2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
日本工業規格 JIS
K1450-1996
水道用硫酸アルミニウム
(水道用硫酸ばんど)
Aluminium sulfate for water works
Al2 (SO4) 3・xH2O
1. 適用範囲 この規格は,水道用硫酸アルミニウムについて規定する。
備考 この規格の引用規格を,付表1に示す。
2. 種類 種類は,次のとおりとする。
(1) 固形
(2) 液体
3. 品質 品質は,5.によって試験したとき,表1のとおりとする。
表1 品質
項目
種類
固形
液体
外観
−
無色〜黄味がかった薄い
褐色の透明な液体
酸化アルミニウム
(Al2O3)
wt %
15.0 以上
8.0〜8.2
pH値
3.0 以上
3.0 以上
不溶分
wt %
0.1 以下
−
アンモニア性窒素(N)
wt ppm 300
以下
100
以下
ひ素 (As)
wt ppm
4
以下
2.0 以下
鉄 (Fe)
wt ppm 600
以下
200
以下
マンガン (Mn)
wt ppm 25
以下
15
以下
カドミウム (Cd)
wt ppm
2.0 以下
1.0 以下
鉛 (Pb)
wt ppm 10
以下
5
以下
水銀 (Hg)
wt ppm
0.2 以下
0.1 以下
クロム (Cr)
wt ppm 10
以下
5
以下
4. 試料採取方法 試料は,全体を代表するように,受渡当事者間の協定に基づく合理的な方法により採
取する。
5. 試験方法
2
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.1
一般事項 試験において共通する一般事項はJIS K 0050,吸光光度法についてはJIS K 0115,発光
分光分析法についてはJIS K 0116,原子吸光分析法についてはJIS K 0121による。
試験において使用する水は,項目によって規定されている場合以外は,JIS K 0557の3.(種別及び質)
に規定するA2以上の質のものとする。
5.2
酸化アルミニウム 酸化アルミニウムの定量は滴定法による。
(1) 要旨 試料中のアルミニウム量に対して過剰のエチレンジアミン四酢酸二水素二ナトリウムを加え,
キレートの生成を完結させた後,キシレノールオレンジ指示薬を加えて亜鉛溶液で逆滴定し,酸化ア
ルミニウムの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 0.05mol/lEDTA溶液 JIS K 8107に規定するエチレンジアミン四酢酸二水素二ナトリウム二水和物
18.61gを量り取り,水1 000mlを加えて溶かした後,ポリエチレン気密容器に入れて保存する。
(b) 酢酸ナトリウム緩衝液 JIS K 8371に規定する酢酸ナトリウム三水和物272gを水に溶かして1
000mlとする。
(c) キシレノールオレンジ溶液(1g/l) JIS K 9563に規定するキシレノールオレンジ0.1gを水に溶かし
て100mlとする。
(d) アルミニウム標準液(1mgAl/ml) JIS K 8069に規定するアルミニウム(純度99.99%以上)1.000gを
量り取り,ビーカー100mlに移し入れ,上部を時計皿で覆い,少量の硝酸(1+1)を注意して加え加熱
溶解する。放冷後,溶液を全量フラスコ1 000mlに移し入れ,硝酸(1+30)を標線まで加える。
(e) 0.02mol/l亜鉛溶液 JIS K 8005に規定する亜鉛1.308gを量り取り,ビーカー100mlに移し入札塩酸
6〜7ml及び少量の水を加え,加熱して溶かす。これを水浴上で乾固近くまで蒸発した後,水を加え
て溶かし,全量フラスコ1 000mlに移し入れ,水を標線まで加える。
ビーカー200mlに0.05mol/lEDTA溶液20mlを採り硝酸(1+12)2mlを加え,以下(3)の(c)及び(d)の
操作を行い,このときの0.02mol/l亜鉛溶液の使用量 (ml) をa1とする。別のビーカー200mlにアル
ミニウム標準液20ml及び0.05mol/lEDTA溶液20mlを採り,硝酸(1+12)2mlを加え,時計皿で覆っ
て1分間煮沸後放冷し,以下(3)の(c)及び(d)の操作を行い,このときの0.02mol/l亜鉛溶液の使用量
(ml) をa2とする。
次の式によって,0.02mol/l亜鉛溶液1mlに相当するアルミニウム量を算出する。
2
1
20
001
.0
a
a
f
−
×
=
ここに, f: 0.02mol/l亜鉛溶液1mlに対応するアルミニウム量 (g)
(3) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約5g,液体では約10gを1mgのけたまで量り取り,ビーカー200mlに移し入れ,
水約100mlを加えて溶かす。必要に応じてろ過し,全量フラスコ500mlに移し入れ,水を標線まで
加える。
(b) この中から20mlを分取し,三角フラスコ200mlに移し入れ,0.05mol/lEDTA溶液20mlを加え,1
分間煮沸し,放冷する。
(c) 酢酸ナトリウム緩衝液約10ml及びキシレノールオレンジ溶液 (1g/l) 2〜5滴を加える。
(d) 0.02mol/l亜鉛溶液で滴定し,溶液の色が薄い赤になったときを終点とする。
(e) 0.05mol/lEDTA溶液20mlを分取し,別の三角フラスコ200mlに移し入れ,水約20ml,硝酸 (1+12) 2ml
を加え,以下(c)及び(d)と同様に操作する。
3
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(4) 計算 次の式によって,試料中の酸化アルミニウムの濃度 (wt%) を算出する。
)
10
9129
.
0
c
(
100
500
20
S
f
)
b
b
(
5
889
.
1
A
4
−
×
×
−
×
×
×
−′
×
=
ここに,
A: 試料中の酸化アルミニウムの濃度 (wt %)
1.889 5: アルミニウム1gから酸化アルミニウムへの換算係数
f: 0.02mol/l亜鉛溶液1mlに対応するアルミニウム量 (g)
b: 試料溶液に消費した0.02mol/l亜鉛溶液の量 (ml)
b': (3)(e)で消費した0.02mol/l亜鉛溶液の量 (ml)
c: 5.7によって求めた鉄の濃度 (wt ppm)
0.912 9: 鉄からアルミニウムへの換算係数
S: 試料の質量 (g)
5.3
pH値
(1) 要旨 試料の水溶液のpH値をガラス電極法によって測定する。
(2) 装置
ガラス電極pH計 JIS Z 8802に規定する形式II
(3) 操作 操作は,次のとおり行う。
(a) 試料を,固形では1.0g,液体では2.0gを量り取り,ビーカーに移し入れ,水約50mlに溶かし,全
量フラスコ100mlに移し入れ,標線まで水を加える。
(b) 約20℃に保ちながら,JIS Z 8802の7.(操作方法)に従いpH値を測定する。
5.4
不溶分
(1) 要旨 試料を水に溶かしてガラスろ過器でろ過し,残分を乾燥して質量を量り,不溶分を求める。
(2) 器具 器具は,次のとおりとする。
(a) ろ過器 るつぼ形ガラスろ過器1G4
(b) 恒温乾燥器
(3) 操作 操作は,次のとおり行う。
(a) 試料10gを1mgのけたまで量り取り,ビーカー200mlに入れ,水約100mlに溶かす。
(b) 溶液のすべてを質量既知のろ過器でろ過し,洗液に硫酸イオンの反応を認めなくなるまで水で洗う。
(c) ろ過器を105〜110℃に調節した恒温乾燥器中で1時間乾燥し,デシケータ中で放冷後,質量を0.1mg
のけたまで量る。
(4) 計算 (3)(b)の操作前のろ過器質量と(3)(c)の質量との差 (g) から,次の式によって不溶分 (wt%) を算
出する。
100
×
=Se
B
ここに,
B: 試料中の不溶分 (wt%)
e: 質量の差 (g)
S: 試料 (g)
5.5
アンモニア性窒素
(1) 要旨 試料に炭酸ナトリウムを加えてアルミニウムを水酸化物として沈殿させ,その上澄液に次亜塩
素酸ナトリウム,次いで1−ナフトールを加え,発色したインドフェノール型色素(青緑色)を波長
720nm付近で吸光度を測定し,アンモニア性窒素の含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
4
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(a) 炭酸ナトリウム溶液 (30g/l) JIS K 8625に規定する炭酸ナトリウム(無水)3gを水に溶かして
100mlとする。
(b) 水酸化ナトリウム溶液 (10g/l) JIS K 8576に規定する水酸化ナトリウム1gを水に溶かして100ml
とする。
(c) 次亜塩素酸ナトリウム溶液(有効塩素1g/l) 次亜塩素酸ナトリウム(市販品)の100/Cml(Cは有
効塩素濃度)及び水酸化ナトリウム15gを水に溶かして1 000mlとする。
(d) EDTA・水酸化ナトリウム混液 JIS K 8107に規定するエチレンジアミン四酢酸二ナトリウム二水
和物0.93gを水酸化ナトリウム溶液 (40g/l) に溶かして250mlとする。
(e) 1−ナフトール溶液 JIS K 8698に規定する1−ナフトール1.6gをアセトン・エチルアルコール溶液
(15 : 85) に溶かして100mlとする。
