K 1570:2010
(1)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
目 次
ページ
序文 ··································································································································· 1
1 適用範囲························································································································· 1
2 引用規格························································································································· 1
3 原理(加圧注入処理) ······································································································· 2
4 用語及び定義 ··················································································································· 3
5 種類······························································································································· 4
6 品質······························································································································· 4
7 性能······························································································································· 8
8 品質の試験方法 ················································································································ 9
8.1 一般事項 ······················································································································ 9
8.2 試料の採取方法 ············································································································· 9
8.3 試験液の調製 ················································································································ 9
8.4 個別成分分析方法 ········································································································· 10
8.5 有効成分の配合比及び合計含有量····················································································· 39
8.6 製品の状態 ·················································································································· 43
8.7 水不溶解分 ·················································································································· 43
8.8 pH ····························································································································· 45
8.9 脂肪酸の酸価 ··············································································································· 45
9 表示······························································································································ 46
K 1570:2010
(2)
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
まえがき
この規格は,工業標準化法第14条によって準用する第12条第1項の規定に基づき,社団法人日本木材
保存協会(JWPA)及び財団法人日本規格協会(JSA)から,工業標準原案を具して日本工業規格を改正す
べきとの申出があり,日本工業標準調査会の審議を経て,経済産業大臣が改正した日本工業規格である。
これによって,JIS K 1570:2004は改正され,この規格に置き換えられた。
この規格は,著作権法で保護対象となっている著作物である。
この規格に従うことは,次の者の有する特許権等の使用に該当するおそれがあるので,留意する。
− 氏名:ランクセスドイツ社 LANXESS Deutschland GmbH
− 住所:LIP Intelectual Property Rights
Building Q18,Room 1457
51369 Leverkusen,Germany
− 氏名:株式会社ザイエンス
− 住所:東京都千代田区丸の内2丁目3番2号
上記の,特許権等の権利者は,非差別的かつ合理的な条件でいかなる者に対しても当該特許権等の実施
の許諾等をする意思のあることを表明している。ただし,この規格に関連する他の特許権等の権利者に対
しては,同様の条件でその実地が許諾されることを条件としている。
この規格に従うことが,必ずしも,特許権の無償公開を意味するものではないことに注意する必要があ
る。
この規格の一部が,上記に示す以外の特許権等に抵触する可能性がある。経済産業大臣及び日本工業標
準調査会は,このような特許権等にかかわる確認について,責任はもたない。
なお,ここで“特許権等”とは,特許権,出願公開後の特許出願,実用新案権又は出願公開後の実用新
案登録出願をいう。
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格
JIS
K 1570:2010
木材保存剤
Wood Preservatives
序文
この規格は,1995年に制定され,その後3回の改正を経て今日に至っている。前回の改正は2004年に,
銅・アゾール化合物系及びクレオソート油の見直しを行い,アゾール・第四級アンモニウム・ネオニコチ
ノイド化合物系を追加し,最近の生産及び使用の実態を踏まえるために行った。
なお,対応国際規格は現時点で制定されていない。
1
適用範囲
この規格は,木質材料に防腐性能及び防ぎ(蟻)性能をもたせるための木材保存剤であって,加圧注入
処理によって用いるものについて規定する。
2
引用規格
次に掲げる規格は,この規格に引用されることによって,この規格の規定の一部を構成する。これらの
引用規格は,その最新版(追補を含む。)を適用する。
JIS A 9002 木質材料の加圧式保存処理方法
JIS B 7525 密度浮ひょう
JIS H 2121 電気銅地金
JIS K 0050 化学分析方法通則
JIS K 0061 化学製品の密度及び比重測定方法
JIS K 0102 工場排水試験方法
JIS K 0114 ガスクロマトグラフ分析通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS K 0124 高速液体クロマトグラフィー通則
JIS K 1503 アセトン
JIS K 1571 木材保存剤−性能基準及びその試験方法
JIS K 2249 原油及び石油製品−密度試験方法及び密度・質量・容量換算表
JIS K 2435-2 ベンゼン・トルエン・キシレン−第2部:トルエン
JIS K 2435-3 ベンゼン・トルエン・キシレン−第3部:キシレン
JIS K 3362 家庭用合成洗剤試験方法
JIS K 8012 亜鉛(試薬)
2
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 8032 アセトニトリル(試薬)
JIS K 8034 アセトン(試薬)
JIS K 8101 エタノール(99.5)(試薬)
JIS K 8102 エタノール(95)(試薬)
JIS K 8117 ジクロロメタン(残留農薬・PCB試験用)(試薬)
JIS K 8125 塩化カルシウム(水分測定用)(試薬)
JIS K 8230 過酸化水素(試薬)
JIS K 8271 キシレン(試薬)
JIS K 8322 クロロホルム(試薬)
JIS K 8361 酢酸エチル(試薬)
JIS K 8464 シクロヘキサン(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8574 水酸化カリウム(試薬)
JIS K 8593 石油エーテル(試薬)
JIS K 8613 炭酸アンモニウム(試薬)
JIS K 8625 炭酸ナトリウム(試薬)
JIS K 8660 銅(試薬)
JIS K 8680 トルエン(試薬)
JIS K 8799 フェノールフタレイン(試薬)
JIS K 8839 2-プロパノール(試薬)
JIS K 8844 ブロモフェノールブルー(試薬)
JIS K 8891 メタノール(試薬)
JIS K 8897 メチレンブルー(試薬)
JIS K 8951 硫酸(試薬)
JIS K 8953 硫酸亜鉛七水和物(試薬)
JIS K 8983 硫酸銅(II)五水和物(試薬)
JIS K 8987 硫酸ナトリウム(試薬)
JIS K 9005 りん酸(試薬)
JIS K 9009 りん酸二水素ナトリウム二水和物(試薬)
JIS K 9702 ジメチルスルホキシド(試薬)
JIS R 3503 化学分析用ガラス器具
JIS R 3505 ガラス製体積計
JIS Z 8401 数値の丸め方
JIS Z 8802 pH測定方法
3
原理(加圧注入処理)
木質材料を入れた加圧処理装置内に木材保存剤(薬剤の種類によっては木材保存剤希釈液)を充満させ,
加圧ポンプによって木質材料内よりも高い圧力で木材保存剤を木質材料内に注入し,内部まで含浸させる
方法で,木質材料に防腐性能及び防ぎ(蟻)性能を付与させる。処理方法は,JIS A 9002に規定している。
3
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
4
用語及び定義
この規格で用いる主な用語及び定義は,JIS K 1571によるほか,次による。
4.1
防腐性能
木材保存剤が,木材腐朽菌による劣化を防止する性能。
4.2
防ぎ(蟻)性能
木材保存剤が,イエシロアリ及びヤマトシロアリによる食害を防止する性能。
4.3
鉄腐食性能
木材保存剤で処理した木材の,鉄に対する腐食性能。
4.4
注入処理
減圧,加圧などの機械的圧力差を用いて,木材中に木材保存剤を浸透させる処理。
4.5
水溶性木材保存剤
水に溶解させて用いる木材保存剤。
4.6
乳化性木材保存剤
水に乳化させて用いる木材保存剤。
4.7
油溶性木材保存剤
有機溶剤に溶解させて用いる木材保存剤。
4.8
油性木材保存剤
原液のままで用いる油状の木材保存剤。
4.9
指定濃度
それぞれの加圧注入処理製品のJIS A 9002の8. a)に規定する製品基準に応じた吸収量を,確保するため
に必要な薬液の濃度。製造業者が指定する。
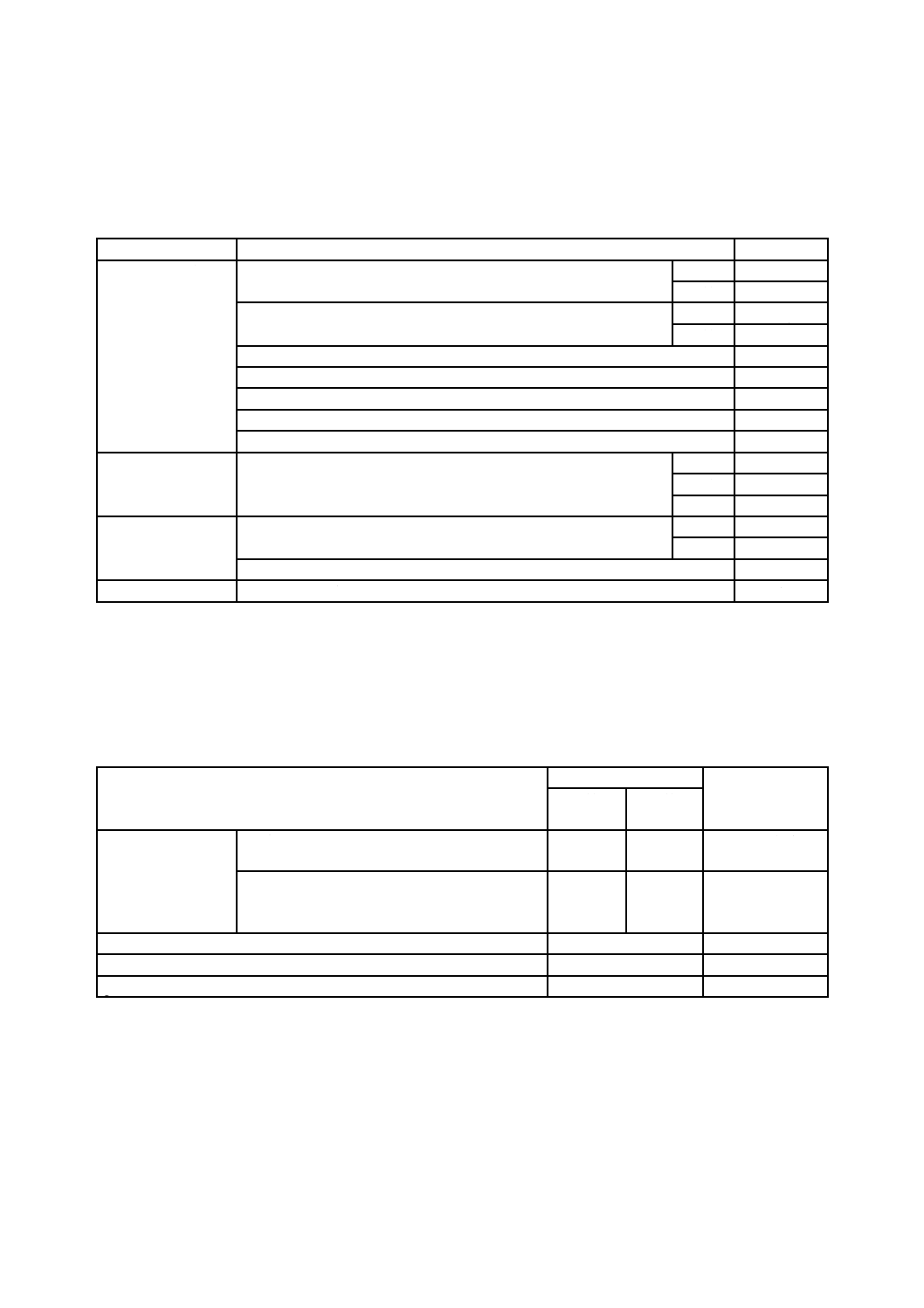
4
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5
種類
木材保存剤の種類は,表1による。
表1−木材保存剤の種類
区分
種類
種類の記号
水溶性木材保存剤
第四級アンモニウム化合物系
1号
AAC-1
2号
AAC-2
銅・第四級アンモニウム化合物系
1号
ACQ-1
2号
ACQ-2
銅・アゾール化合物系
CUAZ
ほう素・第四級アンモニウム化合物系
BAAC
第四級アンモニウム・非エステルピレスロイド化合物系
SAAC
アゾール・第四級アンモニウム・非エステルピレスロイド化合物系
AZAAC
アゾール・第四級アンモニウム・ネオニコチノイド化合物系
AZNA
乳化性木材保存剤
脂肪酸金属塩系
1号
NCU-E
2号
NZN-E
3号
VZN-E
油溶性木材保存剤
ナフテン酸金属塩系
1号
NCU-O
2号
NZN-O
アゾール・ネオニコチノイド化合物系
AZN
油性木材保存剤
クレオソート油
A
6
品質
木材保存剤の品質は,箇条8によって試験したとき,種類に応じて表2〜表12に適合しなければならな
い。
表2−第四級アンモニウム化合物系木材保存剤の品質
項目
品質
試験項目番号
1号
(AAC-1)
2号
(AAC-2)
有効成分の含有量
(質量分率 %)
ジデシルジメチルアンモニウムクロリド(以下,
DDACという。)
30〜55
−
8.3.1及び
8.4.1.1又は8.4.1.2
N,N-ジデシル-N-メチル-ポリオキシエチル-アン
モニウムプロピオネート(以下,DMPAPとい
う。)
−
30〜55
8.3.1及び
8.4.1.1又は8.4.1.2
製品の状態
液状
8.6
水不溶解分(質量分率 %)
1以下
8.7
pH
5.0〜8.0
8.8

5
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表3−銅・第四級アンモニウム化合物系木材保存剤の品質
項目
品質
試験項目番号
1号
(ACQ-1)
2号
(ACQ-2)
有効成分の配合比
(質量分率 %)
銅化合物(CuOとして)
53〜59
62〜71
8.3.2及び
8.4.2.1又は8.4.2.2
N-アルキルベンジルジメチルアンモニウムクロ
リド(以下,BKCという。)
41〜47
−
8.3.2及び8.4.3.1
DDAC
−
29〜38
8.3.2及び8.4.1.1
有効成分の合計含有量a)(質量分率 %)
16以上
−
8.5.1 b)
−
13以上
8.5.2 b)
製品の状態
液状
8.6
水不溶解分(質量分率 %)
1以下
8.7
pH
9.5〜11.0
8.8
注a) 有効成分の合計含有量とは,銅化合物とBKC又はDDACとの合計量をいう。
表4−銅・アゾール化合物系木材保存剤の品質
項目
品質
試験項目番号
CUAZ
有効成分の配合比
(質量分率 %)
銅化合物(CuOとして)
98.6〜99.0
8.3.3及び
8.4.2.1又は8.4.2.2
シプロコナゾールa)
[α-(4-クロロフェニル)-α-(1-シクロプロピルエ
チル)-1H-1,2,4-トリアゾール-1-エタノール]
1.0〜1.4
8.3.3及び
8.4.4.1又は8.4.4.2
有効成分の合計含有量b)(質量分率 %)
11以上
8.5.3 b)
製品の状態
液状
8.6
水不溶解分(質量分率 %)
1以下
8.7
pH
8.7〜11.5
8.8
注a) シプロコナゾールを含むものについては,次の特許がある。
特許登録 第3216946“殺微生物組成物” 登録日 平成13年8月3日
b) 有効成分の合計含有量とは,銅化合物及びシプロコナゾールの合計量をいう。
表5−ほう素・第四級アンモニウム化合物系木材保存剤の品質
項目
品質
試験項目番号
BAAC
有効成分の配合比
(質量分率 %)
ほう素化合物(H3BO3として)
23〜27
8.3.4及び8.4.5.1
DDAC
71〜79
8.3.4及び8.4.1.1
有効成分の合計含有量a)(質量分率 %)
25以上
8.5.4 b)
製品の状態
液状
8.6
水不溶解分(質量分率 %)
1以下
8.7
pH
5.0〜9.0
8.8
注a) 有効成分の合計含有量とは,ほう素化合物及びDDACの合計量をいう。

6
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表6−第四級アンモニウム・非エステルピレスロイド化合物系木材保存剤
項目
品質
試験項目番号
SAAC
有効成分の配合比
(質量分率 %)
DMPAP
99.0〜99.9
8.3.5及び
8.4.1.1又は8.4.1.2
シラフルオフェン
[4-エトキシフェニル[3-(4-フルオロ-3-フェノ
キシフェニル)プロピル]ジメチルシラン]
0.4〜0.6
8.3.5及び
8.4.6.1
有効成分の合計含有量a)(質量分率 %)
35以上
8.5.5 b)
製品の状態
液状
8.6
水不溶解分(質量分率 %)
1以下
8.7
pH
5.0〜8.0
8.8
注a) 有効成分の合計含有量とは,DMPAP及び4-エトキシフェニル[3-(4-フルオロ-3-フェノキシフェニル)プロピル]
ジメチルシランの合計量をいう。
表7−アゾール・第四級アンモニウム・非エステルピレスロイド化合物系木材保存剤の品質
項目
品質
試験項目番号
AZAAC
有効成分の配合比
(質量分率 %)
DMPAP
96.7〜97.3
8.3.6及び
8.4.1.1又は8.4.1.2
シプロコナゾールa)
[α-(4-クロロフェニル)-α-(1-シクロプロピルエ
チル)-1H-1,2,4-トリアゾール-1-エタノール]
0.5〜0.7
8.3.6及び
8.4.4.1又は8.4.4.2
エトフェンプロックス
[2-(4-エトキシフェニル)-2-メチルプロピル-3-
フェノキシベンジルエーテル]
2.2〜2.6
8.3.6及び
8.4.7.1
有効成分の合計含有量b)(質量分率 %)
40以上
8.5.6 b)
製品の状態
液状
8.6
水不溶解分(質量分率 %)
1以下
8.7
pH
5.0〜8.0
8.8
注a) シプロコナゾールを含むものについては,次の特許がある。
特許登録 第3216946“殺微生物組成物” 登録日 平成13年8月3日
b) 有効成分の合計含有量とは,DMPAP,シプロコナゾール及びエトフェンプロックスの合計量をいう。
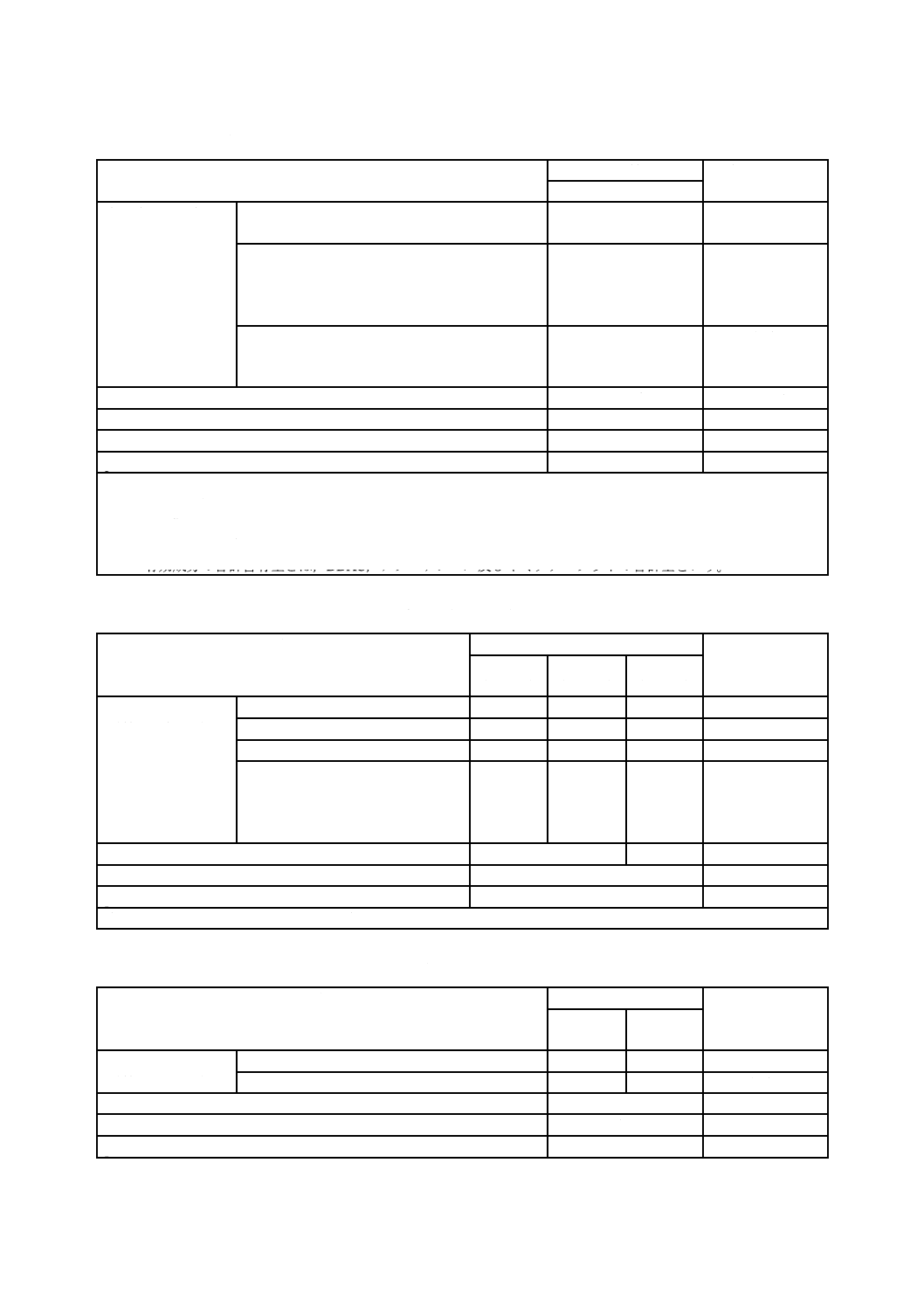
7
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表8−アゾール・第四級アンモニウム・ネオニコチノイド化合物系a) 木材保存剤の品質
項目
品質
試験項目番号
AZNA
有効成分の配合比
(質量分率 %)
DDAC
95.0〜97.5
8.3.7及び
8.4.1.1又は8.4.1.2
テブコナゾール
[α-〔2-(4-クロロフェニル)エチル〕-α-(1,1-ジメ
チルエチル)-1H-1,2,4-トリアゾール-1-エタノー
ル]
2.0〜4.0
8.3.7及び8.4.8.1
イミダクロプリド
[1-[(6-クロロ-3-ピリジニル)メチル]-4.5-ジヒド
ロ-N-ニトロ-1H-イミダゾール-2-アミン]
0.5〜1.0
8.3.7及び8.4.9.1
有効成分の合計含有量b)(質量分率 %)
23以上
8.5.7 b)
製品の状態
液状
8.6
水不溶解分(質量分率 %)
1以下
8.7
pH
4.0〜7.0
8.8
注a) この木材保存剤は,次の特許がある。
特許登録 第4135979“水を基材とする溶媒不含および乳化剤不含の殺微生物物性の,活性物質の組み合わ
せ物”
登録日 平成20年6月13日
b) 有効成分の合計含有量とは,DDAC,テブコナゾール及びイミダクロプリドの合計量をいう。
表9−脂肪酸金属塩系木材保存剤の品質
項目
品質
試験項目番号
1号
(NCU-E)
2号
(NZN-E)
3号
(VZN-E)
有効成分の含有量
(質量分率 %)
ナフテン酸銅(Cuとして)
6.0〜6.6
−
−
8.3.8及び8.4.10.1
ナフテン酸亜鉛(Znとして)
−
8.0〜8.8
−
8.3.8及び8.4.11.1
第三級カルボン酸亜鉛(Znとして)
−
−
5.0〜5.5
8.3.8及び8.4.11.1
ペルメトリン
