出す。直ちに試料カップの
上に薄いプラスチックフィルム(4)を載せ,その上から試料ならし具を押し付けて,できるだけ試料
が試料カップ底部全面に均一な厚さになるように広げる。試料ならし具及びプラスチックフィルム
を取り除き,ふたを閉じてふたどめで固定し,秒時計を始動する。
(e)の操作は,できるだけ迅速に行う。
(f) ふたの上に冷却袋をかぶせて加熱ブロックを保冷し,5分以上加熱ブロックの温度を設定温度又は
設定温度より3℃低い温度の範囲に保つ。
(g) 冷却袋を取り除いた後,ガス調節弁を開き案内炎と試験炎に点火して,試験炎の大きさを直径3.5
±0.5mmに調節する。
(h) 試験器をそのまま放置して,温度計が設定温度を示したら,2.5±0.5秒間シャッタを開いて試験炎
をのぞかせ,引火(5)の有無を観察する。ただし,(e)で秒時計を始動してから試験炎をのぞかせるま
での時間が10分を超えた場合には,(f)の保冷温度を変えて試験をやり直す。
(i) 引火した場合には(7.1)によって“引火”と表し,試験を終了する。未引火の場合には(a)〜(h)の操作
を繰り返し,引火した場合には(7.1)によって“引火”と表す。2回の試験結果がいずれも未引火の
場合には,(7.1)によって“未引火”と表す。
(7) 試験結果及び精度
(7.1) 試験結果 迅速平衡式引火点試験の結果は,(5)で決めた判定温度を整数で,また,判定温度におい
て“引火”又は“未引火”であることを表す。その際,迅速平衡式引火点試験の結果であることを,
例えば略号(RCC)などで記す(例1.及び例2.参照)。
備考 設定温度を補正した場合には,実際に試験を行ったときの温度すなわち補正した設定温度では
8
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
なく,未補正の設定温度で表す。
例1.引火点(RCC)
………
21℃,引火
例2.引火点(RCC)
………
5℃,未引火
参考 RCCは,Rapid equi1ibrium Closed Cupの頭文字を表す。
(7.2) 精度 引火点試験方法の精度は,規定しない。
(8) 試験結果の報告 試験結果には,次の事項を記載する。
(8.1) 試料名,採取場所及び採取年月日
(8.2) JISの規格番号 例 JIS K 2601
(8.3) 試験方法の名称・項番号及び(7.1)によって得られた結果
(8.4) 特記事項
14. 水でい分試験方法
14.1 試験の原理 遠心分離用の目盛試験管2本にそれぞれ試料50m1と水で飽和したトルエン50m1を採
り,完全に混合してから遠心分離器で60℃(7)で10分間の遠心分離操作を2回繰り返し,目盛試験管底部
に沈積した水でい層の容量を読み,水でい分を算出する。
注(7) あらかじめ49℃で沈殿物にワックス状のものが認められないことが分かっている場合には,
49℃で遠心分離を行ってもよい。
備考 この試験方法で得られる結果は,水と沈積物の総量である。したがって,水分だけを正確に測
定するには12.による。
参考 60℃で試験を行った際,沈殿物にワックス状のものが認められた場合には,ワックス状のもの
が認められなくなる温度で試験をやり直す。一例として75℃がよい。ただし,精度は規定しな
い。
14.2 水でい分試験器 水でい分試験器は,次による。
(1) 遠心分離器 試料を入れた規定の目盛試験管2個以上の偶数個を60±3℃(8)の温度に保ち,規定速度
で回転できるもので,かつ,引火性の雰囲気中であっても安全に操作でき,構造が丈夫なもの。目盛
試験管保持管は金属製とし,これに緩衝用としてフェルト,ゴムその他適切なものを入れる。回転部
は破損したとき危険を防止できるように,丈夫な金属製の囲いで覆う。回転速度は,目盛試験管底部
における相対遠心力が600になるように調節する。所要の毎分回転数は,次の式によって算出する。
d
f
R
335
1
=
ここに, R: 毎分回転数(rpm)
f: 相対遠心力
d: 回転直径(回転状態において相対する目盛試験管の両底間の
距離)(mm)
注(8) 49℃で試験を行う場合には,10分間遠心分離した後の目盛試験管内の溶液の温度が46℃以上あ
ればよい。
参考 回転直径と回転数の関係を参考表1に示す。
9
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考表1 遠心分離器の所要回転数
遠心分離器の回転直径
mm
相対遠心力が600となる
毎分回転数 rpm
340
1 770
360
1 720
380
1 680
400
1 640
420
1 600
440
1 560
460
1 520
480
1 490
500
1 460
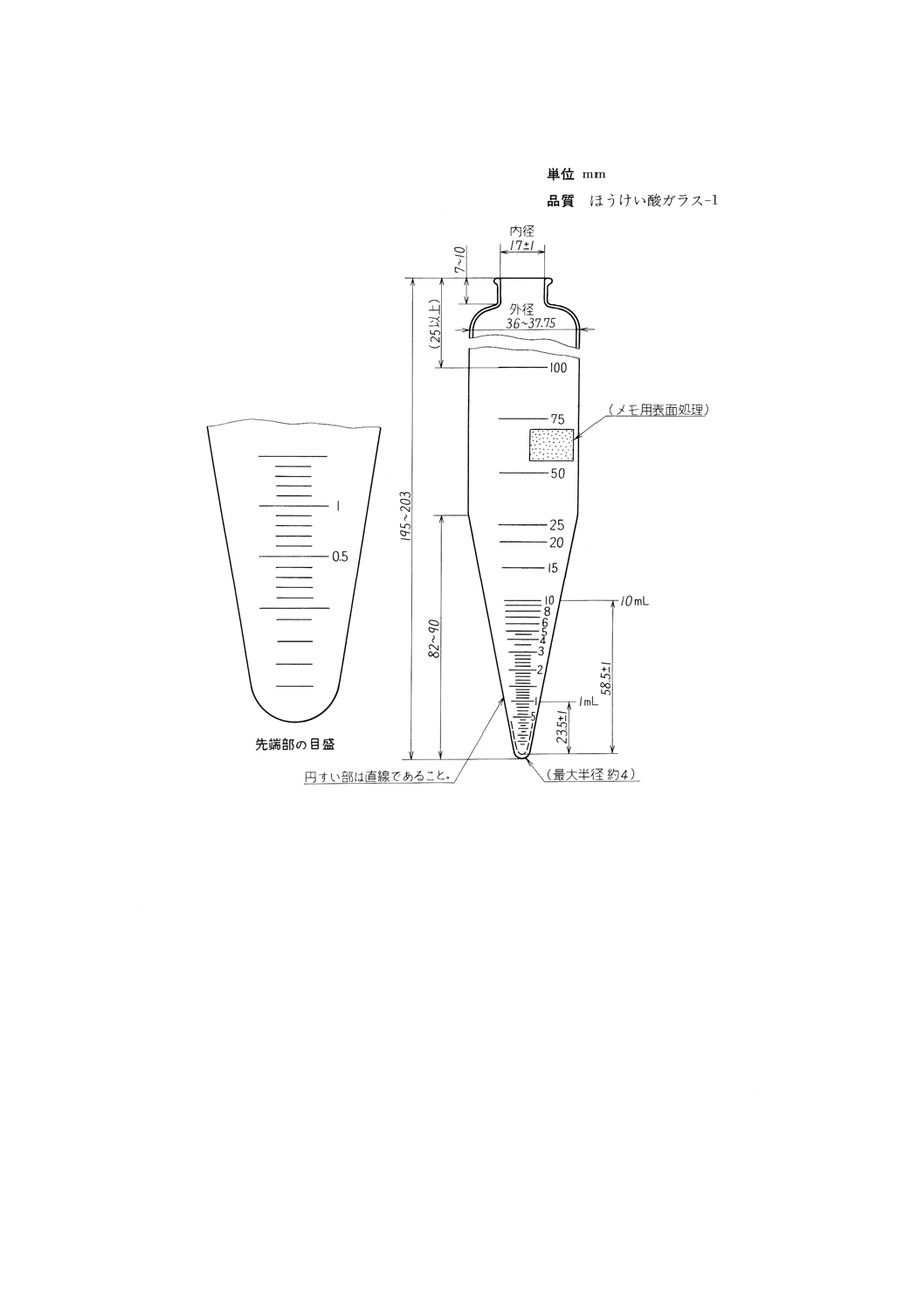
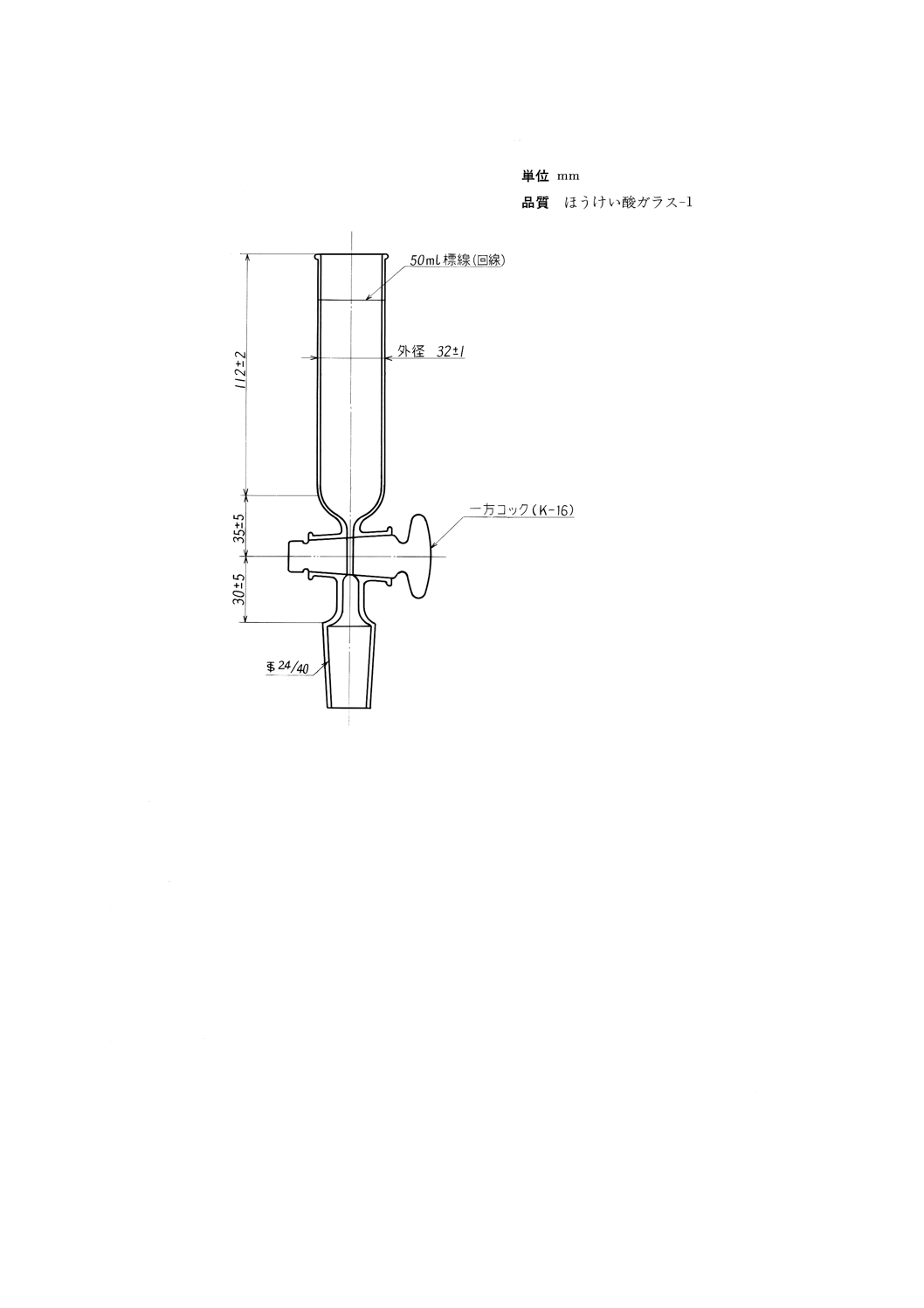
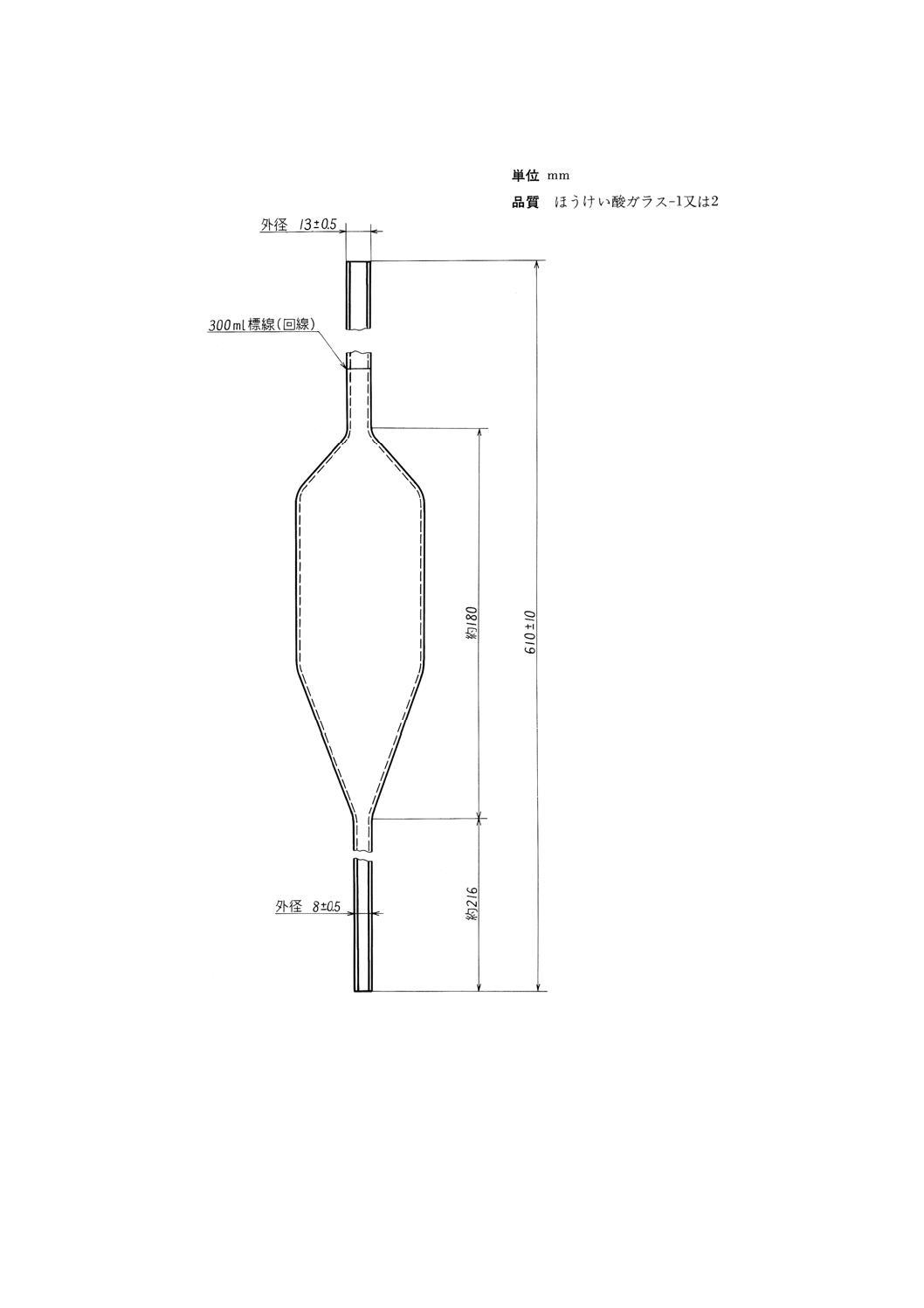
(2) 目盛試験管 目盛試験管は,図4に示す寸法に適合し,ほうけい酸ガラス−1製であること。
各目量及びその許容差については表4に示す。新しい目盛試験管は使用し始める前に,0.25mLまで
の各目盛線(図5参照),及び0.5,1.0,1.5,2.0,50.0,100mLの目盛線の精度を確認する。確認の
方法は,洗浄,乾燥した目盛試験管の質量を正確にはかり,空気を含まない水を20℃において各目盛
線まで加え,それぞれ再度質量を正確にはかる。その差によって求められた水の質量と,そのときの
水の密度を用いて体積に換算し,表4の許容差を超えていないことを確認する。
また,使用する水の密度は,JIS K 2249に規定する試験方法を参照する。
表4 目盛試験管の目盛許容差
単位 mL
範囲
目量
許容差
0〜0.1以下
0.05
±0.02
0.1を超え
0.3以下
0.05
±0.03
0.3を超え
0.5以下
0.05
±0.05
0.5を超え
1.0以下
0.10
±0.05
1.0を超え
2.0以下
0.10
±0.10
2.0を超え
3.0以下
0.20
±0.10
3.0を超え
5.0以下
0.5
±0.20
5.0を超え
10以下
1.0
±0.50
10を超え
25以下
5.0
±1.00
25を超え
100以下
25.0
±1.00
10
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図4 目盛試験管
(3) 加熱浴 金属浴又は液浴とし,目盛試験管を100mLの目盛線まで垂直に浸すことのできる深さで,浴
温を49±3℃及び60±3℃に保つことのできるもの。
14.3 試薬 水でい分試験方法の試薬は,次による。
(1) トルエン JIS K 8680に規定するもの,又はJIS K 2435に規定する純トルエン1号又は純トルエン2
号。
(2) 水飽和トルエン (1)のトルエンに,附属書1(水でい分試験用水飽和トルエン調製法)によって60±
3℃又は49±3℃において水を飽和させたもの。
(3) 解乳化剤溶液 試料中の水の分離を促進し,分離した水分が目盛試験管の内壁に付着するのを防ぐた
め,製油所などで原油の脱塩に通常使用する解乳化剤(フェノール系,塩基性アミン系又はナフテン
酸系)を使用する。
解乳化剤の濃度及び使用量は,水でい分の結果に正の誤差を生じない程度とする。
なお,解乳化剤溶液は濃暗色の瓶に入れ,密栓して貯蔵する。貯蔵中に,解乳化剤が沈降して2層
に分離する場合には,新たに解乳化剤溶液を作り直す。
備考 解乳化剤溶液の濃度は,解乳化剤25容量%及びトルエン75容量%が適切であるが,原油によ
11
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
っては異なる濃度が要求される場合もある。使用する解乳化剤の種類,濃度及び使用量につい
ては,試験結果の報告に付記することが望ましい。
参考 ここで使用する解乳化剤の一例として,N-n-ブチルジエタノールアミンがある。
14.4 試料の採取及び調製 試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法,
又はそれに準じた方法によって採取及び調製し,試験の直前に試料をよく振とうして,試料中の水分及び
でい分を十分に分散させなければならない。
備考1. 試料を均一化するには,附属書2(水でい分試験用試料の均一化方法)によるとよい。
2. 常温で流動しないか,又は粘度の大きい試料は容易にかくはんできる粘度になるように加温
する。ただし,揮発性物質が揮散するおそれがあるため,必要以上に高温度にしたり,長時
間加温したりしてはならない。
14.5 試験の手順 試験の手順は,次による。
(1) 目盛試験管2本それぞれに試料を50mLの標線まで入れ,水飽和トルエン50mLをピペットで加え,
更に解乳化剤溶液0.2mLをメスピペットで加える。次に,密栓して約10回逆さまにし,試料と水飽
和トルエンを完全に混合する。
(2) 試料が非常に高粘度なため,水飽和トルエンとの混合が困難な場合には,混合を容易にするため,目
盛試験管に最初に水飽和トルエンを加えてもよい。この場合には,試料を加える際に100mLの目盛を
超えないように注意する。
(3) 60±3℃(9)の加熱浴に,栓を少し緩めた目盛試験管を100mLの目盛線まで浸し15分以上保持する。再
び目盛試験管に栓をし,逆さまにして試料と水飽和トルエンを完全に混合する。
注(9) あらかじめ49±3℃で沈殿物にワックス状のものが認められないことがわかっている場合には,
49±3℃に加温してもよい。
参考 60±3℃で試験を行った際,沈殿物にワックス状のものが認められた場合には,ワックス状のも
のが認められなくなる温度で(3)〜(6)の操作を繰り返す。一例として75±3℃がよい。
(4) あらかじめ加温した遠心分離器の対称な目盛試験管保持管に2本の目盛試験管を納め(10),相対遠心力
が600となる回転数で10分間回転させる。
注(10) 2本の目盛試験管に質量差がある場合には,目盛試験管保持管に水を入れてバランスをとる。
(5) 遠心分離器の回転が止まったら,直ちに目盛試験管の底部の水分及びでい分の体積の合量を読み取る
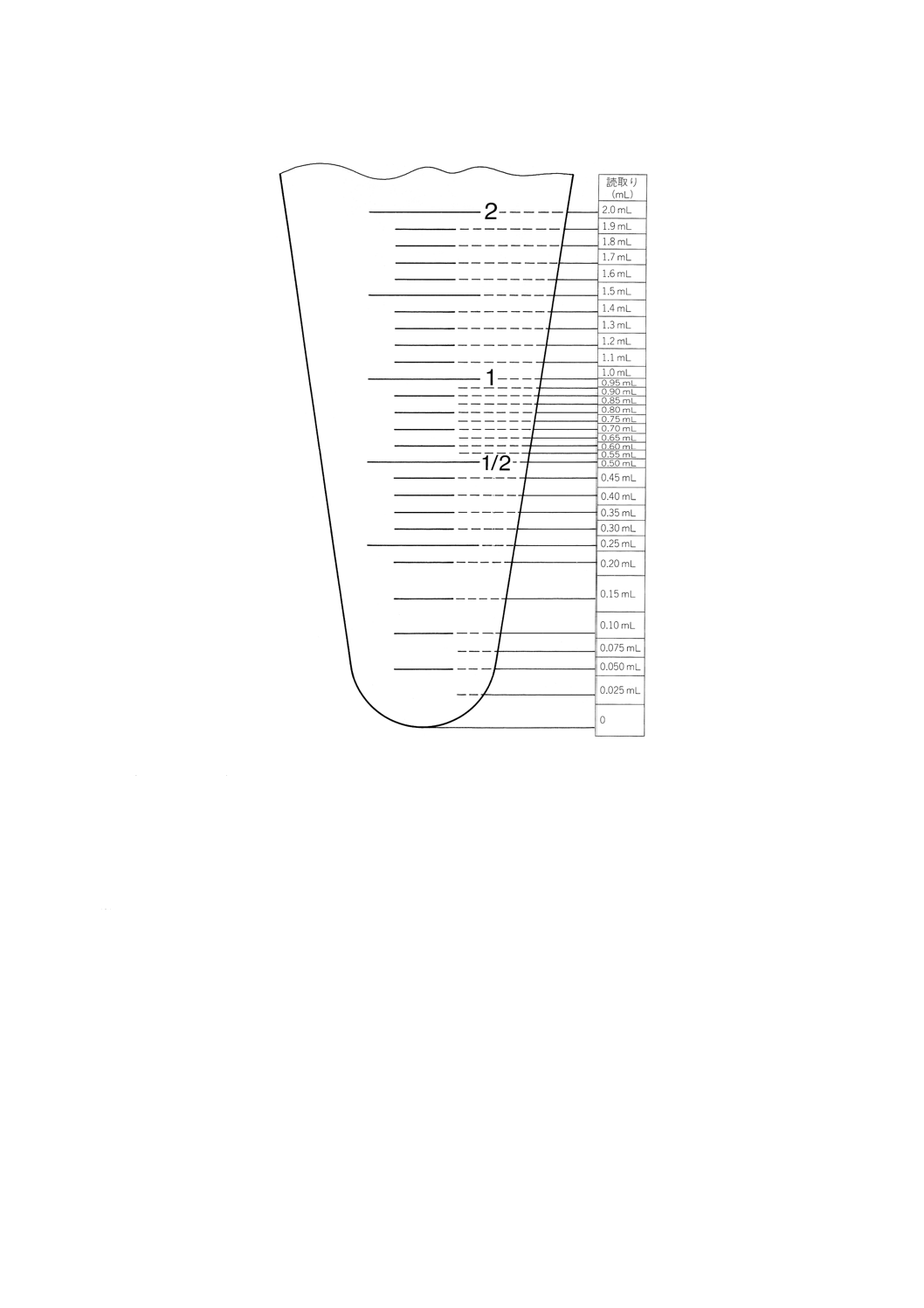
(11)。読取りは,0.1mLから1mLの場合には0.05mLまで,1mL以上の場合には0.1mLまでとし,0.1mL
以下の場合には推定値で0.025mLまでとする。
内容液を振り混ぜないで,再び10分間遠心分離を行う。
注(11) 上澄み液と沈殿物の境界が見にくい場合には,水分及びでい分が流れないように注意しながら,
目盛試験管を静かに傾けて水分及びでい分の体積の合量を読み取るとよい。
49±3℃で試験を行った際,沈殿物にワックス状のものが認められた場合には,試験温度を
60±3℃として,(3)〜(6)の操作を繰り返す。
参考 60±3℃で試験を行った際,沈殿物にワックス状のものが認められた場合には,ワックス状のも
のが認められなくなる温度で(3)〜(6)の操作を繰り返す。一例として75±3℃がよい。
(6) 水分及びでい分の合量の体積が続けて2回同じになるまで,(4)及び(5)の操作を繰り返す。通常は,2
回の操作で十分である。遠心分離操作全体を通じて,試料の温度は,60±3℃(12)を保っていなければ
ならない。
注(12) 49±3℃又は75±3℃で試験を行った場合には,それぞれその温度の範囲内に保っていなければ

12
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ならない。
14.6 試験結果及び精度
14.6.1 試験結果 試験結果は,次による。
(1) それぞれの目盛試験管の最終的な水でい分の体積を記録する。もし二つの読みの差が目盛試験管の最
小目盛を超えるか,又は0.10mL以下のときに0.025mLを超える場合には,試験結果を捨てて試験を
やり直す。
(2) 2本の目盛試験管の最終読取り量の和を水でい分(容量%)とする(表5参照)。
(3) 60℃以外で試験を行った場合は,その温度を明記する。
表5 試験結果の求め方
目盛試験管1の水でい分の体積
mL
目盛試験管2の水でい分の体積
mL
水でい分の結果
%(v/v)
目視で水でい分なし
目視で水でい分なし
0
目視で水でい分なし
0.025
0.025
0.025
0.025
0.05
0.025
0.05
0.075
0.05
0.05
0.10
0.05
0.075
0.125
0.075
0.075
0.15
0.075
0.10
0.175
0.10
0.10
0.20
0.10
0.15
0.25
13
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図5 水でい分体積が低レベルの場合の読取り方
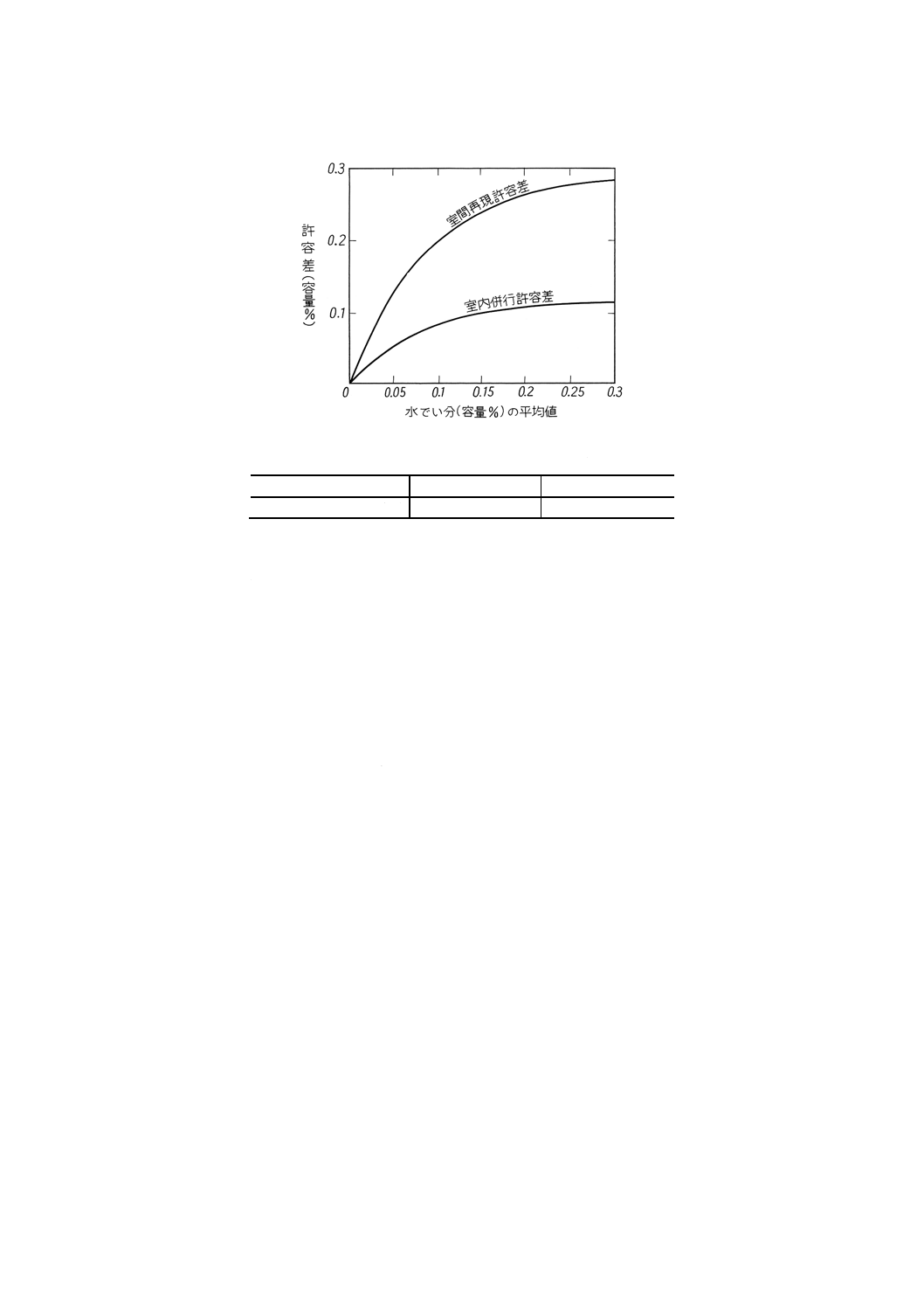
14.6.2 精度 試験温度が60±3℃の場合の,水でい分試験によって得られた試験結果の許容差(確率0.95)
は,次のとおりである。
備考 試験結果が許容差を外れた場合には,JIS Z 8402の規定によって処理する。
(1) 室内併行精度 同一試験室において,同一人が同一試験器で,引き続き短時間内に同一試料を2回試
験したときの試験結果が0.3容量%以下の場合の許容差を図6に示す。
また,試験結果が0.3容量%を超え1.0容量%以下の場合の許容差を表6に示す。
(2) 室間再現精度 異なる試験室において,別人が別の試験器で,同一試料をそれぞれ1回ずつ試験した
ときの2個の試験結果が0.3容量%以下の場合の許容差を図6に示す。
また,試験結果が0.3容量%を超え1.0容量%以下の場合の許容差を表6に示す。
14
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図6 精度
表6 精度
単位 容量%
水でい分
室内併行許容差
室間再現許容差
0.3を超え
1.0以下
0.12
0.28
14.7 試験結果の報告 試験結果には,次の事項を記載する。
(1) 試料名,採取場所及び採取年月日
(2) JISの規格番号 例 JIS K 2601
(3) 試験方法の名称・項番号及び14.6.1(2)によって得られた結果
(4) 特記事項
15. 塩分試験方法(滴定法)
15.1 試験の原理 試料約80gを溶剤及び水と共に煮沸して,試料中の塩化物を水層に抽出分離する。こ
の抽出液を指示薬滴定又は電位差滴定して,塩分を塩化ナトリウムとして定量する。
15.2 滴定法塩分試験器 図7に示す構造のもので,(1)及び(2)からなる。
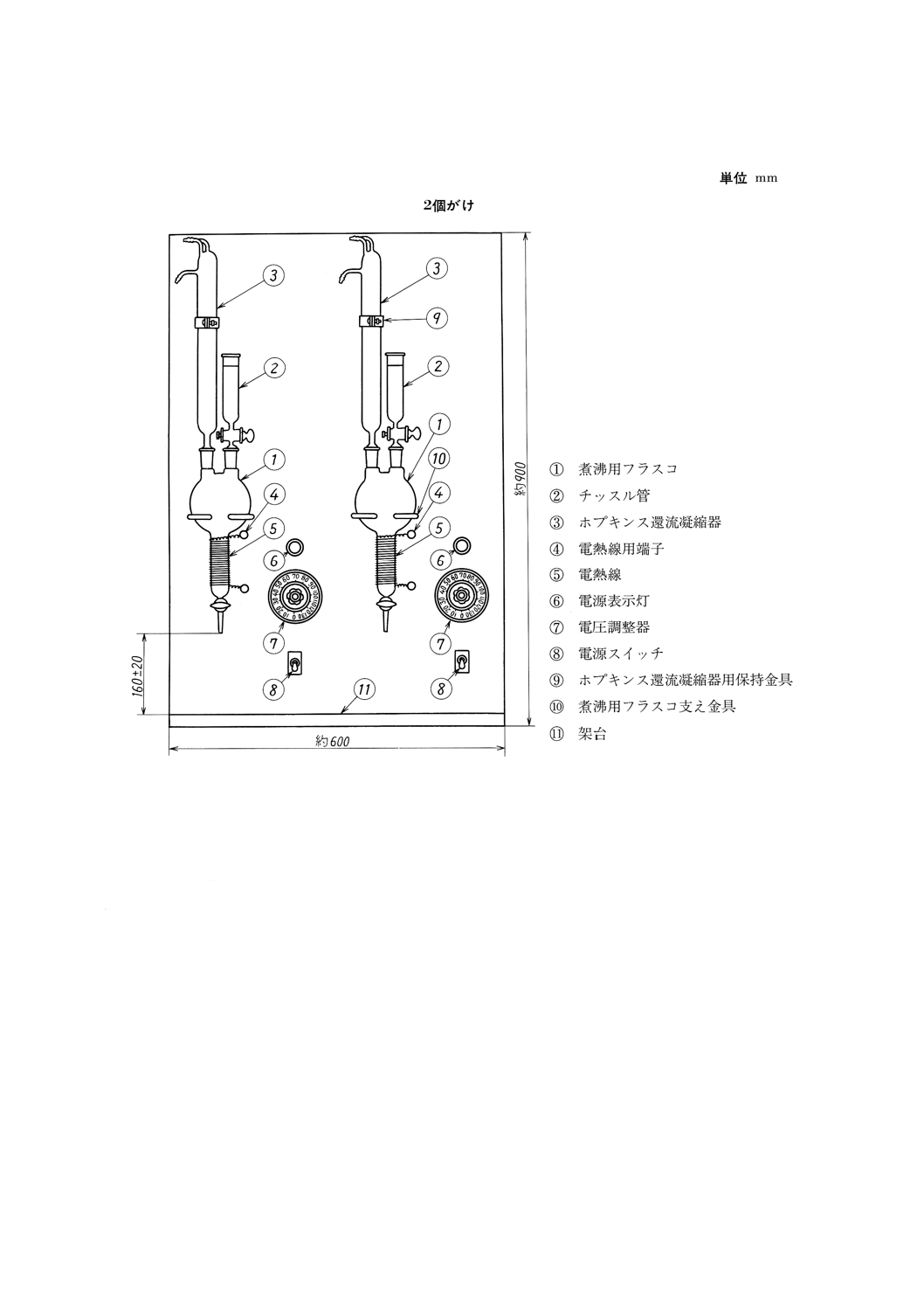
15
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図7 滴定法塩分試験器(一例)
(1) 抽出器 (a)〜(c)からなり,図7のように組み立てる。
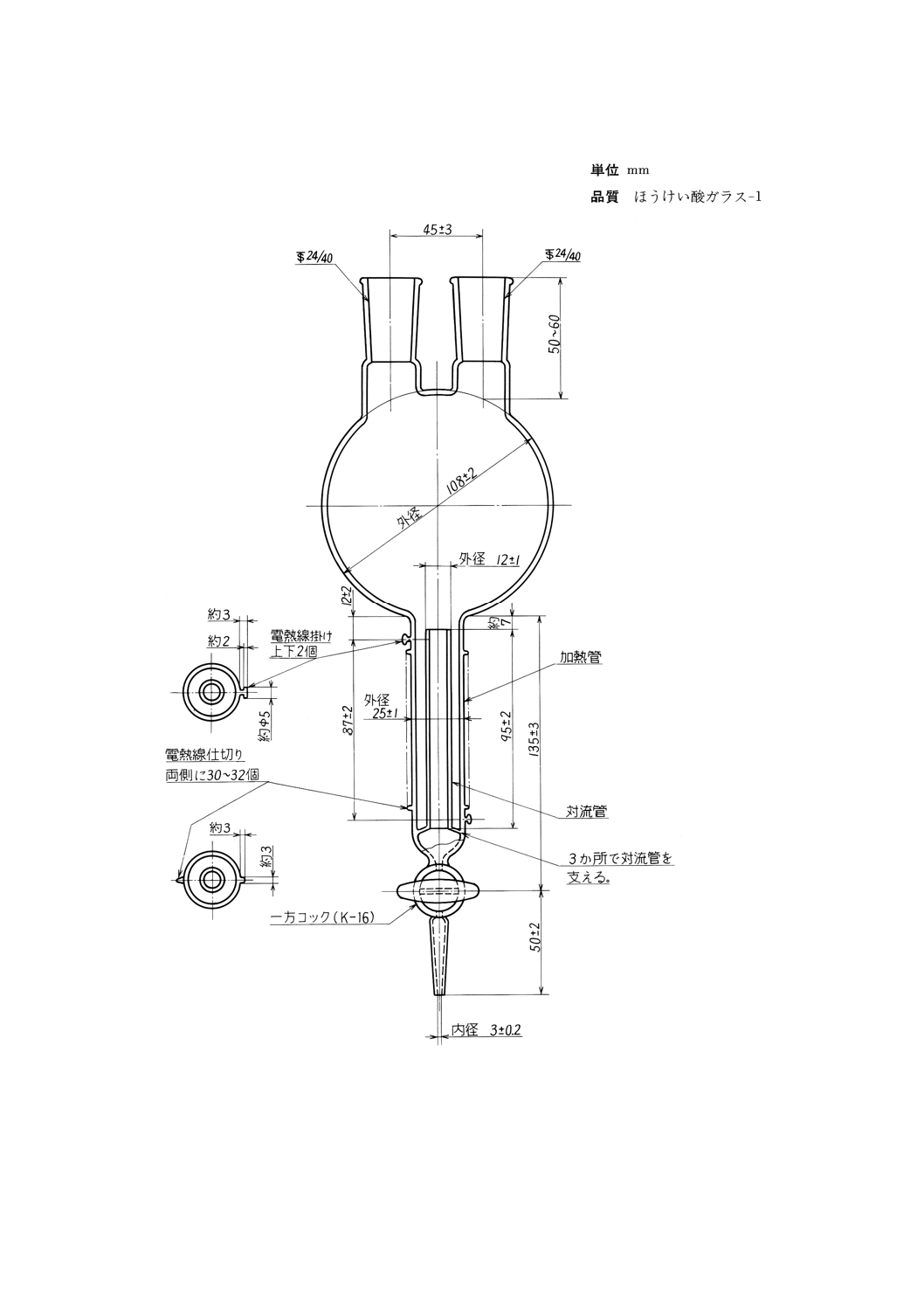
(a) 煮沸用フラスコ 図8に示すもの。ただし,加熱管にはJIS C 2520に規定するニッケルクロム電熱
線1種の,線径0.26mm,長さ約2700mmのものを巻き付ける。
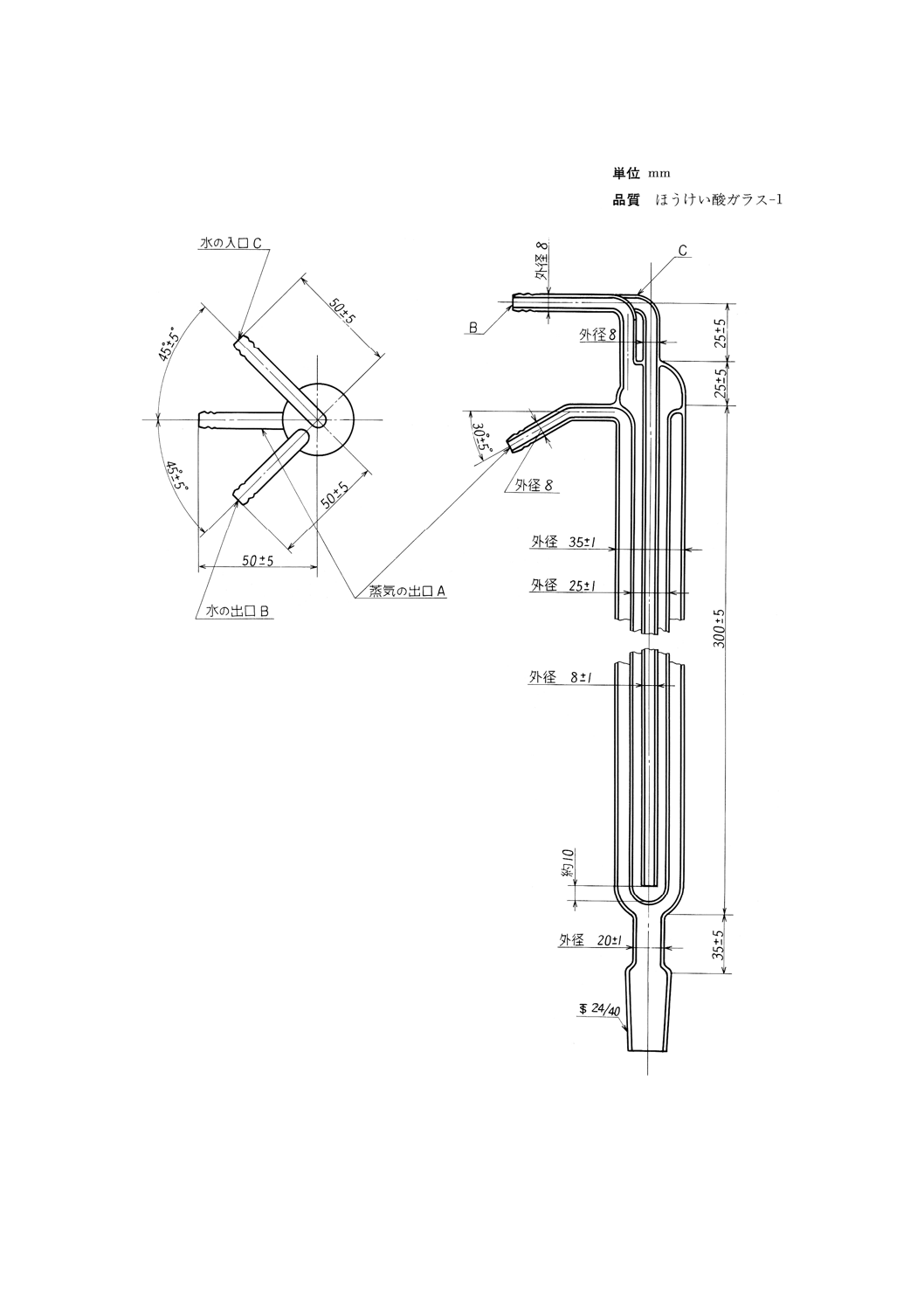
(b) ホプキンス還流凝縮器 図9に示すもの。
(c) チッスル管 図10に示すもの。
(2) 架台 図7に示すように抽出器を保持することができ,かつ,加熱用電圧調整器(0〜100V, 3A以
上)を備えるもの。
16
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図8 煮沸用フラスコ
備考 上下2個の電熱線掛け間の電熱線仕切りの間に電熱線を巻き付ける。
17
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図9 ホプキンス還流凝縮器
18
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図10 チッスル管
15.3 試薬 試薬は,次による。
(1) アセトン JIS K 8034に規定するもの。
(2) エタノール JIS K 8101に規定する特級のもの。
(3) 3−メチル−1−ブタノール JIS K 8051に規定するもの。
(4) トルエン JIS K 8680に規定するもの,又はJIS K 2435に規定する純トルエン1号若しくは純トルエ
ン2号。
(5) 酢酸鉛紙 JIS K 8374に規定する酢酸鉛(II)三水和物19gを水80mLに溶かし,これにろ紙を浸し
たもの。
(6) 硝酸溶液(30%) JIS K 8541に規定する硝酸1容を水1容で薄め,無色になるまで沸騰させ,放冷後空
気を吹き込んで窒素酸化物を除いたもの。
(7) ウラニン指示薬溶液 ウラニン0.2gを水100mLに溶解したもの。
(8) 鉄みょうばん指示薬溶液 JIS K 8982に規定する硫酸アンモニウム鉄(III)・12水28gを水80mLに
溶かし,硝酸溶液(30%)10mLを加えたもの。
(9) 0.02moλ/L硝酸銀標準液 JIS K 8550に規定する硝酸銀約3.4gを水に溶かして1Lとし,着色瓶に入れ
て保存する。
この溶液のモル濃度を,以下によって求める。
19
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(a) JIS K 8005に規定する塩化ナトリウム5g以下を白金るつぼに入れ,600℃で約1時間加熱した後,
無水過塩素酸マグネシウムを乾燥剤としたデシケータ中で0.5〜1時間放冷する。
(b) (a)の塩化ナトウリム1.0〜1.2gを0.1mgのけたまではかり採り,少量の水に溶解する。これを1L全
量フラスコに洗い移し,水を標線まで加える。
(c) (b)の塩化ナトリウム水溶液20mLをピペットで採取し,300mL三角フラスコに入れ,水50mL及び
ウラニン指示薬溶液を加えて,硝酸銀溶液で滴定する。緑色の蛍光が消えて桃色に変わった点を終
点とする。
(d) 硝酸銀溶液のモル濃度は,次の式によって算出し,JIS Z 8401の規定によって小数点以下5けたに
丸める。
100
44
0.058
0.02
P
V
M
C
×
×
×
=
ここに,
C: 硝酸銀溶液のモル濃度(mol/L)
M: (b)ではかり採った塩化ナトリウムの質量(g)
V: 終点までに要した硝酸銀溶液の容量(mL)
P: 塩化ナトウリムの含量(%)
(10) 0.005mol/L硝酸銀標準液 (9)の0.02mol/L硝酸銀標準液を原液として,ピペット及び全量フラスコを
用いて原液を水で4倍に希釈し,着色瓶に入れて保存する。
原液のモル濃度を41倍してモル濃度とする。
(11) 0.02mol/Lチオシアン酸カリウム標準液 JIS K 9001に規定するチオシアン酸カリウム約2gを水に溶
かして1Lとする。
この溶液のモル濃度を,以下によって求める。
(a) 300mL共栓付き三角フラスコにピペットを用いて0.02mol/L硝酸銀標準液25mLを入れ,水25mL,
硝酸溶液(30%)5mL,鉄みょうばん指示薬溶液3mL及びイソアミルアルコール10mLを加える。
(b) よく振り混ぜながらチオシアン酸カリウム標準液で滴定する。終点に近づき,液がやや赤みを帯び
たら三角フラスコに栓をして15秒間激しく振とうし,沈殿物を凝結沈降させた後,栓を取り除く(13)。
注(13) フラスコの栓を取り除く際,振とうしている間に生じた圧力のため,フラスコの口から溶液が
飛び散ることがあるので注意する。
(c) さらに,よく振り混ぜながらゆっくりと滴定して,赤色が消えなくなった点を終点とする。
(d) チオシアン酸カリウム標準液のモル濃度は,次の式によって算出し,JIS Z 8401の規定によって小
数点5けたに丸める。
2
1
V
V
M
C
×
=
ここに,
C: チオシアン酸カリウム標準液のモル濃度(mol/L)
M: 0.02mol/L硝酸銀標準液のモル濃度(mol/L)
V1: 0.02mol/L硝酸銀標準液の採取量(mL)
V2: 滴定に要したチオシアン酸カリウム標準液の量(mL)
15.4 試料の採取及び調製 試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法,
又はそれに準じた方法によって採取及び調製する。
15.5 試験の手順
15.5.1 抽出 抽出は,次による。
(1) 試料約80gを0.1g単位まで250mLビーカーにはかり採り,これを60±5℃まで加熱し,同温度に加熱
20
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
したトルエン40mLを溶液が均一になるようにかき混ぜながらゆっくり加える。この溶液をチッスル
管から煮沸用フラスコに移した後,約60℃に熱したトルエン15mLずつを用いて,ビーカー及びチッ
スル管を2回洗い流す。
煮沸用フラスコ内の溶液が冷えないうちに温エタノール25mL及び温アセトン15mLをチッスル管
から加える。
(2) 煮沸用フラスコ内には突沸防止のため多孔質のゼオライトなどを入れるか,又は窒素ガスの導入など
を行い,溶液を2分間激しく沸騰させる。
次に加熱を止め,沸騰しなくなってから水125mLをチッスル管から加え,更に15分間沸騰させて
放冷する。
(3) 層が分かれたら下層を110mL以上抜き取る。この際必要ならば,ろ紙で抽出液をろ過する。
備考 少量のエタノールとアセトンは試料層に溶解して残るので,有効な下層抽出液量は160mLとす
る。
(4) 抽出液100mLを250mLビーカーにはかり採り,硝酸溶液(30%)5mLを加えてビーカーを時計皿で覆い,
沸騰させる。
蒸気中に硫化水素が存在するか否かを酢酸鉛紙で試験する。
(5) 酢酸鉛紙が黒変しない場合は,15.5.2によって滴定する。
酢酸鉛紙が黒変した場合は,蒸気中に硫化水素がなくなるまで抽出液を沸騰させる。抽出液の沸騰
は,酢酸鉛紙が黒変しなくなった後,更に5分間行う。
15.5.2 滴定 抽出液中の塩分は,(1)又は(2)のいずれかによって測定する。