(f) アンモニア性窒素標準原液 (0.1mgN/ml) JIS K 8116に規定する塩化アンモニウムをデシケータ
[JIS K 8228に規定する過塩素酸マグネシウムを入れたもの]中に16時間以上放置し,その0.382g
を量り取り,ビーカー100mlに移し入れ,少量の水に溶かし,全量フラスコ1 000mlに移し入れ,
水を標線まで加える。
(g) アンモニア性窒素標準液 (5μgN/ml) アンモニア性窒素標準原液(0.1mgN/ml)50mlを分取し,全
量フラスコ1 000mlに移し入れ,水を標線まで加える。使用時に調製する。
(3) 装置 光電分光光度計
(4) 操作 操作は次のとおり行う。
(a) 試料を,固形では約5g,液体では約10gを1mgのけたまで量り取り,ビーカー200mlに移し入れ,
水約100mlを加えて溶かし,全量フラスコ500mlに移し入れ,水を標線まで加える。
(b) この中から25mlを分取し,有栓メスシリンダー100mlに移し入れ,炭酸ナトリウム溶液(30g/l)5ml
を加え,更に水を標線まで加え,静かに振り混ぜる。
(c) 静置して水酸化物を沈殿させ,その上澄液50mlを分取し,共栓比色管100mlに移し入れる。
(d) 水酸化ナトリウム溶液 (10g/l) を加えてpH調整を行い,pH値を約11(1)とする。
(e) 次亜塩素酸ナトリウム溶液 (有効塩素1g/l) 1mlを加えて混和し,2分後に5分以内でEDTA・水酸
化ナトリウム混液1mlを加えて再び混和する。次いで1分後に5分以内(2)で1−ナフトール溶液5ml
を加えて混和し,水を標線まで加え,25〜30℃で15分間静置する。
(f) その一部を吸収セル10mmに採り,波長720nm付近の吸光度を測定する。
(g) 検量線 アンモニア性窒素標準液 (5μgN/ml) 0, 1,〜7mlを段階的に採り,全量フラスコ100mlに移
し入れ,水を加えて約50mlとする。以下(e)及び(f)と同様に操作し,アンモニア性窒素の量と吸光
度との関係から検量線を作成する。
注(1) 検量線用のブランク,標準液は緩衝性か低いので,pH調整を必要としない。
(2) 試薬の添加間隔時間を検量線用も含め正確に行う。
(5) 計算 (4)(g)で作成した検量線から(4)(f)で測定した吸光度に相当するアンモニア性窒素の量 (μg) を
求め,次の式によって試料中のアンモニア性窒素の濃度 (wt ppm) を算出する。
100
50
500
25×
×
=
S
g
C
ここに,
C: 試料中のアンモニア性窒素の濃度 (wt ppm)
g: アンモニア性窒素の量 (μg)
5
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S: 試料の質量 (g)
5.6
ひ素 ひ素の定量は,原子吸光分析法又は吸光光度法のいずれかによる。
5.6.1
原子吸光分析法
(1) 要旨 試料に還元剤を加えてひ素と発生期の水素を反応させ,生成した気体状の水素化ひ素を加熱吸
収セルへ導入してひ素を原子化させ,波長193.7nmで吸光度を測定し,ひ素の含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 塩酸 (1+1) JIS K 8180に規定するひ素分析用の塩酸を用いて調製したもの。
(b) よう化カリウム溶液 (200g/l) JIS K 8913に規定するよう化カリウム20gを水に溶かして100mlと
する。使用時に調製する。
(c) 水素化ほう素ナトリウム溶液 (10g/l) JIS K 8576に規定する水酸化ナトリウム1gと水素化ほう素
ナトリウム10gを水に溶かし1 000mlとする。使用時に調製する。
(d) ひ素標準原液 (0.1mgAs/ml) JIS K 8005に規定する三酸化二ひ素を105℃で約2時間加熱し,デシ
ケータ中で放冷した後,その0.132gを量り取り,ビーカー100mlに移し入れ,上部を時計皿で覆い,
水酸化ナトリウム溶液(4g/l)250mlを加えて加熱溶解する。放冷後,溶液を全量フラスコ1 000mlに
移し入れ,塩酸(1+50)で数回洗い,洗液も全量フラスコに移し入れ,塩酸(1+50)を標線まで加える。
又は,JIS K 0026に規定するひ素標準液のAs100を用いる。
(e) ひ素標準液 (0.05μgAs/ml) ひ素標準原液(0.1mgAs/ml)10mlを分取し,全量フラスコ100mlに移し
入れ,水を標線まで加える。更にこの溶液5mlを分取し,全量フラスコ1 000mlに移し入れ,水を
標線まで加える。
(3) 装置 装置は,次のとおりとする。
(a) 連続式水素化ひ素発生装置
(b) 原子吸光分析装置
(c) ひ素中空陰極ランプ
(d) キャリアーガス JIS K 1105に規定するアルゴン2級又は窒素ガス(99.99vol%以上)
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約5g,液体では約10gを1mgのけたまで量り取り,ビーカー100mlに移し入れ,
水約30mlを加えて溶かす。全量フラスコ100mlに移し入れ,水を標線まで加える。
(b) この中から5mlを分取し,ビーカー100mlに移し入れ,水を加えて約50mlとした後,塩酸 (1+1) 10ml,
よう化カリウム溶液 (200g/l) 5mlを加え,約30分間沸騰しない程度で加熱濃縮する。放冷後,全量
フラスコ50mlに移し入れ,水を標線まで加える。
(c) (b)の操作を行った試料を塩酸 (1+1) 及び水素化ほう素ナトリウム溶液(10g/l)とともに水素化物発
生装置に流しながら反応させ,生成した水素化ひ素をキャリアーガスにより連続的に加熱吸収セル
に導入して,波長193.7nmで吸光度を測定する。
(d) 検量線 ひ素標準液 (0.05μgAs/ml) 0,1,2〜10mlを段階的にビーカー100mlに採り,水を適量加え
て約50mlとする。以下(b)及び(c)と同様に操作し,ひ素の量 (μg) と吸光度との関係から検量線を
作成する。
(5) 計算 (4)(d)で作成した検量線から(4)(c)で測定した吸光度に相当するひ素の量 (μg) を求め,次の式に
よって試料中のひ素の濃度 (wt ppm) を算出する。
6
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
100
5
×
=
S
h
D
ここに,
D: 試料中のひ素の濃度 (wt ppm)
h: ひ素の量 (μg)
S: 試料の質量 (g)
備考 水素化ひ素の発生方法には,このほか亜鉛粉末を用いた非連続式のものがあり,この方法によ
ることもできる。
5.6.2
吸光光度法
(1) 要旨 試料に還元剤を加えてひ素と発生期の水素を反応させ,生成した気体状の水素化ひ素をジエチ
ルジチオカルバミド酸銀のピリジン溶液に吸収・発色させ,波長530nm付近で吸光度を測定し,ひ素
の含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 塩酸 JIS K 8180に規定するひ素分析用のもの。
(b) よう化カリウム溶液 (150g/l) JIS K 8913に規定するよう化カリウム15gを水に溶かして100ml
とする。使用時に調製する。
(c) 塩化すず (II)溶液 JIS K 8136に規定する塩化すず (II) 二水和物40gを塩酸100mlで溶かす。使
用時に調製する。
(d) 酢酸鉛溶液 (85g/l) JIS K 8374に規定する酢酸鉛 (II) 三水和物10gにJIS K 8355に規定する酢酸
(1+2)1滴を加え,水に溶かして100mlとする。密栓して保存する。
(e) 砂状亜鉛 亜鉛(無ひ素)でJIS Z 8801の標準網ふるい目1 410μmを通過し,1 000μmにとどまっ
たもの。
(f) DDTC銀ピリジン溶液 (5g/l) JIS K 9512に規定するN, N−ジエチルジチオカルバミド酸銀1gを
ピリジン200mlに溶かす。褐色瓶に入れて冷暗所に保存すると,約3か月は安定である。
(g) ひ素標準原液 (0.1mgAs/ml) 5.6.1(2)(d)による。
(h) ひ素標準液 (1μgAs/ml) ひ素標準原液 (0.1mgAs/ml) 10mlを分取し,全量フラスコ1 000mlに移し
入れ,水を標線まで加える。
(3) 装置 装置は,次のとおりとする。
(a) 光電分光光度計
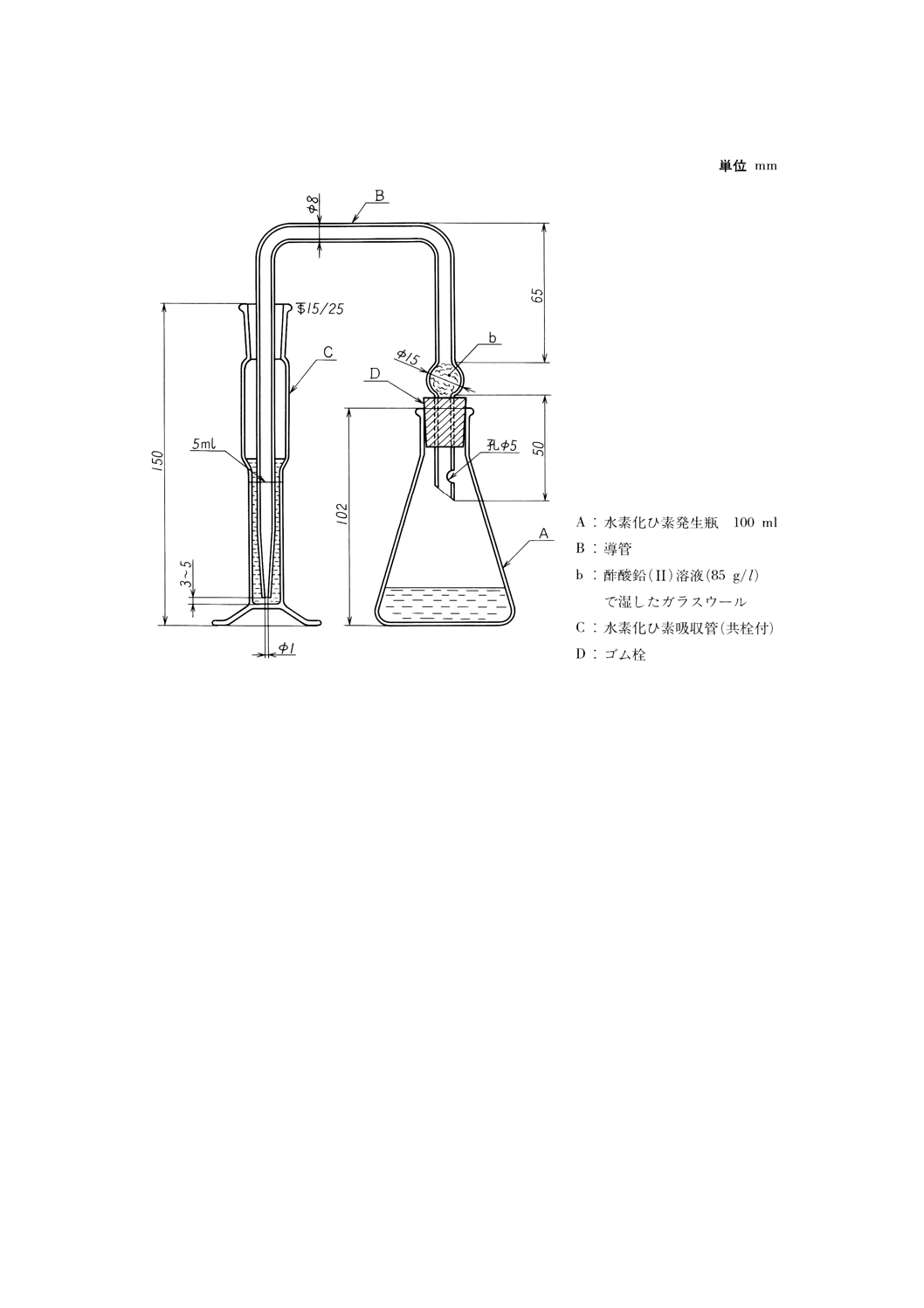
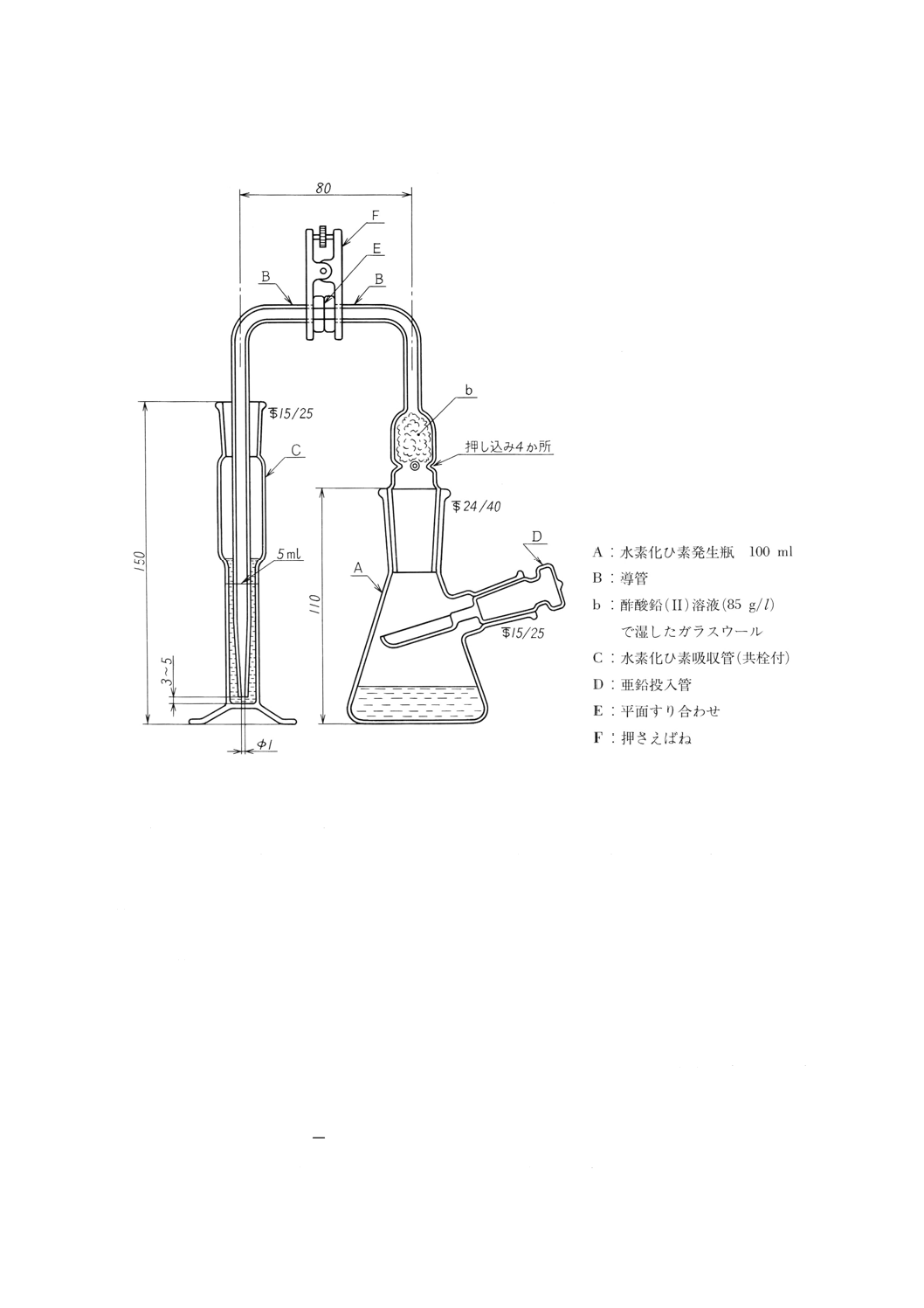
(b) ひ素試験装置 図1〜2に一例を示す。
7
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図1 ひ素試験装置の一例
8
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図2 ひ素試験装置の一例
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約1g,液体では約2gを1mgのけたまで量り取り,ひ素試験装置の発生瓶に入れ,
塩酸5ml及び水を加えて溶かし約40mlとする。
(b) よう化カリウム溶液 (150g/l) 2mlを加えて2〜3分間静置した後,塩化すず (II) 溶液1mlを加えて
混和し,約15分間静置する。