[3-フェノキシベンジル=2-(2,2-ジ
クロロビニル)-3,3-ジメチル-1-シク
ロプロパンカルボキシラート]
−
−
0.50〜0.55 8.3.8及び8.4.12.1
脂肪酸a) の酸価
180〜300
318〜328
8.9
製品の状態
液状
8.6
pH
6.0〜7.0
8.8
注a) 脂肪酸とは,テルペン系のカルボン酸をいう。
表10−ナフテン酸金属塩系木材保存剤の品質
項目
品質
試験項目番号
1号
(NCU-O)
2号
(NZN-O)
有効成分の含有量
(質量分率 %)
ナフテン酸銅(Cuとして)
5.0〜11.0
−
8.3.9及び8.4.13.1
ナフテン酸亜鉛(Znとして)
−
5.0〜11.0
8.3.9及び8.4.14.1
脂肪酸の酸価
180〜300
8.9
製品の状態
液状
8.6
pH
−
−

8
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表11−アゾール・ネオニコチノイド化合物系木材保存剤の品質
項目
品質
試験項目番号
AZN
有効成分の配合比
(質量分率 %)
シプロコナゾールa)
[α-(4-クロロフェニル)-α-(1-シクロプロピルエ
チル)-1H-1,2,4-トリアゾール-1-エタノール]
62〜72
8.3.10及び8.4.4.2
イミダクロプリド
[1-[(6-クロロ-3-ピリジニル)メチル]-4.5-ジヒド
ロ-N-ニトロ-1H-イミダゾール-2-アミン]
28〜38
8.3.10及び8.4.9.1
有効成分の合計含有量b)(質量分率 %)
3.6 以上
8.5.8 b)
製品の状態
液状
8.6
pH
−
−
注a) シプロコナゾールを含むものについては,次の特許がある。
特許登録 第3216946“殺微生物組成物” 登録日 平成13年8月3日
b) 有効成分の合計含有量とは,シプロコナゾール及びイミダクロプリドの合計量をいう。
表12−クレオソート油木材保存剤の品質
項目
品質
試験項目番号
A
密度(g/cm3,40 ℃)
1.01以上
8.4.15.1.1又は
8.4.15.1.2
水分(体積分率 %)
3以下
8.4.15.2
エングラー度a)(40/20 ℃)
2.0以下
8.4.15.3
蒸留試験
(脱水試料)
(体積分率 %)
235 ℃までの留出量
10以下
8.4.15.4
235〜255 ℃の間の留出量
60以上
8.4.15.4
255〜270 ℃の間の留出量
20以下
8.4.15.4
270〜315 ℃の間の留出量
5以上
8.4.15.4
315 ℃までの留出量
85以上
8.4.15.4
235〜315 ℃の留分の密度(g/cm3,40 ℃)
1.02以上
8.4.15.5.1又は
8.4.15.5.2
トルエン不溶分(脱水試料)(質量分率 %)
0.5以下
8.4.15.6
流動性試験
顕著な固形物が生成
しない。
8.4.15.7
ジベンゾ[a,h]アントラセンの含有量(mg/kg)
10以下
8.4.15.8
ベンゾ[a]アントラセンの含有量(mg/kg)
10以下
8.4.15.9
ベンゾ[a]ピレンの含有量(mg/kg)
10以下
8.4.15.10
有効成分の合計含有量(質量分率 %)
規定せず。
−
注a) エングラー度とは,水との相対粘度をいう。
7
性能
木材保存剤の防腐性能,防ぎ(蟻)性能及び鉄腐食性能は,それぞれの木材保存剤の指定濃度において
JIS K 1571の5.2.1.1(注入処理用),5.2.2.1(培養瓶試験)又は5.2.2.2(腐朽槽試験),5.2.3(野外試験),
5.3.1.1(注入処理用),5.3.2.1(注入処理用)及び5.4.1(注入処理用)によって試験し,JIS K 1571の箇条
4(性能基準)に規定する性能基準に適合しなければならない。
9
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8
品質の試験方法
8.1
一般事項
一般事項は,次による。
a) 化学分析方法において共通する事項は,JIS K 0050による。
b) 数値の丸め方は,JIS Z 8401による。
c) 原子吸光分析法は,JIS K 0121による。
d) 吸光光度分析法は,JIS K 0115による。
e) ICP発光分光分析法は,JIS K 0116による。
f)
高速液体クロマトグラフ分析法は,JIS K 0124による。
g) ガスクロマトグラフ分析法は,JIS K 0114による。
h) 試験に用いるガラス器具は,JIS R 3503による。
i)
試験に用いるガラス製体積計は,JIS R 3505による。
8.2
試料の採取方法
試料の採取方法は,ロットを代表する試料を製品の在庫品,試料採取用ノズルなどから行い,試料によ
って,それぞれ適した器具を用いる。
8.3
試験液の調製
8.3.1
第四級アンモニウム化合物系(AAC-1,AAC-2)
8.2で採取した試料約10 gを0.1 mgのけたまではかりとり,全量フラスコ1 000 mLに入れ,蒸留水を標
線まで加えて試験液とする。
8.3.2
銅・第四級アンモニウム化合物系(ACQ-1,ACQ-2)
8.2で採取した試料約0.8 gを0.1 mgのけたまではかりとり,全量フラスコ250 mLに入れ,蒸留水を標
線まで加えて試験液とする。
8.3.3
銅・アゾール化合物系(CUAZ)
8.2で採取した試料約17 gを0.1 mgのけたまではかりとり,全量フラスコ500 mLに入れ,蒸留水を標
線まで加えて試験液とする。
8.3.4
ほう素・第四級アンモニウム化合物系(BAAC)
8.2で採取した試料約13.3 gを0.1 mgのけたまではかりとり,全量フラスコ1 000 mLに入れ,蒸留水を
標線まで加えて試験液とする。
8.3.5
第四級アンモニウム・非エステルピレスロイド化合物系(SAAC)
8.2で採取した試料約10 gを0.1 mgのけたまではかりとり,全量フラスコ1 000 mLに入れ,蒸留水を標
線まで加えて試験液とする。
8.3.6
アゾール・第四級アンモニウム・非エステルピレスロイド化合物系(AZAAC)
8.2で採取した試料約10 gを0.1 mgのけたまではかりとり,全量フラスコ1 000 mLに入れ,蒸留水を標
線まで加えて試験液とする。
8.3.7
アゾール・第四級アンモニウム・ネオニコチノイド化合物系(AZNA)
8.2で採取した試料約4 gを0.1 mgのけたまではかりとり,全量フラスコ100 mLに入れ,蒸留水を標線
まで加えて試験液とする。
8.3.8
脂肪酸金属塩系(NCU-E,NZN-E,VZN-E)
8.2で採取した試料約1 gを0.1 mgのけたまではかりとり,全量フラスコ100 mLに入れ,蒸留水を標線
まで加えて試験液とする。VZN-Eのペルメトリンの場合,8.2で採取した試料をそのまま使用する。
10
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.3.9
ナフテン酸金属塩系(NCU-O,NZN-O)
8.2で採取した試料約0.5 gを0.1 mgのけたまでトールビーカ300 mLにはかりとり,JIS K 8230に規定
する過酸化水素約10 mL及びJIS K 8951に規定する硫酸4 mLを加え,時計皿を載せて,砂浴上で徐々に
加熱分解する。放冷後ろ紙でろ過しながら全量フラスコ500 mLに移し,トールビーカ及び時計皿を蒸留
水でよく洗い,洗液を全量フラスコに加え,蒸留水を標線まで加えて試験液とする。
8.3.10 アゾール・ネオニコチノイド化合物系(AZN)
8.2で採取した試料約1.5 gを0.1 mgのけたまでなす形フラスコ50 mLにはかりとり,JIS K 9702に規定
するジメチルスルホキシドを約2 mL加え,ロータリ・エバポレータを用いて約45 ℃の湯温中で液量が約
2 mLになるまで濃縮する。濃縮液及びなす形フラスコ内壁を少量のJIS K 8101に規定するエタノールで
洗いながら全量フラスコ25 mLに移し入れ,JIS K 8101に規定するエタノールを標線まで加えて,試験液
とする。
8.3.11 クレオソート油(A)
8.2で採取した試料約200 mLを0.1 mgのけたまではかりとり,約50 ℃に加熱し,乾燥剤としてJIS K
8125に規定する塩化カルシウム約60 g又は約120 ℃で1時間以上乾燥した食塩約100 gを加えてよくか
き混ぜた後,約10分間50 ℃に保ち,放冷後,ろ過して脱水試験液とする。
8.4
個別成分分析方法
8.4.1
ジデシルジメチルアンモニウムクロリド(DDAC)及びN,N-ジデシル-N-メチル-ポリオキシエチル
-アンモニウムプロピオネート(DMPAP)
8.4.1.1
滴定法
a) 要旨 カチオンであるDDAC及びDMPAPは,ブロモフェノールブルー(以下,BPBという。)を指
示薬として,水−クロロホルム系において,振り混ぜながらアニオン溶液で滴定する。
b) 試薬 試薬は,次による。
1) アニオン溶液(0.002 mol/L) テトラフェニルほう酸ナトリウム0.684 5 gをとり,蒸留水に溶かし
て1 000 mLとしたもの。
2) 緩衝液(pH 10) JIS K 8625に規定する炭酸ナトリウム100 gと,JIS K 8987に規定する硫酸ナト
リウム100 gとを蒸留水に溶かして1 000 mLとしたもの。
3) BPB溶液(1 g/L) JIS K 8844に規定するBPB 0.1 gを,蒸留水に溶かして100 mLとしたもの。
4) クロロホルム JIS K 8322に規定するもの。
c) 操作 試験液の2 mLを,全量ピペットを用いてはかりとり,三角フラスコ200 mLに入れ,緩衝液
50 mL,クロロホルム50 mL及びBPB溶液(1 g/L)5滴を加え,栓をして振り混ぜる。ミクロビュレ
ットを用いてアニオン溶液(0.002 mol/L)を少量ずつ加え,加えるごとによく振り混ぜ,液の色の変
化を確認する。滴定の終点は,クロロホルム層の青が消えて水層が淡い紫に着色した点とする。
d) 計算 c)によって得たDDAC及びDMPAPの量を各薬剤別に算出し,有効数字2けたに丸める。
1) AAC-1 質量及び含有量は,式(1)及び式(2)による。
2
000
1
000
1
724
.0
1
×
×
×
×
=
f
t
A
······························································· (1)
ここに,
A1: 8.3.1ではかりとった試料中のDDACの質量(g)
t: 滴定に要したアニオン溶液(0.002 mol/L)の量(mL)
f: 0.002 mol/Lアニオン溶液のファクタ
0.724: 0.002 mol/Lアニオン溶液1 mLに相当するDDACの質量(mg)
11
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
1×
=SA
A
············································································· (2)
ここに,
A: 8.3.1ではかりとった試料中のDDACの含有量(質量分率 %)
S: 8.3.1ではかりとった試料の質量(g)
2) AAC-2 質量及び含有量は,式(3)及び式(4)による。
2
000
1
000
1
908
.0
1
×
×
×
×
=
f
t
A
······························································· (3)
ここに,
A1: 8.3.1ではかりとった試料中のDMPAPの質量(g)
t: 滴定に要したアニオン溶液(0.002 mol/L)の量(mL)
f: 0.002 mol/Lアニオン溶液のファクタ
0.908: 0.002 mol/Lアニオン溶液1 mLに相当するDMPAPの質量
(mg)
100
1×
=SA
A
············································································· (4)
ここに,
A: 8.3.1ではかりとった試料中のDMPAPの含有量(質量分率 %)
S: 8.3.1ではかりとった試料の質量(g)
3) ACQ-2 質量は,式(5)による。
2
000
1
250
724
.0
×
×
×
×
=
f
t
D
································································· (5)
ここに,
D: 8.3.2ではかりとった試料中のDDACの質量(g)
t: 滴定に要したアニオン溶液(0.002 mol/L)の量(mL)
f: 0.002 mol/Lアニオン溶液のファクタ
0.724: 0.002 mol/Lアニオン溶液1 mLに相当するDDACの質量(mg)
4) BAAC 質量は,式(6)による。
2
000
1
000
1
724
.0
×
×
×
×
=
f
t
D
······························································· (6)
ここに,
D: 8.3.4ではかりとった試料中のDDACの質量(g)
t: 滴定に要したアニオン溶液(0.002 mol/L)の量(mL)
f: 0.002 mol/Lアニオン溶液のファクタ
0.724: 0.002 mol/Lアニオン溶液1 mLに相当するDDACの質量(mg)
5) SAAC 質量は,式(7)による。
2
000
1
000
1
908
.0
×
×
×
×
=
f
t
D
······························································· (7)
ここに,
D: 8.3.5ではかりとった試料中のDMPAPの質量(g)
t: 滴定に要したアニオン溶液(0.002 mol/L)の量(mL)
f: 0.002 mol/Lアニオン溶液のファクタ
0.908: 0.002 mol/Lアニオン溶液1 mLに相当するDMPAPの質量
(mg)
6) AZAAC 質量は,式(8)による。
000
1
000
1
908
.0
1
×
×
×
=
f
t
A
······························································· (8)
ここに,
A1: 8.3.6ではかりとった試料中のDMPAPの質量(g)
t: 滴定に要したアニオン溶液(0.002 mol/L)の量(mL)
f: 0.002 mol/Lアニオン溶液のファクタ
0.908: 0.002 mol/Lアニオン溶液1 mLに相当するDMPAPの質量
(mg)

12
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
7) AZNA 質量は,式(9)による。
000
1
100
724
.0
1
×
×
×
=
f
t
D
································································ (9)
ここに,
D1: 8.3.7ではかりとった試料中のDDACの質量(g)
t: 滴定に要したアニオン溶液(0.002 mol/L)の量(mL)
f: 0.002 mol/Lアニオン溶液のファクタ
0.724: 0.002 mol/Lアニオン溶液1 mLに相当するDDACの質量
(mg)
8.4.1.2
高速液体クロマトグラフ法
a) 要旨 試料をイオンペア試薬で電気的に中性化し,紫外線吸光光度検出による高速液体クロマトグラ
フで定量する。
b) 試薬 試薬は,次による。
1) DDAC及びDMPAP 定量用に精製されたもので,あらかじめ純度が分かっているもの。
2) 2-ナフタレンスルホン酸ナトリウム溶液 2-ナフタレンスルホン酸ナトリウム575 mgをはかりと
り,全量フラスコ100 mLに入れ,標線まで蒸留水を加えたもの。
3) りん酸溶液 JIS K 9005に規定するりん酸17 mLを全量フラスコ100 mLに入れ,標線まで蒸留水
を加えたもの。
4) メタノール 高速液体クロマトグラフ用のもの。
5) 移動相 JIS K 9009に規定するりん酸二水素ナトリウム二水和物780 mgをはかりとり,全量フラス
コ500 mLに入れ,さらに,2)で調製した溶液を10 mLと3)で調製した溶液を2 mLとをそれぞれ加
え,標線まで蒸留水を加える。この液とメタノールとを体積分率1:4の割合で混合したもの。
6) 検量線用標準溶液 DDAC及びDMPAP約50 mgを0.1 mgのけたまではかりとり,全量フラスコ100
mLに入れ,5)で調製した移動相を標線まで加えたもの。
c) 装置 装置は,高速液体クロマトグラフ分析装置を用いる。
d) 高速液体クロマトグラフ分析条件 高速液体クロマトグラフの分析条件は,表13による。
表13−高速液体クロマトグラフの分析条件
項目
条件
カラム
移動相
移動相流速
カラム温度
測定波長
注入量
ODS系カラム(I.D 4.6 mm×L 150 mm)
8.4.1.2 b) 5)による。
1.0 mL/min
40 ℃
275 nm
10 μL
e) 操作 試験液2 mLを全量ピペットを用いてはかりとり,全量フラスコ100 mLに入れ,移動相を標線
まで入れ,分析試験溶液とする。分析試験溶液の10 μLを高速液体クロマトグラフに注入し,DDAC
及びDMPAPのピーク面積を求める。検量線から分析試験溶液中のDDAC及びDMPAPの濃度を求め
る。
f)
検量線の作成 検量線用標準溶液を移動相で希釈し,約50〜150 mg/LのDDAC及びDMPAPの濃度
になるように段階的に調製する。この溶液を同様に操作して,DDAC及びDMPAPの濃度とピーク面
積との検量線を3点以上作成する。検量線の作成は,その都度行う。
13
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
g) 計算 e)によって得たDDAC及びDMPAPの量を各薬剤別に算出し,有効数字2けたに丸める。
1) AAC-1及びAAC-2 質量及び含有量は,式(10)及び式(11)による。
2
000
000
1
000
1
100
0
1
×
×
×
=D
D
······························································ (10)
ここに,
D1: 8.3.1ではかりとった試料中のDDAC及びDMPAPの質量(g)
D0: 検量線から求めた分析試験溶液中のDDAC及びDMPAPの濃
度(mg/L)
100
1×
=WD
D
············································································(11)
ここに,
D: 8.3.1ではかりとった試料中のDDAC及びDMPAPの含有量(質
量分率 %)
W: 8.3.1ではかりとった試料の質量(g)
2) SAAC 質量は,式(12)による。
000
1
1
100
0
×
×
=M
M
······································································ (12)
ここに,
M: 8.3.5ではかりとった試料中のDMPAPの質量(g)
M0: 検量線から求めた試験液中のDMPAPの濃度(mg/L)
3) AZAAC 質量は,式(13)による。
000
000
1
000
1
100
1
×
×
=A
A
··································································· (13)
ここに,
A1: 8.3.6ではかりとった試料中のDMPAPの質量(g)
A: 検量線から求めた分析試験溶液中のDMPAPの濃度(mg/L)
4) AZNA 質量は,式(14)による。
000
000
1
100
100
1
×
×
=d
D
····································································· (14)
ここに,
D1: 8.3.7ではかりとった試料中のDDACの質量(g)
d: 分析試験液中のDDAC濃度(mg/L)
8.4.2
銅化合物(CuOとして)
8.4.2.1
原子吸光分析法
a) 要旨 原子吸光分析装置によって,銅を定量する。
b) 試薬 試薬は,次による。
1) 硫酸 JIS K 8951に規定するもの。
2) 検量線用銅標準液(Cuとして100 mg/L) JIS K 8660に規定する銅(試薬),JIS H 2121に規定す
る電気銅地金又はJIS K 8983に規定する硫酸銅(II)五水和物を適切な濃度のJIS K 8541に規定す
る硝酸に溶かし,最終の硝酸濃度を0.05〜0.15 mol/Lとした銅標準液(Cuとして1 000 mg/L)10 mL
を全量ピペットを用いてはかりとり,フラスコ100 mLに入れ,標線まで蒸留水を加えたもの。
c) 装置 装置は,原子吸光分析装置を用いる。
d) 操作 試験液をACQは2 mL,CUAZは1 mL全量ピペットを用いてはかりとり,全量フラスコ1 000
mLに入れ,硫酸8 mLを加えた後,蒸留水を標線まで加えて,分析試験液とする。JIS K 0121の8.