(1) 指示薬滴定法
(a) 抽出液を放冷した後,水を用いてこれを300mL共栓付き三角フラスコに洗い流し,3−メチル−1
−ブタノール10mLと鉄みょうばん指示薬溶液3mLを加える。
(b) ビュレットを用いて0.02mol/Lチオシアン酸カリウム標準液0.5mLを300mL共栓付き三角フラスコ
に加えた後,フラスコを激しく振りながら0.02mol/L硝酸銀標準液で無色になるまで滴定する。さ
らに,0.02mol/L硝酸銀標準液5mLを加え,フラスコに栓をして15秒間激しく振り,沈殿物を凝結
沈降させた後,栓を取り除く(14)。
注(14) フラスコの栓を取り除く際,振っている間に生じた圧力のため,フラスコの口から溶液が飛び
散ることがあるから注意すること。
(c) 0.02mol/Lチオシアン酸カリウム標準液でゆっくり滴定し,終点が近づき徐々に赤く着色してきたら
止める。フラスコを激しく振り,赤色が消えれば更に滴定を繰り返し,赤色が消えなくなった点を
終点とする。
(d) 空試験は,試料の変わりにトルエン95mLを用いて,15.5.1及び(a)〜(c)の操作を行う。
(2) 電位差滴定法
(a) 15.5.1(5)で得られた抽出液を5〜50mL(15)に濃縮し,アセトン50mL及び硝酸溶液(30%)2mLを加え
た後,硝酸銀標準液(16)で電位差滴定を行う。
電位差計の読み (mV) に対する硝酸銀標準液の滴定曲線を作図し,その変曲部から滴定終点を求
める(17)。
滴定用試料溶液中にメルカプタンが存在する場合は,塩分の変曲点の前にメルカプタンの変曲点
が現れる。この場合,塩分の変曲点までの滴定量からメルカプタンの変曲点までの滴定量を差し引
いた量を塩分の滴定量とする。
21
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
注(15) 試料の塩分が20質量ppm以下の場合は,5〜10mLに濃縮する。
(16) 試料の塩分が20質量ppmを超える場合は,0.02mol/L硝酸銀標準液を,20質量ppm以下の場
合は0.005mol/L硝酸銀標準液を用いる。
(17) 滴定終点は,JIS K 0113の規定に従って求める。
(b) 空試験は,試料の代わりにトルエン95mLを用いて15.5.1及び(a)の操作を行う。
15.6 計算方法及び精度
15.6.1 計算方法 塩分は次の式によって算出し,JIS Z 8401の規定によって整数位に丸める。ただし,1
〜99質量ppmの場合は整数位(1けた又は2けた)で表示し,100〜999質量ppmの場合は,整数10位(3
けた)に丸めて表示する。
(1) 指示薬滴定法の場合
(
)
(
)
[
]
M
n
v
v
N
V
V
S
B
S
B
S
−
−
−
=
×
×
3
10
5.
93
ここに,
S: 塩分(質量ppm)
VS: 試料の滴定に要した0.02mol/L硝酸銀標準液の量 (mL)
VB: 空試験に要した0.02mol/L硝酸銀標準液の量 (mL)
vS: 試料の滴定に要した0.02mol/Lチオシアン酸カリウム標準
液の量 (mL)
vB: 空試験に要した0.02mol/Lチオシアン酸カリウム標準液の
量 (mL)
N: 0.02mol/L硝酸銀標準液のモル濃度
n: 0.02mol/Lチオシアン酸カリウム標準液のモル濃度
M: 試料採取量 (g)
93.5:
子量)
(塩化ナトリウムの分
(使用した抽出液量)
(有効下層抽出液量)58.44
100
160
×
(2) 電位差滴定法の場合
(
)
M
N
V
V
S
B
S−
=
×
×
3
10
5.
93
ここに,
S: 塩分(質量ppm)
VS: 試料の滴定に要した0.02mol/L,又は0.005mol/L硝酸銀標
準液の量 (mL)
VB: 空試験に要した0.02mol/L,又は0.005mol/L硝酸銀標準液
の量 (mL)
N: 0.02moλ/L,又は0.005mol/L硝酸銀標準液のモル濃度
M: 試料採取量(g)
93.5:
子量)
(塩化ナトリウムの分
(使用した抽出液量)
(有効下層抽出液量)58.44
100
160
×
15.6.2 精度 塩分試験方法によって得られた試験結果の許容差(確率0.95)は,次のとおりである。
備考 試験結果が許容差を外れた場合には,JIS Z 8402の規定によって処理する。
(1) 室内併行精度 同一試験室において,同一人が同試験器で,引き続き短時間内に同一試料を2回試験
したときの試験結果の差の許容差を表7に示す。
(2) 室間再現精度 異なる試験室において,別人が別の試験器で,同一試料をそれぞれ1回ずつ試験した
とき,2個の試験結果の差の許容差を表7に示す。

22
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表7 精度
単位 質量ppm
室内併行許容差
室間再現許容差
20
40
15.7 試験結果の報告 試験結果には,次の事項を記載する。
(1) 試料名,採取場所及び採取年月日
(2) JISの規格番号 例 JIS K 2601
(3) 試験方法の名称・項番号及び15.6.1によって得られた結果
(4) 特記事項
16. 塩分試験方法(導電率法)
16.1 試験の原理 試料10mLをキシレン及び混合アルコール溶剤に溶解して導電率(又は電流値)を測
定し,あらかじめ標準液を用いて作成した検量線から,試料中の塩分濃度を求める。
備考 導電率法によって得られた試験結果に疑義が生じた場合は,15.塩分試験方法(滴定法)に規定
する指示薬滴定法又は電位差滴定法の結果による。
参考1
この試験方法は,ASTM 3230-89,Salts in Crude Oil (Electrometric Method) 及びIP 265/70,Total
Salts Content of Crude Oil Conductivity Methodを参考にして作成した。
2
塩分の測定濃度範囲は,10〜300質量ppmが適切である。
16.2 導電率法塩分試験器 導電率法塩分試験器は,(1)〜(3)からなり,その一例を図11〜13に示す。
(1) 導電率計 0.1〜100μS/cm {0.1〜100μΩ−1/cm} の導電率が測定できるもの。
なお,(2)の測定用セルと組み合わせて,16.6試験の手順の操作によって,試料中の塩分を10〜300
質量ppmの濃度範囲にわたって,規定の精度で測定できるものであれば,電流計を使用してもよい。
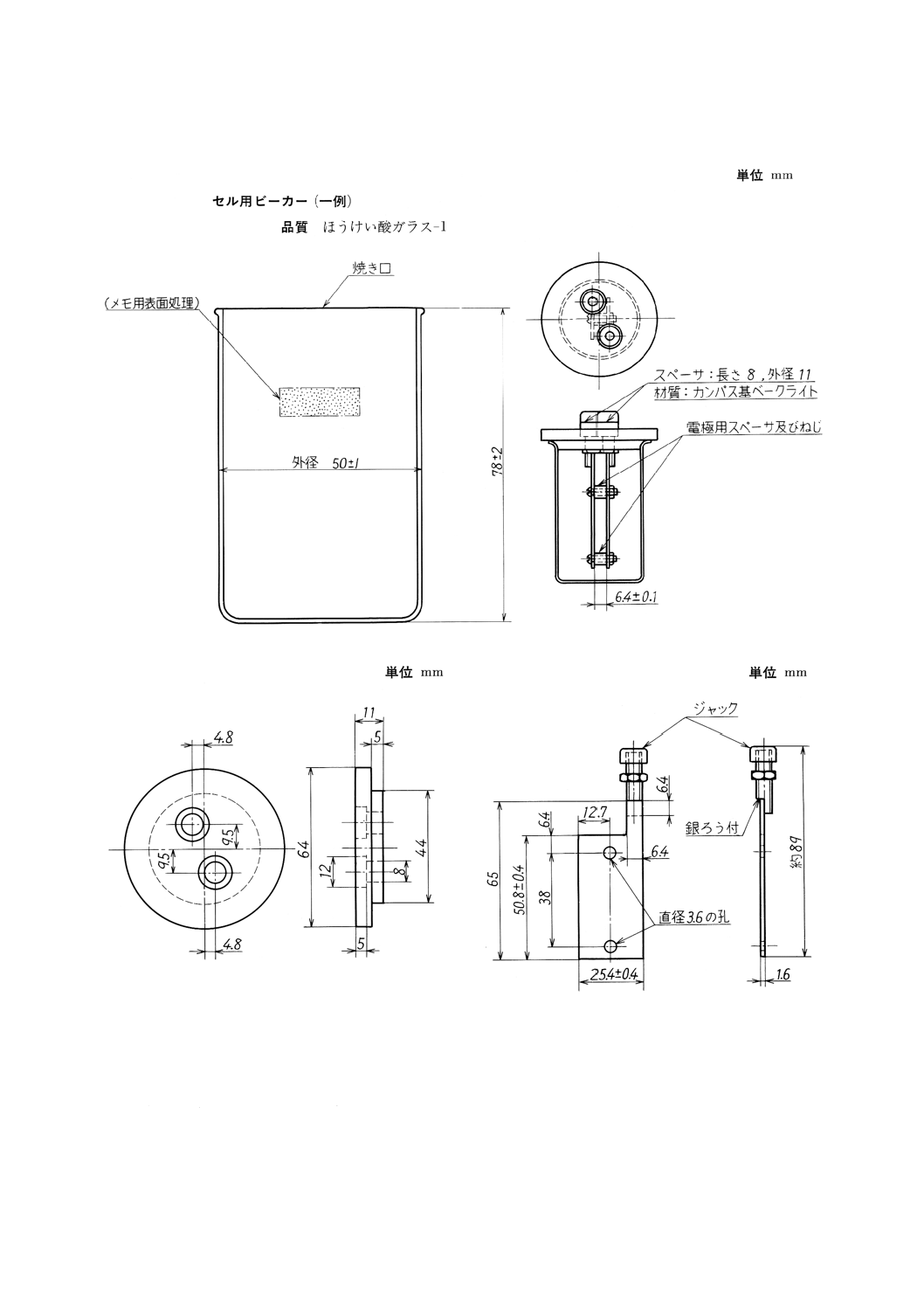
(2) 測定セル (a)〜(c)からなり,図11のように組み立てる。
(a) セル用ビーカー 容量約100mLのトールビーカーで,こぼし口のないもの。
図11に示すものが適切である。
参考 JIS K 2839に規定する図58が相当する。
(b) ふた 図12に示す形状・寸法で,材質はナイロン又はこれと同等以上の耐溶剤性のプラスチックと
する。
(c) 電極 図13に示す形状・寸法で,材質はステンレス鋼 (SUS304) とし,相対して正確に平行で,
6.4mmの間隔になるように(b)のふたに取り付け,ナイロン又は四ふつ化エチレン樹脂製のスペーサ
で絶縁する。なお,曲げたり,位置がずれたりしないように,取扱いには注意する。
(3) 温度計 JIS B 7410に規定する温度計番号 42(SG) のもの。
なお,目量が0.2℃で試験温度の範囲を測定可能なものを使用してもよい。
23
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図11 測定セル
図12 ふた
図13 電極
16.3 試薬 試薬は,次による。
(1) キシレン JIS K 8271に規定するもの,又はJIS K 2435に規定する3度キシレン若しくは5度キシレ
ン。
(2) 1−ブタノール JIS K 8810に規定するもの。
(3) メタノール JIS K 8891に規定するもの。
(4) 混合アルコール溶剤 1−ブタノール63容とメタノール37容を混合し,この混合液1Lにつき3mL
の水を加えたもの。
24
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(5) 基油 40℃における動粘度が約20mm2/s{cSt}の流動パラフィン,又は添加剤を含まない精製鉱油。
(6) エタノール JIS K 8101に規定する特級のもの。
(7) 水 JIS K 0557に規定するA3のもの。
(8) 塩化ナトリウム JIS K 8150に規定する特級のもの。
(9) 塩化カルシウム JIS K 8122又はJIS K 8123に規定するもの。
(10) 塩化マグネシウム JIS K 8159に規定するもの。
(11) ウラニン指示薬溶液 ウラニン0.2gを水100mLに溶解したもの。
(12) 0.1mol/L硝酸銀標準液 JIS K 8550に規定する硝酸銀17gを水に溶かして1Lとし,着色瓶に入れて
保存する。
この溶液のモル濃度を,次によって求める。
(a) JIS K 8005に規定する塩化ナトリウム5g以下を白金るつぼに入れ,600℃で約1時間加熱した後,
無水過塩素酸マグネシウムを乾燥剤としたデシケータ中で0.5〜1時間放冷する。
(b) (a)の塩化ナトリウム0.14〜0.17gを0.1mgのけたまではかり採り,水50mLを加えて溶かした後,
ウラニン指示薬溶液を加えて,硝酸銀標準液で滴定する。緑色の蛍光が消えて桃色に変わった点を
終点とする。
(c) 硝酸銀標準液のモル濃度は,次の式によって算出し,JIS Z 8401の規定によって小数点以下4けた
に丸める。
100
44
058
.0
P
V
M
C
×
×
=
ここに,
C: 硝酸銀標準液のモル濃度(mol/L)
M: (b)ではかり採った塩化ナトリウムの質量(g)
V: 滴定に要した硝酸銀標準液の量(mL)
P: 塩化ナトリウムの含量(質量%)
(13) 塩化ナトリウム溶液(5g/L)
(a) 塩化ナトリウム20±0.1gを,1L有栓メスシリンダに入れ,水を加えて溶解した後,水で希釈して
1Lとする。
(b) 300mL三角フラスコに(a)の溶液5mLを全量ピペットではかり採り,水50mLとウラニン指示薬溶
液を加えて,0.1mol/L硝酸銀標準液で滴定し,次の式によって濃度係数を求め,小数点以下2けた
まで記録する。
2
1
922
.2
)
NaC
(
V
V
M
F
×
×
=
λ
ここに,
M: 0.1mol/L硝酸銀標準液のモル濃度 (mol/L)
V1: 滴定に要した0.1mol/L硝酸銀標準液の容量(mL)
V2: 滴定に用いた塩化ナトリウム溶液の容量(mL)
F (NaCλ): 塩化ナトリウム溶液(20g/L)の濃度係数
(c) 濃度係数が1.00±0.10の範囲であれば,(d)の操作を行う。もし,範囲を外れた場合には,濃度係数
に応じて塩化ナトリウムを加えるか,又は水で希釈して濃度を調整した後,(b)によって濃度係数を
求める。
濃度係数が1.00±0.10の範囲になるまで,(c)及び(b)の操作を繰り返す。
(d) この水溶液25mLを全量ピペットではかり採り100mL全量フラスコに入れ,混合アルコール溶剤で
標線まで希釈する。これを塩化ナトリウム溶液 (5g/L) とする。
25
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(14) 塩化カルシウム溶液 (5g/L)
(a) 塩化カルシウム20±0.1g(18)を1L有栓メスシリンダに入れ,水を加えて溶解した後,水で希釈して
1Lとする。
注(18) 試薬に結晶水が含まれている場合には,その分を加算する。
(b) 300mL三角フラスコに(a)の溶液5mLを全量ピペットではかり採り,水50m1とウラニン指示薬溶液
を加えて,0.1mol/L硝酸銀標準液で滴定し,次の式によって濃度係数を求め,小数点以下2けたま
で記録する。
2
1
2
775
.2
)
CaCl
(
V
V
M
F
×
×
=
ここに,
M: 0.1mol/L硝酸銀標準液のモル濃度 (mol/L)
V1: 滴定に要した0.1mol/L硝酸銀標準液の容量 (mL)
V2: 滴定に用いた塩化カルシウム溶液の容量 (mL)
F (CaCl2): 塩化カルシウム溶液 (20g/L) の濃度係数
(c) 濃度係数が1.00±0.10の範囲であれば,(d)の操作を行う。もし,範囲を外れた場合には,濃度係数
に応じて塩化カルシウムを加えるか,又は水で希釈して濃度を調整した後,(b)によって濃度係数を
求める。
濃度係数が1.00±0.10の範囲になるまで,(c)及び(b)の操作を繰り返す。
(d) この水溶液25mLを全量ピペットではかり採り,100mL全量フラスコに入れ,混合アルコール溶剤
で標線まで希釈する。これを塩化カルシウム溶液 (5g/L) とする。
(15) 塩化マグネシウム溶液 (5g/L)
(a) 塩化マグネシウム20±0.1g(19)を1L有栓メスシリンダに入れ,水を加えて溶解した後,水で希釈し
て1Lとする。
注(19) 試薬に結晶水が含まれている場合には,その分を加算する。
(b) 300mL三角フラスコに(a)の溶液5mLを全量ピペットではかり採り,水50mLとウラニン指示薬溶
液を加えて,0.1mol/L硝酸銀標準液で滴定し,次の式によって濃度係数を求め,小数点以下2けた
まで記録する。
2
1
2
380
.2
)
MgCl
(
V
V
M
F
×
×
=
ここに,
M: 0.1mol/L硝酸銀標準液のモル濃度 (mol/L)
V1: 滴定に要した0.1 mol/L硝酸銀標準液の容量 (mL)
V2: 滴定に用いた塩化マグネシウム溶液の容量 (mL)
F (MgCl2): 塩化マグネシウム溶液 (20g/L) の濃度係数
(c) 濃度係数が1.00±0.10の範囲であれば,(d)の操作を行う。もし,範囲を外れた場合には,濃度係数
に応じて塩化マグネシウムを加えるか,又は水で希釈して濃度を調整した後,(b)によって濃度係数
を求める。
濃度係数が1.00±0.10の範囲になるまで,(c)及び(b)の操作を繰り返す。
(d) この水溶液25mLを全量ピペットではかり採り100mL全量フラスコに入れ,混合アルコール溶剤で
標線まで希釈する。これを塩化マグネシウム溶液 (5g/L) とする。
(16) 混合塩分原液 全量ピペットを用いて,塩化ナトリウム溶液 (5g/L) 70.0mL,塩化カルシウム溶液
(5g/L) 10.0mL及び塩化マグネシウム溶液 (5g/L) 20.0mLを300mL共栓付き三角フラスコに入れ,よく
混合する。これを混合塩分原液とする。
26
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
混合塩分原液の濃度係数は,次の式によって算出し,JIS Z 8401の規定によって小数点以下2けた
に丸める。
100
20
)
MgCl
(
100
10
)
CaCl
(
100
70
)
NaCl
(
)
Mix
(
2
2
×
×
×
F
F
F
F
+
+
=
ここに,
F (NaCl): (13)で求めた塩化ナトリウム溶液 (20g/L) の濃度係数
F (CaCl2): (14)で求めた塩化カルシウム溶液 (20g/L) の濃度係数
F (MgCl2): (15)で求めた塩化マグネシウム溶液 (20g/L) の濃度係数
F (Mix): 混合塩分原液の濃度係数
備考 通常,原油中の塩化ナトリウム,塩化カルシウム及び塩化マグネシウムの比率は,おおよそ
70:10:20であるが,原油中の塩分組成が分かっている場合には,その比率で混合塩分原液を作
るとよい。
(17) 塩分標準液 全量ピペットを用いて混合塩分原液20mLを1L全量フラスコに採り,混合アルコール溶
剤を標線まで加え,よく混合する。これを塩分標準液とする。
16.4 検量線の作成 検量線の作成は,次による。
(1) 100mL共栓付きメスシリンダに,全量ピペットで基油10mLを入れる。少量のキシレンでピペットに
付着した基油を洗い,その洗液をメスシリンダに加えた後,全量が50mLになるようにキシレンを加
え,メスシリンダを激しく振とうし,均一な溶液にする。
(2) 混合アルコール溶剤を加え,全量を100mL(20)として,約30秒間激しく振とうする。振とう終了後,
直ちに秒時計を始動して,約1分間静置する。
注(20) ふたの下面と電極の上縁が密着している試験器を使用する場合には,検量線の作成及び16.6試
験の手順を通して,溶液の容量を200mLとする。したがって,溶液の各成分(基油,キシレン,
混合アルコール溶剤,塩分標準液,試料など)の容量はそれぞれ2倍とする。
(3) セル用ビーカーに溶液を満たし,ふたをする。この際,電極の上縁が少なくとも1.6mm以上液面下に
なるように溶液量を調節する(21)。セルに温度計を挿入して,溶液の温度を測定した後,温度計を抜き
取り,(2)での振とう直後から5分後に導電率を測定し(22),これを空試験値とする。
注(21) ふたの下面と電極の上縁が密着している試験器を使用する場合には,ふたをしたときセル用ビ
ーカーから溶液があふれ出すようにして,電極を完全に浸す。
(22) 電流計を使用した場合には,電流値を測定する。
(4) セル用ビーカー及び電極をキシレン,水,エタノールの順ですすぎ,乾燥させる。
備考 もし,空試験値が0.50μS/cm {0.50μΩ−1/cm} を超える場合は,セルを洗浄して,測定を繰り返
す。再度,高い測定値が得られた場合は,水又は電導性の不純物の存在が考えられるので,原
因となるものを突き止めて,これらを取り除く。
(5) (1)の操作を行った後,表8に従って塩分標準液0.3mLをビュレットで加え,(2)〜(4)の操作によって
導電率を測定する(23)。
(6) 表8に従って,添加する塩分標準液の量を変えて,それぞれ(5)の操作を繰り返す(23)。
注(23) 検量線の作成にかかわる校正操作を通じて,(3)で測定した導電率測定時のそれぞれの溶液の温
度は,溶液の温度の平均値±3℃の範囲に保っていなければならない。

27
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表8 塩分標準液の添加量
塩分標準液添加量
(mL/100mL)
原油中の塩分
(NaCl mg/L)
0.3
3.15
0.6
6.30
1.0
10.5
1.5
15.8
3.0
31.5
5.0
52.5
9.0
94.6
12.0
126
15.0
158
18.0
189
22.0
231
27.5
289
備考 NaCl30mg/L以下の場合には,塩分標準液を1:5又は1:10
に混合アルコール溶剤で希釈して,塩分標準液添加量
の相当量を添加してもよい。
(7) 次の式によって,表8に示す原油中の塩分 (NaCl mg/L) を補正して,X1〜X12を求める。
X1=S1×F (Mix)
X2=S2×F (Mix)
Μ
X12=S12×F(Mix)
ここに, X1〜X12: 原油中の塩分の補正した値 (NaCl mg/L)
S1〜S12: 表8に示す原油中の塩分の値 (NaCl mg/L)
F (Mix): 16.3(16)で求めた,混合塩分原液の濃度係数
(8) 次の式によって,(5)〜(6)で測定した導電率を補正して,Y1〜Y12を求める。
Y1=C1−CB
Y2=C2−CB
Μ
Y12=C12−CB
ここに, Y1〜Y12: 補正した導電率の値
C1〜C12: 導電率の測定値
CB: (3)で測定した空試験値
(9) 両対数目盛のグラフ用紙の縦軸を塩分含有量(NaCl mg/L),横軸を導電率として,(7)で求めたX1〜X12
及び(8)で求めたY1〜Y12の座標をそれぞれプロットする。
備考 各点は直接上に並ぶが,両対数目盛のため読み取り誤差が生じやすいので,通常のグラフ用紙
(両均等目盛のもの)に各点をプロットして検量線を引き,使用してもよい。
16.5 試料の採取及び調製 試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法,
又はそれに準じた方法によって採取及び調製する。
16.6 試験の手順
(1) キシレン40mLを100mL有栓メスシリンダにはかり採る。
(2) 試料を1分以上激しく振とうし,直ちに10mL全量ピペットを先端が油面から100〜120mmの深さに
なるように挿入して,試料を採取する(24)。振とう直後から試料採取までの時間は,30秒以内とする。
注(24) 振とうによって油面が泡立った場合には,真の油面の位置を推定する。
28
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考 泡が消えるのを待って試料採取を行うより,油面の位置の推定を間違えた方が試験結果に与え
る誤差は小さいはずである。
備考 試料の粘度が大きい場合には,同量のキシレンで希釈し激しく振とうして,(2)から操作を続け,
得られた結果を2倍して真の結果とする。
別の方法としては,試料を加温して有栓メスシリンダ中のキシレン中に注ぎ,シリンダの目
盛で10mLをはかり採る。(4)から操作を続け,(5)の洗浄操作は省略する。
(3) ピペット中の試料をメスシリンダに移した後,メスシリンダ中のキシレン・試料混合液を吸い上げて
から,シリンダへ戻す操作を5,6回繰り返してピペット内を洗浄する。ピペットは(5)で再度洗浄す
る。
(4) 混合アルコール溶剤をメスシリンダに加えて全量を100mLとし,栓をして少し振とうする。
(5) メスシリンダ中のキシレン・試料・アルコール混合液を,(3)で洗浄したピペット中に吸い上げてから
戻す。
この操作を5,6回繰り返して,ピペット中に付着した塩水の水滴をシリンダへ移す。
シリンダに栓をして,30秒間激しく振とうした後,約1分間静置する。
(6) セル用ビーカーに(5)の混合液を満たし,ふたをする。この際,電極の上縁が少なくとも1.6mm以上液
面下になるように溶液量を調節する(25)。
セル中に温度計を挿入して,混合液の温度を測定する(26)。
注(25) ふたの下面と電極の上縁が密着している試験器を使用する場合には,ふたをしたときセル用ビ