(c) 吸収管にDDTC銀ピリジン溶液 (5g/l) 5mlを入れ,発生瓶に砂状亜鉛3gを加え速やかに発生瓶,導
管,吸収管を連結し,常温で約1時間静置して,発生した水素化ひ素を吸収させる。
(d) 吸収管の溶液の一部を吸収セル10mmに採り,DDTC銀ピリジン溶液(5g/l)を対照液として波長
530nm付近の吸光度を測定する。
(e) 検量線 ひ素標準液 (1μgAs/ml) 0,1,〜20mlを段階的に発生瓶に採り,塩酸5ml及び水を加えて
約40mlとする。以下(b)〜(d)と同様に操作し,ひ素の量と吸光度との関係から検量線を作成する。
(5) 計算 (4)(e)で作成した検量線から(4)(d)で測定した吸光度に相当するひ素の量 (μg) を求め,次の式に
よって試料中のひ素の濃度 (wtppm) を算出する。
S
h
D=
ここに,
D: 試料中のひ素の濃度 (wt ppm)
9
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
h: ひ素の量 (μg)
S: 試料の質量 (g)
5.7
鉄 鉄の定量は,原子吸光分析法,ICP発光分光分析法又は吸光光度法のいずれかによる。
5.7.1
電気加熱方式原子吸光分析法
(1) 要旨 希釈した試料を電気加熱方式原子吸光分析法により,波長248.3nmで吸光度を測定し,鉄の含
有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 硝酸 (1+1) JIS K 8541に規定する硝酸を用いて調製したもの。
(b) 鉄標準原液 (0.1mgFe/ml) 鉄(99.5wt%以上)0.100gを量り取り,ビーカー100mlに移し入れ,塩
酸 (1+1) 30mlを加え,加熱して溶かし,放冷後,全量フラスコ1 000mlに移し入れ,水を標線ま
で加える。又は,JIS K 0016に規定する鉄標準液のFe100を用いる。
(c) 鉄標準液 (1μgFe/ml) 鉄標準原液(0.1mgFe/ml)10mlを分取し,全量フラスコ1 000mlに移し入れ,
硝酸(1+1)20mlを加え,水を標線まで加える。
(3) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロピペット JIS K 0970に規定するプッシュボタン式液体用微量体積計5〜500μl又は自動注
入装置
(b) 電気加熱方式原子吸光分析装置 電気加熱方式でバックグラウンド補正が可能なもの。
(c) 発熱体 黒鉛製又は耐熱金属製のもの。
(d) 鉄中空陰極ランプ
(e) シースガス JIS K 1105に規定するアルゴン2級
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約5g,液体では約10gを1mgのけたまで量り取り,ビーカー200mlに移し入れ,
水約30ml及び硝酸2mlを加え,時計皿で覆って約1分間煮沸し,放冷後,必要に応じてろ過し,
全量フラスコ100mlに移し入れ,水を標線まで加える。
(b) (a)の操作を行った試料10mlを分取し,全量フラスコ100mlに移し入れ,水を標線まで加える。
(c) (b)の操作を行った試料2mlを分取し,全量フラスコ20mlに移し入れ,硝酸 (1+1) 1mlを加えた後,
水を標線まで加える。
(d) (c)の操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,JIS K
0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,約30秒間)した後,灰化(500〜1 000℃,
約30秒間)し,原子化(3)(2 000〜2 800℃,約4〜6秒間)して,波長248.3nmの吸光度(4)を読み取
る(5)。
(e) 空試験として(a)の操作での試料と同量の水を採り,以下(a)〜(d)と同様に操作し,得られた吸光度
により試料の吸光度を補正する。
(f) 検量線 鉄標準液 (1μgFe/ml) 0,0.2,〜2mlを段階的に全量フラスコ20mlに採り,硝酸 (1+1) 1ml
を加えた後,水を標線まで加える。以下(d)と同様に操作し,鉄の量と吸光度との関係から検量線を
作成する。
注(3) 乾燥,灰化,原子化の条件は装置によって異なる。
(4) 吸光度又はその比例値。
(5) 引き続いて少なくとも(d)の操作を3回繰り返し,指示値が合うことを確認する。
(5) 計算 (4)(f)で作成した検量線から(4)(e)により補正された(4)(d)の吸光度に相当する鉄の量 (μg) を求
10
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
め,次の式によって試料中の鉄の濃度 (wt ppm) を算出する。
1000
2
×
=
S
i
E
ここに,
E: 試料中の鉄の濃度 (wtppm)
i: 鉄の量 (μg)
S: 試料の質量 (g)
5.7.2
ICP発光分光分析法
(1) 要旨 希釈した試料をICP発光分光分析法により,波長238.204nmで発光強度を測定し,内部標準法
により鉄の含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 水 空試験を行って,使用に適することを確認した蒸留水又はイオン交換水を蒸留したもの。
(b) 硝酸 JIS K 8541に規定するもの。
(c) 鉄標準原液 (1mgFe/ml) 鉄(99.5%以上)1.000gを量り取り,ビーカー100mlに移し入れ,塩酸(1
+1) 30mlを加え,加熱して溶かし,放冷後,全量フラスコ1 000mlに移し入れ,水を標線まで加え
る。又は,JIS K 0016に規定する鉄標準液のFe1000を用いる。
(d) 混合標準液 (100μgFe/ml, 10μgMn/ml) 鉄標準原液 (1mgFe/ml) 10ml及びマンガン標準原液
(0.1mgMn/ml) [5.8.1(2)(b)] 10mlをそれぞれ分取し,全量フラスコ100mlに移し入れ,水を標線まで
加える。使用時に調製する。
(e) 内部標準原液 (1mgY/ml) 酸化イットリウム (Y2O3) 0.318gを量り取り,ビーカーに移し入れ,塩
酸3mlと少量の水とを加えて加熱溶解し,放冷後,全量フラスコ250mlに移し入れ,水を標線まで
加える。冷暗所に保存する。
(f) 内部標準液 (10μgY/ml) 内部標準原液 (1mgY/ml) 5mlを分取し,全量フラスコ500mlに移し入れ,
水を標線まで加える。
(g) アルミニウム溶液 (10mgAl/ml) JIS K 8069に規定するアルミニウム(純度99.99%以上)10gを量
り取り,ビーカーに移し入れ,上部を時計皿で覆い,少量の硝酸 (1+1) を注意して加え,加熱溶
解する。放冷後,溶液を全量フラスコ1 000mlに移し入れ,硝酸 (1+30) を標線まで加える。
(3) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(b) キャリアーガス JIS K 1105に規定するアルゴン2級
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約1g,液体では約2gを1mgのけたまで量り取り,ビーカー100mlに移し入れ,
水約50ml及び硝酸4mlを加え,時計皿で覆って静かに1分間煮沸して溶かす。
(b) 放冷後,全量フラスコ200mlにろ入し,内部標準液10mlを加え,水を標線まで加える。
(c) (b)の操作を行った試料をJIS K 0116の5.8(定量分析)に従い,ICP発光分光分析装置に導入し,
鉄の波長238.204nm及びイットリウムの波長371.029nmの発光強度を測定し,イットリウムに対す
る鉄の発光強度比を求める。
(d) 検量線 混合標準液 (100μgFe/ml, 10μgMn/ml) 0,0.2,〜4mlを段階的に全量フラスコ200mlに採り,
それぞれに内部標準液10ml,マトリックスを一致させるため硝酸及びアルミニウムの量が(a)及び
(b)の操作を行った試料と同じになるようそれぞれ加えた後,水を標線まで加える。以下(c)と同様に
11
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
操作して発光強度比を求め,鉄の量と発光強度比との関係から検量線を作成する。
(5) 計算 (4)(d)で作成した検量線から(4)(c)で得られた発光強度比に相当する鉄の量 (μg) を求め,次の式
によって試料中の鉄の濃度 (w tppm) を算出する。
S
i
F=
ここに,
F: 試料中の鉄の濃度 (wt ppm)
i: 鉄の量 (μg)
S: 試料の質量 (g)
5.7.3
吸光光度法
(1) 要旨 試料に塩化ヒドロキシルアンモニウム溶液を加えて鉄イオンを第一鉄イオンに還元し,1, 10−
フェナントロリン溶液を加えて発色させ,波長510nm付近で吸光度を測定し,鉄の含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 塩酸 (1+3) JIS K 8180に規定する塩酸を用いて調製する。
(b) 塩化ヒドロキシルアンモニウム溶液 (100g/l) JIS K 8201に規定する塩化ヒドロキシルアンモニ
ウム10gを水に溶かして100mlとする。褐色瓶中に保存する。