(操作方法)によって,分析試験液をフレーム中に噴霧し,波長324.8 nmで吸光度を読み取り,検量
線から分析試験液中の銅の濃度を求める。
e) 検量線の作成 検量線用銅標準液(Cuとして100 mg/L)0〜5 mLを段階的に全量ピペットを用いては
14
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
かりとり,全量フラスコ100 mLに入れ,分析試験液と同じ条件になるように硫酸を加えた後,蒸留
水を標線まで加える。この溶液について同様に操作して,銅の量と吸光度との検量線を3点以上作成
する。検量線の作成は,その都度行う。
f)
式 d)によって得た銅化合物の量を各薬剤別に算出し,有効数字2けたに丸める。
1) ACQ 質量は,式(15)による。
2
000
000
1
000
1
250
252
.1
×
×
×
×
=c
C
··························································· (15)
ここに,
C: 8.3.2ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
c: 分析試験液中の銅濃度(Cuとして)(mg/L)
1.252: CuをCuOに換算する係数
2) CUAZ 質量は,式(16)による。
000
000
1
000
1
500
252
.1
×
×
×
=c
C
··························································· (16)
ここに,
C: 8.3.3ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
c: 分析試験液中の銅濃度(Cuとして)(mg/L)
1.252: CuをCuOに換算する係数
8.4.2.2
ICP発光分光分析法
a) 要旨 試料を蒸留水に溶かした後,ICP発光分光分析法で銅を定量する。
b) 試薬 試薬は,次による。
1) 硫酸 JIS K 8951に規定するもの。
2) 検量線用銅標準液(Cuとして100 mg/L) 8.4.2.1 b) 2)による。
c) 装置 装置は,ICP発光分光分析装置を用いる。
d) 操作 試験液をACQは2 mL,CUAZは1 mL全量ピペットを用いてはかりとり,全量フラスコ1 000
mLに入れ,硫酸8 mLを加えた後,蒸留水を標線まで加えて,分析試験液とする。ICP発光分光分析
装置を用いて分析試験液の銅の発光光度を測定し,検量線から分析試験液中の銅の濃度を求める。
e) 検量線の作成 検量線用銅標準液(Cuとして100 mg/L)0〜5 mLを段階的に全量ピペットを用いては
かりとり,全量フラスコ100 mLに入れ,分析試験液と同じ条件になるように硫酸を加えた後,蒸留
水を標線まで加える。この溶液について同様に操作して,銅の量と吸光度との検量線を3点以上作成
する。検量線の作成は,その都度行う。
f)
計算 d)によって得た銅化合物の量を各薬剤別に算出し,有効数字2けたに丸める。
1) ACQ 質量は,式(17)による。
2
000
000
1
000
1
250
252
.1
×
×
×
×
=c
C
··························································· (17)
ここに,
C: 8.3.2ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
c: 分析試験液中の銅濃度(mgCu/L)
1.252: CuをCuOに換算する係数
2) CUAZ 質量は,式(18)による。
000
000
1
000
1
500
252
.1
×
×
×
=c
C
··························································· (18)
ここに,
C: 8.3.3ではかりとった試料中の銅化合物の質量(CuOとして)
15
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(g)
c: 分析試験液中の銅濃度(mgCu/L)
1.252: CuをCuOに換算する係数
8.4.3
N-アルキルベンジルジメチルアンモニウムクロリド(BKC)
8.4.3.1
滴定法
a) 要旨 BKCを,メチレンブルーを指示薬として,水−クロロホルム系において,振り混ぜながらラウ
リル硫酸ナトリウム溶液で滴定する。
b) 試薬 試薬は,次による。
1) ラウリル硫酸ナトリウム溶液(0.005 mol/L) あらかじめ,JIS K 3362によって純度を測定したラ
ウリル硫酸ナトリウム約1.5 gを0.1 mgのけたまではかりとり,蒸留水に溶かして1 000 mLとした
もの。
2) メチレンブルー溶液(0.03 g/L) 蒸留水500 mLにJIS K 8951に規定する硫酸12 gをかき混ぜなが
ら徐々に加える。これにJIS K 8897に規定するメチレンブルー(二水塩)0.03 gとJIS K 8987に規
定する硫酸ナトリウム50 gとを加え,かき混ぜて溶かして蒸留水を加えて1 000 mLとしたもの。
3) クロロホルム JIS K 8322に規定するもの。
c) 操作 試験液2 mLを全量ピペットを用いてはかりとり,共栓付き三角フラスコ100 mLに入れ,クロ
ロホルム20 mL,蒸留水20 mL及びメチレンブルー溶液(0.03 g/L)2 mLを加え,振り混ぜながらミ
クロビュレット5 mLを用いてラウリル硫酸ナトリウム溶液(0.005 mol/L)で滴定する。ラウリル硫
酸ナトリウム溶液(0.005 mol/L)を繰り返し少量ずつ加え,加えるごとによく振り混ぜ,液の色の変
化を確認する。滴定の終点は,上層(水層)の青と,下層(クロロホルム層)の青との程度が同じで
あるか,又はわずかに下層がより青い場合とする。別に試験液を加えないで同様に操作し,空試験を
行う。
d) 計算 c)によって得たBKCの量を式(19)によって算出し,有効数字2けたに丸める。
2
000
1
250
005
.0
354
)
(
2
1
×
×
×
×
×
−
=
f
t
t
B
················································ (19)
ここに,
B: 8.3.2ではかりとった試料中のBKCの質量(g)
t1: 滴定に使用したラウリル硫酸ナトリウム溶液(0.005 mol/L)の
量(mL)
t2: 空試験に使用したラウリル硫酸ナトリウム溶液(0.005 mol/L)
の量(mL)
f: ラウリル硫酸ナトリウム溶液(0.005 mol/L)のファクタ
354: BKCの平均分子量
8.4.4
シプロコナゾール
8.4.4.1
ガスクロマトグラフ法
a) 要旨 ガスクロマトグラフによって,試験液中のシプロコナゾールを定量する。
b) 試薬 試薬は,次による。
1) エタノール JIS K 8101に規定するもの。
2) トルエン JIS K 8680に規定するもの。
3) 酢酸エチル JIS K 8361に規定するもの。
4) アセトン JIS K 8034に規定するもの。
5) シクロヘキサン JIS K 8464に規定するもの。
6) 炭酸アンモニウム溶液 JIS K 8613に規定する炭酸アンモニウム80 gを全量フラスコ1 000 mLに

16
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
入れ,蒸留水を加えて溶かし,さらに標線まで蒸留水を加えたもの。
7) 酢酸エチル/シクロヘキサン溶液 JIS K 8361に規定する酢酸エチルと,JIS K 8464に規定するシ
クロヘキサンとを,2:3(体積分率)の比率で混合したもの。
8) 多孔性けい(珪)藻土カラム 内径約2.5 cmカラムに約20 mL保持量のカラムクロマトグラフ用か
粒状けい藻土を充てんしたもの,又はこれと同等の性能をもつもの。
9) シリカゲルミニカラム 内径約1.0 cm,長さ約2.5 cmのカラムにカラムクロマトグラフ用シリカゲ
ル約700 mgを充てんしたもの,又はこれと同等の性能をもつもの。
10) シプロコナゾール溶液 シプロコナゾール標品(純度95 %以上で純度の分かっているもの。)約50
mgを0.1 mgのけたまではかりとり,全量フラスコ50 mLに入れ,JIS K 8101に規定するエタノー
ルを加えて溶かし,更に標線までエタノールを加えたもの。
11) 検量線用溶媒 JIS K 8032に規定する,アセトニトリル60 mLと蒸留水40 mLとを混合したもの。
c) 装置 装置は,ガスクロマトグラフ分析装置を用いる。
d) ガスクロマトグラフ分析条件 ガスクロマトグラフの分析条件は,表14による。
表14−ガスクロマトグラフの条件
項目
条件
固定相液体(膜厚)
カラム用管(内径×長さ)
カラム温度
インジェクション温度
注入量
燃焼ガス
メイクアップガス
キャリヤガス
検出器
[5 % Phenyl]架橋結合型メチルシロキサン(0.25 μm)
溶融シリカ・キャピラリ管(0.32 mm×30 m)
60 ℃,1 min→(20 ℃/min)→240 ℃,10 min→(20 ℃/min)→260 ℃
250 ℃
2 μL
水素30 mL/min,空気370 mL/min
He 30 mL/min
He Split Vent 93 mL/min,Purge Vent 1 mL/min
水素炎イオン化検出器(FID)
e) 操作 操作は,次による。
1) 試験液10 mLを全量ピペットを用いてはかりとり,全量フラスコ50 mLに入れ,炭酸アンモニウム
溶液を標線まで加え,これを希釈試験液とする。
2) 希釈試験液20 mLを全量ピペットではかりとり,多孔性けい藻土カラムに加え,10分間保持する。
3) 多孔性けい藻土カラムに注射器を取り付け,トルエン120 mLで溶出させ,溶出液を丸底フラスコ
200 mLに受け,これをロータリ・エバポレータ(湯浴温度40 ℃)を用いて減圧下で留去する。
4) 残さ(渣)をトルエン10 mLで3回に分けて溶かす。
5) 10 mL容注射器を取り付けたシリカゲルミニカラムを酢酸エチル10 mLで洗浄後,更にトルエン10
mLで洗浄する。洗浄を終えたシリカゲルミニカラムに,10 mL/minの速度で4)の溶液をすべて通液
し,流出液は捨てる。
6) 酢酸エチル/シクロヘキサン溶液5 mLを通液し,流出液は捨てる。
7) 次に酢酸エチル10 mLで溶出させ,溶出液を丸底フラスコ100 mLに受け,これをロータリ・エバ
ポレータ(湯浴温度40 ℃)を用いて減圧下で留去する。
8) 残さ(渣)をアセトンで溶解し,定容(10 mL)として,これを分析試験液とする。
9) 分析試験液の2 μLをガスクロマトグラフ分析装置に注入し,シプロコナゾールのピーク面積を求
め,検量線からシプロコナゾールの濃度を求める。

17
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
f)
検量線の作成 シプロコナゾール溶液を,検量線用溶媒を用いて段階的に10〜50 μg/mLになるように
調製し,2 μLをガスクロマトグラフ分析装置に注入して検量線を3点以上作成する。
g) 計算 e)によって得たシプロコナゾールの量を式(20)によって算出し,有効数字2けたに丸める。
000
1
10
20
000
1
500
50
10
×
×
×
×
×
×
=
y
Y
···························································· (20)
ここに,
Y: 8.3.3ではかりとった試料中のシプロコナゾールの質量(g)
y: 分析試験液中のシプロコナゾール濃度(mg/L)
8.4.4.2
高速液体クロマトグラフ法
a) 要旨 高速液体クロマトグラフによって,試験液中のシプロコナゾールを定量する。
b) 試薬 試薬は,次による。
1) シプロコナゾール溶液 シプロコナゾール標品(純度95 %以上で純度の分かっているもの。)約50
mgを0.1 mgのけたまではかりとり,全量フラスコ50 mLに入れ,JIS K 8101に規定するエタノー
ルを加えて溶かし,更に標線までエタノールを加えたもの。
2) りん酸/エタノール溶液 JIS K 9005に規定するりん酸10 gを全量フラスコ100 mLに入れ,標線
まで蒸留水を加えたものと,JIS K 8101に規定するエタノールとを体積比1:9で混合したもの。た
だし,AZNでは適用しないものとする。
3) 移動相 JIS K 8032に規定するアセトニトリル60 mLと蒸留水40 mLとを混合したもの。
c) 装置 装置は,高速液体クロマトグラフ分析装置を用いる。
d) 高速液体クロマトグラフ分析条件 高速液体クロマトグラフの分析条件は,表15による。
表15−高速液体クロマトグラフの分析条件
項目
条件
カラム
移動相
移動相流速
カラム温度
測定波長
注入量
ODS系カラム(I.D 4.6 mm,L 150 mm)
8.4.4.2 b) 3)による。
1.0 mL/min
40 ℃
221 nm
10 μL
e) 操作 CUAZ及びAZAACの場合,試験液20 mLを全量ピペットを用いてはかりとり,全量フラスコ
50 mLに入れ,りん酸/エタノール溶液を標線まで加え分析試験液とする。AZNの場合は,試験液を
分析試験液とする。分析試験液を孔径0.45 μmのフィルタを用いてろ過する。分析試験液の10 μLを
高速液体クロマトグラフ分析装置に注入し,シプロコナゾールのピーク面積を求め,検量線からシプ
ロコナゾールの濃度を求める。
f)
検量線の作成 シプロコナゾール溶液を,JIS K 8101に規定するエタノールを用いて段階的に10〜50
μg/mLになるように調製し,孔径0.45 μmのフィルタでろ過する。ろ液の10 μLを高速液体クロマト
グラフ分析装置に注入して検量線を3点以上作成する。検量線の作成は,その都度行う。
g) 計算 e)によって得たシプロコナゾールの量を各薬剤別に算出し,有効数字2けたに丸める。
1) CUAZ 質量は,式(21)による。
20
000
1
000
1
500
50
×
×
×
×
=
y
Y
································································· (21)
ここに,
Y: 8.3.3ではかりとった試料中のシプロコナゾールの質量(g)
18
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
y: 分析試験液中のシプロコナゾール濃度(mg/L)
2) AZAAC 質量は,式(22)による。
20
000
1
000
1
000
1
50
1
×
×
×
×
=
y
y
C
C
······························································· (22)
ここに, Cy1: 8.3.6ではかりとった試料中のシプロコナゾールの質量(g)
Cy: 分析試験液中のシプロコナゾールの濃度(mg/L)
3) AZN 質量は,式(23)による。
000
000
1
25
×
=c
Cy
········································································ (23)
ここに,
Cy: 8.3.10ではかりとった試料中のシプロコナゾールの質量(g)
c: 試験液中のシプロコナゾール濃度(mg/L)