ーカーから混合液があふれ出すようにして,電極を完全に浸す。
(26) 混合液の温度は,16.4で求めた溶液の温度の平均値±3℃でなければならない。
なお,この温度範囲を外れる場合には,混合液の温度に合わせて検量線を作り直すか,あら
かじめ異なる温度(例えば,10℃,20℃及び35℃の3点)において検量線を作り,温度による
導電率の変化を補正する。
(7) 温度計を抜き取り,振とうを始めてから正確に5分後に導電率を測定する。
(8) 石油エーテル又はキシレンでセルを洗った後,洗剤で洗い水道水次いで水ですすぎ,エタノールで洗
浄して乾燥する。
(9) キシレン40mL,混合アルコール溶剤50mL及び基油10mLを混合して空試験溶液とし,(6)〜(8)に従
って導電率を測定する。
16.7 計算方法及び精度
16.7.1 計算方法 塩分試験方法(導電率法)の計算方法は,次による。
(1) 試料の導電率の値から,16.6(9)で求めた空試験の導電率の値を差し引く。
(2) 検量線から(1)の値に対応する試料中の塩分含有量 (NaCl mg/L) を,有効数字3けたまで求める。
(3) 次の式によって,試料中の塩分を算出し,JIS Z 8401の規定によって整数位に丸める。ただし,10質
量ppm以上の場合は,有効数字2けたに丸める。
D
C
S=
ここに,
S: 塩分 (質量ppm)
C: (2)で求めた,試料中の塩分含有量(NaCl mg/L)
D: 試料の密度 (g/cm3)(27)
注(27) 16.6(2)で試料をはかり採ったときの試料温度における密度とする。
29
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
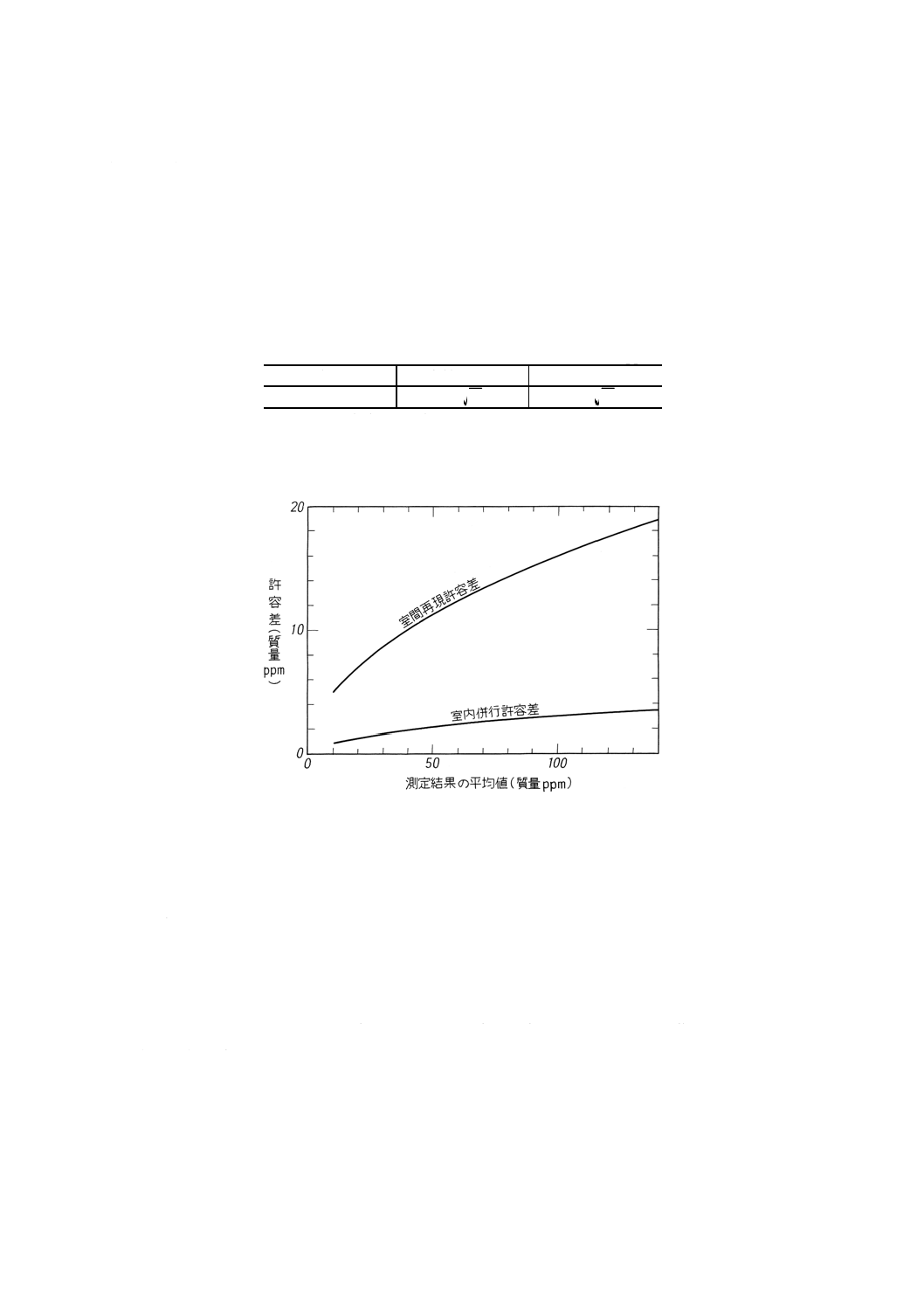
16.7.2 精度 塩分試験(導電率法)の測定結果が10〜140質量ppmの場合,得られた試験結果の許容差
(確率0.95)は,次のとおりである。
備考 試験結果が許容差を外れた場合には,JIS Z 8402の規定によって処理する。
(1) 室内併行精度 同一試験室において,同一人が同一試験器で,引き続き短時間内に同一試料を2回試
験したときの試験結果の差の許容差を表9に示す。
(2) 室間再現精度 異なる試験室において,別人が別の試験器で,同一試料をそれぞれ1回ずつ試験した
ときの2個の試験結果の差の許容差を表9に示す。
表9 精度
単位 質量ppm
塩分
室内併行許容差
室間再現許容差
10以上140以下
A
3.0
A
6.1
備考 A:測定結果の平均値
参考 精度をグラフ化して,参考図1に示す。
参考図1 精度
16.8 試験結果の報告 試験結果には,次の事項を記載する。
(1) 試料名,採取場所及び採取月日
(2) JISの規格番号 例 JIS K 2601
(3) 試験方法の名称・項番号及び16.7.1によって得られた結果
(4) 特記事項
17. 原油常圧法蒸留試験方法
17.1 試験の原理 試料300mLを精留管付き500mLの蒸留フラスコに採り,毎分4〜5mLの留出速度で蒸
留を行い,留出油量5%ごとにその温度を記録する。温度計示度が270℃になるまで蒸留を続け,この留出
油量(容量%)を求める。
備考 自動常圧蒸留試験器を用いてもよい。ただし,この試験方法によって得られた結果との間に有
意差のないことを,JIS Z 8402の規定によって確認すること。
なお,自動試験器によって得られた結果に疑義が生じた場合には,この試験方法によって得
られた結果によって判定する。
30
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考1
この試験方法はASTM D 285-1962,Distillation of Crude Petroleum(1987年に廃止)を参考に
したものである。
2
ISO/DIS 8708-1984,Crude petroleum oil−Determination of distillation characteristics using 15
theoretical plates columnを翻訳した“原油−理論段数15段の精留塔を使用した蒸留試験方法”
を,参考に示す。
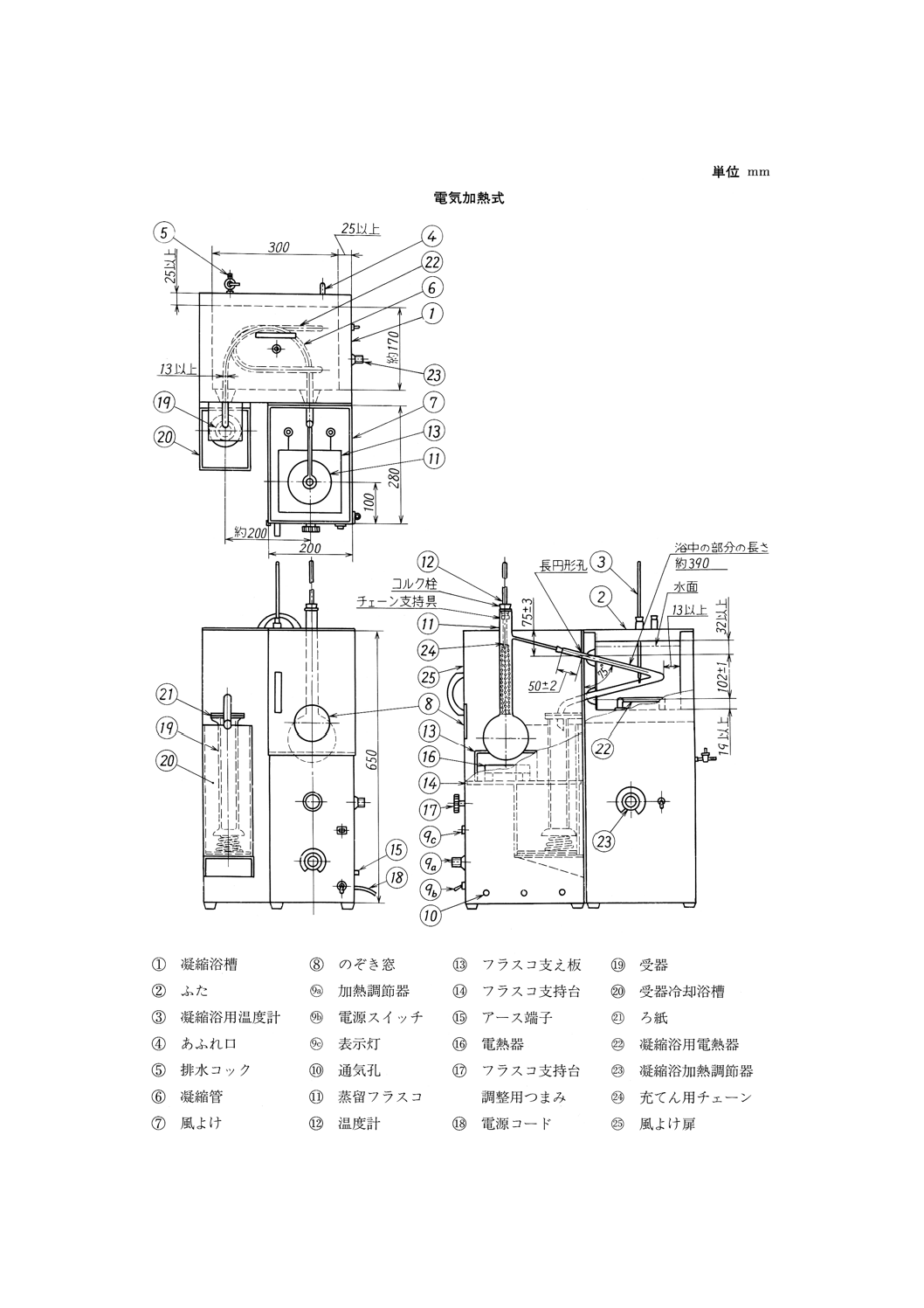
17.2 原油常圧法蒸留試験器 図14及び図15に示す構造のもので,(1)〜(10)からなる。
31
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図14 原油常圧法蒸留試験器(一例)
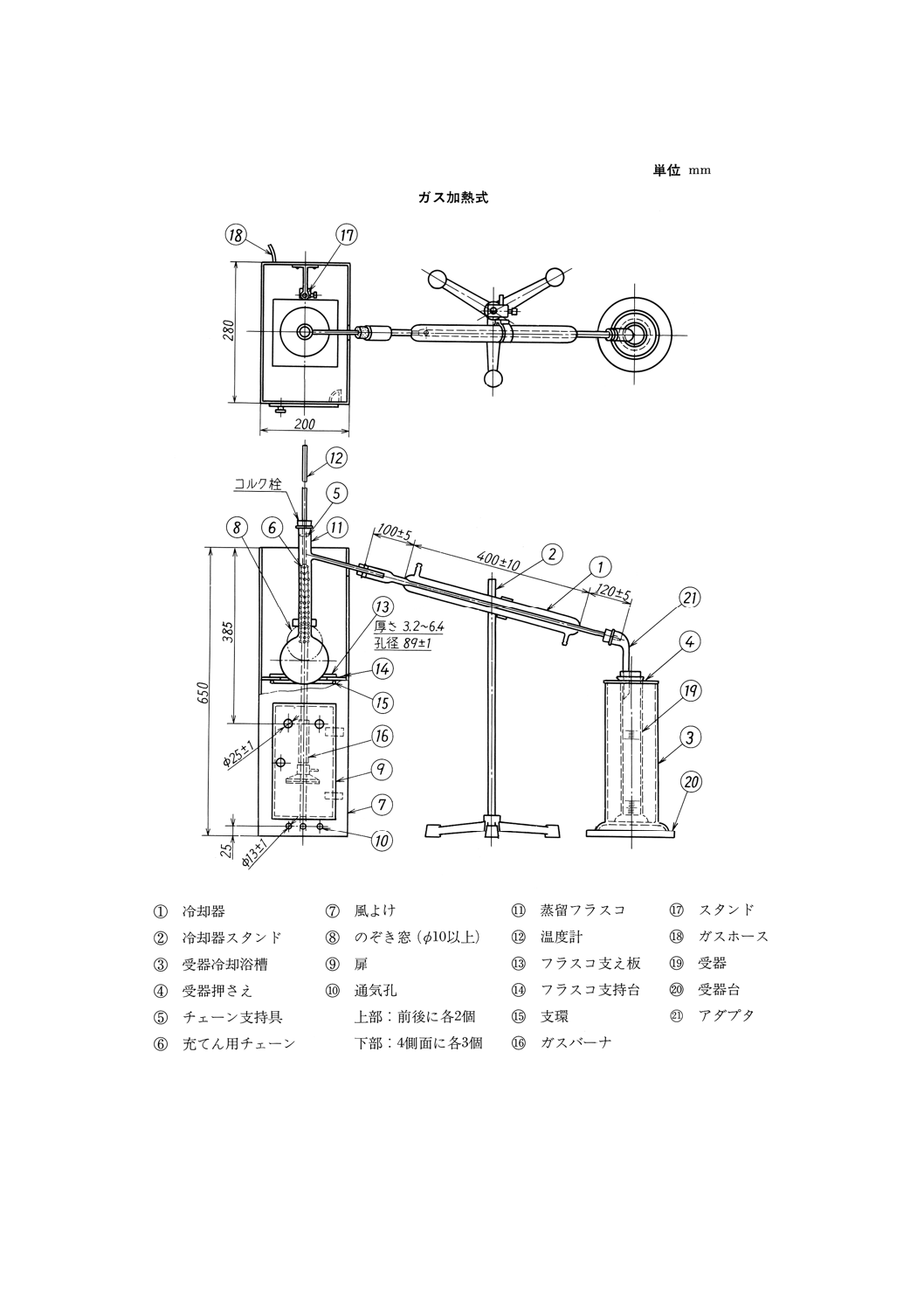
32
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図15 原油常圧法蒸留試験器(一例)
(1) 蒸留フラスコ 図16に示すもの。
参考 JIS K 2839に規定する図178が相当する。
(2) 精留部 図17及び図18に示す充てん用チェーン及びチェーン支持具からなる。
(a) 充てん用チェーン 図17に示す寸法のステンレス鋼線製8の字形リンクをつないだチェーンで,全
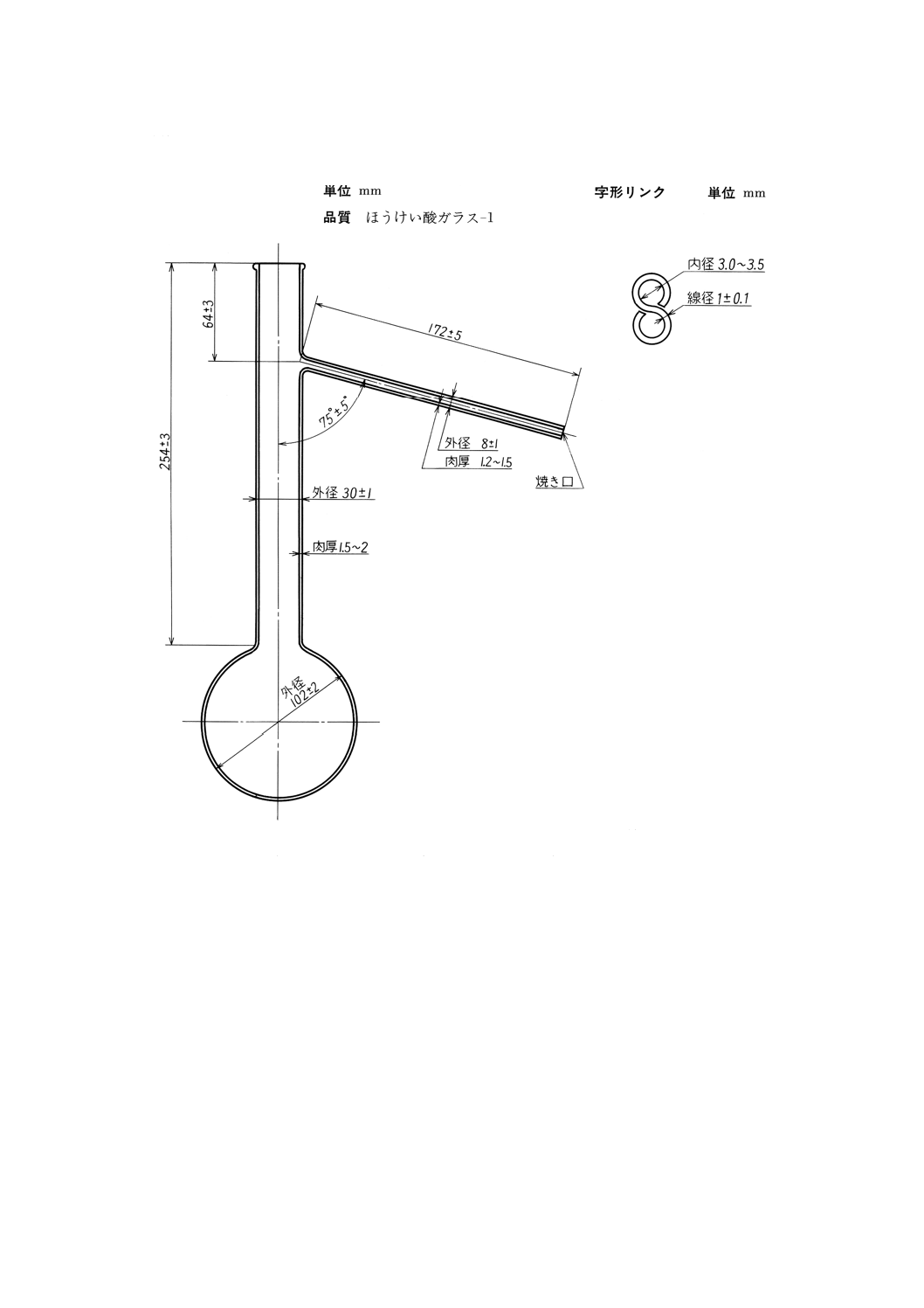
33
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
長約8mのものとする。
図16 蒸留フラスコ
図17 充てん用チェーン8の字形リンク
(b) チェーン支持具 図18に示すように,直径約1mmのステレンス鋼線の先端約150mmを外径約
24mmの渦状に曲げ,残りの部分を,渦巻き面に直角に曲げて約230mmの長さに切り,その先端約
5mmをフック状に曲げる。別に,厚さ0.4mm,長さ72mm,幅9mmのばね鋼板を直径約26mmの
環状に丸めたものの中央部に約1.2mmのあな(孔)をあけ,このあなにフックを通して図18のよ
うに組み立てる。ステンレス鋼線とばね鋼板との接続部は,図18の寸法になるようにスポット溶接
してもよい。これを(1)の蒸留フラスコの首部に入れたとき,環状ばね鋼板の弾力でチェーンを支持
できるものとする。
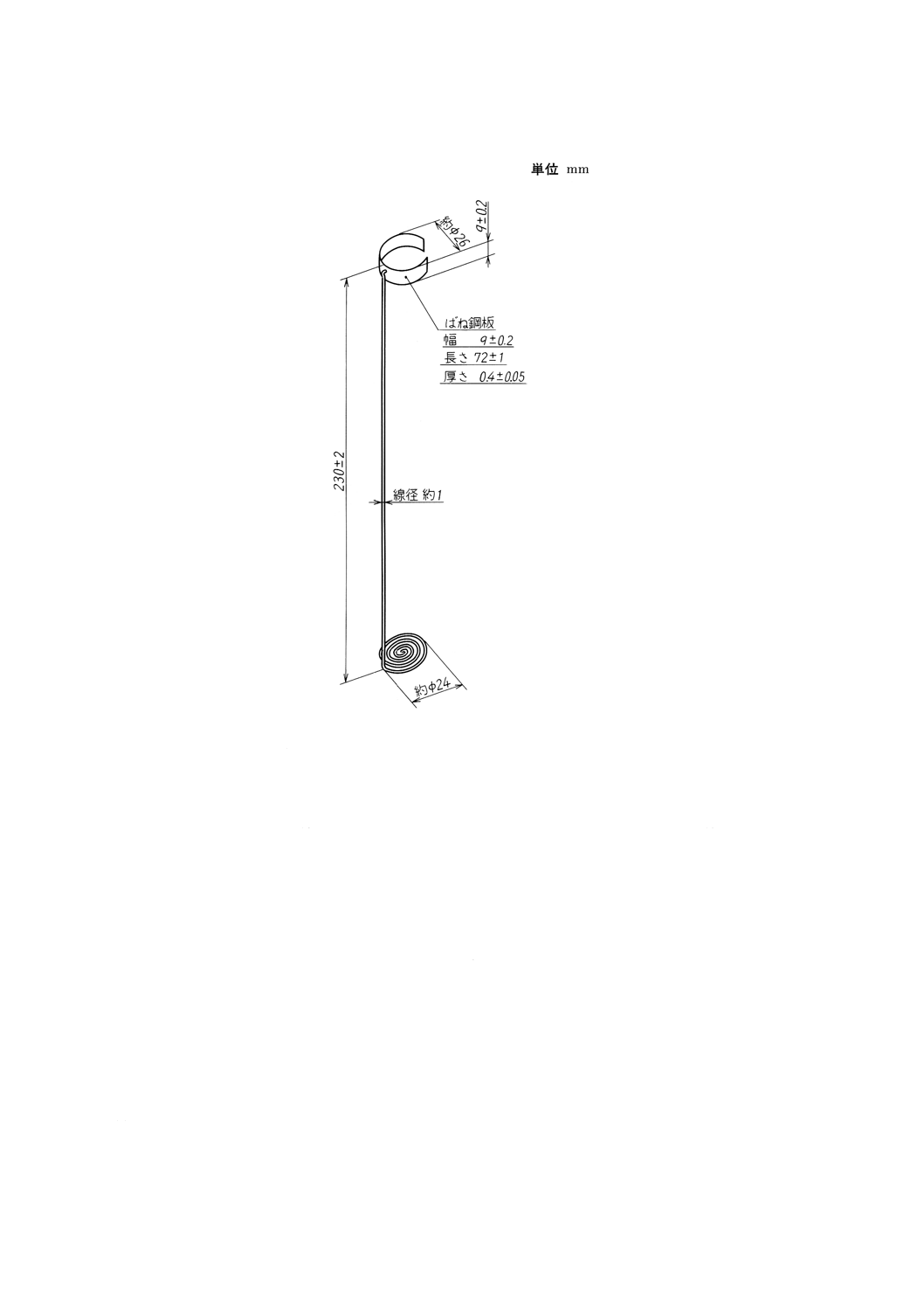
34
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図18 チェーン支持具
(3) 凝縮器 図14に示すような金属製のもの,又は図15に示すようなガラス製のものとする。
(a) 金属製凝縮器 凝縮管,保冷壁をもつ凝縮浴とそのふた,凝縮浴加熱用電熱器などで組み立てたも
のとする。構造,寸法及び材質は,JIS K 2254の凝縮器の規定によるものとし,凝縮浴の上端の高
さは約650mmとする。凝縮浴加熱用電熱器は,消費電力1.5〜2kWのもので,浴の底に近いところ
に取り付け,また,凝縮浴下方の操作しやすい位置に電熱器用スイッチ,加熱調節器などを取り付
ける。
(b) ガラス製凝縮器(28) JIS R 3503に規定する付図37リービッヒ冷却器(外筒長さ40mm)と付図61
アダプタ(頭部外径25mm)とを用い,図15のように組み立てて適切な支持具で冷却器を垂線と
75度の角度に保持できるようにする。
注(28) この規格では特に規定はしないが,ガラス製凝縮器を用いて試験するときは,別に,冷却器に
氷水,水道水及び40〜70℃の温水をそれぞれ切り替えて循環させることができる適当な循環装
置を備えるものとする。
(4) 風よけ 図14又は図15に示すような形状・寸法の金属製の風よけで,いずれも内面には厚さ1.5〜
3.0mmの不燃性断熱材の板を張り,正面には耐熱ガラス板を張った直径70mm以上のフラスコのぞき
窓を付ける。加熱器から上の正面は,扉として開閉できる構造としてもよい。
(a) 電気加熱用風よけ 図14に示すような高さ650mm,幅200mm,奥行き280mm,厚さ0.7mm以上
のもので,背面上方部には(3)(a)に規定した凝縮管上方突出部が適合するような長円形のあなをあけ
35
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
る。四つの各側面の下端から25mm上の箇所に,それぞれ直径13mmの通気孔3個をあけ(29),正面
には加熱調節器及びフラスコ支持台の高さを調節できるつまみなどを付ける。電熱器は,その中心
が長円形あなのある面から180mmの位置になるようにする。
(b) ガス加熱用風よけ 図15に示すような高さ650mm,幅200mm,奥行き280mm及び厚さ0.7mm以
上のもので,フラスコの枝管に適合するような長円形切込みを付け,正面には加熱器及びフラスコ
支持台を操作するための扉を付ける。扉と背面には,上端から385mmの箇所にそれぞれ直径25mm
の通気孔各2個をあける(29)。
注(29) 風よけ下面が開いていて,十分な通気が得られる場合には,この通気孔はあけなくてもよい。
特に電気加熱用の場合には,フラスコ支持台から下の部分の風よけはなくてもよい。
(5) フラスコ支持台及び支え板
(a) 電気加熱用フラスコ支持台 風よけの外部から支持台の高さを調整できる適切な機構に,風よけ内
面に密着する程度の大きさで厚さ5〜8mmの硬質の不燃性断熱材の板を取り付けたもので,不燃性
断熱材の板には,(4)(a)に規定する位置に電熱器を取り付ける。
(b) ガス加熱用フラスコ支持台 直径130mm以上の支環を風よけ内部のスタンドに取り付けて上下に
調整できるようにしたもので,その上部に,風よけ内面に密着する程度の大きさで厚さ5〜8mmの
硬質の不燃性断熱材の板を取り付ける。硬質の不燃性断熱材の板には直径100mmのあなをあけ,
その中心はガスバーナ及び支環の中心と一致し,(c)に規定するフラスコ支え板を載せたとき,透き
間ができないようにする。
(c) フラスコ支え板 厚さ3.2〜6.4mmの磁器板,その他の耐熱性の板で,一辺の長さ約150mmの正方
形とし,その中心に直径89mmのあなをあける。
(6) 加熱器
(a) 電熱器 最大消費電力が0.1〜1.5kWの範囲で,適切に加減できるものとし,フラスコ支え板上面を,
電熱器上面から約30mm以上高くしておくことができるものとする。
(b) ガスバーナ 62.8kJ/min {15kca1/min} 以上の熱量を供給でき,かつ,加熱を自由に加減できるもの
とする。
(7) 温度計 JIS B 7410に規定する温度計番号7(DIST)のもの。
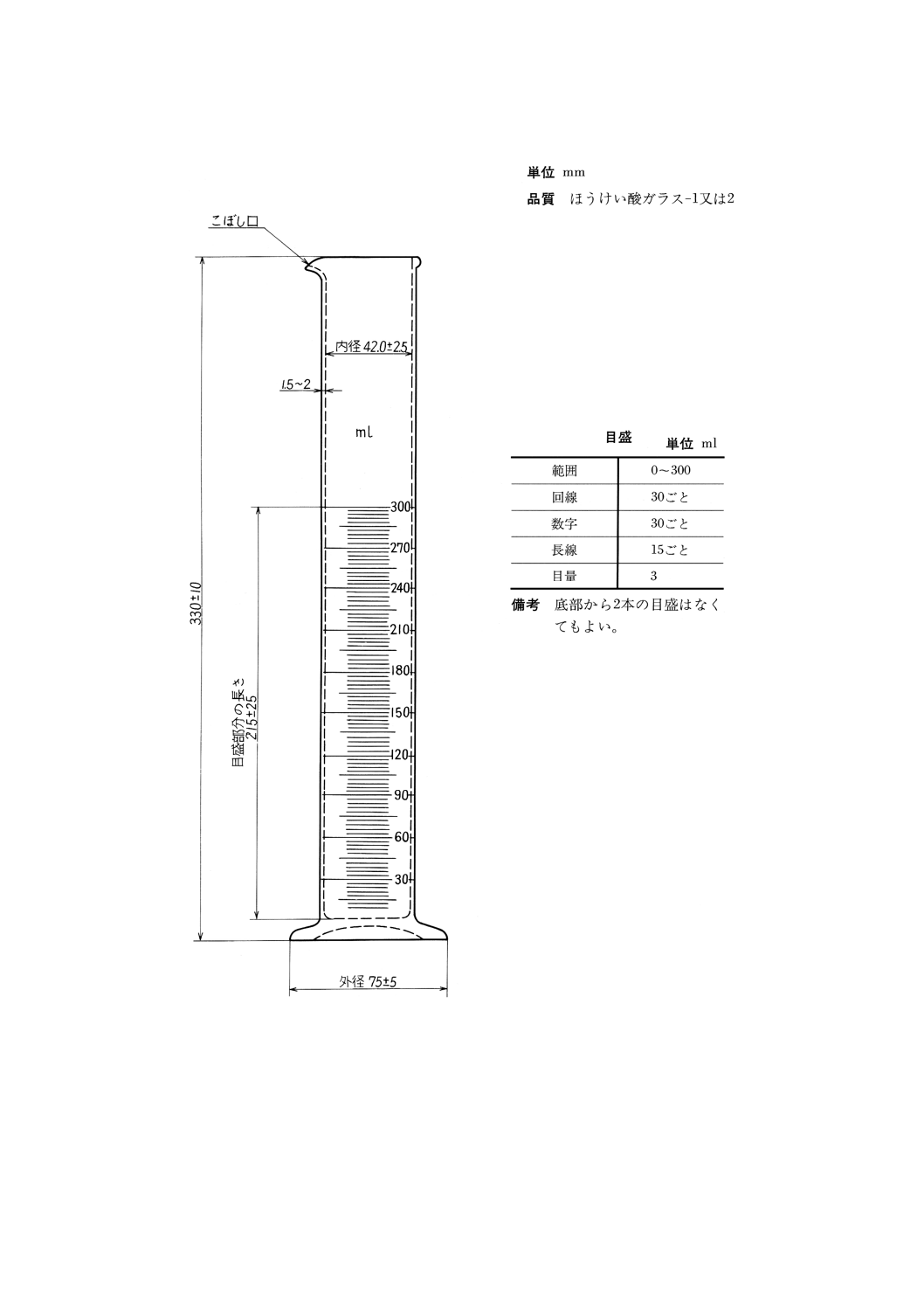
(8) 受器 図19に示すもの。
参考 JIS K 2839に規定する図181が相当する。
(9) 受器冷却槽 (8)に規定する受器を,下端から約270mmの高さまで浸して12〜18℃に保つことができ
る透明な浴槽とし,受器を保持する適切な装置と浴槽の温度が読み取れる適切な温度計とを付けるも
のとする。
(10) 全量ピペット 図20に示すもの。
参考 JIS K 2839に規定する図182が相当する。
36
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図19 受器
37
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図20 全量ピペット
17.3 試料の採取及び調製 試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調製方法,
又はそれに準じた方法によって採取及び調製する。
なお,原油中に水分を含んでいて突沸する場合,又は2%以上の水分が含まれている場合には,次のよ
うな方法で脱水を行う。
(1) 軽質油分の損失が起こらないような温度で遠心分離又は静置によって水を分離する。この際,必要に
応じて解乳化剤を入れる。
38
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(2) JIS K 8124に規定する塩化カルシウム,JIS K 8987に規定する硫酸ナトリウムなど,その他適切な脱
水剤を加えてかくはんした後,ろ紙などを用いてろ過する。
軽質油分が損失するおそれがある場合は,加圧ろ過若しくは試料の冷却を行うか,又はその両方法
を併用する。
(3) 温度計と圧力計を付けた鉄製密閉容器中に試料を約70%入れ,試料の温度が200℃に達するか,又は
圧力計が0.69MPa {7kgf/cm2} になるまで加熱し,放冷後,試料を傾斜法などによって水と分離する。
17.4 試験器の準備 試験器の準備は,次による。
(1) 冷却器に氷水,水道水及び温水を循環することができる適切な装置を用意する。
(2) 保温用として蒸留フラスコ首管部及び球の上半部にグラスウール製のリボンなどを巻く。
17.5 試験の手順 試験の手順は,次による。
(1) 15℃で300mLに相当する試料を適切な方法で蒸留フラスコ中に容量又は質量で採取する(30)。このと
き枝管の方に流れ込まないように注意する。試料を質量ではかり採る場合,密度はJIS K 2249の規定
によって測定する。
注(30) 常温で高粘度であるか,又は流動しない原油は,質量ではかり採る。
(2) 沸騰石を加えた後,蒸留フラスコ首管中にチェーン支持具を適切な位置に入れ,ステンレス鋼製チェ
ーンを一様に詰める。この場合,蒸留フラスコを軽くたたくとチェーンが入りやすいが,これを圧縮
しながら詰めてはならない。
(3) 温度計を付けたコルク栓を蒸留フラスコに温度計の毛管下端が枝管付け根の内壁下面より約1.5mm
下方にあるように差し込む。
(4) このように準備した蒸留フラスコを支持板上に載せ,コルク栓を使って冷却器を蒸留フラスコにつな
ぐ。この際,蒸留フラスコは垂直であるように,しかも枝管は25〜50mmだけ冷却器に入るようにす
る。
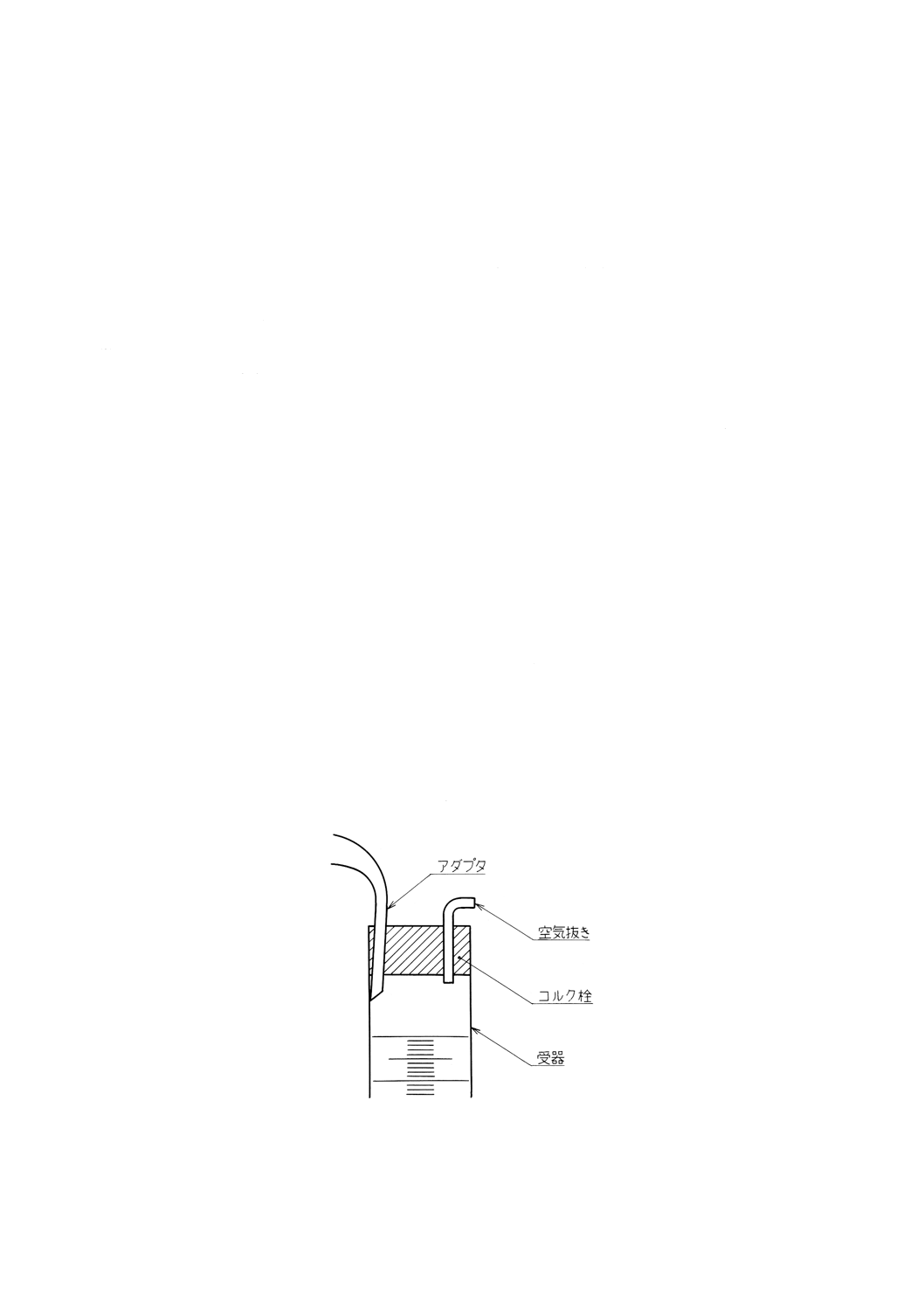
(5) 冷却器の下方にアダプタを取り付け,図21に示すように空気抜き孔を付けたコルク栓で受器の内壁に
アダプタの先端が触れるように接続する。軽質油分の損失を防ぐために,受器は12〜18℃の透明浴槽
中に保つ。
図21 アダプタと受器の接続部
(6) 冷却器に氷水を循環し,加熱を始める。ガスバーナを使用する場合,炎の広がりは,支持板下面で径
約130mm以上にならないようにする。冷却器下端から留出油の初滴が滴下したときの温度計の示度
39
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
を初留点として記録する。初留点から約10mLまでの留分は毎分2〜3mLの速度で,それ以後は毎分
4〜5mLの速度で留出するように加熱を調節し,留分5%ごとに温度計の示度を記録する。
備考 通常は気圧による温度補正は行わないが,必要があればJIS K 2254に規定するシドニーヤング
式によって補正する。
(7) 温度計の示度が80〜90℃に達したら氷水を水道水に切り替えて循環させ,約200℃になったら温水(31)
に切り替え,蒸留を続ける。温度計の示度が270℃に達したら直ちに受器を外し,加熱を止め,その
後の留出油は100mLメスシリンダに受ける。
先に取り外した受器は密栓して,12〜18℃における留出量及び水分量を記録する。
注(31) 温水は40〜50℃が適切であるが,ワックスなどが析出するおそれのある場合は,更に高い温度
の温水,又は熱水を用いる。
17.6 計算方法及び精度
17.6.1 計算方法 原油常圧法蒸留試験の計算方法は,次による。
(1) 270℃までの留出油(容量%)は,次の式によって算出し,0.5容量%の単位に丸める。
100
×
w
M
w
D
A
−
−
=
ここに,
A: 270℃までの留出量 (容量%)
D: 受器中の全留出量 (mL)
w: 受器中の水分量 (mL)
M: 原油はかり採り量 (mL)
(2) 初留点及び留出量5容量%ごとの温度は,0.5℃の単位に丸める。
(3) 試験結果には,原油常圧法蒸留試験で得られた結果であることを明確にするため,原油常圧法と付記
する。
17.6.2 精度 原油常圧法蒸留試験の精度は,規定しない。
17.7 試験結果の報告 試験結果には,次の事項を記載する。
(1) 試料名,採取場所及び採取月日
(2) JISの規格番号 例 JIS K 2601
(3) 試験方法の名称・項番号及び17.6.1によって得られた結果
(4) 特記事項
18. ワックス分試験方法
18.1 試験の原理 試料1又は2gをヘキサンに溶かし,活性白土を用いてアスファルテンを除去した後,
ヘキサンを蒸発させて除く。アスファルテンを除いた試料をアセトン・ヘキサン混合溶剤に溶解し,これ
を−18℃まで冷却して析出したワックスをろ過して分離する。分離したワックスの質量から試料中のワッ
クス分を求める。
備考 この試験方法は,石油学会の技術的検討を経て作成されたものである。
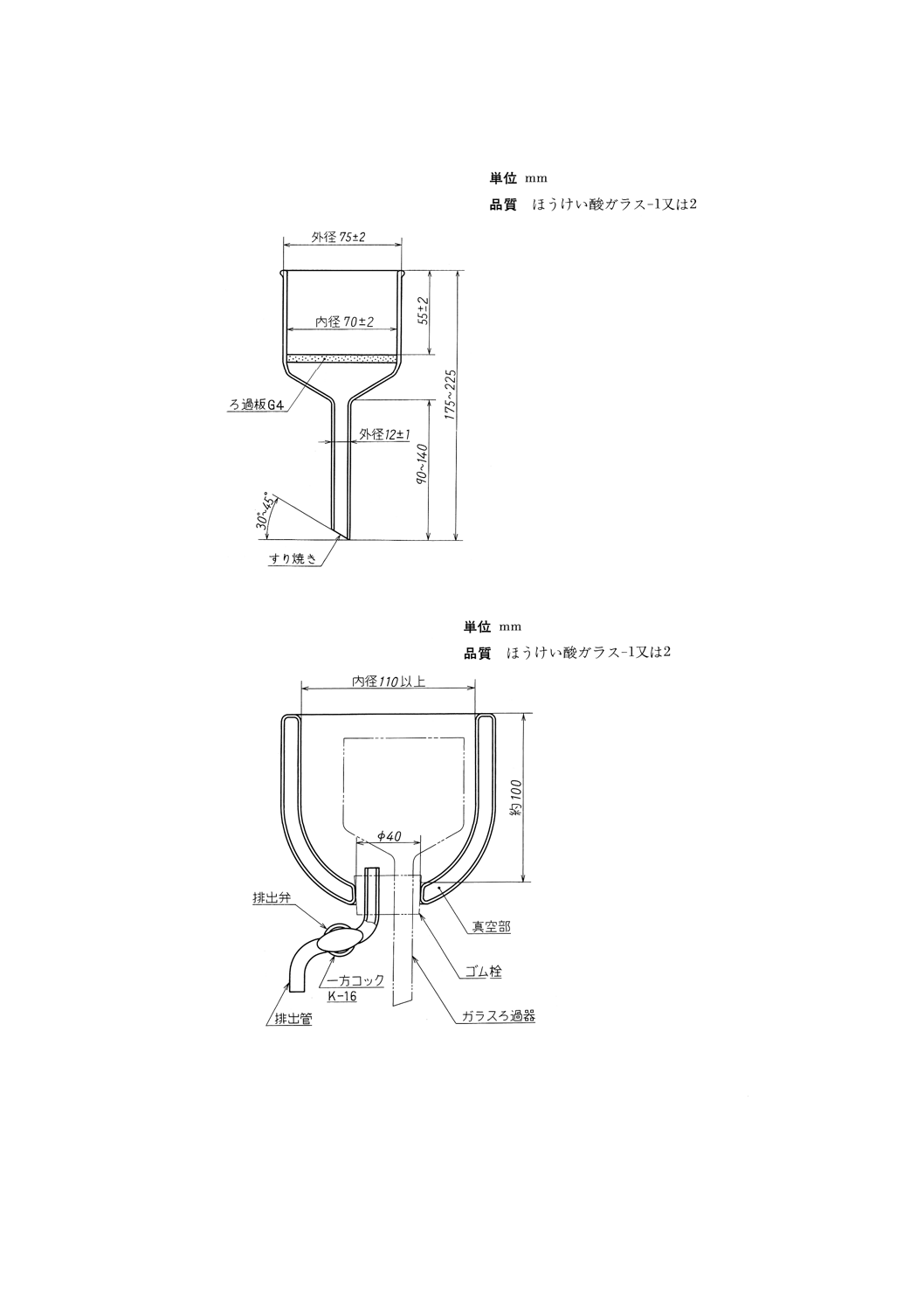
18.2 ワックス分試験器 ワックス分試験器は,次による。
(1) ブフナー漏斗 ろ過面の直径が55mmのもの。
(2) ろ紙 JIS P 3801に規定する5種A,直径55mmのもの。
(3) 水浴 浴温を約95℃に保つことができるもの。
(4) 恒温空気浴 浴温を120±2℃及び105±2℃に保つことができるもの。
40
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(5) デシケータ 乾燥剤としてシリカゲルを入れたもの。
(6) 試料用冷却浴 200mL三角フラスコを2個浸すのに十分な大きさで,冷媒としてエタノールを満たし,
冷却装置又はドライアイスで浴温を−18±1℃に保てるもの。
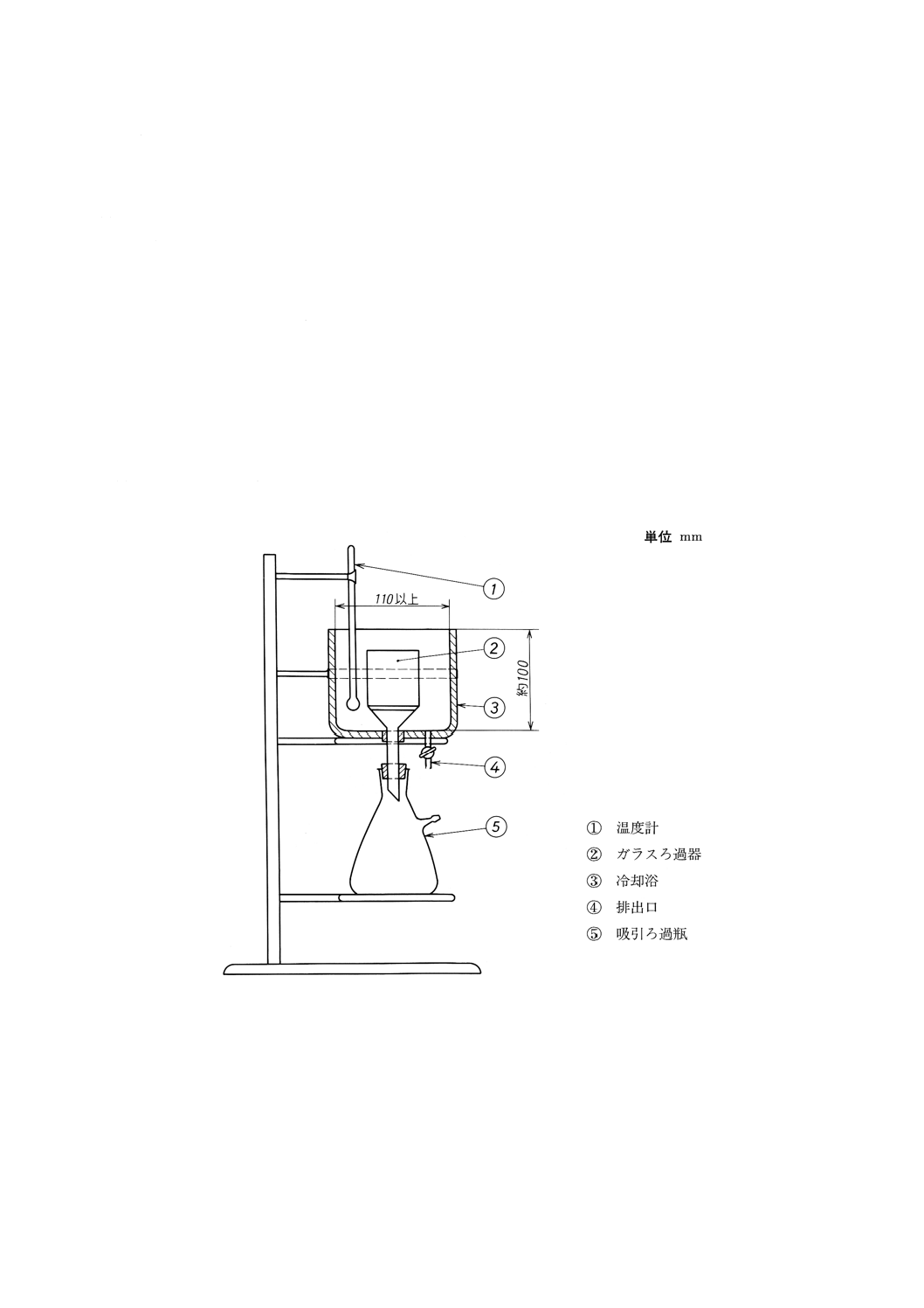
(7) ろ過装置 (a)〜(d)からなり,組立の一例を図22に示す。
(a) ガラスろ過器 図23に示すもの。
参考 JIS K 2839に規定する図242が相当する。
(b) 冷却浴 図24に示すもの。又は金属製とし,外側は適当な断熱材で覆うとよい。底板のほぼ中央に
あなを開け,ゴム栓を介して(a)のガラスろ過器を取り付ける。
また,底部に冷媒の排出口を設ける。内径110mm以上とし,ガラスろ過器の上縁が浴壁上縁の平
面以下となるのに十分な深さとする。冷媒としてエタノールを満たし,冷却装置又はドライアイス
で浴温を−18±1℃に保てるもの。
参考 JIS K 2839に規定する図243が相当する。
(c) 温度計 JIS B 7410に規定する温度計番号60 (TOT) 又は温度計番号61 (TOT) のもの。
(d) 吸引ろ過瓶 JIS R 3503に規定する付図37吸引ろ過瓶で呼び容量1Lのもの。
図22 ろ過装置(一例)
41
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図23 ガラスろ過器
図24 冷却浴
18.3 試薬 試薬は,次による。
(1) ヘキサン JIS K 8848に規定するもの。
(2) 活性白土 厚さを10mm以下として,120±2℃に保った恒温空気浴中で2時間乾燥し,デシケータ内
で放冷したもの。
参考 社団法人日本油化学協会で検定した標準活性白土がある。
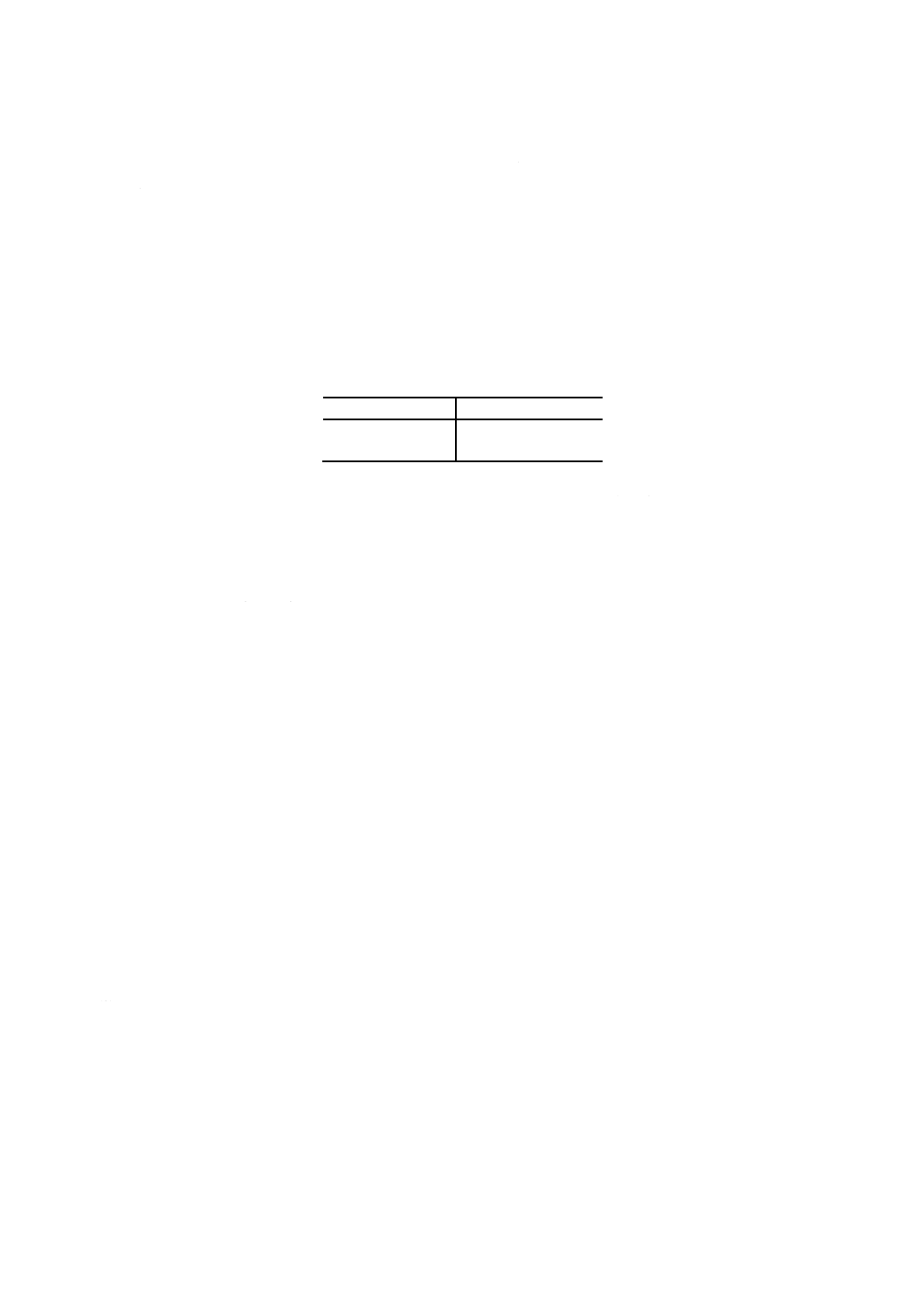
42
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(3) アセトン JIS K 8034に規定するもの。
(4) 混合溶剤 アセトン75容量%とヘキサン25容量%を混合したもの。
(5) ドライアイス
(6) エタノール JIS K 8101又はJIS K 8102に規定する特級又は1級のもの。
18.4 試料の採取及び調製 試験用試料は,JIS K 2251に規定する一次試料の採取方法及び二次試料の調
製方法,又はそれに準じた方法によって採取及び調製する。
18.5 試験の手順 試験の手順は,次による。
(1) ワックス分の含有量に応じて,表10に示す量の試料を1L三角フラスコに0.1mgのけたまではかり採
る。
表10 試料のはかり採り量
ワックス分質量 % 試料はかり採り量 g
10未満
2±0.2
10以上
1±0.1
(2) これにヘキサン500mLを加えて,試料を溶解する。
参考 タンク底部試料,フィルター捕そく物など長鎖のパラフィンを多く含む試料は,パラフィンが
活性白土に吸着され,負の誤差要因となる可能性が考えられる。
このような試料は,(2)〜(4)において試料を60℃に加温したヘキサンに溶解し,脱れき(瀝)
及びろ過操作まで溶液温度を60℃に保持したままで操作するとよい。(5)における繰返しの場合
も同様の溶液温度で行い,(6)におけるヘキサンによる洗浄も60℃に加温したヘキサンで行うと
よい。
(3) 次に活性白土15±0.1gを加え,マグネチックスターラ又は回転翼式スターラで15分間かくはんする。
(4) 吸引ろ過瓶にろ紙をセットしたブフナー漏斗を取り付け,ヘキサン溶液を吸引ろ過して活性白土を分
離する。
さらに,1L三角フラスコ及びブフナー漏斗上の活性白土をヘキサン50mLで洗浄する。
(5) ろ液を新たな1L三角フラスコに移し,ろ液の色が無色又は淡黄色でない場合には,(3)〜(4)の操作を
繰り返す。
(6) 吸引ろ過瓶内を少量のヘキサンで洗浄し,洗液を1L三角フラスコに加える。
(7) 1L三角フラスコを約95℃に保った温浴に載せ,ヘキサン臭がなくなるまでヘキサンを蒸発除去する。
備考 ヘキサンの蒸発除去はドラフト内で行う。
なお,適切な試験器を組み合わせて,ヘキサンの大部分を蒸留して除去した後,適切な大き
さのビーカーに洗い移して蒸発除去を行ってもよい。
(8) 1L三角フラスコに約35℃に加温した混合溶剤50mLを加え,残油を溶解した後,200mL三角フラス
コに移す。1L三角フラスコを混合溶剤50mLを用いて洗浄し,洗液を200mL三角フラスコに加える。
(9) 試料用冷却浴の温度を−18±1℃に保つ。
内部に水分が付着しないようにガラスろ過器の上をガラス板などで覆った後,冷却浴の温度を−18