(c) 1, 10−フェナントロリン溶液 (1g/l) JIS K 8202に規定する塩化1, 10−フェナントロリニウム−
水和物0.12gを水に溶かして100mlとするか,又はJIS K 8789に規定する1, 10−フェナントロリン
一水和物0.10gをJIS K 8102に規定するエタノール (95) 20mlに溶かして水で100mlとする。褐色
瓶中に保存する。
(d) 酢酸ナトリウム−酢酸緩衝液 JIS K 8371に規定する酢酸ナトリウム三水和物450gを水400mlに
溶かし,これにJIS K 8355に規定する酢酸240mlを加え,水で1 000mlとする。
(e) 鉄標準原液 (0.1mgFe/ml) 5.7.1 (2)(b)による。
(f) 鉄標準液(10μgFe/ml) 鉄標準原液 (0.1mgFe/ml) 10mlを分取し,全量フラスコ100mlに移し入れ,
水を標線まで加える。
(3) 装置 光電分光光度計
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約5g,液体では約15gを1mgのけたまで量り取り,ビーカー100mlに移し入れ,
水約30mlを加えて溶かす。全量フラスコ500mlに移し入れ,水を標線まで加える。
(b) この中から20mlを分取し,全量フラスコ100mlに移し入れ,塩酸 (1+3) 3ml及び水を加えて約70ml
とし,塩化ヒドロキシルアンモニウム溶液 (100g/l) 1mlを加えてよく振り混ぜる。
(c) 1, 10−フェナントロリン溶液 (1g/l) 5ml及び酢酸ナトリウム−酢酸緩衝液15mlを加え,水を標線ま
で加えて(6)よく振り混ぜた後,約20分間静置して発色させる。
(d) 発色した溶液の一部を吸収セル10mmに採り,波長510nm付近の吸光度を測定する。
(e) 検量線 鉄標準液 (10μgFe/ml) 0, 1, 〜5mlを段階的に全量フラスコ100mlに採り,塩酸 (1+3) 3ml
及び水を加えて約70mlとし,塩化ヒドロキシルアンモニウム溶液 (100g/l) 1mlを加えてよく振り混
ぜる。以下(c)及び(d)と同様に操作し,鉄の量と吸光度との関係から検量線を作成する。
注(6) このときの溶液のpH値は約4.8である。
(5) 計算 (4)(e)で作成した検量線から(4)(d)で測定した吸光度に相当する鉄の量 (μg) を求め,次の式によ
って試料中の鉄の濃度 (wt ppm) を算出する。
12
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
500
20
×
=
S
i
F
ここに,
F: 試料中の鉄の濃度 (wt ppm)
i: 鉄の量 (μg)
S: 試料の質量 (g)
5.8
マンガン マンガンの定量は,原子吸光分析法,ICP発光分光分析法又は吸光光度法のいずれかによ
る。
5.8.1
電気加熱方式原子吸光分析法
(1) 要旨 希釈した試料を電気加熱方式原子吸光分析法により,波長279.5nmで吸光度を測定し,マンガ
ンの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 硝酸 (1+1) JIS K 8541に規定する硝酸を用いて調製したもの。
(b) マンガン標準原液 (0.1mgMn/ml) JIS K 8247に規定する過マンガン酸カリウム0.288gを量り取り,
ビーカー200mlに移し入れ,硫酸 (1+31) 160mlを加えて溶かし,JIS K 8061に規定する亜硫酸ナト
リウムの溶液(100g/l)をかき混ぜながら滴加して脱色した後,煮沸して二酸化硫黄を追い出す。放冷
後,全量フラスコ1 000mlに移し入れ,水を標線まで加える。又はマンガン(99.9wt%以上)0.100g
を量り取り,ビーカー100mlに移し入れ,硫酸 (1+3) 20mlを加えて溶かし,放冷後,全量フラス
コ1 000mlに移し入れ,水を標線まで加える。又は,JIS K 0027に規定するマンガン標準液のMn100
を用いる。
(c) マンガン標準液 (1μgMn/ml) マンガン標準原液 (0.1mgMn/ml) 10mlを分取し,全量フラスコ1
000mlに移し入れ,硝酸 (1+1) 20mlを加え,水を標線まで加える。
(3) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロピペット 5.7.1 (3)(a)による。
(b) 電気加熱方式原子吸光分析装置 5.7.1(3)(b)による。
(c) 発熱体 5.7.1(3)(c)による。
(d) マンガン中空陰極ランプ
(e) シースガス 5.7.1 (3)(e)による。
(4) 操作 操作は,次のとおり行う。
(a) 5.7.1(4)(a)による。
(b) (a)の操作を行った試料2mlを分取し,全量フラスコ20mlに移し入れ,硝酸 (1+1) 1mlを加えた後,
水を標線まで加える。
(c) (b)の操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,JIS K
0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,約30〜40秒間)した後,灰化(500〜
800℃,約30秒間)し,原子化(7)(2 500〜2 800℃,約4〜6秒間)して,波長279.5nmの吸光度(8)
を読み取る(9)。
(d) 空試験として(a)の操作での試料と同量の水を採り,以下(a)〜(c)と同様に操作し,得られた吸光度に
より試料の吸光度を補正する。
(e) 検量線 マンガン標準液 (1μgMn/ml) 0,0.3,〜3mlを段階的に全量フラスコ20mlに採り,硝酸 (1
+1) 1mlを加えた後,水を標線まで加える。以下(c)と同様に操作し,マンガンの量と吸光度との関
13
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
係から検量線を作成する。
注(7) 5.7.1の注(3)による。
(8) 5.7.1の注(4)による。
(9) 5.7.1の注(5)による。
(5) 計算 (4)(e)で作成した検量線から(4)(d)により補正された(4)(c)の吸光度に相当するマンガンの量
(μg) を求め,次の式によって試料中のマンガンの濃度 (wtppm) を算出する。
100
2
×
=
S
j
G
ここに,
G: 試料中のマンガンの濃度 (wtppm)
i: マンガンの量 (μg)
S: 試料の質量 (g)
5.8.2
フレーム原子吸光分析法
(1) 要旨 試料にジエチルジチオカルバミド酸ナトリウムを加えてマンガンをキレート化し,4−メチル−
2−ペンタノンで抽出する。これをフレーム原子吸光分析法により波長279.5nmで吸光度を測定し,
マンガンの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) くえん酸水素二アンモニウム溶液 (500g/l) JIS K 8284に規定するくえん酸水素二アンモニウム
50gを水に溶かして100mlとする。
(b) 硫酸アンモニウム溶液 (400g/l) JIS K 8960に規定する硫酸アンモニウム40gを水に溶かして
100mlとする。
(c) アンモニア水 (1+5) JIS K 8085に規定するものを用いて調製する。
(d) 塩酸 (1+3) JIS K 8180に規定するものを用いて調製する。
(e) DDTC溶液 (100g/l) JIS K 8454に規定するN, N−ジエチルジチオカルバミド酸ナトリウム三水
和物10gを水に溶かして100mlとする。
(f) 4−メチル2−ペンタノン (MIBK) 使用前に水を飽和させておく。
(g) マンガン標準原液 (0.1mgMn/ml) 5.8.1 (2)(b)による。
(h) 混合標準液 (10μgMn/ml, 1μgCd/ml, 10μgPb/ml) 全量フラスコ500mlに硝酸 (1+12) 約300mlを
入れ,これにマンガン標準原液 (0.1mgMn/ml) 50ml,カドミウム標準原液 (0.1mgCd/ml) [5.9.1(2)(b)]
5ml及び鉛標準原液 [5.10.1(2)(b)] 50mlをそれぞれ分取し,全量フラスコに移し入れ,硝酸 (1+12)
を標線まで加える。使用時に調製する。
(3) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 呼び容量200mlで70mlに標線を付けたもの。
(b) pH計 5.3(2)による。
(c) 原子吸光分析装置
(d) マンガン中空陰極ランプ
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約2g,液体では約4gを1mgのけたまで量り取り,ビーカー200mlに移し入れ,
水約30mlを加えて溶かす。
(b) くえん酸水素二アンモニウム溶液 (500g/l) 3ml及び硫酸アンモニウム溶液 (400g/l) 15mlを加え,ア
14
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
ンモニア水 (1+5) 又は塩酸 (1+3) でpH値を5.0〜5.2とし(pH計で確かめる。),DDTC溶液
(100g/l) 3mlを加えて混和する。
(c) 3分間静置した後,分液漏斗に移し入れ,水を標線まで加える。これに4−メチル−2−ペンタノン