8.4.5
ほう素化合物
8.4.5.1
ICP発光分光分析法
a) 要旨 試験液を誘導結合プラズマ中に噴霧し,ほう素による発光を波長249.773 nmで測定して,ほう
素を定量する。
b) 試薬 試薬は,次による。
ほう素標準液(Bとして1 mg/mL) JIS K 0102に準じて1 mg/mLの標準液を調製する。
c) 装置 装置は,ICP発光分光分析装置を用いる。
d) 操作 試料液4 mLを全量ピペットを用いてはかりとり,全量フラスコ1 000 mLに入れ,標線まで蒸
留水を加え,これを分析試験液とする。分析試験液のほう酸の含有量は,JIS K 0102の47.3(ICP発
光分光分析法)によって求める。
e) 検量線の作成 検量線の作成は,JIS K 0102の47.3 c) 3)による。
f)
計算 d)によって得たほう酸の量を式(24)によって算出し,有効数字2けたに丸める。
4
000
1
000
1/
718
.5
×
×
=b
B
··································································· (24)
ここに,
B: 8.3.4ではかりとった試料中のほう素化合物の質量(H3BO3と
して)(g)
b: 分析試験溶液中のほう素の濃度(mg/L)
5.718: BをH3BO3に換算する係数
8.4.6
シラフルオフェン
8.4.6.1
高速液体クロマトグラフ法
a) 要旨 高速液体クロマトグラフ法によってシラフルオフェンを定量する。
b) 試薬 試薬は,次による。
1) アセトニトリル JIS K 8032に規定するもの(高速液体クロマトグラフ用)。
2) シラフルオフェン標準液(100 mg/L) シラフルオフェン分析用標準品(純度97 %以上)10 mgを
全量フラスコ100 mLに入れ,標線までアセトニトリルを加えたもの。
3) メタノール JIS K 8891に規定するもの(高速液体クロマトグラフ用)。
4) 移動相 JIS K 8032に規定するアセトニトリル65 mL,JIS K 8891に規定するメタノール15 mL及
び蒸留水20 mLを混合したもの。
c) 装置 装置は,高速液体クロマトグラフ分析装置を用いる。
d) 高速液体クロマトグラフ分析条件 高速液体クロマトグラフの分析条件は,表16による。
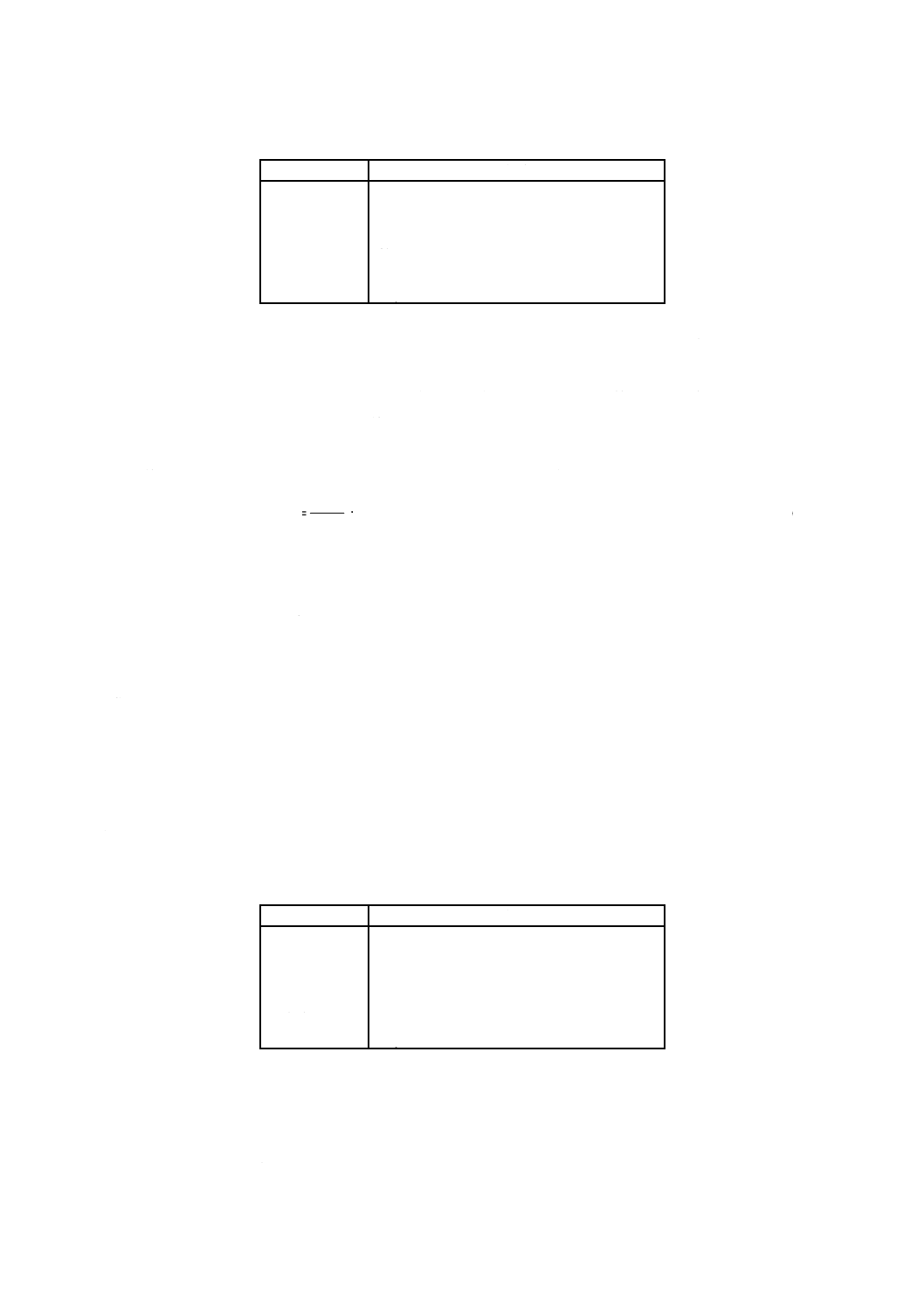
19
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表16−高速液体クロマトグラフの分析条件
項目
条件
カラム
移動相
移動相流速
カラム温度
測定波長
注入量
ODS系カラム(I.D 4.6 mm×L 150 mm)
8.4.6.1 b) 4)による。
2.0 mL/min
40 ℃
230 nm
10 μL
e) 操作 試験液の10 μLを高速液体クロマトグラフ装置に注入し,波長230 nmにおけるシラフルオフェ
ンのピーク面積を求める。検量線から試験液中シラフルオフェンの濃度を求める。
f)
検量線の作成 シラフルオフェン標準液(100 mg/L)5〜15 mLを段階的に正確に全量フラスコ20 mL
にはかりとり,アセトニトリルを標線まで加えたもの。この溶液を高速液体クロマトグラフで分析し,
シラフルオフェン濃度とピーク面積との関係から検量線を3点以上作成する。
g) 計算 e)によって得たシラフルオフェンの量を式(25)によって算出し,有効数字2けたに丸める。
000
1
R
S=
··············································································· (25)
ここに,
S: 8.3.5ではかりとった試料中のシラフルオフェンの質量(g)
R: 試験液中のシラフルオフェン濃度(mg/L)
8.4.7
エトフェンプロックス
8.4.7.1
高速液体クロマトグラフ法
a) 要旨 高速液体クロマトグラフによって,試験液中のエトフェンプロックスを定量する。
b) 試薬 試薬は,次による。
1) エトフェンプロックス溶液 エトフェンプロックス(純度90 %以上で純度の分かっているもの)約
0.1 gを0.1 mgのけたまではかりとり,全量フラスコ100 mLに入れ,JIS K 8891に規定するメタノ
ール50 mLに溶かし,標線までメタノールを加えたもの。
2) 移動相 高速液体クロマトグラフ用のアセトニトリルを用いる。
c) 装置 装置は,高速液体クロマトグラフ分析装置を用いる。
d) 高速液体クロマトグラフ分析条件 高速液体クロマトグラフの分析条件は,表17による。
表17−高速液体クロマトグラフの分析条件
項目
条件
カラム
移動相
移動相流速
カラム温度
測定波長
注入量
ODS系カラム(I.D 4.6 mm×L 150 mm)
8.4.7.1 b) 2)による。
1.0 mL/min
30 ℃
254 nm
10 μL
e) 操作 試験液5 mLを全量ピペットを用いてはかりとり,全量フラスコ25 mLに入れ,JIS K 8891に
規定するメタノールを標線まで加え分析試験溶液とする。これを孔径が0.45 μmのフィルタを用いて
ろ過し,ろ液の10 μLを高速液体クロマトグラフ分析装置に注入し,エトフェンプロックスのピーク
面積を求め,検量線からエトフェンプロックスの量を求める。
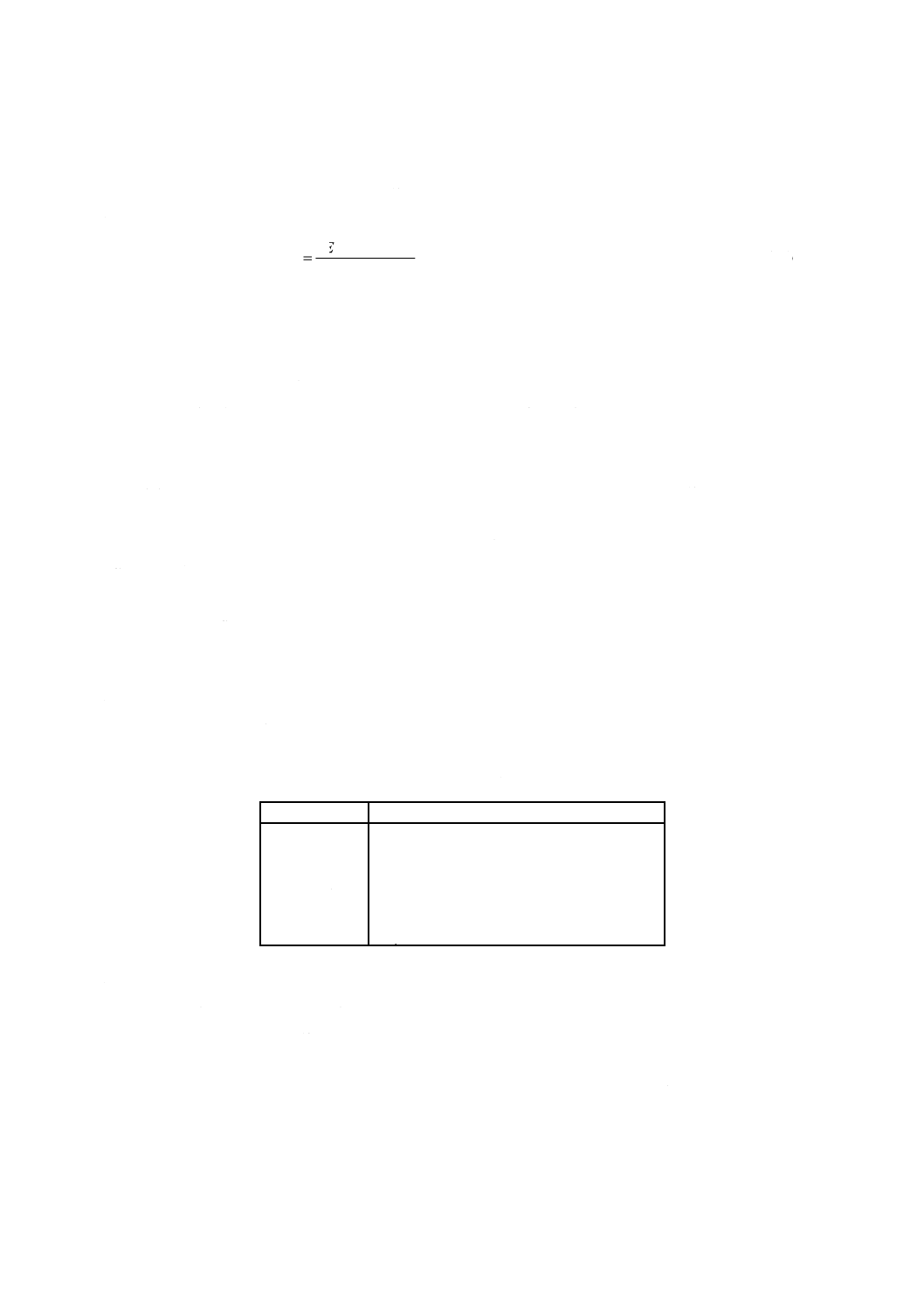
20
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
f)
検量線の作成 エトフェンプロックス溶液を,メタノールを用いて段階的に10〜100 μg/mLになるよ
うに調製し,孔径が0.45 μmのフィルタでろ過する。ろ液の10 μLを高速液体クロマトグラフ分析装
置に注入して,e)と同様に操作し検量線を3点以上作成する。
g) 計算 e)によって得たエトフェンプロックスの量を式(26)によって算出し,有効数字2けたに丸める。
5
000
1
000
1
000
1
25
1
×
×
×
×
=E
E
································································· (26)
ここに,
E1: 8.3.6ではかりとった試料中のエトフェンプロックスの質量
(g)
E: 分析試験液中のエトフェンプロックスの濃度(mg/L)
8.4.8
テブコナゾール
8.4.8.1
高速液体クロマトグラフ法
a) 要旨 高速液体クロマトグラフによって,試験液中のテブコナゾールを定量する。
b) 試薬 試薬は,次による。
1) テブコナゾール溶液 テブコナゾール標品(純度90 %以上で純度の分かっているもの)約0.1 gを
0.1 mgのけたまではかりとり,全量フラスコ100 mLに入れ,アセトニトリル約50 mLに溶かした
後,標線までアセトニトリルを加えたもの。
2) アセトニトリル JIS K 8032に規定するもの(高速液体クロマトグラフ用)。
3) りん酸溶液 JIS K 9005に規定するりん酸17 mLを全量フラスコ100 mLに入れ,標線まで蒸留水
を加えたもの。
4) 移動相 JIS K 9009に規定するりん酸二水素ナトリウム二水和物390 mgをはかりとり,全量フラス
コ500 mLに入れ,3)で調製したりん酸溶液1 mLを加え,標線まで蒸留水を加える。標線まで蒸留
水を加えたもの400 mLとアセトニトリル600 mLとを混合したもの。
c) 装置 装置は,高速液体クロマトグラフ分析装置を用いる。
d) 高速液体クロマトグラフ分析条件 高速液体クロマトグラフの分析条件は,表18による。
表18−高速液体クロマトグラフの分析条件
項目
条件
カラム
移動相
移動相流速
カラム温度
測定波長
注入量
ODS系カラム(I.D 4.6 mm×L 150 mm)
8.4.8.1 b) 4)による。
1.0 mL/min
40 ℃
220 nm
10 μL
e) 操作 試験液5 mLを全量ピペットではかりとり,全量フラスコ50 mLに入れ,移動相を標線まで加
えて分析試験液とする。分析試験液10 μLを高速液体クロマトグラフに注入し,テブコナゾールのピ
ーク面積を求める。検量線から分析試験溶液中のテブコナゾール濃度を求める。
f)
検量線の作成 テブコナゾール溶液を移動相で希釈し,約5〜30 mg/Lになるように段階的に希釈す
る。この溶液を同様に操作して,テブコナゾールの濃度とピーク面積との検量線を3点以上作成する。
検量線の作成は,その都度行う。
g) 計算 e)によって得たテブコナゾールの量を式(27)によって算出し,有効数字2けたに丸める。

21
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
000
000
1
50
5
0
1
×
×
=
T
T
···································································· (27)
ここに,
T1: 8.3.7ではかりとった試料中のテブコナゾールの質量(g)
T0: 検量線から求めた分析試験溶液中のテブコナゾールの濃度
(mg/L)
8.4.9
イミダクロプリド
8.4.9.1
高速液体クロマトグラフ法
a) 要旨 高速液体クロマトグラフによって,試験液中のイミダクロプリドを定量する。
b) 試薬 試薬は,次による。
1) 検量線用標準原液 イミダクロプリド標準品(純度90 %以上で純度の分かっているもの)約0.1 g
を0.1 mgのけたまではかりとり,AZNAの場合は,全量フラスコ100 mLに入れ,AZNの場合は,
全量フラスコ25 mLに入れ,JIS K 8101に規定するエタノールを標線まで加えたもの。
2) 移動相 アセトニトリル600 mLと蒸留水400 mLとを混合したもの。
c) 装置 装置は,高速液体クロマトグラフ分析装置を用いる。
d) 高速液体クロマトグラフ分析条件 高速液体クロマトグラフの分析条件は,表19による。
表19−高速液体クロマトグラフの分析条件
項目
条件
カラム
移動相
移動相流速
カラム温度
測定波長
注入量
ODS系カラム(I.D 4.6 mm×L 150 mm)
8.4.9.1 b) 2)による。
1.0 mL/min
40 ℃
271 nm
10 μL
e) 操作 AZNAの場合は,試験液15 mLを全量ピペットではかりとり,全量フラスコ50 mLに入れ,移
動相を標線まで加えて分析試験液とする。AZNの場合は,試験液をそのまま分析試験液とする。分析
試験液は,0.45 μmのフィルタでろ過し,ろ液の10 μLを高速液体クロマトグラフ分析装置に注入し,
イミダクロプリドのピーク面積を求め,検量線からイミダクロプリドの量を求める。
f)
検量線の作成 AZNAは,イミダクロプリド溶液をJIS K 8101に規定するエタノールで希釈し,約5
〜30 mg/Lになるように段階的に希釈する。AZNは,1〜5 mLを段階的に全量ピペットを用いてはか
りとり,全量フラスコ25 mLに入れ,JIS K 8101に規定するエタノールを標線まで加え希釈する。こ
の希釈溶液を0.45 μmのフィルタでろ過し,ろ液の10 μLを高速液体クロマトグラフ分析装置に注入
しイミダクロプリドの濃度とピーク面積との検量線を3点以上作成する。検量線の作成は,その都度
行う。
g) 計算 e)によって得たイミダクロプリドの量を各薬剤別に算出し,有効数字2けたに丸める。
1) AZNA 質量は,式(28)による。
000
000
1
50
15
0
1
×
×
=
I
I
···································································· (28)
ここに,
I1: 8.3.7ではかりとった試料中のイミダクロプリドの質量(g)
I0: 検量線から求めた分析試験溶液中のイミダクロプリドの濃度
(mg/L)
22
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) AZN 質量は,式(29)による。
000
000
1
25
0
1
×
=I
I
········································································· (29)
ここに,
I1: 8.3.10で採取した試料中のイミダクロプリドの質量(g)
I0: 試験液中のイミダクロプリド濃度(mg/L)
8.4.10 ナフテン酸銅(NCU-E)
8.4.10.1 原子吸光分析法
a) 要旨 試料中の有機物を酸で加熱分解した後,銅を原子吸光分析法によって定量する。
b) 試薬 試薬は,次による。
1) 硫酸 JIS K 8951に規定するもの。
2) 検量線用銅標準液(Cuとして0.1 mg/mL) JIS K 8660に規定する銅(試薬),JIS H 2121に規定す
る電気銅地金又はJIS K 8983に規定する硫酸銅(II)五水和物を適切な濃度のJIS K 8541に規定す
る硝酸に溶かし,最終の硝酸濃度を0.05〜0.15 mol/Lとした銅標準液(Cuとして1 000 mg/L)10 mL
を,全量ピペットを用いてはかりとり,全量フラスコ100 mLに入れ,蒸留水を標線まで加えたも
の。
3) 過酸化水素 JIS K 8230に規定するもの。
c) 装置 装置は,原子吸光分析装置を用いる。
d) 操作 試験液10 mLを,全量ピペットを用いてはかりとり,全量フラスコ50 mLに入れ,標線まで蒸
留水を加え,均一になるまで振り混ぜる。この中から1 mLを,全量ピペットを用いてはかりとり,
トラップ球付丸底フラスコ500 mLに入れて過酸化水素約1 mL及び硫酸4 mLを加え,砂浴上で徐々
に加熱溶解する。放冷後全量フラスコ100 mLに移し,トラップ球付丸底フラスコの内部を蒸留水で
よく洗い,洗液を全量フラスコに加える。蒸留水を標線まで加えて分析試験液とする。JIS K 0121の
8.(操作方法)によって,分析試験溶液をフレーム中に噴霧し,波長324.8 nmでの吸光度を読み取り,
検量線から分析試験液中の銅の濃度を求める。
e) 検量線の作成 検量線用銅標準液(Cuとして0.1 mg/mL)0〜4 mLを,全量ピペットを用いて段階的
にはかりとり,全量フラスコ100 mLに入れ,分析試験液と同じ条件になるように硫酸を加えた後,
蒸留水を標線まで加える。この溶液について同様に操作して,銅の量と吸光度との検量線を3点以上
作成する。検量線の作成は,その都度行う。
f)
計算 d)によって得た銅化合物の量を式(30)及び式(31)によって算出し,有効数字2けたに丸める。
50
0
×
=c
C
············································································· (30)
ここに,
C0: 8.3.8ではかりとった試料中のナフテン酸銅(Cuとして)の質
量(g)
c: 分析液中の銅濃度(Cuとして)(mg/mL)
100
0×
=S
C
C
··········································································· (31)
ここに,
C: 分析液中の銅濃度(Cuとして)(質量分率 %)
S: 試料の質量(g)
8.4.11 ナフテン酸亜鉛(NZN-E)及び第三級カルボン酸亜鉛(VZN-E)
8.4.11.1 原子吸光分析法