±1℃に保つ。
備考 冷却浴に入れるエタノールの量はガラスろ過器が十分に浸る程度とするが,ドライアイスを使
用する場合には泡立ちを考慮してやや少なめとする。
(10) 200mL三角フラスコを試料用冷却浴に入れて冷却する。この際,局部冷却が起きないようにときどき
振り動かす。200mL三角フラスコ内の溶液が−18℃に達してから10分以上経過した後,溶液をガラ

43
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
スろ過器でろ過する(32)。この際,溶液液面が冷媒液面より上にならないように注意する。
注(32) ろ過は,常圧又は減圧のいずれで行ってもよい。
なお,ろ過操作中はなるべく空気を吸い込ませないように注意する。
(11) 200mL三角フラスコに付着したワックスは,混合溶剤10mLを加え,やや加温して溶解させた後,(10)
と同様の操作でガラスろ過器でろ過する。200mL三角フラスコは−18℃に冷却した混合溶剤5〜6mL
で数回洗浄し,同じガラスろ過器でろ過する。
(12) −18℃に冷却した混合溶剤30mLをガラスろ過器に入れ,減圧ろ過してワックスを洗浄する。この操
作を3回繰り返す。
(13) ろ過装置から冷媒を抜き取り,ガラスろ過器の下にあらかじめ質量を0.1mgのけたまではかった
200mLビーカーを置き,ガラスろ過器の温度が室温に達した後,ガラスろ過器上のワックス,及びガ
ラス棒に付着したワックスを約60℃に加温したヘキサン20〜40mLで溶解し,200mLビーカーに移す。
(14) ヘキサン溶液の入ったビーカーを約95℃の水浴に載せ,ヘキサン臭がなくなるまでヘキサンを蒸発除
去する。
備考 ヘキサンの蒸発除去は,ドラフト内で行う。
(15) 200mLビーカーを105±2℃に保った恒温空気浴に15分間入れた後,デシケータ内で1時間以上放冷
し,質量を0.1mgのけたまではかる。
18.6 計算方法及び精度
18.6.1 計算方法 試料中のワックス分は,次の式によって算出し,JIS Z 8401の規定によって0.1質量%
のけたに丸める。
100
×
S
W
C=
ここに,
C: ワックス分(質量%)
W: ワックスの質量(g)
S: 試料採取量(g)
18.6.2 精度 ワックス分試験の結果が2.5〜20質量%の場合,得られた試験結果の許容差(確率0.95)は,
次のとおりである。
備考 試験結果が許容差を外れた場合には,JIS Z 8402の規定によって処理する。
(1) 室内併行精度 同一試験室において,同一人が同一試験器で,引き続き短時間内に同一試料を2回試
験したときの,試験結果の差の許容差を表11に示す。
(2) 室間再現精度 異なる試験室において,別人が別の試験器で,同一試料をそれぞれ1回ずつ試験した
ときの2個の試験結果の差の許容差を表11に示す。
表11 精度
単位 質量%
室内併行許容差
室間再現許容差
0.12±0.08A
1.22+0.15A
備考 A:試験結果の平均値
参考 精度をグラフ化して,参考図2に示す。
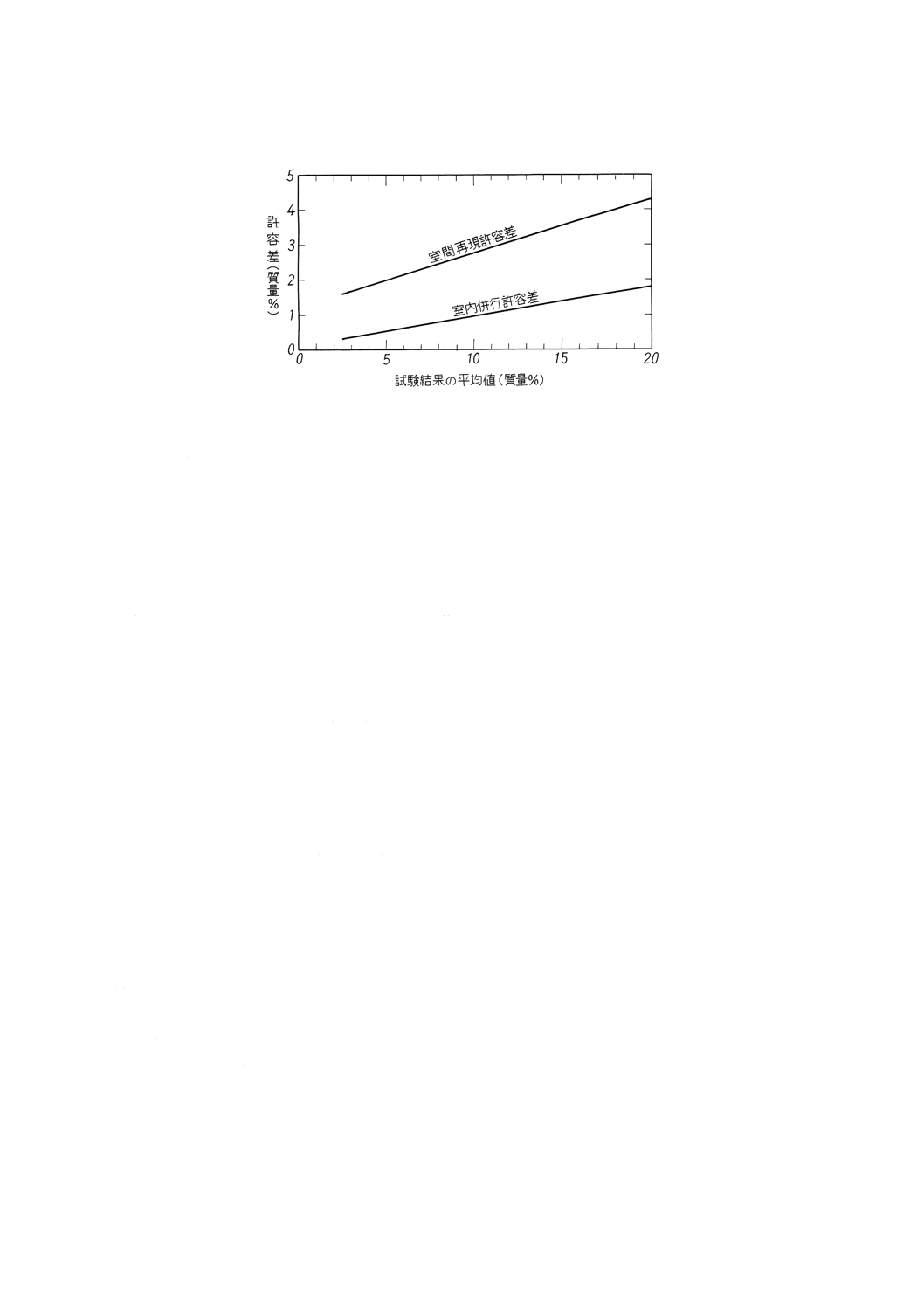
44
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図2 精度
18.7 試験結果の報告 試験結果には,次の事項を記載する。
(1) 試料名,採取場所及び採取月日
(2) JISの規格番号 例 JIS K 2601
(3) 試験方法の名称・項番号及び18.6.1によって得られた結果
(4) 特記事項
19. 硫化水素分試験方法
19.1 試験の原理 試料約100mLを採取し70℃に加熱して,窒素ガスを吹き込み,追い出された硫黄酸化
物を含む硫化水素を酸性過酸化水素水に通して硫黄酸化物を除去した後,亜鉛アミノ錯塩溶液に通して硫
化水素を吸収し,二塩化N,N−ジメチル−p−フェニレンジアンモニウムと塩化鉄(III)を加え,生成し
たメチレンブルーの吸光度を測定して,70℃において発生する硫化水素分を測定する。
備考 この試験方法は,石油学会の技術的検討を経て作成されたものである。
19.2 硫化水素分試験器 硫化水素分試験器は,次による。
(1) 硫化水素吸収装置 (a)〜(f)からなり,組立の一例を図25に示す。
(a) 試験容器 図26に示すもので,主管にはすり合わせによって窒素ガス吸込み管を取り付け,側管に
は温度計を挿入しゴム栓で固定する。
参考 JIS K 2839に規定する図241が相当する。
(b) 温度計 JIS B 7410に規定する温度計番号42 (SG) のもの。
(c) 加熱器 水浴又は適切な電熱器で,窒素ガスを規定の流量で吹き込みながら試験容器内の試料を70
±1℃の温度に保つことができるもの。
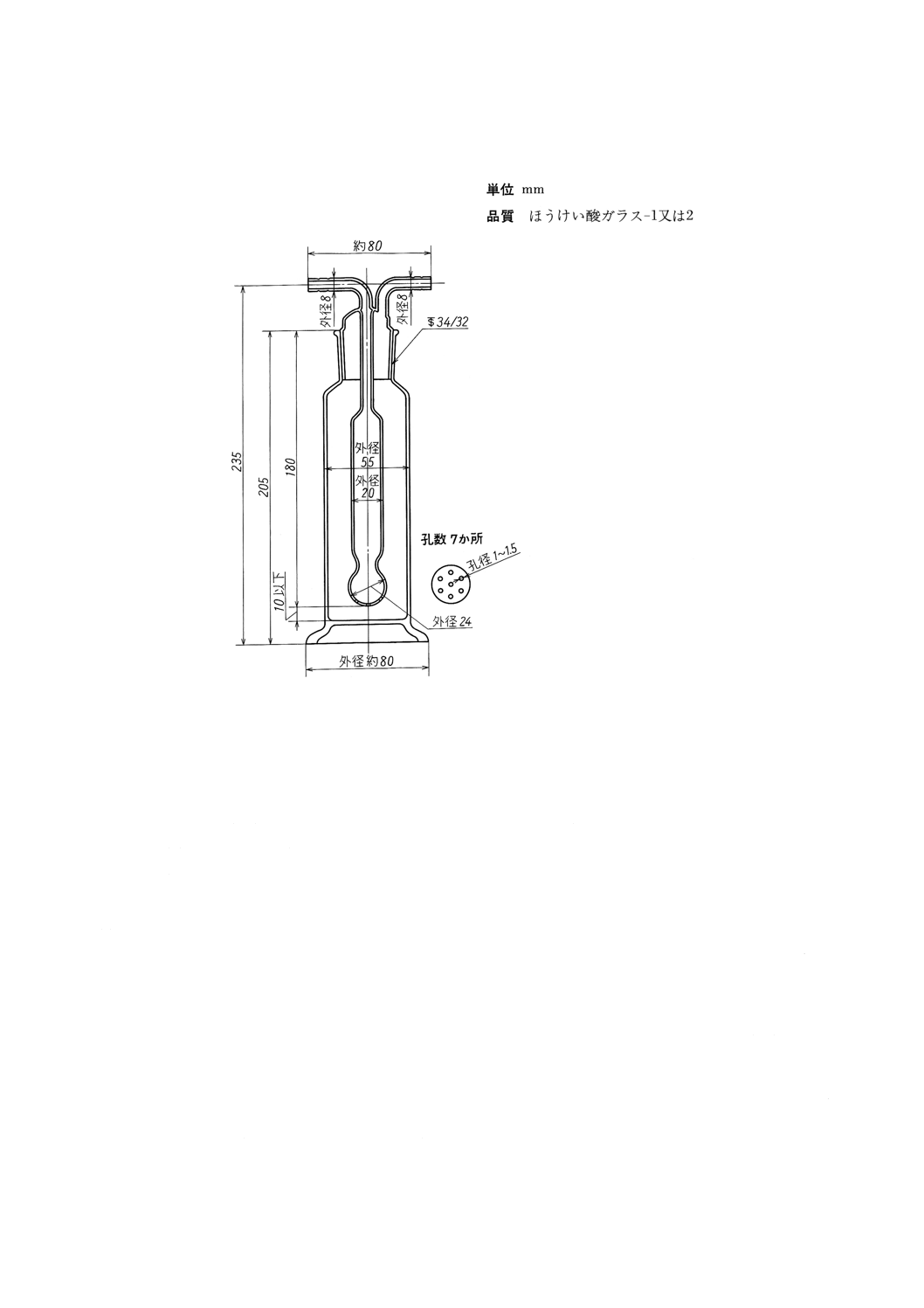
(d) 硫黄酸化物吸収瓶 図27に示す,ミュンケ式ガス洗浄瓶。
参考 JIS K 2839に規定する図240が相当する。
(e) 硫化水素吸収瓶 図27に示す,ミュンケ式ガス洗浄瓶。
参考 JIS K 2839に規定する図240が相当する。
(f) ガス流量計 窒素ガス流量20L/hを測定できるもの。
(2) 全量フラスコ 容量25mL, 50mL, 250mL, 500mL及び1000mLのもの。
(3) 分光光度計又は光電光度計 JIS K 0115に規定する構造のもので,670nmにおける吸光度を測定でき
るもの。
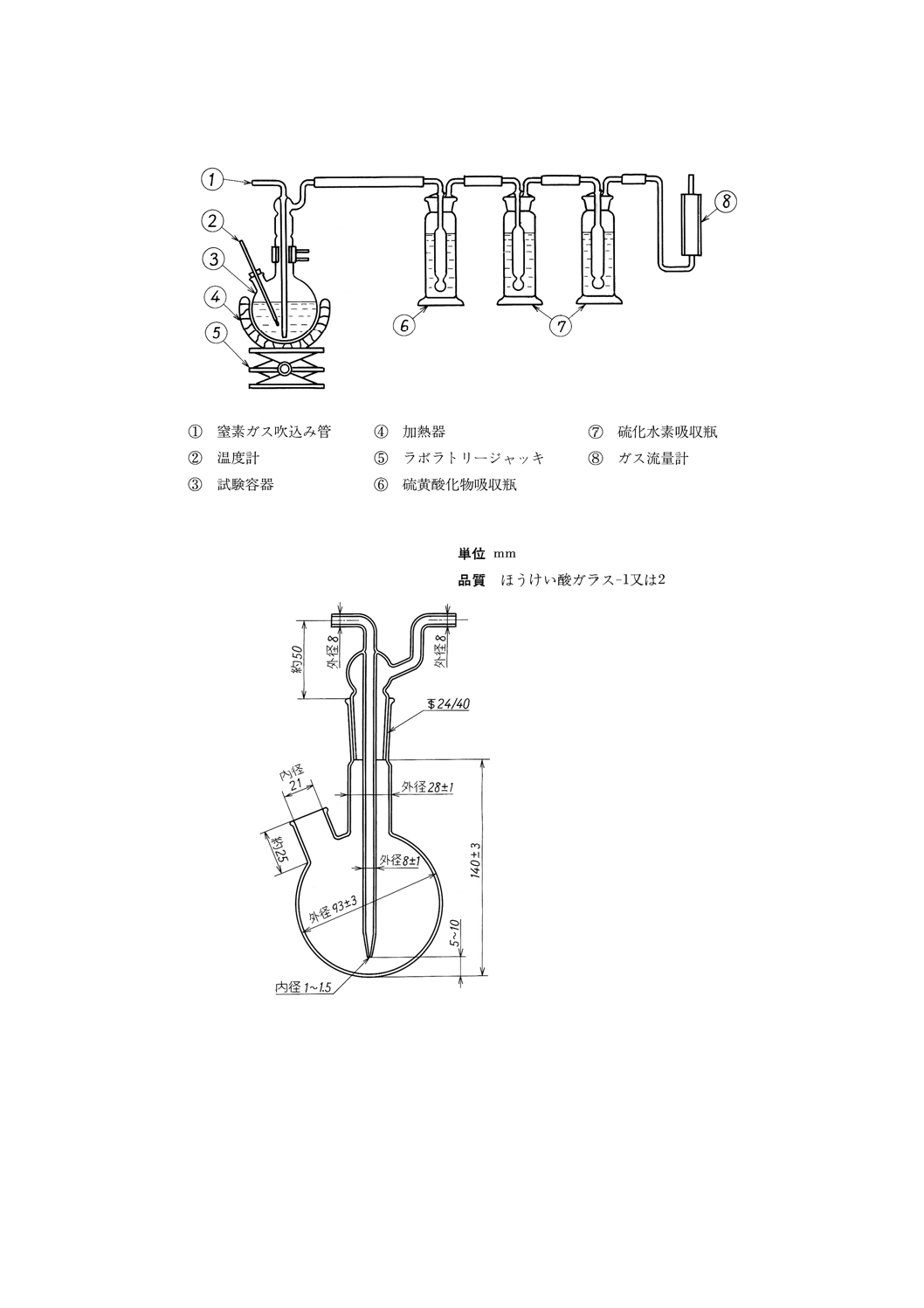
45
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図25 硫化水素吸収装置(一例)
図26 試験容器
46
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図27 硫黄酸化物吸収瓶及び硫化水素吸収瓶
(ミュンケ式ガス洗浄瓶)
19.3 試薬 試薬は,次による。
(1) 硫酸 JIS K 8951に規定するもの。
(2) 塩酸 JIS K 8180に規定する特級のもの。
(3) 水 JIS K 0557に規定するA3のもの。
(4) 1mol/L硫酸 硫酸60mLを水1Lに加えたもの。
(5) 過酸化水素水 (3%) JIS K 8230に規定する過酸化水素水 (30%) 1容に水9容を加えたもの。この溶
液は着色瓶に入れて保存する。
(6) 硫黄酸化物吸収液 1mol/L硫酸50mLに過酸化水素水 (3%) を加え,全量を250mLとしたもの。この
溶液は着色瓶に入れて保存する。
(7) 硫化水素吸収液 JIS K 8953に規定する硫酸亜鉛七水和物5gを水約500mLに溶かし,これにJIS K
8576に規定する水酸化ナトリウム6gを水約300mLに溶解した溶液を加え,更にJIS K 8960に規定す
る硫酸アンモニウム70gをかき混ぜながら加える。水酸化亜鉛の沈殿が溶解した後,水を加えて全量
を1Lとしたもの。
(8) 塩化鉄(III)溶液 JIS K 8142に規定する特級の塩化鉄(III)六水和物1.0gを硫酸水溶液 (1%) 100mL
に溶解したもの。
(9) 0.05mol/Lよう素溶液 JIS K 8913に規定するよう化カリウム40gを水約25mLに溶解した後,JIS K
8920に規定するよう素約13gを加え,更に水約1Lと塩酸3滴を加えたもの。
(10) 二塩化N,N−ジメチル−p−フェニレンジアンモニウム溶液 JIS K 8193に規定する二塩化N,N−
ジメチル−p−フェニレンジアンモニウム0.10gを,水3容と硫酸1容を混合した溶液100mLに溶解
47
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
したもの。
(11) でんぷん溶液 JIS K 8659に規定するでんぷん(溶性)1gを水約30mLと混和し,熱水200mL中にか
き混ぜながら加え約1分間煮沸し,冷却後ろ過したもの。この溶液は使用の都度調製する。
(12) 0.1mol/Lチオ硫酸ナトリウム標準液 JIS K 8637に規定するチオ硫酸ナトリウム五水和物26g及び
JIS K 8625に規定する炭酸ナトリウム0.20gを溶存酸素を含まない水(33)1Lに溶解し,JIS K 8051に規
定する3−メチル−1−ブタノール10mLを加えてよく振り混ぜ,2日間以上放置した後,次によって
標定する。
注(33) フラスコに入れた水の中に,JIS K 1107に規定する窒素ガスを約15分間通気する。
なお,この水は使用時に調製する。
(a) JIS K 8005に規定するよう素酸カリウムを120〜140℃で約2時間乾燥し,無水過塩素酸マグネシウ
ムを入れたデシケータ中で放冷した後,0.9〜1.1gを0.1mgのけたまではかり採り,水に溶解して
250mL全量フラスコに移し,水を標線まで加える。
(b) (a)の溶液25mLをピペットで500mL共栓付き三角フラスコに採り,水約300mL,JIS K 8913に規
定するよう化カリウム2g及び水5容と硫酸1容を混合した溶液5mLを加えた後,直ちに栓をして
静かに振り混ぜ,暗所に5分間放置する。
(c) (b)の溶液をマグネチックスターラでかき混ぜながら,0.1mol/Lチオ硫酸ナトリウム標準液で滴定す
る。
溶液の色が淡い黄色になったら,指示薬としてでんぷん溶液約1mLを加えて滴定を続け,青紫色
が消えた点を終点とする。
(d) よう素酸カリウム溶液を加えないで,(b)〜(c)の操作を行い,空試験値を求める。
(e) 次の式によって,0.1mol/Lチオ硫酸ナトリウム標準液のモル濃度を算出し,JIS Z 8401の規定によ
って小数点以下4けたに丸める。
(
)
250
25
100
667
035
.0
2
1
×
×
×
P
V
V
M
F
−
=
ここに,
F: 0.1mol/Lチオ硫酸ナトリウム標準液のモル濃度
M: (a)ではかり採った,よう素酸カリウムの質量(g)
V1: (c)で滴定に要した0.1mol/Lチオ硫酸ナトリウム標準液の量
(mL)
V2: (d)で求めた空試験値(mL)
P: よう素酸カリウムの含有量(%)
(13) 硫化水素標準液(0.001mgH2S/mL) 硫化水素標準液は,次によって調製する。
(a) JIS K 8949に規定する硫化ナトリウム九水和物約1gを溶存酸素を含まない水(33)に溶解し,100mL
全量フラスコに移し,溶存酸素を含まない水(33)を標線まで加える。
(b) (a)の溶液10mLを,ピペットで200mL共栓付き三角フラスコに採る。
(c) 全量ピペットを用いて0.05mol/Lよう素溶液25mLを加え,更に塩酸1mLを加えた後,栓をして静
かに振り混ぜ暗所に10分間放置する。
(d) 指示薬としてでんぷん溶液約1mLを加え,0.1 mol/Lチオ硫酸ナトリウム標準液で滴定する。終点
は青紫色が消えた点とする。
(e) 水約10mLを200mL共栓付き三角フラスコに採り,(c)〜(d)に従って滴定し,これを空試験値とす
る。
(f) 次の式によって,はかり採り量を算出し,JIS Z 8401の規定によって小数点以下1けたに丸める。
48
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(
)F
A
B
V
×
−
=
11.76
ここに, V: はかり採り量 (mL)
A: (d)で滴定に要した0.1mol/Lチオ硫酸ナトリウム標準液の量
(mL)
B: (e)で求めた空試験値 (mL)
F: 0.1mol/Lチオ硫酸ナトリウム標準液のモル濃度
(g) (f)で求めたはかり採り量の硫化ナトリウム溶液を100mL全量フラスコに入れ,溶存酸素を含まない
水(33)を標線まで加えて混合し,これを原液とする。
(h) 原液5mLを1L全量フラスコに入れ,硫化水素吸収液を標線まで加えて混合する。これを硫化水素
標準液 (0.001mgH2S/mL) とする。
なお,原液及び硫化水素標準液は使用の都度調製する。
19.4 検量線の作成 次の操作はすべて直射日光を避けて行わなければならない。
(1) 25mL全量フラスコに硫化水素吸収液を約20mL入れる。
(2) 3個の25mL全量フラスコに調製直後の硫化水素標準液をピペットでそれぞれ5mL,10mL,15mL入
れ,硫化水素吸収液を加えて液量を約20mLとする。
(3) (1)及び(2)のそれぞれの全量フラスコに二塩化N,N−ジメチル−p−フェニレンジアンモニウム溶液
2mLを加え,続いて塩化鉄(III)溶液1mLを加えた後,栓をして静かに2回転倒させて混合し,硫
化水素吸収液を標線まで加え室温で30分間放置する。これを発色溶液とする。
(4) 分光光度計又は光電光度計と10mmセルを用いて,(1)の全量フラスコの発色溶液を対照液として,(2)
の全量フラスコの発色溶液の670nmにおける吸光度(34)をそれぞれ測定する。
注(34) 吸光度が0.5以上になると読取り誤差が大きくなるので,透過パーセントで読み取り,次の式に
よって吸光度を算出する。
T
E
100
log
=
ここに, E: 吸光度
T: 透過パーセント
(5) グラフ用紙の横軸を硫化水素mg/25mL,縦軸を吸光度としてプロットし,検量線を求める。図28に
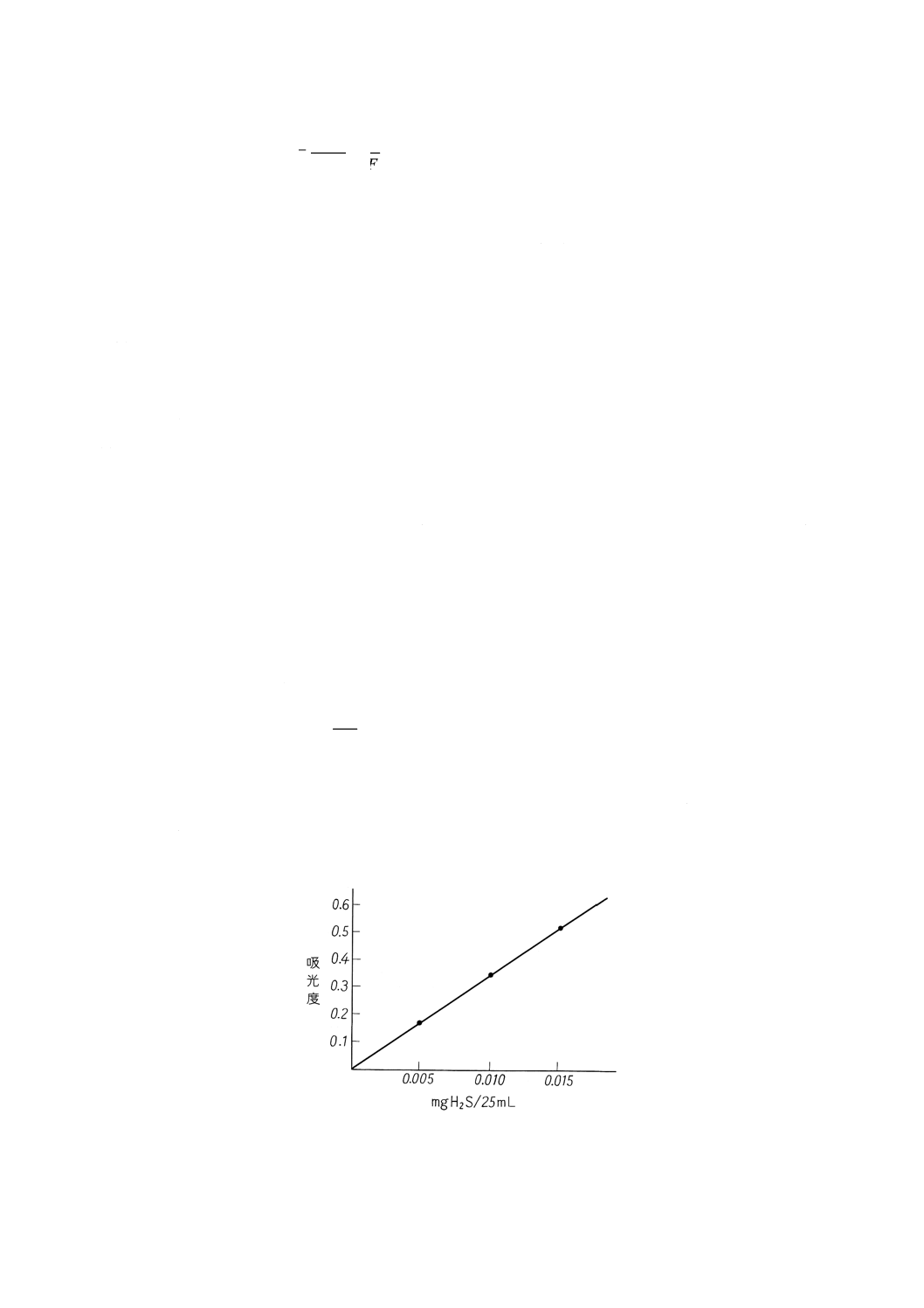
その一例を示す。
図28 検量線(一例)
49
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
19.5 試料の採取及び保存
19.5.1 試料採取方法 試料の採取は,JIS K 2251の規定によるか,又は硫化水素の揮散を減少させるため,
次の方法によることが望ましい。
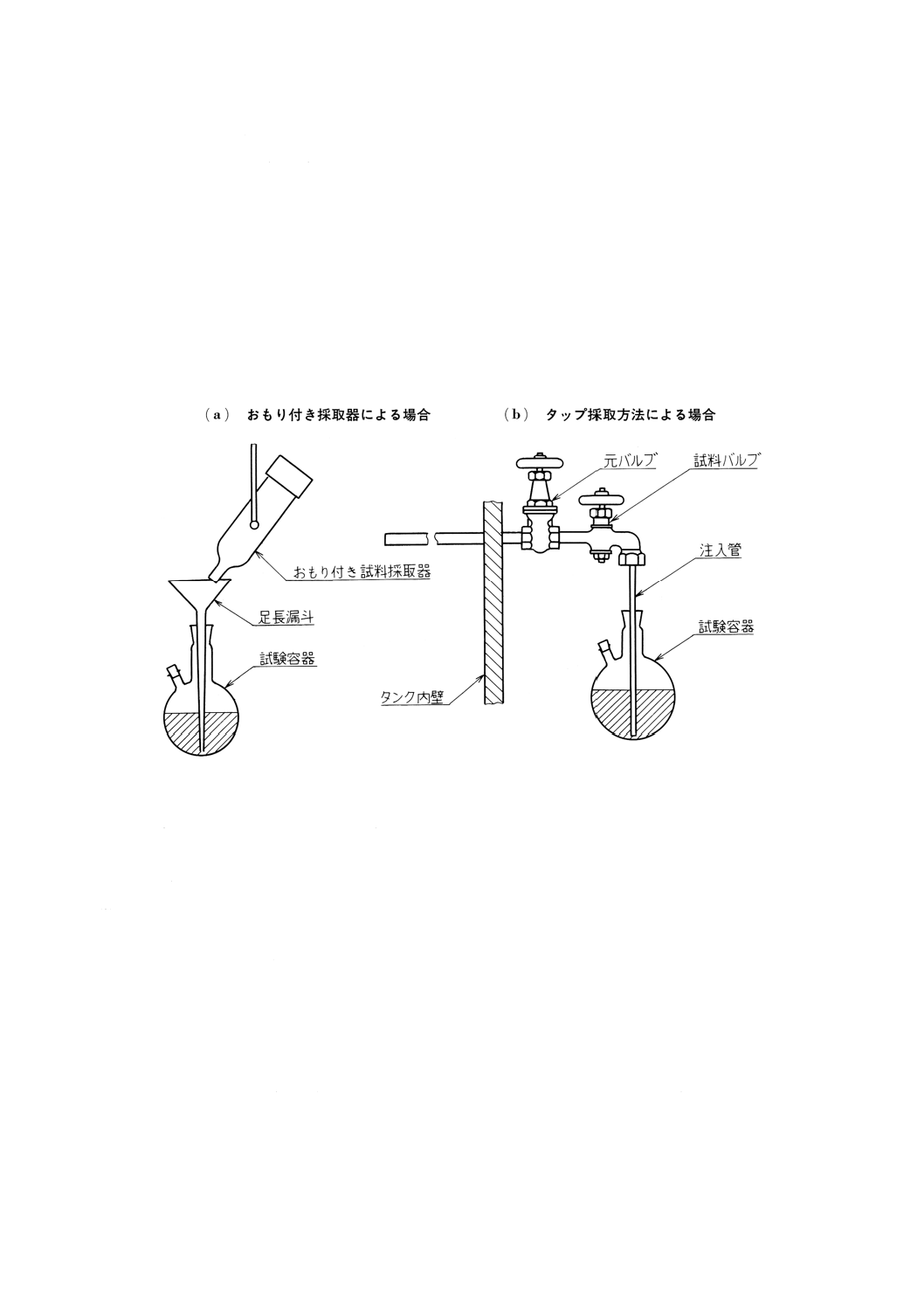
(1) おもり付き採取器による場合は,図29(a)のように適切な口径の足長漏斗を用いる。足長漏斗の足の下
端をあらかじめひょう量した硫化水素試験用試験容器(以下,試験容器という。)の底部にできるだけ
近づけて,おもり付き試料採取器から試料約100mLを直接分取し,直ちに密栓する。
(2) タップ採取方法による場合は,図29(b)のようにタップの出口に試験容器の底まで届くのに十分な長さ
の注入管を取り付ける。あらかじめひょう量した試験容器中に注入管を挿入し,その下端をできるだ
け試験容器底部に近づけて試料約100mLを採取し,直ちに密栓する。
図29 試料採取方法(一例)
19.5.2 試料の保存 採取した試料は直ちに試験することが望ましいが,やむを得ず保存する場合には密栓
して冷暗所に保存する。
19.6 試験の手順 次の操作はすべて直射日光を避けて行わなければならない。
(1) 硫黄酸化物吸収瓶に硫黄酸化物吸収液50mLを入れ,硫化水素吸収瓶2本に硫化水素吸収液100mLを
それぞれ入れる。
(2) 試料の入っている試験容器の質量をはかり,試料はかり採り前後の質量差から試料の質量を求める。
試料量は,約100mLが適切である(35)。
注(35) 硫化水素分が多いことがあらかじめ分かっている場合には,試料はかり採り量を少なくし,キ
シレンで希釈してもよい。
また,流動しにくい試料の場合には,キシレンで希釈するとよい。
(3) 試料容器を室温の加熱器にセットし,試験容器,硫黄酸化物吸収管,硫化水素吸収管2本及びガス流
量計を,図25に示すようにシリコンゴム管で接続する。
(4) 試験容器の窒素ガス吹込み管から窒素ガスを300±50mL/min (18L/h) の流量で吹き込みながら加熱を
始め,試料の温度が70℃に達したら,温度を70±1℃に保つ。
試料の温度が70℃に達してから60分間窒素ガスの吹込みを続ける。
50
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(5) 窒素ガスの吹込みが終了した後,硫化水素吸収瓶2本の吸収液を合わせて500mL全量フラスコに移し,
硫化水素吸収瓶は未使用の硫化水素吸収液の適切量で洗浄して500mL全量フラスコに加える。標線ま
で硫化水素吸収液を加えて混合し,これを比色用原液とする。
(6) 25mL全量フラスコに硫化水素吸収液約20mLを入れる。
(7) 比色用原液1〜20mLの適切量をピペットではかり採り,25mL全量フラスコに入れ,硫化水素吸収液
を加えて全量を約20mLとする。
(8) (6)及び(7)のそれぞれの全量フラスコに二塩化N,N−ジメチル−p−フェニレンジアンモニウム溶液
2mLを加え,更に塩化鉄(III)溶液1mLを加えて,栓をして静かに2回転倒させて混合した後,硫
化水素吸収液を標線まで加えて室温で30分間放置する。これを発色溶液とする。
(9) 分光光度計又は光電光度計と10mmセルを用いて,(6)の全量フラスコの発色溶液を対照液として,(7)
の全量フラスコの発色溶液の670nmにおける吸光度(36)を測定する。
注(36) 吸光度が0.5以上になると読取り誤差が大きくなるので,透過パーセントで読み取り,次の式に
よって吸光度を算出する。
T
E
100
log
=
ここに, E: 吸光度
T: 透過パーセント
(10) 検量線から発色溶液中の硫化水素のmg数を求める。
(11) 吸光度が0.1以下の場合には,(7)での比色用原液のはかり採り量を最大20mLまで増やして,(6)〜(10)
の操作を繰り返す。
また,吸光度が大き過ぎて検量線を外れる場合には,(7)での比色用原液のはかり採り量を最小1mL
まで減らして,(6)〜(10)の操作を繰り返す。
比色用原液のはかり採り量を1mLとしても,検量線を外れる場合には,比色用原液の一部を採り,
硫化水素吸収液で希釈して,これを新たに比色用原液として(6)から(10)の操作を繰り返すか,(2)での
試料採取量を少なくして試験をやり直す。
19.7 計算方法及び精度
19.7.1 計算方法 試料中の硫化水素分は,次の式によって算出し,JIS Z 8401の規定によって整数位に丸
める。
F
M
A
C
×
×000
1
=
ここに,
C: 試料の硫化水素分(質量ppm)
A: 検量線から求めた発色溶液中の硫化水素の量(mg)
M: 試料はかり採り量(g)
F: 発色溶液の希釈係数
500
mL)
原液の容量(
ではかり採った比色用
19.6(7)
(37)
注(37) 19.6(11)で比色用原液を希釈して,新しい比色用原液を作った場合には,その希釈率を発色溶液
の希釈係数に含める。
19.7.2 精度 硫化水素分試験の結果が5〜100質量ppmの場合試験結果の許容差(確率0.95)は,次のと
おりである。
備考 試験結果が許容差を外れた場合には,JIS Z 8402の規定によって処理する。

51
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(1) 室内併行精度 同一試験室において,同一人が同一試験器で,引き続き短時間内に同一試料を2回試
験したときの試験結果の差の許容差を表12に示す。
(2) 室間再現精度 異なる試験室において,別人が別の試験器で,同一試料をそれぞれ1同ずつ試験した
ときの2個の試験結果の差の許容差を表12に示す。
表12 精度
単位 質量%
室内併行許容差
室間再現許容差
0.27+0.14A
1.27+0.32A
備考 A:試験結果の平均値
参考 精度をグラフ化して,参考図3に示す。
参考図3 精度
19.8 試験結果の報告 試験結果には,次の事項を記載する。
(1) 試料名,採取場所及び採取月日
(2) JISの規格番号 例 JIS K 2601
(3) 試験方法の名称・項番号及び19.7.1によって得られた結果
(4) 特記事項
付表1 引用規格
JIS B 7410 石油類試験用ガラス製温度計
JIS C 2520 電熱用合金線及び帯
JIS K 0050 化学分析方法通則
JIS K 0113 電位差・電流・電量・カールフィッシャー滴定方法通則
JIS K 0115 吸光光度分析通則
JIS K 0557 化学分析用の水
JIS K 1107 高純度窒素
JIS K 2249 原油及び石油製品一密度試験方法及び密度・質量・容量換算表
JIS K 2251 原油及び石油製品−試料採取方法
JIS K 2254 石油製品一蒸留試験方法
JIS K 2258 原油及び燃料油蒸気圧試験方法−リード法
JIS K 2265 原油及び石油製品−引火点試験方法
52
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 2269 原油及び石油製品の流動点並びに石油製品曇り点試験方法
JIS K 2270 原油及び石油製品−残留炭素分試験方法
JIS K 2272 原油及び石油製品の灰分並びに硫酸灰分試験方法
JIS K 2275 原油及び石油製品−水分試験方法
JIS K 2279 原油及び石油製品−発熱量試験方法及び計算による推定方法
JIS K 2283 原油及び石油製品−動粘度試験方法及び粘度指数算出方法
JIS K 2435 ベンゼン・トルエン・キシレン
JIS K 2541 原油及び石油製品−硫黄分試験方法
JIS K 2609 原油及び石油製品−窒素分試験方法
JIS K 8005 容量分析用標準物質
JIS K 8034 アセトン(試薬)
JIS K 8051 3−メチル−1−ブタノール(試薬)
JIS K 8101 エタノール (99.5) (試薬)
JIS K 8102 エタノール (95) (試薬)
JIS K 8122 塩化カルシウム二水和物(試薬)
JIS K 8123 塩化カルシウム(試薬)
JIS K 8124 塩化カルシウム(乾燥用)(試薬)
JIS K 8142 塩化鉄(III)六水和物(試薬)
JIS K 8150 塩化ナトリウム(試薬)
JIS K 8159 塩化マグネシウム六水和物(試薬)
JIS K 8180 塩酸(試薬)
JIS K 8193 二塩化N,−ジメチル−p−フェニレンジアンモニウム(試薬)
JIS K 8230 過酸化水素(試薬)
JIS K 8271 キシレン(試薬)
JIS K 8374 酢酸鉛(II)三水和物(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8550 硝酸銀(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8625 炭酸ナトリウム(試薬)