30mlを加え,2分間振り混ぜ,約10分間静置した後,水層を捨て,4−メチル−2−ペンタノン層
を試験液とする。
(d) この試験液について,直ちにJIS K 0121の6.(操作方法)の操作に従い,波長279.5nmの吸光度を
測定する。
(e) 検量線 混合標準液 (10μgMn/ml, 1μgCd/ml, 10μgPb/ml) 0,1,〜10mlを段階的にビーカー200mlに
採り,水を加えて約30mlとする。以下(b)〜(d)と同様に操作し,マンガンの量と吸光度との関係か
ら検量線を作成する。
備考 スロットバーナーを使用して,4−メチル−2−ペンタノン層を噴霧する場合は,次のことに注
意する。
(1) 多燃料炎となるので,炎が飛ばない程度にアセチレンの量をできるだけ少なくする。
(2) ゼロ合わせは(2)(f)の4−メチル−2−ペンタノンで行う。
(5) 計算 (4)(e)で作成した検量線から(4)(d)で測定した吸光度に相当するマンガンの量 (μg) を求め,次の
式によって試料中のマンガンの濃度 (wt ppm) を算出する。
S
j
G=
ここに,
G: 試料中のマンガンの濃度 (wt ppm)
j: マンガンの量 (μg)
S: 試料の質量 (g)
5.8.3
ICP発光分光分析法
(1) 要旨 希釈した試料を,ICP発光分光分析法により波長257.610nmで発光強度を測定し,内部標準法
によりマンガンの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 水 5.7.2(2)(a)による。
(b) 硝酸 JIS K 8541に規定するもの。
(c) 内部標準液 (10μgY/ml) 5.7.2(2)(e)による。
(d) マンガン標準原液 (0.1mgMn/ml) 5.8.1(2)(b)による。
(e) 混合標準液 (0.1mgFe/ml, 10μgMn/ml) 5.7.2(2)(d)による。
(f) アルミニウム溶液 (10mgAl/ml) 5.7.2(2)(g)による。
(3) 装置 5.7.2(3)による。
(4) 操作 操作は,次のとおり行う。
(a)〜(b) 5.7.2の(4)(a)〜(b)による。
(c) (b)の操作を行った試料をJIS K 0116の5.8(定量分析)に従い,ICP発光分光分析装置に導入し,
マンガンの波長257.610nm及びイットリウムの波長371.029nmの発光強度を測定し,イットリウム
に対するマンガンの発光強度比を求める。
(d) 検量線 5.7.2(4)(d)により調製した標準列について,(c)と同様に操作して発光強度比を求め,マンガ
ンの量と発光強度比との関係から検量線を作成する。
(5) 計算 (4)(d)で作成した検量線から(4)(c)で得られた発光強度比に相当するマンガンの量 (μg) を求め,
15
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
5.8.2(5)の式によって試料中のマンガンの濃度 (wt ppm) を算出する。
5.8.4
吸光光度法
(1) 要旨 試料に過よう素酸カリウムを加えてマンガンを過マンガン酸イオンに酸化して発色させ,波長
545nm付近で吸光度を測定し,マンガンの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 硫酸 (1+3) JIS K 8951に規定する硫酸を用いて調製したもの。
(b) 硫酸−りん酸混液 水500mlに,JIS K 9005に規定するりん酸200ml及びJIS K 8951に規定する硫
酸200mlを加え,放冷後,水で1 000mlとする。
(c) 硝酸銀溶液 (10g/l) JIS K 8550に規定する硝酸銀1gを水に溶かして100mlとする。
(d) 過よう素酸カリウム JIS K 8249に規定するもの。
(e) マンガン標準原液 (0.1mgMn/ml) 5.8.1(2)(b)による。
(f) マンガン標準液 (10μgMn/ml) マンガン標準原液 (0.1mgMn/ml) 10mlを分取し,全量フラスコ
100mlに移し入れ,水を標線まで加える。
(3) 装置 光電分光光度計
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約2g,液体では約4gを1mgのけたまで量り取り,ビーカー200mlに移し入れ,
硫酸 (1+3) 10mlを加えて砂浴上で加熱し,硫酸の白煙が発生し,乾固近くなったら加熱をやめて
放冷する。
(b) 水約60ml及び硫酸−りん酸混液5mlを加え,加熱溶解する。放冷後,硝酸銀溶液 (10g/l) 1ml及び
過よう素酸カリウム1g(10)を加え,約2分間煮沸し(11),流水で冷却した後,全量フラスコ100mlに
移し入れ,水を標線まで加える。
(c) 溶液の一部を吸収セル10〜50mmに採り,波長545nm付近の吸光度を測定する(12)。
(d) 検量線 マンガン標準液 (10μgMn/ml) 0,1,〜15mlを段階的にビーカー200mlに採る。以下(b)及
び(c)と同様に操作し,マンガンの量と吸光度との関係から検量線を作成する。
注(10) 有機物を多量に含む場合は,過よう素酸カリウムを更に多く加える。
(11) 白濁を生じたときは,液が透明になるまで煮沸を続ける。
(12) 着色による妨害がある場合には,吸光度を測定した後,直ちにそのセルに過酸化水素水 (30%) 1
滴を加え,残留する色の吸光度を測定して,その数値を前の吸光度から差し引き,これをマン
ガンの吸光度とする。
(5) 計算 (4)(d)で作成した検量線から(4)(c)で測定した吸光度に相当するマンガンの量 (μg) を求め,
5.8.2(5)の式によって試料中のマンガンの濃度 (wt ppm) を算出する。
5.9
カドミウム カドミウムの定量は,原子吸光分析法又はICP発光分光分析法のいずれかによる。
5.9.1
電気加熱方式原子吸光分析法
(1) 要旨 希釈した試料を電気加熱方式原子吸光分析法により,波長228.8nmで吸光度を測定し,カドミ
ウムの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 硝酸 (1+1) JIS K 8541に規定する硝酸を用いて調製したもの。
(b) カドミウム標準原液 (0.1mgCd/ml) カドミウム(99.9%以上)0.100gを量り取り,ビーカー100ml
に移し入れ,硝酸 (1+1) 20mlを加えて溶かし,加熱して窒素酸化物を追い出し,放冷後,全量フ
ラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 0012に規定するカドミウム標準液の
16
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
Cd100を用いる。
(c) カドミウム標準液 (0.1μgCd/ml) カドミウム標準原液 (0.1mgCd/ml) 10mlを分取し,全量フラスコ
1 000mlに移し入れ,硝酸 (1+1) 20mlを加え,水を標線まで加え,更にその10mlを分取し,全量
フラスコ100mlに移し入れ,硝酸 (1+1) 2mlを加え,水を標線まで加える。
(3) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロピペット 5.7.1(3)(a)による。
(b) 電気加熱方式原子吸光分析装置 5.7.1(3)(b)による。
(c) 発熱体 5.7.1(3)(e)による。
(d) カドミウム中空陰極ランプ
(e) シースガス 5.7.1(3)(c)による。
(4) 操作 操作は,次のとおり行う。
(a) 5.7.1(4)(a)による。
(b) (a)の操作を行った試料2mlを分取し,全量フラスコ20mlに移し入れ,硝酸 (1+1) 1mlを加え,水
を標線まで加える。
(c) (b)の操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,JIS K
0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,約30秒間)した後,灰化(300〜600℃,
約30秒間)し,原子化(13)(1 500〜2 300℃,約3〜6秒間)して,波長228.8nmの吸光度(14)を読み
取る(15)。
(d) 空試験として(a)の操作での試料と同量の水を採り,以下(a)〜(c)と同様に操作し,得られた吸光度に
より試料の吸光度を補正する。
(e) 検量線 カドミウム標準液 (0.1μgCd/ml) 0,0.2,〜2mlを段階的に全長フラスコ20mlに採り,硝酸
(1+1) 1mlを加えた後,水を標線まで加える。以下(c)と同様に操作し,カドミウムの量と吸光度と
の関係から検量線を作成する。
注(13) 5.7.1の注(3)による。
(14) 5.7.1の注(4)による。
(15) 5.7.1の注(5)による。
(5) 計算 (4)(e)で作成した検量線から(4)(d)により補正された(4)(c)の吸光度に相当するカドミウムの量
(μg) を求め,次の式によって試料中のカドミウムの濃度 (wt ppm) を算出する。
100
2
×
=
S
k
H
ここに,
H: 試料中のカドミウムの濃度 (wt ppm)
k: カドミウムの量 (μg)
S: 試料の質量 (g)
5.9.2