a) 要旨 試料中の有機物を酸で加熱分解した後,亜鉛を原子吸光分析法によって定量する。
b) 試薬 試薬は,次による。
23
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
1) 硫酸 JIS K 8951に規定するもの。
2) 検量線用亜鉛標準液(Znとして50 mg/L) JIS K 8012に規定する亜鉛(試薬)又はJIS K 8953に
規定する硫酸亜鉛七水和物(試薬)を適切な濃度のJIS K 8541に規定する硝酸に溶かし,最終の硝
酸濃度を0.05〜0.15 mol/Lとした亜鉛標準液(Znとして1 000 mg/L)5 mLを,全量ピペットを用い
てはかりとり,全量フラスコ100 mLに入れ,蒸留水を標線まで加えたもの。
3) 過酸化水素 JIS K 8230に規定するもの。
c) 装置 装置は,原子吸光分析装置を用いる。
d) 操作 操作は,次による。
1) ナフテン酸亜鉛 試験液10 mLを,全量ピペットを用いてはかりとり,全量フラスコ50 mLに入れ,
標線まで蒸留水を加え,均一になるまで振り混ぜる。この中から1 mLを,全量ピペットを用いて
はかりとり,トラップ球付丸底フラスコ500 mLに入れて過酸化水素約1 mL及び硫酸4 mLを加え,
砂浴上で徐々に加熱溶解する。放冷後全量フラスコ100 mLに移し,トラップ球付丸底フラスコの
内部を蒸留水でよく洗い,洗液を全量フラスコに加える。蒸留水を標線まで加えて分析試験液とす
る。JIS K 0121の8.(操作方法)によって,分析試験溶液をフレーム中に噴霧し,波長213.8 nmで
の吸光度を読み取り,検量線から分析試料液中の亜鉛の濃度を求める。
2) 第三級カルボン酸亜鉛 試料液10 mLを,全量ピペットを用いてはかりとり,全量フラスコ50 mL
に入れ,標線まで蒸留水を加え,均一に振り混ぜる。この中から,1 mL全量ピペットを用いてはか
りとり,トラップ球付丸底フラスコ500 mLに入れて過酸化水素約1 mL及び硫酸4 mLを加え,砂
浴上で徐々に加熱溶解する。放冷後全量フラスコ100 mLに移し,トラップ球付丸底フラスコの内
部をよく洗い,全量フラスコに合わせる。蒸留水を標線まで加えて,分析試験液とする。JIS K 0121
の6.(水銀専用原子吸光分析)によって,分析試験液をフレーム中に噴霧し,波長213.8 nmの指示
値を読む。検量線から分析試験溶液中の亜鉛の濃度を求める。
e) 検量線の作成 検量線用亜鉛標準液(Znとして50 mg/L)0〜4 mLを段階的に全量ピペットを用いて
はかりとり,全量フラスコ100 mLに入れ,分析試験液と同じ条件になるように硫酸を加えた後,蒸
留水を標線まで加える。その溶液について同様に操作して,亜鉛の量と指示値との検量線を3点以上
作成する。検量線の作成は,その都度行う。
f)
計算 d)によって得たナフテン酸亜鉛及び第三級カルボン酸亜鉛中の亜鉛の量を式(32)及び式(33)に
よって算出し,有効数字2けたに丸める。
05
.0
0
×
=z
Z
··········································································· (32)
ここに,
Z0: 8.3.8ではかりとった試料中のナフテン酸亜鉛及び第三級カル
ボン酸亜鉛(Znとして)の質量(g)
z: 分析液中の亜鉛濃度(Znとして)(mg/L)
100
0×
=S
Z
Z
··········································································· (33)
ここに,
Z: 分析液中の亜鉛濃度(Znとして)(質量分率 %)
S: 試料の質量(g)
8.4.12 ペルメトリン
8.4.12.1 ガスクロマトグラフ法
a) 要旨 ガスクロマトグラフ法によって試験液中のペルメトリンを定量する。
b) 試薬 試薬は,次による。
1) ペルメトリン標準品 定量用に精製された,純度90 %以上であらかじめ純度が分かっているもの。

24
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) アセトン JIS K 1503に規定するもの。
3) フタル酸ジノルマルオクチル 試薬として市販されているもの。
4) ペルメトリン溶液 ペルメトリン約0.25 gを0.1 mgのけたまではかりとり,全量フラスコ100 mL
に入れ,標線までアセトンを加え,ペルメトリン標準液とする。フタル酸ジノルマルオクチル約0.26
gを0.1 mgのけたまではかりとり,全量フラスコ100 mLに入れ,標線までアセトンを加えて,内
標準液とする。ペルメトリン標準液2 mL及び内標準液2 mL全量ピペットを用いてはかりとり,全
量フラスコ50 mLに入れ,標線までアセトンを加えたもの。
c) 装置 装置は,ガスクロマトグラフ分析装置を用いる。
d) ガスクロマトグラフ分析装置の条件 ガスクロマトグラフの条件は,表20による。
表20−ガスクロマトグラフの条件
項目
条件
固定相液体(保持量)
DEGS(Diethyleneglycol succinate)(2 %)
固定相担体
参考[Chromosorb W(HP)(149〜177メッシュ)]
カラム用管(内径及び長さmm×m)
(ガラス製3×1)
カラム温度(℃)
215
インジェクション温度(℃)
250
水素ガス圧力(kPa)
88.3
空気圧力(kPa)
49.0
窒素ガス流量(mL/min)
50
検出器
水素炎イオン化検出器(FID)
e) 操作 試料約1 gを0.1 mgのけたまではかりとり,全量フラスコ50 mLに入れ,内標準液2 mLを加
え,標線までアセトンを加えて分析試料とし,この2 μLをカラムに注入する。得たクロマトグラムは,
内標準物質,ペルメトリンの順に流出し,ペルメトリンは,二つのピークに分離する。この二つのピ
ークの面積を合計して,分析試験溶液中のペルメトリンのピーク面積を求める。同様の操作によって
求めたペルメトリン溶液のピーク面積との比を求め,ペルメトリン溶液中のペルメトリンの含有量か
ら分析試験液中のペルメトリンの量を求める。
f)
計算 e)によって得たペルメトリンの量を式(34)によって算出し,有効数字2けたに丸める。
Pn
P
St
P
×
=50
0
··········································································· (34)
ここに,
P0: 8.3.8ではかりとった試料中のペルメトリンの質量(g)
P: 分析試料溶液中のペルメトリンと内部標準物質とのクロマト
グラムピーク面積比
St: 標準ペルメトリンの質量(g)
Pn: 標準試験溶液中のペルメトリンと内部標準物質とのクロマト
グラムピーク面積比
8.4.13 ナフテン酸銅(NCU-O)
8.4.13.1 原子吸光分析法
a) 要旨 試料中の有機物を酸で加熱分解した後,銅を原子吸光分析法によって定量する。
b) 試薬 試薬は,次による。
1) 硫酸 JIS K 8951に規定するもの。
2) 硫酸(1+4) JIS K 8951に規定する硫酸を用いて調製したもの。
25
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
3) 硫酸(1+124) JIS K 8951に規定する硫酸を用いて調製したもの。
4) 検量線用銅標準液(Cuとして0.1 mg/mL) JIS K 8660に規定する銅(試薬),JIS H 2121に規定す
る電気銅地金又はJIS K 8983に規定する硫酸銅(II)五水和物を適切な濃度のJIS K 8541に規定す
る硝酸に溶かし,最終の硝酸濃度を0.05〜0.15 mol/Lとした銅標準液(Cuとして1 000 mg/L)10 mL
を,全量ピペットを用いてはかりとり,全量フラスコ100 mLに入れ,硫酸(1+4)を4 mL加え,
蒸留水を標線まで加えたもの。
c) 装置 装置は,原子吸光分析装置を用いる。
d) 操作 試験液4 mLを全量ピペットを用いてはかりとり,全量フラスコ100 mLに入れ,硫酸(1+124)
を標線まで加えて分析試験液とする。JIS K 0121の8.(操作方法)によって,分析試験溶液をフレー
ム中に噴霧し,波長324.8 nmでの吸光度を読み取り,検量線から分析試料液中の銅の濃度を求める。
e) 検量線の作成 検量線用銅標準液(Cuとして100 mg/L)0〜5 mLを段階的に全量ピペットを用いては
かりとり,全量フラスコ100 mLに入れ,分析試験液と同じ条件になるように硫酸(1+124)を標線
まで加える。この溶液について試験液同様に操作して銅の量と吸光度との検量線を3点以上作成する。
検量線の作成は,その都度行う。
f)
計算 d)によって得た銅の量を式(35)及び式(36)によって算出し,有効数字2けたに丸める。
5
012
.0
0
×
=c
C
········································································ (35)
ここに,
C0: 8.3.9ではかりとった試料中の銅の質量(Cuとして)(g)
c: 分析試験溶液中の銅濃度(Cuとして)(mg/L)
100
0×
=S
C
C
··········································································· (36)
ここに,
C: 試料中の銅の含有量(Cuとして)(質量分率 %)
C0: 8.3.9ではかりとった試料中の銅の質量(Cuとして)(g)
S: 試料の質量(g)
8.4.14 ナフテン酸亜鉛(NZN-O)
8.4.14.1 原子吸光分析法
a) 要旨 試料中の有機物を酸で加熱分解した後,亜鉛を原子吸光分析法によって定量する。
b) 試薬 試薬は,8.4.11.1 b)による。ただし,検量線用亜鉛標準液は,次による。
1) 検量線用亜鉛標準液(Znとして100 mg/L) JIS K 8012に規定する亜鉛(試薬),又はJIS K 8953
に規定する硫酸亜鉛七水和物(試薬)を適切な濃度のJIS K 8541に規定する硝酸に溶かし,最終の
硝酸濃度を0.05〜0.15 mol/Lとした亜鉛標準液(Znとして1 000 mg/L)10 mLを全量ピペットを用
いてはかりとり,全量フラスコ100 mLに入れ,硫酸(1+4)を4 mL加え,蒸留水を標線まで加え
たもの。
c) 装置 装置は,原子吸光分析装置を用いる。
d) 操作 試験液4 mLを全量ピペットを用いてはかりとり,全量フラスコ100 mLに入れ,硫酸(1+124)
を標線まで加えて分析試験液とする。JIS K 0121の8.(操作方法)によって,分析試験溶液をフレー
ム中に噴霧し,波長213.9 nmでの吸光度を読み取り,検量線から分析試験液中の亜鉛の濃度を求める。
e) 検量線の作成 検量線用亜鉛標準液(Znとして100 mg/L)0〜5 mLを,段階的に全量ピペットを用い
てはかりとり,全量フラスコ100 mLに入れ,分析試験液と同じ条件になるように硫酸(1+124)を
標線まで加える。この溶液について分析試験液と同様に操作して亜鉛の量と吸光度との検量線を3点
以上作成する。検量線の作成は,その都度行う。
26
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
f)
計算 d)によって得た亜鉛の量を式(37)及び式(38)によって算出し,有効数字2けたに丸める。
5
012
.0
0
×
=z
Z
········································································ (37)
ここに,
Z0: 8.3.9ではかりとった試料中の亜鉛の質量(Znとして)(g)
z: 検量線から求めた分析試験溶液中の亜鉛の濃度(Znとして)
(mg/L)
100
0×
=S
Z
Z
··········································································· (38)
ここに,
Z: 試料中の亜鉛の含有量(Znとして)(質量分率 %)
Z0: 8.3.9ではかりとった試料中の亜鉛の質量(Znとして)(g)
S: 試料の質量(g)
8.4.15 クレオソート油(A)
8.4.15.1 密度測定方法
8.4.15.1.1 I形浮ひょう
a) 要旨 I形浮ひょうによって試料の密度を測定する。
b) 器具 器具は,次による。
1) 浮ひょう JIS K 2249に規定する密度(15 ℃)で目盛られたソーダ石灰ガラス製浮ひょうで,I形
−A,番号9又は番号10のもの。
2) シリンダ 流出口付ガラス製シリンダで,内径約40 mm,長さ約350 mmのもの。
3) 温度計 棒状水銀温度計で,0〜100 ℃は1 ℃ごとに目盛が刻んであり,かつ,10 ℃ごとに目盛を
示す数字を記入したもので,形状は適切なもの。
4) 水槽 恒温槽又は内容積5 000 mL以上の適切なもの。
c) 操作 操作は,水槽に約40 ℃の蒸留水を入れ,約40 ℃に加熱した試料を清浄,乾燥したシリンダに
とり,水槽に入れ,静かに浮ひょう(密度計)を浮かべ,そのまま10分間静置する。このとき,水槽
の温度と試料の温度との差が5 ℃以上にならないようにする。静置後,密度計の示度を読む。密度計
の液外の首部に試料が必要以上に付着しないよう,十分に注意する。
密度計を取り去り,直ちに温度計を入れてかき混ぜた後,温度計の水銀柱先端がわずかに液面上に
現れるようにして,その示度を読み,これを測定時の試料の温度とする。試料の量は,密度計を入れ
たとき,その下端とシリンダ底との間が15 mm以上になるようにはかりとり,測定は35〜45 ℃で行
う。
d) 計算 c)によって得た密度(40 ℃)を式(39)によって算出し,有効数字2けたに丸める。
(
)
5
000
.0
40
7
000
.0
40
−
−
×
+
=
t
Dt
D
·············································· (39)
ここに,
D40: 密度(40 ℃)(g/cm3)
Dt: 測定密度(g/cm3)
t: 測定温度(℃)
0.000 7: 密度の温度換算係数(g/cm3)
0.000 5: I形浮ひょう自身の温度補正値(g/cm3)
8.4.15.1.2 比重浮ひょう
a) 要旨 比重浮ひょうによって試料の密度を測定する。
b) 器具 器具は,次による。
1) 比重浮ひょう JIS B 7525の附属書4(比重浮ひょう)に規定する大形19本組の番号6又は番号7
のもの。比重(15/4 ℃)によって目盛った軟質ガラス製の浮ひょうで,目盛範囲1.000〜1.100で,
0.001ごとに目盛が刻んであり,0.01ごとに目盛を示す数字を記入した,長さ300±20 mmのもの。
27
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
2) シリンダ 流出口付ガラス製シリンダで,内径約40 mm,長さ350 mmのもの。
3) 温度計 棒状水銀温度計で,0 ℃から100 ℃までは1 ℃ごとに目盛が刻んであり,かつ,10 ℃ご
とに目盛を示す数字を記入したもので,形状は適切なもの。
4) 水槽 恒温槽又は内容積5 000 mL以上の適切なもの。
c) 操作 8.4.15.1.1 c)による。
d) 計算 c)によって得た密度を式(40)によって算出し,有効数字2けたに丸める。
(
)
6
000
.0
40
7