JIS K 8637 チオ硫酸ナトリウム五水和物(試薬)
JIS K 8659 でんぷん(溶性)(試薬)
JIS K 8680 トルエン(試薬)
JIS K 8810 1−ブタノール(試薬)
JIS K 8848 ヘキサン(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8913 よう化カリウム(試薬)
JIS K 8920 よう素(試薬)
JIS K 8949 硫化ナトリウム九水和物(試薬)
JIS K 8951 硫酸(試薬)
JIS K 8953 硫酸亜鉛七水和物(試薬)
53
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 8960 硫酸アンモニウム(試薬)
JIS K 8982 硫酸アンモニウム鉄(III)・12水(試薬)
JIS K 8987 硫酸ナトリウム(試薬)
JIS K 9001 チオシアン酸カリウム(試薬)
JIS P 3801 ろ紙(化学分析用)
JIS R 3503 化学分析用ガラス器具
JIS Z 8401 数値の丸め方
JIS Z 8402 分析・試験の許容差通則
関連規格 JIS K 2839 石油類試験用ガラス器具
ASTM D 96-88(1984) Standard Test Method for Water and Sediment in Crude Oil by Centrifuge
Method (Field Procedure)
ASTM D 3230-89 Standard Test Method for Salts in Crude Oil (Electrometric Method)
ASTM D 4007-81(1987) Standard Test Method for Water and Sediment in Crude Oil by the
Centrifuge Method (Laboratory Procedure)
IP 265/70 (1984) TOTAL SALTS CONTENT OF CRUDE OIL CONDUCTIVITY METHOD
ISO/DIS 8708 : 1984 Crude petroleum oil−Determination of distillation characteristics using 15
theoretical plates column
54
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1 水でい分試験用水飽和トルエン調製方法
1. 適用範囲 この附属書1は,水でい分試験方法で使用する水飽和トルエンの調製方法について規定す
る。
備考 附属書1図1に特定の温度範囲におけるトルエンの水溶解度を示す。水の溶解度は,21℃で
0.03%,70℃で0.17%と温度が上昇するにつれて増加する。通常,トルエンはほとんど水分を含
まないので,この状態で使用すると試料中水分の一部又は大部分を溶解してしまうため,見掛
け上,試料の水でい分が減少する。遠心分離法による水でい分の測定を正確に行うためには,
試験温度においてトルエンを水で飽和しておかなければならない。
2. 試験器 試験器は,次による。
(1) 加熱用液浴 容量1Lの瓶を肩まで浸すのに十分な深さの液浴で,60±3℃の温度に保持できるもの。
(2) ガラス瓶 容量1Lでねじぶた付きのもの。
3. 試薬 試薬は,次による。
(1) トルエン 本体14.3(1)に規定するもの。
(2) 水 JIS K 0557に規定するもの又は水道水。
4. 試験の手順 試験の手順は,次による。
(1) 本体14.5で遠心分離操作を行う温度60±3℃(1)に加熱用液浴の温度を合わせる。
注(1) 49±3℃で遠心分離操作を行う場合には,その温度に合わせる。
参考 75±3℃で遠心分離操作を行う場合には,その温度に合わせる。
(2) ガラス瓶にトルエン700〜800mLを入れ,水25mLを加え,ねじぶたをして30秒間激しく振とうする。
(3) ねじぶたを緩めて,ガラス瓶を加熱用液浴に30分間浸した後,ガラス瓶を取り出してねじぶたを閉め,
注意して30秒間振とうする。
参考 60℃におけるトルエンの蒸気圧は38℃のときの約2倍となるので,ねじぶたの密閉度が不完全
な場合には噴出するおそれがある。
(4) (3)の操作を3回繰り返す。
(5) トルエンと分離水との平衡を確実にするため,(4)で得られた水−トルエン混合液は使用前に48時間
以上,加熱用液浴に浸しておき,上澄みを使用する。48時間以前に水飽和トルエンを使用する場合に
は,上澄みを目盛試験管に入れ,試料の遠心分離と同様の条件で遠心分離を行い,目盛試験管の底部
にたまった分離水を乱さないように注意して,上澄みをピペットで採取して使用する。
(6) トルエンの水飽和は時間及び温度に左右されるので,いつでも試験に使用できるように,水−トルエ
ン混合液の瓶は試験温度範囲内に保った加熱用液浴に常時入れておくことが望ましい。
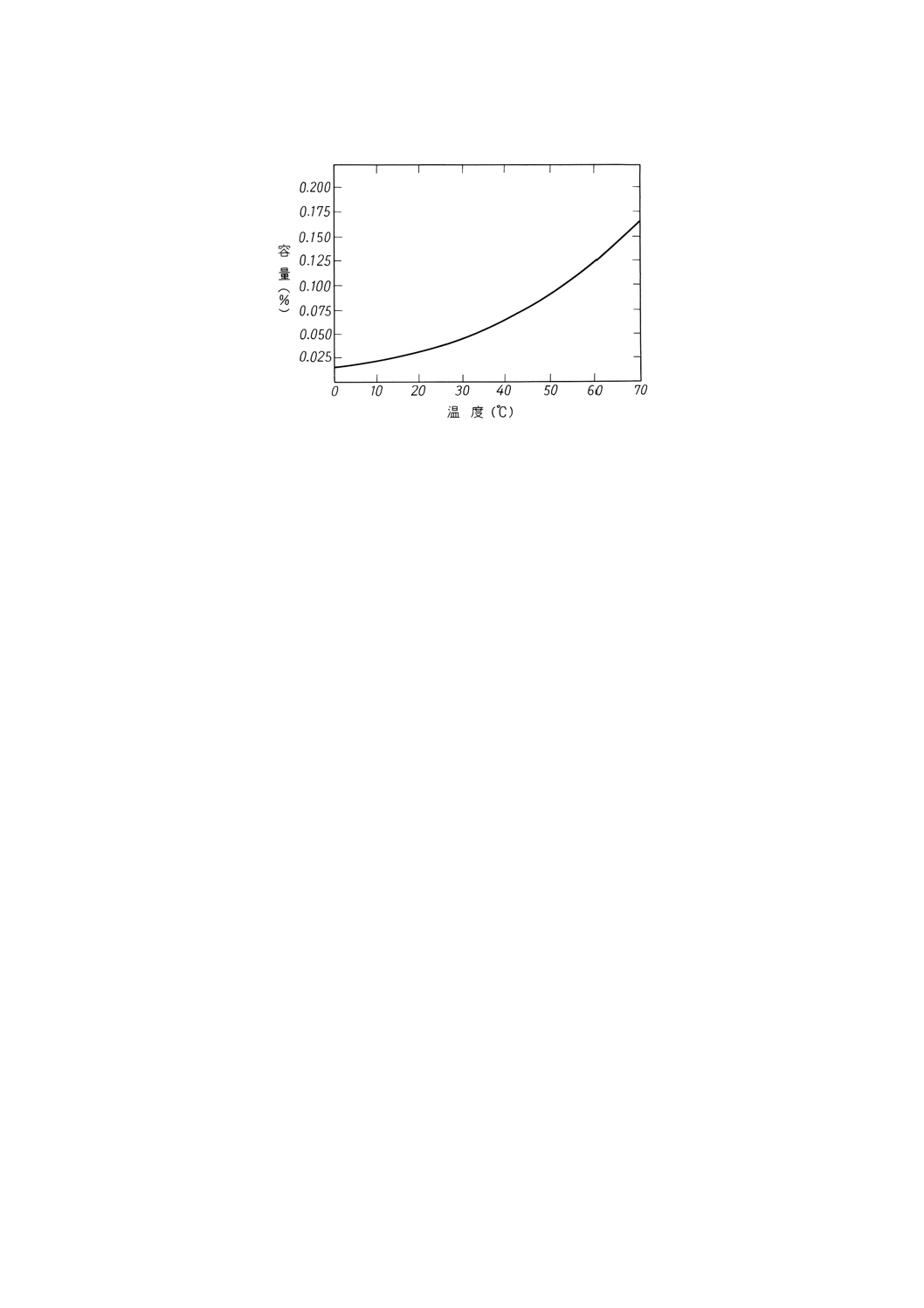
55
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書1図1 トルエンの各温度における水溶解度
56
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
附属書2 水でい分試験用試料の均一化方法
1. 適用範囲 この附属書2は,混和器を使用して水でい分試験用原油試料を均一化する方法について規
定する。
備考 水でい分試験用試料の水でい分の均一性について疑義が生じた場合,この均一化の操作を行っ
てから,試料の水でい分を測定する。
2. 均一化法の原理 あらかじめ低水分原油に,定量の水を加えた試料容器中で,水分が均一になるよう
な混和器の運転条件を求める。ここで求めた混和器の運転条件で試料原油中の水分を均一にする。
3. 器具及び試薬 器具及び試薬は,JIS K 2275の4.(カールフィッシャー式容量滴定法),5.(カールフ
ィッシャー式電量滴定法)又は6.(水素化物反応法)に規定されているものによるほかは,次による。
(1) 低水分原油 JIS K 2275の4.,5.又は6.の規定によって測定した水分が0.1容量%以下の原油。
(2) 試料容器 500mL又は適切な大きさのもの。
(3) 混和器 二重回転翼式のホモジナイザーで回転数約3000rpmのものが適切であるが,満足できる性能
であれば他の型を用いてもよい。
また,揮発性物質を含む試料には密閉式のものがよい。
参考 エアーモータなど引火のおそれのないものがよい。
4. 混和器の効率の測定 新しい混和器を使用する場合,試料容器の形状を変える場合,及び原油の粘度
が大きく変わる場合は,必ず前もってこの測定を行い,均一化を確認する。
(1) 試料容器の質量を0.01 gのけたまではかる。これをM0(g)とする。
(2) 試料容器に低水分原油を約80%満たす。
(3) 混和器の底が試料容器の底から5mm上にくるように,混和器をセットして使用予定の回転数及び時
間で,低水分原油をかき混ぜる。
(4) 直ちに低水分原油の水分をJIS K 2275の4.,5.又は6.の規定によって,2回測定し,2回の結果の平均
値を求める。この水分結果を空試験値として記録する。
備考 2回の測定結果の差が室内併行許容差内であることを確認する。
(5) 低水分原油及び試料容器の質量を0.01gのけたまではかり,これをM1 (g) とする。(3)と同じように混
和器をセットする。
(6) 低水分原油の質量 (M1−M0) から,加えたい水分量に相当する量を求める。
備考 加えたい水分量は,低水分原油の質量の1〜2質量%である。
(7) 注射器に,加えたい水分量に相当する水を満たし,質量を0.1mgのけたまではかる。
(8) 混和器の注ぎ口の近くの低水分原油の表面下に水を注入する。
(9) 針についている油分をすべてふき取り,注射器の質量を再度0.1mgのけたまではかり,加えた水の質
量を求め,(6)の低水分原油量から添加した水分を求める。
(10) (3)と同じ条件で試料をかき混ぜる。
備考 均一化中の温度上昇は,10℃を超えてはならない。
(11) 直ちに液面のすぐ下の試料の水分含量をJIS K 2275の4.,5.又は6.の規定によって1回測定する。
57
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(12) かき混ぜることなく,15分後及び30分後に水分含量をそれぞれ1回測定する。
(13)(11)及び(12)で求めた水分から(4)の空試験値を減じた値と,添加した水分の差が0.05%以内にあること,
及び直後,15分,30分のはかり採り時の水分が0.05%以内にあることを確認する。
(14) もしこれが(13)の条件を満足しない場合は,(3)の混和器回転数及び混和時間を変えて再度(4)から(13)
までの操作をやり直す。
参考 原油によっては,水分の均一化がしにくいことがあるが,この場合は原油を室温以下に冷却し
て行うとよい。
5. 原油試料の均一化方法 試料容器の中の原油の水でい分を測定するに先立って,4.で均一が確認され
た混和器を使用して,同一の条件でかき混ぜる。
備考 この均一化は,水でい分測定の直前(15分以内)に行う。
58
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考 理論段数15段の精留塔を使用した蒸留試験方法
この試験方法は,本体の規定に関係した試験方法を参考として明示したものであって,規格の一部では
ない。
この参考は,1984年に発行されたISO/DIS 8708 (Crude petroleum oil−Determination of distillation
characteristics using 15 theoretical plates column) を翻訳し,技術的内容及び規格票の様式を変更することな
く作成したものである。
なお,この参考で下線(点線)を施した部分及び報告様式の一例は,原国際規格にない事項である。
0. 序文
この参考で規定する方法は,精留の度合いと試験所要時間とを勘案して,試験室間の蒸留データの比較
を容易にするものである。
技術的内容として,15:5(理論段数15段塔,5:1還流比)すなわちT.B.P.(真沸点)による方法を規定し
ている。
したがって,得られる蒸留性状は,原油の商品価値の評価及び石油精製の際の技術的データとして有益
である。
得られる留出油のフラクション又はカットは,混合して分析用及び品質評価用試料とすることができる
が,そのような留出油の混合や評価はこの参考には含まれない。
原油と,この方法で得られた留出油についての試験項目と報告様式の一例を参考表に示す。
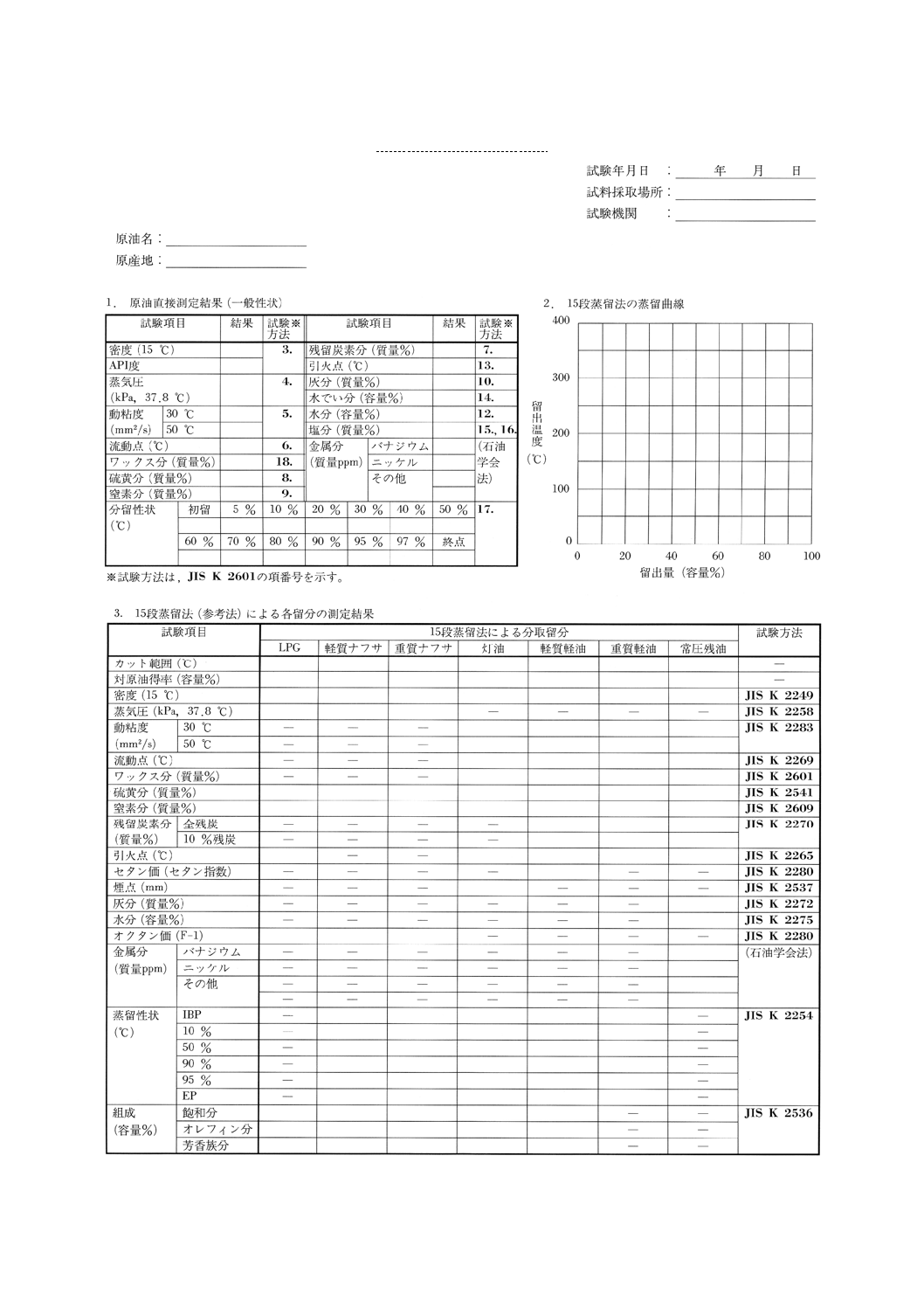
59
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考表 原油分析性状表

60
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
この方法で必要な装置に対する性能基準の測定は,この参考の参考附属書で規定する。参考附属書の一
覧を,参考付表に示す。
参考付表 参考附属書一覧表
表題
参考附属書A
蒸留カラムの効率測定方法
参考附属書B
カラムの動的ホールドアップの測定方法
参考附属書C
蒸留カラムの熱損失の測定方法
参考附属書D
温度検出器の位置の確認方法
参考附属書E
温度応答時間の確認方法
参考附属書F
検出器の校正方法
参考附属書G
還流分割弁の性能検査方法
参考附属書H
含水原油試料の脱水方法
参考附属書I
蒸留温度の計算方法
1. 適用範囲
この参考では,安定化した原油の蒸留試験の手順について規定する。
この方法では,張込み量0.5〜30リットルで,性能が理論段数14〜18段の“精留”カラムを使用し,還
流比5:1で操作することを基本とする。この参考で必要な装置の性能基準を規定し,許容される装置の典
型例を図示する。
この参考は,次の項目について規定する。
− 液化ガス留分と留出油及び残留油の採取。これらに関する分析データを得てもよいが,これら各留分
の分析については記述していない。
− 上述留分の質量及び容量による収率の測定
− 温度 (℃) 対留出油の質量パーセント及び温度対留出油の容量パーセントの蒸留曲線の作成
この方法は,リード蒸気圧が82.7kPa未満の安定化した原油,又は種々の石油系混合物に適用でき
る。
ただし,液化石油ガス,非常に軽質のナフサ及び初留点が400℃を超える留分には適用できない。
2. 引用規格
次の規格は,この参考に関連する規定を含んでいる。国際規格が発行された時点では,次の版が有効で
あった。
規格はすべて改訂されることが多いため,この国際規格に同意する各機関は,次の規格の最新版を採用
する可能性を調査することが望ましい。IEC及びISOのメンバーは,国際規格の現行版を正当なものとし
て保持する。
ISO 3007:Petroleum products−Determination of vapour pressure−Reid method
参考 この項目の内容は,JIS K 2258(原油及び燃料油蒸気圧試験方法−リード法)と技術的内容
は,一致している。
ISO 3675:Crude petroleum and liquid petroleum products−Laboratory determination of density or relative
density−Hydrometer method
参考 この項目の内容は,JIS K 2249(原油及び石油製品−密度試験方法及び密度・質量・容量換
算表)と技術的内容は,一致している。
61
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ISO 3170:Petroleum liquids−Manual samp1ing
参考 この項目の内容は,JIS K 2251(原油及び石油製品−試料採取方法)と技術的内容は,一致
している。
ISO 3171:Petroleum liquids−Automatic pipeline sampling
ISO 3838:Crude petroleum and liquid or soild petroleum products−Determination of density or relative
density−Capillary-stoppered pyknometer and graduated bicapillary pyknometer methods
参考 この項目の内容は,JIS K 2249(原油及び石油製品−密度試験方法及び密度・質量・容量換
算表)と技術的内容は,一致している。
ASTM D-2427:Method for determination of C2 through C5 hydrocarbons in gasoline by gas chromatography
参考 この項目の内容は,JIS K 2536(石油製品−成分試験方法)と技術的内容は,一致している。
3. 用語の定義
3.1
理論段
液体とその蒸気間の熱力学平衡を達成するために必要なカラムの区切り。
充てんカラムでは,1理論段相当高さ(HETP)をミリメートルで表す。実段カラムの効率は,実段1段で
理論段1段の何パーセントを達成できたかで表す。
3.2
還流比 R
カラムの頂部に達した蒸気は完全に凝縮し,その結果生じる液体は二つの部分に分けられる。一つはL
(還流分)としてカラムに戻され,もう一方のD(留出分)は留出油として回収される。還流比はL対D
の比すなわちR=L/Dである。還流比は,L=0すなわち全留出時の0から,D=0すなわち全還流時の無限
大まで変動する。
3.3
内部還流
カラム内を正常に流れ落ちる液体。純粋な化合物を断熱されたカラムを用いて蒸留する場合,内部還流
は頂部から底部まで一定であり,頂部における還流に等しい。原油の場合は,動的ホールドアップ中の混
合物によって温度こう配が生じるため,カラムの底部での還流がより多くなる。
3.4
圧力ドロップ
カラムの底部から頂部へと蒸気を押し上げるのに必要な推進力の目安。充てんカラム長の1m当たりの
mmHg(比圧力ドロップ)か,実段カラムでは全体のmmHgで表される。ある一定の蒸発速度下では,芳
香族の圧力の方がパラフィン族より高く,高分子量物質の方が低分子量物質より高い。
3.5
動的ホールドアップ
正常な操作条件下でカラム内に保持される液体の量。充てんカラムに対しては充てん物容積のパーセン
トとして表されるので,データは互いに比較可能である。
実段カラムでは,1段当たりのmLで,又は理論段1段当たりのmLで表す。トレイの大きさが異なる
ためデータの比較を行えるのは,同じ直径のものどうしだけである。
充てんカラムのデータは,実段カラムのデータとは比較できないが,理論段1段当たりの絶対mL単位
でなら可能である(参考表1参照)。動的ホールドアップは,フラッド点に達するまで蒸留速度が上昇する
につれて増加し,精留塔の種類によって異なる。
3.6
静的ホールドアップ
(“ウエッテイジ”と呼ばれることもある。)。蒸留が終わり,カラムからの液の滴下が止まった後,カラ
ムに残留する液体の量。これは充てん物の特性又は段の設計に特有なものであり,最終カットポイントで
62
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
のカラム中の物質の組成によって決まる。
3.7
蒸発速度
単位時間当たりにカラムに流入する蒸気の量。比較するために,断面積1cm2当たり1時間に流入する量
をmL(比蒸発速度)で表すか,1時間当たりの液体量をmLで表す。
参考 後者の場合,効率評価にはヘプタンとメチルシクロヘキサンの試験混合物を用いて塔の底部で
測定する。ヘプタン・メチルシクロヘキサン試験混合物の最高蒸発量は,クラッディングを起
こさず安定した条件下でカラムが許容できる量である。通常の断熱操作では,蒸発速度を概算
するには留出速度に(還流比+1)を掛けることによって求められる。
3.8
留出速度
還流分割弁から留出油が留出する速度。時間当たりのmLで表す。
3.9
フラッド点
蒸留装置の蒸発速度を増していくと,上昇する蒸気が還流液の流下を妨害して,カラムが突然液体で満
たされる。この現象をフラッディング(あふれ)と呼び,この現象が起こる蒸発速度がフラッド点である。
フラッディングを元に戻すには,ヒータの加熱力,すなわち蒸発速度を低下させる。カラムのフラッド点
は,通常,ヘプタン・メチルシクロヘキサン試験混合物を用いてカラムの効率を調べるときに測定する。
3.10 断熱性
カラムの全体にわたり著しい熱の増加も損失もみられない性質。原油のような混合物を蒸留するときは,
カラムの下方の内部還流が増加するのが普通である。
参考 カラムで熱の損失が起こる場合,内部還流は,頭部の還流よりも異常に多い。過剰加熱したマ
ントルによって,カラムが加熱されたときは,損失と逆の現象が起こる。
3.11 蒸留温度
カラムのすぐ上の頭部で測定される飽和蒸気の温度。頭部温度又は蒸気温度ともいう。
3.12 蒸留圧力
蒸気温度の測定点にできるだけ近いところ,通常は凝縮管の上部で測定する圧力。
3.13 原油の脱ブタン化
ブタンまでの軽質炭化水素を除去し,より重質な炭化水素を保持すること。実際的には,コールドトラ
ップに集められた軽質炭化水素カットに含まれるC2〜C4炭化水素が,元の試料に含まれていた量の95%
を超え,C5炭化水素が元の試料の5%未満であれば,原油は脱ブタン化されたとみなす。
3.14 カット温度
留出油を区分するために,あらかじめ設定する蒸留温度,カットポイントともいう。
通常は,製油所の中間製品の沸点範囲に応じた温度とすることが多い。
3.15 フラクション
あらかじめ定めたカット温度から次のカット温度に達するまで,留出油受器に採り出した個々の留出油
をいう。フラクションの量は,留出油の沸点温度範囲が5℃又は10℃に相当する量か,試料の張込み容量
の2〜5容量%に相当する一定容量とするとよい。
張込み油量に対するフラクションの量から求めた留出量(質量%又は容量%)と対応する留出温度から
試料の蒸留曲線を求めることができる。
3.16 カット(留分)
あるカット温度から次のカット温度に達するまでに留出した各フラクションの全体を,カット又は留分
という。フラクションを採り続けている間に,あらかじめ定めたフラクション量となる前にカット温度に
63
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
達した場合には,留出油受器を付せ替えて,カットの区切りとする。
それぞれのカットについて分析・試験を行うことによって,製油の際の中間製品などの性状を予測する
ことができる。
4. 試験方法の概要
4.1
安定化した原油0.5〜30リットルをはかり採り,最高温度400℃AET(常圧換算温度)まで蒸留する。
使用するカラムは,全還流において理論段数が14段以上18段以下の効率をもつこと。
4.2
すべての操作圧において還流比5 : 1を維持するが,例外として,最低操作圧が0.266kPaの場合には,
還流比2:1で蒸留してもよい。
4.3
温度,圧力及び他の変数の測定値は,一定間隔及び各カット又はフラクションの終了時に記録する。
4.4
各カット又は各フラクションの質量と密度を測定する。蒸留の質量収率は,液化ガスカット及び残
留油を含む全フラクションの質量から算出する。
4.5
蒸留の容量収率は,全フラクション及び残留油の質量と密度から算出するが,その際,温度は15℃
を適用する。
参考 共同試験の場合又は合意が得られていない場合には,蒸留開始に先立って,関係者は,試験操
作条件のすべて,特に,カット温度,最低操作圧及び最低圧下での還流比について合意に達し
ていなければならない。