フレーム原子吸光分析法
(1) 要旨 試料にジエチルジチオカルバミド酸ナトリウムを加えてカドミウムをキレート化し,4−メチル
−2−ペンタノンで抽出する。これをフレーム原子吸光分析法により波長228.8nmで吸光度を測定し,
カドミウムの含有量を求める。
(2) 試薬 5.8.2(2)による。
(3) 器具及び装置 5.8.2(3)による。ただし,マンガン中空陰極ランプの代わりにカドミウム中空陰極ラン
17
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
プを用いる。
(4) 操作 操作は,次のとおり行う。
(a)〜(c) 5.8.2の(4)(a)〜(c)による。
(d) (c)の試験液について,直ちにJIS K 0121の6.(操作方法)の操作に従い,波長228.8nmの吸光度を
測定する。
(e) 検量線 5.8.2の(4)(e)の標準列について,(d)と同様に操作し,カドミウムの量と吸光度との関係か
ら検量線を作成する。
(5) 計算 (4) e)で作成した検量線から(4)(d)で測定した吸光度に相当するカドミウムの量 (μg) を求め,次
の式によって試料中のカドミウムの濃度 (wt ppm) を算出する。
S
k
H=
ここに,
H: 試料中のカドミウムの濃度 (wt ppm)
k: カドミウムの量 (μg)
S: 試料の質量 (g)
5.9.3
ICP発光分光分析法
(1) 要旨 試料にジエチルジチオカルバミド酸ナトリウムを加えてカドミウムをキレート化し,4−メチル
−2−ペンタノンで抽出する。これを水に転溶し,ICP発光分光分析法により波長226.502nmで発光強
度を測定し,内部標準法によりカドミウムの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 硝酸 JIS K 8541に規定するもの。
(b) くえん酸水素二アンモニウム溶液 (500g/l) JIS K 8284に規定するくえん酸水素二アンモニウム
50gを水に溶かして100mlとする。
(c) 硫酸アンモニウム溶液 (400g/l) JIS K 8960に規定する硫酸アンモニウム40gを水に溶かして
100mlとする。
(d) アンモニア水 (1+5) JIS K 8085に規定するものを用いて調製する。
(e) 塩酸 (1+3) JIS K 8180に規定するものを用いて調製する。
(f) DDTC溶液 (100g/l) JIS K 8454に規定するN,N−ジエチルジチオカルバミド酸ナトリウム三水和
物10gを水に溶かして100mlとする。
(g) 4−メチル−2−ペンタノン (MIBK) 使用前に水を飽和させておく。
(h) カドミウム標準原液 (0.1mgCd/ml) 5.9.1(2)(b)による。
(i) 混合標準液 (1μgCd/ml, 5μgPb/ml) カドミウム標準原液 (0.1mgCd/ml) 5ml及び鉛標準原液
(0.1mgPb/ml) [5.10.1(2)(b)] 25mlをそれぞれ分取し,全量フラスコ500mlに移し入れ,水を標線まで
加える。使用時に調製する。
(j) 内部標準液 (5μgY/ml) 5.7.2(2)(d)の内部標準原液 (1mgY/ml) 5mlを分取し,全量フラスコ1 000ml
に移し入れ,水を標線まで加える。使用時に調製する。
(3) 器具及び装置 器具及び装置は,次のとおりとする。
(a) 分液漏斗 呼び容量200mlで70mlに標線を付けたもの。
(b) pH計 5.3(2)による。
(c) ICP発光分光分析装置
(d) キャリアーガス JIS K 1105に規定するアルゴン2級
18
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約5g,液体では約10gを1mgのけたまで量り取り,ビーカー300mlに移し入れ,
水約30ml及び硝酸1mlを加え,時計皿で覆って約1分間煮沸し,放冷する。
(b) くえん酸水素二アンモニウム溶液 (500g/l) 3ml及び硫酸アンモニウム溶液 (400g/l) 15mlを加え,ア
ンモニア水 (1+5) 又は塩酸 (1+3) でpH値を5.0〜5.2とし(pH計で確かめる。),分液漏斗に移
し,水を標線まで加える。
(c) DDTC溶液 (100g/l) 3mlを加えて振り混ぜ,3分間静置した後,4−メチル−2−ペンタノン30mlを
加えて2分間激しく振り混ぜ,約10分間静置した後,水層は別の分液漏斗に移し入れ,4−メチル
−2−ペンタノン層をビーカー200mlに移す。
(d) 水層に4−メチル−2−ペンタノン20mlを加えて2分間激しく振り混ぜ,約10分間静置した後,水
層は捨て,4−メチル−2−ペンタノン層を(c)のビーカーに合わせる。
(e) このビーカーに水50ml,硝酸2ml及び内部標準液10mlを加え,加熱して4−メチル−2−ペンタノ
ンを完全に揮散させ,放冷後,全量フラスコ100mlに移し入れ,水を標線まで加える。
(f) これを,JIS K 0116の5.8(定量分析)に従い,ICP発光分光分析装置に導入し,カドミウムの波長
226.502nm及びイットリウムの波長371.029nmの発光強度を測定して,イットリウムに対するカド
ミウムの発光強度比を求める。
(g) 検量線 混合標準液 (1μgCd/ml, 5μgPb/ml) 0,1,〜10mlを段階的に全量フラスコ100mlに採り,各々
に硝酸2ml及び内部標準液10mlを加え,水を標線まで加える。以下(f)と同様に操作して発光強度
比を求め,カドミウムの量と発光強度比との関係から検量線を作成する。
(5) 計算 (4)(g)で作成した検量線から(4)(f)で測定した発光強度比に相当するカドミウムの量 (μg) を求
め,5.9.2(5)の計算式によって試料中のカドミウムの濃度 (wt ppm) を算出する。
5.10 鉛 鉛の定量は原子吸光分析法又はICP発光分光分析法のいずれかによる。
5.10.1 電気加熱方式原子吸光分析法
(1) 要旨 希釈した試料にマトリックス修飾剤の硝酸パラジウム (II) を加えて,電気加熱方式原子吸光分
析法により,波長283.3nmで吸光度を測定し,標準添加法により鉛の含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 硝酸 (1+1) JIS K 8541に規定する硝酸を用いて調製したもの。
(b) 鉛標準原液 (0.1mgPb/ml) JIS K 8701に規定する鉛(99.9%以上)0.100gを量り取り,ビーカー
100mlに移し入れ,硝酸 (1+3) 40mlを加えて溶かし,加熱して窒素酸化物を追い出し,放冷後,
全量フラスコ1 000mlに移し入れ,水を標線まで加える。又はJIS K 0015に規定する鉛標準液の
Pb100を用いる。
(c) 鉛標準液 (1μgPb/ml) 鉛標準原液 (0.1mgPb/ml) 10mlを分取し,全量フラスコ1 000mlに移し入れ,
硝酸 (1+1) 20mlを加え,水を標線まで加える。
(d) 硝酸パラジウム (II) 溶液 (10mgPd/ml) 硝酸パラジウム (II) 4.331gを量り取り,ビーカー100ml
に移し入れ,硝酸 (1+1) 10mlを加えて溶かし,全量フラスコ200mlに移し入れ,水を標線まで加
える。
(3) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロピペット 5.7.1(3)(a)による。
(b) 電気加熱方式原子吸光分析装置 5.7.1(3)(b)による。
(c) 発熱体 5.7.1(3)(c)による。
19
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(d) 鉛中空陰極ランプ
(e) シースガス 5.7.1(3)(e)による。
(4) 操作 操作は,次のとおり行う。
(a) 5.7.1(4)(a)による。
(b) 4個以上の全量フラスコ20mlそれぞれに,(a)の操作を行った試料2mlずつを採り,1個を除き,他
のものに鉛標準液 (1μgPb/ml) を0.1〜1.0mlの範囲で段階的に添加し,すべての全量フラスコに硝
酸パラジウム (II) 溶液 (10mgPd/ml) 1ml及び硝酸 (1+1) 1mlを加えた後,水を標線まで加える。
(c) (b)の操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,JIS K
0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,約30秒間)した後,灰化(500〜800℃,
約30秒間)し,原子化(16)(1 800〜2 500℃,約3〜6秒間)して,波長283.3nmの吸光度(17)を読み
取る(18)。
注(16) 5.7.1の注(3)による。
(17) 5.7.1の注(4)による。
(18) 5.7.1の注(5)による。
(5) 計算 鉛の添加量と吸光度との関係から計算式を作成し,横軸(鉛の添加量)の切片から鉛の量 (μg)
を求め,次の式により試料中の鉛の濃度 (wt ppm) を算出する。
100
2
×
=
S
m
I
ここに,
I: 試料中の鉛の濃度 (wt ppm)
m: 鉛の量 (μg)
S: 試料の質量 (g)
5.10.2 フレーム原子吸光分析法
(1) 要旨 試料にジエチルジチオカルバミド酸ナトリウムを加えてキレート化し,4−メチル−2−ペンタ