000
.0
40
−
−
×
+
=
t
Dt
D
············································· (40)
ここに,
D40: 密度(40 ℃)(g/cm3)
Dt: 測定密度(g/cm3)
t: 測定温度(℃)
0.000 7: 密度の温度換算係数(g/cm3)
0.000 6: 比重浮ひょう自身の温度補正値(g/cm3)
8.4.15.2 水分定量方法
a) 要旨 水分定量用受器によって,試料中の水分量を測定する。
b) 試薬 試薬は,次による。
1) トルエン JIS K 2435-2又はJIS K 8680に規定するトルエンを,それぞれ脱水したもの。
2) キシレン JIS K 2435-3又はJIS K 8271に規定するキシレンを,それぞれ脱水したもの。
c) 器具 器具は,次による。
1) 水分定量用受器 容量5 mLで,0.05 mLごとに目盛が刻んであり,0.5 mLごとに目盛を示す数字を
記入したもので,図1に示すもの。
なお,水分定量用受器側管の最下部に,液面まで達する長さのらせん状の針金を付けて突沸を避
ける。
2) 丸底フラスコ 容量500 mLのもの。
d) 操作 試料100 mLを乾燥した清浄な丸底フラスコ500 mLにはかりとり,これに,トルエン又はキシ
レン100 mLを加え,振り動かして十分に混合させる。
水分定量用受器を,コルク栓などで丸底フラスコに連結した後,図2に示すように全装置を組み立
て,リービッヒ冷却器に冷却水を通じ,バーナー又は電熱器を用いて毎秒4〜5滴の割合で流出するよ
うに加熱を調節し,水分定量用受器中に集まった水分の量が恒量となったとき加熱を終了する。
なお,水分定量用受器には,あらかじめ0 mL目盛まで水を入れておき,試験を行う。
e) 測定 水分定量用受器中に集まった水分のmL数を読み,その測定値を試料の水分(体積分率 %)
とする。
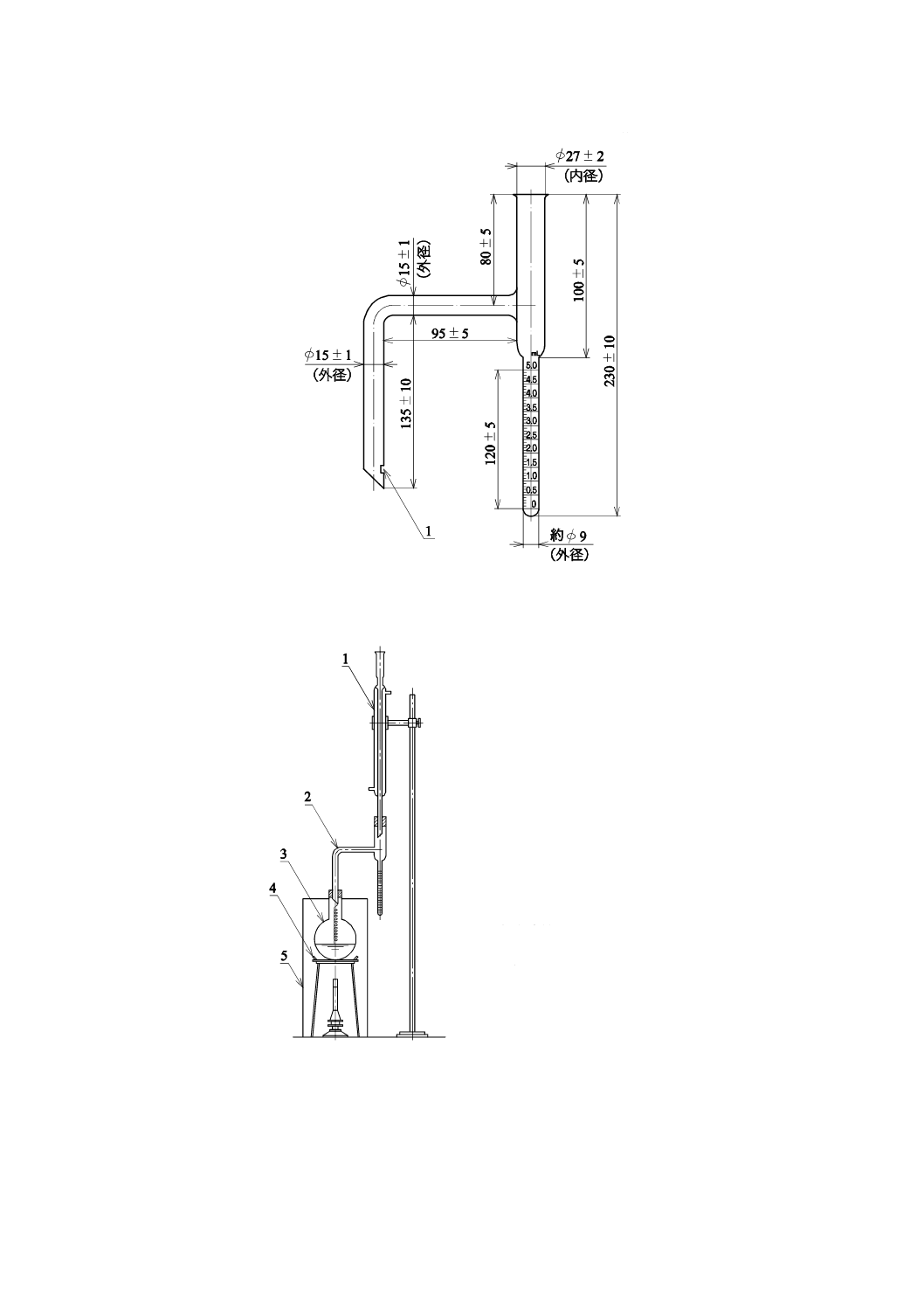
28
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
1
らせん状針金かけ穴
図1−水分定量用受器
1
リービッヒ冷却器
2
水分受器
3
丸底フラスコ500 mL
4
鉄製砂皿
5
風よけ
図2−水分定量装置の例
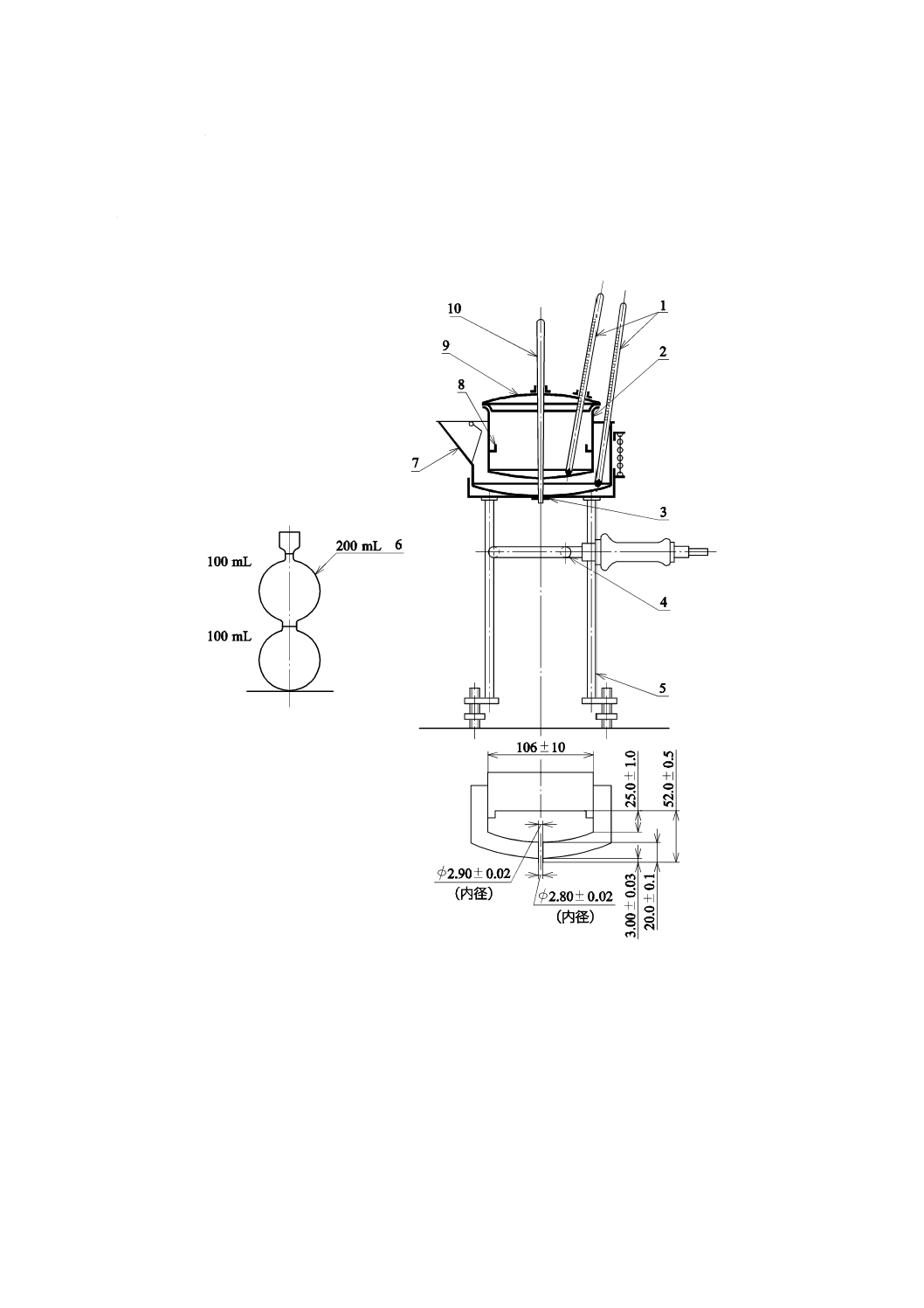
29
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.4.15.3 エングラー度測定方法
a) 要旨 エングラー計によって試料のエングラー度を測定する。
b) 器具 器具は,次による。
1) エングラー計 図3及び表21に示すもの。
単位 mm
1
温度計
2
黄銅製試料容器
3
白金製流出口
4
輪状ガスバーナ
5
三脚架
6
全量フラスコ
7
黄銅製容器
8
標針
9
黄銅製ふた
10 木栓
図3−エングラー計の例

30
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
表21−エングラー計の黄銅製試料容器の寸法
単位 mm
項目
寸法
内径
106±10
流出口の下端から標針までの高さ
52.0±0.5
円筒部分の下端から標針までの高さ
25.0±1.0
流出口の長さ
20.0±0.1
流出口の上端の内径
2.90±0.02
流出口の下端の内径
2.80±0.02
流出口の突出部分の長さ
3.00±0.03
2) 温度計 棒状水銀温度計で,0〜100 ℃は1 ℃ごとに目盛が刻んであり,かつ,10 ℃ごとに目盛を
示す数字を記入したもので,形状は適切なもの。
3) 全量フラスコ 容量200 mLで,図3に示すような適切なもの。
4) ストップウォッチ 適切なもの。
c) 操作 黄銅製試料容器の内部及び流出口を,溶剤(トルエンなど)及び蒸留水で順次洗い,木栓で流
出口をふさぎ,20 ℃の蒸留水を容器内標針の先端まで満たす。このとき,黄銅製容器を同温度の蒸留
水で満たしておく。次に,木栓をわずかに緩めて,蒸留水が流出口の末端から滴下しようとする状態
にとどめ,更に少量の蒸留水を加えて,容器内の水面を正確に標針に一致させる。
次に,全量フラスコ200 mLを流出口の直下に置き,木栓を抜き,全量フラスコの壁に触れないよ
うに蒸留水を流下させ,200 mLの流出秒数をストップウォッチで測定する。3回の測定結果の平均値
を水の流出時間とする。
次に,流出口及び容器をよく乾燥させた後,試料の温度を40±1 ℃とし,これを容器中に標針まで
入れ,黄銅製容器中には41〜42 ℃の蒸留水を入れた後,試料の温度を正確に40 ℃になったときとす
る。全量フラスコ200 mLを用いて,蒸留水の場合と同様の方法で試料200 mLの流出秒数を1回測定
する。
d) 計算 c)によって得たエングラー度を式(41)によって算出し,有効数字2けたに丸める。
2
1
T
T
E=
·················································································· (41)
ここに,
E: エングラー度(40/20 ℃)
T1: 試料の流出秒数(s)
T2: 蒸留水の流出秒数(s)
8.4.15.4 蒸留試験方法
a) 要旨 蒸留試験によって,流出物の質量を測定する。
b) 器具 器具は,次による。
1) 蒸留フラスコ 容量250 mLで,フラスコの首には保温材を巻いてもよい。図4に蒸留フラスコの
例を示す。
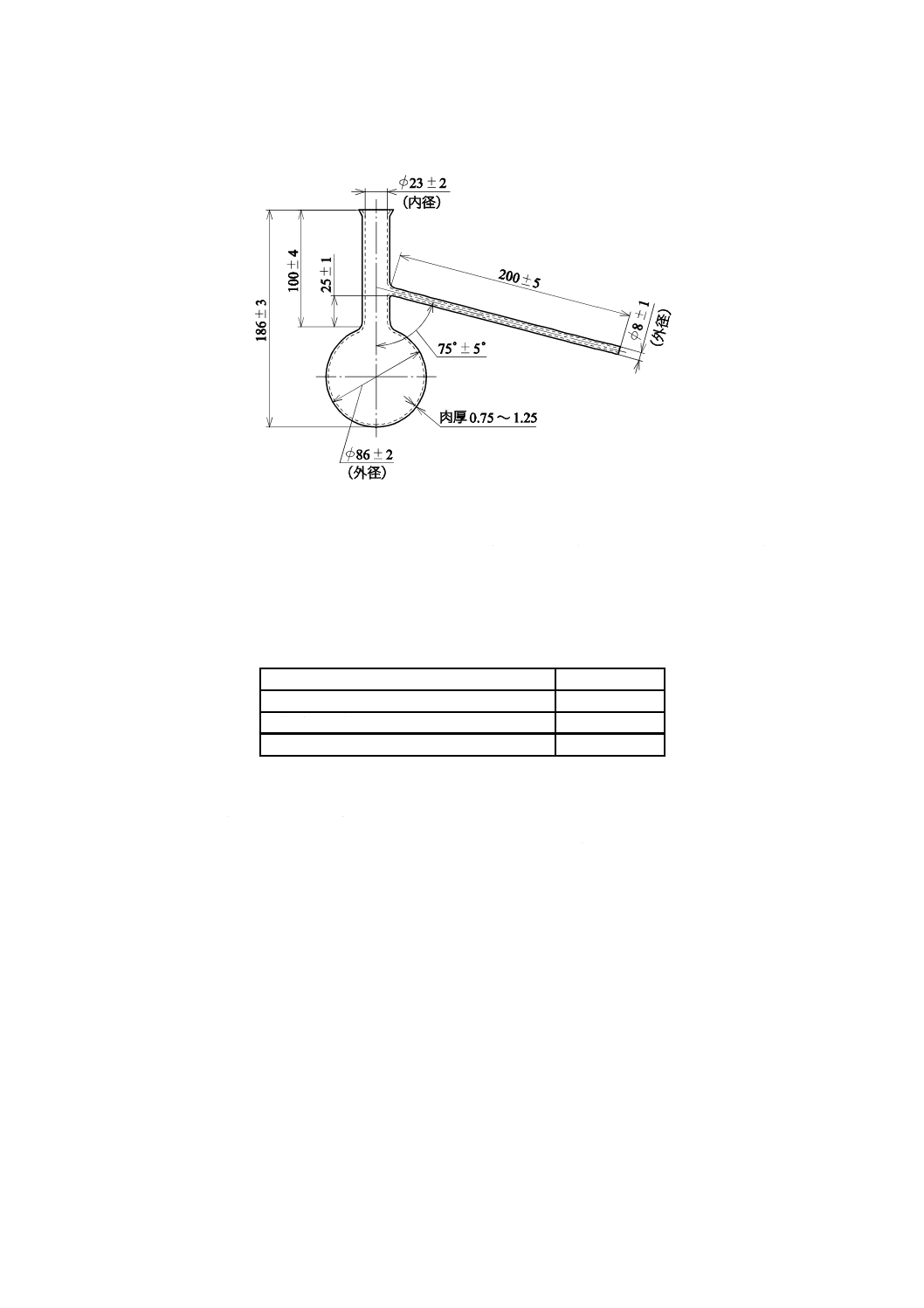
31
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図4−蒸留フラスコの例
2) 温度計 棒状水銀温度計で,100〜360 ℃は1 ℃ごとに目盛が刻んであり,かつ,10 ℃ごとに目盛
を示す数字を記入したもので,表22に示すもの。
表22−温度計の寸法
単位 mm
項目
寸法
幹の外径
5〜 7
水銀球の長さ
10〜 20
水銀球の下端から360 ℃目盛までの距離
280〜350
3) 冷却器 図5に示すもの。
4) 受器(A) 全長約260 mm,容量100 mLのメスシリンダで,0〜7 mLまでは0.2 mLごとに,7〜100
mLまでは1 mLごとに目盛が刻んであり,かつ,目盛を示す数字を0〜10 mLまでは1 mLごとに,
10〜100 mLまでは10 mLごとに記入したもので,図6に示すもの。
32
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
単位 mm
図5−冷却器
図6−受器(A)
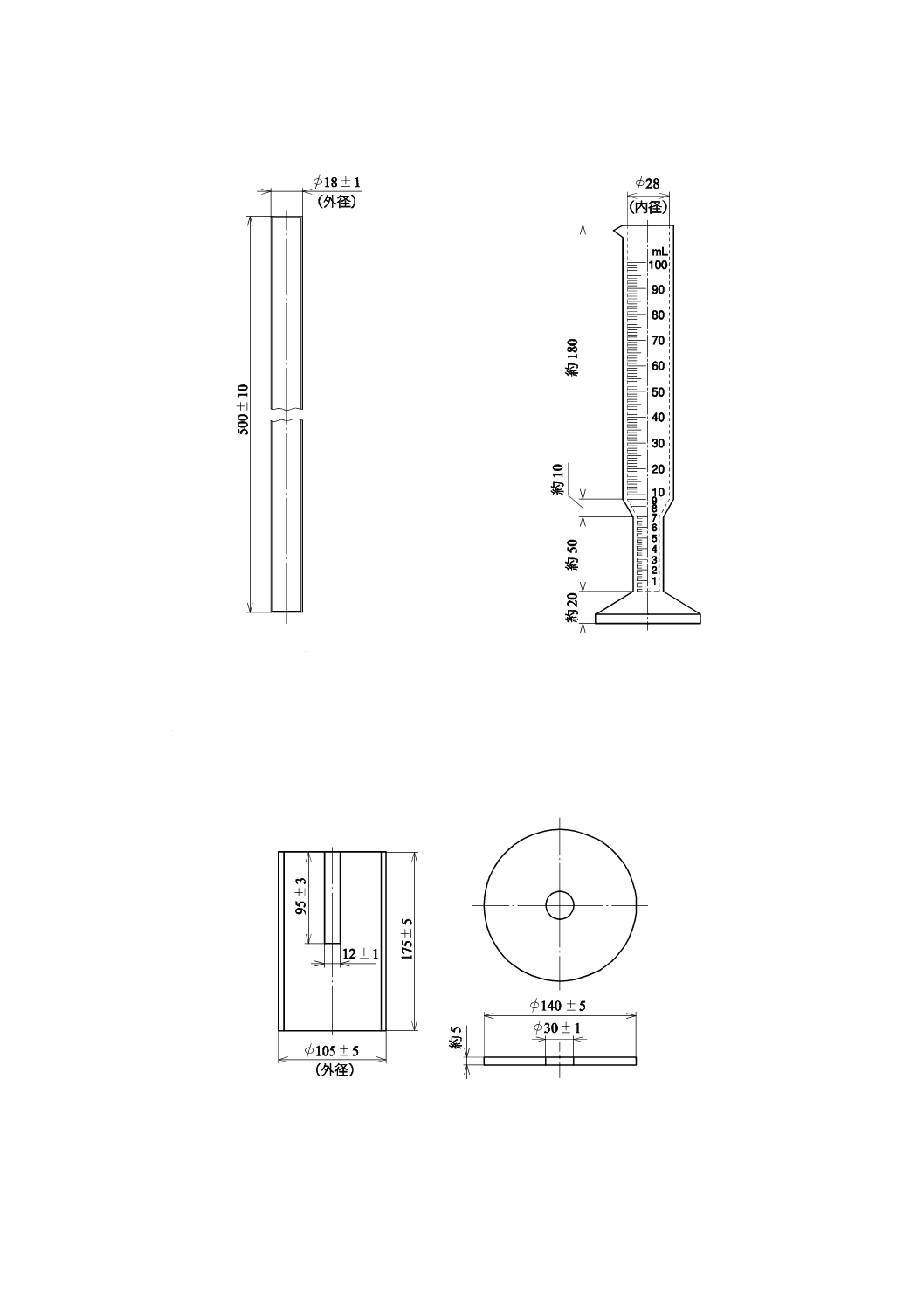
5) 受器(B) 内径約28 mm,全長約220 mm,容量100 mLのメスシリンダで,1 mLごとに目盛を刻
んだもの。
6) 風よけ及び風よけのふた 図7に例を示す。
単位 mm
a) 風よけ
b) 風よけのふた
図7−風よけ及び風よけのふたの例
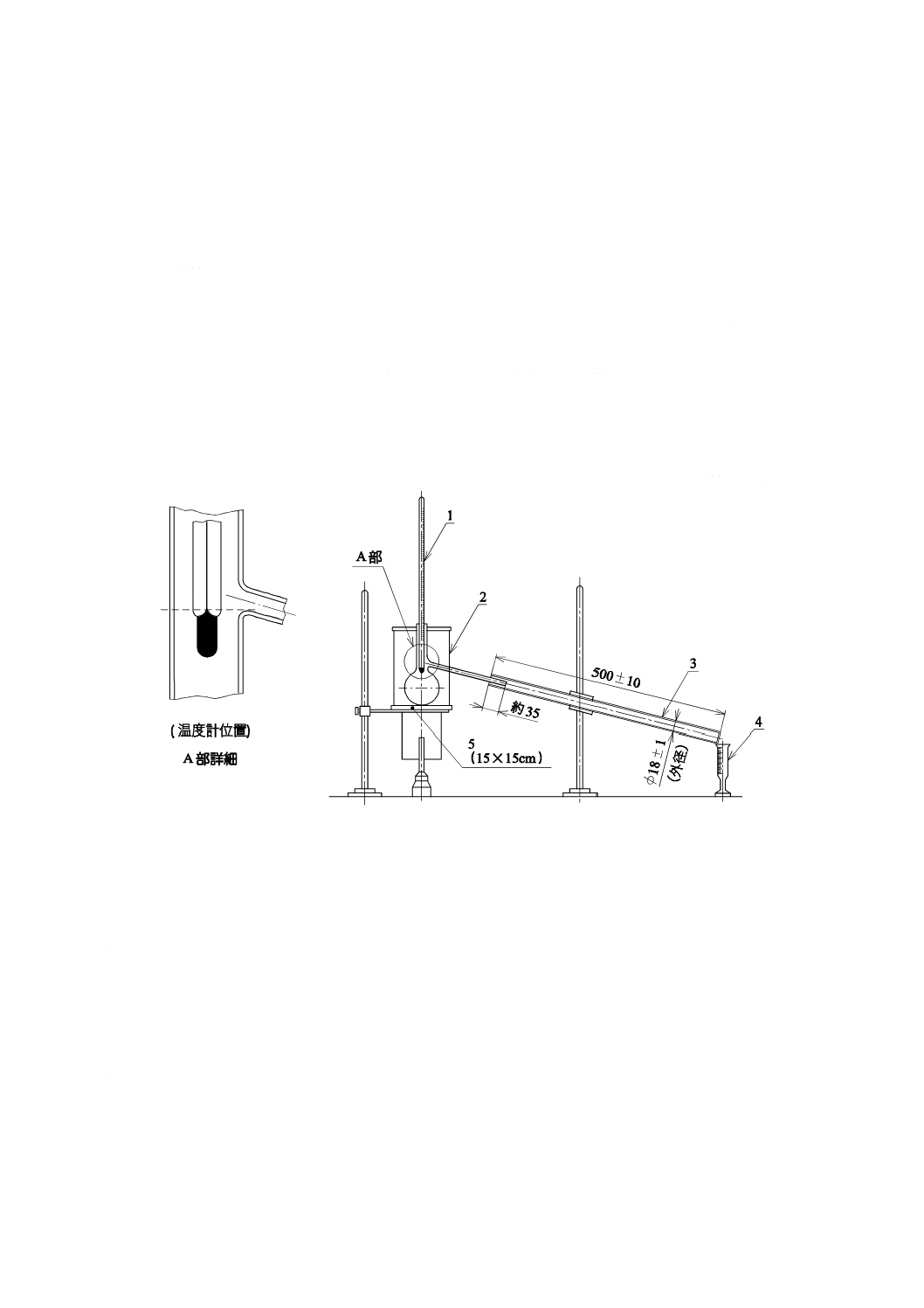
33
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
c) 操作 操作は,次による。
1) 脱水試料(試料中の水分が体積分率0.5 %以下の場合は,そのまま試験に供してもよい。)100 mL
を乾燥した清浄な蒸留フラスコにはかりとり,コルク栓などで温度計を取り付ける。水銀球の上端
が蒸留フラスコの枝管の下部付根に位置するように温度計を取り付けた後,適切な加熱装置に据え
る。枝管は約35 mmだけ冷却器内にはめ込んで連結し,冷却器の末端には受器(A)を置き,全装
置を図8に示すように組み立てる。
留出を始めた後は,毎分60滴の割合で冷却器から受器に留出するように加熱を調節する。蒸留中,
冷却器内に結晶するものがあるときは,適宜に加熱してこれを流下させる。
上記の操作によって,235 ℃に達したときに受器(B)と取り替える。255 ℃,270 ℃,315 ℃
それぞれ達したときに,直ちに加熱をやめ,冷却器内に残留する流出物を受器(B)に流下させ,
ためる。
単位 mm
1
温度計
2
風よけ
3
冷却器(空気)
4
受器
5
金網板2枚
図8−蒸留装置の例
d) 測定 受器(A)及び(B)内の留出物の質量をはかる。
なお,受器内の留出物の液面に結晶が析出している場合は,受器を加熱し,結晶を溶解してから留
出物の質量をはかる。
8.4.15.5 235〜315 ℃留分の密度測定方法
8.4.15.5.1 比重瓶法
a) 要旨 比重瓶によって密度を測定する。
b) 器具 器具は,次による。
比重瓶 図9に示すもの。

34
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
単位 mm
図9−比重瓶
c) 操作 乾燥した清浄な比重瓶の質量を,栓をした状態ではかる。次に,この比重瓶に新しく蒸留した
40 ℃の水をあふれるまで満たし,あらかじめ40 ℃に保ってある水槽内に30分間以上置いた後取り
出す。栓の毛管内の水を紙片などを用いて速やかに吸い取り,液の位置を毛管の標線に一致させる。
外部に付着した水滴を乾燥した布でぬぐい取った後,質量をはかる。
この比重瓶内の水を取り去り,乾燥した後,40 ℃の試料を入れて同様の操作を行う。最後に比重瓶
の外部に付着した水滴,油などを完全にぬぐい取った後,その質量をはかる。
d) 計算 c)によって得た,試料の密度(40 ℃)を式(42)によって算出し,有効数字2けたに丸める。
1
2
1
3
40
2
992
.0
M
M
M
M
D
−
−
×
=
···························································· (42)
ここに,
D40: 密度(40 ℃)(g/cm3)
M1: 乾燥後の比重瓶の質量(g)
M2: (比重瓶+水)の質量(g)
M3: (比重瓶+試料)の質量(g)
0.992 2: 水の密度(40 ℃)(g/cm3)
8.4.15.5.2 振動式密度計による密度測定方法
振動式密度計による235〜315 ℃留分の密度測定は,JIS K 0061の7.3(振動式密度計法)による。
8.4.15.6 トルエン不溶分定量方法
a) 要旨 トルエン不溶分を測定する。
b) 試薬 試薬は,次による。
1) トルエン JIS K 2435-2又はJIS K 8680に規定するトルエン。
2) アセトン JIS K 1503又はJIS K 8034に規定するアセトン。
c) 器具 器具は,次による。
1) 丸底フラスコ 容量125 mLのもの。
2) ガラスろ過器 JIS R 3503に規定するるつぼ形ガラスろ過器1G4。
d) 操作 試料約10 gを丸底フラスコ125 mLに1 mgまではかりとり,これにあらかじめ50〜60 ℃に加

35
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
熱したトルエン約50 mLを加え,十分にかき混ぜた後,還流冷却器を付けて,液が沸騰するまで加熱
する。
加熱後,直ちにろ過器を用いて吸引ろ過する。この間,常にろ床が露出しないように液を注意しな
がら加える。
次に,洗液が無色になるまでろ過ケーキ及びろ過器を加熱したトルエンで洗浄する。さらに,アセ
トン約5 mLずつでろ過ケーキを4回洗浄した後,ろ過器を約110 ℃で60分間乾焼し,デシケータ中
で放冷した後,質量をはかる。
e) 計算 d)によって得た値をもとに不溶分を式(43)によって算出し,有効数字2けたに丸める。
(
)
(
)
3
2
1
100
100
100
M
S
M
M
U
−
×
×
−
=
·················································· (43)
ここに,
U: トルエン不溶分(質量分率 %)
M1: (ろ過器+ろ過ケーキ)の質量(g)
M2: ろ過器の質量(g)
M3: 8.4.15.2によって求めた試料の水分(体積分率 %)
S: 試料の質量(g)
8.4.15.7 流動性試験方法
a) 要旨 下記操作によって,試料の流動性を測定する。
b) 試薬 試薬は,次による。
塩化カルシウム JIS K 8125に規定するもの。
c) 器具 器具は,次による。
1) 三角フラスコ 容量100 mL及び300 mLのもの。
2) ろ過器 JIS R 3503に規定するブフナー漏斗形ガラスろ過器3G3。
3) 棒状水銀温度計 目盛範囲0〜100 ℃で,1 ℃ごとに目盛を刻んだもの。
4) 浸線付二重壁水銀温度計 刻度面は乳白板とし,表23に示すもの。
表23−浸線付二重壁水銀温度計の仕様
項目
仕様
温度範囲
15〜45 ℃
液体
水銀
液上に満たす気体
窒素
最小目盛
0.1 ℃
全長
330〜350 mm
幹の外径
6〜7 mm
大水銀球の外径
5〜6 mm
大水銀球の長さ
10〜15 mm
大水銀球の下端から最低刻度線までの距離
100〜105 mm
温度計の上端から最高刻度線までの距離
20〜30 mm
大水銀球の下端から浸線までの距離
100 mm
許容差
±0.1 ℃
5) 恒温水槽 32.0±0.5 ℃に調節できるもの。
6) 乾燥器 約80 ℃に調節できる恒温乾燥器。
7) 水浴 三角フラスコ300 mLが浴中で十分に振り動かせる形状のもの。
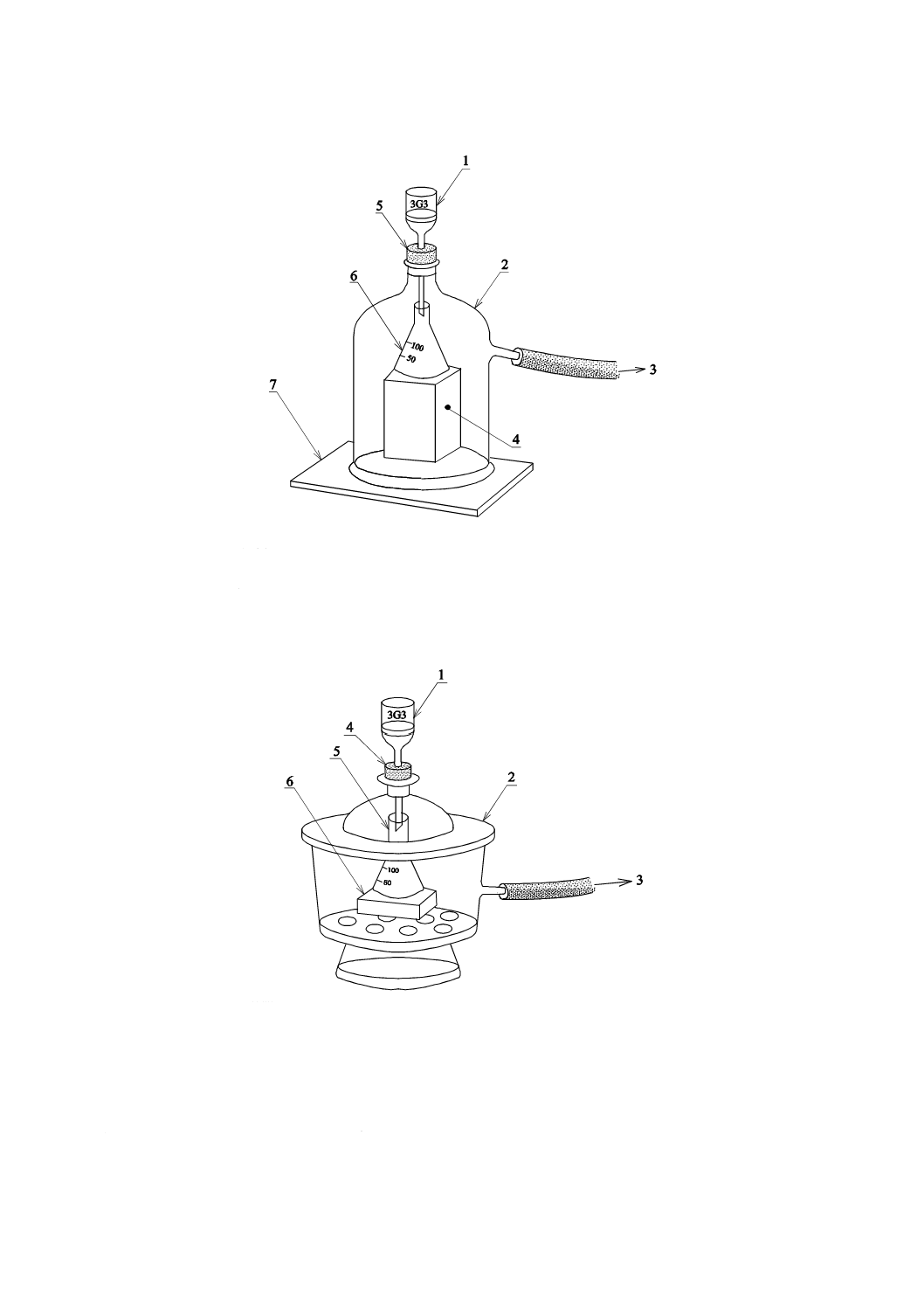
36
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8) 吸引ろ過装置 三角フラスコ100 mLの入るもので,図10又は図11に示すもの。
1
ろ過器
2
吸引瓶
3
アスピレータへ接続
4
台
5
コルク栓
6
三角フラスコ
7
ガラス板
図10−釣鐘状吸引瓶による吸引ろ過装置の例
1
ろ過器
2
真空デシケータ
3
アスピレータへ接続
4
コルク栓
5
三角フラスコ
6
台
図11−真空デシケータによる吸引ろ過装置の例
d) 操作 均一な試料約100 mLを三角フラスコ300 mLにはかりとり,フラスコの首部にコルク栓などを
用いて棒状水銀温度計を取り付ける。フラスコを水浴に浸し,振り動かしながら試料を60〜80 ℃に
37
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
加熱する。乾燥剤約30 gを加えてよく振り混ぜた後,試料を10分間,60〜80 ℃に保つ。
次に,あらかじめ約80 ℃に調節した乾燥器内に5分間以上入れておいたろ過器を取り出し,速や
かにろ過装置を組み立て,試料を傾斜法によってろ過する。ろ液約50 mLが得られたときろ過をやめ,
このろ液を流動性試験試料とする。
流動性試験試料約50 mLを三角フラスコ100 mLにはかりとり,フラスコの首部にコルク栓などを
用いて二重壁水銀温度計を取り付ける。その際,温度計の大水銀球が試料中に浸るようにする。フラ
スコを水浴に浸し,試料を35〜40 ℃に加熱して試料中に固形物のないことを確かめた後,32.0±
0.5 ℃に調節した恒温水槽中にフラスコを入れ,試料の液面が恒温水槽中の水面より下になるように
する。
e) 測定 試料温度が32.0±0.5 ℃になってから,同温度で2時間保持した後,フラスコを取り出し,直
ちに試料の液面に固形物が生成しているか否かを調べる。さらに,フラスコを約45°に傾けてゆっく
り回転させ,フラスコ内壁に遊離した固形物が生成していないことを確認する。
8.4.15.8 ジベンゾ[a, h]アントラセンの試験方法