5. 試料採取
5.1
JIS K 2251に規定する指示に従って,蒸留用試料を採取する。
5.1.1
試料は,漏れのない密閉容器に採取する。試料は開封する前に,冷蔵庫内に数時間(できれば1
夜間)入れ,0〜5℃に冷却する。
5.1.2
もし開封したときに水分が含まれていることが分かったら,試料を使用する前にJIS K 2251の規
定に従って,試料を均一化してから参考附属書Hの指示事項に従って,予備蒸留によって水分を取り除く。
6. 装置
6.1
常圧蒸留 常圧蒸留用の部品は,次のものからなる。
(a) 蒸留フラスコは,沸騰熱の供給源となる加熱装置を備えたもの。
(b) カラム及び還流分割弁は,できれば高反射性真空ジャケットに封じ込められたもの。また必要ならば,
カラムからの熱損失を補償するための熱供給源となる加熱装置に覆われたもの。
(c) 凝縮管頭部は,冷却剤を供給して−20℃まで冷却できること。
(d) 留出油を取り出すための,フラクション採取器及び液体冷却器。
(e) カラム断熱材を使用する場合には,その温度測定器具,蒸気温度,圧力記録器具,カラム内の圧力ド
ロップ調節器具及び蒸留速度の直接測定器具。
(f) 附属品,例えば接続管類,二重壁凝縮管,トラップ及び受器。
蒸留装置系の典型例を参考図1に示す。
参考 すべての部品は,漏れのないように組み立てること。
6.1.1
蒸留フラスコの容量は,張込み量の少なくとも1.5倍はあること。張込み量は0.5〜30リットルの
間で,参考表1に示し,参考附属書Bで説明するカラムのホールドアップ特性によって決まる。蒸留フラ
スコは2本の枝管が付いたものでもよい。
64
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.1.1.1
枝管の1本は,沸騰しているフラスコ内の油の温度測定用センサを挿入するサーモウェルとして
使用する。その終端はフラスコの底から約5mmに位置し,蒸留終了時にも残留油に浸されているように
する。第2の枝管を使用する場合は,圧力ドロップ測定用として用いることができる。その際,窒素の吹
き込み,さらには機械的かくはん装置を併用することによって,特に減圧下における突沸を防ぐことがで
きる。
6.1.1.2
マグネチックスターラを入れて丸底フラスコを使用するときは,マグネチックスターラがガラス
を傷つけずに回転できるように,フラスコの底をわずかに平にするか,くぼみを付けるとよい。この場合,
サーモウェルの終端は中央から40±5mm離して,マグネチックスターラを避けること。代わりに沸騰石
を使用してもよい。
注意
ガラス製蒸留フラスコの,よく見えるという利点は好ましいが,張込み量が多くなればなるほど
危険性も大きくなる。このため,容量が10リットルを超えるガラス製フラスコは勧められない。
6.1.2 加熱装置 すべての圧力レベルにおいて,一定の蒸発速度を保つようにフラスコを加熱できるもの。
これは電気式加熱マントルの使用によって可能で,フラスコの下半分をマントルで覆い,ヒーターエレメ
ントの熱の31が底中央部に,残りの32が残り半球に当たるようにする。比例調節器が好ましいが,加熱用電
力は各回路に設置した可変自動変圧器を使用することによって適切に調節できる。より小さい加熱器は,
差圧マノメータ又は同様の計器に示されたカラム内の圧力ドロップによって自動的に調節される。加熱装
置はまた,蒸留速度直接測定器によっても自動的に調節できる。
6.1.2.1
この手順で原油を完全に沸騰させるのに必要なワット数は,一般的に張込み量の1mL当たり
0.125〜0.2Wであり,原油の性質に依存する。
6.1.2.2
フラスコ加熱器の熱密度は一般的に0.5〜1.2W/cm2であり,十分な耐久性を保つように,石英又
は同等の繊維で被覆したニクロム線を使用すること。
6.1.2.3
浸没式加熱器を同じように使用してもよい。この場合,応答が速いという利点があるものの壊れ
やすく,また,蒸留終了時まで加熱エレメントが浸された状態を保つような特別設計のフラスコが必要で
ある。使用時の熱密度は,約4W/cm2であるのが望ましい。
6.1.2.4
フラスコの上半分は,最高電圧で約0.25W/cm2の電気式加熱器を備えたマントルで覆って,液体
表面からの不必要な熱の損失を防ぐこと。
6.1.3
カラム カラムには,参考表1に要約する性能特性をもつ粒状の充てん物を詰めるか,精留プレー
トを設置する。その典型例を参考図1に示す。カラムは,次の規格に適合しなければならない。
6.1.3.1
内径は,18〜70mm。
6.1.3.2
効率は,参考附属書Aで規定する手順で測定するとき,全還流で14〜18理論段数であること。
6.1.3.3
参考附属書Bに従って最高蒸発速度の75%で操作するときに,動的ホールドアップが張込み量の
1〜4%に等しくなるような張込み量で使用する(参考表1参照)。
6.1.3.4
参考附属書Cに従って試験する場合,基本的に断熱状態で操作すること。カラムと還流分割弁は,
永続的に10-6トール(0.13mPa)未満の真空を保てる高反射性真空ジャケットの内側に完全に封じ込めるのが
望ましい。真空ジャケットにはサーモウェルが備わり,その先端は内壁と外壁の中間にあり,還流分割弁
の真下に位置する。真空ジャケットで囲んだカラムは,加熱式断熱装置で囲んでもよい。その装置の例と
しては,ガラス真空ジャケットの外壁温度を,ディバイディングヘッドの内部蒸気温度に等しく保つガラ
ス繊維マントルがある。
65
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.1.3.5
サーモウェルを備えるが,その先端は,充てん物頂部又は最上部のガラスプレート段の上で,還
流分割弁の上か下に隣接するが,還流液と接触しない所に位置する。設置位置が正しいか否かは,参考附
属書Dに従って検査すること。
6.1.3.6
調節可能な還流分割弁を備えるが,それは充てん物頂部又は最上部ガラスプレートの上からカラ
ムのほぼ直径分だけ上方に位置する。還流分割弁は,参考附属書Gに従って測定した場合,カラムの最高
蒸発速度の25〜95%の範囲にわたって,凝縮液を90%以上の正確さでカラムと取り出しラインに分配でき
ること。
6.1.4
凝縮器 凝縮器は,温度−20℃の冷却剤を使用して,原油に含まれるC4〜C5の全蒸気を凝縮でき
る性能を備えたもの。
6.1.5
コールドトラップ 蒸留開始時のように軽質炭化水素が存在するときには,ドライアイスとアルコ
ールの混合物で冷却した,適切な性能の効率のよいトラップを2個,凝縮管の通気ラインに直列に連結す
る。減圧蒸留の場合には,氷で冷却した同様のトラップを使用して,頭部の蒸気から真空計を保護する。
6.1.6
気体採取器 不凝縮気体を測定するときは,ガスメータをコールドトラップの出口に連結するが,
それらの間に塩化カルシウム乾燥管を取り付けてトラップにたまった水分からの湿気を防ぐ。気体試料を
分析する必要のあるときは,ガスメータの位置か又はそのすぐ後に,適当な大きさの空のプラスチック製
バルーンを取り付けて気体を集める。捕集された気体の容量は,試料を既知容量の減圧容器に入れて膨張
させた後,圧力の上昇から算出することができる。分析用に少量の試料を採取する。
6.1.7
フラクション採取器 装置のこの部分は,大気圧下又は減圧下で受器から留出油を連続的に集める
機能をもつ。また,カラムの状態を乱さずに減圧装置から留出油を取り出すこともできる。
6.1.8
留出油受器 蒸留する原油の量及びその後の分析目的に適した容量のもの。100〜500mL以上の容
量のものが望ましいが,留出油受器総容量の1%のけたまで読み取れるような目盛が付いたものでなけれ
ばならない。
6.1.9
検出器及び記録装置
6.1.9.1
蒸気温度は,頭部にある検出器で0.5℃の単位まで測定できること。この検出器は,参考附属書E
に記すように,応答時間が3分以内であること。温度は手動又は自動のいずれかで記録する。検出器は,
参考附属書Fによって校正する。
6.1.9.2
蒸発速度は,通常留出油採取量から算出するか,又はカラムの圧力ドロップの値から間接的に推
定値を計算する。後者の場合,操作中の圧力ドロップを,フラスコと凝縮管頭部の間に連結したマノメー
タか圧力トランスデューサによって測定する。マノメータとフラスコの間に窒素をごく少量流すと,連結
管での凝縮を防げる。
参考 大気圧下での操作中,U字管マノメータを使用するときは,フラスコに連結していない方の枝
管を大気へ開放する。
6.2
減圧蒸留
減圧下での蒸留に用いる装置では,6.1で述べた装置のほかに,真空ポンプ,1個以上の真空計及び圧力
調節器が必要となる。
6.2.1
真空ポンプ
減圧系はいかなる圧力レベルにおいても,円滑な圧力操作を維持できるもの。また,減圧下で受器を空
にする間,系の乱れを避けるために,受器の圧力を大気圧から0.266kPa {2mmHg}まで30秒以内に低下さ
せる能力をもつもの。代わりに,別に独立したポンプをこの目的のために使用してもよい。
6.2.2
真空計
66
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
6.2.2.1
大気圧以下の圧力測定器は,少なくとも次の精度である。
蒸留圧力
精度
kPa
mmHg
kPa
mmHg
100〜13.3
{760〜100}
±0.13
{±1.0}
13.3〜1.33
{ 99〜 10}
±0.013
{±0.1}
1.33〜0.266
{ 9〜 2}
±0.006
{±0.06}
6.2.2.2
絶対圧力が13.3kPa {100mmHg} 以上のときは,水銀気圧計又は真空マノメータを清浄に,かつ,
適切な減圧度に保っていれば,それらの精度で満足できる。これ以下のときは,マクラウド真空計が唯一
の基本的な計器であり推奨される(参考附属書F参照)。マクラウド真空計は,使用していないときは大
気から隔離して保存するのが望ましい。他の種類の電気式又は機械式真空計は種々の誤差を生じ,したが
って,マクラウド真空計と平行して使用するか,又はマクラウド真空計で定期的に検定する場合にだけ使
用できる。
6.2.2.3
マクラウド真空計の代わりとして,標準温度記録器と共にテンシメータを使用して圧力をアナロ
グで記録することもできる。汚染が疑われるときには,充てん物を取り換えて精度を保つ。
6.2.2.4
真空計と系との接合点は,実用的に可能な範囲で凝縮管頭部に近いこと。接合管の直径は,測定
可能な圧力降下がラインで絶対に起こらないような太さであること。いかなる場合にも,真空計は真空ポ
ンプの近くに接続しないこと。
参考 どのような場合でも,すべての計器を凝縮可能な蒸気,特に水蒸気から注意深く保護するため,
ドライアイスの温度に保たれた冷却トラップを使うこと。
6.2.3
圧力調節器
調節器は,すべての操作圧力において,系の圧力を一定に維持できること。必要に応じてポンプライン
に窒素を流す装置によって,自動調節が可能である。適切な操作を行うためには,真空源と,少なくとも
10リットル容量のサージタンクの間に電磁弁を設置するとよい。電磁弁は圧力検知器によって作動する。
この代わりに,熟練したオペレータが,手動式ブリードバルブを細心の注意を払って操作することによっ
ても維持できる。
7. 装置の準備
蒸留開始前に,蒸留カラム及び全附属ガラス器具を洗浄し乾燥する。系は減圧下での操作に対し漏れが
ないことを確かめる。時計又は他の時間測定器を準備する。すべての加熱器,調節器及び計器が,作動可
能な状態であることを確かめる。
8. 試験の手順
試験室間における試験の場合には,作業開始前に,カット温度,操作圧力及び還流比の詳細について互
いに同意を得ること。
8.1
試料の張込み
試料の密度を,JIS K 2249によって測定する。
8.1.2
突沸を防ぐために,ガラス又は磁器の数片をフラスコに入れる。代わりに,かくはん装置を使用し
てもよい。
67
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.1.3
必要とする張込み容量±5%に相当する原油の質量を算出する。この量の油を1gのけたまではかり
フラスコに入れる。フラスコと試料の両方を0℃以下にはならないように冷やす。試料がワックス状であ
るか粘性が高過ぎるときは,温度をその流動点より5℃高くまで上げてよい。
8.1.4
フラスコをカラムに取り付け,圧力ドロップマノメータ,又は圧力トランスデューサか,もし使用
するなら直接蒸留速度測定系を接続する。加熱系,もし使用するならスターラ,及び支持具を設置する。
注意 原油から毒性の硫化酸素ガスがよく発生するので,予防措置として冷却トラップを通過させて
硫化水素ガスを吸収するか,又は硫化水素ガスを安全な場所に排出すること。
8.2
脱ブタン操作
8.2.1
必要な装置については,6.1.5及び6.1.6を参照する。
8.2.2
凝縮管,留出油冷却器,及び受器が設置されていれば,その中に−20℃以下の温度で冷媒を循環さ
せ始める。
8.2.3
常圧蒸留の間少なくとも2回,大気圧を記録する。
8.2.4
加熱開始後,15〜30分以内に蒸気がカラムの頂部に達するように,フラスコを加熱する。蒸気が
カラムの頂部に達したとき又はその前に加熱を弱め,得られる圧力ドロップは,充てんカラムで
1.0mmHg/m以下,又はオルダーショウ型の実段カラムで0.5mmHg以下であるようにする。自動装置は,
経験に基づいて前もってプログラムする。スターラを使用している場合には,作動させる。
8.2.5
蒸気温度が平衡に達するまで,又は凝縮液の最初の1滴が頭部に現れてから15分間,カラムを全
還流の状態で操作する。
8.2.6
蒸気温度を記録する。
8.2.7
冷媒の循環を止め,蒸気温度を観察する。蒸気温度が15℃に達したら,冷媒の循環を再び始める。
8.2.8
蒸気温度が15℃より低くなった場合は,少なくとも15分間還流を続ける。8.2.7を繰り返す。蒸気
温度が15℃のままか,又はそれ以上に上昇したら,次の操作を続ける。
8.2.9
軽質液体炭化水素を含んだドライアイストラップを取り外し,注意深くふいて乾いた状態にしてか
らひょう量する。
8.2.10 1番目のドライアイストラップの内容物を,最低200mmHgに減圧した10〜50mLの圧力容器に採
取して試料とする。全容器をドライアイスの温度に保ち,揮発物の損失を防ぐ。凝縮管の隣の1番目のト
ラップに全試料が含まれていなければならない。2番目のトラップに凝縮液が含まれるときには,両トラ
ップから試料を採取するか,又は試料採取の前に内容物を混合する。
8.2.11 トラップ試料及び使用した場合には気体バルーンを,ガスクロマトグラフィー又はマススペクトロ
メトリーによる分析に供し,無機ガスを含まない形で報告する。
8.3
常圧蒸留
8.3.1
留出油冷却器,受器及び凝縮管のライン中の温度を−20℃以下に保つ。
8.3.2
加熱を強めて,蒸発速度をその圧力におけるカラムの最高蒸発速度の75%に等しくする。参考図2
及び参考図3はプロパック (Propak) 用の指針として使用できる。他のものに対する速度は,参考表1の
蒸発速度にカラムの横断面積を乗じ,還流比+1で除して概算する。大気圧以下の蒸発速度に対しては,
参考図3で示すこう配を使用する。カラム断熱用加熱器を使用する場合にはそれをオンにし,カラムジャ
ケット温度を蒸気温度より0〜5℃低く保つ。自動式⊿T調節器を使用するほうが,手動式調節より好まし
い。フラクション採取器を大気へ開放する。
8.3.3
還流比5:1で留出を開始する。還流1回にかかる合計時間は30秒以下12秒以上とする。
68
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.3.4
留出油を適切な量のフラクションとして分けて,連続的に取り出す。フラクションの量は,蒸気温
度の5℃又は10℃に対応する量か,試料の張込み量の2〜5容量%に相当する一定の容量とするとよい。65℃
より低い温度で沸騰するフラクションを,0℃以下に冷却した受器に集める。蒸発速度を,参考表1に示し
たカラムの最高蒸発速度の約75%に保つ。⊿Pを測定する場合には,指針として参考図2を使用できるし,
参考図3には還流比5:1でPropakを使用したカラムに対して,種々の圧力下で予期される実用的な留出速
度が図示されている。直接蒸留速度測定器を備えた自動蒸留装置では,留出速度をその期待値に維持しや
すい。
参考 張込み量が1リットルと少量なときは,5℃カットでは試験に必要とする十分な液量が得られな
いこともある。
8.3.5
各フラクションを取り出した時点及び各カットポイントで,次の測定事項を記録する。
8.3.5.1
時刻,時間及び分
8.3.5.2
凝縮液量(mL),室温で測定。
8.3.5.3
蒸気温度 (℃) ,0.5℃の単位まで。補正してから気圧補正したもの。必要ならば参考図4参照。
8.3.5.4
フラスコ内の油の沸騰温度 (℃) ,1℃のけたまで。
8.3.5.5
カラムの圧力ドロップ(mmHg又はkPa),又は平均留出速度。
8.3.6
蒸気温度が65℃に達したら,凝縮管と関連冷却器中の冷媒を止めて,室温の水に替える。凝縮器
及び受器中にかなりの量の水があるときは(受器に集められた水の量が,参考附属書Bに規定の条件下で
決まるカラムの動的内容物の10%を超える場合には),参考附属書Hに記す概要の手順に従い蒸留を続け
試料を脱水してから,次の操作に進む。
8.3.7 フラッディングが起こりそうになったら加熱を弱め,その間安定した条件に戻るまで留出を続ける。
この間に重要なカットポイントが入れば,蒸留を止めて,フラスコ内の油を冷却し,条件外のカットをも
う一度混ぜ合わせた後,15分以内に全還流で再開し,操作条件を正常にした後蒸留を続ける。しかし,開
始の5℃以内で重要なカットは行わない。
8.3.8
カットを取り続け,210℃を超えない範囲で期待最高蒸気温度に達するか,又はフラスコ内の油に
分解が起こりそうになるまで続けるが,いかなる場合にもフラスコ内の油の温度が310℃を超えてはなら
ない。
参考 著しい分解が生じていることは,フラスコが曇った後に頭部が曇ることから分かる。分解は避
けなければならない。
8.3.9
還流弁を閉じ,加熱系の電源を切る。内容物を冷却し,13.3kPa{100mmHg}における蒸留がフラッ
ディングを起こさないで開始できるような温度にまで下げる。この温度は,常圧操作中のカラムの液体と
蒸気温度間の⊿Tを減圧下の期待初期蒸気温度に加えるか,又は⊿Tを,最後に記録したフラスコ内の油
の温度から差し引いて概算できる。
参考 加熱マントルを取り除いた後に,フラスコに圧縮空気を吹きつけてフラスコ内の液体を速く冷
却することを勧めるが,冷たい空気を強く吹きつけるのは避ける。
8.3.10 全フラクションを1gのけたまでひょう量し,その密度を測定する。
8.3.11 最初の留出フラクションについてガスクロマトグラフィー分析を行う (JIS K 2536)。
8.4
13.3kPa {100mmHg} における蒸留
69
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.4.1
より高温でのカットが更に必要なときには,フラスコ内の油が分解しない最高温度まで,減圧下で
の蒸留を続けてもよい。これはほとんどの場合約310℃である。顕著な例外は,熱に不安定な硫黄化合物
を含む原油である。どの場合にも,開始後5℃以内はカラムが平衡状態ではないので,重要なカットを行
ってはならない。
8.4.2
真空ポンプと調節系を蒸留装置に,参考図1に示すように連結する。
8.4.3
真空ポンプを始動して,圧力を徐々に13.3kPa{100mmHg}の値まで下げ,圧力調節器をこの値に設
定する。
参考 フラスコ内の油の温度が,13.3kPa {100mmHg} で沸騰する温度より低いことを確かめる。フラ
スコ内の油がこの圧力に達する前に沸騰し始めるときは,蒸留装置を止め,予定圧力で沸騰し
ないようになるまで,更に冷却する。
8.4.4
フラスコの加熱を開始し,加熱を調節して蒸発速度が最高速度の約75%である状態を維持する。こ
の目的のために必要な圧力ドロップについて,Propakを使用した精留塔の場合を参考図2に示す。カラム
加熱器を使用する場合には,カラム断熱材温度を全操作を通じて蒸気温度より0〜5℃低く保つ。
8.4.5
全還流の安定した条件を少なくとも15分間維持した後に,還流比5:1で留出を開始する。参考図3
には,Propakを使用した内径18〜70mmのカラムの場合の,還流比5:1で予期される留出速度を示す。8.3.4
に示す適切な量のカットを取り出す。蒸留速度直接測定器を備えた自動蒸留装置では,留出速度をその期
待値に容易に維持できる。
8.4.6
各フラクションを取り出した時点及び各カット点で,次の測定事項を記録する。
8.4.6.1
時刻,時間及び分
8.4.6.2
凝縮液量 (mL) ,室温で測定。
8.4.6.3
蒸気温度 (℃) ,0.5℃の単位まで。必要なら補正したもの。
8.4.6.4
フラスコ内の油の沸騰温度 (℃),1℃のけたまで。
8.4.6.5
カラムの圧力ドロップ(mmHg又はkPa),又は平均留出速度。
8.4.6.6
頭部の操作圧力(mmHg又はkPa),必要なら補正したもの。
8.4.6.7
参考図5に示すチャート又は参考表2を用いた大気圧換算温度 (AET)。AETが参考表2から得ら
れないときには,計算式(参考附属書I)から算出する。
8.4.7
カットを取り続け,210℃を超えない範囲で期待最高蒸気温度に達するか,又はフラスコ内の油に
分解が起こりそうになるまで続けるが,いかなる場合にもフラスコ内の油の温度が310℃を超えてはなら
ない。
参考 顕著に分解が起こると,フラスコが曇り,後にヘッドが曇るだけでなく,気体が発生し圧力が
上がって真空ポンプに影響する。このことは,自動調節系の場合早くには気づかれないかもし
れない。
8.4.8
還流弁を閉じ,加熱系の電源を切る。内容物を冷却し,低圧力における蒸留が沸騰せずに開始でき
るような温度まで下げる。この温度は,13.3kPa {100mmHg} 蒸留中のカラムの液体と蒸気温度間の⊿Tを