ノンで抽出する。これをフレーム原子吸光分析法により波長283.3nmで吸光度を測定し,鉛の含有量
を求める。
(2) 試薬 5.8.2(2)による。
(3) 器具及び装置 5.8.2(3)による。ただし,マンガン中空陰極ランプの代わりに鉛中空陰極ランプを用い
る。
(4) 操作 操作は,次のとおり行う。
(a)〜(c) 5.8.2の(4)(a)〜(c)による。
(d) (c)の試験液について,直ちにJIS K 0121の6.(操作方法)の操作に従い,波長283.3nmの吸光度を
測定する。
(e) 検量線 5.8.2(4)(e)の標準列について,(d)と同様に操作し,鉛の量と吸光度との関係から検量線を作
成する。
(5) 計算 (4)(e)で作成した検量線から(d)で測定した吸光度に相当する鉛の量(μg)を求め,次の式によって
試料中の鉛の濃度 (wt ppm) を算出する。
S
m
I=
ここに,
I: 試料中の鉛の濃度 (wt ppm)
m: 鉛の量 (μg)
20
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
S: 試料の質量 (g)
5.10.3 ICP発光分光分析法
(1) 要旨 試料にジェチルジチオカルバミド酸ナトリウムを加えて鉛をキレート化し,4−メチル−2−ペ
ンタノンで抽出する。これを水に転溶し,ICP発光分光分析法により波長220.353nmで発光強度を測
定し,内部標準法により鉛の含有量を求める。
(2) 試薬 5.9.3(2)による。
(3) 器具及び装置 5.9.3(3)による。
(4) 操作 操作は,次のとおり行う。
(a)〜(e) 5.9.3の(4)(a)〜(e)による。
(f) (e)の操作を行った試料を,JIS K 0116の5.8(定量分析)に従い,ICP発光分光分析装置に導入し,
鉛の波長220.351nm及びイットリウムの波長371.029nmの発光強度を測定して,イットリウムに対
する鉛の発光強度比を求める。
(g) 検量線 5.9.3(4)(g)により調製した標準列について,(f)と同様に操作して発光強度比を求め,鉛の量
と発光強度比との関係から検量線を作成する。
(5) 計算 (4)(g)で作成した検量線から(4)(f)で測定した発光強度比に相当する鉛の量 (μg) を求め,
5.10.2(5)の式によって試料中の鉛の濃度 (wt ppm) を算出する。
5.11 水銀 水銀の定量は原子吸光分析法による。
(1) 要旨 試料に塩化すず (II) を加え,水銀化合物を金属に還元し,通気して水銀を気化させ,波長
253.7nmで吸光度を測定し,水銀の含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 硝酸 JIS K 8541に規定するもの。
(b) 過マンガン酸カリウム JIS K 8247に規定するもの(19)。
(c) 塩化ヒドロキシルアンモニウム溶液 (200g/l) JIS K 8201に規定する塩化ヒドロキシルアンモニ
ウム(19)20gを水に溶かして100mlとする。
(d) 硫酸 (1+3) JIS K 8951に規定する硫酸(水銀の含有量が1μg/l以下のもの)を用いて調製したも
の。
(e) 塩化すず (II) 溶液 JIS K 8136に規定する塩化すず (II) 二水和物10gに硫酸 (1+10) 60mlを加え
て溶かし,水で100mlとする。1週間以上経過したものは使用しない。
(f) 水銀標準原液 (0.1mgHg/ml) JIS K 8139に規定する塩化水銀 (II) 0.135gを量り取り,ビーカー
200mlに移し入れ,硝酸10ml及び水約90mlに溶かし,これを全量フラスコ1 000mlに移し入れ,
水を標線まで加える。硬質ガラス瓶中に保存する。
(g) 水銀標準液 (0.05μgHg/ml) 水銀標準原液 (0.1mgHg/ml) 10mlを分取し,全量フラスコ1 000mlに
移し入れ,水を標線まで加える。さらに,この10mlを分取し,全量フラスコ200mlに移し入れ,
水を標線まで加える。使用時に調製する。
注(19) 原子吸光分析用試薬など,水銀含有量の少ないものを用いる。
(3) 装置及び器具 装置及び器具は,次のとおりとする。
(a) 原子吸光分析装置又は水銀用原子吸光分析装置
(b) 水銀中空陰極ランプ又は水銀ランプ
(c) 還元気化装置(20) 原子吸光分析装置と併用する。
なお,各構成部分の詳細は,次のとおりである。
21
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
空気ポンプ 0.5〜3l/minで可変式の送気能力があるダイアフラムポンプ又は同じ性能をもつ空気
ポンプ。水銀蒸気に接する部分が金属製の場合にはコロジオンなどを塗布しておく。開放送気方式
の場合は,調圧した圧縮空気を使用してもよい。
連結管 軟質塩化ビニル管がよい。シリコーンゴム管やゴム管は水銀を吸着するおそれがある。
還元容器 内容200〜300mlのガラス瓶,三角フラスコ又はガラスろ過板付洗気瓶250ml。開放送
気方式の場合は分液漏斗250mlを用いることができる。
乾燥管 乾燥管又はU字管にJIS K 8228に規定する過塩素酸マグネシウム又はJIS K 8124に規
定する塩化カルシウム(乾燥用)を詰める。乾燥剤としてシリカゲルや硫酸を用いると,水銀の吸
着や,吸着した水銀が脱離するおそれがある。
吸収セル 長さ100〜300mm程度の石英ガラス製のもの又はガラス管,塩化ビニル管の両端に石
英ガラス窓を付けたもの。
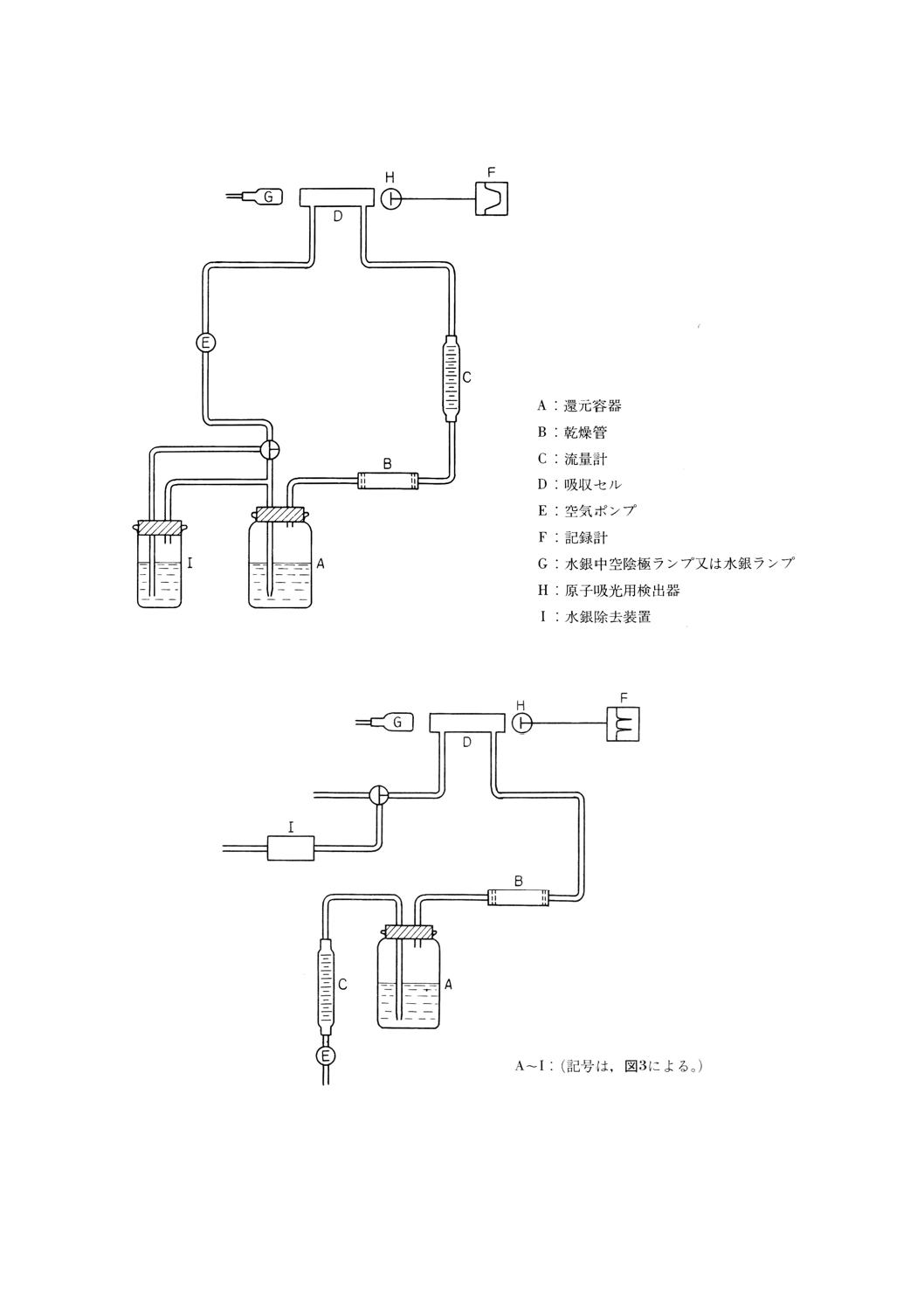
注(20) 還元容器,吸収セル,空気ポンプ,流量計,乾燥管及び連結管から構成される。その一例を図3
〜4に示す。
22
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
図3 密閉循環方式の構成の一例
図4 開放送気方式の構成の一例
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約2.5g,液体では約5gを1mgのけたまで量り取り,耐圧瓶200mlに移し入れ,
23
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
水100ml,硝酸10ml及び過マンガン酸カリウム1gを加えて静かに振り混ぜた後,水浴中で1時間
加熱する。
(b) もし,この間に過マンガン酸カリウムの色が消失する場合には,液温を40℃以下に冷却して過マン
ガン酸カリウム1gを一度に加え,再び加熱して過マンガン酸カリウムの色が10分間以上残るまで,
この操作を繰り返す。
(c) 加熱処理の後,冷却してから還元容器に移し入れ,水で約150mlとする。溶液を振り混ぜながら塩
化ヒドロキシルアンモニウム溶液 (200g/l) を滴加して過マンガン酸カリウムの色を消す。
(d) 硫酸 (1+3) 10mlを加えてよく振り混ぜ,塩化すず (II) 溶液10mlを加え,直ちに還元気化装置に
連結する[開放送気方式の場合は,分液漏斗の通気管にそれぞれコックを付け,硫酸 (1+3) 10ml
及び塩化すず (II) 溶液10mlを加え,密栓して約2分間激しく振り混ぜ,分液漏斗内の空気中の水
銀蒸気が平衡に達した後,送気装置に連結する。]。空気ポンプを作動させて空気を循環させ(開放
送気方式の場合は空気を送り,JIS K 0121の6.(操作方法)に従い水銀中空陰極ランプを用いて波
長253.7nmの吸光度を測定する。
(e) 空試験として,還元容器に水150mlを採り,(a)〜(c)と同量の試薬を加えた後,(d)と同様に操作を
行って得た吸光度により試料の吸光度を補正する。
(f) 検量線 水銀標準液 (0.05μgHg/ml) 0,1,〜20mlを段階的に還元気化装置の還元容器に採り,水を
加えて約150mlとする。以下(d)と同様に操作し,水銀の量と吸光度との関係から検量線を作成する。
(5) 計算 (4)(f)で作成した検量線から(4)(e)により補正された(4)(d)の吸光度に相当する水銀の量 (μg) を
求め,次の式によって試料中の水銀の濃度 (wt ppm) を算出する。
S
n
J=
ここに,
J: 試料中の水銀の濃度 (wt ppm)
n: 水銀の量 (μg)
S: 試料の質量 (g)
5.12 クロム クロムの定量は原子吸光分析法又はICP発光分光分析法のいずれかによる。
5.12.1 電気加熱方式原子吸光分析法
(1) 要旨 希釈した試料にマトリックス修飾剤の硝酸パラジウム (II) を加えて,電気加熱方式原子吸光分
析法により,波長357.9nmで吸光度を測定して,標準添加法によりクロムの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 硝酸 (1+1) JIS K 8541に規定する硝酸を用いて調製したもの。
(b) クロム標準原液 (0.1mgCr/ml) JIS K 8005に規定する容量分析標準物質二クロム酸カリウムを
150℃で約1時間加熱し,デシケータ中で放冷する。その0.283gを量り取り,ビーカー100mlに移
し入れ,少量の水に溶かし,全量フラスコ1 000mlに移し入れ,硝酸 (1+60) を標線まで加える。
又は,JIS K 0024に規定するクロム標準液のCr100を用いる。