a) 要旨 ガスクロマトグラフ法によって試験液中のジベンゾ[a, h]アントラセンを定量する。
b) 試薬 試薬は,次による。
1) ジクロロメタン JIS K 8117に規定するジクロロメタンを用いる。
2) ジベンゾ[a, h]アントラセン標準液 ジベンゾ[a, h]アントラセン0.010 gを正確にはかりとり,ジク
ロロメタンを加えて溶かし,正確に100 mLとする。その1 mLをとり,ジクロロメタンを加えて正
確に100 mLとしたものをジベンゾ[a, h]アントラセン標準液とする。
3) 内部標準液 内部標準物質として,そのモニターイオンが対象物質に含有される他の多環芳香族炭
化水素等のフラグメントイオンとクロマトグラム上で重複しないようなものを選択する。アセナフ
テン−d10,フェナントレン−d10,クリセン−d12などを用いることができる。その内部標準物質
0.010 gを正確にはかりとり,ジクロロメタンを加えて溶かし,正確に100 mLとする。その5〜20 mL
をとり,ジクロロメタンを加えて正確に100 mLとしたものを内部標準液とする。
4) 高純度ヘリウム 純度99.999 %以上のものを用いる。
c) 装置 装置は,次による。
1) 分析装置 ガスクロマトグラフ質量分析計を用いる。
2) カラム管 内径0.25 mm,長さ30 m,膜厚0.25 μmの5 %フェニルメチルポリシロキサンを液相と
するキャピラリカラムを用いる。
d) ガスクロマトグラフの分析装置の条件
1) カラム温度 60 ℃で2分間保持し,その後毎分25 ℃で昇温し,300 ℃に到達後6分間保持する。
2) 試験溶液注入口温度 280 ℃とする。
3) キャリヤガス 高純度ヘリウムを用いる。ジベンゾ[a, h]アントラセンが約15〜16分で流出する流
速に調整する。
4) 注入方法 スプリットレス方式とする。
5) モニターイオン 通常“ジベンゾ[a, h]アントラセン278”を選択すべきであるが,使用する装置,
カラム等によって,対象とする物質に特異性が高く,かつ,イオン強度が高いフラグメントイオン
を適切に選択することが望ましい。
e) 操作 操作は,次による。
1) 試験溶液の調製 試料約0.5 gを正確にはかりとり,これをシリカゲルを充てんしたミニカートリッ
38
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ジカラムに流し込み(カラムへの負荷量を軽減する必要がある試料の場合,ヘキサン/アセトニト
リル分配による精製を行う。),50 mLのなす形フラスコにとる。
さらに,そのミニカートリッジカラムにジクロロメタン10 mLを流し込み,前述のなす形フラス
コに加える。その液について,ロータリエバポレータを用いて50 ℃で約2 mLになるまでジクロロ
メタンを除去し,これを全量フラスコに移し,ジクロロメタンを加えて全量を正確に5 mLとした
ものを試験溶液とする。
2) 試験溶液及びジベンゾ[a, h]アントラセン標準液2 mLをそれぞれ正確に試験管にとり,内部標準液
0.5 mLを加え,それぞれの試験管から1 μLをとり,次の操作条件で試験を行う。
試験溶液を測定し,得たクロマトグラフ上で,標準液のジベンゾ[a, h]アントラセンのモニターイ
オンのピークと保持時間とが一致するピークが存在する場合は,ジベンゾ[a, h]アントラセンに相当
するピーク面積の内部標準物質のピーク面積に対する比(Rt)を求める。同時に,標準液において
得たクロマトグラム上でのジベンゾ[a, h]アントラセンのピーク面積の内部標準物質のピーク面積
に対する比(Rs)を求める。このとき,式(44)によって計算する試料1 gについてのジベンゾ[a, h]
アントラセンの量は,10 μg以下でなければならない。
f)
検量線の作成 マトリックス法の定量に使用する検量線を作成する。
1) 試験溶液及びマトリックス調整用溶液の調製 試料及びジベンゾ[a, h]アントラセンを含まないク
レオソート油約0.5 gを精ひょう(秤)し,各々にn-ヘキサン5 mLを加え,50 mLの分液漏斗に移
す。これにn-ヘキサン飽和アセトニトリル10 mLを加え,5分間振とうし,静置後,下層を200 mL
のなす形フラスコにとる。この操作を3回繰り返し,アセトニトリル層を合わせ,40 ℃以下,減圧
下で約1 mLまでアセトニトリルを除去した後,窒素ガスを噴きつけ乾固する。これらの残留物を
ジクロロメタン5 mLでシリカゲルミニカートリッジカラムに流し込み,流出液を目盛付試験管に
とる。そのカートリッジカラムにジクロロメタン10 mLを加えて,前述の目盛付試験管に合わせる。
この液にジクロロメタンを加えて全量を正確に15 mLとしたものを試験溶液及びマトリックス調整
用溶液とする。
試験溶液1.5 mLを正確に試験管にとり,内部標準液0.1 mL及びジクロロメタン1.5 mLを加える。
同様に,検量線用標準液1.5 mLを正確に試験管にとり,内部標準液0.1 mL及びマトリックス調整
用溶液1.5 mLを加える。
2) ジベンゾ[a, h]アントラセン標準液の調製 ジベンゾ[a, h]アントラセン0.010 gを精ひょうし,ジク
ロロメタン100.0 mLに溶解する(100 μg/mL)。その1.0 mLをとり,ジクロロメタンを加えて100.0
mLとしたもの(1 μg/mL)を標準液とする。
3) 検量線用標準液の調製 ジベンゾ[a, h]アントラセン標準液0 mL,0.5 mL,1.5 mL,2.5 mL及び5.0 mL
(試料を0.5 gとった場合,試料1 g当たり0 μg,1 μg,3 μg,5 μg及び10 μgの濃度に相当する。)
をとり,ジクロロメタンを加えて15.0 mLとする。
4) 内部標準液の調製 内部標準物質としては,b) 3)に記載のアセナフテン−d10,フェナントレン−d10,
クリセン−d12等0.010 gをジクロロメタン100 mLに溶解する(100 μg/mL)。その2 mLをとり,ジ
クロロメタンを加えて20 mLとしたもの(10 μg/mL)を内部標準液とする。選定に当たっては,そ
のモニターイオンが試料中の成分とクロマトグラム上で重複しないようなものを選択する。
g) 計算 e)によって得た,試料1 g中のジベンゾ[a, h]アントラセンの量を式(44)によって算出し,有効数
字2けたに丸める。
39
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
A
Rs
Rt
K
B
1
5×
×
×
=
···································································· (44)
ここに,
B: ジベンゾ[a, h]アントラセンの含有量(μg)
K: 標準液の濃度(μg/mL)
Rt: 試験溶液ピーク面積/内部標準物質のピーク面積
Rs: 標準液ピーク面積/内部標準物質のピーク面積
A: 試料採取量(g)
8.4.15.9 ベンゾ[a]アントラセンの試験方法
ベンゾ[a]アントラセンの試験方法は,8.4.15.8に規定する試験方法に適合しなければならない。ただし,
8.4.15.8において,“ジベンゾ[a, h]アントラセン”とあるのは,“ベンゾ[a]アントラセン”へ,“278”とあ
るのは“228”へ,“約15〜16分”とあるのは“約11〜12分”へと読み替えるものとする。
8.4.15.10
ベンゾ[a]ピレンの試験方法
ベンゾ[a]ピレンの試験方法は8.4.15.8に規定する試験方法に適合しなければならない。ただし,8.4.15.8
において,“ジベンゾ[a, h]アントラセン”とあるのは,“ベンゾ[a]ピレン”へ,“278”とあるのは“252”
へ,“約15〜16分”とあるのは“約13〜14分”へと読み替えるものとする。
8.5
有効成分の配合比及び合計含有量
木材保存剤の中で,有効成分の配合比及び合計含有量を求める必要のあるものは,それぞれの薬剤に応
じて算出する。
8.5.1
ACQ-1
有効成分の配合比及び合計含有量は,8.4.2.1又は8.4.2.2及び8.4.3.1の結果から式(45),式(46)及び式(47)
によって算出する。
a) 有効成分の配合比
100
0
×
+
=
B
C
C
C
······································································ (45)
100
0
×
+
=
B
C
B
B
······································································ (46)
ここに,
C0: 試料の銅化合物の配合比(CuOとして)(質量分率 %)
B0: 試料のBKCの配合比(BKCとして)(質量分率 %)
C: 8.3.2ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
B: 8.3.2ではかりとった試料中のBKCの質量(g)
b) 有効成分の合計含有量
S
B
C
T
+
=
·············································································· (47)
ここに,
T: 有効成分の含有量(質量分率 %)
S: 8.3.2ではかりとった試料の質量(g)
C: 8.3.2ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
B: 8.3.2ではかりとった試料中のBKCの質量(g)
8.5.2
ACQ-2
有効成分の配合比及び合計含有量は,8.4.2.1又は8.4.2.2及び8.4.1.1の結果から式(48),式(49)及び式(50)
によって算出する。
a) 有効成分の配合比
40
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
0
×
+
=
D
C
C
C
······································································ (48)
100
0
×
+
=
D
C
D
D
······································································ (49)
ここに,
C0: 試料の銅化合物の配合比(CuOとして)(質量分率 %)
D0: 試料のDDACの配合比(DDACとして)(質量分率 %)
C: 8.3.2ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
D: 8.3.2ではかりとった試料中のDDACの質量(g)
b) 有効成分の合計含有量
100
×
+
=
S
D
C
T
······································································· (50)
ここに,
T: 有効成分の含有量(質量分率 %)
S: 8.3.2ではかりとった試料の質量(g)
C: 8.3.2ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
D: 8.3.2ではかりとった試料中のDDACの質量(g)
8.5.3
CUAZ
有効成分の配合比及び合計含有量は,8.4.2.1又は8.4.2.2及び8.4.4.1又は8.4.4.2の結果から式(51),式(52)
及び式(53)によって算出する。
a) 有効成分の配合比
100
0
×
+
=
Y
C
C
C
······································································ (51)
100
0
×
+
=
Y
C
Y
Y
······································································· (52)
ここに,
C0: 試料の銅化合物の配合比(CuOとして)
Y0: 試料のシプロコナゾールの配合比
C: 8.3.3ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
Y: 8.3.3ではかりとった試料中のシプロコナゾールの質量(g)
b) 有効成分の合計含有量
100
×
+
=
S
Y
C
A
······································································· (53)
ここに,
A: 有効成分の含有量(質量分率 %)
S: 8.3.3ではかりとった試料の質量(g)
C: 8.3.3ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
Y: 8.3.3ではかりとった試料中のシプロコナゾールの質量(g)
8.5.4
BAAC
有効成分の配合比及び合計含有量は,8.4.5.1及び8.4.1.1の結果から式(54),式(55)及び式(56)によって算
出する。
a) 有効成分の配合比
100
0
×
+
=
D
B
B
B
······································································ (54)
41
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
0
×
+
=
D
B
D
D
······································································ (55)
ここに,
B0: 試料のほう素化合物の配合比
D0: 試料のDDACの配合比
B: 8.3.4ではかりとった試料中のほう素化合物の質量(H3BO3と
して)(g)
D: 8.3.4ではかりとった試料中のDDACの質量(g)
b) 有効成分の合計含有量
100
×
+
=
S
D
B
A
······································································· (56)
ここに,
A: 試料の有効成分の含有量(質量分率 %)
S: 8.3.4ではかりとった試料の質量(g)
B: 8.3.4ではかりとった試料中のほう素化合物の質量(H3BO3と
して)(g)
D: 8.3.4ではかりとった試料中のDDACの質量(g)
8.5.5
SAAC
有効成分の配合比及び合計含有量は,8.4.1.1又は8.4.1.2及び8.4.6.1の結果から式(57),式(58)及び式(59)
によって算出する。
a) 有効成分の配合比
100
0
×
+
=
D
S
S
S
······································································ (57)
100
0
×
+
=
D
S
D
D
······································································ (58)
ここに,
S0: 試料のシラフルオフェンの配合比
D0: 試料のDMPAPの配合比
S: 8.3.5ではかりとった試料中のシラフルオフェンの質量(g)
D: 8.3.5ではかりとった試料中のDMPAPの質量(g)
b) 有効成分の合計含有量
100
×
+
=
T
D
S
A
······································································· (59)