低圧力下の期待初期蒸気温度に加えるか,又は⊿Tを,最後に記録したフラスコ内の油の温度から差し引
いて算出できる。
8.4.9
全フラクションを1gのけたまでひょう量し,その密度を測定する。
8.5
より低い圧力での蒸留
70
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.5.1
最終カット温度に達しない場合には,前と同じ制約事項を条件として(8.4.7を参照)より低い圧
力で蒸留を継続してもよいが,しかし13.3kPa {100 mmHg} より低い圧力で採用できる圧力水準は1水準
だけである。最高カット温度が350℃を超える場合には,0.266kPa {2mmHg} を奨励する。
試験室間における試験の場合は,試験開始前に第二操作圧力及び還流比について特に合意が得られてい
なければならない。
8.5.2
圧力を希望水準に調節し,圧力調節器をこの値に設定する。
参考 フラスコ内の液体温度が,より低い圧力で沸騰する温度より低いことを確かめる。液体がこの
圧力に達する前に沸騰し始めるときは,蒸留装置を止め,予期圧力で沸騰しないようになるま
で,更に冷却する。
8.5.3
凝縮管及び液体冷却器中の冷却水を室温で循環させるか,又は凝縮管若しくは取出しライン中でワ
ックスが結晶しない温度まで暖めて循環させる。その代わりに,冷却コイルに水を満たし循環させずに開
口しておくか,又は冷媒として水の代わりに空気流を循環してもよい。
8.5.4
頭部の圧力を0.266kPa {2mmHg} 以上の適圧にして,8.4.3〜8.4.7のように減圧操作を続ける。こ
の操作の間,蒸留速度を速めるために2: 1の還流比を使用してもよいが,この値は試験室間における試験
には推奨されない。参考図6又は参考表3若しくは参考表4を用いて読み取った後,補正した蒸気温度測
定値を大気圧換算温度に変換する。この操作の間に,凝縮液が凝縮管中を正常に滴り落ち,留出油が取出
しラインへと円滑に流れることを確かめる。結晶化が観察されたら,室温水を30〜40℃の温水に取り換え
る。
8.5.5
フラスコ中の油の温度が230℃より低くなったら,真空ポンプを止めて,蒸留器に窒素又は他の不
活性気体を通す。空気を通してはならない。蒸留終了時のように,まだ熱い蒸留装置に空気を通すと,爆
発を誘発するおそれがある。
8.5.6
凝縮管及び附属装置中の冷媒の循環を止める。フラスコを取り外し,別のフラスコ中の少量の溶媒,
例えばトルエンを沸騰させ,カラム,凝縮器及び取出し系を洗浄して,カラムの静的ホールドアップ(ウ
エッテイジ)を回収する。回収した残留物は,少量の窒素を流して約120℃で溶媒を蒸発させる。意見の
相違がない通常の蒸留,又は相互同意があるときは,ウエッテイジを参考図7と同じようなチャートを作
成して推測し,その密度を前のカットの密度線の外挿によって推測してもよい。静的ホールドアップは別
の小カットとして取り扱うか,又は性状測定前にボトムと混和してもよい。
8.5.7
フラスコ中の残留油をひょう量し,2.に示されている方法のうち,適切な一方法によって密度を測
定した後,15℃に変換する。
9. 計算
9.1
各留出フラクション及び残留油の収率を,張込み量の質量パーセントとして0.1%のけたまで次のよ
うに算出する。
(m/M)×100
ここに,
m= フラクション又は残留油の質量 (g)
M= 原油の張込み質量 (g)
9.1.1
バルーンに集めた気体フラクションが0.1%(質量)以上のときは,これが最初のフラクションで
ある。それ未満のときは,気体を無視する。
71
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9.1.2
ドライアイストラップ中の凝縮液が第二フラクション,又は気体がバルーンに集まらないときには
第一フラクションである。空気を無視した成分分析値から算出した15℃における密度を用いて,その容積
を計算する。
9.2
減失量パーセントを,0.1%(質量)のけたまで算出する。
100-Σ (m/M) ×100
上のように算出した減失量は,0.4%を超えてはならない。超える場合には蒸留をやり直す。減失量が,
0.4%未満ならば32をトラップカットに,31を最初のナフサカットに配分する。トラップカットがないときは,
損失を全カットそれぞれに割り振る。
9.3
原油試料の容積 (mL,15℃) を,次のように算出する。
V=(M/D)
ここに,
M= 張込み質量 (g)
D= 張込み油の15℃における密度
V= 張込み容量 (mL)
9.4
各フラクション及び残留油の15℃における容積 (mL) を,次のように算出する。
v=(m/d)
ここに,
d= フラクション及び残留油の 15℃ における密度
m=
フラクション及び残留油の減失量に対する補正済み質量(g)
9.5
各留出フラクションの収率(容量%)を,0.1%(容量)のけたまで次のように算出する。
(v/V)×100
ここに,
v= 質量を密度 (15℃) で割って得られた,フラクションの容量
9.6
容量パーセントの増加又は減少を,0.1%の精度で次のように算出する。
100−Σ(v/V)×100
通常,上の式は体積の膨張によって負の値を示す。見掛けの膨張及び収縮は,150℃未満で沸騰するフラ
クションの間で,その収率に比例して配分する。
参考 収率を算定する前述の原則から考えると,容積比は十分精密ではないので,各フラクションや
残留油の密度を算出するために使用することはできない。
10. 試験報告書
10.1 蒸留要約報告書には,次の項目を含める。
10.1.1 張り込んだ試料の質量 (g)
10.1.2 試料の密度 (g/mL,15℃)
10.1.3 張り込んだ試料の容量 (mL,15℃)
10.1.4 質量及び容量の増加又は減少を0.1%のけたまで。
10.1.5 各カットの液体容量パーセント及び質量パーセントを0.1%のけたまで。
10.1.6 累積容量及び質量パーセント。
10.2 ガス,脱ブタン化したナフサ,及びそれに続くカットを,沸点が上昇する順序に列記し,残留油を
最後に記録する。
10.3 10.1の各項目に該当する事項を,蒸留操作中の記録(8.3.5,8.4.6及び9.)から求め,第二報告書と
して,通常は要約報告書に添付する。
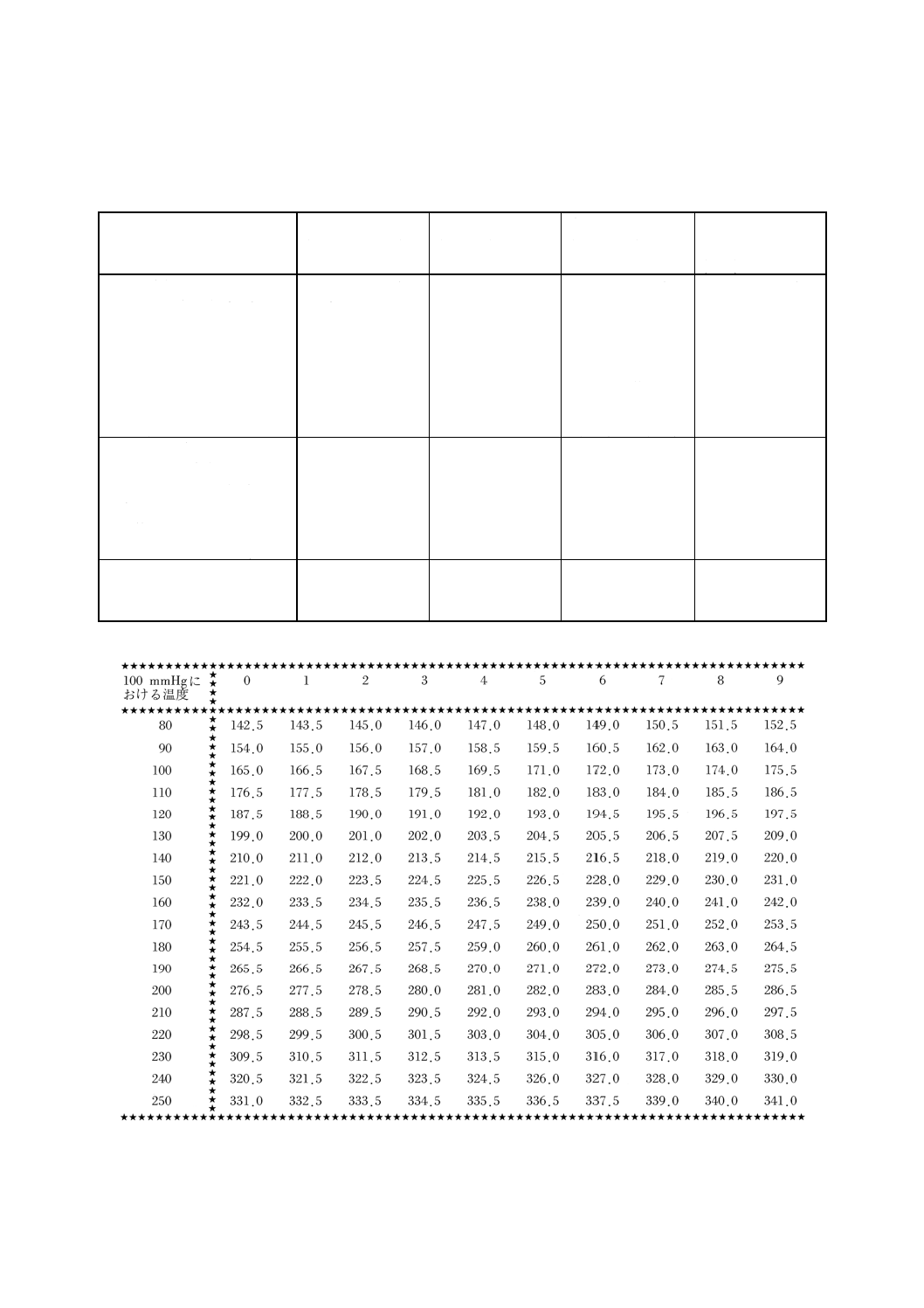
72
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10.4 縦軸にAET温度 (℃),横軸に留出油質量パーセント及び留出油容量パーセントを取って曲線を描く。
これらは,この蒸留における最終的な15/5蒸留曲線である。
参考表1 ヘプタン・メチルシクロヘキサン試験混合物の最高蒸発速度の75%におけるデータ
PROPAK
(1,2,3,4,8)
HELIPAK
(5,6,7)
有孔段
(8,10,11)
GOODLOE WIRE
MESH
(8,9)
カラム直径 (mm)
25
50
25
50
25
50
25
50
充てん物の大きさ (mm)
4
6
n0.2917
n0.2918
蒸発速度 (cm3/h×cm2)
650
670
300
350
640
660
810
1050
動的ホールドアップ (%)
17
15.3
15
14.3
−
8.0
8.0
10.0
(mmHg/理論段)
3.2
16
1.6
8.7
−
12.3
2.0
26
圧力ドロップ (mmHg/m)
9.0
7.9
4
10.6
不明
6.9
5.6
(mL/理論段)
0.34
0.42
0.08
0.34
1.1
0.2
0.35
0.37
HETP (mm/理論段)
38
53
21
32
(60%)
(65%)
48
66
理論段15段搭用:
充てん物の高さ (cm)
57
80
31.5
48
−
−
72
99
充てん物の容積 (mL)
280
1570
155
917
−
−
853
1940
実段
−
−
−
−
25
23
−
−
動的ホールドアップ (mL)
47
240
23
131
−
184
28
194
圧力ドロップ (mmHg)
5.1
6.3
1.3
5.1
16.5
27.6
5.3
5.5
張込み量 (L):
1%ホールドアップ
4.8
24
2.1
13.1
−
18.4
2.8
19.6
4%ホールドアップ
1.2
6.0
0.525
3.3
−
4.6
0.7
4.9
参考表2 100mmHgから大気圧相当への温度変換 (℃)
73
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考表3 1) 10mmHgから大気圧相当への温度変換 (℃)
参考表3 2) 2mmHgから大気圧相当への温度変換 (℃)
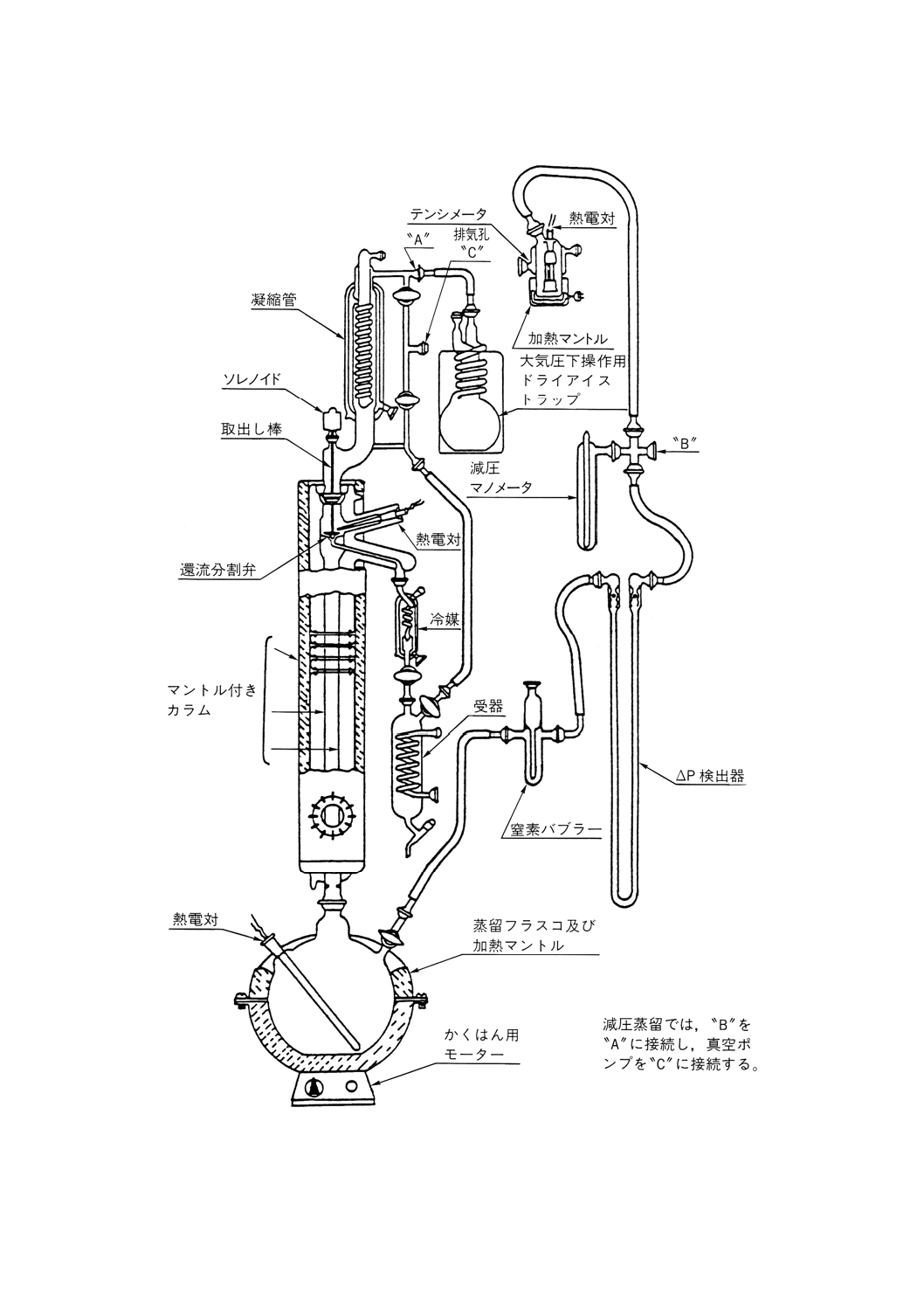
74
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図1-A 原油蒸留試験装置一般図(充てんカラム装置)
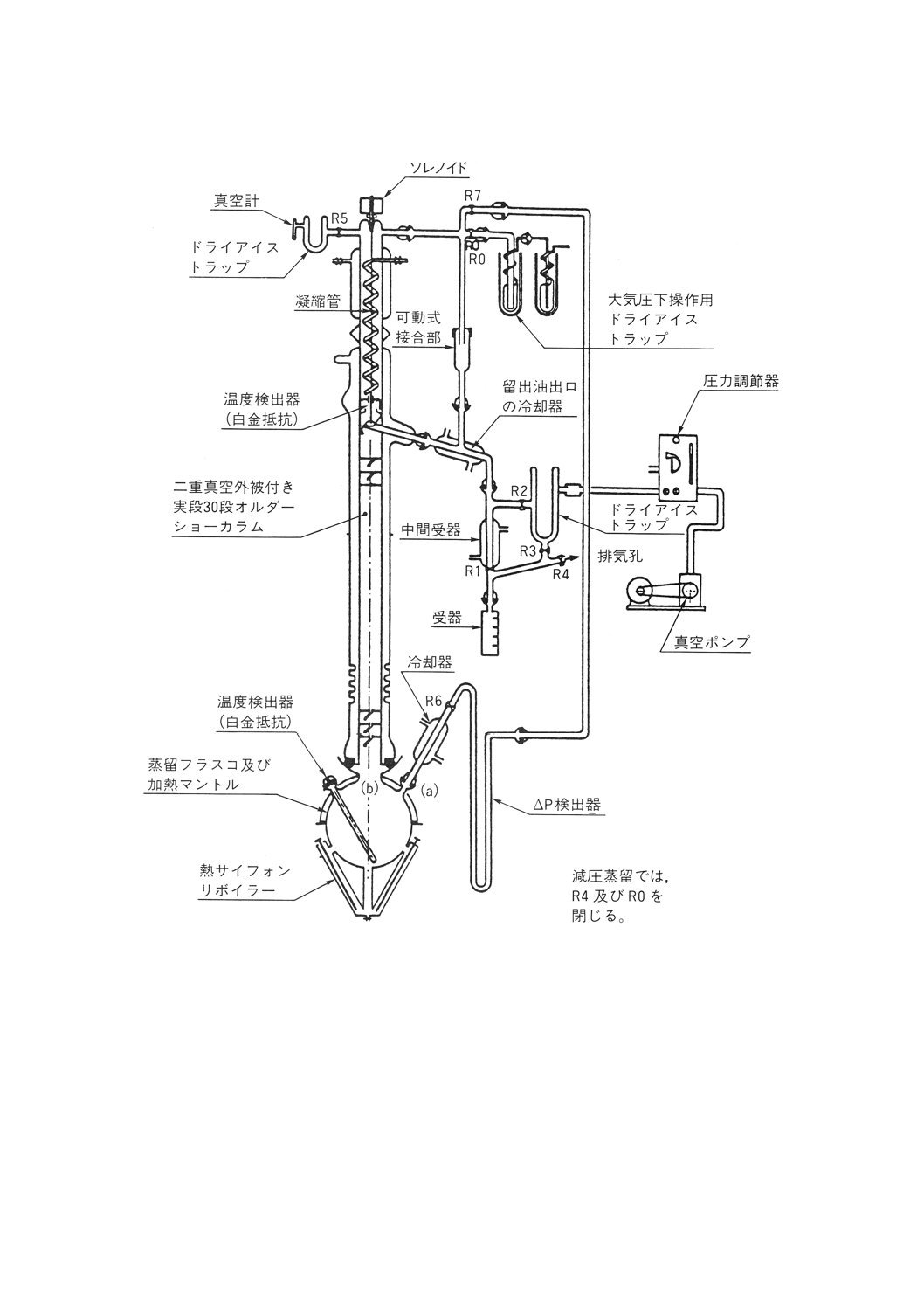
75
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図1-B 原油蒸留試験装置一般図(実段カラム装置)
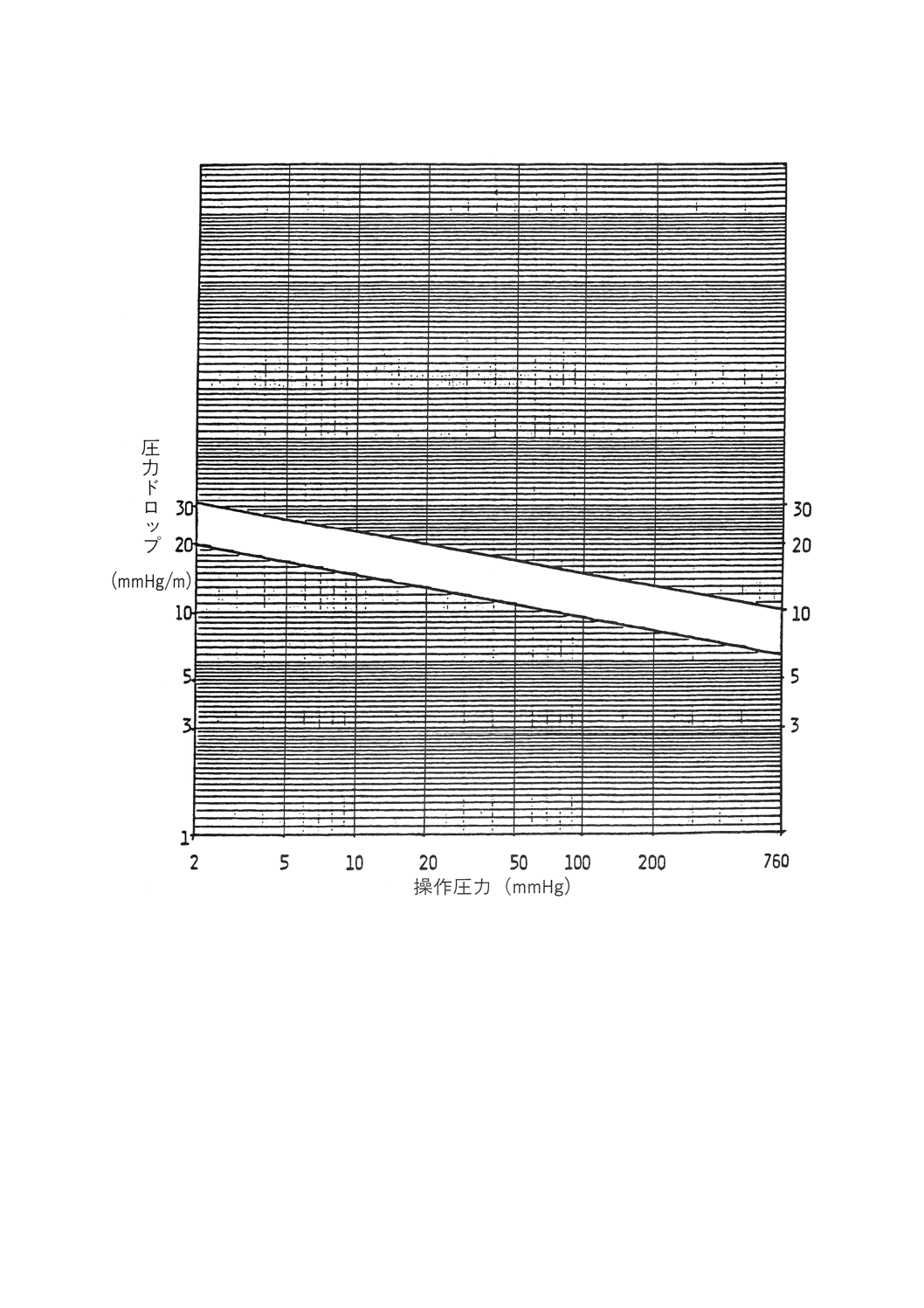
76
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図2 圧力ドロップ近似値(Propak使用の精留塔)
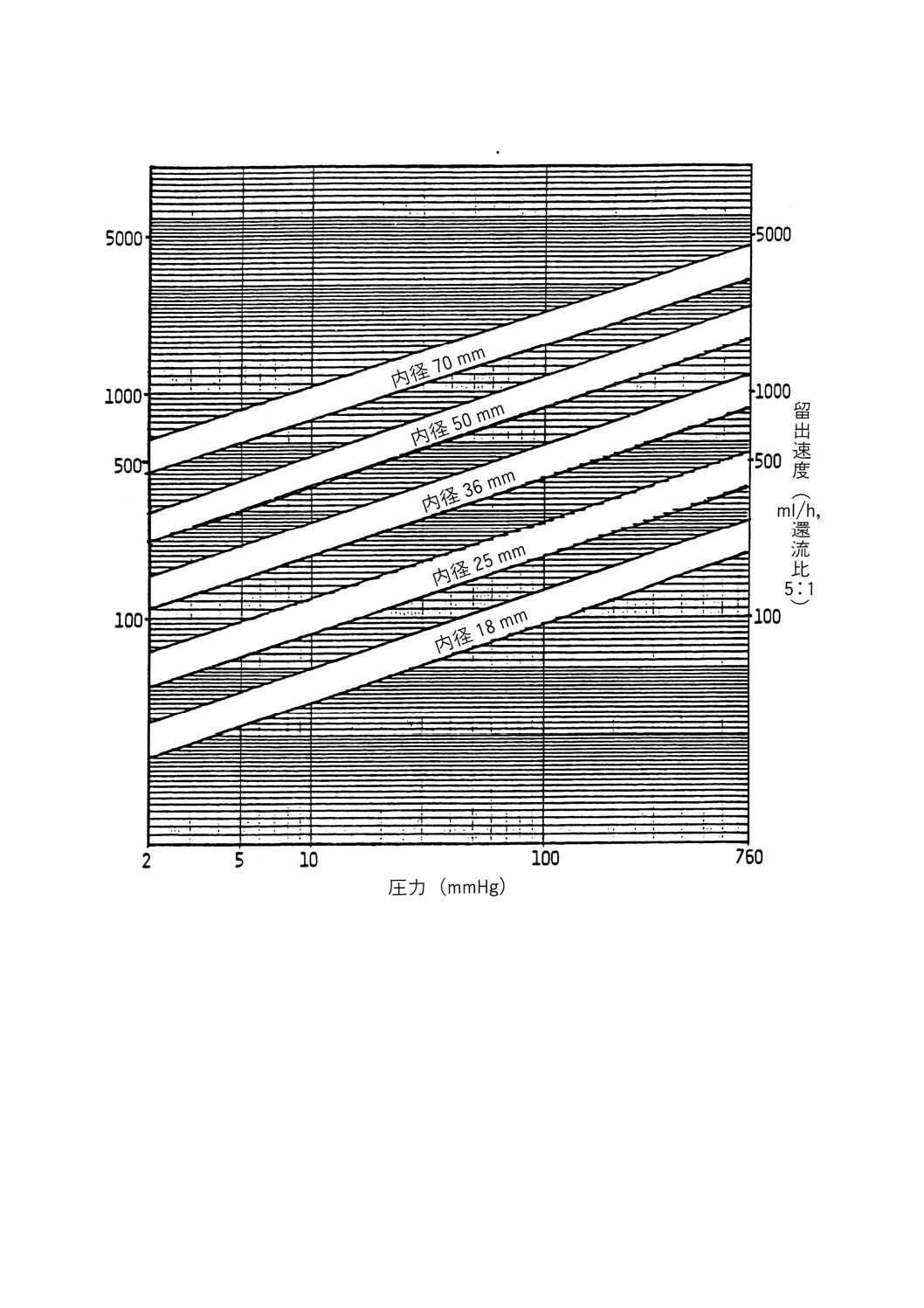
77
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図3 留出速度期待値(還流比5 : 1,Propak使用の精留塔)

78
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図4 温度換算用チャート(圧力範囲730〜790mmHg)

79
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図5 温度換算用チャート(圧力範囲0.01〜100mmHg)

80
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図6 温度換算用チャート(圧力範囲0.01〜10mmHg)
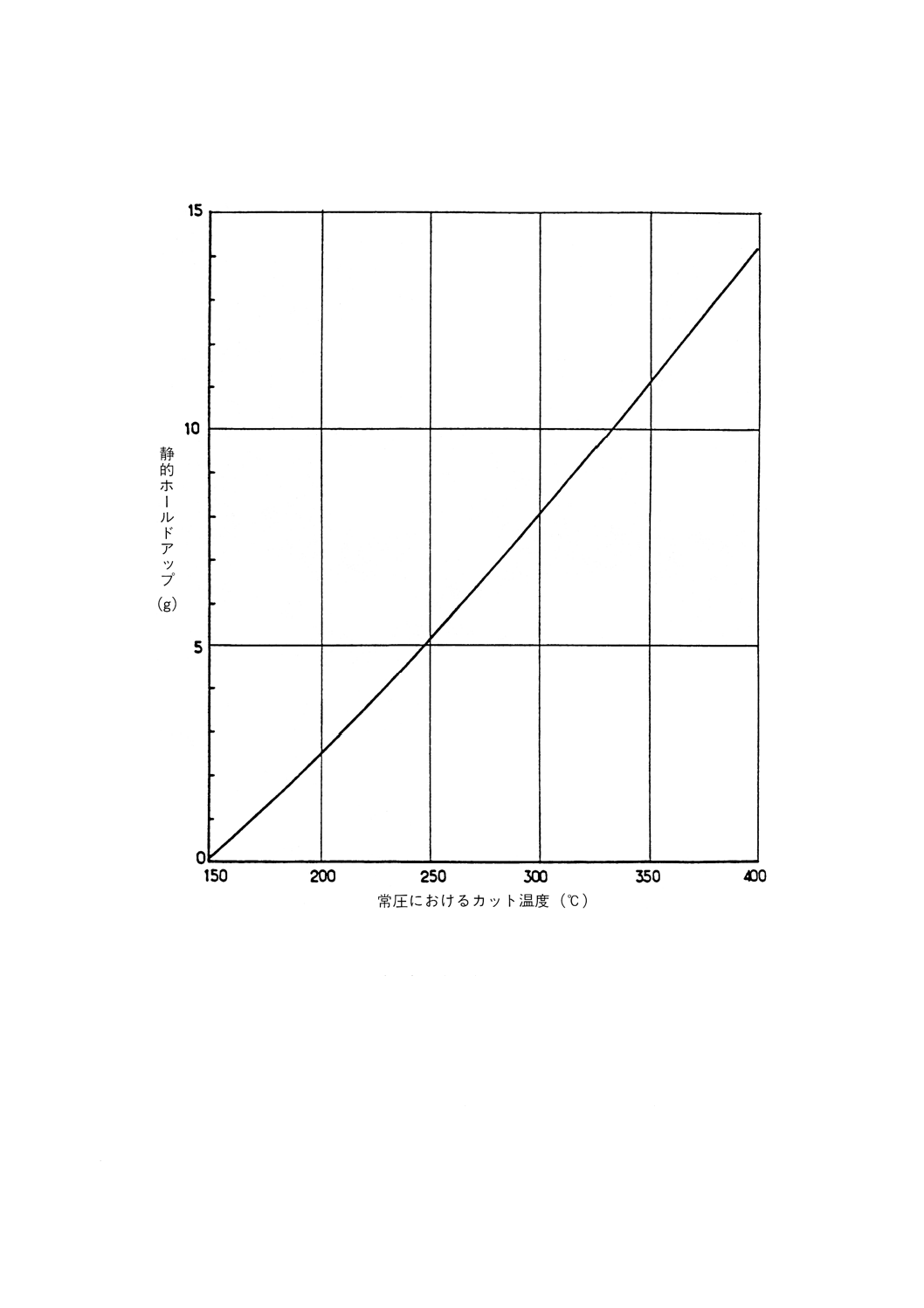
81
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考図7 カット温度に対応する静的ホールドアップ
(静的ホールドアップ近似値,Propak 4×4mmを詰めた内径25mm×560mmのカラムで,平均的原油を蒸留する場
合)
参考文献
1. Cooke,G.M.&Jameson,B.G.,Analytical Chemistry,ANCHA27巻,1955,p.1798
2. Struck,R.T.&Kinney,C.R.,Industrial and Engineering Chemistry, IECHA, 42,1950,p.77
3. Cannon,M.R.,Industrial and Engineering Chemistry,IECHA,41,n°9,1949,p.1953
4. Bulletin 23,Scientific DevelopmentCo.,私書箱795,State College,PA 16801
5. Bulletin,Podbielniak, Reliance Glass Works,私書箱825,Bensenville,Ill,60106
6. Feldman,J.等,Industrial and Engineering Chemistry,IECHA,45,1953年1月,p.214
7. Helipak性能特性,Begeman,C.R.&Turkal,P.J.(Podbielniak社試験室報告書),1950
8. Cooke,G.M.,Ana1ytica1 Chemistry, ANCHA 39巻,1967,P.286
9. Bragg,L.B.,Industrial and Engineering Chemistry,IECHA,49,1947,p.1062
82
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
10. Umholtz,C.L.&Van Winkle,M.,Petroleum Refiner,34巻,1955,p.114,NH: MCH用オルト及びメタ
キシレン,2成分で得られたデータから算出した圧力ドロップ。
11. Oldershaw,C.F.,Industrial and Engineering Chemistry,IECHA,13巻,1941,p.265
83
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書A 蒸留カラムの効率測定方法
A1.1 適用範囲 この参考附属書は,ヘプタン・メチルシクロヘキサン混合液を使用して大気圧で,全還
流条件下の蒸留カラムの効率を測定する方法について規定する。次に規定する大気圧での効率が,このパ
ラメータの唯一の基準として認められる。
A1.2 装置 適切な装置の例を参考附属書A図A1.1に示す。装置の部品は基本的に以下のとおりである。
A1.2.1 校正用フラスコ 適切な大きさで加熱器付き。校正フラスコの一例を参考附属書A図A1.2に示す。
A1.2.2 蒸留カラム及び凝縮管
A1.2.3 マノメータ 又はカラムの圧力ドロップを測定できる同等品。
A1.3 試薬 試験混合液はヘプタンとメチルシクロヘキサンの容量比50: 50の混合液であり,屈折率は次
のとおりである。
ヘプタン
:
64
1.387
20
D=
n
メチルシクロヘキサン
:
12
1.423
20
D=
n
A1.3.1 試験混合液の成分のクロマトグラフィー分析の結果は,軽質不純物が0.01%未満であり,純度は
99.75%を超えること。260及び270nmにおける紫外光で吸収がないこと。すなわち,芳香族を含まないこ
と。
A1.3.2 成分が上の規格に合わないときは,再蒸留若しくはろ(濾)過,又はその両者で精製する。再蒸留
には総効率が理論段30段を超える蒸留カラムを使う。ろ過は200メッシュのシリカゲルに通し溶出液体の
最初の10%を捨てる。ゲルの量は,不純物量に対して十分過剰であることを確かめる。
A1.4 装置の準備
A1.4.1 蒸留カラム及び全ガラス器具は,試験開始前に清浄にして乾燥させること。
清浄にするには,温クロム酸溶液(取扱いに注意)又は強力な工業用洗剤で洗う。十分に水洗し,乾燥
して組み立てる。
A1.4.2 続いて,少量の純ヘプタンを用いて高蒸発速度で少なくとも5分間蒸留して,装置を洗浄する。カ
ラム頂部のサンプリング系から,間隔をおいて一定量を数回取り出す。そこで蒸留フラスコの加熱を止め
て,フラスコを取り除き,塔を熱いうちに風乾する。装置を完全に乾燥状態に保つには,カラム頂部の凝
縮管の出口に水分トラップを接続することが望ましい。
A1.5 試験の手順
A1.5.1 蒸留フラスコに試験混合液を入れるが,その量はカラムの許容最少量(参考表1参照)に等しく,
フラスコ容量の32以下とする。次に,均一に沸騰するように,ガラス又は磁器の数片を入れる。
A1.5.2 参考附属書A図A1.1に示すように,フラスコをカラム及び圧力ドロップマノメータに接続する。
A1.5.3 凝縮管に室温の水を循環させる。
A1.5.4 フラスコを加熱し試験混合液を沸騰させ,加熱をフラッド点に達するまで強める。これは,充てん
物中若しくは段上に少量の液体が見えるか,又はカラム頂部の凝縮管首部が液体で満たされることによっ
て分かる。フラッディング(あふれ)が起こると圧力ドロップ差が突然大きくなる。加熱を弱めてフラッ
ディングを静め,次に加熱を徐々に強めてフラッド点の直前までもってくる。フラッド点の直前の蒸発量
及び⊿P測定値を記録する。これを最高蒸発速度とみなす。
A1.5.5 加熱を調節して,カラムを約200mL/h×cm2に相当する安定した蒸発速度で還流させる。
84
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A1.5.6 カラムを全還流で1時間保つ。圧力ドロップを記録し,校正バルブを満たすのに要する時間をはか
って蒸発速度を測定する。
A1.5.7 フラスコの液体とカラム頂部還流液の必要量を素早く(ほとんど同時に)取り出す。それぞれの取
出し量は,20℃における屈折率を測定するのに十分で,0.5mLを超えない量とする。還流液を取り出す時
間は,2秒を超えてはならない。
A1.5.8 カラム頂部還流液及びフラスコの液体の屈折率を測定する。
A1.5.9 屈折率測定によって最高カラム効率で安定していることが分かるまで,15〜30分間隔ごとに参考
附属書A1.5.7及び参考附属書A1.5.8の操作を,繰り返す。
A1.5.10 フラスコの液体の260〜270nmにおける紫外吸収を調べる。吸収が認められた場合は芳香族不純物
が混入しているので,効率測定に誤差が生じる。この場合には,蒸留装置を洗浄し,試験混合液を精製し
てもう一度やり直す。
A1.5.11 参考附属書A1.5.5〜参考附属書A1.5.10の一連の操作を,フラッディングを起こさずに繰り返して,
蒸発速度200mL/h×cm2と最高蒸発速度の間でほぼ等間隔に,4段階以上の蒸発速度で行う。これらの蒸発
速度のうちの一つは,最高蒸発速度の90%以上でなければならない。
A1.6 計算
A1.6.1 採取した各組の試料のモル組成を,試料の屈折率と次のデータから得られるヘプタンのモル組成対
屈折率の曲線とを比較することによって求める。
ヘプタンの
モル分率
20℃における
屈折率
ヘプタンの
モル分率
20℃における
屈折率
0.00
1.4231
0.60
1.4005
0.10
1.4191
0.70
1.3971
0.20
1.4151
0.80
1.3939
0.30
1.4113
0.90
1.3907
0.40
1.4076
1.00
1.3876
0.50
1.4040
A1.6.2 次のフェンスケの式によって,カラムの理論段数を算出する。
α
log
1
log
1
log
O
O
D
D
X
X
X
X
N
−
−
−
=
−1
ここに,
N: 理論段数
α: ヘプタンのメチルシクロヘキサンに対する比揮発度,1.075
とする。
XD: カラム頂部還流液中のヘプタンのモル分率
XO: フラスコ液中のヘプタンのモル分率
A1.7 結果
A1.7.1 充てんカラムについて,蒸発速度 (mL/h) 及び絶対速度 (mL/cm2×h) の関数としてHETP (mm),
⊿P(mmHgとmmHg/m)及び理論段数の曲線を引く。
実段カラムでは,効率を実段当たりの理論段パーセントとしてプロットする。⊿Pは上述した充てんカ
ラムの蒸発速度と同じ単位を用いて,mmHg/実段及び絶対単位(mmHg/理論段)の両方でプロットす
る。
カラムの効率は,その最高蒸発速度の75%における曲線上のN(理論段数)の値とする。
85
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考 HETPとは,Height Equivalent to one Theoretical Plateの略称であって,理論段数1段に相当する
充てん物の高さ(厚み)をいう。
A1.7.2 一般的な充てんカラム及び実段カラムの典型的な曲線を参考附属書A図A1.4〜参考附属書A図
A1.7に示す。これらの精留塔は,フラッディングを起こさない状態で使用しなければならない。
A1.7.3 グラフ上で最高蒸発速度の75%のところに垂直線を引き,この速度に対応するHETP又は段効率を
読み取る。
A1.7.3.1 充てんカラムの場合には,HETP (mm) に14又は18を乗じて,この参考で使用する充てん物の
必要な高さの範囲を得る。
上述のように測定して,充てんカラムの効率が18段より高ければ,適切な量の充てん物を取り除くこと
によって,性能を損なわずに効率を下げることができる。
A1.7.3.2 実段塔ではその代わりに,14及び18を段効率で除して必要なカラムの実段数を得る(整数に丸
める。)。
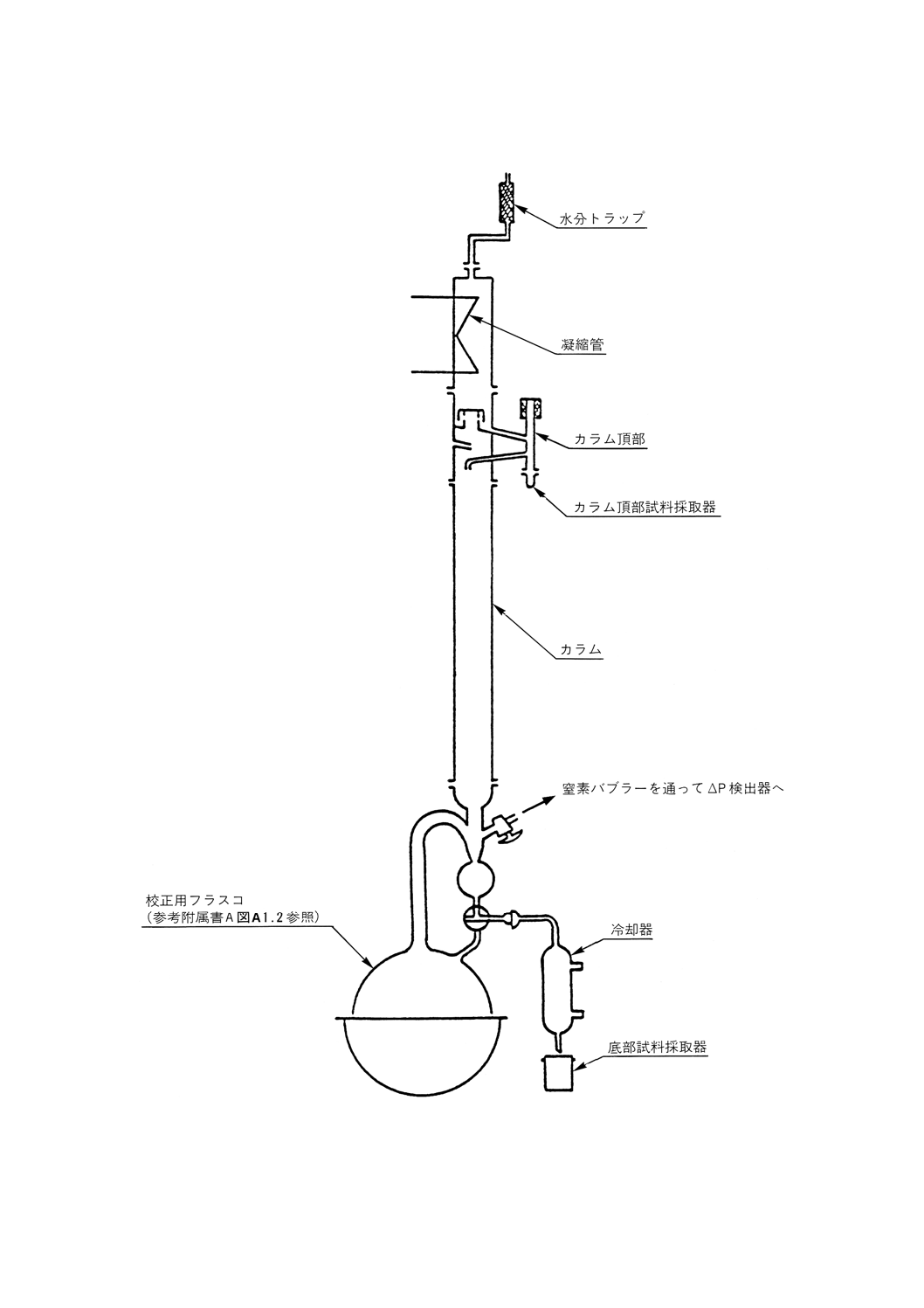
86
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書A図A1.1 効率測定用蒸留装置の一般図
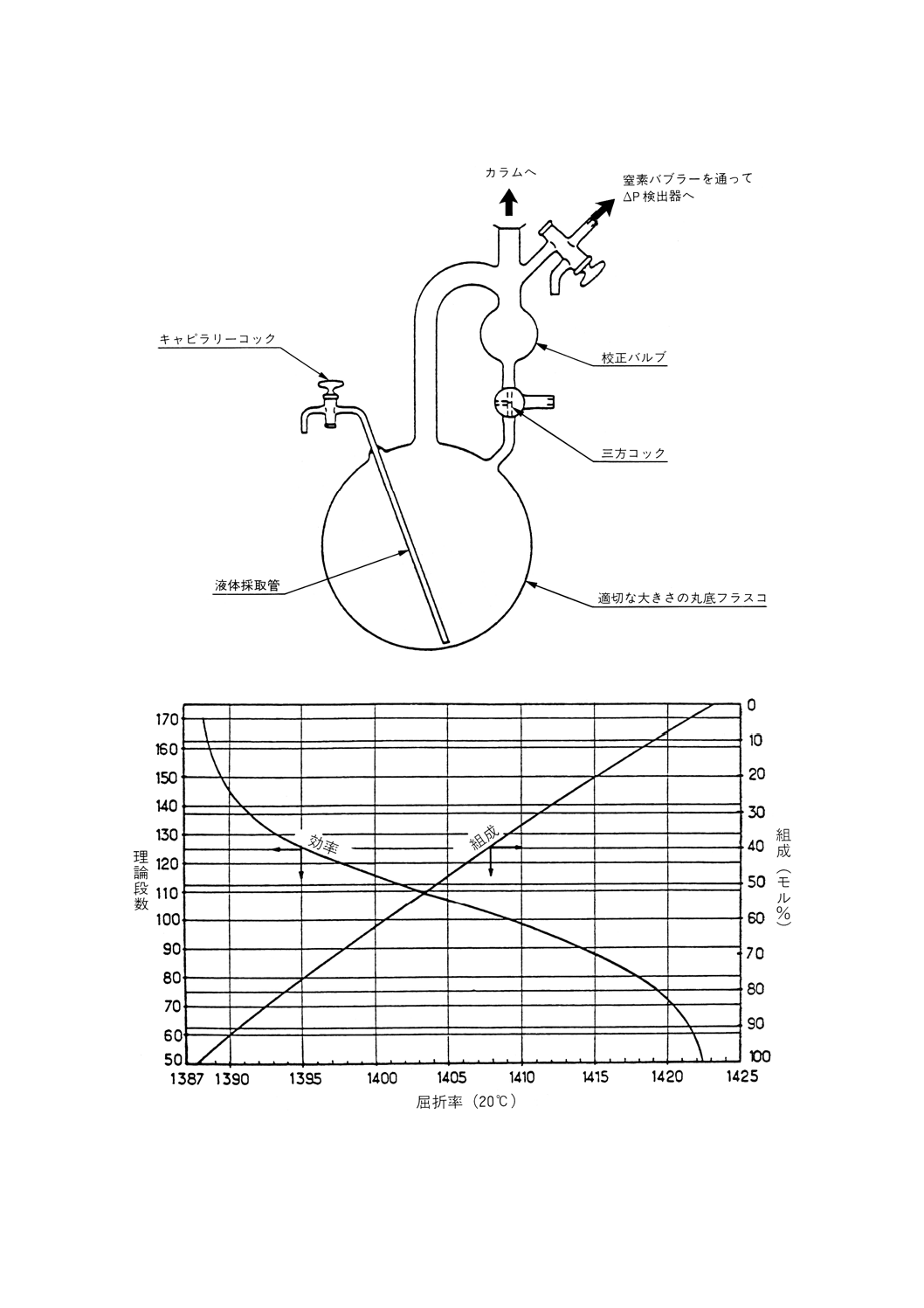
87
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書A図A1.2 蒸留カラムの効率測定用校正フラスコの一般図
参考附属書A図A1.3 n−ヘプタン/メチルシクロヘキサン系の屈折率と理論段数
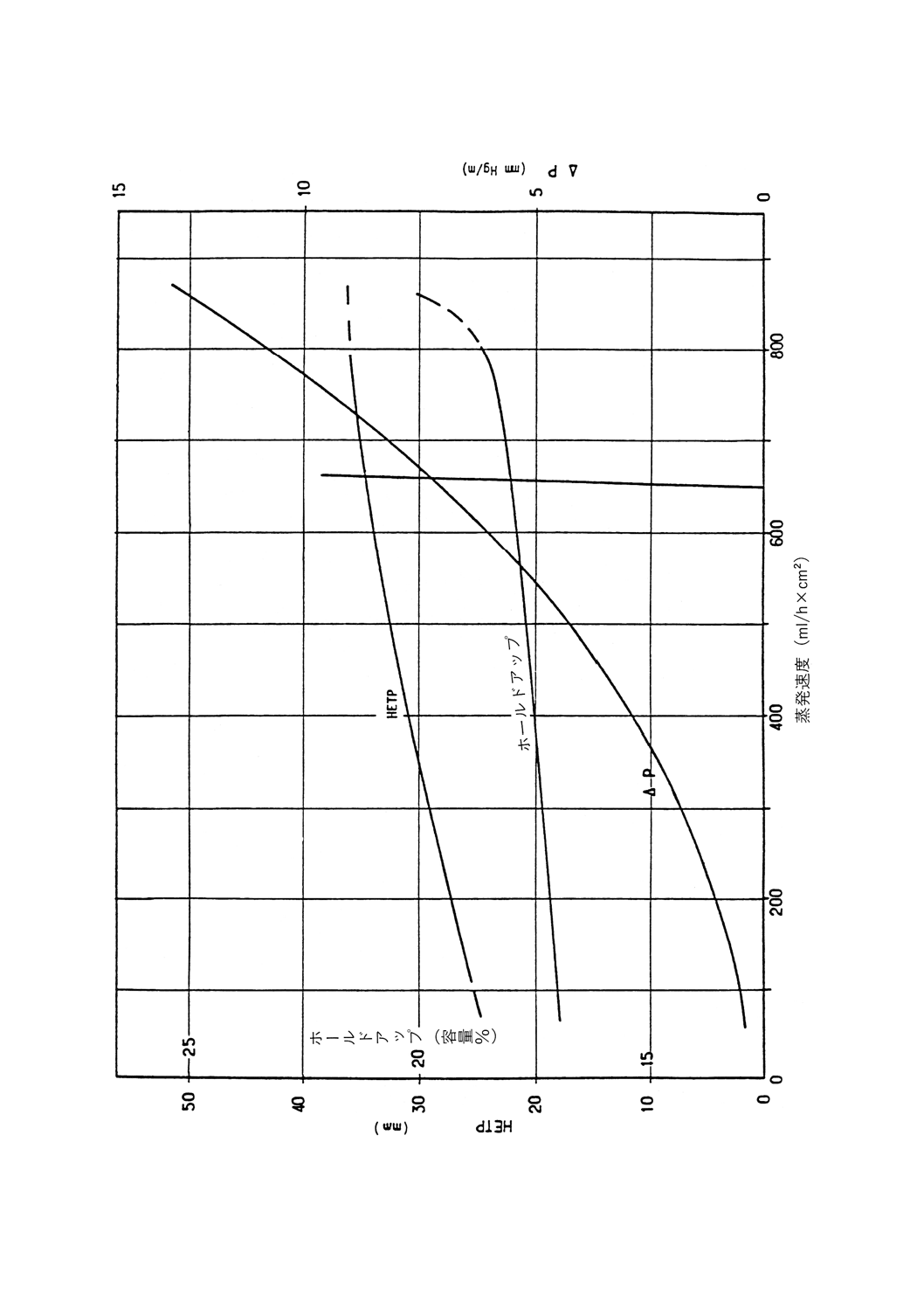
88
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書A図A1.4 Propak (4mm) を充てんした内径25mmカラムの特性
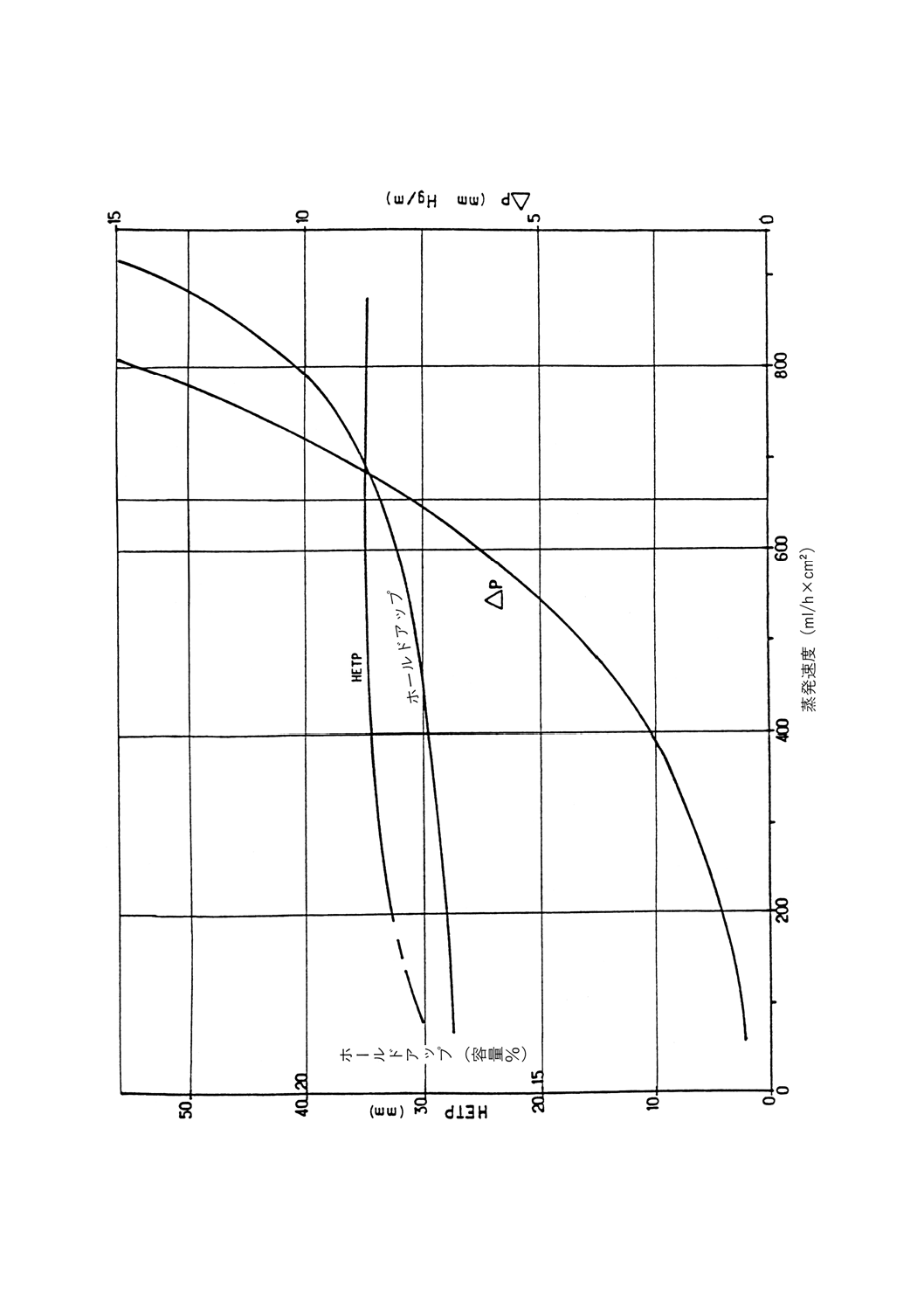
89
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書A図A1.5 Propak (4mm) を充てんした内径50mmカラムの特性
(データ源:参考表1,参考文献1.,4.,8.)
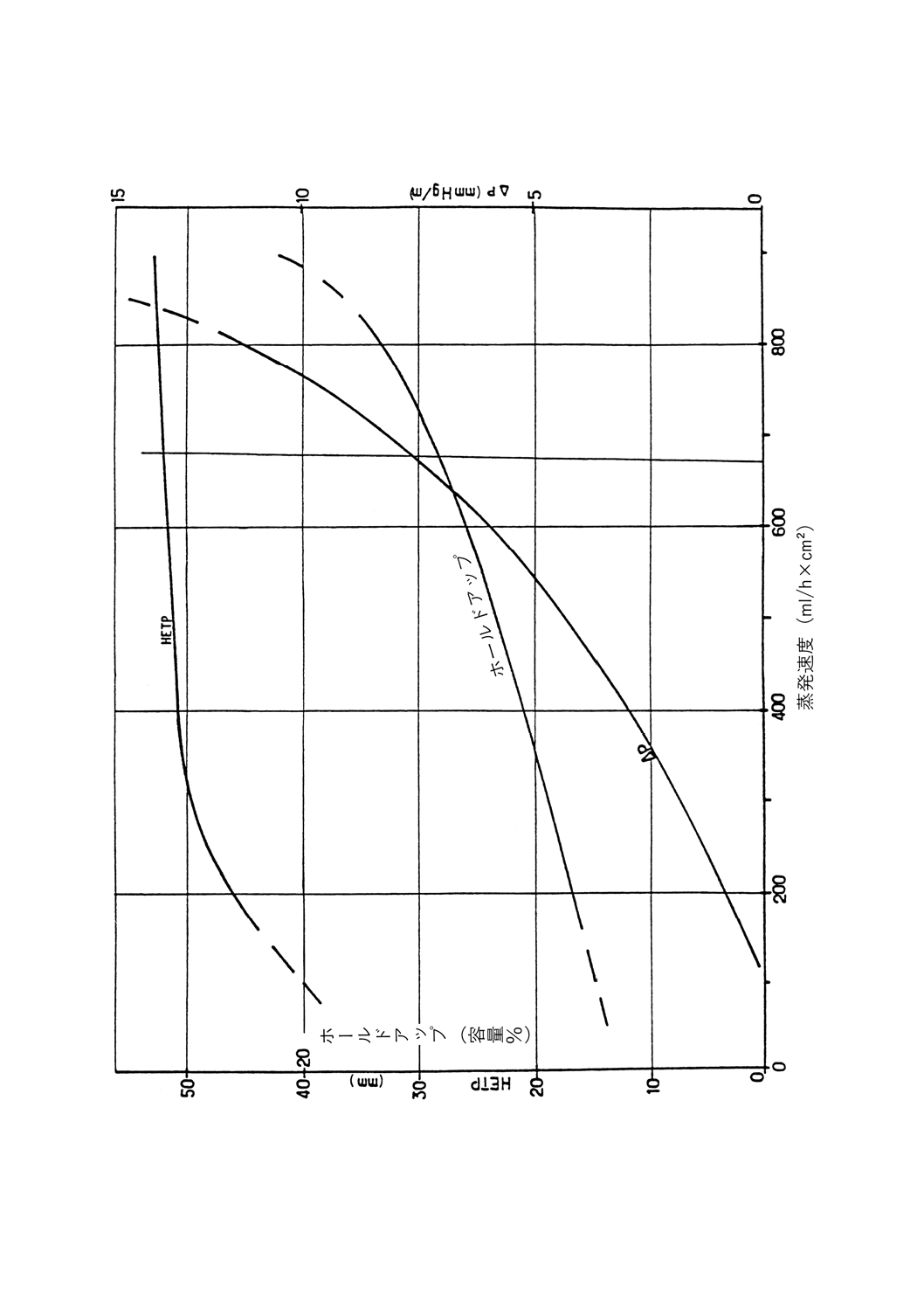
90
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書A図A1.6 Propak (6mm) を充てんした内径50mmカラムの特性
(データ源:参考表1,参考文献1.,4.,8.)