(c) クロム標準液 (1μgCr/ml) クロム標準原液 (0.1mgCr/ml) 10mlを分取し,全量フラスコ1 000mlに
移し入れ,硝酸 (1+1) 20mlを加え,水を標線まで加える。
(d) 硝酸パラジウム (II) 溶液 (10mgPd/ml) 5.10.1(2)(d)による。
(3) 器具及び装置 器具及び装置は,次のとおりとする。
(a) マイクロピペット 5.7.1(3)(a)による。
(b) 電気加熱方式原子吸光分析装置 5.7.1(3)(b)による。
24
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(c) 発熱体 5.7.1(3)(c)による。黒鉛製の場合は,パイロコーティングしたものがよい。
(d) クロム中空陰極ランプ
(e) シースガス 5.7.1(3)(e)による。
(4) 操作 操作は,次のとおり行う。
(a) 5.7.1(4)(a)による。
(b) 4個以上の全量フラスコ20mlそれぞれに,(a)の操作を行った試料2mlずつを採り,1個を除き,他
のものにクロム標準液 (1μgCr/ml) を0.1〜1.0mlの範囲で段階的に添加し,すべての全量フラスコ
に硝酸パラジウム (II) 溶液 (10mgPd/ml) 1ml及び硝酸 (1+1) 1mlを加えた後,水を標線まで加える。
(c) (b)の操作を行った試料の一定量(例えば,10〜50μl)をマイクロピペットで発熱体に注入し,JIS K
0121の6.(操作方法)の操作に従って,乾燥(100〜120℃,約30秒間)した後,灰化(700〜1 000℃,
約30秒間)し,原子化(21)(2 400〜2 900℃,約5〜10秒間)して,波長357.9nmの吸光度(22)を読
み取る(23)。
注(21) 5.7.1の注(3)による。
(22) 5.7.1の注(1)による。
(23) 5.7.1の注(5)による。
(5) 計算 クロムの添加量と吸光度との関係から計算式を作成し,横軸(クロムの添加量)の切片からク
ロムの量 (μg) を求め,次の式により試料中のクロムの濃度 (wt ppm) を算出する。
100
2
×
=
S
o
K
ここに,
K: 試料中のクロムの濃度 (wt ppm)
o: クロムの量 (μg)
S: 試料の質量 (g)
5.12.2 ICP発光分光分析法
(1) 要旨 希釈した試料を,ICP発光分光分析法により波長267.716nmで発光強度を測定して,内部標準
法によりクロムの含有量を求める。
(2) 試薬 試薬は,次のとおりとする。
(a) 水 5.7.2(2)(a)による。
(b) 硝酸 JIS K 8541に規定する硝酸。
(c) クロム標準原液 (0.1mgCr/ml) 5.12.1(2)(b)による。
(d) クロム標準液 (10μgCr/ml) クロム標準原液 (0.1mgCr/ml) 10mlを分取し,全量フラスコ100mlに
移し入れ,水を標線まで加える。
(e) 内部標準液 (10μgY/ml) 5.7.2(2)(f)による。
(f) アルミニウム溶液 (10mgAl/ml) 5.7.2(2)(g)による。
(3) 装置 装置は,次のとおりとする。
(a) ICP発光分光分析装置
(b) キャリアーガス JIS K 1105に規定するアルゴン2級
(4) 操作 操作は,次のとおり行う。
(a) 試料を,固形では約2.5g,液体では約5gを1mgのけたまで量り取り,ビーカー100mlに移し入れ,
水約50ml及び硝酸2mlを加え,時計皿で覆って静かに約1分間煮沸し,放冷する。
25
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
(b) 全量フラスコ100mlにろ入し,内部標準液10mlを加え,水を標線まで加える。
(c) (b)の操作を行った試料をJIS K 0116の5.8(定量分析)に従い,ICP発光分光分析装置に導入し(24),
クロムの波長267.716nm及びイットリウムの波長371.029nmの発光強度を測定し,イットリウムに
対するクロムの発光強度比を求める。
(d) 検量線 クロム標準液 (10μgCr/ml) 0,0.2,〜10mlを段階的に全量フラスコ100mlに採り,それぞ
れに内部標準液10ml,マトリックスを一致させるため硝酸及びアルミニウムの量が(a)及び(b)の操
作を行った試料と同じになるようそれぞれ加えた後,水を標線まで加える。以下(4)(c)と同様に操作
して発光強度比を求め,クロムの量と発光強度比との関係から検量線を作成する。
注(24) 試料中のアルミニウム量が多いのでトーチ先端を詰まらせる可能性があるときは,水によるト
ーチの洗浄時間を通常より長くする。
(5) 計算 (4)(d)で作成した検量線から(4)(c)で得られた発光強度比に相当するクロムの量 (μg) を求め,次
の式によって試料中のクロムの濃度 (wt ppm) を算出する。
S
o
K=
ここに,
K: 試料中のクロムの濃度 (wt ppm)
o: クロムの量 (μg)
S: 試料の質量 (g)
6. 検査 検査は5.によって試験し,表1の規定に適合しなけれはならない。
7. 表示 次の事項を容器の適当な箇所に表示する(タンク車,タンクローリーなどは送り状などに表示
してもよい。)。
(1) 名称
(2) 種類
(3) 正味質量
(4) 製造年月日又はその略号
(5) 製造業者名又はその略号
付表1 引用規格
JIS K 0012 カドミウム標準液
JIS K 0015 鉛標準液
JIS K 0016 鉄標準液
JIS K 0024 クロム標準液
JIS K 0026 ひ素標準液
JIS K 0027 マンガン標準液
JIS K 0050 化学分析方法通則
JIS K 0115 吸光光度分析通則
JIS K 0116 発光分光分析通則
JIS K 0121 原子吸光分析通則
JIS K 0557 化学分析用の水
JIS K 0970 プッシュボタン式液体用微量体積計
26
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 1105 アルゴン
JIS K 8005 容量分析用標準物質
JIS K 8061 亜硫酸ナトリウム(試薬)
JIS K 8069 アルミニウム(試薬)
JIS K 8085 アンモニア水(試薬)
JIS K 8102 エタノール (95) (試薬)
JIS K 8107 エチレンジアミン四酢酸二水素二ナトリウム二水和物(試薬)
JIS K 8116 塩化アンモニウム(試薬)
JIS K 8124 塩化カルシウム(乾燥用)(試薬)
JIS K 8136 塩化すず (II) 二水和物(試薬)
JIS K 8139 塩化水銀 (II) (試薬)
JIS K 8180 塩酸(試薬)
JIS K 8201 塩化ヒドロキシルアンモニウム(試薬)
JIS K 8202 塩化1, 10−フェナントロリニウム一水和物(試薬)
JIS K 8228 過塩素酸マグネシウム(試薬)
JIS K 8247 過マンガン酸カリウム(試薬)
JIS K 8249 過よう素酸カリウム(試薬)
JIS K 8284 くえん酸水素二アンモニウム(試薬)
JIS K 8355 酢酸(試薬)
JIS K 8371 酢酸ナトリウム三水和物(試薬)
JIS K 8374 酢酸鉛 (II) 三水和物(試薬)
JIS K 8454 N, N−ジェチルジチオカルバミド酸ナトリウム三水和物(試薬)
JIS K 8541 硝酸(試薬)
JIS K 8550 硝酸銀(試薬)
JIS K 8576 水酸化ナトリウム(試薬)
JIS K 8625 炭酸ナトリウム(試薬)
JIS K 8698 1−ナフトール(試薬)
JIS K 8701 鉛(試薬)
JIS K 8789 1, 10−フェナントロリン一水和物(試薬)
JIS K 8913 よう化カリウム(試薬)
JIS K 8951 硫酸(試薬)
JIS K 8960 硫酸アンモニウム(試薬)
JIS K 9005 りん酸(試薬)
JIS K 9512 N, N−ジエチルジチオカルバミド酸銀(試薬)
JIS K 9563 キシレノールオレンジ(試薬)
JIS Z 8801 試験用ふるい
JIS Z 8802 pH測定方法
27
K1450-1996
2019年7月1日の法改正により名称が変わりました。まえがきを除き,本規格中の「日本工業規格」を「日本産業規格」に読み替えてください。
JIS K 1450改正原案作成委員会 構成表
氏名
所属
(委員長)
梶 野 勝 司
大阪市水道局
早 川 哲 夫
厚生省生活衛生局水道局環境部水道整備課
増 田 優
通商産業省基礎産業局化学製品課
岡 林 哲 夫
通商産業省工業技術院標準部繊維化学規格課
土 屋 悦 輝
東京都立衛生研究所
中 村 進
通商産業省工業技術院物質工学工業技術研究所
高 橋 孝 一
通商産業省製品評価技術センター
因 幸二郎
財団法人日本規格協会
渡 部 高 弘
仙台市水道局
河 野 恭一郎
東京都水道局
岡 本 文 夫
横浜市水道局
三 品 正 行
名古屋市水道局
小 島 勝 彦
広島市水道局
一 戸 正 憲
社団法人日本水道協会
能 美 邦 昌
浅田化学工業株式会社
河 野 省 三
セントラル硝子株式会社
鶴 本 勍
多木化学株式会社
中 谷 英 夫
大明化学工業株式会社
坂 本 功
日本軽金属株式会社
中 原 茂
ラサ工業株式会社
佐々木 一 郎
日本無機薬品協会
(事務局)
松 原 宏 之
日本無機薬品協会