ここに,
A: 試料の有効成分の含有量(質量分率 %)
T: 8.3.5ではかりとった試料の質量(g)
S: 8.3.5ではかりとった試料中のシラフルオフェンの質量(g)
D: 8.3.5ではかりとった試料中のDMPAPの質量(g)
8.5.6
AZAAC
有効成分の配合比及び合計含有量は,8.4.1.1又は8.4.1.2,8.4.4.1又は8.4.4.2及び8.4.7.1の結果から式(60),
式(61),式(62),及び式(63)によって算出する。
a) 有効成分の配合比
100
1
1
1
1
0
×
+
+
=
E
Cy
A
A
A
····························································· (60)
ここに,
A0: 試料のDMPAPの配合比
A1: 8.3.6ではかりとった試料中のDMPAPの質量(g)
Cy1: 8.3.6ではかりとった試料中のシプロコナゾールの質量(g)
E1: 8.3.6ではかりとった試料中のエトフェンプロックスの質量
(g)
42
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
1
1
1
1
0
×
+
+
=
E
Cy
A
Cy
Cy
···························································· (61)
ここに, Cy0: 試料のシプロコナゾールの配合比
A1: 8.3.6ではかりとった試料中のDMPAPの質量(g)
Cy1: 8.3.6ではかりとった試料中のシプロコナゾールの質量(g)
E1: 8.3.6ではかりとった試料中のエトフェンプロックスの質量
(g)
100
1
1
1
1
0
×
+
+
=
E
Cy
A
E
E
····························································· (62)
ここに,
E0: 試料のエトフェンプロックスの配合比
A1: 8.3.6ではかりとった試料中のDMPAPの質量(g)
Cy1: 8.3.6ではかりとった試料中のシプロコナゾールの質量(g)
E1: 8.3.6ではかりとった試料中のエトフェンプロックスの質量
(g)
b) 有効成分の合計含有量
100
1
1
1
×
+
+
=
S
E
Cy
A
T
······························································· (63)
ここに,
T: 有効成分の含有量(質量分率 %)
S: 8.3.6ではかりとった試料の質量(g)
A1: 8.3.6ではかりとった試料中のDMPAPの質量(g)
Cy1: 8.3.6ではかりとった試料中のシプロコナゾールの質量(g)
E1: 8.3.6ではかりとった試料中のエトフェンプロックスの質量
(g)
8.5.7
AZNA
有効成分の配合比及び合計含有量は,8.4.1.1又は8.4.1.2,8.4.8.1及び8.4.9.1の結果から式(64),式(65),
式(66)及び式(67)によって算出する。
a) 有効成分の配合比
100
1
1
1
1
0
×
+
+
=
I
D
T
T
T
································································ (64)
ここに,
T0: 試料のテブコナゾールの配合比
T1: 8.3.7ではかりとった試料中のテブコナゾールの質量(g)
D1: 8.3.7ではかりとった試料中のDDACの質量(g)
I1: 8.3.7ではかりとった試料中のイミダクロプリドの質量(g)
100
1
1
1
1
0
×
+
+
=
I
D
T
D
D
······························································· (65)
ここに,
D0: 試料のDDACの配合比
T1: 8.3.7ではかりとった試料中のテブコナゾールの質量(g)
D1: 8.3.7ではかりとった試料中のDDACの質量(g)
I1: 8.3.7ではかりとった試料中のイミダクロプリドの質量(g)
100
1
1
1
1
0
×
+
+
=
I
D
T
I
I
································································ (66)
ここに,
I0: 試料中のイミダクロプリドの配合比
T1: 8.3.7ではかりとった試料中のテブコナゾールの質量(g)
D1: 8.3.7ではかりとった試料中のDDACの質量(g)
I1: 8.3.7ではかりとった試料中のイミダクロプリドの質量(g)
b) 有効成分の合計含有量
43
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
1
1
1
×
+
+
=
S
I
D
T
A
································································· (67)
ここに,
A: 有効成分の含有量(質量分率 %)
S: 8.3.7ではかりとった試料の質量(g)
T1: 8.3.7ではかりとった試料中のテブコナゾールの質量(g)
D1: 8.3.7ではかりとった試料中のDDACの質量(g)
I1: 8.3.7ではかりとった試料中のイミダクロプリドの質量(g)
8.5.8
AZN
有効成分の配合比及び合計含有量は,8.4.4.2及び8.4.9.1の結果から式(68),式(69),及び式(70)によって
算出する。
a) 有効成分の配合比
100
0
×
+
=
I
Cy
Cy
Cy
···································································· (68)
ここに, Cy0: 試料のシプロコナゾールの配合比
Cy: 8.3.10ではかりとった試料中のシプロコナゾールの質量(g)
I: 8.3.10ではかりとった試料中のイミダクロプリドの質量(g)
100
0
×
+
=
I
Cy
I
I
······································································ (69)
ここに,
I0: 試料のイミダクロプリドの配合比
Cy: 8.3.10ではかりとった試料中のシプロコナゾールの質量(g)
I: 8.3.10ではかりとった試料中のイミダクロプリドの質量(g)
b) 有効成分の合計含有量 試料の有効成分の含有量は,式(70)によって算出する。
100
)
(
×
+
=
S
I
Cy
A
···································································· (70)
ここに,
A: 試料の有効成分の含有量(質量分率 %)
S: 8.3.10ではかりとった試料の質量(g)
Cy: 8.3.10ではかりとった試料中のシプロコナゾールの質量(g)
I: 8.3.10ではかりとった試料中のイミダクロプリドの質量(g)
8.6
製品の状態
製品の状態は,目視による。
8.7
水不溶解分
8.7.1
操作
試験液をよくかき混ぜ,メスシリンダ100 mLを用い試験液100 mLをはかりとり,質量既知のガラスろ
過器1G4でろ過する。その残分を蒸留水で十分に洗い,105〜110 ℃の乾燥器中で恒量になるまで乾燥し,
デシケータ中で放冷して残分の質量をはかる。
8.7.2
計算
各薬剤の水不溶解分は,次の式によって算出する。
a) AAC-1,AAC-2 8.7.1によって得られた水不溶解分は,式(71)によって算出し,有効数字2けたに丸
める。
100
100
/
10
×
×
×
=
A
S
R
Q
·································································· (71)
ここに,
Q: 水不溶解分(質量分率 %)
R: 残分の質量(g)
A: DDAC及びDMPAPの含有量(質量分率 %)
44
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S: 8.3.1ではかりとった試料の質量(g)
b) ACQ-1,ACQ-2 8.7.1によって得られた水不溶解分は,式(72)によって算出し,有効数字2けたに丸
める。
100
100
)
(
250
×
×
+
×
=
B
C
R
Q
······························································ (72)
ここに,
Q: 水不溶解分(質量分率 %)
R: 残分の質量(g)
C: 8.3.2ではかりとった試料中の銅化合物の質量(CuOとして)
(g)
B: ACQ-1の場合8.3.2ではかりとった試料中のBKCの質量(g)
ACQ-2の場合8.3.2ではかりとった試料中のDDACの質量(g)
c) CUAZ 8.7.1によって得られた水不溶解分は,式(73)によって算出し,有効数字2けたに丸める。
100
100
250
100
/
×
×
×
=
A
S
R
Q
·························································· (73)
ここに,
Q: 水不溶解分(質量分率 %)
R: 残分の質量(g)
A: 試料中の有効成分の含有量(質量分率 %)
S: 8.3.3ではかりとった試料の質量(g)
d) BAAC 8.7.1によって得られた水不溶解分は,式(74)によって算出し,有効数字2けたに丸める。
100
000
1/
×
×
=
A
S
R
Q
································································ (74)
ここに,
Q: 水不溶解分(質量分率 %)
R: 残分の質量(g)
A: 試料の有効成分の含有量(質量分率 %)
S: 8.3.4ではかりとった試料の質量(g)
e) SAAC 8.7.1によって得られた水不溶解分は,式(75)によって算出し,有効数字2けたに丸める。
100
100
/
10
×
×
×
=
A
S
R
Q
·································································· (75)
ここに,
Q: 水不溶解分(質量分率 %)
R: 残分の質量(g)
A: 試料中の有効成分の含有量(質量分率 %)
S: 8.3.5ではかりとった試料の質量(g)
f)
AZAAC 8.7.1によって得られた水不溶解分は,式(76)によって算出し,有効数字2けたに丸める。
100
000
1/
×
×
=
A
S
R
Q
································································ (76)
ここに,
Q: 水不溶解分(質量分率 %)
R: 残分の質量(g)
A: 試料中の有効成分の含有量(質量分率 %)
S: 8.3.6ではかりとった試料の質量(g)
g) AZNA 8.7.1によって得られた水不溶解分は,式(77)によって算出し,有効数字2けたに丸める。
100
100
100
100
/
×
×
×
=
A
S
R
Q
·························································· (77)
ここに,
Q: 水不溶解分(質量分率 %)
R: 残分の質量(g)
A: 試料中の有効成分の含有量(質量分率 %)
S: 8.3.7ではかりとった試料の質量(g)
45
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
8.8
pH
pHは,8.3で調製した各試験液についてJIS Z 8802によって測定する。
8.9
脂肪酸の酸価
8.9.1
滴定法
a) 要旨 試料から脂肪酸を分離し,水酸化カリウム溶液で滴定して酸価を求める。
b) 試薬 試薬は,次による。
1) 2-プロパノール JIS K 8839に規定するもの。
2) 石油エーテル JIS K 8593に規定するもの。
3) 硫酸(1+9) JIS K 8951に規定する硫酸を用いて調製したもの。
4) 水酸化カリウム−エタノール溶液(0.5 mol/L) JIS K 8574に規定する水酸化カリウム(試薬)30 g
をできるだけ少量の蒸留水に溶かし,JIS K 8102に規定するエタノール(95)で薄めて1 000 mLと
し,2〜3日放置後ろ過したもの。
5) 溶剤 体積比1:1若しくは2:1のエチルエーテル−エタノール混液又は体積比1:1若しくは2:
1のベンゼン−エタノール混液を用いる。これらの溶液は,使用直前にフェノールフタレインを指
示薬として上記0.5 mol/L水酸化カリウム溶液で中和しておく。
6) フェノールフタレイン溶液(10 g/L) JIS K 8799に規定するフェノールフタレインとJIS K 8102
に規定するエタノール(95)とを用いて調製したもの。
c) 操作 試料5 gをビーカ400 mLにとり,2-プロパノール50 mLに溶かし,硫酸(1+9)40 mLを加え
てかき混ぜる。これを石油エーテル20 mLで洗いながら分液漏斗250 mLに移し入れ,さらに石油エ
ーテル30 mLを加える。分液漏斗をよく振り混ぜ,静置後下層部を別の分液漏斗250 mLに移し硫酸
(1+9)10 mLと石油エーテル30 mLを加えてよく振り混ぜ,静置して下層部を捨て,上層部は石油
エーテル20 mLで洗いながら先の分液漏斗の上層部と合わせる。この抽出液を再度よく振り混ぜ下層
の分離物を捨てて,ろ紙を用いてろ過しながら三角フラスコ500 mLに移し,水浴上で石油エタノー
ルを蒸発させる。これを石油エーテル10 mLで洗いながら質量既知の蒸発皿に移し,水浴上で石油エ
ーテルを蒸発させて105 ℃の乾燥器中で恒量になるまで乾燥して,質量をはかる。この場合,乾燥器
中には2時間以上置かないようにする。
内容物に中性の溶剤100 mL及びフェノールフタレイン溶液数滴を加え,試料が完全に溶けるまで
十分に振り混ぜる。これを水酸化カリウム溶液で滴定し,指示薬のうすいピンクが30秒間続いたとき
を中和の終点とする。
d) 脂肪酸の酸価の計算 c)によって得られた脂肪酸の酸価は,式(78)によって算出し,有効数字2けたに
丸める。
2
1
5.0
11
.
56
C
C
f
D
A
−
×
×
×
=
······························································· (78)
ここに,
A: 脂肪酸の酸価
D: 水酸化カリウム溶液(0.5 mol/L)使用量(mL)
f: 水酸化カリウム溶液(0.5 mol/L)のファクタ
C1: 残分と蒸発皿の質量の合計(g)
C2: 蒸発皿の質量(g)
46
K 1570:2010
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
9
表示
表示は,次の事項を容器のよく見える場所に記載する。クレオソート油においては,受渡し時に納品書
などに次の事項を記入するものとする。
a) 規格の番号及び名称
b) 種類の記号
c) 有効成分の合計含有量
ただし,脂肪酸金属塩系3号(VZN-E)は,それぞれの有効成分含有量を表示する。
クレオソート油木材保存剤には適用しない。
d) 製造業者名又はその略号
e) 製造年月又はその略号
f)
製造番号又はロット番号