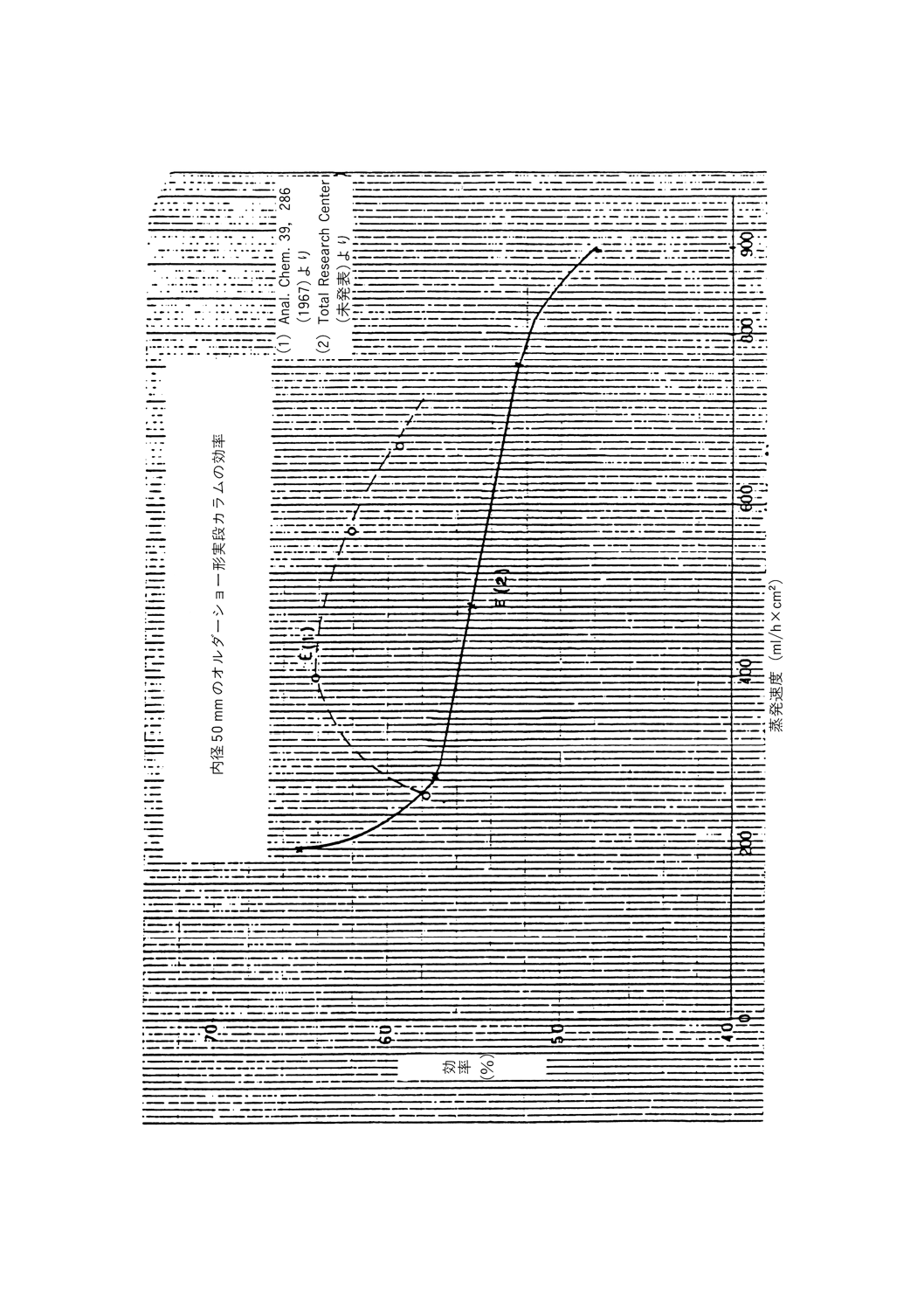
91
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書A図A1.7 内径50mmのオルダーショー形実段カラムの効率
92
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書B カラムの動的ホールドアップの測定方法
B1.1 原理
B1.1.1 不揮発性物質(ステアリン酸又は350ニュートラル原料油のような留出油)を炭化水素(ヘプタン)
に溶解した試験混合物を,参考附属書A図A1.1に示すような装置で全還流の状態で蒸留する。
B1.1.2 初期混合物と還流中の混合物の不揮発性物質濃度の差から,カラムの動的ホールドアップが算出
できる。
B1.2 装置 装置は,参考附属書A図A1.2及び参考附属書A図A1.1に規定する装置と同じものを使用す
る。
B1.3 試験混合物 試験混合物の成分は,20℃の屈折率が1.38764の純ヘプタン,及び純度が95%を超え
融点が68〜70℃のステアリン酸又は留出油,例えば350ニュートラル原料油である。試験混合物は,ステ
アリン酸又は350ニュートラル原料油を約20質量%含むヘプタンから成る。
B1.4 装置の準備(参考附属書A参照)
B1.5 試験の手順
B1.5.1 試験混合物中のステアリン酸又は350ニュートラル原料油の濃度を測定する(例えば,ステアリン
酸は電位差滴定法で,0.1 N NaOH溶液でpH9.0まで滴定するか,350ニュートラル原料油は屈折率測定法
による。)。その結果を不揮発性物質の質量パーセントで記録する(P0)。
B1.5.2 均一に沸騰するように,フラスコにガラス又は磁器の数片を入れるか,適切なスターラを使用する。
B1.5.3 内径25mmのカラムには約1リットル,内径50mmのカラムには約4リットルの試験混合物を1g
のけたまでひょう量しフラスコに入れる。
B1.5.4 フラスコをカラム及び⊿P測定系に連結する。
B1.5.5 凝縮管に室温の水を循環させる。
B1.5.6 フラスコを加熱して試験混合物を沸騰させる。校正バルブを満たすのにかかる時間をはかって,蒸
発速度を約200cm3/h×cm2に調節する。希望速度に達したら,圧力ドロップに注意しながら30分間保つ。
B1.5.7 カラム頂部及びフラスコ中の液体から試料を採取するが,測定に必要な量以上の試料を採らない。
そして直ちに蒸発速度と⊿Pを測定し記録する。
B1.5.8 カラム頂部及びフラスコからの試料を分析し,不揮発性物質の濃度を質量パーセントで求める。
B1.5.9 フラスコ中の不揮発性物質の濃度が一定になるまで,15分間隔でB1.5.7とB1.5.8の操作を繰り返
す。
B1.5.10 約200cm3/h×cm2の単位ごとにフラッド点近くになるまで上記の測定を続け,最大蒸発速度におけ
る一組を含めて,少なくとも四組の測定値を求める。
B1.6 結果
B1.6.1 各測定におけるカラムの動的ホールドアップを,次の式によって算出する。
d
M
P
P
P
H
×
0
−
=
ここに,
H: カラムの動的ホールドアップ(15℃における体積)
M: フラスコ中の試験混合物の質量
P0: 開始時の試験混合物中の不揮発性物質の質量パーセント
P: 蒸留中にフラスコから採取した液体中の不揮発性物質の質
量パーセント
93
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
d: ヘプタンの15℃における密度(0.688kg/L)
B1.6.2 測定を行った各蒸発速度における理論段1段相当の動的ホールドアップを,次の式によって算出す
る。
N
H
h=
ここに, N: その蒸発速度における,全還流状態でのカラム理論段数(参
考附属書A参照)
B1.6.3 蒸発速度の測定値 (L/h) を蒸発速度 (cm3/h×cm2)に換算する。また⊿Pの測定値をmmHg/mに,
ホールドアップの測定値をmL/理論段に換算する。
B1.6.4 縦軸に全データを,横軸に蒸発速度をL/h及びcm3/h×cm2の単位で採りプロットする。ホールド
アップ(mL及びmL/理論段)の点を通る曲線を滑らかに引く。できれば,蒸発速度対⊿P測定値曲線を
効率測定のときに得られる曲線と比較して,それらがほぼ一致することを確かめる。
B1.6.5 最高蒸発速度の75%のところで垂直線を引く。カラムの動的ホールドアップは,ホールドアップ曲
線と最高蒸発速度の75%線との交点から読み取る。張込み量は,ホールドアップ比1〜4%の範囲にあるの
で,この数値の100〜25倍の範囲である。
94
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書C 蒸留カラムの熱損失の測定方法
C1.1 原理
C1.1.1 熱損失は吸収される熱に等しいという原則を,カラムにおける熱損失の試験に使用する。
C1.1.2 カラムの真空ジャケットの外壁を一定の高温に保つ。塔頂部検出器で検出されたカラム内部の温度
は上昇するが,それはカラムが吸収する熱量すなわちカラムによる熱損失量である。
カラムの真空ジャケットの外壁を,室温より100±1℃高く保ち試験を行う。この条件ならば,ガラス器具
に不必要な熱ひずみを与えずに測定を行えるので,適切な温度差が得られる。
C1.1.3 使用前にすべての新ガラス真空ジャケットを試験し,それ以後は少なくとも1年に1回検査する。
C1.2 装置
C1.2.1 できれば,二つのペンを備えたチャート記録計でカラム外壁及び熱検出器の温度を記録することを
推奨する。
C1.2.2 塔壁に熱電対が備わった熱補償マントル中にカラムを封じ込める(6.1.4.6参照)。
C1.3 装置の準備
C1.3.1 熱検出器の位置,応答時間及び校正を,参考附属書D,参考附属書E及び参考附属書Fの規定に
適合するか調べること。
C1.3.2 試験中は,カラム内部の煙突効果を減じるために,凝縮管の頂部を閉じる。
C1.4 試験の手順
C1.4.1 試験の開始時に,次の測定事項を記録する。
C1.4.1.1 時間(時間と分)
C1.4.1.2 室温(℃)
C1.4.2 カラムの真空ジャケットの外壁を加熱し,カラム壁に位置するパッチ温度検出器が室温より100±
1℃高い温度を記録するまで加熱を徐々に強くしながら調節する(30分以内にこの温度に達すること。)。
自動比例制御調節器の使用が望ましい。
C1.4.3 マントル温度が所定の値に達した後,カラムをこの条件下で1時間保つ。
C1.4.4 1時間後に,次の測定項目を記録する。
−
(時間及び分)
−
カラム外壁の温度 (℃)
−
カラム内部の温度 (℃)
C1.5 結果
C1.5.1 カラムが吸収した熱,すなわち失った熱(Q)を次のように算出する。
100
×
A
C
A
B
Q
−
−
=
ここに,
A: 室温(℃)
B: 1時間後のカラム内部温度(℃)
C: カラム外壁温度(℃)
C1.5.2 上記の熱損失が30%未満であれば,そのカラムは原油蒸留に適している。
95
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書D 温度検出器の位置の確認方法
D1.1 目的 次の試験は操作位置にある検出器の性能を調べるために行う。通常は,設計承認のために一
度だけ行い,その後は繰り返す必要はない。
D1.2 原理
D1.2.1 検出器と記録装置を使用して,蒸留装置中の純化合物の蒸気温度を測定し,その化合物の認定され
た沸点と比較する。
D1.2.2 実際に位置の悪い検出器では,蒸気からの熱供給が十分でないために温度誤差を生じる。これは,
蒸気の熱容量が少なくなる,減圧下では特に重要である。
D1.2.3 試験は,大気圧下と減圧 (0.133kPa {1 mmHg} ) 下の両方で行う。
D1.3 試験の手順
D1.3.1 大気圧下での位置の検査
D1.3.1.1 蒸留装置を6.1に規定する大気圧操作用に設定する。室温の冷却水を使用する。通常の張込み量
より少量(0.5〜1リットル)のテトラデカンを使用してもよい。試験用液体は水を含まず,ガスクロマト
グラフィーで軽質不純物含有量が0.1%未満であることが証明されていること。
D1.3.1.2 張込み試料が沸騰するまで加熱し,蒸発量が約400mL/h×cm2で平衡に達するようにする。これ
は充てんカラムに対して約3mmHg/m(実段カラムに対して0.70mmHg/段)の⊿Pに相当する。この定常条
件を少なくとも15分間保つ。蒸気温度の低下が0.2℃を超えるときは,還流比5:1で留出油2%を取り除い
てから,全還流で15分間保つ。蒸気温度が全還流で15分間,0.2℃の範囲内で一定になるまで,上のよう
に2%留出油を取り続ける。
D1.3.1.3 参考附属書Fに従い校正した装置を用いて,沸点を0.5℃のけたまで,大気圧をmmHgのけたま
で測定して記録する。
D1.3.1.4 測定したテトラデカンの沸点が253.5℃±0.5℃の範囲内になければ,検出器の位置が疑わしいの
で,この方法で使用する前に直すこと。
D1.3.2 減圧下での位置の検査
D1.3.2.1 蒸留装置を減圧操作用に設定し,試験用液体をヘキサデカン(沸点286.8℃)に換える。通常の
張込み量より少量(0.5〜1リットル)を使用してもよい。試験用液体は水を含まず,ガスクロマトグラフ
ィーで軽質不純物含有量が0.1%未満であることが証明されていること。温度及び真空測定機器は参考附属
書Fの規定に従って校正する。
D1.3.2.2 塔頂部圧力を定常条件下で1mmHgまで下げる。凝縮管を空にするか冷媒に空気を用いて,凝縮
器中での結晶化を防ぐ。
D1.3.2.3 張込み試料を加熱して,25〜50mL/h×cm2の一定蒸発速度にする。これは,充てんカラムに対し
ては1mmHg/m以下,内径50mmの実段カラムに対しては0.6mmHg/段に相当する。この条件を少なくとも
15分間保つ。
D1.3.2.4 還流比20:1で2%の留出油を取り出し,全還流で5分間沸騰を続ける。参考附属書Fの規定に従
って校正した機器を使って,沸点を0.5℃のけたまで,操作圧力を0.05mmHgのけたまで測定して記録す
る。
96
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
D1.3.2.5 測定した沸点が1mmHgにおいて0.2℃の範囲内に一定していない場合は,還流比5:1で留出油2%
を取り出し,全還流で15分間保つ。全還流条件で一定の温度になるまで,上の操作を続ける。操作の繰返
しは3回以下でよい。
D1.3.2.6 測定したヘキサデカンの蒸気温度が1mmHgにおいて105.2℃±1℃の範囲内になければ,検出器
の位置が疑わしいので,この方法で使用する前に修正すること。
97
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書E 温度応答時間の確認方法
E1.1 目的 この試験の意図は,検出器が温度変化に十分速く応答し,したがって急激に上昇する温度曲
線に,遅れに起因する誤差がもち込まれないことを確かめることにある。減圧状態で,蒸気の熱容量が少
ない場合,この試験の重要性は大となる。
E1.2 原理 この試験は規定条件下での検出器の冷却速度に基づいている。
E1.3 試験の手順
E1.3.1 水1リットルの入ったビーカーをホットプレート上に置き,水中にガラス製サーモウェルを垂直に
保つ。水温は90±5℃に保つ。
E1.3.2 検出器を温度表示器に接続するが,できれば0.1℃単位で温度を表示するデジタル式に読取りでき
るものがよい。
代わりに検出器を適当な範囲のストリップチャート記録計に接続し,1℃目盛に内挿して0.1℃を読みと
ってもよい。チャート速度は30cm/h以上であるとよい。
E1.3.3 検出器を,一辺が約300mmの閉じたカードボードボックスの一辺の中央にあるあな(孔)に差し
込み,平衡温度に達するようにする。あなと検出器のジョイント部をきっちりと接合する。温度が安定し
たら,それを記録する。
E1.3.4 検出器を取り出し,それをビーカー水中の加熱したサーモウェルに挿入する。
E1.3.5 検出器が80℃の温度に達したら,それを取り出し,直ちにボックスのあなに挿入する。
E1.3.6 ストップウオッチを使用するか,ストリップチャート上の記録を見て,E1.3.3で記録した温度より
30℃高い温度から,E1.3.3で記録した温度より5℃高い温度まで,検出器が冷却する時間間隔を測定する。
E1.3.7 E1.3.6での間隔が175秒を超える場合には,検出器は使用できない。
98
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書F 検出器の校正方法
F1.1 原理
F1.1.1 温度測定の校正に際し,考慮しなければならないことが2点ある。すなわち,温度検出器とその附
属器との組合せについての校正,及び検出器が精留塔頂部の飽和蒸気に適切に暴露される位置にあるかを
確認することである。後者の問題は参考附属書Dで取り扱う。
F1.1.2 検出器とその附属器の校正をするには,純化合物又は共融混合物の融点及び沸点を測定し,記録す
る。
F1.1.3 温度の全範囲にわたる蒸気温度検出器の校正は,検出器とその附属器とを組み合わせて,最初に使
用するときに行う。検出器又は附属器の修理をするか手入れをした場合には,再び校正を行う。
F1.1.4 上記の場合以外に,各蒸気温度検出器は,通常使用するカットポイントに近い1点の温度で週に1
回点検を行う。すず(錫)の融点が適切である。
F1.2 装置 これらの要求事項に合う適切な装置を,参考附属書F図F1.1に示す。水の融点については,
粉砕氷と水で満たしたデュアーフラスコで代用してもよい。水の沸点については,平衡蒸留器又はエブリ
オメータを,蒸気・液体平衡測定にはテンシメータ又は他の装置を使用するのがよい。
F1.3 試験の手順
F1.3.1 温度検出器
F1.3.1.1 約0.1mLのシリコン油又は他の不活性液体がサーモウェルの底にあることを確認し,それぞれの
測定器に接続した1個以上の熱電対又は他の検出器を挿入する。
F1.3.1.2 融点浴の内部の金属を融点より約10℃高い温度にまで加熱し,この温度を少なくとも5分間保ち,
金属を完全に融解させる。
F1.3.1.3 融点浴への加熱を止め,冷却曲線を測定し記録する。曲線上に1分間を超える一定温度のプラト
ーが現れたら,記録されたプラトーの温度を校正温度とする。固化プラトーが短すぎるときは,冷却サイ
クル中に熱を加えて延長することもできる。又は,金属浴が汚染されるか過度に酸化されてしまうことも
あるので,その場合には,金属を取り換える。
F1.3.1.4 次の各点で0.5℃のけたまで校正温度を求める。
物質
温度(℃)
水
融点
0.0
水
沸点
100.0
すず−鉛−カドミウム共融混合物
融点
145.0
すず
融点
231.9
鉛
融点
327.4
F1.3.1.5 各校正点における真の温度が得られるように,測定温度に算術的に加えるべき補正値を列記して
補正表を作成する。これら補正値をグラフにプロットし,滑らかな曲線でつなげると,通常の使用に有益
である。
F1.3.2 真空検出器
F1.3.2.1 大気圧と13.3kPa {100mmHg} の間の減圧校正は,真空水銀気圧計又はマノメータで行う。圧力
が13.3kPa未満の場合は,マクラウド真空計が一次標準計であり,注意深く校正する。二次的な真空計は
定期的使用では少なくとも毎週,又は不定期使用では各操作前に検査する。
99
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
F1.3.2.2 13.3kPa {100mmHg} では,13.3kPa用に校正したマクラウド真空計を使用してもよいが,真空マ
ノメータの方が簡単で十分である。参考附属書F図F1.2には,水銀上の内部空気を頂点の細い先端に閉じ
込め,一目で分かるようにした,便利な装置を例示する。適切なマノメータでは,排気したとき,肉眼で
見える空気は先端に存在しない。ガラス内部は化学的に清浄であること。先端に空気が観察された場合に
は,マノメータを次のようにして再び脱気する。
F1.3.2.3 真空接続部を上に向けて,マノメータをほぼ水平に固定する。このとき,上部を底部より低くし
て,中央管底部のあなが水銀面より上になるようにする。水銀が少量,中央管にある状態が望ましい。
F1.3.2.4 0.0133kPa {0.1mmHg} 未満の真空にし,水銀を含むマノメータの端を赤外線加熱ランプ又は熱風
ガンで,水銀が沸騰するまで加熱する。ゆっくり加熱を続け,中央管の水銀の一部を外管に留出させる。
これは,凝縮した水銀が壁につくことで確認できる。そこでマノメータを垂直位置に戻し,ゆっくりと真
空を解除する。十分に排気したとき,中心管の先端に空気は見えない。1.33kPa {10mmHg} より高い圧力
においては,他の真空計をこれと比較して,校正してもよい。
F1.3.2.5 1.33kPa {10mmHg} 以下では,参考附属書F図F1.3で示すような真空マニホールドを組み立てる。
真空マニホールドはどの希望圧力においても,±2%で一定圧力を維持できること。
F1.3.2.6 注意深く校正したマクラウド真空計で,校正希望圧力が目盛の10〜90%の間に納まるような範囲
のものを接続する。マニホールドと真空ポンプの間にドライアイストラップを挿入する。マクラウド真空
計は試験実施が間近になってから熱く焼いた後,大気に触れないように保護しておく。
F1.3.2.7 定常条件を少なくとも3分間維持した後で,校正したマクラウド真空計と比較すべきすべての真
空計の読取りを行う。誤差が±1%以内のものを日常使用する。
F1.3.2.8 テンシメータをマクラウド真空計の代わりに使用できる。テンシメータの校正は,各試験圧力に
おいて純化合物(通常テトラデカン)の沸点を測定して行う。テンシメータの汚染が疑われるときは,そ
れを洗浄し再充てんして正確度を元に戻す。
100
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書F図F1.1 温度検出器用融点浴
101
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書F図F1.2 真空マノメータ
102
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書F図F1.3 真空計の校正
103
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書G 還流分割弁の性能検査方法
G1.1 原理
G1.1.1 この試験は液体分割弁だけに対するものであり,蒸気分割弁は現在までのところ満足できるものは
ない。
G1.1.2 中間密度の炭化水素留出油を用いて,通常の運転方法で還流分割弁を通して流す。還流比は二つの
流れの比によって決まる。
G1.1.3 試験は通常使用する速度範囲にわたって行い,性能が全水準で許容範囲内にあることを確かめる。
G1.2 装置
G1.2.1 装置は,塔及び調節器付き還流分割弁から成り,必要ならば液体が分割弁に入りやすいように凝縮
器を付ける。
G1.2.2 使用する塔における最高蒸発速度の10〜90%にわたる範囲で,試験液を一定速度で導入できるよう
な方法を準備する。この装置はポンプ系でもよく,又は自然流下システム,すなわち一定した注入が可能
な目盛付き容器を用いてもよい。ビーカーとストップウオッチも必要である。
G1.3 試薬 試験液体は,灯油から軽油の範囲の炭化水素留分が適切である。
G1.4 装置の準備
G1.4.1 正しい位置に分割弁を付けて塔を通常どおりに組み立てる。凝縮器を用いる場合には,それを通常
の位置に取り付ける。調節器を分割弁に接続する。
G1.4.2 適当な大きさのビーカーを塔の底部と,抜出しラインの下に置く。
G1.4.3 注入装置を取り付け,必要な範囲の流速が得られるようにする。
G1.5 試験の手順
G1.5.1 分割弁の作動を開始し,調節器をセットして分割弁を2秒以上5秒以下の間開き,その5倍の間閉
じるようにする。
G1.5.2 分割弁に液体を通し始め,速度を試験中の塔の最高蒸発速度の約10%に等しくする。
G1.5.3 2分後に,塔の底部及び抜出しライン下のビーカーを,それぞれひょう量したビーカーに同時に取
り換える。
G1.5.4 5分後に,これらのビーカーを他のひょう量済みビーカー2個とそれぞれ同時に取り換える。
G1.5.5 G1.5.4を繰り返す。
G1.5.6 正確に5分後,この2個のひょう量済みビーカーを残液用ビーカーと取り換え,液体の流れを止め
る。
G1.5.7 各ひょう量済みビーカーの重さをはかり,各ビーカー中の液体の質量を1gのけたまで求める。
G1.5.8 5分間試験を3回行い,各回ごとに,塔底部でビーカー中に回収された油の質量と,抜出しライン
で回収された油の質量との比を算出する。
G1.5.9 得られた比が±5%の範囲内で一定しないときは,参考附属書G1.5.1〜G1.5.8を繰り返す。
G1.5.10 試験中のカラム最高蒸発速度の約30%,60%及び90%に等しい流速で参考附属書G1.5.1〜G1.5.9
を繰り返す。
G1.5.11 分割弁の開閉周期の閉時間が開時間の2倍に等しいバルブ操作で,参考附属書G1.5.1〜G1.5.10の
操作を繰り返す。
104
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
G1.5.12 上述のようにして求めた実際の還流比と希望する比との差が10%を超える場合には,分割弁の操作
は許容できないので,正しく調整する。
105
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書H 含水原油試料の脱水方法
H1.1 原理
H1.1.1 含水原油(すなわち,水分含量が0.1%を超える原油)の十分量を,低効率カラムで大気圧下,還
流比ゼ口(全抜出し)で130℃まで蒸留する。留出油の水分を除去して,フラスコの試料と混合する。
H1.1.2 脱水操作は,軽質ナフサ領域での正確な収率を得るために重要である。
H1.2 装置 含水原油試料の脱水には,参考附属書H図H1.1に示すような装置が必要であり,次の装置
から成り立つ。
H1.2.1 蒸留フラスコ 2本の枝管の付いたもの(差動マノメータの代わりに,液体中に窒素を通すための,
毛細管をつける。)。
H1.2.2 蒸留カラム 適切な効率のもの。普通の15段塔が望ましい。
H1.2.3 残りの装置は,本体に示すものと同じである。ただし,留出油受器の後に軽質炭化水素用トラップ
を取り付ける。
H1.3 装置の準備 試験開始前に,蒸留カラムと全ガラス器具を洗浄する。
H1.4 試験の手順
H1.4.1 所定容量の含水原油を0℃より高い温度に冷やし,ガラスか磁器片を数個入れて冷やした蒸留フラ
スコに含水原油の一定量を1gのけたまではかり入れる。分離した水があれば,静置分離することが望まし
い。
H1.4.2 フラスコをカラムに接続し,窒素をゆっくりと毛細管に通す。
H1.4.3 留出油受器の後に,ドライアイス温度に保った2個のトラップを直列に取り付け,その先を大気に
開放する。
H1.4.4 −20℃の冷媒を凝縮管に循環させる。
H1.4.5 フラスコを加熱し,脱ブタン操作のため8.2.4〜8.2.8に規定した適度の還流速度を得るよう調節す
る。
H1.4.6 蒸気温度が130℃に達するまで,全留出(還流比ゼロ)で留出油をゆっくりと取り出す。このとき,
65℃より低い温度の留出油は,−20℃以下に冷やした受器中に,集めるように注意する。
H1.4.7 加熱と取出しを中止し,得られた留出分をひょう量する。
H1.4.8 フラスコと内容物を室温に冷やし,残留油をひょう量する。
H1.4.9 軽質留出分を含むトラップを,損失がないよう注意しながら外側をふき取った後,ひょう量する。
H1.4.10 各留出油から水を分離するため,−5℃に冷却して炭化水素液体を静置分離する。水分をひょう量
し,凝縮管を取り外し,アルコール又はアセトンですすいで付着している水滴を取り除き,乾燥させてか
ら元どおりに組み立てる。
H1.4.11 トラップ中の留出分を除き,蒸留残留油と静置分離した留出油を混合する。その際,損失がないよ
うに通常の予防措置を取りながらすべてを0℃に冷やしておく。
ただし,残留油はその流動点より5℃高い温度より低く冷やす必要はない。トラップはドライアイス温
度に保つ。
H1.5
結果
H1.5.1 回収した乾燥油量を記録する。乾燥留出油の再混合を元のフラスコで行った場合には,このフラ
スコを引き続き蒸留に使用する。
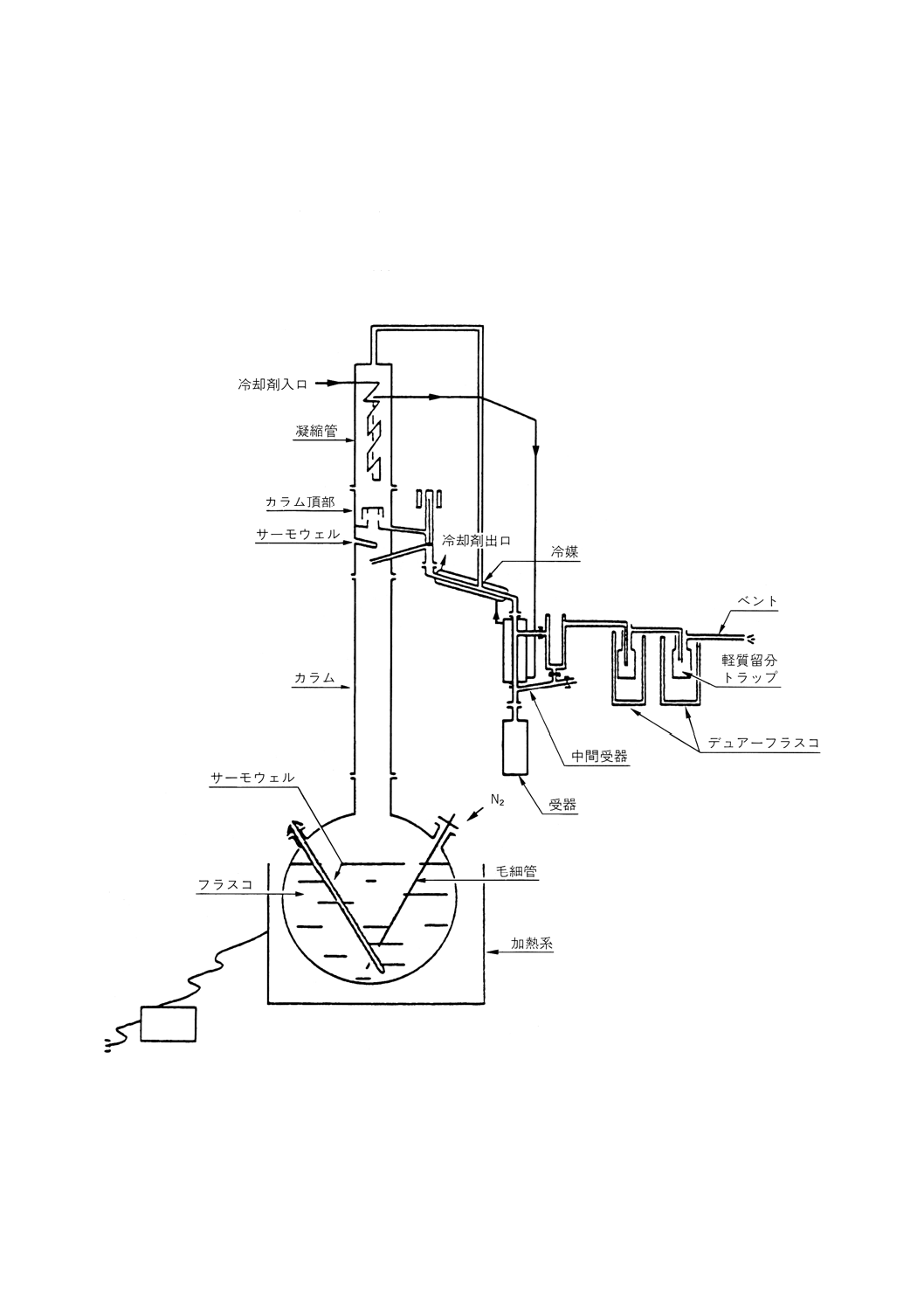
106
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
H1.6 計算
H1.6.1 次の式によって,水分(質量%)を計算する。
W=100(A/B)
ここに,
A: 回収した水の質量 (g)
B: 張り込んだ試料の質量 (g)
W: 水分(質量%)
100: パーセント定数
参考附属書H図H1.1 脱水用蒸留装置の一般図
107
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考付属書I 蒸留温度の計算方法
I1.1 大気圧から0.266kPa {2mmHg} までの測定蒸留温度を,101.3kPa {760mmHg} における相当温度に変
換するためには,参考表2及び参考表3を用いるが,これらの参考表はMaxwell & Bonnellによって導か
れ,IECHA,49巻,1957,p.1187に発表された式に基づいている。一般式は,大気圧〜0.266kPa {2mmHg}
の範囲では次のとおりである。
273
06
516
0.000
1
0.386
273
1
1.
748
−
−
+
+
=
A
T
A
AET
ここに, AET: 大気圧換算温度(℃)
T: 測定した蒸気温度(℃)
ここでPがkPa単位であれば,
P
P
A
log
76
.
95
329
.
579
2
log
546
0.972
324
143
.5
−
−
=
ここでPがmmHg単位であれば,
P
P
A
log
76
.
95
129
.
663
2
log
546
0.972
296
994
.5
−
−
=
グラフ(参考附属書I図I.1又はI.2)は,蒸留中の簡便な指針として記載するものであり,他の目的に
用いてはならない。
I1.2 蒸留する原油の化学的性質を考慮に入れた蒸留温度の補正方法があるが,しかし,これは日常的試
験では適用されない。この方法では,各留出油の特性係数Kを計算し,K値の関数としての温度補正値の
グラフを用いる。カットが芳香族分が主体であるときは,K係数を使用してもよい。
K係数を使用するときは,次の式によって算出する。
(
)
D
B
K
3
273
8.1 +
=
ここに, B: 平均沸点(℃)
D: 15℃における密度
使用上の便宜のため,K計算用の沸点平均値はカットの蒸気温度の中央値か,又はカットのガスクロマ
トグラフィー蒸留の中央値のどちらかであってもよいとの合意があるが,どちらの場合も,その方法は明
記しなければならない。
I1.2.1 K係数概算用チャートを参考附属書I図I.1に示す。
I1.2.2 K係数を求めた後,AETへの補正値は参考附属書I図I.2から求めるか,又は次の式によって計算
する。
t=−1.4 (K−12) [log (Pa/P0)]
ここに,
t: 補正値 (℃)
Pa: 大気圧(kPa又はmmHg)
P0: 測定時の圧力(kPa又はmmHg)
I1.2.3 補正値はグラフを参照して概算できる。

108
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書I図I.1 石油留分のワトソン特性係数
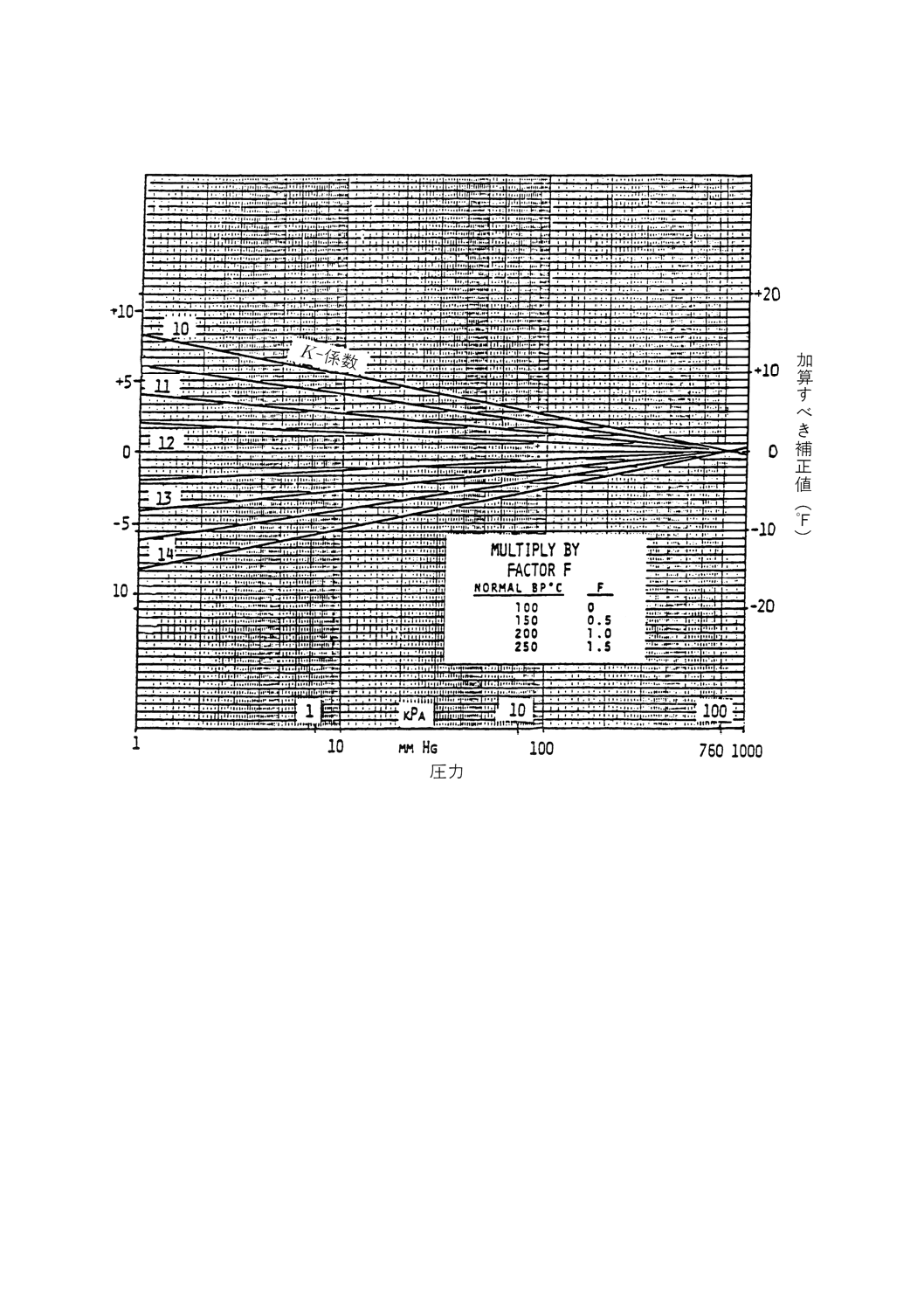
109
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
参考附属書I図I.2 K−係数に対する沸点補正
110
K 2601:1998
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
工業標準原案作成委員会 構成表
氏名
所属
(委員長)
小 西 誠 一
元防衛大学校
天 野 徹
工業技術院標準部
広 田 博 士
資源エネルギー庁石油部
高 橋 千 晴
工業技術院計量研究所
近 藤 輝 男
工業技術院環境技術総合研究所
有 賀 正 夫
社団法人石油学会
中 村 麒久男
社団法人日本海事検定協会理化学研究所
高 野 敏 夫
社団法人自動車技術会
中 西 忠 雄
防衛庁装備局管理調達補給室
福 嶋 信一郎
日本鋼管株式会社本社鉄鋼技術センター環境エネルギー部
中 村 準
菱日エンジニアリング株式会社本牧事業所
吉 田 彰 夫
いすゞ自動車株式会社材料開発部
武 藤 敏 夫
東京電力株式会社火力部
君 島 孝 尚
石川島播磨重工業株式会社技術研究所
加 藤 良 三
東燃株式会社製造計画部
松 崎 昭
日本石油株式会社中央技術研究所
小久保 陽 生
出光興産株式会社製造部
橘 宗 昭
昭和シェル石油株式会社商品技術室
伊 達 和 人
株式会社ジャパンエナジー精製部
下 平 武
日本科学機器団体連合会(田中科学機器製作株式会社)
西 川 輝 彦
石油連盟技術環境部
工業標準原案作成分科会 構成表
氏名
所属
(分科会長)
橘 宗 昭
昭和シェル石油株式会社商品技術室
中 林 賢 司
工業技術院標準部
村 谷 茂 典
財団法人新日本検定協会中央研究所
近 義 彦
社団法人日本海事検定協会理化学研究所
伊 藤 玄
出光興産株式会社製造部
近 藤 修
日本石油株式会社中央技術研究所
番 馬 章
三菱テクノ株式会社業務部
谷 口 宏
三菱石油株式会社開発研究所
大 森 道 昭
日本科学機器団体連合会(株式会社離合社)
鈴 木 繁
東燃株式会社総合研究所
長谷部 好 昭
富士石油株式会社袖ヶ浦製油所生産技術部
広 田 義 則
株式会社コスモ総合研究所分析研究室
高 橋
己
株式会社ジャパンエナジー分析センター
銅 屋 公 一
昭和シェル石油株式会社中央研究所
大 滝 盛 司
ゼネラル石油株式会社中央研究所
林 明
ゼネラル石油株式会社中央研究所
下 平 武
日本科学機器団体連合会(田中科学機器製作株式会社)
橘 田 英 男
日本科学機器団体連合会(吉田科学機器株式会社)
伊 藤 正 保
社団法人全国石油協会品質管理事業部
(事務局)
久保田 亘
石油連盟技術環境部